nu + l nucleophile substrate (leaving group,l)tpatric/ns.pdfaliphatic nucleophilic substitution l nu...
TRANSCRIPT

Aliphatic Nucleophilic Substitution
LNu + products
Nucleophile substrate(Leaving group,L)
conditions
Nucleophiles are chemical species that react with centers of positiveionic character. When the center is an aliphatic carbon, the process is calledaliphatic nucleophilic substitution. Chemical reactions of this type areextremely important for the synthesis of new compounds and forunderstanding the mechanisms in organic chemistry .All nucleophilic substitution reactions may take several reaction courses, butall have similar appearances at the outset.
All reactions have an attacking species, a nucleophile (Nu) that bears apair of electrons either as an anion or as a neutral compound. The organiccompound known as the substrate has a structure that greatly influences theoutcome of the reaction and it contains the leaving group (L) that is lost inthe reaction. The conditions of the reaction, especially solvent andtemperature, are also important contributors to the process.
In order to understand the products of the reaction and how they areformed, the reaction is studied from a mechanistic point of view.
The Nucleophilic Substitution Second-Order reaction (SN2)

The SN2 reaction occurs when a Nucleophile attack a primarysubstrate and sometimes a secondary substrate. The reaction of a secondarysubstrate depends on the nucleophile and the leaving group. Tertiarysubstrates do not undergo reactions by the SN2 mechanism. The overall rateof the reaction depends of the concentration of the nucleophile and theconcentration of the substrate, thus it is called second-order.
The mechanism requires that the nucleophile attack the substrate fromthe backside of the leaving group to give a pentavalent transition state (*).The organic substituents reverse their configuration (an S enantiomer wouldchange into an R enantiomer) The process is sometimes called Waldeninversion. Sometimes when the nucleophile has strongly basic characteristics,such as alkoxides, and alkene is formed also. This product is formed in areaction known as the E2 reaction (Elimination Second-order).
The transition state must be linear as shown from the reactions below.The carbanion must attach a methyl from another molecule (not internally)because a linear attack is not possible internally.
LNu +
Nu
SN2 E2
LNuδ δ+-
*
transition state
Inversion ofconfiguration
Primary (benzylic>aliphatic)>secondary X tertirary
+
reactionrate
= k [Nu][Substrate}
O2S
OCH3
SO2
H3C O2S
O
SO2
H3C
CH3
O2S
H3CO
O2S
CH3
NO
YES
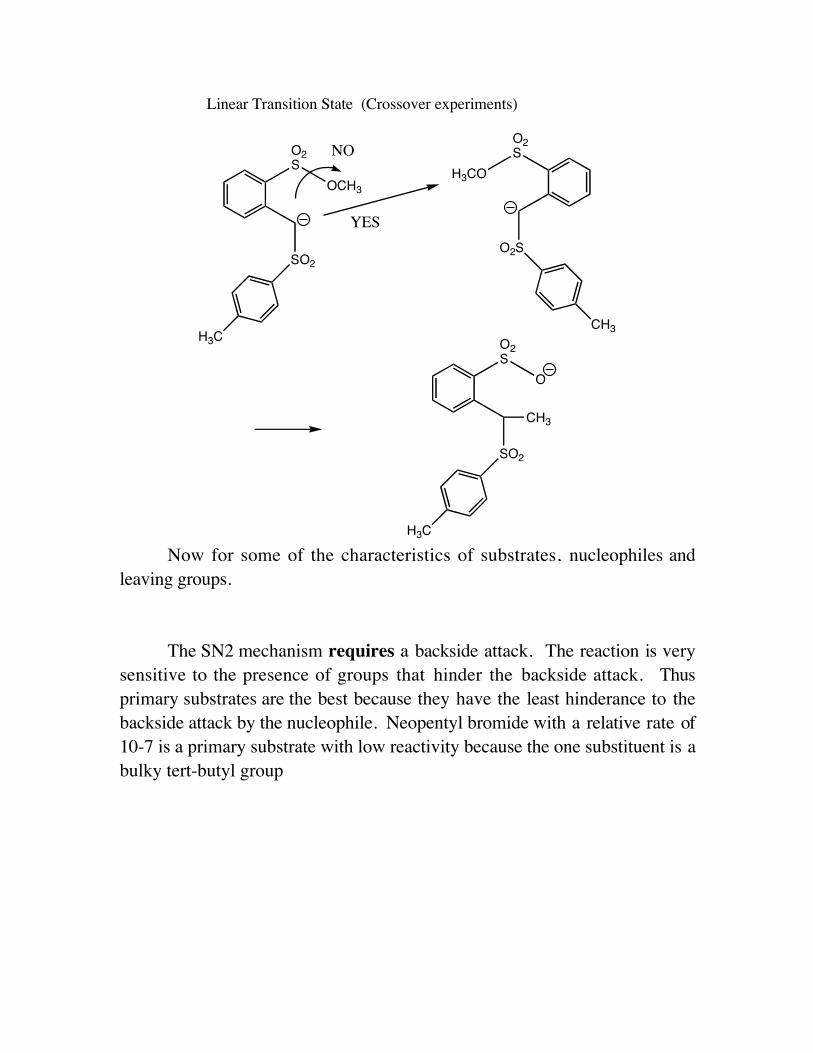
Linear Transition State (Crossover experiments)
Now for some of the characteristics of substrates, nucleophiles andleaving groups.
The SN2 mechanism requires a backside attack. The reaction is verysensitive to the presence of groups that hinder the backside attack. Thusprimary substrates are the best because they have the least hinderance to thebackside attack by the nucleophile. Neopentyl bromide with a relative rate of10-7 is a primary substrate with low reactivity because the one substituent is abulky tert-butyl group

Substrate Reactivity
1o-benzylic > CH3 > 10-aliphatic >> 20 not at all 30
1o-allylic
CH3Br
CH3CH2Br
CH3CHBrCH3
CH3CBrCH3
CH3
CH3C-CH2BrCH3
CH3PhCH2Br
Br
Br
1
.01
10-5
10-7
2.3
4.0
.001
.01
CH2=CHCH2Br
.001
relative reactivity of several aliphatic bromides
Some properties of nucleophiles are give by the Swain-Scott equation.

