photoconjugation and photorelease: an …
TRANSCRIPT

PHOTOCONJUGATION AND PHOTORELEASE: AN INVESTIGATION INTO THE
UTILITY OF NAPHTHOQUINONE METHIDE PHOTOCLICK CHEMISTRY
by
JOSHUA ALLEN VALENCIA
(Under the Direction of Jason Locklin)
ABSTRACT
The fast and effective photogeneration of o-naphthoquinone methides (NQM)
from a naphthoquinone methide precursor (NQMP) has been well studied for its use in
protein labelling and the patterning of surfaces. We have developed a method for using
the fast photogeneration of NQM from NQMP derivatives to synthesize an NQMP-
photobase derivative. This neutral molecule is capable of releasing a mild organic base
under a few minutes of ultraviolet irradiation. Additionally, we have investigated a
method of using the fast Diels-Alder reaction of NQM with enamines to conjugate
enamines generated in-situ from aldehydes or ketones to a surface.
INDEX WORDS: COPPER-FREE CLICK CHEMISTRY, PHOTO-CLICK,
NAPHTHOQUINONE METHIDE, PHOTOBASE, DIELS-
ALDER, ENAMINE, FUNCTIONAL SURFACES, CLICK
CHEMISTRY

PHOTOCONJUGATION AND PHOTORELEASE: AN INVESTIGATION INTO THE
UTILITY OF NAPHTHOQUINONE METHIDE PHOTOCLICK CHEMISTRY
by
JOSHUA ALLEN VALENCIA
B.S., Kennesaw State University, 2011
A Thesis Submitted to the Graduate Faculty of The University of Georgia in Partial
Fulfillment of the Requirements for the Degree
MASTER OF SCIENCE
ATHENS, GEORGIA
2017

© 2017
Joshua Allen Valencia
All Rights Reserved

PHOTOCONJUGATION AND PHOTORELEASE: AN INVESTIGATION INTO THE
UTILITY OF NAPHTHOQUINONE METHIDE PHOTOCLICK CHEMISTRY
by
JOSHUA ALLEN VALENCIA
Major Professor: Jason Locklin
Committee: Vladimir V. Popik
Jin Xie
Electronic Version Approved:
Suzanne Barbour
Dean of the Graduate School
The University of Georgia
August 2017

iv
ACKNOWLEDGEMENTS
I would like to thank my advisors Dr. Vladimir Popik and Dr. Jason Locklin for
their advice, guidance, and eternal patience. In addition to teaching me about research,
chemistry, and proper technique, they have each helped teach me about responsibility and
how to properly guide and teach others. I would also like to thank Dr. Jin Xie for his
time, patience, advice, and guidance throughout my graduate career.
Next, I would like to thank my family. Dad, you’ve always been there when I
needed to talk or just to let me know that you were thinking about me. There are so many
times that you’ve called or messaged me at JUST the right time to get me through the
day. Your endless support is something I can always count on, and I don’t know what I’d
do without it. Mom, even though I moved out almost fourteen years ago, home for me
has always been where you and the kids are. You’re such a huge inspiration to me in so
many ways, and I don’t tell you often enough how much I love you. I also want to thank
Mathew, Angela, Stephanie, Patrick, Hannah, Ryan, Summer, Emma, Jackson, Ronnie,
Nathan, and the countless others I’ve been fortunate enough to call my siblings over my
lifetime. Each one of you has brought something special into my life, and each of you has
no idea how much you mean to me. I’m not the best at expressing myself to you guys, but
I love you all so much and would do anything for you.
I’d also like to thank all the lab mates I’ve been fortunate enough to work with
over my graduate school career. Emmanuel, Matt, Chris, Dewey, Masha, Nannan, Kun,
Chris, and Chen of the Popik lab group are incredible assets for any aspiring organic

v
chemist, and I thank you all for the support through the years. Eric, Evan, Joe, Rachelle,
Jenna, Anandi, Jeremy, Jing, Alex, Karson, Deb, Qiaohong, Li, Yutian, Jess, Grant,
Scott, Jess B., Demichael, and Apisata of the Locklin lab have each taught me so much
about characterization, polymerizations, and how to work well with others. A special
thanks to Dr. Chris McNitt and Dr. Dewey Sutton for their incredible knowledge of
organic chemistry that I was lucky enough to take advantage of during our time together.
Dr. Evan White, who has always been supportive and helpful, and who showed me so
many ways to repurpose lab equipment and even household objects to get data. Dr. Joe
Grubbs, whose infectious laughter and great attitude are second only to his incredible
knowledge and his willingness to help others. Dr. Rachelle Benson, who helped me
understand the importance of balancing fun and work by bringing the fun to work. Dr.
Jeremy Yatvin for introducing and welcoming me to the lab and the entire department.
Your friendship in and out of lab got me through a lot of days of struggle. Karson
Brooks, for helping me to keep my sanity throughout my time here. Sometimes I think
the lab would fall apart if you weren’t around. Thanks for keeping us all in line.
Finally, I would like to thank the wonderful friends I’ve made since coming here.
From our first impromptu road trip to late nights, golf games, last-minute casino trips,
and weddings, I couldn’t have done it without you guys and gals being there for me.
Thanks Henry, Majors, Greg, Jeremy, Walter, Karson, Amy, Mary Elizabeth, Jeff, Dave,
Rachelle, Matt, Morgan, Jiana, Josh, Fabian, Charles, Evan, and Justin. You guys are the
best.

vi
TABLE OF CONTENTS
Page
ACKNOWLEDGEMENTS ............................................................................................... iv
LIST OF TABLES ........................................................................................................... viii
LIST OF FIGURES ........................................................................................................... ix
LIST OF SCHEMES............................................................................................................x
CHAPTER
1 INTRODUCTION .............................................................................................1
1.1 Click Chemistry .....................................................................................1
1.2 Naphthoquinone Methides .....................................................................5
1.3 Conclusion .............................................................................................6
1.4 References ..............................................................................................7
2 Preparation and Characterization of an NQMP-Based Organic Photobase .....11
2.1 Introduction ..........................................................................................11
2.2 Synthesis of NQMP-Photobase............................................................12 "[Click here and type Subheading]" #
2.3 Properties of NQMP-Photobase ...........................................................13
2.4 Characteristics of NQMP-Photobase Release ......................................13
2.5 Conclusion ...........................................................................................17
2.6 Experimental ........................................................................................17
2.7 References ............................................................................................22

vii
3 Using Surface-Tethered NQMP to Conjugate Aldehydes and Ketones ..........24
3.1 Introduction ..........................................................................................24
3.2 Synthesis of Photoactive Surfaces .......................................................25 "[Click here and type Subheading]" #
3.3 Photoconjugation with Aldehydes and Ketones ..................................28
3.4 Characterization of Surfaces ................................................................29
3.5 Conclusion ...........................................................................................32
3.6 Experimental ........................................................................................33
3.7 References ............................................................................................39
APPENDICES
A 1H and 13C NMR Spectra of Essential Compounds .........................................40

viii
LIST OF TABLES
Page
Table 3.01: Thickness and Contact Angle Measurements for Functionalized Slides. .......30

ix
LIST OF FIGURES
Page
Figure 1.01: General Reaction Scheme of CuAAC .............................................................2
Figure 1.02: Naphthoquinone Methide Photogeneration and Reactions .............................5
Figure 2.01: NQMP-Photobase Release of Propylamine ..................................................12
Figure 2.02: Hydrochloric Acid Neutralization by Propylamine Released from NQMP-
Photobase under Constant Irradiation ....................................................................14
Figure 2.03: Hydrochloric Acid Neutralization by Propylamine Released from NQMP-
Photobase under Pulsed Irradiation .......................................................................16
Figure 3.01: NQMP Reaction with Enamines ...................................................................24
Figure 3.02: Preparation of an Enamine from an Aldehyde ..............................................25
Figure 3.03: In Situ Formation and Conjugation of an Enamine onto NQMP ..................25
Figure 3.04: FTIR Spectra of Silicon Substrates with a) Poly(PFPA), b) Poly(Propargyl
Acrylamide), c) Surface-Tethered Protected NQMP, d) Surface-Tethered NQMP,
e) Surface-Tethered NQMP after Isobutyraldehyde Conjugation .........................31

x
LIST OF SCHEMES
Page
Scheme 2.01: Synthesis of NQMP-Photobase ...................................................................13
Scheme 3.01: Synthesis of Azide-Terminated TEG Linker ..............................................26
Scheme 3.02: Synthesis of Protected Azide-Terminated NQMP ......................................27
Scheme 3.03: Preparation of Protected NQMP-Functionalized Slides .............................28
Scheme 3.04: Deprotection of NQMP-Functionalized Slides ...........................................28
Scheme 3.05: Photoconjugation of Isobutyraldehyde onto NQMP-Functionalized Slides29

