overview platelets – normal physiology – categories of thrombocytopenias itp ttp hit...
TRANSCRIPT


Overview
• Platelets– Normal Physiology– Categories of Thrombocytopenias
• ITP• TTP• HIT
• Thrombocytopathy• Thrombocytosis

Normal Physiology-Production and Number
Platelets are normally made in the bone marrow from progenitor cells known as megakaryocytes.
Normal platelet lifespan is 10d. Every day, 1/10 of platelet pool is replenished.
Normal platelet count is between 150 and 450 x 109/l.

Platelet Response
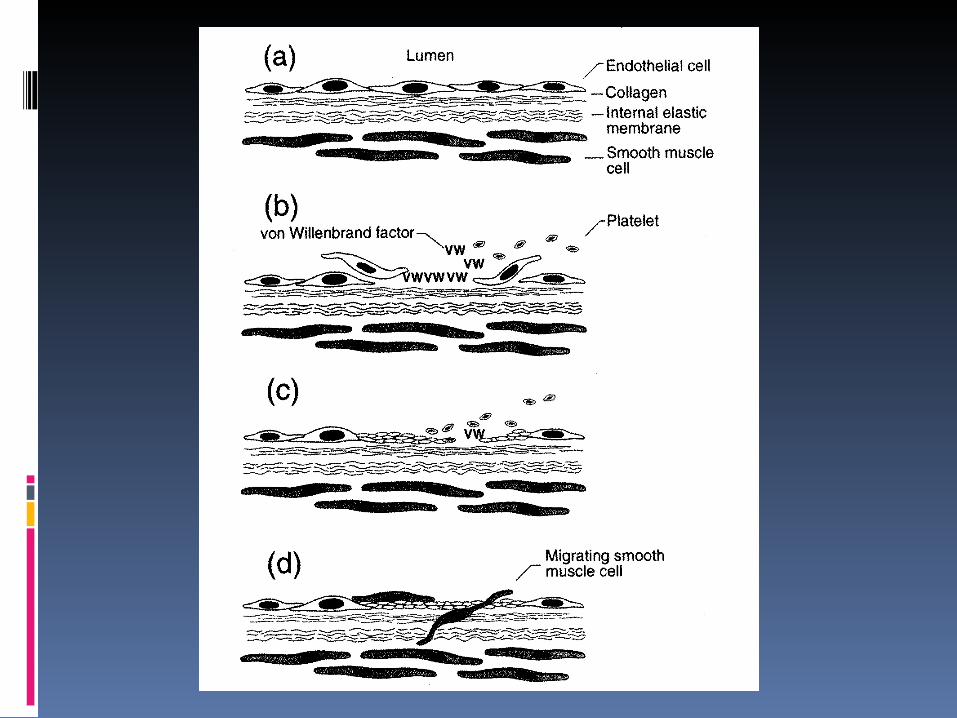
•Platelets provide phospholipid scaffold for thrombin generation.
•Platelets adhere
to vessel wall, then
aggregate, leading
to formation of a
platelet plug


Platelets physiology
PLT contain: mitochondria, glycoprotein containing granules, lysosomes, and:
2 types of granules:
-granules: fibrinogen, von Willebrand f., fibronectin, f. V, PLT f. 4, -thromboglobulin, thrombospondin, PDGF
dense granules: serotonin, calcium, ADP, ATP
PLT surface: GpIb: receptor for vWf
GpIIb-IIIa: receptor for fibrinogen, fibronectin, vitronectin

Platelet disorders
Thrombocytopenia
Thrombocytopathy
Combined
Thrombocytosis
Platelet disorders
Thrombocytopenia
Thrombocytopathy
Combined
Acquired
Congenital/inherited
Platelet disorders
Thrombocytopenia
Thrombocytopathy
Combined
Acquired
Congenital/inherited
Symptoms: Asymptomatic Bleeding Thrombosis

Thrombocytopenias
PLT < lower limit of the range, usualy < 130 x 109/l
ITP definition: PLT < 100 x 109/l Severe thrombocytopenia: PLT ≤ 20 x 109/l
Congenital (rare): Fanconi´s s., Wiskott-Aldrich´s s., May-Hegglin a., Bernard-Soulier´s s., amegakaryocytic trombocytopenia. Often include thrombocytopathy
Acquired: frequent

Thrombocytopenia:5 broad categories of causes
• Pseudothrombocytopenia
• Underproduction (AL, MDS, congenital TP, infiltration of bone marrow)
• Peripheral Destruction or consumption (ITP, TTP, DIC)
• Splenic sequestration (hypersplenism)
• Other: dilution (multiple erytrocyte transfusions etc.)
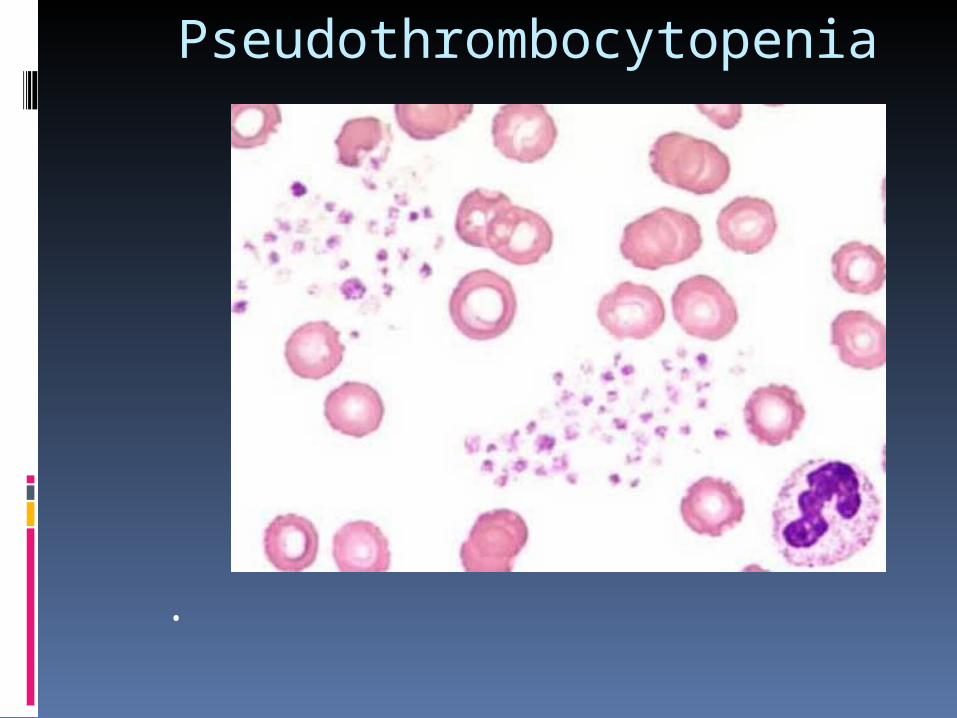
Pseudothrombocytopenia
•

