orgenesis investor corporate presentation
TRANSCRIPT

OTCQB:ORGS
Corporate Presenta4on May 2014
A science-based organization dedicated to curing disease through the development and manufacture of cell-based therapeutics and regenerative medicine

Forward Looking Statements
Forward Looking Statements/ Offer of Securi4es This presenta,on does not cons,tute an offer for the purchase or sale of any securi,es of Orgenesis. You should not make any investment decisions based on this presenta,on. The informa,on in this presenta,on is not a subs,tute for independent professional advice before making any investment decisions. No securi,es regulatory authority has in any way passed on any of the informa,on contained in the presenta,on. Much of this presenta,on includes projec,ons and plans. Our stated plans are subject to known and unknown risks, uncertain,es and other factors that may cause the actual results of Orgenesis to be materially different from those expressed or implied in this presenta,on. Our products may never develop into useful products and even if they do, they may not be approved for sale to the public; we have substan,al hurdles to face in proving our technology, showing it can be safe for medical use and obtaining regulatory approval in each country in which our technology may be sold. We may not be able to fund our plans or keep key employees. We may not be able to protect our intellectual property. Sales projec,ons may not come to frui,on, and our compe,tors may provide beFer or cheaper products or services. Addi,onal informa,on about these and other assump,ons, risks and uncertain,es are set out in the "Risk Factors" sec,on in Orgenesis' most recent 10-‐K filed on EDGAR with the Securi,es and Exchange Commission. Bernard
(OTCQB:ORGS)

§ Company Overview § Business Model § Product Development Plan § Recent Corporate Developments

Orgenesis Inc. is a biotechnology company dedicated to curing Type 1 diabetes through a novel technology, cellular trans-‐differen0a0on, that combines cellular therapy and regenera4ve medicine.
§ Cellular trans-‐differen,a,on is a proven technology that converts autologous liver cells into fully func,onal and physiologically glucose-‐sensi,ve insulin-‐producing cells. § Phase 1 ready with “Fast-‐to-‐Market” clinical development strategy focusing on Ultra-‐Orphan indica,on – leading to accelerated approval § Technology supported with strong IP, and broad patent estate § Recent acquisi,on of MaSTherCell forms a ver,cally integrated, revenue-‐genera,ng business model with focus on rapidly growing cell-‐based therapeu,cs market
Company Snapshot: Symbol: OTCQB:OGRS Headquarters: Gaithersburg, MD Industry: Biotechnology
Market cap.: $38.5M Share Price: $0.70 52 wk High /Low: $1.00 / $0.34
Shares Out./Float: 55.2M / 23.5M Insider Holdings: ~40% Ins,tu,onal Holdings: <10%
Financings YTD: ~$7.5M Available Funds: ~$3.5M September 16th , 2014
Company Overview (OTCQB:ORGS)

(OTCQB:ORGS)
A Science-‐based, Innova4ve and Ver4cally Integrated Business Model
Pioneer and leader in the emerging fields of cell-‐based therapy, regenera,ve medicine and cGMP capabili,es to support each. A science-‐based and innova,ve company with a ver,cally integrated business model:
1. Clinical development of proprietary technology planorm (cellular ‘trans-‐differen,a,on’) – ini,ally targe,ng insulin-‐dependent disorders
2. Revenue genera,on through innova,ve cell-‐based manufacturing capabili,es – cost-‐efficient clinical development of Orgenesis technology while independently posi,oned as CDMO industry partner of choice
Over next 18-‐24 months, main goal is to ensure rapid increase in value by establishing clinical PoC with P1b clinical results in key indica,ons, and securing long-‐term manufacturing service agreements with targeted biotech companies

Orgenesis has performed pre-‐clinical safety and efficacy studies and is moving to:
§ Ini,ate regulatory ac,vi,es in Asia, Europe and U.S.
§ Finalize GMP and complete product scale-‐up (MaSTherCell – Belgium)
§ Transfer technology to US affiliate (Maryland)
§ Move to clinical trials and collaborate with clinical centers.
Product Development Overview
Product Development Timeline 2011 2012 2013 2014 2015
Proof of Principle Pre-‐clinical studies Phase 1b
Trials
Regulatory Plan (Paul Erlich Ins,tute and FDA Engagement)
Develop Produc,on Process Produc,on Scale up Under cGMP
(OTCQB:ORGS)

Recent Corporate Developments
§ Orgenesis acquires MaSTherCell, crea,ng a ver,cally integrated business focusing on the research, development and manufacture of cell-‐based therapeu,cs
Nov 2014
§ ScoF Carmer joins as CEO of Orgenesis North America — led the U.S Specialty Care Division of AstraZeneca PLC (LSE:AZN) Jul 2014
§ Awarded Maryland Stem Cell Research Fund Grant to help fund pre-‐clinical work in prepara,on for Phase I & II clinical trials in the U.S. May 2014
(OTCQB:ORGS)
Over last 12 months, ORGS has had very exci4ng developments. § Funding / Awards & Recogni,on § New Corporate Partnerships / Key Personnel Moves
§ Awarded $3.9M grant from Belgium’s DG06 to complete commercial scale cGMP facility Nov 2014

§ T1D Market Opportunity § Compe,,ve Landscape § Pre-‐clinical data § Differen,a,ng liver-‐derived from stem-‐cell derived IPCs § AIP cells § GMP – Using Advanced Technology & Systems

T1D Market Opportunity
Life-‐threatening and life-‐long disease . § Es,mated 1.5M – 3.0M people with T1D (US)(1); § ~30,000+ new diagnosis per year (US)(2) Significant economic burden to society. § Accounts for $14.9 billion in healthcare costs in the U.S. each year.(3)
US insulin market ~$8.9B in 2013, with forecast 6 Yr. CAGR of 12.4%(4) Daily management includes mul4ple insulin injec4ons, strict blood glucose monitoring, “carb coun4ng” and significant impact on QoL. Despite recent ‘advances’, significant clinical risks remain: § Hypoglycemic episodes: Hypoglycemic unawareness, Diabe,c coma.
§ Hyperglycemic consequences: Ketoacidosis, diabe,c re,nopathy, diabe,c nephropathy, stroke, CV disease.
Currently, no approved therapy for a “Prac4cal Cure”.
(OTCQB:ORGS)
(1) Type 1 Diabetes, 2010: Prime Group for JDRF, Mar 2011 (2) NIDDK: diabetes.niddk.nih.gov/dm/pubs/sta,s,cs/index.htm#i_youngpeople (3) The United States of Diabetes: Challenges and Opportuni,es in the Decade Ahead, 2010: United Health Group (4) Grand View Research, 2014

Paucity of R&D investment dedicated to “Cure”.
Compe44ve Landscape
Disease Management
§ Increase effec,veness of glucose control
— Improved insulin — Ar,ficial pancreas
Disease Progression
§ Maintain beta cell func,on / insulin produc,on
— Autoimmune tolerance
— T-‐cell abla,on
Clinical Cure
§ Long term insulin independence
— Islet cell transplanta,on
Prac4cal Cure
§ Long term insulin independence / no concomitant immunosuppression / normal quality of life
— Encapsula,on of insulin producing cells (Directed Differen,a,on)
— Autologous Insulin Producing Cells (Cellular Trans-‐differen4a4on)
(OTCQB:ORGS)

