parkinson disease (pd) and dementia with lewy bodies (dlb): … · parkinson disease (pd) and...
TRANSCRIPT

Parkinson disease (PD) and Dementia with Lewy bodies (DLB): Lewybody diseases (LBD)
Prof. Isidro Ferrer, Institut Neuropatologia, Servei Anatomia Patològica, IDIBELL-Hospital Universitari de Bellvitge, Universitat de Barcelona, CIBERNED, Hospitalet de LLobregat; Spain
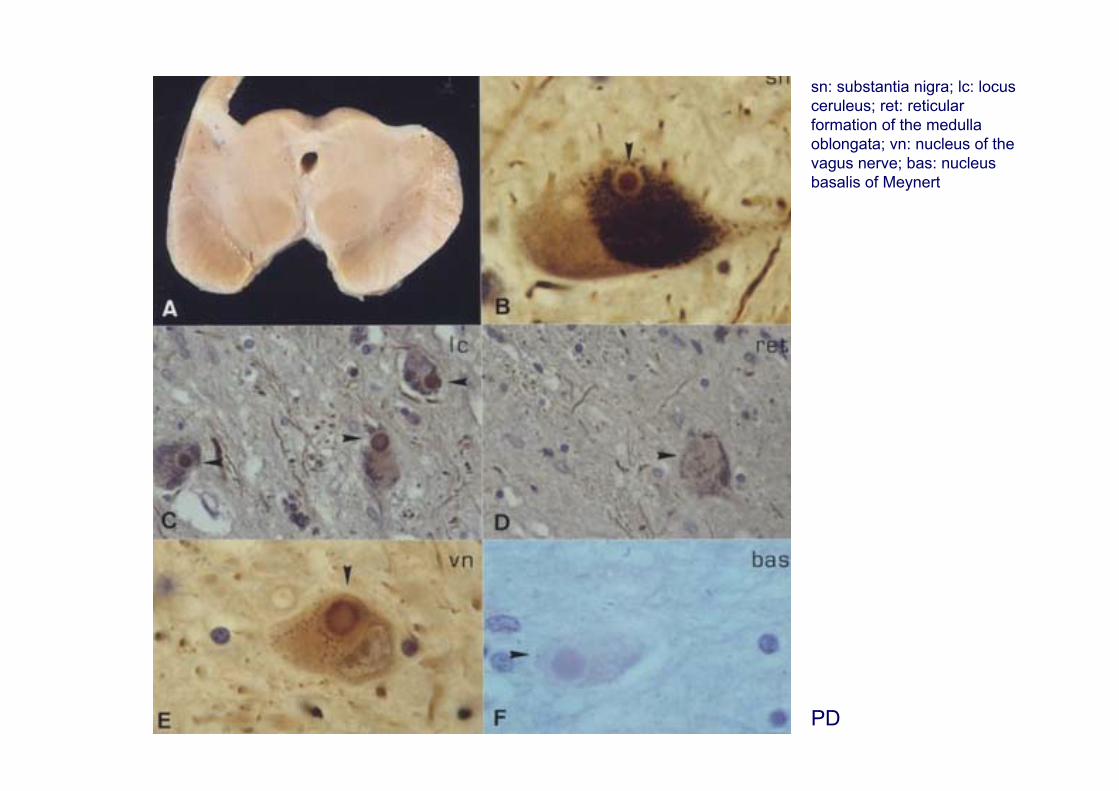
PD
sn: substantia nigra; lc: locus ceruleus; ret: reticular formation of the medulla oblongata; vn: nucleus of the vagus nerve; bas: nucleus basalis of Meynert

Lewy bodiesSpherical intracytoplasmic inclusions with concentric eosinophilic core and peripheral unique ormultilayered narrow, pale-stained, hallo. Diameter ranging from 8 to 30 µm. May be unique or multiple.
The core is formed by densely packed filaments and granular material. The hallo is formed by radially-arranged 7-20 nm intermediate filaments associated with granular electron-dense coating material andvesicular structures.
Composition:
Structural proteins: alpha-synuclein, tail regions of the heavy and medium components ofneurofilaments, tubulin, microtubule-associated proteins, beta-amyloid precursor protein, proteoglycans.
Enzymes associated with phosphorylation: calcium/calmodulin-dependent kinase II (CaMK II), cyclin-dependent kinase 5 (Cdk5), MAPKs: ERK-2, and phosphorylated MAPK/ERK-P, SAPK/JNK andp-38. Casein kinase 1 and 2 (CK-1 and CK-2); CK-2 phosphorylates alpha-synuclein at Ser129 in vitro.
Enzymes associated with ubiquitin-mediated proteolysis and stress: ubiquitin, polyubiquitin chains, ubiquitincarboxyl-terminal hydrolase (PGP9.5), multicatalytic protease (MCP), 26S proteasome, p62, Cu/Zn superoxide dismutase.
Synaptic-associated proteins: synaptophysin, chromogranin A.
Chaperones: alphaB-crystallin, 14-3-3
Schults CW (2006) Lewy bodies. PNAS 103:1661-1668
Wakabayashi K et al. (2007) The Lewy body in Parkinson’s disease: Molecules implicated in the formation and degradation of α-synuclein aggregates. Neuropathology 27: 494-506
Leverenz JB et al. (2007) Proteomic identification of novel proteins in cortical Lewy bodies. Brain Pathol 17: 139-145
Xia et al. (2008) Proteomic identification of novel proteins associated with Lewy bodies. Front Biosci 13: 3850-3856
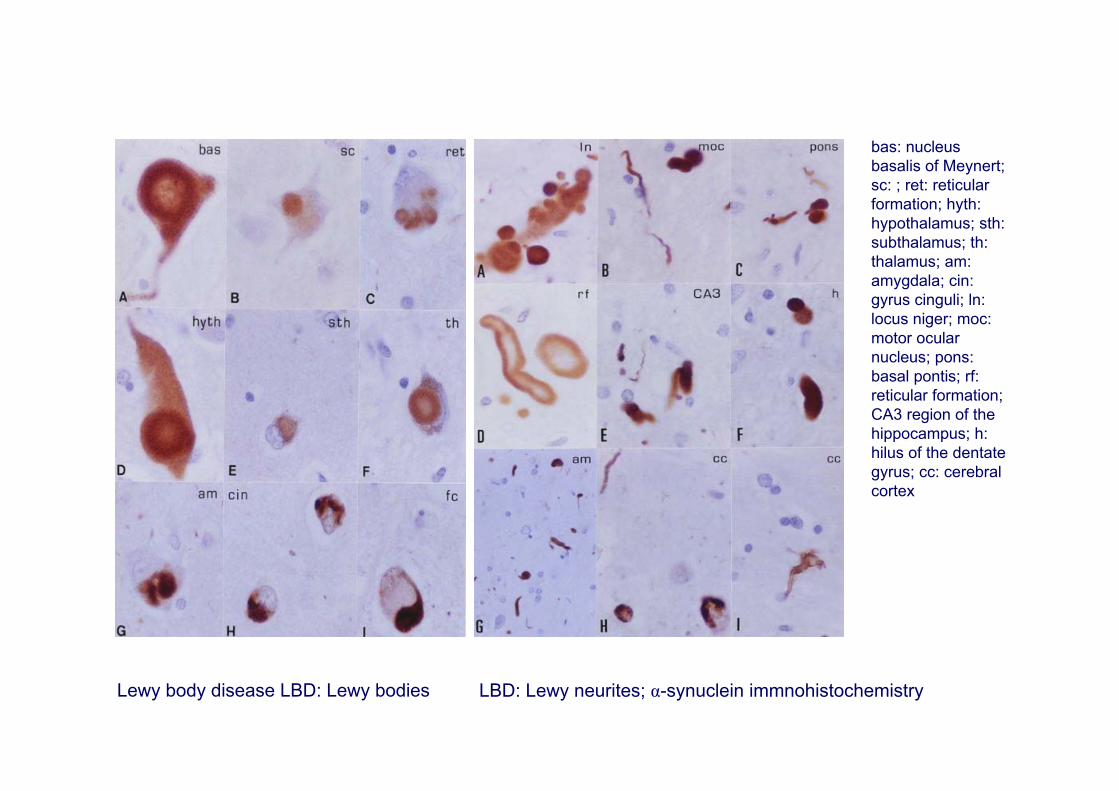
Lewy body disease LBD: Lewy bodies LBD: Lewy neurites; α-synuclein immnohistochemistry
bas: nucleus basalis of Meynert; sc: ; ret: reticular formation; hyth: hypothalamus; sth: subthalamus; th: thalamus; am: amygdala; cin: gyrus cinguli; ln: locus niger; moc: motor ocular nucleus; pons: basal pontis; rf: reticular formation; CA3 region of the hippocampus; h: hilus of the dentate gyrus; cc: cerebral cortex

DLB
pal: globus pallidus; am: amygdala; bas: nucleus basalis of Meynert; lc: locus ceruleus; th: thalamus; cc: cerebral cortex

Staging of brain pathology related to sporadic Parkinson’s disease
Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak ENeurobiology of Aging 24 (2003) 197-211
Stage 1: medulla oblongata: lesions in the dorsal IX/X motor nucleus and/or intermediate reticular zone (plus autonomic system)
Stage 2: medulla oblongata and pontine tegmentum: pathology of stage 1 plus lesions in the caudal raphe nuclei, gigantocellular reticular nucleus, and ceruleus-subceruleus complex (plus olfactory bulb and tract)
Stage 3: midbrain: pathology of stage 2 plus midbrain lesions, particularly in the pars compacta of the substantia nigra
Stage 4: basal prosencephalon and mesocortex: pathology of stage 3 plus prosencephaliclesions: amygdala, nucleus basalis of Meynert. Cortical involvement confined to the temporal mesocortex (transentorhinal region) and allocortex (CA2 plexus)
Stage 5: neocortex: pathology of stage 4 plus lesions in high order sensory association areas of the neocortex and prefrontal neocortex
Stage 6: advanced neocortex: pathology of stage 5 plus lesions in first order sensory association areas of the neocortex and premotor areas, occasionally mild changes in primary sensory areas and the primary motor field

Braak classification correlates with neurological deficits in the majority of patients with early onset andlong duration of the disease:Braak H, Del Tredici K, Bratzke H, Hamm-Clement J, Sandemann-Keil D, Rub U (2002) Staging of the intracerebral inclusion body pathologyassociated with idiopathic Parkinson’s disease (preclinical and clinical stages). J Neurol 249; suppl 3:1-5
Braak H, Rüb U, Jansen Steur EN, Del Treedici K, de Vos RA (2005) Cognitive status correlatesd with neuropathologic state in parkinson disease. Neurology 64: 1404-1410
Braak H, Muller CM, Rüb U, Ackermann H, Bratzke H, de Vos RA, Del Tedici K (2006) Pathology associated with sporadic Parkinson’s disease: where does it end? J Neural Transm Suppl 70: 89-97
Wolters ECh, Braak H (2006) Parkinson’s disease: premotor clinico-pathological correlations. J Neural Transm Suppl 70: 309-319
Przuntek H, Muller T, Riederer P (2004) Diagnostic staging of Parkinson’s disease: conceptual aspects. J Neural Transm 111: 201-216
Jellinger KA (2008) A critical reappraisal of current staging of Lewy-related pathology in human brain. Acta Neuropathol 116: 1-16
Braak classification, or better Lewy pathology, does not correlate with behavioral deficits, cognitiveimpairment and dementia in PD and DLB: Parkkinen L, Pirttila T, Tervahauta M, Alafuzoff I (2005) Widespread and abundant α-synuclein pathology in a neurologically unimpaired subject. Neuropathology 25: 304-314
Parkkinen L, Kauppinen T, Pirttila T, Autere JM, Alafuzoff I (2005) α-synuclein pathology does not predict extrapyramidal symptoms or dementia. Ann Neurol 57: 82-91
Weisman D, Cho M, Taylor C, Adame A, Thai LJ, Hansen LA (2007) In dementia with Lewy bodies, Braak stage determines phenotype, not Lewybody distribution. Neurology 69: 356-359
Parkkinen L, Pirttilä T, Alafuzoff I (2008) Applicability of current staging/categorization of alpha-synuclein pathology and their clinical relevance. ActaNeuropathol115: 399-407
Jellinger KA (2008) A critical reappraisal of current staging of Lewy-related pathology in human brain. Acta Neuropathol 116: 1-16
Atypical forms in about 30% of casesKosaka K (1993) Dementia and neuropathology in Lewy body disease. Adv Neurol 60: 456-463
Braak H, Muller CM, Rüb U, Ackermann H, Bratzke H, de Vos RA, Del Tredici K (2006) Pathology associated with sporadic Parkinson’s disease: where does it end? J Neural Transm Suppl 70: 89-97
Uchikado H, Lin WL, DeLucia MW, Dickson DW (2006) Alzheimer disease with amygdala Lewy bodies: a distinct form of α-synucleinopathy. J Neuropathol Exp Neurol 65: 687-697
Cortical-predominant LBDAmygdala-predominant LBD
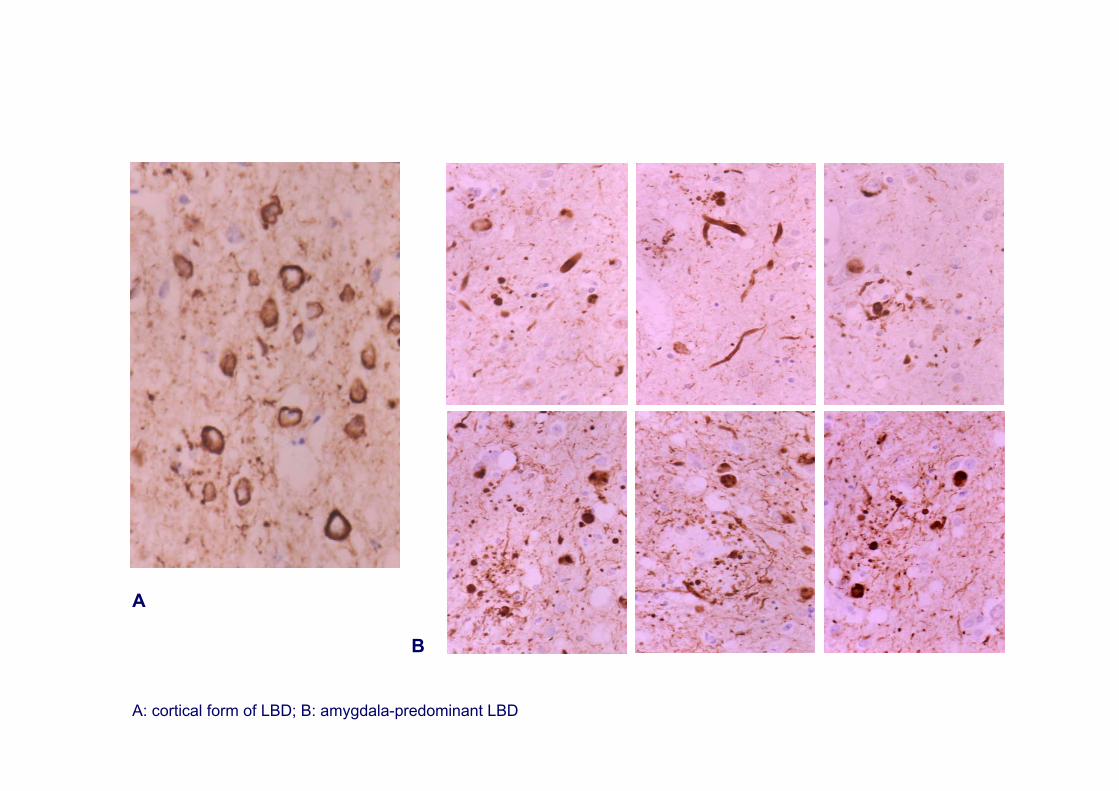
A
B
A: cortical form of LBD; B: amygdala-predominant LBD

