phosphatidylinositol3-kinase signalingthroughprotein ... · phosphatidylinositol3-kinase...
TRANSCRIPT

Phosphatidylinositol 3-Kinase � Signaling through ProteinKinase C� Induces NADPH Oxidase-mediated OxidantGeneration and NF-�B Activation in Endothelial Cells*
Received for publication, August 10, 2005, and in revised form, February 21, 2006 Published, JBC Papers in Press, March 9, 2006, DOI 10.1074/jbc.M508810200
Randall S. Frey‡1, Xiaopei Gao‡, Kamran Javaid‡, Shahid S. Siddiqui‡, Arshad Rahman§, and Asrar B. Malik‡
From the ‡Department of Pharmacology and Center for Lung and Vascular Biology, the University of Illinois College of Medicine,Chicago, Illinois 60612 and the §Departments of Pediatrics and Environmental Medicine, University of Rochester School ofMedicine, Rochester, New York 14642
We addressed the role of class 1B phosphatidylinositol 3-kinase(PI3K) isoform PI3K� in mediating NADPH oxidase activation andreactive oxidant species (ROS) generation in endothelial cells (ECs)and of PI3K�-mediated oxidant signaling in the mechanism ofNF-�B activation and intercellular adhesion molecule (ICAM)-1expression. We used lung microvascular ECs isolated from micewith targeted deletion of the p110� catalytic subunit of PI3K�.Tumor necrosis factor (TNF) � challenge of wild type ECs causedp110� translocation to the plasma membrane and phosphatidyli-nositol 1,4,5-trisphosphate production coupled to ROSproduction;however, this response was blocked in p110��/� ECs. ROS produc-tion was the result of TNF� activation of Ser phosphorylation ofNADPH oxidase subunit p47phox and its translocation to EC mem-branes. NADPHoxidase activation failed to occur in p110��/� ECs.Additionally, the TNF�-activated NF-�B binding to the ICAM-1promoter, ICAM-1 protein expression, and PMN adhesion to ECsrequired functional PI3K�. TNF� challenge of p110��/� ECs failedto induce phosphorylation of PDK1 and activation of the atypicalPKC isoform, PKC�. Thus, PI3K� lies upstream of PKC� in theendothelium,anditsactivationiscrucialinsignalingNADPHoxidase-dependent oxidant production and subsequent NF-�B activationand ICAM-1 expression.
Four mammalian phosphatidylinositol 3-kinase (PI3K)2 type 1 iso-forms, p110�, p110�, p110�, and p110�, have been identified (1), and ofthese, p110� has distinct properties. Type 1A PI3Ks, p110�, p110�, andp110�, associate with one of the five regulatory subunits: p50�, p55�,and p85� (products of alternative splicing of a single gene) and p55� andp85� (2). In contrast, type 1B PI3K (or PI3K�), the catalytic subunitp110� binds to the p101 adaptor molecule (3) or the G��-activatedregulatory subunit p84 (4). Type 1A PI3Ks are activated by interactionswith tyrosine-phosphorylated molecules, whereas p110� is activated by
heterotrimeric G proteins G� and G�� that bind to the pleckstrinhomology domain found in the NH2-terminal region of PI3K� (3, 5).p110� is also activated by pro-inflammatory cytokines such as TNF�
(6). Expression of PI3K� is largely confined to leukocytes, and there is agrowing appreciation of its important role in immunity andhost defense(7–18). Studies also demonstrated the presence of the PI3K� isoform inendothelial cells (ECs) (19, 20), but its function remains unclear.PI3Ks catalyze the conversion of phosphatidylinositol 4,5-bisphos-
phate to phosphatidylinositol 3,4,5-trisphosphate (PIP3), which is involvedin the recruitment and activation of a variety of regulatory proteins viainteractions with their pleckstrin homology and phox homology domains(21). phox domains, present in two subunits of the NADPH oxidasecomplex, p47phox and p40phox, bind to phosphatidylinositol 3,4-bisphos-phate and phosphatidylinositol trisphosphate (both breakdown prod-ucts of PIP3) (21, 22). Degradation of PIP3 occurs by either PTEN(3�-phosphatase and tensin homolog deleted on chromosome 10) orSH2-containing phosphatidyl inositol phosphatases (SHIP-1 andSHIP-2) (7, 23, 24).NADPH oxidase is a tightly regulated membrane-bound enzyme
complex catalyzing the one-electron reduction of oxygen to superoxidewith the simultaneous oxidation of cytosolic NADPH (25). We showedthat TNF�-induced oxidant generation in ECs requires activation ofPKC� (26, 27). PKC� associates with and phosphorylates p47phox, and inturn promotes p47phox association with Nox2 to generate the activeNADPH oxidase complex (28, 29). We also showed that PKC� activa-tion of NADPH oxidase was required for TNF�-induced oxidant gen-eration in ECs (28).In the present study, we addressed a possible role for PI3K� as an
upstream regulator of PKC� activation and thereby in mediating NADPHoxidase assembly and generating the oxidant signaling required for NF-�Bactivation and ICAM-1 expression in ECs. Our results show that TNF�
induces PIP3 production and mediates the PI3K� activation of PKC�. Weshow that PI3K� plays a crucial role in signaling the activation of NADPHoxidase required for NF-�B activation and ICAM-1 expression in ECs.
EXPERIMENTAL PROCEDURES
Materials—The following antibodies were obtained: p110� antibody(catalog numbers SC-7177 and 4252), ICAM-1 (Western blot, catalognumberSC-8439; immunofluorescence, catalognumberSC-1511), SHIP-2(catalog number SC-14502), VE-cadherin (catalog number SC-6458),ICAM-1 fluorescein isothiocyanate (catalog number SC-18853), and actin(catalog number SC-1616) from Santa Cruz Biotechnology (Santa Cruz,CA) and Cell Signaling Technologies (Beverly, MA). Phospho-PDK1(Ser241, catalog number 3061) and phospho-PKC�/� (human, mouse, andrat cross-reactivity) (Thr410/403, catalog number 9378) antibodies wereobtained fromCell Signaling Technologies. Non-phospho-specific rabbit
* This work was supported in part by National Institutes of Health Grants T32 HL07239and HL60678 (to A. B. M.) and HL67424 (to A. R.) and a research grant from the Amer-ican Lung Association (to R. S. F.). The costs of publication of this article were defrayedin part by the payment of page charges. This article must therefore be hereby marked“advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
1 To whom correspondence should be addressed: Dept. of Pharmacology, University ofIllinois College of Medicine, 835 S. Wolcott Ave., E403, Chicago, IL 60612. Tel.: 312-413-3428; Fax: 312-996-1225; E-mail. [email protected].
2 The abbreviations used are: PI3K, phosphatidylinositol 3-kinase; EC, endothelial cell;WT, wild type; MLVEC, mouse lung microvascular EC; PDK, phosphoinositide-depend-ent protein kinase; PKC, protein kinase C; PMN, polymorphonuclear leukocyte; TNF,tumor necrosis factor; SHIP, SH2-containing phosphatidylinositol phosphatase; ICAM,intercellular adhesion molecule; PIP3, phosphatidylinositol 3,4,5-trisphosphate; HPAEC,human pulmonary artery endothelial cell; PBS, phosphate-buffered saline; RT, reversetranscription; MS, mass spectrometry; ESI-MS, electrospray mass spectrometry; HBSS,Hanks’ balanced salt solution.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 281, NO. 23, pp. 16128 –16138, June 9, 2006© 2006 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A.