A unique, proprietary technology that transforms a pa4ent’s liver cells into glucose-‐responsive and func4onally mature Autologous Insulin Producing cells (AIPc).
Cellular Trans-‐Differen4a4on – A Prac4cal Cure (OTCQB:ORGS)
Liver and Pancreas: 1). Derived from same embryonic lineage (endoderm) 2. Share a common progenitor and many transcrip,on factors 3). Both have a built-‐in glucose-‐sensing system . . . Developmentally
related cells show a higher suscep,bility to trans-‐differen,a,on

Pre-‐Clinical Proof-‐of-‐Principal (OTCQB:ORGS)
ORGENESIS -‐ CONFIDENTIAL 5/9/2014
Ectopic PDX-‐1 expression ac,vates insulin produc,on in mice in-‐vivo (Ferber et al Nature Med)
Ectopic PDX-‐1 -‐ short term trigger to an irreversible reprogramming process (Ber et al JBC)
PDX-‐1 treatment in-‐vivo induces an immune modula,on, and ameliorates hyperglycemia in diabe,c NOD mice (Shternhall-‐Ron et al JAI)
Induc,on of pancrea,c lineage in human liver cells in-‐vitro, fetal and adult and the promo,ng effects of soluble factors (Sapir et al PNAS)
The role of hepa,c dedifferen,a,on in the ac,va,on of the alternate pancrea,c repertoire (Meivar-‐Levy et al Hepatology)
The role of Exndin-‐4 in prolifera,on and transdifferen,a,on process (Aviv et al JBC)
NKX6.1 ac,vates PDX-‐1-‐Induced Liver to Pancrea,c Reprogramming (Gefen-‐Halevi et al Cellular Reprograming)
Characteriza,on of adult liver cells reprogramming towards the pancrea,c lineage (Meivar-‐Levy et al J. Transplanta,on)
Methods of human liver cell reprogramming (Meivar-‐Levy et al Methods Mol Biol)
2000
2003
2005
2007
2007
2009
2010
2010
2011
The temporal and hierarchical control of transcrip,on factors-‐induced liver to pancreas transdifferen,a,on (Berneman-‐Zeitouni D, et al PlosOne) 2014
The pre-‐clinical proof-‐of-‐principal has been well established and externally validated.

PDX-‐1 ac4vates a func4onal β-‐cell lineage in liver, in-‐vivo.
First Valida4on of Trans-‐Differen4a4on Hypothesis (OTCQB:ORGS)
Ad-CMV-PDX-1
Ectopic PDX-‐1 expression ac,vates insulin produc,on in mice in-‐vivo (Ferber et al Nature Med)

In Pre-‐clinical model of T1D, PDX-‐1 cells drive ship from from Th1 to Th2 immune response . . . Resul4ng in a state of “tolerance” vs “aqack”
Blun4ng the Auto-‐Immune Response (OTCQB:ORGS)
1 Spleens removed
2 S,mulated with T1D an,gens 3 Studied for cytokine secre,on
Ø Th1 -‐ IFNg (a) Ø Th2 – IL-‐10 (d)
Shternhall Ron K et al, Ectopic PDX-‐1 expression in liver meliorates T1D; Journal of AutoImmunity (2007) doi: 10.1016
groups did not show significant differences in their prolifera-tive responses to Con A.
3.4. Reversal of CAD is associated with a Th1 to Th2shift of the autoimmune T-cell cytokine response
The T cells that mediate the destruction of the insulin-pro-ducing pancreatic b-cells in CAD secrete Th1 cytokines, suchas IFNg [33]. Moreover, immunomodulatory therapies thatarrest the diabetogenic autoimmune process usually lead to
a Th2 shift in the autoimmune T-cell response, marked bythe increased production of IL-10 [27]. To further characterizethe autoimmune response in mice treated with Ad-CMV-PDX-1, we studied IFNg and IL-10 secretion by splenocytes stimu-lated with insulin, GAD, p34, p35, HSP60, p12 or p277. Thesplenocytes taken from the different experimental groups didnot differ in the amounts of IFNg or IL-10 released upon ac-tivation with Con A, and were not stimulated with the controlantigen GST. However, mice that manifested a reversal of hy-perglycemia showed a significant decrease in IFNg secretion
250
200
150
100
50
0
600
1200
1800
GST ConA
0
100
200
300
400
500
600
700
Insulin HSP60 p12 p34 p35
INF
(p
g/m
l)
INF
(p
g/m
l)
INF
(p
g/m
l)
0
200
400
600
800
GAD
b
d
e f
c
a
Untreated
Ad-RIP-b-gal
Ad-CMV-PDX-1
0
100
200
300
400
500
600
700
Insulin HSP60 p277 p12 p34 p35
0
200
400
600
800
1000
GAD
IL-1
0 (p
g/m
l)IL
-10
(pg/
ml)
GST ConA
IL-1
0 (p
g/m
l)
p277
* * * * * *
*
*
*
*
*
*
Fig. 4. Reversal of CAD is associated with a Th1 to Th2 shift of the autoimmune T-cell cytokine response. Twenty to forty days after treatment by recombinantadenoviruses, spleens were removed and studied for the secretion of IFNg (aec) and IL-10 (def) upon stimulation with (a,d) insulin, HSP60, p277, p12, p34, p35,(b,e) GAD, (c,f) GST or Con A. The data are presented as means ! SE for 4e6 individual samples per group (*p < 0.05 compared to the untreated group).
6 K. Shternhall-Ron et al. / Journal of Autoimmunity xx (2007) 1e9
+ MODEL
ARTICLE IN PRESS
Please cite this article in press as: Shternhall-Ron K et al., Ectopic PDX-1 expression in liver ameliorates type 1 diabetes, Journal of Autoimmunity (2007),doi:10.1016/j.jaut.2007.02.010
groups did not show significant differences in their prolifera-tive responses to Con A.
3.4. Reversal of CAD is associated with a Th1 to Th2shift of the autoimmune T-cell cytokine response
The T cells that mediate the destruction of the insulin-pro-ducing pancreatic b-cells in CAD secrete Th1 cytokines, suchas IFNg [33]. Moreover, immunomodulatory therapies thatarrest the diabetogenic autoimmune process usually lead to
a Th2 shift in the autoimmune T-cell response, marked bythe increased production of IL-10 [27]. To further characterizethe autoimmune response in mice treated with Ad-CMV-PDX-1, we studied IFNg and IL-10 secretion by splenocytes stimu-lated with insulin, GAD, p34, p35, HSP60, p12 or p277. Thesplenocytes taken from the different experimental groups didnot differ in the amounts of IFNg or IL-10 released upon ac-tivation with Con A, and were not stimulated with the controlantigen GST. However, mice that manifested a reversal of hy-perglycemia showed a significant decrease in IFNg secretion
250
200
150
100
50
0
600
1200
1800
GST ConA
0
100
200
300
400
500
600
700
Insulin HSP60 p12 p34 p35
INF
(p
g/m
l)
INF
(p
g/m
l)
INF
(p
g/m
l)
0
200
400
600
800
GAD
b
d
e f
c
a
Untreated
Ad-RIP-b-gal
Ad-CMV-PDX-1
0
100
200
300
400
500
600
700
Insulin HSP60 p277 p12 p34 p35
0
200
400
600
800
1000
GAD
IL-1
0 (p
g/m
l)IL
-10
(pg/
ml)
GST ConA
IL-1
0 (p
g/m
l)
p277
* * * * * *
*
*
*
*
*
*
Fig. 4. Reversal of CAD is associated with a Th1 to Th2 shift of the autoimmune T-cell cytokine response. Twenty to forty days after treatment by recombinantadenoviruses, spleens were removed and studied for the secretion of IFNg (aec) and IL-10 (def) upon stimulation with (a,d) insulin, HSP60, p277, p12, p34, p35,(b,e) GAD, (c,f) GST or Con A. The data are presented as means ! SE for 4e6 individual samples per group (*p < 0.05 compared to the untreated group).
6 K. Shternhall-Ron et al. / Journal of Autoimmunity xx (2007) 1e9
+ MODEL
ARTICLE IN PRESS
Please cite this article in press as: Shternhall-Ron K et al., Ectopic PDX-1 expression in liver ameliorates type 1 diabetes, Journal of Autoimmunity (2007),doi:10.1016/j.jaut.2007.02.010

