physically-motivated force fields from symmetry-adapted perturbation theory
TRANSCRIPT

Physically-Motivated Force Fields from Symmetry-AdaptedPerturbation TheoryJesse G. McDaniel and J.R. Schmidt*
Theoretical Chemistry Institute and Department of Chemistry, University of WisconsinMadison, Madison, Wisconsin 53706,United States
*S Supporting Information
ABSTRACT: We present a general methodology for generating accurate andtransferable ab initio force fields, employing the framework of symmetry adaptedperturbation theory (SAPT). The resulting force fields are “physically-motivated” in thatthey contain separate, explicit terms to account for the various fundamentalintermolecular interactions, such as exchange, electrostatics, induction, and dispersion,with each term parametrized to a corresponding term in the SAPT energydecomposition. Crucially, the resulting force fields are largely compatible with existing,standard simulation packages, requiring only minimal modifications. We present severalnovel parametrization techniques that yield robust, physically meaningful atomicparameters that are transferable between molecular environments. We demonstrate theaccuracy and generality of our method by validating against experimental second virial coefficients for a variety of smallmolecules. We then show that the resulting atomic parameters can be combined using physically motivated ansatzes to accuratelypredict arbitrary heteromolecular interaction energies, with example applications including prediction of gas adsorption infunctionalized metal−organic framework materials.
■ INTRODUCTION
Classical molecular dynamics simulations are now routinelyapplied to systems ranging in complexity from neat liquids, tocomplex bulk solids, to biomolecular systems. In conjunctionwith classical empirical force fields, these simulations provide amolecular-level perspective on structure, thermodynamics, andkinetics. However, the accuracy and reliability of thesesimulations is fundamentally dictated by the underlying forcefields. Historically, such intermolecular force fields have beendominated by simple functional forms dictated more bycomputational convenience than by physical significance (e.g.,Lennard-Jones) and empirically parametrized on the basis ofprior experimental data. In contrast, it is generally well acceptedthat accurate, transferable “next generation” force fields must gobeyond standard Lennard-Jones or Buckingham form andshould incorporate functional forms that are intrinsicallyconnected to the fundamental physical interaction(s) theyseek to describe.1−7 Ideally, these next-generation force fieldsshould be “derivable” from rigorous first-principles electronicstructure (i.e., ab initio force fields), free from empiricalparametrization and the associated reliance on prior exper-imental data. Yet despite the long history of high-level quantumchemical calculations in the construction of accurate potentialenergy surfaces (PES), corresponding approaches for generic,transferable force f ields have been much more limited; thetransferability inherent in the latter introduces additionalconstraints related to the “physicality” of the parametersgoverning the various fundamental intermolecular interactionsand the asymptotic correctness of the associated functionalforms. As such, we recently developed a powerful framework
based on symmetry-adapted perturbation theory (SAPT)8 thatallows for the parametrization of accurate and “physically-motivated” ab initio force fields. The aim of the present work isto briefly review these and other related developments and thendemonstrate how our prior methodology can be extended togenerate transferable next generation force field parameters thatare generally applicable to a diverse set of molecular systemsand environments.SAPT (and more recently, density functional theory-based
SAPT [DFT-SAPT]9−20), provides a powerful framework forparametrizing such force fields. In contrast to conventionalempirical force fields, physically motivated force fields describethe intermolecular interactions in terms of individual energycomponents, each described with an appropriate associatedfunctional form. Specifically, we focus on those energycontributions that naturally arise within the context of SAPT:exchange, electrostatic, induction, and dispersion. Thesecomponents are fit individually to separate force field terms.By construction, this approach ensures a proper balancebetween each energy contribution, mitigates spurious errorcancelation, and yields a more robust force field with correctasymptotic behavior. However, because SAPT yields molecularinteraction energies, whereas transferable force fields necessar-ily require atomic interaction parameters, a major challenge isthe partitioning of the molecular interactions into (mostly)pairwise atom−atom interactions.
Received: November 1, 2012Revised: December 21, 2012Published: January 23, 2013
Feature Article
pubs.acs.org/JPCA
© 2013 American Chemical Society 2053 dx.doi.org/10.1021/jp3108182 | J. Phys. Chem. A 2013, 117, 2053−2066

Progress can be made by exploiting monomer properties. It iswell-known that the long-range interactions between twomolecules can be described entirely in terms of monomerparameters, such as the multipole moments and static/frequency dependent polarizability tensors. At close range,however, intermolecular interactions are additionally charac-terized by exchange repulsion and electrostatic chargepenetration effects resulting from the overlap of the monomerelectron densities; these effects cannot be described in terms ofmerely monomer properties. As such, Stone and Misquittaoutline a procedure in which long-range, asymptotic interactionparameters are fit to monomer response calculations, whereasshort-range interaction parameters are fit to dimer SAPTcalculations.1 In the former case, they obtain the necessaryatomic parameters from monomer calculations utilizingdistributed multipole analysis (DMA) and distributed atomicpolarizabilities, methods pioneered by those same authors.These contributions provide an important starting point for ourforce field development methodology.In several recent works, we have shown how ab initio force
fields developed within the framework of SAPT can be utilizedin molecular dynamics (MD) and Monte Carlo (MC)simulations to accurately predict structural, thermodynamic,and dynamic properties in systems ranging in complexity frombulk neat fluids to gas adsorption in nanoporous materials.21−24
For example, we have previously developed the “SYM” forcefield that accurately describes neat CO2, N2, and their mixturesand has been extensively benchmarked to a broad range ofthermodynamic data including virial coefficients, densities,liquid structure, heat capacities, diffusion coefficients, and phasecoexistence (shown in Figure 1). We have also developed a
robust, transferable force field, “ZIF FF”, which accuratelydescribes CO2/N2 adsorption in a variety of zeolitic imidazolateframeworks (ZIFs, a specific type of metal−organic framework)of varying topologies and functionalizations; results from thisforce field are shown in Figure 2. (Note that the isothermsdiffer very slightly from those given in ref 24 due to an error inone of the original reported parameters; see SupportingInformation).As we have shown, and will demonstrate further in this
paper, our SAPT-based approach to force field generation, andthe associated physically motivated force fields, offer several
compelling advantages as compared with traditional empiricalforce fields. First, the force fields are fit to ab initio data and arethus free from any empirical parametrization, allowingapplication to systems for which there is little or no priorexperimental data available. Second, the explicit separation ofthe individual energy components contributes to the robustnessof the parametrization by mitigating spurious error cancellationand facilitates the use of physically appropriate functional formsin describing each of the fundamental intermolecularinteraction types. Furthermore, the associated methodology issystematic and facilitates a clear hierarchy of improvementswithout complete reparameterization. Additionally, due to theexplicit separation of individual energy components byconstruction, the final force field admits a natural energydecomposition that can be exploited during the classicalmolecular simulation to analyze interactions under thermody-namically relevant conditions of finite temperature andpressure. Finally, the resulting force fields not only are accuratebut also (using the modifications described herein) have thepotential for exceptional transferability to diverse chemical andphysical environments.The “physically-motivated” approach described above is not
without precedent, and it is profitable to contrast with severalprevious related approaches. A common theme throughoutthese prior works (as well as ours) is the use of monomercalculations to obtain electrostatic multipoles and static/frequency dependent polarizabilities. Here, we focus specificallyon the methods used to distribute these molecular properties toatomic parameters. These distribution schemes are central toparameter transferability (or lack thereof), and such trans-ferability is a major point of emphasis for the present work. Allof these prior approaches utilize some kind of ab initio energydecomposition (EDA) method for parametrization and/orbenchmarking. Although we utilize SAPT, other methods basedon variational supermolecular wave function calculations(rather than perturbation theory) have been used, and thereader is referred to the literature for descriptions of suchmethods.27
A close parallel between the present approach can be seen inthe SIBFA (sum of interactions between fragments ab initiocomputed) approach of Gresh, Piquemal, and co-work-ers.3,6,28,29 SIBFA is designed to represent the total interactionenergy in terms of five components: electrostatic, repulsion,polarization, charge transfer, and dispersion. The formulas used
Figure 1. Coexistence density for CO2 as predicted by “SYM” forcefield (symbols). This figure was reproduced from ref 21. Copyright2011 American Chemical Society.
Figure 2. CO2 isotherms at 298 K. Solid lines are experimentaldata;25,26 symbols are our simulations.
