potencialização do efeito do quimioterápico mostarda...
TRANSCRIPT

Tatiana Amorim Muniz de Alencar
Potencialização do efeito do quimioterápico
mostarda nitrogenada bifuncional pelo quelante
de ferro 2,2’dipiridil em Escherichia coli
Tese submetida à Universidade Federal do Rio de Janeiro visando
à obtenção do grau de Doutor em Ciências Biológicas (Biofísica).
Universidade Federal do Rio de Janeiro Centro de Ciências da Saúde Instituto de Biofísica Carlos Chagas Filho 2007

Livros Grátis
http://www.livrosgratis.com.br
Milhares de livros grátis para download.

ii
FICHA CATALOGRÁFICA
de Alencar, Tatiana Amorim Muniz
Potencialização do efeito do quimioterápico mostarda nitrogenada bifuncional
pelo quelante de ferro 2,2’ dipiridil em Escherichia coli. Rio de Janeiro, UFRJ, Instituto
de Biofísica Carlos Chagas Filho, Programa de Biologia Molecular e Estrutural, 2007.
XVI, 183 f.
Tese: Doutor em Ciências Biológicas (Biofísica)
1. Escherichia coli
2. Mostarda nitrogenada bifuncional
3. 2,2’ dipiridil
4. Ativação metabólica
I. Universidade Federal do Rio de Janeiro
II. Título

iii
O presente trabalho foi desenvolvido no Laboratório de Radiobiologia Molecular
do Instituto de Biofísica Carlos Chagas Filho da Universidade Federal do Rio de
Janeiro, sob orientação da Profª Drª Claudia de Alencar Santos Lage e co-orientação
do Prof. Dr. Alvaro Augusto da Costa Leitão, com auxílios concedidos pelas seguintes
agências de fomento: CNPq (Conselho Nacional de Desenvolvimento Científico e
Tecnológico), CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível
Superior) e FAPERJ (Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado
do Rio de Janeiro).

iv
O mundo está nas mãos daqueles que têm coragem de sonhar, e correr o risco de
viver seus sonhos.
(Paulo Coelho)

v
AGRADECIMENTOS
Em primeiro lugar a Deus, por se fazer presente em todos os momentos da
minha vida.
A minha família, esta tese é, mais uma vez, dedicada a vocês.
Aos meus pais por tudo que fizeram e ainda fazem por mim. Pela confiança, pelo
incentivo, pelo amor e principalmente por acreditar sempre que eu chegaria lá, muitas
vezes até mais do que eu mesma. Muito obrigada por tudo.
Ao meu irmão Tadeu que, da sua maneira, sempre se preocupou se tudo estava
dando certo.
Ao meu querido Robson pelo seu amor durante todos estes anos, por acreditar
nas minhas idéias e me apoiar de uma maneira que só ele saberia fazer.
A minha filha Lara, um grande presente de Deus, por tudo que sua presença
significa na minha vida, pelo seu sorriso e pelo seu amor. Obrigada por você existir.

vi
À amiga e orientadora Claudia Lage por tudo que me ensinou durante esses 11
anos de convivência. Por todo o incentivo, por acreditar nas minhas idéias, fazendo
com que elas parecessem a mais incríveis do mundo, tudo isso me ensinou a cada dia
gostar mais do meu trabalho.
Ao Alvaro não só pela orientação, pela acolhida no laboratório, pelos
questionamentos, mas principalmente por sempre nos mostrar que é preciso estudar
muito para se chegar a algum lugar e que nada cai do céu.
A minha ex-estagiária Tula Celeste, pela dedicação, pelas discussões, por sua
importância neste trabalho e por me dar a oportunidade de passar os meus
conhecimentos.
Aos meus amigos do Laboratório: Roberto, Ivan, Marcus, Mariana, Larissa,
Luciana, Viviana, Marcelo, por fazerem do nosso laboratório um lugar agradável de se
trabalhar. Obrigada por todos os momentos de discussão de trabalho, por todos os
momentos de descontração. Nunca esquecerei das nossas risadas na hora do almoço.
Aos meus amigos de Doutorado: Deise, Leonardo, Adriana e Silvia. Muito
obrigada por todos os momentos que passamos juntos, até aqueles mais difíceis.
Obrigada pelas discussões de trabalho, pelas opiniões, pelo que aprendemos uns com
os outros. Espero que todos nós tenhamos sucesso nas nossas novas jornadas.
Meninos, valeu.
A todos que não fazem parte oficialmente do laboratório, mas estão sempre por
perto dando uma força.
Ao amigo Leonardo por todos esses anos de convivência, por todas as
experiências que passamos juntos e nos fizeram crescer. Com certeza sua presença
foi muito importante durante todos esses anos.

vii
À comadre Adriana por me ouvir, me incentivar sempre e me mostrar o lado
prático da vida. Obrigada pela nossa amizade.
À amiga Rita por acreditar em mim e me incentivar em todos os momentos.
Muito obrigada pela sua admiração.
À amiga Janine pela sua experiência, por torcer por mim e estar sempre disposta
a me ajudar.
À amiga Carmem Adília pelo incentivo
Aos meus afilhados, Clara, Matheus, Igor e Felipe. Todos vocês são verdadeiros
presentes para mim.
A toda minha família e amigos, que fazem parte da minha grande família, que
sempre acreditaram em mim e me deram muita força para que eu chegasse até aqui.
Aos meus padrinhos, Sérgio e Sônia, por poder contar com eles sempre, e à Tia
Belinha pelo incentivo.
A minha quase cunhada Carol, pela sua admiração
À Mildred, por ser minha grande amiga, vibrar com todas as minhas conquistas,
estar sempre ao meu lado, fazendo parte dos momentos mais importantes da minha
vida. Ogrigada por tudo.
Às amigas Rosane e Rose por sempre torcerem por mim.
A todas as pessoas que mesmo indiretamente contribuíram para o
desenvolvimento dessa tese.

viii
ABREVIATURAS, SÍMBOLOS, UNIDADES E SIGLAS
% - por cento
µg – micrograma
µL – microlitro
ATP – adenosina trifosfato
BER – reparo por excisão de bases – base excision repair
B. subtilis – Bacillus subtilis
cDNA – DNA complementar
Ct – ciclo threshold
Da – Daltons
DEPC - dietilpirocarbonato
Dip – 2,2’dipiridil
DL – dose letal
DNA – ácido desoxirribonucléico
dNTPs – desoxinucleotídeos trifosfato
EAO – espédies ativas de oxigênio
E. coli – Escherichia coli
g – grama
g – aceleração da gravidade
GTP – guanosina trifosfato
HN2 – mostarda nitrogenada bifuncional
HN1 – mostarda nitrogenada monofuncional
H2O – água

ix
H2O2 – peróxido de hidrogênio
kDa – quilodaltons
L - litro
M – molar
mL – mililitro
mM – milimolar
MOPP – mecloretamine, oncovin, procarbazine e prednizone
mRNA – RNA mensageiro
NER – reparo por excisão de nucleotídeos – nucleotide excision repair
PH – potencial hidrogeniônico
PCR – reação em cadeia da polimerase
ºC – graus celsius
RNA – ácido ribonucléico
RT-PCR – reação em cadeia da polimerase em tempo real
rpm – rotações por minuto
Sítio AP – sítio apurínico ou apirimidínico
Tm – temperatura onde 50% dos produtos encontram-se na forma de dupla fita
UV – ultravioleta
UV-C – ultravioleta curto

x
RESUMO
Agentes alquilantes bifuncionais são utilizados em quimioterapia para induzir
morte de células tumorais através do bloqueio da replicação do DNA. Mostardas
nitrogenadas são normalmente utilizadas como agentes quimioterapêuticos, uma vez
que se ligam de maneira mono e bifuncional em guaninas no DNA. A mostarda
nitrogenada HN1 é considerada um análogo monofuncional da mostarda nitrogenada
bifuncional - HN2 (mecloretamine). Cepas mutantes de Escherichia coli K12,
deficientes em reparo por excisão de nucleotídeos (NER) ou reparo por excisão de
bases (BER), foram submetidas a concentrações crescentes de HN2 e HN1, e os
resultados revelam que os danos induzidos por cada agente requer diferentes
caminhos de reparo de DNA. Os danos induzidos por HN2 demandam a atividade do
reparo NER, com um menor requerimento do reparo BER, enquanto os danos
induzidos por HN1dependem da ação de BER, sem nenhum requerimento das funções
NER. HN2 e HN1 também diferem em relação à ativação metabólica intracelular. HN2
tem seu efeito potencializado na presença do quelante de Fe2+ - 2,2’dipiridil,
provavelmente pelo aumento da concentração de radical superóxido, promovido por
este quelante dentro da célula, que parece tornar esta mostarda ainda mais reativa,
efeito que desaparece na presença do captador de radicais livres tiouréia. Entretanto
HN1 não tem seu efeito alterado na presença de 2,2’dipiridil. Ambas as mostardas
precisam da temperatura de 37ºC para se tornarem letais para as células e não são
influenciadas pela presença do quelante de Fe3+ - desferroxiamina. Acreditamos que
HN2 e HN1 induzem lesões diferentes na célula, uma vez que requerem mecanismos
de reparo de DNA diferentes para correção de suas lesões e também se comportam
diferentemente em relação a uma possível ativação metabólica intracelular.

xi
ABSTRACT
Bifunctional alkylating agents are used in tumor chemotherapy to induce the
death of malignant cells through blockage of DNA replication. Nitrogen mustards are
commonly used chemotherapeutic agents that can bind mono- or bifunctionally to
guanines in DNA. Mustard HN1 is considered a monofunctional analog of bifunctional
mustard HN2 (mechlorethamine). Escherichia coli K12 mutant strains deficient in
nucleotide excision repair (NER) or base excision repair (BER) were submitted to
increasing concentrations of HN2 or HN1, and the results revealed that damage
induced by each chemical demands different DNA repair pathways. Damage induced
by HN2 demands the activity of NER with a minor requirement of the BER pathway,
while HN1 damage repair depends on BER action, without any requirement of NER
function. HN2 and HN1 also differ in their intracellular metabolic activation. HN2
presents a more pronunciated effect in presence of 2,2’dipyridyl probably due to the
increase in superoxide concentration. It seems to turn HN2 more reactive, however
this effect can not be found in presence of thiourea. On the other hand, HN1presents
no altered effect in presence of 2,2’dipyridyl. Both nitrogen mustards are lethal to cells
at 37ºC and do not present any influence from desferroxiamine. Taken together, our
data suggest that HN1 and HN2 seem to induce different types of damage, since their
repair depend on distinct pathways in E. coli and they can be metabolically activated
by different ways.

xii
ÍNDICE INTRODUÇÃO
1. Considerações Gerais ................................................................................. 1
2. Câncer e quimioterapia ............................................................................... 2
2.1 – Agentes quimioterápicos .................................................................... 3
2.1.1 – Algumas classes de agentes quimioterápicos............................. 3
3. Metabolismo de ferro................................................................................... 5
4. Agentes alquilantes..................................................................................... 8
4.1 – Agentes formadores de crosslinks...................................................... 10
5. Mostarda nitrogenada bifuncional – HN2.................................................... 12
5.1 – O uso da mostarda nitrogenada bifuncional em quimioterapia............. 20
6. Mostarda nitrogenada monofuncional – HN1.............................................. 23
7. Ativação metabólica das mostardas............................................................ 26
8. O radical superóxido.................................................................................... 27
9. Mecanismos de reparo de DNA em E. coli, envolvidos neste trabalho...... 27
9.1 –Reparo por excisão de bases (BER)................................................... 28
9.2 – Reparo por excisão de nucleotídeos(NER)....................................... 31
9.3 – Reparo de DNA contendo adutos do tipo crosslinks......................... 35
9.4 – Resposta SOS.................................................................................. 38
10. Defesas contra estresse oxidativo........................................................... 41

xiii
........ 10.1 - Regulon SoxRS............................................................................ 41
........ 10.2 - Regulon OxyR………………………………………………………… 44
CARACTERIZAÇÃO DO PROBLEMA 45
OBJETIVOS 46
MATERIAL E MÉTODOS
1.Cepas bacterianas...................................................................................... 47
2. Meios de cultura e tampão........................................................................ 48
2.1 – Meios líquidos.................................................................................. 48
2.2 – Meios sólidos.................................................................................... 49
3. Mostardas nitrogenadas............................................................................ 50
4. 2,2’dipiridil utilizado (quelante de Fe2+)..................................................... 50
5. Desferroxiamina utilizada (quelante de Fe3+)............................................ 50
6. Captador de radicais livres – tiouréia........................................................ 50
7. Manutenção de cepas bacterianas............................................................ . 50
8. Obtenção das culturas bacterianas para os experimentos ...................... 51
9. Sobrevivência bacteriana às mostardas nitrogenadas bifuncional (HN2) e
monofuncional (HN1)...................................................................................
51
10. Sobrevivência bacteriana às mostardas nitrogenadas com pré-tratamento
com quelantes de Fe2+ (2,2’ dipiridil), Fe3+ (desferroxiamina) e captador de
radicais livres (tiouréia)...................................................................................
52
11. Sobrevivência da cepa selvagem a mostarda nitrogenada bifuncional 52

xiv
(HN2) e 2,2’ dipiridil em diferentes condições..............................................
12. Sobrevivência a mostarda nitrogenada bifuncional (HN2) com pré-
tratamento com tiouréia em diferentes condições.........................................
53
13. Sobrevivência a mostarda nitrogenada com pré-tratamento com 2,2’
dipiridil e desferroxiamina...............................................................................
53
14. Sobrevivência bacteriana às mostardas nitrogenadas em diferentes
temperaturas..................................................................................................
54
15. Sobrevivência e mutagênese bacteriana aos tratamentos com HN2 e pré-
tratamento com 2,2’ dipiridil e posterior tratamento com HN2.........................
55
RESULTADOS
1. Sobrevivência celular - Estudos realizados com HN2................................. 58
1.1 – Avaliação da sobrevivência das cepas deficientes em genes
BER.............................................................................................................
58
1.2 – Avaliação da sobrevivência das cepas deficientes em genes NER
SOS............................................................................................................
60
1.3 – Avaliação da sobrevivência das cepas deficientes no gene
recA............................................................................................................
60
1.4 - Avaliação da sobrevivência da cepa deficiente no gene oxyR......... 63
2. Sobrevivência celular - Estudos realizados com HN1.............................. 63
2.1 – Avaliação da sobrevivência das cepas deficientes no gene
recA............................................................................................................
63
2.2 – Avaliação da sobrevivência da cepa deficiente no gene oxyR.......... 66
3. Avaliação da aquisição de resistência ao antibiótico rifampicina –
Mutagênese Rifampicina...............................................................................
66

xv
4. Estudos realizados com HN2.................................................................... 70
4.1 – Utilização do quelante de Fe2+ - 2,2’ dipiridil juntamente a HN2 em
diferentes condições.....................................................................................
70
4.2 – A influência do captador de radicais livres tiouréia nas lesões
causadas por HN2....................................................................................
82
4.3 - Avaliação da aquisição de resistência ao antibiótico rifampicina –
Mutagênese Rifampicina................................................................................
87
4.4 – A influência do quelante de Fe3+ - desferroxiamina nas lesões
causadas por HN2..........................................................................................
89
4.5 – Utilização dos quelantes de Fe2+ e Fe3+, 2,2’ dipiridil e
desferroxiamina..............................................................................................
91
5. Estudos realizados com HN1..................................................................... 91
5.1 – A influência do quelante de Fe2+ - 2,2’dipiridil nas lesões causadas
por HN1......................................................................................................
91
5.2 - A influência do captador de radicais livres tiouréia nas lesões
causadas por HN1..........................................................................................
94
5.3 - A influência do quelante de Fe3+ - desferroxiamina nas lesões
causadas por HN1..........................................................................................
95
6 - Influência da temperatura – HN2............................................................... 98
7 – Influência da temperatura - HN1.............................................................. 103
DISCUSSÃO E CONCLUSÃO 105
HIPÓTESE DA ATIVAÇÃO DE HN2, VIA RADICAL SUPERÓXIDO 121
HIPÓTESE DA GERAÇÃO DO COMPOSTO DIP – HN2 123
PERSPECTIVAS 125

xvi
REFERÊNCIAS BIBLIOGRÁFICAS 127
ANEXO I
ANEXO II

INTRODUÇÃO
1. Considerações Gerais:
Os efeitos deletérios do meio ambiente, através da ação de produtos
químicos naturais ou artificiais, ou mesmo daqueles provenientes do metabolismo
celular, podem gerar conseqüências importantes no que diz respeito à integridade
de qualquer organismo. Lesões ocorridas na molécula de DNA estão intimamente
ligadas à morte ou transformação celular, uma vez que comprometem suas
propriedades codificadoras e podem induzir mutações. Ao longo do tempo, as
células podem acumular mutações, silenciando ou acionando genes específicos, e
a maioria dos cânceres está associada com a inativação de genes supressores de
tumor e ativação de oncogenes.
Seria praticamente impossível a sobrevivência de qualquer organismo, se
lesões ocorridas na molécula de DNA não fossem reparadas. Mecanismos
capazes de eliminar lesões ocorridas no DNA, permitem a sobrevivência celular, e
com isso a manutenção das espécies, através do DNA preservado e transmitido
corretamente de geração a geração. A não reparação dessas lesões pode levar,
não somente a mutagênese e ativação de oncogenes, como também a doenças
degenerativas, envelhecimento e outras conseqüências. Por esta razão se torna
fundamental estudar os mecanismos pelos quais as células e os organismos
reparam ou evitam as lesões causadas por tais agentes físicos e químicos,
mantendo assim as duas propriedades biológicas fundamentais do DNA, que são:
a auto-replicação e a preservação da informação genética.
É importante ressaltar que, em casos de doenças, como o câncer, por
exemplo, as lesões causadas no DNA, por determinadas substâncias, como os
1

quimioterápicos, são vistas de maneira positiva, uma vez que levam à morte das
celulas tumorais. Algumas células tumorais são capazes de reconhecer e eliminar
as lesões causadas por estas substâncias, e com isso tornarem-se resistentes ao
tratamento. Por isso se faz importante estudar os mecanismos pelos quais estas
moléculas (quimioterápicos) se ligam ao DNA, sua metabolização intracelular e o
mecanismo responsável pela reparação dessas lesões.
2. Câncer e Quimioterapia:
O câncer é basicamente uma doença caracterizada por um desvio dos
mecanismos de controle que dirigem a proliferação e a diferenciação celular. As
células que sofreram transformação neoplásica proliferam excessivamente e
formam tumores locais que podem comprimir ou invadir estruturas normais
adjacentes (Oliveira e Alves, 2002).
Células tumorais possuem instabilidade genética característica de distúrbios
(desordens) em vários mecanismos de reparo de DNA. O desenvolvimento de um
câncer geralmente envolve muitos passos cada um deles governado por múltiplos
fatores, alguns dependentes da constituição genética do indivíduo, outros
dependentes do ambiente ou do modo de vida. Estudos indicam que uma única
mutação pode ser suficiente para causar um câncer, por exemplo, no caso de um
gene gatekeeper (Friedberg et al., 2006). A progressão tumoral também parece
envolver sucessivos ciclos de mutação e seleção clonal (Alberts et al., 1994).
A quimioterapia é o tratamento mais efetivo contra tumores metastáticos.
(Gottesman, 2002). Alguns estudos têm enfatizado sistemas de ativação de pró-
drogas, baseados em enzimas do citocromo P-450. Esta abordagem é
extremamente importante no tratamento de tumores, uma vez que algumas drogas
2

anticâncer podem ser ativadas, dessa forma então, passariam de um estado de
pró-droga a droga, uma boa abordagem seletiva para atingir células tumorais
(Jounaidi, 2002).
Entretanto, a habilidade de células tumorais se tornarem resistentes a
diferentes drogas, até mesmo àquelas que nunca tenham sido expostas, através
de diversas exposições a um determinado agente, é caracterizado como
fenômeno de múltipla resistência a drogas (multidrug resistance - MDR) e,
significa um grande impedimento ao sucesso da quimioterapia (Gottesman, 2002).
Há aproximadamente três décadas existem estudos em relação a este
fenômeno, na tentativa de se entender o mecanismo que as células utilizam para
se tornarem resistentes às drogas e com isso também existem estudos para
seleção de novas drogas. O fenótipo MDR de uma célula parece estar associado a
modificações no cariótipo celular, conseqüentemente amplificando seqüências de
genes cujos produtos são membros da superfamília de transportadores ABC, que
previnem o acúmulo intracelular de determinadas drogas, através do efluxo das
mesmas (Alberts et al., 1994).
2.1 - Agentes quimioterápicos
Os quimioterápicos têm como alvo processos necessários ao crescimento e
divisão celular, assim como DNA, RNA, síntese protéica e maquinaria mitótica.
2.1.1 - Algumas classes de agentes quimioterápicos
• Antimetabólitos anti-tumorais - agem através do bloqueio da síntese de DNA,
RNA e precursores de proteínas;
• Antibióticos anti-tumorais - agem por intercalação no DNA, levando à
3

mutagênese, inibição da transcrição e quebras em cadeias de DNA;
• Alcalóides de Vinca - agem inibindo o fuso mitótico e conseqüentemente a
mitose;
• Agentes alquilantes - são compostos eletrofílicos que têm grande afinidade por
algumas seqüências no DNA, podendo se ligar de maneira bi ou
monofuncional, gerando lesões em duas bases nitrogenadas (biadutos intra-
cadeias e crosslinks – ligações covalentes entre cadeias ) ou somente em uma
base nitrogenada do DNA (monoaduto) ou impedindo a replicação do DNA.
São compostos que exibem seletividade para se ligar a determinadas
seqüências de DNA, reagindo de maneira covalente, e por este motivo
continuam sendo importantes alvos para o desenvolvimento de novos agentes
medicinais (Purnell et al., 2006);
• Quelantes de ferro – o desenvolvimento dos quelantes de ferro como agentes
terapêuticos tem focado primariamente seu uso crônico em tratamento
secundário de sobrecarga de ferro. Entretanto, quelantes de ferro podem ser
eficientes como agentes antitumorais, ou por depleção de ferro no tumor ou
por causar estresse oxidativo seletivo no tumor devido a perturbações redox
no seu ambiente (Buss et al., 2003). Alternativamente ou adicionalmente, eles
podem formar complexos com metais, e estes podem ter potencial redox,
causando estresse oxidativo, via produção de espécies ativas de oxigênio
(EAO), danificando alvos intracelulares críticos, e, deste modo, produzindo
uma resposta citotóxica (Buss et al., 2004). Os quelantes de ferro têm sido
testados pela sua atividade antitumoral em experimentos com culturas de
células, modelos animais e triagens clínicas em humanos e podem ser usados
sozinhos ou em conjunto (sinergismo) com outras terapias anti-câncer (Buss et
4

al., 2003). .
Sobre o metabolismo de ferro, este encontra-se alterado em tumores e a
maioria dos estudos tem identificado perturbações consistentes com aumento no
requerimento de ferro para rápida proliferação das células tumorais, portanto, é
evidente que há uma complexidade na relação entre este metal e o câncer. A
dependência aumentada das células tumorais em relação ao ferro sugere que a
depleção deste pode representar uma estratégia para limitar o crescimento
tumoral e é neste momento que os quelantes de ferro podem ser utilizados como
quimioterápicos antineoplásicos (Buss et al., 2003).
Estudos mostram que a expressão de duas moléculas vitais para a célula,
p21 e ciclina D são reguladas por ferro, então o fato dos quelantes de ferro
influenciarem na expressão destas proteínas é importante para o entendimento de
como drogas que se ligam ao ferro inibem o crescimento de células tumorais, além
disso o ferro também regula a expressão do gene NDRG1, responsável pela
diferenciação (processo pelo qual as células se especializam em certa função), e
parece estar ligado ao processo de metástase e crescimento de muitas classes de
células tumorais. Estes estudos facilitam o entendimento do papel do ferro na
carcinogênese e com isso, podem resultar, na geração de quelantes com maior
seletividade para as células tumorais alvo.
Na figura 1 estão os mecanismos básicos de atuação dos quimioterápicos.
3. Metabolismo de ferro
O ferro exibe propriedade iniciadora de tumores, portanto, dietas ricas em ferro
estão correlacionadas com aumento do aparecimento destes tumores, além de
participar da biossíntese de diversas moléculas celulares e apresentar uma
5

ActinomicinaBleomicinaDaunorrubicina Etoposídeo Doxorrubicina Teniposídeo
Figura 1: Mecanismo de atuação dos quimioterápicos (Koland, 2003).
Quebrasde
cadeia
Idorrubicina
Intercalação
Daunorrubicina Doxorrubicina Idorrubicina
Geração de radicais livres
Cisplatina Ciclofosfamida Dacarbazine MECLORETAMINE Melfalan Nitrosoureias Procarbazine
Alquilação Transcrição
MescaptopurinaTioguanina
RNA
Tradução
Asparaginase Protéína
TubulinaPaclitaxelVinblastina Vincristina
Droga que inibe este passo
Droga incorporada na molécula
PurinasFH2
FH4
CMP
dCMP
DNAReplicação
TMP
Fluorouracil
Fluorouracil
Metotrexato
Citarabina
Síntese Caminhode novo de regeneração
6

correlação com o estresse oxidativo (Buss et al., 2004) por isso o metabolismo
de ferro é uma função tão importante para a célula, bem como os meios que a
célula utiliza para manter os níveis deste metal.
O metabolismo de ferro em Escherichia coli (E. coli) está envolvido com a
resposta anti-oxidante. Uma proteína denominada Fur, é o repressor global da
regulação de ferro e controla cerca de 30 genes implicados na regulação deste
metal no ambiente (Braun, 1997, Braun et al, 1998). Esta proteína foi
originalmente descrita em E. coli e é um repressor sensível a ferro que controla a
expressão de genes que codificam para biossíntese de sideróforos (compostos
queladores de Fe3+) e transporte de ferro (Fe2+ é diretamente importado) e ativa a
expressão de muitos genes de maneira direta ou indireta (Lee e Helman, 2007).
Os genes regulados por Fur são desreprimidos em baixas concentrações de íons
ferro e reprimidos em altas concentrações deste íon (Hantke, 1981, Hantke, 2002).
As respostas reguladoras de E. coli, para estresse oxidativo, Oxy R e Sox RS,
ativam a expressão de Fur, sugerindo que o controle do metabolismo de ferro, em
E. coli, é uma parte integrante da resposta antioxidante (Zheng et al, 1999).
A regulação estrita na assimilação e estocagem de ferro em células
procarióticas e eucarióticas, é considerada uma maneira de prevenir a célula do
acúmulo de ferro livre, diminuindo assim sua toxicidade (Touati et al., 1995).
Em células eucarióticas, mecanismos complexos estão envolvidos na
aquisição, transporte e armazenamento deste metal. O maior mecanismo pelo
qual as células humanas adquirem ferro é via endocitose mediada pelo receptor
de transferrina (Touati et al., 1995).
7

4. Agentes Alquilantes:
Esta é a classe mais antiga de drogas anti-câncer (Tortorice, 2002). A
maioria desses agentes originou-se do desenvolvimento racional de drogas, a
partir do conhecimento das propriedades fisiológicas das células tumorais. É a
classe de compostos mais amplamente utilizada, sendo um dos principais
componentes nos regimes combinados de quimioterapia para tumores sólidos
disseminados. Dentre as drogas pertencentes a essa classe, podemos citar: as
mostardas (mecloretamine: alquilante bifuncional), clorambucil e melfalan, a
ciclofosfamida e isofosfamida, o busulfan e as nitrosuréias.
Os agentes alquilantes são compostos eletrofílicos que têm afinidade por
centros nucleofílicos em macromoléculas orgânicas (Friedberg et al., 2006). A
transferência de grupamentos alquil para biomoléculas ocorre a partir de
compostos intermediários reativos, aptos a fazer esta transferência (Merck, 2003).
Dentre estes agentes está uma grande variedade de compostos químicos,
podendo ser alguns mutagênicos e carcinogênicos, devido à sua habilidade em
reagir com estruturas instáveis em anel (Singer e Grunberger, 1983). Podem ser
monofuncionais (Friedberg et al., 2006), ou possuir potencialidade secundária,
levando à formação de crosslinks ou biadutos intra-cadeias de DNA (Singer e
Grunberger, 1983). Muitas dessas lesões, provocadas por alquilação, são
reparadas através da remoção direta do aduto (Mishina et al., 2006).
Alguns agentes alquilantes não só são capazes de se ligar a determinadas
seqüências de DNA, mas também se ligam ao N7 de guaninas, e a centros
nucleofílicos de proteínas, como os átomos de enxofre, pois ambos os centros
8

estão aptos a doar elétrons, permitindo com isso a ligação destes agentes a tais
moléculas. Entretanto existem numerosos sítios específicos com potencial reativo
para sofrer alquilação nas quatro bases nitrogenadas (adenina, guanina, citosina e
timina), porém com menor freqüência (Tortorice, 2002). Os sítios de reação no
DNA para muitos agentes alquilantes monofuncionais são: N1, N3, N6 e N7de
adeninas, N1, N2, N3, N7 e O6 de guaninas; N3, N4 e O2 de citosinas e em N3, O2 e
O4 de timinas. Em geral, os nitrogênios do anel das bases são mais nucleofílicos
que os oxigênios, as posições N7 de guaninas e N3 de adeninas sendo as mais
reativas. A alquilação de oxigênios das ligações fosfodiéster resulta na formação
de fosfotriésteres (Friedberg et al., 2006).
A reatividade dos agentes alquilantes se dá em grupos químicos
particulares no DNA, que são dependentes da natureza deste agente, de forma
que agentes alquilantes mais fortes, são menos seletivos. Então, estudos foram
feitos para se tentar identificar quais os centros nucleofílicos que possuíam maior
reatividade com os agentes alquilantes. Por exemplo, o potencial eletrostático
negativo da posição N7 de guaninas é aumentado se esta guanina estiver
flanqueada por outros resíduos de guanina, gerando um centro nucleofílico capaz
de sofrer mais facilmente um ataque eletrofílico (Singer e Grunberger, 1983). A
reação de agentes alquilantes com sítios específicos no DNA pode ser governada
também por efeitos estéricos. Por exemplo, quando o DNA está na configuração
de hélice B normal (right handed), ambos os átomos O6 e N7 das guaninas são
mais acessíveis no maior sulco, enquanto as posições N3 das adeninas ficam
relativamente menos acessíveis no menor sulco (Friedberg et al., 2006). Estes
sítios que são sensíveis à alquilação são pontos de alta incidência de mutagênese
9

(Friedberg et al., 2006).
Deve ser reforçado que as modificações de base causadas pela alquilação
parecem enfraquecer as ligações N-glicosídicas e, com isso, o tratamento com
muitos agentes alquilantes leva à depurinação ou despirimidinação, gerando sitios
AP (sítios apurínicos ou apirimidínicos), além da aparição de sítios abásicos álcali-
lábeis, produzindo defeitos na estrutura normal da molécula de DNA (dupla
hélice), podendo tomar a forma de quebras nas fitas ou ligações impróprias entre
as fitas que, se reparadas pelos mecanismos de reparo de DNA disponíveis dentro
das células, desencadeiam morte celular por diferentes vias. O tratamento com
agentes alquilantes também pode induzir aberturas de anéis das bases
nitrogenadas (Tortorice, 2002).
A morte celular pode ocorrer quando a célula tentar replicar ou transcrever
o DNA alterado ou mesmo quando enzimas de reparo tentarem corrigir os danos e
acabarem gerando deleções ou correções indevidas, tais como sítios apurínicos,
abertura de anéis ou quebras nas fitas simples ou duplas de DNA. A parte não
alquilante da molécula é que confere as diferenças na farmacocinética, bio-
distribuição e toxicidade dessas drogas. Como o DNA é considerado um alvo não
saturável, é possível usar esses agentes em combinação e se conseguir um efeito
aditivo ou mesmo sinergístico da sua ação citotóxica (Oliveira e Alves, 2002).
Neste particular, os agentes formadores de crosslinks são apontados como os
mais eficazes, como visto a seguir.
4.1 - Agentes formadores de crosslinks:
Os agentes alquilantes bifuncionais podem reagir com dois diferentes
10

centros nucleofílicos no DNA. Se os dois sítios forem em cadeias polinucleotídicas
opostas, resulta em ligações cruzadas entre cadeias do DNA (crosslinks)
(Goldacre et al., 1949, citado em Kohn et al., 1966), e, nesse caso, temos os
exemplos como a cisplatina e mostardas nitrogenadas que se ligam às posições 7
do nitrogênio de guaninas opostas na cadeia de DNA. E outros alquilantes como a
mitomicina C, que se ligam preferencialmente em N2 de guaninas (Friedberg et al.,
2006).
Os crosslinks intercadeias de DNA são uma importante classe de danos
químicos, uma vez que eles impedem a separação das cadeias e podem levar a
um completo bloqueio da sua replicação e transcrição (Larminat et al., 1998). É
justamente por esta razão que um grande número de agentes como: ácido nitroso,
mitomicina C, mostarda nitrogenada e mostarda sulfurada, derivados de platina e
psoralenos fotoativados têm sido usados extensivamente em protocolos de
quimioterapia contra o câncer e uma série de outras doenças crônicas, pois as
lesões do tipo crosslinks, induzidas por estes agentes são classificadas como as
responsáveis pela morte destas células. A radiação UV de 254 nm e a radiação
ionizante também podem resultar na formação de crosslinks de DNA em escala
muito menor (Friedberg et al., 2006).
Um aspecto interessante dos crosslinks, é a presenca de varias vias
atuando para que ocorra sua reparação, tais como reparo por excisão de
nucleotídeos (NER), recombinação homóloga pós-replicativa e o reparo
translesão. É importante ressaltar que um único crosslink pode matar bactérias e
leveduras sensíveis e aproximadamente 20 crosslinks podem matar células de
mamíferos, se igualmente deficientes em reparo (Dronkert e Kanaar, 2001, Noll et
al., 2006).
11

A ligação covalente entre duas cadeias opostas de DNA pode ser detectada
por várias técnicas, como: retardamento da mobilidade em gel de eletroforese (gel
shift assay), eluição alcalina e procedimentos de marcação por densidade.
5. Mostarda nitrogenada bifuncional–HN2
As mostardas emergiram como agentes radiomiméticos, agindo nos
núcleos das células (Lawley e Phillips, 1995).
A mostarda nitrogenada bifuncional (HN2 – mecloretamine) foi o primeiro
agente alquilante introduzido na terapia anti-câncer. Sua ligação com o DNA se dá
através de um rearranjo molecular que esta mostarda sofre quando entra em
solução aquosa, havendo então a perda dos dois cloros das extremidades e se
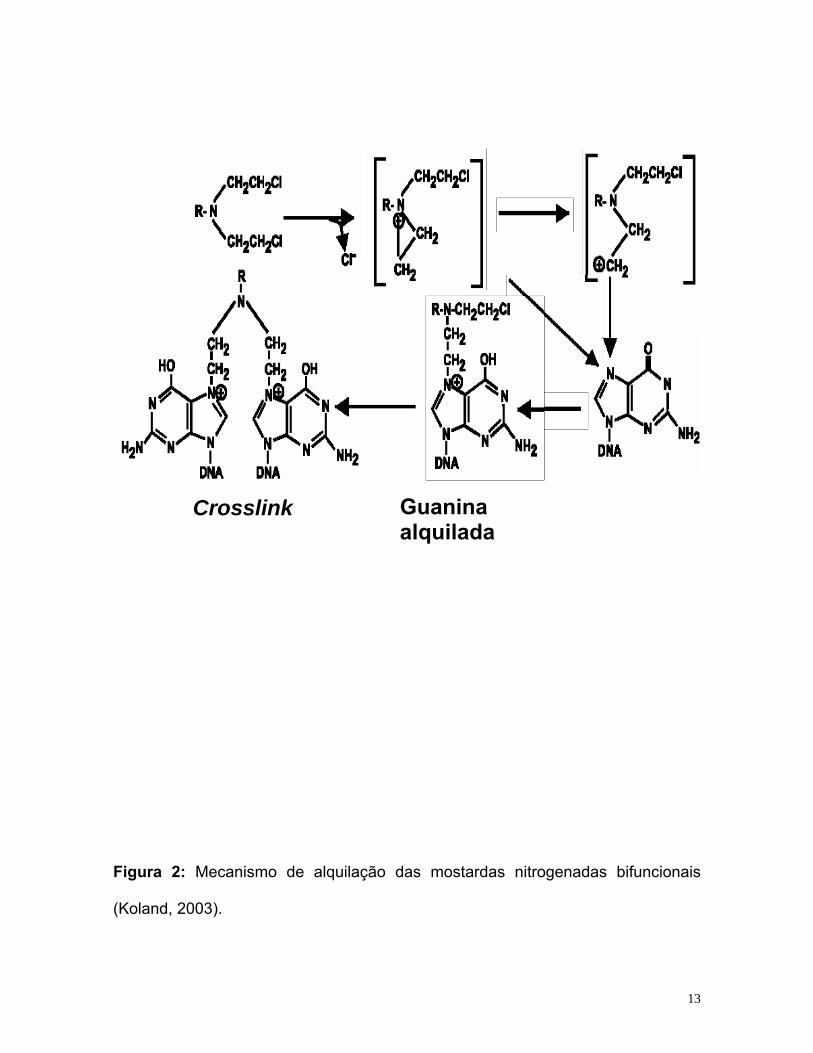
tornando desta forma, reativa (Tortorice, 2002). Na figura 2 encontra-se o modelo
de alquilação das mostardas nitrogenadas bifuncionais.
12
Guanina alquilada
Crosslink
Figura 2: Mecanismo de alquilação das mostardas nitrogenadas bifuncionais
(Koland, 2003).
13

