physikalische chemie 1: das chemische gleichgewicht · physikalische chemie 1: das chemische...
TRANSCRIPT

Physikalische Chemie 1:
Das Chemische Gleichgewicht
F. Temps
Institut für Physikalische ChemieChristian-Albrechts-Universität zu KielOlshausenstr. 40D-24098 KielE-Mail: [email protected]: http://www.temps.phc.uni-kiel.de
Sommersemester 2018
Dieses Skript gehört zur Vorlesung “Physikalische Chemie 1: Das
Chemische Gleichgewicht” im Sommersemester 2018 am Institut für
Physikalische Chemie der Christian-Albrechts-Universität zu Kiel. Die
Vorlesung gibt eine Einführung in die Chemische Thermodynamik. Die
Thermodynamik bietet, obwohl ursprünglich “nur” zum Verständnis
der Wirkungsgrade von Wärmekraftmaschinen entwickelt, auch die
Basis für das Verständnis Chemischer Gleichgewichte; wir verstehen sie
deshalb als die “Lehre vom Chemischen Gleichgewicht”. Die Vorlesung
Physikalische Chemie 1 (PC-1) ist die erste des 3-semestrigen Kieler
Vorlesungszyklus “Physikalische Chemie für Studierende der Chemie
(B.Sc.) und Wirtschaftschemie (B.Sc.)”. PC-1 ist außerdem für
Studierende der Materialwissenschaften (B.Sc.) bestimmt. Der Umfang
beträgt 3 SWS für die Vorlesung und 1 SWS für Übungen; bei Erfüllen
der Anforderungen werden 6 Leistungspunkte vergeben.
c° F. Temps (2006 — 2018) Aktualisiert am: 5. Juli 2018

ii
Inhaltsverzeichnis
Vorwort viii
Einleitung, Organisatorisches, Literaturangaben x
Organisatorisches xi
Literaturangaben xiv
Gegenstand dieser Vorlesung xvi
1 Stoffzustände, Zustandsgleichungen, Zustandsdiagramme 1
1.1 Gase und Gasgesetze 2
1.1.1 Zustandsgleichung idealer Gase 2
1.1.2 Zustandsänderungen idealer Gase 9
1.1.3 Messung der Zustandsgrößen und 10
1.1.4 Molekulare Interpretation des idealen Gasgesetzes (Kinetische Gastheorie) 13
1.1.5 Gasmischungen 16
1.1.6 Reale Gase 19
1.2 Zustandsdiagramme reiner Stoffe 24
1.2.1 Phasenumwandlungen eines Stoffes 24
1.2.2 - -Zustandsdiagramme ( = const.) 25
1.2.3 Beispiele für - -Phasendiagramme 29
1.2.4 - -Zustandsdiagramme ( = const.): 32
1.2.5 Die van der Waals-Gleichung 34
1.2.6 Kombination des - - und des - -Diagramms in einer 3D-Darstellung 36
1.2.7 Das Theorem der übereinstimmenden Zustände 37
1.2.8 Gesetzmässigkeiten für Phasengleichgewichte 38
2 Der Erste Hauptsatz der Thermodynamik 44
2.1 Thermodynamische Systeme 44
2.2 Volumenarbeit 45
2.2.1 Arten von Arbeit 45
2.2.2 Volumenarbeit 45
2.3 Der Erste Hauptsatz der Thermodynamik 49
2.3.1 Experiment von Joule 49
2.3.2 Formulierung des ersten Hauptsatzes der Thermodynamik 50

iii
2.3.3 Messvorschrift für ∆ 50
2.3.4 Messvorschrift für 52
2.3.5 Temperaturabhängigkeit der Inneren Energie: Spezifische Wärme bei konstantem
Volumen 53
2.4 Anwendungen des Ersten Hauptsatzes 54
2.4.1 Gay-Lussac-Versuch 54
2.4.2 Reversible isotherme Expansion eines idealen Gases 56
2.4.3 Irreversible isobare Expansion eines idealen Gases 57
2.4.4 Adiabatische Expansion/Kompression eines idealen Gases: Adiabatengleichung 58
2.5 Die Enthalpie 62
2.5.1 Funktionsprinzip eines Bombenkalorimeters (Reaktionen bei = const) 62
2.5.2 Prozesse bei = const. 63
2.5.3 Die Enthalpie 64
2.5.4 Temperaturabhängigkeit der Enthalpie: Spezifische Wärme bei konstantem Druck 64
2.5.5 Vergleich von Reaktionen bei = const. und = const. 65
2.6 Anwendung auf chemische Reaktionen 68
2.6.1 Hess’scher Satz 68
2.6.2 Standardbildungsenthalpien der Elemente 69
2.6.3 Standardbildungsenthalpien anderer chemischer Verbindungen 70
2.6.4 Standardbildungsenthalpien und Entropien chemischer Verbindungen 71
2.6.5 Standardreaktionsenthalpien 74
2.6.6 Temperaturabhängigkeit der Reaktionsenthalpie 75
2.6.7 Bindungsenthalpien (“Bindungsenergien”) ª298 75
2.6.8 Gitterenthalpie von NaCl 76
2.6.9 Hydratationsenthalpie von NaCl 77
2.6.10 Hydratationsenthalpie des H+- und des Cl−-Ions 77
2.6.11 Verbrennungsenthalpien 78
2.6.12 Berechnung der adiabatischen Flammentemperatur 78
2.7 Der Joule-Thomson-Effekt∗ 81
2.8 Stoßwellen∗ 84
3 Der Zweite Hauptsatz der Thermodynamik 85
3.1 Grundlegende Experimente und Befunde zum 2. Hauptsatz 85
3.2 Der Zweite Hauptsatz der Thermodynamik 87

iv
3.3 Anwendungen des zweiten Hauptsatzes 88
3.3.1 Carnot’scher Kreisprozess 88
3.3.2 Wirkungsgrade verschiedener idealer Wärmemaschinen 91
3.3.3 Gay-Lussac-Versuch 95
3.3.4 Temperaturausgleich 96
3.3.5 Standardbildungsentropien ƻ298 bezogen auf die Elemente 97
3.3.6 -Abhängigkeit von 97
3.3.7 ∆ für Phasenumwandlungen 98
3.3.8 Thermodynamische Wirkungsgrade praktischer Wärmekraftmaschinen* 98
3.4 Der Dritte Hauptsatz der Thermodynamik 109
3.4.1 Formulierungen des dritten Hauptsatzes 109
3.4.2 Absolute Entropien ª298 110
3.4.3 Anwendung auf chemische Reaktionen 110
3.5 Behandlung beliebiger Prozesse nach dem 2. Hauptsatz 112
3.5.1 Beispiel zur Berechnung von ∆gesamt für beliebige Prozesse 113
3.5.2 Gleichgewichtsbedingung für Systeme bei = const. und = const. 115
3.6 Gleichgewichtsbedingungen für verschiedene Prozesse 117
3.6.1 Innere Energie für Prozesse bei = const. 117
3.6.2 Entropie für Prozesse bei = const. 118
3.6.3 Enthalpie für Prozesse bei = const. 118
3.6.4 Gibbs-Energie (= “Freie Enthalpie” ) für Prozesse bei = const. 119
3.6.5 Helmholtz-Energie (= “Freie Energie” ) für Prozesse bei = const. 120
3.6.6 Merkschema (und Merksprüche . . . ) 121
4 Chemisches Gleichgewicht 123
4.1 Partielle molare Größen 123
4.2 Das chemische Potential 124
4.3 Gleichung von Gibbs-Duhem 125
4.4 Thermodynamische Gleichgewichtsbedingung für chemische Reaktionen 126
4.5 Druck-, Konzentrations- und Temperaturabhängigkeit des chemischen Potentials 129
4.5.1 Druckabhängigkeit des chemischen Potentials für Gase ( = Partialdruck) 129
4.5.2 Konzentrationsabhängigkeit des chemischen Potentials in (nichtwässrigen) Lösungen∗130
4.5.3 Konzentrationsabhängigkeit des chemischen Potentials für Elektrolytlösungen
(wässrige Lösung)∗ 131

v
4.5.4 Temperaturabhängigkeit des chemischen Potentials 132
4.6 Homogene Gas-Reaktionsgleichgewichte 133
4.6.1 Beispiel einer homogenen Gasreaktion 133
4.6.2 Allgemeine Ableitung des Massenwirkungsgesetzes für Gasreaktionen 134
4.6.3 Kinetische Ableitung des MWG 135
4.6.4 Dimensionsbetrachtungen 135
4.6.5 Beispiele zur Berechnung homogener Gasgleichgewichte 137
4.7 Bestimmung thermodynamischer Daten aus Gleichgewichtsmessungen 142
4.8 Heterogene Reaktionsgleichgewichte 144
4.8.1 Die Reaktion CaCO3 ()À CaO()+ CO2 () 144
4.8.2 Die Reaktion H2 () +12O2 À (H2O ())À H2O() 146
4.8.3 Die Reaktion NH4Cl() À NH3 () + HCl() 147
4.8.4 Die Reaktion CO2 ()+ C() À 2CO() (Boudouard-Gleichgewicht) 148
4.8.5 Konsequenzen für die Reduktion von FeO zu Fe (Hochofenprozess) 151
4.8.6 Stabilität von Kohlenwasserstoffen 154
4.9 Zusammenstellung wichtiger Gleichungen für homogene und heterogene Gleichgewichte 156
5 Phasengleichgewichte reiner Stoffe 158
5.1 Qualitative Behandlung von Phasengleichgewichten anhand des chemischen Potentials 158
5.2 Die Gleichungen von Clapeyron und Clausius-Clapeyron 161
5.3 Druckabhängigkeit des Dampfdrucks∗ 163
5.4 Dampfdruck kleiner Tröpfchen∗ 164
5.5 Ehrenfest-Klassifikation der Phasenübergänge 166
6 Thermodynamik von Lösungen und Mischungen 167
6.1 Die Grenzgesetze von Raoult und Henry für ideale bzw. ideal verdünnte Lösungen 168
6.1.1 Ideale Lösungen 168
6.1.2 Raoult’sches Gesetz 168
6.1.3 Henry’sches Gesetz 169
6.2 Konzentrationsabhängigkeit des chemischen Potentials in Lösung 170
6.3 Thermodynamische Mischungsgrößen 171
6.4 Kolligative Eigenschaften 174
6.4.1 Rückblick auf die Gesetze von Raoult und Henry 174
6.4.2 Nernst’scher Verteilungssatz 175

vi
6.4.3 Osmotischer Druck 176
6.4.4 Dampfdruckerniedrigung des Lösungsmittels 178
6.4.5 Gefrierpunktserniedrigung: 178
6.4.6 Siedepunktserhöhung 180
6.5 Behandlung realer Lösungen 182
6.5.1 Aktivitäten 182
6.5.2 Chemisches Potential und Exzess-Größen 182
6.5.3 Experimentelle Bestimmung von Aktivitätskoeffizienten 184
6.6 Destillation und Rektifikation 187
6.6.1 Dampfdruckdiagramm 187
6.6.2 Siedediagramm 188
6.6.3 Dampf-Flüssigkeits-Diagramme (0 − 0-Diagramme) 188
6.6.4 Rektifikation 190
6.6.5 Wasserdampfdestillation 193
6.7 Phasendiagramme binärer Mischungen (Beispiele) 195
6.8 Die Gibbs’sche Phasenregel 197
6.9 Berechnung von Reaktionsgleichgewichten in Lösung 198
6.9.1 Löslichkeitsprodukt eines schwerlöslichen Salzes in Wasser 198
6.9.2 Autoprotolyse und Ionenprodukt von H2O 199
6.9.3 Dissoziation einer schwachen Säure 199
6.9.4 pH-Wertberechnungen für schwache Säuren und Basen 200
6.9.5 Puffer 200
7 Gleichgewichtselektrochemie 201
7.1 Beispiele und Konventionen 202
7.1.1 Beispiele für elektrochemische Zellen (elektrochemische Ketten) 202
7.1.2 IUPAC-Konventionen (1953) 205
7.1.3 Durchführung von EMK-Messungen 206
7.1.4 Reversibel arbeitende elektrochemische Zellen 207
7.2 Kurze Zusammenfassung von Ergebnissen der Debye-Hückel-Theorie 208
7.2.1 Thermodynamischer Standardzustand für (wässrige) Elektrolytlösungen 208
7.2.2 Aktivitäten und Aktivitätskoeffizienten 209
7.3 Die elektrochemische Gleichgewichtsbedingung 210

vii
7.3.1 Elektrische und chemische Arbeit 210
7.3.2 Elektrochemische Gleichgewichtsbedingung 211
7.4 Die Nernst’sche Gleichung 212
7.4.1 Allgemeine Herleitung der Nernst’schen Gleichung 212
7.4.2 Daniell-Element 212
7.4.3 Chlor-Wasserstoff-Zelle 213
7.5 Standard-Elektrodenpotentiale 215
7.6 Bestimmung thermodynamischer Daten aus EMK-Messungen 217
7.7 Zusammenstellung elektrochemischer Zellen 218
8 Einführung in die Statistische Thermodynamik 222
8.1 Der Boltzmann-Ausdruck = ln 222
8.1.1 Begriffe 222
8.1.2 Ausgleichsprozesse I: Diffusion in Kristallen 222
8.1.3 Ausgleichsprozesse II: Gase 226
8.2 Die Boltzmann-Verteilung 231
8.2.1 Wahrscheinlichkeit für Makrozustand eines Gases bezgl. Verteilung auf
Energiezustände 231
8.3 Die Zustandssumme 233
8.3.1 Mittlere Energie eines Atoms oder Moleküls 233
8.3.2 Molekülzustandssummen und Systemzustandssummen 235
8.4 Berechnung thermodynamischer Größen aus Zustandssummen 237
8.5 Chemisches Gleichgewicht 238
8.6 Ergebnisse der Quantenmechanik 240
8.6.1 Zustandssumme für elektronische Zustände 240
8.6.2 Zustandssumme für die Schwingungsbewegung 240
8.6.3 Zustandssumme für die Rotationsbewegung 242
8.6.4 Zustandssumme für die Translationsbewegung 243
244
A Das griechische Alphabet 245
B Physikalisch-Chemische Konstanten 246
C Nützliche Umrechnungsfaktoren 247
D Lineare Regression 248
E Die Methode der Lagrange’schen Multiplikatoren zur Bestimmung des Maximums einer
Funktion unter Nebenbedingungen 249

viii
Vorwort
Die Vorlesung “Physikalische Chemie 1: Das Chemische Gleichgewicht” (PC-1) ist dieerste des dreisemestrigen Vorlesungszyklus für Studierende der Chemie (B.Sc.) undWirtschaftschemie (B.Sc.) am Institut für Physikalische Chemie der Christian-Albrechts-Universität zu Kiel. Sie ist außerdem für den Studiengang Materialwissenschaften(B.Sc.) vorgesehen. Der Umfang beträgt 3 SWS für die Vorlesung und 1 SWS fürÜbungen. Bei Erfüllung der Anforderungen werden 6 Leistungspunkte vergeben.
Thema der Vorlesung ist die “Chemische Thermodynamik”, die Lehre vom Chemis-chen Gleichgewicht. Die Thermodynamik wurde ursprünglich zu Beginn der Industri-alisierung im 19. Jahrhundert als “Wärmelehre” zur Beschreibung des Wirkungsgradsvon Wärmekraftmaschinen entwickelt. Schon bald zeigte sich aber, dass der Formalis-mus allgemeingültig ist. In der Chemie interessiert uns dabei besonders die quantitativeBeschreibung der Stoffzustände und Stoffumwandlungen. Welchen Zustand nimmt einStoff im Gleichgewicht ein? Welcher Endzustand stellt sich in einer chemischen Reak-tion ein? Wieviel Energie wird von einem bestimmten Ausgangszustand ausgehend frei,welcher Anteil dieser Energie ist nutzbar, und welcher wird als Wärme an die Umgebungabgegeben? Welches Potential bieten alternative “Energiequellen”? Welche prinzipi-ellen — thermodynamisch nicht überwindbaren — Grenzen sind uns bei ihrer Nutzunggesetzt? Oder wieviel Energie muss aufgewendet werden, um einen gewünschten Zu-stand zu erreichen? Was passiert bei einer Änderung der äußeren Bedingungen, z.B.der Temperatur ( ) oder des Druckes (), welche Auswirkung hat eine Änderung von oder von auf die Lage des Gleichgewichts? Welche Auswirkung hat eine Änderungder chemischen Zusammensetzung? Dies sind zentrale, dem Chemiker im Alltag ständigbegegnende Fragen, auf die die Chemische Thermodynamik Antworten liefert. Sie ver-setzt uns somit in die Lage, den chemischen Gleichgewichtsgewichtszustand quantitativzu beschreiben und vorherzusagen. Die dafür benötigten Kenntnisse und Fähigkeitenzu vermitteln ist das Ziel der Vorlesung und Übungen. Ohne fundierte Kenntnisse derThermodynamik kommt kein Chemiker aus.
Die Physikalische Chemie fällt vielen Studierenden erfahrungsgemäß am Anfang schwer.Sie ist “ungewohnt”, “unanschaulich”, “wirkt fremd”, “man sieht nicht, wozu das gutsein soll”, sie ist “zu formal” oder “zu mathematisch” sind oft gehörte Kommentare. Essei jedoch versichert, dass mit der Zeit eine Gewöhnung an die Vorgehensweise einsetzt.Irgendwann stellen die Studierenden dann auf einmal fest, dass man Chemie “verstehen”und sogar berechnen kann, und nicht nur auswendig lernen muss. Dieses Verständnisder Chemie verlangt allerdings einige mathematische Grundkenntnisse.
Aus Erfahrung fällt die Mathematik vielen Chemiestudierenden schwer. Die benötigtenmathematischen Methoden werden an den entsprechenden Stellen in der Vorlesungund in den Übungen (nicht unbedingt im Skript) kurz wiederholt bzw. mit zunächsteinfachen, dann komplizierteren Beispielen erläutert. Allen, die schnell die ge-rade benötigten mathematischen Grundlagen wiederholen wollen oder die mit derMathematik von jeher Schwierigkeiten hatten, sei nachdrücklich als Ergänzung zueinem ausführlichen Mathematik-Lehrbuch1 das Buch von Barrante empfohlen.2 Zumschnellen Nachschlagen im Internet sei auf die MathWorld-Enzyklopädie verwiesen(http://mathworld.wolfram.com/). Für numerische oder symbolische Rechnungenund schnelle graphische Auftragungen von Funktionen u.v.a.m. bietet Wolfram Alpha
1 z.B. H. G. Zachmann & A. Jüngel, Mathematik für Chemiker, Wiley-VCH, 2007.2 J. R. Barrante, Applied Mathematics for Physical Chemistry, Prentice Hall, 2003.

ix
(http://wolframalpha.com) eine hervorragende Plattform. Außerordentlich nützlich sindzudem Grafik- und Datenauswerteprogramme (z.B. Origin, QtiPlot),3 mit denen SieDatensätze grafisch darstellen und auswerten können, und Computeralgebra-Systeme(z.B. Maxima, Derive, MuPAD, MathCad,4 Maple, Mathematica), mit denen sehr le-icht die Lösungen von Integralen oder Differentialgleichungen, etc., gefunden werdenkönnen oder Funktionen geplottet werden können.
Das vorliegende Skriptum soll den Hörerinnen und Hörern der Vorlesung eine Zusammen-fassung des Vorlesungsstoffs geben, Ihnen in den Vorlesungsstunden die Konzentrationauf den Stoff erlauben und die Vor- und Nachbereitung zu erleichtern. Das Skriptumist aber ausdrücklich kein Ersatz für den Besuch der Vorlesung, in der viele Zusam-menhänge gründlicher erläutert werden. Genauso wird ausdrücklich unterstrichen, dassdas Skriptum kein Ersatz für ein Lehrbuch ist. Das Selbststudium neben der Vorlesungmit Hilfe eines Lehrbuchs ist unverzichtbar! Einige Abschnitte, die in der Vorlesung ausZeitgründen nur knapp oder gar nicht behandelt werden können, sind deshalb absichtlichselbstständig zu erarbeiten (“reading assignments”).5 Fragen zu dem selbstständig zuerarbeitenden Stoff können (sollten!) in den Übungsstunden besprochen werden, dadieser Stoff ebenfalls prüfungsrelevant ist. Stoff ausgegebener “reading assignments”wird in der Vorlesung anschließend als bekannt vorausgesetzt.
Kiel, im April 2018, F. Temps
3 Excel bietet auch einige Möglichkeiten dafür, ist jedoch für ernsthafte wissenschaftliche Anwendun-
gen kaum geeignet.4 MathCAD steht auf Rechnern des Computer-Pools der Chemie zur Verfügung. Für die Vorlesung
wurde Mathematica verwendet.5 Auf die zu lesenden Kapitel oder Unterkapitel wird an den entsprechenden Stellen hingewiesen.

x
Einleitung, Organisatorisches, Literaturangaben
I Abbildung 1: Was ist Physikalische Chemie?
I Abbildung 2: Teilgebiete der Physikalischen Chemie.

xi
I Abbildung 3: Lehrveranstaltungen in der Physikalischen Chemie an der CAU Kiel.
I Modulbeschreibungen: Siehe Webseiten der Sektion Chemie unter
http://www.chemie.uni-kiel.de/de/informationen-fuer-studierende
Organisatorisches
I Tabelle 1: Teilnehmerliste (Teilnehmerstatistik).
Studienfach TeilnehmerInnen
Chemie (B.Sc.) 77Wirtschaftschemie (B.Sc.) 35Biochemie (B.Sc.) —Chemie (B.Sc.-Zweifach) —Materialwissenschaft (B.Sc.) 34Wi-Ing. Materialwissenschaft (B.Sc.) 48Geowissenschaften (B.Sc.) —Physik (B.Sc.) —Andere
Summe 194

xii
I Abbildung 4: Vorlesungs- und Übungstermine.
I Abbildung 5: Leistungsanforderungen.

xiii
I Abbildung 6: ECTS-Kreditpunkte.

xiv
Literaturangaben
I Abbildung 7: Literatur zur Vorlesung (I).
I Abbildung 8: Literatur zur Vorlesung (II).

xv
I Abbildung 9: Computerausstattung, empfohlene Software.
xvi
Gegenstand dieser Vorlesung
I Abbildung 10: Chemische Thermodynamik = Lehre vom Chemischen Gleichgewicht.
I Abbildung 11: Zentrale Fragen in der Chemischen Thermodynamik.

xvii
I Abbildung 12: Physikalisch-Chemische Prozesse.
I Abbildung 13: Übersicht über den Vorlesungsstoff des Moduls PC-1.
xviii
I Quantitatives Verständnis, Vorhersagbarkeit und Berechnung chemischerZustände (Gleichgewichte), chemischer Zustandsänderungen und chemischerReaktionen:
• Wir werden in dieser Vorlesung zahlreiche Formeln und Gleichungen (oft Differ-entialgleichungen) herleiten und auf chemische Prozesse anwenden.
• Vorlesung erfordert mathematische Grundkenntnisse (Modul Mathematik fürChemiker 1 oder vergleichbare Vorlesung)!
• Wiederholen Sie die relevanten Kapitel der Mathematikvorlesung bei Bedarf!
I Abbildung 14: Griechisches Alphabet.
Die Kenntnis des kleinen und großen griechischen Alphabets wird vorausgesetzt!

1
1. Stoffzustände, Zustandsgleichungen, Zustandsdiagramme
Ziel der Physikalischen Chemie ist die quantitative Beschreibung der Zustände derMaterie. Betrachten wir zunächst ganz grob die drei grundsätzlich möglichen Aggre-gatzustände der Materie, so können wir diese wie folgt charakterisieren:
I Gase:
• Zufällige Orts- und Geschwindigkeitsverteilung der Gasatome/-moleküle im Raum,• Keine (ideales Gas) bzw. nur sehr schwache (reale Gase) Wechselwirkungen (WW)der Gasatome/-moleküle miteinander,
• Abstand der Moleküle À Molekul,
• Quantitative Beschreibung mit einfacher Zustandsgleichung (ideales Gasggesetz).
I Flüssigkeiten:
• Abstand der Moleküle ≈ Molekul,
• Anziehende WW halten Moleküle zusammen,
• Nahordnung (nächste Nachbarn), aber keine Fernordnung,• Komplizierte Zustandsgleichung.
I Festkörper:
• Abstand der Moleküle ≈ Molekul,
• Regelmäßige Anordnung der Atome/Moleküle (Kristalle),• Fernordnung.
Wir wenden uns zunächst der physikalisch-chemischen Beschreibung der Zustände vonGasen zu, die wir mit einer einfachen Zustandsgleichung (ideale Gasgleichung) sehr gutbeschreiben können. Die quantitative Beschreibung der Zustände von Festkörpern undvor allem Flüssigkeiten ist demgegenüber sehr viel schwieriger (Struktur und Dynamikvon Flüssigkeiten sind Gegenstand hochaktueller Forschung).

1.1 Gase und Gasgesetze 2
1.1 Gase und Gasgesetze
1.1.1 Zustandsgleichung idealer Gase
I Voraussetzungen für Ideales Gas:
(1) Das Eigenvolumen der Gasteilchen ist vernachlässigbar, d.h. die Gasteilchenwerden als punktförmig angenommen (die allerdings miteinander stoßen könnenund dabei kinetische Energie austauschen können, damit sich thermisches Gleich-gewicht einstellen kann).
(2) Es gibt (außer beim Stoß) keine Wechselwirkung der Gasteilchen un-tereinander, d.h. die Gasteilchen besitzen nur kinetische Energie, aber keinepotentielle Energie (keine anziehenden oder abstoßenden WW zwischen denGasteilchen).
I Von realen Gasen werden diese Bedingungen im Grenzfall p→ 0 und T →∞erfüllt:
→ 0 y Teilchenabstand≫ Teilchenradius (1.1)
→ ∞y kin≫ pot (1.2)
I Das ideale Gasgesetz: Empirisch (durch Messungen) wurde gefunden, dass für einideales Gas gilt
= (1.3)
mit
= Druck,
= Volumen,
= Gastemperatur (absoluteTemperatur)
= Molzahl
= universelle Gaskonstante
Gl. 1.3 ist bekannt als das ideale Gasgesetz.
I Molzahl:
=
=eingewogene Masse
Molmasse(1.4)
I Zustandsgrößen:
• Wir bezeichnen Größen wie , , und , die den Zustand eines Systemsbeschreiben, als Zustandsgrößen.

1.1 Gase und Gasgesetze 3
I Zustandsgleichungen:
• Gleichungen, wie z.B. Gl. 1.3, die den Zusammenhang zwischen den Zustands-größen beschreiben, heißen Zustandsgleichungen.
I Was besagt das ideale Gasgesetz?
• Gl. 1.3 ist die Zustandsgleichung für das ideale Gas. Wir bezeichnen diese Zu-standsgleichung als ideale Gasgleichung oder ideales Gasgesetz.
• Der Zustand des Gases wird beschrieben durch vier Zustandsgrößen: .• Das Gas nimmt das gesamte zur Verfügung stehende Volumen ein.
• Die Zustandsgleichung 1.3 besagt, dass nur drei der vier Größen
voneinander unabhängig einstellbar sind, die vierte ist über die Zustandsgleichungfestgelegt.
• Die ideale Gasgleichung 1.3 ist unabhängig von der Stoffart (chemische Formel)des Gases! D.h., sie gilt (im Rahmen der Näherung des idealen Gases) für alleGase, z.B. He, H2, N2, O2, Xe, CH4, H2O(), UF6, . . .
• Gl. 1.3 ist eine besonders einfache Zustandsgleichung. Wir werden später kom-pliziertere Zustandsgleichungen kennenlernen (z.B. für reale Gase).
• Wert der universellen Gaskonstante : = 8314 462 1 Jmol−1K−1 (1.5)
I Naturkonstanten: Die aktuellen (international akzeptierten) Werte der physikalisch-chemischen Naturkonstanten (“The 2014 CODATA Fundamental Physical Constants”)sowie die akzeptierten Umrechungsfaktoren zwischen SI- und Nicht-SI-Einheiten findetman im WWW unter folgenden Links:
• http://physics.nist.gov/cuu/index.html• http://physics.nist.gov/cuu/units/index.html
In diesem Zusammenhang sei auch eine weitere nützliche Referenzquelle erwähnt, näm-lich das IUPAC Gold Book.6
I Physikalisch-chemische Einheiten: Wir werden uns bemühen, möglichst immer SI-Einheiten zu verwenden. Für die Energie ist die SI-Einheit das Joule:
1 J = 1Nm = 1kgm2 s−2 (1.6)
Allerdings sind mitunter andere Einheiten praktisch, außerdem sind in der angelsächsis-chen Literatur oft Nicht-SI-Einheiten gebräuchlich (z.B kcal statt kJ), sodass wir dieUmrechnung der Einheiten kennen müssen.7
6 IUPAC Compendium on Chemical Terminology, http://goldbook.iupac.org.7 Eine Einführung in die SI-Einheiten gibt folgendes Buch: K.-H. Homann (Ed.), Größen, Ein-
heiten und Symbole in der Physikalischen Chemie, VCH, Weinheim, 1995. Siehe außerdem unter
http://physics.nist.gov/cuu/units/index.html.

1.1 Gase und Gasgesetze 4
I Wichtige Umrechnungsfaktoren:
• Es gibt mehrere verschiedene Definitionen für die Kalorie. Dieser Umrechnungs-faktor gilt für die International Steam Table Calorie (1956):
1 calIT = 41868 J (1.7)
• Umrechungsfaktor für die thermochemische Kalorie:1 calth = 4184 J (1.8)
• SI-Einheit für den Druck:
1Pa = 1Nm−2 = 1kgm
s2× 1
m2= 1kgm−1 s−2 (1.9)
• Andere Druckeinheiten und Umrechnungsfaktoren:1 bar = 105 Pa (1.10)
1 atm = 1013 25× 105 Pa (1.11)
= 1013 25 bar (1.12)
= 760 torr (1.13)
1 torr = 1333 22mbar (1.14)
I Wert der Gaskonstanten in verschiedenen Einheiten:
= 8314 462 1J
molK(1.15)
= 8314 462 1m3 Pa
molK(1.16)
= 83144 621cm3 bar
molK(1.17)
= 82057 4cm3 atm
molK(1.18)
= 62 364cm3 torr
molK(1.19)
= 1987 20cal
molK(1.20)
I Molvolumen des idealen Gases: Als Molvolumen wird das Volumen von 1mol einesStoffes bezeichnet.
Da das ideale Gasgesetz unabhängig von der Stoffart gilt, ist das Molvolumen für alleGase gleich, abhängig nur von und , z.B.:
• = 1mol = 27315K = 1atm = 101325 kPa:
=
= 22414 0
l
mol(1.21)

1.1 Gase und Gasgesetze 5
• = 1mol = 29815K = ª = 1bar = 105 Pa = ª:
ª =
ª
ª= 24789 6
l
mol(1.22)
Letzterer Zustand wird in der Physikalischen Chemie für viele Rechnungenals Referenz- oder Standardzustand (STP) verwendet und mit dem Index ª
gekennzeichnet.8
I Ableitung der idealen Gasgleichung: Die ideale Gasgleichung lässt sich aus dreiempirisch gefundenen einfacheren Gasgesetzen ableiten (⇒ Übungsaufgabe):
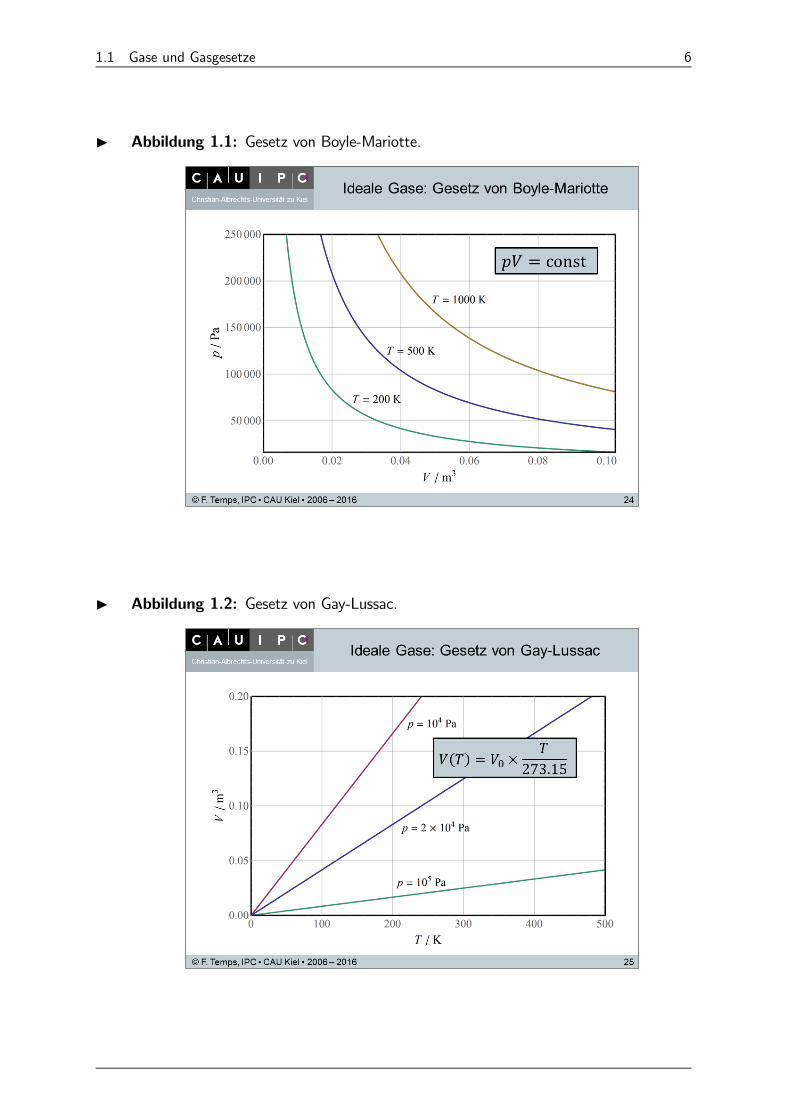
(1) Gesetz von Boyle-Mariotte (1662 von Robert Boyle, 1676 von EdmeMariotte): Für = const gilt (Abb. 1.1)
= const. (1.23)
(2) Gesetz von Gay-Lussac (1787 von Jacques Charles, 1802 von JosephGay-Lussac): Für = const gilt, mit in C (Abb. 1.2),
() = ( = 0)× 27315 +
27315= 0 ×
27315(1.24)
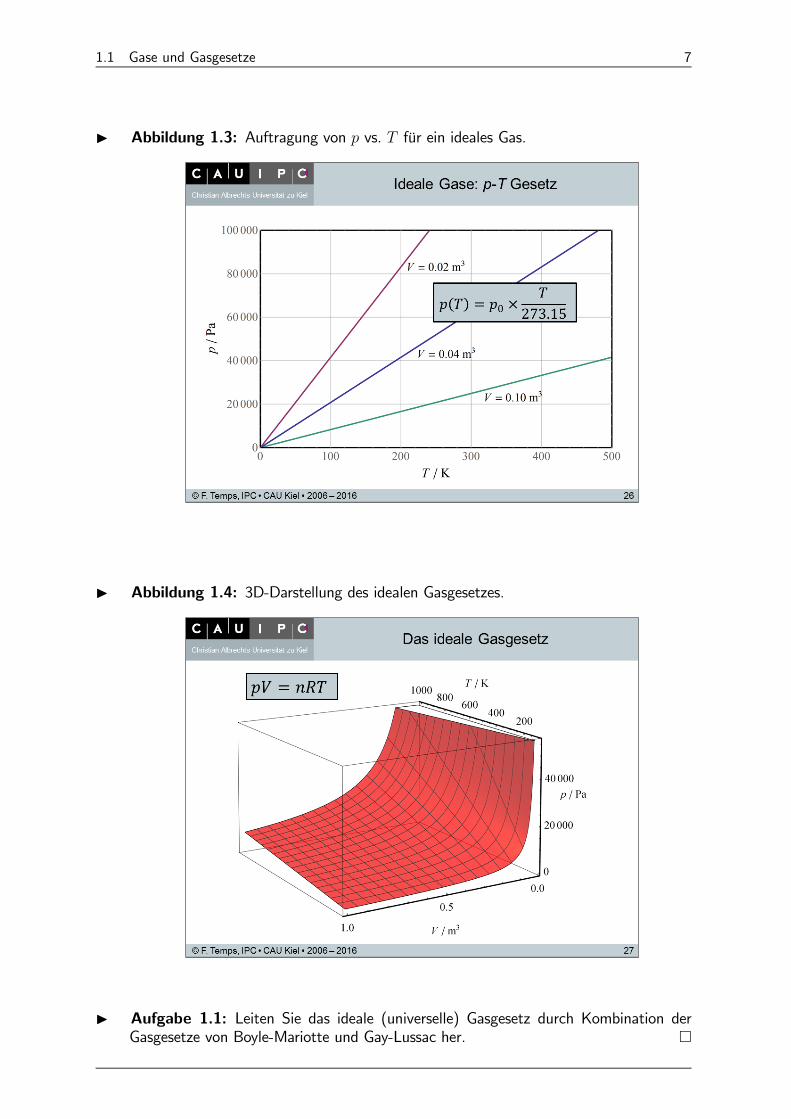
(3) Gesetz für vs. (Guillaume Amontons): Für = const gilt (Abb. 1.3)
() = ( = 0)× 27315 +
27315= 0 ×
27315(1.25)
Abb. 1.4 gibt eine 3D-Darstellung der idealen Gasgleichung.
I Gastemperatur: Aus dem Gay-Lussac’schen Gesetz (Gl. 1.24) resultiert die Defini-tion der Gastemperatur (Einheit = Kelvin, K) als
= ( C+ 27315) K (1.26)
Die Gastemperatur ist, wie wir später sehen werden, identisch mit der absolutenTemperatur.
8 Achtung: In älteren Tabellen, insbesondere in der angelsächsischen Literatur, wird = 1atm als
Standardzustand verwendet. Außerdem werden manchmal andere Temperaturen gewählt; die Tem-
peratur muss daher immer explizit angegeben werden, um Fehler zu vermeiden.
1.1 Gase und Gasgesetze 6
I Abbildung 1.1: Gesetz von Boyle-Mariotte.
I Abbildung 1.2: Gesetz von Gay-Lussac.
1.1 Gase und Gasgesetze 7
I Abbildung 1.3: Auftragung von vs. für ein ideales Gas.
I Abbildung 1.4: 3D-Darstellung des idealen Gasgesetzes.
I Aufgabe 1.1: Leiten Sie das ideale (universelle) Gasgesetz durch Kombination derGasgesetze von Boyle-Mariotte und Gay-Lussac her. ¤

1.1 Gase und Gasgesetze 8
I Lösung 1.1: Wir betrachten = 1mol eines idealen Gases in einem einen beliebi-gen Anfangszustand = . Von ausgehend wollen wir einen beliebigenEndzustand = erreichen. Wir zeichnen zunächst beide Zustände in ein -Diagramm ein (siehe Abb. 1.5). Beide liegen auf jeweiligen Isothermen, die durchdas Boyle-Mariotte’sche Gesetz beschrieben werden. Die Gleichung, die die Zustands-daten von und verknüpft, kennen wir allerdings nicht, da i.A. 6= , 6= , 6= . Zur Lösung des Problems wählen wir deshalb einen Hilfsweg, für den wir dieZustandsänderungen mithilfe der Gesetze von Boyle-Mariotte und Gay-Lussac berechnenkönnen:
(1) Wir führen zunächst eine Isotherme Zustandsänderung vom Zustand zu einemHilfszustand = aus, an dem derselbe Druck wie beim Endzustand herrscht, d.h. = . Diese Zustandsänderung können wir nach Boyle-Mariotteberechnen:
= (1.27)
Da = können wir dies auch schreiben als
=
(1.28)
(2) Vom Punkt erreichen wir den Endzustand durch eine isobare -Erhöhung.Nach Gay-Lussac ( = const für = const) gilt dafür
=
(1.29)
Da = können wir dies auch schreiben als
=
=
(1.30)
(3) Nach Gl. 1.28 gilt damit auch
=
(1.31)
Der Endzustand [ ] kann nun völlig beliebig bewählt werden. Die o.a. Glei-chung 1.31 kann für beliebige Endzustände aber nur dann erfüllt sein, wenn das Ver-
hältnis
eine Konstante ist. Wir nennen diese Konstante :
= (1.32)
d.h. = für 1mol (1.33)
(4) Für mol folgt wegen =
= (1.34)
¥
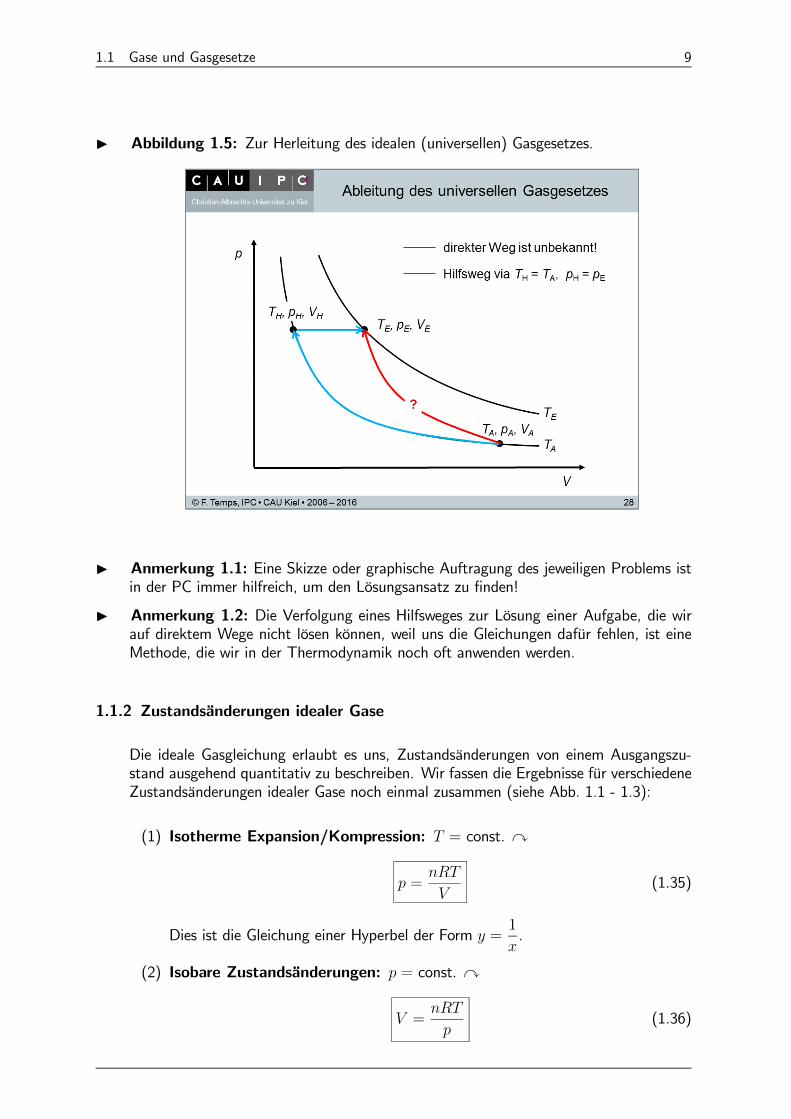
1.1 Gase und Gasgesetze 9
I Abbildung 1.5: Zur Herleitung des idealen (universellen) Gasgesetzes.
I Anmerkung 1.1: Eine Skizze oder graphische Auftragung des jeweiligen Problems istin der PC immer hilfreich, um den Lösungsansatz zu finden!
I Anmerkung 1.2: Die Verfolgung eines Hilfsweges zur Lösung einer Aufgabe, die wirauf direktem Wege nicht lösen können, weil uns die Gleichungen dafür fehlen, ist eineMethode, die wir in der Thermodynamik noch oft anwenden werden.
1.1.2 Zustandsänderungen idealer Gase
Die ideale Gasgleichung erlaubt es uns, Zustandsänderungen von einem Ausgangszu-stand ausgehend quantitativ zu beschreiben. Wir fassen die Ergebnisse für verschiedeneZustandsänderungen idealer Gase noch einmal zusammen (siehe Abb. 1.1 - 1.3):
(1) Isotherme Expansion/Kompression: = const. y
=
(1.35)
Dies ist die Gleichung einer Hyperbel der Form =1
.
(2) Isobare Zustandsänderungen: = const. y
=
(1.36)

1.1 Gase und Gasgesetze 10
(3) Isochore Zustandsänderungen: = const. y
=
(1.37)
(4) Adiabatische Zustandsänderungen: Zustandsänderung mit = 0; d.h. keinWärmeaustausch mit der Umgebung. Dabei ändern sich alle 3 Zustandsgrößen(quantitativer Zusammenhang siehe später); sie gehorchen untereinander aberstets dem idealen Gasgesetz.
1.1.3 Messung der Zustandsgrößen T und p
a) Messung des Volumens V :
Einfach, durch direkte geometrische Längenmessung (Würfel, Quader, Kugel, . . . ). Beiunregelmäßigen Volumina durch Auslitern (Befüllen mit abgemessener Menge Wasser).
b) Temperaturmessung:
Die Messung der Temperatur erfolgt mit einem Thermometer unter Ausnutzung derWärmeausdehnung einer Hg()-Säule.
I Celsius-Temperaturskala:
Fixpunkte definiert durch Phasengleichgewichte von Wasser bei Normaldruck (ª =
105 Pa):
(1) Schmelzpunkt von Eis:Schmelzpunkt = 0
C (1.38)
(2) Siedepunkt von Wasser:Siedepunkt = 100
C (1.39)
I Absolute Temperaturskala (Gastemperatur, Kelvin-Skala):
= ( C+ 27315) K (1.40)
[ ] = K (1.41)
Fixpunkte bei Normaldruck (ª = 105 Pa = 1bar):
(1) Schmelzpunkt von Eis:Schmelz = 27315K (1.42)
(2) Siedepunkt von Wasser:Siede = 37315K (1.43)

1.1 Gase und Gasgesetze 11
I Nullter Hauptsatz der Thermodynamik:
Wenn A mit B im thermischen Gleichgewicht steht und B mit C im thermischenGleichgewicht steht, stehen auch A und C miteinander im thermischen Gleichgewicht.
%. -& (1.44)
À
c) Druckmessung:
Druck =Kraft
Fläche
mitKraft = Masse× Beschleunigung (1.45)
I Definitionsgleichung für den Druck:
=
(1.46)
SI-Einheit für den Druck:
[] = Pa =N
m2=kgm s2
m2=
kg
ms2(1.47)
Nicht-SI-Einheiten für :
1 bar = 105 Pa (1.48)
1mbar = 102 Pa (1.49)
1 atm = 1013 25 bar = 760 torr (1.50)
1 torr = 1333 22mbar (1.51)

1.1 Gase und Gasgesetze 12
I Abbildung 1.6: Hg-Manometer.
Hg() wird verwendet, weil der Dampfdruck (Hg) über der Flüssigkeitssäule sehr klein(und außer bei sehr kleinen Drucken) praktisch vernachlässigbar ist.
I Druckmessung mit Hg-Manometer (Abb. 1.6): Mechanisches Gleichgewicht isteingestellt, wenn der Außendruck gleich ist dem Druck, der von der Hg-Säule auf dieBasisfläche ausgeübt wird.
y =
Hg
(1.52)
mitHg = Hg × = Hg ×× (1.53)
y =
Hg ×× ×
(1.54)
y = × × (1.55)
Hieraus resultiert zunächst die Angabe des Druckes als in “mm Quecksilbersäule”.Da jedoch Hg -abhängig ist, und da die Erdbeschleunigung von der Höhe überdem Meeresspiegel und der geographischen Breite abhängig ist, wird, um vergleichbareWerte zu bekommen, von mm Hg-Säule bei der Messtemperatur auf 0 = 0
C undauf 0m Höhe über dem Meeresspiegel am Erdäquator umgerechnet (Tabellen). Der
daraus resultierende Wert ⊕0 C wird als Druck in torr angegeben:
1 torr = 1mm Hg-Säule @ 0 C, mit = 9806 7m s−2 am Äquator auf Meereshöhe(1.56)

1.1 Gase und Gasgesetze 13
I Standarddruck: Als Bezugspunkt für diverse spätere Definitionen und Konventionenin der Thermodynamik wurde
ª = 105 Pa = 1bar (1.57)
als “Standarddruck” vereinbart.
1.1.4 Molekulare Interpretation des idealen Gasgesetzes (Kinetische Gastheorie)
I Annahmen:
• Ideales Gas aus Teilchen A mit der Teilchendichte =
Teilchen
Volumen (1.58)
• Abstand A−A≫ A,
• potentielle WW-Energie pot = 0,• ungeordnete Teilchenbewegung,• Stöße der Gasteilchen mit Wand und mit anderen Gasteilchen unter Energie- undImpulserhaltung,
• Außerdem vereinfacht:— alle Teilchen haben die gleiche Geschwindigkeit ,
— je1
6der Teilchen fliegen in die Raumrichtungen ±, ±, ±.
I Abbildung 1.7: Zur Berechnung der Anzahl der Wandstöße eines idealen Gases.
Nur die blau gezeichneten Gasteilchen im Volumen treffen in der Zeit auf dieWandfläche .

