organic ion radicals: chemistry and applications elektronike/organic_ion... · common property of...
TRANSCRIPT

TM
Marcel Dekker, Inc. New York • Basel
Zory V. TodresColumbus, Ohio, U.S.A.
ORGANICION RADICALSChemistry and Applications
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

ISBN: 0-8247-0810-5
This book is printed on acid-free paper.
HeadquartersMarcel Dekker, Inc.270 Madison Avenue, New York, NY 10016tel: 212-696-9000; fax: 212-685-4540
Eastern Hemisphere DistributionMarcel Dekker AGHutgasse 4, Postfach 812, CH-4001 Basel, Switzerlandtel: 41-61-260-6300; fax: 41-61-260-6333
World Wide Webhttp://www.dekker.com
The publisher offers discounts on this book when ordered in bulk quantities. For more information,write to Special Sales/Professional Marketing at the headquarters address above.
Copyright © 2003 by Marcel Dekker, Inc. All Rights Reserved.
Neither this book nor any part may be reproduced or transmitted in any form or by any means, elec-tronic or mechanical, including photocopying, microfilming, and recording, or by any informationstorage and retrieval system, without permission in writing from the publisher.
Current printing (last digit):10 9 8 7 6 5 4 3 2 1
PRINTED IN THE UNITED STATES OF AMERICA
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

To my wife, IrinaThe cloudless beauty of her heart, the profundity of her mind, and
the depth of her feelings have always provided reliablesupport for me. For my children, Vladimir and Ellen, their
mother represents a supreme example.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Foreword
This volume presents an overview of the widely varied subject matter that is relevant to un-derstanding the chemistry of organic ion radicals. Detailed consideration is given to all as-pects of ion radical solution chemistry, from discussing how ion radical structures are de-scribed theoretically to presenting the reagents commonly used to generate them, methodsfor their characterization, and synthetic methods based on ion radicals. Less usual topicsare covered in depth in chapters on how to identify and study reactions proceeding throughion radicals, and how to optimize yields for such reactions. A chapter is devoted to solventeffects, which are especially important for such reactions.
Examination of the practical applications of ion radicals includes coverage of theirroles in biological systems as well as in material chemistry, ranging from optoelectronics,organic metals, and magnets to lubricants and the manufacture of paper.
This book gathers into a single place the widely scattered material needed to considerion radicals from an organic chemist’s point of view. A highly useful aspect of this book isits incorporation of relevant studies from the Russian literature, a significant amount of itoriginating from Todres’ own group in Moscow. Little of this material has previously re-ceived proper emphasis, or even citation, in the English-language literature.
Stephen F. Nelsen, Ph.D.Professor
Department of ChemistryUniversity of Wisconsin
Madison, Wisconsin
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Preface
Contemporary organic chemistry puts great emphasis on investigations of the structure andreactivity of intermediate species, originating in the pathway from the starting compoundsto the end products. Knowledge of properties of the intermediate species and penetrationinto the mechanism of reactions open new routes to increase the rate of formation and theyield of the desired final products. Until recently, chemists focused their attention on radi-cals or charged species of the cationic/anionic type. Species of an intermediate nature com-bining ionic and radical properties—ion radicals—remained outside the scope of their in-vestigations. Perfected instrumental techniques markedly advanced fine experiments. As aresult, the species, which were little (if at all) known to the chemists of previous decades,now came to the forefront.
The behavior of organic ion radicals has become an area of current interest. Ion rad-icals arise by one-electron oxidation or one-electron reduction of organic compounds inisolated redox processes and as intermediates along the pathways of reactions. A conver-sion of an organic molecule into an ion radical brings about a significant change in its elec-tron structure and a corresponding alteration in its reactivity. This conversion allows nec-essary products to be obtained under mild conditions with high yields and improvedselectivity of transformation. In addition, there are several reactions that can proceed onlyby the ion radical mechanism and lead to products otherwise inaccessible.
The theme of the book is the formation, transformation, and application of ion radi-cals in typical conditions of organic synthesis. The book presents an overview of organic ionradical reactions and explains the principles of ion radical organic chemistry. Methods ofdetermining ion-radical mechanisms and controlling ion radical reactions are also reviewed.
When applicable, issues relating to ecology and biology are addressed. The inorganicparticipants in the ion radical organic reactions are also considered. One of the chaptersgives representative synthetic procedures and considers the background of related syntheticapproaches.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The book also provides a review of current practical applications as well as an out-look on those predicted to be important in the near future. The reader will learn of theprogress that has been made in technical developments utilizing the organic ion radicals.Electronic and optoelectronic devices, organic magnets and conductors, lubricants, andother applications are considered.
The behavior of organic ion radicals is still not completely understood. Thus, new in-terpretations of scientific data appear frequently in the literature. I have attempted to syn-thesize the ideas from various references that complement one another, although the con-nections among them may not be immediately obvious. (An Author Index is included tohelp readers find such connections in the book.)
Science is a collective affair, and its main task is to produce trustworthy and generalknowledge. My apologies are due to any authors who have contributed significantly to thedevelopment of this vast field but, for various reasons, have not been cited. The many con-tributors who are cited certainly do not reflect my preferences; their publications have beenselected as illustrative examples that may allow the reader to follow evolution of the cor-responding topics.
Every new branch of science passes through several stages in its progress, includingthe latent phase and the phase of increased interest. These initial phases have apparently al-ready passed in organic ion radical chemistry; they spawned decades of heated debates. Inrecent years, however, the boil has cooled to a simmer. The development of organic ion rad-ical chemistry, especially with respect to its practical applications, has allowed chemists tonail down ion radicals more confidently. Having become a regular division of generalknowledge, organic chemistry of ion radicals is now entering the third stage of develop-ment, namely, putting the ideas elaborated into general operation, including them in thecommon property of organic chemistry. It is now necessary to generalize the obtained dataand to treat them comprehensively. Grafting the new branch to the organic chemistry treeis the aim of this book.
I have worked in the field of organic ion radicals and their applications for severaldecades and have become more and more fascinated by the beauty of this area and the di-versity it presents. Understanding the role of ion radicals is as difficult as it is interesting. Ihope that this attempt to graft this branch to the organic chemistry tree will be useful bothfor further advancing basic research and for facilitating new practical applications.
During my entire working life, I, like other researchers, have felt the pressure of thescientific community’s appraisal. Criticism is crucial! The writing of this book was aidedby discussions with my colleagues and friends. I am indebted to all of them for their cor-rections and polemics. At the same time, their support was a great incentive. The scientificatmosphere that surrounded me during my tenure at the Research and Educational Instituteof the Cleveland Clinic Foundation, the time when I was writing the book, also stimulatedmy creative work.
The book is meant for researchers and technologists who are carrying out synthesesand studying principles governing the choice of optimal organic reaction conditions. It willbe useful for physical organic chemists, ecologists, biologists, and specialists in electron-ics of organic materials, as well as professors, researchers, and students. As for students, Ihave assumed that the reader is not acquainted with the field itself but possesses back-ground knowledge in chemistry, both general and organic, a knowledge usually providedduring undergraduate studies in most, if not in all, countries.
Zory V. Todres
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Contents
ForewordPreface
Chapter 1. Nature of Organic Ion Radicals and Their Ground-State ElectronStructure
1.1 Introduction1.2 Unusual Features1.3 Acid–Base Properties of Organic Ion Radicals1.4 Metallocomplex Ion Radicals1.5 Organic Ion Radicals with Several Unpaired Electrons and/or Charges1.6 Polymeric Ion Radicals1.7 Inorganic Ion Radicals in Reactions with Organic Substrates1.8 Conclusion
References
Chapter 2. Formation of Organic Ion Radicals2.1 Introduction2.2 Chemical Methods of Organic Ion Radical Preparation2.3 Equilibria in Liquid-Phase Electron-Transfer Reactions2.4 Electrochemical Methods Versus Chemical Methods2.5 Formation of Organic Ion Radicals in Living Organisms2.6 Organic Ion Radicals in Solid Phases2.7 Isotope-Containing Organic Compounds as Ion Radical Precursors2.8 Conclusion
References
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Chapter 3. Basic Principles of Organic Ion Radical Reactivity3.1 Introduction3.2 Principle of the “Detained” Electron That Controls Ion Radical Reactivity3.3 Principle of the “Released” Electron That Controls Ion Radical Reactivity3.4 Behavior of Organic Ion Radicals in Living Organisms3.5 Conclusion
References
Chapter 4. How to Discern Ion Radical Mechanisms of Organic Reactions4.1 Introduction4.2 Why Does a Reaction Choose the Ion Radical Mechanism?4.3 Chemical Approaches to the Identification of Ion Radical Reactions4.4 Physical Approaches to the Identification of Ion Radical Reactions4.5 Examples of Complex Approaches to the Discernment of the Ion Radical
Mechanism of Particular Reactions4.6 Conclusion
References
Chapter 5. How to Optimize Organic Ion Radical Reactions5.1 Introduction5.2 Physical Effects5.3 Effect of Chemical Additives5.4 Solvent Role5.5 Salt Effects5.6 Conclusion
References
Chapter 6. Organic Ion Radicals in Synthesis6.1 Introduction6.2 Reductive Reactions6.3 Ion Radical Polymerization6.4 Cyclization6.5 Ring Opening6.6 Fragmentation6.7 Bond Formation6.8 Conclusion
References
Chapter 7. Practical Applications of Organic Ion Radicals7.1 Introduction7.2 Organic Ion Radicals in Optoelectronics7.3 Organic Metals7.4 Organic Magnets7.5 Organic Lubricants7.6 Ion Radical Routes to Lignin Treatment7.7 Conclusion
References
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Chapter 8. Concluding Remarks8.1 Introduction8.2 SRN1 Reaction8.3 Stereochemical Aspects of Ion Radical Reactivity8.4 Conclusion
References
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

1
Nature of Organic Ion Radicals andTheir Ground-State Electron Structure
1.1 INTRODUCTION
Organic chemistry represents an extensive body of facts, from which the contemporarydoctrine of reactivity is built. The most important basis of this doctrine is the idea of inter-mediate species that arise along the way from starting material to final product. Dependingon the nature of the chemical transformation, cations, anions, and radicals are created dur-ing an intermediate stage. These species form mainly as a result of bond rupture. Bond rup-ture may proceed heterolytically or homolytically (Scheme 1-1):
R� � X� ← R–X → R� � X� or R–X → R.� X
.
Ions or radicals formed from a substrate further react, with other ions or radicals act-ing as reactants. Such changes in chemical bonds can be accompanied by a one-electronshift. The concept of the one-electron shift (Pross 1985) can be illustrated by Scheme 1-2for nucleophilic substitution:
Nu� � R↑↓Z → Nu↑↓R � Z�
The species Nu↑↓R of Scheme 1-2 is not a radical pair; it is a covalent molecule ofthe product resulting from the SN2 reaction. The process of transfer of the R group to a Nu�
reactant proceeds in synchronicity with a one-electron shift and R–Z bond disruption. Atthat time, two radical particles, Nu
.and R
.(formed in the course of reaction) remain im-
mediately close and therefore unite rapidly. A one-electron shift may or may not lead to theformation of radical particles. There are many reactions that consist not of a one-electronshift, but of a one-electron transfer. The initial results of such one-electron transfers in-volves the formation of ion radicals.
This book concentrates on species of the type (RX)�., i.e., on cation and anion radi-
cals. These species are formed during reaction, when an organic molecule either loses oneelectron from the action of an electron acceptor or acquires one electron from the action of
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

an electron donor, Scheme 1-3:
R–X � e → (R–X)�.or R–X � e → (R–X)�.
Ion radicals differ from their starting molecules only in a change of the total numberof electrons; no bond rupture or bond formation occurs. As will be seen from the followingchapters, after ion radical formation, cleavage and association reactions often occur. Ge-ometry changes upon electron loss or gain can take place. Reactions with the participationof ion radicals bring their own specific opportunities.
The concept of molecular orbital helps explain the electron structure of ion radicals.When one electron abandons the highest occupied molecular orbital (HOMO), a cation rad-ical is formed. If one electron is introduced externally, it takes up the lowest unoccupiedmolecular orbital (LUMO), and the molecule becomes an anion radical.
These ion radicals have a dual character. They contain an unpaired electron and are,therefore, close to radicals. At the same time, they bear a charge and, naturally, are close toions. Being radicals, the ion radicals are ready to react with radicals. Like all other radicals,they can dismutate and recombine. Being ions, the ion radicals are able to react with parti-cles of the opposite charge and are prone to form ionic aggregates. In contrast with radicals,the ion radicals are especially sensitive to medium effects.
As long as an unpaired electron of an ion radical occupies an orbital covering allatoms of a molecule, a definite distribution of spin density occurs between individualatoms. The distribution determines the activity of one or another position of an ion radicalspecies. From the point of view of organic synthesis, such properties of ion radicals as sta-bility, resistance to active medium components, capacity to disintegrate in the needed di-rection, and the possibility of participating in electron exchange are especially important.All these properties become understandable (or predictable in cases of unknown examples)from the organic ion radical electron structure. Therefore, our account will be based on theanalysis of connections between the structure of ion radicals and their reactivity or physi-cal properties. This chapter concerns the peculiarities of conjugation in aromatic ion radi-cals, electron structures, and acid–base properties of ion radicals that have originated frommolecules of different chemical classes.
1.2 UNUSUAL FEATURES
1.2.1 Substituent Effects
The aim of this section is to show that substituent effects for organic ion radicals are quitedifferent from those of their parent, neutral molecules. Amino and nitro compounds aregood examples.
N,N-Dimethylaniline is a molecule with a lone electron pair on the nitrogen atom. Ofcourse, there is a strong interaction between this pair and the �-electron system of the ben-zene ring. We often mark the symbol of the cation radical, i.e. �.
, on the nitrogen atom.However, according to the ab initio Hartree–Fock molecular orbital calculations (Zhang,R., et al. 2000), this nitrogen atom is in fact negatively charged (�0.708), and the positivecharge is distributed on the carbon atoms, especially on the two methyl groups (�0.482 oneach). Influenced by the positive-charge delocalization along the cation radical, the ben-zene ring becomes an electron-deficient unit with a positive charge of �0.744. Summationyields the total charge of �1,000 for the N,N-dimethylamine cation radical. As seen, con-ventional ideas may not be applicable to the chemistry of ion radicals.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

In anion radicals of nitro compounds, an unpaired electron is localized on the nitrogroup, and this localization depends on the nature of the core molecule bearing this nitrosubstituent. The value of the hyperfine coupling (HFC) constant in the ESR spectrum re-flects the extent of localization of the unpaired electron; aN values of several nitro com-pounds are given in Table 1-1.
Let us compare HFC data from Table 1-1. Aliphatic nitro compounds produce anionradicals, in which an unpaired electron spends its time on the nitro group completely. In thenitrobenzene anion radical, an unpaired electron is partially delocalized over the aromaticring due to conjugation, and the nitrogen hyperfine coupling constant decreases by one halfas compared with the aliphatic counterparts. Diminution of the �-conjugation in the PhNO2
system as a result of the nitro group distortion intensifies the localization of the unpairedelectron on the nitro group. In the nitrobenzene anion radical, however, an unpaired elec-tron is not evenly spread between the nitro group and the benzene ring. The calculation(Stone & Maki 1962) of spin distribution in the nitrobenzene anion radical showed that thenitro group retains about 0.65–0.70 of the unit spin density (i.e., an unpaired electron re-sides mainly on the nitro group). These calculations are based upon HFC constants aN andaH taken from the nitrobenzene anion radical ESR spectrum. The same result arises fromthe unpaired electron distribution by the use of the MO Hueckel approximation: 0.31 of theunit spin density over the phenyl nucleus and 0.69 on the nitro group (Todres 1981).
Of course, it is the entire molecule that receives the electron upon reduction. How-ever, the nitro group is the part where the excess electron spends the majority of its time.Consideration of quantum chemical features of the nitrobenzene anion radical is of partic-ular interest. The model for the calculation includes a combination of fragment orbitals forPh and NO2, and the results are represented in Scheme 1-4. The left part of the schemerefers to the neutral PhNO2, and the right part refers to the anion radical, PhNO2
�.(Todres
1981).Some changes in all the orbital energies accompany the placing of an electron on the
lowest unoccupied molecular orbital. According to the calculations, relative energy gapsremain unchanged for the orbitals in the nitrobenzene anion radical compared with those ofthe parent nitrobenzene. For the sake of graphic clearness, Scheme 1-4 disregards the dif-ference mentioned, keeping the main feature of the equality in the energy gaps.
The nitro group in the parent nitrobenzene evidently acts as �-acceptor, which pullsthe electron density out of the aromatic ring. An unpaired electron will obviously occupythe first vacant �-orbital of the nitro fragment (i.e., the lowest-energy-fragment orbital). In-teraction between occupied and vacant orbitals is the most favorable. In the nitrobenzene
TABLE 1-1 Nitrogen HFC Constants (aN) from Experimental ESR Spectra of NitroCompounds
ConstantCompounds aN, mT Reference
Nitroalkanes 2.4–2.5 Stone & Maki (1962),McKinney & Geske (1967)
Nitrobenzene 1.0 Geske & Maki (1960)2-Chloronitrobenzene 0.9 Starichenko et al. (2000)2,6-Dichloronitrobenzene 1.4 Starichenko et al. (2000)Nitrodurene 2.0 Geske & Ragle (1961)
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

anion radical, the one-electron populated fragment orbital of NO2�.
will pump spin densityin the ring. Such interaction should be very advantageous due to the level proximity of thelowest vacant ring orbital and the highest occupied orbital of NO2
�.. Therefore, the nitro
group can, in fact, act as �-donor in the nitrobenzene anion radical. Such prediction is notself-evident, since the nitro group in neutral aromatic nitro compounds is recognized as astrong �-acceptor and, in principle, even as a reservoir for four to six additional electrons.Comparing half-wave potentials of reversible one-electron reduction of meta-dinitroben-zene and other benzene derivatives, one can determine the Hammett constant for the nitrogroup in the nitrobenzene anion radical. When the NO2 group is transformed into the NO2
�.
group, a change in both the sign and the value of the correlation constant is observed (To-dres, Pozdeeva et al. 1972a,b). A formal comparison of the Hammett constants for the NO2,NO2
�., and NH2 groups shows NO2
�.to be close to NH2 in terms of donating ability:
�m(NO2) � �0.71, �m(NO2�.
) � �0.17, �m(NH2) � �0.16.Having captured the single electron, the nitro group then acts as a negatively charged
substituent. Similarly, the stable anion radical resulting from aryl diazocyanides[(ArNBNCN)�] contains a substituent [(NBNCN)�.
] that interacts with the aryl ring asa donor (Kachkurova et al. 1987). Using other nitro derivatives of an aromatic heterocyclicseries, the generality and statistical relevance of the observed �m(NO2
�.) constant were es-
tablished (Todres, Zhdanov et al. 1968; Todres, Pozdeeva et al. 1972a). The sign and ab-solute magnitude of the Hammett constant are invariant regardless of which cation (K�,Na�, or Alk4N�) in the anion radical salts of nitro compounds was studied. Such invari-ance is caused by the linear dependence between electrochemical reduction potentials ofsubstituted nitrobenzenes and the contribution of the lowest vacant �*-orbital of the nitrogroup to the �-orbital of this anion radical, which is occupied by the single electron (Kop-tyug et al. 1988).
Experiments on the reactivity of the anion radicals under consideration are set outlater. The ability of nitrobenzene anion radicals to undergo coupling with benzenediazocations has been studied (Todres, Hovsepyan et al. 1988). This reaction is known to pro-ceed for aromatic compounds having donor-type substituents (NH2, OH). Aromatic com-pounds containing only the nitro group do not participate in azo-coupling. It is also worthnoting that benzenediazo cations are strong electron acceptors. For instance, the interactionbetween benzene- or substituted benzene-diazonium fluoroborates and the sodium salt ofthe naphthalene anion radical results in electron transfer only (Singh et al. 1977). The prod-ucts were naphthalene (from its anion radical) and benzene or its derivatives (from ben-
SCHEME 1-4
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

zene- or substituted benzene-diazonium fluoroborates). The potassium nitrobenzene anionradical appears to react with diazonium cations according to the electron transfer scheme,too (as the naphthalene anion radical does). Products of azo-coupling were not found (To-dres, Hovsepyan et al. 1988).
The potassium salt of the phthalodinitrile (ortho-dicyanobenzene) anion radical alsoreacts with an electrophile according to the electron transfer scheme. If the electrophile istert-butyl halide, the reaction proceeds via the mechanism, including at the first-stage dis-sociative electron transfer from the anion radical to alkyl halide, followed by recombina-tion of the generated tertiary butyl radical with another molecule of the phthalodinitrile anion radical. The product mixture resulting in the reaction includes 4-tert-butyl-1,2-di-cyanobenzene, 2-tert-bytylbenzonitrile, and 2,5-di(tert-butyl)benzonitrile (Panteleeva andco-authors 1998).
An attempt to detain an unpaired electron was made by means of the second nitrogroup in the anion radical of nitrobenzene (Todres, Hovsepyan et al. 1988). The potassiumsalt of the anion radical of ortho-dinitrobenzene did yield an azo-coupled product accord-ing to Scheme 1-5 (the nitrogen oxide evolved was detected).
The reaction leads to a para-substituted product, entirely in accordance with thecalculated distribution of spin density in the anion radical of ortho-dinitrobenzene (To-dres 1990). It was established, by means of labeled-atom experiments and analysis of thegas produced, that azo-coupling is accompanied by conversion of one of the nitro groupsinto the hydroxy group and liberation of nitric oxide. In other words, the initial radicalproduct of azo-coupling is stabilizing by elimination of the small nitrogen monoxide radical to give the stable nonradical final product (Todres, Hovsepyan et al. 1988),Scheme 1-5.
SCHEME 1-5
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

During transformation of the parent molecule to the corresponding anion radical,changes in substituent effects appear to be possible not only for the nitro group, but also forother substituents. We have just observed the opportunity to use the nitro group as a donor(not an acceptor) in the anion radicals of aromatic nitro compounds. In the case of AlkOand AlkS substituents, we have a chance to encounter the donor-to-acceptor transformationof the thioalkyl group after one-electron capture by thioalkylbenzenes (Bernardi et al.1979). Both groups, AlkO and AlkS, are commonly known as electron donors. However,in the anion radical form, these groups exert nonidentical effects. The methoxy group keepsits donor properties, while the methylthio group appears to be an acceptor. This is evidentfrom a comparison of the ESR spectra of the nitrobenzene anion radical and its derivatives,in particular the MeO- and MeS-substituted ones. The introduction of substituents into ni-trobenzene in general affects the value of aN arising from the splitting of an unpaired elec-tron by the nitrogen atom in the anion radical. If the group introduced is a donor, theaN(NO2) value increases; if it is an acceptor, the aN(NO2) value is reduced. As follows fromsuch a comparison of aN constants, the MeO and EtO groups act similarly to the Me and S�
groups (donors). At the same time, the MeS and EtS groups act similarly to CN, SO2Me,and SO2Et groups (acceptors) (Ioffe et al. 1970, Alberti, Martelli, & Pedulli 1977, Alberti,Guerra et al. 1979, Bernardi et al. 1979).
The sharp contrast between the electronic effects exerted by the oxyalkyl andthioalkyl groups in anion radicals was explained by means of group orbital-energy dia-grams. The usual mechanism involving n,�-conjugation requires the MeO or MeS groupto be situated in the same plane as the aromatic ring of the parent (neutral) molecules. Ac-cording to calculations (Bernardi et al. 1979), “the most stable conformation is the planar”for the anion radical of anisol. In the case of the anion radical of thioanisol, however, “thepreferred conformation is orthogonal.” The planar conformation is stabilized by the usualn,�-conjugation between the ring and oxygen or sulfur. Such n,�-conjugation is impossi-ble in the orthogonal arrangement, and only the �-electrons of the sulfur or oxygen appearto be involved. Only the �-orbitals of these atoms are symmetrically available for overlap-ping with the aromatic �-orbitals when fragments of the molecule are oriented perpendic-ularly. However, interaction between the �-electrons of the nucleus and the vacant �-orbitals of the substituent is also possible in principle, because this interaction is sym-metrically allowed. In practice, �,� and �*,� interactions are not too important in the caseof uncharged molecules, since the gap between the aromatic �-orbitals and �/�*-orbitalsof the substituents is too wide. This is obvious from the left part of Scheme 1-6.
Conversion of a neutral molecule into an anion radical leads to occupation of the low-est-energy vacant orbital. The latter is the �-orbital of the benzene ring in both anisole andthioanisole. Charge transfer is possible only by means of an interaction between vacant and occupied orbitals and only if an energy gap between them is not too wide. As the �*-orbitals of the anisole MeO group are too far away from the ring �-orbital occupied bythe single electron, the conjugation conditions in the anion radical compared with the neu-tral molecule remain unchanged. This is evident from the right part of Scheme 1-6.
The thioanisole MeS group differs from the anisole MeO group in the fact that the �*is at a lower energy level (Alberti, Guerra et al. 1979). In this case, population of the low-est vacant aromatic �-orbital by a single electron changes the conjugation conditions.Namely, the �*, � interaction becomes more favorable than the n,� interaction, becausethe energy gap between the �*- and the �-orbitals is narrower. In other words, conditionscreated in the anion radical promote charge transfer from the ring to the substituent ratherthan from the substituent to the ring, as is the case in the neutral molecule. That is why the
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

orthogonal conformation is stabilized instead of the planar one. The conversion ofthioanisole into the anion radical causes the change in the orientation of the MeS group rel-ative to the aromatic ring plane. This is depicted on the right part of Scheme 1-6. Onceagain, not the energy levels but the relative energy gaps remain unchanged for the anionradicals as compared to the parent neutral molecules.
Change in the substituent nature after transformation of neutral aromatics into thecorresponding ion radicals may be used intuitively for preparation of some unusual deriva-tives. One may transform an organic molecule into its anion radical, change the substituenteffect, perform the desired substitution, and, after that, take a surplus electron off by meansof a soft oxidant and eventually obtain the desired unusual derivative in its stable form.Studies along this line are intriguing in the cases of both anion and cation radicals.
1.2.2 Connection Between Ion Radical Reactivity and ElectronStructure of Ion Radical Products
The reaction of aryl and hetaryl halides with the nitrile-stabilized carbanions (RC�–CN)leads to derivatives of the type ArCH(R)CN. Sometimes, however, dimeric products of thetype ArCH(R)CH(R)Ar are formed (Moon et al. 1983). As observed, 1-naphthyl, 2-pyridyl, and 2-quinolyl halides give the nitrile-substituted products, while phenyl halides,as a rule, form dimers. The reason consists of the manner of a surplus electron localizationin the anion radical that arises upon replacing halogen with the nitrile-containing carban-ion. If the resultant anion radical contains an unpaired electron within LUMO coveringmainly the aromatic ring, such an anion radical is stable, with no inclination to split up. Itis oxidized by the initial substrate and gives the final product in the neutral form, Scheme1-7:
[Ar]�.CH(R)CN � e → ArCH(R)CN
If the anion radical formed acquires an unpaired electron on the CN group orbital, thisgroup easily splits off in the form of the cyanide ion. So the dimer is formed as the final
SCHEME 1-6
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

product, Scheme 1-8:
2PhCH(R)[CN]�. → 2CN� � PhCH(R)CH(R)Ph
One-electron reduction of organyl halides often results in the spontaneous elimina-tion of halide and the formation of organyl radicals according to Scheme 1-9:
RX � e → RX�. → R.� X�
The organyl radicals resulting in this cleavage can combine with the nucleophile an-ion, Scheme 1-10:
R.� Y� → RY�.
The anion radical of this substituted product initiates a chain reaction network,Scheme 1-11:
RY�.� RX → RY � RX�.
According to Saveant (1994), an important contribution to the overall efficiency ofthis substitution reaction is given by the step in which the RY�.
anion radical is formed. Inthis step, an intramolecular electron-transfer/bond-forming process occurs when the nu-cleophile Y� attacking the radical R
.begins to form the new species, characterized by an
elongated two-center three-electron C�Y bond. An unpaired electron in this anion radicalis at first allocated on a “low-energy” �C-Y* molecular orbital. With the progress of the for-mation of the C–Y bond, the energy of the �* molecular orbital increases sharply until achangeover occurs. If R is Ar, the �* molecular orbital of the molecule becomes theLUMO. An internal transfer of the odd electron to the LUMO then takes place. So it fol-lows that the substitution under consideration will be easier when the energy of the �*molecular orbital available in the ArY�.
species is lower. Papers by Rossi et al. (1994),Galli with co-workers (1995), and Borosky et al. (2000) have again underlined the rule: Thelower the energy of the LUMO of the RY� (ArY�) species, the easier (faster) the reactionbetween R
.(Ar
.) and Y�.
It is worth noting, however, that primary halide-containing anion radicals may be some-what stable if an aromatic molecule has another electron acceptor group as a substituent—such as the nitro, cyano (Lawless et al. 1969), carbonyl (Bartak et al. 1973), or pyridinyl group(Neta & Behar 1981). In these cases, dehalogenation reactions proceed as intramolecularelectron transfers from the groups NO2
�., CN�.
through the conjugated � system to the car-bon–halogen fragment orbital. After that, the halide ion is eliminated. The splitting rate de-pends on the nature of the halogen (I � Br � CI) and on the position of the halogen with re-spect to another substituent (ortho � para � meta) (Alwair & Grimshaw 1973; Neta & Behar1981; Behar & Neta 1981; Galli 1988). The cleavage proceeds more easily at those positionsthat bear maximal spin density. Change of the nitro group to the nitrile or carbomethyl groupleads to some facilitation of halogen elimination: A greater portion of spin density reachesthe carbon–halogen orbital and the rate of dehalogenation increases. For instance, the anionradical of 4-fluoronitrobenzene is characterized by the aF HFC constant of 0.855 mT and highstability (Starichenko et al. 1981). In contrast, the anion radical of 4-fluorobenzonitrile has asignificantly larger aF HFC constant of 2,296 mT and does readily dissociate into the ben-zonitrile �-radical and the fluoride ion (Buick et al. 1969).
It should be emphasized that the cause of halide mobility in aromatic anion radicalsis quite opposite to that in heterolytic aromatic substitution at the carbon–halogen bond. Inanion radicals, the carbon–halogen bond is enriched with electron density and, after halide
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

ion expulsion, an aromatic �-radical is formed. In neutral molecules, the carbon–halogenbond conjugated with an acceptor group becomes poor with respect to its electron density;a nucleophile attacks a carbon atom bearing a partial positive charge. Some kind of �-bind-ing was established between the nitro group and chlorine through the benzene ring in 4-ni-trochlorobenzene (Geer & Byker 1982). As a result, the inductive effect of chlorine be-comes suppressed in the neutral molecule. In the anion radical, LUMO populated by oneelectron comes into operation. The HOMO role turns out to be insignificant. In anion rad-icals, this orbital can cause only a slight disturbance. The negative charge to a significantdegree moves into the benzene ring, and this movement is enforced at the expense of thechlorine-inductive effect. The carbon–chlorine bond is enriched with an electron. Eventu-ally, Cl� leaves the anion radical species. This event is quite simple, and its simplicity isbased on the �-electron character of HOMO and LUMO.
However, there are some cases when an unpaired electron is localized not on the �-orbital but on the �-orbital of an anion radical. Of course, in such cases a simple molec-ular-orbital consideration based on the � approach does not coincide with experimental data.Chlorobenzothiadiazole may serve as a representative example (Gul’maliev et al. 1975). Al-though the thiadiazole ring is a weaker acceptor than the nitro group, the elimination of thechloride ion from the 5-chlorobenzothiadiazole anion radical does not take place(Solodovnikov & Todres 1968). At the same time, the anion radical of 7-chloroquinolineloses the chlorine anion (Fujinaga et al. 1968). Notably, 7-chloroquinoline is very close to5-chlorobenzothiadiazole in terms of structure and electrophilicity of the heterocycle. To ex-plain this difference, calculations are needed that can clearly take into account the �-elec-tron framework of the molecules compared. Solvation of intermediate states on the way toa final product should be involved in the calculations as well (Parker 1981).
The alkyl halide anion radicals do not have �-systems entirely. Nevertheless, they areable to exist in solutions. The potential barrier for the C–Cl cleavage is estimated to be ca.70 kJ�mol�1 (Abeywickrema & Della 1981; Eberson 1982). The carbon–halogen bondmay capture one electron directly (Casado et al. 1987; Boorshtein & Gherman 1988).
It is interesting to compare SCl and SCN in relation to NO2 as the reference group.Aryl sulfenyl chlorides and thiocyanates were subjected to two independent model reduc-tive cleavage reactions by treatment with (a) cyclooctatetraene dipotassium (C8H8K2) intetrahydrofurane (THF) or (b) HSiCl3 � R3N (R � alkyl) in benzene (Todres & Avagyan1972, 1978). As shown, aromatic sulfenyl chlorides under conditions (a) and (b) producedisulfides or thiols; the presence of the nitro group in the ring does not affect the reaction.Aryl thiocyanates without the nitro group behave in a similar way. However, aryl thio-cyanates containing the nitro group in the ring are converted into anion radicals, with theSCN remaining unchanged. This pathway is represented by Scheme 1-12:
O2NC6H4SCN � 1⁄2C8H8K2 → 1⁄2C8H8 � NCSC6H4NO2�.
K�
Splitting of the SCN group is not observed and, after the one-electron oxidation, theinitial NCSC6H4NO2
�.anion radical produces NCSC6H4NO2. The recoveries are close to
quantitative; disulfides and thiols are not observed. The thiocyanate group (SCN) thus com-petes less successfully with the nitro group (NO2) for the extra electron than the sulfenylchloride group (SCl).
The conclusion just outlined was entirely confirmed by quantum chemical calcula-tions. The results of the calculations are shown on the Table 1-2 (LCAO MO CNDO/2 ap-proach, Todres, Tomilin, & Stankevich 1982). As seen in the table, the SCl group chargedepends slightly on whether or not the NO2 group is present in the benzene ring. In the case
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

of thiocyanate anion radicals, the charge on the SCN group is diminished by 50% if the NO2
group is present in the molecule.Thus, SCl and SCN have different electron-attraction properties. That conclusion
was not predictable a priori. Until recently, the extent of polarization in the SCl group hasbeen considered to be comparable to that in the SCN group, according to the S�–X�
scheme. For instance, Kharash et al. (1953), have pointed out that nitroaryl thiocyanates aswell as nitroaryl sulfenyl chlorides, when dissolved in concentrated sulfuric acid, are con-verted into the same nitroaryl sulfenium ions, O2NArS�. However, the preceding findingsindicate otherwise.
Other pertinent examples include splitting of the anion radicals from p-nitrophenylmetyl sulfone and p-cyanophenyl methyl sulfone (Pilard et al. 2001). The nitrophenylspecies undergoes the preferential cleavage of the Ar–S bond, whereas the cyanophenylspecies expels both CN and CH3SO2 groups in the two parallel cleavage reactions.
All the examples show that foreseeing a splitting direction and extending it from oneparent compound to another is risky, especially in the organic chemistry of radical ions.
1.2.3 Bridge Effect Peculiarities
Let us consider ion radicals of paracyclophanes. As the basis for our consideration, we willchoose the following species, depicted in Scheme 1-13 (a)–(e).
(a) The anion radical of pseudogeminal-[2.2]paracyclophane-4,7,12,15-tetrone, inwhich the 1,4-benzoquinone units lie one underneath the other
(b) The anion radical of syn-[2.2](1,4)naphthalenophane-4,7,14,17-tetrone, inwhich there is the same spatial situation
(c) The anion radical of anti-[2.2](1,4)naphtalenophane-4,7,12,15-tetrone, inwhich the naphthoquinone units are further apart
(d) The cation radical of syn-[2.2](1,4)naphthalenophane-4,714,17-tetramethoxyderivative, which is close to case (b)
(e) The cation radical of anti-[2.2](1,4)naphthalenophane-4,7,14,17-tetramethoxyderivative, which resembles the case (c) structure
As seen, cyclophane structures (b)–(e) have the unique feature that the through-bonddistance within the paracyclophane fragment is held constant while the spatial distance be-tween the ion radicalized and neutral moieties is changed. So the relative importance ofthrough-bond and through-space mechanisms for electron transfer can be learned directlyfrom experimental data on these molecules.
All the depicted compounds (Scheme 1-13) were studied in electrochemical reduction[cases (a) to (c)] or oxidation [cases (d) and (e)]; two one-electron peak potentials were re-
TABLE 1-2 Effective Charges (qi) on R and SXGroups in p-RC6H4SX� Anion Radicalsa
R SX �qR �qsx
H SCI 0.05 0.78NO2 SCI 0.20 0.76H SCN 0.06 0.54NO2 SCN 0.48 0.27
a The rest of the charge, up to unity, is in the benzene rings.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

vealed, with differences significantly higher than 20 mV (Wartini, Valenzuela et al. 1998;Wartini, Staab et al. 1998). A large difference between the first two reductions or oxidationpotentials is indicative of the delocalization of the first (unpaired) electron (Rak & Miller1992). In other words, the two electrochemically active fragments can accept or lose a singleelectron, the second one-electron transfer being markedly hampered. In their turns, ESR andENDOR spectra of the anion radicals under investigation gave evidence of delocalization ofan unpaired electron over the whole molecule in each case. Because of the close spatial con-tact of the quinone units [0.31 nm between the centers of the 1,4-benzoquinone rings, Scheme1-13(a)], one may suppose that the unpaired electron simply jumps over this narrow gap. Ifso, the whole-molecule delocalization would be impossible in the case of the mutual anti-ar-ranged 1,4-naphthoquinone units [see structure (c) in Scheme 1-13]. However, this anti-ar-ranged anion radical shows the full spin electron delocalization. Consequently, �,�-conju-gation is realized in the anion radicals of the paracyclophanes considered.
In the same way, the displacement of the unpaired electron over the whole moleculeswas observed for (d) and (e) cation radicals from Scheme 1-13, in which 1,4-dimethoxy-naphthalene units are syn- or anti-annelated to [2.2]paracyclophane. Taken together, theexperimental results considered provide direct evidence for the through-bond mechanismof electron transfer in these paracyclophane systems. In a recent study, the electron trans-fer between 1,4-dimethoxybenzene and 7,7-dicyanobenzoquinone methide moieties in syn-or anti-cyclophane systems reached the same conclusion: The through-bond mechanism
SCHEME 1-13
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

can remain the dominant reaction pathway at short donor–acceptor distances as well(Pullen et al. 1997).
Other examples of the specified bridge effect deal with anion radicals of aryl deriva-tives of the tri-coordinated boron or tri- and four-coordinated phosphorus. In thetris(pentafluorophenyl)boran anion radical, spin density is effectively transferred from theboron p-orbital to an antibonding �-molecular orbital of the phenyl rings (Kwaan et al.2001). Studies of the phosphorus-containing aromatic anion radicals have been more in-formative. As known, phosphorus interrupts conjugation between aryl fragments in the cor-responding neutral compounds. In contrast to the neutral molecules, the phosphorus atomdoes transmit conjugation in Ar3P and Ar3P(O) anion radicals. At least formally, the P atomappears to be a bridge, not a barrier. Spin delocalization takes place along the wholemolecule in each case. Perhaps, the phosphorus unfilled p- or d-orbitals take part in thistransmission effect (Il’yasov et al. 1980).
The cation radicals of triaryl phosphine and trialkyl phosphites have also been probedto hyperconjugation; photoelectron, ESR spectra, and reactions with aromatic radicals werestudied. As it turned out, the cation radicals of Ar3P are characterized by strong hypercon-jugation (Egorochkin and others 2001). The cation radical [P(OMe/OEt)3]�.
contains an unpaired electron predominantly on the phosphorus atom, which facilitates the radical coupling of this cation radical with the Ar
.radical from aryl diazonium salts. In [PhP(OMe/
OEt)2]�., and [Ph2P(OMe/OEt)]�.
, and [Ph3P]�.cation radicals, an unpaired electron is in-
creasingly shifted from the phosphorus atom to the phenyl ring(s). This reduces the spindensity at the central phosphorus atoms, making the reaction of the mentioned cation radi-cals with Ar
.slower and eventually preventing it altogether (Yasui, Fujii et al. 1994; Yasui,
Shioji, Ohno 1994). Similarly to the cation radicals, the phosphoranyl radicals with andwithout the aryl ligand(s) exhibit small and large values of phosphorus HFC constants, re-spectively, in the ESR spectra (Boekenstein et al. 1974; Davies, Parrot, & Roberts 1974;Davies, Griller, & Roberts 1976). This means that the unpaired electron is located mainlyon the aryl ligands of aryl phosphoranyl radicals or entirely on the central phosphorus inthe case of alkyl phosphoranyl radicals.
Let us direct our attention now to the PBC bond in phosphaalkene ion radicals. Theliterature contains data on two such anion radicals in which a furan and a thiophene ring arebound to the carbon atom, and the 2,4,6-tri(tert-butyl)phenyl group is bound to the phos-phorus atom. According to ESR spectra of the anion radicals, an unpaired electron is delo-calized on a �*-orbital built from the five-membered ring (furanyl or thienyl) and the PBCbond. The participation of the phosphaalkene moiety in this molecular orbital was esti-mated as about 60%. Although the cumbersome tris(tert-butyl)phenyl group is led out ofthe conjugation sterically, some moderate (but sufficient enough) transmission of the spindensity does take place through the PBC bridge (Jouaiti et al. 1997). Scheme 1-14 depictsthe structures under discussion.
The same situation was revealed for the case of the anion radical of the phos-phaalkene containing the phenyl ring linked to the carbon atom and the 2,4,6-tri(tert-butyl)phenyl group linked to the phosphorus atom of the PBC bond. The unpaired elec-tron is delocalized in this anion radical on both the PBC bond and the phenyl ring(Geoffroy et al. 1992). In the benzene and furane derivatives containing two conjugatedArPBC bonds, an unpaired electron is delocalized on the whole bonds of the anion radi-cals, resulting in one-electron reduction. Neither localized structures nor diradical specieswere observed (Al Badri et al. 1999). However, if the carbon atom of this PBC bond is anintegral part of the cyclopentadiene ring, the unpaired electron distribution proceeds ac-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

cording to a scheme of spin-charge scattering (Al Badri et al. 1997). Scheme 1-15 illustratesthis special case.
Hence, the possibility of acquiring aromaticity conferred by the presence of six �-electrons in the five-carbon-membered ring considerably increases the electron affinityof this ring. As a consequence, one of the two �-electrons of the PBC bond remains on thephosphorus atom, and another one combines with the excess electron to create the cy-clopentadienyl �-electron sextet. The situation is analogous to that in the diphenylfulveneanion radical, as analyzed in Chapter 3 (see Section 3.2.2).
In the case of p-phosphaquinone, the 2,4,6-tri(tert-butyl)phenyl group is bound to thephosphorus atom, and the carbon atom of the PBC bridge is an integral part of the 3,5-di(tert-butyl)-4-oxocyclohexa-2,5-dien-1-ylidene moiety. The corresponding anion radical(a structure of the p-benzosemiquinone type) is very similar to diarylphosphoranyl radicals,in the sense of the unpaired electron delocalization (Sasaki et al. 1999). In the phosphoricanalog of biphenyl, namely, 4,4,5,5-tetramethyl-2,2-phosphinine, the corresponding an-ion radical was generated on a potassium mirror. ESR/ENDOR spectra and density func-tional theory calculations show that, in contrast with the neutral species, this anion radicalis planar and that the unpaired electron is delocalized mainly on the phosphorus–car-bon–carbon–phosphorus fragment of the two linked six-membered rings. The phosphorusp�-orbitals play a large part in this delocalization (Choua et al. 2000).
Diphosphaallene derivatives ArPBCBPAr are peculiar compounds because of thepresence of the two orthogonal carbon–phosphorus double bonds. Those compounds weretransformed into cation radicals upon electrochemical or chemical one-electron oxidation.As found, the unpaired electron is located on a molecular orbital constituted mainly of a p-orbital of each phosphorus atom and a p-orbital of the carbon atom (Chentit et al. 1997;Alberti, Benaglia et al. 1999). Upon electrochemical or chemical reduction, aromatic phos-phaallene derivatives yield anion radicals. These species have two equivalent phosphorus
SCHEME 1-14
SCHEME 1-15
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

nuclei. The unpaired electron oscillates between the two phosphorus atoms according toScheme 1-16 (Sidorenkova et al. 1998, Alberti, Benaglia et al. 1999):
ArPBC(�)–P(.)Ar ↔ ArP(
.)–C(�)BPAr
Consequently, the electron structures of the diphosphaallene ion radicals resemble those ofthe allene ion radicals, where the allenic fragment works as a bridge for conjugation (seeSection 3.3.2).
One quite unusual effect deserves to be described as applied to the CBC bridge con-necting two phenyl rings in the 4-nitrostilbene anion radical. This bridge is good at trans-mission of conjugation in neutral stilbenes, but in stilbene anion radicals it can operate asa hollow on the conjugation route. At first glance, the distribution of unpaired electrons inthe nitrostilbene anion radical has to be similar to that in the nitrobenzene anion radical. Theconsensus of opinion is that the LUMO of neutral aromatic nitro derivatives (the orbital thataccommodates the introduced electron) is essentially an orbital of the “free” nitro group.The styryl fragment of neutral 4-nitrostilbene is conjugated with the nitro group and acts asa weak donor. This is indicated by values of the Hammett constants: ��(NO2C6H4–) is�0.23 and ��(–CHBCHPh) is �0.07.
If the styryl substituent retained its donor nature in the anion radical state, an in-crease, not a decrease, in the value of the nitrogen HFC constant [a(N)] would have beenobserved. Experiments show that a(N) values for anion radicals of nitrostilbenes decrease(not increase) in comparison with the a(N) value for the anion radical of nitrobenzene (Todres 1992). Both “naked” anion radicals and anion radicals involved in complexes withthe potassium cations obey such regularity. In cases of potassium complexes with THF asa solvent, a(N) � 0.980 mT for PhNO2 anion radical and a(N) � 0.890 mT forPhCBCHC6H4NO2-4 anion radical. In the presence of 18-crown-6-ether as a decomplex-ing agent in THF, a(N) � 0.848 mT for PhNO2 anion radical and a(N) � 0.680 mT forPhCHBCHC6H4NO2-2 anion radical.
Reduction of nitrobenzene (Grant & Streitwieser 1978; Todres, Dyusengaliev et al.1984) and 4-methoxynitrobenzene (Todres, Dyusengaliev et al. 1984) by uranium-, tho-rium-, and lanthanum-di(cyclooctatetraene) complexes leads to azo compounds. Scheme 1-17 illustrates these reductive reactions using uranium–(C8H8)2 complex as an example.
Under the same conditions, 4-styryl nitrobenzene (4-nitrostilbene) undergoes cis-to-trans isomerization only, with no changes in the nitro group (Todres, Dyusengalier et al.1984, 1985), Scheme 1-18.
Thus, it appears that the focal point of the reaction has transferred. The presence of astyryl (not a methoxyl) group protects the nitro group from reduction. For some reason orother, the styryl group causes a shift of excess electron density from the nitro to the ethy-lene fragment.
SCHEME 1-17
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

This subtle difference between the anion radicals of nitrobenzene and nitrostilbene,observed experimentally, is well reproduced by quantum chemical calculations (Todres etal. 1984). Single-electron wave function analysis of the vacant orbitals in both moleculesshows that one-electron reduction of cis-4-nitrostilbene must be accompanied by the pre-dominant localization of an upaired electron in the region of the ethylene moiety. Partici-pation of the nitro-group atomic orbitals appears to be insignificant. The nitro-group atomiccoefficients in the molecular wave function for cis-4-nitrostilbene are half of those for ni-trobenzene. The excess electron population (q) of the first vacant orbital for the nitro groupsis 0.3832 for the nitrobenzene anion radical and 0.0764 for the nitrostilbene anion radical.The unpaired electron is localized largely on the ethylene fragment of the nitrostilbeneskeleton (q � 0.2629). Moreover, the first vacant level of the cis-4-nitrostilbene moleculehas lower energy than that of the nitrobenzene molecule: 38 and 135 kJ, respectively.
This means that 4-nitrostilbene is a more effective electron acceptor than nitroben-zene. This theoretical conclusion is verified by experiments. The charge-transfer N,N-dimethylaniline complexes formed by nitrobenzene or 4-nitrostilbene have stability con-stants of 0.085 L�mol�1 and 0.296 L�mol�1, respectively. Moreover, the formation of thecharge-transfer complex between cis-4-nitrostilbene and N,N-dimethylaniline indeed re-sults in cis-to-trans conversion (Dyusengaliev et al. 1995). This conversion proceedsslowly in the complex but runs rapidly via the nitrostilbene anion radical (Todres 1992).The cis–trans conversion of ion radicals will be considered in more details later (see Sec-tions 3.2.5 and 7.2.1).
It is interesting to compare the fate of the CBC bridge in the anion radicals of 4-ni-trostilbene (see earlier) and 4-acetyl-�, -diphenylstilbene (Wolf et al. 1996). When treatedwith potassium or sodium in THF and then with water, neutral 4-nitrostibene does not un-dergo a many-electron reduction of the nitro group or the CBC bridge. Under the sameconditions, 4-acetyl-�, -diphenylstilbene produces a pinacol, [Ph2CBC(Ph)C6H4C(OH)CH3]2. As calculations show, the carbonyl of the acetyl group in 4-acetyl-�, -diphenylstilbene is the site of significant reduction. The formal charge on the carbonyl car-bon and oxygen atoms became significantly more negative upon addition of one electron,whereas the olefinic carbons become only slightly more negative (Wolf et al. 1996). It isworth pointing out that the phenylcarbonyl group is a stronger acceptor than the nitrophenylgroup: ��(—COC6H5) � �0.46 [��(—COCH3) � �0.52], whereas ��(NO2C6H4–)� �0.23 [��(—NO2) � �0.78]. In addition, the phenylacetyl group in the molecule underconsideration is conjugated only slightly, if at all, with the CBC bridge, because thismolecule is propeller shaped (Hoekstra & Vos 1975). Although the acetyl group lies in theplane of the phenyl ring attached to it, it remains separated from the CBC bridge.
In contrast, the nitro and ethylenic fragments in trans-4-nitrostilbene form the unitedconjugation system. Such conjugation is a necessary condition for the whole-contour delo-calization of an unpaired electron in arylethylene anion radicals. Whether this condition isthe only one or there is some interval of allowable strength for the acceptor is a questionleft to future experiments.
SCHEME 1-18
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

1.3 ACID–BASE PROPERTIES OF ORGANIC ION RADICALS
1.3.1 Anion Radicals
Let us compare anion radicals with dianions, which are definitely strong bases. For exam-ple, the cyclooctatetraene dianion (C8H8
2�) accepts protons even from such solvents asdimethylsulfoxide and N,N-dimethylformamide. The latter are traditionally qualified asaprotic solvents. In these solvents, the cyclooctatetraene dianion undergoes protonation, re-sulting in the formation of cyclooctatrienes (Allendoerfer & Riger 1965); see Scheme 1-19:
C8H82� � 2H� → C8H10
Hence, such dianions with two introduced electrons are essentially the counterpart or apro-tic equivalent, or base, of the corresponding C–H acid.
1.3.1.A Anion Radical Basicity
Having one excess electron, anion radicals can be considered aprotic “half-equivalents” ofthe corresponding C–H acid. Reacting with protons, anion radicals display some dual be-havior. As bases, they may add protons, get rid of their negative charges, and give radicals.This direction is represented by direction (a) in Scheme 1-20. As radicals with excess elec-trons, they may generate atomic hydrogens from protons and transform into parental un-charged compounds according to direction (b) in Scheme 1-20.
Such dual anion radical reactivity towards protons depends on the difference betweenproton affinity to an electron and the first ionization potential of an anion radical. This dif-ference may be not very strong. The fate of the competition between directions (a) and (b)in Scheme 1-20 also depends on the relative stability of the reaction products. It is reason-able to illustrate the duality with two extreme examples from real synthetic practice.
Direction (a): Alkali metals transform saturated ketones into secondary alcohols.The reaction proceeds in the mixture of ethanol and liquid ammonia in the presence of am-monium chloride as a proton donor and follows Scheme 1-21 (Rautenstrauch et al. 1981).
SCHEME 1-20
SCHEME 1-21
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Controlled one-electron reductions transform 1,2,3,4-tetraphenyl-1,3-cyclopentadi-ene or 1,2,3,4,5-pentaphenyl-1,3-cyclopentadiene into the 1:2 mixtures of the dihydro-genated products and the corresponding cyclopentadienyl anions in the constant ratio 1:2(Farnia et al. 1999). The anion radicals initially formed are protonated by the substratesthemselves. The latter are thermodynamically very strong acids because of their strong ten-dency to aromatization. As to the cyclopentadiene anion radicals, they need two protons togive more or less stable cyclopentaenes. Scheme 1-22 represents the initial one-electronelectrode reduction of C5HAr5, and Scheme 1-23 explains the ratio and the nature of theproducts obtained at the expense of the further reaction in the electrolytic pool.
C5HAr5 � e → (C5HAr5)�� (1-22)
(C5HAr5)�.� C5HAr5 → C5H2Ar5
.� (C5Ar5)�
C5H2Ar5.� (C5HAr5)�. → C5H2Ar5
� � C5HAr5
(1-23)C5H2Ar5
� � C5HAr5 → C5H3Ar5 � (C5Ar5)�
On the whole: C5HAr5 � 2(C5HAr5)�. → C5H3Ar5 � 2(C5Ar5)�
Hence, the case represents a typical example of the so-called “father–son” self-pro-tonation process, provoked by the partial transformation of cyclopentadiene into the anionradical.
Direction (b): The mixture of trichlorosilane and tributylamine in benzene reducesorganic derivatives of valence 2 sulfur such as benzene sulfenates. However, nitrobenzenesulfenates remain intact in this system (Todres & Avagyan 1978), Scheme 1-24.
Introduction of nitrobenzene sulfenates into the same mixture of trichlorosilane andtributylamine results in the evolution of hydrogen. As proven (Todres & Avagyan 1978),trichlorosilane with tributylamine yields the trichlorosilyl anion and tributylammoniumcation. This stage starts the process involving one-electron transfer from the anion to a ni-trobenzene sulfenate. At the time, the sulfenate produces the stable anion radical with the
SCHEME 1-24
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

tributylammonium counterion. The latter anion radical gives off an unpaired electron to theproton from the counterion; see Scheme 1-25.
Returning to direction (a), it is interesting to compare pKa values of aromatic anionradicals after protonation, pKa(ArH
.2), and pKa values of parental aromatics after protona-
tion, pKa(ArH2). As seen from Table 1-3, if the anion radicals accept a proton, they hold itmuch more firmly than the parental neutral molecules (�pKa values are positive).
Contrary to the early indications (Kalsbek & Thorp 1994), the anion radical of C60
fullerene is a very weak base. Acidity of C60H.approaches that of dilute triflic acid; the
solutions in o-dichlorobenzene were compared. In dimethylsulfoxide, the pKa of C60H.is
estimated to be about 9, making it a slightly weaker acid than p-benzoic acid (see the re-view by Reed C., & Bolskar 2000). These data are consistent with the reports that the syn-thesis and ESR spectrum of C60
�.are relatively insensitive to the presence of water. There
are aryl and methyl derivatives of C60�.
that are stable and soluble in water (Sawamura et al.2000). The weak basicity of C60
�.is due to its intrinsically high stability through delocaliza-
tion of the negative charge toward the 50�-electron system. When C60H.comes up, it for-
mally produces a carbon radical � to the site of protonation, and the energetic cost of thislocalization is high. There is no electrochemical evidence for the reasonable expectation ofdimerization of C60H
.radicals (Cliffel & Bard 1994).
The basicity of anion radicals therefore consists of proton landing. The proton land-ing assumes 1:1 stoichiometry with respect to an anion radical and a proton-donor
SCHEME 1-25
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

molecule. For example, in the reaction of the naphthalene anion radical (C10H8C8�.
) withmethanol (Scheme 1-26, see below), this 1:1 stoichiometry should result in the formationof 50:50% mixture of naphthalene (C10H8) and dihydronaphthalene (C10H10).
C10H8�.
� H� → C10H9.
C10H9.� C10H8
�. → C10H9� � C10H8
C10H9� � H� → C10H10
On the whole: 2C10H8�.
� 2H� → C10H8 � C10H10
Surprisingly, Screttas and co-authors (1996) have found that the reaction of thelithium naphthalene anion radical with methanol in THF follows the 2:1 stoichiometry andleads to the C10H8–C10H10 mixture in a 95:5 ratio. The authors proposed an alternative,Scheme 1-27:
C10H8�.
Li� � MeOH → C10H9.
� MeOLi
C10H9.
� C10H8�.
Li� → C10H9�Li� � C10H8
C10H9�Li� → C10H8 � LiH
C10H9� Li� � MeOH → MeOLi � C10H10
LiH � MeOH → MeOLi � H2
The decisive point of the novel scheme is the amortization of C10H9�Li� by the elim-
ination of the metal hydride. The authors admit that the weakness of their scheme is the lackof evidence for the formation of alkali metal hydride and for the formation of H2 from the(supposed) reaction between the protonating agent and the alkali metal hydride. However,the main sense of this scheme consists of its better agreement with the observed stoi-chiometry. As to the first step of Scheme 1-27 (the proton landing), it can certainly have amore intimate mechanism, say, electron transfer from the anion radical to the protonating
TABLE 1-3 Differences in pKa Values Between Aromatic Compounds andTheir Anion Radicals
pKa PKa
ArH (ArH2�) (ArH2
�) �pKa
Benzene �23 �13.5 �36NO2–Benzene �11.3 3.2 14.5Benzonitrile �10.4 7.3 17.7p-Me–Benzoate �7.8 5.5 13.3Benzaldehyde �7.1 8.4–10.5 15.5–17.6Fluorenone �6.6 6.3 12.9Benzophenone �6.2 9.2 15.4Acetophenone �6.2 9.9 16.1Benzamide �1.9 7.7 9.6Benzoate 4.2 12.0 7.8
Source: Bil’kis & Shteingarts 1987.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

agent, followed by hydrogen atom attack on the neutral naphthalene and production of di-hydronaphthyl radical intermediate, according to Scheme 1-28:
C10H8�.
Li� � MeOH ⇔ [C10H8�.
Li�, MeOH] →[C10H8, Li�MeO�, H
.] → MeOLi � C10H9
.
1.3.1.A Pathways of Hydrogen Detachment from Anion Radicals
As a rule, the addition of an extra electron to a parent organic molecule leads to significantweakening of bonds in a forming anion radical, and thereby bond breaking is facilitated.According to Zhao, Y., & Bordwell (1996a, p. 2530), there are three feasible differentpathways of hydrogen detachment from anion radicals (AH�.
). These three pathways arecompared in Scheme 1-29. Path (a) can be demonstrated with examples of anion radicalsof amino and hydroxy derivatives of 2,1,3-benzothiadiazole (Asfandiarov et al. 1998) andthe azafullerene anion radical, C59HN�.
(Keshavarz et al. 1996). Scheme 1-30 describesone of these examples, hydrogen atom detachment from C59HN�.
.
C59HN�. → C59N� � H.
Another example of the path (a) of Scheme 1-29 is the anion radical derived from flu-orene. It undergoes a first-order decay to give the conjugate base (the fluorenide anion) anda hydrogen atom (Casson & Tabner 1969), according to Scheme 1-31.
Pathway (a) of Scheme 1-29 has some kinetic preference, since it can be linked withthe strongly exothermic dimerization of the hydrogen atoms formed by this pathway(Zhang & Bordwell 1992).
As to case (b) of Scheme 1-29, hydride loss from organic anion radicals is generallynot as favorable as the hydrogen atom loss because the solvation of the hydride ion and theorganic anions is similar. Generally, path (a) is favored over path (b) in a wide set of or-ganic anion radicals. Free energies of bond dissociation for the anion radicals to give a hy-dride ion and a radical by path (b) are the highest-energy pathway (Zhao, Bordwell 1996b,p. 6623).
SCHEME 1-29
SCHEME 1-31
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.
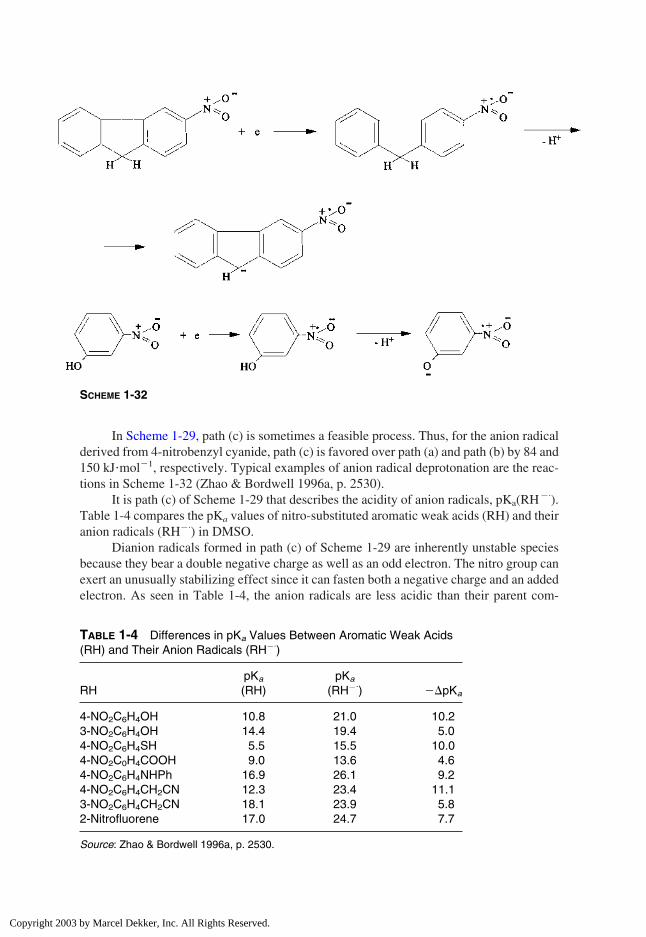
In Scheme 1-29, path (c) is sometimes a feasible process. Thus, for the anion radicalderived from 4-nitrobenzyl cyanide, path (c) is favored over path (a) and path (b) by 84 and150 kJ�mol�1, respectively. Typical examples of anion radical deprotonation are the reac-tions in Scheme 1-32 (Zhao & Bordwell 1996a, p. 2530).
It is path (c) of Scheme 1-29 that describes the acidity of anion radicals, pKa(RH�.).
Table 1-4 compares the pKa values of nitro-substituted aromatic weak acids (RH) and theiranion radicals (RH�.
) in DMSO.Dianion radicals formed in path (c) of Scheme 1-29 are inherently unstable species
because they bear a double negative charge as well as an odd electron. The nitro group canexert an unusually stabilizing effect since it can fasten both a negative charge and an addedelectron. As seen in Table 1-4, the anion radicals are less acidic than their parent com-
SCHEME 1-32
TABLE 1-4 Differences in pKa Values Between Aromatic Weak Acids(RH) and Their Anion Radicals (RH�.
)
pKa pKa
RH (RH) (RH�.) ��pKa
4-NO2C6H4OH 10.8 21.0 10.23-NO2C6H4OH 14.4 19.4 5.04-NO2C6H4SH 5.5 15.5 10.04-NO2C0H4COOH 9.0 13.6 4.64-NO2C6H4NHPh 16.9 26.1 9.24-NO2C6H4CH2CN 12.3 23.4 11.13-NO2C6H4CH2CN 18.1 23.9 5.82-Nitrofluorene 17.0 24.7 7.7
Source: Zhao & Bordwell 1996a, p. 2530.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

pounds. One can expect such weakening in acidity owing to the combined action of twofactors: The negative charge retards the loss of a proton, and the formed dianion radical isinherently unstable.
One very unusual case of prototropic isomerization was revealed for anion radicalsof 1,4-dihydropyridine derivatives (Gavars et al. 1999). These anion radicals transform into4,5-dihydropyridine analogs through proton detachment and addition.
1.3.2 Cation Radicals
1.3.2.A Cation Radical Acidity
Deprotonation is a typical direction of cation radical reactivity. Cation radicals are usuallystrong H acids. (For example, the alkane cation radicals pass their protons on the alcoholmolecules—Sviridenko et al. 2001). Bases that conjugate to these H acids are radicals, seeScheme 1-33:
RH�. ⇔ H� � R.
Scheme 1-34 displays two real cases of such reversible deprotonation (Neugebaueret al. 1972). As can be seen in the scheme, the cation radicals transform into radicals thatare more or less stable and can be protonated reversibly. If the radicals formed are unsta-ble, they perish before protonation. If the “initial” cation radicals have no hydrogen atoms,their stability appears to be higher. Deprotonation is typical for cation radicals that containproton-active hydrogen atoms and form, after deprotonation, either quite stable or, the re-verse, quite unstable radicals.
Formation of radicals having a lower energy than that of the starting cation radicalsis obviously favorable for their deprotonation. The cation radicals of toluene and otheralkylbenzenes are illustrative examples. As shown (Sehested & Holcman 1978), thesecation radicals lose protons even in very acidic aqueous solutions. The deprotonation ratedoes not, in general, depend on the medium acidity. In acetonitrile (AN), the toluene cationradical has high thermodynamic acidity, its pKa is between �9 and �13 (Nicholas &
SCHEME 1-34
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Arnold 1982). In the same solvent (AN), neutral toluene has pKa � �45 (Breslow & Grant1977). So, one-electron oxidation of toluene causes the acidity to grow by 60 pKa units!
One-electron oxidation of toluene results in the formation of a cation radical in whichthe donor effect of the methyl group stabilizes the unit positive charge. Furthermore, theproton abstraction from this stabilized cation radical leads to the conjugate base, namely,the benzyl radical. This radical also belongs to the � type. Hence, there is resonance stabi-lization in the benzyl radical. This stabilization is greater in the benzyl radical than in the� cation radical of toluene. As a result, the proton expulsion appears to be a favorable re-action, and the acid–base equilibrium is shifted to the right. This is the main cause of theacidylation effects that the one-electron oxidation brings.
It is interesting to compare the reactions in Scheme 1-35:
(PhCH3)�. → H� � PhCH.2
(PhCH2CH2Ph)�. → H� � PhCH2CH.Ph
In dimethylsulfoxide, the two starting cation radicals of Scheme 1-35 have pKa values of�20 and �25, respectively (Bordwell & Cheng 1989). It is clear that both species give riseto the stabilized carboradicals after deprotonation. Electron-donating substituents increasethe stability of the arene cation radical and render the odd-electron species less acidic; forexample, the cation radical of hexamethylbenzene has a pKa value of only 2 in AN (Ama-tore & Kochi 1991). The cation radical of tris(bicyclopentyl)annelated benzene is not proneto proton loss, due entirely to the spin-charge location “more or less in the aromatic (nodal)phase” (Rathore & Lindeman et al. 1998), Scheme 1-36.
The tetrahydropyrazine ring in the 5-methyluracil cation-radical is nonaromatic; spindelocalization in the corresponding methylene radical is impossible. Correspondingly, thiscation radical does not expel the proton at all (Rhodes et al. 1988).
Zhang, X.-M., & Bordwell (1994) determined N–H and O–H bond dissociation en-ergies of cation radicals, denoted BDE(AH�.
). They found that the energies are onlyslightly lower than those of their nitrogen or oxygen conjugate acids. However, when theproton being detached is bonded to a carbon atom, the BDE(AH�.
) value is typically lowerthen the BDE(AH) of the corresponding neutral compound.
Medium acidity is not essential for the deprotonation of the toluene cation radical, butmedium basicity provokes the abstraction of the cation radical proton. Although this effectis obvious, special experiments were undertaken to define it more accurately (Bernstein1992 and references therein). The studies show that at least three water molecules are re-
SCHEME 1-36
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

quired to fulfill the deprotonation inside the cluster between the toluene cation radical andthe water molecules. At the same time, only one molecule of ammonia is enough to achievethe deprotonation inside the corresponding cluster. Theoretical calculations for this andother cation radicals are in accord with the experimental results just mentioned (Camanyeset al. 1996).
Cation radicals of weak acids may react either by heterolytic cleavage (loss of a pro-ton to produce a radical) or by homolytic cleavage (loss of a hydrogen to form a carboca-tion). Alkane cation radicals in liquid hydrocarbon solution undergo ion–molecule reac-tions, such as proton transfer on a submillisecond scale (Werst et al. 1990).
In a polar solvent, heterolytic cleavage leading to proton abstraction is usually facil-itated because of the favorable solvation energy of the proton, and cation radicals are ordi-narily much more acidic than the corresponding neutral compounds. Table 1-5 combinesacidity constants of organic compounds (AH) and their cation radicals (AH�.
) calculatedfor their solutions in dimethylsulfoxide (DMSO, a very polar solvent) at 25°C.
The acidity of the phenol-family cation radicals depends on the stability of the cor-responding phenoxyl radical, which formed after the proton abstraction according toScheme 1-37:
(ArOH)� → H� � ArO.
In aqueous solutions, the phenol cation radical has pKa � �2 (Dixon & Murphy1978; Holton & Murphy 1979). Surprisingly, the pKa value of the 2,4,6-triphenylphenolcation radical is equal to �5 only (Land et al. 1961; Land & Porter 1963). A natural ques-tion arises: Why does the stabilization of the phenoxyl radical with the shielding phenylgroups result in such a small increase in acidity?
One can assume that the space shielding with the phenyl groups not only stabilizesthis phenoxyl radical, but to some degree hinders proton removal. In a similar manner, thehydroxy group in 3,5-di(tert-butyl)-4-hydroxybenzylidene malodinitrile is localized in thebenzene-ring plane and clamped between two methyl fragments of the tert-butyl group (forX-ray evidence, see Itoh et al. 1978). If there are two phenyl groups in the ortho positionsof the phenol cation radical, the leaving hydroxylic proton experiences not only steric hin-drance but also stoppage from complexation by the �-system(s) of the phenyl ring(s).
Although the size of the leaving proton is not significant, steric hindrance for depro-tonation can be appreciable. Here’s an additional example: The cumene cation radicalprefers to lose a proton from -carbon rather than �-carbon. This is ascribed to the stereoelectronic effect (Zhao, Ch.-X., et al. 1999).
TABLE 1-5 Differences in pKa Values Between Aromatic Compoundsand Their Cation Radicals
pKa pKa
AH (AH) (AH�) �pKa
Toluene 43.0 �20.0 63Fluorene 22.6 �17.0 39.6Phenylacetonitrile 21.9 �32.0 53.9Phenol 18.0 �8.1 26.1Thiophenol 10.3 �12.0 22.3
Source: Bordwell & Cheng 1989.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Let us now compare the pKa values (in AN) of the cation radicals derived from ani-line, N-methylaniline, and N-phenylaniline: 5.5, 4.2, and 1.8, respectively (Jonsson et al.1996). An additional N-methyl substituent produces only a marginal effect on the pKa ofthe aniline cation radical. At the same time, the effect of a single N-phenyl substituent inaniline is considerable. The phenyl-group effect on the aniline cation radical acidylation ispredictable.
Deprotonation of the adenine cation radical is a convenient starting point for dis-cussing the control of this reaction from the point of view of the relative stability of the rad-ical formed. The proton loss is possible from the N(6)–H and N(9)–H bonds according toScheme 1-38.
It is obvious that one-electron oxidation has to touch the lone electron pair at thegroup NH2. Consequently, the N(6)–H deprotonation is expected. Nevertheless, the realprocess occurs at the expense of both N(6)–H and N(9)–H bonds (Dias & Vieira 1996). Inthe N(9)-deprotonated radical, the unpaired electron is delocalized over the entire purinesystem, incorporating four nitrogen and five carbon atoms; see Scheme 1-39.
SCHEME 1-38
SCHEME 1-39
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

In the N(6)-deprotonated radical, an unpaired electron can be delocalized over onlythree nitrogen atoms. These are one nitrogen of the group NH
.and two nitrogens of the six-
membered ring. An unpaired electron can also be delocalized over two carbon atoms (oneby one of the six- and five-membered rings). This part of delocalization is depicted inScheme 1-40. So the N(9) radical should be more stable than the N(6) one. That is why bothradicals coexist in the system and both N(9) and N(6) deprotonations take place.
A potential problem when discussing the pKa of alkylamine cation radicals is that thedeprotonation can occur on both the nitrogen and the �-carbon, as in Scheme 1-41:
(Et2NH)�. ⇔ H� � Et2N.
(Et2NH)�. ⇔ H� � EtNCH.2Me
In the latter case, the aminomethylene radical is formed upon deprotonation of the cationradical. Unless proton equilibrium for one of these two radical types is much slower thanfor the other type, the radical that corresponds to the lowest pKa should be formed upon de-protonation of the cation radical (Et2NH)�.
. This cation radical has pKa � 5.3 in aqueoussolutions at pH 3–9. Therefore, O2 saturation of the solutions does not affect the determinedpKa (Jonsson et al. 1996). Since O2 reacts more rapidly with carbon-centered radicals thanwith nitrogen-centered radicals, one can conclude that deprotonation of (Et2NH)�.
takesplace at the nitrogen rather than at the �-carbon.
All cation radicals can be categorized as H acids on the basis of the nature of theirconjugate radicals. The benzene cation radical can be named a � acid because it gives thecorresponding �-radical as a result of deprotonation. The toluene cation radical gives a ben-zyl radical of the � type. Therefore, this cation-radical is a � acid. The phenol cation radi-cal should be named a hetero-�-acid (not a hetero-�-acid) because an unpaired electron inits conjugate base—the phenoxyl radical—is delocalized within the aromatic �-system.Some amines can give cation radicals that transform into radicals having three electrons in
SCHEME 1-40
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.
the field of two nuclei according to Scheme 1-42:
R12 N
�.MCH2R2 H�
→←�H�
R12 N
�.MC�HR2 ←→ R1
2 NM� CHR2
Because the three-electron bonded radicals are formed at the cost of the removal ofthe nitrogen p-electron, such cation radicals should considered p acids. Of course, the for-mation and behavior of these p acids have to be dependent on steric factors. Such depen-dence will be considered in Chapter 3 (Section 3.2.3). Works by Tomilin et al. (1996, 2000)and Bieti and co-authors (1998) should be mentioned as describing stereoelectronic requirements to formations and configurational equilibria of N-alkyl-substituted cation radicals.
Deprotonation is essential in some cation radical reactions; the corresponding exam-ples will be described in Chapter 6. Scheme 1-43 depicts a photoreaction between phenan-threne and triethylamine. This reaction includes photoinduced sequential electron-transfer,proton-transfer, and radical-recombination processes (Lawson et al. 1999).
SCHEME 1-43
Many authors note the fact that adsorption of alkenes and alkyl aromatics upon thealumosilicate surfaces leads to a marked increase in their proton acidity (e.g., Farne et al.1972). Formation of cation radicals and their further deprotonation explain many featuresof the hydrocarbon catalytic transformations (Vishnetskaya et al. 1997). Scheme 1-44 (seebelow) represents this phenomenon.
�SiO.� ArCH2R → �SiO� � (ArCH2R)�.
(ArCH2R)�. → H� � ArCH.R
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Vishnetskaya and Romanovskii described the cation radical approach to such cat-alytic reactions in their reviews of 1993 and 1994. It is worth noting here that organic cationradicals are usually very sensitive to surrounding nucleophilic reagents. This often de-presses their deprotonation. In channels of a redox-active catalyst (e.g., in zeolites), thecation radical deprotonation is not aggravated, because competing reactions with the out-side reagents are precluded (Roth et al. 1997; Corma & Garcia 1998).
Concerning catalytic transformation of aliphatic hydrocarbons, an important problemis what positions are active in the deprotonation. In other words, what is a radical generatedby proton abstraction from the initial cation radical? The ESR spectra of alkane cation rad-icals show that the dominant hyperfine coupling is caused by hydrogen atoms of terminalmethyl groups that lie in the plane of the carbon skeleton in its extended conformation. Theproton loss from the cation radical is thought to involve the carbon–hydrogen bond with thehighest unpaired electron density (Toriyama et al. 1982). Calculations show that spin en-richment of the carbon–hydrogen bond leads to its elongation. This results in a weakeningof the carbon–hydrogen binding and facilitates the bond disruption (Shchapin & Chuvylkin1996).
Thus, the fully expanded cation radical of n-heptane undergoes a proton transfer ac-cording to Scheme 1-45:
RH�.� RH → R
.� RH2
�
Reaction 1-45 leads to selective formation of the 1-heptyl radical (Demeyer & Stien-let 1993). In the case of trans-octene-2, the cation radical deprotonation predominantlyleads to the allyl radical formation (Fel’dman et al. 1993, 1996), Scheme 1-46:
[CH3CHBCH(CH2)4CH3]�. → H� � (CH2MCHMCH).–(CH2)4CH3
Like any other rule, the rule of preferential deprotonation at the highest spin densityposition has its exceptions. For example, in the cation radical of silacyclohexane, the max-imal-spin-density atoms are carbons bonded with silicon. Nevertheless, deprotonation (inFresno matrices) occurs preferentially from the �SiH2 (Komaguchi & Shiotani 1997). Theauthors pointed out, that the deprotonation energy from �SiH2 in a silaalkane is a little bitsmaller than that from MCH2 in an alkane. This suggests that formation of the silyl radicalMSiH
.might be energetically more favorable. The solvation energy and the energy of ma-
trix arrangement can also be important factors to explain the observed high selective de-protonation.
Very recently, some attempts were undertaken to uncover the intimate mechanism ofthe cation radical deprotonation. Thus, the reaction of 9-methyl-10-phenylanthracenecation radical with 2,6-lutidine was studied (Lu et al. 2001). The reaction takes place by atwo-step mechanism that involves the intermediate formation of a cation radical/base com-plex prior to unimolecular proton transfer and separation of products. Based on the valueof the kinetic isotope effect observed, it was concluded that extensive proton tunneling isinvolved in the proton-transfer reaction. The assumed structure of the intermediate com-plex involves �-bonding between the unshared electron pair on nitrogen of the lutidine withthe electron-deficient �-system of the cation radical.
1.3.2.B Cation Radical Basicity
At first glance, the question of cation radical basicity seems meaningless: Cation radicalsare usually strong acids. Nevertheless, there are two relevant examples. Corrole and iso-corrole (as the octaethyl derivatives) give cation radicals that are basic in nature. Both neu-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

tral compounds belong to the porphyrin family and are named contracted porphyrins. Theyare typified by their contracted macrocycle, one carbon less than porphyrins, but still con-tain four pyrrole rings (Vogel 1996). One of the pyrrole rings contains nitrogen without theN–H bond. As proven, this nitrogen accepts a proton when the free bases of corrole and iso-corrole are oxidized to the cation radicals by iodine and silver perchlorate in methylenechloride (MC). Solutions of the protonated cation radical are stable for more than a weekat �20°C (Endeward et al. 1998). Notably, the described protonation proceeds at the ex-pense of MC. MC is known as an extremely weak acid. The starting neutral bases are notprotonated in MC in the absence of the oxidants!
Some cation radicals can appear as hydrogen acceptors. Thus, fullerene C60 is oxi-dized to the cation radical at preparative scale by means of photoinduced electron transfer.This cation radical reacts with various donors of atomic hydrogen (alcohols, aldehydes,ethers), yielding the fullerene 1,2-dihydroderivative. In the case of tert-butanol, propionicacid, and glycol, product formation is also initiated by H abstraction from the OH group.The reaction proceeds according to Scheme 1-47 (Siedschlag et al. 2000):
C60 � e → C�.60; C�.
60 � RH → HMC�60 � R
.; HMC�
60 � R.� e → HMC60MR
The naphthalene cation radical can react with the hydrogen atom. The 1-hydronaph-thalene cation resulting from this reaction is more stable than the reactants. This processhas no activation barrier. After its formation, the 1-hydronaphthalene cation can absorb aphoton and lose one of the two hydrogen atoms on carbon 1, since these hydrogens aremore weakly bound than the aromatic hydrogens. Because the C–D bond is slightlystronger than the C–H bond, the reaction mentioned here can lead to some deuterium en-richment (Bauschlicher 1998).
On the other hand, some organic cation radicals can act as hydrogen atom donors. Forexample, one-electron oxidation of 1,3-cyclohexadiene (C6H8) by 3,4-di(tert-butyl)-4-chloro-1,2-benzoquinone leads to the formation of C6H8
�.. In the presence of sulfur, dehy-
droaromatization of the cation radical takes place (Berberova et al. 1998), Scheme 1-48:
C6H8�.
� S → C6H7� � HS
.; 2HS
. →HSSH; C6H7� � C6H8 → C6H
.7 � C6H8
�;
H.� C6H6 ← C6H
.7 → C6H6 � C6H8 etc.
1.4 METALLOCOMPLEX ION RADICALS
1.4.1 Metallocomplex Anion Radicals
Organic anion radicals are odd-electron species. When this odd-electron nature is retainedafter coordination with a metal or it emerges as a result of some redox transformation of aninitial metallocomplex, the newly formed species may be unstable. As to the metallocom-plex anion radical assembling, high basicity and, consequently, high nucleophilicity of thestarting anion radical ligands may play the decisive role in coordination with electrophilicmetal-containing reactants. In other words, the electrostatic stabilization of the formingmetallocomplex may compensate for these unfavorable factors. Besides, relocation of anunpaired electron from the anion radical ligand to the central metal atom may also lead tostabilization of such complexes (see also Section 2.2.1).
For instance, the [(�-C5Ph5)Co(CO)2]�.anion radical complex has the (n � 1)py
metal orbital populated with an unpaired electron, according to calculations (Connelly etal. 1986). On the other hand, reduction of (bpy)Cr(CO)4 (bpy � 2,2-bipyridine) to its an-ion radical is known to occur without any major change in its structure or composition. The
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

ESR spectrum of the [(bpy)Cr(CO)4]�.ion radical resembles that of the uncoordinated
(bpy)�.anion radical, clearly indicating that the unpaired electron is localized on the (bpy)
ligand. This conclusion is fully corroborated by the similarity between the electronic spec-tra of [(bpy)Cr(CO)4]�.
and (bpy)�.(Vlcek et al. 1998 and references therein). Obviously,
the product of one-electron reduction of (bpy)Cr(CO)4 may best be formulated as a for-mally Cr(0) complex with an anion radical ligand, (bpy)�.
Cr(CO)4.There are anion radical metallocomplexes with complete spin retention at the anion
radical ligand (Glockle et al. 2001), as well as those having an unpaired electron on a metalatom entirely, or those that share an unpaired electron with all parts of the complex (Kaim1987 and references therein).
It is clear that the type of spin distribution depends on the participation of fragmentorbitals in such a molecular orbital (MO), which is available to be populated with the in-coming electron. In terms of electron transfer, organometallic chemistry distinguishes theelectron-transfer pathway (if the lowest unoccupied orbital is populated) or the hole-trans-fer pathway (if the highest occupied orbital is fitted with an outgoing electron). For exam-ples, see Andersson et al. (2000).
The lower the MO energy, the more probable the spin density localization in theframework of this MO. At the same time, coordination with a metal often decreases the en-ergy level of the organic ligand MO. This increases the stability of the ligand anion radicalform. For example, 2,1,3-thiadiazole gives an unstable anion radical upon one-electron re-duction. However, this stability sharply increases if the anion radical is coordinated withtwo W(CO)5 fragments (at two nitrogen atoms of the thiadiazole ring) (Bock et al. 1988).If the 2,1,3-thiadiazole ring is condensed with the benzene ring, the anion radical stabilityincreases even without the metal coordination (Todres, Lyakhovetskii, & Kursanov 1969).Meanwhile, complexation with two M(CO)5 fragments (M � Cr, Mo, or W) changes theESR spectra of these anion radicals. This means the metal participates in spin delocaliza-tion (Bock et al. 1988).
The central metal atom in complexes usually has a shell of the nearest noble gas, e.g.,the shell of 18 electrons. Upon one-electron transfer, the electron number turns from aneven into an odd one (e.g., 19 for anion radicals and 17 for cation radicals). The one-unitchange of the electron amount leads to an increase in the complex reactivity. The ligand-substitution reactions are markedly facilitated.
Thus, one-electron reduction of (bpy)Cr(CO)4 results in facilitation of the CO–PPh3
replacement in an electron-transfer-catalyzed reaction (Miholova & Vlcek 1985). Some-times, the catalytic cycles in Scheme 1-49 become available (Kochi 1986).
One-electron reduction of the LM complex starts the reaction cycle in Scheme 1-49. The LM�.
anion radical that is formed reacts with a nucleophile (N) undergoing theL → N ligand change. In so doing, the complex does not lose the unpaired electron; theL ligand is displaced out of the coordination-sphere limits. The anion radical productNM�.
passes its unpaired electron to the initial uncharged complex LM. This results inregeneration of the LM�.
anion radical. The neutral product NM goes out of the catalyticcycle. Scheme 1-49 illustrates the substitution of triphenylphosphine for pyridine as a lig-and in the manganese cyclopentadienyl carbonyl complex. The reaction is initiated elec-trochemically, and only one-electron consumption is sufficient to involve 290 moleculesof the initial LM complex in this transformation. The reaction is completed after half anhour at room temperature. With no electron initiation, prolonged boiling in toluene is re-quired (Kochi 1986).
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Ohst and Kochi (1986) traced changes in the electron structure that take place duringsubstitution of the triethyl phosphine ligand for the carbonyl ligand in the iron–phenyl–phosphine–carbonyl complex; see Scheme 1-50.
The initial anion-radical of Scheme 1-50 is formed from the diamagnetic ternary nu-clear complex upon one-electron reduction. This anion radical undergoes spontaneousbreaking of one of the phosphine–iron bonds. The further substitution restores the Fe–Pbond, which has been opened previously. Such restoration makes the whole reactionmacroscopically reversible (in the sense of the cluster-skeleton preservation).
SCHEME 1-49
SCHEME 1-50
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The main point of the Scheme 1-50 transformation consists of splitting of the bridgebond between the metal and the ligand (not the metal–metal bond). It is important becausethis type of splitting leads to formation of the odd-electron shell around one of the ironatoms. The iron with the odd-electron shell acquires enhanced reactivity and changes oneof its ligands. Without one-electron transfer (in diamagnetic clusters), this transformationdoes not take place at all.
Activation of an M–CO bond for nucleophilic substitution in anion radical metallo-complexes appears to be quite a general effect (Kaim 1987; Mao et al. 1989, 1992; Shut etal. 1995; Klein et al. 1996). Such activation seems to be the basis of metal-cluster catalyticactivity. The iron–sulfur cluster (Bu4N)2Fe4S4(SPh)4 deserves to be mentioned here. Thecluster is considered as a ferredoxin model (Inoue & Nagata 1986); it catalyzes an electrontransfer from n-butyl lithium or phenyl lithium to S-phenyl thiobenzoate or phenylbenzoate(Inoue & Nagata 1986).
Other examples concern the interaction between iron carbonyles and potassiumalkylthiolates that is accompanied by disproportionation. The anion radicals Fe2(CO)8
�.,
Fe3(CO)11�.
, Fe4(CO)13�.
, and Fe(CO)2�.
are formed (Belousov et al. 1987). The interaction ofiron carbonyls Fe(CO)5, Fe2(CO)9, and Fe3(CO)12 with (CH3)3NO occurs according to aone-electron redox–disproportionation scheme, giving rise to iron carbonyl anion radicals:Fe2(CO)8
�.Fe3(CO)11
�., Fe3(CO)12
�., and Fe4(CO)13
�.(Belousov & Belousova 1999).
Sometimes, the ligand substitution in metallocomplexes can also proceed without re-duction. The complex Co(CO)3L2[L2 � bis(diphenylphosphino)maleic anhydride] readilyexchanges one of the carbonyl groups for trialkylphosphine. Kinetic studies showed thatthe substitution goes through the Co–CO bond disruption. The initial complex has tetrago-nal-pyramidal geometry and possesses one unfilled vacancy to coordinate an additional lig-and. Studies of the complex Co(13CO)3L2 by the ESR method revealed that the excess elec-tron presumably populates the antibonding orbital of the Co–CO fragment. This bond isjust weakening. The substitution takes place according to Scheme 1-51:
Co(CO)3L2 � PPh3 → CO � Co(CO)2L2PPh3
The initial 19-electron complex was classified as the 18-electron complex with the anionradical ligand (Mao et al. 1989).
Insertion of new ligands into metallocomplex systems may proceed reversibly. Be-ing reduced in the framework of the complex, these ligands lose the ability to be coordi-nated and leave the coordination sphere as products. One important example of such ligandsliding is the catalytic transformation of CO2 into CO. Rhenium, palladium, platinum, andnickel complexes were recommended to catalyze this process (Hawecker et al. 1986; DuBois & Meidaner 1987). The Ni(II) complex with 1,4,8,11-tetraazacyclotetradecane is pre-ferential (Beley and co-authors 1984).
The preceding examples show that one-electron reduction of metalloorganic com-plexes or coordination between a metal and an anion radical ligand may expand an electronshell of the central metal atom. Sometimes, anion radical metallocomplexes contrast in thisregard with the cation radical ones. Thus, some metalloporphyrins form cation radicalswith charges and unpaired electrons on ligands (Shinomura et al. 1981) and anion radicalswith charges and unpaired electrons on metals (Lexa et al. 1989).
There is some lack in data on anion radical stabilization as a result of binding into ametallocomplex. One specific case was described for bis(trimethylsilyl) diacetylene (Kaim1988). When treated with potassium, this compound gives tetrakis(trimethylsily)butate-traene, probably owing to some transformations of the unstable anion radical. This anion
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

radical is stabilized in the presence of trimethyl aluminum, giving the anion radical metal-locomplex [(Me3Al)(Me3Si)CBCBCBC(SiMe3)(AlMe3)]�.
K�.
1.4.2 Metallocomplex Cation Radicals
There is a large body of literature describing one-electron oxidation of metallocomplexeswith organic ligands, metal–ligand interactions in cation radical products, and structuralchanges on the formation of cation radicals. Such abundance is caused by the chemical na-ture of metallocomplexes, in which the metallic center readily transforms into a higher stateof oxidation. Kochi (1986) and Kaim (1987) have covered many of these problems in theirreviews. Astruc (1995) has done one recent and very important generalization in the field.
One of the most important intricacies in the redox chemistry of organyl metallocom-plexes is that both a metal and a ligand are potential candidates for oxidation. One exam-ple can be taken from the porphyrin (P) family. It is possible to move between the two for-mulations. The porphyrine-oxidized manganese(III) bis-perchlorate, (P�.
)MnIII(ClO4)2,gives the metal-oxidized manganese (IV) bis-chloride, (P)MnIV(Cl)2, when the two per-chlorate ligands have been substituted with the two chloride ligands (Kaustov et al. 1997).
The copper–porphyrin complex gives cation radicals with significant reactivity at themolecular periphery. This reactivity appears to be that of nucleophilic attack on this cationradical, which belongs to the � type (Ehlinger & Scheidt 1999). A new bifunctionaltetrathiafulvalene-type donor molecule (D–�–D) with a copper iodine bridge has recentlybeen synthesized. Its cation radical salt, (D–�–D)�BF4
�, has the hole localized on one ofthe D fragments, although copper is a metal of variable valence (Ramos et al. 1997).
The porphyrin–cobalt complex gives rise to the cation radical with charge spin lo-calization at the nitrogen atom of the porphyrin ring. The cation radical thus formed ac-quires enhanced reactivity and can add tolane (Kochi 1986), Scheme 1-52. The main point
SCHEME 1-52
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

of the transformation in Scheme 1-52 is that the central cobalt atom eventually becomes anelectron reservoir.
Iron complexes with organic ligands are widely used as such reservoirs. For exam-ple, the stable 19-electron complex cyclopentadienyl benzene iron(I) is a clean electronreservoir that has a sufficiently negative oxidation potential and can be easily made, stored,and handled and can be weighed accurately (Alonso & Astruc 2000).
Reversible one-electron oxidation of ferrocene and its derivatives to cation radicals(so-called “ferricenium” cations) is a well-known reaction. The cation radical center is lo-calized at the iron atom. In contrast to this statement, the hole transfers though conjugatedsystems were proven for the bis(ferrocenyl) ethylene cation radical (Delgado-Pena et al.1983) and the cation radical of bis(fulvaleneiron) (LeVanda et al. 1976). Scheme 1-53 de-picts these structures.
Extended Hueckel MO calculations (Kirchner et al. 1976) support the formulationthat the unpaired electron is delocalized over both metallocene residues in the bis(ful-valeneiron) cation radical. Alternatively, very fast intraionic intervalence electron transfermay take place between the formal Fe(II) and Fe(III) atoms.
In general, the ethylene bond in organic cation radicals is weakened, and the barrierto rotation becomes significantly less than that of the neutral ethylene derivative. This par-ticular property of the ethylene bond in cation radicals has been used to probe for the mech-anism of many reactions (Todres 1987).
As revealed, cation radicals of ferrocenyl ethylenes do not undergo the cis-to-transisomerization. Calculations show that the cation radical’s center is located exclusively atthe iron atom, with no participation of the ethylene bond (Todres et al. 1992). Hence, one-electron oxidation of this ferrocenyl ethylene occurs at the iron atom exclusively; anythingelse would be extremely unusual in this case. Thus, the recently described stable enollinked to a ferrocene redox center gives the cation radical upon one-electron oxidation. Thisspecies is better characterized as a ferricenium salt than as an enol cation radical (Schmit-tel & Langels 1998).
Many hydroxyquinones can bind metal cation via chelate formation. Magnesiumchelates of 5,8-dihydroxynaphtho-1,4-quinone and 1,4-dihydroxyanthra-9,10-quinone arecharacterized by large complex-formation constants. These constants become very small incases of the corresponding semiquinones (anion radicals). This apparent paradox is ex-plained by the much stronger intramolecular hydrogen bonding existing in the hydroxy-semiquinones as compared to the neutral quinones (Alegria and co-workers 2000).
SCHEME 1-53
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Recently, variable-temperature absorption data were obtained for oxomolybdenumdithiolate complex [MoVO(qdt)2]1�; here qdt is quinoxaline-2,3-dithiolate. The dataclearly reveal the highly thermochromic nature of this compound. The authors (Helton andothers 2001) explain the observed thermochromism through a thermally induced in-tramolecular electron transfer according to Scheme 1-54:
[(qdt)MoVO(qdt)1� ⇔ [(qdt)MoIVO(qdt)�.]1�
The thermochromism in Scheme 1-54 represents valence tautomerism in molybdenumcomplexes and, at the same time, the inherent redox activity of a dithoilate ligand. This phe-nomenon may be significant with respect to the biological activity of various molybdenumenzymes.
1.5 ORGANIC ION RADICALS WITH SEVERAL UNPAIREDELECTRONS AND/OR CHARGES
In principle, an organic molecule can accept as many electron pairs as it has low-lying vacant orbitals. In the same way, high-lying occupied orbitals can release not a single, butseveral electrons. Such multielectron processes can result in the formation of poly(ion rad-icals). As seen later, the main topic of interest in poly(ion radicals) consists in their spinmultiplicity. Therefore, it is meaningless to divide the material in the anion and cation rad-ical parts. Let us therefore discuss first the reductive electron transfer.
Anion radicals are the first products of reductive electron transfer. Mutual repulsionof the primary and subsequent excess electrons forms the basis of the impediment of poly-electron reduction at the initial step. However, an orbital, which already has one electron,can be populated by another electron if a proper reducer is chosen. This reducer must beable to overcome Coulombic repulsion. As a result, more or less stable dianions are formed.Like anion radicals, dianions can reversibly give excess electrons back. If skeleton rear-rangement is absent, the starting, uncharged molecules are regenerated.
In dianion diradicals, two vacant orbitals are populated, i.e., LUMO and the next one.This next orbital must lay low enough, as well. Such an orbital can be detected in moleculesof unsaturated hydrocarbons of an extensive contour. Within this expanded contour, ex-tensible electron delocalization somewhat decreases the repulsion energy. Several electron-acceptor fragments (substituents with heteroatoms or with high conjugation ability) assistin formation of poly(anion radicals).
Because poly(anion radical)s are polycharged species, they are very prone to asso-ciation with counterions. The association decreases the total energy of the system, in-creases its stability, and facilitates its formation. The formation of poly(anion radical)s isusually successful if alkali metals are used as reducers in DME, THF, or MeTHF as sol-vents. Medium components, e.g., HMPA or cryptands, which weaken ion-pair interac-tion, work unfavorably. Even traces of proton admixtures evoke a transformation of re-ducing compounds into hydrogenated products instead of polycharged anions. As a rule,the higher the charge of a particle to be prepared, the lower must be the temperature andthe longer the contact with a reducer. Working with such species requires an inert atmo-sphere, of course. Poly(ion radical)s excite today’s interest first of all because of theirpossible application in microelectronics. Therefore, the spin distribution is a very impor-tant point for these species. New organic triplet poly(ion radicals), with strong in-tramolecular ferromagnetic coupling and good thermal stability, may serve as buildingblocks for organic magnets.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

For example, sodium reduction of bis(dimesitylboron)-1,3-phenylene in 2-methylte-trahydrofurane-tetrahydrofurane (MeTHF-THF) mixture leads to the corresponding dian-ion diradical as a species of a triplet state (Rajca, Rajca, & Desai 1995).
In the case of 1,8-diphenylnaphthalene, there is an interesting difference in the spinstates of the anion radical on the one hand and of the trianion radical on the other. In the an-ion radical, the main contour for spin distribution is the naphthalene framework. In the tri-anion radical, the naphthalene � system bears two electrons (nonassociated with any bond)while the third (unpaired) electron oscillates between both phenyl substituents at positions1 and 8 (Gerson & Huber 1987).
Teracyanoquinodimethide gives the anion radical and dianion. It is unable to form thetrianion radical. However, with adhered �-electron contour, the trianion radical does ex-ist—such a form was described for tetracyano-2,7-pyrenoquinodimethide (Gerson, Heck-endorn, & Cowan 1983). The anion radical of tetracyano-2,7-pyrenoquinodimethide con-tains “a half” an electron at each dicyanomethide group. The corresponding dianion holds“the whole” electron at the same sites, and the trianion radical has one electron at eachC(CN)2 fragment and one electron even in the arene �-system (Gerson & Huber 1987).This trianion radical was also described as an anion radical bearing two negatively chargeddicyanomethide groups (Rieger et al. 1963).
Several anion biradicals have recently been reported, composed of semiquinone andnitroxide functionalities. The ground-state triplet anion biradicals were derived from one-electron reduction of open-shell acceptors, in which p-benzoquinone (BQ) is substituted bya nitronyl (Shultz & Farmer 1998) or nitronyl-nitroxide (NN) group (Kumai et al. 1994).Scheme 1-55 gives the BQNN example.
The anion diradical (BQNN)�.has two nondegenerated single occupied molecular
orbitals (SOMOs). One is delocalized over the entire molecule and the other (SOMO) islocalized within the NN group.
As a future cation diradical counterpart to this novel class of ground triplet species,the same Japanese crew has designed and prepared the following open-shell donors carry-ing the NN groups (Sakurai et al. 1996, 2000, Kumai et al. 1996). These donors are pre-sented in Scheme 1-56.
All mentioned donors were oxidized by excess I2 in THF or MeTHF. ESR spec-troscopy at cryogenic temperatures (6–110 K) established the paramagnetic nature of theoxidized products. In order to rationalize the ground-state spin multiplicity, the MO calcu-lations were performed on these paramagnetic species. The results strongly suggest that thespecies exist as the cation radicals with triplet ground states. There are two nondegenerated
SCHEME 1-55
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

SOMO and SOMO, too. The delocalized spin on the donor site and the localized spin onthe radical site in these cation diradicals are oriented in a parallel manner. As will be seenin Chapter 7 (Section 7.4), such a spin orientation results in ferromagnetic coupling (tripletinteraction) between the spins. In contrast, an antiparallel type of mutual orientation of thespins results in antiferromagnetic coupling. The parallel type is crucial in the design of or-ganic materials with magnetic properties.
Another motif for preparing magnetic materials based on anion radicals is the so-called “metal radical” approach. The approach consists of designing an extended latticecontaining paramagnetic metal ions whose magnetic interactions are determined by appro-priate bridging radical ligands. This strategy has been applied successfully using as ligandsthe anion radicals of tetracyanoethylene (Brandon et al. 1998) or 1,3-bis(3,4-dihydroxy-5-tert-butylphenyl)-5-tert-butylbenzene (the m-xylylene type of ligand; Caneschi and others2001).
Attempts to build di(anion radicals) with the participation of the fullerene C60 moi-ety deserve a special mention. Reduction of the benzoquinone-linked fullerene C60 firstgives the monoanion containing the semiquinone radical and the C60 moiety. Further re-duction of the monoanion produces the dianion having the semiquinone radical and the C60
anion radical. The ESR spectrum of this di(anion radical) clearly shows the triplet interac-tion between the semiquinone and C60 anion radical (Iyoda et al. 1996). In anion diradicalsof fullerene C60 derivatives bearing a nitroxide group (namely, 2,2,6,6-tetramethylpi-peridyl-1-oxyl), one electron is located in the fullerene moiety and experiences a strong ex-change coupling with the nitroxide unpaired electron (Arena and co-authors 1997). Atroom temperature both triplet and singlet states of this anion diradical turn out to be mixed.
Wienk and Janssen (1996) have designed N,N-bis[4-(diphenylamino)-phenyl]-N,N-diphenyl-1,3-diaminobenzene, Scheme 1-57. This phenylaniline comprises alternat-ing para- and meta-substituted phenylene (C6H4) rings to give the desired chemical stabil-ity from the p-diaminobenzene unit and ferromagnetic coupling from the m-diaminoben-zene unit after two-electron oxidation. The N-phenylaniline oligomer depicted in Scheme
SCHEME 1-56
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

1-57 was synthesized from 4-iodo-N,N-diphenylaminobenzene and N,N-diphenyl-1,3-di-aminobenzene in diphenyl ether at 200°C, using butyllithium and copper(I) iodide as a cat-alyst. Oxidation of the product with more than two equivalents of thianthrenium perchlo-rate resulted in the formation of the dication diradical. This reaction proceeds in thetrifluoroacetic acid (TFA) at room temperature. The product is stable at room temperatureand has a triplet ground state, as evidenced from ESR spectroscopy. Using N,N-bis(p-ani-syl)-1,3-diaminobenzene or N,N-bis(p-anisyl)-2,7-diaminonaphthalene as central units,Blackstock’s group prepared the analogous dication diradicals, which were found to be so-lution-stable triplets (Stickley & Blackstock 1994; Selby Blackstock 1999).
Depending on the amount of thianthrenium perchlorate, N,N,N,N,N�,N�-hexakis(anisyl)-1,3,5-triaminobenzene gives its cation radical, dication diradical, and trication tri-radical as well (Stickley et al. 1997). These species are stable in methylene chloride at lowtemperatures (at 298 K they can exist for several days). Spin and charge are localized ateach oxidized nitrogen atom. The dication diradical and trication triradical structures areground-state triplet and quartet molecules (Sato et al. 1997).
The latter two reactions demonstrate the possibility of using thianthrenium perchlo-rate in preparing magnet-active ion radicals with two or even three unpaired electrons andpositive charges. (At this point one very important caution has to be stated: Thianthreniumperchlorate is a shock-sensitive explosive solid and should be handled with care.)
From the materials just given above, one can conclude that mutual meta orientation(“meta through a benzene”) of the spin-bearing moieties is an indispensable condition forthe existence of triplet states in aromatic di- or tri-(cation radicals). However, in fact, thesesystems have both singlet and triplet forms, and the questions are about what the differenceis in the corresponding energy and which state is the more stable. Indeed, there is some ex-perimental evidence of violation of the meta-topology rule. The following two cases are il-lustrative examples.
1. N,N,N,N-Tetra(anisyl)-3,4-diaminobiphenyl: Being oxidized with dichlorodi-cyanobenzoquinone in trifluoroacetic acid, this compound gives a dication di-
SCHEME 1-57
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

radical according to Scheme 1-58. The di(cation radical) from Scheme 1-58 isstable in solution at room temperature. Studies by cyclic voltammetry and ESRspectroscopy led to the conclusion that the two centers are not wholly indepen-dent and that there is “leakage” of charge and spin between the two halves(Bushby et al. 1997a,b). Hence, the charge and spin distributions substantiallyoverlap. This statement finds its confirmation in cases of other, similar systems(Karafiloglou & Launay 1998; Boman with co-authors 1999).
2. Phenothiazine derivatives: Comparison between the structures depicted inScheme 1-59 can lead to a prediction that hexa(anisyl)-1,3,5-triaminobenzene ismore similar to 10,10-(1,3-phenylene)diphenothiazine than to 10,10-(1,4-phenylene)diphenothiazine. Nevertheless, upon oxidation in concentrated sulfu-ric acid, these two isomeric diphenothiazines give dication diradicals. As shown,m-phenylenediphenothaizine has a singlet state, and p-phenylenediphenoth-iazine has a triplet state (Okada et al. 1996; Baumgarten 1997). Because of ge-ometry peculiarities, the resulting orbital energy splitting between the two SO-MOs is larger in the meta isomer than in the para isomer.
SCHEME 1-58
SCHEME 1-59
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

In these geometries, mixing with �-orbitals of the central benzene ring destabilizeslocal cation radical orbitals (Fang et al. 1995). In particular, the energy difference betweenthe two �-orbitals with different symmetries (S and A) is larger in m-phenylene than in thep-phenylene framework simply because of the smaller number of �-bonds in the m-pheny-lene structure than in the p-phenylene structure. Hence, the triplet state of the para-pheny-lene-bridged dication biradical is more stable than that of the meta-bridged one. Accordingto quantum mechanical calculations performed at distortions close to 90°, para-derivativesmay indeed form more stable triplet states than meta-phenylene-linked structures (Baum-garten 1997).
If extended m-phenylene structures have several spin-bearing centers, their tripletcoupling (at zero field) depends on spatial spin distribution. Namely, the coupling degreeis inversely proportional to R3, where R is the distance between the “unpaired” electrons.
Poly(arylmethyl) anion diradicals are an example of such dependence (Rajca & Raj-ca 1995). When the charge (electron pair) is localized at the terminal site, as in Scheme 1-60, ESR studies indicate ferromagnetic coupling between the remaining “unpaired” elec-trons. However, when the negative charge (electron pair) is localized at the center site, asin Scheme 1-61, antiferromagnetic coupling between the remaining “unpaired” electrons isobserved. The distance between the two spin-bearing centers is much greater in the struc-ture of Scheme 1-61 than in the structure of Scheme 1-60.
Note that in this case a control of spin coupling via control of electron localization isattained. The straightforward control of electron localization was implemented via modifi-cation of the substituents at the center site (see Schemes 1-60 and 1-61). The mentioned an-ion diradicals were prepared by partial oxidation of relative polyanions with iodine (Rajca& Rajca 1995). When the carbanion-stabilizing 4-biphenylyl, in the place of 4-tert-butylphe-nyl, was employed as a substituent at the center site, the negative charge was con-fined to the center site and the spin density to the terminal (nonadjacent) site.
Control of charge/spin localization at the specific sites leads to two different kinds ofspin coupling: (1) When spin sites are adjacent, spin coupling between the “unpaired” elec-trons is ferromagnetic. (2) When spin sites are not adjacent, because the pathway betweenthem includes a site with negative charge (electron pair), spin coupling between the “un-paired” electrons appears to be antiferromagnetic.
SCHEME 1-60
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Distribution of spin density in the framework of trianion radicals allows determiningwhether or not there is some conjugation between substituents at different sites. For exam-ple, reduction of di-nitro-spiro(indoline-benzopyran) with potassium tert-butoxide indimethysulfoxide (DMSO) proceeds according to Scheme 1-62 (Alberti et al. 1995).
As shown by means of electrochemical ESR spectroscopic methods and confirmedby ab initio calculations (Alberti et al. 1995), the first electron is accommodated in the moreaccessible nitrobenzopyran moiety. Next, the second electron fits into the nitroindolinemoiety in virtually the same orbital that in the anion radical of mono-nitro-spiro(indoline-benzopyran), is occupied by the unpaired electron. The mono-nitro anion radical is alsoformed by the aforementioned reaction with tert-BuOK in DMSO, Scheme 1-63.
SCHEME 1-61
SCHEME 1-62
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.
As to the third step of the step-wise one-electron reduction of di-nitro-spiro(indoline-benzopyran), the third extra electron is again accommodated into the nitrobenzopyran unitwithout affecting the spin density distribution on the opposite fragment, which therefore re-mains essentially identical to that of the anion radical of mono-nitro-spiro(indoline-ben-zopyran). Hence, the spiro-nodal carbon atom prevents conjugate interactions between theindoline and benzopyran moieties.
Scheme 1-64 points up one additional and important aspect to the problem of fer-romagnetic coupling in dication diradicals. The stilbenoid compounds of Scheme 1-64give N�.…/…N�.
dication diradicals upon two consecutive one-electron oxidations (an-odic or by means of NOPF6). The compound containing the methoxy group in the stil-benoid fragment has a half-life of up to two months, even under ambient conditions. This
SCHEME 1-63
SCHEME 1-64
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

half-life is five times longer than that of the counterpart with no methoxy group in thestilbenoid moiety. However, introduction of the methoxy group results in torsional twist-ing on the substituted stilbenoid framework. This weakens ferromagnetic coupling in theCH3O-substituted dication diradical as compared with the H (stilbenoid-unsubstituted)analog (Michinobu et al. 2001). So ferromagnetic coupling needs not only the triplet ori-entation of the spins but also the spin-exchanging ability in the framework of a planar di-cation diradical.
1.6 POLYMERIC ION RADICALS
Polymeric ion radicals are usually formed as a result of one-electron redox modificationsof uncharged polymers containing electrochemically active groups. They attract enhancedattention in the sense of possible practical applications. Because polymeric ion radicalscontain many spin-bearing groups, a similarity emerges between polymeric ion radicalsand poly(ion radicals). However, there is some specificity peculiar just to polymers.
For example, NOBF4 oxidation of the triphenylamine and diphenyltetraalkoxy frag-ments in a linear and networked polymer leads to the formation of cation radical centers(Bushby et al. 1997b). As a rule, the quantity of ion radical centers formed is low, not morethan 10–20% of the theoretically possible values. This is a problem that is proving difficultto solve. A feasible explanation is that the effect is not electrostatic but steric. There is acertain difficulty in accommodating large counterions within rather rigid polymer networksof bent polymeric chains.
Anion radicals engrafted to hyperbranched polymers (dendrimers) deserve specialmention. Dendrimers are built up by the sequential synthesis of larger and larger gener-ations, with branch points in each generation. The ground-state spin multiplicity and ge-ometry of the reductively and oxidatively treated homo- or heteronuclear dendrimerswere studied theoretically and deemed technically promising. In particular, poly(ami-doamine) dendrimers were peripherally modified with diimide moieties; see the structurein Scheme 1-65.
After reduction with dithionite, this dendrimer was cast into a film the electrical prop-erties of which were isotropic. (This means that on the molecular and macroscopic levelsthere is a three-dimensional electron delocalization.) The conductivity was humidity de-pendent (water may take part in long-distance electron transfer). At 95% humidity, amixed-valence film (0.55 electron per diimide) showed conduction at room temperaturearound 11 ��1�cm�1 (Miller & Mann 1996). As shown later, partially reduced mixed-va-lence materials are required for organic metals of high electrical conductivity.
The convergent synthesis of a range of aryl ester dendrimers with peripheral tetrathi-afulvalene units was also reported (Devonport et al. 1998). The dendrimers acquire someamount of the cation radical tetrathiafulvalene “tips” upon reaction with iodine in solutions.
We have already pointed out that one of the main reasons for the modern interest inpolymeric and oligomeric ion radicals is the design of organic compounds with magneticproperties (see Section 7.4). Oligomers and polymers allow, in principle, preparing organichigh-spin molecules. A well-established strategy is to connect neutral or charged radicalsas spin-carrying units via organic linkers. These units may act as ferromagnetic couplingparts and provide the desired alignment of electron spins.
High-molecular ion radicals often have 1,3-phenylene (m-phenylene) as a couplingunit too (cf. Section 1.5). This unit couples ion radical centers at the 1 and 3 positions byan in-phase periodicity of the spin polarization. Cooperative interaction of a large number
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

of unpaired electrons may eventually result in high-spin polymers (oligomers) and ulti-mately in ferromagnetic materials.
A interesting theoretical strategy toward high-spin polyradicals/ion radicals was de-veloped (Fukutome et al. 1987). This strategy aimed at the construction of ferromagneticchains in which unpaired electrons can be introduced by reduction or oxidation of �-con-jugated segments. Those ion radicals were supposed to be positioned between the couplingunits. In the language of solid-state physics, the ion radical segments are polarons.
SCHEME 1-65
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Thus, p-phenylene diamine oligomers linked via the benzene ring in the 1,3 or 1,3,5manner have been successfully oxidized to high-spin ground-state oligo(cation radical)sthat are stable at room temperature (Stickley & Blackstock 1994, 1997; Wienk and Janssen1996, 1997). Ferromagnetic coupling occurs between p-phenylene diamine cation radicalsin regioregular substituted �-conjugated chains between neighbors and possibly also be-tween next-nearest neighbors (Meurs & Janssen 2000, 2001). Pendant p-phenylene di-amine cation radicals, regioregularly substituted on a conjugated polymer chain, constitutea promising approach toward stable high-spin polymers.
Poly(aniline-2-chloroaniline)-p-toluenesulfonic acid salt was obtained by oxidativepolymerization of aniline with o-chloroaniline in solutions containing p-toluenesulfonicacid. The copolymer salt was subjected to heat treatment under nitrogen atmosphere at el-evated (about 150°C) temperatures. The heat-treated samples acquired electric conductiv-ity 2.7 � 10�2 ��1�cm�1. According to ESR spectra, the heated poly(aniline-2-chloroani-line)-p-toluenesulfonic salt exists as the poly(semiquinone cation radical), in whichunpaired electrons are localized on or near the nitrogen atoms (Palaniappan 1997).
The polaronic strategy has also been applied to polymers, incorporating m-phenyleneunits as coupling links in �-conjugated polymer chains of poly(thienylene ketone) (ColleDal et al. 1999) as well as of polyarylamine, polyacetylene, polythiophene (Kaisaki et al.1991; Murray et al. 1994; Bushby et al. 1997a). Scheme 1-66 presents these polaronicpolymers.
Oxidation of the polymers from Scheme 1-66 leads to the polymeric cation radicalswith ferromagnetic coupling of spins. Surprisingly, however, the spin concentration inthese polymer networks was extremely low. Only a few percent of the cation radical unitsactually carried an unpaired electron.
Several explanations were proposed for this disappointing result. According toBushby et al. (1997b), for the cross-linked polymers, a more likely interpretation is thesteric difficulty of incorporating counterions into a relatively rigid polymer network. Theauthors consider the PF6
� anion as the counterion to each cation radical part of the cross-linked polymer. If this is so, it is unlikely to be insurmountable. A bulky anion can be re-placed by an anion of small size.
As to the tailor-made polymers, a more serious, perhaps intrinsic, cause is discussed(Haare et al. 1998). These polymeric cation radicals form �-macrodimers according tousual equilibria 2M�. ⇔ (M)2�
2 . The �-dimerization inhibits the formation of high-spinstates. Maybe other �-conjugated ion radicals with a much lower tendency to form �-dimers, such as polymeric cation radicals based on p-phenylenediamine, are a promisingway to construct intrachain ferromagnetic coupling in polymers.
Of course, charge transport in charge-injected polymers is extensively studied be-cause of its importance in practice. Sometimes it is sufficient to introduce a donor or ac-ceptor in a polymer. This leads to the formation of a charge-transfer complex, which trans-forms into an ion radical salt upon physical excitation. Thus, S. A. Lee and co-authors(1997) studied the photoconductivity of pyromellitic dianhydride-diaminodicyclohexyl-methane polyimide and of pyromellitic dianhydride-oxydianiline polyimide in the presenceof electron donors, e.g., N,N,N,N-tetramethyl-p-phenylendiamine. The addition of theelectron donor increased the photocurrent generation of polyimide films by about three or-ders of magnitude. The polyimide from an alicyclic diamine doped with an electron donorshowed a larger enhancement of photocurrent. This occurs due to an intermolecular charge-transfer complex. In the case of the aromatic polyimide, both inter- and intramolecularcharge-transfer complexes can be formed. By and large, any enhancement of the photocur-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

SCHEME 1-66
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

rent by the electron donor is attributed to photoabsorption by the molecular charge-transfercomplex formed between the added electron donor and the pyromellitic imide unit of thepolymer backbone. The photoexcitation provokes the rapid electron transfer from the donorto the pyrromellitic imide. This produces the anion radical of polymer and the cation radi-cal of the donor, resulting in the photoconductivity in the bulk polyimide films.
Localization or delocalization of an excess electron (in anion radicals) and a hole (incation radicals) along the polymer chain determines the electronic conductivity of the poly-meric ion radicals. From this point of view, the cation radical of vinylene-bridged-oc-tithiophene oligomer should be mentioned. Quantum chemical calculation (Casado et al.2000) indicates that the hole localizes in the middle of the molecule. The hole extends overthe central part built from four thyenylene units.
Another representatives of such polymers, polysilanes, also attract significant tech-nical interest. Polysilane is a one-dimensional �-conjugated polymer. In general, the silylgroup can support electron transfer through low-lying-d- and �*-orbitals (see, e.g.,Gherghel et al. 1995). Several scientific groups investigated the localization length (LL)of the electron/hole conduction in polysilanes. Irie and Irie (1997) and then Seki et al.(2001) recorded the absorption spectra of radical ions of polyalkylsilanes. The spectrahad two peaks in the UV and near-infrared regions, and the maxima of the peaks shiftedto longer wavelengths with increasing chain length until about 16 monomer units andclearly showed saturation above this length (i.e., LL � 16). Kumagai et al. (1996;Ichikawa et al. 1999) measured electron spin resonance and the electronic absorptionspectra of anion and cation radicals derived from oligosilanes (up to six monomeric units)or polysilanes (approximately 500 units). The spectra of the polymer ion radicals weresimilar to those of the oligosilane ion radicals. This suggested that both an excess elec-tron and a hole were not delocalized all over the Si–Si main chain of the polymeric ionradical. The excess electron and hole in the polysilane ion radical are confined to only apart of the polymer chain composed of six silicon atoms (LL � 6). Thus, the studies re-ported that excess electrons and holes are localized on a few silicon atoms, although thelocalization length reported in each work is slightly different.
Tachikawa (1999) also analyzed mobilities of carriers along the silicon chain, and hisresults should be mentioned separately. As it turned out, the mobility obtained for a posi-tive charge (hole) was several times larger than that for an excess electron. This result sug-gests that the localization mechanism of a hole and that of an electron are different fromeach other. Probably, an excess electron is trapped in the defect of the main chain, whereasa hole is not trapped. The defects are mainly structural ones, such as branching points andoxidized sites (Seki et al. 1999). This can lead to a different electron conductivity. Contin-uation of the polysilane ion radical studies will hopefully result in some important techni-cal applications.
1.7 INORGANIC ION RADICALS IN REACTIONS WITH ORGANICSUBSTRATES
Practically every organic reaction proceeds with the indispensable participation of inor-ganic substances involved in redox processes. Among these species are oxygen, halogens,compounds of sulfur and nitrogen with oxygen, inorganic ions of the hydroxyl type, andmetallic ions. As a result of electron transfer, the inorganic ions are oxidized, reduced, orsomewhat changed with respect to the central atom’s valence. Many of the electron-trans-fer reactions start from an attack of an organic substrate by an inorganic reactant. The lat-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

ter often reacts in its ion radical state (the SO4�.
anion radical is a typical example). Someion radical transformations of organic compounds take place with the assistance of activecomponents in a gaseous medium (O2, CO2). These components can give rise to ion radi-cals (O2
�., CO2
�.).
Therefore, some concise review of the conditions of formation and the physical andchemical properties of the most important inorganic ion radicals is in order here. Specialattention ought to be given to their reactions with organic compounds, both in an initial un-charged form and in a post-electron-transfer form.
It is also very important to understand the difference between redox reactions of thesame ions acting under different conditions of solvation—or why ions with close redox po-tentials are quite different in their activity in liquid-phase electron transfer. For example,the cupric ion is a weak oxidant in aqueous solutions. In acetonitrile (AN) solutions, thesame ion is a rather strong oxidant (Ahrland et al. 1983). Ions NO� and NO2
� are close intheir E0 values (1.51 and 1.56 V, respectively—Bontempelli et al. 1974; Cauquis & Serve1968). There is a marked solvent dependence of the standard potential E0 for the NO2
�/NO2
couple: 1.56 V in AN, 2.05 V in sulfolane, and 2.32 V in nitromethane (Boughriet & Wartel1989a, 1989b). In other words, the oxidative properties of NO2
� depend on solvating capa-bility. On account of the weak solvation properties of nitromethane, the NO2
� species isfound to be a more powerful oxidizing agent in nitromethane than in sulfolane and partic-ularly in AN.
1.7.1 Superoxide Ion
The standard potential of the O2/O2�.
pair is equal to �0.15 V in water and �0.60 V inDMF. Usually, dioxygen easily captures one and then another electron, Scheme 1-67:
O2 � e ⇔ O2�.
O2�.
� e ⇔ O22�
In DMSO, dioxygen reductions into the superoxide ion and then into the dioxygendianion are characterized by EI
1/2 � �0.5 and EI1/2 � �1.5 V (regarding the saturated
calomel electrode, SCE) (Sawyer & Gibian 1979).The superoxide ion has an intermediate position in the redox triad of Scheme 1-68:
O2 ⇔ O2�. ⇔ O22
2�
In accordance with such a position in the triad, the superoxide ion possesses a dual reac-tivity: Depending on the substrate character, the ion can act as an acceptor or as a donor ofone electron.
For instance, the superoxide ion transfers one electron to quinones, nitro compounds,and transforms them into anion radicals (Sawyer & Gibian 1979). Scheme 1-69 below il-lustrates the reversible transformation of quinone (Q) into semiquinone (SQ).
O2�.
� Q ⇔ SQ � O2
As was found out, the direct and reverse processes are fast but have different rates: k1 � 1.1� 109, and k�1 � 107 L�mole�.1�sec�1 (Morkovnik & Okhlobystin 1979).
According to a widespread opinion, the superoxide ion possesses expressed oxidativeproperties. Thermodynamically, however, it must be a moderately strong reductant and avery weak oxidant (Sawyer & Gibian 1979). This statement refers, of course, to aproticmediums, in which the superoxide ion is stable. In the presence of proton donors, the su-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

peroxide ion undergoes disproportionation according to Scheme 1-70:
O2�.
� H� ⇔ HOO.
O2�.
� HOO. → O2 � HOO�
In all the aprotic solvents (with no proton donor admixtures), the superoxide ion gen-erally cannot act as an oxidant, taking into account a wide range of functionally substitutedcompounds. For example, in dry pyridine, O2
�.does not oxidize 1,2-dimethoxybenzene
(Sawyer & Gibian 1979). This ion, however, reacts with 1,2-dihydroxybenzene. For theOH form, the first step consists of proton transfer from the hydroxyl group to the superox-ide ion. Next, reactions proceed with the participation of HOO
.. The latter is formed ac-
cording to the disproportionation of Scheme 1-70.The literature on the oxidation of hydrazines, thiols, and alcohols with the superox-
ide ion (Sawyer & Gibian 1979) refers to reactions that begin with the formation of theHOO
.radical. It is significant in this respect that there are no reactions between the O2
�.ion
and dialkyl sulfides, dialkyl ethers (esters), or alcoholates. All of these substrates do notcontain a labile proton. The free hydroperoxide radical (HOO
., which is formed from O2
�.
after the proton addition) is a conjugate acid of a base, i.e., of the superoxide ion. It is arather strong acid, with pKa � 4.88 (Sawyer & Gibian 1979). Hence, dissociation of thehydroperoxide radical with the generation of a proton and a peroxide ion is possible evenin acid media. However, the irreversible reaction between HOO
.and O2
�.suppresses the
dissociation. Studies in the chemistry of the O2�.
anion radical have achieved noticeablesuccess during the past two decades only. Convenient preparative methods of electro-chemical reduction of molecular oxygen were developed to obtain solutions of tetraalkylammonium superoxides in aprotic solvents. Usually, oxygen (in a gaseous stream througha phone electrolyte, Alk4NX, solution) is electrolyzed at the controlled potential of �1 V.As solvents, DMF, DMSO, AN, and pyridine are employed. Mercury, platinum, and goldare used as cathodes. The colored solutions formed give an ESR signal at a low tempera-ture. The signal corresponds to the superoxide ion.
A full chemical method has been proposed to obtain the superoxide ion (Miyazawaet al. 1985). In this case, oxygen is bubbled through DMF, THF, DMSO, or CCl4 solutionsof the potassium or tetrabutyl ammonium salt of 4-benzyloxy-1-hydoxy-2,2,6,6-tetram-ethyl piperidine. The resulting products are 4-benzyloxy-2,2,6,6-tetramethyl piperidine-1-oxyl, KO2, or Bu4N�O2
�.in 90% yields. Alkali superoxides are slightly soluble in organic
solvents. Their solubility can be described as moderate only in DMSO. However, in thepresence of crown ether, the solubility of superoxides in DMSO becomes excellent. Thesame applies to benzene as a solvent (Morkovnik & Okhlobystin 1979). Another chemicalreaction of oxygen giving the superoxide ion consists of a single-electron transfer to oxy-gen from Fe1 sandwich cyclopentadienyl (Cp)–arene complexes, Scheme 1-71:
[CpFeIArene]° � O2 → [CpFeIIArene]�, O2�.
[CpFeIIArene]�, O2�.
� M�, X� ⇔ [CpFeIIArene]�, X� � M�, O2�.
In Scheme 1-71, the reversible reaction is shifted to the right when the anion, X�, islarger and the cation, M�, is smaller. For example, this shift to the right is 100% in the pres-ence of Na�, PF6
� and only 35% in the presence of Na�, F� (Hamon & Astruc 1988). Theequilibrium takes place as an exchange reaction between the two ion pairs. Reactions of thistype are based on the symbiotic-effect premise: The interaction between a hard cation and ahard anion or between two soft ions is stronger than that between two ions of different types.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

One method of chemical generation of the superoxide ion deserves a short descrip-tion because of the simplicity of the reagents employed. The ion is generated from oxygenin alkaline aqueous solutions of sodium dithionite, Na2S2O4. The reaction follows Scheme1-72 below; the concentration of O2
�.ion in the oxygenated solutions depends on that of
SO2�.
at any moment of the reaction (Wang & Chen 1997).
S2O42� ⇔ 2SO2
�.
2SO2�.
� O2 ⇔ 2SO2 � O2�.
The reactions of 1-hydroxy- and 1-aminonapthoquinones with O2 open one signifi-cant feature of superoxide ion formation. This ion forms a van der Waals complex with an-other product, a semiquinone. Namely, hydrogen bonds are formed between O2
�.and the
OH and NH2 groups of the corresponding semiquinone. As a result, the reaction equilib-rium is shifted to the right (Liwo et al. 1997).
1.7.1.A Reactions of Superoxide Ion with Organic H Acids
The O2�.
ion in a solution promotes proton abstraction from a substrate or a solvent. Thisresults in the formation of organic bases, which are conjugated with the appropriate Hacids. All H acids with pKa lower than 23 can take part in such proton transfer (Sawyer &Gibian 1979). For this reason, even organic acids (HB), which are weaker than water, en-ter the exothermic reaction expressed by Scheme 1-73:
HB � 2O2�. → O2 � HOO� � B�
If HB is a weak acid, this reaction is rather slow, but it nevertheless takes place. Sub-stances that are unable to react with O2
�.can be involved too when the solvents/reactants
contain water or other proton-donor admixtures. Thus, benzaldehyde is absolutely resistantto the action of the superoxide ion. However, benzaldehyde does transform into benzylicalcohol and benzoic acid upon the action of O2
�.in the presence of moisture. Hence, a “su-
peroxide” variant of the Cannizzaro reaction is realized (Sawyer & Gibian 1979); seeScheme 1-74:
2O2�.
� H2O → O2 � HOO� � OH�
2PhCHO � OH� � H2O → PhCOOH � PhCH2OH
Proton-containing admixtures in a solvent or in benzaldehyde can act like water. Thesuperoxide ion abstracts such labile protons and generates a conjugated base. The base inits turn abstracts protons from the solvent, acetonitrile, for example. If benzaldehyde is pre-sent, it is converted into cinnamyl nitrile according to Scheme 1-75:
PhCHO � �CH2CN � H� → PhCH(OH)CH2CN → H2O � PhCHBCHCN
The treatment of benzaldehyde with potassium hydroxide in acetonitrile results in theformation of the same product, i.e., cynnamyl nitrile (Sawyer & Gibian 1979). Thus, in thepresence of water or other proton sources, the O2
�.ion forms strong bases as well as oxy-
gen and the peroxide anion. Therefore, many reactions that are ascribed to the superoxideion are actually reactions with proton donors. These reactions produce effective oxidants(O2 and HOO�) and strong bases (OH� or B�).
This kind of base generation finds applications in synthetic practice. Thus, ethyl ni-troacetate is a relatively strong acid (in H2O, pKa 5.75; in DMSO, pKa 9.2). Reacting withO�.
2, this compound transforms into the corresponding carbanion needed for several kindsof synthesis (Niyazymbetov & Evans 1993).
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

1.7.1.B Reactions of Superoxide Ion with Organic Electrophiles
As already noted, O2�.
reacts with effective electron acceptors. It is a one-electron reduc-tant of moderate strength. However, the superoxide ion can act as a nucleophile if a sub-strate has a decreased electron affinity. The reactions in Scheme 1-76 between O2
�.and
tetranitromethane or alkylhalides are pertinent:
O2�.
� C(NO2)4 → �C(NO2)3 � NO2.� O2
O2�.
� RHal → ROO.� Hal�
ROO.� O2
�. → ROO� � O2
ROO� � RHal → ROOR � Hal�
As seen from the scheme, the initial step of the reaction between the superoxide ionand alkylhalides is a bimolecular nucleophilic substitution. Reactions of KO2 with opticallyactive alkylhalides proceed with the configurational inversion. Moreover, the reactivitychanges in the same order as that of the usual SN2 reactions (primary � secondary�� ter-tiary; I � Br � Cl) (Sawyer & Gibian 1979). Nevertheless, the SN2 scheme does not ex-clude a possibility of one-electron transfer according to Scheme 1-77:
RHal � O2�. → [Hal����R
.���O2] → ROO
.� Hal�
This mechanism is rather probable: O2�.
is an electron donor, and organyl halogenidescan, in principle, accept one electron. With the one-electron mechanism, the inversion ofconfiguration is determined by O2
�.attack on the side, which is opposite to the halogen atom
in RHal (Morkovnik & Okhlobystin 1979). The direction of this reaction depends on thenature of the solvent. In pyridine, benzene, and DMF, the main product is alkyl peroxide.In DMSO, an alkyl carbinol is the main product (Sawyer & Gibian 1979). Obviously, theaforementioned intermediary product ROO� reacts faster with the solvent Me2SO thanwith the substrate RHal, Scheme 1-78:
ROO� � Me2SO → MeS(O�)(OOR)Me → Me2SO2 � RO�
Alkyl tosylates and mesylates are cleaved upon the action of KO2 in DMSO and giverise to corresponding alcohols. This reaction also proceeds with inversion of configurationat carbon atoms. Such process may be of importance in prostaglandine chemistry(Morkovnik & Okhlobystin 1979).
Reactions of O2�.
with esters and halogenides of carbonic acids also pass nucleophilicsubstitution as an initial step (Sawyer & Gibian 1979). Final products are acyl peroxides orcarbonic acids, Scheme 1-79:
R1COOR2 � O2�. → R1COOO
.� R2O�
R1COOO.� O2
�. → R1COOO� � O2
R1COOO� � R1COOR2 → R1COOOCOR1 � R2O�
R1COO-OOCR1 � 2O2�. → 2R1COO� � 2O2
The reaction of superoxide ion with carbon tetrachloride is important for olefin epox-idations (Yamamoto et al. 1986). The first step of the reaction consists in the formation ofthe trichloromethyl peroxide radical, Scheme 1-80:
O2�.
� CCl4 → Cl� � Cl3COO.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The trichloromethyl peroxide radicals of this scheme oxidize electron-rich olefins. The lat-ter give corresponding epoxides. This peroxide radical is a stronger oxidizing agent thanthe superoxide ion itself (Yamamoto et al. 1986).
Hence, nucleophilic reactions of the superoxide ion are very typical. This ion can becompared with the thiophenoxide and thiocyanate ions with respect to nucleophilicity. Thecause of such high nucleophilicity lies in a so-called � effect: in
.O–O� ion, an attacking
site (O�) adjoins directly to a site (O.) with a significant electronegativity. Such an effect
usually confers special activity to nucleophiles. The effect can be additionally enhanced byincluding the O–O� group in sulfenate, sulfinate, or sulfonate molecules (Oae et al. 1981).Scheme 1-81 presents typical examples:
RSSR � O2�. → RSOO
.� RS�
RSOO� � O2�. → RSOO� � O2
Like the trichloromethyl peroxide radical, peroxothio compounds can perform evennucleophilic oxygenation of substrates that are inert to O2
�.in aprotic solvents. For exam-
ple, stilbene is not changed in dry benzene containing 18-crown-6-ether and KO2. In thepresence of diphenylsulfide, however, the interaction does take place and results in theformation of stilbene epoxide. According to Oae and co-authors (1981), stilbene initiallygives PhCH(OO�)CH
.Ph anion radical adduct. Abstraction of O�.
from the adduct leads tostilbene epoxide with 40% yield (Oae et al 1981).
To sum up, the superoxide ion has high basicity and nucleophilicity. It can react as areducer and cannot serve directly as an oxidant.
1.7.1.C Superoxide Ion in Reactions with Biological Objects
There are several enzymes that convert O2 into O2�.
. The superoxide ion is produced inmany biological reactions and especially in respiration (see the review by Tselinski et al.2001). Also, there are many indications that this anion radical is particularly toxic to cellsand can ultimately have deleterious effects on the health and well-being of certain individ-uals; see reviews by Lang & Wagnerova (1992) and Faraggi & Houee-Levin (1999). More-over, the hydroxyl radical (which can be formed from O2
�.by the well-known Haber-Weiss
reaction) also has very high toxicity (Sies 1986).The superoxide ion in general does not appear to be intrinsically bactericidal (Hurst
1997). However, phagocytic cells have the capacity to enzymatically generate NO.. Nitric
oxide is also not bactericidal at physiologically relevant concentration levels, but reactsvery rapidly with O2
�.to form the peroxonitrite ion (ONO2
�). [The bimolecular rate constantfor this reaction is the largest yet recorded for any reaction of O2
�.(Huie 1993)]. Peroxoni-
trite ion is a powerful, relatively long-lived oxidant with bactericidal capabilities (Hurst1997). Another route of superoxide anion radical transformation lies in its disproportiona-tion upon superoxide dismutase catalysis (2O2
�.� 2H� → O2 � H2O2).
Metal cations are present in many biological objects; O2�.
is known to coordinate tothe metal ion, yielding the corresponding complex. The binding energies of O2
�.with diva-
lent metal ions (including Mg2�) have been determined as 0.5–0.7 eV (Fukuzumi &Ohkubo 2000). Being bonded into the complex, the metal cation is reduced easily at morepositive potentials. This means that O2, the most important biological oxidant, really canact as a catalyst rather than as an oxidant in the biological reaction of electron transfer (seeFukuzumi et al. 2001).
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