Entre as duas grandes Guerras Mundiais, as mostardas nitrogenadas foram
estudadas como agentes de Guerra. A ação citotóxica destes agentes propiciou
estudos em linfosarcomas de ratos (Merck, 2003). Triagens clínicas em humanos
começaram a ser feitas em 1942, estava iniciada então a “Era dos quimioterápicos
antineoplásicos” (Merck, 2003).
A HN2 foi desenvolvida com o interesse em se estudar agentes químicos
com potencial terapêutico contra cânceres (Lawley e Phillips, 1995), e devido à
sua reatividade bifuncional, começou a ser usada como medicamento (Ruhland e
Brendel, 1978), sendo capaz de formar crosslinks, biadutos intra-cadeia de DNA –
utilizando sua capacidade bifuncional, e também monoadutos, quando se liga de
maneira monofuncional. Ainda não é muito bem conhecido qual destes danos é o
mais efetivo no bloqueio da replicação do DNA.
A HN2 é capaz de se ligar ao DNA em através de ligações
com N7 de guaninas em cadeias opostas. A mostarda nitrogenada bifuncional é
capaz de reagir com uma grande variedade de oligonucleotídeos in vitro, porém
seu alvo preferencial são as seqüências 5’-GXC-3’ (visto através de estudos de
modelagem molecular), em que X pode ser qualquer uma das quatro bases
nitrogenadas (Ojwang et al.,1989). Segundo Kohn et al. (1966) estes crosslinks
ocorrem nas seqüências de bases G (3’→5’) C, nesta orientação, uma vez que C
(3’→5’) G nesta orientação, não permite este tipo de crosslink. Estes crosslinks
gerados por HN2 ocorrem no maior sulco do DNA (Kohn et al., 1966) e resultam
na inibição completa da separação das fitas de DNA. Isso ocorre devido à
formação de uma ou mais destas lesões, que ocorrem provavelmente através da
5’ GAC 3’ 3´ CTG 5´
14

alquilação simultânea dos N7 de guaninas em DNA dupla fita (Kohn et al., 1965) e
geram leves distorções na hélice do DNA (Kohn et al., 1966). São (os crosslinks)
completamente instáveis, com uma meia-vida de aproximadamente 2 horas, e sua
toxicidade decresce comparando-se com outros crosslinks mais estáveis, gerados
por outros agentes (Dronkert e Kanaar, 2001). HN2 também é capaz de alquilar o
DNA em N3 de adeninas, que podem resultar na formação de monoadutos, além
dos crosslinks e biadutos intra-cadeias de DNA (Masta et al., 1994).
Os adutos entre N7 de guaninas, formados por mostardas nitrogenadas e
seus derivados, tornam as ligações glicosídicas termolábeis e geram sítios AP, por
este motivo os crosslinks parecem se desfazer (Ojwang et al.,1989).
A molécula da mostarda nitrogenada pode se estender completamente até
uma distância de aproximadamente 7,4Å, havendo deste modo uma distorção na
forma B do DNA, após a formação dos crosslinks acarretados por tal agente
(Ojwang et al.,1989).
No quadro abaixo estão indicadas as distâncias entre as posições N7 de
guaninas em cadeias opostas, no DNA em dupla fita, medidas em Å, feitas através
de modelagem molecular.
15

Quadro 1: Representação da distância entre N7 de guaninas de cadeias
opostas (Arnott et al., 1974, citado em Ojwang et al., 1989).
Seqüência de DNA Distância
5’-GC-3’
3’-CG-5’
7,7Å
5’-CG-3’
3’-GC-5’
8,7Å
5’-GXC-3’
3’-CXG-5’
8,6Å
5’-GXYC-3’
3’-CXYG-5’
10,5Å
A mostarda nitrogenada HN2 também é conhecida pela sua capacidade em
reduzir a eficiência de transformação de DNA (Zamenhof et al., 1956 e Lerman e
Tolmach, 1959, citados em Kohn e Green, 1966), e que tal eficiência de
transformação diminui, à medida em que se aumenta a concentração de HN2.
Estes trabalhos foram realizados em bactérias Bacillus subtilis, e os autores
concluíram que as moléculas de DNA que contêm poucos crosslinks, possuem
ainda alguma atividade de transformação. Além disso, as moléculas de DNA
contendo um maior número de crosslinks são completamente resistentes à
desnaturação (Kohn e Green, 1966). Alguns autores concluem que apenas um
crosslink não leva à inativação da molécula de DNA, entretanto dois já são
16

capazes de abolir a atividade desta molécula.
Apenas uma molécula de HN2, pode alterar o comportamento de um largo
número de moléculas de DNA e torná-las resistente à desnaturação (Kohn et al.,
1966). Este fato dá uma idéia de um tipo de reação particular entre HN2 e DNA
(Kohn et al., 1965). A reação do DNA com concentrações de mostarda de
aproximadamente 5x10-7M é responsável pela alquilação de aproximadamente
0,005% das bases nitrogenadas (Kohn et al., 1966, Friedberg et al, 2006).
Experimentos realizados com HN2 mostraram que apenas uma pequena
fração, aproximadamente 4%, das moléculas de DNA ligadas à mostarda, são
efetivamente crosslinks (Kohn et al., 1966; Friedberg et al., 2006). Assim, um dos
produtos da reação de HN2 com DNA é formado por uma molécula ligada a dois
resíduos de guanina (Kohn et al., 1966). Masta et al., 1994, confirmam como
sendo a 7-alquilguanina o maior produto depurinado resultante da alquilação por
HN2. A mostarda HN2 também pode gerar derivativos diguanil como subprodutos.
Na figura 3 encontra-se a estrutura química da mostarda nitrogenada
bifuncional – HN2 e na figura 4, suas possíveis ligações com o DNA. Estas
modelagens foram feitas para este trabalho
17

Figura 3: Estrutura química da mostarda nitrogenada bifuncional – HN2, (a) visão frontal)
e (b) visão lateral.
18

Figura 4: Possíveis ligações de HN2 com DNA. Seqüências alvo para a ligação entre N7G
– HN2. (a) crosslink e (b) biaduto intra-cadeia.
19

5.1 - O uso da mostarda nitrogenada bifuncional em quimioterapia
HN2 atualmente é utilizada primordialmente em linfomas de Hodgkin
(Tortorice, 2002). A quimioterapia administrada em pacientes com doença de
Hodgkin é feita através de mecloretamine (HN2), que geralmente está associado
com outros quimioterápicos, como – vincristina, procarbazine e prednizone, num
coquetel chamado MOPP (Tortorice, 2002). A quimioterapia parece ser bastante
eficaz nestes casos. Nos pacientes com doença de Hodgkin, o aparecimento de
tumores sólidos, como mielodisplasia secundária e leucemia mielóide parece estar
relacionado com a aplicação de doses cumulativas de agentes alquilantes, como o
mecloretamine. Tais doenças surgem entre dez e quinze anos após o tratamento
quimioterápico (André et al., 1998). Outras aplicações do mecloretamine são para
mycosis fungoides (linfoma de células T cutâneos) e outros tumores de pele
(Tortorice, 2002).
Também pode ser utilizado no tratamento de cânceres de sistema linfático
em geral.
Derivados semelhantes como mostarda fenilalanina (“melfalan”),
clorambucil e ciclofosfamida são usados em muitos tratamentos quimioterápicos,
apesar de possuírem mecanismos de reação não específicos (Masta et al., 1994).
Bendamustine é um derivado de mostarda nitrogenada com alta eficiência
em linfomas malignos e doença de Hodgkin, e é uma mostarda considerada
menos tóxica. A atividade antineoplásica desta mostarda é comparada ao
mecloretamine, que é a mostarda utilizada freqüentemente para este tipo de tumor
(Bremer e Roth, 1996).
A ciclofosfamida é uma mostarda nitrogenada bifuncional, e é efetiva contra
20

uma grande variedade de neoplasias e se acredita que sua atividade anti-tumoral
seja devida à mostarda fosforamida. Entretanto, a marcação de um derivado
conhecido de N7 torna improvável que seu efeito biológico seja devido à formação
de crosslink (Singer e Grunberger, 1983). Mostarda fosforamida é um agente
mutagênico formado pelo metabolismo do agente quimioterápico ciclofosfamida, e
quando reage com guanosina, forma um derivado no N7 altamente instável.
A mostarda nitrogenada HN2 pode assumir outros nomes em quimioterapia,
como: mustargen (Estados Unidos e Canadá), chlormethine, mechloretamine,
mecloretamina (Brasil) e outros. A administração de mostarda nitrogenada como
tratamento só deve ser realizada independentemente se por via oral, uso tópico ou
intra-venoso, com acompanhamento médico, e a indicação do seu uso deve levar
em consideração os riscos e benefícios (United States Pharmacopeia Drug
Information, 2000).
Existem alguns cuidados que devem ser tomados em relação ao seu uso,
por exemplo: não pode ser usado na gravidez, uma vez que pode afetar o bebê,
pode causar esterilidade, não pode ser usado durante a amamentação. Parece
não haver diferenças nos efeitos em relação a aplicação do tratamento entre
crianças, adultos e idosos (United Statés Pharmacopeia Drug Information, 2000).
Na figura 5 encontra-se a estrutura química de algumas mostardas
nitrogenadas
21

Ciclofosfamida
Melfalan
Clorambucil
Figura 5: Estrutura das mostardas nitrogenadas ciclofosfamida, melfalan e
clorambucil, tambem utilizadas em quimioterapia (Koland, 2003).
22

6. Mostarda nitrogenada monofuncional - HN1
Também pode ser chamada de meia mostarda. HN1 é um análogo
monofuncional de HN2. Produz efeitos no DNA, que não crosslinks (não
produzindo resistência à desnaturação), somente monoadutos, uma vez que só
possui um sítio reativo (Kohn et al., 1966).
Segundo Prakash e Strauss (1970, citado em Ruhland e Brendel, 1978), em
experimentos realizados com levedura, as curvas de sobrevivência feitas com
HN1 são capazes de formar “ombros”, e segundo Kohn e Green, 1966, os
tratamentos feitos com B. subtilis com HN1, não são capazes de causar uma
inativação apreciável, o que reflete a tolerância limitada a agentes alquilantes
monofuncionais. As mostardas nitrogenadas monofuncionais foram classificadas
como sendo agentes menos citotóxicos (Lawley e Phillips, 1995).
Na figura 6 encontra-se a estrutura química da mostarda nitrogenada
monofuncional – HN1 e na figura 7, sua possível ligação com o DNA. Estas
modelagens foram feitas para este trabalho.
23
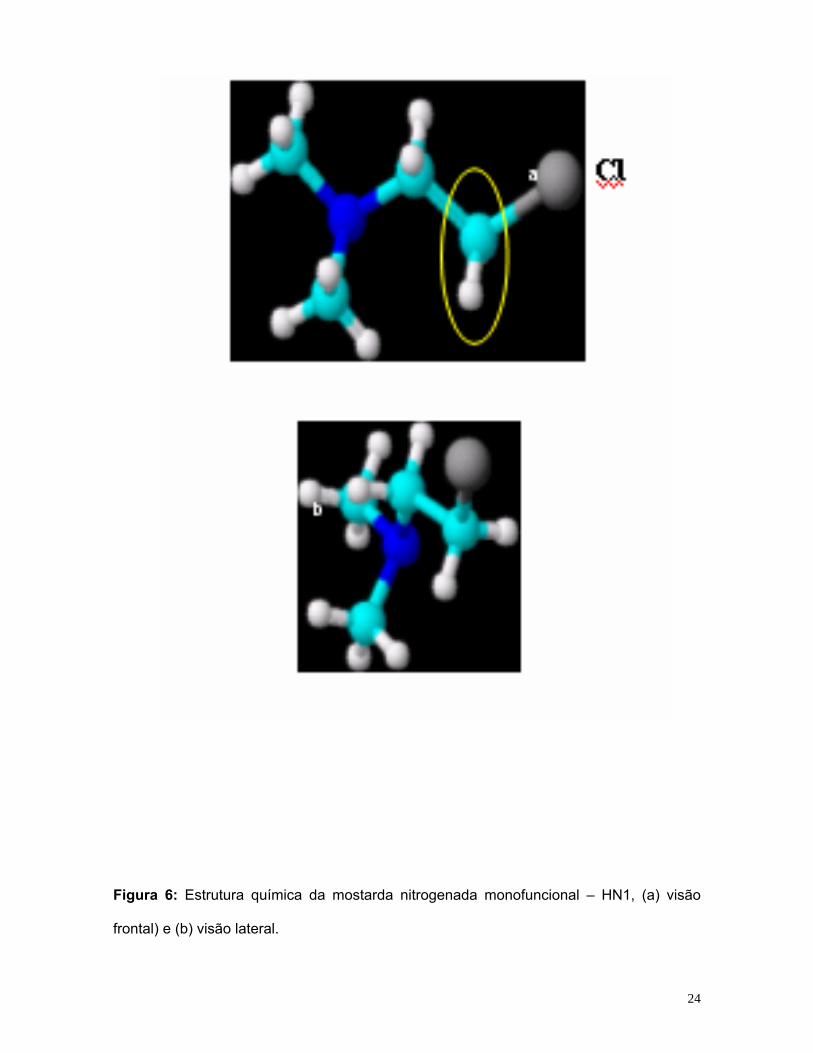
Figura 6: Estrutura química da mostarda nitrogenada monofuncional – HN1, (a) visão
frontal) e (b) visão lateral.
24

Figura 7: Possível ligação de HN1 com DNA – monoaduto. Seqüência alvo para a ligação
entre N7G – HN1.
25

7. Ativação metabólica das mostardas:
Em 1972, Sartorelli e colaboradores levantaram a hipótese de que células
em hipóxia poderiam apresentar maior capacidade de redução do que as células
normalmente oxigenadas. Foi proposto, então, que esta característica das células
em hipóxia poderia ser explorada no desenvolvimento de agentes antineoplásicos,
os quais só se tornariam citotóxicos após ativação metabólica pelas
nitrorredutases celulares. Estes agentes antineoplásicos biorredutíveis são pró-
fármacos, classificados como bioprecursores. Os bioprecursores são substâncias
inativas (pró-fármacos) que, in vivo, sofrem metabolização, geralmente pelo
sistema redox celular, dando origem a uma nova substância na forma ativa
(fármaco) (Oliveira e Alves, 2002). Estes compostos podem se tornar
carcinógenos químicos, uma vez ativados, via transformação metabólica,
causando mutações pela sua reação com o DNA (Alberts et al., 1994), e por isso
muitos destes agentes apresentam sucesso em quimioterapia.
Em relação ao mecanismo de ação das mostardas nitrogenadas, uma das
classes de alquilantes mais utilizadas em quimioterapia, podem agir diretamente,
ou são pró-drogas; algumas requerem atividade enzimática e outras sofrem
alterações químicas, até se tornarem metabólitos ativos. A parte alquilante da
molécula é representada pelo radical (N-CH2-CH2)+ onde o carbono terminal se
liga ao DNA. A ativação dessas moléculas, para se tornarem agentes ativos,
envolve apenas a quebra da molécula, em solução aquosa ou na circulação, com
liberação de cloro e/ou um grupo nitroso, ou a abertura de um anel de 3
componentes, na maioria dos casos. Grande parte desses agentes são moléculas
26

instáveis e devem ser estocadas sob a forma liofilizada. Essas moléculas são
altamente reativas e a sua ligação com o DNA celular é considerado o evento
letal.
8. O radical superóxido
O radical superóxido pode ser gerado como intermediário reativo a partir da
redução do oxigênio molecular e, como os outros intermediários, é um potente
oxidante de lipídeos, proteínas e ácidos nucléicos (Halliwell e Gutteridge, 1984,
Mello-Filho e Meneghini, 1985; Meneghini, 1988, citados em Asad et al., 2004).
Pode ser gerado a partir de alguns compostos, como paraquat e menadiona (Asad
et al., 2004).
Um dos caminhos pelos quais o radical superóxido pode causar toxicidade
celular é através de sua participação na geração de radicais hidroxil (OH.) (Keyer
et al., 1995), via reação de Fenton, uma vez que este radical pode regenerar Fe2+
a partir de Fe3+, através da doação de um elétron. Desta forma o excesso deste
radical apresenta um problema para qualquer organismo, principalmente se este
for deficiente em Superóxido Dismutase (Keyer et al., 1995).
9. Mecanismos de reparo de DNA em E. coli, envolvidos neste trabalho:
Vários são os meios pelos quais as células tentam resgatar a integridade de
seu matérial genético, seja por via direta, retirando enzimaticamente as lesões do
DNA ou por via indireta, através de mecanismos que permitem à célula tolerar as
lesões. Vale lembrar que sistemas enzimáticos diferentes podem agir
coordenadamente.
27

Para reparo das lesões causadas pelas mostardas nitrogenadas quase
nada é sabido quanto aos mecanismos envolvidos. Portanto, serão descritos
aqueles estudados neste trabalho.
9.1 - Reparo por excisão de bases (BER):
O reparo por excisão de bases (BER) é provavelmente o mais importante
sistema de reparação de lesões oxidativas, bases modificadas, sítios com perda
de bases, quebras simples e pequenas lacunas de DNA. Tanto procariotos como
eucariotos possuem uma grande variedade de DNA glicosilases, enzimas
hidrolíticas responsáveis pela retirada de bases alteradas do DNA, cada qual
possuindo especificidade para um ou alguns tipos de lesão (revisto por
Cunningham, 1997).
Uma grande variedade de lesões monoméricas de bases nitrogenadas são
reparadas por BER.
Dependendo da lesão, o reparo pode ser iniciado por enzimas (DNA-N-
glicosilases) que hidrolizam a ligação N glicosídica que liga a base nitrogenada
lesada ao DNA. Como resultado, é gerado um sítio AP. O reparo de sítios AP
pode ser feito de duas formas: a cadeia fosfodiéster pode ser clivada do lado 5’ do
mesmo por uma AP endonuclease de classe II ou verdadeira [Exonuclease III (Exo
III) ou Endonuclease IV (Endo IV)], e o resíduo de desoxirribose 5’ fosfato
remanescente é removido por uma atividade de 5’ desoxirribofosfodiesterase
(dRPase). A outra via de reparo de sítios AP, ocorre pela clivagem da cadeia
fosfodiéster por um mecanismo de β-eliminação promovido por uma AP
endonuclease de classe I ou AP liase [Formamidopirimidina DNA glicosilase (Fpg)
ou Endonuclease III (Endo III)] (esta atividade AP liase está presente em algumas
DNA glicosilases), gerando terminais 5’ fosfato e 3’ contendo um aldeído 2-3
28

insaturado. Nesta segunda via (pelas AP liases), pode ocorrer ainda δ eliminação
do aldeído, mediada pela enzima Fpg, deixando um terminal 5’ fosfato e um
terminal 3’ fosfato. Tanto o terminal 3’ fosfato como o aldeído insaturado, podem
ser removidos pela atividade 3’ fosfodiesterásica presente nas AP endonucleases
de classe II (Demple e Harrison, 1994).
Ambas as vias culminam na excisão da base lesada e do resíduo de
desoxirribose fosfato, deixando uma lacuna que pode ser preenchida pela enzima
DNA polimerase I, e o (s) nucleotídeo (s) incorporado (s) serão ligados ao restante
da cadeia fosfodiéster pela ação da DNA ligase, desta forma, completando o
reparo.
Etapas do processo:
liberação da base lesada ou errada por DNA glicosilase específica;
incisão da cadeia açúcar-fosfato no sítio abásico resultante, por uma
Endonuclease;
remoção do terminal 3’ PO4 , criado pela Endonuclease;
síntese de reparo e ligação do DNA neossintetizado ao pré-existente.
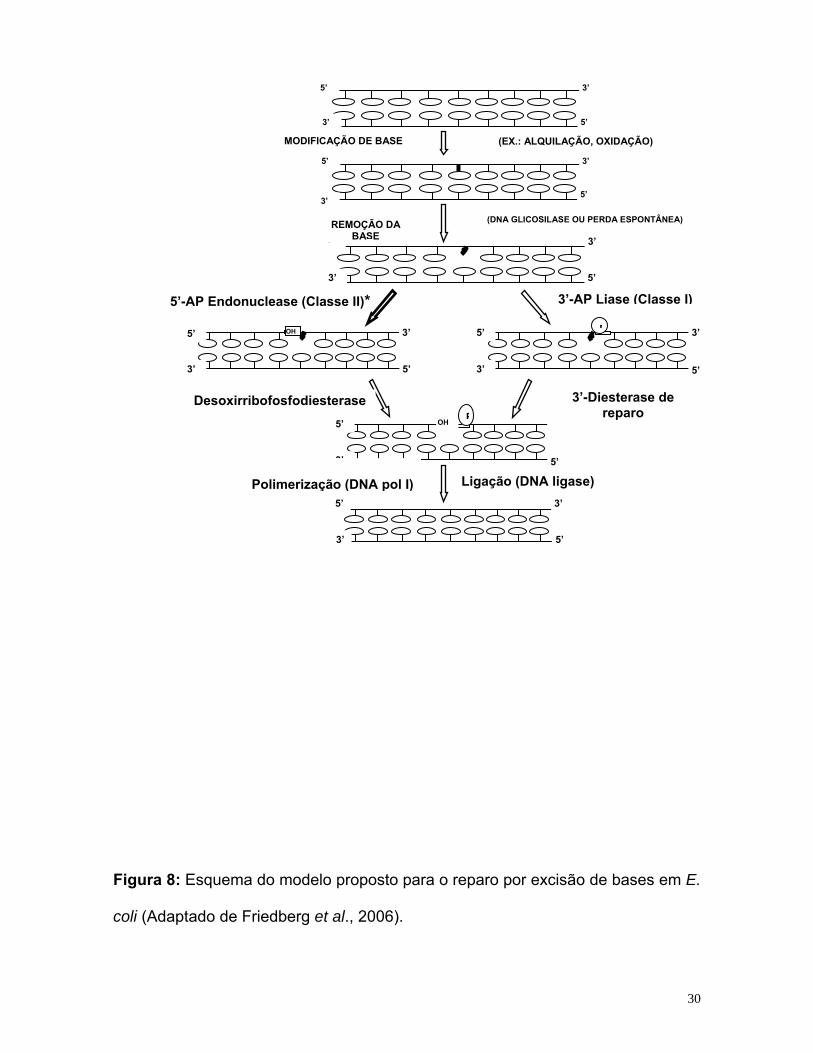
O mecanismo de reparo por excisão de bases está esquematizado na
figura 8.
29
3’ 5’
3’ 5’
Figura 8: Esquema do modelo proposto para o reparo por excisão de bases em E.
coli (Adaptado de Friedberg et al., 2006).
5’-AP Endonuclease (Classe II)*
3’ 5’
5’ 3’
5’
5’ 3’
3’
OH 5’ 3’
3’ 5’
3’
3’
5’
5’
OH
3’
3’
5’
5’
5’
5’
3’
3’
MODIFICAÇÃO DE BASE (EX.: ALQUILAÇÃO, OXIDAÇÃO)
(DNA GLICOSILASE OU PERDA ESPONTÂNEA) REMOÇÃO DA BASE
3’-AP Liase (Classe I)
P
3’-Diesterase de reparo
Desoxirribofosfodiesterase
Ligação (DNA ligase)
P
Polimerização (DNA pol I)
30

9.2 - Reparo por excisão de nucleotídeos (NER):
A lesão-modelo mais extensivamente usada para o estudo de NER foi o
dímero de pirimidina, um fotoproduto produzido no DNA pela radiação UVC
(ultravioleta curto).
Em estudos realizados na década de 60 sobre a reparação de dímeros de
pirimidinas, formados no DNA pelas radiações UV-C, utilizando cepas resistentes
(E. coli B/r e K-12) e os mutantes sensíveis Bs-1 e AB1886 (uvrA), foi mostrado
que os dímeros eram removidos, em ausência de luz, nas cepas resistentes (uvr+),
mas não nos mutantes (uvr-), sendo o processo designado reparo por excisão.
Posteriormente, foi verificado que nenhum dos mutantes uvrA, uvrB ou uvrC era
capaz de remover dímeros de pirimidina do seu DNA e que estes mutantes eram
também sensíveis a outros agentes tais como: alquilantes bifuncionais, ácido
nitroso, mitomicina C e outros (Leitão et al., 2005).
O reparo por excisão de nucleotídeos (NER) consiste em uma série de
reações enzimáticas requeridas para remover virtualmente qualquer lesão do
DNA, incluindo a maioria, senão todas, as lesões removidas por outros sistemas
de reparo.
Etapas do processo:
reconhecimento da lesão;
incisão;
excisão;
síntese de reparo e ligação do fragmento neossintetizado.
As três primeiras etapas deste processo são feitas pelo conjunto das três
31

proteínas UvrA, UvrB e UvrC, denominado complexo UvrABC, que realizam uma
série de reações dependentes da hidrólise de ATP.
O complexo UvrABC, possui um amplo espectro de especificidade de
substratos e foi proposto que ela reconheça modificações da conformação do DNA
e não necessariamente as bases modificadas.
O NER possui um processo chamado de reconhecimento específico da
lesão, que é a sua habilidade em localizar e identificar a lesão no genoma.
Esse reconhecimento é feito pelas interações entre um complexo das
enzimas UvrA e UvrB e suas interações com o DNA lesado (Friedberg et al.,
2006).
Até hoje este modelo do complexo UvrABC, com as três subunidades, só
foi montado in vivo, para as lesões do tipo dímero de pirimidina. Para todas as
outras lesões foi tudo realizado in vitro (Lage et al., 2003).
A proteína UvrA dimeriza-se (UvrA2) e se associa com a proteína UvrB para
formar o complexo (UvrA2)UvrB, e este processo dura em torno de 15 segundos.
Essa interação é estritamente dependente da presença de ATP. A proteína UvrA
ligada ao sítio da lesão possui uma baixa especificidade, a partir do momento que
a proteína UvrB se liga, extrema especificidade é adquirida. A proteína UvrB é
considerada a proteína chave de reconhecimento do NER bacteriano, uma vez
que interage com todos os componentes deste sistema de reparo (Van Houten et
al, 2005).
Este complexo possui uma atividade DNA helicase limitada na presença de
ATP. Essa helicase pode distorcer unidirecionalmente pequenos trechos de DNA
duplex e é inibida pela presença de grandes lesões no DNA (Gordienko e Rupp,
1997). A ATPase que presumivelmente dirige a função de DNA helicase deste
32

complexo, acredita-se ser críptica na proteína UvrB, mas se torna ativada quando
esta sofre uma mudança conformacional associada com a proteína UvrA e DNA
(Friedberg et al., 2005).
Sendo a proteína UvrA uma “casamenteira molecular”, esta leva a UvrB aos
sítios do DNA onde há distorções, formando assim um complexo (UvrA2)UvrB-
DNA que é relativamente estável. Então a proteína UvrB hidrolisa ATP, neste
momento há um pregueamento e desnaturação local do DNA, além de uma
mudança conformacional da proteína UvrB (Shi et al., 1992; Van Houten e
Snowden., 1993).
A partir daí, o DNA em torno do complexo se desenrola (como se estivesse
cheio de helicases) (Oh e Grossman, 1986), e o complexo (UvrA2)UvrB desliza por
esse DNA fita simples, até chegar ao sítio da lesão, sendo assim o complexo de
pré-incisão está formado por DNA lesado-proteína (Gordienko e Rupp, 1997).
Neste momento, UvrA se dissocia do complexo, e um novo complexo UvrB-DNA é
estabelecido, estando o DNA agora preparado para a incisão. Este processo
descrito ocorre em aproximadamente duas horas.
A proteína UvrC se liga ao complexo UvrB-DNA lesado, etapa esta que
precisa de ATP (Friedberg et al., 2005).
Com a chegada de UvrC, podem ser feitas então as incisões (duas) no DNA
lesado, por dois sítios enzimáticos distintos.
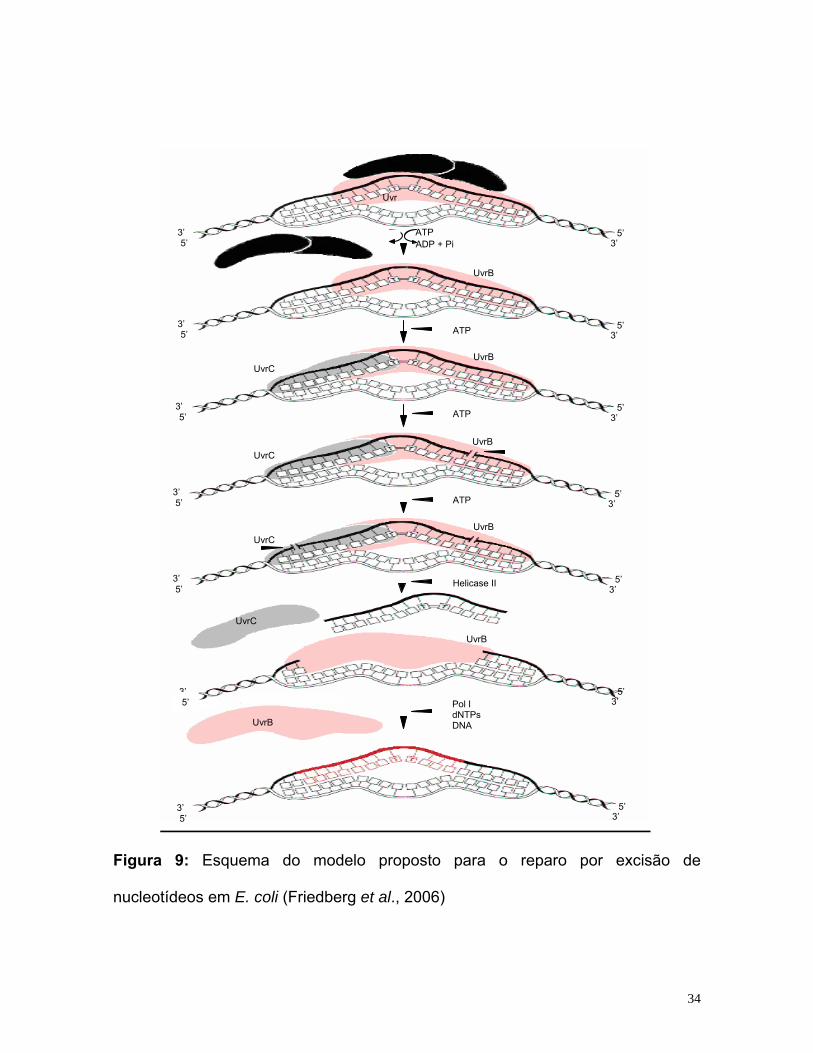
O mecanismo de reparo por excisão de nucleotídeos está esquematizado
na figura 9.
33
UvrUvrA
Figura 9: Esquema do modelo proposto para o reparo por excisão de
nucleotídeos em E. coli (Friedberg et al., 2006)
UvrUvr
UvrB
ATPADP + Pi
UvrB
UvrB
UvrB
UvrB
UvrB
UvrC
UvrC
UvrC
UvrC
3’ 5’
3’ 5’
3’ 5’
3’ 5’
3’ 5’
3’ 5’
3’ 5’
3’ 5’
3’ 5’
3’ 5’
3’ 5’ ATP
3’ 5’
ATP
ATP
3’ 5’
3’ 5’
Helicase II
Pol I dNTPs
Uvr
DNA
34

9.3 - Reparo de DNA contendo adutos do tipo crosslinks:
O reparo das lesões do tipo crosslinks em DNA não é tão bem
caracterizado quanto as outras vias de reparo. Provavelmente o maior obstáculo,
para este tipo de reparo, é o envolvimento de múltiplos processos atuando juntos,
devido à complexidade da lesão.
Em bactérias foram propostas duas vias de reparo de crosslinks, que são
os modelos aceitos até hoje. Estas vias associam o reparo NER ou com
recombinação homóloga, mediada por RecA, que agiria de forma majoritária e a
outra via associa NER com reparo de DNA translesão, dependente da proteína
DNA polimerase II, que agiria de forma minoritária, especialmente quando a
primeira via está impedida de funcionar (Dronkert e Kanaar, 2001)
No primeiro modelo incisional-recombinacional, o complexo UvrABC incisa
o DNA bimodalmente (fazendo os dois cortes a 3’ e a 5’ da lesão) em uma das
fitas, gerando um oligonucleotídeo que permanece ligado covalentemente ao DNA
através do crosslink. A lacuna gerada pelo deslocamento deste oligonucleotídeo,
neste modelo é gerada in vitro, pela atividade exonucleásica 5’→3’ da DNA
polimerase I, levando então à participação do reparo recombinacional, que é feito
pela proteína RecA. Neste momento há então uma estrutura triplex, formada pela
fita de DNA que não foi incisada, pelo oligonucleotídeo incisado (que permaneceu
ligado à outra fita de DNA) e pela proteína RecA. A próxima incisão (da outra fita
de DNA, em que o oligonucleotídeo permaneceu ligado), resultaria em uma
estrutura que seria o oligonucleotídeo contendo o crosslink, e aí então ficaria uma
segunda lacuna a ser reparada pela síntese de reparo convencional e posterior
ligação do DNA, finalizando assim o reparo do crosslink. Bactérias deficientes em
35

uma ou algumas das proteínas de recombinação, como: RecB, C, D, F, G, O, e R,
são mais sensíveis in vivo a agentes formadores de crosslinks, o que indica a
participação destas proteínas no processo (Dronkert e Kanaar, 2001).
No segundo modelo, após a introdução de um crosslink no DNA em cepas
deficientes em RecA, o reparo destes crosslinks se mostra dependente do reparo
de nucleotídeos e da enzima DNA polimerase II. Este reparo é iniciado por
incisões na fita (contendo a lesão) nos lados 5’ e 3’, pelo complexo UvrABC. A
síntese translesão neste caso seria efetuada pela DNA polimerase II (lembrando
que não pode ocorrer recombinação, uma vez que a cepa é deficiente em recA),
que sintetiza DNA através da região deixada livre, usando como a fita
complementar contendo ainda a lesão. Para excisar o oligonucleotídeo contendo o
crosslink, este complexo UvrABC incisa a fita de DNA complementar em ambos os
lados do crosslink. A lacuna de DNA fita simples remanescente é preenchida
então, pela DNA polimerase I e ligada pela DNA ligase, resultando na liberação do
fragmento lesado (com o crosslink), finalizando desta forma o reparo (Berardini et
al., 1999; Dronkert e Kanaar, 2001; Noll et al., 2006).
O modelo proposto para reparo de crosslinks está esquematizado na figura
10.
36
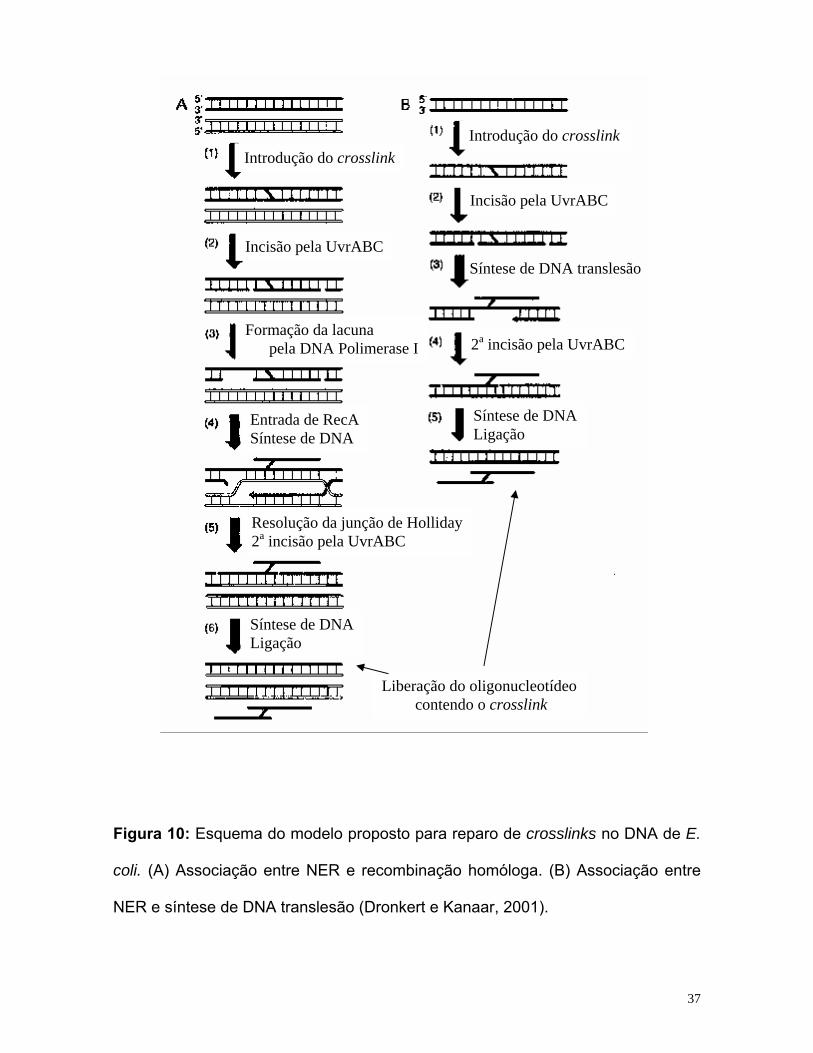
Introdução do crosslink Introdução do crosslink
Incisão pela UvrABC
Incisão pela UvrABCSíntese de DNA translesão
Formação da lacuna 2a incisão pela UvrABC pela DNA Polimerase I
Síntese de DNA Entrada de RecALigação Síntese de DNA
Resolução da junção de Holliday2a incisão pela UvrABC
Figura 10: Esquema do modelo proposto para reparo de crosslinks no DNA de E.
coli. (A) Associação entre NER e recombinação homóloga. (B) Associação entre
NER e síntese de DNA translesão (Dronkert e Kanaar, 2001).
Síntese de DNALigação
Liberação do oligonucleotídeo contendo o crosslink
37