A B
Insulin / Pdx-‐1 / DAPI
C Insulin production and storage in PDX-1 treated liver cells-EM, immuno-
gold IMC
Glucose metabolism is needed for regulated C-peptide
secretion
PDX-1 is delivered using recombinant adenovirus
Sapir et al PNAS 2005 & Berneman-‐Zeituni, PlosOne 2014
AIP cells are “physiologically” glucose-‐sensi4ve: They produce, store and secrete processed insulin in response to elevated glucose concentra4ons
15
C-p
epep
tide
secr
etio
n ng
/m
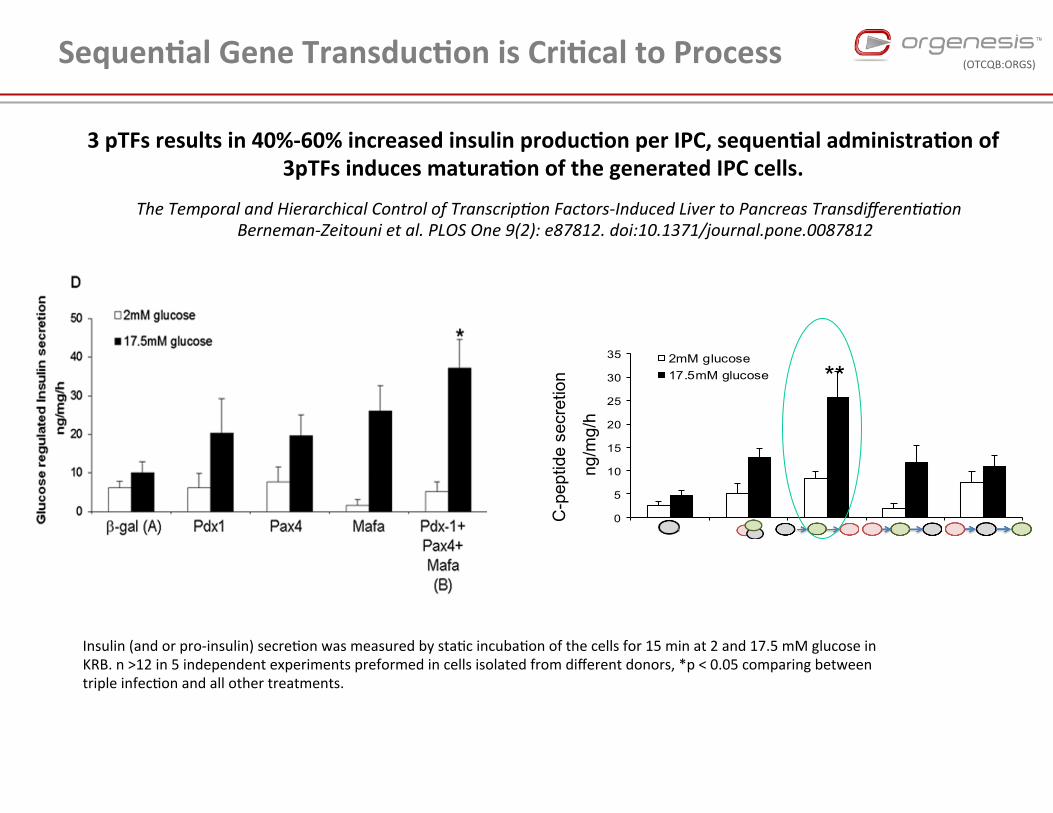
3 pTFs results in 40%-‐60% increased insulin produc4on per IPC, sequen4al administra4on of 3pTFs induces matura4on of the generated IPC cells.
Sequen4al Gene Transduc4on is Cri4cal to Process
The Temporal and Hierarchical Control of Transcrip4on Factors-‐Induced Liver to Pancreas Transdifferen4a4on Berneman-‐Zeitouni et al. PLOS One 9(2): e87812. doi:10.1371/journal.pone.0087812
Insulin (and or pro-‐insulin) secre,on was measured by sta,c incuba,on of the cells for 15 min at 2 and 17.5 mM glucose in KRB. n >12 in 5 independent experiments preformed in cells isolated from different donors, *p < 0.05 comparing between triple infec,on and all other treatments.
(OTCQB:ORGS)
0
5
10
15
20
25
30
35 2mM glucose17.5mM glucose
C-p
eptid
e se
cret
ion
ng/
mg/
h
**

ES and iPS cells giver rise to fetal islets that are not glucose-‐responsive.
Stem Cell-‐Driven Differen4a4on (OTCQB:ORGS)
Differen,ated human stem cells resemble fetal, not adult, β cells Hrva,n et al. PNAS online www.pnas.org/cgi/doi/10.1073/pnas.1400709111
*hPSC refers to human pluripotent stem cells derived from embryonic stem cell or reprogrammed (iPS) cells