Pseudothrombocytopenia• Artificial platelet clumping in the tube with EDTA
• Platelet clumping is of no clinical significance
• No increased risk of bleeding or clotting
• How to confirm:
1. Clumps in light microscopy 2. Repeated blood count test using citrate or Heparin as anticoagulant agent reveal normal PLT count

Thrombocytopenia due to Peripheral Destruction
Non-immune mechanisms: Platelet activation and consumption: e.g. TTP and DIC
Immune Mechanisms: ITP antibody-mediated platelet destruction may be primary, secondary, or drug-
induced.

ITP – Immune Thrombocytopenia (in the past: „Idiopathic Thrombocytopenic Purpura“) Definition: isolated thrombocytopenia with no clinically apparent associated conditions or other causes of thrombocytopenia.
Etiology: autoantibodies directed against platelets coat platelet surface. IgG-coated platelets are taken up by RE system.
Incidence: approximately 100 per million; half of these are children. In adults, two peaks: one are young (<40) with female predominance, one are older (>60), no gender predominance.

ITP – Immune Thrombocytopenia (in the past: „Idiopathic Thrombocytopenic Purpura“) Definition: isolated thrombocytopenia with no clinically apparent associated conditions or other causes of thrombocytopenia. PLT < 100x109/l
Etiology: autoantibodies directed against platelets coat platelet surface. IgG-coated platelets are taken up by RE system.
Incidence: approximately 100 per million; half of these are children. In adults, two peaks: one are young (<40) with female predominance, one are older (>60), no gender predominance.
The most common cause of thrombocytopenia

ITP: terminology
Newly diagnosed ITP: within 3 months from
diagnosis Persistent ITP: between 3 to 12
months from diagnosis. Chronic ITP: lasting for more than 12
months

Severe ITP
Presence of bleeding symptoms at presentation sufficient to mandate
treatment, or occurrence of new bleeding symptoms requiring additional therapeutic intervention with a different platelet-enhancing agent or an increased

Autoantobodies in ITP: 75% GpIIb/IIIa (CD41) (αβ-integriny)
GpIbIX (CD42) HIV positive: anti-IIIa
12- lipooxygenase
Arach. acid
NADPH oxidase
Superoxides + free 02 radicals

Autoantobodies in ITP: 75% GpIIb/IIIa (CD41) (αβ-integriny)
GpIbIX (CD42) HIV positive: anti-IIIa
12- lipooxygenase
Arach. acid
NADPH oxidase
Superoxides + free 02 radicals
OpsonisationADCC
Ab mediated autocytolysis

ITP – pathogenesis
+
differentiation & maturation of MGKC apoptosis of megakaryocyte
Antibodies
T LYMPHOCYTES
Shorter PLT lifespan
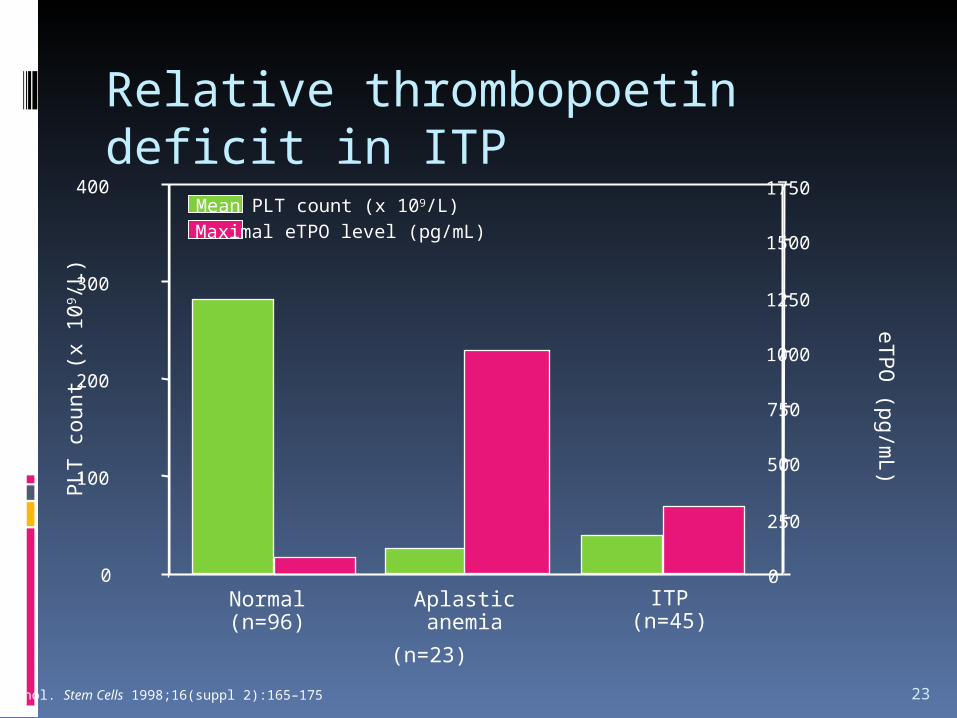
Relative thrombopoetin deficit in ITP
23
Normal(n=96)
Aplasticanemia
ITP(n=45)
0
100
200
300
400
0
250
500
750
1000
1250
1500
1750Mean PLT count (x 109/L)Maximal eTPO level (pg/mL)
PLT
cou
nt (
x 10
9 /L)
eTP
O (pg/m
L)
(n=23)
Podle: Nichol. Stem Cells 1998;16(suppl 2):165–175
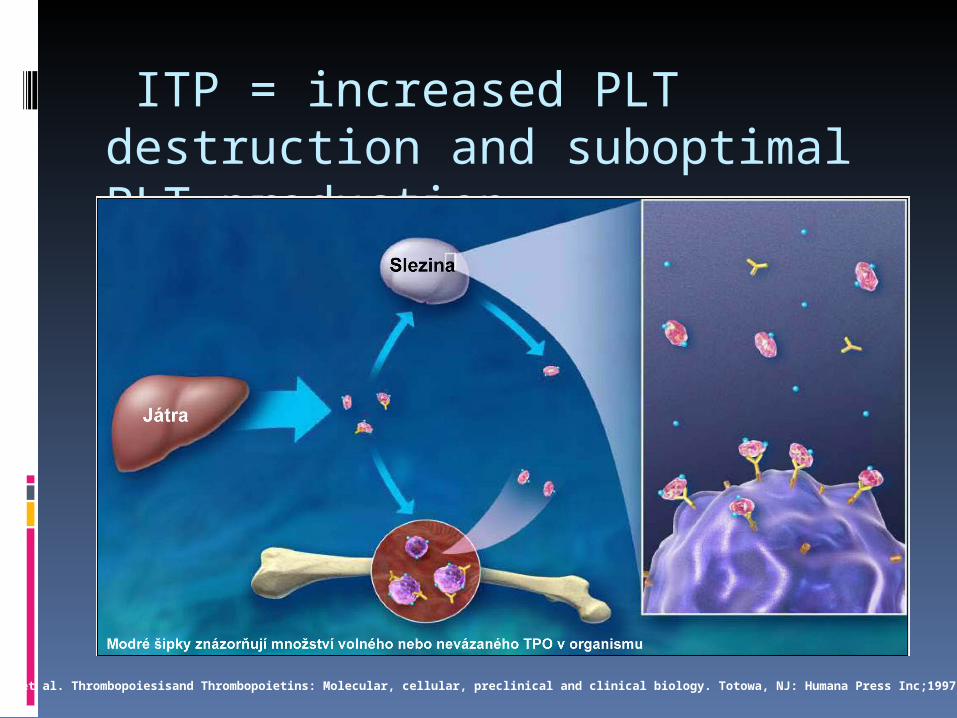
ITP = increased PLT destruction and suboptimal PLT production
Adapted from: Kuter et al. Thrombopoiesisand Thrombopoietins: Molecular, cellular, preclinical and clinical biology. Totowa, NJ: Humana Press Inc;1997

