naglazyme (galsulfase) product...
TRANSCRIPT

NAGLAZYME® (galsulfase) PRODUCT MONOGRAPH
Please see Important Safety Information on pages 2 and 3.
Alena, 17Patient on NAGLAZYME® (galsulfase) since August 2005

2
INDICATIONNAGLAZYME® (galsulfase) is indicated for patients with Mucopolysaccharidosis VI (MPS VI; Maroteaux-Lamy syndrome). NAGLAZYME has been shown to improve walking and stair-climbing capacity.
IMPORTANT SAFETY INFORMATIONLife-threatening anaphylactic reactions and severe allergic reactions have been observed in some patients during NAGLAZYME (galsulfase) infusions and up to 24 hours after infusion. If these reactions occur, immediate discontinuation of NAGLAZYME is recommended and appropriate medical treatment should be initiated, which may include resuscitation, epinephrine, administering additional antihistamines, antipyretics or corticosteroids. In patients who have experienced anaphylaxis or other severe allergic reactions during infusion with NAGLAZYME, caution should be exercised upon rechallenge; appropriately trained personnel and equipment for emergency resuscitation (including epinephrine) should be available during infusions.
As with other enzyme replacement therapies, immune-mediated reactions, including membranous glomerulonephritis have been observed. In clinical trials, nearly all patients developed antibodies as a result of treatment with NAGLAZYME; however, the analysis revealed no consistent predictive relationship between total antibody titer, neutralizing or IgE antibodies, and infusion-associated reactions, urinary glycosaminoglycan (GAG) levels, or endurance measures.
Caution should be exercised when administering NAGLAZYME to patients susceptible to fluid volume overload because congestive heart failure may result. Consider a decreased total infusion volume and infusion rate when administering NAGLAZYME to these patients.
Consideration to delay NAGLAZYME infusion should be given when treating patients who present with an acute febrile or respiratory illness. Sleep apnea is common in MPS VI patients and antihistamine pretreatment may increase the risk of apneic episodes. Evaluation of airway patency should be considered prior to the initiation of treatment. Patients using supplemental oxygen or continuous positive airway pressure (CPAP) during sleep should have these treatments readily available during infusion in the event of an infusion reaction, or extreme drowsiness/sleep induced by antihistamine use.
Pretreatment with antihistamines with or without antipyretics is recommended prior to the start of infusion to reduce the risk of infusion reactions. If infusion reactions occur, decreasing the infusion rate, temporarily stopping the infusion, or administering additional antihistamines and/or antipyretics is recommended.
During infusion, serious adverse reactions included laryngeal edema, apnea, pyrexia, urticaria, respiratory distress, angioedema, and anaphylactoid reaction; severe adverse reactions included urticaria, chest pain, rash, abdominal pain, dyspnea, apnea, laryngeal edema, and conjunctivitis. The most common adverse events (≥10%) observed in clinical trials in patients treated with NAGLAZYME were rash, pain, urticaria, pyrexia, pruritus, chills, headache, nausea, vomiting, abdominal pain and dyspnea. The most common adverse reactions requiring interventions are infusion-related reactions.

3
Spinal/cervical cord compression is a known and serious complication that is expected to occur during the natural course of MPS VI. Signs and symptoms of spinal/cervical cord compression include back pain, paralysis of limbs below the level of compression, and urinary or fecal incontinence. Patients should be evaluated for spinal/cervical cord compression prior to initiation of NAGLAZYME to establish a baseline and risk profile. Patients treated with NAGLAZYME should be regularly monitored for the development or progression of spinal/cervical cord compression and be given appropriate clinical care.
To report SUSPECTED ADVERSE REACTIONS contact BioMarin Pharmaceutical Inc. at 1-866-906-6100, or FDA at 1-800-FDA-1088 or go to www.fda.gov/medwatch.
Please see accompanying full Prescribing Information or visit www.Naglazyme.com.

4 Please see Important Safety Information on pages 2 and 3.
TABLE OF CONTENTS
Section 1: Preface 5
Section 2: About MPS VI 6
2.1: Disease overview 6
2.2: Genetics 8
2.3: Pathophysiology 9
2.4: Clinical presentation 10
2.5: Diagnosis 14
2.6: Management 18
Section 3: NAGLAZYME® (galsulfase) product information 22
3.1: Overview 22
3.2: Pharmacology 24
3.3: Clinical efficacy 26
3.4: Safety 38
3.5: CSP/patient registry 42
Section 4: Indication and clinical usage 44
Section 5: Importance of compliance 46
Section 6: BioMarin Patient and Physician Support (BPPS) 48
Glossary 49
References 50
Full Prescribing Information 52

5
Maroteaux-Lamy syndrome, or mucopolysaccharidosis VI (MPS VI), is a rare, progressive, and multisystemic lysosomal storage disorder characterized by the absence or marked reduction in the enzyme N-acetylgalactosamine-4-sulfatase (arylsulfatase B, or ASB). Care for patients with MPS VI was limited to palliative options until May 2005, when the FDA approved a treatment for MPS VI: NAGLAZYME® (galsulfase), a once-weekly intravenous enzyme replacement therapy (ERT). NAGLAZYME provides a recombinant version of the lysosomal enzyme deficient in individuals diagnosed with MPS VI. It is the only therapy indicated for long-term treatment of patients with MPS VI.1-3
This monograph is intended to provide a comprehensive resource for treating patients with MPS VI with NAGLAZYME. It is also designed to facilitate the diagnosis and management of MPS VI by providing a concise overview of the genetics, pathophysiology, diagnosis, and treatment approaches associated with this disorder and in accordance with the Management Guidelines for mucopolysaccharidosis VI.
PREFACE
SE
CT
ION
1

6 Please see Important Safety Information on pages 2 and 3.
ABOUT MPS VI
2.1 DISEASE OVERVIEW
MPS VI, or Maroteaux-Lamy syndrome, is a devastating, clinically progressive, and heterogeneous disorder with severe pathologies affecting multiple organs. It is part of a broader group of diseases known as lysosomal storage disorders, and it is caused by deficient activity of ASB, the enzyme that catabolizes the glycosaminoglycan (GAG) dermatan sulfate (DS). GAGs are biologically significant and responsible for regulating multiple cellular processes, among other functions (Table 1). Individuals with deficient ASB experience a buildup of GAGs throughout the body. Affected individuals usually appear normal at birth, but intracellular GAG accumulation leads to cellular malfunction in tissues, manifesting through an extensive range of symptoms, such as2,3:
• Short stature
• Skeletal abnormalities
• Respiratory complications
• Cardiac disease
• Corneal clouding
• Hearing loss
• Spinal cord compression
• Reduced physical endurance
Although there is wide variability in the phenotypic presentation of MPS VI, patients are often characterized as having either rapidly or slowly progressing disease. Rapidly progressing MPS VI is typically characterized by onset before 2 or 3 years of age. If untreated, most do not live to adulthood (second and third decade).3,4 Individuals with slowly progressing disease typically develop symptoms in the teenage years.3 Urinary GAG (uGAG) levels >200 μg/mg creatinine are generally associated with rapidly progressing disease and shortened lifespan, while uGAG levels <100 μg/mg creatinine are associated with a slowly progressing clinical course and longer survival.5
Regardless of rate of progression, untreated patients with MPS VI progress over years with skeletal deformities, joint disease, cardiopulmonary disease, blindness, and spinal cord compression. Frequently, patients become wheelchair bound or bedridden. Usually, they eventually succumb to either infection, complications secondary to surgery, or cardiopulmonary failure. MPS VI is not typically associated with progressive impairment of mental status, although physical limitations may impact learning and development.5
SE
CT
ION
2

7
Key points
• MPS VI is a lysosomal storage disorder3
• Affected individuals usually appear normal at birth, but experience progressive development of multisystemic clinical manifestations2,3
• The natural history of MPS VI shows progression over years with skeletal deformities, cardiopulmonary disease, blindness, and spinal cord compression5
A major component of connective tissue
Widely distributed throughout the body
A basic substance of skin, cartilage, and bone
A component of lubricating fluid in joints
Responsible for the regulation of multiple cell processes
A component of proteoglycans
Table 1: Characteristics of GAGs6

8 Please see Important Safety Information on pages 2 and 3.
SECTION 2
2.2 GENETICS
MPS VI is an autosomal recessive disorder resulting from mutations in the chromosomal position 5q13-5q14. Most of the 133 mutations reported by the Human Gene Mutation Database are missense or nonsense mutations (100/133). Additionally, splicing mutations, small deletions, small insertions, a small “indel” (insertion and deletion), and gross deletions have also been documented.3
Each disease-causing mutation impacts the production of a functional ASB enzyme. Disease manifestations are present only in patients with severe deficiency in enzymatic activity—usually below 10% of the lower limit of normal. The greatest severity of symptoms is found in patients who have no detectable enzyme activity.3
MPS VI is expressed only in individuals who are homozygous for the mutant allele. Heterozygous carriers do not develop MPS VI themselves; with 1 functional copy of the gene present, they still produce enough active ASB enzyme to avoid developing clinical disease.7,8
If both parents are carriers, there is a 50% chance with each pregnancy that their child will inherit 1 copy of the disease allele and 1 copy of the normal allele and be a clinically unaffected carrier. Each pregnancy carries a 25% chance that both of the normal alleles will be passed on, producing an unaffected noncarrier, and a 25% chance that the offspring will inherit both disease alleles and have MPS VI (Figure 1).8
As with other autosomal recessive disorders, genetic counseling can be an important component of familial risk assessment and may include7,8:
• Individual counseling, beginning during puberty
• Family risk assessment
• Family screening
• Family planning
Figure 1: MPS VI is an autosomal recessive disorder8

9
2.3 PATHOPHYSIOLOGY
The epidemiological studies of MPS VI are limited to publications describing birth incidence. Several publications provide estimates of MPS VI incidence among different populations and geographic regions, ranging from 1 in 43,261 to as low as 1 in 1.5 million.3
Pathogenic mutations in the arylsulfatase B (ARSB) gene located in chromosome 5 (5q13-5q14) result in reduced or absent activity in the ASB enzyme, also called N-acetylgalactosamine-4-sulfatase, leading to incomplete degradation and cellular accumulation of the GAG DS, resulting in cell injury and death (Figure 2). Clinical manifestations of MPS VI are related to progressive accumulation of DS and sulfated oligosaccharides derived from DS in lysosomes, cells, and tissues.3
Cells without GAG accumulation in the lysosomes
Cells with GAG accumulation in the lysosomes
Figure 2: Normal and abnormal lysosomal enzyme function9
Key points
• Several publications provide estimates of MPS VI incidence among different populations and geographic regions, ranging from 1 in 43,261 to as low as 1 in 1.5 million3
• Pathogenic mutations in the ARSB gene result in reduced or absent activity in the enzyme ASB, leading to incomplete degradation and cellular accumulation of GAGs and cell injury 3

10 Please see Important Safety Information on pages 2 and 3.
2.4 CLINICAL PRESENTATION
MPS VI is a clinically heterogeneous condition in which patients can present with marked disease in the first year of life or disease that progresses more slowly, with symptoms presenting over a longer period of time (Figure 3). Rapidly progressing MPS VI is manifested in the first year of life by nonspecific symptoms. Between years 2 and 3, characteristic features develop, including coarse facial appearance with diffuse and coarse hair, flat bridge of the nose, wide, anteverted nostrils, thick lips, and macroglossia. Patients also present with skeletal and joint deformities, upper airway obstruction, recurrent ear infections, and progressive deceleration of growth rate (Table 2).3,4,10
A. A rapidly progressing patient
B. A slowly progressing patient
newborn 2 years 6 years 10 years 16 years 26 years
7 months 3 years 7 years 10 years 11 years 13 years
Differentiation into rapidly and slowly progressing disease is only apparent in extreme cases.
The entire spectrum of the disease inhibits physical and functional well-being and results in a markedly shortened lifespan.10,11
The heterogeneous, multisystemic, clinical manifestations of MPS VI show a broad range of complications.4,10
Cardiopulmonary disease, blindness, spinal cord compression, and joint disease are the most debilitating effects of this disease. Patients with MPS VI typically die from infection, surgical complications, or cardiopulmonary disease.10
SECTION 2
Figure 3: The progression of MPS VI

11
Regardless of the rate of disease progression, patients with MPS VI express markedly lower growth rates than unaffected age-adjusted peers (Figures 4 and 5). The deviation in growth experienced by patients with MPS VI may be a sign of secondary problems and should prompt clinical surveillance for issues such as malnutrition, endocrine abnormalities, or psychosocial deprivation.11
a For the purpose of this data set, patients with uGAG levels of >200 µg/mg creatinine are considered to have rapidly progressing MPS VI.
Figure 4: Growth comparison between patients with rapidly progressing MPS VI and unaffected age-adjusted peers11,a
Figure 5: Growth comparison between patients with slowly progressing MPS VI and unaffected age-adjusted peers11,b
b For the purpose of this data set, patients with uGAG levels of ≤200 µg/mg creatinine are considered to have slowly progressing MPS VI.