The Journal of Physical Chemistry A Feature Article
dx.doi.org/10.1021/jp3108182 | J. Phys. Chem. A 2013, 117, 2053−20662054

to describe the various energy components are relativelycomplex, incorporating anisotropic interactions, bonding andoff-atom interaction sites, and orbital-dependent repulsion andcharge transfer terms. A DMA is used to describe theasymptotic electrostatics, retaining multipoles up to quadrupoleorder, with sites distributed on atom and bond centers usingthe method of Vigne-Maeder and Claverie.30 The polarizationenergy contribution is computed using distributed anisotropicdipole polarizability tensors. These distributed polarizabilitiesare generated using the procedure of Garmer and Stevens,31 inwhich finite field response calculations are conducted, and thenecessary expectation values of the dipole moment operator arepartitioned into contributions from localized molecular orbitals(LMO). In the SIBFA approach, the repulsion energy isconstructed according to an ansatz that assumes the depend-ence on molecular density overlap can be expressed as a sum ofbond and lone pair contributions, thus requiring approximateformulas for orbital overlap integrals as well as occupationnumbers.3 A complete methodological description of SIBFAcan be found in refs 3 and 6.Comparisons with the present approach can also be drawn
with the effective fragment potential (EFP) method of Gordonand co-workers.4,32−35 EFP was originally developed for use inQM/MM applications, where a solvent environment isrepresented in the QM Hamiltonian by one-electron “effectivefragment” potentials. However, EFPs (and more specifically thenewer, more general EFP2 method) can also be used as “standalone” ab initio force fields for classical molecular simulations;in this respect, recent work has evaluated the accuracy of EFPsvia comparison with SAPT and other high-level ab initiocalculations.36 The energy decomposition inherent in EFPs isessentially identical to that used in SIBFA, namely a sum ofCoulomb, induction, exchange repulsion, dispersion, andcharge transfer components. The Coulomb energy is modeledby distributed multipole expansions (up to octopole) on atomand bond centers, using the DMA method of Stone.37,38 Theinduction energy is modeled using distributed polarizabilitytensors on bonds and lone pairs using a LMO distributionscheme (as in SIBFA, see above). The dispersion energy ismodeled using damped C6/R
6 and C8/R8 terms, which are
computed from frequency-dependent polarizability tensors atthe coupled perturbed Hartree−Fock level (CPHF) anddistributed using the same LMO scheme that is used todistribute the static tensors. Finally, the exchange repulsionenergy is formulated as an expansion of the intermolecularelectron density overlap in the LMO basis.Both SIBFA and EFP employ monomer calculations to
extract information about asymptotic interactions and rely onan anstatz to account for short-range repulsive interactions.Alternatively, Beran and co-workers have devised a hybridmixed QM/MM approach, whereby short-range interactionsare treated with QM, and the long-range interactions with apolarizable force field derived from ab initio monomerproperties.39,40 This approach has been used successfully todiscriminate between lattice energies of crystal polymorphs.However, because it relies on explicit QM calculations (and isnot mapped to a purely classical force field), this hybrid schemewould be prohibitively computationally demanding whenapplied to finite temperate molecular dynamics/Monte Carlosimulations.SAPT (and DFT-SAPT) provide an alternative rigorous
route to QM interaction energies and an associated energydecomposition that is valid at all relevant intermolecular
distances. Furthermore, DFT-SAPT facilitates the use ofcoupled perturbed Kohn−Sham (CPKS) monomer responsecalculations, as opposed to less accurate CPHF, withoutintroducing methodological inconsistences.12,15 Examples ofab initio force field development exploiting the SAPT energydecomposition include the work of Misquitta et al.5 Theyemploy a DMA to describe asymptotic electrostatics and utilizethe Williams−Stone−Misquitta (WSM) method41 to obtaindistributed static and frequency dependent polarizabilitytensors necessary for induction and dispersion.Like many of these prior approaches (and, in particular, the
prior work of Misquitta and Stone), our DFT-SAPT-basedapproach exploits the SAPT energy decomposition andmonomer properties to approximate the intermolecularinteractions in terms of fundamental interaction types such asexchange, electrostatics, polarization, and dispersion. However,there are at least two significant differences that motivate thepresent work. First, we employ a novel fitting procedure toextract truly “physical” and transferable atomic exchange,polarization, and dispersion parameters. As such, the resultingforce f ields are transferable to new molecular systems andenvironments to which they were not specif ically parametrized.Second, although we utilize physically motivated functionalforms for each interaction type, the sum of these terms (i.e., thetotal energy) is in a form that is strikingly similar to thatemployed by standard molecular dynamics (MD) or MonteCarlo (MC) simulation packages, facilitating their implementa-tion with few (or trivial) changes. As a result of these twomodifications, the resulting force fields are accurate, robust andtransferable, with minimal barriers to implementation.Finally, we note that many previous intermolecular potentials
have been derived on the basis of SAPT, often with greatsuccess.2,42−46 Yet with the exception of the work of Li et al.,43
these potentials do not contain an explicit energy decom-position and are fit to total SAPT interaction energies.Furthermore, in many cases (particularly for smaller systems)these potentials were designed to treat rigid-body interactionsvia an expansion of the total molecular interaction energy in thesix rigid-body relative coordinates.42,44 As such, in the parlanceintroduced above, these are more like PESes than transferableatomistic potentials suitable for generic simulation, and theassociated methodologies and objectives differ considerablyfrom the approach we outline here. For larger molecules, asubset of these prior SAPT potentials were fit to functionsconsisting of atomic pairwise contributions.2,45,46 However, asfew techniques were used to decouple the atomic parameters inthe fits, these potentials, while highly accurate, are nottransferable outside of the systems for which they wereparametrized.The remainder of this paper is organized as follows. We
briefly review our existing DFT-SAPT-based force fieldparametrization methodology. We then demonstrate how weare able to extend these efforts to obtain transferable atomicinteraction parameters, including those for dispersion, for awide variety of small molecules and functional groups. We thenbenchmark these force field parameters via second-virialcoefficients for various small organic molecules such as alkanes,alkenes, amines, alcohols, aldehydes, and ketones. Finally, wedemonstrate the transferability of our force field parameters byapplying them to a particularly discriminating exampleapplication: computing the carbon dioxide (CO2) adsorptionisotherms in metal−organic frameworks (MOFs), including IR-MOF1 (MOF5) and amine functionalized IR-MOF3.
The Journal of Physical Chemistry A Feature Article
dx.doi.org/10.1021/jp3108182 | J. Phys. Chem. A 2013, 117, 2053−20662055

■ METHODOLOGYWe briefly recount our established force field developmentmethodology21,23,24 and subsequently describe several enhance-ments that enable the extraction of truly physical, transferableatomic parameters. Our force fields contain separate terms thatrepresent the distinct components of intermolecular inter-actions,
= + + + + δE E E E E Eint elec exch pol disp hf
each with a corresponding partner(s) in the SAPT decom-position:
=E Eelec pol(1)
=E Eexch exch(1)
= + −E E Epol ind(2)
ind exch(2)
= + −E E Edisp disp(2
disp exch(2)
Here, we employ the commonly used groupings of Epol = Eind(2) +
Eind−exch(2) and Edisp = Edisp
(2) + Edisp−exch(2) , motivated (in part) by the
cancelation of artificial mathematical divergences at shortintermolecular distances between the paired terms within theSAPT energy.Each term in the energy decomposition is described by a
corresponding force field term as detailed below:
∑ ∑≅ + −E f B rq q
rA B r( , ) exp( )
i jij ij
i j
ij i jij ij ijelec
,1
,
elec
∑≅ −E A B rexp( )i j
ij ij ijexch,
exch
∑≅ + −E U A B rexp( )i j
ij ij ijpol shell(2)
,
ind
∑ ∑≅ −=
E f B rCr
( , )n i j
n ij ijnij
ijndisp
6,8,10,12 ,
∑≅ + −δδΔE U A B rexp( )
i jij ij ijhf shell
SCF
,
hf
The sum of these terms yields an expression for the overall totalenergy,
∑ ∑
∑
= + −
− +=
⎛⎝⎜⎜⎞⎠⎟⎟
U f B rq q
rA B r
f B rCr
U
( , ) exp( )
( , )
i jij ij
i j
ij i jij ij ij
nn ij ij
nij
ijn
tot,
1,
tot
6,8,10,12shell
Here f n(λ,r) = 1 − e−λrΣm=0n ((λr)m/m!), is the Tang−Toennies
damping function47 and is used to damp both the electrostaticand dispersion multipole expansions. The quantity Ushell = Ushell
(2)
+ UshellΔSCF is the total Drude oscillator polarization energy.21,23
The division of this polarization energy into the quantities Ushell(2)
and UshellΔSCF is discussed later. Aside from the damping functions
and the addition of higher-order van der Waals terms, thefunctional form is compatible with that used in many standardsimulation packages.
Here we describe a comprehensive algorithm for extractingrobust and transferable force field parameters on the basis of abinitio data. As discussed previously, we separate the para-metrization into two parts. We first derive asymptoticparameters from monomer properties: atomic charges, staticpolarizabilities (Drude oscillator charges), and dispersioncoefficients. Subsequently, parameters dependent upon chargedensity overlap, Aij
exch, Aijelec, Aij
ind, Aijδhf, are fit to DFT-SAPT
calculations for appropriate dimer systems. The exponents, Bij,are generated for all pairs of elements based on physicalconsiderations. For each element, we set Bii = 2(2·IPi)
1/2 andform pairwise cross terms using the combination rule Bij = (Bi +Bj)·BiBj/(Bi
2 + Bj2). Here, IPi is the ionization potential of the
element. The motivation for this formula has been described inour previous work.24
The monomer calculations should ideally be conducted at alevel of theory consistent with the dimer DFT-SAPTcalculations. For the dimer SAPT calculations, we havepreviously used the PBE functional48 (or PBE0, as discussedbelow) in conjunction with a dimer centered aug-cc-pVTZ(AVTZ) basis49 augmented with midbond basis functions.21
Due to the asymptotically incorrect behavior of almost all DFTfunctionals, asymptotic corrections18,20 are essential, and wecompute the associated ionization potentials necessary for theseasymptotic corrections at the AVTZ/PBE0 level of theory. Forthe SAPT calculations, we utilize the density fitting DFT-SAPTimplementation in the MolPro 2009 package.50
A particular challenge in maintaining consistency betweenthe monomer and dimer (SAPT) calculations arises in thechoice of basis set. This is particularly true in the case ofmonomer response calculations, which exhibit a strong basis setdependence. (In contrast, the DMA calculations used to deriveatomic charges converge rapidly with respect to basis set.)Ideally, the monomer response calculations would include off-atom basis sites reminiscent of the midbond basis functionsused in the SAPT dimer calculations. As in SAPT, these off-atom sites are effective at describing the diffuse density tails thatmake important contributions to the molecular response.41
Unfortunately, in the absence of a dimer, there is no a prioriobvious location for the off-atom sites and we resort to using adiffuse augmented dAVTZ basis for the monomer responsecalculations,41 applying an additional basis set correction asdescribed later.
