organisches praktikum i für studierende der lehrämter … · aufbau der apparatur begonnen...
TRANSCRIPT

Universität Regensburg WS 2003/2004Institut für Org. ChemieProf. Dr. O. ReiserDr. P. Kreitmeier
Organisches Praktikum I
für Studierende der Lehrämterund der Biologie (Diplom)
Allgemeine Hinweise zum Praktikumund Versuchsvorschriften zum
Apparativem Vorpraktikum
Kurs 1, September/Oktober 2003

-2-
Einige Allgemeine Hinweise
Montag, den 29.09.2003 findet im Rahmen der Vorbesprechung eine Sicherheitseinführung statt. DieAnwesenheit aller Studenten wird durch Unterschrift bestätigt und ist unbedingte Voraussetzung fürdie Teilnahme am Praktikum.Grobe Verstöße gegen die Laborordnung oder allgemeine Sicherheitsvorschriften führen zumsofortigem Entzug des Arbeitsplatzes und Ausschluss aus dem Praktikum!
Vorpraktikum
• Montag, 29.09.2003 ab 13.00 Uhr Übernahme eines Arbeitsplatzes (Saal 6: Nur Biologen!).Dieser Arbeitsplatz ist bis zum Praktikumsende beizubehalten.Zur Platzübernahme füllen Sie die auf den Plätzen liegenden Zettel aus und erhalten damit in derSaalausleihe die Schlüssel für Ihren Arbeitsplatz. Gleichzeitig unterschreiben Sie den öffentlich-rechtlichen Vertrag mit dem Freistaat Bayern über die Überlassung der Arbeitsgeräte. Danachkontrollieren Sie Ihren Arbeitsplatz. Sollten Geräte fehlen oder beschädigt sein, werden diese inder Saalausleihe von Dienstag, 30.09. bis Mittwoch, 1.10. ersetzt bzw. umgetauscht. Dievollständige Übernahme des Platzes wird durch Ihre Unterschrift bestätigt.
Im Vorpraktikum werden Sie die grundlegenden Arbeitstechniken der präparativen OrganischenChemie erlernen.
Hauptpraktikum
• Im Hauptpraktikum sind von jedem Studenten die durch Gruppe und Platznummer zugewiesenenPräparate darzustellen:
Studierende Lehramt (vertieft): 9 Präparate+2 Derivate.Studierende Lehramt (nicht vertieft): 9 Präparate.Studierende der Biologie: 5 Präparate.
• Vor Beginn eines jeden Versuchs muß ein sogenanntes „Vortestat“ beim zuständigen Assistentenabgelegt werden. Inhalt des Vortestats sind Fragen zur Arbeitssicherheit, praktischeDurchführung der Reaktion und grundlegende Fragen zum Reaktionsablauf bzw. Mechanismus.Die zugehörigen „Versuchsbezogenen Betriebsanweisungen“ werden vor Beginn des Haupt-praktikums ausgegeben. Der Assistent bestätigt durch seine Unterschrift auf der „Versuchs-bezogenen Betriebsanweisung“ die Freigabe des Versuchs. Erst nach der Freigabe darf mit demAufbau der Apparatur begonnen werden, nach der Abnahme der Reaktionsapparatur durch denAssistenten darf der Versuch durchgeführt werden.
• Die Präparateausbeuten müssen mindestens 50% der angegebenen Ausbeute betragen, andernfallsist das Präparat zu wiederholen.
• Die angefertigten Präparate sind in Präparategläschen und ausführlicher Beschriftung abzugeben:Etikett mit Name, Vorname, Platznummer, Versuchsnummer, Nomenklatur und phys. Daten!
• Über jeden Versuch ist - auch im Vorpraktikum - genau Protokoll zu führen. Die „Versuchs-bezogenen Betriebsanweisung“ ist Bestandteil des Protokolls und muß mit diesem zusammenabgegeben werden. Die Protokolle werden vom zuständigen Assistent korrigiert und testiert, evtl.auch zur Nachbearbeitung oder Wiederholung des Versuchs zurückgegeben. Erst nach dem Testatgilt der Versuch als erfolgreich durchgeführt.
Verbrauchsmaterial und Chemikalien
Benötigtes Verbrauchsmaterial (Präparategläser, Pipetten, Filterpapier, Schläuche etc.) steht in deneinzelnen Sälen zur Selbstbedienung auf. Für die einzelnen Versuche stehen jeweils alle benötigtenChemikalien auf. Der Verbrauch von Chemikalien und Verbrauchsmaterial ist nicht limitiert, wirdaber anteilig mit den Praktikumskosten verrechnet. Es ist also im Sinne jedes einzelnen, denVerbrauch möglichst gering zu halten.

-3-
Weitere Technische Einzelheiten
• Die Praktikumssäle sind vom 30.09. bis 17.10.2003 täglich von 9.00 Uhr bis 18.00 Uhr geöffnet.Diese Öffnungszeiten sind verbindlich, Mittagspause mit Absprache mit den Saalassistenten.Ab dem 20.10. bis 30.10. täglich von 13.00 bis 18.00 Uhr.
• Geräte, die zur Durchführung von Versuchen benötigt werden und nicht in der Platzausrüstungenthalten sind, können in der Saalausleihe ausgeliehen werden.
• Die Saalausleihe ist an den Praktikumstagen vom 30.09.03 bis 17.10.03 von 9.00 bis 10.00, abdem 20.10.03 von 13.00 bis 14.00 Uhr geöffnet. Eine rechtzeitige Versuchsplanung ist deshalbangebracht.
• Um ein effizientes Arbeiten zu ermöglichen, sollten alle zerbrochenen Glaswaren sofort repariertbzw. ergänzt werden. Dies gilt auch für die abschließende Platzabgabe.
• Am Geräteschalter (Glasgeräte) des Fachbereichs Chemie und Pharmazie (Öffnungszeiten sieheAnschlag) werden unter Nennung des Praktikums (3. Sem. Bio/LA, Prof. Reiser) Einzeleinkaufs-karten für Verbrauchsmaterial angelegt
• Aus Sicherheitsgründen ist es verboten, Chemikalien nach Hause mitzunehmen. Die Präparatesind ebenso wie die Klausuren Bestandteil der Praktikumsleistung und werden zur Nachprüfungaufbewahrt.
Platzabgabe
• Der Praktikumsplatz ist spätestens am 30.10.2003 (Saal 6: 17.10.2003) in sauberem Zustand undmöglichst komplett abzugeben. Fehlende Geräte sind soweit möglich vorher zu ergänzen!
• Zur Platzabgabe zählt auch die vollständige Abgabe der aus der Saalausleihe erhaltenen Gegen-stände.
• Der Verbrauch von Chemikalien und gestelltem Verbrauchsmaterial wird über den Normwert ausMitteln der Universität abgegolten. Darüber hinausgehende Beträge werden auf die Studierendenumgelegt.
Die Rechnung setzt sich folgendermaßen zusammen:
1. Einkauf am Geräteschalter (Glasgeräte) und Skriptkosten2. Individueller Glasbruch (fehlende Geräte bei der Platzabgabe)3. Bei Überschreitung des Normwertes für Chemikalien und Verbrauchsmaterial wird zusätzlich
der anteilige Betrag in Rechnung gestellt.
Manuskripte
Vorpraktikum: Die Versuchsvorschriften werden ausgehändigt. Die begleitende Lektüre des Skripts"Einführung in die apparativen Methoden der Organischen Chemie", AusgabeMärz 2001, wird dringend empfohlen. Das Skript kann über das Internet kostenlosgeladen werden (als Adobe Acrobat PDF-Datei).
Hauptpraktikum: G. Märkl, P. Kreitmeier "Organisches Chemisches Praktikum 1 für Studierende derBiologie und des Lehramts vor dem Vordiplom, 2002"; Das Skript kann über dasInternet kostenlos geladen werden (als Adobe Acrobat PDF-Datei).
Alle Skripten und weitere Informationen sind über die Praktikumsseiten erreichbar:
http://www-oc.chemie.uni-regensburg.de/OCP/la/ocp1/index.html
bzw. http://www-oc.chemie.uni-regensburg.de/OCP/bio/ocp1/index.html