1.7.1.D Superoxide Ion–Ozone Anion Radical Relation
Ozone is a metastable compound, and its decay requires the activation energy of ca. 105kJ�mol� to yield atomic and molecular oxygen. However, the ozone anion radical in its turnsplits up even more readily. Successful alkene ozonolyses are probably initiated by alkene-catalyzed decomposition of O3. Some reversible single-electron transfer to O3 is possibleas a first weakly endothermic step. The ozone anion radical initially formed is the ex-tremely reactive species and should oxygenate alkene cation radicals very quickly andexothermally through mono-oxygen exchange. This process releases the superoxide ion(Schank et al. 2000 and references therein), Scheme 1-82:
�CBC� � O3 ⇔ �C�M.C� � O3
�. → �C�MC(O.)� � O2
�.
Depending on the alkene cation radical nature, open-chain oxygenation and epoxida-tion take place as well as the formation of other trivial ozonolysis products. Alkylaromaticcompounds are also oxidized by ozone via the ion radical mechanism. Ethylbenzene, for ex-ample, undergoes ozone attack on the ring (80%) and on the alkyl group (20%). Accordingto kinetic studies, the ozone consumption obeys the chain law (Galstyan et al. 2001).
The first of the reaction steps in the amine–ozone interaction also consists of one-electron transfer from the amine to the ozone, with the formation of the correspondingcation and anion radicals. The ozone anion radical has been revealed at low temperatures.Formation of the superoxide ion and the amine nitroxide are the understandable results ofthe reaction (Razumovskii & Zaikov 1984 and references therein).
1.7.2 Atomic Oxygen Anion Radical
There are transformations of organic oxygen-containing ion radicals, which developthrough the elimination of an atomic oxygen anion radical. An example of such a transfor-mation is the stilbene epoxidation mentioned earlier. The atomic oxygen anion radical be-comes an acting species when hydrogen peroxide or some other source of hydroxy radicalsis employed in water media with pH � 12 (Vieira et al. 1997). Therefore, properties of theatomic oxygen anion radical should be described, albeit concisely.
The properties of O�., in contrast to these of the conjugated acid HO
., are still poorly
studied. The review by J. Lee and Grabowsky (1992) describes mass spectrometry of thisradical. In conditions that are usual for synthetic organic chemistry, O�.
displays less reac-tivity than its acid
.OH. Oxidation of acetone by radicals O�.
and .OH leads to quite differ-ent products (Morkovnik & Orhlobystin 1979); see combined Scheme 1-83:
MeCOCOOH ← (� HO.)MMe2COM (� O�.
) → MeCOOH
Addition of O�.to double bonds and to aromatic systems was found to be quite slow.
Simic and co-workers (1973) found that O�.reacts with unsaturated aliphatic alcohols, es-
pecially by H atom abstraction. As compared to O�., HO
.reacts more rapidly (by two to
three times) with the same compounds. In the case of 1,4-benzoquinone, the reaction withO�.
consists of the hydrogen double abstraction and leads to the 2,3-dehydrobenzoquinoneanion radical (Davico et al. 1999 and references therein).
Christensen and co-authors (1973) found that O�.reacts with toluene in aqueous so-
lution to form benzyl radical through an H-atom-transfer process from the methyl group. Ingeneral, the O�.
anion radical is a very strong hydrogen atom abstractor, which can with-draw a proton even from organic dianions (Vieira et al. 1997).
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

It is important to emphasize that the atomic oxygen anion radical plays a role in cat-alytic oxidation that occurs on various oxide surfaces. For instance, O�.
reacts with methaneat room temperature over various metal oxides (Lee & Grabowsky 1992). Upon solid catal-ysis, O�.
is more reactive toward alkanes and alkenes than are other ionic oxygen species.Iwamoto and Lunsford (1980) have postulated that O�.
is the active oxygen species that ox-idizes benzene to phenol on V2O5/SiO2 at 550°C with 70% selectivity at 10% conversion.The product is formed just on oxide surfaces, before leaving the solid (Azria et al. 1988).
1.7.3 Molecular Oxygen Cation Radical
Although oxygen has a very high oxidation potential (4.7 V), it can be oxidized to O2�.
un-der special conditions, for example, by means of the ultramicroelectrode technique (Cas-sidy et al. 1985). This cation radical is of course a very strong oxidant. Its stabilized formsare O2
�.SbF6
� and O2�.
AsF6�. At present, however, these salts are not considered to contain
the oxygen cation radical. Although textbooks (e.g., Cotton & Wilkinson 1988 p. 462) de-scribe O2
�.as a firmly established ionic form of oxygen,
this is based on the characteristics of O2MF6 (M � P, As, Sb, or Pt) and the as-sumption that these compounds are salts [(O2
�.)MF6
�]. Such an ionic formulationis unsupported by their chemical and physical properties, and is inconsistent withthe ionization potentials, electron affinities, and electronegativities of the con-stituent atoms. The more accurate description of O2MF6 molecules is a covalentadduct (�OOF)MYF5. (Sawyer 1991)
In any event, O2SbF6 or O2AsF6 act as a very effective one-electron oxidant with re-gard to aromatic amines, nitrogen- and sulfur-containing heterocyclic compounds (Dinno-cenzo and Banach 1986, 1988, 1989), as well as perfluorobenzene, benzotrifluoride, andperfluoronaphthalene (Richardson et al. 1986). The oxidation proceeds in freon (CHCIF2)at temperatures from �115 to �145°C, and the formation of the organic cation radicals hasbeen firmly established.
1.7.4 Carbon Dioxide Anion Radical
Carbon dioxide, CO2, is a typical component of the gaseous environment for reactions inair or in the presence of air traces. Therefore, both interactions between CO2 and organicproducts of an electron transfer as well as reactions of CO2
�.with uncharged molecules of
organic compounds should be considered.Interaction of CO2 with organic anion radicals leads, as a rule, to carboxylic acids;
CO2�.
anion radicals are not formed. Even such a one-electron reductant as O2�.
in aproticmedium simply adds to CO2. The addition product, in turn, accepts one electron from O2
�.
(Sawyer & Gibian 1979). The total result consists of the formation of an aprotic equivalentof peroxycarboxyic acid according to Scheme 1-84:
CO2 � O2�. → .
OO-CO2�.
� O2�. → �OO-CO2
�
Vacuum UV irradiation of aqueous solutions containing formate is one of the meth-ods to generate CO2
�.. However, this method leads to the excited anion radical CO2
�.. Such
an excited anion radical acquires the ability to transfer an unpaired electron to nitroben-zene, benzoic acid, or benzaldehyde (Rosso et al. 2001).
Radiolysis of water (and water solutions) in air produces carbon dioxide anion radi-cals (Morkovnik & Okhlobystin 1979). Hydrated electrons, which are generated during the
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

radiolysis, are captured with CO2. The carbon dioxide anion radical transforms aliphatic al-cohols into �-hydroxyl acids according to Scheme 1-85:
H2O � �-rays → H.�
.OH � eeq � H�; eaq � CO2 → CO2
�.
RCH2OH �.OH → RCH
.OH � H2O
RCH.OH � CO2
�. → RCH(OH)CO2�
Reaction of .OH with either CO or HCOOH in water solution also results in the for-
mation of CO2�.
(Flyuht et al. 2001).Another route to the carbon dioxide anion radical is reduction of hydrogen peroxide
with Ti3� in the presence of formate ions (Morkovnik & Okhlobystin 1979), Scheme 1-86:
Ti3� � H2 O2 → Ti4� �.OH � �OH;
.OH � HCOO� → H2O � CO2
�.
The solvated CO2�.
anion radical has been observed both in the gas phase and in con-densed matter (Holroyd et al. 1997) and has been well characterized by ESR spectroscopy(Knight et al. 1996). Being solvated, CO2
�.anion radicals form complexes that yield quasi-
free electrons upon photoexcitation. Gas-phase studies (Saeki and others 1999) and ab ini-tio calculations (Tsukuda et al. 1999) indicate that static ion–dipole interactions stabilizethe [(CO2)n–m(ROH)m]�.
type of small clusters. In supercritical carbon dioxide, monomersand dimers of water, acetonitrile, and alcohols also form metastable complexes (Shkrob &Sauer 2001a,b). Such complexation should be taken into account in studies of electron-transfer kinetics in reactions with the participation of CO2
�..
Disproportionation of CO2�.
leads to carbon monoxide and a carbonate, while dimer-ization results in oxalate formation; see the left and right directions of Scheme 1-87:
�O2CMCO2� ← 2CO2
�. → CO32� � CO
The carbon dioxide anion radical usually plays a role as one-electron reductant; inDMF its E0 � �1.97 V (Amatore & Saveant 1981). In the gas phase, solitary CO2
�.loses
one electron with an exothermic effect of ca. 45 kJ�mol�1 (Compton et al. 1975).Interactions of CO2
�.with alkyl halogenides or with compounds containing halo-
gen–nitrogen bonds take place as dissociative electron captures. As a result, free radicalsare formed according to Scheme 1-88:
CO2�.
� RHal → CO2 � R.� X�
CO2�.
� MeCONHBr → CO2 � MeCONH.� Br�
The carbon dioxide anion radical was used for one-electron reductions of nitroben-zene diazonium cations, nitrobenzene itself, quinones, aliphatic nitro compounds, ac-etaldehyde, acetone and other carbonyl compounds, maleimide, riboflavin, and certaindyes (Morkovnik & Okhlobystin 1979). This anion radical reduces organic complexes ofCoIII and RuIII into appropriate complexes of the metals in the valence 2 state (Morkovnik& Okhlobystin 1979). In the case of the pentammino-p-nitrobenzoato-cobalt(III) complex,the electron-transfer reaction passes a stage of the formation of the Co(III) complex withthe p-nitrophenyl anion radical fragment. This intermediate complex transforms into the fi-nal Co(II) complex with the p-nitrobenzoate ligand as a result of an intramolecular electrontransfer. Scheme 1-89 illustrates this sequence of transformations:
CO2�.
� [O2NC6H4COOCoIII(NH3)3]2� → CO2 � [.�O2NC6H4COOCoII(NH3)3]� →
[O2NC6H4COOCoII(NH3)3]�
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Hence, despite the presence of the central atom CoIII, an initial electron transfer is directednot to this center, but to the p-nitrophenyl fragment. Obviously, this fragment, and not themetallic ion, provides an orbital that is symmetrically appropriate and low lying. A similarsituation takes place in a reductive cleavage of arylsulfonic salts (see Chapter 3).
A double bond can react with CO2�.
(Morkovnik & Okhlobystin 1979); Scheme 1-90represents one typical example:
CO2�.
� R1R2CBNOH → R1R2C(COO�)NHO.
As seen, the transformation into the anion radical is a very effective way to activate carbondioxide.
1.7.5 Sulfur Dioxide Anion Radical
The couple SO2/SO2�.
is characterized by E0 � �0.26 V (in water—Bradic & Wilkins1984). Such potential falls into the region of redox-protein activity. This suggests that theknown damaging action of gaseous SO2 does not correspond to the sulfuric acid formation,but is stipulated by SO2 participation in biological electron transport. Sulfurous acid alsocauses detrimental effects by irritating the mucous membranes. Interestingly, the resistanceof different organisms to sulfur dioxide correlates with the relative significance of redoxprocesses in these organisms. For instance, plants are among the organisms that suffer themost from the damaging actions of SO2. However, different members of the plant kingdommanifest different levels of sensitivity to the gas. Among trees, the most sensitive are firand pine, while the least sensitive are birch and oak. The highest sensitivity to SO2 amongflowering plants is attributed to the rose.
In humans, inhalation of air containing more than 0.2% of sulfurous gas causes short-ness of breath and confusion. Prolonged and repeated exposure to SO2 leads to loss of ap-petite, constipation, and inflammatory diseases of the respiratory tract. The levels of sensi-tivity to the gas differ from individual to individual. Sensitivity usually diminishes withprolonged exposure. It is explained by the induction of the cytochrome defense that allowsfor a normal electron transport (up to a certain time); see Prousek (1988). It is likely thatthe reductive properties of dithionites (salts of dithionous acid H2S2O4) are partially ex-plained by S2O4
2� ⇔ 2SO2�.
dithionite reversible dissociation. Both the dithionite ion andthe sulfur dioxide anion radical can act as reducers. The former can be oxidized into sulfiteor sulfate; the latter can lose an electron, thus transforming into a volatile dioxide, SO2.This is why the use of a stable sodium dithionite upon dye vatting or dye printing is alwayscomplicated with sulfurous gas evolution.
Dissociation of the dithionite ion can proceed both in aqueous and in nonaqueous me-dia. There is a special equation to determine the average values of the equilibrium constants(Keq) of the formation of SO2
�.from S2O4
2� (Lough & McDonald 1987), namely, Keq
� [SO2�.
]2/{[S2O42�]0 � 0.5 [SO2
�.]}.
In DMF, DMSO, and acetonitrile, the Keq values are 42.4, 11.3, and 40 mM at 25°C,respectively. These values exceed Keq in water (1.4 � 10�6 mM) by seven orders of mag-nitude.
The salt (Et4N)2S2O4 is very soluble in water and in organic solvents. In nonaqueousmedia, its dissociation depends strongly on the moisture content. For the DMF/H2O sys-tem, Keq values fall from 42.4 to ~10�4 mM, along with water content increasing from 0 to20 vol %. Dithionite ions (S2O4
2�) are better stabilized in solvents of high polarity and inwater than in systems of low polarity (Lough & McDonald 1987).
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Organic solvents are typical for organic electron-transfer reactions, and these sol-vents are usually polar ones. The anion-radical SO2
�.is a particularly interesting one-
electron donor. Because this anion radical is formed more easily in less polar solvents, it allows widening the range of solvents acceptable for organic electron-transfer reactions.
1.7.6 Sulfite Anion Radical, SO3�.
The sulfite anion radical (SO3�.
) is obtained from sodium sulfite upon the action of titaniumtrichloride in water in the presence of ethylene diamine tetraacetic acid (as a complexingagent) and hydrogen peroxide (Bradic & Wilkins 1984). Scheme 1-91 below shows thistype of SO3
�.generation.
Ti3� � H2O2 → Ti4� �.OH � �OH;
.OH � SO3
2� → �OH � SO3�.
According to the scheme, the formation and further reactions of anion radical SO3�.
take place in an alkaline medium. Therefore, there are some restrictions to the electron-transfer reactions from this anion radical to acceptors. For instance, aliphatic nitro com-pounds react in alkaline media in aci forms. They add SO3
�.and form new anion radicals;
Scheme 1-92 (Bradic & Wilkins 1984):
RCHBNOO� � SO3�. → RCH(SO3)NO2
�.
In order to generate SO3�.
in acidic media, the reaction between sodium bisulfite andcerium ammonium nitrate should be employed. In (NH4)2Ce(NO3)6, cerium has the oxida-tion state 4�. In acids, valence 4 cerium salts are strong oxidants. With the sulfite ion, theoxidation follows Scheme 1-93:
Ce4� � SO32� → Ce3� � SO3
�.
This method of SO3�.
generation is used to perform the addition of the sulfite anionradical to unsaturated compounds, Scheme 1-94:
RCHBCHR � SO3�. → RCH(SO3
�)CH.R
This addition reaction is also applicable to substrates containing C�C and CBSbonds (Ozawa & Kwan 1985). In cases of nonsymmetrically substituted double bonds, theless shaded carbon atom is attacked by SO3
�.anion-radical. For example, acrylic, 3,3-
dimethyl acrylic, and crotonic acids react with SO3�.
at the expense of the ethylenic carbon,which is remote from the carboxylic group. Because these unsaturated acids are stable inalkali medium, it was possible to examine their anionic forms in the reaction with SO3
�.. It
turned out that the carboxylate group prevents SO3�.
from entering the geminal position. Inalkaline medium, however, a competition is possible for an unsaturated substrate betweenradicals SO3
�.and
.OH (the last originates from hydrogen peroxide, see earlier). The afore-
mentioned restrictions for SO3�.
addition are not relevant to .OH addition. In contrast to an-
ion radical SO3�.
, radical .OH bears no charge. Therefore, there is no electrostatic repulsion
between the entering reactant (the hydroxy radical) and the substrate carboxylate group.For the addition reactions of SO3
�.to CBC bonds (which can be performed both in
alkaline and in acidic solution), the following regularity was established: SO3�.
anion radi-cal is more active in acidic than in alkaline medium (Ozawa & Kwan 1985).
The reactions of SO3�.
anion radicals with organic and inorganic compounds havecommanded considerable interest because of the role of these anion radicals in grievous
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

health and technological problems. Indeed, biochemical and industrial sources produceSO3
2�. SO32� is often transformed into SO3
�.during atmospheric reactions. Both of them are
pollutants whose activity in the environment leads to illness (Reed et al. 1986). Althoughthe toxicological role of inorganic SIV and SV is now accepted, recent studies have shownthat the outer-sphere reactions of electron transfer are not the sole one. There are manycases of oxidative addition of SO3
�.to unsaturated compounds. Recently, even examples of
the substitutional sulfonation have been described for SO3�.
reactivity (Dutta & Ferraudi2001).
1.7.7 Sulfate Anion Radical, SO4�.
Sulfate anion radical is formed during UV irradiation or heating to 80–100°C of persulfatesM2S2O8 in aqueous solutions. This results in S2O8
2� ⇔ 2SO4�.
dissociation (Minisci et al.1983; Dias & Vieira 1996).
Conventional oxidants can, in principle, accept electrons from persulfate ions to formthe sulfate anion radical, but the reaction is extremely slow. For instance, M2S2O8 in H2O2
solution gives M2SO4 for two months at O°C (Skogoreva & Ippolitov 1988).The presence of transition metal salts (for example, salts of Ag� or TI3�) promotes
the reaction (Minisci et al. 1983). In this case, each S2O82� anion generates only one SO�.
4
according to Scheme 1-95:
S2O82� � Ti3� → Ti4� � SO4
2� � SO4�.
The nature of a transition metal is not essential for this redox reaction. However, oneof the reaction products, namely, anion radical SO4
�., can be complexed by a transition
metal in a higher oxidation state. This leads to some stabilization of SO4�.
and increases itseffective concentration. In other words, further reactions with organic substrates are facil-itated (Fristad & Peterson 1984). Cuprous and ferrous salts are preferable.
Sometimes, a transition metal salt is deliberately added to a mixture of a substrate anda persulfate salt (Dobson et al. 1986). The free or metal-coordinated sulfate anion radicalreacts with an organic substrate, giving rise to a substrate cation radical (Minisci et al. 1983;Itahara et al. 1988; Telo & Vieira 1997). One typical example is the reaction betweentoluene and SO2
�.in Scheme 1-96:
ArCH3 � SO4�. → SO4
2� � (ArCH3)�.
Hence, there is a difference between anion radicals SO4�.
and CO2�.
. While the latter is anone-electron reductant (see earlier), the former is a one-electron oxidant.
A substrate cation radical is often able to expel a proton and transforms into a radi-cal. The latter regenerates the starting metallic ion. The whole reaction becomes catalyticin respect of the metal; see Scheme 1-97:
(RCH3)�. → H� � RCH.2
RCH.2 � Cu2� → Cu� � RCH2
�
RCH2� � H2O → H� � RCH2OH
One-electron transfer from a substrate to the sulfate anion radical mostly follows dif-fusion rates. For instance, rate constants of one-electron oxidation of benzene and anisolewith SO4
�.are equal to 3 � 109 and 5 � 109L�mole�1�sec�1, respectively (Goldstein & Mc-
Nelis 1984).
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The sulfate anion radical is not a very strong hydrogen acceptor. It acquires an atomichydrogen from organic substrates at significantly smaller rates a compared with the ratesfor one-electron oxidations. For instance, dehydration rate constants are 107, 106 and 105
L�mole�1�sec�1 for methanol, tert-butanol, and acetic acid, respectively (Goldstein & Mc-Nelis 1984; Zapol’skikh et al. 2001). Such a peculiarity is very important for the selectiv-ity of ion radical syntheses with the participation of SO4
�..
If the sulfate anion radical is bound to the surface of a catalyst (sulfated zirconia),it is capable of generating the cation radicals of benzene and toluene (Timoshok et al.1996). Conversion of benzene on sulfated zirconia was narrowly studied in a batch reactor under mild conditions (100°C, 30-min contact) (Farcasiu and co-workers 1996;Ghenciu and Farcasiu 1996a, 1996b). The proven mechanism consists of one-electrontransfer from benzene to the catalyst, with the formation of the benzene cation radical and the anion radical of sulfate on the catalytic surface. This ion radical pair combines to give a surface combination of sulfite phenyl ester with reduced sulfated zirconia. The ester eventually gives rise to phenol, Scheme 1-98. Coking is not essential for re-action 1-98. To regenerate the catalyst, it is enough just to oxidize the worked-out catalyst.
The described reactions of sulfate anion radical characterize it as a radical of strongoxidation ability (E0 � 2.60 V, in water—Balej 1984). Like organic anion radicals, SO4
�.
has some affinity to the hydrogen radical.With cyclic alkynes, the sulfate anion radical acts as an oxygen-transfer reagent,
transforming the alkyns into �, -epoxy compounds (Wille 2000). In this sense, the sulfateanion radical can also be considered a donor of atomic oxygen in solution. The reactionleads to the release of SO3
�., which is significantly less reactive than SO4
�.(Muller et al.
1997).
SCHEME 1-98
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

In contrast to organic anion radicals, SO4�.
possesses high electrophilicity and isprone to addition reactions (Davies & Gilbert 1985), Scheme 1-99:
CH2BCHCH2OH � SO4�. → �O3SOCH2CH
.CH2OH
MeCHBCHCOOH � SO4�. → MeCH
.CH(OSO3
�)COOH
CH2BCHCH2COOH � SO4�. → �O3SOCH2CH
.CH2COOH
This reaction leads to the adducts that are observable by means of the ESR method.In some cases (maleic, fumaric acids), ESR spectra can be recorded at high-enough pH val-ues only. Being a strong electrophile, the anion radical SO4
�.is more active toward a car-
boxylate ion than to neutral molecules of unsaturated acids.Anions of saturated acids react with sulfate anion radicals, giving rise to carboradi-
cals (Eberson et al. 1968), Scheme 1-100:
Me3CCOO� � SO4�. → SO4
2�.� Me3CCOO
.
Me3COO. → CO2 � Me3C
.
This reaction resembles the decarboxylation of carboxylates during electrode one-electron oxidation. The Kolbe electrochemical reaction also consists of one-electron oxi-dation, decarboxylation, and culminates in dimerization of alkyl radicals formed interme-diately.
In Reaction 1–100, the sulfate anion radical, and not the anode, acts as a one-electronoxidant. In this case, the caboradical dimerization is hampered. The radicals can be used inpreparative procedures. One typical example is alkylation of heterocyclic nitrogen bases(Minisci et al. 1983).
The difference between the Kolbe electrochemical reaction and the reaction with thehelp of a “dissolved” electrode (the sulfate anion radical) deserves some explanation. Theconcentration of the one-electron oxidation products in the electrode vicinity is significantlyhigher than that in the bulk of solution. Therefore, in the case of anode-impelled reactions,the dimerization of radicals produced from carboxylates proceeds easily. Noticeably, SO4
�.
secures the single-electron nature of oxidation more strictly than an anode. In electrode re-actions, radical intermediates can undergo further oxidation if potentials of capture of thefirst and the second electrons are more or less close. In contrast, the homogeneous reactionswith the participation of SO4
�.are characterized by fixed potentials in respect to the reactant
and an organic substrate. The route of homogeneous reactions is strongly dependent uponthe ratio of reagent concentrations. If the ratio is small, the probability of the secondary re-action is low. The distinction can be illustrated with the oxidation of RCOO�: It can give Rand after that, at once, R� in an anodic process. In the case of oxidation with the aid of SO4
�.,
R. becomes the sole product of the reaction (Eberson et al. 1968).Another advantage of the SO4
�.pathway for carboradical generation is its high selec-
tivity. As already noted, this type of oxidation allows one to avoid (practically) the hydro-gen abstraction. In the case of alkyl carboxylates, the rate of one-electron oxidation of thecarboxyl group exceeds the rate of homolytic disruption of the C–H bond in an alkyl restby 100 times (Davies et al. 1985). Cleavage of persulfate in the presence of carboxylates isa simple and reliable method of getting alkyl radical for synthesis.
1.7.8 Hydroxide Anion
This anion is a typical reagent for organic hydroxylation. Many such reactions have ion rad-ical mechanisms according to which an inorganic anion transfers an electron to an organic
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

substrate. It is important to underline the diffuse nature of the excess electron on the �OHion, which allows molecular orbitals of the ion and the substrate to overlap, even with along distance between them (Takahashi et al. 2001). For example, in the reaction betweenthe hydroxyl anion and o-dinitrobenzene, the �OH ion is proved to be an electron donor;see Section 4.3.4.B. When �OH transforms into
.OH, o-dinitrobenzene gives the anion rad-
ical. The o-dinitrobenzene anion radicals are stable. They come out into the reaction vol-ume and meet there with new hydroxyl anions (Abe & Ikegami 1978), Scheme 1-101:
C6H4(NO2)2 � �OH → .OH � C6H4(NO2)2
�.
C6H4(NO2)2�.
� �OH → �C6H4(NO2)(OH)NO2�.
�C6H4(NO2)(OH)NO2�.
� C6H4(NO2)2 → C6H4(NO2)2�.
� �C6H4(NO2)(OH)NO2
�C6H4(NO2)(OH)NO2 → NO2� � C6H4(OH)NO2
There are several cases of hydroxylation according to the “hidden radical” mecha-nism, within a solvent cage. As assumed (Fomin & Skuratova 1978), hydroxylation of theanthraquinone sulfonic acids (AQ–SO3H) proceeds by such a pathway, and
.OH radicals at-
tack the substrate anion radicals in the solvent cage. Anthraquinone hydroxyl derivativesare the final products of the reaction. In the specific case of dimethylsulfoxide as a solvent,hydroxyl radicals give complexes with the solvent and lose their ability to react with theantraquinone sulfonic acid anion radicals (Bil’kis & Shein 1975). The reaction is stoppedjust after anion radical formation, Scheme 1-102:
AQ-SO3� � �OH → .
OH � (AQ-SO3�)�.
Me2SO �.OH → Me2SO
.OH → Me
.� HOSOMe
The hidden-radical mechanism (also dubbed as the mechanism of biradical origin)cannot, however, take into account all the peculiarities of the reaction participants. The re-dox potential of the �OH/
.OH couple is equal to �0.9 V with regard to the saturated
calomel electrode in acetonitrile (Tsang et al. 1987). In conventional reference to the nor-mal hydrogen electrode and in water, this potential had been determined as �1.9 V (Berd-nikov & Bazhin 1970) or as �1.3 V in acetonitrile (Eberson 1987, Chap. 4, p. 62). Thesevalues are too positive to reduce the majority of organic acceptors. Of course, there aresome cases of single-electron transfer from the �OH ion to an organic acceptor. All thesecases, however, refer to substrates with very strong electron affinity, such as tetracya-noethylene and dinitrobenzene (Blumenfel’d et al. 1970). Quinones, ketones, and othersubstrates have less affinity. In the ground (nonexcited) states, they are unable to capturean electron from the �OH ion (Sawyer & Roberts 1988).
The described reactions of quinone hydoxylation, as well as electron transfer fromhydroxyl anion to quinones, are in general accelerated by light irradiation. Wavelengths areup to 500 nm. The degree of acceleration is higher with greater intensity of irradiation andwith an increase in the concentrations of quinone and hydroxide (Blumenfel’d et al. 1970).Direct (outer-sphere) electron transfer takes place from hydroxide to an excited quinonemolecule. In the excited molecule, one of the electrons of a double-occupied orbital popu-lates a vacant orbital. The “derelict” orbital is located at the lower level of energy than thevacant orbital. Therefore, the electron transfer from hydroxide to a quinone becomes ener-getically favorable. This corresponds to an enhanced electron affinity and lifts the prohibi-tion for the electron transfer.
It should be pointed out that the possibility of an �OH → .OH transformation with
one-electron transfer onto a substrate depends on the further fate of the resulting hydroxyl
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

radical. The latter is a strong oxidant, and it must be taken out of the reaction sphere in or-der to shift the electron transfer equilibrium to the right. One opportunity to organize theshift to the right consists of recombination of two
.OH radicals, which results in H2O2 for-
mation. As a rule, hydrogen peroxide is formed in reactions between hydroxide and verystrong acceptors (Endo et al. 1984). However, an attack of hydroxyl anion upon an unsat-urated site of a substrate (an aromatic fragment, the carbonyl group, etc.) is more typicaland more interesting chemically. This results in a �-complex formation. Such a complexcan regroup into a product of hydroxylation. The complex can also decay, with the forma-tion of substrate anion radicals and
.OH radicals. In short, the electron transfer proceeds in
an inner-sphere manner, and the aforementioned restrictions (in potential differences, in.OH oxidation activity) vanish. This inner-sphere mechanism is assumed to be the most fre-quent. One-electron outer-sphere transfer with radical and ion radical formation (on the onehand) and coupling of an activated substrate with hydroxyl anion with no electron transfer(on the other hand) are two rare extremes.
The last but not least peculiarity of the reaction of �OH with electron acceptors con-sists of its dependence on solvation effects. Solvation energy has an influence on the ion-ization potential of �OH and on electron affinity of a substrate as well. For instance, the�OH ion is a stronger base and a more effective electron donor in acetonitrile or dimethyl-sulfoxide than in water. Its solvation energy is lower by 80–100 kJ�mol�1 in these organicsolvents than in water (Sawyer & Roberts 1988). This fall in the solvation energy corre-sponds to a relief of hydroxyl anion oxidation by almost 1 V (Sawyer & Roberts 1988). Asfor the other participants in the equilibrium reaction of electron transfer (a neutral substrate,its anion radical, and the
.OH radical), their solvation susceptibility is not so essential. The
main effect should be attributed to �OH. Of course, the ability of �OH to be an electrondonor depends not only on its solvation energy but also on the energy of covalent bindingof the hydroxyl group in an adduct with a substrate or in a final product of hydroxylation.
1.7.9 Nitrosonium and Nitronium Ions
The nitrosonium ion, �NO, is a strong oxidant, with E0 � 1.51 V vs. the normal hydrogenelectrode. It reacts with organic substrates of E0 � 1.7 V as an outer-sphere acceptor of oneelectron. In contrast, the nitronium ion, NO2
�, is a weaker oxidant than the nitrosonium ion,although its redox potential (E0 � 1.56 V vs. the normal hydrogen electrode) is closeenough to that of the nitrosonium ion. The cause of such a sharp decrease in acceptor prop-erties is the necessity of energy consumption to change the NO2
� structure upon one-elec-tron transfer. The transformation of �NBO into
.NMO results in an elongation of the ni-
trogen–oxygen bond by 0.01 nm, which consumes 42 kJ�mol�1 of energy (Eberson &Radner 1987). Meanwhile, the NO2
� ion is planar, NO.2 is bent (O–N–O angle is equal to
134.3°). The bending consumes considerable energy, estimated to be equal to 218 kJ�mol�1 (Eberson & Radner 1987) or even 587 kJ�mol�1 (Lund & Eberson 1997). It is thefactor that decreases the NO2
� electron affinity. Only very oxidizable substrates can partic-ipate in the outer-sphere electron transfer to NO2
�. The observed cases of the generation ofsubstrate cation radicals and the nitrogen dioxide radicals are stipulated by the inner-spheremechanism. Such a mechanism involves the nucleophilic attack of the nitronium ion upona substrate, the formation of a tetrahedron complex, and the disintegration of the complex.The disintegration results in products, which corresponds formally to one-electron transfer.However, the reorganization energy is lower for the inner-sphere mechanism, since thebending of the nitronium ion is no longer required.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Thus, there is a great difference in electron-transfer reactivity between the nitroniumand the nitrosonium ions, despite the closeness in their redox potentials. Closeness in theelectrode potentials supposedly reflects the peculiarities of electrochemical reactions. Theliquid-phase reactions discussed here have their own string of distinctions. Electrode pro-cesses are heterogeneous. There is a strong electric field in the near-electrode layer. Thefield can have an effect on the behavior of the NO2
� ion. It is probable, under the electrodereaction conditions, that both the linear ion NO2
� and the bent NO.2 radical acquire some-
what similar configurations in the tough part of the double electric layer. Energy con-sumption for such a small bend is revealed in the potential value only vaguely (cf. 1.56 and1.51 V for the couples of NO
.2/NO2
� and NO./NO�, respectively). Some authors suggest
that the experimental values determined for NO.2/NO2
� might be influenced by significantorbital overlap between NO
.2 and the Pt electrode surface (Lee, K.Y., et al. 1991). This
pathway is energetically favored because the outer-sphere reorganization energy is notneeded as much. Of course, a solvent can intervene in all of these phenomena. It should benoted that E0 (NO
.2/NO2
�) in AN has been reported to be over 0.6 V less positive than E0
(NO.2/NO2
�) in sulfolane and nitromethane (Boughriet & Wartel 1989a,b, 1993).In homogeneous liquid-phase reactions, there are no strong electric fields, no influ-
ence of a tough electrode layer. Geometric factors begin to play their independent roles.These factors determine the equilibrium state, which governs the degree of conversion ofthe acceptor (NO2
�, NO�) during one-electron transfer. Moreover, the NO2� ion can form a
complex with a solvent (Ciaccio & Marcus 1962; Hunziker et al. 1971). This causes amarked diminution of the NO2
� reactivity and decreases the nitration rates. The NO2� and
NO� reactivity problem is also considered with respect to aromatic nitration; see Section4.5.4.
1.7.10 Tris(aryl)amine Cation Radicals
After Walter (1966) developed the preparation and redox properties of tris(aryl)aminecation radicals, they have found wide application in organic synthesis. For example, thesalts of the tris(4-bromophenyl)amine and tris(2,4-dibromophenyl)amine cation radicals(occasionally named Magic Blue and Magic Green) with the hexachloroantimonium anionare stable and widely used for the initiation of diverse reactions that begin from cation rad-ical formation. Chapter 6 considers many such reactions. The salts are obtained as a resultof the oxidation of tris(bromophenyl)amines with antimonium pentachloride. The natureand reactivity of inorganic counterions control the stability and redox properties of theseorganic cation radicals. These aspects are analyzed here. Generation of the tris(bro-mophenyl)amine cation radical follows Scheme 1-103:
2 (4-BrC6H4)3N � 3SbCl5 → SbCl3 � 2 (4-BrC6H4)3N�.SbCl6
�
The formation of the SbCl6� anion can be explained by Scheme 1-104:
2SbCl5 ⇔ SbCl6� � SbCl4
�; SbCl4� � e → SbCl
.4; 2SbCl
.4 → SbCl5 � SbCl3
Anion SbCl6� is a good oxidizing agent because it can be reduced from SbV to SbIII.
Organic substrates with E0 up to 1.5 V vs. normal hydrogen electrode transform into cationradicals in the presence of SbCl6
�. Note: tris(4-bromophenyl)amine has E0 � 1.3 V vs. nor-mal hydrogen electrode in acetonitrile (Reynolds et al. 1974). Such acceptor influence ofthe anion in the salt provides high stability for the salt. In cases of other anions, the cationradical component decays gradually (both as a dry solid and in a solution) (Eberson & Lars-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

son 1986). The main role in such degradation belongs to the reaction between the cationradical and water (from air or as solvent moisture). Cation radicals of tris(aryl)amines candecompose water at room temperature. The reaction resembles the process of water de-composition with oxidized forms of chlorophyll “a” and consists of the formation of hy-drogen peroxide and the evolution of oxygen (Pokhodenko et al. 1984); see Scheme 1-105:
Ar3N�. � H2O → Ar3N � H� � 1⁄2H2O2; H2O2 → H2O � 1⁄2O2
The oxygen and hydrogen peroxide evolving in the reaction provoke degradation oftris(4-bromophenyl)amine. Another route of degradation consists of the gradual dimeriza-tion of these cation radicals with bromine depletion (Cowell et al. 1970). However, havingthe hexachloroantimonate counterion, tris(4-bromophenyl)ammoniumyl is stable (Pokho-denko et al. 1984; Cowell et al. 1970). According to IUPAC rules, ammoniumyl is the namefor amine cation radicals.
Like all cation radicals, ammoniumyls are sensitive to nucleophiles (to reactants oradmixtures). At the same time, 1,1,1,3,3,3-hexafluoropropan-2-ol as a solvent drasticallycurtails nucleophilic reactivity and provides good integrity of the tris(4-bromophenyl)am-moniumyl at ambient temperatures (Eberson, Hartshorn et al. 1996).
In cases when tris(4-bromophenyl)ammoniumyl hexachloroantimonate turns out asa weak one-electron oxidant, its 2,4-dibromo- or even 2,3,4,5,6-hexachloro- analogs areemployed (Nelsen et al. 1997). After a one-electron oxidation, these ammoniumyls trans-form into tri(aryl)amines. They are remarkably nonreactive as nucleophiles. After the elec-tron transfer, the hexachloroanytimonate anion begins to serve as a counterion for newlyformed cation radicals of a donor molecule.
Sometimes, the hexachloroantimonate ion of the ammoniumyl salt is changed withhexabromocarborane, which brings the anion’s nucleophilicity to nil. For example, thefullerene carbocation (C�
76) was isolated as the stable salt with the carborane anion after thereaction of C76 with tris(2,4-dibromophenyl)ammoniumyl that had the hexabromocarbo-rane counterion (Bolskar et al. 1996).
1.7.11 Hexachloroantimonate(V) Salts of Trialkyloxonium Cations
These salts belong to a widely used class of alkylating agents; their traditional name isMeerwein’s salts (Meerwein et al. 1937). However, they can be involved in reactions otherthan alkylation (Boettger et al. 1997). One of these reactions is an oxidation of aromaticdonors (Rathore and co-authors 1998a), Scheme 1-106:
2ArH � 3(Et3O�SbCl6�) → 2(ArH�.
SbCl6�) � 3EtCl � 3Et2O � SbCl3
So the triethyloxonium cation is an effective oxidant for the production of aromaticcation radicals, but only as the hexachloroantimonate salt. The analogous (Et3O�BF4
�) or(Bu4N�SbCl6
�) cannot be used to prepare any cation radical. This is consistent with the pre-viously described function of SbCl6
� as an oxidant. The slow release of SbCl5 forms the ba-sis for the efficacy of (Et3O�SbCl6
�) as an aromatic oxidant. Kochi’s group proposedScheme 1-107 (see below) for this slow release (Rathore and co-authors, 1998a).
(Et3O�SbCl6�) → SbCl5 � EtCl � Et2O
The transformations of SbCl5 caused by one-electron transfer from an aromatic com-pound have just been described. If the pure Lewis acid SbCl5 is used, its reactivity is verydifficult to control, and single-electron oxidation as well as chlorinations of various aro-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

matic donors can occur readily (Mori et al. 1998). Meanwhile, in the case of (Et3O�
SbCl6�), the slow release of the active monomer SbCl5 occurs. In the case of SbCl5 as such,
2SbCl5 ⇔ Cl4Sb–Cl2–SbCl4 dimerization takes place (Cotton & Wilkinson 1988, p. 395).The dimeric form may lead to electrophilic chlorination according to Scheme 1-108:
Cl4Sb-Cl2-SbCl4 � 2ArH → 2ArCl � 2HCl � 2SbCl3
Neither Et3O� nor SbCl6� is individually capable of aromatic oxidation. Therefore,
the slow-released monomeric SbCl5 is the active oxidant. This is a reason to consider tri-alkyloxonium hexachloroantimonates together with other inorganic participants of organicion radical reactions.
1.7.12 Transition Metal Ions
In the sense of electron-transfer reactions, transition metal ions can be the one- or two-elec-tron type. The two-electron ions transform into nonstable states upon unit change of themetal oxidation number. In the outer-sphere mechanism, two-electron transfer is a combi-nation of two one-electron steps.
The conditions of the outer-sphere mechanism have been analyzed at length in the lit-erature (Eberson 1987, Chap. 7). An organic substrate must fit a metal ion with respect toredox potentials. The substrate cannot have any hydrogen atom that is prone to depart as aproton. The substrate cannot have any positions that are free from any steric stoppage for ametal attack. The metal ion has to have a relatively high redox potential, and this potentialhas to be away by no less than 0.5 V from potentials of the next redox transformations ofthe ion. Ligands surrounding the metal ion cannot be substituted with the substrate in elec-tron-transfer reactions. Otherwise, the reaction system is directed to the route of inner-sphere electron transfer. The same concern has to do with ligand exchange at the expenseof medium components. Participation of a transition metal ion in electron transfer leads, ofcourse, to some changes in redox properties. For the outer-sphere mechanism, it is impor-tant that the change of redox potential not affect the reorganization energy of the solvateenvironment of the metallic ion. It is better to restrict the metal ion–substrate interactionsterically at both metal and substrate sides.
In all other cases, inner-sphere mechanisms are at work. These mechanisms includeaddition and subsequent dissociation. For C–H dissociation, so-called metallocomplex ac-tivation takes place. This kind of activation consists of inner-sphere electron transfer ac-cording to Scheme 1-109:
R.� CuII(OAc)2 → RCuIII(OAc)2 → R(� H
.) � HOAc � CuIOAc
One example of outer-sphere electron transfer is the reaction between dipotassiumcyclooctatetraene (K2C8H8) and the cobalt complex of bis(salicylidenediamine) (CoIISalen) (Levitin et al. 1971), Scheme 1-110:
K2C8H8 � 2CoIISalen → C8H8 � 2(CoISalen)�K�
Although C8H8K2 is a two-electron donor, only a one-electron transfer takes place.Only this type of transfer is permitted because of a difference between potentials of thedonor and the acceptor. This difference remains a determining factor of electron exchangebecause the cobalt atom firmly reserves the ligand environment for itself. As for cyclooc-tatetraene, it is unable to provide the ligand exchange in this case. The results obtained al-lowed the solving of the reverse problem, namely, the development of an analytical method
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

of determination of Co0Salen2�. This deeply reduced dianionic form of the complex resultsin the metallic sodium reduction of CoIISalen. The dianionic form possesses catalytic prop-erties and is actively used in model studies of biological electron transport. After treatmentof CoIISalen with excess sodium in THF, the reduction product was allowed to react withcyclooctatetraene; the consumption of cyclooctatetraene was quantitatively determined bymeans of the GLC method. If an insufficient amount of C8H8 was used, it transferred intoC8H8Na2 completely. If the amount of C8H8 employed was 1 mole per each 2 moles of CoI-
ISalen used to obtain [(Co0Salen)2� 2Na�], then the degree of transformation of C8H8 intoC8H8Na2 was 70%. This means that the “deeply reduced” sample contained only 70% of[(Co0Salen)2� 2Na�]. In that case, outer-sphere electron transfer looks like a “titration” ofthe double-charged complex with cyclooctatetraene, Scheme 1-111:
C8H8 � 2[(Co0Salen)2� 2Na�] → C8H8Na2 � 2[(CoISalen)�Na�]
Titration according to this scheme showed that the treatment of CoIISalen with ex-cess amounts of Na resulted in the nonquantitative formation of [(Co0Salen)2� 2Na�].Thus, catalytic and, especially, kinetic investigations of such a complex have to take intoaccount the presence of CoIISalen and/or (CoISalen)� in the samples studied. The de-scribed convenient method of quantitative electron transfer in solutions is good at determi-nation of other low-valent metallocomplexes.
Of note is the solvent role for electron-transfer reactions between organic substratesand metal ions. For instance, a transformation of CuII complex into CuI complex proceedsbetter in acetonitrile than in water (Ahrland et al. 1983). Coordination of CuI with acetoni-trile is much better than that with water. In water solutions, with no significant share of or-ganic solvents, CuI salt disproportionates completely and yields CuII salts and Cu0 metal.In aprotic solvents, the CuI ion is much more stable. In dimethylsulfoxide, the dispropor-tionation is not significant as long as the concentration of CuI is low. In pyridine, it doesnot proceed at all, irrespective of the concentration. In acetonitrile, CuI salts are absolutelystable, with no signs of disproportionation or air oxidation. Thus, the role of an organicaprotic solvent consists of the enhancement of stability of a partly reduced form of metal-locomplex or ion. The cause of such enhancement is the more effective solvation than inthe case of water or another proton-donor solvent.
In connection with the data just described, one reasonable question arises: Why doesthe stabilizing role of a nonwater solvent become significant for CuI salts and remain in-significant for CuII salt? As Myagchenko’s group showed (1989), CuII salts (and salts ofHgII, PdII as well) exist in nonwater solutions as polynuclear compounds. Thus, CuCl2forms lamellar lattices with chlorine chains as bridges between copper atoms. By contrast,CuCl does not form such chain structures. CuI ion is easily and effectively solvated by non-water solvents and forms stable solvates. As far as (CuCl2)n associates are concerned, theirinteractions with an organic solvent as well as with an organic substrate in general takesplace at terminal groups that are energetically active (Myagchenko et al. 1989). Naturally,solvation of the associates, in which the metal ion is encapsulated, cannot be effective. Thecopper salts belong to the type of one-electron redox systems. Redox couples CoIII/CoII,CoII/CoI, MinIII/MnII, CeIV/CeIII, AgII/AgI, IrIV/IrIII, FeIII/FeII, CrIII/CrII, and WVI/WV alsobelong to the one-electron string.
Two-electron redox systems are represented by couples TIIII/TII, PbIV/PbII, PdII/Pd0,MgII/Mg0, HgII/Hg0, AuIII/AuI, PtIV/PtII, and PtII/Pt0.
While the action of one-electron redox systems is readily understandable in the con-text of inner- and outer-sphere mechanisms, two-electron redox systems require additional
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

considerations. First of all, if a double one-electron transfer is probable from an organicsubstrate to the same metal ion, does this mean that the same molecule of an organic donorprovides these two electrons, or do two molecules of the substrate act as one-electrondonors?
A study of the kinetics of the oxidation of aromatic hydrocarbons with lead tetraac-etate has shown the second order of the reaction. Hence, the rate-determined stage consistsof the transformation of PbIV into PbII, with the participation of only one molecule of thehydrocarbon (Dessau et al. 1970). The formed hydrocarbon dication can, of course, reactwith the uncharged hydrocarbon according to Scheme 1-112:
ArH2� � ArH → ArH�.� ArH�.
The cation radicals depicted in the scheme were really detected, but they originatefrom the fast reaction of one-electron transfer, which does not affect kinetic constants ofthe oxidation. The rate constant depends linearly on Brown’s �-constants of substituents(Dessau et al. 1970). All these data are in agreement with the formation of the strong po-lar dication of an aromatic hydrocarbon as an intermediate. Because PbII salts (in partic-ular, the diacetate) are not reductants, the two-electron-transfer reaction proceeds irre-versibly.
In some cases, two-electron transfer to a metal ion leads to the formation of a reduc-ing form. This gives the possibility of constructing catalytic cycles. Oxidation of ethylenewith PdII upon catalysis of CuII as in Scheme 1-113 (see below) is a striking example(Denisov 1978).
CH2BCH2 � PdII ⇔ (CH2BCH2)�PdII
(CH2BCH2)�PdII � H2O → Pd0 � 2H� � CH3CHO
Pd0 � 2CuII → PdII � 2CuI
CuI � O2 → CuII � O2�.
, CuI � O2�. → CuII � O2
2�
O22� � 2H� → H2O2; CuI � H2O2 → 2OH� � 2CuII etc.
Metal ion participation in electron transfer reactions can proceed with no changes inits oxidation state, but simply at the expense of complex formations.
Scheme 1-114 (below) illustrates such assistance by 3d metal ions (M � Mn, Co, Ni,Zn) to electron transfer between a reductant (Red) and an oxidant (Ox).
M2� � Red � Ox ⇔ [Red–M2�–Ox] ⇔ Red� � M2�(Ox�)
The reaction depth correlates to the electron donor ability of Red and the stability de-gree of M2�(Ox�) complex. The complexation causes an anodic shift of metal redox po-tentials, which reaches almost 100 mV for transition metal cations (Maletin et al. 1979,1980, 1983).
Alkali, alkaline and rare earth metal cations also catalyze electron-transfer reactions.Thus, in the pair of CoII–tetraphenylporphyrin complex with benzoquinone, no redox re-action takes place, or it goes too slow to be determined accurately. The metal cations pro-mote this reaction. For example, in the presence of Sc(ClO4)3, the corresponding rate con-stant of 2.7 � 105 M�2sec�1 was observed. Benzoquinone transforms into benzosemi-quinone under these conditions (Fukuzumi & Ohkubo 2000).
Zinc perchlorate accelerates the reaction between aromatic amine and quinones(Strizhakova et al. 1985). The reaction results in the formation of charge-transfer com-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

plexes [ArNH2���Q]. The complexes dissociate in polar solvents, giving ion radicals; seeScheme 1-115:
ArNH2 � QK1⇔ [ArNH2
����Q�]K2⇔ ArNH2
�.� Q�.
Equilibrium-constant values are low enough for reactions in Scheme 1-115. For theinteraction of N,N-tetramethylphenylenediamine (ArNH2) with diphenyl benzoquinone(Q) in acetonitrile at 200C, K1 � 4.5 � 10�2, K2 � 1.1 � 10�6 (Levin et al. 1979). How-ever, addition of zinc ions increases sharply the rate of formation of the amine cation radi-cal, leaving the rate of formation of the quinone anion radicals with no change. The influ-ence of zinc ions on the electron transfer from an amine to a quinone can be explained bythe binding of anion radical Q�.
in a complex with ZnII. As a result, the equilibrium maybe shifted to the right. However, the stability constant of another possible complex,[Zn(ArNH2)]2�, is equal to 105 and is much higher than K1 (Strizhakova et al. 1985). Inother words, introduction of ZnII should produce the left, not the right, shift of the equilib-rium. The complexation between Q�.
and ZnII indeed takes place, but it does not preservethese anion radicals from decaying in fast collateral reactions. Moreover, the anion radicaldoes not, prior to its decay, have enough time to leave the inner coordination sphere of thecomplex with zinc. This causes right-shifting of the equilibrium. Meanwhile, the observedresult consists of an increase in the rate of accumulation of cation radicals ArNH2
�, but notthat of anion radicals Q�.
.Complexation between zinc ions and anion radicals of aromatic ketones provides a
basis for simple one-electron reduction of these ketones (Handoo & Gadru 1986). In themethod, dimethylsulfoxide is employed as a solvent, zinc dust serves as a reductant in thepresence of strong alkali (6M, several drops). As widely known, zinc in an alkali mediumis a very active reductant. For example, it reduces nitrates under such conditions to ammo-nia. In the ketone case, electron transfer is stopped at the stage of ketyl formation. Proton-donor treatment leads to the respective alcohols: benzophenone gives benzhydrol, fluo-renone gives fluorenol, etc. Before protonation, the solution contains anion radicals(ketyls), and these anion radicals give the starting neutral molecules after the addition of p-dinitrobenzene. All previously mentioned acceptors have electron affinity values up to�210 kJ�mol�1 (Handoo & Gadru 1986). All of them give anion radicals with practicallyquantitative yields. A breakthrough feature of such method of anion radical generation isthat it does not require anaerobic conditions. The resulting anion radicals (probably becauseof complexation with zinc ions) become stable enough in air. This is in sharp contrast to thehigh sensitivity of the same anion radicals to air when they are obtained with the help of al-kali metals in ether solvents. Hence, the method based on reduction by a Zn/OH system indimethylsulfoxide gives a tempting alternative to common methods of anion radical prepa-ration. Furthermore, this method allows one to perform the transfer of only one electronand, in a sense, has advantages over preparative electrolysis at controlled potentials. At thesame time, the method remains a purely chemical one, which requires no complicated andexpensive instrumentation.
Samarium diiodide (SmI2, the Kagan reagent) represents another principal exampleof concerted reduction and complexation. This salt is very easily prepared in tetrahydrofu-rane from samarium finely ground as a powder and diiodomethane, diiodoethane, or iodine(Krief & Laval 1999). Samarium iodide can be stored as 10�1 M THF solution under atmo-sphere of nitrogen or, better, argon (Kagan et al. 1981). It is well known to have exceptionalqualities as a single-electron-transfer reductant. For species soluble in organic medium, itsubmits a negative-enough reduction potential (E1/2 � �1.62 V in acetonitrile vs. saturated
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

calomel electrode—Kolthoff & Coetzee 1957). Like other lanthanide derivatives, SmI2 pre-sents a high coordination number (usually 7). Therefore, it provides a unique opportunity toinclude molecules of reactants, solvents, and electron-transfer products in the framework ofone subunit (Kagan et al. 1981; Yacovan et al. 1996). This facilitates electron transfer andchanges some of the usual reactions of an intermediate radical R
.. In one example, no cou-
pling product R–R is obtained as a result of the reaction between alkyl halide RX and SmI2
in THF. The intermediate radical R.abstracts hydrogen from THF as the only proceeding re-
action (Kagan et al. 1981). In the second example, SmI2 allows one to perform the reductionof the triselenonium cation radical. Reduction with SmI2 proceeds smoothly and in strictlyone-electron manner (Ogawa et al. 1994). Scheme 1-116 illustrates the reaction.
It would be interesting to examine thulium diiodide in one-electron reduction reac-tions. On the basis of the work of Evans and Allen (2000), TmI2 has the potential to be aneffective replacement for SmI2 when the latter is too weak as a reductant, when subambi-ent reaction temperatures are desirable, etc. Perhaps, TmI2 activity in tetrahydrofurane canbe controlled by the addition of hexamethyl phosphorustriamide in the same manner as itregulates the power and reactivity of SmBr2 (Knettle & Flowers 2001).
1.8 CONCLUSION
The material discussed in this chapter shows that the organic chemistry of ion radicals hasspecific features. The transformation of organic compounds into ion radicals changes or-bital interactions. This causes changes in electron effects of substituents and enhances theability of bridge groups to participate in electron delocalization. Acid–base properties of or-ganic compounds are also changed fundamentally. This opens new ways to widen the mod-ern methodology of organic synthesis. Based on numerous examples, the chapter points outways to enrich the reactivity of metallocomplexes. Comprising an extensive body of workstudying organic poly(ion radical)s of the monomeric nature, the chapter examines cases ofthe separate existence of several unpaired electrons and their ferromagnetic coupling. Theabsence of spin leaking from one ion radical site to another in the framework of the samemolecular skeleton is a very important feature in the design of organic materials with mag-netic properties. On the other hand, the section devoted to polymeric ion radicals describesthe approaches for the design of conductors that the electronics industry relies on.
Every organic reaction proceeds with the participation of inorganic reagents. Ion rad-ical organic reactions also have inorganic participants. The chapter discusses inorganic ionradicals in their reaction with organic substrates. The main aim of this chapter is to providethe foundation for all subsequent chapters.
i, NOPF6, acetonitrile-ether; ii, SmI2, tetrahydrofurane
SCHEME 1-116
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

REFERENCES
Abe, T.; Ikegame, Y. (1978) Bull. Chem. Soc. Jpn. 51, 196.Abeywickrema, R.S.; Della, E.W. (1981) Austr. J. Chem. 34, 2331.Ahrland, S.; Nilsson, K.; Tagesson, B. (1983) Acta Chem. Scand. A37, 193.Al Badri, A.; Chentit, M.; Geoffroy, M.; Jouaiti, A. (1997) J. Chem. Soc., Faraday Trans. 93, 3631.Al Badri, A.; Jouaiti, A.; Geoffroy, M. (1999) Magn. Reson. Chem. 37, 735.Alberti, A.; Barberis, C.; Campredon, M.; Gronchi, G.; Guerra, M. (1995) J. Phys. Chem. 99, 15779.Alberti, A.; Benaglia, M.; D’Angelantonio, M.; Emmi, S.S.; Guerra, M.; Hudson, A.; Macciantelli,
D.; Paolucci, F.; Roffia, S. (1999) J. Chem. Soc., Perkin Trans. 2, 309.Alberti, A.; Guerra, M.; Martelli, G.; Bernardi, F.; Mangini, A.; Pedulli, G.F. (1979) J. Am. Chem.
Soc. 101, 4627.Alberti, A.; Martelli, G.; Pedulli, G.F. (1977) J. Chem. Soc., Perkin Trans. 2, 1252.Alegria, A.E.; Garcia, C.; Santiago, G.; Collazo, G.; Morant, J. (2000) J. Chem. Soc., Perkin Trans.
2, 1569.Allendoerfer, R.D.; Rieger, Ph.H. (1965) J. Am. Chem. Soc. 87, 2336.Alonso, E.; Astruc, D. (2000) J. Am. Chem. Soc. 122, 3222.Alwair, K.; Grimshaw, J. (1973) J. Chem. Soc., Perkin Trans. 2, 1150.Amatore, C.; Kochi, J.K. (1991) Adv. Electron Transfer Chem. 1, 55.Amatore, C.; Saveant, J.-M. (1981) J. Am. Chem. Soc. 103, 5021.Andersson, M.; Linke, M.; Chambron, J.-C.; Davidsson, J.; Heitz, V.; Sauvage, J.-P.; Hammarstrom,
L. (2000) J. Am. Chem. Soc. 122, 3526.Arena, F.; Bullo, F.; Conti, F.; Corvaja, C.; Maggini, M.; Prato, M.; Scorrano, G. (1997) J. Am. Chem.
Soc. 119, 789.Asfandiarov, N.L.; Fal’ko, V.S.; Fokin, A.I.; Lomakin, G.S.; Pozdeev, N.M.; Podkopaeva, O.Yu.;
Chizhov, Yu.V. (1998) Rapid Commun. Mass. Spectrom. 12, 595.Astruc, D. Electron Transfer and Radical Processes in Transition Metal Chemistry. (1995) VCH,
New York.Azria, R.; Parenteau, L.; Sanche, L.; (1988) J. Chem. Phys. 88, 5166.Balej, J. (1984) Electrochim. Acta 29, 1239.Bartak, D.E.; Hoser, K.J.; Rudy, B.C.; Hawley, M.D. (1973) J. Am. Chem. Soc. 95, 6033.Baumgarten, M. (1997) Acta Chem. Scand. 51, 193.Bauschlicher, Ch.W. Astrophys. J. (1998) 509, L125.Behar, D.; Neta, P. (1981) J. Phys. Chem. 85, 690.Beley, M.; Collin, J.-P.; Ruppert, R. (1984) J. Chem. Soc., Chem. Commun., 1315.Belousov, Yu.A.; Belousova, T.A. (1999) Polyhedron 18, 2605.Belousov, Yu.A.; Kolosova, T.A.; Viktorova, E.A.; Babin, V.N. (1987) Izv. AN SSSR, Ser. Khim., 1652.Berberova, N.T.; Letichevskaya, N.N.; Belenko, N.P.; Shinkar’, E.V.; Kolyada, S.A.; Okhlobystin,
O.Yu. (1998) Zh. Obshch. Khim. 68, 1174.Berdnikov, V.M.; Bazhin, N.M. (1970) Zh. Fiz. Khim. 44, 395.Bernardi, F.; Mangini, A.; Guerra, M.; Pedulli, G.F. (1979) J. Phys. Chem. 83, 640.Bernstein, E.R. (1992) J. Phys. Chem. 96, 10105.Bietti, M.; Cuppoletti, A.; Dagostin, C.; Florea, C.; Galli, C.; Gentilli, P.; Petride, H.; Russo Caia, C.
(1998) Eur. J. Org. Chem. 11, 2425.Bil’kis, I.I.; Shein, S.M. (1975) Tetrahedron 31, 969.Bil’kis, I.I.; Shteingarts, V.D. (1987) Izv. SO AN SSSR, Ser. Khim. Nauk, 105.Blumenfel’d, L.A.; Brukhovetskaya, L.A.; Fomin, G.V.; Shein, S.U. (1970) Zh. Fiz. Khim. 44, 931.Bock, H.; Haenel, P.; Niedlein, R. (1988) Phosph. Sulfur 39, 235.Boekenstein, G.; Jansen, E.H.J.M.; Buck, H.M. (1974) J. Chem. Soc., Chem. Commun., 118.Boettger, G.; Geisler, A.; Froehlich, R.; Wuethwein, E.-U. (1997) J. Org. Chem. 62, 6407.Bolskar, R.D.; Mathur, R.S.; Reed, Ch.A. (1996) J. Am. Chem. Soc. 118, 13093.Boman, P.; Eliasson, B.; Grimm, R.A.; Martin, G.S.; Strnad, J.T.; Staley, S.W. (1999) J. Am. Chem.
Soc. 121, 1558.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Bontempelli, G.; Mazzocchin, G.-A.; Magno, F. (1974) J. Electroanal. Chem. 55, 91.Boorshtein, K,Ya.; Gherman, E.D. (1988) Izv. AN SSSR, Ser. Khim., 2051.Bordwell, F.G.; Cheng, J.-P. (1989) J. Am. Chem. Soc. 111, 1792.Borosky, G.L.; Nishimoto, S.; Pierini, A.B. (2000) J. Mol. Struct. 499, 151.Boughriet, A.; Wartel, M. (1989a) J. Chem. Soc., Chem. Commun., 809.Boughriet, A.; Wartel, M. (1989b) J. Electroanal. Chem. Interfacial Electrochem. 262, 183.Boughriet, A.; Wartel, M. (1993) Int. J. Chem. Kinet. 25, 383.Bradic, Z.; Wilkins, R.G. (1984) J. Am. Chem. Soc. 106, 2236.Brandon, E.J.; Kollmar, C.; Miller, J.S. (1998) J. Am. Chem. Soc. 120, 1822.Breslow, R.; Grant, J.L. (1977) J. Am. Chem. Soc. 99, 7745.Buick, A.R.; Kemp, T.J.; Neal, G.T.; Stone, T.J. (1969) J. Chem. Soc. A, 666.Bushby, R.J.; McGill, D.R.; Ng, K.M.; Taylor, N. (1997a) J. Chem. Soc., Perkin Trans. 2, 1405.Bushby, R.J.; McGill, D.R.; Ng, K.M.; Taylor, N. (1997b) J. Mater. Chem. 7, 2343.Camanyes, S.; Lluch, J.M.; Bertran, J. (1996) Chem. Phys. 204, 419.Caneschi, A.; Dei, A.; Lee, H.; Shultz, D.A.; Sorace, L. (2001) Inorg. Chem. 40, 408.Casado, J.; Gallardo, I.; Moreno, M. (1987) Electroanal. Chem. 219, 197.Casado, J.; Maraver Puig, J.J.; Hernandez, V.; Zotti, G.; Lopez Navarrete, J.T. (2000) J. Phys. Chem.
A 104, 10656.Cassidy, J.; Khoo, S.B.; Pons, S.; Fleischmann, M. (1985) J. Phys. Chem. 89, 3933.Casson, D.; Tabner, B.J. (1969) J. Chem. Soc. B, 887.Cauquis, G.; Serve, D. (1968) C. R. Seances Acad. Sci., Ser. C 267, 460.Chentit, M.; Sidorenkova, H.; Jouaiti, A.; Terron, G.; Geoffroy, M.; Ellinger, Y. (1997) J. Chem.
Soc., Perkin Trans. 2, 921.Choua, S.; Sidorenkova, H.; Berclaz, Th.; Geoffroy, M.; Rosa, P.; Mezailles, N.; Ricard, L.; Mathey,
F.; Le Floch, P. (2000) J. Am. Chem. Soc. 122, 12227.Christensen, H.C.; Sehested, K.; Hart, E.J. (1973) J. Phys. Chem. 77, 983.Ciaccio, L.L.; Marcus, R.A. (1962) J. Am. Chem. Soc. 84, 1838.Cliffel, D.E.; Bard, A.J. (1994) J. Phys. Chem. 98, 8140.Colle Dal, M.; Cova, C.; Distefano, G.; Jones, D.; Modelli, A.; Comisso, N. (1999) J. Phys. Chem. A
103, 2828.Compton, R.N.; Reinhardt, P.W.; Cooper, C.D. (1975) J. Chem. Phys. 63, 3821.Connelly, N.G.; Geiger, W.; Lane, G.A. (1986) J. Am. Chem. Soc. 108, 6219.Corma, A.; Garcia, H. (1998) Top. Catal. 6, 127.Cotton, F.A.; Wilkinson, G. Advanced Inorganic Chemistry (1988). 5th ed. Wiley Interscience, New
York.Cowell, G.W.; Ledwith, A.; White, A.C.; Woods, H.J. (1970) J. Chem. Soc. B, 227.Davico, G.E.; Schwartz, R.L.; Ramond, T.M.; Lineberger, W.C. (1999) J. Am. Chem. Soc. 121,
6047.Davies, M.J.; Gilbert, B.C. (1985) J. Chem. Res. Part S, 162.Davies, M.J.; Gilbert, B.C.; Thomas, B.; Young, J. (1985) J. Chem. Soc. Perkin Trans. 2, 1199.Davies, M.J.; Griller, D.; Roberts, B.P. (1976) J. Chem. Soc., Perkin Trans. 2, 993.Davies, A.G.; Parrot, M.J.; Roberts, B.P. (1974) J. Chem. Soc., Chem. Commun., 973.Delgado-Pena, F.; Talham, D.R.; Cowan, D.O. (1983) J. Organometal. Chem. 253, C43.Demeyer, A.; Stienlet, D. (1993) J. Phys. Chem. 97, 1477.Denisov, E.T. Kinetika Gomogennykh Khimicheskikh Reaktsii (1978). Vysshaya Shkola, Moscow.Dessau, R.M.; Shin, S.; Heiba, E.I. (1970) J. Am. Chem. Soc. 92, 412.Devonport, W.; Bryce, M.R.; Marshallsay, G.J.; Moore, A.J.; Goldenberg, L.M. (1998) J. Mater.
Chem. 8, 1361.Dias, R.M.B.; Vieira, A.J.S.C. (1996) Redox. Rep. 2, 279.Dinnocenzo, J.P.; Banach, T.E. (1986) J. Am. Chem. Soc. 108, 6063.Dinnocenzo, J.P.; Banach, T.E. (1988) J. Am. Chem. Soc. 110, 971.Dinnocenzo, J.P.; Banach, T.E. (1989) J. Am. Chem. Soc. 111, 8646.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Dixon, W.T.; Murphy, D. (1978) J. Chem. Soc., Faraday Trans. 74, 432.Dobson, P.; Norman, J.A.; Barry, Th.C. (1986) J. Chem. Soc., Perkin Trans. 1, 1209.Du Bois, D.L.; Meidaner, A. (1987) J. Am. Chem. Soc. 109, 113.Dutta, S.K.; Ferraudi, G. (2001) J. Phys. Chem. A 105, 4241.Dyusengaliev, K.I.; Ermekov, D.S.; Todres, Z.V. (1995) Zh. Obshch. Khim. 65, 858.Eberson, L. (1982) Acta Chem. Scand. B36, 533.Eberson, L. Electron Transfer Reactions in Organic Chemistry (1987). Springer-Verlag, Berlin.Eberson, L.; Graense, S.; Olofsson, B. (1968) Acta Chem. Scand. 22, 2462.Eberson, L.; Hartshorn, M.P.; Persson, O.; Radner, F. (1996) Chem. Commun. (Cambridge) 18, 2105.Eberson, L.; Larsson, B. (1986) Acta Chem. Scand. B40, 210.Eberson, L.; Radner, F. (1987) Acc. Chem. Res. 20, 53.Egorochkin, A.N.; Voronkov, M.G.; Skobeleva, S.E.; Zderenova, O.V. (2001) Russ. Chem. Bull. 50,
35.Ehlinger, N.; Scheidt, W.R. (1999) Inorg. Chem. 38, 1316.Endeward, B.; Plato, M.; Will, S.; Vogel, E.; Szyczewski, A.; Moebius, K. (1998) Appl. Magn. Re-
son. 14, 69.Endo, T.; Miyazawa, T.; Shiihasi, S.; Okawara, M. (1984) J. Am. Chem. Soc. 106, 3877.Evans, W.J.; Allen, N.T. (2000) J. Am. Chem. Soc. 122, 2118.Fang, S.; Lee, M.-S.; Hrovat, D.A.; Borden, W.T. (1995) J. Am. Chem. Soc. 117, 6727.Faraggi, M.; Houee-Levin, C. (1999) J. Chim. Phys. Pys. Chim. Biol. 96, 71.Farcasiu, D.; Ghenciu, A.; Li, J.Q. (1996) J. Catal. 158, 116.Farne, G.; Braggio, F.; Rubino, L. (1972) Ann. Chim. 62, 469.Farnia, G.; Marcuzzi, F.; Sandona, G. (1999) J. Electroanal. Chem. 460, 160.Fel’dman, V.I.; Sukhov, F.F.; Slovokhotova, N.A.; Bazov, V.P. (1996) Radiat. Phys. Chem. 48, 261.Fel’dman, V.I.; Ulyukina, E.A.; Sukhov, F.F.; Slovokhotova, N.A. (1993) Khim. Fiz. 12, 1613.Flyuht, R.; Schuchmann, M.N.; Sonntag von, C. (2001) Chem. Eur. J. 7, 796.Fomin, G.V.; Skuratova, S.I. (1978) Zh. Fis. Khim. 52, 628.Fristad, W.E.; Peterson, J.R. (1984) Tetrahedron 40, 1469.Fujinaga, K.; Takaoka, K.; Nomura, T.; Yoshikava, K. (1968) J. Chem. Soc. Jpn., Pure Chem. Sec.
89, 185.Fukutome, H.; Takahashi, I.; Ozaki, M. (1987) Chem. Phys. Lett. 133, 34.Fukuzumi, Sh.; Ohkubo, K. (2000) Chem. Eur. J. 6, 4532.Fukuzumi, Sh.; Imahori, H.; Yamada, H.; El-Khouly, M.E.; Fujitsuka, M.; Ito, O.; Guldi, D.M.
(2001) J. Am. Chem. Soc. 123, 2571.Galli, C. (1988) Gazz. Chim. Ital. 118, 365.Galli, C.; Gentili, P.; Guarnieri, A. (1995) Gazz. Chim. Ital. 125, 409.Galstyan, G.A.; Degtyaryova, S.I.; Galstyan, S.G.; Mamchur, A.V. (2001) Neftekhimiya 41, 218.Gavars, R.; Baumane, L.; Stradins, Ja.; Vigante, B.; Ozols, J.; Dubars, G. (1999) Khim. Geterotsikl.
Soed., 924.Geer, R.D.; Byker, H.J. (1982) J. Org. Chem. 47, 1662.Geoffroy, M.; Jouaiti, A.; Terron, G.; Cattani-Lorente, M.; Ellinger, Y. (1992) J. Phys. Chem. 96,
8241.Gerson, F.; Heckendorn, R.; Cowan, D.O. (1983) J. Am. Chem. Soc. 105, 7017.Gerson, F.; Huber, W. (1987) Acc. Chem. Res. 20, 85.Geske, D.H.; Maki, A.H. (1960) J. Am. Chem. Soc. 82, 2671.Geske, D.H.; Ragle, J.L. (1961) J. Am. Chem. Soc. 83, 3532.Ghenciu, A.; Farcasiu, D. (1996a) Chem. Commun., 169.Ghenciu, A.; Farcasiu, D. (1996b) J. Mol. Catal. 103, 273.Gherghel, L.; Baumgarten, M.; Declerq, D.; De Schryver, F.C. (1995) Chem. Phys. Lett. 232, 567.Glockle, M.; Hubler, K.; Kummerer, H.-Ju.; Denninger, G.; Kaim, W. (2001) Inorg. Chem. 40,
2263.Goldstein, S.L.; McNelis, E. (1984) J. Org. Chem. 49, 1613.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Grant, C.B.; Streitwieser, A. (1978) J. Am. Chem. Soc. 100, 2433.Gul’maliev, A.M.; Stankevich, I.V.; Todres, Z.V. (1975) Khim. Heterotsikl. Soed., 1055.Haare van, J.A.E.H.; Boxtel van, M.; Janssen, R.A.J. (1998) Chem. Mater. 10, 1166.Hamon, J.R.; Astruc, D. (1988) Organometallics 7, 1037.Handoo, K.L.; Gadru, K. (1986) Tetrahedron Lett. 27, 1371.Hawecker, J.; Lehn, J.-M.; Ziessel, R. (1986) Helv. Chim. Acta 69, 1990.Helton, M.E.; Gebhart, N.L.; Davies, E.S.; McMaster, J.; Garner, C.D.; Kirk, M.L. (2001) J. Am.
Chem. Soc. 123, 10389.Hoekstra, A.; Vos, A. (1975) Acta Crystallogr. B 31, 1716.Holroyd, R.A.; Itoh, K.; Nishikawa, M. (1997) Chem. Phys. Lett. 266, 227.Holton, D.M.; Murphy, D. (1979) J. Chem. Soc., Faraday Trans. 75, 1637.Huie, R.E.; Padmaja, S. (1993) Free Rad. Res. Commun. 18, 195.Hunziker, E.; Penton, J.R.; Zollinger, H. (1971) Helv. Chim. Acta 54, 2043.Hurst, J.K. In: Electron Transfer Reactions: Inorganic, Organometallic, and Biological Applications
(1997). Isied, S.S., ed. Advances in Chemistry Series, No. 253. American Chemical Society,Washington, DC.
Ichikawa, Ts.; Kumagai, J.; Koizumi, H. (1999) J. Phys. Chem. B 103, 3812.Il’yasov, A.V.; Kargin, Yu.M.; Morozova, I.D. (1980) Spektry EPR Organicheskikh Ion-radikalov.
Nauka, Moscow (Russia).Inoue, H.; Nagata, T. (1986) J. Chem. Soc., Chem. Commun., 1177.Ioffe, N.T.; Kalinkin, M.I.; Todres, Z.V. (1970) Izv. AN SSSR, Ser. Khim., 2409.Irie, S.; Irie, M. (1997) Macromolecules 30, 7906.Itahara, T.; Fujii, Yu; Tada, M. (1988) J. Org. Chem. 53, 3421.Itoh, M.; Tanimoto, Y.; Litaka, Y. (1978) J. Chem. Phys. 69, 816.Iwamoto, M.; Lunsford, J.H. (1980) J. Phys. Chem. 84, 3079.Iyoda, M.; Sasaki, Sh.; Yoshida, M.; Kuwatani, Y.; Nagase, Sh. (1996) Teterahedron Lett. 37,
7987.Jonsson, M.; Wayner, D.D.M.; Lustyk, J. (1996) J. Phys. Chem. 100, 17539.Jouaiti, A.; Al Badri, A.; Geoffroy, M.; Bernardinelli, G. (1997) J. Organomet. Chem. 529, 143.Kachkurova, I.Ya.; Ashkinadze, L.D.; Shamsutdinova, M.Kh.; Kazytsina, L.A. (1987) Zh. Org.
Khim. 23, 1831.Kagan, H.B.; Namy, J.L.; Girard, P. (1981) Teterahedron 37-Suppl. 9, 175.Kaim, W. (1987) Coord. Chem. Rev. 76, 187.Kaim, W. (1988) J. Organometal. Chem. 339, 253.Kaisaki, D.A.; Chang, W.; Dougherty, D.A. (1991) J. Am. Chem. Soc. 113, 2764.Kalsbeck, W.A.; Thorp, H.H. (1994) J. Electroanal. Chem. 314, 363.Karafiloglou, P.; Launay, J.-P. (1998) J. Phys. Chem. A 102, 8004.Kaustov, L.; Tal, M.E.; Shames, A.I.; Gross, Z. (1997) Inorg. Chem. 36, 3503.Keshavarz K.M.; Gonzalez, R.; Hicks, R.G.; Srdanov, G.; Srdanov, V.I.; Collins, T.G.; Hummelen,
J.C.; Bellavia-Lund, Ch.; Pavlovich, J.; Wudl, F.; Holczer, K. (1996) Nature 383, 147.Kharash, N.; Buess, G.M.; King, W. (1953) J. Am. Chem. Soc. 75, 6035.Kirchner, R.F.; Loew, G.H.; Mueller-Westerhoff, U.T. (1976) Inorg. Chem. 15, 2665.Klein, A.; Volger, C.; Kaim, W. (1996) Organometallics 15, 236.Knettle, B.W.; Flowers, R.A. (2001) Org. Lett. 3, 2321.Knight, L.B.; Hill, D.; Berry, K.; Babb, R.; Feller, D. (1996) J. Chem. Phys. 105, 5672.Kochi, J.K. (1986) J. Organometal. Chem. 300, 139.Kolthoff, I.M.; Coetzee, J.F. (1957) J. Am. Chem. Soc. 79, 1852.Komaguchi, K.; Shiotani, M. (1997) J. Phys. Chem. A 101, 6983.Koptyug, V.A.; Starichenko, V.F.; Breslav, I.I. (1988) Zh. Org. Khim. 24, 50.Krief, A.; Laval, A.-M. (1999) Chem. Rev. 99, 745.Kumagai, J.; Tashikawa, H.; Yoshida, H.; Ichikawa, T. (1996) J. Phys. Chem. 100, 16777.Kumai, R.; Matsushita, M.M.; Izuoka, A.; Sugawara, T. (1994) J. Am. Chem. Soc. 116, 4523.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Kumai, R. Sakurai, H.; Izuoka, A.; Sugawara, T. (1996) Mol. Cryst. Liq. Cryst. Sci., Technol. Sect. A279, 133.
Kwaan, R.J.; Harlan, C.J.; Norton, J.R. (2001) Organometallics 20, 3818.Land, E.J.; Porter, G. (1963) Trans. Faraday Soc. 59, 2016.Land, E.J.; Porter, G.; Strachan, E. (1961) Trans. Faraday Soc. 57, 1885.Lang, K.; Wagnerova, D.M.; (1992) Chemicke Listy 86, 635.Lawless, J.G.; Bartak, D.E.; Hawley, M.D. (1969) J. Am. Chem. Soc. 91, 7121.Lawson, G.E.; Kitaygorodskiy, A.; Sun, Ya-P. (1999) J. Org. Chem. 64, 5913.Lee, J.; Gralowsky, J.J. (1992) Chem. Revs. 92, 1611.Lee, K.Y.; Amatore, C.; Kochi, J.K. (1991) J. Phys. Chem. 95, 1285.Lee, S.A.; Yamashita, T.; Horie, K. (1997) Polym. J. (Tokyo) 29, 752.LeVanda, C.; Bechgaard, K; Cowan, D.O.; Mueller-Westerhoff, U.T.; Eilbracht, P.; Candela, G.A.;
Collins, R.L. (1976) J. Am. Chem. Soc. 98, 3181.Levin, P.P.; Khudyakov, I.V.; Kuz’min, V.A. (1979) Dokl. AN SSSR 246, 144.Levitin, I.Ya.; Todres, Z.V.; Vol’pin, M.E. (1971) Zh. Obshch. Khim. 41, 193.Lexa, D.; Momenteau, M.; Mispelter, J.; Saveant, J.-M. (1989) Inorg. Chem. 28, 30.Liwo, A.; Jeziorek, D.; Dyl, D.; Ossowski, T.; Chmurzynski, L. (1997) J. Mol. Struct., 398, 445.Lough, S.M.; McDonald, J.W. (1987) Inorg. Chem. 26, 2024.Lu, Y.; Zhao, Y.; Parker, V.D. (2001) J. Am. Chem. Soc. 123, 5900.Lund, T.; Eberson, L. (1997) J. Chem. Soc., Perkin Trans. 2, 1435.Maletin, Yu.A.; Statsyuk, V.N.; Strizhakova, N.G. (1983) Ukrainskii Khim. Zh. 49, 996.Maletin, Yu.A.; Strizhakova, N.G.; Sheka, I.A. (1979) Dokl. AN SSSR 248, 651.Maletin, Yu.A.; Strizhakova, N.G.; Sheka, I.A. (1980) Zh. Obshch. Khim. 50, 702.Mao, F.; Tyler, D.R.; Keszler, D. (1989) J. Am. Chem. Soc. 111, 130.Mao, F.; Tyler, D.R.; Bruce, M.R.M.; Bruce, A.E.; Rieger, A.L.; Rieger, Ph.H. (1992) J. Am. Chem.
Soc. 114, 6418.McKinney, T.M.; Geske, D.H. (1967) J. Am. Chem. Soc. 89, 2806.Meerwein, H.; Hinz, G.; Hoffmann, P.; Kroning, E.; Pfeil, E. (1937) J. pract. Chem. 147, 257.Meurs van, P.J.; Janssen, R.A.J. (2000) J. Org. Chem. 65, 5712.Meurs van, P.J.; Janssen, R.A.J. (2001) Synth. Met. 121, 1832.Michinobu, Ts.; Tsuchida, E.; Nishide, H. (2001) Polyhedron 20, 1147.Miholova, D.; Vlceck, A.A. (1985) J. Organomet. Chem. 279, 317.Miller, L.L.; Mann, K.R. (1996) Acc. Chem. Res. 29, 417.Minisci, F.; Citterio, A.; Giordano, C. (1983) Acc. Chem. Res. 16, 27.Miyazawa, T.; Endo, T.; Okawara, M. (1985) J. Org. Chem. 50, 5389.Moon, M.P.; Komin, A.P.; Wolfe, J.F.; Morris, G.F. (1983) J. Org. Chem. 48, 2392.Mori, T.; Rathore, R.; Lindeman, S.V.; Kochi, J.K. (1998) J. Chem. Soc., Chem. Commun., 927.Morkovnik, A.S.; Okhlobystin, O. Yu. (1979) Usp. Khim. 48, 1968.Muller, J.G.; Hickerson, R.P.; Perez, R.J.; Burrows, C.J. (1997) J. Am. Chem. Soc. 119, 1501.Murray, M.M.; Kaszynski, P.; Kaisaki, D.A.; Chang, W.; Dougherty, D.A. (1994) J. Am. Chem. Soc.
116, 8152.Myagchenko, A.P.; Kolesnik, Yu.R.; Svetkin, Yu.N. (1989) Zh. Obshch. Khim. 59, 146.Nelsen, S.F.; Klein, S.J.; Treiber, D.A.; Rustern, F.I.; Powell, D.R. (1997) J. Org. Chem. 62, 6539.Neta, P.; Behar, D. (1981) J. Am. Chem. Soc. 103, 2280.Neugebauer, F.A.; Fischer, H.; Bamberger, S.; Smith, H.O. (1972) Chem. Ber. 105, 2694.Nicholas, A.N. de P.; Arnold, D.R. (1982) Canad. J. Chem. 60, 2165.Niyazymbetov, M.E.; Evans, D.H. (1993) J. Org. Chem. 58, 779.Oae, Sh.; Takata, T.; Kim, J.H. (1981) Bull. Chem. Soc. Jpn. 54, 2712.Ogawa, S.; Kikuchi, T.; Niizuma, Sh.; Sato, R. (1994) J. Chem. Soc., Chem. Commun., 1593.Ohst, H.H.; Kochi, J.K. (1986) J. Chem. Soc., Chem. Commun., 121.Okada, K.; Imakura, T.; Oda, M.; Murai, H. Baumgarten, M. (1996) J. Am. Chem. Soc. 118, 3047.Ozawa, T.; Kwan, T. (1985) Polyhedron 5, 1531.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Palaniappan, S. (1997) Eur. Polym. J. 33, 1735.Panteleeva, E.V.; Bil’kis, I.I.; Shteingarts, V.D. (1998) Zh. Org. Khim. 34, 1702.Parker, V.D. (1981) Acta Chem. Scand. B35, 595.Pilard, J.-F.; Fourets, O.; Simonet, J.; Klein, L.J.; Peters, D.G. (2001) J. Electrochem. Soc. 148, E171.Pokhodenko, V.D.; Koshechko, V.G.; Titov, V.E.; Golovatyi, V.G.; Shabel’nikov, V.P. (1984) Teor.
Eksperim. Khim. 20, 25.Pross, A. (1985) Acc. Chem. Res. 18, 212.Prousek, J. Reakce iniciovane prenosem electronu (1988). Academia, Praha, Czechoslovakia.Pullen, S.H.; Edington, M.D.; Studler-Martinez, Sh.L.; Simon, J.D.; Staab, H.A. (1997) J. Phys.
Chem. A 103, 2740.Rajca, S.; Rajca, A. (1995) J. Am. Chem. Soc. 117, 9172.Rajca, A.; Rajca, S.; Desai, Sh.R. (1995) J. Chem. Soc., Chem. Commun., 1957.Rak, S.F.; Miller, L.L. (1992) J. Am. Chem. Soc. 114, 1388.Ramos, J.; Yartsev, V.M.; Golhen, S.; Ouahab, L.; Delhaes, P. (1997) J. Mater. Chem. 7, 1313.Rathore, R.; Kumar, A.S.; Lindeman, S.V.; Kochi, J.K. (1998a) J. Org. Chem. 63, 5847.Rathore, R.; Lindeman, S.V.; Kumar, A.S.; Kochi, J.K. (1998) J. Am. Chem. Soc. 120, 6012.Rautenstrauch, V.; Willhalm, B.; Thommen, W. (1981) Helv. Chim. Acta 64, 2109.Razumovskii, S.D.; Zaikov, G.E. Ozone and Its Reactions with Organic Compounds (1984). Else-
vier, Amsterdam.Reed, C.A.; Bolskar, R.D. (2000) Chem. Rev. 100, 1075.Reed, G.A.; Curts, J.F.; Mottley, C.; Eling, T.E.; Mason, R.P. (1986) Proc. Natl. Acad. Sci. U.S.A.
83, 7499.Reynolds, R.; Line, L.L.; Nelson, R.F. (1974) J. Am. Chem. Soc. 96, 1087.Rhodes, Ch.J.; Podmore, I.D.; Symons, M.C.R. (1988) J. Chem. Res., Ser. S., 120.Richardson, T.J.; Tanzella, F.L.; Bartlett, N. (1986) J. Am. Chem. Soc. 108, 4937.Rieger, Ph.H.; Bernal, L.; Reinmuth, W.H.; Fraenkel, G.K. (1963) J. Am. Chem. Soc. 85, 683.Rossi, R.A.; Pierini, A.B.; Borosky, G.L. (1994) J. Chem. Soc., Perkin Trans. 2, 2577.Rosso, J.A.; Bertollotti, S.G.; Braun, A.M.; Martire, D.O.; Gonzalez, M.C. (2001) J. Phys. Org.
Chem. 14, 300.Roth, H.D.; Wong, H.; Zhou, D.; Lakkaraju, P.S. (1997) Acta Chem. Scand. 51, 626.Saeki, M.; Tsukuda, T.; Iwata, S.; Nagata, T. (1999) J. Chem. Phys. Lett. 111, 6333.Sakurai, H.; Izuoka, A.; Sugawara, T. (2000) J. Am. Chem. Soc. 122, 9723.Sakurai, H.; Kumai, R.; Izuoka, A.; Sugawara, T. (1996) Chem. Letters, 879.Sasaki, Sh.; Murakami, F.; Yoshifuji, M. (1999) Angew. Chem., Int. Ed. Engl. 38, 340.Sato, K.; Furuichi, M.; Yano, M.; Shiomi, D.; Abe, K.; Takui, T.; Itoh, K.; Higuchi, A.; Katsuma, K.;
Shirota, Y. (1997) Synth. Met. 85, 1719.Saveant, J.-M. (1994) J. Phys. Chem. 98, 3716.Sawamura, M.; Nagahama, N.; Toganoh, M.; Hackler, U.E.; Isobe, H.; Nakamura, E.; Zhou, Sh.-Q.;
Chu, B. (2000) Chem. Lett., 1098.Sawyer, D.T. Oxygen Chemistry (1991). Oxford University Press, Oxford, UK.Sawyer, D.T.; Gibian, M.J. (1979) Tetrahedron 35, 1471.Sawyer, D.T.; Roberts, J.L. (1988) Accounts Chem. Res. 21, 469.Schank, K.; Beck, H.; Buschlinger, M.; Eder, J.; Heisel, Th.; Pistorius, S.; Wagner, Ch. (2000) Helv.
Chim. Acta 83, 801.Schmittel, M.; Langels, A. (1998) J. Chem. Soc., Perkin Trans. 2, 565.Screttas, C.G.; Ioannou, G.I.; Micha-Screttas, M. (1996) J. Organomet. Chem. 511, 217.Sehested, K.; Holcman, J. (1978) J. Phys. Chem. 82, 651.Seki, Sh.; Yoshida, Y.; Tagawa, S.; Asai, K. (1999) Macromolecules 32, 1080.Seki, Sh.; Yoshida, Y.; Tagawa, S. (2001) Radiat. Phys. Chem. 60, 411.Selby, T.D.; Blackstock, S.C. (1999) J. Am. Chem. Soc. 121, 7152.Shchapin, I.Y.; Chuvylkin, N.D. (1996) Izv. Ross. Akad. Nauk, Ser.Khim., 321.Shinomura, E.T.; Phillippi, M.A.; Goff, H.M. (1981) J. Am. Chem. Soc. 103, 6778.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Shkrob, I.A.; Sauer, M.C. (2001a) J. Phys. Chem. B 105, 4520.Shkrob, I.A.; Sauer, M.C. (2001b) J. Phys. Chem. B 105, 7027.Shultz, D.A.; Farmer, G.T. (1998) J. Org. Chem. 63, 6254.Shut, D.M.; Keana, K.J.; Tyler, D.R.; Rieger, Ph.H. (1995) J. Am. Chem. Soc. 117, 8939.Sidorenkova, H.; Chentit, M.; Jouaiti, A.; Terron, G.; Geoffroy, M.; Ellinger, Y. (1998) J. Chem.
Soc., Perkin Trans. 2, 71.Siedschlag, C.; Dunsch, L.; Ito, O.; Mattay, J. (2000) J. Inf. Rec. 25, 265.Sies, H. (1986) Angew. Chem., Int. Ed. Engl. 25, 1058.Simic, M.; Neta, P.; Hayon, E. (1973) J. Phys. Chem. 77, 2662.Singh, P.R.; Jayaraman, B.; Singh, H.K. (1977) Chem. Ind. (London), 311.Skogareva, L.S.; Ippolitov, E.G. (1998) Zh. Neorg. Khim. 43, 1796.Solodovnikov, S.P.; Todres, Z.V. (1968) Khim. Gheterotsikl. Soed., 360.Starichenko, V.F.; Selivanova, G.A.; Shteingarts, V.D. (1981) Zh. Org. Zhim. 17, 2255.Starichenko, V.F.; Shundrin, L.A.; Shchegoleva, L.N.; Shteingarts, V.D. (2000) Zh. Strukt. Khim. 41,
949.Stickley, K.R.; Blackstock, S.C. (1994) J. Am. Chem. Soc. 116, 11576.Stickley, K.R.; Selby, T.D.; Blackstock, S.C. (1997) J. Org. Chem. 62, 448.Stone, E.W.; Maki, A.H. (1962) J. Chem. Phys. 37, 1326.Strizhakova, N.G.; Maletin, Yu.A.; Sheka, M.A.; Khudyakov, I.V.; De Jonge, C. (1985) Ukr. Khim.
Zh. 51, 148.Sviridenko, F.B.; Stas’, D.V.; Molin, Yu.N. (2001) Dokl. Akad. Nauk 377, 356.Tachikawa, H. (1999) J. Phys. Chem. A 103, 2501.Takahashi, H.; Hori, T.; Wakabayashi, T.; Nitta, T. (2001) J. Phys. Chem. A 105, 4351.Telo, J.P.; Vieira, A.J.S.C. (1997) J. Chem. Soc., Perkin Trans. 2, 1755.Timoshok, A.V.; Bedilo, A.F.; Volodin, A.M. (1996) React. Kinet. Catal. Lett. 59, 165.Todres, Z.V. (1981) Sulfur Reports 1, 133.Todres, Z.V. (1987) Tetrahedron 43, 3839.Todres, Z.V. (1990) Acta Chem. Scand. 44, 535.Todres, Z.V. (1992) J. Organomet. Chem. 441, 349.Todres, Z.V.; Avagyan, S.P. (1972) Int. J. Sulphur Chem. 8, 373.Todres, Z.V.; Avagyan, S.P. (1978) Phosph. Sulphur 4, 223.Todres, Z.V.; Dyusengaliev, K.I.; Sevast’yanov, V.G. (1985) Izv. AN SSSR, Ser. Khim., 1416.Todres, Z.V.; Dyusengaliev, K.I.; Tsvetkova, T.M.; Borisov, Yu.A. (1984) Izv. AN SSSR, Ser.Khim.,
392.Todres, Z.V.; Hovsepyan, G.Ts.; Ionina, Ye.A. (1988) Tetrahedron 44, 5199.Todres, Z.V.; Lyakhovetskii, Yu.I.; Kursanov, D.N. (1969) Izv. AN SSSR, Ser. Khim., 1455.Todres, Z.V.; Pozdeeva, A.A.; Chernova, V.A.; Zhdanov, S.I. (1972a) Izv. AN SSSR, Ser.Khim, 2454.Todres, Z.V.; Pozdeeva, A.A.; Chernova, V.A.; Zhdanov, S.I. (1972b) Tetrahedron Lett. 36, 3835.Todres, Z.V.; Safronov, A.I.; Ermekov, D.S.; Minyaev, R.M. (1992) J. Organometal. Chem. 441,
479.Todres, Z.V.; Tomilin, O.B.; Stankevich, I.V. (1982) Zh. Org. Khim. 18, 2132.Todres, Z.V.; Zhdanov, S.I.; Tsveniashvili, V.Sh. (1968) Izv. AN SSSR, Ser. Khim., 975.Tomilin, O.B.; Konovalova, E.P.; Yuzhalkin, V.N.; Ryabkina, L.V.; Sanaeva, E.P. (1996) Khim.
Gheterotsikl. Soed., 420.Tomilin, O.B.; Shchegolikhin, A.N.; Sanaeva, E.P.; Klyakin, A.N.; Ryabkina, L.B.; Konovalova,
L.B. (2000) Khim. Gheterotsikl. Soed., 1259.Toriyama, K.; Nunome, K.; Iwasaki, M. (1982) J. Chem. Phys. 77, 5891.Tsang, P.K.S.; Cofre, P.; Sawyer, D.T. (1987) Inorg. Chem. 26, 3604.Tselinskii, I.V.; Shugalei, I.V.; Lukogorskaya, S.A. (2001) Ross. Khim. Zh. 45, 35.Tsukuda, T.; Saeki, M.; Kimura, R.; Nagata, T. (1999) J. Chem. Phys. Lett. 111, 7846.Vieira, A.J.S.C.; Telo, J.P.; Dias, R.M.B. (1997) J. Chim. Phys. (Paris) 94, 318.Vishnetskaya, M.V.; Romanovskii, B.V. (1993) Zh. Fiz. Khim. 67, 933.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Vishnetskaya, M.V.; Romanovskii, B.V. (1994) Ross. Khim. Zh. 38, 12.Vishnetskaya, M.V.; Romanovskii, B.V.; Lipovich, V.G. (1997) Neftekhimiya 37, 202.Vicek, A.; Baumann, F.; Kaim, W.; Grevels, F.-W.; Hartl, F. (1998) J. Chem. Soc., Dalton Trans. 2,
215.Vogel, E. (1996) J. Heterocycl. Chem. 33, 1461.Walter, R.I. (1966) J. Am. Chem. Soc. 88, 1293.Wang, Y.; Chen, M. (1997) Xi’an Shiyou Xueyuan Xuebao 12, 49.Wartini, A.; Staab, H.A.; Neugebauer, F.A. (1998) Eur. J. Org. Chem., 1161.Wartini, A.; Valenzuela, J.; Staab, H.A.; Neugebauer, F.A. (1998) Eur. J. Org. Chem., 221.Werst, D.W.; Bakker, M.G.; Trifunac, A.D. (1990) J. Am. Chem. Soc. 112, 40.Wienk, M.M.; Janssen, R.A.J. (1996) Chem. Commun. (Cambridge), 267.Wienk, M.M.; Janssen, R.A.J. (1997) J. Am. Chem. Soc. 119, 4492.Wille, U. (2000) Org. Lett. 2, 3485.Wolf, M.O.; Fox, H.H.; Fox, M.A. (1996) J. Org. Chem. 61, 287.Yacovan, A.; Hoz, S.; Bil’kis, I. (1996) J. Am. Chem. Soc. 118, 261.Yamamoto, H.; Mashino, T.; Nagano, T.; Hirobe, M. (1986) J. Am. Chem. Soc. 108, 539.Yasui, Sh.; Fujii, M.; Kawano, C.; Nishimura, N.; Shioji, K.; Ohno, A. (1994) J. Chem. Soc., Perkin
Trans. 2, 177.Yasui, Sh.; Shioji, K.; Ohno, A. (1994) Tetrahedron Lett. 35, 2695.Zapol’skikh, B.V.; Safiullin, R.L.; Khursan, S.L.; Teregulova, A.N.; Zaripov, R.N.; Komissarov,
V.D. (2001) Khim. Fiz. 20, 58.Zhang, R.; Zhang, G.; Su, Zh.; Qui, Y.; Shen, J. (2000) Macromol. Chem. Phys. 201, 1774.Zhang, X.-M.; Bordwell, F.G. (1992) J. Am. Chem. Soc. 114, 9787.Zhang, X.-M.; Bordwell, F.G. (1994) J. Am. Chem. Soc. 116, 4251.Zhao, Ch.-X.; Gong, Y.-A.; He, H.-Y.; Jiang, X.-K. (1999) J. Phys. Org. Chem. 12, 688.Zhao, Y.; Bordwell, F.G. (1996a) J. Org. Chem. 61, 2530.Zhao, Y.; Bordwell, F.G. (1996b) J. Org. Chem. 61, 6623.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

2
Formation of Organic Ion Radicals
2.1 INTRODUCTION
This chapter describes the preparation of organic ion radicals as well as their properties, i.e.,electronic structure, reactivity, and interaction with counterions.
In the synthetic chemistry of organic compounds, liquid-phase reactions are mosttypical. Under these conditions, ion radical salts appear to be surrounded with solvateshells. Either the solvent can solvate the individual ions (having cation and anion individ-ually surrounded by solvent molecules) or an ion pair may be solvated without solventmolecules between the ions. Further transformations of ion radicals (their disintegration,interaction with reagents directly or after the disintegration) also take place in solvents.Formation of transient states and stabilization of final products occur in solvents, too.Medium effects on the generation and structure of ion radicals are of great experimental andtheoretical interest (see, for example, Orlov et al. 2001). The nature of the solvent deter-mines the efficiency of the chosen method for ion radical generation. That is why this chap-ter examines the peculiarities of organic compounds as ion radical precursors under theconditions of liquid-phase electron transfer.
Ion radicals are widely discussed as intermediate products of electrode reactions.Regularities of the electrode reactions are, undoubtedly, important for organic synthesis. Atthe same time, there are features that are distinctive of the liquid-phase reactions only. Theelectrode reactions are heterogeneous. There is a strong electric field (106–107 V�cm�1) inthe pre-electrode (double-layer) region. Obviously, this may sometimes act on the reactiv-ity of a depolarizer or on an ion radical that originated from it. The solution in the elec-trode/solution interface region has special (double-layer) properties that are significantlydifferent from those in the bulk solution. The electrode practically does not change its prop-erties during a reaction with a depolarizer. At the given potential, the influence uponspecies adsorbed with the electrode surface or located in the framework of the double elec-tric layer remains constant.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

In liquid-phase homogeneous reactions, ion radicals form and react without any in-fluence of either electrode.
Some discrepancy also exists in ion-radical reactions between a donor and an accep-tor in their ground states, on the one hand, and reactions stimulated by photoirradiation orpulse radiolysis, on the other hand. The energy of the incoming electron is much larger inphotochemistry or pulse radiolysis than in homogeneous electron transfer or electrochem-istry. Of course, the problem of organic ion radical formation has both chemical and phys-ical aspects. This book concentrates on chemistry, but includes some physical aspects whennecessary for better understanding. Nevertheless, organic photochemistry and radiolysis oforganic compounds are very specific branches, with their own, very special book reper-toire.
Therefore, this chapter compresses data on the preparation of organic ion radicals asindependent particles that can be free or bound with counterions in ion pairs. It considersliquid-phase equilibria in electron-transfer reactions and compares electrode and liquid-phase processes for the same organic compounds. Because isotope-containing moleculeshave specific features as ion radical precursors, the generation of the corresponding ion rad-icals is considered in a separate section of the chapter. The chapter also pays some atten-tion to the peculiarities of ion radical formation in living organisms.
2.2 CHEMICAL METHODS OF ORGANIC ION RADICALPREPARATION
2.2.1 Anion Radicals
Electrochemical methods to generate anion radicals consist of potential-controlled elec-trolysis. The control of potential allows one to detain reduction practically just after a one-electron transfer to a depolarizer. The one-electron nature of the electron transfer is coinci-dentally studied by means of coulombometry. One molecule must consume one electron.If less than one electron is consumed in the framework of the one-electron reduction, thenthe yield of an anion radical is not quantitative. The electrolysis often occurs in a specialampoule placed into a resonator of the electron spin resonance (ESR) spectrometer. Thispermits one to identify many unstable anion radicals. Electrochemical methods of anionradical generation employ an electrode as an electron donor.
In the case of an alkali metal (M), one electron of an organic molecule follows theensuing scheme (here, ArH is an aromatic hydrocarbon):
M � ArH → ArH�., M�
Solvation of a resulting M� takes place in a liquid phase. The solvation may play a crucial role in the generation of anion radicals. For example, benzene, which is a poorelectron acceptor, forms its anion radical by reacting with a potassium mirror indimethoxyethane (DME). DME solvates the potassium cation; the potassium–chelate com-plex is formed with the participation of two oxygen atoms from the solvent. Solvation ofthe lithium cation proceeds even more effectively because of the smaller ion radius oflithium than that of potassium (0.078 nm for Li�, 0.133 nm for K�). With lithium in DME,the equilibrium concentration of the benzene anion radical turns out to be two orders ofmagnitude higher than that with potassium or sodium (Yakovleva et al. 1960). Without go-ing into detail, the following regularity can be stated: The greater the solvating ability of amedium, the easier is electron transfer from the same metal to the same organic acceptor.In alkali metal reductions, the reducing power follows the order K � Na � Li.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Diethoxymethane (DEM, CH3CH2OCH2CH2CH3) has recently become available incommercial quantities. Although DEM contains a reactive methylene group, it is stable toorganolithium reagents and, supposedly, can be used for preparation of anion radicals. Twoimportant characteristics of DEM that differentiate it from many other ethers are its im-miscibility in water and its nonhygroscopic nature. For example, DEM from a containerthat had been open for over a year performed superbly as a solvent for several water-sensi-tive reactions. With no special drying, it is competitive with “anhydrous-grade” THF,which is usually stored under argon (Boaz & Venepalli 2001).
Another common solvent where oxygen is easily available for coordination with metalcations is tetrahydrofurane (THF). The ability of anion radicals to remove a proton from the2-position of THF is sometimes a problem. Dimethyl ether is more stable as a solvent; itsoxygen atom is also exposed and can coordinate a metal cation with no steric hindrance fromthe framing alkyl groups. An added advantage of dimethyl ether is that, because of its lowboiling point (�22°C), it can be readily removed after reductive metallation and replacedby the desired solvent. The use of aromatic anion radicals in dimethyl ether (instead of THF)is well documented (Cohen and others 2001 and references therein).
Commonly used alkali metals have lower ionization potentials than reduction poten-tials, which are needed for the transformation of an organic molecule into an anion radical.Once a molecule starts getting reduced, it will take up as many electrons as the ionizationpotential of the metal allows. For alkali metals, the scope of application is limited with sub-stances for which the first electron transfer corresponds to enough negative values of ca-thodic potentials and is highly separated at this scale from potentials of the second or, gen-erally, next electron steps. If not, metal reduction proceeds deeper. No delay at anionradical formation takes place. There are reductive reactions that show a clear dependenceon the homogeneity of the metal reducer. For example, during the alkali metal reduction ofbis(gem-dihalocyclopropyl)ethanes in the mixture of liquid ammonia with tetrahydrofuran(THF), the route of any further transformation of primary ion radicals depends on being analkali metal in the homogeneous versus heterogeneous phase (Oku et al. 1983). Lithium isdissolved in this mixture, and sodium forms a liquid suspension in it. This determines thegeneral result of the reduction. The presence of sodium metal in the heterogeneous phaseleads to adsorption of the substrate on the metal surface. The adsorption creates a high con-centration of the reducing metal in the reaction zone. This assists the deeper reduction withthe full loss of a halogen. In a homogeneous medium (lithium as a reducer), the rate of theprimary electron transfer is comparable with the rate of posterior protonation at carbonatoms, and the reaction stops at the stage of elimination of two of the four halogen atomsthat are present.
Colloidal potassium has recently been proved as a more active reducer than the samemetal that has been conventionally powdered by means of shaking it in hot octane (Lucheet al. 1984; Chou & You 1987; Wang et al. 1994). In order to prepare colloidal potassium,a piece of this metal in dry toluene or xylene under an argon atmosphere is submitted to ul-trasonic irradiation at ca. 10°C. A silvery blue color is rapidly developed, and in a few min-utes the metal disappears. A common cleaning bath (e.g., Sonoclean, 35 kHz) filled withwater and crushed ice can be used. A very fine suspension of potassium is thus obtainedthat settles very slowly on standing. In THF, the same method did not work. Attempts todisperse lithium in THF or toluene or xylene were unsuccessful, whereas sodium was dis-persed in xylene but not in THF or toluene (Luche et al. 1984). Ultrasonic waves interactwith the metal via their cavitational effects (see Section 5.2.4). These effects are closely re-lated to the physical constants of the medium, such as vapor pressure, viscosity, and sur-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

face tension (Sehgal et al. 1982). All of these factors have to be taken into account whenone chooses a metal to be ultrasonically dispersed in a given solvent.
Several methods have been reported for the preparation of highly reactive metal pow-ders by means of the reduction of metal salts in ethereal or hydrocarbon solvents. These meth-ods used alkali metals as reducing agents in conjunction with or without an electron carrier ofthe naphthalene type. The reduction of metal salts in this manner produces finely divided metalslurries. These metal slurries are highly reactive and air sensitive and are usually pyrophoricin the absence of solvent. Thus, active magnesium, zinc, indium, copper, and other metals wereprepared (Yus 1996; Rieke & Hanson 1997). The metals were used for the hydrogenation ofalkynes to alkanes (Alonso & Yus 1997) or for the addition of highly reactive zinc to organicbromides (Guijarro et al. 1999). These active metallic forms can, obviously, be utilized in spe-cial cases of anion radical preparation, although the cited works contain no mentions of this.
By and large, a finely divided precipitate of a metal is a very effective one-electronreducer. For example, a finely divided precipitate of Zr(0) was obtained on mixing of thenaphthalene sodium derivative in THF with ZrCl4. The Zr(0) precipitate was dissolved onthe addition of anthracene or benzophenone to form the corresponding zirconium salts ofthe anion radicals (Terekhova et al. 1996).
Neutral organic molecules can also be one-electron donors. For example, tetra-cyanoquinodimethane gives rise to the anion radical on reduction with 10-vinylphenoth-iazine or N,N,N,N-tetramethyl-p-phenylene diamine. Sometimes, alkoxide or phenoxideanions find application as one-electron donors. There is certain dependence between car-banion basicity and their ability to be one-electron donors (Bordwell & Clemens 1981).
Sometimes, anion radicals are formed indirectly, by means of special chemical reac-tions. Photoionization of hydrazine in a mixture of liquid ammonia with THF in the pres-ence of potassium t-butoxide leads to the formation of the diazene anion radical by the se-quence of the following reactions (Brand et al. 1985):
H2NMNH2 � t-BuO� → t-BuOH � H2NMNH�
H2NMNH� � h� → esolv. � H2NMNH.
H2NMNH.� t-BuO� → t-BuOH � �HNMNH
.
�HNMNH. ↔ H2N
.BN�
One unusual case of anion radical preparation is 4- and 3-nitrocatechol or nitrohy-droquinone. Reduction of these nitrocompounds in aqueous solutions with sodium boro-hydride in air leads to the corresponding anion radicals, which are stable at pH 9–12 despitethe presence of water (Grenier et al. 1995).
As already stated, the interaction between the metal cation and the anion radical thatis generated is usually a simple ion association moderated by the coulombic attraction be-tween the two species and the competitive ion-solvating nature of the given solvent. How-ever, interaction between organic one-electron acceptors and some metals results in the for-mation of a special class of organometallic complexes. The metals reduce the acceptormolecules. The anion radical and the cation form a coordination anion radical complex.Thus, when Cd (d10s2) metal reduces benzoquinone in THF, a solvated organometalliccompound is formed (Ch. D. Stevenson, Reiter, Burton, & Halvorsen 1995). According toESR studies, the benzosemiquinone anion radical is ion associated with Cd2� ion or coor-dinated to it (Scheme 2-1).
Exposure of naphthalene dissolved in liquid ammonia to europium metal immedi-ately results in the characteristic green color of the naphthalene anion radical. ESR analy-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.
sis reveals a signal that comes from an unpaired electron interacting with the 151Eu and153Eu nuclei. No hyperfine coupling with the naphthalene protons is observed, althoughtreatment with water leads to 1,4-dihydronaphthalene (Ch.D. Stevenson, Schertz, & Reiter1999). This means that naphthalene has indeed been reduced to its anion radical and un-dergone a normal Birch reaction. These results are consistent with the initial donation ofthe two s electrons to two acceptor molecules. Tight coordination then takes place betweenthe two naphthalene anion radicals and the central atom of Eu2�, presumably through theeuropium s orbital. Since the two electrons, donated by the Eu, are in the same molecularorbital, they are spin paired. Hence, only the remaining seven unpaired f electrons can con-tribute to the ESR signal. Therefore, the resulting ESR spectrum reflects the interaction ofthe europium nucleus with a single electron spin. The structure of the complex is depictedin Scheme 2-2. The structure “is tentative, but it is known that: (1) europium ions prefer anoctahedral geometry, (2) they complex well with ammonia, and (3) (in this case) two naph-thalene units are involved” (Ch.D. Stevenson et al. 1999).
Another rare earth metal cation, Sc3�, catalyzes electron transfer from C60�.
tophenacyl bromide; the effect is supposedly close to the phenomenon just discussed(Fukuzumi et al. 2000).
Benzene and toluene anion radical salts are intriguing examples. Benzene andtoluene anion radicals prepared as Cs� salts were shown to dimerize readily in THF at�70°C. 3,3-Bis(cyclohexa-1,4-diene) and the corresponding toluene 1,1-dimethyl analog
SCHEME 2-1
SCHEME 2-2
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

were isolated on hydrolysis (Grovenstein et al. 1977). In the presence of 18-crown-6-ether,however, toluene gives, with potassium in THF, the anion radical salt as a tight ion pair.The crown ether encapsulates the potassium cation, which is bound in an approximate �6
fashion to the planar six-membered ring in CH3C6H5. Treatment of potassium with 18-crown-6 in a benzene–THF mixture initially yields the very similar benzene-(potassium-18-crown-6) anion radical salt in �6 fashion. This salt is barely soluble in both benzene andTHF. The solid salt transforms into a dimer. The 3,3-bis(cyclohexa-1,4-dienyl) ligand isjust the dianion of the crystalline salt, being �5:�5 bound in a transoid fashion to each ofthe two crown-encapsulated potassium cations (Hitchcock et al. 2001). It unclear whichfactor plays the main role in these different reaction pathways: the low solubility of thecrystalline cyclohexadienyl dimer, or the electronic and steric effects of the methyl groupmaking dimerization of the toluene-(potassium-18-crown-6) anion radical salt unfavorable.
Sometimes, anion radical salts formed in a solution crystallize as aggregates. For in-stance, if anthracene is reduced at an alkali–metal surface, a slight modification in the natureof the solvent leads to various reduction products that were structurally characterized in sin-gle crystals. In the system of Na � triglyme, the bare anthracene anion radical crystallizes outas a crystal solvate containing the sodium cation surrounded by two triglyme molecules. Inthe system of K � tetrahydrofurane, the ion quadruple of two anthracene anion radicals con-nected by a [(K�)2(THF)3] bridge crystallizes (Bock, Gharagozloo-Hubmann et al. 2000b).
Additionally, one specific topic should be underscored: the use of silicon grease forthe fitting of glass in Schlenk vessels, which are usually employed for the preparation ofanion radicals. This is merely an experimental detail, at first glance. However, it has sig-nificant and general importance. Biphenylene reduction by sodium metal mirror in THF so-lution containing [2.2.1]cryptand yields as a structurally characterized product the 9,9-dimethylsilafluorene anion radical salt containing a Me2-Si-expanded cyclobutadiene ring.When the use of any silicon grease is avoided for all glass fittings, the solvent-separated ionpair crystallizes (Bock, Sievert, et al. 1999). Whether or not this reaction is specific for theinitially generated biphenylene anion radical only, the transformation of silicone(Me2SiO)n into the Me2Si-containing reactive species is indisputable. Such species reallyoriginate from trace silicon concentration and the residual oxygen, which is disposed on thesodium mirror surface. If the use of silicon grease is avoided, this difficulty in anion radi-cal preparation is circumvented. The unwanted reaction described took place in THF. Thespecificity of this solvent remains an open question.
2.2.2 Cation Radicals
In order to understand features of oxidative one-electron transfer, it is reasonable to com-pare average energies of formation between cation-radicals and anion-radicals. One-elec-tron addition to a molecule is usually accompanied by energy decrease. The amount of en-ergy reduced corresponds to molecule’s electron affinity. For instance, one-electronreduction of aromatic hydrocarbons can result in the energy revenue from 10 to 100kJ�mol�1 (Baizer & Lund 1983). If a molecule detaches one electron, energy absorptionmostly takes place. The needed amount of energy consumed is determined by molecule’sionization potential. In particular, ionization potentials of aromatic hydrocarbons vary from700 to 1,000 kJ�mol�1 (Baizer & Lund 1983).
Because many electron-deficient cation-radicals are less stable and have much morereactivity, cation-radical studies were harder to do with slower accumulation of the relevantliterature. Thus, the Landolt-Boernstein reference book devotes 149 pages to hydrocarbonanion radicals (1980, vol. 9d1), whereas only 15 pages are reserved for hydrocarbon cation
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

radicals (1980, vol. 9d2). The ratio is 10:1. The recent development of physical methodsdrastically changes this ratio. According to Chemical Abstracts Subject Indices, the ratiowas 1:3 during 1984–1993 and reached 1:5 in 1994–1998.
Studies of cation radical chemistry began with the work of Weiss (1941) andMichaelis and his coworkers (1941). However, intense investigation in this field began af-ter instrumental methods emerged (ESR and optical spectroscopy in particular).
Aromatic hydrocarbons gave products of protonation on dissolving in hydrofluoricacid. Oxidation in aromatic cation radicals did not take place (Kon & Blois 1958). Triflu-oroacetic acid is an effective one-electron oxidant (Eberson & Radner 1991). Meanwhile,sulfuric acid caused not only dissolution and protonation, but also one-electron oxidationof aromatic hydrocarbons. Sulfonation, naturally, proceeded too (Weissmann et al. 1957).
Anodic oxidation in inert solvents is the most widespread method for cation radicalpreparation, with the aim of investigating their stability and electron structure. However,saturated hydrocarbons cannot be oxidized in an accessible potential region. There is oneexception for molecules with the weakened CMH bond, but this does not pertain to thecation radical problem.
Anodic oxidation of unsaturated hydrocarbons proceeds more easily. As usual, thisoxidation is assumed to be a process including one-electron detachment from the �-systemwith cation radical formation. This is the very first step of the oxidation. Certainly, the cationradical formed is not inevitably stable. Under anodic reaction conditions, it can expel thesecond electron and give rise to a dication or lose a proton and form a neutral (free) radical.The latter can either be stable or complete its life at the expense of dimerization, fragmen-tation, etc. Nevertheless, electrochemical oxidation of aromatic hydrocarbons leads tocation radicals, the nature of which is reliably established (Mann & Barnes 1970, Chap. 3).
In the majority of cases, the electrochemical generation of organic cation radicalstakes place in an ampoule lowered into an ESR cavity. Sometimes, however, exhaustive ex-ternal generation and use of a flow system allow one to obtain an ESR spectrum that is farbetter resolved (see, for example, Seo et al. 1966).
Electrochemical methods are very useful in structural studies but are barely applica-ble for preparative aims. The cause is the limited stability of cation radicals. It is difficultto do low-temperature preparative electrolysis, and the main problem is to dispose of thelarge amount of heat generated during the electrode work. That is, not much current can bepassed through an ordinary-sized electrode without generating too much heat. When po-tential and temperature control are necessary, only small quantities of a material can be ob-tained in a reasonable period of time. When potential and temperature control are not nec-essary, as in Kolbe electrolysis, anodic oxidation is indeed useful as a preparative method.
As to chemical routes to cation radical generation, the following oxidants deserve tobe mentioned: concentrated (98%) sulfuric acid (Carrington et al. 1959; Hyde & Brown1962; Carter 1971), persufate (Minisci et al. 1983), iodosobenzene bis(trifluoroacetate)(Alberti et al. 1999), chlorine dioxide (Handoo et al. 1985; Sokolov et al. 1999). The fol-lowing metal ions found application: Tl(III) (Elson & Kochi 1973; McKillop & Taylor1973), Mn(III) (Andrulis et al. 1966), Co(III) (Kochi et al. 1973), Ce(IV) (Norman et al.1973), Ag(I) and Ag(II) (Nyberg & Wistrand 1978), Pd(II) (Eberson & Wistrand 1980).Methods of cation radical generation have also been developed in which oxidants are SbCl5(Ishizu and co-workers 1973), AlCl3 (Forbes & Sullivan 1966), I2 (Stamirez & Turkevich1963), Pb(OAc)4 (Elbl-Weiser et al. 1989), and XeF2 (Shaw et al. 1970). Potassium 12-tungstocobalt(III)ate was also described as a strictly one-electron oxidant for organic sub-stances (Eberson 1983; Baciocchi et al. 1993).
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

This list of oxidants should be accompanied by a consideration of the regularities oftheir actions; see Chapters 1, 4, and 6. Here, the following three oxidants ought to be em-phasized: Ar3N�.
SbCl6�, NO�, and Ag�. Tris(bromophenyl)amine is now available com-
mercially; oxidized with SbCl5 it forms the reagent (see Section 1.7.10). The NO�PF6� and
NO�BF4� salts are also available; they are especially convenient when the aim is the isola-
tion of oxidized products. This is because the reduction product, NO., is a gas that is
evolved (see Section 1.7.9). The Ag� ion is used whenever the compounds are easy enoughto oxidize: the reduced form is separable solid silver (often as a mirror on the reaction ves-sel walls). Just like NO�, Ag� is characterized by a high sensitivity of its standard redoxpotential to the nature of the solvent. (For selecting chemical oxidants and reductants, the1996 review by Connelly and Geiger can be recommended.)
In some cases, cation radicals are formed from neutral organic molecules upon theaction of neutral organic acceptors, such as tetracyanoethylene, tetranitrofluorenone,quinones, and free radicals—aroxyls, nitroxyls, and hydrazyls.
As shown, methods for cation radical generation are very various. Each case needsits own appropriate method. The set of such methods is continuously being supplemented.New methods not claiming to be general ones permit one to prepare nondescribed cationradicals, to strive for more resolved spectra, and so on.
For one example, it was very difficult to prepare the cation radicals of benzenederivatives with the strong acceptor groups. Recently, some progress has been achieved,thanks to the employment of fluorosulfonic acid, sometimes with the addition of antimonypentafluoride, and lead dioxide (Rudenko 1994). As known, superacids stabilize cationicintermediates (including cation radicals) and activate inorganic oxidants. The method men-tioned is effective at �78°C. However, �78°C is the boundary low temperature, becausesolution viscosity increases abruptly. This leads to anisotropy of a sample and sharply de-teriorates an ESR spectrum.
Another example involves the preparation of cation radicals from aromatic hydro-carbons. In concentrated sulfuric acid or in a mixture of antimony pentachloride withmethylene chloride, polyacenes give cation radicals with ill-resolved ESR spectra (Lewis& Singer 1965). Employment of molten antimony trichloride at �80°C results in improvedresolution, diminution of line widths up to the ideal value of 0.005–0.008 mT (Buchananet al. 1980). Under these conditions, even naphthalene gives the cation radical, with an ESRspectrum of very good quality. The mentioned spectrum surprisingly describes the naph-thalene cation radical in its monomeric (not dimeric) form. An essential peculiarity of themethod is its special opportunity to regulate the oxidation capacity of SbCl3: It is sufficientto add several mole percents of an acceptor of chloride ions, e.g., AlCl3, or a donor of theseions, e.g., Me4NCl.
Because the oxidation process is reversible, a measure of its reversibility is the yieldof antimony in the zero-valent state:
SbCl3 � 3ArH ⇔ 3(ArH)�.� 3Cl� � Sb0
Bringing Cl� out of the reaction sphere (by the addition of AlCl3) leads to an increasein (ArH)�.
concentration. Getting Cl� into the reaction sphere (by the addition of Me4NCl)leads to a decrease in (ArH)�.
concentration. As seen, the equilibrium between starting sub-stances and electron-transfer products can be very important, especially for homogeneousreactions. The depicted equilibrium may be more complex in reality and include polyanti-monium polyhalides, but the most important peculiarity is the possibility of regulating theoxidative capacity, as it is shown here.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

A relevant example is a method of indirect generation of cation radicals. Mesoionic5,5-azinobis(1,3-diphenyltetrazole) and related mesoionic compounds give cation radicalson oxidation with lead tetraacetate. The reaction proceeds in the presence of sodiumtetrafluoroborate. The tetrafluoroborate cation radical salt is stable and can even be puri-fied via column chromatography on silica gel and stored under air for several months with-out appreciable decomposition (Araki et al. 1999). The cation radicals of N,N-dimethyl-diazines are usually produced either by reduction of the corresponding diquarternary salts(Schulz et al. 1988) or by oxidation of the parent diazine (Soos et al. 1977).
Lucarini and co-authors (1994) have elaborated a one-pot method for generating suchcation radicals from the corresponding aromatic bases via the action of dimethyl sulfate,Me2SO4, as a methylating agent and zinc powder as reductant. The reaction proceeds inbenzene according to the Scheme 2-3.
To conclude this section, one indirect method of cation radical generation should bementioned. Organic molecules spontaneously form corresponding cation radicals upon in-clusion within activated zeolites (Yoon & Kochi 1988; Yoon 1993; Pitchumani et al. 1997).Zeolites are crystalline alumosilicate minerals that are widely used as sorbents, ion ex-changers, catalysts, and catalyst supports. The cation radicals are formed along with car-bocations, but the carbocation generation can be diminished by selection of the appropriatezeolite and the manner of activation. For instance, Ca(Y) zeolite activated at above 400°Con a high-vacuum line was found to be ideal for generation of the cation radicals from di-arylethenes in cyclohexane solution, even in the absence of light, with minimum interfer-ence from carbocations. Oxygen (not necessarily the free oxygen) plays a crucial role in thegeneration of the cation radicals within the zeolites (Pitchumani et al. 1997).
2.3 EQUILIBRIA IN LIQUID-PHASE ELECTRON-TRANSFERREACTIONS
Let us consider a redox reaction of the following type:
A�.� B ⇔ A � B�.
In this reaction, A�. acts as a one-electron donor and transforms into A. At the same mo-ment, B gives rise to B�.. If the reaction is reversible, the equilibrium is the sum of the fol-lowing two processes:
A� ⇔ A � e and B � e ⇔ B�.
Electrochemically, both processes are expressed by their standard potentials E0(A)and E0(B). In order to consider some polarographic studies, let us mention that the polaro-
SCHEME 2-3
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

graphic half-wave potentials, E1/2(A) and E1/2(B), can, under certain conditions, be used in-stead of E0(A) and E0(B). If both processes are one-electron in nature and they are fast andreversible, their E1/2 values are equal to those of standard redox potentials (referring to theconditions used in a given electrochemical experiment). Values of E1/2 show how easily Aor B accepts an electron, i.e., what the A or B acceptor/donor ability is.
There is a special equation to count equilibrium constants (K) using Faraday’s con-stant (F) and E1/2 values of one-electron reversible polarographic waves for A and B (Gen-naro et al. 1988):
ln K �
The equation can give the E1/2(A) value if E1/2(B) and K are known.This estimation has been made for a reaction between the anion radical of tetra-
cyanoquinodimethide (TCNQ) and 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ) (Iida &Akamatu 1967):
(TCNQ)�.� DDQ ⇔ TCNQ � (DDQ)�.
The equilibrium constant of the reaction was determined spectrometrically in acetonitrile.Having K and E1/2(DCNQ) as the known values, E1/2(TCNQ) was determined to be 38 mV.The experimental (polarographic) value was 40 mV. As seen, the calculated and experi-mental values turned out to be close.
Hence, equilibrium constants of homogeneous electron-transfer reactions between(A)�.
and B are evidently connected to the differences in reduction potentials of A and B.This connection reflects a definite physical phenomenon. Namely, if two redox systems arein the same solution, they react with each other until a unitary electric potential is reached.For the transfer of only one electron at room temperature, the following simplified equa-tion can be employed:
ln K � 2.3[E1/2(A) � E1/2(B)] � 0.059
The applicability of this simple equation has been checked for one particularly im-portant case of the electron exchange between species belonging to quite different classesof chemical compounds. Cyclo-octatetraene dipotassium as a donor and -ferrocenylacry-lonitrile as an acceptor react in THF. The reaction is reversible, and the presence of all fourof the components depicted in Scheme 2-4 has been proved (Todres 1987).
F[E1/2(A) � E1/2(B)]���
RT
SCHEME 2-4
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

According to the yield of C8H8 and the concentrations of the four components, theequilibrium constant of the reaction was determined as 4 � 10�2. The value calculatedfrom the potential difference was 4.4 � 10�2. Consequently, there is coincidence betweenthe calculated equilibrium constant based on the electrode potentials and the equilibriumconstant determined from the liquid-phase experiment. The liquid-phase and electrode pro-cesses are similar.
In all of the examples considered, E1/2 of the acceptor was much more negative thanthat of the donor. However, in liquid phase one-electron transfer from a donor to an accep-tor can proceed even with an unfavorable difference in the potentials if the system containsa third component, the so-called mediator. The mediator is a substance capable of accept-ing an electron from a donor and sending it instantly to an acceptor. Julliard and Chanon(1983), Chanon, Rajzmann, and Chanon (1990), and Saveant (1980, 1993) developed re-dox catalysis largely for use in electrochemistry. As an example, the reaction of ter-achloromethane with N,N,N,N-tetramethyl-p-phenylenediamine (TMPDA) can be dis-cussed. The presence of p-benzoquinone (Q) in the system provokes electron transfer(Sosonkin et al. 1983). Because benzoquinone itself and tetrametyl-p-phenylenediamineinteract faintly, the effect is evidently a result of redox catalysis. The following schemes re-flect this kind of catalysis:
CCl4 � TMPDA → (TNPDA)�.� Cl� �
.CCl3
TMPDA � Q → (TMPDA)�.� Q�.
Q�.� Q�. ⇔ Q � Q2�
Q2� � CCl4 → .CCl3 � Cl� � Q�.
Q�.�
.CCl3 → Q � �CCl3
�CCl3 � H� → CHCl3
The observed catalytic effect can be explained by comparing the redox potentials of thereacting species. The potentials of the reversible electron transfers from Q to Q�.
and then fromQ�.
to Q�.are right in the middle of the gap between the potentials of CCl4 and TMPDA. This
leads to dividing the gap into three components so that each one can be gotten over readily.One curious case of the effect of light on electron-transfer equilibrium involves the
reduction of �, -di(t-butyl)stilbene with potassium in DME. The reaction leads directly toa diamagnetic dianion; a solution of this dianion remains ESR silent unless subjected to ul-traviolet irradiation by a Hg/Xe lamp. The anion radical of �, -di(t-butyl)stilbene thenformed from the dianion by the loss of an electron. The electron reverted within 5–10 minafter ultraviolet irradiation was turned off, transforming the anion radical into the dianion(Gerson et al. 1996). This case deserves to be clarified. Maybe the light effect consists sim-ply in singlet–triplet transformation of the dianion, with the formation of some more or lessstable biradical state of the dianion, which possesses two unpaired electrons and can evenbe a paramagnetic one.
2.4 ELECTROCHEMICAL METHODS VERSUS CHEMICAL METHODS
To date, the results of chemical and electrochemical studies generally have been consideredin isolation. The purpose of this section is to present a digest of data obtained via the twomethods in an effort to demonstrate that a great deal may be gained by both chemical andelectrochemical means rather than solely by one method or the other, as is the currently ac-cepted practice.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

In the electrochemical technique, the electrode provides the source (reduction) orsink (oxidation) for electrons. Variation of the applied potential provides the driving forcethat enables the redox reaction to occur. Organic depolarizers diffuse toward the electrodesurface until they are sufficiently close to the electrode (the double-layer region) to acceptelectrons from the electrode and then to diffuse away from the electrode surface and to be-come involved in the bulk solution.
In chemical redox reactions, organic ion radicals are formed at the final stage ofdonor–acceptor (D-A) interactions:
D � A ⇔ D�, A� ⇔ D�., A�. ⇔ D�.
� A�.
All bimolecular reactions include encounter complexes, which involve as much elec-tron transfer as needed to stabilize the system, the amount varying widely depending onwhat the two molecules are. When there is a lot of charge transfer, it is certainly reasonableto call the complex a charge-transfer complex. The equilibrium constants for complex for-mation is supposed to vary smoothly from small to large, depending on what the moleculesreacting are. It is worthwhile to consider the products of donor–acceptor interaction,namely, charge-transfer complexes and ion radicals.
Regarding electrochemical reactions, ion radicals are well-known primary products.Charge-transfer complexes are more usual for chemical processes, but they have their ownanalogy in electrochemistry: The formation of a charge-transfer complex is in a certain sensesimilar to the formation of an electrode–substrate complex. As an example, aromatic, espe-cially polycyclic, hydrocarbons are adsorbed on platinum and other metal anodes with ad-sorption enthalpies in the range from �25 to �42 kJ�mol�1. As calculated, this adsorptionmust occur with the �-electron contour parallel to the surface. The type of bonding involvedwould be of the same kind as that in a �-complex. The distance between the hydrocarbonmolecule and the surface would probably be of the same magnitude as that in �-donor–�-acceptor complexes, or around 0.35 nm (Baizer & Lund 1983). The adsorption of sup-porting electrolyte or other additives can create a special microenvironment in the reactionlayer near the electrode that can favor distinct pathways. Thus the use of tetra-alkylammo-nium ions as supporting electrolyte depletes the electrode surface of a solvent and sometimesencourages the products of electron transfer to react, with no solvent participation.
On the other hand, the intermediates generated at the surface diffuse into the bulk so-lution. Owing to their high reactivity, the intermediates react in a fairly thin reaction layerthat adheres to the electrode, and thus their concentration is higher than in homogeneousreactions, where they are spread uniformly over the medium. This can influence the natureof the product and its distribution.
The fundamental electrochemical event, i.e., electron transfer, occurs at the electrodesurface. Peculiarities of electrochemical reactions include an electrical field that in a spe-cial way complicates the phenomena of adsorption and desorption at the surface. The firstlayer of the solution, which is in contact with the electrode, possesses a specific structure.It is important for charged particles that the orientation of medium molecules in the vicin-ity of the electrode produces a decrease in dielectric permeability in the compact part of thedouble layer (Damaskin & Kryshtalik 1984).
Substrate and/or intermediate species adsorb on an electrode surface and orient them-selves so that their least hindered sides face the electrode, unless there is another effect,such as polar one. An electrode interface has a layered structure in which a nonuniformelectric field (some slope of potential) is generated by polarization of the electrode. An ex-tremely strong electric field of around 108 V/cm in the innermost layer might cause a vari-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

ety of polar effects. Since not only the electron transfer step but also adsorption and someof the chemical steps involved in an electrode reaction take place in the layer, the wholeprocess should be strongly influenced by polar factors. The orientation of polar adsorbedspecies, such as ion radicals in particular, is electrostatically influenced, and consequentlythe stereochemistry of their reactions is also controlled by this kind of electrostatic factor.
All these phenomena were summarized in several monographs. The collective vol-ume edited by Baizer and Lund (1983) is devoted to organic electrochemistry. This editionis closer to the scope of our consideration than its latest version of 2001 (edited by H. Lundand Hammerich; these editors have changed the authors of the chapters included).
As for chemical reactions, the oxidation-reduction (redox) reactions in homogenousmedium (i.e., in the bulk of the solution) have been experimentally studied with proper in-tensity only in the last two decades. There has been some development of the bulk reac-tions. However, as before, a comparison of one and the same compound in chemical andelectrochemical electron-charge-transfer reactions is still of current interest. Such a com-parison is made in this section. The examples offered are intended to invoke novel inter-pretations or to discover new colors in pictures that have already been drawn.
2.4.1 Charge-Transfer Phenomena
It is worth noting that there is a significant difference between a conversion of one and thesame substrate under one-electron (electrode) transfer and in charge-transfer complexes(homogeneous medium).
Anodic oxidation of tetraphenylethylene at a platinum electrode leads to the productof cyclization, namely, to 9,10-diphenylphenanthrene (Stuart & Ohnesorge 1971). The in-tramolecular coupling reaction does not occur when diphenylethylenes, i.e., stilbene and itsmethyl derivatives, are electrolyzed under the same conditions (Stuart & Ohnesorge 1971).This difference in the anodic behavior of these substances was attributed to the low stabil-ity of the cation radicals of stilbene and its methyl derivatives in comparison to the cationradicals of tetraphenylethylene. The participation of the cation radicals in cyclization oftetraphenylethylene has been unequivocally proved (Svanholm et al. 1974; Steckhan1977).
In homogeneous conditions, when p-chloranil plays the role of electron acceptor, 4-methylstilbene (�-phenyl- -tolylethylene) can, however, be cyclized and converted into3-methylphenanthrene. The reaction takes place via formation of a charge-transfer complexat a very moderate temperature (36°C) and does not require light radiation (Todres, Dyusen-galiev, Buzlanova et al. 1990). Minus details, Scheme 2-5 depicts the transformation.
The behavior of 4,4-dimethoxystilbene on the electrode and with p-chloranil in so-lutions was compared (Todres & Ionina 1992). It was found that the cis and trans isomersof the compound form different charge-transfer complexes with chloranil that are unlike incolor and can be converted; conversion was found to be in the cis-to-trans direction. Re-verse conversion is not observed, and cyclization is also not detected (which takes place inthe case of 4-methylstilbene) (Scheme 2-6).
Formation of charge-transfer complexes was observed in homogeneous solutions.Because of the limited solubility of chloranil, the reaction was performed in a boiling sol-vent. In hexane and methylene chloride (b.p.’s are 69 and 40°C, respectively), the degreeof the cis-to-trans conversion was 50 and 30%, respectively. No conversion was observedin the absence of chloranil when the cis-dimethoxystilbene was kept in those solvents at thetemperatures noted. In the case of benzene (b.p. 80°C), no conversion was observed. This
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

