pharmacokinetics introduction – describes quantitatively the rates of the steps of drug...
TRANSCRIPT

Pharmacokinetics
• Introduction– Describes quantitatively the rates of the steps of drug disposition (i.e.- absorption, distribution, elimination) – encompasses ADME plus clearance– clearance: the removal of a drug in units of volume/time– quantitative data important to detail fate of the drug, but also to be able to predict doses, routes, etc.– allows individual adjustment based on individual pharmacokinetic assessment

Pharmacokinetics
• Relation of dose, plasma drug concentration, and effect
– a specific dose of a drug should produce a specific effect:
• Dosage Conc. in plasma water Conc. At site of action Intensity of effect
– Intensity of effect related to drug conc. at receptor sites– Duration of action related to how long drug conc. at receptor site remains high enough to provide response–Conc. at receptor sites changes as drug enters, distributes, and is eliminated

Pharmacokinetics
• Difficulty in Quantitation– Due to the difficulty of properly modeling so many processes occurring simultaneously– Often make certain assumptions which do not greatly affect the data such as:
• Intensity of effect is correlated to the concentration of free drug in plasma
– not always true – may be very difficult with irreversibly acting drugs, drugs which develop tolerances, or drugs which act synergistically

Pharmacokinetics
• Modeling– Used whenever the fate of a drug is described either qualitatively or quantitatively.– Mathematical model encompassing known factors about drug (such as distribution, etc) hypothesized first, then proven (or modified) by real-life observation.– One-compartment model easiest to use, and many drugs follow this scheme.
• Assumes a single compartment which is in equilibrium which accounts for drug in plasma, and various tissues.
– Two (or more) compartment models more difficult to model.
• Seen when drug moves into tissues and is handled at different rates than central plasma compartment.

Drug Fate in Body
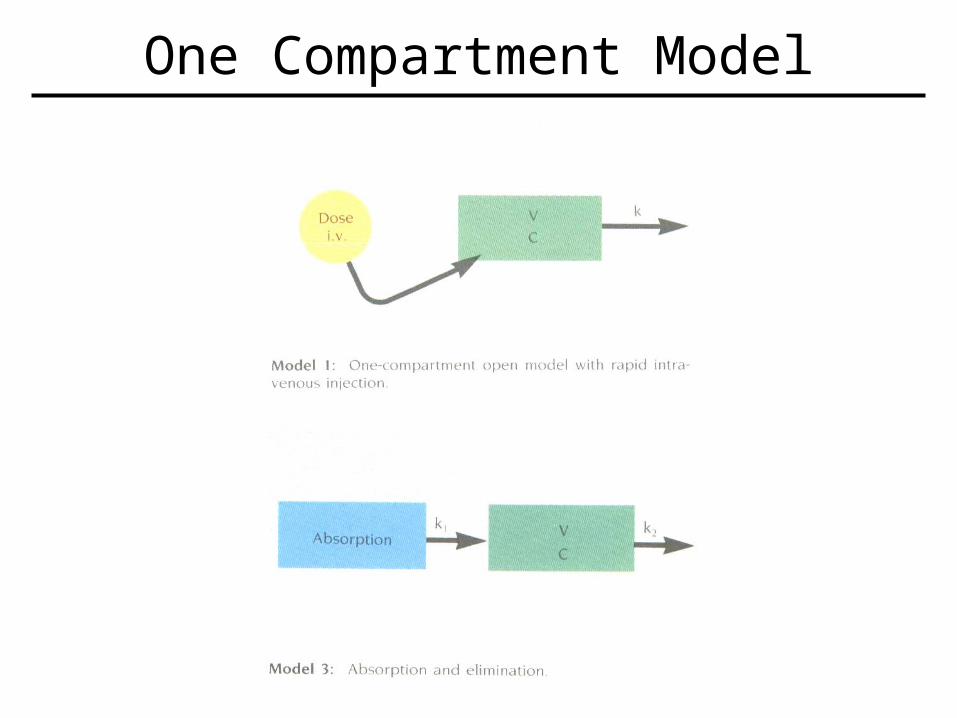
One Compartment Model

One Compartment Model
• Mathematics– Assuming first-order disposition (rate at any time is proportional to concentration of the drug)– Therefore, after IV administration, plasma concentration (Cp) decreases at a rate proportional at all times (t) to the concentration at that time:
• -dCp/dt = kCp (where k = rate constant)
• solving, Cp = C0 e-kt (where C0 is the initial concentration, e is the natural log base, and Cp is the concentration in plasma at any time t).
• OR, log Cp = logC0 – kt/2.303

