mutagenesis by apurinic sites in normal and ataxia telangiectasia human lymphoblastoid cells
TRANSCRIPT

MOLECULAR CARCINOGENESIS 6:32-42 (1992)
Mutagenesis by Apurinic Sites in Normal and Ataxia Telangiectasia Human Lymphoblastoid Cells Donna K. Klinedinst' and Norman R. Drinkwater2 McArdle Laboratory for Cancer Research, University of Wisconsin, Madison, Wisconsin
We used a shuttle vector based on the Epstein-Barr virus origin of plasmid replication (ori f) t o determine the types o f mutations induced by depurination in human cells. Plasmid DNA was incubated a t pH 2 a t 40°C for various times t o induce up to 20 apurinic (AP) sites per 9.7-kb plasmid and electroporated into lymphoblastoid cells derived from either a normal individual o r an ataxia telangiectasia patient. After replication o f t he vector in the human cells, plasmid DNA was isolated and analyzed for mutations induced in the plasmid-encoded herpes simplex virus type I-thymidine kinase gene. Both the frequencies and types o f mutations induced by depurination were essentially identical for normal and ataxia telangiectasia cells. The mutant frequency a t 20 AP siteslplasmid was 10-fold to 13-fold greater than tha t observed for untreated DNA. Deletion and frameshift events accounted for 46-55% of the mutants induced by depurination. The induced deletions were relatively small (median size, 100-1 50 bp) and characterized by short (1-5 bp) regions of sequence homology a t t he end- points. These mutations and the frameshifts, a majority of which occurred in runs of identical nucleotides, are consistent with a model involving AP-site-induced template dislocation during DNA synthesis. A broad spec- trum of base-substitution mutations, which accounted for 19-36% o f the induced mutants, was observed. The apparentpreferenceforinsertionoppositeAPsitesin humancellswasG ( 4 3 4 5 % ) >A-C(18-21%) >T(9-14%). Our results in human cells contrast markedly with those published previously for the mutational specificity of AP sites in Escherichia coli, in which a large majority of t he mutants resulted from insertion of an A opposite the abasic site.
Key words: Depurination, mutational specificity
o 1992 WiIey-Liss, lnc.
INTRODUCTION
Depurination of DNA results from the hydrolysis of the glycosidic bond between the purine base and the deoxy- ribose sugar of the backbone. Loss of purine bases occurs at a relatively high rate, and the apurinic site is likely to be the most frequent single lesion to occur spontaneously in DNA: lo4 normal purines may be lost per mammalian cell per day [I]. Abasic sites are also formed as a result of the actions of various glycosylases, which remove damaged or inappropriate bases (uracil and hypoxanthine) from DNA [2]. In addition, apurinic or apyrimidinic (AP) sites are formed when DNA is modified by chemical carcinogens a t the N3 and N7 positions of purines and the d position of pyrimi- dines, which labilizes the glycosidic bond and increases the rate of base loss [3,4], and by free-radical-mediated processes such as y-irradiation or treatment with antitu- mor antibiotics such as bleomycin and neocarzinostatin [reviewed in 51.
AP sites can have two significant biological effects: lethal- ity and mutagenicity. AP sites have been shown to block DNA synthesis in vitro by Escherichia coli DNA polymerase I [61, E. coli DNA polymerase 111 holoenzyme [71, and Dro- sophila and human DNA polymerase (Y [8,9]. Blockage of the replication fork is probably responsible for the reduced survival of viruses containing AP sites both in f. coli [ lo] and in mammalian cells [I I]. However, bypass of AP sites does occur infrequently in vitro, with a resulting increase
in mutation frequency when the DNA replicated in vitro is introduced into E. coli[ 121.
Mutagenesis by AP sites is SOS-dependent in f. coli in vivo [I 01, requiring the rect recA, and umuC genes [ I 31. Deoxyadenosine is most frequently inserted opposite the presumably noncoding AP site during in vitro replication by DNA polymerase I or DNA polymerase 111 holoenzyme [6,71. The mutational spectrum observed for E. coli after replication in vivo of DNA containing AP sites is consistent with the polymerase insertion data, with preferential inser- tion of an A opposite the AP site [14,151. Data regarding the mutagenicity of AP sites in mammalian cells in vivo is more limited. AP sites were mutagenic in CV1 P African green monkey kidney cells [ I 1 I . Transfection into COS7 cells of a shuttle vector containing a single abasic site resulted in a broader spectrum of base-substitution mutations than that reported for E. coli [ I 61.
There is circumstantial evidence that defects in the repair of AP sites may be related to the hypersensitivity to ioniz-
'Present address: Laboratory of Viral Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892.
'Corresponding author: McArdle Laboratory for Cancer Research, University of Wisconsin, Madison, WI 53706.
Abbreviations: AP, apurinic or apyrimidinic; AT, ataxia telangiecta- sia; HPRT, hypoxanthine phosphoribosyltransferase; HN-tk, Herpes sim- plex virus type 1 -thymidine kinase gene; onP, origin of plasmid replication; d.f., degrees of freedom.
0 1992 WILEY-LISS, INC

AP SITE MUTAGENESlS 33
ing radiation of cells derived from persons afflicted with the cancer-prone, autosomal recessive disease ataxia tel- angiectasia (AT). First, the AP site is the common lesion produced by ionizing radiation and by the radiomimetic drugs to which AT cells are sensitive [3-5,171. Second, defects in several enzymes involved in the repair of AP sites have been demonstrated for some AT cell lines. Two groups have noted subtle differences in the AP endonucleases of some AT cells [ 18,191. Specific AT cell lines were also found to be deficient in an activity that removes base damage that can be recognized by the y-endonuclease of Micrococcus luteus (201, and others were deficient in a "primer- activating" function that acts on some types of radiation- induced damage to enhance the ability of €. coli DNA polymerase I to use it as a substrate for extension [2 1,221. The nature of the lesions recognized by these activities is unknown, but some recent studies of AP site repair in E. coli have provided a candidate for the latter defect. Two groups have independently isolated an activity from E. coli that removes the deoxyribose terminus remaining after cleavage by AP endonucleases [23,24]. This deoxyribose residue must be removed before primer extension or liga- tion or both. If this type of activity were equivalent to the "primer-activating" enzyme that is deficient in some AT cells, then differential repair or mutagenicity by AP sites, or both, might occur in AT cells.
It was shown previously that treatment of the AT cell line GM2783 with ionizing radiation did not yield a higher frequency of mutants at the hprt locus than treatment of normal human lyrnphoblastoid cells [25]. However, this agent induced a high proportion of multilocus lesions that may be lethal when hernizygous targets, such as the X-linked hprt locus, are studied [26]. These results led us to compare the mutant frequency and mutational speci- ficity of AP sites in that AT cell line (GM2783) and in nor- mal cells. We used the Herpes simplex virus type 1 -thymidine kinase (HSV-tk) gene of the oriP-containing shuttle vector pND123 [27] as a target for this study. The shuttle vector is particularly well suited to the study of this lesion because AP sites cannot readily be introduced into the DNA of cells in vivo. We incubated the shuttle plasmid DNA under mild conditions of acid pH and heating to introduce AP sites. Under conditions of acid/heat treatment, the predominant modification to DNA is depurination [ 1,281. Depyrimidina- tion and deamination of cytidine occur at 1 % and 0.2% the rate of depurination, respectively, and deamination of adenine is even less frequent [28-301. The modified shut- tle vector DNA was introduced into the human cells, allowed to replicate to fix mutations, and then recovered for mutant isolation in bacteria.
The mutation rate and mutational spectrum we ob- served in AT cells were similar to those for normal cells, but the spectrum in human cells was much more diverse than that seen in E. coli. Of special interest was the in- creased frequency of frameshift and small deletion mu- tations, indicating that insertional bypass of apurinic sites is not the predominant mechanism for overcoming blocks to replication in human cells as it is in SOS- induced E. coli.
MATERIALS AND METHODS
Cell Culture
All lymphoblastoid cell lines used in our study were rou- tinely maintained in RPMl 1640 medium supplemented with 10% fetal bovine serum (Hyclone Laboratories, Logan, UT) and 50 pg/mL gentamicin. The human B-lymphoblastoid cell line LCL-721 was derived from a normal female donor by immortalization with Epstein-Barr virus [31]. The AT lyrnphoblastoid cell line GM2783 was obtained from the NIGMS Human Genetic Mutant Cell Repository (Camden, NJ). The radiosensitive phenotype of GM2783 was con- firmed by its reduced survival after y-irradiation with a I3'Cs irradiator (Mark I Model 30; JL Shepherd and Assoc., San Fernando, CAI. Postirradiation survival (data not shown) was consistent with previously reported data [25].