-4-
Abschlussklausur und Schein
Um den Schein zu erhalten muss sowohl das Vor- als auch das Hauptpraktikum erfolgreichabgeschlossen sein (mit Testat des Assistenten). Zusätzlich müssen die Klausuren (KlausurOrganische Chemie I, 2. Sem. und die Abschlussklausur zum Praktikum bestanden werden.
Termine:
Die Abschlussklausur zum Organischen Praktikum OC I für Studierende der Lehrämter findet Mittebis Ende Februar 2004 statt, die Abschlussklausur zum Organischen Praktikum OC I für Studierendeder Biologie Ende Dezember 2003. Die genauen Termine werden noch bekanntgegeben.
Regelung für Studierende der Lehrämter:
Die erreichte Punktezahl der Klausur Organische Chemie I (2. Sem.) geht zu 30%, die erreichtePunktezahl der Klausur zum Organischen Praktikum I für Studierende der Lehrämter (3. Sem.) gehtzu 70% in das Gesamtergebnis ein. Insgesamt müssen mindestens 50% der Punkte erreicht werden.
Regelung für Studierende der Biologie (Diplom):
Sowohl die Klausur Organische Chemie I (2. Sem.) als auch die Abschlussklausur zum OrganischenPraktikum OC I für Studierende der Biologie müssen mit jeweils 50% der erreichbaren Punktebestanden werden.
Studierende der Biologie können auch von der Regelung für Lehrämter Gebrauch machen.
-5-
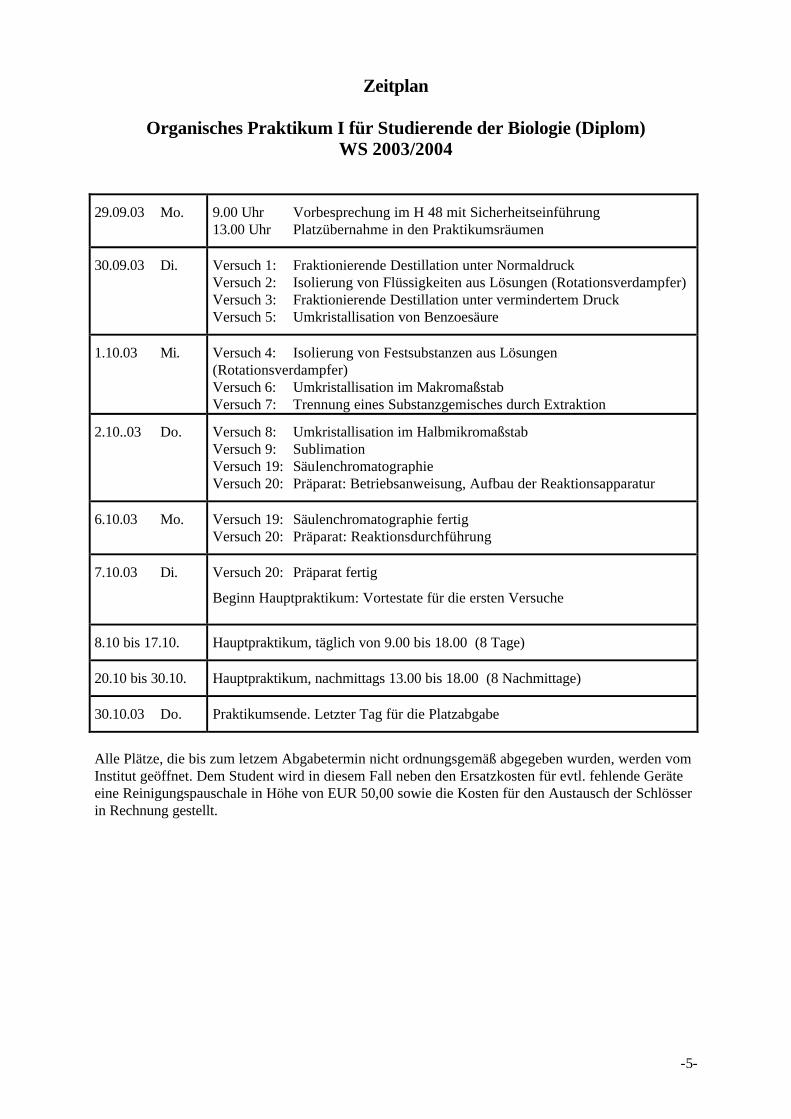
Zeitplan
Organisches Praktikum I für Studierende der Biologie (Diplom)WS 2003/2004
29.09.03 Mo. 9.00 Uhr Vorbesprechung im H 48 mit Sicherheitseinführung13.00 Uhr Platzübernahme in den Praktikumsräumen
30.09.03 Di. Versuch 1: Fraktionierende Destillation unter NormaldruckVersuch 2: Isolierung von Flüssigkeiten aus Lösungen (Rotationsverdampfer)Versuch 3: Fraktionierende Destillation unter vermindertem DruckVersuch 5: Umkristallisation von Benzoesäure
1.10.03 Mi. Versuch 4: Isolierung von Festsubstanzen aus Lösungen(Rotationsverdampfer)Versuch 6: Umkristallisation im MakromaßstabVersuch 7: Trennung eines Substanzgemisches durch Extraktion
2.10..03 Do. Versuch 8: Umkristallisation im HalbmikromaßstabVersuch 9: SublimationVersuch 19: SäulenchromatographieVersuch 20: Präparat: Betriebsanweisung, Aufbau der Reaktionsapparatur
6.10.03 Mo. Versuch 19: Säulenchromatographie fertigVersuch 20: Präparat: Reaktionsdurchführung
7.10.03 Di. Versuch 20: Präparat fertig
Beginn Hauptpraktikum: Vortestate für die ersten Versuche
8.10 bis 17.10. Hauptpraktikum, täglich von 9.00 bis 18.00 (8 Tage)
20.10 bis 30.10. Hauptpraktikum, nachmittags 13.00 bis 18.00 (8 Nachmittage)
30.10.03 Do. Praktikumsende. Letzter Tag für die Platzabgabe
Alle Plätze, die bis zum letzem Abgabetermin nicht ordnungsgemäß abgegeben wurden, werden vomInstitut geöffnet. Dem Student wird in diesem Fall neben den Ersatzkosten für evtl. fehlende Geräteeine Reinigungspauschale in Höhe von EUR 50,00 sowie die Kosten für den Austausch der Schlösserin Rechnung gestellt.

-6-
Universität RegensburgInstitut für Organische Chemie
Versuche zum apparativem Vorpraktikum
für Studierende der Lehrämterund der Biologie (Diplom)
Die Versuche wurden ausgearbeitet und zusammengestellt von P. Kreitmeier,Universität Regensburg, Institut für Organische Chemie
September 2002
Begleitendes Material:
"Einführung in die apparativen Methoden in der Organischen Chemie". Das Skript ist alsOnline-Tutorial oder als ZIP-Archiv über die folgenden Adresse zum Download freiverfügbar:
http://www-oc.chemie.uni-regensburg.de/OCP/methoden/index.html
Allgemeine Hinweise zu den Versuchen zum apparativem Vorpraktikum
Im Rahmen dieses Vorpraktikum sollen die grundlegenden Methoden der präparativen OrganischenChemie in einem Kurspraktikum erlernt werden. Die Beherrschung dieser Methoden ist dieVoraussetzung zur erfolgreichen und sicheren Durchführung von Organisch-Chemischen Präparaten.Dieses Vorpraktikum löst bewusst die einzelnen Arbeitsschritte einzeln heraus. Es ist dadurchmöglich, alle Methoden unter intensiver Anleitung durch den Assistenten zu erlernen und einzuüben.Die Gefahr des Misslingens eines Präparates durch unzureichende Erfahrung - und die Wiederholungdes gesamten Versuchs - besteht somit nicht. Dennoch werden die Zusammenhänge durch dieVerknüpfung von einzelnen Versuchen (Arbeitsschritten) deutlich. Den Abschluss desVorpraktikums bildet ein erstes, einfaches Präparat.Im Rahmen des Vorpraktikums ist es aus Zeitgründen nicht möglich, auf die physikalisch-chemischen Grundlagen der verschiedenen Methoden oder auf spezielle Varianten bzw. Apparatureneinzugehen. Hierzu wird ein Tutorial "Einführung in die apparativen Methoden in der OrganischenChemie" im WWW zur Verfügung gestellt. Die Lektüre dieses Manuskripts wird dringendempfohlen!

-7-
Hinweise zur Sicherheit im Labor
Für das Arbeiten im Chemischen Labor sind ist die Beachtung und Einhaltung der gültigenSicherheitsvorschriften zwingend erforderlich. Eine grobe Mißachtung dieser Vorschriften führt zumsofortigem Ausschluss aus dem Praktikum!Die wichtigsten allgemeinen Sicherheitshinweise werden in der Sicherheitseinführung vor Beginn desPraktikums besprochen. Zur Erinnerung nochmals einige wichtige Punkte:
• Im Labor ist stets die Schutzbrille und ein Laborkittel (100% Baumwolle) zu tragen• Essen, Trinken und Rauchen ist im Labor untersagt.• Die Mitnahme von Lebensmitteln in das Labor ist untersagt• Chemikalien dürfen nur in dafür vorgesehene und vollständig beschriftete Gefäße gelagert
werden. Die Lagerung in Gefäßen, die mit Lebensmittelpackungen verwechselt werden könntenist untersagt!
• Die Mitnahme von Chemikalien aus dem Labor ist verboten.
Darüber hinaus wird nochmals auf die Laboratoriumsordnung der Universität und auf dieeinschlägigen Unfallverhütungsvorschriften hingewiesen. Sie liegen in den Praktikumssälen auf.Spezielle Gefahren der verwendeten Chemikalien und Apparaturen sind unter "Sicherheitshinweise"bei jedem Versuch aufgeführt.

-8-
Versuch 1: Fraktionierende Destillation unter Normaldruck(Abtrennung schwerflüchtiger Verunreinigungen)
Tutorial Kap. 3.2
Apparatur: NS 14-Destillationsapparatur mit 100 ml Destillationskolben mitMagnetrührstab, Claisenbrücke mit Schliffthermometer, Spinne und vier25 ml Vorlagekolben; Magnetrührer mit Ölbad und Ölbadthermometer,Hebebühne.
Aufbau: Die 4 Vorlagekolben werden nummeriert und leer gewogen. DerDestillationkolben wird zusammen mit dem Magnetrührstab gewogen,anschließend werden etwa 30-40 ml einer Verbindung aus Tabelle 1 mitHilfe eines Trichters eingefüllt und der volle Kolben nochmals gewogen.Die Destillationsapparatur wird aufgebaut und auf Spannungsfreiheitüberprüft. Dabei wird der Destillationskolben in einer Höhe befestigt, sodass bei heruntergefahrener Hebebühne der Magnetrührer mit Ölbad geradedaruntergeschoben werden kann. Anschließend werden bei entferntemÖlbad die Wasserschläuche angeschlossen, gesichert und auf Dichtigkeitüberprüft.
Durchführung: Vor Beginn der Destillation muss die Apparatur vom Assistenten abgenommenwerden!Der Destillationskolben wird durch Hochdrehen der Hebebühne möglichst weit in das kalte Ölbadeingetaucht, erst dann wird unter magnetischen Rühren langsam erhitzt, die Ölbadtemperatur sollteetwa 20-30 °C über den Siedepunkt der zu destillierenden Substanz liegen.Wenn der erste Tropfen in den Vorlagekolben fällt wird die Siedetemperatur und dieÖlbadtemperatur abgelesen. Sobald die Siedetemperatur konstant bleibt wird der nächsteVorlagekolben eingedreht und das Destillat so lange gesammelt wie der Siedepunkt konstant bleibt(innerhalb eines Intervalls von 2 °C). Ändert sich die Siedetemperatur muss auf den nächstenVorlagekolben gewechselt werden usw. Dabei wird immer auch die Siede- und Ölbadtemperaturabgelesen.Während der Destillation sollte die Ölbadtemperatur möglichst konstant bleiben, eventuell kann siegegen Ende der Destillation leicht erhöht werden.Die Destillation ist beendet wenn kein Destillat mehr übergeht. Das Ölbad kann dann entfernt und dieWasserkühlung abgestellt werden.Nach dem Abkühlen der Apparatur wird durch Rückwiegen des Destillationskolbens die Masse desDestillationsrückstandes bestimmt. Die Masse der erhaltenen Fraktionen wird ebenfalls durchRückwiegen bestimmt. Von allen Fraktionen wird der Brechungsindex am Refraktometer gemessen.
Protokollführung: Der Aufbau ist kurz zu beschreiben, insbesondere Apparatur- und Kolbengrößen.Die Einwaage wird angegeben, der Destillationsverlauf wird tabellarisch im Destillationsprotokollzusammengestellt. Die Massen und Brechungsindices der erhaltenen Fraktionen werden ebenfalls indas Destillationsprotokoll eingetragen.Aus der Differenz zwischen Einwaage und Summe der Massen aller Fraktionen und desDestillationsrückstandes wird der Destillationsverlust errechnet.Abschließend wird unter Heranziehung aller Daten aus dem Destillationsprotokoll der Hauptlaufbestimmt.