12 Please see Important Safety Information on pages 2 and 3.
ORGAN SYSTEM COMPLICATIONS
Ear, nose, throat, and respiratory12,13
GAG accumulation in the oropharynx and airway, combined with typical dysmorphic features and restrictive lung disease, can cause:
• Thickening of the nose, lips, and tongue
• Severe hearing impairment
• Recurrent otitis media
• Narrow trachea and excessive and thickened secretions
• Obstructive sleep apnea
• Recurrent pulmonary infections and pneumonia
• Skeletal problems and reduced lung function and volume
• The need for a respiratory device such as a continuous positive airway pressure (CPAP) machine or surgical insertion of an endotracheal tube to aid breathing
Cardiovascular3,10,14 Cardiovascular abnormalities are a major cause of morbidity and mortality among patients with MPS VI:
• Heart murmurs
• Mitral and aortic valve degeneration
• Electrocardiographic abnormalities
• Coronary artery disease
• Systemic vascular narrowing and hypertension
• Cardiomyopathy
Skeletal3,10,13 Skeletal deformities and other clinical manifestations are seen in patients with MPS VI:
• Dysostosis multiplex
• Spinal cord or nerve root injury
• Coarse facial features
• Short stature
• Joint abnormalities
• Chest/rib cage restriction
• Growth impairment
• Profound dwarfism
• Limited mobility
• Claw hands
Ophthalmic3,10,15,16 Visual impairment occurs in ~40% of patients with MPS VI:
• Most patients are farsighted
• Corneal clouding occurs in 95% of all patients
• Retinopathy
• Optic nerve abnormalities
• Ocular hypertension and glaucoma
SECTION 2
Table 2: Clinical manifestations of MPS VI

13
ORGAN SYSTEM COMPLICATIONS
Dental17 Dental abnormalities are common in patients with MPS VI and include:
• Mandibular condylar hypoplasia
• Malposition of unerupted teeth
• Large dental follicles
• Anterior open bite
• Maxillary constriction
• Taurodontism
Central nervous system (CNS)/ peripheral nervous system (PNS)3,10
MPS VI involves no direct impairment of CNS activity, such that the patient’s intelligence is typically normal despite the great physical disease burden:
• GAG accumulation causes carpal tunnel syndrome, intracranial pressure, and progressive compressive myelopathy
• Loss of dexterity and fixed flexion
• Cervical stenosis and spinal cord compression
• Severe pain caused by compressed or traumatized nerves and nerve roots
Organ systems3 The abdomen in patients with MPS VI is:
• Large and protruding due to the enlarged liver and spleen, often with the presence of inguinal and/or umbilical hernia
Because of the large number of mutations that can cause MPS VI, clinical manifestations of the disease are heterogeneous, and not all patients will experience all of these symptoms. If untreated, GAGs will build up and patients will experience more symptoms.3,10
Key points
• MPS VI is a clinically heterogeneous condition in which patients can present with marked disease in the first year of life or disease that progresses more slowly over time4,10
• Differentiation into rapidly and slowly progressing disease is only apparent in extreme cases11
• The entire spectrum of the disease inhibits physical and functional well-being10
• Natural progression of the disease shows a broad range of complications4,10

14 Please see Important Safety Information on pages 2 and 3.
2.5 DIAGNOSIS
The onset of MPS VI symptoms usually occurs within the first 2 years of life in rapidly progressing patients.18
Clinicians initiate the diagnostic process upon developing suspicion of MPS VI based on skeletal findings and other physical features (Figure 6). In contrast, patients with slowly progressing phenotypes typically do not develop symptoms until their teens and can present with more subtle disease manifestations.3,18,19 These patients can be particularly challenging to identify as they often present to specialists not accustomed to diagnosing MPS disorders (such as rheumatologists and ophthalmologists) with symptoms such as joint pain and ocular problems. It is recommended that diagnosis of MPS VI be confirmed by a geneticist and a laboratory experienced in diagnosing MPS disorders.19
Laboratory diagnosis of MPS VI
Diagnosis of MPS VI occurs through a combination of clinical findings and laboratory test results, and can be confirmed only through the appropriate lab tests. Diagnosing MPS VI requires the following3:
• The analysis of ASB enzyme activity in isolated leukocytes or cultured skin fibroblasts to demonstrate a large decrease or absence of ASB activity that is diagnostic for MPS VI
» ASB activity in patients diagnosed with MPS VI is generally less than 10% of the lower limit of normal ASB activity
• The identification of normal enzyme activity of a different sulfatase in order to exclude the diagnosis of multiple sulfatase deficiency (MSD)
Additional tests that are supportive of an MPS VI diagnosis include3:
• Evidence of a clinical phenotype (eg, short stature, bone-related dysostosis multiplex, hepatosplenomegaly, macrocephaly, inguinal or umbilical hernia, corneal clouding, or cardiac valve thickening)
» Clinical phenotype may not be evident in newborns or patients with a very mild phenotype
• Demonstration of elevated total uGAG at baseline that decreases significantly within 2 to 3 months of ERT administration, using testing conducted at the same laboratory
» Urinary GAGs are elevated in the newborn period and decrease over the first year, so this sign of response to ERT may not be useful in the newborn
• DS accumulation and absence of chondroitin sulfate, heparan sulfate (HS), keratan sulfate, or hyaluronate using thin-layer chromatography or high-resolution electrophoresis fractionation
• Demonstration of intermediate levels of leukocyte ASB enzyme activity in both parents to support diagnosis of carriers
• Confirmation by mutational analysis of the ARSB gene should be considered if the diagnosis is in question and is important in carrier testing or prenatal diagnosis
It is important to note that mutational analysis will not always detect 2 mutations or may detect novel variants of unknown significance; therefore, molecular analysis alone may result in a false negative.19
SECTION 2

15
Figure 6: To aid in the diagnosis of MPS VI, a diagnostic algorithm has been developed by a consensus of experts19

16 Please see Important Safety Information on pages 2 and 3.
The path to diagnosis is not necessarily linear, and clinicians should maintain an open channel of communication with laboratories and remain vigilant to possible misdiagnoses throughout the diagnostic process. Some clinical/laboratory analyses are not available in all regions of the world and, even if this linear path is followed, cases may be missed. Urinary GAG testing is not always abnormal; enzyme activity analysis has several caveats and requires the appropriate use of reference enzymes; and molecular testing does not always identify 2 previously described pathogenic mutations.19
Delayed or incorrect diagnosis can result from a number of factors including the incorrect interpretation of radiographs, lack of disease awareness, wide phenotypic variability, phenotypic overlap with other disorders, and limitations of uGAG screening. The radiologist can play a critical role in ensuring that an accurate diagnosis is reached expeditiously by raising suspicion of an MPS disorder if dysostosis multiplex changes are evident.18
Differential diagnosis
Differential diagnoses within the MPS and mucolipidoses families of diseases include3:
• MPS I H, MPS I S, MPS I H/S (Hurler syndrome, Scheie syndrome, Hurler-Scheie syndrome, respectively)
• MPS II (Hunter syndrome)
• MPS IV (Morquio syndrome)
• MPS VII (Sly syndrome)
• Multiple sulfatase deficiency (MSD)
• Mucolipidosis I (now known as sialidosis), II, III, and IV
Specific signs that differentiate MPS VI from other MPS/mucolipidoses types include3:
• Corneal clouding–typically will be more severe in MPS VI than MPS I; less common in MPS VII in patients 8 years of age and under
• Absence of corneal clouding and presence of HS and DS in equal amounts in urine–all signs unique to MPS II, an X-linked recessive disease primarily affecting boys
• Ichthyosis and mental deficits–differentiate MSD from MPS VI later in life, although the 2 will appear similar early on
• Clinical manifestations in utero or at birth–more typical in MPS VII
• Absence of increased uGAG–more common in mucolipidosis I, III, and IV than in MPS VI
• Increased uGAG with high levels of beta-hexosaminidase, iduronate sulfatase, and arylsulfatase A at 10 to 20 times the reference range in serum, but deficiency of the same enzyme in cultured fibroblasts–typical in mucolipidosis II
SECTION 2

17
a MED, SEDC, Dyggve–Melchior–Clausen syndrome including Smith-McCort syndrome, and pseudoachondroplasia were among the most common referring diagnoses.
Differential diagnosis of other disorders
Many radiographic and clinical manifestations of MPS can mimic those of other skeletal dysplasias, further complicating the path to diagnosis, which is why it is so important to include MPS in your differential diagnosis (Table 3). Moreover, once clinical suspicion has been established, limitations and complications associated with diagnostic assays may delay definitive confirmation.18
COMMON MISDIAGNOSES SYMPTOMS
Spondyloepiphysealdysplasia congenita (SEDC)
• Short stature
• Abnormal gait
• Skeletal abnormalities
Multiple epiphyseal dysplasia (MED)
• Short stature
• Joint pain and deformity
• Thick ribs and clavicles
Perthes syndrome
• Scoliosis
• Protuberant abdomen
• Hyperreflexia
Table 3: Common misdiagnoses18,a
Key points
• Clinicians initiate the diagnostic process upon developing suspicion of MPS VI based on skeletal findings and other physical features19
• Patients with slowly progressing MPS VI may be challenging to identify, and symptoms may not be apparent until the teenage years18
• Diagnosis of MPS VI occurs through a combination of clinical findings and laboratory test results and requires the following3:
» The analysis of ASB enzyme activity in isolated leukocytes or cultured skin fibroblasts to demonstrate a large decrease or absence of ASB activity
» The identification of normal enzyme activity of a different sulfatase in order to exclude the diagnosis of MSD
• Enzyme activity testing is the gold standard and is necessary to confirm diagnosis19

18 Please see Important Safety Information on pages 2 and 3.
2.6 MANAGEMENT
Over the past decade or so, the management of MPS VI has evolved along with clinicians’ knowledge about the disease, such as its phenotypic variability and progression. Other factors, such as patients living into adulthood, highlight new areas for research and understanding and promise that managing this disease will continue to be a dynamic venture.
Because MPS VI affects multiple body systems, management of a diverse spectrum of disease manifestations is an important part of providing integrated care. Management should include use of adaptive or supportive devices, physical and occupational therapy, symptom-based medications, surgical interventions, and treatment to provide the deficient enzyme.10
ERT is an established treatment for several lysosomal storage diseases (including MPS VI) based on the unique ability of human cells to bind and transport exogenous enzyme into the lysosomal compartment. ERT provides treatment of the underlying pathology of MPS VI by replacing the deficient enzyme activity and reversing the GAG accumulation responsible for the deleterious disease effects.3 ERT is recommended for all patients and should be initiated immediately upon diagnosis of MPS VI.2,14,20
ERT with NAGLAZYME® (galsulfase)
ERT with NAGLAZYME® (galsulfase) provides treatment of the underlying pathology of MPS VI.1 Early treatment initiation with NAGLAZYME immediately upon diagnosis may prevent disease progression and development of severe symptoms of MPS VI.2,14,20
Since the first clinical trial in 2000, several clinical studies have investigated the efficacy and safety of galsulfase ERT for MPS VI. NAGLAZYME has been demonstrated as an effective therapy with a favorable safety profile for all patients with MPS VI. Studies demonstrated that NAGLAZYME reduces uGAG levels and improves walking and stair climbing, 2 key measures of endurance.1 For more information, please refer to the clinical efficacy data in Section 3.3.
Ear, nose, and throat, and respiratory system–related symptoms
Surgical interventions such as adenotonsillectomy are sometimes performed to remove upper airway obstructions. Use of CPAP to maintain the patency of the airway has been beneficial for some patients. Tracheostomy may be necessary for some patients as a treatment for severe obstructive sleep apnea or to facilitate safer anesthesia.10
Given the frequency of hearing loss in patients with MPS VI, regular hearing tests can determine when an individual might benefit from a hearing aid. A minor procedure to place ventilation tubes in the ears can also help reduce the long-term impact of ear infections, and possibly prevent or delay hearing loss.7,10
Cardiac symptoms
In consultation with a cardiologist, treatment may include antibiotic prophylaxis for subacute bacterial endocarditis from dental and other surgical procedures. Additional cardiac medications may be prescribed if patient develops congestive heart failure. Successful replacement of aortic and mitral valves have been reported in patients with MPS VI.10
SECTION 2

19
Skeletal symptoms
Physical therapy and anti-inflammatory medications may provide benefit for some patients. Orthopedic surgical procedures to address/correct hip dysplasia or spinal deformities may significantly improve mobility.10
Ophthalmologic symptoms
Interventions may include corrective lenses, medications, and/or surgery to control increased intraocular pressure, patching for amblyopia, or surgery to correct strabismus, when present. Corneal transplants may be performed to correct severe corneal clouding with vision loss.10
A ventriculoperitoneal shunt may prevent optic atrophy and vision loss in some patients. Decompression of the optic nerve by neurosurgery may also be considered.10
CNS- and PNS-related symptoms
When an MRI of the brain shows dilated ventricles, shunting should be considered if increased intracranial pressure can be documented.10
Surgery can relieve constriction and compression around the spinal cord where it passes through the neck or at other levels of the spinal cord. This can alleviate weakness, paralysis, or spasticity that develops in the arms and legs, and can prevent permanent damage.21
Spinal or cervical cord compression (SCC) with resultant myelopathy is a known and serious complication of MPS VI. SCC is expected to occur in the natural history of the disease. Patients with MPS VI should be monitored for signs and symptoms of SSC, including back pain, paralysis of limbs below the level of compression, and urinary and fecal incontinence, and given appropriate clinical care.1
Surgical decompression of carpal tunnel syndrome, especially when performed in early stages, reduces signs and symptoms of compressive myelopathy and improves chances of preserving hand function.10
Assessment schedule
Patients with MPS VI require early and regular assessments to evaluate disease progression across multiple organ systems and to detect and treat potential complications of the disease.10
Table 4 shows a schedule of assessments for patients with MPS VI as recommended by international experts.

20 Please see Important Safety Information on pages 2 and 3.
INITIAL ASSESSMENTS
EVERY 3 MO EVERY 12 MOAS CLINICALLY
INDICATEDa
Confirmation of MPS VI X
Medical historyb X X
Physical examination X X
Neurologic examination X X
Height, weight X X
Head circumference X Xc
Tanner stage X Xd
Photographs X X
Endurancee
12-minute walk test3-minute stair climb
XX
XX
OphthalmologyVisual acuityCorneal examinationFundoscopic examinationIntraocular pressureRefraction
XXXXX
XXXXX
Audiometry X X
CardiologyEchocardiogramElectrocardiogramBlood pressure
XXX
XXX
ElectrophysiologyNerve conductionf X X
Pulmonary functionForced vital capacity, forced expiratory volume in 1 second, maximum voluntary ventilation9
Sleep study
X
X
X
X
Imaging studiesHip filmsh
Skeletal surveyFlex/ext radiograph of cervical spineMRI of brain and spinei
XXXX
XXXX
Laboratory assessmentsuGAG levels X X
Supplemental assessments for patients on ERTj
Total anti-ASB antibodyk X XYearly after
24 mo
a “As clinically indicated” generally means every 2 to 3 years depending on the rate of disease progression and clinical symptoms.b For infants, more frequent examinations are necessary.c Monitored until head growth has stopped.d Continue assessments until pubertal maturation is completed.e Endurance-testing paradigm before and after ERT: distance walked in 12 minutes (or 6-minute walk test per American Thoracic Society guidelines, but preferably same minute length as completed in previous test); number of stairs climbed in 3 minutes.
f Median nerve conduction measured to evaluate carpal tunnel syndrome.g Pulmonary-function tests are to include forced vital capacity, forced expiratory volume in 1 second, and maximum voluntary ventilation.h Anteroposterior and “frog-leg” lateral views of pelvis.i MRI of brain and spinal cord may require sedation or general anesthesia depending on patient age and cooperation. General anesthesia carries substantial risk for patients with MPS VI.
j For patients on ERT, results should be obtained at baseline, then at months 3, 6, 12, 18, and 24, and then yearly.k Anti-ASB antibody testing is only available for US patients enrolled in the clinical surveillance program.
SECTION 2
Table 4: Recommended schedule of assessments for patients with MPS VI10