Genetics of Parkinson’s disease
PARK1: α-synucleinMutations A53T: Contursi Kindred, Greek families from Patras;A30P: German family; andE46K: Spanish family (Zarranz et al., 2003): Some members with PD others with DLBDuplications and triplications of α-synuclein locusAll cases are LBD
PARK1: parkinDeletions in the parkin geneMutations in the parkin gene have been reported in late-onset PD and in sporadic cases of PDCases with Lewy bodies, cases with phospho-tau inclusions, cases without inclusions
PARK5: UCHL1 coding for ubuiquitin carboxyl-terminal hydrolase L1 (UCHL1)A single family with Lewy body pathology
PARK6: PINK1 coding for PTEN-induced putative kinase 1 (PINK1)No neuropathological studies
PARK7: DJ1 coding for Parkinson disease protein 7 (DJ1)No neuropathological studies
PARK8: LRRK2 coding for Leucine-rich repeat kinase 2: LRRK2, dardarinCases with Lewy bodies, cases with phospho-tau inclusions, cases with ubiquitin-ir inclusions, cases without inclusions. In addition to EOPD, sporadic cases.
PARK13: HTRA2 coding for HtrA serine peptidase 2: HtrA2Neuropathology unknown
Genetics of DLB
PARK1: α-synuclein; PARK8: LRRK2; GBA (glucocerebrosidase); PSN1
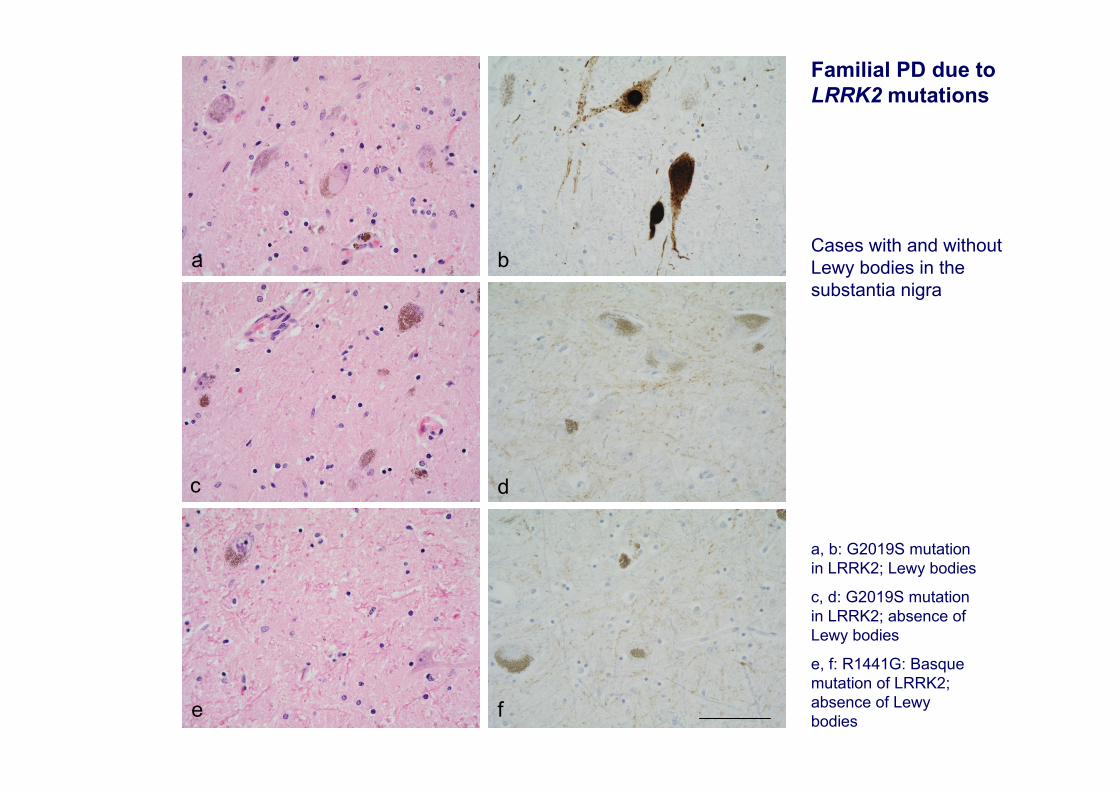
a
fe
dc
b
a, b: G2019S mutation in LRRK2; Lewy bodies
c, d: G2019S mutation in LRRK2; absence of Lewy bodies
e, f: R1441G: Basque mutation of LRRK2; absence of Lewybodies
Familial PD due to LRRK2 mutations
Cases with and without Lewy bodies in the substantia nigra
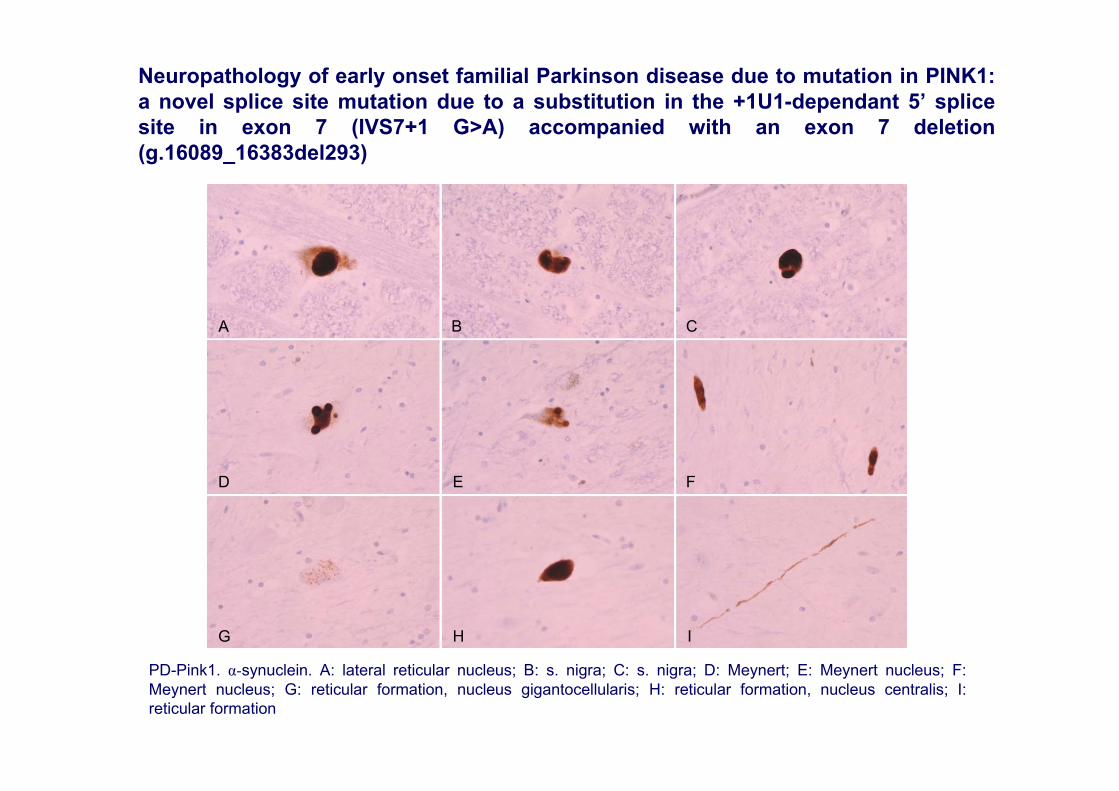
PD-Pink1. α-synuclein. A: lateral reticular nucleus; B: s. nigra; C: s. nigra; D: Meynert; E: Meynert nucleus; F: Meynert nucleus; G: reticular formation, nucleus gigantocellularis; H: reticular formation, nucleus centralis; I: reticular formation
A
ED
I
F
HG
CB
Neuropathology of early onset familial Parkinson disease due to mutation in PINK1: a novel splice site mutation due to a substitution in the +1U1-dependant 5’ splice site in exon 7 (IVS7+1 G>A) accompanied with an exon 7 deletion (g.16089_16383del293)