11/17/14 Confidential Internal Document
18
insulin secretion in vitro, fail to express appropriate b cell markerssuch as NKX6-1 or PDX1, abnormally coexpress other hormoneslike glucagon (GCG), fail to function after transplantation in vivo,or display a combination of these abnormal features (D’Amouret al., 2006; Cheng et al., 2012; Hrvatin et al., 2014; Narayananet al., 2014; Xie et al., 2013; Nostro et al., 2011).Herein, we report the discovery of a strategy for large-scale
production of functional human b cells from hPSC in vitro. Byusing sequential modulation of multiple signaling pathways in athree-dimensional cell culture system, without any transgenesor genetic modification, we generate glucose-responsive,monohormonal insulin-producing cells that show key featuresof a bona fide b cell, including coexpression of key b cell markersand b cell ultrastructure. Furthermore, these cells mimic thefunction of human islets both in vitro and in vivo. Finally, wedemonstrate the potential utility of these cells for in vivo trans-plantation therapy for diabetes.
RESULTS
Generation of Glucose-Sensing Insulin-Secretingb Cells In VitroOur strategy to generate functional b cells from hPSC in vitro isoutlined in Figure 1A. To produce large numbers, we used a scal-
able suspension-based culture system that can generate >108
hPSCs and later differentiated cell types (modified from Schulzet al., 2012). Clusters of cells (!100–200 mm in diameter, eachcluster containing several hundred cells) from a human embry-onic stem cell (hESC) line (HUES8) or two human-induced plurip-otent stem cell (hiPSC) lines (hiPSC-1 and hiPSC-2) wereinduced into definitive endoderm (>95% SOX17+ cells, DE cellsin Figure 1A) and subsequently early pancreatic progenitors(>85% PDX1+ cells, PP1 cells in Figure 1A).Transplantation of pancreatic progenitors expressing PDX1+/
NKX6-1+ (PP2 in Figure 1A) into mice gives rise to functional bcells in vivo after 3–4 months (Kroon et al., 2008; Rezania et al.,2012). And previous studies had shown that these PDX1+/NKX6-1+ pancreatic progenitors (PP2) could be further differen-tiated in vitro into some INS+ cells along with INS+/GCG+ orINS+/SST+ polyhormonal (PH) cells (Nostro et al., 2011; Rezaniaet al., 2012; Thowfeequ et al., 2007; Aguayo-Mazzucato et al.,2013; D’Amour et al., 2006; Hrvatin et al., 2014). We use thenomenclature PH (polyhormonal, Figure 1A) to refer to this cellpopulation of in-vitro-differentiated hPSCs. Transcriptional anal-ysis of in-vitro-differentiated PH cells showed that these cellsresemble human fetal and not adult b cells (Hrvatin et al.,2014). Because these PH cells show neither glucose-stimulatedinsulin secretion (GSIS) nor other key properties of bona fide b
Figure 1. SC-b Cells Generated In VitroSecrete Insulin in Response to MultipleSequential High-Glucose Challenges likePrimary Human b Cells(A) Schematic of directed differentiation from
hPSC into INS+ cells via new or previously pub-
lished control differentiations.
(B–D) Representative ELISA measurements of
secreted human insulin from HUES8 SC-b cells
(B), PH cells (C), and primary b (1"b) cells (D)
challenged sequentially with 2, 20, 2, 20, 2, and
20 mM glucose, with a 30 min incubation for each
concentration (see Experimental Procedures). Af-
ter sequential low/high-glucose challenges, cells
were depolarized with 30 mM KCl.
(E–G) Box and whisker plots of secreted human
insulin from different biological batches of HUES8
(open circles) and hiPSC SC-b (black circles) cells
(E; n = 12), biological batches of PH cells (F; n = 5),
and primary b cells (G; n = 4). Each circle is the
average value for all sequential challenges with
2 mM or 20 mM glucose in a batch. Insulin
secretion at 20 mM ranged 0.23–2.7 mIU/103 cells
for SC-b cells and 1.5–4.5 mIU/103 cells for human
islets, and the stimulation index ranged 0.4–4.1 for
SC-b cells and 0.6–4.8 for primary adult. The thick
horizontal line indicates the median.
SeealsoFiguresS1andS2AandTableS1. *p<0.05
when comparing insulin secretion at 20 mM versus
2 mM with paired t test. Act A, activin A; CHIR,
CHIR99021, aGSK3a/b inhibitor; KGF, keratinocyte
growth factor or FGF family member 7; RA, retinoic
acid; SANT1, sonic hedgehog pathway antagonist;
LDN, LDN193189, a BMP type 1 receptor inhibitor;
PdbU, Phorbol 12,13-dibutyrate, a protein kinase C
activator; Alk5i, Alk5 receptor inhibitor II; T3, triio-
dothyronine, a thyroid hormone; XXI, g-secretase
inhibitor; Betacellulin, EGF family member.
Cell 159, 428–439, October 9, 2014 ª2014 Elsevier Inc. 429
insulin secretion in vitro, fail to express appropriate b cell markerssuch as NKX6-1 or PDX1, abnormally coexpress other hormoneslike glucagon (GCG), fail to function after transplantation in vivo,or display a combination of these abnormal features (D’Amouret al., 2006; Cheng et al., 2012; Hrvatin et al., 2014; Narayananet al., 2014; Xie et al., 2013; Nostro et al., 2011).Herein, we report the discovery of a strategy for large-scale
production of functional human b cells from hPSC in vitro. Byusing sequential modulation of multiple signaling pathways in athree-dimensional cell culture system, without any transgenesor genetic modification, we generate glucose-responsive,monohormonal insulin-producing cells that show key featuresof a bona fide b cell, including coexpression of key b cell markersand b cell ultrastructure. Furthermore, these cells mimic thefunction of human islets both in vitro and in vivo. Finally, wedemonstrate the potential utility of these cells for in vivo trans-plantation therapy for diabetes.
RESULTS
Generation of Glucose-Sensing Insulin-Secretingb Cells In VitroOur strategy to generate functional b cells from hPSC in vitro isoutlined in Figure 1A. To produce large numbers, we used a scal-
able suspension-based culture system that can generate >108
hPSCs and later differentiated cell types (modified from Schulzet al., 2012). Clusters of cells (!100–200 mm in diameter, eachcluster containing several hundred cells) from a human embry-onic stem cell (hESC) line (HUES8) or two human-induced plurip-otent stem cell (hiPSC) lines (hiPSC-1 and hiPSC-2) wereinduced into definitive endoderm (>95% SOX17+ cells, DE cellsin Figure 1A) and subsequently early pancreatic progenitors(>85% PDX1+ cells, PP1 cells in Figure 1A).Transplantation of pancreatic progenitors expressing PDX1+/
NKX6-1+ (PP2 in Figure 1A) into mice gives rise to functional bcells in vivo after 3–4 months (Kroon et al., 2008; Rezania et al.,2012). And previous studies had shown that these PDX1+/NKX6-1+ pancreatic progenitors (PP2) could be further differen-tiated in vitro into some INS+ cells along with INS+/GCG+ orINS+/SST+ polyhormonal (PH) cells (Nostro et al., 2011; Rezaniaet al., 2012; Thowfeequ et al., 2007; Aguayo-Mazzucato et al.,2013; D’Amour et al., 2006; Hrvatin et al., 2014). We use thenomenclature PH (polyhormonal, Figure 1A) to refer to this cellpopulation of in-vitro-differentiated hPSCs. Transcriptional anal-ysis of in-vitro-differentiated PH cells showed that these cellsresemble human fetal and not adult b cells (Hrvatin et al.,2014). Because these PH cells show neither glucose-stimulatedinsulin secretion (GSIS) nor other key properties of bona fide b
Figure 1. SC-b Cells Generated In VitroSecrete Insulin in Response to MultipleSequential High-Glucose Challenges likePrimary Human b Cells(A) Schematic of directed differentiation from
hPSC into INS+ cells via new or previously pub-
lished control differentiations.
(B–D) Representative ELISA measurements of
secreted human insulin from HUES8 SC-b cells
(B), PH cells (C), and primary b (1"b) cells (D)
challenged sequentially with 2, 20, 2, 20, 2, and
20 mM glucose, with a 30 min incubation for each
concentration (see Experimental Procedures). Af-
ter sequential low/high-glucose challenges, cells
were depolarized with 30 mM KCl.
(E–G) Box and whisker plots of secreted human
insulin from different biological batches of HUES8
(open circles) and hiPSC SC-b (black circles) cells
(E; n = 12), biological batches of PH cells (F; n = 5),
and primary b cells (G; n = 4). Each circle is the
average value for all sequential challenges with
2 mM or 20 mM glucose in a batch. Insulin
secretion at 20 mM ranged 0.23–2.7 mIU/103 cells
for SC-b cells and 1.5–4.5 mIU/103 cells for human
islets, and the stimulation index ranged 0.4–4.1 for
SC-b cells and 0.6–4.8 for primary adult. The thick
horizontal line indicates the median.
SeealsoFiguresS1andS2AandTableS1. *p<0.05
when comparing insulin secretion at 20 mM versus
2 mM with paired t test. Act A, activin A; CHIR,
CHIR99021, aGSK3a/b inhibitor; KGF, keratinocyte
growth factor or FGF family member 7; RA, retinoic
acid; SANT1, sonic hedgehog pathway antagonist;
LDN, LDN193189, a BMP type 1 receptor inhibitor;
PdbU, Phorbol 12,13-dibutyrate, a protein kinase C
activator; Alk5i, Alk5 receptor inhibitor II; T3, triio-
dothyronine, a thyroid hormone; XXI, g-secretase
inhibitor; Betacellulin, EGF family member.
Cell 159, 428–439, October 9, 2014 ª2014 Elsevier Inc. 429
Resource
Generation of Functional HumanPancreatic b Cells In VitroFelicia W. Pagliuca,1,3 Jeffrey R. Millman,1,3 Mads Gurtler,1,3 Michael Segel,1 Alana Van Dervort,1 Jennifer Hyoje Ryu,1
Quinn P. Peterson,1 Dale Greiner,2 and Douglas A. Melton1,*1Department of Stem Cell and Regenerative Biology, Harvard Stem Cell Institute, Harvard University, 7 Divinity Avenue, Cambridge,MA 02138, USA2Diabetes Center of Excellence, University of Massachusetts Medical School, 368 Plantation Street, AS7-2051, Worcester, MA 01605, USA3Co-first author*Correspondence: [email protected]://dx.doi.org/10.1016/j.cell.2014.09.040
SUMMARY
The generation of insulin-producing pancreatic bcells from stem cells in vitro would provide an un-precedented cell source for drug discovery and celltransplantation therapy in diabetes. However, insu-lin-producing cells previously generated from humanpluripotent stem cells (hPSC) lack many functionalcharacteristics of bona fide b cells. Here, we reporta scalable differentiation protocol that can generatehundreds of millions of glucose-responsive b cellsfrom hPSC in vitro. These stem-cell-derived b cells(SC-b) express markers found in mature b cells, fluxCa2+ in response to glucose, package insulin intosecretory granules, and secrete quantities of insulincomparable to adult b cells in response to multiplesequential glucose challenges in vitro. Furthermore,these cells secrete human insulin into the serum ofmice shortly after transplantation in a glucose-regu-latedmanner, and transplantation of these cells ame-liorates hyperglycemia in diabetic mice.
INTRODUCTION
The discovery of human pluripotent stem cells (hPSC) openedthe possibility of generating replacement cells and tissuesin the laboratory that could be used for disease treatment anddrug screening. Recent research has moved the stem cell fieldcloser to that goal through development of strategies to generatecells that would otherwise be difficult to obtain, like neurons orcardiomyocytes (Kriks et al., 2011; Shiba et al., 2012; Sonet al., 2011). These cells have also been transplanted into animalmodels, in some caseswith a beneficial effect like suppression ofarrhythmias with stem-cell-derived cardiomyocytes (Shiba et al.,2012), restoration of locomotion after spinal injury with oligoden-drocyte progenitors (Keirstead et al., 2005), or improved visionafter transplantation of retinal epithelial cells into rodent modelsof blindness (Lu et al., 2009).
One of the rapidly growing diseases that may be treatable bystem-cell-derived tissues is diabetes, affecting >300 million peo-ple worldwide, according to the International Diabetes Federa-
tion. Type 1 diabetes results from autoimmune destruction of bcells in the pancreatic islet, whereas themore common type 2 dia-betes results from peripheral tissue insulin resistance and b celldysfunction. Diabetic patients, particularly those suffering fromtype 1 diabetes, could potentially be cured through transplanta-tion of new b cells. Patients transplantedwith cadaveric human is-lets can be made insulin independent for 5 years or longer via thisstrategy, but this approach is limited because of the scarcity andquality of donor islets (Bellin et al., 2012). The generation of anunlimited supply of human b cells from stem cells could extendthis therapy to millions of new patients and could be an importanttest case for translating stem cell biology into the clinic. This isbecause only a single cell type, the b cell, likely needs to be gener-ated, and the mode of delivery is understood: transplantation to avascularized location within the body with immunoprotection.Pharmaceutical screening to identify new drugs that improve b