9.4 - Resposta SOS:
Quando culturas de E. coli são expostas à radiação UV ou agentes
químicos, bem como a fatores que interferem na duplicação do DNA ou causam
lesões nesta molécula, há a indução de um conjunto de funções celulares
denominadas resposta SOS. A indução destas funções acarreta a desrepressão
de aproximadamente 40 genes (Jarosz et al., 2007), capazes de auxiliar na
recuperação celular, através de seus produtos, do reparo livre de erros e outros,
facilitando a replicação do DNA apesar da existência de determinadas lesões
(Myles e Sancar, 1989).
O mecanismo molecular da indução da resposta SOS depende da ação de
duas proteínas, LexA (codificada pelo gene lexA) e RecA (codificada pelo gene
recA).
A proteína LexA é repressora de diversos operons, tais como o do próprio
gene lexA, e também recA, uvrA, uvrB, umuC, umuD, sulA e outros, que estão
envolvidos em vários mecanismos de restauração das funções celulares, tais
como: recombinação genética, reparo de DNA, mutagênese e divisão celular. Uma
vez ocorrendo um sinal indutor, possivelmente trechos de fita simples de DNA
resultantes da presença de uma lesão, da tentativa de correção desta ou da
parada da forquilha de replicação, a proteína RecA interage com o DNA em fita
simples, em presença de ATP, tornando-se “ ativada” (RecA*). Nesta forma, a
proteína RecA* se liga ao repressor LexA, promovendo a auto-clivagem desta
proteína, fazendo com que haja a desrepressão dos genes por ela controlados, os
genes SOS. À medida que os danos no DNA são reparados, o nível do sinal
indutor diminui, acarretando a diminuição da conversão da proteína RecA em
38

RecA*. Há, desta forma, o acúmulo do repressor LexA na célula, voltando os
genes SOS a ficarem reprimidos (Little, 1993). Como resultado da expressão
destas funções podemos verificar, entre outras respostas, o aumento da
sobrevivência e mutagênese de fagos lesados (Weigle reativação e Weigle
mutagênese) (Weigle, 1953), o aumento da mutagênese bacteriana, a
filamentação celular, a indução de profagos em culturas bacterianas lisogênicas,
além de um aumento na atividade dos mecanismos de reparo de DNA, inibição do
processo normal de replicação e parada da respiração celular (Little, 1991;
Fernandez de Henestrosa et al., 2000).
O modelo proposto para indução da resposta SOS está esquematizado na
figura 11.
39

+ 40
Figura 11: Modelo proposto para indução da resposta SOS em E. coli (Adaptado
de Little, 1991).
genes
RecA
Sinal
uvrA;umuDC;sulA
Expressão aumentadaReparo de
outras funções
+ 40
genes recAlexA DNA
Diminuição sinal
Diminuição nível de Ativação da
RecASinal )
Acúmulo repressor Expressão
recAlexA
LexA RecA
40

10. Defesas contra estresse oxidativo
Os organismos vivos têm desenvolvido mecanismos específicos para
prevenir a produção e os efeitos das EAO, tais como: vitaminas e enzimas como
Catalase e Superóxido Dismutase (Asad et al., 2004).
Com respeito às estratégias de defesa mais gerais da célula contra danos
oxidativos em células, estudos em E. coli, revelaram a participação, além de
mecanismos de reparo de DNA, de outras enzimas protetoras anti-oxidantes. Dois
regulons foram descritos como sendo controlados pelos produtos dos genes soxR
e oxyR, e respondem, respectivamente a ânion superóxido e H2O2 (Debenham,
1987).
10.1 - Regulon SoxRS
O sistema regulatorio SoxRS age em dois passos, com a proteína SoxR
agindo como sensora e ativadora. Essas proteínas são ativadoras transcricionais
redox-sensíveis. Então, na presença de ânion superóxido, SoxR sofre uma
oxidação univalente do grupamento 2Fe-2S, sofrendo uma mudança
conformacional desta proteína, induzindo a transcrição de SoxS, um regulador
positivo, que estimula a transcrição de mais de 16 genes ligados à resposta ao
superóxido (Wu e Weiss, 1992, Hidalgo et al, 1995).
Os produtos induzidos pelo regulon soxRS incluem: Manganês Superóxido
Dismutase (sodA) (indução aeróbica, via produção de radical superóxido (Touati,
1988), Endo IV (nfo), Glicose-6-fosfato desidrogenase (zwf) e Fur (fur), uma
proteína ligada a transporte de ferro, dentre outras (Pomposiello e Demple, 2001).
Existe também a proteína SodB (sodB), que é constitutiva e capaz também de
dismutar radical superóxido em H2O2 e oxigênio, e parece não ser regulada via
41

soxRS. Esta enzima é expressa em condições aeróbicas e anaeróbicas (Touati,
1988).
Na figura 12 encontra-se um esquema da indução do sistema SoxRS.
As enzimas Superóxido Dismutases possuem papel chave na defesa contra
a toxicidade mediada pelo oxigênio (Touati et al., 1995). São metaloenzimas cuja
a única função conhecida é a dismutação de radicais superóxido, produzidos em
todos os organismos expostos ao oxigênio (Touati, 1988), através da reação:
2O2.- + 2H → H2O2 + O2
42

SSiisstteemmaa SSooxxRRSS
OXIDAÇÃO O2•-
NO• SoxR SoxS
Figura 12: Esquema da indução do sistema SoxRS.
INDUÇÃO Outros
NITROSILAÇÃO
Diferentes enzimas
Mn-Sod(sodA)
Endo IV (nfo)
43

10.2 - Regulon OxyR.
Bactérias posuem uma resposta adaptativa contra agentes oxidantes,
assim a exposição a baixos níveis de H2O2 permite à célula sobreviver a doses
tóxicas deste mesmo agente (Demple e Halbrook, 1983; Demple, 1991). A
expressão de nove proteínas induzidas pelo tratamento com H2O2, está sob o
controle do gene oxyR (Christman et al., 1985), dentre elas catalase e alquil
hidroperóxido redutase.
H2O2 aumenta a atividade transcricional de oxyR, através da oxidação de
dois resíduos de cisteína (Zheng et al., 1998; Aslund et al., 1999). Quando ativada
OxyR ativa a transcrição de genes que incluem katG (catalase hidroperoxidase I),
ahp (alquilhidroperóxido-NADPH oxido redutase) e fur, além de outros (Martinez e
Kolter, 1997). O número de moléculas de Fur pode ser aumentado de 5000 para
10000, quando a célula está sobre influência de estresse oxidativo e
conseqüentemente sob a atuação dos regulons OxyR e SoxRS (Zheng et al.,
1999).
44

CARACTERIZAÇÃO DO PROBLEMA
Alquilantes bifuncionais são usados em terapia anti-tumoral, porém efeitos
desagradáveis, como comprometimento das propriedades fundamentais da célula
e também o aparecimento de novos tumores, podem acontecer no futuro, uma vez
que estes agentes têm o potencial de danificar tanto células normais quanto
células tumorais.
A mostarda HN2 foi o primeiro agente desta classe utilizado em
quimioterapia, começando desta a forma a “Era dos quimioterápicos
antineoplásicos “ e é utilizada até hoje em quimioterapia, porém pouco se sabe a
respeito das lesões e do reparo associado a esta molécula.
Desta forma é de extrema importância o estudo dos mecanismos de reparo
e a possível metabolização relacionada a estes agentes.
45

OBJETIVOS
Sabendo-se que o modo de atuação de HN2 é via formação de crosslinks
no DNA, porém os mecanismos pelos quais estas podem reagir com tal molécula
(possível metabolização/ativação), os tipos de lesão que são formadas in vivo, os
mecanismos de reparo que atuam sobre estas lesões, ainda não estão bem
esclarecidos, tornaram-se objetivos do nosso trabalho:
• Estudar o reparo, através de experimentos de sobrevivência celular de
diferentes mutantes;
• Analisar a mutagênese provocada pela exposição às mostardas, através da
mutagênese de aquisição de resistência ao antibiótico rifampicina;
• Analisar a expressão dos genes envolvidos neste mecanismo de reparo,
através da determinação da expressão relativa dos genes por RT – PCR
quantitativo em tempo real (Metodologia SyBr Green);
• Estudar o mecanismo pelo qual as mostardas são capazes de lesar as células,
através da utilização de quelantes de metais de transição e captadores de
EAO, em diferentes condições, utilizando diferentes mutantes, uma vez que
grande parte destes alquilantes agem através de metabolização (ativação) e
geração de EAO.
Utilizamos como instrumento do nosso estudo a bactéria E. coli que é um
modelo de estudo já bem conhecido.
_________________________________________________________________
parte dos resultados estão publicados no artigo que se encontra em anexo (Anexo I)
46

MATERIAL E MÉTODOS
1. Cepas Bacterianas:
Foram utilizadas nos experimentos cepas bacterianas de E. coli K12. No
quadro abaixo encontram-se relacionadas todas as cepas, bem como suas
características genéticas mais relevantes.
Quadro 2: Cepas de E. coli K-12
Designação Genótipo e Fenótipo Relevantes
AB1157 Selvagem – proficiente em todos os mecanismos de reparo de DNA
AB1886 [AB1157] uvrA6 – deficiente na proteína UvrA
AB1885 [AB1157] uvrB5 – deficiente na proteína UvrB
AB1884 [AB1157] uvrC34 – deficiente na proteína UvrC
AB2480 [AB1157] uvrA recA – deficiente nas proteínas UvrA e RecA
AB2463 [AB1157] recA13 (Def) – deficiente na proteína RecA
GY4763 [AB1157] recA142 – deficiente em recombinação homóloga
GY2831 [AB1157] recA430 – deficiente na resposta SOS
BH100 [AB1157] nfo nth xthA fpg – deficiente em BER
BW527 [AB1157] nfo – deficiente na proteína Endonuclease IV
BW9091 [AB1157] xthA – deficiente na proteína Exonuclease III
BW544 [AB1157] xthA nfo – deficiente nas proteínas Endo IV e Exo III
OG100 [AB1157] oxyR - deficiente no regulon OxyR
JI130 [AB1157] sodA – deficiente na proteína Mn–Superóxido Dismutase
JI131 [AB1157] sodB – deficiente na proteína Fe–Superóxido Dismutase
47
JI132 [AB1157] sodAsodB - deficiente nas duas Superóxido Dismutases
GC4468 Selvagem
QC1732 [GC4468] fur – deficiente na regulação de ferro
As cepas AB foram obtidas de P. Howard-Flanders (University of Yale, CT-
USA). A cepa BH foi obtida de S. Boiteux (CEA Fontenay-Aux-Roses, França). As
cepas BW foram obtidas de B. Weiss (University of Michigan, USA). As cepas JI
foram obtidas de J.A. Imlay e S. Linn (University of Illinois, USA). As cepas GC e
QC foram obtidas de R. D’ari (Institut Jacques Monod, Paris, França). As cepas
GY foram obtidas de R. Devoret (CNRS, Gif-sûr-Yvette, França). A cepa OG foi
obtida de P. Quillardet (Institut Pasteur, França).
2. Meios de cultura e tampão:
2.1 - Meios líquidos:
Meio LB (Lysogenic Broth) (Miller, 1992) – NaCl 1% (Merck); bacto-triptona
1% (Difco Laboratories); extrato de levedura 0,5% (Difco Laboratories).
Autoclavado por 20 minutos a 120ºC. Utilizado para crescimento das cepas
bacterianas.
Quadro 3: Concentração dos antibióticos acrescentados ao meio de cultura
das cepas bacterianas resistentes.
Antibióticos Quantidade final Fornecedor
Ampicilina 100µg/ml Kodak International Biotechnologies, Inc
Estreptomicina 100µg/ml Sigma Chemical Company
Kanamicina 40µg/ml Sigma Chemical Company
48

Tetraciclina 15µg/ml Sigma Chemical Company
Tampão M9 pH 7,0 (Miller, 1992) – Na2HPO4 anidro 0,6% (QEEL –
Química Especializada Erich LTDA.); KH2PO4 0,3% (Reagen – Quimibrás
Indústrias Químicas S/A); NH4Cl 0,1% (Reagen – Quimibrás Indústrias Químicas
S/A); NaCl 0,05% (Reagen – Quimibrás Indústrias Químicas S/A). Autoclavado por
20’a 120ºC. Após autoclavagem foram acrescentados MgSO4.7H2O 1mM
(Indústrias Químicas Merck) e CaCl2 0,1mM (Indústrias Químicas Merck). Utilizado
para diluição das cepas bacterianas.
Meio M9S pH 7,0 (Miller, 1992) – Tampão M9, suplementado com
casaminoácidos (25µl/ml), tiamina (10µg/ml) e glicose (0,4%). Utilizado para
crescimento e tratamento da cepa BW9091, com H2O2.
Tampão Fosfato pH 7,0 (Miller, 1992) - Na2HPO4 61mM (QEEL),
KH2PO4 39mM (Reagen – Quimibrás Indústrias Químicas S/A). As soluções foram
autoclavadas separadamente a 120ºC por 20 minutos e depois misturadas, e o pH
calibrado para 7,0. Utilizado para o tratamento com as mostardas nitrogenadas.
2.2 - Meios Sólidos:
Meio LB sólido – meio LB solidificado com ágar 1,5% (Difco
Laboratories). Utilizado para semadura das cepas bacterianas.
Para os experimentos de mutagênese a semeadura foi feita em placas
contendo meio LB-Rif, ou seja, meio LB, acrescido de 100µg/ml do antibiótico
rifampicina.
49

3. Mostardas Nitrogenadas:
HN2 (Mechlorethamine hydrochloride – Aldrich), solubilizada em H2O
deionizada estéril. Peso molecular = 192,52.
HN1 (2-Dimethylaminoethyl chloride hydrochloride – Aldrich), solubilizada
em H2O deionizada estéril. Peso molecular = 144,05.
4. 2,2’ dipiridil utilizado (quelante de Fe2+):
Foi utilizado o 2,2’ dipiridil (Sigma Chemical Company), solubilizado em
uma solução de H2O deionizada e etanol absoluto (Reagen) 10%. Peso molecular
= 156,2.
5. Desferroxiamina utilizada (quelante de Fe3+):
Foi utilizada a desferroxiamina (Sigma Chemical Company), solubilizado em
uma solução de H2O deionizada e etanol absoluto (Reagen) 20%. Peso molecular
= 656,8.
6. Captador de radicais livres:
Foi utilizada a tiouréia. Solubilizada em H2O deionizada. Peso molecular =
76,11.
7. Manutenção das cepas bacterianas:
As cepas bacterianas utilizadas neste trabalho foram mantidas em estoque
do tipo frascos de vidro contendo meio LB (Miller, 1992), suplementado com timina
50µg/ml e solidificado com ágar 0,75% à temperatura ambiente. Estes frascos são
denominados slants.
Para utilização freqüente das cepas, as culturas foram mantidas em
criovials contendo a cultura em fase estacionária, em glicerol 50% (Indústrias
Químicas Merck), sob refrigeração a –20ºC ou –80ºC.
50

8. Obtenção das culturas bacterianas para os experimentos:
As culturas bacterianas utilizadas nos experimentos foram obtidas com
auxílio de micropipetas dos estoques a –20ºC ou –80ºC para Erlenmeyers
contendo o meio LB ou M9S com o antibiótico apropriado. Estas culturas cresciam
durante a noite a 37ºC sob agitação (pernoite), aproximadamente 160 rotações
por minuto (rpm), obtendo, desta forma, a cultura em fase estacionária de
crescimento, com aproximadamente 109 células/ml.
As culturas em fase exponencial de crescimento, para realização dos
experimentos, foram obtidas a partir de uma diluição feita da cultura em fase
estacionária (pernoite) na proporção de 1:40 em meio LB sem antibiótico e
incubadas a 37ºC sob agitação (aproximadamente 160rpm), até atingir a fase
exponencial de crescimento(1-2x108 células/ml).
9. Sobrevivência bacteriana às mostardas nitrogenadas bifuncional (HN2) e
monofuncional (HN1):
Após a obtenção das culturas em fase exponencial de crescimento (como
foi descrito anteriormente), as culturas foram centrifugadas duas vezes a 7710 x g
por 10 minutos entre 4–10ºC (rotor Sorvall SS34, Dupont Instruments),
ressuspensas pela primeira vez em tampão M9 e pela segunda vez em tampão
fosfato pH 7,0 (no mesmo volume inicial). Uma alíquota então era retirada para ser
diluída apropriadamente em M9, a fim de se obter o número inicial de células
viáveis, antes do tratamento. As culturas foram divididas em alíquotas contendo
2ml (colocadas em Erlenmeyers) e tratadas com doses crescentes de HN2 e HN1
por 60 minutos sob vigorosa agitação de 240rpm, e após cada dose, alíquotas
51

foram retiradas para determinação da sobrevivência celular. Essas alíquotas,
então, foram diluídas em M9 e semeadas em placas de Petri contendo LB sólido.
As placas foram incubadas a 37ºC por 24 horas e as colônias contadas. A
sobrevivência celular N/No (ordenada) é o quociente obtido pela divisão do
número de células sobreviventes após cada dose (N) pelo número inicial de
células (No), plotada contra concentração de mostardas (abcissa). Cada gráfico
representa a média de três experimentos.
10. Sobrevivência bacteriana às mostardas nitrogenadas com pré-tratamento
com quelantes de Fe2+ (2,2’dipiridil), Fe3+ (deferroxiamina) e captador de
radicais livres (tiouréia):
A partir das culturas em fase exponencial de crescimento e após a segunda
centrifugação, antes do tratamento com as mostardas HN2 e HN1, as culturas
foram divididas em alíquotas de 2ml (colocadas em Erlenmeyers) pré-tratadas
com 2,2’dipiridil (1mM), desferroxiamina (1mM) ou tiouréia (100mM) por 20
minutos a 37ºC sob agitação de aproximadamente 160rpm. Após o pré-
tratamento, uma alíquota era novamente retirada, a fim de se estabelecer o
número de células viáveis após o pré-tratamento e então tratadas com doses
crescentes de HN2 ou HN1 por 60 minutos, conforme descrito no item anterior.
Os controles do 2,2’dipiridl, desferroxiamina e tiouréia foram feitos sempre
utilizando as mesmas condições do tratamento, sem as mostardas.
11. Sobrevivência da cepa selvagem à mostarda nitrogenada bifuncional
(HN2) e 2,2’dipiridil em diferentes condições:
Os experimentos com pré-tratamento utilizando 2,2’ dipiridil (1mM) seguido
52

de HN2 (2mM), ou somente com HN2 (2mM), foram realizados conforme descrito
anteriormente. Nos experimentos com centrifugação após o pré-tratamento com
2,2’ dipiridil (1mM), centrifugamos uma vez a 7710 x g por 5 minutos entre 4-10ºC.
No tratamento simultâneo colocamos o 2,2’ dipiridil (1mM) e HN2 (2mM). E nos
tratamentos de reação entre HN2 e 2,2 dipiridil, estas moléculas foram colocadas
para reagir, previamente, nas mesmas concentrações de tratamento das células,
1mM e 2mM, respectivamente, durante 20 minutos a 37ºC, sendo em seguida
adicionadas a suspensão celular
Em alguns experimentos com as cepas AB1157 (selvagem) e JI132 (sodA
sodB), utilizando o pré-tratamento com 2,2’dipiridil, utilizamos a concentração
máxima de HN2 (2mM) e variamos as rpm - 0, 160 e 240.
Outros experimentos foram realizados utilizando diferentes concentrações do
pré-tratamento com 2,2”dipiridil (0,5; 1,0; 2,0 e 4mM) e fixando a concentração
máxima de HN2 em 2mM.
12. Sobrevivência bacteriana à mostarda nitrogenada bifuncional (HN2) com
pré-tratamento com tiouréia em diferentes condições:
Em alguns experimentos realizados com o pré-tratamento com 2,2-dipiridil e
posterior tratamento com HN2, a adição do captador tiouréia (100mM) foi feita
juntamente com este pré-tratamento, posteriormente seguido da adição de HN2
(2mM), ou após o pré-tratamento com 2,2’dipiridil, juntamente com HN2.
13. Sobrevivência bacteriana à mostarda nitrogenada bifuncional (HN2) com
duplo pré-tratamento com 2,2’ dipiridil e desferroxiamina:
Os experimentos foram realizados utilizando o mesmo protocolo dos
53

tratamentos com apenas um quelante ou captador. A única diferença neste caso,
foi a adição dos dois quelantes simultaneamente, ambos na concentração de
1mM.
14. Sobrevivência bacteriana às mostardas nitrogenadas em diferentes
temperaturas:
Após a obtenção das culturas em fase exponencial de crescimento (como
foi descrito anteriormente), as culturas foram centrifugadas duas vezes a 7710 x g
por 10 minutos entre 4–10ºC (rotor Sorvall SS34, Dupont Instruments),
ressuspensas pela primeira vez em tampão M9 e pela segunda vez em tampão
fosfato pH 7,0 (no mesmo volume inicial). Uma alíquota então era retirada para ser
diluída apropriadamente em M9, a fim de se obter o número inicial de células
viáveis, antes do tratamento. As culturas foram divididas em alíquotas de 2ml
(colocadas em Erlenmeyers) e tratadas com as maiores concentrações utilizadas
para HN2 e HN1 por 60 minutos sob vigorosa agitação de 240rpm a 37ºC, 37ºC
sem agitação ou a 9ºC (geladeira). Após cada dose, alíquotas foram retiradas para
determinação da sobrevivência celular. Essas alíquotas, então, foram diluídas em
M9 e semeadas em placas de Petri contendo LB sólido. As placas foram
incubadas a 37ºC por 24 horas e as colônias contadas. A sobrevivência celular
N/No (ordenada) é o quociente obtido pela divisão do nº de células sobreviventes
após cada dose (N) pelo número inicial de células (No), plotada contra
concentração de mostardas (abcissa). Cada gráfico representa a média de três
experimentos.
Em alguns experimentos foram feitos pré-tratamento com HN2 a 37ºC por
20 minutos, seguidos do restante do tempo a 9ºC, dando um total de 60 minutos
54

de tratamento. Os experimentos realizados com H2O2 (20 mM) foram feitos
com 5 minutos de incubação a 37ºC + 15 minutos a 9ºC, 20 minutos a 9ºC ou 20
minutos a 37ºC. Estes tempos variam de acordo com o composto químico em
questão.
15. Sobrevivência e mutagênese bacteriana aos tratamentos com HN2 e pré-
tratamento com 2,2’ dipiridil e posterior tratamento com HN2:
Após a obtenção das culturas em fase exponencial de crescimento (como
foi descrito anteriormente), as culturas foram centrifugadas a 7710 x g por 10
minutos entre 4–10ºC e ressuspensas em tampão M9. Uma alíquota então era
retirada para ser diluída apropriadamente em M9, a fim de se obter o número
inicial de células viáveis, antes do tratamento. As culturas foram tratadas,
conforme descrito anteriormente, ou somente com HN2 ou pré-tratamento com
2,2’ dipiridil e posterior tratamento com HN2. Essas alíquotas, então, foram
diluídas em tampão M9 e semeadas em placas de Petri contendo LB sólido.
Para determinação da mutagênese induzida, de cada cultura foram
retirados 100µl após cada dose e novamente inoculados em meio LB líquido por
24 horas. Após este período, 100µl foram espalhados em meio LB contendo 100
µg/ml de rifampicina (LB-RIF) em duplicata. As placas foram incubadas a 37ºC por
24 h e as colônias contadas.
A sobrevivência celular N/No (ordenadas) é o quociente obtido pela divisão
do número de células sobreviventes após cada dose (N) pelo número inicial de
células (N0) plotada contra as diferentes concentrações de HN2. Cada curva de
sobrevivência foi obtida a partir da média de três experimentos.
Foram realizados experimentos de mutagênese somente com 2,2’ dipiridil e
55

todas as cepas utilizadas. Não observamos incremento de mutagênese em
nenhuma cepa.
56

RESULTADOS
Reparo de DNA
Sendo as mostardas nitrogenadas capazes de fazer ligações do tipo bi e
monoadutos (HN2) e a tal fato se atribuir a sua grande utilização em
quimioterapia, principalmente devido à formação de crosslinks, resolvemos
estudar os mecanismos pelos quais as células processam as lesões introduzidas
na molécula de DNA por estes agentes. Os estudos realizados com HN1 foram
feitos para traçar uma comparação entre reparo requerido e lesão formada, uma
vez que HN1 pode fazer apenas monoadutos, diferentemente de HN2 que tem
dois sítios ativos.
Nosso trabalho aponta o reparo NER como sendo o reparo principal para
correção das lesões causadas por HN2 (de Alencar et al., 2005) e por isso esta
mostarda foi classificada como um agente UVC–like, pois é o primeiro agente,
além do UVC, que precisa de todo o mecanismo NER para que haja correção de
suas lesões in vivo (Lage et al., 2003). O mecanismo BER participa da correção
das lesões causadas por ambas as mostardas HN2 e HN1 (de Alencar et al.,
2005).
A partir daí se tornou necessário o estudo de outras vias de reparo que
poderiam estar atuando na correção destas lesões, tais como recombinação
homóloga (de Alencar et al., 2005), proteínas envolvidas na reparação de lesões
geradas por estresse oxidativo e uma das vias de BER que elimina qualquer
possibilidade de processamento da lesão do lado 5’, através da eliminação de
duas APendonucleases. Além de outros experimentos que avaliassem a
expressão relativa dos genes NER.
57

Sobrevivência celular
Avaliação dos mecanismos de reparo de DNA envolvidos na correção das
lesões causadas por HN2 e HN1.
1. Sobrevivência celular - Estudos realizados com HN2
1.1 - Avaliação da sobrevivência das cepas deficientes em genes BER
Complementando os dados obtidos no artigo anexado, em relação ao
reparo BER, em que vimos a participação da proteína Exonuclease III (codificada
pelo gene xthA), era importante a análise do duplo mutante xthA nfo, para
sabermos se eliminando a função AP endonucleolitica da célula esta se torna mais
sensível ao tratamento, ou seja, eliminando esta função celular como se
comportaria a cepa em relação ao processamento de tais lesões (Figura 13).
Observamos que o duplo mutante BW544 (xthA nfo) é menos sensível ao
tratamento com HN2 do que o simples mutante xthA, mostrando que há
diminuição na letalidade quando não se tem as duas AP endonucleases.
58
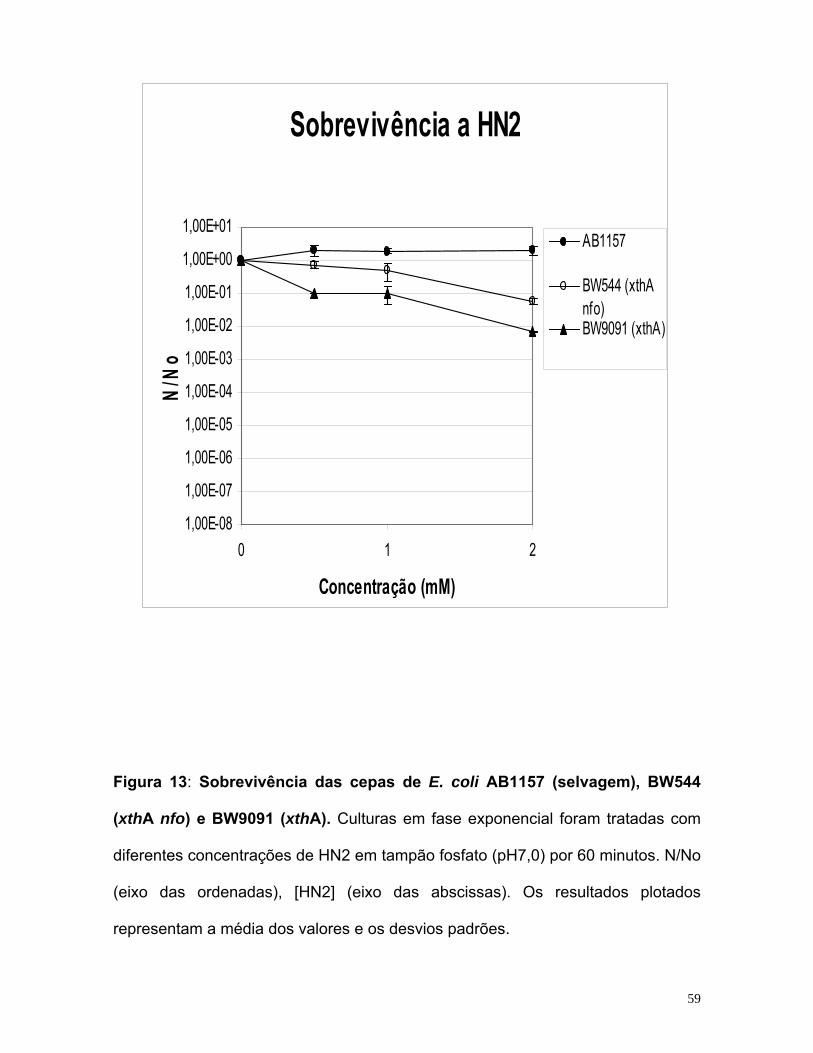
Sobrevivência a HN2
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
0 1 2
Concentração (mM)
N/N
o
AB1157
BW544 (xthAnfo)BW9091 (xthA)
Figura 13: Sobrevivência das cepas de E. coli AB1157 (selvagem), BW544
(xthA nfo) e BW9091 (xthA). Culturas em fase exponencial foram tratadas com
diferentes concentrações de HN2 em tampão fosfato (pH7,0) por 60 minutos. N/No
(eixo das ordenadas), [HN2] (eixo das abscissas). Os resultados plotados
representam a média dos valores e os desvios padrões.
59

1.2 - Avaliação da sobrevivência das cepas deficientes em genes
NER/SOS
Ainda complementando os resultados do trabalho anexado, avaliamos
também, além do simples mutante uvrA, um duplo mutante AB2480 (uvrA recA),
para sabermos se o reparo atua na mesma via.
O duplo mutante uvrA recA apresentou uma sensibilidade maior em relação
aos simples mutantes uvrA e recA, mostrando que estas enzimas podem estar
atuando em vias de reparo diferentes (Figura 14).
1.3 - Avaliação da sobrevivência das cepas deficientes no gene recA
A proteína RecA possui as funções de recombinação homóloga e faz parte
da resposta SOS, fomos analisar se as lesões causadas por HN2 dependiam de
uma destas duas funções isoladamente ou das duas, uma vez que o mutante
recA, que não possui nenhuma destas funções é extremamente sensível ao
tratamento (de Alencar et al., 2005).
As duas funções parecem ser importantes para a correção das lesões
causadas por HN2, pois a presença de uma delas não suprime a necessidade da
outra. Vemos uma grande sensibilidade do mutante GY4763 (recA142 - deficiente
em recombinação homóloga) e também no mutante GY2831 (rec430 - deficiente
na resposta SOS) (Figura 15), nos indicando que as duas funções são importantes
60

Sobrevivência a HN2
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
0 1 2Concentração (mM)
N/N
o
AB1157
AB2480 (uvrArecA)
AB1886 (uvrA)
AB2463 (recA13 - Def)
Figura 14: Sobrevivência das cepas de E. coli AB1157 (selvagem), AB2480
(uvrA recA), AB1886 (uvrA) e AB2463 (recA13 – Def). Culturas em fase
exponencial foram tratadas com diferentes concentrações de HN2 em tampão
fosfato (pH7,0) por 60 minutos. N/No (eixo das ordenadas), [HN2] (eixo das
abscissas). Os resultados plotados representam a média dos valores e os desvios
padrões.
61

Sobrevivência a HN2
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
0 1 2
Concentração (mM)
N/No
AB1157
recA 142
recA 430
AB2463 (recA13 - Def)
Figura 15: Sobrevivência das cepas de E. coli AB1157 (selvagem), GY4763
(recA142 - deficiente em recombinação homóloga), GY2831 (recA430 -
deficiente na resposta SOS) e AB2463 (recA13 – Def). Culturas em fase
exponencial foram tratadas com diferentes concentrações de HN2 em tampão
fosfato (pH7,0) por 60 minutos. N/No (eixo das ordenadas), [HN2] (eixo das
abscissas). Os resultados plotados representam a média dos valores e os desvios
padrões.
62

1.4 - Avaliação da sobrevivência da cepa deficiente no gene oxyR
Avaliando a participação de um possível estresse oxidativo, nas lesões
causadas por HN2, utilizamos a cepa deletada no gene oxyR. Este regulon
responde a altas concentrações de H2O2 e controla a expressão das catalases de
E. coli. Se HN2 fosse capaz de, ao sofrer metabolização (ativação) gerar H2O2, ou
até mesmo reagir com outra molécula gerando peróxido de hidrogênio, este
mutante seria extremamente sensível ao tratamento.
Não observamos letalidade no mutante OG100 (oxyR) (Figura 16), nos
indicando que a HN2 não está envolvida com a produção de H2O2.
2. Sobrevivência celular - Estudos realizados com HN1
2.1 - Avaliação da sobrevivência das cepas deficientes no gene recA
A partir da observação de que o mutante deficiente na proteína RecA (sem
recombinação homóloga e sem resposta SOS) é extremamente sensível ao
tratamento com HN1 (de Alencar et al., 2005), fomos analisar qual ou quais destas
funções seriam importantes para o reparo das lesões causadas por HN1.
Observamos que ambas as funções são importantes para a correção das
lesões causadas por HN1, tanto a recombinação, quanto a resposta SOS, uma
vez que nenhum dos dois mutantes é tão sensível quanto o mutante que possui
deficiência em ambas as funções (Figura 17).
63

Sobrevivência a HN2
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
0 1 2
Concentração (mM)
N/N
o
AB1157
OG100
Figura 16: Sobrevivência das cepas de E. coli AB1157 (selvagem) e OG100
(oxyR). Culturas em fase exponencial foram tratadas com diferentes
concentrações de HN2 em tampão fosfato (pH7,0) por 60 minutos. N/No (eixo das
ordenadas), [HN2] (eixo das abscissas). Os resultados plotados representam a
média dos valores e os desvios padrões.
64

Sobrevivência a HN1
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
0 100 200
Concentração (mM)
N/N
o
AB1157
recA430
recA142
recA13 - Def
Figura 17: Sobrevivência das cepas de E. coli AB1157 (selvagem), GY4763
(recA142 - deficiente em recombinação homóloga), GY2831 (recA430 -
deficiente na resposta SOS) e AB2463 (recA – Def). Culturas em fase
exponencial foram tratadas com diferentes concentrações de HN1 em tampão
fosfato (pH7,0) por 60 minutos. N/No (eixo das ordenadas), [HN1] (eixo das
abscissas). Os resultados plotados representam a média dos valores e os desvios
padrões.
65

2.2 - Avaliação da sobrevivência da cepa deficiente no gene oxyR
Uma vez que a cepa BH100 (deficiente em BER) se mostrou tão sensível a
HN1, fomos avaliar uma cepa deficiente no gene oxyR. Esta cepa possui o gene
oxyR deletado, portanto se HN1 estiver gerando H2O2, observaríamos uma
enorme letalidade, pois não teríamos a indução de catalases induzidas pelo gene
oxyR, para que houvesse a decomposição deste composto. Conforme visto para
HN2, não observamos letalidade no mutante OG100 (oxyR), nos indicando que a
letalidade causada pelas mostardas não passa pela formação de H2O2 (Figura 18).
3. Avaliação de aquisição de resistência ao antibiótico rifampicina –
Mutagênese Rifampicina
Sendo considerada a mostarda nitrogenada bifuncional HN2 como uma
substância fortemente mutagênica, através da formação de crosslinks, e sua
equivalente monofuncional como mais fracamente mutagênica, devido somente à
formação de monoadutos (Wijen et al., 2000) e, por tal motivo, conseqüentemente
agentes bifuncionais possuírem um potencial cancerígeno, fomos avaliar a
mutagênese num sistema de aquisição de resistência ao antibiótico rifampicina, já
bem descrito em E. coli. Através deste mecanismo de avaliação de mutagênese,
podemos usar as cepas deficientes nas enzimas importantes para correção das
lesões causadas pelos agentes estudados, nos dando uma visão da mutagênese
em presença ou ausência de determinado reparo.
66

Sobrevivência a HN1
1,00E-081,00E-071,00E-061,00E-051,00E-041,00E-031,00E-021,00E-011,00E+001,00E+011,00E+02
0 100 200
Concentração (mM)
N/N
o
AB1157
OG100
Figura 18: Sobrevivência das cepas de E. coli AB1157 (selvagem) e OG100
(oxyR). Culturas em fase exponencial foram tratadas com diferentes
concentrações de HN1 em tampão fosfato (pH7,0) por 60 minutos. N/No (eixo das
ordenadas), [HN1] (eixo das abscissas). Os resultados plotados representam a
média dos valores e os desvios padrões.
67

Analisamos a cepa selvagem e as cepas mutantes nos principais genes do
complexo UvrABC. Para tais experimentos utilizamos doses que levassem a célula
a DL10 (Dose letal que leva a 10% de sobrevivência). Todas estas cepas
estudadas possuem deficiência em enzimas que são requisitadas para correção
das lesões causadas por HN2. Observamos que a cepa selvagem apresentou
maior incremento de mutantes em relação aos mutantes nos genes de reparo
NER (Figura 19). Provavelmente a ausência de qualquer uma das enzimas de
NER, diminui a mutagênese.
Estudo da metabolização e letalidade produzidas pelas mostardas no DNA
A partir de dados de sobrevivência celular em relação ao reparo das lesões
causadas por HN2, sugerimos a participação de algumas enzimas que até então
só haviam sido descritas para reparar danos oxidativos, como é o caso da
Exonuclease III e Endonuclease III (de Alencar et al., 2005).
O conceito de bioativação como um mecanismo para a ação de fármacos
vem sendo cada vez mais difundido na busca de fármacos menos tóxicos e mais
seletivos. Uma área particularmente fascinante é o planejamento e o
desenvolvimento de pró-fármacos do tipo bioprecursores biorredutíveis, os quais
só se tornam ativos após redução in vivo. A descoberta da existência de células
em condições de hipóxia nos tumores sólidos pode ser explorada no
desenvolvimento desses pró-fármacos biorredutíveis. Apesar dos vários exemplos
de substâncias que apresentam atividade antitumoral após sofrerem biorredução,
esta é, ainda, uma área pouco estudada, porém muito promissora. Acredita-se que
o ajuste adequado das propriedades eletrônicas e fisico-químicas de tais
68