16128 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 23 • JUNE 9, 2006
by guest on March 13, 2019
http://ww
w.jbc.org/
Dow
nloaded from

polyclonal PKC� antibody (catalog number 9372) and rat monoclonalPKC� antibody (catalog number ALX-804–042) were obtained fromCell Signaling Technologies and Alexis Biochemicals (San Diego, CA),respectively. Alexa Flour 594 and 488 secondary antibodies, carboxy-H2DCFDA (catalog number C-400) cell-permeant indicator for H2O2
that is retained by cells, mouse monoclonal antibody to PIP3 (catalognumber A21328), TRIzol reagent, Taq DNA polymerase and Pro-QDiamond phosphoprotein gel stain (catalog number 33300) wereobtained from Invitrogen. 1,2-Dioctanoyl-sn-glycero-3-[phosphoinosi-tol-3,4,5-trisphosphate] (tetra-ammonium salt) was obtained fromAvanti Polar Lipids, Inc. (Alabaster, AL). Phospho-Ser antibody (catalognumber 61-8100) was obtained from Zymed Laboratories (San Fran-cisco, CA). Anti-p47phox antibody used in this study was a gift from B.Babior and S. Catz (Scripps Research Institute, La Jolla, CA). Fetalbovine serum was from Hyclone (Logan, UT). Endothelial growthmedium (EGM-2) was obtained from BioWhittaker (Walkersville,MD). Primary human pulmonary artery endothelial cells (HPAECs) wereobtained from Clonetics (La Jolla, CA). RAW 264.7 cells were obtainedfromtheAmericanTypeCultureCollection (Manassas,VA).Recombinanthuman TNF� was obtained from Promega (Madison,WI) and R&D Sys-tems (Minneapolis, MN), and recombinant mouse TNF� was obtainedfrom Roche Applied Science.
Cell Culture—HPAECs were cultured in EBM2 (endothelial basalmedium) completemedium in gelatin-coated flasks with bullet kit addi-tives. Mouse lung vascular endothelial cells (MLVECs) from WT(C57BL/6) and p110��/� mice were cultured as described (28, 30).C57BL/6WTmicewere obtained from Jackson Laboratories and p110�
knockout mice were provided by J. Penninger (Amgen Institute,Toronto, Canada). MLVECs were characterized by their cobblestonemorphology, Factor VIII and VE-cadherin staining, and uptake of lowdensity lipoprotein. RAW 264.7 cells are a macrophage-like, Abelsonleukemia virus transformed cell line derived from BALB/c mice. Thecells are grown in Dulbecco’s modified Eagle’s medium supplementedwith 10% fetal bovine serum, 20 mM HEPES, and 2 mM L-glutamine at37 °C in a humidified atmosphere with 5% CO2. Prior to assay, themedium is changed to Dulbecco’s modified Eagle’s medium supple-mented with 20 mM HEPES, 2 mM L-glutamine, and 0.1 mg/ml bovineserum albumin for 2 h. After the addition of zymosanA (125�g/ml), thecells are placed on ice, and the medium is aspirated at the appropriatetime point. The cells are washed with 1.5 ml of ice-cold PBS solution,pelleted, and PBS-aspirated.
PKC� Kinase Assay—PKC� activity was assayed as described (32).The cell lysateswere immunoprecipitatedwith an antibody against PKCusing protein A/G conjugated to agarose. The immunocomplexes werewashed twice with ice-cold PBS and once with kinase buffer (25 mM
Tris-HCl, pH 7.4, 5mMMgCl2, 0.5 mM EGTA, 1mM dithiothreitol) andresuspended in 30 �l of kinase buffer containing 2.5 �g of histone H1,0.5 mM cold ATP, and 20–30 �Ci of [�-32P]ATP. The reaction mixturewas incubated for 20 min at room temperature, and the reaction termi-nated by the addition of SDS sample buffer. The proteins were analyzedby SDS-PAGE, and the phosphorylated formof histoneH1was detectedby autoradiography.
Phosphatidylinositol Extraction—The procedures were conducted asdescribed (31). Briefly, ice-cold 1:1 CHCl3:CH3OH is added to each cellpellet and vortexed for 1 min, samples are centrifuged at 6000 rpm for 5min at 4 °C, and supernatant is discarded. The remaining cell pellet issuspended in 200 �l of 2:1CHCl3:CH3OH containing 0.25% 12 N HCl,vortexed for 5min, and pulse-spun. To the supernatant 40�l of 1 NHClare added, vortexed for 15 s, and centrifuged to separate the phases. Thesolvent from the collected lower layer is evaporated in a vacuum centri-
fuge, and lipid film was rapidly redissolved in 55 �l of 1:1:0.3 CHCl3:CH3OH:H2O. Before analysis, 5 �l of 300 mM piperidine are added, andthe sample is vortexed and pulse-spun.
MS Analysis of PIP3—Mass spectral analysis was performed on aFinnigan TSQ Quantum triple quadrupole mass spectrometer (Thermo-Finnigan, San Jose, CA) as described (31). The samples were analyzed atan infusion rate of 10�l/min in negative ionizationmode over the rangeof m/z 400–1200. Peaks corresponding to known PIP3s were frag-mented and manually inspected for the presence of the identificationpeaks. A confirmed identificationwas achievedwhen key fragmentationpeaks were larger than three times the signal to-noise ratio. The lowerlimit of detection using this method was reported to be less than 9pmol/ml for 38:4 PIP3 (Avanti Polar Lipids).
Immunoblotting—ECs were washed with ice-cold Tris-bufferedsaline and lysed in 10 mmol/liter Tris-HCl, pH 7.5, 5 mM EDTA, 10mM EGTA, 1 mM MgCl2, 50 �g/ml phenylmethylsulfonyl fluoride,and a mixture of protease inhibitors. The lysates were sonicated for10 s and then ultracentrifuged at 100,000 � g for 1 h at 4 °C, and thesupernatants were collected and designated as cytosolic fraction. Toisolate the membrane fraction, the remaining pellet was resuspendedin the above lysis buffer containing 1% Triton X-100, sonicated, andincubated for 30 min at 4 °C (28, 32). These lysates are microcentri-fuged at 4 °C, and the supernatants were designated membrane frac-tion. Immunocomplexes wereWestern blotted as described (32). Foranalysis of Ser-p47phox phosphorylation, total cell lysates wereimmunoprecipitated with p47phox antibody and Western blottedwith phospho-Ser antibody. For analysis of total p47phox phospho-rylation, total cell lysates were immunoprecipitated with p47phox
antibody and subjected to SDS-PAGE, and gels were stained withPro-Q Diamond phosphoprotein gel stain (33).
Confocal Microscopy—HPAECs, grown on gelatin-coated cover-slips, were treated as indicated, washed with HBSS, fixed in 4%paraformaldehyde, and blocked with 5% goat serum containing 0.2%bovine serum albumin, 0.01% NaN3, and 0.1% Triton X-100. There-after, the cells were incubated for 1 h at room temperature with 1 �gof the indicated primary antibody. After three washes in HBSS, 4�g/ml secondary antibodies Alexa Fluor 488 and 594 (MolecularProbes, Eugene, OR) were added for an additional 2 h at room tem-perature. The cells were extensively washed in HBSS and mountedon glass slides with ProLong Antifade mounting medium (MolecularProbes), and the images were acquired with a Zeiss LSM 510 Metaconfocal microscope. For p110�-PKC� co-localization studies, thecells were incubated with TNF�, fixed, and co-incubated with a ratmonoclonal antibody to PKC�, a rabbit polyclonal antibody to p110�
and secondary antibodies Alexa Fluor 488 and 594 IgG as describedabove. For PIP3 detection, WTMLVECs were incubated with TNF�,fixed, and incubated with monoclonal antibody to PIP3 (34) andsecondary Alexa Flour 594 IgM. Appropriate band filters were usedto detect both proteins and PIP3. Fixed cells labeled with p110�-PKC� antibodies were optically sectioned into z-stacks (0.3-�m-thick confocal sections) with the pinhole set to 1 Airy unit. Quanti-fication of co-localization between p110� and PKC� was performedusing the co-localization module of Zeiss LSM 510–3.2 software.