Major proteins linked with familial Parkinson’s disease and dementia with Lewy bodies are related with mitochondria
•Mice lacking α-synuclein exhibit mitochondrial lipid metabolism and electron transport chain impairment (Ellis et al., 2005). •α-synuclein transgenic mice show reduced complex IV activity and mitochondrial DNA damage (Martin et al., 2006). •Mitochondrial associated metabolic proteins are selectively oxidized in α-synuclein transgenic mice (Poon et al., 2005).•Drosophila parkin-null mutants show mitochondrial abnormalities (Greene et al., 2003). •Parkin-deficient mice also show mitochondrial deficits and impaired respiratory function (Palacino et al., 2004). •DJ1 is localized in the mitochondria and modulates responses to oxidative stress (Yang et al., 2005; Ved et al., 2005). •PINK1 is a protein kinase localized in the mitochondria in which mutations in the kinase domain are associated with mitochondrial deficits (Silvestri et al., 2005). •PINK1 is required for mitochondrial function by interacting and complementing parkin (Park et al., 2006; Clark et al., 2006; Poole et al., 2008). •LRRK2 is a kinase localized in the outer mitochondrial membrane which also interacts with parkin (Smith et al., 2005). •HTRA2 is localized at the mitochondria and is involved in apoptosis (Suzuki et al., 2001).
Deficits in mitochondrial function have been identified in patients with DJ1, parkin and PINK1 mutations (Dodson and Guo, 2007)
Mitochondrial abnormalities in the substantia nigra in sporadic Parkinson’s disease
Deficiencies in complex I subunits of the respiratory chain and impaired activity of the electron transport chain in sporadic PD(Mizuno et al., 1989; Parker et al., 1989; Schapira et al., 1989; -90; Schapira, 2008; Onyango 2008). Increased mtDNA deletions in PD nigral neurons when compared with age-matched controls (Bender et al., 2006). Subunits of mitochondrial complex I in PD brains are oxidatively damaged, functionally impaired and misassembled (Keeney
et al., 2006).
Abnormal oxygen tissue and mitochondrial uptake, and reduced complex I activity in the frontal cortex has been demonstrated in DLB (Navarro et al., 2008)
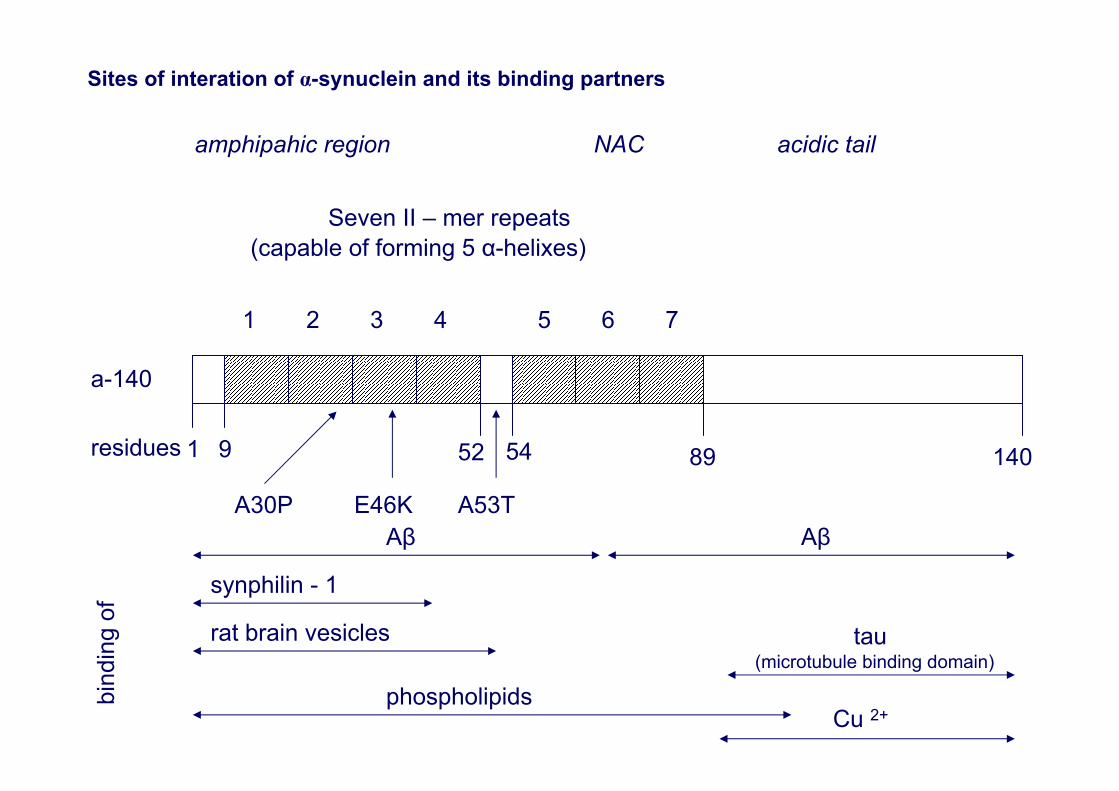
Sites of interation of α-synuclein and its binding partners
amphipahic region NAC acidic tail
Seven II – mer repeats(capable of forming 5 α-helixes)
1 2 3 4 5 6 7
a-140
residues 1 9
A30P
52
A53T
89 140
Aβ Aβ
bind
ing
of
synphilin - 1
rat brain vesicles tau(microtubule binding domain)
phospholipidsCu 2+
E46K
54
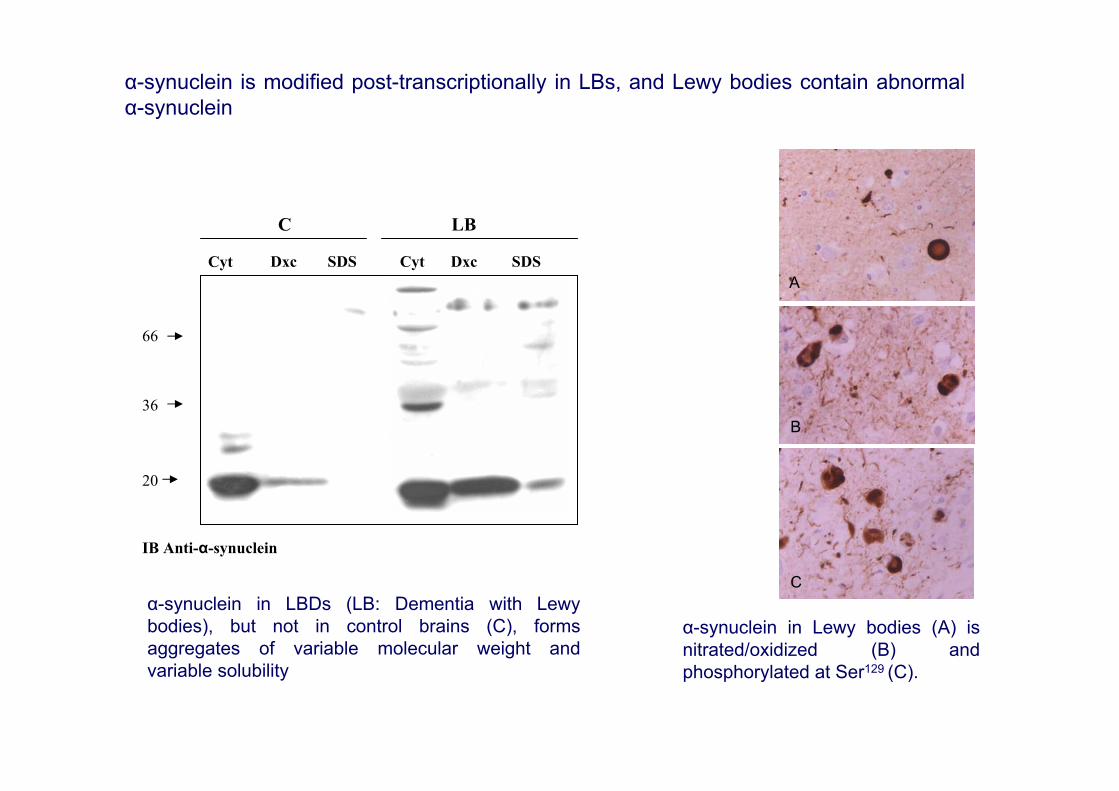
IB Anti-α-synuclein
66
36
20
Cyt Dxc SDS SDSDxcCyt
C LB
α-synuclein in LBDs (LB: Dementia with Lewybodies), but not in control brains (C), formsaggregates of variable molecular weight andvariable solubility
α-synuclein in Lewy bodies (A) isnitrated/oxidized (B) andphosphorylated at Ser129 (C).
A
B
C
α-synuclein is modified post-transcriptionally in LBs, and Lewy bodies contain abnormalα-synuclein
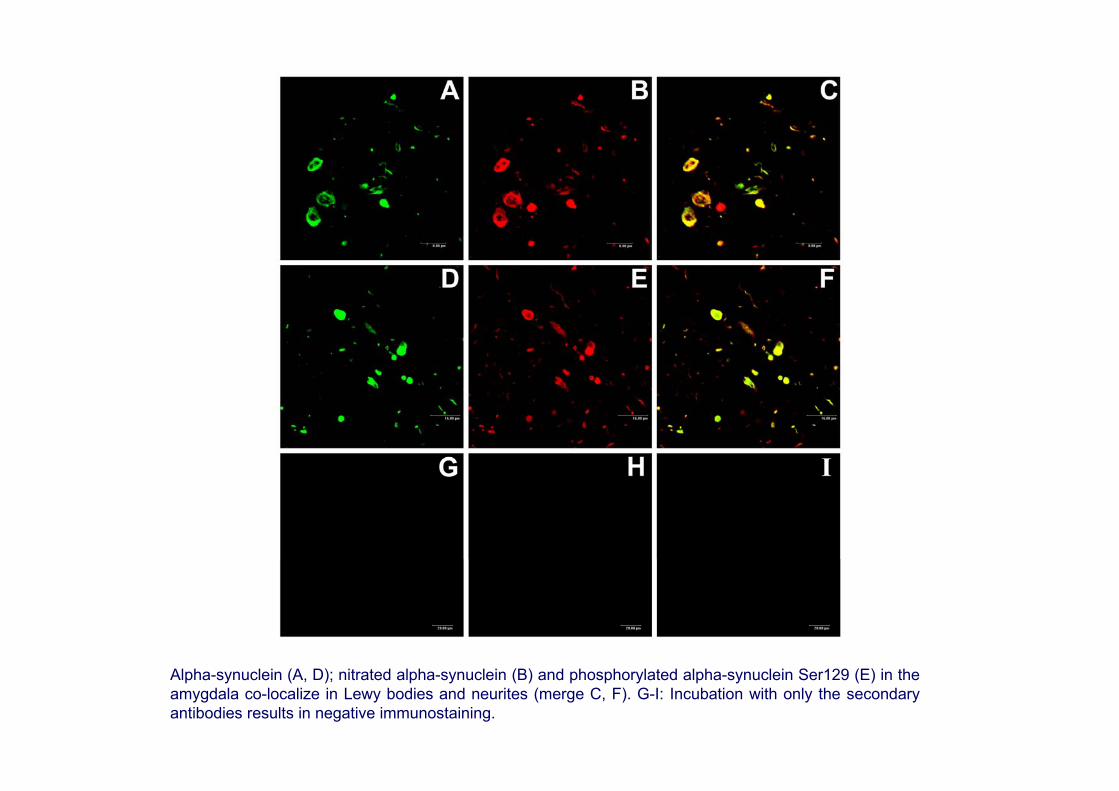
Alpha-synuclein (A, D); nitrated alpha-synuclein (B) and phosphorylated alpha-synuclein Ser129 (E) in the amygdala co-localize in Lewy bodies and neurites (merge C, F). G-I: Incubation with only the secondaryantibodies results in negative immunostaining.
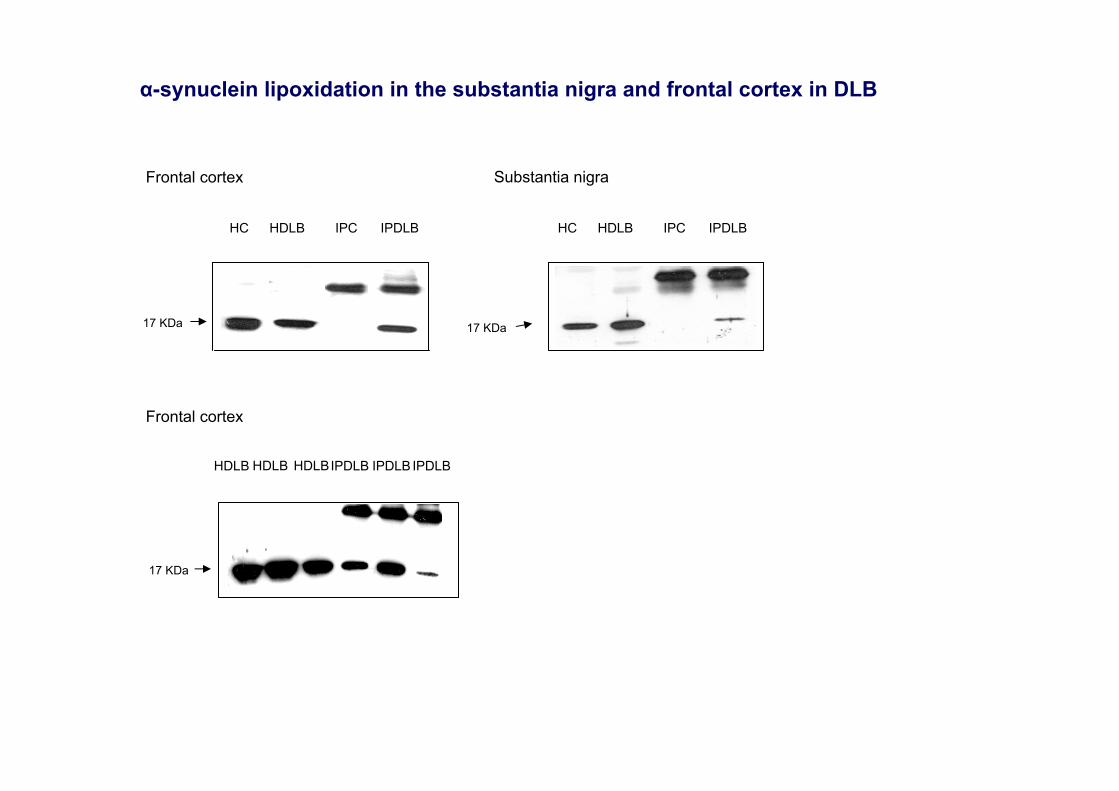
HDLB HDLB HDLBIPDLB IPDLB IPDLB
17 KDa
Frontal cortex
Frontal cortex
HC HDLB IPC IPDLB
17 KDa
HC HDLB IPC IPDLB
Substantia nigra
17 KDa
α-synuclein lipoxidation in the substantia nigra and frontal cortex in DLB
66
45
36
2429
2014
66
45
36
2429
2014
W Tg
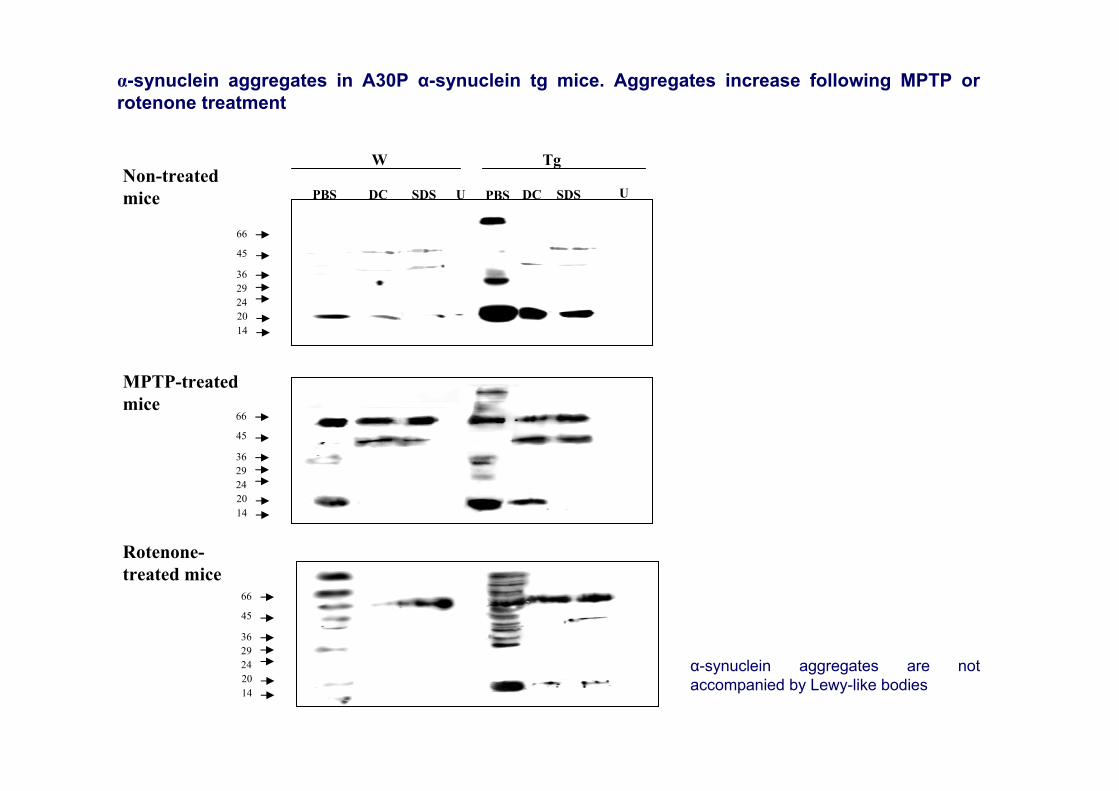
PBS DC SDS PBSU SDS UDCNon-treatedmice
66
45
36
2429
2014
Rotenone-treated mice
MPTP-treatedmice
α-synuclein aggregates in A30P α-synuclein tg mice. Aggregates increase following MPTP orrotenone treatment
α-synuclein aggregates are notaccompanied by Lewy-like bodies

W
14
IB anti-rabphilin
75
19
IB anti-α-syn
ip Rab3a
tTg tW Tg anti-Rab3a+G
24
IB anti-rab3a
IB anti-β-actin
4320
IB anti-α-syn
Wild type α-syn A30P α-syn
Rab5 Rab5 20 10 20 50 502020 10 Ni+ 20
Rab5 pull-down with recombinant α-synuclein

Disorders at the synapses
•α-synuclein is mainly localized at pre-synaptic terminals where it controls synaptic vesicle trafficking (Murphy et al., 2000; Abeliovich et al., 2000; Cabin et al., 2002; Kaplan et al., 2003). •Modified α-synuclein in LBDs and related transgenic mice models abnormally interacts with Rabproteins thus compromising transport, endocytosis and synaptic transmission (Dalfó et al., 2004). •This is associated with altered short-term hippocampal synaptic plasticity in transgenic mice (Steidlet al., 2003). •Irregular α-synuclein/phospholipase C (PLCβ1) interactions undermine abnormal metabotropicglutamate expression and signaling in the cerebral cortex in DLB pure form (Dalfó et al., 2004). This effect further augments in common DLB and correlates with the stage of AD-related changes (Albasanz et al., 2005). •Aberrant α-synuclein oligomers may affect different pathways regulating synaptic protein turnover (Hashimoto and Masliah, 2003).•Pre-synaptic α-synuclein aggregates are, by far, much more extensive than Lewy bodies and aberrant neurites in the cerebral cortex in DLB (Kramer and Schulz-Schaeffer, 2007).
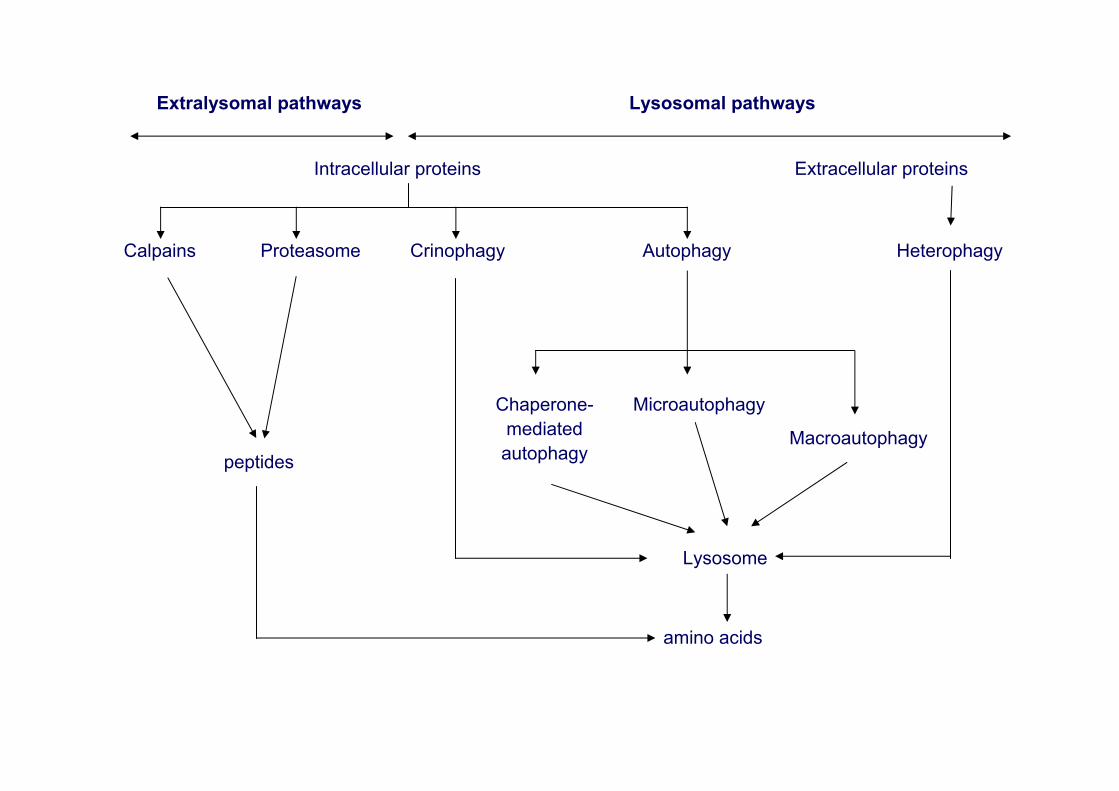
Extralysomal pathways Lysosomal pathways
Intracellular proteins Extracellular proteins
Calpains Proteasome Crinophagy Autophagy Heterophagy
peptides
Chaperone-mediatedautophagy
Microautophagy
Macroautophagy
Lysosome
amino acids
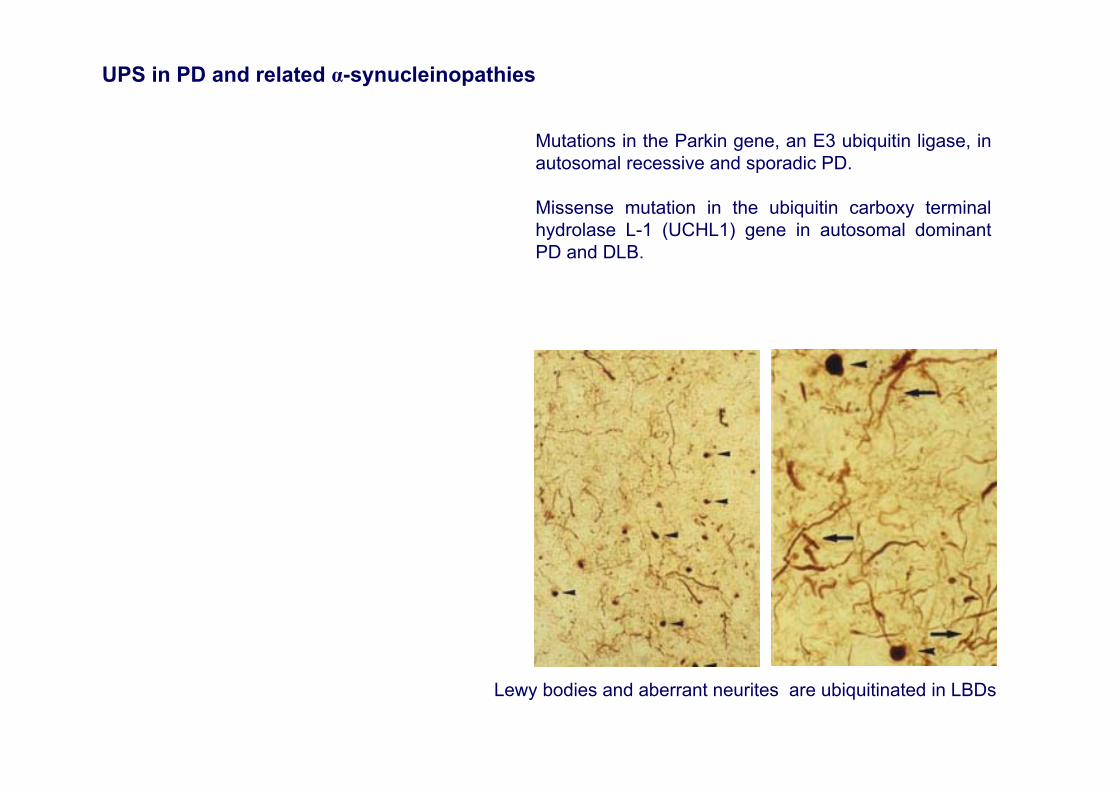
Lewy bodies and aberrant neurites are ubiquitinated in LBDs
Mutations in the Parkin gene, an E3 ubiquitin ligase, in autosomal recessive and sporadic PD.
Missense mutation in the ubiquitin carboxy terminal hydrolase L-1 (UCHL1) gene in autosomal dominantPD and DLB.
UPS in PD and related α-synucleinopathies