Leaving groups are very important in the SN2 process. Some of thevery best leaving groups are not halogens, but instead are esters of sulfonic acidsthat are easily prepared from alcohols and sulfonyl anhydrides or sulfonylchlorides. The use of a triflate or tosylate with a secondary substrate is effectivefor increasing the amount of SN2 reaction when other reactions are compete.
RS > ArS > S2O3 > I > CN > SCN > OH > N3 > ArO > AcO
Nucleophiles
n log(k / kCH3I CH3OH nu= )
n = nucleophilicity constant. Compares nucleophiles in the reactionwith methyl iodide. The standard nucleophile is water.
CH3COO-
N3-
CH3O-
NH2OH-CN
t-BuO-
Ph3P
H2O
n pKa4.35.8
6.3
6.6
6.7
4.0
8.7
0.0
4.84.7
16
5.8
9.3
20
8.7
7

CF3SO3- (Tf, triflate)
CH3PhSO3- (Ts, tosylate)
CH3SO3- (Ms, mesylate)
I-
Br-
CF3COO-
Cl-
F-
CH3COO-
108
105
104
91
14
2
1
10-6
10-6
Leaving Groups
Lrelativerate
The Elimination Reaction
The formation of an alkene can occur as a competing reaction with theSN2 process. The proportion of elimination, E2, depends also on thesubstrate, the basicity of the nucleophile, the leaving group and thetemperature. The E2 reaction rate depends on the concentration of thenucleophile (base in this case) and the concentration of the substrate and istherefore second-order. The elimination requires hydrogens in the betaposition and the hydrogen eliminates best when it is anti-perplaner to theleaving group.

H
L
Base
E2
β α
L
HHH
HH
H
HHH
BE2 only
Good bases for the reaction are alkoxides and nitrogen anions. Thepresence of more beta hydrogens causes more elimination product, thuselimination increases with secondary substrates and is the only product of atertiary substrate with a base. Carbon-hydrogen bonds are stronger thancarbon-leaving group bonds, thus higher temperatures aid the eliminationprocess.

Substitution Nucleophilic First-order (SN1)
Another reaction of organic substrates with leaving groups is a first-order reaction.This means that the rate of the reaction does not depend on the nucleophile concentration,but depends only on the concentration of the substrate. The final outcome of the reactionmay be substitution but elimination and rearrangement are often present.
rate = k [S]The mechanism for the reaction is shown below. In this process the leaving group
departs because of the substrate structure and its interaction with the solvent to form anintermediate called a carbocation. The carbocation is a real-life species with a finite liftimeand thus is not just a transition state.
LSN1 , E1 and rearrangement
a carbocation
Only substrates that can produce a carbocation with reasonable stability canparticipate in the SN1 process. This leaves out methyl and primary substrates as theircations are just too high energy, unless there is some adjacent stabilizing group (such asphenyl or CH3O). Thus the secondary and expecially tertiary substrates are the best. A listof carbocation stability is shown below.
benzylicallylic 3o 2o 1oPh+> > > > >C=C+
SN1 reactions are sometimes referred to as solvolysis reactions, meaning reactioninduced by the solvent. Most solvents are polar to interact with the ionic leaving group andto stabilize the polar carbocation intermediate. The study of the SN1 process centersaround the chemistry of carbocations and factors that influence their stability.
Solvents in SolvolysisThe reactions in this section take place because the substrate forms a relatively stable
carbocation in the presence of the solvent. Thus solvolysis means reaction with the solvent,

and the term implies a carbocation mechanism. The rate of a solvolysis reactio, an Sn1reaction, does not depend on the concentration of any nucleophile and thus the term firstorder, first order in substrate. rate (solvolysis) = k [RX].
Many studies of these reactions in different solvents have led to importantdeterminations of solvent character, or the ability of a solvent to cause a substrate to form acarbocation. An early scale, and still in use, was developed by Grunwald and Winstein in1940. The defined a value Y as the ionizing power of a solvent from the equation below.Experimentally the rate constant for ionization of t-butyl chloride in 80% ethanol-20% wateris determined. Then the rate constant for the ionization of t-butyl chloride in other solventsin determined and the results are substituted in the equation.
lnk(solvent)
k(80% EtOH-20% water)Y =
for t-butyl chloride as substrate
Some values of Y for several solvents are listed below. Thus formation of carbocations bysolvolysis occurs better in solvents with higher Y values.Solvent YCF3COOH 1.84H2O 3.4980% EtOH 0.0HCOOH 2.0520% EtOH 3.05EtOH -2.03
The Y scale was refined to a Y (OTs) scale with the use of another substrate, adamantyltosylate. The new substrate did not permit any backside attack by the solvent on the cationsite and thus is a better measure of pure ionizing ability .
OTs

Some new Y values are for CF3COOH (4.57) and CF3CH2OH (1.8). The higher value oftrifluoroacetic acid with the adamantyl system shows that some backside assistance occureswhen t-buty chloride is the substrate.
Bently and Schleyer in 1972 founded a method to determine the nucleoplicity (N) ofa solvent. The N for several solvents is listed: CF3COOH (-5.6), HCOOH (-3.0), H2O (-.41), 20%EtOH ( -.09). Larger values indicate more solvent nucleophilicity. Here 20%EtOH is the greatest.
Stereochemistry and Ion PairsThe carbocation intermediate is a flat sp2 hybrid with a vacant p orbital.
Nucleophiles could be expected to attact the p orbial equaly from the top or bottom. If achiral substrate were used, the expected product would be racemic as shown below. Theexample shows a secondary substrate with R configuration giving a planar carbocation thatis attacked by the solvent to give the R and S product mixture (after loss of a proton).
