novel bacterial membrane surface display system using cell ... · based on the l-form strains....
TRANSCRIPT

APPLIED AND ENVIRONMENTAL MICROBIOLOGY, Feb. 2002, p. 525–531 Vol. 68, No. 20099-2240/02/$04.00�0 DOI: 10.1128/AEM.68.2.525–531.2002Copyright © 2002, American Society for Microbiology. All Rights Reserved.
Novel Bacterial Membrane Surface Display System Using CellWall-Less L-Forms of Proteus mirabilis and Escherichia coliChristian Hoischen,1* Christine Fritsche,1 Johannes Gumpert,1 Martin Westermann,2
Katleen Gura,1 and Beatrix Fahnert3
Department of Molecular Biology, Institute of Molecular Biotechnology,1 and Department of Applied Microbiology,Hans-Knöll-Institute for Natural Products Research,3 D-07745 Jena, and Department of Ultrastructure Research,
Friedrich-Schiller-University Jena, D-07740 Jena,2 Germany
Received 12 June 2001/Accepted 5 November 2001
We describe a novel membrane surface display system that allows the anchoring of foreign proteins in thecytoplasmic membrane (CM) of stable, cell wall-less L-form cells of Escherichia coli and Proteus mirabilis. Thereporter protein, staphylokinase (Sak), was fused to transmembrane domains of integral membrane proteinsfrom E. coli (lactose permease LacY, preprotein translocase SecY) and P. mirabilis (curved cell morphologyprotein CcmA). Both L-form strains overexpressed fusion proteins in amounts of 1 to 100 �g ml�1, with higherexpression for those with homologous anchor motifs. Various experimental approaches, e.g., cell fractionation,Percoll gradient purification, and solubilization of the CM, demonstrated that the fusion proteins are tightlybound to the CM and do not form aggregates. Trypsin digestion, as well as electron microscopy of immunogold-labeled replicas, confirmed that the protein was localized on the outside surface. The displayed Sak showedfunctional activity, indicating correct folding. This membrane surface display system features endotoxin-poororganisms and can provide a novel platform for numerous applications.
The overexpression of recombinant proteins, which remainbound to the outer surface of the bacterial cells as accessibleand functional active molecules, offers new applications inbiotechnology and medicine. Among these are the develop-ment of diagnostics and vaccines, adhesin-receptor interactionstudies, the generation of peptide libraries, the immobilizationof enzymes, and the expression of heavy metal-binding pep-tides and antibody fragments (6, 10, 25, 33). The surface dis-play systems follow various strategies for anchoring. In gram-negative bacteria, outer membrane proteins (21), pili andflagella (26), modified lipoproteins (6, 7, 11), ice nucleationproteins (17, 23), and autotransporters (19, 27, 35) have beenused as anchors. In gram-positive bacteria, surface anchorshave been derived from lipoproteins, cell wall proteins, orS-layer proteins (24, 32, 33). However, depending on the dis-played protein and the desired application, each system has itsown advantages and disadvantages, such as the size limitationof the displayed protein, mislocalization or formation of inclu-sion bodies, association with lipopolysaccharides (LPS), or de-stabilization of the outer membrane (10, 17, 22). There is stilla need for further developments to increase the repertoire ofapplications of surface display systems (21).
Stable protoplast type L-form bacteria have up to now notbeen considered as a system for surface display, although theyexhibit several interesting features for such applications. Theyhave lost irreversibly the ability to form cell wall structures andperiplasmic compartments, and their cells are surrounded onlyby a cytoplasmic membrane (CM). Cell biological properties ofthe strains and the lipid composition of their CM are well
characterized (13, 16). Furthermore, the L-form strains of Pro-teus mirabilis LVIWEI and Escherichia coli LWF�WEI havebeen used for the efficient overexpression of numerous recom-binant proteins (5, 12, 20, 30). In contrast to surface displaysystems with walled bacteria, both L-form strains lack extra-cellular proteolytic activities (12). Moreover, E. coli LWF�WEI does not synthesize any endotoxic LPS, and only smallquantities of LPS were found in P. mirabilis LVIWEI (O.Holst, unpublished data). Due to the lack of a cell wall, theL-form membrane is easy to isolate and to handle.
We describe here a novel membrane surface display systembased on the L-form strains. Staphylokinase (Sak), a plasmin-ogen activator with potential for medical application, was cho-sen as reporter protein. Sak is functionally and structurally wellcharacterized (31), and it has been shown in previous studies tobe overexpressed and secreted by L-form cells as a solubleprotein (12). Transmembrane domains from integral mem-brane proteins of E. coli (lactose permease LacY, preproteintranslocase SecY) and P. mirabilis (curved cell morphologyprotein CcmA) were selected as membrane anchors. The con-struction and the expression of the fusion proteins (membraneanchor and Sak) in the L-form strains were demonstrated. Thefusion proteins were localized in the CM as surface displayedand functionally active products.
MATERIALS AND METHODS
Bacterial strains, growth conditions, and protein expression. E. coli DH5�was used for cloning and construction of vectors. DH5� and P. mirabilis VI wereused for genomic PCR. Both were cultivated in Luria-Bertani (LB) medium oron LB agar plates. If necessary, the media were supplemented with the appro-priate antibiotics (2).
Surface display of Sak was examined in stable protoplast-type L-form strains ofP. mirabilis LVIWEI and E. coli LWF�WEI (13; J. Gumpert, C. Hoischen, M. J.Kujau, C. Fritsche, G. Elske, B. Fahnert, S. Sieben, and H. Müller, Germanpatent FN 100 11 358.3 [pending]). Cultivation in L-form standard (LFS) me-dium, transformation, selection of transformants, and adaptation to growth in
* Corresponding author. Mailing address: Department of MolecularBiology, Institute of Molecular Biotechnology, Beutenbergstr. 11,D-07745 Jena, Germany. Phone: 49-3641-656305. Fax: 49-3641-656310.E-mail: [email protected].
