new zealand data sheet apo- capecitabine -...
TRANSCRIPT

New Zealand Data Sheet
APO- CAPECITABINE
Capecitabine 150mg/500mg film-coated Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 1
Presentation
APO-CAPECITABINE 150 mg tablets: Light peach, oval, biconvex, film-coated tablet,engraved “APO” on one side, “C150” on the other side.
APO-CAPECITABINE 500 mg tablets: Peach, oval, biconvex, film-coated tablet,engraved “APO” on one side, “C500” on the other side.
Tablet cannot be halved.
Indications
Colon CancerCapecitabine is indicated for the adjuvant treatment of patients with Dukes' stage C andhigh-risk stage B, colon cancer, either as monotherapy or in combination with oxaliplatin.
Colorectal CancerCapecitabine is indicated for first-line treatment of patients with metastatic colorectalcancer.
Oesophagogastric CancerCapecitabine is indicated for the first-line treatment of patients with advancedoesophagogastric cancer in combination with a platinum-based regimen.
Breast CancerCapecitabine is indicated for the treatment of patients with locally advanced ormetastatic breast cancer after failure of taxanes and an anthracycline containingchemotherapy regimen unless therapy with these and other standard agents areclinically contraindicated.
Capecitabine in combination with docetaxel is indicated for the treatment of patients withlocally advanced or metastatic breast cancer after failure of prior anthracyclinecontaining chemotherapy.
Dosage and Administration
Standard DosageCapecitabine tablets should be swallowed with water within 30 minutes after a meal. Dono halve the tablet.
Monotherapy
Colon, colorectal and breast cancer
The recommended monotherapy starting dose of Capecitabine is 1250 mg/m2
administered bd (morning and evening; equivalent to 2500 mg/m2 total daily dose) for 14days followed by a 7-day rest period.

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 2
Combination therapy
Breast cancer
In combination with docetaxel, the recommended starting dose of Capecitabine is 1250mg/m2 bd for 14 days followed by a 7-day rest period, combined with docetaxel at 75mg/m2 as a 1-hour IV infusion every 21 days.
Pre-medication according to the docetaxel labelling should be started prior to docetaxeladministration for patients receiving the Capecitabine plus docetaxel combination.
Colorectal cancer
In combination with oxaliplatin with or without bevacizumab the recommended startingdose of Capecitabine is 1000 mg/m2 bd for 2 weeks followed by a 7-day rest period. Thefirst dose of Capecitabine is given on the evening of day 1 and the last dose is given onthe morning of day 15. Given as a 3 week cycle, on day 1 every 3 weeks bevacizumab isadministered as a 7.5 mg/kg IV infusion over 30 to 90 minutes followed by oxaliplatinadministered as a 130 mg/m2 IV infusion over 2 hours.
Colon cancer
In combination with oxaliplatin the recommended starting dose of Capecitabine is 1000mg/m2 bd for 2 weeks followed by a 7-day rest period. The first dose of Capecitabine isgiven on the evening of day 1 and the last dose is given on the morning of day 15. Givenas a 3 week cycle, on day 1 oxaliplatin is administered as a 130 mg/m2 IV infusion over 2hours. Adjuvant treatment is recommended for a total of 24 weeks.
Oesophagogastric cancer
In triplet combination with epirubicin and cisplatin/oxaliplatin for oesophagogastriccancer, the recommended starting dose of Capecitabine is 625 mg/m2 bd as acontinuous regimen. Epirubicin is administered as a 50 mg/m2 IV bolus on day 1 of a 3week cycle. Platinum therapy should consist of either cisplatin administered at a dose of60 mg/m2 given as a 2 hour IV infusion on day 1 of a 3 week cycle; or oxaliplatinadministered at a dose of 130 mg/m2 given as a 2 hour IV infusion on day 1 of a 3 weekcycle.
In combination with cisplatin (doublet) with or without trastuzumab for gastric cancer, therecommended starting dose of Capecitabine is 1000 mg/m2 bd for 2 weeks followed by a7 day rest period. The first dose of Capecitabine is given on the evening of day 1 and thelast dose is given on the morning of day 15. Cisplatin is administered at a dose of 80mg/m2 as a 2 hour IV infusion on day 1 of a 3-week cycle.
Premedication to maintain adequate hydration and anti-emesis according to the cisplatinand oxaliplatin product information should be started prior to cisplatin/oxaliplatinadministration for patients receiving Capecitabine in combination with these agents.

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 3
Capecitabine dose is calculated according to body surface area. Tables 1 and 2 showexamples of the standard and reduced dose calculations (see Dosage adjustmentsduring treatment) for a starting dose of capecitabine of either 1250 mg/m2 or 1000mg/m2.
Table 1: Standard and reduced dose calculations according to body surface area for astarting dose of Capecitabine of 1250 mg/m
2
Dose level 1250 mg/m2
bd
Full dose
1250 mg/m2
bd
Number of 150 mgtablets and/or 500 mg
tablets peradministration (eachadministration to begiven morning and
evening)
Reduced dose(75%)
950 mg/m2
bd
Reduced dose(50%)
625 mg/m2
bd
BodySurfaceArea (m
2)
Dose peradministration (mg)
150 mg 500 mgDose per
administration(mg)
Dose peradministration
(mg)
<1.26 1500 - 3 1150 800
1.27 - 1.38 1650 1 3 1300 800
1.39 - 1.52 1800 2 3 1450 950
1.53 - 1.66 2000 - 4 1500 1000
1.67 - 1.78 2150 1 4 1650 1000
1.79 - 1.92 2300 2 4 1800 1150
1.93 - 2.06 2500 - 5 1950 1300
2.07 - 2.18 2650 1 5 2000 1300
>2.19 2800 2 5 2150 1450

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 4
Table 2: Standard and reduced dose calculations according to body surface area for astarting dose of Capecitabine of 1000 mg/m
2
Dose level 1000 mg/m2
bd
Full dose
1000 mg/m2
bd
Number of 150 mgtablets and/or 500 mg
tablets peradministration (eachadministration to begiven morning and
evening)
Reduced dose(75%)
750 mg/m2
bd
Reduceddose (50%)
500 mg/m2
bd
BodySurfaceArea (m
2)
Dose peradministration (mg)
150 mg 500 mgDose per
administration(mg)
Dose peradministration
(mg)
<1.26 1150 1 2 800 600
1.27 - 1.38 1300 2 2 1000 600
1.39 - 1.52 1450 3 2 1100 750
1.53 - 1.66 1600 4 2 1200 800
1.67 - 1.78 1750 5 2 1300 800
1.79 - 1.92 1800 2 3 1400 900
1.93 - 2.06 2000 - 4 1500 1000
2.07 - 2.18 2150 1 4 1600 1050
> 2.19 2300 2 4 1750 1100
Dosage Adjustment during Treatment
General
Toxicity due to capecitabine administration may be managed by symptomatic treatmentand/or modification of the capecitabine dose (treatment interruption or dose reduction).Once the dose has been reduced it should not be increased at a later time.
For those toxicities considered by the treating physician to be unlikely to become seriousor life threatening, treatment can be continued at the same dose without reduction orinterruption.
Dosage modifications are not recommended for Grade 1 events. Therapy withcapecitabine should be interrupted if a Grade 2 or 3 adverse experience occurs. Oncethe adverse event has resolved or decreased in intensity to Grade 1, capecitabinetherapy may be restarted at full dose or as adjusted according to Table 4. If a Grade 4experience occurs, therapy should be discontinued or interrupted until the experiencehas resolved or decreased to Grade 1, and therapy can then be restarted at 50% of theoriginal dose. Patients taking capecitabine should be informed of the need to interrupttreatment immediately if moderate or severe toxicity occurs. Doses of capecitabineomitted for toxicity are not replaced. For examples of toxicity grades for some relevantadverse events, please refer to Table 3.