One Compartment Model

One Compartment Model
• Mathematics– From the dose given, the volume of distribution can also be calculated:
• Vd = D0/C0
– The elimination half-life would then be:
• t1/2 = 0.693/k ( 0.693 = ln 2)

Two Compartment Model
• Involves both distributive and elimination phases normally.
• Log plot does not give a single straight line, but instead shows two phases.
• So now have a central compartment (ex.- plasma), and another compartment (ex.- tissue).
• Can be described mathematically by two differential equations.
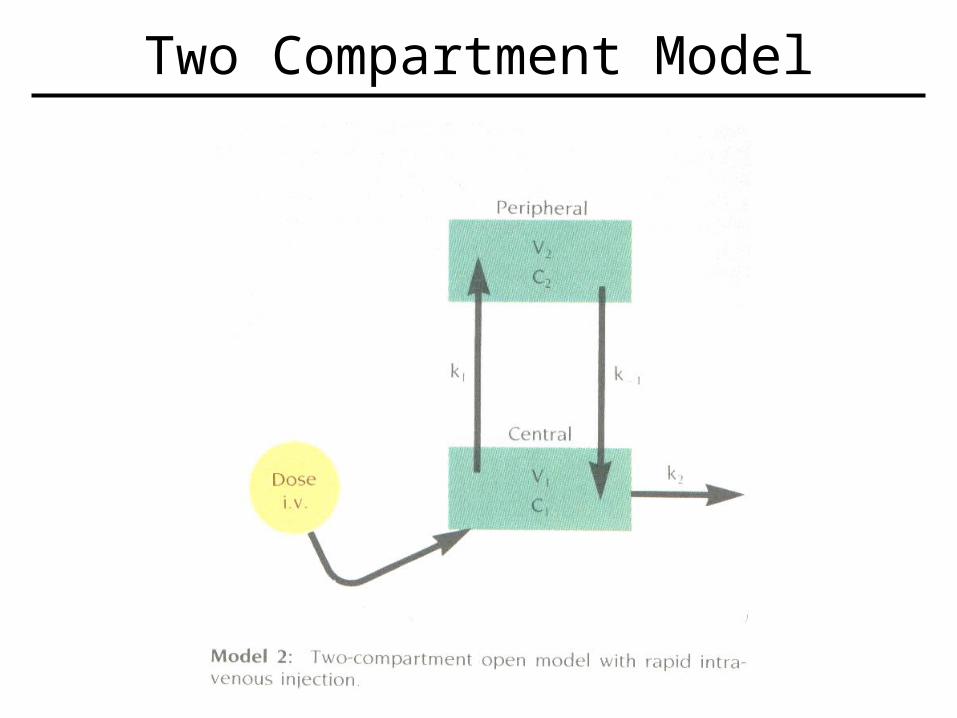
Two Compartment Model
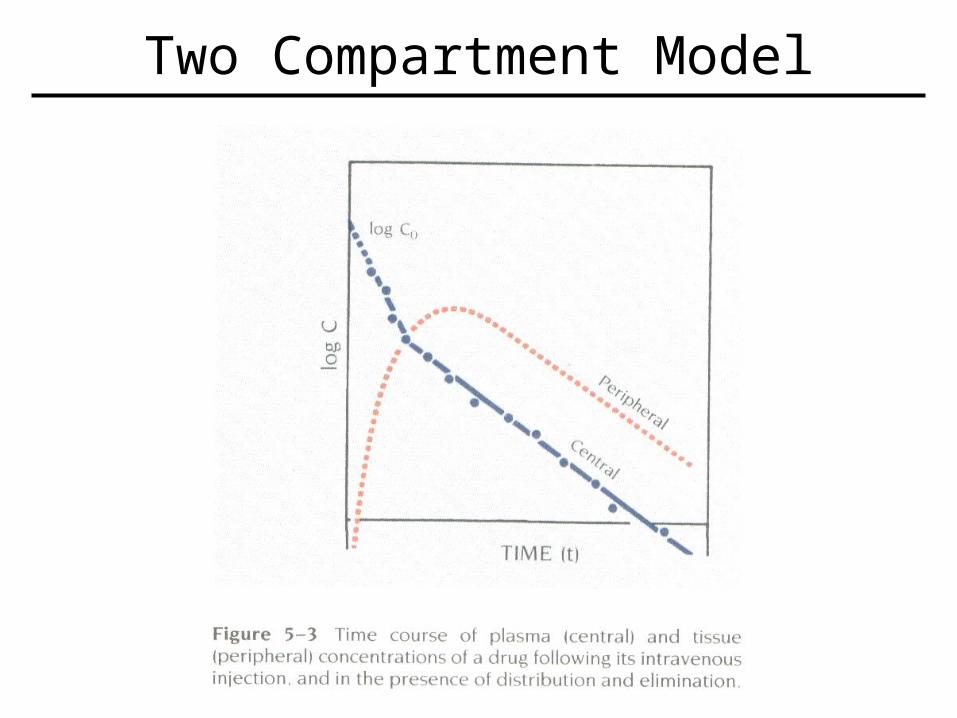
Two Compartment Model