525
on August 27, 2019 by guest
http://aem.asm
.org/D
ownloaded from

liquid medium were carried out as described previously (14). For the expressionof fusion proteins, 100-ml bottles containing 30 ml of LFS medium, supple-mented with kanamycin (10 to 20 �g ml�1), were inoculated with 1 to 2 ml of anovernight culture. After 3 h of cultivation on a rotary shaker (220 rpm, 26°C) atan optical density at 540 nm of 0.4 to 0.8, protein expression was induced by theaddition of IPTG (isopropyl-�-D-thiogalactopyranoside) to a final concentrationof 1 mM. Cells were generally analyzed for the expression of fusion proteins 20 hafter induction.
Construction of expression plasmids. The DNA cloning procedures wereperformed by standard methods (2). For the fusion of Sak with the relevanttransmembrane protein domains, a vector, pF003, was constructed. This vectorwas derived from plasmid pGEX-4T-2 (Amersham Pharmacia Biotech), whichwas digested with FspI-SapI and from a modified version (M. Hartmann, unpub-lished data) of pMEX6 (U.S. Biochemicals), which was cleaved with Eco47III-SapI. The isolated fragments FspI-SapI containing the lacIq gene and fragmentEco47III-SapI containing promoter P tac, the terminator of the rrnB operon(rrnBT), an ampicillin resistance region (bla), and the ColE1ori were ligated,resulting in plasmid pBF003 (4,245 bp). After ScaI digestion of the single ScaIrestriction site of pBF003 and treatment of the blunt ends with alkaline phos-phatase, a SmaI fragment from vector pACK02sckan (20) was inserted, whichcontained a kanamycin resistance region (kan). pF003 harbored a multicloningsite containing the restriction sites NdeI, PstI, and HindIII for insertion ofexpression cassettes under the control of the tac promoter. The DNA sequencecoding for the mature Sak (amino acids 28 to 163) without signal peptide (3) wasamplified by overlap PCR with the upstream primer 5�-CGCCTGCAGTCAAGTTCATTCGACAAAGGA-3� and the downstream primer 5�-CGCCTGCAGTCAAGTTCATTCGACAAAGGA-3� from vector pSIS2 (S. Sieben, unpublisheddata). After PstI-HindIII restriction of the PCR product, the resulting fragmentwas cloned into the PstI-HindIII fragment of pF003, resulting in plasmid pFsak.The introduction of the PstI site at the 5� end caused the insertion of two linkeramino acids (Leu-Gln) at the N terminus of Sak. The coding DNA of thefollowing transmembrane domains of membrane proteins was amplified by over-lap PCR from genomic DNA: helix 1 (amino acids 1 to 41; LacYH1) and helices1 to 3 (amino acids 1 to 101; LacYH1-3) of E. coli LacY (8, 18), helix 1 (aminoacids 1 to 74; SecYH1) and helices 1 to 3 (amino acids 1 to 153; SecYH1-3) ofE. coli SecY (1), and helix 1 (amino acids 1 to 34; CcmAH1) of the P. mirabilisCcmA (15). The following upstream primers were used: 5�-GGAATTCCATATGTACTATTTAAAAAACACAAACTTTT-3� for LacYH1 and LacYH1-3, 5�-CGCGCATATGGCTAAACAACCGGGATTAGATTTCAAA-3� for SecYH1and SecYH1-3, and 5�-CGCCATATGGATAATAAGCGAACACAGCGG-3�for CcmAH1. The following downstream primers were used: 5�-CGCCTGCAGGCTGATATGGTTGATGTCATG-3� for LacYH1, 5�-CGCCTGCAGGTATTGTAACAGTGGCCCGAAGATAAA-3� for LacYH1-3, 5�-CGCCTGCAGACGGCTGAGAGCACCACCAGAGAACATGTT-3� for SecYH1, 5�-CGCCTGCAGGCCCGGGTTAATCACCAGGCCTTGCATACC-3� for SecYH1-3, and 5�-CGCCTGCAGTTCTCTGGTGGTGCTCTCACCAGCTATTTGATATCG-3�for CcmAH1. The primers introduced to all amplified DNA fragments an NdeIsite at the 5� end and a PstI site at the 3� end, respectively. In addition, a DNAsequence coding for a linker of five amino acids (Ser-Thr-Thr-Arg-Glu) wasintroduced to the 5� end of CcmAH1. After the NdeI-PstI restrictions, theamplified fragments were cloned into the NdeI-PstI fragment of pFsak, resultingin the expression plasmids pFLacYH1-Sak, pFLacYH1-3-Sak, pFSecYH1-Sak,pFSecYH1-3-Sak, and pFCcmAH1-Sak.
Fractionation of cells. Cells were separated from growth medium by centrif-ugation at 14,000 rpm in an Eppendorf centrifuge (5415C) for 10 min. Thesupernatants were directly used for the detection of soluble proteins. The pelletscontaining the cells were washed once in sucrose (0.4 M) and resuspended in anequal volume of buffer A (20 mM Tris-HCl, 15 mM MgCl2; pH 7.2). Formembrane preparation, the washed cells were disrupted by ultrasonication witha Branson 250 Sonifier (50 kHz, two 2-min intervals; Emerson Technologies,Dietbach, Germany). Membranes were isolated by ultracentrifugation (100,000� g, 30 min). The resulting supernatants, which were almost depleted in mem-branes and lipids, represented the cytosolic fraction. It should be noted that the100,000 � g supernatant can still contain certain amounts of membrane enzymessuspended as small particles. Membranes were used for analysis after two washesin buffer A and resuspension in the initial volume of buffer A.