21
Surgical considerations
Patients with MPS VI often require surgical intervention to address the multisystemic complications of the disease. This surgical care is complicated by the nature of the disease. Patients with MPS VI suffer from multiple factors that can dramatically increase surgical risk and the need for monitoring, including reduced respiratory capacity, impaired cardiovascular function, skeletal morphology, and complex airway structure. These factors complicate surgical and anesthetic care, require preplanning, and necessitate disease-specific techniques to increase optimal outcomes. Because of these factors, the benefits of a procedure in patients with MPS VI should always be balanced against the associated risks. Specialized perioperative procedures during anesthesia, such as intubation and extubation, and using an intraoperative neuromonitoring checklist, are essential to successful surgical interventions. An integrated surgical team consisting of MPS VI specialists is critical for positive, durable outcomes.22
Transitioning to adult care
As patients with MPS VI reach adulthood, their relationship with their medical team will change. To help manage this transition, individual plans are required to minimize treatment interruptions, extend support beyond the scope of pediatric care and parental support, and ensure adult patients are knowledgeable in managing MPS VI.23
These transition plans should be tailored to each patient’s specific needs, so that those who can take over their own care will have the tools they need, and those who are more limited will have the appropriate care and services in place to support them. The plans should include an assessment to determine the patient’s capacity as well as his or her ability and knowledge to communicate information about his or her condition.23
Key points
• Because MPS VI affects multiple body systems, management of a diverse spectrum of disease manifestations is an important part of providing integrated care10
• ERT provides treatment of the underlying pathology of MPS VI by replacing the deficient enzyme activity and reversing the GAG accumulation responsible for the deleterious disease effects3
• Patients with MPS VI require early and regular assessments to evaluate disease progression across multiple organ systems and to detect and treat potential complications of the disease10

22 Please see Important Safety Information on pages 2 and 3.
NAGLAZYME® (galsulfase)PRODUCT INFORMATION
3.1 OVERVIEW
NAGLAZYME® (galsulfase) is the only therapy indicated for long-term ERT in patients with MPS VI.1,2
Pivotal studies
Four clinical trials involving a total of 56 patients with MPS VI, ages 5 to 29 years, have been performed in order to assess the efficacy, safety, and tolerability of NAGLAZYME (Table 5). The majority of patients had severe manifestations of the disease as evidenced by poor performance on the 6- or 12-minute walk tests, assessments of physical endurance.1
NAGLAZYME demonstrated efficacy across 3 key end points:
• Significantly improved endurance on the 12-minute walk test (P=0.025)24
• Improved rate of stair climbing in the 3-minute stair climb test (approached statistical significance; P=0.053)1,24
• Reduced uGAG levels (75% reduction)24
Detailed efficacy data can be found in Section 3.3.
Observed adverse events with NAGLAZYME in 4 open-label studies (up to 261 weeks of treatment) were not different in nature or severity from those observed in the placebo-controlled study. No patients discontinued during open-label treatment with NAGLAZYME due to adverse events.1 Find detailed information about safety and tolerability results in Section 3.4.
SE
CT
ION
3

23
Abbreviation: rhASB, recombinant human arylsulfase B.a Two patients were transitioned from o.2 mg/kg to 1.0 mg/kg dose at weeks 59 and 69 after the double-blind analysis.
STUDY Phase 1/2 Phase 2 Phase 3 Phase 4
STUDY DESIGN
Double-blind, randomized, dose- comparison/open-
label extension
Open-label, nonrandomized
Double-blind, placebo-controlled, randomized/open-
label extension
Open-label, randomized, 2-dose level
in infants
WEEKS OF STUDY: EFFICACY (SAFETY)
240 (260) 144 (214) 96 (159) 52
NO. OF PATIENTS ENROLLED (COMPLETED)
7 (5) 10 (10) 39 (38) 4 (4)
DOSE OF rhASB0.2 mg/kga or
1.0 mg/kg1.0 mg/kg 1.0 mg/kg
1.0 mg/kg or 2.0 mg/kg
AGE IN YEARS MEAN ± SD (RANGE YEARS)
12.0 ± 3.8 (7-16) 12.1 ± 5.3 (6-21)
13.7 ± 6.47 (rhASB)
10.7 ± 4.35 (5-29) (placebo)
9.23 mo (3.30-12.7 mo)
SEX (M/F) 4/3 7/3 13/26 4/0
HEIGHT (cm) MEAN ± SD
107.5 ± 21.5 103.7 ± 14.4
104.4 ± 12.87 (rhASB)
100.3 ± 13.54 (placebo)
N/A
ETHNICITY 6 White, 1 Black 10 White24 White, 3 Black,
4 Hispanic, 2 Asian, 6 Other
N/A
NO. OF PATIENTS WITHDRAWN AND TIME OF WITHDRAWAL
1 after week 3, 1 after week 32
01 (placebo group)
after week 50
BASELINE uGAG (µg/mg CREATININE) MEAN ± SD
365 ± 148 336 ± 116346 ± 128 (rhASB)
330 ± 114 (placebo)870
Table 5: Clinical trials: design and study populations25,26

24 Please see Important Safety Information on pages 2 and 3.
3.2 PHARMACOLOGY
Mechanism of action
Mucopolysaccharide storage disorders are caused by the deficiency of specific lysosomal enzymes required for the catabolism of GAGs. MPS VI is characterized by the absence or marked reduction in N-acetylgalactosamine-4-sulfatase. This deficiency leads to accumulation of DS throughout the body. The result is widespread cellular, tissue, and organ dysfunction.1
NAGLAZYME® (galsulfase) treats the underlying pathology of MPS VI by replacing the deficient ASB enzyme activity and reversing the GAG accumulation responsible for the deleterious disease effects. It provides an exogenous enzyme that will be taken up into lysosomes and increase the catabolism of GAGs (Figure 7). NAGLAZYME uptake by cells into lysosomes is most likely mediated by the binding of mannose-6-phosphate–terminated oligosaccharide chains of NAGLAZYME to specific mannose-6-phosphate receptors.1,3
Pharmacodynamics
The responsiveness of uGAG to dosage alterations of NAGLAZYME is unknown, and the relationship of uGAG to other measures of clinical response has not been established.1
No association was observed between development of antibodies to NAGLAZYME and uGAG levels.1
Pathological GAG accumulation in lysosome
Increased enzyme levels due to NAGLAZYME therapy
Increased GAG degradation
SECTION 3
Figure 7: NAGLAZYME® (galsulfase) reduces GAG accumulation1,3

25
Table 6: Pharmacokinetic parameters at week 1 and week 241
Pharmacokinetics
The pharmacokinetic parameters of galsulfase were evaluated in 13 patients with MPS VI who received 1 mg/kg of NAGLAZYME® (galsulfase) as a weekly 4-hour infusion for 24 weeks.1 The pharmacokinetic parameters at week 1 and week 24 are shown in the table below.
Pharmacokinetic parameters (median, range)
Galsulfase pharmacokinetic parameters listed in Table 6 require cautious interpretation because of large assay variability. Development of anti-galsulfase antibodies appears to affect galsulfase pharmacokinetics; however, the data are limited.1
Nonclinical toxicology
Long-term studies in animals to evaluate carcinogenic potential or studies to evaluate mutagenic potential have not been performed with galsulfase. At intravenous doses up to 3.0 mg/kg (about 0.5 times the recommended human dose of 1 mg/kg based on body surface area), galsulfase was found to have no effect on the fertility and reproductive performance of male and female rats.1
PHARMACOKINETIC PARAMETER WEEK 1 WEEK 24
Cmax (mcg/mL) 0.8 (0.4-1.3) 1.5 (0.2-5.5)
AUC0-t (hr•mcg/mL)a 2.3 (1.0-3.5) 4.3 (0.3-14.2)
Vz (mL/kg) 103 (56-323) 69 (59-2799)
CL (mL/kg/min) 7.2 (4.7-10.5) 3.7 (1.1-55.9)
Half-life (min) 9 (6-21) 26 (8-40)
a Area under the plasma galsulfase concentration-time curve from start of infusion to 60 minutes post-infusion.
Key points
• NAGLAZYME treats the underlying pathology of MPS VI by replacing the deficient ASB enzyme activity and reversing the GAG accumulation responsible for the deleterious disease effects1,3
• NAGLAZYME provides an exogenous enzyme that is taken up into lysosomes and increases the catabolism of GAGs1
• Galsulfase uptake by cells into lysosomes is most likely mediated by the binding of mannose-6-phosphate–terminated oligosaccharide chains of galsulfase to specific mannose-6-phosphate receptors1

26 Please see Important Safety Information on pages 2 and 3.
Table 7: List of studies to evaluate the impact of galsulfase treatment on MPS VI2,5,25-27
3.3 CLINICAL EFFICACY
Studies have shown that long-term ERT with NAGLAZYME® (galsulfase) slows or stabilizes disease progression and improves endurance, uGAG levels, and pulmonary function.2
Since the first clinical trial in 2000, BioMarin Pharmaceutical Inc has sponsored several clinical studies that have investigated the natural history of MPS VI and efficacy and safety of NAGLAZYME for MPS VI (Table 7).
STUDY TYPE YEARNO. OF PATIENTS
ENROLLED (COMPLETED)
DURATION REFERENCE
Phase 1/2a
Randomized, double-blind,
dose-comparison/open-label extension
2000-2005 7 (5) 240 weeksHarmatz et al, J Pediatr, 2004
Phase 2a Open-label 2002-2006 10 (10) 144 weeksHarmatz et al, J Pediatr, 2006
Phase 3a
Randomized, double-blind,
placebo-controlled/open-label extension
2003-2006 39 (38) 96 weeksHarmatz et al, J Pediatr, 2008
Phase 4Open-label, dose-
comparison (age 3.3-12.7 mo)
2012 4 (4) 1-3 yearsHarmatz et al,
J Inherit Metab Dis, 2013
Clinical Surveillance Program (CSP)
Observational 2006-ongoing 132b OngoingHendriksz et al,
J Inherit Metab Dis, 2011 (5-year data)
SurveyStudy
Cross-sectional 2002 121 (121) BaselineSwiedler et al,
Am J Med Genet A, 2005
ResurveyStudy
10-year follow-up of Survey Study
201259 (for efficacy) 117 (for survival)
10-year follow-up
Giugliani et al, Am J Med Genet A,
2014
a Phase 1/2, phase 2, and phase 3 trials had open-label extension phases up to 240 weeks.b Number of patients enrolled as of publication of 5-year data.
SECTION 3
27
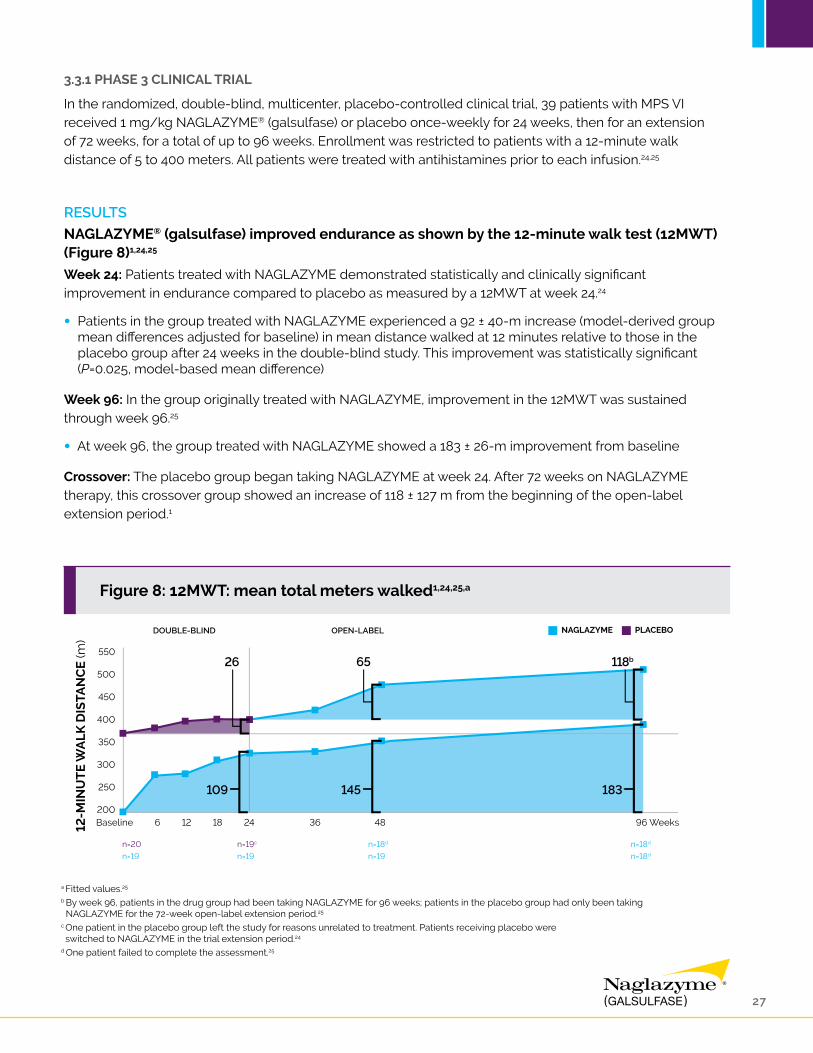
3.3.1 PHASE 3 CLINICAL TRIAL
In the randomized, double-blind, multicenter, placebo-controlled clinical trial, 39 patients with MPS VI received 1 mg/kg NAGLAZYME® (galsulfase) or placebo once-weekly for 24 weeks, then for an extension of 72 weeks, for a total of up to 96 weeks. Enrollment was restricted to patients with a 12-minute walk distance of 5 to 400 meters. All patients were treated with antihistamines prior to each infusion.24,25
RESULTS
NAGLAZYME® (galsulfase) improved endurance as shown by the 12-minute walk test (12MWT) (Figure 8)1,24,25
Week 24: Patients treated with NAGLAZYME demonstrated statistically and clinically significant improvement in endurance compared to placebo as measured by a 12MWT at week 24.24
• Patients in the group treated with NAGLAZYME experienced a 92 ± 40-m increase (model-derived group mean differences adjusted for baseline) in mean distance walked at 12 minutes relative to those in the placebo group after 24 weeks in the double-blind study. This improvement was statistically significant (P=0.025, model-based mean difference)
Week 96: In the group originally treated with NAGLAZYME, improvement in the 12MWT was sustained through week 96.25
• At week 96, the group treated with NAGLAZYME showed a 183 ± 26-m improvement from baseline
Crossover: The placebo group began taking NAGLAZYME at week 24. After 72 weeks on NAGLAZYME therapy, this crossover group showed an increase of 118 ± 127 m from the beginning of the open-label extension period.1
a Fitted values.25
b By week 96, patients in the drug group had been taking NAGLAZYME for 96 weeks; patients in the placebo group had only been taking NAGLAZYME for the 72-week open-label extension period.25
c One patient in the placebo group left the study for reasons unrelated to treatment. Patients receiving placebo were switched to NAGLAZYME in the trial extension period.24
d One patient failed to complete the assessment.25
Figure 8: 12MWT: mean total meters walked1,24,25,a

