potentiation cytotoxicity poly(adp-ribose) polymerase ... of anti...potentiation...
TRANSCRIPT

Brvsah Joumal of Cancer (1998) 78(10). 1269-1277C 1998 Cancer Research Campaign
Potentiation of anti-cancer agent cytotoxicity by thepotent poly(ADP-ribose) polymerase inhibitors NU1025and NU1064*
KJ Bowman', A White2, BT Golding2, RJ Griffin2 and NJ Curtin"*
Cancer Research Unit and 2Departmnent of Chemistry. University of Newcastle upon Tyne. Medical School. Framlington Place. Newcaste upon Tyne NE2 4HH. UK
Summary The ability of the potent poly(ADP-rbose) polymerase (PARP) inhibitor. NU1025 (8-hydroxy-2-methyl-quinazolin-4-[3H]one) topotentiate the cytotoxicity of a panel of mechanistically diverse anti-cancer agents was evaluated in L1210 cells. NU1025 enhanced thecytotoxicity of the DNA-methylating agent MTIC, y-irradiation and bleomycin 3.5-, 1.4- and 2-fold respectively. The cytotoxicities of thethymidylate synthase inhibitor, nolatrexed, and the cytotoxic nucleoside, gemcitabine, were not increased. Potentiation of MTIC cytotoxicity bya delayed exposure to NU1025 was equally effective as by a simultaneous exposure to NU1025, indicating that the effects of NU1025 weremediated by an inhibition of the cellular recovery. The recovery from potentially lethal ;--irradiation damage cytotoxicity in plateau-phase cellswas also inhibited by NU1025. Investigation of DNA strand breakage and repair in t-irradiated cells by alkaline elution demonstrated thatNU1025 caused a marked retardation of DNA repair. A structurally different PARP inhibitor, NU1064 (2-methylbenzimidazole-4-carboxamide).also potentiated the cytotoxicity of MTIC. to a similar extent to NU1025. NU1064 potentiated a sublethal concentration of a DNA methylatingagent in a concentration-dependent manner. Collectively, these data suggest that the most suitable cytotoxic agents for use in combinationwith PARP inhibitors are methylating agents, bleomycin and ionizing radiation, but not anti-metabolites.
Keywords: poly(ADP-rbose) polymerase; DNA repair; cytotoxicity: PARP inhibitors; DNA-alkylating agents:y-irradiation
Polv(ADP-ribose) polxmerase (PARP: EC 2.4.2.30) is anabundant 1 16-kDa nuclear enzyme with approximately 2 millionmolecules per (HeLa) cell: equi-alent to I molecule for every kbof DNA. The enzyme comprises an N-tenminal DNA-bindingdomain (DBD) containing tw o zinc fingers. w-hich recoglnize DNAstrand breaks. an automodification domain and a C-terminalcatalv tic domain. PARP can bind to undamaged DNA but has anabsolute requirement for catalx tic activation on DNA breaks.Ahen activated. PARP catalyses the formation of long, homo-poly mers of ADP-ribose on nuclear proteins usin, NAD- as asubstrate. The main protein acceptor is PARP itself (automodifica-tion). but the enzN-me has also been shown to modify histones.HMG proteins. topoisomerases. DNA polymerases and ligases(see reviews by Cleaver and Morgan. 1991: Lautier et al. 1993: deMurcia and Menissier de Murcia. 1994: Lindahl et al. 1995 andreferences therein). The ADP-ribose polymers formed by PARPare degraded by ADP-ribose gly cohy drolase. and under conditionsof PARP stimulation by DNA damage a dy-namic sysstem of rapidsynthesis and deggradation exists causing rapid NAD- depletion(see Boulikas. 1991 and references therein).The activation of PARP by DNA strand breaks implies that it is
in-olved in the repair of such lesions. although the precise role ofthe enzy-me remains to be elucidated. It has been proposed that theADP-ribose polI mer causes relaxation of chromatin at the site ofthe DNA strand break. allowing access of repair enzy mes (Althauset al. 1993 . Alternatively. poly(ADP-ribose) synthesized on PARP
Received 15 January 1998Revised 30 March 1998Accepted 15 Apnl 1998
Correspondence to: NJ Cumn
bound to nicked DNA miaht stabilize histone residues and main-tain nucleosomal structure. thereby keeping the txwo DNA endscorrectly positioned for subsequent rejoining (Lindahl et al. 1995).