-9-
Tabelle 1: Substanzen für die Destillation unter Normaldruck.
Verbindung Sdp. [°C] nD20 Gefahren-
symbolSicherheitsdaten
(R/S-Sätze)1-Propanol(n-Propanol)
97 1.3850 F, Xi R 11-41-67S 7-16-24-26-39
2-Propanol(iso-Propanol)
82 1.3792 F, Xi R 11-36-67S 7-16-24/25-26
2-Butanol 99 1.3978 Xi R 10-36/37-67S 7/9-13-24/25-26-46
Destillationsprotokoll:
Destillation von: ____________________________
Eingewogene Masse: ________ g
Fraktion Zeit Ölbad Siedetemperatur Masse nD20
1234
Destillationrückstand: _________ gSumme der Massen: _________ gDestillationsverlust: _________ g
Recycling und Entsorgung:
Alle erhaltenen Fraktionen werden wieder in die entsprechenden Flaschen zurückgegeben. DerDestillationsrückstand und in der Apparatur verbliebene Reste werden mit wenig Aceton in denSammelbehälter für halogenfreien organischen Sondermüll A2 gespült.
Sicherheitshinweise:
Vor Beginn des Versuchs muss die Apparatur vom Assistenten abgenommen werden.Alle verwendeten Substanzen sind entzündlich oder leichtentzündlich. Alle Substanzen können dieAugen und die Atmungsorgane reizen, 1-Propanol kann zu ernsten Augenschäden führen. Dämpfekönnen Schläfrigkeit und Benommenheit verursachen.Mögliche Zündquellen entfernen, keine offenen Flammen! Schutzbrille tragen, alle Gefäße sofortwieder verschließen, Substanzen nicht in offenen Gefäßen erhitzen. Hautkontakt vermeiden.Bei Berührung mit den Augen sofort mit der Augendusche spülen (mindestens 15 Minuten). Assistentverständigen und Arzt konsultieren. Beim Verschlucken von Substanzen ebenfalls Assistentverständigen und sofort Arzt hinzuziehen und die Vorschrift mit diesen Hinweisen vorzeigen.

-10-
Versuch 2: Isolierung schwerflüchtiger Flüssigkeiten aus Lösungen(Abdestillation von Lösungsmittel am Rotationsverdampfer)
Tutorial Kap. 3.2.4
Apparatur: Rotationsverdampfer, 250 ml NS 29 Destillationskolben
Aufbau: Der Rotationsverdampfer ist fest aufgebaut.Das Heizbad (Wasserbad) wird auf 60 °C eingestellt. Am Vakuumcontrollerwird der Solldruck so gewählt, dass das zu destillierende Lösungsmitteleinen Siedepunkt von etwa 40 °C erreicht.
Durchführung: Vor der erstmaligen Benutzung des Rotationsverdampfers erfolgt eine Einweisungdurch den Assistenten. Der Destillationskolben darf keine Sprünge oder Schläge (Sternchen)aufweisen, Implosionsgefahr im Vakuum!Ein 250 ml NS 29-Rundkolben wird leer gewogen. Anschließend werden 100 ml einer Lösung ausTabelle 2 mit Hilfe eines Trichters in den Kolben gefüllt (mit Messzylinder abmessen!). Es ist daraufzu achten, dass keine Substanzreste auf die Schlifffläche gelangen.Der Auffangkolben des Rotationsverdampfers muss vor Beginn der Destillation leer sein, um dieVermischung der destillierten Lösungsmittel zu verhindern.Der Destillationskolben wird am Steigrohr des Rotationsverdampfers mit einer Schliffklemmebefestigt (Schliff nicht fetten!), der Motor eingeschaltet und am Vakuumcontroller der gewünschteSolldruck eingestellt. Danach wird der Controller gestartet und der Destillationskolben in das Heizbadabgesenkt.Das Ende der Destillation ist erreicht wenn kein Destillat mehr übergeht.Zum Abschalten wird zuerst der Destillationskolben aus dem Heizbad gefahren, der Motorabgeschaltet und der Vakuumcontroller gestoppt. Anschließend wird das Belüftungsventileingeschaltet. Nach vollständigem Druckausgleich (Druckanzeige beachten!) wird derDestillationskolben abgenommen und der Auffangkolben geleert.Der Destillationskolben wird zurückgewogen und die erhaltene Masse berechnet.
Protokollführung: Die Kolbengröße, eingesetzte Menge (hier in ml) sowie die relevanten Parameterder Destillation (Badtemperatur, Druck) sind anzugeben.Vergleichen Sie die erhaltene Ausbeute mit der aus der Gehaltsangabe zu erwartenden Menge!Folgerungen? Warum darf der Druck nicht niedriger eingestellt werden?
Tabelle 2: Lösungen zur Destillation am Rotationsverdampfer.
Verbindung Sdp. [°C]bei Normaldruck
Gefahren-symbol
Sicherheitsdaten(R/S-Sätze)
Benzylalkohol 205 Xn R 20/22, S 26in Cyclohexan 81 F, Xn, N R 11-38-50/53-65-67
S 9-16-33-60-61-62Benzoesäureethylester 214 - -in Ethanol 78 F R 11, S 7-16
Recycling und Entsorgung:
Das abdestillierte Lösungsmittel wird in in den entsprechenden Sammelbehälter für Recycling-Lösungsmittel gegeben. Der erhaltene Destillationsrückstand wird für Versuch 3 aufbewahrt.

-11-
Sicherheitshinweise:
Vor Beginn des Versuchs muss eine Einweisung in die Bedienung des Rotationsverdampfers durchden Assistent erfolgen. Der verwendete Glaskolben darf keine Sprünge oder Schläge aufweisen:Gefahr der Implosion beim Arbeiten unter Vakuum!Alle verwendeten Lösungsmittel sind leichtentzündlich. Cyclohexan reizt die Haut, ist sehr giftig fürWasserorganismen und kann in Gewässern längerfristig schädliche Wirkungen haben. Cyclohexankann beim Verschlucken Lungenschäden verursachen, seine Dämpfe können Schläfrigkeit undBenommenheit verursachen. Benzylalkohol ist gesundheitsschädlich beim Einatmen und beiBerührung mit der Haut.Mögliche Zündquellen entfernen, keine offenen Flammen! Schutzbrille tragen, alle Gefäße sofortwieder verschließen, Substanzen nicht in offenen Gefäßen erhitzen. Hautkontakt vermeiden.Cyclohexan darf auf keinen Fall in das Abwasser gelangen.Bei Berührung mit den Augen sofort mit der Augendusche spülen (mindestens 15 Minuten). Assistentverständigen und Arzt konsultieren. Beim Verschlucken von Substanzen ebenfalls Assistentverständigen und sofort Arzt hinzuziehen und die Vorschrift mit diesen Hinweisen vorzeigen.

-12-
Versuch 3: Fraktionierende Destillation unter vermindertem Druck
Tutorial Kap. 3.2.3
Apparatur: NS 14-Destillationsapparatur mit 50 ml Destillationskolben mitMagnetrührstab, Claisenbrücke mit Schliffthemometer, Spinne und vier25 ml Vorlagekolben; Magnetrührer mit Ölbad und Ölbadthermometer,Hebebühne, Vakuummessgerät im Seitenschluss.
Aufbau: Die 4 Vorlagekolben werden nummeriert und leer gewogen. DerDestillationkolben wird zusammen mit dem Magnetrührstab gewogen,anschließend wird die aus Versuch 2 erhaltene Flüssigkeit mit Hilfe einesTrichters eingefüllt und der volle Kolben nochmals gewogen.Die Destillationsapparatur wird aufgebaut und auf Spannungsfreiheitüberprüft. Dabei wird der Destillationskolben in einer Höhe befestigt, sodass bei heruntergefahrener Hebebühne der Magnetrührer mit Ölbad geradedaruntergeschoben werden kann. Anschließend werden bei entferntemÖlbad die Wasserschläuche angeschlossen, gesichert und auf Dichtigkeitüberprüft.Anschließend wird die Destillationsapparatur mit einem Vakuumschlauch andie Vakuumleitung angeschlossen (Sicherheitswaschflasche mitBelüftungshahn dazwischenschalten, Vakuummessgerät mit Hilfe eines T-Stückes im Seitenschluss).
Durchführung: Zuerst wird das kalte Ölbad an den Destillationskolben herangefahren und derMagnetrührer eingeschaltet. Erst dann wird der Absperrhahn zur Vakuumleitung langsam geöffnet.Das Destillationsgut darf nicht zu heftig aufschäumen (Lösungsmittelreste!). Erst wenn das Sieden imDestillationskolben nachlässt und sich ein Enddruck < 25 hPa einstellt wird der Destillationskolbenvollständig in das Ölbad getaucht und langsam erhitzt.Die Ölbadtemperatur sollte etwa 20-30 °C über den nach der Faustregel abgeschätzen Siedepunkt derzu destillierenden Substanz liegen.Wenn der erste Tropfen in den Vorlagekolben fällt wird die Siedetemperatur und dieÖlbadtemperatur abgelesen. Sobald die Siedetemperatur konstant bleibt wird der nächsteVorlagekolben eingedreht und das Destillat so lange gesammelt wie der Siedepunkt konstant bleibt(innerhalb eines Intervalls von 2 °C). Ändert sich die Siedetemperatur muss auf den nächstenVorlagekolben gewechselt werden usw. Dabei wird immer auch die Siede- und Ölbadtemperatursowie der Druck abgelesen.Während der Destillation sollte die Ölbadtemperatur möglichst konstant bleiben, eventuell kann siegegen Ende der Destillation leicht erhöht werden. Druckschwankungen führen zu schwankendenSiedepunkten, im Zweifelsfall wird eine neue Fraktion geschnitten.Die Destillation ist beendet wenn kein Destillat mehr übergeht. Das Ölbad kann dann entfernt und dieWasserkühlung abgestellt werden.Nach dem Abkühlen der Apparatur wird durch Rückwiegen des Destillationskolbens die Masse desDestillationsrückstandes bestimmt. Die Masse der erhaltenen Fraktionen wird ebenfalls durchRückwiegen bestimmt. Von allen Fraktionen wird der Brechungsindex am Refraktometer gemessen.
Protokollführung: Der Aufbau ist kurz zu beschreiben, insbesondere Apparatur- und Kolbengrößen.Die Einwaage wird angegeben, der Destillationsverlauf wird tabellarisch in einemDestillationsprotokoll zusammengestellt. Die Massen und Brechungsindices der erhaltenen Fraktionenwerden ebenfalls in das Destillationsprotokoll eingetragen.Aus Differenz zwischen der Einwaage und der Summe der Massen aller Fraktionen und desDestillationsrückstandes wird der Destillationsverlust errechnet.Abschließend wird unter Heranziehung aller Daten aus dem Destillationsprotokoll der Hauptlaufbestimmt.