Alterations inCell Surface ICAM-1—MLVECs fromWTandp110��/�
micewere grownon gelatin-coated coverslips, incubatedwithTNF�, fixedin 3.7% formaldehyde, permeabilized in 0.4% Triton X-100/PBS, blockedwith PBS containing 0.1%TritonX-100, 5% bovine serum albumin, 0.5%gelatin, and incubated for 16 h at 4 °C with ICAM-1 antibody, or for 1 hwith anti-ICAM IgG-fluorescein isothiocyanate and goat polyclonalVE-cadherin antibodies. The cells were washed three times with PBS,
Oxidant Generation and NF-�B Activation via PI3K�
JUNE 9, 2006 • VOLUME 281 • NUMBER 23 JOURNAL OF BIOLOGICAL CHEMISTRY 16129
by guest on March 13, 2019
http://ww
w.jbc.org/
Dow
nloaded from
incubated with Alexa Fluor 594 IgG, and mounted on glass slides withProLong Antifade mounting medium (Molecular Probes). The imageswere acquired with a Zeiss LSM 510Meta confocal microscope. Quan-tification of ICAM-1 staining intensity in VE-cadherin-stained plasmamembranes was from three to four images/coverslip, with each imagecontaining an average of eight cells using ImageJ software (NationalInstitutes of Health, Bethesda, MD).
RT-PCR—All of themethods were performed byACGT Inc. (Wheel-ing, IL). Total RNA was isolated and reversed-transcribed using theInvitrogen Super-Script RT-PCR kit. All of the following human genespecific primers for p110� amplification were used for PCR (35, 36):forward, 5�-GCTTGAAAACCTGCAGAATTCTCAAC-3�; reverse,5�-CGTCTTTCACAATCTCGATCATTCC-3�. Mouse specific prim-ers designed to span exons 3 to 6 of the p110� gene were as follows:forward, 5�-AGAGAAGTATGACGTCAGTTCC-3�; reverse 5�-TT-GAGCCATCGTTGTGGCATCC-3�. The cDNA obtained from RT-PCR was PCR-amplified using Invitrogen Platinum Taq, and the entirePCR product was sequenced in double strand and compared with theexpected reference sequence. The human sequence is deposited inGen-BankTM (accession number AY496423), and the mouse sequence isdeposited in GenBankTM (accession number AY831679).
OxidantGeneration—Oxidant generation inMLVECswasmeasuredas described (28). The cells were loaded with the fluorescent dye car-
boxy-H2DCFDA (10 �M; Molecular Probes) for 1 h. After treatmentwith TNF�, the cells were washed twice with HBSS and fixed in 4%paraformaldehyde for 20 min at room temperature. The cultures werethen viewed with fluorescence microscopy.
Electrophoretic Gel Mobility Shift Assay—Nuclear protein extractswere prepared as describedwith the addition of protease inhibitors (37).The extracted protein was quantified, aliquoted (40–50 �g/aliquot),and stored at �70 °C until use. The oligonucleotide was radiolabeledwith T4 polynucleotide kinase. Nuclear protein (10–15 �g) was incu-bated for 15 min at room temperature with labeled oligonucleotide.Incubation mixtures were separated on a 5% nondenaturing polyacryl-amide gel. The following oligonucleotides were used for gel shift analy-sis: ICAM-1 NF-�B: 5�-AGCTTGGAAATTCCGGAGCTG-3� andIg-�B: 5�-AGTTGAGGGGACTTTCCCAGGC-3�. The oligonucleo-tide designated as ICAM-1 NF-�B represents the 21-bp sequence ofICAM-1 promoter encompassing NF-�B binding site located 223 bpupstream of translation initiation site. The Ig-�B oligonucleotide con-tains the consensus NF-�B binding site present in the immunoglobulingene. Sequence motifs within the oligonucleotides are underlined.
PMNAdhesion Assay—PMNadhesion to endothelial cells was deter-mined with the modifications as described below (38). IsolatedMLVECs from WT and p110��/� mice were grown to confluence in96-well gelatin-coated plates. Mouse PMNs were isolated from whole
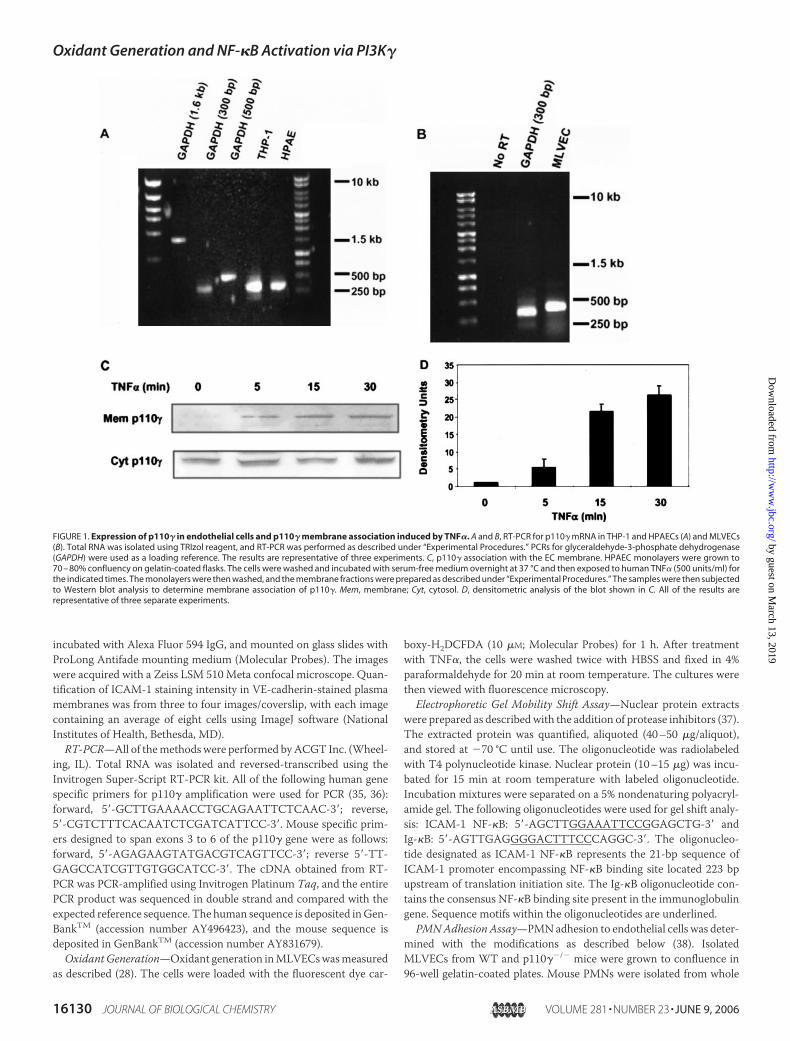
FIGURE 1. Expression of p110� in endothelial cells and p110� membrane association induced by TNF�. A and B, RT-PCR for p110� mRNA in THP-1 and HPAECs (A) and MLVECs(B). Total RNA was isolated using TRIzol reagent, and RT-PCR was performed as described under “Experimental Procedures.” PCRs for glyceraldehyde-3-phosphate dehydrogenase(GAPDH) were used as a loading reference. The results are representative of three experiments. C, p110� association with the EC membrane. HPAEC monolayers were grown to70 – 80% confluency on gelatin-coated flasks. The cells were washed and incubated with serum-free medium overnight at 37 °C and then exposed to human TNF� (500 units/ml) forthe indicated times. The monolayers were then washed, and the membrane fractions were prepared as described under “Experimental Procedures.” The samples were then subjectedto Western blot analysis to determine membrane association of p110�. Mem, membrane; Cyt, cytosol. D, densitometric analysis of the blot shown in C. All of the results arerepresentative of three separate experiments.
Oxidant Generation and NF-�B Activation via PI3K�
16130 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 23 • JUNE 9, 2006
by guest on March 13, 2019
http://ww
w.jbc.org/
Dow
nloaded from

blood as described (38); PMN purity was �95%, and the viabilityassessed by trypan blue exclusion was �98%. PMNs labeled with cal-cein-AM (Molecular Probes) were added to MLVECs pretreated withTNF� (40 ng/ml) for the times indicated at 37 °C. Naı̈ve PMNs wereincubated with MLVECs for 2 h and washed six times with EBM2medium, and the fluorescence readings were obtained using the PTIspectrofluorometer (Photon Technology International, MonmouthJunction, NJ) with detection at 494 and 517 nm, respectively.