CH3
IPhH CH3
HPh
a carbocation
CH3OH
R enantiomerCH3
H3CO PhH
R enantiomer
CH3
OCH3PhH
S enantiomer
The example above is often presented as the final story in undergraduate organicchemistry. But, many carbocation reactions with chiral substrates give products that showsome of inversion of configureation and sometimes retention of configuration during theprocess. These observations give rise to a more detailed description of the carbocationintermediate as existing as a series of ion pairs. The substrate first solvates into tight ionpairs, then to solvated ion pairs, and finally to the free carbocation. It is only the freecarbocation (shown above) that gives the racemization reaction. The ion pairs requiredifferent stereochemical outsomes depending on the reaction condition, and many SN1reactions occur from the ion pairs before the free ion is formed.
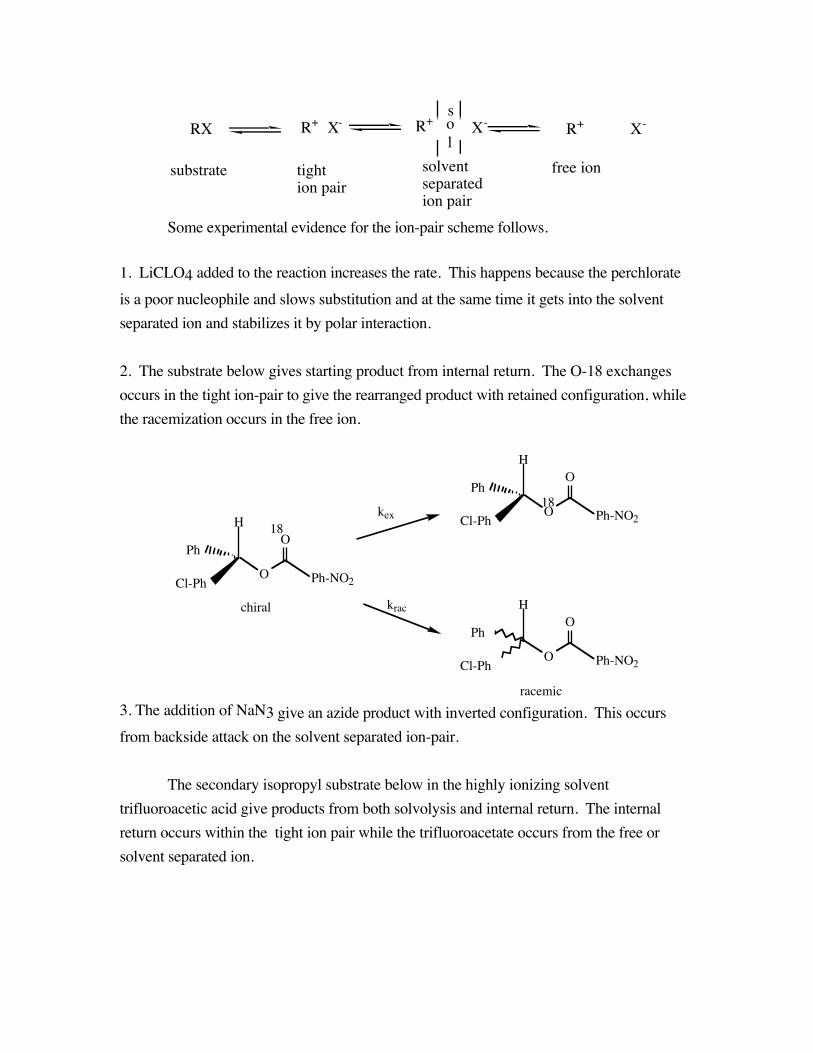
RX R+ X- R+ X- R+ X-sol
substrate tightion pair
solvent separatedion pair
free ion
Some experimental evidence for the ion-pair scheme follows.
1. LiCLO4 added to the reaction increases the rate. This happens because the perchlorateis a poor nucleophile and slows substitution and at the same time it gets into the solventseparated ion and stabilizes it by polar interaction.
2. The substrate below gives starting product from internal return. The O-18 exchangesoccurs in the tight ion-pair to give the rearranged product with retained configuration, whilethe racemization occurs in the free ion.
OCl-Ph
Ph
HO
Ph-NO2
18O
Cl-Ph
Ph
HO
Ph-NO218
OCl-Ph
Ph
HO
Ph-NO2
kex
kracchiral
racemic3. The addition of NaN3 give an azide product with inverted configuration. This occursfrom backside attack on the solvent separated ion-pair.
The secondary isopropyl substrate below in the highly ionizing solventtrifluoroacetic acid give products from both solvolysis and internal return. The internalreturn occurs within the tight ion pair while the trifluoroacetate occurs from the free orsolvent separated ion.

O S Ph
O
O
18
18
O S Ph
O
O
18
18O C-CF3
O
ks = 36 x 10-4 s-1ki = 8 x 10-4 s-1
ks / ki = 4.5
CF3COOH
The Chemistry of CarbocationsReactions that proceed by SN1 or E1 mechanisms are reactions that involve
carbocation intermediates. The study of carbocation intermediates involves learning aboutthe factors that influence the stability (or instability) of the intermediate. In some cases thisbecomes interesting from the academic view of learning about reaction mechanisms and insome cases practical synthetic procedures result.
Substituent Effects on CarbocationsThe cationic charge is stabilized by substituents that can donate electrons to help
reduce the localized charge. Thus any substituent or function that contains non-bonding pelectrons or contains pi electrons can stabilize a carbocation. The structures below showhow different functions can stabilize a carbocation.
A fluorine atom alpha to a carbocation center is stabilizing. The 6 non-bondingelectrons on the F are in 2p orbitals that are of the correct size to overlap with the 2p cationicsite. The stabilizing effect is more noted as a directing effect than as a rate enhancing effect.Fluorine atoms beta to a carbocation center are strongly destabilizing and simple betafluoroalkyl carbocations are unknown. Fluorne atoms more remote from the cationic center,especially in the beta position are very destabilizing.Thus fluorine atoms destabilze thetripheny cation system and slow the solvolysis of benzylic tosylates as shown below. Butexamples are alpha stablization are well know and are used to control some carbocationreactions as is also shown below. (Banks, Smart, Tatlow, Organofluorine Chem. 1994)..