cell function, survival, or proliferation is also hindered by limitedsupplies of islets and high variability due to differential causesof death, donor genetic background, and other factors in theirisolation. A consistent, uniform supply of stem-cell-derived b cellswould provide a unique and valuable drug discovery platform fordiabetes. Additionally, genetically diverse stem-cell-derived bcells could be used for disease modeling in vitro or in vivo.Studies on pancreatic development in model organisms
(Gamer and Wright, 1995; Henry and Melton, 1998; Ninomiyaet al., 1999; Apelqvist et al., 1999; Kim et al., 2000; Hebroket al., 2000; Murtaugh et al., 2003) identified genes and signalsimportant for the pancreatic lineage, and these have been effec-tively used to form cells in the b cell lineage in vitro from hPSC.Definitive endoderm and subsequent pancreatic progenitorscan now be differentiated with high efficiencies (Kroon et al.,2008; D’Amour et al., 2005, 2006; Rezania et al., 2012). Thesecells can differentiate into functional b cells within 3–4 monthsfollowing transplantation into rodents (Kroon et al., 2008; Rezaniaet al., 2012), indicating that some cells in the preparation containthe developmental potential to develop into b cells if providedenough time and appropriate cues. Unfortunately, the months-long process the cells undergo in vivo is not understood, and itis unclear whether this process of in vivo differentiation wouldalso occur in human patients. Attempts to date at generatinginsulin-producing (INS+) cells from human pancreatic progeni-tors in vitro have generated cells with immature or abnormalphenotypes. These cells either fail to perform glucose-stimulated
428 Cell 159, 428–439, October 9, 2014 ª2014 Elsevier Inc.
Resource
Generation of Functional HumanPancreatic b Cells In VitroFelicia W. Pagliuca,1,3 Jeffrey R. Millman,1,3 Mads Gurtler,1,3 Michael Segel,1 Alana Van Dervort,1 Jennifer Hyoje Ryu,1
Quinn P. Peterson,1 Dale Greiner,2 and Douglas A. Melton1,*1Department of Stem Cell and Regenerative Biology, Harvard Stem Cell Institute, Harvard University, 7 Divinity Avenue, Cambridge,MA 02138, USA2Diabetes Center of Excellence, University of Massachusetts Medical School, 368 Plantation Street, AS7-2051, Worcester, MA 01605, USA3Co-first author*Correspondence: [email protected]://dx.doi.org/10.1016/j.cell.2014.09.040
SUMMARY
The generation of insulin-producing pancreatic bcells from stem cells in vitro would provide an un-precedented cell source for drug discovery and celltransplantation therapy in diabetes. However, insu-lin-producing cells previously generated from humanpluripotent stem cells (hPSC) lack many functionalcharacteristics of bona fide b cells. Here, we reporta scalable differentiation protocol that can generatehundreds of millions of glucose-responsive b cellsfrom hPSC in vitro. These stem-cell-derived b cells(SC-b) express markers found in mature b cells, fluxCa2+ in response to glucose, package insulin intosecretory granules, and secrete quantities of insulincomparable to adult b cells in response to multiplesequential glucose challenges in vitro. Furthermore,these cells secrete human insulin into the serum ofmice shortly after transplantation in a glucose-regu-latedmanner, and transplantation of these cells ame-liorates hyperglycemia in diabetic mice.
INTRODUCTION
The discovery of human pluripotent stem cells (hPSC) openedthe possibility of generating replacement cells and tissuesin the laboratory that could be used for disease treatment anddrug screening. Recent research has moved the stem cell fieldcloser to that goal through development of strategies to generatecells that would otherwise be difficult to obtain, like neurons orcardiomyocytes (Kriks et al., 2011; Shiba et al., 2012; Sonet al., 2011). These cells have also been transplanted into animalmodels, in some caseswith a beneficial effect like suppression ofarrhythmias with stem-cell-derived cardiomyocytes (Shiba et al.,2012), restoration of locomotion after spinal injury with oligoden-drocyte progenitors (Keirstead et al., 2005), or improved visionafter transplantation of retinal epithelial cells into rodent modelsof blindness (Lu et al., 2009).
One of the rapidly growing diseases that may be treatable bystem-cell-derived tissues is diabetes, affecting >300 million peo-ple worldwide, according to the International Diabetes Federa-
tion. Type 1 diabetes results from autoimmune destruction of bcells in the pancreatic islet, whereas themore common type 2 dia-betes results from peripheral tissue insulin resistance and b celldysfunction. Diabetic patients, particularly those suffering fromtype 1 diabetes, could potentially be cured through transplanta-tion of new b cells. Patients transplantedwith cadaveric human is-lets can be made insulin independent for 5 years or longer via thisstrategy, but this approach is limited because of the scarcity andquality of donor islets (Bellin et al., 2012). The generation of anunlimited supply of human b cells from stem cells could extendthis therapy to millions of new patients and could be an importanttest case for translating stem cell biology into the clinic. This isbecause only a single cell type, the b cell, likely needs to be gener-ated, and the mode of delivery is understood: transplantation to avascularized location within the body with immunoprotection.Pharmaceutical screening to identify new drugs that improve b
cell function, survival, or proliferation is also hindered by limitedsupplies of islets and high variability due to differential causesof death, donor genetic background, and other factors in theirisolation. A consistent, uniform supply of stem-cell-derived b cellswould provide a unique and valuable drug discovery platform fordiabetes. Additionally, genetically diverse stem-cell-derived bcells could be used for disease modeling in vitro or in vivo.Studies on pancreatic development in model organisms
(Gamer and Wright, 1995; Henry and Melton, 1998; Ninomiyaet al., 1999; Apelqvist et al., 1999; Kim et al., 2000; Hebroket al., 2000; Murtaugh et al., 2003) identified genes and signalsimportant for the pancreatic lineage, and these have been effec-tively used to form cells in the b cell lineage in vitro from hPSC.Definitive endoderm and subsequent pancreatic progenitorscan now be differentiated with high efficiencies (Kroon et al.,2008; D’Amour et al., 2005, 2006; Rezania et al., 2012). Thesecells can differentiate into functional b cells within 3–4 monthsfollowing transplantation into rodents (Kroon et al., 2008; Rezaniaet al., 2012), indicating that some cells in the preparation containthe developmental potential to develop into b cells if providedenough time and appropriate cues. Unfortunately, the months-long process the cells undergo in vivo is not understood, and itis unclear whether this process of in vivo differentiation wouldalso occur in human patients. Attempts to date at generatinginsulin-producing (INS+) cells from human pancreatic progeni-tors in vitro have generated cells with immature or abnormalphenotypes. These cells either fail to perform glucose-stimulated
428 Cell 159, 428–439, October 9, 2014 ª2014 Elsevier Inc.
Resource
Generation of Functional HumanPancreatic b Cells In VitroFelicia W. Pagliuca,1,3 Jeffrey R. Millman,1,3 Mads Gurtler,1,3 Michael Segel,1 Alana Van Dervort,1 Jennifer Hyoje Ryu,1
Quinn P. Peterson,1 Dale Greiner,2 and Douglas A. Melton1,*1Department of Stem Cell and Regenerative Biology, Harvard Stem Cell Institute, Harvard University, 7 Divinity Avenue, Cambridge,MA 02138, USA2Diabetes Center of Excellence, University of Massachusetts Medical School, 368 Plantation Street, AS7-2051, Worcester, MA 01605, USA3Co-first author*Correspondence: [email protected]://dx.doi.org/10.1016/j.cell.2014.09.040
SUMMARY
The generation of insulin-producing pancreatic bcells from stem cells in vitro would provide an un-precedented cell source for drug discovery and celltransplantation therapy in diabetes. However, insu-lin-producing cells previously generated from humanpluripotent stem cells (hPSC) lack many functionalcharacteristics of bona fide b cells. Here, we reporta scalable differentiation protocol that can generatehundreds of millions of glucose-responsive b cellsfrom hPSC in vitro. These stem-cell-derived b cells(SC-b) express markers found in mature b cells, fluxCa2+ in response to glucose, package insulin intosecretory granules, and secrete quantities of insulincomparable to adult b cells in response to multiplesequential glucose challenges in vitro. Furthermore,these cells secrete human insulin into the serum ofmice shortly after transplantation in a glucose-regu-latedmanner, and transplantation of these cells ame-liorates hyperglycemia in diabetic mice.
INTRODUCTION
The discovery of human pluripotent stem cells (hPSC) openedthe possibility of generating replacement cells and tissuesin the laboratory that could be used for disease treatment anddrug screening. Recent research has moved the stem cell fieldcloser to that goal through development of strategies to generatecells that would otherwise be difficult to obtain, like neurons orcardiomyocytes (Kriks et al., 2011; Shiba et al., 2012; Sonet al., 2011). These cells have also been transplanted into animalmodels, in some caseswith a beneficial effect like suppression ofarrhythmias with stem-cell-derived cardiomyocytes (Shiba et al.,2012), restoration of locomotion after spinal injury with oligoden-drocyte progenitors (Keirstead et al., 2005), or improved visionafter transplantation of retinal epithelial cells into rodent modelsof blindness (Lu et al., 2009).
One of the rapidly growing diseases that may be treatable bystem-cell-derived tissues is diabetes, affecting >300 million peo-ple worldwide, according to the International Diabetes Federa-
tion. Type 1 diabetes results from autoimmune destruction of bcells in the pancreatic islet, whereas themore common type 2 dia-betes results from peripheral tissue insulin resistance and b celldysfunction. Diabetic patients, particularly those suffering fromtype 1 diabetes, could potentially be cured through transplanta-tion of new b cells. Patients transplantedwith cadaveric human is-lets can be made insulin independent for 5 years or longer via thisstrategy, but this approach is limited because of the scarcity andquality of donor islets (Bellin et al., 2012). The generation of anunlimited supply of human b cells from stem cells could extendthis therapy to millions of new patients and could be an importanttest case for translating stem cell biology into the clinic. This isbecause only a single cell type, the b cell, likely needs to be gener-ated, and the mode of delivery is understood: transplantation to avascularized location within the body with immunoprotection.Pharmaceutical screening to identify new drugs that improve b
cell function, survival, or proliferation is also hindered by limitedsupplies of islets and high variability due to differential causesof death, donor genetic background, and other factors in theirisolation. A consistent, uniform supply of stem-cell-derived b cellswould provide a unique and valuable drug discovery platform fordiabetes. Additionally, genetically diverse stem-cell-derived bcells could be used for disease modeling in vitro or in vivo.Studies on pancreatic development in model organisms
(Gamer and Wright, 1995; Henry and Melton, 1998; Ninomiyaet al., 1999; Apelqvist et al., 1999; Kim et al., 2000; Hebroket al., 2000; Murtaugh et al., 2003) identified genes and signalsimportant for the pancreatic lineage, and these have been effec-tively used to form cells in the b cell lineage in vitro from hPSC.Definitive endoderm and subsequent pancreatic progenitorscan now be differentiated with high efficiencies (Kroon et al.,2008; D’Amour et al., 2005, 2006; Rezania et al., 2012). Thesecells can differentiate into functional b cells within 3–4 monthsfollowing transplantation into rodents (Kroon et al., 2008; Rezaniaet al., 2012), indicating that some cells in the preparation containthe developmental potential to develop into b cells if providedenough time and appropriate cues. Unfortunately, the months-long process the cells undergo in vivo is not understood, and itis unclear whether this process of in vivo differentiation wouldalso occur in human patients. Attempts to date at generatinginsulin-producing (INS+) cells from human pancreatic progeni-tors in vitro have generated cells with immature or abnormalphenotypes. These cells either fail to perform glucose-stimulated
428 Cell 159, 428–439, October 9, 2014 ª2014 Elsevier Inc.