1
CHAPTER 1
INTRODUCTION
1.1 Click Chemistry
Background
The term “click” chemistry was coined in 1998 by K. Barry Sharpless to refer to
chemistry that modular, wide in scope, high-yielding, stereospecific, easily purified,
create few or benign byproducts, and use no solvent, or a benign or easily removed
solvent.1
For many applications, whether in the biomedical or material field, the use of volatile
organic solvents, transition metal catalysts, or small molecule byproducts can render an
otherwise robust and versatile chemistry commercially useless. To remedy this, recent
research has turned to various “click” chemistries to solve some of the more taxing
problems in the biomedical and materials field.
While there are many chemistries that have come to be known as “click”
chemistries, some of the most studied and useful examples are the various
cycloadditions.2 These most commonly involve an alkyne or strained alkene, and have
been utilized for everything from drug discovery to protein tagging to surface
modification.2-5

2
Copper(I)-Catalyzed Alkyne-Azide Cycloaddition (CuAAC)
One of the earliest examples of click chemistry is copper(I)-catalyzed alkyne-
azide cycloaddition (CuAAC). This reaction takes place between a terminal alkyne and
an azide to form a 1,2,3-triazole species (Figure 1.1).
Figure 1.01. General Reaction Scheme of CuAAC
These reactions have been carried out with many variations. Studies have been
done involving using a solid support resin for a copper acetylide to react with a terminal
azide to yield a mixture 1,4- and 1,5-disubstituted [1,2,3]-triazoles in good yield under
mild conditions.6 It was also found that this cycloaddition could take place in the
presence of a copper (II) species and a reducing agent such as sodium ascorbate.7 Under
relatively mild conditions, the reaction between an alkyne and an azide would take place
to give predominantly the 1,4-disubstituted [1,2,3]-triazoles in very good yield.7 One of
the biggest drawbacks to this method is its reliance on high catalyst loading and its
limited use outside of terminal alkynes. To address this, studies have recently been done
to address these issues and have found that 1-iodoalkynes are readily accessible internal
alkynes that show an improved reactivity over terminal alkynes, leading to a lowered
reliance on copper catalyst loading.8

3
Strain-Promoted Alkyne-Azide Cycloaddition (SPAAC)
Another method of reducing the reliance on copper catalysts involves the use of a
strained 8-membered cyclic alkyne. Also known as “copper-free click” chemistry,
SPAAC uses the ring strain to increase the reactivity of an internal alkyne to achieve
cycloaddition without the use of a cytotoxic copper catalyst. By using a cyclooctyne, it
was found that the alkyne-azide cycloaddition could take place under mild conditions
without affecting live cell viability.9
Further developments in cyclooctynes has led to the creation of
dibenzocyclooctynes (DIBO)10, difluorinated cyclooctynes (DIFO)11, aza-
dibenzocyclooctynes (ADIBO)12, and oxa-dibenzocyclooctynes (ODIBO)13. Each of
these derivatives shows increased reactivity and kinetics over the original cyclooctyne
moiety when reacting with azides while retaining the same biocompatibility10-13.
Strain-Promoted Alkyne-Nitrone Cycloaddition (SPANC)
Another example of strain-promoted click cycloaddition is strain-promoted
alkyne-nitrone cycloaddition (SPANC). Similar to SPAAC, SPANC does not involve the
use of a copper catalyst to perform the cycloaddition. Instead, SPANC relies on the ring
strain of the alkyne and the increased reactivity of a nitrone over an azide to achieve a
high rate of reaction without the need for a copper catalyst.14 Several studies have found
that it is fairly straightforward to convert various peptides to nitrones, which allow the
use of SPANC to quickly and efficiently label various biological materials.14
Additionally, cyclic nitrones have an even greater reactivity toward strained alkynes,
further increasing the rate.15-17

4
Diels-Alder Cycloaddition
In addition to strained alkyne systems, there are several examples of strained
alkene click chemistry. The most well-known is perhaps Diels-Alder cycloaddition.
Through the use of a diene and an electron-rich dienophile, a [4+2]-cycloaddition can
occur with great speed and without any byproducts or cytotoxic catalysts.
Some of the recent advancements in Diels-Alder cycloaddition include the use of
tetrazines as a diene to increase the rate of the reaction.18-19 These reactions can be used
for protein tagging,18 as well as live cell tagging.19 One advantage that Diels-Alder
cycloaddition has over other forms of click chemistry involves the aqueous environments
in which most biological applications take place. For biological applications, the low
solubility of many of the strained alkynes can limit their usefulness, while the Diels-
Alder reaction with tetrazines not only has increased solubility, but increased rate in
aqueous systems.19
Photoclick
Photochemistry involves using high-energy light to induce a chemical change in a
molecule. The use of light allows a researcher to exert spatial and temporal control over a
reaction. By choosing when and where the reaction takes place, there are many
applications such as in patterning and targeted reaction where photochemistry is the ideal
method. However, photochemistry is not without its drawbacks. The major drawback for
photochemistry in biological systems is the limited penetration depth of most
wavelengths of light. While this limits the usefulness for in-vivo tagging, the control
gained over the reaction is a reasonable tradeoff.

5
There have been several studies that have combined photochemistry with click
chemistry. By utilizing a photolabile cyclopropenone protecting group, ODIBO can be
used in the patterning of surfaces and multifunctional surfaces.3, 13, 20 Additionally, very
reactive Diels-Alder dienes can be prepared photochemically from stable naphthol
derivatives.21
1.2 Naphthoquinone Methides
Naphthoquinone methides (NQMs) are highly reactive molecules under Diels-
Alder and Michael addition conditions. These molecules can be photochemically
generated from naphthol derivatives known as “naphthoquinone methide precursors”
(NQMP) as shown in Figure 1.02.21 Once the NQM is generated, it reacts in a rapid
manner with vinyl ethers and enamines in a Diels-Alder fashion, or in Michael addition
fashion in the presence of thiols.21-22
Figure 1.02. Naphthoquinone Methide Photogeneration and Reactions.
While the Diels-Alder reaction between NQM and enamines and vinyl ethers
proceeds in a non-reversible fashion, the Michael addition between the reactive alkene of
the NQM and a thiol is reversible. This means that the reaction is highly dependent on the

6
concentration of thiol relative to the water in the system.22 Additionally, the NQMP can
be derivatized to release a “payload” of sorts under UV irradiation. By taking advantage
of the reversibility of the reaction and the rapid hydration of NQM back to NQMP, these
derivatives can be used to release a functional molecule or create/remove a pattern from a
surface with temporal and spatial control.21, 23 These reactions allow NQMP to be used as
a versatile caging group for the release of alcohols, thiols, or other groups21, 23, or as a
versatile group for the patterning of surfaces.23-25
1.3 Conclusion
There has been a boom in the past decade of new chemistries that rely on strained
alkynes and alkenes to perform conjugations and surface modifications in a manner that
is fast, simple, clean, and effective. These “click” chemistries show particular promise in
the fields of bioconjugation and materials science. Particularly when these “click”
chemistries are combined with photochemistry, we can begin to achieve a level of control
and functionality far beyond what has been seen in the past.
Of particular interest is the naphthoquinone methide precursor molecule. Through
the rapid generation of the highly reactive naphthoquinone methide moiety under UV
irradiation, we can envision using this molecule for the targeted release of various
functionalities into a system or the targeted capture of various molecules onto a surface
using the rapid Diels-Alder cycloaddition.