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 5
Table 3: Toxicity Grades for Some Relevant Adverse Events
Intensity of Clinical Adverse Events*
AdverseEvents
Mild(Grade 1)
Moderate(Grade 2)
Severe(Grade 3)
Life-threatening(Grade 4)
Diarrhoea Increase of 1 - 3stools/day
Increase of 4 - 6stools/day, ornocturnal stools
Increase of7 - 9 stools/dayorincontinence andmalabsorption
Increase of ≥ 10 stools/dayor grossly bloodydiarrhoeaor the need forparenteralsupport
Nausea No significanteffect on foodintake
Food intakesignificantlydecreased butableto eatintermittently
Unable to eat
Vomiting 1 episode in a24-hour period
2 - 5 episodes ina24-hour period
6 - 10 episodesin a24-hour period
> 10 episodes ina 24-hourperiod or theneed forparenteralsupport
Hand-footsyndrome++(clinicaldomain)
Numbness,tingling, painlesserythema, andswelling
Painful erythemaand swelling
Moistdesquamation,ulceration,blistering,and severe pain
Hand-footsyndrome++(functionaldomain)
Discomfortwhich does notdisrupt normalactivities
Discomfort whichaffects activitiesofdaily living
Severediscomfort,unable to work orperform activitiesofdaily living
Stomatitis Erythemawithout pain
Painfulerythema,oedema, orulcers,but able to eat
Painful erythemarestricting abilityto eat
*The intensity of clinical adverse events was graded on a 4-point scale following the National CancerInstitute of Canada Common Toxicity Criteria (NCIC CTC).++
Criteria as established by Roche protocol.

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 6
Haematology
Patients with baseline neutrophil counts of <1.5 x 109 /L and/or thrombocyte counts of<100 x 109 /L should not be treated with capecitabine. If unscheduled laboratoryassessments during a treatment cycle show grade 3 or 4 haematologic toxicity,treatment with capecitabine should be interrupted.
The following table shows the recommended dose modifications following toxicity withcapecitabine:
Table 4 Capecitabine dose reduced Schedule
Toxicity NCICGrades#
Dose changes within a treatmentcycle
Dose Adjustment for NextCycle (% of starting dose)
Grade 1 Maintain dose level Maintain dose level
Grade 2
1st appearance
2nd appearance
3rd appearance
4th appearance
Interrupt until resolved to Grade 0-1
Interrupt until resolved to Grade 0-1
Interrupt until resolved to Grade 0-1
Discontinue treatment permanently
100%
75%
50%
Not applicable
Grade 3
1st appearance
2nd appearance
3rd appearance
Interrupt until resolved to Grade 0-1
Interrupt until resolved to Grade 0-1
Discontinue treatment permanently
75%
50%
Not applicable
Grade 4
1st appearance Discontinue permanently
or
If physician deems it to be in thepatient’s best interest to continue,
interrupt until resolved to Grade 0-1
50%
2nd appearance Discontinue permanently Not applicable#
According to the National Cancer Institute of Canada Clinical Trial Group (NCIC CTG) Common ToxicityCriteria or the Common Terminology Criteria for Adverse Events (CTCAE) of the Cancer Therapy EvaluationProgram, US National Cancer Institute version 3.0. For hand-foot syndrome and hyperbilirubinaemia seeWarnings and Precautions.
General combination therapyDose modifications for toxicity when capecitabine is used in combination with othertherapies should be made according to Table 4 for capecitabine and according to theappropriate Prescribing information for the other agent(s).
At the beginning of a treatment cycle, if a treatment delay is indicated for eithercapecitabine or the other agent(s), then administration of all agents should be delayeduntil the requirements for restarting all medicines are met.

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 7
During a treatment cycle for those toxicities considered by the treating physician not tobe related to capecitabine, capecitabine should be continued and the dose of the otheragent adjusted according to the appropriate Prescribing Information.
If the other agent(s) have to be discontinued permanently, capecitabine treatment canbe resumed when the requirements for restarting capecitabine are met.
This advice is applicable to all indications and to all special populations.
Dosage Adjustments in Special Populations
Patients with hepatic impairment due to liver metastases
In patients with mild to moderate hepatic impairment due to liver metastases, no startingdose adjustment is necessary. However, such patients should be carefully monitored(see Pharmacokinetic Properties and Warnings and Precautions). Patients with severehepatic impairment have not been studied.
Patients with renal impairment
In patients with moderate renal impairment (creatinine clearance 30 – 50 mL/min[Cockroft and Gault]) at baseline, a dose reduction to 75% for a starting dose of 1250mg/m2 is recommended. In patients with mild renal impairment (creatinine clearance51 - 80 mL/min) no adjustment in starting dose isrecommended. Careful monitoring andprompt treatment interruption is recommended if the patient develops a Grade 2, 3, or 4adverse event with subsequent dose adjustment as outlined in Table 4 above (seePharmacokinetics in special populations). If the calculated creatinine clearancedecreases during treatment to a value below 30 mL/min, capecitabine should bediscontinued. The dose adjustment recommendations for patients with moderate renalimpairment apply both to monotherapy and combination use. For dosage calculations,see Tables 1 and 2.
Children
The safety and efficacy of Capecitabine in children have not been established.
Elderly
For Capecitabine monotherapy, no adjustment of the starting dose is needed. However,severe Grade 3 or 4 treatment-related adverse events were more frequent in patientsover 80 years of age compared to younger patients. When Capecitabine was used incombination with other agents, elderly patients (≥ 65 years) experienced more Grade 3 and Grade 4 adverse reactions and adverse reactions that led to discontinuation, thanyounger patients. Careful monitoring of elderly patients is advisable.
In combination with docetaxel, an increased incidence of Grade 3 or 4 treatment-relatedadverse events and treatment-related serious adverse events was observed in patients60 years of age or more. For patients 60 years of age or more treated with the

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 8
combination of capecitabine plus docetaxel, a starting dose reduction of capecitabine to75% (950 mg/m2 bd) is recommended. For dosage calculations, see Table 2.
In combination with irinotecan: for patients 65 years of age or more, a starting dosereduction of capecitabine to 800mg/m2 bd is recommended.
Maximum Tolerated Daily DoseNIL.
Contraindications
Capecitabine is contraindicated in patients with known hypersensitivity to capecitabine orto any of its components.
Capecitabine is contraindicated in patients who have a history of severe and unexpectedreactions to fluoropyrimidine therapy or with known hypersensitivity to fluorouracil.
As with other fluoropyrimidines, capecitabine is contraindicated in patients with knowndihydropyrimidine dehydrogenase (DPD) deficiency.
Capecitabine should not be administered concomitantly with sorivudine or its chemicallyrelated analogues, such as brivudine (see Interactions with other Medicinal Products andother Forms of Interaction).
Capecitabine is contraindicated in patients with severe renal impairment (creatinineclearance below 30 mL/min).
If contraindications exist to any of the agents in a combination regimen, that agentshould not be used.
Warnings and Precautions
Diarrhoea
Capecitabine can induce diarrhoea, which can sometimes be severe. Patients withsevere diarrhoea should be carefully monitored and, if they become dehydrated, shouldbe given fluid and electrolyte replacement. Standard anti-diarrhoea treatments (e.g.loperamide) should be initiated, as medically appropriate, as early as possible. Dosereduction should be applied as necessary (see Dosage and Administration).
Dehydration
Dehydration should be prevented or corrected at the onset. Patients with anorexia,asthenia, nausea, vomiting or diarrhoea may rapidly become dehydrated. If Grade 2 (orhigher) dehydration occurs, capecitabine treatment should be immediately interruptedand the dehydration corrected. Treatment should not be restarted until the patient is

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 9
rehydrated and any precipitating causes have been corrected or controlled. Dosemodifications should be applied for the precipitating adverse event as necessary (seeDosage and Administration).
Dehydration may cause acute renal failure, especially in patients with pre-existingcompromised renal function or when capecitabine is given concomitantly with knownnephrotoxic agents. Fatal outcome of renal failure has been reported in these situations(see Undesirable Effects, Post-marketing experience).
Precautions
The spectrum of cardiotoxicity observed with capecitabine is similar to that of otherfluorinated pyrimidines. This includes myocardial infarction, angina, dysrhythmias,cardiac arrest, cardiac failure and electrocardiographic changes. These adverse eventsmay be more common in patients with a prior history of coronary artery disease.
Rarely, unexpected, severe toxicity (e.g. stomatitis, diarrhoea, neutropenia andneurotoxicity) associated with 5-FU has been attributed to a deficiency ofdihydropyrimidine dehydrogenase (DPD) activity. A link between decreased levels ofDPD and increased, potentially fatal toxic effects of 5-FU therefore cannot be excluded.
Capecitabine can induce severe skin reactions such as Stevens-Johnson syndrome andToxic Epidermal Necrolysis (TEN). Capecitabine should be permanently discontinued inpatients who experience a severe skin reaction possibly attributable to capecitabinetreatment (see Undesirable Effects, Post-marketing experience).
Capecitabine can induce hand-foot syndrome (palmar-plantar erythrodysesthesia orchemotherapy-induced acral erythema), which is a cutaneous toxicity. For patientsreceiving capecitabine monotherapy in the metastatic setting, the median time to onsetwas 79 days, range from 11 to 360 days), with a severity range of Grades 1 to 3.
Grade 1 hand-foot syndrome is defined by numbness, dysaesthesia/paraesthesia,tingling, or erythema of the hands and/or feet and/or discomfort which does not disruptnormal activities. Grade 2 is defined as painful erythema and swelling of the handsand/or feet and/or discomfort affecting the patient’s activities of daily living. Grade 3 isdefined as moist desquamation, ulceration, blistering or severe pain of the hands and/orfeet and/or severe discomfort that causes the patient to be unable to work or performactivities of daily living.
If Grade 2 or 3 hand-foot syndrome occurs, administration of capecitabine should beinterrupted until the event resolves or decreases in intensity to Grade 1. Following Grade3 hand-foot syndrome, subsequent doses of capecitabine should be decreased (seeDosage and Administration). When capecitabine and cisplatin are used in combination,the use of vitamin B6 (pyridoxine) is not advised for symptomatic or secondaryprophylactic treatment of hand–foot syndrome, because of published reports that it maydecrease the efficacy of cisplatin. There is some evidence that dexpanthenol is effectivefor hand-foot syndrome prophylaxis in patients treated with capecitabine.