■ MONOMER PROPERTIESWithin our model, electrostatic interactions are treated viadamped atomic point charges. The atomic point charges(dictating the asymptotic electrostatic interaction) arecomputed from the monomers as follows. First, we obtain aDMA representation (Stone’s method37,38) of the monomerelectron density computed with either the PBE or PBE0functional, and either a double or triple-ζ basis set, dependingon the size of the monomer. We find that the DMA is relativelyinsensitive to the choice of functional/basis set. Although, inprinciple, we could use the results of this DMA directly withinour force field,4,5,40 we seek a simpler point charge model that ismore amenable to standard MD/MC simulation packages. Assuch, we subsequently apply the method of Ferenczy51 to fitatom-centered point charges to reproduce the electrostaticpotential (ESP) of this DMA. This two-step procedure hasdistinct advantages over simply directly fitting point charges tothe total ESP. Here, in contrast, point charges are fit toreproduce the ESP separately from the multipoles on each site.
The Journal of Physical Chemistry A Feature Article
dx.doi.org/10.1021/jp3108182 | J. Phys. Chem. A 2013, 117, 2053−20662056

Therefore, unlike other ESP fitting methods, this procedure isrobust for providing physical charges on “buried” atoms. Arecent paper by Totton et al. has shown that this method offitting point charges is superior in accuracy and transferability ascompared to other ESP type approaches.52
The remaining monomer properties include the distributedstatic and frequency-dependent polarizabilities, from which weobtain Drude oscillator and dispersion parameters, respec-tively.23,24 The distributed polarizabilities are derived via anovel extension of the Williams−Stone method,41,53 whichcomputes the static and frequency dependent linear molecularresponse over a grid of point charge perturbations. Thesemolecular responses are computed at the CPKS level of theory,using the asymptotically corrected PBE0 functional with anALDAX + CHF kernel, and a dAVTZ basis set.24 We use theCAMCASP program41,54−56 to compute these responses, anduse the CAMCASP default grid point number and positionsettings. To extract transferrable distributed polarizabilities, weextend the standard Williams-Stone method by introducingsystematic constraints in a self-consistent iterative procedure, asdescribed in detail below.The Williams−Stone method is based on computing the ESP
at grid points Q, due to the linear molecular response inducedby point charge perturbations at grid points P. We fit thesepoint-to-point responses to a local, isotropic model:
∑α α̃ = − T Tat
tPa
ta
taQ
PQ 0 0
(modified from the more general eq 2 of ref 53). Here, T0tPa and
Tt0aQ are electrostatic interaction functions, αt
a are the polar-izability tensor components to be fit, and α̃PQ is the modelpredicted point-to-point response at grid points P and Q.53 Thesum is over all sites, a, and all tensor components t up to aspecified rank (note that although the sum is over all tensorcomponents t, there is only one unique component per rankbecause the model is isotropic).This procedure can be used for both static and frequency-
dependent responses, thus generating distributed induction anddispersion parameters. However, when the process is applied tostatic polarizabilities, we introduce several straightforwardmodifications to account for the fact that Drude oscillators
are finite (rather than point) dipoles and also exhibitintramolecular interactions among themselves.21 For fre-quency-dependent responses, we fit tensors up to rank 3,resulting in dispersion coefficients up to C12, whereas for staticresponses, we only fit the equivalent of rank 1 tensors for theDrude oscillator models. For hydrogen atoms, although we fitrank 3 frequency-dependent polarizability tensors, we neglectC12 dispersion coefficients on these atoms as we find thatdamping for these terms becomes problematic due to the shortinteratomic distances. A similar “special case” treatment ofhydrogen atoms has been used previously.41 We note that thenecessity (or lack thereof) of including dispersion coefficientsthrough C12 terms has been thoroughly investigated byMisquitta and Stone.41
We have recently described a self-consistent, iterativeextension of the Williams−Stone method, fitting the point-to-point molecular responses for a “library” of molecules withsimilar functional groups.24 One of the principle goals of thepresent work is to demonstrate that this strategy yields a robustset of transferable atomic parameters. Our iterative approachinvolves a systematic sequence of fits to the point to pointresponse of each molecule, with self-consistent constraintsintroduced at each fitting step. This constrained, iterative fittingis necessary, as coupling between the various αi
a in theunconstrained Williams−Stone procedure can lead to un-physical atomic parameters,41 resulting in poor transferability.The first step in our procedure is to calculate the point-to-pointmolecular response (either static or frequency-dependent) for acollection of molecules sharing common functional groups. Ifthe collection of molecules contains atom types present in apreviously fit “library”, then the polarizability tensors of theseatom types are constrained to those values throughout theentire fitting process. Next, reasonable zeroeth order values forthe αi
a(ω) (explicitly writing the frequency dependence) of thetarget atom types are obtained through unconstrainedimplementation of the Williams−Stone method on simplesmall molecules (e.g., homonuclear diatomics, etc.), where wefind that the standard Williams−Stone method often yieldsreasonable results. An iterative fitting of the point-to-pointresponse of every molecule in the collection is then carried out,in which all molecules are fit separately, but the αi
a(ω) are only
Figure 3. Schematic depiction of iterative fitting procedure for obtaining atomic static/frequency dependent polarizabilities.
The Journal of Physical Chemistry A Feature Article
dx.doi.org/10.1021/jp3108182 | J. Phys. Chem. A 2013, 117, 2053−20662057

fit for a single atom type in each iteration; all other atom typesare constrained to the αi
a(ω) values obtained from previousiterations. At the end of the iteration, the new αi
a(ω)parameters for the fit atom type are averaged over all of themolecules in the collection, and these averaged values are usedas constraints in subsequent iterations. In subsequent iterations,a new target atom type is chosen, and the procedure isrepeated. Iterations proceed by sequentially looping over alltarget atom types until convergence. This iterative fittingprocedure is shown schematically in Figure 3.We conducted this iterative fitting procedure for three
collections of molecules, for which various functional groupdispersion parameters have been obtained. These collectionswere fit sequentially, with parameters from earlier fits added tothe atom type “library” and used for subsequent constraints. Wefirst fit a collection of 23 alkanes and alkenes, consisting oflinear, branched, and cyclic molecules, using only three atomtypes: hydrogen, sp3 hybridized carbon, and sp2 hybridizedcarbon. We subsequently fit a collection of 24 moleculesconsisting of alcohols and primary, secondary, and tertiaryamines, using only three new atom types: alcohol oxygen,amine nitrogen, and one type (total) for hydrogen bonding N−H/O−H hydrogens; all necessary alkane/alkene parameters forthese molecules were constrained to the previously fitparameters. The third collection of molecules includes a mixof 23 acids, ketones, and aldehydes, again fit using only threenew atom types: carbonyl carbon and oxygen and aldehydehydrogen. The oxygen and hydrogen of the acid group wereconstrained to those parameters obtained from the alcohols,and all alkane/alkene/amine constraints were imposed usingprevious parameters. The resulting “library” of polarizabilitytensor parameters for these nine atom types is given in theSupporting Information. The atomic dispersion parameters aregenerated via numerical quadratures involving these frequencydependent polarizability tensors.41,53,57
We show that these parameters are accurate and transferableby comparing the RMS error of the fitted point-to-pointmolecular response using parameters from the nine-atom type“library” to that of corresponding unconstrained fits. Becauseour iterative self-consistent procedure introduces additionalconstraints, the RMS error must be larger than that of thestandard Williams−Stone approach; the ratio of these twoerrors is given in the Supporting Information for all moleculesin the library. Focusing on the smallest nonzero frequency, theaverage ratio of RMS errors are 1.02, 1.02, and 1.01 for thealkane/alkene, alcohol/amine, and acid/ketone/aldehyde sets,respectively. The fact that these ratios are very close to 1.0indicates that the atom-type definitions are chemically relevantand transferable among environments, faithfully reproducingthe molecular response while enforcing physical, transferableparameters. We recently utilized a very similar approach toobtain transferrable parameters for functionalized imidazolatederivatives.24
It is well-known that dispersion energies converge slowlywith respect to basis set size, and this is the primary reason forusing midbond basis functions in a SAPT calculation.58
Unfortunately, such midbond basis functions are not presentin the monomer response calculations, and thus thesecalculations are likely not as converged with respect to basisset as the corresponding SAPT calculations.41 Previously, wetested this basis set effect for an imidazolate−CO2 dimersystem by comparing dispersion energies from monomer-centered dAVTZ basis (consistent with our monomer response
calculations) and dimer-centered AVTZ + midbond (consistentwith our SAPT dimer calculations) SAPT calculations.24 Wefound that the monomer-centered dAVTZ SAPT calculationswere not fully converged with respect to basis set size,underestimating dispersion by up to 10% for certainconfigurations. We thus estimated and applied a 1.07 scalefactor “basis set correction” to all dispersion coefficientscalculated via monomer response calculations. We employ asimilar basis set correction scheme in this work. However, wenote the prior basis set correction was deduced fromcalculations involving an aromatic molecule (imidazolate),potentially leading to a higher than typical dispersioncorrection. Therefore, we use the same analysis,24 this timeexamining an ethane dimer, which we take as representative ofa small, nonaromatic dimer. For this dimer, we find a “basis setcorrection” of ∼1.03, which we use to scale all dispersioncoefficients determined from molecular response calculations inthis work.Finally, we comment on the use of combination rules to form
C6−C12 cross term parameters. These cross term parameterscan be rigorously calculated from the atomic frequencydependent polarizability tensors,41 and therefore formally noadditional combination rules are necessary. However, the use ofthese explicit algorithms requires many force field parametersand involves multiple numerical quadratures. As such, weinvestigate the accuracy (or inaccuracy) of using the commonlyemployed combination rule: Cn
ij = (CniiCn
jj)1/2. In the SupportingInformation, we compare the cross term Cn
ij parametersgenerated using this rule, to the exact (calculated) crossparameters. Considering all possible cross term parametersbetween the 9 atom types, we find average absolute errors of0.7%, 3%, 5%, and 8% for the C6, C8, C10, and C12 crossparameters respectively. Because these errors are likely withinthe accuracy of our method and are mostly nonsystematic, wedeem them acceptable and worth the resulting simplification.We employ the combination rule Cn
ij = (CniiCn
jj)1/2 for theremainder of this work.