AIP cells are meaningfully differen4ated from any other current (or future) compe4tor.
AIP Cells (OTCQB:ORGS)
Insulin Independence
Therapy & Quality of Life
Tissue Availability
Quality Control
Ini4a4ng clinical trials within 12 – 15 months
§ Glucose-‐responsive insulin produc,on within one week of AIP cell transplanta,on
§ Insulin-‐independence within one month
§ Single course of therapy (5-‐10 years insulin independence) § No need for concomitant immunosuppressive therapy § Return to (near) normal quality of life for pa,ents
§ Single liver biopsy supplies unlimited source of therapeu,c ,ssue (bio-‐banking of ,ssue for future use if needed)
§ Highly controlled and ,ghtly closed GMP systems § QC of final product upon release and distribu,on

Orgenesis’ GMP systems improve quality and speed while decreasing costs.
GMP – Using Advanced Technology & Systems
Cell Culturing Propaga4on Packaging Trans-‐differen4a4on
Liver Biopsy Mechanic & enzyma,c Isola,on
Transplanta4on Liver biopsy
Cell expansion – Single use bioreactors
Trans -‐differen,a,on via
pTFs
Washing, QA/QC, Packaging, Release and distribu,on
AIP cells transplanted into liver via infusion
(OTCQB:ORGS)
5-6 weeks