28 Please see Important Safety Information on pages 2 and 3.
NAGLAZYME® (galsulfase) improved the rate of stair climbing (Figure 9)1,24,25
Week 24: Patients treated with NAGLAZYME® (galsulfase) demonstrated improvement in the 3-minute stair climb compared to placebo at week 24, though statistical significance was not reached (P=0.053).1,24
• The rate in the group treated with NAGLAZYME was 19.4 ± 12.9 stairs/min at baseline, increasing to 26.9 ± 16.8 stairs/min at week 24
• In the longitudinal analysis, the difference between the mean change in the rates for patients treated with NAGLAZYME and placebo was 5.7 ± 2.9 stairs/min and approached statistical significance (P=0.053, model-based mean difference)
• The placebo group showed a near-constant rate climb of 31.0 ± 18.1 stairs/min at baseline and 32.6 ± 19.6 stairs/min at week 24
Week 96: In the group originally treated with NAGLAZYME, improvement in the rate of stair climbing was sustained through week 96.25
• At week 96, the group treated with NAGLAZYME showed an improvement of 13.1 ± 2.0 stairs/min over baseline
Crossover: The placebo group began taking NAGLAZYME at week 24. After 72 weeks on NAGLAZYME therapy, this crossover group showed an increase of 11.1 ± 10.0 stairs/min in the rate of stair climbing from the beginning of the open-label extension period.1
a Fitted values.25
b By week 96, patients in the drug group had been taking NAGLAZYME for 96 weeks; patients in the placebo group had only been taking NAGLAZYME for the 72-week open-label extension period.25
c One patient in the placebo group left the study for reasons unrelated to treatment. Patients receiving placebo were switched to NAGLAZYME in the trial extension period.24
d One patient failed to complete the assessment.25
SECTION 3
Figure 9: 3-minute stair climb test: mean stairs per minute1,24,25,a

29
NAGLAZYME® (galsulfase) reduced uGAG levels (Figure 10)24,25
Week 24: In patients treated with NAGLAZYME® (galsulfase), mean uGAG levels were significantly reduced by 75% at week 24.24
• Urinary GAG excretion was selected as a secondary efficacy variable based on the association of reduction in lysosomal storage with reduction in uGAG observed during rhASB treatment of felines with MPS VI
Week 96: In the group originally treated with NAGLAZYME, the reduction in uGAG levels was sustained through week 96. Urinary GAG levels observed in the NAGLAZYME group were similar at week 48 and week 96.24,25
Crossover: The placebo group began taking NAGLAZYME at week 24. At week 36 mean uGAG level in this crossover group decreased to a level similar to that of patients originally in the NAGLAZYME treatment group.24
Over 95% of all patients showed at least a 50% reduction in uGAG levels after 72 weeks of treatment with NAGLAZYME.1
Figure 10: uGAG levels in patients taking NAGLAZYME vs placebo24,25

30 Please see Important Safety Information on pages 2 and 3.
Figure 11: Mean percent change in height and pulmonary function by treatment week and age group over all available patient data28
3.3.2 COMBINED ANALYSIS OF CLINICAL TRIALS
Combined data from 3 clinical studies using NAGLAZYME ERT consist of a phase 1/2 and a phase 2 study, both demonstrating that weekly infusions of 1 mg/kg NAGLAZYME were well tolerated, produced a rapid reduction in uGAG levels, and improved endurance; and a phase 3, double-blind, placebo-controlled study that demonstrated significantly greater improvement in endurance on the 12MWT after 24 weeks of treatment as compared to the placebo group. Results were extended from 97 to 260 weeks in all studies.25
Patients treated with NAGLAZYME® (galsulfase) for 96 weeks demonstrated improvement in pulmonary function (Figure 11)28
In patients with MPS VI <12 years of age treated with NAGLAZYME® (galsulfase), both forced expiratory volume in 1 second (FEV1) and forced vital capacity (FVC) showed little change from baseline during the first 24 weeks of treatment. By 96 weeks of treatment, these parameters showed meaningful improvement from baseline, with increases in FEV1 and FVC averaging approximately 10% and 13%, respectively, for patients <12 years of age. For patients ≥12 years of age, FEV1 and FVC did not improve relative to baseline in the first 24 weeks but showed meaningful improvement in subsequent weeks. At 96 weeks of treatment, FEV1 and FVC improved from baseline by approximately 13% and 23%, respectively.28
SECTION 3

31
Figure 12: Mean change from pre-ERT baseline in distance walked in 6MWT and 12MWT27
3.3.3 ADDITIONAL CLINICAL STUDIES
Additional studies have been conducted to further determine the effects of MPS VI disease progression and the benefit of long-term NAGLAZYME® (galsulfase) treatment.
The ongoing MPS VI Clinical Surveillance Program and 2 other natural history studies that were conducted 10 years apart with the same population further demonstrated disease progression of MPS VI and the long-term efficacy and safety of NAGLAZYME.2,5,27
The Clinical Surveillance Program (CSP)
In September 2005 a multicenter, multinational survey was opened to track the long-term clinical outcomes of patients with MPS VI. Its objectives are to characterize the natural progression of MPS VI, to provide a comprehensive resource of data for physicians on the management of patients with MPS VI, to optimize patient care, and to evaluate the long-term efficacy and safety of ERT with NAGLAZYME. Published data show that NAGLAZYME is well tolerated and can result in overall improvements in endurance, hepatosplenomegaly, and pulmonary function, and suggest that treatment may promote growth if initiated early in childhood.27
RESULTS
The CSP demonstrates improved endurance following up to 4 years of NAGLAZYME® (galsulfase) ERT (Figure 12)27
Pre- and post-ERT 6-minute walk test (6MWT) data were available for 25 patients (18.9%); for 17 of these patients, pre- and post-ERT 12MWT data were available as well. The overall mean distance walked during the 6MWT increased from 332.6 m (SD 162.1) before the start of ERT to 421.4 m (SD 131.8) at last follow-up, and during the 12MWT, distance increased from 750.5 m (SD 268.5) to 893.3 m (SD 206.6).

32 Please see Important Safety Information on pages 2 and 3.
Figure 13: Mean uGAG and percent change from pre-ERT baseline by uGAG level measured before the start of ERT27
The CSP demonstrates improved pulmonary function following 3 years of NAGLAZYME® (galsulfase) ERT27
The MPS VI CSP was opened in 2005 to collect observational data for at least 15 years from standard clinical and laboratory assessments of patients with MPS VI. The study is ongoing.
The first report from the CSP was published in 2011. Twenty-eight patients (21.2%) had pre- and post-ERT pulmonary function test data, including 27 with data for FVC and 26 with FEV1 measurements. Baseline and follow-up data of other pulmonary function measurements were not reported in a fashion consistent enough to allow a meaningful analysis. The percent improvement in both FEV1 and FVC ranged between 10% and 30% during the first 3 years of follow-up when data from an acceptable number of patients (n=11 at 3-year follow-up) were available.
The CSP demonstrates continued reduction in uGAG (Figure 13)27
Pre- and post-ERT data for uGAG were available for 59 patients (44.7%). Urinary GAG decreased by a mean of at least 65%, and up to 92% in patients with a pre-ERT baseline uGAG >200 μg/mg creatinine, and by around 50% in patients with a baseline uGAG ≤200 μg/mg creatinine for the duration of the study. The mean percent change in uGAG level from pre-ERT baseline to last follow-up was -78.5% (SD 18.2) in patients with baseline uGAG >200 μg/mg creatinine and -53.3% (SD 41.7) in patients with baseline uGAG ≤200 μg/mg creatinine.
SECTION 3

33
The Survey Study
From 2001 to 2002 the multicenter, multinational, natural history Survey Study was conducted to establish demographics and clinical progression of patients with MPS VI in correlation with uGAG levels.2,5
RESULTS5
• As the disease progresses, patients experience significant disability
• Multisystemic manifestations such as cardiopulmonary complications and abnormal skeletal growth were experienced
• Endurance was shown to decline across all age groups
• Endurance was also shown to decrease with age as measured by the 6MWT, a standard clinical tool
» Increased GAG accumulation may be associated with a greater rate of disease progression
• Disease progression in certain patients with MPS VI tends to be positively correlated with higher levels of accumulated GAG

34 Please see Important Safety Information on pages 2 and 3.
Figure 14: Improvements in endurance were sustained in patients who completed the walk test at follow-up2
The Resurvey Study
The Resurvey Study was performed to obtain 10-year follow-up data on medical histories and clinical assessments (n=59), and survival status of NAGLAZYME® (galsulfase)-treated and untreated patients (n=117) who participated in the original Survey Study. Throughout the duration of the study period, NAGLAZYME-treated patients had an average of 6.8 years of ERT. The findings were consistent with the results of clinical trials of long-term use of NAGLAZYME in the treatment of MPS VI.2
RESULTS
Patients treated with NAGLAZYME® (galsulfase) maintained increased endurance on the 6MWT after 10-year follow-up (Figure 14)2
The mean 6-minute walk test distance for the NAGLAZYME ERT group (n=54) changed from 304.0 ± 108.4 m at baseline to 320.4 ± 195.7 m at 10 years, an increase of 16.4 ± 155.9 m. When excluding 8 patients who could not attempt the walk test, 7 of whom were wheelchair bound, there was a mean increase of 65.7 ± 100.6 m from a baseline of 310.4 ± 111.8 m. These data demonstrate sustained improvement in endurance with continuing NAGLAZYME ERT.
SECTION 3

35
Pulmonary function increased over baseline at 10-year follow-up in NAGLAZYME® (galsulfase)-treated vs untreated patients (Figure 15)2
The mean change in FVC from baseline was 0.37 L (P<0.0001) in the NAGLAZYME® (galsulfase)-treated group (n=48) and -0.70 L in the untreated group (n=3). The mean change in FEV1 from baseline was 0.21 L (P=0.001) in the NAGLAZYME-treated group (n=47) and -0.60 L in the untreated group (n=3).
Figure 15: Pulmonary function across all age groups in the ERT-treated patients2

36 Please see Important Safety Information on pages 2 and 3.
Urinary GAG levels in NAGLAZYME® (galsulfase)-treated patients decreased more than in untreated patients (Figure 16)2
Urinary GAG levels at follow-up were <100 μg/mg creatinine in all patients (n=55) who were exposed to NAGLAZYME® (galsulfase) ERT, including 33 of 55 (60%) patients whose uGAG levels were >200 μg/mg creatinine at baseline. Urinary GAG levels decreased by 87.9% in NAGLAZYME-treated patients (n=55)compared to 49.8% in untreated patients (n=3).
a Urinary GAG levels ›200 µg/mg are associated with rapidly progressing disease; uGAG levels ≤200 µg/mg are associated with slowly progressing disease.5
SECTION 3
Figure 16: Urinary GAG levels in all ERT-treated patients2

37
Key points
• In the phase 3 pivotal trial, patients treated with NAGLAZYME® (galsulfase) demonstrated statistically and clinically significant improvement in endurance as measured by a 12MWT at week 24 when compared to placebo24
• Patients treated with NAGLAZYME in the phase 3 trial demonstrated numerical improvement in the rate of stair climbing compared to placebo at week 24, though statistical significance was not reached1,24
• In patients treated with NAGLAZYME in the phase 3 trial, mean uGAG levels were reduced by 75% at week 2424
• A 10-year follow-up study of patients treated with NAGLAZYME demonstrated2:
» Maintained improvement in endurance on the 6MWT
» Increased pulmonary function
• Results of multiple studies have shown that long-term ERT with NAGLAZYME may slow or stabilize disease progression25

38 Please see Important Safety Information on pages 2 and 3.
3.4 SAFETY
CONTRAINDICATIONS1
None.
WARNINGS AND PRECAUTIONS1
Anaphylaxis and Allergic Reactions:Anaphylaxis and severe allergic reactions have been observed in patients during and up to 24 hours after NAGLAZYME® (galsulfase) infusion. Some of the reactions were life-threatening and included anaphylaxis, shock, respiratory distress, dyspnea, bronchospasm, laryngeal edema, and hypotension. If anaphylaxis or other severe allergic reactions occur, NAGLAZYME should be immediately discontinued, and appropriate medical treatment should be initiated. In patients who have experienced anaphylaxis or other severe allergic reactions during infusion with NAGLAZYME, caution should be exercised upon rechallenge; appropriately trained personnel and equipment for emergency resuscitation (including epinephrine) should be available during infusion.
Immune-mediated Reactions:Type III immune complex-mediated reactions, including membranous glomerulonephritis have been observed with NAGLAZYME, as with other enzyme replacement therapies. If immune-mediated reactions occur, discontinuation of the administration of NAGLAZYME should be considered, and appropriate medical treatment initiated. The risks and benefits of re-administering NAGLAZYME following an immune-mediated reaction should be considered. Some patients have successfully been rechallenged and have continued to receive NAGLAZYME under close clinical supervision.
Risk of Acute Cardiorespiratory Failure:Caution should be exercised when administering NAGLAZYME to patients susceptible to fluid volume overload; such as in patients weighing 20 kg or less, patients with acute underlying respiratory illness, or patients with compromised cardiac and/or respiratory function, because congestive heart failure may result. Appropriate medical support and monitoring measures should be readily available during NAGLAZYME infusion, and some patients may require prolonged observation times that should be based on the individual needs of the patient.
Acute Respiratory Complications Associated with Administration:Sleep apnea is common in MPS VI patients and antihistamine pretreatment may increase the risk of apneic episodes. Evaluation of airway patency should be considered prior to initiation of treatment. Patients using supplemental oxygen or continuous positive airway pressure (CPAP) during sleep should have these treatments readily available during infusion in the event of an infusion reaction, or extreme drowsiness/sleep induced by antihistamine use.
Consider delaying NAGLAZYME infusions in patients who present with an acute febrile or respiratory illness because of the possibility of acute respiratory compromise during infusion of NAGLAZYME.
SECTION 3

