point scattering: a new geometric invariant with ... · pdf filepoint scattering: a new...
TRANSCRIPT

Point Scattering: A New Geometric Invariant with
Applications From (Nano)Clusters to Biomolecules
ERNESTO ESTRADA
Complex Systems Research Group, X-ray Unit, RIAIDT, Edificio CACTUS, University of Santiagode Compostela, 15782 Santiago de Compostela, Spain
Received 2 May 2006; Revised 10 August 2006; Accepted 16 August 2006DOI 10.1002/jcc.20541
Published online 16 January 2007 in Wiley InterScience (www.interscience.wiley.com).
Abstract: A new geometric invariant is defined from ‘‘first principles’’ for a point ensemble, which can represent
clusters, molecules, crystals, and biomolecules. The scattering of a point ensemble is defined in terms of the Euclidean
distance matrix and a vector measuring the weighted departure of the points from the cluster centre. Using the Rayleigh–
Ritz theorem this function is maximized obtaining the point scattering of the ensemble. The point scattering shows
several properties which are useful for studying clusters, molecules, crystals, and biomolecules. We examined different
natural clusters of hard spheres such as colloidal particles and fullerenes, as well as protein–peptide complexes and the
effect of temperature on protein structure. In all cases point scattering differentiates point ensembles with different
structures, which are not distinguished by other geometric invariants, such as the second moment of mass distribution,
surface areas, and volumes. Point scattering also shows better correlation with thermodynamic parameters of binding
and describes the interior cavities of hollowed ensembles better than the other geometric measures.
q 2007 Wiley Periodicals, Inc. J Comput Chem 28: 767–777, 2007
Key words: Euclidean distance; discrete geometry; molecular geometry; protein packing; fullerenes; clusters
Introduction
A geometric invariant is a quantity which remains unchanged
under certain classes of transformation, such as the group of
translations or rotations.1 They are useful for comparing objects
because they usually reflect intrinsic properties of objects. Among
the most well-known geometrical invariants we can mention the
perimeter, second moment of mass distribution, surface area, and
volume.1 The last two are particularly of great interest in chemis-
try because of their relationship with the concept of packing,
which is a fundamental and essential characteristic of natural sys-
tems.2,3 In particular these concepts are necessary in understand-
ing protein structure and for uncovering the relationship between
packing and stability as well as for the study of solute hydropho-
bicity.4 A very well known example is provided by the average
packing density inside proteins,5–8 which is as high as in crystal-
line solids. It is known that a number of oligomeric proteins,
including, acetylcholine receptors, aquaporins, tight junction
occluding, and claudins, and gap junction channels arrange into
densely packed clusters, arrays or strands in the plasma mem-
brane.9–13 The optimal packing of tubes has also received atten-
tion as a model for understanding the way in which a DNA mole-
cule could be packed within a small virus.14–17 Packing is also an
important characteristic of clusters, such as colloidal materials
like photonic crystals and macroporous media.18,19
In macromolecules, such as proteins and nucleic acids, the sur-
face area and volume are commonly used as geometric invari-
ants,6,20,21 which are related to various molecular properties, such
as stability, solubility, crystal packing or molecular recognition.
Some of these measures, such as the hydrophobic surface area,
excluded volume, and radius of gyration, have been incorporated
into energy minimization algorithms used in studies of the three-
dimensional (3D) structure of proteins.22–24 The development of
global optimization procedures for clusters, crystals, and biomole-
cules is of great importance in fields ranging from protein struc-
ture prediction to the design of microprocessor circuitry.25 In
computational geometry, the second moment of mass distribution
is often used as a geometric invariant which is related to packing
density26–30 and has been found to be of great utility in the study
of colloidal clusters obtained from densely packed micro-
spheres.18,19
In this article, we introduce a new measure of packing that
accounts for the scattering of points in a discrete ensemble. This
measure solves some of the problems found with other packing
measures and reveals several other useful properties with which
to study clusters, molecules, crystals, and biomolecules.
Contract/grant sponsor: Ramon y Cajal, Spain.
Correspondence to: E. Estrada; e-mail: [email protected]
q 2007 Wiley Periodicals, Inc.

Theoretical Approach
Some ‘‘Classical’’ Geometric Invariants
Some of the most widely used geometric invariants are related to
the area and volume of surfaces. There are three definitions of
‘‘surface’’ which are widely used as geometric invariants of
points, particularly molecules, as well as for the definition of
packing measures.31–33 They are the van der Waals (vdW) sur-
face, the solvent accessible surface (SA), and the molecular sur-
face (MS). The first is defined as the surface of what is covered
by the points, i.e., atoms, which are represented by spherical balls
with radii equal to their vdW radii. The second surface is gener-
ated by the center of the solvent, which is modeled by a rigid
sphere, when rolling over the vdW surface of the cluster or mole-
cule. The third surface is generated by the front of the same sol-
vent sphere. The area and volume of such surfaces can then be
determined by approximation techniques or by analytic methods.
One of these methods, which treats volume and area overlaps
fully and accurately, is the ‘‘alpha shape method.’’20
The second moment of mass distribution is defined as: M2 ¼Si¼1n kri � r0k2, where ri is the centre coordinate of the ith vertex,
e.g., centre of cluster or atom, and r0 ¼ n�1 Si¼1n ri is the centre
of mass of the object, e.g., cluster or molecule, subject to the con-
straint kri � rjk � 2 for i = j. (Here k. . .k denotes the usual Eu-
clidean distance). M2 is the sum, divided by n, of the squared dis-
tances between all pairs of points. That is, M2 ¼ 1n
Pi>jðrijÞ2,
which geometrically represents the sum of the areas of the squares
formed by the edges of length rij.26 M2 has also been interpreted as
an energy function in determining clusters of hard spheres26 as
well as in communications, where the points represent a constella-
tion of n signals with total energyM2.34,35 It is known that the clus-
ters obtained using this energy function are quite different from
those using other potentials, such as Lennard–Jones for n � 8. The
second moment is intimately related to the radius of gyration, Rgyr,
another parameter which provides information regarding the global
conformation of a system, which is widely used in polymer statis-
tics.36 It can be calculated from the second moment M2 as follows:
Rgyr ¼ (M2/n)1/2.