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 10
Capecitabine can induce hyperbilirubinaemia. Administration of capecitabine should beinterrupted if treatment-related elevations in bilirubin of > 3.0 x ULN or treatment-relatedelevations in hepatic aminotransferases (ALT, AST) of > 2.5 x ULN occur. Treatmentmay be resumed when bilirubin decreases to ≤3.0 x ULN or hepatic aminotransferases decrease to ≤2.5 x ULN.
In a medicine interaction study with single-dose warfarin administration, there was asignificant increase in the mean AUC (+ 57%) of S-warfarin. These results suggest aninteraction, probably due to an inhibition of the cytochrome P450 2C9 isoenzyme systemby capecitabine. Patients receiving concomitant capecitabine and oral coumarin-derivative anticoagulant therapy should have their anticoagulant response (INR orprothrombin time) monitored closely and the anticoagulant dose adjusted accordingly(see Interactions with other Medicinal Products and other Forms of Interaction).
General
Patients treated with capecitabine should be carefully monitored for toxicity. Mostadverse events are reversible and do not require permanent discontinuation of therapy,although doses may have to be withheld or reduced (see also Dosage andAdministration).
Interactions with other Medicinal Products and other Formsof Interaction
Coumarin anticoagulants
Altered coagulation parameters and/or bleeding have been reported in patients takingCapecitabine concomitantly with coumarin-derivative anticoagulants such as warfarinand phenprocoumon. These events occurred within several days and up to severalmonths after initiating capecitabine therapy and, in a few cases, within one month afterstopping capecitabine. In a clinical interaction study, after a single 20 mg dose ofwarfarin, capecitabine treatment increased the AUC of S-warfarin by 57% with a 91%increase in INR value. Patients taking coumarin-derivative anticoagulants concomitantlywith capecitabine should be monitored regularly for alterations in their coagulationparameters (PT or INR) and the anticoagulant dose adjusted accordingly.
Cytochrome P-450 2C9 substrates
No formal interaction studies with capecitabine and other medicines known to bemetabolised by the cytochrome P450 2C9 isoenzyme have been conducted. Careshould be exercised when capecitabine is co-administered with these medicines.
Phenytoin
Increased phenytoin plasma concentrations have been reported during concomitant useof Capecitabine with phenytoin. Formal interaction studies with phenytoin have not beenconducted, but the mechanism of interaction is presumed to be inhibition of the CYP2C9isoenzyme system by capecitabine (see Courmarin anticoagulants). Patients taking

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 11
phenytoin concomitantly with capecitabine should be regularly monitored for increasedphenytoin plasma concentrations.
Medicine-food interaction
In all clinical trials, patients were instructed to take capecitabine within 30 minutes after ameal. Since current safety and efficacy data are based upon administration with food, itis recommended that capecitabine be administered with food.
Antacid
The effect of an aluminium hydroxide and magnesium hydroxide-containing antacid onthe pharmacokinetics of capecitabine was investigated in cancer patients. There was asmall increase in plasma concentrations of capecitabine and one metabolite (5'-DFCR);there was no effect on the 3 major metabolites (5'-DFUR, 5-FU and FBAL).
Leucovorin (folinic acid)
The effect of leucovorin on the pharmacokinetics of capecitabine was investigated incancer patients. Leucovorin has no effect on the pharmacokinetics of capecitabine andits metabolites. However, leucovorin has an effect on the pharmacodynamics ofcapecitabine and its toxicity may be enhanced by leucovorin.
Sorivudine and analogues
A clinically significant interaction between sorivudine and 5-FU, resulting from theinhibition of dihydropyrimidine dehydrogenase by sorivudine, has been described in theliterature. This interaction, which leads to increased fluoropyrimidine toxicity, ispotentially fatal. Therefore, capecitabine should not be administered concomitantly withsorivudine or its chemically related analogues, such as brivudine (seeContraindications). There must be at least a 4-week waiting period between the end oftreatment with sorivudine or its chemically related analogues, such as brivudine and thestart of capecitabine therapy.
Oxaliplatin
No clinically significant differences in exposure to capecitabine or its metabolites, freeplatinum or total platinum occur when capecitabine and oxaliplatin were administered incombination, with or without bevacizumab.
Bevacizumab
There was no clinically significant effect of bevacizumab on the pharmacokineticparameters of capecitabine or its metabolites.

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 12
Use in Special Populations
PregnancyPregnancy Category D
There are no studies in pregnant women using capecitabine; however, based on thepharmacological and toxicological properties of capecitabine, it can be assumed thatcapecitabine may cause foetal harm if administered to pregnant women. In reproductivetoxicity studies in animals, capecitabine administration caused embryolethality andteratogenicity. These findings are expected effects of fluoropyrimidine derivatives.capecitabine should be considered a potential human teratogen. Capecitabine shouldnot be used during pregnancy. If capecitabine is used during pregnancy, or if the patientbecomes pregnant while receiving this medicine, the patient must be apprised of thepotential hazard to the foetus. Women of childbearing potential should be advised toavoid becoming pregnant while receiving treatment with capecitabine.
LactationIt is not known whether capecitabine is secreted in human milk. In a study of single oraladministration of capecitabine to lactating mice, a significant amount of capecitabinemetabolites was detected in the milk. Breast-feeding should be discontinued duringcapecitabine treatment.
Geriatric UseAmong patients with colorectal cancer aged 60 – 79 years receiving capecitabinemonotherapy in the metastatic setting, the incidence of gastrointestinal toxicity wassimilar to that in the overall population. In patients aged 80 years or older, a largerpercentage experienced reversible Grade 3 or 4 gastrointestinal adverse events, suchas diarrhoea, nausea and vomiting (see Special dosage instructions). Whencapecitabine was used in combination with other agents elderly patients (≥ 65 years) experienced more Grade 3 and Grade 4 adverse reactions and adverse reactions thatled to discontinuation than younger patients. An analysis of safety data in patients equalto or greater than 60 years of age treated with capecitabine plus docetaxel combinationtherapy showed an increase in the incidence of treatment-related Grade 3 and 4 adverseevents, treatment-related serious adverse events and early withdrawals from treatmentdue to adverse events compared to patients less than 60 years of age.
Renal ImpairmentPhysicians should exercise caution when capecitabine is administered to patients withimpaired renal function. As seen with 5-FU the incidence of treatment-related Grade 3 or4 adverse events was higher in patients with moderate renal impairment (creatinineclearance 30-50 mL/min) (see Special dosage instructions).
Hepatic ImpairmentPatients with hepatic impairment should be carefully monitored when capecitabine isadministered. The effect of hepatic impairment not due to liver metastases or severehepatic impairment on the disposition of capecitabine is not known (seePharmacokinetics in special populations and Special dosage instructions).