Ph-X
Ph-XC
X-Ph X pKR
H
CH3
OCH3
NO 2
(CH3)2N
-6.6
-3.6
+0.8
-16.2
+9.4
PhCH2
Ph2CH
-20
larger positive pKRis more stable cation
benzylicallylic 30
NH2
10Ph>
CH3O
20
OH
-13.3
CH3C=C> > >XX
Ph CH3 F CN C=O NO2;> > > > > > > > >
C6F4-X
C6F4-XC
X-C6F4 F
CF3
-17.7
-26
Ph-C-CH3
H
OTsPh-C-CH3
CF3
OTs
rel rate 105 rel rate 1
C-CF3
unfavorable β influence of fluorine atoms
CH3O-CH2 CH3O=CH2
CH3CH-F CH3CH=F
Fluorine alpha to a carbocation stabilized the carbocationin a manner similar to oxygen stabilization.
CH-C=O CH=C-O+ not good
H
OTs
CN
OTs
H
OTs
CN
1 10-3 10-5
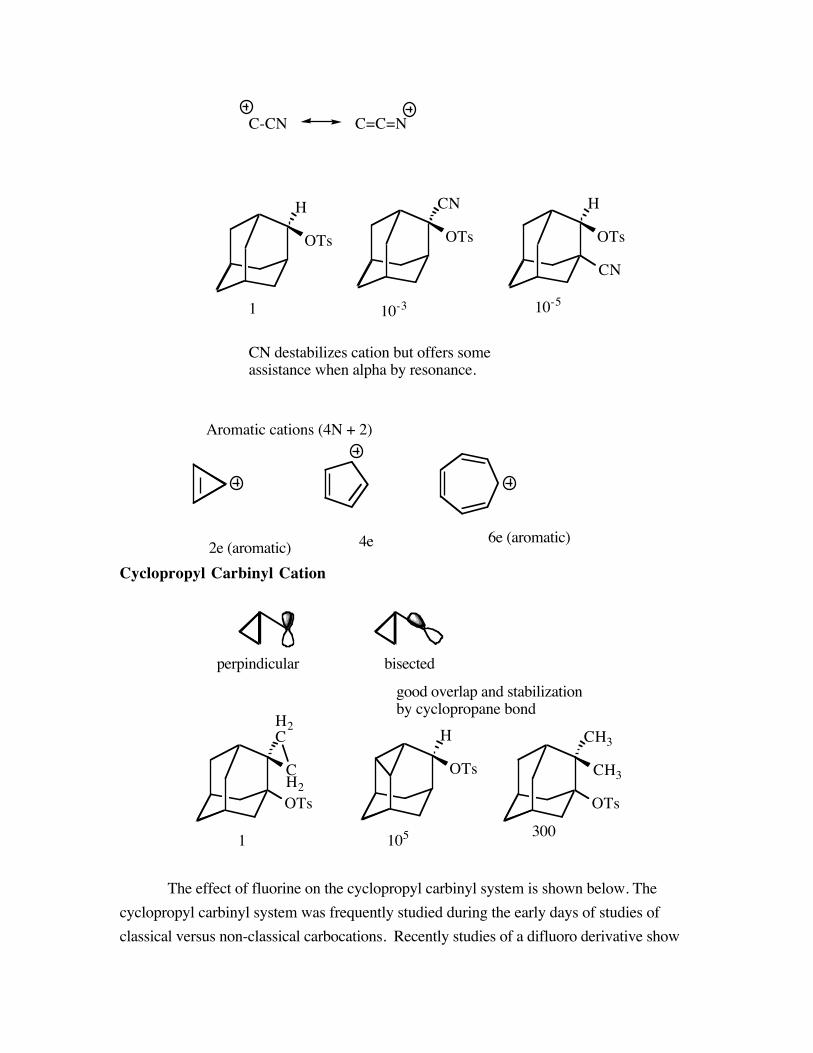
C-CN C=C=N
CN destabilizes cation but offers someassistance when alpha by resonance.
2e (aromatic) 4e 6e (aromatic)
Aromatic cations (4N + 2)
Cyclopropyl Carbinyl Cation
H
OTs
CH3
CH3
OTs
H2C
CH2OTs
3001051
perpindicular bisected
good overlap and stabilizationby cyclopropane bond
The effect of fluorine on the cyclopropyl carbinyl system is shown below. Thecyclopropyl carbinyl system was frequently studied during the early days of studies ofclassical versus non-classical carbocations. Recently studies of a difluoro derivative show

some interesting results. The relative rates of solvolysis of the parent system, the isobutylsystem and the difluoro system show that the two fluorine atoms on the cyclopropane ringslow the rate of solvolysis.
In the parent system usually three products are observed as shown below.
In the difluoro system only two products are observed and the one with the cyclopropanering intact occurs mainly from SN2 attack on the primary carbon. The difluoro alkeneshows the strong directing effect of the fluorine atom on the carbocation, but the first orderrate constant for solvolysis is about 2 x 10 -5 sec-1, which is about105 smaller than theparent hydrogen system. The absence of the cyclobutyl product shows the strongdestabilization of the fluoine atoms on the beta or gamma carbocation.