§ Cell Therapy Market § Cell Therapy CDMO Market § Business and Expansion Strategies § Ra,onale suppor,ng acquisi,on
MaSTherCell . . . The Perfect Fit

The Cell Therapy Market – Clinical Trials
10 Source: Culme-‐Seymour EJ, Davie NL, Brindley DA, Edwards-‐Parton S, Mason C: A decade of cell therapy clinical trials (2000-‐2010). Regenera4ve medicine 7,4 (2012); ClinicalTrials.gov (www.clinicaltrials.gov)
• 22,500+ Clinical Trials
• 2800 “new” Cell Therapies
• 560 in PIII/Pivotal Trials
• Most therapies developped in US & EU

Global Cell Therapy Market -‐ Value & Forecasts
• Cell therapy products market set to grow to nearly 32B$ by 2018.
• Global cell therapy market expected to grow exponen,ally
• Organ replacement/transplant is playing an increasingly large role1
• Key Market Drivers: BeFer treatment outcomes and reduc,on of the direct costs associated with chronic diseases (by ~250B$ annually in the U.S.)²
1) MedMarket Diligence, Oct. 2012 2) Alliance for Regenera4ve Medicine Annual Report 2012-‐2013 11
2.5X
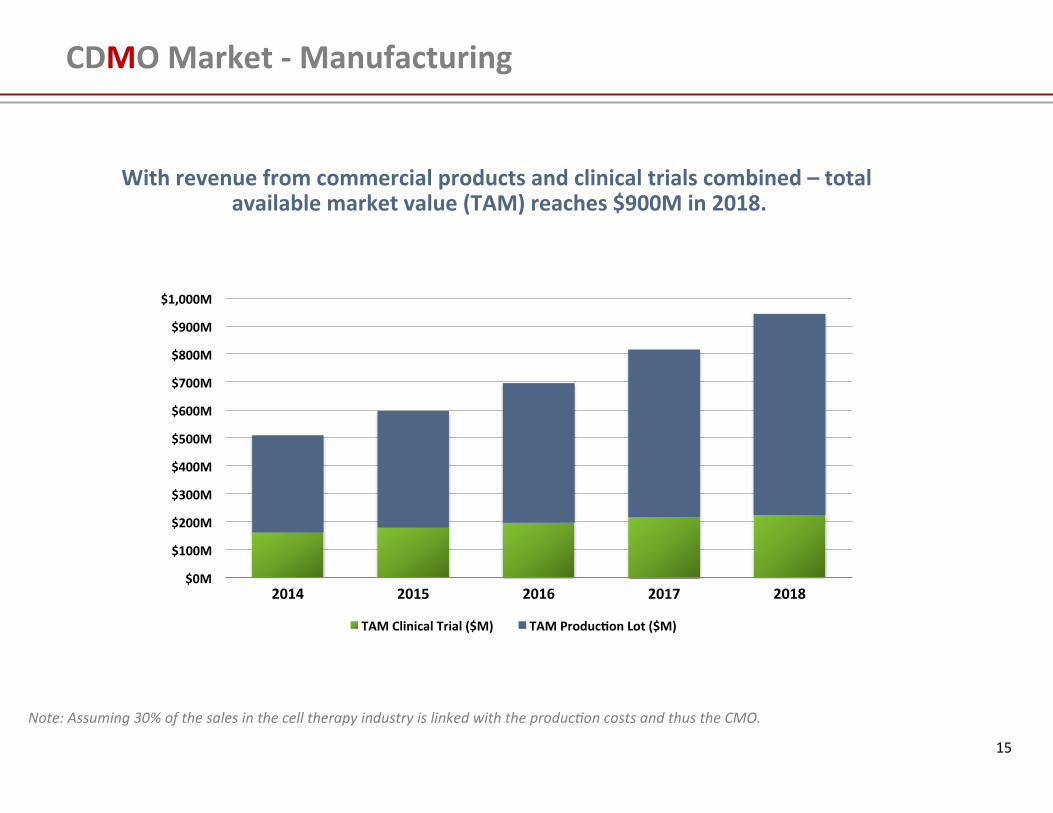
CDMO Market -‐ Manufacturing
With revenue from commercial products and clinical trials combined – total available market value (TAM) reaches $900M in 2018.
Note: Assuming 30% of the sales in the cell therapy industry is linked with the produc4on costs and thus the CMO.
$0M
$100M
$200M
$300M
$400M
$500M
$600M
$700M
$800M
$900M
$1,000M
2014 2015 2016 2017 2018
TAM Clinical Trial ($M) TAM Produc4on Lot ($M)
15

Tech transfert
Process development
CoGs / Batch
Total out of the pocket expense for customer
R&D Phase I Phase II Phase III Commercial
Year 1 Year 2 Year 3 Year 4 Year 5
Customer acquired in R&D / phase I
Customer acquired in phase II/III
AFract customers during early stage • to minimize tech transfer costs • to enable process development… • to provide cost efficient
manufacturing… • throughout en,re product lifecylce
Process development tools & exper,se are key
Business Strategy: Lock IN Early
34
Total out of pocket customer expense
Development Manufacturing

• Rental of exis,ng US cost-‐efficient clean room and high quality infrastructure o Hospital o Cluster o Local incubators / cell therapy ecosystem
• Deploy qualified work force in strategic
area(s) o Washington, DC o Boston
• Final stage nego,a,ons with MAJOR US Cancer Center – JV partnership o Late stage research thru P2 clnical trials o Companies exit to MaSTherCell for P3
and Commercial scale manufacture o Leverage JV and Brand for EU expansion /
compe,,ve advantage
Expansion Strategy – US Market Entry
36