Data Analysis—Data are expressed as the mean � S.E. Statistical anal-ysis was performed using two-way analysis of variance. The numbers ofexperiments in the different groups are given in the figure legends. A valueof p � 0.05 was used as the criterion for significance. The ECL signal wasquantitated with Scion Image 4.02 software (Scion Image Corp., Frederick,MD). Carboxy-H2DCFDA fluorescence was quantified using Image Pro-Plus 1.3 software (MediaCybernetics, Silver Spring,MD), and ICAM-1 cellsurface expression was quantified using ImageJ software (National Insti-tutes of Health, Bethesda, MD). The data for MS were collected with theXcalibur software package (Thermo, San Jose, CA).
RESULTS
PI3K� Expression in Endothelial Cells—We initially determined theexpression profile of p110� mRNA and protein in primary human andmouse ECs. RT-PCR was carried out using a set of primers specific forhuman and mouse p110� mRNA on total RNA from HPAECs, humanmonocytic cell line THP-1, and MLVECs (35, 36). As shown in Fig. 1A,p110� mRNA was detected in HPAECs, and it was identical to THP-1p110� mRNA with a 316-bp PCR fragment. Sequencing and BLASTanalysis indicated that the PCR fragment was 100% identical to human
p110� (5, 39). p110� was also detected in MLVECs with a 350-bp PCRfragment (Fig. 1B). Sequencing and BLAST analysis indicated that thisPCR fragment was 100% identical to mouse p110� (39). We next deter-mined the effects of TNF� in inducing themembrane association of p110�
because this is a requirement for activation of PI3K� (40, 41). Westernblotting showed thatTNF� exposure ofHPAECs increased themembranelocalization of p110�within 5min, and the response remained elevated upto 30min (Fig. 1,C andD). TNF� challenge ofMLVECs induced a similarp110� translocation to the membrane (data not shown).
p110� Co-localizes with PKC�—To examine the relative distributionof p110� in endothelial cells, HPAECs were treated with TNF�, fixed,and double stained for p110� and PKC� using Alexa Flour 594 and 488secondary antibodies, respectively (Fig. 2, A and B). In the absence ofTNF� (control), PKC� and p110� were primarily localized in the cyto-plasm and perinuclear regions. TNF� induced accumulation of PKC�
and p110� at the plasma membrane (Fig. 2, A and B). To assess co-localization of PKC� and p110�, we optically sectioned cells into 0.3-�msections, and the plasma membrane and perinuclear regions wereselected for each cell.Weobserved a striking overlap of p110� andPKC�
after TNF� exposure (Fig. 2C). Co-localization coefficient in controlcells was 0.34 comparedwith 0.90 after TNF� treatment.We also deter-mined co-localization of p110� with p120-catenin, a juxta-EC plasmamembrane protein. In control and TNF�-treated cells, we did notobserve any pattern of co-localization of p110� with p120-catenin (datanot shown), suggesting the specificity of p110�-PKC� co-localization. Inanother experiment, we determined the association between p110� andPKC� by immunoprecipitation using an antibody to PKC� andWesternblotting for p110� (Fig. 2D).Membranes isolated from control HPAECs
FIGURE 2. TNF� induces association of p110� and PKC�. HPAECs were left untreated (control) or stimulated with TNF� (500 units/ml) for 30 min. The cells were fixed with 4%paraformaldehyde, stained with a rabbit polyclonal p110� antibody (red, Cell Signaling, catalog number 4252, dilution 1:50) in combination with a rat monoclonal PKC� antibody(green, Alexis Biochemicals, dilution 1:100) and 4�,6�-diamino-2-phenylindole (blue, 1 �g/ml) to view the nucleus. The slides were mounted and analyzed by confocal microscopy. A,xy confocal images of HPAECs using 63� objective. B, xy confocal images of HPAECs using �-plan-fluor 100�/1.45 objective. C, xz orthogonal confocal images of cells in B; arrowsindicate regions of intense p110�-PKC� co-localization (yellow). D, association of PKC� and p110�. The membrane fractions were isolated from HPAEC, and equal amounts of proteinwere immunoprecipitated (IP) with PKC� antibody and immunoblotted (IB) with an antibody to p110�. All of the results are representative of three experiments.
Oxidant Generation and NF-�B Activation via PI3K�
JUNE 9, 2006 • VOLUME 281 • NUMBER 23 JOURNAL OF BIOLOGICAL CHEMISTRY 16131
by guest on March 13, 2019
http://ww
w.jbc.org/
Dow
nloaded from

(0 min) showed that p110� and PKC� were found in the same immuno-complex, and TNF� challenge increased the appearance of p110� inimmunoprecipitates of PKC� (Fig. 2D).
TNF� Induces Activation of p110� and PIP3 Production in Endothe-lial Cells—Activationof PKB/Akt is dependent onPDK1phosphorylation,whichrequires activationofPI3Ks (1).Thisproduces the secondmessengerPIP3,whichbinds to thepleckstrinhomologydomainofPDK1, recruiting itto theplasmamembranewherePDK1 is activatedbyphosphorylationof itsactivation loop residue Ser241 (42). To determine whether TNF� inducedmembrane translocation of p110� in ECs and resulted in PI3K activation,we carried out immunoblot analysis of PDK1. Challenge of HPAECs withTNF� induced the phosphorylation of the membrane-associated PDK1within 5min, and the response wasmaintained up to 30min (Fig. 3A).Wealso observed that challenge ofMLVECs with TNF� resulted in phospho-rylation of membrane-associated PDK-1 (Fig. 3B). However, this responsewas inhibited in p110��/� MLVECs (Fig. 3B), indicating that p110� isrequired for TNF�-induced PDK-1 activation.Fig. 4A shows confocalmicroscopy results of PIP3 staining before and
after TNF� challenge using a monoclonal antibody specific for PIP3(34). TNF� increased PIP3 staining within 1 min, and the response wassustained for 30 min and localized mainly in the cytoplasm and perinu-clear regions. We used electrospray mass spectrometry (ESI-MS) todetermine PIP3 lipid species produced in ECs. Fig. 4B shows a typicalpositive control ESI-MS profile of control and zymosan-treated RAW264.7 macrophages demonstrating an increase in PIP3 with a atomicmass of 1101m/z. Using an internal PIP3 standard (31), we determinedthat ESI-MS correctly identified PIP3 (data not shown). To confirm theconfocal immunofluorescence results for PIP3, we determined whetherTNF� could stimulate PIP3 production as measured using ESI-MS. Weobserved that TNF� induced the production of PIP3with atomicmassesof 1075 and 1101 (Fig. 4B), similar to those reported for zymosan-treated RAW 264.7 cells (31). We did not detect any effect of TNF� onPIP3 production in the p110��/� MLVECs (Fig. 4B).We next examined themembrane localization of the nonhematopoietic
cell SH2-containing phosphatidyl inositol phosphatase SHIP-2 (43–45)becauseSHIPactivation isdependenton its associationwith themembrane(46).MLVECmembrane fractions obtained fromWTand p110��/�micewere Western blotted for SHIP-2. In WT MLVECs, in the basal state,SHIP-2 was present in the membrane, and its distribution was not furtheraltered after TNF� challenge (Fig. 4C). In contrast, SHIP-2 was undetect-able inmembranes isolated fromp110��/�ECs (Fig. 4C) butwaspresent inthe cytosolic fraction, indicating that p110� is responsible for EC mem-brane association of SHIP-2 in the basal state.
p110� Deletion Prevents TNF�-induced Phosphorylation and Mem-brane Translocation of p47phox—We determined the phosphorylationof p47phox, a requirement for the activation of NADPH oxidase (47, 48).TNF� challenge of WT cells induced time-dependent phosphorylationof p47phox as assessed by Pro-Q Diamond staining; the phosphorylatedform of p47phox increased 2.8-fold at 5 min after TNF� challenge andwas sustained for 30 min at the fold level (Fig. 5A). We also observedthat TNF� challenge of WT cells reduced the electrophoretic mobil-ity of p47phox, indicative of increased phosphorylation. However,TNF�-induced phosphorylation of p47phoxwas significantly reduced inp110��/� ECs, ranging from 1.2-fold at 5 min to 0.9-fold at 30min (Fig.5A). A number of earlier studies in leukocytes have shown that p47phox
is phosphorylated on several serine residues located between aminoacids 303 and 379, and p47phox serine phosphorylation is crucial for theassembly of NADPH oxidase in phagocytes (47); thus, we determinedp47phox Ser phosphorylation using immunoprecipitation and Westernblotting. We observed that TNF� induced Ser phosphorylation ofp47phox in WT cells similar to the phosphorylation shown above usingPro-Q Diamond staining (Fig. 5A), but this response was absent inp110��/� cells (Fig. 5, B and C).