The ubiquitin-proteasome system (UPS) in PD and DLB
•Parkin is an E3 ubiquitin ligase and α-synuclein is one of the few substrates of parkin (Zhang et al., 2000; Shimura et al., 2000)
•Parkin mutations may interfere with α-synuclein degradation by UPS
•The I93M mutation in UCHL1 is associated with familial PD (Leroy et al., 1998), which is neuropathologicallymanifested as LBD (Auburger et al., 2005). This mutation leads to a 50% reduction in catalytic UPS activity in vitro(Lansbury and Brice, 2002)
•S18Y UCHL1 polymorphism variant confers antioxidant function to neuronal cells and may protect individuals bearingthis polymorphism from PD (Kyratzi et al., 2008)
•UCHL1 mRNA reduced in the striatum and substantia nigra in PD (Miller et al., 2006)
•Reduced UCHL1 mRNA and protein expression levels are found in the substantia nigra in PD and DLB, and frontal cortex in DLB
•Certain α-synuclein mutations may interfere with the proteasome (Stefanis et al., 2001)
•α-synuclein protofibrils inhibit 26S proteasome-mediated protein degradation (Zhang et al., 2008)
•Proteasomal dysfunction in the brains of patients with sporadic PD (McNaught et al., 2000)
•Proteasome inhibition induces a parkinsonian syndrome accompanied by biochemical and pathological features in rats (McNaught and Olanow, 2006).
•Wild and mutant α-synuclein expression levels do not affect proteasome function and expression in mice and stably transfected PC12 cells (Martín-Clemente et al., 2004)
•Expression levels of proteasome subunits are not modified in the cerebral cortex in DLB (Barrachina et al., 2006).
•The proteasome-inhibition model of parkinsonism has not been reproduced in rats, mice and monkeys (Bové et al., 2006; Kordower et al., 2006)
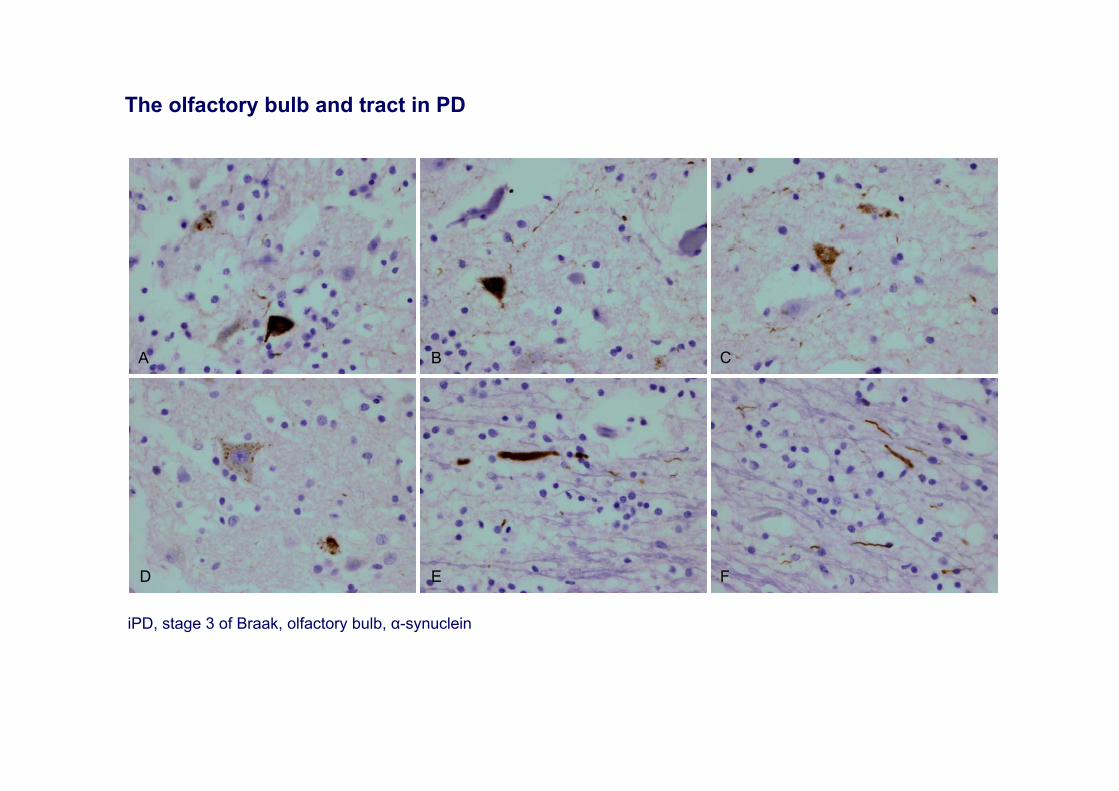
A
FED
B C
iPD, stage 3 of Braak, olfactory bulb, α-synuclein
The olfactory bulb and tract in PD

Clinical symptoms and signs, and neuropathological findings outside the substantia nigra in pre-motor stages of PD
Autonomic dysfunction in patients with PD has been recognized since the original description by James Parkinson in 1817. More recently, olfactory dysfunction, constipation, sleep fragmentation, lethargy, rapid eye movement (REM) behavior disorder, pain, mood and anxiety disorders, and depression have been noticed as common symptoms in PD. Most importantly, these manifestations appear at the pre-motor phase of PD and they may worsen with disease progression.
Lewy body dysphagia has been reported in association with Lewy body pathology restricted to the dorsallvagal motor nuclei.
Lewy bodies have been found in selected nuclei of the brain stem, sympathetic ganglia, epicardial plexus and autonomic nerves in the muscularis of the urinary bladder in one patient with symptoms of orthostatic hypotension and urinary dysfunction for 15 years.
Lewy body pathology restricted to selected nuclei of the brain stem occurred in an old man with a 20-year history of nocturnal violent behavior sleep and polysomnographic evidence of REM sleep behavior disorder.
Progressive autonomic failure and lethargy in the absence of parkinsonism has been reported in another patient with brainstem-type LBD

Lewy body pathology is present in the olfactory bulb and related olfactory nuclei at very early stages of PD. It also occurs in cases of AD with amygdala-predominant Lewy pathology.
Lewy bodies are consistently found in the hypothalamus, sympathetic (intermediodorsal nucleus of the thoracic cord and sympathetic ganglia), and parasympathetic system (dorsal vagal and sacral parasympathetic nuclei, and peripheral parasympathetic ganglia); and enteric plexus.
Neuropathological studies in large cohorts of neurologically unimpaired aged individuals have shown that the autonomic nuclei of the spinal cord and the peripheral autonomic nervous system are early affected by Lewy body pathology.
α-synuclein-immunoreactive inclusions are seen in neurons of the Meissner’s and Auerbach’s plexuses and in the corresponding axons projecting into the mucosa.
The cardiovascular autonomic system is also affected in iPD and PD, and alterations implicate both tyrosine hydroxylase-positive (extrinsic) and negative (intrinsic) nerves of the cardiac plexus. Functional studies have also demonstrated cardiac involvement in PD. [1231] meta-iodobenzylguanidine (MIBG) myocardial scintigraphy has shown reduced MIBG uptake in PD and DLB. Decreased MIBG uptake precedes neuronal loss in the sympathetic ganglia.
Involvement of the adrenal gland is constant in PD. However, the adrenal gland seems spared in cases with amygdala-predominant LBD.