Absorption and Elimination

Absorption Curve

Absorption and Elimination

Rates of Processes
• Have been assuming first-order rate kinetics so far.
• This is usually ok, but what happens if a process (ex.- elimination) is dependant on a carrier or enzyme that may become saturated?
– Rate now no longer dependant on concentration, but instead becomes constant, at least until concentration falls below saturation.– This is termed zero-order kinetics, where the rate is independent of the concentration.

Rates of Processes
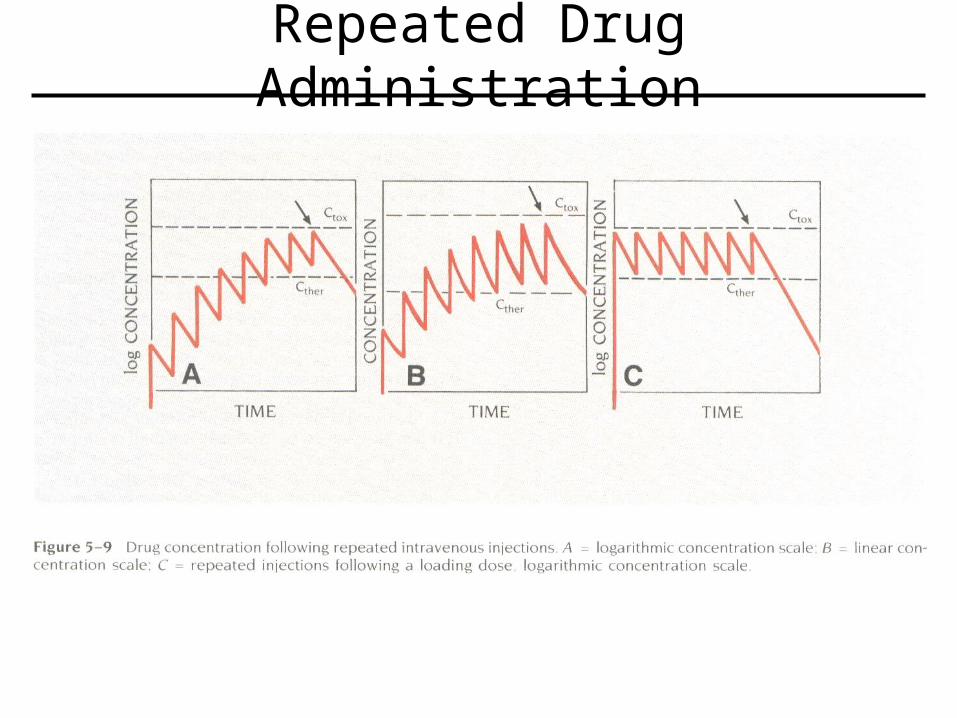
Repeated Drug Administration