Mutagenesis
We performed kinetic studies of the rate of depurination of plasmid DNA under various pH and temperature con- ditions to select appropriate treatment conditions for our mutagenesis studies. Our depurination buffer was 0.01 M NaCI, 0.01 M sodium citrate, and 0.01 M Na3P04 adjusted to the appropriate pH with NaOH. Reactions were stopped by neutralization with HEPES, pH 7.4 (final concentration 50 mM), and NaOH, and then incubated in 50 m M Tris, pH 8.0, 5 mM MgCI2, 5 m M dithiothreitol, and 50 U/pg DNA E. coli exonuclease 111 for 15 min a t 37°C to cleave the AP sites. Samples were loaded on a 1 YO agarose gel containing 0.1 kg/mL ethidium bromide to separate forms I, 11, and 111. Soft-laser densitometry was performed on a photographic negative to determine the relative propor- tion of each form. The number of nicks per molecule was expected to be distributed according to a Poisson distri- bution; the mean number of AP sites per plasmid for each treatment condition was determined by the equation Po=e-m, where PO was the proportion of DNA in the supercoiled band 141. Depurination buffer a t pH 2 and 40°C was found to cause rapid depurination (0.1 AP siteis) with- out excessive cleavage of phosphodiester backbones.
These conditions were used to introduce up to 20 AP sites/plasmid for our mutagenicity studies. The buffer was prewarmed to 40°C. plasmid DNA was added and mixed briefly, and the mixture was incubated for various times to induce AP sites. Reactions were stopped by adding HEPES, pH 7.4, to 50 mM and NaOH to 46 mM, then incu- bating on ice. Electroporation [32] into the human cells was carried out immediately after each treatment to reduce the possibility of spontaneous backbone hydrolysis at the AP sites. An aliquot of each sample was then saved and analyzed by gel electrophoresis to determine the amount of backbone hydrolysis. A reaction mixture that was expected to yield 2.5 AP sites/molecule was cleaved with E. coli exonuclease 1 1 1 to quantitate the actual number of AP sites introduced in each experiment. As a control for the effect of backbone hydrolysis on mutagenesis, a simi- larly treated sample was also electroporated into human cells.
The electroporated cells were diluted in 10 mL of com- plete RPMl medium at room temperature and then incu-

34 KLlNEDlNSTAND DRINKWATER
bated at 37°C in a humidified 5% COz atmosphere. Cell number and viability were monitored every 2-3 d. When the cell concentration reached 1 06/mL with a viability of at least 70%, hygromycin was added to the medium at a concentration of 100 (GM2783) to 300 (LCL-72 1) pg/mL. Plasmid-bearing cells were selected by growth in hygromycin to yield 2 x 1 O8 viable cells.
Isolation and Characterization of Mutant Plasmids
Plasmid DNA was isolated from human cells by an alka- line extraction method (331, ethanol precipitated, redis- solved in TE (10 m M Tris, pH 8.0, and 1 m M EDTA), and washed extensively with TE in a Centricon-30 microcon- centrator (Amicon, Beverly, MA). The recovered DNA was used to transform bacterial strain FT334 (supE44, hsdS2O (rB-,mB-), recA13, ara-14, proA2, lacY1, galK2, rpsL20, xyl-5, mtl-1, upp, tdk) [34] or DK502, a upp-, tdk- deriv- ative of CAG 1 574. CAG 1 574 (kindly provided by C.A. Gross, University of Wisconsin, Madison, WI) is a recA- deriva- tive of MC1061 that has the genotype araD139, A (ara,/eu) 7697, AlacX74, galU-, galK-, hsr , hsm+, strA [351, mcrA-, mcrl- [36]. Selection of HSV-tkmutants was per- formed as previously described [27,37]. The approximate location of each HSV-tk mutation was determined by a recombinational method [371, and DNA sequencing was carried out by the dideoxy-chain termination method [381 with Sequenase (United States Biochemicals Corp., Cleve- land, OH). The locations of the mutations are indicated using the guanine base in the unique Bglll site of pND123 as position 1 [271.
Computer Simulation of Deletion Endpoints
A computer model was used to determine whether the structures of deletions we observed in the plasmid pNDl23 differed, either in size or in the degree of homology observed at the endpoints, from those that would be observed if the deletions were randomly generated. We recognized that not all deletions that are generated in vivo are recovered owing to the loss of essential genetic loci. Thus, we designed the model so that the computer- generated deletions had the same limits as those imposed by our selection in vivo. In addition, each deletion gener- ated by the computer was required to have at least one endpoint 3’ to position 9555, the -35 sequence in the tet promoter that drives expression of the HSV-tk gene in E. coli. Twenty thousand deletions that met these criteria were generated; and the sizes, the distributions of end- points, and the amount of homology at the endpoints were tabulated.
Statistical Analyses
Either chi-square tests or Fisher’s exact test were used to compare the observed and expected contexts for base- substitution mutations, the distributions of mutations among classes of mutagenic events, and the homology of deletion endpoints. Wilcoxon’s rank sum test was used to compare the distributions of the sizes of deletion muta- tions in AT and normal cells.
RESULTS
Determination of AP Site-Induced Mutant Frequencies
Plasmid pNDl23 DNA was electroporated into normal (LCL-721) or AT (GM2783) cells after the DNA was first incubated in pH 2.0 buffer a t 40°C for various lengths of time to induce up to 20 apurinic sites per 9.7-kb mole- cule. Two independent experiments were performed for each cell type. Plasmid DNA was recovered after 3-4 wk, and mutants in the HSV-tk gene were selected by trans- formation of the €. colistrain FT334. During the course of the experiments, we observed that the total number of bacteria transformed with DNA recovered from the GM2783 cells was very low. Transformation of an mcrA- strain of E. coli (CAG1574) with DNA from the GM2783 cells resulted in a transformation frequency approximately 30-fold higher than that with the mcrA+ strain FT334, whereas the transformation frequency of DNA derived from LCL-721 or of bacterially derived DNA was the same for the two bacterial strains (data not shown). The most likely explanation for the differential ability of the GM2783-derived DNA to transform the two strains of E. coli is methylation of plasmid DNA during passage through the AT cell line. The methylated DNA would be restricted by the mcrA gene product of FT334. To select and characterize tk- mutants from experiments involving GM2783, a upp-, tdk- deriv- ative of strain CAGI 574 was isolated; this newly isolated strain was designated DK502. For plasmid DNA recovered from LCL-72 1 cells, virtually identical mutant frequencies were obtained after transformation of FT334 or DK502 cells.
A dose-dependent increase in HSV-tk mutant frequency was observed with plasmids derived from both LCL-721 and GM2783 cells (Table 1). For LCL-721 cells, the mutant frequency increased linearly with dose up to 20 AP sites/plasmid. No increase in mutant frequency was observed for GM2783 cells between 10 and 20 AP siteslmolecule. The mutant frequencies at the highest dose tested were 13- and 1 I-fold greater than those for control DNA for LCL-72 1 and GM2783 cells, respectively.
Although the depurinated DNA was electroporated into the human cells immediately after treatment, some hydrol-
Table 1. Frequency of HSV-tk Mutants Induced by Depurination in GM2783 and LCL-721 Cells
Dose (AP sites/ plasmid) LCL-721 GM2783
Frequency ( x 1 04)*
Experiment 1 0 4 6 2 3 2 5 7 8 5 0
10 20 24 20 44 27
Experiment 2 0 4 6 2 2 2 5 13 8 3 5 15 12
10 35 32 20 78 26
*These values are the mutant frequencies estimated from a minimum of 70,000 transformants for each point
AP SITE MUTAGENESIS 35
ysis of the AP sites leading to single-strand breaks occurred. Plasmid DNA treated to produce 20 AP sites/molecule con- tained an average of approximately 0.7 strand breakslplas- mid, as measured by comparing the amounts of supercoiled and nicked plasmid DNA. As a control to demonstrate that the AP sites, rather than strand breaks formed before electroporation, were responsible for the induced muta- tions, plasmid DNA with 2.5 AP sites/molecule was incu- bated with exonuclease Ill to convert the AP sites to single-strand breaks and was electroporated into both types of lymphobiastoid cells. Although the exonuclease Ill- treated DNA contained a fourfold greater number of strand breaks than the DNA containing 20 AP sitesimolecule, the observed mutant frequency for the enzyme-treated DNA fell between those for undigested DNA containing 0 and 2.5 AP sitesholecule. This result indicates that single-strand breaks occurring before electroporation do not contrib- ute disproportionately to the mutant frequency.