■ DIMER SAPT CALCULATIONSThe remaining force field parameters depend explicitly onelectron density overlap between dimers (Aij
exch, Aijelec, Aij
ind, Aijδhf),
and as such are not derivable in terms of monomer properties.These parameters are fit to distinct interaction energycomponents from DFT-SAPT calculations on appropriatedimers. Here, important considerations for obtaining accurateparameters include the selection of the dimer system and themethod of choosing dimer configurations for the SAPTcalculations. For force fields specific to a single system orapplication, the choice of dimer is straightforward. For example,in our previous work on constructing force fields to study CO2capture in zeolitic imidazolate frameworks (ZIFs), we chosedimers consisting of a CO2 molecule and representative ZIFfunctional groups.23,24 However, for more general force fielddevelopment, it is desirable to avoid any parametrization biastoward specific heteromolecular interactions, and may bepreferable to fit homomolecular dimers. This later choice isexplored in this work, and we show how heteromolecularinteractions can be accurately modeled from parametersderived using homomolecular SAPT parametrization.The methods we use to generate dimer configurations
depend on whether we are parametrizing homomolecular orheteromolecular dimers, although both methods should beequally robust. For homomolecular dimers, we conduct
The Journal of Physical Chemistry A Feature Article
dx.doi.org/10.1021/jp3108182 | J. Phys. Chem. A 2013, 117, 2053−20662058

molecular dynamics (MD) simulations of the correspondingneat fluid, at temperatures and pressures corresponding to aliquid-like phase. After equilibration, we sample dimerconfigurations randomly from the trajectory and group themaccording to closest-contact distances. We have found thatinteractions closer than ∼0.8 times the approximate van derWaals contact distance (henceforth σ) are typically toorepulsive to use for fitting, whereas interactions farther than∼1.2σ do not exhibit much charge penetration and thusbecome useless for fitting such parameters. Therefore, wechoose dimer configurations spanning the range from 0.8 to1.2σ for most of our SAPT fits. For heteromolecular dimers,similar MD sampling could be used for miscible mixtures, butuniform sampling could be difficult for mixtures that areimmiscible. Therefore, we use a different method in which wefix one of the monomers, generate a grid of angular pointsaround the monomer, and then position the second monomerin a random orientation at a fixed distance from the firstmonomer at each of these angular points. We choose the samerange of 0.8−1.2σ to generate these configurations. In additionto this choice of configurations, for all SAPT fits we employ aweighting function to make the fits insensitive to high-energy,very repulsive configurations, as described previously.23
Exchange Component. The exchange energy is entirelydependent on charge overlap, and therefore this term is fitsolely to the corresponding SAPT energy, using the functionalform as previously given:
∑≅ −E A B rexp( )i j
ij ij ijexch,
exch
As the Bij exponents are determined a priori, the onlyparameters fit are atomic exchange parameters, Aii
exch; thecombination rule Aij
exch = (AiiexchAjj
exch)1/2 is used to generate crossterms.Electrostatic Component. The electrostatic energy is
composed of an asymptotic term consisting of point chargeinteractions previously determined from a DMA, as well as ashort-range component describing charge penetration effects:
∑ ∑≅ + −E f B rq q
rA B r( , ) exp( )
i jij ij
i j
ij i jij ij ijelec
,1
,
elec
The monomer charge penetration parameters, Aiielec, are fit to
differences between the SAPT electrostatic energies and theCoulombic “point charge” contribution of the force field; thecombination rule Aij
elec = −(AiielecAjj
elec)1/2 is used for crossterms.23
Induction Component. The induction energy consists of apolarizable Drude oscillator59 component as well as a short-range charge penetration component:
∑≅ + −E U A B rexp( )i j
ij ij ijpol shell(2)
,
ind
Here, Ushell(2) is the second-order Drude oscillator energy, i.e., the
energy of the drude oscillators in the electrostatic field due toall static charges (besides the intramolecular static charges). Inother words, to compute this second-order energy, the Drudeoscillators do not interact between molecules, and the positionsof the Drude oscillators on each molecule are determined as theminimum energy configuration in the field due to the fixed(nonpolarizable) charges of all other molecules. Here we fit tothe second-order Drude energy for logical consistency with thesecond-order SAPT; subsequent molecular simulations using
our force fields employ the fully converged, self-consistentpolarization. Intramolecular Drude oscillator interactions arecomputed using a screening function, and the spring constantand Thole parameter were set to 0.1 au and 2.0, respectively, asin our prior work.21
As the Drude oscillator charges are determined via molecularresponse calculations, the atomic induction penetrationparameters, Aii
ind, are the only parameters fit to the SAPTcalculations. Here, the penetration parameters are fit to thedifference between the SAPT polarization energy, Epol, and theDrude oscillator computed Ushell
(2) energy. The combination ruleAijind = −(Aii
indAjjind)1/2 is used to generate the pairwise
parameters.23
δHF Component. The delta Hartree−Fock energy, Eδhf, hasbeen described previously.20,21 It is fit to a functional formgiven by
∑≅ + −δδΔE U A B rexp( )
i jij ij ijhf shell
SCF
,
hf
Here, UshellΔSCF is the Drude oscillator energy above second order,
i.e., the difference between the fully converged, self-consistentDrude energy (Ushell) and second-order Drude energy (Ushell
(2) );Ushell
ΔSCF = Ushell − Ushell(2) . Note that this extended decomposition
need not be computed in actual simulations (only Ushell needsto be computed) but is computed for parameter fitting so thatthe higher order induction effects included in Eδhf are notdouble counted.The penetration component of the δHF energy is
subsequently fit to the difference between the ab initiocomputed Eδhf and Ushell
ΔSCF. The associated atomic penetrationparameters, Aii
δhf, are explicitly fit, and cross terms are formedusing the combination rule Aij
δhf = −(AiiδhfAjj
δhf)1/2. Note that thiscombination rule produces strictly attractive δHF penetrationterms, in contrast to our previous combination rule.23 Thisstrictly attractive combination rule was adopted to allow forgreater parameter transferability.
■ HYDROGEN BONDING/CLOSE POLARINTERACTIONS