Alternatively, the membranes obtained by ultracentrifugation were furtherpurified by Percoll gradient ultracentrifugation (28) in buffer A containing 30%(vol/vol) Percoll. Membranes from 10 ml of a CcmAH1-Sak-expressing P. mira-bilis LVIWEI culture were isolated as described above and resuspended in 28 mlof buffer A supplemented with 30% (vol/vol) Percoll. After centrifugation (30min, 15,000 rpm) in a Beckman 50.2Ti rotor at 4°C, a self-generating buoyantdensity gradient was established. Eight fractions with increasing buoyant densi-
ties were collected by pipetting 3.5-ml aliquots from the top of the centrifugationtubes. The NADH-oxidase activity of the resulting fractions was monitored at30°C in buffer A supplemented with 1 mM dithiothreitol and 0.2 mM NADH2 at340 nm by using a Spekol 1100 spectrophotometer (Zeiss, Jena, Germany).
For solubilization of the fusion proteins, membranes or cells from 1 ml ofculture were washed and resuspended in 1 ml of buffer A supplemented with 0.5or 1% (vol/vol) Triton X-100 and then gently shaken for 60 min at 30°C. Thesolution was subsequently centrifuged at 14,000 � g for 30 min to separateinsoluble material.
Protein methods. Proteins of culture supernatants, washed cells, cytosolicfractions, and washed membranes were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on a 15% polyacrylamide geland then visualized by Coomassie brilliant blue. For Western blot analysis, thegels were electroblotted to polyvinylidene difluoride membranes (Millipore,Bedford, Mass.). To compare the amounts of fusion proteins in the fractions, allsamples were equalized for the same volumes of the initial culture. The amountsof applied proteins per lane were determined by the method of Bradford (4). Theblotted membranes were incubated in TTBS (Tris-HCl-buffered saline [TBS]containing 1% [vol/vol] Tween 20) supplemented with 5% (wt/vol) skim milk andthen washed four times with TTBS (2). For immunostaining the membranes wereincubated in TTBS containing purified diluted (1:2,500) rabbit anti-Sak as pri-mary antibody. The membranes were rinsed four times with TTBS prior to theaddition of the secondary alkaline phosphatase-conjugated anti-rabbit antibody(Bio-Rad) diluted 1:5,000 in TTBS. Antigen-antibody conjugates were visualizedby the color reaction recommended by the manufacturer. The intensity of thebands was determined by densitometry with Molecular Analyst Software (Bio-Rad).
Functional activity of Sak. In the standard assay for soluble Sak, 50-�l aliquotsof Sak-containing samples and defined standards of purified Sak (1 to 100 �gml�1) were applied to holes (9 mm in diameter) in agar layers (2% [wt/vol]),which contained skim milk (1% [wt/vol]) and plasminogen (10 �g ml�1), andthen incubated for 3 to 8 h at 37°C. The Sak molecules activate plasminogen,which causes digestion of milk casein and the formation of clearing zones (3). Todetermine the amount of functional active Sak, the diameters of the clearingzones were compared with those of the standards. However, it was impossible toquantify the activity of the cell-bound Sak with the standard assay because wholecells could not diffuse freely in agar layers. To facilitate the diffusion, the CM wassolubilized with Triton X-100 (0.5% [vol/vol]) prior to the functionality assays.The obtained values demonstrate that the fusion proteins were functionallyactive. The determined functional activities corresponded to the activity of sol-uble purified Sak, but they did not reflect an accurate estimation of the activitybecause even in the presence of a detergent the fusion proteins still have lipidsattached to their membrane anchors, which might hinder the diffusion in the agarlayer.
Trypsin digestion of displayed fusion proteins. Culture aliquots of 900 �l weremixed with 100 �l of buffer B (200 mM Tris-HCl, 150 mM MgCl2; pH 7.5) andsupplemented with trypsin to a final concentration of 2 mg ml�1. After incuba-tion for 60 min at 37°C, trypsin inhibitor (8 mg ml�1) was added, and the sampleswere analyzed by SDS-PAGE and Sak-specific immunoblotting. To digest non-accessible (intracellular) fusion protein, aliquots were treated with ultrasonica-tion or 0.5% (vol/vol) Triton X-100 prior to trypsin digestion.
Freeze-fracturing, replica immunogold-labeling, and electron microscopy. Ata temperature of 30°C, cells were separated from growth medium, washed, andresuspended in 0.4 M sucrose (10% of the initial volume). Aliquots were en-closed between two 0.1-mm copper profiles as used for the sandwich double-replica technique. The sandwiches were rapidly frozen in liquid propane cooledby liquid nitrogen. Freeze fracturing was performed in a BAF400T freeze-fracture unit (BAL-TEC; Liechtenstein) at �150°C by using a double-replicastage. The samples were shadowed without etching and with a 2- to 2.5-nm-thicklayer of Pt/C at an angle of 35°.
For immunogold labeling the replicas were transferred to an SDS digestionsolution (2.5% [wt/vol] SDS in 10 mM Tris-HCl buffer [pH 8.3] and 30 mMsucrose) and incubated overnight at room temperature (9). After four washes inTBS and 30 min of incubation in TBS plus 1% (wt/vol) bovine serum albumin(TBS–1% BSA), the replicas were incubated for 1 h in TBS–0.5% BSA plusSak-specific primary antibody (diluted 1:50). Subsequently, the replicas werewashed four times in TBS and incubated for 1 h in TBS–0.5% BSA plus sec-ondary antibody (1:50-diluted goat anti-rabbit immunoglobulin G conjugatedwith 10-nm gold [British Biocell International, Cardiff, United Kingdom]). Afterfour washes in TBS and fixation with 0.5% (vol/vol) glutaraldehyde in TBS, thereplicas were washed twice in distilled water and picked onto Formvar-coatedgrids for observation in an EM902 electron microscope (Zeiss, Oberkochen,Germany).