Role of lymphocytes in pathogenesis of ITP
• Cytokine profile TH1/Th0 corelates with disease activity
• Regulatory T cells reduced + not functional
• T cells can directly influence MGKCs and decrease PLT production
• 25% ITP without detected antibodies: „T cell - mediated toxicity“

B lymfocytes in ITP
• Autoantibodies production
BAFF (B cell activating factor): TNF family
- Produced and released by monocytes a T lymphocytes
- increases lifespan of B and T lymphocytes decreases apoptosis
- BAFF itself increases apoptosis in
thrombocytes - mRNA BAFF in ITP patients s ITP is increased
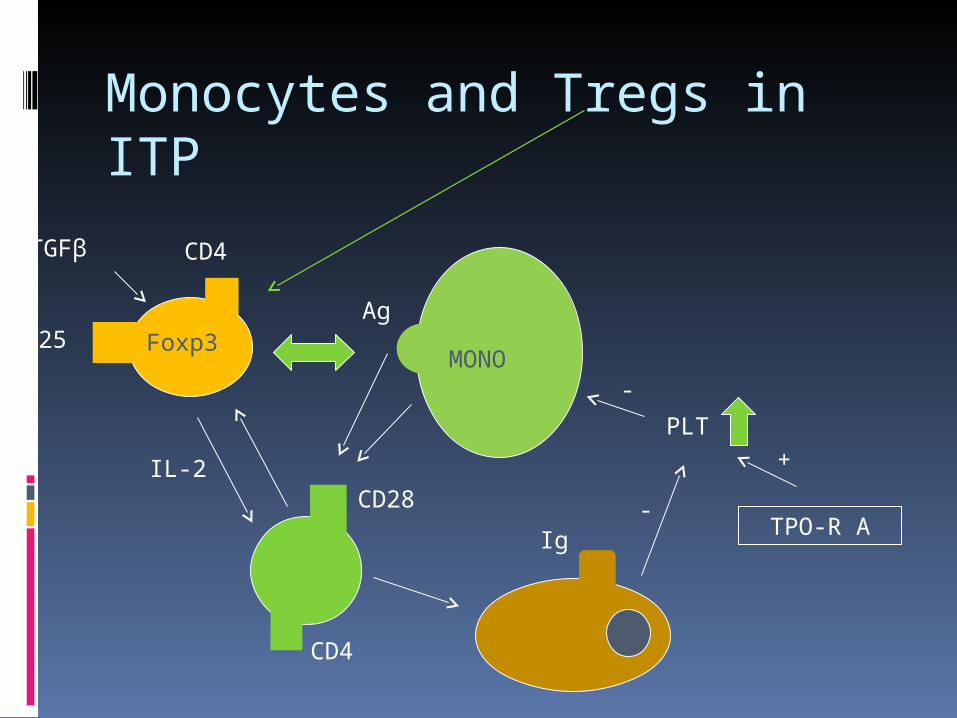
Monocytes and Tregs in ITP
CD4
CD25 Foxp3
CD28
Ag
IL-2
TGFβ
MONO
TPO-R A
CD4
PLT -
Ig-
+