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 13
Effects on ability to drive and use machinesPresumed to be safe or unlikely to produce and effect on the ability to drive oruse machinery.
OtherNIL.
Adverse Effects
Clinical trialsAdverse reactions considered by the investigator to be possibly, probably, or remotelyrelated to the administration of capecitabine have been obtained from clinical studiesconducted with capecitabine monotherapy (in adjuvant therapy of colon cancer, inmetastatic colorectal cancer and metastatic breast cancer), and clinical studiesconducted with capecitabine in combination with different chemotherapy regimens formultiple indications. Adverse reactions are added to the appropriate category in thetables below according to the highest incidence from the pooled analysis of sevenclinical trials. Within each frequency grouping, adverse reactions are listed in descendingorder of seriousness. Frequencies are defined as very common ≥ 1/10, common ≥ 5/100 to < 1/10 and uncommon ≥ 1/1000 to < 1/100.
Capecitabine monotherapy
Safety data of capecitabine monotherapy were reported for patients who receivedadjuvant treatment for colon cancer and for patients who received treatment formetastatic breast cancer or metastatic colorectal cancer. The safety information includesdata from a phase III trial in adjuvant colon cancer (995 patients treated withcapecitabine and 974 treated with IV 5-FU/LV) and from 4 phase II trials in femalepatients with breast cancer (n = 319) and 3 trials (1 phase I and 2 phase III trials) in maleand female patients with colorectal cancer (n = 630). The safety profile of capecitabinemonotherapy is comparable in patients who received adjuvant treatment for coloncancer and in those who received treatment for metastatic breast cancer or metastaticcolorectal cancer. The intensity of adverse reactions were graded according to thetoxicity categories of the NCIC CTC Grading System.
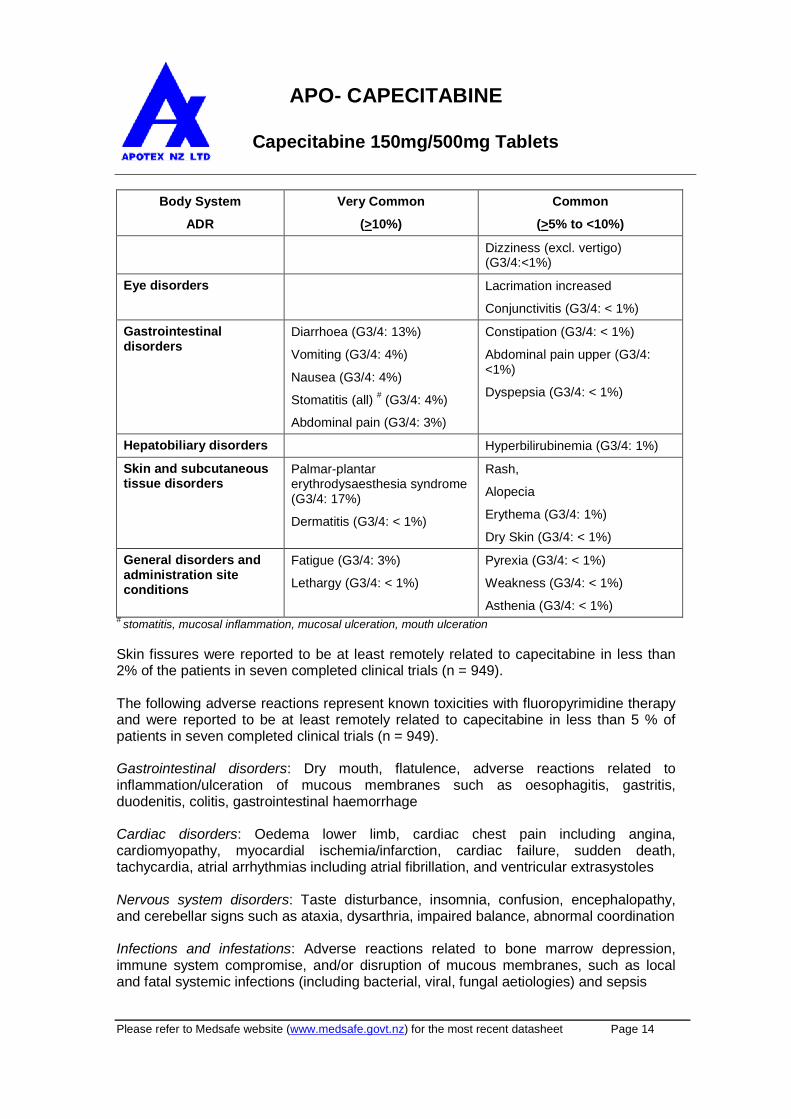
Table 5: Summary of adverse reactions reported in >5% of patients treated withcapecitabine monotherapy
Body System
ADR
Very Common
(>10%)
Common
(>5% to <10%)
Metabolism andnutrition disorders
Anorexia (G3/4: 1%) Dehydration (G3/4: 3%)
Appetite decreased (G3/4: <1%)
Nervous systemdisorders
Paraesthesia
Dysgeusia (G3/4: <1%)
Headache (G3/4: <1%)
APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 14
Body System
ADR
Very Common
(>10%)
Common
(>5% to <10%)
Dizziness (excl. vertigo)(G3/4:<1%)
Eye disorders Lacrimation increased
Conjunctivitis (G3/4: < 1%)
Gastrointestinaldisorders
Diarrhoea (G3/4: 13%)
Vomiting (G3/4: 4%)
Nausea (G3/4: 4%)
Stomatitis (all)#
(G3/4: 4%)
Abdominal pain (G3/4: 3%)
Constipation (G3/4: < 1%)
Abdominal pain upper (G3/4:<1%)
Dyspepsia (G3/4: < 1%)
Hepatobiliary disorders Hyperbilirubinemia (G3/4: 1%)
Skin and subcutaneoustissue disorders
Palmar-plantarerythrodysaesthesia syndrome(G3/4: 17%)
Dermatitis (G3/4: < 1%)
Rash,
Alopecia
Erythema (G3/4: 1%)
Dry Skin (G3/4: < 1%)
General disorders andadministration siteconditions
Fatigue (G3/4: 3%)
Lethargy (G3/4: < 1%)
Pyrexia (G3/4: < 1%)
Weakness (G3/4: < 1%)
Asthenia (G3/4: < 1%)#
stomatitis, mucosal inflammation, mucosal ulceration, mouth ulceration
Skin fissures were reported to be at least remotely related to capecitabine in less than2% of the patients in seven completed clinical trials (n = 949).
The following adverse reactions represent known toxicities with fluoropyrimidine therapyand were reported to be at least remotely related to capecitabine in less than 5 % ofpatients in seven completed clinical trials (n = 949).
Gastrointestinal disorders: Dry mouth, flatulence, adverse reactions related toinflammation/ulceration of mucous membranes such as oesophagitis, gastritis,duodenitis, colitis, gastrointestinal haemorrhage
Cardiac disorders: Oedema lower limb, cardiac chest pain including angina,cardiomyopathy, myocardial ischemia/infarction, cardiac failure, sudden death,tachycardia, atrial arrhythmias including atrial fibrillation, and ventricular extrasystoles
Nervous system disorders: Taste disturbance, insomnia, confusion, encephalopathy,and cerebellar signs such as ataxia, dysarthria, impaired balance, abnormal coordination
Infections and infestations: Adverse reactions related to bone marrow depression,immune system compromise, and/or disruption of mucous membranes, such as localand fatal systemic infections (including bacterial, viral, fungal aetiologies) and sepsis

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 15
Blood and lymphatic system disorders: Anaemia, bone marrow depression, ancytopenia
Skin and subcutaneous tissue disorders: Pruritus, localised exfoliation, skinhyperpigmentation, nail disorders, photosensitivity reactions, radiation recall syndrome
General disorders and administration site conditions: asthenia, pain in limb, lethargy,chest pain (non-cardiac)
Eye: Eye irritation
Respiratory: Dyspnoea, cough
Musculoskeletal: Back pain, myalgia, arthralgia
Psychiatric disorders: Depression
Hepatic failure and cholestatic hepatitis have been reported during clinical trials andpost-marketing exposure. A causal relationship with capecitabine treatment has notbeen established.
Capecitabine in combination therapy
Table 6 lists adverse reactions associated with the use of capecitabine in combinationtherapy with different chemotherapy regimens in multiple indications and occurred inaddition to those seen with monotherapy and/or at a higher frequency grouping. Thesafety profile was similar across all indications and combination regimens. Thesereactions occurred in ≥ 5% of patients treated with capecitabine in combination with other chemotherapies. Adverse reactions are added to the appropriate category in thetable below according to the highest incidence seen in any of the major clinical trials.Some of the adverse reactions are reactions commonly seen with chemotherapy (e.g.peripheral sensory neuropathy with docetaxel or oxaliplatin, hypertension seen withbevacizumab); however, an exacerbation by capecitabine therapy cannot be excluded.
Table 6: Very common and common adverse reactions for capecitabine in combinationwith different chemotherapies in addition to those seen for capecitabine monotherapy
Body System
Adverse Event
Very Common
>10%
Common
>5% to <10%
Infections and Infestations Infection+
Oral candidiasis
Blood and lymphaticsystem disorders
Neutropenia+
Leukopenia+
Febrile neutropenia+
Thromboyctopenia+