Types of HSV-tk Mutations Induced by AP Sites in Normal and AT Cells
All mutants were initially characterized by restriction- digest analysis 132) to identify plasmids with deletions (> 30 bp) or other rearrangements in the HN-tk gene. The proportions of mutants with visible alterations of the restric- tion pattern were 25% and 40% for the AT and normal cells, respectively, Between 5% (normal cells) and 14% (AT cells) of mutants with normal restriction-digest patterns could not be mapped to a specific location within the gene because they survived poorly in the tk+-selective medium. These "leaky" mutants were classified as base substitu-
Table 2. Types of Mutations Induced by Depurination*
Type of change LCL-72 1 + GM2783+
Base substitution 15 [ I 11 (0.19) 28 [I41 (0.36)
Multiple5 5 (0.06) 3 (0.04) Deletion 35 [23]11(0.45) 26 [ I 11 (0.34) Complexn 14 [6](0.18) 10 [71(0.13) Total 77 [53] 76 [431
Frameshift* 8 (0.10) 9 (0.12)
*Shuttle-vector DNA was depurinated by treatment a t 40°C at pH 2 for various lengths of time; the DNA sample was then divided and electroporated into the normal (LCL-721) and AT (GM2783) human cells. Two independent experiments were performed for each cell line. Only treatment groups with a 10-fold or greater increase in mutant fre- quency were analyzed. 'Number of mutants observed [number of mutants sequenced1 (pro- portion of total number of mutants). The structures of unsequenced mutants included in the total were inferred from restriction-digest analysis. Mutants with no change in restriction pattern and a "leaky" phenotype that could not be mapped with our genetic system were assumed to be base substitutions, since small deletions or frameshift mutations would be expected to result in a null phenotype. *Deletion or addition of 1 or 2 bp. 51ncludes the following types of changes: two or more base substi- tutions and tandem - 1 frameshift with transversion. "Number in brackets indicates the number sequenced. The total num- ber obsewed is the sum of those mutants that were sequenced and those whose structure was inferred from restriction-digest analysis only. "Includes deletions with insertions of nucleotides, deletions with base substitutions, duplications, and inversions.
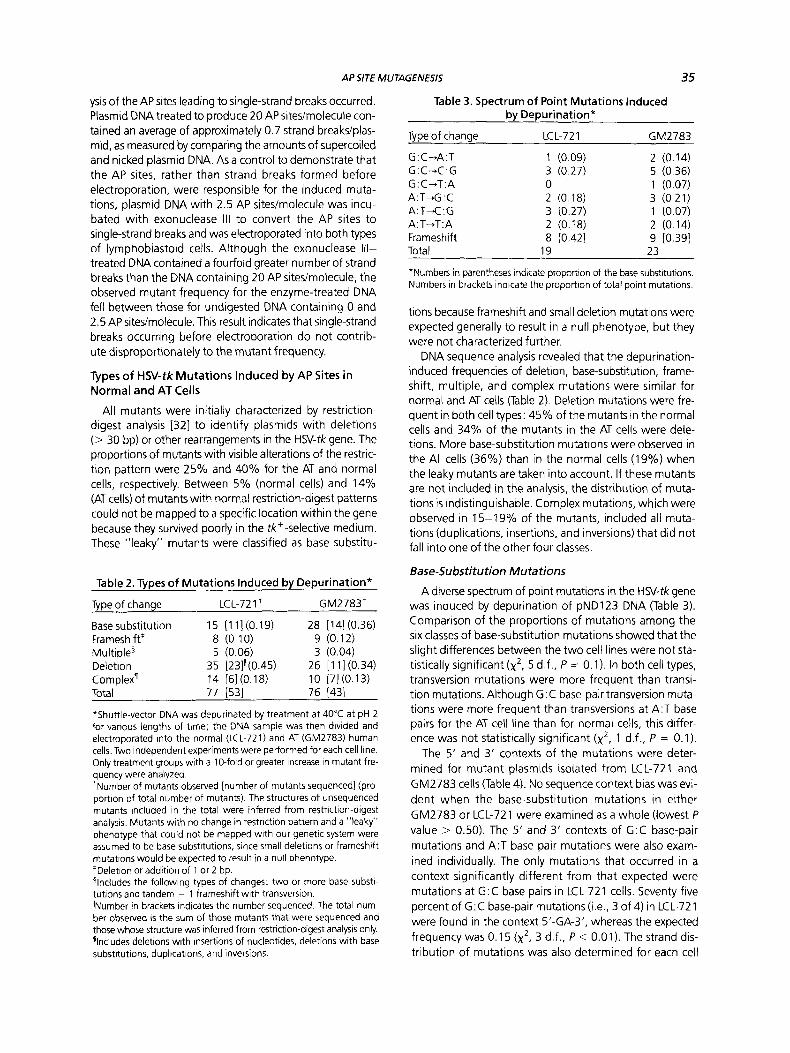
Table 3. Spectrum of Point Mutations Induced by Depurination*
GM2783
G:C-A:T 1 (0.09) 2 (0.14) G : C-C : G 3 (0.27) 5 (0.36) G:C+T:A 0 1 (0.07)
A:T-.C:G 3 (0.27) 1 (0.07) A: T+T: A 2 (0.18) 2 (0.14) Frameshift 8 10.421 9 10.391 Total 19 23
*Numbers in parentheses indicate proportion of the base substitutions. Numbers in brackets indicate the proportion of total point mutations.
tions because frameshift and small deletion mutations were expected generally to result in a null phenotype, but they were not characterized further.
DNA sequence analysis revealed that the depurination- induced frequencies of deletion, base-substitution, frame- shift, multiple, and complex mutations were similar for normal and AT cells (Table 2). Deletion mutations were fre- quent in both cell types: 45% of the mutants in the normal cells and 34% of the mutants in the AT cells were dele- tions. More base-substitution mutations were observed in the AT cells (36%) than in the normal cells (199'0) when the leaky mutants are taken into account. If these mutants are not included in the analysis, the distribution of muta- tions is indistinguishable. Complex mutations, which were observed in 15-19% of the mutants, included all muta- tions (duplications, insertions, and inversions) that did not fall into one of the other four classes.
Base-Substitution Mutations
A diverse spectrum of point mutations in the HN-tkgene was induced by depurination of pND123 DNA (Table 3). Comparison of the proportions of mutations among the six classes of base-substitution mutations showed that the slight differences between the two cell lines were not sta- tistically significant (x2, 5 d.f., P = 0.1 ). In both cell types, transversion mutations were more frequent than transi- tion mutations. Although G: C base-pair transversion muta- tions were more frequent than transversions at A:T base pairs for the AT cell line than for normal cells, this differ- ence was not statistically significant (x', 1 d.f., P = 0.1).