526 HOISCHEN ET AL. APPL. ENVIRON. MICROBIOL.
on August 27, 2019 by guest
http://aem.asm
.org/D
ownloaded from

RESULTS
Construction of fusion proteins and expression in L-forms.In comparison to currently used surface display systems, whichanchor the exposed proteins in the outer membrane of gram-negative bacteria or in the cell wall of gram-positive bacteria,the strategy employed here was the anchoring in the CM ofstable protoplast type L-forms (Fig. 1). Transmembrane do-mains of integral proteins of the CM were used as membraneanchors, i.e., helix 1 (LacYH1) or helices 1 to 3 (LacYH1-3) ofE. coli LacY (8, 18), helix 1 (SecYH1) or helices 1 to 3(SecYH1-3) of E. coli SecY (1), and helix 1 (CcmAH1) of P.mirabilis CcmA (15). The reporter protein Sak (31) was fusedto the C termini of external regions of the various anchor motifs.
We intended to express all five constructs in the L-formstrains E. coli LWF�WEI and P. mirabilis LVIWEI by usingexpression plasmids that bear the fusion proteins under thecontrol of the tac promoter (Fig. 2A). In E. coli LWF�WEI,the fusion proteins LacYH1-Sak, LacYH1-3-Sak, andSecYH1-Sak were overexpressed (Fig. 2B), whereas CcmAH1-Sak with a membrane anchor derived from P. mirabilis andSecYH1-3-Sak were not expressed (data not shown). P. mira-
bilis LVIWEI overexpressed CcmAH1-Sak with a homologousmembrane anchor and SecYH1-Sak with an anchor derivedfrom E. coli (Fig. 2B). The molecular masses of the fusionproteins corresponded to the values calculated from the aminoacid sequences (LacYH1-Sak, 20.5 kDa; LacYH1-3-Sak, 27.9kDa; SecYH1-Sak, 23.7 kDa; CcmAH1-Sak, 20.5 kDa). Thegrowth rate of the cells was not affected by the expression ofthe fusion protein, and the cells remained viable during pro-longed cultivation over 48 h (data not shown).
Quantification of fusion proteins. Synthesized fusion pro-teins were quantified by densitometry of Sak-specific immuno-stained Western blots. Different amounts of overexpressingcells and defined amounts of purified Sak were blotted, and thesignal intensities were compared. If we assume that the fusionproteins and the soluble reference protein were transferredand detected equally, the following approximate molar yieldsand corresponding weight yields were calculated: (i) �1 �gml�1 for SecYH1-Sak, 1 to 5 �g ml�1 for LacYH1-3-Sak, and5 to 15 �g ml�1 for LacYH1-Sak in E. coli LWF�WEI and (ii)�1 �g ml�1 for SecYH1-Sak and 70 to 100 �g ml�1 forCcmAH1-Sak in P. mirabilis LVIWEI.
FIG. 1. Schematic illustration of the L-form membrane surface display system and commonly used bacterial systems. In endotoxin-poor L-formcells the protein is anchored in the CM by transmembrane domains of integral proteins of the CM, whereas in gram-negative bacteria the proteinsare anchored in the outer membrane or bound to surface components such as pili or flagella. For surface display in gram-positive bacteria, theproteins are bound to protein components of the cell wall.
VOL. 68, 2002 L-FORMS OF P. MIRABILIS AND E. COLI 527
on August 27, 2019 by guest
http://aem.asm
.org/D
ownloaded from

Detection of fusion proteins in cells and membranes. Thefusion proteins were determined in the culture supernatant, inthe washed cells, in the membrane fraction, and in the 100,000� g supernatant representing the cytosol. Figure 3A comparesthe band intensities of CcmAH1-Sak in the fractions of P.mirabilis LVIWEI. The total recombinant CcmAH1-Sak wascell bound, and only traces were found in the culture superna-tant. The band intensity of the purified membranes was equalto the signal of the total cell-bound fusion protein, and onlyminor amounts were detected in the 100,000 � g supernatant.This demonstrates the exclusive location of CcmAH1-Sak inthe CM. In the case of LacYH1-Sak and SecYH1-Sak ex-pressed in E. coli LWF�WEI, 80 to 90% of the total fusionprotein was located in the membrane fraction, and the remain-ing 10 to 20% was found in the culture supernatant (data notshown). LacYH1-3-Sak was located exclusively in the mem-brane fractions.
Overexpressed membrane proteins or proteins with hydro-phobic domains often tend to form insoluble self-aggregates ofhigh density when they are not properly integrated into the CM(34). These aggregates cannot be separated from cells or mem-branes by conventional centrifugation. Therefore, the mem-branes, which were prepared by conventional ultracentrifuga-tion, were purified further by Percoll density gradientcentrifugation (Fig. 3B, C, and D). Among the eight fractionscollected after centrifugation, fraction 2, with a buoyant den-sity of 1.03 g ml�1, contained ca. 70% of the total recoveredNADH-oxidase activity, a marker enzyme for the CM (Fig.3B). This demonstrates a concentration of the CM in fraction2, which also contained by far the majority of the totalCcmAH1-Sak, as shown by immunoblot analysis of the frac-tions (Fig. 3C). The corresponding Coomassie blue-stainedSDS-PAGE gels showed that CcmAH1-Sak is a major protein
of the purified CM fraction (Fig. 3D). The similar band pat-terns of the different fractions in Fig. 3D can be explained bythe fact that the applied membranes consist of vesicles andmembrane fragments of different sizes. The membranes arenot completely concentrated in one fraction, and certain minorpopulations with different sedimentation properties, whichcannot be concentrated by prolonged centrifugation, are alsofound in the other fractions. A second reason might be theoccurrence of proteins, which are loosely bound to the mem-brane. They can be released during the membrane prepara-tions and are found evenly distributed in the gradient (28).
Another indication for the localization of fusion proteins inthe CM is their solubilization by detergents. It was found thatall expressed fusion proteins could be solubilized from washedmembranes or cells by using the nonionic detergent TritonX-100 (data not shown). These findings show that the fusionproteins are embedded in the lipid bilayer of the membrane.