-13-
-14-
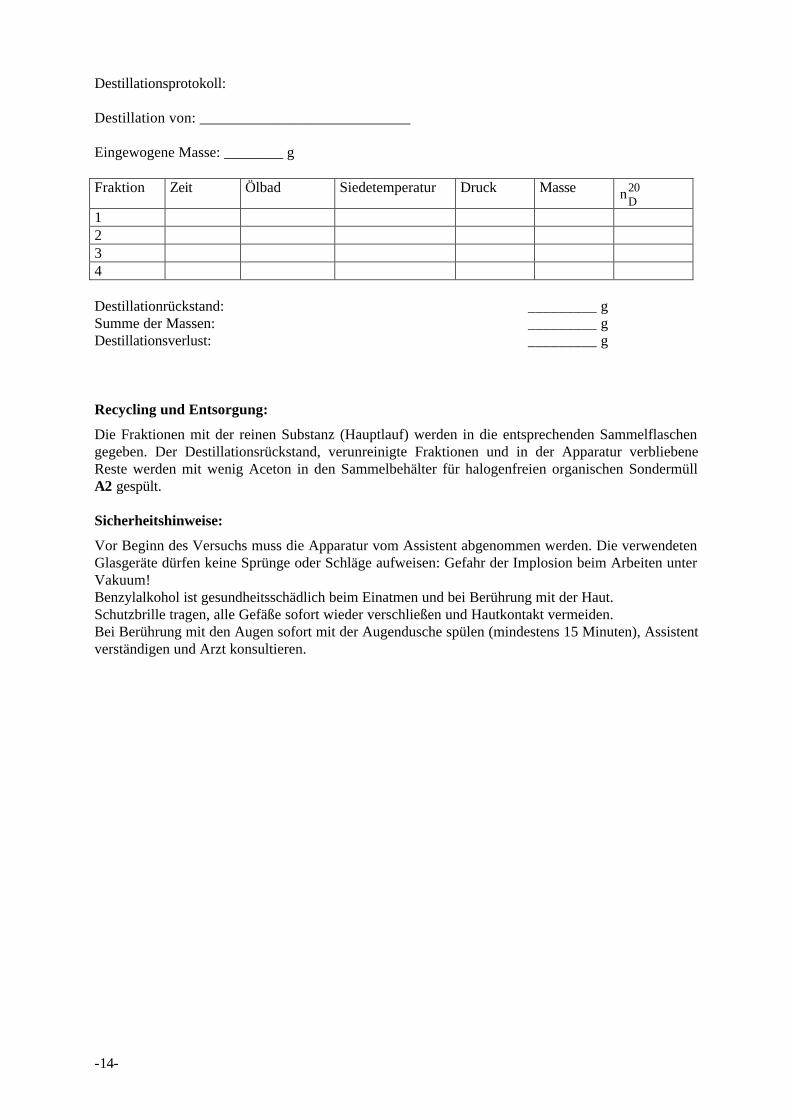
Destillationsprotokoll:
Destillation von: ____________________________
Eingewogene Masse: ________ g
Fraktion Zeit Ölbad Siedetemperatur Druck Masse nD20
1234
Destillationrückstand: _________ gSumme der Massen: _________ gDestillationsverlust: _________ g
Recycling und Entsorgung:
Die Fraktionen mit der reinen Substanz (Hauptlauf) werden in die entsprechenden Sammelflaschengegeben. Der Destillationsrückstand, verunreinigte Fraktionen und in der Apparatur verbliebeneReste werden mit wenig Aceton in den Sammelbehälter für halogenfreien organischen SondermüllA2 gespült.
Sicherheitshinweise:
Vor Beginn des Versuchs muss die Apparatur vom Assistent abgenommen werden. Die verwendetenGlasgeräte dürfen keine Sprünge oder Schläge aufweisen: Gefahr der Implosion beim Arbeiten unterVakuum!Benzylalkohol ist gesundheitsschädlich beim Einatmen und bei Berührung mit der Haut.Schutzbrille tragen, alle Gefäße sofort wieder verschließen und Hautkontakt vermeiden.Bei Berührung mit den Augen sofort mit der Augendusche spülen (mindestens 15 Minuten), Assistentverständigen und Arzt konsultieren.

-15-
Versuch 4: Isolierung von Festsubstanzen aus Lösungen(Abdestillation von Lösungsmittel am Rotationsverdampfer)
Tutorial Kap. 3.2.4
Apparatur: Rotationsverdampfer, 250 ml NS 29 Destillationskolben
Aufbau: Der Rotationsverdampfer ist fest aufgebaut.Das Heizbad (Wasserbad) wird auf 60 °C eingestellt. Am Vakuumcontrollerwird der Solldruck so gewählt, dass das zu destillierende Lösungsmitteleinen Siedepunkt von etwa 40 °C erreicht.
Durchführung: Ein 250 ml NS 29-Rundkolben wird leer gewogen. Anschließend werden 100 mleiner Lösung aus Tabelle 3 mit Hilfe eines Trichters in den Kolben gefüllt (mit Messzylinderabmessen!). Es ist darauf zu achten, dass keine Substanzreste auf die Schlifffläche gelangen.Der Auffangkolben des Rotationsverdampfers muss vor Beginn der Destillation leer sein, um dieVermischung der destillierten Lösungsmittel zu verhindern.Der Destillationskolben wird am Steigrohr des Rotationsverdampfers mit einer Schliffklemmebefestigt (Schliff nicht fetten!), der Motor eingeschaltet und am Vakuumcontroller der gewünschteSolldruck eingestellt. Danach wird der Controller gestartet und der Destillationskolben in das Heizbadabgesenkt.Das Ende der Destillation ist erreicht wenn kein Destillat mehr übergeht.Zum Abschalten wird zuerst der Destillationskolben aus dem Heizbad gefahren, der Motorabgeschaltet und der Vakuumcontroller gestoppt. Anschließend wird das Belüftungsventileingeschaltet. Nach vollständigem Druckausgleich (Druckanzeige beachten!) wird derDestillationskolben abgenommen und der Auffangkolben geleert.Zur Entfernung von Lösungsmittelresten wird der Destillationskolben am Arbeitsplatz direkt an dieVakuumleitung angeschlossen und der Destillationsrückstand etwa 1 h im Vakuum getrocknet.Anschließend wird der Kolben zurückgewogen und Masse der erhaltenen Substanz berechnet.Bestimmen Sie den Schmelzpunkt der erhaltenen Festsubstanz!
Protokollführung: Die Kolbengröße, eingesetzte Menge (hier in ml) sowie die relevanten Parameterder Destillation (Badtemperatur, Druck) sind anzugeben.Vergleichen Sie die erhaltene Ausbeute mit der aus der Gehaltsangabe zu erwartenden Menge!Vergleichen Sie den Schmelzpunkt Ihres Produktes mit den Literaturangaben. Folgerungen?
Tabelle 3: Lösungen zur Destillation am Rotationsverdampfer.
Verbindung Sdp., Schmp.bei Normaldruck
Gefahren-symbol
Sicherheitsdaten(R/S-Sätze)
9-Fluorenon Schmp. 81-83 °C R -, S -in Essigsäureethylester(Ethylacetat)
Sdp. 77 °C F, Xi R 11-36-66-67S 16-26-33
Benzil Schmp. 94-95 °C Xi R 36/38in Essigsäureethylester(Ethylacetat)
Sdp. 77 °C F, Xi R 11-36-66-67S 16-26-33

-16-
Recycling und Entsorgung:
Das abdestillierte Lösungsmittel wird in den entsprechenden Sammelbehälter für Recycling-Lösungsmittel gegeben. Die erhaltene Festsubstanz wird für Versuch 6 aufbewahrt.
Sicherheitshinweise:
Vor Beginn des Versuchs muss eine Einweisung in die Bedienung des Rotationsverdampfers durchden Assistent erfolgen. Der verwendete Glaskolben darf keine Sprünge oder Schläge aufweisen:Gefahr der Implosion beim Arbeiten unter Vakuum!Essigsäureethylester ist leichtentzündlich, reizt die Augen und kann beim VerschluckenLungenschäden verursachen, seine Dämpfe können Schläfrigkeit und Benommenheit verursachen.Benzil reizt die Augen und die Haut.Mögliche Zündquellen entfernen, keine offenen Flammen! Schutzbrille tragen, alle Gefäße sofortwieder verschließen, Substanzen nicht in offenen Gefäßen erhitzen. Hautkontakt vermeiden,gegebenenfalls Einmal-Schutzhandschuhe verwenden.Bei Berührung mit den Augen sofort mit der Augendusche spülen (mindestens 15 Minuten). Assistentverständigen und Arzt konsultieren. Beim Verschlucken von Substanzen ebenfalls Assistentverständigen. Sofort Arzt hinzuziehen und die Vorschrift mit diesen Hinweisen vorzeigen.