1.1 Gase und Gasgesetze 14
I Berechnung des Gasdrucks:
• Anzahl der Wandstöße A in der Zeit :Auf die Wandfläche treffen in der Zeit alle Teilchen im Volumen
1
6×× (1.59)
Dies sind, mit der Teilchendichte = , gerade
A = × 16×× (1.60)
Teilchen.
• Impulsänderung der Gasteilchens beim Stoß auf die Wand:
= 2A ×1
6 (1.61)
• Resultierende Kraft: =
(1.62)
• Druck: =
=1
=1
× 2A ×
1
6 (1.63)
y =
1
3A
2 (1.64)
• Für 1mol ( =
mit = 6022 141 29×1023mol−1) folgt mit = 1
22
=1
3A
2 =2
3 (1.65)
Mit = als der mittleren Energie von einem Mol Gasteilchen ergibtsich somit
=2
3 (1.66)
• Der Vergleich mit dem idealen Gasgesetz für = 1mol = (1.67)
liefert für die mittlere Energie von 1mol Gasteilchen das Ergebnis
2
3kin = (1.68)
y
kin =3
2 (1.69)

1.1 Gase und Gasgesetze 15
• Pro Freiheitsgrad (Raumrichtung) nach Gleichverteilungssatz:
(1)
kin =1
2 (1.70)
• Mittlere kinetische Energie eines Gasteilchens:
kin =3
2 =
1
22 (1.71)
mit der Boltzmann-Konstanten
=
= 1380 648 8× 10−23 J−1 (1.72)
y2 =
3
(1.73)
bzw. p2 =
r3
(1.74)
I Achtung, Fehlerkompensation: In der o.a. Ableitung haben wir zwei Fehlergemacht:
(1) Es ist nicht richtig, dass je1
6der Teilchen Geschwindigkeiten in ±-, ±-, ±-
Richtung haben. In Wirklichkeit liegt eine Verteilung in 3D vor!
(2) Anstelle der angenommenen mittleren Geschwindigkeit von liegt eineGeschwindigkeitsverteilung () vor.
Diese zwei in der o.a. Ableitung gemachten Fehler kompensieren sich gerade, sodass wirdas korrekte Ergebnis erhalten haben.
I Kinetische Gastheorie: Eine korrekte Berücksichtigung der Geschwindigkeits-verteilung und Herleitung der Zahl der Wandstäße gelingt im Rahmen der kinetischenGastheorie und der statistischen Thermodynamik (siehe z.B. Scriptum zur VorlesungPC-3: “Chemical Kinetics”). Sie liefert
=
r8
(1.75)
und p2 =
r3
(1.76)

1.1 Gase und Gasgesetze 16
1.1.5 Gasmischungen
Wir betrachten zwei (. gleiche oder . verschiedene) Gase mit den Molzahlen 1 und2 in zwei Volumina 1 und 2, die durch eine bewegliche (und entfernbare) Trennplattevoneinander getrennt sind (Abb. ??) und nehmen an, dass sowohl thermisches Gleich-gewicht (durch Temperaturangleichung) als auch mechanisches Gleichgewicht (durchvertikales Verschieben der Trennplatte) eingestellt ist.
I Abbildung 1.8: Zum Verhalten von Gasmischungen (1).
I Mischung zweier gleicher Gase im thermischen und mechanischen Gleich-gewicht (Abb. 1.8):
• Ausgangszustand:Thermisches Gleichgewicht:
1 = 2 (1.77)
Mechanisches Gleichgewicht:1 = 2 (1.78)
• Endzustand bei Herausnehmen der Trennplatte (oder Öffnen eines Ventils in derTrennplatte):
= 1 = 2 (1.79)
= 1 = 2 (1.80)
= 1 + 2 (1.81)
= 1 + 2 (1.82)

1.1 Gase und Gasgesetze 17
• Wir stellen fest, dass die allgemeine Gasgleichung sowohl für die einzelnen Anteile11 = 1 = 1 (1.83)
22 = 2 = 2 (1.84)
als auch für die Summe der Volumina gilt:
(1 + 2)| z =
= (1 + 2)| z =
(1.85)
y =
(1 + 2)
=
(1.86)
I Abbildung 1.9: Zum Verhalten von Gasmischungen (2).
I Mischung zweier verschiedener Gase (Abb. 1.9): Wir nehmen nun an, dass diebeiden Teilvolumina 1 und 2 zwei verschiedene Gase (Gas 1 und Gas 2) enthaltenund betrachten den Zustand, der nach Erreichen des thermischen (Temperaturangle-ichung bis 1 = 2 = ) und mechanischen (Verschieben des Kolbens bis 1 = 2)Gleichgewichts bei Entfernen (Öffnen) der Trennscheibe erreicht wird:
• Ausgangszustand:Im thermischen und mechanischen Glgew. vor Entfernen der Trennscheibe giltgasartunabhängig
11 = 11 (1.87)
22 = 22 (1.88)
mit
= 1 = 2 (1.89)
= 1 = 2 (1.90)

1.1 Gase und Gasgesetze 18
• Endzustand #1a bzw. #1b, wenn sich die beiden Gase jeweils einzeln auf dasGesamtvolumen = 1 + 2 ausbreiten würden:
01 (1 + 2) = 1 (1.91)
bzw.02 (1 + 2) = 2 (1.92)
mit
= 1 = 2 (1.93)
= 1 + 2 (1.94)
y10 =
1
(1.95)
und
20 =
2
(1.96)
• Endzustand #2 der Gasmischung nach Entfernen der Trennfläche:
=
=(1 + 2)
=
1
+
2
= 1
0 + 20 (1.97)
In der Gasmischung nach Entfernen der Trennfläche stellen sich die gleichen Par-tialdrücke 01 und
02 ein, wie bei getrennter Expansion der einzelnen Gase in das
Gesamtvolumen, denn das ideale Gasgesetz gilt gasartunabhängig.
I Partialdruck: Allgemein ist der Partialdruck eines Gases in einer Gasmischunggegeben durch
=
(1.98)
Der Partialdruck ist ein geeignetes Maß für den Anteil eines Gases in einer Gasmis-chung.
I Dalton’sches Gesetz: Die Summe der Partialdrucke in einer Gasmischung ergibt denGesamtdruck:
=X
=X
(1.99)
d.h. die Partialdrucke in einer Gasmischung sind additiv.
Entsprechendes gilt für die Gesamtmolzahl:
=X
(1.100)

1.1 Gase und Gasgesetze 19
I Molenbruch: Die o.a. Beziehungen lassen sich noch einfacher ausdrücken, wenn wirden Molenbruch einführen.
I Definition 1.1: Molenbruch:
=P
=
(1.101)
Aus der Definition folgt X
=X
= 1 (1.102)
Für ideale Gase kann zur Berechnung von anstatt direkt der Partialdruck ver-wendet werden, da ∝ :
=
(1.103)
y = (1.104)
I Beispiel 1.1: Die Molenbrüche der Komponenten einer Mischung von 20mol O2 und80mol N2 (= 100mol synthetische Luft) sind
O2 =O2
O2 + N2= 020 (1.105)
undN2 =
N2O2 + N2
= 080 = 1− O2 (1.106)
¤
1.1.6 Reale Gase
Für das ideale Gas (punktförmige Teilchen ohne WW) ergibt die ideale Gasgleichung
= 1 (1.107)
Das Verhältnis
wird für reale Gase jedoch immer stärker von 1 abweichen, je
näher man an den Kondensationspunkt (Siedepunkt der Flüssigkeit) kommt, da dieWW der Gasteilchen untereinander zunimmt, bis es schließlich zur Kondensation desGases kommt.
I Der Realfaktor z: Ein Maß für die Abweichung eines realen Gases vom idealen Ver-halten ist der Realfaktor
=
(1.108)
1.1 Gase und Gasgesetze 20
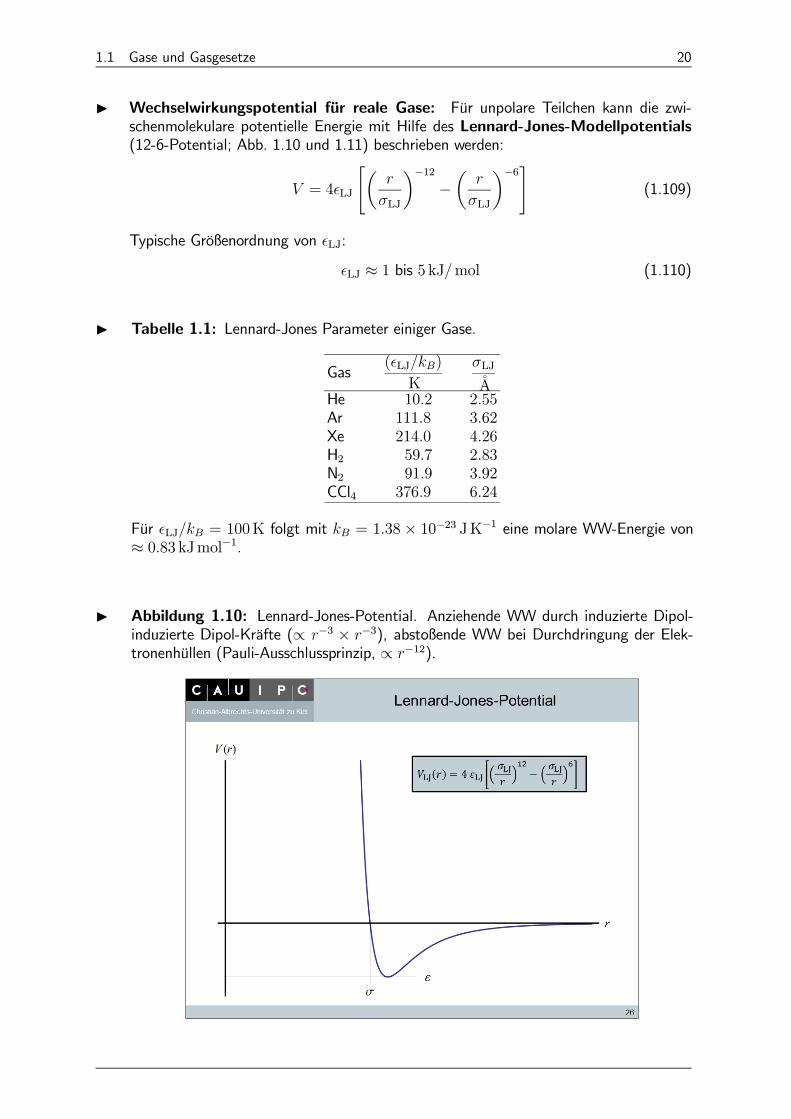
I Wechselwirkungspotential für reale Gase: Für unpolare Teilchen kann die zwi-schenmolekulare potentielle Energie mit Hilfe des Lennard-Jones-Modellpotentials(12-6-Potential; Abb. 1.10 und 1.11) beschrieben werden:
= 4LJ
"µ
LJ
¶−12−µ
LJ
¶−6#(1.109)
Typische Größenordnung von LJ:
LJ ≈ 1 bis 5 kJmol (1.110)
I Tabelle 1.1: Lennard-Jones Parameter einiger Gase.
Gas(LJ)
K
LJ
AHe 102 255
Ar 1118 362
Xe 2140 426
H2 597 283
N2 919 392
CCl4 3769 624
Für LJ = 100K folgt mit = 138× 10−23 JK−1 eine molare WW-Energie von≈ 083 kJmol−1.
I Abbildung 1.10: Lennard-Jones-Potential. Anziehende WW durch induzierte Dipol-induzierte Dipol-Kräfte (∝ −3 × −3), abstoßende WW bei Durchdringung der Elek-tronenhüllen (Pauli-Ausschlussprinzip, ∝ −12).
1.1 Gase und Gasgesetze 21
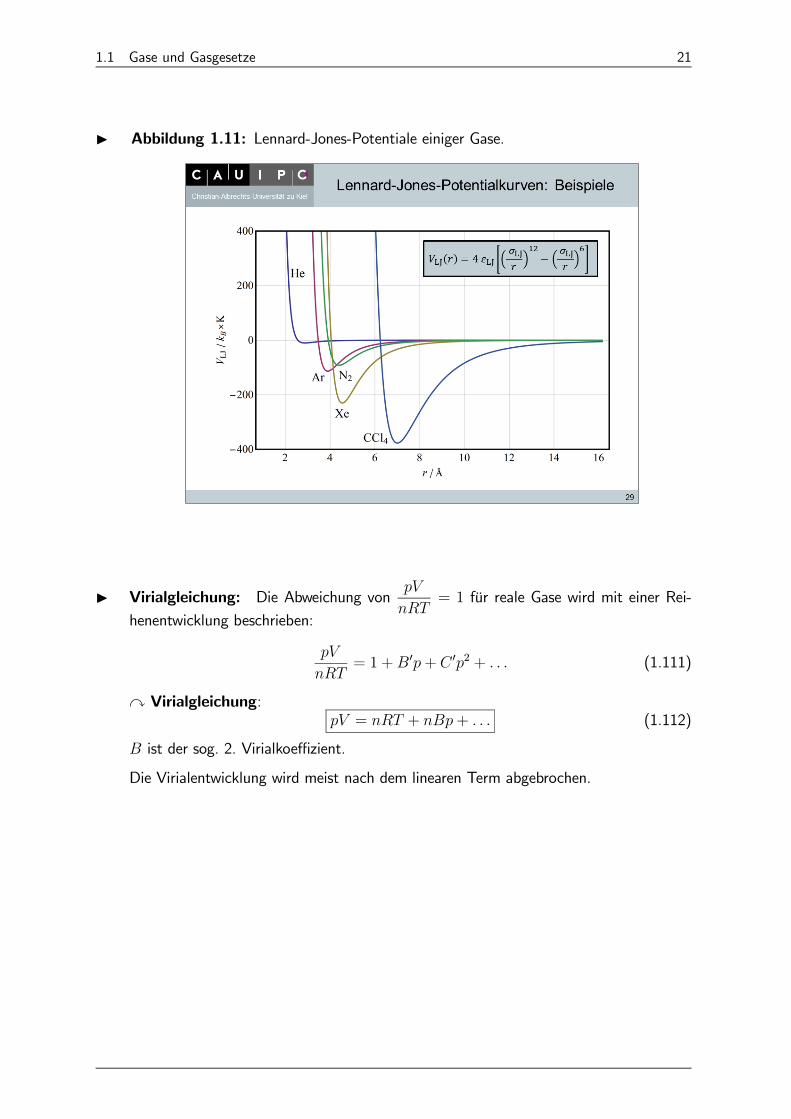
I Abbildung 1.11: Lennard-Jones-Potentiale einiger Gase.
I Virialgleichung: Die Abweichung von
= 1 für reale Gase wird mit einer Rei-
henentwicklung beschrieben:
= 1 +0+ 02 + (1.111)
y Virialgleichung: = + + (1.112)
ist der sog. 2. Virialkoeffizient.
Die Virialentwicklung wird meist nach dem linearen Term abgebrochen.
1.1 Gase und Gasgesetze 22
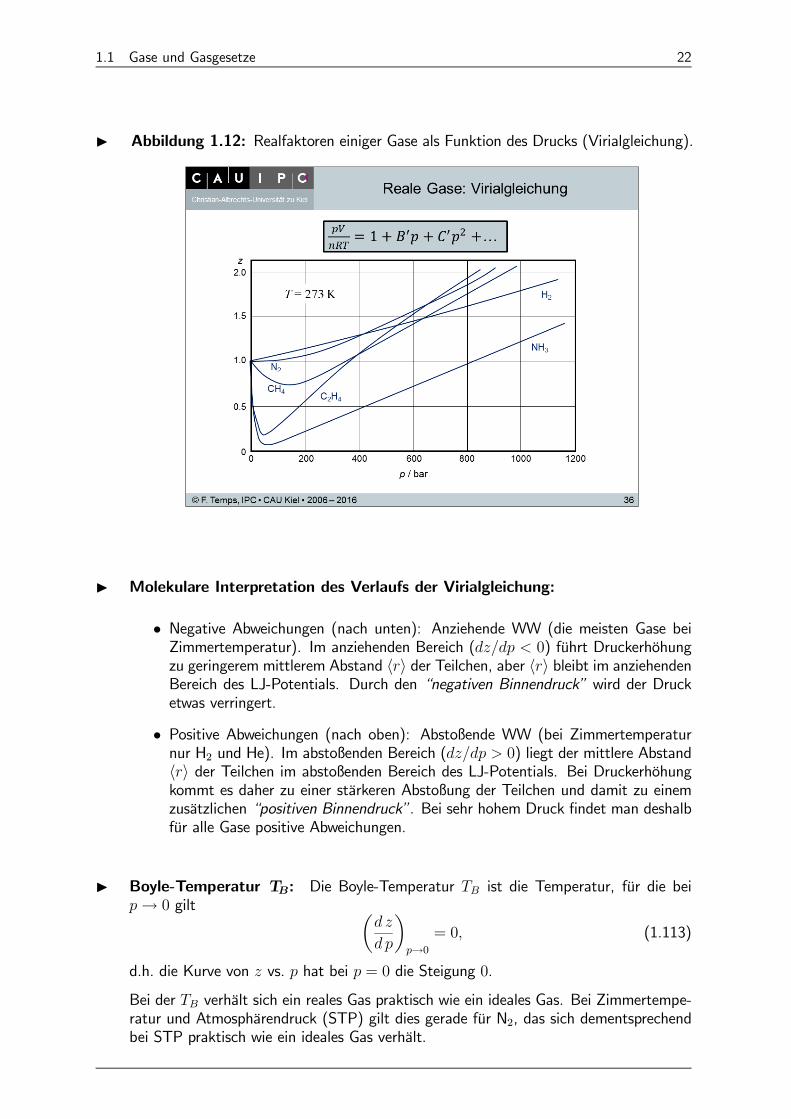
I Abbildung 1.12: Realfaktoren einiger Gase als Funktion des Drucks (Virialgleichung).
I Molekulare Interpretation des Verlaufs der Virialgleichung:
• Negative Abweichungen (nach unten): Anziehende WW (die meisten Gase beiZimmertemperatur). Im anziehenden Bereich ( 0) führt Druckerhöhungzu geringerem mittlerem Abstand hi der Teilchen, aber hi bleibt im anziehendenBereich des LJ-Potentials. Durch den “negativen Binnendruck” wird der Drucketwas verringert.
• Positive Abweichungen (nach oben): Abstoßende WW (bei Zimmertemperaturnur H2 und He). Im abstoßenden Bereich ( 0) liegt der mittlere Abstandhi der Teilchen im abstoßenden Bereich des LJ-Potentials. Bei Druckerhöhungkommt es daher zu einer stärkeren Abstoßung der Teilchen und damit zu einemzusätzlichen “positiven Binnendruck”. Bei sehr hohem Druck findet man deshalbfür alle Gase positive Abweichungen.
I Boyle-Temperatur T: Die Boyle-Temperatur ist die Temperatur, für die bei→ 0 gilt µ
¶→0
= 0 (1.113)
d.h. die Kurve von vs. hat bei = 0 die Steigung 0.
Bei der verhält sich ein reales Gas praktisch wie ein ideales Gas. Bei Zimmertempe-ratur und Atmosphärendruck (STP) gilt dies gerade für N2, das sich dementsprechendbei STP praktisch wie ein ideales Gas verhält.

1.1 Gase und Gasgesetze 23
I Anwendung der Virialgleichung zur Molmassenbestimmung eines Gases nachRegnault:
= ( +) =
( +) (1.114)
y
=1
+
(1.115)
I Experiment: Für ein Gas in einem Kolben des Volumens werden Datensätze von
, , und gemessen. Trägt man dann
vs. auf, ergibt sich nach Gl. 1.115
eine Gerade mit dem Ordinatenabschnitt1
und der Steigung
.
I Anmerkung 1.3: Die gerade durchgeführte Umformung einer Gleichung in eine lineareForm ist eine typische Vorgehensweise, die wir wenn irgend möglich immer zur Auswer-tung von Experimenten durchführen werden. Die graphische Auftragung in Form einerGeraden erlaubt eine schnelle Abschätzung der Qualität von Messdaten und durch lin-eare Regression (siehe Anhang D) eine schnelle Bestimmung der Parameter Ordinaten-abschnitt und Steigung (siehe Übungen).
Wenn Sie in einer Übungsaufgabe oder in einem Experiment einen Satz von mehr alszwei Datenpunkten zur Auswertung erhalten, sollten Sie immer prüfen, ob Sie einegeeignete graphische Auftragung in Form einer Geraden machen können, dann durchlineare Regression die Geradenparameter ermitteln, und ggf. daraus die gewünschtenAngaben bestimmen!
I Van der Waals-Gleichung: Die van der Waals-Zustandsgleichung (1873)³+
2´ ¡
− ¢= (1.116)
mit den zwei van der Waals Parametern und erlaubt eine physikalisch begründeteZustandsbeschreibung vom idealen Gas über reale Gase bis hin zu Flüssigkeiten (sieheAbschnitt 1.2.5).

1.2 Zustandsdiagramme reiner Stoffe 24
1.2 Zustandsdiagramme reiner Stoffe
Bis hierher haben wir Stoffe nur in einem Aggregatzustand betrachtet. Wir wollen jetztStoffe in mehreren Aggregatzuständen gleichzeitig und Phasenumwandlungen zwischenden Aggregatzuständen betrachten. Wir suchen dazu sog. Zustandsdiagramme, ausdenen wir unmittelbar ablesen können, unter welchen Bedingungen ( ) ein Stoffwelchen Aggregatzustand ( ) einnimmt.
1.2.1 Phasenumwandlungen eines Stoffes
I Definition 1.2: Phase:
Unter einer Phase verstehen wir einen homogenen Bereich der Materie.
I Definition 1.3: Gleichgewichtszustand:
Der Gleichgewichtszustand ist der Zustand eines Systems, der sich nach∞ langer Zeiteinstellt.
Bei Störungen des Gleichgewichtszustands kehrt das System von allein in den Gleichge-wichtszustands zurück.
I Anmerkung 1.4: Die Thermodynamik kann keine Aussage darüber machen, wie langes dauert, bis sich das Glgew. einstellt, aber der Glgew.-zustand ist eindeutig von bestimmt.
I Experimentell beobachtetes Temperatur-Zeit-Profil bei Phasenumwandlun-gen: Wir starten mit einem Festkörper (z.B. Eis) in einem geschlossenen System undführen Energie (Wärme) zu. Dabei beobachten wir einen Temperaturanstieg bis zumSchmelzpunkt. Am Schmelzpunkt bleibt konstant, bis alles geschmolzen ist (⇒Wasser). Dann steigt wieder an, bis zum Siedepunkt. Am Siedepunkt bleibt erneut konstant, bis aller Stoff in die Gasphase übergegangen ist (⇒ Wasserdampf)).Erst danach steigt wieder an. Abbildung 1.13 zeigt den -Verlauf. Der Prozess istreversibel, d.h. verläuft entsprechend umgekehrt, wenn Wärme entzogen wird.
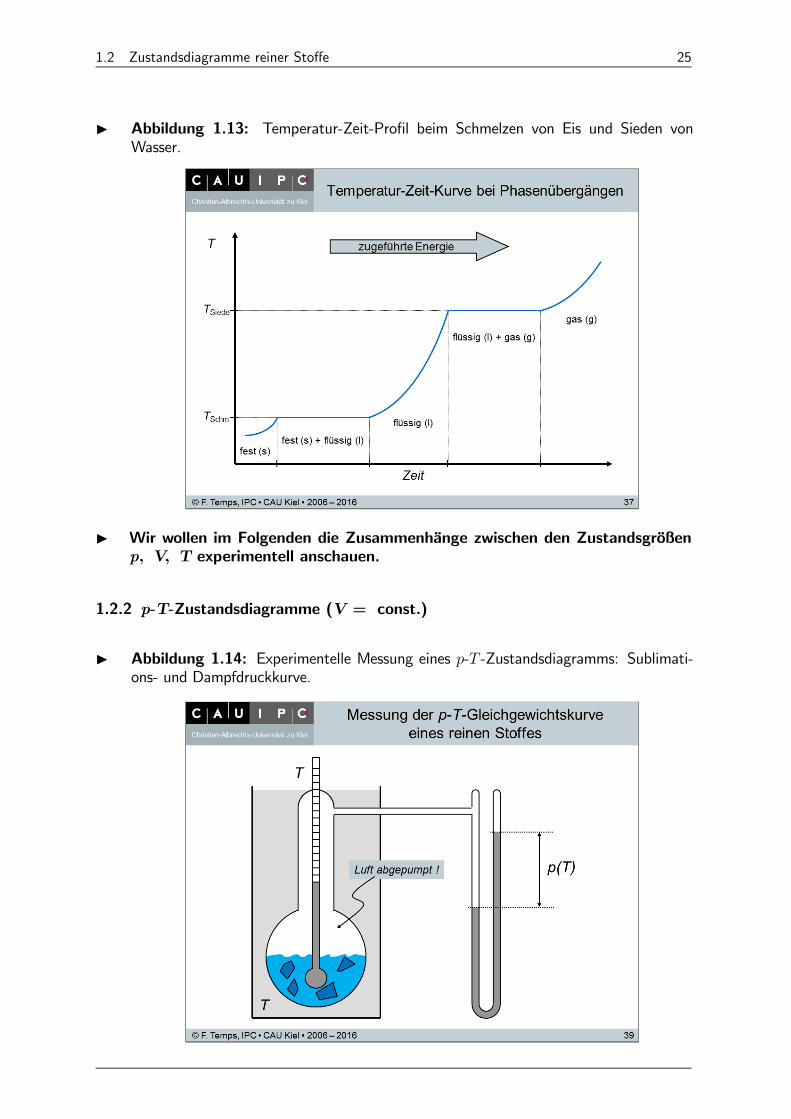
1.2 Zustandsdiagramme reiner Stoffe 25
I Abbildung 1.13: Temperatur-Zeit-Profil beim Schmelzen von Eis und Sieden vonWasser.
I Wir wollen im Folgenden die Zusammenhänge zwischen den Zustandsgrößenp, V, T experimentell anschauen.
1.2.2 p-T -Zustandsdiagramme (V = const.)
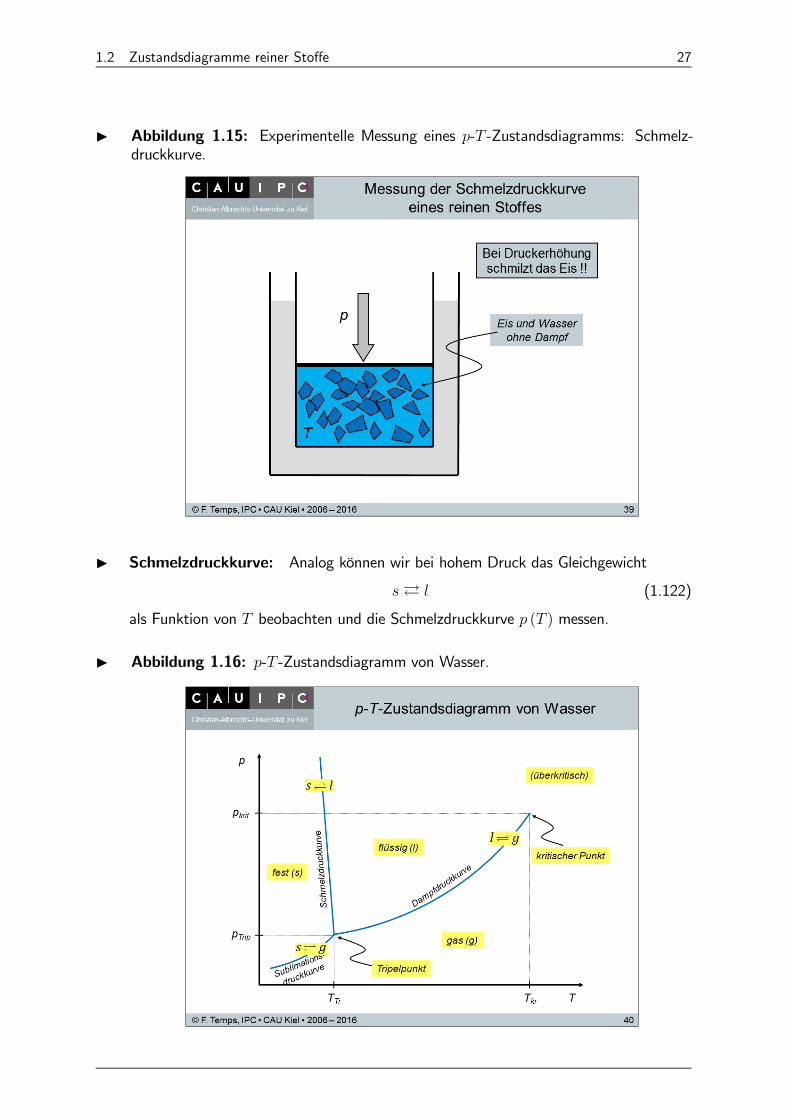
I Abbildung 1.14: Experimentelle Messung eines - -Zustandsdiagramms: Sublimati-ons- und Dampfdruckkurve.

1.2 Zustandsdiagramme reiner Stoffe 26
I Sublimations- und Dampfdruckkurve: Beobachtungen bei langsamer Wärmezu-fuhr:
(1) Nur Eis: Es stellt sich ein bestimmter Druck ein (= Sublimationsdruck).9
Vorliegendes Gleichgewicht:À (1.117)
Der Sublimationsdruck ist von abhängig (nur von !):
= ( ) (1.118)
⇒ - -Diagramm.
(2) Bei einer bestimmten Temperatur tritt auch Flüssigkeit auf (⇒ Tripelpunkt, =Tr, = Tr, = Tr).
Vorliegendes Gleichgewicht:À À (1.119)
Am Tripelpunkt stehen alle drei Phasen (s, l, g) im Gleichgewicht.
(3) Oberhalb des Tripelpunktes: Nur flüssiges Wasser und Wasserdampf.
Vorliegendes Gleichgewicht:À (1.120)
Der Dampfdruck ist nur von abhängig:
= ( ) (1.121)
⇒ - -Diagramm.
(4) Am kritischen Punkt ( = kr, = kr, = kr) verschwinden alle Unterschiedezwischen Flüssigkeit und Dampf.
(5) Oberhalb des kritischen Punkts spricht man von überkritischen (oder fluiden)Zuständen bzw. Phasen.
9 Draußen aufgehängte Wäsche trocknet auch im Winter, wenn es friert.
1.2 Zustandsdiagramme reiner Stoffe 27
I Abbildung 1.15: Experimentelle Messung eines - -Zustandsdiagramms: Schmelz-druckkurve.
I Schmelzdruckkurve: Analog können wir bei hohem Druck das Gleichgewicht
À (1.122)
als Funktion von beobachten und die Schmelzdruckkurve ( ) messen.
I Abbildung 1.16: - -Zustandsdiagramm von Wasser.

1.2 Zustandsdiagramme reiner Stoffe 28
I Abbildung 1.17: - -Diagramm von Wasser mit Siede- und Schmelzpunkt.
I Anomalie des Wassers:
• Die negative Steigung der Schmelzdruckkurve von Wasser ist der Ausnah-mefall. Normalerweise ist die Steigung der Schmelzdruckkurve positiv, d.h. beiDruckerhöhung kristallisiert der Festkörper aus.
• Wasser schmilzt bei Druckerhöhung, da es eine höhere Dichte als Eis hat (Eisschwimmt oben auf Wasser, Wasser hat seine größte Dichte bei 4 C). Die neg-ative Steigung der Schmelzdruckkurve von Wasser erlaubt das Schlittschuhlaufenund den Gletscherfluss.
1.2 Zustandsdiagramme reiner Stoffe 29
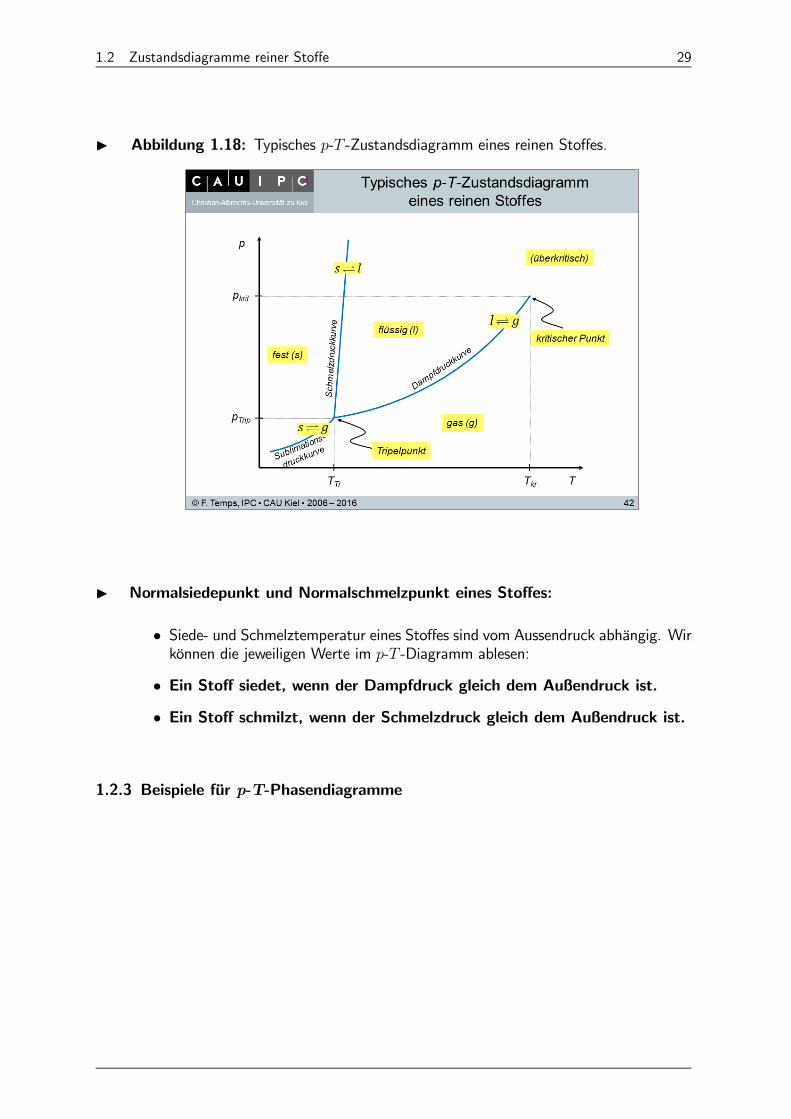
I Abbildung 1.18: Typisches - -Zustandsdiagramm eines reinen Stoffes.
I Normalsiedepunkt und Normalschmelzpunkt eines Stoffes:
• Siede- und Schmelztemperatur eines Stoffes sind vom Aussendruck abhängig. Wirkönnen die jeweiligen Werte im - -Diagramm ablesen:
• Ein Stoff siedet, wenn der Dampfdruck gleich dem Außendruck ist.
• Ein Stoff schmilzt, wenn der Schmelzdruck gleich dem Außendruck ist.
1.2.3 Beispiele für p-T -Phasendiagramme
1.2 Zustandsdiagramme reiner Stoffe 30
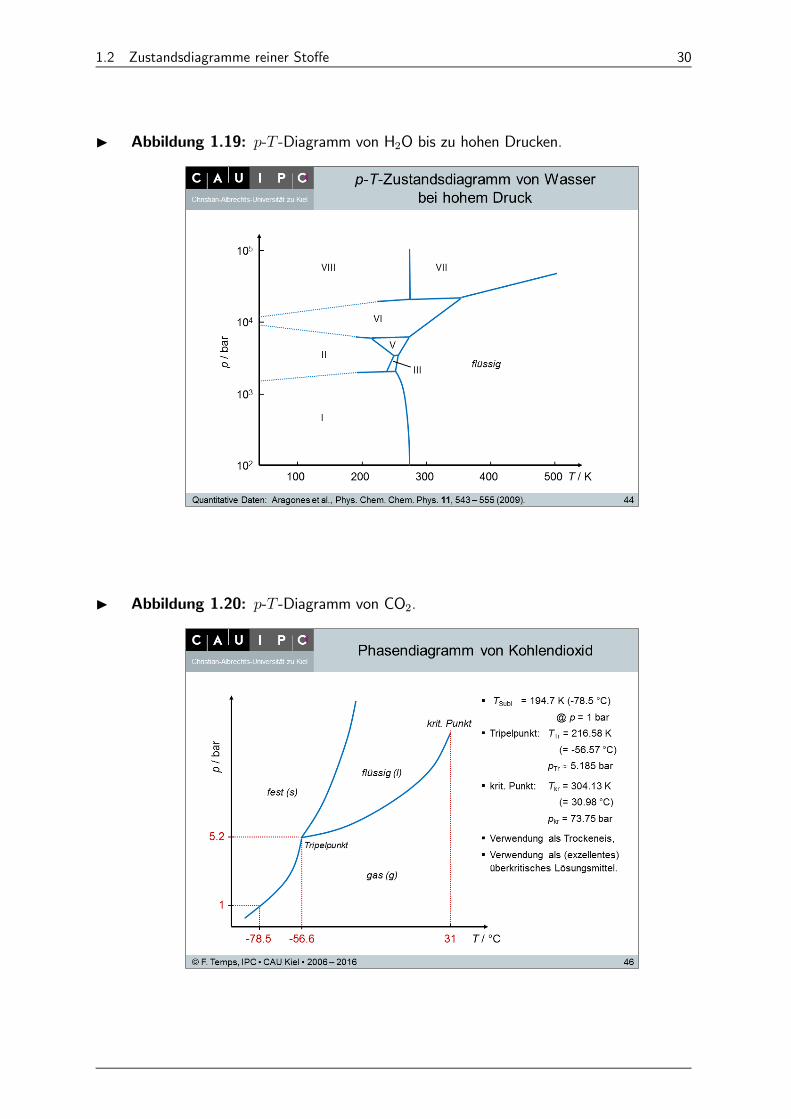
I Abbildung 1.19: - -Diagramm von H2O bis zu hohen Drucken.
I Abbildung 1.20: - -Diagramm von CO2.
1.2 Zustandsdiagramme reiner Stoffe 31
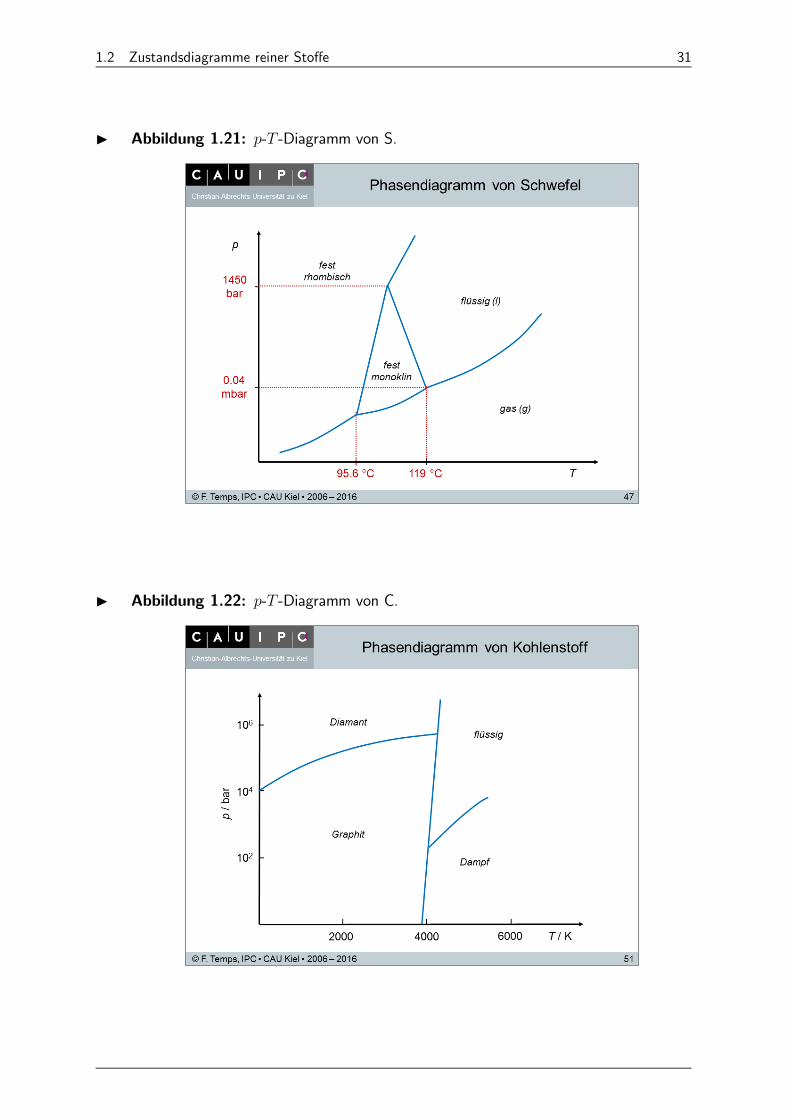
I Abbildung 1.21: - -Diagramm von S.
I Abbildung 1.22: - -Diagramm von C.

1.2 Zustandsdiagramme reiner Stoffe 32
I Abbildung 1.23: - -Diagramm von He.
1.2.4 p-V -Zustandsdiagramme (T = const.):
I Abbildung 1.24: Experimentelle Messung des - -Diagramms eines Stoffes.
1.2 Zustandsdiagramme reiner Stoffe 33
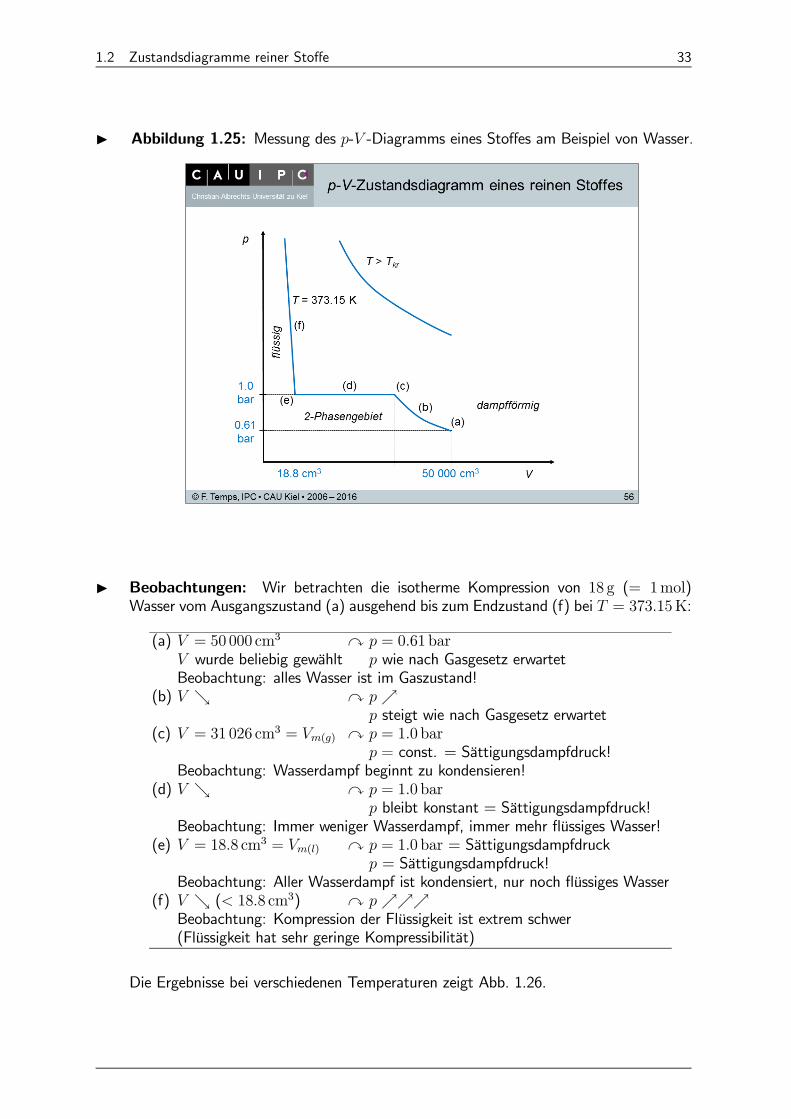
I Abbildung 1.25: Messung des - -Diagramms eines Stoffes am Beispiel von Wasser.
I Beobachtungen: Wir betrachten die isotherme Kompression von 18 g (= 1mol)Wasser vom Ausgangszustand (a) ausgehend bis zum Endzustand (f) bei = 37315K:
(a) = 50 000 cm3 y = 061 bar
wurde beliebig gewählt wie nach Gasgesetz erwartetBeobachtung: alles Wasser ist im Gaszustand!
(b) & y % steigt wie nach Gasgesetz erwartet
(c) = 31 026 cm3 = () y = 10 bar
= const. = Sättigungsdampfdruck!Beobachtung: Wasserdampf beginnt zu kondensieren!
(d) & y = 10 bar
bleibt konstant = Sättigungsdampfdruck!Beobachtung: Immer weniger Wasserdampf, immer mehr flüssiges Wasser!
(e) = 188 cm3 = () y = 10 bar = Sättigungsdampfdruck = Sättigungsdampfdruck!
Beobachtung: Aller Wasserdampf ist kondensiert, nur noch flüssiges Wasser(f) & ( 188 cm3) y %%%
Beobachtung: Kompression der Flüssigkeit ist extrem schwer(Flüssigkeit hat sehr geringe Kompressibilität)
Die Ergebnisse bei verschiedenen Temperaturen zeigt Abb. 1.26.

1.2 Zustandsdiagramme reiner Stoffe 34
I Abbildung 1.26: - -Diagramms eines Stoffes mit Isothermen bei verschiedenen Tem-peraturen und Zweiphasen-Koexistenzbereich.
Die Steigung der Isothermen im - -Diagramm wird durch die reziproke Kom-pressibilität angegeben:
I Definition 1.4: Kompressibilität.
= − 1
µ
¶
(1.123)
I Anmerkung 1.5: Im Zweiphasengebiet ist = 0y =∞.
1.2.5 Die van der Waals-Gleichung
I Frage: Gibt es eine geeignete Zustandsgleichung, die das beobachtete - -Verhaltenbeschreibt?
I Antwort: ⇒ van der Waals-Gleichung.
I Plausibilitätsbetrachtung zum Verständnis der van der Waals-Gleichung:
(1) Ideales Gas: = (1.124)
(2) Berücksichtigung des Eigenvolumens der Gasmoleküle:
→ ( − ) (1.125)
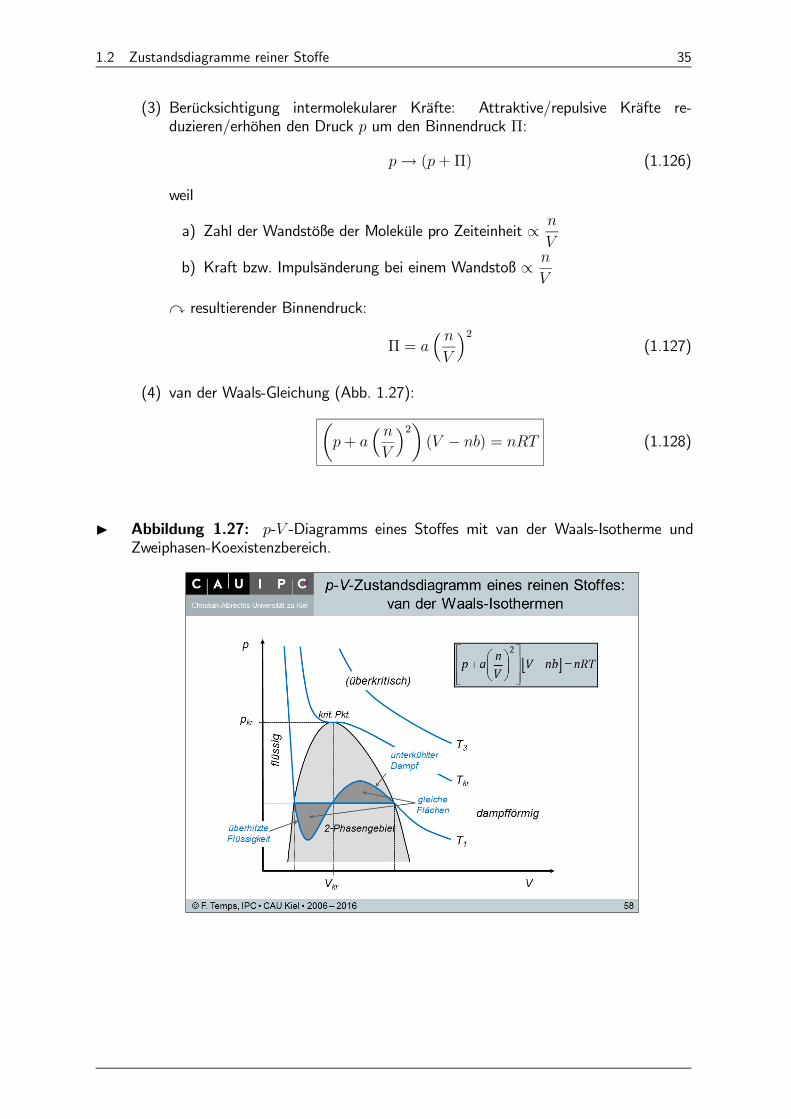
1.2 Zustandsdiagramme reiner Stoffe 35
(3) Berücksichtigung intermolekularer Kräfte: Attraktive/repulsive Kräfte re-duzieren/erhöhen den Druck um den Binnendruck Π:
→ (+Π) (1.126)
weil
a) Zahl der Wandstöße der Moleküle pro Zeiteinheit ∝
b) Kraft bzw. Impulsänderung bei einem Wandstoß ∝
y resultierender Binnendruck:
Π = ³
´2(1.127)
(4) van der Waals-Gleichung (Abb. 1.27):µ+
³
´2¶( − ) = (1.128)
I Abbildung 1.27: - -Diagramms eines Stoffes mit van der Waals-Isotherme undZweiphasen-Koexistenzbereich.

1.2 Zustandsdiagramme reiner Stoffe 36
I Bemerkungen und Hinweise zur van der Waals-Gleichung:
• Die vdW-Gl. ist eine Gleichung 3. Grades. Eine solche Gl. hat deshalb i.a. 2Extremalwerte (1 Minimum + 1 Maximum).
• Das 2-Phasengebiet wird so konstruiert, dass die Flächen unter der Kurve gleichgroß sind (Energieerhaltung).
• Im Zweiphasengebiet beschreiben die Bereiche der -Kurve mit negativer Stei-gung überhitzte Flüssigkeit (links) bzw. unterkühlten Dampf (rechts). Beide sind“metastabil”).
• Der Bereich mit 0 ist physikalisch unsinnig.
• Am kritischen Punkt liegt ein Sattelpunkt vor, mitµ
¶=
= 0 (1.129)
und µ2
2
¶=
= 0 (1.130)
Beide Gleichungen stellen die Verbindung zwischen den vdW-Parametern undden kritischen Daten , , her (siehe Übungsaufgabe).
1.2.6 Kombination des p-T - und des p-V -Diagramms in einer 3D-Darstellung
- - und - -Diagramm sind jeweils 2D-Schnitte durch den dreidimensionalen - - -Raum, die erhalten werden, wenn man das 3D-Diagramm auf die entsprechendenFlächen projiziert.
I Abbildung 1.28: 3D-Zustandsdiagramm eines Stoffes und Projektionen auf die - -und p- -Ebenen (gelöscht aus Copyright-Gründen).

1.2 Zustandsdiagramme reiner Stoffe 37
I Abbildung 1.29: 3D-Zustandsdiagramm von Wasser und Projektionen auf die - -und p- -Ebenen (gelöscht aus Copyright-Gründen).
1.2.7 Das Theorem der übereinstimmenden Zustände
Aus der van der Waals-Gleichung erhalten wir unter Verwendung der o.a. Bedingungenfür den kritischen Punkt
=1
3 (1.131)
=9
8 (1.132)
und
=8
3
(1.133)
Einsetzen dieser Ergebnisse in die van der Waals-Gleichung liefert (Übungsaufgabe)Ã
+ 3
µ
¶2!µ3
− 1¶= 8
(1.134)
In dieser Gleichung treten die Zustandsgrößen , und nur noch in ihrer reduziertenForm
=
(1.135)
=
(1.136)
=
(1.137)

1.2 Zustandsdiagramme reiner Stoffe 38
auf. Geschrieben mit den reduzierten Zustandsgrößen wird die van der Waals-Gleichungvon der Gasart unabhängig, und wir erhalten die universell gültige reduzierte Zustands-gleichung µ
+3
2
¶(3 − 1) = 8 (1.138)
Trägt man für verschiedene Gase gegen auf, so fallen die Zustandsdatenzusammen (Theorem der übereinstimmenden Zustände).
1.2.8 Gesetzmässigkeiten für Phasengleichgewichte
Quantitative Gesetzmässigkeiten und Regeln für Phasengleichgewichte werden hierzunächst als empirische Befunde angegeben, die wir vorerst ohne Prüfung akzeptieren.Wir werden die Gesetze später herleiten.
a) Zusammenfassung der Beobachtungen
(1) - - und - -Diagramm ergeben sich durch Projektion des 3D - - -Diagrammsauf die entsprechenden 2D Flächen.
(2) Die Sättigungsdampfdruckkurve 0 für das Gleichgewicht À (wie auch dieGleichgewichtskurven für À und À ) sind von abhängig:
0 = ( ) (1.139)
Wir schreiben für den (uns noch unbekannten) Zusammenhang 0 ( ) oder, wennder Zusammenhang klar ist, auch einfach ( ).
(3) Am Siedepunkt einer Flüssigkeit ist
0 ( ) = Aussen (1.140)
(4) Im Bereich koexistierender Phasen ist = const., aber die Mengen der Phasen() und () ändern sich (⇒ Hebelgesetz der Phasenmengen, siehe später).
(5) Am kritischen Punkt verschwinden die Unterschiede zwischen Gas und Flüssigkeit,und die Molvolumina10 und Dichten von Gas und Flüssigkeit werden gleich:
= (1.141)
= (1.142)
= − 1
µ
¶=
=∞ (1.143)
(6) Für sprechen wir von einer “überkritischen” oder “fluiden” Phase.
10 Molare Größen, wie z.B. das Molvolumen, kennzeichnen wir entweder mit dem Index (z.B. )
oder durch Überstreichen, z.B. . Streng genommen werden in der Thermodynamik durch Über-
streichen vereinbarungsgemäß partielle molare Größen gekennzeichnet; für Reinstoffsysteme ist aber
natürlich = , etc.

1.2 Zustandsdiagramme reiner Stoffe 39
b) Gleichung von Clausius-Clapeyron
Quantitativ werden die - -Phasengleichgewichtskurven (d.h. 0 ( )) durch die Glei-chung von Clausius-Clapeyron beschrieben. Diese Gl. werden wir später herleiten;hier akzeptieren wir sie zunächst als empirischen Befund:
I Gleichung von Clausius und Clapeyron: Für das Phasengleichgewicht
À (1.144)
gilt µ
¶Glgew.
=∆
( − )(1.145)
mit:∆ = ∆À = molare Umwandlungsenthalpie (1.146)
und∆ = ( − ) = molare Volumendifferenz (1.147)
I Anmerkungen:
• Die Clausius-Clapeyron’sche Gleichung 1.145 beschreibt die Steigung der - -Gleichgewichtskurve bei der Temperatur .
• Für die Werte gilt:∆ ≥ 0 (1.148)
∆
0 (1.149)
• Am kritischen Punkt ist(∆)=krit = 0 (1.150)
c) Anwendung auf H2O(s) À H2O(l):
I Gleichung von Clausius-Clapeyron:µ
¶À
=∆Schm
À ( − )(1.151)

1.2 Zustandsdiagramme reiner Stoffe 40
I Zahlenwerte @ T = 273.15K (Schmelzen von Eis):
∆Schm = +60 kJmol (1.152)
= 18004 cm3mol (1.153)
=
=18 gmol
18004 cm3mol= 0999 78
g
cm3(1.154)
= 1960 cm3mol (1.155)
= 0916 8g
cm3(1.156)
y (1.157)
∆Schm = − = −163 cm3 (1.158)
y für H2O: µ
¶À
0 (1.159)
I Anmerkung 1.6: Typisch für die meisten Stoffe ist allerdings
µ
¶À
0 denn i.a.
ist ∆ = − 0.
d) Anwendung auf H2O(l) À H2O(g):
I Gleichung von Clausius-Clapeyron:µ
¶À
=∆Verd
À ( − )(1.160)
I Zahlenwerte @ T = 373.15K (Verdampfung):
∆Verd = +4069 kJmol (1.161)
= 30 100 cm3mol (1.162)
= 188 cm3mol (1.163)
∆Verd = − = 30 081 cm3 ≈ (1.164)
I Näherungen: Der Fehler bei Vernachlässigung von y ist 0.063 %, also sehrklein. Wir können daher zwei Näherungen machen:
(1) À y − ≈ (1.165)
(2) = (1.166)
y µ
¶À
(1)≈ ∆Verd
À
(2)≈ ∆Verd ×
2(1.167)

1.2 Zustandsdiagramme reiner Stoffe 41
Mit11
=
ln
(1.169)
folgt für À in guter Näherung:
ln
=
∆Verd
2(1.170)
e) Bestimmung der Verdampfungsenthalpie aus experimentellen Daten für p (T ):
I Umformung von Gl. 1.170 (zur Bestimmung von ∆VerdHVerd):
µ1
¶
= (−1)
= −−2 = − 1 2
(1.171)
y = − 2 ×
µ1
¶(1.172)
Einsetzen ergibt:
ln
(1 )= −∆Verd
(1.173)
I Graphische Auftragung: Diese Gleichung erlaubt eine Bestimmung von ∆Verd
durch Auftragung von experimentellen Daten für ln gegen 1 , denn:
(1) Auftragung von gegen ergibt (Exponential)kurve (ungeeignet zur Auswertungexpt. Daten),
(2) Auftragung von ln gegen ergibt Kurve mit positiver 2. Ableitung (konvex),aber leider keine Gerade (ebenfalls ungeeignet zur Auswertung expt. Daten).
(3) Aber: Auftragung von ln gegen 1 ergibt Gerade mit Steigung:
= −∆Verd (1.174)
⇒ Ausdruck 1.173 ist geeignet zur Auswertung expt. Daten.
11 Folgt aus ln
=1
(1.168)

1.2 Zustandsdiagramme reiner Stoffe 42
I Abbildung 1.30: Graphische Auftragung zur Bestimmung der Verdampfungsenthalpie.
I Integration der Gleichung von Clausius-Clapeyron: Unter der Annahme, dass∆Verd ≈ const. (nur in kleinem -Intervall gültig), können wir die Clausius-Clapeyron’sch Gl. integrieren:
2Z1
ln = −2Z
1
∆Verd
µ1
¶= −∆Verd
2Z1
µ1
¶(1.175)
y
ln 2 − ln 1 = ln 21= −∆Verd
µ1
2− 1
1
¶(1.176)
I Energieerhaltung bei Kreisprozess um den Tripelpunkt (Abb. 1.31):
0 = ∆Schm +∆Verd + (−∆Subl) (1.177)
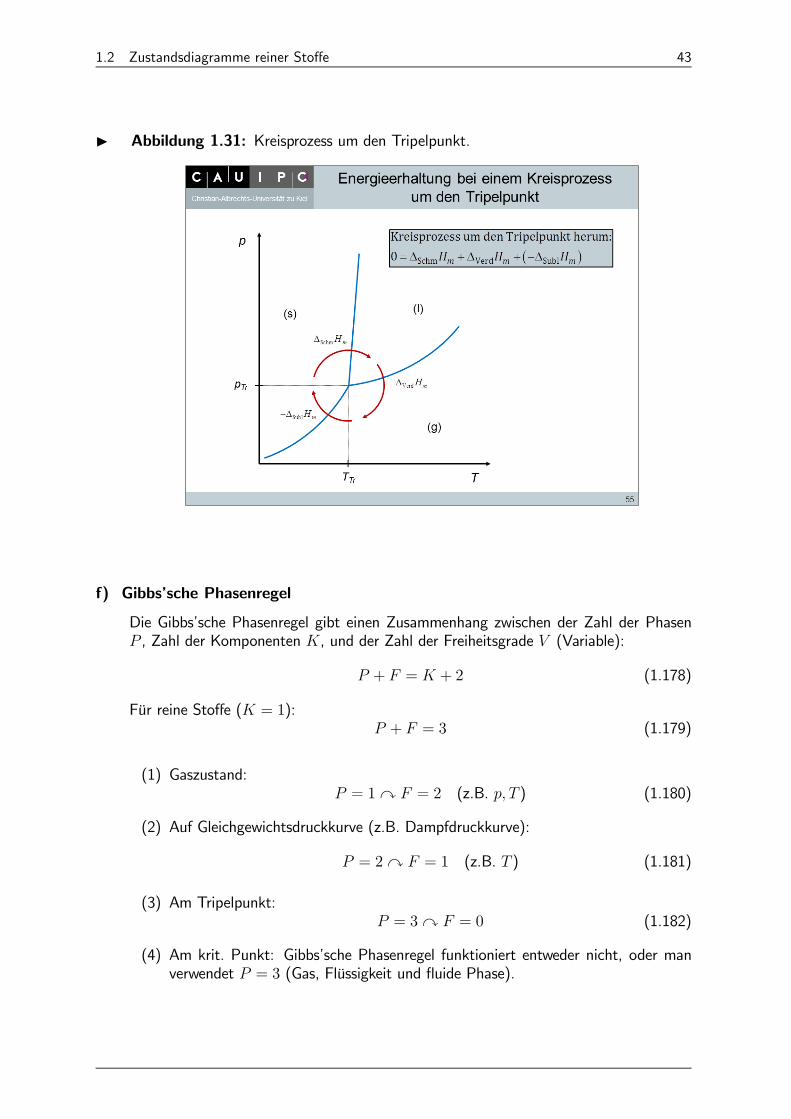
1.2 Zustandsdiagramme reiner Stoffe 43
I Abbildung 1.31: Kreisprozess um den Tripelpunkt.
f) Gibbs’sche Phasenregel
Die Gibbs’sche Phasenregel gibt einen Zusammenhang zwischen der Zahl der Phasen , Zahl der Komponenten , und der Zahl der Freiheitsgrade (Variable):
+ = + 2 (1.178)
Für reine Stoffe ( = 1): + = 3 (1.179)
(1) Gaszustand: = 1y = 2 (z.B. ) (1.180)
(2) Auf Gleichgewichtsdruckkurve (z.B. Dampfdruckkurve):
= 2y = 1 (z.B. ) (1.181)
(3) Am Tripelpunkt: = 3y = 0 (1.182)
(4) Am krit. Punkt: Gibbs’sche Phasenregel funktioniert entweder nicht, oder manverwendet = 3 (Gas, Flüssigkeit und fluide Phase).