ITP - Diagnosis
ITP is a Diagnosis of Exclusion (diagnosis per exclusionem)
No specific laboratory test can confirm diagnosis of the ITP
Need to exclude other causes of thrombocytopenia
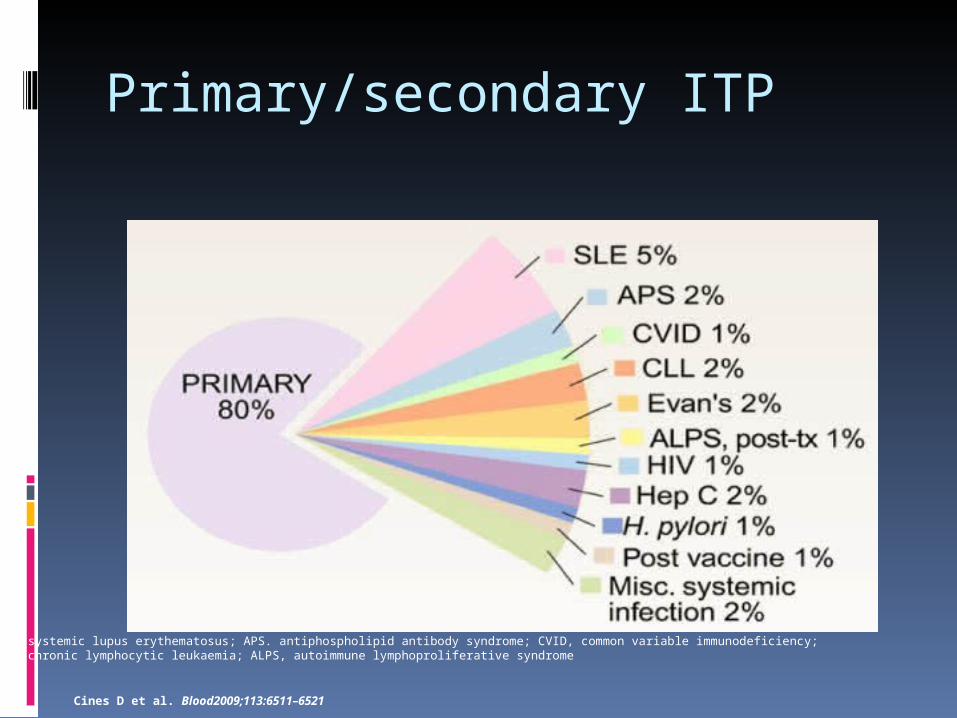
Primary/secondary ITP
SLE, systemic lupus erythematosus; APS. antiphospholipid antibody syndrome; CVID, common variable immunodeficiency; CLL, chronic lymphocytic leukaemia; ALPS, autoimmune lymphoproliferative syndrome
Cines D et al. Blood2009;113:6511–6521

Primary/secondary ITP
SLE, systemic lupus erythematosus; APS. antiphospholipid antibody syndrome; CVID, common variable immunodeficiency; CLL, chronic lymphocytic leukaemia; ALPS, autoimmune lymphoproliferative syndrome
Cines D et al. Blood2009;113:6511–6521

Evaluation of Patient with Low Platelets
History Has the patient ever had a normal platelet count?
Family history Carefully review medications, including Over the
counter meds. Antibiotics, quinine, anti-seizure medications
Ask about other conditions which may be associated with low platelets Liver Disease/hepatitis Thyroid Disease - both hypo- and hyper- Infections: viral, rickettsial Pregnancy
Consider other conditions which may be associated with ITP Lupus erythematosus, CLL, lymphoma,
antiphospholipid syndrome
Evaluation of Patient with Low Platelets
Physical Evaluate for lymphadenopathy and splenomegaly Look for signs of bleeding Blood blisters and oral petechiae, ie “Wet Purpura”
best harbinger of intracranial hemorrhage Laboratory Data
Other blood counts should be normal. Check B12 and folate levels. Look at peripheral blood smear to exclude
pseudothrombocytopenia, exclude TTP (especially if anemia also present.)
Send coagulation screens (PT/PTT) to exclude DIC Send HIV, hepatitis serologies and TSH H. pylori: antigen in stools or breath urease test
Consider doing a bone marrow biopsy and smear Obligatory in pts >60 years of age
Megakaryocytes should be present.

Thrombocytopenia-How low is too low?
100,000 - 50,000: no symptoms No treatment generally required.
50,000 - 20,000: first symptoms Generally need to begin therapy
20,000-10,000: may be life-threatening Sometimes requires admission
<10,000: risk for spontaneous intracranial hemorrhage, usually requires inpatient treatment

Need for PLT counts if normal PLT function
Dental prophylaxis (descaling, deep cleaning) ≥20–30 × 109/l
Simple extractions ≥30 × 109/lComplex extractions ≥50 × 109/lRegional dental block ≥30 × 109/lMinor surgery ≥50 × 109/lMajor surgery ≥80 × 109/lMajor neurosurgery ≥100 × 109/lVaginal delivery: 50 x 109/l
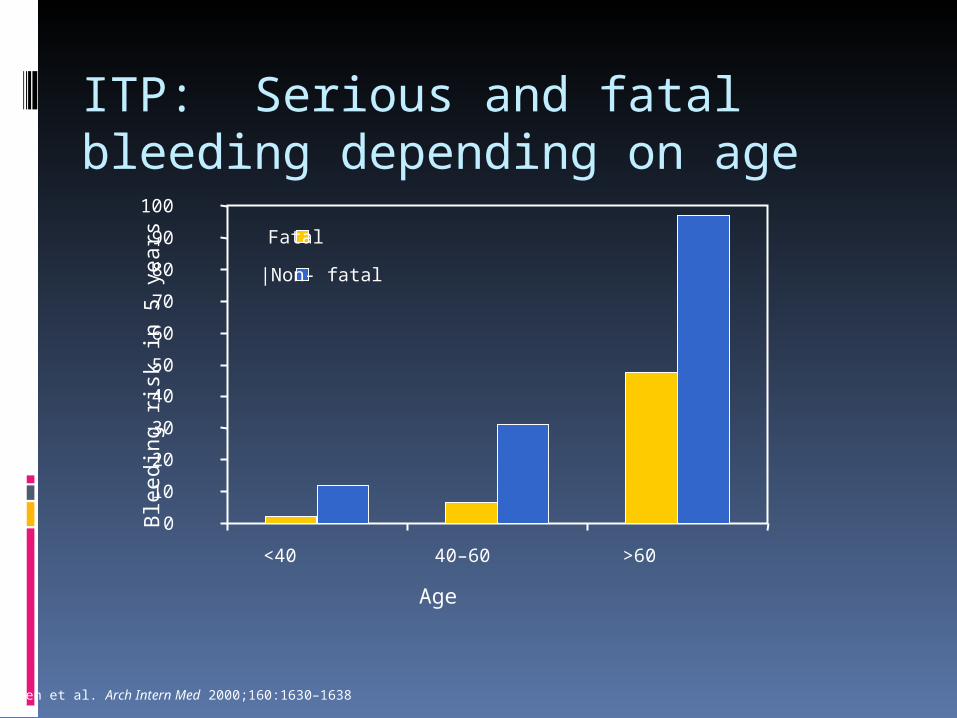
ITP: Serious and fatal bleeding depending on age
Podle: Cohen et al. Arch Intern Med 2000;160:1630–1638
Age
Ble
edin
g ris
k in
5 y
ears
0
10
20
30
40
50
60
70
80
90
100
<40 40–60 >60
Fatal
|Non- fatal