The possible involvement of PARP in DNA repair has stimu-lated an interest in its role in determining the response to anti-cancer therapies that damage DNA. The availabilitv of inhibitorsof PARP has greatly aided these studies (Shall. 1984). most ofwhich has-e investigated the potentiation of monofunctional alky l-atina agents and ionizing, radiation as these therapies are the mostpotent actixators of PARP. althoug!h some other agents have alsobeen evaluated (reviewed in Griffin et al. 1995).
Studies of the potentiation of DNA-damaging agents by PARPinhibition have primarilx used the benzamide inhibitors. How ever.the benzamides are not very potent and. although their PARPinhibitonr IC- concentrations are in the low micromolar range.millimolar concentrations are invariably required to achievepotentiation in cvtotoxicitv assays. Furthermore. the benzamidesalso have other effects. most notably on de novo purine biosyn-thesis (Cleaver. 1984: Hunting et al. 1985: Milam et al. 1986).Nevertheless. trans-dominant inhibition of PARP. through over-expression of the DBD. also sensitizes cells to y-irradiation andalkylating acgents (Molinete et al. 1993: Kupper et al. 1995). anobservation that is consistent with the effects of the benzamidesbeing primarily caused by PARP inhibition. However. given thehigh concentrations of benzamides required in cy totoxicitystudies. more potent and specific inhibitors of PARP are requiredin order to assess the clinical potential of PARP inhibitors toimprove current cancer therapy.
Using rational drug desicgn. two novel series of PARP inhibitorshave been developed: the benzimidazole4-carboxamides and'T'his paper is part 4 of the .senies Resistance Modift-ing Agents.: for par .seeGriffin et al 1996h Pharn Si 2: 43-7.
1269

prepared as previously described (Grnffin et al 1995. 1996). weredissolved in DMSO at 100 imM and stored at -20°C. Gemcitabine(a gift from Eli Lilly. Indianapolis. IN. USA) was dissolved inwater at 10 mnm and stored at -20°C. MTIC [5-(3-methyltriazen-l-yl)imidazole-4-carboxamide: a gift from Dr C Bleasdale.University of Newcastle upon Tyne. UK] and bleomycin(Lundbeck. Milton Keynes. UK) were dissolved in DMSO andused immediately. 3-Aminobenzamide (3AB; Pfaltz and Bauer.Phase Separations. Deeside. UK) was dissolved in tissue culturemedium (see below) on the day of use by stirring for 4 h and filtersterilized. Drugs were added to cell cultures so that the finalDMSO concentration was always 1% (v/v). All other chemicalswere obtained from Sigma (Poole. UK) unless stated otherwise.
Cells
B
NH2
HN
CH3
Figure 1 Chemical strctures of NU1025 (A) and NU1064 (B)
quinazolin4-[3H]-ones. which exhibit much greater potency thanthe benzamides (Griffin et al. 1995. 1996). The potentiation ofDNA methylating agent-induced cytotoxicity and DNA strandbreakage by one of these novel PARP inhibitors (NU1025:8-hydroxy-2-methylquinazolin4-[3H]one. Figure IA) has beendescribed previously (Boulton et al. 1995). The aims of the studiesreported here were to investigate the effects of NU1025 (IC,o forPARP inhibition = 0.4 .LM) on the cytotoxicity of a range of anti-cancer agents, and to compare the effects of quinazolinone(NU 1025) and benzimidazole (NU 1064: 2-methylbenzimiidazole-4-carboxamide: IC;,, for PARP inhibition = 1 Mk. Figure 1B)PARP inhibitors on methylating agent-induced cytotoxicity. Thecytotoxic treatment studied were methylating agents (MTIC andtemozolomide). y-irradiation. bleomycin and anti-metabolites(nolatrexed and gemcitabine). Studies with topoisomerase I and Hinhibitors are described elsewhere (Bowman et al. 1996 and manu-
script in preparation)
MATERIALS AND METHODS
Drugs
Nolatrexed (a gift from Agouron Pharmaceuticals. San Diego. CA.USA) and temozolomide (a gift from the Cancer ResearchCampaign. London. UK) were dissolved in dimethyl sulphoxide(DMSO) at O nmi and stored at -200C. NU1025 and NU1064.