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 16
Anaemia+
Metabolism and nutritiondisorders
Appetite decreased Hypokalaemia
Weight decreased
Psychiatric disorders Insomnia
Nervous system disorders Neuropathy peripheral
Peripheral sensory neuropathy
Neuropathy
Taste disturbance
Paraesthesia
Dysgeusia
Dysaesthesia
Headache
Hypoaesthesia
Eye disorders Lacrimation increased
Vascular Disorders Thrombosis/embolismHypertension
Lower limb oedema
Respiratory Dyseasthesia pharynx
Sore throat
Epistaxis
Dysphonia
Rhinorrhoea
Dyspnoea
Gastrointestinal disorders Constipation
Dyspepsia
Dry mouth
Skin and subcutaneoustissue disorders
Alopecia
Nail disorder
Musculoskeletal andconnective tissuedisorders
Arthragia
Myalgia
Pain in extremity
Pain in jaw
Back Pain
General disorders andadministration siteconditions
Pyrexia
Asthenia
Weakness
Temperature intolerance
Fever+
Pain
Frequencies based on all grades except those denoted with +, which are based on G3/4Adverse reactions only.

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 17
Hypersensitivity reactions (2%) and cardiac ischaemia/infarction (3%) have beenreported commonly for capecitabine in combination with other chemotherapy but in lessthan 5% of patients.
Rare or uncommon adverse reactions reported for capecitabine in combination withother chemotherapy are consistent with the adverse reactions reported for capecitabinemonotherapy or the combination product monotherapy.
Laboratory Abnormalities
The following table displays laboratory abnormalities observed in 995 patients (adjuvantcolon cancer) and 949 patients (metastatic breast and colorectal cancer), regardless ofrelationship to treatment, with Capecitabine.
Table 7: Laboratory abnormalitiesa: Capecitabine monotherapy in adjuvant coloncancer and in metastatic breast and colorectal cancer
Parametera
Capecitabine 1250 mg/m2
bd
Intermittent
Patients with Grade 3 / 4 abnormality
(%)
Increased ALAT (SGPT)
Increased ASAT (SGOT)
Increased alkaline phosphatase
Increased calcium
Decreased calcium
Decreased granulocytes
Decreased haemoglobin
Decreased lymphocytes
Decreased neutrophils
Decreased neutrophils/granulocytes
Decreased platelets
Decreased potassium
Increased serum creatinine
Decreased sodium
Increased bilirubin
Hyperglycaemia
1.6
1.1
3.5
1.1
2.3
0.3
3.1
44.4
3.6
2.4
2.0
0.3
0.5
0.4
20
4.4a
Laboratory abnormalities were graded according to the categories of the NCIC CTC Grading System.

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 18
Post-Marketing Experience
The following adverse reactions have been identified during post-marketing exposure:Rare: Acute renal failure secondary to dehydration including fatal outcome (seeWarnings and Precautions)Very rare: lacrimal duct stenosis NOS, corneal disorders including keratitis.Very rare: hepatic failure and cholestatic hepatitis have been reported during clinicaltrials and postmarketing exposure.Very rare: Cutaneous lupus erythematosus, Severe skin reactions such as Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis (TEN) (see Warnings andPrecautions).
Overdose
The manifestations of acute overdose include nausea, vomiting, diarrhoea, mucositis,gastrointestinal irritation and bleeding and bone marrow depression.
Medical management of overdose should include customary therapeutic and supportivemedical interventions aimed at correcting the presenting clinical manifestations andpreventing their possible complications.
Pharmacodynamics
Mechanism of actionCapecitabine is a fluoropyrimidine carbamate derivative, which was designed as anorally administered, tumour-activated and tumour-selective cytotoxic agent.
Capecitabine is non-cytotoxic in vitro. However, in vivo, it is sequentially converted to thecytotoxic moiety, 5-fluorouracil (5-FU), which is further metabolised.
Formation of 5-FU is catalysed preferentially at the tumour site by the tumour-associatedangiogenic factor thymidine phosphorylase (dThdPase), thereby minimising theexposure of healthy tissues to systemic 5-FU.
The sequential enzymatic biotransformation of capecitabine to 5-FU leads to higherconcentrations of 5-FU within tumour tissues. Following oral administration ofcapecitabine to patients with colorectal cancer (n = 8), the ratio of 5-FU concentration incolorectal tumours to adjacent tissues was 3.2 (range, 0.9 to 8.0). The ratio of 5-FUconcentration in tumour to plasma was 21.4 (range, 3.9 to 59.9) whereas the ratio inhealthy tissues to plasma was 8.9 (range, 3.0 to 25.8). Thymidine phosphorylaseactivity was four times greater in primary colorectal tumour than in adjacent normaltissue.
Several human tumours, such as breast, gastric, colorectal, cervical and ovariancancers, have a higher level of thymidine phosphorylase (capable of converting 5'-DFUR[5'-deoxy-5-fluorouridine] to 5-FU) than corresponding normal tissues.

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 19
Normal and tumour cells metabolise 5-FU to 5-fluoro-2-deoxyuridine monophosphate(FdUMP) and 5-fluorouridine triphosphate (FUTP). These metabolites cause cell injuryby two different mechanisms. First, FdUMP and the folate cofactor N5-10-methylenetetrahydrofolate bind to thymidylate synthase (TS) to form a covalently boundternary complex. This binding inhibits the formation of thymidylate from uracil.Thymidylate is the necessary precursor of thymidine triphosphate, which is essential forthe synthesis of DNA, so that a deficiency of this compound can inhibit cell division.Second, nuclear transcriptional enzymes can mistakenly incorporate FUTP in place ofuridine triphosphate (UTP) during the synthesis of RNA. This metabolic error caninterfere with RNA processing and protein synthesis.
Clinical/efficacy studies
Colon and Colorectal cancer
Monotherapy - adjuvant colon cancer
Data from one multicentre, randomised, controlled phase 3 clinical trial in patients withDukes Stage C colon cancer supports the use of capecitabine for the adjuvant treatmentof patients with colon cancer (XACT Study: M66001). In this trial, 1987 patients wererandomised to treatment with capecitabine (1250mg/m2 bd for 2 weeks followed by a 1-week rest period and given as 3-week cycles for 24 weeks) or 5- FU and leucovorin(Mayo regimen: 20 mg/m2 leucovorin IV followed by 425 mg/m2 IV bolus 5-FU, on days 1to 5, every 28 days for 24 weeks). Capecitabine was at least equivalent to IV 5-FU/LV indisease-free survival (DFS) (p = 0.0001, non-inferiority margin 1.2). In the all-randomised population, tests for difference of capecitabine vs. 5-FU/LV in DFS andoverall survival (OS) showed hazard ratios of 0.88 (95% CI 0.77 – 1.01; p = 0.068) and0.86 (95% CI 0.74 – 1.01; p = 0.060), respectively. The median follow up at the time ofthe analysis was 6.9 years.
Study M66001 did not include patients with Dukes Stage B disease, however, thefindings of the study are considered to support the use of CAPECITABINE as adjuvanttherapy in patients with high-risk stage B disease, such as those with inadequatelysampled nodes, T4 lesions, perforation or poorly differentiated histology.
Combination therapy - adjuvant colon cancer
Data from a multicentre, randomised, controlled phase III clinical trial in patients withDukes’ Stage C colon cancer supports the use of capecitabine in combination withoxaliplatin (XELOX) for the adjuvant treatment of patients with colon cancer (NO16968).In this trial, 944 patients were randomised to 3 week cycles for 24 weeks withcapecitabine (1000 mg/m2 bd for 2 weeks followed by a 7-day rest period) incombination with oxaliplatin (130 mg/m2 IV infusion over 2 hours on day 1 every 3weeks); 942 patients were randomised to bolus 5-FU and leucovorin. In the primaryanalysis (ITT population), median observation time was 57 months for DFS and 59months for OS. XELOX was shown to be significantly superior to 5-FU/LV (HR = 0.80[95% CI: 0.69; 0.93]; p =0.0045). The 3 year DFS rate was 71% for XELOX versus 67%for 5-FU/LV. The analysis for the secondary endpoint of relapse free survival (RFS)