H
H
OTsH
H
OTs
F
FH
H
OTs
105 1.0 1.8
H
H
OTsH
H
H
HHOAc
H
H
OAc
OAc
OAc+ +
H
H
OTs
F
F H
HF
F
F
F
H
HF
FOAc
F
FAcO
OAc
FF
39 % SN2 56 %0 %
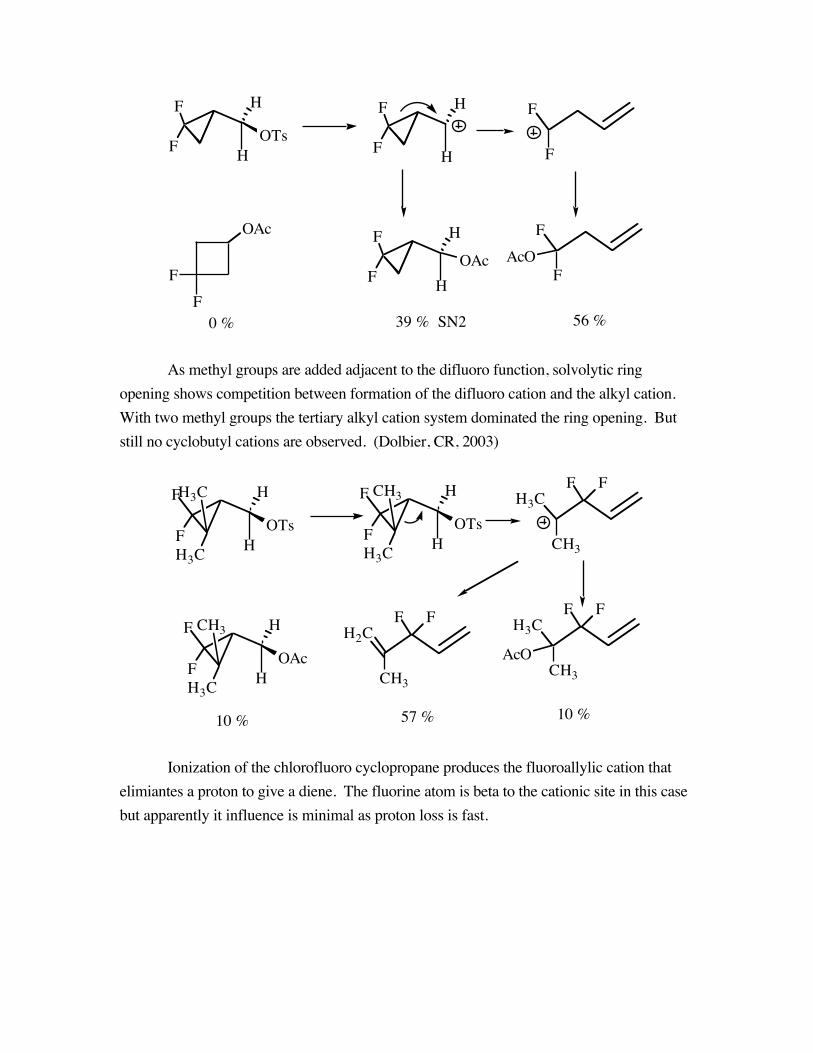
As methyl groups are added adjacent to the difluoro function, solvolytic ringopening shows competition between formation of the difluoro cation and the alkyl cation.With two methyl groups the tertiary alkyl cation system dominated the ring opening. Butstill no cyclobutyl cations are observed. (Dolbier, CR, 2003)
H
H
OTs
F
F
H3C
CH3H3C
H3C F F
H
H
OTs
F
FH3C
CH3
H
H
OAc
F
FH3C
CH3 H3C
CH3
F F
AcOH2C
CH3
F F
10 % 57 % 10 %
Ionization of the chlorofluoro cyclopropane produces the fluoroallylic cation thatelimiantes a proton to give a diene. The fluorine atom is beta to the cationic site in this casebut apparently it influence is minimal as proton loss is fast.
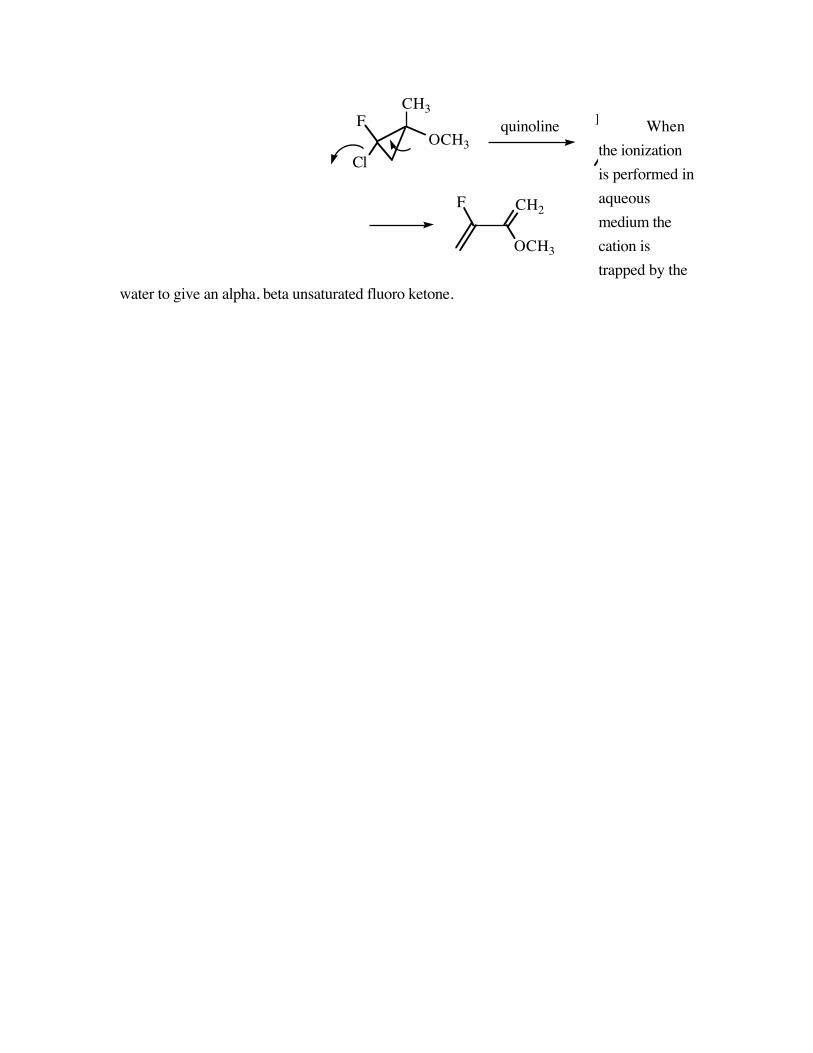
Whenthe ionizationis performed inaqueousmedium thecation istrapped by the
water to give an alpha, beta unsaturated fluoro ketone.