-17-
Versuch 5: Umkristallisation von Benzoesäure aus Wasser
Tutorial Kap. 5.1
Apparatur: Erlenmeyerkolben mit Uhrglas, elektrische Heizplatte.
Durchführung: In einem 250 ml-Erlenmeyerkolben werden 6.70 gBenzoesäure eingewogen, 3-5 Siedesteinchen dazugegeben und mit etwa 100ml Wasser versetzt. Der Erlenmeyerkolben wird mit einem Uhrglasabgedeckt und im Abzug auf einer elektrischen Heizplatte erwärmt. In derSiedehitze löst sich schließlich die gesamte Benzoesäure auf, es entsteht eineklare Lösung.Der Erlenmeyerkolben wird von der Heizplatte genommen und zumAbkühlen auf einen Korkring gestellt. Die gesättigte, heiße Lösung lässt manlangsam auf Raumtemperatur abkühlen, dabei kristallisiert die Benzoesäurewieder aus. Die Kristallisation wird vervollständigt, indem man denErlenmeyerkolben mit der auskristallisierten Benzoesäure noch einige Zeit inein Eisbad stellt.Der erhaltene Kristallbrei wird hierauf auf einem Büchnertrichter(Ø ~5 cm) aus Porzellan mit eingelegtem Rundfilter (vorher mit etwasWasser anfeuchten!) mit Hilfe einer Absaugflasche am Vakuum abgesaugt,portionsweise mit zweimal je 10 ml eiskaltem Wasser gewaschen undschließlich gut trockengesaugt. Die erhaltenen Kristalle werden möglichstquantitativ in eine tarierte Porzellanschale überführt und gründlich imExsikkator über Kieselgel (mit Feuchtigkeitsindikator, "Orange-Gel") überNacht getrocknet.Bestimmen Sie die Ausbeute und den Schmelzpunkt der umkristallisiertenBenzoesäure.
Protokollführung: Die eingesetzten Mengen (Substanz in g, Lösungsmittel in ml), Beobachtungensowie erhaltene Masse und Schmelzpunkt der umkristallisierten Benzoesäure sind anzugeben.Vergleichen Sie die erhaltene Ausbeute mit der eingesetzten Substanzmenge, berechnen Sie dieAusbeute der Umkristallisation. Wo befindet sich die verlorene Substanz? Was könnten Sie bei zuwenig Ausbeute tun?Vergleichen Sie den Schmelzpunkt Ihres Produktes mit den Literaturangaben. Folgerungen?
Recycling und Entsorgung:
Die Mutterlauge (= Filtrat beim Absaugen) wird in den Sammelbehälter für wässrigen organischenRecycling-Lösungsmittel gegeben. Die erhaltene Benzoesäure wird in die Vorratsflaschezurückgegeben.
Sicherheitshinweise:
Die Benzoesäure-Wasser-Mischung darf nur im Abzug erhitzt werden.Benzoesäure ist gesundheitsschädlich beim Verschlucken und reizt die Augen. Berührung mit derHaut vermeiden.

-18-
Versuch 6: Umkristallisation im Makromaßstab
Tutorial Kap. 5.2
Apparatur: 100 ml NS 29 Rundkolben, NS 29 Rückflusskühler,Magnetrührstab und Magnetrührer mit Hebebühne.
Aufbau: Der Rundkolben mit Magnetrührstab wird in einer Höhe befestigt, sodass bei heruntergefahrener Hebebühne der Magnetrührer mit Ölbad geradedaruntergeschoben werden kann. Anschließend wird mit Hilfe einesPulvertrichters die abgewogene Festsubstanz aus Versuch 4 eingefüllt. DerRückflußkühler wird aufgesetzt und bei entferntem Ölbad die Wasserschläucheangeschlossen, gesichert und auf Dichtigkeit überprüft.
Durchführung: Über einen Trichter wird etwa zwei Drittel der voraussichtlichbenötigten Menge an Lösungsmittel zugegeben (siehe Tab. 4). Das Heizbadwird unter die Apparatur geschoben und der Kolben bis knapp unter denFlüssigkeitsspiegel eingetaucht. Das Heizbad wird bis etwa 20 °C über denSiedepunkt des Lösungsmittels erhitzt, das Lösungsmittel muss refluxieren. Hatsich auch nach 5-10 Minuten unter Rückfluss noch keine klare Lösung gebildetwird weiteres Lösungsmittel in kleinen Portionen zugegeben. Dazwischen mussstets einige Minuten refluxiert werden. Wenn sich alles gelöst hat notiert mandie Menge an benötigten Lösungsmittel. Das Heizbad wird abgeschaltet undentfernt. Die heiße Lösung läßt man ohne Rühren langsam aufRaumtemperatur abkühlen, dabei scheiden sich Kristalle ab. Die Kristallisationwird durch etwa halbstündiges Kühlen im Eisbad vervollständigt.Der erhaltene Kristallbrei wird auf einem Büchnertrichter (Ø ~ 5 cm) ausPorzellan mit eingelegtem Rundfilter (vorher mit etwas Lösungsmittelanfeuchten!) mit Hilfe einer Absaugflasche am Vakuum abgesaugt, nochmalsmit wenig eiskaltem Lösungsmittel gewaschen und trocken gesaugt. DieMutterlauge wird zunächst aufbewahrt (Beschriftet und Verschlossen!) und erstnach der Bestimmung der Ausbeute verworfen! Die erhaltenen Kristallewerden möglichst quantitativ in eine tarierte Porzellanschale überführt undgründlich im Vakuum-Exsikkator bis zur Gewichtskonstanz getrocknet. ZumSchluss wird der Schmelzpunkt des umkristallisierten Feststoffs bestimmt.
Protokollführung: Die verwendeten Geräte (Art und Größe), die eingesetzten Mengen (Substanz ing, benötigtes Lösungsmittel in ml), erhaltenen Mengen, Farbe und Aussehen sowie der Schmelzpunktdes umkristallisierten Produkts sind anzugeben.Entscheiden Sie anhand der Massenbilanz, ob eine weitere Aufarbeitung der Mutterlauge nötig wäre.Wie würden Sie diese Aufarbeitung durchführen?Vergleichen Sie den Schmelzpunkt der Substanz vor und nach der Umkristallisation!
Recycling und Entsorgung:
Die Mutterlauge (= Filtrat beim Absaugen) wird in den Sammelbehälter für wässrigen organischenSonderabfall gegeben. Die erhaltenen reinen Feststoffe werden in die aufstehenden Sammelflaschengegeben.

-19-
Tabelle 4: Substanzen und Lösungsmittel zur Umkristallisation im Makromaßstab.
Verbindung Schmp. Solvens,benötigte Menge
Sdp. des Solvens
9-Fluorenon Schmp. 81-83 °C Ethanol,ca. 2 ml/g
78 °C
Benzil Schmp. 94-95 °C Cyclohexan,ca 2 ml/g
81 °C
Sicherheitshinweise:
Vor Beginn des Versuchs muss die Apparatur vom Assistenten abgenommen werden. Keine offenenFlammen in der Umgebung. Lösungsmittel dürfen nur Apparaturen mit Rückflusskühler erhitztwerden.Alle verwendeten Lösungsmittel sind leichtentzündlich. Cyclohexan reizt die Haut und ist sehr giftigfür Wasserorganismen, es kann in Gewässern längerfristig schädliche Wirkungen haben. Benzil reiztdie Augen und die Haut.Mögliche Zündquellen entfernen, keine offenen Flammen! Schutzbrille tragen, alle Gefäße sofortwieder verschließen, Substanzen nicht in offenen Gefäßen erhitzen. Hautkontakt vermeiden,gegebenenfalls Einmal-Schutzhandschuhe verwenden.Bei Berührung mit den Augen sofort mit der Augendusche spülen (mindestens 15 Minuten). Assistentverständigen und Arzt konsultieren. Beim Verschlucken von Substanzen ebenfalls Assistentverständigen und sofort Arzt hinzuziehen. und die Vorschrift mit diesen Hinweisen vorzeigen.Cyclohexan darf auf keinen Fall in das Abwasser gelangen.

-20-
Versuch 7: Trennung eines Substanzgemisches durch Extraktion,Trocknen von organischen Lösungen
Tutorial Kap. 7.2
Apparatur: Scheidetrichter, Becherglas, Erlenmeyerkolben, 250 ml NS 29Rundkolben, Büchnertrichter mit Absaugflasche und Rotationsverdampfer.
Die aufstehende Lösung enthält je 2.00 g 4-Methylbenzoesäure und rac.Borneol pro 100 ml Essigsäureethylester (Ethylacetat).
Durchführung: Von der aufstehenden Lösung des Substanzgemischeswerden 100 ml Lösung abgemessen und über einen Trichter in einen 250 mlScheidetrichter gegeben. Anschließend werden 25 ml 2 M Natronlaugehinzugegeben, der Scheidetrichter mit einem Kunststoffstopfen verschlossenund gut geschüttelt. Man wartet die Phasentrennung ab und trennt diewässrige Phase (untere Phase) in ein 250 ml Becherglas ab. Die imScheidetrichter verbliebene organische Phase wird nochmals mit 25 ml 2 MNatronlauge extrahiert und die wässrige Phase abgetrennt.Die organische Phase wird in einen Erlenmeyerkolben abgelassen und mitNatriumsulfat getrocknet. Dazu wird portionsweise solange wasserfreiesNatriumsulfat zugegeben bis sich beim Umschütteln keine Klumpen bilden.Der Erlenmeyerkolben wird mit einem Uhrglas abgedeckt, beschriftet undetwa 2 h zum Trocknen beiseite gestellt, gelegentlich kann umgeschütteltwerden.
Die wässrigen Phasen werden vereinigt und vorsichtig mithalbkonzentrierter Salzsäure angesäuert, dabei bildet sich ein farbloserNiederschlag. Zur Vervollständigung der Abscheidung wird etwa 30Minuten im Eisbad gekühlt, anschließend wird der Niederschlag über einenHirschtrichter abgesaugt, mit wenig Eiswasser gewaschen und nochmals guttrockengesaugt. Der Filterrückstand (die rohe 4-Methylbenzoesäure) wird ineine tarierte Porzellanschale überführt und im Exsikkator über Kieselgel(Orange-Gel) bis zur Gewichtskonstanz getrocknet.
Von der organischen Phase (sie darf nicht mehr trübe sein) wird mit Hilfeeines Alihn'schen Rohrs und Witt'schen Topfes vom Trockenmittel direkt ineinen tarierten 250 ml Rundkolben abfiltriert und anschließend dasLösungsmittel am Rotationsverdampfer abdestilliert.Zur Entfernung von Lösungsmittelresten wird der Destillationskolben amArbeitsplatz direkt an die Vakuumleitung angeschlossen und derDestillationsrückstand etwa 1 h im Vakuum getrocknet. Anschließend wirdder Kolben zurückgewogen und Masse der erhaltenen Substanz (= rohesBorneol) berechnet.Von den beiden erhaltenen Produkten wird jeweils der Schmelzpunktbestimmt. Die 4-Methylbenzoesäure wird durch Umkristallisation weitergereinigt (Versuch 8), das Borneol wird im Versuch 9 durch Sublimationgereinigt.
Protokollführung: Die verwendeten Geräte (Art und Größe), die eingesetzten Mengen (Substanz ing, benötigtes Lösungsmittel in ml), erhaltenen Mengen, Farbe und Aussehen sowie der Schmelzpunktder erhaltenen Produkte sind anzugeben.Entscheiden Sie anhand der Massenbilanz, ob die Trennung erfolgreich war. Erklären Sie den Ablaufder Trennung. Wie würden Sie vorgehen, wenn die Mischung als weitere Komponente eine basischeSubstanz (z.B. ein Amin) enthalten würde?