The 5' and 3' contexts of the mutations were deter- mined for mutant plasmids isolated from LCL-721 and GM2783 cells (Table 4). No sequence context bias was evi- dent when the base-substitution mutations in either GM2783 or LCL-721 were examined as a whole (lowest P value > 0.50). The 5' and 3' contexts of G : C base-pair mutations and A:T base-pair mutations were also exam- ined individually. The only mutations that occurred in a context significantly different from that expected were mutations at G :C base pairs in LCL-721 cells. Seventy-five percent of G:C base-pair mutations (i.e., 3 of 4) in LCL-721 were found in the context 5'-GA-3', whereas the expected frequency was 0.1 5 (x', 3 d.f., P < 0.01). The strand dis- tribution of mutations was also determined for each cell
Type of change LCL-721
A : T 4 : C 2 (0.18) 3 (0.21)
36 KLlNEDlNSTAND DRlNKWATER
G:C-T:A
A:T-.C:G
A :T-T: A
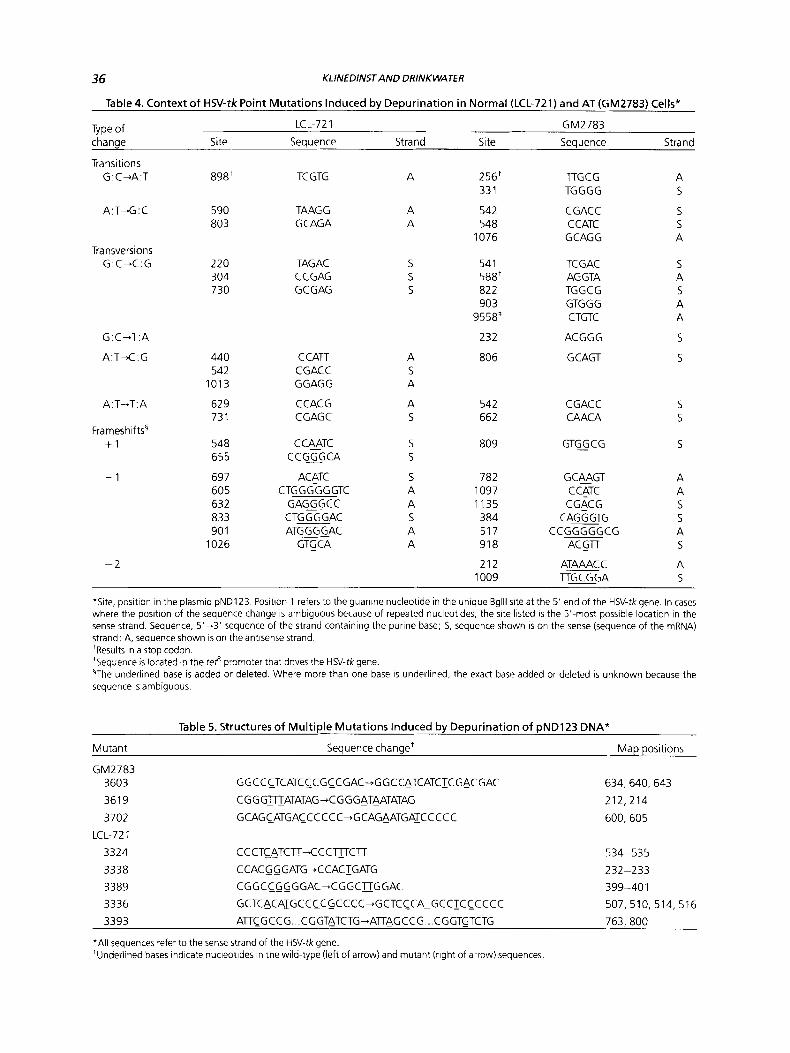
Table 4. Context of HSV-tk Point Mutations Induced by Depurination in Normal (LCL-721) and AT (GM2783) Cells*
Type of LCL-721 GM2783
change Site Sequence Strand Site Sequence Strand
Transitions G:C+A:T 89Eit TC GTG A 256' TTGCG A
33 1 TGGGG 5
A:T-G:C 590 TAAG G A 542 CGACC 5 803 G CAGA A 548 C CATC 5
1076 G CAG G A
G: C-C:G 220 TAGAC S 54 1 TC GAC 5 304 CCGAG S 588' AGGTA A 730 GCGAG S 822 TGGCG 5
903 GTG G G A 9558* CTGTC A
232 ACGGG 5
440 C CATT A 806 GCAGT 5 542 CGACC S
1013 GGAGG A
629 C CAC G A 542 C GAC C 5 73 1 CGAGC S 662 CAACA 5
Transversions
Frameshifts' +I
- 1
548 C CLATC S 809 GTG-G C G 5 655 C C GGG C A S
697 ACATC S 782 GC@GT A 605 CTG GG GG GTC A 1097 C CATC A 632 G AGLG C C A 1135 CGACG 5 833 CTG G G GAC S 384 CAG G GTG 5 90 1 ATG G G G AC A 51 7 CCGGGGGCG A
1026 GTSCA A 918 AC STT 5
A ATAAACC - 2 212 1009 T T B G A 5
*Site, position in the plasmid pND123 Position 1 refers to the guanine nucleotide in the unique Bglll site at the 5' end of the HSV-tkgene In cases where the position of the sequence change is ambiguous because of repeated nucleotides, the site listed is the 5' most possible location in the sense strand Sequence, 5'-3' sequence of the strand containing the purine base, 5, sequence shown is on the sense (sequence of the mRNA) strand, A, sequence shown is on the antisense strand 'Results in a stop codon 'Sequence IS located in the re$ promoter that drives the HN-tk gene §The underlined base is added or deleted Where more than one base is underlined. the exact base added or deleted is unknown because the sequence is ambiguous
Table 5. Structures of Multiple Mutations Induced by Depurination of pND123 DNA*
Mutant Sequence changet Map positions
GM2783 3603 GGCC C_TCATCC_CGC_CGAC -G GC CATCATCIC GAC GAC 634,640,643 361 9 212,214
3702 GCAGC_ATGAC_CCCCC-GCAGAATGAJCCCCC C 600,605
3324 CC CTC_ATCTT-.CC C m C l l 534-535
C G G Gm-ATATAG +C G G G ATAATATAG
LCL-72 1
3338 CCACGGGATG -CCACIGATG 232-233 3389 CGGCCGGGGAC-CGGClJGGAC 399-40 1
3336 GCTCACAIGCC C_C GCCCC -GCTCC_CA-GCCICC_CCCC 507.510,514, 516 3393 ATTCGCCG CGGTATCTG+AllAGCCG . CGGTGTCTG 763,800
*All sequences refer to the sense strand of the HSV-tk gene 'Underlined bases indicate nucleotides in the wild-type(1eft of arrow) and mutant (right of arrow) sequences
A P SITE MUTAGENESiS 37
type. Assuming all mutations occurred directly opposite the depurinated base, six of 1 1 mutations in LCL-721 cells resulted from loss of a purine in the sense strand of the plasmid DNA, whereas in GM2783, nine of 13 mutations contained the targeted base in the sense strand. Thus, no apparent strand bias for mutation induction was observed given the purine/pyrimidine ratios of the sense and anti- sense strands.
A small number of multiple base substitutions occurred in both the LCL-721 cells and in the GM2783 cells (Table 5). Three GM2783-derived mutants and two LCL-721- derived mutants contained two or more base substitutions within 20 nucleotides.
Deletion Mutations
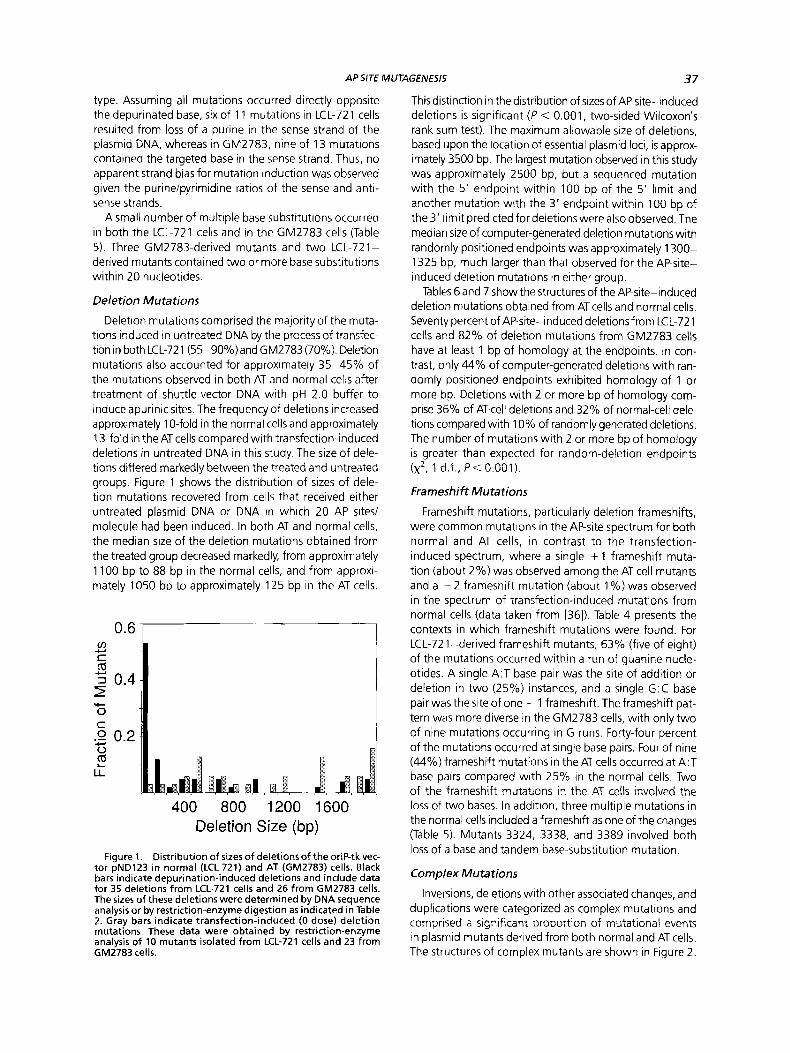
Deletion mutations comprised the majority of the muta- tions induced in untreated DNA by the process of transfec- tion in both LCL-721 (55-90Y0)and GM2783 (70%). Deletion mutations also accounted for approximately 35-45% of the mutations observed in both AT and normal cells after treatment of shuttle-vector DNA with pH 2.0 buffer to induce apurinic sites. The frequency of deletions increased approximately 10-fold in the normal cells and approximately 13-fold in the AT cells compared with transfection-induced deletions in untreated DNA in this study. The size of dele- tions differed markedly between the treated and untreated groups. Figure 1 shows the distribution of sizes of dele- tion mutations recovered from cells that received either untreated plasmid DNA or DNA in which 20 AP sites/ molecule had been induced. In both AT and normal cells, the median size of the deletion mutations obtained from the treated group decreased markedly, from approximately 1 100 bp to 88 bp in the normal cells, and from approxi- mately 1050 bp to approximately 125 bp in the AT cells.