7
1.4 References
1. Kolb, H. C.; Finn, M. G.; Sharpless, K. B., Click Chemistry: Diverse Chemical
Function from a Few Good Reactions. Angewandte Chemie International Edition
2001, 40 (11), 2004-2021.
2. Kolb, H. C.; Sharpless, K. B., The growing impact of click chemistry on drug
discovery. Drug Discovery Today 2003, 8 (24), 1128-1137.
3. Arumugam, S.; Popik, V. V., Sequential “Click” – “Photo-Click” Cross-Linker
for Catalyst-Free Ligation of Azide-Tagged Substrates. The Journal of Organic
Chemistry 2014, 79 (6), 2702-2708.
4. Agard, N. J.; Baskin, J. M.; Prescher, J. A.; Lo, A.; Bertozzi, C. R., A
Comparative Study of Bioorthogonal Reactions with Azides. ACS Chemical
Biology 2006, 1 (10), 644-648.
5. Lang, K.; Chin, J. W., Bioorthogonal Reactions for Labeling Proteins. ACS
Chemical Biology 2014, 9 (1), 16-20.
6. Tornøe, C. W.; Christensen, C.; Meldal, M., Peptidotriazoles on Solid Phase:
[1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar
Cycloadditions of Terminal Alkynes to Azides. The Journal of Organic
Chemistry 2002, 67 (9), 3057-3064.
7. Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B., A Stepwise
Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation”
of Azides and Terminal Alkynes. Angewandte Chemie International Edition
2002, 41 (14), 2596-2599.

8
8. Hein, J. E.; Tripp, J. C.; Krasnova, L. B.; Sharpless, K. B.; Fokin, V. V.,
Copper(I)-Catalyzed Cycloaddition of Organic Azides and 1-Iodoalkynes.
Angewandte Chemie International Edition 2009, 48 (43), 8018-8021.
9. Agard, N. J.; Prescher, J. A.; Bertozzi, C. R., A Strain-Promoted [3 + 2]
Azide−Alkyne Cycloaddition for Covalent Modification of Biomolecules in
Living Systems. Journal of the American Chemical Society 2004, 126 (46),
15046-15047.
10. Ning, X.; Guo, J.; Wolfert, M. A.; Boons, G.-J., Visualizing Metabolically
Labeled Glycoconjugates of Living Cells by Copper-Free and Fast Huisgen
Cycloadditions. Angewandte Chemie International Edition 2008, 47 (12), 2253-
2255.
11. Codelli, J. A.; Baskin, J. M.; Agard, N. J.; Bertozzi, C. R., Second-Generation
Difluorinated Cyclooctynes for Copper-Free Click Chemistry. Journal of the
American Chemical Society 2008, 130 (34), 11486-11493.
12. Kuzmin, A.; Poloukhtine, A.; Wolfert, M. A.; Popik, V. V., Surface
Functionalization Using Catalyst-Free Azide−Alkyne Cycloaddition.
Bioconjugate Chemistry 2010, 21 (11), 2076-2085.
13. McNitt, C. D.; Popik, V. V., Photochemical generation of oxa-
dibenzocyclooctyne (ODIBO) for metal-free click ligations. Organic &
Biomolecular Chemistry 2012, 10 (41), 8200-8202.
14. Ning, X.; Temming, R. P.; Dommerholt, J.; Guo, J.; Ania, D. B.; Debets, M. F.;
Wolfert, M. A.; Boons, G.-J.; van Delft, F. L., Protein Modification by Strain-

9
Promoted Alkyne–Nitrone Cycloaddition. Angewandte Chemie International
Edition 2010, 49 (17), 3065-3068.
15. McKay, C. S.; Chigrinova, M.; Blake, J. A.; Pezacki, J. P., Kinetics studies of
rapid strain-promoted [3 + 2]-cycloadditions of nitrones with biaryl-aza-
cyclooctynone. Organic & Biomolecular Chemistry 2012, 10 (15), 3066-3070.
16. MacKenzie, D. A.; Sherratt, A. R.; Chigrinova, M.; Cheung, L. L. W.; Pezacki, J.
P., Strain-promoted cycloadditions involving nitrones and alkynes—rapid tunable
reactions for bioorthogonal labeling. Current Opinion in Chemical Biology 2014,
21, 81-88.
17. Friscourt, F.; Fahrni, C. J.; Boons, G.-J., Fluorogenic Strain-Promoted Alkyne–
Diazo Cycloadditions. Chemistry – A European Journal 2015, 21 (40), 13996-
14001.
18. Blackman, M. L.; Royzen, M.; Fox, J. M., Tetrazine Ligation: Fast
Bioconjugation Based on Inverse-Electron-Demand Diels−Alder Reactivity.
Journal of the American Chemical Society 2008, 130 (41), 13518-13519.
19. Devaraj, N. K.; Weissleder, R.; Hilderbrand, S. A., Tetrazine-Based
Cycloadditions: Application to Pretargeted Live Cell Imaging. Bioconjugate
Chemistry 2008, 19 (12), 2297-2299.
20. Brooks, K.; Yatvin, J.; McNitt, C. D.; Reese, R. A.; Jung, C.; Popik, V. V.;
Locklin, J., Multifunctional Surface Manipulation Using Orthogonal Click
Chemistry. Langmuir 2016, 32 (26), 6600-6605.

10
21. Kulikov, A.; Arumugam, S.; Popik, V. V., Photolabile Protection of Alcohols,
Phenols, and Carboxylic Acids with 3-Hydroxy-2-Naphthalenemethanol. The
Journal of Organic Chemistry 2008, 73 (19), 7611-7615.
22. Arumugam, S.; Popik, V. V., Photochemical Generation and the Reactivity of o-
Naphthoquinone Methides in Aqueous Solutions. Journal of the American
Chemical Society 2009, 131 (33), 11892-11899.
23. Arumugam, S.; Popik, V. V., Attach, Remove, or Replace: Reversible Surface
Functionalization Using Thiol–Quinone Methide Photoclick Chemistry. Journal
of the American Chemical Society 2012, 134 (20), 8408-8411.
24. Arumugam, S.; Orski, S. V.; Locklin, J.; Popik, V. V., Photoreactive Polymer
Brushes for High-Density Patterned Surface Derivatization Using a Diels–Alder
Photoclick Reaction. Journal of the American Chemical Society 2012, 134 (1),
179-182.
25. Arumugam, S.; Popik, V. V., Patterned Surface Derivatization Using Diels–Alder
Photoclick Reaction. Journal of the American Chemical Society 2011, 133 (39),
15730-15736.

11
CHAPTER 2
PREPARATION AND CHARACTERIZATION OF AN NQMP-BASED
ORGANIC PHOTOBASE
2.1 Introduction
One of the key strengths of the NQMP photochemistry is the level of spatial and
temporal control that is afforded. There are many situations where a particular chemical
functionality is desired under specific circumstances in a system. One instance where this
may be advantageous involves pH. There are many biological systems that are heavily
dependent on the pH of the surrounding environment.
The adhesive plaques of mussels relies on 3,4-dihydroxyphenylalanine (DOPA)
residues to adhere to iron on ships and rocks.1 This DOPA residue relies on the local pH
of its surroundings to bind or release from the iron. This biological system has been
mimicked to create a hydrogel that can undergo a sol-gel transition based on the pH of
the system.1 To achieve a level of spatial and temporal control over this transition, both a
photoacid generator and a photobase generator are needed.
While there are numerous studies involving the synthesis and characterization of
photoacid generators2-4, there are relatively few organic photobase generators.5 To this
end, we have designed a molecule that will take advantage of the fast photogeneration of
the NQM from NQMP and its derivatives. By derivatizing NQMP with a carbamate
functionality tied into the photocleavage site, we hope to be able to synthesize and

12
characterize a purely organic photobase that can rapidly alter the pH of a system at our
direction (Figure 2.01).
Figure 2.01. NQMP-Photobase Release of Propylamine
2.2 Synthesis of NQMP-Photobase
The target NQMP-photobase 2.04 (Scheme 2.01) was prepared from
commercially available methyl 3-hydroxy-2-naphthoate in four steps. The naphthol was
reacted in a Williamson ether synthesis with sodium hydride (NaH) and chloromethyl
methyl ether in dimethyl formamide to give the protected naphtholic product 2.01. Next,
compound 2.01 was reduced with lithium aluminum hydride (LAH) in tetrahydrofuran to
yield 2.02. Next, compound 2.02 was reacted with propyl isocyanate in dichloromethane
to afford compound 2.03. Finally, compound 2.03 was deprotected with Amberlyst-15
proton exchange resin in 90% methanol in water to give the NQMP-photobase 2.04. In
four steps with a total yield of 53% NQMP-photobase 2.04 can be synthesized in gram-
scale quantities.