PD is a systemic disease of the central and peripheral nervous system
• Variegated non-motor deficits and neurological symptoms, covering impaired olfaction, sleep disorders, gastrointestinal and urinary abnormalities and cardiovascular dysfunction, in addition to other symptoms and signs as pain, depression and mood disorders, may occur at early stages of the disease.
• Olfactory tests, polysomnographic studies and MIBG myocardial scintigraphy in combination may be used to discover early signatures of the disease.
However,
• Autonomic symptoms are not always present in PD.
• Lewy pathology in autonomic peripheral ganglia and plexus is not always associated with clinical symptoms.
• Little is known about the nature and composition of Lewy bodies in peripheral autonomic nervous system.
• No data are available about molecular changes preceding, or associated with, early and late stages of Lewy pathology in the autonomic peripheral nervous system.
• Little is known about early molecular changes in the olfactory bulb.
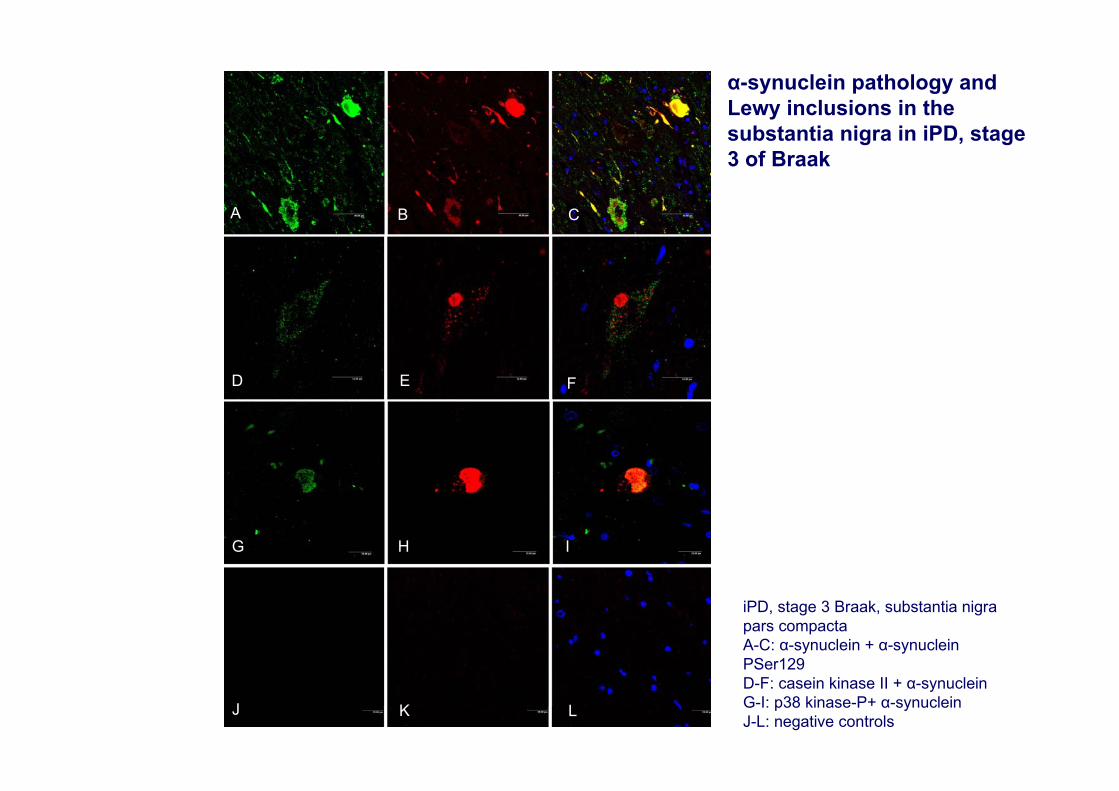
A
FED
IHG
LKJ
B C
iPD, stage 3 Braak, substantia nigra pars compactaA-C: α-synuclein + α-synuclein PSer129D-F: casein kinase II + α-synucleinG-I: p38 kinase-P+ α-synucleinJ-L: negative controls
α-synuclein pathology and Lewy inclusions in the substantia nigra in iPD, stage 3 of Braak
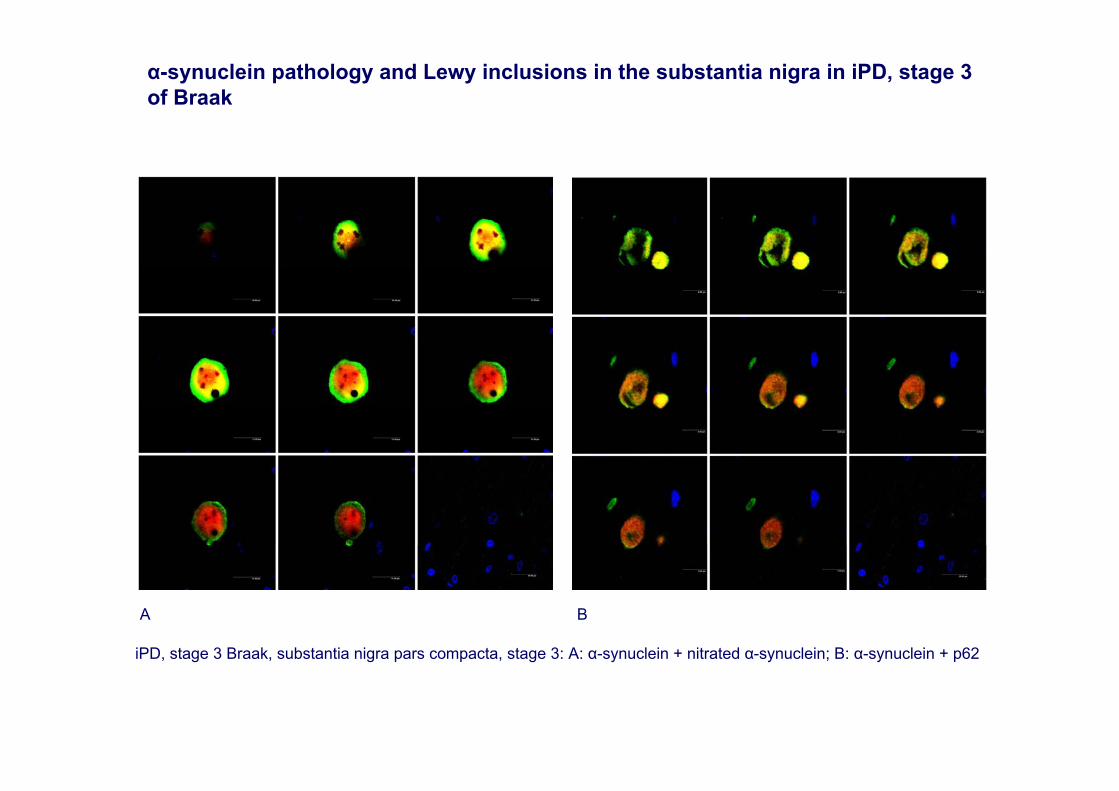
iPD, stage 3 Braak, substantia nigra pars compacta, stage 3: A: α-synuclein + nitrated α-synuclein; B: α-synuclein + p62
A B
α-synuclein pathology and Lewy inclusions in the substantia nigra in iPD, stage 3 of Braak
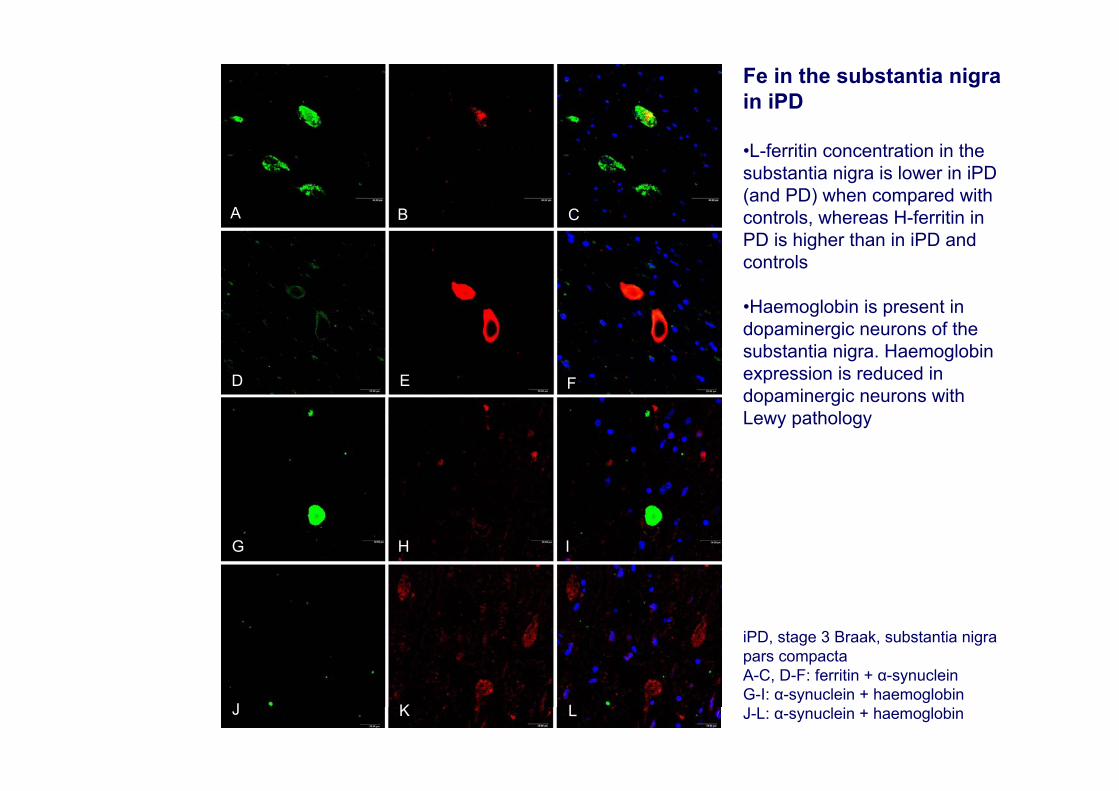
A
FED
IHG
LKJ
B C
iPD, stage 3 Braak, substantia nigra pars compactaA-C, D-F: ferritin + α-synuclein G-I: α-synuclein + haemoglobinJ-L: α-synuclein + haemoglobin
Fe in the substantia nigra in iPD
•L-ferritin concentration in the substantia nigra is lower in iPD(and PD) when compared with controls, whereas H-ferritin in PD is higher than in iPD and controls
•Haemoglobin is present in dopaminergic neurons of the substantia nigra. Haemoglobinexpression is reduced in dopaminergic neurons with Lewy pathology
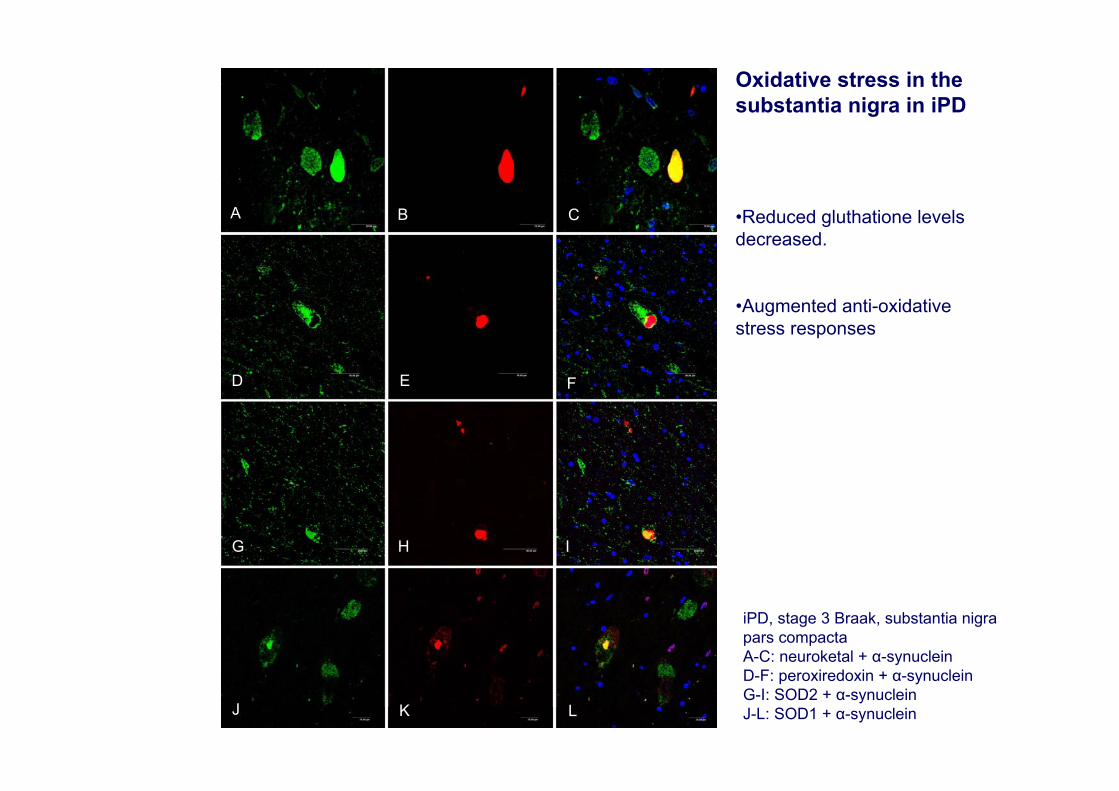
iPD, stage 3 Braak, substantia nigra pars compactaA-C: neuroketal + α-synucleinD-F: peroxiredoxin + α-synucleinG-I: SOD2 + α-synucleinJ-L: SOD1 + α-synuclein
A
A
FED
IHG
LKJ
B C
Oxidative stress in the substantia nigra in iPD
•Reduced gluthatione levels decreased.
•Augmented anti-oxidative stress responses
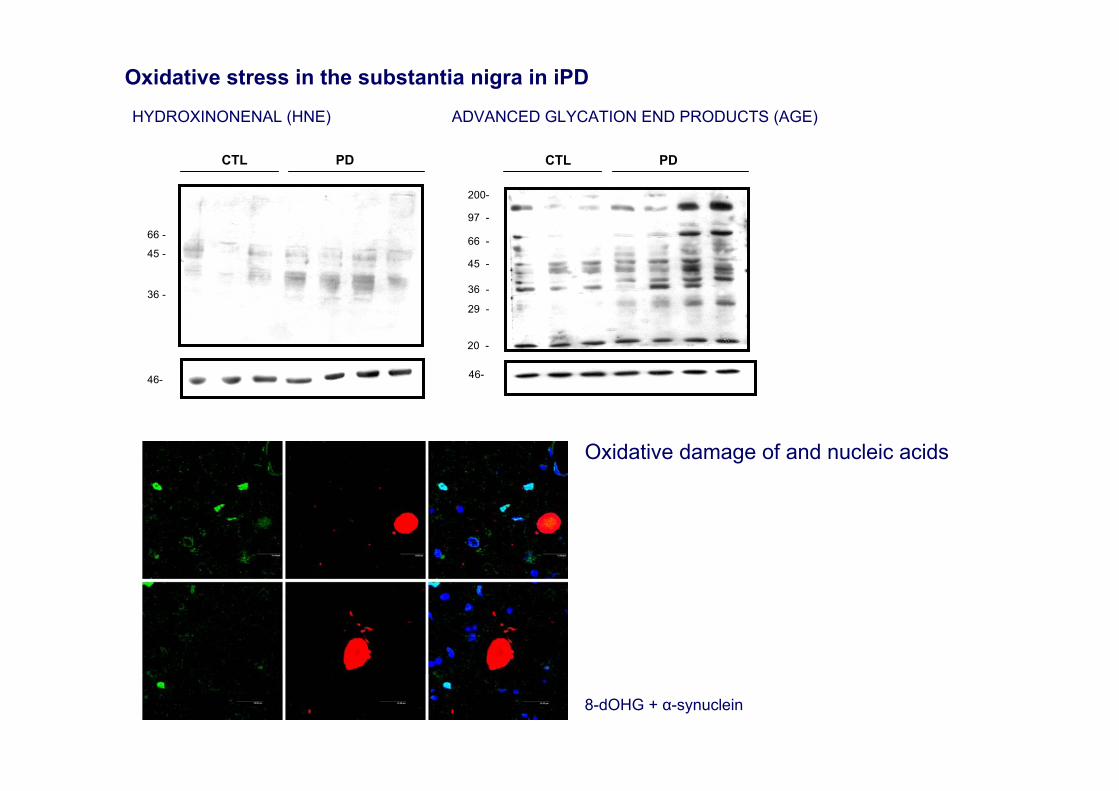
8-dOHG + α-synuclein
Oxidative stress in the substantia nigra in iPD
Oxidative damage of and nucleic acids
66 -
45 -
36 -
46-
CTL PD
6
HYDROXINONENAL (HNE) ADVANCED GLYCATION END PRODUCTS (AGE)
200-
97 -
66 -
45 -
36 -
29 -
20 -
CTL PD
46-
A
FED
IHG
LKJ
B C
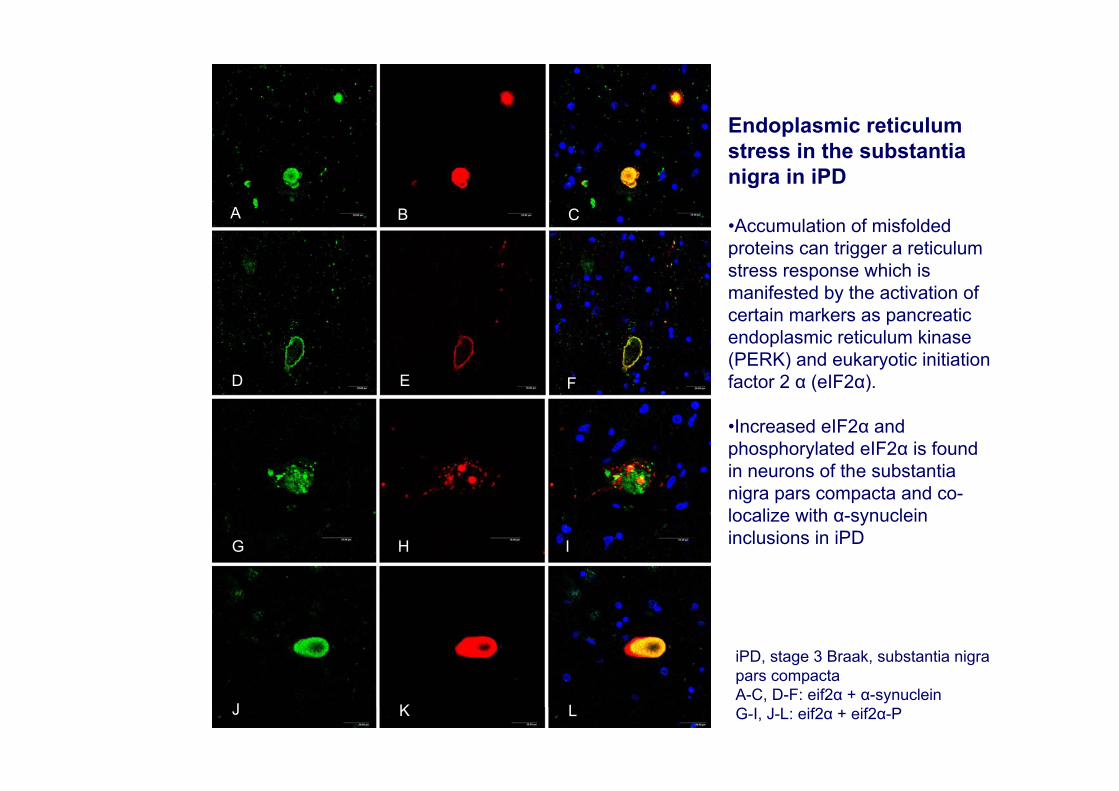
iPD, stage 3 Braak, substantia nigra pars compactaA-C, D-F: eif2α + α-synuclein G-I, J-L: eif2α + eif2α-P
Endoplasmic reticulum stress in the substantia nigra in iPD
•Accumulation of misfoldedproteins can trigger a reticulum stress response which is manifested by the activation of certain markers as pancreatic endoplasmic reticulum kinase(PERK) and eukaryotic initiation factor 2 α (eIF2α).
•Increased eIF2α and phosphorylated eIF2α is found in neurons of the substantia nigra pars compacta and co-localize with α-synuclein inclusions in iPD
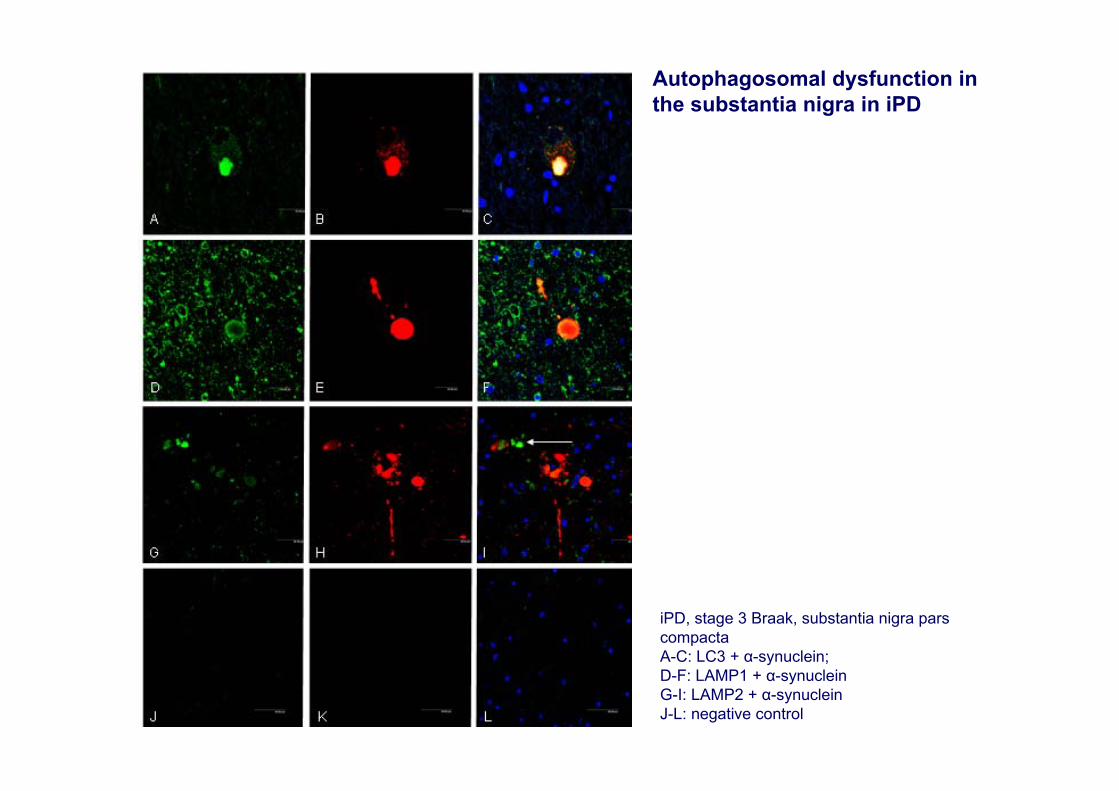
Autophagosomal dysfunction in the substantia nigra in iPD
iPD, stage 3 Braak, substantia nigra pars compactaA-C: LC3 + α-synuclein; D-F: LAMP1 + α-synucleinG-I: LAMP2 + α-synucleinJ-L: negative control
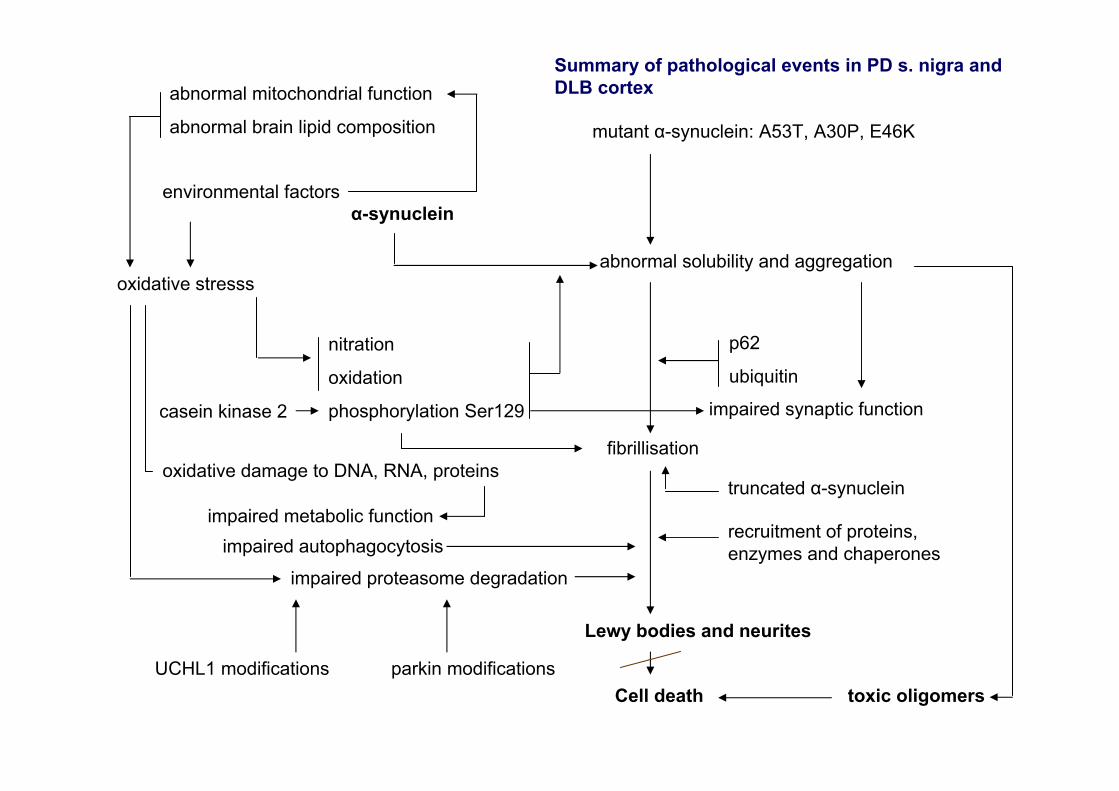
mutant α-synuclein: A53T, A30P, E46K
α-synuclein
nitration
oxidation
phosphorylation Ser129casein kinase 2
oxidative stresssabnormal solubility and aggregation
p62
ubiquitin
fibrillisation
recruitment of proteins, enzymes and chaperones
Lewy bodies and neurites
impaired proteasome degradation
UCHL1 modifications parkin modifications
abnormal mitochondrial function
abnormal brain lipid composition
truncated α-synuclein
Cell death toxic oligomers
impaired autophagocytosis
environmental factors
oxidative damage to DNA, RNA, proteins
impaired metabolic function
impaired synaptic function
Summary of pathological events in PD s. nigra and DLB cortex