1.2 Zustandsdiagramme reiner Stoffe 44
2. Der Erste Hauptsatz der Thermodynamik
1. Hauptsatz der Thermodynamik = Energieerhaltungssatz
2.1 Thermodynamische Systeme
Vor dem Einstieg in die Behandlung des 1. Hauptsatzes der Thermodynamik haben wireinige Begriffe zu klären. Es ist wichtig zu wissen, wovon wir reden. In der Thermo-dynamik heißt dies, dass wir die Systeme spezifizieren müssen, von denen wir reden.Ohne Angabe des Systems, für die wir eine Aussage machen, ist die Aussage meistenswertlos.
I Definition 2.1: Unter einem System verstehen wir einen Bereich des Weltalls, der unsinteressiert.
I Definition 2.2: Die Umgebung des Systems ist der Bereich, der das System umgibt.
I Wir unterscheiden folgende Arten von Systemen:
(1) Offenes System: Energie- und Stoffaustausch mit der Umgebung sind möglich.
(2) Geschlossenes System: Nur Energieaustausch mit der Umgebung möglich, keinStoffaustausch.
(3) Adiabatisch geschlossenes System: Energieaustausch mit der Umgebung nurin Form von Arbeit möglich, aber kein Wärmeaustausch.
(4) Abgeschlossenes System: Keinerlei Austausch mit der Umgebung, wederWärme noch Arbeit.

2.2 Volumenarbeit 45
2.2 Volumenarbeit
Arbeit ist eine Form von Energie, die einem System zugeführt werden kann oder vomSystem geleistet werden kann.
2.2.1 Arten von Arbeit
• Hubarbeit (physikalischer Prozess des Transports eines Systems auf eine andereHöhe),
• Volumenarbeit,• Oberflächenarbeit,• elektrische Arbeit,• magnetische Arbeit.
Wir interessieren uns in dieser Vorlesung vor allem für Volumenarbeit, im Kapitel Elek-trochemie später auch für elektrische Arbeit, und bei der Behandlung der PhysikalischenChemie von Grenzflächen für Oberflächenarbeit.
2.2.2 Volumenarbeit
Wir betrachten ein Gas in einem Kolben und berechnen die bei Kompression oder Ex-pansion des Kolbens geleistete Volumenarbeit (Abb. 2.1):
• Druck: =
(2.1)
• Kräftegleichgewicht: = × = Stange + Luft × = × (2.2)
• Zur Kompression des Kolbens um das Wegstück müssen wir Arbeit leisten (dem Gas im Kolben zuführen):
= − = − = − (2.3)
• Thermodynamische Vorzeichenkonvention:— Dem System zugeführte Arbeit wird + gezählt.
— Vom System geleistete Arbeit wird − gezählt.• Zugeführte Volumenarbeit:12
= − (2.4)
Für zugeführte Arbeit ist 0, daher wird positiv!
12 Achtung: Wir verwenden das kleine für die differentielle Größe , da die Arbeit keine Zus-
tandsgröße ist, sondern vom Weg abhängig ist, auf dem wir den Endzustand vom Anfangszustand
ausgehend erreichen.
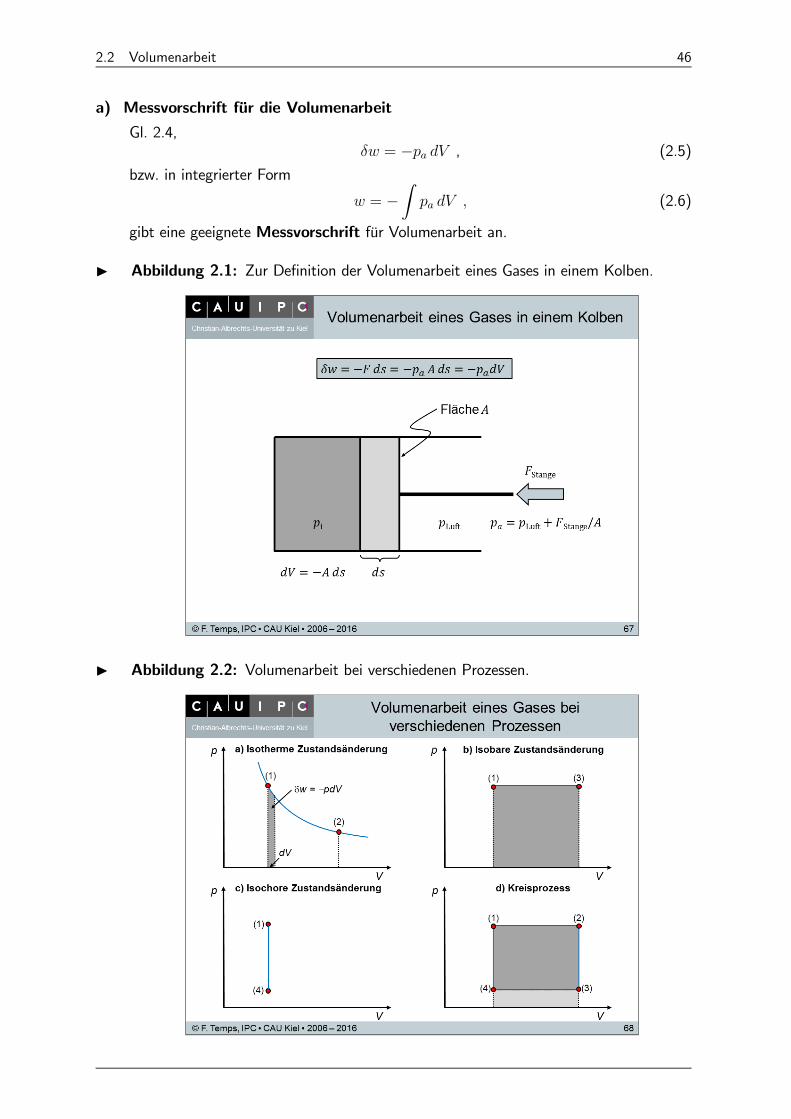
2.2 Volumenarbeit 46
a) Messvorschrift für die Volumenarbeit
Gl. 2.4, = − , (2.5)
bzw. in integrierter Form
= −Z
(2.6)
gibt eine geeignete Messvorschrift für Volumenarbeit an.
I Abbildung 2.1: Zur Definition der Volumenarbeit eines Gases in einem Kolben.
I Abbildung 2.2: Volumenarbeit bei verschiedenen Prozessen.

2.2 Volumenarbeit 47
b) Volumenarbeit für eine reversible (p = p) isotherme Zustandsänderung einesidealen Gases (Abb. 2.2a):
Für = 12 = const. gilt für ein ideales Gas:
1→2 =Z 2
1
=
Z 2
1
− = −Z 2
1
12
(2.7)
= −12Z 2
1
= −12
Z 2
1
ln = −12 ln 21
(2.8)
c) Volumenarbeit für eine isobare Zustandsänderung (Abb. 2.2b):
Für = const. folgt
= 0; =
(2.9)
y1→3 =
Z 3
1
= −Z 3
1
= −Z 3
1
= −13 (3 − 1) (2.10)
d) Volumenarbeit für eine isochore Zustandsänderung (Abb. 2.2c):
= const.y = 0 (2.11)
y1→4 =
Z 4
1
= −Z 4
1
= 0 (2.12)
e) Volumenarbeit für einen Kreisprozess im pV -Diagramm (Abb. 2.2d):
Wir kombinieren die Prozesse in Abb. 2.2b und Abb. 2.2c in Abb. 2.2d so, dass wir wiederzum Ausgangspunkt zurückkehren, führen also einen Kreisprozess 1→ 2→ 3→ 4→ 1
aus:
1→2 = −Z 2
1
= −12 (2 − 1) (2.13)
2→3 = 0 (2.14)
3→4 = −Z 4
3
= −34 (4 − 3) = −34 (1 − 2) (2.15)
4→1 = 0 (2.16)
=X
= −12 (2 − 1) + 0− 34 (1 − 2) + 0 (2.17)
= (34 − 12) (2 − 1) 6= 0 (2.18)
I Ergebnisse:
(1) Die Gesamtarbeit ist die im -Diagramm beschriebene Fläche!
(2) Beim Durchlaufen des vollständigen Kreisprozesses wird Arbeit zugeführt, obwohlam Ende derselbe Zustand ( ) erreicht wird, wie er am Anfang vorlag.
(3) Wenn 3 = 4 (beide 1 2) höher werden, wird weniger Arbeit umgesetzt.

2.2 Volumenarbeit 48
I Schlussfolgerungen:
• Die umgesetzte Arbeit hängt vom Weg ab. y Arbeit ist keine Zustandsgröße.
• Aber sind Zustandsgrößen, sie hängen nicht vom Weg ab, auf dem einZustand erreicht wird.

2.3 Der Erste Hauptsatz der Thermodynamik 49
2.3 Der Erste Hauptsatz der Thermodynamik
2.3.1 Experiment von Joule
I Abbildung 2.3: Versuch von Joule (Äquivalenz von Arbeit und Wärme).
I Beobachtungen:
• Mechanische Arbeit: = × × (2.19)
• Wärme: = ∆Wasser (2.20)
• Beobachtung: Beim Fallen des Gewichtes Temperaturerhöhung.• Schlussfolgerung:
|| = × ×∆ = || (2.21)
y
Arbeit und Wärme sind ineinander umwandelbare Formen von Energie. (2.22)
⇓

2.3 Der Erste Hauptsatz der Thermodynamik 50
2.3.2 Formulierung des ersten Hauptsatzes der Thermodynamik
Der 1. HS (= Energieerhaltungssatz) besteht aus drei zusammen gehörenden Teilaus-sagen:
(1) Es gibt eine Innere Energie
= Innere Energie (2.23)
ist eine Zustandsgröße, d.h. H = 0 (2.24)
(2) Die Innere Energie kann verändert werden. Die Änderung setzt sich dabei ausdem Anteil Arbeit und aus dem Anteil Wärme zusammen:
∆ = + (2.25)
bzw. differentiell geschrieben13
= + (2.26)
(3) In einem abgeschlossenen System ist die Innere Energie konstant (Energieerhal-tung!):
Im abgeschlossenen System ist: = const d.h. = 0 (2.27)
I Andere Formulierungen:
• Energie ist eine Erhaltungsgröße.• Es gibt keine Maschine, die aus dem Nichts Arbeit schafft.• Es gibt kein Perpetuum Mobile 1. Art.
2.3.3 Messvorschrift für ∆U
Nachdem wir eine Messvorschrift für schon kennen (über − R , siehe Abschnitt2.2.2), suchen wir Messvorschriften für und :
13 Wir schreiben und um anzudeuten, dass und keine Zustandsgroßen sind. Für differentielle
Änderungen einer Zustandsgröße, z.B. , schreiben wir dagegen .

2.3 Der Erste Hauptsatz der Thermodynamik 51
I Abbildung 2.4: Die Messvorschrift für ∆ wird über die Kompression oder Expansioneines idealen Gases in einem adiabatischen System ( = 0, aber 6= 0) festgelegt.
I Messvorschrift für ∆U : Für = 0 folgt aus dem 1. HS:
= (2.28)
bzw.∆ = (2.29)
• Adiabatische Kompression: ⇒ -Erhöhung, da zugeführte Arbeit in InnererEnergie gespreichert wird.
• Adiabatische Expansion: ⇒ -Erniedrigung, da Arbeit auf Kosten der InnerenEnergie geleistet wird.
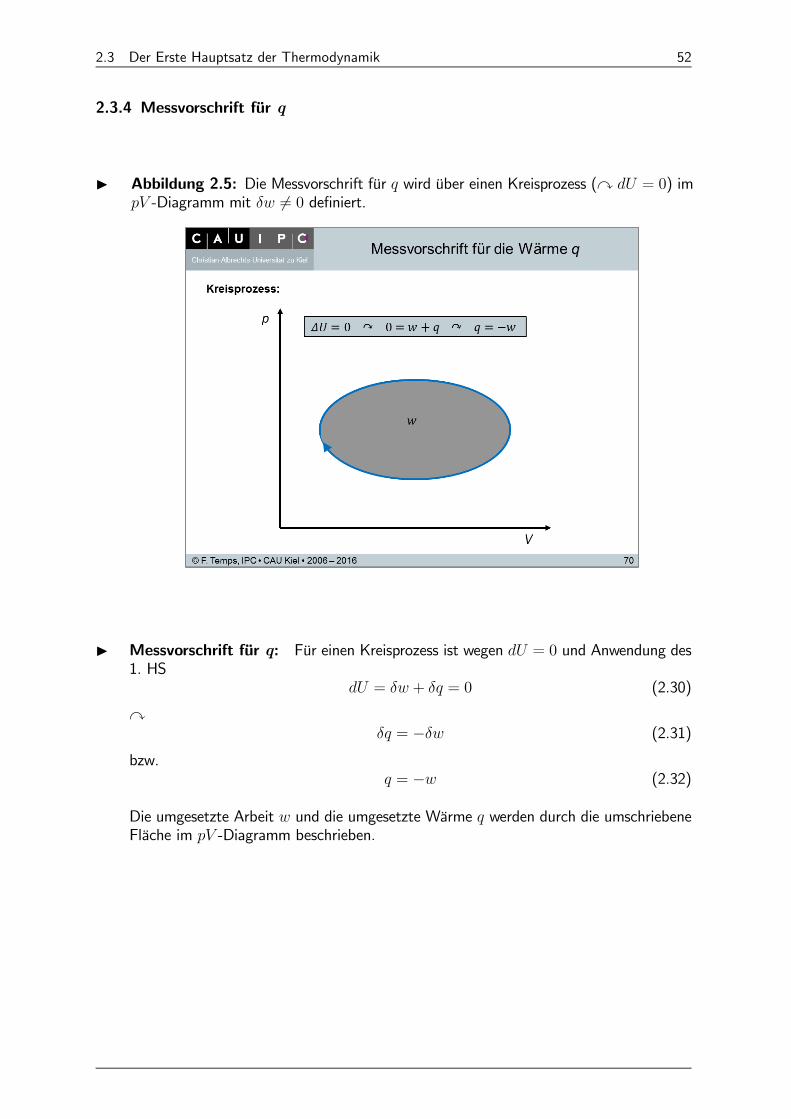
2.3 Der Erste Hauptsatz der Thermodynamik 52
2.3.4 Messvorschrift für q
I Abbildung 2.5: Die Messvorschrift für wird über einen Kreisprozess (y = 0) im -Diagramm mit 6= 0 definiert.
I Messvorschrift für q: Für einen Kreisprozess ist wegen = 0 und Anwendung des1. HS
= + = 0 (2.30)
y = − (2.31)
bzw. = − (2.32)
Die umgesetzte Arbeit und die umgesetzte Wärme werden durch die umschriebeneFläche im -Diagramm beschrieben.

2.3 Der Erste Hauptsatz der Thermodynamik 53
2.3.5 Temperaturabhängigkeit der Inneren Energie: Spezifische Wärme beikonstantem Volumen
Energiezufuhr in Form von Wärme oder in Form von Arbeit führt zu einer Tempera-turerhöhung des Systems an. Die Größe des -Anstiegs für einen Stoff hängt ab vonseiner spezifischer Wärme .
I Definition 2.3: Spezifische Wärme bei konstantem Volumen.
=
µ
¶
(2.33)
I Änderung der Inneren Energie bei einer Änderung der Temperatur:
∆ = 2 − 1 =
2Z1
(2.34)

2.4 Anwendungen des Ersten Hauptsatzes 54
2.4 Anwendungen des Ersten Hauptsatzes
2.4.1 Gay-Lussac-Versuch
I Abbildung 2.6: Irreversible Expansion eines idealen Gases in einem abgeschlossenenSystem in ein Vakuum (Gay-Lussac-Versuch): Anfangszustand
I Abbildung 2.7: Irreversible Expansion eines idealen Gases in einem abgeschlossenenSystem in ein Vakuum (Gay-Lussac-Versuch): Endzustand

2.4 Anwendungen des Ersten Hauptsatzes 55
• Abgeschlossenes System: = 0 (2.35)
= 0 (2.36)
= 0 (2.37)
• Versuchsdurchführung: Irreversible Expansion eines idealen Gases ins Vakuum:
— Ausgangszustand:
: 1 6= 0 2 = 0 (2.38)
= (2.39)
= (2.40)
= (2.41)
— Endzustand:
= (2.42)
= = 1 + 2 (2.43)
= = (2.44)
• Beobachtung: = = const (2.45)
• Schlussfolgerungen:
(1) Die Innere Energie bleibt konstant (1. HS für ein abgeschlossenesSystem!), obwohl sich und ändern, d.h. für das ideale Gas mussgelten µ
¶
= 0 (2.46)
und µ
¶
= 0 (2.47)
(2) Für ein ideales Gas hängt die Innere Energie nur von der Temperaturab, nicht von oder :
Ideales Gas = ( ) (2.48)
• Anmerkung: Die beiden Schlussfolgerungen gelten nur für ein ideales Gas. Fürein reales Gas würde sich bei der irreversiblen Expansion in ein Vakuum die Tem-peratur etwas ändern, da die zwischenmolekularen Anziehungs-/Abstoßungskräfteüberwunden werden müssen. Für ein reales Gas (und für Flüssigkeiten, Festkör-per) ist also allgemein = ( ).

2.4 Anwendungen des Ersten Hauptsatzes 56
2.4.2 Reversible isotherme Expansion eines idealen Gases
I Abbildung 2.8: Reversible isotherme Expansion eines idealen Gases.
I Reversible Prozesse: Reversible (“umkehrbare”) Prozessführung bedeutet, dassdie Zustandsänderung immer über Gleichgewichtszustände erfolgt (also hinreichendlangsam, sodass immer = gilt).
I Reversible isotherme Expansion eines idealen Gases:
• Für ideales Gas: = ( ) (2.49)
y∆ = + = 0 (2.50)
y = − (2.51)
• Vom System wird Arbeit geleistet. Damit = const bleibt, muss man Wärmezuführen.
• Dabei wurde in diesem Fall die gesamte zugeführte Wärme in Arbeit umgewandelt( = −).
• Die bei reversibler Prozessführung (d.h. System ist immer im Glgew.)geleistete Arbeit ist die maximal mögliche!!
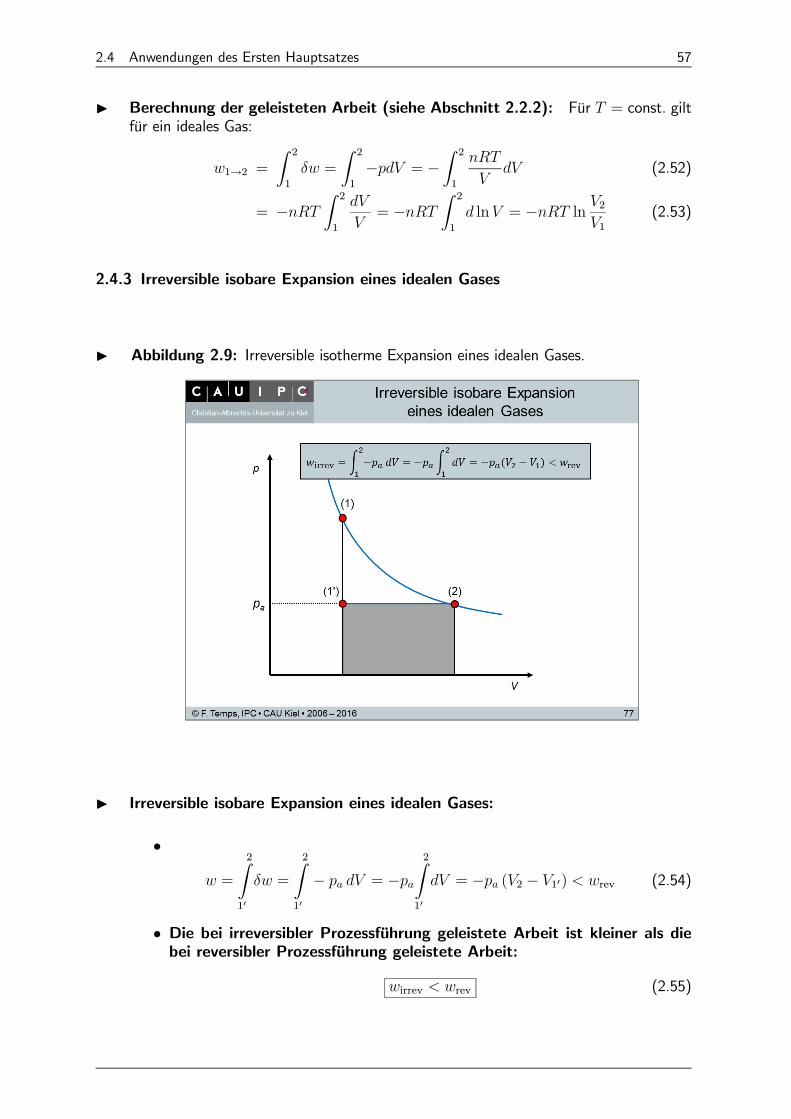
2.4 Anwendungen des Ersten Hauptsatzes 57
I Berechnung der geleisteten Arbeit (siehe Abschnitt 2.2.2): Für = const. giltfür ein ideales Gas:
1→2 =Z 2
1
=
Z 2
1
− = −Z 2
1
(2.52)
= −Z 2
1
= −
Z 2
1
ln = − ln 21
(2.53)
2.4.3 Irreversible isobare Expansion eines idealen Gases
I Abbildung 2.9: Irreversible isotherme Expansion eines idealen Gases.
I Irreversible isobare Expansion eines idealen Gases:
• =
2Z10
=
2Z10
− = −2Z
10
= − (2 − 10) rev (2.54)
• Die bei irreversibler Prozessführung geleistete Arbeit ist kleiner als diebei reversibler Prozessführung geleistete Arbeit:
irrev rev (2.55)
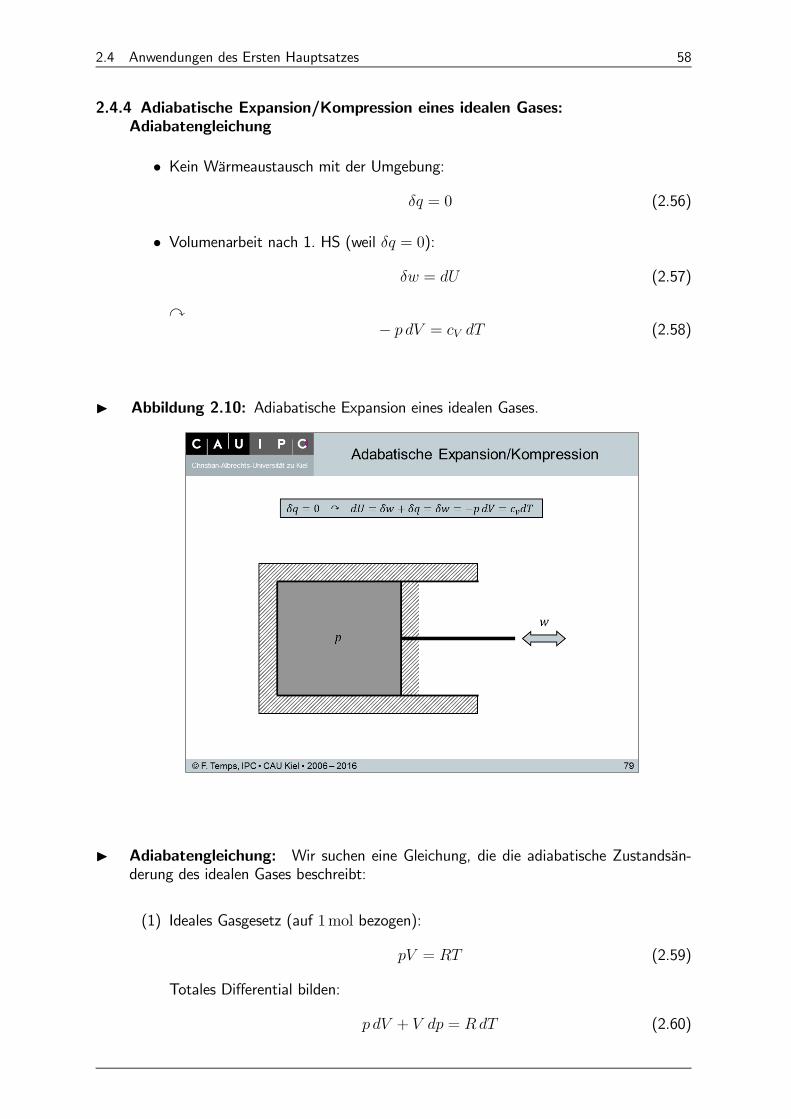
2.4 Anwendungen des Ersten Hauptsatzes 58
2.4.4 Adiabatische Expansion/Kompression eines idealen Gases:Adiabatengleichung
• Kein Wärmeaustausch mit der Umgebung: = 0 (2.56)
• Volumenarbeit nach 1. HS (weil = 0): = (2.57)
y− = (2.58)
I Abbildung 2.10: Adiabatische Expansion eines idealen Gases.
I Adiabatengleichung: Wir suchen eine Gleichung, die die adiabatische Zustandsän-derung des idealen Gases beschreibt:
(1) Ideales Gasgesetz (auf 1mol bezogen):
= (2.59)
Totales Differential bilden:
+ = (2.60)

2.4 Anwendungen des Ersten Hauptsatzes 59
(2) 1. HS (siehe oben):− = (2.61)
(3) Division von Gl. 2.60 durch Gl. 2.61:
− 1−
=
(2.62)
y1 +
+
= 0 (2.63)
y + +
= 0 (2.64)
y µ1 +
¶| z
≡
+ = 0 (2.65)
Mit der Abkürzung:
= 1 +
(2.66)
folgt + = 0 (2.67)
y
+
= 0 (2.68)
y ln = − ln (2.69)
(4) Integration von 0 bis bzw. 0 bis :
ln
0= − ln
0(2.70)
y µ
0
¶
=0
(2.71)
y = 0
0 = const (2.72)
(5) bzw. mit = :
−1 = 0−10 (2.73)
Diese Gleichung (bzw. Gl. 2.72) heißt Adiabatengleichung.
2.4 Anwendungen des Ersten Hauptsatzes 60
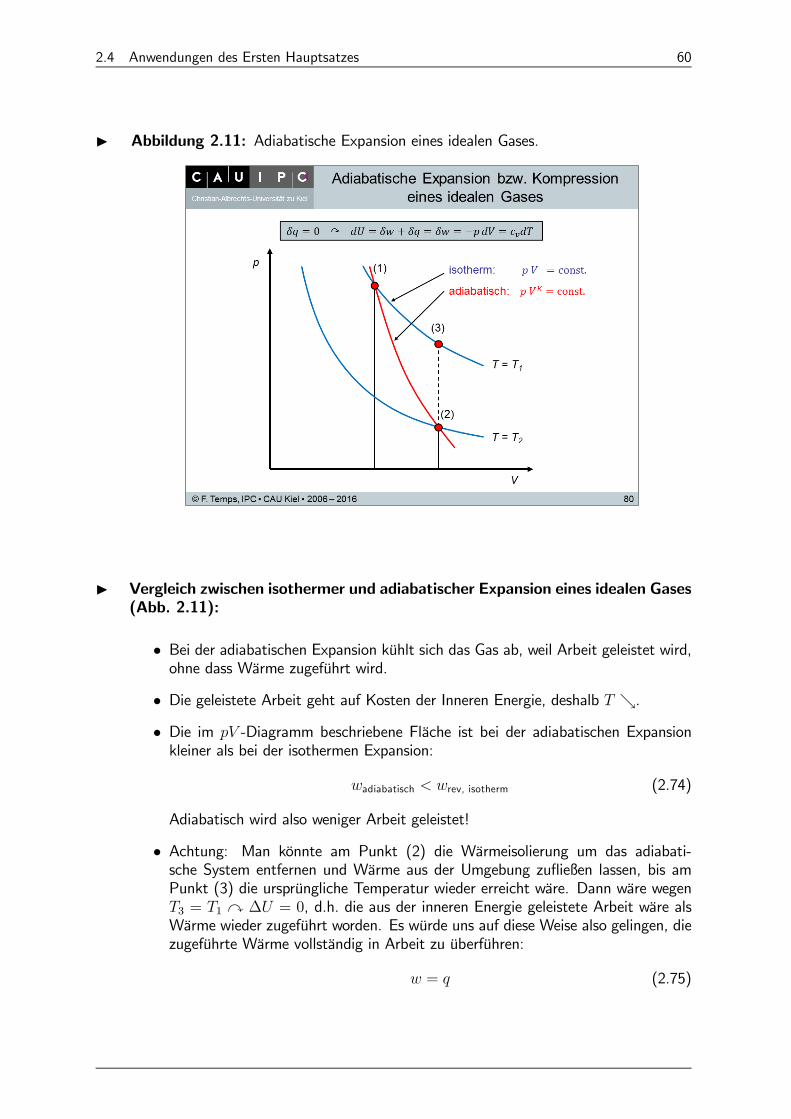
I Abbildung 2.11: Adiabatische Expansion eines idealen Gases.
I Vergleich zwischen isothermer und adiabatischer Expansion eines idealen Gases(Abb. 2.11):
• Bei der adiabatischen Expansion kühlt sich das Gas ab, weil Arbeit geleistet wird,ohne dass Wärme zugeführt wird.
• Die geleistete Arbeit geht auf Kosten der Inneren Energie, deshalb &.• Die im -Diagramm beschriebene Fläche ist bei der adiabatischen Expansionkleiner als bei der isothermen Expansion:
adiabatisch rev, isotherm (2.74)
Adiabatisch wird also weniger Arbeit geleistet!
• Achtung: Man könnte am Punkt (2) die Wärmeisolierung um das adiabati-sche System entfernen und Wärme aus der Umgebung zufließen lassen, bis amPunkt (3) die ursprüngliche Temperatur wieder erreicht wäre. Dann wäre wegen3 = 1 y ∆ = 0, d.h. die aus der inneren Energie geleistete Arbeit wäre alsWärme wieder zugeführt worden. Es würde uns auf diese Weise also gelingen, diezugeführte Wärme vollständig in Arbeit zu überführen:
= (2.75)

2.4 Anwendungen des Ersten Hauptsatzes 61
I Bei adiabatischer Expansion geleistete Arbeit:∗
= − (2.76)
mit
=0
0
(2.77)
y = −0
0
(2.78)
Integration vom Punkt (1) zum Punkt (2) liefert
1→2 = −1 1
2Z1
= +1
1 ×
1
− 1 × −(−1)¯21
(2.79)
= 11 ×
1
− 1 ×h−(−1)2 −
−(−1)1
i(2.80)
2.5 Die Enthalpie 62
2.5 Die Enthalpie H
2.5.1 Funktionsprinzip eines Bombenkalorimeters (Reaktionen bei V = const.)
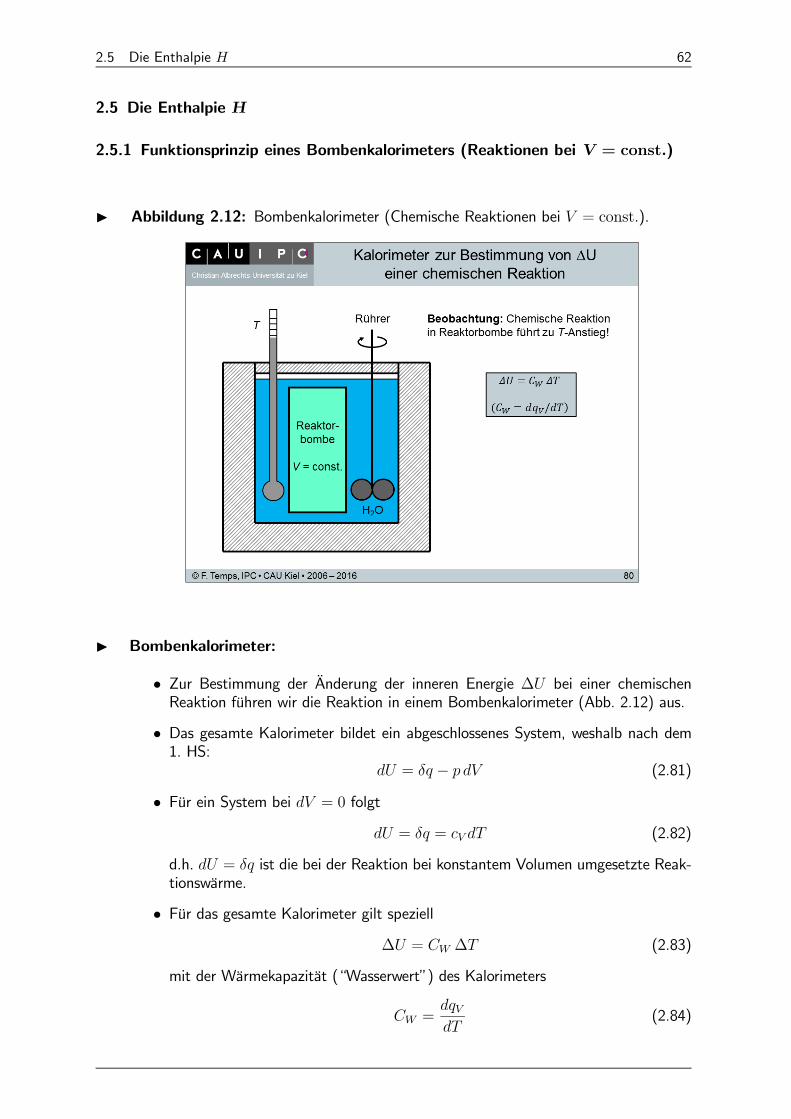
I Abbildung 2.12: Bombenkalorimeter (Chemische Reaktionen bei = const).
I Bombenkalorimeter:
• Zur Bestimmung der Änderung der inneren Energie ∆ bei einer chemischenReaktion führen wir die Reaktion in einem Bombenkalorimeter (Abb. 2.12) aus.
• Das gesamte Kalorimeter bildet ein abgeschlossenes System, weshalb nach dem1. HS:
= − (2.81)
• Für ein System bei = 0 folgt = = (2.82)
d.h. = ist die bei der Reaktion bei konstantem Volumen umgesetzte Reak-tionswärme.
• Für das gesamte Kalorimeter gilt speziell∆ = ∆ (2.83)
mit der Wärmekapazität (“Wasserwert”) des Kalorimeters
=
(2.84)

2.5 Die Enthalpie 63
I Bestimmung von C : Messung von ∆ bei Zufuhr von elektrischer Energie × × , oder Messung von ∆ für eine Reaktion mit bekanntem ∆ .
I Ergebnis:
Die Innere Energie gibt den Wärmeumsatz chemischer Reaktionen bei kon-stantem Volumen an.
(2.85)
2.5.2 Prozesse bei p = const.
Die meisten chemischen Prozesse laufen jedoch bei konstantem Druck (z.B. = 1atm)ab. Zur Beschreibung der energetischen Verhältnisse von Prozessen bei = const istdie Innere Energie nicht die geeignete Größe.
I Frage: Warum?
I Antwort: Bei der Reaktion bei = const. wird bei einer Änderung des Volumens Arbeitgegen die Umgebung geleistet, die somit nicht mehr für die abgeführte Reaktionswärme bei konstantem Druck zur Verfügung steht.
I Beispiel:
CO+1
2O2 → CO2 (2.86)
Änderung der Molzahl ∆ bei der Reaktion⇒ Volumenausdehnung (oder Volumenver-ringerung; je nach ∆) ⇒ Arbeit gegen Atmosphärendruck!
I Abbildung 2.13: Zur Definition der Enthalpie .

2.5 Die Enthalpie 64
2.5.3 Die Enthalpie H
Statt der Inneren Energie ist die Größe = + besser zur Beschreibung vonProzessen bei konstantem Druck geeignet.
I Definition 2.4: Enthalpie. = + (2.87)
I Totales Differential von H:
= + ( ) (2.88)
= + + (2.89)
= + + + (2.90)
= − + + (2.91)
= + (2.92)
y Bei = const (d.h. = 0): = (2.93)
I Schlussfolgerungen:
(1) Bei = const (d.h. = 0) gibt (und nicht ) die umgesetzte Wärmean!
(2) ist wie eine Zustandsgröße, berücksichtigt aber die gegen geleis-tete Arbeit!
2.5.4 Temperaturabhängigkeit der Enthalpie: Spezifische Wärme bei konstantemDruck
I Definition 2.5: Spezifische Wärme bei konstantem Druck.
=
µ
¶
(2.94)
I Enthalpieänderung bei Änderung der Temperatur:
∆ = 2 −1 =
2Z1
(2.95)

2.5 Die Enthalpie 65
I Differenz von c und c für ideale Gase:
− =
µ
¶
−µ
¶
(2.96)
=
µ +
¶
−µ
¶
(2.97)
=
µ +
¶
−µ
¶
(2.98)
=
µ
¶+−
µ
¶(2.99)
y − = (2.100)
I Allgemeingültiger Ausdruck für c − c :14
− = × 2
(2.101)
mit
=1
µ
¶
(2.102)
= − 1
µ
¶
(2.103)
2.5.5 Vergleich von Reaktionen bei V = const. und p = const.
a) Fest-Fest- oder Fest-Flüssig-Reaktionen
I Beispiel 2.1: 15
CaCO3(Calcit) → CaCO3(Aragonit) (2.104)
y∆ = ∆ +∆
¡¢
(2.105)
yfür = const.: ∆ = ∆ + ∆ (2.106)
Zahlenwerte:
Aragonit = 34 cm3mol (2.107)
Calcit = 37 cm3mol (2.108)
14 Herleitung siehe Wedler, S. 27ff.15 Der Index (verwendet wird manchmal auch ) steht für “reaction”, d.h. der Wert bezieht sich auf
die angegebene Reaktion.

2.5 Die Enthalpie 66
@ = 1bar = 105 Pa:∆ = −03 Jmol (2.109)
Zum Vergleich:∆
ª298 = −021 kJmol (2.110)
y ∆ ist außer bei sehr hohen Drucken (Geophysik!) vernachlässigbar. ¤

2.5 Die Enthalpie 67
b) Fest-Gas- oder Flüssig-Gas-Reaktionen
I Beispiel 2.2:
CO +1
2O2 → CO2 (2.111)
Berechnung von ƻ298:
∆ = ∆ +∆ ( ) (2.112)
= ∆ +∆ ( ) (2.113)
= ∆ + ∆ (2.114)
@ = 298K:
∆× = −12 = −124 kJmol (2.115)
Zahlenwerte für ∆ª298 und ∆
ª298:
∆ª298 = −28177 kJmol (2.116)
y∆
ª298 = ∆
ª298 −
1
2 = −28301 kJmol (2.117)
¤

2.6 Anwendung auf chemische Reaktionen 68
2.6 Anwendung auf chemische Reaktionen
2.6.1 Hess’scher Satz
Der Hess’sche Satz erlaubt die Berechnung von Reaktionsenthalpien für Reaktionen, dienicht direkt gemessen werden können, aus Daten für bekannte Reaktionen der beteiligtenReaktionspartner.
I Beispiel:
• Die Enthalpieänderungen für die folgenden beiden Reaktionen können direkt (z.B.in einem Bombenkalorimeter) bestimmt werden:
(R1) CGraphit + O2 → CO2 ∆(1) = −39351 kJmol
(R2) CO +1
2O2 → CO2 ∆(2) = −28301 kJmol
(2.118)
• Die Enthalpieänderungen für die Reaktion
(R3) CGraphit +1
2O2 → CO ∆(3) = ? (2.119)
kann dagegen nicht direkt gemessen werden, da die Reaktion normalerweise nichtbeim CO stehen bleibt, sondern weiterläuft. Bei hohen Temperaturen (Kohle-verbrennung) führt die Reaktion in der primären Oxidationszone einer Flammejedoch tatsächlich zunächst zu CO; CO2 entsteht erst bei der Weiteroxidation ineiner höheren Flammenzone. Die Kenntnis von ∆(3) ist sehr wichtig.
• Eine Berechnung von∆(3) gelingt uns jedoch nach dem Hess’schen Satz, indemwir über einen Umweg gehen:
C + O2(1)−→ CO2
(3)
&(2)
%CO +
1
2O2
Da eine Zustandsgröße ist, ist der Weg, auf dem wir von Reaktanden zu Pro-dukten gehen, gleichgültig. Durch Addition der Reaktionen (2) und (3) erhaltenwir
(R3) CGraphit + O2 → CO +1
2O2 ∆(3)
(R2) CO +1
2O2 → CO2 ∆(2)
(R3) + (R2) C + CO +3
2O2 → CO + CO2 +
1
2O2 ∆(3) +∆(2)
= (R1) C + O2 → CO2 = ∆(1)
(2.120)

2.6 Anwendung auf chemische Reaktionen 69
• Ergebnis:∆(1) = ∆(2) +∆(3) (2.121)
y
∆(3) = ∆(1) −∆(2) (2.122)
= −39351 kJmol− (−28298) kJmol (2.123)
= −11053 kJmol (2.124)
I Hess’scher Satz:
Die Enthalpieänderung der Gesamtreaktion setzt sich aus den Enthalpieän-derungen der einzelnen Teilreaktionen zusammen.
(2.125)
2.6.2 Standardbildungsenthalpien der Elemente
Es ist uns nicht möglich, absolute Werte von oder anzugeben. Dies ist jedoch fürdie Chemie kein Problem, da wir uns immer nur für die Änderungen von oder beieiner chemischen Reaktion interessieren. Wir können daher die∆ - bzw.∆-Werte fürchemische Reaktionen auf einen (per Konvention “willkürlich”) gewählten Bezugspunkt(= Referenz- oder Standardzustand, gekennzeichnet mit dem Index ª) beziehen.
I Standardzustand:ª = 1bar (2.126)
ª = 29815K (2.127)
Achtung: Ältere Tabellen benutzen als Standarddruck noch = 1atm. Außerdemwerden verschiedene Temperaturen verwendet, ist daher bei allen Werten unbedingtimmer anzugeben.
Eine neben ª = 29815K häufig verwendete Temperatur für den Standardzustand ist = 0K.
I Konvention:
ƻ298K (Elemente in physikalisch stabilster Modifikation) = 0 (2.128)
Der Index steht dabei für “formation”, die entsprechenden Enthalpien im Standard-zustand ª werden als Standardbildungsenthalpien bezeichnet.

2.6 Anwendung auf chemische Reaktionen 70
I Beispiele:
ƻ298 (H2298K) = 0 (2.129)
ƻ298 (O2298K) = 0 (2.130)
ƻ298 (N2298K) = 0 (2.131)
ƻ298
¡Hg298K
¢= 0 (2.132)
ƻ298 (Fe298K) = 0 (2.133)
ƻ298 (CGraphit298K) = 0 (2.134)
Aber:∆
ª298 (CDiamant298K) 6= 0 (2.135)
I Ionen: Ionen entstehen immer paarweise, z.B.:
H2O → H+ +OH− (2.136)
Hierbei bedeutet die wässrige Lösung (“hydratisiert”). Wegen der paarweisen Bildungwird eine weitere Standardbildungsenthalphie auf Null gesetzt:
ƻ298
³H+=1mol kg Lösungsmittel
´= 0 (2.137)
Bezugszustand (= “Standardzustand”) ist dabei die hypothetische “ideal verdünnte”Lösung der Molalität 1mol kg Lösungsmittel.
Wegen der bei konzentrierteren ionischen Lösungen (schon ab & 10−3mol kg Lm)großen Abweichungen vom idealen Verhalten muss dabei die Aktivität
= × (2.138)
verwendet werden ( = Aktivitätskoeffizient, = Molalität).
2.6.3 Standardbildungsenthalpien anderer chemischer Verbindungen
I Bestimmung der Standardbildungsenthalpien chemischer Verbindungen: DieStandardbildungsenthalpien anderer chemischer Verbindungen als der Elemente sind gle-ich den Reaktionsenthalpien für die Bildung der Verbindungen aus den chemischen Ele-menten, z.B.
CGraphit298K1 bar +O2298K1 bar → CO2298K1 bar (2.139)
y
∆ª298 = ∆
ª298 (CO2298K1 bar)| z
=
−∆ª298 (CGraphit298K1 bar)| z
=0
−∆ª298 (O2298K1 bar)| z
=0
(2.140)
= −39351 kJmol (2.141)
y = ∆
ª298 (CO2298K1 bar) = ∆
ª298 = −39351 kJmol (2.142)
Die Werte der Standardbildungsenthalpien der interessierenden chemischen Verbindun-gen sind tabelliert (siehe z.B. Anhang Atkins).

2.6 Anwendung auf chemische Reaktionen 71
I Beispiel für die Berechnung von Standardreaktionsenthalpien aus Standardbil-dungsenthalpien:
H2298K1 bar +1
2O2298K1 bar → H2O298K1 bar (2.143)
y
∆ª298 = ∆
ª298 (H2O298K1 bar)
−∆ª298 (H2298K1 bar)−
1
2∆
ª298 (O2298K1 bar) (2.144)
= −24182 kJmol− 0 kJmol− 0 kJmol (2.145)
= −24182 kJmol (2.146)
Aber: H2O298K1 bar entspricht nicht dem stabilsten Zustand.16 Deshalb ist zuberücksichtigen, dass
H2O298K1 bar → H2O298K1 bar (2.147)
mit
∆ª298 = ∆
ª298 (H2O298K1 bar)−∆
ª298 (H2O298K1 bar) (2.148)
= −∆Verdª298 (2.149)
= −44 kJmol (2.150)
Für die Reaktion
H2298K1 bar +1
2O2298K1 bar → H2O298K1 bar (2.151)
ergibt sich somit
∆ª298 = −241832 kJmol− 44 kJmol = −28582 kJmol (2.152)
2.6.4 Standardbildungsenthalpien und Entropien chemischer Verbindungen
Umfangreiche Sammlungen der aktuellen Werte der Standardbildungsenthalpien und derEntropien17 zahlreicher chemischer Verbindungen finden sich im Internet unter:
• http://webbook.nist.gov/• http://www.update.uu.se/~jolkkonen/pdf/CRC_TD.pdf .
Alle im folgenden tabellierten Werte gelten für ª = 1bar und = 29815K.
16 Bei der Verbrennung von H2 entsteht Wasserdampf bei hoher Temperatur. Die Temperaturab-
hängigkeit wird weiter unten in Abschnitt 2.6.6 betrachtet.17 Für die Angabe der absoluten Entropien beötigen wir den dritten Hauptsatz der Thermodynamik
(siehe Abschnitt 3.4).
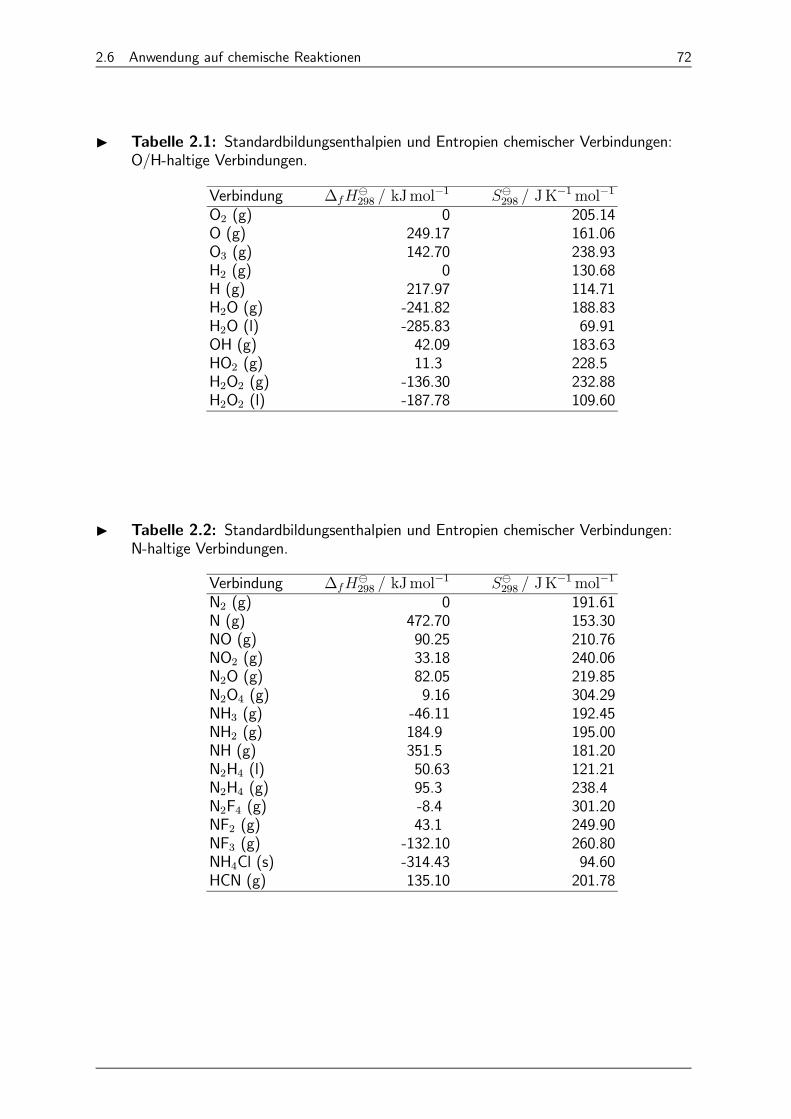
2.6 Anwendung auf chemische Reaktionen 72
I Tabelle 2.1: Standardbildungsenthalpien und Entropien chemischer Verbindungen:O/H-haltige Verbindungen.
Verbindung ƻ298 kJmol
−1 ª298 JK−1mol−1
O2 (g) 0 205.14O (g) 249.17 161.06O3 (g) 142.70 238.93H2 (g) 0 130.68H (g) 217.97 114.71H2O (g) -241.82 188.83H2O (l) -285.83 69.91OH (g) 42.09 183.63HO2 (g) 11.3 228.5H2O2 (g) -136.30 232.88H2O2 (l) -187.78 109.60
I Tabelle 2.2: Standardbildungsenthalpien und Entropien chemischer Verbindungen:N-haltige Verbindungen.
Verbindung ƻ298 kJmol
−1 ª298 JK−1mol−1
N2 (g) 0 191.61N (g) 472.70 153.30NO (g) 90.25 210.76NO2 (g) 33.18 240.06N2O (g) 82.05 219.85N2O4 (g) 9.16 304.29NH3 (g) -46.11 192.45NH2 (g) 184.9 195.00NH (g) 351.5 181.20N2H4 (l) 50.63 121.21N2H4 (g) 95.3 238.4N2F4 (g) -8.4 301.20NF2 (g) 43.1 249.90NF3 (g) -132.10 260.80NH4Cl (s) -314.43 94.60HCN (g) 135.10 201.78

2.6 Anwendung auf chemische Reaktionen 73
I Tabelle 2.3: Standardbildungsenthalpien und Entropien chemischer Verbindungen:C/H/O-haltige Verbindungen.
Verbindung ƻ298 kJmol
−1 ª298 JK−1mol−1
C (s, Diamant) 1.90 2.38C (s, Graphit) 0 5.74CO (g) -110.53 197.67CO2 (g) -393.51 213.74CH4 (g) -74.81 186.26C2H6 (g) -84.68 229.60C2H4 (g) 52.26 219.56C2H2 (g) 226.73 200.94C3H8 (g) -103.85 269.91C6H12 (l) -156.0 204.40C6H12 (g) -123.2 298.3C6H6 (g) 82.93 269.31CH3OH (g) -200.66 239.81C2H5OH (g) -235.10 282.70CH3 (g) 145.69 194.17CH2 (g) 386.4 193.9CH (g) 594.1 183.0CH2OH (g) -7.8 254.CH3O (g) 20.5 227.6CH2O (g) -108.60 218.80CHO (g) 43.1 224.7
I Tabelle 2.4: Standardbildungsenthalpien und Entropien chemischer Verbindungen:Halogen-haltige Verbindungen.
Verbindung ƻ298 kJmol
−1 ª298 JK−1mol−1
F2 (g) 0 202.78F (g) 78.99 158.75HF (g) -271.10 173.78Cl2 (g) 0 223.07Cl (g) 121.68 165.20HCl (g) -92.31 186.91Br2 (l) 0 152.23Br2 (g) 30.91 245.46Br (g) 111.88 175.02HBr (g) -36.40 198.70I2 (g) 62.44 260.69I2 (s) 0 116.14I2 (l) 13.52 150.36I (g) 106.76 180.80HI (g) 26.48 206.59

2.6 Anwendung auf chemische Reaktionen 74
I Tabelle 2.5: Standardbildungsenthalpien und Entropien chemischer Verbindungen:S-haltige Verbindungen.
Verbindung ƻ298 kJmol
−1 ª298 JK−1mol−1
S (s, rhomb.) 0 31.80S (s, monokl.) 0.33 32.60S (g) 278.81 167.82S2 (g) 128.37 228.18S8 (g) 102.40 430.87SO2 (g) -296.83 248.22SO3 (g) -395.72 256.76H2S (g) -20.63 205.79SF6 (g) -1209 291.82
I Tabelle 2.6: Standardbildungsenthalpien und Entropien chemischer Verbindungen:Ionen in wässriger Lösung.
Verbindung ƻ298 kJmol
−1 ª298 JK−1mol−1
H+ (aq) 0 0OH− (aq) -229.99 -10.75Cl− (aq) -167.16 56.50Na+ (aq) -240.12 59.00
Cu2+ (aq) 64.77 -99.60
SO2−4 (aq) -909.27 20.10HSO−4 (aq) -887.34 131.80NH+4 (aq) -132.51 113.40NO−3 (aq) -205.0 146.40
I Aufgabe 2.1: Berechnen Sie aus den Angaben in Tab. 2.3 die Standard-Reaktions-enthalpie für die Reaktion
CH4 +CO2 → CH3OH+CO (2.153)
¤
2.6.5 Standardreaktionsenthalpien
I Allgemeine Vorschrift zur Berechnung von Standardreaktionsenthalpien ausStandardbildungsenthalpien: Allgemein gilt
ƻ =
P∆
ª (Produkte)−P∆ª (Edukte) (2.154)
Für eine allgemeine Reaktion vom Typ
1B1 + 2B2 + → B + +1B+1 + (2.155)
ist
∆ª = ||∆
ª (B) + |+1|∆ª (B+1) +
− ¡|1|∆ª (B1) + |2|∆
ª (B2) + ¢
(2.156)

2.6 Anwendung auf chemische Reaktionen 75
I Abgekürzte Schreibweise:
ƻ =
P ∆
ª (B) (2.157)
mit den folgenden Konventionen:
= positiv für Produkte (2.158)
= negativ für Edukte (2.159)
I Exotherme und endotherme Reaktionen:
ƻ 0 : exotherm (2.160)
ƻ = 0 : thermoneutral (2.161)
ƻ 0 : endotherm (2.162)
2.6.6 Temperaturabhängigkeit der Reaktionsenthalpie
µ
¶
= (2.163)
y
∆ª ( ) = ∆
ª (0) +
Z0
³X (Produkte)−
X (Edukte)
´(2.164)
= ƻ (0) +
Z0
∆ (2.165)
Genaue Werte für ( ) (in Form von Potenzreihenentwicklungen) finden sich inTabellen.
I Häufig ausreichende Näherung für chemische Reaktionen: Wegen Kompensa-tion der Beiträge der Produkte und Edukte kann man oft annehmen, dass
∆ =X
(Produkte)−X
(Edukte) ≈ 0 (2.166)
2.6.7 Bindungsenthalpien (“Bindungsenergien”) Dª298
Bezug auf die Bildung der Atome aus den Elementen:
AB→ A+ B (2.167)
y
∆Dissª298 () = ∆
ª298 (AB)−∆
ª298 (A)−∆
ª298 (B) (2.168)
= ª298 (AB) (2.169)

2.6 Anwendung auf chemische Reaktionen 76
I Tabelle 2.7: Bindungsenergien einiger Moleküle.
Molekül → Atome ∆ª /
¡kJmol−1
¢H2 () → H + H +43584 = 2× 21792− 0O2 () → O + O 49834
N2 () → N + N 9454
F2 () → F + F 15798
CGraphit → C() 7173 (pro C-Atom)H2O() → H + OH 50188
OH → H + O 42505
H2O() → H + H + O 92693 (926932 = 46347)
2.6.8 Gitterenthalpie von NaCl
I Frage: Wie groß ist die Gitterenthalpie für die Bildung von NaCl() aus Na+()und Cl−
()
nachNa+() + Cl
−() → NaCl() (2.170)
I Antwort: Aufstellen eines Kreisprozesses für die Reaktionsenthalpie.
I Abbildung 2.14: Kreisprozesse zur Bestimmung der Gitterenthalpie und Hydratation-senthalpie von NaCl.