Definition of Point Scattering
In this work we will use the term ‘‘point’’ to designate a body
whose spatial extent and internal motion and structure, if any, are
irrelevant to the specific problem under study, which is the pack-
ing of points in the discrete object. Consequently, a point here
can be a sphere, colloidal particle or an atom which is part of a
cluster or a molecule, which represents the discrete object.
We start by considering an object O formed by n points. Let us
represent the object by means of a column vector x, whose kthentry captures the relative departure of point k from the geometri-
cal centre, o, of the object. The entries of the vector x, xi, take val-ues between 0 and 1. They represent a sort of weighted distance
from the center of the object in which the outlying points receives
more weights than the points closest to the center. We impose the
restriction that the norm of this weight vector x be one or xxT ¼ 1.
Then, based on the Euclidean distances between the pairs of
points in the object, rij, we can define a measure for the spreading
of the points in O in a similar way as used in spectral clustering
techniques37–39:
SðOÞ ¼Xni¼1
Xnj¼1
rijxixj ¼ xTDx (1)
where D is the Euclidean distance matrix of the points in the
object. The function S(O) increases with the increase in the sepa-
ration between points as well as with the departure of the points
from the centre of the object. Consequently, we consider S(O) asa measure of the scattering of the points in the discrete object,
which will take minimum values for the least scattered objects.
We are interested in finding the maximum value of the scatter-
ing function, which can be obtained by maximizing the expres-
sion (2). Let {�1, �2, . . . , �n} be the nondecreasing order of the
eigenvalues of D and let xi be the orthonormal eigenvector corre-
sponding to the ith eigenvalue:kxk2 ¼ xxT ¼ 1.40 Then, according
to the Rayleigh–Ritz theorem,41 we have:
SðOÞ ¼ maxx
xTDxjxTx ¼ 1� � ¼ �1ðDÞ (2)
where �1(D) is the spectral radius or largest eigenvalue of D. Thismaximum is attained when x ¼ x1, where x1 is the principal eigen-
vector of D. It is clear that �1(D), which is our measure of point
scattering, remains unchanged under translation and rotation. This
geometric measure can also be invariant to scaling by normalizing
the distances by using a canonical representation of the object.
However, we will not consider this item in the current work.
The interpretation of the principal eigenvector of D as a rela-
tive weighted measure of the departure of a point from the centre
of the object can be understood by means of the following analy-
sis. It is known that the principal eigenvector is proportional to
the row sum of a matrix M formed by summing all powers of the
distance matrix, weighted by corresponding powers of the recip-
rocal of the principal eigenvalue:
M ¼ limn!1
1
nDþ ��1
1 D2 þ ��21 D3 þ � � � þ ��n
1 Dnþ1� �� �
: (3)
Then, we can consider an object formed by three points placed
on a straight line, in such a way that point i is equidistant from
points j and k, i.e., i is placed at the centre of the object. Let
�a ¼ Pb rab be the sum of distances for the point a in the discrete
object, i.e., the sum of the ath row or column of D. It is obvious
that the minimal value of �a will be obtained for the point which is
at the centre of the object, which has the lowest entries in D: min
�a ¼ �i. The same is also true for the different powers of D, which
makes that the lowest value of the row sum for the matrixM corre-
sponds to the point at the centre of the object. As the ith row sum
of this matrix is proportional to the ith entry of the principal eigen-
vector of D we have that the lowest entry of xl corresponds to the
point at the center of the object. As far as we add more points, for
instance in different orbits from the center, the points more distant
from the center will have larger values of their sum rows in M and
of its components in xl. Then, it is clear that xl is the relative depar-
ture of the points from the centre of the cluster weighted in a way
that the most distant points are weighted more strongly than the
768 Estrada • Vol. 28, No. 4 • Journal of Computational Chemistry
Journal of Computational Chemistry DOI 10.1002/jcc

points closest to the centre. This measure is not appropriate for
comparing points in different clusters because points which are
equidistant from the centre in two different clusters will give the
same values of xl. For instance, the value of xl for the arrangement
of unit spheres in a square and in a tetrahedron are identical, e.g.,
x1 ¼ (0.5 0.5 0.5 0.5) for both objects. In these objects the points
are equidistant from the respective centers, but the distances from
the center to the points in both objects are different, which makes
xl inappropriate for comparing points in the square to points in the
tetrahedron. Consequently, the use of the following measure is
more appropriated for comparing points in different objects. Since
xl is a �1-eigenvector, one has x1(i) ¼ Sjrijx1(j)/�1, so that
SðiÞ � x1 ið Þ�1ðDÞ (4)
can be considered as a measure of local geometric invariant for
the points of the cluster.
