new insights into hydride bonding, dynamics, and migration ... · new insights into hydride...
TRANSCRIPT

New Insights into Hydride Bonding, Dynamics, and Migration inLa2LiHO3 OxyhydrideØystein S. Fjellvåg,† Jeff Armstrong,*,‡ Ponniah Vajeeston,† and Anja O. Sjåstad†
†Centre for Materials Science and Nanotechnology, Department of Chemistry, University of Oslo, P.O. Box 1033, N-0315 Oslo,Norway‡ISIS Facility, Rutherford Appleton Laboratory, Harwell Oxford, Didcot, Oxfordshire OX11 0QX, United Kingdom
*S Supporting Information
ABSTRACT: Hydride anion-conducting oxyhydrides have recently emerged as a brandnew class of ionic conductors. Here we shed a first light onto their local vibrations,bonding mechanisms, and anion migration properties using the powerful combination ofhigh-resolution inelastic neutron scattering and a set of rigorously experimentally validateddensity functional theory calculations. By means of charge-density analysis we establishthe bonding to be strongly anisotropic; ionic in the perovskite layer and covalent in therock salt layer. Climbing nudged elastic band calculations allow us to predict the hydridemigration paths, which crucially we are able to link to the observed exotic ionic−covalenthybrid bonding nature. In particular, hydride migration in the rock salt layer is seen to begreatly hindered by the presence of covalent bonding, forcing in-plane hydride migrationin the perovskite layer to be the dominant transport mechanism. On the basis of thismicroscopic insight into the transport and bonding, we are able to propose futurecandidates for materials that are likely to show enhanced hydride conductivity.
Over the years, an array of ions such as O2−, H+, Li+, andNa+ have shown themselves to be excellent charge
carriers in a variety of ionic conductors used within the field ofenergy materials (e.g., in fuel cells and batteries). However, anew exotic class of potential ionic conductors has recentlyemerged from the discovery of oxyhydrides, where the anionicsublattice is formally composed of both O2− and H− ions.1
Recently Kobayashi et al.2 demonstrated pure H− conductivityin La2−x−ySrx+yLiH1−x+yO3−y. The hydride conductivity wasfound to depend on composition (x, y), with the highestconductivity (3.2 × 10−5 S cm−1 at 300 °C) observed forSr2LiH3O. Excitingly, they further documented the H−
transport properties of La2LiHO3 through a prototype solid-state Ti/La2LiHO3/TiH2 battery, demonstrating the potentialapplications of such a class of materials.2 To progress theserapid developments and to arrive at more viable devices it isessential that we develop a general understanding of thestructure, bonding, and diffusion mechanism of this new class ofmaterials.The existence of La2LiHO3 was first reported in 1991 by
Schwarz.3 The compound was synthesized by reacting La2O3
and LiH in a LiCl salt melt under an inert atmosphere at 750°C. La2LiHO3 has a K2NiF4-type structure (Ruddlesden−Popper type, RP; n = 1) which is composed of perovskite layersseparated by a half rock salt layer, which crystallizes in theorthorhombic space group Immm with an ordered distributionof the O2− and H− anions.3 Kobayashi et al. synthesizedLa2−x−ySrx+yLiH1−x+yO3−y by direct solid-state reaction betweenLa2O3, SrO, and LiH at high pressures (1 to 2 GPa) andintermediate temperatures (650−750 °C).2 Depending on the
reaction conditions, they concluded that La2LiHO3 can appearin either of two variants of the RP type, n = 1: orthorhombic(Immm) or tetragonal (I4/mmm). In the tetragonal variantreported by Kobayashi et al., the distribution of the equatorialO2− and H− is disordered. In addition, it is nonstoichiometricand partially contains anion vacancies with a nominal formulaof La2Li(H0.53O1.21V0.26)O2.
2
When considering potential applications of this new class ofmaterials, knowledge of the thermal stability becomes crucial.By means of combined thermogravimetric-differential scanningcalorimetry-mass spectroscopy, quasi-elastic neutron scattering(QENS), and in situ synchrotron powder X-ray diffraction(XRD), Fjellvåg et al. found the orthorhombic La2LiHO3 phaseto be remarkably stable and to first decompose at ∼450 °C toLa2LiO3.5 in an oxygen atmosphere without any signs of thetetragonal polymorph.4 Furthermore, QENS revealed thathydride ion diffusion was the driving force behind thedecomposition.There have been many insightful density functional theory
(DFT) studies of ion vibration/migration, with, in particular,BaTiO3−xHx being a widely studied compound. Zhang et al.calculated that the activation energy for migration between twointerstitial sites is 0.18 eV, comparable to the activation energiesfor proton conductors.5 The activation energy for hydridemigration from a hydride anion on an oxygen position to anoxygen vacancy in BaTiO3−xHx was calculated to be 0.28 eV.6
Received: November 22, 2017Accepted: January 3, 2018Published: January 3, 2018
Letter
pubs.acs.org/JPCLCite This: J. Phys. Chem. Lett. 2018, 9, 353−358
© 2018 American Chemical Society 353 DOI: 10.1021/acs.jpclett.7b03098J. Phys. Chem. Lett. 2018, 9, 353−358