2.6 Anwendung auf chemische Reaktionen 77
I Tabelle 2.8: Kreisprozess zur Bestimmung der Gitterenthalpie von NaCl.
Reaktion ∆ª298 kJmol−1
NaCl() → Na() + 0.5 Cl2() −∆ª298
¡NaCl()
¢= +41115
Na() → Na() ∆Sublª298 (Na) = +10732
Na() → Na+()+ e− ∆IE
ª298 (Na) = +4983
0.5 Cl2() → Cl() 05 ∆ª298 (Cl2) = +12168
Cl() + e− → Cl−
()∆EA
ª298 (Cl) = −3512
Σ : NaCl() → Na+()+ Cl−
()−∆
ª298 = +78725
2.6.9 Hydratationsenthalpie von NaCl
I Frage: Wie groß ist die Hydratationsenthalpie von NaCl(), d.h. die Reaktionsenthalpiefür
Na+() + Cl−() → NaCl() (2.171)
I Antwort: Aufstellen eines Kreisprozesses für die Reaktionsenthalpie.
I Tabelle 2.9: Kreisprozess zur Bestimmung der Hydratationsenthalpie von NaCl.
Reaktion ∆ª298 kJmol−1
NaCl() → NaCl() −∆ª298
¡NaCl()
¢= − 388
NaCl() → Na() + 0.5 Cl2() −∆ª298
¡NaCl()
¢= +41115
Na() → Na() ∆ª298 (Na) = +10732
Na() → Na+()+ e− ∆
ª298 (Na) = +4983
0.5 Cl2() → Cl() 05 ∆ª298 (Cl2) = +12168
Cl() + e− → Cl−
()∆
ª298 (Cl) = −3512
Σ : NaCl() → Na+()+ Cl−
()−∆
ª298
¡NaCl()
¢= +7834
yNa+() + Cl
−() → NaCl() ∆
ª298 = −7834 kJmol (2.172)
2.6.10 Hydratationsenthalpie des H+- und des Cl−-Ions
H+() → H+() ∆ª298 = −1090 kJmol (2.173)
Cl−() → Cl−() ∆ª298 = − 364 kJmol (2.174)

2.6 Anwendung auf chemische Reaktionen 78
2.6.11 Verbrennungsenthalpien
I Tabelle 2.10: Verbrennungswärmen einiger Brennstoffe (Werte in kJ/mol).
Reaktanden → Produkte ∆Verbrª298 ∆Verbr
ª298
pro Formelumsatz pro O2H2 + 0.5 O2 → H2O() − 24183) −48366
→ H2O() − 28583) −57166CH4 + 2 O2 → 2 H2O() + CO2 − 80232 −40116C2H6 + 3.5 O2 → 3 H2O() + 2 CO2 −142783 −40795C2H4 + 3 O2 → 2 H2O() + 2 CO2 −132326 −44299C2H2 + 2.5 O2 → H2O() + 2 CO2 −125558 −50223C6H6 + 7.5 O2 → 3 H2O() + 6 CO2 −316948 −42260CH3OH + 1.5 O2 → 2 H2O() + CO2 − 6765 −4510C() + O2 → CO2 − 39351 −39351CO + 0.5 O2 → CO2 − 28301 −56602
)Unterer Heizwert. )Oberer Heizwert.
• Man beachte, dass bei der Verbrennung in Luft pro O2 immer 4 N2 beteiligtsind, die frei gewordene Verbrennungswärme aufnehmen und damit also die Flam-mentemperatur im Vergleich zur Verbrennung in reinem O2 herabsetzen.
I Schlussfolgerungen auf die Stabilitäten der Brennstoffe:
(1) CH4 hat unter den KW die höchste CH-Bindungsenergie (ist am stabilsten).
(2) C2H4 und C2H2 sind vergleichsweise instabil (⇒ große Verbrennungsenthalpien)
(3) Bei C6H6 sieht man den Effekt der Aromatizität.
2.6.12 Berechnung der adiabatischen Flammentemperatur
I Beispiel:CH4 (298K) + 2 O2 + 8 N2 → CO2 + 2 H2O+ 8 N2 (2.175)
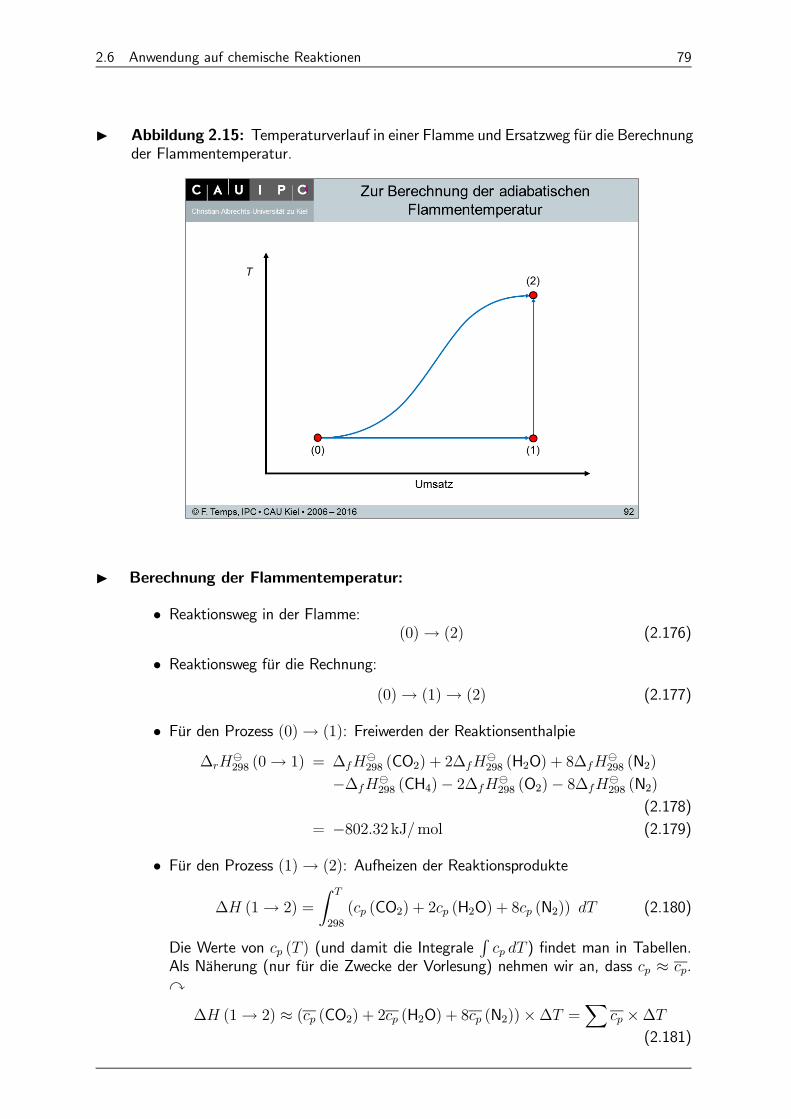
2.6 Anwendung auf chemische Reaktionen 79
I Abbildung 2.15: Temperaturverlauf in einer Flamme und Ersatzweg für die Berechnungder Flammentemperatur.
I Berechnung der Flammentemperatur:
• Reaktionsweg in der Flamme:(0)→ (2) (2.176)
• Reaktionsweg für die Rechnung:(0)→ (1)→ (2) (2.177)
• Für den Prozess (0)→ (1): Freiwerden der Reaktionsenthalpie
∆ª298 (0→ 1) = ∆
ª298 (CO2) + 2∆
ª298 (H2O) + 8∆
ª298 (N2)
−∆ª298 (CH4)− 2∆
ª298 (O2)− 8∆
ª298 (N2)
(2.178)
= −80232 kJmol (2.179)
• Für den Prozess (1)→ (2): Aufheizen der Reaktionsprodukte
∆ (1→ 2) =
Z
298
( (CO2) + 2 (H2O) + 8 (N2)) (2.180)
Die Werte von ( ) (und damit die IntegraleR ) findet man in Tabellen.
Als Näherung (nur für die Zwecke der Vorlesung) nehmen wir an, dass ≈
y
∆ (1→ 2) ≈ ( (CO2) + 2 (H2O) + 8 (N2))×∆ =X
×∆
(2.181)

2.6 Anwendung auf chemische Reaktionen 80
• Ergebnis (mit Zahlenwerten für aus Tabellen):−∆
ª298(0→ 1) = ∆ (1→ 2) (2.182)
y∆ ≈ 1900K (2.183)

2.7 Der Joule-Thomson-Effekt∗ 81
2.7 Der Joule-Thomson-Effekt∗
Die bislang betrachteten Phänomene konnten wir als chemische Reaktionen schreiben.Dies geht jedoch nicht immer. Der Joule-Thomson-Effekt, ein bei der Gasverflüssigungausgenutzter Realgaseffekt, ist ein Beispiel.
I Abbildung 2.16: Joule-Thomson-Effekt.
I Beobachtung:2 6= 1 (2.184)
Meist:2 1 (2.185)
⇒ Gasverflüssigung.
I Die adiabatische Entspannung des Gases durch eine Drossel ist ein isenthalpis-cher Prozess:
• Bei adiabatischer Entspannung des Gases durch die Drossel geleistete Arbeit: = − (22 − 11) (2.186)
• Anwendung des 1. HS, für = 0:∆ = 2 − 1 = = − (22 − 11) (2.187)
y2 + 22 = 1 + 11 (2.188)
y2 = 1 (2.189)

2.7 Der Joule-Thomson-Effekt∗ 82
I Definition 2.6: Joule-Thomson-Koeffizient. Die erreichte Temperaturänderung ist vonder Druckdifferenz ahängig. Wir definieren deshalb den Joule-Thomson-Koeffizientenals
=
µ
¶
(2.190)
I Berechnung des Joule-Thomson-Koeffizienten:∗
• Drücken wir als Funktion von und aus, erhalten wir für beim JT-Effekt( = 0)
=
µ
¶| z
=
+
µ
¶| z
=?
= 0 (2.191)
y µ
¶
= −µ
¶
(2.192)
• Generell gilt außerdem die zyklische Beziehungµ
¶
×µ
¶
×µ
¶
= −1 (2.193)
• Der 2. HS liefert für = + = 0 (2.194)
y mit =. . .
=
"µ
¶
+
µ
¶
#+ (2.195)
=
µ
¶
+
∙ ×
µ
¶
+
¸ (2.196)
Dabei sind µ
¶
=
(2.197)
und, wegen = − und dem Schwarz’schen Satz für die gemischtenzweiten Ableitungen, µ
¶
= −µ
¶
= −× (2.198)
mit dem thermischen Ausdehnungskoeffizienten
=1
µ
¶
(2.199)
• Ergebnis für : = + [ − × ] = + (1− ) = 0 (2.200)

2.7 Der Joule-Thomson-Effekt∗ 83
• Ergebnis für : µ
¶
=
( − 1) = (2.201)
y Der JT-Koeffizient kann größer oder kleiner als 0 sein.
• für ein ideales Gas:
=
(2.202)
y =
1
µ
¶
=1
=
=1
(2.203)
y =
( − 1) =
µ1
− 1
¶= 0 (2.204)
Der JT-Effekt tritt also nur bei realen Gasen auf!
• für ein reales Gas (van der Waals-Gas, bezogen auf 1mol):³+
2
´( − ) = (2.205)
y − +
−
2= (2.206)
Hieraus folgt unter Vernachlässigung des kleinen Terms
2bei kleinen Drucken
= + −
≈ + −
(2.207)
y ≈
+³−
´(2.208)
y µ
¶
=
+
2(2.209)
Einsetzen dieses Ergebnisses in die Gl. 2.201 für , die wir schreiben könnenals
=
µ
¶
−
(2.210)
liefert uns einen Wert für abhängig vom van der Waals Parameter
=
+
−
(2.211)
y wegen ≈
=
(2.212)
• Anmerkung: Über Messungen des JT-Efffektes kann man sehr genaue Zustands-gleichungen ableiten.

2.7 Der Joule-Thomson-Effekt∗ 84
2.8 Stoßwellen∗
I Abbildung 2.17: Stoßwellen.
Bei einer Stoßwelle, bei der sehr hohe Gasgeschwindigkeiten (Überschallgeschwindigkeit)
auftreten, ist zusätzlich zur Enthalpie die kinetische Energie2
2des Gases zu berück-
sichtigen.
• Bezogen auf die Masseneinheit ist deshalb anzusetzen
1 +212= 2 +
222
(2.213)
• Ausserdem ist der Druck:1 + (11)| z
= Massenfluss
1 = 2 + (22) 2 (2.214)
• Kontinuitätsgleichung: Massenstrom durch die Querschnittsfläche an denPunkten (1) und (2):
1 = 111 = 111 (2.215)
y ·1 = 111 = 222 =
·2 (2.216)
• Impulserhaltungssatz:11 = 22 (2.217)

2.8 Stoßwellen∗ 85
3. Der Zweite Hauptsatz der Thermodynamik
3.1 Grundlegende Experimente und Befunde zum 2. Hauptsatz
Es gibt eine lange Reihe von experimentellen Befunden, die mit dem 1. HS im Einklangstehen, durch diesen aber nicht erklärt werden:
• Versuch von Joule (Äquivalent von Arbeit und Wärme): Ein Gewicht fällt stetsbergab (nie bergauf).
• Versuch von Gay-Lussac: Ein Gas füllt spontan das gesamte zur Verfügung ste-hende Volumen aus (nie umgekehrt).
• Temperaturausgleich: Werden zwei unterschiedlich heiße Körper in Kontakt ge-bracht, kühlt sich der heißere ab, der kältere erwärmt sich (nie umgekehrt).
• Diffusion zweier Stoffe ineinander (nie spontane Trennung der gemischten Stoffe).• Lösung eines Salzes in Wasser.• Chemische Reaktionen laufen spontan in eine bestimmte Richtung ab, nichtumgekehrt.
I Abbildung 3.1: Experimentelle Beobachtungen zum 2. Hauptsatz.

3.1 Grundlegende Experimente und Befunde zum 2. Hauptsatz 86
I Irreversible Prozesse: Die genannten Prozesse könnten nach dem 1. HS auch inumgekehrter Richtung ablaufen (Energieerhaltung gilt in beide Richtungen), sie tun esvon selbst (spontan) aber nicht. Man sagt deshalb, die Prozesse sind irreversibel.
Zur Umkehrung irreversibler Prozesse muss Aufwand geleistet werden, der zu einer Än-derung an der Umgebung führt.
I Erklärung für spontanen Verlauf in eine bestimmte Richtung: ⇒ ZweiterHauptsatz der Thermodynamik.

3.2 Der Zweite Hauptsatz der Thermodynamik 87
3.2 Der Zweite Hauptsatz der Thermodynamik
I Drei Teilaussagen:
• Es gibt eine Zustandsfunktion genannt Entropie ,18
= Entropie (3.1)
die definiert ist über
=
(3.2)
wobei die auf reversiblem Weg bei der Temperatur umgesetzte Wärmeist.
• In einem abgeschlossenen System gilt
irrev.≥rev.
0 (3.3)
• Im Glgew. ist = 0 und hat ein Maximum.
I Andere Formulierungen:
• Für einen beliebigen Prozess in einem nichtabgeschlossenen System ist
gesamt = System + Umgebungirrev.≥rev.
0 (3.4)
• Es gibt keine periodisch arbeitende Maschine, die Wärme vollständig in Arbeitumwandelt.
• Es gibt kein Perpetuum Mobile 2. Art.
18 Von griech. = in Umwandlung.
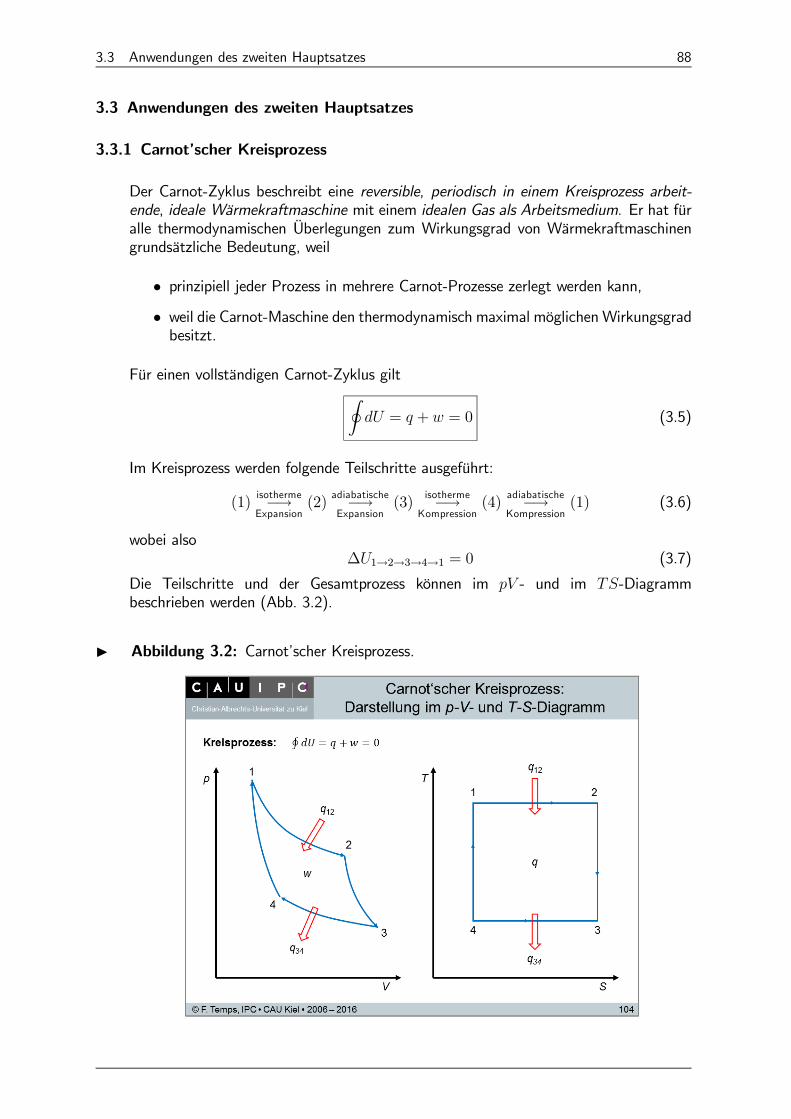
3.3 Anwendungen des zweiten Hauptsatzes 88
3.3 Anwendungen des zweiten Hauptsatzes
3.3.1 Carnot’scher Kreisprozess
Der Carnot-Zyklus beschreibt eine reversible, periodisch in einem Kreisprozess arbeit-ende, ideale Wärmekraftmaschine mit einem idealen Gas als Arbeitsmedium. Er hat füralle thermodynamischen Überlegungen zum Wirkungsgrad von Wärmekraftmaschinengrundsätzliche Bedeutung, weil
• prinzipiell jeder Prozess in mehrere Carnot-Prozesse zerlegt werden kann,• weil die Carnot-Maschine den thermodynamisch maximal möglichenWirkungsgradbesitzt.
Für einen vollständigen Carnot-Zyklus giltI = + = 0 (3.5)
Im Kreisprozess werden folgende Teilschritte ausgeführt:
(1)isotherme−→Expansion
(2)adiabatische−→Expansion
(3)isotherme−→Kompression
(4)adiabatische−→Kompression
(1) (3.6)
wobei also∆1→2→3→4→1 = 0 (3.7)
Die Teilschritte und der Gesamtprozess können im - und im -Diagrammbeschrieben werden (Abb. 3.2).
I Abbildung 3.2: Carnot’scher Kreisprozess.

3.3 Anwendungen des zweiten Hauptsatzes 89
I Betrachtung der Teilschritte: ( = 1mol)
(1) Reversible isotherme Expansion (1)→ (2):
= 0y = 0 (3.8)
12 = −Z 2
1
= −Z 2
1
= −
Z 2
1
ln = −12 ln 21
(3.9)
12 = −12 = 12 ln2
1(3.10)
∆12 =
Z 2
1
=
Z 2
1
rev
12=
Z 2
1
−12
= ln2
1(3.11)
Da 2 1 y∆12 0 (3.12)
(2) Reversible adiabatische Expansion (2)→ (3):
23 = 0 (3.13)
y∆23 = 0 (3.14)
Die reversible adiabatische Expansion ( = 0) ist isentropisch.
(3) Reversible isotherme Kompression (3)→ (4):
= 0y = 0 (3.15)
34 = = −34 ln 43
(3.16)
34 = −34 = 34 ln4
3(3.17)
∆34 =
Z 4
3
=
Z 4
3
rev
34= ln
4
3(3.18)
Da 4 3 y∆34 0 (3.19)
(4) Reversible adiabatische Kompression (4)→ (1):
41 = 0 (3.20)
y∆41 = 0 (3.21)

3.3 Anwendungen des zweiten Hauptsatzes 90
I Verhältnis der Temperaturen T12 und T34 und der entsprechenden Volumina:Anwendung der Adiabatengleichung (Gl. 2.73):
12
34=
µ2
3
¶−1=
µ1
4
¶−1(3.22)
y2
3=
1
4(3.23)
y2
1=
3
4(3.24)
I Zusammenfassung pro Umlauf:
(1) Entropieänderung pro Umlauf:
∆ª = ∆12 +∆23 +∆34 +∆41 (3.25)
= ln2
1+ 0 + ln
4
3+ 0 (3.26)
= ln2
1− ln
3
4(3.27)
= ln2
1− ln
2
1(3.28)
= 0 (3.29)
Dieses Ergebnis wird für einen Kreisprozess auch genau so erwartet, da eineZustandsgröße sein soll.
(2) Umgesetzte Wärme pro Umlauf:
umgesetzte Wärme = umschriebene Fläche im -Diagramm (3.30)
ª =
Zª (3.31)
= 12 + 23 + 34 + 41 (3.32)
= 12 ln2
1+ 0 +34 ln
4
3+ 0 (3.33)
= 12 ln2
1−34 ln
2
1(3.34)
= + (12 − 34) ln2
1(3.35)
0 (3.36)
(3) Geleistete Arbeit pro Umlauf:
geleistete Arbeit = umschriebene Fläche im -Diagramm (3.37)

3.3 Anwendungen des zweiten Hauptsatzes 91
ª = −Zª = ∆ª − ª = −ª (3.38)
= − (12 − 34) ln2
1(3.39)
0 (3.40)
I Ergebnisse:
• Wir sehen, dass bei einem Umlauf ein Teil der bei der hohen Temperatur 12 = (“hot”) aufgenommenen Wärme 12 = bei der niedrigen Temperatur 34 = (“cold”) als Wärme q34 = an die Umgebung abgegeben wird.
• Nur der Differenzbetrag || = | |− || wird in Arbeit umgewandelt.
• Der Wirkungsgrad =¯
¯jeder Carnot-Maschine ist begrenzt!!
• Die Carnot-Maschine hat den maximal möglichen Wirkungsgrad aller Wärmekraft-maschinen!
• Jede praktische, reale periodisch arbeitende Wärmekraftmaschine hat einen gerin-geren Wirkungsgrad.
3.3.2 Wirkungsgrade verschiedener idealer Wärmemaschinen
I Definition 3.1: Wirkungsgrad:
=Nutzen
Aufwand(3.41)

3.3 Anwendungen des zweiten Hauptsatzes 92
I Abbildung 3.3: Wirkungsgrade verschiedener Wärmemaschinen.
a) Carnot’sche Wärmekraftmaschine (Abb. 3.3 a)
I Arbeitsschema:
• arbeitet reversibel,• entnimmt aus einem heißen Reservoir = Brenner (“hot”; Temperatur ) dieWärme ( 0),
• verrichtet die Arbeit || ( 0),
• gibt an ein kälteres Reservoir = Umgebung (“cold”; Temperatur = ) dieWärme || = − || ab ( 0).
I Wirkungsgrad η:
=||
=
(12 − 34) ln2
1
12 ln2
1
=12 − 34
12(3.42)
y
= −
1 (3.43)

3.3 Anwendungen des zweiten Hauptsatzes 93
I Ergebnisse:
• Der Wirkungsgrad einer Wärmekraftmaschine ist immer 1.• Die (reversibel arbeitende) Carnot-Maschine hat den maximal erreichbarenWirkungsgrad (siehe Abschnitt Allgemeine Prozesse weiter unten).
• Der real erreichbare Wirkungsgrad ist immer kleiner als der theoretische Wirkungs-grad der Carnot-Maschine (Reibung, unvollständige Reversibilität, . . . )
I Vergleich der Wirkungsgrade verschiedener Systeme:
• Dampfmaschine:
= 1−
= 1− 300
930= 068 = max (3.44)
• Motor mit Wasserkühlung ( = 400K):
= 1−
= 1− 400
930= 057 = max (3.45)
• Solarzellen: ≈ 025 (3.46)
• Brennstoffzellen: ≈ 10 (3.47)
Im Idealfall kann eine Brennstoffzelle sogar einen Wirkungsgrad 100% haben(siehe Kapitel 7).
b) Wärmepumpe (Abb. 3.3 b)
I Arbeitsschema: Umgekehrter Carnot-Prozess.
• entnimmt aus einem kalten Reservoir (Temperatur ) die Wärme .• es wird Arbeit zugeführt.
• gibt an ein wärmeres Reservoir (Temperatur ) die Wärme | | = + ab.
I Anwendung: Heizzwecke.
I Wirkungsgrad η:
=| ||| =
1
−
=
− 1 (3.48)

3.3 Anwendungen des zweiten Hauptsatzes 94
c) Kühlschrank (Abb. 3.3 d)
I Arbeitsschema: Umgekehrter Carnot-Prozess, aber mit q als Nutzen.
• arbeitet reversibel.• entnimmt aus einem kalten Reservoir = Kühlschrank (Temperatur ) die Wärme.
• es wird Arbeit zugeführt.
• gibt an ein wärmeres Reservoir = Umgebung (Temperatur = ) die Wärme| | = + ab.
I Wirkungsgrad η:
=
=
− (3.49)
I Achtung: → 0 wenn → 0 (3.50)
⇒ Dritter Hauptsatz der Thermodynamik: Es ist unmöglich, den absoluten Tempera-turnullpunkt zu erreichen.
d) Kalte Wärmekraftmaschine (Abb. 3.3 c)
I Arbeitsschema:
• arbeitet analog zur normalen Wärmemaschine.• entnimmt aus einem heißen Reservoir = Umgebung (Temperatur = ) dieWärme ( 0).
• verrichtet die Arbeit || ( 0).
• gibt an ein kälteres Reservoir (Temperatur ) die Wärme || = − || ab( 0).
I Wirkungsgrad η :
max =
=
−
=
−
1 (3.51)

3.3 Anwendungen des zweiten Hauptsatzes 95
e) Zusammenfassung
(1) Jeder reversible Prozess ist in eine Reihe von Carnot-Prozessen zerlegbar.
(2) Der Carnot-Prozess gibt den maximal möglichen Wirkungsgrad an.
(3) Die angestellten Überlegungen gelten auch für andere Arbeitsmedien als ein ide-ales Gas, da diese immer mit einer Carnot-Maschine mit idealem Gas als Ar-beitsmedium gekoppelt werden können.
(4) In der Praxis interessiert der effektive Wirkungsgrad bezogen auf den thermody-namisch maximal möglichen Wirkungsgrad, z.B.
=nutzbareArbeit
maximal nutzbareArbeit(3.52)
3.3.3 Gay-Lussac-Versuch
Für den Gay-Lussac-Versuch kann die Entropieänderung nicht direkt berechnet werden,da die Expansion des Gases in ein Vakuum irreversibel ist. Aber da eine Zustandsgrößeist, spielt der Weg, auf dem der Endzustand erreicht wird, keine Rolle. Man kann dahereinen reversiblen Ersatzweg suchen, auf dem der gleiche Endzustand erreicht wird, und
die Entropieänderung dafür überR =
R rev
berechnen.
I Abbildung 3.4:

3.3 Anwendungen des zweiten Hauptsatzes 96
• Irreversible Expansion des idealen Gases in einem abgeschlossenen System insVakuum vom Volumen = 1 auf das Volumen = 21:
∆ = 0 (3.53)
= 0 (3.54)
= 0 (3.55)
∆ =
Z
= ?? (3.56)
• Reversibler Ersatzweg: Isotherme reversible Expansion des idealen Gases vom Vol-umen = 1 auf das Volumen = 21 unter Arbeitsleistung und Wärmezu-fuhr:
∆ = + = 0 (3.57)
= − = − (3.58)
y
∆ =
Z
=
Z
rev
=
Z
=
Z
= ln
0 (3.59)
3.3.4 Temperaturausgleich
Temperaturausgleich zwischen zwei gleich großen, identischen Körpern (jeweils 1, 2 =1mol) mit verschiedenen Anfangstemperaturen führt zur mittleren Temperatur:
=1 + 2
2(3.60)
I Abbildung 3.5:

3.3 Anwendungen des zweiten Hauptsatzes 97
I Berechnung der Entropieänderung: Der Wärmeübergang muss langsam, immer imGlgew. und reversibel erfolgen, damit wir ∆ berechnen können:
∆ = ∆1 +∆2 =
Z1
1
+
Z2
2
=
Z1
+
Z2
(3.61)
=
µln
1+ ln
2
¶(3.62)
= ln 212
= ln(1 + 2)
2
412(3.63)
Um zu sehen, ob ∆
0, müssen wir untersuchen, ob (1 + 2)
2
412:
(1 + 2)2= 21 + 212 + 22 (3.64)
= 21 + 412 + 22 − 212 (3.65)
= (1 − 2)2+ 412 (3.66)
412 (3.67)
y 0 (3.68)
3.3.5 Standardbildungsentropien ∆Sª298 bezogen auf die Elemente
I Frage: Wie können wir die Entropieänderung bei einer chemischen Reaktion bestim-men?
I Antwort:∆
ª298 (Elemente in stabilstem Zustand) = 0 (3.69)
y∆
ª298 (Chemische Verbindungen) = (3.70)
analog zu ƻ298.
I Anmerkung 3.1: Wir werden später allerdings anders vorgehen, da wir mithilfe des3. Hauptsatzes für die Entropien Absolutwerte (ª298) angeben können (siehe Abschnitt3.4).
3.3.6 T -Abhängigkeit von S
µ
¶
=
µrev
¶
= y rev = (3.71)
y µ
¶
=
(3.72)
y ( ) = 0 +
Z
0
(3.73)
3.3 Anwendungen des zweiten Hauptsatzes 98
3.3.7 ∆S für Phasenumwandlungen
Phasenumwandlungen sind reversibel, außerdem ist = const. und = const. undU = ∆U.
y
∆U =∆U
U(3.74)
3.3.8 Thermodynamische Wirkungsgrade praktischer Wärmekraftmaschinen*
a) Stirling’scher Kreisprozess und Stirling-Motor
In der Praxis werden Carnot-Maschinen aufgrund hoher Reibungs- und anderer Verlustenicht realisiert. Eine praktisch eher realiserbare verwandte Maschine ist die Stirling-Maschine. Es laufen Versuche, einen Verbrennungsmotor als Stirling-Motor zu betreiben.
I Arbeitsmedium: Gas (Luft, Stickstoff, Helium).
I Abbildung 3.6: Stirling’scher Kreisprozess.
I 1→ 2: Isotherme Kompression.
∆ = 0 (3.75)
• Wärmeabgabe an kaltes Reservoir.

3.3 Anwendungen des zweiten Hauptsatzes 99
I 2→ 3: Isochore Kompression durch Wärmeaufnahme aus Speicherreservoir.
∆ = 0 (3.76)
• Wärmeaufnahme =R aus einem Speicherreservoir ⇒ -Anstieg.
I 3→ 4: Isotherme Expansion.∆ = 0 (3.77)
• Wärmeaufnahme aus heißem Reservoir.
I 4→ 1: Isochore Abkühlung durch Wärmeabgabe an Speicherreservoir.
∆ = 0 (3.78)
• Wärmeabgabe 0 =R0 an Speicherreservoir ⇒ -Abnahme
I Speicherreservoir: Z 3
2
≈Z 4
1
0 (3.79)
y ≈ 0 (3.80)
0 kann in einem Speicher geparkt werden und im nächsten Zyklus wieder zugeführtwerden.
I Entropieänderung: Aus dem gleichen Grund giltZ 3
2
≈
Z 4
1
0
y3 − 2 = 4 − 1 (3.81)
bzw.4 − 3 = 1 − 2 (3.82)
I Arbeit:
=
I = −
Z 4
3
−Z 2
1
= −( − ) ln4
3(3.83)
I Wärme:
=
Z 4
3
= ln4
3(3.84)
=
Z 2
1
= ln2
1(3.85)

3.3 Anwendungen des zweiten Hauptsatzes 100
I Wirkungsgrad:
=
=
−
= max (3.86)
• Wie beim Carnot’schem Kreisprozess wird der thermodynamisch maximaleWirkungsgrad erreicht! D.h., der Stirling-Motor ist ein “praktisch realisierbarerMotor” mit maximalem Wirkungsgrad!
b) Rankine’scher Kreisprozess (Dampfturbine)
I Abbildung 3.7: Wirkungsprinzip einer Dampfmaschine bzw. Dampfturbine.
I 1 → 2: Adiabatische Kompression von H2O(). Flüssiges Wasser wird vomUmgebungsdruck (Druck = ) unter Aufbringung einer Pumparbeit in einenHochdruck-Boiler (Druck ) verbracht.
I 2→ 3: Isobare Wärmezufuhr von q.
(1) isobares Aufheizen des flüssigen Wassers bis zum Siedepunkt unter dem Druck ,
(2) isobares Sieden (Verdampfen) des Wassers,
(3) weiteres Aufheizen des Dampfes.

3.3 Anwendungen des zweiten Hauptsatzes 101
I 3→ 4: Adiabatische Expansion des Dampfes in Turbine (Dampfturbine) oderKolben (Dampfmaschine).
• Dabei Arbeitsleistung
I 4→ 1: Hinter der Turbine erfolgt Kondensation des Wassers.
• Dabei Abgabe der Wärmemenge an die Umgebung.• Alternativ kann statt der Kondensation des Wasserdampfes in einem offenen Kreis-prozess auch frisches Wasser zugeführt werden.
I Enthalpie-Änderungen:
•∆12 =
Z 2
1
= 1(2 − 1) = 12
• Da ∆34 = 0 (adiabatische Expansion):
∆34 = ∆34 = 34 (3.87)
I Arbeit:|| = |34|− |12| = |∆34|− |∆12|
I Wärme:
= ∆23 0 (3.88)
= ∆41 0 (3.89)
I Wirkungsgrad:
=|34|− |12|
| | (3.90)
• sollte möglichst hoch sein, damit 34 möglichst hoch werden kann (y hoherDruck 2 und hohes 3).
3.3 Anwendungen des zweiten Hauptsatzes 102
c) Gasturbine (Brayton-Kreisprozess)
• Brayton-Kreisprozess = offener Kreisprozess
I Abbildung 3.8: Funktionsprinzip der Gasturbine (I).
I Abbildung 3.9: Funktionsprinzip der Gasturbine (II).

3.3 Anwendungen des zweiten Hauptsatzes 103
I a→ b: Adiabatische Kompression von Luft und Brennstoff (gekoppelt an dieTurbine).
=
Z
(3.91)
I b→ c: Isobare Verbrennung in der Brennkammer.
= ∆ (3.92)
I c→ d: Adiabatische Expansion in der Turbine.
=
Z
(3.93)
I d → a: Isobare Abkühlung des Abgases in der Umgebung (offener Kreis-prozess).
• Ggf. Einsatz eines Wärmetauschers zum Vorheizen des Luft/Brennstoff-Gemisches.
I Wirkungsgrad:
= −
=
−
∆
(3.94)
• Zur Erhöhung des Gesamtwirkungsgrades wird das heiße Abgas einer Dampftur-bine zugeführt.
d) Otto-Motor
• Otto-Motor = offener Kreisprozess.
3.3 Anwendungen des zweiten Hauptsatzes 104
I Abbildung 3.10: Funktionsprinzip des Otto-Motors.
I 1→ 2: Adiabatische Kompression von p1 = p auf p2:
= 0y ∆ = 0 (3.95)
y -Anstieg gemäß1
−11 = 2
−12 (3.96)
y
1 = 2
µ2
1
¶−1(3.97)
mit =
.
I 2→ 3: Wärmezufuhr durch Verbrennungswärme q = ∆U ⇒ isochoreKompression mit T -Anstieg.
3 = 2 (3.98)
= (3 − 2) = ∆ (3.99)

3.3 Anwendungen des zweiten Hauptsatzes 105
I 3→ 4: Adiabatische Expansion des Kolbens.
= 0y ∆ = 0 (3.100)
y -Abnahme gemäß3
−13 = 4
−14
bzw. wegen 3 = 2 und 1 = 4:
3−12 = 4
−11 (3.101)
y
4 = 3
µ2
1
¶−1(3.102)
I 4→ 1: Isochore Dekompression des Abgases mit T -Abnahme.
• Verbranntes Gas wird bei bei = durch Öffnen des Auslassventils auf4 = entspannt y -Abnahme.
1 = 4 (3.103)
= (1 − 4) (3.104)
• Abgas wird ausgestossen y kein geschlossener, sondern offener Kreisprozess.
I Wirkungsgrad:
=
=
− ||
= 1− ||
(3.105)
= 1− (4 − 1)
(3 − 2)≈ 1− 4 − 1
3 − 2(3.106)
= 1−3
µ2
1
¶−1− 2
µ2
1
¶−1
3 − 2(3.107)
= 1−(3 − 2)×
µ2
1
¶−1
3 − 2(3.108)
y
≈ 1− 1
−1(3.109)
mit
=1
2= Kompressionsverhältnis (3.110)
Typische Werte des Kompressionsverhältnisses:
= 10− 13 (3.111)
• Größere Werte für führen zu einem besseren Wirkungsgrad.• Aber: Problem der Selbstzündung (Klopfen) ⇒ Zerstörung des Motors.
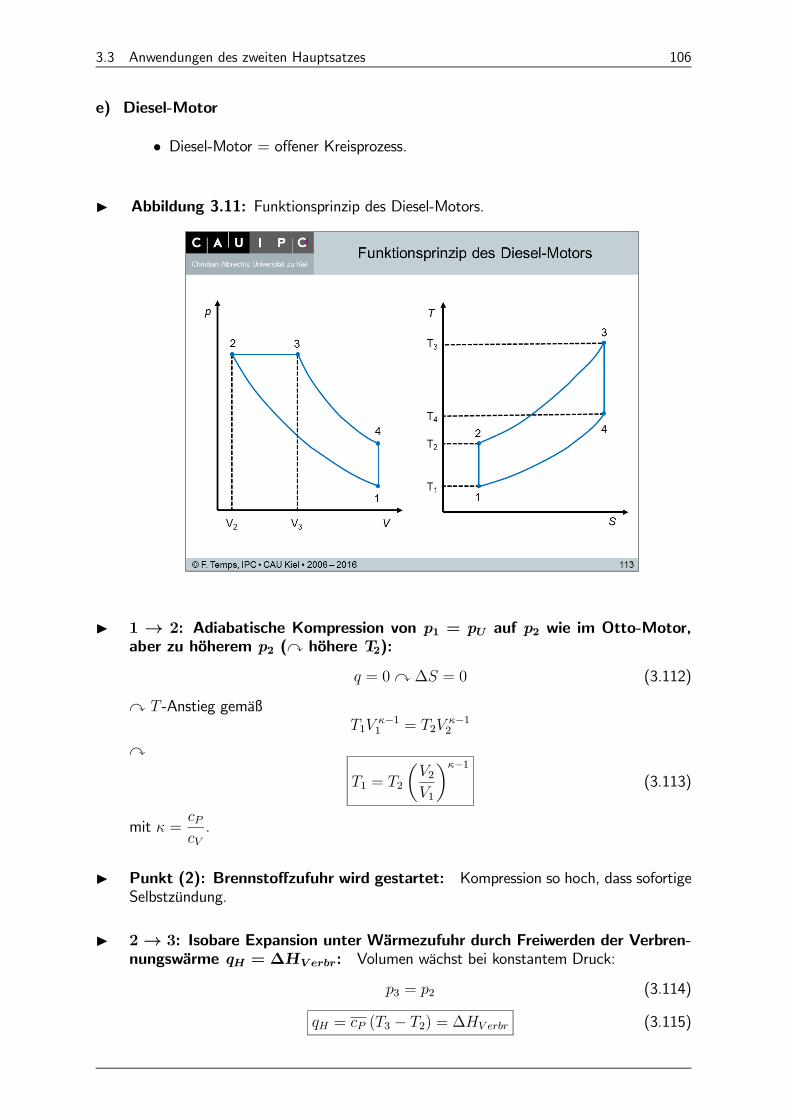
3.3 Anwendungen des zweiten Hauptsatzes 106
e) Diesel-Motor
• Diesel-Motor = offener Kreisprozess.
I Abbildung 3.11: Funktionsprinzip des Diesel-Motors.
I 1 → 2: Adiabatische Kompression von p1 = p auf p2 wie im Otto-Motor,aber zu höherem p2 (y höhere T2):
= 0y ∆ = 0 (3.112)
y -Anstieg gemäß1
−11 = 2
−12
y
1 = 2
µ2
1
¶−1(3.113)
mit =
.
I Punkt (2): Brennstoffzufuhr wird gestartet: Kompression so hoch, dass sofortigeSelbstzündung.
I 2→ 3: Isobare Expansion unter Wärmezufuhr durch Freiwerden der Verbren-nungswärme q = ∆H : Volumen wächst bei konstantem Druck:
3 = 2 (3.114)
= (3 − 2) = ∆ (3.115)

3.3 Anwendungen des zweiten Hauptsatzes 107
I Punkt (3): Brennstoffzufuhr wird gestoppt y Stopp der isobaren Expansion.
=3
2= Cut-Off-Verhältnis (3.116)
I 3→ 4: Adiabatische Expansion des Kolbens.
= 0y ∆ = 0 (3.117)
y3
−13 = 4
−14
y
4 = 3
µ3
4
¶−1= 3
µ3
1
¶−1(3.118)
= 3
µ2
1
¶−1(3.119)
y
4 = 3 −1
µ2
1
¶−1(3.120)
I 4 → 1: Isochore Dekompression des verbrannten Gases durch Öffnen desAuslassventils auf p4 = p mit T -Abnahme:
1 = 4 (3.121)
= (4 − 1) (3.122)
• Abgas wird ausgestossen y kein geschlossener sondern offener Kreisprozess.
I Wirkungsgrad:
=
=
− | |
= 1− | |
(3.123)
= 1− (4 − 1)
(3 − 2)= 1− 1
4 − 1
3 − 2(3.124)
= 1− 1
3 −1
µ2
1
¶−1− 2
µ2
1
¶−1
3 − 2(3.125)
= 1− 1
µ2
1
¶−1 3 −1 − 2
3 − 2(3.126)
= 1− 1
1
−13
−1 − 2
3 − 2(3.127)

3.3 Anwendungen des zweiten Hauptsatzes 108
y
≈ 1− 1
1
−1 − 1 − 1 (3.128)
mit dem Kompressionsverhältnis
=1
2(3.129)
• Typische Werte von : = 18− 25 (3.130)
⇒ höherer Wirkungsgrad des Diesel-Motors.
• Vergleich mit Otto-Motor: Extrafaktor1
− 1 − 1 (3.131)
als Gewinn.
• In der Praxis müssen und beide zusammen optimiert werden.

3.4 Der Dritte Hauptsatz der Thermodynamik 109
3.4 Der Dritte Hauptsatz der Thermodynamik
3.4.1 Formulierungen des dritten Hauptsatzes
Der 3. HS erlaubt die Angabe von Absolutwerten von , da er eine Aussage über denWert von lim→0 macht. Aus demWert von =0 können wir ( ) für eine beliebigeandere Temperatur wie oben beschrieben berechnen.
I Abbildung 3.12: Verschiedene Formulierungen des 3. Hauptsatzes der Thermody-namik.
I Formulierung von Nernst: Nernst untersuchte das Verhalten von ∆ und ∆
chemischer Reaktionen für → 0:
• Es gibt eine Funktion = − (3.132)
y∆ = ∆ − ∆ (3.133)
• Steigung von ∆ vs. : µ∆
¶
= −∆ (3.134)
• Experimentelle Beobachtungen für → 0 (Abb. 3.12):
(1)∆→ ∆ (3.135)
(2)
lim→0
µ∆
¶
= lim→0
(−∆)→ 0 (3.136)

3.4 Der Dritte Hauptsatz der Thermodynamik 110
I Formulierung von Fowler und Guggenheim: Für jeden isothermen Prozess, andem nur Phasen im inneren Gleichgewicht beteiligt sind, oder eingefrorene Phasen,deren inneres Gleichgewicht nicht gestört wird, gilt
lim→0
∆ → 0 (3.137)
I Formulierung von Planck: Für reine kristalline Stoffe im inneren Gleichgewicht kannman sagen, dass
lim→0
→ 0 (3.138)
• Vorsicht: Gilt nicht für amorphe Festkörper, Gläser, . . .• Damit kann man absolute Entropien von Stoffen angeben!
3.4.2 Absolute Entropien Sª298
Für reine kristalline Stoffe im inneren Gleichgewicht ist
( ) = 0 +
1Z0
1
+
∆Schm
Schm+
2Z1
2
+
∆Verd
Verd+
Z2
3
(3.139)
3.4.3 Anwendung auf chemische Reaktionen
• Die Entropien ª298 zahlreicher chemischer Verbindungen finden sich in Tabellen(vgl. Tab. 2.1 — 2.6).
• Für Ionen muss ein zusätzlicher Wert fixiert werden. Vereinbarungsgemäß setzenwir:
ª298¡H+¢= ∆
ª298
¡H+¢= 0 (3.140)
I Beispiel 3.1: Berechnung der Reaktionsenthalpie und -entropie für die Bildung von NOin Flammen:
N2 + O2 → 2 NO (3.141)
∆ª298 = 2∆
ª298 (NO)−∆
ª298 (N2)−∆
ª298 (O2) (3.142)
= (2× 9019− 0− 0) kJmol−1 = +18038 kJmol−1 (3.143)
∆ª298 = 2ª298 (NO)− ª298 (N2)− ª298 (O2) (3.144)
= (2× 21096− 1915− 20502) Jmol−1K−1 (3.145)
= +254 Jmol−1K−1 (3.146)
¤

3.4 Der Dritte Hauptsatz der Thermodynamik 111
I Beispiel 3.2: Vorgriff auf die Berechnung der Gleichgewichtskonstanten (MWG):
∆ª298 = ∆
ª298 − ∆
ª298 (3.147)
Massenwirkungsgesetz (MWG):
2NON2 O2
= exp
µ−∆
ª
¶= ( ) (3.148)
Zahlenwerte:
∆ª ∆
ª ∆ª ∆
ª ( )
K kJmol−1 Jmol−1K−1 kJmol−1 kJmol−1
298 +18038 +254 76 172 7× 10−312000 508 129 4× 10−4
(3.149)
¤

3.5 Behandlung beliebiger Prozesse nach dem 2. Hauptsatz 112
3.5 Behandlung beliebiger Prozesse nach dem 2. Hauptsatz
I Behandlung reversibler Prozesse in einem abgeschlossenen System: Eine di-rekte Anwendung des 2. HS nach
=
(3.150)
ist ohne weiteres möglich.
I Behandlung irreversibler Prozesse in einem abgeschlossenen System: Da
eine Zustandsgröße ist, ist der Weg, auf dem wir ∆ berechnen, egal. Damit folgt fürdie Berechnung der Entropieänderung bei irreversiblen Prozessen folgende Strategie:
(1) Wir suchen einen reversiblen Ersatzweg, der vom Ausgangszustand zu demselbenEndzustand führt.
(2) Wir berechnen∆ für den reversiblen Ersatzweg unter Verwendung der Definitionvon .
I Behandlung beliebiger Prozesse in einem nicht abgeschlossenen System:Reale chemische Prozesse (Beispiel: Reaktion in einem Becherglas auf einem Wasser-bad) laufen meist in nicht-abgeschlossenen Systemen statt, für die der 2. HS keinedirekte Aussage macht.
Um dennoch den 2. HS anwenden zu können, betrachten wir die gesamte Entropieän-derung ∆gesamt im aus dem eigentlich interessierenden System (Becherglas) und demrelevanten Teil der Umgebung (Wasserbad, ggf. mit Heizung, etc.) gebildeten “Super-system”.
Dazu gehen wir folgendermaßen vor:
(1) Wir ergänzen das System unter Hinzunahme eines Teils der Umgebung so, dassein abgeschlossenes System erreicht wird.
(2) Wir berechnen ∆gesamt wie üblich nach:
∆gesamt = (∆System +∆Umgebung)irrev, natürlich≥rev, im Glgew.
0 (3.151)
I Allgemeine Schreibweise: Für gesamt hat sich in der irreversiblen Thermodynamikin der Literatur die Bezeichnung eingebürgert. Wir schreiben deshalb
=P
≥ 0 (3.152)

3.5 Behandlung beliebiger Prozesse nach dem 2. Hauptsatz 113
3.5.1 Beispiel zur Berechnung von ∆Sgesamt für beliebige Prozesse
I Abbildung 3.13: Beispiel zur Berechnung von ∆gesamt.
I Frage: Ein System nehme aus der Umgebung (Wärmebad ) Wärme auf (siehe Abb.3.13). Wie groß ist die gesamte Entropieänderung?
I Antwort:
(1) Wärmebad gibt Wärme ab: 0 (3.153)
(2) System nimmt Wärme auf: 0 (3.154)
(3) Bilanz: = − = (3.155)
(4) Gesamtentropieänderung:
=
+
=
−
irrev.≥rev.
0 (3.156)
(5) Damit Gl. 3.156 erfüllt ist, muss gelten:
≥ (3.157)

3.5 Behandlung beliebiger Prozesse nach dem 2. Hauptsatz 114
I Schlussfolgerung: Wärme fließt immer vom heißeren Körper (in diesem Fall Wärme-bad (= Umgebung)) zum kälteren Körper (in diesem Fall System ).
I Frage:Wieviel Wärme wird vom System bei reversibler bzw. bei irreversibler Prozess-führung aufgenommen, wenn in beiden Fällen derselbe Endzustand erreicht wird?
I Antwort:
= + = −
= −
(3.158)
Wir schreiben für abgekürzt , weil uns hier und im Folgenden nur die Entropieän-derung des Systems interessiert.
y
(1) Wärme bei reversibler Prozessführung:
= −
= 0 y rev = (3.159)
(2) Wärme bei irreversibler Prozessführung:
= −
0 y irrev (3.160)
y
I Ergebnis:
irrev rev = (3.161)
I Frage: Was heißt das für den Absolutbetrag der vom System aufgenommenen bzw.abgegebenen Wärme?
I Antwort:
(1) Bei Wärmeaufnahme ( 0) gilt:
|irrev| |rev| (3.162)
(2) Bei Wärmeabgabe ( 0) gilt:
|irrev| |rev| (3.163)
I Schlussfolgerung: Bei irreversibler Prozessführung wird vom System½weniger
mehr
¾Wärme
½aufgenommen
abgegeben
¾als bei reversibler Prozessführung!