Management of ITP: Asymptomatic Adult
If platelet count is >40-50 x109/l, no therapy is required. Check platelet counts at designated intervals.
If platelet count is < 20-30 x109/l , begin therapy with corticosteroids.
Stop all NSAIDS and ASA to improve platelet function.

Corticosteroids in ITP
1. Prednison: 0,5 – 2mg/kg of BW/day
1. Or: Methylprednisolone i.v. 1g/D for 3-5 days followed by oral Prednison
2. Or: Dexamethason 40mg p.o.(i.v.) D1-4
CAVE: in pregnancy only Prednison 20mg/day and less and No corticosteroids in the 1st trimester!!

Initial Management of ITPAdult with Symptomatic Purpura
If platelet count is >10, treat with prednisone alone
If suspicious intracranial or GI bleeding (ulcer etc.), treat with itravenous immunoglobulins IVIg 1g/kg/d x 2d. - may require admission
If patient has severe bleeding and life is in danger: may need platelet transfusions
Along with prednisone, add Calcium and Vitamin D to prevent bone loss.

Subsequent Management of ITPAdult with Symptomatic Purpura
Follow platelet counts daily until >20, then can d/c patient with close follow-up
Once platelet count normalizes, commence a slow steroid taper over 6-8 weeks.
1/3 of adults will go into remission. 2/3 of patients will relapse during or
after steroid taper.
Management of Relapsed ITP• Once the patient relapses, may need to use
steroids to increase the platelet count out of the danger range, but THIS CANNOT SUBSTITUTE FOR DEFINITIVE THERAPY.
• Prednisone is now a crutch to support a dangerously low platelet count. Long corticosteroid treatment should be avoided.
• Options today include: 1. splenectomy, if contraindicated (including
patient´s refusal): 2. TPO mimetics (Romiplostim, Eltrombopag). 3. In the past: intermittent treatment with anti-
D immune globulin.

Management of Relapsed ITP: Splenectomy Splenectomy is effective in 2/3 of
patients, leading to normal platelet counts.
Can be performed via open method or laparoscopically.
Need to vaccinate against encapsulated bacteria 2 weeks before procedure.
May need steroids and/or IVIg before procedure to boost platelet counts preoperatively.

Management of Relapsed ITPAnti-D Immune Globulin
Can be used as a substitute for IVIg for maintenance therapy
Especially useful in patients with contraindications to splenectomy.
Coats red cells with IgG and allows red cells to serve as decoy for splenic macrophages.
Patient must be Rh positive. Not effective after splenectomy. Designed to cause hemolytic anemia--Hgb
may drop as much as 3g/dl. Intermittent dosing may allow patients to
avoid splenectomy.