Mutagênese Rif - DL10
1,00E+00
1,00E+01
1,00E+02
1,00E+03
1Cepas
Incr
emen
to n
a fr
eqüê
ncia
de
m
utan
tes
AB1157
AB1886
AB1885
AB1884
Figura 19: Mutagênese Rif das cepas de E. coli AB1157 (selvagem), AB1886
(uvrA), AB1885 (uvrB) e AB1884 (uvrC). Culturas em fase exponencial foram
tratadas com concentrações de HN2 que levassem a DL10, em tampão fosfato
(pH7,0) por 60 minutos. O incremento na freqüência de mutantes (eixo das
ordenadas), e as diferentes cepas (eixo das abscissas). Os resultados plotados
representam a média dos valores e os desvios padrões.
69

substâncias possa levar ao desenvolvimento de novos fármacos
antitumorais úteis na quimioterapia dos tumores sólidos (Oliveira e Alves, 2002).
De acordo com a literatura, alguns agentes formadores de crosslinks precisam de
ativação metabólica para se ligar ao DNA, como é o caso da mitomicina C e do
benzo-[a]-pireno, dentre outros.
Sendo a mostarda nitrogenada bifuncional um quimioterápico, da classe
dos agentes alquilantes, formador de crosslinks no DNA, fomos investigar uma
possível participação de EAO na metabolização (ativação) de HN2 e HN1, e
conseqüentemente nas lesões causadas por ambas as mostardas e a formação
de EAO a partir destas moléculas. Esta ativação metabólica, que algumas drogas
necessitam, pode ser através de nitroredutases celulares (Oliveira e Alves, 2002)
e também podem gerar EAO que, teoricamente são capazes de potencializar o
efeito destes agentes, uma vez que alteram o metabolismo redox celular.
4. Estudos realizados com HN2
4.1 - Utilização do quelante de Fe2+ - 2,2 dipiridil juntamente a HN2 em
diferentes condições
Começamos os estudos de metabolização utilizando um quelante de Fe2+ -
2,2’ dipiridil (Dip), baseados em dados da literatura, de que o pré-tratamento com
quelantes de ferro protege células procarióticas e eucarióticas contra os efeitos do
H2O2 (Asad e Leitão, 1991), uma vez que diminuindo a concentração de ferro do
meio intracelular, ocorreria uma inibição da reação de Fenton, demonstrada
abaixo, e com isso diminui a formação de radicais hidroxil (OH.).
Fe2+ +H2O2 Fe3+ + OH- + OH. (Reação de Fenton – Ciclo de Harber Weiss)
Então, caso as mostardas dependessem da formação de EAO, para
70

ocasionar letalidade (via formação de EAO), produzindo ou potencializando seu
efeito, com este protocolo conseguiríamos diminuir o número de lesões no DNA,
vendo portanto uma proteção conferida pelo pré-tratamento com Dip, em relação
aos tratamentos unicamente com mostardas.
Foram feitos os primeiros experimentos utilizando a cepa selvagem
(AB1157), como um critério de padronização, uma vez que já conhecíamos o perfil
de sobrevivência dela, e sabíamos que o tratamento com HN2 não acarretava
queda significativa na sobrevivência celular, quando comparada a outros mutantes
(Figura 20a) e os mutantes AB1886 (uvrA), (Figura 20b) e BH100 (nfo nth xthA
fpg) (Figura 20c), mutantes nas principais vias de reparo requisitadas para HN2.
Ao contrário do esperado, além de não observarmos nenhuma proteção
conferida por Dip, ainda observamos um aumento de letalidade conferido pela
presença do Dip (de Alencar, 2002).
Com isso algumas hipóteses sobre este fenômeno de maior letalidade
foram questionadas, tais como:
Geração de um composto mais letal, formado entre o quelante Dip e
HN2;
Situação metabólica deixada pelo Dip, que contribuísse para o aumento de
letalidade induzida por HN2. É esperado que células tratadas com Dip, em que
temos a diminuição de ions Fe2+ no meio, sofram uma explosão respiratória,
aumentando com isso os níveis de radical superóxido. Já foi observado que o
tratamento com quelantes de ferro é capaz de induzir o aumento de expressão da
enzima Superóxido Dismutase (codificada pelo gene sodA) (Touati, 1988). Este
fato é de extrema importância para a célula, pois com este aumento do radical
superóxido, há maior doação de elétrons para os íons Fe3+, que são reduzidos a
71

Sobrevivência a HN2 com pré-tratamento com 2,2' dipiridil
1,00E+00
1,00E+01
N/N
o
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
0 1 2
Concentração (mM)
AB1157(selvagem) (HN2)
AB1157(selvagem) (Pre-tratamento + HN2)
Sobrevivência a HN2 com pré-tratamento com 2,2'dipiridil
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
0 1 2
Concentração (mM)
N/N
o
AB1886 (uvrA)(HN2)
AB1886 (uvrA)(Pre-tratamento Dip+ HN2)
Sobrevivência a HN2 com pré-tratamento com 2,2' dipiridil
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
0 1 2
Concentração (mM)
N/N
o
BH100 (quadruplomutante) (HN2)
BH100 (quadruplomutante) (Pre-tratamento Dip +HN2)
Figura 20: Sobrevivência das cepas de E. coli (a) AB1157 (selvagem), (b)
AB1886 (uvrA) e (c) BH100 (nfo nth xthA fpg). Culturas das cepas em fase
exponencial pré-tratadas ou não com 2,2’ dipiridil e posteriormente tratadas com
diferentes concentrações de HN2, em tampão fosfato (pH7,0) por 60 minutos.
N/No (eixo das ordenadas), [HN2] (eixo das abscissas). Os resultados plotados
representam a média dos valores obtidos e os desvios padrões.
72

Fe2+ (reação demonstrada abaixo) (Touati et al., 1995), que é o ferro
metabolicamente envolvido nas reações celulares.
Fe3+ + O2-.→ Fe2+ + O2 (equação 1 do Ciclo de Harber Weiss)
O radical superóxido poderia então, de alguma maneira influenciar no
aumento de letalidade causado por HN2.
Outras hipóteses seriam a possível intercalação do quelante no DNA,
ataque ao núcleo Fe-S de algumas proteínas, formação de um complexo redox
que sensibilizasse ainda mais a célula do que somente HN2, dentre outras.
Para testarmos algumas destas hipóteses (situação metabólica e geração
de um composto mais letal) realizamos experimentos com a cepa selvagem
(AB1157), com Dip e HN2 em diferentes situações, alem do pré-tratamento com
20 minutos 1mM de Dip e posterior tratamento com concentrações crescentes de
HN2. Fixamos a maior concentração de HN2 em 2mM, a fim de produzirmos maior
número de lesões e realizamos experimentos com pré-tratamento com Dip (1mM)
e outros com centrifugação após o tratamento com Dip, para garantir que não
teríamos mais o quelante na célula e fizemos também um tratamento simultâneo
Dip e HN2.
Observamos que dentre todos os tratamentos realizados o mais letal para a
célula é quando esta é pré-tratada com o quelante e posteriormente tratada com
HN2 (Figura 21). No tratamento em que há uma centrifugação pós-tratamento com
Dip, vemos ligeiro aumento no nível de sobrevivência, seguido de um maior
aumento no tratamento simultâneo Dip e HN2 (Figura 21).
Para testar a hipótese da formação de um composto mais letal para a
célula, gerado entre Dip e HN2, fizemos outros experimentos reagindo estas
73
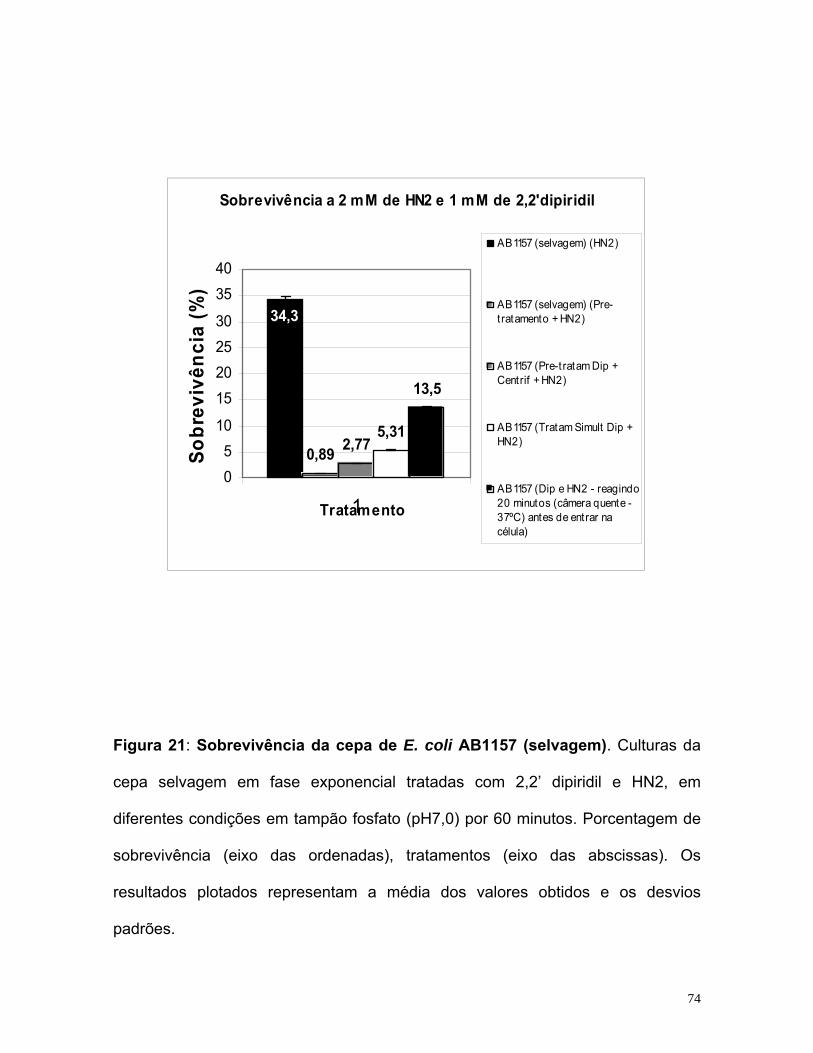
Sobrevivência a 2 mM de HN2 e 1 mM de 2,2'dipiridil
0,89 2,775,31
13,5
34,3
05
10
15202530
3540
1Tratamento
Sobr
eviv
ênci
a (%
)
AB1157 (selvagem) (HN2)
AB1157 (selvagem) (Pre-tratamento + HN2)
AB1157 (Pre-t ratam Dip +Centrif + HN2)
AB1157 (Tratam Simult Dip +HN2)
AB1157 (Dip e HN2 - reagindo20 minutos (câmera quente -37ºC) antes de entrar nacélula)
Figura 21: Sobrevivência da cepa de E. coli AB1157 (selvagem). Culturas da
cepa selvagem em fase exponencial tratadas com 2,2’ dipiridil e HN2, em
diferentes condições em tampão fosfato (pH7,0) por 60 minutos. Porcentagem de
sobrevivência (eixo das ordenadas), tratamentos (eixo das abscissas). Os
resultados plotados representam a média dos valores obtidos e os desvios
padrões.
74

substâncias a 37º C por 20 minutos, nas mesmas concentrações que
utilizamos nas células (1mM de Dip + 2mM de HN2). O que observamos foi que
não parece haver uma reação que gere um composto ainda mais letal para a
célula, do que as duas moléculas sozinhas (Figura 21), pois neste caso teríamos
uma extrema sensibilidade quando reagíssemos as duas moléculas e tratássemos
as células com este produto da reação.
Para teste da hipótese do Dip estar gerando radical superóxido, a partir da
quelação de Fe2+, avaliamos a sobrevivência celular do mutante JI132 (sodA
sodB). A ausência das enzimas Superóxido Dismutases, tanto a induzida - SodA,
quanto a constitutiva – SodB, faz com esta bactéria não dismute radical
superóxido (via enzimática), sendo este produzido espontaneamente ou como
conseqüência de algum tratamento, então os níveis deste radical nesta cepa, são
bem maiores, quando comparados a uma cepa selvagem. Então, utilizamos esta
cepa, além da cepa AB1157 (selvagem), como controle para estabelecermos um
background de altas e baixas concentrações deste radical, para avaliar se HN2 se
tornaria ainda mais letal. Os experimentos foram realizados sob diferentes
agitações, para que hovesse maior difusão de oxigênio e com isso o tratamento
com Dip gerasse mais superóxido, e com a concentração máxima usada para HN2
– 2 mM (Figura 22).
Para a cepa AB1157 (selvagem) não observamos diferença em relação às
diferentes condições de agitação utilizadas, ou seja, tanto em 0, 165 ou 240 rpm, o
efeito lesivo é o mesmo, tanto para a sobrevivência só com HN2, quanto para
Dip+ HN2. Em relação à cepa JI132 (sodA sodB), o tratamento feito somente com
HN2, não foi capaz de causar queda acentuada, como esperado para uma cepa
75

Sobrevivência a 2 mM de HN2 e 1 mM de 2,2'dipiridil
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
0 50 100 150 200 250
Rpm
N/N
o
AB1157 (selvagem) (HN2)
AB1157 (selvagem) (pre-t rat ament o Dip + HN2)
JI132 (sodAsodB) (HN2)
JI132 (sodAsodB) (Pre-t rat ament o Dip + HN2)
Figura 22: Sobrevivência das cepas de E. coli AB1157 (selvagem) e JI132
(sodA sodB). Culturas das cepas em fase exponencial pré-tratadas ou não com
2,2’ dipiridil e posteriormente tratadas com a concentração de 2mM de HN2, em
tampão fosfato (pH7,0) por 60 minutos em diferentes rpm. N/No (eixo das
ordenada), rpm (eixos das abscissas). Os resultados plotados representam 0, 165
e 240 rpm, respectivamente, e são a média dos valores obtidos e os desvios
padrões.
76

selvagem para reparo de DNA, entretanto, o tratamento realizado com o
pré-tratamento com Dip + HN2, causou uma considerável queda na sobrevivência
celular.
Podemos destacar também que para a cepa sodA sodB, o pré-tratamento
com Dip + tratamento com HN2 levou a uma queda muito maior em relação ao
tratamento somente com HN2, do que para as outras cepas testadas, AB1157
(selvagem), AB1886 (uvrA) e BH100 (nfo nth xthA fpg).
Realizamos experimentos com a cepa sodA sodB, fixando uma única
concentração de pré-tratamento com Dip (1mM) e tratando posteriormente com
concentrações crescentes de HN2, como foi feito anteriormente para as outras
cepas, com a finalidade de sabermos se em uma cepa que não dismuta o radical
superóxido, este radical pode aumentar progressivamente a letalidade da HN2
(Figura 23).
Observamos que a letalidade induzida por HN2 vai sendo aumentada de
acordo com o aumento da sua concentração, tanto para o tratamento somente
com HN2, quanto para o tratamento com HN2, precedido de Dip. Na cepa sodA
sodB, o fenômeno de maior letalidade que ocorre com o pré-tratamento com Dip,
também ocorre, em todas as concentrações estudadas, e neste caso, em maior
proporção, do que para as cepas selvagem uvrA e BH100. Tal fato, sugere mais
uma vez que o radical superóxido presente em altas concentrações na célula,
torna a HN2 ainda mais reativa.
Para testar se o possível aumento de radical superóxido era capaz de
sensibilizar a célula, na mesma concentração de HN2, mostrando que o aumento
77

Sobrevivência a HN2 com pré-tratamento com 2,2' dipiridil
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
0 1 2
Concentração (mM)
N/N
o
JI132 (sodAsodB)(HN2)
JI132 (sodAsodB)(Pre-tratamento comDip HN2)
Figura 23: Sobrevivência da cepa de E. coli JI132 (sodA sodB). Culturas das
cepas em fase exponencial pré-tratadas ou não com 2,2’ dipiridil e posteriormente
tratadas com diferentes concentrações de HN2, em tampão fosfato (pH7,0) por 60
minutos. N/No (eixo das ordenadas), [HN2] (eixo das abscissas). Os resultados
plotados representam a média dos valores obtidos e os desvios padrões.
78

destes radicais no meio intracelular, torna a mesma concentração de HN2
mais reativa, foram feitos experimentos com a cepa AB1157 (selvagem) e JI132
(sodA sodB), definindo assim um background de baixa e alta concentração de
radical superóxido, com diferentes concentrações de Dip, a fim de aumentarmos a
produção de superóxido, e a célula foi posteriormente tratada com a mesma
concentração de HN2 (Figuras 24a e 24b). Podemos observar que para ambas as
cepas, conforme aumentamos a concentração de Dip e, conseqüentemente o
nível de radical superóxido, a mesma concentração de mostarda se torna mais
reativa, observando-se uma diminuição na sobrevivência celular. Este fenômeno é
mais acentuado no mutante sodA sodB, provavelmente pela não dismutação do
radical superóxido, indicando mais uma vez, que tal radical de alguma forma
interage com HN2, aumentando com isso o número de lesões na célula.
Foram realizados experimentos com simples mutantes nas duas
Superóxido Dismutases A e B – JI130 (sodA) e JI131 (sodB) presentes em E. coli,
a fim de sabermos se na falta de uma delas e na presença de outra teríamos a
concentração de radical superóxido diminuída e com isso o efeito do Dip diminuiria
(Figuras 25a e 25b).
Nenhuma das duas cepas testadas apresentou sensibilidade acentuada ao
pré-tratamento com Dip e posterior tratamento com HN2. Podemos dizer que
praticamente não houve sensibilização com o tratamento com Dip, quando
comparado com os resultados obtidos com o duplo mutante sodA sodB. Neste
caso, a presença de uma Superóxido Dismutase parece suprir (ser suficiente) a
falta da outra, diminuindo com isso as lesões causadas pelo duplo tratamento Dip
+ HN2.
79

Sobrevivência a 2 mM de HN2 e diferentes concentrações de 2,2' dipiridil
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
0 2 4
Concentração Dip (mM)
N/N
o
Sobrevivência a 2mM de HN2 e diferentes concentrações de 2,2' dipiridil
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
0 2 4
Concentração Dip (mM)
N/N
oJI132 (sodAsodB)
AB1157 (selvagem)
a
b
Figura 24: Sobrevivência das cepas de E. coli (a) AB1157 (selvagem) e (b)
JI132 (sodA sodB). Culturas das cepas em fase exponencial pré-tratadas com
diferentes concentrações de 2,2’ dipiridil e posteriormente tratadas 2mM de HN2,
em tampão fosfato (pH7,0) por 60 minutos. N/No (eixo das ordenadas), [2,2’
dipiridil] (abscissas). Os resultados plotados representam a média dos valores
obtidos e os desvios padrões.
80

Sobrevivência a HN2 com pré-tratamento com 2,2' dipiridil
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
0 1 2
Concentração (mM)
N/N
o
JI130 (sodA)(HN2)
JI130 (sodA) (Pre-tratamento)HN2)
Sobrevivência a HN2 com pré-tratamento com 2,2' dipiridil
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
0 1 2
Concentração (mM)
N/N
o
JI131 (sodB)(HN2)
JI131 (sodB) (Pre-tratamento)HN2)
Figura 25: Sobrevivência das cepas de E. coli (a) JI130 (sodA) e (b) JI131
(sodB). Culturas das cepas em fase exponencial pré-tratadas ou não com 2,2’
dipiridil e posteriormente tratadas com diferentes concentrações de HN2, em
tampão fosfato (pH7,0) por 60 minutos. N/No (eixo das ordenadas), [HN2] (eixo
das abscissas). Os resultados plotados representam a média dos valores obtidos
e os desvios padrões.
81

Como já foi dito anteriormente o ferro é um elemento essencial para a
célula. Bactérias utilizam o ferro para transporte de oxigênio, sintese de DNA,
transporte de elétrons, alem de fazer parte da metabolização de peróxidos.
Alterações na concentração de ferro do meio intracelular causam uma perturbação
redox na célula. Na tentativa de diminuir a concentração de Fe3+ (lembrando que o
Fe2+ já estava sendo quelado) no meio, para com isso haver um aumento da
concentração de radical superóxido, promovido pelo pré-tratamento com Dip, uma
vez que este teria menos ferro para reduzir e com isso se tornar mais disponível
para reagir com HN2, foram realizados experimentos com uma cepa deletada no
gene fur - QC1732 (∆fur), que codifica a proteína responsável pela regulação de
ferro no meio intacelular, e a cepa da qual foi originada (GC4468 – selvagem)
(Figura 26). Os tratamentos geraram níveis de sobrevivência semelhantes.
Provavelmente a quelação de Fe3+ não foi significativa a ponto de deixar uma
situação em que grande parte do radical superóxido doasse o seu elétron para
HN2.
A deleção no gene fur não aumentou a sensibilidade ao pré-tratamento com
Dip e posterior tratamento com HN2, pois este mutante é tão sensível quanto a
sua cepa selvagem, em ambos os tratamentos.
4.2 - A influência do captador de radicais livres tiouréia nas lesões
causadas por HN2
Como todos os indícios apontam para a geração de radical superóxido,
durante o tratamento com Dip e que este radical, de alguma maneira torna HN2
ainda mais reativa, sensibilizando ainda mais as células, realizamos experimentos
com o captador de radicais livres, tiouréia.
82
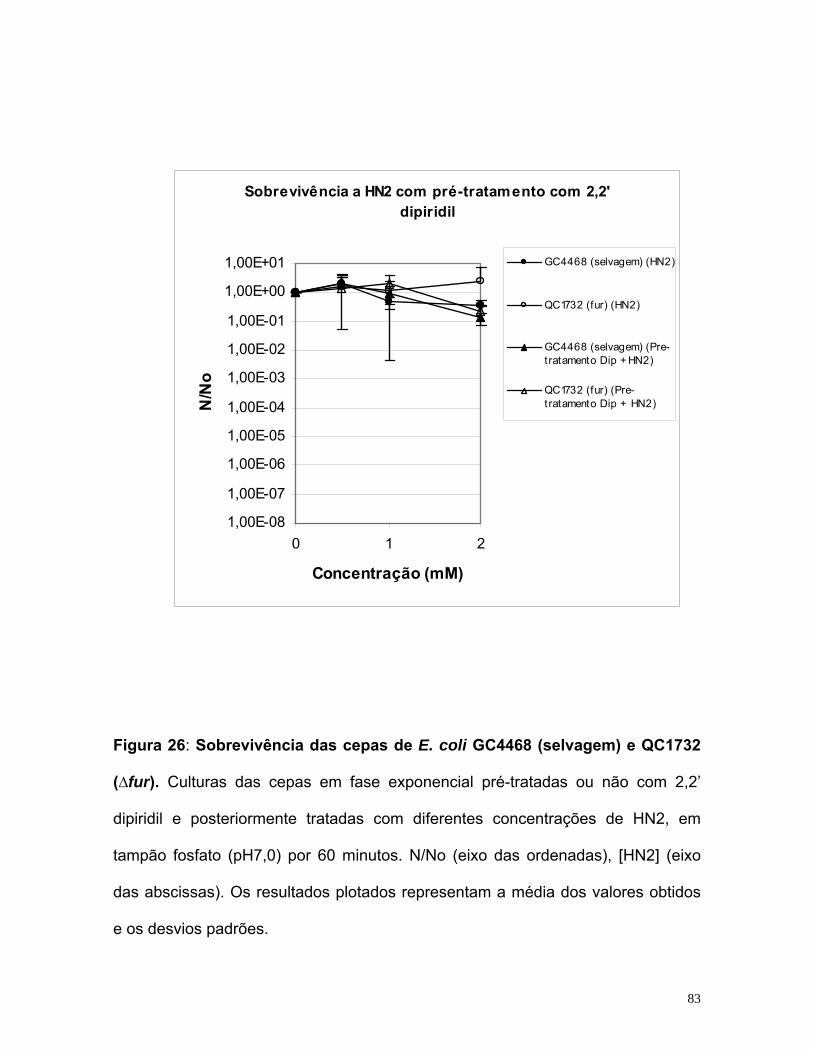
Sobrevivência a HN2 com pré-tratamento com 2,2' dipiridil
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
0 1 2
Concentração (mM)
N/N
o
GC4468 (selvagem) (HN2)
QC1732 (fur) (HN2)
GC4468 (selvagem) (Pre-tratamento Dip + HN2)
QC1732 (fur) (Pre-tratamento Dip + HN2)
Figura 26: Sobrevivência das cepas de E. coli GC4468 (selvagem) e QC1732
(∆fur). Culturas das cepas em fase exponencial pré-tratadas ou não com 2,2’
dipiridil e posteriormente tratadas com diferentes concentrações de HN2, em
tampão fosfato (pH7,0) por 60 minutos. N/No (eixo das ordenadas), [HN2] (eixo
das abscissas). Os resultados plotados representam a média dos valores obtidos
e os desvios padrões.
83

A tiouréia tem sido freqüentemente utilizada em experimentos in vivo e in
vitro, na tentativa de capturar radicais livres gerados a partir de reações de
degradação do H2O2 e outros agentes oxidativos (Asad e Leitão, 1991). É um forte
captador de radicais OH., porém não é um captador específico, podendo capturar
outros radicais livres presentes no meio, além de ser um inibidor de Xantina
Oxidase, reagir diretamente com o H2O2, se ligar a proteínas de membrana e
também a íons metálicos, em caminhos que formam radicais OH.(Wasil et al.,
1987).
Os experimentos foram realizados com a cepa AB1157 (selvagem) e a
cepa JI132 (sodA sodB), novamente na tentativa do pré-tratamento com Dip gerar
uma situação de diferentes concentrações de superóxido. A tiouréia foi adicionada
juntamente com o Dip (no pré-tratamento), ou imediatamente após o pré-
tratamento com este quelante, junto com HN2 (Figuras 27a e 27b). A utilização
deste captador promoveu um aumento nos níveis de sobrevivência de ambas as
cepas, selvagem e sodA sodB, elevando a sobrevivência a níveis similares aos
observados somente com HN2, provavelmente pela captura do superóxido que
está sendo gerado a partir do tratamento com Dip.
Utilizamos a tiouréia também em experimentos com as principais cepas
envolvidas na correção de HN2 – AB1157 (selvagem) (Figura 28a), como controle,
AB1886 (uvrA) (Figura 28b) e BH100 (nfo nth xthA fpg) (Figura 28c), na tentativa
de sabermos se somente o tratamento com HN2 era capaz de gerar EAO.
84

Sobrevivência a 2 mM de HN2 com pré-tratamentos com 2,2' dipiridil e tiouréia
8,00E-01
1,36
E-02
9,76
E-01
1,02
E+00
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
1
Tratamento
N/N
o
AB1157 (selvagem) (HN2)
AB1157 (selvagem) (Pre-t rat ament o Dip + HN2)
AB1157 (selvagem) (Pre-t rat ament o duplo + HN2)
AB1157 (selvagem) (Pre-t rat ament o Dip + Tioureia e HN2)
Sobrevivência a 2 mM de HN2 com pré-tratamentos com 2,2' dipiridil e tiouréia
1,19E-02
9,85
E-07
4,04
E-01
6,38
E-01
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
1
Tratamento
N/N
o
JI132 (sodAsodB) (HN2)
JI132 (sodAsodB) (Pre-t ratamento Dip + HN2)
JI132 (sodAsodB) (Pre-t ratamento duplo + HN2)
JI132 (sodAsodB) (Pre-t ratamento Dip + Tioureia e HN2)
Figura 27: Sobrevivência das cepas de E. coli (a) AB1157 (selvagem) e (b)
JI132 (sodA sodB). Culturas das cepas em fase exponencial tratadas com 2,2’
dipiridil, tiouréia e HN2 em diferentes concentrações, em tampão fosfato (pH7,0)
por 60 minutos. N/No (eixo das ordenadas), tratamentos (eixo das abscissas). Os
resultados plotados representam a média dos valores obtidos e os desvios
padrões.
85
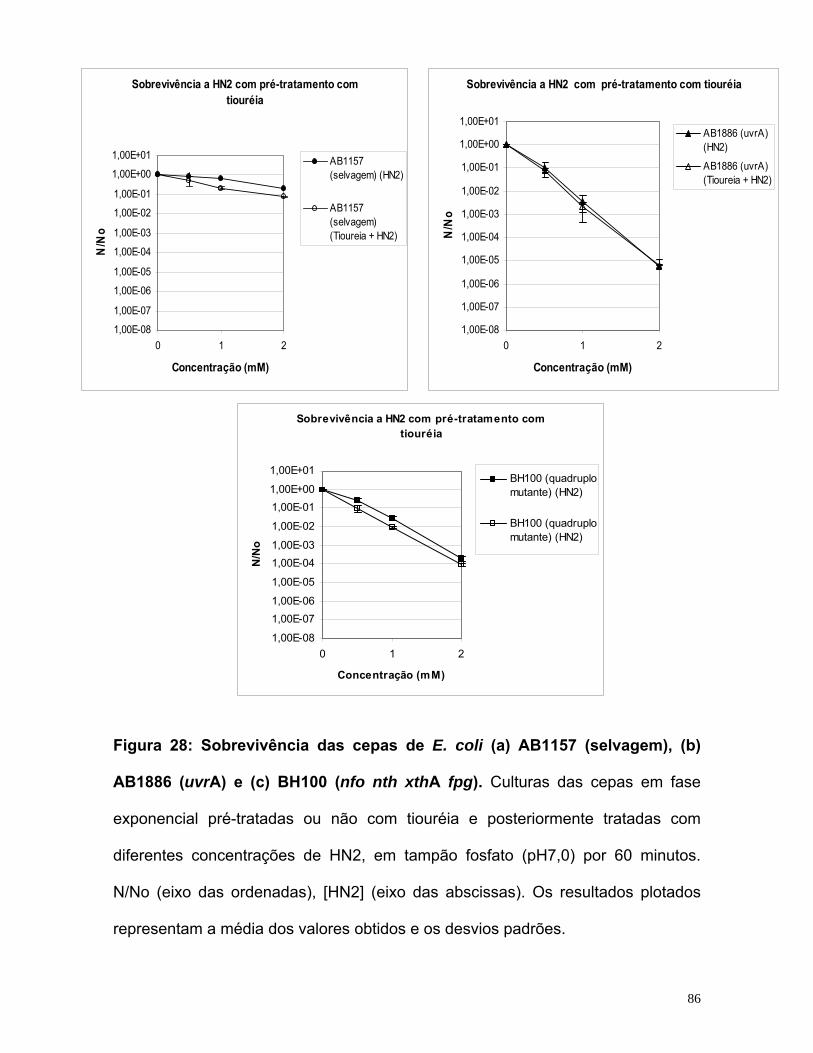
Sobrevivência a HN2 com pré-tratamento com tiouréia
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
0 1 2
Concentração (mM)
N/N
o
AB1157(selvagem) (HN2)
AB1157(selvagem)(Tioureia + HN2)
Sobrevivência a HN2 com pré-tratamento com tiouréia
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
0 1 2
Concentração (mM)
N/N
o
AB1886 (uvrA)(HN2)
AB1886 (uvrA)(Tioureia + HN2)
Sobrevivência a HN2 com pré-tratamento com tiouréia
1,00E-08
1,00E-071,00E-06
1,00E-05
1,00E-041,00E-03
1,00E-02
1,00E-011,00E+00
1,00E+01
0 1 2
Concentração (mM)
N/N
o
BH100 (quadruplomutante) (HN2)
BH100 (quadruplomutante) (HN2)
Figura 28: Sobrevivência das cepas de E. coli (a) AB1157 (selvagem), (b)
AB1886 (uvrA) e (c) BH100 (nfo nth xthA fpg). Culturas das cepas em fase
exponencial pré-tratadas ou não com tiouréia e posteriormente tratadas com
diferentes concentrações de HN2, em tampão fosfato (pH7,0) por 60 minutos.
N/No (eixo das ordenadas), [HN2] (eixo das abscissas). Os resultados plotados
representam a média dos valores obtidos e os desvios padrões.
86

A tiouréia não foi capaz de proteger a célula das lesões causadas por HN2,
em nenhuma concentração utilizada. Provavelmente, HN2 não é capaz de gerar
radicais livres, sem o auxílio de um componente que altere o sistema redox da
célula.
4.3 - Avaliação de aquisição de resistência ao antibiótico rifampicina –
Mutagênese rifampicina.
Sendo o pré-tratamento com Dip seguido da adição de HN2, o tratamento
mais letal, em termos de sobrevivência, em relação a todos os tratamentos já
descritos, envolvendo estes dois agentes e provavelmente sendo o radical
superóxido, devido a todas as descrições já feitas anteriormente, o grande
potencializador deste efeito, fomos estudar a mutagênese ocasionada por este
tratamento, comparando com o tratamento realizado somente com HN2. Para tais
experimentos utilizamos novamente as cepas AB1157 (selvagem) e o duplo
mutante JI132 (sodA sodB) (Figura 29).
Utilizamos a mutagênese de aquisição de resistência ao antibiótico
rifampicina, descrita anteriormente. Observamos que, o incremento no número de
mutantes no pré-tratamento com Dip e HN2, na cepa sodA sodB, apresenta-se
maior, em relação ao tratamento somente com HN2, mostrando que as lesões
feitas na presença de altas concentrações de superóxido são mais mutagênicas.
Na cepa selvagem a diferença não parece ser significativa entre os dois
tratamentos, provavelmente pelo fato desta cepa possuir Superóxido Dismutase.
87

Mutagênese Rif - DL10
1,00E+00
1,00E+01
1,00E+02
1,00E+03
1Tratamento
AB1157 -HN2AB1157 -Dip + HN2JI132 - HN2
JI132 - Dip+ HN2
Figura 29: Mutagênese Rif da cepa de E. coli AB1157 (selvagem) e JI132
(sodA sodB). Culturas em fase exponencial foram tratadas com concentrações de
HN2 que levassem a DL10, em tampão fosfato (pH7,0) por 60 minutos. O
incremento na freqüência de mutantes (eixo das ordenadas), tratamentos (eixo
das abscissas). Os resultados plotados representam a média dos valores e os
desvios padrões.
88

4.4 - A influência do quelante de Fe3+ - desferroxiamina nas lesões
causadas por HN2
Ainda na investigação do mecanismo sobre metabolização das mostardas,
através do uso de doadores de elétrons, utilizando quelantes de ferro como
promotores de tal situação, utizamos a desferroxiamina, que é um quelante de
Fe3+. Com isso, poderíamos tirar a dúvida se a letalidade ocasionada pelo duplo
tratamento com Dip e HN2 poderia estar somente relacionada com a produção de
radical superóxido ou também ser promovida pela escassez de ferro na célula,
fato que altera o metabolismo redox no meio intracelular.
A desferroxiamina tem sido um dos quelantes utilizados em quimioterapia,
uma droga aprovada para o tratamento de sobrecarga de ferro. Estudos têm
mostrado que este quelante pode retardar o crescimento tumoral em muitos
contextos experimentais diferentes. Entretanto a atividade da desferroxiamina é
moderada. Existem outros quelantes de ferro que tem mostrado uma atividade
antitumoral promissora (Buss et al., 2003).
O pré-tratamento com desferroxiamina (1mM) e posterior tratamento com
HN2 (2mM) não sensibiliza tanto a célula, quanto o pré-tratamento com Dip e
posterior tratamento com HN2, não mostrando uma diferença significativa entre
este (desferroxiamina + HN2) e somente HN2 em nenhuma das cepas testadas
(Figuras 30a, 30b e 30c). Provavelmente o processo de quelação de ferro, não é o
causador da maior letalidade ocasionada nos tratamentos com Dip e HN2.
89
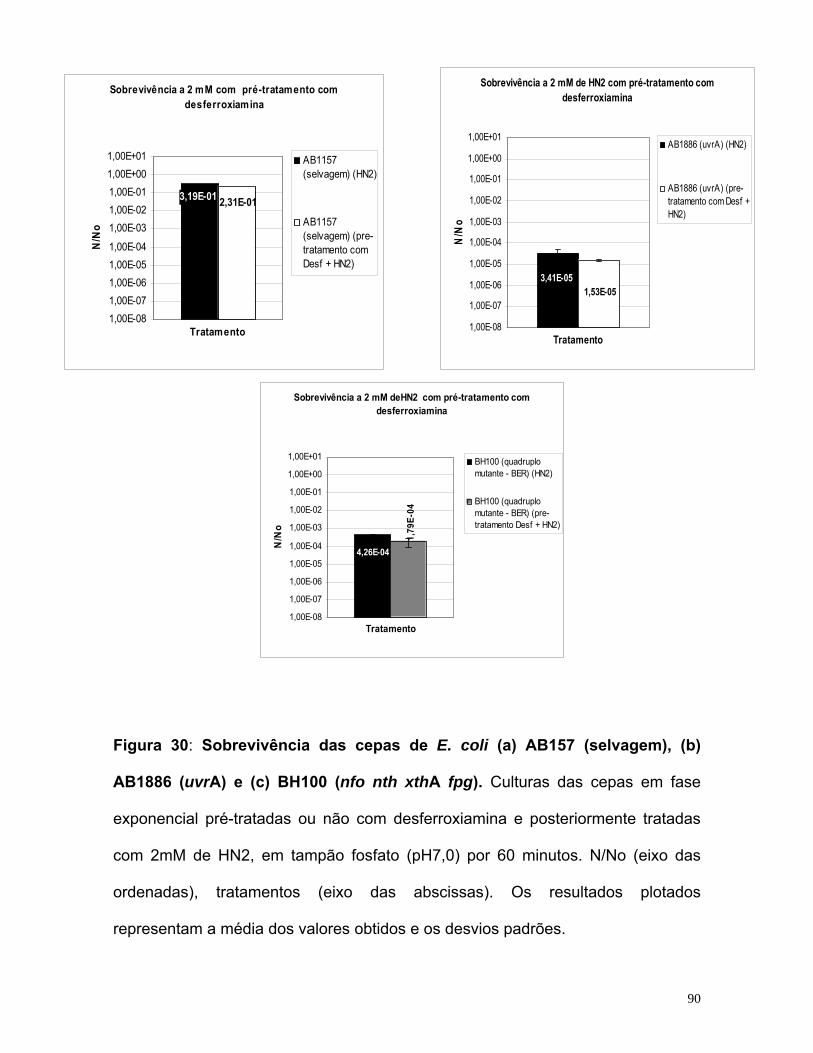
Sobrevivência a 2 mM de HN2 com pré-tratamento com desferroxiamina
3,41E-051,53E-05
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
1
Tratamento
N/N
o
AB1886 (uvrA) (HN2)
AB1886 (uvrA) (pre-tratamento com Desf +HN2)
Sobrevivência a 2 mM com pré-tratamento com desferroxiamina
3,19E-01 2,31E-01
1,00E-081,00E-071,00E-061,00E-051,00E-04
1,00E-031,00E-021,00E-011,00E+001,00E+01
1
Tratamento
N/N
o
AB1157(selvagem) (HN2)
AB1157(selvagem) (pre-tratamento comDesf + HN2)
Sobrevivência a 2 mM deHN2 com pré-tratamento com
desferroxiamina
4,26E-04
1,79
E-04
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
1
Tratamento
N/N
o
BH100 (quadruplomutante - BER) (HN2)
BH100 (quadruplomutante - BER) (pre-tratamento Desf + HN2)
Figura 30: Sobrevivência das cepas de E. coli (a) AB157 (selvagem), (b)
AB1886 (uvrA) e (c) BH100 (nfo nth xthA fpg). Culturas das cepas em fase
exponencial pré-tratadas ou não com desferroxiamina e posteriormente tratadas
com 2mM de HN2, em tampão fosfato (pH7,0) por 60 minutos. N/No (eixo das
ordenadas), tratamentos (eixo das abscissas). Os resultados plotados
representam a média dos valores obtidos e os desvios padrões.
90