Digestion by trypsin. Trypsin does not penetrate or destroyL-form cells (data not shown). Therefore, the proteolytic di-gestion of fusion proteins was used as an indication of theirsurface exposure and accessibility. Western blot analysis oftrypsin-treated and untreated whole cells of CcmAH1-Sak-expressing P. mirabilis LVIWEI revealed that the fusion pro-tein disappeared almost completely after trypsin treatment(Fig. 4), indicating that it was located on the outside surface ofthe cells. Analysis of the signal intensities revealed that 95%of the Sak signal disappeared after trypsin treatment. Thefusion protein, which was not accessible to trypsin, could bedigested when the cells were sonicated or solubilized withTriton X-100 prior to trypsin addition (Fig. 4). Comparableresults were obtained with E. coli LWF�WEI expressingLacYH1-Sak, SecYH1-Sak, and LacYH1-3-Sak (data notshown).
FIG. 2. Principle of vector constructs and expression of fusion proteins. (A) Plasmid pFCcmAH1-Sak illustrates the expression vectors used inthis study. It carries DNA sequences encoding for ColE1 origin, lacIq repressor, kanamycin resistance (kan), terminator (rrnBT), and the fusionprotein (CcmAH1-Sak) under control of the tac promoter (P tac). (B) Synthesis of fusion proteins by L-form cells. Shown are a Sak-specificimmunoblot of purified mature Sak (lane 1); washed cells of E. coli LWF�WEI expressing LacYH1-Sak (lane 2), LacYH1-3-Sak (lane 3), andSecYH1-Sak (lane 4); and cells of P. mirabilis LVIWEI expressing CcmAH1-Sak (lane 5) and SecYH1-Sak (lane 6). The following total proteinamounts were applied: 2 �g, lane 1; 31 �g, lane 2; 26 �g, lane 3; 32 �g, lane 4; 27 �g, lane 5; and 35 �g, lane 6.
528 HOISCHEN ET AL. APPL. ENVIRON. MICROBIOL.
on August 27, 2019 by guest
http://aem.asm
.org/D
ownloaded from

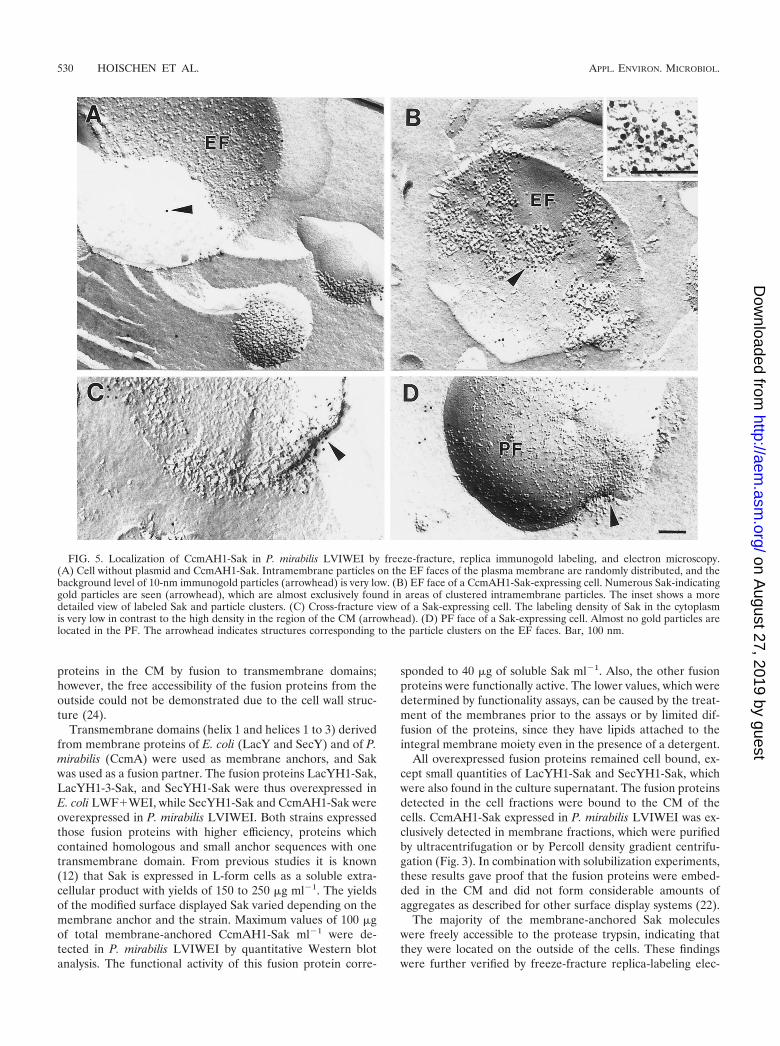
Localization of CcmAH1-Sak by electron microscopy. Thesurface display was verified further by freeze-fracturing, replicaimmunogold labeling, and electron microscopy. It was demon-strated by using P. mirabilis LVIWEI that CcmAH1-Sak waslocalized on the exoplasmic fracture (EF) face of the CM. Alarge number of gold particles indicating Sak molecules werefound on this EF face (Fig. 5B), whereas almost no gold par-ticles were located on the protoplasmic fracture (PF) face (Fig.5D). Cross-fractures (Fig. 5C) showed only a few particles inthe cytoplasm, but a high concentration of Sak molecules wasobserved in defined areas on the outside of the CM. Theanchored proteins were clustered in domains of the CM (Fig.
5B), which were not found in nonexpressing cells (Fig. 5A).The background labeling in cells without expression plasmidwas rather low (Fig. 5A).
The distribution of gold particles was determined for thevarious surfaces of expressing cells and also for the nonexpress-ing cells. For nonexpressing cells, an average of three particlesper square micrometer was determined (minimum of two,maximum of four) out of five frames. This value represents theunspecific background. A total of 15 frames from nine express-ing cells were analyzed, showing an average density of 670particles �m�2 (minimum of 520, maximum of 960) in thedomains containing protein clusters of the EF. Outside of theclusters was found an average of 10 particles �m�2 (minimumof 4, maximum of 20), as calculated for 20 frames from 15 cells.Analysis of 20 frames from 15 cells resulted in an average of 7particles �m�2 (minimum of 4, maximum of 16) on the PF.