SCHEME 2-5
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

SCHEME 2-6
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

is perhaps because benzene competes with dimethoxystilbene in binding with chloranil ina charge-transfer complex much more successfully than the molecules of the other solventsused. This hypothesis is totally logical if we assume that the chloranil acceptor binds in acharge-transfer complex with dimethoxystilbene due to the benzene ring and not becauseof the ethylene bond.
During oxidation on a platinum anode, the same 4,4-dimethoxystilbene yields thecation radical at the first step. These cation radicals have a sufficient lifetime and dimerizeeven in the presence of nucleophiles (Parker & Eberson 1969; Steckhan 1978; Burgbacher& Schaefer 1979):
2 AnCHBCHAn � 2e → 2 (AnCHBCHAn)�. → �CH(An)CH(An)CH(An)(An)CH�
�CH(An)CH(An)CH(An)(An)CH� � 2 Nu� → NuCH(An)CH(An)CH(An)(An)CHNu
An � 4-MeOC6H4, Nu � OH, etc.
Another example concerns the initial electronic reduction of �-nitrostilbene (Todres,Dyusengaliev, & Ustynyuck 1982; Todres, Dyusengaliev, & Sevast’yanov 1985; Todres &Tsvetkova 1987; Charoenkwan and others, in preparation). The reduction develops as inprocess (a) in Scheme 2-7 if the mercury cathode as well as the cyclooctatetraene dianionare electron sources, and as in process (b) if the same stilbene enters the charge-transfercomplexes with bis(pyridine)-tungsten tetra(carbonyl) or uranocene. For (b), the charge-transfer bands in the electronic spectra are fixed. So the mentioned data reveal a great dif-ference in electrochemical and chemical reduction processes (a) and (b).
The difference between the last two reactions may be also considered in terms of thecomplete electron transfer in both cases. If the �-nitrostilbene anion radical and the metal-locomplex cation radical are formed as short-lived intermediates, then the dimerization ofthe former becomes doubtful. The dimerization under electrochemical conditions may be aresult of the increased concentration of reactive anion radicals near the electrode. This con-centration is simply much higher in the electrochemical reaction because all of the stuff isbeing formed at the electrode, so there is more dimerization. Such a difference betweenelectrode and chemical reactions should be kept in mind too.
Interestingly, treatment of �-nitrostilbene in a water–ethyl acetate mixture (the two-phase system) by the cation radical from N,N-dioctyl-4,4-bipyridinium leads to productsderived from the nitro-group reduction (Tomioka et al. 1986).
Being involved in a charge-transfer complex with N,N-dimethylaniline, cis-�, -dini-trostilbene undergoes a conversion into its trans form with no changes in the nitro groups(Todres et al. 1986) (Scheme 2-8).
One-electron polarographic reduction of �, -dinitrostilbene yields an anion radicalthat is stabilized in a nitronic form with a carboradical center. These radicals possess an en-hanced electron affinity and are prone to capture of the second electron at the first-wave po-tential, with the formation of a stable dinitronic dianion. It is impossible to stop the cathodicreduction at the one-electron step (Scheme 2-9).
2.4.2 Template Effects
A similar situation occurs during electrochemical reduction of azoxybenzene: At the firstpolarographic wave, azoxybenzene forms azobenzene, which is more readily reduced thanis the initial substrate; the first and the second one-electron transfers are merged. In the ab-sence of protons, a single polarogaphically irreversible four-electron wave is observed.Whatever the nature of the supporting electrolyte (with cations of Bu4N� or K�), only a
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

SCHEME 2-7
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

combined reduction process takes place (Lipsztain et al. 1974). Scheme 2-10 illustrates thiselectrochemical reaction (the azoxybenzene cis structure was chosen arbitrarily).
The behavior of the same azoxybenzene was studied under homogeneous condi-tions—when the dipotassium salt of cyclo-octatetraene dianion (C8H8K2) acts as a “dis-solved electrode.” In this case the reduction of azoxybenzene stops at the first stage, that is,after the transfer of one electron only (Todres, Avagyan, & Kursanov 1975). This producesthe azoxybenzene anion radicals, which are not reduced further despite the presence of theelectron donor in the solution. The ESR method does not reveal these anion radicals, al-though one-electron oxidation by phenoxyl radicals quantitatively generates azoxybenzeneand produces the corresponding potassium phenolate molecules in a quantitative yield.Treatment with water leads to the 100% yield of azobenzene (Scheme 2-11).
The logical conclusion reached from consideration of these data is as follows. In liq-uid phase (THF), under the conditions of a regular volume continuum without gradients ofconcentration and potential, all anion radicals of azoxybenzene can be stabilized just afterformation due to their bonding with potassium cations. This yields the coordination com-plex. The complex is diamagnetic, and therefore the azoxybenzene anion radicals cannotbe revealed by ESR spectroscopy (Scheme 2-12).
SCHEME 2-8
SCHEME 2-9
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

SCHEME 2-10
SCHEME 2-11
SCHEME 2-12
SCHEME 2-13
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The diamagnetic complex is not reduced further by the cyclo-octatetraene dianion.This prevents conversion of the azoxybenzene anion radicals into azodianions. Potassium-cations play an important role in this limitation of the reduction process, which, in general,proceeds easily (the electrode reduction takes place as a merged four-electron wave; seeearlier). It is very probable that potassium cations serve as bridges for the excess electrons,placing their low-lying orbitals at such �-electron disposal. There are theoretical (Meccoziet al. 1996) and experimental (De Wall and others 1999) proofs of the potassium cation in-teraction with �-electrons.
The removal of potassium cations from the reaction sphere can be accomplished bytheir binding with 18-crown-6-ether (Scheme 2-13). The removal of potassium cationsmakes the results of the liquid-phase and electrode reactions similar. In the presence of thecrown ether, the liquid-phase process also leads to the azodianion. The azodianion was in-deed identified via benzidine after protonation and rearrangement (Scheme 2-14).
Of course, the azoxybenzene anion radical can form a pair with the potassium cationduring an electrode reaction too. This stabilizes the one-electron reduced form and hamperselectron back-transfer. The electrode reaction is favored and proceeds further. Saveant(2001) gave complete thermodynamic reasons for this phenomenon.
2,5-Di(thiocyano)thiophene also exhibits a different behavior at a mercury-droppingelectrode and in the case of treatment by dipotassium salt of cyclooctatetraene dianion;THF is the same solvent in both cases (Todres, Furmanova et al. 1979). The reaction be-tween the di(thiocyano) derivative and C8H8K2 taken in equimolecular amounts leads to thepotassium salt of 2-mercapto-5-thiacyanothiophene (potassium mercaptide), potassiumcyanide, and cyclooctatetraene (see Scheme 2-15). Potassium mercaptide is stable in THF.It was characterized through a monobenzoyl derivative. If water is added to THF, polysul-fide is formed. X-ray analysis has unambiguously proven the cyclic structure of polysifide.The same product was formed in experiments when enhanced concentrations of thereagents were used. The cyclization most probably proceeds via intermediate disufides.
Once again, the potassium cation acts as a coordination center that ensures the tem-plate organization of the anion fragments during the homogeneous reduction. The cavitydiameter of the obtained multithiaheterocycle and the doubled radius of the potassiumcation match.
When 2,5-di(thiocyano)thiophene reacts with C8H8K2 in the presence of 18-crown-6 ether, the other conditions being equal, the product formed was found to be a linear poly-
SCHEME 2-14
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

sulfide. The crown ether combines with the potassium cation; the condensation changes itsdirection and leads to the formation of a linear rather than a cyclic product (Scheme 2-16).
The linear product is obviously formed according to the sequence 2,5-di(thio-cyano)thiophene → potassium 2-mercaptido-5-thiacyanonothiophene → tristhiomaleic an-hydride ⇔ thiophene-2,5-disulfenyl biradical (the diradical valence tautomer) → the de-picted linear polymer, in which the thiophene rings are connected via dusulfide bridges. Itwas recently confirmed that tristhiomaleic anhydride is unstable and polymerizes just at themoment of its formation (see Paulssen et al. 2000 and Ref. 15 therein).
At a mercury electrode, 2,5-di(thiocyano)thiophene undergoes reduction, with thecleavage of both SCN groups within one four-electron wave. In the case of electrolysis, alinear polysufide can be obtained (Scheme 2-17). As seen from the scheme, the electrodeand crown-modified homogeneous reactions give the same product (although their mecha-nisms are obviously different). In this case too, the noncrown reaction in the THF solution,performed at high concentrations of the reagent, does not result in formation of the linearpolysulfide. Hence, the possibility of the cyclic S–S bond cleavage by the intermediary thi-olate anion can be ruled out. The exhaustive reduction of 2,5-di(thiocyano)thiophene inparticular can be influenced by the thiocyano-group affinity for the mercury of the elec-trode. The waves observed, however, have diffusion characteristics. So this example,though not connected with the discrete formation of ion radicals, has two interesting fea-tures: On the one hand, it demonstrates the significance of the template effects in organic
SCHEME 2-15
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

electron-transfer reactions that proceed under the conditions that favor involving the alkalimetal cation. On the other hand, it suggests that just diffusion waves may sometimes reflectreduction of depolarizers after their adsorption on electrodes. This phenomenon deservesseparate consideration, because it is very unusual for organic electrochemistry. As com-pared with homogeneous electron-transfer reactions, such “hidden” adsorption alters thegeneral picture of ion radical formation.
2.4.3 “Hidden” Adsorption Phenomena
A comparison of the reactions of specially selected substrates at the electrode with those inthe liquid phase reveals differences associated with the influence of the electrode. These dif-ferences cannot be established by direct analysis of the polarograms if there are no adsorp-tion waves of the test substances. Let us consider the aromatic derivatives of divalent sulfur.
SCHEME 2-16
SCHEME 2-17
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The reduction of the benzenosulfene derivatives is independent of the nature of theelectron donor and leads to the phenylthiolate ion (Todres 1980) (Scheme 2-18). However,the analogs containing the nitro group exhibit different behavior in reactions at the surfaceof the electrode and upon reductions by the cyclo-octatetraene dianion (Scheme 2-19).
The addition of mercury in reaction mixtures of nitroarylsulfenates with C8H8K2 inTHF did not change the reaction results. Disintegration of the XSC6H4NO2
�K� ion pairs(controlled via ESR) does not affect the reaction results either.
The polarographic reduction of the nitro analogs proceeds with the primary cleavage ofthe sulfur-containing group (two-electron irreversible diffusion wave). The primary productsof the homogeneous reaction are stable anion radicals that can exist under air-free conditionsfor a long time. The cyclo-octatetraene dianion, taken in excess, is capable of reducing the sul-fur-containing group too. However, the primary and detectable product of the homogeneousreaction is the anion radical, which is not detected in reduction at the electrode. Electrochem-ical studies were carried out in aprotic solvents, and no evidence for the adsorption of the sul-fur-containing substances on the electrode was found. Only diffusion waves were observed.
The difference found between the homogeneous and heterogeneous reduction pro-cesses can be attributed to a specific interaction of the sulfur-containing groups with thematerial of the electrode.
Some literature supporting this point of view should be mentioned. An exchange re-action of the two diorganyl disulfide moieties results in the formation of the assymetricdisulfide RSSR (R and R are different alkyls):
RSSR � RSSR → 2RSSR
SCHEME 2-18
SCHEME 2-19
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

This well-known reaction is strongly accelerated by mercury metal (Hoffmann et al.1999).
Polarographic studies of diphenyl disulfide or methylphenyl sulfenate (Persson &Nygard 1974; Persson & Lindberg 1977) reveal that the interaction of these substances withmercury precedes the electron transfer in dimethylformamide. Namely, both PhS-SPh andPhSH are reduced cojointly, in the framework of the same polarographic wave. It was alsoshown that E1/2 values of the first wave of PhS-SPh and the second wave of PhS-Hg-SPhcoincide. Therefore, the formation of PhS-Hg-SPh or PhS-Hg-Hg-SPh can really precedethe electron-transfer reaction from the mercury-dropping electrode to PhS-SPh.
There are other examples of organic calomel formation during polarographic reduc-tion (Reutov & Butin 1975). The lifetimes of dialkyl and diaryl calomels do not exceed10�2 and 10�4s, respectively.
The reduction of arenesulfenates at the mercury electrode can proceed via the for-mation of intermediate arenethiomercuric derivatives. Such derivatives are reduced just af-ter their formation and more easily than the initial arenesulfenates. In line with this argu-ment it logically follows that the limiting currents of the polarographic waves woulddepend solely on the diffusion of the substances at the electrode. In fact, diffusion currentshave been observed experimentally. Attempts at using, not mercury, but platinum orgraphite glass electrodes were ineffective because of the nonexpressed character of thewaves. Experiments with electrolysis on mercury (a preparative scale) confirmed the gen-eral conclusion (Todres 1988).
As an analogous example, the behavior of sulfonium salts can be mentioned. At amercury electrode, both trialkyl (Colichman & Love 1953) and triaryl (Matsuo 1958) sul-fonium salts can be reduced, with the formation of a sulfur-centered radical. This radical isadsorbed at the mercury surface and then eliminates the carboradical. The latter, just afterits abstraction, captures one more electron and transforms into carboanion. This is the finalstage of reduction. The mercury surface cooperates in both successive one-electron steps(Luettringhaus & Machatzke 1964) (Scheme 2-20). This scheme is important for the prob-lem of hidden adsorption, but it cannot to be generalized in the sense of a stepwise vs. concerted mechanism of dissociative electron transfer. As shown, the reduction of some
SCHEME 2-20
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

sulfonium salts does follow the stepwise mechanism, but others are reduced according tothe concerted mechanism (Andrieux et al. 1994).
It is worthwhile underlining the selectivity of carbon–sulfur bond splitting: Themethyl group is preferentially cleaved, and in the case of (CH3)2(PhCH2)S�Br� the quan-tity of CH4 formed is ten times higher than that of PhCH3 (Luettringhaus & Machatzke1964). The affinity of the radical for mercury is well known, and small-size radicals (CH3
.)
must be adsorbed more strongly than large-size radicals (PhCH.2). Scheme 2-20 illustrates
this peculiarity. In this scheme, the incipient radical is not exactly the more steadfast one(benzylic stabilization), but the one more comfortably placed on the electrode surface(small-size effect).
In general, for chemical adsorption a significant overlap between substrate and elec-trode orbitals is needed; i.e., a weak chemical bond has to be established. This implies thatthere must be an orientation effect, depending on the symmetry of orbitals involved, andthat the substrate molecule must be fairly close to the electrode surface.
2.4.4 Stereochemical Phenomena
It has already been mentioned that an electrode reaction implies some orientation effect be-cause a substrate molecule must be fairly close to the electrode surface. The following pairsof the cis and trans isomers were reported to exhibit identical reduction potentials: 1,2-dimethyl-1,2-diphenylethylenes (Weinberg & Wienberg 1968), 1,2-bis(4-cyanophenyl)-1,2-bis(4-methoxyphenyl) ethylenes (Leigh & Arnold 1981), 1,2-bis(4-acetylphenyl)-1,2-diphenylethylenes (Wolf et al. 1996). In particular, the trans isomer of 1,2-dimethyl-1,2-diphenylethylene is coplanar and the cis isomer is noncoplanar. However, both isomersare oriented in an identical manner within the electrode space and electric field (Horner &Roder 1969). The energy needed for such an orientation is not markedly reflected in thevalue of a potential.
Analogously, 1,2-dicyano-1,2-diphenylethylene, which is free from steric strains,and its strained isomer 1,1-dicyano-2,2-diphenylethylene are reduced at practically thesame potential (Ioffe et al. 1971; Todres & Bespalov 1972). In DMF with the support ofEt4NI, the reversible two-step one-electron reductions are characterized by the followingpotentials (mercury pool as a reference electrode): �0.48 and �0.98 V for the 1,2-dicya-noethylene, �0.50 and �1.07 V for the 1,1-dicyano isomer. Thus, electrochemical reduc-tion does not fix the difference in isomer structures.
This difference is clearly displayed in homogeneous electron transfers (Ioffe & To-dres et al. 1971; Todres & Bespalov 1972). When cyclo-octatetraene dipotassium is usedas an electron donor in a THF solution, the mentioned isomers react in an unlikely way. The1,2-1,2-isomer takes the dianion’s electrons off completely and irreversibly (Scheme 2-21).On the same conditions, the reaction of 1,1-2,2-isomer is reversible (Scheme 2-22). Withrespect to the ethylene bond plane, the phenyl rings deviate only 30° in the 1,2-1,2-isomerand 90° in 1,1-2,2-isomer (Wallwork 1961; Todres & Bespalov 1972).
SCHEME 2-21
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Properly speaking, the conjugation chain includes all the structure elements in theformer isomer. In the latter isomer, at least one of the phenyl group is led out of the conju-gation. According to calculations (Todres & Bespalov 1972), the aforementioned differ-ence in bend angles is supposed to be the same in cases of corresponding dianions. There-fore, such an explanation of the reversibility for the 1,1-2,2-isomer is reasonable: Becauseof steric hindrances in the geminal node, the conjugation chain is broken (is shortened). Itleads to some diminution in the degree of excess electron delocalization. This means thatthe stability of the electron-transfer products decreases. The electron-transfer reaction be-comes less favorable and the degree of transformation declines.
Hence, the space strain is indicative of the stability of the electron-transfer products.Electrode reactions fail to reveal such an effect. In liquid-phase processes, however, this ef-fect plays a decisive role.
As Baizer and Lund’s book (1983) underlines (p. 907):
When the stereochemistry of an electrochemical reaction is discussed it isnormally assumed that the geometry of the molecule in question remains essen-tially unchanged until bond breaking or bond transformation occurs. It should berecognized, however, that an electron transfer might entail significant changes inthe geometry and bond strengths of a molecule with concomitant implications forthe stereochemistry of its reaction (Todres 1974). Unfortunately, this importantarea has not been extensively investigated.
From this point of view, a brief comparison of the acyloxylation of cis or trans stil-benes under electrochemical and chemical conditions is also relevant. Anodic oxidation (Pt)of cis or trans stilbenes in the presence of acetic or benzoic acid gives predominantly mesodiacylates of hydroxybenzoin or, if some water is present, threo monoacylate. None ofstereoisomeric erythro monoacylate and rac diacylate was obtained in either case. There wasno evidence of isomerization of cis-to-trans stilbene under the electrolytic conditions em-ployed (Mango & Bonner 1964; Koyama et al. 1969). The sequence of reaction steps inScheme 2-23 was proposed. Adsorption-controlled one-electron oxidation of the substratetakes place. Then an adsorption-controlled rotation proceeds of cis stilbene monoadduct intothermodynamically more stable trans benzoxonium ion. The trans benzoxonium ion is thecommon intermediate for conversions of both cis and trans stilbenes and, of course, for allthe final products (Scheme 2-24). Hence, oxidized molecules of stilbene are, at the electrode,involved in a reaction with acylate ions. There is no passing into the solution volume, withthe following electron being exchanged there with unoxidized molecules of stilbene.
The chemical oxidation of cis or trans stilbenes was also investigated (Vinogradov etal. 1976). The oxidants were cobalt or manganese acetates, and, in separate experiments,thallium trifluoroacetate. Acetic or trifluoroacetic acid was used as a solvent. The results ofsuch chemical oxidation were considered from the standpoint of the geometry of the re-covered (nonreacted) part of the initial substrate and of the stereoisomeric composition of
SCHEME 2-22
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.
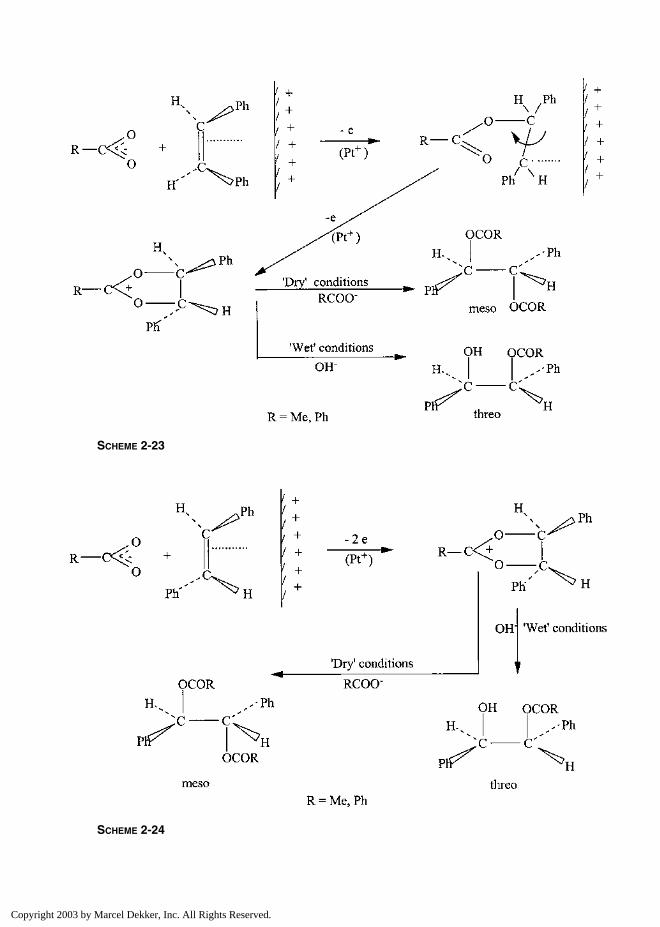
SCHEME 2-23
SCHEME 2-24
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

the products obtained. This allowed the desirable comparison of electrochemical and chem-ical reactions to be made.
By means of ESR spectroscopy, the cation radicals of stilbene have been detected.These cation radicals are accumulated and then consumed in the course of the consecutivereactions. The stereoisomeric composition of the final products appears to be constant andnot to depend on the configuration of the initial substrate. Acetoxylation of the olefinicbond in cis stilbene is almost one order of magnitude slower than in trans stilbene. This ki-netic feature deserves a special explanation, because cis stilbene is less stable thermody-namically than trans stilbene and should react faster. The products obtained are depicted inScheme 2-25.
When the initial compound was trans stilbene, the unconsummated part was recov-ered, with no change in configuration. When cis stilbene was employed as the initialreagent, the recovered olefin was a mixture of trans and cis isomers. Hence, the trans con-
SCHEME 2-25
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

figuration is more favorable for oxidative acetoxylation than is the cis configuration. In ac-cordance with this conclusion the mechanism in Scheme 2-26 can be proposed.
It can be seen that the cation radical of stilbene, but not stilbene itself, is subjected toacetoxylation. Stilbene in trans form yields the trans form of the cation radical, which un-dergoes further reaction directly. Stilbene in cis form gives the cation radical with the cisstructure. Such a cis cation radical at first acquires the trans configuration and only afterthat adds the acetate ion. It is the isomerization that causes the observed retardation of thetotal reaction. It is the absence of adsorption at the electrode surface that allows the nonace-toxylated part of cis stilbene to isomerize and to turn into the more rich stereoisomeric setof final products. To support this point of view, one can mention the cation radical epoxi-dation and cylopropanation of stilbenes. In the aminiumyl ion–catalyzed reactions, cis stil-benes react about 2.5 times slower than trans stilbenes, whereas in electrophilic oxidationsthe cis isomers are more reactive (Kim et al. 1993; Bauld & Yeuh 1994; Mirafzal et al.1998; Adamo et al. 2000).
2.4.5 Formation of Ion Aggregates in Solutions
Organic ion radicals bear charges that must be compensated. In solutions, the ion radicalsexist in the form of salts. Under certain conditions, interaction between the counterions ofthe salt can be very strong. So-called ion pairs are formed. An ion pair is two touching, op-positely charged ions that are held together by the attraction between the unlike charges.Ion-pair formation defines the energies of an ion radical salt and, consequently, the energyof a reaction that leads to ion radical formation. In order to study the role of ion pairs, it isnecessary to investigate electron-transfer reversibility in a solution and to compare the re-
SCHEME 2-26
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

sults obtained with the redox potentials of a donor and an acceptor. As a rule, ion-pairingphenomena define electrode processes too (Baizer & Lund 1983). However, the knownequations for equilibrium calculations cannot take ion pairing into consideration becausethe equations do not contain “ion-pair” terms. One has to rely on experiments, which areable to take into account the equilibrium of electron transfer in solutions.
2.4.5.A Direct Influence on Electron-Transfer Equilibrium
The reaction between cyclo-octatetraene dipotassium and 2,4,6-tri(tert-butyl)nitrobenzenein THF is irreversible. The acceptor gives rise to an anion radical salt. An unpaired electronof the organic anion radical is localized predominantly within the nitro group. The potas-sium cation is coordinated with the “anion radical” nitro group. Such coordination also en-hances this localization. So the reaction leads to a very stable species that is unable to re-verse the one-electron transfer to cyclo-octatetraene. Being a very strong dissociatingsolvent, hexamethyl phosphorotriamide (HMPA) destroys the K� coordination complexwith ArNO2
�., and the reaction becomes reversible (Todres 1980) (Scheme 2-27).
Now let us focus on an important group of ion radical reactions, i.e., recombinations.They may proceed either as disproportionation or as dimerization. It is of interest to com-pare the ion radical and “uncharged” radical recombination. As known, the “uncharged”radicals recombine at zero activation energy. Ion radicals have a dual nature: As radicalsthey are highly reactive, and as ions they attract particles of opposite charge and repel thoseof the same charge. Disproportionation is the interaction of ion radicals of equal signs. Itdepends on two factors: charge-repulsion energy and spin-pairing energy. A decrease incharge-repulsion energy, naturally, promotes recombination. This energy is large whennaked ions react. This energy decreases significantly when tight pairs with counterions areinvolved.
Understandably, the state of ion pairs depends on the nature of the solvent, particu-larly on its dissociating ability. For example, in HMPA (a strong dissociating solvent), thepotassium salt dissociates giving the anion radical of tetraphenylethylene and potassiumcation. In THF (a nondissociating solvent), ion pairs (Ph2CBCPh2)2�, 2K� or(Ph2CBCPh2)�.
, K� are the main particles acting (Czezhegyi et al. 1969). The most im-portant effect of this aggregation between the counterions consists of the diminution of theresulting negative charge of the anion radicals. This means that electrostatic repulsion be-tween these anion radicals is decreasing. This favors the recombination of the anion radi-cals. So in THF, (Ph2CBCPh2)�.
, K� disproportionates rapidly. In HMPA, naked(Ph2CBCPh2)�.
anion radicals exist for a long time and the disproportionation equilibriumis strongly shifted to the left (Todres 1985):
(Ph2CBCPh�., K�)�THF � (Ph2CBCPh2
�., K�)�THF ←→
Ph2CBCPh2 � (Ph2CBCPh2���, 2K�)�THF
SCHEME 2-27
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Hence, one very important factor of preferential dianion formation is a decrease inthe electrostatic repulsion between anion radicals. By changing the ion-pair stability in par-ticular by means of solvent selection, one can manage equilibrium of liquid-phase electron-transfer reactions.
The polarographic reduction of tetraphenylethylene in dimethylformamide with n-Bu4NClO4 as a supporting electrolyte is an irreversible two-electron process. Preparativeelectrolysis in the same solution resulted in the formation of the dianion. The exocyclicbond of this dianion can be disrupted homolytically. Therefore, water treatment of the elec-trolyzed products leads to diphenylmethane (Wawzonek et al. 1965).
One special case of disproportionation as a consequence of ion association should bementioned in conclusion. If a suspension of triethoxy hexachlorantimonate and tetra(ani-syl)ethylene is stirred in dichloromethane, the corresponding cation radical salt formsquantitatively (Rathore and co-authors 1998):
2 An2CBCAn2 � 3 Et3OSbCl6 → 2 (An2CBCAn2)�.SbCl6�
� 3 EtCl � 3 Et2O � SbCl3
An � p-MeOC6H5
Slow crystallization of the salt from the reaction solution layered with diethyl etherleads to a mixture of the tetrakis(anisyl)ethylene cation radical and dicationic salts alongwith tetrakis(anisyl)ethylene. All the compounds precipitate from the solution containingthe cation radical salt only:
2 (An2CBCAn2)�.SbCl6
� ⇔ (An2CBCAn2)2�(SbCl6�)2 � An2CBCAn2
Hence, two possible causes of this disproportionation can be considered: changes in thesolution parameters during slow mixing of the solvent layers (dichloromethane and diethylether) and, on the other hand, requirements with respect to the closest packing in crystals.
2.4.5.B Electron-Transfer Reactions with the Participation of Ion-Polymeric Aggregates
The disodium salt of diphenylacetylene dianion is stable in THF solution at �78°C.Methanol acts as a proton source toward the salt and causes the formation of a mixture of1,2-diphenylethane with diphenylacetylene and small amounts of trans stilbene (Chang &Johnson 1965, 1966). It seems logical that the reaction between (PhC�CPh)2�, 2 Na�, andMeOH leads at first to PhCHBCHPh. The second step is supposed to consist of the furtherreduction of PhCHBCHPh at the expense of electrons from the nonreacted part of the ini-tial dianion. In principle, the electron transfer may proceed faster than the reaction of theinitial dianion with protons. As a result, the latter has got to discharge into diphenylacety-lene, whereas the stilbene dianion has got to form diphenylethane:
(PhC�CPh)2�, 2Na� � PhCHBCHPh → PhC�CPh � (PhCHBCHPh)2�, 2Na�
(PhCHBCHPh)2�, 2Na� � 2 MeOH → 2 MeONa � PhCH2CH2Ph
Hence, one could predict that being mixed with the methanol solution of PhCHBCHPh,(PhC�CPh)2�, 2Na� in THF might give PhCH2CH2Ph and PhC�CPh in quantitativeyields.
This obvious supposition turned out to be false: The addition of stilbene causes nochanges in the results of the reaction (Chang & Johnson 1965, 1966). It follows that an ef-fective contact between PhCHBCHPh and (PhC�CPh)2�, 2Na� is less than probable. As
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

known, (PhC�CPh)2�, 2Na� is a polymeric aggregate [(PhC�CPh)2�, 2Na�]n. Bulk stil-bene molecules are incapable of diffusing inside the aggregate. At the same time, the pen-etration of proton (or methanol) appears to be possible.
It seems that (PhCHBCHPh)2�, 2Na� is also capable of existing in the polymericform in a THF solution. This salt is a typical initiator in the polymerization of unsaturatedcompounds such as isoprene and methylstyrene. In THF such polymerization can includethe detachment or fixation (chemisorption) of a framing (terminal) link from the polymericaggregate [(PhCHBCHPh)2�, 2Na�]n. Methylstyrene captures the “come-off” link, start-ing the polymerization process. The link and, probably, [(PhCHBCHPh)2�, 2Na�]n�1
oligomer are included in a polymeric globule. The situation resembles a snake in a cage. Inexperiments (Podol’sky and others 1982), �-methylstyrene was polymerized in THF uponthe action of [(PhCHBCHPh)2�, 2Na�]n labeled with 3H, 14C in the presence of nonla-beled PhCHBCHPh. The authors obtained a polymer that contains the whole amount ofthe label. In the case of the mixture of nonlabeled [(PhCHBCHPh)2�, 2Na�]n with labeledPhCHBCHPh in THF, the polymer obtained did not contain any label.
These chemical electron-transfer reactions are in contrast with electrode reactions.For instance, stilbene gives a mixture of 1,2-diphenylethylene with 1,2,3,4-tetraphenylbu-tane upon electrolysis (Hg) in dimethylformamide at potentials around that of the first one-electron wave. This solvent has faint proton-donor properties. Stilbene anion radical is sta-ble under these conditions; it has enough time to diffuse from the electrode into the solventand to dimerize therein (Wawzonek et al. 1965).
The phenomena enumerated in Section 2.4 do not, of course, fully describe all the dif-ferences between chemical and electrode processes of ion radical formation. From time totime, effects are found that cannot be clearly interpreted and categorized. For instance, onepaper should be mentioned. It bears the symbolic title “�- and �-Diazo Radical Cations: Elec-tronic and Molecular Structure of a Chemical Chameleon” (Bally et al. 1999). In this work,diphenyldiazomethane and its 15N2, 13C, and D10 isotopomers, as well as the CH2–CH2
bridged derivative, 5-diazo-10,11-dihydro-5H-dibenzo[a,d]cycloheptene, were ionized viaone-electron electrolytic or chemical oxidation. Both reactions were performed in the samesolvent (dichloromethane). Tetra-n-butylammonium tetrafluoroborate served as the support-ing salt in the electrolysis. The chemical oxidation was carried out with tris(4-bromophenyl)-or tris(2,4-dibromophenyl)ammoniumyl hexachloroantimonates. Two distinct cation radi-cals that corresponded to �- and �-types were observed in both types of one-electron oxida-tion. These electromers are depicted in Scheme 2-28 for the case of diphenyldiazomethane.
SCHEME 2-28
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The electromers differ drastically in their optical and ESR spectra. There is arather small energy difference between them (ca. 1 eV). The authors conclude that “theexperimentally found energetic proximity of the two states is not an intrinsic propertyof the diaryldiazo cation radicals, but must be due to some solvent and/or counterioneffects acting to preferentially stabilize the �-state by about 1 eV.” These effects, how-ever, failed to be identified in the quoted paper. We await further development of theproblem.
2.5 FORMATION OF ORGANIC ION RADICALS IN LIVINGORGANISMS
One-electron transfer reactions are typical in living organisms. Ion radicals are acting par-ticipants in metabolism. Of course, such ion radicals are instantly included in further bio-transformations. Therefore, it is reasonable to consider the problem of ion radical forma-tion together with data on their behavior in biosystems. Section 3.4 covers this topic.However, the issue of competition for an electron during ion radical formation deserves tobe mentioned here.
Sometimes mesonidazole and AF-2 medication are administered jointly. Mesonida-zole is 1-(2-nitroimidazole-1-yl)methoxypropane-2-ol, and AF-2 is 2-(2-furyl)-3-(5-nitro-2-furyl)acrylamide. It is clear that the latter is a stronger acceptor than the former. Whenanion radicals of mesonidazole are formed, they pass their unpaired electrons to moleculesof AF-2. One medication cancels the action of the other (Clarke et al. 1984a,b).
Ion radical generation is a result of the interaction between some organic species andenzymes or other participants in metabolism. First of all we should note that so-called cellconditions can determine the very possibility of electron transfer. For example, there is awell-founded assumption that the reduction potential of colchicine is lowered enough oncontact with proteins, particularly in tumor cells, to accept an electron (Cavazza and others1999). The cell conditions can also determine whether an enzyme acts as an oxidative or areductive agent. Cytochrome P-450 enzymes are hemoproteins of significant importance inthe oxidation of a broad variety of drugs, pesticides, carcinogens, steroids, and fat-solublevitamins. They contain an Fe3� center, which transforms into an Fe2� kernel. These en-zymes are well-known biological oxidants. However, they can also catalyze the reduction,cycling between Fe2� and Fe3� forms; see Section 3.4.
Electron-transfer chains in plants differ in several striking aspects from their mam-malian counterparts. Plant mitochondria are well known to contain an alternative oxidasethat couples the oxidation of hydroquinones (e.g., ubiquinol) directly to the reduction ofoxygen. Semiquinones (anion radicals) and superoxide ions are formed in such reactions.The alternative oxidase thus provides a bypass to the conventional cytochrome electron-transfer pathway and allows plants to respire in the presence of compounds such ascyanides and carbon monoxide. There are a number studies on the problem; for example,see Affourtit and co-authors’ paper (2000) and references therein.
Besides the enzyme, the superoxide ion can also be an electron donor. The ion arisesas a result of detoxification of xenobiotis (xenobiotics are outsiders, which are involved inthe chain of metabolism). Xenobiotics yield anion radicals upon the neutralizing influenceof redox proteins. Oxygen (inhaled with air) takes an unpaired electron off from a part ofthese anion radicals and forms the superoxide ion. The latter plays its own active role inbiochemical reactions (see Sections 1.7.1.C. and 3.4.5).
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

2.6 ORGANIC ION RADICALS IN SOLID PHASES
2.6.1 Organic Ion Radicals in Frozen Solutions
Many organic compounds form ion radicals that are unstable in the liquid phase. In suchcases, pulse radiolysis in frozen solutions is used, although the energy of the incoming elec-tron is much larger than in the liquid-phase reactions of one-electron transfer. Sometimesthe sample is included in the rare gas and nitrogen matrices (this technique is described byKnight; for example, see Knight & Ebner 1976). More often, organic solvents are used. Ir-radiation drives electrons out from a solvent. An organic precursor (P) transforms into anion radical. At first glance, two reactions might be expected to take place: electron capture(P � e → P�.
) and electron detachment (P � e → P�.� 2e). As a matter of fact, an indi-
rect redox process takes place, with solvent participation. The example in Scheme 2-29 in-volves 2-methyltetrahydrofuran (MeTHF) participation in the redox process when P is asubstance of electron affinity higher than that of the solvent.
If P is a substance of electron affinity lower than that of the solvent (e.g., AlkHal inSF6), the process leads to formation of the cation radical. If P is a substance of electronaffinity higher than that of a solvent (e.g., AlkHal in CH3OH), the process leads to forma-tion of the anion radical. These possibilities are depicted in the following schemes, basedon alkyl halides:
AlkHal → (AlkHal)�.� e
AlkHal � e → (AlkHal)�. → Alk.� Hal�
(AlkHal)�.� P → AlkHal � P�.
The generation of ion radicals usually proceeds in frozen diluted solutions at 77 Kand under irradiation for not more than half an hour. An ion radical concentration of10�4–10�3 is reached, which is sufficient to record optical or ESR spectra of the ion radi-cals. The solvents must form transparent matrices as they get cold. For example, THF gives
SCHEME 2-29
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

opaque glasses, which prevents any spectral studies, whereas MeTHF (C5H10O) formsideal glacial matrices at 77 K. However, these samples contain the radical
.C5H9O (see
Scheme 2-29), and its ESR signal is often superimposed over that of the dissolved anionradical P�.
. In other glass-making solvents, good spectra with superfine structures areachievable: Tetramethylsilane for anion radicals and trichlorofluoromethane for cation rad-icals are recommended (Shida et al. 1984). The latter solvent (CFCl3) can be substituted forCF2ClCFCl2; in CFCl3 glasses, even such unstable species as the 3-methylpentane and 3-methylhexane cation radicals give excellent ESR spectra (Ohta & Ichkawa 1987). The aro-matic cation radicals in the CFCl3 matrix have, however, a tendency to form partly orientedsamples in which the amount of order/disorder depends on the concentration of the soluteand on the freezing rate. This brings its own obstacles to spectral studies. Meanwhile, thearomatic cation radicals in CF3CCl3 matrix form only glassy samples (Kadam et al. 1999).
One simple method for the production and cryogenic trapping of ion radicals has re-cently been devised. The technique, acronymed CWRD (cold window radical discharge),enables the isolation in rare gas matrixes of short-lived species like the p-dichlorobenzenecation radical. These species are formed within discharge plasmas, close to the trappingsurface (Kolos 1995).
Let us consider several examples of the application of the glass method.Cation radicals of substituted benzene were prepared upon �-radiation of dilute frozen
solutions in CFCl3 at 77 K (Ramakrishna Rao & Symons 1985). According to ESR spectraand quantum mechanical calculations, an unpaired electron is preferentially localized in theframework of the phenyl ring of the cation radicals of benzalehyde or phenyl methyl ketone.Styrene and its derivatives give cation radicals in which an unpaired electron spends 30% ofits time in the vinyl group. The same spin distribution was displayed in 2-vinylpyridinecation radicals. Surprisingly, the 4-vinylpyridine cation radical has an unpaired electron, tobe held with the heterocyclic nitrogen atom and not with the vinyl group. Both vinyl pyri-dine cation radicals were generated under the same conditions (Eastland et al. 1984).
The cation radical of 1,3-dioxolane (i.e., of 1,3-dioxacylopentane) was obtained byirradiation from 60Co in CFCl3 at 77 K. Its ESR spectrum shows an unusually strong split-ting from protons of the OCH2O group, namely, 15.3 mT. Interestingly, proton splitting forthe CH2 group in the THF cation radical is equal to 8.9 mT only. For the 1,3-dioxolanecation radical, the splitting enhancement is explained by a one-electron shift from the
..O
atom to O�.through the CH2 group (Symons & Wren 1984); see Scheme 2-30.
This also proves an earlier conclusion on hyperconjugation in an OCH2O fragmentof the 1,3-dioxolane cation radical; this conclusion was based on mass spectrometry (To-dres, Kukovitskii et al. 1981). As calculated, the carbon–hydrogen bonds corresponding toOCH2O in the radical cation are weaker than those in the neutral molecule. For this reason,this site exhibits a maximal probability that deprotonation will result in the formation of the2-yl radical (Belevskii et al. 1998). In experiments, photoirradiation of 1,3-dioxolane solu-tions in sulfur hexafluoride at 77 K really leads to formation of the cation radical of 1,3-dioxolane and the 1,3-dioxolan-2-yl radical as a result of deprotonation. Consecutive ring
SCHEME 2-30
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

opening, isomerization into linear products, and then ring closure reactions were registeredby the ESR method for this cation radical (Baskakov et al. 2000).
The following two examples concern organic anion radicals. Radiolysis of 4-ni-trobenzyl chloride or bromide (60Co, MeTHF, 77 K) leads to the corresponding anion rad-icals with no C–Hal cleavage (Symons, Bowman 1984). It is worth noting that all attemptsto fix halonitrobenzyl anion radicals in liquid phase were unsuccessful. Indeed, the glasstechnique allows one to record ESR spectra, observe splitting at nitrogen and at halogen,and, as a result, establish the principal ability of these anion radicals to exist.
The acetylene anion radical was successfully generated in a glassy MeTHF matrixvia ionizing irradiation (60Co) at 77 K. Upon illumination with light 430 nm, the followingisomerization takes place (Itagaki & Shiotani 1999):
(CH�CH)�. → CH2BC�..
When 3-methylpentane was used as a matrix molecule, instead of MeTHF, however,no such isomerization reaction was observed. This could be due to the nature of 3-methylpentane, which gives a nonpolar and soft matrix at 77 K. MeTHF is a polar moleculethat forms a rigid glassy matrix at 77 K. The electrons generated by ionizing irradiation arestabilized in MeTHF. This polar and rigid nature of the MeTHF matrix can be responsiblefor the phenomenon: Both anion radicals are thermally stable, so the photoisomerization re-action shown was observed.
2.6.2 Organic Ion Radicals in Solid Salts
The problem of preparation of pure ion radical salts in the solid state is very important tech-nically. This problem is decisive in such new application fields as organic conductors andorganic magnets. Sections 7.3 and 7.4 are devoted to the methods for preparing the solidion radicals salt for these materials.
2.7 ISOTOPE-CONTAINING ORGANIC COMPOUNDS AS IONRADICAL PRECURSORS
Isotope substitution aims to ascertain whether a bond labeled with an isotope takes part ina certain reaction. One then either looks at the products, noting the label distribution, or at-tempts to determine the kinetic isotope effect. The latter can be detected as the change inthe rate constant of the reaction upon putting an isotope in the place of an atom taking partin the reaction of interest.
When applied to electron-transfer reactions, this kinetic-isotope-effect technique canprovide information on the real reaction pathway leading to the product. Frequently, spec-troscopic detection of species or identification of products is indicative of radical interme-diates. The formation of the intermediates could simply be a blind step.
Isotope-containing organic compounds as ion radical precursors have important sig-nificance for ion radical organic chemistry. The link between the isotopic substitution at thereaction center and the change in the kinetic properties is not so obvious for one-electrontransfer. In that case, the highest occupied molecular orbit (MO) of a donor loses one elec-tron. This electron is then shuttled to the lowest unfilled MO of an acceptor.
Therefore, one should expect to see the isotopic effect in electron-transfer reactionsas being dependent on the change in the corresponding orbital energy levels of the donorand the acceptor as a result of the isotopic substitution. Theory shows that electron transfer
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

causes the perturbation in the vibronic levels of a molecule (Efrima & Bixon 1974). As forthe reactions in solutions (the most typical conditions in synthetic organic chemistry), thereis an observed disturbance in vibronic levels of a complex system, consisting of a donorand an acceptor (reagents) as well as a solvent itself. The transfer of an electron is definedwith a difference between the electron affinity of the acceptor (EA) and the ionization po-tential of the donor (ID). The solvent effect on this process is chiefly dependent on its reor-ganization energy, on its way from reagents to products (�ES). It should be noted that thetransfer of an electron brings about a sudden change in substrate polarity and results in acardinal change of a solvate shell (see also Section 5.4). Hence, isotopic substitution canhave an effect on the electron-transfer reaction only if the following two conditions are ob-served: (1) EA or ID changes and (2) this change is greater than that of �ES. In other words,�ID � �ES or �EA � �ES (Johnansen & Schoen 1980).
Concerning the change in EA or ID for isotopic substitution, currently there is notmuch data and the pattern of the phenomenon is still not known. Nevertheless it seems use-ful to compare the existing data both for consideration of the isotopic effects in reactionsand for information on the possible new ways of using electron transfer for a more effec-tive enrichment of the isotopic mixtures with one specific isotope form.
2.7.1 Kinetic Isotope Effects in Electron-Transfer Reactions
In the presence of dimethylsulfide, the dissociation of tert-butylperoxybenzoate is acceler-ated (Pryor & Hendrickson 1983). Dimethylsulfide acts as a donor, whereas the peroxideacts as an acceptor. In the same solvent, the rate of the dissociation under the action of Me2Sis almost 1.5 times greater than that under the action of (CD3)2S. The deuterated compoundpossesses a higher ionization potential, so for the Me2S/(CD3)2S pair, �ID � 3.3 kJ/mole(Pryor & Hendrikson 1983).
Assuming that this difference is exhibited during the conversion of the reagents intothe products, and that it is greater than the reorganization energy of the solvate shell, theisotopic effect Hk/Dk of this particular reaction at 80°C should be approximately equal to1.4. This value matches closely the experimental magnitude (Pryor & Hendrikson 1983).Analogous similarities are also found for phenylhydroxylamine oxidation with diben-zoylperoxide (Hk/Dk � 1.53) and trimethylamine oxidation with chloroperoxide (Hk/Dk� 1.3) (Pryor & Hendrikson 1983).
The oxidation reaction of alkylaromatic compounds with cerium-ammonium nitratein acetic acid medium is crucial for understanding the problem (Baciocchi, Rob, & Man-dolini 1980):
ArMe � Ce(IV) ⇔ ArMe�.� Ce(III)
Hexamethylbenzene and 4-methoxytoluene are oxidized easily. However, the reac-tion is slowed down considerably upon the addition of Ce(III), and the equilibrium is left-shifted (toward the reagents), as expected. The abrupt increase in the Hk/Dk ratio is never-theless not expected and is due to the concomitant deprotonation of ArMe�.
by thefollowing mechanism:
ArCH3�.
� AcO� → ArCH.2 � AcOH
This last reaction is not significant when the electron-transfer equilibrium is shiftedto the right. Because acetic acid is weakly dissociated, the binding of acetate by an “alien”proton from ArMe� leaves its “own” proton free to suppress the acid dissociation further.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

This results in a deficit of acetate ions. However, when the electron-transfer equilibrium isshifted to the left, there is a decrease in the number of ArMe�.
radicals. So a relative excessof the acetate ions is formed. This makes deprotonation of the radical a rate-limiting step.That the 1H being transferred is substituted for 2H is the reason for the growth of the Hk/Dkratio, i.e., an increase in the kinetic isotopic effect. This demonstrated mechanism of“switching from one rate-limiting step to another” is also used in cases of oxidation ofalkylbenzenes with tris(phenanthrolene)iron (III) (Schlesener et al. 1984; Schlesener &Kochi, 1984) and with manganese triacetate in acetic acid (Andrulis, Dewar et al. 1966;Eberson 1967). If the rate-limiting step is the electron transfer, then the kinetic isotope ef-fect cannot be very significant.
As assumed, the small and positive value of the H/D kinetic isotope effect may be usedas a criterion for an electron-transfer pathway. For example, anion radicals of �-benzoyl-�-haloalkanes can react along two routes (Kimura & Takamuku 1994). The first route is thecommon one: An electron is transferred from the carbonyl oxygen anion to a terminal halo-gen. The transfer provokes fission of the carbon–halogen bond. The second route is the SN2reaction, leading to a cyclic product according to Scheme 2-31. In the scheme, SH is a sol-vent (HMPT). For the cases of halogen-hydrogen substitution in �OCH
.–(CH2)n�1CH2Cl
and �OCH.–(CH2)n�1CD2Cl, the magnitudes of Hk/Dk are calculated as 1.059 at n � 2 and
1.141 at n � 3. The calculated magnitudes of Hk/Dk for the cyclization route are 0.996 (n � 2) and 0.992 (n � 3) (Sastry et al. 1995).
Interestingly, the anion radical from RC6H4CO(CH2)4CH2Br reacts according to thehalogen-hydrogen substitution, while the anion radical from RC6H4CO(CH2)3CH2Br takesthe cyclization route (Kimura & Takamuku 1994). The route choice is dependent on thespatial structure of the transition state. This means dependence on a distance between �Oand CH2–Hal fragments and an orientation of the orbitals of a radical pair formed by theconcerted electron-transfer and bond-breaking process.
For the reactions of MeLi with benzophenone and benzophenones labeled at the car-bon atom of the carbonyl group, 12k/14k ratio is 1.000 in ether at 0°C. Under the same con-ditions, this ratio is 1.029 for Me2CuLi and 1.050 for MeMgBr (Hiroshi et al. 1987; Ya-mataka, Matsuyama, & Hanafusa 1987, 1989; Yamataka, Fujimara et al. 1987; Matsuyamaet al. 1988).
For MeMgBr in ether at 20°C, the 12k/13k ratio was found to be 1.030 (Holm 1993).The magnitudes of 12k/14k and 12k/13k are in accord, “since the origin of kinetic isotope ef-fects lies exclusively in the difference in molecular mass and 13C effect should be on theorder of one-half of the 14C effect” (Holm 1993). Hence, in the case of MeLi, one-electrontransfer is a rate-limiting step; and for both Me2CuLi and MeMgBr, a rate-limiting step is
SCHEME 2-31
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

the ion radical pair recombination by the following mechanism:
Ph2CBO � MeMgBr → [Ph2C.MO� ...(MeMgBr)�.
] → Ph2C(Me)OMgBr
One should be aware, however, that none of the Grignard reactions of benzophenoneproceeds through a complete free coupling process of the benzophenone anion radicalswith alkyl radicals. For example, the portion of the electron-transfer pathway in the Grig-nard reactions of benzophenone with isomeric C4H9MgCl was estimated to be 65, 61, and26% for (CH3)3C–,CH3CH2CH(CH3)–, and CH3CH2CH2CH2–, respectively (T. Lund et al.1999).
According to the kinetic-isotope-effect test, the reaction of benzaldehyde withlithium pinacolone enolate proceeds through the polar addition mechanism (Yamataka,Sakaki et al. 1997). As it turned out, the mechanistic switching relates to the stability of thereagents measured by the intrinsic acidity of the conjugate acids RH of R� anions in thegas phase. The reagent whose conjugate acid is most acidic reacts with benzaldehyde viathe polar addition mechanism, whereas the reagents whose conjugate acids are less acidicgo through the electron-transfer pathway (Yamataka, Sasaki et al. 2001).
As logical as this diagnostic method is, one needs to recognized its lack of absoluteapplicability: The observed magnitude of the kinetic isotopic effect is not great, and theaforementioned statement of independence of electron affinity from the increase in themolecular mass of the substrate is not obvious. This postulate should be proven in eachcase. Benzophenone, taken as an isotopic mixture of 12CBO and 13CBO gives a mixtureof anion radicals with a decreased proportion of 13CBO isotomer when reduced with potas-sium in HMPA (G.R. Stevenson, Reiter, Espe, & Bartmess 1987). In effect, this means thatfor the heavier isotopomer of benzophenone, the electron affinity is smaller.
Until now, the isotopic effect has been discussed only in relation to the reactants. Inelectron-transfer reactions, the solvent plays an equally important role. As mentioned, dif-ferent solvate forms are possible for reactants, transition states, and products. Therefore, itseems significant to find a reaction where the kinetic effect resulting from the introductionof an isotope would be present for the solvent but absent for the reagents. There is publishedwork concerning this problem (Yusupov & Hairutdinov 1987). In this work, the authorsstudied photoinduced electron transfer from magnesium ethioporphyrin to chloroform fol-lowed by dark recombination of ion radicals in frozen alcohol solutions. It was determinedthat deuteration of chloroform does not affect the rate of the transfer, whereas deuterationof the solvent reduces it. The authors correlate these results with the participation of the sol-vent vibrational modes in the manner of energy diffraction during electron transfer.
Electron-transfer reactions deal with ionic participants. It might therefore be worth-while to bear in mind that replacing the solvent with its deuterated analog may also resultin some perturbations in the delicate interactions between the anion radical and the coun-terion. As shown, substitution of the hydrogens with deuteriums results in a slight increasein the polarity of the tetrahydrofurane (THF) (G.R. Stevenson, Ballard, & Reiter 1991). Theeffect is understandable due to the electron-donating nature of deuterium relative to hydro-gen. This increased solvent polarity augments the solvent ability of the charge separation.The charge separation may change the anion radical reactivity, of course.
2.7.2 Behavior of Isotope Mixtures in Electron-Transfer Reactions
It is clear that deuterium as a substituent has an electron-donating effect. In other words, itcan decrease the electron affinity of the whole molecule. Potentials of reversible one-elec-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

tron reduction for naphthalene, anthracene, pyrene, perylene, and their perdeuterated coun-terparts indicate that the counterparts exhibit slightly more negative potentials (Goodnow& Kaifer 1990; Morris & Smith 1991). For example, the measurable differences in the re-duction potentials are equal to �13 mV for the pair of naphthalene-naphthalene-d8, or �12mV for the pair of anthracene-anthracene-d10. The possible experimental error does not ex-ceed 2 mV (Morris & Smith 1991). For another example, in dimethylformamide (DMF)with 0.1 M n-Bu4NPF6, the deuterated pyrenes were invariably found to be more difficultto reduce than pyrene itself. The largest difference observed, �12.4 mV, was that betweenperdeuterated pyrene and pyrene bearing no deuterium at all, with standard deviations be-tween 0.2 and 0.4 mV (Hammerich et al. 1996).
A comparative analysis of vibrational modes of perdeuteriobenzene, benzene, andtheir anion radicals shows that deuteration leads to an increase in the level of zero-point en-ergy. Other energy levels of the perdeutero derivatives are also shifted, and the electronaffinity is reduced by almost 2 kJ (G.R. Stevenson & Alegria 1976; G.R. Stevenson, Espe,& Reiter 1986; G.R. Stevenson, Espe, Reiter & Lovett 1986). Similarly, the electron affin-ity is reduced by 1 kJ upon the change from 12C6H6 to 13C6H6. A comparable change of theelectron affinity should be expected for other classes of organic compounds as well.
To verify these estimations experimentally, an analysis of electron spin resonance(ESR) spectra resulting from an incomplete reduction of the precisely measured amountsof benzene isotopomers with potassium in THF was conducted (G.R. Stevenson, Espe, &Reiter 1986). That approach had been outlined by Chang and Coombe in 1971. The super-imposed spectra were analyzed to find the intensity ratio of the signals belonging to the iso-topomeric anion radicals. That ratio produced the concentration ratio of the anion radicalsunder consideration.
For example, a 4.5:1 mixture of 13C6H6 � 12C6H6 forms the 2:1 mixture of(13C6H6)�.
� (12C6H6)�.upon reduction at �100°C by the half-stoichiometric amount of
potassium in THF. So the heavier anion radical amount is less than the amount of the heav-ier substrate in the initial mixture. The ESR data were reproduced by means of NMR andmass spectral methods after transformation of the anion radical mixtures into the mixturesof corresponding neutral compounds and removal of the solvent.
For the equation (C6H6)�.K� � C6D6 ↔ C6H6 � (C6D6)�.
K�, the ratio of iso-topomers before and after the electron transfer can be recalculated into the correspondingequilibrium constants. Table 2-1 gives the order of these constants and the experimentalconditions typical for their study.
The equilibrium constant for the reaction of the electron transfer from the anion rad-ical salts of aromatic compounds (with the usual isotope content) to neutral molecules ofthe same compounds containing heavier isotopes is less than unity (entries 1–10 in Table2-1). This means that for heavier compounds (enriched with neutrons), the electron affin-ity is smaller. This difference is conserved at different temperatures and reaction mediums(including those favorable to the destruction of ionic pairs—in HMPA and in THF con-taining 18-crown-6).
With respect to ionic pairs, perdeuteration in organic anion radicals alters the zero-point energies of the vibrational modes involving the interaction of a metal cation with anorganic counterpart. As shown, the anion radical of perdeuterionaphthalene associates withsodium cations in THF more tightly than the anion radical of perprotionaphthalene underthe same conditions (Ch.D. Stevenson, Wagner, & Reiter 1993).
The solution electron affinities of a series of monosubstituted benzenes were mea-sured by means of ESR (G.R. Stevenson, Wehrmann, & Reiter 1991). The equilibrium con-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

stant for the electron transfer XC6H5�.
� XC6D5 ↔ XC6H5 � XC6D5�.
, where X � H, t-Bu,OMe, Ph, CN, was found to be less than unity for all cases (entries 1–6 in Table 2-1). Theseequilibrium constants are in linear correlation with the appropriate constants � of the sub-stituents.
It would be interesting to explore the same correlation for cation radicals. It is logi-cal to expect the deuterated compounds with decreased electron affinities to also have de-creased ionization potentials. Koopmans theorem states that the ionization potential of amolecule is equal to the negative energy of its highest occupied molecular orbital. How-ever, there are numerous examples where this theorem is not applicable. Therefore, it is notclear whether the theorem is valid for neutron enrichment of the cation radical system. Thecase, noted earlier, where the ionization potential increased upon switching from Me2S to(CD3)2S shows either that Koopmans theorem is not applicable here or that this exampledoes not fit the general trend. 15N-labeling of Me2N-C6H4-NMe2-p decreases appreciablythe ionization potential of the molecule, making it easier to lose an electron forming the cor-responding cation radical in acetonitrile solution (Lue et al. 2001). Even though the prob-lem is still new and there have not been many examples studied, some electron-transfer re-actions between an anion radical and a heavy molecule were found in which the equilibriumconstant is greater than 1. One can see this in entry 11 in Table 2-1. The behavior of ben-zophenone was a part of the general trend. Fluorenone (G.R. Stevenson, Espe, & Reiter1986) gives the anion radical mixture in which there is more of 13C
.–O� (heavier) iso-
topomer than in the 12CBO and 13CBO mixture before reduction with a small amount ofmetallic sodium in HMPA. The equilibrium constant of this reaction at 25°C is 2.74 versusthe analogous equilibrium constant of 0.58 for the benzophenone reaction under the sameconditions. The more recent publications on the subject, by Holm (1994) and Yamataka,
TABLE 2-1 Equilibrium Constants (Keq) of Electron-Transfer Reactions
Startingaromatic Starting anion
No. compound radical salt �C Solvent Keq Ref.a
1 C6D6 (C6H6)��K� �100 THF 0.27 SER ’86�25 THF 0.46 SWR ’91
2 t-BuC6D5 (t-BuC6H5)��K� �25 THF 0.36 SWR ’913 MeOC6D5 (MeOC6H5)��K� �25 THF 0.39 SWR ’914 C6H5MC6D5 (C6H5MC6H5)��K� �25 HMPA 0.51 SWR ’915 NBMCC6D5 (NBMCC6H5)��K� �25 HMPA 0.70 SWR ’916 O2NC6D5 (O2N C6H5)��K� �25 NH3(liq) 0.86 SWR ’917 Naphth.-d8 (Naphthalene)��K� �100 THF 0.26 SS ’908 Anthracene-d10 (Anthracene)��K� �100 THF 0.30 SS ’909 Pyrene-d10 (Pyrene)��K� �100 THF 0.37 SS ’90
10 Perylene-d10 (Perylene)��K� �100 THF 0.40 SS ’9011 (C6D5)2CBO [(C6H5)2CBO]��K� �75 NH3(liq) 0.52 LPR ’88
SHK ’9312 [(C6H5)2
13CBO [(C6H5)2CBO]��K� �25 HMPA 0.58 SER ’8613 Benzoquinone (OBC6H4BO)��Na� �75 THF� 0.66 SHR ’93
(13CBO) HMPA14 Benzoquinone (OBC6H4BO)��Na� �75 THF� 1.00 SHR ’93
(OBC6D4BO) HMPA
a SER ’86—G.R. Stevenson, Espe, & Reiter 1986; SWR ’91—G.R. Stevenson, Wehrmann, & Reiter 1991; SS’90—G.R. Stevenson & Sturgeon 1990; LPR ’88—Luaricella, Pescatore, Reiter et al. 1988; SHK ’93—Ch.D.Stevenson, Halvorsen, Kage et al. 1993; SHR ’93—Ch.D. Stevenson, Halvorsen, & Reiter 1993.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.
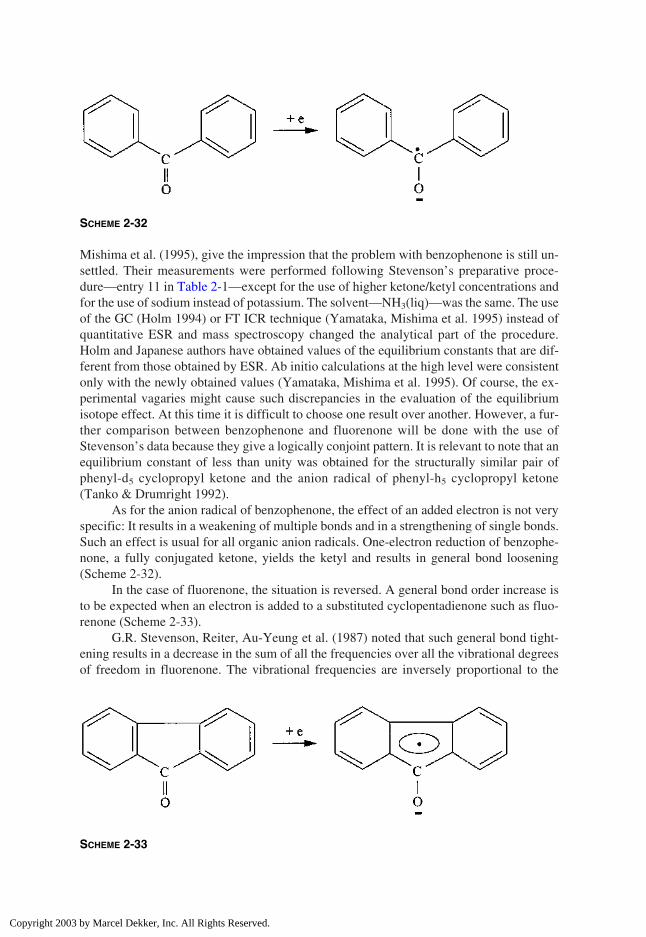
Mishima et al. (1995), give the impression that the problem with benzophenone is still un-settled. Their measurements were performed following Stevenson’s preparative proce-dure—entry 11 in Table 2-1—except for the use of higher ketone/ketyl concentrations andfor the use of sodium instead of potassium. The solvent—NH3(liq)—was the same. The useof the GC (Holm 1994) or FT ICR technique (Yamataka, Mishima et al. 1995) instead ofquantitative ESR and mass spectroscopy changed the analytical part of the procedure.Holm and Japanese authors have obtained values of the equilibrium constants that are dif-ferent from those obtained by ESR. Ab initio calculations at the high level were consistentonly with the newly obtained values (Yamataka, Mishima et al. 1995). Of course, the ex-perimental vagaries might cause such discrepancies in the evaluation of the equilibriumisotope effect. At this time it is difficult to choose one result over another. However, a fur-ther comparison between benzophenone and fluorenone will be done with the use ofStevenson’s data because they give a logically conjoint pattern. It is relevant to note that anequilibrium constant of less than unity was obtained for the structurally similar pair ofphenyl-d5 cyclopropyl ketone and the anion radical of phenyl-h5 cyclopropyl ketone(Tanko & Drumright 1992).
As for the anion radical of benzophenone, the effect of an added electron is not veryspecific: It results in a weakening of multiple bonds and in a strengthening of single bonds.Such an effect is usual for all organic anion radicals. One-electron reduction of benzophe-none, a fully conjugated ketone, yields the ketyl and results in general bond loosening(Scheme 2-32).
In the case of fluorenone, the situation is reversed. A general bond order increase isto be expected when an electron is added to a substituted cyclopentadienone such as fluo-renone (Scheme 2-33).
G.R. Stevenson, Reiter, Au-Yeung et al. (1987) noted that such general bond tight-ening results in a decrease in the sum of all the frequencies over all the vibrational degreesof freedom in fluorenone. The vibrational frequencies are inversely proportional to the
SCHEME 2-32
SCHEME 2-33
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

square root of the reduced masses of the atoms involved. It means that the difference be-tween the mentioned sums is greater for a 13C-substituted system than it is for the 12C sys-tem. Since bonds are tightening upon one-electron addition, they are tightened more for13C-substituted material than for the fluorenone itself. From this, the authors predicted that“the zero-point energy effects would result in fluorenone having a lower electron affinitythan fluorenone substituted at the carbonyl position with a 13C.” The experiment confirmedthose predictions.
For the p-benzoquinone series (Table 2-1, lines 13 and 14), the equilibrium isotopeeffect (conformed via physical separation of the isotopic isomers involved) was observedby means of ESR analysis (Stevenson, Halvorsen, Kage et al. 1993; Stevenson, Halvorsen,& Reiter 1993). In this case, entry 13, the equilibrium constant is equal to 0.66. An analo-gous competition for an electron between p-benzoquinone and perdeuterated p-benzo-quinone (entry 14) shows no equilibrium isotope effect. For benzoquinone (13CBO), the13C effect is attributed almost exclusively to the carbonyl group. The constant of electronexchange equilibrium between benzoquinone OB13C6H4BO and (OBC6H4BO)�.
Na�
is 0.63 in THF � HMPA at �75°C. The low spin density on the noncarbonyl positions ren-ders ineffective the substitution of the protons by deuteriums and the substitution of thenoncarbonyl carbons by 13C in altering the solution electron affinity. The most significantdecrease in vibrational frequency upon electron addition was indeed observed in the CBOstretch (Chipman & Prebenda 1986).
Nevertheless, recent rigorous and detailed calculations (Jacob et al. 1999) did notsupport the large heavy-atom isotope effect determined from ESR experiments on electrontransfer with the participation of benzoquinone. Equilibrium constants involving 13C-,17O-, and 18O-labeled species were predicted not to deviate significantly from unity. At thesame time, a large isotope effect is expected for the process with deuterium substitution, re-flecting the importance of the large change in reduced masses. (The deuterium mass is dou-ble with respect to the protium mass.) The discrepancy between theory and experiment can-not be attributed to the role of the counterion. A redetermination of the equilibrium isotopeeffects using alternative experimental techniques would be of interest. The case with p-ben-zoquinone is very important in terms of the regioselectivity of the effect discussed.
In the case of cyclo-octatetraene, an electron “prefers” the isotopically heavier mate-rial in the following equilibrium:
C8H8 � C8D8�. ⇔ C8H8
�.� C8D8
However, when this anion radical reacts with cyclo-octatetraene dianion (not with the an-ion radical), the transferred electrons “prefer” the isotopically lighter material (G. R.Stevenson, Peters et al. 1990, 1992):
C8H8�.
� C8D82� ⇔ C8H8
2� � C8D8�.
The semiempirical quantum chemical consideration led to the conclusion that the dis-crepancy can be a consequence of ion-pair formation (Zuilhof & Lodder 1995). In the caseof cyclo-octatetraene, the ion-pairing phenomenon deserves more detailed explanation.While the dianion of cyclo-octatetraene is completely planar and meets all the requirementsof aromaticity, the anion radical of cyclo-octatetraene is a nonaromatic species and is notcompletely planar. In the equilibria just considered, both the anion radical and the dianionhad alkali cations as counterparts. The dialkali salt of the dianion has two cations symmet-rically located over and beneath the octagonal plane. Distortions from ion pairing betweenthe dianion plane and these two alkali cations are reciprocally compensated. With the
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

slightly puckered anion radical of cyclo-octatetraene, the rigid ion pairing is possible. Anal-ysis of vibrational fine structure in the absorption and emission spectra of the potassium saltof the cyclo-octatetraene anion radical in a matrix has shown the formation of a rigid ionpair (Dvorak & Michl 1976). The rigid ion pairing restricts ring breathing and out-of-planebending vibrations, which contribute significantly to the overall isotope effect (Zuilhof &Lodder 1992).
The solution electron affinity of C8D8 proved to be slightly greater than that for C8H8.Cyclo-octatetraene is [8]annulene. For [16]annulene, the solution electron affinity ofC16D16 is by the normal manner lower than that of C16H16. NMR experiments reveal thatthe barrier to ring flattening is greater in the C16D16 system than in the C16H16 system. Cou-pled with the prediction of density functional theory that C16D�.
16 is nearly planar, the dataaccount for the normal equilibrium isotope effect observed in the electron transfer (Kurthet al. 1999; Ch.D. Stevenson & Kurth 1999).
One interesting but still unexplained case involves nitrobenzene. The reversible elec-tron exchange between nitrobenzene-15N and the sodium salt of the nitrobenzene-14N an-ion radical is characterized by the usual constant of 0.40. G.R. Stevenson, Reiter, Espe, andBartmess (1987) used NH3(liq) as a solvent for those measurements at �75°C. Under thesame conditions, they obtained the equilibrium constant of 2.1(!) for the electron exchangebetween nitrobenzene-15N and the potassium salt of the nitrobenzene-14N anion radical.Perhaps the difference between ion radii of sodium and potassium cations is crucial for thestability of the corresponding ion pair with the nitrobenzene anion radical. Such diversitycan be pivotal when the electron prefers the “heavy” or “light” nitrobenzene.
The future will supposedly bring more precise description of the trends consideredhere, and the reasons for the exceptions will be clearer. It is quite possible that in this field,too, the exceptions will only confirm the rule. For now, it is worth concluding that all theseregularities have very real practical applications. It is a fact that the equilibrium constantsof the previously described reactions differ from unity. This provides an opportunity to sep-arate and to enrich isotopic mixtures.
The most interesting possibility consists of separation of the natural mixture of ni-trobenzenes containing 14N and 15N isotopomers (G.R. Stevenson, Lovett, & Reiter 1986).A solution of 0.03 mmol of Ph15NO2 and 0.08 mmol of PhNO2 were treated with 0.01mmol of potassium metal in dehydrated liquid NH3. After the evaporation of NH3, the mix-ture of the initial compounds with their anion radical potassium salts remains for furthertreatment. The neutral nitrobenzenes are liquid; they are distilled out under high vacuum.The rest is solids. The solid remainder is oxidized with the solution of iodine in ether:
PhNO2�.
K� � 1/2(I2) → KI � PhNO2
The resulting mixture contains twice as much Ph15NO2 as the starting mixture (theequilibrium constant of electron transfer is approximately 2). Upon repetition of the proce-dure, the Ph15NO2 content in the mixture is increased twofold, and so on. The natural nitrobenzene sample with Ph15NO2 content of 0.37% only was enriched up to the ni-trobenzene-15N of 99% purity after 16 repetitions of the treatment just described.
This kind of physical separation of neutral molecules and anion radicals is, of course,one very unusual and effective way to enrich isotopic mixtures.
In the case of benzene, the potassium salt of its anion radical can be separated as aprecipitate after the benzene reduction by potassium in the presence of low concentrationsof 18-crown-6 ether. For benzene, the heavy-form content is greatest in the solution, not inthe precipitate. It is the solution where most of the nonreduced neutral molecules remain.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Since the neutral molecules are inert toward protons, the anion radicals combine with theprotons to give dihydro derivatives (products of the Birch reaction). Therefore, it is possi-ble to conduct the separation chemically. The easiest way is to protonate a mixture after theelectron transfer and then to separate the aromatic compounds from the respective dihy-droaromatics (cyclohexadiene, dihydronaphtalene, etc.) (Chang & Coombe 1971; G.R.Stevenson & Alegria 1976; G.R. Stevenson, Espe, & Reiter 1986; G.R. Stevenson, Lovett,& Reiter 1986; G.R. Stevenson, Sturgeon, Vines, & Peters 1988).
This chemical way to enrich the isotope-containing mixtures is easier and more ef-fective than many other methods currently in use. The formation of dihydro derivatives inthe Birch reaction can, obviously, occur not only via protonation of an anion radical butalso that of a dianion. For example, the anion radical C10H8
�.. can easily acquire another
electron to give the dianion C10H8�.
. Then a two-step protonation gives dihydronaphtalene:
C10H28� � H� → C10H9
� � H� → C10H10
This is why an important question should be raised: Can the direction of isotopic enrich-ment as outlined for anion radicals be extended to apply to dianions as well?
Experiments concerning the reduction of a mixture of anthracenes C14H10 � C14D10
and perylenes C20H12 � C20D12 with excess amounts of potassium in THF give an answerto that question (G.R. Stevenson, Espe, & Reiter 1986). The reduction of these 1:1 mixturesresults predominantly in nondeuterated anion radicals. However, upon subsequent reduc-tion (with the transfer of the second electron), the relative concentration of deuterated an-ion radicals increases, and these particles are the main paramagnetic products after the dis-solution of 2 moles of potassium per mole of the hydrocarbon. These reactions are listedhere with the corresponding constants, which were determined at �100 °C:
C14H102� 2K� � C14D10
�.K� ⇔ C14H10
�.K� � C14D10
2� 2K� Hk/hDk � 0.74
C20H122� 2K� � C20H�.
12 K� ⇔ C20H�.12 K� � C20D12
2� 2K� Hk/hDk � 0.65
By comparing these constants with the equilibrium constants of the anion radical re-actions (entries 7–10 in Table 2-1) one can conclude that in the case of dianions, the equi-librium is definitely shifted to the right. The equilibrium constants of ca. 0.35 are increasedalmost twofold but remain less than unity. This means that even with the formation of di-anions, the possibility of isotopic enrichment still remains quite feasible, even though theprocess becomes less favorable than that in the previous case of nitrobenzene.
The difference in electron affinity between light and heavy isotopic isomers is, inother words, the difference in the stability of their anion radicals. Such difference gives avaluable tool for use in probing the chemistry of anion radicals. The difference in the sta-bility of the ring-deuterated and ring-nondeuterated arene anion radicals has been em-ployed to examine the transition states for the one-electron-promoted cleavage of naph-thylmethyl phenyl ether and naphthyl benzylether (Guthrie & Shi 1990). In the reaction, thepotassium salt of fluoranthene anion radical was an electron donor:
C6H5OCH2C10H7 � e → C6H5OCH2C10H7�. → C6H5O� �
.H2CC10H7
C6H5CH2OC10H7 � e → C6H5CH2OC10H7�. → C6H5CH
.2 � �C10H7
The starting material C6H5OCH2C10H7 reacted more easily than did C6H5OCH2C10D7.Equimolar mixtures of D- and H-naphthylmethyl phenyl ethers were reduced by the one-elec-tron donor. The product of this cleavage was found to contain an excess of nondeuteratedmethylnaphthalene. Conversely, the remaining unchanged material contains an excess of na-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

phyhalene-ring-deuterated ether. Obviously, in the rate-determining step (i.e., scission of theCH2—O bond), the corresponding transition state resembles a naphthalene anion radical. Inthe transition state, the added electron remains localized in a �* molecular orbital. Clearly,to the extent that naphthalene-ring-deuteration raises the energy of the transition state rela-tive to that of its unsubstituted isotopic isomer, fractionation must occur on proceeding fromthe ether (reactant) to methylnaphthalene (product).
Meanwhile, the starting materials C6H5CH2OC10H7 and C6H5CH2OC10D7 wereequal in their reactivity. The cleavage of C6H5CH2OC10H7
�.is better viewed as involving
a �*-like transition state.Isotope effects on redox reactions of the type considered in Section 2.7 are of inter-
est for a number of reasons. At a fundamental level, the magnitude of the effect providesan important clue to the electronic structure and the vibrational properties of the species in-volved. From a practical point of view, a large deviation from unity for the equilibrium con-stant offers a convenient procedure for enrichment of the isotopomer mixtures.
2.8 CONCLUSION
Data on ion radical formation show wide diversity of preparative methods. A variety ofmethods are available, and the choice between them is still largely empirical.
Liquid-phase electron-transfer reactions that lead to ion radicals can be reversible.The equilibria of these reactions can be managed to obtain the desired results. This chap-ter considers methods for such management. Electrochemical methods of ion radical gen-eration are given in comparison with chemical ones. Chemically generated ion radicalscan exist in solutions or, in some special cases, as solids. The peculiarities of all themethods used for ion radical generation are essential in understanding of ion radical re-activity.
Generally speaking, the experimental results presented emphasize some distinctionbetween chemical and electrochemical electron-transfer reactions. At the same time, bothkinds of reactions share a fair number of features. A greater combination of these two meth-ods in the organic chemistry of ion radicals would seem to be fruitful.
Isotope-containing organic compounds participate in electron-transfer reactions in aspecific manner. This chapter gives a concise review of relevant data, especially concern-ing the enrichment of the isotopomer mixtures of organic compounds by means of theirtransformation into ion radicals.
REFERENCES
Adamo, M.F.A.; Aggarwal, V.K.; Sage, M.A. (2000) J. Am. Chem. Soc. 122, 8317.Affourtit, Ch.; Whitehouse, D.G.; Moore, A.L. (2000) IUBMB Life 49, 533.Alberti, A.; Benaglia, M; D’Angelantonio, M.; Emmi, S.S.; Guerra, M.; Hudson, A.; Macciantelli,
D.; Paolucchi, F.; Roffia, S. (1999) J. Chem. Soc., Perkin Trans. 2, 309.Alonso, F.; Yus, M. (1997) Tetrahedron Lett. 38, 149.Andrieux, C.P.; Robert, M.; Saeva, F.D.; Saveant, J.-M. (1994) J. Am. Chem. Soc. 116, 7864.Andrulis, P.J.; Dewar, M.J.S.; Dietz, R.; Hunt, L. (1966) J. Am. Chem. Soc. 88, 5473.Araki, Sh.; Yamamoto, K.; Inoue, T.; Fujimoto, K.; Yamamura, H.; Kawai, M.; Butsugan, Ya.; Zhou,
J.; Eichhorn, E.; Rieker, A.; Huber, M. (1999) J. Chem. Soc., Perkin Trans. 2, 985.Baciocchi, E.; Bietti, M.; Mattioli, M. (1993) J. Org. Chem. 58, 7106.Baciocchi, E.; Rob, C.; Mandolini, L. (1980) J. Am. Chem. Soc. 102, 7597.Baizer, M.M.; Lund, H., Eds. (1983) Organic Electrochemistry, 2nd ed. Marcel Dekker, New York.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Bally, Th.; Carra, C.; Matzinger, S.; Truttmann, L.; Gerson, F.; Schmidlin, R.; Platz, M.S.; Admasu,A. (1999) J. Am. Chem. Soc. 121, 7011.
Baskakov, D.V.; Baranova, I.A.; Fel’dman, V.I.; Mel’nikov, M.Ya. (2000) Dokl. Akad. Nauk 375, 56.Bauld, N.L.; Yueh, W. (1994) J. Am. Chem. Soc. (1994) 116, 8845.Belevskii, V.N.; Belopushkin, S.I.; Chuvylkin, N.D. (1998) High-Energy Chem. 32, 171.Boaz, N.W.; Venepalli, B. (2001) Org. Process Res. Develop. 5, 127.Bock, H.; Gharagozloo-Hubmann, K.; Holl, S.; Sievert, M. (2000a) Z. Naturforsch. B: Chem. Sci. 55,
1163.Bock, H.; Gharagozloo-Hubmann, K.; Sievert, M.; Prizner, T.; Havlas, Z. (2000b) Nature 404, 267.Bock, H.; Sievert, M.; Bogdan, C.L.; Kolbesen, B.O.; Wittershagen, A. (1999) Organometallics 18,
2387.Bordwell, F.G.; Clemens, A.H. (1981) J. Org. Chem. 46, 1035.Brand, J.C.; Roberts, B.P.; Strube, R. (1985) J. Chem. Soc. Perkin Trans. 2, 1659.Buchanan, A.C.; Livingston, R.; Dvorkin, A.S.; Smith, G.P. (1980) J. Phys. Chem. 84, 423.Burgbacher, G.; Schaefer, H.J. (1979) J. Am. Chem. Soc. 101, 7590.Carrington, A.; Dravnieks, F.; Symons, M.C.R. (1959) J. Chem. Soc., 947.Carter, M.K. (1971) J. Phys. Chem. 75, 902.Cavazza, M.; Nucci, L.; Pannocchia, E.; Pardi, L.; Pergola, P.; Pinzino, C.; Pietra, F. (1999) Tetra-
hedron 55, 11601.Chang, R.; Coombe, R. (1971) J. Phys. Chem. 75, 447.Chang, R.; Johnson, Ch.S. (1965) J. Chem. Phys. 43, 3183.Chang, R.; Johnson, Ch.S. (1966) J. Am. Chem. Soc. 88, 2338.Chanon, M.; Rajzmann, M.; Chanon, F. (1990) Tetrahedron 46, 6193.Charoenkwan, K.; Todres, Z.V.; Evans, D.H. (in preparation).Chipman, D.M.; Prebenda, M.F. (1986) J. Phys. Chem. 90, 5557.Chou, T.Sh.; You, M.L. (1987) J. Org. Chem. 52, 2224.Clarke, E.D.; Wardman, P.; Wilson, I. (1984a) Biochem. Pharmacol. 33, 83.Clarke, E.D.; Wardman, P.; Wilson, I.; Tatsumi, K. (1984b) J. Chem. Soc. Perkin Trans. 2, 1155.Cohen, Th.; Kreethadumrongdat, Th.; Liu, X.; Kulkarni, V. (2001) J. Am. Chem. Soc. 123, 3478.Colichman, E.L.; Love, D.L. (1953) J. Organ. Chem. 18, 40.Connelly, N.G.; Geiger, W.E. (1996) Chem. Rev. 96, 877.Czezhegyi, A; Jagur-Grodzinski, J.; Szwarc, M. (1969) J. Am. Chem. Soc. 91, 1892.Damaskin, B.B.; Kryshtalik, L.I. (1984) Electrokhimiya 20, 291.De Wall, S.L.; Barbour, L.J.; Gokel, G.W. (1999) J. Am. Chem. Soc. 121, 8405.Dvorak, V.; Michl, J. (1976) J. Am. Chem. Soc. 98, 1080.Eastland, G.W.; Kurita, Y.; Symons, M.C.R. (1984) J. Chem. Soc., Perkin Trans. 2, 1843.Eberson, L. (1967) J. Am. Chem. Soc. 89, 4669.Eberson, L. (1983) J. Am. Chem. Soc. 105, 3192.Eberson, L.; Radner, F. (1991) Acta Chem. Scand. 45, 1093.Eberson, L.; Wistrand, L.-G. (1980) J. Electrochem. Soc. 127, 33.Efrima, S.; Bixon, M. (1974) Chem. Phys. Lett 25, 34.Elbl-Weiser, K.; Neugebauer, F.A.; Staab, H.A. (1989) Tetrahedron Lett. 30, 6161.Elson, J.; Kochi, J.K. (1973) J. Am. Chem. Soc. 95, 5060.Forbes, W.F.; Sullivan, P.D. (1966) J. Am. Chem. Soc. 88, 2862.Fukuzumi, Sh.; Ohkubo, K.; Suenobu, T.; Ito, O.; Fujitsuka, M.; Shao, J.; Kadish, K.M. (2000) Proc.
Electrochem. Soc., 2000–10, 68.Gennaro, A.; Maye, A.; Maran, F.; Vianello, E. (1988) Electrochem. Acta 33, 167.Gerson, F.; Lamprecht, A.; Scholtz, M.; Troxler, H.; Lenoir, H. (1996) Helv. Chim. Acta 79, 307.Goodnow, T.T.; Kaifer, A.E. (1990) J. Phys. Chem. 94, 7682.Grenier, J.L.; Cotelle, P.; Cotelle, N.; Catteau, J.P. (1995) J. Chim. Phys. Phys. Chim. Biol. 92, 97.Grovenstein, E.; Longfield, T.H.; Quest, D.E. (1977) J. Am. Chem. Soc. 99, 2800.Guijarro, A.; Rosenberg, D.M.; Rieke, R.D. (1999) J. Am. Chem. Soc. 121, 4155.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Guthrie, R.D.; Shi, B. (1990) J. Am. Chem. Soc. 112, 3136.Hammerich, O.; Nielsen, M.F.; Zuilhof, H.; Mulder, P.P.J.; Lodder, G.; Reiter, R.C.; Kage, D.E.;
Rice, Ch.V.; Stevenson, Ch.D. (1996) J. Phys. Chem. 100, 3454.Handoo, K.L.; Handoo, S.K.; Gadru, K.; Kaul, A. (1985) Tetrahedron Lett., 26, 1765.Hiroshi, Ya.; Fijimura, N.; Kawafuji, Yu.; Hanafusa, T. (1987) J. Am. Chem. Soc. 109, 4305.Hitchcock, P.B.; Lappert, M.F.; Protchenko, A.V. (2001) J. Am. Chem. Soc. 123, 189.Hoffmann, G.G.; Steinfatt, I.; Brockner, W.; Kaiser, V. (1999) Z. Naturforsch. 54b, 887.Holm, T. (1993) J. Am. Chem. Soc. 115, 916.Holm, T. (1994) J. Am. Chem. Soc. 116, 8803.Horner, L.; Roder, H. (1969) Liebigs Ann. Chem. 723, 11.Hyde, J.S.; Brown, H.W. (1962) J. Chem. Phys. 37, 368.Iida, Y.; Akamatu, H. (1967) Bull. Chem. Soc. Japan 40, 231.Ioffe, N.T.; Todres, Z.V.; Tsyryapkin, V.A.; Kursanov, D.N. (1971) Zh. Obshch. Khim. 41, 372.Ishizu, K.; Ochichi, M.; Nemoto, F.; Suga, M. (1973) Bull. Chem. Soc. Japan 46, 2932.Itagaki, Y.; Shiotani, M. (1999) J. Phys. Chem. 103, 5189.Jacob, R.; Puranik, M.; Chandrasekhar, Ja. (1999) Chem. Phys. Lett. 301, 498.Johnansen, H.; Schoen, J. (1980) Z. Phys. Chem. 261, 282.Julliard, M.; Chanon, M. (1983) Chem. Rev. 83, 425.Kadam, R.M.; Itagaki, Y.; Erickson, R.; Lund, A. (1999) J. Phys. Chem. A 103, 1480.Kim, T.; Mirafzal, G.A.; Liu, J.; Bauld, N.L. (1993) J. Am. Chem. Soc. 115, 7653.Kimura, N.; Takamuku, S. (1994) J. Am. Chem. Soc. 116, 4087.Knight, L.B.; Ebner, M.A. (1976) J. Mol. Spectrosc. 61, 412.Kochi, J.K.; Tang, R.T.; Bernath, T.J. (1973) J. Am. Chem. Soc. 95, 7114.Kolos, R. (1995) Chem. Phys. Lett. 247, 289.Kon, H.; Blois, M.S. (1958) J. Chem. Phys. 23, 743.Koyama, K.; Ebara, T.; Tani, T.; Tsutsumi, Sh. (1969) Can. J. Chem. 47, 2484.Kurth, T.L.; Brown, E.C.; Smirnov, A.I.; Reiter, R.C.; Stevenson, Ch.D. (1999) J. Phys. Chem. A 103,
8566.Landolt-Boernstein, Numerical Data and Fundamental Relationships in Science and Technology
(1980) 9d1, 708–856; 9d2, 6–20. Springer Verlag, Berlin.Lauricella, T.L.; Pescatore, J.A.; Reiter, R.C.; Stevenson, R.D.; Stevenson, G.R. (1988) J. Phys.
Chem. 92, 3687.Leigh, W.J.; Arnold, D.R. (1981) Can. J. Chem. 59, 3061.Lewis, I.C.; Singer, L.S. (1965) J. Chem. Phys. 43, 2712.Lipsztain, M.; Krygowskii, T.M.; Laren, E.; Galus, E. (1974) J. Electroanal. Chem. 54, 313.Lucarini, M.; Pedulli, G.F.; Valgimigli, L. (1994) Gazz. Chim. Ital. 124, 455.Luche, J.L.; Petrier, C.; Dupuy, C. (1984) Tetrahedron Lett. 25, 753.Lue, F.-U.; Wen, X.-L.; Wu, L.-U.; Liu, Y.-Ch.; Liu, Zh.-L. (2001) J. Phys. Chem. A 105, 6971.Luettringhaus, A.; Machatzke, H. (1964) Liebigs Ann. Chem. 671, 165.Lund, H.; Hammerich, O., Ed. (2001) Organic Electrochemistry, 4th ed. Marcel Dekker, New York.Lund, T.; Ohlrich, D.; Borling, P. (1999) Acta Chem. Scand. 53, 932.Mango, J.D.; Bonner, W.A. (1964) J. Org. Chem. 29, 1367.Mann, Ch.K.; Barnes, K.K. (1970) Electrochemical Reactions in Nonaqueous Systems. Marcel
Dekker, New York.Matsuo, H. (1958) J. Sci. Hiroshima Univ. Ser. A 22, 281.Matsuyama, T.; Yamataka, H.; Hanafusa, T. (1988) Chem. Lett. 8, 1367.McKillop, A.; Taylor, E.C. (1973) Adv. Organomet. Chem. 11, 147.Meccozi, S.; West, A.P.; Dougherty, D.A. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 10566.Michaelis, L.; Granick, M.; Schulbert, L. (1941) J. Am. Chem. Soc. 63, 351.Minisci, J.; Citterio, A.; Giordano, C. (1983) Acc. Chem. Res. 16, 27.Mirafzal, G.A.; Loseva, A.M.; Olson, J.A. (1998) Tetrahedron Lett. 39, 9323.Morris, D.E.; Smith, W.H. (1991) J. Electrochem. Soc. 138, 1351.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Norman, R.O.C.; Thomas, C.B.; Ward, P.J. (1973) J. Chem. Soc., Perkin Trans. 1, 2914.Nyberg, K.; Wistrand, L.-G. (1978) J. Org. Chem. 43, 2613.Ohta, N.; Ichikawa, T. (1987) J. Phys. Chem. 91, 3736.Oku, T.; Yoshiura, N.; Okuda, T. (1983) J. Org. Chem. 48, 617.Orlov, V.Yu.; Kotov, A.D.; Rusakov, A.I.; Begunov, R.S.; Khyen, N.D. (2001) Izv. Vyssh.
Uchebn.Zaved. Khim. Khim. Tekhnol. 44, 127.Parker, V.D.; Eberson, L. (1969) J. Chem. Soc. Chem. Commun., 340.Paulssen, H.; Haitjema, H.; Van Asselt, R.; Mylle, P.; Adriaensens, P.; Gelan, J.; Vanderzande, I
(2000), Polymer 41, 3121.Persson, B.; Lindberg, B. (1977) J. Electroanal. Chem. 78, 123.Persson, B.; Nygard, B. (1974) J. Electroanal. Chem. 56, 373.Pitchumani, K.; Lakshminarasimhan, P.H.; Presvost, N.; Corbin, D.R.; Ramamurthy, V. (1997)
Chem. Commun. 181.Podol’sky, A.F.; Boldyrev, A.G.; Sapurina, I.Yu. (1982) Acta Polimerica 33, 181.Pryor, W.A.; Hendrickson, W.H. (1983) J. Am. Chem. Soc. 105, 7114.Ramakrishna Rao, D.N.; Symons, M.C.R. (1985) J. Chem. Soc. Perkin Trans. 2, 991.Rathore, R.; Lindeman, S.V.; Kumar, A.S.; Kochi, J.K. (1998) J. Am. Chem. Soc. 120, 6931.Reutov, O.A.; Butin, K.P. (1975) J. Organomet. Chem. 99, 171.Rieke, R.D.; Hanson, M.V. (1997) Tetrahedron 53, 1925.Rudenko, A.P. (1994) Zh. Org. Khim. 30, 1847.Sastry, N.; Reddy, A.Ch.; Shaik, S. (1995) Angew. Chem. Int. Ed. Engl. 34, 1495.Saveant, J.-M. (1980) Acc. Chem. Res. 13, 323.Saveant, J.-M. (1993) Acc. Chem. Res. 23, 455.Saveant, J.-M. (2001) J. Phys. Chem. B, 105, 8995.Schlesener, C.J.; Kochi, J.K. (1984) J. Org. Chem. 49, 3142.Schlesener, C.J.; Amatore, C.; Kochi, J.K. (1984) J. Am. Chem. Soc. 106, 3567.Schulz, A.; Kaim, W.; Hausen, H.D. (1988) J. Chem. Soc. Faraday Trans. 84, 3207.Sehgal, C.; Yu, T.J.; Sutherland, R.G.; Verrall, R.E. (1982) J. Phys. Chem. 86, 2982.Seo, E.T.; Nelson, R.F.; Fritsch, J.M; Marcoux, L.S.; Leedy, D.W.; Adams, R.N. (1966) J. Am. Chem.
Soc. 88, 3498.Shaw, M.J.; Wiel, J.A.; Hyman, H.H.; Filler, R. (1970) J. Am. Chem. Soc. 92, 5096.Shida, T.; Haselbach, E.; Bally, Th. (1984) Acc. Chem. Res. 17, 180.Sokolov, V.I.; Bashilov, V.V.; Timerghazin, Q.K.; Avzyanova, E.V.; Khalizov, A.F.; Shishlov, N.M.
Shereshovets, V.V. (1999) Mendeleev Commun., 54.Soos, Z.G.; Keller, H.J.; Moroni, W.; Noethe, D.J. (1977) J. Am. Chem. Soc. 99, 5040.Sosonkin, I.M.; Kalb, G.L.; Yur’ev, V.P. (1983) Dokl. AN SSSR 270, 340.Stamirez, D.N.; Turkevich, J. (1963) J. Am. Chem. Soc. 85, 2557.Steckhan, E. (1977) Electrochim. Acta 22, 395.Steckhan, E. (1978) J. Am. Chem. Soc. 100, 3526.Stevenson, Ch.D.; Halvorsen, T.D.; Kage, D.E.; Reiter, R.C.; McElhemy, D.J. (1993) J. Org. Chem.
58, 4634. [Regardless of the initials (G.R.—Gerald R. or Ch.D.—Cheryl D.), ProfessorStevenson is the same author, see this reference.]
Stevenson, Ch.D.; Halvorsen, T.D.; Reiter, R.C. (1993) J. Am. Chem. Soc. 115, 12405.Stevenson, Ch.D.; Kurth, T.L. (1999) J. Am. Chem. Soc. 121, 1623.Stevenson, Ch.D.; Reiter, R.C.; Burton, R.D.; Halvorsen, T.D. (1995) Inorg. Chem. 34, 1368.Stevenson, Ch.D.; Schertz, T.; Reiter, R.C. (1999) J. Am. Chem. Soc. 121, 6964.Stevenson, Ch.D.; Wagner, E.P.; Reiter, R.C. (1993) Inorg. Chem. 32, 2480.Stevenson, G.R.; Alegria, A.E. (1976) J. Phys. Chem. 80, 69.Stevenson, G.R.; Ballard, M.K.; Reiter, R.C. (1991) J. Org. Chem. 56, 4070.Stevenson, G.R.; Espe, M.P.; Reiter, R.C. (1986) J. Am. Chem. Soc. 108, 5760.Stevenson, G.R.; Espe, M.P.; Reiter, R.C.; Lovett, D.J. (1986) Nature 323, 522.Stevenson, G.R.; Lovett, D.J.; Reiter, R.C. (1986) J. Phys. Chem. 90, 4461.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Stevenson, G.R.; Peters, S.J.; Reidy, K.A. (1990) Tetrahedron Lett. 31, 6151.Stevenson, G.R.; Peters, S.J.; Reidy, K.A.; Reiter, R.C. (1992) J. Org. Chem. 57, 1877.Stevenson, G.R.; Reiter, R.C.; Au-Yeung, W.; Pescatore, J.A.; Stevenson, R.D. (1987) J. Org. Chem.
52, 5063.Stevenson, G.R.; Reiter, R.C.; Espe, M.E.; Bartmess, J.E. (1987) J. Am. Chem. Soc. 109, 3847.Stevenson, G.R.; Sturgeon, B.E. (1990) J. Org. Chem. 55, 4090.Stevenson, G.R.; Sturgeon, B.E.; Vines, K.Sh.; Peters, S.J. (1988) J. Phys. Chem. 92, 6850.Stevenson, G.R.; Wehrmann, G.C.; Reiter, R.C. (1991) J. Phys. Chem. 95, 901.Stuart, J.D.; Ohnesorge, W.E. (1971) J. Am. Chem. Soc. 93, 4531.Svanholm, U.; Ronlan, A.; Parker, V.D. (1974) J. Am. Chem. Soc. 96, 5108.Symons, M.C.R.; Bowman, W.R. (1984) J. Chem. Soc. Chem. Commun., 1445.Symons, M.C.R.; Wren, B.W. (1984) J. Chem. Soc. Perkin Trans. 2, 511.Tanko, J.M.; Drumright, R.E. (1992) J. Am. Chem. Soc. 114, 1844.Terekhova, M.I.; Kron, T.E.; Petrov, E.S. (1996) Zh. Obshch. Khim. 66, 339.Todres, Z.V. (1970) Izv. AN SSSR, Ser. Khim., 1749.Todres, Z.V. (1974) Russian Chem. Rev. (English transl.) 43, 1099.Todres, Z.V. (1980) Zh. Fis. Khim. 54, 1097.Todres, Z.V. (1985) Tetrahedron 41, 2771.Todres, Z.V. (1987) Tetrahedron 43, 3839.Todres, Z.V. (1988) Elektrokhimiya, 24, 563.Todres, Z.V.; Avagyan, S.P.; Kursanov, D.N. (1975) Zh. Org. Khim. 11, 2457.Todres, Z.V.; Bespalov, V.Ya. (1972) Zh. Org. Khim. 8, 19.Todres, Z.V.; Ionina, E.A. (1992) Izv. Ross. AN Ser. Khim., 1219.Todres, Z.V.; Tsvetkova, T.M. (1987) Izv. AN SSSR Ser. Khim., 1553.Todres, Z.V.; Dyusengaliev, K.I.; Buzlanova, M.M.; Shklover, V.E.; Struchkov, Yu.T. (1990) Zh.
Org. Khim. 26, 845.Todres, Z.V.; Dyusengaliev, K.I.; Garbuzova, I.A. (1986) Zh. Org. Khim. 22, 370.Todres, Z.V.; Dyusengaliev, K.I.; Sevast’yanov, V.G. (1985) Izv. AN SSSR Ser. Khim., 1416.Todres, Z.V.; Dyusengaliev, K.I.; Ustynyuk, N.A. (1982) Izv. AN SSSR Ser. Khim., 1389.Todres, Z.V.; Furmanova, N.G.; Avagyan, S.P.; Struchkov, Yu. T.; Kursanov, D.N. (1979) Phosph.
Sulfur 6, 309.Todres, Z.V.; Kukovitskii, D.M.; Zorin, V.V.; Zlotskii, S.S.; Rahmankulov, D.L. (1981) Izv. AN
SSSR Ser. Khim., 1577.Tomioka, H.; Ueda, K.; Ohi, H.; Izawa, Ya. (1986) Chem. Lett., 1359.Vinogradov, M.G.; Todres, Z.V.; Il’ina, G.P.; Rutavichus, A.I.; Kursanov, D.N.; Nikishin, G.I.
(1976) Izv. AN SSSR Ser.Khim., 1331.Wallwork, S. (1961) Acta Crystallogr. 14, 375.Wang, L.X.; Shi, Y.Z.; Yao, Y.M.; Hu, H.W. (1994) Chinese Chem. Lett. 5, 825.Wawzonek, S.; Blaha, E.; Berkey, R.; Runner, M. (1965) J. Electrochem. Soc. 102, 235.Weinberg, N.L.; Weinberg, H.R. (1968) Chem. Rev. 68, 470.Weiss, J. (1941) Nature 147, 512.Weissmann, S.T.; Boer de, T.; Couradi, J.J. (1957) J. Chem. Phys. 26, 963.Wolf, M.O.; Fox, H.H.; Fox, M.A. (1996) J. Org. Chem. 61, 287.Yakovleva, E.A.; Petrov, E.S.; Solodovnikov, S.P.; Shatenshtein, A.I. (1960) Dokl. AN SSSR 133, 645.Yamataka, H.; Fujimara, N.; Kawafuji, Yu.; Hanafusa, T. (1987) J. Am. Chem. Soc. 109, 4305.Yamataka, H.; Matsuyama, T.; Hanafusa, T. (1987) Chem. Lett., 647.Yamataka, H.; Matsuyama, T.; Hanafusa, T. (1989) J. Am. Chem. Soc. 111, 4912.Yamataka, H.; Mishima, M.; Kuwatani, Y.; Tsuno, Y. (1995) J. Am. Chem. Soc. 117, 5829.Yamataka, H.; Sakaki, D; Kuwatani, Y.; Mishima, M.; Tsuno, Y. (1997) J. Am. Chem. Soc. 119,
9975.Yamataka, H.; Sasaki, D.; Kuwatani, Y.; Mishima, M.; Shimizu, M.; Tsuno, Y. (2001) J. Org. Chem.
66, 2131.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Yoon, K.B. (1993) Chem. Rev. 93, 321.Yoon, K.B.; Kochi, J.K. (1988) J. Am. Chem. Soc. 110, 6586.Yus, M. (1996) Chem. Soc. Rev., 155.Yusupov, R.G.; Hairutdinov, R.F. (1987) Dokl. AN SSSR 295, 665.Zuilhof, H.; Lodder, G. (1992) J. Phys. Chem. 96, 6957.Zuilhof, H.; Lodder, G. (1995) J. Phys. Chem. 99, 8033.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

3
Basic Principles of Organic Ion Radical Reactivity
3.1 INTRODUCTION
Organic ion radicals carry a charge and an unpaired electron. Their distribution along amolecular contour determines ion radical reactivity. That is why it is so important to eluci-date the principles that control organic ion radical reactivity.
This chapter considers ion radicals with “detained” and “released” unpaired elec-trons. Some ion radicals contain fragment orbitals that suspend an unpaired electron pref-erentially. Other ion radicals are characterized by delocalization of an unpaired electronalong orbitals that are more or less evenly populated with an unpaired electron. This chap-ter considers the material from this point of view, using the terms detained and releasedelectron. Such abstraction helps us to analyze these two intrinsic features of organic ionradical reactivity.
Section 3.2 (Principle of the “Detained” Electron That Controls Ion Radical Reactiv-ity) includes an extensive body of phenomena, from the formation of three-electron bonds,to ion pairing, to the distonic stabilization of ion radicals at the expense of separation be-tween their spins and charges.
Section 3.3 (Principle of the “Released” Electron that Controls Ion Radical Reactiv-ity) deals with ion radicals from the better-known group of species. However, the readerwill find newly developed versions of the principle of the “released” electron, concerningspread conjugation and the fates of ion radical precursors with increased dimensionality. Itis obvious that any categorization tends to name the main trait of the phenomenon underconsideration. That is useful. At the same time, the categorization ought not to be under-stood literally, because each effect possesses multiple characteristics. However, it is im-possible to study anything without at least a minimal classification.
Section 3.4 is devoted to organic ion radical behavior in living organisms. In partic-ular, consideration of an ion radical mechanism of carcinogenesis reflects a point of viewthat is new and, in many respects, promising.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

3.2 PRINCIPLE OF THE “DETAINED” ELECTRON THAT CONTROLSION RADICAL REACTIVITY
3.2.1 Frontier-Orbital Control
In the donor–acceptor interaction, the acceptor provides its lowest unoccupied molecularorbital (LUMO) and the donor participates at the expense of its highest occupied molecu-lar orbital (HOMO). These orbitals are frontier orbitals. In the corresponding ion radicals,the distribution of an unpaired electron proceeds, naturally, under frontier-orbital control.This definitely is reflected in ion radical reactivity.
A reaction between methoxythioanisole and metallic sodium in HMPA results in theformation of an initial anion radical. This anion radical contains an unpaired electron at thethiomethyl group. Scission of the thiomethyl group is the next step of the reaction. Theproduct obtained is reduced no further. Such selective dealkylation proceeds according toScheme 3-1:
Ar(OMe)SMe � Na → Ar(OMe)SMe�.Na� → Ar(OMe)SNa � Me
.
The strict selectivity of the reaction is explained by the electroacceptor properties of sulfurd-orbitals. These orbitals are not as high in energy as oxygen d-orbitals are (Testaferi et al.1982).
However, if a sulfur-containing group is conjugated with the greater acceptor sub-stituent, this substituent, though not a sulfur-containing group, accepts an unpaired elec-tron. This protects the sulfur-containing group from scission. For example, anion radicalsof benzene thiocyanates, sulfenamides, and alkylsulfenates are not stable. They are cleavedto give diphenyl disulfides. The analogs with the nitro group in the ortho or para positionof the benzene ring give stable anion radicals with no scission of the enumerated sulfur-containing groups (Todres & Avagyan 1972, 1978). Similarly, in anion radicals ofthioamides of nitrobenzoic acids, the nitro group prevails over the thioamide group[C(S)NR2] in competition for the unpaired electron (Ciureanu 1987).
If the nitro group is located at the ethylenic fragment, one-electron transfer initiatesdimerization of the developing anion radicals. �-Nitrostilbene, �-methyl-�-nitrostyrene,and �-nitro- -ferrocenylethylene give anion radicals that dimerize spontaneously. It is in-teresting to compare reactions of cyclo-octatetraene dipotassium (C8H8K2) with �-nitro
SCHEME 3-2
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

and �-cyano ferrocenylethylenes (Todres & Tsvetkova 1987; Todres & Ermekov 1989),Scheme 3-2. When X � CN, a stable anion radical is obtained, as is evident from the ESRspectrum. In the case of X � NO2, the ESR signal cannot be observed. This anion radicalis unstable and gives rise to the dimer, Scheme 3-3.
It is interesting that reduction of cinnamonitrile, related aromatic nitriles, and acry-lonitrile gives dimers, Scheme 3-4:
2CH2BCHCN �2e→ 2[CH2BCHCN]�. ↔ 2�CH2C
�HCN H�
→ NC(CH2)4CN
In contrast to acrylonitrile and the related nitriles, ferrocenylacrylonitrile does notproduce a dimer upon one-electron reduction. The ferrocenyl moiety is the constant frag-ment of the nitro and cyano ferrocenylethylene anion radicals. Hence, the shielding effectof the ferrocenyl group is evidently not the determining factor (in spite of the bulky size ofthe group). In the nitro-group case, the ferrocenyl moiety cannot compete with the nitrogroup for an unpaired electron. [From infrared spectra and ab initio molecular orbital cal-culations, “the main holder of the anionic charge in the 1-cyano-4-nitrobenzene anion rad-ical is the nitro group” (Tsenov et al. 1998)]. In the cyano substituent case, the ferrocenylmoiety can compete for an unpaired electron. The ability of the ferrocenyl group to partic-ipate in unpaired electron delocalization is documented (Todres, Safronov, et al. 1992).
Competition between the ferrocenyl and cyano acceptors leads to strong delocaliza-tion of unpaired electron throughout a molecular skeleton. This prevents dimerization ofanion radicals. Meanwhile, if an unpaired electron is suspended in the framework of theNOO� group, a radical center is formed at the -position, causing the mentioned dimer-ization.
Comparing the electrochemical behavior and biological transformations of purinebases, Japanese chemists have considered the anion radicals of purine, its 8-deuterio- and 6,8-dideuterioderivatives (Yao & Musha 1974; Ohya-Nishiguchi et al. 1980). As it turned out, upto 40% of the total spin density is localized in position 6 of the purine anion radical (see
SCHEME 3-3
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Scheme 3-5). Ohya-Nishiguchi et al. (1980) noted that such a large localized spin density isvery rare in a �-electron system of purine’s size and should have important application to itschemical reactivity. Reactions such as protonation should take place preferentially at position6. This was deduced from the result of molecular orbital calculations (Nakajima & Pullman1959). According to Fukui’s frontier electron theory (Fukui et al. 1952), such a reaction shouldtake place at the position where the frontier electron density is the largest. The calculationsclearly indicate that the large electron density is at position 6. Scheme 3-5 describes the pro-tonation of the purine anion radical (Yao & Musha 1974). Protonation indeed takes place atposition 6. After that, the radical center appears at the cyclic nitrogen in the vicinal 1 position.
In the pyridine-N-oxide anion radical, the greatest term of the LUMO belongs to thecarbon atom in position 2 (Chaha 1986). In accordance with that, a reaction between thepyridine-N-oxide anion radical and the benzophenone metal-ketyl yielded preferentially 2-diarylcarbinole derivatives (Kurbatova et al. 1980; Turaeva et al. 1993), Scheme 3-6.
SCHEME 3-5
SCHEME 3-6
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The reaction depicted also proceeds between pyridine oxide in its neutral form andthe double amount of benzophenone metal-ketyl. Pyridine bases (not N-oxides) do not re-act with the ketyls. The N-oxides of pyridine and �-picoline give both the N-oxide of thepyridyl carbinol and the pyridyl carbinol without the N-oxide oxygen. Yields can be 70 and80%, respectively (depending on the metal nature in the metal-ketyl). Having pronouncedphysiological activity, these compounds are the key materials in syntheses of atropine-likedrugs.
The reaction found has features that are significant for the organic chemistry of an-ion radicals. Firstly, one-electron transfer from a reactant (the ketyl) to the substrate (thepyridine N-oxide) proceeds against the gradient of electrochemical potentials. Secondly,there is doubling of the two anion radicals (which belong to different classes) despitecharge repulsion. It follows that one-electron transfer can also take place at a nonfavorabledifference between redox potentials if the transfer is reversible but one of the formed prod-ucts is going out of the equilibrium swiftly and irreversibly. Another conclusion is thatdimerization of different anion radicals can prevail over dimerization of identical anionradicals in cases where a “different-ligand” dimer is promptly stabilized into a substitutionproduct. The presence of a less thermodynamically stable but more reactive compound de-rived from the starting material makes a crucial contribution to the reaction.
With respect to the aromatic N-oxide cation radicals, one unusual aspect of theirbehavior should be noted. Having been formed, they are prone to form dimers with a pro-ton bridging the two N-oxide oxygens of two of these cation radicals. Such structures areformed, e.g., in the reactions between N-oxides of substituted quinolines and tetra-cyanoquinodimethide, TCNQ (Alekseeva and others 1999). The origin of the proton ofthe bridge deserves some additional study, perhaps using deuterated participants. Proba-bly, TCNQ, being a strong acceptor, is capable of abstracting an electron from the donor,quinoline N-oxide. The latter thus converted into a cation radical, which takes up a hy-drogen atom from the solvent (anhydrous acetonitrile) to form an N-hydroxylquinoliniumcation. The latter acts as a protonodonor for other N-oxide cation radicals in the solution.N-oxide cation radicals were shown to abstract hydrogen even from alkanes (Geletii etal. 1986).
Doubling of identical ion radicals seems to be the most effective way to preparestereochemically identical dimers (Kise et al. 2001). Analyzing the dimer structure, onecan distinguish a position of preferential localization of spin density. Thus, since the sin-gle-occupied molecular orbital coefficient is greater at the �-position than at the -posi-tion of the thiophene cation radicals, only �,�-coupling occurs. This is in accord withthe experimental spin density distribution obtained from the ESR spectrum. Moreover,polymerization of the thophene cation radicals also proceeds at the expense of the �-po-sitions. The reaction leads to polythiophene. Polythiophene is of interest as a stable elec-trical conductor and semiconductor, especially when doped. This material may be ofvalue for “plastic electronics,” in which lightweight, processable, deformable plastics re-place metals. There is a special review detailing the properties of the oligothiophenecation radicals (Glass 1999) as well as two recent original papers on the oligothiophene-dendron cation radicals and the fullerene-oligothiophene-fullerene dumbbell cation radi-cals (Apperloo and co-authors 2000a,b).
Dimerization of pyrazolines-2 upon the action of oxidants includes the formation anddoubling of cation radicals (Morkovnik & Okhlobystin 1979). The doubling process is usu-ally characterized by the terms head and tail. The term head is applicable to the positionbearing a polar fragment, while the term tail is adopted for an unsaturated molecular site.The reaction under consideration follows the “head-to-head” order. This means that the
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

doubling cation radical has only a single position with the maximal density of an unpairedelectron, Scheme 3-7.
In contrast, if two positions in a cation radical have the increased density of an un-paired electron, the dimerization understandably follows a “head-to-tail” pattern. This isthe case of 2,3-diphenyl indole (Check & Nelson 1978), Scheme 3-8.
SCHEME 3-7
SCHEME 3-8
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The anion-radical of 1,1-binaphthyl acts as the single �-system with homogeneousspin distribution. Comparison between 1,1-binaphthyl and biperylenyl in Scheme 3-9shows their structural similarity.
It is therefore somewhat surprising that in the biperylenyl anion radical Baumgartenet al. (1993) observed a localization of the unpaired electron in one moiety only, althoughthe steric situation is anticipated to be similar to that in the binaphthyl anion radical. Thisis explained by the fact that the atomic orbital coefficients of the single-occupied molecu-lar orbital for the bridgehead positions are larger for the binaphthyl anion radical than forthe biperynyl anion radical. Therefore, the interaction of the separate entities is more de-coupled in the biperynyl case.
3.2.2 Steric Control over Spin Delocalization
A typical example of steric control over spin delocalization in cation radicals has been de-scribed for permethylated dithia[6]radialene (Gleiter et al. 1996). As shown, the unpairedelectron is delocalized only in one half of this cation radical, within the limits of the 2,3-dithiatetramethyl-butadiene unit. Owing to the steric demand of the isopropilidene groups,two of the four-methylene groups are twisted, while the other two are coplanar; the authorsgive Scheme 3-10.
The equilibrium shown in the Scheme 3-10 calls for a high activation energy due tothe steric requirements. Therefore, the inner reorganization energy is high when inner elec-tron transfer occurs. Of course, the outer reorganization energy of the solvent cage (see Sec-
SCHEME 3-9
SCHEME 3-10
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

tion 5.4.2) must be high in this case, too. The sum of the energies is responsible for the lo-calized cation radical structure.
Diphenyl fulvene is another example of steric control over spin delocalization in an-ion-radicals. The anion radical is a result of the reduction of diphenyl fulvene (Todres &Bespalov 1972) or the oxidation of diphenyl cyclopentadienyl methane (Camaggi et al.1971). The ESR spectrum of this anion radical allows it to be viewed as a triarylmethyl rad-ical in which one of the aryl groups is substituted for the anion of cyclopentadienyl (Cam-aggi et al. 1971). The radical center (the site of an unpaired electron) is shielded with twophenyl rings. The situation resembles that in triphenylmethyl radical, where stabilization ofthe radical center is secured with steric shielding and spin delocalization over and throughphenyl rings. The cyclopentadienyl ring is stabilized just like an aromatic anion. The anionradical Ph2C
.–C5H5
� is stable; it is not involved in dimerization or disproportionation andcan be produced by the inverted reaction of the preliminary prepared dianion with the neu-tral molecule (Todres & Bespalov 1972), Scheme 3-11.
Hence, the anion radical of diphenyl fulvene acquires a spin-charge distribution dic-tated by steric shielding at the Ph2C
.node and six-�-electron delocalization in the C5H5
ring. Anion radicals of sterically congested stilbenes represent examples that are quite dif-ferent but at the same time are similar in principle. Let us compare the E structures of stil-bene and its congested derivative, �, -di(tert-butyl)stilbene in their neutral and anion rad-ical forms (Scheme 3-12).
E-Stilbene is planar in crystalline form, in gas phases, and presumably in solution, al-though the phenyl group may be rotated as much as 32° to reduce nonbonded repulsionsbetween hydrogen atoms (Waldek 1991; Meier 1992). This modest twisting still allows suf-ficient �-overlap for a continuous, conjugated �-system between the phenyl groups and thecentral double bond.
The introduction of two tert-butyl groups onto the central double bond in stilbenecauses the phenyl groups to rotate out of the molecular plane so that their planes becomeperpendicular and their �-systems become orthogonal to the central double bond (Gano,Park, et al. 1991). There is no conjugation between the phenyl groups through the ethylenic�-system. In spite of the apparent lack of conjugation, photoelectron spectroscopy estab-lishes that there is still orbital interaction between the �-systems of the phenyl groups(Gano, Jacob, et al. 1996).
A stilbene anion radical undergoes only moderate additional twisting about the cen-tral double bond so as to retain much of the conjugation throughout the �-system, while the�, -di(tert-butyl)stilbene anion radical appears to be more twisted as compared to its pre-cursor. In the latter anion radical, the phenyl group is coplanar, with the adjacent carbonatom creating the resonance-stabilized benzyl group. Evidently, when an electron is trans-ferred to the dibutyl stilbene, conjugation is lost by twisting about the CBC bond as con-jugation is gained by twisting about the Ph–C bond (Gano, Jacob, et al. 1996).
The concluding principal example is the anion-radical of 9,9-bianthryl. In the neu-tral molecule of bianthryl, the anthracene units are arranged nearly orthogonally. Such an
SCHEME 3-11
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

arrangement keeps in the anion radical, in which the unpaired electron is localized in onesubunit. In this example, the conjugative interaction between the two halves of themolecule is inhibited by the strong torsion about the connecting bond (Baumgarten et al.1992). This, however, is not generally true for biaryl systems. As will be shown later, theanion radical of binaphthyl appears to be free of such torsion. This anion radical excels ata complete delocalization of the unpaired electron over the whole molecular framework.
3.2.3 Localization of an Unpaired Electron in a Field of Two or MoreAtoms
This section is devoted to two-center three-electron bonds. Pauling first described thesebonds in 1931. In the years since the first description by Pauling, a great deal of interest hasbeen expressed in such systems. Leading references are enumerated later.
Hoefelmeyer and Gabai (2000) have synthesized 1,8-bis(diphenylboryl)naphthaleneand compared the structures of this molecule and the anion radical prepared from it. In theparent neutral molecule, the boron centers are separated by 3 nm. In the anion radical, theboron–boron distance is approximately 1 nm longer than that observed in compounds withsingle bonds between four-coordinated boron atoms. According to the ESR spectrum of theanion radical, the unpaired electron is preferentially suspended in the field of the two boronatoms. The one-electron �-bond emerges. This one-electron �-bond can obviously beviewed as resulting from the overlap of the formerly vacant and parallel p-orbitals of two
SCHEME 3-12
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

boron atoms. In the anion radical, both orbitals are simultaneously populated with the un-paired electron.
Anion-radicals of alkylphenyldiazirines have been prepared electrochemically (El-son et al. 1986). Their ESR spectra show that the anion radicals persistently hold the intactdiazirine rings. A large portion of the unpaired spin density resides on the nitrogen atoms.In the three-membered diazirine ring the sp3 carbon atom is bound with two nitrogen atomshaving a double bond between them. Hence, some kind of valence shell expansion takesplace. The phenomenon develops, however, in agreement with a simple valence bond pic-ture of the molecules. By and large, the azo group is considered a fairly good electron ac-ceptor favorable for the accommodation of the negative charge in anion radicals (Gerson,Lamprecht, et al. 1996).
Apparently, a similar valence bond picture holds true for 4-alkylidene pyrazoline an-ion radicals. These radicals were prepared by reducing (potassium mirror in DME) of thepyrazolines depicted in Scheme 3-13. According to ESR spectra and MO calculations forthese anion radicals, the unpaired electron is indeed associated with the nitrogen–nitrogen�*-orbital (Bushby & Ng 1996).
Anion radicals of compounds with the azo and diphosphene central bonds(R1NBNR2 and R1PBPR2) have three �-electrons between two nitrogen or phosphorusatoms (Stagko et al. 1998; Binder et al. 1996; Shah et al. 1997).
Gebicki’s group compared absorption spectra of the cation radicals of N,N-dimethylethane diamine, N,N-dimethyl piperazine, and hexamethylene diamine (Scheme3-14) with those of other �,�-alkyldiamines cation radicals (Gebicki et al. 1990; Marcineket al. 1990). The absorption spectra of diamine cation radicals with three or fewer methy-lene groups separating the nitrogen atoms show a visible transition, in contrast to those withfour and six methylene groups, where only UV transition is seen. As the length of the poly-methylene chain increases, the probability of achieving the conformation favorable for the(N�N)� bond formation decreases sharply. Hence, cation radicals generated from �,�-di-aminobutane or -diaminohexane possess a charge localized on only one of the two nitrogenatoms, similar to a monoamine cation radical.
SCHEME 3-13
SCHEME 3-14
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

In the case of two pairs of the nitrogen atoms within a six-member saturated ring, acation radicals is formed whose ESR spectrum contains signals of two only (not of four) ni-trogen atoms (Nelsen, Hintz, Buschek, & Weisman 1975). In other words, the unpairedelectron is localized in one hydrazine unit of tetramethyl- or tetraethyl-sym-hexahydrote-trazine. Intramolecular electron exchange between the two hydrazine halves of the cationradical cyclic molecule either does not take place at all or goes out of the ESR time scale.Hence, one-electron oxidation of the tetrazines gives rise to the cation radicals of a specificconformation. In this conformation, only two neighboring nitrogen atoms are capable ofdelocalizing the unpaired electron. Scheme 3-15 explains this inference; the important fea-ture is a suspension of three electrons in the field of two nitrogen atoms.
Removal of an electron from a hydrazine unit changes the lone pair orbital occupancyfrom four to three, which has a large effect on the preferred geometry with respect to thenitrogens. The formation of a three-electron bond has also been demonstrated in the cationradical of octamethyl-1,2,4,5-tetra-aza-3,6-disilacyclohexane. In this cation radical, thespin density is distributed between only two of the four nitrogen atoms. There is a pro-nounced interaction of the unpaired electron with protons of the methyl groups joined tothese two nitrogen atoms.
These conformational changes, which take place during the transformation of neutralhydrazines or bicyclic azo compounds into cation radicals, have also been observed in awide range of derivatives. These changes seemingly have a general character (Williams etal. 1988; Nelsen, Rumack, & Meot-Ner 1988).
The conformational changes also explain the slow electron transfer between two hy-drazine moieties separated by a bridge (Gleiter et al. 1996; Nelsen, Hintz et al. 1975, 1976;Nelsen, Ismagilov, & Trieber 1997). In all these cases, the inner reorganization energy ishigh when electron transfer occurs. In particular, when the unpaired electron is transferredfrom the planar hydrazine cation radical unit to the tetrahedral neutral hydrazine unit, re-ally large conformational changes take place. It is noteworthy to mention that although theelectron transfer barrier is sensitive to the inner reorganization energy upon electron re-moval, it is especially sensitive to the substituents attached to nitrogen. Solvation of thebridge between two hydrazine units is also too large to be ignored (Nelsen, Trieber, & co-authors 2001). Slight structural modifications result in changes of intramolecular electrontransfer constants, and these changes are large enough to be measured by dynamic ESR.The three structures in Scheme 3-16 illustrate this statement.
Even changing from the N-methyl substitution (the first structure in the row ofScheme 3-16) to the N-isopropyl substitution (the second one) lowers the electron transferbarrier (Nelsen 1997, p. 171). The p,p-phenylene-linked system (see the third structure in
SCHEME 3-15
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Scheme 3-16) features fast electron transfer under comparable conditions. The cation rad-ical from the last bis-hydrazine is instantaneously localized, in contrast to the cation radi-cal from tetramethyl-p-phenylenediamine, which is delocalized (Nelsen, Ismagilov, &Powell 1996, 1997; Nelsen, Tran, & Nagy 1998).
The fast electron transfer mentioned for the p-phenylene-linked system results in thetransfer of the three-electron two-nitrogen unit from the one-hydrazine ring to the other.Such a “superexchange” transfer is a specific phenomenon. As found (Nelsen & Ismagilov1999), this bis(hydrazine) cation radical is significantly ion paired with the PF6
� counterionin methylene chloride. The counterion “touches” both hydrazine fragments, but the distancebetween the N�N linkage and PF6
� is permanently shorter than that between N–N and PF6�.
One-electron oxidation of 1,6-diazabicyclo[4.4.4]tetradecane proceeds at a remark-ablys low rate. The cation radical obtained contains a three-electron �-bond between thetwo nitrogen atoms (Alder & Sessions 1979). In this case, the three-electron bond links thetwo nitrogens that are disjoint in the starting neutral molecule, at the expense of the oneelectron that remains from the lone electron pair of the first nitrogen and the two electronsof the second nitrogen, which last as if being unchangeable. The authors name such a phe-nomenon “strong inward pyramidalization of the nitrogens” with “remarkable flexibilityfor the N–N interaction.” This interaction results in 2�–1�* bond formation, Scheme 3-17.
Such “strong inward pyramidalization of the nitrogens” is stabilized by the overlap-ping of the nitrogen electrons forming a two-center three-electron bond in the cation radi-
SCHEME 3-16
SCHEME 3-17
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

cal just considered and even a covalent bond in the corresponding dication (Zwier et al.2001). The initial neutral compound of Scheme 3-17 is very sensitive as a solid to air andis stable for only a few hours. The fluoroborate salt of the 1,6-diazabicyclo[4.4.4]tetrade-cane cation radical can be isolated as dark red solid. The solid is indefinitely stable and isstable for months in organic or even aqueous solution in the absence of a base. The dica-tion is also stable as a solid in acidic aqueous solutions (Alder & Sessions 1979).
Three-electron N–N-bound cation radicals are known in a wide range of structures.Most examples are, however, unstable outside the glassy or solid state (for a review, seeAlder 1983). It is important that the tricyclic cation radical just portrayed be structurallyprevented from cleaving the (N�N)� bond and that it has a long solution lifetime (Nelsen,Alder, et al. 1980).
Transannular cation radicals with the intramolecular sulfur–sulfur bond of the2�–1�* type generated from medium-ring disulfides like 1,5-dithiacyclooctane are an ex-ception in terms of their stability, although they are not resistant to water (Musker 1980).ESR and resonance Raman spectroscopy studies revealed the existence of the 1,5-dithia-cyclooctane cation radical, with substantial bonding between the sulfur atoms (T. Brown etal. 1981; Tamaoki et al. 1989). Computations confirmed this statement and pointed out thatthe chair-boat conformer has the lowest energy as compared to other possible conformers(Stowasser et al. 1999).
The molecular geometry, which allows optimal p-orbital interaction to yield a three-electron bond, presumes an orientation of p-orbitals belonging to each sulfur atom alongthe S���S axis. This is the case for the chair-boat conformer of the 1,5-dithiacyclooctanecation radical, Scheme 3-18. In the 1,3-dithiacyclopentane cation radical, the sulfur p-or-bitals are aligned almost perpendicular to the ring plane, and this prevents stabilization bythe transannular interaction between the two sulfur atoms in the cycle. Therefore, the five-membered structure in Scheme 3-18 cannot exist.
The open-chain MSM(CH2)nMSM, MSeM(CH2)nMSeM, and MSM(CH2)n
MSeM compounds give cation radicals upon one-electron oxidation. These open-chainspecies with n from 3 to 5 can also form the two-center three-electron bonds (Muller &Henze 1998a,b). Two of them are depicted in Scheme 3-19. While the 2,6-dithiaheptane
SCHEME 3-18
SCHEME 3-19
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

cation radical can assume a five-member ring configuration and gives a more or less stablethree-electron bond, the cation radical of 2,5-dithiahexane cannot overcome inner stericstrains (Asmus 1979; Drewello et al. 1989). Therefore, such quasididthiacyclobutanecation radicals cannot be formed.
It should be noted, however, that the bond strength of the three-electron sulfur–sul-fur bond is inevitably lower than that of a normal sulfur–sulfur bond in an organic disul-fide. The theoretical results indicate that the bond is localized between the two sulfur atoms,that both sulfur atoms have equal unpaired electron density, and that the S�S bond lengthis about 30% longer than an average single-bond length. The bond strength is about 45%of an S–S single-bond energy. The bond order is 1/2 (James et al. 1996 and Refs. 2–5therein). Cation radicals containing such 2�–1�* bonds are unexpectedly ready for one-electron oxidation. In these cation radicals, a p-orbital of the sulfur atoms combines in a �-fashion to generate �-bonding and �*-antibonding orbitals (Baird 1977; Gill & Radom1988). Two electrons are accommodated in the �-bonding MO (2�) and one in the �*-an-tibonding MO (1�*). The electron to be removed from the cation radical upon oxidation isof course the antibonding �*-electron. In this case, only an increase in the bonding betweenthe sulfur atoms can take place. For example, the cation radical 1,5-dithiacyclooctane inScheme 3-18 undergoes oxidation in acetonitrile at a potential less positive than that for1,5-dithiacycooctane itself (Wilson et al. 1979; Ryan et al. 1981). Its two-electron oxida-tion proceeds reversibly and in the framework of a merged process. As opposed to thisdithia compound, 2,3,7,8-tetramethoxythianthrene, which cannot form a cation radical witha three-electron bond, gives two separate reversible one-electron steps under the same con-ditions of electrochemical oxidation.
It is interesting to compare S�S and Se�Se cation radicals in terms of oxidation. Ingeneral, oxidation of selenium requires considerably less energy than oxidation of sulfur.This means that Se�Se species are more stable thermodynamically than their S�S analogs.Nevertheless, Se�Se species are readily oxidized to the corresponding dications. The reac-tion in Scheme 3-20 is an illustrative example (Asmus 2000):
(Me2Se�SeMe2)� � FeIII(CN)63� → FeII(CN)6
4� � (Me2SeMSeMe2)2�
As for 2�–1�* dithia cation radicals, they do not react readily with O2, despite thegreat inclination to oxidation. Aliphatic thioether cation radicals in Scheme 3-21 become ca-pable of reacting with O2 only after the addition of hydroxyl anion (Schoeneich et al. 1993).
SCHEME 3-21
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

One note is necessary at this point. As Nelsen (1987) stated, even when carbon–oxy-gen bonds are formed by a reaction of an olefin cation radical with O2, the reaction israpidly reversible and endothermic. Structures of observable products depend upon the rateof reactions other than the interaction with O2. This does not mean that adducts do not form,but that the R�MOO
.product is thermodynamically unstable. The same should be taken
into account for 2�–1�* dithia cation radicals.There are three possible types of three-electron bonds. Oxidation of a �-bond results
in a cation radical with a 2�–1� three-electron bond. This bond contains no antibondingelectrons, and the total bond strength exceeds that of a 2�-bond by the energy of half a �-bond. Olefins can acquire the 2�–1� bond upon one-electron oxidation. Oxidation of or-ganic disulfides, RSSR, to their cation radicals (RSSR)�.
yields species in which the un-paired electron from the oxidized sulfur interacts with the unbound p-electron pair of thesecond sulfur (Glass 1999). This establishes a 2�–1�* bond on top of the already existing�-bond. The overall bond strength of this five-electron (2�–2�–1�*) bond also exceedsthat of the normal 2s bond by ca. half of a �-bond. In other words, the (RSSR)�.
assumesa partial �-character. The 2�–1�* bond (with a formal bond order of 0.5) is formed fromtwo electrons located in an �-orbital and one electron in an antibonding �*-orbital in thecase of no �-bond between the atoms participating in formation of 2�–1�* bond.
In contrast to (CH3SSCH3)�., [(CH3)2S�S(CH3)2]�.
undergoes a rearrangement inwhich a C–C bond is formed (Goslish et al. 1985). The arrangement includes �-deproto-nation at one of the (CH3)2S fragments. This protonation produces ilyde containing a[M�S(CH3)CH2M] fragment. Further regrouping leads to formation of the SCH2CH3 moi-ety (Glass 1999).
The well-known instability of the disulfide anion radicals, (R1SSR2)�., is apparently
explained by the antibonding electron population, presumably in the framework of thedisulfide bond. In some cases, however, these anion radicals turned out to be more or lessstable (Breitzer et al. 2001). Two examples in Schemes 3-22 and 3-23 deserve to be distin-guished. Firstly, one-electron reduction of naphthalene-1,8-disulfide using sodium indimethoxyethane generates the corresponding anion radical, Scheme 3-22. Second, the ox-idation of a [1,n]-dithiol by Ti(III)–H2O2 at pH 7 produces the cyclic disulfide according toScheme 3-23:
SMS�.
HS(CH2)nSH pH 7→ �S(CH2)nS. →
Ti(III) CH2CH2
n � 3, 4 H2O2 \ /(CH2)n�2
The final product of the dithiol oxidation forms rather stable anion radicals. There aretwo possible locations for the unpaired electron: Either it resides in a sulfur 3d-orbital or it
SCHEME 3-22
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

resides in the S–S �*3p antibonding orbital. Considering ESR spectra of cyclic disulfide an-ion radicals, Bassindale and Iley (1990) note that the orbital containing the unpaired elec-tron has almost entirely a p character. In particular, the naphthalene-1,8-disulfide anion rad-ical has an unpaired electron highly localized on sulfur, with little interaction between thiselectron and the �-system of the aryl rings. Whereas this is in accord with a �*-orbital,d-orbital participation would be expected to interact strongly with the �-system and cantherefore be ruled out. Disulfide anion radicals ought to be considered species of the2�–1�* type.
Let us now consider the formation of three-electron bonds between different atoms.Stabilization of an oxidized sulfur atom can, in principle, be achieved in cases of its inter-action with other heteroatoms if they provide free (preferably p-) electron pairs. Nitrogen,oxygen, and halogens (except fluorine) can be mentioned as such heteroatoms (Anklam etal. 1988; Carmichael 1997). The stability of these bonds is generally not as high as that ofa symmetric S�S system. An important reference for the enhanced stability of symmetri-cal three-electron bonds is Clark’s (1988) calculations.
Because of a difference in electronegativity, electron density is distributed unequallybetween two diverse heteroatoms. The bond strength for such a bond depends on the dif-ference between ionization potentials of these two atoms: the smaller the difference, thestronger the bond. In concrete language, the following sulfur neighbors are capable offorming three-electron bonds: oxygen of the hydroxy or carboxylate group, nitrogen of theamino group, phosphorus of various phosphine moieties (Glass 1995). The examples inScheme 3-24 represent more or less stable cation radicals with three-electron bonds.
The cation radicals depicted in Scheme 3-24 form upon oxidation ([which, accordingto Asmus (1990), takes place at the sulfur atom]) of endo-2-(2-hydroxy-2-methyl-ethyl)-endo-6-(methylthio)-bicyclo[2.2.1]heptane and endo-2-(carboxyl)-endo-6-(methylthio)-bicyclo[2.2.1]heptane, respectively. When geometric constraints preclude neighboring-group participation, the three-electron bond is not formed. Scheme 3-25 gives one suchexample, namely, (exo-2-(carboxyl)-endo-6-(methylthio)-bicyclo[2.2.1]heptane).
Scheme 3-26 depicts one intriguing case, when one-electron oxidation of conforma-tionally constrained exo-2-(carboxyl)-endo-2-(amino)-endo-6-(methylthio)-bicyclo[2.2.1]heptane gives rise to a cation radical in which an amino but not a carboxylate group partic-ipates in the three-electron bond with sulfur (Glass 1995).
The S�N bond formation can serve as a driving force of the conformational transi-tion. Thus, the 2-amino-4-(methylthio)butanoic acid (methionine) cation radical exists in
SCHEME 3-24
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

several forms: open-chain protonated, open-chain deprotonated, and cyclic with the two-center three-electron bond between sulfur and nitrogen. All the forms are in equilibrium,and the participation of the cyclic form is predominant. At a high pH of the medium, all thecation radicals exist in the cyclic form. Evaluation of the polarization pattern yields the fol-lowing spin-density distribution (�i) in the cyclic cation radical: �N � 0.36, and �S � 0.64(Goez & Rozwadowski 2000).
In sulfenamides (R1SMNR2R3), the cation radicals keep an unpaired electron occu-pying a �*-orbital. This orbital is localized between the sulfur and nitrogen atoms. In otherwords, somewhat SBN double-bond character exists in such species. A consequence ofthis “double-bond” character is an increase in the energy barrier to rotation about the S–Nbond. Restricted rotation about the S–N bond is known for the neutral sulfenamides (Kost& Raban 1990). The energy barrier to this rotation is greater for the derived cation radicalsthan for the parent compounds (Bassindale & Iley 1990).
In the cation radicals, the N–S fragments with their nearest environments are planar(Zverev, Musin, & Yanilkin 1997): N–X rotation disrupts the 3e–� bond. The resonancestabilization between N and X disappears.
By and large, the highest resonance stabilization should occur when N and X areequivalent, leading to the prediction of the highest rotational barrier for N-centered 3e–�bonds being for hydrazine cation radicals (Nelsen 1967). Barriers will, of course, be low-ered by delocalization onto substituents and by steric strain in the nearly planar, most sta-ble form for species, which bear N-containing substituents.
N-Acetylmethionine amide [CH3SCH2CH2CH(NHCOCH3)CONH2] gives the cationradical with the 3e-bond between atoms S and O belonging to the amide function, but notbetween S and N. Specifically, one-electron oxidation of a methionine part in -amyloidpeptide has been associated with the neurotoxicity of these sequences (Schoeneich et al.2000). This peptide is the major constituents of senile plaques in Alzheimer decease.
SCHEME 3-25
SCHEME 3-26
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Cation radicals containing three electrons in the field of four atoms or anion radicalswith five electrons retained by four atoms represent a special group of multicentered ionradicals. Thus, nonclassical, cyclically delocalized 3e/4C cation radicals and 2e/4C (dica-tion) radicals of substituted cyclobutadienes are known (Allen and Tidwell 2001). The3e/4N cation radical and the 5e/4N anion radical depicted in Scheme 3-27 have also beendiscovered (Exner et al. 1998, 1999, 2000). The reactions in Scheme 3-27 illustrate suchspecies as well as the corresponding dianion and acetylated products of the latter.
In the case of tetra(N-oxide) of the starting bis(diazene), one-electron oxidation inScheme 3-28 leads to a novel, O-stabilized, 3e/4N cation radical with three-dimensionaldelocalization of the unpaired electron (Exner et al. 1999).
SCHEME 3-27
SCHEME 3-28
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