13
Scheme 2.01
Reagents and Conditions: a) NaH, chloromethyl methyl ether, DMF, 90%; b) LAH, THF,
94.6%; c) Propyl isocyanate, DMAP, triethylamine, CH2Cl2, 78%; d) Amberlyst 15,
methanol/water 9:1, 80%.
2.3 Properties of NQMP-Photobase
NQMP-photobase 2.04 is an off-white crystalline compound that is shelf stable
for long periods of time if stored away from light. It is stable in water, methanol, and
solutions of the two. Prior to irradiation, the molecule is neutral in a 75% methanol/water
mixture.
2.4 Characteristics of NQMP-Photobase Release
The efficiency and speed of the NQMP-photobase release was measured by pH
probe. NQMP-photobase 2.04 in a tenfold excess was dissolved in a 0.125 mM solution
of hydrochloric acid in a 75% methanol/water mixture. The pH of these solutions was
monitored before, during, and after irradiation to track the release of propylamine by the
neutralization of hydrochloric acid.

14
While under constant irradiation at 310nm, the pH of the solution was checked
every sixty seconds. The solution was initially at pH 4, indicating a fairly acidic solution.
During irradiation, the pH of the solution continually rose, reaching a neutral pH within
ten minutes, and a pH of 8 within twenty minutes. At this point it is assumed that the
released propylamine (pKb = 3.3) is in a buffered equilibrium with the slightly acidic
naphthol on the NQMP (pKa = 9.01) (Figure 2.02).6
Figure 2.02. Hydrochloric Acid Neutralization by Propylamine Released from NQMP-
Photobase under Constant Irradiation.

15
To determine whether the photorelease of the carbamate from the NQMP or the
degradation of the carbamic acid was the rate-limiting factor in the neutralization of acid,
a solution of 1.25 mM NQMP-photobase 2.04 was dissolved in a 0.125 mM solution of
hydrochloric acid in 75% methanol/water. The solution was subjected to one-minute
pulses of irradiation at 310 nm, then the pH was monitored for five minutes. After the
first minute of irradiation, the pH climbed from 4.5 to 5.2. The rate of change of the pH
was found to slow over the five minutes of monitoring after the first minute of irradiation.
After the second minute of irradiation, the pH climbed at a much faster rate, and reached
a pH of 6.6 after five minutes. Unlike the initial irradiation, there was no observable
slowing of the rate of pH change. Subsequent irradiation cycles followed a similar
smooth curve to a final pH of 8.3 after five irradiation cycles (a total of five minutes of
irradiation)(Figure 2.03).

16
Figure 2.03. Hydrochloric Acid Neutralization by Propylamine Released from NQMP-
Photobase under Pulsed Irradiation.
This change in the rate of pH change leads to the conclusion that the degradation
of the carbamic acid is a slower process than the photorelease of the carbamic acid.
Additionally, the steady rate of pH change after two minutes of irradiation suggests that
only two minutes of irradiation is needed to fully convert the NQMP-photobase to
NQMP and carbamic acid. The carbamic acid then degrades into CO2 and propylamine
over the course of twenty minutes.
Further investigation into this phenomenon indicates that this is most likely the
case. Studies have indicated that the degradation of carbamic acids into linear

17
alkylamines takes place on the order of 102-103 M-1s-1.7 The photochemical conversion of
NQMP derivatives into NQM occurs on the order of 104-105 M-1s-1.8 These rates support
the conclusion that the initial release of the carbamic acid from the NQMP-photobase
takes place rapidly under only a few minutes of irradiation, while the degradation of the
released carbamic acid into CO2 and propylamine is a much slower process that takes
several minutes to reach an equilibrium.
2.5 Conclusion
The development of a purely organic photobase using a naphthoquinone methide
photocore was achieved. The NQMP-photobase could be prepared quickly and in high
yields and quantities. The NQMP-photobase is remarkably stable, remaining pure by
NMR after three years of storage on a shelf away from light. The NQMP-photobase
reacts very quickly under 310 nm UV light to release a propyl carbamic acid which
degrades into CO2 and propylamine over a short period of time. The future goals of this
work will be to prepare NQMP-photobase with a synthetic handle that will allow its
integration into materials and surfaces. Additionally, there may be other “payloads” that
can be delivered using this system of photocleavage from an NQMP derivative.
2.6 Experimental
Synthetic Methodology. All syntheses were carried out under an inert atmosphere of
nitrogen using standard Schlenk techniques unless otherwise noted. Unless otherwise
noted, all data was worked up using Origin 9.0.0 SR2 software from OriginLab.
NMR spectroscopy. NMR spectra were recorded using a Varian Mercury 400 NMR
working at 400 MHz for proton spectra and 100 MHz for carbon spectra. Chemical shifts

18
are reported relative to an internal tetramethylsilane standard. The data was worked up
using MNova 10.0.2 software from Mestrelab Research S. L.
UV-Vis measurements. UV-vis spectra were obtained on a Cary 50 Bio UV-vis
spectrophotometer from Varian.
PH measurements. PH measurements were taken on a FiveEasy FE20 pH probe from
Mettler Toledo that was calibrated by two-point calibration using 4.00 and 10.00 buffer
solutions from VWR.
Synthesis of methyl 3-(methoxymethoxy)-2-naphthoate. In a dry round-bottom flask
under positive N2 pressure containing 15 mL dry DMF at 0 °C was added sodium hydride
(NaH) (1.48 g, 37 mmol). To this was added a solution of methyl 3-hydroxy-2-napthoate
(5g, 24.73 mmol) in 10 mL dry DMF dropwise. The solution was allowed to stir under N2
at 0 °C for one hour. While stirring, chloromethyl methyl ether (2.81 mL, 37 mmol) was
added dropwise. The solution was allowed to come to room temperature and stir for five
hours. The solution was then cooled to 0 °C and quenched with water (30 mL) and the
product was extracted with diethyl ether. The organic layer was then washed with water,
brine, and dried over MgSO4. The crude mixture was purified by silica column using
20% ethyl acetate in hexanes mixture. 1H NMR analysis indicated a pure product (5.48 g,
22.25 mmol, 90%). 1H NMR (400 MHz, CDCl3)
Synthesis of (3-(methoxymethoxy)naphthalen-2-yl)methanol. In a dry round-bottom
flask under positive N2 pressure containing 50 mL dry THF at 0 °C was added lithium
aluminum hydride (LAH) (0.927 g, 24.4 mmol). To this was added a solution of methyl
3-(methoxymethoxy)-2-naphthoate (5.47 g, 22.21 mmol) in 20 mL dry THF. The solution
was allowed to come to room temperature and react for three hours while stirring. The