3.5 Behandlung beliebiger Prozesse nach dem 2. Hauptsatz 115
I Frage: Was heißt das für die geleistete Arbeit?
I Antwort: Dairrev rev = (3.164)
y = −
irrev.≤rev.
(3.165)
folgt für die geleistete Arbeit ( 0):
− = || irrev.≤rev.
− (3.166)
I Ergebnisse:
(1) Bei irreversibler Prozessführung wird vom System weniger Arbeit geleistet als beireversibler Prozessführung, d.h.
|rev| = | − | (3.167)
|irrev| | − | (3.168)
(2) Bei reversibler Prozessführung wird die maximal mögliche Nutzarbeit erzielt!
3.5.2 Gleichgewichtsbedingung für Systeme bei V = const. und S = const.
Mit Gl. 3.166
− irrev.≤rev.
− (3.169)
y
+ − irrev.≤rev.
0 (3.170)
haben wir folgende Gleichgewichtsbedingung für Systeme bei = const. und =
const. gefunden:
I Gleichgewichtsbedingung für Systeme bei V = const. und S = const.:
= const.y ≤ 0 (3.171)
y
(1) strebt ein Minimum an ( ≤ 0)!(2) ist die geeignete Zustandsfunktion zur Beschreibung von Gleichgewichtszustän-
den bei = const.

3.5 Behandlung beliebiger Prozesse nach dem 2. Hauptsatz 116
I Anwendung der Gleichgewichtsbedingung für reversible Prozesse:
• 1. und 2. HS kombiniert: = − (3.172)
• Da ein totales Differential ist, schließen wir, dass
= ( ) (3.173)
d.h. ist nur eine Funktion von und !

3.6 Gleichgewichtsbedingungen für verschiedene Prozesse 117
3.6 Gleichgewichtsbedingungen für verschiedene Prozesse
Wir wollen die Gleichgewichtsbedingungen für verschiedene Arten von Prozessen sam-meln:
3.6.1 Innere Energie U für Prozesse bei S, V = const.
irrev≤rev.
− (3.174)
y
I Gleichgewichtsbedingung für S, V = const.:
= const.y ≤ 0 (3.175)
yGlgew → Minimum (3.176)
y ist die geeignete Zustandsfunktion zur Beschreibung des angestrebten Gleich-gewichtszustands für Prozesse bei = const.
I Totales Differential von U : Aus Gl. 3.174 folgt, dass
= ( ) (3.177)
y =
µ
¶
+
µ
¶
(3.178)
I Koeffizientenvergleich: Mit =rev
− folgt
µ
¶
= (3.179)
µ
¶
= − (3.180)
I Anwendung des Schwarz’schen Satzes für die gemischten 2. Ableitungen:µ
¶
=
µ2
¶= −
µ
¶
(3.181)

3.6 Gleichgewichtsbedingungen für verschiedene Prozesse 118
3.6.2 Entropie S für Prozesse bei U, V = const.
irrev.≥ rev
=
+
(3.182)
y
≥
+
(3.183)
y
I Gleichgewichtsbedingung für U, V = const.:
= const.y ≥ 0 (3.184)
yGlgew → Maximum (3.185)
y ist die geeignete Zustandsfunktion zur Beschreibung von Prozessen bei =
const.
I Totales Differential von S: Aus Gl. 3.183 folgt, dass
= ( ) (3.186)
y =
µ
¶
+
µ
¶
(3.187)
I Koeffizientenvergleich: µ
¶
=1
(3.188)
µ
¶
=
(3.189)
I Anwendung des Schwarz’schen Satzes für die gemischten 2. Ableitungen:µ(1 )
¶
=
µ2
¶= +
µ( )
¶
(3.190)
3.6.3 Enthalpie H für Prozesse bei S, p = const.
I Definition: = + (3.191)

3.6 Gleichgewichtsbedingungen für verschiedene Prozesse 119
I Totales Differential:
= + + (3.192)
= − + + (3.193)
y = + (3.194)
y
I Gleichgewichtsbedingung für reversible Prozesse bei S, p = const.:
= const. y = 0 (3.195)
Glgew → Minimum (3.196)
y ist die geeignete Zustandsfunktion zur Beschreibung von Prozessen bei =const.
I Totales Differential von H: = ( ) (3.197)
y = (3.198)
3.6.4 Gibbs-Energie G (= “Freie Enthalpie” ) für Prozesse bei p, T = const.
I Definition 3.2: Gibbs-Energie.
= − (3.199)
I Totales Differential:
= − () (3.200)
= + + − − (3.201)
= − + + − − (3.202)
y = − (3.203)
y

3.6 Gleichgewichtsbedingungen für verschiedene Prozesse 120
I Gleichgewichtsbedingung für reversible Prozesse bei p, T = const.:
= const. y = 0 (3.204)
Glgew → Minimum (3.205)
y ist die geeignete Zustandsfunktion zur Beschreibung von Prozessen bei =
const.
I Totales Differential von G: Aus Gl. 3.203 folgt, dass
= ( ) (3.206)
y =
µ
¶
+
µ
¶
(3.207)
I Koeffizientenvergleich: µ
¶
= (3.208)
µ
¶
= − (3.209)
I Anwendung des Schwarz’schen Satzes für die gemischten 2. Ableitungen:µ
¶
=
µ2
¶= −
µ
¶
(3.210)
3.6.5 Helmholtz-Energie A (= “Freie Energie” ) für Prozesse bei V, T = const.
I Definition 3.3: Helmholtz-Energie.
= − (3.211)
I Totales Differential:
= − () (3.212)
= − − (3.213)
= − − − (3.214)
y = − − (3.215)
y

3.6 Gleichgewichtsbedingungen für verschiedene Prozesse 121
I Gleichgewichtsbedingung für reversible Prozesse bei p, T = const.:
= const. y = 0 (3.216)
Glgew → Minimum (3.217)
y ist die geeignete Zustandsfunktion zur Beschreibung von Prozessen bei =
const.
I Totales Differential von G: Aus Gl. 3.203 folgt, dass
= ( ) (3.218)
y = (3.219)
I Koeffizientenvergleich: µ
¶
= (3.220)
µ
¶
= (3.221)
I Anwendung des Schwarz’schen Satzes für die gemischten 2. Ableitungen:
(3.222)
I Anmerkung 3.2: Für Festkörper und Flüssigkeiten bei niedrigen Drucken ist ≈ .Bei hohem Druck und für Gase muss ( ) berücksichtigt werden.
3.6.6 Merkschema (und Merksprüche . . . )
I Tabelle 3.1: Zusammenstellung der Gleichgewichtsbedingungen für verschiedeneProzesse.
Größe totales Differential Gleichgewichtsbedingung
= ( ) = + − = 0 wenn = 0 = ( ) = − − = 0 " = 0
= ( ) = + + = 0 " = 0
= ( ) = + − = 0 " = 0

3.6 Gleichgewichtsbedingungen für verschiedene Prozesse 122
I Merkschema:− +
− +(3.223)
• Die wichtigen Zustandsgrößen stehen jeweils in der Mitte einer Zeile bzw. Spalte,• Links und rechts daneben (bzw. darüber und darunter) stehen die Größen, vondenen die Zustandsgröße abhängig ist, z.B. ( ),
• Diagonal gegenüber stehen die Faktoren, mit den Vorzeichen als hochgestelltenIndices.

3.6 Gleichgewichtsbedingungen für verschiedene Prozesse 123
4. Chemisches Gleichgewicht
4.1 Partielle molare Größen
I Intensive Zustandsgrößen: Sind nicht von der Stoffmenge abhängig.
Beispiele: (4.1)
I Extensive Zustandsgrößen: Sind von der Stoffmenge abhängig.
Beispiele: (4.2)
y = ( 1 2 3 ) (4.3)
I Partielle molare Größen: Werden definiert, um für Mehrstoffsysteme die chemischenGleichgewichtsbedingungen herzuleiten.
I Definition 4.1: Partielle molare Größen.
=
µ
¶ 6=
(4.4)
I Beispiel 4.1: Partielles molares Volumen:
=
µ
¶ 6=
(4.5)
¤
• Das partielle molare Volumen für einen reinen Stoff ist gleich dem Molvolumen.• Das partielle molare Volumen eines Stoffes in einer Mischung entspricht dem“Molvolumen in der Mischung”.
I Beispiel 4.2: Partielle molare Enthalpie:
=
µ
¶ 6=
(4.6)
¤
• Die partielle molare Enthalpie eines reinen Stoffes ist gleich seiner Bildungsen-thalpie.

4.2 Das chemische Potential 124
4.2 Das chemische Potential
Das chemische Potential ist die für uns wichtigste partielle molare Größe.
I Definition 4.2: Chemisches Potential
=
µ
¶ 6=
(4.7)
• ist die Gibbs’sche Energie von 1mol des Stoffes in der Mischung.
I Vollständiges Differential von G für eine Mischung:
=
µ
¶
+
µ
¶
+X
µ
¶ 6=
(4.8)
y = − +
P (4.9)
y
I Gleichgewichtsbedingung für Mischungen:
= − +P
= 0 (4.10)
• Für = const.: =
P = 0 (4.11)

4.3 Gleichung von Gibbs-Duhem 125
4.3 Gleichung von Gibbs-Duhem
Integration von Gl. 4.11 bei = const. liefert
=X
(4.12)
y im Glgew. für = const.:
=X
| z =0 (Gl. 4.11)
+X
= 0|zim Glgew.
(4.13)
y P = 0 für = const. (4.14)
Diese Gleichung wird als Gibbs-Duhem-Gleichung bezeichnet. Sie findet Anwendung u.a.zur Konsistenzprüfung von Gleichgewichtsmessungen (siehe Kapitel 6 und Versuche imPC-Grundpraktikum).

4.4 Thermodynamische Gleichgewichtsbedingung für chemische Reaktionen 126
4.4 Thermodynamische Gleichgewichtsbedingung für chemische Reaktionen
I Chemische Reaktion:
1B1 + 2B2 + À B + +1B+1 + (4.15)
mit
= stöchiometrische Koeffizienten
½ 0 : Produkte 0 : Reaktanden
(4.16)
∆ª = ∆
ª (B) + +1∆
ª (B+1) +
− |1|∆ª (B1)− |2|∆
ª (B2)− (4.17)
ƻ =
ª (B) + +1
ª (B+1) +
− |1|ª (B1)− |2|ª (B2)− (4.18)
∆ª = ∆
ª − ∆
ª (4.19)
I Reaktionsfortschrittskoordinate ξ: Die GleichgewichtsbedingungP
= 0
(Gl. 4.11) enthält die Änderungen der Molzahlen aller beteiligten Stoffe. Diesesind jedoch nicht unabhängig voneinander, sondern über die Stöchiometrie der Reaktionverknüpft.
Der Fortschritt der Reaktion (Gl. 4.15) von der Reaktanden- zur Produktseite kann mitHilfe der Reaktionsfortschrittskoordinaten beschrieben werden.
→ + y → + = + (4.20)
Die Molzahl der Produkte nimmt mit dem Reaktionsfortschritt zu:
= (4.21)
+1 = +1 (4.22)
(4.23)
Die Molzahl der Reaktanden nimmt mit dem Reaktionsfortschritt ab:
1 = − |1| = 1 (4.24)
2 = − |2| = 2 (4.25)
(4.26)

4.4 Thermodynamische Gleichgewichtsbedingung für chemische Reaktionen 127
I Gleichgewichtsbedingung für chemische Reaktionen: Einsetzen von =
in Gl. 4.11 ergibt, für = const.,
0 =X
=X
(4.27)
Im Glgew. muss ein Minimum haben, d.h.
= 0 (4.28)
y X
= 0 (4.29)
Diese Gleichung werden wir im Folgenden als Gleichgewichtsbedingung für chemischeReaktionen verwenden.
I Beispiel 4.3: Chemische Reaktion:
2 H2 +O2 À 2 H2O (4.30)
H2O = +2 (4.31)
O2 = −1 (4.32)
H2 = −2 (4.33)
y =
X
=¡2H2O − O2 − 2H2
¢ = 0 (4.34)
y X
= 2H2O − O2 − 2H2 = 0 (4.35)
y Im Gleichgewicht:2H2O − O2 − 2H2 = 0 (4.36)
¤I Beispiel 4.4: Phasenübergang:
H2O()À H2O() (4.37)
Anwendung von X
= 0 (4.38)
liefertH2O() − H2O() = 0 (4.39)
y Im Gleichgewicht:H2O() = H2O() (4.40)
¤

4.4 Thermodynamische Gleichgewichtsbedingung für chemische Reaktionen 128
I Allgemeine Anwendung von Gl. 4.29 auf Phasengleichgewichte: Für
A() À A() À A() (4.41)
yA() = A() = A() (4.42)
Im Gleichgewicht ist das chemische Potential eines reinen Stoffes in allen beteiligtenPhasen gleich!

4.5 Druck-, Konzentrations- und Temperaturabhängigkeit des chemischen Potentials 129
4.5 Druck-, Konzentrations- und Temperaturabhängigkeit des chemischenPotentials
Allgemein gilt wegen µ
¶
= und
µ
¶
= − (4.43)
für das chemische Potential µ
¶
= (4.44)
und µ
¶
= − (4.45)
mit den entsprechenden partiellen molaren Größen und .
4.5.1 Druckabhängigkeit des chemischen Potentials für Gase (p = Partialdruck)
I Chemisches Potential für ideale Gase:
Für = const. folgt = (4.46)
yZ
ª
=
Zª
=
Zª
=
Zª
ln (4.47)
y ( ) = ª ( ) + ln
ª(4.48)
⇒ Gl. 4.48 zerlegt die Temperatur- und Druckabhängigkeit des chemischen Potentialsfür ein ideales Gas in zwei Terme, von denen einer nur -abhängig ist (ª ( )) undeiner die -Abhängigkeit beschreibt ( ln (
ª)).
I Anmerkung 4.1: Oft schreiben wir einfach
( ) = ª ( ) + ln (4.49)
wobei wir implizit wissen, dass in den Einheiten von ª (also bar) anzugeben ist.
I Berechnung von μª (T ) im Standardzustand (pª = 1bar): ⇒ Gibbs-Energiefür 1mol:
ª ( ) = ∆ª ( )− ª ( ) (4.50)

4.5 Druck-, Konzentrations- und Temperaturabhängigkeit des chemischen Potentials 130
I Chemisches Potential für reale Gase: Statt des Partialdrucks ist die Fugazität zu verwenden, mit dem Fugazitätskoeffizienten . y
= (4.51)
y
( ) = ª ( ) + ln
ª ª(4.52)
= ª ( ) + lnª+ ln
ª(4.53)
Unter Verwendung der Virialgleichung
= + (4.54)
als Zustandsgleichung des realen Gases erhalten wirµ
¶
= =
+ (4.55)
yZ
ª
=
Zª
=
Zª
µ
+
¶ (4.56)
y
= ª +
Zª
ln +
Zª
(4.57)
= ª + ln
ª+
¡ − ª
¢(4.58)
= ª + ln
ª+ ln
ª(4.59)
yln =
(4.60)
bzw.
= exp
µ
¶(4.61)
4.5.2 Konzentrationsabhängigkeit des chemischen Potentials in (nichtwässrigen)Lösungen∗
Im Vorgriff seien an dieser Stelle auch die Konzentrationsabhängigkeiten des chemischenPotentials in Lösung angeführt.
I Standardzustand: Reiner Stoff bei
() = 1 = 1bar ( = 298K) (4.62)

4.5 Druck-, Konzentrations- und Temperaturabhängigkeit des chemischen Potentials 131
I Chemisches Potential (für ideale Lösung bzw. Mischung):
³()
´= ∗ ( ) + ln
() (4.63)
da (Raoult’sches Gesetz):
= ∗ () (4.64)
mit ∗ = Dampfdruck der reinen Flüssigkeit bei () = 1.
I Reale Lösungen: Verwendung der Aktivitäten
= () (4.65)
y
³()
´= ∗ ( ) + ln
() (4.66)
4.5.3 Konzentrationsabhängigkeit des chemischen Potentials fürElektrolytlösungen (wässrige Lösung)∗
I Standardzustand: Hypothetische ideal verdünnte Lösung der Molalität
= 1mol
kgLm= ª = 1bar ( = 298K) (4.67)
Zusätzlich setzen wir fest:
ƻ298(H
+) = 0 (4.68)
ª298(H+) = 0 (4.69)
I Chemisches Potential für ideale Lösung:
( ) = ∗ ( ) + ln
ª (4.70)
I Reale Lösungen: Verwendung der Aktivitäten
=
ª (4.71)
y ( ) = ∗ ( ) + ln (4.72)

4.5 Druck-, Konzentrations- und Temperaturabhängigkeit des chemischen Potentials 132
4.5.4 Temperaturabhängigkeit des chemischen Potentials
µ
¶
= − (4.73)
Für = const. y Z = −
Z (4.74)
y
( ) = (0)−Z
0
(4.75)
mit
( ) = (0) +
Z
0
(4.76)

4.6 Homogene Gas-Reaktionsgleichgewichte 133
4.6 Homogene Gas-Reaktionsgleichgewichte
4.6.1 Beispiel einer homogenen Gasreaktion
I Reaktionsgleichung:H2 + Cl2 À 2 HCl (4.77)
I Thermodynamische Gleichgewichtsbedingung für chemische Reaktionen:X
= 0 (4.78)
y
0 = 2HCl − H2 − Cl2 (4.79)
= 2ªHCl + ln
µHCl
ª
¶2− ªH2 − ln
H2ª− ªCl2 − ln
Cl2ª
(4.80)
Sammeln der ª- und ln -Terme:
2ªHCl − ªH2 − ªCl2| z =∆ª( )
= − lnµHCl
ª
¶2+ ln
H2ª
+ lnCl2ª
(4.81)
= − ln (HClª)2
(H2ª) (Cl2ª)
(4.82)
= − ln 2HClH2 Cl2
(4.83)
y
I Massenwirkungsgesetz:
ln2HCl
H2 Cl2= −∆
ª ( )
(4.84)
oder2HCl
H2 Cl2= exp
µ−∆
ª ( )
¶| z
= Konstante( )
= ( ) (4.85)
Diese Gleichung ist als Massenwirkungsgesetz bekannt. ist die Massenwirkungskon-stante, die nur von abhängig ist.
I Anmerkungen:
• Da der Ausdruck expµ−∆
ª ( )
¶eine nur von abhängige Konstante ist, ist
auch2HCl
H2 Cl2eine nur von abhängige Konstante, die wir = Gleichgewichts-
konstante nennen.

4.6 Homogene Gas-Reaktionsgleichgewichte 134
• ∆ª ( ) ist die Änderung der Gibbs-Energie für den Formelumsatz unter Stan-
dardbedingungen. I.a. ist ƻ ( ) 6= 0.19
• Der Wert von ( ) kann vorhergesagt werden, da wir ∆ª leicht gemäß
∆ª ( ) = ∆
ª ( )− ∆ª ( ) (4.86)
berechnen können:
∆ª = 2∆
ª (HCl)−∆ª (H2)−∆
ª (Cl2) (4.87)
= 2∆ª (HCl)−∆
ª (H2)−∆ª (Cl2)
− ¡2ª (HCl)− ª (H2)− ª (Cl2)¢
(4.88)
=P
ª (4.89)
• Achtung: Wenn wir bei = 298K starten, muss die -Abhängigkeit von ∆ª,
∆ª bzw. ∆
ª für die genaue Berechnung von berücksichtigt werden.
Nur für kleine Temperaturintervalle kann für eine Überschlagsrechnung aufgrunddes “Kompensationseffekts” in
R∆ auf diese Korrektur verzichtet werden.
• Wir kennen natürlich die -Abhängigkeit von ∆ª:
ƻ
= −∆
ª (4.90)
y∆
ª ( ) = ∆ª (0)−
Z
0
ƻ (4.91)
4.6.2 Allgemeine Ableitung des Massenwirkungsgesetzes für Gasreaktionen
I Thermodynamische Gleichgewichtsbedingung für chemische Reaktionen beip, T = const.:
0 =X
(4.92)
=X
ª +
X
ln
ª(4.93)
= ƻ + ln
Y
µ
ª
¶
(4.94)
ylnY
µ
ª
¶
= −∆ª
= ln (4.95)
bzw. Q
µ
ª
¶
= exp
µ−∆
ª
¶= ( ) (4.96)
19 ∆ª für den Formelumsatz unter Standardbedingungen ist zu unterscheiden von der Gleich-
gewichtsbedingung ∆ = 0.

4.6 Homogene Gas-Reaktionsgleichgewichte 135
4.6.3 Kinetische Ableitung des MWG
I Reaktion: A+ BÀ C+ D (4.97)
I Reaktionsgeschwindigkeit:
≡ − 1
|| [A]
= − 1
|| [B]
= +
1
|| [C]
= +
1
|| [D]
(4.98)
= 0 im Glgew. (4.99)
H2 + Cl2 À 2HCl (4.100)
y+1
2
[HCl]
= 1 [H2] [Cl2]− −1 [HCl]
2= 0 (4.101)
y1 [H2] [Cl2] = −1 [HCl]
2(4.102)
y[HCl]
2
[H2] [Cl2]=
1
−1= (4.103)
Einsetzen von =
=
(4.104)
y2HCl
H2Cl2=
1
−1= = −∆
ª (4.105)
I Probleme bei der kinetischen Ableitung: Viele Reaktionen laufen nicht so wieaufgeschrieben ab, sodass die Reaktionsgeschwindigkeitskonstanten 1 oder −1 nichtgemessen werden können. Oder die Reaktionsgeschwindigkeiten können oft unmessbarklein sein. Die thermodynamische Ableitung bietet dagegen eine fundierte Basis, undsie erlaubt quantitative Vorhersagen der Gleichgewichtskonstanten.
4.6.4 Dimensionsbetrachtungen
• Die thermodynamische Gleichgewichtskonstante (Gl. 4.96)
( ) =Y
µ
ª
¶
= exp
µ−∆
ª
¶(4.106)
ist dimensionslos.
• Häufig (vor allem im Ingenieursbereich) gibt man dennoch mit Dimension an:
0 ( ) =
Y
() = exp
µ−∆
ª
¶×Y
¡ª¢
(4.107)
Die dimensionsbehaftete Glgew.-konstante 0 ist auf den entsprechenden Stan-
darddruck bezogen.
• Nützlich sind mitunter auch , und .

4.6 Homogene Gas-Reaktionsgleichgewichte 136
I Beispiel:N2O4 À 2NO2 (4.108)
I Dimensionslose Gleichgewichtskonstante K:
=(2
ª)2
(24ª)= exp
µ−∆
ª
¶(4.109)
I Dimensionsbehaftete Gleichgewichtskonstante K0:
0 =
(2)2
24= exp
µ−∆
ª
¶× ª = × ª (4.110)
dim¡ 0
¢= barΣ (4.111)
I K: = × ges (4.112)
y
2 = 2 × ges (4.113)
24 = 24 × ges (4.114)
y 0
=(2 × ges)
2
(24 × ges)=(2)
2
(24)× ges = × ges (4.115)
y =
0
ges= × ª
ges(4.116)
mitdim () = 1 (4.117)
I K:
=
= (4.118)
y 0
=(2 )
2
24=(2)
2
24× = × (4.119)
dim () =
µmol
l
¶Σ
(4.120)

4.6 Homogene Gas-Reaktionsgleichgewichte 137
I K:
=
(4.121)
y
0 =
µ2
¶2µ24
¶ =(2)
2
(24)×
= ×
(4.122)
dim () = molΣ (4.123)
I Berücksichtigung von realem Verhalten:
=(2)
2
(24)(4.124)
I Anmerkung 4.2: Abhängig vom zu lösenden Problem kann es günstig sein, statt
einen der o.a. anderen Ausdrücke für die Gleichgewichtskonstante zu nutzen.
4.6.5 Beispiele zur Berechnung homogener Gasgleichgewichte
I Vorgehensweise (Schema):
(1) Reaktionsgleichung aufstellen
(2) MWG formulieren:
= exp
µ−∆
ª
¶=Y
µ
ª
¶
(4.125)
(3) Berechnung von ∆ª ( ), ∆
ª ( ) und ∆ª ( ) (Tabellen)
(4) Berücksichtigung weiterer Bedingungen (Atombilanzen, Elektroneutralität,vorgegebener Gesamtdruck, . . . )
I Beispiel 4.5: 20
N2 +O2 À 2NO (4.126)
=(
ª)2
(2ª) (2ª)
= exp
µ−∆
ª
¶(4.127)
20 Für volumenständige Reaktionen ist die Gleichgewichtskonstante immer dimensionslos.

4.6 Homogene Gas-Reaktionsgleichgewichte 138
∆ª298 = 2∆
ª298 (NO)−∆
ª298 (N2)−∆
ª298 (O2) (4.128)
= (2× 909− 0− 0) kJmol−1 = +18038 kJmol−1 (4.129)
∆ª298 = 2ª298 (NO)− ª298 (N2)− ª298 (O2) (4.130)
= (2× 21096− 1915− 20502) Jmol−1K−1 (4.131)
= +254 Jmol−1K−1 (4.132)
∆ª298 = ∆
ª298 − ∆
ª298 (4.133)
∆ª ∆
ª ∆ª ∆
ª
K kJmol−1 Jmol−1K−1 kJmol−1 kJmol−1
298 +18038 +254 76 172 7× 10−312000 ... ... 508 129 4× 10−4
(4.134)
¤I Beispiel 4.6:
CO+1
2O2 À CO2 (4.135)
=(2
ª)
(ª) (2ª)12= exp
µ−∆
ª
¶(4.136)
0 =
(2)
() (2)12= exp
µ−∆
ª
¶× ¡ª¢−12 (4.137)h
bar−12i
(4.138)
∆ª298 = ∆
ª298 (CO2)−∆
ª298 (CO)−
1
2∆
ª298 (O2) (4.139)
= (−39351− (−11050)− 0) kJmol−1 (4.140)
= −28301 kJmol−1 (4.141)
∆ª298 = ª298 (CO2)− ª298 (CO)−
1
2ª298 (O2) (4.142)
=
µ21368− 19753− 1
220502
¶Jmol−1K−1 (4.143)
= −8636 Jmol−1K−1 (4.144)
y∆
ª298 = ∆
ª298 − ∆
ª298 (4.145)
y
∆ª298 = −257 kJmol−1: 0
298 =(2)
() (2)12= 1× 1045 bar−12 (4.146)
∆ª2000 = −110 kJmol−1: 0
2000 =(2)
() (2)12= 760 bar−12 (4.147)
¤

4.6 Homogene Gas-Reaktionsgleichgewichte 139
I Beispiel 4.7:H2 () + I2 ()À 2HI () (4.148)
Anfangsbedingungen: Gegeben seien 0 mol H2 und 0 mol I2 im Volumen .
Frage: Wie groß sind die Molzahlen 2 2 im Glgew.?
Berechnung der Gleichgewichtskonstanten:
=()
2
(2) (2)= exp
µ−∆
ª
¶(4.149)
∆ª298 = 2∆
ª298 (HI)−∆
ª298 (H2)−∆
ª298 (I2) (4.150)
= (2× 2648− 0− 6243) kJmol−1 (4.151)
= −947 kJmol−1 (4.152)
∆ª298 = 2ª298 (HI)− ª298 (H2)− ª298 (I2) (4.153)
= (2× 20648− 13058− 26058) Jmol−1K−1 (4.154)
= +218 Jmol−1K−1 (4.155)
∆ª298 = ∆
ª298 − ∆
ª298 (4.156)
y
∆ª298 = −160 kJmol−1 y 298K = 613 (4.157)
∆ª1000 = −310 kJmol−1 y 1000K = 42 (4.158)
Berechnung der Lage des Gleichgewichts:
Anfang:
02 = 02 = 0 (4.159)
0 = 0 (4.160)
Atomerhaltung:2 = 2 (4.161)
y
2 +1
2 = 0 (4.162)
2 +1
2 = 0 (4.163)
Gleichgewichtskonstante:
=()
2
(2) (2)(4.164)

4.6 Homogene Gas-Reaktionsgleichgewichte 140
Mit =
y
=()
2
(2) (2)(4.165)
Mit 2 = 0 −
2und 2 = 0 −
2y
=()
2³0 −
2
´³0 −
2
´ (4.166)
=4 ()
2
(20 − )2
(4.167)
=4 (20)
2
(1− 20)2
(4.168)
=42
(1− )2
(4.169)
mit =
20(4.170)
y p =
2
1− (4.171)
Auflösung nach =
20: p
− p = 2 (4.172)p
=³2 +
p
´ (4.173)
y
=
20=
p
2 +p
(4.174)
und
= 20 ×p
2 +p
(4.175)
¤
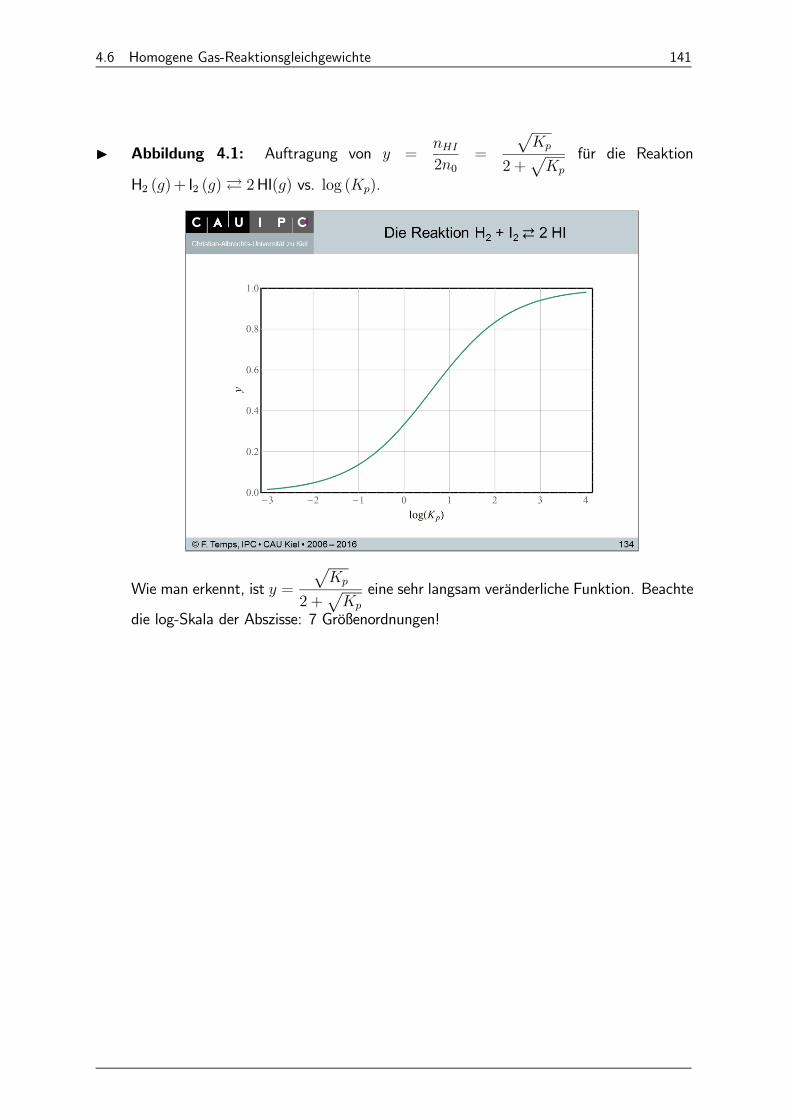
4.6 Homogene Gas-Reaktionsgleichgewichte 141
I Abbildung 4.1: Auftragung von =
20=
p
2 +p
für die Reaktion
H2 ()+ I2 ()À 2HI() vs. log ().
Wie man erkennt, ist =
p
2 +p
eine sehr langsam veränderliche Funktion. Beachte
die log-Skala der Abszisse: 7 Größenordnungen!

4.7 Bestimmung thermodynamischer Daten aus Gleichgewichtsmessungen 142
4.7 Bestimmung thermodynamischer Daten aus Gleichgewichtsmessungen
I Bestimmung von ∆Gª: Gleichgewichts-Partialdrücke bzw. Konzentrationen
messen y y∆
ª = − ln (4.176)
I Bestimmung von ∆Hª (van’t Hoff-Gleichung):
= −∆ª (4.177)
ln = −∆ª
(4.178)
ln
= −
µ∆
ª
¶(4.179)
= − 1
µ∆
ª
−∆
ª¶
(4.180)
= − 1
⎛⎜⎝ × ∆ª
−∆
ª
2− ∆
ª
⎞⎟⎠ (4.181)
= − 1
µ∆
− ∆
ª
2− ∆
¶(4.182)
y van’t Hoff-Gleichung:
ln
=
ƻ
2(4.183)
Umformung mit (1 )
= − 1
2ergibt
ln
(1 )= −∆
ª
(4.184)
y Auftragung von ln vs. −1 ergibt Gerade mit Steigung
= −∆ª
(4.185)

4.7 Bestimmung thermodynamischer Daten aus Gleichgewichtsmessungen 143
I Abbildung 4.2: van’t Hoff-Auftragung zur Bestimmung der Reaktionsenthalpie ausGleichgewichtsmessungen.
I Bestimmung von ∆Sª:
(1)∆
ª = ∆ª − ∆
ª (4.186)
y∆
ª = −∆ª −∆
ª
(4.187)
(2)
ln = −∆ª
= −∆
ª
+
ƻ
(4.188)
y Tangente an Auftragung von ln vs. −1 ergibt ∆
ª durch Extrapolationfür →∞.
I Temperaturabhängigkeiten:
ƻ =
X
ª =
X∆
ª − X
ƻ (4.189)
∆ª ( ) = ∆
ª (0) +Z
0
∆ (4.190)
∆ª ( ) = ∆
ª (0) +Z
0
∆
(4.191)

4.8 Heterogene Reaktionsgleichgewichte 144
4.8 Heterogene Reaktionsgleichgewichte
Wir wollen heterogene Reaktionsgleichgewichte anhand einiger Beispiele kennenlernen.
4.8.1 Die Reaktion CaCO3 (s) À CaO(s)+ CO2 (g)
I Gleichgewichtsbedingung für Zersetzungsgleichgewicht beim Kalkbrennen:
CaCO3 ()À CaO () + CO2 () (4.192)
X = 2() + () − 3() = 0 (4.193)
I Chemische Potentiale:
(1) CO2:
2() = ª2 + ln2ª
(4.194)
(2) Entsprechend könnten wir auch schreiben (in Anführungsstrichen):
“ 3() = ª3()
+ ln3ª
” (4.195)
(analog für CaO). Leider kennen wir allerdings die Dampfdrucke von CaCO3 (undebenso von CaO) nicht — sie sind viel zu klein, als dass man sie messen könnte.
(3) Aber: Solange festes CaCO3 (und festes CaO) im Gleichgewicht vorliegen, ist (daPhasengleichgewichte)
3() = 3() (4.196)
und() = () (4.197)
Wir können also, da CaCO3 und CaO keine Mischkristalle bilden, die Wertevon 3() und () auf der Gleichgewichtskurve (Sublimationsdruckkurve)
nehmen.21
I Formulierung der Gleichgewichtsbedingung:
ª2()| z bei ª
+ ln2ª
+ ∗(Glgew)| z bei Glgew
− ∗3(Glgew)| z bei Glgew
= 0 (4.198)
Achtung: (Glgew) und 3(Glgew) sind die Werte bei dem im Gleichgewichtherrschenden Druck (nicht der Standardzustand), wir schreiben deshalb dafür ∗()und ∗3()!
21 Dies geht nur, solange die beteiligten Feststoffe keine Mischkristalle bilden!!

4.8 Heterogene Reaktionsgleichgewichte 145
I (Partielle) Gleichgewichtskonstante:
ln2ª
= −³ª2()
+ ∗() − ∗3()´
= −∆
∗
(4.199)
mit der Reaktions-Gibbsenergie ∆∗ beim herrschenden Reaktionsdruck.
y2ª
= exp
µ−∆
∗
¶= (4.200)
I Berechnung von∆G∗: Aus Tabellen für∆
ª und ª finden wir ª()
, ª()
und ª3()
. Für ∆∗ benötigen wir allerdings ∗() und ∗3() beim Reak-
tionsdruck. Die notwendigen Korrekturen erhalten wir gemäßµ
¶
= (4.201)
yZ
ª
=
Zª
(4.202)
y
() = ¡ª¢+
Zª
(4.203)
≈ ¡ª¢+
¡− ª
¢(4.204)
I Zahlenwerte für p = 10bar, T = 298K:
= 165 cm3mol (4.205)
@ = 10bar = 298K:
(10 bar) ≈ ¡ª¢+ 165 cm3mol−1 (10− 1)× 105 Pa (4.206)
= ¡ª¢+ 148
J
mol(4.207)
Aber:
ª()
= ∆ª − × ª
()= 604 kJmol−1 − 298K× 40 Jmol−1K−1
(4.208)
= 592 kJmol−1 (4.209)
Die Druckkorrektur von ª auf ∗ ist im Beispiel praktisch vernachlässigbar, obwohl wirmit = 10bar einen relativ hohen Druck gewählt haben. Für niedrigere Drucke wäre sienoch viel kleiner und im Rahmen der Genauigkeit von ∆
ª und ª()
vollständig
vernachlässigbar. Wir können für ∗() und ∗3() also beruhigt die Werte imStandardzustand (ª = 1bar) einsetzen.
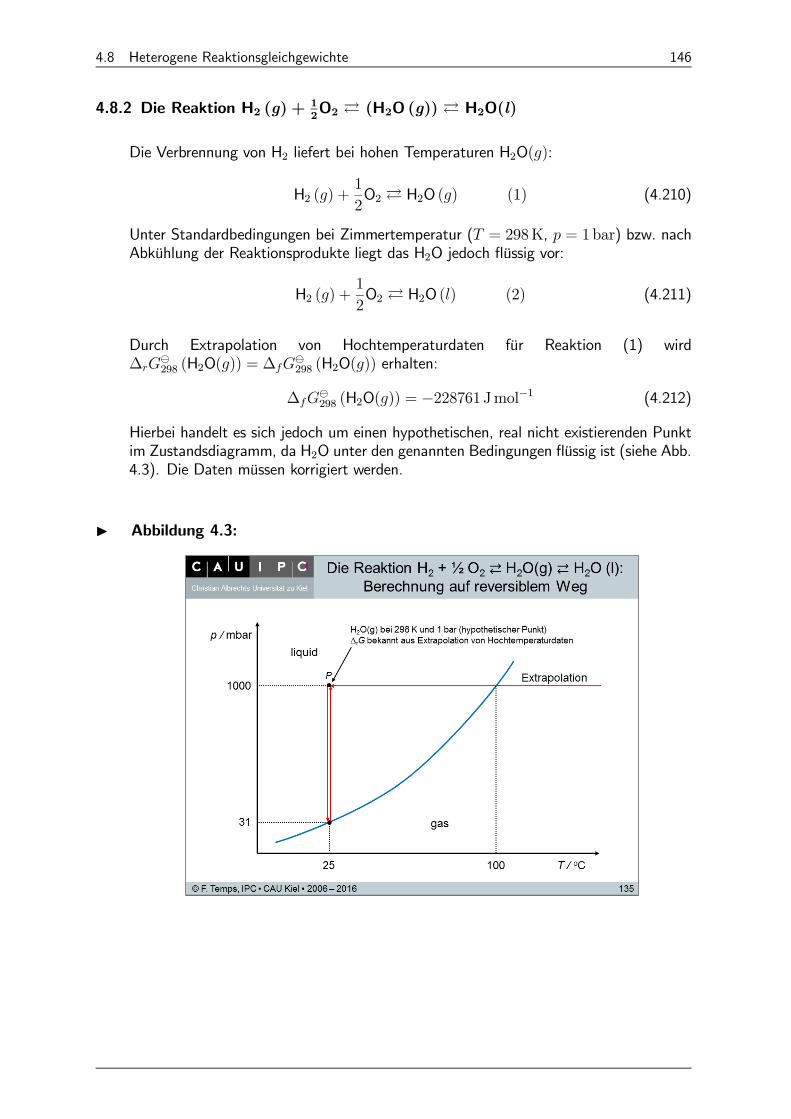
4.8 Heterogene Reaktionsgleichgewichte 146
4.8.2 Die Reaktion H2 (g) +12O2 À (H2O (g)) À H2O(l)
Die Verbrennung von H2 liefert bei hohen Temperaturen H2O():
H2 () +1
2O2 À H2O () (1) (4.210)
Unter Standardbedingungen bei Zimmertemperatur ( = 298K, = 1bar) bzw. nachAbkühlung der Reaktionsprodukte liegt das H2O jedoch flüssig vor:
H2 () +1
2O2 À H2O () (2) (4.211)
Durch Extrapolation von Hochtemperaturdaten für Reaktion (1) wird∆
ª298 (H2O()) = ∆
ª298 (H2O()) erhalten:
∆ª298 (H2O()) = −228761 Jmol−1 (4.212)
Hierbei handelt es sich jedoch um einen hypothetischen, real nicht existierenden Punktim Zustandsdiagramm, da H2O unter den genannten Bedingungen flüssig ist (siehe Abb.4.3). Die Daten müssen korrigiert werden.
I Abbildung 4.3:

4.8 Heterogene Reaktionsgleichgewichte 147
I Korrektur unter Zerlegung in Teilschritte:
Ausgangspunkt:∆
ª298 (H2O()) = −228761 Jmol−1 (4.213)
(1) H2O() als ideales Gas bei 298K von 1000mbar auf 31mbar (= Glgew.-druck)expandieren:
∆1 =
Z 31mbar
1000mbar
= ln31
1000= −8639 Jmol−1 (4.214)
(2) H2O() auf der Gleichgewichtskurve zu H2O() kondensieren:
∆2 = 0 (4.215)
(3) H2O() bei 298K von 31mbar auf 1000mbar komprimieren:
∆3 =
Z 1000mbar
31mbar
= 18cm3
mol(1000− 31) mbar = 18 Jmol−1 (4.216)
Ergebnis:
∆ª298K (H2O ()) = ∆
ª298 (H2O()) +∆1+∆2+∆3 (4.217)
= (−228761− 8639 + 0 + 18) Jmol−1 (4.218)
= −237398 Jmol−1 (4.219)
4.8.3 Die Reaktion NH4Cl(s) À NH3 (g) + HCl(g)
I Gleichgewichtsreaktion:
NH4Cl ()À NH3 () + HCl () (4.220)
I Anwendung der Gleichgewichtsbedingung:
3+ − 4
= 0 (4.221)
yª3
+ ln¡3
ª¢+ ª + ln¡
ª¢− 4= 0 (4.222)
yln(3)
(ª)2= −
ª3
+ ª − 4
(4.223)
bzw.(3)
(ª)2= exp
µ−∆
∗
¶(4.224)
mit∆
∗ = ª3+ ª − 4
(4.225)
und22
4≈ ª4
(4.226)
y∆
∗ ≈ ∆ª (4.227)
22 Gilt solange der Druck nicht zu groß wird.

4.8 Heterogene Reaktionsgleichgewichte 148
I Berechnung der Gleichgewichtskonstanten (T = 298K):
∆ª (298K) = ∆
ª − ∆ª (4.228)
= (−46190− 92300 + 314430) Jmol−1−298K× (19234 + 18677− 9456) Jmol−1K−1 (4.229)
= 91144 Jmol−1 (4.230)
y
0 = 3 = exp
µ−∆
ª
¶× ¡ª¢2 = −367 bar2 = 10−16 bar2 (4.231)
y Das Gleichgewicht liegt weit auf der Seite des NH4Cl.
I Konsequenzen bei verschiedenen NH3 bzw. HCl Partialdrucken:
[NH3] = 10−13mol cm−3 y 3 = 243× 10−9 bar (4.232)
(1)[HCl] = 10−13mol cm−3 y = 3 = 243× 10−9 bar (4.233)
y3 =
¡243× 10−9 bar¢2 = 58× 10−18 bar2 0
(4.234)
(2)
[HCl] = 17× 10−12mol cm−3 y = 3 = 41× 10−8 bar (4.235)
y3 =
¡243× 10−9 bar¢2 = 1× 10−16 bar2 ≈ 0
(4.236)
y Bei höheren HCl-Drucken schneit festes NH4Cl aus (Analogie zu Löslichkeits-produkt).
I Anmerkung: Bei Anwesenheit von flüssigem NH3 wird die Chemie komplizierter, dadas Gleichgewicht 4.220 mit den Ionenreaktionen
2 NH3 À NH−2 + NH+4 (4.237)
NH4Cl À NH+4 + Cl− (4.238)
gekoppelt ist.
4.8.4 Die Reaktion CO2 (g)+ C(s) À 2CO(g) (Boudouard-Gleichgewicht)
I Boudouard-Reaktion:CO2 () + C ()À 2 CO () (4.239)

4.8 Heterogene Reaktionsgleichgewichte 149
I Gleichgewichtskonstante: Da es sich bei dem Boudouard-Glgew. um eine technischsehr wichtige Reaktion handelt (Hochofenprozess, siehe Abschnitt 4.8.5), liegen sehrgenaue Messungen der Gleichgewichtskonstanten vor:
( ) =(
ª)2
(2ª)
(4.240)
mit (van’t Hoff-Auftragung):23
lg 0
bar= −8916K
+ 918 (4.242)
I Einsetzen von T = 1000K in Gl. 4.242:
0 = 185 bar (4.243)
I Berechnung der Lage des Gleichgewichtes:
+ 2 = 1 (4.244)
y
= × (4.245)
2 = (1− )× (4.246)
0 =
2
(1− )(4.247)
y 0
=
2(1− )
(4.248)
2 + 0
− 0
= 0 (4.249)
y
= − 0
2±↑
nur +
sµ 0
2
¶2+
0
(4.250)
23 lg steht für den dekadischen Logarithmus:
lg () = log10 () (4.241)

4.8 Heterogene Reaktionsgleichgewichte 150
I Prinzip vom kleinsten Zwang (Le Chatelier):
• Bei Verringerung des Druckes wird das Glgew. auf die Seite von 2 CO verschoben:
& y 0
% y % ; 2 & (4.251)
• Bei Druckerhöhung wird das Gleichgewicht in Richtung C + CO2 verschoben(Prinzip vom kleinsten Zwang (Le Chatelier), das System weicht dem äußerenZwang aus):
% y 0
& y & ; 2 % (4.252)
I Abbildung 4.4: Zur Lage des Boudouard-Gleichgewichtes.
I Ergebnisse:
• Tiefe : Gleichgewicht liegt auf der Seite des CO2,
• Hohe : Gleichgewicht liegt auf der Seite des CO.• Oben: Zustandsfeld des C + CO2 (stabil),• Unten: Zustandsfeld des CO (stabil).• = 1bar: Vom Ausgangspunkt (A) (C + CO + CO2 bei 1020K) ausgehenderfolgt Reaktion in der Richtung C + CO2 → 2 CO, bis der Punkt (B) erreichtist.
• Ausgehend von einem Punkt (C) oberhalb von (B) erfolgt Reaktion in der Rich-tung 2 CO → C + CO2, bis (B) wieder erreicht ist.
• Bei Druckerhöhung von = 1 auf = 10 bar verschiebt sich die Lage desGleichgewichts auf die für 10 bar eingezeichnete Kurve.

4.8 Heterogene Reaktionsgleichgewichte 151
I Anwendungen des Boudouard-Glgew.:
• Reduktion von Oxiden (Hochofenprozess)• Vergasung fossiler Brennstoffe (Kohlevergasung)
4.8.5 Konsequenzen für die Reduktion von FeO zu Fe (Hochofenprozess)
I Hochofen: Zwei umgekehrt aufeinandergesetzte Kegelstümpfe, bis 30 m hoch,Durchmesser bis 7 m. Beschickung mit Erz und Koks von oben.
I Abbildung 4.5: Schematischer Aufbau eines Hochofens.
I Gekoppelte Gleichgewichte:
2 FeO+ 2 CO → 2 Fe+ 2 CO2 (4.253)
CO2 + C → 2 CO (4.254)
I Summenreaktion:2 FeO+ C→ 2 Fe+ CO2 (4.255)
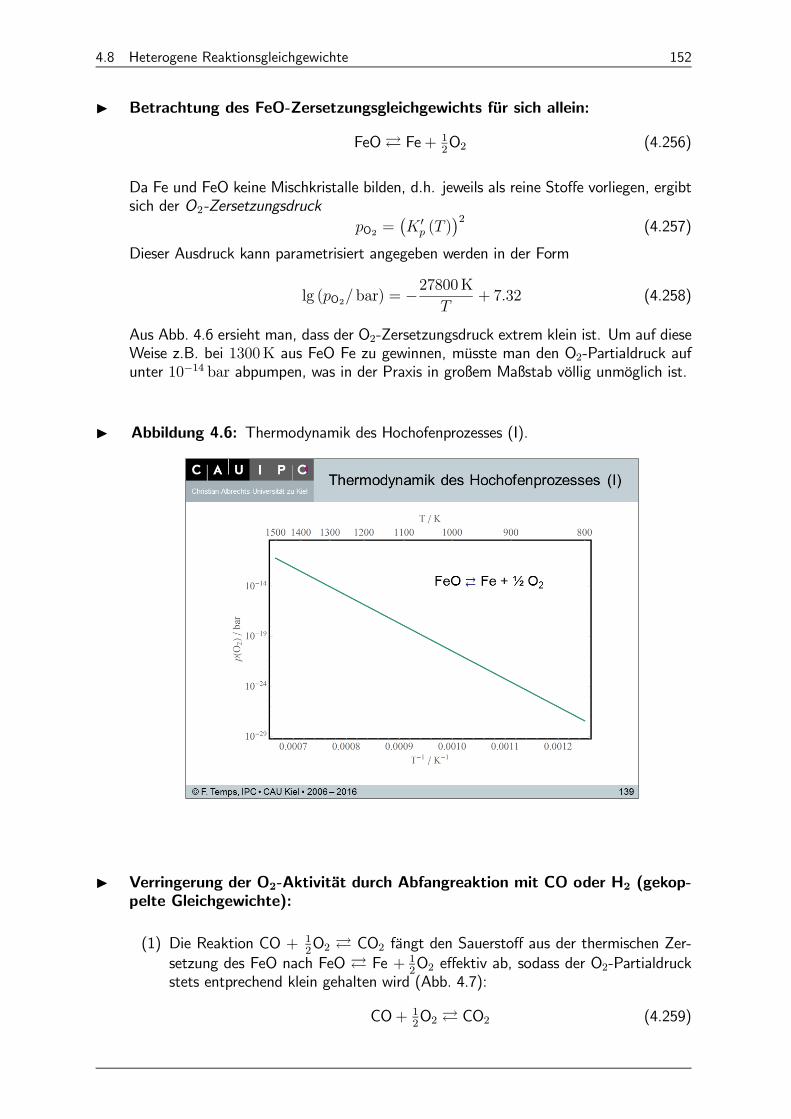
4.8 Heterogene Reaktionsgleichgewichte 152
I Betrachtung des FeO-Zersetzungsgleichgewichts für sich allein:
FeOÀ Fe+ 12O2 (4.256)
Da Fe und FeO keine Mischkristalle bilden, d.h. jeweils als reine Stoffe vorliegen, ergibtsich der O2-Zersetzungsdruck
O2 =¡ 0
( )¢2
(4.257)
Dieser Ausdruck kann parametrisiert angegeben werden in der Form
lg (O2bar) = −27800K
+ 732 (4.258)
Aus Abb. 4.6 ersieht man, dass der O2-Zersetzungsdruck extrem klein ist. Um auf dieseWeise z.B. bei 1300K aus FeO Fe zu gewinnen, müsste man den O2-Partialdruck aufunter 10−14 bar abpumpen, was in der Praxis in großem Maßstab völlig unmöglich ist.
I Abbildung 4.6: Thermodynamik des Hochofenprozesses (I).
I Verringerung der O2-Aktivität durch Abfangreaktion mit CO oder H2 (gekop-pelte Gleichgewichte):
(1) Die Reaktion CO + 12O2 À CO2 fängt den Sauerstoff aus der thermischen Zer-
setzung des FeO nach FeO À Fe + 12O2 effektiv ab, sodass der O2-Partialdruck
stets entprechend klein gehalten wird (Abb. 4.7):
CO+ 12O2 À CO2 (4.259)
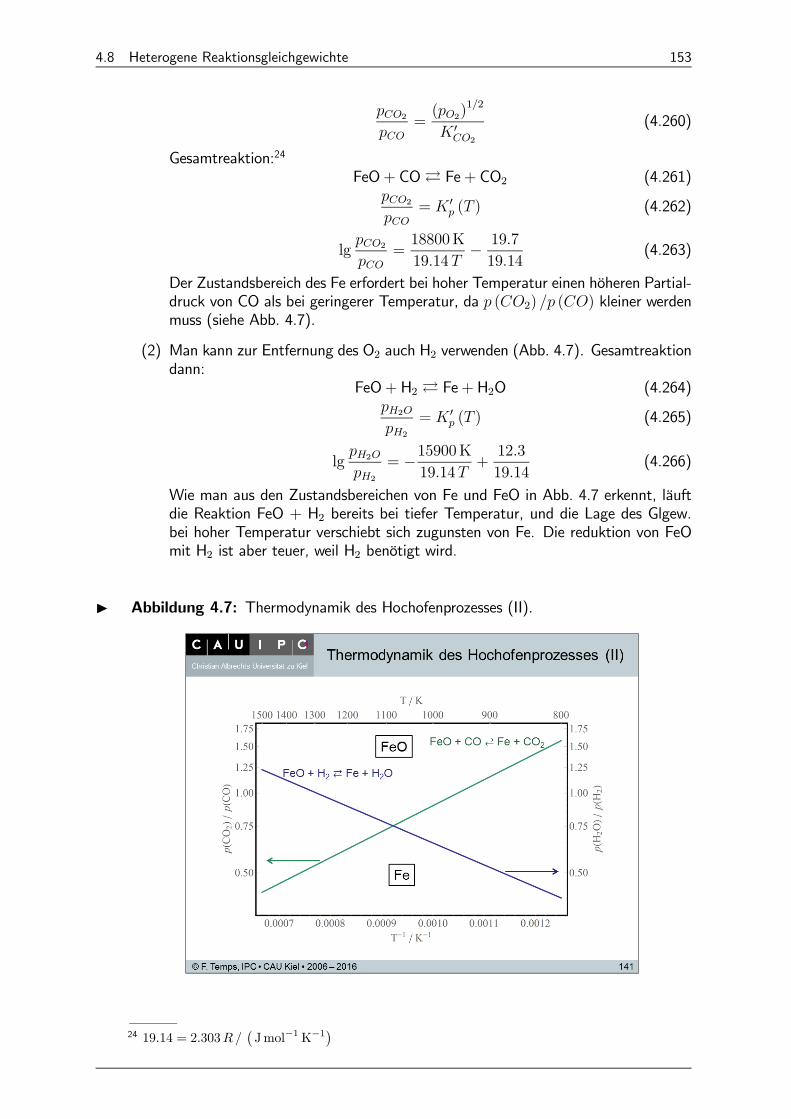
4.8 Heterogene Reaktionsgleichgewichte 153
2
=(2)
12
02
(4.260)
Gesamtreaktion:24
FeO+ COÀ Fe+ CO2 (4.261)
2
= 0 ( ) (4.262)
lg2
=18800K
1914− 197
1914(4.263)
Der Zustandsbereich des Fe erfordert bei hoher Temperatur einen höheren Partial-druck von CO als bei geringerer Temperatur, da (2) () kleiner werdenmuss (siehe Abb. 4.7).
(2) Man kann zur Entfernung des O2 auch H2 verwenden (Abb. 4.7). Gesamtreaktiondann:
FeO+ H2 À Fe+ H2O (4.264)
2
2
= 0 ( ) (4.265)
lg2
2
= −15900K1914
+123
1914(4.266)
Wie man aus den Zustandsbereichen von Fe und FeO in Abb. 4.7 erkennt, läuftdie Reaktion FeO + H2 bereits bei tiefer Temperatur, und die Lage des Glgew.bei hoher Temperatur verschiebt sich zugunsten von Fe. Die reduktion von FeOmit H2 ist aber teuer, weil H2 benötigt wird.
I Abbildung 4.7: Thermodynamik des Hochofenprozesses (II).
24 1914 = 2303¡Jmol−1K−1
¢
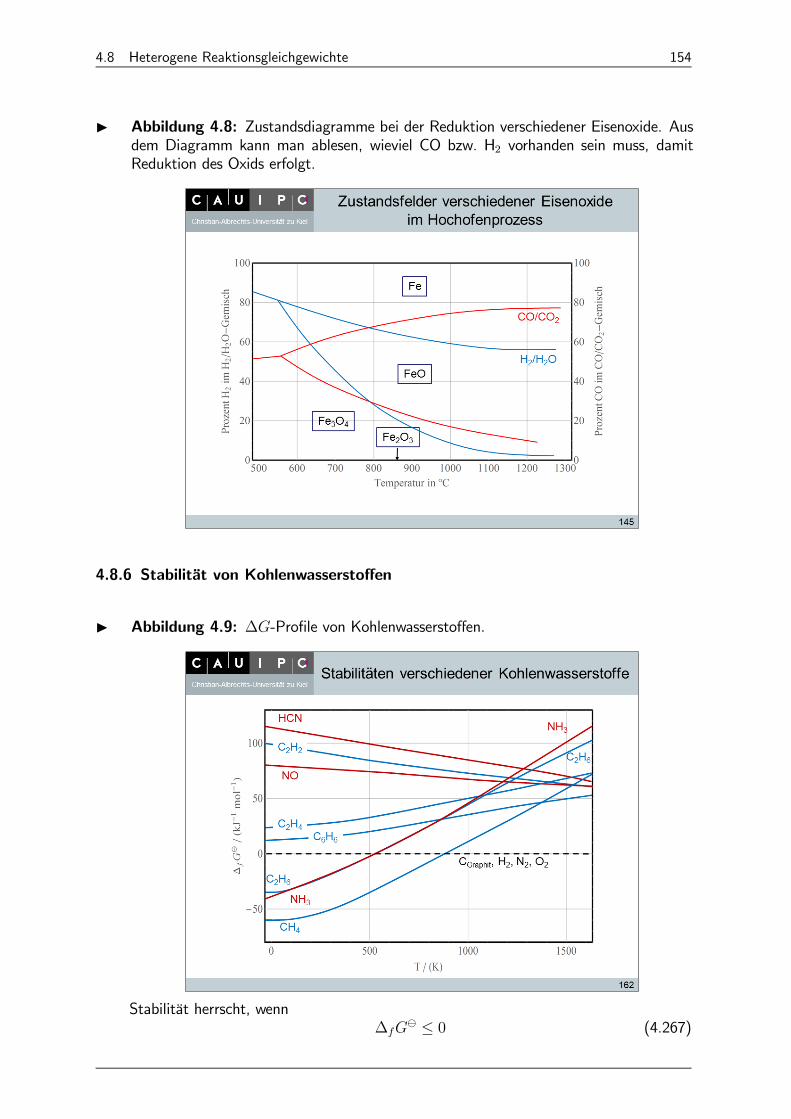
4.8 Heterogene Reaktionsgleichgewichte 154
I Abbildung 4.8: Zustandsdiagramme bei der Reduktion verschiedener Eisenoxide. Ausdem Diagramm kann man ablesen, wieviel CO bzw. H2 vorhanden sein muss, damitReduktion des Oxids erfolgt.
4.8.6 Stabilität von Kohlenwasserstoffen
I Abbildung 4.9: ∆-Profile von Kohlenwasserstoffen
Stabilität herrscht, wenn∆
ª ≤ 0 (4.267)

4.8 Heterogene Reaktionsgleichgewichte 155
I Ergebnisse:
• Bei niedrigen Temperaturen sind CH4 und NH3 stabil (allerdings ist die NH3-Bildung aus N2 und H2 kinetisch gehemmt).
• Mit steigenden Temperaturen wird CH4 zunehmend instabiler, C2H2 und HCNwerden stabiler.
• C6H6 wird bei hohen bezüglich 3 C2H2 instabil.