Another consequence of the current representation of discrete
objects is that M2 can also be calculated from the spectrum of D. It
can be shown thatM2 is equal to the half sum of the diagonal entries
of D2, i.e., M2 ¼ 12nTrðD2Þ ¼ 1
2n
Pni¼1ðD2Þii, where Tr stands for
the trace of the matrix. It is well-known that Tr Dk� � ¼ Pn
j¼1 �j
� �k,
from which it follows that M2 ¼Pn
j¼1 �j
� �2. The radius of gyra-
tion, Rgyr is then easily expressed in terms of the spectrum of D
as:Rgyr ¼ ½1n
Pnj¼1 �j
� �2�1=2.Computational Methods
The calculations of the point scattering measure for the different
objects described in this work were carried out using a Matlab1 pro-
gram developed in-house. The input for the program is the distance
matrix, which is previously obtained from the Cartesian coordinates of
the points using an implementation in MODESLAB (www.mode-
slab.com). The output of the Matlab1 program are the principal eigen-
value and eigenvector of the distance matrix, which correspond to the
geometric invariants introduced here. Other geometric invariants were
also calculated in this work for the sake of comparison. The first of
them is the second moment of mass distribution, which was calculated
using the squared eigenvalues of the distance matrix obtained from the
Matlab1 program according to: M2 ¼P
nj¼1 �j
� �2. Surface areas and
volumes (SA or vdW) were calculated from the grid method
according to the implementation of the Bodor et al.42 approach
used in HyperchemTM. This method uses the atomic radii of Gave-
zotti.43 In SA calculations the solvent probe radius used was 1.4 A
and we always used 50 points on cube side. The calculations of SA
surface area and volumes of the square grid with holes were car-
ried out using the same grid approach but using hydrogen atoms
with radius equal to 1 A (unit radius spheres) instead of using the
Gavezotti radius for hydrogen, which is 1.17 A (this value can be
modified in the file VDWGRID.TXT of HyperchemTM).
Results and Discussion
Point Scattering in Clusters and Crystals
Clusters of hard spheres can represent a variety of structures
found in nature ranging from pollen grains to crystals and virus
capsids. When applicable, the examples of clusters studied here
can also be considered as crystals in which the spheres are occu-
pying the lattice points of space lattice.44 When considering clus-
ters of hard spheres we will take the centers of the spheres as the
points forming the object. Here we will consider hard spheres,
i.e., noninterpenetrating spheres, of unit radius. Thus, rij � 2 for
every pair of nodes in G.
Scattering in Clusters with Degenerated Second Moments
As a first example we will consider the optimal clusters in two-
dimensions (2D). Graham and Sloane conjectured 15 years ago
that for n = 4 every optimal packing is (up to limits imposed by
symmetry) a subset of the hexagonal lattice A2, which is gener-
ated by (1,0) and �1=2;ffiffiffi3
p=2
� �.29
The exclusion of the four point cluster appears to be due to the
well known fact that the second moment of mass distribution is
unable to differentiate a square and a rhombic arrangements of
points as they both have: M2 ¼ 8a2, where a is the radius of the
circles. This result contradicts the intuitive idea that the rhombus,
which is a subset of A2, is more tightly packed than the square,
which is not a subset of the hexagonal lattice. This situation is found
not only for this pair of clusters but for several other pairs of clus-
ters. In Figure 1 we illustrate some of these examples. On the third
and fourth lines of Figure 1 we show two examples that extend this
observation to 3D clusters. They correspond to a pair of clusters of
six spheres forming a rhombic bipyramid (rhombic octahedron) and
a triangular prism. The last example is provided by the pair of clus-
ters with eight spheres forming a cube and a triangular biprism.
The consideration of S(O) clearly differentiates all these pairs
of different clusters according to their packings. First, S(O) indi-cates that the rhombus has lower point scattering than the square
as expected from the fact that the first is a subset of the hexagonal
lattice. It also indicates that the prism and the biprism are less
scattered than the rhombic pyramid and the cube, respectively
(Fig. 1). In all cases the clusters with optimal packing (according
to second moment of mass distribution)26 having 4 (tetrahedron),
5 (triangular bipyramid), 6 (octahedron or square bipyramid), and
8 (dodecadeltahedron or snub disphenoid) nodes show the lowest
values of S(O) (see last column of Fig. 1).
Square Lattices with Holes
As a second example we build a toy model consisting of square
lattice of 16 spheres in which we remove two spheres at random.
In total there are 21 different configurations of 14 spheres plus
two holes in this square lattice. In Figure 2 we illustrate the
square lattice with sphere numbering as used here and three con-
figurations of 14 spheres and two holes. In Table 1 we give the
values of the several geometric invariants: second moment, SA sur-
face areas and volumes and the values of point scattering, for these
21 configurations of 14 spheres. The vdW surface area and the vol-
umes are the same for all these objects: 175.80 A2 and 58.91 A3,
respectively. As can be seen, there are four pairs and a triple of
clusters having identical values of SA surface areas, one pair, two
triples, and one quadruple of clusters with identical values of the