4.5 - Utilização dos quelantes de Fe2+ e Fe3+ , 2,2 dipiridil e
desferroxiamina
Utilizamos uma estratégia experimental de utilizar um pré-tratamento duplo
com Dip + desferroxiamina e posterior tratamento com HN2, a fim de sabermos se
na situação de alto nível de superóxido, e diminuição de Fe3+, para quem
teoricamente este radical doaria elétrons, potencializaríamos o efeito de HN2,
através da doação deste elétron, utilizando para isso, além da cepa selvagem
(Figura 31a) o mutante mais sensível para as lesões causadas por HN2 – AB1886
(uvrA) (Figura 31b).
O duplo pré-tratamento Dip (1mM) + desferroxiamina (1mM) e posterior
tratamento com HN2 não se apresentou mais letal do que o pré-tratamento com
Dip e posterior tratamento com HN2, ou seja adição de desferral não potencializa
o efeito do Dip.
5. Estudos realizados com HN1
Ainda que HN1 (análoga monofuncional de HN2) não seja utilizada em
quimioterapia, as lesões causadas por HN1 (monoadutos) constituem um bom
modelo para o estudo comparativo entre HN2 e HN1, uma vez que HN2 pode
causar bi e monoadições no DNA, tanto em termos de reparo, como também para
se conhecer melhor o mecanismo de atuação destas mostardas.
5.1 - A influência do quelante de Fe2+ - 2,2’dipiridil nas lesões
causadas por HN1
Realizamos experimentos utilizando a cepa AB1157 (selvagem) (Figura
32a), como um critério de padronização, uma vez que já conhecíamos o perfil de
91

Sobrevivência a 2 mM de HN2 com pré-tratamento 2,2' dipiridil e desferroxiamina
9,63E-01
2,40
E-02
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
1
Tratamento
N/N
o
AB1157 (selvagem)(HN2)
AB1157 (selvagem)(pre-tratamento Dip+ Desf + HN2)
Sobrevivência a 2 mM de HN2 com pré-tratamento 2,2' dipiridil e desferroxiamina
7,32E-06
1,47
E-07
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
1Tratamento
N/N
oAB1886 (uvrA) (HN2)
AB1886 (uvrA) (pre-tratamento Dip + Desf+ HN2)
Figura 31: Sobrevivência das cepas de E. coli (a) AB1157 (selvagem) e (b)
AB1886 (uvrA). Culturas das cepas em fase exponencial pré-tratadas ou não com
2,2 dipiridil e desferroxiamina e posteriormente tratadas com 2mM de HN2, em
tampão fosfato (pH7,0) por 60 minutos. N/No (eixo das ordenadas), tratamentos
(eixo das abscissas). Os resultados plotados representam a média dos valores
obtidos e os desvios padrões.
92

Sobrevivência a HN1 com pré-tratamento com 2,2' dipiridil
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
0 100 200
Concentração (mM)
N/N
o
AB1157(selvagem) (HN1)
AB1157(selvagem) (Pre-tratamento Dip +HN1)
Sobrevivência a HN1 com pré-tratamento com 2,2' dipiridil
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
0 100 200
Concentração (mM)
N/N
o
BH100(quadruplomutante) (HN1)
BH100(quadruplomutante) (Pre-tratamento +HN1)
Sobrevivência a HN1 com pré-tratamento com 2,2' dipiridil
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
0 100 200
Concentração (mM)
N/N
o
AB1886 (uvrA)(HN1)
AB1886 (uvrA)(Pre-tratamento +HN1)
Figura 32: Sobrevivência das cepas de E. coli (a) AB1157 (selvagem), (b)
BH100 (nfo nth xthA fpg) e (c) AB1886 (uvrA). Culturas das cepas em fase
exponencial pré-tratadas ou não com 2,2 dipiridil e posteriormente tratadas com
diferentes concentrações de HN1, em tampão fosfato (pH7,0) por 60 minutos.
N/No (eixo das ordenadas), [HN1] (eixo das abscissas). Os resultados plotados
representam a média dos valores obtidos e os desvios padrões.
93

sobrevivência dela, e, conhecíamos também que o tratamento com HN1 não
acarretava queda significativa na sobrevivência celular, quando comparada aos
demais mutantes. Utilizamos além da cepa BH100 (nfo nth xthA fpg) (Figura 32b),
que é deficiente no principal reparo associado as lesões causadas por HN1 (de
Alencar et al., 2005), a cepa AB1886 (uvrA) (Figura 32c), para averiguarmos se o
pré-tratamento com Dip era capaz de sensibilizar esta cepa a HN1, uma vez que
esta é mutante em uma das principais vias de reparo de DNA da célula, NER. Os
tratamentos foram semelhantes aos primeiros experimentos realizados para HN2,
com a mesma dose de Dip e aumentando as concentrações de HN1.
Não observamos o mesmo fenômeno que ocorreu com HN2, ou seja, não
houve diferença entre o pré-tratamento com Dip e posterior tratamento com HN1
(diferentes doses por 60 minutos), para o tratamento somente com HN1. O
quelante de Fe2+ - Dip não foi capaz de aumentar a letalidade promovida por HN1,
como visto anteriormente para HN2. Provavelmente o radical superóxido esteja
influenciando na formação (favorecendo o aumento) somente da segunda ligação
ao DNA, feita por HN2, ou até mesmo ativando o segundo lado da molécula, uma
vez que HN1 não é capaz de fazer esta ligação. Talvez por isso não sofra a
influência deste quelante.
5.2 - A influência do captador de radicais livres tiouréia nas lesões
causadas por HN1.
Utilizamos o captador de radicais livres tiouréia, em experimentos utilizando
os principais mutantes nas enzimas envolvidas na correção das lesões causadas
94

por ambas as mostadas – HN2 e HN1, AB1157 (selvagem) (controle)
(Figura 33a), AB1886 (uvrA) (Figura 33b) e BH100 (nfo nth xthA fpg) (Figura 33c),
na tentativa de sabermos se somente o tratamento com HN1 era capaz de gerar
EAO.
A tiouréia não foi capaz de proteger a célula das lesões causadas por HN1,
em nenhuma concentração utilizada. HN1 não parece gerar radicais livres no seu
processo de metabolização (ativação).
5.3 - A influência do quelante de Fe3+ - desferroxiamina nas lesões
causadas por HN1
Ainda na tentativa de se estabelecer uma comparação entre as lesões
causadas por ambas as mostardas, com isso tentando entender melhor
metabolização e o reparo destes agentes, estudamos a influência da
desferroxiamina nas lesões causadas por HN1.
O pré-tratamento com desferroxiamina (1mM) não sensibiliza as células ao
posterior tratamento com HN1, nas cepas AB1157 (selvagem) e AB1886 (uvrA)
(Figuras 34a e 34b), conforme visto anteriormente para HN2. Entretanto para a
cepa BH100 (nfo nth xthA fpg), parece haver uma pequena sensibilização
ocasionada pela desferroxiamina (Figura 34c).
Para as lesões causadas por HN1, não observamos influência de nenhum
quelante de ferro e nem nenhum captador de radicais livres. Provavelmente a
metabolização (ativação) desta mostarda não requer e nem gera EAO para
provocar letalidade e por isso, não parece ser potencializada por nenhum doador
de elétrons.
95

Sobrevivência a HN1 com pré-tratamento com e sem tiouréia
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
0 1 2
Concentração (mM)
N/N
o
AB1886 (uvrA)(HN1)
AB1886 (uvrA)(Tioureia + HN1)
b
Sobrevivência a HN1 com pré-tratamento com tiouréia
1,00E+02
1,00E-081,00E-071,00E-061,00E-051,00E-041,00E-031,00E-021,00E-011,00E+001,00E+01
0 1 2
Concentração (mM)
N/N
oAB1157(selvagem) (HN1)
AB1157(selvagem)(Tioureia + HN1)
a
Sobrevivência a HN1 com pré-tratamento com tiouréia
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
0 1 2
Concentração (mM)
N/N
o
BH100 (quadruplomutante) (HN1)
BH100 (quadruplomutante) (Tioureia+ HN1)
c
Figura 33: Sobrevivência das cepas de E. coli (a) AB1157 (selvagem), (b)
AB1886 (uvrA) e (c) BH100 (nfo nth xthA fpg). Culturas das cepas em fase
exponencial pré-tratadas ou não com tiouréia e posteriormente tratadas com
diferentes concentrações de HN1, em tampão fosfato (pH7,0) por 60 minutos.
N/No (eixo das ordenadas), [HN1] (eixo das abscissas). Os resultados plotados
representam a média dos valores obtidos e os desvios padrões.
96
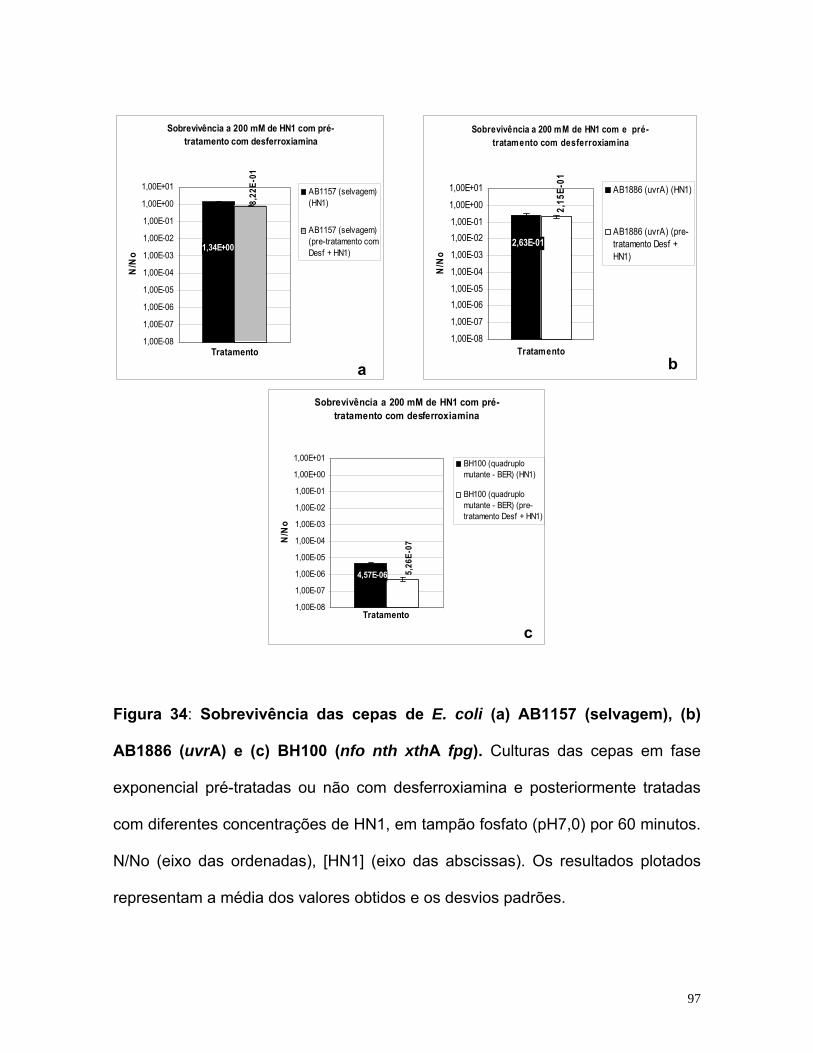
Sobrevivência a 200 mM de HN1 com pré-tratamento com desferroxiamina
1,34E+008,
22E-
01
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
1
Tratamento
N/N
o
AB1157 (selvagem)(HN1)
AB1157 (selvagem)(pre-tratamento comDesf + HN1)
a
Sobrevivência a 200 mM de HN1 com e pré-tratamento com desferroxiamina
2,63E-01
2,15
E-01
1,00E-08
1,00E-07
1,00E-061,00E-05
1,00E-04
1,00E-03
1,00E-021,00E-01
1,00E+00
1,00E+01
1
Tratamento
N/N
o
AB1886 (uvrA) (HN1)
AB1886 (uvrA) (pre-tratamento Desf +HN1)
b
Sobrevivê ia a 200 mM de HN1 com pré-trata nto com desferroxiamina
ncme
4,57E-06 5,26
E-07
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
1Tratamento
N/N
o
BH100 (quadruplomutante - BER) (HN1)
BH100 (quadruplomutante - BER) (pre-tratamento Desf + HN1)
c
Figura 34: Sobrevivência das cepas de E. coli (a) AB1157 (selvagem), (b)
AB1886 (uvrA) e (c) BH100 (nfo nth xthA fpg). Culturas das cepas em fase
exponencial pré-tratadas ou não com desferroxiamina e posteriormente tratadas
com diferentes concentrações de HN1, em tampão fosfato (pH7,0) por 60 minutos.
N/No (eixo das ordenadas), [HN1] (eixo das abscissas). Os resultados plotados
representam a média dos valores obtidos e os desvios padrões.
97

Influência da temperatura na metabolização (ativação) das mostardas
que em termos de metabolização a temperatura exerce uma
grande
- Influência da temperatura – HN2
sensíveis ao tratamento com HN2, para
vermo
ra 35a) a diferença
entre a
nitrogenadas
É sabido
influência. Em relação às mostardas não há nada na literatura em termos
de que temperatura elas se ligariam melhor ao DNA (na célula). Fizemos um
estudo para avaliar se as lesões causadas por estes agentes ocorriam somente a
37ºC ou também são capazes de acontecer em baixas temperaturas, onde
acontece diminuição no ritmo metabólico celular. Para isso utilizamos além de
cepa AB1157 (selvagem), cepas bem sensíveis ao tratamento para cada uma das
mostardas, a fim de sabermos se em baixas temperaturas o mesmo fenômeno de
letalidade acontece. A agitação também foi controlada, a fim de avaliarmos se, na
mesma temperatura, maior difusão de oxigênio influencia na formação das lesões.
6
Utilizamos os mutantes mais
s se a letalidade ocorre em altas e baixas temperaturas.
Observamos que para a cepa AB1157 (selvagem) (Figu
lta e baixa temperatura ocorre em pequeno grau, uma vez que esta cepa é
altamente resistente ao tratamento, porém podemos observar que há dois padrões
de letalidade um a 37ºC e outro na geladeira, em torno de 9ºC, nos indicando que
em baixas temperaturas o número de lesões é muito menor do que em altas
temperaturas, independente da agitação. Para os mutantes AB1886 (uvrA) (Figura
35b) e BH100 (nfo nth xthA fpg) (Figura 35c), vemos estes padrões de letalidade
98

Sobrevivência a 2 mM de HN2 em diferentes temperaturas
1,32
E-06
1,33
E-01
4,75
E-06
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
1Tratamento
N/N
o
AB1886 (uvrA) (37ºC comalta agitacao)
AB1886 (uvrA) (37ºC semagitacao)
AB1886 (uvrA) (baixatemperatura)
Sobrevivência a 2mM de HN2 em diferentes temperaturas
2,80
E-01
6,28
E-01
8,53
E-01
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
1
Tratamento
N/N
oAB1157 (selvagem)(37ºC com altaagitacao)
AB1157 (selvagem) (37ºC sem agitacao)
AB1157 (selvagem) (baixa temperatura)
Sobrevivência a 2 mM de HN2 em diferentes temperaturas
3,09
E-05
1,78
E-01
5,33
E-06
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
1
Tratamento
N/N
o
BH100 (quadruplomutante) (37ºC com altaagitacao)
BH100 (quadruplomutante) (37ºC semagitacao)
BH100 (quadruplomutante) (baixatemperatura)
c
Figura 35: Sobrevivência das cepas de E. coli (a) AB1157 (selvagem), (b)
AB1886 (uvrA) e (c) BH100 (nfo nth xthA fpg). Culturas das cepas em fase
exponencial tratadas com 2mM de HN2, em diferentes temperaturas, em tampão
fosfato (pH7,0) por 60 minutos. N/No (eixo das ordenadas), tratamentos (eixo das
abscissas). Os resultados plotados representam a média dos valores obtidos e os
desvios padrões.
99

muito mais acentuados, ou seja a temperatura de 37ºC faz com que a HN2
se torn
a temperatura a molécula de
HN2 n
N2 entrando na célula, quando a
tempe
agente
e muito mais letal para a célula, do que a temperatura em torno de 9ºC. As
cepas quando tratadas na geladeira são extremamente resistentes ao tratamento,
se comportando como cepas selvagens, mesmo sendo estas extremamente
sensiveis a HN2 (de Alencar et al., 2005). Podemos observar também que a falta
de letalidade em baixas temperaturas não é devida à falta de metabolismo celular
uma vez que nestas temperaturas as bactérias continuam se divivindo. A agitação
parece não influenciar nas lesões geradas por HN2.
Diante deste resultado fomos avaliar se nest
ão estava entrando na célula, ou se realmente, esta temperatura influencia
na letalidade. Realizamos experimentos fazendo um pré-tratamento de 20 minutos
com HN2 a 37ºC, para garantirmos que tal agente entrava, e depois colocamos
em baixas temperaturas (Figuras 36a e 36b).
O que observamos é que mesmo H
ratura baixa, HN2 não é mais capaz de fazer lesões na célula, quando
comparado à letalidade na temperatura de 37ºC, dados sugeridos a partir da
análise da cepa AB1886 (uvrA).
Experimentos com H2O2 foram realizados para avaliarmos se com um
que conhecidamente precisa sofrer uma metabolização celular, a mesma
influência da temperatura ocorre. Para isso, foi utilizado uma cepa extremamente
sensível ao tratamento com H2O2 – BW9091 (xthA), e a estratégia experimental foi
igual a realizada anteriormente, somente mudando os tempos de pré- incubação e
incubação, que são específicos para cada tratamento (Figura 37).
100

igura 36: Sobrevivência das cepas de
Sobrevivência a 2 mM de HN2 em diferentes temperaturas
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
0 20 40 60
Tempo (minutos)
N/N
o
AB1157 (selvagem)(37ºC com altaagitacao)
AB1157 (selvagem)(20 minutos a37ºC +40 minutos a baixatemperatura)
AB1157 (selvagem)(baixa temperatura)
Sobrevivência a 2 mM de HN2 em diferentes condições de temperatura
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
0 20 40 60
Tempo (minutos)
N/N
o
AB1886 (uvrA) (37ºC comalt a agit acao)
AB1886 (uvrA) (20 minut osa37ºC + 40 minut os a baixat emperat ura)
AB1886 (uvrA) (baixat emperat ura)
F E. coli (a) AB1157 (selvagem) e (b)
AB1886 (uvrA). Culturas das cepas em fase exponencial tratadas com 2mM de
HN2, em temperaturas e tempos de exposição ao agente diferentes, em tampão
fosfato (pH7,0) por 60 minutos. N/No (eixo das ordenadas), tempo de exposição a
HN2 (eixo das abscissas). Os resultados plotados representam a média dos
valores obtidos e os desvios padrões.
101

Sobrevivência a 20 mM de H2O2 em diferentes temperaturas
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
0 10 20
Tempo (minutos)
N/N
o
BW9091 37ºC (comagitacao normal)
BW9091 (5 min 37º +baixa temperatura)
BW9091 (baixatemperatura)
Figura 37: Sobrevivência da cepa de E. coli BW9091 (xthA). Cultura das cepa
em fase exponencial tratada com 20mM de H2O2, em temperaturas e tempos de
exposicao ao agente diferentes, em tampão fosfato (pH7,0) por 60 minutos. N/No
(eixo das ordenadas), tempo de exposição a H2O2, (eixo das abscissas). Os
resultados plotados representam a média dos valores obtidos e os desvios
padrões.
102

O mesmo que ocorre para HN2 ocorre para H2O2, nos indicando que HN2
não se
7 – Influência da temperatura - HN1
zados com HN1. Utilizamos o mutante mais
sensív
e ocorre para HN2, ocorre para HN1,
mostra
liga simplesmente ao DNA, precisa de uma ativação metabólica, também
prevista para H2O2, que ocorre melhor a de 37ºC, que é a temperatura ideal para
que ocorra a maior parte das reações celulares.
Os mesmos estudos foram reali
el ao tratamento com HN1, para avaliarmos se num background de
deficiência, só que agora em baixas temperaturas, além da influência deste fator,
o perfil de reparo mudaria (Figura 38).
Observamos que o mesmo qu
ndo que na geladeira a cepa se comporta como uma cepa selvagem, muito
resistente a este tratamento e o mesmo não ocorre a 37ºC, com ou sem agitação.
103

Sobrevivência a 200 mM de HN1 em diferentes
temperaturas
1,88
E-06
1,28
E-05
7,56
E-01
1,00E-08
1,00E-07
1,00E-06
1,00E-05
1,00E-04
1,00E-03
1,00E-02
1,00E-01
1,00E+00
1,00E+01
1
Tratamento
N/N
o
BH100 (quadruplo mutante)(37ºC com alta agitacao)
BH100 (quadruplo mutante)(37ºC sem agitacao)
BH100 (quadruplo mutante)(baixa temperatura)
Figura 38: Sobrevivência da cepa de E. coli BH100 (nfo nth xthA fpg). Cultura
da cepa em fase exponencial tratada com 2mM de HN1, em diferentes
temperaturas, em tampão fosfato (pH7,0) por 60 minutos. N/No (eixo das
ordenadas), tratamentos (eixo das abscissas). Os resultados plotados
representam a média dos valores obtidos e os desvios padrões.
104

DISCUSSÃO E CONCLUSÃO
trabalho é estudar caminhos pelos quais as
mostar
nteriormente, é capaz de
se liga
A proposta geral deste
das nitrogenadas bi (HN2) e monofuncional (HN1), geram lesões no DNA e
como estas lesões são “vistas” e reparadas pelas células. Para tal objetivo
utilizamos cepas deficientes nos principais mecanismos de reparo requisitados
para correção destes danos impostos ao DNA e algumas abordagens
experimentais que pudessem nos informar como estas moléculas se ligam aos
seus alvos, provocando desta forma a letalidade celular.
A mostarda nitrogenada bifuncional, como já dito a
r de maneira bi e monofuncional no DNA, gerando com isso além de
biadutos intra-cadeias e crosslinks, uma diversidade de lesões do tipo
monofuncional. Era esperado que esses danos disparassem variados mecanismos
de reparo nas células, uma vez que há uma diversidade de lesões. Avaliamos os
mecanismos de reparo envolvidos nas lesões de ambas as mostardas citadas (de
Alencar et al., 2005) e apontamos NER como reparo majoritário na correção das
lesões causadas por HN2, e por este motivo, em outros trabalhos, classificamos
este reparo UVC-like (Lage et al., 2003). Entretanto, outros agentes formadores de
adutos no DNA, tais como: mitomicina C e PUVA (psoralenos + UVA), em certas
condições, requerem apenas uma das proteínas do complexo UvrABC, a UvrB,
para a correção das lesões (Vidal et al., 2006) e (Gonçalves, 2007; Alves, 2007).
O reparo por excisão de nucleotídeos também parece estar envolvido nas lesões
causadas por HN2 em mamíferos (De Silva et al., 2000). Em conjunto, estes
dados configuram as lesões induzidas por HN2, pertencentes a um grupo de
lesões de características diferentes das mostradas para outros agentes químicos
105

formadores de adutos.
Já para a mostarda monofuncional HN1 (considerada uma análoga
monof
rmente, fomos avaliar a
particip
mutante uvrA recA maior, em
relação
uncional de HN2), inesperadamente os danos causados por esta mostarda
não são substratos para NER (de Alencar et al., 2005).
Em adição a esse trabalho discutido anterio
ação de mais algumas vias de reparo, possivelmente envolvidas na
correção das lesões causadas por HN2 e HN1.
A sensibilidade apresentada pelo duplo
aos simples mutantes uvrA e recA, nos indica que estes reparos podem
estar atuando em vias diferentes, ou seja, uma parcela do reparo sendo efetuada
por NER, tendo como iniciadora do processo a proteína UvrA e a outra parte pela
recombinação efetuada pela proteína RecA. Nossos resultados concordam com o
modelo proposto por Dronkert e Kanaar, para crosslinks, in vitro, em que eles
sugerem a participação do NER associado a RecA (Dronkert e Kanaar, 2001).
Entretanto, a participação de diferentes vias de reparo nos sugere, que estas
podem estar atuando na correção de lesões do tipo monoadutos, ou biadutos
intra-cadeias. A proteína RecA é de extrema importância para o reparo das lesões
causadas por HN2, tanto na sua porção recombinacional, quanto na participação
da resposta SOS. Tal importância se deve ao fato de que a proteína RecA, como
uma das reguladoras da resposta SOS, influencia na expressão das proteínas
UvrA e UvrB, de extrema importância para reparação de tais lesões, e sua função
na recombinação já foi prevista por Dronkert e Kanaar, em 2001, que sugerem a
recombinação, como um dos passos do reparo de crosslinks. Em células de
mamíferos também foi observada a participação de recombinação nas lesões
causadas por HN2 (De Silva et al., 2000). Para HN1, ambas as funções da
106

proteína RecA também são importantes. Nenhum dos dois mutantes é tão
sensível quanto o mutante que possui deficiência em ambas as funções, indicando
que a uma parcela de reparo tanto recombinacional, quanto de expressão das
funções SOS.
Um dos aspectos conferidos às mostardas em geral, é o caráter
mutag
observamos que a cepa selvagem
aprese
ênico e potencialmente carcinogênico destas substâncias. Os riscos de
aparecimento de novos cânceres no futuro são reais, para pacientes tratados com
quimioterapia. Esta relação entre a utilização de agentes alquilantes e geração de
novos tumores é complexa e envolve vários fatores (Boffetta e Kaldor, 1994). Não
só o mecloretamine (HN2), como também outras mostardas nitrogenadas, são
capazes de induzir mutagênese, e esta indução depende de vários fatores como a
formação de monoadutos e seqüências ricas em G-C (Kunz e Mis 1989). Melfalan
e clorambucil, outros tipos de mostarda nitrogenada bifuncional, são capazes,
através de monoalquilações feitas em adeninas, de gerar mutações do tipo
transversões em estudos com plasmídeos (Charles et al., 1997). Alguns outros
trabalhos também sugerem aparecimento de mutações em células CHO,
induzidas por melfalan e outros alquilantes bifuncionais que se ligam em N7 de
guaninas, e tais mutações acontecem em seqüências G-G-C (Bauer e Povirk.,
1997).
Nos nossos estudos de mutagênese,
ntou maior incremento de mutantes em relação às outras cepas estudadas
deficientes em NER. Provavelmente a ausência de qualquer uma das subunidades
do complexo e, por conseqüência, a ausência de todo reparo NER, diminui a
mutagênese, nos sugerindo que quando o reparo está atuando, temos uma
mutagênese aumentada, sugerindo um reparo mutagênico. Também foi observado
107

um aumento de mutagênese, de aproximadamente 32 vezes, em linfócitos
humanos tratados com diferentes doses de HN2, in vitro, em relação às células
não expostas (Olsen et al., 1997).
O reparo por excisão de nucleotídeos também foi estudado através da
avaliaç
2, a expressão levemente
aumen
ão da expressão relativa dos genes uvrA, uvrB, uvrC, cho e recA, pois este
reparo é requerido para HN2 e não para HN1 (de Alencar et al., 2005). Dessa
forma, fomos analisar se os genes NER estavam sendo expressos nos
tratamentos com HN2 e não nos tratamentos com HN1, e por isso neste último
agente não veríamos a participação deste reparo.
Observamos, nos tratamentos com HN
tada dos genes de reparo NER, em relação ao tratamento com 5 minutos,
na DL90, dando ênfase à maior expressão do gene recA . Quando as células são
expostas a maior concentração de HN2, chegando à sua DL10, continua havendo
o aumento de expressão de todos os genes estudados. Através da análise destes
experimentos, sugerimos que o aumento da expressão desses genes, de acordo
com o tempo e com a concentração de HN2, seja devido a um aumento na
formação de lesões no DNA, que sinalizam para a expressão destes genes, que,
de acordo com os dados de sobrevivência celular participam do reparo destas
lesões. Quando chegamos a um nível de sobrevivência em torno daquele induzido
pela DL10, o número de lesões no DNA já é o suficiente para aumentar a
expressão de todos os genes, em igual proporção. Provavelmente nesta faixa de
sobrevivência a célula precisa não só do reparo NER, como também das funções
desempenhadas pela proteína RecA, até mesmo da sua função como reguladora
da expressão dos genes uvrA e uvrB, e talvez por isso mesmo a expressão do
gene recA seja a primeira a ser aumentada.
108

Nos experimentos realizados com HN1, observamos uma expressão
leveme
bém é requerido para a correção das
lesões
nte aumentada dos genes de reparo uvrA e uvrB, em relação aos 5
minutos de tratamento, na DL90. Considerando que estes dois genes compõem o
complexo de reconhecimento do sistema NER - UvrA2B, sugerimos que
provavelmente está ocorrendo um reconhecimento que não é especifico,
requisitando então um outro reparo mais específico, possivelmente BER, pois nos
dados de sobrevivência celular não vemos a participação deste complexo. É
sabido que o mecanismo NER, através do seu complexo de reconhecimento é
capaz de detectar teoricamente quase todos as lesões na molécula de DNA, pois
este complexo percorre a cadeia polinucleotídica, até encontrar uma lesão. Então
neste caso, é possível que NER esteja sendo expresso, porém como BER é um
reparo mais específico para as lesões causadas por HN1, o reparo NER não atue
sobre tais lesões (de Alencar et al., 2005). Pode estar havendo também alguma
modificação pós transcricional que justifique o aumento da expressão destes
genes e a ausência de participação deles no reparo para HN1. Comparando os
dados obtidos na DL10, com os obtidos na DL90, não vemos a expressão
aumentada dos genes NER, somente um pequeno aumento na expressão de
recA. Provavelmente altas concentrações sejam um sinal indutor específico para
outros mecanismos de reparo, que não NER. Sugerimos que a indução seja do
sistema BER, uma vez que este está envolvido na reparação das lesões causadas
por HN1 e o pequeno aumento de recA, pode ser um reflexo do que ocorre na
sobrevivência com o mutante recA e HN1.
Em relação ao reparo BER, este tam
causadas por HN2 (de Alencar et al., 2005). A participação desses reparos
nos confirma mais uma vez as características particulares das lesões produzidas
109

por HN2 no DNA, principalmente do tipo monofuncional, uma vez que lesões do
tipo crosslinks só são geradas em aproximadamente 4% (Kohn et al., 1996,
Friedberg et al., 2006), embora o sucesso desta mostarda em quimioterapia ainda
seja atribuído à formação de crosslinks no DNA (Bramson et al., 1995).
A análise do duplo mutante xthA nfo, se tornou importante, para sabermos
se elim
sos resultados, nos quais a ausência de
nfo su
asos de supressão da sensibilidade, por ausência de um
inando a função AP endonucleolítica da célula esta se torna mais sensível
ao tratamento, uma vez que observamos a participação da enzima Exonuclease III
na correção das lesões causadas por HN2. Nós observamos que como o duplo
mutante BW544 (xthA nfo) foi menos sensível ao tratamento com HN2 do que o
simples mutante BW9091 (xthA). A enzima Endo IV pode reconhecer alguns
produtos que não são substrato para DNA glicosilases sugerindo uma nova via de
reparo das lesões oxidativas, na qual Endo IV funcionaria auxiliando as DNA
glicosilases, porém como esta é uma AP endonuclase, incisa o DNA do lado 5’da
base lesada (Cunningham et al., 1986).
Esta via de reparo explicaria nos
prime a sensibilidade de xthA, indicando que esta supressão poderia estar
ligada à diminuição na geração de quebras simples adjacentes a uma base
lesada, provocadas pela Endo IV. A alta letalidade do mutante xthA pode ser
explicada pelo fato de Endo IV gerar quebras que necessitariam da atividade 3’→
5’ exonucleolítica da Exo III (produto do gene xthA). Provavelmente o duplo
mutante xthA nfo é menos sensível (mais resistente) porque não possui as duas
enzimas Exo III e Endo IV, então não gera quebras e por isso não necessita de
Exo III para reparar. Este fato já foi observado para a radiação ultravioleta B
(Souza, 2004).
Outros c
110

determ
ssível participação do estresse
oxidati
o trabalho se constituiu na avaliação de quais
mecan
l,
influen
inado gene, já foram vistos na literatura. Galhardo e colaboradores em
2000, observaram que para 5mM de H2O2 a ausência da proteína Fpg no duplo
mutante xthA nfo, suprime a sensibilidade deste mutante ao H2O2, provavelmente
porque a proteína Fpg gera produtos que não podem ser reconhecidos pelas
enzimas ExoIII e Endo IV, então a sua ausência diminui a letalidade no triplo
mutante, pois não há a presença destes produtos.
Em adição a esta análise, enfocando a po
vo, possivelmente gerado pelas mostardas, avaliamos a sensibilidade da
cepa oxyR, que é sensivel a altas concentrações de H2O2. Nenhuma das
mostardas foi capaz de sensibilizar este mutante. Em termos de formação de
lesões, HN2 e HN1 se assemelham, ou seja, não parecem gerar peróxidos
durante o seu metabolismo.
A segunda parte d
ismos as mostardas poderiam utilizar para ligação ao DNA. Sendo a
mostarda nitrogenada bifuncional HN2 uma das classes de alquilantes mais
utilizadas em quimioterapia, podendo agir diretamente no DNA, através da
geração de crosslinks, fomos investigar a participação de EAO na ativação de
HN2 e, conseqüentemente, HN1, uma vez que várias substâncias utilizadas em
quimioterapia têm sido classificadas como pró-drogas, sofrendo alterações
enzimáticas ou químicas (Oliveira e Alves, 2002) e, também de acordo com os
nossos resultados anteriores, em que vimos a participação do reparo BER, através
de enzimas até então requeridas para danos oxidativos (de Alencar et al., 2005).
É sabido que o processo de quelação de ferro, de uma maneira gera
cia no potencial redox da célula (Buss et al., 2003), e que o pré-tratamento
com quelantes de ferro protege células procarioticas e eucarioticas contra os
111

efeitos do H2O2 (Asad e Leitão, 1991), uma vez que diminuindo a concentração de
ferro do meio intracelular, ocorre uma inibição da reação de Fenton, via Fe2+, e
com isso diminuição da formação de radicais hidroxil.
Começamos os estudos de metabolização utilizando um quelante de Fe2+ -
Dip, na
m a cepa AB1157 (selvagem), quanto com os
mutan
letalida
Geração de um composto mais letal, formado entre o quelante Dip e
HN od
o Dip, que contribuísse para o
aum to
tentativa de avaliarmos se os radicais OH. poderiam estar envolvidos nas
lesões causadas por HN2, pois, conforme dito anteriormente, algumas EAO estão
envolvidas na ativação de drogas.
Nossos resultados tanto co
tes AB1886 (uvrA) e BH100 (nfo nth xthA fpg), nos apontaram exatamente o
contrário do esperado. Nos tratamentos em que a HN2 é precedido pelo
tratamento com Dip, obsevamos uma sensibilização de 100 vezes – selvagem e
uvrA, e para o mutante BH100, deficiente em BER, observamos uma sensibilidade
ainda maior, ocasionada pelo pré-tratamento.
A partir daí, algumas hipóteses foram levantadas para explicar essa maior
de de HN2, na presenca de Dip. Algumas delas foram analisadas neste
trabalho
2. P e haver a formação de um complexo redox que sensibilizasse ainda
mais a célula do que somente HN2. Isto foi sugerido em quimioterapia de
células tumorais. Alguns complexos formados entre derivados de Dip e ferro,
podem gerar EAO (Richardson et al., 2006).
Situação metabólica deixada pel
en de letalidade induzida por HN2. A quelação de Fe2* é capaz de
ocasionar um aumento da respiração celular, aumentando a produção de
radical superóxido para que a célula possa regenerar o ferro disponível a partir
112

da doação de um elétron para o Fe3+, que é reduzido a Fe2+, metabolicamente
envolvido nas reações celulares (Keyer et al., 1995).
Fe3+ + O2-. Fe2+ + O2 (equação 1 do Ciclo de Harber Weiss)
O ciar no
aumen
el intercalação do
que te
a cepa AB1157
(selvag
radical superóxido poderia então, de alguma maneira influen
to de letalidade causado por HN2, uma vez que o pré-tratamento com este
quelante é capaz de aumentar os níveis de radical superóxido.
Outras hipóteses foram questionadas - possív
lan Dip no DNA, modificando assim a angulação desta molécula,
permitindo com isso maior número de ligações de HN2 ao DNA e
conseqüentemente maior número de eventos letais. Mesmo alguns autores já
tendo sugerido que um outro quelante de Fe2+, a fenantrolina, possivelmente
era capaz de se intercalar no DNA (Furtado et al., 1997), isso nunca foi visto
para Dip. Foi postulada a hipótese do Dip, através do possível aumento de
radical superóxido, atacar o núcleo Fe-S (Keyer et al., 1995) de algumas
proteínas, como por exemplo, Endo III (nth), Exo III (xthA), e Superóxido
Dismutase (sodA sodB) o que possivelmente não está acontecendo, uma vez
que observamos uma extrema sensibilidade produzida pelo tratamento com Dip
no mutante BH100 e sodA sodB, e estes são deficientes nestas enzimas, então
provavelmente esta não é a causa desta maior sensibilidade.
Para tentar desvendar algumas destas hipóteses tratamos
em) em diferentes situações com HN2 e Dip. De todos os tratamentos
realizados o do pré-tratamento com Dip seguido de HN2, continuou sendo o mais
letal para a célula, porém observamos que a retirada, através da centrifugação do
Dip da célula, não fez com que esta voltasse ao nível de sobrevivência induzido
somente por HN2. Adicionalmente, o tratamento simultâneo com o quelante mais
113