-21-
Recycling und Entsorgung:
Das abdestillierte Lösungsmittel wird in den Sammelbehälter für Recycling-Ethylacetat gegeben. DasTrockenmittel wird in den Sonderabfallbehälter für Feststoffe gegeben (blaues Fass im Labor). DieMutterlauge (= Filtrat beim Absaugen) wird in den Sammelbehälter für wässrigen organischenSonderabfall gegeben. Die erhaltenen Feststoffe werden in den Versuchen 8 und 9 weiter verwendet.
Tabelle 5: Substanzen zur Extraktion.
Verbindung Gefahrensymbol R- und S-Sätze Schmp. , Sdp.
4-Methylbenzoesäure Xn R 22-36S -
Schmp. 180-183 °C
rac-Borneol - R -, S - Schmp. 205-208 °CEssigsäureethylester F, Xi R 11-26-66-67
S 16-26-33Sdp. 77 °C
Natronlauge (2 M) C R 35S 26-36/37/39-45
Salzsäure (halbkonz.) C R 34-37S 26-36/37/39-45
Sicherheitshinweise:
Das Ausschütteln erfolgt im Abzug. Beim Belüften des Scheidtrichters den Auslauf immer gegen diePrallwand des Abzugs richten. Der verwendete Glaskolben zum Abrotieren darf keine Sprünge oderSchläge aufweisen: Gefahr der Implosion beim Arbeiten unter Vakuum!Essigsäureethylester ist leicht entzündlich, reizt die Augen und kann beim VerschluckenLungenschäden verursachen, seine Dämpfe können Schläfrigkeit und Benommenheit verursachen. 4-Methylbenzoesäure ist gesundheitsschädlich beim Verschlucken und reizt die Augen. Die verwendete2 M Natronlauge und die halbkonzentrierte Salzsäure sind ätzend.Mögliche Zündquellen entfernen, keine offenen Flammen beim Umgang mit Essigsäure-ethylester! Schutzbrille tragen, alle Gefäße sofort wieder verschließen, Substanzen nicht inoffenen Gefäßen erhitzen. Hautkontakt vermeiden, gegebenenfalls Einmal-Schutzhandschuheverwenden.Bei Berührung mit den Augen sofort mit der Augendusche spülen (mindestens 15 Minuten). Assistentverständigen und Arzt konsultieren. Beim Verschlucken von Substanzen ebenfalls Assistentverständigen und sofort Arzt hinzuziehen. und die Vorschrift mit diesen Hinweisen vorzeigen.

-22-
Versuch 8: Umkristallisation im Halbmikromaßstab
Tutorial Kap. 5.3
Apparatur: 25 ml NS 14 Rundkolben, NS 14 Rückflusskühler,Magnetrührstab und Magnetrührer mit Hebebühne.
Aufbau: Der Rundkolben mit Magnetrührstab wird in einer Höhe befestigt,so dass bei heruntergefahrener Hebebühne der Magnetrührer mit Ölbadgerade daruntergeschoben werden kann. Anschließend wird mit Hilfe einesPulvertrichters die abgewogene rohe 4-Methylbenzoesäure aus Versuch 7eingefüllt. Der Rückflußkühler wird aufgesetzt und bei entferntem Ölbad dieWasserschläuche angeschlossen, gesichert und auf Dichtigkeit überprüft.
Durchführung: Über einen Trichter wird etwa zwei Drittel dervoraussichtlich benötigten Menge an Ethanol zugegeben (benötigte Menge:ca 3 ml/g Rohprodukt). Das Heizbad wird unter die Apparatur geschobenund der Kolben bis knapp unter den Flüssigkeitsspiegel eingetaucht. DasHeizbad wird bis etwa 20 °C über den Siedepunkt des Lösungsmittelserhitzt, das Lösungsmittel muss refluxieren. Hat sich auch nach 5-10Minuten unter Rückfluss noch keine klare Lösung gebildet wird weiteresLösungsmittel in kleinen Portionen zugegeben. Dazwischen muss stets einigeMinuten refluxiert werden. Hat sich alles gelöst notiert man die Menge anbenötigten Lösungsmittel. Das Heizbad wird abgeschaltet und entfernt. Dieheiße Lösung läßt man ohne Rühren langsam auf Raumtemperaturabkühlen, dabei scheiden sich Kristalle ab. Die Kristallisation wird durchetwa halbstündiges Kühlen im Eisbad vervollständigt.
Der erhaltene Kristallbrei wird auf einem Hirschtrichter aus Porzellan miteingelegtem Rundfilter (vorher mit etwas Lösungsmittel anfeuchten!) mitHilfe einer Absaugflasche am Vakuum abgesaugt, nochmals mit wenigeiskaltem Lösungsmittel gewaschen und trocken gesaugt. Die Mutterlaugewird zunächst aufbewahrt (Beschriftet und Verschlossen!) und erst nach derBestimmung der Ausbeute verworfen! Die erhaltenen Kristalle werdenmöglichst quantitativ in eine tarierte Porzellanschale überführt und gründlichim Vakuum-Exsikkator bis zur Gewichtskonstanz getrocknet. Zum Schlusswird der Schmelzpunkt der umkristallisierten 4-Methylbenzoesäurebestimmt.
Protokollführung: Die verwendeten Geräte (Art und Größe), die eingesetzten Mengen (Substanz ing, benötigtes Lösungsmittel in ml), erhaltenen Mengen, Farbe und Aussehen sowie der Schmelzpunktdes umkristallisierten Produkts sind anzugeben.Entscheiden Sie anhand der Massenbilanz, ob eine weitere Aufarbeitung der Mutterlauge nötig wäre.Wie würden Sie diese Aufarbeitung durchführen?Vergleichen Sie den Schmelzpunkt der Substanz vor und nach der Umkristallisation!

-23-
Recycling und Entsorgung:
Die Mutterlauge (= Filtrat beim Absaugen) wird in den Sammelbehälter für organischen Sonderabfallgegeben. Die erhaltene reine 4-Methylbenzoesäure wird in den aufstehenden Sammelbehältergegeben.
Sicherheitshinweise:
Ethanol ist leicht entzündlich. 4-Methylbenzoesäure ist gesundheitsschädlich beim Verschlucken undreizt die Augen.Mögliche Zündquellen entfernen, keine offenen Flammen beim Umgang mit Ethanol! Schutzbrilletragen, alle Gefäße sofort wieder verschließen, Substanzen nicht in offenen Gefäßen erhitzen.Hautkontakt vermeiden, gegebenenfalls Einmal-Schutzhandschuhe verwenden.Bei Berührung mit den Augen sofort mit der Augendusche spülen (mindestens 15 Minuten). Assistentverständigen und Arzt konsultieren. Beim Verschlucken von Substanzen ebenfalls Assistentverständigen und sofort Arzt hinzuziehen. und die Vorschrift mit diesen Hinweisen vorzeigen.

-24-
Versuch 9: Sublimation
Tutorial Kap. 6.3
Apparatur: Sublimationsapparatur, Heizbad mit Hebebühne.
Aufbau: In das tarierte Sublimationsgefäß wird das aus Versuch 7 erhaltene roheBorneol eingewogen und mit etwas Glaswolle abgedeckt. Das Sublimationsgefäßwird in einer Höhe befestigt, so dass bei heruntergefahrener Hebebühne derMagnetrührer mit Ölbad gerade daruntergeschoben werden kann. Der Kühlfingerwird eingesetzt. Achten Sie darauf, dass der Kühlfinger möglichst weit in das dasSublimationsgefäß ragt, die Glaswolle aber nicht berührt! Bei entferntem Ölbadwerden die Wasserschläuche angeschlossen, gesichert und auf Dichtigkeitüberprüft.
Anschließend wird die Destillationsapparatur mit einem Vakuumschlauch an die Vakuumleitungangeschlossen (Sicherheitswaschflasche mit Belüftungshahn dazwischenschalten, Vakuummeßgerätmit Hilfe eines T-Stückes im Seitenschluss).
Durchführung: Der Absperrhahn zur Vakuumleitung wird langsam geöffnet. Erst wenn sich einEnddruck < 25 hPa einstellt hat wird das Sublimationsgefäß soweit wie möglich vollständig in dasÖlbad getaucht und langsam erhitzt. Die Ölbadtemperatur darf auf keinen Fall den Schmelzpunkt derzu sublimierenden Substanz erreichen.Im Verlauf der Sublimation scheidet sich die zu sublimierende Substanz kristallin am Kühlfinger ab.Die Sublimation ist beendet, wenn sich im Sublimationsgefäß nur noch Verunreinigungen befinden.Das Heizbad wird entfernt und die Sublimationsapparatur nach dem Abkühlen vom Vakuum getrenntund vorsichtig belüftet. Der Sublimationsfinger wird vorsichtig herausgezogen und die abgeschiedeneSubstanz mit Hilfe eines Spatels sorgfältig in eine tarierte Porzellanschale abgeschabt. Von dergereinigten Substanz wird die Ausbeute und der Schmelzpunkt bestimmt. Das Sublimationsgefäßwird zusammen mit dem verbliebenen Rückstand zurückgewogen und eine Massenbilanz derSublimation erstellt.Ist der Endpunkt der Sublimation nur schwer zu erkennen wird die Sublimation nach einiger Zeitunterbrochen, die bis dahin abgeschiedene Substanz isoliert und weiter sublimiert. Die Sublimationist beendet, wenn sich keine weitere Substanz mehr abscheidet.
Protokollführung: Die verwendeten Geräte (Art und Größe), die eingesetzten Mengen (Substanz ing, benötigtes Lösungsmittel in ml), erhaltenen Mengen, Farbe und Aussehen sowie der Schmelzpunktdes sublimierten Produkts sind anzugeben.Vergleichen Sie den Schmelzpunkt und Aussehen der Substanz vor und nach der Sublimation!
Recycling und Entsorgung:
Das erhaltene reine Borneol wird in den aufstehenden Sammelbehälter gegeben. DerSublimationsrückstand wird mit etwas Aceton in den Sammelbehälter für organischen Sonderabfallgespült.
Sicherheitshinweise:
Vor Beginn des Versuchs muss die Apparatur vom Assistent abgenommen werden. Die verwendetenGlasgeräte dürfen keine Sprünge oder Schläge aufweisen: Gefahr der Implosion beim Arbeiten unterVakuum!