4.9 Zusammenstellung wichtiger Gleichungen für homogene und heterogene Gleichgewichte 156
4.9 Zusammenstellung wichtiger Gleichungen für homogene und heterogeneGleichgewichte
(1) Chemisches Potential:
=
µ
¶3
(4.268)
a) Druckabhängigkeit für ideale Gase:
( ) = ª ( ) + ln
ª(4.269)
b) Abhängigkeit vom Molenbruch (für ideale Flüssigkeiten)
( ) = ∗ ( ) + ln (4.270)
c) Abhängigkeit von der Aktivität (reales Verhalten):
( ) = ∗ ( ) + ln (4.271)
Gase: Fugazität statt
= ¡
ª¢ (4.272)
Flüssigkeiten: = (4.273)
(2) Thermodynamische Gleichgewichtsbedingung für chemische Reaktionen bei = const.:
∆ =X
= 0 (4.274)
(3) Thermodynamische Gleichgewichtskonstante:
a) Homogene Reaktionen: Gleichgewichtskonstante
= exp
µ−∆
ª
¶(4.275)
mit
ƻ ( ) =
X
ª ( ) = ∆
ª ( )− ×∆ª ( ) (4.276)
b) Heterogene Reaktionen: Partielle Gleichgewichtskonstante
∗ = exp
µ−∆
∗
¶(4.277)
mit25
∆∗ ( ) =
X
∗ ( ) = ∆
∗ ( )− ×∆∗ ( ) (4.278)
25 Bei idealen Gasen setze man ª für ∗. Bei realen Gasen ist die Aktivität zu berücksichtigen.

4.9 Zusammenstellung wichtiger Gleichungen für homogene und heterogene Gleichgewichte 157
(4) = ( ): = − (4.279)
a) Umrechnung von ∆ auf andere Temperaturen:µ∆
¶
= −∆ (4.280)
b) Umrechnung von ∆ auf andere Drucke:µ∆
¶
= ∆ (4.281)
y∆ = ∆ª +
Z
ª∆ (4.282)
ln
=− (∆
∗ )
= −∆
(4.283)
(5) = ( ):
=
µ
¶
+
µ
¶
(4.284)
=
−
µ
¶
(4.285)
=
− (4.286)
Dabei haben wir folgende Beziehung benutzt:
= − (4.287)
y nach Schwarz’schem Satz:
−µ
¶
=
µ
¶
= × (4.288)
mit = thermischer Ausdehnungskoeffizient bei konstantem Druck.
(6) = ( ):
= + (4.289)
= ³ −
´+ (4.290)
= + (1− ) (4.291)
Für und können experimentelle Werte verwendet werden; man muss alsonicht explizit die Zustandsgleichungen kennen, um die o.a. Beziehungen verwendenzu können.
4.9 Zusammenstellung wichtiger Gleichungen für homogene und heterogene Gleichgewichte 158
5. Phasengleichgewichte reiner Stoffe
Wir können das Phasengleichgewichte zwischen verschiedenenen Phasen einesreinen Stoffes A wie eine chemisch Reaktionen schreiben:
A À A À A (5.1)
Anwendung der chemischen GleichgewichtsbedingungP
= 0 (für = const.)liefert dann für die chemischen Potentiale
= = (5.2)
Die Zustandsbereiche, in denen die verschiedenen Phasen vorliegen, stellen wir imPhasendiagramm dar (siehe Abschnitt 1.2.2):
I Abbildung 5.1: Typisches - -Zustandsdiagramm eines reinen Stoffes.
I Frage: Können wir die Lage und den Verlauf der Phasengrenzkurven im -Diagrammquantitativ vorhersagen?
5.1 Qualitative Behandlung von Phasengleichgewichten anhand des chemischenPotentials
(1) µ
¶= − (5.3)
y % y % y & (5.4)
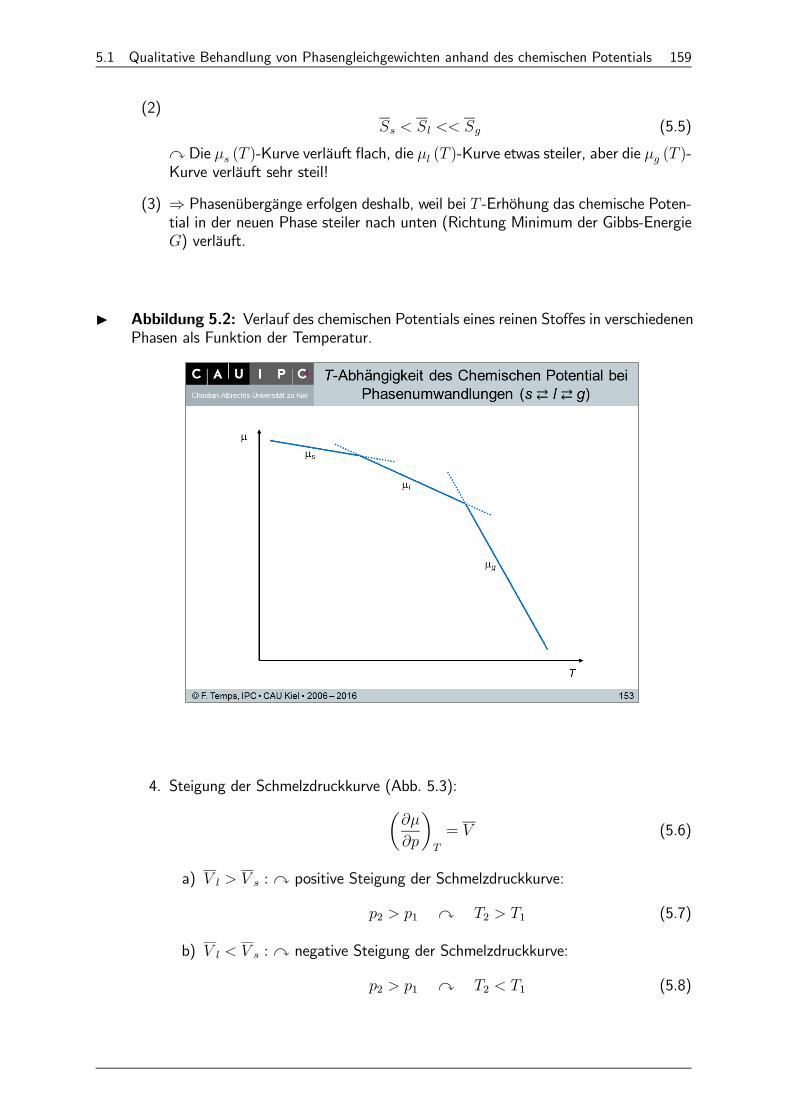
5.1 Qualitative Behandlung von Phasengleichgewichten anhand des chemischen Potentials 159
(2) (5.5)
y Die ( )-Kurve verläuft flach, die ( )-Kurve etwas steiler, aber die ( )-Kurve verläuft sehr steil!
(3) ⇒ Phasenübergänge erfolgen deshalb, weil bei -Erhöhung das chemische Poten-tial in der neuen Phase steiler nach unten (Richtung Minimum der Gibbs-Energie) verläuft.
I Abbildung 5.2: Verlauf des chemischen Potentials eines reinen Stoffes in verschiedenenPhasen als Funktion der Temperatur.
4. Steigung der Schmelzdruckkurve (Abb. 5.3):µ
¶
= (5.6)
a) : y positive Steigung der Schmelzdruckkurve:
2 1 y 2 1 (5.7)
b) : y negative Steigung der Schmelzdruckkurve:
2 1 y 2 1 (5.8)
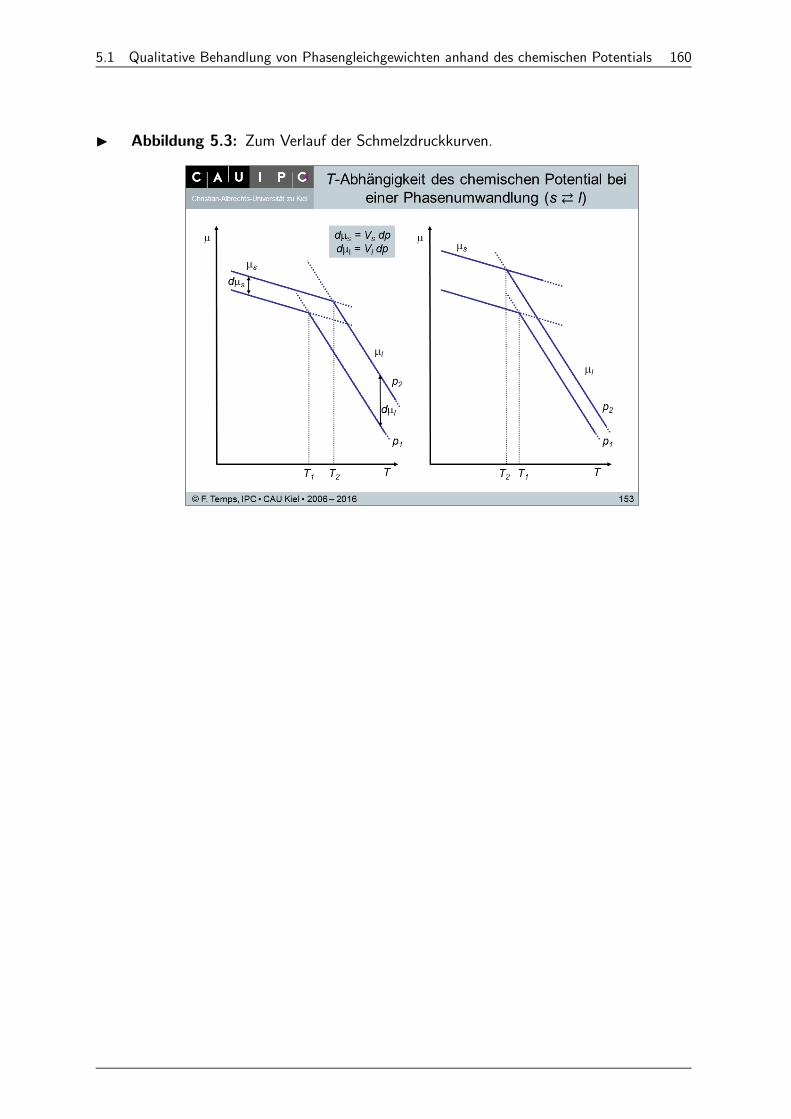
5.1 Qualitative Behandlung von Phasengleichgewichten anhand des chemischen Potentials 160
I Abbildung 5.3: Zum Verlauf der Schmelzdruckkurven.

5.2 Die Gleichungen von Clapeyron und Clausius-Clapeyron 161
5.2 Die Gleichungen von Clapeyron und Clausius-Clapeyron
I Herleitung der Clapeyron’schen Gleichung:
= (5.9)
y = (5.10)
− + = − + (5.11)¡ −
¢ =
¡ −
¢ (5.12)
=
−
−
(5.13)
y Gleichung von Clapeyron:
=
∆
∆=
∆
∆(5.14)
• Anwendung auf Schmelzdruckkurve (Gleichgewicht À ):
∆ 0 (5.15)
(1)
∆ 0 y
0 (5.16)
(2)
∆ 0 y
0 (5.17)
I Gleichung von Clausius-Clapeyron. Bei Anwendung der Clapeyron’schen Gleichungauf Verdampfungsgleichgewichte ( À ) und Sublimationsgleichgewichte ( À )können zwei Vereinfachungen gemacht werden:
(1)∆ = − ≈ (5.18)
(2)
=
(5.19)
y
=
∆ ×
2(5.20)
y ln
=
∆
2(5.21)
Diese Gleichung wird als Clausius-Clapeyron’sche Gleichung bezeichnet.
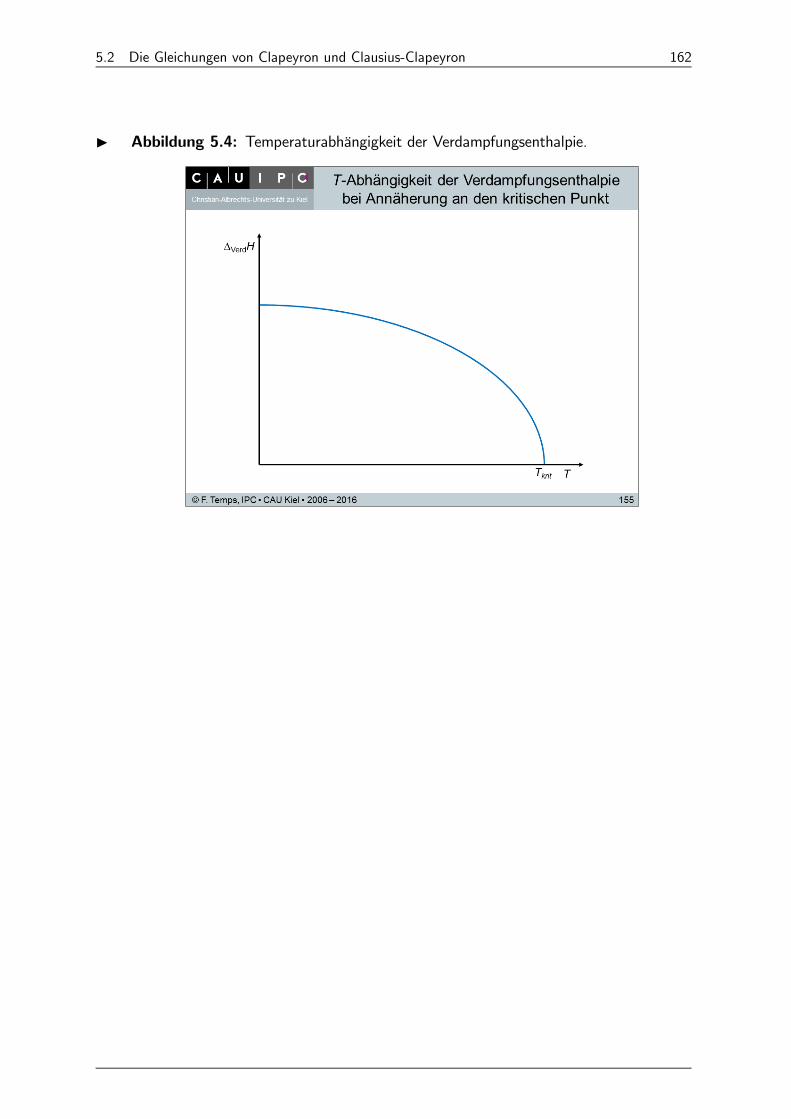
5.2 Die Gleichungen von Clapeyron und Clausius-Clapeyron 162
I Abbildung 5.4: Temperaturabhängigkeit der Verdampfungsenthalpie.

5.3 Druckabhängigkeit des Dampfdrucks∗ 163
5.3 Druckabhängigkeit des Dampfdrucks∗
I Frage: Was passiert, wenn auf einer Flüssigkeit zusätzlich zum eigenen Dampfdrucknoch der Druck eines Inertgases oder der Druck der Erdkruste (z.B. in Geysiren, Vulkanenlastet)?
I Antwort: Es muss die Druckabhängigkeit des chemischen Potentials der Flüssigkeit inRechnung gestellt werden.
= (5.22)
y = (5.23)
mit = (5.24)
und26
= (5.25)
y = (5.26)
=
(5.27)
= ln (5.28)
y∗+∆Z
∗
=
Z∗
ln (5.29)
mit ∗ = Dampfdruck der reinen Flüssigkeit.
I Ergebnis 5.1:
∆ = ln
∗(5.30)
y
∗= exp
µ ∆
¶(5.31)
I Beispiel 5.1: Änderung des Dampfdruckes von H2O bei = 10bar gegenüber =1bar ( = 298K):
≈ 1 + für ¿ 1 (5.32)
y ≈ ∗ ×
µ1 +
∆
¶(5.33)
Mit = 181 cm3mol, = 298K folgt
∗≈ 10073 (5.34)
¤Der Effekt der Druckabhängigkeit auf den Dampfdruck ist also klein.
26 Wir vernachlässigen hier die Löslichkeit des Inertgases in der Flüssigkeit.

5.4 Dampfdruck kleiner Tröpfchen∗ 164
5.4 Dampfdruck kleiner Tröpfchen∗
I Frage: Man beobachtet, dass die Regentropfen bzw. Wassertröpfchen im Nebel prak-tisch alle gleich groß sind. Insbesondere fällt auf auf, dass sehr kleine Tropfen fehlen.Warum?
I Antwort: Bei kleinen Tropfen muss die benötigte Oberflächenspannung (benötigteOberflächenarbeit bei der Bildung der Tropfen aus der Gasphase) in Rechnung gestelltwerden.
I Gleichgewichtsbedingung unter Berücksichtigung von Oberflächenarbeit:
• Für eine Phase: = ( − ) (5.35)
= | z =−+
+ + − − = 0 (5.36)
y = − + = 0 (5.37)
Für = folgt = (5.38)
• Für Flüssigkeit und Dampf im Gleichgewicht (2 Phasen): = + + = 0 (5.39)
wobei = − (5.40)
y = +
µ
¶
(5.41)
• Wir betrachten die Oberfläche und das Volumen von Tröpfchen mit Radius:
= × 42 (5.42)
= = × 433 =
×
3(5.43)
y
=3
(5.44)
bzw.
=3
(5.45)
• Ableitung nach Quotientenregel, weil = ():
µ
¶
=
× 3 − 3 ×µ
¶2
(5.46)
=3
− 3
2
µ
¶(5.47)

5.4 Dampfdruck kleiner Tröpfchen∗ 165
wobei nach Gl. 5.43
µ =
× 433¶
µ
¶=
µ
× 433¶=
× 42 =
=3
(5.48)
y µ
¶=
3(5.49)
y
• µ
¶
=3
− 3
2
µ
¶(5.50)
=3
− 3
2
3(5.51)
=3
−
=2
(5.52)
y = +
µ
¶
(5.53)
y = +
2
(5.54)
• Schreiben wir für die -Abhängigkeit des chemischen Potentials der Flüssigkeit
den Ansatz ln
∗=∞, so erkennen wir dass
ln
∗=∞=
2
(5.55)
y
∗=∞= exp
µ2
×
¶(5.56)
I Schlussfolgerung 5.1: Der Dampfdruck kleiner Tröpfchen ist gegenüber der ho-mogenen makroskopischen Phase erhöht. Bei Abwesenheit von sonstigen Kondensa-tionskeimen muss Dampf übersättigt sein, um die Oberflächenspannung zu überwinden.
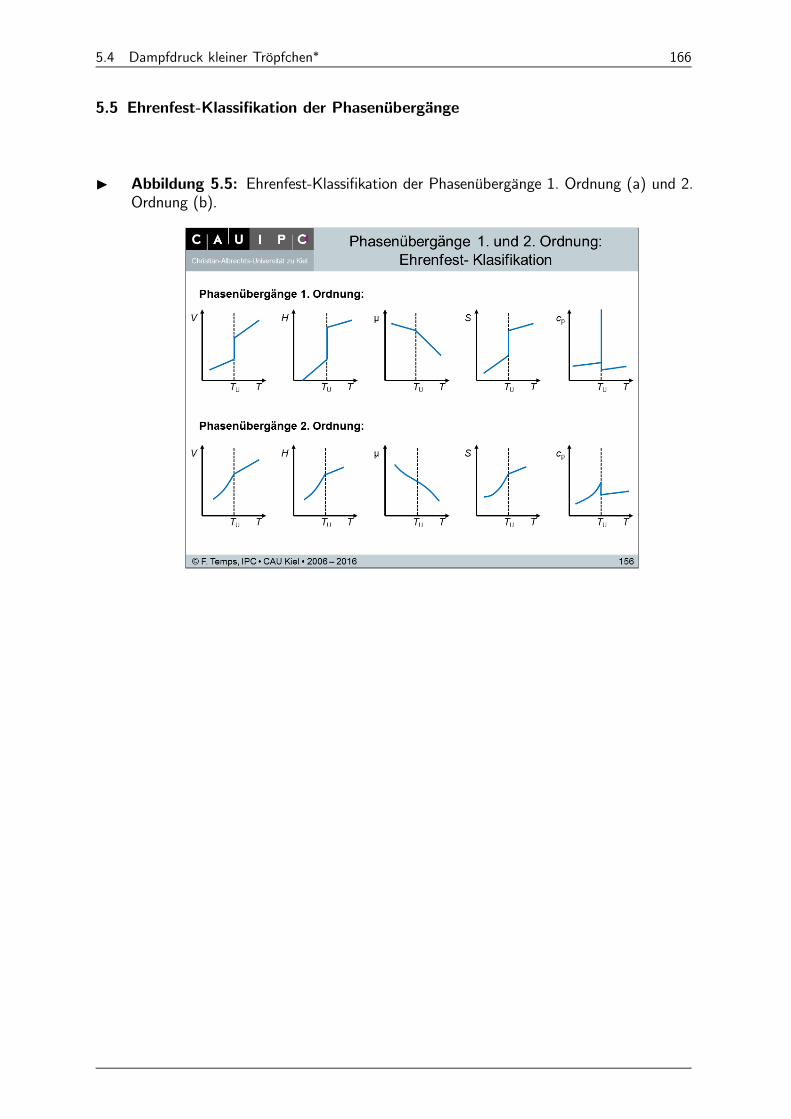
5.4 Dampfdruck kleiner Tröpfchen∗ 166
5.5 Ehrenfest-Klassifikation der Phasenübergänge
I Abbildung 5.5: Ehrenfest-Klassifikation der Phasenübergänge 1. Ordnung (a) und 2.Ordnung (b).

5.5 Ehrenfest-Klassifikation der Phasenübergänge 167
6. Thermodynamik von Lösungen und Mischungen
Im Mittelpunkt dieses Kapitels steht die thermodynamische Beschreibung binärer Mis-chungen und Lösungen (Mischphasenthermodynamik). In den ersten Abschnitten wer-den wir uns auf ideale bzw. ideal verdünnte Mischungen/Lösungen konzentrieren und diewichtigen thermodynamischen Grenzgesetze für ideale bzw. ideal verdünnte Lösungenbehandeln. Die Erweiterung auf reale Mischungen wird in einem späteren Unterabschnittdieses Kapitels vorgenommen.
Die Begriffe Mischung und Lösung werden wir synonym behandeln; es ist nur ein se-mantisches Problem, ob wir von einer Lösung sprechen sollten (wenn ein Stoff - das Lö-sungsmittel - deutlich überwiegt) oder von einer Mischung (z.B. bei zwei Flüssigkeitenmit vergleichbaren Konzentrationen oder über einen weiten Konzentrationsbereich)
I Typen von Lösungen:
(1) Lösungsmittel mit Dampfdruck + nichtflüchtiger Stoff (vernachlässigbarerDampfdruck), z.B.
a) molekular:Zucker() → Zucker() (6.1)
b) ionisch:NaCl() → Na+() + Cl
−() (6.2)
Meist beobachtet man nur eine begrenzte Löslichkeit der Stoffe in einem Lö-sungsmittel. Wenn die Löslichkeit überschritten ist, bleibt ein ungelöster Bo-denkörper zurück; es liegt dann eine gesättigte Lösung vor.
(2) Lösungsmittel mit Dampfdruck + Flüssigkeit mit Dampfdruck, z.B.
a) vollständig mischbar:H2O/C2H5OH (6.3)
Benzol/Toluol (6.4)
b) wenig mischbar:H2O/CCl4 (6.5)
(3) Lösungsmittel + Gas, z.B.
a) gut löslich:NH3/H2O (6.6)
b) schlecht löslich:O2/H2O (6.7)

6.1 Die Grenzgesetze von Raoult und Henry für ideale bzw. ideal verdünnte Lösungen 168
I Geeignete Konzentrationsangaben:
• Molenbruch: =
P
(6.8)
• Konzentration (Liter-Molarität):
=mol gelöster Stoff
l Lösung(6.9)
Nachteil: Schwer zu behandeln, wenn reale Lösungen mit ∆ 6= 0, da sichdie Menge des benötigten Lösungsmittels mit ändert.
• Molalität (Kilogramm-Molarität):
=mol gelöster Stoff
kg Lösungsmittel(6.10)
6.1 Die Grenzgesetze von Raoult und Henry für ideale bzw. ideal verdünnteLösungen
6.1.1 Ideale Lösungen
I Ideale Lösungen:
• Für ideale Mischungen/Lösungen gilt∆ = 0 und ∆ = 0 (6.11)
• Dies ist dann der Fall, wenn für die intermolekularen Wechselwirkungen giltWW (−) =WW (−) =WW ( −) (6.12)
• Es gilt das Raoult’sche Gesetz (siehe unten).
I Ideal verdünnte Lösungen: Reale Lösungen verhalten sich, was die Eigenschaftendes Lösungsmittels anbetrifft, wie ideale Lösungen, wenn sie hoch verdünnt sind:
reale Lösung∞ Verdünnung−→ ideale Lösung (6.13)
6.1.2 Raoult’sches Gesetz
I Frage: Ändert sich der Dampfdruck des Lösungsmittels, wenn kleine Mengen einesgelösten Stoffes hinzugegeben werden?
I Antwort: Der Dampfdruck des Lösungsmittels (1) wird dem Raoult’schen Gesetz zu-folge erniedrigt.

6.1 Die Grenzgesetze von Raoult und Henry für ideale bzw. ideal verdünnte Lösungen 169
I Raoult’sches Gesetz:1 = 1 × ∗1 (6.14)
mit
1 = Dampfdruck des Lösungsmittels (1) über der Lösung
∗1 = Dampfdruck des reinen Lösungsmittels (1)
1 = Molenbruch des Lösungsmittels (1) in der Lösung
Das Raoult’sche Gesetz (Abb. 6.1) gilt
• für ideale Lösungen über den gesamten Bereich von 1,• für reale Lösungen im Grenzfall 1 → 1.
6.1.3 Henry’sches Gesetz
Das Henry’sche Gesetz beschreibt den Dampfdruck eines gelösten Gases (2) im Grenzfall2 → 0 (Abb. 6.1):
2 = × 2 (6.15)
mit der Henry-Konstanten .
I Abbildung 6.1: Raoult’sches und Henry’sches Gesetz.

6.2 Konzentrationsabhängigkeit des chemischen Potentials in Lösung 170
6.2 Konzentrationsabhängigkeit des chemischen Potentials in Lösung
I Gleichgewichtsbedingung: Wir betrachten das chemische Potential einer Flüssigkeit(“Lösungsmittel”) 1 einmal in der reinen Flüssigkeit und einmal in einer Lösung mit demMolenbruch 1 jeweils im Gleichgewicht mit der Gasphase. Anwendung der Gleichge-wichtsbedingung
1 = 1 (6.16)
liefert dann
(1) für das reine Lösungsmittel
∗1 ( ) = ª1 ( ) + ln∗1ª
(6.17)
mit ∗1 = Dampfdruck der reinen Flüssigkeit,
(2) für 1 in der Lösung
1 ( 1) = ª1 ( ) + ln1
ª(6.18)
mit 1 = Dampfdruck von 1 über der Lösung.
Subtraktion der Gl. 6.17 von Gl. 6.18 ergibt
1 ( )− ∗1 ( ) = ln1
∗1(6.19)
Mit dem Raoult’schem Gesetz (1∗1 = 1) folgt daraus
1 ( 1) = ∗1 ( ) + ln1 (6.20)
I Allgemein:
( ) = ∗ ( ) + ln (6.21)
mit dem chemischen Potential der reinen Flüssigkeit ∗ ( ) (Referenzzustand bei ∗ ,
) und dem Molenbruch von in der Lösung .27
27 Einen geeigneten Standardzustand für gelöste Elektrolyte werden wir später definieren.

6.2 Konzentrationsabhängigkeit des chemischen Potentials in Lösung 171
6.3 Thermodynamische Mischungsgrößen
I Gibbs-Energie der Mischung für ideale Mischungen:
=X
∗ ( ) (6.22)
=X
( ) (6.23)
=X
(
∗ ( ) + ln) (6.24)
y∆ = − =
X ln =
X ln (6.25)
Für 1mol der Mischung folgt
∆
= X
ln (6.26)
I Mischungsentropie für ideale Mischungen:
∆ = −µ∆
¶
(6.27)
y∆
= −
X ln (6.28)
I Mischungsenthalpie für ideale Mischungen:
∆
= ∆+ ∆ = 0 (6.29)
I Mischungsvolumen für ideale Mischungen:
∆ =
µ∆
¶
(6.30)
y∆
= 0 (6.31)

6.3 Thermodynamische Mischungsgrößen 172
I Mischungs-Gibbs-Energie und Mischungs-Entropie idealer Mischungen alsFunktion des Molenbruches für zwei Komponenten:
(1)
∆
= 1 ln1 + (1− 1) ln (1− 1) (6.32)
a) Da 1 1 und (1− 1) 1 folgt ln1 0 und ln (1− 1) 0 und damit
∆
0 (6.33)
b) Das Minimum von ∆
wird erreicht, wenn 1 = 2 = 05. y
∆
min =
µ1
2ln1
2+1
2ln1
2
¶= − ln 2 (6.34)
(2)
∆
= −1 ln1 − (1− 1) ln (1− 1) 0 (6.35)
Die Mischungsentropie erreicht ihr Maximum bei 1 = 2 = 05.
(3) Steigung bei 1 → 0: Für 1 → 0 wird28
∆ = (1 ln1 + (1− 1) ln (1− 1)) (6.36)
≈ (1 ln1 + (1− 1) (−1)) (6.37)
= ¡1 ln1 + 21 − 1
¢(6.38)
Steigung von ∆ vs. 1:
∆
1=
µ11
1+ ln1 + 21 − 1
¶(6.39)
= (ln1 + 21) (6.40)
Steigung bei 1 → 0: µ∆
1
¶1=0
→ ln1 →−∞ (6.41)
28 Man betrachte den Verlauf von ln und von ln
=1
in der Umgebung von = 1.
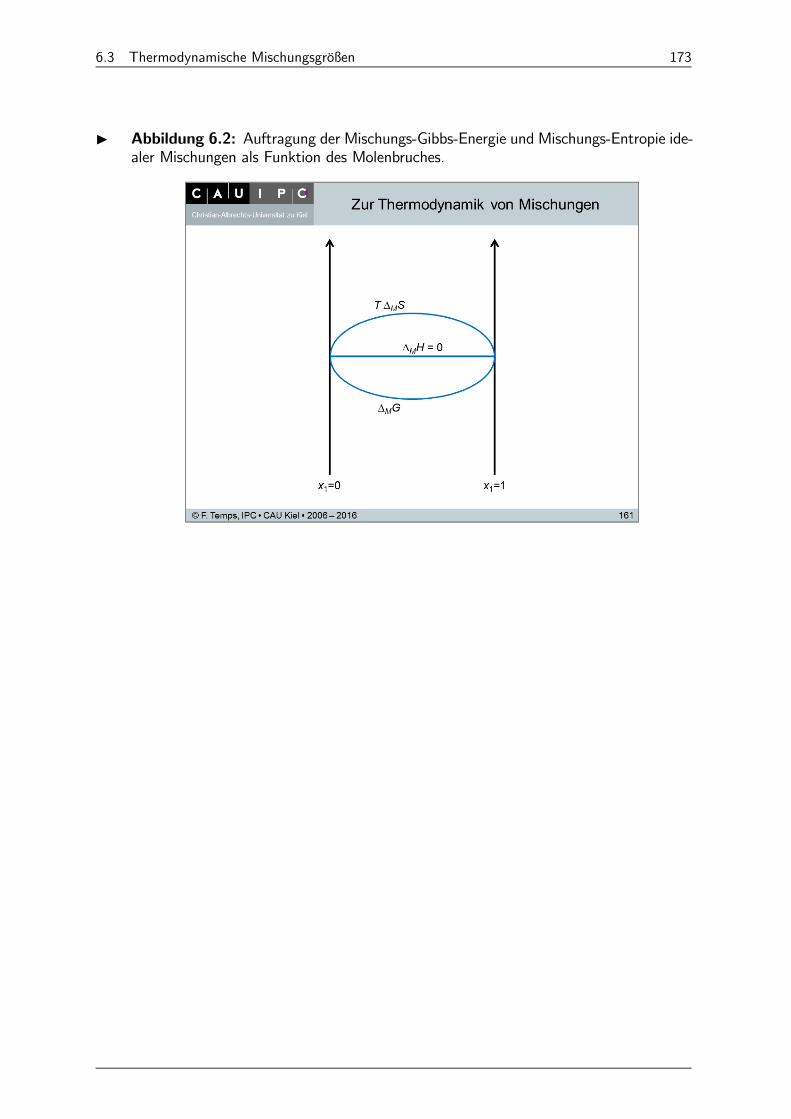
6.3 Thermodynamische Mischungsgrößen 173
I Abbildung 6.2: Auftragung der Mischungs-Gibbs-Energie und Mischungs-Entropie ide-aler Mischungen als Funktion des Molenbruches.

6.4 Kolligative Eigenschaften 174
6.4 Kolligative Eigenschaften
Als kolligative Eigenschaften bezeichnet man solche Eigenschaften von Lösungen, dienur von der gelösten Stoffmenge (Molzahl!), nicht aber von der Stoffart abhängig sind.
6.4.1 Rückblick auf die Gesetze von Raoult und Henry
Aus der Gleichgewichtsbedingung = (6.42)
mit = ∗ ( ) + ln (6.43)
und = ª ( ) + ln
ª(6.44)
folgt
∗ ( ) + ln = ª ( ) + ln
ª(6.45)
yln
ª × = −
ª ( )− ∗ ( )
(6.46)
oder
ª= exp
Ã−
ª ( )− ∗ ( )
!× (6.47)
Hieraus folgt:
I Raoult’sches Gesetz: Für → 1, und daher → ∗ , folgt aus Gl. 6.47 selbstver-ständlich, dass
∗ª= exp
Ã−
ª ( )− ∗ ( )
! (6.48)
y
∗= für → 1 (6.49)
I Henry’sches Gesetz: Für 2 → 0 können wir i.a. nicht davon ausgehen, dass sichder gelöste Stoff (2), der praktisch vollständig nur von Lösungsmittel (1) umgeben ist,ideal verhält. Es ist daher angezeigt, 2 im Ausdruck für das chemische Potential durchdie Aktivität (vgl. Abschnitt 6.5) von (2) zu ersetzen:
2 = ∗2 ( ) + ln 2 (6.50)
mit2 = 2 2 . (6.51)

6.4 Kolligative Eigenschaften 175
2 ist der Aktivitätskoeffizient. Gleichung 6.47 lautet damit
2
ª= exp
Ã−
ª2 ( )− ∗2 ( )
!× 2 (6.52)
= exp
Ã−
ª2 ( )− ∗2 ( )
!× 2 × 2 (6.53)
y2 = H × 2 (6.54)
mit
H = 2 exp
Ã−
ª2 ( )− ∗2 ( )
!ª (6.55)
6.4.2 Nernst’scher Verteilungssatz
I Beispiel: Verteilung von I2 zwischen zwei flüssigen Phasen und (z.B. H2O undCCl4).
I Abbildung 6.3: Zum Nernst’schen Verteilungssatz.
I Herleitung:∗2 + ln2 =
∗2+ ln
2
(6.56)
y2
2= exp
Ã−
∗2− ∗2
!= const (6.57)
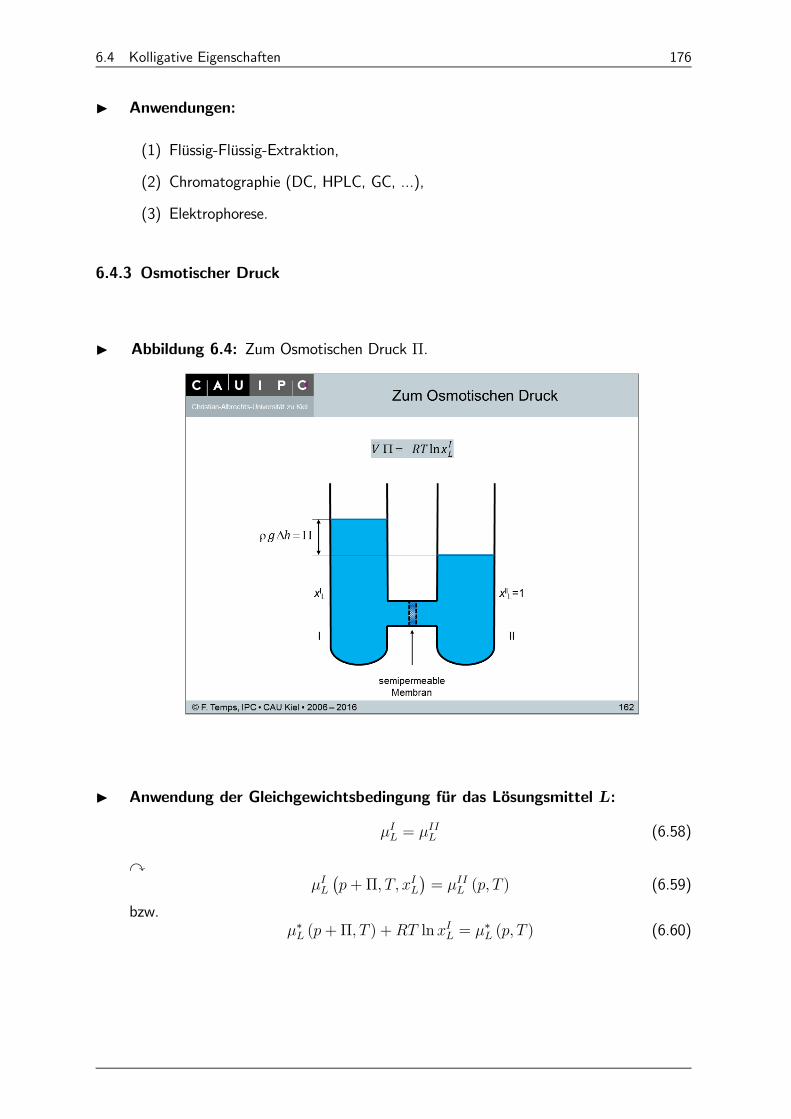
6.4 Kolligative Eigenschaften 176
I Anwendungen:
(1) Flüssig-Flüssig-Extraktion,
(2) Chromatographie (DC, HPLC, GC, ...),
(3) Elektrophorese.
6.4.3 Osmotischer Druck
I Abbildung 6.4: Zum Osmotischen Druck Π.
I Anwendung der Gleichgewichtsbedingung für das Lösungsmittel L:
= (6.58)
y¡+Π
¢= ( ) (6.59)
bzw.∗ (+Π ) + ln = ∗ ( ) (6.60)

6.4 Kolligative Eigenschaften 177
I Taylor-Reihenentwicklung um p für Π¿ p:
(0 + ) = (0) + 0 (0)1!
+ 00 (0)2
2!+ (6.61)
y
∗ (+Π ) = ∗ ( ) +µ∗
¶×Π| z
=×Π
+1
2
µ2∗2
¶×Π2 + | z
vernachlässigbar
(6.62)
y
I Gesetz vom osmotischen Druck:
∗ ( ) + ×Π+ ln = ∗ ( ) (6.63)
×Π = − ln (6.64)
I Näherungen:
(1) Mit = 1− und ln¡1−
¢→− für → 0 (verdünnte Lösung) folgt
×Π = − ln ¡1− ¢ ≈ − ¡−¢ = (6.65)
(2)
=
+ ≈
(6.66)
und = (6.67)
I Ergebnis:
Π =
= (6.68)
mit = Konzentration des gelösten Stoffes in mol l (unabhängig von der Stoffart).
I Achtung: Bei Dissoziation des gelösten Stoffes in Ionen entsteht ein entsprechend derhöheren Teilchenzahl höherer osmotischer Druck!
I Anwendungen:
(1) Molmassenbestimmung von Makromolekülen,
(2) physiologische Kochsalzlösung,
(3) Meerwasserentsalzung durch umgekehrte Osmose.
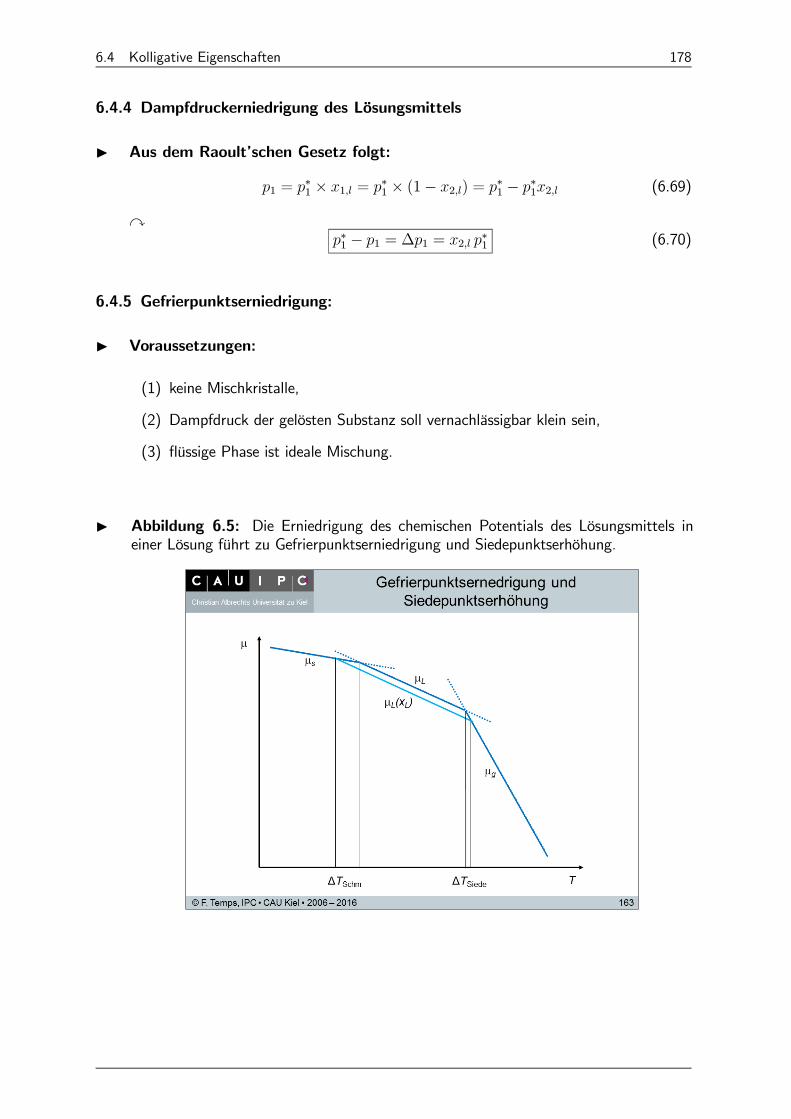
6.4 Kolligative Eigenschaften 178
6.4.4 Dampfdruckerniedrigung des Lösungsmittels
I Aus dem Raoult’schen Gesetz folgt:
1 = ∗1 × 1 = ∗1 × (1− 2) = ∗1 − ∗12 (6.69)
y∗1 − 1 = ∆1 = 2
∗1 (6.70)
6.4.5 Gefrierpunktserniedrigung:
I Voraussetzungen:
(1) keine Mischkristalle,
(2) Dampfdruck der gelösten Substanz soll vernachlässigbar klein sein,
(3) flüssige Phase ist ideale Mischung.
I Abbildung 6.5: Die Erniedrigung des chemischen Potentials des Lösungsmittels ineiner Lösung führt zu Gefrierpunktserniedrigung und Siedepunktserhöhung.
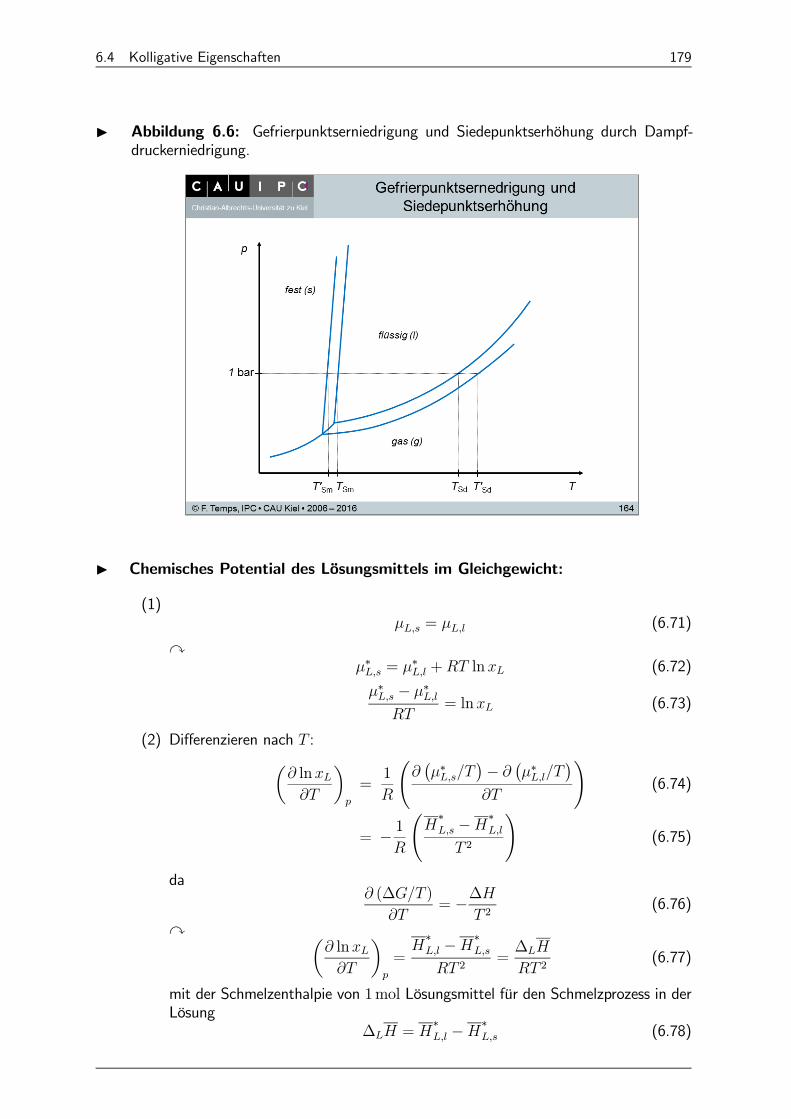
6.4 Kolligative Eigenschaften 179
I Abbildung 6.6: Gefrierpunktserniedrigung und Siedepunktserhöhung durch Dampf-druckerniedrigung.
I Chemisches Potential des Lösungsmittels im Gleichgewicht:
(1) = (6.71)
y∗ = ∗ + ln (6.72)
∗ − ∗
= ln (6.73)
(2) Differenzieren nach :µ ln
¶
=1
á∗
¢− ¡∗
¢
!(6.74)
= − 1
Ã∗ −
∗
2
!(6.75)
da (∆ )
= −∆
2(6.76)
y µ ln
¶
=∗ −
∗
2=
∆
2(6.77)
mit der Schmelzenthalpie von 1mol Lösungsmittel für den Schmelzprozess in derLösung
∆ = ∗ −
∗ (6.78)

6.4 Kolligative Eigenschaften 180
(3) Integration von = 1 (Gefriertemperatur ∗) bis (Gefriertemperatur ):
Z1
ln =
Z∗
∆
2 (6.79)
y
ln = −∆
µ1
− 1
∗
¶(6.80)
(4) Näherungen (Vereinfachungen) für verdünnte Lösungen:
ln = ln (1− ) ≈ − für → 0 (6.81)
y ≈ ∆
µ1
− 1
∗
¶=
∆
∗ −
∗≈ −∆
∆
∗2(6.82)
mit der Gefrierpunkserniedrigung ∆ = − ∗.
(5) Ergebnis:
≈ −∆
∗2∆ (6.83)
I Anwendung zur Molmassenbestimmung: Für ¿ 1 folgt
≈
=
1 kg= × (6.84)
y
∆ = −∗2 ×
∆× = − × (6.85)
ist die kryoskopische Konstante oder molale Gefrierpunktserniedrigung.
I Anmerkung 6.1: ∆ hängt nur von der Menge (Molalität, d.h. Molzahl) des gelöstenStoffes ab, nicht von der Stoffart. Hierauf beruht die Anwendung zur Molmassenbes-timmung.
I Anmerkung 6.2: Achtung bei Salzen!
6.4.6 Siedepunktserhöhung
Unter der Voraussetzung, dass der Dampfdruck der gelösten Substanz vernachlässigbarist, folgt analog zur Gefrierpunktserniedrigung aus
= (6.86)
die Siedepunktserhöhung
∆ = ∗2
∆ × (6.87)
bzw.
∆ = ∗2 ×
∆ × = × (6.88)
mit der ebullioskopischen Konstante oder molalen Siedepunktserhöhung .

6.4 Kolligative Eigenschaften 181
I Tabelle 6.1: Molale Gefrierpunktserniedrigungen bzw. Siedepunktserhöhungen einigerLösungsmittel.
Lösungsmittel molale Siedepunktserhöhung molale Gefrierpunktserniedrigung
Kkgmol−1 Kkgmol−1
H2O 0.521 1.858CHCl3 3.80Benzol 2.54 5.065Campher 6.09 40.