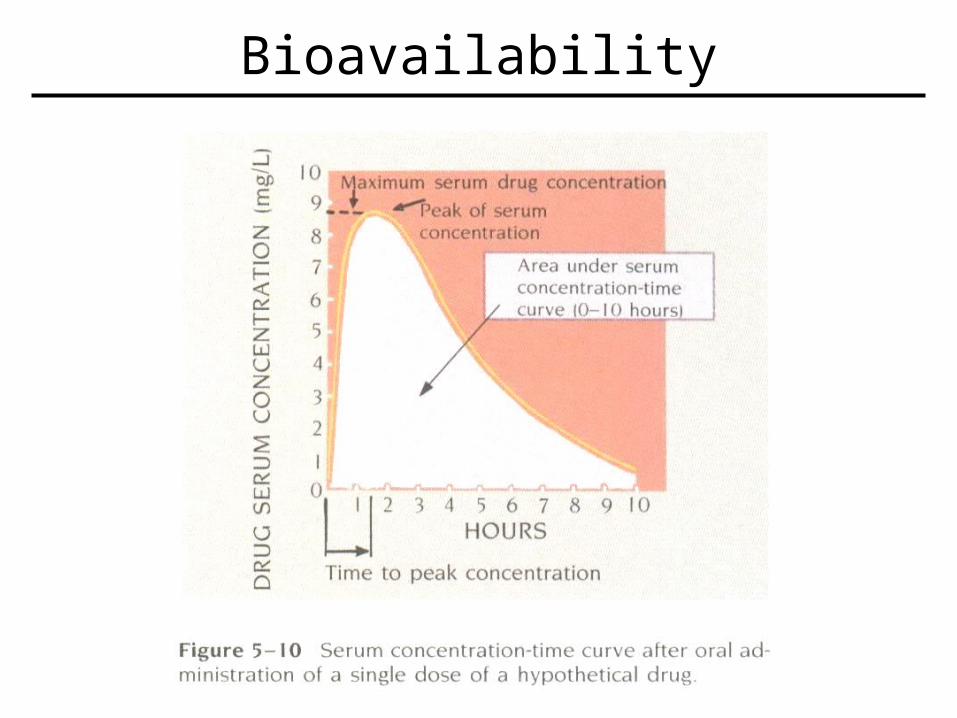
Bioavailability
• Definitions:–Bioavailability – percentage of a drug or drug product that enters the general systemic circulation.
•Includes not only amount entering body, but also rate of entry
–Bioequivalence – comparable bioavailability between drugs.–Therapeutic equivalence – comparable clinical effectiveness and safety between similar drugs.
• Mathematics–Bioavailability = F = AUC (oral) / AUC (IV)
Bioavailability

Volume of Distribution
• Vd = D/C0
• D = amount of Drug in the body
• C0 = initial plasma concentration
• The volume of distribution, Vd, is the apparent or “virtual” volume into which a drug distributes.

Volume of Distribution
• Can Vd be larger than the total plasma volume in the body?
– heparin = 5 liters (plasma only)– chlordiazepoxide = 28 liters (extracellular water)– imipramine = 1600 liters(highly lipid soluble)
•Note: knowledge of the Vd is also important in estimating the loading dose.

Clearance
• Introduction– Quantitative measure of the removal of endogenous or exogenous substances from the body or a specific organ.– Examples include:
•Hepatic biotransformation•Renal excretion•Fecal excretion•Lung exhalation
– Can be mathematically modeled to help define proper dosing regimes.

Total Body Clearance
• Cltot = k Vd , where:
– Cltot = total body clearance
– k = first order elimination rate constant
– Vd = apparent volume of distribution
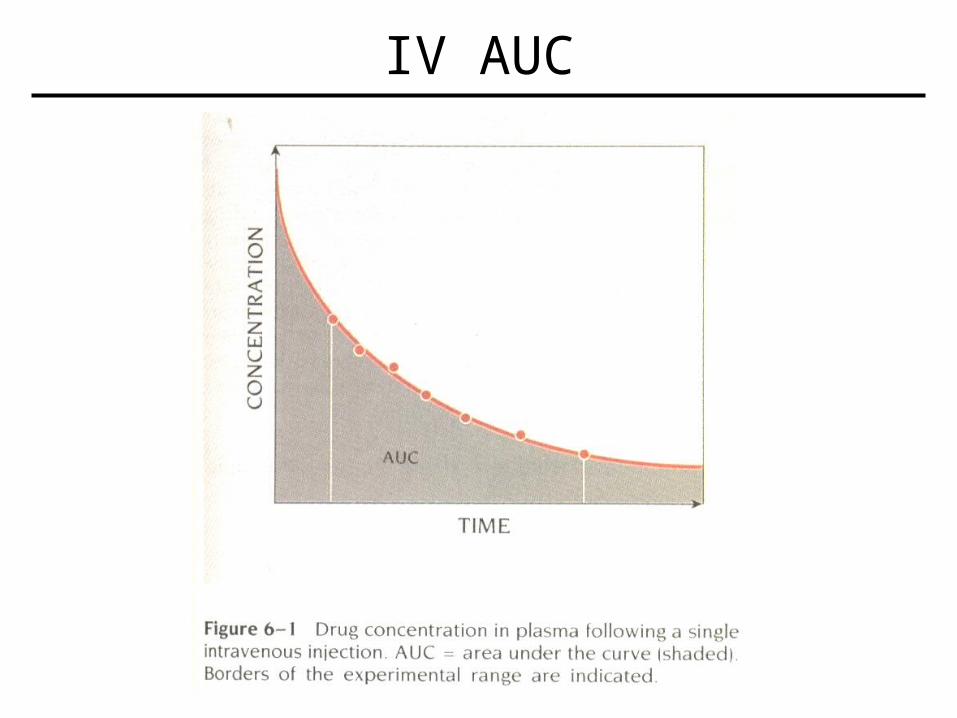
• Cltot = D / AUC – Assumes drug is completely absorbed (IV)– D = dose of drug– AUC = area under plasma conc. (y) vs time curve (x)
– If not completely absorbed, Cltot = F D / AUC, where F is the fraction ‘absorbed’ (bioavailability)
IV AUC