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 20
supports these results with a HR of 0.78 ([95% CI: 0.67; 0.92]; p =0.0024) for XELOX vs.5-FU/LV. XELOX showed a trend towards superior OS with a HR of 0.87 ([95% CI: 0.72;1.05]; p=0.1486). The 5 year OS rate was 78% for XELOX versus 74% for 5-FU/LV.
Monotherapy – metastatic colorectal cancer
Data from two identically designed, multicentre, randomised, controlled phase 3 clinicaltrials support the use of capecitabine for first-line treatment of metastatic colorectalcancer (SO14695; SO14796). In these trials, 603 patients were randomised to treatmentwith capecitabine (1250 mg/m2 bd for 2 weeks followed by a 1-week rest period andgiven as 3-week cycles) and 604 patients were randomised to treatment with 5-FU andleucovorin (Mayo regimen: 20 mg/m2 leucovorin IV followed by 425 mg/m2 IV bolus 5-FU, on days 1 to 5, every 28 days).
The overall objective response rates in the all-randomised population (investigatorassessment) were 25.7% (Capecitabine) vs. 16.7% (Mayo regimen); p < 0.0002. Themedian time to progression was 140 days (Capecitabine) vs. 144 days (Mayo regimen).Median survival was 392 days (Capecitabine) vs. 391 days (Mayo regimen).
Combination therapy – first-line treatment of colorectal cancer
Data from a multicentre, randomised, controlled phase III clinical study (N016966)support the use of capecitabine in combination with oxaliplatin or in combination withoxaliplatin and bevacizumab (Avastin) for the first-line treatment of metastastic colorectalcancer. The study contained two parts: an initial 2-arm part in which patients wererandomised to two different treatment groups, including XELOX or FOLFOX-4, and asubsequent 2 x 2 factorial part with four different treatment groups, includingXELOX + placebo (P), FOLFOX-4+P, XELOX+ Avastin (XELOX-A), and FOLFOX-4+Avastin. The treatment regimens are summarised in the table below.
Table 8: Treatment regimens in study NO16966
Treatment Starting Dose Schedule
FOLFOX-4
or
FOLFOX-4 +Avastin
Oxaliplatin
Leucovorin
5-Fluorouracil
85 mg/m2
IV 2 hr
200 mg/m2
IV 2 hr
400 mg/m2
IV bolus600 mg/m
2IV 22 hr
Oxaliplatin on Day 1, every 2weeks
Leucovorin on Day 1 and 2, every2 weeks
5-fluorouracil IV bolus/infusion,each on Days 1 and 2, every 2weeks
Placebo orAvastin
5 mg/kg IV 30 – 90m
Day 1, prior to FOLFOX-4, every2 weeks

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 21
Treatment Starting Dose Schedule
XELOX
or
XELOX +Avastin
(XELOX-A)
Oxaliplatin
Capecitabine
130 mg/m2
IV 2 h
1000 mg/m2
oral bd
Oxaliplatin on Day 1, every 2weeks
Capecitabine oral bd for 2 weeks(followed by 1 week off treatment)
Placebo orAvastin
7.5 mg/kg IV30 - 90 min
Day 1, prior to XELOX, every 3weeks
5-Fluorouracil: IV bolus injection immediately after leucovorin
Non-inferiority of the XELOX-containing arms compared with the FOLFOX-4-containingarms in the overall comparison was demonstrated in terms of progression-free survival(PFS) in the eligible patient population and the intent-to-treat population (see Table 9below). The results indicate that XELOX is equivalent to FOLFOX-4 in terms of overallsurvival (OS). A comparison of XELOX-A versus FOLFOX-4 + Avastin was a pre-specified exploratory analysis. In this treatment subgroup comparison, XELOX-A wassimilar compared to FOLFOX-4 + Avastin in terms of PFS (hazard ratio 1.01 [97.5% CI0.84, 1.22]). The median follow up at the time of the primary analyses in the intent-to-treat population was 1.5 years; data from analyses following an additional 1 year offollow up are also included in the table below.
Table 9: Key non-inferiority results for the primary analysis and 1 year follow-up data(EPP and ITT population, Study NO16966)
PRIMARY ANALYSIS
XELOX/XELOX+P/XELOX-A
(EPP: n = 967;ITT; n=1017)
FOLFOX-4/FOLFOX-4+P/FOLFOX-4+Avastin
(EPP: n = 937;ITT:n=1017)
Population Median Time to Event (Days)HR
(97.5% CI)
Parameter: Progression-free Survival
EPPITT
241244
259259
1.05 (0.94, 1.18)
1.04 (0.93,1.16)
Parameter: Overall Survival
EPPITT
577581
549553
0.97 (0.84, 1.14)
0.96 (0.83;1.12)
ADDITIONAL 1 YEAR FOLLOW UP
Population Median Time to Event (Days)HR
(97.5% CI)
Parameter: Progression-free Survival
APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 22
EPPITT
242244
259259
1.02 (0.92; 1.14)1.01 (0.91; 1.12)
Parameter: Overall Survival
EPPITT
600602
594596
1.00 (0.88; 1.13)0.99 (0.88; 1.12)
EPP=eligible patient population; ITT=intent-to-treat population
The CAIRO study was a randomised, controlled phase III trial to study the use ofcapecitabine at a starting dose of 1000 mg/m2 for 2 weeks every 3 weeks in combinationwith irinotecan for the first-line treatment of patients with metastatic colorectal cancer.The efficacy in terms of Overall Response Rate (ORR), Progression Free Survival (PFS)and Overall Survival (OS) was similar to that reported in pivotal studies of 5-FU,leucovorin, and irinotecan (FOLFIRI).
Data from an interim analysis of a multicentre, randomised, controlled phase II study(AIO KRK 0604) support the use of capecitabine at a starting dose of 800 mg/m2 for 2weeks every 3 weeks in combination with irinotecan and bevacizumab for the first-linetreatment of patients with metastatic colorectal cancer. In this study, 115 patients wererandomised to treatment with Capecitabine combined with irinotecan (XELIRI) + Avastin(XELIRI-A): Capecitabine (800 mg/m2 bd for two weeks followed by a 7-day rest period),irinotecan (200 mg/m2 as a 30 minute infusion on day 1 every 3 weeks) and Avastin (7.5mg/kg as a 30 to 90 minute infusion on day 1 every 3 weeks); a total of 118 patientswere randomised to treatment with capecitabine combined with oxaliplatin + Avastin(XELOX-A): Capecitabine (1000 mg/m2 bd for two weeks followed by a 7-day restperiod), oxaliplatin (130 mg/m2 as a 2 hour infusion on day 1 every 3 weeks) and Avastin(7.5 mg/m2 as a 30 to 90 minute infusion on day 1 every 3 weeks). PFS at 6 months inthe intent-to-treat population was 80% (XELIRI-A) versus 74% (XELOX-A). Overallresponse rate (complete response plus partial response) was 47% (XELIRI-A) versus45% (XELOX-A).
Combination therapy – Second-line treatment of colorectal cancer
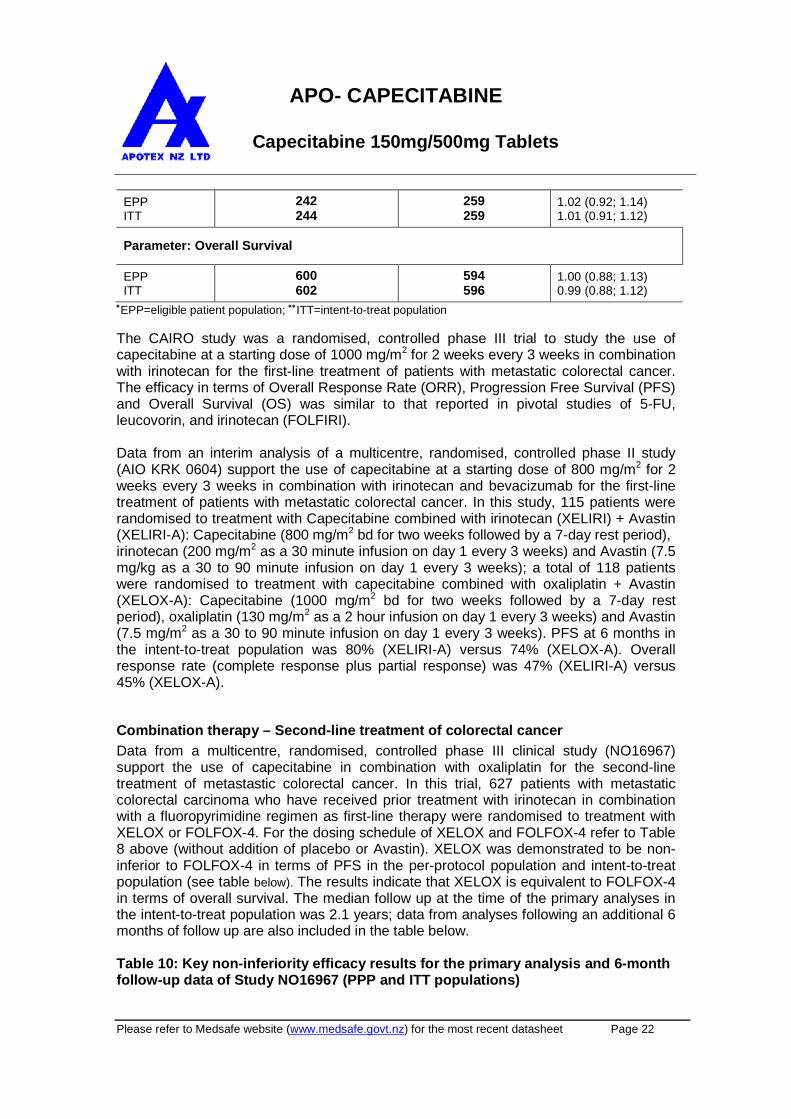
Data from a multicentre, randomised, controlled phase III clinical study (NO16967)support the use of capecitabine in combination with oxaliplatin for the second-linetreatment of metastastic colorectal cancer. In this trial, 627 patients with metastaticcolorectal carcinoma who have received prior treatment with irinotecan in combinationwith a fluoropyrimidine regimen as first-line therapy were randomised to treatment withXELOX or FOLFOX-4. For the dosing schedule of XELOX and FOLFOX-4 refer to Table8 above (without addition of placebo or Avastin). XELOX was demonstrated to be non-inferior to FOLFOX-4 in terms of PFS in the per-protocol population and intent-to-treatpopulation (see table below). The results indicate that XELOX is equivalent to FOLFOX-4in terms of overall survival. The median follow up at the time of the primary analyses inthe intent-to-treat population was 2.1 years; data from analyses following an additional 6months of follow up are also included in the table below.
Table 10: Key non-inferiority efficacy results for the primary analysis and 6-monthfollow-up data of Study NO16967 (PPP and ITT populations)