3.2.4 Spin-Charge Separation (Distonic Stabilization of Ion Radicals)
Localization of an unpaired electron in the framework of a definite molecular fragment cansometimes lead to ion radicals with spatially separated charge and radical sites. They canbe considered free radicals with an appended, remote charge. These species form a partic-ular class of distonic ion radicals. Distonic is from the Greek diestos and the Latin distans,meaning “separate.” Yates, Bouma, and Radom introduced this term in 1984 for ions thatformally arise by removal of an electron from a zwitterion or a biradical.
For instance, oxidation of most neutral closed-shell hydrocarbons generates cationradicals that possess nearly coincident spin and positive-charge distribution. This distribu-tion is approximated by the spatial density of the singly occupied molecular orbital. How-ever, cation radicals derived from certain neutral organic biradicals can possess a morenovel electronic structure, namely, ones in which spin and charge distributions are reallynoncoincident. In principle, this is possible because a biradical contains its two frontierelectrons in a set of two degenerate or nearly degenerate molecular orbitals. Electron lossfrom a biradical can thus give a cation radical whose spin distribution is a function of oneorbital space and whose charge distribution is a function of another orbital space. If thesetwo orbitals differ spatially, then the resultant cation radical will be distonic (Radom et al.1986). Such cation radicals will possess different spin and charge sites, even within a fullyconjugated �-system.
Radom and co-workers used this term for systems containing a charge and an elec-tron in two vicinal (side-by-side) positions (see, for example, Gauld & Radom 1997). Nev-ertheless, the whole idea of “distonic” ion radicals as used by most of its practitioners isthat the electron and the charge are separated by at least two heavy atoms between thecharge and the radical sites. Such systems are also known (Kenttaemaa 1994; Gronert2001). In this section we will treat “truly distonic” systems, i.e., systems in which there aresp3-hybridized atoms between the radical and charge centers (or they do not overlap signifi-cantly). Such a definition is relevant to the principle of the detained electron and can helpto distinguish ion radicals with really separated charged and radical sites. Several examplesfrom anion radicals and cation radicals illustrate the problem.
3.2.4.A Distonic Stabilization of Anion Radicals
As ESR spectra testify, cyclobutanone and tetramethylene sulfone undergo dissociativeelectron capture process (Scheme 3-29) in argon matrices and yield distonic anion radicals(Kasai 1991; Koeppe & Kasai 1994).
SCHEME 3-29
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

One-electron reduction of 1,1-dimethyl-5,7-di(tert-butyl)spiro[2,5]octa-4,7-diene-6-one leads to formation of the anion radical that undergoes ring opening; see Scheme 3-30.Relief of the cyclopropane ring strain and generation of an aromatic ring provide the ther-modynamic driving force for this ring opening. It results in the formation of two anion rad-icals in which the charged and radical centers are not in direct polar conjugation (Tanko &Philips 1999; Tanko and co-authors 1994).
Sometimes distonic anion radicals can be recognized among aromatic derivatives inwhich a radical center is strongly localized according to the method of its formation. In thesodium reduction of o-diazobenzophenone (Scheme 3-31), the formation of the radical cen-ter is a result of nitrogen elimination. At the same time, the carbonyl group transforms intoan aprotic equivalent of the hydroxyl group. Analysis of the ESR spectrum of argon-ma-trix-isolated anion radical confirms its distonic nature (Hacker & Kasai 1993).
Calculations (Hacker & Kasai 1993) in the framework of extended Hueckel theoryfind the LUMO of o-diazobenzoquinone to be mainly (up to 60%) localized at the N–N sec-tor. The electronegativity of the conjugated carbonyl oxygen enhances the acceptor prop-erties the diazo group. Charge-spin separation is an understandable consequence of the sit-uation.
SCHEME 3-30
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Scheme 3-32 portrays the gas-phase generation of the m-benzyne anion (the distonicanion biradical) from isophthalic acid (Reed and co-workers 2000) or m-bis(trimethylsilyl)benzene (Wenthold et al. 1994, 1996; Wenthold & Squires 1998). According to Wentholdet al., the reaction of m-bis(trimethylsilyl) benzene with fluoride ion, followed by treatmentof the formed trimethylsilyl phenyl anion with fluorine in helium, produces the anion bi-radical mentioned. The latter is transformed into the corresponding nitrobenzoate anionthrough additions of CO2 and NO2, Scheme 3-32.
Interestingly, the stabilizing reagents (CO2 and.NO2) cannot be added in reverse or-
der (.NO2 first and CO2 second) because the anionic site undergoes an electron-transfer re-
action with .NO2 faster than the radical site can add
.NO2. Conversion of the phenyl anion
into the carboxylate eliminates the electron-transfer pathway, because the carboxylate hasa higher electron-binding energy. This behavior again points to the dual reactivity of dis-tonic species.
Theoretical studies of the m-benzyne anion led to the inference that it holds a pair ofweakly interacting orbitals. One orbital contains the odd-spin density, whereas another con-tains two electrons that are responsible for the negative charge of this species (Nash &Squires 1996).
Also to be mentioned are the reactions of oxygen atom radical anions with organicsubstrates. These reactions were reviewed recently (Gronert 2001). For the reactivity of theatomic oxygen anion radical, see Section 1.7.2 of this book. The atomic oxygen anion rad-ical reacts with benzene, tetramethylene ethane, or cyclopentadienylidene trimethylen-
SCHEME 3-31
SCHEME 3-32
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

emethane. The reactions consist of abstraction of H2�, with the formation of H2O and the
corresponding distonic anion radicals as products.
3.2.4.B Distonic Stabilization of Cation Radicals
According to mass spectrometric studies by Bigler and Hesse (1995), �,�-diaminoalkanecation radicals undergo intramolecular hydrogen atom rearrangements leading to distoniccation radicals, Scheme 3-33:
H2N(CH2)nNH2 � e → 2e � [H2N(CH2)nNH2]�.
[H2N(CH2)nNH2]�. → H3N�(CH2)nNH.
The example of glycine is much more descriptive. The spin and charge separationhave been found at the C-terminus and the N-terminus, respectively (Rodriguez-Santiagoet al. 2000), Scheme 3-34:
[H2NCH2COOH]�. → [H3N�CH2COO�]�. → H3N�CH2COO.
The activation energy for this separation is 0.4 eV in the ground state, with no barrier in thefirst excited state (Nielsen et al. 2000). The driving force is the high proton affinity of theamino group. This leads to formation of such stabilized distonic cation radicals.
A similar explanation helps us to understand the separate existence of the conven-tional and distonic forms of the benzonitrile cation radical, cf. (H5C6C�N)�.
and.H4C6C�NH� (Flammang et al. 2001). Although the distonic cation radicals are less sta-ble than the classical ones by ca. 45–50 kJ�mol�1, they are protected against isomerizationby a relatively high energetic barrier.
The example of the ethyl acetate cation radical is also noteworthy; see Scheme 3-35.According to an ESR study (Rhodes 1988), ethyl acetate upon one-electron oxidation givesnot the carbonyl (conventional) but the onium (distonic) cation radical. The onium form ofthe ethylacetate cation radical is more stable than the corresponding carbonyl form by al-most 50 kJ�mol�1 (Rhodes 1988). By and large, OH bonds are stronger than CH bonds. TheCH
.2 fragment is stabilized by the three-electron bonding with the neighboring oxygen in
the manner of MO�CH2. Carbonyl-group oxidation is very difficult, while oxidation of thehydroxyalkyl moiety is very easy. On the other hand, the carbonyl oxygen atom can pre-serve its electron surroundings if converted into the onium state. In this case, however, anunpaired electron is localized on the methylene carbon atom and separated from the onium
SCHEME 3-35
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

one with other heavy atoms. Therefore, the total stability of the distonic cation radical be-comes higher than that of the conventional one.
Distonic cation radicals with onium ethereal oxygens are also known. For instance,addition of tert-butoxy radicals to ethylenic or acetylenic compounds was described(Bloodworth et al. 1988), Scheme 3-36 (below). The butoxy radicals are added in their pro-tonated forms (CF3COOH is the proton source):
(t-Bu)2O2h�
MM→ 2(t-BuO.) �2H�
MMM→ 2(t-BuOH�
)
t-BuOH�
� CH2BCHMe → t-BuO�
CH2CHMeH
t-BuOH�
� CR�CR → t-BuO�
CRB CRH
Studies of distonic ion radicals have been performed in recent years with an empha-sis on theoretical approaches. From the experimental point of view, the presence of ionicmoieties makes free radicals, which would not normally be investigated by mass spec-trometry, amenable to detection in the gas phase. A lot of experiments were carried out toprove their existence and to observe their behavior in mass spectrometers; see reviews(Kenttaemaa 1994; Hammerum 1988) and, for example, one recent experimental work(Polce & Wesdemiotis 1996). At the next stage, syntheses of distonic ion radical organicsalts stable under common conditions will likely be developed. These salts would be usedto create magnetic, conductive, and other materials of practical use. In a chemical sense, theespecial strength of distonic organic ion radicals is that they can, in principle, enter reac-tions of the ionic type at the charged center and reactions of the radical type at the radicalcenter.
For example, the distonic anion radical of cyclopentadienylidene trimethylen-emethane reacts under mass spectrometer gaseous-phase conditions with carbon disulfideby sulfur abstraction and with nitric oxide by NO-radical addition. The first reaction char-acterizes the distonic anion radical mentioned as a nucleophile bearing a negative chargedmoiety. The second reaction describes the same anion radical as a species having a groupwith radical unsaturation (Zhao et al. 1996).
For another example of the strong duality in the chemical behavior of distonic cationradicals, the work of Moraes and Eberlin (1998) should be mentioned. In the gaseousphase, m- and p-dehydrobenzoyl cation radicals react selectively as either free radicals oracylium ions, depending on the choice of the neutral reaction partner. Transacetalizationwith 2-methyl-1,3-dioxolane, ketalization with 2-methoxyethanol, and epoxide ring ex-pansion with epichlorohydrin demonstrate their acylium ion reactivity. Abstraction of SMefrom MeSSMe demonstrates their free radical reactivity. In one-pot reactions with gaseousmixtures of epichlorohydrin and dimethyldisulfide, the m- and p-dehydrobenzoyl cationradicals react selectively at either site to form the two monoderivatized ions. Further reac-tion at either the remaining radical or the acylium charge site leads to a single biderivatizedion as the final product.
It would be very important to find the reactions of such type in the liquid phase, too.Successes in studies of such dual reactivity in liquid phase may seriously widen the possi-bilities of organic synthesis. There are only few works of this sort, and they are of impor-tance.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Octamethoxytetrabenzo[a,c,e,g]cyclooctatetraene gives the corresponding cationradical upon one-electron oxidation. The latter undergoes an intramolecular C–C bond for-mation to yield the distonic cation radical containing two fused five-membered centralrings and four condensed dimethoxyphenyl moieties. This distonic cation radical is readilyoxidized into the dication with the same rearranged structure (Rathore et al. 2000). Impor-tantly, it is the distonic nature of this rearranged cation radical that facilitates the removalof a second electron at a much lower potential and leads to the formation of stable dications.The dication structure was established by the X-ray method.
In simple olefins of the stilbene type, cis → trans isomerization in the cation radicalstate is known to proceed readily. However, in the cation radical of 2,6-di(tert-butyl)ful-vene there is no isomerization (Abelt & Roth 1985). According to the authors, this stabil-ity is a result of the existence of this cation radical not in an orthogonal but in a twisted formof the distonic type. In this distonic cation radical, a spin density is distributed within thecyclopentadyenyl ring and a positive charge localized at the central carbon of themono(tert-butyl) out-of-ring fragment. Experimental studies involving CIDNP have alsogenerated evidence for distonic cation radical intermediates in cation radical Diels–Alderreactions (Roth and co-workers 1986). Stereochemical studies of the final products formedin such reactions also confirm their stepwise/distonic mechanism (Bauld & Yang 1999a,b;Bauld et al. 2000).
Methyl tricyclo[4.1.0.02,7]heptane-1-carboxylate gives the cation radical in whichthe spin density is almost completely localized on C1 and the positive charge is on C7. Therevealed structural feature of the intermediate cation radical explains fairly well the re-gioselectivity of the N,N-dichlorobenzenesulfonamide addition to the molecular precursorof this cation radical. In this reaction, the nucleophilic nitrogen atom of the reagent adds toelectrophilic C7, and the chlorine radical adds to C1, whose spin population is a maximum(Zverev & Vasin 1998, 2000).
3.2.5 Ion-Pair Formation
Organic ion radicals exist together with counterions and often form ion pairs. Since the pi-oneering works of Grunwald (1954), Winstein with co-authors (1954) and Fuoss and Sadek(1954), the terms contact, tight, or intimate ion pair and solvent-separated or loose ion pairhave become well known in the chemical world. More recently, Marcus (1985) and Boche(1992) introduced other colloquial expressions, the solvent-shared ion pair and the pene-trated ion pair.
In solvent-separated ion pairs, the solvation shells of the cation and the anion toucheach other; in solvent-bridged ion pairs, the ions share solvent molecules. In contact ionpairs, the cation and the anion are bound directly to each other and are surrounded by acommon solvation shell. In penetrated ion pairs, an empty space between edge groups inone ion of a salt is occupied to a certain degree by a counterion. The two latter types of ionpair may have quite a different electronic distribution than the corresponding “naked” ions.The following examples show the influence of ion-pair formation.
3.2.5.A Detention of an Unpaired Electron in the Framework of OneSpecific Molecular Fragment
A single electron transfer from cyclo-octatetraene dipotassium (C8H8K2) to 2- and 4-nitro-stilbenes in THF leads to the formation of paramagnetic potassium salts of the anion radi-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

cals. In this solvent, the salts exist as coordination complexes, Scheme 3-37:
C6H5HCBCHC6H4NO2
��12
� C8H8K2 (THF)MMMMMMM→
��12� C8H8 (THF)
C6H5HCBCHC6H4NO�.2 ,K�
The complexes can be destroyed by the addition of 18-crown[6]ether or HMPA to theTHF solution, Scheme 3-38. Such dissociation has been proven by ESR spectra (Todres1992).
Upon one-electron reduction followed by one-electron oxidation, cis isomers of 2-and 4-nitro stilbenes turn into the neutral nitrostilbene molecules, but in the trans forms.Upon oxidation of the “naked” anion radical, the neutral trans forms are the only products(cis → trans conversion degrees were 100%). In case of the coordination complexes, thetrans isomers are formed only up to 40% (Todres 1992). Scheme 3-39 describes these trans-formations.
Thus, one-electron transfer causes cis → trans isomerization. It is quite effective withfree migration of an unpaired electron over the molecular framework. It is less effectivewhen the unpaired electron is confined within the limits of a coordination complex. Suchfixation of the unpaired electron hinders the rotation around the CBC bond.
In the same manner, a ball tied to a finger with a rubber band flies upon being struckuntil the rubber band stretches to its utmost, and then the ball abruptly returns to the finger,to the ball’s initial point of movement.
The isomerization is more effective when the (nitroanion radical � alkali counterion)ion pair does not exist. This underlining is necessary: It is assumed that intimate ion pairspossess a larger electron affinity and are characterized by a larger contribution to the freeenergy of electron transfer than free ions (Grigoriev and co-authors 2001). In this case, the
SCHEME 3-38
SCHEME 3-39
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

ion pair considered could be reduced by more than one electron, and the degree of isomer-ization would be larger in the ion pair than in the free anion radical.
Now let us compare 4,4- and 2,4-dinitrostilbenes with respect to their relative capa-bilities to rotate around the CBC bond upon electron transfer. The difference betweenthem is shown in Scheme 3-40 (Todres, Kuryaeva, et al. 1980). It is clear that an unpairedelectron oscillates between two nitro groups in the 4,4-dinitro derivative and is retained inthe dinitrophenyl ring in the 2,4-dinitro compound. The potassium salt of the 2,4-dinitros-tilbene anion radical does not change the initial cis geometry, even in the presence of crownether. It is known, however, that trans-2,4-dinitrostilbene is more stable than the cis isomer(Pfeifer and others 1915), so once again cis-to-trans isomerization is highly correlated withthe spin population of the CBC bond. The quantum chemical considerations unambigu-ously establish such correlation (Dyusengaliev et al. 1981).
The potassium salt of the 2,2-dipyridyl acetylene anion radical represents anotherimportant example. In this case, the spin and charge are localized in the framework of theNMCMC�CMCMN fragment. The atomic charge on each nitrogen atom is �0.447, i.e.,close to unity in total. The energy of this ion pair is minimal when the potassium counte-rion is located midway between the two rather close nitrogen lone pairs. Such a structure isconsistent with the fact that the ESR spectrum of this species is almost insensitive to tem-perature. It means that the counterion does not hop between two remote sites of the anionradical (Choua et al. 1999).
3.2.5.B Formation of a Closed Contour for Unpaired ElectronDelocalization
This phenomenon can efficiently restrain an unfavorable configuration. It is well knownthat in 1,2-semidione anion radicals, the trans isomer has a higher stability, owing to theminimization of dipole–dipole repulsions. Nevertheless, the presence of a counterion maychange the isomer distribution by favoring the cis structure, owing to the two negativelycharged oxygen atoms.
As calculations (Calle et al. 1992) show, the preferential approach of an alkalinecation to a 1,2-semidione is indeed in the middle of the two oxygen atoms. A cation-semid-
SCHEME 3-40
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

ione chelate (or a penetrated ion pair) is formed, the equilibrium distance being around 0.23nm. This conformation is 120 kJ�mol�1 more stable than the corresponding trans isomer.Divalent cations must be closer to the semidiones than the monovalent ones (Be2� andMg2� were calculated to be closer than Li� and Na�, respectively). Both Be2� and Mg2�
cations stabilize the cis isomer better than Li� and Na� cations (Calle et al. 1992). This factis due to a stronger electrostatic interaction associated with the charge.
The one-electron reduction of 3,4,5-trimethoxyphenyl glyoxal with potassium tert-butoxide in dimethylsulfoxide gives rise mainly to the cis-semidione, while upon electrol-ysis in dimethylformamide, in the presence of tetraethyl ammonium perchlorate as the car-rier electrolyte, the main product is the trans isomer (Sundaresan & Wallwork 1972),Scheme 3-41.
The ESR spectrum of the sodium salt of dibenzoyl ketyl in THF shows splitting dueto Na�, while in the ESR spectrum of the sodium salt of the ketyl of 2,2,6,6-tetramethyl-hexane-3,4-dione in THF there is no splitting whatever due to Na� (Luckhurst & Orgel1963). Whereas dibenzoyl ketyl can give rise to the contact (or penetrate) ion pair, the for-mation of such a species type from the 3,4-dione is difficult, owing to steric hindrance. Thissemidione exists in the form of a free ion (or an ion pair separated by solvent molecules);compare the two structures in Scheme 3-42.
If the dibenzoyl ketyl ion is not included into the ion-pair complex with potassiumcation, it exists in the trans form. The potassium salt does exist in the cis form, in whichboth charged oxygen atoms are in the vicinity of the metal cation (Bauld 1965). On ben-zoylation, the cis ion radical pair gives rise mainly to cis-dibenzoyl stilbene. Meanwhile,the reduction of dibenzoyl under conditions that do not stabilize the contact ion pair is not
SCHEME 3-41
SCHEME 3-42
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

stereospecific (Bauld 1965; Thiele 1899). Scheme 3-43 compares results of these two dif-ferent reactions for one and the same substrate.
We are considering here the relation between the distribution of spin density in theproducts of electron transfer to organic molecules and the behavior of these products in re-actions. This relation is important but has so far been little investigated. It would be espe-cially useful to know the reason for the particular sequence of chemical changes in thegroups that are present simultaneously in the molecule. Let us compare the results of one-electron and multielectron reduction of 4,4-dinitro dibenzoyl.
The one-electron reduction leads to paramagnetic species whose ESR spectra dependon temperature and concentration. As shown (Maruyama & Otsuki 1968), such electron-transfer products can exist in the monomeric or dimeric form. The monomer is present athigh dilution. The dimer exists at an increased concentration or at a low temperature; seeScheme 3-44.
In the monomeric form, the unpaired electron is delocalized mainly over the dicar-bonyl moiety and in the dimeric form over the nitro groups. Under the conditions favoringthe dimer formation, 4,4-dinitrobenzophenone gives rise to anion radicals in which the ni-tro group and not the carbonyl group carries the maximum electron density (Maruyama &Otsuki 1968).
SCHEME 3-43
SCHEME 3-44
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

These results of the analysis of the ESR spectra are consistent with the chemical datadepicted in Scheme 3-45 (below). Namely, upon reduction with tin in hydrochloric acid,4,4-dinitrobenzophenone forms 4,4-diaminobenzophenone (only the nitro groups are re-duced, Staedel 1883). Under the same conditions, 4,4-dinitrodibenzoyl gives 4,4-diamin-odesoxybenzoin (both nitro and carbonyl groups are reduced, Golubeff 1873).
4-O2NMC6H4MCOMC6H4MNO2-4 → 4-H2NMC6H4MCOMC6H4MNH2-4
4-O2NMC6H4MCOMCOMC6H4MNO2-4 →4-H2NMC6H4MCOMCH2MC6H4MNH2-4
The metals Li, Na, K, Cs, and Hg-amalgam of Mg have been employed as reducingagents for trans and cis 1,4-diones; see combined Scheme 3–46 (Lazana et al. 1993). It wasfound that cis isomeric ion pairs are favored under conditions involving greater ion associ-ation, for example, in systems with small counterions, low solvating power of the solvent,and at high temperatures. Isomerization of the cis → trans type occurs at �80°C, even forlithium ion pairs in DME. As will be seen in Chapter 5, contact (tight) ion pairs tend totransform into solvent-separated (loose) ion pairs with a temperature decrease. Except forthe small Li� and Mg� counterions associated with a 1,4-dione, the ion pairs with the transconfiguration undergo cyclization, with the formation of a new five-member ring (the bot-tom of Scheme 3-46).
The formation of the bottom structure in Scheme 3-46 seems surprising. The relatedsemidione from 1,4-dimesityl-buta-1,2,3,4-tetraone (dimesityl tetraketone) forms ion pairswith Na�, Cs�, and Ba2� but exhibits no chemical reactions (Bock et al. 1990).
In all the cases considered, stabilization of the cis isomers of semidiones is observedwhen the cation is present. As a rule, for the nonchelated semidione the trans isomer is morestable than the cis isomer. However, the interaction with cations produces the opposite ef-fect, and the cis isomer appears to be more stable than the trans form. Carbonyl compoundscapture an electron and are converted into ketyls, which contain a negatively charged oxy-gen atom. This atom is a particularly powerful proton acceptor. Therefore, a proton, whichsteps forward as a cation, can provide the contour closure for “ion-pair” formation.
In the case of alkali metals, ion pairing can be visualized as hyperfine couplings fromparamagnetic nuclei of the metal cations associated with organic anion radicals in etherealsolvents. In this respect, alkali metal cations associated with the anion radical of o-dimesi-toylbenzene in dimethoxyethane or tetrahydrofurane served as a paradigm (Herold and co-workers 1965).
All the examples just considered have carbonyl (chelating) groups. Therefore, alkalisalts of anion radicals without chelating groups would be particularly important.
Casado and co-workers (2000) considered such a situation. They found that the po-sition of the counterion (together with the relative orientation of the alkyl ether bond withrespect to the benzene ring plane) is a main factor governing the regioselectivity of anionradical fragmentation in alkyl aryl ethers. The ethers can undergo both dealkylation anddealkoxylation. The dealkoxylation occurs when an alkali metal cation is coordinated withthe oxygen atom of the ether bond. If such coordination is destroyed (e.g., in the presenceof crown ethers), significant amounts of dealkylation product are obtained. A free-energycalculation was performed for C–O bond fragmentation in alkyl aryl ether anion radicals(Casado et al. 2000). This calculation shows that such an anion radical system, when no ex-ternal influences are present, has a significant thermodynamic driving force in favor ofdealkylation. Consequently, the formation of an oxygen–metal ion pair allows overcomingof the thermodynamic restriction.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

It seems that some strong association with the countercation should be favored by thesmallness of the �-system in the organic anion radical. Two main factors can be seen asdefining this association. Firstly, the lower electron affinity of a small �-system is expectedto ease a partial retro-transfer of an unpaired electron to the countercation. Secondly, thehigher degree of localization of the negative charge in such a �-system should strengthenthe association of the cation with the anion radical.
These assumptions suggest that the anion radical of ethene, a two-center �-system,would be the best candidate for such a purpose. Not ethene, with an isolated �-system, but1,3-butadiene and its derivatives give observable anion radicals (Levy & Myers 1964,1966).
Of course, the reaction of 1,3-butadiene and its derivatives with the alkali metal in anethereal solvent fails to yield a persistent anion radical, because of rapid polymerization to
SCHEME 3-46
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

rubberlike products. This process is, however, hindered by bulky alkyl substitution (Ger-son et al. 1998). The ion pairs between the anion radical of 2,3-di(tert-butyl)-1,3-butadieneand cations of potassium, rubidium, or cesium deserve our consideration in this part of thebook.
Both theory and experiment point to an almost perpendicular orientation of the twobutadiene H2CBC(t-Bu)M moieties; see Scheme 3-47. On passing from the neutralmolecule to its anion radical, this orthogonal orientation should flatten, because the LUMOof 1,3-butadiene is bonding between C(2) and C(3). So this bond should be considerablystrengthened after formation of the anion-radical. The anion radical will acquire a cisoidalconformation. This conformation places two bulky tert-butyl substituents on “one side” ofthe molecule, so the alkali metal counterion (M�) can closely approach the anion radicalfrom the “other side.” In this case, the cation will detain spin density in the localized partof the molecular skeleton. A direct transfer of the spin population from the singly occupiedMO of the anion radical into the alkali cation has been proven (Gerson et al. 1998).
Low-lying vacant orbitals of alkali metal cations can, consequently, accept an un-paired electron density even if it is delocalized over an extended �-system of carbon chains.The anion radical of 1,4-diphenylbutadiene can exist in s-trans and in s-cis forms. The rel-ative amounts of these geometrical isomers appear to depend highly on the counterion/sol-vent system. As counterions, Li� and K� were studied; tetrahydrofurane, 2-methyltetrahy-drofurane, and dimethoxyethane were employed as solvents (Schenk et al. 1991).Interaction between the anion radical and the cation contributes to a stabilization of the s-cis arrangement “if the cation is close to the carbons C-1 and C-4.” This stabilization ismost pronounced in an ion pair with a tight interaction of the countercation and the �-sys-tem, and indeed, the s-cis form of the anion radical is only observed under those experi-mental conditions that favor tight ion pairing.
There is already an extensive body of work studying the role of the proton in meet-ing ring ends. Therefore, only the most prominent example will be considered here. It wasshown that in pyridine, dimethylsulfoxide, and dimethylformamide, dialkyl fumarates ordialkyl maleates are reduced in stages (Takahashi & Elving 1967; Nelsen 1967; Il’yasov,Kargin, & Kondranina 1971). Anion radicals capable of undergoing one-electron oxidationare formed at the first stage. The half-wave potentials for the reduction of maleate and fu-marate differ, but the one-electron oxidation of the resulting anion radicals is characterizedby the same half-wave potentials. The earlier-described relations between E1/2 for the ca-thodic and anodic waves also hold at a very high alternating frequency, up to 500 Hz(Il’yasov, Kargin, & Kondranina 1971). Yeh and Bard (1977) obtained the same results.This implies that the anion radicals formed from maleate and fumarate have similar ener-
SCHEME 3-47
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

gies. The ESR spectra of the products of the electrochemical reduction of dimethyl fu-marate and maleate are identical at the potential of the limiting currents of the first waves,regardless of the temperature at which they are recorded (Il’yasov, Kargin, Sotnikova, et al.1971). According to the authors, it is unlikely that the ESR spectra of the cis and the transanion radicals are completely identical. Thus, it is assumed that the ESR spectra of the cisand trans anion radicals should differ, and the preceding observation indicates a rapid andalmost complete trans isomerization of the cis anion radicals. At the same time, if the an-ion radicals are obtained in a proton-donating medium, the cis and trans isomers give riseto different ESR (Anderson et al. 1971) and electronic (Hayon & Simic 1973) spectra. Un-der these conditions, maleates evidently give rise to unusually stable anion radicals whosestructure is fixed by the formation of a ring with participation of a proton from the medium.Scheme 3-48 illustrates the phenomenon.
The coordination of the potassium cation to two oxygen atoms in the anion radical ofortho-dinitrobenzene leads to the stabilization of an unusual configuration in which the ni-tro groups are spread out in a single plane. In neutral ortho-dinitro benzene, the coplanarityof the two nitro groups is ruled out both by calculation (Ghirvu et al. 1971) and by experi-ment (Calderbank et al. 1968; Myagi 1971). In the potassium derivative of ortho-dini-trobenzene, the coordination of the metal cation to both nitro groups leads to the formationof a condensed “ring” in which there is the possibility of an additional delocalization of theunpaired electron. Indeed, the ESR spectra in dimethoxyethane or in tetrahydrofurane showthat the unpaired electron in this species is equally delocalized over the two nitro groups(the constants for the splitting by the nitrogen atoms aN
1 � aN2 � 0.32 mT, Ward 1961).
Meanwhile, in the potassium salt of the anion radical of meta-dinitro benzene, the delocal-ization is essentially over only one of the two groups (aN
1 � 0.93 mT, aN2 � 0.02 mT, Ward
1961; Gol’teuzen et al. 1972). This constrasts sharply with ESR spectroscopic data for theanion radicals obtained by electrolysis in acetonitrile in the presence of tetra (n-propyl) am-monium perchlorate as the supporting electrolyte: aN
1 � aN2 � 0.32 mT for the ortho-dinitro
benzene anion radical and aN1 � aN
2 � 0.47 mT for the meta-dinitro benzene anion radical(Maki & Geske 1960). The difference can be understood if we take into account the natureof the counterion. In the case of the potassium cation, the formation of a closely knit ionpair is possible. The K� ion can then provide its low-lying vacant-d and p-orbitals to bepopulated with the �-electrons of the “condensed” anion radical system.
The idea of cation–� interactions is receiving more and more acceptance. The inter-action of an aromatic �-system with an alkali metal cation has been known for two decades(Sunner et al. 1981). Recently, it has been confirmed in several experimental (De Wall etal. 1999) and theoretical works (Kumpf & Dougherty 1993; Dougherty 1996; Nicholas et al. 1999).
When a tetra-alkylammonium cation is used as a counterion in solvents of high po-larity, such as acetonitrile and dimethylformamide, the alkyl groups hinder the mutual ap-
SCHEME 3-48
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

proach of species with different charges. Ion pairs with the potassium cation are stable, i.e.,exist longer than is required for the detection of the ESR signal. This follows from a com-parison of the polarographic behavior of the three isomeric dinitrobenzenes in the samesolvent (dimethylformamide) using tetraethyl ammonium or potassium perchlorate as thecarrier electrolyte (Todres 1970). The half-wave potentials corresponding to the conversionof para- and meta-dinitrobenzenes into anion radicals are independent of whethertetra(ethyl) ammonium or potassium counterions are employed. The anion radical isformed from ortho-dinitrobenzene at a potential less negative by almost 100 mV whentetra(ethyl) ammonium perchlorate is replaced by potassium or sodium perchlorate. Underthese conditions, changes in the composition and structure of the electrical double layer donot influence the reduction mechanism, which has been shown by a special study (Todres,Pozdeeva, et al. 1972) to consist of the reversible transfer of one electron. Since the trans-fer of one electron to ortho-dinitrobenzene in the presence of potassium perchlorate as thecarrier electrolyte proceeds at a less negative potential than in the presence of tetra(ethyl)ammonium perchlorate, it is clear that the potassium salt does indeed have the minimal en-ergy.
It was ion pairing between the potassium cation and the ortho-dinitrobenzene an-ion radical that gave the first successful example of anion radical azo coupling. Perhapsazo coupling is the most characteristic and practically used reaction in aromatic range. Anindispensable requirement for this reaction is the presence of the amino or hydroxylgroups in an aromatic substrate. Azo-coupling reactions with aromatic compounds con-taining only the nitro groups were deemed impossible. As was established (Todres, Hovsepyan, & Ionina, et al. 1988), the anion radical of ortho-dinitrobenzene reacts withbenzene diazo cations in THF undergoing azo coupling at the para position; see Section1.2.1.
The azo coupling just mentioned is accompanied by the conversion of one of the ni-tro groups into the hydroxyl group. Hence, the radical product is stabilized by eliminationof the nitrogen monoxide radical. All the radicals are prone to stabilize, expelling a smallradical particle. This is the case too. And nitrogen mono-oxide was established as a gas-phase product of the reaction (Todres, Hovsepyan, & Ionina 1988).
As a result of quantum chemical calculations on the ortho-dinitrobenzene anion rad-ical (Todres 1990), the attack of cation-type electrophiles is predicted to take place at thetwo para positions of the benzene ring. ortho Substitutions seem to be impossible. The ex-periments have confirmed this theoretical prediction.
An azo coupling could be prevented by changing tetrahydrofurane as a solvent todimethylsulfoxide or by adding 18-crown-6 ether to the THF-reaction mixture. The split-ting of the coordination complex follows Scheme 3-49.
SCHEME 3-49
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

When the coordination complex is destroyed, no azo coupling takes place. In thiscase, only electron-transfer products are formed: ortho-dinitrobenzene, a benzene deriva-tive RC6H5 from RC6H4N2BF4, gaseous nitrogen, and KBF4.
From this point of view, the studies of the interaction of benzene diazo cations withthe potassium salt of the para-dinitrobenzene anion radical should be mentioned (Todres,Hovsepyan, lonina, et al. 1988). The potassium cation in such anion radical salts is knownto be located near one of the nitro groups. This lone nitro group bears the main part of thespin density (Ling & Gendell 1967). In a solvent of low dissociating ability, this asymmet-ric coordination complex reacts with benzene diazo compounds through one-electron trans-fer only, without any azo coupling. By using tetrahydrofurane as a solvent for both anionradicals of ortho- and para-dinitrobenzenes, i.e., under comparable experimental condi-tions, quite different products are obtained. As mentioned, the ortho isomer gives rise to theazocoupling product, whereas the para isomer is discharged according to Scheme 3-50.
As seen, the formation of a closed contour for unpaired electron delocalization is awidespread phenomenon that detains the electron in the framework of a definite molecularfragment and really defines the physical and chemical properties of ion radicals.
3.3 PRINCIPLE OF THE “RELEASED” ELECTRON THAT CONTROLSION RADICAL REACTIVITY
Ion radicals of conjugated acyclic or aromatic hydrocarbons (butadiene or naphthalene) aretypical examples of the species with a released unpaired electron. They are named �-elec-tron ion radicals and have a spin distribution along the whole molecular contour. An im-portant feature of such species is that all the structural components are coplanar or almostcoplanar. In this case, spin density appears to be uniformly or symmetrically distributedover the molecular framework. Spin-density distribution has a decisive effect on the ther-modynamic stability of ion radicals. In general, the stability of ion radicals increases withan enhancement in delocalization and steric shielding of the reaction centers bearing themaximal spin density.
SCHEME 3-50
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Blockading of positions with high spin densities diminishes the reactivity of ion rad-icals and to some extent keeps them safe from undesirable transformations. For instance,the substitution of hydrogen atoms with the phenyl or methyl groups in the active 9 and 10positions of anthracene is a method for stabilizing the cation radicals of 9, 10-diphenyl- or9,10-dimethylanthracene. Blockading of only one active position leads to a rather stable 9-phenylanthracene cation radical but to an completely unstable 9-methylanthracene cationradical (Phelps et al. 1967). The phenyl group can delocalize a spin density more efficientlythan the methyl group. It is worth noting, however, that the formation of hydrocarboncation radicals is rather difficult and demands high anodic potentials.
In comparison with hydrocarbons, aromatic amines transform into cation radicalsmore readily. The stable cation radical of N,N,N,N-tetramethyl-p-phenylenediamine (so-called Wuerster’s Blue) was one of the first ion radicals studied via ESR spectroscopy(Weissmann et al. 1953). Its stability is ascribed to the electron delocalization in itssemiquinone structure (Elbl-Weiser et al. 1989). Recently, the use of this cation radical asa spin-containing unit for high-spin molecules has been reported (Ito et al. 1999). Chemi-cal oxidation of N,N-bis[4-(dimethylamino)-phenyl-N,N-dimethyl-1,3-phenylenedi-amine with thianthrenium perchlorate in n-butyronitrile in the presence of trifluoroaceticacid at �78°C led to the formation of the di(cation radical) depicted in Scheme 3-51. Asseen, the spin-charge systems are disjoint in this paramagnetic species. The following twofeatures of this di(cation radical) are worth noting: It was found to be a ground-state tripletand to be unstable at ambient temperature.
The methoxy group usually acts as a stabilizing substituent on amine cation radials.For example, N,N-dimethyl anisidine gives a stable cation radical, whereas the N,N-dimethyl-p-toluidine cation radical is short lived (although its ESR spectrum can berecorded). Deblockading of the para position sharply diminishes the cation radical stabil-ity: N,N-dimethylaniline gives N,N,N,N-tetramethyl benzidine instantly at the very be-ginning of anodic oxidation (Mann & Barnes 1970, Chap. 9).
Applying the concept of isoelectronic compounds and redox series (Kaim 1980), onecan point out the existence of analogous redox systems based on–BR2/–BR2
�.instead
of–NR2/–NR2�.
as essential end groups of conjugated �-systems. The corresponding redoxseries then involve anionic charges instead of cationic ones, as can be seen in Scheme 3-52.
In the nitrogen and boron analogs depicted in Scheme 3-52, two methyl groups pro-vide a sufficient shielding at the NR2
�.centers (R � Me), while two mesityl groups are
needed for protection of the BR2�.
centers (R � 2,4,6-trimethyl phenyl). Electrochemicalstudies of 1,4-bis(dimesitylboryl)benzene have shown two well-separated one-electron re-duction processes, with the formation of the corresponding anion radicals and dianions, re-spectively (Fiedler et al. 1996). According to UV/vis/near-IR and ESR spectroscopic data
SCHEME 3-51
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

that were confirmed with quantum mechanical calculations, the unpaired electron popu-lates primarily a �(C6H4)–pz(B)–based MO (Fiedler et al. 1996). Resonance formulationforesees the participation of the central benzene ring (denoted in Scheme 3-53 as �) andboth boron atoms in delocalization of an unpaired electron (a delocalazed BIII/BII mixed-valence model). Scheme 3-53 (below) visualizes this delocalization:
[�BIIIM�°MBIII�] � e → [�BIIIM�°M�BII�] ↔[�BIIIM��.
MBIII] ↔ [��BIIM�MBIII�]
Naturally, the product of two-electron reduction of the para derivative can be de-picted (Scheme 3-54) as an anionic diborataquinoid system. In contrast with the para-dib-orataquinoid dianion, an anionic meta-quinoid system is impossible. Indeed, the meta-sub-stituted isomer depicted in Scheme 3-54 has been characterized as a spin-unpaired tripletspecies with boron-centered spins (Rajca et al. 1995). This dianion diradical can be viewedas two stable borane anion radicals linked with a “ferromagnetic coupling unit,” i.e., 1,3-phenylene; see Chapter 1.
3.3.1 Effects of Spread Conjugation in Ion Radicals Derived fromMolecules with Large Contours of Delocalization
Attention should also be paid to the fact that the electron motion from a donor to an accep-tor or within spread-conjugated ion radicals generates measurable electromagnetic radia-tion. In other words, there is one specific way to reveal intramolecular electron motion orintermolecular electron transfer in charge-transfer complexes. In order to probe electrontransfer in this manner, the only fundamental requirements are that the molecule can be ori-ented in the external magnetic field. This takes place most easily if the species consideredhas a ground-state dipole moment. Upon photoexcitation, there is a change in the dipolemoment along the molecular axis of orientation. The sign of the radiated electromagnetic
SCHEME 3-52
SCHEME 3-54
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

field depends on whether the change in dipole is positive or negative. For example, uponphotoexcitation at 400 nm of 4-dimethylamino-4-nitrostilbene dissolved in toluene, thedipole moment of the stilbene increases. This corresponds to the amino-to-nitro electrontransfer. Thus the direction of the electron transfer can be determined directly from theelectromagnetic measurement (Beard et al. 2000).
The rather clear difference between homologs of para- and meta-phenylenevinylenesin anion radical forms is depicted in Scheme 3-55.
The anion radicals in Scheme 3-55 were investigated by ESR and electron adsorptionspectroscopy (Gregorius et al. 1992). The para isomer appears to behave completely dif-ferent from the meta isomer. The authors conclude, and this is in full agreement with theresults from MO theoretical calculations, that the unpaired electron is delocalized over thewhole para isomer but confined to a stilbene unit in the meta isomer. The remaining partsin the meta isomer are uncharged. This spontaneous charge localization is not a conse-quence of steric hindrance, but follows from the role of the meta-phenylene unit as a con-jugational barrier.
In a similar manner, para-bis(9-anthryl) phenylene gives a mono(anion radical) or amono(cation radical) under reductive or oxidative conditions with spin delocalizationaround the whole molecular framework. In the case of meta-bis(9-anthryl) phenylene, re-duction or oxidation leads to formation of dianion or dication diradicals. Based on ESR ex-periments at cryogenic temperatures (6.5–85 K), these species contain two separated ionradical moieties. They have parallel alignment of their spins (Tukada 1994). The workgives clear experimental evidence for so-called ferromagnetic interaction between theseion radical substituents; see Chapter 7.
In some cases, electron releasing depends on temperature. An example is the anionradical of Scheme 3-56. The stable anion-radical of Scheme 3-56 contains two perchlorot-riphenylmetyl radical units linked by an all-trans-p-divinylbenzene bridge. At 200 K, theunpaired electron of the anion radical is localized on the ESR time scale on one stilbene-like moiety only. At 300 K, thermal activation forces the unpaired electron at one strongelectrophilic center to move to another one. Such electron transfer takes place between two
SCHEME 3-55
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

equivalent redox sites (Bonvoisin et al. 1994). In contrast to this situation, no electron trans-fer was observed for the anion radical that contains two perchlorotriphenylmetyl radicalunits linked by an all-trans-m-divinylbenzene bridge (Rovira et al. 2001). Such a result canbe ascribed to the localization of frontier orbitals in the m-isomeric anion radical becauseof the meta connectivity of this non-Kekule structure.
There is one special case when the ring contour of delocalization is organized just af-ter transformation of neutral molecules into ion radicals. The case is depicted in Scheme 3-57 as cyclo[3.3.3]azine and its analogs. In these condensed systems, the central nitrogenatom bears a lone electron pair. All of these condensed compounds are readily oxidizedwith silver perchlorate or silver fluoroborate in dimethylformamide and give cation radi-cals. It is logical to suppose that the maximal spin density in these cation radicals should belocalized at the central nitrogen. As a matter of fact, however, the formation of a peripheral�-electron system takes place. Now then, the central nitrogen atom presumably keeps theperipheral system planar, and its unshared electrons contribute very little to the peripheralconjugation. By means of SCF-MO calculations and ESR experiments, both cation radicalsand anion radicals of cyclo[3.3.3] azine are aromatic with no essential double-bond local-ization (symmetry D3h). This is in contrast to the neutral compound, which exhibits alter-nating essential single and double bonds (symmetry C3h). In the ion radicals, the central ni-trogen atom is presumably coplanar with the 12 membered rings (Dewar & Trinajstic 1969;Gerson et al. 1973). The electron distribution in these cation radicals and anion radicals isnot time dependent, and the peculiarities just discussed should not be understood as in-volving an electron coming off the nitrogen atom.
SCHEME 3-56
SCHEME 3-57
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Related compounds, cyclo[2.2.3]azine and 1,2,3,4-dibenzocyclo[2.2.3]azine, alsogive ion radicals with peripheral �-electron conjugate systems (Gerson et al. 1973; Mat-sumoto et al. 1996). The difference in conjugation between neutral molecules and their ionradicals can be additionally traced in the case of keto-enol tautomerizm. As a rule, enols areusually less stable than ketones. Under equilibrium conditions, enols exist only at very lowconcentration. However, the situation is different in the corresponding cation radicals,where gas-phase experiments have shown that enol cation radicals are usually more stablethan their keto tautomers. This is due to the fact that enol cation radicals profit from allylicresonance stabilization that is not available to ketones (see Bednarek et al. 2001 and refer-ences therein); see Scheme 3-58:
(R2CHMCHBCHMC(R)BO)�. → (R2CBCHMCHBC(R)MOH)�.
3.3.2 Spin Delocalization in Ion Radicals Derived from Molecules withIncreased Dimensionality
Sometimes transformation of aromatic compounds into ion radicals sets stereochemicallyunusual forms. For instance, the enantiomeric binaphthyls racemize, being converted intothe anion radical state and then being oxidized. Let us examine causes for this racemization.
In a neutral 1,1-binaphthyl, the most stable conformation is that in which the naph-thalene rings form an angle of 70°. This leads to a very small �-overlap and to the existenceof two enantiomers (atropoisomers) differing in the arrangement of one naphthyl nucleusabove or below the plane of the other. Following transformation into the anion radical state,1,1-binaphthyl acquires a stereochemically unusual form in which the torsion angle is re-duced to about 50° (Baumgarten et al. 1995). This permits complete delocalization of theunpaired electron and leads to a loss of chirality. Steric strains inherent in the conformationare compensated for by the gain in the delocalization energy: The unpaired electron may bedistributed on two naphthyl nuclei instead of one. Upon one-electron oxidation, the anionradical of 1,1-binaphthyl loses planarity and should produce a mixture of enantiomers. Asthe calculations (Eisenstein et al. 1977) show, when the anion radical of binaphthyl ac-quires an almost planar conformation, the number of most reactive sites decreases fromthree (positions 4,5, and 8) to one (position 4). Carbon-4 has the greatest electron density.This has been confirmed by ESR data (Eisenstein et al. 1977; Baumgarten et al. 1993).Hence, the ion radical reactions of binaphthyl must proceed nonstereospecifically but re-gioselectively.
And indeed, treating one of the enantiomers (in tetrahydrofurane) with lithium andthen with aminonitrile results in aminoalkylation strictly in position 4 of the naphthalenefragment. The part of binaphthyl that does not participate in alkylation returns in the opti-cally inactive form (Eisenstein et al. 1977). Scheme 3-59 presents the whole process.
The last stage of the reaction in Scheme 3-59 involves protonation, yielding thederivative of 1,4-dihydronaphthalene. The oxidation may produce a 4-substituted binaph-thyl not contaminated with the isomeric products. It is worth noting here that the describedion radical method of introduction of the alkyl group into the aromatic nucleus has an ad-vantage over radical or heterolytic alkylation, since the neutral substrate may produce acomposite mixture of isomeric products. The binaphthyl anion radical reaction proceeds re-gioselectively and nonstereospecifically.
The twist angle in the ethylene cation radical is about 40°. The twisting is necessaryfor an overlap to occur between the ethylenic bond and the p-orbital bearing a spin (and a
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.
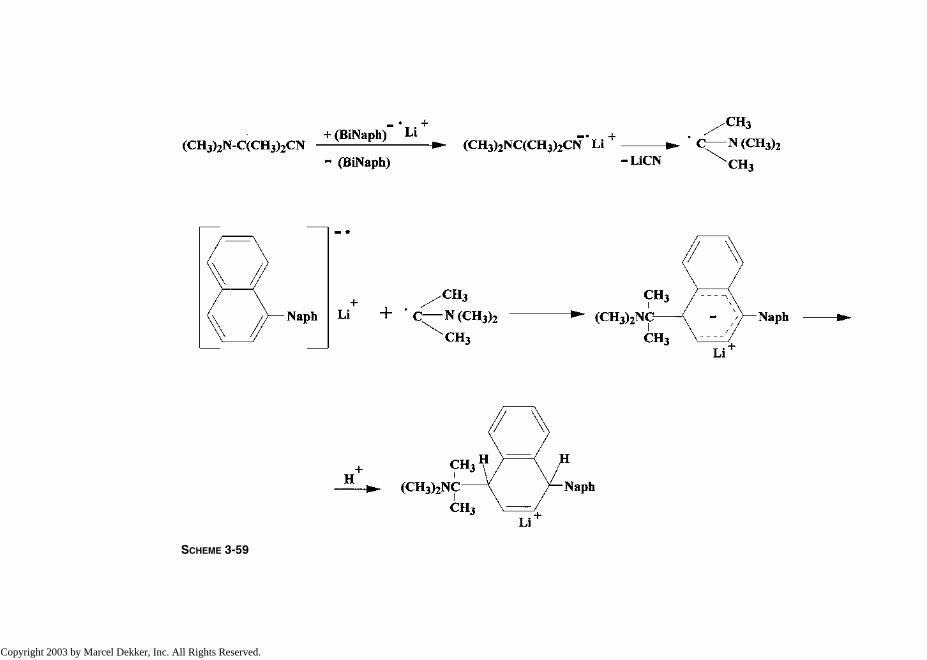
SCHEME 3-59
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

charge). The untwisted system has no hyperconjugative stabilization. There is basically nobarrier for the twist in the ethylene cation radical, but simply alkylating to the 2-butenecation radical makes its cis and trans isomers different, and gives a high barrier to inter-conversion (Clark & Nelsen 1988).
The allene molecule resembles binaphthyl in terms of its two cumulated �-systemsand the orthogonality of these system planes. (A quantum mechanical model of cumulativedouble bonds in allenes forecasts an sp2 electron state for both terminal carbon atoms. Thecentral carbon atom has sp hybridization. Therefore, the bond system, which proceeds fromthis central atom, is linear. Both double-bond and terminal substituents are located in mu-tually perpendicular planes.) Both the neutral allene and its cation radical are depicted inScheme 3-60. In the allene cation radical, the angle between the double-bond planes is di-minished and reaches 30–40° (Takemura & Shida 1980). According to calculations (Has-selbach 1970), a multiatomic linear cation radical of allene has a degenerate electron state.Indeed, in accordance with the Jahn–Teller theorem, this cation radical should undergo dis-tortion of the geometry in order to acquire a less symmetric form. As follows from calcu-lations (Takemura & Shida 1980; Somekawa et al. 1984), the most favorable form is theone with an angle between the ethylene bond planes of ca. 40°. The allenic group(MCBCBCM) is a moderate �-donor (Nagase et al. 1979).
The allenes bearing four tert-butyl or four trimethylsilyl groups at the terminal car-bons react in methylene chloride with antimony pentachloride. The cation radicals formedcontain an unpaired electron delocalized along the neighbouring �-bonds. The conclusionis based on the analysis of 1H, 13C, and 29Si ESR spectra (Bolze et al. 1982) as well as onphotoelectron spectra (Elsevier et al. 1985; Kamphius et al. 1986). These data have foundcorroboration in a recent study (Werst & Trifunac 1991). The tetramethylallene cation rad-ical spectrum was observed by fluorescent-detected magnetic resonance. The well-resolvedmultiplet due to this cation radical consists of a binomial 13-line pattern owing to 12 equiv-alent methyl protons. This is in full accord with Scheme 3-60.
As proven, the fundamental difference was found to be between allene (cumulateddouble bonds are separated in the space) and its cation radical (cumulated double bonds areconjugated and lay at the same plane).
Spiro compounds are known to have joined rings in two perpendicular planes with anodal common atom. This atom (the quaternary carbon) can prevent conjugative interac-tions between the two joined rings. In the case of two identical perpendicular �-networksjoined by a spiro atom, the orbitals of the “halves” may interact only if they possess thesame symmetry. Such interactions lead to pairs of delocalized orbitals encompassing the
SCHEME 3-60
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

entire molecule. This is material realization of the spiroconjugation phenomenon (for a re-view of spiroconjugation, see Durr & Gleiter 1978).
Spirobis(indandione), from Scheme 3-61, is one example of this kind of conjugation.Moreover, its frontier orbital (LUMO) also satisfies the symmetry requirement mentionedearlier. Hence, it can be formally considered as a bonding combination of the “half-molecule” orbitals (Maslak et al. 1990).
Spirobis(indanedione) was reduced, with anion radicals of polyaromatic compoundsserving as electron donors at low temperatures (195–173 K). An anion radical of the spiro-compound was formed. When treated with dioxygen, this anion radical gave the starting,unchanged dione, Scheme 3-61.
The spiro(indanedione) anion radical in Scheme 3-61 was studied via ESR andUV/visible spectroscopy (Maslak et al. 1990). The spectra clearly indicated delocalizationof the unpaired spin density over the entire molecular framework. The unpaired electronundergoes simultaneous delocalization between the halves (in the ESR time scale). The ob-served spectra were independent of the counterion (Li�, Na�, and K�), thus excluding anyion-pairing complication. As a general inference, an unpaired electron spends its time onboth half-shaped orbitals, with no geometrical changes in the molecular skeleton of this an-ion radical.
Simultaneous delocalization of an unpaired electron over two molecular halves iscaused by rapid electron transfer. Ballester and co-authors (1991) described another exam-ple of such a transfer with no geometrical changes in the molecular skeleton. In the per-chloro-�,�,�,�-tetraphenyl bis(p-tolyl)-�-yil-�-ylium anion radical or cation radical,(C6Cl5)2C
.MC6Cl4MC6Cl4MC�.
(C6Cl5)2, there is a very rapid intramolecular electrontransfer between radical and ion radical sites. Repulsion among the four chlorine atoms or-tho with respect to the central liaison (MC6Cl4MC6Cl4M) causes the two phenyl rings ofthe biphenyl system to be perpendicular to each other. The authors conclude, on account ofthis perpendicularity, that “the overlap involving the higher-energy bonding �-orbitals ispractically nonexistent in these ion radicals. It is doubtful that any through-�-bond mecha-nism might be involved in the spin-charge exchanges, since it hardly provides a low-energypath for them. Therefore, at least some spin charge interchange here described takes placealong the �-path.”
A significant subject is the involvement of the ion radicals’ counterions [SbCl6� or(n-Bu)4N�] in the exchange process. According to the experimental conditions employed,the authors had the free ion radicals exclusively. Hence, the counterion did not appear toplay any important role in the spin-charge alternation.
It is some kind of �-conjugation that also plays a role in delocalization (or releasing)of an unpaired electron in ion radicals.
SCHEME 3-61
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

3.4 BEHAVIOR OF ORGANIC ION RADICALS IN LIVING ORGANISMS
3.4.1 Cation Radical Damage in DNA
A substantial body of experimental evidence indicates that the formation of a covalent bondbetween chemical carcinogens and cellular macromolecules represents the first critical stepin the multistage process eventually leading to tumor formation (see Geacintov et al. 1997and references therein).
Most chemical carcinogens are not active on their own, but require metabolic activa-tion to produce reactive intermediates capable of binding covalently to target macro-molecules, in particular DNA, and thereby initiate cancer.
The most widespread environmental carcinogens are the polycyclic aromatic hydro-carbons (PAHs), which are found, among other places, in automobile exhaust, cigarettesmoke, and broiled meats. PAHs undergo two main pathways of bioactivation: one-elec-tron oxidation and oxygenation. The former yields cation radicals; the latter produces hy-droxyl derivatives.
One-electron oxidation to form cation radicals is the major pathway of activation forthe most potent carcinogenic PAHs, whereas oxygenation is generally a minor pathway.For benz[a]pyrene and 7,12-dimethyl benz[a]anthracene, 80% and 90%, respectively, ofthe DNA adducts formed by rat liver microsomes or in mouse skin arise via the cation rad-icals (Cavaleri & Rogan 1992).
One-electron oxidation of PAHs to form cation radicals is a coexisting mechanism ofactivation that can account for the binding of the most potent hydrocarbons to DNA andpresumably for their carcinogenic activity. These reactive electrophiles are produced by re-moval of a �-electron by the biological oxidant P450 (Cavalieri, Devanesan, & Rogan1988; Cavalieri, Rogan, Devanesan, et al. 1990) and peroxidases, such as prostaglandin Hsynthase and horseradish peroxidase (Cavaleri, Devanesan, & Rogan 1988; Rogan, Cava-leri, et al. 1988; Rogan, Katomski, et al. 1979).
The reason that one-electron oxidation is suggested as playing a central role in themetabolic activation of polycyclic aromatic hydrocarbons derives from certain features ofthe radical cations that are common to the most potent carcinogens of the family:
1. Relatively low ionization potential (IP), which allows the metabolic removal ofone electron, with the formation of a relatively stable cation radical
2. Charge localization in the cation radical that renders this intermediate specifi-cally and efficiently reactive toward nucleophiles
3. Optimal geometric configuration that allows the formation of appropriate inter-calating physical complexes with DNA, thus favoring metabolic activation andthe formation of covalent bonds with DNA nucleophiles.
All of these factors are important in carcinogenesis. Therefore it is worthwhile con-sidering them here, separate from the general regularities of the formation and stability oforganic ion radicals.
3.4.1.A Ionization Potentials (IPs) of Carcinogens
The ease of formation of PAH cation radicals is related to their IP. Above a certain IP, ac-tivation by one-electron oxidation becomes unlikely due to the more difficult removal ofone electron by the active forms of P450 or peroxidases. A cutoff IP above which one-elec-tron oxidation is not likely to occur was tentatively proposed to be about 7.35 eV (Cava-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

lieri & Rogan 1995). For example, 7,12-dimethylbenz[a]-anthracene has an IP of 7.22 eVand is extremely carcinogenic. Benz[a]anthracene has an IP of 7.54 eV and is very weaklyactive in this sense. The active carcinogenicity of dibenz[a,h]anthracene (IP of 7.61 eV) isnot attributable to the one-electron mechanism. It is worth noting that one-electron transferis not only one of the operating mechanisms of carcinogenesis.
3.4.1.B Localization of Charges and Spins in Cation Radicals of Carcinogens
A relatively low IP is a necessary, but not sufficient, prerequisite for activating PAHs byone-electron oxidation. Another important factor that must be combined with IP to predictcarcinogenic activity via this mechanism is charge localization in the PAH cation radical.Specificity in cation radical reactivity derives from the relative localization of charge at oneor a few carbon atoms.
In situ generation of those cation radicals has been used in their identification as bi-ological metabolites. A valid alternative was to synthesize the cation radical as a solid salt,isolate it, and investigate its reactions with various nucleophiles, including DNA. Suchstudies were conducted to better understand the role of those intermediates in biologicalsystems (Murata & Shine 1969; Ristagno, & Shine 1971; Cremonesi et al. 1994; Stack etal. 1995).
For instance, oxidation of benz[a]pyrene (BP) by NOBF4 in the mixture of methy-lene chloride with acetonitrile generates the BP�.
BF4� salt. When this cation radical salt is
attacked with nucleophiles of various strengths, the pattern of nucleophilic substitution re-flects the distribution of a positive charge in the cation radical part of the salt. For PAHs, apositive charge is localized mainly at the meso-anthracenic position, i.e., at the C-6 atom.Nucleophiles (Nu�) such as OH�, AcO�, and F� enter that position, Scheme 3-62.
The reaction of BP�.with DNA produces two DNA-depurinating adducts, BP-6-
N7Gua and BP-6-C8Gua (Stack et al. 1995), as in Scheme 3-63.When binding of the uncharged BP to DNA is catalyzed by horseradish peroxidase
(Cavalieri, Rogan, et al. 1988) or rat liver microsomes (Cavalieri, Devanesan, & Rogan1988), the same pattern of the major DNA-depurinating adducts was obtained. As men-
SCHEME 3-62
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

tioned earlier, horseradish peroxidase activates BP via one-electron oxidation; hence thisresult is expected (Rogan et al. 1979, 1988).
When a carbon atom bearing a positive charge is bound with the methyl group, thereaction path consists of a methyl proton loss. The deprotonation generates a benzylic rad-ical that is rapidly oxidized to a benzylic carbenium ion that reacts with a nucleophile ac-cording to Scheme 3-64. In accordance with Scheme 3-64, BP-6-methyl is characterized asan active carcinogen (Cavalieri & Rogan 1995).
When the positive charge in the cation radical is localized adjacent to the ethyl group,such as in 6-ethyl-BP, a reaction cannot occur at the benzylic methylene group. The sourceof this is that the C–H bond is less favorably aligned with the �-system to cause deproto-nation as compared to the C–H bond of the methyl group. This ethyl-substituted BP is notcarcinogenic at all (Cavalieri & Rogan 1995).
SCHEME 3-63
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

In addition to charge localization, another important factor is nucleophile activity inthe interception of a cation radical. For example, 7,12-dimethyl-benz[a]anthracene(DMBA) is oxidized with iodine in the presence of pyridine, yielding pyridinium deriva-tives at the methyl groups in positions 5, 7, and 12. Acetate ion is a weaker nucleophile thanpyridine. Thus, when DMBA is oxidized with manganese acetate in acetic acid, only theacetoxy derivatives at the 7-CH3 and 12-CH3 groups are obtained. The same exclusivemethyl substitutions occur with such weak nucleophiles as adenine and guanine to produceadducts when DMBA is electrolytically oxidized in the presence of deoxyadenosine or de-oxyguanine (Cavalieri & Rogan 1995). Thus, BP cation radicals show little selectivity withstronger nucleophiles, but higher selectivity with weaker nucleophiles. This feature is es-pecially important for biochemical reactions.
3.4.2 On Geometrical and Spatial Factors Governing the Behavior ofIon Radicals in Biological Systems
Another important factor affecting the ability of PAH cation radicals to elicit carcinogenicactivity is their shape and size. In general, carcinogenic activity is found in PAHs that con-tain from three to seven condensed rings. A more stringent requirement concerning geo-metric features of PAHs is the presence of an angular ring in the benz[a]anthracene series.This ring is necessary for eliciting the carcinogenic activity, regardless of whether the ringis aromatic or alicyclic. One-electron oxidation of PAHs gives cation radicals with nochanges in precursor geometry. As noted (Cavalieri & Rogan 1995), the DNA adducts withthe cation radicals of BP and DMBA can be obtained only from double-stranded DNA (an-imal DNA) but not from single-stranded DNA (bacterium DNA). According to Geacintovand co-workers (1997), metabolites of PAHs bind to the purine bases in DNA to formadducts that intercalate between bases in the DNA double helix.
By locally stretching and unwinding the helix, the metabolite can slip between pairsof bases in the DNA double helix without displacing any of these bases. DNA treated with
SCHEME 3-64
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

PAH cation radicals contains relatively large amounts of apurinic sites as well as some sta-ble adducts. Enzymes, many of which move along the polymeric strand in a particular di-rection, process DNA. Thus the enzymes will see damaged parts of DNA (depurinated orstretched and unwound) and provoke tumorigenic replications. Of course, particular DNAsequences are more prone to mutation than others. These “hot spots” vary in different or-ganisms. PAH cation radicals can serve as a useful tool in studying this particular problem.
At this point it is expedient to give some intermediate inference. As mentioned in theintroduction to this section, there are two principal mechanisms of bioactivation: oxygena-tion and one-electron oxidation. Recently, the scientific community has considered oxy-genation to be the exclusive mechanism of carcinogenesis of PAHs (see, e.g., Hall &Grover 1990). The arranged material involves one more and, obviously, main mechanismof activation of PAHs. The formation of PAH cation radicals plays the central role in acti-vation of the most potent carcinogenic PAHs. These PAHs generally have relatively lowionization potentials. They give cation radicals with expressed localization of a spin and acharge. Oxygenation to form bay-region diol epoxides is generally a minor, reserve path-way. For instance, benz[c]phenanthrene cannot effectively generate its cation radical in thecommon biological condition of oxidation. It has a relatively high IP. In this case the onlypossible pathway of carcinogenic activation is the formation of its diol derivative.
It is worth mentioning that the cation radical mechanism of carcinogenesis was es-tablished for readily oxidized PAHs that belong to a series of alternant aromatics. Nonal-ternant PAHs remain an attractive field for future exploration. Hydrocarbons with con-densed aromatic rings containing at least one ring with an odd number of skeletal atoms canbe pinpointed as possible candidates for such study. Benz[ j]fluoranthene, benz[k]fluoran-thene, and benz[c,g]carbazole were named as desirable examples (Cavalieri & Rogan1992).
One very intriguing work on benz[c]acridines and benzo[a]phenothiazines estab-lishes a connection between their carcinogenicity and radical production (Kurihara et al.1998). It would be interesting to check for a connection between the carcinogenicity andelectron structures of the corresponding cation radicals too.
The aforementioned behavior of P450 as one-electron oxidant deserves some expla-nation. Since P450 is overall a two-electron oxidant, it is a priori unbelievable that this en-zyme would release a highly reactive cation radical from its active site. The authors citednext considered different aspects of such unexpected resistivity of cation radicals to furtheroxidation.
Rogan, Cavalieri, et al. (1988) and Cavalieri, Rogan, Devanesan, et al. (1990) sug-gest that the cation radical pathway may represent a nonactive-site, “heme-edge” oxidativeprocess that is akin to that observed for reactions between peroxidases and PAHs havinglow oxidation potentials.
As already mentioned, double-helix DNA is able to stabilize PAH cation radicals. Itis also capable of stabilizing cation radicals of other xenobiotics. For instance, chloropro-mazine is also oxidized to its cation radical form by peroxidases. Double-helix DNA stabi-lizes this cation radical due to intercalation into the helix (Kelder et al. 1991). The interca-lation enhances the stability of the chloropromazine cation radical. It is suggested (Piette etal. 1964) that this inner-cell intercalation prevents further oxidation of the cation radical.
Chloropromazine and related phenazines have significant physiological activity(mostly due to their neuroleptic properties) and are used as tranquilizers. They have lowionization potentials and can act as good electron donors. For example, the cation radicalsof these compounds are easily generated in the cavity of cyclodextrins as a result of one-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

electron oxidation by iodine in aqueous solutions. Such ease of cation radical generation isconnected to the host (cyclodextrin) and guest (phenazine) association (Guo et al. 1992).
The problem of spatial specificity in the ion radical binding in biological surround-ings also deserves separate consideration. Thus, phenothiazines may act in living organ-isms as electron-transfer donors at drug receptor sites. Some mechanisms of biological ac-tivity directly involve stable cation radicals of the chloropromazine-type drugs (Gooley etal. 1969). As shown, the cation radicals of this type have a more planar structure than thecorresponding neutral molecules. Therefore, the cation radicals have increased aromaticresonance stabilization (Pan & Phillips 1999).
Being more planar than the corresponding neutral molecules, these cation radicals re-main bent some in their skeleton. The chlorine atom at the 2 position in chloropromazineappears to be in noticeable conjugation and/or in through-bond interactions with the cen-tral-ring heterocycle (Pan and co-authors 1999, 2000). This seemingly affects the donorability of the drug and its pharmacological activity. Even though chloropromazine is aboutan order of magnitude more toxic than promazine (Merville et al. 1984), chloropromazineis still used as a psychotropic drug. What is important for our consideration is that the vari-ation of the dihedral angle accompanying the transformation of neutral molecules into theircation radicals may play an important role in the pharmacological activity of the drugs.
Anion radical generation is also accompanied by conformational changes; the anionradicals of the ubiquinone family are illustrative. Ubiquinones belong to Qn-coenzymes;they are 2,3-dimethoxy-5-methyl-6-poly(prenyl)benzoquinones. The prenyl group meansan isoprene (3-methylbut-2-en-iso-yl) unit; the n-index shows the amount of these prenylunits. The Q6–Q10 coenzymes are prevalent in nature; all of them have the E-configurationof the prenyl units. The Q10-coenzyme is found in humans. The main biological function ofQn-coenzymes consists of an electron and proton transfers from different substrates to cy-tochromes during respiration (breathing) and oxidative phosphorylation. As shown, afterelectron uptake, both methoxy groups in positions 2 and 3 align in out-of-plane orientationand reside on the same side of the six-membered ring plane (Himo et al. 1999).
So the C(2) methoxy group in ubiquinone is found to orient differently in the neutraland in the anion radical molecular system. This electron-induced conformational modifi-cation may cause a modified influence from surrounding hydrogen-bonding groups to theanion radical. Proteins bind enzyme by means of hydrogen bonding. Formation of hydro-gen bonds modifies the quinone electron affinity (Eriksson et al. 1997). As a result, it mod-ifies the quinone ability for electron donation/uptake and proton uptake relative to the neu-tral system. Calvo et al. (2000) studied exchange interaction and the electron-transfer ratebetween two anion radicals of ubiquinone, considering them as participants at photosyn-thetic reaction centers in Rhodobacter sphaeroides. The protein medium surrounding thesesemiquinones appeared to be especially favorable for the electron exchange. The mediumextends the distance for which the electron transfer remains possible.
In principle, quinones can generate both cation and anion radicals being involved inredox cycles of living organisms. The question of what reactive species—cation or anionradical—is responsible for genotoxic activity has been the subject of recent investigations.For example, 3,4-estrone quinone is capable of inducing single-strand DNA breaks inMCF-7 breast cancer cells. Akanni and Abul-Hajj (1999) carried out studies on the reac-tivity of the quinone, its cation and anion radicals, with calf thymus DNA under differentpH conditions. Their results suggest that the reactive species responsible for adduct forma-tion under physiological conditions is most likely to be the 3,4-estrone quinone anion rad-ical (semiquinone).
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

3.4.3 Ion Radical Repair of Damaged DNA
Some ion radical mechanisms are implicated in DNA damage; others can be described asrepairing the damage. In normal DNA, bases belonging to the two opposite strands are tiedup by relatively weak hydrogen bonds. In the case of complementary base pairs, several hy-drogen bonds are formed; they are depicted in Scheme 3-65.
As shown in Scheme 3-65, there are two hydrogen bonds between the thymine andadenine bases. Upon photoirradiation (in sunlight, for instance), specific damage occursthrough cycloaddition between the neighboring thymine bases to create a cyclobutylthymine dimer. Common solar light quanta in the range of 250–300 nm evoke formation ofthe depicted thymine dimers (containing cyclobutane rings) in suitable regions of duplexDNA. This lesion is potentially very dangerous. Fortunately, these regions in double-stranded DNA with dangerous defects are readily recognized by a number of DNA repairenzymes. A cell possesses ways to deal with the dimers (to repair this DNA damage). Somecells simply excise the dimers from their DNA strands. Other cells use DNA photolyase en-zymes that simply break the cyclobutane structure, restoring the two adjacent, undamaged,thymine units. This requires photoexcitation (330–400 nm) of the enzyme, which is thenthought to transfer one electron to the dimer, and thus can repair the dimer by electron re-duction; see Schemes 3-65 and 3-66. The resulting anion radical then breaks open, and theelectron is returned to the enzyme. DNA strands were recently prepared containing a cy-clobutane-thymidine dimer lesion and a flavin-building block. These doubly modifiedDNA strands show light-induced self-repairing properties (Schwogler et al. 2000). Flavinacts as an electron donor to the dimer acceptor in this DNA strand.
The basis of the repair reaction, which rescues many insects, fish, amphibians, andplants from UV-induced cell death and mutagenesis, is a light-induced electron transferfrom a reduced and deprotonated flavin coenzyme to the DNA lesion. Despite the wide oc-currence of this repair process in nature, it is not clear whether it is used in the human body.This is a highly controversial area at present, with strong views on both sides (Podmore etal. 1994; de Grujil & Roza 1991). What is crucial for our consideration is the rapid break-down of the dimer following electron addition before electron return to the enzyme can oc-cur. A special study of the anion radical of the thymine dimer revealed that it gives themonomer and the monomer anion radical rapidly, even at 77 K. Splitting follows Scheme3-66 (Pezeshk et al. 1996).
An ab initio study on the structure and splitting of the uracil dimer anion radical (seeScheme 3-66 and keep R � H) gives preference to the one-step mechanism (Voityuk &Roesch 1997). Anion radical anions of the pyrimidine dimers cleave with rate constants inexcess of 106 sec�1 (Yeh & Falvey 1991). However, the cyclobutyl dimer of a quinone,dithymoquinone, also cleaves upon single-electron reduction but much more slowly thanthe pyrimidine dimers (Robbins & Falvey 1993). It is truly an unresolved issue as to whythe anion radical cleavage depicted in Scheme 3-66 is so facile. Water participation canprobably decrease the barrier of the cycloreversion on physiological conditions (Saettel &Wiest 2001).
Oxidative repair of DNA damage is also possible, since the radical cation of thethymine cyclobutane dimer is similarly unstable (Kruger & Wille 2001). Employing cat-alytic rhodium complexes attached to the DNA strands, Dandliker, Holmlin, and Barton(1997) have shown that the duplex strand can shuttle a positive charge to a damaged siteover distance of 0.16–0.26 nm. The charge is transferred to the dimer, generating the dimercation radical. This cation radical rearranges back into a normal thymine doublet. After
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

SCHEME 3-65
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

SCHEME 3-66
that, the positive charge returns to the rhodium complex. The reparative reaction occurs insunlight or at 400 nm.
Hence, the DNA duplex is a good medium for electron-transfer reactions even overlong distances (according to Abdou et al. 2001, over distances greater than 30 nm). Theconcept of DNA as “an electron-conductive wire” works in the case of guanine doubletstoo (Arkin et al. 1996). This long-range charge migration can promote a repair of a com-mon photochemical lesion in DNA. Such an opportunity to repair the lesion has opened anew and interesting approach to genetic engineering.
There is another important aspect of DNA damages. A unique feature of many can-cerous tumors is the existence of hypoxic regions, that is, regions of oxygen-poor cells(J.M. Brown 1999). Such cells are often resistant to more conventional forms of antitumortreatment, such as radiotherapy and chemotherapy (Denny & Wilson 2000). There has beenconsiderable effort to identify potential antitumor drugs that specifically target such cells.One such class of potential hypoxia-specific drugs is the benzotriazine N,N-dioxides, of
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

which a particularly promising candidate is 3-amino-1,2,4-benzotriazine-1,4-dioxide, ortirapazamine (J.M. Brown & Wang 1998). The latter causes DNA cleavage in hyopoxyc tu-mor cells. This dioxide undergoes an enzymatic one-electron reduction to form the corre-sponding anion radical. Then hydrogen abstraction from DNA takes place, eventually re-sulting in the killing of cancer cells. The ion radical hypothesis seems to be promising inthe search for new anticancer medications.
3.4.4 Cation Radical Intermediates in the Metabolism of Furan Xenobiotics
One compound of interest originating from natural sources is 3-methylfuran. This pneu-motoxic compound was identified in smog and is believed to arise from photodecomposi-tion of naturally occurring terpenoids (Saunders et al. 1974).
As mentioned earlier, cytochrome P-450–catalyzed oxidation may proceed by dis-crete, one-electron steps. When microsome suspensions containing 2- or 3-methylfuranwere incubated in the presence of semicarbazone, ene-dial products (acetylacrolein or 2-methylbutenedial) were isolated as bis(semicarbazones). The authors (Ravindranath et al.1984) have given (on the basis of solid and diverse proofs) the following sequence of trans-formations: P-450 FeVBO gives P-450 FeIVBO, which has a radical character, and furangives its cation radical. The latter is stable enough to leave the site of its formation, but itis too reactive to migrate far. Radical–radical interaction between these two products ofone-electron transfer leads to butenedial. This sequence is shown in Scheme 3-67, with un-substituted furan as a participant of the biological reaction. The final enedial is quite reac-tive and is the cause of an eventual damage in living organisms.
3.4.5 Behavior of Anion Radicals in Living Organisms
The majority of the enzyme-catalyzed reactions discussed thus far are oxidative ones. How-ever, reductive electron transfer reactions take place as well. Diaphorase, xanteneoxidase,and other enzymes can act as electron donors. Another source of an electron is the super-oxide ion. It arises after detoxification of xenobiotics, which are involved in the metabolicchain. Under the neutralizing influence of redox proteins, xenobiotics yield anion radicals.Oxygen, which is inhaled with air, strips unpaired electrons from these anion radicals andproduces the superoxide ions (Mason & Chignell 1982).
Some biologically important o-quinones can react with the superoxide ion, givingcatechol derivatives that may play a role in many diseases. For example, compounds bear-
SCHEME 3-67
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

ing a nitrocatechol moiety have been claimed to be efficient catechol-O-methyl transferaseinhibitors (Suzuki et al. 1992; Perez et al. 1992). The transferase is the first enzyme in themetabolism of catecholamine; a hyperactivity of this enzyme leads to Parkinson’s disease.Therefore, the prediction of biological activity and antioxidant properties of quinones is animportant challenge for researchers. Smertenko and co-authors (2000) have proposed aspecial index for this purpose based on differences in redox potentials between a quinoneand oxygen. This index takes into account the peak merging for electrochemical reductionsof the quinone and oxygen and, according to first estimations, works well.
Biotransformation pathways of nitroaromatic compounds are believed to originatefrom nitroreductases, which have the ability to use nitro aromatics as either one- or two-electron acceptors. One-electron acceptance by the nitro compounds results in the produc-tion of the nitro anion radical. This anion radical becomes one of the most aggressivespecies in biological systems because of its reaction on endogenous molecules and its well-known catalytic ability to transfer one electron to molecular oxygen, with formation of thesuperoxide. Thus, activation of nitrofurantoin—the medication used against urethral de-cease—proceeds via one-electron reduction of the nitro group (Yongman et al. 1982).Nimesulide [N-(4-nitro-2-phenoxyphenyl)methanesulfonamide, one of calcium ion antag-onists] forms the anion radical that is actively scavenged by endobiotics like adenine(Squella and co-authors 1999). Importantly, in this group of drugs (alongside nifedipine,nimodipine, and nicardipine) the nimesulide nitro anion radical has a weaker reactivity thananion radicals of the other representatives. This could be considered an advantage inchronic treatment, where the appearance of toxicity is more possible.
The biological oxidant P-450 can also catalyze reduction, cycling between the FeII
and FeIII forms (Guengerich & Macdonald 1993). The preponderance of oxidation reac-tions is not surprising because, in the sequence of oxygen activation steps, ferrous P-450binds O2 rapidly and tightly. Scheme 3-68 shows this feature; it should be compared toScheme 3-68:
P-450 [Fe]III � e → P-450 [Fe]II � O2 → P-450 [FeO2]II
As Guengerich and Macdonald (1993) pointed out, P-450 reduction must competi-tively involve binding of the substrate in a manner that is analogous to that of molecularoxygen. For the P-450 binding with oxygen, the oxygen-expansive force should be signif-icant. It means that the oxygen pressure must be marked. As for a substrate, it must have aredox potential sufficient to perform easy reductase-mediated reduction. Since the oxygen-expansive force is sometimes rather low in cells of the liver and other tissues, reductive re-actions of substrates do proceed in some cases. All of the reductions must be one-electronprocesses (note the sense of Scheme 3-68: Fe2�/Fe3� couple transition).
Reductive dehalogenation reactions catalyzed by P-450 have been studied exten-sively, primarily because of the interest in these compounds as anesthetics, pesticides, andpotentially toxic industrial solvents. An anesthetic named halothane gives an anion radicalthat undergoes dehalogenation according to Scheme 3-69:
CF3MCHBrCl � e → [CF3MCHBrCl]�. → CF3MCH.Cl � Br�
CF3MCH.Cl → CF2BCHCl � F
.
The primary anion radical of Scheme 3-69 produces the 1-chloro-2,2,2-trifluoroethylradical. Having been spin-trapped in vivo, this radical was detected by the ESC method(Poyer et al. 1981). Ahr et al. (1982) has presented additional evidence for the formation ofthe radical as an intermediate in halothane metabolism and identified 2-chloro-1,1-difluo-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

roethene as a product of radical stabilization. Metabolytic transformations of 2-chloro-1,1-difluoroethene lead to acyl halides, which are relevant to halothane biotoxicity (Guengerich& Macdonald 1993).
The metabolism of carbon tetrachloride (a chemical solvent that was formerly incommon use) attracts attention as well. Its bioactivation appears to involve consecutiveone-electron reduction and the formation of chloride ion and the trichloromethyl radical.The latter radical then reacts with oxygen, giving rise to an oxygenated radical and, even-tually, to highly toxic phosgene (Mico & Pohl 1983). Scheme 3-70 (below) describes thesereactions:
CCl4 � e → [CCl4]�. → .CCl3 � Cl�
.CCl3 � O2 → CCl3OO
. → → COCl2
Probably, the spontaneous degradation of methyl bromide in the oceans reported re-cently (Butler 1997) has a similar mechanism. Both nature and humans contribute to theworld production of methyl bromide. This compound is thought to be responsible for a sig-nificant fraction of the ozone-destroying bromine that reaches the stratosphere. As a resultof the chemical degradation in the oceans, the danger of methyl bromide in the higher lay-ers of the atmosphere becomes less significant. Research from the National Oceanic andAtmospheric Administration (USA) reported that previously unrecognized biologicaldegradation of atmospheric methyl bromide is comparable with its oxidation with ozoneparticipation. This statement appears to be important from the point of view of the propo-sition of the European Commission, part of the European Union, to phase out the manu-facture and use of methyl bromide by year 2001. Up to now, several other chemicals andmethods have been tested in the search for replacements for methyl bromide, but most costmore, require longer treatment periods, or are effective on fewer pests. Methyl bromide isstill used widely to limit the spread of damaging insect populations around the world.
For example, the lack of good alternatives pushed the U.S. Department of Agricul-ture in 1998 to recommend use of methyl bromide to treat wood packaging materials fromChina. A voracious, non-native insect pest had been found in such packaging materials in14 states around the United States. The Asian beetle had no known U.S. predators and couldcost the United States more than $41 billion in lost forest product, commercial fruit, maplesyrup, nursery, and tourist industries. This beetle was an extremely serious insect, andmethyl bromide was the only known effective insecticide. Heat-treatment was also sug-gested, but it appeared to be more difficult and expensive (Morse 1998). Therefore, methylbromide as an insecticide may be in for reprieve.
3.5 CONCLUSION
This chapter set out approaches for binding the electron structure of organic ion radicalswith their reactivity. Of course, the amount of structural data is much greater than the dataon reactivity. It is commonplace in this field that structural (instrumental) data are accu-mulated more rapidly than data on reactivity from chemical experiments.
The systematization provided here is useful for the future development of ion radicalorganic chemistry. (Systematization is fruitful for every field of science). It is worthwhileto underline several aspects of the considered materials that can enhance their possible ap-plications.
Research on spin-charge separation in organic ion radicals has been carried out in re-cent years with an emphasis on theoretical calculations. Experiments were performed to
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

prove their existence and to observe their behavior in a mass spectrometer chamber. Thenext step is likely to emphasize the synthesis of distonic ion radical salts, that are stable un-der common conditions. Applications of the salts will be possible in creating magnetic,conductible media and other materials that possess useful properties. The attractivestrength of distonic ion radicals is that they can enter ionic reactions at the charged site andradical reactions at the radical site. Success in this direction can open a new window interms of organic reactivity.
Understanding ion radical transformations in living organisms is very important too.The problem is intriguing in general. Our aim was to create some incentives for overcom-ing ecological obstacles and designing new and effective drugs.
REFERENCES
Abdou, I.M.; Sartor, V.; Cao, H.; Schuster, G.B. (2001) J. Am. Chem. Soc. 123, 6696.Abelt, Ch.J.; Roth, H.D. (1985) J. Am. Chem. Soc. 107, 6814.Ahr, H.J.; King, L.J.; Nastaiczyk, W.; Ulrich, V. (1982) Biochem. Pharmacol. 31, 391.Akanni, A.; Abul-Hajj, Yu.J. (1999) Chem. Res. Toxicol. 12, 1247.Alder, R.W. (1983) Acc. Chem. Res. 16, 321.Alder, R.W.; Sessions, R.B. (1979) J. Am. Chem. Soc. 101, 3651.Alekseeva, O.O.; Ryzhalov, A.V.; Rodina, L.L. (1999) Zh. Org. Khim. 35, 1873.Allen, A.D.; Tidwell, Th.T. (2001) Chem. Rev. 101, 1333.Anderson, N.H.; Dobbs, A.J.; Edge, A.J.; Norman, R.O.C.; West, P.R. (1971) J. Chem. Soc. B, 1004.Anklam, E.; Mohan, H.; Asmus, K.-D. (1988) J. Chem. Soc. Perkin Trans. 2, 1297.Apperloo, J.J.; Janssen. R.A.J.; Malenfant, P.R.L.; Groenendaal, L.; Frechet, J.M.J. (2000a) J. Am.
Chem. Soc. 122, 7042.Apperloo, J.J.; Langeveld-Voss, B.M.W.; Knol, J.; Hummelen, J.C.; Janssen, R.A.J. (2000b) Adv.
Mater. 12, 908.Arkin, M.R.; Stemp, E.D.A.; Holmlin, R.E.; Barton, J.K.; Hormann, A.; Olson, E.J.C.; Barbara, P.F.
(1996) Science 273, 475.Asmus, K.-D. (1979) Acc. Chem. Res. 12, 436.Asmus, K.-D. (1990). In: Chatgilialoglu, C.; Asmus, K.-D., eds. Sulfur-Centered Reactive Interme-
diates in Chemistry and Biology. Plenum Press, New York.Asmus, K.-D. (2000) Nukleonika 45, 3.Baird, C.N. (1977) J. Chem. Ed. 54, 291.Ballester, M.; Pascual, I.; Riera, J.; Castaner, J. (1991) J. Org. Chem. 56, 217.Bassindale, A.R.; Iley, J (1990). In: Patai, S., ed. The Chemistry of Sulphenic Acids and Their Deriva-
tives, Chap. 4. Wiley, New York.Bauld, N.L. (1965) J. Am. Chem. Soc. 87, 4788.Bauld, N.L.; Yang, J. (1999a) Org. Lett. 1, 773.Bauld, N.L.; Yang, J. (1999b) Tetrahedron Lett. 40, 8519.Bauld, N.L.; Yang, J.; Gao, D. (2000) J. Chem. Soc., Perkin Trans. 2, 207.Baumgarten, M.; Mueller, U.; Bohnen, A.; Muellen, K. (1992) Angew. Chem., Int. Ed. Engl. 31, 448.Baumgarten, M.; Anton, U.; Gherghel, L.; Muellen, K. (1993) Synth. Met. 57, 4807.Baumgarten, M.; Gherghel, L.; Karabunarliev, S. (1995) Synth. Metals 69, 633.Beard, M.C.; Turner, G.M.; Schmuttenmaer, C.A. (2000) J. Am. Chem. Soc. 122, 11541.Bednarek, P.; Zhu, Z.; Bally, Th.; Filipiak, T.; Marcinek, A.; Gebicki, J. (2001) J. Am. Chem. Soc.
123, 2377.Bigler, L.; Hesse, M. (1995) J. Am. Soc. Mass Specrtom. 6, 634.Binder, H.; Riegel, B.; Heckmann, G.; Moscherosch, M.; Kaim, W.; Schnering von, H.-G.; Hoenle,
W.; Flad, H.-Ju.; Savin, A. (1996) Inorg. Chem. 35, 2119.Bloodworth, A.J.; Davies, A.G.; Hay-Motherwell, R.S. (1988) J. Chem. Soc. Chem. Commun, 862.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Boche, G. (1992) Angew. Chem. Int. Ed. Engl. 31, 731.Bock, H.; Haenel, P.; Hermann, H.-F. (1990) Z. Naturforsch. B: Chem. Sci. 45, 1197.Bolze, R.; Eierdanz, H.; Schlueter, K.; Massa, W.; Grahn, W.; Berndt, A. (1982) Angew. Chem. Int.
Ed. Engl. 21, 924.Bonvoisin, J.; Launay, J.-P.; Rovira, C.; Veciana, J. (1994) Angew. Chem. Int. Ed. Engl. 33, 2106.Breitzer, J.G.; Smirnov, A.I.; Szczepura, L.F.; Wilson, S.R.; Rauchfuss, Th.B. (2001) Inorg. Chem.
40, 1421.Brown, J.M. (1999) Cancer Res. 59, 5863.Brown, J.M.; Wang, L.H. (1998) Anti-Cancer Drug Des. 13, 529.Brown, T.; Hirschon, A.; Musker, W.K. (1981) J. Phys. Chem. 85, 3767.Bushby, R.J.; Ng, K.M. (1996) J. Chem. Soc., Perkin Trans. 2, 1053.Butler, J.H. (1997) Geophys. Res. Lett. 24, 171 and 1227.Calderbrank, K.E.; Le Fevre, R.J.W.; Ritchie, G.L.D. (1968) J. Chem. Soc. B, 503.Calle, P.; Martin, J; Garcia de la Vega, J.M.; Sieiro, C. (1992) J. Mol. Struct. 269, 163.Calvo, R.; Abresch, E.C.; Bittl, R.; Feher, G.; Hofbauer, W.; Isaacson, R.A.; Lubitz, W.; Okamura,
M.Y.; Paddock, M.L. (2000) J. Am. Chem. Soc. 122, 7327.Camaggi, C.M.; Perkins, M.J.; Ward, P. (1971) J. Chem. Soc. B, 2416.Carmichael, I. (1997) Acta Chem. Scand. 51, 567.Casado, F.; Pisano, L.; Farriol, M.; Gallardo, I.; Marquet, J.; Melloni, G. (2000) J. Org. Chem. 65,
322.Cavalieri, E.L.; Devanesan, P.; Rogan, E. (1988) Biochem. Pharmacol. 37, 2183.Cavalieri, E.L.; Rogan, E.G. (1992) Pharmacol. Ther. 55, 183.Cavalieri, E.L.; Rogan, E.G. (1995) Xenobiotica 25, 677.Cavalieri, E.L.; Rogan, E.G.; Cremonesi, P.; Devanesan, P.D. (1988) Biochem. Pharmacol. 37, 2173.Cavalieri, E.L.; Rogan, E.G.; Devanesan, P.D.; Cremonesi, P.; Cerny, R.L.; Gross, M.L.; Bodell,
W.J. (1990) Biochem. 29, 4820.Chaha, R. (1986) Indian J. Chem. A 25, 7.Check, G.T.; Nelson, R.F. (1978) J. Org. Chem. 43, 1230.Choua, S.; Jouati, A.; Geoffroy, M. (1999) Phys. Chem. Chem. Phys. 1, 3557.Ciureanu, M.; Hillebrand, M.; Volanschi, E.; Ghetu, D. (1987) Rev. Roum. Chim. 32, 467.Clark, T. (1988) J. Am. Chem. Soc. 110, 1672.Clark, T.; Nelsen, S.F. (1988) J. Am. Chem. Soc. 110, 868.Cremonesi, P.; Stack, D.E.; Rogan, E.G.; Cavalieri, E.L. (1994) J. Org. Chem. 59, 7683.Dandliker, P.J.; Holmlin, R.E.; Barton, J.K. (1997) Science 275, 1465.De Wall, S.L.; Meadows, E.S.; Babour, L.J.; Gokel, G.W. (1999) J. Am. Chem. Soc. 121, 5613.Denny, W.A.; Wilson, W.R. (2000) Expert Opin. Invest. Drug. 9, 2889.Dewar, M.J.S.; Trinajstic, N. (1969) J. Chem. Soc. A, 1754.Dougherty, D. (1996) Science 271, 1163.Drewello, Th.; Lebrilla, C.B.; Asmus, K.-D.; Schwarz, H. (1989) Angew. Chem. Int. Ed. Engl. 28,
1275.Durr, H.; Gleiter, R. (1978) Angew. Chem. Int. Ed. Engl. 17, 559.Dyusengaliev, K.I.; Borisov, Yu.A.; Todres, Z.V. (1981) Izv. AN SSSR Ser. Khim., 1159.Eisenstein, O.; Mazaleyrat, J.-P.; Tordeux, M.; Welvart, Z. (1977) J. Am. Chem. Soc. 99, 2230.Elbl-Weiser, K.; Neugebauer, F.A.; Staab, H.A. (1989) Tetrahedron Lett. 30, 6161.Elsevier, C.J.; Vermeer, P.; Gedanken, A.; Runge, W. (1985) J. Am. Chem. Soc. 107, 2537.Elson, C.M.; Liu, M.T.H.; Mailer, C. (1986) J. Chem. Soc., Chem. Commun., 504.Eriksson, L.A.; Himo, F.; Siegbahn, P.E.M.; Babcock, G.T. (1997) J. Phys. Chem. A 101, 9496.Exner, K.; Hunkler, D.; Gescheidt, G.; Prinzbach, H. (1998) Angew. Chem. Int. Ed. Engl. 37, 1910.Exner, K.; Prinzbach, H.; Geschedt, G.; Grossmann, B.; Heinze, Ju. (1999) J. Am. Chem. Soc. 121,
1964.Exner, K.; Cullman, O.; Voegte, M.; Prinzbach, H.; Grossmann, B.; Heinze, Ju.; Liesum, L.; Bach-
mann, R.; Schweiger, A.; Gescheidt, G. (2000) J. Am. Chem. Soc. 122, 10650.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Fiedler, J.; Zalis, S.; Klein, A.; Hornung, F.M.; Kaim, W. (1996) Inorg. Chem. 35, 3039.Flammang, R.; Barbieux-Flammang, M.; Gualano, E.; Gerbaux, P.; Le, H.Th.; Nguyen, M.Th.; Ture-
cek, F.; Vivekananda, Sh. (2001) J. Phys. Chem. A, 105, 8579.Fukui, K.; Yonezawa, T.; Shingu, H. (1952) J. Chem. Phys. 20, 722.Fuoss, R.M.; Sadek, H. (1954) J. Am. Chem. Soc. 76, 5897 and 5905.Gano, J.E.; Jacob, E.J.; Sekher, P.; Subramaniam, G.; Eriksson, L.A.; Lenoir, D. (1996) J. Org.
Chem. 61, 6739.Gano, J.E.; Park, B.S.; Pinkerton, A.A.; Lenoir, D. (1991) Acta Crystallogr. Sect. C.: Cryst. Struct.
Commun. 47, 162.Gauld, J.W.; Radom, L. (1997) J. Am. Chem. Soc. 119, 9831.Geacintov, N.E.; Cosman, M.; Hingerty, B.E. (1997) Chem. Res. Toxicol., 10, 111.Gebicki, J.; Marcinek, A.; Stradowski, C. (1990) J. Phys. Org. Chem. 3, 606.Geletii, Yu. V.; Lavrushko, V.V.; Shilov, A.E. (1986) Dokl. AN SSSR 288, 139.Gerson, F.; Jachimowicz, J.; Leaver, D. (1973) J. Am. Chem. Soc. 95, 6702.Gerson, F.; Lamprecht, A.; Scholtz, M.; Troxler, H.; Lenoir, H. (1996) Helv. Chim. Acta 79, 307.Gerson, F.; Hopf, H.; Merstetter, P.; Mlynek, C.; Fischer, D. (1998) J. Am. Chem. Soc. 120, 4815.Ghirvu, C.I.; Gropen, O; Skancke, P.N. (1971) Acta Chem. Scand. 25, 2023.Gill, P.M.W.; Radom, L. (1988) J. Am. Chem. Soc. 110, 4931.Glass, R.S. (1995) Xenobiotica 25, 637.Glass, R.S. (1999) Top. Curr. Chem. 205 (Organosulfur Chemistry II), 1.Gleiter, R.; Roeskel, H.; Neugebauer, F.A. (1996) J. Org. Chem. 61, 1739.Goez, M.; Rozwadowski, J. (2000) J. Inf. Rec. 25, 301.Gol’teuzen, E.E.; Todres, Z.V.; Kaminskii, A.Ya.; Gitis, S.S.; Kursanov, D.N. (1972) Izv. AN SSSR
Ser. Khim., 1083.Golubeff, H. (1873) Ber. Dtsch. Chem. Ges. 6, 1252.Gooley, C.M.; Keyzer, H.; Setchel, F. (1969) Nature 223, 80.Goslish, R.; Weiss, J.; Moeckel, H.J.; Moenig, J.; Asmus, K.-D. (1985) Angew. Chem. Int. Ed. Engl.
24, 73.Gregorius, H.; Baumgarten, M.; Reuter, R.; Tyutyulkov, N.; Muellen, K. (1992) Angew. Chem. Int.
Ed. Engl. 31, 1653.Grigoriev, V.A.; Cheng, D.; Hill, C.L.; Weinstock, I.A. (2001) J. Am. Chem. Soc. 123, 5292.Gronert, S. (2001) Chem. Rev. 101, 329.Grujil, de, F.R.; Roza, L. (1991) J. Photochem. Photobiol. B: Biol. 10, 367.Grunwald, E. (1954) Anal. Chem. 26, 1696.Guengerich, F.P.; Macdonald, T.L. (1993). In: Mariano, P., ed. Advances in Electron Transfer Chem-
istry, Vol. 3. JAI Press.Guo, Q.; Huan, P.; Liu, B.; Liu, Y. (1992) Chin. Chem. Lett. 3, 53.Hacker, N.P.; Kasai, P.H. (1993) J. Am. Chem. Soc. 115, 5410.Hall, M.; Grover, P.L. (1990) In: Cooper, C.S.; Grover, P.L., eds. Chemical Carcinogenesis and Mu-
tagenesis. I. Springer-Verlag, Berlin.Hammerum, S. (1988) Mass Spectrom. Rev. 7, 123.Hasselbach, E. (1970) Chem. Phys. Lett., 428.Hayon, E.; Simic, M. (1973) J. Am. Chem. Soc. 95, 2433.Herold, B.J.; Neiva Correia, A.F.; Santos Veiga dos, J. (1965) J. Am. Chem. Soc. 87, 26661.Himo, F.; Babcock, G.T.; Eriksson, L.A. (1999) J. Phys. Chem. A 103, 3745.Hoefelmeyer, J.; Gabbai, F.P. (2000) J. Am. Chem. Soc. 122, 9054.Il’yasov, A.V.; Kargin, Yu.M.; Kondranina, V.Z. (1971) Izv. AN SSSR Ser. Khim., 927.Il’yasov, A.V.; Kargin, Yu.M.; Sotnikova, N.N.; Kondranina, V.Z.; Mel’nikov, B.V.; Vafina, A.A.
(1971) Izv. AN SSSR Ser. Khim., 932.Ito, A.; Taniguchi, A.; Yamabe, T.; Tanaka, K. (1999) Org. Lett. 1, 741.James, M.A.; McKee, M.L.; Illies, A.J. (1996) J. Am. Chem. Soc. 118, 7836.Kaim, W. (1980) Angew. Chem. Int. Ed. Engl. 19, 911.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Kamphius, J.; Bos, H.J.; Worell, C.W.; Runge, W. (1986) J. Chem. Soc. Perkin Trans. 2, 1509.Kasai, P.H. (1991) J. Am. Chem. Soc. 113, 3317.Kelder, P.P.; Fisher, M.J.E.; Mol de, N.J.; Janssen, L.H.M. (1991) Arch. Biochem. Biophys. 284, 313.Kenttaemaa, H.I. (1994) Org. Mass Spectrom. 29, 1.Kise, N.; Iwasaki, K.; Tokieda, N.; Ueda, N. (2001) Org. Lett. 3, 3241.Koeppe, R.; Kasai, P.H. (1994) J. Phys. Chem. 98, 12904.Kost, D.; Raban, M. (1990). In: Patai, S., ed. The Chemistry of Sulphenic Acids and Their Deriva-
tives, Chap. 2. Wiley, New York.Kruger, O.; Wille, U. (2001) Org. Lett. 3, 1455.Kumpf, R.A.; Dougherty, D.A. (1993) Science 261, 1708.Kurbatova, A.S.; Kurbatov, Yu.V.; Niyazova, D.A. (1980) Zh. Org. Khim. 16, 648.Kurihara, T.; Watanabe, T.; Yoshikawa, K.; Motohashi, N. (1998) Anticancer Res. 18, 429.Lazana, M.C.R.L.R.; Franco, M.L.T.M.B.; Herold, B.J. (1993) J. Chem. Soc. Faraday Trans. 89,
1327.Levy, D.H.; Myers, R.J. (1964) J. Chem. Phys. 41, 1062.Levy, D.H.; Myers, R.J. (1966) J. Chem. Phys. 44, 4177.Ling, C.-Y.; Gendell, J. (1967) J. Chem. Phys. 47, 3475.Luckhurst, C.R.; Orgel, L.E. (1963) Mol. Phys. 7, 297.Maki, A.; Geske, D. (1960) J. Chem. Phys. 33, 825.Mann, Ch.K.; Barnes, K.K. (1970). Electrochemical Reactions in Nonaqueous Systems. Marcell
Dekker, New York.Marcinek, A.; Gebicki, J.; Plonka, A. (1990) J. Phys. Org. Chem. 3, 757.Marcus, Y. (1985) “Ion Solvation”. Wiley, Chichester, UK.Maruyama, K.; Otsuki, T. (1968) Bull. Chem. Soc. Jpn. 41, 444.Maslak, P.; Augustine, M.P.; Burkey, J.D. (1990) J. Am. Chem. Soc. 112, 5359.Mason, R.P.; Chignell, C.F. (1982) Pharmacolog. Rev. 33, 189.Matsumoto, K.; Katsura, H.; Yamaguchi, J.; Uchida, T.; Aoyama, K.; Machiguchi, T. (1996) Bull.
Soc. Chim. Fr. 133, 891.Meier, H. (1992) Angew. Chem. Int. Ed. Engl. 31, 1399.Merville, M.P.; Pitte, J.; Lopez, M.; Decuyper, J.; Van de Vorst, A. (1984) Photochem. Photobiol.
39, 57.Mico, B.A.; Pohl, L.R. (1983) Arch. Biochem. Biophys. 225, 596.Moraes, L.A.B.; Eberlin, M.N. (1998) J. Am. Chem. Soc. 120, 11136.Morkovnik, A.S.; Okhlobystin, O.Yu. (1979) Khim. Gheterots. Soed., 128.Morse, P. (1998) Chem. Eng. News 76, 9.Muller, R.; Heinze, Ju. (1998a) Phosphorus, Sulfur, Silicon, Relat. Elem. 136, 137, & 138, 557.Muller, R.; Heinze, Ju. (1998b) Phosphorus, Sulfur, Silicon, Relat. Elem. 141, 111.Murata, Y.; Shine, H.J. (1969) J. Org. Chem. 34, 3368.Musker, W.K. (1980) Acc. Chem. Res. 13, 200.Myagi, M. (1971) Izv. AN Est. SSR, Khim. Geol. 20, 364.Nagase, S.; Fueno, T.; Morokuma, K. (1979) J. Am. Chem. Soc. 101, 5849.Nakajima, T.; Pullman, B. (1959) J. Am. Chem. Soc. 81, 3876.Nash, J.J.; Squires, R.R. (1996) J. Am. Chem. Soc. 118, 11872.Nelsen, S.F. (1967) Tetrahedron Lett., 3795.Nelsen, S.F. (1987) Accounts Chem. Res. 20, 269.Nelsen, S.F. (1997) In: Patai, S., ed. The Chemistry of the Hydrazo, Azo and Azoxy Groups. Suppl. 2,
Chap. 6. Wiley, New York.Nelsen, S.F.; Alder, R.W.; Sessions, R.B.; Asmus, K.-D.; Hiller, K.-O.; Goebl, M. (1980) J. Am.
Chem. Soc. 102, 1429.Nelsen, S.F.; Hintz, P.J.; Buschek, J.M.; Weisman, G.R. (1975) J. Amer. Chem. Soc. 97, 4933.Nelsen, S.F.; Hintz, P.J.; Buschek, J.M.; Weisman, G.R. (1976) J. Am. Chem. Soc. 98, 3281.Nelsen, S.F.; Ismagilov, R.F. (1999) J. Phys. Chem. A 103, 5373.Nelsen, S.F.; Ismagilov, R.F.; Powell, D.R. (1996) J. Am. Chem. Soc. 118, 6313.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Nelsen, S.F.; Ismagilov, R.F.; Powell, D.R. (1997) J. Am. Chem. Soc. 119, 10213.Nelsen, S.F.; Ismagilov, R.F.; Trieber, D.A. (1997) Science 278, 846.Nelsen, S.F.; Rumack, D.T.; Meot-Ner (Mautner), M. (1988) J. Am. Chem. Soc. 110, 7945.Nelsen, S.F.; Tran, H.Q.; Nagy, M.A. (1998) J. Am. Chem. Soc. 120, 298.Nelsen, S.F.; Trieber, D.A.; Ismagilov, R.F.; Teki, Y. (2001) J. Am. Chem. Soc. 123, 5684.Nicholas, J.B.; Hay, B.P.; Dixon, D.A. (1999) J. Phys. Chem. A 103, 1394.Nielsen, M.L.; Budnik, B.A.; Haselman, K.F.; Olsen, J.V.; Zubarev, R.A. (2000) Chem. Phys. Lett.
330, 558.Ohya-Nishiguchi, H.; Shimizu, Y.; Hirota, N.; Watanabe, K. (1980) Bull. Chem. Soc. Jpn. 53, 1252.Pan, D.; Phillips, D.L. (1999) J. Phys. Chem. A 103, 4737.Pan, D.; Shoute, L.C.T.; Phillips, D.L. (1999) J. Phys. Chem. A 103, 6851.Pan, D.; Shoute, L.C.T.; Phillips, D.L. (2000) J. Phys. Chem. A 104, 4140.Pauling, L. (1931) J. Am. Chem. Soc. 53, 3225.Perez, R.A.; Ferrandez-Alvarez, E.; Nieto, O.; Piedrafita, F.J. (1992) J. Med. Chem. 35, 4584.Pezeshk, A.; Podmore, I.D.; Heelis, P.F.; Symons, M.C.R. (1996) J. Phys. Chem. 100, 19714.Pfeifer, P.; Braude, S.; Keller, J.; Marcon, G.; Wittcop, P. (1915) Ber. Dtsch. Chem. Ges. 48, 1777.Phelps, J.; Santhanam, K.S.V.; Bard, A.J. (1967) J. Am. Chem. Soc. 89, 1752.Piette, L.H.; Bulow, G.; Yamazaki, I. (1964) Biochem. Biophys. Acta 88, 120.Podmore, I.D.; Heelis, P.F.; Symons, M.C.R.; Pezeshk, A. (1994) Chem. Commun., 1005.Polce, M.J.; Wesdemiotis, Ch. (1996) Rapid Commun. Mass Spectrom. 10, 235.Poyer, J.L.; McCay, P.B.; Weddle, C.C.; Downs, P.E. (1981) Biochem. Pharmacol. 30, 1517.Radom, L.; Yates, B.F.; Bouma, W.J. (1986) Tetrahedron 42, 6225.Rajca, A.; Rajca, S.; Desai, S.R. (1995) J. Chem. Soc., Chem. Commun., 1957.Rathore, R.; Le Magueres, P.; Linderman, S.V.; Kochi, J.K. (2000) Angew. Chem. Int. Ed. 39, 809.Ravindranath, V.; Burka, L.T.; Boyd, M.R. (1984) Science 224, 884.Reed, D.R.; Hare, M.; Kaas, S.R. (2000) J. Am. Chem. Soc. 122, 10689.Rhodes, Ch.J. (1988) J. Chem. Soc. Chem. Commun., 1477.Ristango, C.V.; Shine, H.J. (1971) J. Org. Chem. 36, 4050.Robbins, R.J.; Falvey, D.E. (1993) J. Org. Chem. 58, 3616.Rodriguez-Santiago, L.; Sodupe, M.; Oliva, A.; Bertran, J. (2000) J. Phys. Chem. A 104, 1256.Rogan, E.G.; Cavalieri, E.L.; Tibbels, S.R.; Cremonesi, P.; Warner, C.D.; Nagel, D.L.; Tomer, K.B.;
Cerny, R.L.; Gross, M.L. (1988) J. Am. Chem. Soc. 110, 4023.Rogan, E.; Katomski, P.A.; Roth, R.; Cavalieri, E. (1979) J. Biol. Chem. 254, 7055.Roth, H.D.; Schilling, M.L.M.; Abelt, Ch.J. (1986) J. Am. Chem. Soc. 108, 6098.Rovira, C.; Ruiz-Molina, D.; Elsner, O.; Vidal-Gancedo, J.; Bonvoisin, J.; Launay, J.-P.; Veciana, J.
(2001) Chem. Eur. J. 7, 240.Ryan, M.D.; Swanson, D.D.; Glass, R.S.; Wilson, G.S. (1981) J. Phys. Chem. 85, 1069.Saettel, N.J.; Wiest, O. (2001) J. Am. Chem. Soc. 123, 2693.Saunders, R.; Griffith, J.; Saalfeld, F. (1974) Biomed. Mass Spectrom. 1, 192.Schenk, R.; Gregorius, H.; Meerholz, K.; Heinze, J.; Muellen, K. (1991) J. Am. Chem. Soc. 113, 2634.Schoeneich, Ch.; Aced, A.; Asmus, K.-D. (1993) J. Am. Chem. Soc. 115, 11376.Schoeneich, Ch.; Pogocki, D.; Wisnowski, P; Hug, G.L.; Bobrowski, K. (2000) J. Am. Chem. Soc.
122, 10224.Schwogler, A.; Burgdorf, L.T.; Carell, Th. (2000) Angew. Chem., Int. Ed. 39, 3918.Shah, Sh.; Burdette, Sh.C.; Swavey, Sh.; Urbach, F.L.; Protasiewicz, J.D. (1997) Organometallics
16, 3395.Smertenko, E.A.; Staninets, V.I.; Datsenko, S.D.; Ignat’ev, N.V. (2000) Teor. Eksper. Khim. 36, 37.Somekawa, K.; Haddaway, K.; Mariano, P.S.; Tossell, J.A. (1984) J. Am. Chem. Soc. 106, 3060.Squella, J.A.; Gonzalez, P.; Bollo, S.; Nunez-Vergara, L.J. (1999) Pharmaceutic. Res. 16, 161.Stack, D.E.; Cremonesi, P.; Hanson, A.; Rogan, E.G.; Cavalieri, E.L. (1995) Xenobiotica 25, 755.Staedel, W. (1883) Ann. 218, 344.Stagko, A.; Erentova, K.; Rapta, P.; Nuyken, O.; Voit, B. (1998) Magn. Reson. Chem. 36, 13.Stowasser, R.; Glass, R.S.; Hoffmann, R. (1999) J. Chem. Soc. Perkin Trans. 2, 1559.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Sundaresan, T.; Wallwork, S.C. (1972) Acta Crystall. B28, 3507.Sunner, J.; Nishizawa, K.; Kebarle, P. (1981) J. Phys. Chem. 85, 1814.Suzuki, Y.J.; Tsuchiya, M.; Safadi, A.; Kagan, V.E.; Packer, L. (1992) Free Radical Biol. Med. 13,
517.Takahashi, K.; Elving, P. (1967) Electrochim. Acta 12, 213.Takemura, Y.; Shida, T. (1980) J. Chem. Phys. 73, 4133.Tamaoki, M.; Serita, M.; Shiratori, Y.; Itoh, K. (1989) J. Phys. Chem. 93, 6052.Tanko, J.M.; Phillips, J.P. (1999) J. Am. Chem. Soc. 121, 6078.Tanko, J.M.; Brammer, L.E.; Hervas, M.; Campos, K. (1994) J. Chem. Soc. Perkin Trans. 2, 1407.Terabe, S.; Konaka, R. (1971) J. Am. Chem. Soc. 93, 4306.Terabe, S.; Kumura, K.; Konaka, R. (1973) J. Chem. Soc. Perkin Trans. 2, 1252.Testaferi, L.; Tiecco, M.; Tingoli, M. (1982) Tetrahedron 38, 2721.Thiele, J. (1899) Ann. Chem. 306, 142.Todres, Z.V. (1970) Izv. AN SSSR Ser. Khim., 1749.Todres, Z.V. (1990) Acta Chem. Scand. 44, 535.Todres, Z.V. (1992) J. Organomet. Chem. 441, 349.Todres, Z.V.; Avagyan, S.P. (1972) Int. J. Sulphur Chem. 8, 373.Todres, Z.V.; Avagyan, S.P. (1978) Phosph. Sulfur 4, 223.Todres, Z.V.; Bespalov, V.Ya. (1972) Zh. Org. Khim. 8, 19.Todres, Z.V.; Ermekov, D.S. (1989) J. Organomet. Chem. 369, 371.Todres, Z.V.; Hovsepyan, G.T.; Ionina, E.A. (1988) Tetrahedron 44, 5199.Todres, Z.V.; Hovsepyan, G.T.; Ionina, E.A.; Kosnikov, A.Y.; Lindeman, S.V.; Struchkov, Y.T.
(1988) Izv. AN SSSR Ser. Khim., 1845.Todres, Z.V.; Kuryaeva, T.T.; Ryzhova, G.L.; Kursanov, D.N. (1980) Izv. AN SSSR Ser. Khim., 1602.Todres, Z.V.; Pozdeeva, A.A.; Chernova, V.A.; Zhdanov, S.I. (1972) Izv. AN SSSR Ser. Khim., 2454.Todres, Z.V.; Safronov, A.I.; Ermekov, D.S.; Minyaev, R.M. (1992) J. Organomet. Chem. 441, 479.Todres, Z.V.; Tsvetkova, T.M. (1987) Izv. AN SSSR Ser. Khim., 1553.Tsenov, J.A.; Binev, I.G.; Juchnovski, I.N. (1998) Dokl. Bulg. Akad. Nauk 51, 57.Tukada, H. (1994) J. Chem. Soc. Chem. Commun., 2293.Turaeva, D.A.; Kurbatova, A.S.; Kurbatov, Yu.V. (1993) Zh. Org. Khim. 29, 1588.Voityuk, A.A.; Roesch, N. (1997) J. Phys. Chem. A 101, 8335.Waldek, D.H. (1991) Chem. Rev. 91, 415.Ward, R.L. (1961) J. Am. Chem. Soc. 83, 1296.Weissmann, S.T.; Townsend, J.; Paul, D.E.; Pack, G.E. (1953) J. Chem. Phys. 21, 2227.Wenthold, P.G.; Squires, R.R. (1998) Int. J. Mass Spectrom. Ion Processes 175, 215.Wenthold, P.G.; Hu, J.; Squires, R.R. (1994) J. Am. Chem. Soc. 116, 6961.Wenthold, P.G.; Hu, J.; Squires, R.R. (1996) J. Am. Chem. Soc. 118, 11865.Werst, D.W.; Trifunac, A.D. (1991) J. Phys. Chem. 95, 3466.Williams, F.; Guo, Q.-X.; Petillo, P.A.; Nelsen, S.F. (1988) J. Am. Chem. Soc. 110, 7887.Wilson, G.S.; Swanson, D.D.; Klug, J.T.; Glass, R.S.; Ryan, M.D.; Musker, W.K. (1979) J. Am.
Chem. Soc. 101, 1040.Winstein, S.; Clippinger, E.; Fainberg, A.H.; Robinson, G.C. (1954) J. Am. Chem. Soc. 76, 2597.Yao, T.; Musha, S. (1974) Bull. Chem. Soc. Jpn. 47, 2650.Yates, B.F.; Bouma, W.J.; Radom, L. (1984) J. Am. Chem. Soc. 106, 5805.Yeh, S.-R.; Bard, A.J. (1977) J. Electrochem. Soc. 124, 189.Yeh, S.-R.; Falvey, D.E. (1991) J. Am. Chem. Soc. 113, 8557.Yongman, R.J.; Osswald, W.F.; Elstner, E.F. (1982) Biochem. Pharmacol. 31, 3723.Zhao, J.; Dowd, P.; Grabowski, J.J. (1996) J. Am. Chem. Soc. 118, 8871.Zverev, V.V.; Vasin, V.A. (1998) Russ. J. Gen. Chem. 68, 1828.Zverev, V.V.; Vasin, V.A. (2000) Russ. J. Gen. Chem. 70, 921.Zverev, V.V.; Musin, B.M.; Yanilkin, V.V. (1997) Russ. J. Gen. Chem. 67, 1254.Zwier, J.; Hoeth, J.W.; Brouwer, A.M. (2001) J. Org. Chem. 66, 466.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

4
How to Discern Ion RadicalMechanisms of Organic Reactions
4.1 INTRODUCTION
A reaction mechanism is a sum of the perceived elementary stages taken in their sequenceand relative rate. A knowledge of the properties of the intermediate particles and a study ofthe mechanism of the reactions open up possibilities of increasing the rate of formation andthe yield of the desired final product. Transformations, including the formation of ion rad-icals in the intermediate step, require special approaches to optimize them. In other words,it is necessary to determine whether the reaction proceeds through the formation of ion rad-icals and, if so, which radicals originate and at what stages. It is essential to learn whetherthese stages belong to the main or side routes of the reactions.
This chapter is devoted to just such problems. The chapter describes how one can re-veal ion radical conversions by means of physical methods and kinetic approaches and onpurely chemical information, including data on material balance and the nature of the endproducts. These methods will be woven together to form a cohesive unit. All the methodswill be considered separately, and then complex approaches will be described in their ap-plications to several representative reactions.
4.2 WHY DOES A REACTION CHOOSE THE ION RADICAL MECHANISM?
The functional groups in a molecule determine, to a considerable extent, whether its ionradical form can be the leading particle in the course of the reaction. The effect of sub-stituents on the properties of the ion radical molecule has not yet been elucidated ade-quately. The main problem is to find out what groups should be introduced into themolecule to direct the reaction to the ion radical mechanism. At first, these groups shouldimpart pronounced acceptor or donor properties to the molecule. Then these groups mustbe able to stabilize the ion radical produced. In case of anion radicals, such groups are ni-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

tro, cyano, carbonyl and sulphonyl, and trifluoromethyl (when more than one CF3 group ispresent in the molecule). This list, however, is not final; it will be completed as researchyields new data. In the present state of the art, such strong donors as the methyl and aminogroups are considered stabilizers for cation radicals. Cation radicals are more reactive thananion radicals, and the cation radical center must be shielded. Therefore, scientists preferto use donor substituents loaded with different fragments, such as the N,N-dialkylaminogroup or the vinyl group carrying alkyl substituents. The following examples illustratethese regularities.
The reaction of o,p-dicyano-�-phenylsulphonyl cumene with sodium thiophenolatein dimethylformamide produces o,p-dicyano-�-phenylthiocumene. A product of similarsubstitution is obtained with the potassium salt of diethyl malonate as a reactant (Kornblum& Fifolt 1980) (Scheme 4-1).
Irradiation accelerates the reactions, and the substitution products are formed in70–80% yields. Acceptors of radicals (e.g., di-tert-butylnitroxyl) or acceptors of electrons(e.g., m-dinitrobenzene) completely inhibit the substitution even if they are present in the
SCHEME 4-1
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

reaction mixture in small amounts. The mentioned substitution reactions do not take placewhen no cyano groups are present in the initial �-phenylsulphonyl cumene. Hence, thecyano groups send the reaction via the ion radical pathway. Even one cyano group in thepara position of the aromatic nucleus can initiate the reaction. Like the nitro group, thecyano group promotes the formation of the anion radical, which originates upon one elec-tron transfer from the thiophenolate or malonate ions to the substrate.
Of course, the very direction of the anion radical disintegration depends on the sub-stituents. Let us compare the anion radicals from p-cyano-�-nitrocumene and p-nitro-�-ni-trocumene. As was established by electrochemical study (Zheng et al. 1999), there is onesubtle but important difference in their cleavage. In the p-cyano-�-nitro case, the charge islocated mainly on the �-nitro group. This charge remains there when the cleavage has com-pleted. Nitrite and the cumyl radical are formed. In the p-nitro-�-nitro case, the charge andunpaired electron are originally located in the nitrophenyl portion of the anion radical, butthe charge moves to the leaving nitrite ion. The spin density (an unpaired electron) remainswithin the nitrophenyl group.
Now then, a substrate molecule should carry groups that could promote the forma-tion and stabilize the ion radical produced. As for a reactant molecule, it should give or trapan electron, otherwise the substrate would not be able to form an anion radical or a cationradical. A radical produced from an ion radical after the cleavage of a leaving group shouldpossess electrophilicity with respect to the anion reactants and nucleophilicity with respectto the cation reactants. For example, radicals bearing electron-acceptor groups should addnegatively charged reactants more easily than radicals devoid of such groups. For a chainprocess to develop, the molecule of the initial substrate should enter into electron transfermore readily (be more of an acceptor or a donor) than the product molecule. Only in thiscase may the unpaired electron density move from the product ion radical to the initial un-charged substrate (this requirement will be detailed later). And finally, the reactant shouldeffectively capture radicals formed from the substrate. Chapters 5 and 8 detail these re-quirements in a series of examples.
In the range of anions PhZ� (Z � O, S, Se, Te), for example, the thiophenolate ion(PhS�) effectively traps aryl radicals (Ar
.), whereas the anion of phenyl selenide (PhSe�)
is 20 times less active, and the phenolate anion (PhO�) is absolutely inactive. The reactionof aryl radicals with phehyltelluride ions (PhTe�) proceeds in an abnormal fashion: Bothasymmetrical and symmetrical tellurides are produced (Rossi & Pierini 1980):
Ar.� PhTe� → Ph2Te � ArTePh � Ar2Te
As molecular orbital calculations show (Vilar et al. 1982), the energy levels for pairs(Ar
.� �TePh) and (ArTe� � Ph
.) are equal. This makes a dual direction of decomposition
possible for the intermediate anion radical:
Ar.� �TePh ⇔ (Ar
.�TePh ↔ ArTe�.Ph) ⇔ ArTe� �
.Ph
Many reactions demonstrate a high activity of the phenylthiolate ion in trapping arylradicals and the inability of the phenolate ion to do so. Thus, the phenylthiolate ion actingon 5-chloro-2H,3H-benzo[b]thiophenedione-2,3 produces the substitution product (in theanion radical form), while the phenolate anion initiates only the reductive dechlorination(Ciminale et al. 1978) (Scheme 4-2).
To disrupt the carbon—chloride bond at position 5 of the substrate anion radical,population of this bond with an unpaired electron should be increased. However, if a spindensity at the carbon-carrying chlorine is too great, the initial chlorine-containing anion
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

radicals dimerize instead of cleaving the chloride ion. Thus, in the isomeric 6-chloro-2H,3H-benzo[b]thiophenedione-2,3 anion radical, unpaired electron density at carbon-6 isfive times greater than at carbon-5, and the dimerization proceeds much more rapidly thanthe cleavage of the carbon–chlorine bond (Alberti et al. 1981).
The bond dissociation energy of the bond being broken is an additional crucial fac-tor for the ion radical pathway. In a series of �-halosubstituted acetophenones, for exam-ple, the fluoro substituent provides an out-of-row example of anion radical cleavage reac-tivity. This is a general rule: Aromatic fluorine–containing anion radicals do not readilydissociate into aryl radicals and fluoride ions (Denney et al. 1997). The strength of the car-bon–fluorine bond significantly contributes to slowing down the cleavage, as opposed tothe other substituents in all of the reactions where solvent reorganization (see Chapter 5) islargely predominant (Andreiux, Saveant, et al. 1997).
A solvent may play a decisive role when a reaction chooses the ion radical pathway.For instance, the solvent effect on the thermodynamic contribution to the activation free en-ergy causes an increase in the cleavage rate constant for the chloroanthracene anion radicalwith an increase in the solvent acceptor number. There are examples of similar solvent ef-fects in a review by Jaworski (1998).
Sometimes researchers change solvent polarity deliberately, in order to discern theorigin of the reaction products. A representative example is the photoreaction depicted inScheme 4-3. The reaction gives rise to the three products.
Whereas the yields of the first two products are insensitive to changes in solvent po-larities, the yield of the third increases with changing a solvent from CD2Cl2 to CD3CN
SCHEME 4-2
SCHEME 4-3
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

and, especially, with the addition of LiClO4 to the CD3CN reaction medium. We concludethat the third product is formed on a parallel reaction having an ion radical mechanism (fordetails, see Painter & Blackstock 1995).
The dependence of ionic strength on reaction rate can supposedly be an indicator ofion radical participation in rate control.
Attractive interactions between ion radicals and their environment may lower the en-ergy of the corresponding molecular orbital populated with an unpaired electron. This en-hances the stability of ion radicals and favors the stepwise pathway of the reaction; e.g.,
RX � e ⇔ (RX)�. ⇔ R.� X�
Contrary to the stepwise mechanism, a concerted mechanism of dissociative electrontransfer is also possible, e.g.,
RX � e ⇔ R.� X�
The competition between these two reaction pathways depends upon both the driv-ing forces and the intrinsic barrier factors. As proved in several cases of anion radicals,there is a rule for a reaction to proceed down the stepwise or concerted mechanism. Thehigher the energy of the lowest unoccupied molecular orbital and the weaker the bondstrength between R and X in the starting molecule, the greater is the tendency for the con-certed mechanism to prevail over the stepwise mechanism, and vice versa (Andrieux,Robert, et al. 1994); see also Section 8.2.
Thus, if the concerted pathway has a stronger driving force than that for the first stepof the stepwise pathway, the cleavage of the anion radical is thermodynamically favorable.Stepwise mechanisms are considered viable when the intermediate (the anion radical in thepreceeding schemes) has a lifetime that is longer than the time for a bond vibration (ca.10�13 sec). Concerted mechanisms would occur under the opposite conditions, i.e., whenthe intermediate “does not exist” (Eldin & Jencks 1995; Speiser 1996). However, anotherpoint of view has been developed, according to which the concerted character of the reac-tion results from an energetic advantage rather than from the “nonexistence” of the ion rad-ical intermediate (see, for example, Andreiux, Saveant et al. 1997).
Considering energy factors that govern the reaction pathway, it is worthwhile notingthe mode of electron injection (abstraction). For instance, the energy of the incoming elec-tron is much larger in pulse radiolysis than in electrochemistry. The additional driving forcethus offered may stimulate a transition from a concerted to a stepwise mechanism (An-drieux, Robert, Saveant 1995).
As to the further fate of the formed radical R., it may be trapped with the reactant ion,
say Z�, and transformed into the (RZ)�.anion-radical. The latter will pass an unpaired
electron to RX to start the new reaction sequence. Therefore, the starting compound musthave a greater electron affinity than the substitution product. This is necessary to developthe described chain process.
Another pathway to stabilize the radical R.consists in its dimerization:
R.� R
. → R � R
This route leads to termination of the chain process. Large concentrations of the substrateand a small concentration of the nucleophile are factors favorable for the formation of thedimer. This statement concerns only the reaction in which the attack of the nucleophileupon the radical R
.is not too slow (Ettaeb et al. 1992).
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

4.3 CHEMICAL APPROACHES TO THE IDENTIFICATION OF IONRADICAL REACTIONS
4.3.1 Identification According to the Structure of the Final Products
The interaction of alkyl halide with mercaptans or alkali mercaptides produces thioalkylderivatives. This is a typical nucleophilic substitution reaction, and one cannot tell by thenature of products whether or not it proceeds through the ion radical stage. However, theversion of the reaction between 5-bromo-5-nitro-1,3-dioxan and sodium ethylmercaptidemay be explained only by an intermediate stage involving electron transfer. As has beenfound (Zorin et al. 1983), this reaction in dimethylsulfoxide leads to diethyldisulfide (yield95%), sodium bromide (quantitative yield), and 5,5-bis(5-nitro-1,3-dioxanyl) (yield 90%).
Ultraviolet irradiation markedly accelerates the reaction, while benzene nitro deriva-tives decelerate it. The result obtained shows that the process begins with the formation ofethylthiyl radicals and anion radicals of the substrate. Ethylthiyl radicals dimerize (di-ethyldisulfide is obtained), and anion radicals of the substrate decompose monomolecu-larly to give 5-nitro-1,3-dioxa-5-cyclohexyl radicals. The latter radicals recombine andform the final dioxanyl (Scheme 4-4).
Another representative and important example consists of thioarylation of 9-bromo-9-phenylfluorene. The reaction yields sodium bromide and 9-phenylthio-9-phenylfluorene(Singh & Jayaraman 1974) (Scheme 4-5). In this reaction, sodium bromide is produced inquantitative yield, but the yield of the product of the nucleophilic substitution is only 42%.The substrate and the nucleophile also enter into other conversions, leading to 9,9-
SCHEME 4-4
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

diphenylbifluorenyl (yield 33%) and diphenyldisulfide (yield 30%). The formation of thesesubstances contradicts common ideas on nucleophilic substitution. The presence of radicaltraps (oxygen or tetrabromobenzoquinone) decelerates the formation of both unexpectedcompounds and the product of thioarylation. Consequently, this stage produces thephenylthiyl radical and the anion radical of the substrate. Both electron-transfer productsundergo further conversions: The phenylthiyl radical gives diphenyldisulfide, and the an-ion radical of the substrate produces the 9-fluorenyl radical. The latter reacts in two direc-tions: Dimerizing, it forms bifluorenyl; and reacting with the nucleophile, it gives the an-ion radical of the substitution product. The chain continues because the electron from theanion radical is transferred to the unreacted molecule of the substrate. The latter losesbromine and then reacts with the nucleophile, and so on (Scheme 4-6).
The mechanism of the reaction depicted in Scheme 4-6 differs from the SN1 or SN2mechanism in that it involves the stage of one-electron oxidation reduction. The impetus ofthis stage may be the easy detachment of the bromine anion followed by the formation ofthe fluorenyl radical. The latter is unsaturated at position 9, near three benzene rings thatstabilize the radical center. The radical formed is intercepted by the phenylthio anion. Thatleads to the anion radical of the substitution product. Further electron exchange producesthe substrate anion radical and the final product in its neutral state. The reaction takes placeand consists of radical (R) nucleophilic (N) monomolecular (1) substitution (S), with thecombined symbol of SRN1. Reactions of SRN1 type may have both branch-chain and non-chain character.
The chain mechanism is, apparently, more common than the nonchain one. The veryconception of the SRN1 reaction was formulated as a result of the development of chain-radical theory. In 1964, alkylation of a saturated carbon atom bearing a good leaving groupwas found to be proceeding through one-electron transfer, resulting in the formation of an-ion radicals and radicals. Kornblum and his colleagues were the first ones to discover sucha mechanism (Kerber et al. 1964, 1965). In 1966, Kornblum, Michel, and Kerber and Rus-sell and Danen had independently shown that such substitution develops as the chain pro-cess. As found later, the chain radical mechanism, with anion radical participation, takesplace in some cases of aromatic nucleophilic substitution, and subsequently the symbol ofSRN1 was proposed (Kim & Bunnett 1970).
The SRN1 mechanism has been proven via many different approaches. Russell andco-authors (1971) reconstructed the stage of the nucleophilic substitution of 4-iodonitrobenze with the cyanide ion (Scheme 4-7).
One-electron reduction at the cathode in the presence of cyanide leads to the anionradical of 4-iodonitrobenzene. Like other halide derivatives, 4-iodonitrobenzene in the an-ion radical state easily expels the halide anion and converts into the 4-nitrophenyl radical.The latter reacts with the cyanide ion and produces the anion radical of 4-cyanonitroben-zene. The same anion radical can be obtained by reducing the 4-cyanobenzenediazonium
SCHEME 4-5
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