19
solution was then returned to 0 °C and quenched with ethyl acetate (20 mL). The crude
product was filtered through celite with dichloromethane, and the solvent was reduced
under vacuum. The crude product was purified through a silica column with a
dichloromethane/methanol solution (9:1 to 8:2). The solvent was removed under vacuum
to give (3-(methoxymethoxy)naphthalen-2-yl)methanol as a yellow oil (4.583 g, 21.01
mmol, 94.6%). 1H NMR (400 MHz, CDCl3) δ 7.76-7.72 (m, 3H), 7.45-7.42 (t, 1H), 7.40
(s, 1H), 7.38-7.34 (t, 1H), 5.33 (s, 2H), 4.85 (s, 2H), 3.51 (s, 3H).
Synthesis of (3-(methoxymethoxy)naphthalen-2-yl)methyl propylcarbamate. A 15
mL solution of (3-(methoxymethoxy)naphthalen-2-yl)methanol (4.58 g, 21 mmol) in
dichloromethane at 0 °C was added 4-dimethylaminopyridine (0.051 g, 0.42 mmol) and
triethylamine (9 mL, 63 mmol). The solution was allowed to stir for 30 minutes at 0 °C.
To this solution was then added propyl isocyanate (2 mL, 25.2 mmol) dropwise. The
solution was allowed to come to room temperature and stir for four hours. The solution
was then washed with 1M hydrochloric acid, sodium bicarbonate, water, and brine,
passed through celite, and dried under MgSO4 and the solvent was reduced under
vacuum. The crude product mixture was purified through silica with a 75% ethyl
acetate/hexanes solution. The solvent was reduced under vacuum to give (3-
(methoxymethoxy)naphthalen-2-yl)methyl propylcarbamate as a yellow oil (4.985 g,
16.44 mmol, 78 %). 1H NMR (400 MHz, cdcl3) δ 7.79 (s, 1H), 7.75-7.70 (m, 2H), 7.43-
7.39 (m, 2H), 7.35-7.32 (t, 2H), 5.31 (s, 4H), 4.97 (s, 1H), 3.50 (s, 3H), 3.20-3.15 (m,
2H), 1.57-1.48 (m, 2H), 0.94-0.90 (t, 3H). 13C NMR (101 MHz, CDCl3) δ 156.57,
153.00, 134.17, 129.00, 128.46, 127.68, 126.94, 126.80, 126.47, 124.26, 108.85, 94.35,
62.35, 56.17, 42.88, 23.27, 11.27.

20
Synthesis of (3-hydroxynahthalen-2-yl)methyl propylcarbamate. To a round-bottom
flask containing (3-(methoxymethoxy)naphthalen-2-yl)methyl propylcarbamate (4.01 g,
13.22 mmol) in 90% methanol/water (50 mL) was added amberlyst-15 exchange resin
(12 g). The solution was stirred for 48 hours under reflux. The solution was passed
through a paper filter to remove the remaining resin, then extracted with diethyl ether,
washed with water, dried under NaSO4, and the solvent was reduced under vacuum. The
crude product was purified through silica with an ethyl acetate/hexanes solvent system
(60% to 100% EtOAc) and the solvent was removed under vacuum. The remaining
orange semisolid was recrystallized in hot dichloromethane to give (3-
hydroxynaphthalen-2-yl)methyl propylcarbamate as off-white crystals (2.74 g, 10.58
mmol, 80%). 1H NMR (400 MHz, dmso) δ 10.06 (s, 1H), 7.76-7.73 (m, 2H), 7.69-7.67
(d, 1H), 7.40-7.36 (t, 1H), 7.33-7.30 (t, 1H), 7.28-7.25 (t, 1H), 7.18 (s, 1H), 5.16 (s, 2H),
3.02-2.97 (m, 2H), 1.49-1.40 (m, 2H), 0.87-0.84 (t, 3H). 13C NMR (101 MHz, dmso) δ
156.69, 153.86, 134.46, 128.13, 127.86, 127.00, 126.51, 126.08, 123.36, 108.98, 61.59,
42.56, 23.12, 11.66. ESI HRMS: calcd. (M+H+): C15H18NO3 260.1281, found 260.1291.
UV photorelease general sample preparation. To a 0.125 mM solution of hydrochloric
acid in 75% methanol/water (6 mL) in a quartz cell was added (3-hydroxynaphthalen-2-
yl)methyl propylcarbamate (1.9 mg, 0.0075 mmol) to give a 1.25 mM solution of
photobase.
UV photorelease of propylamine. The solution was stirred and irradiated with a hand
lamp equipped with a 300 nm bulb (~1.00 mW/cm2) at a distance of one inch above the
surface of the solution. The pH of the solution was measured prior to the addition of the
photobase, after the addition of the photobase, and every sixty seconds during irradiation.

21
Pulsed UV photorelease of propylamine. The solution was stirred and irradiated in a
UV reactor equipped with eight 300 nm bulbs (~1.00 mW/cm2). The solution was
irradiated for one minute, then removed from the reactor and the pH was monitored for
five minutes before repeating this process for a total of five minutes irradiation. The pH
data was collected prior to irradiation and every fifteen seconds following each
irradiation cycle.

22
2.7 References
1. White, E. M.; Seppala, J. E.; Rushworth, P. M.; Ritchie, B. W.; Sharma, S.;
Locklin, J., Switching the Adhesive State of Catecholic Hydrogels using
Phototitration. Macromolecules 2013, 46 (22), 8882-8887.
2. Shirai, M.; Tsunooka, M., Photoacid and photobase generators: Chemistry and
applications to polymeric materials. Progress in Polymer Science 1996, 21 (1), 1-
45.
3. Li, R.; Nakashima, T.; Kanazawa, R.; Galangau, O.; Kawai, T., Efficient Self-
Contained Photoacid Generator System Based on Photochromic Terarylenes.
Chemistry – A European Journal 2016, 22 (45), 16250-16257.
4. Nakashima, T.; Tsuchie, K.; Kanazawa, R.; Li, R.; Iijima, S.; Galangau, O.;
Nakagawa, H.; Mutoh, K.; Kobayashi, Y.; Abe, J.; Kawai, T., Self-Contained
Photoacid Generator Triggered by Photocyclization of Triangle Terarylene
Backbone. Journal of the American Chemical Society 2015, 137 (22), 7023-7026.
5. Suyama, K.; Shirai, M., Photobase generators: Recent progress and application
trend in polymer systems. Progress in Polymer Science 2009, 34 (2), 194-209.
6. Arumugam, S.; Popik, V. V., Photochemical Generation and the Reactivity of o-
Naphthoquinone Methides in Aqueous Solutions. Journal of the American
Chemical Society 2009, 131 (33), 11892-11899.
7. Caplow, M., Kinetics of carbamate formation and breakdown. Journal of the
American Chemical Society 1968, 90 (24), 6795-6803.

23
8. Kulikov, A.; Arumugam, S.; Popik, V. V., Photolabile Protection of Alcohols,
Phenols, and Carboxylic Acids with 3-Hydroxy-2-Naphthalenemethanol. The
Journal of Organic Chemistry 2008, 73 (19), 7611-7615.

24
CHAPTER 3
USING SURFACE-TETHERED NQMP TO CONJUGATE ALDEHYDES AND
KETONES
3.1 Introduction
One of the most common goals in materials science and surface chemistry is the
fast and versatile conjugation of materials to a surface. NQMP has been shown to be a
powerful tool for conjugating thiols and vinyl ethers to a surface in the past.1-3 However,
the fast Diels-Alder reaction with enamines has not been studied in-depth in the past
(Figure 3.01).4 This is partially due to the relative instability of enamines. They are very
reactive molecules, and their method of preparation is reversible, so they often can
rapidly hydrolize to an inactive state.
Figure 3.01. NQMP Reaction with Enamines.
The standard method of preparing enamines involves reacting an aldehyde or
ketone with an amine (typically a secondary cyclic amine) in the presence of an acid
catalyst (Figure 3.02).5 While this method can give good product yield, the molecule is
not necessarily stable, and preparing and storing large amounts of it may be troublesome.
However, if we were to perform the preparation of an enamine from an aldehyde or

25
ketone in situ while irradiating an NQMP-functionalized surface, we may be able to
“trap” the molecule in its reactive enamine state with our highly reactive NQM. In this
manner, we hope to be able to use the high relative concentration of NQM on a surface to
rapidly and easily conjugate any aldehyde or ketone to a surface using an in situ enamine
generation.
Figure 3.02. Preparation of an Enamine from an Aldehyde.
Figure 3.03. In Situ Formation and Conjugation of an Enamine onto NQMP.
3.2 Synthesis of Photoactive Surfaces
The target azide-terminated triethylene glycol linker was prepared from
commercially available 2-(2-(2-chloroethoxy)ethoxy)ethanol in two steps with a total
yield of 56%. The 2-(2-(2-chloroethoxy)ethoxy)ethanol was reacted with sodium azide in
dimethylformamide to give the azide-terminated triethylene glycol product 3.01. Next,
compound 3.01 was reacted with p-toluenesulfonyl chloride and pyridine in
dichloromethane to give the tosylated triethylene glycol azide product 3.02.