400 800 1200 1600 Deletion Size (bp)
Figure 1. Distribution of sizes of deletions of the oriP-tk vec- tor pND123 in normal (LCL-721) and AT (GM2783) cells. Black bars indicate depurination-induced deletions and include data for 35 deletions from LCL-721 cells and 26 from GM2783 cells. The sizes of these deletions were determined by DNA sequence analysis or by restriction-enzyme digestion as indicated in Table 2. Gray bars indicate transfection-induced (0 dose) deletion mutations. These data were obtained by restriction-enzyme analysis of 10 mutants isolated from LCL-721 cells and 23 from GM2783 cells.
This distinction in the distribution of sizes of AP-site-induced deletions is significant ( P < 0.001, two-sided Wilcoxon's rank sum test). The maximum allowable size of deletions, based upon the location of essential plasmid loci, is approx- imately 3500 bp. The largest mutation observed in this study was approximately 2500 bp, but a sequenced mutation with the 5' endpoint within 100 bp of the 5' limit and another mutation with the 3' endpoint within 100 bp of the 3' limit predicted for deletions were also observed. The median size of computer-generated deletion mutations with randomly positioned endpoints was approximately 1300- 1325 bp, much larger than that observed for the AP-site- induced deletion mutations in either group.
Tables 6 and 7 show the structures of the AP-site-induced deletion mutations obtained from AT cells and normal cells. Seventy percent of AP-site-induced deletionsfrom LCL-72 1 cells and 82% of deletion mutations from GM2783 cells have at least 1 bp of homology a t the endpoints. In con- trast, only 44% of computer-generated deletions with ran- domly positioned endpoints exhibited homology of 1 or more bp. Deletions with 2 or more bp of homology com- prise 36% of AT-cell deletions and 32% of normal-cell dele- tions compared with 10% of randomly generated deletions. The number of mutations with 2 or more bp of homology is greater than expected for random-deletion endpoints (x', 1 d.f., P < 0.001).
Frameshift Mutations
Frameshift mutations, particularly deletion frameshifts, were common mutations in the AP-site spectrum for both normal and AT cells, in contrast to the transfection- induced spectrum, where a single + 1 frameshift muta- tion (about 2%) was observed among the AT cell mutants and a -2 frameshift mutation (about 1 %)was observed in the spectrum of transfection-induced mutations from normal cells (data taken from 1361). Table 4 presents the contexts in which frameshift mutations were found. For LCL-72 1 -derived frameshift mutants, 63% (five of eight) of the mutations occurred within a run of guanine nucle- otides. A single A:T base pair was the site of addition or deletion in two (259'0) instances, and a single G:C base pair was the site of one - 1 frameshift. The frameshift pat- tern was more diverse in the GM2783 cells, with only two of nine mutations occurring in G runs. Forty-four percent of the mutations occurred a t single base pairs. Four of nine (44%) frameshift mutations in the AT cells occurred at A:T base pairs compared with 25% in the normal cells. Two of the frameshift mutations in the AT cells involved the loss of two bases. In addition, three multiple mutations in the normal cells included a frameshift as one of the changes (Table 5). Mutants 3324, 3338, and 3389 involved both loss of a base and tandem base-substitution mutation.
Complex Mutations
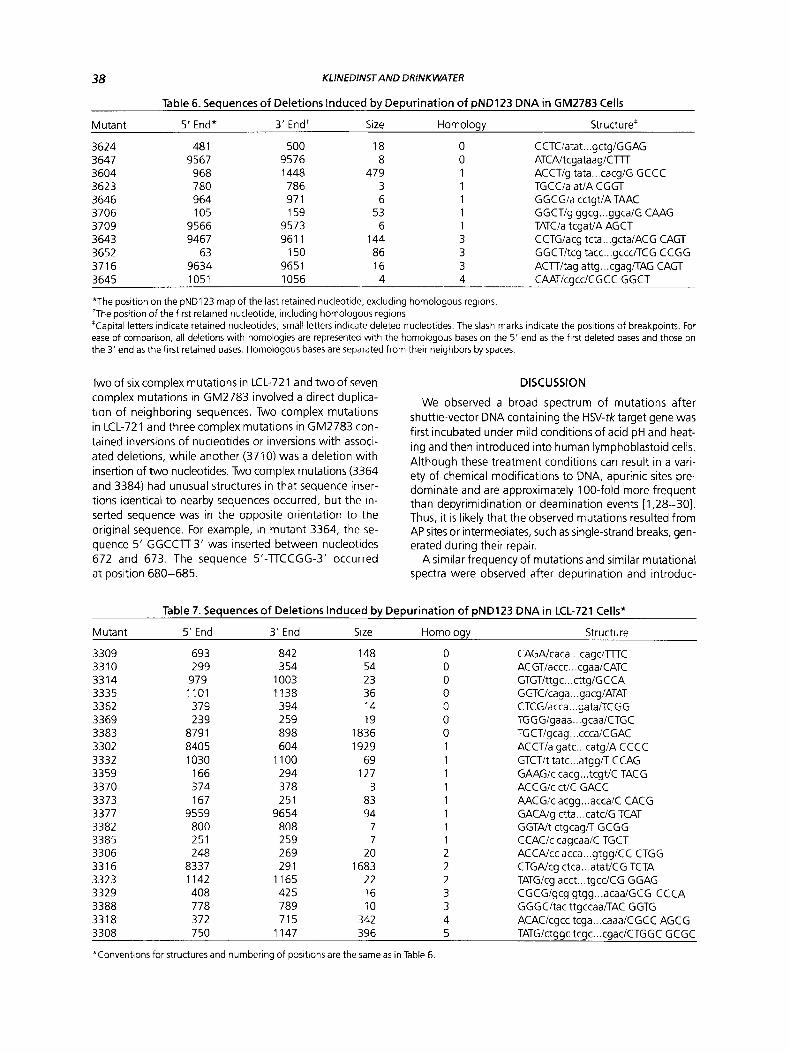
Inversions, deletions with other associated changes, and duplications were categorized as complex mutations and comprised a significant proportion of mutational events in plasmid mutants derived from both normal and AT cells. The structures of complex mutants are shown in Figure 2.
38 ~ l / N f ~ / N S r A N ~ DRfNKWATER
Table 6. Seauences of Deletions Induced bv DeDurination of ~ND123 DNA in GM2783 Cells
Mutant
3624 3647 3604 3623 3646 3706 3709 3643 3652 3716 3645
5’ End*
48 1 9567
968 780 964 105
9566 9467
63 9634 1051
3’ End’
500 9576 1448 786 97 1 159
9573 961 1
150 9651 1056
Size
18 8
479 3 6
53 6
144 86 16 4
Homology
0 0 1 1 1 1 1 3 3 3 4
Structure’
CCTClatat.. .gctg/GGAG ATCNtcgataagIClT ACCTlg tata.. .cacg/G GCC C TGCCla at1A CGGT GGCGla cctgt/ATAAC GGCTIg ggcg ...gg ca1G CAAG TATCIa tcgat/A AGCT CCTGlacg tcta ...g cta/ACG CAGT GGCTItcg tacc.. .g ccciTC G CCGG ACTItag attg ... cgag/TAG CAGT CAATIcacdCGCC GGCT
*The position on the pND123 map of the last retained nucleotide, excluding homologous regions. ‘The position of the first retained nucleotide, including homologous regions *Capital letters indicate retained nucleotides; small letters indicate deleted nucleotides. The slash marks indicate the positions of breakpoints. For ease of comparison, all deletions with homologies are represented with the homologous bases on the 5’ end as the first deleted bases and those on the 3’ end as the first retained bases. Homologous bases are separated from their neighbors by spaces.