SCHEME 4-6
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.
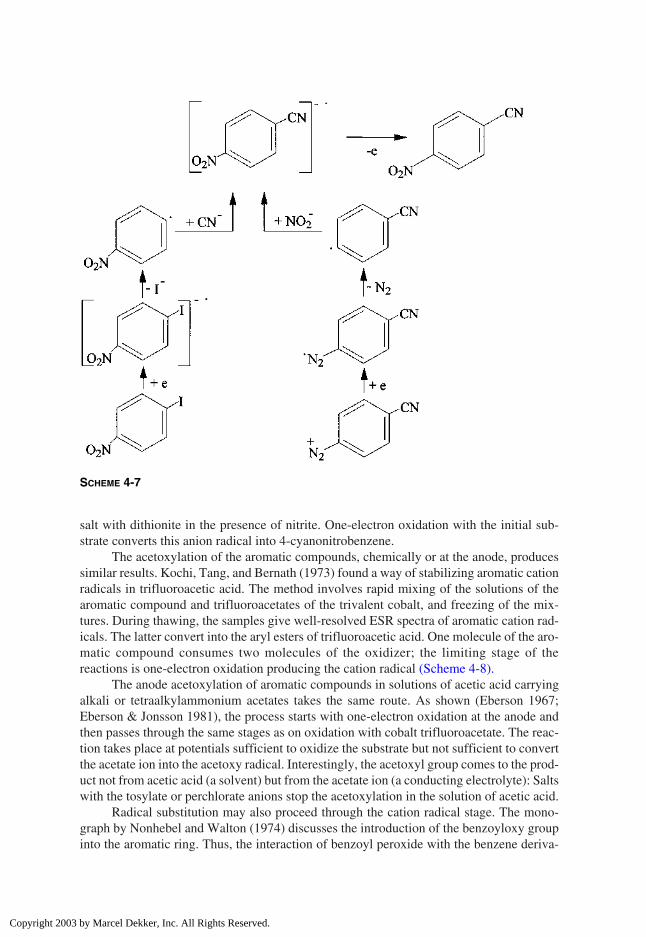
salt with dithionite in the presence of nitrite. One-electron oxidation with the initial sub-strate converts this anion radical into 4-cyanonitrobenzene.
The acetoxylation of the aromatic compounds, chemically or at the anode, producessimilar results. Kochi, Tang, and Bernath (1973) found a way of stabilizing aromatic cationradicals in trifluoroacetic acid. The method involves rapid mixing of the solutions of thearomatic compound and trifluoroacetates of the trivalent cobalt, and freezing of the mix-tures. During thawing, the samples give well-resolved ESR spectra of aromatic cation rad-icals. The latter convert into the aryl esters of trifluoroacetic acid. One molecule of the aro-matic compound consumes two molecules of the oxidizer; the limiting stage of thereactions is one-electron oxidation producing the cation radical (Scheme 4-8).
The anode acetoxylation of aromatic compounds in solutions of acetic acid carryingalkali or tetraalkylammonium acetates takes the same route. As shown (Eberson 1967;Eberson & Jonsson 1981), the process starts with one-electron oxidation at the anode andthen passes through the same stages as on oxidation with cobalt trifluoroacetate. The reac-tion takes place at potentials sufficient to oxidize the substrate but not sufficient to convertthe acetate ion into the acetoxy radical. Interestingly, the acetoxyl group comes to the prod-uct not from acetic acid (a solvent) but from the acetate ion (a conducting electrolyte): Saltswith the tosylate or perchlorate anions stop the acetoxylation in the solution of acetic acid.
Radical substitution may also proceed through the cation radical stage. The mono-graph by Nonhebel and Walton (1974) discusses the introduction of the benzoyloxy groupinto the aromatic ring. Thus, the interaction of benzoyl peroxide with the benzene deriva-
SCHEME 4-7
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.
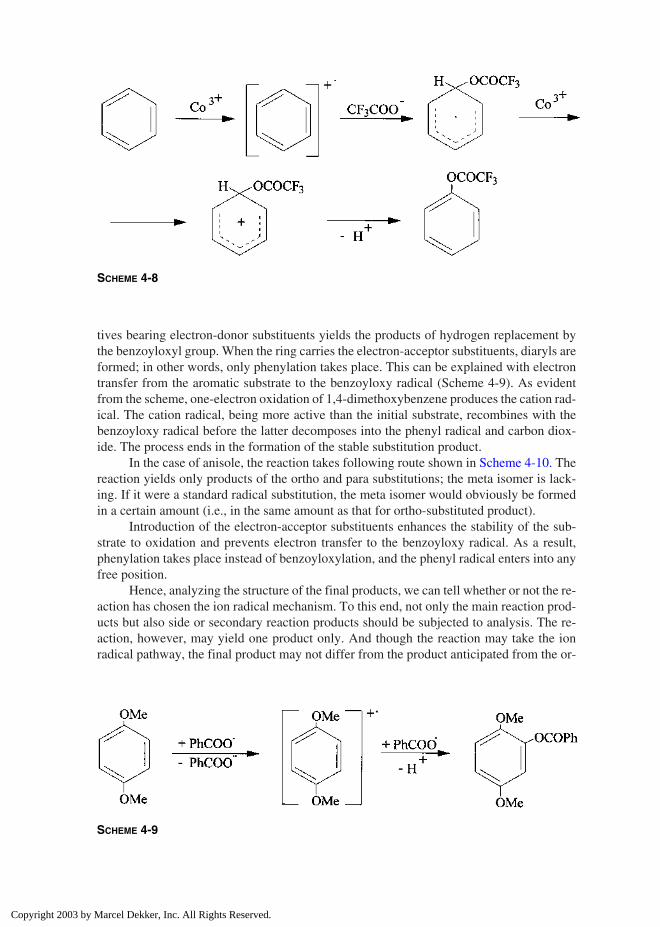
tives bearing electron-donor substituents yields the products of hydrogen replacement bythe benzoyloxyl group. When the ring carries the electron-acceptor substituents, diaryls areformed; in other words, only phenylation takes place. This can be explained with electrontransfer from the aromatic substrate to the benzoyloxy radical (Scheme 4-9). As evidentfrom the scheme, one-electron oxidation of 1,4-dimethoxybenzene produces the cation rad-ical. The cation radical, being more active than the initial substrate, recombines with thebenzoyloxy radical before the latter decomposes into the phenyl radical and carbon diox-ide. The process ends in the formation of the stable substitution product.
In the case of anisole, the reaction takes following route shown in Scheme 4-10. Thereaction yields only products of the ortho and para substitutions; the meta isomer is lack-ing. If it were a standard radical substitution, the meta isomer would obviously be formedin a certain amount (i.e., in the same amount as that for ortho-substituted product).
Introduction of the electron-acceptor substituents enhances the stability of the sub-strate to oxidation and prevents electron transfer to the benzoyloxy radical. As a result,phenylation takes place instead of benzoyloxylation, and the phenyl radical enters into anyfree position.
Hence, analyzing the structure of the final products, we can tell whether or not the re-action has chosen the ion radical mechanism. To this end, not only the main reaction prod-ucts but also side or secondary reaction products should be subjected to analysis. The re-action, however, may yield one product only. And though the reaction may take the ionradical pathway, the final product may not differ from the product anticipated from the or-
SCHEME 4-8
SCHEME 4-9
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

dinary reaction. Fortunately, there are also other ways to discern the ion radical mechanismif the reaction has really chosen it.
4.3.2 Identification Through Changing the Reaction Direction:Violation of a Hammett-Type Correlation When the ReactionPasses from the “Classical” to the Ion-Radical Mechanism
The Hammett-type (�,�) analysis of a broad range of organic reactions has demonstratedthat within the framework of one mechanism, a definite characteristic of a reaction cen-ter is linearly dependent on the constants of the substituents in the same molecule. Thislinear free energy–structure relationship certainly works when the thermodynamic con-tribution to the activation barrier of the given reaction is strongly dependent on the sub-stituent. If the linear correlation is disturbed, or if the slope of the �,� line changes, or ifthe reaction takes another route, then the compounds begin to react according to anothermechanism. Various causes can produce changes in the mechanism; however, the movefrom the “classical” to the ion radical route has special manifestations. Let us illustratethis with several examples.
While interacting with the anion of 2-nitropropane, benzyl chloride and its deriva-tives bearing groups CN, CF3, Me2N�, Me, and Br in the para position yield products of O-alkylation (route a in Scheme 4-11). Meanwhile, when the nitro group occupies the paraposition instead of these substituents, the reaction gives the products of C-alkylation (route
SCHEME 4-10
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

b). With the nitro group, the anion radicals are more easily produced, they occur to be morestable, and the reaction takes the ion radical pathway (Haas & Bender 1949a, 1949b).
In a similar way, Kim and Bunnett (1970) have demonstrated that the substitution ofthe amino group for iodine in iodotrimethylbenzene proceeds via the ion radical mecha-nism, in contrast to the bromo- and chloro-analogs. The reaction of 5- and 6-halo-1,2,4-trimethylbenzenes with potassium amide in liquid ammonia gives rise to 5- and 6-amin-oderivatives. This is the cine substitution reaction (Scheme 4-12).
As seen, the reaction yields both isomeric amines regardless of the halide position (5or 6) in the initial molecule. In the cases of chloro- and bromo-analogs the ratio of 5- and6-amino-derivatives is irrespective of the halide nature and is always close to 1.5. The re-actions with the chloro- and bromo-analogs proceed according to the same mechanism. Forthe analogs bearing iodine, this 1.5 ratio decreases to 0.63 in the case of 5-iodo-derivativeas a starting material and rises to 5.86 in the case of 6-iodo-isomer. This means that the iodo
SCHEME 4-11
SCHEME 4-12
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

derivatives react not only via the mechanism of cine substitution but also via some othermechanism, the contribution of which differs for 5- and 6-isomers. The iodo derivatives,however, produce the amino derivatives in the same ratio (1.5) if the reaction is conductedin the presence of tert-butylnitroxide or tetraphenylhydrazine. Because these agents scav-enge radicals, Kim and Bunnett (1970) concluded that the iodo derivatives react via themechanism of cine substitution and at the same time via the ion radical mechanism. Ion rad-icals iodotrimethylbenzenes cleave the halide ion more readily than bromo- and chloro-analogs. This makes the ion radical route coincident with the cine mechanism:
ArI → [ArI]�. → Ar.� I�
Ar.� NH�
2 → [ArNH2]�.
[ArNH2]�.� ArI → ArNH2 � [ArI]�. etc.
The ion radical pathway of a reaction is sometimes reflected in changes in the coef-ficient �, i.e., in the slope of the relationship connecting the substituent constants (�’s) andthe reaction rates. This may be used frequently when determining the mechanism of the re-action. Let us illustrate this.
The rates of formation of aryl iodide from aryl diazonium salts and potassium iodidein methanol depend on the nature of the substituent conjugated with the diazo group (Singh& Kumar 1972a, 1972b). Electron-donor substituents decelerate the process as comparedwith benzene diazonium (the substituent is hydrogen), whereas electron-acceptor sub-stituents accelerate it. Oxygen inhibits the reaction, and photoirradiation speeds it up. Asthe authors point out, in the case of 4-nitrobenzene diazonium, the reaction leads not onlyto 4-iodonitrobenzene but also to nitrobenzene, elemental iodine, and formaldehyde. All ofthese facts support the following mechanism:
ArN2� � I� → ArN2
.� I
.
I.�
.I → I2
ArN2. → N2 � Ar
.
Ar.� I� → [ArI]�.
[ArI]�.� ArN2
� → ArI � ArN2� etc.
The nitrophenyl radical can react with the iodide ion and the solvent, methanol, aswell. Transference of the hydrogen radical from methyl alcohol to the nitrophenyl radicalgives rise to nitrobenzene and formaldehyde (CH3OH → CH2O). Though carefully soughtamong the products of the reaction, 3-iodonitrobenzene and 4-nitroanisole were lacking.This completely rejects another possible mechanism of the reaction, cine substitution,which involves the formation of dehydrobenzene, as was described earlier.
Recently, the distinction between electrophilic and ion radical (electron-transfer)mechanisms of addition reactions to the vinyl double bond of aryl vinyl sulfides andethers has been achieved by studying substituent effects (Aplin & Bauld 1997). Specifi-cally, the effects of meta and para substituents on the rates of electrophilic addition cor-relate with Hammett � values, while ionization of the substrates to the correspondingcation radicals correlates with ��. The significance of the respective correlations wasconfirmed by statistical tests. The application of this criterion to the reaction of aryl vinylsulfides and ethers with tetracyanoethylene revealed that formation of cyclobutanes oc-curs via direct electrophilic addition to the electron-rich alkene and not via an electron-transfer mechanism.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

4.3.3 Identification Through Disturbance of the “Leaving-GroupStrength” Correlation
Classical (standard) nucleophilic substitutions show a clear-cut dependence of the rate of aprocess on the strength of the leaving group. In the literature, the leaving group with itselectron pair is sometimes called the nucleofuge (from the Latin words nucleus and fugio,meaning “I am running away”). The inclination of the nucleofuge to cleave is called nu-cleofugicity.
For instance, piperidine replaces a substituent in a series of 4-substituted nitroben-zenes with the following relative rates (dimethylsulfoxide, 50°C): 1 (Cl), 412 (F), 1.17 (Br),0.26 (I), 0.01 (SPh) (Suhr 1964). Similar reactions of amination proceed in liquid ammo-nia at photostimulation, but the foregoing sequence is disturbed. Bunnett (1978) supportedthe ion radical mechanism of the reaction and showed that at the same dose of irradiation,iodides appear to be more active than bromides, while chlorides and fluorides react withgreat difficulty, if at all. Bunnett and Creary (1974a,b) studied the photostimulated reactionof aryl halides as substrates with sodium thiophenolate instead of piperidine as a reagent(liquid ammonia, �45°C). As shown, aryl iodides undergo substitution, aryl chloridesprobably do not react, and aryl bromides are represented by only one example of success-ful substitution with the yield of arylphenylsulfide of about 20%. It is of interest that tri-ethylamine and tetrabutylammonium hydroxide facilitate substitution and PhSNa producesdiphenyl disulfide when acting both on bromobenzene (yield 65–70%) and on chloroben-zene (yield 30–33%) (Rybakova et al. 1982). These bases (triethylamine and tetrabutylam-monium hydroxide) act as catalysts. The catalysts probably act as additional electrondonors and initiate the formation of the primary anion radical of the substrate. The anionradical may originate in the donor–acceptor complex between the substrate and the base.This should also promote the decomposition of the anion radical under photoirradiation.
Thus, disturbance of the correlation between the ease of the reaction and the strengthof a leaving group definitely supports the ion radical mechanism. This, however, cannot bea sufficient proof because, as pointed out earlier, cine substitution through arynes also dis-turbs a common correlation. Besides, the nature of X affects only the rate of fragmentationArX�. → Ar
.� X�. Indeed, the rate of fragmentation of the anion radicals increases in the
series F, Cl, Br, I (Tremelling & Bunnett 1980). If we pump a strong energy up, say, by anincrease in the dose of irradiation, the rates of fragmentation of ArX�.
will equalize. Thecapture of Ar
.by a nucleophile will become a limiting stage, and it cannot depend on the
nature of the leaving group. In fact, the reactivity of phenyl iodide, phenyl chloride, phenylfluoride, phenyltrimethylammonium iodide, and diphenyl sulfide toward a pair of nucle-ophiles (EtO)2PO�K� (p) and tBuC(O)CH2
�K� (c) under conditions of sufficient irradia-tion are very close. For all the substrates, kp/kc (liquid ammonia, nitrogen atmosphere,�40°C) is equal on average to 1.4; the deviation does not exceed 8–10% (Galli & Bunnett1979, 1981). The rate constant for the reaction with (EtO)2PO�K� (p) is denoted as kp, andkc is the rate constant of the reaction with t-BuC(O)CH2
�K� (c).The usually considered monomolecular mechanism of substitution implies that one-
electron reduction activates a substrate sufficiently so that it could dissociate with no fur-ther assistance from a nucleophile. The next steps of the reaction consist of transformationsof the resultant radical. However, in substrates having sp3 carbon as a reaction center, theinfluence of the leaving group has been fixed (Russell & Mudryk 1982a, 1982b). This ledto the formulation of the SRN2 bimolecular mechanism of radical–nucleophilic substitution.In this mechanism, the initial products of single-electron transfer are combined to form the
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

product anion radicals without the formation of intermediate radicals, for example:
X O� X O.
Me2CNO2 � R1R2C � CR3 → �Me2CNO2
��.
� R1R2C � CR3 →
NO2 O
X� � �Me2CMCR1R2MCR3��.
NO2 O X �Me2CMCR1R2MCR3�
�.
� Me2CNO2 →
NO2 O X
Me2CMCR1R2MCR3 � �Me2CNO2�
�.
etc.
Other reactions with unusual effects of leaving groups (see, e.g., Amatore et al. 1982)probably also proceed according to SRN2 mechanisms. The SRN2 formulation and the con-cerned SRN1 formultaion are very close. What is important with respect to SRN2 reactionsis that the stage of free radical formation is absent. Since free radicals are not present, sol-vents are acceptable even if they are good hydrogen donors with no risk of breaking the rad-ical chain.
4.3.4 Kinetic Approaches
4.3.4.A Kinetic Isotopic Effects
The rate constant of a reaction changes when an isotope replaces the atom participating inthe reaction. Deuterium and tritium are the most commonly used isotopes. The quantitativemeasure of the kinetic isotopic effect is the ratio of the reaction rate constants, for example,kH/kD. This effect proves conclusively that a particular atom is subject to attack and that alabeled bond participates in the limiting stage of a complex chemical reaction. The kineticisotopic effects as applied to ion radical reactions have the following peculiarities. If the ionradical reactions proceed through cleavage of a leaving group, then the kinetic isotopic ef-fect can be observed, but it may not be addressed to the ion radical nature of the reactionstudied. On the other hand, the ion radical reactions assume an electron transfer from adonor to an acceptor. An electron leaves the highest occupied molecular orbital (HOMO)belonging to the whole donor molecule and populates the lowest unoccupied molecular or-bital (LUMO) of the whole acceptor molecule. This process certainly differs from the sub-stitution of atoms and groups. Thus, the direct connection between the “heaviness” of aleaving group and the rate of its cleavage cannot be the primary cause of the effect. At thesame time, introducing a “heavy” atom into a donor or acceptor molecule changes its elec-tron affinity and has a definite influence over the electron-transfer rate constant. The phe-nomenon is detailed from the theoretical and experimental standpoints in Section 2.7, “Iso-tope-Containing Organic Compounds as Ion Radical Precursors.”
It is worthwhile mentioning here as a reminder that the reactions of electron transferproceed through a polar transition state. Therefore, kH/kD for them depends above all on thedielectric constant of the solvent. This feature of the kinetic isotopic effect can really beused to establish the ion radical stage, as has been proven for reactions of phenylhydrazones
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

with quinones (the solvents employed were dimethylsulfoxide, acetonitrile, THF, anddioxan; see Rykova et al. 1980).
4.3.4.B Other Kinetic Approaches
A thorough investigation into the kinetics of a chemical reaction usually, though not al-ways, determines its mechanism. With respect to ion radical reactions, the substitution ofthe nitro group in o-dinitrobenzene (o-DNB) by the hydroxy group is a relevant example(Scheme 4-13).
The following kinetic features have been established:
1. o-DNB reacting with OH� in aqueous dimethylsulfoxide produces only o-nitro-phenolate and nitrite (Abe & Ikegame 1976, 1978).
2. Mixing of the reagents quantitatively yields long-lived anion radicals of o-DNB(Bil’kis & Shein 1975; Abe & Ikegami 1976, 1978).
3. Ion OH� is indeed the donor of an electron within the system, and it converts intoa short-lived radical OH
.(Bil’kis & Shein 1975).
4. The pathway to o-nitrophenol (in the phenolate form) proceeds through a � com-plex carrying the hydroxy group at a tetrahedral carbon (Artamkina et al. 1982).
5. The initial o-DNB and the � complex do not exchange an electron (Abe &Ikegame 1976, 1978).
6. The first stage of electron transfer, yielding the anion radical, in the interactionof o-DNB with OH� proceeds with the participation of the uncharged o-DNB(Abe & Ikegami 1978).
7. The kinetic curve of the accumulation and consumption of the o-DNB anion rad-icals is S-shaped and that of the accumulation of nitrophenolate is parabolic. Thecurve of the anion radical starts to fall at the maximum of the curve of the finalproduct (Abe & Ikegami 1978).
8. Kinetic calculations accord well with the experimental data when the anion rad-ical of o-DNB is considered as the starting compound in the substitution of OH� for the nitro group (Abe & Ikegami 1978).
These results unambiguously support the ion radical mechanism for this reaction. De-spite the complexity of the problem, the kinetic approaches make its solution quite possible.
Now let us turn to the stages of ion radical conversion and try to estimate the activa-tion barriers for each of the stages.
Initiation of ion radical conversion
In the general case, this is the interaction of a donor (D) and an acceptor (A) involving thetransfer of one electron. The probability of one-electron transfer is determined by thermo-dynamics, namely, the positive difference between the acceptor electron affinity and the
SCHEME 4-13
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

donor ionization potential. The electron transfer is accompanied by a change in the solvatesurroundings: Charged particles are formed, and the solvent molecules (the solvent is usu-ally polar) create a sphere around the particles, thereby promoting their formation. Elevatedtemperatures destroy the solvate shell and hinder the conversion. Besides, electron transferis often preceded by the formation of the charge-transfer complexes by the scheme:
D � A → D���A → (D�, A�) → (D�., A�.
) → D�.� A�.
The formation of the charge-transfer complexes presumes that donor and acceptormolecules are held in place by some forces. Here too, an increase in temperature hindersthe formation of complexes because it enforces the disorder of molecules in the solution.To summarize, the stage of origin of ion radical conversion has, as a rule, a negative tem-perature coefficient or, in any event, does not increase the total activation energy of the ionradical reaction.
Development of the ion radical reaction
A profound rebuilding of a molecule, which is associated with bond cleavage, cannot takeplace at a negative or zero activation energy. Thus, disintegration of the anion radicals of 4-iodo (or 4-bromo) nitrobenzene in acetonitrile or dimethylformamide proceeds with positiveactivation energy of 70–85 kJ�mol�1 (Parker 1981). The disintegration follows the scheme:
4-O2NC6H4I → 4-O2NC6H4.� I�
As to the next step, namely, the reaction of aryl radicals with nucleophiles, we shouldtake into account the fact that a �* molecular orbital, which initially accommodates the in-coming electron, is available in the aryl halide. The electron is subsequently transferred in-tramolecularly from the �* to the �* molecular orbital of the carbon–halogen bond. Arylradicals effectively scavenge H atoms. Therefore, an abstraction of a hydrogen atom fromthe solvent may occur. However, in the case of nucleophiles that can act as effective trapsof aryl radicals, the addition of a nucleophile to the phenyl radical takes place. At this point,let us focus on the step of addition of the nucleophile (Y�) to the intermediate radical (Ph
.).
When a new � bond begins to form between the sp3 carbon-centered radical (H5C6.) and
Y�, this bond is at first going to be a two-center, three-electron bond (H5C6�Y). This pro-cess of nucleophile addition can and must be viewed as an inner-sphere electron transfer.In other words, formation of the new bond is connected with a transfer of the electron tophenyl radical. Therefore, the good electron donor character of Y� is necessary for this stepeventually to lead to the efficient formation of (H5C6Y)�.
, i.e., the product anion radical.Galli and Gentili (1998) have evaluated the thermodynamic driving force (TDDF) of thenucleophile/radical addition step. With respect to this step, they classified a nucleophile Y� as good (Y� � �CH2COCH3, TDDF � �79.6 kJ�mol�1), weak (Y� � �SPh, TDDF� �15.5 kJ�mol�1), nonreactive (Y� � �OPh, TDDF � �21.4 kJ�mol�1), and no-returnnucleophile (Y� � �Br, TDDF � �55.5 kJ�mol�1). It is obvious that the addition stepswith negative TDDF values are thermodynamically favored.
When ion radical substitution proceeds, it has only moderate activation energy and,if permitted thermodynamically, proceeds under mild conditions.
4.3.5 Positional Reactivity and the Distribution of Spin Density inSubstrate Ion Radicals
Electron spin resonance spectra of ion radicals reveal a quantitative distribution of the spindensity. The ESR spectrum determines the hyperfine coupling (HFC) constant for the ith
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

hydrogen, aiH. The constant is directly proportional to the spin density at the ith carbon car-
rying the ith hydrogen.Of course, any correlation between ai
H and the direction of, say, substitution does notprove that the reaction necessarily takes the ion radical pathway. This means that the cor-relation may represent the relationship, for example, between the orientation and the ten-dency of a substrate to locate a charge, or between the electronic structure of the transitionstate and the distribution of the spin density in the substrate ion radical, etc. Nevertheless,such correlation deserves to be considered; it can serve as one, though not single and self-sufficient, proof in favor of the ion radical pathway.
Let us first consider the intramolecular selectivity of substitution at 2 and 1 methylgroups in substituted 1,2,3-trimethylbenzenes. The k2/k2 ratio in Table 4-1 correlates withaH
Me of the corresponding cation radicals. The following scheme compares cation radicaland pure radical pathways of substitution in 1,2,3-trimethylbenzenes bearing a Z group inposition 5 (Baciocchi, 1995) (Scheme 4-14).
Table 4-1 weighs the positional selectivity of the side-chain cation-radical acetoxy-lation against the side-chain pure radical bromination.
Table 4-1 compares two different reactions, namely, anode oxidation and oxidationwith cerium ammonium nitrate (which are bona fide electron-transfer processes) andbromination by N-bromosuccinimide in the presence of azobis(iso-butyro)nitrile (which isbona fide hydrogen-atom-transfer process). Both electron-transfer and hydrogen-atom-transfer processes have the benzylic radical as a common intermediate, but positional se-lectivity is stronger for electron-transfer processes. Another important point is the prefer-ence of the 2-positioned methyl group over the 1-positioned group, in terms of selectivity.For 1,2,3-tetramethylbenzene, such a preference reaches values from 16 to 55, and it is over200 for 5-methoxy-1,2,3-tetramethylbenzene.
As a second point of the comparison, the selectivity factors mentioned are in accordwith values of hyperfine coupling (HFC) constants in ESR spectra of the correspondingcation radicals. Thus, for the cation radical of 1,2,3,5-tetramethylbenzene, the HFC con-stant aH
1-Me � 0.3 mT, whereas aH2-Me � 1.7 mT (Dessau et al. 1970). For the p-methy-
lanisole cation radical, aHm � 0.02 mT and aH
p-Me � 1.5 mT; for the anisole cation radical,
TABLE 4-1 Positional Selectivity (k2/k1)a in Cation Radical and Pure Radical Side-ChainSubstitution of 5-Z-1,2,3-trimethylbenzenes
Z � Me, Z � t-Bu, Z � Br, Z � OAc, Z � OMe,Reaction k2/k1 k2/k1 k2/k1 k1/k2 k1/k2
CANb in AcOH 55 24 42 100 �200Anode oxidation 37 16 31 59
in AcOH/BF4�c
Anode oxidation 16 8 41 �200in AcOH/AcO�c
NBS�AIBNd 5 4 5 8 11in CCl4
a k2/k1 is the reactivity ratio (statistically correct) of the 2- and 1-methyl groups in a substrate.b CAN � cerium ammonium nitrate, (NH4)2Ce(NO3)6; products are benzylic acetates.c Products are benzylic acetates.d NBS � N-bromosuccinimide; AIBN = azo-iso-butyronitrile; products are benzyl bromides.Source: Baciocchi 1995.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.
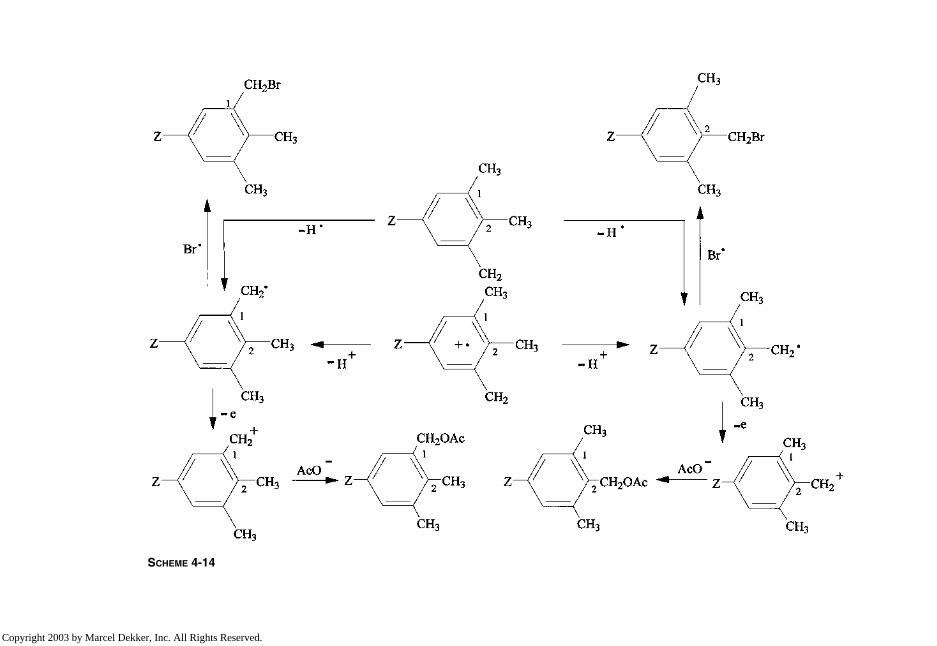
SCHEME 4-14
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

aHm � 0.02 mT and ap
H � 1.06 mT (Dixon & Murphy 1976). Obviously, proton abstractionproceeds more easily from that position of a cation radical when the spin (hole) density ismaximal. This predetermines intramolecular selectivity in the cases of acetoxylation dis-cussed. Interesting data can also be cited on nucleophilic reactions. The relative rates ofchlorine substitution in nitrochlorobenzenes under the action of different nucleophilicreagents accord well with ai
H of the anion radicals. It is true that one is forced to rely on thevalues of ai
H not carrying chlorine. The spin of the chlorine nucleus is 3/2 and that of a pro-ton is 1/2. As a result, coupling at the chlorine is 1/10 of that at the proton at the same spindensity on the nucleus. In passing from nitrochlorobenzene to nitrobenzene, the HFC con-stants of the corresponding anion radicals do not really change; see Table 4-2. In Table 4-2, the papers by Fujunaga et al. (1964) and Freed and Fraenkel (1964) are the sources forthe HFC constants. Chapman et al. (1954) reported the values of Eact.
As can be seen from Table 4-2, the relative rates of chlorine substitution in ni-trochlorobenzenes under the action of different nucleophilic reagents are in agreement withai
H of the anion radicals. The constants a2H and a6
H of the 4-chloronitrobenzene anion radi-cal are close to the a2
H and a6H constants of the nitrobenzene anion radical. The pair of an-
ion radicals of 2-chloronitrobenzene and nitrobenzene show the same agreement betweena6
H and a4H. In the anion radical of nitrobenzene, a4
H is larger than a2H. The substitution of
ethoxyl for chlorine in 4-chloronitrobenzene proceeds much more easily and requires alower activation energy than the same substitution in 2-chloronitrobenzene. The spin den-sity in position 4 of the anion radical of 1,3-dinitrobenzene is greater than that in position2 (a4
H � a2H). Therefore, 1,3-dinitro-4-chlorobenzene is more active in nucleophilic substi-
tution than 1,3-dinitro-2-chlorobenzene.For the anion radical of 3-methylnitrobenzene, a6
H � 0.330 mT and a4H � 0.488 mT
(Geske et al. 1964). For the 3-nitrochlorobenzene anion radical, a6H � 0.320 mT and a4
H
� 0.407 mT (Freed & Fraenkel 1964).Table 4-3 presents the relative rate constants of chlorine-to-methoxy substitution in
the nitrobenzene series (Epiotis 1973). The methoxide ion replaces chlorine in 4-chloro-3-methylnitrobenzene more rapidly than in 6-chloro-3-methylnitrobenzene (Table 4-3, nos.1 and 2). This agrees with the fact that the anion radical of 3-methylnitrobenzene has agreater spin density in position 4 than in position 6. The aforementioned HFC constants ofthe 3-nitrochlorobenzene anion radical and the relative rate constants (krel) for the substitu-tion of methoxyl for chlorine at the i th carbons (Table 4-3, nos. 3 and 4) correlate in thesame way.
TABLE 4-2 HFC Constants and Activation Energy in Ethoxyl Group Substitution forChlorine When Treating Nitro- and Dinitrochlorobenzenes with a Mixture of Ethyl Alcoholand Piperidine
Eact ,Compound HFC Constant, mT kJ·mol�1
Nitrobenzene a2H � a6
H � 0.330; a4H � 0.382 —
2-Nitro-1-chlorobenzene a6H � 0.330; a4
H � 0.392 75.64-Nitro-1-chlorobenzene a2
H � a6H � 0.342 71.5
1,3-Dinitrobenzene a2H � 0.277; a4
H � a6H � 0.449 —
1,3-Dinitro-2-chlorobenzene — 51.11,3-Dinitro-4-chlorobenzene — 44.7
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Nucleophilic substitution reactions proceeding via the chain ion radical mechanismalso demonstrate some correlation between the distribution of electron density in ion radi-cals and the reactivity of the corresponding uncharged substrates. As reported previously(Bunnett & Creary 1974b; Bunnett & Traber 1978), m-chloroiodobenzene reacts with suchnucleophiles (Nu�) as diethylphosphite and thiophenolate upon photoirradiation in liquidammonia. Under similar reaction conditions, monosubstitution yielding diethyl-m-chlorobenzene phosphonate prevails in the first case, whereas in the second case the mainproduct is m-bis(phenylthio)benzene (the product of disubstitution). When the reaction involves the diethylphosphite ion, a certain degree of disubstitution can be achieved bylowering the concentration of the substrate. Thus, when the concentration of m-dichlorobenzene is 0.1 M, the reaction gives rise to the product of monosubstitution only.However, when this concentration is lowered to 0.008 M, disubstitution (up to 15%) pro-ceeds concurrently with monosubstitution.
It has been shown (Bunnett & Shafer 1978) that the products of thiophenolate and di-ethylphosphite disubstitution do not form from monosubstituted derivatives. The mono-substituted product is 20 times less active than the initial substrate with regard to thiophe-nolate. The SRN1 model takes all these facts into consideration (Scheme 4-15). The schemeshows that the course of the reaction is determined by the fate of the monosubstituted prod-uct in the anion radical form. Because (EtO)2P(O) is a stronger acceptor than the PhS group,anion radicals of [m-ClC6H4SPh]�.
decompose much more easily than anion radicals of[m-ClC6H4P(O)(OEt)2]�.
. A more stable anion radical exists until the moment when itmeets the initial substrate and gives the electron to it, thus producing a monosubstitutedderivative. By contrast, a less stable anion radical cleaves the chloride ion before this an-ion radical is subjected to one-electron oxidation, and the process tends toward disubstitu-tion. The frequency of collisions of the chlorine-bearing anion radical with the initial sub-strate is greater when the concentration of the substrate is higher. Therefore, diluting thesolution promotes disubstitution. The ion radical disubstitution also proceeds stepwise.What is important is that the second step involves the product in the anion radical form,which is far more reactive than the corresponding uncharged molecule. Considered to-gether, all these simple observations support the SRN1 mechanism for the reaction.
In conclusion, it should be noted that it is necessary to check whether the SRN1 mech-anism is operative when passing from one reaction system to another, even if they are sim-ilar.
Thus, ketone enolates easily substitute chlorine in position 2 of the electrophilic nu-cleus of pyrazine (1,4-diazabenzene), and even in the dark the reaction proceeds via theSRN1 mechanism (Carver, Komin, et al. 1981). It might be expected that the introductionof the second chlorine in the ortho position to another nitrogen in the electrophilic nucleusof pyrazine promotes the ion radical pathway even more effectively. However, 2,6-
TABLE 4-3 Relative Rate Constants for Methoxy Ion Substitution for Chlorine(25�C, MeOH)
No. Nitrobenzene Final nitrobenzene krel
1 6-Chloro-3-methyl- 6-Methoxy-3-methyl- 12 4-Chloro-3-methyl- 4-Methoxy-3-methyl- 3.63 3,6-Dichloro- 3-Chloro-6-methoxy- 14 3,4-Dichloro- 3-Chloro-4-methoxy- 2.6
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

SCHEME 4-15
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

dichloropyrazine in the dark or subjected to light reacts with the same nucleophiles by theSN2 mechanism and not by the SRN1 mechanism (Carver, Greenwood, et al. 1983). The au-thors are of the opinion that two halogens in the pyrazine cycle facilitate the formation ofthe � complex to the extent that dehalogenation of the anion radicals in the solution and asubsequent nucleophilic attack of free pyrazine radical become virtually impossible. Thus,the reaction may proceed via either of the mechanisms involving or excluding the interme-diate � complex, and only special identification experiments can tell which is the true one.
It is worth noting that the correlation between the spin densities in ion radicals andthe selectivity in their reaction of substitution has no value unless combined with otherproof. However, such correlations may contribute to elucidation of the real mechanism.
4.3.6 Chemical Probing of Ion Radical Reactions
4.3.6.A Initiation of Polymerization of Vinyl Compounds
If free radicals are formed during a reaction including an electron transfer stage, they canbe revealed through their initiation of polymerization of vinyl compounds. The vinyl com-pounds act, therefore, as chemical probes. Thus, the reaction of sodium thiophenolate withalkyl or benzyl halides proceeds through a stage of electron transfer producing thephenylthiyl radical. This radical is unstable, but it can be trapped by introducing styreneinto the reaction system. Even minute quantities of the phenylthiyl radical cause styrenepolymerization. The polymerization proceeds with the insertion of the phenylthiyl frag-ments. The sulfur-containing oligomer was separated and characterized (Flesia et al. 1978).The oligomer does not form when thiophenolate is mixed with styrene in the absence of theacceptor component (alkyl or benzyl halide). The introduction of the radical trap—phenyl-tert-butyl nitrone—decelerates the reaction of the acceptor component with thiophenolate.ESR spectroscopy registers the product of the addition of the phenylthiyl radical to nitrone.This indicates that the phenylthiyl radicals form on the main pathway of the reaction:
PhSNa � PhCH2Hal → PhS.� [PhCH2Hal]�.
Na� → NaHal � PhSCH2Ph
In the same way, the presence of the phenyl radicals in the substitution of the nitrogroup for the diazonium group has been demonstrated in the reaction of benzene diazoniumsalts with sodium nitrite. Acrylonitrile introduced into the reaction mixture polymerizes;the polymerization takes place in nitrogen, and oxygen inhibits it (Singh, Khumar, &Khanna 1982). This supports the ion radical route of the reaction. However, the initiationalone (of polymerization of vinyl compounds) cannot be regarded as sufficient proof of theion radical pathway of the reaction. In the same paper, Singh, Khumar, and Khanna (1982)reported that benzene- and p-nitrobenzene diazonium fluoroborates convert into nitro- andp-dinitrobenzene under the action of sodium nitrite in methanol, whereas p-methoxyben-zene diazonoium does not produce p-nitroanisole. The fact that p-methoxybenzene diazo-nium falls out of the series is easily explained: The p-methoxyphenyl radical is incapableof coupling with the nitrite ion, and the sequence of stages needed for the ion radical reac-tion is interrupted.
Now then, the separation of polymers from the reaction mixture containing the vinyladditive indicates that the substrate produces a radical at the intermediate stage. The radi-cal produced adds to a “probe” molecule and forms an adduct with the vinyl monomer, i.e.,initiates the monomer polymerization.
Sometimes, however, the polymerization does not start, but the reaction yields a low-molecular-weight individual substance containing fragments of substrate, monomer, and
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

reagent. To illustrate, let us consider the reaction of perfluoroalkyl iodide (substrate) withthe nitropropenide salt (reagent) in the presence of the monomer probe (vinyl acetate,methylmethacrylate, styrene) (Feiring 1983):
C6F13I � [Me2CNO2]� Bu4N� → Bu4NI � (C6H.13, Me2C
.NO2)
(C6H.13, Me2C
.NO2) → C6H13C(NO2)Me2
(C6H.13, Me2C
.NO2) � PhCHBCH2 → C6F13CH2CH(Ph)C(NO2)Me2
In the latter case, the initial donor–acceptor interaction yields radicals of identical ac-tivity. The presence of a styrene gives rise not to a polymer but to a low-molecular-weightindividual compound containing fragments of the probe and of both radicals formed.
4.3.6.B Method of Inhibitors
Each ion radical reaction involves steps of electron transfer and further conversion of ionradicals. Ion radicals may either be consumed within the solvent cage or pass into the sol-vent pool. If they pass into the solvent pool, the method of inhibitors may determinewhether the ion radicals were produced on the main pathway of the reaction, i.e., whetherthese ion radicals are necessary to obtain the final product. Depending on its nature, the in-hibitor may oxidize the anion radical or reduce the cation radical.
It is quite evident that both anion and cation radicals cannot always leave the solventcage and exist together in the bulk solution for a long time. One such rare example is thenucleophilic substitution of chlorine in 2,4-dinitrochlorobenzene (substrate) by the diethy-lamino group from triethylamine (reactant) (Scheme 4-16).
The anion radical of the 2,4-dinitrochlorobenzene and the cation radical of the tri-ethylamine pass into the solvent volume. In this case, both acceptors of an electron (p-ben-zoquinone, tetracyanoethylene, tetracyanoquinodimethane) and donors of an electron(potassium iodide, ferrous sulfate, N,N-tetramethyl-p-phenylenediamine) inhibit the sub-stitution (Shein 1983).
Sometimes a substrate anion radical quickly decomposes, giving off the organic rad-ical, and only then converts into the final product. In this case, the usual inhibitors of a rad-ical reaction can be employed and the reaction mechanism can be disclosed from the natureof the products. Thus, the transfer of an electron from the anion radical of naphthalene toorganomercury halides gives naphthalene and the substrate anion radical. The latter de-composes in two stages:
[RHgHal]�. → Hal� � RHg.
RHg. → Hg � R
.
SCHEME 4-16
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Then symmetrization proceeds via the following scheme:
R.� RHgHal → [R2HgHal]
.
[R2HgHal].� e → [R2HgHal]�
[R2HgHal]� → Hal� � R2Hg
Cumene (H.donor) inhibits the symmetrization. The main direction becomes the reductive
demercurization, because the R.radicals controlling the process leave the sphere of the re-
action (Singh & Khanna 1983):
R.� H
. → RH
Widely encountered are the reactions that produce unstable radicals of the .OH type,
.OAlk from reagents �OH, �OAlk and rather stable anion radicals from substrates, say,quinones. The radicals are revealed by means of radical interceptors, and the anion radicalsare disclosed with the help of inhibitors (oxidizers). Radical interceptors will be consideredin Section 4.3.6.C; here we draw our attention to inhibitors.
Sodium methylate acting on 2-chloroanthraquinone substitutes the methoxy groupfor chlorine and produces anion radicals of the substrate (Shternshis et al. 1973). The studyof kinetics has demonstrated that the amount of the substrate anion radical first increasesand then sharply decreases. The inhibitor (p-benzoquinone) decelerates the formation ofthe anion radicals. The rate of formation of 2-methoxyanthraquinone also decreases. If theanion radicals were produced on the side pathway, the rate of formation of the end productupon the introduction of the inhibitor should not have decreased. Moreover, it should evenrise, because oxidation of the anion radicals regenerates the uncharged molecules of thesubstrate. Hence, the anion radical mechanism controls this reaction.
As has been reported, other nucleophilic reactions in the anthraquinone series also in-volve the production of anion radicals. These reactions are the following: hydroxylation of9,10-anthraquinone-2-sulfonic acid (Fomin & Gurdzhiyan 1978), hydroxylation, alkoxyla-tion, and cyanation in the homoaromatic ring of 9,10-anthraquinone condensed with the2,1,5-oxadiazole ring at positions 1 and 2 (Gorelik & Puchkova 1969). These studies sug-gest that one-electron reduction of quinone proceeds in parallel with the main nucleophilicreaction. The concentration of the anthraquinone-2-sulfonate anion radicals, for example,becomes independent of the time of duration of the reaction with an alkali hydroxide, andthe total yield of the anion radicals does not exceed 10%. Inhibitors (oxygen, potassium fer-ricyanide) prevent formation of the anion radicals, and the yield of 2-oxyanthraquinoneeven increases somewhat. In this case, the anion radical pathway is not the main one. Thesame conclusion was drawn in the case of oxadiazoloanthraquinone.
Therefore, only when it is strongly grounded may the anion radical stage be includedin the mechanism of a reaction. At this point, it is interesting that another representative ofsulfonated quinones, sodium 3-methyl-1,4-naphthoquinone-2-sulfonate, substitutes the hy-droxy group for the sulfonate fragment, mainly via the ion radical mechanism. The initialstage involves formation of the substrate anion radicals, and the end product is 2-hydroxy-3-methyl-1,4-naphthoquinone. As the reaction proceeds, the quantity of the anion radicalsreaches a maximum and then abruptly drops. At that moment, the concentration of the endproduct starts to grow. Upon addition of inhibitors (oxygen, potassium ferricyanide), theanion radicals do not accumulate, and the end products do not form. This indicates that thehydroxylation of 3-methyl-1,4-naphthoquinone-2-sulfonate proceeds at the expense of theion radicals via the chain mechanism (Fomin & Gurdzhiyan 1970) (Scheme 4-17).
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The method of inhibitors has demonstrated that substitution of chlorine in triph-enylchloromethane by the tert-butoxy anion does not follow the anion radical mechanism.This mechanism was widely accepted for the reaction (Bielevich et al. 1968; Ashby et al.1981). (Scheme 4-18). This widely accepted scheme had postulated the formation of theradical pair followed by the formation of the carbon–oxygen bond within the pair. Whenthe solution of triphenylmethyl chloride in THF was mixed with potassium tert-butylate inthe radiospectrometer cavity, the ESR spectrum visualized the presence of the triphenyl-methyl radical. The intensity of this signal first increased, reaching a maximum, and thendecreased to an equilibrium value. In the opinion of Bielevich and co-authors (1968), thesuperequilibrium concentration of the radicals agreed well with their generation at the pri-mary stage of one-electron transfer. In other words, the substitution product supposedlyformed at the expense of the primary generated radicals. The ESR spectrum fixed thosetriphenylmethyl radicals that failed to recombine with tert-butoxyl radicals prior to theirpassage into the solvent pool.
However, when m-dinitrobenzene was added to a solution of triphenylchloromethaneand potassium tert-butylate in 2,2-dimethoxypropane, the yield of the substitution productmarkedly increased and the yield of the dimer of the triphenylmethyl radical decreased(Simig & Lempert 1979). Therefore, the main pathway of the reaction does not involve theion radical step. Those authors suggested an alternative pathway, which is confirmed by athorough structural analysis of the secondary products formed along with the tert-butyl es-
SCHEME 4-17
SCHEME 4-18
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

ter of triphenylcarbinole (Huszthy, Lempert, & Simig 1982; Huszthy, Lempert, Simig,Vekey 1982) (Scheme 4-19).
4.3.6.C Method of Radical and Spin Traps
The aim of this section is to give a concise description of the use of traps, to note the mostpopular traps, and mainly to underline the possible artifacts connected with the applicationof traps to ion radical reactions. The problem of radical trapping is also relevant becauseradicals are often the primary products of ion radical transformations. The radicals are, asa rule, not stable, and special traps—radical and spin traps—are used to reveal them.
Radical traps belong to the class of stable free radicals, e.g., of the nitroxyl or phe-noxyl type. Interacting with radicals produced by the reaction, radical traps give diamag-netic compounds. One can follow the progress of the reaction by a decreasing intensity ofthe ESR spectrum of the radical trap.
Nitroso compounds, nitrones, and other diamagnetic molecules can be used as spintraps. Capturing radicals produced in the reaction, spin traps form so-called spin adducts—stable nitroxyl radicals easily detected via ESR spectroscopy. In other words, the progressof the reaction can easily be followed by an increasing intensity of the spin-adduct signal.By and large, the method of traps reveals radicals by the disappearance (or appearance) ofthe ESR signal.
Radical and spin traps may inhibit the observed conversions, and this is their com-mon drawback. Chain reaction may be inhibited either at the stage of generation or at thestage of branching. A portion of the radicals, which combines with trap molecules, leavesthe sphere of a reaction. Even small amounts of a trap can affect the kinetics of reaction ifit proceeds via a chain mechanism. Traps can exchange electrons with initial anion radicalsor with the anion radicals of a product. As a result, spin traps convert to paramagneticspecies, which distort the spectral picture. In case of radical traps, capturing an electronproduces diamagnetic compounds prior to combining with radicals, and ESR spectra can-not be observed. Even when a radical trap has an additional group with strong electronaffinity, a biradical is not formed, and so no ESR signal can be generated. Thus, piperidonenitroxyl in the presence of cyclo-octatetraene dianion undergoes one-electron reduction atthe site of a free valence without the participation of the carbonyl group. In other words,the anion of corresponding hydroxylamine is formed instead of the nitroxyl bearing theketyl group (Todres 1970).
Let us now consider some features of the use of traps in cases that are free of mask-ing effects.
Radical traps. The kinetics of a decrease in the ESR signal intensity represents thekinetics of radical generation in the reaction mixture, because the adduct forms rapidly at a
SCHEME 4-19
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

rate of diffusion. Later we will cite several examples of application of radical traps in ki-netic studies.
Spin traps. Nitroso compounds and nitrons interact with radicals to form nitroxylradicals:
R1.R1 � R2NBO → NMO
.
R2
R1.R1 � R2CHBN→ O → CHMNMO
.
R2 R3 R3
The nitroxyl radicals produced from these traps give ESR spectra differing markedlyin nitrogen hyperfine coupling. The coupling constant, aN, depends on the nature of thegroups bonded to the nitrogen. Coupling constants for the nitroxyl radicals vary over a widerange from 0.4–0.5 mT for diacylnitroxyls (Lemaire et al. 1965) to 2.5–2.8 mT foralkoxynitroxyls (Rehorek 1980). Changes in these coupling constants after formation of thespin adduct provide information on the nature of a short-lived radical fixed by a trap.
Types of spin traps
1. The most universal and therefore most commonly used traps are nitroso-tert-butaneand di(tert-butyl)nitroxide (cf. Wajer et al. 1967; Arguello and others 2000). Usually, radicalscombining with these traps produce stable and easily identifiable spin adducts. When study-ing ion radical reactions that involve prolonged heating or light irradiation, one should keep inmind that the trap decomposes upon those conditions according to the following scheme:
Me3CNBO → Me3C.�
.NO
Me3C.� Me3CNBO → (Me3C)2NMO
.
As can be seen from this scheme, the tert-butyl radical adds to the initial nitroso trap, pro-ducing the nitroxyl radical. A triplet in ESR spectra with aN � 1.5–1.7 mT (depending onthe solvent) corresponds to the radical. The triplet is often so intense that it can lap over sig-nals corresponding to other spin adducts (Forshult et al. 1969).
2. 2-Methyl-2-nitrosobutanone-3 exists in the nonactive form of a dimer. Thedimer dissociates in solution to give a monomer. Light irradiation or heating promotes thedissociation, causing at the same time the decomposition of the monomers into radicals ac-cording to the following scheme:
O
→ .NO � Me2C.MCMe
NO O
MeCMCMe NO OMe
→ MeC.�
.CMe
Me
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Thus the radicals produced add to the initial molecule of a trap, giving spin adducts. Thenitrogen coupling constants of these spin adducts lie in the ESR regions of 1.4–1.5 and0.7–0.8 mT (Torsell 1970; Janzen 1971; Lagercrantz 1977).
3. C-Phenyl-N-tert-butylnitrone [PhCH � N�(tBu)O�] is rather stable. The for-mation of its spin adducts is illustrated with the following scheme (Sang et al. 1996 and ref-erences therein):
R
� PhCHBNCMe3 � R
. → PhCHMN CMe3
O�
.O
In the spin adducts mentioned (this is an essential feature), the unpaired electron interactsnot only with the nitrogen nucleus but also with the nucleus of the hydrogen of the CH frag-ment neighboring the nitrogen [R1R2CHMN(R3)MO] (Janzen & Blackburn 1969). Thisextends the identification possibilities of the nitrone as a spin trap.
In this type of spin trap, 5,5-dimethyl-1-pyrroline-N-oxide deserves mention. The ox-ide is a particularly reactive radical scavenger and as a nitrone shows a wide reactivity to-ward radical additions to give persistent spin adducts, with useful information derived fromthe nitrogen and -hydrogen splittings. These splittings appear within a broad range, facil-itating the spectral interpretation (Cano et al. 1999).
4. Nitrosobenzenes are commonly used as spin traps. They are stable and are usedto identify radicals (Zuman & Shah 1994). Most often, however, not nitrosobenzene itselfbut its 2,4,6-trimethyl and 2,4,6-tritertbutyl derivatives are utilized for this purpose; some-times 2,3-dichloro- and 2,6-dichloronitrosobenzenes may be used. Nitrosobenzenes have awider application than do other traps. This is explained by the fact that the structure of thespin adducts depends strongly on the nature of the added radical, cf. this scheme:
ArMN.MOR ← R
.� ArNBO → ArN(O
.)R
The main advantage of nitrosobenzenes as compared to nitrones is that a radical is addedto the nitrogen rather than to the carbon, as in the nitrones. This gives more direct infor-mation on the structure of the radical trapped.
Spin adducts with nitrosobenzenes differ in the g factor and can be distinguished (Ter-abe & Konaka 1971; Terabe and co-authors 1973). The primary alkyl radicals, aryl and arylthioradicals, form spin adducts with a free valence at nitrogen; the tertiary radicals produce spinadducts with a free valence at oxygen; and the secondary radicals give both types of adducts.
The application of nitrosobenzenes has a number of peculiarities. First, nitrosoben-zene may add a nucleophilic reagent (Nu�). The product of this addition easily oxidizes togenerate the radical PhN(Nu)O
.. This causes errors in assigning the reaction to an ion rad-
ical type. To avoid this, Lagercrantz (1977) suggested the use of derivatives in which thenitrogen of the nitroso group is sterically hidden. In these spin traps only oxygen of the ni-troso group can react, and only when attacked by radicals.
Second, nitrosobenzenes may give spin adducts interacting with solvents without theparticipation of reagents or substrates. Compounds of the nitrosobenzene series react insuch solvents as n-decane, ethylbenzene, iso-propylbenzene, and o-xylene. The followingscheme depicts these reactions, taking n-decane as an example (Simon et al. 1980):
C10H22 � ArNBO → C10H.21 � ArN(O
.)H
C10H.21 � ArNBO → ArN(O
.)C10H21
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The reaction proceeds at room temperature or at moderate heating up to 60°C without lightexcitation and in the absence of oxygen, i.e., in conditions common to electron-transfer re-actions. (Some of these reactions take place only in nonpolar solvents of the decane or xy-lene type.) Hence, the application of nitrosobenzene as a spin trap may be complicated bysolvent participation.
The method of spin traps can be used to perform not only qualitative but also quan-titative measurements. For quantitative determinations the spin trap method can be appliedif the rate constant of radical initiation is 107–108 L�mol�1�sec�1 and the trap concentra-tion is not below 10�2 M (Freidlina et al. 1979).
Participation of traps in redox reactions
This problem has two aspects: consumption of spin traps in one-electron oxidation/reduc-tion either of a free radical or of an initial ion radical. An electron exchange between a trapand a radical depends on the relative rate of the exchange as compared to the rates of theaddition reactions considered. An electron exchange between a trap and an ion radical isrepresented by the following scheme (Nu� is a nucleophile):
RX � Nu� → (RX)�.� Nu
.
(RX)�.� AlkNBO → RX � (AlkNBO)�.
(RX)�. → R.� X�
R.� Nu� → (RNu)�.
(RNu)�.� AlkNBO → RNu � (AlkNBO)�.
(i) Electron exchange between a trap and a free radical
A trap and an unstable radical may, in general, undergo addition with the formation of anadduct (see earlier). Electron transfer may yield a pair (a cation from a radical � an anionradical from a trap) or (an anion from a radical � a cation radical from a trap). All of thesereactions are possible and, indeed, take place under certain conditions. Some radicals eitherdo not form adducts with traps or form these adducts in extremely low yields. Therefore,the method fails to give information in cases where it should be effective. For example,Sosonkin et al. (1982) failed to reveal RCH
.OH radicals by means of PhNBO. This result
becomes understandable when comparing the rate constants of the corresponding pro-cesses. Namely, the rate constant of the addition of the radical to the trap is 103–108
L�mol�1�sec�1, and the rate constant of electron transfer from RCH.
OH to PhNBO is109–1010 L�mol��sec�1. It is readily apparent that the addition of the radical cannot com-pete with one-electron transfer. Sosonkin et al. compared the redox potentials of a numberof free radicals and spin traps; they demonstrated that the nitroso compounds can capturequantitatively only radicals having oxidation potentials below—0.6 V. Those are radicalsAlk
., Ph
., RO
., HO
.,
.CH2COOH,
.CH2COR and some others. Nitrones, however, are re-
duced at extremely negative potentials and cannot capture electrons, even from thestrongest reducers—ketyl anion radicals (R1R2CBO)�.
. Consequently, nitrones can beused widely for quantitative interception of radicals even if they are liable to one-electronoxidation.
So far we have considered the acceptor properties of spin traps. Their donor proper-ties are also known, although they have been studied to a lesser extent. The literature dataare scarce, and only several examples of one-electron oxidation of traps can be cited. Ni-trosodurene forms stable cation radicals upon photolysis in the presence of Ce4� and U6�
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

in CF3COOH (Rehorek 1979). Murabayashi et al. (1979) observed the formation of cationradicals from 2,4,6-tris(tert-butyl)nitrosobenzene subjected to the action of the 3-methylpentane molecular cation.
While using the trap method, one should take into account the oxidation properties ofa trap with respect to radicals or to other electron donors that are present in the system. Be-cause spin traps can be electron donors themselves, their oxidation potentials should bemore positive than the oxidation potential of the reagents.
(ii) Electron exchange between a trap and the initial ion radical
The previous material has illustrated the ability of spin traps to act as one-electron oxidiz-ers. This property is even more pronounced in the interactions of traps with anion radicals.Traps can block the ion radical pathway. In other words, they inhibit the whole reaction, in-cluding the ion radical step. This may be explained by both oxidation of the substrate an-ion radical and chain termination due to oxidation of the product anion radical. An illus-trative example is the inhibition of SRN1 nucleophilic substitution of 2-chloroquinoxalineby the radical trap bis(tert-butyl)nitrone (Carver, Hubband, & Wolfe, 1982).
The yields of reaction products from thermal nucleophilic substitution reactions indimethylsulfoxide of o- and p-nitrohalobenzenes with the sodium salt of ethyl �-cyanoac-etate were found to be markedly diminished from the addition of small amounts of strongelectron acceptors (nitrobenzenes). At the same time, little or no diminution effects on theyields of the reaction products were observed from the addition of radical traps such as ni-troxyls. These results are consistent with the conclusion that such reactions proceed via anonchain radical nucleophilic substitution mechanism (Zhang et al. 1993) (Scheme 4-20).
This nonchain mechanism explains the failure of the radical traps to affect the yieldof the reaction. According to the mechanism, the reactive radical intermediates are heldwithin the solvent cage. Evidently, the radical traps cannot penetrate the solvent cage likean electron can. In other words, the radical traps present in the solution, even in great ex-cess, cannot intercept the radicals in the framework of the nonchain radical mechanism.The literature contains other, similar examples of this phenomenon (e.g., Galli 1988a).
4.4 PHYSICAL APPROACHES TO THE IDENTIFICATION OF IONRADICAL REACTIONS
Previous parts of this chapter treated the substances entering into the ion radical conver-sions and analyzed the resulting products. However, in order to gain deeper insight into thecourse of the process, it is necessary to reveal intermediate species participating in conver-sions. When ion radicals are formed, they can be discovered via numerous physical meth-ods.
4.4.1 Radiospectroscopy
4.4.1.A Electron Spin Resonance (ESR)
This method unambiguously establishes the presence of species bearing unpaired electrons(ion radicals and radicals). The ESR spectrum quantitatively characterizes the distributionof the electron density within the paramagnetic particle by hyperfine ESR structure. Thisestablishes the nature and electronic configuration of the particle. The ESR method domi-nates in ion radical studies. Its modern modifications, namely, electron–nuclear double res-onance (ENDOR) and electron–nuclear–nuclear triple resonance (TRIPLE), and special
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.
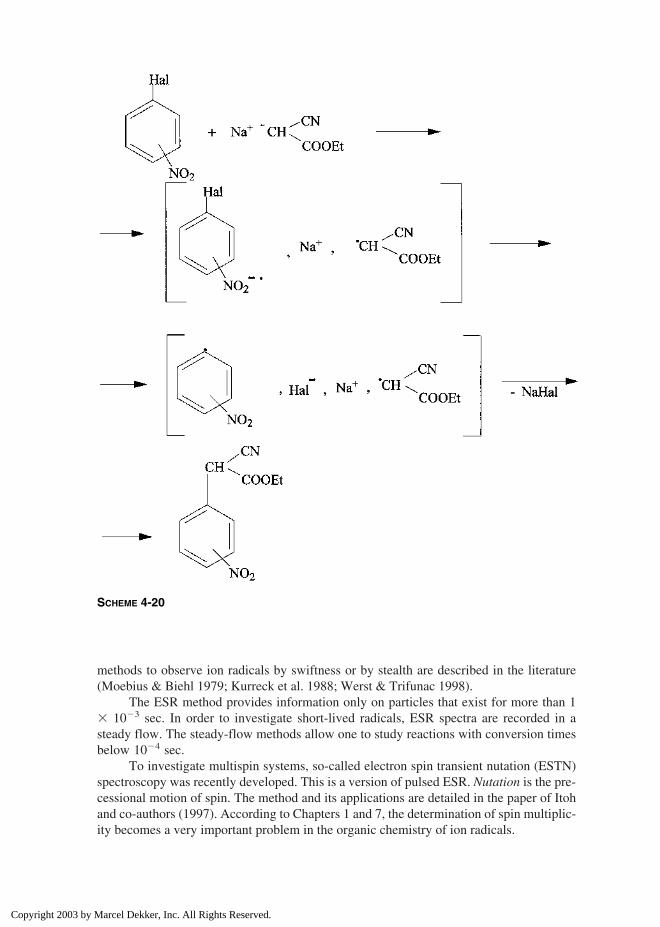
methods to observe ion radicals by swiftness or by stealth are described in the literature(Moebius & Biehl 1979; Kurreck et al. 1988; Werst & Trifunac 1998).
The ESR method provides information only on particles that exist for more than 1� 10�3 sec. In order to investigate short-lived radicals, ESR spectra are recorded in asteady flow. The steady-flow methods allow one to study reactions with conversion timesbelow 10�4 sec.
To investigate multispin systems, so-called electron spin transient nutation (ESTN)spectroscopy was recently developed. This is a version of pulsed ESR. Nutation is the pre-cessional motion of spin. The method and its applications are detailed in the paper of Itohand co-authors (1997). According to Chapters 1 and 7, the determination of spin multiplic-ity becomes a very important problem in the organic chemistry of ion radicals.
SCHEME 4-20
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

4.4.1.B Nuclear Magnetic Resonance (NMR) and Chemically InducedDynamic Nuclear Polarization (CIDNP)
The NMR methods ascertain the concentration of ion radicals and sometimes establish theirstructure. The concentration of ion radicals in solution may be determined either directlyby the intensity of the ion radical signals or indirectly by splitting the standard signal (Ma-lykhin et al. 1975). Sometimes a chemical shift of the solvent signal can be observed in thepresence of the ion radical (Screttas & Micha-Screttas 1982, 1983). It is important, then, tounderstand the mechanism by which a solvent receives spin density. One pertinent exam-ple is the interaction of the sodium fluorenone anion radical, (C6H4)2C
.MONa, as a para-
magnetic species, with hexamethylphosphorotriamide, HMPTA, i.e., (Me2N)3P → O, as asolvent (Screttas et al. 1998 and references therein). The mutual complex can be written as(C6H4)2C
.MONa���O←P(NMe2)3. Usually spin density is transferred through covalent
bonds. In the case considered, the phosphorus atom of HMPTA, though located a bondaway from the paramagnetic center, still receives some spin density. Let us refer to the crys-tal structure of the tetrameric sodium fluorenone anion radical complex with HMPTA(Scheme 4-21).
The complex possesses a Na4O4 cubic core in which each ketyl oxygen is bonded tothree sodiums and each sodium to three ketyl oxygens. The fourth coordination site on themetal is occupied by the HMPTA ligand. So the metal can receive some spin densitythrough the ketyl oxygen and transfer a part of this density to the phosphorus-containingligand. This results in chemical shifts of the ligand and opens up the possibility of usingchanges in the solvent (31P)NMR pattern for ion radical studies.
One very important variety of the NMR method consists of detection of chemicallyinduced dynamic nuclear polarization (CIDNP), a method far more sensitive than ESR withregard to the detection of the ion radical stages of reactions. Particles with uniformly pop-ulated Zeeman levels give normal NMR spectra. Molecules produced as a result of radi-cal–radical recombination may have nonuniformly populated Zeeman levels. This leads tothe abnormal NMR behavior called chemically induced dynamic nuclear polarization: en-hanced absorption at positive polarization, and emission at negative polarization. TheCIDNP signals are observed immediately after particle formation within the period of timenecessary for nuclear relaxation. This time is 1–30 sec. The NMR spectra often visualizethe multiplet effect. The effect is observed when the lines in spin multiplet in high and lowfields have opposite signs, revealing both emission and absorption.
SCHEME 4-21
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The CIDNP method proves that the reaction proceeds through intermediate param-agnetic particles (ion radicals, radicals, biradicals, etc.). A pair of any spin carriers is a mul-tispin system with a manifold of spin states. Chemical reaction selects those of them thatare spin-allowed. Any radical pair (R
., R
.) needs to undergo triplet–singlet spin conversion
in order to recombine and to produce a zero-spin diamagnetic molecule RMR. Thus, chem-ical reaction leaves spin-forbidden states unbound. These forbidden states undergo mag-netically induced spin conversion. Therefore, any multispin pair is a spin-selective mi-croreactor and a potential source of magnetic effects. The CIDNP method establisheswhich radical pair gave rise to a molecule and determines the spin multiplicity of the re-acting particles forming a radical pair. And CIDNP evaluates (by the kinetics of nuclear polarization) the rate constants of the ion radical conversions and their activation energies.
As with any other physical methods, CIDNP is not universal. It has certain draw-backs: The polarization is weak and is hardly detected in reactions involving extremelyshort-lived radicals, and, if so, it disappears quickly. It is often difficult to attribute the po-larization to the products of the main conversions rather than the side or reverse conver-sions. The latter threat is most serious for reactions involving the participation of ion radi-cals: The formation of end products often proceeds concurrently with the restoration of theinitial neutral molecules, due to a reverse electron transfer:
→ R1X � R2Y
R1X � YR2 → (R1X�.,
.�YR2) → (R1X)�.� (R2Y)�.
→ (.R1,
.R2)
The scheme shows that not only the end products, but also the initial molecules couldbe polarized. Besides, one of the ion radicals formed may exchange electrons with the neu-tral starting molecule. These phenomena and others, which can be attributed to electron ex-change, lead to a loss of memory in nuclear spin states. In addition, the initial polarizationmay be “scattered” as a result of a chain ion radical process. This is illustrated in the fol-lowing scheme (Chanon & Tobe 1982):
R1Li � R2X → (R1Li)�.� (R2X)�.
�Li� �X�
↓ ↓.R1 .
R2
.R2 � R1Li → �R2 � (R1Li)�.
↓
�Li�
R2Li.R1 � R2X → �R1 � (R2X)�.
↓
�X�
R1X
Buchachenko (1974) has advanced another theory. He based his reasoning on the ab-sence of the CIDNP signals for the reaction of n-butyl iodide with t-butyl lithium conductedin ether at �70°C. The halogen and metal quickly exchange under these conditions, but theCMC bond does not form. In contrast to the preceding scheme, Buchachenko’s theory as-sumes that the radicals produced form complexes with the alkyl lithium associates. Alkyl
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

lithium forms stable hexamers (two pyramids having a common base) and tetramers (tetra-hedrons). These associates exist even in the gas phase and are revealed by mass spec-troscopy. A radical bonded in such a cluster produces a paramagnetic widening of the NMRsignals. This makes them nonobservable long before the end of the reaction. Therefore, inthis case we should speak not about the absence of the CIDNP effect but about its masking.
Another word of caution may be in order. The spectra may visualize nuclear polar-ization of the products due to polarization of the initial substances. Bubnov and co-work-ers (1972) recorded 15N-NMR spectra to investigate azo-coupling of benzene diazoniumtetrafluoroborate with sodium phenolate in methanol. Benzene diazonium was preparedfrom aniline-15N and H15NO2. The spectrum demonstrated a strong polarization of signalsfrom the azo dye just after mixing the solutions of the diazo compound and phenol. The sig-nal from the initial diazonium salt was also polarized. The researchers concluded that theazo dye is produced via Scheme 4-22, and the nuclear polarization of the diazonium nitro-gen was regarded as evidence for the reversibility of the electron stage.
Such treatment of the CIDNP results produced serious objections. Lippmaa et al.(1973), investigating the same reaction, revealed a strong 15N, 13C, and 1H CIDNP effect.The 13C nuclei in the phenoxyl C6-ring of the azo dye were not polarized. At the same time,the polarization of 15N nuclei of the azo bond and 13C nuclei at positions 1 and 2 of thephenyl ring connected with the diazo link was an exact replica of the polarization of thesame nuclei in the diazonium salt. This has led to the conclusion that the diazo componentpolarizes as a result of the side reactions and that it is the diazo component that brings it tothe azo dye. Thus, the CIDNP effect does not support the ion radical mechanism presentedearlier. Several explanations for the observed CIDNP effect have been proposed. We wantto discuss one of them here because it seems to explain a whole range of interactions of di-azonium salts with oxyanions, an interaction that is accompanied by a pronounced polar-ization of nuclei.
The reaction of diazo cation with phenolate yielding the azo dye may proceedthrough the formation of the diazo ether. Kekule came to this conclusion in 1870. Zollinger(1958), considering this conclusion, proposed and explained the mechanism by which thediazo ethers convert into the C-diazo compounds, that is, into the hydroxyazo dyes. The di-azo ether preliminarily dissociates into the phenolate ion and the diazonium ion; i.e., a two-stage intermolecular reaction takes place. The CIDNP effect suggests that the diazo ethermay reversibly convert into the radical pair:
ArN2� � �OPhD ArNBNMOPhD ArNBN
.,
.OPh
While interacting with the alkoxyl anions, the diazonium cation also produces the pri-mary diazo ether, although it can give no azo dye. 15N and 13C nuclei of aryldiazonium
SCHEME 4-22
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

tetrafluoroborate enriched with 15N in both nitrogen positions become polarized in the pres-ence of sodium alcoholates (Levit et al. 1971). The reaction yields benzene, showing nu-clear polarization. The data on kinetics and the CIDNP results (Lewis & Chalmers 1971;Levit et al. 1972) agree well with the following scheme:
ArN2� � �ORD ArNBNORD ArNBN
.,
.OR
ArNBN. → N2 � Ar
.
Ar.� H(SolH) → ArH
Schemes on interactions between the aryazo cation and the phenolate or alkoxy an-ions have much in common. There is a very clear reason to consider them together.
As follows from the foregoing, the CIDNP method is of great help to the chemist.However, it cannot always give straightforward information, first because the CIDNP ef-fect may be masked, and second because errors may creep into its interpretation. TheCIDNP method requires strong chemical and physical professional skills (which are usefulfor all the methods considered here). However, using CIDNP, a researcher can be com-pensated by the reliability of the conclusions.
4.4.2 Optical Spectroscopy
4.4.2.A Electron Spectroscopy
Ion radicals have, as a rule, a deeper coloring than the initial neutral molecules. An unpairedelectron on the molecular orbital increases the molecule’s polarizability and facilitates itsexcitation by light. This enhances the intensity of absorption and shifts it to the region ofhigher wavelength. Therefore, ion radicals can be quite easily revealed via electron spec-troscopy. This method is often applied to investigate the kinetics of the ion radical reactionand to establish the significance of the ion radical pathway.
For example, the ESR method has revealed that methoxylation of 4-nitro-1-chlorobenzene produces ion radicals of the initial substance. The presence of oxygen pro-motes the reaction. It has also been shown that besides the main product, 4-nitroanilsole,the reaction yields the side product 4-nitrophenol (up to 15%) (Shein et al. 1973).
Solodovnikov (1976) studied the kinetics of the interaction of the 4-nitro-1-chlorobenzene with sodium methylate in dimethylsulfoxide in air via the method of spec-trophotometry. Kinetic calculations were made in an assumption that all the anion radicalsof 4-nitro-1-chlorobenzene are converted into 4-nitrophenolate. The calculations gave asum of rate constants for formation of 4-nitroanisole and of the 4-nitro-1-chlorobenzene an-ion radicals close to the rate constant for the consumption of 4-nitro-1-chlorobenzene.Solodovnokov (1976) concluded that the anion radicals of 4-nitro-1-chlorobenzene are pro-duced by a reaction parallel to substitution. Then it should be assumed that the reaction pro-ceeds either by a nonradical mechanism or by a “hidden radical” mechanism, which impliesthat particles of a radical nature are produced and unite in a solvent cage without passinginto a solvent pool. This conclusion generated objections (Shein 1983). The discussion de-serves our consideration because it reveals features and limitations of the method for dis-cerning the ion radical nature of a reaction.
Shein (1983) pointed out that Solodovnikov’s (1976) kinetic result may also be causedby a sequential reaction that led to the formation of the anion radicals of 4-nitro-1-chloroben-zene and then of 4-nitroanosole. This is really possible when, say, the formation of the an-ion radical of the initial substrate as a result of its interaction with the methylate anion is the
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

limiting stage. Abe and Ikegame (1976, 1978), investigating the kinetics of the reaction be-tween p-dinitrobenzene and alkali, demonstrated that the formation of anion radical is thelimiting stage. The assumption that all the anion-radicals of 4-nitro-1-chlorobenzene con-vert into 4-nitrophenolate is, according to Shein (1983), invalid because these anion radicalsmay be consumed in other reactions. If the presence of oxygen leads only to 4-nitropheno-late (the side product), then oxygen would hardly promote the nucleophilic substitution ofmethoxyl for chlorine (see earlier). The discussion of experimental errors (which could becommitted by Solodovnikov) is of a special methodological interest.
Shein (1983) paid attention to the following facts. Dimethylsulfoxide used as a sol-vent may contain water and MeSNa. Water may hydrolyze the initial 4-nitro-1-chloroben-zene (spectrophotometry uses solutions extremely diluted with respect to the substrate).The presence of MeSNa may cause the formation of 4-nitrophenylmethyl sulfide and its an-ion radical, and this was not included in the kinetic equations. Solodovnikov (1976) con-siders neither the production of 4-nitroanisole nor the formation of the other products of adeeper reduction of the substrate.
From the discussion cited, the following inferences can be drawn. When the kineticsof ion radical reactions is investigated spectrophotometrically, solvents should be analyzedfor purity; reagents and products should be checked against the material balance. These re-quirements are not simple, but they are essential in order to obtain adequate kinetic data.
Electron spectroscopy is also applicable to structural studies of ion radicals. There isan extensive body of work studying this matter. Spectrophotometry is indispensable inthose cases where an equilibrium exists between the paramagnetic and diamagnetic formsof an ion radical salt. For instance, ketyl dimers can be paramagnetic and diamagnetic. Thedimers of the sodium ketyls of fluorene or benzoquinone are, in the main, paramagnetic inether solvents. In hydrocarbon solvents such as toluene and cyclohexane, a portion of theparamagnetic species becomes smaller, and color intensity changes decrease markedly.Spectrophotometry fixes such a change clearly (Rao et al. 1972). The drop in absorptivityis a consequence of the formation of the diamagnetic dimer:
ArC.MO� Na� � Na� �OM
.CArD
(ion radical salt)
Na�
D Ar2C.MO� �OM
.CArD
Na�
(paramagnetic dimer)
Ar2CMO� Na�
D (diamagnetic dimer)
Ar2CMO� Na�
The equilibrium is reversible at the whole steps. Both spectrometry and ESR are applica-ble to studying the equilibrium, but the first method is more accurate.
It is interesting to compare electron spectroscopy and ESR with respect to substituenteffects on electron systems of organic ion radicals. The nitrobenzene anion radical has acharacteristic band in its ultraviolet spectrum at 440 nm. The band position is not changedmarkedly with the presence of electron donor substituents (OMe, NR2) in the para positionof the nitrobenzene anion radical. Electron-acceptor groups (p-NO2, p-CN, p-COMe)cause a very strong shift of the band toward larger wavelengths. Analogous effects are char-acteristic of the anion radicals of pyridine and azobenzene (Rao et al. 1972). In ultraviolet
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

spectra, consequently, the influence of electron-donor substituents is practically impercep-tible, whereas electron-acceptor substituents cause strong bathochromic shifts. The ESRspectra, however, fix the influence of both substituent types: In anion radicals of nitro-phenyl derivatives, donor substituents increase the values of the hyperfine coupling con-stants of the nitro group at the nitrogen atom. In contrast, acceptor substituents diminishthese constants (Todres 1981).
Disturbance of the conjugation system of the nitrophenyl anion radical caused by ac-ceptor substituents verges on the formation of quinoid structures. Donor substituents can-not participate in the formation of such structures. As for electron spectroscopy, it is verysensitive to changes in a chromophore system structure. The influence of acceptor groupsis, therefore, stronger than that of donor groups. If changes in chromophore systems are absent, spectrophotometry remains relatively less informative.
4.4.2.B Vibration Spectroscopy
When the solvents used are not masking the bands of the ion radical particles and when theparticles are stable, the infrared spectroscopy may also be employed. It gives some advan-tage in identifying the ion radical structure (by a change in the number of absorption bandsin ion radical spectra or by a different pattern of the band distribution as compared to theinitial neutral molecule). Moreover, vibration spectroscopy can determine the localizationof spin density; i.e., it can answer the key question concerning the structure of ion radicals.For example, infrared (IR) spectra of the metalloporphyrine cation radicals have estab-lished that spin density and positive charge are localized not on the metal (iron) but ratheron the porphyrine ligand (Shinomura et al. 1981).
Vibration spectroscopy is also able to measure the concentration of ion radicals (byestimation of the band intensities). Moreover, the IR intensities of some bands in the fin-gerprint region for organic ion radicals may be much larger than the intensities of the bandsfor the neutral parent molecules. The examples are polycyclic aromatic hydrocarbons orlinear polyenes and their ion radicals. The vibration patterns of the intensity-carryingmodes are closely related to the electronic structure of the ion radicals (Torii et al. 1999 andreferences therein).
4.4.3 Other Physical Methods
4.4.3.A Magnetic Susceptibility
The magnetic susceptibility of paramagnetic particles is used to determine the concentra-tion of ion radicals, but it yields no structural information. The method often demandssolid samples of ion radical salts. Many ion radical salts are unstable in the solid state,and this requirement turns out to be a decisive limit. Fortunately, there are special waysto determine the magnetic susceptibility of paramagnetic particles in solution (Selwood1958). However, instruments for such measurements are only rarely used in chemical lab-oratories. Besides, special devices should be used to conduct investigations at differenttemperatures.
4.4.3.B Mass Spectrometry
The behavior of ion radicals in the mass spectrometer chamber opens up principal venuesfor their alteration. The liquid-phase chemical reaction, however, has many peculiarities,and mass spectrometric methods of ion radical transformation are not inevitably repro-ducible. This is quite evident and needs no further comment.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

4.4.3.C Electrochemical Modeling of Ion Radical Reactions
By the traditional understanding, electrochemistry is a science that considers transforma-tions caused by electrochemical reactions strictly, i.e., by electrode depolarization. Or-ganic depolarizers are involved in electron transfer that, however, may be caused not onlyby electrode discharge but also by simple interactions between donors and acceptors in apool, i.e., by purely chemical reactions. Contemporary organic electrochemistry is widen-ing its scope every day. Accordingly, more and more attempts appear to perform elec-trochemical modeling of ion radical reactions. The differences and similarities betweenpurely chemical and electrochemical (electrode) methods for studying ion radical trans-formations were analyzed in Section 2.4. Therefore, we can now directly enumerate therequirements for accuracy in the electrochemical modeling of organic ion radical reac-tions.
The following approach can be recommended:
1. By means of electrochemical methods and the method of ESR, establish the be-havior of every reaction participant and ascertain the sequence of its transforma-tion, up to the formation of a stable final product.
2. Determine the potentials of reversible one-electron waves for each participant.Comparing the potentials, estimate the probability of the whole ion radical routefor the reaction studied.
3. Based on the data obtained, make an electrochemical model: Use an electrode in-stead of a donor or an acceptor, and employ solutions containing a supportingelectrolyte, another reagent (an acceptor or a donor, correspondingly), and stableproducts that this reagent produces as a result of the ion radical reaction.
4. Draw an analogy between the chemical and the electrochemical reactions. Suchan analogy is correct: (1) if the principal intermediate products and final prod-ucts of the chemical and electrochemical reactions are identical; (2) if a specificinteraction of particles with an electrode material is absent; (3) if both systems(chemical and electrochemical) respond equally to changes in the process con-ditions.
Obviously, the heterogeneous character of the electrochemical process can in somecases lead to essential differences between electrode and homogeneous reaction pathways.Therefore, one needs eventually to verify the results by means of homogeneous donors andacceptors of an electron. In other words, the problem of the correctness of the electro-chemical modeling should be analyzed for each reaction anew and at the same time bechecked chemically, i.e., in purely liquid-phase conditions.
4.5 EXAMPLES OF COMPLEX APPROACHES TO THEDISCERNMENT OF THE ION RADICAL MECHANISM OFPARTICULAR REACTIONS
4.5.1 Oxidative Polymerization of Aniline
As a conducting polymer, polyaniline has many electronics-related applications, such asrechargeable batteries (Tsutsumi et al. 1995), multilayer heterostructure light-emittingdiode devices (Onoda & Yoshino 1995), biosensors (Bartlett & Whitaker 1987), elec-trochromic windows (Nguyen & Dao 1989), and nonlinear optical materials (Papacostadi-nou & Theophilou 1991). Polyaniline may be prepared from aniline by both electrochemi-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

cal and chemical methods. The chemical method is considered more useful for mass pro-duction than the electrochemical method.
Although polyaniline synthesis has gained commercial importance, its polymeriza-tion mechanism is a subject of debate. Wei and co-authors (1989, 1990) as well as Ding etal. (1999) carried out the oxidative polymerization of aniline in aqueous acidic solutions,adding ammonium persulfate at 0°C and trapping agents. Ding et al. (1999) used a varietyof organic compounds as traps. For instance, hindered phenols and electron-rich alkenes in-hibited the polymerization, being traps for cation radicals. All of the results obtained haveled to a cation radical polymerization mechanism for aniline, in which the polymerizationis a chain growth reaction through the combination of a polymeric cation radical and an ani-line cation radical. This mechanism in the main resembles the one discussed by Percec andHill (1996). The formation of the aniline cation radicals and their dimerization are the ini-tial steps of the polymerization (Scheme 4-23).
The attack of the monomeric cation radical of aniline on the oligomeric amine cationradical leads to the chain growth. The chain growth may also proceed as copolymerizationof the two oligomeric species (Scheme 4-24).
4.5.2 Reactions of Hydroperoxides with Phosphites and Sulfides
It is well known that phosphites or sulfides added to stabilizers of polymeric materials con-siderably enhance the stabilizing effects. These additives decompose hydroperoxides ac-cording to the following equations:
R1MOOH � (R2O)3P → (R2O)3PBO � R1MOH
R1MOOH � R2R3S → R2R3SBO � R1MOH
SCHEME 4-23
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

While investigating the mechanism, Pobedimskii and Buchachenko (1968a, 1968b)concluded that these reactions have an ion radical nature and consist of electron transferfrom phosphites or sulfides (denoted further as D) to hydroperoxides according to the fol-lowing scheme:
R1MOOH � D → [(R1MOOH)�., D�.
] → [R1MO., �OH, D�.
]
The reaction products of the scheme depicted are retained in the solvent cage. Benzene wasused as a solvent in the experiments. The cage complex [R1�O
., �OH, D�.
] decays eitherupon disproportionation in the cage or upon dissociation. Disproportionaton leads to thephosphinoxides or sulfoxides mentioned earlier. Dissociation results in the passage of theradicals out of the cage into the solvent pool.
In order to check for the possibility of primary electron transfer, hydroperoxides wereallowed to react with the donors that contained the stabilizing groups, namely, 4,4-di(ani-syl) sulfide or 1,2-dihydroxyphenyl-bis[2,4,6-tri(tert-butyl)phenyl]phosphite. The ESRmethod revealed the formation of corresponding cation radicals.
The radical trap 2,2,6,6-tetramethylpiperidine-1-oxyl (RNO.) decayed when intro-
duced into the reaction mixture. The rate of the RNO.decay was determined by ESR spec-
SCHEME 4-24
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

troscopy, while the rate of the ROOH consumption in the reaction with an additive was dis-covered through polarography (by evaluating the residual part of ROOH). The rate con-stants for both processes were proven to be practically the same. According to the authors,this decay proceeds only at the expense of the ion radical reaction. [It is worthwhile noting,however, that another trap, 5,5-dimethyl-2-phenyl-1-pyrroline-N-oxide, reacts in benzenewith perbenzoic or perpropionic acids to produce a significant amount of aminoxy radicalsas a result of the addition of a peracid to the trap (Sang et al. 1996).] In the Buchachenkopapers cited, the kinetics of decay fully correspond to those of the bimolecular reaction(ROOH � D). The radicals formed are extremely active, and the rate of RNO
.consump-
tion is almost independent of its concentration in solution. In the presence of oxygen, how-ever, the rate of RNO
.consumption markedly decreases (when [O2]0 �� [RNO
.]0). This is
explained by the fact that oxygen converts a significant amount of active radicals into per-oxide radicals, which do not react with RNO
.. The rate of hydroperoxide consumption is
independent of whether the foreign radical RNO.
is introduced into the system or not.Hence, the reaction is not a chain reaction. When the reaction is conducted in alcohol di-luted with H2
18O, the donor (phosphite) and hydroperoxide produce phosphate and alcoholnot bearing the label. This means that the reaction considered either does not produce free�OH ions or does not exchange them with the medium. According to the ion radical equa-tion, the isotope exchange should not take place if the ion radical complex monomolecu-larly disproportionates. The rate constant of disproportionation is independent of solventviscosity, whereas the rate constant of RNO
.consumption decreases as the viscosity of the
medium rises. This decrease corresponds to the Stokes–Einstein law. This is typical for re-actions occurring in the solvent cage.
The next step was to replace benzene as a solvent by styrene or methylmethacrylate.These solvents contain the ethylenic bond, which is attractive for the radicals. This gener-ates a driving force for the radicals for leaving the solvent cage. The general rate of the pro-cess in the vinyl-containing solvents remains the same as that in benzene, while the rate ofRNO
.consumption increases. Naturally, more radicals leave the cage and pass into the pool
due to their affinity for the solvent molecules. So the inference follows that the reaction pro-ceeds by a radical mechanism. However, the amount of radicals leaving the cage is smallbecause they disproportionate inside the cage at such a high rate that even the rate of radi-cal addition to the ethylenic bond cannot compete with it.
This inference can be checked stereochemically. If hydroperoxide produces alcoholat the expense of disproportionation of the radicals not leaving the cage, the enantiomerichydroperoxide should give the alcohol that retains its optical activity. And this is actuallywhat takes place (Davies & Feld 1958).
The material considered here allows us to understand the influence of sterically hin-dered amines, which act as stabilizers against the light-induced degradation of polyolefines.The sterically hindered amines easily undergo oxidation after cation radical formation:
ROO.� �NH → [ROO� � �NH�.
] → ROOH � �N.
This equation seems to be a key reaction point in the antioxidant action of these amines:�N
.radicals in the presence of oxygen are transformed via peroxy radical intermediates
into nitroxy radicals. The nitroxy radicals are very persistent and react efficiently with rad-icals produced on polyolefine degradation. Such radical interception blocks the chain inradical oxidation and therefore causes the actual antioxidant phenomenon of sterically hin-dered amines (Brede et al. 1998).
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

4.5.3 ter Meer Reaction
The ter Meer reaction consists of the production of 1,1-dinitro compounds from 1-halo-1-nitroalkanes. The reaction proceeds under the action of alkali metal nitrites in a basicmedium:
Hal NO2
RCHMNO2 � NO2
� � :B → Hal� � HB � RCMNO2
This reaction is used to synthesize 1,1-dinitroalkanes, which find wide application as in-termediate products in preparing drugs, biologically active substances, and high-energycompositions.
It had been established several decades ago that the reaction of 1-chloro-1-ni-troethane with sodium nitrite in aqueous-alcohol medium is second order overall and firstorder in each reactant (Hawthorne 1956). 1-Deutero-1-chloro-1-nitroethane reacts moreslowly than its lighter isotopomer. This means that the kinetic isotopic effect is observed.The reaction proceeds only in moderately alkaline media; in strongly alkaline media it doesnot take place. Only those geminal halo nitro compounds, which carry hydrogen in thegeminal position, can undergo the conversion. Based on these facts, Hawthorne (1956) sug-gested the SN2 substitution preceded by the isomerization of the initial substrate into theaci-nitro form:
Cl Cl
MeCHMNO2 � NO2�DMeCBNOO� � HNO2D
Cl NO2
DMeCBNOOH � NO2
� �Cl�→ MeCBNOOH D
NO2
DMeC� � H�
NO2
Recent research into the ter Meer reaction (Shugalei & Tselinskii 1994) has demon-strated that it actually chooses the chain ion radical mechanism. Chain branching is at-tributed to air oxygen, after its transformation in the superoxide ion (O2
�.; see Section
1.7.1). The whole process of substitution in the aqueous-alkaline buffer medium is ex-pressed by a 14-step sequence:
Cl Cl
1. MeCHMNO2 � �OH → MeCBNOO� � H2O
Cl Cl Cl
2. MeCBNOO� � MeCHMNO2 → MeC.� MeCH� � Cl�
NO2 NO2
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Cl Cl
3. MeC.� NO2
� → �MeCMNO2��.
NO2 NO2
Cl NO2
4. �MeCMNO2�
�.
→ MeC.� Cl�
NO2 NO2
NO2 Cl NO2 Cl
5. MeC.� MeCBNOO� → MeC� � MeC
.etc.
NO2 NO2 NO2
NO2
6. MeC� � H� →
NO2
Cl �.Cl
7. MeCMNO2 � O2 → MeCMNO2 � O2
�.
�NO2
�NO2
8. O2�.
� H2OD HO.2 � �OH
9. HO.2 � NO2
� → HO2� �
.NO2
10. HO2� � H�D H2O2
11. H2O2 � NO2� → .
OH � �OH �.NO2
12. MeCH.�
.NO2 →
NO2
Cl OH
13. MeCMNO2 � �OH → MeCMNO2 �H�
→ MeCHO � 2 HNO2
NO2 NO2
NO2
MeCH
NO2
NO2
MeCH
NO2
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

ONO
14. MeCH.�
.NO2 → MeCH → MeCHO � N2O3
NO2 NO2
The 14-step scheme takes into account the data obtained by Hawthorne (1956) andaccords well with later results. The mechanism depicted has been supported as follows. Ina moderately alkaline medium, the substrate ionization at stage 1 completely governs thekinetics of the reaction (Shugalei et al. 1978). This agrees with the kinetic isotopic effectand the essential presence of the hydrogen atom in the geminal node. For a chain ion radi-cal process to originate at stage 2, the reaction mixture should contain both the neutral sub-strate and the corresponding anion. This explains why the ter Meer reaction does not occurwith excess alkali: All the initial molecules convert into anions, and electron exchange be-comes impossible because there is no neutral substrate—an acceptor of an electron—in thereaction mixture. It has been revealed that the lack of alkali also decelerates the conversion:In the acetate-buffer solution the rate of the process drops and kinetic characteristics ceaseto obey the chain process laws. Under these conditions the reaction remains a radical one,and the introduction of 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl (ROHNO
.) inhibits
the conversion. The aci-nitro anion is required not only for the origin (stage 2) but also forthe development of the chain process (stage 5).
The presence of halonitro compounds, which are incapable of producing carbanion,such as 2-chloro-2-nitropropane and trichloronitromethane, considerably increases the rate ofchain origin in an alkaline medium, pH � 8.0. These compounds accept electrons instead ofnonionized 1-chloro-1-nitroethane, the concentration of which is small at a high alkalinity.
The chain propagation (stage 3) involving the addition of the 1-chloro-1-nitroethylradical to the nucleophilic nitrite ion has been supported by a number of works devoted toradical interaction with anions; see, for example, Chawla and Fessenden (1975). Stage 4presupposes that the anion radical of 1-chloro-1,1-dinitroethane is only slightly stable andthat it decomposes into the radical and the anion. This agrees with the results of investiga-tions of the polarographic behavior of geminal halonitroalkanes conducted with the helpESR spectroscopy (Shapiro et al. 1969). And finally, stage 5 of one-electron oxidation ofaci-nitro anion by the 1,1-dinitroethyl radical is a well-known process. It regenerates themain particle, the chloronitroethyl radical, which acts as an initial species for the new chain.
In a moderately alkaline medium, the ter Meer reaction proceeds through a consider-able induction period; the kinetic curves are S-shaped. Peroxide compounds and ultravio-let irradiation accelerate the process (Bazanov et al. 1978). Radical traps inhibit the reac-tion; this was discussed earlier. This indicates the radical nature of the process. The rate offormation of active radical centers obeys the second-order equation in the total concentra-tion of chloronitroethane introduced into the reaction. With respect to the nonionized sub-strate and the anion conjugated with it, the reaction is first-order one. The rate of the wholereaction is independent of nitrite concentration.
The superoxide ion or its protonated form (hydroperoxy radical HO.2) is produced in
the system (Shugalei & Tselinskii 1993, 1994). Hydroquinone, which is known to interacteffectively with the superoxide ion (Afanas’ev & Polozova 1978), exerts a fairly strong in-hibiting effect on the reaction. Addition to the system of potassium ferricyanide has, in con-trast, an accelerating effect. The cause of the effect consists in the transformation of the fol-lowing type: Fe3� � HO2
� → Fe2� � HO..
The chain ion radical mechanism of the ter Meer reaction has been supported by a thor-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

ough kinetic analysis. The reaction is well described by the standard equation of chain rad-ical processes (with square-law chain termination) (Shugalei et al. 1981). This mechanismalso explains the nature of the side products: aldehydes (see steps 13 and 14) as well as vic-inal dinitroethylenes. The following scheme explains the formation of vic-dinitroethylenes:
Cl Cl Cl Cl �.
RCBNOO� � RC
. → �RCMMCR � �Cl�
→
.NO2 NO2 NO2
Cl
→ RC.MMCR
�Cl�→ RCBBCR
NO2 NO2 NO2 NO2
The data of kinetics of parallel reactions permitted Shugalei and co-authors (1981) tocalculate the rate constants for competing pathways, which are essentially the constants ofthe conversion selectivity. The analysis of the constant allowed the scientists to formulatethe optimal conditions of the ter Meer synthesis of 1,1-dinitroalkanes.
They suggested conducting the reaction at concentrations of the initial reagents, 1-halo-1-nitroalkane and sodium nitrite, exceeding 1 mol�L�1. Then, because of the low sol-ubility of molecular oxygen in water (about 10�4 mol�L�1), the presence of oxygen wouldnot affect the yield of the target product. An increase in concentration of the nitrite ion pro-motes the ter Meer reaction.
Increasing the concentration of alkali up to a certain limit also accelerates the reac-tion; above this limit, however, alkali has an adverse effect on the reaction. Therefore, theoptimal concentration of alkali should be determined in each particular case. Bazanov andothers (1980a,b) suggested the following solution to this problem: They recommended us-ing a strongly alkaline medium and sodium persulfate. 1-Halo-1-nitroethane does not reactwith sodium nitrite in 0.01 N aqueous sodium hydroxides (the substrate converts into theanion entirely, and the system has no electron acceptor). The persulfate dianion performsthe acceptor function (see Section 1.7.7), and 1,1-dinitroethane forms in 80–90% yield. Asknown (Pagano & Shechter 1970), the persulfate dianion oxidizes nitro carbanions to thenitro alkyl radicals. The chain ion radical nature of the reaction involving persulfate wasproved with the help of the stable radical ROHNO
., as described earlier. The rate of chain
origin in the “persulfate” version of the ter Meer reaction depends on the concentrations ofhalonitroethane, nitrite, and persulfate. Changing the concentration of hydroxide and re-moving molecular oxygen from the reaction mixture do not really affect the rate of chaininitiation. It has been established (Bazanov et al. 1980a, 1980b; Shugalei et al. 1981) thatthe persulfate dianion initiates chains while oxidizing both aci-nitro anions and nitrite, seeSteps 1–10 of the following scheme:
1. S2O82� → 2 SO4
�.
X X
2. SO4�.
� MeCBNOO� → MeC.� SO4
2�
NO2
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

3. SO4�.
� NO2� → .
NO2 � SO42�
X X �.
4. MeC
.� NO2
� → �MeCMNO2 � NO2 NO2
X �.NO2
5. �MeCMNO2� → MeC
.� X�
NO2 NO2
NO2 X NO2 X
6. MeC.� MeCBNOO� → MeC� � MeC
.etc.
NO2 NO2 NO2
Cl Cl Cl
7. MeCBNOO� � MeCHMNO2 → MeC.� MeCH
.� Cl�
NO2 NO2
8. MeCH.�
.NO2 →
NO2
NO2
9. MeC� � H� →
NO2
X X X X
10. MeC.�
.CMe → MeCMMC Me
NO2 NO2 NO2 NO2
X � Cl, Br, I
Step 2 represents the chain origin; step 3 is nitrite ion oxidation; steps 4–6 are thechain propagation; and steps 8 and 10 show chain termination at the expense of radical
NO2
MeCH
NO2
NO2
MeCH
NO2
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

dimerization. Steps 4 and 5 can probably be united into one stage:
X Me NO2
MeC
.� NO2
� → X�C�NO2� → X� � MeC
.
NO2 NO2 NO2
X � Cl, Br, I
The anion radical of 1-halo-1,1-dinitroethane is not a kinetically independent parti-cle, because of its instability (Shapiro et al. 1969). Therefore, the addition of the nitrite ionto the halonitro alkyl radical and the decomposition of the anion radical of 1-halo-1,1-dini-troethane may proceed in one stage, as shown earlier. As follows from this scheme, the ef-fect of the leaving group—the halide ion—depends on the halogen’s affinity to electronsand the energy of disruption of the carbon–halogen bond. The difference between these twoenergies increases in the series chlorine, bromine, iodine derivatives (10; 14.7; 60kJ�mol�1, respectively). The coupling-cleaving merged scheme obviously demonstratesthat 1-fluoro-1-nitroethane is incapable of entering into the ter Meer reaction: the C–F bondin the anion radicals of fluoronitro alkanes is extremely stable (Shapiro et al. 1969). Thisexcludes the possibility of fluoride ion cleavage. The difference between the fluorine affin-ity to electrons and the energy of the CMF bond disruption is �127.5 kJ�mol�1 (Bazanovet al. 1980b, p. 910).
The revealed mechanism of the ter Meer reaction is rather illustrative. It helps us un-derstand the peculiarities of the nucleophilic substitution reactions having the chain ionradical mechanism and involving the interaction of radicals with anions at the chain prop-agation steps. The example also demonstrates how the knowledge of kinetics and the mech-anism can be used to find new ways of initiating and optimizing important practical reac-tions. The ter Meer reaction turns out to be a reaction having one name and one mechanism.This differs from, say, aromatic nitration, which has one name but different mechanisms.
4.5.4 Aromatic Nitration
In the past two decades there has been an increasing recognition that ion radicals play a veryimportant role in many organic reactions. Eventually, a situation has arisen where, for prac-tically every reaction between a donor and an acceptor, an ion radical mechanism has to becarefully considered in addition to the classical polar pathway. The very subject of thisbook directs attention to cases when ion radical formation is the obvious effect in play.
Aromatic nitration is the prime example of an established mechanism. A substantialbody of data concerning reaction kinetics, structure–reactivity relationships, and chemi-cally induced dynamic nuclear polarization (CIDNP) has permitted a thorough under-standing of the electron-transfer step in aromatic nitration. The following review materialscan be addressed: Todres 1985; Morkovnik 1988; Ridd 1991, 1998; Eberson et al. 1994,1995; Kochi 1990, 1992; Cardoso & Mesquita Caneiro 2001; and the monograph by Olahand co-authors of 1989. In principle, the general mechanism of nitration can include the fol-lowing distinct steps:
1. Generation of the electrophile
2H2SO4 � HNO3 ⇔ NO2� � 2HSO4
� � H3O�
or
HNO3 ⇔ NO2� � NO3
� � H2O
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

2. Attack on the aromatic ring
NO2� � ArH → Ar�(H)NO2 (� complex)
or
NO2� � ArH → .
NO2, ArH�. → Ar�(H)NO2 (� complex)
3. Deprotonation
Ar�(H)NO2 (� complex) → ArNO2 � H�
According to present knowledge, the one-electron transfer of stage 2 (the outer-sphere transfer) is hardly probable from the energetic point of view (see Chapter 1). Mean-while, cation radical formation frequently takes place at aromatic nitration (Morkovnik1988). Positional selectivity depends on spin-density distributions in these cation radicals.In principle, the attack of the
.NO2 radical is probably at the position of the aromatic cation
radical, which bears the maximal spin density.For instance, nitration of naphthalene, azulene, biphenylene, and triphenylene pro-
ceeds preferentially in positions with the greatest constant of hyperfine splitting at the hy-drogen atom in ESR spectra of the corresponding cation radicals. The constant is known tobe proportional to the spin density on the carbon atom bearing the mentioned hydrogen. Itis important, however, that the same orientation is also observed in the “classical” mecha-nism of nitration in the cases of naphthalene, azulene, and biphenylene but not of triph-enylene (see Todres 1985).
According to calculations (Dewar et al. 1956), triphenylene itself has position 2 asmost reactive for “classical” reactions. For its cation radical, the most reactive is position 1at the C atom, alongside with the same position 2 (Perrin 1977). These predictions and ex-periments are in line with triphenylene cation radical participation in nitration (Baker et al.1955).
For aromatic electrophilic substitution, positional selectivity is defined in terms ofthe ability of one atom in an aromatic ring to give the most stable bond upon �-complexformation. In the dibenzofuran case, calculations predict C-2 � complex to be more stablethan C-3 � complex by 7.25 kJ for nitration and by 12.5 kJ for acetylation. Experimentaldata are in accord with this estimation only for acetylation: 2 �� 3 � 4 � 1 (MeCOClreagent, AlCl3 catalyst, CH2Cl2 solvent, 0°C reaction temperature). As for nitration, posi-tion 3 turns out to be the most active: 3 �� 2 � 1 � 4 (99% HNO3, CH2Cl2, �45°C). Thepreferential nitration in position 3 is seemingly not accidental. The maximal spin density islocalized in just the same position of the dibenzofuran cation radical. The authors of thecited work conclude that acetylation follows the “classical” scheme, and the nitration pro-ceeds through cation radical formation (Keumi et al. 1988).
On one hand, there is a strong presumption against the cation radical mechanism of�-complex formation (see Eberson et al. 1994). On the other hand, there is a strong corre-lation between selectivity in nitration and spin distribution in substrate cation radicals.There were attempts (see, e.g., Morkovnik 1988) to reconcile such a contradiction bymeans of the following mechanism. The electron transfer is in fact an inner-sphere trans-fer, in the framework of a � complex. In this case the energetic restrictions are removed.The � complex gives a substrate cation radical and
.NO2 radical. These species form a new
complex (�) that is isomeric to the � complex with respect to a position of the nitro group.The � complex expels a proton and gives the final product:
NO2� � ArH → Ar�(H)NO2 (� complex) → .
NO2, ArH�. →Ar�(H)NO2 (� complex) → ArNO2 � H�
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

This mechanism seems to imply that if some substitution occurs at the “classical” po-sition of attack (the first � complex), then such a substitution should show the deuterium iso-tope effect for proton loss from this position. The deuterium effect is absent in the majorityof nitration cases, except for nitration in sterically shielded positions (Schoffield 1980). Per-haps a systematic investigation of kinetic isotopic effects would be useful in a much widerrange of substrates in comparison with their ionization potentials and nitration conditions.
Feng and others (1986) have performed quantum chemical calculations of aromaticnitration. The results they obtained were in good accord with the ionization potentials of.NO2 and benzene and its derivatives. The radical pair recombination mechanism is favoredfor nitration whenever the ionization potential of the aromatic is much less than that of.NO2. According to the calculations, nitration of toluene and xylene with NO2
� most prob-ably proceeds according to the ion radical mechanism. Nitration of nitrobenzene and otherbenzene derivatives with electroacceptor substituents can proceed through the “classical”polar mechanism only. As for benzene itself, both mechanisms (ion radical and polar) arepossible.
Substituents or reaction conditions that raise the ionization potential of an aromaticmolecule to a value higher than that of
.NO2 prevent formation of this radical pair (one-
electron transfer appears to be forbidden). This forces the “classical” mechanism. As for re-action conditions, a solvent plays the decisive role in nitration.
In comparison with gas-phase conditions, the calculated ionization potential of .NO2
in a solvent with a dielectric strength of 78.5 (such as that in water) is reduced significantly.This diminution is stronger than that of either benzene or toluene (Feng et al. 1986). This isa consequence of the fact that NO2
� ion is preferentially stabilized in dielectric media. Thenature of the solvent is, therefore, quite important in deciding which mechanism is operative.
Examples of the multiplicity of nitration mechanisms that depend on the ionizationpotentials of substrates are the nitration of naphthalene (NaphH � NO2
� scheme) and ofperylene (PerH � NO2
� scheme) (Scheme 4-25).In the cases of both NaphH � NO2
� and PerH � NO2�, the transition state of the het-
erolytic reaction lies energetically lower than the transition state of the electron-transfer re-action. However, the ionization potential of perylene is significantly less than that of naph-thalene. Therefore, the PerH� cation radical has the smaller fund of energy than the NaphH�
cation radical. In other words, the products of the reaction PerH � NO2� → PerH� �
.NO2
find themselves in an energetically permitted zone (which is lower than that of the initiallevel). Meanwhile, the products of the reaction NaphH � NO2
� → NaphH� �.NO2 find
themselves in an energetically forbidden zone (which is higher than that of the initial level).Neutral perylene reacts with NO2
�, giving the cation radical. However, its formationis, in principle, a result of �-complex splitting. Another possible route of �-complex split-ting consists of proton elimination and nitro perylene formation. As experiments show, thenitration of perylene is accompanied by collateral reactions of PerH�.
, such as recombina-tion and interaction with solvent molecules (Eberson & Radner 1985). This testifies to therelease of cation radical.
According to Scheme 25, the NaphH�.cation radical cannot be formed as a result of
the interaction of NaphH with NO2�. In this sense, Ridd’s work is relevant (Johnston et al.
1989, 1991). The work shows that, to some extent, the nitration of naphthalene can indeedproceed through an outer-sphere electron transfer. Naphthalene, durene, and mesitylenewere compared in their reactions with H15NO3. The reactions were performed in a solventcontaining trifluoroacetic acid (49%), nitromethane (50%), and water (1%) (weight-to-weight). The reaction mixture also contained some sodium azide and methanesulphonic
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

acid. Sodium azide is needed to stop the nitrous acid–catalyzed nitration (see later), and themethanesulphonic acid is used to bring the reaction rate to the required value. The role of ni-tromethane deserves a special explanation. According to electrochemical data (Boughriet &Wartel 1989), log K for the reaction NaphH � NO2
� ⇔ NaphH�.� NO2 has the following
values in different solvents: �8.67 (acetonitrile); 0.8 (sulfolane); 6.75 (nitromethane).These results imply that the boundary between the classical and electron-transfer mecha-nisms of nitration is likely to be very dependent on the solvent used, with nitromethane fa-voring the electron-transfer process.
The aforementioned reaction was monitored by NMR (15N). Under these conditions,nitromesitylene showed no effect of nuclear polarization. Nitronaphthalene revealed aslightly but trustworthily enhanced absorption of the 15N signal. Because the conditions ofnitration were strictly the same for both substrates, artifacts from the solvent or other com-ponents of the mixture were excluded. The observed enhanced absorption could not becaused by the catalytic influence of HNO2. When perdeuterionaphthalene was used as a sub-strate, the nitro product obtained gave no further enhancement of the NMR absorption. Theauthors concluded that a small part (a few percent) of the reaction of naphthalene with thenitronium ion does involve direct electron transfer between the reactants before the forma-tion of � complex. Under the experimental conditions, an encounter, or � complex (a pre-decessor to � complex), is distinguished by such a strong interaction among its componentsthat bending of the nitronium cation occurs at even this initial stage. Some rather small partof the encounter complex gives electron-transfer products, i.e., naphthalene cation radicaland nitrogen dioxide. The latter form the polarized nitronaphthalene after recombination.Hence, one cannot “completely rule out some contribution from electron transfer in the ni-tration of naphthalene, particularly if the initial �-complex interaction is considered to besufficient to treat the electron transfer as an inner-sphere reaction” (Ridd 1991).
SCHEME 4-25
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The reaction of durene with H15NO3 under the conditions just mentioned is anotherrather important case. The reaction is accompanied by strongly enhanced absorption in theNMR (15N) spectrum with respect to a signal belonging to the product, i.e., nitrodurene.Durene and naphthalene have very similar standard potentials (respectively, 2.07 V and2.08 V, in acetonitrile; see Ridd 1991). One significant difference between them is that,with durene, much of the nitration supposedly arises from ipso attack followed by rear-rangement (Scheme 4-26).
The CIDNP studies show that a significant part of the overall reaction can involve thecorresponding durene cation radical.
It is reasonable that the radical cation should be formed in the rearrangementstage rather than in the initial substitution, for the different geometry of the nitro-gen dioxide (bent) and the nitronium ion (linear) lead to a high reorganization en-ergy for electron transfer to the nitronium ion. Since the OMNMO bond in the ni-tro group is bent, the change in geometry is less when the nitrogen dioxidemolecule is formed by homolysis of a CMNO2 bond. Of course, the direct reac-tion [the first step of the preceding scheme] also involves a major change in thegeometry of the OMNMO group, but the energy terms involved in bond forma-tion should then stabilize the transition state (Ridd 1998).
When considering aromatic nitration, it seems reasonable to examine other nitratingagents (besides the nitrating mixture leading to formation of the nitronium ion) and to out-line the scope of the ion radical mechanism in each case.
SCHEME 4-26
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

4.5.4.A System of HNO3, H2SO4 with Catalytic Amounts of HNO2
The mixture of HNO3 and H2SO4 is a classical tool for aromatic nitration. However, thepresence of nitrous acid is sometimes necessary. For instance, naphthalene-1,3,5-trisul-fonic acid gives 8-nitro-naphthalene-1,3,5-trisulfonic acid upon reaction with a technicalnitrating mixture. However, this starting trisulfonic acid remains unchanged if pure sul-furic acid and nitric acid that is free of nitrogen oxides are employed (Ufimtsev 1983).The addition of NaNO2 is needed to nitrate naphthalene with nitric acid in sulfuric acidof 56% concentration (Ross et al. 1983). Nitrous acid has a catalytic effect on the nitra-tion of activated aromatic compounds. The typical examples refer to aniline, N,N-di-alkylanilines, phenols, anisole, mesitylene (Brickman & Ridd 1965; Giffney & Ridd1979, Gorelik and others 1995). The role of nitrous acid was initially interpreted as “ni-tration through nitrosation” when nitric acid oxidizes a primary formed nitroso com-pound. Such a mechanism is presently admitted for the nitration of phenols and N,N-di-alkyl aromatic amines. Thus, in the nitrous acid–catalyzed nitration of anisole in 43–47%sulfuric acid (Dix & Moodie 1986), the intermediate p-nitrosoanisole undergoesdemethylation to give p-nitrosophenol. The latter gives rise to p-nitrophenol as the pre-dominant final product.
As to anisole itself, nitration in the mixture of nitric, sulfuric, and acetic acids at roomtemperature leads to 2-nitroanisole (25%) and 4-nitroanisole (65%), with some by-productsof unidentified structures. In the presence of sodium azide as a scavenger for nitrous acid(which is usually contained in nitric acid), no reaction takes place. So the process proceedsthrough a nitrous acid–catalyzed reaction (see later). During the nitration of anisole with15N-enriched nitric acid in the sulfuric and acetic acid mixture, the (15N)-NMR signals of2- and 4-nitroanisole exhibit emissions indicating their formation by recombination of theanisole cation radical with the nitrogen dioxide radical. It is concluded from the magnitudeof the (15N)-CIDNP effect that 2-nitroanisole is formed only via coupling of the ion radi-cal with the radical, whereas 3-nitroanisole may also be formed via a nonradical pathwaythrough a direct attack of the nitronium cation on neutral anisole. The probability of the lat-ter pathway is estimated to be 60% (Lehnig 1997).
Aromatic N,N-dialkylamines react rapidly with HNO2 and undergo ring nitration andnitrosative dealkylation; both reactions are linked through the formation of a nitrosammo-nium ion R1R2
2N�MNBO (R1 � Ar, R2 � Alk). This nitorosoammonium ion then under-goes reversible homolysis to NO and a cation radical (Loeppky et al. 1998).
Nitrous acid catalysis is also exhibited in the nitration of compounds (naphthalene)that are unable to undergo nitrosation under the given conditions or whose nitrosation pro-ceeds more slowly than nitration. As accepted, the nitrosonium ion is formed from HNO2
in acid media. The nitrosonium ion oxidizes an aromatic substrate into a cation radical, giv-ing nitric oxide. The latter reduces nitronium cation to nitrogen dioxide, which gives a �complex with the aromatic cation radical:
HONO � H� → H2O � NO�
ArH � NO� → ArH�.� NO
.
(NO.� NO2
� → NO� � NO.2)
ArH�.� NO
.2 → Ar�(H)NO2 → H� � ArNO2
The third reaction is recommended (Ridd 1998) to be put in parentheses to imply thatit illustrates the stoichiometry of the process, not the mechanism. The mechanism must be
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

more complex because the rate of these nitrous acid–catalyzed nitrations can greatly exceedthe rate of formation of nitronium ions in the solution.
The above mentioned sequence of mechanistic steps has been experimentally proven fornitration of a wide range of organic compounds (Ridd 1991; Ridd & Sandall 1981; Clemens,Ridd, & Sandall 1984, 1985; Clemens, Helsby, Ridd, et al. 1985; Lehnig 1996, 1997; Giffney& Ridd 1979; Powell et al. 1990). Some examples deserve special consideration.
A kinetic study of nitrous acid–catalyzed nitration of naphthalene with an excess ofnitric acid in an aqueous mixture of sulfuric and acetic acids (Leis et al. 1988) showed atransition from first-order to second-order kinetics with respect to naphthalene. (At thatacidity, the rate of reaction through the nitronium ion was too slow to be significant; theamount of nitrous acid was sufficient to make one-electron oxidation of naphthalene themain reaction path.) The reaction that initially had the first order respecting naphthalene be-comes the second-order reaction. The electron transfer from naphthalene to NO� has anequilibrium (reversible) character. At an excess of the substrate, the equilibrium shifts tothe right. A cause of the shift is stabilization of the cation radical by uncharged naphtha-lene. The stabilized cation radical dimer (NaphH)2
�.is just involved in nitration:
NaphH � NaphH � NO� ⇔ (NaphH)2�.
� NO.
(NO.� NO2
� ⇔ NO� � NO.2)
(NaphH)2�.
� NO.2 → NaphNO2 � NaphH � H�
Therefore, one-electron oxidation of naphthalene by NO� is the rate-determiningstage at low naphthalene concentrations (⇔ means equilibrium of this oxidation). At highnaphthalene concentrations, the rate of the process no longer depends on the rate of accu-mulation of the cation radical species. In this case the rate depends on recombination of thespecies with NO2 radical. The authors point out that “for many of the more reactive aro-matic compounds, reaction paths involving electron transfer in nitration will become moreimportant as the concentration of the aromatic compound is increased, irrespective of theconcentration of the species accepting the electron” (Leis et al. 1988).
Nitration of mesitylene by means of H15NO3 in the mixture of CF3COOH with 10%H2O yields to formation of nitro mesitylene with no nuclear polarization (Clemens, Helsby,Ridd, et al. 1985). However, the same product with very strong 15N polarization can be ob-tained after addition of nitrous acid. Nitrosonium cation oxidizes mesitylene; then the ionradical pair ArH�.
NO2 is formed. There is evidence (Eberson & Radner 1980) that the re-action of the naphthalene cation radical with nitrogen dioxide is slower than a diffusion-controlled process. Hence, ArH�.
NO.2 is formed at the expense of components coming up
to each other, and the pair sometimes moves independently. Without such separation of theion radical pair, no nuclear polarization effect can be observed. This is an appropriate mo-ment to emphasize that when these pairs have already been formed, they (or some part ofthem) do not pass immediately on to form the nitro compound (Ridd 1991). The mesitylenecation radical, which exists as an intermediate in the reaction under consideration, gives by-products, too: methyl derivatives of nitro diphenylmethane and compounds bearing the ni-tro group in the side chain (Clemens, Helsby, Ridd, et al. 1985).
4.5.4.B System of HNO3, (CH3CO)2O
In this case, nitration takes place in solutions prepared by dissolving nitric acid in acetic an-hydride. Acetyl nitrate is formed in these solutions:
HNO3 � (CH3CO)2O → CH3COONO2 � CH3COOH
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Such solutions are very potent nitrating mixtures and effect nitrations at higher rates thansolutions of nitric acid in inert organic solvents. There are scarce data on the nature of ni-trating species formed from acetylnitrate in the presence of aromatics. The data existingpermit one to conclude that cation radicals of xylenes play a role in the nitration.
For the nitration of aromatic hydrocarbons with acetylnitrate, there is a clearly linearcorrelation between the ionization potentials of these hydrocarbons and the rate constantsrelative to benzene (Pedersen and others 1973). Table 4-4 juxtaposes spin densities of thecation radicals and the partial rate factors of ring attacks in the case of the nitration of iso-meric xylenes by means of the (nitric acid–acetic anhydride) mixture.
Table 4-4 helps us understand why 1,4- and 1,2-xylenes undergo the substitution inthe ipso positions (the positions where the methyl groups are located). The electrophilicreagents obviously have to attack the substrate at the position with maximal electron den-sity. These are the ipso positions for the cation radicals of the 1,4- and 1,2-isomers. (For theneutral isomers, these are positions where the methyl groups are absent!) The substitutionis directed into the ipso positions of 1,4-xylene up to 76% and into the ring for 24% only.In the case of 1,2-xylene, the substitution takes place for 60% at the ipso positions and for40% in the ring. In the 1,3-xylene cation radical, spin density is maximal in the nonmethy-lated positions 4 and 6, which are involved in the substitution for 84% (Fischer & Wright1974).
4.5.4.C System of NaNO2, CF3SO3H
The title system in acetonitrile forms a homogeneous solution. The generation of NO�
cation takes place. As already known, NO� is a remarkable, diverse reagent, not only fornitrosation and nitration but also for oxidation. Kochi and co-workers recently christened anew general mechanism oxidative aromatic substitution to describe aromatic substitutionreactions (Kochi 1990; Bosch & Kochi 1994). The mechanism incorporates ground-stateelectron transfer prior to the substitution step (see also Skokov & Wheeler 1999).
Tanaka and others (1996, 2000) studied the behavior of a series of naphthalene deriva-tives in acetonitrile solution containing NaNO2 and CF3SO3H at 0°C in air. Naphthaleneshowed very low reactivity, and most of that starting material was recovered after the reac-tion. In the case of 1-methylnaphthalene, a coupling reaction took place to produce 4,4-dimethyl-1,1-binaphthyl in 97% yield alongside mononitro derivatives of that dimer in1.5% yield. However, when the reaction was carried out under the same conditions but un-
TABLE 4-4 Calculated Spin-Density Distribution (�i) in Xylene Cation Radicals andDetermined Partial Factors (�i) of Ring-Attack Rates for Nitrations of Neutral IsomericXylenes with Nitric Acid in Acetic Anhydride
Xylene �1 (�1) �2 (�2) �3 (�3) �4 (�4) �5 (�5) �6 (�6)
1,2 0.450 0.450 �0.310 0.360 0.358 �0.308(30) (30) (7) (14) (14) (7)
1,3 0.241 �0.152 0.241 0.539 �0.416 0.539(0.5) (15) (0.5) (42) (0) (42)
1,4 0.482 0.060 0.060 0.482 0.060 0.060(38) (6) (6) (38) (6) (6)
Sources: Calculated spin-density distribution (�i) from Feng et al. 1986; partial factors (�i) from Fisher& Wright 1974.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

der a N2 atmosphere, the yield of the dimer decreased from 97% to 15%, and no mononitroderivatives were formed. Therefore, the oxidation of NO with O2 to form NO2 (after the elec-tron transfer to NO� from 1-methylnaphthalene) is an obvious step of (Scheme 4-27).
It is interesting to compare these results with those for the reactions of the same sub-strate with NOBF4 in the presence or absence of CF3SO3H. In the presence of CF3SO3H,4,4-dimethyl-1,1-binaphthyl was obtained as the sole product with excellent yield (morethan 90%). However, in the absence of CF3SO3H, only a small amount of 1-methylnaph-thalene was converted to mononitro 4-methylnaphthalene (5%) and 4,4-dimethyl-1,1-bi-naphthyl (up to 1%). The unreacted substrate was recovered (Tanaka and others 1996).
Hence, CF3SO3H plays two roles in the reactions considered: (1) It produces NO�
from NaNO2, and (2) inhibits the nitration by trapping NO2 (or N2O4, which is NO�NO3):
NaNO2 � 2CF3SO3H → CF3SO3NO � CF3SO3Na � H2O
CF3SO3NO ⇔ CF3SO3� � NO�
NO� � e → NO.; 2 NO
.� O2 → 2 NO
.2
2 NO.2 ⇔ N2O4 ⇔ NO�NO3
�
NO�NO3� � 2CF3SO3H → CF3SO3NO � CF3SO3NO2 � H2O
In terms of nitration, the system (NaNO2 � CF3SO3H) is of no interest. At the sametime, dimerization in this system can be attractive. For the last direction, CF3SO3H (orFSO3H) is necessary to produce binaphthyl derivatives more preferentially than nitro com-pounds (Tanaka et al. 1996). This work was preceded with the observation by Russianchemists that the reaction of NO�AlCl4
� with 1-methyl-, 1,2-dimethyl-, 1,3-dimethyl-, or
SCHEME 4-27
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

1,8-dimethylnaphthalenes in liquid SO2 leads to a partial �,�-dimerization (Borodkin andco-authors 1993). Another Russian group later published the dimerization of 1,8-N,N-bis(dimethylamino)naphthalene upon the action of
.NO2 in CHCl3. The reaction is accom-
panied by the formation of 4-nitro-1,8-N,N-bis(dimethylamino)naphthalene (Ozeryanskiiet al. 1998). Both groups consider cation radicals of the initial substrates as intermediatespecies.
Tanaka and co-workers (2000) reported that the NO2� nitration of 1,8-dimethylnaph-
thalene leads to 2-nitro and 4-nitro products. For the 2-nitro products, the reaction proceedsas electrophilic substitution: The nitro group comes into the ipso position and then migratesto position 2, thus giving the final product. For the 4-nitro product, the process develops ac-cording to the electron-transfer route. The spin density of the 1,8-dimethylnaphthalenecation radical is highest at position 4 (or the same at position 5). It is the para nitration thattakes place in the experiment.
4.5.4.D Systems of Metallic Nitrites with Oxidizers
Aromatic cation radicals can also react with NO2�, giving nitro compounds. Such reactions
proceed either with a preliminary prepared cation radical or with a starting uncharged com-pound if iodine and silver nitrite are added. As for mechanisms, two of them seem feasible:single electron transfer from the nitrite ion to a cation radical and/or nitration of ArH withthe NO
.2 radical. This radical is quantitatively formed from iodine and silver nitrite in car-
bon tetrachloride (Neelmeyer 1904).Cation radicals of naphthalene and its homologues, pyrene or perylene, react with
NO2� ion in acetonitrile, giving electron-transfer products, i.e., ArH and NO
.2. The latter
radical is not very active in these reactions, and nitration takes place only with extremelyreactive compounds such as perylene (Eberson & Radner 1985, 1986). This mechanism isseemingly distinctive of compounds with E0 less than or equal to 1 V in acetonitrile (or inother solvents, solvating NO2
� ions sparingly).An attempt to combine electrochemical and micellar-catalytic methods is interesting
from the point of view of the mechanism of anode nitration of 1,4-dimethoxybenzene withsodium nitrite (Laurent et al. 1984). The reaction was performed in a mixture of water within 2% surface-active compounds of cationic, anionic, or neutral nature. It was establishedthat 2,5-dimethoxy-1-nitrobenzene—the product—was formed only in the region of po-tentials corresponding to simultaneous electro-oxidation of the substrate to the cation rad-ical and of the nitrite ion to the nitrogen dioxide radical (1.5 V versus saturated calomelelectrode).
At potentials of oxidation of the sole nitrite ion (0.8 V), no nitration was observed.Consequently, radical substitution in the neutral substrate does not take place. Two feasi-ble mechanisms remain for addition to the cation radical form, as follows:
ArH�.� NO2
� → [Ar(H)NO2].
↓
�e
[Ar(H)NO2]� → H� � ArNO2
↑ArH�.
� NO.2
Micellar catalytic methods were used to make a choice between these two mecha-nisms. When an ion radical has a charge opposite to that of the micelle surface, it is trappedby the micelle (Okamoto and co-workers 2001). In the presence of a surface-active com-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

pound, the aromatic substrate is nitrated in the very depth of a micelle, and the reaction ratedepends on a local concentration of the nitrating agent on the phase boundaries between themicelle and the solution. A positively charged micelle will have the higher concentrationof the nitrite anion. However, such a micelle is less likely to include the substrate cationradical, which also bears a positive charge. A negatively charged micelle should assist inthe insertion of the cation radical and repel the nitrite ion. If nitration proceeds with the par-ticipation of the neutral radical NO
.2, the sign of the micelle charge cannot be significant.
(For instance, chemical treatment of 1,4-dimethoxybenzene with gaseous nitrogen dioxide,by bubbling through the micellar system, gives rise to the nitro product in the same yields,irrespective of the micelle charge.)
As for the anode process at comparable conditions, the yield of 2,5-dimethoxy nitrobenzene depends distinctly on the electrical nature of the micelle. Namely, the yields areequal to 30% for the positively charged micelle, 40% for the negatively charged micelle,and 70% for the neutral charged micelle. The observed micellar effect corroborates themechanism, including the dimethoxy benzene cation radical and the nitrogen dioxide rad-ical as reacting species.
4.5.4.E Systems of Metallic Nitrates with Oxidizers
As already known (Addison & Logan 1964), anhydrous nitrates exhibit oxidizing proper-ties. Their oxidizing activity increases from ionic nitrates with alkali and alkaline earthmetal cations to covalent nitrates with transient metal cations. Oxidation reactions result inthe formation of nitrogen-containing oxides. Depending on the kind of nitrate salt and onthe reaction conditions, one of these oxides can be predominant. Organic substrates can ev-idently serve as reductant.
One promising practical variant of such a process is aromatic nitration by the use ofsolid supports. Microporous solids, such as silica, alumina, and alumosilicates, offer a widerange of active sites for catalysis, and most of them can be regenerated if deactivated dur-ing a reaction. According to Delaude et al. (1993), the following features of the method areimportant. By constraining an organic molecule onto a solid surface, a support reduces thedimensionality of an adsorbate. Three-dimensional reactions change to surface-contactones. Thereby the frequency of diffusion encounters increases. The adsorbates also migrateto catalytic sites. This reduces the activation energy. The geometric consequence of an-choring onto a solid is to restrict the angles of attack, i.e., to enhance selectivity. The nitra-tion of phenols is an example of such enhanced selectivity.
Conventional nitration of phenol results in the formation of ca. 67% ortho and 33%para nitrophenols. This corresponds to a statistical distribution in the substitution. (It isworth noting that only one of these isomers, namely p-nitrophenol is the desired product.)Thus, the challenge is to reverse the distribution into predominantly para preference. Theion radical route of nitration approaches this. As Dewar and co-workers (1985) haveshown, the phenol cation radical differs from the uncharged phenol molecule with the en-hanced reactivity of the para position. In the cation radical, the unpaired spin density ap-pears to be greater in the para than in the ortho position. To guide the system along thisroute, Cornelis and Laszlo (1985) have devised a rather efficient heterogeneous nitrationprocedure. The main ingredient is clayfen, i.e., montmorillonite clay (K10) supported fer-ric nitrate. The authors point out that the clay, the ferric ions with which it is doped, and thenitrosonium ions that it evolves are all oxidants cooperating to preoxidize the substrate intothe corresponding cation radical. Indeed, the phenol clayfen nitration led to significant im-provements in yields and to the desired selectivity with respect to common procedures.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