Fc carrier - part of Ig
Podle: Bussel et al. N Engl J Med 2006;355:1672–1681
Peptide domain
Romiplostim: structure
Bussel et al. N Engl J Med;355:1672–1681. Copyright © 2006 Massachusetts Medical Society. All rights reserved.
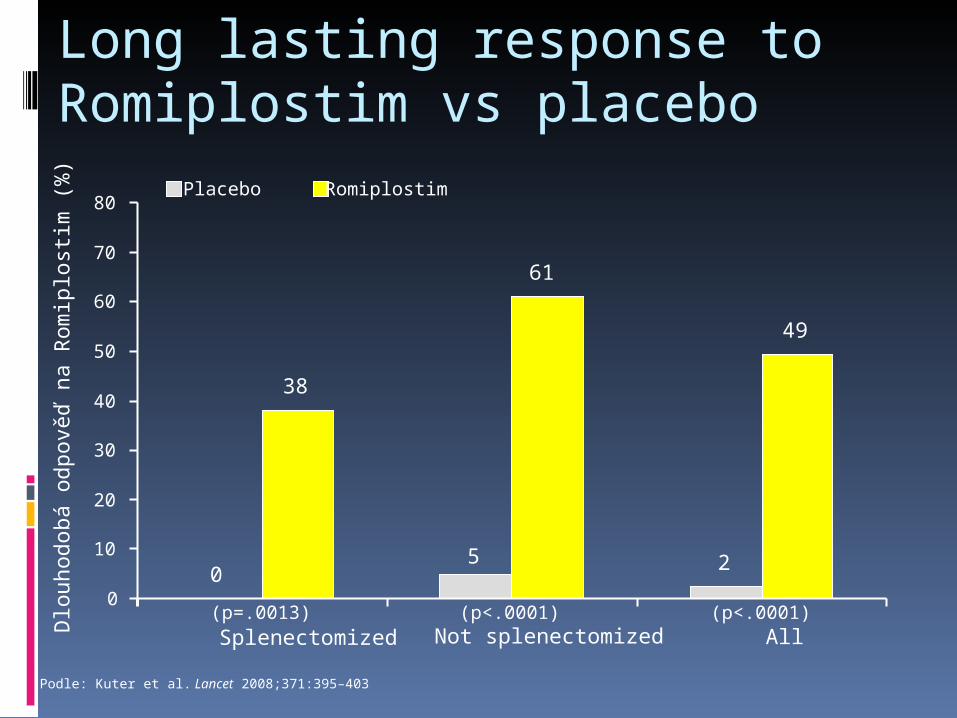
Long lasting response to Romiplostim vs placebo
Dlo
uhod
obá
odpo
věď
na
Rom
iplo
stim
(%
)
(p=.0013) (p<.0001) (p<.0001)
05 2
38
61
49
0
10
20
30
40
50
60
70
80
Splenectomized Not splenectomized All
Placebo Romiplostim
Podle: Kuter et al. Lancet 2008;371:395–403
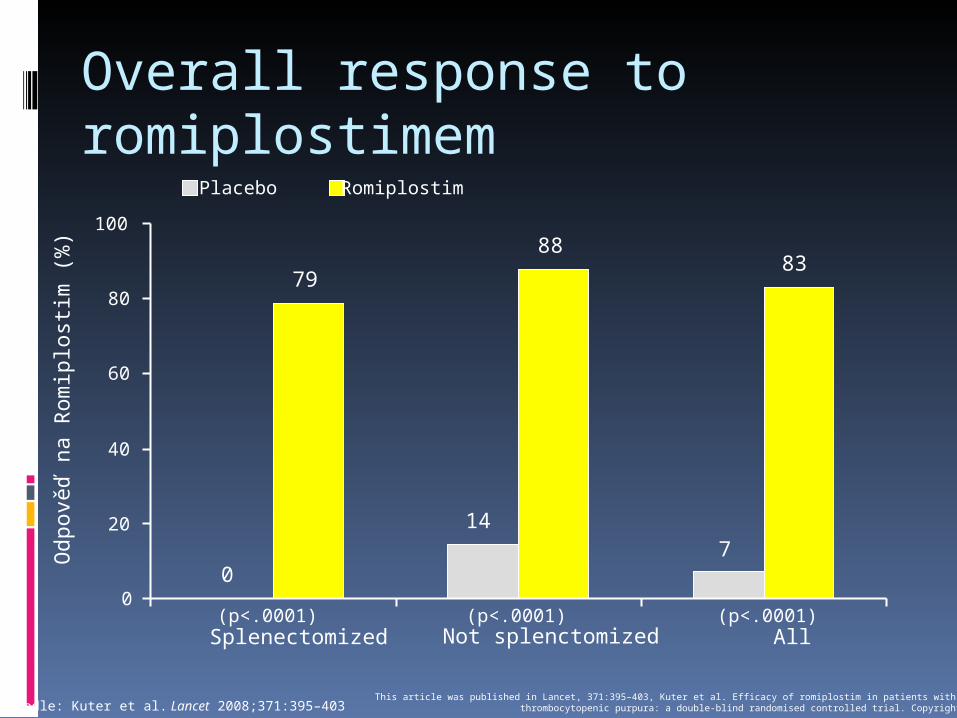
Overall response to romiplostimem
Odp
ověď
na
Rom
iplo
stim
(%
)
(p<.0001) (p<.0001) (p<.0001)
0
147
79
8883
0
20
40
60
80
100
Splenectomized Not splenctomized All
Placebo Romiplostim
Podle: Kuter et al. Lancet 2008;371:395–403This article was published in Lancet, 371:395–403, Kuter et al. Efficacy of romiplostim in patients with chronic immune
thrombocytopenic purpura: a double-blind randomised controlled trial. Copyright Elsevier 2008
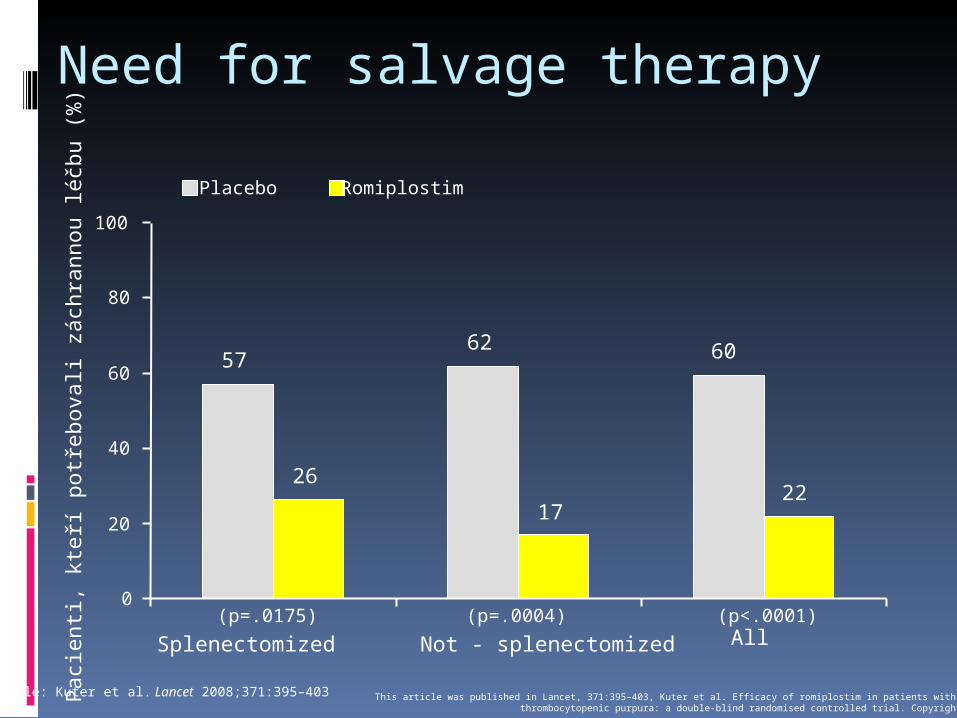
Need for salvage therapyP
acie
nti,
kteř
í pot
řebo
vali
zách
rann
ou lé
čbu
(%)
(p=.0175) (p=.0004) (p<.0001)
5762 60
26
1722
0
20
40
60
80
100
Splenectomized Not - splenectomized All
Placebo Romiplostim
Podle: Kuter et al. Lancet 2008;371:395–403 This article was published in Lancet, 371:395–403, Kuter et al. Efficacy of romiplostim in patients with chronic immunethrombocytopenic purpura: a double-blind randomised controlled trial. Copyright Elsevier 2008

Management of Relapsed ITPAnti-D Immune Globulin Can be used as a substitute for IVIg for
maintenance therapy Especially useful in patients with
contraindications to splenectomy. Coats red cells with IgG and allows red cells
to serve as decoy for splenic macrophages. Patient must be Rh positive. Not effective after splenectomy. Designed to cause hemolytic anemia--Hgb
may drop as much as 3g/dl. Intermittent dosing may allow patients to
avoid splenectomy.
No more available: induced several cases of fatal haemolytic anaemia