SA volumes and three pairs of clusters with degenerate values of
M2. However, there is not a single pair of clusters with identical
values of S(O). The geometric invariants show good linear correla-
769New Geometric Invariant with Applications From Clusters to Biomolecules
Journal of Computational Chemistry DOI 10.1002/jcc
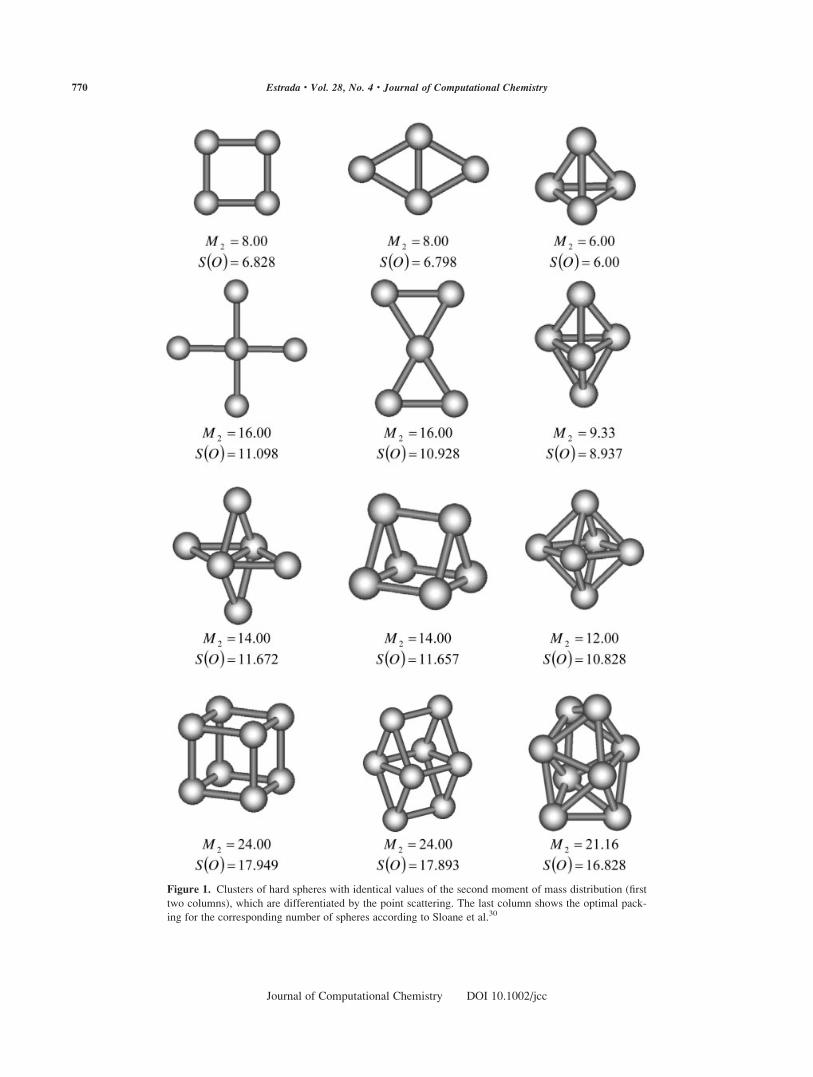
Figure 1. Clusters of hard spheres with identical values of the second moment of mass distribution (first
two columns), which are differentiated by the point scattering. The last column shows the optimal pack-
ing for the corresponding number of spheres according to Sloane et al.30
770 Estrada • Vol. 28, No. 4 • Journal of Computational Chemistry
Journal of Computational Chemistry DOI 10.1002/jcc

tions between each other. The best linear correlation is observed for
M2 and S(O) with a correlation coefficient > 0.99. In general, S(O)shows excellent correlations with the other geometric invariants
(Table2). For instance, S(O) has larger correlation coefficients with
the SA surface areas and the volumes thanM2.
Hollowed Clusters: Fullerenes and Virus Capsids
The third example of clusters to be studied here is that of carbon
clusters or fullerenes. A fullerene is a closed cage formed by ncarbon atoms distributed in a Degree-3 network of pentagons and
hexagons on the surface of a spheroid.45 Consequently, fullerenes
are good representatives of hollow clusters in which the carbon
atoms on the surface create nonpolar cavities of different sizes in
the interior of the cluster.46 In fact, these cavities have been used
to model the penetration of water molecules to nonpolar interior
of proteins. Vaitheeswaran et al. have shown that up to nine water
molecules can be easily accommodated in the internal cavity of
C180.47 They have shown that the stability of encapsulated water
clusters depends critically on cavity size. The existence of other
endohedral complexes with fullerenes has been observed experi-
mentally. Among the most significant, one can cite the existence
of ‘‘bucky-onions,’’ in which one fullerene is encapsulated inside
another one, such as C60 @ C240 @ C540 . . . ,48 or the existence
of endohedral metal encapsulated fullerenes, such as Sc2 @ C84
or Sc3 @ C82.49 Thus, the study of the geometric invariants of
these carbon clusters is of great importance for understanding
these inclusion phenomena, which critically depend on the rela-
tion between the size of the cluster and the size of the interior cavity.
Fullerenes also represent interesting models of viral protective pro-
Figure 2. Square grids of spheres of unit radius numbered from 1 to 16 (A). Three clusters of 14
spheres, which are subsets of the square grid, having the lowest (B) (1,4-cluster), intermediate (C) (1,7-
cluster), and the largest scattering (D) (6,11-cluster) among the 21 clusters. The numbers indicate the
spheres which were removed according to the numbering given in A.
771New Geometric Invariant with Applications From Clusters to Biomolecules
Journal of Computational Chemistry DOI 10.1002/jcc
tein shells (capsids), as it is well known that there are numerous
roughly spherical viruses whose capsids display perfect icosahedral
symmetry.50 For instance, the tobacco ringspot virus capsid is com-
posed of 60 copies of a 513-amino-acid capsid protein, each of which
corresponds to one of the atoms in C60 ‘‘buckminsterfullerene.’’51
Here we study the areas and the volumes (vdW and surface
accessible) as well as the second moment of mass distribution
and point scattering of 19 fullerenes ranging from C20 to C540. In
Table3 we give the values of the geometric invariants for all these
carbon clusters.
In general there is very good linear correlation between the
different pairs of measures. However, plots of the geometric
invariants versus the size of the carbon clusters reveal interesting
characteristics of the different invariants. In Figure 3 we illustrate
these plots for S(O) and the vdW area and volume.
The behavior of M2 is similar to that of S(O). The surface acces-sible area and the volume plots are similar to those of the vdW ana-
logues. As can be seen in Figure 3, surface area and volume increase
linearly with the size of the clusters (these values are plotted on an
inverse scale in Fig. 3). This means that the size of the interior cav-
ities of these fullerenes increase linearly with the size of the cluster.
However, a different picture is provided by S(O) (as well as by M2)
which shows a nonlinear increment of the scattering as a function of
the cluster sizes. This indicates that according to S(O) the size of theinterior nonpolar cavity of a fullerene increases nonlinearly with the
increase of the number of carbon atoms—an observation with im-
portant consequences for the study of inclusion complexes in fuller-
ene cavities. For instance, while the optimal number of water mole-
cules that can be encapsulated in C140 is four, giving (H2O)4 @
C140, this number increases up to eight for C180, which leads to
(H2O)8 @ C180.47 Torrens has calculated the volume and area of the
internal cavities of smaller fullerenes, C60, C70, and C82, in which it
is observed that the volume of the internal cavity increases nonli-
nearly with the size of the carbon cluster in agreement with our cur-
rent findings even for these small fullerenes.52 These results illus-
trate the utility of the point scattering as a geometric invariant which
contains information not duplicated by other invariants.
