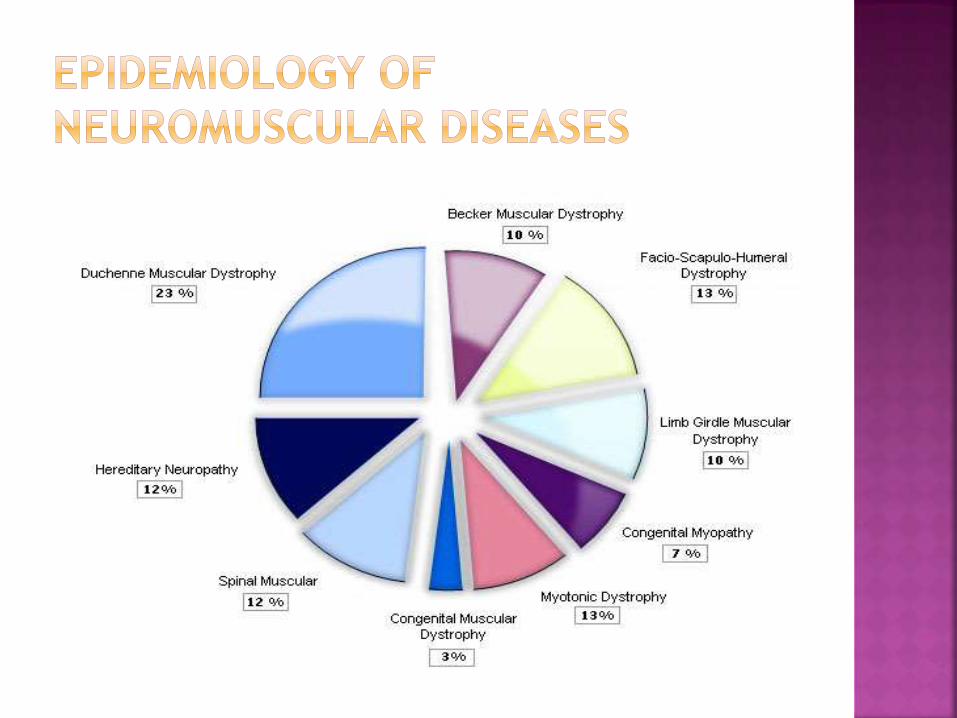
neuromuscular disorders in children (2)
TRANSCRIPT

BY
BHAVYA SHARMA

The term neuromuscular
disease defines disorders of
the motor unit and excludes
influences on muscular
function from the brain, such
as spasticity.

The motor unit has 4 components:
Motor neuron in the brainstem or anterior horn of
the spinal cord;
Axon(with other axons forming the peripheral
nerve);
Neuromuscular junction disorder; and
Myopathies (all muscle fibers innervated by a
single motor neuron).
The motor unit is influenced by suprasegmental or
upper motor neuron.

Muscular Dystrophies
Congenital and Metabolic Myopathies
Anterior Horn Cell Disorders
Neuromuscular Junction Diseases

Weakness, poor cough, retained airway
secretions
Inability to lift extremities against gravity
Muscle wasting
Low muscle tone (hypotonia)
Poor feeding, swallowing dysfunction
Failure to thrive
Increased respiratory rate
Use of accessory muscles of respiration
Recurrent infections
Night sweats

Weakness results either from -:
Upper motor neuron lesion e.g cerebral palsy causing increased tone,
brisk reflexes and extensor plantar response.
Lower motor neuron lesion causing hypotonia, depressed reflexes and
flexor plantar response.
Tone and strength should not be confused: Passive tone is range of
motion around a joint; Active tone is physiologic resistance to
movement.
Involvement of the face, tongue, palate, and extraocular muscles provides
an important distinction in the differential diagnosis.
Prenatal history
Acute flaccid paralysis

It’s a common sign of neuromuscular disorders.
CAUSES OF HYPOTONIA
Central Hypotonia-chromosome disorders, static insult, infections
(hyperactive reflexes)
Peripheral Hypotonia (distal weakness and wasting)
Neuromuscular junction (Myasthenia Gravis leads to fluctuating
wekness)
Muscular dystrophies(proximal weakness)
Anterior horn cells- spinal muscular atrophy (asso. with wasting,
hyporeflexes)
Neurometabolic condition- deficiencies


Serum Enzymes- creatine kinase
MM for skeletal muscle
MB for cardiac muscle
BB for brain
Mainly elevated in Duchenne muscular dystrophy.
Molecular Genetic Markers
Nerve Conduction Velocity
Electromyography- insertion of needle into belly of muscle → recording electric potentials in various states of contraction.
Imaging of Muscle
Muscle Biopsy- most important (vastus lateralis is sampled)
ECG

Classification-
Infectious - poliomyelitis
Motor neuron disease - amyotrophic lateral
sclerosis
Spinal muscular atrophy (SMA)

Autosomal Recessive disease
Type 1(Werdnig-hoffmann disease) present with profound hypotonia
and areflexia, respiratory weakness, poor swallowing and tongue
fasciculation. Children never learn to sit and Aspiration pneumonia
cause of death.
Type 2(Dubowitz disease) onset at 6-18 months and usually able to sit
unaided but may develop kyphoscoliosis, tremors.
Type 3(Kugelberg-Welander disease) onset >18 months and usually
able to walk
Treatment is supportive and respiratory care, management of feeding
and swallowing and provide adequate nutrition.