OCH3F
Cl
CH3quinoline
OCH3
F CH3
OCH3
F CH2
OCH3F
Cl
CH3
OCH3
F CH3
OCH3
F CH3H20
H20
OHO
F CH3
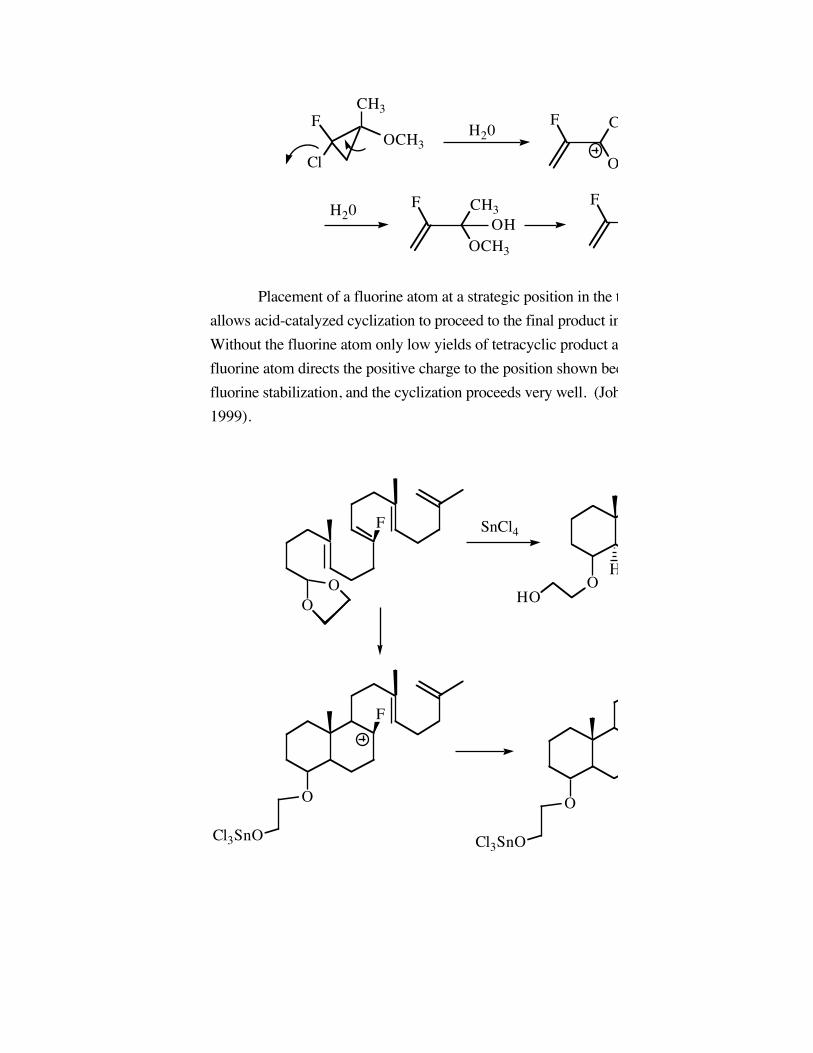
Placement of a fluorine atom at a strategic position in the trienes system belowallows acid-catalyzed cyclization to proceed to the final product in 86 % yield.Without the fluorine atom only low yields of tetracyclic product are obtained. Thefluorine atom directs the positive charge to the position shown because of alphafluorine stabilization, and the cyclization proceeds very well. (Johnson, JACS 493,1999).
O
F
H H
H
F
OO
HO
SnCl4
86 %F = H, low yield
F
O
Cl3SnO
F
O
Cl3SnO
H2O

Neighboring groups and RearrangementsCase 1. The exo bicyclic tosylate undergoes solvolysis 1011 times faster than the endoisomer. The happens because the pi bond assists the formation of the carbocation. Theassistance is much better from the backside of the tosyl group.
TsO OTs
10111
OTs
Case 2. The trans tosylate undergoes solvolysis 1000 times faster than the cis isomer. thenon bonding electrons of the carbonyl group assist the formation of the developing charge,and again the assistance is much better from the backside of the leaving group.
OO
CH3OTs
O-C-CH3
OO-C-CH3
O
OTs
10001Case 3. Although primary cations are not formed in solvolysis reactions, neighboringgroups can assist the solvolysis of a primary substrate in a synchronous step to form astable carbocation. Rearrangement occurs in beta pheny substrates because the phenylgroup donates electrons, from the backside, to give a stable cyclic phenonium ion.
CH2CH2-OTs
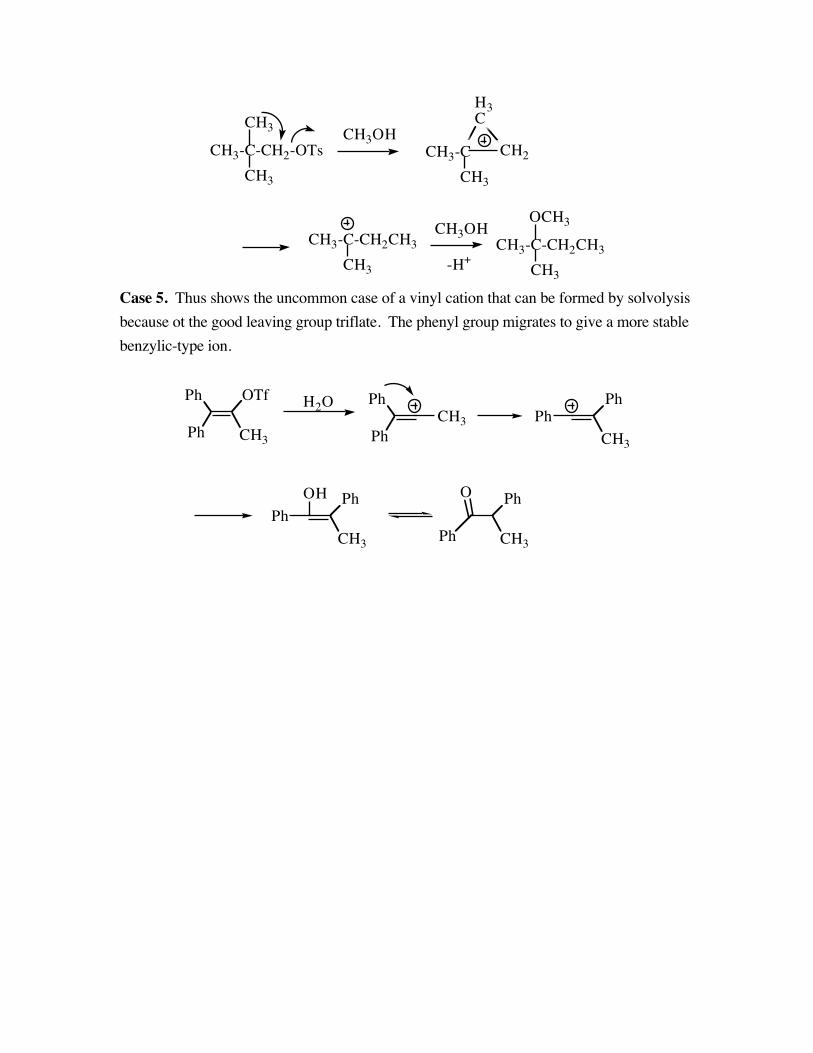
Case 4. The neopentyl system , another primary substrate, rearranges rapidly to give theproduct shown. The methyl group assists the formation of a charge in a cyclic ion to givethe more stable tertiary carbocation.