Functional activity of Sak. In order to test whether themembrane-bound fusion proteins were correctly folded, thefunctional activity of Sak was determined. A modified versionof the standard assay, which is based on the diffusion of Sak inagar layers, was used. After solubilization of the CM from thevarious L-form cells the functional activities corresponded toat least 40 �g of soluble Sak per ml of culture for CcmAH1-Sakexpressed in P. mirabilis LVIWEI and 1 to 5 �g ml�1 forLacYH1-Sak and LacYH1-3-Sak expressed in E. coli LWF�WEI. The functional activities determined for SecYH1-Sakand SecYH1-3-Sak expressed in E. coli LWF�WEI and in P.mirabilis LVIWEI were below values sufficient for quantifica-tion.
DISCUSSION
Two novel approaches for bacterial surface display havebeen introduced: (i) protoplast type, endotoxin-poor L-formbacteria, which lack the cell wall and the periplasmic compart-ment (13), and (ii) transmembrane domains of integral pro-teins of the CM as membrane anchors. Integral membraneproteins from the CM or their transmembrane domains havenot hitherto been successfully used as anchor motifs. In gram-negative bacteria the proteins are usually anchored in the outermembrane or bound to surface proteins (6, 10, 25, 33) (Fig. 1).In gram-positive bacteria there have been attempts to anchor
FIG. 3. Localization of CcmAH1-Sak synthesized by P. mirabilisLVIWEI. (A) Sak-specific immunoblot of culture supernatant (lane 1),washed cells (lane 2), 100,000 � g supernatant representing the cytosol(lane 3), and washed membranes (lane 4). The following proteinamounts were applied: 18 �g, lane 1; 58 �g, lane 2; 31 �g, lane 3; and24 �g, lane 4. (B) Density gradient centrifugation of washed mem-branes. Eight fractions with increasing buoyant densities were col-lected. The densities of the eight fractions (F) and the correspondingrelative amounts of totally recovered NADH-oxidase activity (E),which serves as the marker enzyme for the CM, are presented. (C) Sak-specific immunoblot analysis of the eight fractions collected after den-sity gradient centrifugation. Equal volumes of each fraction wereapplied, and, therefore, the intensity of the immunostained CcmAH1-Sak bands represents the amounts of fusion protein in each fraction.The majority of CcmAH1-Sak is located in fraction 2 (density, 1.03 gml�1). This finding correlates with the occurrence of NADH-oxidaseactivity, which was also mainly found in fraction 2 (70% of totalactivity). (D) Coomassie blue-stained SDS-PAGE gels of the eightfractions. The arrow indicates CcmAH1-Sak in lane 2 that correspondsto fraction 2. Equal volumes of each fraction were applied.
FIG. 4. Accessibility of CcmAH1-Sak for trypsin digestion. TheSak-specific immunoblot shows cells of P. mirabilis LVIWEI expressingCcmAH1-Sak: without trypsin treatment (lane 1), after treatment withtrypsin (lane 2), after ultrasonication prior to trypsin treatment (lane3), and after Triton X-100 addition prior to trypsin digestion (lane 4).To lane 1 was added 28 �g of protein.
VOL. 68, 2002 L-FORMS OF P. MIRABILIS AND E. COLI 529
on August 27, 2019 by guest
http://aem.asm
.org/D
ownloaded from
proteins in the CM by fusion to transmembrane domains;however, the free accessibility of the fusion proteins from theoutside could not be demonstrated due to the cell wall struc-ture (24).
Transmembrane domains (helix 1 and helices 1 to 3) derivedfrom membrane proteins of E. coli (LacY and SecY) and of P.mirabilis (CcmA) were used as membrane anchors, and Sakwas used as a fusion partner. The fusion proteins LacYH1-Sak,LacYH1-3-Sak, and SecYH1-Sak were thus overexpressed inE. coli LWF�WEI, while SecYH1-Sak and CcmAH1-Sak wereoverexpressed in P. mirabilis LVIWEI. Both strains expressedthose fusion proteins with higher efficiency, proteins whichcontained homologous and small anchor sequences with onetransmembrane domain. From previous studies it is known(12) that Sak is expressed in L-form cells as a soluble extra-cellular product with yields of 150 to 250 �g ml�1. The yieldsof the modified surface displayed Sak varied depending on themembrane anchor and the strain. Maximum values of 100 �gof total membrane-anchored CcmAH1-Sak ml�1 were de-tected in P. mirabilis LVIWEI by quantitative Western blotanalysis. The functional activity of this fusion protein corre-
sponded to 40 �g of soluble Sak ml�1. Also, the other fusionproteins were functionally active. The lower values, which weredetermined by functionality assays, can be caused by the treat-ment of the membranes prior to the assays or by limited dif-fusion of the proteins, since they have lipids attached to theintegral membrane moiety even in the presence of a detergent.
All overexpressed fusion proteins remained cell bound, ex-cept small quantities of LacYH1-Sak and SecYH1-Sak, whichwere also found in the culture supernatant. The fusion proteinsdetected in the cell fractions were bound to the CM of thecells. CcmAH1-Sak expressed in P. mirabilis LVIWEI was ex-clusively detected in membrane fractions, which were purifiedby ultracentrifugation or by Percoll density gradient centrifu-gation (Fig. 3). In combination with solubilization experiments,these results gave proof that the fusion proteins were embed-ded in the CM and did not form considerable amounts ofaggregates as described for other surface display systems (22).