HN2, também mostrou sensibilidade, porém menor do que a vista após a
centrifugação. Então, podemos sugerir que mesmo Dip não precisa estar presente
na célula, basta entrar em contato com ela, pois é capaz de deixar uma situação
metabólica, que permita maior letalidade produzida por HN2 e, é importante
ressaltar que quanto maior o tempo de exposição a este quelante, maior a
letalidade produzida por HN2.
Uma possível interação entre Dip e HN2, não foi descartada, pois as
molécu
expressão da enzima
Superó
las de Dip que estavam presentes na célula, em todos os tratamentos,
poderiam interagir com HN2 e gerar um composto, podendo haver então lesões
geradas pela situação metabólica deixada pelo Dip e também por este possível
composto. Entretanto, quando reagimos Dip + HN2 in vitro e tratamos a célula com
este produto da reação não observamos maior letalidade que nos indicasse a
formação de um composto mais letal. Provavelmente não está acontecendo a
formação de um complexo mais letal para a célula, uma vez que a maior letalidade
produzida ainda é o tratamento com HN2 precedido do pré-tratamento com o
quelante. Quando reagimos as duas moléculas e tratamos a célula, não houve
tempo, antes da célula entrar em contato com HN2, do Dip aumentar os níveis de
radical superóxido, por isso não observamos uma letalidade tão grande quanto a
do pré-tratamento. Então a letalidade que vemos ocasionada pelo Dip e HN2 na
célula atuando ao mesmo tempo. A letalidade não é tão grande quanto a
observada para o tratamento simultâneo, pois, provavelmente, está acontecendo
alguma degradação ou alguma interação destas moléculas.
A partir de dados da literatura em que há aumento na
xido Dismutase, em resposta a tratamentos com quelantes de ferro (Touati,
1988), e especificamente o aumento na Superóxido Dismutase dependente de
114

manganês (codificada pelo gene sodA), em experimentos realizados com quelante
de Fe2+ - Dip (Moody e Hassan, 1984), sendo a indução desta enzima feita de
maneira aeróbica, via produção de radicais superóxido (Touati, 1988), sendo seu
substrato este mesmo radical, sugerimos então, que esta situação metabólica
deixada pelo Dip que aumenta a letalidade de HN2 em todas as cepas tratadas
com Dip e posteriormente HN2, principalmente na cepa JI132 (sodA sodB), é o
aumento do radical superóxido, uma vez que o tratamento somente com HN2 não
sensibilizou consideravelmente esta cepa, conforme o esperado, já que em termos
de reparo de DNA, esta é uma cepa selvagem.
O superóxido parece aumentar progressivamente a letalidade da HN2, pois
nos tratamentos com a cepa sodA sodB, conforme aumentamos a concentração
de HN2 e mantemos a mesma concentração deste radical, aumentamos a
letalidade, provocada pelo pré-tratamento com Dip. Então há uma relação entre
concentração tanto de radical superóxido quanto de HN2, fato demonstrado
também, quando observamos que conforme aumentamos a concentração de pré-
tratamento com Dip (e conseqüentemente a concentração de superóxido
intracelular) e fixamos a mesma concentração de HN2, há uma diminuição da
sobrevivência celular, dose dependente, e este resultado é mais evidenciado no
mutante sodA sodB. De maneira geral, os agentes alquilantes são divididos em
dois grupos – SN1 e SN2, de acordo com a cinética de reação destes (Holand e
Frei, 2000). As mostardas nitrogenadas são consideradas agentes do tipo SN1 e,
portanto, sua atuação é dependente somente da concentração de intermediários
reativos formados (Holand e Frei, 2000), então é possível que o radical superóxido
que está formado no meio, interagindo com HN2, gere um intermediário reativo e
com isso a letalidade de HN2, seja aumentada conforme aumentamos a
115

concentração de HN2 no meio.
Através dos resultados obtidos com os simples mutantes JI130 (sodA) e
JI131
+ na célula não é um fator
import
lo pré-tratamento com Dip +
HN2, c
(sodB), podemos observar que a presença de apenas uma enzima
Superóxido Dismutase, é capaz de diminuir a letalidade ocasionada por HN2.
Acreditamos que haja uma diminuição dos níveis de radical superóxido
intracelulares, através da dismutação realizada pela Superóxido Dismutase, e com
isso há diminuição da letalidade ocasionada por HN2. No mutante sodA, em que
temos a ausência de SodA, e a presença de SodB, ainda vemos uma letalidade
(pré-tratamento com Dip + HN2) que não observamos no mutante sodB, em que
temos a ausência de SodB e a presença de SodA. Isto provavelmente ocorre
porque a enzima SodA, codificada pelo gene sodA, é induzida pela presença de
radicais superóxido na célula, fato que não ocorre com SodB, que é constitutiva e
não induzida. Então, o pré –tratamento com Dip elevando os níveis deste radical,
aumenta a produção desta enzima, que então diminui os níveis deste radical na
célula, diminuindo a letalidade ou reatividade de HN2.
Provavelmente a regulação na entrada de Fe3
ante para aumentar a letalidade produzida por HN2, na presença de Dip,
pois a diminuição deste íon na célula não acrescentou nada, em termos de
letalidade celular. Com certeza, mesmo com a deleção da proteína Fur, ainda
exista Fe3+ na célula e por isso os radicais superóxido que estão sendo formados
tem uma preferência na reação com este íon, através da doação de elétrons, ao
invés da reação que possivelmente ocorre com HN2.
O fenômeno de maior letalidade ocasionado pe
onsegue ser revertido quando os radicais livres que estão sendo formados
pelo pré-tratamento, são capturados pelo captador inespecífico de radicais livres –
116

tiouréia. Esta captura dos radicais livres só se faz importante quando a célula é
pré-tratada com o quelante Dip e posteriormente tratada com HN2, mostrando que
HN2 sozinha não gera radicais livres para a sua ligação ao DNA, e o pré-
tratamento é capaz de gerar estas EAO possibilitando maior reatividade de HN2,
gerando com isso maior número de eventos letais, e conseqüentemente uma
queda na sobrevivência celular, independente do background genético. A tiouréia,
sendo um captador inespecífico, pode estar capturando os radicais superóxido
que estão sendo formados, pelo tratamento com Dip e isso se torna mais uma
evidência de que este radical é capaz de promover uma ativação de HN2. A
tiouréia sendo adicionada antes ou junto com HN2, mostra efeito protetor contra
as lesões deste agente, o importante é que esta seja adicionada ao sistema após
ou junto com o Dip, diminuindo conseqüentemente os efeitos gerados por ele.
Existem estudos mostrando que algumas mostardas podem sofrer ativação
redutiv
ovida pelo radical superóxido não parece acontecer com
HN1,
a em determinados grupamentos da molécula, gerando com isso outros
compostos que se ligam ao DNA (Oliveira e Alves, 2002). A ativação da mostarda
2-nitroimidazólica ocorre a partir de uma fragmentação do ânion nitro radical
formado pela redução monoeletrônica, originando um radical do tipo benzílico, via
transferência de elétron intramolecular. Após sofrer redução, esta mostarda libera
então o mecloretamine (mostarda nitrogenada bifuncional – HN2), altamente
tóxica para as células tumorais (Oliveira e Alves, 2002). Além das mostardas
nitroaromáticas, outras mostardas biorredutíveis vêm sendo desenvolvidas
(Oliveira e Alves, 2002).
Esta ativação prom
pois o pré-tratamento com Dip não alterou em nada o comportamento
celular, nos experimentos realizados com esta mostarda. De posse destes dados,
117

sugerimos que a participação deste radical seja na ativação que leva à formação
da segunda ligação ao DNA. Tal ativação pode acontecer no sentido de gerar um
maior número de lesões ou ainda lesões mais sérias, diminuindo com isso a
capacidade da célula sobreviver, devido à diminuição de reparo destas lesões,
quando comparado aos tratamentos com HN2 somente.
Estas lesões ocorridas no tratamento Dip + HN2 são mais mutagênicas na
cepa J
quando a
célula
I132 (sodA sodB), onde não temos a dismutação do radical superóxido,
quando comparado ao tratamento realizado somente com HN2. Provavelmente
este radical e mais HN2, com o aumento do potencial deste último, sejam os
promotores de uma lesão mais mutagênica. Os dados obtidos com a cepa AB1157
(selvagem), em que observamos que praticamente não há diferença no
incremento de mutagênese nos dois tratamentos, nos levam a crer que a
dismutação do radical superóxido influencia na geração de lesões mutagênicas.
Então, provavelmente as lesões formadas na ausência e presença de Dip são
diferentes, e este fato pode ser visto não só em termos de mutagênese, como
através dos dados de sobrevivência celular, na qual a cepa sodA sodB é mais
sensível ao duplo tratamento com Dip + HN2 do que a cepa selvagem.
Essas lesões ou esse fenômeno de maior letalidade, adquirido
é submetida ao pré-tratamento com Dip e posterior tratamento com HN2,
não é observado quando a célula é exposta previamente a desferroxiamina. O
intuito na utilização deste quelante era saber se essa maior letalidade era obtida
apenas quando tínhamos a geração de radical superóxido, via exposição ao Dip,
ou era um processo conseguido às custas da quelação de ferro na célula.
Quelantes de ferro apresentam sucesso na quimioterapia, uma vez que diminuem
os níveis de um dos elementos essencias para a célula, envolvido na liberação de
118

oxigênio, transferência de elétrons, metabolismo energético e síntese de DNA
(Buss et al., 2003) além do ferro fazer parte dos sítios ativos de uma grande
variedade de proteínas envolvidas na respiração (Richardson, 2001). Além da
desferroxiamina, outros quelantes, naturais ou sintéticos têm mostrado atividade
promissora em vários estágios do desenvolvimento, tais como: O-trensox,
desferritiocina, tachpiridine e outros Um grupo de quelantes conhecidos como
tiosemicarbazones também apresentam pronunciada atividade antitumoral, devido
à sua habilidade em inibir Ribonucleotídeo Redutases (Lovejoy e Richardson,
2002).
Apesar de sua crescente utilização em quimioterapia, a desferroxiamina,
não foi capaz de promover aumento de sensibilidade ao quimioterápico HN2. Os
quelantes, em geral, podem ser utilizados como agentes únicos ou podem
apresentar efeitos sinergísticos com outras terapias anti–câncer. Eles podem
esgotar/reduzir o ferro ou causar estresse oxidativo nos tumores devido a
perturbações redox no seu ambiente. Entretanto, parece que a quelação de ferro
neste caso não é tão importante quanto comparada à formação de intermediários
reativos que possam ativar HN2 e nem proporciona letalidade adicional, pois as
células pré-tratadas com Dip e desferroxiamina se apresentam tão sensíveis
quanto as pré-tratadas somente com Dip. Para a mostarda nitrogenada
monofuncional, também não houve aumento do número de lesões causadas pelo
pré-tratamento com desferroxiamina, indicando que este quelante não está
envolvido nas lesões causadas por mostardas nitrogenadas, provavelmente não
influencia no mecanismo básico de ligação das mostardas ao DNA,
diferentemente do que ocorre para outros quimioterápicos como mitomicinaC
(Vidal, 2007).
119

Além do estresse oxidativo, a temperatura também parece influenciar no
mecanismo de formação de lesão das mostardas nitrogenadas no DNA. Neste
aspecto HN2 e HN1 se parecem uma vez que as duas só geram lesões à
temperatura de 37ºC. Quando comparamos HN2 e H2O2, percebemos que a
temperatura exerce um papel extremamente importante para ambas, e, estando a
temperatura diretamente ligada ao metabolismo celular, podemos inferir que HN2
e HN1 precisam de metabolização celular para provocar letalidade, ou a saída dos
cloros e conseqüentemente sua posterior ligação ao DNA, acontece melhor nesta
temperatura, fator que justificaria a maior letalidade produzida a 37ºC, ou ainda,
talvez a 37ºC, as lesões são mais instáveis, gerando substratos mais difíceis de
serem reparados pelas células.
120

HIPÓTESE DA ATIVAÇÃO DE HN2, VIA RADICAL SUPERÓXIDO
ssos estudos
sugere
lante e, teoricamente sua ação no DNA
seja a
ão da HN2,
via rad
Baseados nos resultados discutidos neste trabalho, em que no
m como sendo o radical superóxido o grande potencializador dos efeitos
causados por HN2, e uma vez que estes efeitos não são observados para HN1,
propomos um modelo em que a HN2 pode ser reduzida/ativada através da doação
de elétrons via radical superóxido, gerando com isso um subproduto – mostarda
ligada ao DNA em um dos lados reativos e do outro lado o radical superóxido. Na
figura 39 encontra-se o modelo proposto.
Mesmo sendo HN2 um agente alqui
través da perda dos cloros, quando entra em solução, existem algumas
mostardas que podem ser reduzidas gerando outras, que então, se ligariam ao
DNA. Este é o caso da mostarda 2-nitroimidazólica que após sofrer redução
monoeletrônica gera o mecloretamine (HN2) (Oliveira e Alves, 2002).
Este produto, gerado teoricamente a partir da reação de reduç
ical superóxido, pode não só se ligar ao DNA, como sofrer ataque de outras
EAO, gerando com isso lesões do tipo oxidativas e isso corroboraria mais uma vez
nossos dados de proteção, conferidos pela tiouréia.
121
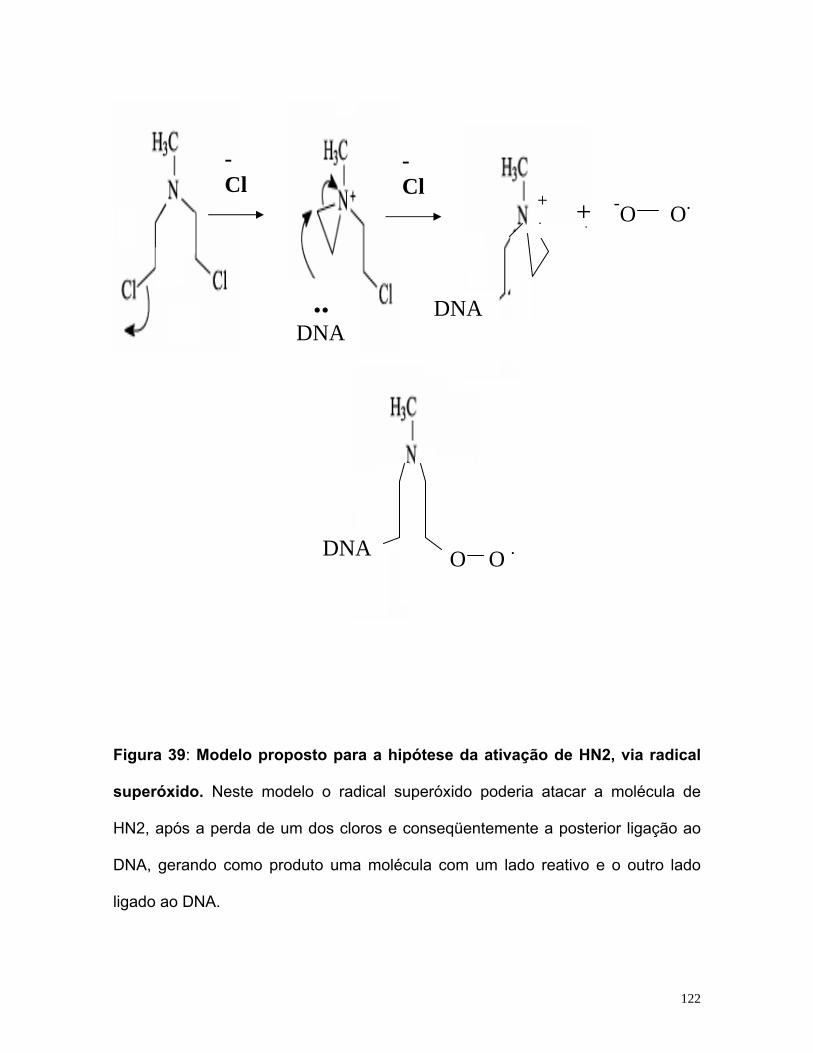
Figura 39: Modelo proposto para a hipótese da ativação de HN2, via radical
superóxido. Neste modelo o radical superóxido poderia atacar a molécula de
HN2, após a perda de um dos cloros e conseqüentemente a posterior ligação ao
DNA, gerando como produto uma molécula com um lado reativo e o outro lado
ligado ao DNA.
-O O.
+
A
+
DNA
DN
- l
- lC C
.. DNA
+
DNA
+
O O .
+ +
122

HIPÓTESE DA GERAÇÃO DO COMPOSTO DIP – HN2
os entre o tratamento
simultâ
Com base na diferença dos resultados obtid
neo e o tratamento em que as duas moléculas Dip e HN2 reagem antes de
entrar na célula, não esta descartada a possível geração de um complexo Dip +
HN2, que ocorreria concomitantemente a hipótese de ativação de HN2 - via radical
superóxido. Neste caso o Dip se ligaria a molécula de HN2, após a perda dos dois
cloros, o que também seria extremamente letal para a célula uma vez que esta
molécula resultante seria um potente agente alquilante, devido a suas cargas
positivas, sem precisar de ativação para que a ligação ao DNA ocorresse. Na
figura 40 encontra-se o modelo proposto
123
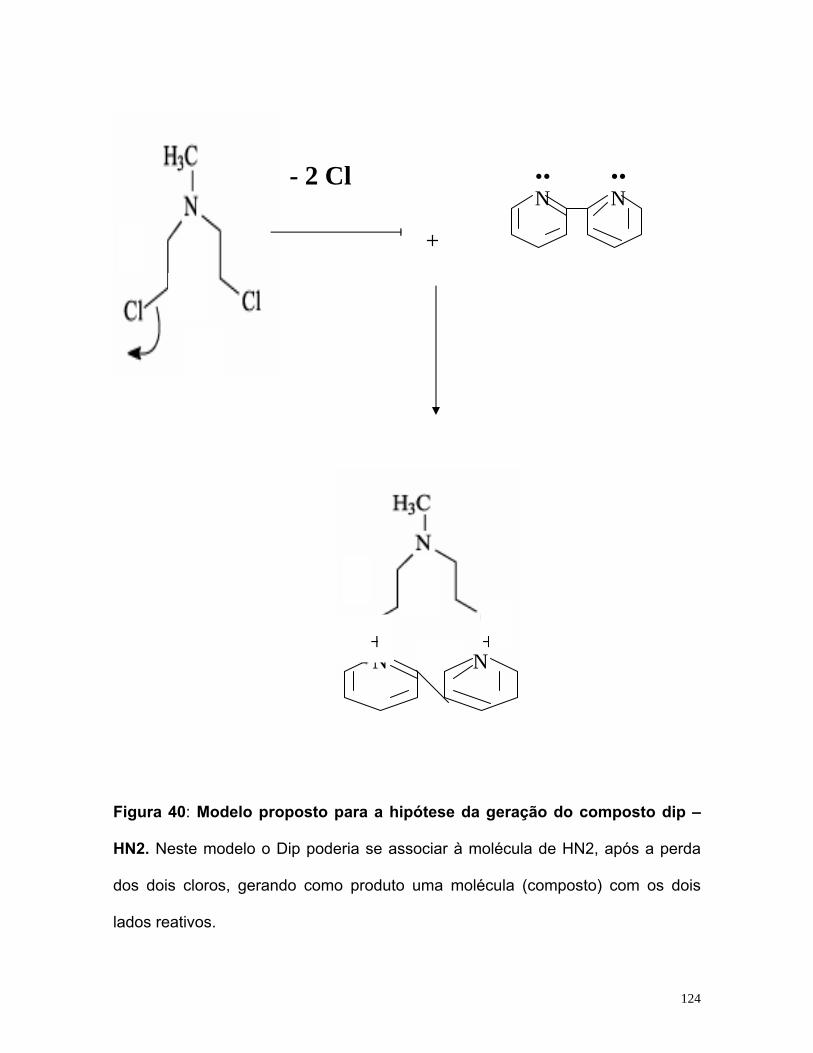
- 2 Cl
+
.. .. N N
.. N N
+ +
Figura 40: Modelo proposto para a hipótese da geração do composto dip –
HN2. Neste modelo o Dip poderia se associar à molécula de HN2, após a perda
dos dois cloros, gerando como produto uma molécula (composto) com os dois
lados reativos.
124

PERSPECTIVAS
ivo
da cepa sodA sodB, com o duplo tratamento Dip + HN2,
com m p
Experimentos de RT-PCR em tempo real → análise da expressão de
out tra
eróxido, tais como:
me ion
egulação de ferro →
inc ent
tro quelante de Fe2+ → mesmo fenômeno Dip.
xperimentos in vitro
s lesões produzidas no DNA extraído de E. coli, após o
tra ent
Experimentos in v
Tratamento
u lasmídeo que expresse Superoxido Dismutase → volta ao fenótipo
HN2;
ros nscritos gênicos → duplo tratamento Dip + HN2; tratamentos somente
com HN2 [(BER) e aumentando o tempo de recuperação];
Utilização de geradores químicos de radical sup
nad a e paraquat → semelhança com o fenômeno provocado pelo Dip;
Dosagem colorimétrica de radical superóxido;
Utilização de outras cepas deficientes na r
rem o na letalidade;
Utilização de ou
E
Análise da
tam o com HN2 e com o pré-tratamento Dip + HN2, bem como a análise
das lesões remanescentes no DNA, após recuperação de 1 hora, em cepas
selvagem e sodA sodB (diferentes concentrações de radical superóxido
intracelular), através da Espectrometria de Massas. Além das outras situações
experimentais já testadas in vivo.
125

Além disso, elaboração de técnicas in vitro, que nos permitam analisar se
há form
ação destas moléculas propostas nas hipóteses de redução de HN2 - via
superóxido e formação do composto a partir da reação entre Dip e HN2 →
Espectrometria de Massas e Ressonância Magnética Nuclear.
126

REFERÊNCIAS BIBLIOGRÁFICAS
M., Roberts, K. e Watson, J.D. (1994)
Alv
And mbat, P., Fleuty, J., Milpied, N.,
Asa s of metal íon chelators on DNA strand
Asa ,
Asl eng, M., Beckwith, J. e Storz, G. (1999) Regulation of OxyR
Bau Kinetics of interstrand and
Alberts, B., Bray, D., Lewis, J., Raff,
Molecular biology of the cell. Garland Publishing, Inc., 3rd ed.
es, A.M. (2007) Caracterização das lesões em DNA in vitro através da
utilização de espectrometria de massas e estudo dos mecanismos de
reparação envolvidos na correção das lesões PUVA in vivo (Psoralenos +
luz ultravioleta A). Tese de Doutorado. Instituto de Biofísica Carlos
Chagas Filho. UFRJ. Rio de Janeiro.
ré, M., Henry-Amar, M. Blaise, D.Colo
Cahn, J.Y., Pico, J.L., Bastion, Y., Kuentz, M., Nedellec, G., Attal, M.,
Ferm é, C. e Gisselbrecht, C. (1988) Treatment-related deaths and
second risk after autologous stem-cell transplantation for Hodgkin ’s
disease. Blood, 92(6): 1933-1940.
d, N.R. e Leitão, A.C. (1981) Effect
breaks and inactivation produced by hydrogen peroxide in Escherichia
coli: detection of iron-dependenty lesions. J. Bacteriol., 173: 2562-2568.
d, N.R., Asad, L.M.B.O., Almeida, C.E.B., Felzenszwalb, I., Cabral-Neto
J.B. e Leitão, A.C. (2004) Several pathways of hydrogen peroxide action
that damage the E. coli genome. Genetics and Molecular Biology, 27(2):
291-303.
und, F. Zh
transcriptional factor by hydrogen peroxide and the celular thiol-disulfide
status. Proc. Natl. Acad. Sci., 96: 6161-6165.
er, G.B. e Povirk, L.F. (1997) Specificity and
127

intrastrand bifunctional alkylation by nitrogen mustards at a G-G-C
sequence. Nucleic Acids Res., 25(6): 1211-1218.
ardini, M., Foster, P.L. e Loechler, L. (1999) DNABer polymerase II (polB) is
Bof gnancies following cancer-
Bra ijamali, M., Batist, G.,
Bra al iron
Bra (1998) Bacterial ion transport :
Bre m gamma-irradiated
Bre oxic nitrogen-mustard
Buss, J.L., Greene, B.T., Turner, J., Torti, F.M. e Torti, S.V. (2004) Iron
involved in a new DNA repair pathway for DNA interstrand cross-links in
Escherichia coli. J. Bacteriol., 181: 2878-2882
fetta, P. e Kaldor, J.M. (1994) Secondary mali
chemotherapy. Acta Oncologica, 33(6): 591-598
mson, J., Mcquillan, A. , Aubin, R., Alaou
Christodoulopoulos, G. e Panasci, L.C. (1995) Nitrogen-mustard drug-
resistance B-cell chronic lymphocytic-leukemia as in-vivo model for cross-
linking agent resistance. Mut. Res. – DNA Repair, 336(3): 269-278
un, V. (1997) Avoidance of iron toxicity throughregulation of bacteri
transport. Biol. Chem., 378: 779-786.
un, V. Hantke, K. e Koster, W.
mechanisms, genetics and regulation. In Sigel A and Sigel H (eds) Metal
ions in biological systems. Marcel Dekker, 67-145.
imer, L.H. (1984). Enzymatic excision fro
deoxiribonucleotides of adenine residues whose imidazole rings have
been ruptured. Nucleic Acids Res., 12: 6359-6367
mer, K. e Roth, W. (1996) Bendamustine, a low t
derivative with high efficacy in malignant lymphomas. Tumordiagnostik
and Therapie, 17(1): 1-6.
chelators in cancer chemotherapy. Curr. Top. Med. Chem., 4(15): 1623-
1635.
128

Buss, J.L., Torti, F.M. e Torti, S.V. (2003) The role of iron chelator in cancer
Cha Monofunctional adenine N-3
Cho d of RNA isolation by
Christman, M.F., Morgan, R.W., Jacobson, F.S. e Ames, B.N. (1985) Positive
Cun
ts of
da nes de reparo
de . M. (2002) Estudo do mecanismo de reparo das lesões
de Alencar, T.A.M., Leitão, A.C. e Lage, C. (2005) Nitrogen mustard and half-
therapy. Curr. Med. Chem., 10: 1021-1034.
rles, K, Bauer, G.B. e Povirk, L.F. (1997)
adducts of melphalan: Ocurrence at a mutational hotspot sequence and
resistance to removal by AlkA protein. Interscience.
mczynski, P. e Sacchi, N. (1987) Single-step metho
acid guanidinium thiocyanate-phenol-chloroform extraction. Analytical
Biochem., 162: 156-159
control of a regulation for defenses against oxidative stress and some
heat-shock proteins in Salmonella typhimurium. Cell, 41: 753-762.
ningham, R. P.(1997) DNA glycosylases. Mutat. Res. 383: 189-196
Cunningham, R.P & Weiss, B. (1986). Endonuclease III (nth) mutan
Escherichia coli. Proc. Natl. Acad. Sci. USA., 82: 474-478
Silva, R.I. (2006) Estudo da regulação e da expressão de ge
de DNA de Escherichia coli em resposta a quimioterápicos anti-tumorais.
Tese de Mestrado. Instituto de Biofísica Carlos Chagas Filho. UFRJ. Rio
de Janeiro.
Alencar, T.A
causadas por mostardas nitrogenadas em Escherichia coli in vivo. Tese
de Mestrado. Instituto de Biofísica Carlos Chagas Filho. UFRJ. Rio de
Janeiro.
mustard induced damage in Escherichia coli requires different DNA repair
pathways. Mut. Res. /Genet. Toxicol. and Envirom Mutagen. 582: 105-
129

115.
de Silva, I.U., McHugh, P.J., Clingen, P.H. e Hartley, J.A. (2000) Defining the
Debenham, P. G., Webb, M. B. T., Jones, N. J. e Cox, R. (1987) Molecular
Dem es. Annu. Ver.
Demple, B. e Halbrook, J. (1983) Inducible repair of oxidative DNA damage in
Dem r of oxidative damage to DNA:
Dro cross-links.
Fernandez de Henestrosa, A. R., Ogi, T., Aoyagi, S., Chafin, D., Hayes, J.J.,
Friedberg, E.C., Walker, G.C., Siede, W., Wood, R.D., Schultz, R.A. e
Fur 1997) Effects of 1,10-phenanthroline
roles of nucleotide excision repair and recombination in the repair of DNA
interstrand cross-links in mammalian cells. Molecular and Celular Biology,
7980-7990.
studies on the nature the repair defect in ataxia telangiectasia and their
implications for radiobiology. J. Cell Sci. Supll. 6:179-189.
ple, B. (1991) Regulation of bacterial oxidative stress gen
Genet., 25: 315-337.
Escherichia coli. Nature, 304: 466-468.
ple, B. e Harrison, L. (1994) Repai
enzimology and biology. Annu. Rev. Biochem., 63: 915-948.
nkert, M.L.G. e Kanaar, R. (2001) Repair of DNA interstrand
Mutat. Res., 486:217-247.
Ohmori, H. e Woodgate, R. (2000) Identification of additional genes
belonging to the LexA regulon in Escherichia coli. Mol. Microbiol., 35:
1560-1572.
Ellenberger, T. (2006) DNA repair and mutagenesis. Ed. ASM Press,
Washington DC, USA – 2nd edition.
tado, F.A., Asad, N.R. e Leitão, A.C. (
and hydrogen peroxide in Escherichia coli: lethal interaction. Mutat.
130

Res., 385: 251-258.
Galhardo, R.S., Almeida, C.E.B., Leitão, A.C. e Cabral-Neto, J.B. (2000)
Go canismos envolvidos na reparação
Gordienko, I. e Rupp, W.D. (1997) The limited strand-separating activity of
Gottesman, M.M., Fojo, T. e Bates, S.E. (2002) Multidrug resistance in
Hantke, K. (1981) Regulations of ferric ion transport in Escherichia coli K12:
Han d zinc
Hid .M., Walsh, C.T. e Demple, B.
Holand e Frei (2000) Cancer Medicine. B.C. Decker Inc. 5 ed.
Repair of DNA lesions induced by hydrogen peroxide in the presence of
iron chelators in Escherichia coli: participation of Endonuclease IV and
Fpg. J. Bacteriol., 182(7): 1964-1968.
nçalves, S.R.F. (2007) Estudo dos me
das lesões causadas por PUVA (psoralenos mais UV-A) em Escherichia
coli. Tese de Doutorado. Instituto de Biofísica Carlos Chagas Filho.
UFRJ. Rio de Janeiro.
the UvrAB protein complex and its role in the recognition of DNA damage.
EMBO J., 16(4):889-895.
cancer: role of ATP-dependent transporters. Nature Reviews Cancer, 2:
48-58.
Isolation of a constitutive mutant. Mol. Gen. Genet., 182: 288-292.
tke, K. (2002) Members of the Fur protein Family regulat iron an
transport in E. coli and characteristics of the Fur-regulated fhuF protein. J.
Mol. Microbiol. Biotechnol., 4: 217-222.
algo, E., Bollinguer, Jr.J.M., Bradley, T
(1995) Binuclear [2Fe-2S] clusters in Escherichia coli SoxR protein and
role of the metal centers in transcription. J. Biol. Chem. 270: 20908-
20914.
131

Jarosz, D.F., Beuning, P.J., Cohen, S.E. e Walker, G.C. (2007) Y-family DNA
Jou for cancer
Keyer, K., Gort, A.S. e Imlay, J.A. (1995) Superoxide and the production of
Koh ogen mustard-
Koh er-strand crosslinking of DNA
Koh ) Cross-linking and repair
Kol
n of excision repair
Kunz, B.A. e Mis, J.R. (1989) Mutational specificities of 1,3-bis(2
Lag onçalves, S.R.,
polimerases in E. coli. Trends in Microbiology, 15(2): 70-77.
naidi, Y. (2002) Cytochromo P450-based gene therapy
treatment : from concept to the clinic. Curr. Drug Metabolism, 14: 609-
622.
oxidative DNA damage. J. Bacteriol., 177(23): 6782-6790.
n, K. W. e Green, D. M. (1966) Transforming activity of nitr
crosslinked DNA. J. Mol. Biol., 19: 289-302.
n, K. W., Spears, C. L. e Doty, P. (1966) Int
by nitrogen mustard. J. Mol. Biol., 19: 266-288.
n, K. W., Steigbigel, N. H. e Spears, C. L. (1965
of DNA in sensitive and resistant strains of E. coli treated with nitrogen
mustard. Proc. Natl. Acad. Sci USA, 53: 1154-1161.
and, J.G. (2003) Antineoplasic agents I and II, 71:111
Kow, Y. W. e Van Houten, B. (1990) Mechanism of actio
enzimes. In: Ionizing radiation damage to DNA: Molecular Aspects. Wiley-
Liss, Inc. 81-89.
chloroethyl)-1-nitrosourea and nitrogen mustard in the SUP4-o gene of
Saccharomyces cerevisae. Cancer Res., 49(2): 279-283.
e, C., de Padula, M., de Alencar, T.A.M., da Fonseca G
Vidal, L.S., Cabral-Neto, J. e Leitão, A.C. (2003) New insights on how
nucleotide excision repair could remove DNA adducts induced by
chemotherapeutic agents and psoralens plus UV-A (PUVA) in Escherichia
132

coli cells. Mutat. Res., 544: 143-157 – Review
minat, F., Cambois G. , Zdizienicka, M.Z. e Lar Defais, M. (1998) Lack of
Lawley, P. D. e Phillips, D. H. (1995) DNA adducts from chemotherapy
Lee unctional specialization within the Fur
Lei e Alcantara Gomes,
Litt ssing reactions. J.
Little, J.W. (1991) Mechanism of specific LexA cleavage: autodigestion and
Lov n chelators derived
Ma A during oxidative stress by
Ma
correlation between repair of DNA interstrand cross-links and
hypersensitivy of hamster cells toward mitomicyn C and cisplatin. FEBS
Lett., 437: 97-100
agents. Mutat. Res., 335: 13-40.
, J.W. e Helmann J.D. (2007) F
family of metalloregulators. Biometals. 1:14853-8101
tão, A. C., Lage, C., Cabral-Neto, J.B., de Padula, M.
R. (2005) Radiobiologia e Fotobiologia. Respostas celulares às lesões
induzidas por agentes físicos e químicos. Apostila didática. Instituto de
Biofísica Carlos Chagas Filho. UFRJ. Rio de Janeiro.
le, J. W. (1993) LexA cleavage and other self-proce
Bacteriol., 175 (16): 4943-4950.
the role of RecA coprotease. Biochimie, 73:411-422.
ejoy, D.B. e Richardson, D.R. (2002) Novel “hybrid” iro
aroylhydrazones and thiosemicarbazones demonstrate selective
antiproliferative activity against tumor cells.
rtinez, A. e Kolter, R. (1997) Protection of DN
the nonespecific DNA-binding protein Dps. J. Bacteriol., 179: 5188-5194.
sta, A., Peter, J.G. e Phillips, R. DE. (1994) Molecular basis of nitrogen
mustard effects on transcription processes: role of depurination. Nucleic
Acids Res., 22 (19): 3880-3886.
133