-25-
Versuch 19: Säulenchromatographische Trennung einesFarbstoffgemisches
Tutorial Kap. 8.3
Eine Mischung von 100 mg Tetraphenylcyclopentadienon und 100 mg 2,4-Dinitrophenol, gelöst in 2ml Ethylacetat soll chromatographisch getrennt werden.
Verwendete Chemikalien
2,4-Dinitrophenol R 23/24/25-33, S 28-36/37. Schmp. 114 °CTetraphenylcyclopentadienon R -, S -. Schmp. 219-221 °CCyclohexan R 11-38-50/53-65-67, S 9-16-33-60-61-62
Sdp. 81 °CEthylacetat (Essigsäureethylester) R 11-36-66-67, S 16-26-33. Sdp. 77 °C
Vorbereitung: Die Chromatographiesäule mit Hahn (Ø 2cm, Länge 30 cm) wird mit einer NS 14-Klammer senkrecht mit dem Hahn nach unten so an einer Stativstange befestigt, dass man nochbequem einen 250 ml Erlenmeyerkolben darunter stellen kann. In die Verjüngung der Säule zumHahn wird ein kleiner Wattebausch eingebracht, der das Auslaufen der Säulenfüllung verhindern soll.Dazu wird der Wattebausch oben mit einem Glasstab in die Säule gestopft und durch kurzes Anlegeneines Wasserstrahlvakuums über den geöffneten Hahn und verschließen der oberen Öffnung mit demHandballen, der dann mehrmals ruckartig entfernt wird, nach unten gebracht. Danach wird derAuslaufhahn geschlossen und die Säule zu etwa 1/3 mit der aufstehenden Laufmittelmischung(Cyclohexan/Ethylacetat 1:1) gefüllt und ein leerer 250 ml Erlenmeyerkolben untergestellt. Es mußdarauf geachtet werden, daß sich keine Luftblase im Wattebausch oder am Auslauf festsetzt.In einem 250 ml Becherglas wird nun aus 30 g Kieselgel (=75 ml, mit dem Meßzylinder abmessen!)und 70 ml der Laufmittelmischung (Cyclohexan/Ethylacetat 1:1) durch Rühren mit dem Glasstab eineSuspension hergestellt.Dieser dünne Brei wird nun langsam durch einen Trichter (mit breitem Auslauf) in die zu 1/3 gefüllteSäule eingegossen, dabei wird der Auslaufhahn soweit geöffnet, daß das Laufmittel in dem Maßausläuft, wie die Suspension oben zufließt. Dabei darf die Säule nie trocken laufen! Dasausgeflossene Laufmittel kann zum Ausspülen des Becherglases verwendet werden.Um Risse und Luftblasen in der Säule zu verhindern, wird während des Absetzens der Säulenfüllunggleichmäßig von allen Seiten an die Säule geklopft.Zum Schluß wird die Säulenfüllung vorsichtig mit einer etwa 1 cm dicken Seesandschicht abgedeckt.
Durchführung: Zur eigentlichen Chromatographie wird das Laufmittel in der Säule langsam soweitabgelassen, daß die Oberkannte des Flüssigkeitsspiegels bündig mit der Oberkannte der Seesand-schicht ist. Danach werden 2 ml des aufstehenden Farbstoffgemisches mit der Meßpipetteentnommen, in ein Präparategläschen gegeben und von dort mit einer Tropfpipette vorsichtig rundumentlang der Innenwand der Säule auf die Seesandschicht aufgetragen Das Präparateglas und dieTropfpipette werden zum Schluß mit etwas Laufmittel nachgespült.Nun wird der Säulenhahn vorsichtig geöffnet und das Laufmittel tropfenweise abgelassen bis derFlüssigkeitsspiegel gerade wieder die Seesandoberkannte erreicht hat. Dann wird mit etwa 2Pipettenfüllungen nachgespült, wieder abgelassen und nochmals nachgespült, bis die Substanz völligauf die Säule aufgezogen ist.Zur Entwicklung des Chromatogramms werden nun etwa 75 ml Laufmittel (Cyclohexan/ Ethylacetat1:1) portionsweise nachgefüllt und ein konstanter Auslauf von etwa 2 - 3 ml/ Minute eingestellt.Dabei darf die Säule nie trockenlaufen!Die auf der Säule zuerst laufende weinrote Bande enthält das Tetraphenylcyclopentadienon. Daszunächst farblose Eluat wird in einem Erlenmeyerkolben aufgefangen und verworfen. Sobald die

-26-
weinrote Zone in die Nähe des Auslaufs kommt, wird auf ein neues Auffanggefäß gewechselt unddiese Fraktion aufgefangen.Das 2,4-Dinitrophenol wird gleichzeitig als gelbe Bande entwickelt: Sobald eine Farbänderung amAuslauf beobachtet werden kann (nach etwa 50 ml weinroter Fraktion), wechselt man auf ein neuesAuffanggefäß und eluiert das Dinitrophenol mit ca. 200 ml reinem Ethylacetat. Bei Unsicherheit beimFraktionswechsel kann auch ein "Zwischenlauf" geschnitten werden. Nach dem Verbrauch der 200ml Ethylacetat wird die Entwicklung der Chromatographie abgebrochen.
Die beiden erhaltenen Fraktionen werden in je einen tarierten NS 29-Rundkolben geeigneter Größe(100 oder 250 ml) gefüllt und jeweils das Lösungsmittel am Rotationsverdampfer abdestilliert. ZurEntfernung von Lösungsmittelresten wird der Destillationskolben am Arbeitsplatz direkt an dieVakuumleitung angeschlossen und der Destillationsrückstand etwa 1 h im Vakuum getrocknet.Anschließend werden die Kolben zurückgewogen und Masse der erhaltenen Produkte berechnet. Vonbeiden Produkten werden zur Reinheitsbestimmung und Charakterisierung die Schmelzpunktebestimmt.
Protokollführung: Die verwendeten Geräte und Mengen (Substanz, Kieselgel, Laufmittel),erhaltenen Mengen, Farbe und Aussehen sowie der Schmelzpunkt des getrennten Produkts sindanzugeben. Beschreiben Sie den Verlauf der Trennung.Vergleichen Sie die Schmelzpunkte der erhaltenen Substanzen mit den Literaturangaben. Ist eineweitere Reinigung notwendig? Welche Reinigungsmethode würden Sie wählen?
Recycling und Entsorgung:
Die abdestillierten und übriggebliebenen Lösungsmittel (Etylacetat und die Mischung Cyclo-hexan/Ethylacetat) werden in den aufgestellten Recycling-Sammelbehälter "LösungsmittelChromatographie" gegeben.Die Chromatographiesäule läßt man trockenlaufen und stellt sie mit geöffneten Hahn nach obensenkrecht in den Abzug, darunter wird eine Kristallisierschale gestellt. Das belegte Kieselgel rieseltbeim Trocknen in die Kristallisierschale und kann danach in den aufgestellten Sammelbehälter für"Kieselgelabfall aus der Chromatographie" gegeben werden. Das Azobenzol und Nitrophenol wirdmit etwas Aceton in den Sammelbehälter für halogenfreien organischen Sondermüll A2 gespült.
Sicherheitshinweise:
Vor Beginn des Versuchs muß darauf geachtet werden, daß alle offenen Flammen (Bunsenbrenner)abgeschaltet werden! Die Rundkolben zum Abdestillieren am Rotationsverdampfer dürfen keineSprünge oder Schläge aufweisen: Gefahr der Implosion beim Arbeiten unter Vakuum!Cyclohexan und Ethylacetat ist leichtentzündlich, sie können die Atmungsorgane reizen. 2,4-Dinitrophenol ist giftig beim Einatmen, Verschlucken und Berührung mit der Haut. Es besteht dieGefahr kummulativer Wirkungen. Cyclohexan und Ethylacetat können beim VerschluckenLungenschäden verursachen, die Dämpfe können Schläfrigkeit und Benommenheit verursachen.Wiederholter Hautkontakt mit Ethylacetat kann zu spröder und rissiger Haut führen. Cyclohexan istsehr giftig für Wasserorganismen und kann in Gewässern längerfristig schädliche Wirkungen haben.Beim Arbeiten mit 2,4-Dinitrophenol Einmal-Schutzhandschuhe tragen. Hautkontakt mit Ethylacetatebenfalls vermeiden. Alle Vorratsflaschen sofort nach Gebrauch wieder verschließen. Die Substanzenund Lösungsmittel dürfen auf keinen Fall in das Abwasser gelangen.Bei Berührung von Substanzen mit den Augen sofort mit viel Wasser ausspülen (Augendusche).Wenn 2,4-Dinitrophenol auf die Haut gelangt sofort mit viel Wasser abwaschen. Beim Verschluckenvon Substanzen kein Erbrechen auslösen. Bei Unfall sofort Arzt hinzuziehen und diese Vorschriftvorzeigen.
-27-
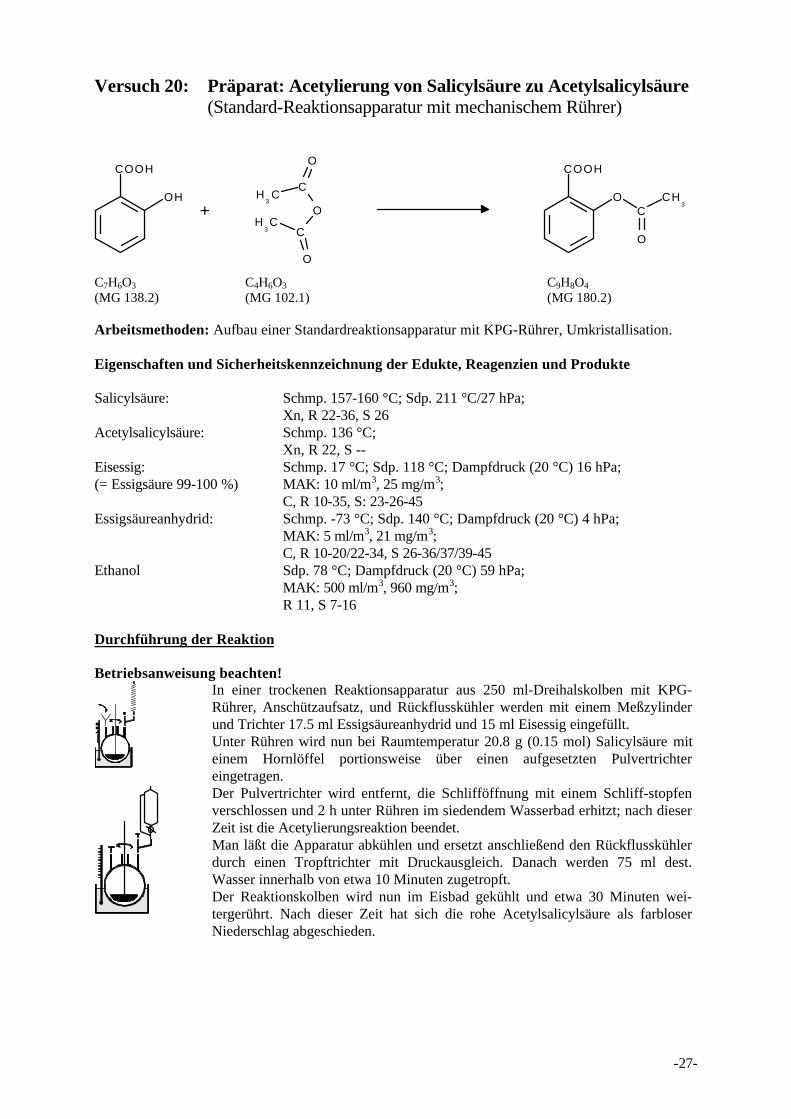
Versuch 20: Präparat: Acetylierung von Salicylsäure zu Acetylsalicylsäure(Standard-Reaktionsapparatur mit mechanischem Rührer)
+
COOH
OH
COOH
OC
CH3
O
H3C
C
O
CH
3C
O
O
C7H6O3 C4H6O3 C9H8O4
(MG 138.2) (MG 102.1) (MG 180.2)
Arbeitsmethoden: Aufbau einer Standardreaktionsapparatur mit KPG-Rührer, Umkristallisation.
Eigenschaften und Sicherheitskennzeichnung der Edukte, Reagenzien und Produkte
Salicylsäure: Schmp. 157-160 °C; Sdp. 211 °C/27 hPa;Xn, R 22-36, S 26
Acetylsalicylsäure: Schmp. 136 °C;Xn, R 22, S --
Eisessig: Schmp. 17 °C; Sdp. 118 °C; Dampfdruck (20 °C) 16 hPa;(= Essigsäure 99-100 %) MAK: 10 ml/m3, 25 mg/m3;
C, R 10-35, S: 23-26-45Essigsäureanhydrid: Schmp. -73 °C; Sdp. 140 °C; Dampfdruck (20 °C) 4 hPa;
MAK: 5 ml/m3, 21 mg/m3;C, R 10-20/22-34, S 26-36/37/39-45
Ethanol Sdp. 78 °C; Dampfdruck (20 °C) 59 hPa;MAK: 500 ml/m3, 960 mg/m3;R 11, S 7-16
Durchführung der Reaktion
Betriebsanweisung beachten!In einer trockenen Reaktionsapparatur aus 250 ml-Dreihalskolben mit KPG-Rührer, Anschützaufsatz, und Rückflusskühler werden mit einem Meßzylinderund Trichter 17.5 ml Essigsäureanhydrid und 15 ml Eisessig eingefüllt.Unter Rühren wird nun bei Raumtemperatur 20.8 g (0.15 mol) Salicylsäure miteinem Hornlöffel portionsweise über einen aufgesetzten Pulvertrichtereingetragen.Der Pulvertrichter wird entfernt, die Schlifföffnung mit einem Schliff-stopfenverschlossen und 2 h unter Rühren im siedendem Wasserbad erhitzt; nach dieserZeit ist die Acetylierungsreaktion beendet.Man läßt die Apparatur abkühlen und ersetzt anschließend den Rückflusskühlerdurch einen Tropftrichter mit Druckausgleich. Danach werden 75 ml dest.Wasser innerhalb von etwa 10 Minuten zugetropft.Der Reaktionskolben wird nun im Eisbad gekühlt und etwa 30 Minuten wei-tergerührt. Nach dieser Zeit hat sich die rohe Acetylsalicylsäure als farbloserNiederschlag abgeschieden.