To the best to our knowledge no mechanistic insight into H−
migration pathway for La2LiHO3 or in fact any otheroxyhydride is yet at hand. In this work we report the hydridedynamics in the hydride-conducting oxyhydride La2LiHO3 viainelastic neutron scattering (INS) and DFT calculations. Weverify our DFT model by comparing the in silico spectra withINS and Raman spectra. From the accepted model we thenperform a comprehensive charge distribution analysis tounderstand the bonding, before performing a migrationpathway analysis by climbing nudged elastic band (cNEB)calculations to shed light on the hydride transport mechanism.Structural and Vibrational Properties. La2LiHO3 has hydrogen
as its most dominant source of incoherent scattering, and thusINS is a very well-suited technique to study the localenvironment and bonding of its hydride anions. The INSspectra of La2LiHO3 (Figure 1, top) show three distinctivehigh-energy peaks. To uncover the atomic nature of thesevibrations, DFT phonon calculations were performed. Wedeveloped a DFT model starting from the reported structure ofLa2LiHO3 (Immm) and find the relaxed structure to be inexcellent agreement with these experimental reports (TableS1).3 The structure is visualized in Figure S1. The phonondispersion were calculated from DFT (Figure 1, bottom) andthe neutron-weighted projected density of states (Figure 1,middle) was then calculated and compared with theexperimental INS spectra (Figure 1, top). The in silico spectra(Figure 1, middle) show good agreement with the INS spectra,with the specific shape of each peak being very well reproduced.A systematic shift in the position of each peak is, however, seen,which is commonly observed when calculating vibrationalspectra by DFT.7−9 After considering several different func-tionals we concluded that PBE reproduced all of the essentialfeatures required for our study. For this reason PBE functionalwas therefore used for further calculations.
Decomposition of the DFT phonon spectra into incoherentand coherent contributions showed that the three vibrationalmodes at high-energy transfers originate from the incoherentscattering of the hydride anions (Figure 1, middle). Thecoherent contribution is negligibly small for these three modes.The DFT additionally reveals that the vibrations originate fromthe hydride vibrations along the x (1665 cm−1), y (1191 cm−1),and z axes (986 cm−1), with the relation νz< νy< νx (Figure 1,middle). Closer examination of the highest energy vibrationreveals that it corresponds to the vibration of the hydride aniontoward the lithium cation to which it is bonded. The resultantbonding along this direction therefore forms the most tightlybound local potential. The second highest hydride vibration cansimilarly be shown to correspond to the hydride anion vibratingin the y direction, which is in the direction of the neighboringhydride anion. In this direction there are no bonds. This isconsistent with a looser local potential and lower vibrationalfrequency than that along the x direction. The lowest of thethree high-energy peaks originates from vibration in the zdirection (i.e., the direction of the rock salt layer). Thisdirection has no direct neighbors. Thus one could anticipatethe local potential to be the lowest of the three spatialdimensions, which is indeed consistent with the observation ofit being the lowest frequency incoherent hydride mode.Neutron diffraction has previously been used to determine
the crystal structure of La2LiHO3,2−4 giving information on the
average positions of the constituents of the compound. BecauseLa2LiHO3 is reported to be a hydride conductor, the hydrideanions display mobility. In such cases, INS provides a moreconvincing confirmation of the local environment of thehydride anions, including validating the dynamics of thestructure. First, the general good agreement between INS andour DFT model (based on the average structure fromdiffraction experiments) validates the hydride position from
Figure 1. Top: INS spectra (black) of La2LiHO3 collected on TOSCA at 10 K and Raman spectra (dark red) with a laser wavelength of 532 nm. Theball−stick models [Li(O/H)6, lithium (purple), oxygen (red) and hydrogen (white)] illustrate the direction of the hydride vibration. Middle:Calculated Raman spectra (dark red) from DFT with the PBE functional. The peaks are assigned to the corresponding labeled modes. Totalscattering contribution calculated from DFT, weighted with the neutron cross section (black line). The incoherent scattering contribution fromhydrogen is represented by the shaded gray region. Bottom: Calculated phonon dispersion from DFT.
The Journal of Physical Chemistry Letters Letter
DOI: 10.1021/acs.jpclett.7b03098J. Phys. Chem. Lett. 2018, 9, 353−358
354

diffraction experiments. Additionally, as discussed above, thepeak positions of the incoherent hydride modes are perfectlyconsistent with the local structure/potential, which providesgreater assurance that the reported hydride positions fromprevious diffraction studies are indeed correct.2−4
The hydride anion vibrations are well separated from thevibrational modes of the other elements, as one would expectfrom the mass disparities present. At these lower energytransfers Raman spectroscopy is a well-suited technique,capable of giving better contrast toward the heavier elementsthan INS. The features in the experimental Raman spectra arewell accounted for in the in silico Raman spectra (Figure 1, topand middle). This gives extra confidence in our DFT modelbecause the modes from Raman and INS span a great range offrequencies and result from both coherent and incoherentmotions. All modes are identified to originate at low energiesfrom lanthanum and at higher energies from the axial oxideanion vibrations in the x, y, and z directions (Table S2). The insilico spectra show that the B2g and Ag modes have merged toform one peak from the lanthanum vibrations in the z directionand axial oxide anion in x direction, respectively, in contrastwith the experimental spectra where the peaks are separated.Previous experimental literature has reported two poly-
morphs of La2LiHO3, an orthorhombic and a tetragonalvariant.2,3 To our knowledge the stability of the two phases hasnot been investigated by DFT. We have therefore calculated thetotal energy versus unit cell volume for both phases (Figure 2,
Table S1). The calculations show that the orthorhombicpolymorph is the lower energy structure with a calculated cellvolume of 88 Å3 f.u.−1. Correspondingly, Rietveld refinementsof the same phase give a cell volume of 87.06 Å3 f.u.−1 (FigureS2 and Table S3).Interestingly, at larger cell volumes the DFT model predicts
that the tetragonal variant becomes energetically morefavorable than the orthorhombic. Both orthorhombic andtetragonal polymorphs were synthesized by Kobayashi et al.2 athigh pressures. The tetragonal sample was not stoichiometricwith a reported composition of La2Li(H0.53O1.21V0.26)O2.Similarly, Fjellvåg et al.4 observed by means of in situsynchrotron powder XRD no sign of the orthorhombic totetragonal phase transition before the material decomposes at450 °C. However, a trend toward a more tetragonal cell wasobserved. Anion vacancies are larger than the correspondinganion species in a structure. An increased vacancy concen-tration would therefore increase the cell volume. Consequentlythe tetragonal structure should become more favorable withincreased vacancy concentration. This result is therefore indirect support of the idea that a vacancy-filled La2LiHO3
sample, such as the tetragonal sample synthesized byKobayashi, should adopt the tetragonal structure.Bonding in La2LiHO3. With confidence in the comprehen-
sively validated DFT model, we may ask fundamental questionsabout the electronic structure through the use of Born-effective-charge (BEC) analysis. For an ideal ionic system, the ions havea spherically characterized charge distribution. We find forLa2LiHO3 that the BEC for all ions except the hydride anionand the axial oxygen anion (O2) deviates <15% from a perfectspherical charge distribution (Table 1). Most prominently thehydride anion is strongly deformed to be undersized in Zzz
(44% difference, Zzz < Zyy ≈ Zxx) making the hydride aniondisc-shaped, while the axial oxide anion has the relation Zyy <Zxx < Zzz. Consequently, the BEC predicts that the bonding inLa2LiHO3 deviates from the ideal ionic picture with thepresence of a sizable covalent contribution.In an attempt to quantify the bonding and estimate the
amount of electrons on and between the atomic sites, we haveperformed a Bader charge analysis and a Mulliken populationanalysis. Commonly, in near-pure ionic compounds, the Badercharge analysis and the Mulliken effective charges (MEC) areclose to their ideal ionic charges (La3+, Li+, H−, and O2−). Boththe Bader charge analysis and the MEC show deviations fromthe expected charge values for an ideal ionically bonded system(Table 1). A covalent character to the bonding would accountfor the deviations in charge for the ions, and hence the Baderanalysis and MEC point toward some degree of covalentbonding.
Figure 2. Calculated total energy versus unit-cell volume for the twodifferent polymorphs of La2LiHO3 [orthorhombic (Immm) andtetragonal (I4/mmm)]. The arrow indicates the theoretical equilibriumat 88 Å3 f.u.−1.
Table 1. Calculated Born-Effective-Charges, Bader Charges, Mulliken Population Analysis for La2LiHO3, and the Bond OverlapPopulation (BOP) between Constituents of La2LiHO3
a
Born-Effective-Charge Bader Mulliken population
xx yy zz charges (e) charges (e) bonds BOP
La (4i) 4.18 3.56 3.70 1.92 1.2 La−O1/O2 0.9/0.52Li (2a) 0.89 0.79 0.92 0.8 0.98 La−H 0.13H (2b) −0.95 −0.89 −0.53 −0.6 −0.59 Li−O1/O2 0.1/−0.25O1 (4i) −2.89 −2.47 −2.5 −1.65 −0.86 Li−H −0.2O2 (2d) −2.5 −2.1 −2.81 −1.72 −0.93
aFor the BEC, xx, yy, and zz correspond to charge distribution along the x, y, and z directions, respectively.
The Journal of Physical Chemistry Letters Letter
DOI: 10.1021/acs.jpclett.7b03098J. Phys. Chem. Lett. 2018, 9, 353−358
355