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 23
PRIMARY ANALYSIS
XELOX(PPP: n = 251; ITT: n = 313)
FOLFOX-4(PPP: n = 252; ITT: n = 314)
Population Median Time to Event (Days)HR
(95% CI)
Parameter: Progression-free Survival
PPPITT
154144
168146
1.03 (0.87, 1.24)
0.97 (0.83; 1.14)
Parameter: Overall Survival
PPP
ITT388363
401382
1.07 (0.88, 1.31)
1.03 (0.87; 1.23)
ADDITIONAL 6 MONTHS OF FOLLOW UP
Population Median Time to Event (Days)HR
(95% CI)
Parameter: Progression-free Survival
PPPITT
154143
166146
1.04(0.87, 1.24)0.97 (0.83; 1.14)
Parameter: Overall Survival
PPPITT
393363
402382
1.05 (0.88, 1.27)1.02 (0.86; 1.21)
PPP=per-protocol population; ITT=intent-to-treat population
A pooled analysis of the efficacy data from first-line (study NO16966; initial 2-arm part)and second line treatment (study NO16967) further support the non-inferiority results ofXELOX versus FOLFOX-4 as obtained in the individual studies: PFS in the per-protocolpopulation (hazard ratio 1.00 [95% CI: 0.88; 1.14]) with a median PFS of 193 days(XELOX; 508 patients) versus 204 days (FOLFOX-4; 500 patients). The results indicatethat XELOX is equivalent to FOLFOX-4 in terms of OS (hazard ratio 1.01 [95% CI: 0.87;1.17]) with a median OS of 468 days (XELOX) versus 478 days (FOLFOX-4).
Combination therapy - Oesophagogastric cancer
Two multicentre, randomised, controlled phase III clinical trials were conducted toevaluate the safety and efficacy of capecitabine in patients with previously untreatedadvanced or metastatic oesophagogastric.
Data from a multicentre, randomised, controlled phase III clinical trial (ML17032) inpatients with advanced or metastatic gastric cancer supports the use of capecitabine inthis setting. In this trial, 160 patients were randomised to treatment with capecitabine(1000 mg/m2 bd for 2 weeks followed by a 7-day rest period) and cisplatin (80 mg/m2 asa 2-hour infusion every 3 weeks). A total of 156 patients were randomised to treatmentwith 5-FU (800 mg/m2 per day, continuous infusion on days 1 to 5 every 3 weeks) and

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 24
cisplatin (80 mg/m2 as a 2-hour infusion on day 1, every 3 weeks). The primary objectiveof the study was met, capecitabine in combination with cisplatin was at least equivalentto 5-FU in combination with cisplatin in terms of PFS in the per-protocol analysis. Theresult for duration of survival (overall survival) was similar to the result for PFS (Table11).
Table 11: Summary of results for key efficacy parameters (PPP, Study ML17032)
Median (Months) (95% Cl)
ParameterCapecitabine/cisplatin
n = 1395-FU/cisplatin
n = 137Hazard Ratio
(95% CI)
Progression-Free Survival5.6 [4.9, 7.3] 5.0 [4.2, 6.3] 0.81 [0.63, 1.04]
Duration of Survival10.5 [9.3, 11.2] 9.3 [7.4, 10.6] 0.85 [0.64, 1.13]
Unadjusted treatment effect in Cox proportional model.
Data from a randomised multicentre, phase III study (REAL-2) comparing capecitabineto 5-FU and oxaliplatin to cisplatin in patients with advanced oesophagogastric cancersupports the use of capecitabine for the first-line treatment of advancedoesophagogastric cancer. In this trial, 1002 patients were randomised in a 2x2 factorialdesign to one of the following 4 arms:
- ECF: epirubicin (50 mg/m2 as a bolus on day 1 every 3 weeks), cisplatin (60mg/m2 as a 2 hour infusion on day 1 every 3 weeks) and 5-FU (200mg/m2 daily given by continuous infusion via a central line).
- ECX: epirubicin (50 mg/m2 as a bolus on day 1 every 3 weeks), cisplatin (60mg/m2 as a 2 hour infusion on day 1 every 3 weeks), and capecitabine(625 mg/m2 bd continuously).
- EOF: epirubicin (50 mg/m2 as a bolus on day 1 every 3 weeks), oxaliplatin (130mg/m2 given as a 2 hour infusion on day 1 every three weeks), and 5-FU(200 mg/m2 daily given by continuous infusion via a central line).
- EOX: epirubicin (50 mg/m2 as a bolus on day 1 every 3 weeks), oxaliplatin (130mg/m2 given as a 2 hour infusion on day 1 every three weeks), andcapecitabine (625 mg/m2 bd continuously).
The primary efficacy analyses in the per protocol population demonstrated non-inferiorityin OS for capecitabine- vs. 5-FU-based regimens (hazard ratio 0.86, 95% CI: 0.8 to 0.99)and for oxaliplatin- vs. cisplatin-based regimens (hazard ratio 0.92, 95% CI: 0.8 to 1.1).The median OS was 10.9 months in capecitabine-based regimens and 9.6 months in 5-FU based regimens. The median OS was 10.0 months in cisplatin-based regimens and10.4 months in oxaliplatin-based regimens.
Capecitabine has also been used in combination with oxaliplatin for the treatment ofadvanced gastric cancer. Studies with capecitabine monotherapy indicate thatcapecitabine has activity in advanced gastric cancer.