FIGURE 3. p110� is required for TNF�-induced PDK1 phosphorylation in endothe-lial cells. A, HPAECs were grown on gelatin-coated flasks and challenged with TNF� forthe indicated times. The membrane and cytosolic fractions were separated by SDS/PAGEand immunoblotted with an antibody against the phosphorylated form (Ser241) of PDK1.B, MLVECs were isolated from WT and p110��/� mice and challenged with TNF� for 5min. The cells were lysed, and the membrane fractions were prepared. The lysates wereseparated by SDS/PAGE and immunoblotted with an antibody against the phosphoryl-ated form (Ser241) of PDK1. All of the results are representative of three experiments.Mem, membrane; Cyt, cytosol.
FIGURE 4. TNF� induces PIP3 production. A, MLVECs were isolated from WT mice, and the cells were treated with TNF� for the indicated times, fixed with 4% paraformaldehyde, andstained with a mouse monoclonal PIP3 antibody (red) in combination and 4�,6�-diamino-2-phenylindole (blue, 1 �g/ml) to view the nucleus. The slides were mounted and analyzedby confocal microscopy. B, positive control; mass spectrometry of lipid present in RAW 264.7 macrophages before and after zymosan (125 �g/ml) treatment for 15 min. Massspectrometry of lipids present in MLVECs untreated or treated with TNF� for 5 and 30 min was performed. After 5 min TNF�, we observed a lipid corresponding to PIP3 with an atomicmass of 1075 m/z and PIP3 corresponding to a mass of 1101 m/z at 30 min after TNF�. We did not detect any TNF�-induced PIP3 production in p110��/� MLVEC. C, membrane andcytosolic fractions were isolated from MLVECs from WT and p110� �/� mice and Western blotted with an antibody against SHIP-2. Mem, membrane; Cyt, cytosol. The results arerepresentative of three experiments.
Oxidant Generation and NF-�B Activation via PI3K�
16132 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 23 • JUNE 9, 2006
by guest on March 13, 2019
http://ww
w.jbc.org/
Dow
nloaded from

FIGURE 4 —continued
Oxidant Generation and NF-�B Activation via PI3K�
JUNE 9, 2006 • VOLUME 281 • NUMBER 23 JOURNAL OF BIOLOGICAL CHEMISTRY 16133
by guest on March 13, 2019
http://ww
w.jbc.org/
Dow
nloaded from

We determined the consequences of deletion of p110� on the mem-brane translocation of p47phox, another requirement for activation ofNADPH oxidase (47). TNF� challenge of WT cells induced time-de-pendent translocation of p47phox (Fig. 5, D and E). However, TNF�-induced p47phox translocation was significantly reduced in p110��/�
ECs (Fig. 5, D and E).We used the fluorescent redox-sensitive dye carboxy-H2DCFDA to
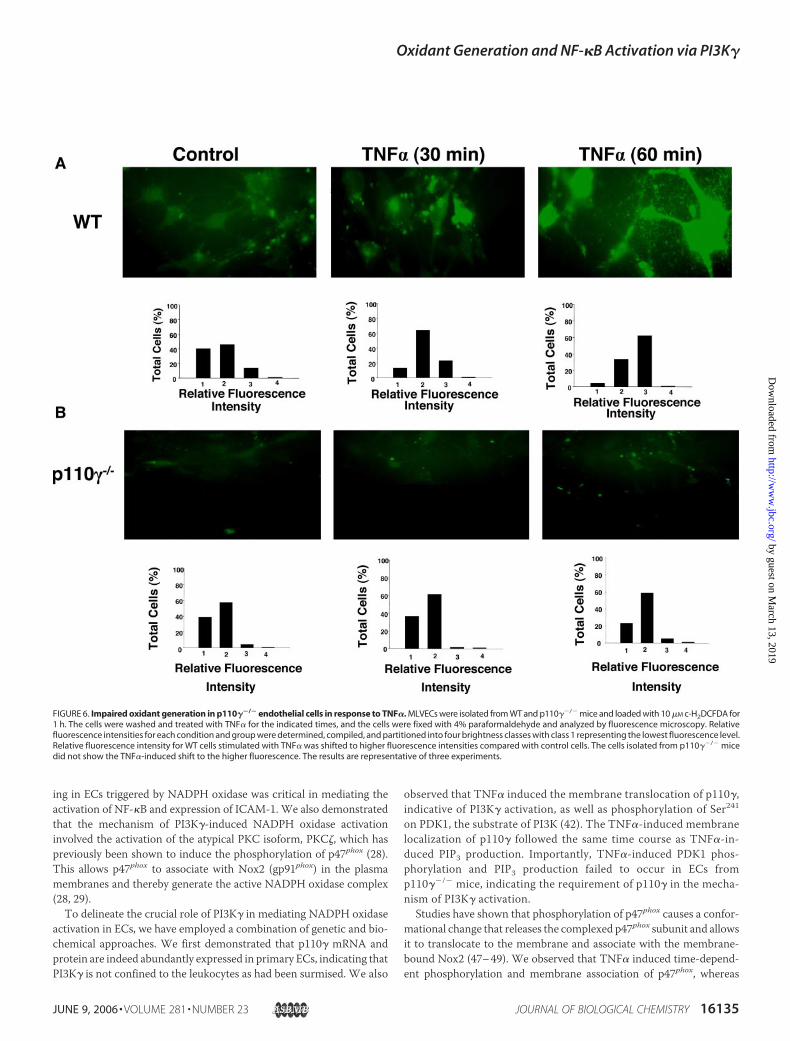
determine the role of p110� in mediating TNF�-induced oxidant gen-eration. WT ECs challenged with TNF� for 30 and 60 min showedmarked oxidant generation, whereas the response was absent inp110��/� ECs (Fig. 6), consistent with the impairment of NADPH oxi-dase activation shown above.Because PKC� is required for signaling TNF�-induced NADPH oxi-
dase and NF-�B activation in ECs (27, 28), we examined the possiblerelationship between p110� and PKC�. TNF� stimulation of WT ECsresulted in phosphorylation and membrane translocation of PKC�
within 5min, and the response was sustained up to 30min (Fig. 7,A andB). However, TNF� failed to induce PKC� phosphorylation and trans-location in p110��/� ECs (Fig. 7, A and B), indicating that PI3K� isrequired for TNF�-induced activation phosphorylation and transloca-tion of PKC�. To determine whether p110� is required for TNF�-in-duced PKC� activation, PKC� from control and TNF�-treatedWT andp110��/� MLVECs was used in an in vitro kinase assay. PKC� activitywas increased 7–8-fold after 5 min of TNF� challenge (Fig. 7C), aresponse sustained for 30min. In contrast, TNF� did not increase PKC�
activity in ECs isolated from p110��/� mice (Fig. 7C), indicating thatp110� is required for TNF�-induced PKC� activation.
p110� Regulates NF-�B Activation and ICAM-1 Expression—Be-cause p47phoxmembrane translocation is necessary forNADPHoxidase
activation and oxidant signaling in ECs (47), we next determined therole for PI3K� in signaling TNF�-induced NF-�B activation andICAM-1 expression. MLVECs isolated from WT and p110��/� micewere challenged with TNF� for various times, and the nuclear proteinswere isolated. We observed that TNF� induced NF-�B DNA bindingand ICAM-1 protein expression in WT ECs, whereas both responseswere blocked in p110��/� ECs (Fig. 8).