39
Infusion Reactions:Because of the potential for infusion reactions, patients should receive antihistamines with or without antipyretics prior to infusion. Despite routine pretreatment with antihistamines, infusion reactions, some severe, occurred in 33 of 59 (56%) patients treated with NAGLAZYME® (galsulfase). Serious adverse reactions during infusion included laryngeal edema, apnea, pyrexia, urticaria, respiratory distress, angioedema, and anaphylactoid reaction. Severe adverse reactions included urticaria, chest pain, rash, dyspnea, apnea, laryngeal edema, and conjunctivitis.
The most common symptoms of drug-related infusion reactions were pyrexia, chills, rash, urticaria, dyspnea, nausea, vomiting, pruritus, erythema, abdominal pain, hypertension, and headache. Respiratory distress, chest pain, hypotension, angioedema, conjunctivitis, tremor, and cough were also reported. Infusion reactions began as early as Week 1 and as late as Week 146 of NAGLAZYME treatment. Twenty-three of 33 patients (70%) experienced recurrent infusion reactions during multiple infusions though not always in consecutive weeks.
Symptoms typically abated with slowing or temporary interruption of the infusion and administration of additional antihistamines, antipyretics, and occasionally corticosteroids. Most patients were able to complete their infusions. Subsequent infusions were managed with a slower rate of NAGLAZYME administration, treatment with additional prophylactic antihistamines, and, in the event of a more severe reaction, treatment with prophylactic corticosteroids.
If severe infusion reactions occur, immediately discontinue the infusion of NAGLAZYME and initiate appropriate treatment. The risks and benefits of re-administering NAGLAZYME following a severe reaction should be considered.
No factors were identified that predisposed patients to infusion reactions. There was no association between severity of infusion reactions and titer of anti-galsulfase antibodies.
Spinal or Cervical Cord Compression:Spinal or cervical cord compression (SCC) with resultant myelopathy is a known and serious complication of MPS VI. SCC is expected to occur in the natural history of the disease, including in patients on NAGLAZYME. There have been post-marketing reports of patients treated with NAGLAZYME who experienced the onset or worsening of SCC requiring decompression surgery. Patients with MPS VI should be monitored for signs and symptoms of spinal/cervical cord compression (including back pain, paralysis of limbs below the level of compression, urinary and fecal incontinence) and given appropriate clinical care.
40 Please see Important Safety Information on pages 2 and 3.
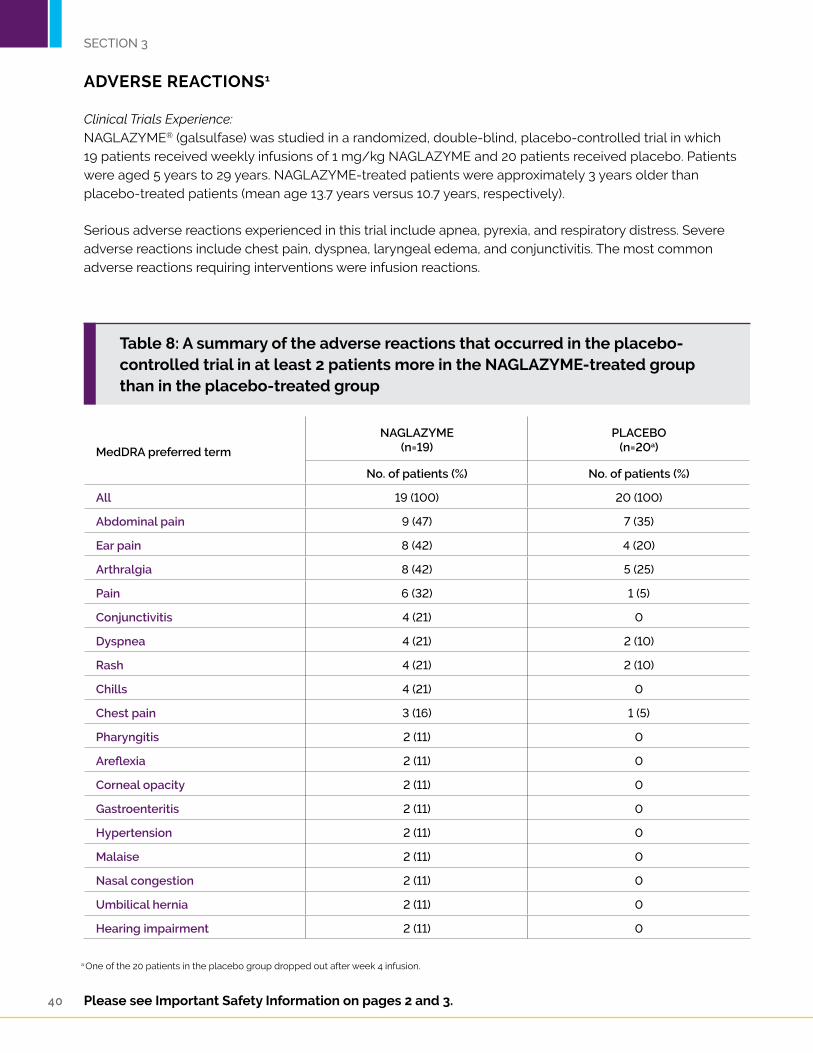
ADVERSE REACTIONS1
Clinical Trials Experience:NAGLAZYME® (galsulfase) was studied in a randomized, double-blind, placebo-controlled trial in which 19 patients received weekly infusions of 1 mg/kg NAGLAZYME and 20 patients received placebo. Patients were aged 5 years to 29 years. NAGLAZYME-treated patients were approximately 3 years older than placebo-treated patients (mean age 13.7 years versus 10.7 years, respectively).
Serious adverse reactions experienced in this trial include apnea, pyrexia, and respiratory distress. Severe adverse reactions include chest pain, dyspnea, laryngeal edema, and conjunctivitis. The most common adverse reactions requiring interventions were infusion reactions.
MedDRA preferred term
NAGLAZYME(n=19)
PLACEBO(n=20a)
No. of patients (%) No. of patients (%)
All 19 (100) 20 (100)
Abdominal pain 9 (47) 7 (35)
Ear pain 8 (42) 4 (20)
Arthralgia 8 (42) 5 (25)
Pain 6 (32) 1 (5)
Conjunctivitis 4 (21) 0
Dyspnea 4 (21) 2 (10)
Rash 4 (21) 2 (10)
Chills 4 (21) 0
Chest pain 3 (16) 1 (5)
Pharyngitis 2 (11) 0
Areflexia 2 (11) 0
Corneal opacity 2 (11) 0
Gastroenteritis 2 (11) 0
Hypertension 2 (11) 0
Malaise 2 (11) 0
Nasal congestion 2 (11) 0
Umbilical hernia 2 (11) 0
Hearing impairment 2 (11) 0
SECTION 3
Table 8: A summary of the adverse reactions that occurred in the placebo-controlled trial in at least 2 patients more in the NAGLAZYME-treated group than in the placebo-treated group
a One of the 20 patients in the placebo group dropped out after week 4 infusion.

41
In addition to those listed in Table 8, common adverse reactions observed in the open-label trials include pruritus, urticaria, pyrexia, headache, nausea, and vomiting. The most common adverse reactions requiring interventions were infusion reactions. Serious adverse reactions included laryngeal edema, urticaria, angioedema, and other allergic reactions. Severe adverse reactions included urticaria, rash, and abdominal pain.
Observed adverse reactions in four open-label studies (up to 261 weeks treatment) were not different in nature or severity to those observed in the placebo-controlled study. No patients discontinued during open-label treatment with NAGLAZYME® (galsulfase) due to adverse reactions.
USE IN SPECIFIC POPULATIONS1
Pregnancy Category B:Adequate and well-controlled studies have not been conducted with NAGLAZYME in pregnant women. Reproduction studies have been performed in rats at intravenous doses up to 3 mg/kg/day (about 0.5 times the recommended human dose of 1 mg/kg based on the body surface area) and in rabbits at intravenous doses up to 3 mg/kg/day (about 0.97 times the recommended human dose of 1 mg/kg based on the body surface area) and have revealed no evidence of impaired fertility or harm to the fetus due to NAGLAZYME. NAGLAZYME should be used during pregnancy only if clearly needed.
Pregnant women with MPS VI who are treated with NAGLAZYME should be encouraged to enroll in the MPS VI Clinical Surveillance Program at 800-983-4587.
Nursing Mothers:It is not known whether NAGLAZYME is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when NAGLAZYME is administered to a nursing mother. Nursing mothers with MPS VI who are treated with NAGLAZYME should be encouraged to enroll in the MPS VI Clinical Surveillance Program at 800-983-4587.
Pediatric Use:Clinical studies with NAGLAZYME were conducted in 56 patients, ages 5 to 29 years, with the majority of these patients in the pediatric age group. In addition, an open-label study was conducted in four infants (3 months to 12.7 months) treated with 1 mg/kg (n = 2) or 2 mg/kg (n = 2) of NAGLAZYME. Safety results in infants were consistent with results observed in patients 5 to 29 years old.
Geriatric Use:Clinical studies of NAGLAZYME did not include patients older than 29 years of age. It is not known whether older patients respond differently from younger patients.

42 Please see Important Safety Information on pages 2 and 3.
3.5 CSP/PATIENT REGISTRY
BioMarin encourages participation in data collection for the MPS VI CSP. The MPS VI CSP is an ongoing observational database tracking the medical history and outcomes of patients with MPS VI.
Advancing MPS VI treatment and research:• Provides a resource to physicians and patients for optimizing patient care
• Evaluates long-term safety and efficacy of NAGLAZYME® (galsulfase)
• Establishes the first comprehensive resource for understanding the variability and progression of MPS VI, as well as the long-term safety and efficacy of the only ERT indicated for its treatment
• Collects data from multiple centers around the world to compile in an observational database of longitudinal data, available to participating physicians
Optimizing patient care:• Consolidates data to develop new patient-monitoring standards
• Reports patient outcomes
• Provides new understanding for optimizing patient care
• Recommended assessments reflect a high standard of patient care based on feedback from geneticists who have treated patients with MPS VI
Easy participation for both physician and patient:• Participation in the CSP is voluntary
• No experimental treatments or assessments
• The only requirements to enroll a patient are a confirmed diagnosis of MPS VI, appropriate institutional approval, and patient or legal guardian consent
Inform your patients of the opportunity to participate in the MPS VI CSP. For more information, please call 1-866-906-6100 or visit www.MPSVI.com/CSP.
SECTION 3

43

44 Please see Important Safety Information on pages 2 and 3.
Indication1
NAGLAZYME® (galsulfase) is indicated for patients with Mucopolysaccharidosis VI (MPS VI, Maroteaux-Lamy syndrome). NAGLAZYME has been shown to improve walking and stair-climbing capacity.
Precautions1
• Pretreatment with antihistamines with or without antipyretics is recommended 30 to 60 minutes prior to the start of the infusions
• Evaluate airway patency prior to initiation of treatment with NAGLAZYME
• Patients using supplemental oxygen or CPAP should have these available during infusion
• Consider delaying infusion in patients who present with an acute febrile or respiratory illness
How supplied1
NAGLAZYME is supplied as a sterile injection in clear, type 1, glass, 5-mL vials containing 5 mg galsulfase per 5 mL solution.
Supplies needed1
• NAGLAZYME 5-mL, single-use vials
• 0.9% Sodium Chloride Injection, USP, infusion bag (100 mL or 250 mL)
• Syringes for dilution
• Low-protein–binding infusion set with a 0.2-μm in-line filter
Dosage recommendations1
The recommended dosage regimen of NAGLAZYME is 1 mg per kg of body weight administered once weekly as an intravenous infusion over no less than 4 hours.
Storage1
Store NAGLAZYME vials under refrigeration at 2°C to 8°C (36°F to 46°F)
• DO NOT FREEZE OR SHAKE
• DO NOT USE AFTER EXPIRATION DATE ON VIAL
• Protect from light
• NAGLAZYME contains no preservatives and should be used immediately following preparation
• Prepared NAGLAZYME must be refrigerated at 2°C to 8°C (36°F to 46°F) and administered within 48 hours from the time of preparation to completion of administration
SE
CT
ION
4 INDICATION AND CLINICAL USAGE

45
Preparation1
• Determine the number of vials to be used based on the patient’s weight and the recommended dose of 1 mg per kg: Patient’s weight (kg) × 1 mL/kg of NAGLAZYME® (galsulfase) = Total mL of NAGLAZYME
• Total mL of NAGLAZYME ÷ 5 mL per vial = Total number of vials
• Round up to the next whole vial. Remove the required number of vials from the refrigerator to allow them to reach room temperature. Do not allow vials to remain at room temperature longer than 24 hours prior to dilution. Do not heat or microwave vials
• Before withdrawing the NAGLAZYME solution from the vial, visually inspect each vial for particulate matter and discoloration. The NAGLAZYME solution should be clear to slightly opalescent and colorless to pale yellow. Some translucency may be present in the solution. Do not use if the solution is discolored or if there is particulate matter in the solution
• From a 250-mL infusion bag of 0.9% Sodium Chloride Injection, USP, withdraw and discard a volume equal to the volume of NAGLAZYME solution to be added. If using a 100-mL infusion bag, this step is not necessary
• Slowly withdraw the calculated volume of NAGLAZYME from the appropriate number of vials using caution to avoid excessive agitation. Do not use a filter needle, as this may cause agitation. Agitation may denature NAGLAZYME, rendering it biologically inactive
• Slowly add the NAGLAZYME solution to the 0.9% Sodium Chloride Injection, USP, using care to avoid agitation of the solutions. Do not use a filter needle
• Gently rotate the infusion bag to ensure proper distribution of NAGLAZYME. Do not shake the solution
• Administer the diluted NAGLAZYME solution to patients using a low-protein–binding infusion set equipped with a low-protein–binding 0.2-μm in-line filter
Administration rate examples1
• 250 mL: 6 mL/h for the first hour. If the infusion is well tolerated, the infusion rate can be increased to 80 mL/h for approximately 3 hours
• 120 mL: 3 mL/h for the first hour. If the infusion is well tolerated, the infusion rate can be increased to 39 mL/h for approximately 3 hours
• Patient vital signs should be monitored for signs of infusion reactions
46 Please see Important Safety Information on pages 2 and 3.
STUDY DOSE n TOTAL INFUSIONS(actual/possible)
OVERALL COMPLIANCE (%)b
24 Weeks Overalla
Phase 1/20.2 mg/kg
0.2/1.0 mg/kg1.0/1.0 mg/kg
2c
2d
3
27/2748/4872/72
34/35477/485740/750
97.198.498.7
Phase 2 1.0 mg/kg 10 240/240 1771/1812 99.8
Phase 3 1.0 mg/kg 19e 455/456 - 98.9
Phase 3 extension
1.0 mg/kg 38f _ 5036/5134 98.1
Studies have shown that long-term ERT with NAGLAZYME has been shown to slow or stabilize disease progression and improve endurance, uGAG levels, and pulmonary function.2 However, patients and families may have unrealistic expectations about treatment options.
• It is important to help them understand that results vary and some improvements occur over a long period of time1
• Efficacy of ERT for MPS VI seen in the pivotal trial was measured over a 6-month period1
a Total infusions were calculated based on treatment periods of 25 to 260 weeks for phase 1/2 study, 214 weeks for phase 2 study, and 135 weeks for phase 3 extension study. After the first 24 weeks in the phase 1/2 study, patients 45 and 41 continued on 0.2 mg/kg until weeks 59 and 69, respectively.
b Patients were excluded from compliance calculation from time of exit from study.c n=4 patients were randomized to 0.2 mg/kg dose group; patient 40 withdrew after week 3; patient 50 withdrew after week 32.d Patients 45 and 41, who had been randomized to the 0.2 mg/kg dose group for the first 24 weeks of the phase 1/2 study, were transitioned to 1.0 mg/kg
at weeks 59 and 69, respectively.e For the phase 3 study, overall percent compliance for the 24-week treatment period examines only the 19 patients randomized to rhASB.f Nineteen of the 20 patients randomized to the placebo group in the phase 3 study completed the study and initiated rhASB treatment in the phase 3
extension. Patient 020-006, who had been randomized to the placebo group, withdrew after 4 infusions.
SE
CT
ION
5
ERT is a lifelong commitment. In order for the patient to benefit from the treatment, NAGLAZYME® (galsulfase) must be infused on a weekly basis. If therapy stops, GAGs will build up, and symptoms may return.5 The table below shows the compliance rates for the clinical trial program for NAGLAZYME.
IMPORTANCE OF COMPLIANCE
Table 9: Compliance with NAGLAZYME® (galsulfase) treatment regimens25