The majority of the membrane-anchored Sak moleculeswere freely accessible to the protease trypsin, indicating thatthey were located on the outside of the cells. These findingswere further verified by freeze-fracture replica-labeling elec-
FIG. 5. Localization of CcmAH1-Sak in P. mirabilis LVIWEI by freeze-fracture, replica immunogold labeling, and electron microscopy.(A) Cell without plasmid and CcmAH1-Sak. Intramembrane particles on the EF faces of the plasma membrane are randomly distributed, and thebackground level of 10-nm immunogold particles (arrowhead) is very low. (B) EF face of a CcmAH1-Sak-expressing cell. Numerous Sak-indicatinggold particles are seen (arrowhead), which are almost exclusively found in areas of clustered intramembrane particles. The inset shows a moredetailed view of labeled Sak and particle clusters. (C) Cross-fracture view of a Sak-expressing cell. The labeling density of Sak in the cytoplasmis very low in contrast to the high density in the region of the CM (arrowhead). (D) PF face of a Sak-expressing cell. Almost no gold particles arelocated in the PF. The arrowhead indicates structures corresponding to the particle clusters on the EF faces. Bar, 100 nm.
530 HOISCHEN ET AL. APPL. ENVIRON. MICROBIOL.
on August 27, 2019 by guest
http://aem.asm
.org/D
ownloaded from

tron microscopy. The Sak molecules were mainly found in theEF face, and only traces could be detected in the cytoplasm orin the PF face. Obviously, the Sak moieties of the fusion pro-teins were translocated immediately after translation to theouter surface of the CM, where they remained fixed by theirmembrane anchors. Furthermore, comparison of the EF facesfrom overproducing cells and those which did not synthesizefusion proteins indicated that overexpression of CcmAH1-Sakcaused the formation of protein clusters in the EF face, whichconsisted of overexpressed fusion proteins and endogenousmembrane proteins. The molecules remained bound tightly tothe CM, even after repeated washes and ultrasonication. Themembrane-anchored Sak was functionally active. This indi-cated that the Sak molecules, when fused to anchor peptides,were correctly folded after translocation, although the cellslacked a periplasmic compartment and associated folding factors.
The L-form strains are free of or reduced in surface com-ponents, such as endotoxic LPS, pili, flagella, adhesins, andproteases, which are responsible for numerous interactions atthe cellular level, among them the binding to receptors, as wellas the induction of inflammatory processes or immune re-sponses (25, 29). This novel membrane surface display systemtherefore has potential in applications, such as the develop-ment of diagnostics and vaccines, and specific adhesin-receptorinteraction studies between bacterial and eukaryotic cells atthe cellular and molecular level (Gumpert et al., patent FN 10011 358.3 [pending]).
ACKNOWLEDGMENTS
C.F. and C.H. contributed equally to this study.We gratefully thank B. Küntzel, G. Elske, and S. Pfeiffer for excel-
lent technical assistance; M. Hartmann and S. Sieben for plasmids;E. J. Allan and J. Rippmann for critical reading of the manuscript; andStephan Diekmann for support.
This work was supported by a grant from the Deutsche Forschungs-gemeinschaft (Sonderforschungsbereich 197, TP A2).
REFERENCES
1. Akiyama, Y., and K. Ito. 1987. Topology analysis of the SecY protein, anintegral membrane protein involved in protein export in Escherichia coli.EMBO J. 6:3465–3470.
2. Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A.Smith, and K. Struhl (ed.). 1994. Current protocols in molecular biology.John Wiley & Sons, Inc., New York, N.Y.
3. Behnke, D., and D. Gerlach. 1987. Cloning and expression in Escherichia coli,Bacillus subtilis, and Streptococcus sanguis of a gene for staphylokinase—abacterial plasminogen activator. Mol. Gen. Genet. 210:528–534.
4. Bradford, M. M. 1976. A rapid and sensitive method for the quantification ofmicrogram quantities of proteins utilizing the principle of protein-dye bind-ing. Anal. Biochem. 72:248–254.
5. Bushueva, A. M., A. B. Shevelev, J. Gumpert, G. G. Chestukhina, A. V.Serkina, C. Hoischen, M. V. Matz, M. V. Kuryatova, and V. M. Stepanov.1998. Expression of the carboxypeptidase T gene from Thermoactinomycesvulgaris in stable cell wall-less L-forms of Proteus mirabilis. FEMS Microbiol.Lett. 159:145–150.
6. Cornelis, P. 2000. Expressing genes in different Escherichia coli compart-ments. Curr. Opin. Biotechnol. 11:450–454.
7. Cote-Sierra, J., E. Jongert, A. Bredan, D. Gautam, M. Parkhouse, P. Cor-nelis, P. De Baetselier, and H. Revets. 1998. A new membrane-bound OprIlipoprotein expression vector. High production of heterologous fusion pro-teins in gram-negative bacteria and the implications for oral vaccination.Gene 221:25–34.
8. Foster, D. L., M. Boublik, and H. R. Kaback. 1983. Structure of the laccarrier protein of Escherichia coli. J. Biol. Chem. 258:31–34.
9. Fujimoto, K. 1997. SDS-digested freeze-fracture replica labeling electronmicroscopy to study the two-dimensional distribution of integral membraneproteins and phospholipids in biomembranes: practical procedure, interpre-tation, and application. Histochem. Cell Biol. 107:87–96.
10. Georgiou, G., C. Stathopoulos, P. S. Daugherty, A. R. Nayak, B. L. Iverson,
and R. Curtiss III. 1997. Display of heterologous proteins on the surface ofmicroorganisms: from the screening of combinatorial libraries to live recom-binant vaccines. Nat. Biotechnol. 15:29–34.
11. Georgiou, G., D. L. Stephens, C. Stathopoulos, H. L. Poetschke, J. Menden-hall, and C. F. Earhart. 1996. Display of �-lactamase on the Escherichia colisurface: outer membrane phenotypes conferred by Lpp�-OmpA�-�-lacta-mase fusions. Protein Eng. 9:239–247.
12. Gumpert, J., and C. Hoischen. 1998. Use of cell wall-less bacteria (L-forms)for efficient expression and secretion of heterologous gene products. Curr.Opin. Biotechnol. 9:506–509.