Case 1
A 18 y.o. female high school student presents with a rash over her lower extremities. She had a viral illness with pharyngitis 3 weeks ago. She has no other medical problems, and she takes no medications.
Physical examination reveals petechiae over the shins.
Platelet count is 1 x 109/l (!). The patient was admitted to the hospital
and was given DEX 40mg i.v. D1-4

Case 2 A 40 y.o. man presents with epistaxis to
the ER. He has no medical problems, and he takes no medications. He works as a big truck driver and has no occupational exposures. He is married and has 2 children.
Physical examination is remarkable only
for epistaxis and scattered petechiae.
The platelet count is 28 x109/l The patient is given PDN 1mg/kg/D as
outpatient but not able to drive a truck

Case 3
A 46 y.o. woman is found to have a platelet count of 20 x109/l on routine laboratory testing. She has some easy bruising and gum bleeding, but admits to not flossing.
She has no PMHx, and is on no medications. She works as gym instructor in a fitness center.
She is started on 1 mg/kg of prednisone. Adviced not to do gym lessons.

Management of Refractory ITP One third of patients will have an inadequate
response to splenectomy. Management of these patients is a chalange
and involves accepting that they have a chronic, incurable condition.
Target platelet counts should be lower--aim for about 30K or absence of bleeding.
Today:
Agonists of TPO receptor (TPO agonists):ROMIPLOSTIM, ELTROMBOPAG
In the past:

Management of Refractory ITP One third of patients will have an inadequate
response to splenectomy. Management of these patients is a chalange
and involves accepting that they have a chronic, incurable condition.
Target platelet counts should be lower--aim for about 30K or absence of bleeding.
Today:
Agonists of TPO receptor (TPO agonists):ROMIPLOSTIM, ELTROMBOPAG If not effective
In the past:

Treatment of Refractory ITP Immunosuppressive agents
Cyclophosphamide Azathioprine Rituximab (anti-CD20) – off label Cyclosporin A Mycofenolate mofetil
Adjunct agents Danazol Examine & Treat Helicobacter pylori
Other - Vincaalkaloids - Alemtuztumab (anti-CD52)
Stem cell transplant

Drugs Commonly Implicated in Thrombocytopenia
Beta-lactam antibiotics. Trimethoprim-sulfamethoxazole and
other sulfa drugs. Vancomycin. Quinine/quinidine. Heparin. Abciximab (ReoPro®). H2 blockers If a patient’s platelets fall, ALL
unnecessary drugs need to be stopped.

Case 4
A 60 y.o. woman presented with bleeding from her nose and mouth and gums.
PMHx – Hypertension, DM Medications - glucotrol, glucophage,
HCTZ, quinine for leg cramps PEx - petechiae over limbs and trunk,
blood blisters in mouth, epistaxis. Platelet count 2 x109/l

Case 4
Pt admitted to hospital, quinine stopped, patient treated with platelet transfusions and IVIg.
Platelet count rose to normal over the next 5-6 days.
Eight months later, thrombocytopenia recurred, and patient admitted to taking quinine again for recurrent leg cramps.

Drug Induced ITP
Usually, removing the offending agent is enough to allow the platelets to rise on their own.
If platelets are severely low, platelet transfusions may be required.
IVIg is particularly helpful in quinine-induced ITP.

Case 5 A 65 y.o. male smoker with a h/o
peripheral vascular disease presented to the ER with unstable angina. He was admitted to the hospital and placed on heparin. Platelet count on admission was 450. Cardiac catheterization showed severe 3-vessel coronary disease, and the patient was scheduled for CABG which occurred on hospital day #7.
Pre-op platelet count was 200. Post-op platelet count was 90.

Case 5 On hospital day #12, the patient
developed acute left leg swelling and a DVT was diagnosed by ultrasound. Platelet count was 150. The patient was started on IV heparin. The next day, he developed a pulseless left leg and had a platelet count of 30.
While in vascular radiology, he developed acute chest pain and suffered a cardiac arrest and subsequently died. Autopsy showed occlusion of all of his bypass grafts.

Evan´s syndrome
Immune thrombocytopenia (ITP)
+
Autoimmune hemolytic anemia (AIHA)

Heparin-Induced Thrombocytopenia (HIT) Seen in 1-3% of patients treated with
heparin Usually, 7-10 d after heparin started,
platelets fall by at least 1/3 to 1/2. Can occur earlier in patients who have been
previously exposed to heparin, even as SQ injections.
Less often but still risk exists with low molecular weight heparin
Caused by antibodies against the complex of heparin and PF4. These antibodies activate platelets.
Can lead, paradoxically, to THROMBOSIS, in up to half of patients.
More common in patients with vascular disease

Alternate Presentations of HIT/T Small drop in platelet count
(especially with skin necrosis) Earlier onset thrombocytopenia with
heparin re-exposure Delayed-onset thrombocytopenia/
thrombosis after stopping heparin Thrombosis after heparin exposure

HIT/T treatment
1. IF PLATELETS FALL ON HEPARIN, STOP HEPARIN IMMEDIATELY.
2. Stop heparin3. Stop heparin4. Use a different anticoagulant
1. Fondaparinux (Arixtra®)2. Lepirudin3. Argatroban

Case 6 A 36 y.o.woman presented in the summer with
fever, cefalea, nausea, vomiting and ppsychomotoric deterioration. Admitted to ICU of Neurology dept with provisional dg. Viral encephalitis – tickborn. On Hospital day 2, hemoglobin and platelet count both noted to drop. By hospital day 4, Hgb 70 g/l, Plts 12 x109/l. PT/PTT normal. Bilirubin 87 umol/l.
CSF clear from infection The patient recovered after 20
plasmaexchanges.