Fajans’ rules10−12 predict that a cation with high chargedensity will polarize a bond more than a cation with low chargedensity. On this basis one would expect La2LiHO3 to have astrong ionic character as the cations have low polarizing powerdue to the small charge of lithium and the large size oflanthanum. The bond overlap population (BOP) suggestsrather surprisingly the lanthanum−oxygen bonds to be covalentin character as BOP values larger than 0.4 are consistent withsignificant covalent character. This effect is attributed to lithiumpulling weakly on the electrons of oxygen and hydrogen,allowing lanthanum to form covalent bonding. Hence the BOPis closer to zero for the lithium−oxygen and lithium−hydrogenbonds. The negative value for the Li−H and Li−O2 bondsrepresents an ionic character of the bonds.To gain more information about the bonding character in
La2LiHO3, we turned our attention to charge-density andcharge-transfer analysis (Figure 3). The nonspherical chargedistribution around La and O1 again clearly supports anoticeable covalent character for the bond. On the contrary,the spherical charge distribution around Li, O2, and H indicatesa more “ideal” form of ionic bonding. The charge-transfer plot(the difference between the electron density of the compoundand the electron density of the constituent atoms) provides aclear visualization of how the chemical bonding is formed inreal space (Figure 3). It is clear that electrons are transferredfrom lithium and lanthanum to oxygen and hydrogen, andhence there is a considerable ionic bonding component presentbetween lithium and oxygen/hydrogen. Because of the sizablecovalent bonding, the charge depletion at the lanthanum site isnot spherically symmetric and finite electron density is presentin between lanthanum and oxygen, and hence the highestdegree of covalent bonding is to be found in the rock salt layer.In conclusion, the bonding character in La2LiHO3 is highlyanisotropic. The rock salt layer and the perovskite layer have acovalent and ionic character, respectively, giving mixed polar-covalent bonding situation.Hydride Anion Migration. Our DFT model was further used
to perform cNEB calculations to study the migration pathwaysin La2LiHO3. Several migration pathways for hydride and oxideanion migration have been considered, and the barrier heightsare listed in Table 2 and illustrated in Figure 4, whereas theminimal energy paths are visualized in Figure 5 and Figure S3.The lowest barrier height for hydride migration was found to bebetween the O1 and the H position, that is, in-plane migration,with a barrier height of 0.68 eV. Migration directly between two
H positions has a higher barrier height of 8.10 eV, largely dueto the long distance between these positions. Hydride anionsdiffusing in-plane will then not move directly between twohydride positions but rather by the corner-sheared positionswhere the octahedrons are connected.The lowest energy pathway for hydride migration was seen
to be between the O1 and H positions in the perovskite layer.This is also consistent with the fact that Fjellvåg et al. foundthat all vacancies in the oxidized derivative of the oxyhydride,La2LiO3.5, were positioned at the equatorial oxygen position,with none being present on the axial oxygen position.4 Thisindicates that during oxidation, the hydride anions weretransported through the material by this path, forming anionvacancies.The rock salt layer in La2LiHO3 has vacant tetrahedral sites;
therefore, hydride migration into and within the rock salt layerhas been considered as a potential pathway. In particular,previous reports have suggested that interstitial oxide positionsin the rock salt layer are the dominant transport mechanismsfor oxide anions migration in the two RP n = 1 oxides La2CoO4and La2NiO4
13−15 and in the RP n = 3 La4Co3O10.16 This
migration pathway was anticipated to be favorable due to theopen space in the structure and the similar atomic arrangementin the rock salt layer as in La2CoO4, La2NiO4, and La4Co3O10.
Figure 3. Calculated valence electron volumetric charge density (a), charge-density distribution (b, d), and charge-transfer (c, e) plot for La2LiHO3(all units are in e).
Table 2. Considered Migration Pathwaysa
pathnumber
distance(Å)
migrationion transport sites
barrier height(eV)
1 2.593 H H−O1 0.682 3.764 H H−H 8.13 2.927 H H−O2 8.524 2.202 H H−Hi into RS layer 2.95 3.575 H Hi−Hi
(along x in RS)2.0
6 3.781 H Hi−Hi(along y in RS)
1.93
7 2.593 O O1−H 1.28 3.568 O O1−O1 5.959 3.568 O O2−O2 4.810 3.764 O O2−O2 3.111 3.211 O O2−O2 4.06
aMigration distances, involved sites, and calculated migration barrierheights (in eV) for migration in the La2LiHO3 lattice are listed. Thepath number corresponds to the paths illustrated in Figure 4. RS refersto the rock salt layer.
The Journal of Physical Chemistry Letters Letter
DOI: 10.1021/acs.jpclett.7b03098J. Phys. Chem. Lett. 2018, 9, 353−358
356