Point Scattering in Proteins
The previous analysis of S(O) in clusters of hard spheres provides
several suggestions as to how this measure quantifies the scatter-
ing of a set of points. Models based on sphere packing have been
used for studying optimal properties of proteins.53 In this sense,
the study of the toy model of spheres in a square lattice with two
holes clearly indicates that the least scattered structures are those
in which the holes are as far as possible from the centre of the
cluster. On the contrary, the structure with the largest scattering
(see D in Fig. 2) corresponds to the one in which the two holes
are adjacent to the middle point of the cluster. This is an impor-
tant characteristic for the study of proteins, which are believed to
have efficiently packed interiors. The existence of internal cav-
ities has also been considered to be an important characteristic of
these macromolecules, which in general are well accounted for
by the S(O) index (see structure B in Fig. 2). Consequently, we
will study here the relationships between S(O), as well as other
geometric invariants, and the thermodynamic parameters of bind-
ing between peptides and proteins in the complex RNase-S. In
this case, point scattering can be referred to as the atomic scatter-
ing as it is calculated for all atoms in the proteins.
The protein–peptide complex RNase-S is obtained by cleavage
of bovine pancreatic ribonuclease A (RNase A) with subtilisin to
give an ‘‘S protein’’ and an ‘‘S peptide.’’ These two fragments can
be reconstituted to give rise to RNase-S, which is catalytically
active with a structure very similar to that of ribonuclease-A
(RNase-A).54,55 The S peptide consists of the first 20 amino acids
of RNase-A, but it has been shown that a truncated version formed
by Residues 1–15 forms a complex with protein S which is struc-
turally identical with RNase-S. There are two hydrophobic resi-
dues in the S peptide that contribute significantly to the stability of
RNase-S. These residues, methionine 13 (M13) and phenylalanine
8 (F8), are buried inside the RNase-S core.55,56 Mutation experi-
ments have led to their replacement by several other smaller
hydrophobic amino acids of different sizes resulting in mutated S
peptides of the type F8X and M13X, where X represents alanine
(A), methionine (M), norleucine (Nle), �-aminobutyric acid
(ANB), valine (V), leucine (L) or isoleucine (I). The structures of
Table 1. Values of Geometric Invariants for the 21 Clusters of
Spheres of Unit Radius Which are Subsets of the Square Grid
as Explained in Figure 2.
Cluster S(O) M2 SASA (A2) VSA (A3)
1,4 53.239 121.43 301.68 386.13
1,16 53.633 123.99 306.13 388.62
1,2 54.587 128.28 301.69 386.13
1,3 54.927 129.14 316.03 399.08
1,8 55.254 130.85 315.52 399.08
1,12 55.362 131.71 315.52 399.08
2,3 56.620 137.43 323.42 402.26
1,6 56.648 137.71 316.38 404.09
1,7 56.704 138.57 316.40 404.09
2,5 56.738 137.71 329.87 412.01
1,11 56.823 139.43 316.40 404.09
2,8 56.937 138.57 330.40 412.05
2,12 57.063 139.43 330.40 412.05
2,14 57.098 139.71 329.85 412.05
2,15 57.129 139.99 329.85 412.05
2,6 58.339 146.57 332.28 415.51
2,7 58.422 146.85 330.73 417.06
2,10 58.529 147.43 330.73 417.06
2,11 58.568 147.71 330.73 417.06
6,7 59.980 155.71 332.19 420.54
6,11 60.042 155.99 331.06 422.08
Table 2. Correlation Coefficients for the Pairs of Geometric Invariants
Analyzed for the 21 Clusters of 14 Spheres of Unit Radius Which are
Subsets of the Square Grid.
M2 SASA VSA
S(O) 0.998 0.867 0.952
M2 0.838 0.936
SASA 0.963
SASA, solvent accessible solvent area; VSA, solvent accessible volume.
772 Estrada • Vol. 28, No. 4 • Journal of Computational Chemistry
Journal of Computational Chemistry DOI 10.1002/jcc

the RNase-S complexes with S peptide mutants were determined
by X-ray crystallography and the free energies (DG8) and enthal-
pies (DH8) of the S-peptide-protein binding were determined using
titration calorimetry.57 In Table 4 we show the values of the differ-
ences in the thermodynamic parameters upon mutation: DDG8 andDDH8, e.g., DDG8 ¼ DG8 (mutant) � DG8 (wild type).
We calculated the values of S(O) as well as areas and vdW
and SA volumes for eight protein–peptide complexes. The values
are given in Table 4 where we also provide the correlation coeffi-
cients of the linear fits between the thermodynamic parameters
and the geometric measures. As can be seen S(O) gives the best
linear correlations for both thermodynamic properties, which has
Table 3. Values of Geometric Invariants for Several Fullerenes Studied Here.