To elevate p-selectivity in the nitration of toluene is another important task. Com-mercial production of p-nitrotoluene heretofore leads with a twofold amount to the un-wanted o-isomer. This stems from the statistical percentage of o:m:p nitration (63:3:34). De-laude and co-authors (1993) enumerate such a relative distribution of the unpaired electrondensities in the toluene cation radical: ipso 1/3, ortho 1/12, meta 1/12, and para 1/3. As seen,the para position is the one favored for nitration by the attack of NO
.(or NO
.2) radical. A
procedure was described (Delaude et al. 1993) that used montmorillonite clay–supportedcopper (cupric) nitrate (abbreviated as claycop) in the presence of acetic anhydride (to re-move excess humidity) and with carbon tetrachloride as a solvent, at room temperature. Ni-trotoluene was isolated almost quantitatively with a 23:1:76 ratio of ortho/meta/para monon-itrotoluene.
Using acyl nitrates as nitrating agents (cf. section 4.5.4.B) and zeolite H-ZSM-11treated with tributylamine, Nagy and co-workers (1991, 1994) were able to nitrate toluenewith an even more impressive percentage of the isomers obtained: ortho 2–3%: meta 1–2%:para 95–98%!
4.5.4.F Systems with Tetranitromethane as a Nitrating Agent
Nitration with tetranitromethane proceeds along the ion radical route. Tetranitromethane isa smooth nitrating agent and a mild oxidizer. It is convenient for the nitration of highly ac-tivated substrates such as phenols, azulene, heterocycles in the presence of pyridine, N,N-dialkylaniline, and other bases. As shown (Morkovnik 1988), these reactions include theone-electron transfer:
ArH � C(NO2)4 → [ArH���C(NO2)4] → [ArH�.C(NO2)�.
4 ] →[ArH�.
, NO.2, C(NO2)�
3 ]
The photochemical addition of tetranitromethane to aromatic compounds under con-ditions of excitation of the [ArH���C(NO2)4] charge-transfer complex by light matching thewavelength of the charge-transfer band results in a recombination within the [ArH�.
, NO.2,
C(NO2)3] triad.The destiny of the triad depends on the nature of the solvent (Sankararaman et al.
1987; Sankararaman & Kochi 1991). In dissociating solvents, radical substitution is pre-dominant, leading to nitro products and trinitromethane:
[ArH�., NO
.2, C(NO2)3
�] → ArNO2 � HC(NO2)3
In nondissociating solvents, the main process consists of the ion-pair reaction that re-sults in the disengagement of nitrous acid and the formation of trinitromethylated products:
[ArH�., NO
.2, C(NO2)3
�] → HNO2 � ArC(NO2)3
The addition of a low concentration of trifluoroacetic acid leads to fast protonationof C(NO2)3
�. The major follow-up reaction now becomes the slower reaction betweenArH�.
and NO.2 (Eberson et al. 1996).
The activity of the NO.2 radical is quite moderate. The leading role belongs to the an-
ion C(NO2)3�. Upon disintegration of the triple complex, this anion departs with the proton
(the light particle). During the reaction within the triad, ArH�.couples predominantly with
C(NO2)3� rather than with NO
.2. Addition of tetrabutyl ammonium perchlorate results in the
binding of the C(NO2)3� anion with the Bu4N� cation. This binding entirely suppresses
alkylation, and nitration remains the sole direction.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Studies of interactions between tetranitromethane and aryl derivatives of magnesium,tin, and mercury in sulfolane (Shevelyov et al. 1974, 1975) confirm that the NO
.2 radical
has some slight activity. For example, the reaction of diarylmercury with tetranitromethanepasses through a transient step, with the formation of the radicals Ar
.and NO
.2. These rad-
icals almost do not interact:
ArMHgMAr � C(NO2)4 → [Ar., ArHg�, NO
.2, C(NO2)3
�] →Ar
.� ArHgC(NO2)3 � NO
.2
The major route of Ar.transformation is H
.abstraction from the solvent, with formation of
ArH.
4.5.4.G Systems with the Participation of Nitrogen Dioxide
The paramagnetic dioxide .NO2 is in equilibrium with the diamagnetic dimer N2O4. At nor-
mal pressure, the percentage of .NO2 in the equilibrium mixture is 31 at 40°C, 88 at 100°C,
and 100 above 140°C. The liquid mixture contains mainly N2O4, and the solid is the dimerentirely. Because of the equilibrium, reaction paths through the N2O4 lead to the same prod-ucts as reaction through
.NO2; see, for example, Chaterjee et al. (1995).
In 1,1,1,3,3,3-hexafluoropropan-2-ol, the reaction of 1,4-dimethoxy-2,3-dimethyl-benzene with a deficit of nitrogen dioxide gives a high concentration of the aromatic cationradical, which lives long enough and can be detected spectroscopically. In the presence ofexcessive amounts of
.NO2, this cation radical decays rapidly, giving the 5-nitro derivative
of the starting compound (Eberson et al. 1996).Kinetic characteristics were obtained for the reaction between several polycyclic aro-
matic hydrocarbons with nitrogen dioxide in dichloromethane at 25°C. They are in accordwith the intermediate formation of the cation radicals (Pryor et al. 1984).
Considering nitration with the help of NO2/N2O4 in an aprotic medium, one shouldavoid a simplified approach to its mechanism. Of course, radical dissociation of dinitrogentetroxide is clear and usual. However, two ionic routes of dissociation of N2O4 are also pos-sible in aprotic media:
NO� � NO3� ⇔ N2O4 ⇔ NO2
� � NO2�
In the presence of water (even in traces), acids are generated:
N2O4 � H2O ⇔ HNO3 � HNO2
The acidity constants of HNO3 and HNO2 are as follows: pKH�(HNO3) � 16.0 andpKH�(HNO2) � 20.6 at 30°C in sulfolane (Boughriet et al. 1987). The authors deduced theequilibrium constants (K) for N2O4 and N2O3 reactions:
N2O4 � H� ⇔ HNO3 � NO� (K � 6.3 � 108)
N2O4 � H� ⇔ HNO2 � NO2� (K � 4.0 � 10�2)
N2O4 � HNO2 ⇔ HNO3 � N2O3 (K � 2.5 � 10�1)
N2O3 � H� ⇔ HNO2 � NO� (K � 2.5 � 109)
Hence, all the reactions are probable and each takes place in the corresponding proportion.Dinitrogen trioxide undergoes both radical and ion dissociation in aprotic solvents
(e.g., in sulfolane):
N2O3 ⇔ NO.� NO
.2
N2O3 ⇔ NO� � NO2�
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Nitration of naphthalene (NaphH) by means of dinitrogen tetroxide can be describedas follows:
NaphH � 2N2O4 → NaphNO2 � HNO3 � N2O3
This reaction is irreversible, but it is attended with the following equilibrium:
N2O3 � HNO3 ⇔ HNO2 � N2O4
Consequently, this nitration follows this scheme:
NaphH � N2O4 → NaphNO2 � HNO2
In reality, nitration of naphthalene with dinitrogen tetroxide in an aprotic medium isa complex process. The leading role belongs to the nitrosyl cation. This species is the strongoxidant acting according to the outer-sphere mechanism (compare with section 1.7.9):
NaphH � �NBO → NaphH����NBO → NaphH�.�
.NMO, etc.
4.5.4.H Nitration and Hydroxylation by Peroxynitrite
Peroxynitrite (ONOO�) is a cytoxic species that is considered to form nitric oxide (NO)and superoxide (O2
�.) in biological systems (Beckman et al. 1990). The toxicity of this com-
pound is attributed to its ability to oxidize, nitrate, and hydroxylate biomolecules. Tyrosineis nitrated to form 3-nitrotyrosine (Ramazanian et al. 1996). Phenylalanine is hydroxylatedto yield o-m-, and p-tyrosines. Cysteine is oxidized to give cystine (Radi et al. 1991a). Glu-tathione is converted to S-nitro or S-nitroso derivatives (Balazy et al. 1998). Cate-cholamines are oxidatively polymerized to melanin (Daveu et al. 1997). Lipids are also ox-idized (Radi 1991b), and DNA can be cut by peroxynitrite (Szabo & Ohshima 1997).
Despite a considerable literature on the various modes of reactions induced by per-oxynitrite, the kinetic and mechanistic aspects of these transformations has been clarifiedonly quite recently (Nonoyama et al. 1999). The authors give the following picture of per-oxynitrite chemical behavior. Peroxynitrite is a stable anionic species in alkaline solutions.At physiological pH, it is rapidly protonated to form peroxynitrous acid (ONOOH):
ONOO� � H� → ONOOH
This acid undergoes homolytic decomposition to .OH and
.NO2 radical species:
ONOOH ⇔ .OH �
.NO2
Heterolytic decomposition of the acid is also possible; Pryor and Squadrito (1995)connect this direction with the generation of a high-energy intermediate [ONOOH]*:
NO2� � �OH ⇔ [ONOOH]* ⇔ NO� � �OOH
Interactions among species thus generated eventually lead to the formation of NO�
and NO3� ions:
ONOO� � NO� → 2NO2
2NO2 ⇔ N2O4 ⇔ NO� � NO3�
In order to gain insight into how peroxynitrite attacks activated aromatic substrates,Nonoyama and co-authors (1999) examined the kinetic features of the reaction of perox-ynitrite with para-substituted phenols. The authors used ONOONa as a reagent. The latterwas prepared by ozonolysis of sodium azide in aqueous solution at pH 12. The reactions
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

with substituted phenols were performed in aqueous phosphate buffer or in acetonitrile, andthe nature and yields of the resulting products were determined. The major products werethe corresponding 2-nitro and 2-hydroxy derivatives of the starting phenols. Kinetic studyshowed a good correlation with Hammett �p
� parameters and reduction potentials of thesubstrates. The cation radical mechanism was proposed involving the following keyspecies: the nitrosonium ion (NO�) as the initial electrophile generated from the peroxyni-trous anion, the cation radicals formed as a result of the oxidative action of NO� on the phe-nolic substrates, and the
.NO2 radical as a nitrating agent to those cation radicals (Scheme
4-28).
4.5.4.I Gas-Phase Nitration
Gas-phase nitration is important from the theoretical and practical points of view. In solu-tion, the solvation of the small nitronium ion should exceed that of the large aromatic cationradical, and hence electron transfer should be less probable. In the gas-phase process thesolvation is absent and only inner reorganization energy remains significant.
Gas-phase nitration of aromatic compounds with nitrogen dioxide or the nitratingmixture is a serious ecological problem. It proceeds simultaneously in the atmosphere andresults in the formation of cancer-producing components in air (Warnek 1988).
Aromatic compounds are the products of incomplete combustion of fuels. Aromaticsare the usual pollutants of the chemical, petroleum, and coal-pyrogenic industries. Thesecompounds are permanently present in air. As for NO2 in air, its presence is always suffi-cient. Air-sols of HCl and H2SO4 are also present. In addition to sulfuric acid, nitric acid(the second component of the nitrating mixture) is formed as a result of the following re-action:
NO2 � 2O3 � HCl → ClO � 2O2 � HNO3
Adsorption of nitric and sulfuric acids on ice particles provides a sol of the nitratingmixture. An important catalyst of aromatic nitration, nitrous acid, is typical for polluted at-mospheres. Combustion sources contribute to air pollution via soot and NOx emissions.The observed formation of HNO2 results from the reduction of nitrogen oxides in the pres-
SCHEME 4-28
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

ence of water by CMO and CMH groups in soot (Ammann et al. 1998). As can be seen,gas-phase nitration is important ecologically.
Up to now, experimental studies of this kind of nitration have been performed viamass spectroscopy and ion cyclotron resonance methods. This work deals with the originsof � complexes only.
A gaseous mixture of nitrogen dioxide with perdeuteriobenzene gives (C6D6NO2)�
� complex. Its origin was studied via ion cyclotron resonance (Benezra et al. 1970). Therate of the �-complex formation depends only on the rate of formation of perdeuterioben-zene cation radicals, but not nitronium ions. Hence, � complex has its origin from the pairof NO
.2 � (C6D6)�.
and not from the pair of NO2� � C6D6. If ionization is performed in va-
por compositions containing pyridine or THF, protonation of these additives occurs. De-crease of the (C6D6NO2)�.
ion, but not the (C6D6)�.ion, takes place. Studies of benzene
homologues produce analogous results (Schmitt et al. 1984). This confirms the structure of(C6D6NO2)� complex with an easily ionizing deuteron at the tetrahedral carbon atom(Scheme 4-29).
According to the work of Ausloos and Lias (1978), the main process of the NO2�.
� C6H6 interaction leads to NO.2 � (C6H6)�.
for 70%. The collateral reaction affects30% and produces NO
.� C6H5(H)O�.
Obviously, intermediates, which are formed in the gaseous phase, arise as a result ofthe strong electrostatic attraction of species with no solvation. Calculations predict thatmixing of the nitronium ion with benzene causes an interaction of frontal molecular or-bitals. As a result, their energetic levels are changed. At a distance between the reagents offrom 0.025 to 0.015 nm, an electron transfer is possible, even from benzene to the nitron-
SCHEME 4-29
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

ium ion (D’yachenko & Ioffe 1976). Such a transfer is energetically allowed: The energyof the lowest unoccupied orbital of the nitronium ion (�11 eV) is below that of the highestoccupied orbital of benzene (�9.24 eV). In principle, one �-electron can be moved frombenzene to the nitronium ion (Nagakura & Tanaka 1954). This point of view was developedin their later publications (Nagakura & Tanaka 1959; Nagakura 1963) and by other authors(Brown 1959; Takabe et al. 1976). “The importance of the electron-transfer step in suchgas-phase reactions is not in dispute” (Ridd 1991). As a result, the nitronium cation givesa radical species, i.e., nitrogen dioxide, and benzene transforms into its cation radical. Thisredox process takes place in the gaseous phase. Conjunction of both the particles, which areradicals in their nature, leads to the � complex and then to the substitution product.
We may infer that although aromatic nitration is a typical reaction for a wide numberof substrates, it cannot be considered a process with a single mechanism in all cases. Theion radical mechanism is characteristic in cases of substrates that are ready for one-electronoxidation and capable of giving stable cation radicals in appropriate solvents. As the citedexamples showed, such a mechanism can be revealed. However, very rapid transformationsof aromatic cation radicals can mask the ion radical nature of many other reactions and cre-ate an illusion of their nonradical character. At the same time, the ion radical mechanismdemands its own attempts to further optimization of commercially important cases of ni-tration. This mechanism deserves our continued attention.
4.5.5 Meerwein and Sandmeyer Reactions
Reduction of arenediazonium salts provides the basis for a substantial number of chemicalreactions. One notable application is the Sandmeyer reaction, which utilizes the diazo moi-ety to facilitate functionalization of aromatic systems and remains one of the most reliabletransformations in organic chemistry. The general reaction involves the addition of thecuprate salt of the desired moiety to the diazonium species:
ArN2� � CuX → ArX
A large body of literature exists investigating the mechanism of the Sandmayer reac-tion, and as early as 1942 ion radical mechanisms were thought to be applicable (Waters1942). Kochi furthered this mechanistic interpretation in 1957, and the current consensussupports that opinion (Galli 1988b; Weaver et al. 2001).
Meerwein reactions consist of condensation of ethylenic compounds with aryldiazo-nium salts in the presence of cupric and cuprous salts:
ArN2Cl � �CBC� → �C(Ar)C(Cl)�
Ganushchak and co-workers proposed to perform the Meerwein chloroarylation ofethylenic compounds using preliminary prepared aryldiazonium tetrachlorocuprates (1972,1984). They found that methyl, ethyl, butyl acrylate, methyl methacrylate, and acrylonitritein the polar solvent reacted with the tetrachlorocuprate. Chloroarylation products were ob-tained with better yields than when using traditional Meerwein reaction conditions.
The interaction of aryldiazonium tetrachlorocuprates [Cu(II)] with olefins has beenstudied by ESR spectroscopy using the spin-trapping technique (Lyakhovich et al. 1991).The radicals ArCH2CH(
.)Ph and ArCH2CH(
.)CN have been detected in mixtures of the
aryldiazonium tetrachlorocuprates [Cu(II)] with styrene and acrylonitrile using nitroso-durene as a spin adduct. However, aryl radical signals were not detected under those con-ditions. Obviously, aryl radicals react with the nearby ethylenic bond within the activated
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

ternary complex without leaving the solvent cage (Ganushchak et al. 1972, 1984). Note thatCu(II) salts in the modification of the Meerwein reaction considered are not reduced toCu(I) salts by solvents used in the reaction. At the same time, reduction of CuCl2 by ace-tone plays an important role in the mechanistic description of Meerwein arylation becausethe resulting Cu(I) is able to generate aromatic radicals (Kochi 1955):
CuCl2� CH3COCH3 → CH3COCH2Cl � CuCl
ArN2� � Cu(I) → Ar
.� Cu(II) � N2
There is every reason to explain the catalytic activity of copper(II) in terms of a cationradical mechanism. This mechanism is confirmed by the unusual direction of the Meerweinreaction in some cases, for example, when the replacement of halogen by an aryl radical oc-curs in the reaction of halostyrenes with aryldiazonium salts (Obushak et al. 1991). A cationradical in the system [olefin–Cu(II)] has been detected by UV spectroscopy (Obushak et al.1991). In the case of cis isomers of benzylidenacetone (Allard & Levisalles 1972) andmaleic esters (Isayev et al. 1972), the unreacted part of the olefin comes back in the corre-sponding trans form. This cis–trans isomerization is understandable if the olefin goesthrough the cation radical state during the Meerwein reaction (Todres 1974).
Using the language of chemical symbols, the following schematic equations bringstogether the results of the mechanistic studies (Obushak et al. 1998):
ArN2Cl � Cu(I) → Ar.� N2 � Cu(II) � Cl�
Cu(II) � �CBC� → Cu(I) � [�CBC�]�.
[�CBC�]�.� Ar
. → �C(Ar)MC.�
Cl� � Cu(II) → Cl.� Cu(I)
�C(Ar)MC.� � Cl
. → �C(Ar)M(Cl)C�
In sum, the cupric ion transfers an electron from the unsaturated substrate to the diazo-nium cation, and the newly formed diazonium radical quickly loses nitrogen. The aryl radi-cal formed attacks the ethylenic bond within the active complexes that originated from the[aryldiazonium tetrachlorocuprate(II)–olefin] or [initial arydiazonium salt–catalyst–olefin]associate and yields the �C(Ar)MC
.� radical. The latter was detected by ESR spectroscopy.
The formation of both the cation radical [�CBC�]�.and the radical �C(Ar)MC
.� as in-
termediates indicates that the reaction involves two catalytic cycles. In the other case, the rad-ical �C(Ar)MC
.� will not be formed, being consumed in the following reaction:
Ar.� [�CBC�]�. → �C(Ar)MC��
The radical �C(Ar)MC.� is oxidized by ligand transfer, as Jenkins and Kochi indi-
cated (1972). If the cation radical [�CBC�]�.obtained as a result of the initial electron
transfer is not fully consumed in the reaction, it is reduced by Cu(I) and returns in the formof its geometrical isomer. In the olefin cation radical state, cis–trans conversion has to takeplace, and it indeed takes place in the systems considered.
4.6 CONCLUSION
Our analysis of different ways to identify ion radical reactions leads to the following im-portant conclusions.
1. None of the approaches can, by themselves and taken alone, identify an ion rad-ical conversion.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

2. A set of suitable methods gives reliable information on the role that the ion rad-ical stage plays in the net mechanism of a reaction.
3. A choice between the conventional (or classical) and ion radical mechanism is avery important issue. The ion radical pathway can lead to products of the desiredstructure or can make the conversion conditions more mild or can change the re-activity of the secondary intermediate particles. If ion radicals form and react ina solvent cage, the reaction proceeds rapidly, and product yields are indistin-guishable from “standard” schemes, then the role that ion radicals play is only oftheoretical interest and is, thus far, not essential for practical organic synthesis.
4. The ion radical mechanism should be checked with respect to every individualreaction. Compounds of even one class may behave differently: Some of themreact by the ion radical mechanism, while others may take quite another path-way. At this point, it is very important to guess why the reaction chooses one orthe other mechanism.
5. The results obtained by different methods for revealing a mechanism shouldagree. Only such an approach might uncover the mechanism chosen by a reac-tion.
REFERENCES
Abe, T.; Ikegame, Y. (1976) Bull. Chem. Soc. Jap. 49, 3227.Abe, T.; Ikegame, Y. (1978) Bull. Chem. Soc. Jap. 51, 196.Addison, C.C.; Logan, N. (1964) Adv. Inorg. Chem. Radiochem. 6, 72.Afanas’ev, I.B.; Polozova, N.I. (1978) Zh. Org. Khim. 14, 1013.Alberti, A.; Ciminale, F.; Pedulli, G.F.; Testaferri, L.; Tiecco, M. (1981) J. Org. Chem. 46, 751.Allard, M.; Levisalles, J. (1972) Bull. Soc. Chem. Fr., 1921.Amatore, Ch.; Pinson, J.; Saveant, J.-M.; Thiebault, A. (1982) J. Am. Chem. Soc. 104, 817.Ammann, M.; Kalberer, M.; Jost, D.T.; Tobler, L.; Roessler, E.; Piguent, D.; Gaeggeler, H.W.;
Baltensperger, U. (1998) Nature 395, 157.Andrieux, C.P.; Robert, M.; Saeva, F.D.; Saveant, J.-M. (1994) J. Am. Chem. Soc. 116, 7864.Andrieux, C.P.; Robert, M.; Saveant, J.-M. (1995) J. Am. Chem. Soc. 117, 9340.Andrieux, C.P.; Saveant, J.-M.; Tallec, A.; Tardivel, R.; Tardy, C. (1997) J. Am. Chem. Soc. 119,
2420.Aplin, J.T.; Bauld, N.L. (1997) J. Chem. Soc. Perkin Trans. 2, 853.Arguello, J.E.; Penenory, A.B.; Rossi, R.A. (2000) J. Org. Chem. 65, 7175.Artamkina, G.A.; Egorov, M.P.; Beletskaya, I.P. (1982) Chem. Rev. 82, 427.Ashby, E.C.; Goel, A.B.; De Priest, R.N. (1981) J. Org. Chem. 46, 2429.Ausloos, P.; Lias, S.G. (1978) Int. J. Chem. Kinet. 10, 657.Baciocchi, E. (1995) Xenobiotica 25, 653.Baker, C.C.; Emmerson, R.G.; Periam, J.D. (1955) J. Chem. Soc., 4482.Balazy, M.; Kaminski, P.M.; Mao, K.; Tan, J.; Wolin, M. S. (1998) J. Biol. Chem. 273, 32009.Bartlett, P.N.; Whitaker, R.G. (1987) Biosensors 3, 1987.Bazanov, A.G.; Shugalei, I.V.; Tselinskii, I.V. (1980a) Zh. Org. Khim. 16, 905.Bazanov, A.G.; Shugalei, I.V.; Tselinskii, I.V. (1980b) Zh. Org. Khim. 16, 910.Bazanov, A.G.; Tselinskii, I.V.; Shugalei, I.V. (1978) Zh. Org. Khim. 14, 901.Beckman, J.S.; Beckman, T.W.; Chen, J.; Marshall, P.A.; Freeman, B.A. (1990) Proc. Natl. Acad.
Acad. Sci. USA 87, 1620.Benezra, S.A.; Hoffman, M.K.; Bursey, M.M. (1970) J. Am. Chem. Soc. 92, 7501.Bielevich, K.A.; Bubnov, N.N.; Okhlobystyn, O. Yu (1968) Tetrahedron Lett. 31, 3465.Bil’kis, I.I.; Shein, S.M. (1975) Tetrahedron 31, 969.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Borodkin, G.I.; Elanov, I.R.; Shakirov, M.M.; Shubin, V.G. (1993) J. Phys. Org. Chem. 6, 153.Bosch, E.; Kochi, J.K. (1994) J. Org. Chem. 59, 5573.Boughriet, A.; Wartel, M. (1989) J. Chem. Soc., Chem. Commun., 809.Boughriet, A.; Bremard, C.; Wartel, M. (1987) New J. Chem. 11, 245.Brede, O.; Beckert, D.; Windolph, C.; Goettinger, H.A. (1998) J. Phys. Chem. A 102, 1457.Brickman, M.; Ridd, J.H. (1965) J. Chem. Soc., 6845.Brown, R.D. (1959) J. Chem. Soc., 2224.Bubnov, N.N.; Bielevitch, K.A.; Polyakova, L.A.; Okhlobystin, O.Yu. (1972) J. Chem. Soc. Chem.
Commun., 1058.Buchachenko, A.L. Khimicheskaya polyarizatsiya electronov i yader (1974). Nauka, Moscow.Bunnett, J.F. (1978) Acc. Chem. Res. 11, 413.Bunnett, J.F.; Creary, X. (1974a) J. Org. Chem. 39, 3173.Bunnett, J.F.; Creary, X. (1974b) J. Org. Chem. 39, 3611.Bunnett, J.F.; Shafer, Sh.J. (1978) J. Org. Chem. 43, 1873.Bunnett, J.F.; Traber, R.P. (1978) J. Org. Chem. 43, 1867.Cano, M.; Quitana, J.; Julia, L.; Camps, F.; Joglar, J. (1999) J. Org. Chem. 64, 5096.Cardoso, Sh. P.; de Mesquita Caneiro, J.W. (2001) Quim. Nova 24, 381.Carver, D.R.; Greenwood, Th.D.; Hubburd, J.S.; Komin, A.P.; Sachdeva, Ye.P.; Wolfe, J.F. (1983)
J. Org. Chem. 48, 1180.Carver, D.R.; Hubburd, J.S.; Wolfe, J.F. (1982) J. Org. Chem. 47, 1036.Carver, D.R.; Komin, A.P.; Hubburd, J.S.; Wolfe, J.F. (1981) J. Org. Chem. 46, 294.Chanon, M.; Tobe, M. (1982) Angew. Chem. Int. Ed. Engl. 21, 1.Chapman, N.B.; Parker, R.E.; Soames, P.W. (1954) J. Chem. Soc., 2109.Chatterjee, J.; Coombes, J.R.; Fildes, M.J. (1995) J. Chem. Soc. Perkin Trans. 2, 1031.Chawla, O.P.; Fessenden, R.W. (1975) J. Phys. Chem. 79, 2693.Ciminale, F.; Bruno, G.; Testaferri, L.; Tiecco, M.; Martelli, G. (1978) J. Org. Chem. 43, 4509.Clemens, A.H.; Helsby, P.; Ridd, J.H.; Al-Omran, F.; Sandall, J.P.B. (1985) J. Chem. Soc. Perkin
Trans. 2, 1217.Clemens, A.H.; Ridd, J.H.; Sandall, J.P.B. (1984) J. Chem. Soc. Perkin Trans. 2, 1659.Clemens, A.H.; Ridd, J.H.; Sandall, J.P.B. (1985) J. Chem. Soc. Perkin Trans. 2, 1227.Cornelis, A.; Laszlo, P. (1985) Synthesis, 909.Daveu, C.; Servy, C.; Dendane, M.; Marin, P.; Ducrocq, C. (1997) Nitric Oxide 1, 234.Davies, A.; Feld, R. (1958), J. Chem. Soc., 4637.Delaude, L.; Laszlo, P.; Smith, K. (1993) Acc. Chem. Res. 26, 607.Denney, D.B.; Denney, D.Z.; Fenelli, S.P. (1997) Tetrahedron 53, 9835.Dessau, R.M.; Shih, S.; Heiba, E.I. (1970) J. Am. Chem. Soc. 92, 412.Dewar, M.J.S.; Mole, T.; Warford, E.W.T. (1956) J. Chem. Soc., 3581.Dewar, M.J.S.; Zoebisch, E.G.; Healey, E.F.; Stewart, J.J.P. (1985) J. Am. Chem. Soc. 107, 3902.Ding, Y.; Padias, A.B.; Hall, H.K. (1999) J. Polym. Sci. Part A: Polym. Chem. 37, 2569.Dix, L.R.; Moodie, R.B. (1986) J. Chem. Soc. Perkin Trans. 2, 1097.Dixon, W.T.; Murphy, D. (1976) J. Chem. Soc. Perkin Trans. 2, 1823.D’yachenko, A.I.; Ioffe, A.I. (1976) Izv. AN SSSR, Ser. Khim., 1160.Eberson, L. (1967) J. Am. Chem. Soc. 89, 4669.Eberson, L.; Jonsson, L. (1981) J. Chem. Soc. Chem. Commun., 133.Eberson, L.; Radner, F. (1980) Acta Chem. Scand. Ser. B. 34, 739.Eberson, L.; Radner, F. (1985) Acta Chem. Scand. Ser. B. 39, 343.Eberson, L.; Radner, F. (1986) Acta Chem. Scand. Ser. B. 40, 71.Eberson, L.; Hartshorn, M.P.; Radner, F. (1994) Acta Chem. Scand. 48, 937.Eberson, L.; Hartshorn, M.; Radner, F. (1995) Adv. Carbocation Chem. 2, 207.Eberson, L.; Hartshorn, M.P.; Persson, O.; Radner, F. (1996) Chem. Commun., 2105.Eldin, S.; Jencks, W.P. (1995) J. Am. Chem. Soc. 117, 9415.Epiotis, N.D. (1973) J. Am. Chem. Soc. 95, 3188.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Ettaeb, R.; Saveant, J.-M.; Thiebault, A. (1992) J. Am. Chem. Soc. 114, 10990.Feiring, A.E. (1983) J. Org. Chem. 48, 347.Feng, J.; Zheng, X.; Zerner, M.C. (1986) J. Org. Chem. 51, 4531.Fisher, A.; Wright, G.J. (1974) Austral. J. Chem. 27, 217.Flesia, E.; Crozet, M.P.: Surzur, J.-M.; Jauffred, R.; Ghiglione, C. (1978) Tetrahedron 34, 1699.Fomin, G.V.; Gurdzhiyan, L.M. (1970) Zh. Fiz. Khim. 44, 1809.Fomin, G.V.; Gurdzhiyan, L.M. (1978) Zh. Fiz. Khim. 52, 628.Forshult, S.; Lagercrantz, C; Torsell, K. (1969) Acta Chem. Scand. 23, 522.Freed, J.F.; Fraenkel, G.K. (1964) J. Chem. Phys. 41, 699.Freidlina, R.Kh.; Kandror, I.I.; Gasanov, R.G. (1979) Usp. Khim. 47, 508.Fujinaga, T.; Deguchi, Y.; Umemoto, K. (1964) Bull. Chem. Soc. Jpn. 37, 822.Galli, C. (1988a) Tetrahedron 44, 5205.Galli, C. (1988b) Chem. Rev. 88, 765.Galli, C.; Bunnett, J.F. (1979) J. Am. Chem. Soc. 101, 6137.Galli, C.; Bunnett, J.F. (1981) J. Am. Chem. Soc. 103, 7140.Galli, C.; Gentili, P. (1998) Acta Chem. Scand. 52, 67.Ganushchak, N.I.; Golik, V.D.; Migaychuk, I.V. (1972) Zh. Org. Khim. 8, 2356.Ganushchak, N.I.; Obushak, N.D.; Kovalchuk, E.P.; Trifonova, G.V. (1984) Zh. Obshch. Khim.
54, 2334.Geske, D.H.; Ragle, J.L.; Bambenek, U.A.; Balch, A.L. (1964) J. Am. Chem. Soc. 86, 87.Giffney, J.C.; Ridd, J.H. (1979) J. Chem. Soc. Perkin Trans. 2, 618.Gorelik, M.V.; Puchkova, V.V. (1969) Zh. Org. Khim. 5, 1695.Gorelick, M.V.; Lomzakova, V.I.; Khadimova, E.A.; Steiman, V.Ya.; Kuznetsova, M.G. (1995)
Rus. J. Org. Chem. 31, 371.Haas, H.B.; Bender, M.L. (1949a) J. Am. Chem. Soc. 71, 1767.Haas, H.B.; Bender, M.L. (1949b) J. Am. Chem. Soc. 71, 3482.Hawthorne, M.F. (1956) J. Am. Chem. Soc. 78, 4980.Huszthy, P; Lempert, K.; Simig, G. (1982) J. Chem. Res. Synop. 5, 126.Huszthy, P; Lempert, K.; Simig, G.; Vekey, K. (1982) J. Chem. Soc. Perkin Trans. 1, 3021.Isayev, S.D.; Yurchenko, A.G.; Chernova, Y.S.; Mramornova, S.A. (1972) Zh. Org. Khim. 8, 2054.Itoh, K.; Nakazava, Sh.; Yano, M.; Fu-Ruichi, M.; Sato, K.; Shiomi, D.; Kinoshita, T.; Abe, K.;
Takui, T. (1997) Mol. Cryst. Liq. Cryst. Sci. Technol. Sec. A 305, 515.Janzen, E.G. (1971) Accounts Chem. Res. 4, 31.Janzen, E.; Blackburn, B. (1969) J. Am. Chem. Soc. 91, 4481.Jaworski, J.S. (1998) Wiad. Chem. 52, 367.Jenkins, C.L.; Kochi, J.K. (1972) J. Am. Chem. Soc. 94, 856.Johnston, J.F.; Ridd, J.H.; Sandall, J.P.B. (1989) J. Chem. Soc., Chem. Commun., 244.Johnston, J.F.; Ridd, J.H.; Sandall, J.P.B. (1991) J. Chem. Soc. Perkin Trans. 2, 623.Kekule, A.; Hidegh, C. (1870) Ber. Dtsch. Chem. Ges. 3, 233.Kerber, R.C.; Urry, G.W.; Kornblum, N. (1964) J. Am. Chem. Soc. 86, 3904.Kerber, R.C.; Urry, G.W.; Kornblum, N. (1965) J. Am. Chem. Soc. 87, 4520.Keumi, T.; Hamanaka, K.; Hagesawa, H. (1988) Chem. Lett., 1285.Kim, J.K.; Bunnett, J.F. (1970) J. Am. Chem. Soc. 92, 7463.Kochi, J.K. (1955) J. Am. Chem. Soc. 77, 5090.Kochi, J.K. (1957) J. Am. Chem. Soc. 79, 2942.Kochi, J.K. (1990) Acta Chem. Scand. 44, 409.Kochi, J.K. (1992) Acc. Chem. Res. 25, 39.Kochi, J.K.; Tang, R.T.; Bernath, T.J. (1973) J. Am. Chem. Soc. 95, 7114.Kornblum, N.; Fifolt, M.J. (1980) J. Org. Chem. 45, 360.Kornblum, N.; Michel, R.E.; Kerber, R.C. (1966) J. Am. Chem. Soc. 88, 5660.Kurreck, H.; Kirste, B.; Lubitz, W. Electron Nuclear Double Resonance Spectroscopy of Radicals in
Solutions (1988). VCH, New York.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Lagercrantz, C. (1977) J. Phys. Chem. 75, 3466.Laurent, E.; Rauniyar, G.; Tomalla, M. (1984) Bull. Soc. Chim. France, 78.Lehnig, M. (1996) J. Chem. Soc. Perkin Trans. 2, 1943.Lehnig, M. (1997) Acta Chem. Scand. 51, 211.Leis, J.R.; Pena, M.E.; Ridd, J.H. (1988) J. Chem. Soc., Chem. Commun., 670.Lemaire, H.; Marechal, Y.; Ramasseul, R.; Rassat, A. (1965) Bull. Soc. Chim. France, 372.Levit, A.P.; Gragerov, I.P.; Buchachenko, A.L. (1971) Doki. AN SSSR 201, 897.Levit, A.F.; Buchachenko, A.L.; Kiprianova, L.A.; Gragerov, I.P. (1972) Doki. AN SSSR 203, 628.Lewis, E.S.; Chalmers, D.J. (1971) J. Am. Chem. Soc. 93, 3267.Lippmaa, E.T.; Pehk, T.J.; Saluvere, T.A. (1973) Org. Magn. Reson. 5, 595.Loeppky, R.N.; Singh, S.P.; Elomari, S.; Hastings, R.; Theiss, Th.E. (1998) J. Am. Chem. Soc. 120,
5193.Lyakhovich, M.B.; Gasanov, R.G.; Obushak, N.D.; Ganushchak, N.I.; Todres, Z.V. (1991) Izv. AN
SSSR, Ser. Khim., 1214.Malykhin, E.V.; Mamatyuk, V.I.; Shteingarts, V.D. (1975) Izv. Sib. Otd. AN SSSR Ser. Khim. Nauk
12, 145.Moebius, K.; Biehl, R. In: Dorio, M.M.; Freed, J.H., eds. Multiple Electron Resonance Spectroscopy
(1979). Plenum Press, New York.Morkovnik, A.S. (1988) Usp. Khim. 57, 254.Murabayashi, S.; Shiotany, M.; Sohma, J. (1979) J. Phys. Chem. 83, 844.Nagakura, S. (1963) Tetrahedron 19, 361.Nagakura, S.; Tanaka, J. (1954) J. Chem. Phys. 22, 563.Nagakura, S.; Tanaka, J. (1959) Bull. Chem. Soc. Jpn. 32, 734.Nagy, S.M.; Yarovoy, K.A.; Shakirov, M.M.; Shubin, V.G.; Vostrikova, L.A.; Ione, K.G. (1991)
J. Mol. Catal. 64, L31.Nagy, S.M.; Yarovoy, K.A.; Shubin, V.G.; Vostrikova, L.A. (1994) J. Phys. Org. Chem. 7, 385.Neelmeyer, W. (1904) Ber. Dtsch. Chem. Ges. 37, 1386.Nguyen, M.T.; Dao, L.H. (1989) J. Electrochem. Soc. 136, 2131.Nonhebel, D.; Walton, G. Free-radical Chemistry (1974). Cambridge University Press, London.Nonoyama, N.; Chiba, K.; Hisatome, K.; Suzuki, H.; Shintani, F. (1999) Tetrahedron Lett. 40, 6933.Obushak, N.D.; Bilaya, E.E.; Ganushchak, N.I. (1991) Zh. Org. Khim. 27, 2372.Obushak, N.D.; Lyakhovich, M.B.; Ganushchak, N.I. (1998) Tetrahedron Lett. 39, 9567.Okamoto, K.; Hirota, N.; Tominaga, T.; Terazima, M. (2001) J. Phys. Chem. A, 105, 6586.Olah, G.A.; Malhotra, R.; Narang, S.C. Nitration: Methods and Mechanisms (1989). VCH, New
York.Onoda, M.; Yoshino, K. (1995) Jpn. Appl. Phys. Part A 34, 260.Ozeryanskii, V.A.; Pozharskii, A.F.; Fomenkov, A.M. (1998) Russ. Chem. Bull. 47, 313.Pagano, A.H.; Shechter, H. (1970) J. Org. Chem. 35, 295.Painter, Sh.L.; Blackstock, S.C. (1995) J. Am. Chem. Soc. 117, 1441.Papaconstadinou, S.; Theophilou, N. (1991) Synth. Met. 41, 1.Parker, V.D. (1981) Acta Chem. Scand. B35, 655.Pedersen, E.B.; Petersen, T.E.; Torsell, K.; Lawesson, S.-O. (1973) Tetrahedron 29, 579.Percec, V.; Hill, D.H. In: Matyjaszewski, K.; Pugh, C., eds. Cationic Polymerization: Mechanism,
Synthesis, and Applications (1996) Marcel Dekker, New York.Perrin, Ch.L. (1977) J. Am. Chem. Soc. 99, 5516.Pobedimskii, D.G.; Buchachenko, A.L. (1968a) Izv. AN SSSR Ser. Khim., 1181.Pobedimskii, D.G.; Buchachenko, A.L. (1968b) Izv. AN SSSR Ser. Khim., 2720.Powell, J.L.; Ridd, J.H.; Sandall, J.P.B. (1990) J. Chem. Soc. Chem. Commun., 402.Pryor, W.A.; Squadrito, G.L. (1995) Am. J. Physiol. 268, L699.Pryor, W.A.; Gleicher, G.J.; Cosgrove, J.P.; Church, D.F. (1984) J. Org. Chem. 49, 5189.Radi, R.; Beckman, J.S.; Bush, K.M.; Freeman, B.A. (1991a) J. Biol. Chem. 266, 4244.Radi, R.; Beckman, J.S.; Bush, K.M.; Freeman, B.A. (1991b) Arch. Biochem. Biophys. 288, 481.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Ramazanian, M.S.; Padmaja, S.; Koppenol, W.H. (1996) Chem. Res. Toxicol. 9, 232.Rao, S.N.R.; Kalianaraman, V.; George, M.V. (1972) Usp. Khim. 41, 940.Rehorek, D. (1979) Zeitschr. Chem. (DDR) 19, 227.Rehorek, D. (1980) Zeitschr. Chem. (DDR) 20, 325.Ridd, J.H. (1991) Chem. Soc. Rev. 20, 149.Ridd, J.H. (1998) Acta Chem. Scand. 52, 11.Ridd, J.H.; Sandall, J.P.B. (1981) J. Chem. Soc., Chem. Commun., 402.Ross, D.S.; Moran, K.D.; Malhotra, R. (1983) J. Org. Chem. 48, 2118.Rossi, R.A.; Pierini, A.B. (1980) J. Org. Chem. 45, 2914.Russell, G.A.; Danen, W.C. (1966) J. Am. Chem. Soc. 88, 5663.Russell, G.A.; Mudryk, B. (1982a) Tetrahedron 38, 1059.Russell, G.A.; Mudryk, B. (1982b) J. Org. Chem. 47, 1879.Russell, G.A.; Panck, E.J.; Makosza, M.; Metkalfe, A.R.; Norris, P.K.; Pecararo, J.; Reinolds, J. In:
XXIIIrd Int. Congress of Pure Appl. Chem. (1971). Vol. 4, 67. Butterworth, London.Rybakova, I.A.; Shekhtman, R.I.; Prilezhaeva, E.N. (1982) Izv. AN SSSR Ser. Khim., 2414.Rykova, L.A.; Kiprianova, L.A.; Gragerov, I.P. (1980) Theor. Eksp. Khim. 16, 825.Sang, H.; Janzen, E.G.; Lewis, B.H. (1996) J. Org. Chem. 61, 2358.Sankararaman, S.; Kochi, J.K. (1991) J. Chem. Soc. Perkin Trans. 2, 1.Sankararaman, S.; Haney, W.A.; Kochi, J.K. (1987) J. Am. Chem. Soc. 109, 5235.Schmitt, R.J.; Ross, D.S.; Buttrill, S.E. (1984) J. Am. Chem. Soc. 106, 926.Schoffield, K. Aromatic Nitration (1980). Cambridge University Press, London.Screttas, C.G.; Micha-Screttas, M. (1982) J. Chem. Soc. Chem. Commun., 1168.Screttas, C.G.; Micha-Screttas, M. (1983) J. Org. Chem. 48, 252.Screttas, C.G.; Heropoulos, G.A.; Steele, B.R.; Bethell, D. (1998) Magn. Reson. Chem. 36, 656.Selwood, P. Magnetochemistry (1958). Interscience, New York.Shapiro, B.I.; Kozakova, V.M.; Syrkin, Ya.K.; Khutoretskii, V.M.; Okhlobystina, L.V. (1969) Izv.
AN SSSR Ser. Khim., 458.Shein, S.M. (1983) Izv. Sib. Otd. AN SSSR Ser. Khim. Nauk, 20.Shein, S.M.; Brukhovetskaya, L.V.; Ivanova, T.I. (1973) Izv. AN SSSR Ser. Khim., 1594.Shevelyov, S.A.; Kolesnikov, R.V.; Beletskaya, I.P. (1974) Zh. Org. Khim. 9, 1793.Shevelyov, S.A.; Kolesnikov, R.V.; Fainzil’berg, A.A.; Beletskaya, I.P. (1975) Izv. AN SSSR Ser.
Khim., 1368.Shinomura, E.T.; Phillippi, M.A.; Goff, H.M. (1981) J. Am. Chem. Soc. 103, 6778.Shternshis, M.V.; Brukhovetskaya, L.V.; Katkova, N.M.; Bilkis, I.I.; Shein, S.M. (1973) Zh. Org.
Khim. 9, 1242.Shugalei, I.V.; Tselinskii, I.V. (1993) Zh. Obshch. Khim. 63, 1884.Shugalei, I.V.; Tselinskii, I.V. (1994) Zh. Obshch. Khim, 64, 309.Shugalei, I.V.; Bazanov, A.G.; Tselinskii, I.V. (1978) Zh. Org. Khim. 14, 1577.Shugalei, I.V.; Bazanov, A.G.; Tselinskii, I.V. (1981) Zh. Org. Khim. 17, 1837.Simig, G.; Lempert, K. (1979) Acta Chim. Sci. Hung. 102, 101.Simon, P.; Sumegi, L.; Rockenbuer, A.; Nemes, I.; Tudos, F. (1980) React. Kinet. Catal. Lett.
15, 493.Singh, P.R.; Jayaraman, B. (1974) Indian J. Chem. 12, 1306.Singh, P.R.; Khanna, R.K. (1983) Tetrahedron Lett. 24, 973.Singh, P.R.; Kumar, P. (1972a) Tetrahedron Lett. 7, 613.Singh, P.R.; Kumar, P. (1972b) Austr. J. Chem. 25, 2133.Singh, P.R.; Kumar, R.; Khanna, R.K. (1982) Tetrahedron Lett. 23, 5191.Skokov, S.; Wheeler, R.A. (1999) J. Phys. Chem. A 103, 4261.Solodovnikov, S.P. (1976) Izv. AN SSSR Ser. Khim., 996.Sosonkin, I.M.; Belevskii, V.N.; Strogov, G.N.; Domarev, A.N.; Yarkov, S.P. (1982) Zh. Org. Khim.
18, 1504.Speiser, B. (1996) Angew. Chem., Int. Ed. Engl. 35, 2471.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Suhr, H. (1964) Chem. Ber. 97, 3268.Szabo, C.; Ohshima, H. (1997) Nitric Oxide 1, 373.Takabe, T.; Takenaka, K.; Yamaguchi, K.; Fueno, T. (1976) Chem. Phys. Lett. 44, 65.Tanaka, M.; Nakanishi M.H.; Fujiwara, M.; Ando, H.; Souma, Y. (1996) J. Org. Chem. 61, 788.Tanaka, M.; Muro, E.; Ando, H.; Xu, Q.; Fujiwara, M.; Souma, Y.; Yamaguchi, Y. (2000) J. Org.
Chem. 65, 2972.Terabe, S.; Konaka, R. (1971) J. Am. Chem. Soc. 93, 4306.Terabe, S.; Kumura, K.; Konaka, R. (1973) J. Chem. Perkin Trans. 2, 1252.Todres, Z.V. (1970) Izv. AN SSSR Ser. Khim., 1749.Todres, Z.V. (1974) Usp. Khim., 43, 2274.Todres, Z.V. (1981) Sulfur Reports 1, 133.Todres, Z.V. (1985) Tetrahedron 41, 2771.Torii, H.; Ueno, Yu.; Sakamoto, A.; Tasumi, M. (1999) J. Phys. Chem. A 103, 5557.Torsell, K. (1970) Tetrahedron 26, 2759.Tremelling, M.J.; Bunnett, J.F. (1980) J. Am. Chem. Soc. 102, 7375.Tsutsumi, H.; Fukuzawa, S.; Ishikawa, M.; Morita, M.; Matsuda, Y. (1995) J. Electrochem. Soc. 142,
L168.Ufimtsev, V.N. (1983) Zh. Org. Khim. 19, 1930.Vilar, H.; Castro, E.A.; Rossi, R.A. (1982) Canad. J. Chem. 60, 2525.Wajer, A.J.W.; Mackor, A.; Boer de, Th.J.; Voorst van, J.D.W. (1967) Tetrahedron 23, 4021.Warnek, P. Chemistry of Natural Atmosphere (1988). Academic Press, San Diego, CA.Waters, W.A. (1942) J. Am. Chem. Soc. 64, 266.Weaver, M.N.; Janicki, S.Z.; Petillo, P.A. (2001) J. Org. Chem. 66, 1138.Wei, Y.; Tang, X.; Sun, Y.; Focke, W. (1989) J. Polym. Sci. Polym. Chem. Ed. 27, 2385.Wei, Y.; Jang, G.-W.; Chan, C.-C.; Hsueh, K.F.; Hariharan, R.; Patel, S.A.; Whitecar, C.R. (1990) J.
Phys. Chem. 94, 7716.Werst, D.W.; Trifunac, A.D. (1998) Acc. Chem. Res. 31, 651.Zhang, X.-M.; Yang, D.-L.; Liu, Y.-C. (1993) J. Org. Chem. 58, 224.Zheng, Z.-R.; Evans, D.H.; Chan-Shing, E.S.; Lessard, J. (1999) J. Am. Chem. Soc. 121, 9429.Zollinger, H. Chemie der Azofarbstoffe (1958). Birkhauser, Basel, Switzerland.Zorin, V.V.; Kukovitskii, D.M.; Zlotskii, S.S.; Todres, Z.V.; Rakhmankulov, D.L. (1983) Zh. Ob-
shch. Khim. 53, 906.Zuman, P.; Shah, B. (1994) Chem. Rev. 94, 1621.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

5
How to Optimize Organic Ion Radical Reactions
5.1 INTRODUCTION
Ion radical reactions require special methods to stimulate or impede them. The specificityof these methods is determined with particular properties of ion radicals. Many ion radi-cal syntheses are highly selective, yielding products unattainable by other methods or un-der mild conditions. The aim of this chapter is to analyze the phenomena that determinethe ways to optimize ion radical reactions. The chapter considers factors governing thedevelopment of reactions with proven ion radical mechanisms. Two groups of optimiz-ing factors will be discussed: physical and purely chemical ones. Such factors as solventchange and salt addition are certainly in the borderline between chemical and physical ef-fects.
5.2 PHYSICAL EFFECTS
5.2.1 Effect of Light
There is clear evidence that excitation of the electrons of reacting molecules accelerates theprocesses involving electron transfer (Endicott & Ramasami 1982; Juillard, Chanon 1983).Having absorbed a quantum of light, a molecule becomes excited. Electronically excitedstates of organic molecules are richer in energy by 2–4 eV than the corresponding groundstates. Thus, photochemical reactions begin at an energy level much higher than that ofthermal reactions. Upon photoirradiation, an electron in a donor occupies a higher orbital.The energy of the “external” electron increases, and so the donating properties of themolecule increase, too. In other words, electron transfer to an acceptor becomes more prob-able. Of course, the acceptor molecule demonstrates the same change in electron configu-ration as a result of photoirradiation. After binary ionization in solution, solvent-separatedion pairs of reactants are formed. These pairs then undergo some association and enter fur-ther transformations (Burstein 1999). Reactions may be photoinitiated if the difference be-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

tween the ionization potential of a donor and the electron affinity of an acceptor increasessignificantly. When the irradiation wavelength is chosen correctly, only donors out of touchwith acceptors may be excited. In this case the difference increases. This is often achievedby using irradiation in the spectral region corresponding to a charge-transfer band of a com-plex produced by a donor and an acceptor (Fox et al. 1983). Sometimes, substances thatnever react with each other become capable of forming charge-transfer complexes uponelectron excitation and then to yield ion radicals. However, we must also take into accountthat photoexcitation also frequently involves significant electron redistribution. In photo-chemical processes, ion radical reactivity can be different from that in “normal” (chemicalor electrochemical) reactions.
Excited molecular complexes of the donor–acceptor type are called excimers ifformed from identical molecules and exiplexes if they originate from different molecules.As follows from theory, photochemical influences will more readily accelerate electrontransfer in a weak donor–acceptor pair than in a strong pair (Juillard & Chanon 1983). Anorganic molecule in an electron-excited state is a more active oxidant or stronger reducerthan the same molecule in a ground state.
Photoelectron transfer is usually described by the so-called Foerster’s cycle: Upontransformation of a molecule into the excited state, the donor’s ionization potential is re-duced by the value of the donor’s excitation energy, and the acceptor’s electron affinity in-creases by the value of the acceptor’s excitation energy.
Compounds of enhanced excitability are used as sensitizers. When introduced into areaction system of a donor and an acceptor, the sensitizer absorbs light. The sensitizer it-self does not undergo bond breaking or isomerization and acts only as an oxidant or a re-ducer with regard to the substrate.
For instance, 1-methoxynaphthalene is active as a reducer, while 1-cyanonaphtha-lene plays a role as an oxidant. A sensitizer’s reducing ability increases when its valenceelectron is transferred from a relatively low energy level to a rather high energy level,which corresponds to the lowest unoccupied molecular orbital. The enforced oxidizing ac-tivity is also conditioned by a one-electron shift to the higher level, with the synchronousformation of a hole at the orbital left. For 1-cyanonaphthalene and related molecules, how-ever, this energy increase is less significant than is the growth of the electron affinity of thearising electron hole. In these sensitizers, the “abandoned” orbital is located low enough tomake an electron transfer energetically favorable.
In general, a sensitizer transforms into its excited state, passes an excited electron (orhole) to a substrate, and then remains in the reaction sphere. Thus, electron back-transfer ischaracteristic of the whole process. During its lifetime, a substrate ion radical is often ableto undergo some chemical transformation, especially when the transformation proceedswithin a solvent cage. Examples are given at the end of Section 5.2.1.
Of course, there are photoinduced electron-transfer reactions that proceed with nosensitizers. Thus, nitro aromatic anion radicals can be generated by photoirradiation of themixture of a parent nitro compound and sodium dithionite in strongly alkaline solution (pHclose to 13). Importantly, this method gives a strong ESR signal of the resulting anion rad-ical. The spectra have extremely narrow line widths (typically �0.01 mT), largely becauseoxygen (which can cause line broadening) is scavenged by excess dithionite. The anionradical generated in this way typically persists for 5–15 min (Corrie et al. 2000).
Rossi and de Rossi (1983) and Rossi et al. (1999) cited a substantial body of pho-toinduced ion radical reactions of aromatic substitution. It is interesting here to comparephotoinduction and electrochemical stimulation of ion radical reactions. As pointed out in
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Chapter 2, a mediator is used in order to take an electron from the cathode and transfer itonto a substrate. It is reasonable to do so if the substrate itself is not reduced at the cathodeat the given potential. The same concern holds with anode reactions that a mediator partic-ipates in. As seen, photosensitizing and electrocatalysis are quite close, differing in theirelectron origins or electron suckers. Electrochemical reduction or oxidation takes place atthe electrode, which can be described as a concentrated, long-living surface with an excessof electrons or electron holes. As for the photochemical excited state, it can be consideredas containing a local “microelectrode” in an organic molecule. In comparison with a macro-electrode (cathode, anode), this “microelectrode” has a shorter lifetime and an essentiallylower concentration. Despite that, the “microelectrode” appears to be very effective be-cause it is distributed between all solvent cages and occasionally is connected directly witha substrate in a charge-transfer complex.
Hence, the influence of light initiates a one-electron transfer between a reactant anda substrate. This results in the formation of a substrate ion radical. Further reactions includethe generation of a radical that interacts with the second molecule of the reagent. The prod-uct of this step is in ion radical form, and it starts another cycle of substrate conversion inthe newly formed ion radical, at the expense of electron transfer.
Two general advantages of photoinduced redox processes deserve to be emphasized.First, they can be carried out in neat organic solvents, not requiring the use of a conductingsalt as in electrochemistry and not encountering the problems of limited solubility typicalof the use of inorganic compounds. Second, the active redox reagent is in the excited stateformed by light absorption and present at a very low steady-state concentration; this avoidscompetition with overreduction or overoxidation. The latter is an understandable problemwhen the ion radicals are formed in the vicinity of an electrode or in the presence of a sig-nificant concentration of the inorganic reagent.
Attempts to use a heterogeneous rather than a homogeneous sensitizer in the photo-chemical stimulation of ion radical reactions are relevant. Sensitizers can be used as suchor as covalently linked to the surface of silica beads. Supported photosensitizers used as asuspension in liquid offer obvious advantages: relief in the separation of photoproducts,simplicity of analysis, recycling of the sensitizer, and circumventing of poor solubility inthe reaction medium. As an example, the photoreactive deprotection of sulfonamides pro-ceeded smoothly with the participation of the immobilized sensitizer 4-methoxynaphtholcovalently grafted on silica. The sensitizer was filtered after the reaction, washed, and suc-cessfully recycled: The efficiency of the grafted sensitizer remained unchanged (Ayadim etal. 1999). One can use a polymer incorporating the chromophore (Albini & Spreti 1986) orone can place a sensitizer on the semiconductor surface (Fox 1991).
Also worth mentioning is an attempt to use solar light rather than excitation via spe-cial lamps (Cermenati et al. 1998).
One important discrepancy should be noted between photochemical and chemical ionradical reactions. In the photochemical mode, an oxidized donor and a reduced acceptor re-main in the same cage of a solvent and can interact instantly. In the chemical mode, theseinitial products of electron transfer can come apart and react separately in the bulk solvent.For example, one-electron oxidation of phenylbenzyl sulfide results in formation of thecation radical both in the photoinduced reaction with nitromethane and during treatmentwith ammoniumyl species. Sulfide cation radicals undergo fragmentation in the chemicalprocess, but they form phenylbenzyl sulfoxide molecules in the photochemical reaction.The sulfoxide is formed at the expense of the oxygen atom donor. The latter comes fromthe nitromethane anion radical and is directly present in the solvent cage. As for the am-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

moniumyl reaction, the sulfide cation radical reacts as the separated species undergoingfragmentation and then oxidation (Adam et al. 1998).
Here’s another example: The “normal” electrochemical reduction of 4-nitrobenzylthiocyanate leads to the 4-nitrobenzyl radical and thiocyanate. The only products are 4,4-dinitrobibenzyl and 4-nitrotoluene arising from the 4-nitrobenzyl radical (Bartak et al.1971):
4-O2NC6H4CH2SCN � e →(4-O2NC6H4CH2SCN)�. → 4-O2NC6H4CH2
.� �SCN
4-O2NC6H4CH2CH2C6H4NO2-4 ← 4-O2NC6H4CH2. → 4-O2NC6H4CH3
Photoinduced electron transfer in the presence of a sensitizer (9,10-diphenylan-thracene) also generates the same anion radical. However, its disintegration proceedswithin the solvent (acetonitrile) cage. Inside the cage, 4-nitrobenzyl radical and thiocyanateion unite anew, but in this case by their soft-to-soft ends. This nucleophilic reaction takesplace faster than the electron back-transfer occurs. The final, stable product of the wholeprocess is 4-nitrobenzyl-iso-thiocyanate (Wakamatsu et al. 2000):
4-O2NC6H4CH2SCN � e → (4-O2NC6H4CH2SCN)�.→{4-O2NC6H4CH
.2...�SCN} → {4-O2NC6H4CH
.2...�NCS}
→ 4-O2NC6H4CH2NCS � e
One interesting feature of anion radical photochemical behavior has been describedrecently by Camps et al. (2001). The anion radical of bromoadamantane loses bromide andgives the adamantyl radical. This photoactivated radical undergoes 1,5-hydrogen shift. Re-actions of such kinds were not observed before.
5.2.2 Effect of Electric Current
Certain ion radical reactions can be stimulated by means of direct potential imposition,without mediators. In these reactions, the substrate is a depolarizer, and the reagent is aconducting electrolyte. Here are two significant examples demonstrating that thioarylationmay be facilitated electrochemically.
The nucleophilic substitution of bromine by the thiophenolate ion in 4-bromoben-zophenone requires extremely rigid conditions. When a difference in electric potentials isset up, the reaction proceeds readily and gives products in a high yield (80%). It is suffi-cient to set up only the potential difference necessary to ensure the formation of the sub-strate anion radical (with the potential and current strictly controlled). The chemical reac-tion takes place in the bulk solution and yields 4-bromobenzophenone (Pinson & Saveant1974) (Scheme 5-1).
1-Bromonaphthalene does not react with benzenethiol (thiophenol) salts. However,if electric current is passed through a solution containing 1-bromonaphthalene, the tetra-butylammonium salt of thiophenol, and dimethylsulfoxide, then 1-(phenylthio)naphthaleneis produced in 60% yield. When the reaction is conducted in acetonitrile, it leads to naph-thalene above all (Pinson & Saveant 1978; Saveant 1980; Amatore et al. 1982). In the electrochemically provoked reaction, it is sufficient to set up the potential difference cor-responding to the initial current of the reduction wave to transform 1-bromonaphtahaleneinto 1-naphthyl radical. The difference in the consumption of electricity is rather remark-able: In the absence of thiophenolate, bromonaphthalene is reduced, accepting two elec-trons per single molecule; in the presence of thiophenolate, 1-bromonaphthalene is re-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

duced, accepting two electrons per 10 molecules. The reaction with the thiophenolate ionis catalyzed by electric current and takes reaction path shown in Scheme 5-2. This reactionproceeds through the formation of the 1-bromonaphthalene anion radical, which rapidlyconverts into the naphthyl radical. Thiophenolate intercepts the naphthyl radical and formsthe anion radical of 1-phenylthionaphthalene. The reaction takes place in the pre-electrodespace. It competes with the formation of the unsubstituted naphthalene as a result of hy-drogen abstraction from the solvent (SolH). The neutral molecules of the substrate oxidizethe anion radicals formed. The product neutral molecules and the anion radicals of 1-bromonaphthalene are formed. This reaction takes place in the bulk solution. It is the keypoint for the chain propagation.
SCHEME 5-1
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

In addition to bromonaphthalene, bromobenzophenone (see earlier) or 4-bromoben-zonitrile may be used as a substrate (Pinson & Saveant 1978). The latter two compoundscarry not only bromine but also other electrochemically active groups, i.e., CBO or CBMN.Along with the thiophenyl, the thiomethyl or thio-tert-butyl groups can be introduced assubstituting fragments. The yields of the substitution products are high (from 60 to 95%),and one electron is consumed per 20–30 molecules of substrate. The reactions proceed atambient temperature and do not proceed at all when a potential difference is not set up.
Cleavage of a CMS bond in the initially formed anion radical has been shown to be thefirst chemical step in the electrochemically induced rearrangement of S,S-diarylbenzene-1,2-dicarbothioates to 3,3-bis(arylthio)phthalides in dimethylformamide. The reaction can be ef-fected with 0.1 F/mol and is considered a kind of internal SRN1 reaction (Praefke et al. 1980).
The effect of electric current has the following features:
1. The reactions are selective and give products in high yields. They need no acti-vation of a substrate by electron-accepting substituents.
2. The starting compounds have a greater electron affinity than the substitutionproducts. Therefore, the substrate easily accepts an unpaired electron belongingto the product anion radical. This creates conditions needed for the developmentof the chain process, and the reaction becomes catalytic with respect to the cur-rent passed.
SCHEME 5-2
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

3. The electrode potential may be such as to initiate the substrate substitution with-out reducing the product of substitution.
The method of electrochemical initiation of these reactions has some limitations, themain being that the probability of substitution of a leaving group by a nucleophile dependson the nature of the substrate. Let us compare two reactions similar in the solvent employed(dimethylsulfoxide) and the nucleophile used (Bu4NSPh), but different in the chosen sub-strate (4-bromobenzophenone or bromobenzene) (Pinson & Saveant 1978; Swartz & Sten-zel 1984).
Upon electrochemical initiation (Hg cathode), 4-bromobenzophenone gives rise to 4-(phenylthio)benzophenone in 80% yield, while bromobenzene yields diphenyldisulfide witha yield of only 10% and unsubstituted benzene with a yield of more than 95%. In the boro-mobenzene case, this means that the substitution is a minor reaction, while the main route isordinary debromination. As shown (Swartz & Stenzel 1984), the cause of the observed dif-ference consists merely in the specificity of the electrochemical influence when the initial an-ion radicals of the substrate are formed in the pre-electrode space. The less stable anion rad-icals of bromobenzene do not have enough time to go out into the catholyte pool. They giverise to the phenyl radicals in the vicinity of the cathode. The phenyl radicals are instantly re-duced to phenyl anions. They tear protons from the solvent and yield benzene.
The much steadier anion radicals of 4-bromobenzophenone are able to diffuse intothe volume without disintegration. Being in the volume, they lose the bromine anion andform the corresponding radicals, which are intercepted with the thiophenolate anions. Theproduct of this so-to-speak nucleophilic substitution is formed.
Swartz and Stenzel (1984) proposed an approach to widen the applicability of the cath-ode initiation of nucleophilic substitution, consisting of the use of a catalyst to facilitate one-electron transfer. Thus, in the presence of PhCN, the cathode-initiated reaction betweenPhBr and Bu4NSPh leads to diphenydisulfide in such a manner that the yield increases from10% to 70%. Benzonitrile captures an electron and diffuses into the pool, where it meets bro-mobenzene. The latter is converted into the anion radical. The next reaction consists of thegeneration of the phenyl radical, with the elimination of the bromine anion. Since genera-tion of the phenyl radical takes place far from the electrode, this radical is attacked with theanion of thiophenol faster than it is reduced to the phenyl anion. As a result, instead of de-bromination, substitution develops in its chain variant. In other words, the problem is tochoose a catalyst such that it would be reduced more easily than the substrate. Of course, thecatalyst anion radical should not decay spontaneously in solution.
The rate of an electron transfer from the reduced catalyst to the substrate is also im-portant. If the rate is excessively high, the electron exchange will occur within the pre-elec-trode space and the catalytic effect will not be achieved. If the rate is excessively low, toohigh a concentration of the catalyst will be needed. However, at high concentration, the an-ion radicals of the catalyst will reduce the phenyl radicals. Naturally, this will be unfavor-able for the chain process of the substitution. As catalysts, substances should be chosen thatcould be reduced at potentials 50 mV less negative than those of the substrates. The opti-mal concentration of the catalyst should be an order lower than that of the substrate (Swartz& Stenzel 1984).
5.2.3 Effect of Magnetic Field
The effect of a magnetic field on the rate of ion radical reactions has a physical background(Buchachenko 1976; Salikhov 1996). It can be explained not by a change in the energy of
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

the reactions in a magnetic field but rather by the effect of the field on the probability of el-ementary chemical actions. The effect of a magnetic field on processes involving radicalshas been thoroughly discussed (Sagdeev et al. 1977). Increasing the magnetic field strengthfrom 0.398 A�cm�1 (0.5 Oe) to 15,920 A�cm�1 (20,000 Oe) considerably changes the ra-tio of products resulting from the interaction of pentafluorobenzyl chloride with n-butyllithium; in high magnetic fields, the yield of a substitution product, namely, n-pentylpentafluorobenzene, rises considerably. The whole set of products obtained can beseen from the following scheme:
C6F5CH2Cl � BuLi → (C6F5CH2Cl)�.Li� � Bu
.
(C6F5CH2Cl)�.Li� → C6F5CH
.2 � LiCl
C6F5CH.2 �
.H2CC6F5 → C6F5CH2CH2C6F5
Bu.� Bu
. → Bu-Bu
C6F5CH.2 � Bu
. → C6F5CH2Bu
Obviously, the process leading to the substitution product is a chain process:
C6F5CH.2 � BuLi → (C6F5CH2Bu)�.
Li�
(C6F5CH2Bu)�.Li� � C6F5CH2Cl → (C6F5CH2Cl)�.
Li� � C6F5CH2Bu etc.
Sensitivity to the magnetic field intensity has also been observed in the process ofphotoisomerization of trans-stilbene (tS) into cis-stilbene (cSt) in the presence of pyrene(P). The reaction was run in solution (acetonitrile, dimethylsulfoxide, or hexafluoroben-zene as a solvent). During the reaction, spin polarization was established (Lyoshina et al.1980). The spin polarization was explained by the following sequence of elementary ac-tions:
tS � P � hv → tS � P1S → (tS)�.
� (P+.)S ⇔ (tS)�.
� (P+.)T → (tS � PT) � (cS � P)
According to this sequence, formation of cis- and trans-stilbenes is preceded by for-mation of a magnetosensitive ion radical by a singlet–triplet conversion. This means thatspin polarization must be observed in cis- and trans-stilbenes, and the isomerization ratemust depend on the intensity of the magnetic field. These predictions were confirmed ex-perimentally (Lyoshina et al. 1980). Hence, the ion radical route for trans/cis conversion isthe main one under photoirradiation conditions. Until now, the mechanisms assumed forsuch processes have involved energy transfer and did not take into account donor–acceptorinteraction. This interaction makes the process energetically more favorable.
Many organic ion radical reactions are initiated electrochemically. Recent reportsfrom several laboratories have demonstrated that magnetic forces, generated by the inter-action of an external field with current-carrying ions or with redox molecules that possesslarge intrinsic magnetic moments, can lead to significant increases in the rate of the elec-trochemical reaction. For example, the transport of a paramagnetic ion away from the elec-trode surface is greatly facilitated by an externally applied nonuniform magnetic field. Themagnetic field effect results in current enhancements as large as 400% at disk-shaped plat-inum microelectrodes. On the other hand, experiments have been reported in which anonuniform magnetic field generated internally by magnetization of the disk-shaped ironor nickel microelectrodes was used to focus electrochemically generated ion radicals to-ward the electrode surface. In particular, a magnetic field effect has been demonstrated onthe freedom of diffusion of the nitrobenzene anion radical generated electrochemically(Grant et al. 1999 and references therein). The uniform magnetic field of 1 T (generated by
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

an external electromagnet) was applied orthogonally to the exposed surface of the micro-electrode, resulting in magnetization of ferromagnetic (iron, nickel) material. The ion rad-ical, originating near the electrode, possesses a magnetic dipole moment. The redox reac-tion creates a thin layer in the electrode vicinity. It is clear that the thin layer has a volumemagnetic susceptibility different and more than the corresponding susceptibility in the bulksolution. Paramagnetic species have a strong tendency to move toward a region of highermagnetic field (Griffith 1999). If the magnetic force is large enough to overcome thermaldiffusion, then the ion radical will be effectively “trapped” at the electrode surface. Theproof-of-concept experiment described suggests that magnetic forces can be employed tocontrol the spatial position of redox molecules in electrochemical cells.
The concept of electromagnetic electron transfer acceleration is being discussedmore and more with respect to biochemical reactions. Thus, electromagnetic field activa-tion of genes and the synthesis of stress proteins are supposedly initiated through the mag-netic field effect on moving electrons in DNA. This idea is supported by studies showingthat the magnetic fields increase electron transfer rates in cytochrome oxidase. Electro-magnetic fields accelerate electron transfer and appear to compete with the intrinsic chem-ical forces driving the reactions. The “moving charge interaction” model provides a rea-sonable explanation of these effects (Blank & Soo 2001).
5.2.4 Effect of Ultrasound
The ultrasonic irradiation of a solution induces acoustic cavitation, a transient process thatpromotes chemical activity. Acoustic cavitation is generated by the growth of preexistingnuclei during the alternating expansion and compression cycles of ultrasonic waves. Forexample, in aqueous liquid, temperatures as high as 4300 K and pressures over 1000 atmare estimated to exist within each gas- and vapor-filled microbubble following an adiabaticcollapse (Didenko et. al. 1999).
If the yield of a silent reaction is n% after a specific period of time while the yield ofthe corresponding sonochemical reaction is m%, the ratio m/n higher than 1 is described asthe effect of ultrasound. Since its beginning, ultrasound effects have been considered tooriginate in the general phenomenon of cavitation, which generates high temperatures,pressures, and shock waves.
According to the hot-spot theory (Neppiras & Noltingk 1950), the homogeneous ul-trasound reaction takes place in the collapsing cavitation bubble and in the superheated (ca.2,000 K) liquid shell around it. Species with sufficient vapor pressure diffuse into the cav-ity, where they undergo the effect of adiabatic collapse.
However, this commonly accepted theory is incomplete and applies with much diffi-culty to systems involving nonvolatile substances. The most relevant example is metals.For a heterogeneous system, only the mechanical effects of sonic waves govern the sono-chemical processes. Such an effect as agitation, or “cleaning” of a solid surface, has a me-chanical nature. Thus, ultrasound transforms potassium into its dispersed form. This trans-formation accelerates electron transfer from the metal to the organic acceptor; see Chapter2. Of course, ultrasonic waves interact with the metal by their cavitational effects.
It seems reasonable to note that the micro-jet stream generated by the ultrasonic cav-itation promotes mass transport. Such an effect was discussed for proton transport in aque-ous solutions (Atobe et al. 1999). Understandably, a proton moves in the solution as a hy-drated particle. Nevertheless, we should pay attention to the similarity between proton andelectron, in the sense that both are essentially quantum particles. A solvated electron, there-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

fore, can be considered as a species similar to a hydrated proton. Hence, the micro-jetstream can promote electron transfer.
Stripping of an electron from the metal surface and electron transfer from the high-est occupied molecular orbital belonging to an organic donor are similar phenomena. It issignificant that sonication promotes electron-transfer reactions, while ionic processes areessentially insensitive to cavitational phenomena (Luche et al. 1990). Ionic species are notproduced by ultrasound. By and large, sonochemical reactions, either homogeneous or het-erogeneous, correspond to processes in which the production of the reactive intermediate,an ion radical or a radical, is stimulated by ultrasound effects. Although a fully physical ex-planation of such stimulation has not been elaborated yet, there is a set of ultrasonically in-fluenced reactions. The following examples illustrate several principal directions of the ul-trasonic effect.
Sonolysis provokes electron-transfer reactions in which hindered phenols act asdonors (Aleksandrov et al. 1995). The primary importance of this example consists in thehindrance to the donor–acceptor approach, which prevents or significantly hampers over-lapping of the corresponding orbitals.
The lithium salt of 2-nitropropane reacts with 4-nitrobenzyl bromide according toboth ionic and ion radical mechanisms (Russell & Danen 1968). The ionic mechanismleads to the O-alkylation product and, eventually, to 4-nitrobenzaldehyde:
Me2CBNOOLi � BrCH2C6H4NO2-4 → LiBr � Me2CBN(O)MOCH2C6H4NO2-4
Me2CBN(O)-OCH2C6H4NO2-4 → Me2CBNOH � OBCHC6H4NO2-4
The ion radical route leads to the C-alkylated product, namely, 2-(4-nitrobenzyl)-2-nitro propane:
Me2CBNOOLi � 4-O2NC6H4CH2Br → Me2CNO2 � (4-O2NC6H4CH2Br)�.Li�
(4-O2NC6H4CH2Br)�.Li� → LiBr � [4-O2NC6H4CH
.2]
[4-O2NC6H4CH.2] � Me2CBNOOLi → [4-O2NC6H4CH2C(Me2)NO2]�.
Li�
[4-O2NC6H4CH2C(Me2)NO2]�.Li�
� 4-O2NC6H4CH2Br → LiBr � [4-O2NC6H4CH.2]
� 4-O2NC6H4CH2C(Me2)NO2
[4-O2NC6H4CH.2] � Me2CBNOOLi → [4-O2NC6H4CH2C(Me2)NO2]�.
Li� etc.
Experiments in deoxygenated ethanol solution in the dark showed that, by stirring, 4-nitrobenzaldehyde is obtained in 60% yield, accompanied by 13% 2-(4-nitrobenzyl)-2-ni-tro propane. Under the same conditions, but with sonication, the yields are 23% 4-ni-trobenzaldehyde and 48% 2-(4-nitrobenzyl)-2-nitropropane (Einhorn et al. 1990). Theimportance of this result consists in the following conclusion: The ultrasonic irradiation hasa marked influence on the relative rates of the competing reactions and stimulates just theion radical one.
Romanian scientists compared one-electron transfer reactions from triphenylmethylchloride to nitrobenzene in thermal (210°C) conditions and upon ultrasonic stimulation at50°C (Iancu et al. 1992; Vinatoru et al. 1994). In the first step, the triphenylmethyl chlo-ride cation radical and the nitrobenzene anion radicals are formed. In both cases theirtransformations yield the same set of products, with one important exception: The ther-mal reaction leads to formation of HCl, whereas ultrasonic stimulation results in Cl2 evo-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

lution. At present, it is difficult to elucidate the mechanisms behind these two reactions.As an important conclusion, the sonochemical process goes through inner-sphere electrontransfer. The outer-sphere electron-transfer mechanism is operative in the thermally in-duced process.
According to Ando and co-authors (2000), the sonolytic acetoxylation of styrene bylead tetraacetate follows the ion radical mechanism. Lead tetraacetate was not subject to thesonication influence. The ultrasonic effect facilitates electron transfer from styrene (thenonmetallic donor) to lead tetraacetate.
5.3 EFFECT OF CHEMICAL ADDITIVES
Some chemical additives can induce ion radical formation and direct the reaction along theion radical route. The effect was discovered and studied in cases of nucleophilic substitu-tions of cumene derivatives (Kornblum 1975, 1982). Cumyl radicals are formed at the firststep of substitution irrespective of whether a dissociative or homolytic cleavage takes placeas a result of electron transfer to the cumene derivatives (Zheng et al. 1999).
As it turned out, the cumyl radical could be trapped not only by those nucleophilicions, part of which were spent to generate the initial anion radical, but also with other an-ions. Hence, the products of substitution may also be formed with anions that either do notenter into a common reaction with the substrate or react with it slowly. In other words, thevery small amount of a reactive nucleophile may induce the reaction.
Thus, sodium azide and �,p-dinitrocumene do not react unless subjected to the actionof light (48-hr control period). In contrast to sodium azide, the lithium salt of 2-nitro-propane reacts with �,p-dinitrocumene in the dark for 3 hr, giving the product of �-substi-tution in 87% yield. When �,p-dinitrocumene (1 mole) is treated with sodium azide (2moles) in the presence of the lithium salt of 2-nitropropane (only 0.1 mole), the initial �,p-dinitrocumene quantitatively converts into p-nitrocumyl azide for 3 hours. The product isextremely pure, and the reaction requires no UV irradiation (Kornblum et al. 1970)(Scheme 5-3). Typical one-electron donors, e.g., sodium naphthalene, also induced the re-action of p-nitrocumyl chloride with sodium nitrite (Kornblum et al. 1970).
Zoltewicz and Oestrich (1973) employed sodium methylate to accelerate the reactionbetween 4-bromo-iso-quinoline and sodium thiophenolate. In this case, the CH3O� ion actsas a competing electron donor with respect to the PhS� ion. Upon electron transfer to thesubstrate, thiophenolate converts into the phenylthiyl radical and then to diphenyldisul-phide. Diphenyldisulfide is inactive in further transformations. The methylate ions gener-
SCHEME 5-3
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

ate the anion radicals of the substrate, thus preserving the greater part of the thiophenolatefor use in substitution. The observed rate of thioarylation and the yield of 4-phenylthio-iso-quinoline increase in the presence of sodium methylate. Azobenzene inhibits the action ofsodium methylate. Scheme 5-4 summarizes what has been said.
It is important to note that sodium methylate initiates only the formation of 4-phenylthio-iso-quinoline; the product of the competing substitution, 4-methoxy-iso-quino-line, is produced only in traces. The methylate ion, however, converts a part of the iso-quinoline �-radicals into the unsubstituted iso-quinoline and produces formaldehyde.
Let us now consider the conversion of nitro compounds into mercaptanes. Nitro com-pounds were treated with a mixture of sodium sulfide with sulfur and then reduced with alu-minum amalgam (Kornblum & Widmer 1978) (Scheme 5-5).
The first stage of the synthesis involves the interaction of a nitro compound withsodium sulfide. When used alone, sodium sulfide is only slightly effective: The reactionsproceed slowly and the yields of mercaptanes are small. If elemental sulfur is added, theconversion accelerates markedly and the yield increases to 75–80%. The promoting effectof elemental sulfur can easily be explained by the radical-chain mechanism. The reactionstarts with one-electron transfer from the nucleophile to the nitro compound; further con-
SCHEME 5-4
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

versions resemble other chain ion radical substitutions:
S2� � S → �SMSn�
R3MNO2 � �SMSn� → (R3CMNO2)�.
�.SMSn
�
R3CMNO2)�. → NO2� � R3C
.
R3C.� �SMSn
� → R3CMS�MSn�
R3CMS�MSn� � R3CMNO2 → (R3CMNO2)�.
� R3CMSMSn�
R3CMSMSn� Al/Hg→ R3CMS� � Sn
�
Obviously, the donor activity of the nucleophile, i.e., the sulfide ion, is enhanced asthe negative charge is dispersed along the polysulfide ion produced from the sulfide uponthe addition of elemental sulfur. This increases the mobility of the electrons and facilitateselectron transfer. That is why this reaction can be initiated in such a simple way as the ad-dition of elemental sulfur.
The chemical entrainment method was used by Ono et al. (1979) to eliminate the ni-tro group in nitroalkene derivatives. Upon simple mixing with thiophenol and sodium sul-fide in dimethylformamide, nitro aryl olefins substitute hydrogen for the nitro group:
The authors hold to the opinion that the thiophenolyc moiety adds to the olefin bondand an electron adds to the nitro group. Hence, the anion radical R1R2C(SPh)CH(R3)NO2
�.
controls the reaction. The final product is formed as a result of the cleavage of the latter an-ion radical with the expulsion of the nitrite ion and the phenylthiyl radical. The radical nor-mally transforms into diphenyldisulfide. The yields of the denitrated olefines are high andreach 80–95%.
Na2S � PhSH12
R1 R3
R2 NO2
C C �BJ
JJ
J
R1 R3
R2 H
C CBJ
JJ
JPhS1
2MSPh � S � NaNO2 �
→
SCHEME 5-5
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The production of olefines from -nitrosulfones provides another example of the ef-fect of chemical additives. The reaction is conducted in dimethylformamide without lightirradiation. Usually, sodium sulfide is used as a reagent, but the sodium salt of thiophenolmay be used instead of sodium sulfide. In the case of Na2S, the olefin forms in 97% yield(Ono et al. 1980, 1983):
Me CN Me CN
Na2S � MeMCMCMBu → MeC6H4SO2Na � S � NaNO2 � MeCBCBu NO2SO2C6H4Me
This process is of interest because the reaction reveals an unexpected effect of suchadditives, which usually hinder anion radical reactions. According to the authors, the reac-tion has the following ion radical mechanism:
Me CN Me CN
MeMCMCMBu �e→ �MeMCMCMBu ��.
→ NO2SO2C6H4Me NO2SO2C6H4Me
Me CN
NO.2 � MeC6H4SO2
� � MeMCBCMBu
The authors attribute the olefin formation to elimination of the sufinate ion and ni-trogen dioxide. Small amounts of di-tert-butylnitroxide (5 mol. %) completely inhibit theproduction of olefin. This points to the chain free-radical nature of the process. Aromaticnitro compounds, which remove electrons from the anion radical participating in the reac-tion, usually inhibit ion radical conversions. In the reaction scheme just depicted, m- and p-dinitrobenzene (in amounts not exceeding 10 mol. %) markedly accelerate the reaction.Three acceptor groups (NO2, CN, SO2Ar) in the anion radical seemingly keep an unpairedelectron from being removed with the leaving group. Probably, upon addition of aromaticnitro compounds into the reaction sphere, a donor–acceptor complex is formed with thesubstrate anion radical. The unpaired electron shifts to a �-acid (dinitrobenzene) in theframework of the complex. This increases electron mobility and catalyzes elimination of aleaving group taking the superfluous electron away.
Consequently, in ion radical reactions, inhibitors may become promoters. Thisshould also be taken into account when developing ways to stimulate reactions of an ionradical nature.
The reactions discussed so far involved anion radicals. Now we want to turn to con-versions of the cation radical type. One of these reactions is anisylation of the thiantrenecation-radical (Svanholm et al. 1975; Hammerich and Parker 1982) (Scheme 5-6). Thereaction gives the sulfonium salt (anion ClO4
�) in 90% yield (route a). One-electron re-duction of the thianthrene cation radical by anisole is the side reaction (route b). Route bleads to products with a 10% total yield. Addition of the dibenzodioxine cation radicalaccelerates the reaction 200 times. The cation radicals of thianthrene and dibenzodioxineare stable. Having been prepared separately, they are introduced into the reaction as per-chlorate salts.
When the concentration of the thianthrene cation radical drops from 10�3–10�4 M to10�5 M, the reaction results are changed: The sulfonium salt is not produced at all (route a
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.
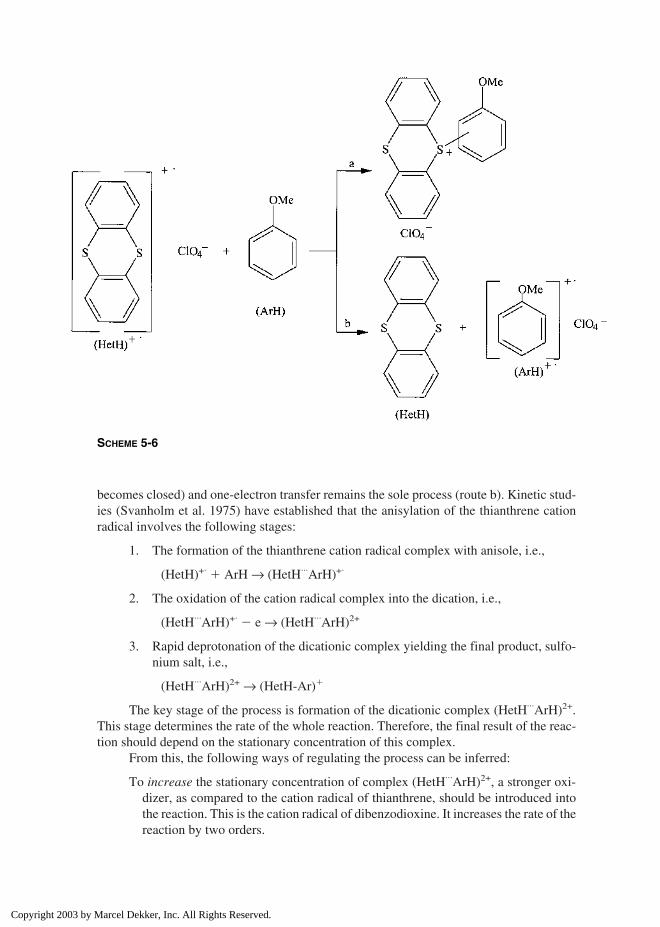
becomes closed) and one-electron transfer remains the sole process (route b). Kinetic stud-ies (Svanholm et al. 1975) have established that the anisylation of the thianthrene cationradical involves the following stages:
1. The formation of the thianthrene cation radical complex with anisole, i.e.,
(HetH)+.� ArH → (HetH
...ArH)+.
2. The oxidation of the cation radical complex into the dication, i.e.,
(HetH...
ArH)+.� e → (HetH
...ArH)2+
3. Rapid deprotonation of the dicationic complex yielding the final product, sulfo-nium salt, i.e.,
(HetH...
ArH)2+ → (HetH-Ar)�
The key stage of the process is formation of the dicationic complex (HetH...
ArH)2+.This stage determines the rate of the whole reaction. Therefore, the final result of the reac-tion should depend on the stationary concentration of this complex.
From this, the following ways of regulating the process can be inferred:
To increase the stationary concentration of complex (HetH...
ArH)2+, a stronger oxi-dizer, as compared to the cation radical of thianthrene, should be introduced intothe reaction. This is the cation radical of dibenzodioxine. It increases the rate of thereaction by two orders.
SCHEME 5-6
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

To decrease the stationary concentration of complex (HetH...
ArH)2+, it will suffice tolower the concentration of the oxidizer, i.e., substrate (HetH)+.
. This also decreasesthe equilibrium concentration of the cation radical complex (HetH
...ArH)+.
. The rateof anisylation—the main process—drops sharply. The side process, one-electrontransfer from anisole to the cation radical of thianthrene, also decelerates, but notso markedly. So this side process (route b on Scheme 5-6) remains the only one.
Shono et al. (1979) recommend the use of thioanisole as a catalyst that allows low-ering the electrode potential in the oxidation of the secondary alcohols into ketones. Thecation radical of thioanisole is generated at a potential of up to �1.5 V in acetonitrile con-taining pyridine (Py) and a secondary alcohol. (The background electrolyte was tetraethy-lammonium p-toluene sulfonate.) Thioanisole is recovered and therefore a ratio ofR1(R2)CHOH:PhSMe � 1:0.2 is sufficient. The yield of ketones depends on the nature ofthe alcohol and varies from 70% to 100%:
PhSMe �e→ (PhSMe)+.
R1(R2)CHOH � (PhSMe)+. → R1(R2)CHOMS�Ph � H�
Me
Py � H� → PyH�
R1(R2)CHMOMS�Ph → PhSMe � R1(R2)CBOMe
Let us now turn to the role that oxygen plays in ion radical conversions. As a com-ponent of air, oxygen is a typically an active part of the medium in which chemical con-versions mainly proceed.
Cation radicals are often unstable and dissociate. If the dissociation is an equilibriumprocess, oxygen promotes it, because oxygen reacts with fragment ions and radicals pro-duced in the decomposition. Therefore scientists prefer to conduct reactions with the par-ticipation of cation radicals in an inert atmosphere, although oxygen (air) is, strictly speak-ing, inert with respect to primary cation radicals. Exceptions to this are only those reactions(e.g., biological ones) where oxygen is required to obtain cation radicals from neutralmolecules.
For anion radicals, air (i.e., oxygen, carbon dioxide, and air), on the whole, is an ac-tive component of the medium and so it should be removed prior to conducting reactions.
Understandably, air inhibits anion radical reactions: the anion radicals primarilyformed are consumed at the expense of oxidation, carboxylation, and protonation. Cer-tainly, oxidation can take place only if the acceptor organic molecule possesses a loweraffinity for an electron than does oxygen or if one-electron oxidation of the anion radicalby oxygen proceeds more rapidly than the decomposition of the anion radical into radicaland anion (RX�. → R
.� X�.
).If the oxidation proceeds more slowly than the decomposition, oxygen may affect the
nature of the reaction products. Thus, treating p-nitrocumyl chloride with sodium malonateester in a flow of pure dry nitrogen yields a product of C-alkylation (route a in Scheme 5-7); the yield is 90%. Oxygen completely inhibits the C-alkylation, and the reaction gives p-nitrocumyl alcohol in the same yield (Kornblum et al. 1968).
When the malonate is absent, oxygen is incapable of converting p-nitrocumyl chlo-ride into the alcohol (Kornblum et al. 1968). In the presence of oxygen, the reaction devel-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

ops in two directions, with different rates. The reaction first produces anion radicals of p-nitrocumyl chloride (the sources of electrons are the malonate anions), and then chlorideions cleave from these anion radicals. Nitrocumyl radicals accumulate at a faster rate thanthe initial anion radicals perish under the effect of oxygen. Therefore oxygen may trap onlycumyl radicals and give cumyl peroxide radicals, which convert into the hydroperoxidewhen abstracting hydrogen from the solvent. The hydroperoxide decomposes and formscumyl alcohol. Because the yield of the alcohol reaches 90%, the conclusion follows thatthat the conversion is highly selective. It follows the scheme:
4MO2NC6H4C(Me)2Cl NaCH(COOEt)2→ [4MO2NC6H4C(Me)2Cl]�. →
�Cl�→ 4MO2NC6H4C.Me2 �O2→ 4MO2NC6H4CMe2 →
OO
.
→ 4MO2NC6H4CMe2 �O→ 4MO2NC6H4CMe2
O OH OH
Then the case is possible when the acceptor ability of oxygen is lower than that of thesubstrate but greater than that of the charged (intermediate or final) products of anion rad-ical conversions. The superoxide ion is produced in the final stages of the process and actsas an electron carrier with respect to the substrate, thus branching the chain process. Inother words, oxygen promotes rather than inhibits these reactions. This case is especiallyimportant for the practice of organic synthesis. As has been reported (Omelechko et al.1982), the reaction between 1-nitroanthraquinone and sodium methylate in the mixture ofdimethylsulfoxide with methanol accelerates when conducted in air and not under argon.In the absence of air, anion radicals of 1-nitroanthraquinone obtained in dimethylsulfoxideelectrolytically do not change upon the addition of MeOH or MeONa. When air gains ac-cess to the system, the anion radicals are consumed completely and produce 1-methoxyan-thraquinone (the main product) and 1-hydroxyanthraquinone (the admixture). Oxygentherefore promotes methoxylation via the anion radical mechanism. An earlier study re-vealed that methoxylation of 2,4-dinitrochlorobenzene accelerates by an order of magni-tude when conducted in air rather than in nitrogen and the following mechanistic scheme
SCHEME 5-7
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

was proposed (Blumenfel’d et al. 1970) (Scheme 5-8).As seen from the scheme, not the initial substrate but rather its anion radical under-
goes methoxylation. This produces the anion radical of the �-complex, which is oxidizedby oxygen into the conventional anionic �-complex carrying no unpaired electron. Thisstage generates the superoxide ion, which later competes advantageously with the methox-ide ion for the starting 2,4-dinitrochlorobenzene to reduce it into the anion radical.
The main point with respect to the catalytic effect of oxygen is the ability of the su-peroxide ion to transfer electrons to strongly accepting molecules of the substrate. This wasconfirmed by electrochemical generation of the superoxide ion: o- and p-nitrochloroben-zenes and o-nitrobromobenzene react with these ions in dimethylformamide, giving o- andp-nitrophenols (Sagae et al. 1980). Frimer and Rosenthal (1976) treated 2,4-dinitrobro-mobenzene with K18O2 in the presence of dicyclohexane-18-crown-6-ether (the solventwas benzene saturated with 16O2) and obtained 2,4-dinitrophenol carrying practically no18O. According to mass-spectrometric data (Frimer & Rosenthal 1976), the content of thelabel in phenol was below 10%. Hence, superoxide ion 18O2
�.transfers electrons only to the
substrate, and phenol is produced as a result of the reaction between the anion radical of2,4-dinitrobromobenzene or 2,4-dinitrophenyl radical and oxygen-16O.
It should be underlined that reactions conducted in traditional ways in inert atmospheremay sometimes fail. Oxygen accelerates the reactions involving strongly acceptor substrate.This is similar to a promoting effect of active organic oxidizers of the dinitrobenzene type.
The first example of such catalytic reactions was described almost half a century ago(Russell 1954). A carbanion (R�) reacts with O2 according to the mechanism of catalysiswith a one-electron transfer:
R� � O2 → R.� O2
�.
R.� O2 → ROO
.
ROO.� R� → ROO� � R
.
ROO� → RO� � O
O � O → O2
R.� O2 → ROO
.etc.
SCHEME 5-8
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Electron-transfer catalytic cycles with oxygen were also discovered in photochemi-cal reactions with the participation of an excited sensibilizer (9,10-dicyanoanthracene,DCNA) and stilbene. The sensitizer assists an electron transfer from the substrate to oxy-gen. Oxygen transforms into the superoxide ion. Stilbene turns into benzaldehyde. In theabsence of the sensitizer, this reaction does not take place, even upon photoirradiation(when oxygen exists in the first singlet state). In the singlet state, oxygen acquires enhancedoxidative ability, but only to a limited degree. At the same time, the superoxide ion reactswith olefin cation radicals 30 times faster (Julliard & Chanon 1983):
DCNA → (DCNA)*
PhCHBCHPh � (DCNA)* → (DCNA)�.� (PhCHBCHPh)+.
(DCNA)�.� O2 → DCNA � O2
�.
(PhCHBCHPh)+.� O2
�. → PhCHMCHPh → 2PhCHO OMMO
DCNA → (DCNA)* etc.
In the absence of oxygen, photoirradiation of stilbene with the same sensitizer(DCNA) provokes cis–trans isomerization of the olefine. The reaction is initiated with 365-nm light and proceeds at 25°C (Lewis et al. 1985):
DCNA → (DCNA)*H H H H
PhCBCPh � (DCNA)* → (DCNA)�.�
�PhCBCPh��.
H H H �PhCBCPh�
+.
→ �PhCBCPh��.
H
Η Η Η Η Η Η �PhCHBCHPh�
�.
� PhCBCPh → �PhCHBCHPh��.
� PhCBCPh H H
(DCNA)�.� (PhCHBCHPh)+. → DCNA � PhCHBCHPh
DCNA → (DCNA)* etc.
This reaction gives us an opportunity to consider the roles of the salt additive, the solventpolarity, the stilbene concentration, the temperature level, and the intensity of photoirradi-ation. The reaction is facilitated by the replacement of the nonpolar solvent (benzene) by apolar one (acetonitrile), by a rise in the reaction temperature, by an increase in the stilbeneconcentration, by a decrease in the irradiation intensity, or by the addition of alkali metalsalts. All these factors intensifying the process are related directly to the mechanism justdescribed. It is substantial enough to analyze the effects of these factors on the efficiencyof the photoreaction.
An increase in the cis-stilbene concentration favors the chain propagation and de-creases the probability of termination when the dicyanoanthracene anion radicals react withthe stilbene cation radicals. A decrease in the irradiation intensity has a similar effect: Thechain propagation is the first-order process, whereas termination of the chains is the sec-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

ond-order process. Temperature rise accelerates the accumulation of the stilbene cationradicals. In this system the free energy of electron transfer is �53 � �44 kJ�mol�1 (thecation radical generation is in fact an endothermal process). If a polar solvent is substitutedfor a nonpolar one, the conversion of the cis-stilbene cation radical into the trans-stilbenecation radical deepens because free cation radicals isomerize more easily than the ion rad-ical pairs. The stilbene cation radical not shielded with a counterion has a more positivecharge and therefore becomes stabilized in the more polar solvent. In other words, isomer-ization is less effective when (cis-stilbene)+.
forms an ion pair with (DCNA)�.. When Na-
ClO4 (alkali metal salt) is added, (DCNA)�.becomes bonded in another (more compact and
therefore more stable) ion pair, namely, (DCNA)�.Na�. As a result, the cation radical of
cis-stilbene is liberated. In addition, chain termination caused by the interaction of the stil-bene cation radicals with the dicyanoanthracene anion radicals becomes less probable be-cause the cation radical and the anion radicals are not as close to each other as in a unitedion radical pair.
This example shows the important role of the solvent’s nature and the salt additive inion radical transformations. These two significant factors are examined in the followingtwo sections.
5.4 SOLVENT ROLE
Organic ion radical reactions in solution are among the most important reactions in chem-istry and biology. A particularly significant question in chemical reaction dynamics in so-lution is the influence of the solvent on the direction and rate of the reaction. Medium ef-fects on electron-transfer reactions (which lead to ion radical formation) are usuallyclassified as static or dynamic interactions between the solvent and reacting solutes. Staticeffects refer to the stabilization of reactants, transition states, and products, that is, how thesolvent affects the free energies of these species and the activation energy of the reaction.This interpretation of solvent effects on all kinds of chemical reactions is well established.There is an extensive body of reviews and monographs on the topic. A more recent devel-opment is the investigation of the influence of solvent dynamics on the reaction rates. Ar-ticles by Heitele (1993), Drago and Ferris (1995), and Leite (1999) have described thesemodern developments in the theory and experimental study of electron-transfer processesespecially. The transfer of an electron is triggered by a fluctuation of the dielectric polar-ization in the surrounding solvent. Formation of ion radicals, which are a more polar com-ponent of the solution than the starting neutral molecules, can lead to nonspecific (non-bonding) solvation. The term specific solvation is meant to describe some selectivity ininteraction between a solvent and a solute skeleton fragment, in contrast to a general (bulk)dielectric effect.
5.4.1 Static Effects
The fewer factors that lower ion radical stability, the more easily ion radical organic reac-tions proceed. Because ion radicals are charged species with unpaired electrons, solventsfor the ion radical reactions have to be polar too, incapable of expelling cationic or anionicgroups that the ion radical bears as well as chipping off radicals from it (especially to ab-stract the hydrogen atom). Several examples are discussed next.
Photoinduced electron transfer from 4-chlorophenylthiolate to 2-nitro-2-thiocyanatopropane leads to the formation of the following ion radical pair, IRP (Al-Khalil & Bowman
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

1984):
4-ClC6H4S� � Me2C(NO2)SCN → [4-ClC6H4S., Me2C(NO2)SCN�.
]
(IRP)
The final destiny of IRP depends on the nature of the solvent. In dimethylsulfoxide(DMSO), the main process is an inner-cage recombination (route a), a minor process con-sists of IRP disintegration after its diffusion into volume (route b):
a. IRP → 4-ClC6H4SC(NO2)Me2 � �SCNb. IRP → 4-ClC6H4SSC6H4Cl-4 � �SCN � Me2CBNOO�
Both a and b are nonchain processes because the yields of the final products (54 and35%, respectively) do not change upon addition of p-dinitrobenzene, tert-butylnitrone aswell as in the presence of oxygen. Substitution of MeOH for DMSO as the solvent sup-presses route a entirely. As suggested (Al-Khalil & Bowman 1984), the presence of a pro-ton in the methanol solvent cage stabilizes the nitro thiocyanate anion radical. Such stabi-lization prevents inner-cage recombination and contributes to diffusion of theelectron-transfer products into the solution volume.
Solvent stabilization can often determine the initial steps of ion radical generation.Hence, alkali metal hydroxides are highly stabilized in water and in aqueous solvents, andtherefore their reactivities in simple one-electron processes are either very low or practi-cally nonexistent. Nevertheless, polar solvents, such as DMSO, hexamethylphosphotri-amide (HMPA), and THF, in which alkali metal hydroxides are at least somewhat soluble,particularly in the presence of water, diminish drastically the HO� solvation (Popovich &Tomkins 1981). As a result, reactions of one-electron transfer from the hydroxy anion tothe substrate can take place (Ballester & Pascual 1991).
Potassium hydroxide when merely dissolved in methanol is not effective in the elec-tron-transfer reduction of 9-diazofluorene and fluoren-9-ylides. Addition of DMSO to thesystem makes a drastic change, with the highest efficiency in pure DMSO (Handoo & Kaul1992; Handoo et al. 1983).
At this point a common method of conversion of tertiary aliphatic nitro compoundsinto nitromethyl derivatives and, further, into aldehydes deserves mention (Kornblum &Erickson 1981). According to this method, the reagent NaCH2NO2 is used. To prepare thisreagent, sodium hydride reacts with nitromethane. Then a tertiary aliphatic nitro compoundis introduced into the solution formed. Several organic solvents were probed. The reactionconsidered proceeds most effectively in DMSO. Kornblum and Erickson (1981) attributedthis feature to small amounts of NaCH2SOCH3 (sodium dimsyl) produced in DMSO at theexpense of its reaction with sodium hydride. Sodium dimsyl acts as a powerful one-elec-tron reducer that induces the following chain anion radical process:
Me2RCNO2 � NaCH2NO2 → Me2RCH2NO2 → Me2RCHO
Me2RCNO2 � NaCH2SOCH3 → (Me2RCNO2)�.Na� � CH3SOCH
.2
(Me2RCNO2)�.Na� → NaNO2 � Me2RC
.
Me2RC.� NaCH2NO2 → (Me2RCNO2)�.
Na� etc.
CH3SOCH.2 � H
. → CH3SOCH3
These examples show how a solvent changes the direction of an ion-radical reaction.Another important problem is to choose a solvent that is capable of making ion radicals livelonger or even of detaining certain reactions at the ion radical stage. For cation radicals,
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

1,1,1,3,3,3-hexafluoropropan-2-ol (HFP) has proven (Eberson et al. 1996) to be an excel-lent solvent. It has a rare combination of low nucleophilicity, high hydrogen bonding donorstrength, low hydrogen bonding acceptor strength, high polarity, and high ionizing power.It allows the use of mild conditions for the generation of radical cations from chemicallysensitive compounds and avoids the need for cooling or the use of flow systems. In com-parison to trifluoroacetic acid (a common solvent for the generation of cation radicals, seechapter 2), HFP is 109 times less acidic, although it does have the proton donor hydroxylgroup. Therefore, it is also convenient for generating cation radicals that are acid sensitive.It solvates anions extensively but cations poorly. Being a weak hydrogen bond acceptor,HFP is uniquely strong as a hydrogen bond donor. As illustrative examples, it is worth not-ing its very slight ability to form the self-associated dimers (Murto et al. 1971) and, on theother hand, its potential to form an azeotropic mixture with THF (Middleton & Lindsey1964). Thus, HFP is just the solvent for polar compounds. The scope and limitations of HPFapplicability as a solvent for ESR spectral observation of cation radicals as well as a sol-vent for anode electrochemistry are detailed by Eberson and co-authors (1995, 1996); seealso Tabakovich et al. (1996) and Barbosa et al. (1998).
By and large, high hydrogen bonding donor strength of a solvent can influence thefate of ion radicals formed in a reaction. Workentin and co-authors (1994) described an in-teresting solvent effect on the competition between electron transfer and the addition reac-tion between organic cation radicals and azides. 2,2.2-Trfluoroethanol (TFE) and acetoni-trile (AN) were compared as solvents. In TFE the cation radicals of 4-methoxystyrene, -methyl-4-methoxystyrene, or , -dimethyl-4-methoxystyrene react with the azide ionaccording to the following scheme:
(CH3OC6H4CHBCR1R2)+.� N3
� → CH3OC6H4CH.C(N3)R1R2
Rate constants (k � 10�9 M�1sec�1) were determined to be 7.0, 3.5, and 1.0 forthe enumerated substrates, respectively. The change in kinetics for the three cation radi-cals with increasing steric hindrance at the -carbon is in accordance with the depictedaddition reaction. In contrast with that, a reaction of the azide ion with these three cationradicals in acetonitrile proceeds with rate constants that are the same in all three cases(~3 � 109 M�1sec�1). In acetonitrile, the reaction consists of one-electron transfer fromthe azide ion to a cation radical. As a result, a neutral styrene and the azidyl radical areformed. The azidyl radical reacts with the excess azide ion, and the addition reaction doesnot take place:
(CH3OC6H4CHBCR1R2)+.� N�
3 → CH3OC6H4CHBCR1R2 � N.3
N.3 � N�
3 ⇔ N�.6
The change in mechanism from electron transfer in acetonitrile to addition in TFE isdetermined by the relative oxidation potentials of the styrenes and the azide ion in these twosolvents. For example, in acetonitrile the oxidation potential of the azide ion is approxi-mately 0.6 eV lower than that of the styrenes, indicating that electron transfer should occurat the diffusion-controlled rate, as observed. The oxidation potentials of styrenes in TFE donot differ substantially from those in acetonitrile. However, the oxidation potential of theazide anion increases drastically in TFE, presumably due to stabilization of the anion byhydrogen bonding. The shift in oxidation potential means that electron transfer is not suf-ficiently exergonic to occur at a diffusion-controlled rate, allowing addition to compete ef-fectively with electron transfer. The results demonstrate, on the one hand, the importanceof the hydrogen bonding activity of a solvent and, on the other hand, the importance of re-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

dox properties (which reflect relative solvation energies) in determining the selectivity ofion radical reactions.
Another example involves alkylation of the lithium salt of the anthracene anion rad-ical by 2-octyl fluoride (Herbert et al. 1985). The akylation does not occur in dimethylfor-mamide (which strongly solvates Li�), whereas it is facilitated and becomes a quantitativereaction in diethyl ether. Coordination of the type ( CHMF....Li�), which is possible in aslightly basic solvent such as diethyl ether, seems to be the decisive factor in the reaction.
It is worthwhile mentioning that there are some solvents that combine good solvencypower with coordinating properties. The most salient example is 1,2-dimethoxyethane(DME), which can form chelate complexes with alkali cations. This makes easier one-elec-tron reduction of organic substances by means of alkali metals, with the formation of anionradicals and alkali cations.
Another point of such coordination activity is the specific interaction of the solventscontaining heteroatoms with cation radicals having a suspended unpaired electron (cf.Chapter 3). A more pertinent example in this context is the interaction between the di-alkylsulfide cation radicals and the oxygen belonging to the water molecule. Such interac-tion enhances the stability of the “coordinated” cation radical:
R2S+.� H2O ⇔ (R2S�OH2)�
Of course, the strength of the intermolecular three-electron bond depicted here islower than that of the intramolecular three-electron bond (see section 3.2.3). Interestingly,no dimer complex (t-Bu2S � St-Bu2)� was formed. The strength of the S � S bond in thisdimer is smaller than the strength of the S � O bond in the (t-Bu2S � OH2)� complex (As-mus 1990).
Certainly, cases are possible of a specific interaction between a solvent and an ionradical. The qiunone ion radicals are examples. The cation radicals of 1,4-benzoqinonealkoxy derivatives form very stable O–D bonds when frozen in the D2O–D2SO4 solventmixture (Spoyalov et al. 1992). The phenomenon is close to redox-enhanced hydrogenbonding and stabilization of the ubiquinone anion radical through complexation withthiourea (Greaves et al. 1999). Hydrogen bonding was established between 1,4-bis(dihy-droquininyl)-anthraquinone and the liquid ammonia solvent. This anion radical is prone torearrangement, but the hydrogen bonding conserves the initial anion radical structure (Kim& Stevenson 1999). Generation of hydroxide and the protonated quinone dianion duringelectron transfer to 3,5-di(tert-butyl)-1,2-benzoquinone also can be explained by the coor-dination of water with the quinone anion radical (Lehmann & Evans 2001).
It should be also mentioned that there are some cases when the cation selective sol-vation promotes greater �-electron delocalization in remaining “free” anion radicals; seeSection 3.2.5. Staley and co-workers (1999 and Staley and Kehlbeck 2001) studied the phe-nomenon for the organic dianion too. Such a solvent effect leads to changes in the reactiv-ity of these species.
5.4.2 Dynamic Effects
An electron transfer occurs momentarily between neighboring molecules. No nuclear mo-tions in the molecules occur during this transfer. [The Frank–Condon principle Transfer ofan electron takes place much faster than nuclear movements]. Therefore, such a rapid elec-tron transfer may take place only when the geometry of the ion radical and the parentmolecule are similar (Warman 1982). Solvent relaxation is assumed to be much faster than
M
M
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

electron transfers (Heitele 1993). This means small-amplitude motions of an individual sol-vent molecule that experiences only weak friction with the surrounding liquid. Once anelectron transfer has occurred, excess energy is dissipated fast enough to trap the system onthe product side. For weak frictional or random forces, the system undergoes several oscil-lations between the reactant and the product wells before finally setting down in one ofthem. The reaction rate is limited by how fast the necessary activation energy is acquiredand how fast excess energy in the course of the reaction is dissipated to trap the system ineither well.
5.4.2.A Solvent Reorganization
Bimolecular electron-transfer reactions in solution frequently have rates limited by the dif-fusion of the donor and acceptor molecules, because one or both of the reactant species isusually at a low concentration relative to the solvent. To obtain a detailed mechanistic andkinetic understanding of electron-transfer reactions in solution, chemists have devised in-genious schemes in which the two reactants, the donor and acceptor, are held at a fixed dis-tance and orientation so that diffusion will not complicate the study of the intrinsic elec-tron-transfer rates. Recent developments, however, have led to theoretical models in whichthe orientation and distance are changeable (see Rubtsov et al. 1999).
In order for an electron transfer to take place, the energy levels of the donor and theacceptor must match. In other words, the donor, the acceptor, and their solvate shells musthave a configuration that fits the equilibrium state of the product system (for examples, seeGuldi et al. 2001). According to Marcus theory (1964), such a balance is attained by bondand solvent reorganization. The bond and solvent reorganization energies are denoted as �i
and �o (after inner and outer). The sum of �i and �o is �, the reorganization energy. Bondreorganization involves bond stretching and/or compression, angle deformation, and thechanging of torsion moments. Solvent reorganization involves induced changes in the elec-trostatic environment around the reactants. Marcus theory was crucial in the developmentof electron-transfer studies. It created indispensable demands for reliable values of standardpotentials and reorganization energies. It allowed calculating electron-transfer rate con-stants and comparing them with experimental values. In Marcus theory, the solvent is de-scribed by using the dielectric continuum model. Without doubt, the dielectric continuummodel is an oversimplification. It does not take into account the molecular nature of the sol-vent. In this connection, attempts to generalize the theory are noteworthy (Tachiya 1993;Leite 1999). The approaches mentioned take into account that solvent dynamics happenson a very high-dimensional surface, and this complex landscape is populated by a largenumber of minimums. Models including energy correlation lead to a single collective re-action coordinate. These models recover successful Marcus theory when they are combinedwith the potential difference distribution calculated on the basis of the dielectric continuummodel.
Depending on the solvent polarity and redox potentials of donor and acceptor, theions resulting from electron transfer may remain associated either as a contact ion radicalpair or as a solvent-separated ion radical pair. In the contact pair electron back-transfer cantake place. For such electron back-transfer, the solvent reorganization energy is less than5% of the total reorganization energy (Serpa & Arnaut 2000).
In general, however, solvent polarity is favorable for electron transfer. The solventreorganization energy is particularly large for small ions in polar solvents and is usuallylow in low-polarity solvents. Therefore, the low polarity of a solvent cannot prevent elec-tron transfer in all the cases when there is a significant difference between the donor ion-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

ization potential and the acceptor electron affinity. Moreover, fragmentation of an ion rad-ical results in the formation of a neutral radical and a small ion (e.g., H3CBr�. → H3C
.�
Br�). The size of Br� is smaller than that of the parent anion radical, H3CBr�.. The smaller
size of the charged species is favorable for solvation. Solvation accelerates the fragmenta-tion reaction. As a result, a decrease in the solvent polarity increases anion radical stabil-ity. For comparison between calculation and experimental data on anion radicals of halo-genated compounds in nonpolar solvents, see Matchkarovskaya et al. (1995) andShimamori et al. (1993).
Solvent reorganization energy should be taken into account when, during the reac-tion, a charge moves from one to another portion of the molecule. For instance, in the ace-tophenone series, such charge transfer takes place according to the following scheme:
RC(BO)CH2X � e → RC.(MO�)CH2X ⇔ RC(BO)CH2X�. → RC(BO)CH
.2 � X�
Once the initial anion radical is formed, the solvent is organized around a negativecharge that is concentrated mostly on the carbonyl oxygen. Then the solvent has to reorga-nize around the negative charge that develops on the leaving group during the reaction. Ac-cording to estimations by Andrieux et al. (1996), the solvent reorganization contributes75–100% to the total intrinsic energy barrier for the reaction.
5.4.2.B Solvent Polarizability
One possible solvent interaction with solute molecules or ion radicals includes nonspecific(nonbonding) solvation and electron donor–acceptor or ion-induced-dipole interactions. Todiscern these interactions, one can compare such solvents as toluene and n-hexane. At tem-peratures when all these solvents have comparable viscosity, the reaction between 1,2,4,5-tetrafluorobenzene and its anion radical proceeds in different ways (Werst 1993).
In toluene, the reaction consists of electron exchange:
C6H2F4 � (C6H2F4)�. → (C6H2F4)�.� C6H2F4
In n-hexane, dimer formation takes place:
C6H2F4 � (C6H2F4)�. → (C6H2F4)�.2
Hence, in toluene, no dimer anion radicals are observed. Encounters between anion radi-cals and neutral molecules result in electron transfer instead. Charge transfer from the so-lute anion radicals to toluene is unlikely, since toluene is a poor electron acceptor. Consis-tent with this fact, no differences were observed in the anion radical ESR couplingconstants in toluene compared to those in n-hexane (Werst 1993). Charge (not electron)transfer from the toluene molecules to the neutral tetrafluorobenzene is more plausible.However, spectroscopic studies show that such charge transfer is weak (Werst 1993).Ion–dipole interactions are insignificant, since the dipole moment of n-hexane is near zeroand the dipole moment of toluene is negligibly small (0.36 D, Werst 1993). The author sur-mises that the described solvent dependence is attributable to the greater polarizability ofthe ring electron system in the toluene solvent molecules in the presence of the anion radi-cals, to compare favorably with the situation in n-hexane (Werst 1993).
The solvent effects in nonpolar solvents described earlier are not limited to anion rad-icals solely. A cation radical example that closely parallels the anion radical one is thetetramethylethyene cation radical (Me2CBCMe2)+.
. Like (C6H2F4)�., (Me2CBCMe2)+.
reacts with the neutral Me2CBCMe2 molecule to form the ion dimer (and also high-orderaggregates) in the alkane solutions but not in toluene. Electron donor–acceptor interactions
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

between (Me2CBCMe2)� and the solvent molecule can be ruled out, because the ESR pa-rameters of this cation radical are the same in the alkanes and in toluene (Werst 1993).Now, then, the greater polarizability of arenes as compared to alkanes leads to a strongerinteraction between ion radicals and solvent molecules. This in turn prevents dimer forma-tion between neutral molecules and their ion radicals in arene solvents. Electron-exchangereactions between these solutes take place instead of dimer formation.
5.4.2.C Solvent Internal Pressure
At least in outer-sphere ion radical reactions, a reagent and a substrate form a charge-trans-fer complex. Then ion radicals are formed, chemistry takes place between them, and prod-ucts are released. In other words, there are several intermediate states in these reactions andin each state the size of the structure is not small. Therefore each of them needs an appro-priate hole in the bulk solvent. Some authors indicate that the mechanism of electron trans-fer involves a gradual electron shift from electron donor to acceptor. This shift is conju-gated with a simultaneous reorganization of the reactants and media, rather than an electronjump after preliminary reorganization (see, for example, Kuzmin 1996). It is obvious thatfor the dissolution of a substance in a solvent, a hole (cavity) must be created within the sol-vent to accommodate the new solute. This process certainly requires the expenditure of en-ergy. The amount of energy expended will depend on the magnitude of the intermolecularforces of attraction between the solvent molecules on the one hand and between the solventand solute molecules on the other. In effect, the solvent exerts “pressure” on the solute. In-ternal pressure can affect the liquid-phase ion radical reactions and deserves special study.For instance, such pressure can determine the selectivity and even the stereochemistry ofthese reactions. Thus, inversion selectivity was established in free radical bromination ofphenyl cyclopropane as a result of solvent (internal) pressure (Tanko et al. 1993).
5.4.2.D Conformational Dynamics in Solvents During Electron Transport
Some organic ion radical reactions can be initiated by laser photolysis or pulse radiolysis.The main result of these processes consists of the generation of the thermalized electron (et)and the solvent hole (RH+.
). The et is usually scavenged by means of small amounts of ad-ditives, such as haloalkanes (e.g., carbon tetrachloride), or electrophilic gases (carbon diox-ide, nitrous oxide, etc.). The solvent hole, i.e., RH+.
, transfers charge to an organic sub-strate. The latter plays a role as a starting material for further reactions. Hence, electrontransport through the solvent takes place. Let us consider two conventional solvents forsuch reactions, cyclohexane and decalin. According to Shkrob, Sauer, and Trifunac (1996,1999), Shkrob, Sauer, Yan, and Trifunac (1996), and Sauer et al. (1996), these two solventsproduce quite different kinetics of electron-transfer reactions.
At 25°C, the cyclohexane molecules mainly have the chair form. The equilibriumconcentration of the isomeric twist form is ~10�4 mol�dm�3. On ionization, the solventcation radicals in chair form are predominant. Electron transfer between the chair form ofthe cyclohexane cation radicals and the chair-shaped surrounding cyclohexane moleculesis very fast, since it requires minimum reorganization energy. However, the chair-formcation radical sometimes approaches a minor part of the neutral molecules in twist form.Because the twist cyclohexane has lower ionization potential, the twist-shaped moleculesscavenge the cation radicals in chair form.
Once formed, the twist cation radical become surrounded by the chair-form neutralmolecules. It does not transfer the charge to the neighboring chair-form molecules until thecation radical returns chair form (in a spontaneous manner or by charge transfer). As a re-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

sult, fast migration of the hole, i.e., the one-electron transfer from the neutral solventmolecule to its cation radical, is detained. In 1986 Hummel and Luthjens considered theidea that conformation dynamics is involved in electron transfer. Scheme 5–9 illustratesthis dynamic.
The twist conformer, which has a lower ionization potential, rapidly scavenges thechair form of the cation radical. Being endothermic, the backward transfer is relativelyslow, and equilibrium is reached in 20–30 ns. Thus, the electron transport can be describedas a series of periods of very fast hole migration between the chair forms and intermittentmigration with participation of the twist forms.
It is important that both twist and chair forms of the cylclohexane cation radical re-act with a solute, for example, with perylene. Therefore, generation of the perylene cationradical is characterized by bimodal kinetics under these conditions.
No such confusion arises for decalins. In decalin mixtures, a reversible electron-transfer reaction takes place:
cis-decalin � (trans-decalin)+. ⇔ (cis-decalin)+.� trans-decalin
Because of the high concentration of isomer molecules (�0.1 mol�dm�3), the equi-librium depicted is established instantaneously. The ionization potential of trans-decalin is0.02 eV lower than the ionization potential of cis-decalin (9.24 eV vs. 9.26 eV). Therefore,the electron-transfer equilibrium is shifted slightly to the left side. Thus, in terms of charge-transfer kinetics, the two ions behave as a single species.
Hence, both cyclohexane and decalin are a binary mixture of conformers. From thematerial just discussed it is obvious that conformers of the decalin cation radical are actingin parallel, whereas conformers of the cyclohexane cation radical have quite different ki-netics of their reactions with uncharged molecules of a solvent and a solute.
Considering nonequilibrium configurations of the solvent and the solute in electrontransfer, some authors note that the solvent hole should be suitable to accommodate an ionradical salt in the case where the counterions are kept with each other. For example, the in-termolecular electron transfer between the coumarin 337 solute and the N,N-dimethylani-line solvent results in solution anisotropy. The anisotropy indicates that the donor and ac-ceptor molecules are aligned with their long axes roughly perpendicular. The donor(dimethylaniline) and the acceptor (coumarin) have 1.6-D and 10-D ground-state dipolemoments, respectively, and align roughly along their long axes. Electron transfer is ratecontrolled by a favorable overlap of the corresponding highest occupied molecular orbitals.The mutual orientation of the counterions dictates hole size and shape in the solvent(Akhremitchev and others 1999).
SCHEME 5-9
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Solvent polarizability can also play a role in the stabilization of conformationallynonuniform particles. For example, 4-(1-phenylpiperidin-4-ylidene) cyclohexylidenepropanedinitrile transforms into the fully charge-separated species on photoirradiation.This species contains the CBC bond and bears two ion radical centers: N+.
and C�.. As re-
vealed (Hoogesteger et al. 2000), the species formed keeps a folded conformation in cy-clohexane and a stretched conformation in benzene.
5.4.2.E Temperature Dynamics of Ion Pairs
Karasava et al. established in 1971 that the anion radical of biphenyl or naphthalene formsan ion pair in THF solution in the presence of the sodium cation. THF has a 7.6 dielectricconstant (i.e., � � 10) and is a nondissociating solvent in which ion radicals are essentiallyassociated with their counterions. At 20°C, tight pairs of the biphenyl and naphthalene an-ion radicals with sodium cations dominate, while the loose ion pairs are most abundant atthe lowest temperatures. Hence, lowering the temperature decreases the stability of tightion pairs containing organic ion radicals. This unusual regularity is explained (Smid 1972)by certain thermochemical effects. In ethers, the formation of separated ion pairs is usuallyexothermic. Selective ion–solvent interaction contributes significantly to the exothermicityof the ion-pair separation in ethereal solutions. A temperature decrease assists in heat dis-sipation and facilitates this ion-pair separation. In addition, a temperature decrease causesan increase in the dielectric constants of polar solvents. Polar solvent molecules have dipolemoments. A dipole order–disorder transition takes place in the course of thermal move-ment. The lower the temperature, the greater the regularity of disposition of solventmolecules and the higher the dielectric constant. This phenomenon results in ion-pair loos-ening. Association into an ion pair tends to reduce the reactivity of the ion. The higher theloosening of ion pairs, the lower the reduction in their reactivity. The rate and state of equi-librium of electron transfer reactions also depend on the association of the resulting ion rad-icals with counterions.
So the temperature decrease results in an increase in the dielectric constant of the liq-uid polar solvent. However, freezing a solvent has the opposite effect. Upon freezing withglass formation, the effective solvent dielectric constant decreases drastically, because thesolvent dipoles cannot reorient. The frozen glass, therefore, cannot stabilize the newlyformed ions (Liddell and others, 1997).
5.5 SALT EFFECTS
5.5.1 Salt Effect That Results in Redistribution of Spin Density
Upon one-electron reduction, aldehydes and ketones give anion radicals. It is the carbonylgroup that serves as a reservoir for the unpaired electron: Ketones yield pinacols exclu-sively. Thus, acetophenone forms 2,3-diphenylbutan-2,3-diol as a result of electrolysis atthe potential of the first one-electron transfer wave (nonaqueous acetonitrile as a solvent,with tetraalkylammonium perchlorate as a supporting electrolyte) (van Tilborg & Smit1977). In contrast, calculations have shown that the spin densities on the carbonyl groupand in the para position of the benzene ring are equal (Mendkovich et al. 1991). This meansthat one should wait for the formation of three types of dimer products, “head-to-head,”“tail-to-tail,” and “head-to-tail”; cf. Section 3.2.1. For the anion radical of acetophenone,all three possible dimers are depicted in Scheme 5-10.
For this section, the yield of the “head-to-head” product is relevant. In dimethylfor-mamide with 0.1 M of tetrabutylammonium perchlorate, electrolysis of acetophenone at the
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

potential of the first one-electron wave produces this dimer in 30% yield. This is in accordwith the earlier-discussed prediction that all three directions of this dimerization are equallyprobable. If lithium perchlorate is the supporting electrolyte in the same solution, the“head-to-head” dimer yield rises to 70% (Gul’tyai, Mendkovich, & Rubinskaya 1987).Hence, “head-to-head” coupling becomes the main route of dimerization.
In the case of the “free” 9-acetylanthracene anion radical, the spin density on the car-bonyl group is lower than that in the para position (position 10) by a factor of 5. The for-mation of the “tail-to-tail” dimer should be expected. Actually, preparative reduction of 9-acetylanthracene in dimethylformamide against the background of a tetrabutylammoniumsalt results in just such a dimer, with a yield of 70%. Addition of a lithium salt, however,decreases the dimer yield to 45% (Mendkovich et al. 1991; Gul’tyai, Rubinskaya, Mend-kovich, & Rusakov 1987).
It is interesting to compare yields of the “head-to-head” diol from 2-acetylthiophenein their dependence on the composition of the reaction medium (dimethylformamide as a
SCHEME 5-10
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

solvent). This diol is formed in 10–15% yield with 0.1 M of Bu4NClO4, in 42% yield with0.1 M of Bu4NClO4 � 0.01 M of LiClO4, and in 62% yield with 0.1 M of LiClO4 (Gul’tyai,Korotaeva, & Rubinskaya 1988). For 2-acetylthiophene, Scheme 5-11 demonstrates theformation of the “head-to-head” and “head-to-tail” dimers.
As follows from all of the quoted data, the addition of lithium salt results in the for-mation of ion pairs, i.e., lithium-ketyls. This helps displace the spin density toward the car-bonyl group. The lithium cation has a high affinity to the “hydroxy anion” in the ketyl frag-ment (interaction between two hard ions, O� and Li�). Such affinity prevails over theion-pair disrupting power of dimethylformamide, which has a high dielectric constant (� �37, very far from the threshold of � � 10 for nondissociating solvents). On the other hand,the marked dependence of yields of the “head-to-head” product on the concentration of thelithium salt means that the tight ion pair with spin localization on CBO is in equilibriumwith the more or less free anion radicals. According to the usual rule of equilibrium shift,the percentage of (ThMC
.MO�, Li�), i.e., thiophene lithium-ketyl tight pair, should in-
crease as the concentration of LiClO4 increases:
ThMC.MO�//Bu4N� � Li�ClO�
4 ⇔ Th-C.-O�, Li� � Bu4N�ClO�
4
The addition of the sodium salt (NaI) to a solution of the camphoroquinone anion rad-ical sodium salt results in enhanced ion association. This association pulls the spin densityonto the oxygen. Stevenson et al. (1998) used the camphoroquinone R- or S-conformer foranion radical preparation in the chiral solvent [SS- or RR-2,3-dimethoxy-1,4-bis(dimethy-lamino)butane]. In this case, the interaction of the chiral-solvated cation with the chiral an-
SCHEME 5-11
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

ion radical (semidione) results in specific line broadening in ESR spectra. This opens a newapproach to the ESR chiral discrimination of the semidiones in a chiral solvent(solvent–ion-pair chiral recognition). Diastereotopic-solvated ion pairs that form and dis-sociate rapidly on the ESR time scale produce some asymmetry in the spectrum. As known,chiral-shift reagents perturb the NMR chemical shifts of each enantiomer differently andallow for chiral resolution. A significant difference in the ESR experiments described byStevenson with her co-workers (1998) and the use of NMR chiral-shift reagents come fromthe fact that the ESR chiral-shift reagent is the solvent itself. Obviously, similar effects canbe observed for pairs of other ion radicals with counterions surrounded by other chiral sol-vents.
5.5.2 Salt Effect That Suppresses Ion Radical Reactions
Another example concerning the reduction of carbonyl compounds also relates to the salteffect theme. Shaefer and Peters (1980), Simon & Peters (1981, 1982, 1983, 1984), Rudzkiet al. (1985), and Goodman and Peters (1986) described photoreductions of aromatic ke-tones by amines. In this case, the addition of excess NaClO4 results in considerable retar-dation, even prevention, of final product formation. The two fundamental steps in this pho-toreduction consist of rapid electron transfer from the amine to the photoactivated ketone(in its triplet state), followed by the slow transfer of proton from the amine cation radicalto the carbonyl anion radical:
CBO � N..MCH ⇔ C
.MO� � N+.
MCH → C.MOH � N
..MC
.
↓Products
The formation of a contact ion pair between the anion radical and the cation radicalfacilitates the proton transfer. Addition of sodium perchlorate breaks this ion pair up ac-cording to the following scheme:
[ C.MO
., N+.
MCH ] � M�, ClO�4 ⇔ C
.MO�, M� � N+.
MCH , ClO�4
The rate of this ion-pair exchange could be measured in various solvents using picosecondlaser absorption spectroscopy. The rate decreases as the ability of the solvent to solvate themetal cation increases. It even falls to “zero rate” in the presence of appropriate crownethers.
5.5.3 Salt Effect That Hinders Coupling Between Ions and RadicalsInside Solvent Cages
Each cation radical has a positive charge and an unpaired electron. In the simultaneouspresence of an anion (A�) and a free radical (R.), both of them can attack a cation radicalcompetitively. These two possibilities are shown for the example of an aromatic cation rad-ical ArH+.
:
ArH+.� A� → Ar
.(H)A → ArA � H
.
ArH+.� R
. → Ar�(H)R → ArR � H�
The driving force of the reaction between a radical and an aromatic cation radical isbased on their mutual affinity, because both species possess unpaired electrons. There is nocoulombic attraction, no coulombic repulsion in this case. The reaction between an anionand an aromatic cation radical does involve coulombic attraction as the driving force. It
M
MM
M
M
M
M
M
M
M
M
M
M
MM
M
M
M
M
M
M
M
M
M
M
MM
MM
M
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

necessarily proceeds through a contact (intimate) ion pair Ar+., A�. It is obvious that dis-
integration of the mentioned ion pair has to retard or even prevent ArA formation.Kochi and his co-workers studied factors that lead to the predominance of the radical
or the anionic reaction. Three distinct classes of substitution reactivity can be discerned inthe halogenation of methyl-substituted methoxybenzenes (ArH) by iodine monochloride(ICl): exclusive iodination, exclusive chlorination, and mixed iodination-chlorination (Hu-big, Jung, & Kochi 1994). Spectral studies established the prior formation of the charge-transfer complex [ArH, ICl]. The complex suffers electron transfer to produce the reactivetriad [ArH+.
, I., Cl�]. Chlorination and iodination result from the quenching of the aromatic
cation radical by chloride ions and iodine atoms, respectively. Iodination vs. chlorinationthus represents the competition between radical-pair and ion-pair collapses from the reac-tive triad, and it is predictably modulated by the dissociating ability of a solvent and by theaddition of a “foreign” salt. In nondissociating solvents (CCl4 and CH2Cl2), the collapse ofthe destabilized ion pair is enhanced and leads to a higher proportion of the chlorinatedproduct. In contrast, in polar solvents such as acetonitrile, the ions are readily separated andstabilized by solvation to allow the radical collapse. Iodination becomes competitive. Theratio [Arl]/[ArCl] is equal to 90/10 in CH3CN (� � 36) or 60/40 in CH2Cl2 (� � 8.9). IfCH2Cl2 contains 0.2 M Bu4N�PF�
6, the ratio changes to 40/60. Tetrabutylammonium hex-afluorophosphate liberates chloride ions from the reactive triad by preferential ion pairing.Hence, the cation radical remains approachable for the radical attack.
Similar results were obtained for the reaction between ArH and C(NO2)4
(Sankarararaman et al. 1987; Masnovi et al. 1985, 1989). After photostimulated electrontransfer, the related triad was formed:
ArH � C(NO2)4 → [ArH+.,.NO2,�C(NO2)3]
In dissociating solvents, ArNO2 is obtained almost exclusively. In solvents of low di-electric constant, the main product is ArC(NO2)3. Formation of the latter product is com-pletely prevented on the addition of Bu4N�ClO�
4 to nondissociating CH2Cl2, whereas for-mation of ArNO2 is unaffected. Here again, this effect is due to the added salt breaking upthe ion pair via an ion-pair exchange process:
ArH�,�C(NO2) � Bu4N�ClO�4 → ArH+.
,ClO�4 � Bu4N�,�C(NO2)3
5.5.4 Salt Effect That Suppresses Electron Back-Transfer During IonRadical Formation
One of the most prominent manifestations of this effect consists of the oxidation of 1,2-di-arylcyclopropane (CP) with oxygen in the presence of photoexcited 9,10-dicyanoan-thracene (DCNA)* in acetonitrile (Mizuno et al. 1987) (Scheme 5–12).
SCHEME 5-12
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

If the reaction proceeds in the presence of Mg(CIO4)2, the product yield becomes sig-nificantly better. The added salt suppresses electron back-transfer:
CP � (DCNA)* ⇔ [CP+., DCNA�.
]
2 [CP+., DCNA�.
] � Mg(CIO4)2 ⇔ 2 [CP+.,ClO�
4] � [2 DCNA�.Mg2�]
After all, the released cation radical reacts with oxygen according to Scheme 5-13. This re-action was performed in acetonitrile, a solvent of high polarity. For solvents with moderatepolarities, the addition of salts, i.e., electrolytes, increases their polarity (Thompson & Si-mon 1993). With increased solvent polarity, electron-transfer rates increase too.
The oxidative photocleavage of 1,2-bis(4-methoxyphenyl)cyclobutane is also accel-erated in the presence of Mg(ClO4)2. However, no salt effect was found for 1,2-diphenyl-cyclobutane because the very fast electron back-transfer prevents interaction of the cagedion pair with the added salt (Pac et al. 1987).
It is important to differentiate between the effects of a non-nucleophilic salt, such asMg(ClO4)2, on the one hand, and a weak nucleophilic salt, such as Et4NOAc, on the other.The effect of non-nucleophilic salts on photo-oxygenation via electron transfer can be un-derstood as the stabilization of ion radicals by coulombic interaction, resulting in the sup-pression of an electron back-transfer between ion radicals. The weak nucleophilic saltscause unusual effects. Nucleophilic addition to the cation radical and complexation with theion radicals bring these effects about.
5.5.5 Salt Effect That Creates an Interface to Electron-TransferPathways
The salt effects just considered are counterion effects. Sometimes, however, an added saltcan induce electron transfer from a donor to an acceptor. Here are several examples.
Ruiz et al. (1989) studied substitution reactions of transition metal complexes withorganic ligands. They found that the presence of a simple sodium salt completely changesthe course of these reactions. One of the cases consists of the disproportionation of Fe(I)sandwiches with PMe3. This reaction proceeds only in the presence of a sodium salt. With-out addition of this salt, the 19-electron (19e) complex [(cp)Fe(C6H6)], cp � �5-C5H5, re-acts with PMe3 in THF at �15°C according to the following scheme:
[(cp)Fe(C6H6)] � PMe3 → [(cp)Fe(PMe3)2(H)]
In the presence of NaPF4 (1 equivalent), disproportionation occurs under the same re-action conditions. The two products, depicted here, are formed in equimolecular amounts:
[(cp)Fe(C6H6)]/NaPF6 � PMe3 → [(cp)Fe(PMe3)3]� PF�6 � (PMe3)3Fe(PMe2CH2)(H)
SCHEME 5-13
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The replacement of the benzene ligand with PMe3 results in the formation of the 17-electron (17e) complex and then of the 19-electron complex:
[(cp)Fe(C6H6)] � PMe3 → [(cp)Fe(PMe3)2](17e) � PMe3 ⇔ [(cp)Fe(PMe3)3(19e)]
Abstraction of a H atom from the medium is one way to stabilize the 17-electroncomplex; cf. the reaction of [(cp)Fe(C6H6)] with PMe3 in the absence of NaPF6:
[(cp)Fe(PMe3)2](17e) � H → [(cp)Fe(PMe3)2(H)]
It is worth noting at this point that THF and PMe3 are good H donors.The 19-electron species (cp)Fe(PMe3)3 is extremely electron rich. It readily reduces
the initial complex [(cp)Fe(C6H6)]:
[(cp)Fe(PMe3)3] � [(cp)Fe(C6H6)] ⇔ [{(cp)Fe(PMe3)3}�, {(cp)Fe(C6H6)}�]
In the presence of NaPF6, the large ion pair undergoes metathesis and transforms intothe two smaller ion pairs:
{[(cp)Fe(PMe3)3]�, [(cp)Fe(C6H6)]�} � [Na�, PF�6 ]
⇔ [(cp)Fe(PMe3)� PF�6] � [(cp)Fe(C6H6)� Na�]
The (20e) anion [(cp)Fe(C6H6)]� becomes much more unstable when incorporatedinto the salt with Na� than in the salt with (cp)Fe(PMe3)�
3. The sodium salt is decomposedeasily:
[(cp)Fe(C6H6)� Na�] � PMe3 → (cp)� Na� � C6H6 � (PMe3)3Fe(PMe2CH2)(H)
The decomposition of the salt [(cp)Fe(C6H6)� Na�] displaces this equilibrium to the right.Hence, the difference in stability of the (20e) anion (cp)Fe(C6H6)� is the major factor re-sponsible for the salt effect. The NaPF6 salt can very efficiently induce electron transfer be-tween neutral organometallic species.
The next example deals with the effect adding ferrous chloride (Galli & Gentili1993). Phenyl iodide reacts with the potassium derivative of 3,3-dimethyl-2-butanone(pinacolin) in dimethylsulfoxide according to the following scheme:
CH3COCMe3 � t-BuOK → t-BuOH � KCH2COCMe3
PhI � KCH2COCMe3 → PhCH2COCMe3 � KI
Without illumination, the reaction proceeds slowly, but by no means negligibly: 8%of phenylpinacolin is formed, without regard to the prolonged duration. The addition of fer-rous chloride in amounts of 40% to molar equivalent of PhI results in an incisive accelera-tion of this reaction. The disappearance of PhI is observed within 20 min, replaced by 74%of the substitution product, PhCH2COCMe3, and ca. 10% of the disubstitution product,Ph2CHCOCMe3. The authors cite diverse data, theorizing that iron(II), associated with theenolate ion, acts as an electron-transfer relay between the enolate and phenyl iodide(Scheme 5–14). Hence, the addition of FeCl2 improves the ease of the electron transfer. Asalready mentioned, this electron-transfer reaction occurs spontaneously without the addi-tion of the salt, but with lower efficiency. Ferrous chloride bridges these difficulties. It isinteresting that the analogous reaction of PhCBMCBr with PhCOCH2K in dimethylsulfox-ide gives rise to PhCBMCH only, in quantitative yield. A transfer of one electron initiatesthe initial debromination, after which further reduction of radical PhCBMC into anionPhCBMC� takes place. In spite of the mentioned difference in the resulting products, the
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

common feature of these two reactions consists of the necessity in iron(II) ion catalysis forthe initial electron transfer (Galli & Gentili 1994).
One surprising example is how ferric (not ferrous) chloride catalyzes the formationof ketyl radicals in a Gringnard-type reaction between (CH3)3CCl, Mg, and (CH3)2CBO(Ashby & Wiesemann 1978). As the authors theorized, the initially formed species isformed according to the following scheme:
(CH3)3CMgCl � FeCl3 → MgCl2 � {(CH3)3CFeIIICl2}
The product in braces decomposes, giving an intermediate iron hydride species at the ex-pense of -hydrogen elimination:
{(CH3)3CFeIIICl2} → (CH3)2CBCH2 � [HFeIIICl2]
It is likely that such iron hydride species is capable of reducing acetone:
[HFeIIICl2] � (CH3)2CBO → [(CH3)2CHOFeCl2]
[(CH3)2CHOFeCl2] � MgCl2 → FeCl3 � (CH3)2CHOMgCl
(CH3)2CHOMgCl � H2O → Mg(OH)Cl � (CH3)2CHOH
As previously shown, N,N-dimetylaniline acts as an electron donor toward the elec-tronically excited Ru(II) tris(dipirydyl) complex (Bock et al. 1979). Recently, this complexwas juxtaposed to N,N-dimethylaniline via a salt bridge (Deng et al. 1997; Kirby et al.1997; Roberts et al. 1997). Supramolecular assemblies have been prepared in which anelectron may transfer from N,N-dimethylaniline to the electronically excited Ru(II)tris(dipyridyl) acceptor through the intervening amidinium–carboxylate salt bridge inter-face (Scheme 5–15).
SCHEME 5-14
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