47
Table 10: Suggestions for proactively addressing situations that may lead to compliance issues
Clinicians can aid compliance by helping patients and caregivers manage key treatment obstacles, such as IV pain as well as logistical issues (Table 10).
Patients with MPS VI may have difficulty making infusion appointments due to work and/or school obligations or frequent illness or hospitalization
Be as flexible as possible when scheduling or rescheduling appointments
Painful IV insertion can be a cause for anxiety, which could reduce compliance
Consider the use of topical anesthetics and other strategies to reduce the pain of IV insertion
Transportation and other problems may arise Utilize social services or call BioMarin Patient and Physician Support for advice
Sitting still for the length of the infusion may be difficult for young children
Have entertainment options available that may help the time pass more quickly

48 Please see Important Safety Information on pages 2 and 3.
Individualized support to help patients with MPS VI gain access to NAGLAZYME® (galsulfase)
BioMarin is committed to working with patients, healthcare providers, and insurance companies to access NAGLAZYME® (galsulfase) through BPPS. BPPS helps patients get access to and maintain NAGLAZYME treatment and provides assistance to patients, treating physicians, and their staff.
Support for physicians
BPPS is available to assist treating physicians and their staff with a variety of tasks, including:
• Access to laboratory testing around diagnosis confirmation
• Coordination with site for drug delivery logistics
• Infusion day coordination and logistics
• Patient support (see below)
Support for patients
BPPS provides a wide variety of support services for all patients with MPS VI. It’s confidential and customized to each patient’s unique needs.
MPS VI and NAGLAZYME education
Case managers can connect patients with the MPS specialist in their area to help them understand the clinical benefits of NAGLAZYME, and can provide valuable MPS VI and NAGLAZYME resources.
Access to treatment Case managers can help patients and treating physicians navigate the reimbursement process as it pertains to necessary tests and treatment. If a patient has limited or no insurance, BPPS will work with them to find out if they qualify for an assistance program or whether alternative ways to cover infusions need to be explored. If needed, case managers can direct patients to third-party, nonprofit organizations such as the National Organization for Rare Disorders (NORD) that can potentially help pay for NAGLAZYME.
Treatment support Treatment support is offered for patients receiving infusions at a clinic.
• For patients receiving infusions at a clinic, case managers can take care of details such as arranging for transportation to and from the clinic, if necessary, and ensuring NAGLAZYME is delivered to the clinic prior to infusion
Starting and staying on NAGLAZYME Long-term commitment to treatment can be an issue with chronic therapies such as NAGLAZYME. Case managers are available to help patients make necessary plans to maintain their weekly infusion schedule, especially in the face of changes such as a new job, new insurance, a move to a different location, or going off to college.
Enrolling in BPPS The patient needs to sign and return the Health Insurance Portability and Accountability Act of 1996 consent forms. These forms can be obtained by contacting BPPS or by downloading from www.Naglazyme.com.
Please contact BPPS at 1-866-906-6100 or [email protected] to learn more.
BIOMARIN PATIENT AND PHYSICIAN SUPPORT (BPPS)S
EC
TIO
N 6

49
Autosomal recessive—One of the ways a trait, disorder, or disease (such as MPS VI) can be passed down through families. For an autosomal recessive disorder to manifest, 2 copies of an abnormal gene must be present.
Enzyme replacement therapy (ERT)—A type of treatment for disorders such as MPS VI, in which the enzyme deficiency seen with the disorder is corrected/partially corrected by an IV infusion of the missing enzyme. NAGLAZYME® (galsulfase) is the only ERT available for MPS VI.
Glycosaminoglycans (GAGs)—Also known as mucopolysaccharides, GAGs are long, unbranched polysaccharides consisting of a repeating disaccharide unit. In MPS VI, GAGs are not catabolized within the cells as they otherwise would be; therefore, they accumulate, causing damage at the cellular, tissue, and organ levels.
Lysosomal enzymes—Enzymes within a cell’s lysosome that are responsible for the digestion/ degradation of complex waste molecules. Diseases such as MPS VI are the result of abnormal lysosomal enzyme function.
Lysosomal storage disorders (LSDs)—A group of approximately 40 rare, inherited, metabolic disorders that result from defects in lysosomal function. MPS VI is an LSD.
Lysosomes—Cellular organelles that contain enzymes for the breakdown of waste materials and cellular debris.
MPS VI—Also known as mucopolysaccharidosis VI (Maroteaux-Lamy syndrome), MPS VI is a progressive, multisystemic, autosomal recessive disorder in which a lysosomal enzyme is not functioning properly. This is an MPS disorder and, more broadly, an LSD. ERT, in the form of NAGLAZYME, is available for patients with MPS VI.
Mucopolysaccharidosis (MPS) disorders—A group of disorders in which a lysosomal enzyme needed for the breakdown of GAGs is deficient/absent.
NAGLAZYME® (galsulfase)—A systemic ERT indicated for patients with MPS VI. NAGLAZYME can modify disease progression by replacing deficient enzyme activity, clinically reducing GAG accumulation, to minimize the effects of the disease.
Six- or twelve-minute walk test (6MWT/12MWT)—The distance that can be walked by a test subject in the specified time. Used as a surrogate marker to assess functional capacity/status of patients. The 12MWT was the primary efficacy end point in the NAGLAZYME pivotal phase 3 trial.
Three-minute stair climb test (3MSCT)—The number of stairs that can be climbed by a test subject in 3 minutes. Like the 6MWT, this test assesses the functional capacity/status of patients. The 3MSCT was a secondary efficacy end point in the NAGLAZYME pivotal phase 3 trial.
GLOSSARY

50 Please see Important Safety Information on pages 2 and 3.
REFERENCES
1. NAGLAZYME [prescribing information]. Novato, CA: BioMarin; 2013.
2. Giugliani R, Lampe C, Guffon N, et al. Natural history and galsulfase treatment in mucopolysaccharidosis VI (MPS VI, Maroteaux–Lamy syndrome)—10-year follow-up of patients who previously participated in an MPS VI Survey Study. Am J Med Genet A. 2014;164A(8):1953-1964. doi:10.1002/ajmg.a.36584.
3. Valayannopoulos V, Nicely H, Harmatz P, Turbeville S. Mucopolysaccharidosis VI. Orphanet J Rare Dis. 2010;5:5-20. doi:10.1186/1750-1172-5-5.
4. Thümler A, Miebach E, Lampe C, et al. Clinical characteristics of adults with slowly progressing mucopolysaccharidosis VI: a case series. J Inherit Metab Dis. 2012;35(6):1071-1079. doi:10.1007/s10545-012-9474-1.
5. Swiedler SJ, Beck M, Bajbouj M, et al. Threshold effect of urinary glycosaminoglycans and the walk test as indicators of disease progression in a survey of subjects with mucopolysaccharidosis VI (Maroteaux–Lamy syndrome). Am J Med Genet A. 2005;134A(2):144-150. doi:10.1002/ajmg.a.30579.
6. Islam T, Linhardt RJ. Chemistry, biochemistry, and pharmaceutical potentials of glycosaminoglycans and related saccharides. In: Wong C-H, ed. Carbohydrate-based Drug Discovery. Vol 1. Weinheim, Germany: Wiley-VCH; 2003:407-439. doi:10.1002/3527602437.ch15.
7. Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease. Vol 3. 8th ed. New York, NY: McGraw-Hill; 2001:2465-2494.
8. National MPS Society: A Guide to Understanding MPS VI. Available at: http://mpssociety.org/wp-content/uploads/2011/07/booklet_MPS_VI_v5.pdf. Accessed February 9, 2015.
9. Kakkis ED. Enzyme replacement therapy for the mucopolysaccharide storage disorders. Expert Opin Investig Drugs. 2002;11(5):675-685. doi:10.1517/13543784.11.5.675.
10. Giugliani R, Harmatz P, Wraith JE. Management guidelines for mucopolysaccharidosis VI. Pediatrics. 2007;120(2):405-418. doi:10.1542/peds.2006-2184.
11. Quartel A, Hendriksz CJ, Parini R, Graham S, Lin P, Harmatz P. Growth charts for individuals with mucopolysaccharidosis VI (Maroteaux-Lamy syndrome) [published online December 18, 2014]. JIMD Rep. doi:10.1007/8904_2014_333.
12. Lin H-Y, Chen M-R, Lin C-C, et al. Polysomnographic characteristics in patients with mucopolysaccharidoses. Pediatr Pulmonol. 2010;45(12):1205-1212. doi:10.1002/ppul.21309.
13. Muhlebach MS, Wooten W, Muenzer J. Respiratory manifestations in mucopolysaccharidoses. Paediatr Respir Rev. 2011;12(2):133-138. doi:10.1016/j.prrv.2010.10.005.
14. Kampmann C, Lampe C, Whybra-Trümpler C, et al. Mucopolysaccharidosis VI: cardiac involvement and the impact of enzyme replacement therapy. J Inherit Metab Dis. 2014;37(2):269-276. doi:10.1007/s10545-013-9649-4.
15. Willoughby CE, Ponzin D, Ferrari S, Lobo A, Landau K, Omidi Y. Anatomy and physiology of the human eye: effects of mucopolysaccharidoses disease on structure and function—a review. Clin Experiment Ophthalmol. 2010;38:2-11. doi:10.1111/j.1442-9071.2010.02363.x.

51
16. Ganesh A, Bruwer Z, Al-Thihli K. An update on ocular involvement in mucopolysaccharidoses. Curr Opin Ophthalmol. 2013;24(5):379-388. doi:10.1097/ICU.0b013e3283644ea1.
17. Kantaputra PN, Kayserili H, Güven Y, et al. Oral manifestations of 17 patients affected with mucopolysaccharidosis type VI. J Inherit Metab Dis. 2014;37(2):263-268. doi:10.1007/s10545-013-9645-8.
18. Lachman RS, Burton BK, Clarke LA, et al. Mucopolysaccharidosis IVA (Morquio A syndrome) and VI (Maroteaux-Lamy syndrome): under-recognized and challenging to diagnose. Skeletal Radiol. 2014;43(3):359–369. doi:10.1007/s00256-013-1797-y.
19. Wood T, Bodamer OA, Burin MG, et al. Expert recommendations for the laboratory diagnosis of MPS VI. Mol Genet Metab. 2012;106(1):73-82. doi:10.1016/j.ymgme.2012.02.005.
20. Braunlin E, Rosenfeld H, Kampmann C, et al; MPS VI Study Group. Enzyme replacement therapy for mucopolysaccharidosis VI: long-term cardiac effects of galsulfase (Naglazyme®) therapy. J Inherit Metab Dis. 2013;36(2):385-394. doi:10.1007/s10545-012-9481-2.
21. Vougioukas VI, Berlis A, Kopp MV, Korinthenberg R, Spreer J, van Velthoven V. Neurosurgical interventions in children with Maroteaux-Lamy syndrome: case report and review of the literature. Pediatr Neurosurg. 2001;35(1):35-38. doi:10.1159/000050383.
22. Walker R, Belani KG, Braunlin EA, et al. Anaesthesia and airway management in mucopolysaccharidosis. J Inherit Metab Dis. 2013;36(2):211-219. doi:10.1007/s10545-012-9563-1.
23. American Academy of Pediatrics, American Academy of Family Physicians, American College of Physicians, Transitions Clinical Report Authoring Group, Cooley WC, Sagerman PJ. Supporting the health care transition from adolescence to adulthood in the medical home. Pediatrics. 2011;128(1):182-200. doi:10.1542/peds.2011-0969.
24. Harmatz P, Giugliani R, Schwartz I, et al; MPS VI Phase 3 Study Group. Enzyme replacement therapy for mucopolysaccharidosis VI: a phase 3, randomized, double-blind, placebo-controlled, multinational study of recombinant human N-acetylgalactosamine 4-sulfatase (recombinant human arylsulfatase B or rhASB) and follow-on, open-label extension study. J Pediatr. 2006;148(4):533-539.e6. doi:10.1016/j.jpeds.2005.12.014.
25. Harmatz P, Giugliani R, Schwartz IVD, et al; MPS VI Study Group. Long-term follow-up of endurance and safety outcomes during enzyme replacement therapy for mucopolysaccharidosis VI: final results of three clinical studies of recombinant human N-acetylgalactosamine 4-sulfatase. Mol Genet Metab. 2008;94(4):469-475. doi:10.1016/j.ymgme.2008.04.001.
26. Harmatz PR, Garcia P, Guffon N, et al. Galsulfase (Naglazyme®) therapy in infants with mucopolysaccharidosis VI. J Inherit Metab Dis. 2014;37(2):277-287. doi:10.1007/s10545-013-9654-7.
27. Hendriksz CJ, Giugliani R, Harmatz P, et al; CSP Case Study Group. Design, baseline characteristics, and early findings of the MPS VI (mucopolysaccharidosis VI) Clinical Surveillance Program (CSP). J Inherit Metab Dis. 2013;36(2):373-384. doi:10.1007/s10545-011-9410-9.
28. Harmatz P, Yu Z-F, Giugliani R, et al. Enzyme replacement therapy for mucopolysaccharisdosis VI: evaluation of long-term pulmonary function in patients treated with recombinant human N-acetylgalactosamine 4-sulfatase. J Inherit Metab Dis. 2010;33(1):51-60. doi:10.1007/s10545-009-9007-8.