Because ICAM-1 expression in ECs results in the firm adhesion ofPMNs, we determined the role of p110� in the mechanism of TNF�-induced ICAM-1 cell surface expression.Weobserved that TNF� expo-sure of WT MLVECs induced ICAM-1 expression on the cell surface,whereas this was blocked in p110��/� MLCECs (Fig. 9A). Studies alsodetermined the adhesion of WT mouse PMNs to WT or p110��/�
MLVECs. We observed that TNF� exposure of MLVECs induced adhe-sion of naı̈ve PMNs toMLVECs, whereas adhesion of PMNs to p110��/�
MLVECs similarly challenged with TNF� was blocked (Fig. 9B).
DISCUSSION
The activation of PI3K is an important step in the host defenseresponse involving PMNandmacrophagemigration in response to che-motactic stimuli and chemokine-induced oxidative burst of PMNs(7–14). The chemoattractant-stimulated p110��/� PMNs failed to pro-duce PIP3 (17) and activate Akt/protein kinase B (16) and showedimpaired respiratory burst and motility responses (15). Because thedescribed role of PI3K� in immunity is largely restricted to leukocytes(15), in the present studywe investigated the possibility that PI3K� playsa potentially important role in regulating the immune response in thevessel wall, specifically the vessel wall lining ECs.We demonstrated thatin response to stimulation with TNF�, PI3K�-induced oxidant signal-
FIGURE 5. p110� regulates TNF�-induced p47phox phosphorylation and translocation to endothelial membrane. A, MLVECs were isolated from WT and p110��/� mice andcells were treated with TNF� for the indicated times, whole cell lysates were prepared, and p47phox was immunoprecipitated (IP). IB, immunoblot. Immunoprecipitates were separatedby 7.5% SDS-PAGE, and gels were stained with Pro-Q Diamond phosphoprotein stain (top panel) or Western blotted for p47phox (bottom panel). B, MLVECs were isolated from WT andp110��/� mice and cells were treated with TNF� for the indicated times and whole cell lysates were prepared. p47phox was immunoprecipitated and Western blotted for phospho-Ser. C, densitometry analysis of the blot shown in B. D, MLVECs were isolated from WT and p110��/� mice, the cells were challenged with TNF� for the indicated times, and membraneand cytosolic fractions were prepared and subjected to Western blot analysis to determine p47phox translocation to the membrane. Mem, membrane; Cyt, cytosolic. E, densitometricanalysis of the blot shown in D. The results are representative of three experiments.
Oxidant Generation and NF-�B Activation via PI3K�
16134 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 23 • JUNE 9, 2006
by guest on March 13, 2019
http://ww
w.jbc.org/
Dow
nloaded from
ing in ECs triggered by NADPH oxidase was critical in mediating theactivation of NF-�B and expression of ICAM-1. We also demonstratedthat the mechanism of PI3K�-induced NADPH oxidase activationinvolved the activation of the atypical PKC isoform, PKC�, which haspreviously been shown to induce the phosphorylation of p47phox (28).This allows p47phox to associate with Nox2 (gp91phox) in the plasmamembranes and thereby generate the active NADPH oxidase complex(28, 29).To delineate the crucial role of PI3K� in mediating NADPH oxidase
activation in ECs, we have employed a combination of genetic and bio-chemical approaches. We first demonstrated that p110� mRNA andprotein are indeed abundantly expressed in primary ECs, indicating thatPI3K� is not confined to the leukocytes as had been surmised. We also
observed that TNF� induced the membrane translocation of p110�,indicative of PI3K� activation, as well as phosphorylation of Ser241
on PDK1, the substrate of PI3K (42). The TNF�-induced membranelocalization of p110� followed the same time course as TNF�-in-duced PIP3 production. Importantly, TNF�-induced PDK1 phos-phorylation and PIP3 production failed to occur in ECs fromp110��/� mice, indicating the requirement of p110� in the mecha-nism of PI3K� activation.
Studies have shown that phosphorylation of p47phox causes a confor-mational change that releases the complexed p47phox subunit and allowsit to translocate to the membrane and associate with the membrane-bound Nox2 (47–49). We observed that TNF� induced time-depend-ent phosphorylation and membrane association of p47phox, whereas
FIGURE 6. Impaired oxidant generation in p110��/� endothelial cells in response to TNF�. MLVECs were isolated from WT and p110��/� mice and loaded with 10 �M c-H2DCFDA for1 h. The cells were washed and treated with TNF� for the indicated times, and the cells were fixed with 4% paraformaldehyde and analyzed by fluorescence microscopy. Relativefluorescence intensities for each condition and group were determined, compiled, and partitioned into four brightness classes with class 1 representing the lowest fluorescence level.Relative fluorescence intensity for WT cells stimulated with TNF� was shifted to higher fluorescence intensities compared with control cells. The cells isolated from p110��/� micedid not show the TNF�-induced shift to the higher fluorescence. The results are representative of three experiments.
Oxidant Generation and NF-�B Activation via PI3K�
JUNE 9, 2006 • VOLUME 281 • NUMBER 23 JOURNAL OF BIOLOGICAL CHEMISTRY 16135
by guest on March 13, 2019
http://ww
w.jbc.org/
Dow
nloaded from

these responseswere absent in p110��/�MLVECs.We showed that theTNF�-induced PKC� Thr410 phosphorylation, and PKC� membranetranslocation and activation were also blocked in MLVECs fromp110��/� mice. These findings demonstrate that p110� lies upstreamof PKC�, and p110� signals PKC� activation.Confocal immuofluorescence data obtained using HPAECs showed
that TNF� induced the membrane translocation of PKC�. We alsoobserved thatTNF� increased the phosphorylated PKC� in themembranefractionofMLVECs,whereas the total amountofPKC� recovered inmem-brane immunoprecipitates increased slightly. This finding suggests that theactivated PKC� is present in themembrane. Because themembrane asso-ciation of PKC� after TNF� challenge decreased in a time-dependentmanner, there is the possibility that PKC�mayhave translocated back tothe cytosol and degraded. However, it is important to note that the
results showing the time course of the TNF�-induced PIP3 productionoccurring between 15 and 30 min corresponds well with increasedmembrane association of phosphorylated PKC�, further suggesting animportant relationship between PI3K� and PKC�.The role of the PI3K� 3 PKC� signaling pathway in the mecha-
nism of NADPH oxidase assembly in ECs seen in the present studiesis consistent with the function of PIP3 in activating PKC� (47, 51).Themechanism of PI3K� activation of PKC� involves PDK1, a kinaseknown to phosphorylate PKC� (52). Studies have shown that PDK1induced the activation of PKC� in a PIP3-dependent manner and thatPDK1 and PKC� became associated in response to platelet-derivedgrowth factor exposure (53). These findings are in agreement withour evidence that PI3K� plays a crucial role in signaling PKC� acti-vation in ECs via PDK1.
FIGURE 7. p110� is required for TNF�-inducedPKC� activation and translocation to endothe-lial membranes. A, MLVECs were isolated fromWT and p110��/� mice, the cells were treatedwith TNF� for the indicated times, and the mem-brane and cytosolic fractions were prepared. PKC�phosphorylation was determined using a phos-pho-specific PKC� Thr410 antibody and Westernblotted. B, densitometric analysis of the blotshown in A (top panel). C, MLVECs from WT andp110��/� mice were stimulated with TNF� (500units/ml) as indicated. The cell lysates were immu-noprecipitated with an antibody against PKC�,and an in vitro kinase assay was carried out onimmunoprecipitates using histone H1 as an exog-enous substrate. The proteins were analyzed bySDS-PAGE, and a phosphorylated form of histoneH1 was detected by autoradiography. Mem, mem-brane; Cyt, cytosolic. The results are representa-tive of three experiments.