13. Gumpert, J., and U. Taubeneck. 1983. Characteristic properties and biolog-ical significance of stable protoplast type L-forms. Experientia Suppl. 46:227–241.
14. Gumpert, J., H. Cron, R. Plapp, H. Niersbach, and C. Hoischen. 1996.Synthesis and secretion of recombinant penicillin G acylase in bacterialL-forms. J. Basic Microbiol. 36:89–98.
15. Hay, N. A., D. J. Tipper, D. Gygi, and C. Hughes. 1999. A novel membraneprotein influencing cell shape and multicellular swarming of Proteus mirabi-lis. J. Bacteriol. 181:2008–2016.
16. Hoischen, C., K. Gura, C. Luge, and J. Gumpert. 1997. Lipid and fatty acidcomposition of cytoplasmic membranes from Streptomyces hygroscopicus andits stable protoplast-type L-form. J. Bacteriol. 179:3430–3436.
17. Jung, H.-C., J.-M. Lebeault, and J.-G. Pan. 1998. Surface display of Zy-momonas mobilis levansucrase by using the ice-nucleation protein of Pseudo-monas syringae. Nat. Biotechnol. 16:576–580.
18. King, S. C., C. L. Hansen, and T. H. Wilson. 1991. The interaction betweenaspartic acid 237 and lysine 358 in the lactose carrier of Escherichia coli.Biochim. Biophys. Acta 25:177–186.
19. Konieczny, M. P., M. Suhr, A. Noll, I. B. Autenrieth, and A. M. Schmidt.2000. Cell surface presentation of recombinant (poly)peptides includingfunctional T-cell epitopes by the AIDA autotransporter system. FEMS Im-munol. Med. Microbiol. 27:321–332.
20. Kujau, M. J., C. Hoischen, D. Riesenberg, and J. Gumpert. 1998. Expressionand secretion of functional miniantibodies McPC603scFvDhlx in cell wall-less L-form strains of Proteus mirabilis and Escherichia coli. Appl. Microbiol.Biotechnol. 45:51–58.
21. Lång, H. 2000. Outer membrane proteins as surface display systems. Int.J. Med. Microbiol. 290:579–585.
22. Lång, H., M. Mäki, A. Rantakari, and T. K. Korhonen. 2000. Characteriza-tion of adhesive epitopes with the OmpS display system. Eur. J. Biochem.267:163–170.
23. Lee, J.-S., K.-S. Shin, J.-G. Pan, and C.-J. Kim. 2000. Surface-displayed viralantigens on Salmonella carrier vaccine. Nat. Biotechnol. 18:645–648.
24. Leenhouts, K., G. Buist, and J. Kok. 1999. Anchoring of proteins to lacticacid bacteria. Antonie Leeuwenhoek 76:367–376.
25. Liljeqvist, S., and S. Ståhl. 1999. Production of recombinant subunit vac-cines: protein immunogens, live delivery systems and nucleic acid vaccina-tion. J. Biotechnol. 73:1–33.
26. Lu, Z., K. S. Murray, V. Van Cleave, E. R. LaVallie, M. L. Stahl, and J. M.McCoy. 1995. Expression of thioredoxin random peptide libraries on theEscherichia coli cell surface as functional fusions to flagellin: a system de-signed for exploring protein-protein interactions. Bio/Technology 13:366–372.
27. Maurer, J., J. Jose, and T. F. Meyer. 2000. Autodisplay: one-componentsystem for efficient surface display and release of soluble recombinant pro-teins from Escherichia coli. J. Bacteriol. 179:794–804.
28. Morein, S., D. Henricson, and L. Riffors. 1994. Separation of inner and outermembrane vesicles from Escherichia coli in self-generating percoll gradients.Anal. Biochem. 216:47–51.
29. Oelschlaeger, T. A., and J. Hacker. 2000. Bacterial invasion into eukaryoticcells. Plenum Publishing Co., New York, N.Y.
30. Rippmann, J. F., M. Klein, C. Hoischen, B. Brocks, W. J. Rettig, J. Gumpert,K. Pfizenmaier, R. Mattes, and D. Moosmayer. 1998. Expression and secre-tion of recombinant scFv antibodies in L-form cells lead to active productand overcomes the limitations of periplasmic expression in E. coli. Appl.Environ. Microbiol. 64:4862–4869.
31. Schlott, B., K. H. Gührs, M. Hartmann, A. Röcker, and D. Collen. 1998.NH2-terminal structural motifs in staphylokinase required for plasminogenactivation. J. Biol. Chem. 273:22346–22350.
32. Sleytr, U. B., H. Bayley, M. Sára, A. Breitwieser, S. Küpcü, C. Mader, S.Weigert, F. M. Unger, P. Messner, B. Jahn-Schmid, B. Schuster, D. Pum, K.Douglas, N. A. Clark, J. T. Moore, T. A. Winningham, S. Levy, I. Frithsen,J. Pankovc, P. Beale, H. P. Gillis, D. A. Choutov, and K. P. Martin. 1997.Applications of S-layers. FEMS Microbiol. Rev. 20:151–175.
33. Ståhl, S., and M. Uhlén. 1997. Bacterial surface display: trends and progress.Trends Biotechnol. 15:185–192.
34. Tadayyon, M., J. R. Gittins, J. M. Pratt, and J. K. Broome-Smith. 1994.Expression of membrane proteins in Escherichia coli, p. 29–83. In G. W.Gould (ed.), Membrane protein expression systems: a user’s guide. PortlandPress, London, United Kingdom.
35. Valls, M., S. Atrian, V. de Lorenzo, and L. A. Fernández. 2000. Engineeringa mouse metallothionein on the cell surface of Ralstonia eutropha CH34 forimmobilization of heavy metals in soil. Nat. Biotechnol. 18:661–665.
VOL. 68, 2002 L-FORMS OF P. MIRABILIS AND E. COLI 531
on August 27, 2019 by guest
http://aem.asm
.org/D
ownloaded from