26
Scheme 3.01
Reagents and Conditions: a) NaN3, DMF, 68%; b) p-toluenesulfonyl chloride, pyridine,
CH2Cl2, 82%.
The target azide-terminated protected NQMP derivative was prepared from
commercially available 5-dihydroxy-2-naphthoic acid in four steps with a total yield of
46%. The naphthoic acid was reacted in a Fischer esterification with sulfuric acid
(H2SO4) and methanol to give the methyl ester 3.03. Next, compound 3.03 was reduced
by diisobutylaluminum hydride (DIBAL) in tetrahydrofuran to yield the diol 3.04. Next,
compound 3.04 was reacted with 2,2-dimethyoxypropane and p-toluenesulfonic acid in
acetone to yield the protected diol 3.05. Finally, compound 3.05 was reacted with 3.02
and potassium carbonate in dimethyl formamide to give the azide-termined protected
NQMP derivative 3.06.

27
Scheme 3.02
Reagents and Conditions: a) H2SO4, methanol, 97%; b) DIBAL, THF, 91%; c) 2,2-
dimethoxypropane, p-toluenesulfonic acid, acetone, 95%; d) 3.02, K2CO3, DMF, 55%.
The protected NQMP-derivatized slides were prepared in two steps from
previously prepared grafted-from films of poly(pentafluorophenyl acrylate) (pPFPA) on
quartz, glass, and silicon slides in two steps. The poly(propargyl acrylamide) slides were
prepared by reacting the pPFPA slides with propargyl amine and triethylamine in
dimethylformamide to give the slides functionalized with 3.07. The functionalized slides
3.07 were then reacted with copper (i) bromide, N,N,N’,N”,N”-
pentamethyldiethylenetriamine (PMDETA), and 3.06 in dimethylformamide to give
slides functionalized with 3.08.

28
Scheme 3.03
Reagents and Conditions: a) propargyl amine, triethylamine, DMF; b)CuBr, PMDETA,
3.06, DMF.
The deprotected surface-bound NQMP slides 3.09 were prepared in one step. The
surface-bound protected NQMP slides 3.08 were reacted with hydrochloric acid in
methanol to give the deprotected surface-bound NQMP slides 3.09.
Scheme 3.04
Reagents and Conditions: a) HCl, MeOH.
3.3 Photoconjugation with Aldehydes and Ketones
The conjugation of isobutyraldehyde to the surface-bound NQMP slides was
perfomed in one step. The surface-bound NQMP slides 3.09 were reacted with

29
isobutyraldehyde, morpholine, and p-toluenesulfonic acid in a 50:50 solution of
acetonitrile and water to give conjugate 3.10.
Scheme 3.05
Reagents and Conditions: a) isobutyraldehyde, morpholine, p-toluenesulfonic acid,
310nm hν, acetonitrile/water 1:1.
3.4 Characterization of Surfaces
The functionalized surfaces were characterized by spectroscopic ellipsometry,
drop-shape analysis (DSA), and FTIR. When looking at the change in film thickness and
contact angle (Table 3.01) the initial pPFPA brushes were 44 nm in thickness, and
relatively hydrophobic with a contact angle of 91° as measured by DSA. After
conversion to 3.07, the thickness decreased by 17.5 nm to 26.5 nm. This is expected due
to the much smaller size of the propargyl group compared to the pentafluorophenyl
group. The contact angle also decreased by 22° to 73°, indicating a more hydrophilic
surface. After the reaction with 3.06, the thickness increased by 75.5 nm to 102 nm. This
is due to the much larger size of the tethered protected NQMP compared to the relatively
small propargyl group. The contact angle slightly increased by 10° to 83°, in accordance
with previously published data.1 After deprotection to yield 3.09, the thickness decreased
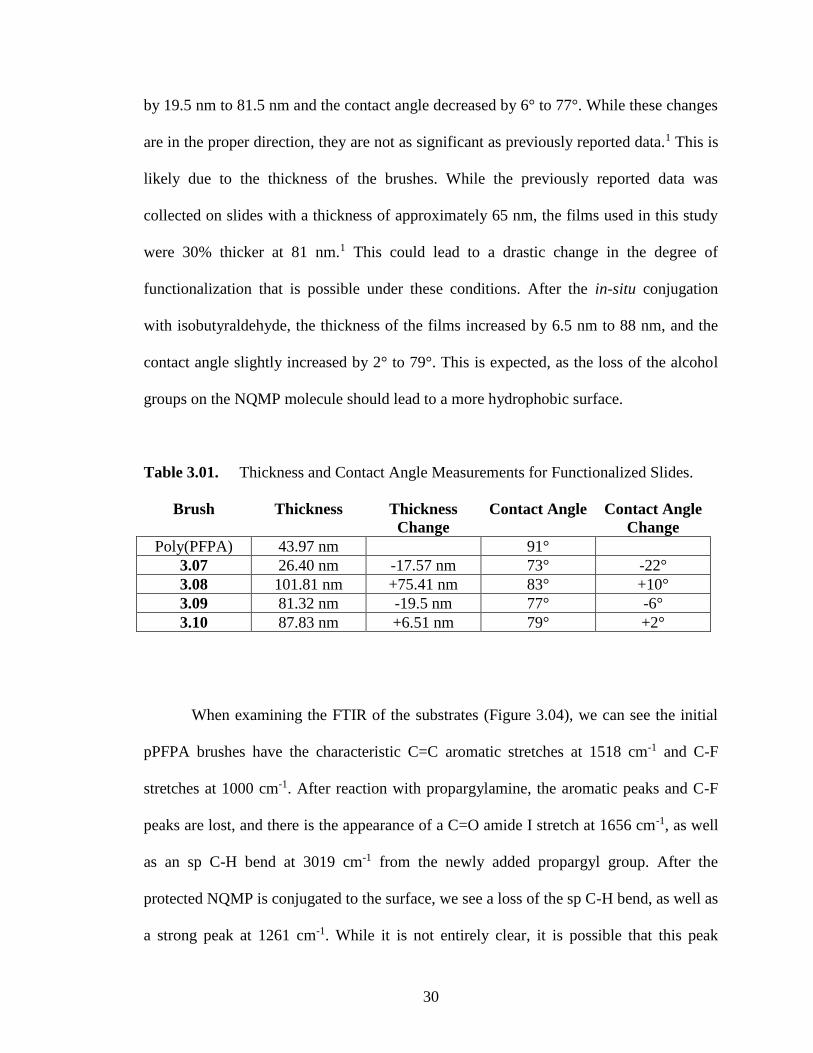
30
by 19.5 nm to 81.5 nm and the contact angle decreased by 6° to 77°. While these changes
are in the proper direction, they are not as significant as previously reported data.1 This is
likely due to the thickness of the brushes. While the previously reported data was
collected on slides with a thickness of approximately 65 nm, the films used in this study
were 30% thicker at 81 nm.1 This could lead to a drastic change in the degree of
functionalization that is possible under these conditions. After the in-situ conjugation
with isobutyraldehyde, the thickness of the films increased by 6.5 nm to 88 nm, and the
contact angle slightly increased by 2° to 79°. This is expected, as the loss of the alcohol
groups on the NQMP molecule should lead to a more hydrophobic surface.
Table 3.01. Thickness and Contact Angle Measurements for Functionalized Slides.
Brush Thickness Thickness
Change
Contact Angle Contact Angle
Change
Poly(PFPA) 43.97 nm 91°
3.07 26.40 nm -17.57 nm 73° -22°
3.08 101.81 nm +75.41 nm 83° +10°
3.09 81.32 nm -19.5 nm 77° -6°
3.10 87.83 nm +6.51 nm 79° +2°
When examining the FTIR of the substrates (Figure 3.04), we can see the initial
pPFPA brushes have the characteristic C=C aromatic stretches at 1518 cm-1 and C-F
stretches at 1000 cm-1. After reaction with propargylamine, the aromatic peaks and C-F
peaks are lost, and there is the appearance of a C=O amide I stretch at 1656 cm-1, as well
as an sp C-H bend at 3019 cm-1 from the newly added propargyl group. After the
protected NQMP is conjugated to the surface, we see a loss of the sp C-H bend, as well as
a strong peak at 1261 cm-1. While it is not entirely clear, it is possible that this peak