Several additional factors need to be accounted for in the caseof strongly hydrogen bonding systems, such as amines/alcohols. The first consideration involves the density functionalused in the SAPT calculations. It has been found previouslythat DFT-SAPT calculations are relatively insensitive to thechoice of density functional so long as appropriate asymptoticcorrections are employed.16 Indeed, in our experience, we havefound that asymptotically corrected PBE and PBE0 functionalsgive very similar results when used with SAPT. However,calculations on methylamine dimer suggest that for hydrogenbonding systems, the PBE functional may not be sufficientlyaccurate. For these dimers, we compared interaction energiescomputed at the CCSD(T), SAPT/PBE, and SAPT/PBE0levels of theory, with all calculations using the same AVTZ+midbond basis. We find that whereas the SAPT/PBE0interaction energies agree very well with the CCSD(T)benchmark, the SAPT/PBE method systematically andsubstantially underpredicts the attraction (see SupportingInformation). Comparing the energy decomposition betweenSAPT/PBE and SAPT/PBE0 suggests that the overpredictionof exchange repulsion energy by SAPT/PBE is the main causeof the decreased attraction. Therefore, we use the asymptoti-cally corrected PBE0 functional for all calculations involving
The Journal of Physical Chemistry A Feature Article
dx.doi.org/10.1021/jp3108182 | J. Phys. Chem. A 2013, 117, 2053−20662059
strongly polar systems (amines/alcohols/acids/aldehydes/ketones). (Note that as implemented in MolPro 2009,50 hybridfunctionals like PBE0 use local effective Hartree−Fockexchange when used with density fitting DFT-SAPT.).The second (related) consideration for these systems
involves the equilibrium separation of the dimers. Asmentioned above, whereas “typical” dimers exhibit highlyrepulsive interactions at separations closer than ∼0.8σ,hydrogen bonding dimers (amines/alcohols) form hydrogenbonds at distances as close as ∼0.6σ. These systems thereforerequire accurate modeling of the important (attractive)interactions at distances exhibiting very high charge pene-tration. We have noted before that our decomposed force fieldsare not well-suited to describe interactions at these shortdistances, due to breakdown of the multipole expansions andthe relatively simple description of charge penetration terms(single exponential, isotropic Born−Mayer functions).23,24
Furthermore, the energy decomposition for these configu-rations becomes less meaningful, because the total interactionenergy is mostly a sum of highly canceling charge penetrationcontributions. We previously proposed that for such short-range interactions, a continuous, piecewise force field could beused that smoothly switches between a single “short-range”repulsive function, and the “long range” force field of separateinteraction terms.23 Although such a piecewise force fieldrequires additional parameters and the short-range part isunlikely to be transferrable, the corresponding short-range termneeds to be fit/used only for specif ic atom pairs that exhibitespecially close contact distances (i.e., hydrogen bonding pairs).The functional form of this piecewise force field is
∑
λ
=
+ −
−
≥
+ −
+ Δ
<
=
⎧
⎨
⎪⎪⎪⎪⎪
⎩
⎪⎪⎪⎪⎪
U
f B rq q
rA B r
f B rCr
r r
f B rq q
rA B r r r
( , ) exp( )
( , )
( , ) exp( )
ij
ij iji j
ijij ij ij
nn ij ij
nij
ijn
ij ij
ij iji j
ijij ij ij ij
ij
ij ij
1tot
6,8,10,12
switch
1pen switch
Although three new parameters, Aijpen, λij, Δij, are introduced for
the short-range force field, there is only one degree of freedomonce we apply constraints of continuity and differentiability;Drude polarizability is added in both cases, withoutmodification. The choice of rij
switch remains somewhat arbitrary,although limited by the functional form and the continuity anddifferentiability constraints. We pick rij
switch as the distance atwhich the pairwise potential is zero, changing from a repulsiveto attractive interaction. For the hydrogen-bonding atom pairs,we then fit corresponding dimer SAPT energies (total energies)for configurations with short-range attractive character (i.e.,∼0.6−0.8σ). We note that these parameters are very systemspecific, and are not meant to be transferable.For these hydrogen bonding systems, we also find that the
dispersion expansion is fairly underdamped, yielding dispersionenergies much too attractive at close distances as compared toSAPT. This suggests that the exponents, Bii, determined forhydrogen atoms, are not appropriate in the context of hydrogenbonding. Employing the accurately determined value of Bii =1.666 a.u. (determined for H2 molecule47) for hydrogen-bonding H atoms, we find a much better dampedapproximation to the SAPT dispersion energies for these
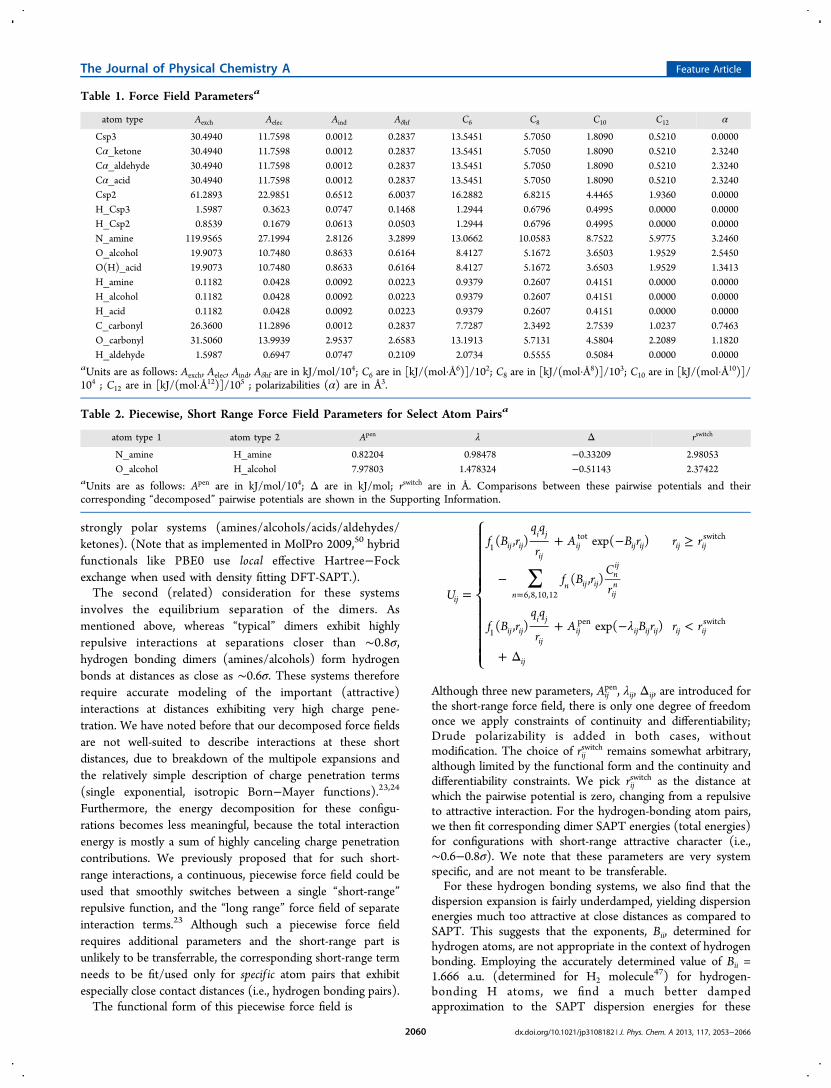
Table 1. Force Field Parametersa
atom type Aexch Aelec Aind Aδhf C6 C8 C10 C12 α
Csp3 30.4940 11.7598 0.0012 0.2837 13.5451 5.7050 1.8090 0.5210 0.0000Cα_ketone 30.4940 11.7598 0.0012 0.2837 13.5451 5.7050 1.8090 0.5210 2.3240Cα_aldehyde 30.4940 11.7598 0.0012 0.2837 13.5451 5.7050 1.8090 0.5210 2.3240Cα_acid 30.4940 11.7598 0.0012 0.2837 13.5451 5.7050 1.8090 0.5210 2.3240Csp2 61.2893 22.9851 0.6512 6.0037 16.2882 6.8215 4.4465 1.9360 0.0000H_Csp3 1.5987 0.3623 0.0747 0.1468 1.2944 0.6796 0.4995 0.0000 0.0000H_Csp2 0.8539 0.1679 0.0613 0.0503 1.2944 0.6796 0.4995 0.0000 0.0000N_amine 119.9565 27.1994 2.8126 3.2899 13.0662 10.0583 8.7522 5.9775 3.2460O_alcohol 19.9073 10.7480 0.8633 0.6164 8.4127 5.1672 3.6503 1.9529 2.5450O(H)_acid 19.9073 10.7480 0.8633 0.6164 8.4127 5.1672 3.6503 1.9529 1.3413H_amine 0.1182 0.0428 0.0092 0.0223 0.9379 0.2607 0.4151 0.0000 0.0000H_alcohol 0.1182 0.0428 0.0092 0.0223 0.9379 0.2607 0.4151 0.0000 0.0000H_acid 0.1182 0.0428 0.0092 0.0223 0.9379 0.2607 0.4151 0.0000 0.0000C_carbonyl 26.3600 11.2896 0.0012 0.2837 7.7287 2.3492 2.7539 1.0237 0.7463O_carbonyl 31.5060 13.9939 2.9537 2.6583 13.1913 5.7131 4.5804 2.2089 1.1820H_aldehyde 1.5987 0.6947 0.0747 0.2109 2.0734 0.5555 0.5084 0.0000 0.0000
aUnits are as follows: Aexch, Aelec, Aind, Aδhf are in kJ/mol/104; C6 are in [kJ/(mol·Å6)]/102; C8 are in [kJ/(mol·Å8)]/103; C10 are in [kJ/(mol·Å10)]/104 ; C12 are in [kJ/(mol·Å12)]/105 ; polarizabilities (α) are in Å3.
Table 2. Piecewise, Short Range Force Field Parameters for Select Atom Pairsa
atom type 1 atom type 2 Apen λ Δ rswitch
N_amine H_amine 0.82204 0.98478 −0.33209 2.98053O_alcohol H_alcohol 7.97803 1.478324 −0.51143 2.37422
aUnits are as follows: Apen are in kJ/mol/104; Δ are in kJ/mol; rswitch are in Å. Comparisons between these pairwise potentials and theircorresponding “decomposed” pairwise potentials are shown in the Supporting Information.
The Journal of Physical Chemistry A Feature Article
dx.doi.org/10.1021/jp3108182 | J. Phys. Chem. A 2013, 117, 2053−20662060

systems and use this value for the remainder of our work. Asthe damped dispersion energies seem to be very accurate for allother systems previously studied, this seems to be anexceptional case associated with unusually short intermoleculardistances.