Thrombotic thrombocytopenic purpura (TTP)
Microangiopathic Hemolytic Anemia (MAHA) Elevated LDH, elevated bilirubin Schistocytes on the peripheral smear MUST BE PRESENT
Low platelets - MUST BE PRESENT
Fever
Neurologic Manifestations - headache, sleepiness, confusion, stupor, stroke, coma, seizures
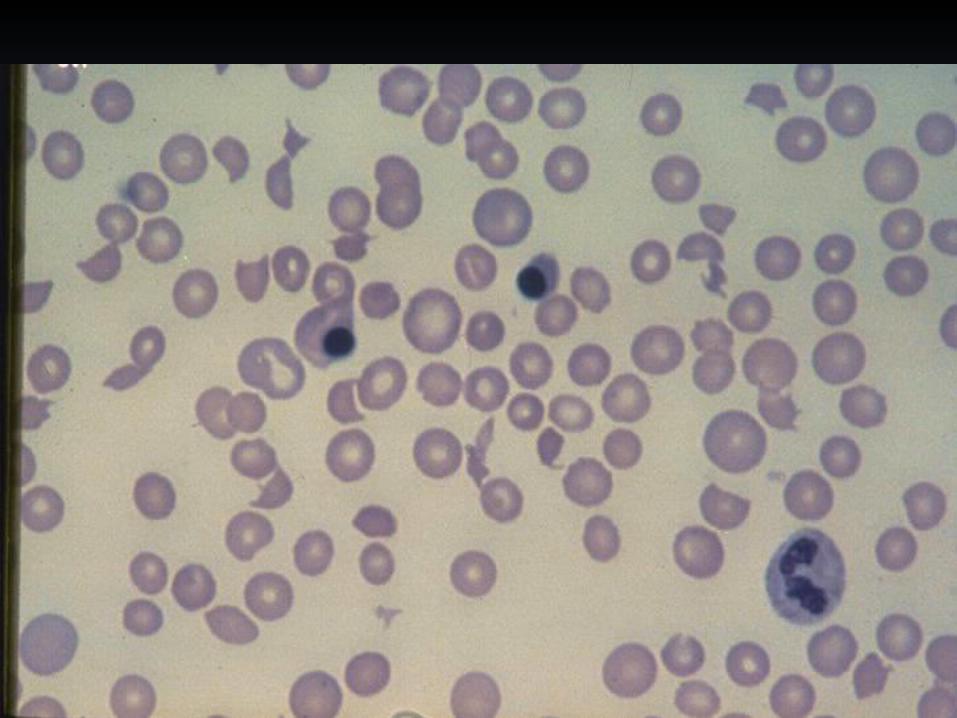

TTP - etiology
May be associated with an antibody against or a deficiency of the protease which cleaves the ultra-high molecular weight multimers of von Willebrand’s factor. These very high molecular weight vWF multimers cause abnormal platelet activation.
Can be induced by drugs, including ticlopidine, quinine, cyclosporine, tacrolimus, mitomycin C.
Increased incidence with pregnancy or HIV

TTP - Course and Prognosis 95% fatal prior to therapy, now 5% fatal.
Treatment relies on PLASMA EXCHANGE + corticosteroids Plasma exchange is superior to plasma infusion,
but if PLEX is delayed, give FFP. Remove all inciting agents. Platelet transfusions contra-
indicated. Multiple case reports of stroke and/or death
during or immediately after platelet transfusion. Can consider giving if life-threatening
hemorrhage is present, but avoid routine platelet transfusions.
Secondary measures if no response to plasma exchange include splenectomy, vincristine
HUS - Hemolytic Uremic Syndrome Usually classified along with TTP as
“TTP/HUS” Has fewer neurologic sequelae, more
renal manifestations. More abdominal pain in symptoms Usually precipitated by diarrheal illness,
especially E. coli O157:H7 („Shigatoxin“) or Shigella
Seen more in pediatric patients, usually has better prognosis. May respond less well to plasma exchange.

Thrombocytopathy
Congenital/inherited: rare
Acquired: frequent

Congenital/inherited thrombocytopathy Bernard – Soulier´s syndrome AR inehritance GP Ib/IX defect von Willebrand´s factor not
bound adhesivity defectiveClinically: bleeding of „platelet type“, may be
dengerous and fatalDg lab: giant PLT in the blood smear, often
thromocytopenia, abnormal PLT aggregation with Ristocetin. Negative reaction with anti GP Ib/IX Ab
Therapy: Symptomatic + DDAVP, rF VIIa

Glanzman´s thrombastheniaAR inheritanceDefect ib GPIIb/IIIa fibrinogen not
bound aggregation defectiveClinically: no spontenous bleeding but
after invasion: surgery, dental, delivery, etc.
Dg lab: morfology and No. of PLT normal but aggregation with ADP, epineprine, kolagen and thrombin affected
Th: symptomatic + rFVIIa

PLT secretory dysfunction Chediak-Higashi´s sy, Wiskott-Aldrich
´s sy, Hermansky-Pudlak´s sy Defects of δ and α granules Aspirine-like disease: realease from
granules defectDg lab: pathology in PLT aggregation
with colagen, arachidonic acid, ADP.Th: Symptomatic (thrombocytes),
DDAVP, rFVIIa

Acquired thrombocytopathy
1. Diseases accociated with thrombocytopathy: Myeloprolipherative diseases Myelodysplastic syndrome Leukemias + Lymphoprolipherative disease Paraproteinemia Renal insufficiency Liver failure Autoimmune diseases Excorporeal circulation

2. Medication-related thrombocytopathy
Met. arach. acid involved: acetylsalicylic ac., corticosteroids, NSA (ibuprofen, indomethacine, phenylbutasone, thromboxane antagonists, etc.)
PLT cAMP: dipyridamol, theofylin, phospohodiesterase inhibitors,
Clopidogrel (Plavix, Thrombex) ADP receptor blocade GPIIb/IIIa inactivation aggregation
Other: penicillin, cephalosporins, dextran, tricyclic antidepressants, antihistamins, betablockers, Ca antagonists, ticlopidin, vitE, UFH, etc.

Thrombocytosis PLT > 450 x109/l Clonal thrombocytosis = ET (essential
thrombocythaemia, PMF, PV, CML) Reactive thrombocytosis: - Bacterial infections: osteomyelitis,
pneumonia, TBC, - Postsplenectomy thrombocytosis- Iron defficiency- Bleeding- Th: if >700 x109/l consider antiaggregation

Thank You very much