52 Please see Important Safety Information on pages 2 and 3.
1 INDICATIONS AND USAGENAGLAZYME® (galsulfase) is indicated for patients with Mucopolysaccharidosis VI (MPS VI, Maroteaux- Lamy syndrome). NAGLAZYME has been shown to improve walking and stair-climbing capacity.
2 DOSAGE AND ADMINISTRATION2.1 Recommended Dose The recommended dosage regimen of NAGLAZYME is 1 mg per kg of body weight administered once weekly as an intravenous infusion.
Pretreatment with antihistamines with or without antipyretics is recommended 30 to 60 minutes prior to the start of the infusion [see Warnings and Precautions (5.2)].
The total volume of the infusion should be delivered over a period of time of no less than 4 hours. NAGLAZYME should be diluted with 0.9% Sodium Chloride Injection, USP, to a final volume of 250 mL and delivered by controlled intravenous infusion using an infusion pump. The initial infusion rate should be 6 mL per hour for the first hour. If the infusion is well tolerated, the rate of infusion may be increased to 80 mL per hour for the remaining 3 hours. The infusion time can be extended up to 20 hours if infusion reactions occur.
For patients 20 kg and under or those who are susceptible to fluid volume overload, physicians may consider diluting NAGLAZYME in a volume of 100 mL [see Warnings and Precautions (5.1) and Adverse Reactions (6.3)]. The infusion rate (mL per hour) should be decreased so that the total infusion duration remains no less than 4 hours.
Each vial of NAGLAZYME provides 5 mg of galsulfase (expressed as protein content) in 5 mL of solution and is intended for single use only. Do not use the vial more than one time. The concentrated solution for infusion must be diluted with 0.9% Sodium Chloride Injection, USP, using aseptic techniques. Prepare Naglazyme using low-protein-binding containers and administer the diluted NAGLAZYME solution to patients using a low-protein-binding infusion set equipped with a low-protein-binding 0.2 μm in-line filter. There is no information on the compatibility of diluted NAGLAZYME with glass containers.
2.2 Instructions for Use Prepare and use NAGLAZYME according to the following steps. Use aseptic techniques.
a. Determine the number of vials to be used based on the patient’s weight and the recommended dose of 1 mg per kg:
Patient’s weight (kg) × 1 mL/kg of NAGLAZYME = Total number of mL of NAGLAZYME
Total number of mL of NAGLAZYME ÷ 5 mL per vial = Total number of vials
Round up to the next whole vial. Remove the required number of vials from the refrigerator to allow them to reach room temperature. Do not allow vials to remain at room temperature longer than 24 hours prior to dilution. Do not heat or microwave vials.
b. Before withdrawing the NAGLAZYME solution from the vial, visually inspect each vial for particulate matter and discoloration. The NAGLAZYME solution should be clear to slightly opalescent and colorless to pale yellow. Some translucency may be present in the solution. Do not use if the solution is discolored or if there is particulate matter in the solution.
FULL PRESCRIBING INFORMATION

53
c. From a 250 mL infusion bag of 0.9% Sodium Chloride Injection, USP, withdraw and discard a volume equal to the volume of NAGLAZYME solution to be added. If using a 100 mL infusion bag, this step is not necessary.
d. Slowly withdraw the calculated volume of NAGLAZYME from the appropriate number of vials using caution to avoid excessive agitation. Do not use a filter needle, as this may cause agitation. Agitation may denature NAGLAZYME, rendering it biologically inactive.
e. Slowly add the NAGLAZYME solution to the 0.9% Sodium Chloride Injection, USP, using care to avoid agitation of the solutions. Do not use a filter needle.
f. Gently rotate the infusion bag to ensure proper distribution of NAGLAZYME. Do not shake the solution.
g. Administer the diluted NAGLAZYME solution to patients using a low-protein-binding infusion set equipped with a low-protein-binding 0.2 μm in-line filter.
NAGLAZYME does not contain preservatives; therefore, after dilution with saline, the infusion bags should be used immediately. If immediate use is not possible, the diluted solution must be stored refrigerated at 2ºC to 8ºC (36ºF to 46ºF) and administered within 48 hours from the time of dilution to completion of administration. Other than during infusion, do not store the diluted NAGLAZYME solution at room temperature. Any unused product or waste material must be discarded and disposed of in accordance with local requirements.
NAGLAZYME must not be infused with other products in the infusion tubing. The compatibility of NAGLAZYME in solution with other products has not been evaluated.
3 DOSAGE FORMS AND STRENGTHSInjection; 5 mL vials (5 mg per 5 mL).
4 CONTRAINDICATIONSNone.
5 WARNINGS AND PRECAUTIONS5.1 Anaphylaxis and Allergic Reactions Anaphylaxis and severe allergic reactions have been observed in patients during and up to 24 hours after NAGLAZYME infusion. Some of the reactions were life-threatening and included anaphylaxis, shock, respiratory distress, dyspnea, bronchospasm, laryngeal edema, and hypotension. If anaphylaxis or other severe allergic reactions occur, NAGLAZYME should be immediately discontinued, and appropriate medical treatment should be initiated. In patients who have experienced anaphylaxis or other severe allergic reactions during infusion with NAGLAZYME, caution should be exercised upon rechallenge; appropriately trained personnel and equipment for emergency resuscitation (including epinephrine) should be available during infusion [see Adverse Reactions (6)].

54 Please see Important Safety Information on pages 2 and 3.
5.2 Immune-mediated Reactions Type III immune complex-mediated reactions, including membranous glomerulonephritis have been observed with NAGLAZYME, as with other enzyme replacement therapies. If immune-mediated reactions occur, discontinuation of the administration of NAGLAZYME should be considered, and appropriate medical treatment initiated. The risks and benefits of re-administering NAGLAZYME following an immune-mediated reaction should be considered. Some patients have successfully been rechallenged and have continued to receive NAGLAZYME under close clinical supervision [see Adverse Reactions (6.3)].
5.3 Risk of Acute Cardiorespiratory Failure Caution should be exercised when administering NAGLAZYME to patients susceptible to fluid volume overload; such as in patients weighing 20 kg or less, patients with acute underlying respiratory illness, or patients with compromised cardiac and/or respiratory function, because congestive heart failure may result. Appropriate medical support and monitoring measures should be readily available during NAGLAZYME infusion, and some patients may require prolonged observation times that should be based on the individual needs of the patient [see Adverse Reactions (6.3)].
5.4 Acute Respiratory Complications Associated with Administration Sleep apnea is common in MPS VI patients and antihistamine pretreatment may increase the risk of apneic episodes. Evaluation of airway patency should be considered prior to initiation of treatment. Patients using supplemental oxygen or continuous positive airway pressure (CPAP) during sleep should have these treatments readily available during infusion in the event of an infusion reaction, or extreme drowsiness/sleep induced by antihistamine use.
Consider delaying NAGLAZYME infusions in patients who present with an acute febrile or respiratory illness because of the possibility of acute respiratory compromise during infusion of NAGLAZYME.
5.5 Infusion Reactions Because of the potential for infusion reactions, patients should receive antihistamines with or without antipyretics prior to infusion. Despite routine pretreatment with antihistamines, infusion reactions, some severe, occurred in 33 of 59 (56%) patients treated with NAGLAZYME. Serious adverse reactions during infusion included laryngeal edema, apnea, pyrexia, urticaria, respiratory distress, angioedema, and anaphylactoid reaction. Severe adverse reactions included urticaria, chest pain, rash, dyspnea, apnea, laryngeal edema, and conjunctivitis [see Adverse Reactions (6)].
The most common symptoms of drug-related infusion reactions were pyrexia, chills, rash, urticaria, dyspnea, nausea, vomiting, pruritis, erythema, abdominal pain, hypertension, and headache. Respiratory distress, chest pain, hypotension, angioedema, conjunctivitis, tremor, and cough were also reported. Infusion reactions began as early as Week 1 and as late as Week 146 of NAGLAZYME treatment. Twenty-three of 33 patients (70%) experienced recurrent infusion reactions during multiple infusions though not always in consecutive weeks.
Symptoms typically abated with slowing or temporary interruption of the infusion and administration of additional antihistamines, antipyretics, and occasionally corticosteroids. Most patients were able to complete their infusions. Subsequent infusions were managed with a slower rate of NAGLAZYME administration, treatment with additional prophylactic antihistamines, and, in the event of a more severe reaction, treatment with prophylactic corticosteroids.

55
If severe infusion reactions occur, immediately discontinue the infusion of NAGLAZYME and initiate appropriate treatment. The risks and benefits of re-administering NAGLAZYME following a severe reaction should be considered.
No factors were identified that predisposed patients to infusion reactions. There was no association between severity of infusion reactions and titer of anti-galsulfase antibodies.
5.6 Spinal or Cervical Cord Compression Spinal or cervical cord compression (SCC) with resultant myelopathy is a known and serious complication of MPS VI. SCC is expected to occur in the natural history of the disease, including in patients on NAGLAZYME. There have been post-marketing reports of patients treated with NAGLAZYME who experienced the onset or worsening of SCC requiring decompression surgery. Patients with MPS VI should be monitored for signs and symptoms of spinal/cervical cord compression (including back pain, paralysis of limbs below the level of compression, urinary and fecal incontinence) and given appropriate clinical care.
6 ADVERSE REACTIONS6.1 Clinical Trials Experience Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates observed in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
NAGLAZYME was studied in a randomized, double-blind, placebo-controlled trial in which 19 patients received weekly infusions of 1 mg/kg NAGLAZYME and 20 patients received placebo; of the 39 patients 66% were female, and 62% were White, non-Hispanic. Patients were aged 5 years to 29 years. NAGLAZYME- treated patients were approximately 3 years older than placebo-treated patients (mean age 13.7 years versus 10.7 years, respectively).
Serious adverse reactions experienced in this trial include apnea, pyrexia, and respiratory distress. Severe adverse reactions include chest pain, dyspnea, laryngeal edema, and conjunctivitis. The most common adverse reactions requiring interventions were infusion reactions.
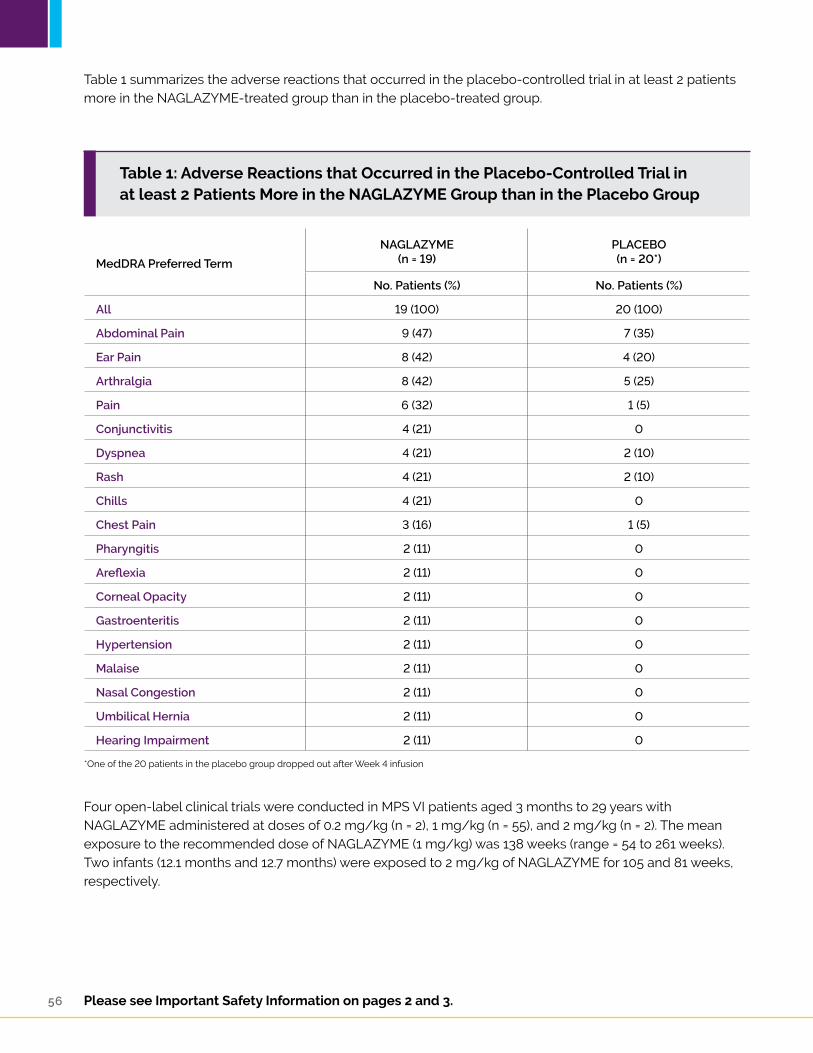
56 Please see Important Safety Information on pages 2 and 3.
Table 1: Adverse Reactions that Occurred in the Placebo-Controlled Trial in at least 2 Patients More in the NAGLAZYME Group than in the Placebo Group
Table 1 summarizes the adverse reactions that occurred in the placebo-controlled trial in at least 2 patients more in the NAGLAZYME-treated group than in the placebo-treated group.
Four open-label clinical trials were conducted in MPS VI patients aged 3 months to 29 years with NAGLAZYME administered at doses of 0.2 mg/kg (n = 2), 1 mg/kg (n = 55), and 2 mg/kg (n = 2). The mean exposure to the recommended dose of NAGLAZYME (1 mg/kg) was 138 weeks (range = 54 to 261 weeks). Two infants (12.1 months and 12.7 months) were exposed to 2 mg/kg of NAGLAZYME for 105 and 81 weeks, respectively.
MedDRA Preferred Term
NAGLAZYME(n = 19)
PLACEBO(n = 20*)
No. Patients (%) No. Patients (%)
All 19 (100) 20 (100)
Abdominal Pain 9 (47) 7 (35)
Ear Pain 8 (42) 4 (20)
Arthralgia 8 (42) 5 (25)
Pain 6 (32) 1 (5)
Conjunctivitis 4 (21) 0
Dyspnea 4 (21) 2 (10)
Rash 4 (21) 2 (10)
Chills 4 (21) 0
Chest Pain 3 (16) 1 (5)
Pharyngitis 2 (11) 0
Areflexia 2 (11) 0
Corneal Opacity 2 (11) 0
Gastroenteritis 2 (11) 0
Hypertension 2 (11) 0
Malaise 2 (11) 0
Nasal Congestion 2 (11) 0
Umbilical Hernia 2 (11) 0
Hearing Impairment 2 (11) 0
*One of the 20 patients in the placebo group dropped out after Week 4 infusion