Remarkably, however, this pathway was found to be lessfavorable with barrier heights of 2.9 eV for migration into therock salt layer and 2.0 eV (x direction) and 1.93 eV (ydirection) for migration within the rock salt layer. We believethat the unexpectedly high barrier heights for migration intoand within the rock salt layer for the hydride anion can beexplained by the covalent character of the lanthanum−oxygenbonds. The covalent character of these bonds creates channelsof electron density in the structure, preventing hydride anionsfrom migrating into and within the rock salt layer. Thishindrance for migration into the rock salt layer results in the in-plane migration becoming the more favorable energy pathwayfor hydride migration.This striking finding has led us to question what the main
differences between La2LiHO3 and the oxide-conductingLa2NiO4, La2CoO4, and La4Co3O10 are, and what aspectspromote a covalent character in the rock salt layer (thushindering ion mobility). The first clear difference is in thecharge and electronegativity of the octahedral-coordinated B-site cation (lithium versus nickel and cobalt). The second is inthe basal plane of the octahedron in La2LiHO3 there are twohydride anions, partly binding to the lanthanum cations. Themore ionic lithium−oxygen bonding may strengthen covalent
lanthanum−oxygen interactions in the rock salt layer. Likewisethe more ionic lanthanum−hydrogen interactions may have asimilar impact on the lanthanum−oxygen bonding. Theseconsiderations are strong indications that the hydride/oxideanion conductivity in the rock salt layer may be tuned byadequate choices of A and B site cations to give lower degree ofcovalent bonding in the rock salt layer. This idea can act as theguiding principle for the synthesis of novel functionaloxyhydrides. From these guidelines we thus proposeBa2MgH2O2 and Sr2MgH2O2 as promising oxyhydridecandidates for improved hydride conductivity. We justify thisstatement on the basis that magnesium at the B-site providessimilar bonding conditions as nickel and cobalt in La2NiO4 andLa2CoO4. This action should tune the bonding in the rock saltlayer to be more ionic due to higher charge density ofmagnesium compared with lithium and ergo facilitate transportin the layer.Finally, the migration properties for the oxide anions have
been considered. The lowest barrier height migration pathwayfor the oxide anions was found to be 1.20 eV for the in-planemigration between the H and O1 positions, similar to thefavorable hydride migration pathway (0.68 eV). The fact that allbarrier heights for oxide migration are higher than that forhydride migration is compelling evidence that La2LiHO3 isindeed a hydride conductor, where the migration of hydrideanions is the dominant anionic transport mechanism.In summary, we have discussed the microscopic under-
pinnings of the vibrations, bonding, and migration through theuse of an experimentally validated DFT model. Furthermore,these microscopic features provide a fully consistent story ofthe structure and dynamics, which gives us great confidence inthe chosen model. Several different charge-density analyses ofthe bonding have all shown the perovskite layer to bedominated by ionic bonding, while the rock salt layer ischaracterized by covalent bonding. Crucially, this covalentcharacter of the bonding in the rock salt layer is seen to hinderhydride migration into and within the rock salt layer, which inother similar structures has been shown to be the favorableanion migration path. The result of this revelation is that in-plane migration of hydride anions becomes the favorablemigration pathway and the dominant anionic transportmechanism with a barrier height of 0.68 eV. This insight intothe precise bonding mechanisms and its resultant effects on thehydride migration provide us with an intelligent designprinciple that has allowed us to propose Ba2MgH2O2 and
Figure 4. Sketch of the considered migration pathways in La2LiHO3. The atoms are lanthanum (green), lithium (purple), hydrogen (white), andoxygen (red). Distances and barrier heights are summarized in Table 2 and minimal energy pathways are illustrated in Figure 5 and Figure S3. Thedashed circles are interstitial hydride anions in the rock salt layer (both interstitial positions in the Figure are equivalent).
Figure 5. Minimal energy path from cNEB calculation of selectedmigration paths;, the other paths are illustrated in Figure S3. The pathsare described in Table 2 and illustrated in Figure 4.
The Journal of Physical Chemistry Letters Letter
DOI: 10.1021/acs.jpclett.7b03098J. Phys. Chem. Lett. 2018, 9, 353−358
357

Sr2MgH2O2 as new materials, which we anticipate to havedrastically improved hydride-transport properties.
■ ASSOCIATED CONTENT
*S Supporting InformationThe Supporting Information is available free of charge on theACS Publications website at DOI: 10.1021/acs.jpclett.7b03098.
Experimental methods, including sample synthesis,structural and spectroscopic characterization, as well ascomputational methods. List of calculated Ramanfrequencies, crystallographic information from Rietveldrefinement, calculated formation energies, calculatedBEC, complete list of Bader analysis, structuralparameters from DFT, comparison of calculated INSspectra with different functionals, Rietveld refinement ofLa2LiHO3, calculated total and site-projected phonondensity of states, and electron density map. (PDF)
■ AUTHOR INFORMATION
Corresponding Author*E-mail: [email protected].
ORCIDØystein S. Fjellvåg: 0000-0003-0215-5260Jeff Armstrong: 0000-0002-8326-3097NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTS
This work is a part of the FOXHOUND project funded by TheFaculty of Mathematics and Natural Sciences, University ofOslo via the Strategic Research Initiative program. Wegratefully acknowledge the use of the Norwegian Center forX-ray Diffraction, Scattering and Imaging (RECX) andgratefully acknowledge ISIS neutron and muon source forbeamtime (TOSCA instrument). P.V. and Ø.S.F. acknowledgethe Research Council of Norway for providing the computertime (under the project number NN2875k) at the Norwegiansupercomputer.
■ REFERENCES(1) Hayward, M. A.; Cussen, E. J.; Claridge, J. B.; Bieringer, M.;Rosseinsky, M. J.; Kiely, C. J.; Blundell, S. J.; Marshall, I. M.; Pratt, F.L. The Hydride Anion in an Extended Transition Metal Oxide Array:LaSrCoO3H0.7. Science 2002, 295, 1882−1884.(2) Kobayashi, G.; Hinuma, Y.; Matsuoka, S.; Watanabe, A.; Iqbal,M.; Hirayama, M.; Yonemura, M.; Kamiyama, T.; Tanaka, I.; Kanno,R. Pure H− conduction in oxyhydrides. Science 2016, 351, 1314−1317.(3) Schwarz, H. Neuartige Hydrid-Oxide der Seltenen Erden:Ln2LiHO3 mit Ln = La, Ce, Pr und Nd. Dissertation. University ofKarlsruhe, 1991.(4) Fjellvåg, Ø. S.; Armstrong, J.; Sławinski, W. A.; Sjåstad, A. O.Thermal and Structural Aspects of the Hydride-Conducting Oxy-hydride La2LiHO3 Obtained via a Halide Flux Method. Inorg. Chem.2017, 56, 11123−11128.(5) Zhang, J.; Gou, G.; Pan, B. Study of Phase Stability and HydrideDiffusion Mechanism of BaTiO3 Oxyhydride from First-Principles. J.Phys. Chem. C 2014, 118, 17254−17259.(6) Liu, X.; Bjorheim, T. S.; Haugsrud, R. Formation and migrationof hydride ions in BaTiO3-xHx oxyhydride. J. Mater. Chem. A 2017, 5,1050−1056.
(7) Ke, X.; Kuwabara, A.; Tanaka, I. Cubic and orthorhombicstructures of aluminum hydride AlH3 predicted by a first-principlesstudy. Phys. Rev. B: Condens. Matter Mater. Phys. 2005, 71, 184107.(8) Vajeeston, P.; Ravindran, P.; Fjellvåg, H. Phonon, IR, and RamanSpectra, NMR Parameters, and Elastic Constant Calculations for AlH3Polymorphs. J. Phys. Chem. A 2011, 115, 10708−10719.(9) Wang, C.-Z.; Yu, R.; Krakauer, H. First-principles calculations ofphonon dispersion and lattice dynamics in La2CuO4. Phys. Rev. B:Condens. Matter Mater. Phys. 1999, 59, 9278−9284.(10) Fajans, K. II. Die Eigenschaften salzartiger Verbindungen undAtombau. Z. Kristallogr. - Cryst. Mater. 1924, 61, 18−48.(11) Fajans, K. Struktur und Deformation der Elektronenhullen inihrer Bedeutung fur die chemischen und optischen Eigenschaftenanorganischer Verbindungen. Naturwissenschaften 1923, 11, 165−172.(12) Fajans, K.; Joos, G. Molrefraktion von Ionen und Molekulen imLichte der Atomstruktur. Eur. Phys. J. A 1924, 23, 1−46.(13) Minervini, L.; Grimes, R. W.; Kilner, J. A.; Sickafus, K. E.Oxygen migration in LaNiO. J. Mater. Chem. 2000, 10, 2349−2354.(14) Kushima, A.; Parfitt, D.; Chroneos, A.; Yildiz, B.; Kilner, J. A.;Grimes, R. W. Interstitialcy diffusion of oxygen in tetragonalLa2CoO4+δ. Phys. Chem. Chem. Phys. 2011, 13, 2242−2249.(15) Cleave, A. R.; Kilner, J. A.; Skinner, S. J.; Murphy, S. T.; Grimes,R. W. Atomistic computer simulation of oxygen ion conductionmechanisms in La2NiO4. Solid State Ionics 2008, 179, 823−826.(16) Nagell, M. U.; Sławinski, W. A.; Vajeeston, P.; Fjellvåg, H.;Sjåstad, A. O. Temperature induced transitions in La4(Co1−xNix)3O10+δ; oxygen stoichiometry and mobility. Solid State Ionics2017, 305, 7−15.
The Journal of Physical Chemistry Letters Letter
DOI: 10.1021/acs.jpclett.7b03098J. Phys. Chem. Lett. 2018, 9, 353−358
358