(OTCQB:ORGS) Strategic and Financial Ra4onale for Merger
Complimentary management teams combine to strengthen overall company leadership
Leverages exis,ng collabora,ons (Orgenesis, MaSTerCell, ATMI) to realize $25M -‐ $50M in opera,onal synergies:
• Revenue cycle management
• Overhead efficiencies
• Supply chain management
• 3rd party collabora,on / partnerships
Increase scale, expand geographic footprint, and enhance technology planorm
COGs efficiencies to Orgenesis make acquisi,on accre,ve in Yr 1 of product launch
Orgenesis revenue genera,on expedites MaSTherCell ,me to profitability by 2 years
Individual Corporate Brands retained to drive diversified business strategy, while maximizing technical support and P&L efficiencies

§ Future Growth Drivers / Next Steps

Future Growth Drivers / Next Steps
Opera4ons § Complete European GMP manufacturing of clinical grade cells § Expand U.S. opera,ons (clinical, manufacturing, commercial) § Submit IND § Ini,ate Phase 1b trials in U.S. and EU Complete near-‐term financing § Complete $10M financing of current opera,ng plan -‐ $3.5M already raised
Execute medium-‐term Capital Markets plan § Ini,ate roadshow with US investment bank consor,um to raise capital for P1 expansion § File S-‐1 § Up-‐list to NASDAQ
(OTCQB:ORGS)

Management Team
Vered Caplan – Chairman and Interim Chief Execu4ve Officer, Orgenesis Ltd § Formerly served as CEO of GammaCan, a company focused on the use of immunoglobulins for the treatment of cancer. § Serves as director of various companies including: Op,cul Ltd., a company involved with op,c based bacteria classifica,on; Inmo,on Ltd., a company focused on self-‐propelled disposable colonoscopies; Nehora Photonics Ltd., a company involved with a non-‐invasive blood monitoring; Ocure Ltd., a company focused with wound management; Eve Medical Ltd., a company involved with hormone therapy for Menopause and PMS; and Biotech Investment Corp., a company focused on prostate cancer diagnos,cs. Scoq Carmer – Chief Execu4ve Officer (Orgenesis Inc, North America) § Led the U.S Specialty Care Division of AstraZeneca PLC (LSE:AZN), a $93 billion pharmaceu,cals company; responsible for the company’s pornolio of specialty care biopharmaceu,cal products. § Former EVP, Commercial Opera,ons of MedImmune -‐ acquired by AstaZeneca (LSE:AZN) for ~$16 billion. § Served as VP, Immunology for Genentech, Inc.; responsible for the U.S. launches of Rituxan and ACTEMRA in Rheumatoid Arthri,s. § Served as Global Therapeu,c Area Head for Bone and Metabolic Disorders at Amgen, Inc.; responsible for global development and commercializa,on strategies for denosumab (Xgeva and Prolia). § Began his career at GlaxoSmithKline plc (LSE:GSK), where he held various posi,ons of increasing responsibility in sales, marke,ng, strategic pricing and business development.
(OTCQB:ORGS)

Management Team (con0nued)
Hugues Bultot – Chief Execu4ve Officer (MaSTherCell); Member, Orgenesis BoD Serial life sciences entrepreneur, ‘from science to business’. • Former CEO of Artelis, a company focused on disposable bioreactors and now integrated into Pall
Life Sciences. Co-‐founded company in 2005, developed the business model, helped acquire ini,al customer base, ini,ated diversifica,on in Cell Therapy area, and eventually nego,ated trade sale in 2010.
• Successfully developed a global network in the bio-‐process industry – big pharma, academic ins,tu,ons, equipment supplier, NGO…
• Recently co-‐founded Univercells, a company focused on low-‐cost biopharmaceu,cals – vaccines, mAbs and recombinant proteins – bringing further depth to his industry-‐leading knowledge and exper,se in low-‐cost manufacturing to the benefit of MaSTherCell.
• Served as Director of various companies during his career as a private equity manager, as a tech transfer manager and as a corporate finance specialist.
Sarah Ferber, Ph.D. – Chief Scien4fic Officer & Founder • Studied biochemistry at the Technion under the supervision of Professor Avram Hershko and
Professor Aaron Ciechanover, winners of the Nobel Prize in Chemistry in 2004. • Completed a post-‐doctoral fellowship at the Joslin Diabetes Center at Harvard Medical School.
Her breakthrough discovery suggested that humans carry their own 'stem-‐cells' throughout adulthood, thus obvia,ng the need for embryonic stem cells for genera,ng an organ in need.
• Most of the research was conducted in Prof. Ferber’s lab, in the Endocrine Research Lab at the Sheba Medical Center, and currently employs 11 scien,sts.
• Received TEVA, LINDNER, RUBIN and WOLFSON awards for this research. • Research work has been funded over the past 10 years by the Juvenile Diabetes Research
Founda,on (JDRF), the Israel Academy of Science founda,on (ISF) and D-‐Cure, a non-‐profit organiza,on.
(OTCQB:ORGS)

OTCQB:ORGS | Company Presenta4on – October 2014
Company Contact:
ScoF Carmer Chief Execu,ve Officer Orgenesis Maryland, Inc.
Headquarters: Orgenesis, Inc. Germantown Innova,on Center 20271 Goldenrod Lane Germantown, MD 20876
Telephone: 301.204.1983 Email: [email protected] Corporate Website: (www.orgenesis.com)
Investor Rela4ons:
Tobin Smith
NBT Capital Markets 240-‐483-‐4629 (office) Email: [email protected]

Appendix

Extensive Patent Porvolio
IP Pornolio Patent Title Pub. Date US 2012/0329710 A1 patent applica,on
Methods of Inducing Regulated Pancrea,c Hormone Produc,on in Non-‐Pancrea,c Islet Tissues
December 27, 2012
US 8119405 granted patent
Methods of Inducing Regulated Pancrea,c Hormone Produc,on in Non-‐Pancrea,c Islet Tissues
February 21, 2012
AU 2004/236573 B2 grandet patent
Methods of Inducing Regulated Pancrea,c Hormone Produc,on in Non-‐Pancrea,c Islet Tissues
October 22, 2009
EP 1354942 B1 granted patent
Induc,on of insulin-‐producing cells January 30, 2008
EP 1180143 B1 granted patent
IN Vitro Methods of Inducing Regulated Pancrea,c Hormone Produc,on in Non-‐Pancrea,c Islet Tissues, Pharmaceu,cal Composi,ons Related Thereto
May 9, 2007
US 2005/0090465 A1 patent applica,on
Methods of Inducing Regulated Pancrea,c Hormone Produc,on in Non-‐Pancrea,c Tissues
April 28, 2005
AU 779619 B2 grandet patent
Methods of Inducing Regulated Pancrea,c Hormone Produc,on in Non-‐Pancrea,c Tissues
February 3, 2005
AU 2004/236573 A1 patent applica,on
Methods of Inducing Regulated Pancrea,c Hormone Produc,on in Non-‐Pancrea,c Tissues
November 18, 2004
WO 2004/098646 A1 patent applica,on
Methods of Inducing Regulated Pancrea,c Hormone Produc,on in Non-‐Pancrea,c Islet Tissues
November 18, 2004
WO 2004/098646 A1R1 patent applica,on
Methods of Inducing Regulated Pancrea,c Hormone November 18, 2004
(OTCQB:ORGS)
“Methods Of Inducing Regulated Pancrea4c Hormone Produc4on In Non-‐Pancrea4c Islet Tissues” § Patent granted in U.S. & Australia § Published in Europe & Japan
“Methods Of Inducing Regulated Pancrea4c Hormone Produc4on” § Patent granted in Australia & Europe § Published in Japan & Canada Currently filing third family of patents protec4ng produc4on process