CH3
CH3
H3C
CH3
CH2
CH3 CH3
OCH3
CH3-C-CH2-OTs CH3-C
CH3-C-CH2CH3 CH3-C-CH2CH3-H+
CH3OH
CH3OH
Case 5. Thus shows the uncommon case of a vinyl cation that can be formed by solvolysisbecause ot the good leaving group triflate. The phenyl group migrates to give a more stablebenzylic-type ion.
Ph
Ph
OTf
CH3
Ph
PhCH3 Ph
Ph
CH3
PhPh
CH3
OH
Ph
Ph
CH3
O
H2O

Norbornyl Systems
OBs H OBs
OBsH H350 1 0.3
classical non-classical
chiral achiral
Observed byC NMR
Chiral substrate give achiral product.
exo endo
Only exo products are obtained on solvolysis.
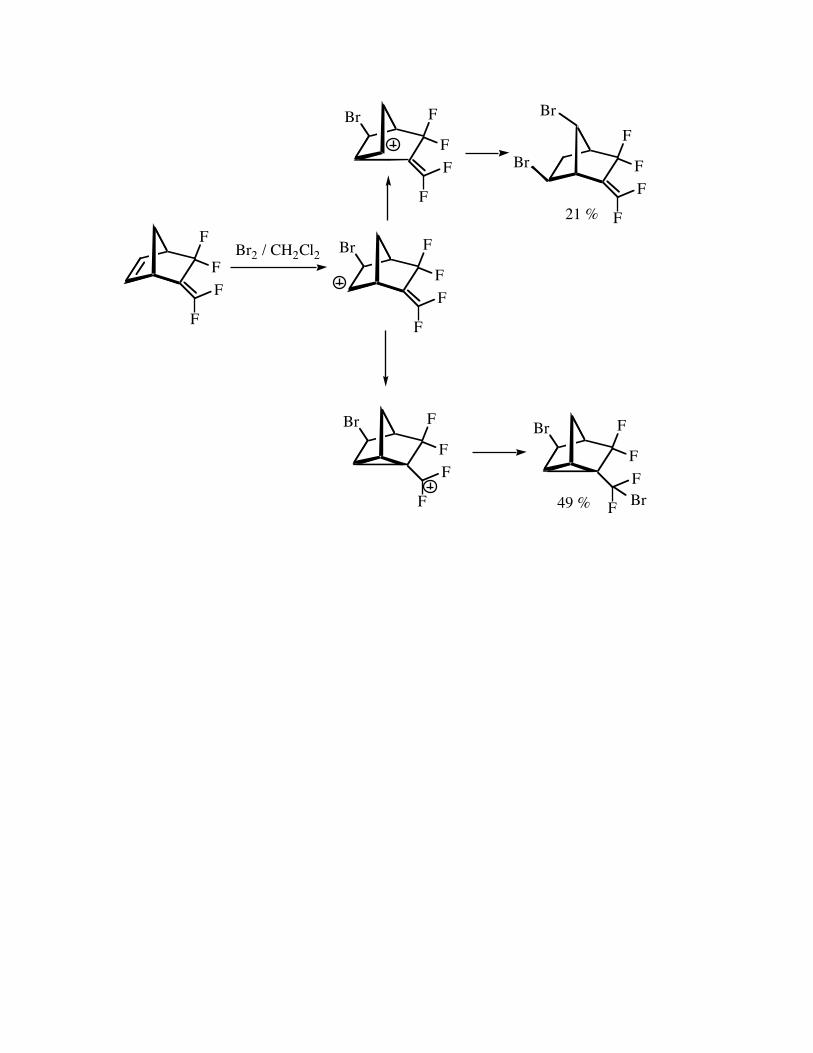
The fluorinated norbornyl system shown below undergoes rearrangement ontreatment with Br2 under ionic conditions. The product produced in 49% yield shows theinfluence of the fluorine atoms of the exocyclicdifluoro ethylene function by donating the pielectrons in order to achieve a fluorine stabilized carbocation that reacts with bromide ion.(Smart, JOC, 831, 1974).