1
New Insights into Hydride Bonding, Dynamics, and
Migration in La2LiHO3 Oxyhydride
Øystein S. Fjellvåg,† Jeff Armstrong,
‡* Ponniah Vajeeston,
† and Anja O. Sjåstad
†
† Centre for Materials Science and Nanotechnology, Department of Chemistry, University of Oslo, P.O.
Box 1033, N-0315 Oslo, Norway
‡ ISIS Facility, Rutherford Appleton Laboratory, Harwell Oxford, Didcot, Oxfordshire OX11 0QX, U.K.
*E-mail: [email protected]

2
Experimental Methods
La2LiHO3 samples were synthesized using the LiCl halide flux method described on ref1 by
reacting La2O3 and LiH (1:4 molar ratio) in LiCl (60 wt.%) at 750 °C for 48 hours. The flux was
removed by washing with methanol. Samples were stored in an argon filled glovebox after
preparation. Sample purity was assured from Rietveld refinements using the TOPAS2 software of
XRD patterns collected on a Bruker AXS D8 Discover instrument with Cu Kα1 radiation from a
Ge(111) Johansson monochromator and a LynxEye strip detector.
High-resolution INS measurements on a 5 g powder sample in a flat aluminum cell of cross-
sectional area 4×4.8 cm2 were performed at the inverted-geometry neutron spectrometer
TOSCA3-5
, spanning energy transfers up to 4000 cm–1
, at the ISIS Pulsed Neutron & Muon
Source, Rutherford Appleton Laboratory, United Kingdom. The spectral resolution of TOSCA is
similar to that of infrared and Raman techniques, approximately 2 % of the energy in question.
The sample was studied at a temperature of ~10 K using a dedicated closed-cycle helium
refrigerator, so as to minimize the effect of the Debye Waller factor, thus allowing us to fully
distinguish the individual peaks. Data were reduced with the Mantid software.6
Raman spectroscopy was performed using a Bruker Senterra high performance Raman
microscope spectrometer with a 532 nm laser. Measurements were performed at room
temperature.
Computational Methods
The phonon dispersion spectra, phonon density of states and Raman spectra for La2LiHO3
obtained from density functional perturbation theory as implemented in the CASTEP package.7
For the CASTEP computation we have used the optimized VASP structures with a similar k-
point mesh as input with Norm-conserving pseudopotentials (energy cut-off 800 eV) and the

3
GGA exchange correlation functional proposed by PBE. In addition to the GGA-PBE functional,
the GGA-PW91 and LDA functionals were tested and gave similar lattice parameters, atomic
positions and vibrational spectra parameters as GGA-PBE, Table S1 and Figure S4. The PBE
functional was further used as it reproduced all the essential features required for our study.
Phonon calculation was weighted with the neutron scattering cross sections in the Mantid
software using an algorithm that outputs a DOS spectra that can be directly can compared to
measured INS spectra at TOSCA according to the instrument resolution.6
Total energies were calculated by the projected augmented plane-wave (PAW) implementation
of the Vienna ab initio package (VASP) 8-9
. All calculations were performed with the PBE10
exchange correlation functional. The ground-state geometries were determined by minimizing
stresses and Hellman-Feynman forces using the conjugate-gradient algorithm with force
convergence < 10–3
eV Å–1
. Brillouin zone integration was performed with a Gaussian
broadening of 0.1 eV during all relaxations. 512 k points in the whole Brillouin zone for the
structure with a 600 eV plane-wave cutoff were found sufficient to ensure the required accuracy.
The k-points were generated using the Monkhorst-Pack method with a grid size of 88×8 for
structural optimization. A similar density of k-points and energy cutoffs were used to estimate
total energy as a function of volume curve for Immm and I4/mmm phases. In the I4/mmm phase
O1 and H partially occupy the 4c site with the occupancy 0.5 and 0.5, respectively. In order to
model this system with the La2LiHO3 composition a supercell with the 332 dimension was
constructed from the experimental structure11
and the considered occupancy is 0.5 and 0.5 for H
and O1, respectively. Iterative relaxation of atomic positions was stopped when the change in
total energy between successive steps was < 1 meV/cell.