6.5 Behandlung realer Lösungen 182
6.5 Behandlung realer Lösungen
6.5.1 Aktivitäten
In realen Lösungen treten Abweichugen vom idealen Verhalten auf. Diese werden durchEinführung der Aktivitäten bzw. Aktivitätskoeffizienten
= × (6.89)
berücksichtigt.
6.5.2 Chemisches Potential und Exzess-Größen
I Chemisches Potential:
( ) = ∗ ( ) + ln (6.90)
= ∗ ( ) + ln + ln (6.91)
= + ()
(6.92)
I Thermodynamische Mischungsgrößen:
• ∆:
∆ = X
ln (6.93)
= X
ln +X
ln (6.94)
= ∆
+∆()
(6.95)
y∆
=
P ln (6.96)
• ∆:
∆= −
Ã∆
!
= −P ln −P
µ ln
¶
(6.97)
• ∆:
∆= ∆
+ ∆
= − 2P
µ ln
¶
6= 0 (6.98)
• ∆:
∆=
Ã∆
!
= P
µ ln
¶
6= 0 (6.99)
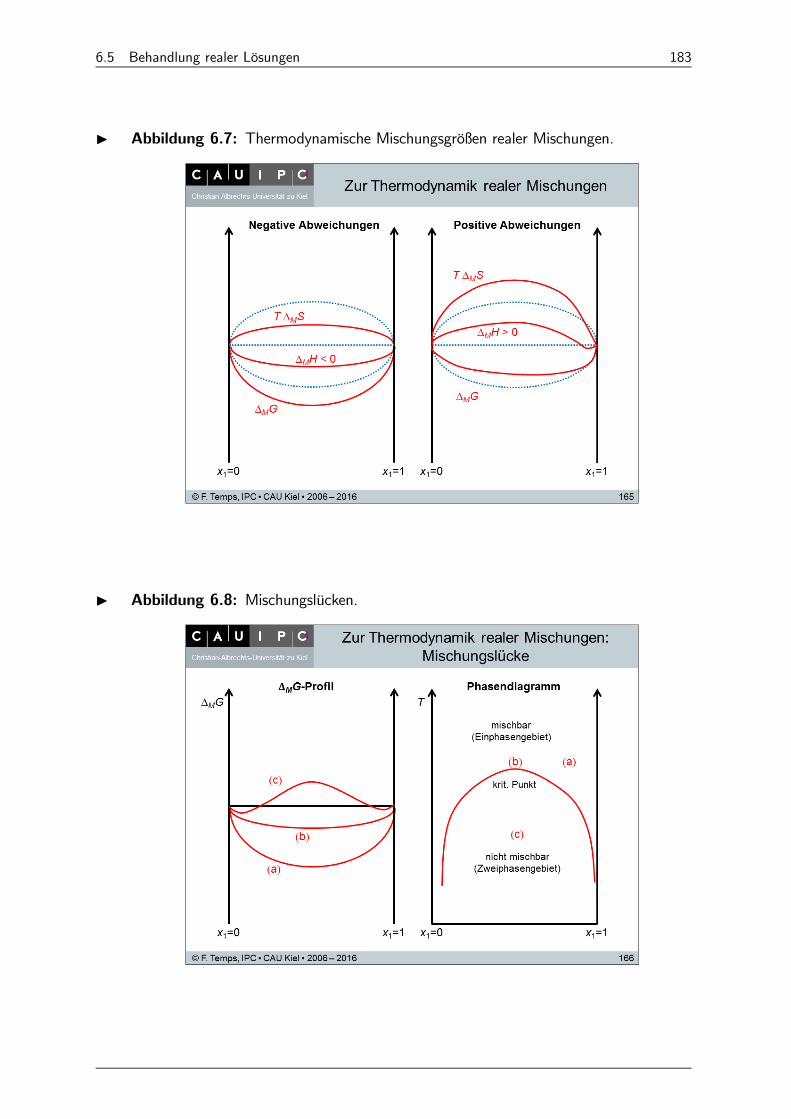
6.5 Behandlung realer Lösungen 183
I Abbildung 6.7: Thermodynamische Mischungsgrößen realer Mischungen.
I Abbildung 6.8: Mischungslücken.

6.5 Behandlung realer Lösungen 184
6.5.3 Experimentelle Bestimmung von Aktivitätskoeffizienten
I Anwendung des Raoult’schen Gesetzes auf die beiden Komponenten einerbinären Mischung:
= ∗ = ∗ = (6.100)
y =
∗
(6.101)
Man misst , 1, 2 sowie 1 und 2 und bestimmt daraus 1 und 2. Dazuentnimmt man aus Flüssigkeit und (verflüssigtem) Dampf Proben, die mit Standard-methoden auf ihre Zusammensetzung analysiert werden (z.B. Brechungsindex). Mitden Ergebnissen ist das System jedoch überbestimmt, da die chemischen Potentiale derbeiden Komponenten und damit 1 und 2 über die Gibbs-Duhem-Gleichung gekoppeltsind. Dies kann nach Herington zur Konsistenzprüfung der Messungen genutzt werden.
I Konsistenzprüfung mithilfe des Herington-Kriteriums:*
(1) Die Gibbs-Duhem-Gleichung für = X = 0 (6.102)
liefert für 1mol einer binären Mischung
11 + 22 = 0 (6.103)
y1
µ11
¶1 + 2
µ22
¶2 = 0 (6.104)
Differentiation des chemischen Potentials
1 = ∗1 + ln1 + 1 = ∗1 + ln1 + ln 1 (6.105)
führt zu µ11
¶= 0 +
1+
µ ln 1
1
¶(6.106)
(analog für 2).
Einsetzen in Gl. 6.104 liefert
1
µ
1+
µ ln 1
1
¶¶1+2
µ
2+
µ ln 2
2
¶¶2 = 0 (6.107)
und mit 2 = −1 y
1 + 1
µ ln 1
1
¶1 −1 + (1− 1)
µ ln 2
1
¶1 = 0
(6.108)y
1
µ ln 1
1
¶1 + (1− 1)
µ ln 2
1
¶1 = 0 (6.109)
bzw.
1
µ ln 1
1
¶1 + (1− 1)
µ ln 2
1
¶1 = 0 (6.110)

6.5 Behandlung realer Lösungen 185
(2) Die Excess-Gibbs-Energie für eine binäre Mischung ist
∆= (1 ln 1 + (1− 1) ln 2) (6.111)
y∆
1= (ln 1 − ln 2)+
µ1 ln 1
1+ (1− 1)
ln 2
1
¶| z
=0 nach Gl. 6.110
= ln1
2
(6.112)
Integration liefert das Konsistenzintegral
∆(1=1)Z
∆(1=0)
∆=
1=1Z1=0
ln1
21 = 0 (6.113)
da
∆(1=1)Z
∆(1=0)
∆= ∆
(1 = 1)| z
=0 für reinen Stoff 1
−∆(1 = 0)| z
=0 für reinen Stoff 2
= 0 (6.114)
Das Konsistenzintegral besagt, dass eine Auftragung von ln1
2gegen 1 eine
Kurve mit gleich großen positiver und negativer Fläche unter der Kurve gebenmuss (Abb. 6.9 a):
1=1Z1=0
ln1
21 = 0 (6.115)
I Abbildung 6.9: Auftragung von ln1
2gegen 1 zur Konsistenzprüfung nach Herington
(a) allgemein und (b) nach dem Porter’schen Ansatz.

6.5 Behandlung realer Lösungen 186
I Porter’scher Ansatz:* Der Konzentrationsverlauf von ∆als Funktion von
1 lässt sich bei nicht allzu großer Abweichung vom idealen Verhalten mithilfe desPorter’schen Ansatzes ( = experimentell angepasster Parameter) beschreiben:
∆= × 12 = × 1 (1− 1) = × ¡1 − 21
¢(6.116)
y
ln1
2=
1
∆
1=
1
1
¡× ¡1 − 21
¢¢(6.117)
y
ln1
2=
(1− 21) (6.118)
D.h., Auftragung von ln1
2gegen 1 im Bereich 0 bis 1 liefert eine Gerade mit Achsen-
abschnitt +
bei 1 = 0 und Achsenabschnitt −
bei 1 = 1 (Abb. 6.9 b).
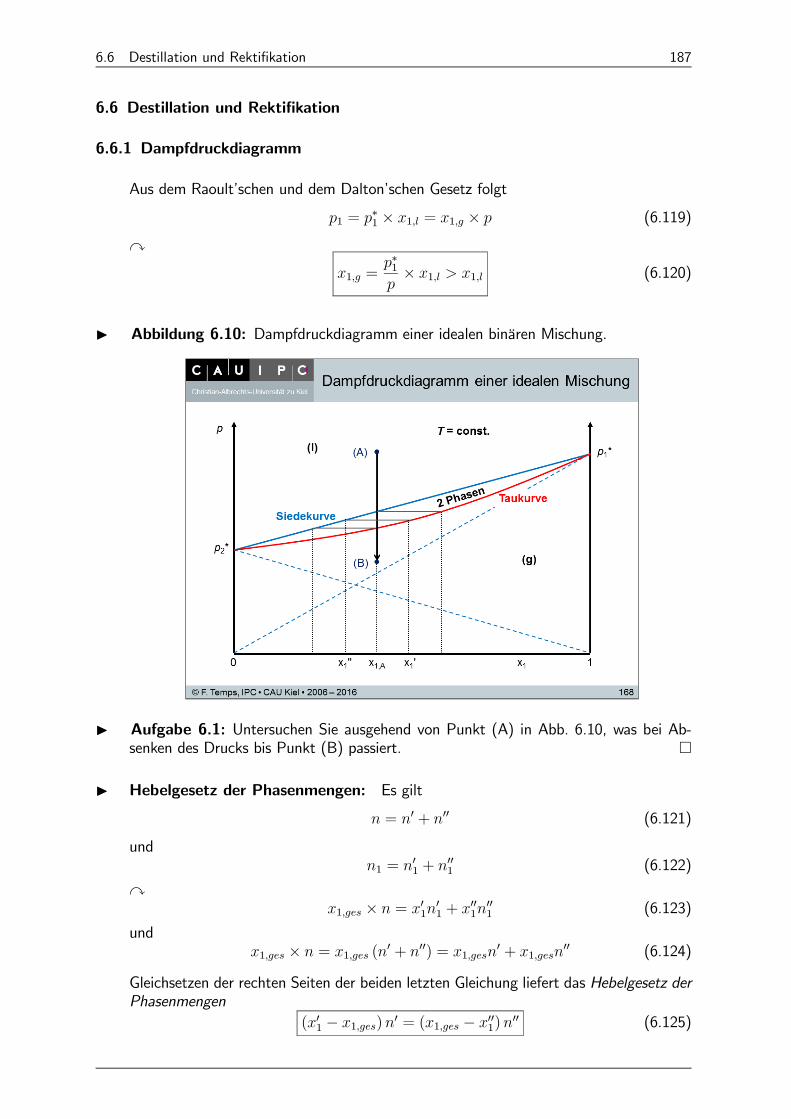
6.6 Destillation und Rektifikation 187
6.6 Destillation und Rektifikation
6.6.1 Dampfdruckdiagramm
Aus dem Raoult’schen und dem Dalton’schen Gesetz folgt
1 = ∗1 × 1 = 1 × (6.119)
y
1 =∗1× 1 1 (6.120)
I Abbildung 6.10: Dampfdruckdiagramm einer idealen binären Mischung.
I Aufgabe 6.1: Untersuchen Sie ausgehend von Punkt (A) in Abb. 6.10, was bei Ab-senken des Drucks bis Punkt (B) passiert. ¤
I Hebelgesetz der Phasenmengen: Es gilt
= 0 + 00 (6.121)
und1 = 01 + 001 (6.122)
y1 × = 01
01 + 001
001 (6.123)
und1 × = 1 (
0 + 00) = 10 + 1
00 (6.124)
Gleichsetzen der rechten Seiten der beiden letzten Gleichung liefert das Hebelgesetz derPhasenmengen
(01 − 1)0 = (1 − 001)
00 (6.125)
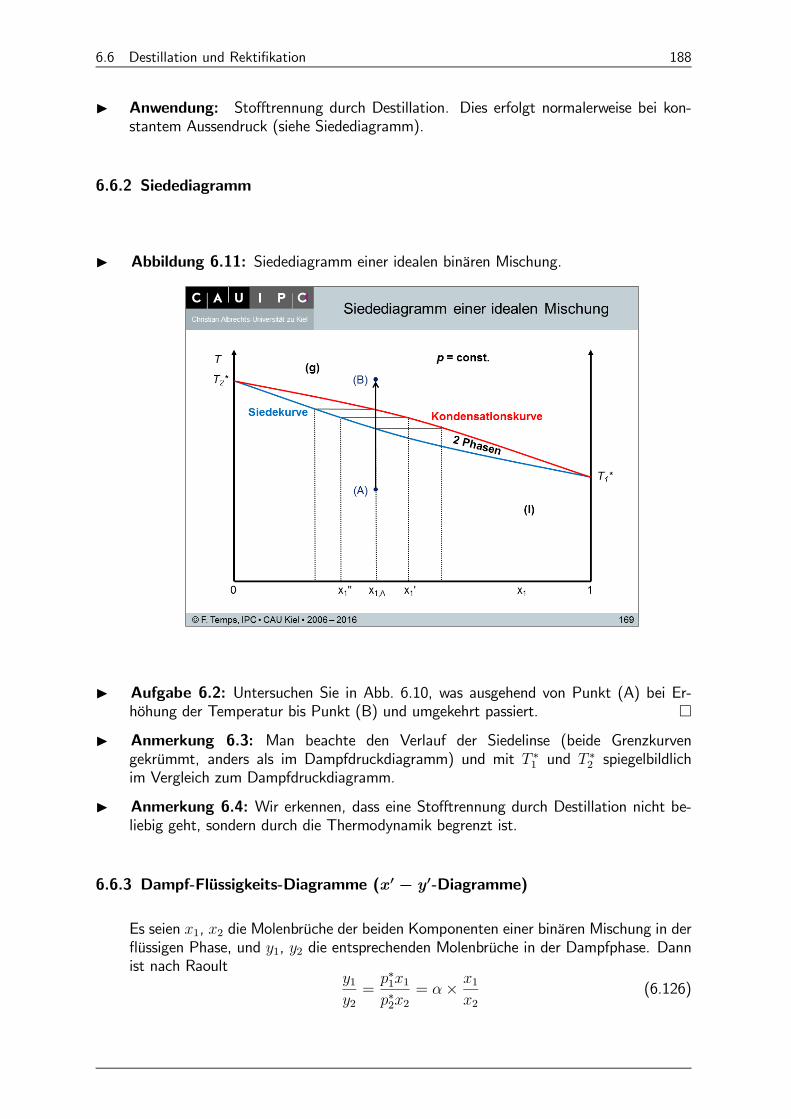
6.6 Destillation und Rektifikation 188
I Anwendung: Stofftrennung durch Destillation. Dies erfolgt normalerweise bei kon-stantem Aussendruck (siehe Siedediagramm).
6.6.2 Siedediagramm
I Abbildung 6.11: Siedediagramm einer idealen binären Mischung.
I Aufgabe 6.2: Untersuchen Sie in Abb. 6.10, was ausgehend von Punkt (A) bei Er-höhung der Temperatur bis Punkt (B) und umgekehrt passiert. ¤
I Anmerkung 6.3: Man beachte den Verlauf der Siedelinse (beide Grenzkurvengekrümmt, anders als im Dampfdruckdiagramm) und mit ∗1 und ∗2 spiegelbildlichim Vergleich zum Dampfdruckdiagramm.
I Anmerkung 6.4: Wir erkennen, dass eine Stofftrennung durch Destillation nicht be-liebig geht, sondern durch die Thermodynamik begrenzt ist.
6.6.3 Dampf-Flüssigkeits-Diagramme (x0 − y0-Diagramme)
Es seien 1, 2 die Molenbrüche der beiden Komponenten einer binären Mischung in derflüssigen Phase, und 1, 2 die entsprechenden Molenbrüche in der Dampfphase. Dannist nach Raoult
1
2=
∗11∗22
= × 1
2(6.126)

6.6 Destillation und Rektifikation 189
I Trennfaktor: ist der Trennfaktor. Für reale Mischungen ist er gegeben als
=∗1 × 1
∗2 × 2(6.127)
mit = ( ).
I Trenndiagramm (Dampf-Flüssigkeits-Diagramm): Wir lösen
1
2= × 1
2(6.128)
nach 1 auf, indem wir 2, 2 ersetzen:
1
1− 1= × 1
1− 1(6.129)
y1 =
1
1 + (− 1)1 (6.130)
Dies ist die Gleichung einer Hyperbel, die um so flacher verläuft, je näher an 1 ist.
I Abbildung 6.12: Dampf-Flüssigkeits-Diagramm mit drei Stufen.
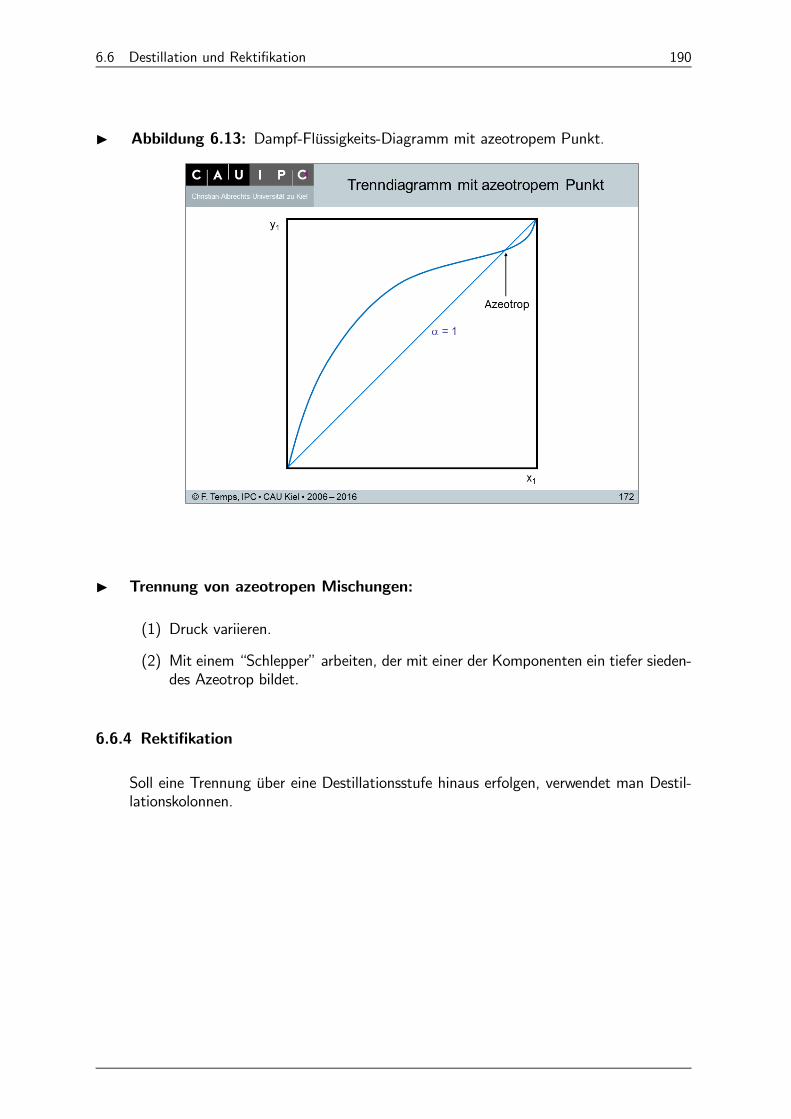
6.6 Destillation und Rektifikation 190
I Abbildung 6.13: Dampf-Flüssigkeits-Diagramm mit azeotropem Punkt.
I Trennung von azeotropen Mischungen:
(1) Druck variieren.
(2) Mit einem “Schlepper” arbeiten, der mit einer der Komponenten ein tiefer sieden-des Azeotrop bildet.
6.6.4 Rektifikation
Soll eine Trennung über eine Destillationsstufe hinaus erfolgen, verwendet man Destil-lationskolonnen.

6.6 Destillation und Rektifikation 191
I Abbildung 6.14: Aufbau einer Destillationskolonne.
I Funktionsprinzip:
• Gegenstromverfahren (vgl. Funktionsprinzip des Wärmeaustauschers),• ständiger Stoffaustausch zwischen Dampf- und Flüssigkeitsstrom,• adiabatische Betriebsweise,• Kolonnenkopf nicht vergessen (ein Teil muss zurücklaufen, sonst keine Trennung),• Zwischen Flüssigkeit und Dampf herrscht kein Gleichgewicht !!
I Varianten:
• Glockenbodenkolonne (in der Technik, z.B. Raffinierien),• Füllkörperkolonne,• Vigreux-Kolonne.

6.6 Destillation und Rektifikation 192
I Herleitung der Rücklaufgeraden:
(1) Massenbilanz insgesamt und für jede Komponente an jeder beliebigen Stelle inder Kolonne:
= + (6.131)
= + (6.132)
mit
• = Molenbruch der Komponenten 1 in Flüssigkeit,
• = Molenbruck der Komponenten 1 im Dampf.
(2) Auflösung nach :
=
+
=
++
+ (6.133)
=
++
+ (6.134)
(3) Einführung des Rücklaufverhältnisses
=
(6.135)
liefert
() =
+ 1 () +
1
+ 1 (6.136)
(4) Grenzfall des vollständigen Rücklaufs:
=
→∞ y
→ 1 (6.137)
y () = () (6.138)
Man erkennt dass zwischen Dampf und Flüssigkeit an der Stelle ( beliebig)kein Gleichgewicht vorliegt!
(5) Aber (), () ändern sich natürlich mit . Das Verhältnis
( + ) = () (6.139)
entspricht dem Verhältnis zwischen und im Gleichgewicht.
(6) Für ein gegebenes Trennproblem ist die Einhaltung eines minimalen Rücklaufver-hältnisses erforderlich, ansonsten ist die gewünschte Reinheit nicht erreichbar.
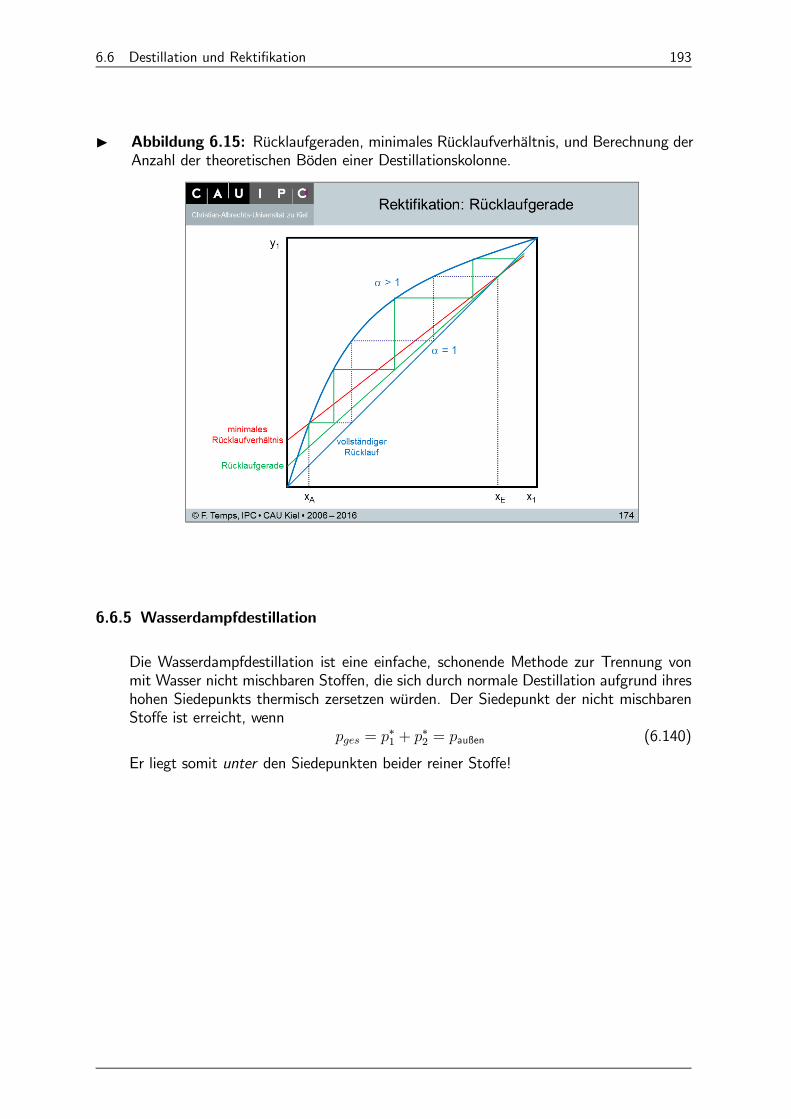
6.6 Destillation und Rektifikation 193
I Abbildung 6.15: Rücklaufgeraden, minimales Rücklaufverhältnis, und Berechnung derAnzahl der theoretischen Böden einer Destillationskolonne.
6.6.5 Wasserdampfdestillation
Die Wasserdampfdestillation ist eine einfache, schonende Methode zur Trennung vonmit Wasser nicht mischbaren Stoffen, die sich durch normale Destillation aufgrund ihreshohen Siedepunkts thermisch zersetzen würden. Der Siedepunkt der nicht mischbarenStoffe ist erreicht, wenn
= ∗1 + ∗2 = außen (6.140)
Er liegt somit unter den Siedepunkten beider reiner Stoffe!
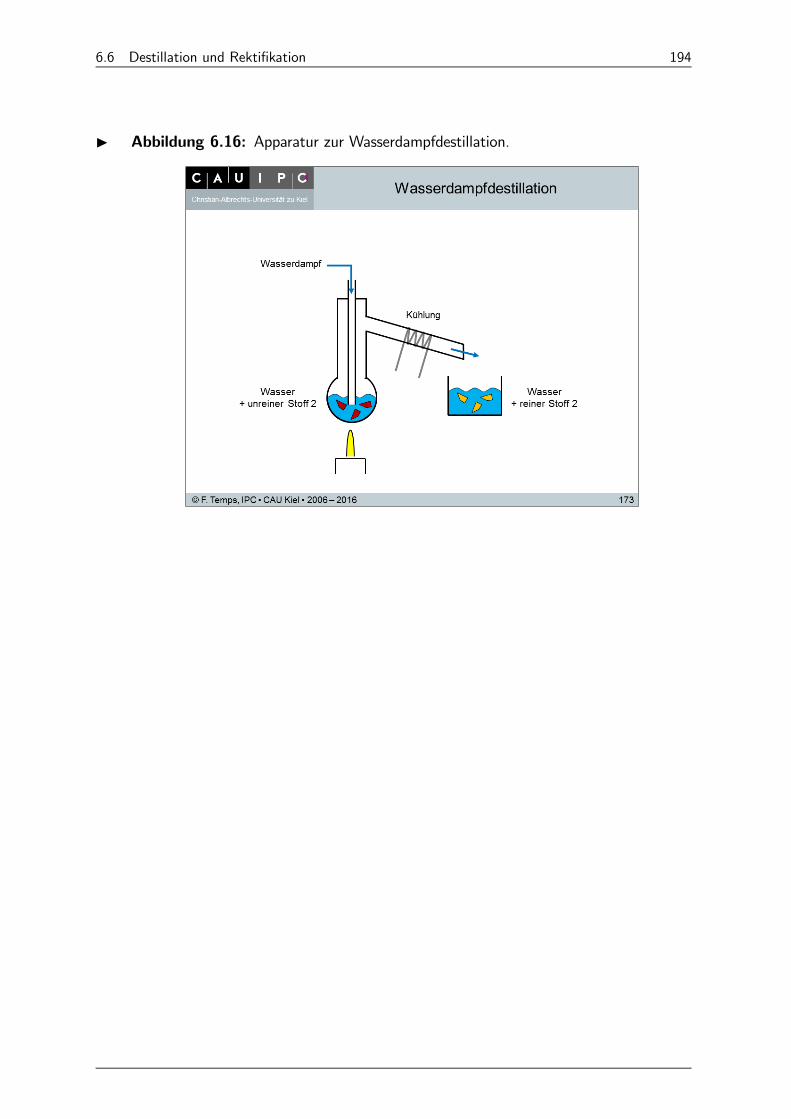
6.6 Destillation und Rektifikation 194
I Abbildung 6.16: Apparatur zur Wasserdampfdestillation.
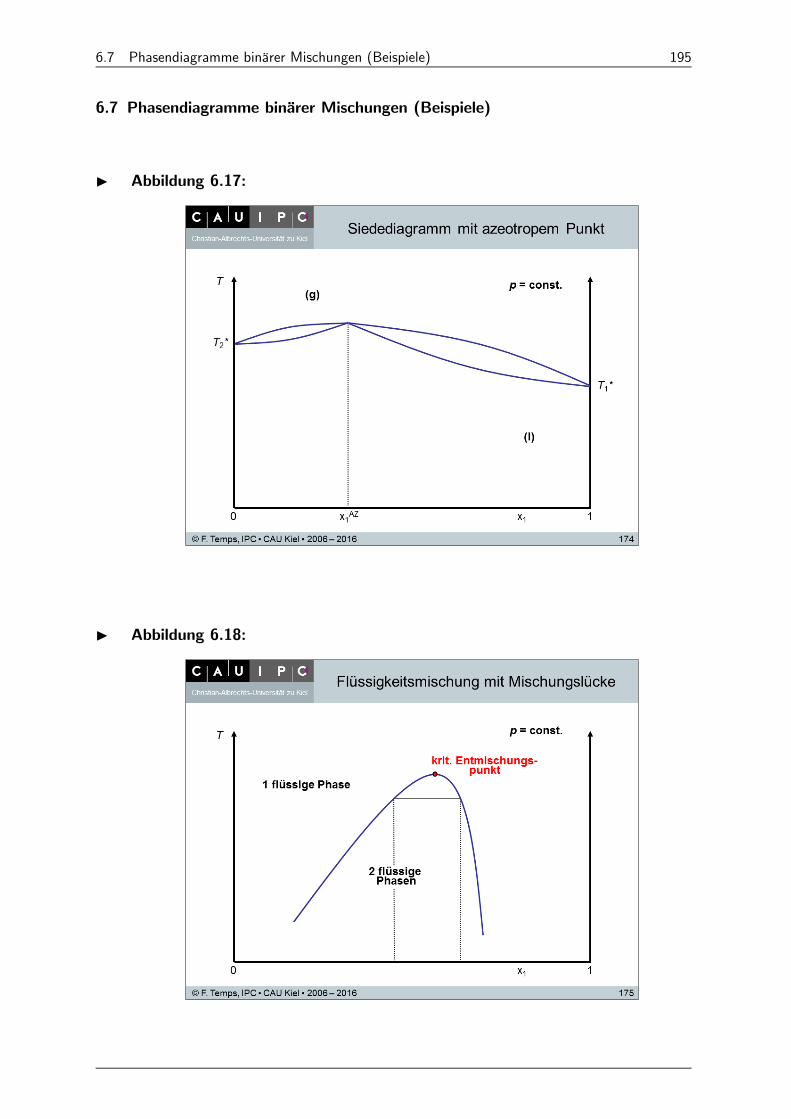
6.7 Phasendiagramme binärer Mischungen (Beispiele) 195
6.7 Phasendiagramme binärer Mischungen (Beispiele)
I Abbildung 6.17:
I Abbildung 6.18:

6.7 Phasendiagramme binärer Mischungen (Beispiele) 196
I Abbildung 6.19:

6.8 Die Gibbs’sche Phasenregel 197
6.8 Die Gibbs’sche Phasenregel
= − + 2 (6.141)
mit
: Zahl der Freiheitsgrade (6.142)
: Zahl der unabhängigen Komponenten (6.143)
: Zahl der Phasen (6.144)
⇒ Erläuterung an den behandelten Phasendiagrammen.
I Herleitung der Gibbs’schen Phasenregel:
(1) Zahl der Unbekannten:
, , ,. . . , Unbekannte, , ,. . . , Unbekannte
1 , 2 ,
3 , . . . ,
− 1 Unbekannte)
1 ,
2 ,
3 , . . . ,
− 1 Unbekannte)
. . .
1 , 2 ,
3 , . . . ,
− 1 Unbekannte)
Summe: 2 + × ( − 1) Unbekannte) ( − 1) Unbekannte daP = 1
(6.145)
(2) Zahl der Gleichungen:
= = = = − 1 Gleichungen = = = = − 1 Gleichungen1 =
1 =
1 = = 1 − 1 Gleichungen
2 = 2 =
2 = = 2 − 1 Gleichungen
= =
= = − 1 Gleichungen
2 ( − 1) + ( − 1) Gleichungen
(6.146)
(3) Zahl der Freiheitsgrade:
= (2 + ( − 1))− (2 ( − 1) + ( − 1)) (6.147)
= (2 + − )− (2 − 2 + −) (6.148)
= − + 2 (6.149)
(4) Bei Vorhandensein von Reaktionsgleichgewichten zwischen den Phasen und von zusätzlichen Bedingungen (z.B. fest vorgegebenen Parametern):
a) ⇒ zusätzliche Gleichungen der ArtP
= 0
b) ⇒ zusätzliche Gleichungen
⇒ Anzahl der unabhängigen Komponenten:
= −− (6.150)

6.9 Berechnung von Reaktionsgleichgewichten in Lösung 198
6.9 Berechnung von Reaktionsgleichgewichten in Lösung
6.9.1 Löslichkeitsprodukt eines schwerlöslichen Salzes in Wasser
I Beispiel 6.1:AgI À Ag+ + I
− (6.151)
Gleichgewichtsbedingung: X = 0 (6.152)
y∗Ag+
+ lnAg+
ª + ∗I− + ln
I−
ª − ∗AgI = 0 (6.153)
yAg+
ª × I−
ª = expµ−∗Ag+
+ ∗I− − ∗AgI
¶= exp
µ−∆
∗
¶= (6.154)
Der Wert for ƻ und damit folgt aus den Werten der Standardbildungsenthalpen
und -entropien (siehe Tabellen). ¤I Beispiel 6.2:
HgS À Hg2+ + S2− (6.155)
Hg2+
ªS2−
ª = exp
µ−∗Hg2+
+ ∗S2− − ∗HgS
¶= exp
µ−∆
∗
¶= (6.156)
Mit∆
∗ = ∆∗ − ∆
∗
∆∗ = ∆
∗Hg2+
+∆∗S2−−∆
∗HgS
(6.157)
y∆
∗298K = (+1711 + 331− (−536)) kJmol = 2578 kJmol (6.158)
∆∗ = ∗
Hg2++ ∗
S2−− ∗HgS (6.159)
y∆
∗298K = (−322− 146− 883) JKmol = −1351 JKmol (6.160)
y∆
∗298 = 2578 kJmol− 298K (−1351 JKmol) (6.161)
= 2981 kJmol (6.162)
y298K = exp
µ−∆
∗
¶= 56× 10−53 (6.163)
yHg2+ = S2− = 7483 3× 10−27
mol
kgH2O 1
Molekül
kgH2O(6.164)
Für
S2− = 1mol
kgH2O(6.165)
yHg2+ = 56× 10−53
mol
kgH2O(6.166)
¤

6.9 Berechnung von Reaktionsgleichgewichten in Lösung 199
6.9.2 Autoprotolyse und Ionenprodukt von H2O
H2O À H+ +OH− (6.167)
Gleichgewichtsbedingung: X = 0 (6.168)
y∗H++ ln
H+
ª + ∗OH− + ln
OH−
ª − ∗H2O = 0 (6.169)
y
H+
ª × OH−
ª = exp
µ−
∗H++ ∗
OH− − ∗H2O
¶= exp
µ−∆
∗
¶= (6.170)
Der Wert forƻ und damit folgt aus den Werten der Standardbildungsenthalpen
und -entropien (siehe Tabellen). Für = 29815K und = 1bar finden wir
∗H+()
= 0 + 298K× 0 (6.171)
∗OH−() = −230 kJmol−1 − 29815K×
¡−1075 Jmol−1K−1¢ = −22679 kJmol−1(6.172)
∗H2O() = −28583 kJmol−1 − 29815K× 6991 Jmol−1K−1 = −30667 kJmol−1(6.173)
y
∆∗ = (0− 22679 + 30667) kJmol−1 (6.174)
= 7988 kJmol−1 (6.175)
y
= exp
µ−7988 kJmol
−1
× 298K¶
(6.176)
= exp (−32239) (6.177)
= 9972 0× 10−15 ≈ 10−14 (6.178)
6.9.3 Dissoziation einer schwachen Säure
HAc À H+ + Ac− (6.179)
Gleichgewichtsbedingung: X = 0 (6.180)
y
∗H++ ln
H+
ª + ∗Ac− + ln
Ac−
ª − ∗HAc − lnHAc
ª = 0 (6.181)
y
(H+ª)× (Ac−
ª)(HAcª)
= exp
µ−
∗H++ ∗
OH− − ∗HAc
¶= exp
µ−∆
∗
¶=
(6.182)

6.9 Berechnung von Reaktionsgleichgewichten in Lösung 200
I Anmerkung: Wenn höhere Konzentrationen behandelt werden, bzw. für reale Lösun-gen höhere Genauigkeit gewünscht wird, sollte anstatt mit den Molalitäten mit denentsprechenden Aktivitäten gerechnet werden.
6.9.4 pH-Wertberechnungen für schwache Säuren und Basen
⇒ Siehe diverse Übungsaufgaben
6.9.5 Puffer
⇒ Siehe diverse Übungsaufgaben

6.9 Berechnung von Reaktionsgleichgewichten in Lösung 201
7. Gleichgewichtselektrochemie
Bis hierher haben wir normale chemische Reaktionen in homogener Phase oder hetero-genen Phasen betrachtet und gesehen, wie die Gleichgewichtskonstanten mit thermody-namischen Größen zusammenhängen und wie man thermodynamische Größen bestim-men kann (z.B. Kalorimetrie, van’t Hoff-Auftragung, Gleichgewichtskonstante, . . . ).
In diesem Kapitel wollen wir elektrochemische Reaktionen — Oxida-tions/Reduktionsreaktionen — betrachten, bei denen die Teilreaktionen an zweigetrennten Elektroden stattfinden, die elektrisch leitend verbunden werden. Zwischenden beiden Elektroden misst man eine Spannung, die elektromotorische Kraft (EMK).
I Frage: Wie hängt die EMK im elektrochemischen Gleichgewicht mit thermodynamis-chen Größen zusammen?
I Antwort: ⇒ Gleichgewichtselektrochemie.
I Bedeutung der Gleichgewichtselektrochemie:
• Energie“quellen” und Energiespeicher (Batterien, Brennstoffzellen, . . . )• Elektrochemische Sensoren (Gassensoren, Biochips, . . . )• Herstellung von Metallen (z.B. Aluminium) und Materialien, Stoffreinigung und-trennung, Materialbeschichtung, . . .
• Bestimmung präziser thermodynamischer Daten aus EMK-Messungen.
I Modul chem5016: Elektrochemie Wahlpflichtmodul im 4. FS (oder 6. FS) mit 4LP
• Vorlesung (2 SWS): Von Batterien zu Brennstoffzellen,• Übungen (1 SWS),• Dozentin: Prof. Dr. Melanie Schnell.

7.1 Beispiele und Konventionen 202
7.1 Beispiele und Konventionen
7.1.1 Beispiele für elektrochemische Zellen (elektrochemische Ketten)
a) Daniell-Element ohne Salzbrücke
I Abbildung 7.1: Daniell-Element ohne Salzbrücke.
I Kurzschreibweise:Zn | Zn2+ | Cu2+ | Cu (7.1)
I Elektrodenreaktionen: Die beiden Halbzellen sind getrennt durch eine Glasfritte,durch die die Ionen hindurchtreten können, damit der Stromkreis geschlossen ist.
• links:Zn ()→ Zn2+ () + 2− (7.2)
• rechts:Cu2+ () + 2− → Cu () (7.3)
I Gesamtreaktion:
Zn () + Cu2+ ()→ Zn2+ () + Cu () (7.4)

7.1 Beispiele und Konventionen 203
I Elektromotorische Kraft: EMK = Zellpotential (Spannungsdifferenz zwischen denElektroden) im elektrochemischen Gleichgewicht (bei stromloser Messung!)
b) Daniell-Element mit Salzbrücke
Um Fehler durch Diffusion der Salzlösungen ineinander zu vermeiden, verwendet maneine Salzbrücke mit einer Elektrolytlösung mit hoher Konzentration (die damit denStromfluss durch die Phasengrenzfläche dominiert). Kation und Anion des Elektrolytenin der Salzbrücke sollen gleichermaßen zum Stromfluss beitragen, d.h. gleiche Über-führungszahlen haben:
− = + (7.5)
Gut geeignet sind z.B. KNO3 oder KCl.
I Abbildung 7.2: Daniell-Element mit Salzbrücke.
I Kurzschreibweise:
Zn () | Zn2+ () | KNO3 (4M) | Cu2+ () | Cu () (7.6)
Oft lässt man die Salzbrücke auch weg und deutet sie nur durch einen Doppelstrich an:
Zn () | Zn2+ () || Cu2+ () | Cu () (7.7)
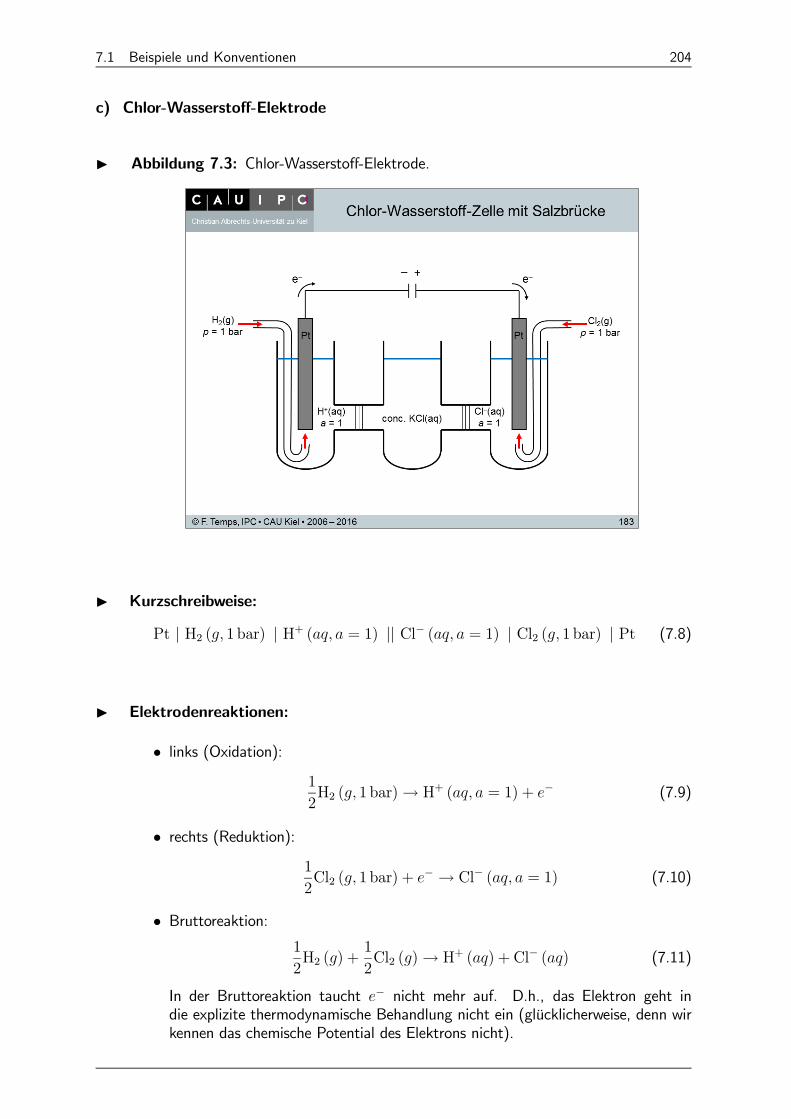
7.1 Beispiele und Konventionen 204
c) Chlor-Wasserstoff-Elektrode
I Abbildung 7.3: Chlor-Wasserstoff-Elektrode.
I Kurzschreibweise:
Pt | H2 ( 1 bar) | H+ ( = 1) || Cl− ( = 1) | Cl2 ( 1 bar) | Pt (7.8)
I Elektrodenreaktionen:
• links (Oxidation):1
2H2 ( 1 bar)→ H+ ( = 1) + − (7.9)
• rechts (Reduktion):1
2Cl2 ( 1 bar) + − → Cl− ( = 1) (7.10)
• Bruttoreaktion:1
2H2 () +
1
2Cl2 ()→ H+ () + Cl− () (7.11)
In der Bruttoreaktion taucht − nicht mehr auf. D.h., das Elektron geht indie explizite thermodynamische Behandlung nicht ein (glücklicherweise, denn wirkennen das chemische Potential des Elektrons nicht).

7.1 Beispiele und Konventionen 205
7.1.2 IUPAC-Konventionen (1953)
(1) = − (7.12)
(2) Wahl von rechts/links so, dass 0 (außer im Fall der Normalwasserstoffelek-trode)
a) Elektrodenreaktion links: Oxidation, z.B.
Zn→ Zn2+ + 2− (7.13)
b) Elektrodenreaktion rechts: Reduktion (ReRe-Regel), z.B.
Cu2+ + 2− → Cu (7.14)
(3) Allgemeine Kurzschreibweise für eine elektrochemische Kette (A | A+ = An-odenpaar, K+ | K = Kathodenpaar):
a) ohne Salzbrücke:
A() | A+ ( = ) | K+ ( = ) | K() (7.15)
b) mit Salzbrücke (z.B. KCl):
A | A+ | KCl | K+ | K (7.16)
oderA | A+ || K+ | K (7.17)
(4) Normalwasserstoffelektrode = Ausnahme von der Rechts/Links-Regel!
Die Normalwasserstoffelektrode wird immer links geschrieben (Reaktionsrichtung12H2 ( 1 bar)→ H+ ( = 1) + −):
Pt | H2 ( 1 bar) | H+ ( = 1) | | (7.18)
Dadurch erhalten “unedlere” Elektroden eine negative EMK und “edlere” Elek-troden eine positive EMK (siehe Spannungsreihe).
(5) Anode/Kathode:
a) Die Elektrode, an der die Oxidationsreaktion abläuft, wird immer als Anodebezeichnet.
b) Die Elektrode, an der die Reduktionsreaktion abläuft, wird immer als Kath-ode bezeichnet.

7.1 Beispiele und Konventionen 206
7.1.3 Durchführung von EMK-Messungen
I Messung muss reversibel, immer im elektrochem. Glgew. und stromlos erfol-gen:
• reversible Bedingungen,• im elektrochemischen Gleichgewicht,• stromlos.
I Außerdem zu beachten:
• Vermeidung von Spannungsabfall duch Innenwiderstand der Zelle (d.h. stromlos),• Vermeidung von Polarisationseffekten an den Elektroden,• Vermeidung von Überspannungen,• Es muss Glgew.-Thermodynamik gelten.
Bei nicht-reversiblen Zellen ist die Glgew.-Thermodynamik nicht mehr anwendbar.
I Praktische Realisierung:
(1) Hochohmiges Spannungsmessgerät,
(2) Poggendorf’sche Kompensationsschaltung.
I Abbildung 7.4: Poggendorf’sche Kompensationsschaltung zur stromlosen EMK-Messung.
Wenn = 0y =
1
1 +20 = (7.19)

7.1 Beispiele und Konventionen 207
7.1.4 Reversibel arbeitende elektrochemische Zellen
Reversible elektrochemische Zellen zeichnen sich durch folgende Eigenschaften aus:
(1)| −|→ 0 (7.20)
d.h. → .
(2) Umkehrung des Vorzeichens gemäß
| −|→ − | −| (7.21)
durch Änderung der Gegenspannung bewirkt schon bei → 0 eineUmkehrung der Zellreaktion.
(3) Beispiel Daniell-Element:
©Zn () + Cu2+
ª
À
©Zn2+ + Cu ()
ª(7.22)
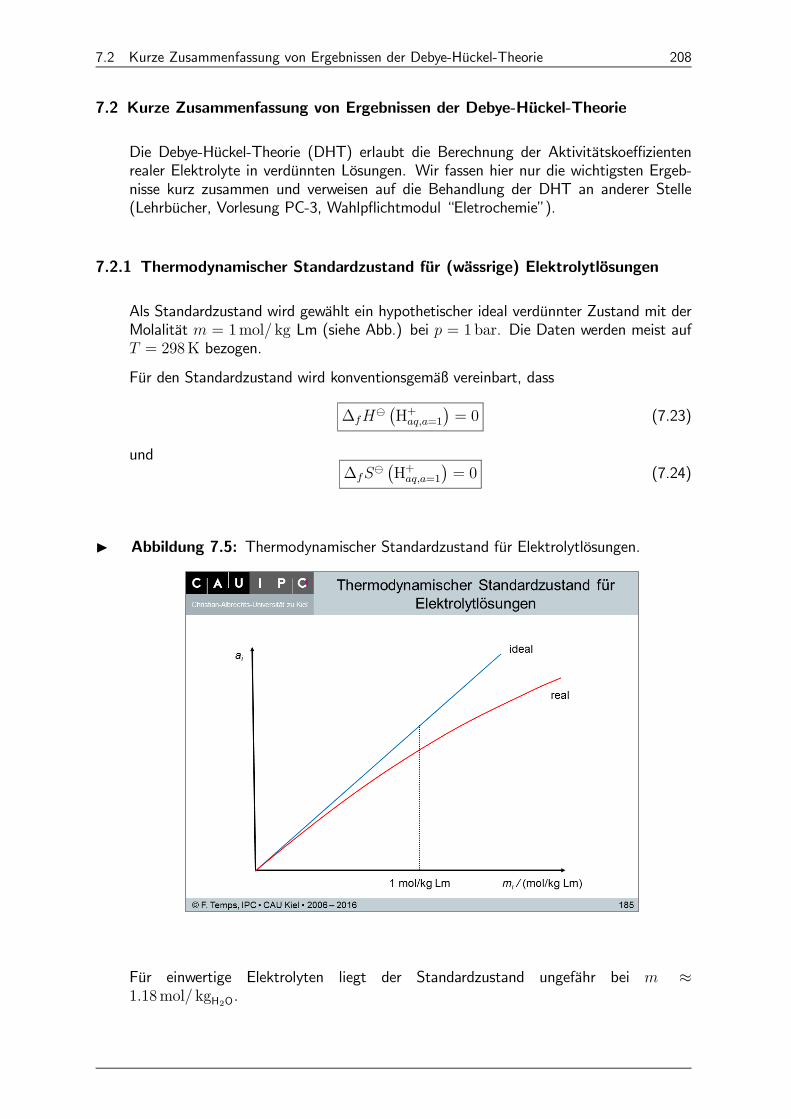
7.2 Kurze Zusammenfassung von Ergebnissen der Debye-Hückel-Theorie 208
7.2 Kurze Zusammenfassung von Ergebnissen der Debye-Hückel-Theorie
Die Debye-Hückel-Theorie (DHT) erlaubt die Berechnung der Aktivitätskoeffizientenrealer Elektrolyte in verdünnten Lösungen. Wir fassen hier nur die wichtigsten Ergeb-nisse kurz zusammen und verweisen auf die Behandlung der DHT an anderer Stelle(Lehrbücher, Vorlesung PC-3, Wahlpflichtmodul “Eletrochemie”).
7.2.1 Thermodynamischer Standardzustand für (wässrige) Elektrolytlösungen
Als Standardzustand wird gewählt ein hypothetischer ideal verdünnter Zustand mit derMolalität = 1mol kg Lm (siehe Abb.) bei = 1bar. Die Daten werden meist auf = 298K bezogen.
Für den Standardzustand wird konventionsgemäß vereinbart, dass
∆ª ¡H+=1¢ = 0 (7.23)
und∆
ª ¡H+=1¢ = 0 (7.24)
I Abbildung 7.5: Thermodynamischer Standardzustand für Elektrolytlösungen.
Für einwertige Elektrolyten liegt der Standardzustand ungefähr bei ≈118mol kgH2O.

7.2 Kurze Zusammenfassung von Ergebnissen der Debye-Hückel-Theorie 209
7.2.2 Aktivitäten und Aktivitätskoeffizienten
In Elektrolytlösungen treten aufgrund der Coulomb-WW zwischen den Ionen schonbei sehr niedrigen Konzentrationen ( 10−4mol kg Lm) merkliche Abweichungenvom Verhalten idealer Lösungen auf. Diese können näherungsweise (bei nicht zu ho-hen Konzentrationen) im Rahmen der Debye-Hückel-Theorie beschrieben werden (sieheKapitel über Transportprozesse in der Vorlesung Reaktionskinetik bzw. Vorlesung Elek-trochemie).
I Debye-Hückel-Theorie: Die DHT liefert für die gemäß29
= ± (7.25)
mit± = (
++ ×
−− )
1(++−) (7.26)
definierten mittleren Aktivitätskoeffizienten das Debye-Hückel’sche Wurzelgesetz
lg ± = − |+−| ×√
1 +√
(7.27)
mit3031
=3 (2)
12
8 × 2303 (0 )32und =
µ2
2
0
¶12(7.28)
Für sehr verdünnte Lösungen kann √ im Nenner vernachlässigt werden:
lg ± ≈ − |+−|√ (7.29)
ist die Ionenstärke:
=1
2
X2 ≈ 1
2
X2 (7.30)
I Zahlenwerte für H2O als Lm: Für H2O, = 300K:
= 7854 (7.31)
= 05091
µkg Lm
mol
¶12(7.32)
lg ± = −05091 |+−|√ (7.33)
Allgemein (für andere Temperaturen und andere Lösungsmittel):
lg ± ∝r
3 3(7.34)
29 Wir verzichten in diesem Abschnitt aus Gründen der Übersichtlichkeit der Gleichungen auf die ex-
plizite Berücksichtigung der korrekten Dimension und schreiben stattª und statt ª.
30 ist die Elementarladung, Lm ist die Dichte des Lösungsmittels.31 ist der effektive Ionenradius einschliesslich der 1. Solvathülle ( kristallographischer Radius).

7.3 Die elektrochemische Gleichgewichtsbedingung 210
7.3 Die elektrochemische Gleichgewichtsbedingung
7.3.1 Elektrische und chemische Arbeit
I Elektrische Arbeit: Die elektrische Arbeit wird berechnet nach32
= × × = × (7.35)
Beim Umsatz von || mol − in einer elektrochemischen Zelle mit der EMK =
− wird somit die Arbeit
= − || ( − ) (7.36)
y = − || (7.37)
mit =
geleistet.
= 1602 2× 10−19C× 6022 1× 1023mol−1 = 96 486Cmol−1 (7.38)
• Das Vorzeichen folgt aus der thermodynamischen Vorzeichenkonvention.• Die elektrochemische Zelle ist nicht im elektrischen Gleichgewicht, da 6= 0.
I Vergleich mit chemischer Arbeit: Die chemische Arbeit ergibt sich aus der freienEnthalpieänderung pro Formelumsatz:
chemrev = ƻ =
X
ª (7.39)
I Die elektrochemische Zelle ist nicht im chemischen Gleichgewicht: Die elektro-chemische Zelle ist nicht im chemischen Gleichgewicht, da ∆
ª 6= 0. Im chemischenGleichgewicht wäre µ
∆
¶
= 0 yX
= 0 (7.40)
32 = Spannung, = × = Ladung.