Fullerene S M2 SAvdW (A2) SASA (A2) VvdW (A3) VSA (A3)
C20 (Ih) 53.68 82.22 170.27 318.28 186.06 525.78
C24 (D6) 71.51 121.32 195.21 353.05 221.84 598.12
C26 (D3h) 80.61 142.18 205.91 361.59 238.09 630.73
C28 (Td) 89.81 163.53 216.32 376.62 254.00 660.92
C30 (D5h) 100.68 192.98 227.75 391.77 270.24 696.19
C32 (D3) 109.86 213.92 239.76 398.30 287.05 726.42
C36 (D6h) 131.20 271.05 262.47 425.65 320.10 791.65
C50 (D5h) 214.86 521.84 342.04 500.42 436.83 1,016.75
C60 (Ih) 280.98 742.29 397.31 552.89 516.99 1,169.50
C76 (D2) 405.93 1,223.88 499.18 637.41 662.44 1,453.72
C78 (D3h) 420.25 1,276.38 507.84 651.25 676.12 1,478.92
C78 (D3h) 421.07 1,283.57 508.42 648.06 676.15 1,477.48
C78 (D3) 421.14 1,283.94 508.27 648.89 675.92 1,478.12
C78 (C2v) 420.40 1,277.84 508.27 648.89 675.92 1,478.12
C80 (Ih) 438.35 1,353.59 522.74 668.90 697.89 1,522.54
C180 (Ih) 1,470.1 6,756.38 1,097.36 1,200.40 1536.2 3,139.81
C240 (Ih) 2,277.8 12,161.70 1,472.53 1,542.21 2,070.28 4,177.71
C320 (Ih) 3,476.6 21,244.27 1,917.71 1,952.61 2,722.84 5,430.73
C540 (Ih) 7,596.0 60,084.84 3,196.6 3,131.93 4,571.25 9,004.91
Figure 3. Plot of geometric measures (in reverse scale) versus number of carbon atoms in fullerenes.
[Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
773New Geometric Invariant with Applications From Clusters to Biomolecules
Journal of Computational Chemistry DOI 10.1002/jcc

correlation coefficients identical to those of VSA. The best linear
correlation previously obtained for DDH8 was by using the
occluded surface area, which measures the internal packing of a
protein, and shows a correlation coefficient identical to those
obtained here with S(O) and VSA, i.e., 0.98. Occluded surface
area shows a correlation coefficient of 0.95 for DDG8, which is
slightly lower than the one obtained here by using S(O) and VSA.
In contrast, other geometric invariants, such as cavity volumes
and SA area, show very poor correlations, while the packing mea-
sure called ‘‘depth’’ shows correlation coefficients of approxi-
mately 0.90 for both thermodynamic parameters of binding.
As previously indicated, the values of S(i) can be used as a
local scattering parameter for the points of a cluster. In the partic-
ular case of proteins, S(i)corresponds to atomic contributions to
protein scattering. It is also possible to consider the average val-
ues of S(i) for the atoms in an amino acid as the scattering of
amino acid residues in the protein: S(res). Using this approach we
have calculated the values of S(res) for all the amino acids in the
wild type RNAse A solved at 1.6 A (2rns). In Figure 4 we plot
the values of S(res) for all residues in 2rns, where the lowest val-
ues of S(res) correspond to the least scattered residues and the
higher values to the most scattered ones. In the same figure we
illustrate the ten least scattered residues as well as the ten most
scattered ones. As can be seen, the least scattered residues corre-
spond to those which are located in the interior of the protein in a
region close to the centre of the structure, which is commonly
identified as the most packed region. The most scattered residues
are located far away from the middle of the protein, in the periph-
eral regions of the protein where residues 1, 68, 88–94, and 113
are found.
In a similar way S(res) can be used to analyze the changes on
residue scattering produced by external factors such as temperature.
As an example we calculate the values of S(res) for all the amino
acids in the RNAse-A, which is a kidney-shaped monomeric
enzyme of 124 residues. The structure of this protein has been
determined by X-ray diffraction at a resolution of 1.5 A at nine dif-
ferent temperatures ranging from 98 to 320 K.53 In previous studies
it has been shown that the protein molecule expands slightly with
increasing temperature which affects principally the degree of fold-
ing of the main backbone of the protein.6,58–60 It was also previ-
ously observed that the 3D structure of RNAse-A undergoes no
dramatic change over the range of temperatures analyzed. Here we
analyze the changes in scattering of individual amino acids with
changing temperature. In Figure 5 we plot the change in the values
of S(res) during step-by-step ‘‘heating’’ of RNAse-A. The first plot
shows the difference in S(res) for the amino acids in the structures
determined at 98 and 130 K, i.e., DS(res) ¼ S130 K(res) �S98 K(res). As can be seen in this figure, most of the amino acids
suffer small changes in their scattering when analyzed in this step-
by-step method. However, a dramatic change in scattering is
observed for residue Gln101, which appears as an intense peak in
most of the plots. For instance, in the 130–98 K plot this residue
appears with an intense positive peak, which indicates that Gln101
is least scattered at 98 K than at 130 K. However, this situation is
inverted in the next step, which indicates that Gln101 returns to a
least scattered conformation when passing from 130 to 160 K. In
Figure 6 we illustrate these two conformations for Gln101. This
alternating change of conformation of Gln101 is observed for all
RNAse-A structures below 220 K and it still appears for the changes
at the highest temperatures, i.e., 120–260 K and 320–280 K. This
residue is located in one of the extended loops of the protein, which
have been previously observed to undergo the most intense move-
ments. In general, the most packed regions at the interior of the pro-
tein structure do not suffer large variations in the scattering com-
pared to the atoms which are in the protruding loops. An exception
is the 180–220 K transition where the scattering of most of the resi-
dues is altered. This change coincides with the known fact that in
the neighborhood of 200 K changes occur in the dynamic properties
of many proteins in solutions and in the crystal state.58 These
dynamic changes are believed to be produced primarily in the coor-
dination shells of water that are bound to the surface of the protein,
which in this case is expressed as an increment in the packing
changes of amino acid residues in the whole protein.
These results clearly indicate that the point scattering appears
to be a convenient geometric invariant, which can account for the
vdW interactions between peptides and proteins as well for the
effects of temperature on protein structure and dynamics. In con-
sequence, this measure can be useful as a geometric parameter
for empirical potentials of energy minimization algorithms for the
study of the 3D structure of proteins.
Table 4. Values of Geometric Invariants as well as Thermodynamic Binding Parameters for the Protein–Peptide Complexes in RNAse-S.