F
F
F
F
F
F
F
F
Br
F
F
F
F
Br Br
Br
F
F
F
F
F
F
F
F
Br
F
F
F
F
Br
Br
Br2 / CH2Cl2
49 %
21 %
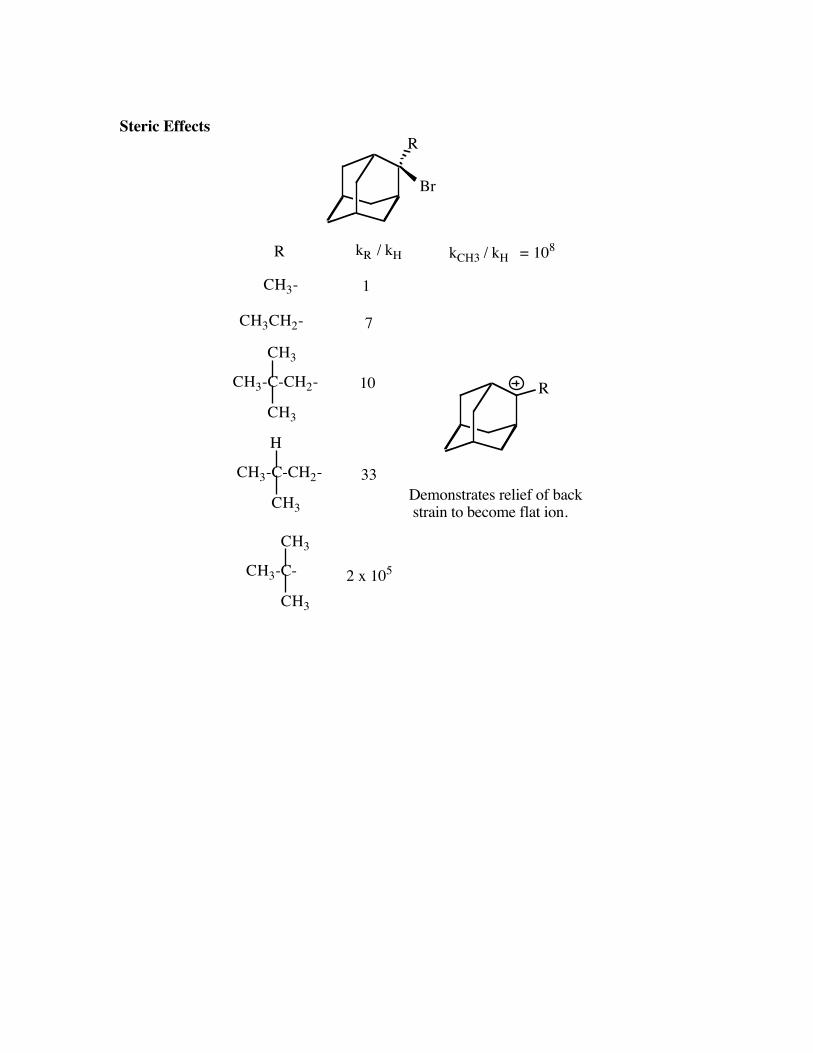
Steric EffectsR
Br
CH3
CH3
H
CH3
CH3
CH3
R kR / kH
CH3-
CH3CH2-
CH3-C-CH2-
CH3-C-CH2-
CH3-C-
R
1
7
10
33
2 x 105
kCH3 / kH = 108
Demonstrates relief of back strain to become flat ion.
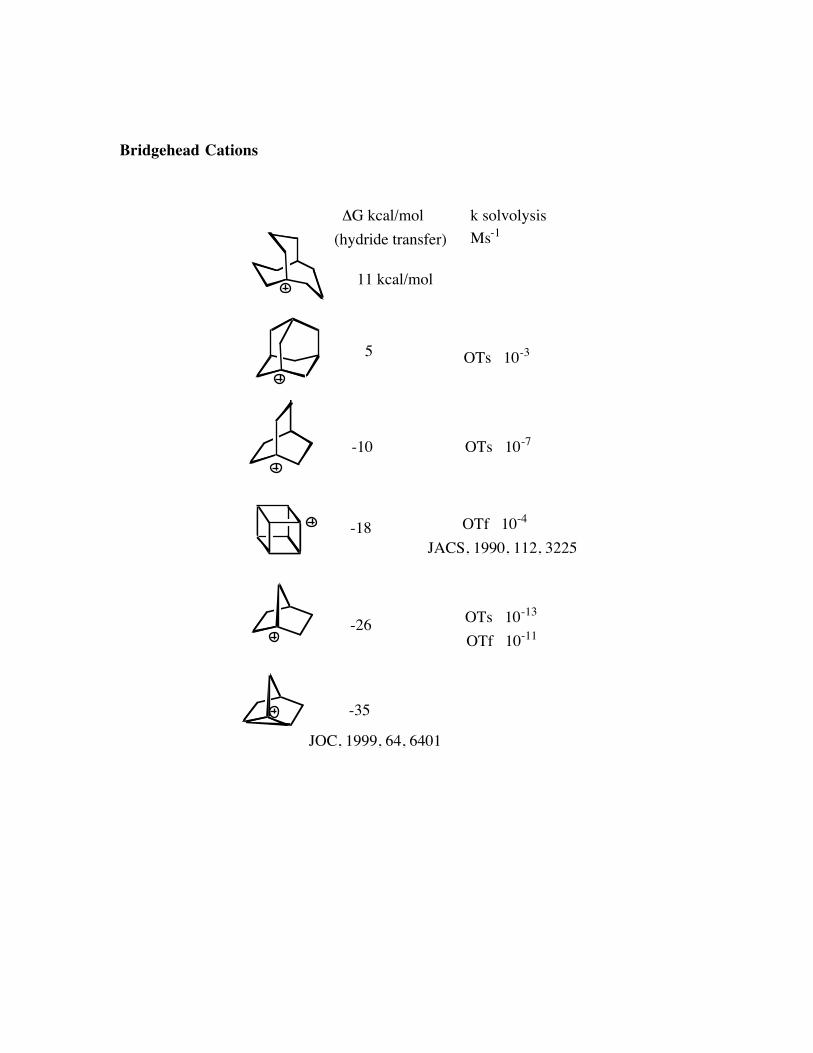
Bridgehead Cations
-18
-10
5
-26
-35
11 kcal/mol
JOC, 1999, 64, 6401
ΔG kcal/mol(hydride transfer)
OTs 10-3
OTs 10-7
OTf 10-4
OTs 10-13
OTf 10-11
k solvolysisMs-1
JACS, 1990, 112, 3225