7.3 Die elektrochemische Gleichgewichtsbedingung 211
7.3.2 Elektrochemische Gleichgewichtsbedingung
Im elektrochemischen Gleichgewicht müssen wir bei der Untersuchung von die elek-trische Arbeit = − mit der elektrischen Spannung berücksichtigen:33
= − () (7.41)
= + ( )− () (7.42)
= − − + + − − (7.43)
= − − (7.44)
Bei = y = − (7.45)
oder nach Integration für || mol Ladung ( = ||):∆ = − || (7.46)
I Elektrochemische Gleichgewichtsbedingung:
∆ = − || (7.47)
Hierbei ist || die pro Formelumsatz umgesetzte Ladungszahl (|| = ++ = −−).
I Beispiel Daniell-Element:
Zn () + CuSO4 ( = 1)→ ZnSO4 ( = 1) + Cu () (7.48)
EMK-Messung liefert = 1100V (7.49)
y∆ = − || (7.50)
y
∆ = − || = −2× 96485Cmol−1 × 1100V = −2123 kJmol
(7.51)
I Aber die gemessene EMK ist i.a. abhängig von den Cu2+ und Zn2+ Konzen-trationen: ⇒ Nernst’sche Gleichung!
33 Wir verwenden den Buchstaben für die EMK (= Spannung) anstelle des üblichen , da wir in
der Thermodynamik schon für die Innere Energie verbraucht haben.

7.4 Die Nernst’sche Gleichung 212
7.4 Die Nernst’sche Gleichung
7.4.1 Allgemeine Herleitung der Nernst’schen Gleichung
I Anwendung der elektrochemischen Gleichgewichtsbedingung: Aus der elektro-chemischen Gleichgewichtsbedingung
∆ = − || (7.52)
folgt mit ∆ =P
34X
=X
∗ +
X ln
ª= − || (7.53)
y = −
P
∗
|| −
|| lnY
³ ª
´(7.54)
y
= −∆∗
|| −
|| lnY
³ ª
´(7.55)
y
I Nernst’sche Gleichung:
= 0 −
|| lnY
³ ª
´(7.56)
mit
0 = −∆∗
|| (7.57)
7.4.2 Daniell-Element
I Elektrodenreaktion:
Zn () + Cu2+ ( = 1M)→ Zn2+ ( = 1M) + Cu () (7.58)
yZn () + CuSO4 ( = 1M)→ ZnSO4 ( = 1M) + Cu () (7.59)
34 Analog zur Behandlung von Gasen ( = ª + ln (ª)) beziehen wir die dimensionsbehafteten
Aktivitäten auf die jeweiligen Referenzzustände ª.

7.4 Die Nernst’sche Gleichung 213
I Thermodynamische Daten:
∆∗298K = ∆
∗298K (Cu) +∆
∗298K
¡Zn2+
¢−∆∗298K (Zn)−∆
∗298K
¡Cu2+
¢(7.60)
= (0− 15389− 0− 6477) kJmol−1 (7.61)
= −21866 kJmol−1 (7.62)
∆∗298K = ∗298K (Cu) + ∗298K
¡Zn2+
¢− ∗298K (Zn)− ∗298K¡Cu2+
¢(7.63)
= (3315− 1121− 4163− (−996)) JK−1mol−1 (7.64)
= −2098 JK−1mol−1 (7.65)
y
∆∗298 = −21866 kJmol−1 − 298K
¡−2098 JK−1mol−1¢ (7.66)
= −21241 kJmol−1 (7.67)
y
I EMK:
0 = −∆∗298
|| =21241 kJmol−1
2× 96485Cmol−1 = +1100 7V (7.68)
I Nernst’sche Gleichung mit Aktivitäten:
= −∆∗
|| −
|| ln4
S4(7.69)
mit, z.B. für ZnSO4:4 =
++ ×
−− = ± (7.70)
mit = 1 und± = ±4 (7.71)
7.4.3 Chlor-Wasserstoff-Zelle
I Elektrodenreaktion:
1
2H2 ( 2) +
1
2Cl2 ( 2)→ H+ ( +) + Cl− ( −) (7.72)

7.4 Die Nernst’sche Gleichung 214
I Anwendung der ellektrochemischen Gleihgewichtsbedingung:
− || = ¡H+¢+
¡Cl−
¢− 12 (H2)− 1
2 (Cl2) (7.73)
= ∗++ ln
+
ª+ ∗
−+ ln
−
ª(7.74)
−12ª2
− ln
µ2
ª
¶12− 122 − ln
µ2ª
¶12(7.75)
= ∆∗ + ln
(+ª) (−ª)
(2ª)12 (2ª)
(7.76)
y
= −∆∗
|| −
|| ln(+ª) (−ª)
(2ª)12 (2ª)
(7.77)
bzw.
= 0(2|+|−|2) −
|| ln(+ª) (−ª)
(2ª)12 (2ª)
(7.78)
I Berechnung von E0 @ T = 298K:
∆ª = ∆
ª ¡H+¢+∆ª ¡Cl−¢− 12∆
ª (H2)− 12∆
ª (Cl2)
(7.79)
= (0− 16716− 0− 0) kJmol (7.80)
= 16716 kJmol (7.81)
∆ª = ª
¡H+¢+ ª
¡Cl−
¢− 12ª (H2)− 1
2ª (Cl2) (7.82)
=
µ0 + 5648− 1
2× 13058− 1
2× 22297
¶JmolK (7.83)
= −12030 JmolK (7.84)
∆ª = ∆
ª − ∆ª = −13131 kJmol (7.85)
y0 = −∆
ª
|| = 136V (7.86)
I Normalelektroden: Für = 1mol kg, 2 = 1bar, 2 = 1bar folgt
= 0 −
|| ln 1 = 0 (7.87)

7.5 Standard-Elektrodenpotentiale 215
7.5 Standard-Elektrodenpotentiale
I Einführung des elektrochemischen Potentials eμ: Mit ∆ =P
und =
− können wir auch schreibenX = − || ( − ) (7.88)
oder X( + ) = 0 (7.89)
bzw. Pe = 0 (7.90)
mit dem elektrochemischen Potentiale = + || (7.91)
ist das elektrische Potential im Inneren der jeweiligen Phase (Galvani-Potential).Das Galvani-Potential ist für sich nicht messbar. Messbar ist die Potentialdifferenz zwischen zwei Phasen.
Gl. 7.90 ist eine andere Schreibweise für die elektrochemische Gleichgewichtsbedingung.
I Elektrodenpotentiale werden gemessen gegen die Normal-Wasserstoff-Elektrode. Dabei steht die Normalwasserstoffelektrode immer links (Oxidation), unddie Elektrode, für die das Elektrodenpotential zu estimmen ist, immer rechts (Reduk-tion):
Pt | H2 ( 1 bar) | H+ ( = 1) || Cu2+ ( 2+) | Cu () | (7.92)
I Elektrodenreaktionen und Gesamtreaktion:
• links:1
2H2 ( 1 bar)→ H+ ( = 1) + − (7.93)
mit = 0000 (7.94)
• rechts:(1)
Cu2+ + 2− → Cu (7.95)
mit
= −∗ − ∗
2+
|| +
|| ln2+
ª= 0|2+ +
|| ln2+
ª(7.96)
(2) bzw. analogZn2+ + 2
− → Zn (7.97)
mit
= −∗ − ∗
2+
|| +
|| ln2+
ª= 0|2+ +
|| ln2+
ª(7.98)

7.5 Standard-Elektrodenpotentiale 216
I Nernst’sche Gleichung für Daniell-Element:
= − (7.99)
= 0|2+ +
|| ln2+
ª−µ0|2+ +
|| ln2+
ª
¶(7.100)
= 0|2+ −0|2+ +
|| ln2+
2+(7.101)
I Standard-Elektrodenpotentiale: Für = 1 folgen die Standard-Elektrodenpotentiale, die wir in der Spannungsreihe anordnen können:
I Tabelle 7.1: Beispiele für Standard-Elektrodenpotentiale (ausführliche Tabellen siehez.B. Atkins).
Reaktion Kurzschreibweise 0 V
Li+ + − À Li Li+|Li −3045Zn2+ + 2 − À Zn Zn2+|Zn −0763Cd2+ + 2 − À Cd Cd2+|Cd −0403H+ + − À 1
2H2 H+|H2|Pt 0000
Cu2+ + 2 − À Cu Cu2+|Cu +0337
Hg+ + − À Hg Hg+|Hg +0799
Au3+ + 3 − À Au Au3+|Au +1498

7.6 Bestimmung thermodynamischer Daten aus EMK-Messungen 217
7.6 Bestimmung thermodynamischer Daten aus EMK-Messungen
EMK-Messungen sind wichtige Quellen für thermodynamische Daten, z.B.
(1)∆ = − || und ∆ª = − ||0 (7.102)
(2)
−∆ =
µ∆
¶
y ∆ = ||µ
¶
(7.103)
(3)
∆ = ∆+ ∆ = ||"
µ
¶
−
#(7.104)
(4)
∆ =
µ∆
¶
yµ
¶
= − ∆
|| (7.105)

7.7 Zusammenstellung elektrochemischer Zellen 218
7.7 Zusammenstellung elektrochemischer Zellen
a) Zusammenstellung diverser elektrochemischer Halbzellentypen
• Normalwasserstoffelektrode (Referenzelektrode):1
2H2 ( = 1bar)→ H+ ( = 1) + − : 0 = 0000V (7.106)
Anwendung: Messung von£H+¤
• Metall-Metallion-Elektroden (wie z.B. im Daniell-Element):Zn2+ () + 2 − À Zn () : 0 = −076V (7.107)
Cu2+ () + 2 − À Cu () : 0 = +034V (7.108)
• Metall-Metalloxid-Elektrode:Sb2O3 () + 3 H2O+ 6
− À 2 Sb () + 6 OH− () (7.109)
Anwendung: Messung von von£OH−
¤• Elektroden zweiter Art:
AgCl () + − → Ag () + Cl− () : 0 = +022V (7.110)
Teilreaktionen:
1. Ag+ () + − À Ag ()
2. AgCl ()À Ag+ () + Cl− ()P: AgCl () + − À Ag () + Cl− ()
(7.111)
Hg2Cl2 () + 2 − → 2 Hg () + 2 Cl− () : 0 = +027V (7.112)
Anwendung: konstantes Elektrodenpotential, unabhängig von£M+ ()
¤bei Ver-
wendung hoher£Cl−¤-Konzentrationen
• Redox-Elektroden:Fe3+ () + − À Fe2+ () : 0 = +077V (7.113)
MnO−4 ()+8 H+ ()+5 − ÀMn2+ ()+4 H2O : 0 = +151V (7.114)
Cr2O2−7 ()+14 H++6 − À 2 Cr3+ ()+7 H2O : 0 = +133V (7.115)
Potentielle Anwendung: Energiespeicherung, da Lösungen in großen Tanks ge-speichert werden können.

7.7 Zusammenstellung elektrochemischer Zellen 219
b) Zusammenstellung diverser elektrochemischer Zellen
• Membranelektroden:Na+ () + H+ ( )À H+ () + Na+ () (7.116)
Anwendung: z.B. Glaselektrode zur pH-Messung.
• Elektrolyt-Konzentrationszetten:Pt | H2
¡ ª
¢ | HCl ¡ª¢ | HCl () | H2 ¡ ª¢ | Pt (7.117)
Teilreaktionen:
li : 12H2¡ª¢→ H+
¡ª¢+ − (7.118)
re : H+ () + − → 12H2¡ª¢
(7.119)
Σ: H+ ()→ H+¡ª¢
(7.120)
y = −
ln
ª
=
ln
ª=
2302pH (7.121)
Anwendung: Messung von + (d.h. von pH)
• Gas-Konzentrationszellen:Pt | H2 ( 0) | HCl () | H2 ( 00) | Pt (7.122)
Teilreaktionen:
li: 12H2 (
0)→ H+ () + − (7.123)
re: H+ () + − → 12H2 (
00) (7.124)
Σ : 12H2 (
0)→ 12H2 (
00) (7.125)
y = −
2ln
00
0(7.126)
• Elektroden-Konzentrationszellen:Hg () Na (0) | Na2SO4 () | Na (00) Hg () (7.127)
Gesamtreaktion: Na (0)→ Na (00) (7.128)
y = −
ln
00
0(7.129)
• Weston-Normalelement:Hg | Cd (Hg) | CdSO4 · 83 H2O | CdSO4 ( gesattigt) | HgSO4 | Hg (7.130)Anwendung: Liefert konstante EMK, wenig -abhängig.

7.7 Zusammenstellung elektrochemischer Zellen 220
• Blei-Akku:Pb | PbSO4 | H2SO4 () | PbSO4 | PbO2 (7.131)
Teilreaktionen:
li: Pb + SO2−4 → PbSO4 + 2 − : 0 = −0356V (7.132)
re: PbO2 + 4 H+ + SO2−4 + 2 − → PbSO4 + 2 H2O : 0 = +1685V
(7.133)
Σ : Pb + PbO2 + 2 H2SO4 → 2 Pb2SO4 + 2 H2O : 0 = 2041V
(7.134)
• Ni/Cd-Zelle:Cd | Cd (OH)2 | KOH() | Ni (OH)2 | NiOOH (7.135)
Teilreaktionen:
li: Cd + 2 OH− → Cd (OH)2 + 2 − : 0 = −0809V (7.136)
re: 2 NiOOH+ 2 H2O+ 2 − → 2 Ni (OH)2 + 2 OH
− : 0 = +0450V
(7.137)
Σ : Cd + 2 NiOOH+ 2 H2O→ 2 Ni (OH)2 +Cd (OH)2 : 0 = 1259V
(7.138)
• Ni/Metallhydrid-Zelle:LaNi5 | LaNi5H6 | KOH() | Ni (OH)2 | NiOOH (7.139)
Teilreaktionen:
li : NiH +OH− → Ni + H2O+ − : 0 = −0809V (7.140)
re : NiOOH+H2O+ − → Ni (OH)2 +OH− (7.141)
Σ : NiH + NiOOH→ Ni + Ni (OH)2 : 0 = 135V (7.142)
• Ni/Zn-Zelle:Zn | Zn (OH)2 | KOH() | Ni (OH)2 | NiOOH (7.143)
Teilreaktionen:
li : Zn + 2 OH− → Zn (OH)2 + 2 − : 0 = −125V (7.144)
re : 2 NiOOH+ 2 H2O+ 2 − → 2 Ni (OH)2 + 2 OH
− : 0 = +0450V
(7.145)
• Zn/Luft-Zelle:Zn | Zn (OH)2 | KOH | O2 (7.146)
Teilreaktionen:
li : Zn + 2 OH− → Zn (OH)2 + 2 − (7.147)
re : 12O2 +H2O+ 2
− → 2 OH−
Σ : Zn+12O2 +H2O→ Zn (OH)2 (7.148)

7.7 Zusammenstellung elektrochemischer Zellen 221
• Zn/MnO2-Zelle:Zn | Zn2+ | NH4Cl |MnOOH | MnO2 | C (7.149)
Teilreaktionen:
li : Zn→ Zn2+ + 2 − (7.150)
re : 2 MnO2 + 2 H2O+ 2 − → 2 MnOOH+ 2 OH−
(7.151)
Σ : 2 MnO2 + Zn+2 NH4Cl + H2O→ 2 MnOOH+ Zn (NH3)2Cl2
(7.152)
Anwendung: Standardbatterie, = 15V
• Na/S-Akku:li : 2 Na→ 2 Na+ + 2 − (7.153)
re : 5 S + 2 − → S2−5 (7.154)
Σ : 2 Na + 5 S→ Na2S5 : 0 = 2076V (7.155)
• Li-Ionenakku (auf Basis einer Li-C-Interkalationsverbindung):li : LiC6 → 6 C + Li+ + − (7.156)
re : CoO2 + Li+ + − → LiCoO2 (7.157)
Σ : LiC6 +CoO2 → 6 C + LiCoO2 (7.158)
• Brennstoffzellen (FCs):
— Brennstoffe: H2, CH4, CH3OH, HCOOH, C
— Oxidationsmittel: O2 (rein oder Luftsauerstoff),
— Elektrolyte: KOH(), Polymerelektrolytmembrane (PEMFC), Photospho-rsäure (PAFC), melted Alkalicarbonat (MCFC), solid Oxydkeramik (SOFC),
— Probleme: CO- oder S-haltige Verunreinigungen wirken als Eletrodengifte.

7.7 Zusammenstellung elektrochemischer Zellen 222
8. Einführung in die Statistische Thermodynamik
Ziel der statistischen Thermodynamik ist die Deutung der makroskopischen thermody-namischen Eigenschaften der Stoffe vom mikroskopischen, atomistischen Standpunkther. Wir wollen in der statistischen Thermodynamik die Gleichgewichtseigenschafteneines makroskopsichen Systems (Makrozustand) ausgehend von seinen atomistischenbzw. molekularen Eigenschaften her (Mikrozustände) verstehen. Auf dem Weg dort-hin werden wir sehen, dass es immer darum gehen wird, die Wahrscheinlichkeit einesMakrozustands zu bestimmen. Dazu dienen statistische Überlegungen (Kombinatorik).
Es zeigt sich, dass die uns interessierenden makroskopischen Prozesse als Ausgleichs-prozesse (im weitesten Sinne ) verstanden werden können, z.B.
(1) Materie-, Massenausgleich (Verteilung von Teilchen im Raum):
a) Irreversible Expansion eines Gases ins Vakuum,
b) Diffusion und Vermischung;
(2) Energieausgleich (Verteilung von Teilchen auf die möglichen Energiezustände):
a) Wärmeleitung,
b) chemische Reaktionen.
I Erfahrung: Makroskopische Prozesse verlaufen so, dass Gleichverteilungenangestreben werden.
8.1 Der Boltzmann-Ausdruck S = k lnW
8.1.1 Begriffe
I Mikrozustand: Unter einem Mikrozustand verstehen wir eine unterscheidbare,abzählbare Anordnung bzw. Konfiguration von Atomen auf Gitterplätzen bzw. Quantenauf Energiezuständen.
I Makrozustand: Ein Makrozustand setzt sich aus Mikrozuständen zusammen, wobeies nur auf die Zahl der Atome pro Gitterplatz bzw. Phasenraumzelle (siehe Abschnitt8.1.3) und die Zahl der Quanten insgesamt (gesamte Energie) ankommt.
I Postulat der statistischen Thermodynamik:
Jeder Mikrozustand ist gleich wahrscheinlich. (8.1)
8.1.2 Ausgleichsprozesse I: Diffusion in Kristallen

8.1 Der Boltzmann-Ausdruck = ln 223
a) Beispiel
I Diffusion der Atome aus zwei Kristallen bestehend aus unterschiedlichen Iso-topen (A = ¤, B = ¥) eines Elements, z.B.:
¥ ¥¥ ¥
¤ ¤¤ ¤ → ¥ ¤
¥ ¥¥ ¤¤ ¤ (8.2)
I Annahmen:
• Die Teilchen der Sorte und seien für sich jeweils ununterscheidbar,
• Bei der Diffusion gibt es keine energetischen Effekte.
I Berechnung der Zahl der Mikrozustände:
⇒ Wir untersuchen die Verteilung von 4 Teilchen auf 8 Kästen.
I Tabelle 8.1: Verteilung von 4 Teilchen auf 8 Kästen.
Beispiel für MikrozustandZahl der Mikrozustände =Zahl der Verteilungen = Zahlder Realisierungsmöglichkeiten
Wahrscheinlichkeit
¥ ¥¥ ¥
¤ ¤¤ ¤ Ω40 = 1 40 =
1
70= 1
¥ ¤¥ ¥
¥ ¤¤ ¤ Ω31 = 4× 4 = 16 31 =
16
70= 161
¥ ¤¤ ¥
¥ ¥¤ ¤
Ω22 =4× 32
× 4× 32
=
µ4× 32
¶2= 6× 6 = 36
22 =36
70= 361
¥ ¤¤ ¤
¥ ¥¤ ¥
Ω13 =4× 3× 22× 3 × 4× 3× 2
2× 3=
µ4× 3× 22× 3
¶2= 4× 4 = 16
13 =16
70= 161
¤ ¤¤ ¤
¥ ¥¥ ¥ Ω04 = 1 04 =
1
70= 1
PΩ = Ω = 70
P = 1

8.1 Der Boltzmann-Ausdruck = ln 224
I Zahl der Mikrozustände = Zahl der Realisierungsmöglichkeiten: Abzählenzeigte, dass wir für die Anordnung von 2×4 jeweils nicht unterscheidbaren Teilchen auf8 Plätzen 70 Realisierungsmöglichkeiten haben.
Berechnung mit Permutationsformel:
Ω = 70 =8!
4!× 4! (8.3)
y
Ω =Zahl der Vertauschungen insgesamt
Zahl der jeweils nicht unterscheidbaren Vertauschungen(8.4)
Unter Berücksichtigung des Grundpostulats der statistischen Thermodynamik (Gl. 8.1)ergibt sich für die Wahrscheinlichkeit einer der ( )-Kombinationen
=ΩPΩ
(8.5)
Die Wahrscheinlichkeit irgendeiner der (3 1)-Anordnungen ist somit 16× größer als dieWahrscheinlichkeit der (4 0)-Anordnung:
22 31 = 13 40 = 04 (8.6)
I Allgemeiner Ausdruck für die Zahl der Vertauschungsmöglichkeiten von N =(N1 +N2) Teilchen, wobei die N1 Teilchen A und N2 Teilchen B jeweilsununterscheidbar sind:
Ω =(1 +2)!
2!×2!=
!
2!×2!(8.7)
I Näherung für große Zahlen (N À 1): Für À 1 folgt mit der Stirling’schenNäherung35
Ω ≈µ1 +2
¶1+2
×µ
1
¶1
×µ
2
¶2
(8.11)
y
Ω ≈ (1 +2)1+2
11 ×2
2
(8.12)
35 Die Stirling’sche Näherung lautet (ln = 1)
ln ! ≈ ln − (8.8)
= ln − ln (8.9)
y
! ≈µ
¶(8.10)

8.1 Der Boltzmann-Ausdruck = ln 225
b) Boltzmann-Ausdruck S = k lnW
In der phänomenologischen Thermodynamik hatten wir Ausgleichsprozesse mit der Zu-nahme der Entropie erklärt. Das o.a. Beipiel zeigt offensichtlich, dass Ausgleichsprozessewie Diffusion in Kristallen deshalb irreversibel ablaufen, weil der Endzustand im Ver-gleich zum Ausgangszustand eine viel höhere Zahl von Realisierungsmöglichkeiten hat(im Beispiel 70 gegenüber 1). In der statistischen Thermodynamik wollen wir Aus-gleichsprozesse deshalb mit der Zunahme der thermodynamischen Wahrscheinlichkeiterklären.
I Irreversible Prozesse:Ω À Ω (8.13)
I Prinzip der maximalen Wahrscheinlichkeit:
• Die Triebkraft für Ausgleichsprozesse ist um so größer, je größer die Wahrschein-lichkeit des Zustands ist, der erreicht wird.
• Der wahrscheinlichste Makrozustand ist derjenige, für den es eine maximale Zahlvon Realisierungsmöglichkeiten gibt.
• Thermodynamische Wahrscheinlichkeit: = Ω (8.14)
I Boltzmann-Ausdruck für die Entropie:
= ln (8.15)
I Warum?
(1) Sowohl als auchΩ sind Zustandsfunktionen, die durch und die bestimmtsind.
(2) Beide ( und Ω) nehmen bei irreversiblen Prozessen zu.
(3) Wenn das System vergrößert wird, gilt
=1 ×2 (8.16)
aber = 1 + 2 (8.17)
Da aberln = ln1 + ln2 (8.18)
können wir mit Boltzmann ansetzen, dass
∝ ln (8.19)
(4) Die Proportionalitätskonstante zwischen und ln ist die nach Boltzmann be-nannte Konstante , sodass
= ln (8.20)

8.1 Der Boltzmann-Ausdruck = ln 226
c) Beispiel: Berechnung der Mischungsentropie
Berechnung der Mischungsentropie bei der Diffusion von zwei Atomsorten und :
∆ = Ende − Anfang (8.21)
wobeiAnfang = lnΩAnfang = ln 1 = 0 (8.22)
und
Ende = lnΩEnde = ln(1 +2)
1+2
11 ×2
2
(8.23)
y
∆ = ln(1 +2)
1+2
11 ×2
2
(8.24)
= (1 +2) ln (1 +2)−1 ln1 −2 ln2 (8.25)
= −µ1 ln
1
1 +2
+2 ln2
1 +2
¶(8.26)
Für 1+2 = Teilchen ergibt sich mit 1 (1 +2) = 1, 2 (1 +2) = 2der bekannte Ausdruck für die Mischungsentropie pro mol
∆ = − (1 ln1 + 2 ln2) = −P
ln (8.27)
8.1.3 Ausgleichsprozesse II: Gase
Gase erfordern eine etwas andere Behandlung als Kristalle, da jede “Zelle” im Raummit mehr als einem Teilchen besetzt werden kann.
a) Der Phasenraum
Für jedes Atom eines Gases benötigen wir zur Beschreibung seines Zustands 3 Ortsvari-able und 3 konjugierte Impulsvariable, d.h. pro Atom benötigen wir 6 Angaben.
• Die 3 Ortskoordinaten und 3 Impulskoordinaten eines Atoms spannen den 6-dimensionalen sogenannten Phasenraum auf.
• Der Quantenmechanik zufolge können die wegen der Heisenberg’schenUnschärferelation
∆×∆ ≈ (8.28)
nicht beliebig gewählt werden, sondern der Phasenraum erhält eine “Körnigkeit”.Im Ergebnis bedeutet die Unschärferelation, dass wir den Phasenraums pro Atomunterteilen können in Zellen vom Volumen
(1)
PR = 3 (8.29)
• Innerhalb einer Phasenraumzelle können die Teilchen nicht unterschieden werden,da sie quantenmechanisch betrachtet denselben Ort und denselben Impuls haben.

8.1 Der Boltzmann-Ausdruck = ln 227
I Abbildung 8.1: Verteilung von Gasatomen im Phasenraum.
Die Wahrscheinlichkeit für einen Makrozustand eines Gases können wir wie folgt berech-nen:
b) Wahrscheinlichkeit für Makrozustand eines Gases bezgl. Verteilung im Raum
Wie durch die QM vorgegeben teilen wir das zur Verfügung stehende Phasenraumvolu-men in Zellen ein.
(1) Wahrscheinlichkeit für einen Mikrozustand:
Frage: Wie lassen sich unterscheidbare Teilchen (Gasatome mit unterscheid-baren ) in Zellen im Phasenraum unterbringen, wenn wir jede Zelle miteiner beliebigen Zahl von Teilchen belegen dürfen?36
Antwort:1. Teilchen: → Möglichkeiten → 1
2. Teilchen: → Möglichkeiten → 2
3. Teilchen: → Möglichkeiten → 3
. Teilchen: → Möglichkeiten →
(8.30)
⇒ Resultierende Wahrscheinlichkeit für einen Mikrozustand:
=1
(8.31)
36 Achtung: Dies gilt nicht für Elektronen. Elektronen können eine Zelle maximal zu zweit besetzen
(mit antiparallelem Spin).

8.1 Der Boltzmann-Ausdruck = ln 228
(2) Zahl der Mikrozustände zu einem Makrozustand:
Frage: Wie groß ist die Zahl der Mikrozustände Ω, die zu einem Makrozustandgehören?
Antwort: Ω = Zahl der Vertauschungen der Gasatome ( !), ohne die Ver-tauschungen der Gasatome innerhalb der Phasenraumzellen 1!×2!×3!× =
Q(!), d.h.
⇒ Zahl der Mikrozustände zu einem Makrozustand:
Ω = !Q(!)
(8.32)
(3) Wahrscheinlichkeit für einen Makrozustand ?
Frage: Wie groß ist dann die Wahrscheinlichkeit für einen Makrozustand?
Antwort:
= (Anzahl Mikrozust. zum Makrozust.)×(Wahrscheinlichkeit der Mikrozust.)(8.33)
⇒ Wahrscheinlichkeit für einen Makrozustand:
= ×Ω =1
Ω =
1
× !Q
(!)(8.34)
Gleichung 8.34 müssen wir nun unter verschiedenen Nebenbedingungenauswerten, denn wir wollen wissen, welcher Makrozustand sich als wahrschein-lichster einstellt. Wie gehen wir vor?
(4) Frage: Welcher Makrozustand stellt sich ein?
Antwort: Es stellt sich der Makrozustand mit der höchsten Wahrscheinlichkeitein.
Frage: Wie finden wir den wahrscheinlichsten Makrozustand?
Antwort: Wir müssen nach dem Maximum von suchen!
Vorgehensweise: Wir fragen, für welche Besetzungverteilung
die Funktion
ein Maximum hat und suchen dazu wie folgt:
max ( )→ max (Ω)→ max (lnΩ) mit Ω = !
Q=1
(!)
(8.35)
c) Verteilung der Gasatome im Raum (ausgearbeitete Lösung)*
Im nächsten Abschnitt (8.2) untersuchen wir die Verteilung der Atome im Raum undauf ihre Energiezustände (Energiezustände siehe Ergebnisse der Quantenmechanik).
Als einfacheres Beispiel suchen wir hier zunächst das Maximum der Wahrscheinlichkeit(Gl. 8.34) für die Verteilung der Atome im Raum. Hierfür wenden wir die Methode derLagrange’schen Multiplikatoren (siehe Anhang E) für die Bestimmung des Maximums

8.1 Der Boltzmann-Ausdruck = ln 229
einer Funktion unter Nebenbedingungen (= konstante Teilchenzahl) an. Das Ergebnissei hier vorweggenommen:
=1
(8.36)
Das heißt, die Verteilung der Atome im Raum hat die maximale Wahrscheinlichkeit, beider sich alle Teilchen gleichmäßig auf alle Zellen des Phasenraums verteilen (“gleich-mäßige Verteilung der Teilchen im Raum”).
I Aufgabe 8.1: Bestimmung des Maximums von
=1
Ω =
1
!Q(!)
(8.37)
unter der NebenbedingungP
=1
= (8.38)
¤I Lösung 8.1: Wir suchen das Maximum der Funktion
ln = ln !
Q
=1
()!
(8.39)
bzw. (da = const für eine vorgegebene Teilchenzahl ) der Funktion lnΩ
lnΩ = ln !−P
=1
ln! (8.40)
≈ ln − −P
=1
( ln −) (8.41)
= (ln − 1)−P
=1
(ln − 1) (8.42)
Bedingung für das Maximum von lnΩ:
lnΩ =P
=1
µ lnΩ
¶ = 0 (8.43)
y
lnΩ =
∙P
=1
∙ (ln − 1)−
P=1
(ln − 1)¸¸
(8.44)
=
∙0−
P=1
µ1
¶−
P=1
(1 (ln − 1))¸ (8.45)
=
∙0− −
P=1
ln +
¸ (8.46)
= −P
=1
ln = 0 (8.47)
Nebenbedingung:
=P
=1
= (8.48)

8.1 Der Boltzmann-Ausdruck = ln 230
y =
P=1
=P
=1
= 0 (8.49)
Lagrange-Trick: Multiplikation von Gl. 8.49 mit beliebigem Parameter :
=P
=1
= 0 (8.50)
und Addition von Gl. 8.47 und 8.50:
P=1
(− ln + ) = 0 (8.51)
Lösung: Damit die beliebig sein können, muss der Ausdruck (− ln + ) ver-schwinden, d.h.
− ln + = 0 (8.52)
yln = (8.53)
y = (8.54)
Bestimmung von durch Ausnutzen der Nebenbedingung:
=P
=1
=P
=1
= (8.55)
y
=
=1
(8.56)
wie oben angegeben. ¥

8.2 Die Boltzmann-Verteilung 231
8.2 Die Boltzmann-Verteilung
8.2.1 Wahrscheinlichkeit für Makrozustand eines Gases bezgl. Verteilung aufEnergiezustände
Tatsächlich ist die Verteilung der Atome bzw. Moleküle im Raum und ihre Verteilungauf die einzelnen Energiezustände . Die erlaubten Werte für die Energiezustände von Atomen und Molekülen ergeben sich aus der Quantenmechanik (siehe Vorlesung“Physikalische Chemie 2: Aufbau der Materie“.
I Ergebnis: Für die Verteilung der Atome bzw. Moleküle auf ihre Energiezustände gilt die Boltzmann-Verteilung:
=
−P
−=
−
(8.57)
mit =
X
− (8.58)
und
=1
(8.59)
I Achtung: Wir gehen in dieser Einführung nicht auf die Spezialfälle der Fermi-Dirac-Statistik und der Bose-Einstein-Statistik ein.
I Herleitung der Boltzmann-Verteilung:
• Wir müssen das Maximum der Funktion
=1
×Ω =
1
× !Q
(!)(8.60)
(≡ Gl. 8.34) unter den Nebenbedingungen der TeilchenerhaltungP
= 0 (8.61)
und Energieerhaltung P
= 0 (8.62)
suchen. Hierzu verwenden wir die Methode der Lagrange’schen Multiplikatoren(siehe Anhang E).
• Zur Lösung suchen wir statt max ( ) das Maximum der Funktion
ln = ln1
Ω = ln
1
!Q
()!(8.63)

8.2 Die Boltzmann-Verteilung 232
bzw. (da = const für eine vorgegebene Teilchenzahl ) der Funktion lnΩ
lnΩ = ln !−P ln! (8.64)
≈ ln − −P ( ln −) (8.65)
= (ln − 1)−P (ln − 1) (8.66)
Bedingung für das Maximum von lnΩ:
lnΩ =P
µ lnΩ
¶ = 0 (8.67)
y
lnΩ =
∙P
∙ (ln − 1)−P
(ln − 1)¸¸
(8.68)
=
∙0−P
µ1
¶−P
(1 (ln − 1))¸ (8.69)
=
∙0− −P
ln +
¸ (8.70)
y lnΩ = −P
ln = 0 (8.71)
• Bedingung für Maximum und Nebenbedingungen mit zwei Lagrange-Multiplikatoren ( und −):
−P
ln = 0 (8.72)
×P
= 0 (8.73)
− ×P
= 0 (8.74)
y P
[− ln + − ] = 0 (8.75)
y− ln + − = 0 (8.76)
y = − (8.77)
• Ausnutzung der Nebenbedingungen zur Bestimmung von : =
P =
P
− = ×P − (8.78)
• Division von Gl. 8.77 durch Gl. 8.78 liefert die Boltzmann-Verteilung
=
−P
−=
−
(8.79)
Den Wert für (= 1 ) bestimmen wir weiter unten (Abschnitt 8.3.1).

8.3 Die Zustandssumme 233
8.3 Die Zustandssumme
I Definition: Die Größe =
P
− (8.80)
wird Zustandssumme genannt. Sie ist die zentrale Größe der statistischen Thermody-namik. Denn, wie wir sehen werden, erlaubt sie die Berechnung der thermodynamischenZustandsfunktionen.
I Boltzmann-Verteilung mit Berücksichtigung des Entartungsfaktors. Üblicher-weise werden in der Zustandssumme mehrfach auftretende Energiezustände durch ihrenEntartungsgrad faktorisiert ausgewiesen:
=
−P
−=
−
(8.81)
Mit =1
(siehe Abschnitt 8.3.1 unten) wird daraus
=
−P
−=
−
(8.82)
8.3.1 Mittlere Energie eines Atoms oder Moleküls
I Allgemeiner Ausdruck für die mittlere Energie eines Atoms oder Moleküls beiGültigkeit der Boltzmann-Verteilung:
=X
=1
X
− (8.83)
= − 1
X
¡−
¢(8.84)
= − 1
X¡−
¢= − 1
(8.85)
y
= − ln
(8.86)
Für (1mol) Atome oder Moleküle, mit = :
= −
ln
(8.87)

8.3 Die Zustandssumme 234
I Bestimmung von β:
• Die Quantenmechanik liefert (Vorgriff auf PC-2) für die Energiezustände einesTeilchens in einem 1D-Potentialtopf der Länge mit unendlich hohen Wändenden Ausdruck
=2
8
2
2(8.88)
Dabei ist = 1, 2, 3, . . . die Translationsquantenzahl. Für = 1 ist
1 =2
8
1
2 (8.89)
sodass die Energiezustände bezogen auf 1 als Nullpunktsenergie also gegebensind durch
=2
82
¡2 − 1¢ = ¡2 − 1¢ (8.90)
mit =2
82.
• Translationszustandssumme in 1D:37
trans =
µ212
22¶12
(8.93)
• Mittlere Translationsenergie für 1D:
= − ln
= −
ln
µ212
22¶12
=1
2
ln =
1
2(8.94)
• Mittlere kinetische Energie eines Gasatoms pro Freiheitsgrad aus kinetischer Gas-theorie:
=1
2 (8.95)
• Vergleich:1
2=1
2 (8.96)
y =
1
(8.97)
37 Wir berechnen die Translationszustandssumme gemäß
trans =∞X=
−(2−1) =
∞Z1
−(2−1) ≈
∞Z0
−2 (8.91)
Dieses Integral ergibt mit der Substitution = ()12 y = ()
12
trans =
µ1
¶12 ∞Z0
−2
=
µ1
¶12µ12
2
¶=
µ212
22¶12
(8.92)

8.3 Die Zustandssumme 235
• Wir erhalten für die Zustandsumme somit: =
P
− (8.98)
• Anschaulich steht = ( ) für die Zahl der im thermischen Gleichgewicht imMittel besetzten Energiezustände.
8.3.2 Molekülzustandssummen und Systemzustandssummen
I Der Begriff des Ensembles: Wir betrachten ein abgeschlossenes System bei konstan-tem Volumen, konstanter Zusammensetzung und konstanter Energie bzw. konstanterTemperatur. Ein Ensemble erhalten wir, wenn wir uns N Replikationen dieser Systemedenken, die wir unter Einhaltung bestimmter Regeln erhalten (Regel = Kanon). AlleEinzelsysteme in dem Ensemble sollen im thermischen Gleichgewicht miteinander stehen(sie dürfen zwischen einander Energie austauschen).
Es werden verschiedene Sorten von Ensembles unterschieden:
• Mikrokanonisches Ensemble: Ensemble von Systemen (z.B. Atomen,Molekülen) mit konstanter Teilchenzahl, konstantem Volumen, konstanter En-ergie.
• Makrokanonisches Ensemble (kurz kanonisches Ensemble genannt): Ensemblevon Systemen (z.B. Atomen, Molekülen) mit konstanter Teilchenzahl, konstan-tem Volumen, konstanter Temperatur.
• Großkanonisches Ensemble: Ensemble von offenen Systemen (z.B. Teilchen,Molekülen) mit konstantem Volumen, konstanter Temperatur, die zwischeneinan-der Teilchen austauschen können.
I Die Molekülzustandssumme: Gleichung 8.98 = Zustandssumme für ein Molekül:
=X
alle Molekülzustände
− (8.99)
I Die Systemzustandssumme: Für ein System von Teilchen müssen wir die Sys-temzustandssummen nehmen. Dabei kommt es darauf an, ob die Teilchen unter-scheidbar sind (z.B. Atome in einem Kristall) oder ob sie ununterscheidbar sind (z.B.Gasteilchen).
=X
alle Systemzustände
− (8.100)
• Systemzustandssumme für unterscheidbare Teilchen (z.B. in Kristall): =
Q = (8.101)

8.3 Die Zustandssumme 236
• Systemzustandssumme für nicht unterscheidbare Teilchen (z.B. Gas):
=
!(8.102)
• Bei mehreren Teilchensorten (A, B, C, . . . ): Zustandsummen sind multip-likativ, da die Energien additiv sind, daher
= × × × (8.103)

8.4 Berechnung thermodynamischer Größen aus Zustandssummen 237
8.4 Berechnung thermodynamischer Größen aus Zustandssummen
I Innere Energie:
• Systemzustandssumme für 1mol () unterscheidbare Teilchen (z.B. Kristall):
= (8.104)
y
= 2 ln
= 2
ln
= 2
ln
(8.105)
• Systemzustandssumme für nicht unterscheidbare Teilchen (z.B. Gas):
=
!(8.106)
y für 1mol ():
= 2 ln
= 2
ln
!= 2
ln
(8.107)
I Helmholtz-Energie: Aus der Defintion = − folgt
=
− (8.108)
y
µ
¶
=
µ
¶
−
2−µ
¶
(8.109)
= −
2−
= −
2= − ln
(8.110)
y
= − ln (8.111)
y = − ln (8.112)
I Entropie:
= −µ
¶
= −
= (8.113)
I Druck:
= −µ
¶
=
µ ln
¶
= (8.114)

8.5 Chemisches Gleichgewicht 238
8.5 Chemisches Gleichgewicht
+ À (8.115)
y = exp
µ−∆
ª
¶(8.116)
bzw.∆
ª = − ln (8.117)
I Gleichgewichtskonstante: Wir berechnen zunächst
= − ln , (8.118)
indem wir =
!etc.. . . einsetzen38 und die Stirling’sche Näherung ln ! =
ln − anwenden:
= − [ln + ln + ln ] (8.119)
= −"ln
!+ ln
!+ ln
!
#(8.120)
= − [( ln − ln!)
+ ( ln − ln!)
+ ( ln − ln !)] (8.121)
= − [( ln − ln +)
+ ( ln − ln +)
+ ( ln − ln +)] (8.122)
I Gleichgewichtsbedingung: Im chemischen Gleichgewicht muss geltenµ
¶ 6=
= 0 (8.123)
38 , , . . . steht hier für die Anzahl der Moleküle A, B, . . .

8.5 Chemisches Gleichgewicht 239
y
0 =
[( ln − ln +)
+ ( ln − ln +)
+ ( ln − ln +)] (8.124)
=
µln −
1
− ln + 1
¶+
µln −
1
− ln + 1
¶−µln −
1
− ln + 1
¶(8.125)
= (ln − ln) + (ln − ln)− (ln − ln) (8.126)
= ln
+ ln
(8.127)
y
=
(8.128)
Übergang auf Teilchenzahldichten und volumenbezogene Zustandssummen:
( )
( ) ( )=
( )
( ) ( )(8.129)
y =
( )
( ) ( )=
∗∗
∗
(8.130)
Bezug auf die getrennten molekularen Nullpunktsenergien:
=0
0
0
× −∆0 (8.131)
mit 0 = volumenbezogene Molekülzustandssummen auf jeweilige Nullpunktsenergiender Moleküle:
0 =
× × × (8.132)
I Endergebnis: Die Indices ∗ und 0 wurden hier zur Klarheit benutzt. Der Einfachheithalber werden diese üblicherweise weggelassen.
y
=
× −∆0 (8.133)

8.6 Ergebnisse der Quantenmechanik 240
8.6 Ergebnisse der Quantenmechanik
8.6.1 Zustandssumme für elektronische Zustände
Die elektronische Zustandssumme muss unter Einschluss aller elektronischen Entartun-gen explizit berechnet werden:
=X
− (8.134)
Speziell sind zu berücksichtigen:
• Spinentartung (Spinmultiplizität ): = 2 + 1 (8.135)
• elektronischer Bahndrehimpuls ( (Atome) bzw. Λ (lin. Moleküle) bzw. Symme-trierasse oder für nichtlineare Moleküle):
• elektronische Feinstrukturkomponenten (Index im Atom-Termsymbol): = 2 + 1 (8.136)
• Nur niedrig liegende Elektronenzustände liefern einen nennenswerten Beitrag.• Die elektronische Zustandsumme ist explizit auszurechnen.
8.6.2 Zustandssumme für die Schwingungsbewegung
I Harmonischer Oszillator:
= × mit = 0 1 2 (8.137)
I Schwingungszustandssumme pro Oszillator:
=
∞X=0
− =∞X=0
−· mit =
(8.138)
=©1 + − + −2 + −3 +
ªmit = − 1 (8.139)
= 1 + + 2 + 3 + (8.140)
=1
1− für 1 (8.141)
y
=1
1− −(8.142)

8.6 Ergebnisse der Quantenmechanik 241
I s Oszillatoren:
=
Y=1
∙1− exp
µ−
¶¸−1(8.143)
I typische Werte bei Zimmertemperatur:
≈ 1 10 (8.144)
I Mittlere Energie und spezifische Wärme eines harmonischen Oszillators: MitBerücksichtigung der Nullpunktsenergie erhalten wir pro Schwingungsfreiheitsgrad
= 2 ln
= 2
ln
µ−2
1− −
¶(8.145)
= 2
µ−2
− ln ¡1− −¢¶
(8.146)
y
=
2+
1− −(8.147)
I Spezifische Wärme pro Schwingungsfreiheitsgrad:
=
µ
1− −
¶(8.148)
I Boltzmann-Verteilung:
=
=
−
(8.149)
(Auftragung an Tafel.)
I Grenzwert der Schwingungszustandssumme für T →∞ bzw. für ν → 0: Für
→∞ bzw. für → 0 kann exp
µ−
¶entwickelt werden:
exp
µ−
¶≈ 1−
+ (8.150)
y =
1
1− expµ−
¶ = 1
1−µ1−
+
¶ (8.151)
y ≈
(8.152)
Für klassische Oszillatoren:
≈Y
=1
(8.153)

8.6 Ergebnisse der Quantenmechanik 242
I Abweichungen vom harmonischen Oszillator:
• Für anharmonische Oszillatoren ist die Zustandssumme am besten durch explizitenumerische Berechnung nach
∞P=0
− zu ermitteln.
• Torsionsschwingungen können bei tiefen Temperaturen als harmonische Oszilla-toren beschrieben werden, bei hohen Temperaturen als freie 1D-Rotatoren. ImZwischenbereich muss die Zustandssumme ebenfalls explizit berechnet werden(wobei die Energiezustände für den gehinderten 1D-Rotator zugrundegelegt wer-den müssen).
8.6.3 Zustandssumme für die Rotationsbewegung
I lineare Moleküle:
=~2
2( + 1) = 0 1 2 (8.154)
~2
2= (8.155)
I Rotationszustandssumme:
(2) =1
∞X=0
− (8.156)
=1
∞X=0
(2 + 1) −~2(+1)2 (8.157)
=1
∞Z=0
(2 + 1) −~2(+1)2 (8.158)
y
(2) =2
~2=
(8.159)
I Symmetriezahl: (8.160)
I Rotationszustandssumme für nichtlineare Moleküle: ist nur für symmetrischeKreisel analytisch darstellbar, nicht für asymmetrische Kreisel. Für nicht zu tiefe Tem-peraturen ist
(3) =12
µ2
~2
¶32()
12=
12
( )
32()
−12(8.161)

8.6 Ergebnisse der Quantenmechanik 243
I typische Werte bei Zimmertemperatur:
≈ 100 1000 (8.162)
8.6.4 Zustandssumme für die Translationsbewegung
I Teilchen im 1D-Kasten:
=2
8׳
´2 = 1 2 3 (8.163)
I 1D-Translationszustandssumme:
=
∞X
− =
∞Z1
−2282 (8.164)
=
µ2
2
¶12×
¡in cm−1
¢(8.165)
y
=
µ2
2
¶12(8.166)
I Teilchen im 3D-Kasten:
=2
8×"µ
¶2+
µ
¶2+
µ
¶2#(8.167)
I 3DTranslationszustandssumme:
=
µ2
2
¶32×
¡in cm−3
¢(8.168)
y
=
µ2
2
¶32(8.169)
I typische Werte: ≈ 1024 × (8.170)
y
≈ 1024 cm−3 (8.171)

Anhang A 244
Anhang:
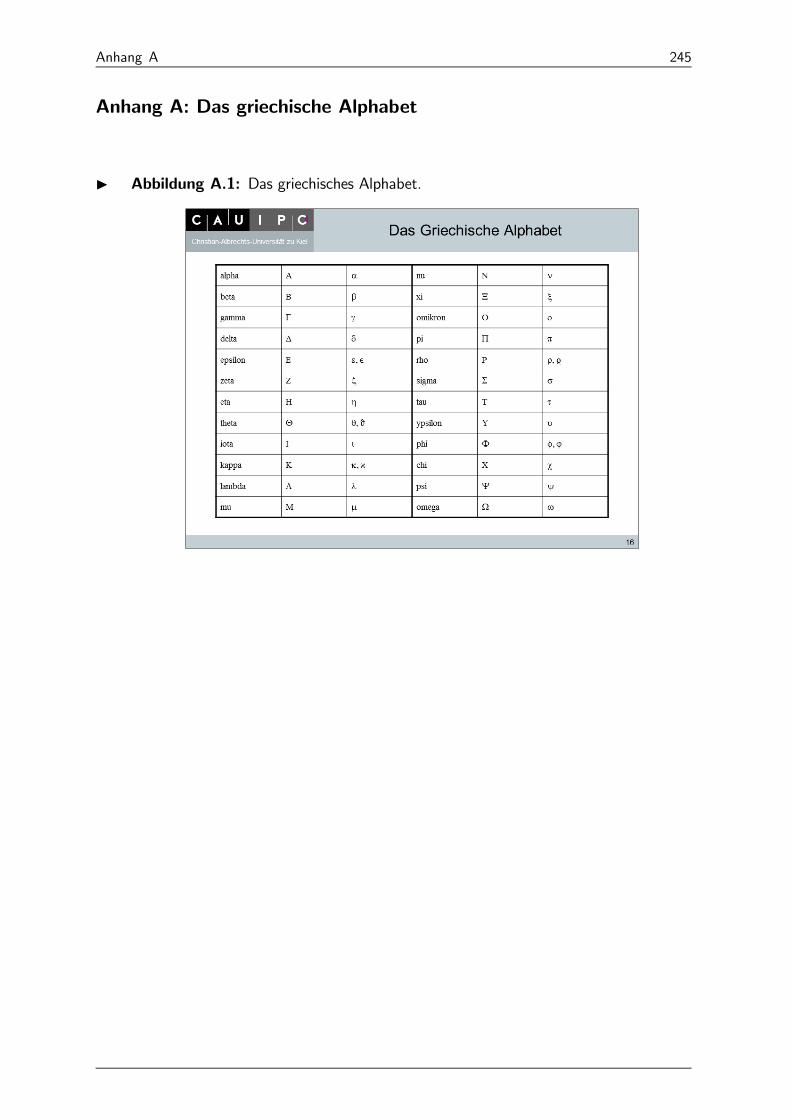
Anhang A 245
Anhang A: Das griechische Alphabet
I Abbildung A.1: Das griechisches Alphabet.

Anhang B 246
Anhang B: Physikalisch-Chemische Konstanten
Empfohlene Werte für physikalisch-chemische Konstanten wurden von der CODATA-Evaluierungsgruppe zusammengetragen.
I Tabelle B.1: Nützliche Physikalisch-Chemische Konstanten.ab
Physikalische Konstante Symbol Wert
Avogadro-Konstante = 6022 141 29× 1023mol−1 X(Loschmidt-Konstante) = 6022 141 29× 1023mol−1 Xuniverselle Gaskonstante = = 83144621 Jmol−1K−1 XBoltzmann-Konstante = 1380 648 8× 10−23 JK−1 Xelektrische Elementarladung = 1602 176 565× 10−19C XFaraday-Konstante = = 96 485336 5Cmol−1 XLichtgeschwindigkeit im Vakuum = 2997 924 58× 108ms−1 XPlanck’sches Wirkungsquantum = 6626 069 57× 10−34 J s X
~ ~ = 1054 571 726× 10−34 J s XMasse des Elektrons = 9109 382 91× 10−31 kg XMasse des Protons = 1672 621 777× 10−27 kg XMasse des Neutrons = 1674 927 351× 10−27 kg Xatomare Masseneinheit = 1660 538 921× 10−27 kg X
Stefan-Boltzmann-Konstante = 5670 373× 10−8Wm−2K−4 XWien displacment law constant = 2897 772 1× 10−3mK XRydberg-Konstante e e = 1097 373 156 853 9× 107m−1 XBohr’scher Radius 0 0 = 0529 177 210 92× 10−10m X
elektrische Feldkonstante 0 0 = 1(02) X
0 = 8854 187 817× 10−12AsV−1m−1 Xmagnetische Feldkonstante 0 0 = 4 × 10−7V sA−1m−1 X
-Faktor des Elektrons = 2002 319 304 361 53 XBohr’sches Magneton =
~2
= 9274 009 68× 10−24 JT−1 X = 0466 864 498 cm
−1T−1 XKernmagneton =
~2
= 5050 783 53× 10−27 JT−1 X = 7622 593 57MHzT
−1 X
Erdbeschleunigung = 9806 65m s−2 X
a The 2010 CODATA Adjustment of the Fundamental Constants,
http://physics.nist.gov/cuu/constants.b Table last checked on 10 April 2012.

Anhang D 247
Anhang C: Nützliche Umrechnungsfaktoren
I SI-Einheiten: Exzellente Überblicke über die im SI-System empfohlenen Maßeinheitenund Symbole geben die Bücher von Homann39 or Mills.40
I Umrechnungsfaktoren für oft benutzte “Nicht-SI”-Einheiten:
1 Bohr = 0529177249 A (C.1)
1 Hartree =2
400= 43597482× 10−18 J (C.2)
1 eV = 160217733× 10−19 J (C.3)
= 96495309 kJmol (C.4)
1 Debye = 333564× 10−30Cm (C.5)
39 K.-H. Homann (Ed.), Größen, Einheiten und Symbole in der Physikalischen Chemie, VCH, Weinheim,
1995.40 I. M. Mills et al., Units and Symbols in Physical Chemistry, Blackwell, Oxford, 1988.

Anhang E 248
Anhang D: Lineare Regression
I Kriterium für Güte der Anpassung einer Geraden y = ax+ b:
2 =X
( ()− )2=X
( + − )2
(D.1)
soll ein Minimum haben, d.h.
2
2 = 0 und
2
2 = 0 (D.2)
I Daraus ergeben sich die Bestimmungsgleichungen für a und b:
= (D.3)
und = (D.4)

Anhang E 249
Anhang E: Die Methode der Lagrange’schen Multiplikatoren zur
Bestimmung des Maximums einer Funktion f unter
Nebenbedingungen g
I Aufgabe: Wir suchen das Maximum einer Funktion
= (1 2 ) (E.1)
I Totales Differential:
=X
µ
¶ (E.2)
I Im Maximum gilt: = 0 (E.3)
y X
µ
¶ = 0 (E.4)
I Nebenbedingungen: Wir haben zu berücksichtigen, dass für die Funktion
= (1 2 ) (E.5)
gelten soll: = const (E.6)
y = 0 (E.7)
y X
µ
¶ = 0 (E.8)
Z.B. kann gefordert sein, dass die Zahl der Teilchen erhalten bleibt:
1 +2 + = (E.9)
y =
X = (E.10)
I Trick: Wir multiplizieren Gl. E.8 mit einem beliebigen Parameter
=X
µ
¶ = 0 (E.11)
Addieren wir dazu die Gl. E.4, erhalten wirX
∙µ
¶+
µ
¶¸ = 0 (E.12)
I Lösung: Der Parameter muss so gewählt werden, dass [ ] = 0, d.h.:∙µ
¶+
µ
¶¸= 0 (E.13)
Diese Gleichung dient zur Bestimmung von