4
The diffusion barriers for the La2LiHO3 phase was investigated with the cNEB method in a
supercell approach 12-13
. A supercell with 3×3×1 dimensions was used to ensure that the atoms
are separated from their periodic image, providing a more accurate answer for the activation
barrier in the diluted limit. The formation energy is calculated by using periodically repeated
finite-sized supercells, artificial long-range elastic and electrostatic interactions between the
periodic defect images can be introduced. However, when supercells of sufficiently large sizes
are used, the interaction of the defect with the spurious periodic images and with the jellium
background will become negligible. The formation energy of O2–
, Li+, H– defects are calculated
in the following manner.
∆𝐻𝑑𝑒𝑓 = (𝐸𝑡𝑜𝑡𝑑𝑒𝑓 − 𝐸𝑡𝑜𝑡
𝑏𝑢𝑙𝑘 ) ± ∑ 𝑛𝑖𝜇𝑖𝑖
where 𝐸𝑡𝑜𝑡𝑑𝑒𝑓
is the total energy of the supercell containing O/Li/H defect and 𝐸𝑡𝑜𝑡𝑏𝑢𝑙𝑘
is the total
energy of the ideal supercell. µi indicates the number of atoms of type i that have been added to
(ni > 0) or removed from (ni < 0) the supercell when the defect is created. The μi is the
corresponding chemical potentials of the defect species (O/Li/H atom) that determine the flow of
atoms between the atomic reservoirs and the host crystal. To determine the minimum energy
path (MEP) through the cNEB method, six replicas of the system were created, in each of which
the diffusing O atom was moved by equidistant steps to positions between the initial and final
states for the path obtained by linear interpolation.
5
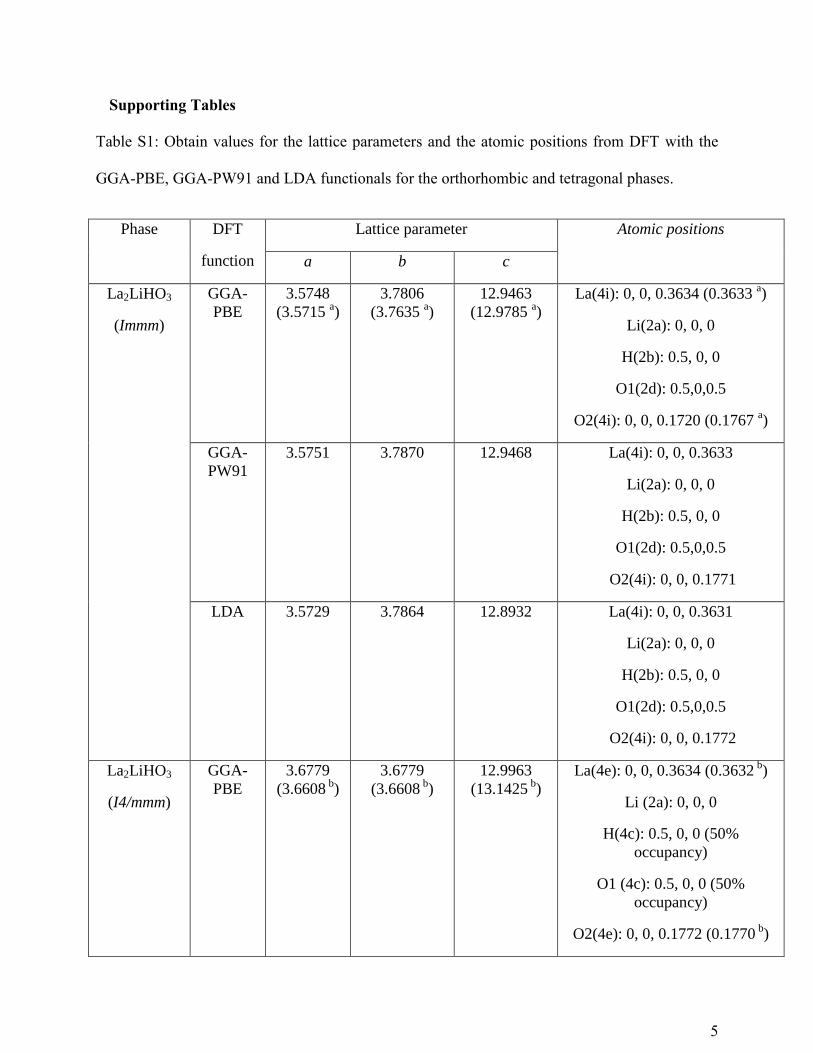
Supporting Tables
Table S1: Obtain values for the lattice parameters and the atomic positions from DFT with the
GGA-PBE, GGA-PW91 and LDA functionals for the orthorhombic and tetragonal phases.
Phase DFT
function
Lattice parameter Atomic positions
a b c
La2LiHO3
(Immm)
GGA-
PBE
3.5748
(3.5715 a)
3.7806
(3.7635 a)
12.9463
(12.9785 a)
La(4i): 0, 0, 0.3634 (0.3633 a)
Li(2a): 0, 0, 0
H(2b): 0.5, 0, 0
O1(2d): 0.5,0,0.5
O2(4i): 0, 0, 0.1720 (0.1767 a)
GGA-
PW91
3.5751 3.7870 12.9468
La(4i): 0, 0, 0.3633
Li(2a): 0, 0, 0
H(2b): 0.5, 0, 0
O1(2d): 0.5,0,0.5
O2(4i): 0, 0, 0.1771
LDA 3.5729 3.7864 12.8932
La(4i): 0, 0, 0.3631
Li(2a): 0, 0, 0
H(2b): 0.5, 0, 0
O1(2d): 0.5,0,0.5
O2(4i): 0, 0, 0.1772
La2LiHO3
(I4/mmm)
GGA-
PBE
3.6779
(3.6608 b
)
3.6779
(3.6608 b
)
12.9963
(13.1425 b
)
La(4e): 0, 0, 0.3634 (0.3632 b
)
Li (2a): 0, 0, 0
H(4c): 0.5, 0, 0 (50%
occupancy)
O1 (4c): 0.5, 0, 0 (50%
occupancy)
O2(4e): 0, 0, 0.1772 (0.1770 b
)

6
GGA-
PW91
3.6792 3.6792 13.0032 La(4e): 0, 0, 0.3631
Li (2a): 0, 0, 0
H(4c): 0.5, 0, 0 (50%
occupancy)
O1 (4c): 0.5, 0, 0 (50%
occupancy)
O2(4e): 0, 0, 0.1774
LDA 3.6520 3.6520 12.8637 La(4e): 0, 0, 0.3633
Li (2a): 0, 0, 0
H(4c): 0.5, 0, 0 (50%
occupancy)
O1 (4c): 0.5, 0, 0 (50%
occupancy)
O2(4e): 0, 0, 0.1764
a from ref.
1
b from ref.
11
Table S2: Calculated vibrational frequencies for Raman at the Γ point of the Brillouin Zone for
La2LiHO3. O2 refers to the axial oxygen.
Energy (cm-1
) Mode Raman intensity Vibration
132.8801 B1g 2.395793 La in y
174.9199 B2g 0.581638 La in x
295.9512 B2g 26.50779 La in z
300.6237 Ag 66.07859 O2 in x
341.4462 B1g 64.53504 O2 in y
603.8718 Ag 176.1462 O2 in z