Cerebral cortex in PD
Cognitive impairment in PD•changes in personality and moderate or mild cognitive debilitation •often subtle at the beginning and difficult to detect without neuropsychological tests. •mainly affect executive function including working memory and visuospatial capacity. •often accompanied by anxiety and depression, and excessive daytime sleepiness probably related with sleep disturbances. •hallucinations and slowly progressive dementia may appear with disease progression. •behaviour and cognitive impairment in PD differs from that occurring in mild cognitive impairment connected to AD-related pathology •abnormal behaviour and cognitive impairment in PD has aimed at developing a new scale, the Parkinson’s Disease-Cognitive Rating Scale (PD-CRS) to cover the full spectrum of cognitive deficits associated with PD
Complementary methods•cerebral glucose metabolism is reduced in the cerebral cortex in PD patients suffering from cognitive impairment•limited, mainly posterior, blood flow reductions in PD cases with mild cognitive deficits •decreased prefrontal and parietal 18F-fluorodeoxyglycose uptake in PD cases with mild cognitive deficits•MRI: T1-weighed images and mean diffusitivity and fractional anisotropy values were increased in the frontal cortex in PD cases
Role of neurotransmitters in PD behaviour•Involvement of noradrenergic, serotoninergic and cholinergic systems does not justify cognitive decline
Limitations of current neuropathological methods in the study of the cerebral cortex in PD•Cognitive impairment and dementia do not correlate with Lewy pathology in cerebral cortex•Association of AD pathology may explain, in part, cognitive deficits in a subset of patients•Novel α-synuclein antibodies have shown abundant striatal pathology in LBDs, thus suggesting that α-synuclein pathology exceeds Lewy body pathology in PD and DLB (Duda et al., 2002).
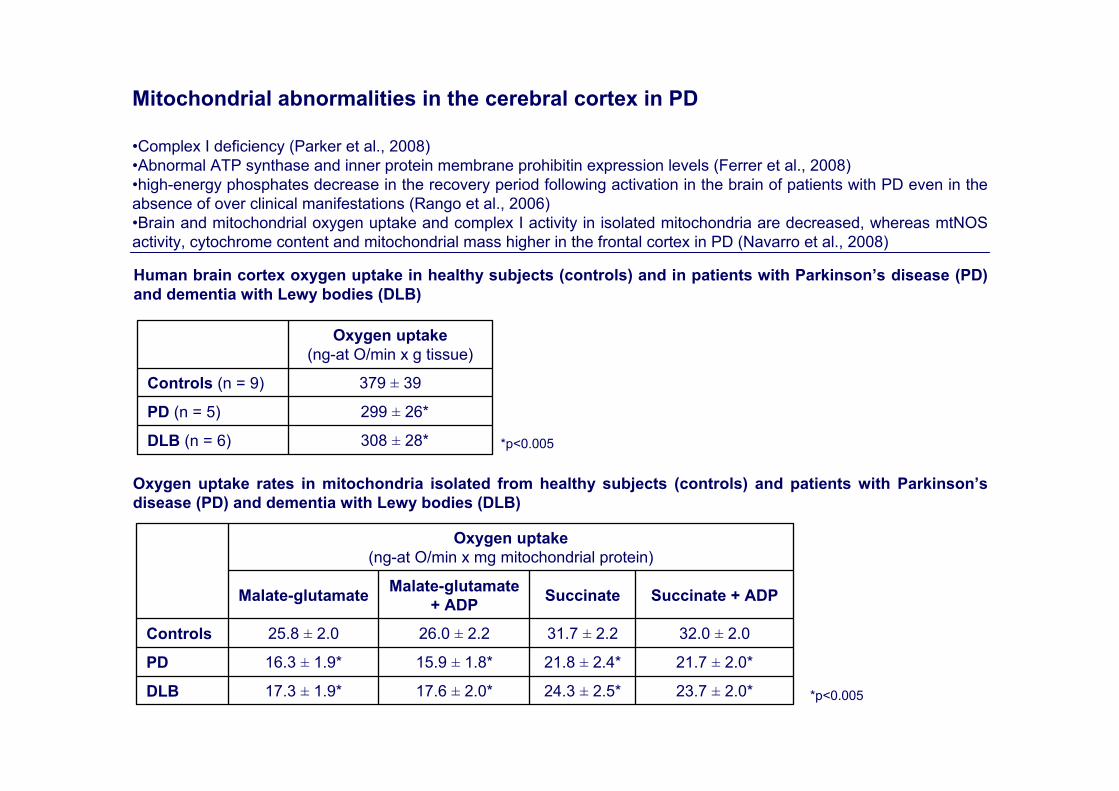
Mitochondrial abnormalities in the cerebral cortex in PD
•Complex I deficiency (Parker et al., 2008)•Abnormal ATP synthase and inner protein membrane prohibitin expression levels (Ferrer et al., 2008)•high-energy phosphates decrease in the recovery period following activation in the brain of patients with PD even in the absence of over clinical manifestations (Rango et al., 2006) •Brain and mitochondrial oxygen uptake and complex I activity in isolated mitochondria are decreased, whereas mtNOSactivity, cytochrome content and mitochondrial mass higher in the frontal cortex in PD (Navarro et al., 2008)
308 ± 28*DLB (n = 6)
299 ± 26*PD (n = 5)
379 ± 39Controls (n = 9)
Oxygen uptake (ng-at O/min x g tissue)
23.7 ± 2.0*24.3 ± 2.5*17.6 ± 2.0*17.3 ± 1.9*DLB
21.7 ± 2.0*21.8 ± 2.4*15.9 ± 1.8*16.3 ± 1.9*PD
32.0 ± 2.031.7 ± 2.226.0 ± 2.225.8 ± 2.0Controls
Succinate + ADPSuccinateMalate-glutamate + ADPMalate-glutamate
Oxygen uptake (ng-at O/min x mg mitochondrial protein)
Human brain cortex oxygen uptake in healthy subjects (controls) and in patients with Parkinson’s disease (PD) and dementia with Lewy bodies (DLB)
*p<0.005
Oxygen uptake rates in mitochondria isolated from healthy subjects (controls) and patients with Parkinson’s disease (PD) and dementia with Lewy bodies (DLB)
*p<0.005
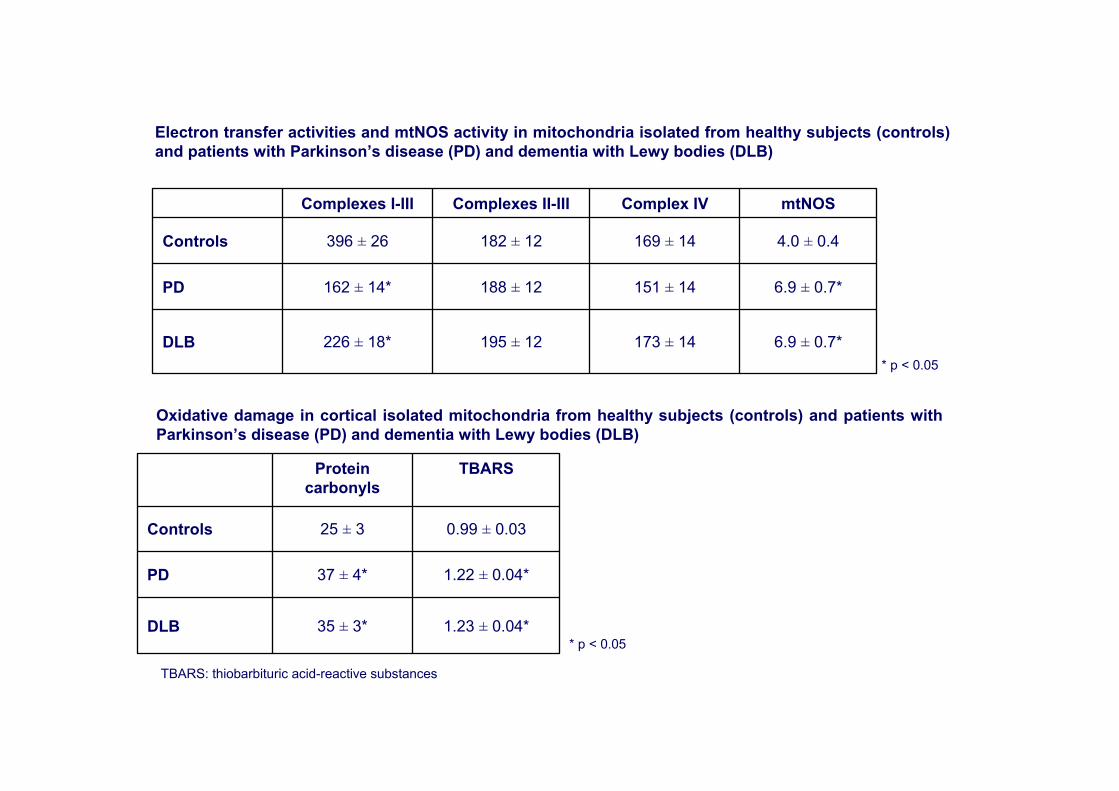
6.9 ± 0.7*173 ± 14195 ± 12226 ± 18*DLB
6.9 ± 0.7*151 ± 14188 ± 12162 ± 14*PD
4.0 ± 0.4169 ± 14182 ± 12396 ± 26Controls
mtNOSComplex IVComplexes II-IIIComplexes I-III
Electron transfer activities and mtNOS activity in mitochondria isolated from healthy subjects (controls) and patients with Parkinson’s disease (PD) and dementia with Lewy bodies (DLB)
Oxidative damage in cortical isolated mitochondria from healthy subjects (controls) and patients with Parkinson’s disease (PD) and dementia with Lewy bodies (DLB)
1.23 ± 0.04*35 ± 3*DLB
1.22 ± 0.04*37 ± 4*PD
0.99 ± 0.0325 ± 3Controls
TBARSProtein carbonyls
* p < 0.05
* p < 0.05
TBARS: thiobarbituric acid-reactive substances
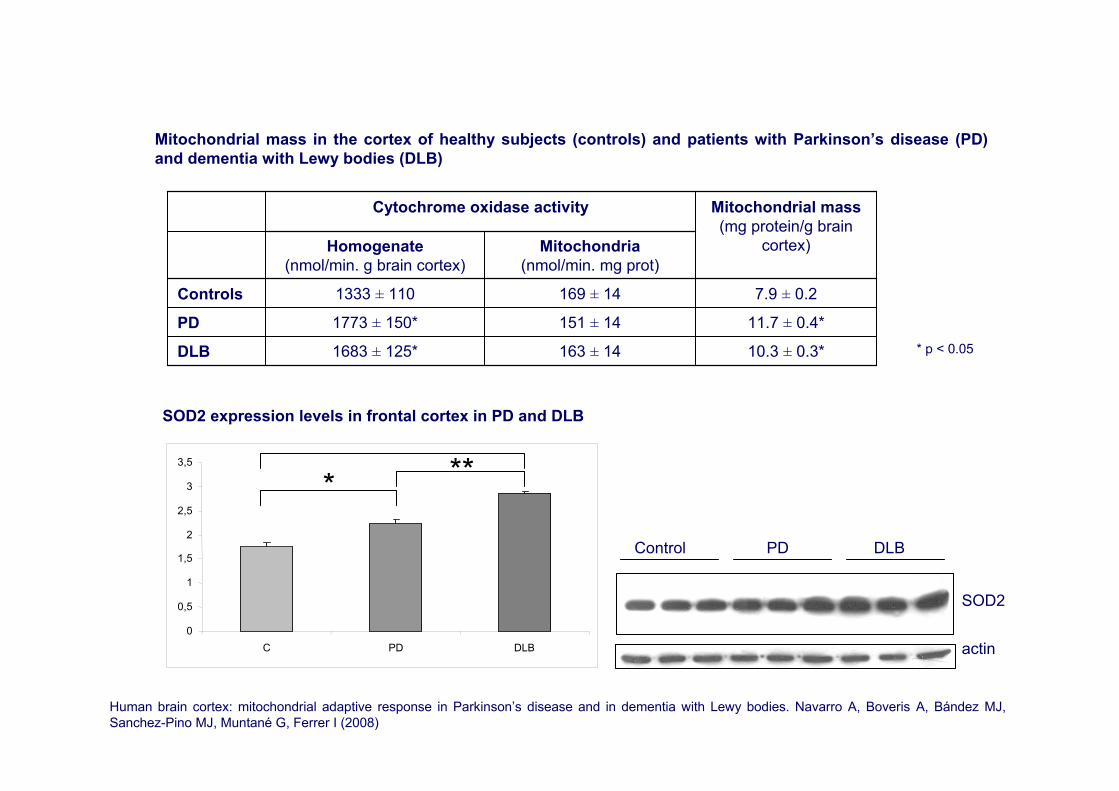
10.3 ± 0.3*163 ± 141683 ± 125*DLB
11.7 ± 0.4*151 ± 141773 ± 150*PD
7.9 ± 0.2169 ± 141333 ± 110Controls
Mitochondria(nmol/min. mg prot)
Homogenate(nmol/min. g brain cortex)
Mitochondrial mass(mg protein/g brain
cortex)
Cytochrome oxidase activity
Mitochondrial mass in the cortex of healthy subjects (controls) and patients with Parkinson’s disease (PD) and dementia with Lewy bodies (DLB)
* p < 0.05
Human brain cortex: mitochondrial adaptive response in Parkinson’s disease and in dementia with Lewy bodies. Navarro A, Boveris A, Bández MJ, Sanchez-Pino MJ, Muntané G, Ferrer I (2008)
0
0,5
1
1,5
2
2,5
3
3,5
C PD DLB
***
SOD2
actin
Control PD DLB
SOD2 expression levels in frontal cortex in PD and DLB
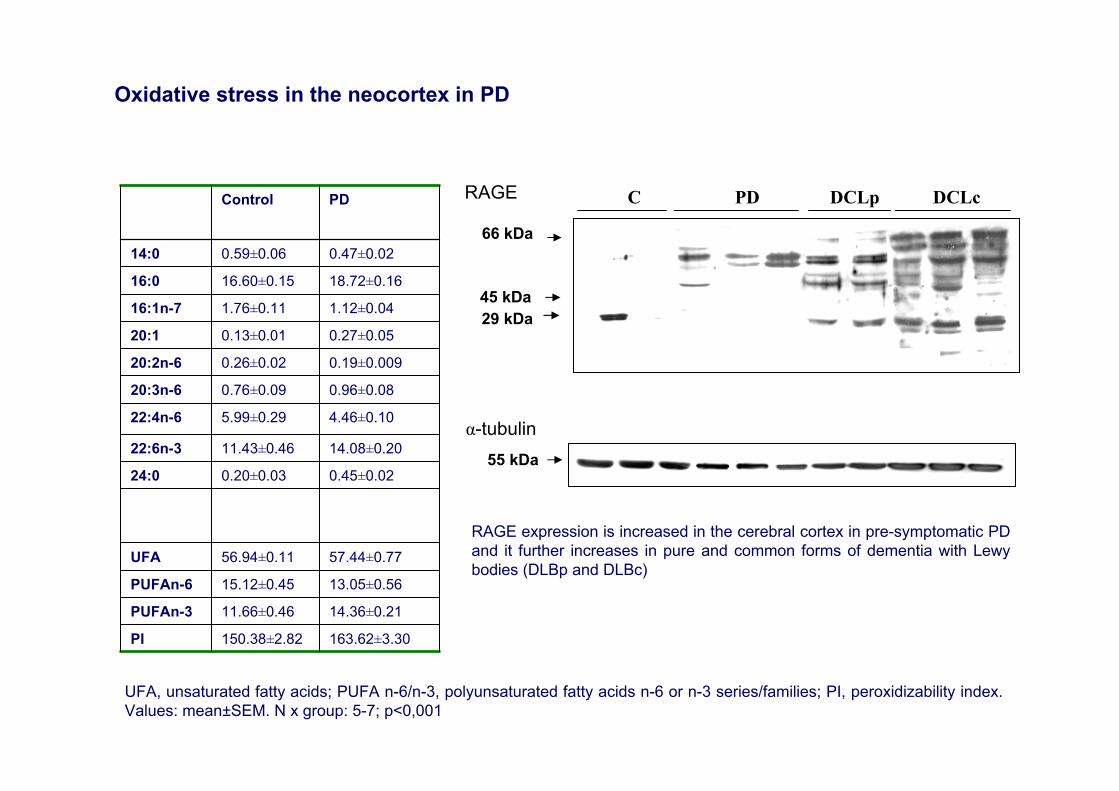
163.62±3.30150.38±2.82PI
14.36±0.2111.66±0.46PUFAn-3
13.05±0.5615.12±0.45PUFAn-6
57.44±0.7756.94±0.11UFA
0.45±0.020.20±0.0324:0
14.08±0.2011.43±0.4622:6n-3
4.46±0.105.99±0.2922:4n-6
0.96±0.080.76±0.0920:3n-6
0.19±0.0090.26±0.0220:2n-6
0.27±0.050.13±0.0120:1
1.12±0.041.76±0.1116:1n-7
18.72±0.1616.60±0.1516:0
0.47±0.020.59±0.0614:0
PDControl
UFA, unsaturated fatty acids; PUFA n-6/n-3, polyunsaturated fatty acids n-6 or n-3 series/families; PI, peroxidizability index. Values: mean±SEM. N x group: 5-7; p<0,001
Oxidative stress in the neocortex in PD
45 kDa
C PD DCLp DCLcRAGE
α-tubulin55 kDa
29 kDa
66 kDa
RAGE expression is increased in the cerebral cortex in pre-symptomatic PD and it further increases in pure and common forms of dementia with Lewybodies (DLBp and DLBc)
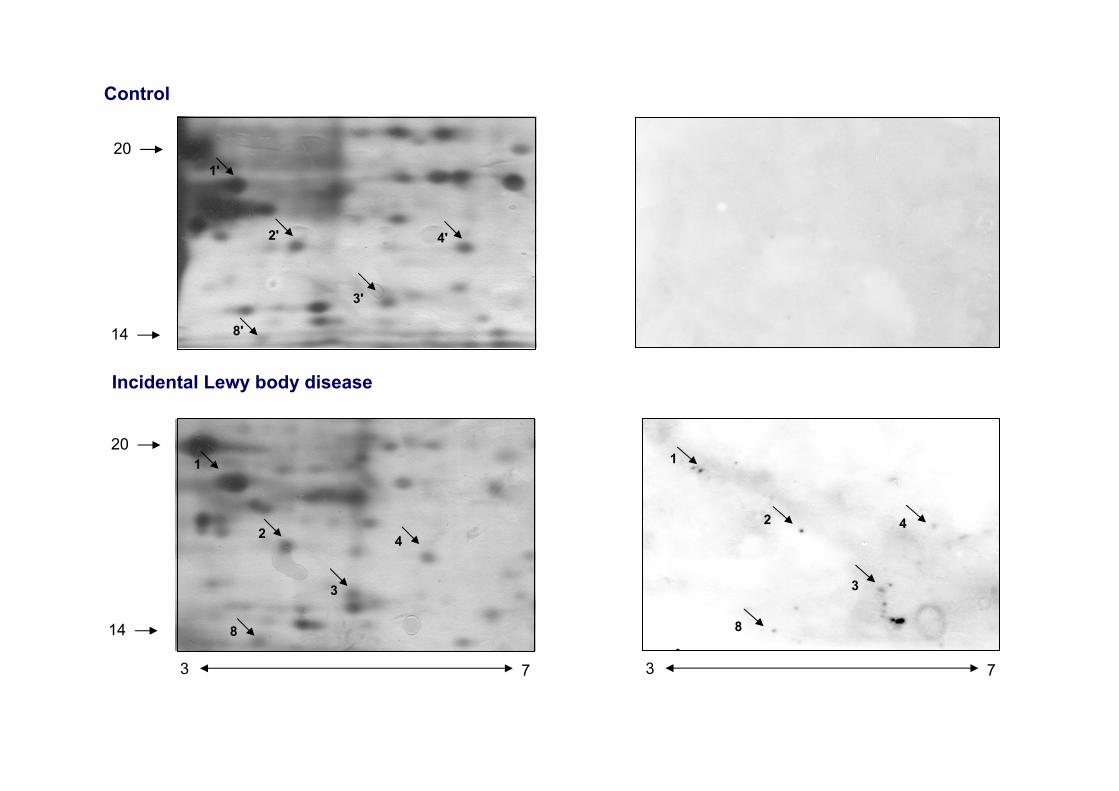
Control
4
3
2
1
8
3 7
1
2
3
4
20
14
3 7
8
Incidental Lewy body disease
8'
1'
2'
3'
4'
20
14