31
corresponds to the C-O stretch from the acetonide protecting group on the NQMP. The
deprotection of the NQMP is also difficult to trace by FTIR. While there is a loss of the
C-O stretch at 1261 cm-1, it cannot be definitively stated whether this corresponds to a
successful deprotection. After the in-situ conjugation with isobutyraldehye, there is
virtually no change in the FTIR of the substrates. The primary difficulty with using FTIR
for this characterization is the thickness of the films. Since the GATR attachment for the
FTIR requires intimate contact with the surface films, very thick films (more than 75 nm)
can be very difficult to analyze using FTIR.
Figure 3.04. FTIR Spectra of Silicon Substrates with a) Poly(PFPA), b) Poly(Propargyl
Acrylamide), c) Surface-Tethered Protected NQMP, d) Surface-Tethered NQMP, e)
Surface-Tethered NQMP after Isobutyraldehyde Conjugation.

32
The glass and quartz substrates that were prepared according to the previously
mentioned procedure were analyzed by UV-Vis spectroscopy to determine the extent of
functionalization with NQMP. Since NQMP has a strong absorbance at 323 nm, it was
expected that this could be used to track the attachment of NQMP to the surface as well
as the extent of conjugation with isobutyraldehyde. Unfortunately, there was little change
in the UV spectrum for either reaction. Again, this is likely due to the thickness of the
films. Since the NQMP moiety is only a fraction of the bulk material on the surface of the
substrates, it is difficult to tease out any small changes in the spectra.
3.5 Conclusion
The development of a new method of conjugating aldehydes and ketones to
surfaces using in-situ enamine formation and trapping via Diels-Alder reaction with
NQM experienced several setbacks. While the protected NQMP could successfully be
attached to the surface, we were unable to determine the success of the deprotection step,
as well as the subsequent conjugation with isobutyraldehyde.
There are several methods that can be used to overcome these difficulties. Thinner
pPFPA films prepared via grafting-to method may make it easier to see the changes in
our substrates by FTIR and UV-Vis. Additionally, other aldehydes or ketones can be used
to determine the success of the conjugation to the deprotected NQMP. An aldehyde with
a long alkyl chain should impart a significant hydrophobicity and increase in contact
angle when it conjugates to the surface. Conversely, a hydrophilic aldehyde or ketone,
such as one based on polyethylene glycol, should show a strong decrease in contact angle
upon conjugation. While the idea of attaching an aldehyde or ketone dye to the surface

33
using this chemistry is an enticing proposition, unfortunately most dyes have a strong
enough absorbance that they may hinder the photoconversion of NQMP to NQM.
If the conjugation of simple aldehydes and ketones onto the NQMP surface can be
performed simply under UV light with mildly acidic conditions, it is not unreasonable to
assume that we can use this method to conjugate several aldehydes or ketones to a
surface using a photomask. With this method, we may be able to have a single surface
that is capable of reacting with a very common functional group, and we can control
when and where these common groups are conjugated to the surface.
3.6 Experimental
Synthetic Methodology. All syntheses were carried out under an inert atmosphere of
nitrogen using standard Schlenk techniques unless otherwise noted. Unless otherwise
noted, all data was worked up using Origin 9.0.0 SR2 software from OriginLab.
NMR spectroscopy. 1H NMR spectra were recorded using a Varian Mercury 300 NMR
working at 300 MHz. 13C NMR spectra were recorded using a [MODEL] 500 NMR
operating at 120 MHz. Chemical shifts are reported relative to an internal
tetramethylsilane standard. The data was worked up using MNova 10.0.2 software from
Mestrelab Research S. L.
Thickness measurements. The film thickness was measured using a J. A. Woolam M-
2000V spectroscopic ellipsometer with a white light source at three angles of incidence
(65°, 70°, and 75°) to the slide normal. The film thickness was fit using a Cauchy model.
Contact angle measurements. The contact angle measurements were obtained using a
Krüss DSA 100 using a 1 μL drop of deionized water. For each measurement, three drops
were recorded and the readings were averaged.

34
UV-Vis measurements. UV-vis spectra were obtained on a Cary 50 Bio UV-vis
spectrophotometer from Varian.
FTIR spectroscopy. Infrared spectra of the surfaces were obtained from a Thermo-
Nicolet Model 6700 spectrometer equipped with a variable angle grazing angle
attenuated total reflection (GATR-ATR) accessory and processed using the Omnic 8.0
software suite.
Synthesis of 2-(2-(2-azidoethoxy)ethoxy)ethanol. In a round-bottom flask containing 2-
(2-(2-chloroethoxy)ethoxy)ethanol (2.27 mL, 15 mmol) in 50 mL dry DMF was sodium
azide (1.463 g, 22.5 mmol) slowly added. The reaction was heated slowly to 100 °C and
stirred for twelve hours. The reaction was cooled to 25 °C, diluted with 100 mL water
and extracted with ethyl acetate. The organic layer was washed with brine, dried with
sodium sulfate, and concentrated under vacuum to give 2-(2-(2-
azidoethoxy)ethoxy)ethanol (1.79 g, 10.2 mmol, 68%) as a colorless oil. 1H NMR (300
MHz, CDCl3) δ 3.73-3.70 (t, 2H), 3.68-3.65 (m, 6H), 3.61-3.58 (t, 2H), 3.39-3.36 (t, 2H),
2.45 (s, 1H).
Synthesis of 2-(2-(2-azidoethoxy)ethoxy)ethyl 4-methylbenzenesulfonate. To a round
bottom flask containing 2-(2-(2-azidoethoxy)ethoxy)ethanol (0.5 g, 2.85 mmol) in 12 mL
dichloromethane was added p-toluenesulfonyl chloride (0.653 g, 3.42 mmol) and
pyridine (0.460 mL, 5.71 mmol). The reaction was allowed to stir at room temperature
for 8 hours. The solution was diluted with 20 mL dichloromethane, washed with water,
washed with 10% copper sulfate solution, dried over magnesium sulfate, and
concentrated under vacuum. The residue was purified through a silica column with 50%
ethyl acetate in hexanes to give 2-(2-(2-azidoethoxy)ethoxy)ethyl 4-

35
methylbenzenesulfonate as a colorless oil (0.770 g, 2.34 mmol, 82%). 1H NMR (300
MHz, CDCl3) δ 7.80-7.77 (d, 2H), 7.35-7.32 (d, 2H), 4.17-4.14 (t, 2H), 3.71-3.67 (t, 2H)
3.65-3.61 (t, 2H), 3.59 (s, 4H), 3.37-3.34 (t, 2H), 2.44 (s, 3H).
Synthesis of methyl 3,5-dihydroxy-2-naphthoate. In a dry round-bottom flask
containing 50 mL methanol was added 5-dihydroxy-2-naphthoic acid (3g, 14.69 mmol).
To this was added concentrated sulfuric acid (0.1 mL, 1.876 mmol). The solution was
allowed to stir under reflux at 67 °C for sixteen hours. The reaction was quenched in 100
mL ice water and extracted with ethyl acetate. The organic layers were dried with
magnesium sulfate and the solvent was removed under vacuum. The remaining dark
yellow solid was dried under vacuum for twelve hours to yield methyl 3,4-dihydroxy-2-
naphthoate (3.12 g, 14.30 mmol, 97%) 1HNMR (300 MHz, CDCl3) δ 10.43 (s, 1H), 8.47
(s, 1H), 7.63 (s, 1H), 7.43-7.40 (d, 1H), 7.19-7.13 (t, 1H), 6.86-6.84 (d, 1H), 5.22 (s, 1H),
3.98, (s, 3H).
Synthesis of 6-(hydroxymethyl)naphthalene-1,7-diol. In a round-bottom flask
containing dry THF at 0 °C was dissolved methyl 3,4-dihydroxy-2-naphthoate (1.09 g, 5
mmol). To this was added dropwise a 1M solution of diisobutyl aluminum hydride (30
mL, 30 mmol) over one hour. The reaction was heated slowly to 45 °C and stirred for
sixteen hours. The reaction was quenched with 50 mL ethyl acetate dropwise at 0 °C. To
remove the remaining aluminum salts, a 300 mg/mL solution of potassium tartrate (150
mL) was added to the flask and the reaction was stirred at 25 °C for fifteen minutes. The
product was extracted with ethyl acetate, washed with water, dried with magnesium
sulfate, and concentrated under vacuum. The residue was recrystallized in chloroform to
yield 6-(hydroxymethyl)naphthalene-1,7-diol as a white solid (0.862 g, 4.5 mmol, 90.6%)