■ SAPT FORCE FIELD FITS
We apply our comprehensive force field parametrizationmethodology to self-consistently fit force fields for a “library”of small molecules and functional groups, including alkanes,alkenes, amines, alcohols, acids, ketones, and aldehydes. We useSAPT calculations on small, homomolecular dimers to fit theassociated charge penetration terms. The use of homomolec-ular dimers is a simple and natural choice, and we demonstratelater on that the resulting atomic penetration parameters areextremely transferable to arbitrary heteromolecular systems.The force field parameters for these molecules/functionalgroups are listed in Tables 1 and 2 (the charges used for secondvirial calculations are given in the Supporting Information), andthe corresponding fits to the SAPT energy decompositions areshown in the Supporting Information.It is important to analyze the extent to which our chosen
functional forms, motivated by the underlying physics, canaccurately reproduce the SAPT dimer interaction energy data.An immediate question concerns the neglect of explicitanisotropic terms (higher order multipoles, anisotropicBorn−Mayer terms) in our force field functional form, andthe effect on the SAPT energy fits. In general, we find that theerrors due to this approximation are system dependent, as onemight expect. For alkanes and ketones, explicit isotropic termsseem to describe the data quite well, whereas there is significantscatter in the SAPT fits for alcohols and amines. The lattermight be anticipated due to anisotropic charge distributionsassociated with oxygen and nitrogen nonbonding lone pairs.These systems likely could be described better with the use ofhigher-order electrostatic multipoles and anisotropic Born−Mayer charge penetration terms. However, as our focus is onparameter transferability, simplicity, and consistency, we foregosuch an approach for now.Similarly, the effect of utilizing isotropic dispersion
coefficients (rather than anisotropic models) has beenpreviously studied.41 As evidenced by the SAPT fits, we findthat the neglect of explicit anisotropic dispersion terms seemsto introduce relatively little error for medium to long-rangedimer separations. The isotropic C6, C8, C10, and C12 dispersionparameters derived using our self-consistent, iterative fits, areevidently highly accurate, with relatively little scatter present inthe SAPT energy fits at such separations. We note that there isan important distinction between representing atomic aniso-tropy compared to molecular anisotropy. Alcohols and aminesexhibit atomic anisotropy due to the charge distributions in thevicinity of the oxygen and nitrogen atoms. However, moleculessuch as benzene and imidazoles exhibit molecular anisotropy,due to the arrangement of atoms in these molecules. Thismolecular anisotropy can be accurately captured by our forcefields, as evidenced by our previous highly accurate SAPTenergy fits for benzene/CO2, imidazolate/CO2 dimer systems(which utilize the same functional forms).23,24
■ SECOND VIRIAL COEFFICIENTS
In the previous sections, we outlined a comprehensivemethodology for parametrizing ab initio force fields based on
DFT-SAPT interaction energies, focusing specifically on noveltechniques to extract robust and transferable force fieldparameters. Recently, we used a similar approach, with greatsuccess, to accurately model the adsorption of CO2 innanoporous zeolitic imidazolate frameworks (see ref 24 andFigure 2). Here we demonstrate the generality of this approach,and the transferability of the resulting force fields outside thesystems for which they were parametrized. Second virialcoefficients provide an appealing initial benchmark againstwhich to test force fields for many reasons, including theirexperimentally ubiquity,60 and the fact that they provide anexplicit measure of two-body interactions. As such, they are astringent test of our two-body force field terms. It should benoted that, in the context of our physically motivated force fieldparametrization, additional many-body interactions aside frompolarization (which accounts for electrostatic many-bodyeffects), such as three-body dispersion, exchange, etc., arelikely necessary to accurately describe condensed-phasesystems; these can be added separately to these pairwiseforce fields, when appropriate, using related techniques we havedescribed before.22 Nonetheless, for many applications in thelow- to medium-density limit, including gas-sorption, theseadditional many-body terms can be omitted without significantloss in accuracy.We compute second virial coefficients according to the
following formula:61
∫π β ξ ξ ξ= − ⟨ − ⟩α αB U2 (1 exp[ ( )] ) d2 inter2
1 2
Here, ξ is the distance between the centers of mass of the twomolecules, and ⟨...⟩α1α2 denotes an average over the config-
urations and orientations of the two molecules, weighted by aBoltzmann factor that depends only on the intramolecularHamiltonians.61 For longer-chain molecules, it is important tosample intramolecular configurations, and this was done usingthe OPLS-AA force field for all bonded interactions.62,63
Although OPLS-AA certainly does not yield quantitativelycorrect conformation energies, the impact of molecularflexibility is modest and the configurations are likelyqualitatively representative. GROMACS64,65 was used to runshort NVT trajectories at conditions corresponding to a low-density gas phase, and molecular configurations were pulledfrom this trajectory and stored. Then, to compute the average⟨...⟩α1α2, ∼10
5 interactions were computed (for each ξ at spacing
of ∼0.01 Å) by choosing two molecular configurations atrandom, generating random orientations, and then placing themolecules at a fixed separation, ξ.Computed second virial coefficients are shown in Figure 4−
Figure 7. In general, our computed virials are in excellentagreement with experimental data. The alcohol/amine virialcoefficients are fairly sensitive to the short-range portion of thepiecewise force field used to describe the hydrogen bonding(N−H, O−H) interactions, due to the dominance of the low-energy, attractive regions in the virial calculation. These short-range force field components are highly specific for the pairwiseinteractions for which they were parametrized, and they are notrequired (nor intended) to be used in describing general, non-hydrogen bonding heteromolecular interactions (as shown inour acetic acid/CO2, methylamine/CO2 dimer calculationsbelow).
The Journal of Physical Chemistry A Feature Article
dx.doi.org/10.1021/jp3108182 | J. Phys. Chem. A 2013, 117, 2053−20662061

■ PARAMETER TRANSFERABILITYWe now investigate the transferability of the force fieldparameters derived using our methodology. There are twodistinct aspects of parameter transferability, each with its ownimportant consequences. The first aspect involves whether ornot functional group parameters can be transferred to differentmolecular environments; e.g., can nitro parameters fit usingnitromethane calculations be used to accurately describe thenitro group in a nitrobenzene molecule? This type oftransferability has been investigated and verified in our previouswork.24 The second aspect involves specifically the chargepenetration parameters of our force field. As these parameterscannot be derived using strictly monomer calculations, they arefit to dimer interaction calculations. In this case, thetransferability concerns the extent to which the parametersare biased toward the specific dimer interaction for which theywere fit. We address this question using two specific dimers asexamples: acetic acid/CO2 dimer and methylamine/CO2 dimer.For acetic acid and methylamine, we use the force fieldparameters developed in this work, and for CO2, we use theparameters from our previous work (modified to include C12dispersion coefficients).24 We note that all of the (chargepenetration) parameters for these molecules have been derivedstrictly from homomolecular calculations, and thereforereproducing said heteromolecular interactions is a stringenttest of transferability. Comparisons between cross terminteractions predicted by our force field and those calculatedfrom SAPT are shown in the Supporting Information. Ingeneral, the fits are very satisfactory, and the RMS errors in thetotal interaction energy are comparable to the homomoleculardimer systems (amines, acids) for which the force fieldparameters were fit. This is a crucial result, as it suggests apromising methodology based on relatively simple, homo-molecular calculations, for generating force field parametersthat are general and may be applicable for describing arbitraryheteromolecular interactions.We believe that this transferability is a direct result of the
physically motivated approach taken to model and fit theassociated parameters, thus ensuring the extraction of trulymeaningful atomic parameters. By fitting each term in theSAPT energy decomposition individually to an appropriatefunctional form, we ensure by construction that each parameteris capturing a specific, well-defined fundamental interaction. As
Figure 4. Second virial coefficients for alkanes and alkenes. Symbolsare the results from simulations, and the corresponding lines are theexperimental data from ref 60.
Figure 5. Second virial coefficients for amines and alcohols. Symbolsare the results from simulations, and the corresponding lines are theexperimental data. Experimental data for methylamine and ethylaminewere taken from ref 60, data for methanol was taken from ref 66, anddata for ethanol was taken from refs 67 and 68.
Figure 6. Second virial coefficients for acetone. Symbols are the resultsfrom simulations, and the corresponding line is the experimental datafrom refs 60 and 69.
Figure 7. Second virial coefficients for acetaldehyde and propanal.Symbols are the results from simulations, and the corresponding linesare the experimental data from refs 60 and 70.
The Journal of Physical Chemistry A Feature Article
dx.doi.org/10.1021/jp3108182 | J. Phys. Chem. A 2013, 117, 2053−20662062

such, simple combination rule ansatz are successful in providingaccurate cross term parameters. This is in contrast to traditional(i.e., nonpolarizable, Lennard-Jones) force fields in whichparameters generally describe multiple types of physicalinteractions simultaneously, and thus the magnitude of theparameter reflects a complicated superposition of differentfundamental interactions. In this latter case, generating crossterms via simple combination rules is not generally robust,particularly in regimes very different from the originalparametrization. In the next section, we will show an exampleof how our force fields can be applied to just such a case.
■ APPLICATION TO PREDICTING CO2 ADSORPTIONIN IRMOF-1 AND IRMOF-3
We have previously developed and applied ab initio, SAPT-based force fields in the study of CO2/N2 gas adsorption inmetal−organic frameworks (MOFs), and specifically zeoliticimidazolate frameworks (ZIFs).23,24 Here, we examine theapplication of our newly developed force field parameters,developed on the basis of monomer properties and homomo-lecular diatomic interactions, to a completely unrelated chemicalenvironment: gas adsorption in the isoreticular MOF series,IRMOF. As in our prior work, we are motivated in part by thepractical applications of these nanoporous systems in terms offlue gas separation/CO2 sequestration. We focus on theubiquitous IRMOF-1 (MOF5), and its amine functionalizedversion, IRMOF-3. IRMOF-1 is composed of Zn4O inorganicclusters bridged by 1,4-benzenedicarboxylate (BDC) linkergroups. IRMOF-3, an isoreticular analog of IRMOF-1, insteademploys amine-functionalized BDC linkers. We approximatethe “buried” inorganic Zn4O cluster using only point charges(note that although this may be a good approximation for CO2,it certainly would fail for small gas molecules such as H2 wherethis inorganic cluster is an important, accessible, binding site71),and employ our derived force fields to model the organic linkergroups, consistent with our previous work.23,24
Additional force field parameters are needed for the BDCand amine−BDC linkers. For the BDC linker, in addition topreviously derived benzene parameters (modified to includeC12 parameters),24 we fit carboxylate parameters to SAPTcalculations employing Li-capped, methyl carboxylate/CO2dimers, consistent with our previous treatment of inorganic−organic linker atoms.23,24 The SAPT fits for this dimer (as wellas test calculations on the full BDC linker) are shown in theSupporting Information. For the amine functional group inIRMOF-3, the parameters previously derived in this work were
used. The force field parameters for these systems are shown inTable 3.Grand canonical Monte Carlo (GCMC) simulations were
conducted to calculate CO2 adsorption isotherms for IRMOF-1and IRMOF-3. The “SYM” force field,21,22 which has beenextensively benchmarked to thermodynamic data, was used todescribe adsorbate−adsorbate interactions. In addition to ourpreviously reported GCMC simulation details,23 a particle-mesh Ewald (PME)72 treatment of long-range dispersioninteractions was employed (the simulation code is available fordownload on our Web site73). The IRMOF-1 and IRMOF-3frameworks were treated as rigid, with experimental crystalstructures taken from the Cambridge Structural Database(CSD). For the IRMOF-3 crystal structure, amine functionalgroup positions are indeterminate from the crystal structure.Due to the relative distance between the BDC linker groups, weexpect little to no correlation between amine positions onadjacent linker groups, and therefore these positions werechosen randomly. In choosing random site occupancies, thequestion arises of what size supercell is needed to sample allpossible important random arrangements of functional groups.We have conducted simulations for IRMOF-3 using a singleunit cell as well as a 2 × 2 × 2 supercell and have found thatboth frameworks lead to very similar isotherms.The CO2 isotherms for IRMOF-1 and IRMOF-3 are shown
in Figure 8. The simulated isotherm for IRMOF-1 is inexcellent agreement with the experimental data, verifying theaccuracy of the SAPT-based force field. For IRMOF-3, thesimulated isotherm is in semiquantitative agreement withexperiment, overpredicting uptake at higher pressures by 10−15%. Some of this discrepancy may arise from the quasi-disordered nature of the IRMOF-3 amine groups. Thesimulations reproduce the qualitative trend of higher uptakeat lower pressures in IRMOF-3 (due to attractive interactionsintroduced by the amines) and lower uptake at higher pressuresin IRMOF-3 (due to less free volume from introduction ofamines) compared to IRMOF-1. As the amine functionalizationis the only difference between the systems, the fact that thesetrends are well-reproduced highlights the transferability of theamine force field parameters to systems dissimilar to that forwhich they were parametrized.