Oxidant Generation and NF-�B Activation via PI3K�
16136 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 23 • JUNE 9, 2006
by guest on March 13, 2019
http://ww
w.jbc.org/
Dow
nloaded from

We also observed that the 5� lipid phosphatase SHIP-2 was basallyexpressed in WT MLVEC membranes, whereas SHIP-2 was not local-ized in the membranes of ECs isolated from p110��/� mice. SHIPsregulate the activation of several PKC and PI3K isoforms by controllingthe phosphorylation of PIP3 (23). SHIPs mediate the inhibitory effect bylocalizing at sites of synthesis of PIP3 (46, 54). Because SHIP-2 was notassociated withMLVECmembranes in the absence of p110�, it appearsthat the PIP3 produced by PI3K� is a requirement for the membraneassociation of SHIP-2. The failure of SHIP-2 to localize to the mem-brane in p110��/� ECs also raises the possibility that this may beanother factor responsible for the impairment of PKC� activation andphosphorylation of p47phox seen in p110��/� ECs.To address the functional implications of PI3K� in the mechanism of
NF-�B activation and ICAM-1 expression, we studied the effects ofTNF� in p110��/� MLVECs. Deletion of p110� prevented TNF�-ac-tivated oxidant generation as well as NF-�B activation and ICAM-1expression. Because TNF� mediates ICAM-1 expression in ECs andresults in the adhesion of naı̈ve PMNs to ECs, we determined whetherPI3K�-induced oxidant signaling is capable of promoting the binding ofPMNs to MLVECs, the initial step in the transendothelial PMNmigra-tion response. We observed that adhesion of WT mouse PMNs to WTMLVECs was increased �5-fold in response to 4 h of treatment withTNF�; however, PMN failed to adhere to p110��/� MLVECs similarlychallenged with TNF�. This finding demonstrates that PI3K� is essen-tial for PMN adhesion to TNF�-activated MLVECs; however, only acomponent of endothelial adhesivity activated by PI3K� may bedependent on ICAM-1. Because E-selectin-mediated adhesion was alsodecreased in p110��/� mice (20), PI3K� likely plays a critical role in theexpression of multiple NF-�B-regulated adhesive proteins, includingICAM-1 and E-selectin.In summary, we show that PI3K� is required for the TNF�-induced
oxidant generation in ECs, and it does so through activation of PKC�. Dataobtained using ECs from p110��/� mice showed that PKC� was not acti-vated in the absence of p110�, andp47phox failed to translocate toECmem-brane, thereby preventing NADPH oxidase-induced reactive oxidant spe-cies production. Deletion of p110� expression also prevented the TNF�
activation ofNF-�Band ICAM-1 expression, and the adhesion of PMNs toECs. Because PI3K� mediates endothelial adhesiveness through its abilityto promoteNADPHoxidase assembly required for the activationofNF-�B
and expression of ICAM-1, our results suggest a key role of PI3K� in regu-lating vessel wall innate immune responses.
Acknowledgment—We thank the Research Resource Center at the Universityof Illinois for assisting in the mass spectral analysis.
REFERENCES1. Vanhaesebroeck, B., Leevers, S. J., Ahmadi, K., Timms, J., Katso, R., Driscoll, P. C.,
Woscholski, R., Parker, P. J., and Waterfield, M. D. (2001) Annu. Rev. Biochem. 70,535–602
2. Koyasu, S. (2003) Nat. Immunol. 4, 313–3193. Stephens, L. R., Eguinoa, A., Erdjument-Bromage, H., Lui, M., Cooke, F., Coadwell, J.,
Smrcka, A. S., Thelen,M., Cadwallader, K., Tempst, P., andHawkins, P. T. (1997)Cell89, 105–114
4. Suire, S., Coadwell, J., Ferguson, G. J., Davidson, K., Hawkins, P., and Stephens, L.(2005) Curr. Biol. 15, 566–570
5. Stoyanov, B., Volinia, S., Hanck, T., Rubio, I., Loubtchenkov, M., Malek, D., Stoy-anova, S., Vanhaesebroeck, B., Dhand, R., Nurnberg, B., Gierschik, P., Seedorf, K.,Hsuan, J. J., Waterfield, M. D., and Wetzker, R. (1995) Science 269, 690–693
6. Cadwallader, K. A., Condliffe, A. M., McGregor, A., Walker, T. R., White, J. F.,Stephens, L. R., and Chilvers, E. R. (2002) J. Immunol. 169, 3336–3344
FIGURE 8. Signaling function of p110� in TNF�-induced activation of NF-�B andICAM-1 expression in endothelial cells. MLVECs were isolated from WT and p110��/�
mice and treated with TNF� for the indicated times. A, nuclear extracts were assayed forNF-�B binding activity by electrophoretic mobility shift assay using radiolabeled oligo-nucleotide containing the ICAM-1 �B site. ICAM-1 (B) was determined by Western blot-ting. The results are representative of two experiments.
FIGURE 9. TNF� induces endothelial adhesiveness toward naı̈ve PMNs in p110�-de-pendent manner. A, quantification of cell surface ICAM-1 expression. MLVECs isolatedfrom WT and p110��/� mice were treated with TNF� for the indicated times, fixed, andstained for cell surface ICAM-1 as described under “Experimental Procedures.” The val-ues represent the means (n � three to four images/sample containing an average ofeight cells/sample) ICAM-1 staining intensity with VE-cadherin staining in mergedimages. VE-cadherin staining intensity was not different between WT and p110��/�
MLVECs in the presence or absence of TNF�. Error bars, S.E. B, confluent MLVEC mono-layers from WT and p110��/� mice were treated with TNF� for different periods, andadhesion of WT PMNs was assayed as described under “Experimental Procedures.” Flu-orescence units were the absolute numbers of PMNs adhering to MLVECs. The results arethe means of three experiments.
Oxidant Generation and NF-�B Activation via PI3K�
JUNE 9, 2006 • VOLUME 281 • NUMBER 23 JOURNAL OF BIOLOGICAL CHEMISTRY 16137
by guest on March 13, 2019
http://ww
w.jbc.org/
Dow
nloaded from

7. Okkenhaug, K., and Vanhaesebroeck, B. (2003) Nat. Rev. Immunol. 3, 317–3308. Wymann, M. P., and Pirola, L. (1998) Biochim. Biophys. Acta 1436, 127–1509. Katso, R., Okkenhaug, K., Ahmadi, K., White, S., Timms, J., and Waterfield, M. D.
(2001) Annu. Rev. Cell Dev. Biol. 17, 615–67510. Cantley, L. C. (2002) Science 296, 1655–165711. Fan, J., and Malik, A. B. (2003) Nat. Med. 9, 315–32112. Strassheim, D., Asehnoune, K., Park, J. S., Kim, J. Y., He, Q., Richter, D., Kuhn, K.,
Mitra, S., and Abraham, E. (2004) J. Immunol. 172, 5727–573313. Yum, H. K., Arcaroli, J., Kupfner, J., Shenkar, R., Penninger, J. M., Sasaki, T., Yang,
K. Y., Park, J. S., and Abraham, E. (2001) J. Immunol. 167, 6601–660814. Yang, K. Y., Arcaroli, J., Kupfner, J., Pitts, T.M., Park, J. S., Strasshiem, D., Perng, R. P.,
and Abraham, E. (2003) Cell Signal. 15, 225–23315. Sasaki, T., Irie-Sasaki, J., Jones, R. G., Oliveira-dos-Santos, A. J., Stanford, W. L.,
Bolon, B., Wakeham, A., Itie, A., Bouchard, D., Kozieradzki, I., Joza, N., Mak, T. W.,Ohashi, P. S., Suzuki, A., and Penninger, J. M. (2000) Science 287, 1040–1046
16. Hirsch, E., Katanaev, V. L., Garlanda, C., Azzolino, O., Pirola, L., Silengo, L., Sozzani,S., Mantovani, A., Altruda, F., and Wymann, M. P. (2000) Science 287, 1049–1053
17. Li, Z., Jiang, H., Xie, W., Zhang, Z., Smrcka, A. V., and Wu, D. (2000) Science 287,1046–1049
18. Hannigan, M., Zhan, L., Li, Z., Ai, Y., Wu, D., and Huang, C. K. (2002) Proc. Natl.Acad. Sci. U. S. A. 99, 3603–3608
19. Go, Y. M., Park, H., Maland, M. C., Darley-Usmar, V. M., Stoyanov, B., Wetzker, R.,and Jo, H. (1998) Am. J. Physiol. 275, H1898–H1904
20. Puri, K. D., Doggett, T. A., Huang, C. Y., Douangpanya, J., Hayflick, J. S., Turner, M.,Penninger, J., and Diacovo, T. G. (2005) Blood 106, 150–157
21. Ellson, C. D., Gobert-Gosse, S., Anderson, K. E., Davidson, K., Erdjument-Bromage,H., Tempst, P., Thuring, J.W., Cooper,M. A., Lim, Z. Y., Holmes, A. B., Gaffney, P. R.,Coadwell, J., Chilvers, E. R., Hawkins, P. T., and Stephens, L. R. (2001) Nat. Cell Biol.3, 679–682
22. Ponting, C. P. (1996) Protein Sci. 5, 2353–235723. Krystal, G. (2000) Semin. Immunol. 12, 397–40324. Clement, S., Krause, U., Desmedt, F., Tanti, J. F., Behrends, J., Pesesse, X., Sasaki, T.,
Penninger, J., Doherty, M., Malaisse, W., Dumont, J. E., Le Marchand-Brustel, Y.,Erneux, C., Hue, L., and Schurmans, S. (2001) Nature 409, 92–97
25. Babior, B. M., Lambeth, J. D., and Nauseef, W. (2002) Arch. Biochem. Biophys. 397,342–344
26. Rahman, A., Anwar, K. N., and Malik, A. B. (2000) Am. J. Physiol. 279, C906–C91427. Rahman, A., Bando,M., Kefer, J., Anwar, K. N., andMalik, A. B. (1999)Mol. Pharma-
col. 55, 575–58328. Frey, R. S., Rahman, A., Kefer, J. C., Minshall, R. D., and Malik, A. B. (2002) Circ. Res.