Merck Vet. Edition (2003) Antineoplasic agents.
Mid chemotherapy
Miller, J.H. (1992) A short course bacterial genetics – 456pp. – Cold Spring
Mis rsal of DNA alkylation
Moody, C.S. e Hassan, H.M. (1984) Anaerobic biosynthesis of the
Myles, G.M. e Sancar, A. (1989) DNA repair. Chem. Res. Toxicol., 2(4):197-
Noll, D.M., Mason, T.M. e Miller, P.S. (2006) Formation and repair of
Ojw Synthesis of a
Oliv ineoplásicos biorredutíveis:
Olsen, L.S., Korsholm, B., Nexo, B.A e Wassermann, K. (1997) Nitrogen
http://www.merckvetmanual.com/mvm/htm/bc/191600.htm.
dleton, M.R. e Margison, G.P. (2003) Improvement of
efficacy by inactivation of DNA-repair pathway. Lancet. Oncol., 4(1): 37-
44.
Harbor Laboratory Press – cold Spring Harbor, NY
hina, Y., Duguid, E.M. e He, C. (2006) Direct reve
damage. Chem. Rev., 106:215-232.
manganese-containing superoxide dismutase in Escherichia coli. J. Biol.
Chem., 259(20):12821-12825
226.
interstrand cross-links in DNA. Chem. Rev., 106:277-301.
ang, J. O., Grueneberg, D. A. e Loecher, E. L. (1989)
duplex oligonucleotide containing a nitrogen mustard interstrand DNA-
DNA cross-link. Cancer Res., 49: 6529-6537.
eira, R.B. e Alves, R.J. (2002) Agentes ant
uma nova alternativa para o tratamento de tumors sólidos. Quim. Nova,
25(6):976-984.
mustard-mediated mutagenesis in human T-lymphocytes in vitro. Archives
of Toxicology, 71(3): 198-201
134

Pom 001) Redox-operated genetic switches: the
Pur S.,
o
Richardson, C.C. & Kornberg, A. (1961). A deoxyribonucleic acid
Ric
Richardson, D.R., Sharpe, P.C., Lovejoy, D.B., Senaratne, D., Kalinowski,
Ruh tic alkylating agents
Sam ning: a
San A novel repair enzyme: UvrABC excision
posiello, P,J, e Demple, B. (2
SoxR and OxyR transcription factors. Trends Biotechnol., 19: 109-114
nell, B., Sato, A., O'kelley, A., Price, C., Summerville, K., Hudson,
O'hare, C., Kiakos, K., Asao, T., Lee, M., Hartley, J.A. (2006) DNA
interstrand crosslinking agents: synthesis, DNA interactions, and
cytotoxicity f dimeric achiral seco-amino-CBI and conjugates of achiral
seco-amino-CBI with pyrrolobenzodiazepine (PBD). Bioorg Med Chem
Lett., 16(21): 5677-5681.
phosphatase-exonuclease from Escherichia coli. Purification and
characterization of the phosphatase activity. J. Biol. Chem., 239: 242-255.
hardson, D.R. (2001) Iron Chelators as therapeutic agents for the
treatment of cancer. Critical Reviews in Oncology/Hematology, 42: 267-
281
D.S., Islam, M. e Bernardt, P.V. (2006) Dipyridyl thiosemicarbazone
chelators with potent and selective antitumor activity form iron complexes
with redox activity. J. Med. Chem., 49(22):6510-6521
land, A. e Brendel, M. (1978) Mutagenesis by cytosta
in yeast strains of differing repair capacities. Genetics, 92: 83-97.
brook, J., Fritsch, E.F. e Maniatis, T. (1995) Molecular clo
laboratory manual, 3 vols 2nd ed. – Cold Spring Harbor Laboratory Press,
Cold Spring Harbor, New York
car, A. e Rupp, W.D. (1983)
nuclease of Escherichia coli cuts a DNA strand on both sides of the
135

damaged region. Cell, 33:249-260.
li, H., Karadenizli, A., Kolayli, F., Sav Gundes, U. e Vahaboglu, H. (2003)
Shi icroscopic
Sin olecular biology of mutagens and
Sou as pela radiação
Tor y of
Tou nscriptional regulation of
Tou
United States Pharmacopeia Drug Information, 2000.
ism of action of the
Expression stability of six housekeeping genes: a proposal for
resistancegene quantification studies of Pseudomonas aeruginosa by
real-timequantitative RT-PCR. J. Med. Microbiol., 52: 403-408
, Q., Thresher, R., Sancar, A. e Griffith, J. (1992) Electron m
study of (A)BC excinuclease DNA is sharply bent in the UvrB-DNA
complex. J. Mol. Biol., 226: 425-432.
ger, B. e Grunberger, D. (1983) M
carcinogens. Ed. Plenum Press, New York. Capítulo IV.
za, L.L. (2004) Estudo da reparação das lesões induzid
ultravioleta B (UV-B) no DNA de Escherichia coli. Tese de Mestrado.
Instituto de Biofísica Carlos Chagas Filho. UFRJ. Rio de Janeiro.
torice, P. (2002) Principles of Chemotherapy: Pharmacolog
Chemotherapeutic drugs. CancerSource.com
ati, D. (1988) Transcriptional and posttra
manganese Superoxide Dismutase biosynthesis in Escherichia coli,
studied with operon and protein fusions. J. Bacteriol., 170(6): 2511-2520.
ati, D., Jacques, M., Tardat, B., Bouchard, L. e Despied, S. (1995) Lethal
oxidative damage and mutagenesis are generated by iron in ∆fur mutants
of Escherichia coli: protective role of Superoxide Dismutase. J. Bacteriol.,
177(9): 2305-2314.
Van Houten, B. e Snowden, A. (1993) Mechan
Escherichia coli UvrABC nuclease: clues to the damage recognition
136

problem. Bioessays, 15: 51-59
Van Houten, B., Croteau, D.L., Della Vecchia, M.J. Wang, H.E. e Kisker, C.
Vid reparo das lesões causadas
Vid nsitivity
Wa . Biochem.,
Wasil, M., Halliwell, B., Grootveld, M., Moorehouse, C.P., Hutchison, D.C.S. e
Weigle, J. J. (1953) Induction of mutations in a bacterial virus. Proc. Natl.
Wij , E.W. (2000) The in vivo genetic activity
Wu induction of the soxRS (superoxide
Zhe ional
(2005) “Close-fifting” sleeves: DNA damage recognition by the UvrABC
nuclease system. Mutat. Res., 577: 92-117.
al, L.S. (2007) UvrB: uma proteína chave no
por mitomicina C no DNA de Escherichia coli. Tese de Doutorado.
Instituto de Biofísica Carlos Chagas Filho. UFRJ. Rio de Janeiro.
al, L.S., Santos, L.B., Lage, C. e Leitão, A.C. (2006) Enhenced se
of Escherichia coli uvrB mutants to mitomycin C pointsto a UV-C distinct
repair for DNA adducts. Chem. Res. Toxicol., 19: 1351-1356
lker, G.C. (1985) Inducible DNA repair systems. Annu. Rev
54:425-457.
Baum, H. (1987) The specificity of thiourea, dimethylthyourea and
dimethyl sulphoxide as scavengers of hydroxyl radicals. Biochem. J., 243:
867-870
Acad. Sci. USA, 39: 628-636.
en, J.P., Nivard, M.J. e Vogel
profile of the monofuncional nitrogen mustard 2-chloroethylamine differs
drastically from its bifunctional counterpart mechlorethamine.
Carcinogenesis. 21(10): 1859-1867
, J. e Weiss, B. (1992) Two stage
response) regulon of Escherichia coli . J. Bacteriol., 174: 3915-3920.
ng, M., Aslund, F. e Sorz, G. (1998) Activation of OxyR transcript
137

factor by reversible disulfide bond formation. Science, 279: 1718-1721.
ng, M., Doan, B., Schneider, T.D. e Storz, G. (1999) OxyR and SoxRZhe S
regulation of fur. J. Bacteriol., 181: 4639-4643.
138

ANEXO I

Mutation Research 582 (2005) 105–115
Nitrogen mustard- and half-mustard-induceddamage inEscherichia colirequires different DNA
repair pathways
T.A.M. De Alencar, A.C. Leitao, C. Lage∗
Lab. Radiobiologia Molecular, Programa de Biologia Molecular e Estrutural, Instituto de Biof´ısica Carlos Chagas Filho—UFRJ,Centro de Ciencias da Sa´ude, Bloco G, 21941-540 Rio de Janeiro, RJ, Brazil
Received 18 October 2004; received in revised form 10 January 2005; accepted 14 January 2005
Abstract
Bifunctional alkylating agents are used in tumor chemotherapy to induce the death of malignant cells through blockage of DNAreplication. Nitrogen mustards are commonly used chemotherapeutic agents that can bind mono- or bifunctionally to guaninesin DNA. Mustard HN1 is considered a monofunctional analog of bifunctional mustard HN2 (mechlorethamine).Escherichiacoli K12 mutant strains deficient in nucleotide excision repair (NER) or base excision repair (BER) were submitted to increasingconcentrations of HN2 or HN1, and the results revealed that damage induced by each chemical demands different DNA repairpathways. Damage induced by HN2 demands the activity of NER with a minor requirement of the BER pathway, while HN1damage repair depends on BER action, without any requirement of NER function. Taken together, our data suggest that HN1and HN2 seem to induce different types of damage, since their repair depends on distinct pathways inE. coli.© 2005 Elsevier B.V. All rights reserved.
Keywords:Nitrogen mustard;Escherichia coli; Nucleotide excision repair; Base excision repair
1. Introduction
Chemotherapeutic agents are used in tumor therapy,but undesirable effects may occur later on, since theyhave the potential to damage normal cells as well. This
∗ Corresponding author. Tel.: +55 21 2590 7147/2562 6577;fax: +55 21 2280 8193.
E-mail address:[email protected] (C. Lage).
is a matter of concern to many researchers, since theseagents can damage DNA and compromise its funda-mental properties of self-replication and preservationof genetic information. These agents are usually bi-functional, generating crosslinks between the two DNAstrands[1].
Mustard compounds emerged as radiomimeticchemical agents acting in the cell nucleus[2]. Nitrogenmustard mechlorethamine hydrochloride (HN2) was
1383-5718/$ – see front matter © 2005 Elsevier B.V. All rights reserved.doi:10.1016/j.mrgentox.2005.01.004

106 T.A.M. De Alencar et al. / Mutation Research 582 (2005) 105–115
the first drug used in cancer chemotherapy[3]. Thisand other mustard derivatives are used in chemotherapyagainst Hodgkin’s disease[4], cutaneous T-cell lym-phomas[5], and brain tumors[6], just to mention a fewexamples.
Bifunctional nitrogen mustard HN2 crosslinksDNA, but monoadducts and intrastrand biadducts mayalso occur[7]. HN2 binds to N7 positions in gua-nines[8], but the possibility of interaction with N3 inadenines has been suggested as well[7,9].
In vitro, HN2 can react with several targets in DNA,but the preferential binding sites are the 5′-GXC-3′sequences[1]. The molecular size of HN2 favorscrosslinks mostly within the major groove[10], andeven though only slight distortions of the DNA occur,they seem to block strand separation[11]. HN2-induced crosslinks are highly unstable lesions, with ahalf-life of about 2 h, since nitrogen mustards boundto N7 positions in guanines are supposed to inducelability of the glycosylic bonds, thus spontaneouslyconverting part of them into apurinic (AP) sites[1].Consequently, HN2-induced toxicity is lower whencompared to that observed for agents predicted toinduce more stable crosslinks[12]. Nevertheless,crosslinks are by far considered the main inactivatingdamage induced in the DNA by bifunctional alkylatingagents, and DNA function is expected to be inactivatedif there are at least two of them in the same genome[12].
Monofunctional nitrogen mustard 2-dimethyl-aminoethyl chloride hydrochloride (HN1) is also calledhalf-mustard and is considered a monofunctional ana-log of HN2 [10]. It is classified as a monofunctionalalkylating agent because when its single reactive siteloses one atom of chloride, it becomes able to reactwith one base, and to generate only monoadducts[10].According to Prakash and Strauss (1970 cited in[13]),HN1 has a low ability to kill yeast cells. This is sug-gested by the presence of shoulders in the survivalcurves of yeast that underwent HN1 treatment. Addi-tionally,B. subtiliscells can develop tolerance to treat-ment with HN1, resulting in some resistance againstthis agent[14]. Thus, monofunctional mustard HN1was classified as a weaker cytotoxic agent than HN2[2].
The use of HN2 as a chemotherapeutic agent inpatients may result in the unpredictable induction ofnew tumors in the future. There is lacking information
concerning the DNA repair mechanisms that removeHN1 or HN2 lesions in vivo, or the kind of lesions pro-duced by treatment with half-mustard. This promptedus to study the mechanisms involved in the repair ofHN2- and HN1-induced damage in the main DNA re-pair mutants ofEscherichia coli. Both nucleotide ex-cision repair (NER) and base excision repair (BER)mutants ofE. coli were used. The repair of most ofthe chemically induced damage in DNA seems to de-pend on the sequential action of NER proteins[8].Damage is recognized by the UvrA2B complex, fol-lowed by the release of the UvrA subunit, the endonu-cleolytic incisions by the UvrBC complex, the resyn-thesis of the remaining gap and the ligation by DNALigase. However, BER enzymes may remove somespecific base damage. The damaged base can be re-leased by theN-glycosylase activity of DNA glyco-sylases such as 3-methyl-adenine DNA glycosylase(e.g.,tagA or alkA), Endonuclease III (EndoIII;nth) orFormamido-pyrimidine DNA glycosylase (Fpg;fpg).The resulting AP site may then be processed by the3′-AP Lyase activity present in Endo III or Fpg, orby 5′-AP Endonucleases: Exonuclease III (Exo III;xthA) or Endonuclease IV (Endo IV;nfo). Completerepair is achieved after insertion of new nucleotidesby DNA Polymerase I and ligation by DNA Lig-ase.
Crosslinks represent only a small fraction of all le-sions induced by bifunctional chemotherapeutic agents[8], but if unrepaired, can lead to cell death. Their re-moval seem to depend on the sequential action of NERproteins performing incisions in one damaged strand,followed by the displacement of the incised base-linkedfragment. The resulting gap can be filled either bymeans of homologous recombination or translesionsynthesis. The connected fragment may be then sub-jected to a second incision by NER proteins, releasinga duplex fragment containing the crosslinking agent[8].
We show herein that the survival ofE. coliafter bi-functional mustard-induced lesions requires the pro-ficiency of both NER and BER mechanisms. Half-mustard lesions seem to be different, since we showthat BER plays an important role whereas NER appearsnot to be involved. Our results suggest that, in terms ofDNA damage and repair, the half-mustard agent is nota monofunctional version of the bifunctional nitrogenmustard HN2.

T.A.M. De Alencar et al. / Mutation Research 582 (2005) 105–115 107
2. Material and methods
2.1. Bacterial strains
TheE. coliK-12 strains used in this study are listedin Table 1.
2.2. Nitrogen mustards
Bifunctional nitrogen mustard HN2 (mechloret-hamine hydrochloride, MW 192.52) was purchasedfrom Sigma (catalogue – 12,256-4). Monofunctionalnitrogen mustard HN1 (2-dimethylaminoethyl chlo-ride hydrochloride, MW 144.05) was purchased fromAldrich (catalogue – D14,120-8). Both were solubi-lized in sterile deionized water and used at the indicatedconcentrations.
2.3. Growth conditions
Bacterial cultures were obtained by transfer-ring 50�L aliquots from 50% glycerol stocks at
−20◦C to 10 mL LB (Lysogeny Broth) medium[15]containing the appropriated antibiotics. These cul-tures were grown overnight at 37◦C with shak-ing (150 rpm), and then kept at 4◦C for up to 1week. For survival experiments, fresh overnight cul-tures were obtained from this refrigerated stock,and diluted 1:40 in fresh LB medium to growup to the mid-exponential growth phase (ca.2× 108 cells/mL).
2.4. Treatment conditions
Cultures in the mid-exponential growth phase werewashed twice (7710×g, 10 min each, 4–10◦C), andresuspended in 0.1 M PB (phosphate buffer), pH 7.0[15]. After washing, one initial aliquot was takenfor titration. Aliquots of 2 mL from each culturewere treated, in Erlenmeyer flasks, with the indi-cated doses of HN2 or HN1. Samples were incubatedat 37◦C for up to 60 min under vigorous shaking(240 rpm).
Table 1E. coliK12 strains used in this study
Designation Relevant genotype Source
AB1157 Wild type P. Howard-Flanders, Yale University, USAAB1886 [AB1157]uvrA6 P. Howard-Flanders, Yale University, USAAB1885 [AB1157]uvrB5 P. Howard-Flanders, Yale University, USAAB1884 [AB1157]uvrC34 P. Howard-Flanders, Yale University, USAAB2463 [AB1157]recA13 (Def) P. Howard-Flanders, Yale University, USAJC3890 [AB1157]�uvrB301 A.J. Clark (Through R. Devoret, CNRS, France)RJF603 [AB1157]�uvrA �uvrB Our laboratoryRJF604 [AB1157]�uvrA uvrC34 Our laboratoryRJF605 [AB1157]�uvrB �uvrC Our laboratoryBH20 [AB1157]fpg S. Boiteux, CNRS, FranceBH50 [AB1157]�xthA fpg S. Boiteux, CNRS, FranceBH100 [AB1157]nfo nth∆xthA fpg S. Boiteux, CNRS, FranceBH160 [AB1157]nth fpg S. Boiteux, CNRS, FranceBW375 [AB1157]nth B. Weiss, Emory University, USABW527 [AB1157]nfo B. Weiss, Emory University, USABW9091 [AB1157]xthA B. Weiss, Emory University, USABW9116 [AB1157]�xthA::Tn10 B. Weiss, Emory University, USARJF626 [AB1157]nth�xthA our laboratoryPQ65 Wild type P. Quillardet, Institut Pasteur, FrancePQ236 [PQ65]alkA1 tagA1 P. Quillardet, Institut Pasteur, FranceN3137 uvrA::Tn10 R.G.Loyd, Queens Med. Center, Nottingham, UKN3124 uvrC::Tn10 R.G.Loyd, Queens Med. Center, Nottingham, UK
The RJF strains used in this study were constructed through of standard P1vir transduction techniques[14], as follows: RJF603 and RJF604 wereconstructed by transduction with P1 obtained from N3137 (uvrA::Tn10) into JC3890 strain (�uvrB) and AB1884 (uvrC34) strains, respectively.RJF605 was constructed by transduction with P1 obtained from N3124 (uvrC::Tn10) into JC3890 strain (�uvrB). RJF626 was constructed bytransduction with P1 obtained from BW9116 (�xthA::Tn10) into BW375 (nth) strain.

108 T.A.M. De Alencar et al. / Mutation Research 582 (2005) 105–115
2.5. Measurement of bacterial inactivation bynitrogen mustards
After treatment with mustard compounds at the in-dicated concentrations, aliquots were diluted in M9buffer [15] without prior washing of the cultures, andplated in LB containing 1.5% agar. Plates were incu-bated at 37◦C for 24 h, and the colonies were scored.Survival was calculated by dividing the number of re-maining viable cells at each dose (N) by those of thetiter (No). The results are expressed as the mean andstandard errors of at least three independent experi-ments.N/No was plotted in semi-logarithmic graphs asa function of the mustard compounds’ concentrations(mM).
3. Results
3.1. Cell survival for bifunctional HN2 andmonofunctional HN1 treatments
The initial experiments with HN2 were designed todefine the incubation time necessary to obtain the max-imum lethality for each dose used (death rate is not in-creased when cells are incubated for longer periods oftime). The incubation time was defined as 60 min forthe wild type strain AB1157 (data not shown) and wasalso used in the protocol for the treatment of the DNArepair-deficient strains. Each culture was aliquoted into2 mL samples, and exposed to increasing concentra-tions of HN2 for 60 min. These experimental condi-tions allowed us to distinguish among resistant, sen-sitive, and hypersensitive strains. A similar procedurewas used for testing the same strains to HN1, unlessfor concentrations, increased 100-fold in this case.
3.2. Inactivation of uvr mutants by nitrogenmustards
The first indication that anuvr mutant was sensi-tive to HN2 was obtained with an unidentifieduvrmu-tant strain compared to a parentaluvr+ [8]. Our presentstudy was done with both single and double mutantsin uvrA, uvrB, anduvrC genes. Single mutantsuvrA6(AB1886),uvrB5 (AB1885),uvrC34 (AB1884), andthe double mutant�uvrA �uvrB (RJF603), were allsimilarly sensitive to HN2 (Fig. 1). �uvrA uvrC34
Fig. 1. Survival ofE.coli wild type and NER-deficient strains toHN2. Cultures in the mid-exponential phase of growth were treatedwith different concentrations of HN2 in phosphate buffer (pH 7.0) for60 min. (�) Wild type (AB1157), (�) uvrA6 (AB1886), (�) uvrB5(AB1885), (�)uvrC34 (AB1884), and (©)�uvrA �uvrB (RJF603).Plots represent mean values± standard errors.
(RJF604) and�uvrB �uvrC (RJF605) double mutantswere as sensitive as the�uvrA �uvrB (data not shown)one, and all of them were as sensitive as it was theuvrA6mutant.
Wild type and singleuvrmutants were exposed toHN1 under the same experimental conditions estab-lished for HN2. All strains are extremely resistant tothis agent, even when subjected to 100-fold higherdoses of HN1 (Fig. 2).
3.3. Participation of RecA-mediated repair insurvival against nitrogen mustard treatments
The recA (Def) mutant strain (recA13) was testedfor sensitivity to nitrogen mustards since the RecA pro-tein is involved in both the regulation ofuvrA anduvrBexpression and in homologous recombination. TherecA-defective strain was as sensitive to HN2 (Fig. 3),as it was theuvrA6 mutant (seeFig. 1). The recA13(Def) mutant is also sensitive to HN1 (Fig. 4), althoughnot to the same extent as it was to HN2 (seeFig. 3).Thus, it appears that RecA functions are involved inthe repair of lesions induced by both mustards.
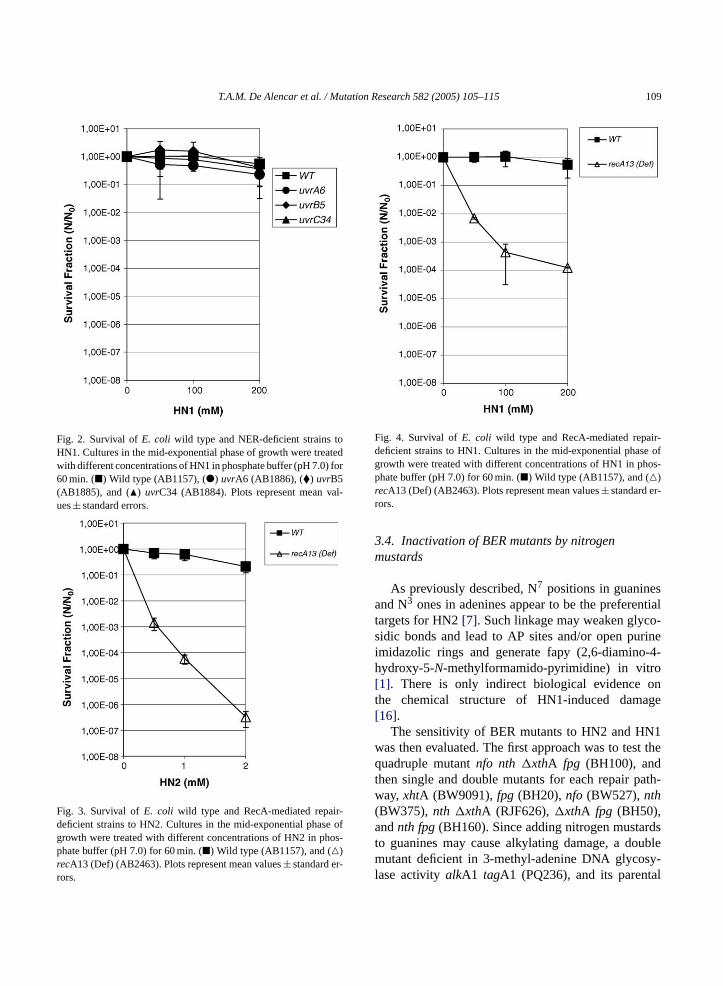
T.A.M. De Alencar et al. / Mutation Research 582 (2005) 105–115 109
Fig. 2. Survival ofE. coli wild type and NER-deficient strains toHN1. Cultures in the mid-exponential phase of growth were treatedwith different concentrations of HN1 in phosphate buffer (pH 7.0) for60 min. (�) Wild type (AB1157), (�) uvrA6 (AB1886), (�) uvrB5(AB1885), and (�) uvrC34 (AB1884). Plots represent mean val-ues± standard errors.
Fig. 3. Survival ofE. coli wild type and RecA-mediated repair-deficient strains to HN2. Cultures in the mid-exponential phase ofgrowth were treated with different concentrations of HN2 in phos-phate buffer (pH 7.0) for 60 min. (�) Wild type (AB1157), and (�)recA13 (Def) (AB2463). Plots represent mean values± standard er-rors.
Fig. 4. Survival ofE. coli wild type and RecA-mediated repair-deficient strains to HN1. Cultures in the mid-exponential phase ofgrowth were treated with different concentrations of HN1 in phos-phate buffer (pH 7.0) for 60 min. (�) Wild type (AB1157), and (�)recA13 (Def) (AB2463). Plots represent mean values± standard er-rors.
3.4. Inactivation of BER mutants by nitrogenmustards
As previously described, N7 positions in guaninesand N3 ones in adenines appear to be the preferentialtargets for HN2[7]. Such linkage may weaken glyco-sidic bonds and lead to AP sites and/or open purineimidazolic rings and generate fapy (2,6-diamino-4-hydroxy-5-N-methylformamido-pyrimidine) in vitro[1]. There is only indirect biological evidence onthe chemical structure of HN1-induced damage[16].
The sensitivity of BER mutants to HN2 and HN1was then evaluated. The first approach was to test thequadruple mutantnfo nth�xthA fpg (BH100), andthen single and double mutants for each repair path-way, xhtA (BW9091), fpg (BH20),nfo (BW527),nth(BW375), nth �xthA (RJF626),�xthA fpg (BH50),andnth fpg(BH160). Since adding nitrogen mustardsto guanines may cause alkylating damage, a doublemutant deficient in 3-methyl-adenine DNA glycosy-lase activityalkA1 tagA1 (PQ236), and its parental

110 T.A.M. De Alencar et al. / Mutation Research 582 (2005) 105–115
Fig. 5. Survival ofE. coli wild type and BER-deficient strains toHN2. Cultures in the mid-exponential phase of growth were treatedwith different concentrations of HN2 in phosphate buffer (pH 7.0) for60 min. (�) Wild type (AB1157), (�) nfo nth�xthA fpg (BH100),(©) nth (BW375), and (♦) xthA (BW9091). Plots represent meanvalues± standard errors.
wild type strain (PQ65), were also exposed to HN2and HN1.
The quadruple mutant BH100 is sensitive to HN2(Fig. 5) and its sensitivity is intermediate between thatof the wild type and theuvr mutant strains (Fig. 1).The fpg and nfo mutant strains, in which only oneBER enzyme was absent, were as resistant to HN2as their parental wild type strain (data not shown).Single mutantsnth and xthA were slightly sensitiveto HN2 when compared to their parental wild typestrain, but not as sensitive as the quadruple mutantBH100 (Fig. 5). The quadruple mutant BH100 is alsosensitive to HN1 (Fig. 6), and except for the slightsensitivity of thenth single mutant to HN1 (Fig. 6),all other single mutant strains were as resistant toHN1 as it was the wild type strain (data not shown).Several combinations of BER enzymes possibly in-volved in the repair of nitrogen mustards HN2 andHN1 were then investigated by using double mu-tant strains in which both one DNA glycosylase/3′-AP lyase and one 5′-AP endonuclease activity weremissing. For this purpose,fpg �xthA, nth fpg and
Fig. 6. Survival ofE. coli wild type and BER-deficient strains toHN1. Cultures in the mid-exponential phase of growth were treatedwith different concentrations of HN1 in phosphate buffer (pH 7.0) for60 min. (�) Wild type (AB1157), (�) nfo nth�xthA fpg (BH100),and (©) nth (BW375). Plots represent mean values± standarderrors.
nth �xthA double mutants were tested in terms oftheir sensitivity to HN2 and HN1. All double mutantswere nearly as sensitive to HN2 as the quadruple mu-tant BH100, with all 3′-AP lyase and/or 5′-AP en-donuclease combinations being significantly relevant(Fig. 7).
Removal of 3-Me-alkyl-purines by DNA glycosy-lases is, conceptually, an important activity to repairalkylation damage, being absent in thealkA1 tagA1double mutant. Nevertheless, the inactivation of thisdouble mutant strain (Fig. 8) matched the survival ratesshown by the tested BER mutants for the same concen-trations of HN2 (seeFig. 5for survival curves of otherBER mutants to HN2).
Concerning HN1, wheneverfpg is absent, the corre-spondent double mutant is slightly more sensitive thanthe wild type strain. It is noteworthy that the doublemutantnth�xthA is as resistant to HN1 as the wildtype strain (Fig. 9). However, the absence of DNA gly-cosylase activity to repair alkylation damage in doublemutantalkA1 tagA1 did not result in increased sensi-tivity to HN1 (Fig. 10).
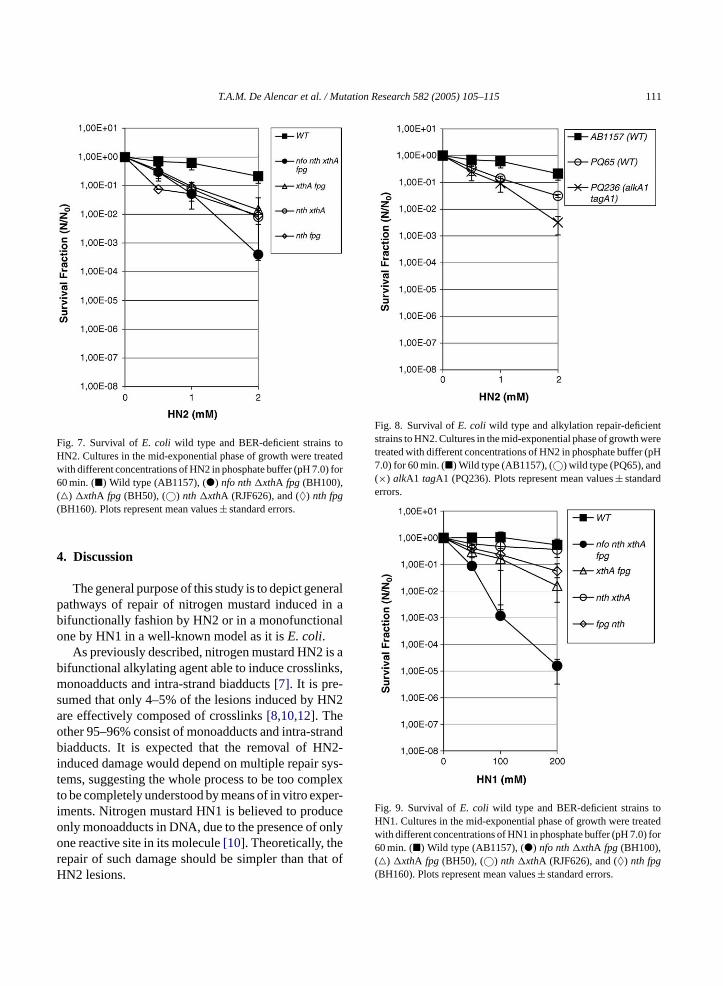
T.A.M. De Alencar et al. / Mutation Research 582 (2005) 105–115 111
Fig. 7. Survival ofE. coli wild type and BER-deficient strains toHN2. Cultures in the mid-exponential phase of growth were treatedwith different concentrations of HN2 in phosphate buffer (pH 7.0) for60 min. (�) Wild type (AB1157), (�) nfo nth�xthA fpg (BH100),(�) �xthA fpg (BH50), (©) nth�xthA (RJF626), and (♦) nth fpg(BH160). Plots represent mean values± standard errors.
4. Discussion
The general purpose of this study is to depict generalpathways of repair of nitrogen mustard induced in abifunctionally fashion by HN2 or in a monofunctionalone by HN1 in a well-known model as it isE. coli.
As previously described, nitrogen mustard HN2 is abifunctional alkylating agent able to induce crosslinks,monoadducts and intra-strand biadducts[7]. It is pre-sumed that only 4–5% of the lesions induced by HN2are effectively composed of crosslinks[8,10,12]. Theother 95–96% consist of monoadducts and intra-strandbiadducts. It is expected that the removal of HN2-induced damage would depend on multiple repair sys-tems, suggesting the whole process to be too complexto be completely understood by means of in vitro exper-iments. Nitrogen mustard HN1 is believed to produceonly monoadducts in DNA, due to the presence of onlyone reactive site in its molecule[10]. Theoretically, therepair of such damage should be simpler than that ofHN2 lesions.
Fig. 8. Survival ofE. coliwild type and alkylation repair-deficientstrains to HN2. Cultures in the mid-exponential phase of growth weretreated with different concentrations of HN2 in phosphate buffer (pH7.0) for 60 min. (�) Wild type (AB1157), (©) wild type (PQ65), and(×) alkA1 tagA1 (PQ236). Plots represent mean values± standarderrors.
Fig. 9. Survival ofE. coli wild type and BER-deficient strains toHN1. Cultures in the mid-exponential phase of growth were treatedwith different concentrations of HN1 in phosphate buffer (pH 7.0) for60 min. (�) Wild type (AB1157), (�) nfo nth�xthA fpg (BH100),(�) �xthA fpg (BH50), (©) nth�xthA (RJF626), and (♦) nth fpg(BH160). Plots represent mean values± standard errors.