■ SUMMARY AND CONCLUSIONS
We have presented a comprehensive methodology forgenerating ab initio, transferable force fields on the basis ofthe SAPT energy decomposition. Although the accuracy of this
Table 3. Atom Type Parameters Used for CO2/IRMOF-1 and CO2/IRMOF-3 interactionsa
atom type q Aexch Aelec Aind Aδhf C6 C8 C10 C12
C_CO2 0.65738 7.4377 2.4130 1.2513 0.0795 11.4742 6.3294 2.9660 1.8355O_CO2 −0.32869 35.4373 10.8209 0.2545 −1.8179 8.6728 4.2665 2.8761 1.3258C_benzene −0.02144 42.3935 20.4172 1.8741 4.4968 16.7742 6.6332 6.4248 2.6799H_benzene 0.06202 1.0600 0.2749 0.0892 0.0971 1.3439 0.7056 0.5186 0.0000C_carboxylate 0.85741 26.3600 11.2896 0.0012 −0.2837 7.7287 2.3492 2.7539 1.0237O_carboxylate −0.82111 65.6757 17.4054 4.5411 −3.3954 17.9956 11.5761 5.0165 0.0000N_amine −0.65314 119.9565 27.1994 2.8126 −3.2899 13.0662 10.0583 8.7522 5.9775H_amine 0.35758 0.1182 0.0428 0.0092 −0.0223 0.9379 0.2607 0.4151 0.0000Zn 1.53100 0.0000 0.0000 0.0000 0.0000 0.0000 0.0000 0.0000 0.0000O (Zn4O) −1.77346 0.0000 0.0000 0.0000 0.0000 0.0000 0.0000 0.0000 0.0000
aUnits are the same as in Table 1. All framework polarizabilities are set to zero. For the Aδhf combination rule to be consistent within this work aswell as previous works, we use |Aij
δhf| = |AiiδhfAjj
δhf|1/2, with the sign of Aijδhf determined to be negative if either Aii
δhf or Ajjδhf is negative, and positive
otherwise.
The Journal of Physical Chemistry A Feature Article
dx.doi.org/10.1021/jp3108182 | J. Phys. Chem. A 2013, 117, 2053−20662063

methodology has been previously shown,21,23,24 here we havefocused on the generality of the method and transferability ofthe resulting force field parameters. While we outlineapproaches to obtain physically meaningful and transferableexchange, polarization, charge penetration, and dispersionparameters, we find that the latter are both particularlyimportant, and particularly challenging, in terms of bothaccuracy and transferability. Using a novel, self-consistentiterative extension of the Williams−Stone method, we are ableto extract truly physical and transferable atomic dispersionsparameters on the basis of monomer calculations; very similarapproaches can also be applied to static polarizabilities.Crucially, this approach yields dispersion parameters for thegiven atom within its unique molecular environment, thuscapturing the subtle (but often important and systematic)differences from one chemical environment to another. Weanticipate that this procedure may also have related applicationsoutside the present work, including generating dispersioncoefficients for use in DFT-D calculations, or developingaccurate and transferable polarizability parameters to be used inconjunction with existing, nonpolarizable force fields.When considering general methods for force field develop-
ment, it is essential to weigh the benefits of more accurate,sophisticated functional forms versus the associated complexityand practical barriers to adoption. As such, it is important tonote that the functional forms employed in our force fieldspresent few implementation challenges in standard molecularmechanics programs (a generic, fully functional Monte Carloimplementation is also available from our Web site). Althoughour force fields are polarizable, the use of Drude oscillators isnow a standard practice in conventional simulation packages.Beyond the damping functions employed for electrostatic anddispersion interactions, the remaining energy expression is inrelatively standard form. We have neglected explicit anisotropicterms in our force field, for reasons of simplicity andtransferability. However, even for anisotropic environments(amines, alcohols, ...), the correct physics is captured in anaverage sense, as evidenced by the SAPT energy fits and thecorrect description of second virial coefficients. Overall, webelieve our physically motivated ab initio force field method-ology and associated functional form represent a significant
(and practical) advance over generic Lennard-Jones andBuckingham force fields, and a solid step toward thedevelopment of practical “next generation” force fields formolecular simulation.
■ ASSOCIATED CONTENT
*S Supporting InformationTable showing correction of CO2 parameters for computingadsorption isotherms as published in ref 24. Tables offrequency dependent polarizability tensors and correspondingRMS errors for self-consistent, molecular response fits foralkanes/alkenes, amines/alcohols, and acids/aldehydes/ke-tones. Figures depicting accuracy of C6, C8, C10, and C12 crossterms given by combination rules compared to exact crossterms from frequency dependent polarizability tensors. Figurescomparing accuracy of interaction energies for methylaminedimer calculated with SAPT(PBE), SAPT(PBE0), and CCSD-(T). Table giving charges and exponents used for organicmolecule force fields and SAPT energy fits. Figures showingSAPT fits for homomolecular dimers: Ethane, butane, ethene,propene, methylamine, methanol, acetone, acetic acid, andacetaldehyde. Figures depicting piecewise pair potentials foramine (N−H) and alcohol (O−H) hydrogen bonding pairs.Figures showing SAPT fits for heteromolecular dimers: aceticacid/CO2 dimer and methylamine/CO2 dimer. Figuresshowing SAPT parameter fits for IRMOF-1 and IRMOF-3systems. This material is available free of charge via the Internetat http://pubs.acs.org.
■ AUTHOR INFORMATION
Corresponding Author*E-mail: [email protected].
NotesThe authors declare no competing financial interest.
Biographies
Jesse McDaniel earned his B.A. in chemistry in 2008 from Washington
University in St. Louis. He is currently a Ph.D. student under the
supervision of J. R. Schmidt, at the University of WisconsinMadison.
His research interests include the development of ab initio force fields
for use in molecular simulation, with particular focus on the study of
gas adsorption in metal organic frameworks.
Figure 8. CO2 excess adsorption isotherms for IRMOF-1 (red) andIRMOF-3 (green). The experimental data from ref 74 are shown aslines, and the simulation data are shown as triangles. The simulatedCO2 uptake has been converted to excess adsorption, consistent withthe experimental data,75 using the method described in ref 76.
The Journal of Physical Chemistry A Feature Article
dx.doi.org/10.1021/jp3108182 | J. Phys. Chem. A 2013, 117, 2053−20662064

J. R. Schmidt is an Assistant Professor of Chemistry at the Universityof WisconsinMadison, and a member of the Theoretical ChemistryInstitute. He earned his B.S. from Hope College in 2001, and a Ph.D.in Physical Chemistry from the University of Wisconsin (with JimSkinner) in 2006. After postdoctoral work at Yale (with John Tully),he returned to UW in 2008. His current research interests involvemodeling complex materials of energy relevance, including nano-porous materials and heterogeneous catalysts, as well as a strong focuson “enabling technologies” via the development of novel computa-tional approaches. Beyond research, he is a strong proponent of theintegration of computational chemistry into the graduate andundergraduate curriculum, and the principal developer of the web-based WebMO software package.
■ ACKNOWLEDGMENTS
This work was supported by Chemical Sciences, Geosciencesand Biosciences Division, Office of Basic Energy Sciences,Office of Science, U.S. Department of Energy, under award DE-FG02-09ER16059. Additional computational resources wereprovided via the Center for High Throughput Computing atthe University of Wisconsin77 and by National ScienceFoundation Grant CHE-0840494.
■ REFERENCES(1) Stone, A. J.; Misquitta, A. J. Int. Rev. Phys. Chem. 2007, 26, 193−222.(2) Bukowski, R.; Szalewicz, K.; Chabalowski, C. F. J. Phys. Chem. A1999, 103, 7322−7340.(3) Gresh, N. J. Phys. Chem. A 1997, 101, 8680−8694.(4) Gordon, M. S.; Freitag, M. A.; Bandyopadhyay, P.; Jensen, J. H.;Kairys, V.; Stevens, W. J. J. Phys. Chem. A 2000, 105, 293−307.(5) Misquitta, A. J.; Welch, G. W. A.; Stone, A. J.; Price, S. L. Chem.Phys. Lett. 2008, 456, 105−109.(6) Gresh, N.; Cisneros, G. A.; Darden, T. A.; Piquemal, J.-P. J. Chem.Theory Comput. 2007, 3, 1960−1986.(7) Dzubak, A. L.; Lin, L.-C.; Kim, J.; Swisher, J. A.; Poloni, R.;Maximoff, S. N.; Smit, B.; Gagliardi, L. Nat. Chem. 2012, 4, 810−816.(8) Jeziorski, B.; Moszynski, R.; Szalewicz, K. Chem. Rev. 1994, 94,1887−1930.(9) Bukowski, R.; Podeszwa, R.; Szalewicz, K. Chem. Phys. Lett. 2005,414, 111−116.(10) Hesselmann, A.; Jansen, G. Chem. Phys. Lett. 2002, 362, 319−325.(11) Hesselmann, A.; Jansen, G. Chem. Phys. Lett. 2002, 357, 464−470.(12) Hesselmann, A.; Jansen, G. Chem. Phys. Lett. 2003, 367, 778−784.(13) Hesselmann, A.; Jansen, G. Phys. Chem. Chem. Phys. 2003, 5,5010−5014.