Spot Calculated PI
Mr (kDa) (Nominal mass)
Protein Subcellular location Score Number of peptides
Gi accession Protein Identification Method
1 4.67 14451 Alpha-synuclein isoformNACP140
Cytosol 219 6 gi|4507109 MS/MS
2 4.81 17410 Phosphoprotein enriched in astrocytes 15
Cytosol 160 3 gi|55957227 MS/MS
3 5.22 12766 SH3 domain binding glutamic acid-rich protein like
Cytosol (?) 96 2 gi|57160805 MS/MS
4 5.66 17366 Ubiquitin-conjugating enzyme E2N-like
Cytosol 99 1 gi|6941766 MS/MS
5 5.57 29843 Prohibitin Mitochondria 174 4 gi|30583661 MS/MS
6 4.80 25527 Proteasome subunit Y 121 2 gi|558528 MS/MS
7 5.78 49845 Enolase Mitochondria 55 1 gi|31179 MS/MS
8 4.82 11985 Thioredoxin Cytosol (?) 63 1 gi|231098 MS/MS
9 8.35 24891 Manganese superoxidedismutase (MnSOD)
Mitochondria 57 1 gi|34709 MS/MS
10 4.41 14279 Beta-synuclein Cytosol 261 5 gi|30582093 MS/MS
MDAL-modified proteins in cerebral cortex of incidental Parkinson’s disease

A GEC
B HFD
α-synuclein in the substantia nigra (A-G) and frontal cortex (B-H) in control (A, B), incidental PD (C,D), PD (E, F) and dementia with Lewy bodies (G, H)
Early α-synuclein lipoxidation in neocortex in Lewy body diseases
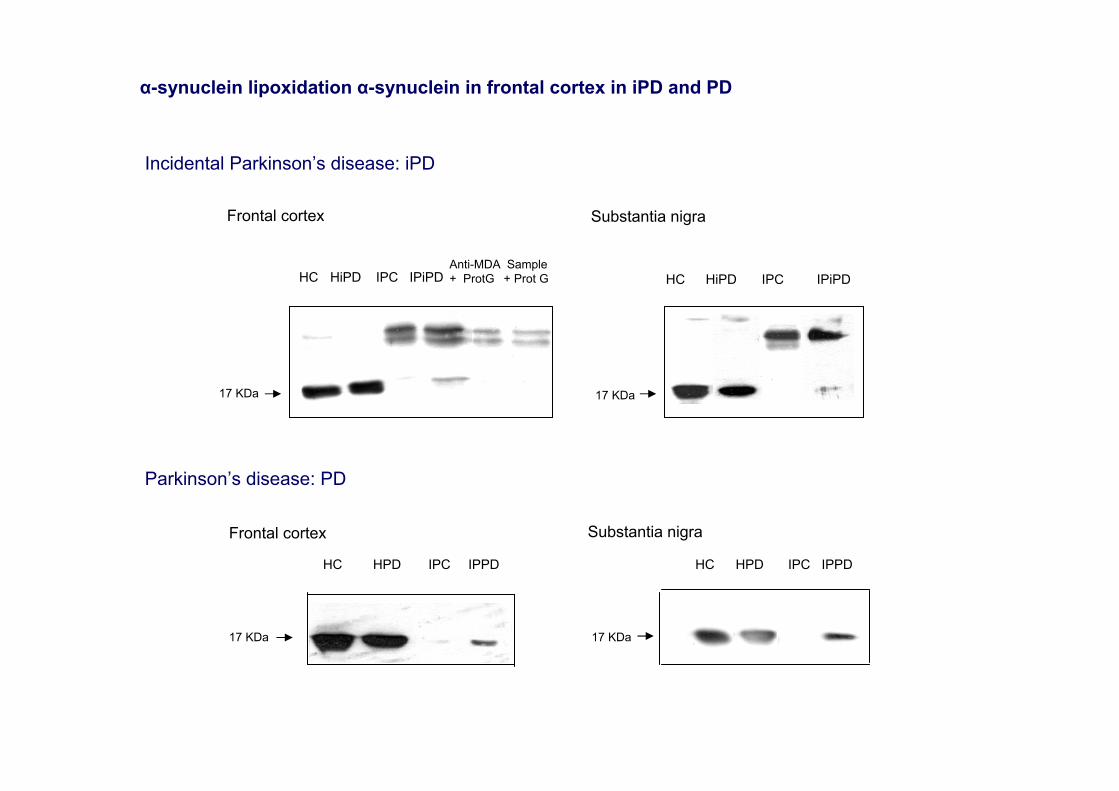
Frontal cortex Substantia nigra
17 KDa
HC HiPD IPC IPiPDAnti-MDA + ProtG
Sample+ Prot G HC HiPD IPC IPiPD
17 KDa
Incidental Parkinson’s disease: iPD
HC HPD IPC IPPD
17 KDa
Frontal cortex
HC HPD IPC IPPD
17 KDa
Substantia nigra
Parkinson’s disease: PD
α-synuclein lipoxidation α-synuclein in frontal cortex in iPD and PD

6 pH 11 pH
50 -
40 -
30 -
85 -
11 pH
PD
6 pH
50 -
85 -
30 -
40 -
11 pH
Anti-HNE Anti-HNE Anti-HNE
DLB
6 pH
50 -
30 -
85 -
40 -
1 2 34 5
6 776
1 32 54
iPD
35 -
25 -20 -
40 -50 -
85 -
6 pH
Anti-HNE
1 2 3
6 7
4
Control
6 pH 11 pH
50 -
40 -
85 - 4 5
321
76
Control
Oxidation of glycolysis and energy metabolism enzymes in the frontal cortex in Lewy body diseases
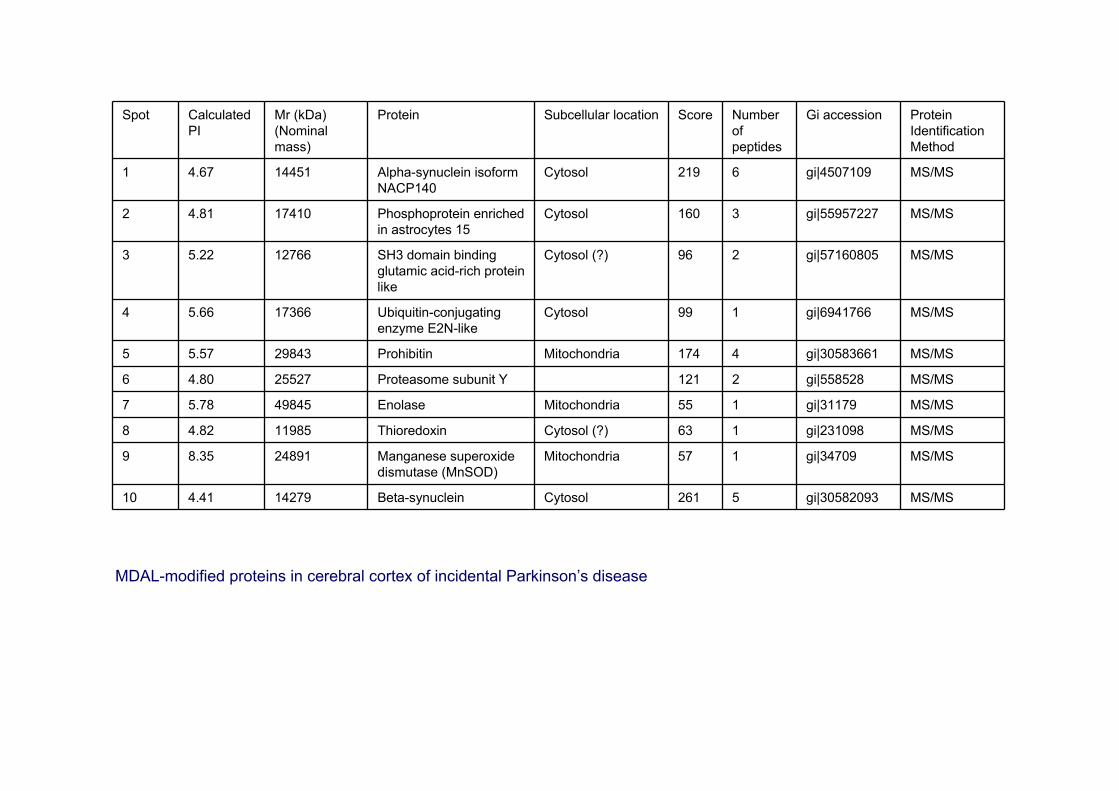
Spot Calculated PI
Mr (kDa) (Nominal mass)
Protein Subcellular location Score Number of peptides
Gi accession Protein Identification Method
1 4.67 14451 Alpha-synuclein isoformNACP140
Cytosol 219 6 gi|4507109 MS/MS
2 4.81 17410 Phosphoprotein enriched in astrocytes 15
Cytosol 160 3 gi|55957227 MS/MS
3 5.22 12766 SH3 domain binding glutamic acid-rich protein like
Cytosol (?) 96 2 gi|57160805 MS/MS
4 5.66 17366 Ubiquitin-conjugating enzyme E2N-like
Cytosol 99 1 gi|6941766 MS/MS
5 5.57 29843 Prohibitin Mitochondria 174 4 gi|30583661 MS/MS
6 4.80 25527 Proteasome subunit Y 121 2 gi|558528 MS/MS
7 5.78 49845 Enolase Mitochondria 55 1 gi|31179 MS/MS
8 4.82 11985 Thioredoxin Cytosol (?) 63 1 gi|231098 MS/MS
9 8.35 24891 Manganese superoxidedismutase (MnSOD)
Mitochondria 57 1 gi|34709 MS/MS
10 4.41 14279 Beta-synuclein Cytosol 261 5 gi|30582093 MS/MS
MDAL-modified proteins in cerebral cortex of incidental Parkinson’s disease