PDB S SAvdW (A2) SASA (A2) VvdW (A3) VSA (A3) DDH8 (258 C) DDG8 (kcal mol�1)
2rln 18,137 11,753.2 6,653.4 10,343.8 22,508.9 �7.9 �0.8
1rbg 18,029 11,758.4 6,586.4 10,324.9 22,391.9 �2.5 �0.7
1rbh 18,030 11,755.7 6,596.6 10,324.9 22,380.3 �2.2 �0.5
1rbi 18,031 11,745.4 6,577.7 10,311.2 22,307.4 �1.9 �0.2
1rbd 18,019 11,719.5 6,539.8 10,292.7 22,294.0 1.3 0.7
1d5e 17,618 11,438.7 6,653.3 10,113.1 22,117.1 10.0 2.9
1d5d 17,554 11,384.2 6,674.8 10,083.6 22,073.1 14.7 3.6
1d5h 17,543 11,447.9 6,593.7 10,117.4 22,037.2 17.7 5.1
r(DDH8) �0.98 �0.94 �0.28 �0.96 �0.98
r(DDG8) �0.96 �0.94 �0.31 �0.95 �0.96
The correlation coefficients between the geometric invariants and thermodynamic parameters are given in the last two rows.
774 Estrada • Vol. 28, No. 4 • Journal of Computational Chemistry
Journal of Computational Chemistry DOI 10.1002/jcc

Conclusions
We have analyzed the main deficiencies of some of the most
widely used ‘‘classical’’ geometric invariants for the analysis of
clusters, molecules, and macromolecular systems, such as pro-
teins. This analysis has led to the introduction of a new measure,
which is based on first principles, and resolves the deficiencies
previously observed associated with other geometric measures,
such as second moment of mass distribution, surface areas, and
volumes. This new geometric invariant accounts for the scattering
of points from the center of the object, e.g., cluster, molecule,
protein, etc. We have proved that the principal eigenvalue of the
Figure 4. Values of residues scattering in ribonuclease-S (PDB 2rns). The largest values of the scatter-
ing correspond to most packed residues, e.g., 88–94 and the lowest values of scattering correspond to
the least scattered residues, e.g., 44–47.
775New Geometric Invariant with Applications From Clusters to Biomolecules
Journal of Computational Chemistry DOI 10.1002/jcc

Euclidean distance matrix represents the point scattering of the
object studied. The Euclidean distance matrix is built by consider-
ing the distances between all pairs of points in the discrete object,
such as the center of spheres in clusters or interatomic distances
in molecules. This matrix can be obtained experimentally, for
example by X-ray diffraction, or by empirical or ab initio optimi-
Figure 5. Change of residues scattering in RNAse-A as an effect of temperature.
Figure 6. Change in conformation of residue Gln101 at two different temperatures which produces the
change in scattering observed in Figure 5. At 98 K this residue is least scattered than at 130 K, which
produces a large positive peak in the first plot of Figure 5. [Color figure can be viewed in the online
issue, which is available at www.interscience.wiley.com.]
776 Estrada • Vol. 28, No. 4 • Journal of Computational Chemistry
Journal of Computational Chemistry DOI 10.1002/jcc

zation procedures, such as molecular mechanics or quantum
chemical calculations.
In this work we have illustrated some of the advantages of the
point scattering for studying hypothetical and real clusters, such as
those that can arise from dense packings of colloidal microspheres
as well as carbon clusters or fullerenes. We have also studied
‘‘atomic’’ scattering in proteins by illustrating the relationship
between this measure and the binding energetics of peptide–pro-
tein interaction as well as the effects of temperature on protein
structure and dynamics. In all cases point scattering appears to be
a convenient geometric invariant, which is easily and exactly com-
puted for any discrete configuration of points, accounting for im-
portant structural characteristics of the objects studied, which are
invariants to the group of rotation and translation of the object.
One of the most significant applications of geometric invari-
ants in chemistry is that they are used in measuring packing.61
For instance, packing efficiency of a given atom is simply defined
as the ratio of the space it could minimally occupy to the space
that it actually does occupy.61 In a similar way as volume and
surface area are applied to define packing efficiency in different
ways, the new geometric invariant we have introduced here can
be used for similar purposes giving it a wide spectrum of applica-
tions in different areas of research.
Acknowledgments
The author thanks Ms. Y. Gutierrez for assistance in developing a
computer program to calculate the point scattering. Prof. D. J.
Klein and Dr. J. A. Rodrıguez-Velazquez are also acknowledged
for useful comments and clarifications.
References
1. Mumford, D.; Forgarty, J.; Kirwan, F. Geometric Invariant Theory;
Springer: New York, 1994.
2. Thompson, D. On Growth and Form; Cambridge University Press:
Cambridge, 1961.
3. Tarnai, T. Struct Topol 1984, 9, 39.
4. Hofinger, S.; Zerbetto, F. Chem Soc Rev 2005, 34, 1012.
5. Liang, J.; Dill, K. A. Biophys J 2001, 81, 751.
6. Fleming, P. J.; Richards, F. M. J Mol Biol 2000, 299, 487.
7. Tsai, J.; Taylor, R.; Chothia, C.; Gerstein, M. J Mol Biol 1999, 290, 253.
8. Pintar, A.; Carugo, O.; Pongor, S. Biophys J 2003, 84, 2553.
9. Knupp, C.; Squire, J. M. Adv Protein Chem 2005, 70, 375.
10. Niu, S. L.; Mitchell, D. C. Biophys J 2005, 89, 1833.
11. Tsukita, S.; Furuse, M. Trends Cell Biol 1999, 149, 268.
12. Tsukita, S.; Furuse, M. J Cell Biol 2000, 149, 13.
13. Yang, B.; Brown, D.; Verkman, A. S. J Biol Chem 1996, 271, 4577.
14. Odijk, T. Biophys J 1998, 75, 1223.
15. Maritan, A.; Mincheletti, C.; Trovato, A.; Banavar, R. B. Nature
2000, 406, 287.