90, 1012–101929. Dang, P. M., Fontayne, A., Hakim, J., El Benna, J., and Perianin, A. (2001) J. Immunol.
166, 1206–121330. Fan, J., Frey, R. S., and Malik, A. B. (2003) J. Clin. Investig. 112, 1234–124331. Milne, S. B., Ivanova, P. T., Decamp, D., Hsueh, R. C., and Brown, H. A. (2005) J. Lipid
Res. 46, 1796–180232. Javaid, K., Rahman, A., Anwar, K. N., Frey, R. S., Minshall, R. D., and Malik, A. B.
(2003) Circ. Res. 92, 1089–109733. Martin, K., Steinberg, T.H., Cooley, L. A., Gee, K. R., Beechem, J.M., and Patton,W. F.
(2003) Proteomics 3, 1244–125534. Chen, R., Kang, V. H., Chen, J., Shope, J. C., Torabinejad, J., DeWald, D. B., and
Prestwich, G. D. (2002) J. Histochem. Cytochem. 50, 697–70835. Krymskaya, V. P., Ammit, A. J., Hoffman, R. K., Eszterhas, A. J., and Panettieri, R. A.,
Jr. (2001) Am. J. Physiol. 280, L1009–L101836. Ho, L. K., Liu, D., Rozycka, M., Brown, R. A., and Fry, M. J. (1997) Biochem. Biophys.
Res. Commun. 235, 130–13737. Fan, J., Frey, R. S., Rahman, A., andMalik, A. B. (2002) J. Biol. Chem. 277, 3404–341138. Lo, S. K., Janakidevi, K., Lai, L., andMalik, A. B. (1993)Am. J. Physiol. 264, L406–L41239. Strausberg, R. L., Feingold, E. A., Grouse, L. H., Derge, J. G., Klausner, R. D., Collins,
F. S., Wagner, L., Shenmen, C. M., Schuler, G. D., Altschul, S. F., Zeeberg, B., Buetow,K. H., Schaefer, C. F., Bhat, N. K., Hopkins, R. F., Jordan, H., Moore, T., Max, S. I.,Wang, J., Hsieh, F., Diatchenko, L., Marusina, K., Farmer, A. A., Rubin, G. M., Hong,L., Stapleton,M., Soares, M. B., Bonaldo,M. F., Casavant, T. L., Scheetz, T. E., Brown-stein,M. J., Usdin, T. B., Toshiyuki, S., Carninci, P., Prange, C., Raha, S. S., Loquellano,N. A., Peters, G. J., Abramson, R. D., Mullahy, S. J., Bosak, S. A., McEwan, P. J.,McKernan, K. J., Malek, J. A., Gunaratne, P. H., Richards, S., Worley, K. C., Hale, S.,Garcia, A. M., Gay, L. J., Hulyk, S. W., Villalon, D. K., Muzny, D. M., Sodergren, E. J.,Lu, X., Gibbs, R. A., Fahey, J., Helton, E., Ketteman, M., Madan, A., Rodrigues, S.,Sanchez, A., Whiting, M., Young, A. C., Shevchenko, Y., Bouffard, G. G., Blakesley,R.W., Touchman, J.W., Green, E. D., Dickson,M. C., Rodriguez, A. C., Grimwood, J.,Schmutz, J., Myers, R. M., Butterfield, Y. S., Krzywinski, M. I., Skalska, U., Smailus,D. E., Schnerch, A., Schein, J. E., Jones, S. J., andMarra, M. A. (2002) Proc. Natl. Acad.Sci. U. S. A. 99, 16899–16903
40. Gillham, H., Golding, M. C., Pepperkok, R., and Gullick, W. J. (1999) J. Cell Biol. 146,869–880
41. Brock, C., Schaefer,M., Reusch,H. P., Czupalla, C.,Michalke,M., Spicher, K., Schultz,G., and Nurnberg, B. (2003) J. Cell Biol. 160, 89–99
42. Casamayor, A., Morrice, N. A., and Alessi, D. R. (1999) Biochem. J. 342, 287–29243. Damen, J. E., Liu, L., Rosten, P., Humphries, R. K., Jefferson, A. B., Majerus, P.W., and
Krystal, G. (1996) Proc. Natl. Acad. Sci. U. S. A. 93, 1689–169344. Lioubin, M. N., Algate, P. A., Tsai, S., Carlberg, K., Aebersold, A., and Rohrschneider,
L. R. (1996) Genes Dev. 10, 1084–109545. Pesesse, X., Deleu, S., De Smedt, F., Drayer, L., and Erneux, C. (1997) Biochem. Bio-
phys. Res. Commun. 239, 697–70046. Phee, H., Jacob, A., and Coggeshall, K. M. (2000) J. Biol. Chem. 275, 19090–1909747. Babior, B. M. (1999) Blood 93, 1464–147648. Quinn, M. T., and Gauss, K. A. (2004) J. Leukocyte Biol. 76, 760–78149. Nauseef, W. M. (2004) Histochem. Cell Biol. 122, 277–29150. Deleted in proof51. Wang, Y. X., Dhulipala, P. D., Li, L., Benovic, J. L., and Kotlikoff, M. I. (1999) J. Biol.
Chem. 274, 13859–1386452. Balendran, A., Hare, G. R., Kieloch, A., Williams, M. R., and Alessi, D. R. (2000) FEBS
Lett. 484, 217–22353. Chou, M. M., Hou, W., Johnson, J., Graham, L. K., Lee, M. H., Chen, C. S., Newton,
A. C., Schaffhausen, B. S., and Toker, A. (1998) Curr. Biol. 8, 1069–107754. Wang, J., Keogh, R. J., Hunter,M.G.,Mitchell, C. A., Frey, R. S., Javaid, K.,Malik, A. B.,
Schurmans, S., Tridandapani, S., andMarsh, C. B. (2004) J. Immunol.173, 6820–6830
Oxidant Generation and NF-�B Activation via PI3K�
16138 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 23 • JUNE 9, 2006
by guest on March 13, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Asrar B. MalikRandall S. Frey, Xiaopei Gao, Kamran Javaid, Shahid S. Siddiqui, Arshad Rahman and
Endothelial CellsB Activation inκNADPH Oxidase-mediated Oxidant Generation and NF-
Inducesζ Signaling through Protein Kinase CγPhosphatidylinositol 3-Kinase
doi: 10.1074/jbc.M508810200 originally published online March 9, 20062006, 281:16128-16138.J. Biol. Chem.
10.1074/jbc.M508810200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/281/23/16128.full.html#ref-list-1
This article cites 50 references, 25 of which can be accessed free at
by guest on March 13, 2019
http://ww
w.jbc.org/
Dow
nloaded from