36
1HNMR (300 MHz, DMSO-d6) δ 9.71 (s, 1H), 9.64 (s, 1H), 7.70 (s, 1H), 7.36 (s, 1H),
7.21-7.18 (d, 1H), 7.05-7.00 (t, 1H), 6.72-6.70 (d, 1H), 5.12-5.08 (t, 1H), 4.61 (d, 2H).
Synthesis of 2,2-dimethyl-4H-naphtho[2,3-d][1,3]dioxin-9-ol. In a round-bottom flask
containing 6-(hydroxymethyl)naphthalene-1,7-diol (0.25 g, 1.31 mmol) in 50 mL dry
acetone was added 2,2-dimethoxypropane (0.0.484 mL, 3.94 mmol). To this solution was
added p-toluenesulfonic acid monohydrate (0.011 g, 0.066 mmol) and the reaction was
heated to 50 °C and stirred for five hours. The reaction was quenched with water,
extracted with dichloromethane, washed with water, dried with sodium sulfate, and
concentrated under vacuum to yield 2,2-dimethyl-4H-naphtho[2,3-d][1,3]dioxin-9-ol as a
dark oil (0.302 g, 1.24 mmol, 95%) 1HNMR (300 MHz, CDCl3) δ 7.58 (s, 1H), 7.41 (s,
1H), 7.28-7.25 (d, 1H), 7.14-7.09 (t, 1H), 6.76-6.74 (d, 1H), 5.05 (s, 2H), 1.59, (s, 6H).
Synthesis of 9-(2-(3-(2—azidoethoxy)propoxy)ethoxy)-2,2-dimethyl-4H-naphtho[2,3-
d][1,3]-dioxine. To a round bottom flask containing 2,2-dimethyl-4H-naphtho[2,3-
d][1,3]dioxin-9-ol (0.175 g, 0.76 mmol) and potassium carbonate (0.252 g, 1.824 mmol)
in 3 mL DMF was added a solution of 2-(2-(2-azidoethoxy)ethoxy)ethyl 4-
methylbenzenesulfonate (0.30 g, 0.91 mmol) in 2 mL DMF. The reaction was heated to
60 °C and stirred overnight. The solution was then cooled to room temperature, diluted
with 20 mL diethyl ether, washed with water, dried with magnesium sulfate, and
concentrated under vacuum. The residue was purified through an alumina column with
50% ethyl acetate in hexanes to give 9-(2-(3-(2—azidoethoxy)propoxy)ethoxy)-2,2-
dimethyl-4H-naphtho[2,3-d][1,3]-dioxine as a yellow oil (0.162 g, 0.418 mmol, 55%). 1H
NMR (300 MHz, CDCl3) δ 7.66 (s, 1H), 7.42 (s, 1H), 7.31-7.29 (d, 1H), 7.22-7.17 (t,

37
1H), 6.74-6.72 (d, 1H), 5.06 (s, 2H), 4.29-4.26 (t, 2H), 4.00-3.96 (t, 2H), 3.82-3.79 (t,
2H), 3.72-3.69 (m, 4H), 3.41-3.37 (t, 2H), 1.60 (s, 6H).
Preparation of alkyne functionalized slides. Quartz, glass, and silicon substrates with a
44 nm film of grafted-from poly(pentafluorophenyl acrylate) were reacted overnight into
a solution of 40 mM propargyl amine and 80 mM triethylamine in DMF (2mL). After
reaction the thickness of the film decreased to 29 nm, and infrared spectroscopy indicated
the disappearance of the PFPA peaks and the appearance of two amide peaks. The
contact angle of the slides decreased from 91° to 73°, indicating a slightly more
hydrophilic surface.
Preparation of protected-NQMP-functionalized slides. To a dry, air-free Schlenk flask
containing alkyne-functionalized slides and 16.7 mg CuBr was added a degassed solution
of 13 mM 9-(2-(3-(2—azidoethoxy)propoxy)ethoxy)-2,2-dimethyl-4H-naphtho[2,3-
d][1,3]-dioxine and 24.4 μL PMDETA in DMF. The solution was allowed to stir
overnight at room temperature. The thickness of the films increased to 102 nm, and
infrared spectroscopy indicated the loss of the alkyne peak. The contact angle of the
slides increased to 83°, indicating a slightly more hydrophobic surface.
Deprotection of NQMP-functionalized slides. To a Schlenk flask containing 0.1 M
hydrochloric acid in methanol was placed the protected NQMP-functionalized slides. The
flask was allowed to stir for twelve hours. The thickness of the films decreased to 82 nm.
There was no significant change on the infrared spectrum. The contact angle of the slides
decreased to 77°, indicating a slightly more hydrophilic surface.
Photoconjugation of isobutyraldehyde onto NQMP-functionalized slides. To a
solution of 40 mM isobutyraldehyde, 40 mM morpholine, and 40 mM p-toluenesulfonic

38
acid in 50% acetonitrile in water was placed a glass slide functionalized with NQMP. The
solution was stirred and irradiated under 300 nm light for 20 minutes. The thickness of
the films increased slightly to 88 nm. There was no significant change on the infrared
spectrum. UV-Vis indicated no significant difference in the slide. Contact angle
measurements also indicated no significant difference in the hydrophilicity of the
surfaces.

39
3.7 References
1. Arumugam, S.; Orski, S. V.; Locklin, J.; Popik, V. V., Photoreactive Polymer
Brushes for High-Density Patterned Surface Derivatization Using a Diels–Alder
Photoclick Reaction. Journal of the American Chemical Society 2012, 134 (1),
179-182.
2. Arumugam, S.; Popik, V. V., Attach, Remove, or Replace: Reversible Surface
Functionalization Using Thiol–Quinone Methide Photoclick Chemistry. Journal
of the American Chemical Society 2012, 134 (20), 8408-8411.
3. Arumugam, S.; Popik, V. V., Patterned Surface Derivatization Using Diels–Alder
Photoclick Reaction. Journal of the American Chemical Society 2011, 133 (39),
15730-15736.
4. Arumugam, S.; Popik, V. V., Light-Induced Hetero-Diels−Alder Cycloaddition:
A Facile and Selective Photoclick Reaction. Journal of the American Chemical
Society 2011, 133 (14), 5573-5579.
5. Noack, M.; Göttlich, R., Iodide-Catalysed Cyclization of Unsaturated N-
Chloroamines: A New Way to Synthesise 3-Chloropiperidines. European Journal
of Organic Chemistry 2002, 2002 (18), 3171-3178.

40
APPENDIX A
1H AND 13C NMR SPECTRA OF ESSENTIAL COMPOUNDS

41
Figure A-1. 1HNMR of (3-(methoxymethoxy)naphthalen-2-yl)methyl propylcarbamate.

42
Figure A-2. 13CNMR of (3-(methoxymethoxy)naphthalen-2-yl)methyl propylcarbamate.

43
Figure A-3. 1HNMR of (3-hydroxynaphthalen-2-yl)methyl propylcarbamate.

44
Figure A-4. 13CNMR of (3-hydroxynaphthalen-2-yl)methyl propylcarbamate.