Two of six complex mutations in LCL-721 and two of seven complex mutations in GM2783 involved a direct duplica- tion of neighboring sequences. Two complex mutations in LCL-721 and three complex mutations in GM2783 con- tained inversions of nucleotides or inversions with associ- ated deletions, while another (3710) was a deletion with insertion of two nucleotides. Two complex mutations (3364 and 3384) had unusual structures in that sequence inser- tions identical to nearby sequences occurred, but the in- serted sequence was in the opposite orientation to the original sequence. For example, in mutant 3364, the se- quence 5’-GGCCTT-3’ was inserted between nucleotides 672 and 673. The sequence 5’-TTCCGG-3’ occurred a t position 680-685.
DISCUSSION
We observed a broad spectrum of mutations after shuttle-vector DNA containing the HSV-tk target gene was first incubated under mild conditions of acid pH and heat- ing and then introduced into human lymphoblastoid cells. Although these treatment conditions can result in a vari- ety of chemical modifications to DNA, apurinic sites pre- dominate and are approximately 100-fold more frequent than depyrimidination or deamination events [ 1,28-301. Thus, it is likely that the observed mutations resulted from AP sites or intermediates, such as single-strand breaks, gen- erated during their repair.
A similar frequency of mutations and similar mutational spectra were observed after depurination and introduc-
Table 7. Sequences of Deletions Induced by Depurination of pND123 DNA in LCL-721 Cells*
Mutant 5’ End 3’ End Size Homology Structure
3309 693 842 148 0 CAGAIcaca ... cagcTmC 3310 299 3 54 54 0 ACGTlaccc.. .cgaalCATC 3314 979 1003 23 0 GTGTittgc.. .cttg/GCCA 3335 1101 1138 36 0 GGTClcaga .. .gacgIATAT 3362 379 394 14 0 CTCGlacca.. .gata/TCGG 3369 239 2 59 19 0 TGGGlgaaa.. .gcaa/CTGC 3383 879 1 898 1836 0 TGCTlgcag.. .ccca/CGAC 3302 8405 604 1929 1 ACCTla gatc ... catglA CCCC 3332 1030 1100 69 1 GTCTA tatc ... atgg/T CCAG 3359 166 294 127 1 GAAGIc cacg ... tcgt/C TACG 3370 374 378 3 1 ACCGIc ct/C GACC 3373 167 251 83 1 AACGIc acgg ... acca/C CACG 3377 9559 9654 94 1 GACAIg ctta ... catc1G TCAT 3382 800 808 7 1 GGTNt ctgcag/T GCGG 3385 251 259 7 1 CCACIc cagcaa1C TGCT 3306 248 269 20 2 ACCAlcc acca ...g tgglCC CTGG 3316 8337 29 1 1683 2 CTGAlcg ctca ... atat/CG TCTA 3323 1142 1165 22 2 TATGlcg acct ... tgcdCG GGAG 3329 408 42 5 16 3 CGCGIgcg gtgg ... acaa1GCG CCCA 3388 778 789 10 3 GGGCltac ttgccaalTAC GGTG 3318 372 71 5 342 4 ACAClcg cc tcga . . . caaaK GC C AG C G 3308 750 1147 396 5 TATGlctggc tcgc ... cgac1CTGGC GCGC
*Conventions for structures and numbering of positions are the same as in Table 6.

AP SITE MUTAGENESIS 39
A LCL-721
G12783 B Inversion
3606 (deletion with inversion, homology present at both endpoints of the inversion)
468 779 I I
CGCC- TAC
Inversion
3303 (double inversion with deletion)
608 864 806 I 1 inv I
CCCC,€€iXEl€ ,ELG.GG.. .CACT gcag \ R;. .? cctaoqgg ccc AGG
I 709 614 866
3387 (inversion with deletion)
991 1114 I I
GCCT tgga cgtc ACCA \ I
cage AGG . . : . . . . . AGG ccca I inv I
1076 989
Duplication
3361 (duplication of base pairs 464-467)
GTGACCG [ACGC] ACGC CGTT
3396 (deletion with duplication)
694 871 I I
AGAC [ GCCG] GCCG AGGC
Insertion of nearby sequence in reverse orientation
3364 (insertion between positions 672 and 673)
TGCTT[GGCCTT]GGGGCCCTTCCGG
3384 (2 bp deletion with insertion of nearby sequence)
AATGG ( GC ) ATGC + AATGG [ CGTA] ATGC CTTATGC I I 1 I
442 445 442 445
Figure 2. Structure of depurination-induced complex muta- tions derived from (A) LCL-721 (normal) or (6) GM2783 (AT) cells. For inversion and deletion mutations, the retained nucleotides are capitalized; slash marks joining capital letters are included to aid the reader in following the mutant nucleotide sequence. Lower case letters flanking the capital letters represent adja- cent nucleotides that were eliminated in the deletion and are
tion of shuttle-vector DNA into normal and AT cells. The spectrum of deletion mutations and base-substitution mutations were virtually identical; only minor differences in the context of frameshift mutations were found in com- paring the two types of human cells. Although these data demonstrate that the mutagenic processing of AP sites is not aberrant for at least one AT cell line, this result may not be generally applicable to all of the complementation groups of this disease [40].
The results of this study in human cells contrast mark- edly with those obtained for the mutational specificity of AP sites in E. coli. Deletion mutations were predominant in the human cell spectrum (35-45'/0), followed by base- substitution mutations (1 5-22%). Frameshift mutations,
361 1 (inversion with no loss of bases) 9508 288 291
I I 1 ATAAg L& GCA
\ /
I I I 288
ATCGT . . . T T T T W - 9507 9504
Deletion with insertion
3639 608 617 608 617
I I I I CCCC (CCAGGCCG) TGCTGG C -3 CCCC i CTGG] TGCTGG CGTT
Duplication
3642 (inverted duplication and insertion) 211 211 212
I I I ACTGCG GGTTTATA + ACTGCG GGLCGCAGITTTATA
3627 341
1 TCCGAGACAAT + TCCG [AGACAATI AGACAAT
3661 928
I TATTTACCCTGTT T --f TATTTACC [ CTGTTI CTGTFI
Deletion with insertion
371 0 481 502
I I GGCTCCTC [CCI AGCTCACA
shown so that homologies can be assessed. Homologous se- quences are underlined. For those mutants that involve only short deletions or insertions, sequences surrounded by brackets are insertions, and sequences surrounded by parentheses are dele- tions. Letters are depicted in bold italics to call attention to simi- larities in nearby nucleotides.
multiple mutations, and complex mutations were also observed. In contrast, the mutational specificity induced by depurination of single-stranded M I 3mp2 DNA followed by transformation of SOS-induced E. coli was more lim- ited [14]; base substitutions made up 62% of the spec- trum of lacZ mutations in this virus. Twenty-five percent of the mutants contained an identical 93-bp deletion that resulted from a recombinational event between the tacZ gene in the virus and the lacZ deletion allele in the E. coli genome; if these mutants are excluded, the base-substi- tution predominance is even more striking. Similarly, in 93-97% of the mutants observed after introduction of a single-stranded vector containing a single abasic site into SOS-induced E. coli, base substitutions occurred [ I 51. In

40 KLlNEDlNSTAND DRINKWATER
both studies, f rames hift mutations corn prised approxi- mately 5% of the E. colispectrum, half the proportion seen for human cells.