112 T.A.M. De Alencar et al. / Mutation Research 582 (2005) 105–115
Fig. 10. Survival ofE. coliwild type and alkylation repair-deficientstrains to HN1. Cultures in the mid-exponential phase of growth weretreated with different concentrations of HN1 in phosphate buffer (pH7.0) for 60 min. (�) Wild type (AB1157), (©) wild type (PQ65), and(×) alkA1 tagA1 (PQ236). Plots represent mean values± standarderrors.
The first approach was to check for the sensitivity ofNER mutants. NER participation in the repair of dam-age produced by several chemical agents is inferred bytesting onlyuvrA mutants[17]. However, evaluationof the sensitivity of anuvrA mutant does not implythat NER participates in the repair of these lesions[8].The participation of NER in vivo was demonstratedonly for Ultraviolet C-induced cyclobutane pyrimidinedimers (CPD)[18] and for 6–4 photoproducts[19]. Ad-ditionally, most of the lesions believed to be substratesfor NER were demonstrated by in vitro experimentswith isolated damaged DNA and purified enzymes[8].This paper brings evidence that all the enzymes be-lieved to be involved in NER are necessary to repairHN2-induced damage inE. coli. The finding of similarsensitivity to the drug in single mutants does not mean,however, that all of them act through the same pathway.The implication of NER was further ascertained bystudying the inactivation produced in double mutantsconstructed by deletions in twouvr genes (helicase-deficientuvrA uvrB, nuclease-deficientuvrBuvrC, andUvrB recognition-proficientuvrA uvrC). Their sensi-tivity to HN2 was nearly identical to that obtained for
their parental single mutants. Therefore, this approachwas used to test if alluvrA, uvrB, anduvrC geneswere necessary for the repair of HN2 lesions. It is stillunder investigation whether this similarity in require-ment for NER in an UV-C-like manner may arise fromsimilarity in the induced damages, for instance, CPDand intrastrand biadducts. In support of this similar-ity, Hanawalt and Haynes have already demonstratedthat the pattern of DNA incision and resealing weresimilar for both HN2 and UV-C induced lesions ([20],reviewed in Lage et al., 2003[21]).
It has been accepted that the fundamental reason forthe broad activity of NER enzymes upon quite differ-ent substrates is that all of the lesions are able to inducevariable degrees of topological DNA distortions, whichshould be recognized by the UvrA2B complex[22,23].If both mustards induce similar DNA distortions thatare able to be recognized by NER, there should be an-other explanation for the results obtained with HN1.HN1-induced damage may be recognized by anotherspecific repair mechanism. This supposition led us tosearch for the involvement of other repair pathways inthe removal of HN1-induced damage.
The participation of all NER enzymes in the repairof damage induced by HN2 was otherwise expectedand confirmed, differently from some other crosslink-ing agents (mitomycin C and psoralen + UV-A light)[21]. Structural modifications of the double helix mayarise because of the induction of a significant numberof biadducts. Molecular measurements predict that theHN2 molecule can span 7.4A when completely ex-panded inside the helix[1]. It appears that some DNAsequences are frequently involved in linkage to HN2because of the distance between adjacent N7 positionsin guanines[1]. The crosslinks formed appear to be un-stable, with a reported half-life of about 2 h. Thus, HN2was assumed to be a less toxic agent than other bifunc-tional ones that may induce more stable crosslinks[12].A single crosslink in the genome of repair-deficientbacteria is lethal, but proficient bacteria can easily re-move about 20 crosslinks through excision repair[2].Berardini et al.[24] pointed out the participation ofNER in the repair of HN2-induced damage in a plas-mid, when analyzing its replication in bothuvrA anduvrB hosts.
Unexpectedly, the repair of HN1-induced damagedoes not seem to involve any of the NER proteins, sinceall NER mutants tested,uvrA6, uvrB5, anduvrC34

T.A.M. De Alencar et al. / Mutation Research 582 (2005) 105–115 113
were as resistant as it was their parental wild type strain.This is a novel finding since virtually all lesions testedup to date were repaired in vitro by NER enzymes[8,25].
RecA protein is involved in both regulatinguvrAanduvrB expression and in homologous recombina-tion. Since the repair of lesions induced by HN2 de-mands the functionality ofuvrA anduvrB gene prod-ucts, we studied the effect of arecA mutation on sur-vival to nitrogen mustard challenge. A role for RecA inthe repair of a crosslink induced by PUVA was previ-ously inferred[12], while the SOS response was shownto be induced by other alkylating agents inE. coli tagmutants[26]. So, RecA functions are considered es-sential[12] to relieve the blocking effect of crosslinksduring replication[1]. Accordingly, therecA (Def) mu-tant is highly sensitive to HN2.
The blocking replication effect associated withHN1-induced adducts was also verified in terms ofthe high sensitivity to HN1 found in therecA13 (Def)mutant. AsuvrA anduvrB gene products were not re-quired to repair HN1-induced damage, this implies thatRecA may participate in the repair of HN1-inducedlesions through RecA-mediated homologous recombi-nation. In considering RecA-mediated repair a mech-anism of damage tolerance, Ruhland and Brendel[13]had already noticed shoulders in survival curves to HN1in yeast, indicating the existence of a somehow lim-ited cell tolerance against monofunctional alkylatingagents.
The unstable crosslinks were described as leadingto AP sites in vitro. This specific information directedour study towards the evaluation of sensitivity of dis-tinct BER mutant strains. BER enzymes, especiallyEndonuclease III and Exonuclease III, were found torepair HN2-induced damage, thus reflecting a joint ac-tion of the DNA glycosylase-3′-AP lyase activity ofEndonuclease III and the 5′-AP endonuclease activ-ity of Exonuclease III. The double mutantnth�xthAis as sensitive as the original single mutant strains toHN2 treatment. Thenfomutant was resistant to HN2,probably because Exonuclease III’s 5′-AP endonucle-ase activity may substitute it.
In fact, thexthA mutant is sensitive to HN2. SinceN-glycosidic bonds seem to be weakened through thelinkage of HN2[27], the resulting AP sites might berecognized by Exonuclease III, suggesting a direct rolefor this enzyme in the repair of this kind of damage.
The required 3′-AP lyase activity of Endonuclease IIImay also be important because of its direct action onbase residues. Such 3′-AP lyase activity would demandthe subsequent action of Exonuclease III.
The 3′-AP lyase activity of Fpg protein was alsoinvestigated. However, the singlefpgmutant was notsensitive to HN2, while double mutantsnth fpgand�xthA fpgwere more sensitive to HN2 than their sin-gle parental mutant strainsnthandxthA, respectively.In the case of HN2 induced lesions, there may be nofunction predicted for Fpg protein alone. These resultsdisagree with those of Brooks et al.[28], who couldnot detect any participation of the Fpg enzyme in therepair of damage induced by bifunctional benzoic acidmustard (BAM).
Considering the results obtained with BER mutants,it seems plausible to assume that all enzymes appear tobe important and concurrent, i.e., there may be a com-mon substrate recognized by more than one enzyme.Anyway, according to the hypothesis raised by Mastaet al. [7], failure in the repair of AP sites should leadto accumulation of potentially mutagenic and carcino-genic damage.
Of greater remark than what was observed for HN2,there is an important demand for BER enzymes to re-move HN1-induced damage, although it was not pos-sible to detect other specific activities besides that ofEndonuclease III. Different from what was observedfor HN2, double mutants of BER in which one of thedeficient genes wasfpgappeared not to be so sensitiveto HN1 as their parental single mutants.
The enzymes AlkA and TagA fromE. coli (3-methyl-adenine DNA glycosylases) are described asDNA glycosylase enzymes that remove alkylatedpurines from DNA. Their absence in thealkA tagAdouble mutant consequently led to a significant sen-sitivity to HN2, probably due to the presence of un-repaired adenine adducts, as described by Balcome etal. [9] who detected DNA adenine–adenine crosslinksinduced by nitrogen mustards in vitro. These find-ings were interpreted as the basis to explain the en-hanced cytotoxicity and mutagenicity of nitrogen mus-tards found by other authors in cells lacking 3-methyl-adenine DNA glycosylase activity. However, the ob-served inactivation was nearly the same as that foundin the above-discussed BER mutants. This suggeststhat enzymes such as Exonuclease III could be re-sponsible for the finalization of the primary actions

114 T.A.M. De Alencar et al. / Mutation Research 582 (2005) 105–115
Table 2Summary ofE. coliDNA Repair functions relevant to repair HN2- and HN1-induced damage, as depicted from our data
The shadowed region in the table represents DNA damage predicted to be absent in HN1-treated cells; ND: not detected in our conditions.
of DNA glycosylases. The survival of cells to HN1-induced lesions is not dependent onalkA and tagA,thus suggesting that HN1 might produce chemicallydistinct alkylated guanines, what appears to differ froma previously observed mutation spectrum inDrosophilasuggesting that HN1 should induce N7-alkyl-guanines[16].
The analysis of survival of DNA repair mutants tothe chemical analogous nitrogen mustards HN2 andHN1 implies that, despite predicted to mostly targetguanines, the chemical nature of the induced lesionsappears to be different. Once the repair of HN1-inducedmonoadducts depends essentially on BER enzymes, itis then inferred that that the sensitivity of BER mutantsto HN2 probably derives from their inability to repairHN2-induced monoadducts and AP-sites. Unlikely tooccur in the HN1-induced damage pool, crosslinks andintrastrand biadducts may thus be the substrates forNER. Thus, HN1 cannot be simply considered a mono-functional analog of HN2, since repair of DNA dam-age induced by each agent demand specific DNA re-pair pathway. For simplification, a summary of the dis-cussed data can be viewed onTable 2.
Experiments are being carried out to chemically de-fine the induced damage in the DNA after treatmentwith both mustards.
Acknowledgements
This work was supported by CNPq, CAPES andFAPERJ. We thank J.S. Cardoso and A.B. Silva fortheir expert technical support. We are grateful to M.Padula, D.P. Carvalho and F.A.C. Furtado for criti-cal reading of this manuscript. We also thank Dr. E.C.Friedberg for his helpful comments.
References
[1] J.O. Ojwang, D.A. Grueneberg, E.L. Loechler, Synthesisof a duplex oligonucleotide containing a nitrogen mustardinterstrand DNA–DNA cross-link, Cancer Res. 49 (1989)6529–6537.
[2] P.D. Lawley, D.H. Phillips, DNA adducts from chemotherapeu-tic agents, Mutat. Res. 355 (1996) 13–40.
[3] A. Gilman, Fr.S. Philips, The biological actions and therapeuticapplications of B-chloroethyl amines and sulfides, Science 103(1946) 409–416.
[4] M. Andre, M. Henry-Amar, D. Blaise, P. Colombat, J. Fleury, N.Milpied, J.-Y. Cahn, J.-L. Pico, Y. Bastion, M. Kuentz, G. Nedel-lec, M. Attal, C. Ferme, C. Gisselbrecht, Treatment–relateddeaths and second risk after autologous stem-cell transplan-tation for Hodgkin’s disease, Blood 92 (6) (1998) 1933–1940.
[5] E. Esteve, M. Bagot, P. Joly, P. Souteyrand, M. Beylot–Barry,L. Vaillant, M. Delaunay, M.F. Avril, L. Laroche, F. Grange,

T.A.M. De Alencar et al. / Mutation Research 582 (2005) 105–115 115
E. Thomine, J. Wechsler, A prospective study of cutaneous in-tolerance to topical mechlorethamine therapy in patients withcutaneous T-cell lymphomas. French Study Group of Cuta-neous Lymphomas, Arch. Dermatol. 135 (11) (1999) 1349–1353.
[6] J.L. Ater, J. van Eys, S.Y. Woo, B. Moore III, D.R. Copeland,J. Bruner, MOPP chemotherapy without irradiation as primarypostsurgical therapy for brain tumors in infants and young chil-dren, Neurooncology 32 (3) (1997) 243–252.
[7] A. Masta, P.J. Gray, D.R. Phillips, Molecular bases of nitrogenmustard effects on transcription processes: role of depurination,Nucleic Acids Res. 22 (19) (1994) 3880–3886.
[8] E.C. Friedberg, G.C. Walker, W. Siede (Eds.), DNA Repair andMutagenesis, ASM Press, Washington, DC, USA, 1995.
[9] S. Balcome, S. Park, D.R. Quirk Dorr, L. Hafner, L. Phillips,N. Tretyakova, Adenine-containing DNA–DNA cross-links ofantitumor nitrogen mustards, Chem. Res. Toxicol. 17 (2004)950–962.
[10] K.W. Kohn, C.L. Spears, P. Doty, Inter-strand crosslinkingof DNA by nitrogen mustard, J. Mol. Biol. 19 (1966) 266–288.
[11] K.W. Kohn, N.H. Steigbigel, C.L. Spears, Cross-linking andrepair of DNA in sensitive and resistant strains ofE. coli treatedwith nitrogen mustard, Biochemistry 53 (1965) 1154–1161.
[12] M.L.G. Dronkert, R. Kanaar, Repair of DNA interstrand cross-links, Mutat. Res. 486 (2001) 217–247.
[13] A. Ruhland, M. Brendel, Mutagenesis by cytostatic alkylatingagents in yeast strains of differing repair capacities, Genetics92 (1979) 83–97.
[14] K.W. Kohn, D.M. Green, Transforming activity of nitrogenmustard-crosslinked DNA, J. Mol. Biol. 19 (1966) 289–302.
[15] J. Miller, A Short Course in Bacterial Genetics, Cold SpringHarbor Laboratory Press, Cold Spring Harbor, N.Y., 1992.
[16] J.P.H. Wijen, M.J.M. Nivard, E.W. Vogel, The in vivogenetic activity profile of the monofunctional mustard 2-chloroethylamine differs drastically from its bifunctionalcounterpart mechlorethamine, Carcinogenesis 21 (10) (2000)1859–1867.
[17] M. Cupido, B.A. Bridges, Uvr-independent repair of 8-methoxypsoralen crosslinks inEscherichia coli: evidence fora recombination process, Mutat. Res. 146 (1985) 135–141.
[18] P. Howard–Flanders, R.P. Boyce, L. Theriot, Threeloci inEscherichia coliK12 that control the excision of pyrimidinedimers and certain other mutagen products from DNA, Genet-ics 53 (1966) 1119–1136.
[19] W.A. Franklin, W.A. Haseltine, Removal of UV light-inducedpyrimidine-pyrimidone (6-4) products fromEscherichia coliDNA requires theuvrA, uvrB, anduvrC gene products, Proc.Natl. Acad. Sci. U.S.A. 81 (12) (1984) 3821–3824.
[20] P.C. Hanawalt, R.H. Haynes, Repair replication of DNA in bac-teria: Irrelevance of chemical nature of base defect, Biochem.Biophys. Res. Commun. 19 (4) (1965) 462–467.
[21] C. Lage, M. de Padula, T.A.M. de Alencar, S.R.F. Gonc¸alves,L.S. Vidal, J. Cabral-Neto, A.C. Leitao, New insights on hownucleotide excision repair could remove DNA adducts inducedby chemotherapeutic agents and psoralens plus UV-A (PUVA)in Escherichia coli, Mutat. Res. Rev. 544 (2003) 143–157.
[22] E.Y. Oh, L. Grossman, Helicase properties of theEscherichiacoliUvrA2B protein complex, Proc. Natl. Acad. Sci. U.S.A. 84(1987) 3638–3642.
[23] Y. Zou, B. Van Houten, Strand opening by the UvrA2B complexallows dynamic recognition of DNA damage, Embo J. 18 (1999)4889–4901.
[24] M. Berardini, W. Mackay, E.L. Loechler, Evidence for arecombination-independent pathway for the repair of DNA in-terstrand cross-links based on a site-specific study with nitrogenmustard, Biochemistry 36 (1997) 3506–3513.
[25] A. Snowden, Y.W. Kow, B. Van Houten, Damage repertoire oftheEscherichia coliUvrABC nuclease complex includes abasicsites, base-damage analogues, and lesions containing adjacent5′ or 3′ nicks, Biochemistry 29 (31) (1990) 7251–7259.
[26] R.C. de Oliveira, J. Laval, S. Boiteux, Induction of SOS andadaptive responses by alkylating agents inEscherichia colimu-tants deficient in 3-methyladenine-DNA glycosylase activities,Mutat. Res. 183 (1986) 11–20.
[27] B. Singer, D. Grunberger, Molecular Biology of Mutagens &Carcinogens, Plenum Press, New York, USA, 1983.
[28] N. Brooks, P.J. McHugh, M. Lee, J.A. Hartley, The role of baseexcision repair in the repair of DNA adducts formed by a seriesof nitrogen mustard-containing, analogues of distamycin of in-creasing binding site size, Anti-Cancer Drug Design 14 (1999)11–18.

ANEXO II

ANEXO II
Determinação da expressão relativa dos genes NER por RT-PCR quantitativo
em tempo real (Metodologia SyBr Green):
Realizamos experimentos com RT-PCR em tempo real a fim de sabermos
se no caso de HN1 o reparo NER não participa porque não há sinalização para
sua possível atuação, ou se realmente mesmo UvrABC sendo induzido, não é tão
específico para as lesões do tipo HN1. Os experimentos com HN2 foram
realizados, neste caso, como controle, uma vez que acreditamos ser NER o
principal reparo associado a esta mostarda, devido aos resultados de
sobrevivência celular, e portanto, teoricamente, deve haver sinal indutor para a
participação deste mecanismo (de Alencar et al., 2005).
Para verificar se o sistema de reparo de DNA por excisão de nucleotídeos,
como caracterizado por Sancar e Rupp (1983) era requerido em sua integridade
para indução da resposta as mostardas nitrogenadas (bi- HN2 e monofuncional –
HN1), foram realizados experimentos para quantificar os transcritos mensageiros
dos genes NER nas células tratadas.
O RT-PCR quantitativo em tempo real é uma metodologia que permite
quantificar o RNA mensageiro que está sendo expresso durante cada tratamento.
Este conteúdo de RNA é normalizado em relação à expressão de RNA da
situação sem tratamento (controle) e também em relação à expressão do gene
escolhido como normalizador (gene constitutivo).
A metodologia aplicada nesta técnica consiste na discriminação da
quantidade de determinada seqüência de ácido nucléico presente em uma dada
amostra tendo como instrumento de processividade o monitoramento de
2

fluorescência emitida por fluoróforos presentes em moléculas fluorescentes que se
intercalam inespecificamente entre os pares de base do DNA. A metodologia SyBr
Green consiste no emprego de um corante análogo ao brometo de etídeo que se
associa preferencialmente aos produtos de ácidos nucléicos dupla fita que estão
sendo gerados a partir do processo de amplificação especificados pelos
iniciadores. Uma vez complexado aos produtos gerados a cada ciclo de
amplificação, este corante emite uma fluorescência que é detectada pelo
termociclador onde um software, estruturado dentro de um modelo matemático
apropriado, registra a intensidade de fluorescência captada e denuncia o ciclo
onde o número de cópias do alvo gerado, atinge uma reprodução acima de uma
faixa basal (este ciclo recebe o nome de Ct).
Finda a reação, uma curva de desnaturação do DNA é executada com a
finalidade de averigüar a especificidade da amplificação. O SyBr Green
indiscriminadamente interage com qualquer ácido nucléico dupla fita, neste
sentido, inespecificidades advindas de hibridizações parciais dos iniciadores, por
exemplo, podem interferir no processo quantitativo conduzindo a erros de
interpretação. Um procedimento de averigüação é realizado e consiste em
observar o Tm (temperatura onde 50% dos produtos encontram-se na forma de
dupla fita) dos produtos gerados, submetendo-os a uma elevação da temperatura
dentro de uma faixa apropriada.
A modalidade RT-PCR quantitativa consiste em detectar o mRNA ao invés
do DNA tendo como requisito prévio, sua conversão a cDNA através de atuação
da transcriptase reversa.
3

METODOLOGIA
1 - Desenho dos oligonucleotídeos iniciadores
A expressão dos seguintes transcritos foi analisada: uvrA, uvrB, uvrC, cho,
recA, e rpoD.
As seqüências codificantes dos genes correspondentes foram obtidas no
banco de seqüência genômica de E. coli K12 no seguinte endereço eletrônico:
http://cmr.tigr.org/tigr-scripts/CMR/GenomePage. Tais seqüências foram usadas
como molde em um programa de construção de oligonucleotídeos – Primer
ExpressR Software 2.0, Applied Biosystems.
2 - Isolamento de RNA
Para a extração de RNA foi utilizada água estéril previamente tratada com
dietilpirocarbonato – DEPC (Sigma), um inibidor de RNAse com a finalidade de
evitar a degradação do RNA (nesta etapa 0,1% de DEPC eram dissolvidos em
1000 ml de água milliQ sendo incubado a temperatura ambiente por uma noite.
Logo após, tal solução era autoclavada a 100ºC (para inativar seus efeitos
tóxicos).
A bancada de trabalho foi tratada com álcool 70%, e os equipamentos de
manipulação das amostras, tratados com NaOH 0,1N com o intuito de eliminar
qualquer RNAse ou fragmentos de RNA remanescente de experimentos
anteriores. Controles negativos, destituídos de RNA ou cDNA das amostras
analisadas, foram utilizados em cada etapa como parâmetro de monitoramento de
possíveis contaminações nos reagentes utilizados (na etapa de extração de RNA,
o controle negativo foi constituído pela adição dos reagentes de extração exceto o
RNA).
O isolamento de RNA foi realizado pelo tratamento das amostras com Trizol
4

(Chomczynski e Sacchi, 1987).
Inicialmente, para cada tratamento, 15ml de cultura em fase exponencial
eram centrifugados (10.000rpm/10’/4ºC, Sorvall RC5B rotor SS-34) e o sedimento
celular ressuspendido em 15ml de tampão M9. Esta suspensão por sua vez, era
subdividida em três alíquotas de 5ml cada e submetida ao tratamento.
Uma vez tratadas, as amostras eram centrifugadas
(10.000rpm/5minutos/4ºC, Sorvall RC5B F28/micro) e o sedimento resultante,
ressuspenso com 300µl de TrizolR (Invitrogen) seguido de incubação por 5
minutos à temperatura ambiente. Posteriormente, 200µl de clorofórmio eram
adicionados e a mistura resultante era submetida à agitação brusca por 2 minutos
e então centrifugada (15.000rpm/15’/4ºC, Sorvall RC5B F28/micro).
Três fases distintas eram observadas, a aquosa contendo o RNA, era
transferida para outro tubo. O RNA era então precipitado pela adição do dobro do
volume da fase aquosa de isopropanol, por 10 minutos à temperatura ambiente, e
então centrifugado (8000rpm/15minutos/4ºC, Sorvall RC5B F28/micro). O
sobrenadante resultante desta última era descartado e o sedimento lavado com
etanol 75% (7500rpm/5minutos/4ºC, Sorvall RC5B F28/micro) e ressuspenso em
50µL de água milliQ estéril previamente tratada com DEPC.
Em seguida, com o intuito de eliminar resquícios de DNA recuperados
durante o processo anterior, a amostra de RNA era tratada com
deoxirribonuclease I (DNAseI, Invitrogen). Assim, para cada 1µg de RNA
adicionavam-se 1U da DNAse, 1/10 do volume final de reação de tampão de
reação 10X (fornecido pelo fabricante, Invitrogen), água bidestilada estéril tratada
com DEPC. A reação ocorria à temperatura ambiente por 15 minutos. Ao final,
prosseguia-se com a inativação da DNAseI pela adição de 1/10 do volume da
5

reação de EDTA 25mM e aquecimento a 65ºC por 10 minutos.
3 - Avaliação da quantidade e qualidade do RNA
O RNA era quantificado espectrofotometricamente pela determinação da
D.O.260nm e D.O.280nm e aplicação da equação (Sambrook et al., 1989):
D.O.260nm X 40 X fator de diluição = Y µg/ml RNA.
A concentração de proteínas era determinada pela fórmula:
D.O.280nm X 50 X fator de diluição = Z mg/ml de proteínas.
A razão entre concentrações de RNA/proteínas constitui-se num requisito
indicador da qualidade do RNA isolado e em nossos trabalhos consideramos
aceitáveis valores entre 1,6 – 2,0.
A qualidade do RNA foi avaliada por eletroforese em mini-géis de agarose
(1%) em tampão TAE 1X (Sambrook et al., 1995) em amostras contendo 1µL da
preparação de RNA, 2µl de solução de aplicação e 7µl de água bidestilada.
Amostras de boa qualidade apresentavam bandas discretas correspondentes a
RNA ribossômicos 16S e 23S de aproximadamente 3,7kb.
4 - Síntese de cDNA
Inicialmente as amostras de RNA tiveram suas concentrações ajustadas
para 0,5 µg/ µl com água/DEPC. Quanto a síntese de cDNA, foi feita como abaixo:
A um volume de solução de RNA a 0,5µg/µl eram adicionadas 2,5µl de
iniciadores oligonucleotídicos randômicos (Invitrogen) na concentração de
50pM/µl, 5µl de dNTP a 10mM e água bidestilada totalizando 32,5µl. A mistura foi
aquecida a 65ºC por 5 minutos (para eliminar possíveis estruturas secundárias
6

resultantes da auto-hibridização do RNA). Imediatamente, 5,0µl de tampão 10X,
5,0µl de DTT (0,1M) e 2,5µl da enzima Superscript (200U/µl, Invitrogen) eram
acrescentados, seguido de incubação da amostra a 50ºC por 40 minutos. Ao final,
a enzima era inativada a 70ºC por 15 minutos.
No que concerne aos controles instituídos, estes se caracterizaram pela
recuperação de uma alíquota da solução proveniente do controle negativo do
processo de extração do RNA com sua subseqüente submissão às etapas da
síntese de cDNA. Possíveis ocorrências de contaminação em nível de transcrição
reversa foram avaliadas com a formação de um controle inerente a esta etapa
resultante da adição dos reagentes empregados na mesma e água DEPC.
Previamente à reação de amplificação, todas as amostras contendo o
cDNA sintetizado foram expostas à atuação da ribonuclease H para evitar que
moléculas de RNA interferissem nos processos subseqüentes (RNase H constitui-
se numa endorribonuclease que especificamente degrada o RNA estando
complexado ou não a um DNA que lhe é complementar). A cada amostra, 5µl de
RNAse (2U/µl, Invitrogen) eram adicionados sendo a reação de degradação
efetivada com incubação a 37ºC por 20 minutos.
5 - RT PCR em tempo real (Real time RT-PCR)
Para padronização da técnica, três experimentos foram executados: (a)
Determinação da concentração ideal de oligonucleotídeos de modo que a reação
não fosse limitada por este fator. (b) Escolha do gene constitutivo a ser utilizado
para normalizar a quantificação do gene alvo. (c) Avaliação de expressão dos
genes NER induzidos pelo tratamento das células com radiação UV-C (controle
positivo).
7

(a) Determinação da concentração ideal de oligonucleotídeos - RNA de
células de uma cultura de E. coli AB1157 (3ml) em fase de crescimento
exponencial foi preparado e submetido a transcrição reversa. Uma amostra de
50ng do cDNA foi submetida à reação de amplificação com concentrações de
oligonucleotídeos de 50nM, 100nM, 200nM, 300nM e 400nM, para se
determinar uma concentração não limitante à eficiência da reação.
Amplificações dos genes alvo e do constitutivo normalizador foram analisados
nestas condições.
(b) Determinação do gene constitutivo normalizador - Uma vez estabelecida a
concentração ideal de oligonucleotídeos, o passo seguinte foi a escolha do gene
endógeno que seria usado como referência para normalizar os resultados
quantitativos obtidos no estudo dos genes alvos. O critério para a escolha do gene
referência é que sua expressão não deve ser fortemente alterada pelos
tratamentos e tenha a mesma eficiência de amplificação dos alvos. Além disto, ele
deve funcionar como um controle interno de quantidade, integridade do cDNA e de
possível inibição. Um recurso a esta análise consistiu em gerar uma curva, através
de uma diluição seriada previamente estabelecida dos respectivos cDNAs, que
representasse a diferença entre o ciclo threshold (Ct) referente à amostra e o
respectivo ciclo do gene de referência ( Ct Ctamostra – Ctreferência). Uma vez
construída, o valor da inclinação da reta deve ser menor que 0,1. rpoD [gene do
fator sigma 70 (σ70) da RNA polimerase de E. coli) foi selecionado e testado como
um possível controle de referência endógeno pelo fato de já ter sido utilizado neste
aspecto em trabalhos anteriores (Savli et al., 2003). Assim, inicialmente foram
8

feitas diluições seriadas de 10-1 da preparação do cDNA (de 0,0005 a 50ng) e
curvas de amplificação foram obtidas. Valores de Ct foram determinados para
cada diluição do cDNA. Um gráfico de valores de Ct x diluição foi utilizado para
verificar a linearidade, que deveria apresentar um coeficiente de correlação
aceitável ≥ 0.950 para que o gene em análise pudesse ser usado como referência.
(c) Análise de expressão dos genes NER induzidos por UV-C - Uma calibração
foi executada analisando-se a expressão dos genes NER (uvrA, uvrB e uvrC) em
células tratadas com UV-C. As culturas foram irradiadas com doses de letalidade
de 90 e 10%, as células foram recuperadas, seus RNA foram extraídos e cDNAs
foram preparados, como previamente descrito. Após a amplificação as
quantidades dos cDNAs dos genes uvrA, uvrB e uvrC nas amostras tratadas em
relação ao do gene referência rpoD foram determinados. Esta análise funcionou,
tal como um controle positivo validando as condições utilizadas no experimento.
Os mesmos procedimentos foram aplicados à análise das células após
tratamento com concentrações de mostarda nitrogenada bifuncional HN2 de 10%
e 90% da população. Esta foi determinada após submeter a cepa selvagem –
AB1157 ao tratamento com concentrações diversas e determinar a sobrevivência.
Simultaneamente um controle positivo de expressão dos genes NER foi realizado
tratando-se as células com UV-C nas doses de letalidades correspondentes.
Adicionalmente ao controle endógeno citado acima, os seguintes controles
foram empregados em cada reação de amplificação:
(d) Referência Passiva - É representado por uma glicina complexada com um
composto fluorogênico que não interage com os fatores de amplificação com o
9

intuito de avaliar e normalizar a fluorescência não relacionada à amplificação. No
presente estudo, o fluoróforo Rox foi utilizado para esta função, e
(e) Calibrador – Amostras que servem para averiguar alterações na expressão
dos transcritos induzidas por algum outro fator. Neste estudo foram as amostras
não submetidas aos tratamentos.
As reações de PCR foram realizadas em poços de uma placa opticamente
apropriada (96 – Well Optical Reaction Platé/Optical Caps – Applied Biosystems).
Em cada poço eram adicionados 25µl, com a seguinte composição:
1 12,5 µL de tampão PlatinumR SyBrR Green qPCR Super Mix–UDG [Tris-
HCl 20mM (pH 8,4), KCl 50mM, MgCl2 3mM, 200µM de GTP, 200µM dATP,
200µM de CTP, 400µM dUTP, 1U UDG, 1,5U PlatinumR Taq DNA
Polimerase], (Invitrogen);
2 1µl de primer Forward (10µM);
3 1µl de primer Reverse (10µM);
4 1µl de ROX Reference Dye;
5 água bidestilada estéril qsp 20µl;
6 5µl (50ng) do cDNA em água bidestilada estéril.
A placa era centrifugada (2.000rpm por 2 minutos – Centrifuga B4i Jouan) e
posteriormente submetida termociclador ABI Prism Sequence Detector 7700/7000.
Peculiaridades desta reação são: uridina trifosfatadas (dUTP) ao invés de
timidina trifosfatadas (dTTP) e a presença da Uracil-N-glicosilase (UDG) que
remove resíduos de ácidos nucléicos contendo uracil, provenientes de reações de
PCR prévias, funcionando como uma etapa de descontaminação que antecede a
amplificação.
10

Aos controles negativos, o cDNA não era adicionado, mas alíquotas das
amostras dos controles negativos de reações precedentes ou água estéril
bidestilada.
Os parâmetros de termociclagem para amplificação dos genes analisados
foram:
1 50ºC/2’, temperatura ótima da UDG, para inativar RNA remanescente e
ácidos nucléicos contendo uracil;
2 95ºC/10’, para inativação da UDG e concomitante ativação da polimerase;
50 ciclos de 95ºC por 1’, 59ºC/30’’
3 72ºC/1’ extensão final de 72ºC/2’.
Finda a reação, avaliava-se os Tms dos produtos amplificados para verificar
se correspondiam aos esperados, como um controle da especificidade do
processo. Para isto fazia-se inicialmente uma desnaturação do produto de
amplificação a 95ºC/15” seguido de um desaquecimento a 60ºC/20” e a partir daí,
reeleva-se a temperatura gradualmente de 60ºC a 95ºC por 19’ e 59”: à medida
que o cDNA vai desnaturando, a fluorescência inerente ao SyBr Green em sua
forma complexada ao cDNA vai decaindo até que atinja um ponto onde
corresponda a metade da fluorescência inicial. O software de análise gera um
gráfico que mostra no eixo das abscissas a variação da temperatura e no eixo das
ordenadas a intensidade de fluorescência correspondente. A especificidade da
reação é indicada pelo aparecimento de um pico único de fluorescência na
temperatura correspondente ao Tm do produto.
6 - Quantificação dos produtos do RT-PCR em tempo real
O modelo matémático aplicado para este fim se baseia na comparação dos
Cts (cycle threshold; o número do ciclo onde a amplificação de uma determinada
11

molécula alvo começa a ser perceptível pelo aumento da fluorescência gerada
pela intercalação do SyBr Green na dupla fita de DNA) e requer que a eficiência
de amplificação do gene alvo e do constitutivo normalizador sejam
aproximadamente equivalentes. Além desta normalização em relação à referência
endógena, deve haver uma correlação entre a quantidade de alvo amplificado
numa amostra tratada e o calibrador (amostra não tratada).
A expressão aritmética inerente ao processo de quantificação relativa no
método mencionado, encontra-se assim especificada:
Xn = X0 (1+Ex)n
Sendo Xn o número de moléculas alvos em um determinado ciclo; X0 =
número inicial de moléculas alvos; Ex = eficiência de amplificação e n = número de
ciclo no qual a reação se encontra.
Se considerarmos o Ct como sendo o número fracional do ciclo que indica o
ponto constante onde a amplificação de uma determinada molécula alvo passa a
ser perceptível, a fórmula Xn = X0 (1+Ex)n pode assim ser redescrita:
XT = X0 (1+Ex)Ct ,X = KX
Sendo XT = moléculas alvos produzidas no Ct; Ct,X = referente à
amplificação do alvo e Kx = constante threshold para a molécula alvo.
Prosseguindo, uma equação similar pode ser obtida para o Ct referente à
amplificação do gene constitutivo:
RT = R0 (1 + ER)Ct,R = KR
Sendo RT = número de moléculas do gene referência; Ct,R = Ct para
amplificação referência; Kr = constante threshold para a molécula de referência.
Assim, ao normalizarmos XT por RT, matematicamente isto se comporta da
seguinte maneira:
12

XT/XR = X0 (1 + EX) Ct,X/R0(1+ER)CT,R = KX/KR = K
Assumindo que as eficiências de amplificação para molécula alvo e
referência sejam as mesmas, tem-se que:
EX=ER=E (X0/R0 (1+E)CT,X-CT,R = K ou K = XN (1+E)∆Ct
Sendo Xn equivalente ao número de moléculas iniciais; X0/R0, quantidade inicial
de moléculas alvos normalizadas; ∆Ct = Ct,X – Ct, R (correspondendo a diferença
no ciclo Ct onde foi detectado o primeiro ciclo fracional onde um sinal de
fluorescência indicando amplificação foi encontrado entre referência e controle).
Finalmente, ao normalizar com Xn advindo do calibrador tem-se que:
K = Xn(1+E)∆Ct ∆ Xn = K(1+E)-∆Ct
Xn,q/Xn,cb = k(1+E)∆Ct,q/k(1+E)∆Ct,cb = (1+E)- ∆∆Ct
Sendo, -∆ ∆Ct = ∆ Ct,q - ∆ Ct,cb ∆ Ctcb = Ct do calibrador; Ct,q = Ct de
uma amostra tratada.
Como se econtra estipulado que para amplicons desenhados e otimizados
de acordo com Primer Express (Applied Biosystems), a eficiência de amplificação
é próxima de 1, logo têm-se que a quantidade de molécula alvo, normalizado por
uma referência endógena e relativa a um calibrador, equivale a seguinte
expressão:
XN,q/SN,cb = 2-∆ ∆Ct.
13

RESULTADOS
Estudos realizados com HN2.
Avaliamos a expressão relativa dos genes uvrA, uvrB, uvrC, cho e recA.
Todos estes genes estão envolvidos na reparação das lesões causadas por HN2
(o gene cho foi usado por ser um gene homólogo ao gene uvrC), sendo, portanto,
de extrema importância o estudo da transcrição destes genes, quando as células
são expostas a diferentes concentrações de HN2.
Observamos uma expressão levemente aumentada dos genes de reparo
NER, em relação ao tratamento com 5 minutos, na DL90 (Dose letal que leva a
90% de sobrevivência), dando ênfase à maior expressão do gene recA (Figura
1a). Quando as células são expostas a uma maior concentração de HN2,
chegando à sua DL10, chegamos a um nível de formação de lesão, em que
vemos a expressão aumentada de praticamente todos os genes estudados, em
igual nível (Figura 1b).
Estudos realizados com HN1
Mesmo os genes uvrA, uvrB e uvrC não estando envolvidos na correção
das lesões causadas pela HN1 (de Alencar et al., 2005), se tornou necessário
avaliar a transcrição destes genes, a fim de esclarecer se os genes do complexo
UvrABC eram induzidos e não reparavam estas lesões, ou se nem eram induzidos
por este agente, uma vez que, teoricamente, o mecanismo de reparo NER é capaz
de reconhecer praticamente a maioria dos danos que acontecem na molécula de
DNA, inclusive aqueles que não introduzem nenhuma deformação nesta molécula.
14

Figura 1: Quantificação relativa dos transcritos dos genes uvrA, uvrB, uvrC, cho e recA, normalizados com o
transcrito do gene rpoD – experimentos com HN2. Culturas da cepa selvagem (AB1157) em fase exponencial foram
tratadas com concentrações de HN2 que levem a DL90 (a) e a DL10 (b), em tampão fosfato (pH7,0) por 60 minutos,
posteriormente o RNA foi extraído e normalizado como descrito anteriormente. Quantidade relativa dos transcritos (eixo
das ordenadas) e transcritos (eixo das abscissas). Os resultados plotados representam a média dos valores obtidos *.
1 * Estes resultados foram primeiramente publicados na dissertação de mestrado “Estudo da regulação e da
expressão de genes de reparo de DNA de Escherichia coli em resposta a quimioterápicos anti- tumorais”(da Silva, 2006) e
também fazem parte do conjunto deste trabalho.
15

Em relação a recA, a análise foi devido à requisição desta proteína no
reparo das lesões causadas pela HN1.
Observamos uma expressão levemente aumentada dos genes de reparo
NER, na DL90 (Figura 2a), considerando que o maior aumento acontece nos
genes uvrA e uvrB. Este fato já era esperado, uma vez que UvrA2B, compõem o
complexo de reconhecimento do NER. Para a DL10 (Figura 2b), observamos uma
manutenção na expressão dos genes NER e um aumento na expressão do gene
recA. A expressão deste gene, pode ser devida a ambas as funções exercidas
pela proteína codificada por recA. Nos resultados de curvas de sobrevivência
bacteriana, podemos ver a participação da proteína RecA, conforme a dose de
HN1 aumenta, obtendo-se uma curva dose-resposta (de Alencar et al., 2005).
16

Figura 2: Quantificação relativa dos transcritos dos genes uvrA, uvrB, uvrC, cho e recA, normalizados com o
transcrito do gene rpoD – experimentos com HN1. Culturas da cepa selvagem (AB1157) em fase exponencial foram
tratadas com concentrações de HN1 que levem a DL90 (a) e a DL10 (b), em tampão fosfato (pH7,0) por 60 minutos,
posteriormente o RNA foi extraído e normalizado como descrito anteriormente. Quantidade relativa dos transcritos (eixo
das ordenadas) e transcritos (eixo das abscissas). Os resultados plotados representam a média dos valores obtidos.
1 * Estes resultados foram primeiramente publicados na dissertação de mestrado “Estudo da regulação e da
expressão de genes de reparo de DNA de Escherichia coli em resposta a quimioterápicos anti- tumorais” (da Silva, 2006)
e também fazem parte do conjunto deste trabalho.
17
Livros Grátis( http://www.livrosgratis.com.br )
Milhares de Livros para Download: Baixar livros de AdministraçãoBaixar livros de AgronomiaBaixar livros de ArquiteturaBaixar livros de ArtesBaixar livros de AstronomiaBaixar livros de Biologia GeralBaixar livros de Ciência da ComputaçãoBaixar livros de Ciência da InformaçãoBaixar livros de Ciência PolíticaBaixar livros de Ciências da SaúdeBaixar livros de ComunicaçãoBaixar livros do Conselho Nacional de Educação - CNEBaixar livros de Defesa civilBaixar livros de DireitoBaixar livros de Direitos humanosBaixar livros de EconomiaBaixar livros de Economia DomésticaBaixar livros de EducaçãoBaixar livros de Educação - TrânsitoBaixar livros de Educação FísicaBaixar livros de Engenharia AeroespacialBaixar livros de FarmáciaBaixar livros de FilosofiaBaixar livros de FísicaBaixar livros de GeociênciasBaixar livros de GeografiaBaixar livros de HistóriaBaixar livros de Línguas

Baixar livros de LiteraturaBaixar livros de Literatura de CordelBaixar livros de Literatura InfantilBaixar livros de MatemáticaBaixar livros de MedicinaBaixar livros de Medicina VeterináriaBaixar livros de Meio AmbienteBaixar livros de MeteorologiaBaixar Monografias e TCCBaixar livros MultidisciplinarBaixar livros de MúsicaBaixar livros de PsicologiaBaixar livros de QuímicaBaixar livros de Saúde ColetivaBaixar livros de Serviço SocialBaixar livros de SociologiaBaixar livros de TeologiaBaixar livros de TrabalhoBaixar livros de Turismo