Guillain-Barrẽ syndrome is the most common.
Clinical patterns to a demyelating process include-
Presence of global areflexia
Moderate to severe muscle weakness with bulk
Motor symptoms
Hypertrophy of nerves
Guillain- Barrẽ Syndrome
Common cause of AFP
Immune mediated, rapidly progressive, predominantly motor, symmetric polyradiculoneuropathy
Condition can occur at any age within six weeks prior to symptom

Clinically the respiratory illness and weakness(2-
4weeks after onset) along with tachycardia, arrhythmia,
bladder dysfunction, labile blood pressure and
impaired thermoregulation.
Diagnosis
TREATMENT
IVIG at 2g/kg over 2-5 days or plasma exchanges if
child presents within 2-4weeks.
Donot respond give second course
General supportive care
Cardiorespiratory care and nutritional management.

MYASTHENIA GRAVIS
Autoimmune and autosomal recessive trait disorder
characterized by rapid fatigability of striated muscle.
Three clinical varieties-
Juvenile in late infancy and childhood showing extraocular
muscle weakness.
Congenital
Transient neonatal-symptoms arises after birth till 3rd day.
Occasionally associated with hypothyroidism, systemic
lupus erythematosus.
Acetylcholine receptor antibodies may be positive.
Electromyography show increases jitter or blocking.

Diagnostic test done by Edrophonium chloride(0.1-0.2mg/kg IV) and effects of weakness as distance b/w upper and lower lid is seen in 10sec.
TREATMENT
Mild myasthenia gravis require no treatment.
Cholinesterase inhibiting drugs such as
Neostigmine methylsulfate (0.4mg/kg) I/M every 4-6hr or oral neostigmine bromide.
Pyridostigmine (1-7mg/kg/day) in 4 divided doses
Prednisone(0.5mg/kg/day)
Thymectomy, Plasmapheresis, IVIG is effective in high circulating levels of anti-A ch receptor antibodies.

A group of genetically
determined, progressive primary
myopathy characterized by
degeneration and death of
muscle fibres, occurring at some
stage of the disorder.

–Duchenne muscular dystrophy
–Becker muscular dystrophy
–Myotonic muscular dystrophy
–Congenital muscular dystrophy
- Glycogenoses
- Mitochondrial
–Distal muscular dystrophy
–Others- Polymyositis
Dermatomyositis
inclusion body myositis

X-linked recessive
Absence of dystrophin protein
Slow to reach motor milestones
Symptoms appear in 2nd year, with clumsy walking or falling on walking or running on an uneven ground.
Muscles replaced by fat may appear hypertrophic
Child is mentally retarded
Life expectancy < 20 years with death related to respiratory failure or cardiomyopathy.
Gower Sign : Evident by 6 years of age-child trying to get up from squatting position, turns to side, lifts his trunk by supporting his weight on his arms and then stands up supporting the body with his hands.

Peudohypertrophy
of Calf Muscles in
Duchenne
Muscular
Dystrophy


Serum CPK elevated upto 15,000-20,000 U/L (normal
2000U/L)
EMG rarely necessary but useful in doubtful cases.
Histopathology of muscle show diffuse changes of
degeneration & regeneration.
Advanced genetic studies can find gene deletion.
Physiotherapy, exercises including walking & cycling
useful; tenotomy may be required for contractures.
Prednisone (0.75mg/kg/day) for 10days of each month to
avoid chronic complications.

Nemaline myopathy
Myotubular/Centronuclear myopathy
Central core disease
Multiminicore disease
Congenital fiber-type disproportion
myopathy

Genetics Autosomal recessive and dominant forms
First discovered in 1956 by Dr. Reyes
1/50,000 births
6 different mutations identified
Onset Infancy and early childhood
Clinical presentation Face, neck and proximal muscle
weakness
Absent deep tendon reflexes (DTR), normal creatinine
kinase

A form of centronuclear myopathy
Genetics X-linked recessive
Autosomal recessive and dominant
Onset Birth for X-linked recessive
Infancy and childhood for autosomal recessive
Adult for autosomal dominant
X-linked is most common form and most severe
Clinical Hypotonia, respiratory pump failure,
scaphocephaly