16. Stasiak, A.; Maddocks, J. H. Nature 2000, 406, 251.
17. Banavar, J. R.; Maritan, A. Rev Mod Phys 2003, 75, 23.
18. Manoharan, V. N.; Elsesser, M. T.; Pine, D. J. Science 2003, 301, 483.
19. Yi, G.-R.; Manoharan, V. N.; Michel, E.; Elsesser, M. T.; Yang, S.-M.;
Pine, D. J. Adv Mater 2004, 16, 1204.
20. Liang, J.; Edelsbrunner, H.; Fu, P.; Sudhakar, P. V.; Subramanian, S.
Proteins: Struct Funct Genet 1998, 33, 1.
21. Voss, N. R.; Gerstein, M. J Mol Biol 2005, 346, 477.
22. Kuszewski, J.; Gronenborn, A. M. M.; Clore, G. M. J Am Chem Soc
1999, 121, 2337.
23. Baker, B. M.; Murphy, K. P. Methods Enzymol 1998, 295, 294.
24. Luque, I.; Freire, E. Methods Enzymol 1998, 295, 100.
25. Wales, D. J.; Scheraga, H. A. Science 1999, 285, 1368.
26. Conway, J. H. M.; Sloane, N. J. A. Discrete Comput Geom 1995, 13,
282.
27. Conway, J. H.; Sloane, N. J. A. Sphere Packing, Lattices and Groups,
3rd ed.; Springer: New York, 1999.
28. Chow, T. Y. Combinatorica 1995, 15, 151.
29. Graham, R. L.; Sloane, N. J. A. Discrete Comput Geom 1990, 5, 1.
30. Sloane, N. J. A.; Hardin, R. H.; Duff, T. D. S.; Conway, J. H. Discrete
Comput Geom 1995, 14, 237.
31. Richards, F. M. Ann Rev Biophys Bioeng 1977, 6, 151.
32. Lee, B.; Richards, F. M. J Mol Biol 1971, 55, 379.
33. Connolly, T. J Appl Cryst 1985, 16, 548.
34. Campopiano, C. N.; Blazer, B. G. IEEE Trans Commun 1962, 10, 90.
35. Foschini, G. J.; Gitlin, R. D.; Weinstein, S. B. IEEE Trans Commun
1974, 22, 28.
36. Flory, P. J. Statistical Mechanics of Chain Molecules. Interscience:
New York, 1969.
37. Hotta, S.; Inoue, K.; Urahama, K. Electron Commun Jpn 2003, 86,
80.
38. Inoue, K.; Urahama, K. Pattern Recognit Lett 1999, 20, 699.
39. Ng, A. Y.; Jordan, M. I.; Weiss, Y. In Advances in Neural Informa-
tion Processing Systems; Dietterich, T. G.; Becker, S.; Ghahramami,
Z., Eds.; MIT Press: Cambridge, MA, 2002; Vol. 14.
40. Bogomolny, E.; Bohigas, O.; Schmit, C. J Phys A: Math Gen 2003,
36, 3595.
41. Horn, R. A.; Johnson, C. R. Matrix Analysis; Cambridge University
Press: Cambridge, 1990.
42. Bodor, N.; Gabanyi, Z.; Wong, C. J Am Chem Soc 1989, 111,
3783.
43. Gavezotti, A. J Am Chem Soc 1983, 100, 5220.
44. Patterson, A. L. Rev Sci Instrum 1941, 12, 206.
45. Kroto, H. W.; Walton, A. R. M. The Fullerenes: New Horizons for
the Chemistry, Physics and Astrophysics of Carbon; Cambridge Uni-
versity Press: Cambridge, 1993.
46. Cioslowski, J. Electronic Structure Calculations on Fullerenes and
Their Derivatives; Oxford University Press: Oxford, 1995.
47. Vaitheeswaran, S.; Yin, H.; Rasaiah, J. C.; Hummer, G. Proc Natl
Acad Sci USA 2004, 101, 17002.
48. Ugarte, D. Nature 1992, 359, 707.
49. Shinohara, H. Prog Phys 2000, 63, 843.
50. Richman, D. D.; Whitley, R. J.; Hayden, F. G. Clinical Virology, 2nd
ed.; ASM Press: Washington, DC, 2002.
51. Chandrasekar, V.; Johnson, J. E. Structure 1998, 6, 157.
52. Torrens, F. Int J Mol Sci 2001, 2, 72.
53. Shen, M.; Davis, F. P.; Sali, A. Chem Phys Lett 2005, 405, 224.
54. Raines, R. T. Chem Rev 1998, 98, 1045.
55. Richards, F. M.; Wyckoff, H. W. Enzymes 1971, 4, 647.
56. Hearn, R. P.; Richards, F. M.; Sturtevant, J. M.; Watt, G. D. Biochem-
istry 1971, 10, 806.
57. Ratnaparkhi, G. S.; Varadarajan, R. Biochemistry 2000, 39, 12365.
58. Tilton, R. F.; Dewan, J. C.; Petsko, G. A. Biochemistry 1992, 31,
2469.
59. Estrada, E. Proteins: Struct Funct Bioinformat 2004, 54, 727.
60. Estrada, E. J Chem Inf Comput Sci 2004, 44, 1238.
61. Gerstein, M.; Richards, F. M. In the International Tables for Crystal-
lography; Rossman, M.; Arnold, A., Eds.; Kluwer: Dordrecht, 2001;
Vol. F, Ch. 22.1.1, p. 531.
777New Geometric Invariant with Applications From Clusters to Biomolecules
Journal of Computational Chemistry DOI 10.1002/jcc