The introduction of vectors containing a single abasic site into both E. co/i[l5] and mammalian cells [ I 61 resulted in the induction of base-substitution mutations targeted to the position of the abasic site, indicating that these muta- tions arose by mutagenic bypass of the lesion rather than by error-prone repair. In both human and E. colicells, trans- versions predominated among the base substitutions. How- ever, only 18-2 l % of the transversions in human cells were A:T-tT:Aor G :C+T:A mutations, which presumably result from adenine insertion opposite the depurinated residue. In E. coli, adenine insertion gave rise to 54-80% [ 14, 151 of the mutants, depending on the target site. The relative insertion frequencies of the other nucleotides in E. coliwere asfolIows:T(4-28%),G(l 1-20%),andC(1%)[14,15]. In our study of human cells, the apparent order of prefer- ence for insertion opposite the AP site was G (43-55%, fornormalandATcells),AorC(18-21 %),andT(9-14%), assuming all mutations occurred opposite a depurinated base. Our results are similar to the mutational specificity reported by Gentil et al. [ 161 for introduction of a shuttle vector containing a single abasic site into COS7 cells in that approximately equal frequencies of A, C, and T inser- tion were observed. Because of the construction of the modified vector in that study [ 161, G insertions could not be detected.
The preference for insertion of a base opposite an AP site can be mediated by several factors, including the intrinsic bias in dNTP binding to the polymerase molecule, stack- ing interactions with neighboring nucleotides, and dif- ferential ability of the polymerase to extend the nucleotide chain from different unmatched 3' termini. The mutational spectrum obtained in E. coliwas consistent with the inser- tion preference of purified polymerases on a substrate con- taining abasic sites in vitro [6-81. Preferential insertion of adenine was also noted for eukaryotic DNA polymerase cx in vitro [8,9]. Thus, the mutational spectrum we observed does not reflect the intrinsic insertional preference of poly- merase a opposite a noncoding lesion, implying that other factors must be operating to determine the mutational spectrum in human cells in vivo. Accessory factors of the replication complex could modify the specificity of inser- tions at the AP site.
The structures of other mutations induced by depurination in human cells, including frameshift, deletion, and multi- ple or complex mutations, are in many cases consistent with the template-dislocation model [41], an extension of the Streisinger slippage model [42] of frameshift mutagen- esis. In our studies, 12 of 17 frameshift mutations occurred within runs of repeated nucleotides. Among the nonrun frameshift mutations, one (at nucleotide 1097) may be explained by quasi-palindrome formation [43]. In that case, one half of the palindrome could have served as a tem- plate for the other half, either by strand switching during DNA synthesis or by repair while in the palindromic sec- ondary structure. The structure of the frameshift muta- tion at base pair 1097 is ACCATCCCGGAGGT, where the
underlined base is deleted and the italicized bases could have served as the template for the deletion.
The most frequent class of mutations observed after heat and acid treatment of shuttle-vector DNA to produce AP sites and after introduction of the DNA into human cells was the deletion mutation. Although deletion mutations are also recovered at high frequency after transfection of untreated DNA (this study) [27,291, two lines of evidence indicate that most of the deletions analyzed in this study were induced by depurination. First, the frequency of dele- tion mutations recovered from the cells that received treated DNA was increased 10-fold to 13-fold over that observed with untreated DNA. Thus, of the 58 deletion mutations analyzed at the level of restriction patterns, only five to six would be expected to be transfection-induced mutations. Second, the sizes of the deletion mutations in the treated groupsaresignificantlysmaller (P< 0.001) than the mutations seen in the transfection-induced groups. A majority of the deletions (24 of 33) were characterized by the loss of bases between short, direct repeats with reten- tion of one of the repeats in the mutant. This type of dele- tion, which has also been observed for spontaneous and ionizing-radiation-induced deletions in mammalian cells and Drosophi/a [44-461, is also consistent with the template- dislocation model. Although the studies of Gentil et al. [I 61 (see above) did not demonstrate the induction of dele- tions by an abasic site in mammalian cells, their colony hybridization method for identifying mutants could not detect deletions.
Two types of multiple mutations were noted in response to depurination in human cells. One class was seen only in normal cells and involved tandem frameshift and trans- version events. The other class (three AT-cell mutants and two normal-cell mutants) consisted of HW-tk mutants with two to four clustered base substitutions occurring within 37 bases of each other. Such multiple base-substitution mutations may also be generated by direct repeats via a mechanism involving template/primer misalignment [41,47]. One mutant in our collection of AP-site mutants can be explained by this mechanism. Mutant 3619 has the struc- ture GGgTAATATAG; the wild-type sequence is GGGD- IATATAG. This mutant sequence could result from the loop- ing out of the AAA sequence on the antisense strand, replication of the subsequent ATA sequence, realignment, and continued replication of the ATATAG sequences. In an alternative model, the multiple mutations could result from an error-prone repair pathway [48].
Much of the foregoing discussion is based on the assumption that the depurination-induced mutations resulted from the interaction of the replication complex with the abasic site. As noted above, the studies of Gentil et al. [I 61 provide good evidence that the single-base sub- stitutions were targeted to AP sites in mammalian cells. However, it is possible that some of the mutations observed in our studies resulted from error-prone gap filling during the repair of AP sites. It is of interest to note that polymer- ase @, which has been suggested to fulfill this role in mam- malian cells [49], produces errors during replication in vitro

APSITE MUT
[41,501 that are characterized by a broad specificity for base substitutions, frequent frameshifts, and deletions via a template-dislocation mechanism.
This study of the mutagenicity of AP sites in human cells produced surprising results. In E. coli, SOS induction is required for mutagenesis, indicating that the cells cannot bypass this blocking lesion in the DNA. When the 5 0 5 sys- tem is induced, the majority of mutations are base-sub- stitution mutations, resulting from insertional bypass of the lesion. The mutational spectrum is consistent with the preference of bacterial polymerases for adenine insertion opposite the AP site. A smaller proportion of mutations induced in human cells were base substitutions, and the mutational spectrum was more complex than in E. coli, not simply a reflection of polymerase bias for nucleotide insertion. In contrast, the introduction of depurinated DNA into human cells resulted in a large proportion of deletion mutations, frameshift mutations, and complex mutations. This result has several possible implications: (i) The ability of human cells to bypass a strongly blocking lesion by inser- tion opposite the lesion and extension from an unpaired template may be lower than that for SOS-induced E. coli. (ii) Human cells may more readily "bypass" noncoding lesions by looping out the strand containing the lesion, thus forming a deletion or a frameshift mutation. (iii) Repair replication, presumably by DNA polymerase p, and cellu- lar DNA replication by polymerases a and 6 may play com- plex, interacting roles in determining the mutagenic outcome of a oarticular AP site.
ACKNOWLEDGMENTS
We thank D. Davido and T. Francisco for their skilled tech- nical assistance. This work was supported by U.S. Public Health Service grants CA37166, CA07175, and CA09135. DKK was supported by a fellowship from I. E. du Pont De Nemours and Company.
Received September 12, 1991; revised March 9, 1992; accepted March 13, 1992.
REFERENCES
1. Loeb LA, Preston BD. Mutagenesis by apuriniciapyrimidinic sites.
2. Lindahl T DNA repair enzymes. Annu Rev Biochem 51 :61-87,1982, 3. Lawley PD. Brookes P Further studies on the alkylation of nucleic
acids and their constituent nucleotides. J Mol Biol 89: 127- 139,1963.
4. Drinkwater NR, Miller EC, Miller JA. Estimation of apurinici apyrimidinic sites and phosphotriesters in deoxyribonucleic acid treated with electrophilic carcinogens and mutagens. Biochem- istry 19:5087-5092, 1980.
5. Povirk L, Steighner RJ. Oxidized apurinidapyrimidinicsites formed in DNA by oxidative mutagens. Mutat Res 214:13-22. 1989.
6. Sagher D. Strauss B. Insertion of nucleotides opposite apurinid apyrimidinic sites in deoxyribonucleic acid during in vitro synthe- sis: Uniqueness of adenine nucleotides. Biochemistry 22:4518- 4526, 1983
7. Hevroni D, Livneh 2. Bypass and termination at apurinic sites dur- ing replication of single-stranded DNA in vitro: A model for apurinicsite mutagenesis. Proc Natl Acad Sci USA85-5046-5050, 1988.
8. Randall SK. Eritja R. Kaplan BE, Petruska J, Goodman MF. Nucle- otide insertion kinetics opposite abasic lesions in DNA. J Biol Chern 262:6864-6870.1987.
9. Takeshita M, Chang CN, Johnson F, Will 5, Grollman AP Oligonu-
Annu RevGenet20:201-230, 1986.
4GENESlS 41
cleotides containing synthetic abasic sites: Model substrates for DNA polymerases and apurinidapyrimidinic endonucleases. J Biol Chem 2 1 : 101 71 -1 01 79,1987.