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 25
Colon, colorectal and advanced oesophagogastric cancer: meta-analysis
A meta-analysis of six clinical trials (studies SO14695, SO14796, M66001, NO16966,NO16967,M17032) supports capecitabine replacing 5-FU in mono- and combinationtreatment in gastrointestinal cancer. The pooled analysis includes 3097 patients treatedwith capecitabine-containing regimens and 3074 patients treated with 5-FU-containingregimens. The hazard ratio for OS was 0.94 (95% CI: 0.89; 1.00,p = 0.0489) indicatingthat capecitabine-containing regimens are comparable to 5-FU –containing regimens.
Monotherapy - breast cancer
Data from two multicentre phase 2 clinical trials support the use of capecitabinemonotherapy for treatment of patients with locally advanced or metastatic breast cancerafter failure of a taxane and an anthracycline-containing chemotherapy regimen or forwhom further anthracycline therapy is not indicated. In these trials, a total of 236 patientswere treated with capecitabine (1250 mg/m2 bd for 2 weeks followed by 1-week restperiod). The overall objective response rates (investigator assessment) were 20% (firsttrial) and 25% (second trial). The median time to progression was 93 and 98 days.Median survival was 384 and 373 days.
Combination therapy - breast cancer
Data from one multicentre, randomised, controlled phase 3 clinical trial support the useof capecitabine in combination with docetaxel for treatment of patients with locallyadvanced or metastatic breast cancer after failure of cytotoxic chemotherapy, includingan anthracycline. In this trial, 255 patients were randomised to treatment withCapecitabine (1250 mg/m2 bd for 2 weeks followed by a 1-week rest period) anddocetaxel (75 mg/m2 as a 1 hour IV infusion every 3 weeks). A total of 256 patients wererandomised to treatment with docetaxel alone (100 mg/m2 as a 1 hour IV infusion every3 weeks). Survival was superior in the capecitabine + docetaxel combination arm (p =0.0126). Median survival was 442 days (capecitabine + docetaxel) vs. 352 days(docetaxel alone). The overall objective response rates in the all-randomised population(investigator assessment) were 41.6% (capecitabine + docetaxel) vs. 29.7% (docetaxelalone); p = 0.0058. Time to disease progression or death was superior in thecapecitabine +docetaxel combination arm (p < 0.0001). The median time to progressionwas 186 days (capecitabine + docetaxel) vs. 128 days (docetaxel alone).
Pharmacokinetic Properties
AbsorptionAfter oral administration, capecitabine is rapidly and extensively absorbed, followed byextensive conversion to the metabolites 5'-deoxy-5-fluorocytidine (5'-DFCR) and 5'-DFUR. Administration with food decreases the rate of capecitabine absorption, but hasonly a minor effect on the areas under the curve (AUC) of 5'-DFUR and the subsequentmetabolite 5-FU. At the dose of 1250 mg/m2 on day 14 with administration after foodintake, the peak plasma concentrations (Cmax in mcg/mL) for capecitabine, 5'-DFCR, 5'-DFUR, 5-FU and FBAL were 4.47, 3.05, 12.1, 0.95 and 5.46 respectively. The times to

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 26
peak plasma concentrations (Tmax in hours) were 1.50, 2.00, 2.00, 2.00 and 3.34. TheAUC0-∞ values in mcg•h/mL were 7.75, 7.24, 24.6, 2.03 and 36.3.
Distribution
Protein Binding
In vitro human plasma studies have determined that capecitabine, 5'-DFCR , 5'-DFURand 5-FU are 54%, 10% , 62% and 10% protein bound, mainly to albumin.
MetabolismCapecitabine is first metabolised by hepatic carboxylesterase to 5'-DFCR, which is thenconverted to 5'-DFUR by cytidine deaminase, principally located in the liver and tumourtissues.
Formation of 5-FU occurs preferentially at the tumour site by the tumour-associatedangiogenic factor dThdPase, thereby minimising the exposure of healthy body tissues tosystemic 5-FU.
The plasma AUC of 5-FU is 6 to 22 times lower than that following an IV bolus of 5-FU(dose of 600 mg/m2 ). The metabolites of capecitabine become cytotoxic only afterconversion to 5-FU and anabolites of 5-FU (see Pharmacological Properties andEffects).
5-FU is further catabolised to the inactive metabolites dihydro-5-fluoruracil (FUH2), 5-fluoroureidopropionic acid (FUPA) and a–fluoro-ß-alanine (FBAL) via dihydropyrimidinedehydrogenase (DPD), which is rate limiting.
EliminationThe elimination half lives (t½ in hours) of capecitabine, 5'-DFCR, 5'-DFUR, 5-FU andFBAL were 0.85,1.11, 0.66, 0.76 and 3.23 respectively. The pharmacokinetics ofcapecitabine have been evaluated over a dose range of 502 - 3514 mg/m2 /day. Theparameters of capecitabine, 5'-DFCR and 5'-DFUR measured on days 1 and 14 weresimilar. The AUC of 5-FU was 30% - 35% higher on day 14, but did not increasesubsequently (day 22). At therapeutic doses, the pharmacokinetics of capecitabine andits metabolites were dose proportional, except for 5-FU.
After oral administration capecitabine metabolites are primarily recovered in the urine.Most (95.5%) of administered capecitabine dose is recovered in urine. Faecal excretionis minimal (2.6%). The major metabolite excreted in urine is FBAL, which represents57% of the administered dose. About 3% of the administered dose is excreted in urineas unchanged medicine.
Combination therapyPhase I studies evaluating the effect of capecitabine on the pharmacokinetics of eitherdocetaxel or paclitaxel and vice versa showed no effect by capecitabine on thepharmacokinetics of docetaxel or paclitaxel (Cmax and AUC) and no effect by docetaxelor paclitaxel on the pharmacokinetics of 5’-DFUR (the most important metabolite ofcapecitabine).

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 27
Other
Pharmacokinetics in special populationsA population pharmacokinetic analysis was carried out after capecitabine treatment of505 patients with colorectal cancer dosed at 1250 mg/m2 bd. Gender, presence orabsence of liver metastasis at baseline, Karnofsky Performance Status, total bilirubin,serum albumin, ASAT and ALAT had no statistically significant effect on thepharmacokinetics of 5'-DFUR, 5-FU and FBAL.
Patients with hepatic impairment due to liver metastasesNo clinically significant effect on the bioactivation and pharmacokinetics of capecitabinewas observed in cancer patients with mildly to moderately impaired liver function due toliver metastases (see Special dosage instructions).
There are no pharmacokinetic data on patients with severe hepatic impairment.
Patients with renal impairmentBased on a pharmacokinetic study in cancer patients with mild to severe renalimpairment, there is no evidence for an effect of creatinine clearance on thepharmacokinetics of capecitabine and 5-FU. Creatinine clearance was found to influencethe systemic exposure to 5'-DFUR (35% increase in AUC when creatinine clearancedecreases by 50%) and to FBAL (114% increase in AUC when creatinine clearancedecreases by 50%). FBAL is a metabolite without antiproliferative activity; 5'-DFUR is thedirect precursor of 5-FU (see Special dosage instructions).
ElderlyBased on a population pharmacokinetic analysis that included patients with a wide rangeof ages (27 to 86 years) and included 234 (46%) patients greater than or equal to 65years, age has no influence on the pharmacokinetics of 5'-DFUR and 5-FU. The AUC ofFBAL increased with age (20% increase in age results in a 15% increase in the AUC ofFBAL). This increase is likely due to a change in renal function (see Special dosageinstructions and Pharmacokinetics in special populations subsection; Patients with renalimpairment).
RaceIn a population pharmacokinetic analysis of 455 white patients (90.1%), 22 blackpatients (4.4%) and 28 patients of other race or ethnicity (5.5%), the pharmacokinetics ofCapecitabine in black patients were not different compared to white patients.
Chemical Structure
Chemical Name: Capecitabine
Structural Formula:

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 28
List of ExcipientsContains lactose, gluten free.
anhydrous lactose croscarmellose sodium methylcellulose magnesium stearate silica - colloidal anhydrous Hypromellose Hydroxypropylcellulose Macrogol 8000 Titanium dioxide Iron oxide yellow CI77492 Iron oxide red CI77491
Pharmaceutical Precautions
Instructions for HandlingNIL
IncompatibilitiesNIL
Shelf Life2 years from date of manufacture
Special Precautions for Storage
Store at or below 25ºC.
Package Quantities
APO-CAPECITABINE tablets are available in the following presentations:Blister Pack: 60 tablets or 120 tabletsBottles: 500 tablets or 1000 tablets

APO- CAPECITABINE
Capecitabine 150mg/500mg Tablets
Please refer to Medsafe website (www.medsafe.govt.nz) for the most recent datasheet Page 29
Medicine Schedule
Prescription Medicine
Sponsor Details
Name and AddressApotex NZ Ltd32 Hillside RoadGlenfieldPrivate Bag 102-995North Shore Mail CentreAucklandTelephone: (09) 444 2073Fax: (09) 444 2951
Date of Preparation 24 March 2016