7
Table S3: Crystallographic data of La2LiHO3 in space group Immm [a = 3.5684(5) Å, b =
3.7642(6) Å and c = 12.963(2) Å] from powder XRD. The cell volume is 87.06 Å3 f.u.
–1. The
same thermal displacement parameter Uiso was used for all atoms.
Atom Wyckoff x y y Occupancy Uiso
La 4i 0 0 0.3635(9) 1 0.004(5)
Li 2a 0 0 0 1 0.004(5)
H 2b 0.5 0 0 1 0.004(5)
O1 2d 0.5 0 0.5 1 0.004(5)
O2 4i 0 0 0.179(7) 1 0.004(5)
Table S4: Calculated formation energies (Eform) for the considered defects and interstitial atoms
in the La2LiHO3 lattice. RS refers to rock salt layer.
Eform (eV)
Hydrogen Lithium Oxygen
VH = 0.804 VLi = 3.47 VO1 = 6.04
Hi = 1.5 (Hi in RS) VO2 = 6.59
Table S5: Calculated Born-Effective-Charge tensor elements (Z) for the constituents of
La2LiHO3.
Born-Effective-Charge
Position xx yy zz xy xz yx yz zx zy
La (4i) 4.18 3.56 3.70 0 0 0 0 0 0
Li (2a) 0.89 0.79 0.92 0 0 0 0 0 0

8
H (2b) -0.95 -0.89 -0.53 0 0 0 0 0 0
O1(4i) -2.89 -2.47 -2.5 0 0 0 0 0 0
O2(2d) -2.5 -2.1 -2.81 0 0 0 0 0 0
Table S6: Complete list of calculated values from Bader analysis.
Atom x y z VORONOI BADER % MIN DIST
La 7.3127 0.0000 0.0000 8.2509 9.0934 10.8254 1.29
La 4.1640 0.0000 0.0000 8.2508 9.0964 10.8291 1.2948
La 1.5065 1.6304 1.6314 7.9616 9.2970 11.0678 1.0937
La 9.9702 1.6304 1.6314 7.9632 9.2776 11.0447 1.0937
O 5.7384 0.0000 1.6314 7.2922 7.3582 8.7598 1.2585
O 9.6796 0.0000 0.0000 7.6188 7.3560 8.7571 0.9824
O 1.7971 0.0000 0.0000 7.6188 7.3555 8.7565 0.9824
O 3.5859 1.6304 1.6314 7.7362 7.2918 8.6808 0.8961
O 7.8908 1.6304 1.6314 7.7362 7.2918 8.6808 0.8961
O 5.7384 1.6304 0.0000 7.2303 7.3376 8.7353 1.2585
Li 0.0000 0.0000 0.0000 0.8933 0.8000 0.0000 0.0000
Li 5.7384 1.6304 1.6314 0.9360 0.8300 0.0000 0.0000
H 11.4767 0.0000 1.6314 2.2749 1.6176 1.9257 0.9012
H 11.4767 1.6304 0.0000 2.2368 1.6271 1.9370 0.9012
9
Supporting Figures
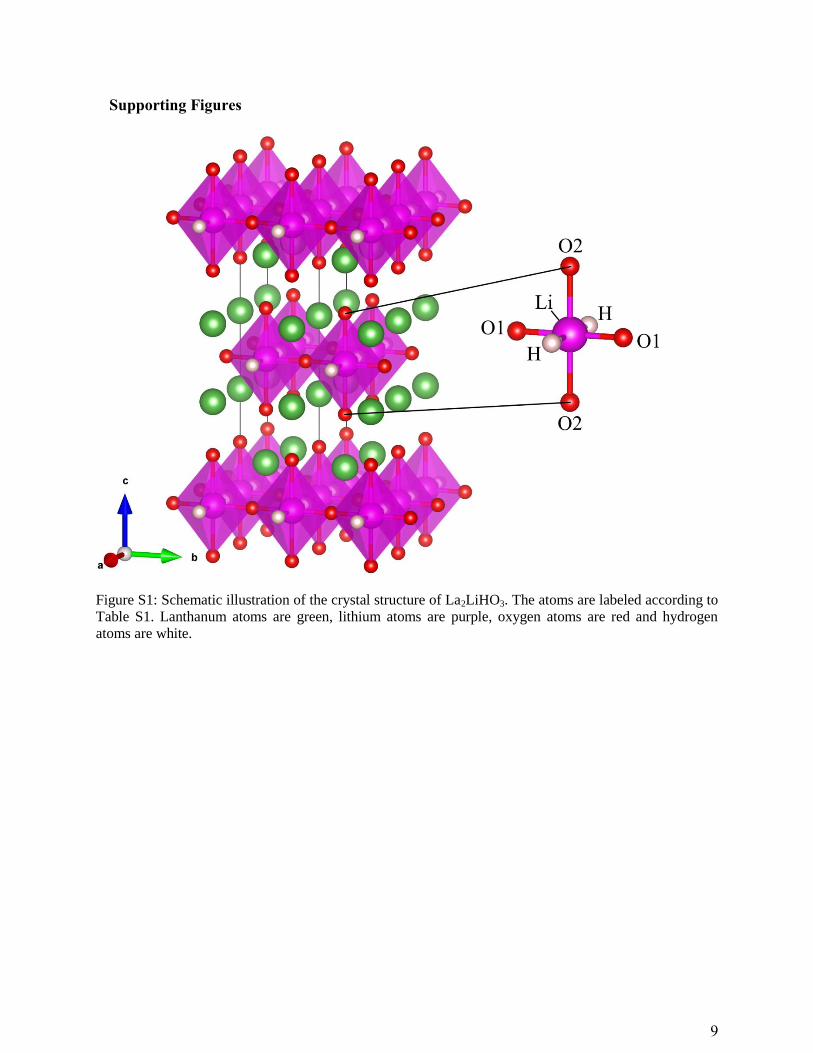
Figure S1: Schematic illustration of the crystal structure of La2LiHO3. The atoms are labeled according to
Table S1. Lanthanum atoms are green, lithium atoms are purple, oxygen atoms are red and hydrogen
atoms are white.

10
Figure S2: Observed (black), calculated (red) and difference (blue) intensity profiles based on
Rietveld refinements of powder XRD of La2LiHO3 in space group Immm. Ticks indicate the
position of the Bragg reflections originating from La2LiHO3.

11
Figure S3: Minimal energy path from cNEB calculation of selected migration paths, the other
paths are illustrated in Figure 5. The paths are described in Table 2 and visualized Figure 4.