SCHEME 5-15
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The amidinium–carboxylate interface is apparently exceptionally stable. This asso-ciation persists in solution, even when the dielectric constant of the solvent is high. Suchstructures open up the possibility to consider the orientation of the salt bridge relative to thedirection of the electron transfer from the dimethylaniline donor to the metallocomplex ac-ceptor. The rate of electron transfer along the [donor–(carboxylate–amidinium)–acceptor]route is significantly faster than that along the route when the interface is switched, i.e., forthe case of [donor–(amidinium–carboxylate)–acceptor]. In the carboxylate–amidinium ori-entation, the electron transfer obeys the direction of the permanent dipole of the salt bridge.Moreover, the proton already resides on the acceptor site and hence is likely to remain onthe arrival of the electron. Franck–Condon factors arising from proton motion within thesalt bridge are therefore minimized. In the reaction considered, the cation component is in-corporated in the donor molecule. The anion component becomes incorporated in the form-ing acceptor metallocomplex. Nevertheless, they play the role of a typical salt bridge.Therefore, this example is relevant.
Incorporation of an ionic component into a donor/acceptor molecule is a very effec-tive way of suppressing electron back-transfer. One interesting example consists of thephoto-oxidation of leuko crystal violet (LCV) to crystal violet (CV, the dye) by benzophe-none bearing a quaternary ammonium ion (Tazuke & Kitamura 1984). In this case, thecation radical of LCV formed is repulsed by the ammonium positive charge. At the sametime, the benzophenone anion radical remains stabilized by the attached cationic atmo-sphere (Scheme 5–16). As shown in the scheme, two favorable results are achieved: the sta-bilization of an ion radical pair by counterion exchange and the charge separation bycoulombic repulsion between the two positive charges. This leads to 100% efficiency of thephoto-oxidation. With unsubstituted benzophenone itself, the efficiency does not exceed20%.
Photoinduced electron transfer rates can be considerably reduced when the counte-rion X� is changed from chloride to bromide. Charge transfer between the cationic part ofa molecule and the bromide ion may be responsible to the reduction of photoinduced elec-tron-transfer rates. Such a counterion effect on the photoinduced electron transfer and thereverse process has been demonstrated for examples of porphyrin-viologen–linked com-pounds (Mitsui et al. 1989).
SCHEME 5-16
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The salt effects considered are obviously capable of suppressing electron back-trans-fer during ion radical formation. Sometimes, however, there is a necessity not to suppressbut only to reduce the rate of electron back-transfer. The goal can be achieved by a specialmolecular design with no salt participation. In connection with such a goal, the photoin-duced electron transfer should be mentioned within the molecular architectural creationbased on benzene bearing in all three meta positions 4-(N-piperidinyl)naphthalene–1,8-car-boximide moieties. In this case, optical switching was achieved between two long-lived ionradical pair states (Lukas et al. 2001).
5.6 CONCLUSION
This chapter discusses the methods for regulating ion radical transformations with respectto the nature of reacting species. These species possess both an ionic and a radical charac-ter at the same time. Being charged, the species demand specific solvents and salt additivesand obey laws on ion-pair behavior. Having an unpaired electron, these species are sus-ceptible to magnetic effects, sometimes to light or ultrasound irradiation, and are especiallysensitive to factors of spin-density distribution. Coexistence of these ionic and radical prop-erties determines a rather wide set of methods for suppressing undesirable processes or forstimulating reactions leading to the needed final products.
Meanwhile, ion radicals differ from ions and neutral molecules in their lower stabil-ity. As a rule, ion radicals exist at lower temperatures. Therefore, heating is not a typicalway to stimulate ion radical reactions. Such reactions often require inert gaseous media, ap-paratus with polished walls, and so on. In general, approaches to the stimulation of ion rad-ical reactions seem not to be quite regular or usual for organic chemists. Nevertheless, thehigh activity of ion radicals permits different kinds of directed influence over the reactions,which follow ion radical mechanisms.
Two questions are inseparable: how to optimize ion radical reactions, and how to fa-cilitate electron transfer. As noted in the preceding chapters, electron transfers betweendonors and acceptors can proceed as outer-sphere or inner-sphere processes. In this con-nection, the routes to distinguish and regulate one and another process should be men-tioned. The brief statement by Hubig, Rathore, and Kochi (1999) seems to be appropriate:Outer-sphere electron transfers are characterized by (a) bimolecular rate constants that aretemperature dependent and well correlated by Markus theory; (b) no evidence for the for-mation of (discrete) encounter complexes; (c) high dependence on solvent polarity; (d) en-hanced sensitivity to kinetic salt effects.
Inner-sphere electron transfers are characterized by (a) temperature-independent rateconstants that are greatly higher and rather poorly correlated by Marcus theory; (b) weakdependence on solvent polarity; (c) low sensitivity to kinetic salt effects. This type of elec-tron transfer does not produce ion radicals as observable species but deals with the pre-equilibrium formation of encountered complexes with the charge-transfer (inner-sphere)nature (see also Rosokha & Kochi 2001).
REFERENCES
Adam, W.; Arguello, J.E.; Penenory, A.B. (1998) J. Org. Chem. 63, 3905.Akhremitchev, B.; Wang, Ch.; Walker G.C. (1999) Laser Chem. 19, 403.Al-Khalil, S.I.; Bowman, R. (1984) Tetrahedron Lett. 25, 461.Albini, A.; Spreti, S. (1986) J. Chem. Soc., Chem. Commun. 1426.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Aleksandrov, A.I.; Prokof’ev, A.I.; Metlenkova, I.Yu.; Bubnov, N.N.; Tipkin, D.S.; Perekhodtsev,G.D.; Lebedev, Ya.S. (1995) Zh. Fiz. Khim. 69, 739.
Amatore, Ch.; Pinson, J.; Saveant, J.-M.; Thiebault, A. (1982) J. Am. Chem. Soc. 104, 817.Ando, T.; Kimura, T.; Leveque, J.-M.; Fujita, M.; Luche, J.-L. (2000) Chem. Lett., 1124.Andrieux, C.P.; Saveant, J.-M.; Tallec, A.; Tardivel, R.; Tardy, C. (1996) J. Am. Chem. Soc. 118,
9788.Ashby, E.C.; Wiesemann, T.L. (1978) J. Am. Chem. Soc. 100, 189.Asmus, K.-D. (1990) In: Chatgilialogly C., Asmus, K.-D., eds. Sulfur-Centered Reactive Intermedi-
ates in Chemistry and Biology. Plenum Press, New York.Atobe M.; Tonoi, T.; Nonaka, Ts. (1999) Electrochem. Commun. 1, 593.Ayadim, M.; Jiwan, J.L.H.; Soumillion, J.Ph. (1999) J. Am. Chem. Soc. 121, 10436.Ballester, M.; Pascual, I. (1991) J. Org. Chem. 56, 841.Barbosa, F.; Peron, V.; Gescheidt, G.; Fuerstner, A. (1998) J. Org. Chem. 63, 8806.Bartak, D.E.; Shields, T.M.; Hawley, M.D. (1971) J. Electroanal. Chem. Interfac. Electrochem. 30,
289.Blank, M.; Soo, L. (2001) J. Cell. Biochem. 81, 278.Blumenfel’d, L.A.; Brukhovetskaya, L.V.; Fomin, G.V.; Shein, S.M. (1970) Zh. Fiz. Khim. 44,
931.Bock, C.R.; Connor, J.A.; Gutierrez, A.R.; Meyer, T.J.; Whitten, D.G.; Sullivan, B.P.; Nagle, J.K.
(1979) J. Am. Chem. Soc. 101, 4815.Buchachenko, A.L. (1976) Usp. Khim. 45, 761.Burstein, A.I. (1999) Chem. Phys. 247, 275.Camps, P.; Lukach, A.E.; Rossi, R.A. (2001) J. Org. Chem. 66, 5366.Cermenati, L.; Richter, C.; Albini, A. (1998) J. Chem. Soc. Chem. Commun. 805.Corrie, J.E.T.; Gilbert, B.C.; Munasinghe, V.R.N.; Whitwood, A.C. (2000) J. Chem. Soc. Perkin
Trans. 2, 2483.Deng, Y.; Roberts, J.; Peng, S.-M.; Chang, C.K.; Nocera, D.G. (1997) Angew. Chem. Int. Ed. Eng.
36, 2124.Didenko, Y.T.; McNamara, W.B.; Suslick, K.S. (1999) J. Phys. Chem. A 103, 10783.Drago, R.; Ferris, D. (1995) J. Phys. Chem. 99, 6563.Eberson, L.; Hartshorn, M.P.; Persson, O. (1995) J. Chem. Soc. Perkin Trans. 2, 1735.Eberson, L.; Hartshorn, M.P.; Persson, O.; Radner, F. (1996) Chem. Commun. (Cambridge), 2105.Einhorn, C.; Einhorn, J.; Dickens, M.J.; Luche, J.L. (1990) Tetrahedron Lett. 31, 4129.Endicott, J.F.; Ramasami, T. (1982) J. Am. Chem. Soc. 104, 2252.Fox, M.A. (1991) In: Mariano, P.S., ed. Advances in Electron Transfer Chemistry, Vol. 1, JAI Press,
Greenwich, CT.Fox, M.A.; Younathan, J.; Fryxell, G. (1983) J. Org. Chem. 48, 3109.Frimer, A.; Rosenthal, I. (1976) Tetrahedron Lett. 32, 2809.Galli, C.; Gentili, P. (1993) J. Chem. Soc. Perkin Trans. 2, 1135.Galli, C.; Gentili, P. (1994) J. Chem. Soc. Chem. Commun., 2013.Goodman, J.L.; Peters, K.S. (1986) J. Am. Chem. Soc. 108, 1700.Grant, K.M.; Hemmert, J.W., White, H.S. (1999) Electrochem. Commun. 1, 319.Greaves, M.D.; Niemz, A.; Rotello, V.M. (1999) J. Am. Chem. Soc. 121, 266.Griffith, D.J. (1999) Introduction to Electrodynamics. Prentice-Hall, Upper Saddle River, NJ.Guldi, D.M.; Luo, Ch.; Prato, M.; Troisi, A.; Zerbetto, F.; Scheloske, M.; Dietel, E.; Bauer, W.;
Hirsch, A. (2001) J. Am. Chem. Soc. 123, 9166.Gul’tyai, V.P.; Korotaeva, L.M.; Rubinskaya, T.Ya. (1988) Izv. AN SSSR, Ser. Khim., 1121.Gul’tyai, V.P.; Mendkovich, A.S.; Rubinskaya, T.Ya. (1987) Izv. AN SSSR, Ser. Khim., 1576.Gul’tyai, V.P.; Rubinskaya, T.Ya.; Mendkovich, A.S.; Rusakov, A.T. (1987) Izv. AN SSSR, Ser.
Khim., 2812.Hammerich, O.; Parker, V.D. (1982) Acta Chem. Scand. B 36, 421.Handoo, K.L.; Kaul, A. (1992) Indian J. Chem. B 31, 561.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Handoo, K.L.; Gadru, K.; Jain, C.K. (1983) Indian J. Chem. B 22, 526.Heitele, H. (1993) Angew. Chem. Int. Ed. Eng. 32, 359.Herbert, E.; Mazaleyrat, J.P.; Welvart, Z.; Nadjo, L.; Saveant, J.-M. (1985) Nouv. J. Chem. 9, 75.Hoogesteger, F.J.; Walree van, C.A.; Jenneskens, L.W.; Roest, M.R.; Verhoeven, J.W.; Schudde-
boon, W.; Piet, J.J.; Warman, J.M. (2000) Chem. Eur. J. 6, 2948.Hubig, S.M.; Jung, W.; Kochi, J.K. (1994) J. Org. Chem. 59, 6233.Hubig, S.M.; Rathore, R.; Kochi, J.K. (1999) J. Am. Chem. Soc. 121, 617.Hummel, A.; Luthjens, L.H. (1986) Radioanal. Nucl. Chem. 101, 293.Iancu, A.I.; Oniciu, D.C.; Plugariu, C.; Bartha, E.; Vinatoru, M.; Badea, F. (1992) Rev. Rouman.
Chem. 37, 1275.Juillard, M.; Chanon, M. (1983) Chem. Rev. 83, 425.Karasawa, Y.; Levin, G.; Szwarc, M. (1971) J. Am. Chem. Soc. 93, 4614.Kim, Y.S.; Stevenson, Ch. D. (1999) J. Phys. Chem. A 103, 7139.Kirby, J.P.; Roberts, J.A.; Nocera, D.G. (1997) J. Am. Chem. Soc. 119, 9230.Kornblum, N. (1975) Angew. Chem., Int. Ed. Eng. 87, 797.Kornblum, N. (1982) In: Patai, S., ed. Supplement for the Chemistry of Amino, Nitroso and Nitro
Compounds and Their Derivatives. (1982). Wiley, New York.Kornblum, N.; Erickson, A.S. (1981) J. Org. Chem. 46, 1037.Kornblum, N.; Widmer, J. (1978) J. Am. Chem. Soc. 100, 7086.Kornblum, N.; Earl, G.W.; Holy, N.L.; Manthey, J.W.; Musser, M.T.; Snow, D.H.;Swiger, R.T. (1968) J. Am. Chem. Soc. 90, 6221.Kornblum, N.; Swiger, R.T.; Earl, G.W.; Pinnick, H.W.; Stuchal, F.W. (1970) J. Am. Chem. Soc. 92,
5513.Kuzmin, M.G. (1996) J. Photochem. Photobiol. Ser.A. 102, 51.Lehman, M.W.; Evans, D.H. (2001) J. Phys. Chem. B 105, 8877.Leite, V.B.P. (1999) J. Chem. Phys. 110, 10067.Lewis, F.D.; Petisce, J.R.; Oxman, J.D.; Nepras, M.J. (1985) J. Am. Chem. Soc. 107, 203.Liddell, P.A.; Kuciauskas, D.; Sumida, J.P.; Nash, B.; Nguen, D.; Moore, A.L.; Moore, Th.A.; Gust,
D. (1997) J. Am. Chem. Soc. 119, 1400.Luche, J.L.; Einhorn, G.; Einhorn, J.; Sinisterra-Gago, J.V. (1990) Tetrahedron Lett. 31, 4125.Lukas, A.S.; Bushard, P.J.; Wasielewski, M.R. (2001) J. Am. Chem. Soc., 123, 2440.Lyoshina, T.V.; Belyaeva, S.G.; Mar’yasova, V.I.; Sagdeev, R.Z.; Molin, Yu. N. (1980) Dokl. Akad.
Nauk SSSR 255, 141.Marcus, R.A. (1964) Ann. Rev. Phys. Chem. 15, 155.Masnovi, J.M.; Levine, A.; Kochi, J.K. (1985) J. Am. Chem. Soc. 107, 4356.Masnovi, J.M.; Sankararaman, S.; Kochi, J.K. (1989) J. Am. Chem. Soc. 111, 2263.Matchkarovskaya, I.A.; Burshtein, K.Ya.; Faustov, V.I. (1995) J. Mol. Struct. 338, 101.Mendkovich, A.S.; Churilina, A.P.; Rusakov, A.I.; Gul’tyai, V.P. (1991) Izv. AN SSSR Ser. Khim.,
1777.Middleton, W.J.; Lindsey, R.V. (1964) J. Am. Chem. Soc. 86, 4948.Mitsui, A.; Uehata, A.; Nakamura, H.; Matsuo, T. (1989) Chem. Lett., 1445.Mizuno, K.; Ichinose, N.; Otsuji, Y. (1987) Stud. Org. Chem. (Amsterdam) 33, 79.Murto, J.; Kivinen, A.; Strandman, L. (1971) Suomen Kemistilethti B 44, 308.Neppiras, E.A.; Noltingk, B.E. (1950) Proc. Phys. Soc. (London) B 63, 1032.Omelechko, E.N.; Ryabinin, V.A.; Shein, S.M. (1982) Zh. Org. Khim. 18, 1123.Ono, N.; Kawai, Sh.; Tanaka, K.; Kaji, A. (1979) Tetrahedron Lett. 19, 1733.Ono, N.; Tamura, R.; Nakatsuka, T.; Hayami, Yu.; Aritsune, K. (1980) Bull. Chem. Soc. Jpn. 53,
3295.Ono, N.; Tamura, R.; Eto, H. (1983) J. Org. Chem. 48, 3678.Pac, C.; Fukunaga, T.; Go-An, Y.; Sakae, T.; Yanejida, S. (1987) Photochem. Photobiol. A.: Chem.
41, 37.Pinson, J.; Saveant, J.-M. (1974) J. Chem. Soc. Chem. Commun., 933.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Pinson, J.; Saveant, J.-M. (1978) J. Am. Chem. Soc. 105, 1506.Popovich, O.; Tomkins, R.P.T. (1981) Nonaqueous Solution Chemistry. Wiley, New York.Praefke, K.; Weichsel, C.; Falsig, M.; Lund, H. (1980) Acta Chem. Scand. B 34, 403.Roberts, J.A.; Kirby, J.P.; Wall, S.T.; Nocera, D.G. (1997) Inorg. Chim. Acta 263, 395.Rosokha, S.V.; Kochi, J.K. (2001) J. Am. Chem. Soc. 123, 8985.Rossi, R.A.; Rossi de, R.H. (1993) Aromatic Substitution by the SRN1 Mechanism. American Chem-
ical Society, Washington, DC.Rossi, R.A.; Pierini, A.B.; Santiago, A.N. (1999) Org. React. (New York) 54, 1.Rubtsov, I.V.; Shirota, H.; Yoshihara, K. (1999) J. Phys. Chem. A 103, 1801.Rudzki, J.; Goodman, J.L.; Peters, K.S. (1985) J. Am. Chem. Soc. 107, 7849.Ruiz, J.; Lacoste, M.; Astruc, D. (1989) J. Chem. Soc. Chem. Commun., 813.Russel, G.A. (1954) J. Am. Chem. Soc. 76, 1595.Russel, G.A.; Danen, W.C. (1968) J. Am. Chem. Soc. 90, 347.Sagae, H.; Fujihara, M.; Komasawa, K.; Lund, H.; Osa, T. (1980) Bull. Chem. Soc. Jpn. 53, 2188.Sagdeev, R.Z.; Salikhov, K.M.; Molin, Yu.N. (1977) Usp. Khim. 46, 569.Salikhov, K.M. (1996) Magnetic Isotope Effect in Radical Reactions: An Introduction. Springer-Ver-
lag, New York.Sankararaman, S.; Haney, W.A.; Kochi, J.K. (1987) J. Am. Chem. Soc. 109, 7824.Sauer, M.C.; Shkrob, I.A.; Yan, J.; Schmidt, K.; Trifunac, A.D. (1996) J. Phys. Chem. 100, 11325.Saveant, J.-M. (1980) Acc. Chem. Res. 13, 323.Serpa, C.; Arnaut, L.G. (2000) J. Phys. Chem. A 104, 11075.Shaefer, C.G.; Peters, K.S. (1980) J. Am. Chem. Soc. 102, 7566.Shimamori, H.; Hanamuro, K.; Tatsumi, Y. (1993) J. Phys. Chem. 97, 3545.Shkrob, I.A.; Sauer, M.C.; Trifunac, A.D. (1996) J. Phys. Chem. 100, 7237.Shkrob, I.A.; Sauer, M.C.; Trifunac, A.D. (1999) J. Phys. Chem. B 103, 4773.Shkrob, I.A.; Sauer, M.C.; Yan, J.; Trifunac, A.D. (1996) J. Phys. Chem. 100, 6876.Shono, T.; Matsumura, Y.; Mizoguchi, M.; Hayashi, Ju. (1979) Tetrahedron Lett. 40, 3861.Simon, J.D.; Peters, K.S. (1981) J. Am. Chem. Soc. 103, 6403.Simon, J.D.; Peters, K.S. (1982) J. Am. Chem. Soc. 104, 6142.Simon, J.D.; Peters, K.S. (1983) J. Am. Chem. Soc. 105, 4875.Simon, J.D.; Peters, K.S. (1984) Acc. Chem. Res. 17, 277.Smid, J. (1972) In: Szwarc, M., ed. Ions and Ion Pairs in Organic Reactions. Vol. 1. Wiley, New
York.Spoyalov, A.P.; Samoilova, R.; Tyryshkin, A.M.; Dikanov, S.A.; Liu, B.L.; Hoff, A.J. (1992) J.
Chem. Soc. Perkin Trans. 2, 1519.Staley, S.W.; Kehlbeck, J.D. (2001) J. Am. Chem. Soc. 123, 8095.Staley, S.W.; Grimm, R.A.; Boman, P.; Eliasson, B. (1999) J. Am. Chem. Soc. 121, 7182.Stevenson, Ch.D.; Wilham, A.L.; Brown, E.C. (1998) J. Phys. Chem. A. 102, 2999.Svanholm, U.; Hammerich, O.; Parker, V.D. (1975) J. Am. Chem. Soc. 97, 101.Swartz, J.E.; Stenzel, T.T. (1984) J. Am. Chem. Soc. 106, 2520.Tabakovich, I.; Maki, T.; Miller, L.; Yu, Y. (1996) Chem. Commun. (Cambridge), 1911.Tachiya, M. (1993) J. Phys. Chem. 97, 5911.Tanko, J.M.; Suleman, N.K.; Hulvey, G.A.; Park, A.; Powers, J.E. (1993) J. Am. Chem. Soc. 115,
4520.Tazuke, Sh.; Kitamura, N. (1984) Pure Appl. Chem. 56, 1269.Thompson, P.A.; Simon, J.D. (1993) J. Am. Chem. Soc. 115, 5657.Tilborg van, W.J.; Smit, C.J. (1977) Tetrahedron Lett. 41, 3651.Vinatoru, M.; Iancu, A.; Bartha, E.; Petride, A.; Badescu, V.; Niculescu-Duvaz, D.; Badea, F. (1994)
Ultrason. Sonochem. 1, S27.Wakamatsu, K.; Dairiki, Ju.; Etoh, T.; Yamamoto, H.; Yamamoto, S.; Shigetomi, Ya. (2000) Tetra-
hedron Lett. 41, 365.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Warman, J.M. (1982) In. Baxendale, J.H.; Busi, F., eds. The Study of Fast Processes and TransientSpecies by Electron-Pulse Radiolysis. Reidel, Dordrecht (The Netherlands).
Werst, D.W. (1993) Chem. Phys. Lett. 202, 101.Workentin, M.S.; Schepp, N.P.; Johnston, L.J.; Wayner, D.D.M. (1994) J. Am. Chem. Soc. 116, 1141.Zhang, Z.; Evans, D.H.; Chan-Shing, E.S.; Lessard, J. (1999) J. Am. Chem. Soc. 121, 9425.Zoltewicz, J.A.; Oestreich, T.M. (1973) J. Am. Chem. Soc. 95, 6863.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

6
Organic Ion Radicals in Synthesis
6.1 INTRODUCTION
This chapter discusses typical concrete syntheses based on ion radical transformation. Theexamples provided follow these guidelines:
1. The procedures given must have definite (and tangible) advantages.2. Yields of the products must be high enough.3. The products themselves must be of practical interest.4. The synthesis procedures included sometimes are the only ones available for a
certain compound.
At this point, the author would like not to lose any readers interested in the mecha-nistic aspect of the book. For them, this truism can be offered: A chemist’s heart is devotedto mechanisms, but public demands for the chemist originate from needs for new sub-stances and new reactions. Necessity is the mother of invention! Therefore, the focus of thischapter is to express the general ideology of pursuits in the area of ion radical organicchemistry and to examine the methodologies that have evolved in the search for solutionsto synthetic problems. The chapter details achievements in ion radical organic syntheses,not only for their scientific and practical merits, but also for the aesthetic appeal of the ex-amples chosen and the intrinsic beauty of the solutions that have emerged.
The goal of this chapter is more to describe general approaches to ion radical syn-theses, the general synthetic methodology, than to provide a detailed account of each reac-tion. The choice of examples was aimed at illustrating the usefulness of ion radical synthe-ses.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

6.2 REDUCTIVE REACTIONS
6.2.1 Transformation of Ethylenic Ion Radicals
6.2.1.A Specificity of Anion Radical Reduction
Stilbene derivatives can be reduced with alkali metals in liquid ammonia. The reaction isusually performed in a homogeneous medium to give substituted diphenylethane com-pounds as a mixture of the stereoisomer forms. However, there are compounds (in particu-lar, biologically active ones) for which stereospecificity of the synthesis has decisive im-portance. A simple modification of the reduction method with an alkali metal in liquidammonia was found (Collins & Hobbs 1983) that makes it possible to perform the processstereoselectively. The metal is not predissolved, as is usual, but is added in small portionswithout trying to make the reaction medium homogeneous. Stereoselectivity is ensured byhaving the reduction proceed not in the solution bulk but on the surface of the metal.
Adsorption forces cause �-methyl- -isopropyl stilbene to be arranged in such a waythat the substituents at the ethylenic bond deviate from the metal surface. The reaction un-der consideration consists of the two consecutive one-electron steps resulting in the forma-tion of the dianion. The dianion accepts two protons, from the metal side. Therefore, re-gardless of the configuration of the initial stilbene, the reduction proceeds with apredominant formation of the erythro product (Scheme 6-1).
Eisch and Im (1977) found another simple example of a technique controlling thestereoisomeric composition of the reaction product. The technique consists of varying thetime of contact between the reagents. Scheme 6-2 illustrates the transformation of -(trimethylsilyl)styrene oxide into -(trimethylsylil)styrene under the action of complexesof zero-valent nickel; the reaction involves oxidation of the complex-bonded metal. As canbe seen from the scheme, the epoxy ring is cleaved simultaneously via nickel oxidation.The nickel–oxygen bond formed results in the formation of the carbon–nickel biradical inwhich the PhMCH fragment can rotate freely. The cleavage of the (NiO)MC bond leads toa mixture of the styrenes. At early reaction stages (30 min), cis- and trans-olefins areformed in a 50:50 ratio. After prolonged contact (30 hr.), when all possible transformationsshould be complete, the trans isomer becomes the main product and the cis:trans ratio be-comes 5:95. This enrichment of the mixture with the trans isomer follows from the forma-
SCHEME 6-1
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

tion of the cis- -(trimethylsilyl)styrene anion radical and its isomerization. The styreneformed interacts with an excess of the nickel complex. The trans isomer remains un-changed, whereas the cis isomer is converted into the trans form. The mixture thus becomesenriched with the molecules having the trans configuration. In a reference experiment, thetreatment of pure cis- -(trimethylsilyl)styrene with the same zero-valent nickel complexresults in 95% conversion to the trans isomer.
To isomerize, the anion radical’s integrity must be maintained. Therefore, the degreeof isomerization depends on the gas medium of the reaction. Let us take an example. Ul-trasonically dispersed potassium promotes the extrusion of SO2 from disubstituted cyclic3-sulfolenes to give the corresponding dienes as a mixture of the E,Z and E,E forms. TheE,Z isomer is the primary products, and the E,E isomer is the secondary product of the re-action (the E,E form is more stable thermodynamically). In the dry, oxygen-free nitrogen,this ratio is 1:20. Air is chemically aggressive with respect to anion radicals. If the reactionproceeds in air, the (E,Z)/(E,E) ratio is 1:8. Consumption of the E,E form is much greaterthan that of the E,Z counterpart, because the E,E form was contained in the higher concen-tration. Scheme 6-3 depicts the extrusion reaction under consideration (Chou & You 1987).
SCHEME 6-2
SCHEME 6-3
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Fraser and Taube, in their pioneer work of 1959, studied the interaction of Cr(2�) orV(2�) ions with complex salts containing a cation of methylmaleatopentamminocobalt. Inacid medium (HClO4 � H2O), a redox reaction takes place. As a result of this reaction, thepositive charge in the complex cation decreases by unity and the complex cation exchangeswater for the methylmaleate ligand:
[Co(NH3)5(CHCOOMe)2]3+ � Cr2+(or V2+) � H2OHClO4 � H2O→
[Co(NH3)5�H2O]2+ � Cr3+(or V3+) � (CHCOOMe)2
The liberated esteric ligand is hydrolyzed in the acid medium to give ethylene-1,2-dicarboxylic acid. However, not all the diacid formed has the cis configuration corre-sponding to the ligand in the initial complex. Along with maleic (cis) acid, fumaric (trans)acid is also formed. The higher the concentration of perchloric acid in the reaction mixture,the greater the amount of fumaric acid. The reduction of the methylfumaratopentam-minocobalt ion in D2O (see the preceding scheme) yields only fumaric acid, without maleicacid. Interestingly, this fumaric acid does not contain CMD bonds. The electron transferfrom Cr(2�) or V(2�) to the complex ion is accomplished by the so-called double-exchangemechanism. The reductant [Cr(2�) or V(2�)] transfers an electron to the bridge group(maleate) and the latter to the oxidant [Co(3�)]. In this way, the bridge MeOCOCHBCH-COOMe is reduced and oxidized alternately. This is accompanied by isomerization of cis-ethylenedicarboxylic ester into its transform. When an electron is localized at the ethylenicbond, the addition of a deuteron from the medium takes place. After removal of the elec-tron from the ethylenic bond, a D� ion is also detached. Redox changing of the bridge pro-ceeds faster than H/D isotope exchange can take place. The conversion of Co(3�) intoCo(2�) proceeds simultaneously with the replacement of the ethylenedicarboxylic acid di-ester with a water-d2 molecule in the inner sphere of the complex.
6.2.1.B Specificity of Cation Radical Reduction
Ethylenic cation radicals are also capable of rotating around the “double” bond. At the sametime, the main specificity of ethylenic cation radical reduction consists of the high selec-tivity of the reaction. One-electron oxidation was developed as a strategy for selective andefficient reduction of relatively ionizable functionalities, including conjugated dienes,styrenes, and vinyl sulfides (Mirafzal et al. 1993). Reduction is highly sensitive to substrateionizability and permits selective reduction of the more ionizable function in a difunctionalcompound.
Ionization of the substrates to cation radicals is effected by means of tris(4-bro-mophenyl) ammoniumyl hexachloroantimonate (see Section 1.7.10). Subsequent reductionof the cation radicals is accomplished by tributyltin hydride. For example, 1,1-di(anisyl)ethylene was efficiently (93%) reduced essentially in the time of mixing (lessthan 1 min).
The case of anethole, depicted next, is interesting because of the relationship betweenthe cation radical cyclodimerization and reduction processes. While the rapid cation radi-cal dimerization of trans-anethole is strongly predominant, the cyclobutadimer formed isalso ionized. Therefore, reductive cleavage of the dimer proceeds efficiently (80%):
An2CBCH2 � 2Bu3SnH2Ar3N+. SbCl6�→ An2CHCH3
93%
An � 4MMeOC6H4
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Simple alkenes such as 1-octene are completely resistant to this cation radical hy-drogenation. This makes it possible to reduce a more ionizable double bond selectively, inthe presence of a simple alkene moiety, as is illustrated for 1,1-bis(anisyl)hexa-1,5-dieneScheme 6-4.
Among the electron-rich alkenes, vinylsulfides are especially amenable to cation rad-ical reduction; one attractive feature is the absence of hydrogenolysis of carbon–sulfurbonds. The reduction of [(phenylthio)methylene]cyclohexane is efficient (88%), and the re-tention of the phenylthio group clearly contrasts with catalytic hydrogenation (Mirafzal etal. 1993). This provides versatile functionality for further synthetic operations.
The tris(4-bromophenyl)ammoniumyl hexachloroantimonate salt is not a rare reac-tant. It is available commercially. It can be readily prepared in quantity from the recoveredtris(4-bromophenyl)amine (Bell et al. 1969). It is also probably the most shelf stable of allthe stable cation radical salts; see Section 1.7.10.
6.2.2 Reduction of Ketones into Alcohols
Methods to transform ketones into alcohols are based on using of alkali metals as electrondonors. One of these methods is the Bouveault–Blanc reaction: A ketone is dissolved in analcohol and boiled with an excess of sodium. Sometimes, a ketone–alcohol mixture israpidly added to molten sodium. Sodium reacts with both ketone and alcohol.
The ketone gives the desired products; the reaction with the alcohol is undesirable:
R1R2CBO � 2Na � R3OH → R1R2CHONa � R3ONa
2 R3OH � 2Na → 2 R3ONa � H2
In a mixture of liquid ammonia with the alcohol, ketoenols and pinacols are stillformed, along with sec alcohols. Process selectivity was enhanced on the basis of mecha-nistic studies (Rautenstrauch et al. 1981). The initial stages of the reaction include the for-mation of ketone anion radicals and their dimerization with metal cation participation. Thisdimerization results in pinacol formation (Scheme 6-5).
In order to prevent the dimerization and by-product formation, Rautenstrauch and co-authors proposed to protonate the ketone anion radicals just at the moment of their forma-tion. These anion radicals contain the negatively charged oxygen atom. They can be proto-nated faster than they undergo the dimerization. The resulting hydoxyl-containingcarboradicals accept electrons faster then they undergo disproportionation or recombina-tion. This leads to suppression of ketoenol and pinacol formation.
The solvent mixture (alcohol � liquid ammonia) is not a good medium for protona-tion. Ammonia has pKa � 34; alcohols are characterized with pKa values of 16–19. Hence,an outside protonodonor should be introduced. Water is not effective as a protonatingagent; its pKa is 15.74 and close to that of alcohol. As for the ammonium cation, its pKa is
SCHEME 6-4
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

equal to 9.24. Therefore, ammonium chloride was proposed as a protonating additive. Be-sides, the addition of ammonium chloride assists buffer formation in the reaction solution.The authors used a small concentration of ammonium chloride because the metal can pref-erentially reduce the ketone, not the ammonium ion (NH4
� � e → NH3 � 1⁄2H2). Despitelow concentrations of ammonium chloride, the protonation of the ketone anion radical pro-ceeds rapidly. In the presence of ammonium chloride, the reaction results are independentof the nature of the metal (Li, Na, K). According to the authors, in the presence of ammo-nium ions, the reaction proceeds as in Scheme 6-6.
It is important that this process results in the preferential formation of thermody-namically stable alcohol diastereomer. The anion radicals almost undoubtedly contain pla-nar �CMO� and give rise to pyramidal hydroxy carboradicals. The latter form pyramidalhydroxy carbanions, which cannot exist in the presence of the ammonium cation for long.Therefore, the equilibrium, including pyramidal inversion, probably takes place at the time
SCHEME 6-5
SCHEME 6-6
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

of carboradical formation rather than carbanion formation. Transformation of the carbo-radical into the carbanion obviously proceeds faster than its dimerization or disproportion-ation. As a consequence, the reduction of an optically active ketone into an alcohol goeswithout racemization (Rautenstrauch et al. 1981).
Stereospecific ketone reduction was also observed (Giordano et al. 1985) with potas-sium, rubidium, and cesium (but not with sodium) in tertiary alcohols (but not in secondaryor primary alcohols). The undesirable dimerization probably proceeds more readily in thecase of sodium. Tertiary alcohols are simply more acidic than primary or secondary alco-hols. It is reasonable to point out that the ketone-to-alcohol reduction of 3�-hydroxy-7-oxo-5 -cholic acid by alkali metals is a key step in the industrial synthesis of 3�,7 -dihydroxy-5 -cholic acid.
The dispersity or homogeneity of the reductant in the reaction system sometimesplays a decisive role. It is also important for synthetic practice. Crandall and Muala (1986)compared the reduction of nona-5,6-diene-2-one (MeCBCBCHCH2CH2COMe) in THFupon the action of naphthalene-sodium on the one hand and, on the other, by means of son-ically activated sodium. In both cases, one-electron transfer yields the anion radical salt ofthe allenic ketone with sodium. However, only in the case of the sonicated sodium is thissalt stabilized, eventually giving MeCBCBCHCH2CH2C(OH)Me alongside cyclic prod-ucts (1-methyl-2-isopropylidene cyclopentanol and 1-methyl-2-isopropylcyclopent-2-enol). If naphthalene sodium is used, only the cyclic alcohols mentioned are obtained.
6.2.3 Preparation of Dihydroaromatics
Dihydroaromatics find diverse applications. The main way to prepare them is via Birch reduction of aromatic compounds (Birch 1944; Wooster, Godfrey 1937; Hueckel &Bretschneider 1939). Aromatic compounds are hydrogenated in diethyl ether or in liquidammonia, with alkali metals as reductants and alcohols as proton sources.
One important application of the Birch reaction is the synthesis of steroids contain-ing the keto group (Scheme 6-7). Though about 60 years old, the reaction is still widelyused. It has also attracted significant effort to elucidate its mechanism and establish its reg-ularities (see, for example, Birch et al. 1980a, 1980b, 1981; Tomilin et al. 1980; Zimmer-man & Wang 1990, 1993).
It was realized that the mechanism of the Birch reduction involves protonation of theanion radical formed by addition of one electron to the reacting aromatic compound. This
SCHEME 6-7
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

is followed by the rapid addition of a second electron and protonation of the forming car-banion to yield unconjugated alicyclic products. Protonation of the anion radical via addedalcohol is the rate-limiting stage. Recent calculations show that the ortho and meta posi-tions in anisole are most enhanced in density by electron introduction. The para site is notappreciably affected (Zimmerman & Alabugin 2001) (Scheme 6-8).
The regularities of the Birch reaction are a problem relevant to the aim of this chap-ter; bringing them together is useful in making the right decision when planning synthesis.
1. There is a kinetic preference in obtaining such regioisomers that will contain themaximum number of alkyl and/or alkoxy group on the residual double bond.
2. It is the position ortho to the maximum number of substituents that is most elec-tron-rich in an anion radical of a starting aromatic compound. Being less basicspecies, anion radicals exhibit a more selective primary isotope effect than theirmore basic (and therefore more reactive) carbanion counterparts. As a conse-quence, the deuterium enrichment in the meta position is usually higher than inthe ortho position.
3. In one of the more frequently utilized Birch reactions—the reduction ofalkyl/alkoxy-substituted naphthalenes—two reduction products are obtained inthe presence of tBuOH or tBuOD (Scheme 6-9). The acidity of the alcohol em-ployed for protonation determines the ratio of these two products. For example,the ratio of the product hydrogenated in the substituted fused ring to the product
SCHEME 6-8
SCHEME 6-9
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

hydrogenated in the unsubstituted fused ring was compared for methanol andtert-butanol. The ratio was found to be 2:1 in the case of methanol and 1.3:1.0 iftert-butanol was used (Zimmerman & Wang 1993).
4. While donor substituents assist the ortho and meta protonation, acceptor sub-stituents direct the protonation of the primary anion radicals to the ipso and parapositions.
One case of anion radical protonation—the protonation of the nitrobenzene anionradical—deserves special consideration. The main part of the electron density is concen-trated within the nitro group of this anion radical (see Section 1.2.1). It is the nitro groupthat the proton attacks. Such an attack helps enhance the population of the nitro group withan unpaired electron. As a result, ring protonation becomes prohibited. The nitro groupbearing the unpaired electron interacts with a proton. At first glance, this should lead to re-duction of the NO2
�� group. Unexpectedly, the proton captures an electron and goes awayas hydrogen. Alkali salts of the nitrobenzene anion radical almost quantitatively recover ni-trobenzene upon protonation (Russell & Bemis 1967). The evolution of hydrogen undersuch conditions is also documented (see Section 1.3.1).
This specific direction of “protonation” might be caused by the inclusion of a protonin the chelate between the two oxygen atoms of the NO2
�� group. The negative charge ofthe NO2
�� group attracts a proton. Being included in the unpaired electron delocalizationwithin the chelate, the proton seizes an electron and departs as a small radical particle �H.
In o-nitrophenol one of these chelating oxygen atoms is immobilized at the expenseof the intramolecular hydrogen bond between the neighboring nitro and hydroxy groups.Protonation of the o-nitrophenol anion radicals does result in reduction of the nitro group.The presence of oxygen significantly aids this reduction (Bil’kis & Shteingarts 1982):
ArMNO2�� � O2 → O2� ��O2NAr → ArMNO2 � O2
��
OH
O2�� � H2O � ArMNO2
�� → O2 � �OH � ArMNJ
O�
OH
ArMN → �OH � ArMNOJ
O�
This scheme describes protonation of the o-nitrophenol anion radical. If the nitro-bearing aromatic anion radical also contains another electron-withdrawing group of com-parable strength, then oxygen as a proton simply takes an unpaired electron off. The rele-vant neutral nitro compound is regenerated. Substituents neighboring NO2
� as CHBCHPh,SCl, SCN, SOR, SNR2, or another NO2 become these groups. Sections 1.2.2 and 1.3.1 in-clude the corresponding examples.
6.3 ION RADICAL POLYMERIZATION
6.3.1 Anion Radical Polymerization
Ethylene and propylene episulfides polymerize in THF at 0–70°C in the presence of sodiumnaphthalene, and the polymer contains no naphthalene residues. The reaction involves one-
J
J
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

electron transfer followed by dimerization of the resulting radical to give a dithiolate ion.This ion then polymerizes an episulfide via an anionic mechanism (Boileau et al. 1967)(Scheme 6-10). This example demonstrates the well-known polymerization initiated by an-ion radicals. Next we consider the more unusual cases of such initiation.
6.3.1.A Mechanochemically Initiated Polymerization
Polymers can indeed be made by vibromilling some monomers with steel balls. No initia-tors are needed. Such polymerization is initiated under the action of the electronic streamdeveloped by mechanoemission on conditions of vibratory milling. Mechanoemission ofexo-electrons is known as Kramer’s effect; this effect is examined in Section 7.5.
Upon vibratory milling, acryl and methacryl amides give anion radicals that are keyspecies in the reaction (Simonescu et al. 1983):
CH2BCRCONH2 � e → (CH2BCRCONH2)��
(CH2BCRCONH2)� � CH2BCRCONH2 → �CH2MCH(CONH2)MCH2MCR�MCONH2
R � H, CH3
Further growth of the polymeric chain proceeds in the usual manner. Compared tothe polymeric materials obtained via conventional methods, the mechanochemically syn-thesized polyacryl and polymethacryl amides have lower molecular weights (Simonescu etal. 1983). Acrylonitrile, styrene, �-caprolactam, isoprene as well as aryl- and methacrylamides have a special optimal duration of the polymerization upon grinding (Oprea & Popa1980). In the case of the aryl- and metacrylamides, the polymerization proceeds slowly,usually between 24 and 72 hr. After that, some acceleration takes place and the process iscompleted within 96 hr (total).
Chain growth is predominant at the beginning of the process, when there is mainlyunreacted monomer in the reaction medium and the synthesized polymers have not reachedsizes (“critical length”) sufficient to concentrate the mechanical energy. Molecular weightis also increased near the maximum conversion, when most monomer is consumed duringthe period of acceleration. When this maximum is reached, degradation takes effect and re-sults in a decrease in molecular weight to a limiting value of 103–104. Hence, the fixed andeven reduced molecular weight of polymers is the specific feature of this kind of polymer-ization. Another peculiarity consists of the amorphous character of the synthesized prod-ucts (Simonescu et al. 1983).
SCHEME 6-10
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

This type of polymerization can be of interest if amorphous polymers with moderatemolecular weights are needed.
6.3.1.B Fullerene Anion Radical Polymerization
Intercalation of fullerenes by metals results in formation of metal derivatives. Paramagneticmetallofullerenes (anion radicals) are the fullerenes doped with the endohedral metal. Ac-cording to calculations and structural studies, LaC82, for example, contains La in the cen-ter of one hexagonal ring of the fullerene cage (Akasaka and co-authors 2000; Nishiboriwith co-workers 2000; Nomura et al. 1995). Intrafullerene electron transfer in metallo-fullerenes is possible (Okazaki et al. 2001).
It was found that intercalation of C60 fullerene by alkali metals in stoichiometric ra-tio (1:1) gives rise to the formation of the anion radical salts KC60, RbC60, and CsC60 (Bom-meli et al. 1995; Btouet et al. 1996). On slow cooling of the intercalation products, [2 � 2]cycloaddition of the fullerene species neighboring in a crystal lattice occurs. Linear chainfullerenic polymers are formed. These polymers are stable in air and insoluble in THF andpossess metallic conductivity. They depolymerize only on heating above 320°C.
One of possible explanation of this conductivity assumes that polymerization offullerene anon radicals results in the formation of a long conjugated chain. That is why theconduction electrons can move along the chain.
Interestingly, if the C60 fullerene doped by alkali metals is rapidly cooled to the tem-perature of liquid nitrogen, polymerization does not occur. Only monomeric anion radicalsalts are obtained. Heating these monomers to 80–160 K results in dimerization; polymer-ization does not take place. The dimer (KC60)2 is a dielectric (Pekker et al. 1995).
It has been shown that the tris(anion)-radical C360�� can polymerize too. In particular,
Na2CsC60 forms a polymer that maintains superconducting properties (Mizuki et al. 1994).
6.3.2 Cation Radical Polymerization
6.3.2.A Linear Chain Formation
The linear cation radical polymerization of diazoacetophenone was described two decadesago (Jones 1981). The reaction proceeds with the evolution of nitrogen and attracts someinterest as a route to porous plastic materials.
Tris(4-bromophenyl)ammoniumyl hexafluoroantimonate was used as an oxidant.Reacting with a great excess of diazoacetophenone in dichloromethane at room tempera-ture, the ammoniumyl transforms into tris(4-bromophenyl)amine. This means that one-electron oxidation of the substrate takes place. Diazoacetophenone transforms into thecation radical, loses nitrogen instantaneously, and gives a polymer containing only thephenylcarbonyl side groups (Scheme 6-11).
This linear polymerization represents one unusual case of diazoacetophenone oxida-tion. For instance, upon the action of copper oxide, diazoacetophenone gives the ketocar-bene, which is involved in typical carbene reactions—dimerization, addition to olefins, in-sertion in the OMH bonds of alcohols, etc. If the amine cation radical is used as an oxidantinstead of copper oxide, only the polymer is formed. The ketocarbene was not observed,despite careful search (Jones 1981).
The synthesis of polyaniline and copolymers of aniline with o-nitroaniline is aimedat obtaining an electroactive material. This material can be used, for example, as an elec-trode in junction with magnesium to construct chemical power sources. The polymers wereprepared by oxidation of aniline or its mixture with nitroaniline; ammonium persulfate in
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

aqueous hydrochloric acid was the oxidation system. The yields of high-polymer productswere in the range of 50–80%. The first stage of the polyaniline synthesis was found to bethe formation of PhNH2
�� cation radical. The mechanism of the step-by-step condensationof aniline includes quinoidization of this cation radical and its head-to-tail coupling withthe initial cation radical. Later on, dimer deprotonation and chain lengthening take place(Koval’chuk et al. 2001).
6.3.2.B Cyclic and Branched Chain Formation
In the presence of catalytic amounts of tris(4-bromophenyl)ammoniumyl hexachloroanti-monate in methylene chloride at 0°C trans-anethole is smoothly converted in less than 5min to the cyclobutane dimer (Bauld, Aplin, et al. 1996) (Scheme 6-12).
Under the same conditions, a bis(ethylenic) close analog of anethole gives rise to acyclobutapolymer according to Scheme 6-13.
SCHEME 6-11
SCHEME 6-12
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

SCHEME 6-13
The degree of polymerization depends closely on the duration of the process. After 7min, the molecular mass is equal to 9,400 (the polydispersity index is 5.30). When the re-action is carried out for 15 min, the molecular mass of the polymer increases to 37,000 andthe polydispersity index reaches 7.31 (Bauld, Aplin, et al. 1996). Depending on whethercation radical centers arise at the expense of intramolecular electron transfer or in a step-wise intermolecular lengthening, polymerization can occur, respectively, via a chain or astep-growth process. In the present instance, the authors assume that both chain and step-growth propagations are involved.
Alongside homopolymerization, copolymerization has been studied in the frame-work of the initiation by tris(4-bromophenyl)ammoniumyl hexachloroantimonate (Bauld,Aplin, et al. 1998). In general, cation radical cycloaddition occurs more efficiently whenthe reactive cation radical is the ionized dienophile (Bauld 1989, 1992). In the cited workon copolymerization, the bi(diene) was chosen to be resistant to ionization by the initiatorused. As to dienophile functionality, propenyl rather than vinyl moieties were selected be-cause terminal methyl groups greatly enhance the ionizability of the alkene functions. Thepolymerization shown in Scheme 6-14 was performed in dichloromethane at 0°C.
The final copolymer was obtained in 90% yield; it had a molecular weight of 10,800and a polydispersity index of 2.1. In this case Diels–Alder copolymerization dominatesover the cyclobutane homopolymerization of a bi(dienophile). This means that theDiels–Alder addition of the dienophile to the diene is substantially faster than the compet-ing addition of the dienophile cation radical to the neutral dienophile.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

SCHEME 6-14
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Hence, cation radical Diels–Alder copolymerization leads to the polymer of a lowermolecular weight and lower polydispersity index than does cation radical polymerization-homocyclobutanation. Nevertheless, the copolymerization occurs under very mild condi-tions and is regio- and stereospecific (Bauld, Aplin, et al. 1998). This reaction appears tooccur by a step-growth mechanism rather than by the more efficient cation radical chainmechanism proposed for the poly(cyclobutanation). As the authors concluded, “the appar-ent suppression of the chain mechanism is viewed as an inherent problem with the copoly-merization format of cation radical Diels–Alder polymerization.”
Generally speaking, at least in theory, one attractive aspect of cation radical poly-merization, from a commercial standpoint, is that either catalysts or monomer cation radi-cals can be generated electrochemically. Such an approach deserves special treatment. Thescope of cation radical polymerization appears likely to be very substantial. A variety ofcation radical pericyclic reaction types can potentially be applied, including cyclobutana-tion, Diels–Alder addition, and cyclopropanation. The monomers most effectively em-ployed in the cation radical context are diverse and distinct from those that dominate stan-dard polymerization methods (i.e., vinyl monomers). Consequently, the obtained polymersare structurally distinct from those available via conventional methods, although the molec-ular masses observed thus far are still modest. Further development in this area would bepromising.
6.4 CYCLIZATION
Among the methods of organic synthesis, the Diels–Alder reaction holds an important po-sition. The cycloaddition of 1,3-dienes with olefins is one of the most thoroughly studiedreactions of organic chemistry.
The ion radical Diels–Alder reactions represent a new development (see, for exam-ple, the reviews by Hintz et al. 1996 and Berger & Tanko 1997). These reactions initiatedby ion radicals proceed faster by several orders of rate magnitude than the correspondingneutral (conventional) reactions. Section 6.4 presents the most important cyclizations de-veloping through cation radical and anion radical schema and the scheme that includes bothcation and anion radicals.
6.4.1 Cation Radical Cyclization
The cation radicals of ethenes, which are the primary products of one-electron oxidation,differ in their reactivity from the corresponding neutral compounds. This widens the pos-sibilities of syntheses on the basis of ethenes. Primary oxidation of ethenes (photochemi-cally, with salts of transition metals or by means of ammonlumyl salts) makes it possibleto obtain cation radicals, which initiate reactions that are unusual for ethenes in an un-charged state. For instance, the cation radical of phenylvinyl ether initiates head-to-headcyclodimerization according to Scheme 6-15 (Ledwith 1972; Farid & Shealer 1973;Kuwata et al. 1973).
Dimerization is attained via the addition of an olefin cation radical to an olefin in itsneutral form; one chain ends by a one-electron reduction of the cyclic dimer cation radical.Unreacted phenylvinyl ether then acts as a one-electron donor and the transformation con-tinues. Up to 500 units fall per cation radical. The reaction has an order of 0.5 with respectto the initiator and an order of 1.5 with respect to the monomer (Bauld, Bellville, et al.1987). Such orders are usual for branch-chain reactions. In this case, cyclodimerization in-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

volves the following steps:
1. Initiation by means of one-electron oxidation of the monomer (M):
M � Ar3N�� → M�� � Ar3N
2. Formation of the dimer (D) at the expense of the association between the cationradical obtained and the initial monomer:
M�� � M → D��
3. The chain delivery:
D�� � M → D � M��
4. The chain termination:
2M�� → (MMM)2�
Similar to other branch-chain processes, cation radical dimerization is characterizedby not too high an activation enthalpy. These magnitude are under 20 kJ�mol-1 for cyclo-hexadiene and trans-anethole (p-MeOC6H4CHBCHCHMe), respectively (Lorenz &Bauld, 1987). It is clear that the cation radical variant of cyclodimerization differs in its ad-mirable kinetic relief. For cyclohexadiene and trans-anethole, catalytic factors are 1023 and1049, respectively (Bauld, Bellville, et al. 1987).
Even dienes with shielded double bonds can be involved in diene synthesis. The pres-ence of donor groups at the double bond normally prevents its involvement in conventionalDiels–Alder condensations. With the cation radicals, these reactions do take place. Cyclicadducts are formed in high yields (80–90%) and under mild conditions. Polymerization thatusually decreases the yield is inhibited completely in the framework of the cation radicalvariant (Bellville et al. 1981). The stereoselectivity of the addition, which is usually typi-cal for diene condensation, does not change in the cation radical version and even increases.The position selectivity also increases. The regioselectivity is enhanced, as well. Bauld’sgroup has discovered and explained these effects (Bellville & Bauld 1982; Bellville et al.1981, 1983; Bauld, Bellville, et al. 1983; Bauld & Pabon 1983; Pabon & Bauld 1984).
The position selectivity is observed in those cases where a nonsymmetrically substi-tuted diene acts as a dienophile. The more substituted double bond is involved in an ion rad-ical reaction, which develops according to Scheme 6-16. This scheme makes understand-able the regioselectivity observed. Such regioselectivity is possible when both the dieneand the dienophile are nonsymmetrically substituted. Then dimerization can be of the head-to-head type, with the formation of 1,2-disubstituted derivatives of cyclohexene, or the
SCHEME 6-15
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.
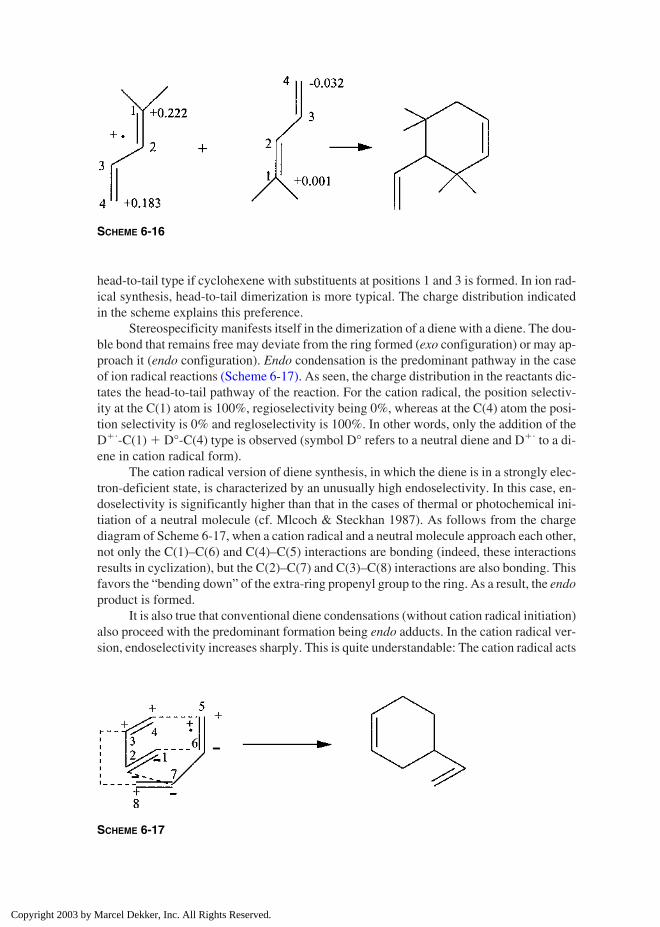
head-to-tail type if cyclohexene with substituents at positions 1 and 3 is formed. In ion rad-ical synthesis, head-to-tail dimerization is more typical. The charge distribution indicatedin the scheme explains this preference.
Stereospecificity manifests itself in the dimerization of a diene with a diene. The dou-ble bond that remains free may deviate from the ring formed (exo configuration) or may ap-proach it (endo configuration). Endo condensation is the predominant pathway in the caseof ion radical reactions (Scheme 6-17). As seen, the charge distribution in the reactants dic-tates the head-to-tail pathway of the reaction. For the cation radical, the position selectiv-ity at the C(1) atom is 100%, regioselectivity being 0%, whereas at the C(4) atom the posi-tion selectivity is 0% and regloselectivity is 100%. In other words, only the addition of theD��-C(1) � D°-C(4) type is observed (symbol D° refers to a neutral diene and D�� to a di-ene in cation radical form).
The cation radical version of diene synthesis, in which the diene is in a strongly elec-tron-deficient state, is characterized by an unusually high endoselectivity. In this case, en-doselectivity is significantly higher than that in the cases of thermal or photochemical ini-tiation of a neutral molecule (cf. Mlcoch & Steckhan 1987). As follows from the chargediagram of Scheme 6-17, when a cation radical and a neutral molecule approach each other,not only the C(1)–C(6) and C(4)–C(5) interactions are bonding (indeed, these interactionsresults in cyclization), but the C(2)–C(7) and C(3)–C(8) interactions are also bonding. Thisfavors the “bending down” of the extra-ring propenyl group to the ring. As a result, the endoproduct is formed.
It is also true that conventional diene condensations (without cation radical initiation)also proceed with the predominant formation being endo adducts. In the cation radical ver-sion, endoselectivity increases sharply. This is quite understandable: The cation radical acts
SCHEME 6-16
SCHEME 6-17
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

as an independent particle, and the role of the suitable molecular orbital of the cation radi-cal turns out to be determinant.
The publications of Bauld’s group, which were reviewed earlier, deal only with re-actions in which a cation radical (readymade) acted as the reactant. However, there can becases where a cation radical is formed in the course of donor–acceptor interaction betweeninitially neutral molecules. Then the rigid or sharply enhanced selectivity of the reaction ac-quires diagnostic significance. For such cases, this is the general rule: Condensation is per-missible only for the (dienophile cation radical � diene) pair and forbidden for the(dienophile � diene cation radical) pair.
This rule can be understood from the orbital correlation diagram of Scheme 6-18,where the symbols S and A denote symmetric and antisymmetric orbitals, respectively(Bellville & Bauld 1982; Bauld, Bellville, et al. 1983). The interaction between orbitals ofequal symmetry is the indispensable condition of the condensation under consideration. Asseen from the scheme, the condensation becomes possible only when the diene suppliesfour electrons and the dienophile provides one electron. Bauld and co-authors denote suchinteraction as [4 � 1]. If the diene supplies three electrons and the dienophile provides twoelectrons (in the manner of [3 � 2] electrons), no cyclic adduct can be formed.
If cation radicals of both the diene and the dienophile can be formed upon the actionof the cation radical initiator, some kind of separation operates. Each of these cation radi-cals can exchange an electron with any participant in the reaction. However, since only thediene cation radical is consumed, the equilibrium of the electron transfer is graduallyshifted toward this particular cation radical.
SCHEME 6-18
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The diene depicted in Scheme 6-18 enters the reaction in its s-cis form. If the dienecation-radical is in the s-trans form, a cylobutane product forms (Reinolds et al. 1989;Botzem et al. 1998).
Naturally, the question arises as to whether the diene component really has to be inits s-cis form for the cation radical Diels–Alder reaction. According to calculations (Hof-mann & Schaefer 1999), the s-trans-butadiene is more stable than its s-cis isomer by 12kJ�mol�1, and for the cation radicals the trans preference is even slightly more pronounced(16 kJ�mol�1).
However, the vinylcyclobutane cation radical can rearrange to the cyclohexenecation radical (Bauld & Yang 1999); the latter is more stable than the former by about 120kJ�mol�1 (Haberl et al. 1999). Of course, such rearrangement is possible only when thevinyl group in the vinylcyclbutane structure has transformed from the exo to the endo con-figuration through rotation around the (vinyl)-(cyclobutane) bond. The rotation barrier isnot high, so “the cyclohexene cation radical can be formed from ethylene and the trans-bu-tadiene cation radical as easily as from ethylene and the cis-butadiene through a cation rad-ical vinylcyclobutane/cyclohexene rearrangement” (Hofmann & Schaefer 1999). It wasshown experimentally that vinylcyclobutane can be converted into cyclohexene under elec-tron-transfer conditions (Haberl et al. 1999).
It is very important to consider trienes of the general formula MeCHBCHMCHBCHMCH2MCH2MCH2MCHBCHR. Such trienes contain moieties of both the di-ene and dienophile types in the framework of the same molecule. Upon initiation by theAr3N� cation radical, cyclization takes place. Tetrahydroindanes are formed. The trienesof R � PhS, p-MeOC6H4CH2, and H have been studied. The third triene, R � H, containsthe dienophile fragment of lowered oxidizability; it is not cyclized at all. The diene syn-thesis proceeds in the cases of the first two trienes only (Harirchian & Bauld 1987) (Scheme6-19).
As it has already been noted, the reaction is initiated by an arylamine cation radical,and dichloromethane is commonly used as a solvent. Recently, a simple but valuable tech-nique has been elaborated for minimizing acid-catalyzed side reactions under aminiumyl
SCHEME 6-19
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

salt conditions (Bauld, Yang, & Gao 2000). The technique consists of using a two-phasesystem of dichloromethane with water instead of just dichloromethane. The use of water asa second phase was meant to remove any inorganic acid, which may initially have been pre-sent in the salt catalyst, and also to continue to extract these acids from the dichloromethanesolution as they are formed. Since the salt catalyst is rather insoluble in water and since thetypical cation radical cycloaddition reactions are completed within 1–3 minutes or less, re-action efficiency is diminished only moderately by the presence of water. Under the two-phase conditions, the cyclopentadiene cycloaddition to N-vinylcarbazol is achieved, eventhough this dienophile is extremely prone to acid-catalyzed polymerization.
As was noted in Section 1.3.2.A, one-electron oxidation causes deprotonation ofcation radicals. Cation radicals bearing protons that are to a site of charge/spin densityare superacids. Because of this feature, attention must be given to the distinction betweencation radical and H-acid catalysis of cycloaddition. Bauld’s group has elaborated a set ofcriteria that allow one to differentiate these mechanisms one from another (Reinolds et al.1987).
For H-acid catalysis: stereospecifity is lowered and appears to be the same as in thereactions initiated with trifluroacetic acid instead of the ammoniumyl salt. For the cation-radical mechanism: the sterically hindered base 2,6-bis(tert-butyl)pyridine does not inhibitthe cyclization; triarylamine retards this reaction; photosensibilized one-electron oxidationof a diene leads to the same products, which are formed in the presence of the ammoniumylsalt. As shown, in the majority of cases only the cation radical chain mechanism of the diene–diene cyclization is feasible (Bauld, Bellville, & Harirchian 1987). Meanwhile, cy-clodimerizations of 2,4-dimethylpenta-1,3-diene (Gassman & Singleton 1984) and 1,4-dimethylcyclohexa-1,3- or -1,4-diene (Davies et al. 1985) proceed through both mecha-nisms.
Steckhan’s group described a wide range of cycloaddition reactions between 2-vinylindoles acting as heterodienes and cyclic or acyclic enamines bearing acceptor groups in positions (Guertler et al. 1996). The reaction was induced by formation of the 2-vinylindolecation radicals via anodic oxidation. The synthesis of 4a-carbomethoxy-6-cyano-5,7-dimethylindolo[1,2-a]-1,2,3,4,4a,12a-hexahydro-1,8-naphthyridine can serve as an exam-ple (Scheme 6-20).
The known tendency of the 2-vinylindole to homopolymerize is sufficiently low onthe electrode surface. Therefore, an electrochemical initiation of the reaction under poten-tiostatic control is very favorable. The yield is high (90%), and the product formed is es-pecially interesting because it incorporates the skeleton of the indole alkaloid goniomitine.
SCHEME 6-20
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Intramolecular cyclization of o-diethynylbenzene gives us an opportunity to comparethe results of the thermal and cation radical variants of the reaction. There are three possi-ble modes of cyclization (Scheme 6-21). While 1,6 cyclization takes place in the thermalprocess, cation radical initiation leads to 1,5 cylization (Ramkumar et al. 1996). Chemicaloxidation of o-diethynylbenzene bearing two terminal phenyl groups by tris(p-bro-mophenyl)aminiumyl hexachloroatimonate as the catalytic oxidizing agent in the presenceof oxygen yields 3-benzoyl-2-phenylindenone in 70% yield (Scheme 6-22).
The initial step of this reaction consists of the one-electron oxidation of the substrate.The resulting cation radical of o-diethynylbenzene cyclizes to the fulvenyl form, which fur-ther reacts with the neutral substrate to yield the fulvenyl diradical and the substrate cationradical. The latter is the chain carrier. The fulvenyl diradical adds oxygen and transformsinto the final product (Scheme 6-23).
According to the authors, the 1,5-cyclization mode of the o-diethynylbenzene is de-termined by electron state symmetry, which is different from that of the neutral moleculeof o-diethynylbenzene (Ramkumar et al. 1996).
SCHEME 6-21
SCHEME 6-22
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Cation radical cyclization can be performed (and accelerated significantly) undersonication. Thus, the reaction of methyl vanillate with phenyliodonium bis(acetate) is ini-tiated by ultrasonic irradiation and gives rise to the corresponding o-quinone monoketal,which is trapped by a series of furanes. Monoadducts are formed; reactions can be con-ducted at room temperature and are essentially complete within 15–50 min (Avalos et al.2000).
Sonochemically induced cation radical intramolecular cyclization upon the action ofan iodonium salt was also demonstrated (Arizawa and others 2001). Being oxidized withphenyliodonium bis(trifluoroacetate), 1-(3-anisyl)-2-(1,3-cyclohexadien-2-yl) ethaneforms the cation-radical and then 5-methoxyspiro[cycloxehane-1,1-indan]-2,6-dione.The yield of this final product is high enough.
Iodonium salts are at present receiving much attention because of their similar reac-tivity to those of heavy metal reagents. The results of the application of salt are close toelectrochemical oxidation. Added advantages that render them synthetically more attrac-tive are their low toxicity, ready availability, and easy handling. In contrast to the anodicversion, the iodonium reactions are amendable to large-scale synthesis.
Turning from the intramolecular process to intermolecular ones, we now extend ourcomparison of the thermal and cation radical cyclizations. It is also interesting to take son-ication into account as a route to initiate cyclizations. The reaction between 2-butenal N,N-dimethylhydrazone (a diene) and 5-hydroxy-1,4-naphthoquinone (a dienophile) gives suchan opportunity. In toluene at 20°C, the reaction follows Scheme 6-24 (Nebois et al. 1996).
Without ion radical initiation, the yield of the resulting product reaches 50% for 24hr. Practically the same yield can be achieved for the same time in the presence of tris(4-bromophenyl)ammoniumyl hexachloroantimonate and for only 6 hr upon sonication(Nebois and associates 1996). Sonication accelerates the rate-determining formation of the
SCHEME 6-23
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

diene cation radical. Of course, hydroxynapthoquinone is strong enough as an electron ac-ceptor with respect to 2-butenal N,N-dimethylhydrazone. Therefore, the question remainswhether sonication is a more or less general method for initiation of ion radical cycloaddi-tion. A possible role of sonication in the optimization of ion radical reactions is consideredin Section 5.2.4.
Additionally, we should mention the photoinitiated reaction between the diphenylbu-tadiene cation radical and acetonitrile (Mattes & Farid 1980) (Scheme 6-25). Lijser andArnold have given a theoretical explanation for this reaction (1998). As to potoinitiation ingeneral, it is important to be very careful in one’s choice of sensitizers. For example, at-tempts to initiate the cyclization of homobenzylic ethers failed if 1,4-dicyanobenzene wasused as a sensitizer. Rapid regeneration of the starting material by return electron transferfrom the dicyanobenzene anion radical to the substrate cation radical was the cause of cy-clization inefficiency. To slow this unproductive process, a mixture of N-methylquinolin-ium hexafluorophosphate (sensitizer), solid sodium acetate (buffer), and tert-butylbenzene(cosensitizer) in 1,2-dichloroethane was employed. This dramatically increased the effi-ciency of the reaction, providing cyclic product yields of more than 90% in only 20 min(Kumar & Floreancig 2001).
6.4.2 Anion Radical Cyclization
As was seen in Section 6.4.1, one-electron oxidation brings certain advantages to diene per-icyclic reactions. One-electron reduction of unsaturated �,-diketones may sometimes gen-erate a diene structure. This may start condensation. Thus, triazoledions (TAD) pass intotetra-azabicyclooctanes (TABO). Naphthalene-sodium (ca. 1 mol.%), or sodium metal, oreven sodium iodide efficiently catalyzes the reaction. Tetracyanoethyene and lead tetra-ac-etate retard it. Consequently, the condensation has a chain character (Borhani & Greene1986) (Scheme 6-26).
SCHEME 6-24
SCHEME 6-25
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

SCHEME 6-26
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Several variants of Sml2-induced cyclizations have attained synthetic importanceowing to their high stereoselectivity (for reviews see Molander & Harris 1996, 1998).Samarium iodide is a very promising one-electron transfer reagent in organic chemistry(Section 1.7.12). Frequently, ketyl anion radicals generated by electron transfer are the re-active species that intramolecularly add to a multiple bond offered at an appropriate dis-tance. These ketyl anion radicals can also attack an aryl moiety in an intramolecular fash-ion and, after a second electron transfer, lead to 1,4-cyclohexadiene derivatives. Let us takeas an example the reaction of the N,N-dibenzyl-substituted amino ketone, which producesan isomeric mixture of the iso-quinoline derivatives, with total yield being 90% (Dinesh &Reissig 1999) (Scheme 6-27).
The mechanism shown in Scheme 6-28 seems likely. The first (reversible) electrontransfer generates the ketyl anion radical. It then attacks the aryl group in the ortho posi-tion. The resulting cyclohexadienyl radical is reduced to a cyclohexadienyl anion by a sec-ond electron transfer, and the anion is finally protonated. HMPA as a cosolvent can be re-placed with noncarcenogenic N-methylpyrrolidone (Dinesh & Reissig 1999). The newreaction mode of samarium ketyls undoubtedly has a synthetic perspective.
Tetrathiafulvalene can also provoke cyclizations. This can be seen from Scheme 6-29 (Lampard et al. 1993). The final cyclic alcohol is formed almost quantitatively, and only0.2 equivalent of tetrathiafulvalene is needed.
Another, practically attractive example of intermolecular anion radical cyclization isthe zipper reaction of cyclic o-ethynylbenzenes (Bradshaw et al. 1994) (Scheme 6-30). The
SCHEME 6-27
SCHEME 6-28
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

SCHEME 6-29
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

SCHEME 6-30
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

final product, 9,8-bis(trimethylsilyl)diindeno[2,3-g:23-p]chrysene (60% yield) has a he-lical geometry in the crystal state. The trimethylsilyl group is very reactive and can be in-volved in further reactions. Hence, this zipper cyclization offers a route to helical ribbonoligomers. It may be applicable to syntheses of helical ribbon polymers from o-ethynyl-benzene monomers. Such polymers are attractive as conducting and nonlinear optical ma-terials because of their �-electron conjugation, environmental stability, high mechanicalstrength, and enhanced threshold to laser damage. The most serious problem associatedwith such applications is that of poor solubility. However, helical aromatic ribbon poly-mers, in which the helical axes extend down the aromatic chains, usually have lower latticeenergies and therefore higher solubilities than planar systems of comparable molecularweights. The reaction depicted foresees the possibility of extending the helical axes at theexpense of the trimethylsilyl groups in the cyclic product.
Ion radical reactions also open convenient routes to fused benzoheterocycles as the re-sults of intramolecular cyclization. The fused heterocycles are interesting as compounds of po-tential physiological activity. Many of them are used as medications. Certainly, only those syn-theses that do not change the functional groups needed to provide or enforce the curative effectare of interest. At the same time, it is desirable to exclude acidic agents or reagents leading tosplitting of the final heterocycles, to decreasing yields, or to contamination of the desired prod-ucts. Certainly, direct nonmultistep syntheses, which can proceed in mild conditions, are at-tractive. Many of these problems can be solved in the framework of ion radical reactions.
Thus, direct SRN1(Ar) substitution reactions allow preparing isoquinolines (Beugel-mans et al. 1984), indoles (Beugelmans & Roussi 1979), and benzothiazoles (Boujlel et al.1982) via “one-pot” syntheses. The photoinitiated synthesis of 2-methylindole shown inScheme 6-31 is a representative example.
The synthesis of 4-azaindole is also a photochemical synthesis and is very simple. 3-Amino-2-chloropyridine and acetaldehyde are the starting materials (Fontan et al. 1981)(Scheme 6-32).
With conventional methods, the formation of indole derivatives proceeds at the expenseof both unsubstituted otho positions in the phenyl ring. This leads to undesirable by-products.In particular, the formation of the by-products takes place during Fisher’s synthesis of benzo-heterocycles. In the previously described ion radical variant of the synthesis, only one indole iso-mer is formed, the isomer that corresponds strictly to the structure of the starting haloaniline.
SCHEME 6-31
SCHEME 6-32
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The anion radical mechanism for these syntheses is based on the following facts. Thereactions require photo- or electrochemical initiation. Oxygen inhibits the reactions totally,even with photoirradiation. Indoles are formed from o-iodoaniline only, the meta isomerdoes not give rise to indole. Hence, the alternative aryne mechanism (cine substitution) isnot valid. What remains as a question is the validity of the ion radical mechanism only forsubstitution of the acetonyl group for the halogen atom in o-haloareneamine or for the in-tramolecular condensation.
At this point, one unsuccessful attempt deserves to be mentioned. Creating anonacidic procedure for benzothiazole syntheses, Bowman and co-authors (1982) tried toperform intramolecular cyclization according to the top line of Scheme 6-33. However, thecyclization did not take place, irrespective of solvent polarity or the strength of the base.UV irradiation did not help either. Nevertheless, the cyclization appears to be successful inthe presence of acetone; see the bottom reaction in Scheme 6-33 (Bowman et al. 1982). Asusual, inhibitors stop this anion radical reaction; 3-iodothiobenzanilide does not experiencethe cyclization.
The phototransformation of 1,2-bis(phenylsulfonyl)-3,4,5,6-tetramethylbenzene into2,3,4,5-tetramethyldibenzothiophene-S,S-dioxide upon the action of arylthiolates shouldalso be mentioned. The yield of dibenzothiophene-S,S-dioxide is more than 90%. The ad-dition of m-dinitrobenzene prevents the cyclization. The reaction proceeds as shown inScheme 6-34 (Novi et al. 1982).
As has been found (Engman et al. 1999), sodium alkyltelluroates are excellentreagents for the generation of aryl radicals from the corresponding iodides in the dark. Ac-cordingly, 1-(2-iodophenyl)-1-methyloxirane reacts with two equivalents of sodium n-butyltelluroate, giving 2,3-dihydro-3-hydroxy-3-metylbenzo[b]tellurophene, which wasisolated in 62% yield. The latter was readily converted into 2,3-dihydro-3-methylbenzo[b]tellurophene by treatment with a catalytic amount of p-toluenesulfonic acid (94% yield)(Scheme 6-35).
SCHEME 6-33
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.
The reaction develops according to typical SRN1 mechanism and proceeds withoutphotoirradiation (Scheme 6-36). The practical significance of this reaction consists in thedevelopment of novel antioxidants carrying chalcogen atoms. Divalent organochalcogencompounds react readily with many types of oxidants (peroxide, peroxyl radicals, perox-ynitrite, singlet oxygen, and ozone). The tetravalent organylchalcogenides formed are re-duced by many mild reducing agents. Therefore, compounds of this sort have the potentialto act as catalytic antioxidants.
6.4.3 Cyclizations Involving Cation Radicals and Anion Radicals inLinked Molecular Systems
If a carbon chain separates the electron donor (D) and electron acceptor (A) sites in thesame molecule, an exiplex-resembling state may originate upon photoirradiation. Such anexiplex state represents some resonance hybrid of the electron-transfer configuration[D��M(CH2)nMA��] mixed with the locally excited configuration [D*M(CH2)nMA] or[DM(CH2)nMA*].
The photolysis of donor–acceptor systems provides unique synthetic opportunities.Direct irradiation of the donor–acceptor systems, such as systems containing arene and
SCHEME 6-34
SCHEME 6-35
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

amine components, leads to intramolecular electron transfer, i.e., to amine cation radicaland arene anion radical moieties.
Having been generated, these moieties undergo cyclization reactions providing effi-cient synthetic routes to N-heterocycles with a variety of ring sizes. Thus, direct irradiationof secondary aminoethyl and aminopropyl stilbenes leads to benzazepines in improvedyields (Hintz et al. 1996). As known, benzazepines are used in medicine as antidepressants.Scheme 6-37 illustrates ion radical cyclization with the formation of the benzazepinederivative (65% yield).
One representative example belongs to the systems linked topologically, in singlecrystals. The solid-state cycloaddition of bis(N-ethylimino)-1,4-dithiin to anthracene pro-ceeds at the expense of the 9, 10 positions. This is usual for the anthracene cycloadditions.In the single crystal the reaction begins with the formation of the intermolecular (1:1) com-plex. The latter is formed as a result of electron (charge) transfer. The thermal hetero-molecular solid-state condensation involves the entire crystal. And this rare crystallineevent is under topochemical control during the entire cycloaddition. As a result, a specialcrystalline modification of the cycloaddition product is formed, with crystal packing simi-lar to that of the starting charge-transfer crystal but very different from that of the (ther-modynamically favored) modification product obtained from solution-phase crystalliza-tion. Such a single-phase transformation was readily monitored by X-ray crystallography
SCHEME 6-36
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

at various conversion stages, and the temporal changes in crystallographic parameters werecorrelated with the temperature-dependent (solid-state) kinetic data by 1H-NMR spec-troscopy at various reaction times. Thus, an acceleration of the solid-state reaction overtime was found. The acceleration results from a progressive lowering of the activation bar-rier for cycloaddition in a single crystal as it slowly and homogeneously converts from thereactant to the product crystal lattice (Kim and co-authors 2001).
6.5 RING OPENING
The three-membered ring as a part of metal-ketyl anion radicals is readily opened. Insteroid synthesis, this reaction is a classic procedure for introducing angular methyl groups(Dauben & Deviny 1966) (Scheme 6-38).
The ring opening of 2-acylaziridines can be performed upon the action of Sml2 in aTHF-MeOH mixture. The final product—N-tosyl- -aminoketone—is formed in 95% yield(Molander & Stengel 1995) (Scheme 6-39).
The reaction of 1,3-disubstituted bicyclo[2.1.0]pentanes with tris(4-bromophenyl)ammoniumyl hexachloroantimonate (the latter in catalytic amounts) leads to the corre-sponding cyclopentene after 1,2-hydrogen or 1,2-alkyl migration in the intermediary 1,3-cation radicals (Adam & Sahin 1994) (Scheme 6-40).
SCHEME 6-37
SCHEME 6-38
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Adam and Heidenfelder (1998, 1999) analyzed rearrangement of the regio- and stere-oselctivity of cyclopenta-1,3-diyl cation radicals. The regioselectivity of the migration maybe tuned through the electronic character of the substituents on the diyl sites, which was ra-tionalized in terms of a single molecular orbital interaction diagram. The diastereoselectiv-ity of the 1,2-shift is controlled by the steric factors in the intermediary 1,3-cation radicals.The chemical one-electron oxidation of the initial fused structures with ammoniumyl saltshas two advantages. On the one hand, electron back-transfer to the cyclopentadiene-1,3-ylcation radical is minimized; on the other hand, this reaction may be run on a preparativescale (Adam, et al. 1995).
The ammoniumyl oxidant induces the quantitative conversion of a cage compound tothe corresponding diene; the reaction proceeds at room temperature for 1 min (Hasegawa& Mukai 1985) (Scheme 6-41). The addition of tetramethoxybenzene (a donor compound)retards the reaction. In the presence of this electron donor, the degree of conversion is di-minished up to 30%. Addition of alkylamines, such as trietylamine, diethylamine, or 1,4-diazabicyclo[2,2,2]octane, completely quenches the ring opening in this case. By contrast,triphenylamine does not quench the reaction (Hasegawa & Mukai 1985). These observa-tions are consistent with the fact that alkylamine cation radicals are less stable than ary-lamine cation radicals (Chow et al. 1978). The ammoniumyl cation radical converts triph-enylamine into its cation radical. The lifetime of the latter is relatively long, so this cationradical can really interact with the starting cage compound. This results in the conversionof the cage cation radical into the cation radical of the diene. One-electron reduction of thiscation radical by a neutral amine (triphenylamine of its tribromoanalog) leads to the finaldiene compound. At this stage, triarylamine transforms into the corresponding cation radi-cal and returns to the catalytic cycle.
Takahashi and co-authors (1996) described another case of cation radical cyclore-version. Benzocyclobutenols undergo ring opening induced by electron transfer to gener-ate quinodimethane intermediates, which then tautomerize to benzenoids. The reaction pro-ceeds upon irradiation in the presence of tetracyanoanthracene (� � 350 nm). Yields (basedon PMR analyses) are quantitative (Scheme 6-42).
SCHEME 6-39
SCHEME 6-40
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

6.6 FRAGMENTATION
6.6.1 Hydrogen Abstraction
6.6.1.A Selective Oxidation of p-Cresol and p-Xylene
Benzoic and hydroxybiphenyl carboxylic acids are widely used in the synthesis of drugs,biologically active compounds, and heat-resistant polymers. The known methods forpreparation of aromatic hydroxy carboxylic acids include many stages, require not readilyaccessible starting compounds, and provide poor yields of the target products.
A method was proposed for preparation of p-hydroxybenzoic acid by oxidation of p-cresol with atmospheric oxygen in an acetic acid–acetic anhydride mixture with the cat-alytic action of cobalt acetate, manganese(II) acetate, and sodium bromide (Litvintsev et al.1994). This procedure ensures a 60% yield of p-acetoxybenzoic acid and 100% conversionof the initial p-cresol.
In contrast to para-cresol, ortho-cresol does not undergo oxidation under these con-ditions. The same restriction holds in the case of 4-hydroxy-3,4-dimethylbiphenyl: Onlyone methyl group undergoes oxidation, the one in position 4 (Koshel’ et al. 1997). Themethyl group that is in position 3—ortho with respect to the hydroxy group—remains in-tact. Such inactivity is explained in terms of the cation radical mechanism (Scheme 6-43).
It is known that oxidation of alkyl-substituted aromatic hydrocarbons in acetic acidupon the action of a metal bromide catalyst follows the one-electron transfer mechanism(Sheldon & Kochl 1981). The rate-determining stage is the one-electron transfer from thesubstrate to the metal ion in the highest oxidation state (Digurov et al. 1986). As a result, anunstable cation radical is formed that loses a proton to give more stable hydroxybenzyl rad-ical. The latter reacts with oxygen, yielding at first aldehyde and then carboxylic acid.Methyl hydrogen atoms in the cation radical are more acidic than in the initial molecule (see
SCHEME 6-41
SCHEME 6-42
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Section 1.3.2.A). The loose proton of the methyl group in cation radicals is capable of form-ing an intramolecular hydrogen bond with the oxygen atom of the ortho-hydroxy group. Thishydrogen bonding keeps the proton within the “fused” cycle, blocks the formation of the or-tho-hydroxybenzyl radical, and retards or prevents the consequent oxidation.
Such an explanation seems logical. A newly developed infrared (IR) spectroscopictechnique, called autoionization-detected IR (ADIR) spectroscopy, was applied for a studyon hydroxy–alkyl interaction in the o-cresol and o-ethyl phenol cation radicals. The re-markable low-frequency shift of the O–H vibration was observed for these o-derivativesonly, not for the m- or p-analogues (Fujii et al. 1998; Fujimaki and others 2000). However,ab initio geometry optimization and topological electron-density analysis led Vank and co-authors (2001) to the conclusion that there is “no evidence for this hydrogen bond” and theADIR experimental data “should be explained without reference to a hydrogen bond” at all.According to Trindle (2000), the perturbative admixture of antibonding orbitals with thehydroxyl fragment orbital provides an effective rationale for the o-cresol cation radicalphenomenon. Future events will obviously uncover the truth; that is the way of science.
The selective oxygenation of ring-substituted toluenes to aromatic aldehydes hasbeen one of the most important organic reactions in industrial chemistry because of the use-ful applications of aromatic aldehydes as key intermediates for the production of pharma-ceuticals, dyes, pesticides, and perfumes. The known methods of aldehyde synthesis havebeen limited because of low yield and poor selectivity as well as the generation of copiousamounts of inorganic waste. Ohkubo and Fukuzumi (2000) discovered a cation radicalmethod of 100% selective oxidation of p-xylene to p-tolualdehyde. The reaction is initiatedby photoinduced electron transfer from p-xylene to the singlet excited state of 10-methyl-9-phenylacridinium ion under visible light irradiation (in air). The yield of p-tolualdehydeis quantitative. The reason for such high selectivity lies in the fact that the primary cationradical readily loses protons and transforms into p-methyl benzyl radical. This radical istrapped by air oxygen and eventually gives rise to p-tolualdehyde:
[p-H3CC6H4CH3]�� → H� � p-H3CC6H4CH�2
p-H3CC6H4CH�2 � O2 → p-H3CC6H4CH2OO�
p-H3CC6H4CH2OO� � H� → p-H3CC6H4CH2OOH → H2O � p-H3CC6H4CHO
SCHEME 6-43
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

6.6.1.B Selective Oxidation of 4,5-Dimethylimidazole
Methylated aromatic heterocycles (HetCH3) form cation radicals that are typical �-acidsand expel a proton. Methylene radicals are formed. These radicals give rise to the corre-sponding carbocations if an oxidant was taken in excess. Nucleophiles attack the ions, com-pleting the reaction. If water is the reaction medium (the hydroxyl anion is a nucleophile),an alcohol is formed. The alcohol rapidly transforms into the aldehyde upon action of thesame oxidant:
HetCH3 � e → HetCH3�� → HetCH�
2 � e → HetCH2�
HetCH2� � OH� → HetCH2OH → HetCHO
During oxidation of 4,5-dimethylimidazol by peroxydisulfate in water, the reactionis stopped just at the alcohol-formation step. The alcohol is stabilized at the expense of anintramolecular hydrogen bond. This keeps it safe from further oxidation (Citterio & Minisci1982) (Scheme 6-44).
The final product, 4-hydroxymethyl-5-methylmidazol, is a key intermediate in an in-dustrial synthesis of cimetidine, the effective antiulcer drug. The cation radical route to themethylimidazol carbinol is practically waste free with respect to the organic substance: Thetarget product is formed in 70% yield, and the starting material returns (30%) (Citterio &Minisci 1982). It is worth noting that the starting material is easily obtained from cheapmethylethyl ketone. Other methods for carbinol preparation are less effective: Hydrox-ymethylation of 4-methylimidazole is less selective, and reduction of 4-methylimidazol-5-carboxyethylate is characterized by lowered yields of the target product.
6.6.2 Cation Radical Route to Group Deprotection
The protection of certain functional groups and the deprotection of the protected deriva-tives constitute important processes in the synthetic organic chemistry of polyfunctionalmolecules, including the total synthesis of natural products. Following are examplesdemonstrating some important improvements to the conventional methods for defensegroup deprotections.
6.6.2.A Removal of Butoxycarbonyl Defense
The tert-butoxycarbonyl (t-BOC) group is often used for protection of the amino, hydroxy,and sulfhydryl groups. This kind of defense is usual in the protection of amino acids in pep-tide synthesis.
Reagents to remove the t-BOC group include hydrochloric acid in ethyl acetate (Stahlet al. 1978), sulfuric acid in dioxan (Houghton et al. 1986), anhydrous hydrogen fluoride
SCHEME 6-44
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

(Yamashiro et al. 1972), boron trifluoride etherate in acetic acid (Schnabel et al. 1971),trimethylsylil triflate (Schmidt et al. 1987), trimethylsilyl perchlorate (Vorbrueggen &Krolikiewicz 1975), and, most frequently, trifluoroacetic acid (Farowicki & Kocienski1995 and references therein). Deprotection of the t-BOC group under neutral conditionswas not described until recently, yet it is highly desirable. Now it has been found that thetert-butoxycarbonyl protecting group for amines, alcohols, or thiols is removed efficiently(90–99% yield) with use of 0.2 equivalent of cerium ammonium nitrate in acetonitrile at80°C (Hwu et al. 1996):
OR � But Ce(NH4)2(NO3)6 (0.20 equiv)→
MeCNRXH
J J (90–99%)X O
X � NH, NR, O, S
The proposed mechanism includes transformation of the carbonyl group into thecation radical state while reduction of Ce(IV) to Ce(III) takes place. The cation radical thenundergoes fragmentation to give the tert-butyl cation and the carboxylic radical. Regener-ation of Ce(IV) from Ce(III) during reduction of the carboxylic radical to the carboxylateion allows using cerium ammonium nitrate in catalytic amounts for the entire deprotectingprocess. Finally, extrusion of CO2 from the carboxylate ion followed by protonation givesthe free amine (Scheme 6-45).
Hence, the method has been worked out to remove the tert-butoxycarbonyl group thatprotects amines, alcohols, or thiols under neutral conditions rather than the conventionalstrongly acidic conditions. The work reports that high-yield deprotection can be achievedby using catalytic amounts of ceric ammonium nitrate in refluxing acetonitrile. The data aregiven on a variety of the t-BOC-protected substrates, including ones containing acid-labilegroups.
6.6.2.B Removal of Methoxybenzyl Defense
The methoxybenzyl group is often employed for protection of the carboxyl function. Let usconsider completely protected phenylalanine as an example.
JJ
SCHEME 6-45
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

In complicated syntheses of natural or bilogically active compounds, it is very im-portant to remove protection for one functional group and to keep protection for anothergroup in a polyfunctional molecule. Such a molecule is the phenylalanine derivative, withthe NH2 and COOH functions having been protected with the tert-butoxycarbonyl- and p-methoxybenzyl groups, respectively. Both protective groups are saponified in acid media.Therefore, the usual methods are not applicable for the selective deprotection. However,these two functions differ by their electron-donor capabilities. Therefore, single-electronoxidation by tris(4-bromophenyl)ammoniumyl hexachloroantimonate leads to the forma-tion of a cation radical with spin-positive density centered on the benzylic fragment. Theone-electron-oxidized benzylic group expels its proton. A radical center emerges, and thiscenter is adjacent to the oxygen atom bearing an electron pair. The electron pair and the un-paired electron of the CH�
2 group form a three-electron bond, and the oxygen-benzylic spinlocalization becomes even more pronounced. It is the OMCH2 bond that is eventually split.The breakdown proceeds purely selectively; the N-acylated phenylalanine is formed withmore than 90% yield. The following scheme illustrates the transformations described (Dap-perheld & Steckhan 1982):
O CH2Ph�
Me3CMOMCMNHMCHMCMOMCH2C6H4OMeM4 →�O
O CH2Ph�
→ Me3CMOMCMNHMCHMCMOMC��H2C6H4OMeM4 →
�O
O CH2Ph�
→ Me3CMOMCMNHMCHMCMO��MC
�HC6H4OMeM4 →
�O
O CH2Ph O� �
→ Me3CMOMCMNHMCHMCMOH � HMCMC6H4OMeM4�O
Because the compared protective groups in phenylalanine also differ in their electron-acceptor activity, one can expect that anion radical generation would result in a selective re-moval of the amine defense. This assumption is supported by ESR studies of some peptidecation radicals (Lin et al. 1998). For instance, neat histon (a protein from cell nuclei) givesan anion radical in which the unpaired electron is localized at the amidocarbonyl function.
6.6.2.C Removal of Trimethylsilyl Defense
The trimethylsilyl moiety is now widely used as a protecting group for alcohols (throughthe formation of trimethylsilyl ethers) or for aldehydes and ketones (through the formationof trimethylsilyl enol ethers). The trimethylsilyl-protecting group has routinely been re-moved with fluoride ion, acids, or bases. Unfortunately, these reagents offer little in theway of selectivity between trimethylsilyl ethers and trimethylsilyl enol ethers. Gassman
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

and Bottorff (1988) proposed a selective method for the deprotection of the trimethylsilylenol ether defense in the presence of the trimethylsilyl ethereal moiety. The method is ele-gant, although it is presently unacceptable in laboratory practice. The initial step of this in-teresting reaction consists of photoinduced one-electron oxidation of the trimethylsilyl enolethereal moiety. The trimethylsilyl ethereal group remains intact.
In the protocol, diprotected 4-hydroxycylohexanone was irradiated in the presence ofbiphenyl and 1-cyanonaphthalene (1-CN) for 5 hours (300 nm, 40:60 methanol/acetoni-trile) (Scheme 6-46). The half-deprotected product was obtained in 65% yield. Of course,such a yield is insufficient from a synthetic point of view, the photovariant of the redox re-action is not simple instrumentally, and the duration of the reaction (5 hr) is too long. Nev-ertheless, this approach is promising; it deserves attention and development. Thus, the pho-tochemical method was shown to be successful in the removal of protecting groups basedon covalently linked donor–acceptor systems (Lee & Falvey 2000).
The economical, practical, and environmentally acceptable procedure was elaboratedfor oxidative deprotection of trimethylsilyl ethers to their corresponding carbonyl com-pounds. The reaction proceeds in a solventless system in a short time, and yields are good.Upon 30 sec of irradiation in a conventional microwave, PhCH2OSiMe2 in the presence ofmontmorilonite K10 and finely grounded Fe(NO3)3�9H2O gave rise to PhCHO in 95%yield. The applicability of this method was tested with several aromatic, alicyclic, andaliphatic trimethylsilyl ethers. Duration did not exceed 1 min; yields were no lower than80% (Mojtahedi et al. 1999).
6.6.3 Scission of Carbon–Carbon Bonds
When considering the general aspects of cation radical acidity in Section 1.3.2.A, we in-tentionally postponed discussion of fragmentation. However, this problem is also of gen-eral importance.
One example is deprotonation of the cation radical of 9-benzyl-sym-nonahydro-10-selenaanthracene (Blinokhvatov et al. 1991). Upon dissolution in trifluoroacetic acid, the
SCHEME 6-46
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.
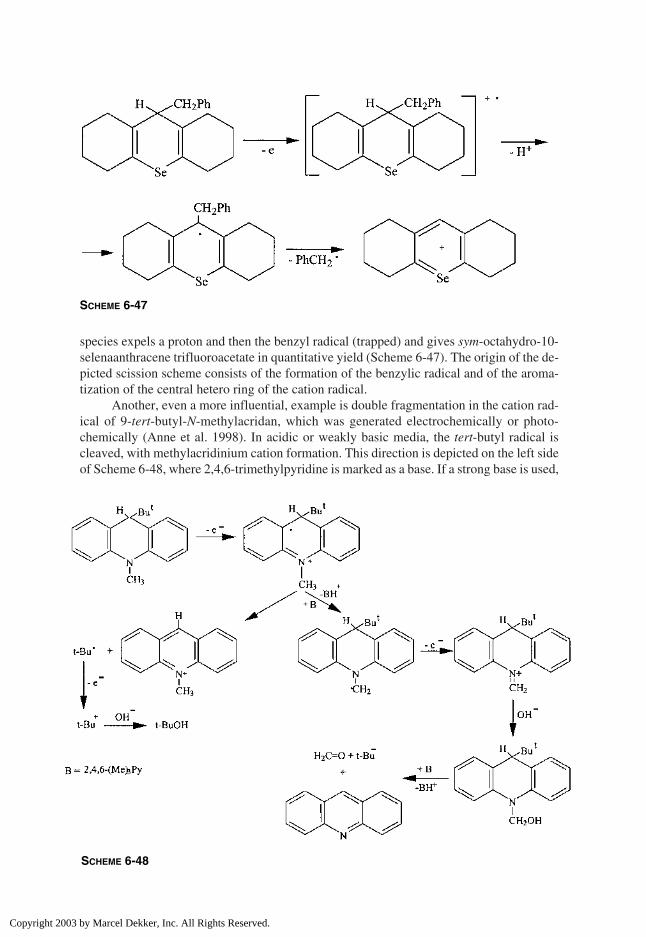
species expels a proton and then the benzyl radical (trapped) and gives sym-octahydro-10-selenaanthracene trifluoroacetate in quantitative yield (Scheme 6-47). The origin of the de-picted scission scheme consists of the formation of the benzylic radical and of the aroma-tization of the central hetero ring of the cation radical.
Another, even a more influential, example is double fragmentation in the cation rad-ical of 9-tert-butyl-N-methylacridan, which was generated electrochemically or photo-chemically (Anne et al. 1998). In acidic or weakly basic media, the tert-butyl radical iscleaved, with methylacridinium cation formation. This direction is depicted on the left sideof Scheme 6-48, where 2,4,6-trimethylpyridine is marked as a base. If a strong base is used,
SCHEME 6-47
SCHEME 6-48
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

the direction chosen is the one that leads to acridine, formaldehyde, and tert-butyl anion.This direction is depicted on the right side of the scheme. The origin of such dual behaviorresides in the prior deprotonation of the methyl group borne by the nitrogen atom that out-runs the usual deprotonation at the 9-carbon atom. The deprotonation at the 9-carbon isslowed by the steric hindrance, due to the presence of the tert-butyl group. The steric hin-drance effect on the cation, radical deprotonation was already noted in Section 1.3.2.A.
This example is important because carbon bond fragmentations occur at the firststages of the cation radical evolutions in cases of tert-butylated analogues of NADH(Fukuzumi et al. 1993). Scheme 6-48 describes this double-way fragmentation of the 9-tert-butyl-N-methylacridan cation radical. Examples of the cation radical deprotonation-fragmentation of alcohols also deserve consideration. Hammerum and Audier described thereactions in 1988:
PhCH2CH2OH → PhCH2CH2O��H → H� � PhCH�2 � CH2BO
Me3CCH2OH → Me3CCH2O��H → H� � Me3CH�2 � CH2BO
The carboradicals formed act as effective alkylating agents.The driving forces behind these reactions consist of formations of rather stable radi-
cals (PhCH�2 and Me3CH�
2) after deprotonation. A proton is a stable particle too. The for-mation of formaldehyde also helps to drive the reactions.
In this connection, it is interesting to compare the cation radicals of 1,2-diphenyl- and1,1,2,2-tetraphenylethanes in their ability to expel a proton or to cleave the exocyclic CMCbond. The former cation radical preferentially reacts by deprotonation (Camaioni & Franz1984; Baciocchi et al. 1986). The CMC bond strength of 121.5 kJ�mol�1 in the 1,2-diphenylethane cation radical is too high. At the same time, the CMC scission induced byelectron transfer is feasible only if the strength of this bond is less than 42 kJ�mol�1. In thecase of the 1,1,2,2-tetraphenylethane cation radical, this is equal to 38 kJ�mol�1 only, andthe CMC scission indeed takes place (Arnold & Lamont 1989):
(Ph2CHMCHPh2)�� → Ph2CH� � Ph2CH�
It would be interesting to consider the same reaction with unsymmetrically substi-tuted arylethanes. Maslak and Asel (1988) showed that in a series of bis(cumene) cationradicals, p-(Me2NMC6H4)��MCMe2MCMe2MC6H4MXMp, in which the unpaired elec-tron is localized on the p-(Me2NMC6H4)�� moiety, the rates of the CMC bond cleavagewell correlate with the �� values of substituent X.
Upon adding the hydroxyl substituent to the CH2CH2 unit, the barrier for CMC scis-sion is lowered because of more favorable thermodynamics (Albini & Spreti 1987; Bartonet al. 1996). However, the hydroxyl substituent becomes effective only after its deprotona-tion. Cation radicals of 2-, 3-, and 4-arylalkanols, all of them, undergo C(1)–H deprotona-tion at pH 4. At pH 10, they display a different behavior: The 2-(4-methoxybenzene)ethanolcation radical experiences C(2)–C(1) scission, resulting in the formation of formaldehydeand the 4-methoxybenzyl radical; the 3-(4-methoxybenzene)propanol cation radical givesrise to 3-(4-methoxybenzene) propanal; the 2-(4-methoxybenzene)butanol cation radicalbehaves as the C�–H acid, both in acidic and in basic solution (Baciocchi and co-authors1996, 1999a).
In the 1-arylakanol cation radicals, only C(1)–H deprotonation takes place at pH 4;at pH 10, C(1)–C(2) bond cleavage proceeds. Replacement of a 1-OH group by OMe has avery slight effect on the decay rate when the cation radical undergoes deprotonation, buthas a very large and negative effect in the case of C(1)–C(2) bond cleavage (Baciocchi et
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

al. 1999b). It is suggested that hydrogen bonding of the 1-OH group with water (the sol-vent) stabilizes the transition state of the C(1)–C(2) bond fragmentation reaction but notthat of the deprotonation process. Of course, other factors could also contribute to this phe-nomenon.
The unpaired electron and positive charge in arylalkane cation radicals are localizedessentially on aromatic rings. At first glance, the CMC bond in arylalkanes should there-fore be largely unaffected by one-electron oxidation. What happens in the course of theCMC bond cleavage? How is the CMC bond activated in the cation radicals? Chanon withco-authors (1990), as well as Takahashi and Kikuchi (1991), pointed out that transforma-tion of a neutral arylalkane to the cation radical evokes some bond elongation in the alkylfragment. Thus, in the phenylethane cation radical, the unpaired electron is highly (96%)localized on the phenyl ring, and so is the positive charge (Takahashi & Kikuchi 1991). Adrastic change in the electronic structure occurs if the CMC bond between the methyleneand methyl groups is elongated. The cation radical changes its character from an “aromaticcation radical” to an “ethane” cation radical. The unpaired electron density sharply de-creases on the phenyl ring and increases in the CH2MCH3 framework. The latter becomesready to cleave.
6.6.4 Synthone-Influential Bond Scission
The generation of active radicals as a result of bond breakage makes cation radicals usefulas synthones. For example, arylsulfenamide cation radicals may be used as sources ofsulfenyl radicals. The reaction of 4-nitrobenzenesulfenanilide with Lewis acids, such asBF3 and AlCl3, results in the formation of the sulfenamide cation radical. The latter appearsto be an active sulfenyl transfer species. In the presence of anisol, ethenes, or ethynes, itgives phenylthiyl derivatives (Benati et al. 1990; Grossi & Montevecchi 1993).
The benzenediazene cation radicals easily give benzenediazonium cations, eliminat-ing the MNBNM fragment together with an unpaired electron (Speiser & Stahl 1992):
(p-Me2NMC6H4NBNMNMe2)�� → N2 � (p-Me2NMC6H4MNMe2)�
This type of fragmentation is probably important in the biochemical sense because tri-azenes are regarded as carcinogenic compounds.
The decomposition of organic nitroso compounds with the generation of nitrogen ox-ide attracts the attention of biochemists as well. Initiating many important reactions in liv-ing organisms, nitrogen oxide acts as a “biochemical synthone.” Note that the CMN bondbreaking for t-BuNO�� is roughly 165 kJ�mol�1 easier than for neutral t-BuNO (Greer etal. 1995). Therefore, one can expect that the CMN bond in t-BuNO�� to be extremely weakor nonexistent. In this sense, the behavior of 1-nitrosoadamantane (1-Ad-NO) upon one-electron oxidation is interesting. The CMN bond of 1-nitrosoadamantane is no doubtstronger than that of t-BuNO. Chemical oxidation of this nitrosocompound by tris(2,4-di-bromophenyl)ammmoniumyl hexachloroantimonate in acetonitrile results in denitrosation.The reaction stoichiometry requires a two-mole equivalent of the oxidant. Upon aqueousworkup, 1-adamantylacetamide is formed. The following scheme shows a proposed mech-anism for this oxidative CMN bond cleavage (Greer et al. 1995):
1-Ad-NO � e → (1-AdNO)�� → NO� � [1-Ad�]
[1-Ad�] � MeCN → [1-Ad-N�BMC-Me]
[1-Ad-N�BMC-Me] � H2O → 1-Ad-NHCOMe
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

6.7 BOND FORMATION
Nucleophilic substitutions in anion radicals and electrophilic substitutions in cation radi-cals have been considered throughout the book, including the problem of choosing betweenaddition and electron-transfer reactions. Therefore, only some unusual cases will be dis-cussed here.
One interesting process of CMC bond formation is represented by the auto-oxidationof the Mercurials perennis L. plant alkaloid hermidin. The reaction proceeds through theformation of the transient blue anion radical, which dimerizes with the transfer of the reac-tion center to give, eventually, chrysohermidin as a dimeric hexaketone (Wasserman et al.1993) (Scheme 6-49).
The reaction between the diazocation from anthranylamide and tetrathiafulvalene re-sults in the formation of the �-radical from the diazocation and the cation radical of tetrathi-afulvalene. These two initially formed radicals combine to give salts of the S-arylatedtetrathiafulvalene (a minor product) and of the C-arylated tetrathiafulvalene (the mainproduct). This last one demonstrates an unprecedented carbon–carbon bond formation withthe cation radical of tetrathiafilvalene; the structure depicted was confirmed by single-crys-tal X-ray analysis (Begley et al. 1994) (Scheme 6-50).
SCHEME 6-49
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

As for the radical participant in this coupling reaction, the main product surely isformed as a result of the radical translocation. As for the cation radical participant, the po-sition of the coupling is explained as follows (Begley et al. 1994):
Calculations indicate that the unpaired electron density in the cation radicalof tetratiafulvalene resides principally on sulfur, but with the internal carbon be-ing the site of second-highest density. The product of coupling of an �-carbonylradical to sulfur, an �-carbonyl-sulfonium salt, would be destabilized by the adja-cent dipoles. The transition state would be expected to mirror this, thus slowingdown the CMS coupling and permitting the observed coupling to the carbon oftetrathiafulvalene.
This is not only unprecedented, but it is also a promising feature for the preparationof organic metals (see Chapter 7). The CMC bond formation consists of coupling the rad-ical with the cation radical.
SCHEME 6-50
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The coupling between two different anion radicals can also lead to CMC bond for-mation; Section 3.2.1 mentioned one such example. Another example, more attractive as asynthetic method, is the following. Anion radicals prepared via the addition of alkali met-als to aromatic hydrocarbons in THF react with alkanoates or formate esters to give the cor-responding acylated or formylated products, with good yields (Periasamy et al. 1999).Thus, the reaction of sodium naphthalene, sodium anthracene, or sodium phenathrene withethyl formate yields the corresponding aldehydes. Sodium naphthalene gives n-propyl-1-naphthyl ketone or methyl-1-naphthyl ketone on reaction with methyl butanoate or ethylacetate, respectively. All the aromatic anion radicals yield the corresponding aldehydes ontreatment with N,N-dialkylformamides. The authors rationalized the results through themechanistic pathway shown in Scheme 6-51. As the authors mentioned, use of more thantwo equivalents of sodium is necessary to obtain the products in reasonable yields: Oneequivalent is needed for the formation of an aromatic anion radical, and another is used upfor the formation of an anion radical derived from ester or amide.
Substituted naphthalenes (acenaphthene, 2-methylnaphthalene) are also formylatedby N,N-dialkylformamides, but in low (20, 26%) yields. Generally speaking, several othermethods of formylation and acylation exist in the organic synthesis repertoire. Neverthe-less, the procedure described here is useful, for it uses inexpensive starting materials.
Nucleophilic reactions between various carbanion nucleophiles and heteroaromaticcompounds are used in syntheses of physiologically active compounds (Wong et al. 1997).Let us consider as an example the photoassisted reaction between 2-bromopyridine and thecarbanions that are stabilized by the cyano group. The potassium derivatives of acetonitrileand phenylactonitrile were compared (Moon et al. 1983). To obtain the corresponding car-banion, a nitrile is treated with potassium amide in liquid ammonia. Phenylacetonitrile is astronger CH acid than acetonitrile. Phenylacetonitrile forms the potassium derivative quan-titatively, whereas acetonitrile reacts with potassium amide reversibly. Interacting with 2-bromopyridine, phenylacetonitrile-potassium gives solely pyridyl phenyl acetonitrile,whereas acetonitrile-potassium produces a 3:1 mixture of pyridyl acetonitrile withaminopyridine (Scheme 6-52).
If di(tert-butyl)nitroxide (a radical trap) is present, the reaction with phenylacetoni-trile-potassium does not proceed entirely. Acetonitrile-potassium (which is in equilibrium
SCHEME 6-51
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

with potassium amide) forms only aminopyridine in the trap presence (Moon et al. 1983).Consequently, amination is a classical nucleophile reaction, and the formation of pyridylacetonitrile is a reaction of the SRN1 type. These two reactions are quite different. Astronger CH acid leads to a well-defined synthesis.
The results obtained in the photostimulated SRN1 reaction between carbanions from2,4,4-trimethyl-2-oxazoline or 2,4-dimethylthiazole and 2-bromopyridine are also consis-tent with the incomplete formation of the carbanions in the KNH2–NH3(liq.) system. In thesecases, 2-aminopyridine is formed alongside the corresponding pyridyl-2-methylene oxa-zolinyl or thiazolyl substitution products (Wong et al. 1997). When the SRN1 pathway isimpeded by conducting the reaction in the dark or in presence of di(tert-butyl) nitroxide,the ionic amination reaction dominates.
The synthesis of pyridines containing an arylated methylene group belongs to thesame SRN1 type of substitution reactions. These derivatives serve as starting materials inthe manufacture of various medications. Methylpyridines can easily be transformed intomethylencarbanions upon metallation. The carbanions were commonly used in reactionswith compounds bearing strong departing groups, such as bromide or trialkyl ammonium.These reactions were recognized as typically nucleophilic ones. To promote them, theusual methods were employed (e.g., increase in temperature or pressure). The synthesisof dipyridyl methane based on 4-chloropyridine and the sodium derivative of 4-methylpyridine can serve as an example (Wiberg & Lewis 1970; Gaus et al. 1977). Theyields were usually not high and were nonreproducible. The yield turned out well whenion radical mechanism of the transformation was revealed. An inert atmosphere is neededfor the reaction; the transformation should be stimulated by photoirradiation instead ofcondition toughening. That resulted in stable and high yields (Moon et al. 1983; Bunnett& Glooz 1974).
The electrochemical behavior of 1-fluoro-2,4-dinitrobenzene, 1-chloro-2,4-dini-trobenzene, and 1-bromo-2,4-dinitrobenzene in dimethylformamide was described (Gal-lardo et al. 2000). The 1-fluoro-2,4-dinitrobenzene anion radical dimerizes before cleav-ing, whereas 1-chloro-2,4-dinitrobenzene and 1-bromo-2,4-dinitrobenzene anion radicalsdimerize after cleavage. The preliminary cleavage leads to the formation of 2,4-dini-trobenzene from the chloro- and bromoderivatives. In the case of the fluoroderivative, the change in mechanism allows obtaining 2,2,4,4-tetranitrobiphenyl selectively andwith good efficiency. In a synthetic context, the dimerization of anion radicals previousto carbon–halogen bond breaking in these substrates can be very significant since the Ull-mann method, the classical synthesis of biphenyls, has not been described for 2,2,4,4-tetranitrobiphenyl. The latter is obtained alternatively by nitration of biphenyl (Conforth1996). The ion radical route opens a new, convenient, and safe way to prepare this nitrocompound important for polymer chemistry. The energy of carbon–halogen bond disso-ciation is the dominant factor responsible for the difference uncovered (Gallardo et al.2000).
SCHEME 6-52
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

6.8 CONCLUSION
Within our previously stated restrictions, this chapter has considered some selected syn-theses. Possible generalizations were made as well. This material should be consideredalong with data from other chapters of the book. The recent development of mild reagents,reaction conditions, and new methodologies promise forthcoming breakthroughs in thearea of ion radical synthesis. Their utility in synthetic strategies is only now beginning tobe exploited.
Organic chemistry knows many diversified synthetic methods; the ion radical meth-ods are among them. Of course, the book considers the organic chemistry of ion radicals.However, when the only tool you have is a hammer, every problem begins to look like anail. This chapter showed the features and specific advantages of ion radical approaches toneeded compounds, with no intention to eliminate other synthetic routes.
REFERENCES
Adam, W.; Heidenfelder, Th. (1998) J. Am. Chem. Soc. 120, 11858.Adam, W.; Heidenfelder, Th. (1999) Chem. Soc. Rev. 28, 359.Adam, W.; Sahin, C. (1994) Tetrahedron Lett. 35, 9027.Adam, W.; Heidenfelder, Th., Sahin, C. (1995) Synthesis, 1163.Akasaka, T.; Wakahara, T.; Nagase, Sh.; Kobayashi, K.; Waelchli, M.; Yamamoto, K.; Kondo, M.;
Shirakura, Sh.; Okuba, Sh.; Maeda, Yu.; Kato, T.; Kako, M.; Nakadaira, Ya.; Nagahata, R.;Gao, X.; Van Caemelbecke, E.; Kadish, K. (2000) J. Am. Chem. Soc. 122, 9316.
Albini, A.; Spreti, S. (1987) J. Chem. Soc. Perkin Trans. 2, 1175.Anne, A.; Fraoua, S.; Moiroux, J.; Saveant, J.-M. (1998) J. Phys. Org. Chem. 11, 774.Arizawa, M.; Ramesh, N.G.; Nakajima, M.; Tohma, H.; Kita, Ya. (2001) J. Org. Chem. 66, 59.Arnold, D.R.; Lamont, L.J. (1989) Can. J. Chem. 67, 2119.Avalos, M.; Babiano, R.; Bravo, J.L.; Cabello, N.; Cintas, P.; Hursthouse, M.B.; Jimenez, J.L.; Light,
M.E.; Palacios, J.C. (2000) Tetrahedron Lett. 41, 4101.Baciocchi, E.; Bartoli, D.; Rol, C.; Ruzziconi, R.; Sebastiani, G.V. (1986) J. Org. Chem. 51, 3587.Baciocci, E.; Bietti, M.; Putignani, L.; Steenken, S. (1996) J. Am. Chem. Soc. 118, 5952.Baciocchi, E.; Bietti, M.; Manduchi, L.; Steenken, S. (1999a) J. Am. Chem. Soc. 121, 6624.Baciocchi, E.; Bietti, M.; Steenken, S. (1999b) Chem. Eur. J. 5, 1785.Barton, R.D.; Bartberger, M.D.; Zhang, Y.; Eyler, J.R.; Schanze, K.S. (1996) J. Am. Chem. Soc. 118,
5655.Bauld, N.L. (1989) Tetrahedron 45, 5307.Bauld, N.L. (1992) In: Mariano, P.S., ed. Advances in Electron Transfer Chemistry. Vol. 2. JAI Press,
Greenwich, CT.Bauld, N.L.; Pabon, R.A. (1983) J. Am. Chem. Soc. 105, 633.Bauld, N.L.; Yang, J. (1999) Org. Lett. 1, 773.Bauld, N.L.; Aplin, J.T.; Yueh, W.; Sarker, H.; Bellville, D.J. (1996) Macromolecules 29, 3661.Bauld, N.L.; Aplin, J.T.; Yueh, W.; Endo, S. (1998) J. Phys. Org. Chem. 11, 825.Bauld, N.L.; Bellville, D.J.; Harirchian, B. (1987) Acc. Chem. Res. 20, 371.Bauld, N.L.; Bellville, D.J.; Pabon, R.A.; Chalsky, R.G.G. (1983) J. Am. Chem. Soc. 105, 2378.Bauld, N.L.; Yang, J.; Gao, D. (2000) J. Chem. Soc. Perkin Trans. 2, 207.Begley, M.J.; Murphy, J.A.; Roome, S.J. (1994) Tetrahedron Lett. 35, 8679.Bell, F.A.; Ledwith, A.; Sherrington, D.C. (1969) J. Chem. Soc. C., 2719.Bellville, D.J.; Bauld, N.L. (1982) J. Am. Chem. Soc. 104, 2665.Bellville, D.J.; Wirth, D.D.; Bauld, N.L. (1981) J. Am. Chem. Soc. 103, 718.Bellville, D.J.; Bauld, N.L.; Pabon, R.A.; Gardner, S.A. (1983) J. Am. Chem. Soc. 105, 3584.Benati, L.; Montevechi, P.C.; Spagnolo, P. (1990) J. Chem. Soc., Perkin Trans. 1, 1691.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Berger, D.J.; Tanko, J.M. (1997) In: Patai, S., ed. Chem. Double-Bonded Funct. Groups. Vol. 3, Ch.22. Wiley, Chichester, England.
Beugelmans, R.; Roussi, G. (1979) J. Chem. Soc. Chem. Commun., 950.Beugelmans, R.; Chastanet, J.; Roussi, G. (1984) Tetrahedron 40, 311.Bil’kis, I.I.; Shteingarts, V.D. (1982) Zh. Org. Khim. 18, 359.Birch, A.J. (1944) J. Chem. Soc., 430.Birch, A.J.; Hinde, A.L.; Radom, L. (1980a) J. Am. Chem. Soc. 102, 3370.Birch, A.J.; Hinde, A.L.; Radom, L. (1980b) J. Am. Chem. Soc. 102, 4074.Birch, A.J.; Hinde, A.L.; Radom, L. (1981) J. Am. Chem. Soc. 103, 284.Blinokhvatov, A.F.; Berberova, N.T.; Archegova, A.S.; Klimov, E.S.; Shpakov, A.V.; Okhlobystin,
O.Yu. (1991) Khim. Geterotsikl. Soedin., 900.Boileau, S.; Champetier, G.; Sigwalt, P. (1967) J. Polym. Sci., Polym. Symp. No. 16, Pt. 5, 3021.Bommeli, F.; Degiory, L.; Wacher, P.; Legeza, O.; Janossy, A.; Oszlanyi, G.; Chauvet, O.; Forro, L.
(1995) Phys. Rev. B. Solid State 51, 794.Borhani, D.W.; Greene, F.D. (1986) J. Org. Chem. 51, 1563.Botzem, J.; Haberl, U.; Steckhan, E.; Blechert, S. (1998) Acta Chem. Scand. 52, 175.Boujlel, K.; Simonet, J.; Roussi, G.; Beugelmans, R. (1982) Tetrahedron Lett. 23, 173.Bowman, W.R.; Heaney, H.; Smith, Ph.H.G. (1982) Tetrahedron Lett. 23, 5093.Bradshaw, J.D.; Solooki, D.; Tessier, C.A.; Youngs, W.J. (1994) J. Am. Chem. Soc. 116, 3177.Btouet, V.; Alloul, H.; Yoshinary, Y.; Forro, L. (1996) Phys. Rev. Lett. 76, 3638.Bunnett, J.F.; Glooz, B.F. (1974) J. Org. Chem. 39, 382.Camaioni, D.M.; Franz, J.A. (1984) J. Org. Chem. 49, 1607.Chanon, M.; Rajzmann, M.; Chanon, F. (1990) Tetrahedron 46, 6193.Chou, T.S.; You, M.L. (1987) J. Org. Chem. 52, 2224.Chow, Y.L.; Danen, W.C.; Nelsen, S.F.; Rosenblatt, D.H. (1978) Chem. Rev. 78, 243.Citterio, A.; Minisci, F. (1982) Chim. Ind. (Italy) 64, 320.Collins, D.J.; Hobbs, J.J. (1983) Austral. J. Chem. 36, 619.Conforth, J. (1996) J. Chem. Soc. Perkin Trans. 1, 2889.Crandall, J.K.; Mualla, M. (1986) Tetrahedron Lett. 27, 2243.Dapperheld, S.; Steckhan, E. (1982) Angew. Chem. Intl. Ed. Engl. 21, 780.Dauben, W.G.; Deviny, E.J. (1966) J. Org. Chem. 31, 3796.Davis, A.G.; Hay-Motherwell, R.S.; Evans, J.S.; Rowlands, C.C. (1985) J. Chem. Soc. Chem. Com-
mun., 1513.Digurov, N.G.; Bukharkina, T.V.; Lebedev, N.N. (1986) Neftekhimiya 26, 787.Dinesh, Ch.U.; Reissig, H.-U. (1999) Angew. Chem. Int. Ed. Eng. 38, 789.Eisch, J.J.; Im, K.R. (1977) J. Organometal. Chem. 139, C45.Engman, L.; Laws, M.J.; Malmstrom, J.; Schiesser, C.H.; Zugaro, L.M. (1999) J. Org. Chem. 64,
6764.Farid, S.; Shealer, S.E. (1973) J. Chem. Soc. Chem. Commun. 677.Fontan, R.; Galves, C.; Viladoms, P. (1981) Heterocycles 16, 1473.Fraser, R.T.M.; Taube, H. (1959) J. Am. Chem. Soc. 81, 5514.Fujii, A.; Fujimaki, E.; Ebata, T.; Mikami, N. (1998) J. Am. Chem. Soc. 120, 13256.Fujimaki, E.; Fujii, A.; Ebata, T.; Mikami, N. (2000) J. Chem. Phys. 112, 137.Fukuzumi, S.; Tokuda, Y.; Kitano, T.; Okamoto, T.; Otega, J. (1993) J. Am. Chem. Soc. 115, 8960.Gallardo, I.; Guirado, G.; Marquet, J. (2000) J. Electroanal. Chem. 488, 64.Gassman, P.G.; Bottorff, K.J. (1988) J. Org. Chem. 53, 1097.Gassman, P.G.; Singleton, D.A. (1984) J. Am. Chem. Soc. 106, 7993.Gaus, P.L.; Hain, A.; Johnson, F. (1977) J. Org. Chem. 42, 564.Giordano, C.; Perdonicn, G.; Castaldi, G. (1985) Angew. Chem. Int. Ed. Engl. 24, 499.Greer, M.L.; Sarker, H.; Mendicino, M.E.; Blackstock, S.C. (1995) J. Am. Chem. Soc. 117, 10460.Grossi, L.; Montevecchi, P.C. (1993) Tetrahedron 49, 9095.Guertler, Ch.F.; Steckhan, E.; Blechert, S. (1996) J. Org. Chem. 61, 4136.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Haberl, U.; Wiest, O.; Steckhan, E. (1999) J. Am. Chem. Soc. 121, 6730.Hammerum, S.; Audier, H.E. (1988) J. Chem. Soc. Chem. Commun., 860.Harirchian, B.; Bauld, N.L. (1987) Tetrahedron Lett. 28, 927.Hasegawa, E.; Mukai, T. (1985) Bull. Chem. Soc. Jpn. 58, 3391.Hintz, S.; Heidbreder, A.; Mattay, J. (1996) Top. Curr. Chem. 177, 77.Hofmann, M.; Schaefer, H.F. (1999) J. Am. Chem. Soc. 121, 6719.Houghton, R.A.; Beckman, A.; Ostresh, J.M. (1986) Int. J. Pept. Protein Res. 27, 653.Hueckel, W.; Bretschneider, H. (1939) Libigs Ann. Chem. 540, 157.Hwu, J.R.; Jain, M.L.; Tsay, Sh.-Ch.; Hakimelahi, G.H. (1996) Tetrahedron Lett. 37, 2035.Jarowicki, K.; Kocienski, P. (1995) Contemp. Org. Synth. 2, 315.Jones, C.R. (1981) J. Org. Chem. 46, 3370.Kim, J.H.; Hubig, S.M.; Lindeman, S.V.; Kochi, J.K. (2001) J. Am. Chem. Soc. 123, 87.Koshel’, S.G.; Voronenkov, V.V.; Koshel’, G.N.; Rudkovskii, E.K.; Krestinina, T.B. (1997) Zh. Org.
Khim. 33, 1114.Koval’chuk, E.P.; Whittingham, S.; Skolozdra, O.M.; Zavalij, P.Y.; Zavaliy, I.Yu.; Reshetnyak,
O.V.; Seledets, M. (2001) Mater. Chem. Phys. 69, 154.Kumar, V.S.; Floreancig, P.E. (2001) J. Am. Chem. Soc. 123, 3842.Kuwata, S.; Shigemitsu, Y.; Odaira, Y. (1973) J. Org. Chem. 38, 3803.Lampard, Ch.; Murphy, J.A.; Lewis, N. (1993) J. Chem. Soc. Chem. Commun., 295.Ledwith, A. (1972) Acc. Chem. Res. 5, 133.Lee, K.; Falvey, D.E. (2000) J. Am. Chem. Soc. 122, 9361.Lijser de, H.J.P.; Arnold, D.R. (1998) J. Phys. Chem. A. 102, 5592.Lin, W.; Tu, T.; Dong, J.; Zhang, J.; Lin, N. (1998) Radiat. Phys. Chem. 53, 651.Litvintsev, I. Yu.; Sapunov, V.N.; Koshel’, G.N. (1994) Russ. Pat. 20021250.Lorenz, K.T.; Bauld, N.L. (1987) J. Am. Chem. Soc. 109, 1157.Maslak, P.; Asel, S.L. (1988) J. Am. Chem. Soc. 110, 8260.Mattes, S.L.; Farid, S. (1980) J. Chem. Soc. Chem. Commun., 126.Mirafzal, G.A.; Liu, J.; Bauld, N.L. (1993) J. Am. Chem. Soc. 115, 6072.Mizuki, J.; Takai, M.; Takahashi, H.; Mori, N.; Tanigaki, K.; Hirisawa, I.; Prasides, K. (1994) Phys.
Rev. B. Solid State 50, 3466.Micoch, J.; Steckhan, E. (1987) Tetrahedron Lett. 28, 1081.Mojtahedi, M.M.; Saidi, M.R.; Bolourtchian, M.; Heravi, M.M. (1999) Synth. Commun. 29, 3283.Molander, G.A.; Harris, C.R. (1996) Chem. Rev. 96, 307.Molander, G.A.; Harris, C.R. (1998) Tetrahedron 54, 3321.Molander, G.A.; Stengel, P.J. (1995) J. Org. Chem. 60, 6660.Moon, M.P.; Komin, A.P.; Wolfe, J.P.; Morris, G.F. (1983) J. Org. Chem. 48, 2392.Nebois, P.; Bouaziz, Z.; Fillion, H.; Moeini, L.; Piquer, J.A.; Luche, J.-L.; Riera, A.; Moyano, A.;
Pericas, M.A. (1996) Ultrason. Sonochem. 3, 7.Nishibori, E.; Takata, M.; Sakata, M.; Tanaka, H.; Hasegawa, M.; Shinohara, H. (2000) Chem. Phys.
Lett. 330, 497.Nomura, M.; Nakao, Y.; Kikuchi, K.; Achiba, Y. (1995) Physica B 208 & 209, 539.Novi, M.; Dell’Erba, C.; Gabarino, G. (1982) J. Org. Chem. 47, 2292.Ohkubo, K.; Fukuzumi, Sh. (2000) Org. Lett. 2, 3647.Okazaki, T.; Suenaga, K.; Lian, Y.; Gu, Z.; Sinohara, H. (2001) J. Mol. Graphics Modell. 19, 244.Oprea, C.V.; Popa, M. (1980) Angew. Makromol. Chem. 90, 13.Pabon, R.A.; Bauld, N.L. (1984) J. Am. Chem. Soc. 106, 1145.Pekker, S.; Granasy, L.; Oszlanyi, G.; Bortel, G.; Faigel, G.; Tegze, M.; Chauvet, O.; Forro, L.;
Stephens, P.W.; Janossy, A. (1995) Proc. Electrochem. Soc., 95–10.Periasamy, M.; Reddy, M.R.; Bharathi, P. (1999) Synth. Commun. 29, 677.Ramkumar, D.; Kalpana, M.; Varghese, B.; Sankararaman, S.; Jagadeesh, M.N.; Chandrasekhar, J.
(1996) J. Org. Chem. 61, 2247.Rautenstrauch, V.; Willhalm, B.; Thommen, W. (1981) Helv. Chim. Acta 64, 2109.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Reinolds, D.W.; Lorenz, K.T.; Chiou, H.-S.; Bellville, D.J.; Pabon, R.A.; Bauld, N.L. (1987) J. Am.Chem. Soc. 109, 4960.
Reinolds, D.W.; Harirchian, B.; Chiou, H.-S.; Marsh, B.K.; Bauld, N.L. (1989) J. Phys. Org. Chem.2, 57.
Russell, G.; Bemis, A. (1967) Inorg. Chem. 6, 403.Schmidt, U.; Utz, R.; Lieberknecht, A.; Criesser, H.; Potzolli, B.; Bahr, J.; Wagner, K.; Fisher, P.
(1987) Synthesis, 236.Schnabel, E.; Klostermeyer, H.; Berndt, H. (1971) Liebigs Ann. 749, 90.Sheldon, R.A.; Kochi, J.K. (1981) Metal-Catalyzed Oxidation of Organic Compounds. Academic
Press, New York.Simonescu, C.I.; Oprea, C.V.; Nicoleanu, J. (1983) Eur. Polym. J. 19, 525.Speiser, B.; Stahl, H. (1992) Tetrahedron Lett. 33, 4429.Stahl, G.L.; Walter, R.; Smith, C.W. (1978) J. Org. Chem. 43, 2285.Takahashi, O.; Kikuchi, O. (1991) Tetrahedron Lett. 32, 4933.Takahashi, Ya.; Miyamoto, K.; Sakai, K.; Ikeda, H.; Miyashi, Ts.; Ito, Yo.; Tabohashi, K. (1996)
Tetrahedron Lett. 37, 5547.Tomillin, O.B.; Stankevich, I.V.; Surin, N.G. (1980) Izv. AN SSSR Ser. Khim. 4, 936.Trindle, C. (2000) J. Phys. Chem. A 104, 5298.Vank, J.C.; Knak Jensen, S.J.; Tang, T.-H.; Csizmadia, I.G. (2001) J. Mol. Struct. 537, 189.Vorbrueggen, H.; Krolikiewicz, K. (1975) Angew. Chem. Int. Ed. Engl. 14, 818.Wasserman, H.H.; DeSimone, R.W.; Boger, D.L.; Baldino, C.M. (1993) J. Am. Chem. Soc. 115,
8457.Wiberg, K.B.; Lewis, T.P. (1970) J. Am. Chem. Soc. 92, 7154.Wong, J.-W.; Natalie, K.J.; Nwokogu, G.C.; Pisipati, J.S.; Flaherty, P.T.; Greenwood, Th.D.; Wolfe,
J.F. (1997) J. Org. Chem. 62, 6152.Wooster, C.B.; Godfrey, K.L. (1937) J. Am. Chem. Soc. 59, 596.Yamashiro, D.; Blake, J.; Li, C.H. (1972) J. Am. Chem. Soc. 94, 2855.Zimmerman, H.E.; Alabugin, I.V. (2001) J. Am. Chem. Soc. 123, 2265.Zimmerman, H.E.; Wang, P.A. (1990) J. Am. Chem. Soc. 112, 1280.Zimmerman, H.E.; Wang, P.A. (1993) J. Am. Chem. Soc. 115, 2205.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

7
Practical Applications of Organic Ion Radicals
7.1 INTRODUCTION
The aim of this part of the book is to look at current practical applications of organic ionradical chemistry. Chapter 7 examines patents and original (experimental) papers that of-fer commercial advantages when compared to conventional approaches. It pays special at-tention to newly developed branches of material science that may become technically im-portant in the near future. Ion radical organic chemistry opens new possibilities in thesynthesis of materials for molecular electronic applications, including the construction oforganic metals and magnets. One section of the chapter is devoted to a mechanism of lu-brication during the rubbing together of metallic surfaces; it explains lubrication effects andproposes new approaches to the synthesis of new additives and lubricating compositions.Another section considers the behavior of lignin during the industrial cooking of woods.Redox reactions play the decisive role in delignification. Some commercial advantagesbased on ion radical participation in paper fabrication are also analyzed.
7.2 ORGANIC ION RADICALS IN OPTOELECTRONICS
The creation of new systems to record, store, and organize information is one of the maintasks in the development of microelectronics. At present, there are working systems that or-ganize information from optical or electrical signals (J.K. Fisher et al. 1976). However, itis reasonable to use one of these signals for recording and another for organizing the infor-mation accepted. This avoids the use of complicated electronic and optical switches and al-lows use of two separate sources of signals, electronic and optical. As a result, the deviceis simplified significantly.
7.2.1 Ion Radical Approach to Molecular Switches and Modulators
Accumulation and organization of information can be achieved on the base of cis–trans iso-merization of olefins in their ion radical states. In preceding chapters, the phenomenon was
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

examined closely (see also Todres 2001). Those studies were used as the basis for select-ing samples for applicability in real electronic memory systems. For the neutralarylethylenes, conversion from cis to trans results in a bathochromic shift in their absorp-tion bands of 20–25 nm and in a 100- to 250-fold increase in absorptivity. For the neutralolefins, trans-to-cis transformation can occur only upon light irradiation. So materialsbased on these photochromic compounds exhibit enhanced sensitivity to light. They alsoacquire a sensitivity to electric impulse and an ability to keep the information recorded.
If cis-arylethylenes are used as electrochromic substances (in media capable of con-ducting electric current), applying a 10- to 12-V cyclically changing voltage enhances thespectral absorptivity as just described. These changes are readily registered and remainconstant for at least two years. To return to the initial state of the system, it is sufficient toapply a near-UV light impulse for just seconds. The record–erase cycles can be repeatedmore than 1000 times with the same capacity.
Exploiting arylethylenes as bearers of electron memory has the following advan-tages: First, the compounds are commercially available and inexpensive. Second, theywork for very long time. Finally, the information recorded can be transformed simply andwith little energy consumption. In other words, the ion radical route to creating electronmemory systems is fruitful.
Electron transfer is often associated with the conformation changes. For organic ionradicals the inner reorganizational energies remain modest, allowing a fast passage be-tween the different conformations during the electron transfer (for example, see Bellec andothers 2000).
Let us now consider molecular switches based on intramolecular electronic transi-tion. Generally speaking, transfer of energy or an electron within a molecule proceeds infemtoseconds. The aim is to produce molecular electronic devices that respond equallyrapidly. Molecular switches that employ optically controlled, reversible electron-transferreactions sometimes bring both speed and photostability advantages over molecularswitches, which are usually based on photochemical changes in their molecular structure.
Important examples are the molecular switches in Scheme 7-1 (Debreczeny et al.1996). These switches work as a two-pulse electronic pump. In toluene, the first laserpulse (at 416 nm) instantly produces a one-electron shift, forming the amine cation radi-cal part and the di-imide anion radical part. The second laser pulse (at 480 nm) rapidly(3 ns) switches the ion radical separated state back to the initial form. Substitution of themethoxy group for the methyl group in the benzenoid bridge represents a useful expan-sion of the switch-molecule series. The dimethoxy derivative exhibits increased rates ofcharge separation and recombination relative to those of the corresponding dimethyl-sub-stituted bridge. The effect is also particularly strong in toluene (S.E. Miller et al. 2000).The molecular arrays described provide a basis for the design of molecular electronic de-vices.
Electro-optical modulators are other examples whose efficiency is enhanced in thepresence of ion radicals. These devices are based on sandwich-type electrode structurescontaining organic layers as the electron-hole-injecting layers at the interface between theelectrode and the emitter layer. The presence of ion radicals lowers the barrier heights forthe electron or hole injection. Anion radicals (e.g., anion radicals from 4,7-diphenyl-1,10-phenanthroline—Kido & Matsumoto 1998; from tetra-arylethynyl-cyclo-octatetraenes—Lu and co-workers 2000) or cation radicals (e.g., cation radicals from �-sexithienyl—Ku-rata et al. 1998; 1,1-diphenyl-2-[phenyl-4-N,N-di(4-methylphenyl] ethylene—Umeda andco-workers 1990, 2000), all of them used as electron or hole carriers.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