40 -
40 -
anti-aldolase A
Control iPD
6 pH 11 pH
6 pH 11 pH
40 -
6 pH 11 pH
PD DLB
6 pH 11 pH
anti-aldolase A
Anti-aldolase A
40 -
anti-aldolase A
Control iPD
anti-enolase
6 pH 11 pH 6 pH 11 pH
6 pH 11 pH 6 pH 11 pH
50 - 50 -
50 -50 -
PD DLB
anti-enolase
anti-enolaseanti-enolase
Fig. 4
A B CiPD
anti-DPNH anti-DPNH
control
45 -
3 pH 10 pH
Verification of ms data by aldolase A, enolase 1 and glyceraldehyde 3-phosphate dehydrogenase blotting (upper panel); andDPNH derivation (lower panel: A-C) in iPD
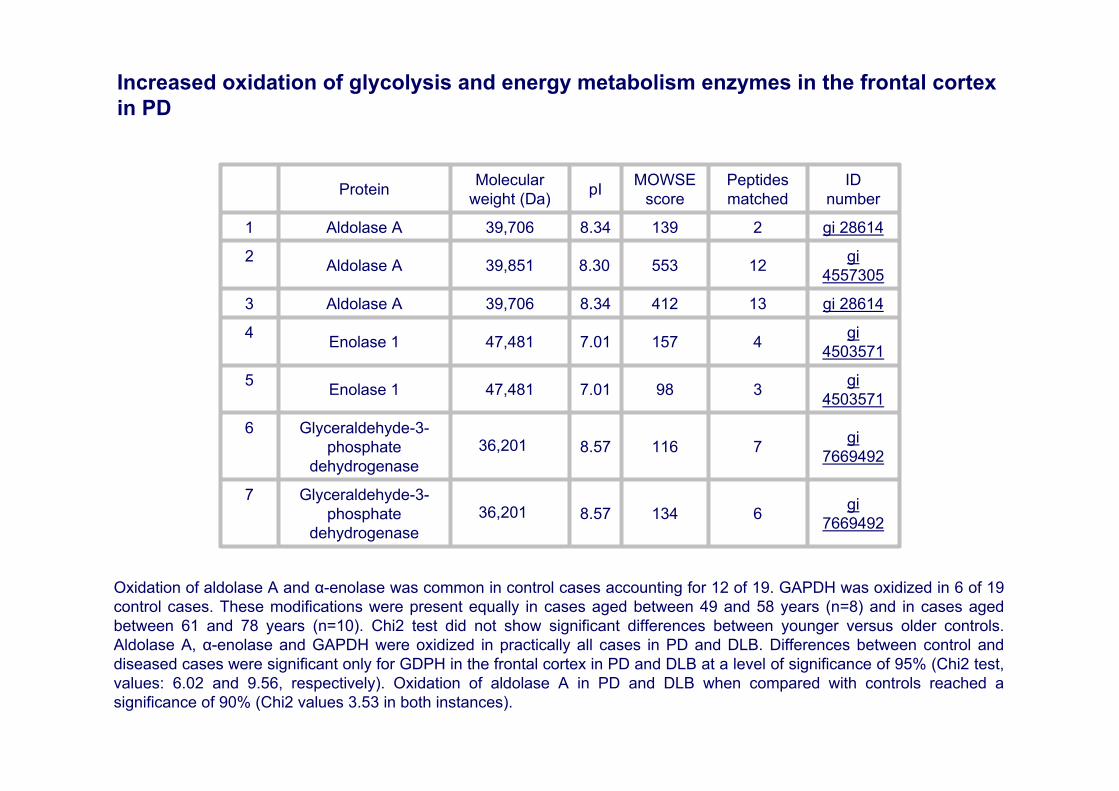
gi766949261348.5736,201
Glyceraldehyde-3-phosphate
dehydrogenase
7
gi766949271168.5736,201
Glyceraldehyde-3-phosphate
dehydrogenase
6
gi45035713987.0147,481 Enolase 15
gi450357141577.0147,481 Enolase 14
gi 28614134128.3439,706 Aldolase A3
gi4557305125538.3039,851 Aldolase A2
gi 2861421398.3439,706 Aldolase A1
ID number
Peptides matched
MOWSE scorepIMolecular
weight (Da)Protein
Oxidation of aldolase A and α-enolase was common in control cases accounting for 12 of 19. GAPDH was oxidized in 6 of 19 control cases. These modifications were present equally in cases aged between 49 and 58 years (n=8) and in cases aged between 61 and 78 years (n=10). Chi2 test did not show significant differences between younger versus older controls. Aldolase A, α-enolase and GAPDH were oxidized in practically all cases in PD and DLB. Differences between control and diseased cases were significant only for GDPH in the frontal cortex in PD and DLB at a level of significance of 95% (Chi2 test, values: 6.02 and 9.56, respectively). Oxidation of aldolase A in PD and DLB when compared with controls reached a significance of 90% (Chi2 values 3.53 in both instances).
Increased oxidation of glycolysis and energy metabolism enzymes in the frontal cortex in PD

iPD
PD
DLBp
AD I-II
AD III-IV
AD V-VI
DLBc+ AD IIControl50
1 3 5 7 9 11 13 15 17 19
50
50
50
50
50
50
50
50
50
50
50
50
50
1 3 5 7 9 11 13 15 17 19
50
50
1 3 5 7 9 11 13 15 17 191 3 5 7 9 11 13 15 17 19
Phospho-tauSer396Total tau: tau-13
Phosphorylation of tau and α-synuclein in synaptic-enriched fractions of the frontal cortex in Alzheimer’s disease, and in Parkinson’s disease and related α-synucleinopathies
tg A53Twt 12m1 3 5 7 9 11 13 15 17 191 3 5 7 9 11 13 15 17 19
synaptophysin
phospho-tauSer396
α-synuclein
50
20
40
50
tau-13
Control
iPD
PD
DLBp
AD V-VI
DLBc+AD II
30
20
30
1 3 5 7 9 11 13 15 17 19
30
30
30
30
20
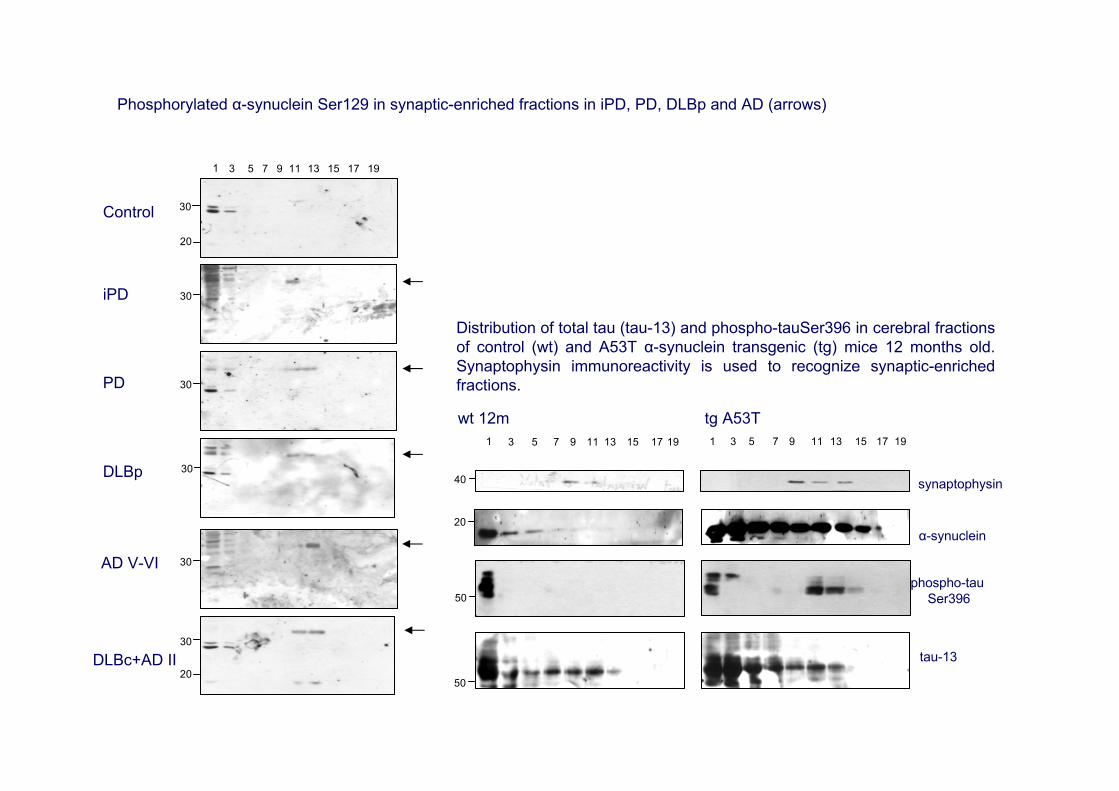
Distribution of total tau (tau-13) and phospho-tauSer396 in cerebral fractions of control (wt) and A53T α-synuclein transgenic (tg) mice 12 months old. Synaptophysin immunoreactivity is used to recognize synaptic-enriched fractions.
Phosphorylated α-synuclein Ser129 in synaptic-enriched fractions in iPD, PD, DLBp and AD (arrows)

7. Oxidative damage in the cerebral cortex in iPD and PD
•Indices of oxidative stress in iPD and PD (Dexter et al., 1994)•Oxidative DNA damage, as revealed by increased levels of 8-hydroxyguanine, occurs in PD (Sanchez-Ramos et al., 1994)•Increased levels of lipoxidation and glycoxidation markers, lipid peroxidation, and increased expression of RAGE (AGE receptor) in iPD and PD (Dalfó et al., 2005)•Abnormal levels of fatty in the frontal cortex in iPD and PD; docosahexaenoic acid (DHA) significantly increased in in the frontal cortex in iPD and PD (Dalfó et al., 2005) •Increased protein carbonyls and augmented expression levels of SOD2 (Navarro et al., 2008)•SOD2 and γ-synuclein identified as protein targets of oxidative damage in the frontal cortex in PD (Dalfó et al., 2005)•Oxidative damage and aggregation of Cu,Zn-superoxide dismutase (SOD1) (Choi et al., 2005) •α-synuclein is damaged by lipoxidation in the frontal cortex in iPD and PD (Dalfó and Ferrer, 2008)•increased lipoxidation of aldolase A, enolase 1 and glyceraldehyde dehydrogenase (GAPDH) (Gómez and Ferrer, 2008) •Oxidative damage of UCHL1 (Choi, 2004) •DJ1 is oxidized by carbonylation and by methionine oxidation to methionine sulfone in the frontal cortex in PD (Choi et al., 2006)
Carbonic anhydrase, α-enolase and lactate dehydrogenase 2 are selectively oxidized in A30P α-synuclein transgenic mice, and this is accompanied by decreased enzymatic functions (Poon et al., 2005)
Several PD-key proteins are oxidatively damaged in the frontal cortex in PD, and some of them are also oxidized in the frontal cortex in iPD. In addition to α-synuclein and γ-synuclein, proteins related with mitochondria, glycolysis and energy metabolism, oxidative stress responses and ubiquitin-proteasomesystem are oxidatively damaged and, as a consequence, functionally altered in the frontal cortex in PD.

8. α-synuclein modifications
•Oxidative damage of α-synuclein in the frontal cortex in PD and in certain iPD cases (Dalfó and Ferrer, 2008)•No significant α-synuclein aggregation in total homogenates in PD in contrast to DLB (Dalfó et al., 2005). No apparent increase in phosphorylated synuclein at Ser129 in total brain homogenates. •Increased expression levels of P-SynSer129 in the enriched-synaptic fractions in PD and DLB pure form, as well as in advanced AD and DLB common form (Muntané et al., 2008).
9. Disorders at the synapses
•increased phosphorylated Ser129 synuclein at the synapses•phospho-tauSer396 in synaptic-enriched fractions in the frontal cortex in PD and DLB pure and common. Between 20% and 40% phospho-tauSer396, in relation with tau-13 in PD and DLB; the percentage reaches about 95% in AD stage V and DLB common form•tau phosphorylation characteristic of neurofibrillary tangles, as revealed with the AT8 antibody, only at advanced stages of AD, but not in PD and DLB pure forms (Muntané et al., 2008)
Further evidence of the association of tau pathology in α-synucleinopathies relies on the observation of phosphorylated tau in transgenic mice over-expressing the A30P and A53T α-synuclein mutations (Frasier et al., 2005; Muntané et al., 2008)

Cerebral cortex in PD. Possible neuropathological bases of impaired behaviour and cognitive impairment.
•Clinical, neuroimaging, biochemical and neuropathological studies are rarely available in the same individual
•No relationship between advanced Braak LB stages and behavioural and cognitive deficits
•Complex and overlapping biochemical abnormalities and impaired metabolic pathways that may undermine normal brain physiology
•Abnormal brain mitochondria and function
•Oxidative damage on DNA, RNA and proteins
•Increased RAGE and abnormal stress responses
•Modifications of α-synuclein as a seed of abnormal folding, oligomerization and aggregation.
•Enzymes involved in mitochondria, as DJ1 and parkin, oxidatively damaged thus compromising mitochondrial function.
•Enzymes involved in the ubiquitin-proteasome system, as parkin and UCHL1, oxidatively damaged thus compromising the degradation of abnormal proteins.
•Oxidative damage of enzymes involved in glycolysis and energy metabolism, as enlnolase 1, aldolase A and glyceraldehyde-3-phosphate dehydrogenase
•Abnormalities at the synapses: phosphorylated tau Ser396 and phosphorylated α-synuclein Ser 129 that may impair normal synaptic function

α-synuclein
phosphorylation Ser129casein kinase 2
oxidative stresssabnormal solubility and aggregation
impaired proteasome degradation
UCHL1 oxidation parkin oxidation
abnormal mitochondrial function
abnormal brain lipid composition
toxic oligomers
impaired autophagocytosis ?
environmental factors
oxidation
impaired synaptic function
oxidative damage to DNA, RNA, proteins
impaired metabolic function
fibrillisation
recruitment of proteins, enzymesand chaperones
Lewy bodies and neurites
truncated α-synuclein
p62
ubiquitin