12
Figure S4: Comparison of the calculated INS spectra from the GGA-PBE, GGA-PW91 and LDA
functional. The phonon spectra are weighted with the neutron scattering cross sections to
compare with INS measurements. All the three functionals give good agreement with the
measured INS spectra.
13
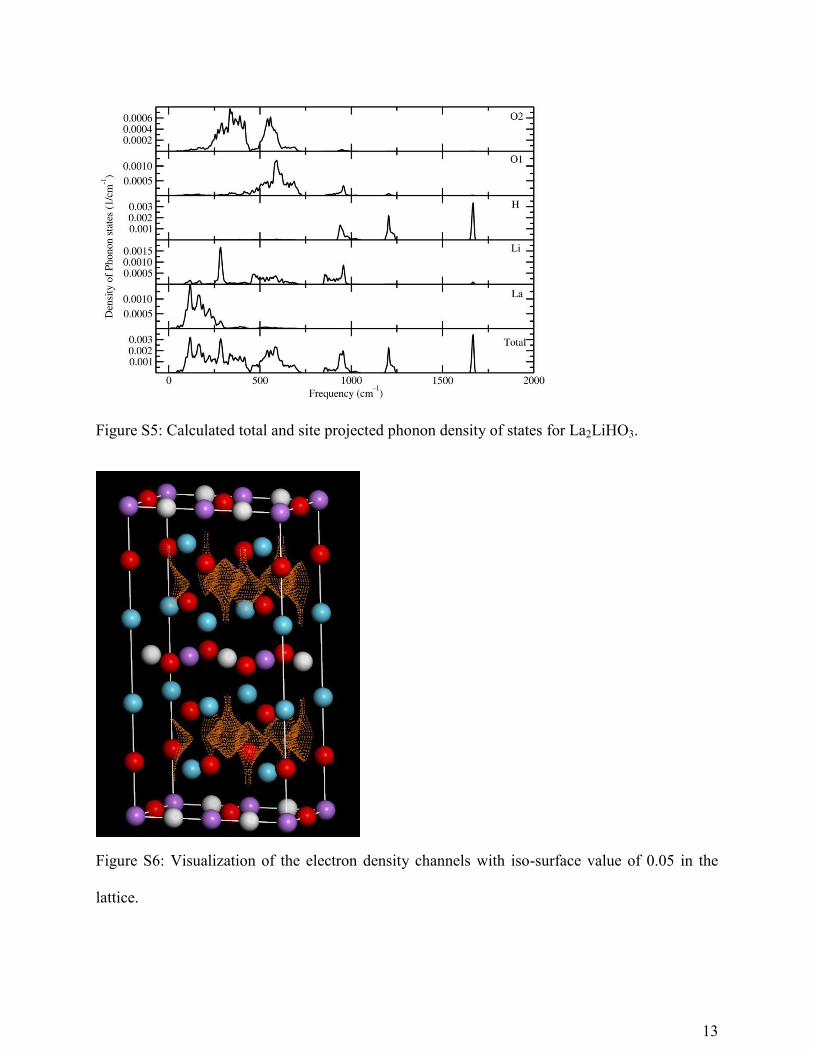
Figure S5: Calculated total and site projected phonon density of states for La2LiHO3.
Figure S6: Visualization of the electron density channels with iso-surface value of 0.05 in the
lattice.

14
References
1. Fjellvåg, Ø. S.; Armstrong, J.; Sławiński, W. A.; Sjåstad, A. O., Thermal and Structural
Aspects of the Hydride-Conducting Oxyhydride La2LiHO3 Obtained via a Halide Flux Method.
Inorg. Chem. 2017, 56, 11123-11128.
2. Bruker-AXS TOPAS V5: General profile and structure analysis software for powder
diffraction data. - User's Manual, Bruker AXS, Karlsruhe, Germany., 2013.
3. Pinna, R. S.; Rudić, S.; Capstick, M. J.; McPhail, D. J.; Pooley, Daniel E.; Howells, G.
D.; Gorini, G.; Fernandez-Alonso, F., Detailed characterisation of the incident neutron beam on
the TOSCA spectrometer. Nuclear Instruments and Methods in Physics Research Section A:
Accelerators, Spectrometers, Detectors and Associated Equipment 2017, 870, 79-83.
4. Pinna, R. S.; Rudić, S.; Parker, S. F.; Gorini, G.; Fernandez-Alonso, F., Monte carlo
simulations of the TOSCA spectrometer: Assessment of current performance and future
upgrades. EPJ Web of Conferences 2015, 83, 03013.
5. Stewart, F. P.; Felix, F.-A.; Anibal, J. R.-C.; John, T.; Svemir, R.; Roberto, S. P.;
Giuseppe, G.; Javier Fernández, C., Recent and future developments on TOSCA at ISIS. Journal
of Physics: Conference Series 2014, 554, 012003.
6. Arnold, O.; Bilheux, J. C.; Borreguero, J. M.; Buts, A.; Campbell, S. I.; Chapon, L.;
Doucet, M.; Draper, N.; Ferraz Leal, R.; Gigg, M. A.; Lynch, V. E.; Markvardsen, A.;
Mikkelson, D. J.; Mikkelson, R. L.; Miller, R.; Palmen, K.; Parker, P.; Passos, G.; Perring, T. G.;
Peterson, P. F.; Ren, S.; Reuter, M. A.; Savici, A. T.; Taylor, J. W.; Taylor, R. J.; Tolchenov, R.;
Zhou, W.; Zikovsky, J., Mantid—Data analysis and visualization package for neutron scattering

15
and SR experiments. Nuclear Instruments and Methods in Physics Research Section A:
Accelerators, Spectrometers, Detectors and Associated Equipment 2014, 764, 156-166.
7. Refson, K.; Tulip, P. R.; Clark, S. J., Variational density-functional perturbation theory
for dielectrics and lattice dynamics. Phys. Rev. B 2006, 73, 155114.
8. Kresse, G.; Furthmüller, J., Efficient iterative schemes for <i>ab initio</i> total-energy
calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169-11186.
9. Kresse, G.; Furthmüller, J., Efficiency of ab-initio total energy calculations for metals
and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15-50.
10. Perdew, J. P.; Burke, K.; Ernzerhof, M., Generalized Gradient Approximation Made
Simple. Phys. Rev. Lett. 1996, 77, 3865.
11. Kobayashi, G.; Hinuma, Y.; Matsuoka, S.; Watanabe, A.; Iqbal, M.; Hirayama, M.;
Yonemura, M.; Kamiyama, T.; Tanaka, I.; Kanno, R., Pure H– conduction in oxyhydrides.
Science 2016, 351, 1314-1317.
12. Jonsson, H.; Mills, G.; Jacobsen, K. W., Nudged elastic band method for finding
minimum energy paths of transitions. In Classical and Quantum Dynamics in Condensed Phase
Simulations, WORLD SCIENTIFIC: 2011; pp 385-404.
13. Henkelman, G.; Jónsson, H., Improved tangent estimate in the nudged elastic band
method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000, 113, 9978-
9985.