-28-
Isolierung und Reinigung
Der Niederschlag wird auf einem Büchnertrichter abgesaugt und auf demTrichter zweimal mit je 50 ml kaltem dest. Wasser nachgewaschen; das Filtratwird verworfen (→ E1). Der Rückstand wird mit einem breiten Spatel oder mitdem flachen Kopf eines Glasstöpsels fest auf die Filterplatte gepreßt und durchlängeres Durchsaugen von Luft weitgehend getrocknet. Danach wird dasRohprodukt in eine tarierte Porzellanschale überführt und nach dem Trocknenim Exsikkator die Rohausbeute (in g und %) sowie der Schmelzpunktbestimmt.Zur Umkristallisation wird die rohe Acetylsalicylsäure in einem 500 ml-NS-29-Rundkolben mit aufgesetztem NS-29-Rückflusskühler unter Erhitzen in wenigEthanol (für 10 g Rohprodukt etwa 20 ml Ethanol) gelöst, danach wirdvorsichtig über den Rückflusskühler heißes dest. Wasser zugegeben (für 10 gRohprodukt etwa 40 ml Wasser). Nun wird das Heizbad entfernt und langsamabgekühlt, zur Vervollständigung der Kristallisation wird zum Schluß noch miteinem Eisbad gekühlt (etwa 15 Minuten).Das Kristallisat wird wieder über einen Büchnertrichter abgesaugt, mit etwa 50ml kalter Mischung Ethanol/Wasser (1:2) nachgewaschen und scharf abgepreßt(→ R1). Nach dem Trocknen werden Ausbeute und Schmelzpunkt bestimmt.
Reinprodukt
Ausb. 19.7 g , 0.11 mol (73%), Schmp. 136 °C.
Recycling und Entsorgung
E1: Die Waschwasser enthalten im wesentlichen verdünnte Essigsäure und können mit Wasser inden Abfluß gespült werden.
R1: Die Mutterlauge der Umkristallisation (wässrige ethanolische Lösung) sowie übriggebliebenerwässriger Ethanol wird zur Redestillation in den Sammelbehälter für "Recycling-Ethanol(wässrig)" gegeben.
Das erhaltene Reinprodukt wird zusammen mit dem Protokoll dem Assistenten übergeben.
Protokollführung: Zeichnen Sie den Formelkopf mit Summenformeln und Molmassen. BeschreibenSie kurz den Apparaturaufbau (mit Kolbengrößen) und alle verwendeten Chemikalien mit genauenMengenangaben. Beschreiben Sie die Durchführung des Versuchs. Geben Sie dabei alle Arbeitschrittemit Zeit- und Temperaturangaben sowie allen Beobachtungen an. Protokollieren Sie die erhaltenenMengen an Rohprodukt zusammen mit dem Schmelzpunkt. Geben Sie auch die Art und Weise derTrocknung (Trockenmittel, Zeitangabe) an.Protokollieren Sie die Reinigung des Rohprodukts: Menge an eingesetzter Substanz, benötigtesLösungsmittel, Isolierung und Trocknung des Kristallisats. Erstellen Sie eine Massenbilanz.Geben Sie abschließend die Ausbeute an Reinprodukt in g, mol und % der Theorie sowie diephysikalischen Daten des Produkts (Schmp., Aussehen) an. Vergleichen Sie die Daten mit derLiteratur (= Vorschrift)

-29-
Hinweise:
Bei diesem Präparat wird mit einem "KPG-Rührer" (kerngezogenes Präzisions-Glasgerät) gearbeitet(siehe Tutorial Kap. 1.2.5) Gegenüber einem Magnetrührer besitzt der KPG-Rührer eine deutlichhöhere Durchzugskraft. Daraus ergeben sich folgende Einsatzgebiete:
• Intensive Durchmischung bei Mehrphasensystemen
• Durchmischung von schwer rührbaren Mischungen (unlösliche Feststoffe in Suspension)
• Bei der Notwendigkeit eines intensiven Wärmeaustausches (z.B. exotherme Reaktionen)
• Durchmischen großer Reaktionsvolumina.
Ein KPG-Rührer besteht aus einem Glasrohr, das im Mittelteil einen etwa 10 - 20 cm langengenormten Präzissions-Zylinderschliff besitzt und drehbar in einer dazu exakt passend geschliffenenHülse (Schaft) sitzt. Die Rührhülse besitzt im allgemeinen einen NS 29-Kern-Schliff und kann somiteinfach auf den zentralen Schliff eines Kolbens aufgesetzt werden.
KPG-Rührer können mit verschiedenen Rührblättern ausgestattet sein, in der Regel werden heuteauswechselbare Teflon- oder Porzellanrührblätter eingesetzt.
Der Antrieb erfolgt durch einen Motor, dessen Antriebswelle über eine flexible Kupplung (imeinfachsten Fall ein Stück dickwandiger Gummischlauch) mit dem oberen Ende des KPG-Rührstabesverbunden wird. Der Gummischlauch wird durch Schlauchschellen gegen ''Durchrutschen' gesichert.
Rührer und Rührmotor müssen unbedingt in Achse liegen, um das "Ausschlagen" des Rührers undder Hülse zu vermeiden. Durch Vibrationen kann sich die Rührhülse während des Versuchs lockernund muß deshalb festgeklammert werden.
Sehr wichtig ist auch die richtige Schmierung des Kernschliffes: am besten eignet sich dazu "Stirr-Ol". Auf keinen Fall geeignet ist Schliffett, Exsikkatorfett oder Glycerin!
Lassen Sie Ihre Apparatur vor der Inbetriebnahme von Ihrem Assistenten überprüfen!
Der Anschützaufsatz in der oben genannten Reaktionsapparatur kann auch entfallen wenn derRückflusskühler und Tropftrichter auf den seitlichen Schliff aufgesetzt und geklammert werdenkönnen, ohne dass der Rührmotor stört.