10. Schaaper RM, Loeb LA. Depurination causes mutations in SOS- induced cells. Proc Natl Acad Sci USA 78: 1773-1 777, 1981.
11. Gentil A, Margot A, Sarasin A. Apurinic sites are mutagenic in simianvirus40. Mutat Res 129:141-147, 1984.
12. Kunkel TA, Shearman CW, Loeb LA. Mutagenesis in vitro by depurination of 6x174 DNA. Nature 291 :349-351,1981,
13. Schaaper RM, Glickman BW, Loeb LA. Mutagenesis resulting from depurination is an SO5 process. Mutat Res 106: 1-9, 1982.
14. Kunkel TA. Mutational specificity of depurination. Proc Natl Acad SciUSA81:1494-1498,1984.
15. Lawrence CW, Borden A, Banerjee SK, LeClerc JE. Mutation fre- quency and spectrum resulting from a single abasicsite in a single- stranded vector. Nucleic Acids Res 18~2153-2157, 1990.
16. Gentil A, Renault G. Madrak C, et al Mutagenic properties of a unique abasic site in mammalian cells. Biochem Biophys Res Commun 173:704-710,1990,
17. Shiloh Y, Tabor E, Becker Y. Abnormal response of ataxia telangi- ectasia cells to agents that break the deoxyribose moiety of DNA via a targeted free radical mechanism. Carcinogenesis 4: 1317- 1322, 1983.
18. Sheridan RB Ill, Huang PC. Apurinic and/or apyrimidinic endonu- clease activity in ataxia telangiectasia cell extracts. Mutat Res 52:129-136, 1978.
19. Kuhnlein U. Comparison of apurinic DNA-binding protein from an ataxia telangiectasia and a HeLa cell line: Evidence for an altered processing of apurinidapyrimidinic endonuclease J Biol Chem 260: 1491 8-14924,1985,
20. Paterson MC, Smith BF: Lohman PHM, Anderson AK, Fishman L. Defective excision of gamma-ray-damaged DNA in human (ataxia telangiectasia) fibroblasts. Nature 260:444-446, 1976.
21. lnoue T, Yokoiyarna A, Kada T. DNA repair enzyme deficiency and in vitro complementation of the enzyme activity in cell-free extra- from ataxia telangiectasia fibroblasts. Biochim Biophys Acta 655:49-53, 1981.
22. Edwards MJ, Taylor AMR, Duckworth G. An enzyme activity in normal and ataxia telangiectasia cell lines which is involved in the repair of y-irradiation-induced DNA damage. Biochem J 188:677-682, 1980.
23. Bernelot-Moens C, Demple B. Multiple DNA repair activities for 3'-deoxyribose fragments in Ocherichia cofi. Nucleic Acids Res 17 587-600. 1989.
24 Franklin WA, Lindahl T DNA deoxyribosephosphodiesterase EMBO J 7 3617-3622,1988
25. Tatsumi K, Takebe H. Gamma-irradiation induces mutation in ataxia-telangiectasia lymphoblastoid cells. Gann 75: 1040- 1043, 1984.
26. Bradley WEC, Belouchi A, Messing K. The a p t heterozygotei hemizygote system for screening mutagenic agents allows detec- t ionof largedeletions. Mutat Res 199:131-138, 1988.
27. lngle CA, Drinkwater NR. Mutational specificities of 1 '-acetoxy- safrole, N-benzoyloxy-N-methyI-4-arninoazobenzene, and ethyl methanesulfonatein humancells. MutatRes220:133-142, 1989.
28. Lindahl T, Karlstrom 0. Heat-induced depyrimidination of deoxy- ribonucleic acid in neutral solution. Biochemistry 12:5151- 5154, 1973.
29. Lindahl T, Nyberg 8. Heat-induced deamination of cytosine resi- duesindeoxyribonucleicacid. Biochemistry 13,3405-3410,1974,
30. Shapiro R, Danzig M. Acidic hydrolysis of deoxycytidine and deoxyuridine derivatives: The general mechanism of deoxyribo- nucleoside hydrolysis. Biochemistry 1 1 :23-29, 1972.
31. Kavathas F: Bach FH, DeMars R. Gamma ray-induced lossof expres- sion of HLA and glyoxylase I alleles in lymphoblastoid cells. Proc Natl Acad Sci USA 77:4251-4255, 1980.
32. Drinkwater NR, Klinedinst DK. Chemically induced mutagenesis in a shuttle vector with a low-background mutant frequency. Proc Natl Acad Sci USA 83:3402-3406. 1986.
33. Griffin BE, Bjorck E, Bjursell G, Lindahl T. Sequence complexity of circular tpstein-Barr virus DNA in transformed cells. J Virol 4O:l l-19, 1981.
34. Eckert KA, Drinkwater NR. recA-dependent and recA-independent N-ethyl-N-nitrosourea mutagenesis at a plasmid-encoded herpes simplex virus thymidine kinase gene in Ochenchia coli. Mutat Res 178:l-10, 1987
35. Casabadan MJ, Cohen SN. Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J Mol Biol 138: 179-207,1980.
36. Raleigh EA, Murray NE. Revel H, et al. McrA and McrB restriction

42 KLINEDINSTAND DRINKWATER
phenotypes of some E. colistrains and implications for gene clon- ing. NucleicAcids Res 16:1563-1575, 1988.
37. Eckert KA, lngle CA, Klinedinst DK, Drinkwater NR. Molecular anal- ysis of mutations induced in human cells by N-ethyl-N-nitrosourea. Mol Carcinog 1 :50-56, 1988.
38. Sanger F, Nicklen 5, Coulson AR. DNA sequencing with chain- terminating inhibiton. Proc Natl Acad Sci USA 74:5463-5467,1977.
39. Drinkwater NR, Eckert KA, lngle CA, Klinedinst DK. Shuttle plas- mids derived from Epstein-Barr virus for the molecular analysis of mutagenesis in human cells. In. Moses RE, Summers WC (eds), DNA Replication and Mutagenesis. American Society for Micro- biology, Washington, DC, 1988, pp 416-422.
40. Jaspers NGJ, Gatti RA, Baan C, Linssen PCML, Bootsma D. Genetic complementation analysis of ataxia telangiectasia and Nijmegen breakage syndrome: A survey of 50 patients. Cytogenet Cell Genet 49:259-263,1988.
41. Kunkel TA. Misalignment-mediated DNA synthesis errorj Biochem- istry29:8003-8011,1990
42. Streisinger G, Okada Y, Ernrich J, et al. Frameshift mutations and the genetic code. Cold Spring Harbor Symp Quant Biol 31 : 77-84, 1966.
43. Ripley LS. Model for the participation of quasi-palindromic DNA sequences in frameshift mutation. Proc Natl Acad Sci USA 79:4128-4132,1982.
44. Grosovsky AJ, de Boer JG, de Jong PJ, Drobetsky EA, Glickman BW. Base substitutions, frarneshifts, and small deletions consti- tute ionizing radiation-induced mutations in mammalian cells. Proc Natl Acad Sci USA 85: 185-188, 1986.
45. Pastink A, Vreeken C, Schalet AP! Eeken JCJ. DNA sequence anal- ysis of X-ray-induced deletions at the white locus of Drosophila melanogaster. Mutat Res 207.23-28. 1988.
46. Nalbantoglu J, Hartley D, Phear G, Tear G, Meuth M. Spontane- ous deletion formation at the aprt locus of hamster cells: The presence of short sequence homologies and dyad symmetries at deletion termini. EMBO 16: 1199-1204, 1986.
47. Hampsey DM, Ernst JF, Stewart JW, Sherman F. Multiple base- pair mutations in yeast. J Mol 8iol201 :471-486, 1988.
48. Seidman MM, Bredberg A, Seetharam 5, Kraemei KH. Multiple point mutations in a shuttle vector propagated in human cells: Evidence for an error-prone DNA polymerase activity. Proc Natl Acad Sci USA 84:4944-4948, 1987.
49. Mosbaugh DW, Linn 5. Excision repair and DNA synthesis with a combination of HeLa DNA polymerase p and DNase V. I Biol Chem 258:108-118, 1983.
50. Kunkel TA, Alexander PS. The base substitution fidelity of eucary- otic DNA oolvmerases: Misoairina freauencies. site Dreferences. insertion preferences. and base sibstitbtion by'dislobation. J Bioi Chem 261:160-166,1986.