(14) Hesselmann, A.; Jansen, G.; Schutz, M. J. Chem. Phys. 2005, 122,14103−14119.(15) Misquitta, A. J.; Jeziorski, B.; Szalewicz, K. Phys. Rev. Lett. 2003,91, 33201−33204.(16) Misquitta, A. J.; Podeszwa, R.; Jeziorski, B.; Szalewicz, K. J.Chem. Phys. 2005, 123, 214103−214116.(17) Misquitta, A. J.; Szalewicz, K. Chem. Phys. Lett. 2002, 357, 301−306.(18) Misquitta, A. J.; Szalewicz, K. J. Chem. Phys. 2005, 122, 214109−214127.(19) Podeszwa, R.; Bukowski, R.; Szalewicz, K. J. Chem. TheoryComput. 2006, 2, 400−412.(20) Williams, H. L.; Chabalowski, C. F. J. Phys. Chem. A 2001, 105,646−659.(21) Yu, K.; McDaniel, J. G.; Schmidt, J. R. J. Phys. Chem. B 2011,115, 10054−10063.(22) Yu, K.; Schmidt, J. R. J. Chem. Phys. 2012, 136, 34503−34509.(23) McDaniel, J. G.; Yu, K.; Schmidt, J. R. J. Phys. Chem. C 2011,116, 1892−1903.(24) McDaniel, J. G.; Schmidt, J. R. J. Phys. Chem. C 2012, 116,14031−14039.(25) Morris, W.; Leung, B.; Furukawa, H.; Yaghi, O. K.; He, N.;Hayashi, H.; Houndonougbo, Y.; Asta, M.; Laird, B. B.; Yaghi, O. M. J.Am. Chem. Soc. 2010, 132, 11006−11008.(26) Banerjee, R.; Furukawa, H.; Britt, D.; Knobler, C.; O’Keeffe, M.;Yaghi, O. M. J. Am. Chem. Soc. 2009, 131, 3875−3877.(27) Gordon, M. S.; Fedorov, D. G.; Pruitt, S. R.; Slipchenko, L. V.Chem. Rev. 2011, 112, 632−672.(28) Gresh, N.; Claverie, P.; Pullman, A. Theor. Chem. Acc. 1984, 66,1−20.(29) Piquemal, J.-P.; Williams-Hubbard, B.; Fey, N.; Deeth, R. J.;Gresh, N.; Giessner-Prettre, C. J. Comput. Chem. 2003, 24, 1963−1970.(30) Vigne-Maeder, F.; Claverie, P. J. Chem. Phys. 1988, 88, 4934−4948.(31) Garmer, D. R.; Stevens, W. J. J. Phys. Chem. 1989, 93, 8263−8270.(32) Adamovic, I.; Gordon, M. S. Mol. Phys. 2005, 103, 379−387.(33) Mullin, J. M.; Roskop, L. B.; Pruitt, S. R.; Collins, M. A.;Gordon, M. S. J. Phys. Chem. A 2009, 113, 10040−10049.(34) Jensen, J. H.; Gordon, M. S. J. Chem. Phys. 1998, 108, 4772−4782.(35) Day, P. N.; Jensen, J. H.; Gordon, M. S.; Webb, S. P.; Stevens,W. J.; Krauss, M.; Garmer, D.; Basch, H.; Cohen, D. J. Chem. Phys.1996, 105, 1968−1986.(36) Smith, Q. A.; Gordon, M. S.; Slipchenko, L. V. J. Phys. Chem. A2011, 115, 4598−4609.(37) Stone, A. J. Chem. Phys. Lett. 1981, 83, 233−239.(38) Stone, A. J.; Alderton, M. Mol. Phys. 1985, 56, 1047−1064.(39) Wen, S.; Beran, G. J. O. J. Chem. Theory Comput. 2011, 7,3733−3742.(40) Wen, S.; Nanda, K.; Huang, Y.; Beran, G. J. O. Phys. Chem.Chem. Phys. 2012, 14, 7578−7590.(41) Misquitta, A. J.; Stone, A. J. Mol. Phys. 2008, 106, 1631−1643.(42) Bukowski, R.; Sadlej, J.; Jeziorski, B.; Jankowski, P.; Szalewicz, K.J. Chem. Phys. 1999, 110, 3785−3803.(43) Li, X.; Volkov, A. V.; Szalewicz, K.; Coppens, P. Acta Crystallogr.Sect. D 2006, 62, 639−647.(44) Mas, E. M.; Szalewicz, K.; Bukowski, R.; Jeziorski, B. J. Chem.Phys. 1997, 107, 4207−4218.(45) Podeszwa, R.; Bukowski, R.; Rice, B. M.; Szalewicz, K. Phys.Chem. Chem. Phys. 2007, 9, 5561−5569.(46) Podeszwa, R.; Bukowski, R.; Szalewicz, K. J. Phys. Chem. A 2006,110, 10345−10354.(47) Tang, K. T.; Toennies, J. P. J. Chem. Phys. 1984, 80, 3726−3741.(48) Perdew, J. P.; Burke, K.; Ernzerhof, M. Phys. Rev. Lett. 1996, 77,3865−3868.(49) Dunning, T. H. J. Chem. Phys. 1989, 90, 1007−1023.
The Journal of Physical Chemistry A Feature Article
dx.doi.org/10.1021/jp3108182 | J. Phys. Chem. A 2013, 117, 2053−20662065

(50) Werner, H.-J.; Knowles, P. J.; Lindh, R.; Manby, F. R.; Schutz,M.; Celani, P.; Korona, T.; Mitrushenkov, A.; Rauhut, G.; Adler, T. B.;et al. MOLPRO, a package of ab initio programs, version 2009.1; seehttp://www.molpro.net.(51) Ferenczy, G. G. J. Comput. Chem. 1991, 12, 913−917.(52) Totton, T. S.; Misquitta, A. J.; Kraft, M. Chem. Phys. Lett. 2011,510, 154−160.(53) Williams, G. J.; Stone, A. J. J. Chem. Phys. 2003, 119, 4620−4628.(54) Misquitta, A. J.; Stone, A. J. J. Chem. Theory Comput. 2008, 4, 7−18.(55) Misquitta, A. J.; Stone, A. J.; Price, S. L. J. Chem. Theory Comput.2008, 4, 19−32.(56) See also http://www-stone.ch.cam.ac.uk/programs.html#CamCASP.(57) Stone, A. J. The Theory of Intermolecular Forces; ClarendonPress: Oxford, U.K., 1996.(58) Williams, H. L.; Mas, E. M.; Szalewicz, K.; Jeziorski, B. J. Chem.Phys. 1995, 103, 7374−7391.(59) Rick, S. W.; Stuart, S. J. Rev. Comput. Chem. 2002, 18, 89−146.(60) Dymond, J. H.; Smith, E. B. The Virial Coefficients of Pure Gasesand Mixtures; Oxford University Press: New York, 1980.(61) Harismiadis, V. I.; Szleifer, I. Mol. Phys. 1994, 81, 851−866.(62) Kaminski, G.; Duffy, E. M.; Matsui, T.; Jorgensen, W. L. J. Phys.Chem. 1994, 98, 13077−13082.(63) Jorgensen, W. L.; Maxwell, D. S.; Tirado-Rives, J. J. Am. Chem.Soc. 1996, 118, 11225−11236.(64) Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H. J. C. J. Comput. Chem. 2005, 26, 1701−1718.(65) Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. J. Chem.Theory Comput. 2008, 4, 435−447.(66) Massucci, M.; du’Gay, A. P.; Diaz-Laviada, A. M.; Wormald, C. J.J. Chem. Soc., Faraday Trans. 1992, 88, 427−432.(67) Tsonopoulos, C.; Dymond, J. H.; Szafranski, A. M. Pure Appl.Chem. 1989, 61, 1387−1394.(68) Boublikova, L.; Lu, B. C. Y. J. Appl. Chem. 1969, 19, 89−92.(69) McElroy, P. J.; Hashim, H.; Tatt, W. L. AIChE J. 1983, 29,1007−1010.(70) Coto, B.; Pando, C.; Rubio, R. G.; Renuncio, J. A. R. J. Chem.Soc., Faraday Trans. 1995, 91, 273−278.(71) Mueller, T.; Ceder, G. J. Phys. Chem. B 2005, 109, 17974−17983.(72) Essmann, U.; Perera, L.; Berkowitz, M. L.; Darden, T.; Lee, H.;Pedersen, L. G. J. Chem. Phys. 1995, 103, 8577−8593.(73) https://schmidt.chem.wisc.edu/.(74) Millward, A. R.; Yaghi, O. M. J. Am. Chem. Soc. 2005, 127,17998−17999.(75) Walton, K. S.; Millward, A. R.; Dubbeldam, D.; Frost, H.; Low,J. J.; Yaghi, O. M.; Snurr, R. Q. J. Am. Chem. Soc. 2008, 130, 406−407.(76) Myers, A. L.; Monson, P. A. Langmuir 2002, 18, 10261−10273.(77) Litzkow, M.; Livney, M.; Mutka, M. Condor - A Hunter of IdleWorkstations; IEEE: New York, 1988.
The Journal of Physical Chemistry A Feature Article
dx.doi.org/10.1021/jp3108182 | J. Phys. Chem. A 2013, 117, 2053−20662066