SCHEME 7-1
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

7.2.2 Cation Radicals of Triarylamines in Optical Recording Media
Organic dyes of a particular structure change their physical properties upon irradiation withlight of a relatively long wavelength. Such dyes can be used in recording layers. If a poly-methine dye, which exhibits appropriate sensitivity to exposure by a laser beam, is used ina thin film, this film forms an optical recording medium that permits both laser recordingand reflection reading.
In the recording layer, however, the organic dye tends to deteriorate after repeated ir-radiation with reproduction light, and the reproduction properties of the optical recordingmedium thereby also deteriorate.
As claimed (Mihara et al. 1997), it is possible to improve significantly the light re-sistance of the organic dye used by combining the organic dye with a nitrogen-containingcation radical salt with metal complex anion. One such cation radical salt is shown inScheme 7-2.
The mentioned patent enumerates many cation radical salts, dyes, and polymeric ma-terials for layers. The problem of salt solubility represents a bottleneck in applications likethis. It is the salt of the metal complex anion and the cation radical of a triarylamine thateasily dissolves in the typically used organic solvents that do not affect plastics (the baseof the layer). At the same time, the productivity of the optical recording media is signifi-cantly increased. It is possible to obtain an optical recording medium exhibiting excellentpreservation stability under conditions of high temperature and high humidity. It is alsopossible to obtain an optical recording medium having a distinct threshold value for laserpower without degrading the high reflectance and high sensitivity of the organic dye used.The mechanism of cation radical action remains unclear. Probably, it is caused by the re-dox activity of the cation radical additives.
7.2.3 Ladder Polymerization of Fluoranthene-Based Cation Radicalsas a Route to Electrochromic Materials
Fluoranthene derivatives transform into cation radicals upon one-electron oxidation. Thesespecies are not stable and quickly undergo a further oxidation. For example, 7,14-dipheny-lacenaphtho[1,2-k]fluoranthene gives a ladder polymer according to Scheme 7-3 (Debad &Bard 1998.) As a result, an insoluble transparent blue polymer film forms on the electrode.Electrochemical oxidation of the film in acetonitrile initiates a rapid color change from blue
SCHEME 7-2
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

to pale gray, while reduction to the first or second cathodic waves causes the film to be-come pale green or orange, respectively. These electrochromic effects are stable and re-versible when air and water are excluded, even after 30,000 reduction cycles. The materialhas potential uses in electrochromic or electroluminescent devices.
Stable ladder oligosilanes are also of interest. The potassium metal reduction of theneutral ladder oligosilanes containing 6–12 silicon atoms fully substituted by isopropylgroups leads to the corresponding anion radicals that are highly stable at room temperature(Kyushin et al. 2000). These silanes can be used as electric switchers.
7.3 ORGANIC METALS
Organic ion radicals exist as salts with counterions. As seen from the preceding chapters,neutral molecules with strong acceptor/donor properties form rather stable ion radical salts.Under certain conditions (see later), components of an ion radical salt pack up in a crystal
SCHEME 7-3
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

lattice in a special manner. Different ion radical parts (cations and anions separately) line upone over another in the form of endlessly long, practically linear, one-dimensional piles-chains. These piles-chains form a crystal. Such crystals are dubbed quasi-one-dimensionalcrystals.
This term reflects the structural motif of their arrangement. At the same time, thename also underlines the “one-dimensionality” of the charge-bearer (electron-bearer) shiftin such crystals. Electron delocalization occurs in the framework not only for the one-molecule contour but also in an ensemble of many molecules. As a consequence, electronsshift along chains of organic ions. Collective conductivity arises. Naturally, this conduc-tivity is large along the chain axes and insignificantly small in all other directions.
The electric conductivity of such organic materials makes them related to metals.This was the reason conductive ion radical salt were named “organic metals” or “organicconductors.” Some organic conductors can become superconductors under certain condi-tions. Superconductivity is a disappearance of electrical impedance that allows electric cur-rent to flow with no loss of energy.
Organic metals have been reviewed in many sources, for example, Khidekel’ andZhilyaeva (1978), Andre et al. (1976), and, more recently, Farges (1994) as well as Grosseland Weston (1994).
The most prospective donors are those with ionization potentials of ID � 6.6 eV. Ac-ceptors with electron affinities of EA � 2.6 eV are suitable. When Id � EA � 4 eV, donor–ac-ceptor interaction leads to strong molecular complexes with a charge-transfer degree �0.5.Donor–acceptor charge transfer often results in the formation of ion radical salts havingmetallic conductivity. In terms of charge-transfer degree, ion radical salts have values �0.7.
One important condition for achieving the metallic state is the maximal close mutualdisposition of components of an ion radical salt in a crystal (within and between stacks ofcation and anion components). For charge-transfer or electron-transfer interactions, thespecies contacts must be equal to or shorter than the van der Waals distances (Le Marguereset al. 2000). Such short contact means maximal density in the components’ packing, theirintense interaction, and the transference of electrons along a conductive chain. The electron(charge) transfer can, in principle, be implicated in the interstack hydrogen bonds, and acharge-transfer increase strengthens these hydrogen bonds (Oison et al. 2001).
The last phenomenon was never considered until Oison’s work of 2001. It deservesexplanation in greater detail. In each molecule, the molecular orbitals are coupled via re-pulsive electron–electron interactions. A charge variation in the frontier molecular orbitalis associated with both acceptor and donor. This leads to dilation or contraction of the elec-tronic cloud formed by lower molecular orbitals, essentially in the molecular plane. Thisleaves an excess of charges of the same sign on the periphery of each molecule. This in-creases the reactivity of the molecules in their plane. This effect is enhanced in the crystalsby the mutual influences of neighboring molecules. Hence the effects along �- and hydro-gen-bond chains are intimately coupled in the charge (electron) movement with respect tomolecular ensembles.
The following compounds are the most popular as donors for organic metals (seeScheme 7-4): tetrathiafulvalene (TTF), tetraselenafulvalene (TSeF), tetrathiatetracene(TTT), tetraselenatetracene (TSeT), tetramethyl tetraselenafulvalene (TMeTSeF),bis(thiadimethylene) tetrathiafulvalene (BTDM-TTF), bis(ethylenedithia) tetrathiafulva-lene (BEDT-TTF or ET), tetrakis(methyltelluro) tetrathiafulvalene (TTeC1-TTF), tetram-ethylbis(ethylenedithia) tetrathiafulvalene (TMET), 2,2-(2,6-naphthalenediylydene)bis(1,3-dithiole) (NBDT). All the abbreviations are those used in the current literature.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Organic donors must exhibit not only a desirable electron-donating capability, butalso strong intermolecular interactions. The latter property, which permits the delocaliza-tion of an unpaired electron along the uniform stack, is achieved by increasing the numberof relatively voluminous heteroatoms. To date, it has been shown that multisulfur hetero-cycles and their selena and tellura analogues are the most promising candidates as compo-nents for organic metals. The introduction of chalcogen atoms in organic unsaturatedderivatives decreases their ionization potential and stabilizes the corresponding cation rad-icals, as well as enhancing intermolecular interactions in the solid state.
BDTT is one of the most powerful donors of the TTF class. Alkylthio-substitution,such as in BEDT-TTF (ET), leads to a slight reduction in its electron-donor capability.Nevertheless, salts of this cation radical with tetracyanoquinodimethane (TCNQ) and its
SCHEME 7-4
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

derivatives show significantly higher conductivity (by ca. 102–103) than the analogousBDTT salts (Grossel & Weston 1994). This reflects some enhancement of intra- and inter-stack interactions produced by the sulfur atoms at the edges of the donor skeleton.
Thus, the ion radical salt of BTDM-TTF (ET) with TCNQ has a high conductivity of1.3 � 102 ��1�cm�1 at room temperature with metallic behavior down to 26 K (Rovira etal. 1994). (The unit of conductance is denoted as ��1, or ohm�1, or even mho, but mostlyas S, i.e., siemens: 1 S � 1 ��1). The striking observation was made that the nature andmolecular size of donor or acceptor have no substantial effect on the molecular distancewithin a stack (from 0.32 to 0.35 nm). Meanwhile, increasing the volume of a heteroatomleads to improving intermolecular interactions within given stacks. The enhanced inter-molecular interactions were indeed observed with the substitution of selenium for sulfur.The aforementioned orbital dispersion is more pronounced in the selenium-containing rep-resentatives. This arises from the more extensive overlap properties of selenium (Beer et al.2001).
Whereas various organic salts of tetramethyltetrathiafulvalene never exhibited su-perconductivity, those of tetramethyltetraselenatetrathiafulvalene, known as the Bechgaardsalts, were the first organic superconductors to be discovered (Jerome et al. 1980). The se-lena analog of BEDT-TTF has been synthesized (Lee and co-authors 1983). However, thiscompound appeared to have very limited solubility, even to the extent that its oxidation po-tential could not be determined. In general, the solubility problem implies severe limita-tions in choosing the proper components for organic metals.
Another important way to increase the intermolecular interactions is to use the effectof a “molecular fastener.” This effect has been observed in tetrakis(alkylthio)tetrathiaful-valenes (the alkyl groups were longer than C8). A relatively high conductivity was observedeven in neutral donors (Inokuchi et al. 1986). This phenomenon was explained through theenhancement of intermolecular contacts induced by van der Waals interactions of the alkylside chains. Importantly, a high intrinsic conductivity has also been observed fortetrakis(methyltelluro)tetrathiafulvalene. In this case, tellurium atoms were claimed to playthe role of molecular fasteners (Inokuchi et al. 1987). The authors explain this effect interms of the manner of molecular packing and stacking within the crystal. The centralskeleton of this compound, the tetratelluro-tetrathiafulvalene moiety (C6Te4S4) is almostplanar and is regularly stacked in the form of column. Along the stacking axis, telluriumatoms in neighboring columns come close to each other and form zigzag columns. The dis-tance between those Te atoms is 0.364 nm, which is significantly shorter than the van derWaals distance (0.412 nm). The zigzag chalcogen chains are supposedly formed fromquasi-covalent bonds (Inokuchi et al. 1987) (Scheme 7-4).
As acceptors for organic metals, the following compounds are important (see Scheme7-5): tetracyanoquinodimethane (TCNQ), tetracyanoperfluoroquinodimethane (TCNQF4),tetracyano-2,5-diiodoquinodimethane (TCNQI2), N,N-dicyanoquinodiimine (DCNQDIm),2,5-dimethyl-N,N-dicyanoquinodiimine (DMDCNQDIm), tetracyanoethylene (TCNE),hexacyanobutadiene (HCNB), bis(thiadiazole) tetracyanoquinodimethane (BDTA-TCNQ), bis(selenadiazole) tetracyanoquinodimethane (BSeDA-TCNQ), 1,4,5,8-tetra-chloro-9,10-anthraquinodimethane (TCCAQ), fullerenes Cxy, e.g., C60 or C80). The abbre-viations used here are also generally found in the current literature.
Most organic acceptors used in the preparation of organic conducting materials be-long to a class of polycyano compounds. The reasons are quite obvious if one takes into ac-count the unique properties of the cyano group. It is a very strong acceptor and has a verysmall size, with minimal steric strain. (Note the rod-shaped form of C�N group).
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The TCNQ anion radical also has a high thermal stability. Ring substitution in thesix-membered ring in TCNQ allows the modifying of properties of the corresponding ionradicals. Grossel and Weston (1994) pointedly mention that the incorporation of het-eroatoms or heterocyclic rings into the TCNQ skeleton leads to greater intra- and interstackinteractions. This increases dimensionality, stabilize conductivity, and enhances the metal-lic state. Selective deuteration of dimethyl-TCNQ is also a way to change its properties inthis sense. Interestingly, selective deuteration of the methyl groups plays a much greaterrole than deuteration of the ring protons (Grossel & Weston 1994) (Scheme 7-5).
In creating an organic metal, one should avoid transforming a conductor into a di-electric. Such a transformation can occur with decreasing temperature. The conductivity ofmany organic metals grows upon cooling from �70 to �100°C, after which temperature itdrops abruptly. Organic metals turn into insulators and cease conducting electric current.The cause is Peierls instability or Peierls distortion, which is typical for one-dimensionalsystems. According to the so-called Peierls theorem for long chains, irregular domains arepreferentially stable. At certain critical temperatures, ion chains become sectioned. At theends of each section, the distances between ions are shortened, while those between the sec-tions are lengthened. This creates high barriers to electron jumps along the chain and abol-ishes its electrical conductivity.
SCHEME 7-5
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

In this sense, the salt of the TTF cation radical with the TCNQ anion radical is an out-standing example. The electrical conductivity of the salt is 103–104 ��1�cm�1 in the entiretemperature interval from �40 to �170°C (Khidekel’ & Zhilyaeva 1978). By comparison,metallic copper has an electrical conductivity of 106 ��1�cm�1 at 27°C.
Attempts to convert organic metals into superconductors have also been undertaken.The first organic superconductor was (TMeTSeF)��
2 PF6�. The transformation took place at
0.9 K and 1 hPa. Substitution of ClO4� for PF6
� led to a superconductor even at normal pres-sure and temperature of 1.2 K. In addition, the salt of the ET cation radical with iodide wasprepared. It also showed superconductivity at normal pressure. Superconductivity temper-atures increased up to 7 K with iodine contents. It was established that a semiconductingsample of (ET)2
��I3� (the so-called �-phase) transformed into the superconducting -phase
upon heating (Baram and others 1986).Fullerenes, Cxy, have internal cavities that can act as containers for metal atoms. The
metal reduces Cxy and is trapped in a fullerene cage (Saalfrank 1996). For example, Buck-minsterfullerene C60 can react with two potassium atoms, producing a dianion diradical thatis a triplet in its ground state. Alkali metal salts of Buckminsterfullerene C60 possess thehighest superconductivity transition temperatures (Tc) to date for organic materials, e.g.,K3C60 with Tc � 19 K, Rb3C60 with Tc � 29 K, and Cs2RbC60 with Tc � 33 K (Haddon1992). Localization of the unpaired electrons at definite sites on the fullerene internalsphere is a promising property of these alkali metal salts.
All these successes were important, but their significance was lessened because in-organic ceramics had been discovered. The ceramics displayed superconductivity with nocooling at all (Vanderah, 1992). Nevertheless, organic metals and superconductors havetheir advantages, and investigations in this direction continue.
As starting materials for organic metals, organic substances must correspond to thefollowing general requirements: Both acceptors and donors must form redox systems withthe participation not only of ion radicals but also of double charged ions:
A � e → A�� � e → A2� or D � e → D�� � e → D2�
Ion radicals play a role as mediators in these two-electron transfers. Each one-elec-tron step achieves a maximal rate, and both rate constants become close. Coulombic repul-sion of positive (or negative) charges makes the double-charged ion formation difficult.Therefore, donors (or acceptors) are preferable for which some possibility exists to dispersethe charge. Extension of the �-system reduces intramolecular coulombic repulsion in thedianion state. Electron-donor (or electron-acceptor) substituents should be located at dia-metrically opposite sites of the molecule. Examples are 11,11,12,12-tetracyano-9, 10-an-thraquinodimethane, TCNQ, DCNQI, and tetracyanobenzene.
In TCNQ, the cyano groups are in the most remote (face-to-face) positions. This di-minishes coulombic repulsion of two surplus electrons in the dianion and, consequently, fa-cilitates an electron transfer along the conductive chain. In tetracyanobenzene, the ring sub-stituents are close to each other. The (anion radical)-to-(dianion) transformation energyappears to be larger than that for TCNQ. Tetracyanobenzene forms only dielectric salts. Incontrast, TCNQ is able to give salts with high conductivity.
Donors and acceptors should acquire and keep a planar or almost planar structure inan ion radical state. This ensures the maximal density in a crystal lattice and the spacemonotony in the arrangement of stacks.
This requirement for planarity is important but not very strict. In other words, it nar-rows the search for new molecular systems unabruptly. The salt [(TTF)��(TCNQ)��] is an
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

example. It possesses metallic conductivity. However, its fulvalene rings are not strictlycoplanar, and the cyano groups are, to a certain degree, bent with respect to the quinonering. Moreover, one organic metal has been prepared on the basis of the principally non-planar chiral ion radical (TMET)2
�� PF6�. Its conductivity is equal to 5 ��1�cm�1 at ambi-
ent conditions (Wallis et al. 1986).By and large, donor and acceptor components for a planned ion radical salt should
have a symmetrical molecular geometry. However, if substituents are not very bulky,they can be located unsymmetrically without detriment to conductivity (Tatemitsu et al.1985).
The coexistence of both ion radicals and molecules with no formal charge is neces-sary to build conducting stacks of quasi-one-dimensional organic metals. For instance, infilms of the highly conducting salt [(TTF)�� (TCNQ)��], 20–50% of the neutral TCNQmolecules are at the surface. Meanwhile, there are no neutral and separately charged com-ponents in a stack. Bond lengths are equal in all TCNQ molecules, and all of them are in-volved in complexation (Khidekel’ & Zhilyaeva 1978). In this material, the degree ofcharge transfer between the TTF donor and the TCNQ acceptor is different from unity andequal to 0.59 (Van Duyne et al. 1986; Tanaka and co-authors 1986). In the case of 2,5-dimethyltetracyanoquinodimethane (DMTCNQ) as the donor, [Me4N�
(DMTCNQ)��]�1/2DMTCNQ exhibits a conductivity of 1.5 ��1�cm�1, and [Me4P�
(DMTCNQ)��]�1/2DMTCNQ shows a conductivity of 3.7 ��1�cm�1 (both magnitudes at280–310 K). Besides this high electrical conductivity, the salts exhibit ferromagnetic be-havior (see Section 7.4). Apparently, these mixed salts provide the first example of amolecular organic system that can exhibit both electrical conductivity and ferromagneticbehavior at room temperature (Sugimoto et al. 1998). Obviously, for these mixed salts acarrier s/p electron and a local s/p spin coexist, which is expected to contribute to high elec-trical conductivity and ferromagnetic behavior, respectively. Ferromagnetic behavior isdiscussed in Section 7.4.
All these data verify that in real systems, the rate of electron transfer between com-ponents of a conductive chain is high. There are states of mixed valence. Enhanced electri-cal conductivity and other unusual physical properties are widespread among those inor-ganic or coordination compounds that contain metals in “intermediate”-valence states. Incases of organic metals, nonstoichiometric donor/acceptor ratios provide even better re-sults. For example, the salt of (TTF)1(Br)0.7 composition displays an electrical conductiv-ity of 2 � 102 ��1�cm�1, while (TTF)1(Br)1 salt conducts electricity almost not at all(Khideckel’ & Zhilagaeva 1978).
Increasing the polarizability of components facilitates the collective shift of electronsand the stabilization of the material’s metallic state. Thus, substituting selenium for sulfur(changing from TTF to TSeF) allows one to obtain organic metals that do not transform intodielectrics up to very low temperatures. Chloride and bromide of tetraselenatetracene,(TSeT)2(Cl)1 and (TSeT)2(Br)1, have the same conductivity at room or low temperatures.
Preparative methods in organic metal chemistry differ from those in organic ion rad-ical chemistry, the main difference being the necessity of constructing conductive stacks.Stacks must be built with components that are in the mixed-valence state. The reviews citedearlier describe these methods in detail.
The majority of ion radical salts having structures with regular endless chains are ob-tained by means of the direct interaction of donors and acceptors in solution. To achievethese mixed-valence compounds, nonstoichiometric relations of components are em-ployed. The rates of cooling of the resulting mixture are very important.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

For example, at a ratio of molecular iodine to tetrathiatetracene (TTT) of 1:2, mix-tures of iodides (TTT)1(I)1 and (TTT)2(I)3 can be obtained via fast cooling. Slow coolingof the same component mixture in the same proportion leads to the single product(TTT)2(I)3. The latter is the product that possesses metallic properties. Sometimes, even atnonstoichiometric ratio, salts of the 1:1 composition are formed. Interaction between TTFand TCNQ can be exemplified. In order to obtain the 2:1 composition, an indirect synthe-sis is appropriate. The synthesis is based on an exchange reaction of the following type:
(TTF)8(BF4)5 � (Bu4N)2(TCNQ)1 → (Bu4N)(BF4) � (TTF)2(TCNQ)1
In individual cases, it is expedient to perform the interaction of a donor and an ac-ceptor in gaseous phase by means of joint vacuum sublimation of the components. Such amethod was employed in the case of (TTT)(TCNQ)2. In this example, solvent admixturesare absolutely absent in the conductive material.
Superconducting films of C60 compounds with alkali metals (M3C60) were first pre-pared at AT&T Bell Laboratories by means of this vapor diffusion method (Haddon andothers 1991). Meanwhile, pure K3C60 can be prepared simply by mixing powdered potas-sium metal and C60 in toluene (Wang et al. 1991).
The physical properties of crystals depend on admixtures; this is common knowl-edge. The admixtures can be present in the starting solution. Solvent molecules included ina crystal during its formation should be considered a contamination, too. In this sense, thehighest attention should be paid both to the purification of the starting materials and to im-proving the crystal-growing methods. The main purpose is to obtain a crystal lattice withminimal defects. For the already-mentioned salt [(TTF)��(TCNQ)��], electrical conductiv-ity depends more on the lattice defects than on the degree of purity that can usually beachieved in chemical syntheses. As the same time, the indices of magnetic susceptibility,heat absorption capacity, and paramagnetism are extremely subject to the influence of ad-mixtures. In the sense of molecular properties, admixtures (disturbing the laws of contin-uum) lower the appropriate indices.
As a rule, recrystallization cannot be used for the purification of organic metals. Re-crystallization is usually performed under definite thermal influence and leads to dirtied,“imperfect” crystals. Ion radical salts are not thermally stable in solution. The direct donor-to-acceptor interaction is the best way to limit chemical impurities. In this case, the reac-tion mixture contains minimal amounts of substances that are not included in the structureof a given ion radical salt. The oxidation of donors in the presence of anions or ion ex-change usually results in the formation of less pure crystals.
The correct choice of the method for growing crystals is very important. Slow crys-tallization of a reaction mixture at a fixed diffusion rate can be achieved by means of an H-shaped tube. Separate flasks with the solutions of a donor and an acceptor are joined via theoverturned H-shaped tube. The tube is filled with a solvent. A donor and an acceptor slowlydiffuse together, react, and form a crystalline product. As a rule, this process proceeds atroom temperature. The methods leads to the most perfect crystals of an ion radical salt.
In the H-shaped tube, all processes take place in a slow, controlled way. One simplemodification consists of separating the two halves of the H-shaped tube with a fitted glassdisk (medium, fine, or ultrafine porous) to slow the diffusion down. Another modificationinvolves diffusion inside a reaction solvent containing a polymer. In this case, diffusion isretarded due to an increase in solution viscosity (Scott et al. 1974; Berg et al. 1976). Some-times, the synthesis of ion radical salts is conducted ultrasonically if starting materials areinsoluble in the solvents desired (see, for example, Neilands et al. 1997).
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Electrocrystallization is also an effective method. It is used for preparation of an or-ganic metal containing an organic ion radical and an inorganic counterion. The techniquegives rise to high-quality organic molecular conductors and superconductors. In this ap-proach, donors or acceptors are oxidized or reduced electrochemically to form cation rad-icals or anion radicals. Crystal formation takes place at the working electrode when theradical cations/anions combine with suitable counterions that are furnished by the sup-porting electrolyte. Moderately polar organic solvents are generally employed (THF,methylene chloride, chlorobenzene, benzonitrile). For instance, electrochemical oxidationof DCNQDIm and CuBr2 leads to the formation of black crystals of the composition DCNQDIm�2Cu. Electrical conductivity of this material reaches even 103 ��1�cm�1 atroom temperature (Huenig et al. 1988). The crystal structure of the salt consists of seg-regated columns formed from the quinone imine and the copper ion in which the copperchains are surrounded by four quinone imine stacks (Aumueller et al. 1986).
Electrocrystallization can be conducted under conditions of constant current or con-stant voltage. Under constant-current conditions, the initial current density should be lowand then increased as required. Optimum current densities are usually in the range 0.1–0.5�A�cm�2. The influence of current density and voltage on the sizes, quality, phase states,and stoichiometry of the crystals obtained has been discussed (Ward 1989; Faulmann andothers 1993).
Although the electrochemical method is widely employed for producing monocrys-talline nonstoichiometric salts, it does have some disadvantages, such as long reactiontimes (up to several weeks) and drastic limitations imposed on the solvents and the amountof the target product (a few milligrams). In contrast, the chemical approach does not sufferfrom these restrictions and can theoretically afford unlimited quantities of materials.
Recently, one general route was proposed that involves oxidation of the starting neu-tral molecule by (diacetoxyiodo)benzene in the presence of strong acids. The resulting saltis highly soluble. Thus, oxidation of TTF in acetonitrile follows this scheme (Giffard et al.2001):
R4TTF � 12Phl(OAc)2 � CF3SO3H → R4TTF��CF3SO3
� � 12Phl � AcOH
R4TTF��CF3SO3� � nR4TTF � Q�X� → Q�CF3SO3
� � (R4TTFn�1)�X�
Stoichiometric triflates are convenient precursors for this conversion depicted. On the ad-dition of suitable onium (Q � R4N or R4P) salt in acetonitrile solution, either stoichiomet-ric (n � 0) or nonstoichiometric (n ! 0) can be obtained. These target salts precipitate in avery pure state.
Widening of the technical applications of organic metals depends in many respectson the development of methods for forming molecular structures with designed architec-ture. The preparation of organic metals is as much an art as a science.
7.4 ORGANIC MAGNETS
The electrical conductivity of ion radical salts springs from the mobility of their unpairedelectrons. At the same time, each of the unpaired electrons possesses a magnetic moment.This small magnetic moment is associated with the electron quantum mechanical “spin.”
Spin-originated magnetism as a phenomenon is described in many sources (see, forexample, monographs by Khan 1993; Bauld 1997; Itokh & Kinoshita 2001; and reviews by
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

J. S. Miller 2000; J.S. Miller & Epstein 1994, 1995; Wudl & Thompson 1992). This sec-tion is, naturally, devoted to the organic magnets based on ion radicals.
Any (cation-anion) radical salt has two unpaired electrons: One belongs to the cationradical and the other to the anion radical. If the spins of these two electrons have an an-tiparallel orientation, the corresponding magnetic moments are compensating. If both spinsare in parallel orientation, their magnetic moments are added together.
In parallel orientation, unpaired electrons set up according to ferromagnetic type. Theantiferromagnetic type of electron setup corresponds to the antiparallel spin configuration.When the spins are coupling in parallel manner, the material response to an applied mag-netic field—termed its magnetization—is enhanced. When the spins are coupling in an-tiparallel fashion, the magnetization is suppressed. As an example of the ferromagnetictype of material, (FeCp*2)��(TCNE)�� ion radical salt can be mentioned (J.S. Miller and co-authors 2001 and references therein). In this structure FeCp*2 means decamethylferrocene.
The problem of the ferromagnetism of a solid ion radical salt has two features. Oneis the ferromagnetism at the level of a salt as the molecule, which consists of two param-agnetic species. Another is the ferromagnetism at the level of a solid sample formed fromassemblies of the spin-bearing molecules. These molecules may contain one or more mag-netic centers.
Assemblies of molecules are most often found in molecular crystals with very weakinteractions between the molecular entities. They can also be found in extended systems,built from molecular or ion radical precursors, in a way that maximizes the interactions be-tween the precursors and, hopefully, yields bulk magnetic properties.
In order to understand the sample (material) magnetism, we need to understand thenature of molecular magnetism. The two paramagnetic components of the salt can take partin electron exchanges. One type of electron exchange is D�� ← A�� and the other is D�� →A��. These types of electron interactions are depicted in Scheme 7-6 “EE 1–5.”
In accordance with the Pauli exclusion principle, electron exchange can take placeonly between electrons with antiparallel spins. Therefore, from schemes EE1 and EE2, onlythe EE2 type is permitted. Interactions according to schemes EE3 and EE4 are real, as well.Scheme EE3 corresponds to the ferromagnetic orientation of spins. Scheme EE5 (one-elec-tron transfer from the cation radical to the anion radical) is energetically forbidden.
According to the Hund rule, only the states with maximal spin multiplicity have thelowest energy among all possible states of a given electron configuration. As for multi-plicity, it is defined with parallelism in spin orientations. Therefore, the interaction depictedin scheme EE3 is the most probable, because it, in contrast to that of schemes EE2 and EE4,leads to the state with the maximal spin multiplicity. That is the picture of microscopic fer-romagnetism of an ion radical salt.
In the macroscopic sense, spin-bearing molecules or ions are usually far enough apartthat their spin-coupling energy is small compared with the coupling-breaking thermal en-ergy. Such spins do not couple; instead they form a very weak type of a magnet called aparamagnet. When spins are closer together, the spin-coupling energy increases and maybecome large enough to enable an effective ferromagnetic or antiferromagnetic coupling.
The ferromagnetic coupling observed for (FeCp*2)��(TCNE)�� leads to a long-rangeorder throughout the solid below a critical temperature (Tc � 4.8 K; see later for what Tc
is). Substitution of a bulkier anion radical component, (TCNQ)��, for the smaller anion rad-ical component, (TCNE)��, makes the magnetic behavior worse. In other words, an in-crease in spin density in the smaller acceptor portion of the salt enhances spin coupling andstabilizes the ferromagnetic behavior of the material.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

If the number of unpaired electrons on the cation rises from one to two, such as in(MnCp*2)��(TCNE)��, Tc increases to 8.8 K; but in the case of (CrCp*)��(TCNE)��, withthree unpaired electrons, Tc reduces to 3.65 K. Ferromagnetic ordering has also been foundin (MnCp*2)��(TCNQ)�� salt. The trend in the magnetic properties resembles that for thesimilarly structured (TCNE)�� salt, although Tc is lower.
Replacing (FeCp*2)�� with nonspin (CoCp*2)� eliminates magnetic coupling. Thisdemonstrates that both the cation and the anion of these salts need to have unpaired elec-trons to stabilize ferromagnetic coupling. In other words, spin sites on both the cation rad-ical and anion radical parts of the salt contribute to the material’s magnetic properties.
Long-range antiferromagnetic order can, nevertheless, result in the formation of aferrimagnet below Tc. In the ferrimagnetic state, neighboring electron spins are oriented inopposite directions, but their magnetic moments do not cancel out, because the total of themoments oriented in one direction are larger than the total of the moments oriented in theother. Ferrimagnets, such as (MnIIITPP)�(TCNE)�� (TPP is meso-tetraphenylporphyrin),has a net magnetic moment. The material is characterized with a Tc of 14 K.
Hence, the ferromagnetic or ferrimagnetic behavior of a material is not a property ofthe molecule or ion. Like electrical conductivity, it is a cooperative property seen only inthe solid state, in assembles of molecules.
Magnets can have behaviors other then ferrimagnetic, ferromagnetic, and antiferro-magnetic. For example, metamagnetism is the transformation from an antiferromagneticstate to a ferromagnetic state by application of an external field. The (MnCp*2)��(TCNQ)��
salt just mentioned is an example of metamagnetics. Its transformation into a ferromagneticoccurs at 127.36 A�cm�1 (1,600 oersted) and at a Tc below 2.55 K.
SCHEME 7-6
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

A canted or weak ferromagnet can be formed if spins, which couple ferromagneti-cally, are only partially aligned. When Buckmingsterfullerene C60 reacts with tetrakis(dimethylaminoethylene), (Me2N)2CBC(NMe2)2, the salt of C60 anion radical and(Me2N)2CBC(NMe2)2 cation radical is formed to produce a semiconducting materialwith magnetic ordering at 16 K. However, this material does not have all the propertiesexpected for a ferromagnet. This suggests that the material may be either a canted ferro-magnet or a spin glass, in which nearby spins align but their direction shifts in differentparts of the material. Salts resulting from interactions between tetrakis(dimethy-lamino)ethylene and larger fullerenes (those with 70, 84, 90, or 96 carbon atoms) do notexhibit magnetic ordering (Wudl & Thompson 1992; Khan 1993; J.S. Miller & Epstein1995).
It would be interesting to test the magnetic properties of C60-based materials cocrys-tallized with donors. Pure C60 crystallizes as a cubic close-packed arrangement of spheresin a face-centered cubic lattice that contains large voids. However, the molecules can bepacked more efficiently (and particularly in a one-dimensional manner) by using a cocrys-tallization agent. For example, it is possibly to arrange a supramolecular assembly of C60
molecules with p-bromocalix[4]arene propyl ether. In such an assembly there are closelypacked columns that are perfectly ordered in a linear fashion (Barbour et al. 1998). Theone-dimensional strands of the fullerene molecules are separated from each other by cal-ixarene molecules. The calixarenes bearing strong donor fragments could, therefore, pre-sent some interesting opportunities.
The theoretical design of donor oligomers that gives parallel spins upon electrontransfer has been reported (Mizouchi et al. 1995). TTF and TSeF were used as the donorunits, and they linked with �CBY [Y’s are CH2, O, S, or C(CN)2]. Ferromagnetic inter-action between the spins is possible in these systems.
Magnetic interaction between the spins has to be more significant than the thermalmovement of atoms or ions. When the energy of electron exchange (J) becomes positive,a paramagnetic material is turned into a ferromagnetic one. If J � kT (k is the Boltzmannconstant), the perfect order occurs in electron orientation. If, in the sense of absolute val-ues, J � kT, thermal movement destroys this order and chaos arises. The point of conver-sion from order to chaos corresponds to some critical temperature called the Curie temper-ature (Tc). The equality J � kTc meets the boundary condition of order–chaos conversion.The stronger the interaction is, the higher is the temperature of order loss. In metallic iron,for example, the exchange interaction of electrons is positive and so strong that loss of fer-romagnetism takes place only at very high temperature (1043 K, or 770°C). Magnets areuseful only below their Tc, which for most purposes must be substantially above room tem-perature.
Miller and Epstein (1995) developed an organic-based material that retains magneticproperties up to its decomposition temperature of 350 K (75°C). They have made this mag-net by reacting TCNE with V(C6H6)2 or, better, with V(CO)6. As a result, TCNE gives theanion radical coordinated to vanadium, the nonmagnetic metal. Each [TCNE]�� binds upto four vanadium atoms. The vanadium atoms, in turn, are each surrounded by up to six lig-ands, usually nitrogens from different [TCNE]�� species. The [TCNE]� moieties in the as-sembly are most probably planar, but some of them may be twisted. Molecules of the sol-vent, such as methylene chloride, may also coordinate to the vanadium. The ability of[TCNE]�� to bind more than one vanadium results in the formation of a three-dimensionalnetwork. This network can support the strong spin coupling necessary for such a high Tc.Other solvents, such as THF and acetonitrile, produce materials with lower Tc, typically
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

about 200 K with THF and 140 K with acetonitrile. When the solvent molecules coordinateto the vanadium, they may block [TCNE]�� sites, thereby reducing the number of connec-tions between spin sites and increasing structural disorder. These changes lead to local ran-dom differences in the magnetic interactions.
The molecule-based magnet V(TCNE) y(CH2Cl2), sealed in ampoules with an inertgas, can deflect the magnetic field of another magnet. Regrettably, widespread applicationof this vanadium-TCNE compound will have to wait. It reacts readily with water and ex-plosively with oxygen. Therefore, it is impossible to exploit it in ambient conditions of airand atmospheric moisture.
Ferromagnetic organic materials capable of being processed “above room tempera-ture” are very attractive technically. Organic ferromagnetics could easily be deposited onother materials. Organic magnets should bend and spread more easily than magnetic met-als. They might also be cheaper than metal magnets, which are typically produced at vul-canian temperatures. Flexible magnetic coatings and systems for storage of magnetic dataare two obvious possible applications. The densities of organic ferromagnetics are lowerthan those of metals and metal oxides. Hence applications in medical devices would be pos-sible (the magnetic valves in artificial hearts, etc.). Organic magnets could also supplymagnetic memory. The point is that they possess properties of so-called spin glasses.Namely, their characteristics can change depending on the parameters of the magnetic fieldthey have been in. Among other organic magnetic materials (for instance, based on radicalsor carbenes), ion radicals have a particular advantage. Anion radicals can be oxidized andcation radicals can be reduced (reversibly in both cases) into uncharged molecules with nounpaired electrons. In principle, this peculiarity permits molecular magnetism to beswitched on or off and turned up or down by application of an external electric potential tocontrol the redox state (and spin state) of a molecule or, ultimately, of a bulk material.
Coronado et al. (2000) described a hybrid material that opens a new frontier in molec-ular electronics. They created an ion radical salt that can behave simultaneously as amagnet and as an electrical conductor. Crystals of composition [BEDT-TTF]3
��
[MnIICrIII (C2O4)3]� were prepared by electrocrystallization of a methanol/benzonitrile/dichloromethane solution containing chromium oxalate [Cr(C2O4)3], Mn(II) ion, and a sus-pension of bis(ethylenedithio)tetrathiafulvalene [BEDT-TTF]. The structure of the materialprepared consists of layers of [BEDT-TTF]3
�� cation radicals alternating with honeycomblayers of [MnIICrIII(C2O4)3]� bimetallic oxalato complex anions. The [BEDT-TTF]3
�� com-ponents are tilted at an angle of 45° respect to the [MnIICrIII(C2O4)3]� inorganic layer. Thisopens up the possibility for the two components to act independently: The cation radicalplays the role of an organic metal, whereas the bimetallic complex acts as an artificial mag-net. The hybrid material becomes a magnet only below 5.5 K, so cooling with liquid heliumis necessary. Such materials are now primarily of theoretical interest. Nevertheless, this ionradical salt gives an attractive example of future materials that can be both ferromagneticand electrically conductive. This unique feature, which is made possible by the molecularnature of the material, may yield unforeseen physical behavior. One can imagine, for ex-ample, materials that will really deliver smaller and smaller devices offering more than onefunction. Ferromagnetism and superconductivity are considered properties that, like oil andwater, usually do not mix.
The synthesis, structure, and electrical and magnetic characteristics were recently described for two other salts formed with BEDT-TTF and hexacyanoferrate, (Et4N�)3
[Fe(CN)6]3� or nitroprusside, K2�[Fe(CN)5NO]2�. The study sheds light on the general pe-
culiarities of making two-network solids by combining magnetic anions, which provide lo-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

calized magnetic moments, with partially oxidized organic donor molecules, which supportelectronic conduction (Clemente-Leon et al. 2001).
The two salts mentioned present very different stoichiometry: [(BEDT-TTF)4(NEt)2][Fe(CN)6] and [(BEDT-TTF)2] [Fe(CN)5NO]. They also present different packing motifsand the oxidation states of the BEDT-TTF moiety. For the first salt, the molecular ar-rangement in the organic layer is centrosymmetrical eclipsed dimers. The organic networkhas a two-dimensional character: Each dimer of a given layer is orthogonal to the closestdimer on the next layer. Each BEDT-TTF moiety bears one-fourth of the unit positivecharge, whereas the whole BEDT-TTF ensemble exists in a mixed-valence state. Thatgives rise to a high room-temperature electrical conductivity and a temperature-indepen-dent paramagnetism. The inorganic anion also contributes to the paramagnetism.
The second salt presents a structure where the anions occupy the tunnels formed byBEDT-TTF dimers. BEDT-TTF bears the unit positive charge. This salt behaves as a semi-conductor with a very low room-temperature electrical conductivity. The unpaired electronson the organic cation radicals are strongly antiferromagnetic coupled, giving rise to a dia-magnetic behavior of the second salt, because the nitroprusside anion is also diamagnetic.
These results support the view that magnetism and superconductivity in ion radicalsalts have a common electronic origin (Wilhelm et al. 2001).
By and large, molecular magnetism is a fresh research area, and it has grown rapidlyin the past two decades (see a review by J.S. Miller 2000). Among industrial corporationselaborating organic magnets, the largest and the most powerful are American DuPont andIBM. Data from existing publications are still not numerous. However, it is obvious thatthis is a promising field and that modern scientists have declared the basic ideas on the fer-romagnetism of organic ion radical salts correctly.
7.5 ORGANIC LUBRICANTS
7.5.1 Boundary Lubrication and Electron Transfer
Boundary lubrication is defined as the type of lubrication in which the friction between twosurfaces in relative motion is determined by the properties of the surfaces and by the prop-erties of the lubricant (other than viscosity). Boundary lubrication occurs when there is ahigh loading (and usually high temperatures) between two rubbing surfaces. This forces outthe bulk fluid, leaving a thin residual or surface film remaining to provide the needed lu-brication. In boundary lubrication the kinetic coefficient of friction may range from 0.03 to1.00, with wear occurring because of solid-to-solid contact during sliding (Godfrey 1968).
The ability of lubricating oil to reduce wear and prevent damage of interacting solidsis the crucial factor controlling lubricant formulations. Chemical reactions of lubricantcomponents, especially of so-called antiwear and extreme-pressure additives, occur duringfriction. These reactions involve the formation of a film on the contact surface. The film al-ters the surface’s character and thus protects it.
Under these temperature and pressure conditions, extreme-pressure additives in theresidual film chemically react with the metal surfaces to form a surface coating that allowsmetal-to-metal contact without causing any scuffing or wear. This surface coating acts likea “solid lubricant” and, hopefully, provides needed lubrication under these conditions.
The mechanical action at solid surfaces tends to promote chemical reactions and pro-duce a surface chemistry that may be entirely different from that observed under static con-ditions. Friction initiates and accelerates chemical reactions that otherwise would takeplace at much higher temperatures or will not proceed at all.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The behavior of mechanically activated metal can be generally attributed to the fol-lowing factors:
1. High temperature and high pressure at the point of working contact (Thiessen etal. 1966)
2. The newly formed (nascent) surface (Saint-Pierre 1964)3. Lattice disorder (Schroeder et al. 1969)4. The so-called Kramer effect (Kramer 1950)
The Kramer effect consists of the emission of electrons from certain freshly abradedor exposed metal surfaces. The effect has been supported by the measurement of self-gen-erated voltages between two metallic surfaces under boundary lubrication (Anderson et al.1969; Adams & Foley 1975). Since the exoelectrons mentioned have a kinetic energy ofabout 1–4 eV (Kobzev 1962), they may involve in some chemical reactions. It is pertinentto mention the electrochemical reduction of arydiazonium salts (in acetonitrile or in acidicaqueous medium) on an iron or mild steel surface (Adenier et al. 2001). This electrochem-ical (not frictional) process results in the strong bonding (which resisted an ultrasoniccleaning) of aryl groups on these surfaces. The attachment of the aryl groups is exactly toan iron and not to an oxygen atom of the embedded iron oxide. The aryl–iron bond is co-valent. This grafting process can evidently be assigned to the chemical bonding of the ironsurface with the very reactive aryl radicals produced upon one-electron reduction of thearyldiazonium cations.
The following three examples illustrate the Kramer effect during friction betweentwo surfaces.
Example 1 (Kuzuya et al. 1993)
When mechanical vibration of dipyridinium dications (see Scheme 7-7) was conductedwith a stainless steel ball in a stainless steel twin-shell blender at room temperature under
SCHEME 7-7
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

strictly anaerobic conditions, the powdery white surface of the dication salts turned a deepblue-purple. Nearly isotropic broad single-line ESR spectra were observed in the resultingpowder. No ESR spectra were observed in any of the dipyridinium salts when mechanicalvibration was conducted with a Teflon ball in a Teflon blender under otherwise identicalconditions.
When observed, the ESR signals were quickly quenched on exposure to air to recoverthe starting dications.
To identify the structure of the cation radicals formed, each of the resulting powderswas dissolved in air-free acetonitrile, and the ESR spectra of the solutions were recordedunder anaerobic conditions. Analysis of superfine structures of the spectra confirmed theformation of the corresponding cation radicals according to Scheme 7-7.
Example 2 (Mori et al. 1982)
No reaction of unmilled aluminum powder with alkyl halides was observed during 10 hoursof contact. When aluminum was milled with stainless steel balls in a stainless steel pot un-der helium at room temperature in the presence of butyl iodide for 8 min, an exothermic re-action was initiated and no more activation was required for the continuation of the reac-tion.
If additional butyl iodide was injected into the mixture, the reaction continued with-out milling until aluminum was exhausted. Little gaseous product was evolved. The distil-late of liquid product was colorless.
NMR measurement confirmed that the C–Al bond did exist in the distillate content.When the distillate was hydrolyzed with water, butane was evolved. The amount of butanewas nearly equivalent to that of the reacted butyl iodide. The equivalent amount of iodideion was detected in aqueous solution. From the results of these analyses, the liquid productwas determined to be butyl aluminum iodide, probably tributylaluminum sesqui-iodide:
3C4H9I � 2Al → (C4H9)3Al2I3
As already mentioned, once the reaction was initiated by mechanical activation, itcontinued without additional activation. This phenomenon suggests that the reaction is au-tocatalytic. As known, activators such as iodine and alkylaluminum halide initiate the con-ventional thermal reaction of aluminum with alkyl halides. In the case discussed, the reac-tion was initiated by mechanical working without any activator.
As for the fate of butyl bromide, 1.1 mmole of a gaseous product was evolved for thereaction of the starting material (4.6 mmole) after 12 min of milling. The main componentof that gas was butane. Butene was a minor product. The residue was a viscous and darkbrown material. Bromide ion (4.6 equivalents) was detected in the residue. The residue wasa mixture of aluminum bromide and a polymerized matter. The reaction with aluminum ac-tivated by vibromilling may be described with these equations:
3C4H9Br � Al → AlBr3 � 3(C4H�9)
2(C4H�9) → C4H10 � C4H8
C4H8 → polymer
The mechanochemical reactions of aluminum were investigated under two reactionconditions, namely, during and after the milling. The active source may be different in thetwo reactions. However, high temperature, high pressure, and nascent surface appeared notto be active factors in this case, because preactivated aluminum was observed to react withbutyl bromide even after the termination of milling.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Obviously, the lattice disorder and the Kramer effect remain to be analyzed. An X-ray study showed that the lattice disorder in aluminum increased slightly when milled anddid not change with time. Consequently, lattice disorder is not the main cause of themechanochemical activity.
In the meantime, the reactivity of milled aluminum correlated well with the intensityof exoelectron emission. Such an emission decayed with time after termination of milling,along with the suppression of the chemical reaction. The aluminum, which had entirely lostelectron emission activity, did not react with butyl bromide at all. Alkyl halides capture freeelectrons. The emission intensity of the free (unused) electrons under butyl bromide “at-mosphere” was less than 20% of that under benzene “atmosphere.” In other words, exo-electrons are captured with butyl bromide more easily than with benzene. Butyl bromidehas much stronger electron affinity than benzene.
Thus, the exoelectrons that result from the vibromilling of aluminum initiate the re-action between aluminum and organic acceptors.
Example 3 (Ikeda et al. 1998)
Copper(I) oxide catalyzes the splitting of water into H2 and O2 in visible light upon mag-netically stirring the suspension of Cu2O powder in H2O. The reaction temperature was ator somewhat lower than room temperature. The gases continue to evolve in the dark forseveral hundred hours after the light is turned off. The amounts of evolved H2 and O2 even-tually exceeded the amount of Cu2O used. H2 and O2 kept evolving in exactly the stoi-chiometric ratio of water decomposition. Another noticeable feature of this reaction is themarked dependence of the H2 and O2 evolution on the rate of stirring. The rate of H2O de-composition increases monotonically with the rate of rotation of the stirring rod. Withoutstirring, no H2 and O2 evolution occurred. A mechanical effect such as the rubbing of thecatalyst powder by the stirring rod is essential for the decomposition of water. Magneticfield plays no role. Similar results are observed with other oxides, such as NiO, Co3O4, andFe3O4. The authors conclude that the mechanical energy supplied by stirring is convertedto chemical energy, with the oxide functioning as a mediator or catalyst. Electrical charg-ing of the powders by mechanical friction followed by local discharge may cause the wa-ter cleavage.
7.5.2 Anion Radical Concept of Boundary Lubrication
Materials such as graphite, molybdenum disulfide, and Teflon emit few or no electronswhen disturbed. Meanwhile, fresh surfaces of such metals as aluminum and steel producea large number of emitted electrons (Connely & Rabinowicz 1983). This emission occurswhen plastic deformation, abrasion, or fatigue cracking disturbs a material’s surface. Elec-tron emission from freshly formed surface reaches a maximum immediately and then de-cays with time. Emission has been observed for both metals and metal oxides. There isstrong evidence that the existence of oxides is necessary. The exoelectron emission occursfrom a clean, stain-free metallic surface upon adsorption of oxygen (Ferrante 1977).
Goldblatt (1971) explained the lubricating properties of polynuclear aromatics by as-suming that anion radicals are generated at the freshly abraded surface. As a matter of fact,a low-energy electron (1–4 eV) emission process (exoemission) creates positively chargedspots on a surface, generally on top of surface asperities. Naturally, these asperities havegained positive charges after exoemission. At the same time, the exoemission producesnegatively charged anion radicals of lubricant components. The anion radical formed un-dergoes chemisorption on the metallic surface.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

In reality, the atmosphere of the tribological system is of particular importance. Twousual components of the atmosphere are substantial for boundary lubrication, i.e., oxygenand water vapor. The following sequence of chemical transformations is obvious:
RX � e → (RX)��
(RX)�� → R� � X�
R� � O2 → ROO�
ROO� � H2O → ROOH � HO�
ROOH → RO� � HO� etc.
The radicals formed are involved in further reactions that result in the formation ofpolymers and organometallics. Whereas radical reactions within organic additive mixtureslead to polymeric films, organometallic compounds are understandable as products of theinteraction between the metallic surface and the radicals. Both polymeric films andorganometallic species can protect the rubbing surface from wear.
Appeldorn and Tao (1968) and Goldblatt (1971) showed that boundary lubrication isnot effective in dry argon under the steel ball-on-cylinder test. In the presence of methyl-naphthalene or indene, wear scar diameters (in mm) are 0.82 or 0.93. Remarkably, these di-ameters are 0.33 or 0.72 mm in dry air and 0.36 or 0.33 mm in wet air.
In contrast with these unsaturated hydrocarbons, saturated hydrocarbons performedbetter in dry argon than in the presence of water or oxygen. For instance, decalin displaysthe following wear scar diameters (also in mm): 0.26 in dry argon, 0.35 in dry air, 0.42 inwet air (Appeldorn & Tao 1968).
Saturated hydrocarbons have lower electron affinity than unsaturated hydrocarbons.In dry argon, the decalin anion radicals are formed, if at all, in extremely low concentra-tion. It suggests that their further reactions are insignificant. In the presence of decalin, nowear occurs. In dry or wet air, decalin works worse, but still effectively. The anion radicalsof oxygen, which are formed in greater concentration than that of the anion radicals of de-calin, can initiate decalin oxidation. Oxidation products were capable of accepting exo-electrons and were involved in further reactions, with the formation of polymeric ororganometallic lubricants.
In the case of thyil radicals, however, the polymeric products can be formed withoutthe participation of oxygen or water. These radicals dimerize rapidly:
2 RS� → RS-SR
For example, a poly(disulfide) was obtained as a result of a two-electron transfer to2,5-dithiocyanatothiophene in dry argon (Todres et al. 1979) (Scheme 7-8). This reactionprovides a good reason to probe such very simple compounds as sources of lubricatingfilms for rubbing metallic surfaces.
SCHEME 7-8
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

It has been generally accepted that the extreme-pressure performance of disulfides isbetter than that of monosulfides (Allum & Ford 1965; Forbes 1970). The difference wasexplained simply with the anion radical lubrication model (Kajdas 1994). Monosulfides arereduced less readily than disulfides. Reductive cleavage of disulfides with the generationof active species RS� and RS� proceeds more easily than that of monosulfides. Accord-ingly, disulfides exhibit more efficient load-carrying properties.
An application of the anion radical model to the polymerization of vinyl monomersduring boundary friction has also been discussed (Kajdas 1994). The formation of anionradicals gives rise to the species that contain one radical end and one anion end:
(CH2BCHX)�� � CH2BCHX → �CH2MCHXMCH2MCH�X
Such species can add monomer from the two ends by different mechanisms. Two radicalends may dimerize, leaving anion ends to propagate. The formation of key anion radical re-active intermediates (CH2BCHX)�� is caused by the reaction of low-energy electronsemitted during friction with the monomers. Hence, it has to be related to the work functionof the rubbing surfaces and to the electron affinity of the monomers involved. This mech-anism has been discussed in terms of tribopolymerization models as a general approach toboundary lubrication. To evaluate the validity of the anion radical mechanism, two metalsystems were investigated: a hard steel ball on a softer steel plate, and a hard ball on an alu-minum plate. Both metals emit exoelectrons under friction, but aluminum was found to pro-duce more exoelectrons than steel (Connely & Rabinowicz 1983). With aluminum on steel,the addition of 1% styrene to hexadecane reduced the wear volume of the plate by over65%. Conclusive evidence of polystyrene was found by means of FTIR microspectrometryon the plates lubricated with styrene-containing solutions. It was additionally found thatlauryl methacrylate, diallyl phthalate, and vinyl acetate reduced aluminum wear in a pin-on-disc test by 60–80% (Kajdas 1994).
The anion radical concept of boundary lubrication provides a good understanding ofthe wear behavior of bearing steels with the perfluoro polyalkyl ether (PFPAE) fluids.These fluids are being considered for practical applications. For instance, PFPAEs are ofinterest as high-temperature gas turbine oils, owing to their oxidative stability and perfectviscosity. As a lubricant for spaceships, PFPAEs offer an excellent wide-temperature, us-able-liquid range, and extremely low volatility. These properties are important for the ul-tralow vacuum of outer space. Moreover, PFPAEs are relatively inert to systems materials(Gschwender et al. 1996).
Among these useful materials, one major commercial product, named PFAE-D, hasthis structure: CF3(OCF2)x(OCF2CF2)y(OCF2CF2CF2)zOCF3. It contains the OCF2O unitsresulting from the manufacturing process. These units have been attributed to the lowerthermal stability of the materials as compared with other commercial fluids. To gain insightinto the decomposition mechanism of the polymer, Matsunuma and co-authors (1996) se-lected a compound containing five (CF2O) units as a model for the commercially equiva-lent fluid. They performed a semiempirical molecular orbital calculation, coupling opti-mized structures with the energies of bond breaking. According to the calculations, theanion radical has a lower heat of formation than even the neutral species. Electron attach-ment to the neutral species reduces the C–O bond order. Cleavage of the weakest C–O bondof the anion radical produces the anion and the neutral radical:
F(CF2O)5CF3 � e → [F(CF2O)5CF3]�� → F(CF2O)3� � F(CF2O)2CF�
2
The shortened anion formed here also has a weak C–O bond with the reduced bond order.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The degradation of this anion proceeds in a stepwise manner:
F(CF2O)3� → F(CF2O)2
� � CF2O↑F(CF2O)2
� → CF3O� � CF2O↑
Favorable factors for this successive degradation are the low estimated activation en-ergy and the vaporization of the carbonyl fluoride product at room temperature. Strom etal. (1993) observed degradation of PFPAEs during friction tests on magnetic disks using amass spectrometer. The successive degradation of PFPAEs, which produces carbonyl flu-oride, is well known as an unzipping mechanism (Kasai 1992; Strom et al. 1993). Helmickand co-workers (1997) explored the wear behavior of steel bearings at various humidities,with PFPAE-D of the CF3(OCF2CF2CF2)xOCF3 structure. PFPAE-D does not contain di-fluoroacetal groups. It apparently generates acyl fluoride during the friction. The authorsfound that wear-reducing activity of additives to PFPAE-D was based on their possibilityof reacting with acyl fluoride to prevent its interaction with the metal specimens.
One important point for understanding the lubrication behavior of zinc dialkyldithio-phosphates consists of their participation in electron-transfer reactions. As shown, anionradicals of these salts are cleaved according to this scheme (Kajdas et al. 1986):
(RO)2P(S)SMZnMSP(S)(RO)2 � e → (RO)2P(S)S� � [ZnSP(S)S(RO)2]�
Polymers originating from the [ZnSP(S)(RO)2]. radical form lamellar aggregates thatcontain P, S, O, C, and C but not Fe (Berndt & Jungmann 1983; Sheasby & Rafael 1993).These aggregates transform into zinc sulfide and nonmetallic polymers at high tempera-tures. Interestingly, the effectiveness of zinc organodithiophosphates as the antiwear andfriction-reduced agents is enhanced by applying an external electric field. The coatingsformed on sliding in situ charged steel surfaces reduce friction by up to 35% in comparisonwith uncharged surfaces (Tung & Wang 1991).
Under conditions of electron transfer, thione-thiol rearrangement of the type P(S)OR→ P(O)SR takes place (Kajdas et al. 1986). The rearrangement changes the bond polariz-ability of a metal dialkyldithioposphates and enhances the forces driving all the chemicalprocesses described earlier.
Hence, to understand the chemical behavior of lubricant components during bound-ary lubrication, a general concept of anion radical reactive intermediate formation for thesecomponents has been put for word. The concept is based on the ionization mechanism ofthese compounds caused by the action of electrons of low energy (1–4 eV). Electrons ofsuch energy (exoelectrons) are emitted spontaneously from the fresh surfaces formed dur-ing friction. The principal thesis of the model is that lubricant components form anion rad-icals, which are then chemisorbed on the positively charged areas of rubbing surfaces. Themodel encompasses the following major stages:
1. The low-energy process of electron emission and the creation of positivelycharged spots
2. The interaction of emitted electrons with lubricant components and the genera-tion of anion radicals, anions, and radicals
3. The reactions of these anion radicals and anions with positively charged metalsurfaces, forming films protecting the surface from wear
4. The cracking of chemical bonds, producing other radicals
The model explains many lubrication phenomena in which antiwear and extreme-pres-sure additives are involved. It spurs the design of new additives and lubricating compositions.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

7.6 ION RADICAL ROUTES TO LIGNIN TREATMENT
7.6.1 Paper Fabrication
Paper products (newsprint, tissue, packaging, etc.) are made from pulps, which consist ofnatural fibers derived from vascular plants such as trees, sugar cane, bamboo, and grass.The vascular fiber walls are composed of bundles of cellulose polymeric filaments. Thislong, linear glucose polymer is what paper is made from. The polymer has the structureshown in Scheme 7-9.)
Nature rigorously shields the glucose polymer from harm by enclosing it in a mixtureof barrier polymers. In wood, cellulose fibers are protected in a matrix of lignin. Plants syn-thesize it, mainly to provide strength and protection. In the sense of protection, lignin is justthe wrapping material that protects cellulose from fungal damage. What the fungus reallywants is the cellulose, which has an ordered structure and gives them the same piece eachtime the fungus cuts it. Lignin is an amorphous, three-dimensional, and water-insolublepolymer. It is stereo-irregular, and there are no large pores in it. Lignin is formed via a peroxydase-catalyzed polymerization of the methoxy-substituted 4-hydroxycinnamyl alco-hols. Scheme 7-10 depicts these alcohols, from left to right: 4-hydroxy-cynnamyl- (or p-coumaryl-) alcohol, 4-hydroxy-3-methoxy-cynnamyl- (or coniferyl-) alcohol, and 4-hy-droxy-3,5-dimethoxy-cynnamyl- (or sinapyl-) alcohol.
SCHEME 7-9
SCHEME 7-10
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The initially formed phenoxy radicals randomly combine to form a variety of bonds.Scheme 7-11 shows major linkages between units in softwood lignin. Hardwood lignins aresimilar, but contain varying quantities of the 3,5-dimethoxylated aromatic rings.
In lignin, there are attendant ketone (sometimes quinone) chromophoric moieties thatgive it a yellow/brown color. Cellulose is a white substance that provides the desiredstrength and brightness properties of paper products. The strength and whiteness of a paperproduct depends on the extent of removal of lignin from the pulp fibers. Raw woods con-tain as much as 20% to 30% lignin.
Various modifications have been made to the conventional chemical pulping processto increase the extent and selectivity of delignification. Delignification occurs through the
SCHEME 7-11
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

fragmentation of the lignin polymer, followed by the ionization and solubilization of themolecular fragments. This degradation process is characterized by cleavage of the aryl-propane side chains, ether-bond cleavage, aromatic-ring opening, hydroxylation, and theformation of carboxylic acids. With the possible exception of the carboxylic-acid-produc-ing reaction, most of the reactions have been adequately understood as the formation of ionradicals (Schoemaker 1990; Schoemaker and Piontek 1996).
The following schemes present some examples. As for demethoxylation, it is knownthat 1,4-dimethoxybenzene is oxidized to the corresponding benzoquinone. The reactiontakes place under conditions of bio-oxidation and proceeds via the intermediacy of an ESR-detectable cation radical (Kersten et al. 1985) (Scheme 7-12).
As for -O-4 ether bond cleavage, reaction of the primary cation radical with solventwater under the same conditions of bio-oxidation was shown to lead to an arylglycerol andthe corresponding phenoxy radical (Kirk et al. 1986) (Scheme 7-13). Since the -O-4 ether
SCHEME 7-12
SCHEME 7-13
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

bond is the most abundant type of interunit linkage in the lignin polymer, this ether bondcleavage represents an important depolymerization reaction.
One modification that pertains to the present book is the role of anthraquinone (AQ)in the pulping process. Under conditions of alkaline pulping, carbohydrates in the wood re-duce AQ into an anion radical (AQ�.
). Experiments with lignin quinone methide as a modelcompound showed that the AQ anion radical caused fragmentation of the quinone methide(Scheme 7-14). The reaction results in the formation of a substituted phenolate ion and aradical. The radical is further reduced by the next AQ��(Dimmel et al. 1985). The ketogroups in lignin can be involved in the analogous reaction with AQ��. This leads to for-mation of the ketyl functions, which originate analogous fragmentation processes.
Several pulp mills are now using AQ on a commercial basis (Oloman 1996). Ac-cording to Oloman, the addition of AQ to pulping liquor at the rate of 1 kg AQ per ton ofpulp increases the pulp yield from starting woods by about 1%. This complementary actiongives rise to increased revenue of about $2 million per year for a pulp mill with a daily pro-ductivity of 1,000 tons.
The pulp industry also uses another anion radical precursor, 1,4,4a,9a-tetrahydroan-thraquinone. The product is easily obtained from cheap starting materials—1,4-naphtho-
SCHEME 7-14
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

quinone and butadiene—in the Diels–Alder reaction. The mixture of tetrahydroan-thraquinone with sulfite is very effective. The main role of sulfite in delignification duringalkaline sulfite-quinone pulping is to decrease the condensation reactions of lignin by sul-fonation. Sulfite does not play an important role in cleaving the -O-4 ether bonds of lignin(Ohi et al. 1994).
Quite recently, fungi were discovered with a special ability to chew up lignin in a va-riety of woods (Srebotnik et al. 1997). Apparently these fungi do not produce lignin per-oxidase (Srebotnik et al. 1994). The discovery opened a simple and time-controlled biop-ulping process: Wood chips are steamed, to rid them of native microorganisms, and thencooled and inoculated with a suspension of the white rot fungus Ceriporiopsis subvermis-pora in a mixture of water and corn steep liquor. The liquor, a by-product of the starch in-dustry, furnishes a growth medium for the fungus. Only 0.25 g of fungus is needed to treat1 ton of wood chips.
To explore the mode of action of C. subvermispora, Hammel’s group synthesizedvarious models of lignin, labeling functional groups with carbon-13. After incubating thepolymer with fungus, they used NMR spectroscopy to examine changes in the labeledgroups. The results indicate that the organism uses the one-electron oxidation mechanismto cleave the lignin substrate. The white rot fungus is thought to break the bond betweenthe side chain �- and -carbons as well as the bond between the side-chain -carbon andthe aryl oxygen. The unidentified one-electron oxidant forms cation radical subunits inlignin. Undoubtedly, the fungus produces this oxidant. For instance, cation radicals wereshown to be intermediates in the C�–C oxidative-cleavage reaction of lignin with the lign-inase from the white rot fungus Phanerochaete chrysosporium (Hammel et al. 1985; Ker-sten et al. 1985).
The ensuing reactions of the cation radicals include C�–C cleavage, demethoxyla-tion and other ether-bond cleaving reactions, hydroxylation of benzylic methylene groups,oxidation of benzylic alcohols, decarboxylation, the formation of phenols and quinones,and aromatic ring cleavage. In these processes, the reaction of oxygen with intermediateradicals occurs, resulting either in oxygen incorporation or in oxygen activation (i.e., re-duction to the superoxide ion; see Section 1.7.1). Incorporation of oxygen from the ambi-ent water is also possible (in the presence of an oxidant).
During the two-week biopulping process, the linkages that connect lignin subunitsare being cleaved, that is, making the wood soft. Biopulping is stopped well before the fun-gus can attack cellulose, which takes place no sooner than six weeks after inoculation. Pa-per produced from biopulped chips is stronger than paper derived from conventional pulp.Mills that produce paper for magazine printing could save $13 per ton of pulp and retainpaper quality by shifting to biopulping. Fungal pretreatment of wood chips would cut theenergy cost of conventional pulping by 30%, a savings of $10 per ton of pulp produced.Certainly, holding wood pulp for two weeks in a reactor is unacceptable for mills produc-ing 1,000 tons of pulp per day. Hence, systems are needed that would be stable at high tem-peratures and oxidize lignin in an hour or two. The use of hyperthermophilic bacteria andsome of their cloned modifications is promising (Brennan 1998).
7.6.2 Related Problems of Lignin Degradation
The alkaline oxidative cleavage of softwood lignin to vanillin is an other important reac-tion of wood chemistry. Such typical one-electron oxidants as nitrobenzene, cupric salts,and cerium ammonium nitrate are used. The reaction includes a cation radical step and pro-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

ceeds through the cleavage of the side-chain bonds (T. Fisher et al. 1988). The cleavage ismade possible by the significant weakening of the C�–C bond induced by the formationof cation radical fragments.
Oxidative degradation of spruce lignin by nitrobenzene in alkaline aqua media wasdiscovered in 1940 by Freudenberg et al. During this oxidation process, the lignin biopoly-mer is degraded into low-molecular-weight phenolic compounds that also contain cabonylgroups: ketones, aldehydes, and carboxylic acids. For example, Leopold (1950) reportedthat guaiacol derivatives were oxidized to give high yields of vanillin under the rather dras-tic conditions of alkaline nitrobenzene oxidation. The method requires a reaction tempera-ture of 170–190°C, a pressure of 10–12 atm, and a pH of 13–14. However, by introducingthe more powerful oxidant 1,3-dinitrobenzene, the oxidation process proceeds smoothly atatmospheric pressure and the much lower reaction temperature of 100°C (Bjorsvik and co-authors 2001). According to the authors, the dinitrobenzene anion radicals are the drivingspecies of the reaction. Based on oxidation of mandelic acid as an example, the followingmechanism was proposed (all the one-electron oxidation steps include transformation ofdinitrobenzene into its anion radical):
PhC(BO)COO� → PhC(BO)COO� → PhC.BO → PhC�BO
PhC�BO � �OH → PhC(BO)OH
In studies on the oxidation of lignin that had alternately been methylated at the p-hy-droxyl and benzylic hydroxyl groups, Leopold (1952) concluded that methylation causedthe low yield of vanillin obtained in the oxidation. As mentioned in Section 6.6.3, the re-placement of the OH group by OMe seriously impedes C–C bond cleavage in the water re-action medium.
The chemical and enzymatic oxidative degradation of lignin (and coal) is used to ob-tain not only vanillin but also other aromatics (Baciocchi et al. 1999 and references therein).
In principle, lignin could be a major nonfossil and renewable source of aromaticcompounds, a feedstock for the synthesis of useful products. The problem deserves findingnew ion radical routes to cleave lignin. However, wider practical applications of ion radi-cal lignin chemistry are not anticipated in the near future. Oil, gas, and coal are ready avail-able sources of aromatics. In the long term, however, biomass may (and must) replace fos-sil-originated materials in the manufacture of commercial carbon-based products.
An anion radical mechanism has recently been proposed as one of the chemical re-actions contributing to the photo-induced degradation of residual lignin leading to pho-toyellowing of paper (Andersen & Wayner 1999). Such a process leads to ketyl anion rad-ical formation. (Ketones of complex structures are also present in lignin.) Because papertends to be somewhat basic, the ketyl anion radicals can be deprotonated to form the anionradicals followed by their fragmentation to phenoxide. The phenoxide ion is further oxi-dized in air to form the phenoxyl radicals and ultimately ortho quinones. The latter are thespecies responsible for the yellow color. Andersen and Wayner concluded: “It is likely thatthe strategies to prevent or inhibit photoyellowing of paper will have to be reevaluated totake these mechanistic possibilities into account.”
7.7 CONCLUSION
Although ion radical organic chemistry is a new branch, its development reaches an “out-let” stage in the sense of practical applications. Some of them have already brought com-mercial advantages; others remain as promising fields with attractive prospects. The further
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

development of ion radical organic chemistry will surely proceed along with the search fornew applications.
In general, why do scientists call their job research when they are looking for some-thing new? Because they want to proceed on the basis of their previous achievements. Inaddition, every researcher wants to realize practical applications for results already ob-tained. On the other hand, this author knew one mathematician who had created an elegantand quite general theory, of which he was justifiably proud. When asked about possible ap-plications of the theory, he answered, “How can you talk about that? My theory is so gen-eral that any particular use is impossible.” Pure mathematics probably tolerates such an ap-proach—maybe it is even acceptable. However, chemistry is different; both scientificinterest and practical necessity are the driving forces for development.
The example of an anion radical concept of lubrication is symptomatic. Current in-terest in biological electron transfer will undoubtedly result in new ways to design drugs.Data on reversible isomerization of ion radical species will lead to a widening of mate-rial choice for “electron memory” systems. Many other fields of applications are alsopossible. Knowledge of the laws of ion radical chemistry opens a way to optimize manyorganic reactions, as was shown in Chapter 5. Currently, such a chemical process is ap-plicable in industry, justified by economics, including product yields, time for their man-ufacture, costs of reagents and energy bearers. All these points are the subjects of ion rad-ical chemistry.
REFERENCES
Adams, A.A.; Foley, R.T. (1975) Corrosion 31, 84.Adenier, A.; Bernard, M.-C.; Chehimi, M.; Cabet-Deliry, E.; Desbat, B.; Fagebaume, O.; Pinson, J.;
Podvorica, F. (2001) J. Am. Chem. Soc. 123, 4541.Allum, K.G.; Ford, J.F. (1965) J. Inst. Petrol. 51, 145.Andersen, M.L.; Wayner, D.D.M. (1999) Acta Chem. Scand. 53, 830.Anderson, T.N.; Anderson, J.L.; Eyring, H. (1969) J. Phys. Chem. 73, 3652.Andre, J.-J.; Bieber, A.; Gautier, F. (1976) Ann. Phys. 1, 145.Appeldorn, Y.K.; Tao, F.F. (1968) Wear 12, 117.Aumueller, A.; Erk, P.; Klebe, G.; Huenig, S.; Schuetz von, J.M.; Werner, H.P. (1986) Angew. Chem.,
Int. Ed. Engl. 25, 740.Baciocchi, E.; Bietti, M.; Lanzalunga, O.; Steenken, S. (1999) Atual. Fiz.-Quim. Org. [Lat. Am. Conf.
Phys. Org. Chem.] 4th, 21.Baram, G.O.; Buravov, L.I.; Degtyaryov, L.S. (1986) Pis’ma Zh. Eksper. Teor. Fiz. 44, 293.Barbour, L.J.; Orr, W.G.; Atwood, J.L. (1998) Chem. Commun., 1901.Bauld, N.L. (1997) Radicals, Ion Radicals, and Triplets: The Spin-Bearing Intermediates. Wiley-
VCH, New York.Beer, L.; Britten, J.F.; Cordes, A.W.; Clements, O.P.; Oakley, R.T.; Pink, M.; Reed, R.W. (2001) In-
org. Chem. 40, 4705.Bellec, N.; Boubekeur, K.; Carlier, R.; Hapiot, P.; Lorcy, D.; Tallec, A. (2000) J. Phys. Chem. A 104,
9750.Berg, C.; Bechgaard, K.; Andersen, J.R.; Jacobsen, C.S. (1976) Tetrahedron Lett., 1719.Berndt, H.; Jungmann, S. (1983) Schmier.-techn. 14, 306.Bjorsvik, H.-R.; Liguori, L.; Minisci, F. (2001) Org. Process Res. Develop. 5, 136.Brennan, M.B. (1998) Chem. Eng. News 76, 39.Clemente-Leon, M.; Coronado, E.; Galan-Mascaros, J.R.; Gimenes-Saiz, C.; Gomez-Garcia, C.J.;
Ribera, E.; Vidal-Gancedo, J.; Rovira, C.; Canadell, E.; Laukhin, V. (2001) Inorg. Chem. 40,3256.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Connely, M.; Rabinowicz, E. (1983) ASLE Trans. 26, 139.Coronado, E.; Galan-Mascaros, J.R.; Gomes-Garcia, C.L.; Laukhin, V. (2000) Nature 408, 447.Debad, J.D.; Bard, A.J. (1998) J. Am. Chem. Soc. 120, 2476.Debreczeny, M.P.; Swec, W.A.; Marsh, E.M.; Wasielewski, M.R. (1996) J. Am. Chem. Soc. 118,
8174.Dimmel, D.R.; Perry, L.F.; Palasz, P.D.; Chum, H.L. (1985) J. Wood Chem. Technol. 5, 15.Farges, J.-P., ed. (1994) Organic Conductors. Fundamentals and Applications. Marcel Dekker, New
York.Faulmann, C.; Cassoux, P.; Yagubskii, E.B.; Vetoshkina, L.V. (1993) New J. Chem. 17, 385.Ferrante, J. (1977) ASLE Trans. 20, 328.Fisher, J.K.; Brunning von, D.M.; Labhard, H. (1976) Appl. Optics 15, 2812.Fisher, T.; Dershem, S.M.; Schultz, T.P. (1988) J. Org. Chem. 53, 1504.Forbes, E.S. (1970) Wear 15, 87.Freudenberg, K.; Lautsch, W.; Engler, K. (1940) Ber. 73, 167.Giffard, M.; Mabon, G.; Leclair, E.; Mercier, N.; Allain, M.; Gorgues, A.; Molinie, Ph.; Neilands, O.;
Krief, P.; Khodorkovsky, V. (2001) J. Am. Chem. Soc. 123, 3852.Godblatt, I.L. (1971) Ind. Eng. Chem., Prod. Res. Div. 10, 270.Godfrey, D. (1968) In: O’Connor, J.J., ed. Standard Handbook of Lubrication, Ch. 2. McGraw-Hill,
New York.Grossel, M.C.; Weston, S.C. (1994) Contemp. Org. Synth. 1, 367.Gschewender, L.; Snyder, C.E., Jr.; Oleksiuk, M.; Kochler, M. (1996) Tribology Trans. 39, 368.Haddon, R.C. (1992) Acc. Chem. Res. 25, 127.Haddon, R.C.; Hebard, A.F.; Rosseinsky, M.J.; Murphy, D.W.; Duclos, S.J.; Lions, K.B.; Miller, B.;
Rosamilia, J.M.; Fleming, R.M.; Kortan, A.R.; Glarum, S.H.; Makhija, A.V.; Muller, A.J.; Er-ick, R.H.; Zahurak, S.M.; Tycko, R.; Dabbagh, G.; Thiel, F.A. (1991) Nature 350, 320.
Hammel, K.E.; Tien, M.; Kalyanaraman, B.; Kirk, T.K. (1985) J. Biol. Chem. 260, 8348.Helmick, L.S.; Gschwender, L.J.; Sharma, S.K.; Snyder, C.E.; Liang, J.C.; Fultz, G.W. (1997) Tri-
bology Trans. 40, 393.Huenig, S.; Aumueller, A.; Erk, P.; Meixner, H.; Schuetz von, J.U.; Gross, H.J.; Langhor, U.; Werner,
H.P.; Wolf, H.C.; Burschka, C.; Klebe, G.; Peters, K.; Schnering von, H.C. (1988) Synth. Met.27, B181.
Ikeda, Sh.; Takata, Ts.; Kondo, T.; Hitoki, G.; Hara, M.; Kondo, J.N.; Domen, K.; Hosono, H.; Kawa-zoe, H.; Tanaka, A. (1998) Chem. Commun., 2185.
Inokuchi, H.; Saito, G.; Wu, P.; Seki, K.; Tang, T.B.; Mori, Y.; Imaeda, K.; Enoki, T.; Higuchi, Y.;Inaka, K.; Yasupka, N. (1986) Chem. Lett., 1263.
Inokuchi, H.; Imaeda, K.; Enoki, T.; Mori, T.; Maruyama, Y.; Saito, G.; Okada, N.; Yamochi, H.;Seki, K.; Higuchi, Y.; Yasuoka, N. (1987) Nature, 329, 39.
Itokh, K.; Kinoshita, M. (2001) Molecular Magnetism: New Magnetic Materials. Gordon & Breach,Amsterdam.
Jerome, D.; Mazaud, A.; Ribault, M.; Bechgaard, K. (1980) J. Phys. Lett., 41, L195.Kajdas, Cz. (1994) Lubrication Science 6, 203.Kajdas, Cz.; Tuemmler, R.; Ardenne von, H.; Schwartz, W. (1986) Zeitschr. fuer Industrie (Leipzig)
115, 107.Kasai, P.H. (1992) Macromolecules 25, 6791.Kersten, P.J.; Tien, M.; Kalyanaraman, B.; Kirk, T.K. (1985) J. Biol. Chem. 260, 2609.Khan, O. (1993) Molecular Magnetism. VCH, New York.Khidekel’, M.L.; Zhilyaeva, E.I. (1978) Zh. Vses. Khim. Obshch. im. D.I. Mendeleeva 23, 506.Kido, J.; Matsumoto, T. (1998) Appl. Phys. Lett. 73, 2866.Kirk, T.K.; Tien, M.; Kersten, P.J.; Mozuch, M.D.; Kalyanaraman, B. (1986) Biochem. J. 236, 279.Kobzev, N.I. (1962) Exoelectronic Emission. Foreign Publishing House, Moscow.Kramer, J. (1950) Der Metallische Zustand Vandenhock und Ruprecht, Goettingen (Germany).Kurata, T.; Fukada, Ch.; Fuchigami, H.; Hamano, K.; Tsunoda, S. (1998) Thin Solid Films 331, 55.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Kuzuya, M.; Kondo, Sh.-i.; Murase, K. (1993) J. Phys. Chem. 97, 7800.Kyushin, S.; Miyajima, Yo.; Matsumoto, H. (2000) Chem. Lett. 1420.Le Magueres, P; Lindeman, S.V.; Kochi, J.K. (2000) Org. Lett. 2, 3567.Lee, V.Y.; Engler, E.M.; Schumaker, R.R.; Parkin, S.S.P. (1983) J. Chem. Soc., Chem. Commun.,
235.Leopold, B. (1950) Acta Chem. Scand. 4, 1523.Leopold, B. (1952) Acta Chem. Scand. 6, 1294.Lu, P.; Hong, H.; Cai, G.; Djurovich, P.; Weber, W.; Thompson, M.E. (2000) J. Am. Chem. Soc. 122,
7480.Matsunuma, S.; Toshimasa, M.; Kataoka, H. (1996) Tribology Trans. 39, 380.Mihara, Ch.; Sugata, Y.; Santoh, T. (1997) U.S. Pat. 5,605,732 (to Canon Kabushiki Kaisha, Tokyo).Miller, J.S. (2000) Inorg. Chem. 39, 4392.Miller, J.S.; Epstein, A.J. (1994) Angew. Chem. Int. Ed. Engl. 33, 385.Miller, J.S.; Epstein, A.J. (1995) Chem. Eng. News 73, 30.Miller, J.S.; Glatzhofer, D.T.; Vazquez, C.; McLean, R.S.; Calabrese, J.C.; Marshall, W.J.; Raebiger,
J.W. (2001) Inorg. Chem. 40, 2058.Miller, S.E.; Lukas, A.S.; Marsh, E.; Bushard, P.; Wasielewski, M.R. (2000) J. Am. Chem. Soc. 122,
7802.Mizouchi, H.; Ikawa, A.; Fukutome, H. (1995) J. Am. Chem. Soc. 117, 3260.Mori, S.; Kuriyama, O.; Maki, Y.; Tamai, Y. (1982) Z. anorg. allg. Chem. 492, 201.Neilands, O.; Tilika, V.; Sudmale, I.; Grigorjeva, I.; Edzina, A.; Fonavs, E.; Muzikante, I. (1997) Adv.
Mater. Opt. Electron. 7, 39.Ohi, H.; Meshitsuka, G.; Ishizu, A. (1994) Mokuzai Gakkaishi 40, 440.Oison, V.; Katan, C.; Koenig, Ch. (2001) J. Phys. Chem. A 105, 4300.Oloman, C. (1996) Electrochemical Processing for the Pulp and Paper Industry. Electrochemical
Consultancy, Hants, England.Rovira, C.; Veciama, J.; Santalo, N.; Tarres, J.; Cirujeda, J.; Molins, E.; Llorca, J.; Espinosa, E.
(1994) J. Org. Chem. 59, 3307.Saalfrank, R.W. (1996) Nature 383, 124.Saint-Pierre, L.E.; Owens, R.S. (1964) Nature 202, 1204.Schoemaker, H.E. (1990) Rec. Trav. Chim. Pays-Bas 109, 255.Schoemaker, H.E.; Piontek, K. (1996) Pure Appl. Chem. 68, 2089.Schroeder, R.; Nobst, P.; Tetzner, G.; Petzold, D. (1969) Z. anorg. allg. Chem. 365, 255.Scott, J.C.; Garito, D.F.; Heeger, A.J. (1974) Phys. Rev. B 10, 3131.Sheasby, J.S.; Rafael, Z.N. (1993) Tribology Trans. 36, 399.Srebotnik, E.; Jensen, K.A.; Hammel, K.E. (1994) Proc. Natl. Acad. Sci. USA 91, 12794.Srebotnik, E.; Jensen, K.A.; Kawai, S.; Hammel, K.E. (1997) Appl. Environ. Microbiol. 63, 4435.Strom, B.D.; Bogy, D.B.; Walmsley, R.G.; Brandt, J.; Singh, B.C. (1993) Wear 168, 31.Sugimoto, T.; Ueda, K.; Endo, S.; Toyota, N.; Tada, T.; Nishimura, K.-i.; Kohama, M.; Shiwaku, K.;
Yamamoto, K.; Yamaguchi, T.; Suenaga, Y.; Munakata, M. (1998) Chem. Phys. Lett. 288, 767.Tanaka, M.; Shimizu, M.; Saito, Y.; Tanaka, J. (1986) Chem. Phys. Lett. 125, 594.Tatemitsu, H.; Nishikata, E.; Sakata, Yo. (1985) J. Chem. Soc., Chem. Commun., 106.Thiessen, P.A.; Meyer, K.; Heinicke, G. (1966) Grundlagen der Tribochemie. Akademie Verlag,
Berlin.Todres, Z.V. (2001) Chem. Innovation 31, 26.Todres, Z.V.; Furmanova, N.G.; Avagyan, S.P.; Struchkov, Yu, T.; Kursanov, D.N. (1979) Phosph.
Sulfur 6, 309.Tung, S.C.; Wang, S.S. (1991) Tribology Trans. 34, 479.Umeda, M.; Niimi, T.; Hashimoto, M. (1990) Jpn. J. Appl. Phys. 29, 2746.Umeda, M.; Nishizawa, M.; Itoh, T.; Selman, J.R.; Uchida, I. (2000) Chem. Phys. Lett. 326, 219.Vanderah, T.A.; ed. (1992) Chemistry of Superconductor Materials: Preparation, Chemistry, Char-
acterization and Theory. Noyes Publications, Park Ridge, NJ.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Van Duyne, R.P.; Cape, T.W.; Suchanski, M.R.; Siedle, A.R. (1986) J. Phys. Chem. 90, 739.Wallis, J.D.; Karrer, A.; Dunitz, J.D. (1986) Helv. Chim. Acta 69, 69.Wang, H.H.; Kini, A.M.; Savall, B.M.; Carlson, K.D.; Williams, J.M.; Lykke, K.R.; Wurz, P.; Parker,
D.H.; Pellin, M.J.; Gruen, D.M.; Welp, U.; Kwok, W.-K; Fleshler, S.; Crabtree, G.W. (1991)Inorg. Chem. 30, 2838, 2962.
Ward, M.D. (1989) J. Electroanal. Chem. 273, 79.Wilhelm, H.; Jaccard, D.; Duprat, R.; Bourbonnais, C.; Jerome, D.; Moser, J.; Carcel, C.; Fabre, J.M.
(2001) Eur. Phys. J. 21, 175.Wudl, F.; Thompson, J.D. (1992) J. Phys. Chem. Solids 53, 1449.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

8
Concluding Remarks
8.1 INTRODUCTION
The aim of this chapter is to underline some general and important topics of ion radical or-ganic chemistry and to formulate some current problems still awaiting solution. It is advis-able to scrutinize the topics that could open up new chemical routes but whose generalityand, sometimes, applicability are at present unclear.
Single-electron transfers to or from electronically neutral molecules result in the for-mation of anion radicals and cation radicals, respectively. The unpaired spin and the chargecan delocalize within the molecular carcass, can be located on the same fragment or evenatom, or can be spatially separated (distonic species). Each type has its own synthetic op-portunities, which were discussed in the previous chapters. All of that material shows thatthese ion radicals cannot be treated either as conventional radical species or even as theirionic counterparts. They are characterized by unique behavior.
8.2 SRN1 REACTION
This type of reaction is important and deserves some comment. The addition of one elec-tron to a molecule generates an anion radical. This results in an increase in its reactivity. Inparticular, the bond dissociation energies in anion radicals are much smaller than those inthe corresponding neutral molecules. Thus, substitution reactions proceed more easily inthe case of anion radicals. The reactions of the SRN1 type are good examples of this feature.Two possible schemes (a and b) for the reaction course are listed here:
Scheme a
(RX)�� → X� � R�; R� � Nu� → (RNu)��;
(RNu)�� � RX → RNu � (RX)�� etc.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Scheme b
(RX)�� → X� � R�; R� � Nu� → (RNu)��;
(RNu)�� � RX → RNu � X� � R� etc.
Substitution of type a forms a well-documented class of reactions; see these very recentpapers and the references therein: Costentin et al. 1999, 2000; Rossi 1999; Corsico & Rossi2000; Adcock and co-authors 2001. In contrast with conventional nucleophilic substitution,the nucleophile, Nu�, reacts not with the substrate, RX, to give a product but with the radicalR. The latter emerges as a result of the RMX bond cleavage. Substituent X is very often ahalogen atom, but other leaving groups can also be used; see later. In the majority of aromaticSRN1 reactions, the anion radical RX�� (R � Ar) is the observable intermediate. It is depictedin scheme a. With aliphatic substrates, SRN1 substitution indeed takes place rather than SN2or SN1 substitutions and the concerted mechanism depicted in scheme b is feasible.
In most cases, the coupling reaction between the radical and nucleophile species isthe rate-determining step in the dark (see, for example, Tamura et al. 1991; Azuma et al.1992). This step leads to RNu��, the product of real substitution. The chain process is com-pleted by a reaction in which one electron is transferred from the product anion radical tothe substrate. A neutral substitution product is formed; the propagation loop is closed. Thissequence of steps has been mentioned in Chapters 4 and 5.
It is logical to consider the nucleophile, Nu�, as a source of the electron to be trans-ferred onto the substrate molecule, RX. However, in most cases, the nucleophile is such apoor electron donor that electron transfer from Nu� to RX is extremely slow, if it is possi-ble at all. In most cases, the reaction requires an external stimulation in which a catalyticamount of electrons is injected. We have already pointed out such kinds of assistance to thereaction from photochemical and electrochemical initiations or from solvated electrons inthe reaction solvent. Alkali metals in liquid ammonia and sodium amalgam in organic sol-vents can serve as the solvated electron sources. Light initiation is also used widely. How-ever, photochemical initiation complicates the reaction performance.
Next we give a concise description of the requirements concerning the structure ofthe substrate, the nature of the introducing group, and the reaction medium.
8.2.1 Substrate Structure
The groups to be substituted as a result of the reaction must be stable in the form of the cor-responding ion. Other substituents (not participants in the transformation) must be inertelectrochemically; i.e., they must be insensitive to electron transfer.
Halogens are the substituent with good fugacity. Iodine and bromine demonstrateclose activity during photostimulated substitution. Therefore, the more accessible deriva-tives can be used.
Such groups as SPh, Me3N�, and OP(O)(OEt)2 are also substituted readily, espe-cially in aromatic substrates. The following are substituent effects on the SRN1 replacementat the reaction center: alkyl, alkoxy do not prevent the substitution. Steric shielding of alkylgroups is insignificant. For instance, the reaction rates of o- and p-halotoluenes are practi-cally equal. The reaction does not proceed in the case of hydroxy or dialkylamino deriva-tives of halobenzenes (Wolfe & Carver 1978). There are many excellent examples of theeasy preparation of hindered compounds with significantly screened centers.
A �-acceptor (carbonyl, phenyl) assists the reaction via an intramolecular electrontransfer to the leaving group. This intramolecular redox catalysis is well illustrated by com-
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

paring 1-chloronorbonane and its 2-carbonyl analog. The former is nonreacting towardPh2P� under photoirradiation; the latter does react, to give, after oxidation, substitutionproduct in 93% yield (Rossi 1999) (Scheme 8-1).
The ketone receives an electron in the antibonding �* molecular orbital to form �*anion radical that is transformed into a (MCMCl)� particle by an intramolecular electrontransfer to the antibonding �* molecular orbital of the MCMCl bond. The (MCMCl)�
bond then fragments to form a chloride ion and the corresponding radical (Scheme 8-2).The photostimulated reaction of 4-chlorocamphor (in which the spatial distance be-
tween the leaving group Cl and the CBO function is increased with an extra CMC bond)yields, after oxidation, the organophosphoric product in 80% yield (Rossi 1999) (Scheme8-3). The reactions depicted for the norbormane and camphor derivatives do not proceedwithout photostimulation.
Unlike conventional nucleophile substitution, the cyclopropane ring does not cleaveduring a SRN1 reaction.
The influence of the nitro group depends on the nature of the substrate. The presenceof a nitro group is sometimes unfavorable. However, the substitution at a saturated carbonatom is strongly facilitated if the nitro group is bonded with the same carbon atom that bearsa leaving group. In this case, either X or NO2 can come off (Scheme 8-4). The nature of Xgoverns the spitting preference. If X � I, Br, Cl, S(O)2R, S(O)R, SR, or SCN, the leavinggroup is X. NO2 is eliminated when X � C(O)R, C(O)OR, RPO3, NO2, CN, N3, or Alk(Bowman 1988). In the substrates under consideration, both X and NO2 are located at thesame tetrahedral carbon atom. The first step of the reaction consists in the formation of thesubstrate anion radical. The unpaired electron apparently occupies the orbital centered onthe nitrogen atom of the nitro group. Substituent X is in the zone of influence of this orbital.Being in this zone, substituent X can recapture the unpaired electron of this orbital. Favor-able factors are the CMX bond lability and X� fugacity. In contrast, if the X� fugacity issmall and the CMX bond is strong, then NO2
� is the leaving group.A cation arriving with a nucleophile is another important factor. The nature of the
cation determines the active form of the nucleophile in a given solvent. The nucleophile can
SCHEME 8-1
SCHEME 8-2
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

attack the substrate in the form of a free ion or in the form of an ionic pair. As a rule, lithiumsalts are less reactive than sodium and potassium salts. Russell and Mudryk (1982) reportedseveral examples of this. The sodium salt of ethyl acetylacetate reacts with 2-nitro-2-chloropropane in dimethylformamide, yielding ethyl 2-iso-propylidene acetylacetate. Un-der the same conditions, the lithium salt does not react. Potassium diethyl phosphite inter-acts with 1-methyl-1-nitro-1-(4-tolylsulfonyl)propane in THF and gives diethyl-1-methyl-1-nitro-1-phosphite. The lithium salt of the same reactant does not react with the same substrate in the same solvent.
Transformation of a substrate into its ion radical enhances the species’ reactivity.Sometimes, this can overcome steric encumbrance of the substituent to be removed. Thus,1,4-di-iodo-2,6-dimethybenzene expels only one, sterically congested, iodine from posi-tion 1 upon the action of the tributylstannyl radical. Upon the action of the enolate ion ofpinacolone (Me3CCOCH2
�) in the photoinitiated SRN1 process, both iodines (from posi-tions 1 and 4) are substituted (Branchi and co-authors 2000).
8.2.2 Nature of the Introducing Groups (New Substituents)
There are two important requirements regarding reagents for SRN1 reactions: They have tobe electronodonors with respect to the substrates or active interceptors of intermediary rad-icals. For example, the phenyl thiolate and diphenylphosphite ions are active in such inter-ception; the phenolate ion is inactive. The introducing group sometimes exerts an influenceon chain branching. All of these peculiarities have already been detailed in Chapters 3, 4,and 5.
8.2.3 Reaction Medium
The reactions under consideration often require an inert atmosphere. However, some ex-amples can be found in Chapters 4 and 5 when air and even pure oxygen atmospheres areoptimal.
As for solvents, liquid ammonia and dimethylsulfoxide are those most often used.There are some cases when tert-butanol is used as a solvent. In principle, ion radical reac-tions need aprotic solvents of expressed polarity. This facilitates the formation of such po-
SCHEME 8-3
SCHEME 8-4
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

lar forms as ion radicals are. Meanwhile, the polarity of the solvent assists in ion-pair dis-sociation. This enhances the reactivity of organic ions, sometimes to an unnecessary de-gree.
Certainly, a decrease in the permissible limit of the solvent’s polarity widens the pos-sibilities for ion radical synthesis. Interphase catalysis is a useful method to circumvent thesolvent restriction. Thus, 18-crown-6-ether assists in anion radical formation in the reac-tion between benzoquinone and potassium triethylgermyl in benzene (Bravo-Zhivotovskiiet al. 1980). In the presence of tri(dodecyl)methylammonium chloride, fluorenylpinacolineforms the anion radical upon the action of calcium hydroxide octahydrate in benzene. Thecation of the onium salts stabilizes the anion radical (Cazianis & Screttas 1983). Surpris-ingly, anion radicals are stable even in the presence of water.
Similarly, interphase catalysis permits carbonylation of haloaryls and halovinyls byNaCo(CO)4 with CO in water. The reaction proceeds according to the SRN1 mechanism andleads to high enough yields of carboxylic acids. Because the CArMCl bond is inert underthese conditions, the direct and selective synthesis of chlorocarboxylic acids becomes pos-sible. Thus, 1-bromo-4-chlorobenzene gives 4-chlorobenzoic acid only (Brunet et al. 1983)(Scheme 8-5).
At initiation with metal ions, a solvent can play the role of a donor ligand to the metalion and thus enforce its activity. One example comes from SmI2 chemistry: In mixtures ofhexamethylphosphorotriamide and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H )pyrimidinonewith acetonitrile (but not with THF), the rate of the reductive cyclization of o-ally-loxyiodobenzene with SmI2 sharply increases (Hasegawa & Curran 1993).
8.2.4 Dark SRN1 Reactions
Photostimulation increases the number of reagents and substrates that can be involved inSRN1 reactions. However, it complicates their performance; see the review by Ivanov(2001). Therefore, the dark reactions are of special interest.
The photoirradiation effect can be replaced by copper salt catalysis. The catalyzed re-actions go rapidly and result in a high degree of transformation. Interestingly, the ESRmethod reveals no paramagnetic particles in the course of the reaction between haloarylsand phenyl thiolates. The addition of oxidants (oxygen, dinitrobenzene) or radical accep-tors [di(tert-butyl)nitroxide] does not inhibit the substitution. These facts are understand-able from Scheme 8-6 (Bowman et al. 1984; Liedholm 1984).
Several substitution reactions are catalyzed by iron ions (Galli & Bunnett 1984). Adetailed preparative study was recently reported on the ferrous ion–initiated SRN1 reactionsof haloarenes with the sodium enolates of tert-butyl acetate, N-acetylmorpholine and anumber of higher N-acylmorpholines. Smooth and rapid substitution occurs and good to ex-cellent yields were obtained of arylacetic esters, arylacetamides, and arylalkyl amides. The
SCHEME 8-5
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

catalytic activities of cuprous chloride, ruthenium trichloride, mercuric sulfate, cobalt sul-fate, and dicerium trisulfate were also mentioned (van Leeuven & McKillop 1993).
Some SRN1 reactions can take place in the dark and with no catalyst. For example,the interaction of freons with nucleophiles in dimethylformamide at 20°C proceeds with-out photoirradiation. The chain process begins when the system pressure reaches 2 atm, inother words, when the concentration of the gaseous reagent becomes sufficient (Waksel-man & Tordeux 1984) (Scheme 8-7).
There is a favorable difference between the ionization potential of the nucleophile(PhS�) and the electron affinity of the substrate (CF3Br); the expressed bromide fugacityis also a favorable factor.
The Kornblum reactions at the tertiary carbon atoms also belong to the dark SRN1substitutions. Let us compare the reactions of �-cumyl chloride and 4-nitro-�-cumyl chlo-ride with the phenylthiolate ion (Kornblum 1975) (Scheme 8-8). As seen, the substitutionof the arylthio moiety for chlorine at the former position of the chlorine is observed onlyfor the 4-nitroderivative. The optically active substrate gives the racemic substitution prod-uct upon reaction with the phenylthiolate ion (Scheme 8-9).
Radical inhibitors and electron acceptors prevent the formation of the substituentproduct. Oxygen drastically retards the substitution but facilitates p-nitrocumyl peroxideformation. Photoirradiation accelerates the reaction. Consequently, the reaction belongs tothe ion radical type. The nucleophile (PhS�) is sufficiently active as an electron donor andas a radical interceptor.
The reaction of the same 4-nitro-�-cumyl chloride under the same conditions butwith another nucleophile, the 2-nitropropanide ion, leads principally to the same result. Theonly species that might provide simulating electrons to the system is the nucleophile (thenitropropenide ion). However, this nucleophile is such a poor donor that electron transferfrom this ion to 4-nitro-�-cumyl chloride is expected to be extremely slow, apparently tooslow to serve as a viable initiation step. Saveant’s group (Costentin et al. 1999) tried to
SCHEME 8-6
SCHEME 8-7
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

solve such an enigma. Their strategy was to identify the reaction product, measure the re-action kinetics, and determine all of the thermodynamic and kinetic factors of initiation,propagation, and termination. This allowed them to predict what the reaction kineticsshould be if the nucleophile were the electron-donor initiator, and to compare the resultswith the experimentally obtained data.
They carried this reaction out under pseudo-first-order conditions (excess of 2-nitro-propanate ions) in acetonitrile at 25°C, under argon atmosphere in a light-protecting ves-sel. The 2-nitropropanate ion was introduced as the tetramethylammonium salt. Two prod-ucts were formed (Scheme 8-10). One of the products was the expected C-substitutedcompound. The other was an unstable species, which decomposed into the 4-nitrocumyl al-cohol during workup and was ascribed to O-substitution. In 1975, Kornblum had obtainedthe same products. He considered the C-substitution as an SRN1 reaction and the O-substi-
SCHEME 8-8
SCHEME 8-9
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

tution as an SN2 substitution. Since steric hindrance at the reacting carbon prevents the SN2reaction from occurring with 4-nitrocumyl chloride, Saveant’s group concluded that boththe C- and the O-substitution products result from an SRN1 reaction. (Dual reactivity of nu-cleophiles is well known.)
The SRN1 character of the reaction was ascertained by the effect of light irradiationand the addition of a radical trap. Namely, under light irradiation, the half-reaction time wasconsiderably shortened (3 instead of 41 min). Addition of di-tert-butyl nitroxide com-pletely quenched the reaction: Neither the C-substitution nor the O-substitution was ob-served after 4 hr. The radical trap may only react with the R� radicals that escaped the sol-vent cage where R�, Nu�, and X� have been formed. This means that, in the absence of thetrap, the R� radical does react with Nu� before diffusing out of the cage. The fact that theradical trap quenches the formation of both the C- and O-substitution products confirmsthat both species result from an SRN1 reaction.
The outer-sphere electron-transfer initiation mechanism cannot account for the ob-served kinetics, the half-reaction time being more than 100 times larger than that observed.The chain process considerably enhances the global rate of the reaction (without a chainprocess, the half-reaction time would be three centuries).
The solution of the riddle posed by Kornblum’s dark SRN1 reaction is as follows. Thenucleophile does work as a single electron-transfer initiator of the chain process. However,the mechanism of initiation does not consist of a mere outer-sphere electron transfer fromthe nucleophile to form the anion radical of the substrate. Rather, it involves a dissociativeprocess in which electron transfer and bond breaking are connected (Costentin & Saveant2000). Scheme b at the beginning of Section 8.2 (p. 387) illustrates this mechanism.
The passage from a stepwise (the phenylthiolate ion as a nucleophile) to a concertedmechanism (the nitropropanate ion as a nucleophile) is a consequence of the low drivingforce offered by the poor electron-donor properties of the nucleophile.
SCHEME 8-10
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

8.3 STEREOCHEMICAL ASPECTS OF ION RADICAL REACTIVITY
8.3.1 Problem of Steric Restrictions
Reactions of the SRN1 type are less sterically restricted than classical SN reactions. In gen-eral, the nucleophilic (not SRN) reactivity varies with the steric demand at the reaction cen-ter. The electron-transfer reactivity does not depend on steric effects. To illustrate, one cancompare electron-transfer and nucleophilic reactivity of ketene silyl acetals with cationicelectrophiles (Fukuzumi et al. 2001). Nevertheless, space strains may determine the over-all results of the reactions if either intermediate radicals or forming products are stericallyhindered. Scheme 8-11 shows the case in which the intermediate radical is sterically hin-dered while the reagent is strongly active with respect to the radical (Norris & Smyth-King1982). The case represents the SRN1 substitution that takes place when sodium thiopheno-late acts on e,4-tert-butyl-e,2-methyl-a,4-nitro-e,4-(4-nitrophenyl) cyclohexane. Light ir-radiation stimulates the reaction. It is carried out under nitrogen in hexamethylposphoro-triamide. The ion radical type of process has been established by means of inhibitors. Ashas been found, the stereochemical outcome of the reaction depends on the concentrationof the PhSNa nucleophile.
At a low concentration of PhSNa, the reaction leads to a mixture of phenylthiylderivatives; the content of a,SPh-substituted product is higher by 20% than that of e,SPhproduct. At a high concentration of PhSNa, the reaction produces practically the singlestereoisomer bearing the a-PhS group.
SCHEME 8-11
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

In the course of the reaction, the nitrite ion leaves the primary anion radical. This pro-duces the cyclohexyl radical in a pyramidal configuration. The vicinal methyl group steri-cally hinders the conversion of the pyramidal radical into the planar one. With a high con-centration of the nucleophile, the rate of addition exceeds the rate of conversion; i.e. radd
� rconv. Then the entering PhS group occupies the axial position. With a low concentrationof the nucleophile, the conversion occurs earlier than the addition (radd �� rconv) and theplanar radical center is attacked from both the axial and equatorial sides. This results in theformation of the isomer mixture.
Certainly, the simultaneous formation of the planar and pyramidal radicals and theirmutual interconversions is possible. Nevertheless, the earlier explanation of the stere-ospecificity seems sufficient.
The stereospecific substitution produces a sterically less strained product when amore bulky p-nitrophenyl substituent occupies the equatorial position and the less bulkyphenylthiyl substituent is located in the axial position. Steric limitations of the nucleophileattack caused by strains in the final product are thus removed. This is why the ion radicaladdition of the p-nitrobenzyl radical to the lithium salts of 5-nitro-1,3-dioxanes proceedsstereospecificially (Zorin et al. 1983) (Scheme 8-12).
Steric encumbrance in the attacking reactant blocks the SRN1 reaction in a standardmanner (Look & Norris 1999). It is more interesting to follow the role of steric hindrancein a forming product on the course of the SRN1 process, according to Kornblum and Erick-son (1981) as well as Akbulut et al. (1982). Scheme 8-13 clearly demonstrates the effect;here, all of the constituent reactions were performed under the same conditions (hexam-ethylphosphorotriamide as a solvent, at 25°C). The SRN1 nature was proven for all of thecases. The intermediary cumyl radical reacts with the nitroalkane anion, but this reaction isretarded with an increase in the size of the alkyl anion. The significance of the cumyl rad-ical dimerization grows accordingly.
SRN1 reactions are, in general, eased up sterically. This feature is employed for syn-thetic purposes. For instance, the cyanoacetate substituent was inserted in the stericallyshielded position of a benzene ring (Suzuki et al. 1983) (Scheme 8-14). The reaction pro-ceeds in hexamethylphosphorotriamide. Photoirradiation results in the formation of unde-sirable by-products, but initiation with cuprous iodide leads to the target substance, at morethan 60% yield.
SCHEME 8-12
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

One important aspect of steric restriction concerns the difference in internal energiesbetween neutral and ion radical states of a molecule. Thus, in its neutral state, cis-1,3,5-hex-atriene is free to adopt three conformers, cis-cis-cis, cis-cis-trans, and trans-cis-trans, by rotation around a single bond with a barrier of only 4 kJ�mol�1. For the cation radical state,this barrier height increases up to 46 kJ�mol�1, thus making the interconversion of the conformers a slow process compared to that of the neutral molecule. The trans-cis-transconformer of the cation radical has the lowest internal energy. Being involved in the cation-radical state, 1,3,5-hexatriene does not undergo ring closure. The origin of such inertnessis the rapid formation of the trans-cis-trans conformer where the carbon termini are too faraway from each other to close the possible six-member ring (Radosevich & Wiest 2001).
SCHEME 8-13
SCHEME 8-14
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

8.3.2 Reflection of Electron-Transfer Step in ReactionStereochemistry
The chiral nucleophiles C6H13C*H(Me)X (X � Br, OSO2Me) alkylate benzophenonemetal ketyl at one of the rings as well as at the carbonyl carbon atom. Both pathways leadto the corresponding products in approximately equal amounts (Hebert et al. 1983)(Scheme 8-15).
The ketone is formed as a racemate (with respect to the alkyl substituent). Conse-quently, the main route includes an electron-transfer step (Scheme 8-16).
SCHEME 8-15
SCHEME 8-16
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The carbinol is optically active, and a configuration inversion takes place partially.Obviously, a dual reactivity is observed. Accordingly, the electron transfer route can berepresented by the following scheme:
Me Me MeM*CHMC6H13
*
PhMCMO�M��CHX �MX→ PhMCMOMCH e→ PhMCMO�
* Ph C6H13 Ph C6H13 Ph
As emphasized (Bakken et al. 2001), the observation of an excess of inversion of theconfiguration in the CMC alkylated products produced in this reaction may well be the re-sult of a single transition state that partitions into the electron-transfer product and the sub-stitution product. In fact, the amount of enantiomeric excess in such a reaction can serve asa measure of the bonding in the electron-transfer transition state.
The following scheme depicts the SN2 route:
Me MeCC6H13| |
PhMCMO�M� � �CH → PhMCMO�Me| | |
Ph C6H13 Me
The nature of X in the starting C6H13C*H(Me)X determines the relation between these tworoutes. If X � Br, the configuration inversion does not exceeds 8%. If X � OSO2Me, theinversion takes place for 25%. The bromide ion is more active in elimination than themethyl sulfonate ion. Therefore, the electron-transfer contribution to the substitution reac-tion is higher.
The alkylation of ketones by haloalkyl in the presence of alkali metals is known as theBarbier reaction. Luche and Cintas (1999) compared the stereochemical results of the reac-tion with the participation of cycohexanone, the homochiral 2-bromooctane, and lithiummetal in the typical conditions of the Barbier reaction (0°C, stirring) and on ultrasonic stim-ulation (at �50°C). As has already been noted (see Section 5.2.4), rate-determining elec-tron-transfer steps are dependent on the sonication effect. From C6H13C*H(Me)Br, the re-active entity is the anion radical generated on the activated metal surface. Under sonicationconditions, the reaction is completed in 1.5 hr and leads to 98% yield of the product. The lat-ter is formed in a partially inverted configuration. The inversion degree is 19% at medium-energy sonication and increases up to 24% for high-energy sonication. Under conditions forthe Barbier reaction, the yield barely reaches 50% in 7 hr, and the degree of configurationalinversion does not exceed 6%. According to Luche and Cintas (1999), sonication increasesthe concentration of the primary haloalkane anion radical and accelerates its addition to thecarbonyl group, in a direction anti to the leaving bromide ion. The conventional reaction(without sonication) provides much lower yields of a practically racemic product. The over-all sonicated reaction proceeds as in Scheme 8-17.
It should be noted that nonmetallic redox reactions also experience the sonication in-fluence. The preparation of �-lactons from olefines upon manganese triacetate oxidation isan example. The reaction with monomethyl malonate in acetic acid, which does not occurat 0–10°C, proceeds smoothly when sonication is applied (Allegretti et al. 1993). From cy-clohexene, only the cis ring fusion in the bicyclic lactone is observed; the product is formedat 80% yield for 15 min at 10°C. The overall scheme of the reaction, without the detailedmechanism is shown in Scheme 8-18.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

The stereoselectivity of the sonochemical process probably reflects the enhanced re-action rate, which does not allow equilibration processes to take place.
8.3.3 Conformational Behavior of Ion Radicals
This problem has attracted considerable attention, and intriguing results have been ob-tained. It would be important, however, to develop some application of the results. Thistask awaits solution.
The anion radicals from aromatic nitro compounds preserve the second-order axis ofsymmetry. The analysis of the superfine structure of the ESR spectrum of the nitrobenzeneanion radical reveals equivalency of the ortho and meta protons (Ludwig et al. 1964; Levy& Myers 1965).
With the anion radical of nitrosobenzene, the situation is quite different, as evidencedby ESR data (Levy & Myers 1965; Geels et al. 1965). Following electron transfer, the ni-troso group fixes in the plane of the benzene ring to a certain extent. This produces five dif-ferent types of protons, since both meta and both ortho protons become nonequivalent. Thenonequivalence of the ortho and meta protons has also been established for the anion rad-icals of acetophenone (Dehl & Fraenkel 1963) and S-methylthiobenzoate (Debacher et al.1982) (Scheme 8-19).
SCHEME 8-17
SCHEME 8-18
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.
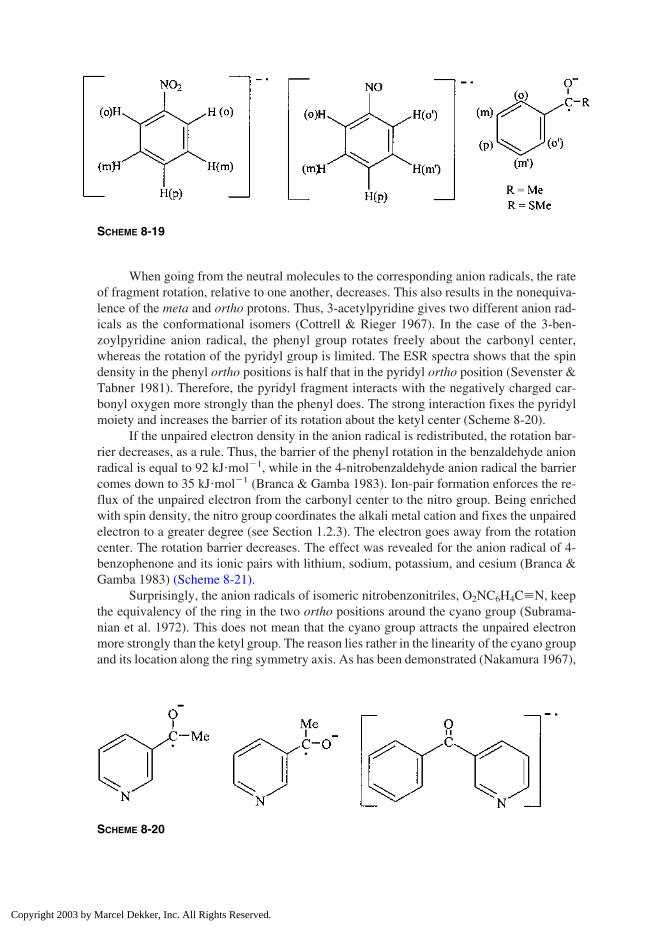
When going from the neutral molecules to the corresponding anion radicals, the rateof fragment rotation, relative to one another, decreases. This also results in the nonequiva-lence of the meta and ortho protons. Thus, 3-acetylpyridine gives two different anion rad-icals as the conformational isomers (Cottrell & Rieger 1967). In the case of the 3-ben-zoylpyridine anion radical, the phenyl group rotates freely about the carbonyl center,whereas the rotation of the pyridyl group is limited. The ESR spectra shows that the spindensity in the phenyl ortho positions is half that in the pyridyl ortho position (Sevenster &Tabner 1981). Therefore, the pyridyl fragment interacts with the negatively charged car-bonyl oxygen more strongly than the phenyl does. The strong interaction fixes the pyridylmoiety and increases the barrier of its rotation about the ketyl center (Scheme 8-20).
If the unpaired electron density in the anion radical is redistributed, the rotation bar-rier decreases, as a rule. Thus, the barrier of the phenyl rotation in the benzaldehyde anionradical is equal to 92 kJ�mol�1, while in the 4-nitrobenzaldehyde anion radical the barriercomes down to 35 kJ�mol�1 (Branca & Gamba 1983). Ion-pair formation enforces the re-flux of the unpaired electron from the carbonyl center to the nitro group. Being enrichedwith spin density, the nitro group coordinates the alkali metal cation and fixes the unpairedelectron to a greater degree (see Section 1.2.3). The electron goes away from the rotationcenter. The rotation barrier decreases. The effect was revealed for the anion radical of 4-benzophenone and its ionic pairs with lithium, sodium, potassium, and cesium (Branca &Gamba 1983) (Scheme 8-21).
Surprisingly, the anion radicals of isomeric nitrobenzonitriles, O2NC6H4C�N, keepthe equivalency of the ring in the two ortho positions around the cyano group (Subrama-nian et al. 1972). This does not mean that the cyano group attracts the unpaired electronmore strongly than the ketyl group. The reason lies rather in the linearity of the cyano groupand its location along the ring symmetry axis. As has been demonstrated (Nakamura 1967),
SCHEME 8-19
SCHEME 8-20
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

in the anion radicals of para-dicyanobenzene the cyano group and all the unsubstitutedfour-ring protons are equivalent (Scheme 8-22).
Benzene para-dialdehyde (terephthalic aldehyde) produces the anion radical at thepotential of the limiting current of its first reduction wave in dimethylsulfoxide withtetrapropylammonium salt as a supporting electrolyte. The anion radicals demonstrate twoESR spectra. One of them is superimposed on the other. They have the same g-factor butdiffer considerably in the splitting constants (Stone & Maki 1963). If the rate of rotation ofthe aldehyde groups were many times greater than the frequency difference in the splittingconstants of cis and trans rotamers, the ESR spectra of the anion radicals of terephthalicaldehyde would be averaged. If the rate of mutual rotation of the groups were comparablewith the difference in the splitting constants of the rotamers, the lines of the spectra wouldwiden. The spectra overlap one another; this means that the frequency of rotation is muchlower than the frequency difference in splitting constants. It seems improbable that the for-mation of different ionic pairs with one and the same anion radical with the countercationis responsible for such a superposition of spectra: tetra-alkylammonium was used as thecation, and the latter does not form arbitrarily stable ionic pairs. According to calculations(Todres 1974), the neutral cis and trans rotamers of terephthalic aldehyde differ energeti-cally by not more than 1.3 J�mol�1. The trans anion radical of this aldehyde is more stablethan the cis anion radical by 8.5 J�mol�1. The analysis of the ESR line intensities revealsthat the cis and trans forms of this anion radical are in the ratio 1:1.4 (Stone & Maki 1963).Hence, electron transfer to terephthalic aldehyde yields two conformers (Scheme 8-23).
Summarizing his group studies on thienyl ion radicals, Pedulli (1993) described thebehavior of oligothiophenes upon one-electron transfer. For the anion radical of 2,2-bithienyl, the ESR spectrum shows the presence of two species with relative concentrationsof 4:1 identified as the rotational isomers of this anion radical. According to ESR (Pedulli
SCHEME 8-21
SCHEME 8-22
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

1993) and Raman (Keszthelyi et al. 1999, 2000) spectra, the cation radical of 2,2-bithienylalso exists as a mixture of at least two rotamers. The derivatives of 2,2-bithienyl contain-ing substituents capable of stabilizing the corresponding anion radicals were also studied.In every case ESR detected both conformational isomers. No exchange broadening tookplace, even at temperatures as high as 100°C. The internal barrier is very high, about 70kJ�mol�1. Meanwhile, for 2,2-bithienyl (the neutral molecule), the magnitude of the rota-tion barrier is about 20 kJ�mol�1. Restricted rotation was also observed in both anion andcation radicals of terthienyl, whose ESR spectra showed signals from two of the three pos-sible conformational isomers up to room temperature. The rotamers are formed as a mix-ture, containing the S-trans, trans and S-cis, trans forms in 1.8:1 ratio (Scheme 8-24).
As has been established, 4,5,9,10-tetrahydropyrene yields the cation and anion radicalsin the form of an equilibrium mixture of conformers (Iwaizumi & Isobe 1975) (Scheme 8-25).
The activation energy of the mutual interconversion depends on the charge nature ofthe ion radical. For the cation radical, it is half of that for the anion radical. The authors em-phasize that the reaction proceeds through an intermediary planar structure in which themethylene groups of the saturated ring are hyperconjugated with the neighboring aromaticrings. The methylene groups stabilize cations to a greater extent than anions. Therefore, thehyperconjugation lowers the system energy in cations far more than in anions. This is thereason why the temperature dependence of the ESR spectra point to a greater facility ofconversion of one isomer into the other for the cation radical than for the anion radical.
The same was observed in the series of the 1,2,3,6,7,8-hexahydropyrene cation andanion radicals (Pijpers et al. 1971). For example, at very low temperatures, ENDOR spec-tra reveal the formation of two cation radical isomers and the formation of only one anionradical isomer (Maekelae & Vuolle 1985).
A complete silicon analog of cyclohexane, cyclohexapermethyl silane, exists in amobile chair conformation (Bock et al. 1979). In the cation radical, the inversion rate de-creases, while in the anion radical this rate sharply increases. The reason for such behavior
SCHEME 8-23
SCHEME 8-24
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

seemingly lies in the electron properties of the ion radicals. The one-electron oxidationtouches the highest occupied molecular orbital of the neutral molecule and evidently en-hances the initial overlapping of silicon orbitals. In the one-electron reduction, the incom-ing electron populates the lowest orbital, probably composed of earlier unoccupied datomic orbitals of silicon. This leads to ring conjugation of the d,� type, and the systemtends to flatten. The conformation that flattens is the middle one between the invertomers.
It is also worthwhile to compare the ferrocenyl ethylene (vinylferrocene) anion radi-cals and cation radicals. For the cyano vinylferrocene anion radical, the strong delocaliza-tion of an unpaired electron was observed; see Section 1.2.2. This is accompanied by ef-fective cis → trans conversion (the barrier of rotation around the MCBCM bond islowered). As for the cation radicals of the vinylferrocene series, a single electron remainsin the highest molecular orbital formerly occupied by two electrons. According to photo-electron spectroscopy and quantum mechanical calculations, the highest occupied molecu-lar orbital is mostly or even exclusively the orbital of iron (Todres et al. 1992). This orbitalis formed without participation of the ethylenic fragment. The situation is quite differentfrom the arylethylene radical cations, in which all �-orbitals overlap.
After one-electron oxidation of ferrocenyl ethylene, an unpaired electron and a positivecharge are centered on iron. The MCBCM bond does not share the �-electron cloud with theFe�� center. As a result, cis → trans conversion never occurs (Todres et al. 1980; Todres 2001).
Unexpectedly, the analysis of the molecular orbitals of the cation radical from -cyano vinylferrocene reveals the possibility for cis → trans conversion if more than one-electron oxidation takes place. Namely, the cation radical has a molecular orbital four lev-els higher in energy than the one occupied by the single electron, which is centered on thecyanoethylene fragment (Todres et al. 1992).
In a similar manner, introduction of the ferrocenyl group drastically changes the re-activity of the mesityl enol ester cation radicals. These cation radicals undergo a bond scis-sion in solution according to the following scheme (Mes � mesityl):
[Mes2CBC(R1)O−C(O)R2]�� → Mes2CBC(R1)O� � �C(O)R2
The reaction takes place if R1 is tert-Bu and R2 isMC6H4N(CH3)2-p. If R1 is still tert-Bubut R2 is Fc (ferrocenyl), no cleavage occurs (Schmittel et al. 2001). The ferrocenyl-con-taining cation radical is apparently a persistent ferricenium type of species. Intramolecularactivation of the scission with ferrocene as a redox unit is not operative.
8.3.4 On the Relationship Between Ion Radical Stereochemistry andValence Isomerization
Reduction of poly(butyl)naphthalenes with a sodium–potassium alloy in ether causes theirisomerization (Goldberg et al. 1976). The reduction of 1,3,6,8-tetra(tert-butyl)naphthalene
SCHEME 8-25
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

produces the anion radical that is stable in the temperature range from �10 to �60°C. At�60°C, the anion radical disproportionates, yielding the initial tetrabutylnaphthalene andthe corresponding dianion. The initial ESR signal disappears. Repeated cooling of the so-lution restores the ESR spectrum but with other characteristics. The newly recorded spec-trum corresponds to the anion radical of the isomerization product (Scheme 8-26).
We can see that the isomerization consists not in a direct conversion of the initial an-ion radical into the final one but rather involves the two intermediary dianions. However,what is the driving force for this isomerization? The molecule of 1,3,6,8-tetra(tert-butyl)naphthalene is nonplanar: The tert-butyl group and the carbon atom at position 1 ofthe naphthalene skeleton lie off the plane of the molecule. The corresponding anion radical
SCHEME 8-26
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

has the same stereochemical peculiarity (Goldberg 1973). Such bending removes the stericstrain but, naturally, decreases the degree of the �-electron delocalization over the neutralmolecule. As for the anion radical, its unpaired electron delocalizes less effectively than inthe anion radical of the unsubstituted naphthalene. Bending of the naphthalene skeleton in-creases the overlapping of orbitals of atoms 1 and 4. The addition of two extra electrons in-creases the order of the ordinary bonds. Atoms 1 and 4 come closer together. Their bond-ing becomes possible. This facilitates the formation of the dianion and then the anionradical in the form of the valence isomer.
One-electron reduction of a norcaradiene derivative produces the corresponding an-ion radical. The conditions of the odd-electron delocalization in this anion radical are lessfavorable than in its valence isomer. According to calculations, the unpaired electron in-corporation in the nonatetraenyl �-system lowers the energy content by 0.62 . However,the anion radical initially formed is less stable than the benzotropylidene anion radical. Thelatter is the end product of the isomerization (Gerson et al. 1978) (Scheme 8-27).
This conversion is directed so as to create the most favorable conditions for the de-localization of the unpaired electron within the aromatic nucleus. It is worth noting herethat thermal treatment (150–190°C) also initiates isomerization of the initial neutralmolecule of norcaradiene into the benzotropylidene system. At the same time, the reduc-tive transformation described earlier proceeds smoothly even at negative temperatures. Un-der comparable reaction conditions (25°C), the rate of conversion of the neutral moleculeis 15 orders lower than that of the anion radical.
Tetra(tert-butyl)tetrahedrane converts into terta(tert-butyl)cyclobutadiene only whenheated in vacuum up to 140°C. The barrier of 170 kJ�mol�1 separates these two valenceisomers (Heilbronner et al. 1980). In the cation radical state, the tetrahedrane structure con-verts into the cyclobutadiene structure without heating (Bock et al. 1980; Fox et al. 1982).As one can see from Scheme 8-28, upon the action of aluminum chloride in methylenechloride, tetrahedrane forms not the cation radical of the same skeleton but the cation rad-ical of its valence isomer, the cyclobutadiene cation radical. The latter is more stable thanthe former because of the more effective delocalization of the unpaired electron and thepositive charge. Both tetrahedrane and cyclobutadiene give the same adduct with di-cyanoacetylene, which acts as an “adding oxidant” (Maier et al. 1982).
8.3.5 On the Possible Significance of Ion Radical Stereochemistry
The material in Section 8.3 should be understood along with the already-discussed data inChapters 1, 2, and 3. There are many examples in which the ion radical stage affects thestereochemical outcome of entire reactions. The stereochemistry of intermediary ion radi-cals obviously correlates with the steric characteristics of final products. Nevertheless, it
SCHEME 8-27
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

would be incorrect to generalize such a correlation. It is clear now that an important con-sequence of ion radical formation is the Z/E isomerization of olefines, stereochemicalchanges accompanying the formation of the three-electron bonds, and valence isomeriza-tion in ion radicals. The change in the steric structure governed by the conversion into theion radical state sometimes makes a decisive contribution to the reaction kinetics. Certainreactions involving the ion radical stage become less sensitive to steric factors, and this mayresult in highly branched compounds. The direction of a reaction along the ion radicalcourse may also be used to provoke conversions between streoisomers under mild condi-tions.
The stereochemical opportunities of the ion radical reactions deserve further study,since they may produce many results important for science and commercial applications.
Although this branch of organic ion radical chemistry is presently insufficiently de-veloped, its consideration have may attract attention to this promising problem.
8.4 CONCLUSION
Throughout this book, the author has presented numerous examples. However, his purposehas never been to write a comprehensive review of the field or to recount the historical de-velopment of organic ion radical chemistry. The examples, selected from of a large num-ber, were to a large extent chosen to describe not species but phenomena. Such an approachmade it impossible to reference all the papers dealing with the organic chemistry of ion rad-icals or even to cite all the good papers.
SCHEME 8-28
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

An analysis of the literature included in the References for every chapter leads tothe following estimations: The average time of publication redoubles every 4 years,rather than the normal value of 10 years. The number of references to work from the ionradical field in papers on general organic chemistry doubles every 2 years, on the aver-age. As to the individual authors, the most productive creators are, as a rule, also the mostcited. The information noise generated is considerably lower here than in other fields.The frequency of the transfer of concepts and terms from ion radical chemistry to gen-eral organic chemistry quadruples every 10 years. More and more new groups get in-volved in the field, with the groups bringing their own ideas and discussing earlier resultsfrom new viewpoints.
It is obvious that ion radical organic chemistry attracts much attention. It has devel-oped intensively. Achievements in this field have their echoes in and are rapidly drawn intothe general disciplinary ownership of organic chemistry.
Of course, this book emphasizes organic ion radicals and their reactivity. However,readers should be cautioned against considering all organic reactions as including ion rad-ical mechanisms. Such attempts have already been undertaken in organic chemistry (Biele-vich & Okhlobystin 1968), but they could not eliminate the diversity of the chemistry. Or-ganic chemistry, living and varying, requires no preconceived theories or approaches.
Scientific research has led to significant success in the practical applications of or-ganic ion radicals. Such applications should widen in the future. It is very important to con-centrate effort on the elaboration of preparative methods of ion radical organic chemistry,including stereospecific methods. The correct choice of new developmental proceduresopens up a wealth of new directions. It is hoped that interest in this area will continue to en-sure the flow of new ideas and reactions, particularly in the area of organic synthesis in liq-uid phase and without light irradiation.
REFERENCES
Adcock, W.; Clark, Ch. I.; Trout, N.A. (2001) J. Org. Chem. 66, 3362.Akbulut, U.; Toppare, L.; Utley, H.R. (1982) J. Chem. Soc. Perkin Trans. 2, 391.Allegretti, M.; D’Annibale, A.; Trogolo, C. (1993) Tetrahedron 49, 10705.Azuma, N.; Ozawa, T.; Yamawaki, K.; Tamura, R. (1992) Bull. Chem. Soc. Jpn. 65, 2860.Bakken, V.; Danovich, D.; Shaik, S.; Schlegel, H.B. (2001) J. Am. Chem. Soc. 123, 130.Bielevich, K.A.; Okhlobystin, O. Yu. (1968) Uspekhi Khimii 37, 2163.Bock, H.; Kaim, W.; Kira, M.; West, R. (1979) J. Am. Chem. Soc. 101, 7667.Bock, H.; Roth, B.; Maier, G. (1980) Angew. Chem., Int. Ed. Eng. 19, 209.Bowman, W.R. (1988) Chem. Soc. Rev. 17, 283.Bowman, W.R.; Heaney, H.; Smith, Ph. H. G. (1984) Tetrahedron Lett. 25, 5821.Branca, M.; Gamba, A. (1983) Chem. Phys. 75, 253.Branchi, B.; Galli, C.; Gentill, P.; Martinelli, M.; Mencarelli, P. (2000) Eur. J. Org. Chem., 2663.Bravo-Zhivotovskii, D.A.; Kruglaya, O.A.; Vyazankin, N.S. (1980) Zh. Org. Khim. 50, 2144.Brunet, J.-J.; Sidot, Ch.; Caubere, P. (1983) J. Org. Chem. 48, 1166.Cazianis, C.T.; Screttas, C.G. (1983) Tetrahedron 39, 165.Corsico, E.F.; Rossi, R.A. (2000) Molecules 5, 431.Costentin, C.; Saveant, J.-M. (2000) J. Am. Chem. Soc. 122, 2329.Costentin, C.; Hapiot, Ph.; Medebielle, M.; Saveant, J.-M. (1999) J. Am. Chem. Soc. 121, 4451.Costentin, C.; Hapiot, Ph.; Medebielle, M.; Saveant, J.-M. (2000) J. Am. Chem. Soc. 122, 5623.Cottrell, P.T.; Rieger, Ph.H. (1967) Mol. Phys. 12, 149.Debacher, U.; Schmuser, W.; Voss, Ju. (1982) J. Chem. Res. S., 74.Dehl, R.; Fraenkel, G.K. (1963) J. Chem. Phys. 39, 1793.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.

Fox, M.A.; Campbell, K.A.; Huenig, S.; Berneth, H.; Maier, G.; Schneider, K.-A.; Malsch, K.-D.(1982) J. Org. Chem. 47, 3408.
Fukuzumi, Sh.; Ohkubo, K.; Otera, Ju. (2001) J. Org. Chem. 66, 1450.Galli, C.; Bunnett, J.F. (1984) J. Org. Chem. 49, 3041.Geels, E.J.; Konaka, R.; Russell, G.A. (1965) Chem. Commun., 13.Gerson, F.; Huber, W.; Muellen, K. (1978) Angew. Chem., Int. Ed. Eng. 17, 208.Goldberg, I.B. (1973) Org. Magn. Reson. 5, 29.Goldberg, I.B.; Growe, H.R.; Frank, R.W. (1976) J. Am. Chem. Soc. 98, 7641.Hasegawa, E.; Curran, D.P. (1993) J. Org. Chem. 58, 5008.Hebert, E.; Mazaleyrat, J.P.; Welvart, Z. (1983) Nouv. J. Chim. 7, 55.Heilbronner, E.; Jones, T.B.; Krebs, A.; Maier, G.; Malsch, K.-D.; Pocklington, J.; Schmebzer, A.
(1980) J. Am. Chem. Soc. 102, 564.Ivanov, V.L. (2001) Vestn. Mosk. Univ., Ser. 2: Khim. 42, 172.Iwaizumi, M.; Isobe, T.E.S. (1975) Mol. Phys. 29, 549.Keszthelyi, T.; Grage, M.M.-L.; Wilbrandt, R.; Svendsen, Ch.; Mortensen, O.S. (1999) Laser Chem.
19, 393.Keszthelyi, T.; Grage, M.M.-L.; Offersgaard, J.F.; Wilbrandt, R.; Svendsen, Ch.; Mortensen, O.S.;
Pedersen, J.K.; Jensen, H.J.A. (2000) J. Phys. Chem. A 104, 2808.Kornblum, N. (1975) Angew. Chem., Int. Ed. Engl. 87, 797.Kornblum, N.; Erickson, A.S. (1981), J. Org. Chem. 46, 1037.Levy, D.H.; Myers, B.J. (1965) J. Chem. Phys. 42, 3731.Liedholm, B. (1984) Acta Chem. Scand. B. 10, 877.Look, K.; Norris, R.K. (1999) Austral. J. Chem. 52, 1047.Luche, J.-L.; Cintas, P. (1999) Adv. Sonochem. 5, 147.Ludwig, P.; Layoff, T.; Adams, R.N. (1964) J. Am. Chem. Soc. 86, 4568.Maekelae, R.; Vuolle, M. (1985) Magn. Reson. Chem. 23, 666.Maier, G.; Schneider, K.-A.; Malsch, K.-D.; Irngartinger, H.; Lenz, A. (1982) Angew. Chem., Int. Ed.
Eng. 21, 437.Nakamura, K. (1967) Bull. Chem. Soc. Jpn. 40, 1019.Norris, R.K.; Smyth-King, R.J. (1982) Tetrahedron 38, 1051.Pedulli, G.F. (1993) Res. Chem. Intermed. 19, 617.Pijpers, F.W.; Arick, M.R.; Hendricks, B.M.P.; Boer de, E. (1971) Mol. Phys. 22, 781.Radosevich, A.T.; Wiest, O. (2001) J. Org. Chem. 66, 5808.Rossi, R.A. (1999) In: Atual. Fis.-Quim. Org. [Lat. Am. Conf. Phys. Org. Chem.] 4th, 72.Russell, G.A.; Mudryk, B. (1982) Tetrahedron 38, 1059.Schmittel, M.; Peters, K.; Peters, E-M.; Haeuseler, A.; Trenkle, H. (2001) J. Org. Chem. 66, 3265.Sevenster, A.J.L.; Tabner, B.J. (1981) J. Chem. Soc. Perkin Trans. 2, 1148.Stone, E.W.; Maki, A.H. (1963) J. Chem. Phys. 38, 1999.Subramanian, J.; Copinathan, M.S.; Narasimhan, P.T. (1972) J. Magn. Reson. 7, 388.Suzuki, H.; Kobayashi, Ts.; Yoshida, Y.; Osuka, A. (1983) Chem. Lett., 193.Tamura, R.; Yamawaki, K.; Azuma, N. (1991) J. Org. Chem. 56, 5743.Todres, Z.V. (1974) Rus. Chem. Rev. Engl. Ed. 43, 1099.Todres, Z.V. (2001) Chem. Innovation 31, 26.Todres, Z.V.; Chernyshova, T.M.; Koridze, A.A.; Denisovich, L.I. (1980) Dokl. Akad. Nauk SSSR
254, 902.Todres, Z.V.; Safronov, A.I.; Ermekov, D.S.; Minyaev, R.M. (1992) J. Organomet. Chem. 441, 479.van Leeuwen, M.; McKillop, A. (1993) J. Chem. Soc. Perkin Trans. 1, 2433.Wakselman, C.; Tordeux, M. (1984) J. Chem. Soc. Chem. Commun., 793.Wolfe, J.F.; Carver, D.R. (1978) Org. Prep. Proced. Int. 10, 225.Zorin, V.V.; Kukovitskii, D.M.; Zlotskii, S.S.; Todres, Z.V.; Rakhmankulov, D.L. (1983) Zh. Org.
Khim. 19, 426.
Copyright 2003 by Marcel Dekker, Inc. All Rights Reserved.