57
In addition to those listed in Table 1, common adverse reactions observed in the open-label trials include pruritus, urticaria, pyrexia, headache, nausea, and vomiting. The most common adverse reactions requiring interventions were infusion reactions. Serious adverse reactions included laryngeal edema, urticaria, angioedema, and other allergic reactions. Severe adverse reactions included urticaria, rash, and abdominal pain.
Observed adverse events in four open-label studies (up to 261 weeks treatment) were not different in nature or severity to those observed in the placebo-controlled study. No patients discontinued during open-label treatment with NAGLAZYME due to adverse events.
6.2 Immunogenicity Ninety-eight percent (53/54) of patients treated with NAGLAZYME and evaluable for the presence of antibodies to galsulfase developed anti-galsulfase IgG antibodies within 4 to 8 weeks of treatment (in four clinical studies). In 19 patients treated with NAGLAZYME from the placebo-controlled study, serum samples were evaluated for a potential relationship of anti-galsulfase antibody development to clinical outcome measures. All 19 patients treated with NAGLAZYME developed antibodies specific to galsulfase; however, the analysis revealed no consistent predictive relationship between total antibody titer, neutralizing or IgE antibodies, and infusion-associated reactions, urinary glycosaminoglycan (GAG) levels, or endurance measures. Antibodies were assessed for the ability to inhibit enzymatic activity but not cellular uptake.
The data reflect the percentage of patients whose test results were considered positive for antibodies to galsulfase using specific assays and are highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibodies in an assay may be influenced by several factors including sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to galsulfase with the incidence of antibodies to other products may be misleading.
6.3 Postmarketing Experience The following adverse reactions have been identified during postapproval use of NAGLAZYME. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
In addition to infusion reactions reported in clinical trials, serious reactions which occurred during NAGLAZYME infusion in the worldwide marketing experience include anaphylaxis, shock, hypotension, bronchospasm, and respiratory failure.
Additional infusion reactions included pyrexia, erythema, pallor, bradycardia, tachycardia, hypoxia, cyanosis, tachypnea, and paresthesia.
During postmarketing surveillance, there has been a single case of membranous nephropathy and rare cases of thrombocytopenia reported. In the case of membranous nephropathy, renal biopsy revealed galsulfase-immunoglobulin complexes in the glomeruli. With both membranous nephropathy and thrombocytopenia, patients have been successfully rechallenged and have continued to receive NAGLAZYME.

58 Please see Important Safety Information on pages 2 and 3.
8 USE IN SPECIFIC POPULATIONS8.1 Pregnancy Pregnancy Category B. Adequate and well-controlled studies have not been conducted with NAGLAZYME in pregnant women. Reproduction studies have been performed in rats at intravenous doses up to 3 mg/kg/day (about 0.5 times the recommended human dose of 1 mg/kg based on the body surface area) and in rabbits at intravenous doses up to 3 mg/kg/day (about 0.97 times the recommended human dose of 1 mg/kg based on the body surface area) and have revealed no evidence of impaired fertility or harm to the fetus due to NAGLAZYME. NAGLAZYME should be used during pregnancy only if clearly needed.
Pregnant women with MPS VI who are treated with NAGLAZYME should be encouraged to enroll in the MPS VI Clinical Surveillance Program at 800-983-4587 [see Patient Counseling Information (17.2)].
8.3 Nursing Mothers It is not known whether NAGLAZYME is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when NAGLAZYME is administered to a nursing mother. Nursing mothers with MPS VI who are treated with NAGLAZYME should be encouraged to enroll in the MPS VI Clinical Surveillance Program at 800-983-4587 [see Patient Counseling Information (17.2)].
8.4 Pediatric Use Clinical studies with NAGLAZYME were conducted in 56 patients, ages 5 to 29 years, with the majority of these patients in the pediatric age group [see Clinical Studies (14)]. In addition, an open-label study was conducted in four infants (3 months to 12.7 months) treated with 1 mg/kg (n = 2) or 2 mg/kg (n = 2) of NAGLAZYME. Safety results in infants were consistent with results observed in patients 5 to 29 years old [see Adverse Reactions (6)].
8.5 Geriatric Use Clinical studies of NAGLAZYME did not include patients older than 29 years of age. It is not known whether older patients respond differently from younger patients.
11 DESCRIPTIONNAGLAZYME is a formulation of galsulfase, which is a purified human enzyme that is produced by recombinant DNA technology in a Chinese hamster ovary cell line. Galsulfase (glycosaminoglycan N–acetylgalactosamine 4-sulfatase, EC 3.1.6.12) is a lysosomal enzyme that catalyzes the cleavage of the sulfate ester from terminal N–acetylgalactosamine 4-sulfate residues of glycosaminoglycans (GAG), chondroitin 4-sulfate and dermatan sulfate.
Galsulfase is a glycoprotein with a molecular weight of approximately 56 kDa. The recombinant protein consists of 495 amino acids and possesses six asparagine-linked glycosylation sites, four of which carry a bis-mannose–6–phosphate residue for specific cellular recognition. Post-translational modification of Cys53 produces the catalytic amino acid residue, Ca-formylglycine, which is required for enzyme activity. NAGLAZYME has a specific activity of approximately 70 units per mg of protein content. One activity unit is defined as the amount of enzyme required to convert 1 micromole of 4-methylumbelliferyl sulfate to 4-methylumbelliferone and free sulfate per minute at 37ºC.

59
NAGLAZYME is intended for intravenous infusion and is supplied as a sterile, nonpyrogenic, colorless to pale yellow, clear to slightly opalescent solution that must be diluted with 0.9% Sodium Chloride Injection, USP, prior to administration. NAGLAZYME is supplied in clear Type I glass 5 mL vials. Each vial provides 5 mg galsulfase, 43.8 mg sodium chloride, 6.20 mg sodium phosphate monobasic monohydrate, 1.34 mg sodium phosphate dibasic heptahydrate, and 0.25 mg polysorbate 80 in a 5 mL extractable solution with pH of approximately 5.8. NAGLAZYME does not contain preservatives. Each vial is for single use only.
12 CLINICAL PHARMACOLOGY12.1 Mechanism of Action Mucopolysaccharide storage disorders are caused by the deficiency of specific lysosomal enzymes required for the catabolism of GAG. MPS VI is characterized by the absence or marked reduction in N–acetylgalactosamine 4-sulfatase. The sulfatase activity deficiency results in the accumulation of the GAG substrate, dermatan sulfate, throughout the body. This accumulation leads to widespread cellular, tissue, and organ dysfunction. NAGLAZYME is intended to provide an exogenous enzyme that will be taken up into lysosomes and increase the catabolism of GAG. Galsulfase uptake by cells into lysosomes is most likely mediated by the binding of mannose-6-phosphate-terminated oligosaccharide chains of galsulfase to specific mannose-6-phosphate receptors.
12.2 Pharmacodynamics The responsiveness of urinary GAG to dosage alterations of NAGLAZYME is unknown, and the relationship of urinary GAG to other measures of clinical response has not been established. No association was observed between antibody development and urinary GAG levels [see Adverse Reactions (6.2)].
12.3 Pharmacokinetics The pharmacokinetic parameters of galsulfase were evaluated in 13 patients with MPS VI who received 1 mg/kg of NAGLAZYME as a weekly 4-hour infusion for 24 weeks. The pharmacokinetic parameters at Week 1 and Week 24 are shown in Table 2.
Galsulfase pharmacokinetic parameters listed in Table 2 require cautious interpretation because of large assay variability. Development of anti-galsulfase antibodies appears to affect galsulfase pharmacokinetics, however, the data are limited.
Pharmacokinetic Parameter Week 1 Week 24
Cmax (mcg/mL) 0.8 (0.4 to 1.3) 1.5 (0.2 to 5.5)
AUC0-t (hr•mcg/mL)* 2.3 (1.0 to 3.5) 4.3 (0.3 to 14.2)
Vz (mL/kg) 103 (56 to 323) 69 (59 to 2,799)
CL (mL/kg/min) 7.2 (4.7 to 10.5) 3.7 (1.1 to 55.9)
Half-life (min) 9 (6 to 21) 26 (8 to 40)
*Area under the plasma galsulfase concentration-time curve from start of infusion to 60 minutes post infusion.
Table 2: Pharmacokinetic Parameters (Median, Range)

60 Please see Important Safety Information on pages 2 and 3.
13 NONCLINICAL TOXICOLOGY13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility Long-term studies in animals to evaluate carcinogenic potential or studies to evaluate mutagenic potential have not been performed with galsulfase.
Galsulfase at intravenous doses up to 3.0 mg/kg (about 0.5 times the recommended human dose of 1 mg/kg based on body surface area) was found to have no effect on the fertility and reproductive performance of male and female rats.
14 CLINICAL STUDIESA total of 56 patients with MPS VI, ages 5 years to 29 years, were enrolled in four clinical studies. The majority of patients had severe manifestations of the disease as evidenced by poor performance on a test of physical endurance.
In the randomized, double-blind, multicenter, placebo-controlled clinical trial, 38 patients with MPS VI received 1 mg/kg NAGLAZYME or placebo, once-weekly for 24 weeks. The patients’ ages ranged from 5 to 29 years. Enrollment was restricted to patients with a 12-minute walk distance of 5 to 400 meters. All patients were treated with antihistamines prior to each infusion.
The NAGLAZYME-treated group showed greater mean increases in the distance walked in 12 minutes (12-minute walk test, 12-MWT) and in the rate of stair climbing in a 3-minute stair climb test, compared with the placebo group (Table 3).
NAGLAZYME Placebo NAGLAZYME vs.
Placebo
Baseline Week 24 Change Baseline Week 24 Change Difference in Changes
N 19 19 19 20 19* 19
Results from the 12-Minute Walk Test (Meters)
Mean ± SD
MedianPercentiles (25th, 75th)
227 ± 170
21090, 330
336 ± 227
316125, 483
109 ± 154
487, 183
381 ± 202
365256, 560
399 ± 217
373204, 573
26 ± 122
34–3, 89
83 ± 45† 92 ± 40‡
(p = 0.025)‡,§
Results from 3-Minute Stair Climb Test (Stairs/Minute)
Mean ± SD
MedianPercentiles (25th, 75th)
19.4 ± 12.9
16.710.0, 26.3
26.9 ± 16.8
22.814.8, 33.0
7.4 ± 9.9
5.22.2, 9.9
31.0 ± 18.1
24.718.1, 51.5
32.6 ± 19.6
29.014.2, 57.9
2.7 ± 6.9
4.31.0, 6.2
4.7 ± 2.8†
5.7 ± 2.9‡
(p = 0.053)‡,§
*One patient in the placebo group dropped out after 4 weeks of infusion†Observed mean of NAGLAZYME - Placebo ± SE‡Model-based mean of NAGLAZYME - Placebo ± SE, adjusted for baseline§p-value based on the model-based mean difference
Table 3: Results from Placebo-Controlled Clinical Study

61
Following the 24-week placebo-controlled study period, 38 patients received open-label NAGLAZYME for 72 weeks. Among the 19 patients who were initially randomized to NAGLAZYME and who continued to receive treatment for 72 weeks (total of 96 weeks), increases in the 12-MWT distance and in the rate of stair climbing were observed compared to the start of the open-label period (mean [ ± SD] change): 72 ± 116 meters and 5.6 ± 10.6 stairs/minute, respectively). Among the 19 patients who were randomized initially to placebo for 24 weeks, and then crossed over to treatment with NAGLAZYME, the increases after 72 weeks of NAGLAZYME treatment compared to the start of the open-label period, (mean [ ± SD] change): were 118 ± 127 meters and 11.1 ± 10.0 stairs/minute, for the 12-MWT and the rate of stair climbing, respectively.
Bioactivity was evaluated with urinary GAG concentration. Overall, 95% of patients showed at least a 50% reduction in urinary GAG levels after 72 weeks of treatment with NAGLAZYME. No patient receiving NAGLAZYME reached the normal range for urinary GAG levels [see Clinical Pharmacology (12.2)].
In an additional open-label extension study, patients receiving NAGLAZYME showed maintenance of initial improvement in endurance for approximately 240 weeks.
16 HOW SUPPLIED/STORAGE AND HANDLINGNAGLAZYME is supplied as a sterile injection in clear Type I glass 5 mL vials, containing 5 mg galsulfase (expressed as protein content) per 5 mL solution. The closure consists of a siliconized chlorobutyl rubber stopper and an aluminum seal with a plastic flip-off cap.
NDC 68135-020-01, 5 mL vial
Store NAGLAZYME under refrigeration at 2°C to 8°C (36°F to 46°F). Do not freeze or shake. Protect from light. Do not use NAGLAZYME after the expiration date on the vial. This product contains no preservatives.
17 PATIENT COUNSELING INFORMATION17.1 Infusion Reactions Patients and caregivers should be counseled that reactions related to administration and infusion may occur during NAGLAZYME treatment, including life-threatening anaphylaxis. Premedication and reduction of infusion rate may alleviate those reactions associated with the infusion [see Warnings and Precautions (5.4)].
Patients should be advised to report any adverse reactions experienced while on NAGLAZYME treatment.
17.2 Clinical Surveillance Program Patients should be informed that a Clinical Surveillance Program has been established in order to better understand the variability and progression of the disease in the population as a whole, and to monitor and evaluate long-term treatment effects of NAGLAZYME. The Clinical Surveillance Program will also monitor the effect of NAGLAZYME on pregnant women, nursing mothers and their offspring, and determine if NAGLAZYME is excreted in breast milk. Patients should be encouraged to participate and advised that their participation is voluntary and may involve long-term follow-up. For more information call 1-800-983-4587.
NAGLAZYME is manufactured and distributed by: BioMarin Pharmaceutical Inc. Novato, CA 94949 US License Number 1649 1-866-906-6100 (phone)
NAGLAZYME® is a registered trademark of BioMarin.

©2015 BioMarin Pharmaceutical Inc. All Rights Reserved. USMPS1226ENFEB2015