Dosage for IV Infusion
• Goal is to provide a constant plasma level while supplying drug at the same rate as elimination.
• Q = k Vd Cpss , where:– Q = amount of drug supplied per unit time– k = elimination rate constant
– Vd = apparent volume of distribution
– Cpss = plasma concentration at steady state
• Since Cltot = k Vd, then Q = Cltot Cpss

Loading Dose
• Used to reach steady state plasma concentration (Cpss) immediately, instead of waiting the normal 5 half-lives.
• L = Vd Cpss , where:– L = loading dose (amount of drug)
– Vd = apparent volume of distribution
– Cpss = plasma concentration desired at steady state

Repeated IV Dosing
• Can not maintain a constant Cpss, but instead maintain an average Cpss.
• Dosing interval will determine severity of fluctuation above and below average Cpss.
• Cpss = (Dm / Tm) / Cl , where:– Dm = maintenance dose (amount)
– Tm = maintenance dose interval
• Dm = k Vd Cpss Tm (rearranging by Cl = k Vd)

Ideal Dosing Regimen
• Determine the maintenance dose which will keep plasma level in therapeutic window:
– Dm = (Cptox – Cpther) Vd
– Tm = (ln Cptox – ln Cpther) / k
= (2.3 / k) log (Cptox / Cpther)
= 3.32 t1/2 log (Cptox / Cpther)
• To determine a loading dose:– L = Vd Cptox

IV AUC

Practical Dosing Regimen
• Maintenance doses are frequently given at intervals equal to their t1/2 , but must also be given at manageable times (ex. – q4h, q6h, q12h, qd).
•Mathematically, giving a dose at intervals of it’s t1/2 yields a Cpmax = 2Cpmin, which is a 100% fluctuation.
• Obviously more frequent dosing would be preferred to diminish fluctuations, but may not be critical.

Practical Dosing Regimen
• Drugs with a t1/2 less than 6 hours require a very wide therapeutic window to use them in repeated doses.
• Drugs with narrow therapeutic windows should be given by continuous infusion.
•To utilize repeated oral dosing, one must take bioavailability (F, the fraction entering general systemic circulation) into all calculations.

Clearance By Specific Organs
• Clearances are additive.– Cltot = ClH + ClR + …
• Amount of drug removed by an organ dependant on perfusion and extraction ratio:
– E = (Ca – Cv) / Ca , where:•E = extraction ratio
•Ca = concentration in arterial inflow
•Cv = concentration in venous outflow

Clearance By Specific Organs
• By taking blood flow into account, – Cltissue = Qtissue E , where Q = tissue blood flow
– Then, ClH = QH (Ca – Cv) / Ca
• First pass effect can also be described as:– FH = 1 – E , where:
•FH = the bioavailability fraction due to first pass
• First pass may be saturable, and like all liver metabolism may increase due to enzyme induction.

Clearance By Specific Organs
• High Extraction Ratio–ClH controlled by blood flow rate
–Strong first-pass effect–Plasma protein binding may facilitate clearance
• Low Extraction Ratio–ClH controlled by intrinsic clearance
–Biotransformation limited by diffusion–Plasma protein binding reduces clearance–Sensitive to enzyme inhibition and induction

Clearance By Specific Organs
• For Kidneys:–Rate of renal excretion = Rate of filtration + Rate of secretion – Rate of reabsorption
–ClR = (Excreted amount / Time Interval)
Plasma Concentration
• For Biliary Excretion:–ClB = (Conc. in bile / Conc. In plasma) Bile flow
–Bile flow normally 0.5 – 0.8 ml/min.