Murine leukaemia L 12 10 cells were used to allow comparison
with the PARP inhibitory potency of the compounds that had beendetermined previously using permeabilized L 1210 cells (Griffin etal 1995. 1996). Cells were maintained as exponentially growing
cultures (<8xlI0 cells ml-') in RPMI-1640 medium supplementedwith 10% (v/v) fetal calf serum (Sigma. Poole. UK). at 37°C in an
atmosphere of 5% carbon dioxide in air. The cell doubling timewas approximately 12 h. For experiments with the thymidylatesynthase inhibitor. nolatrexed. cells were adapted to grow in RPMImedium supplemented with 10% (v/v) dialysed serum (to removethymidine) and maintained in this medium for at least 4 weeksbefore cytotoxicity assays. when the cell doubling time was alsoapproximately 12 h. Cells were tested to exclude mycoplasmacontamination (Chen. 1977) every 4-8 weeks.
Cytotoxicity assays
L12 10 cells were diluted to l0 ml-' in medium containing thedesired concentration of the cytotoxic drug. with or without thePARP inhibitor, in duplicate wells of a six-well plate. After theselected exposure period. cells were harvested by centrifugation(200 g. room temperature) to remove the drugs. resuspended infresh medium and counted (Coulter Counter Z1: CoulterElectronics. Luton. UK). Cell suspensions were then furtherdiluted and dispensed into sterile polyurethane tubes (Falcon.Becton Dickinson. Oxford. UK) in triplicate at a suitable densityin 1 ml of culture medium (estimated to give 10-60 colonies). and5 ml of 0.15% (w/v) agarose (SeaKem ME Agarose. Flowgen.Sittingbourne. UK) in culture medium was added. The tubes were
incubated for 1-2 weeks to allow colonies to appear and thecontents placed in dishes containing 1 ml 0.5 mg ml-' MTT (3-[4.5-dimethylthiazol-2-yl]-2.5-diphenyltetrazolium bromide) tostain the viable colonies. After 4 h colonies were counted and theplating efficiency. relative to the appropriate control (DMSO aloneor PARP inhibitor alone). was calculated. DMSO control incuba-tions gave approximately 100% plating efficiency.
In order to investigate if the potentiation of a cytotoxic agent byNU1025 was dependent on the simultaneous presence of theinhibitor. or if NU1025 would be equally effective when adminis-tered after the cytotoxic agent had been removed. the followingprotocol was employed: cells were exposed to varying concentra-tions of MTIC for 20 min. duplicate samples were then harvestedby centrifugation and resuspended in drug-free medium or
medium containing 200 gm NU1025 for a further 16 h. In addi-tion. cells were exposed to vary ing concentrations of MTIC in the
British Joumal of Cancer (1998) 78(10), 1269-1277
1270 KJ Bowman et al
A
a
NH
CH3
OH
0 Cancer Research Campaign 1996
Potentiation of anti-cancer agents by potent PARP inhibitors 1271
were exposed to a `Cs source (Gammacell 1000 Elite. NordianInternational. Canada) then incubated at 37C. in the presence orabsence of 200 im Ni 1025 for 2 h before determining colonyformation as described above. To insvestigate the repair of poten-tially lethal damage. duplicate cultures of cells were held at plateauphase by maintaining, them at a density of 106 cells ml-' in condi-tioned medium from plateau-phase cells (which ensured acomplete cessation of cell division without a decrease in cellviability durinc the exposure period: data not shown) and exposedto 8 Gy of y-irradiation. Cells were kept on ice immediately beforeand after irradiation. then incubated at 37C in the presence orabsence of 200 .iM NUT1025 for the time period indicated in theresults. counted and seeded for colony formation in 0.15%c (w/s)agarose as above.The degree of potentiation [enhancement factor (EF)] produced
bv the PARP inhibitor was calculated by comparing the IC9, orID90 [the concentration (C) of druc or dose (D) of radiation causin,90% cell death. i.e. a 10% relative plating efficiency] of the crto-toxic agent alone with the ICg) or ID, in the presence of PARPinhibitor.
EFI,, = IC., or ID., controlfIC,, or ID., + PARP inhibitor
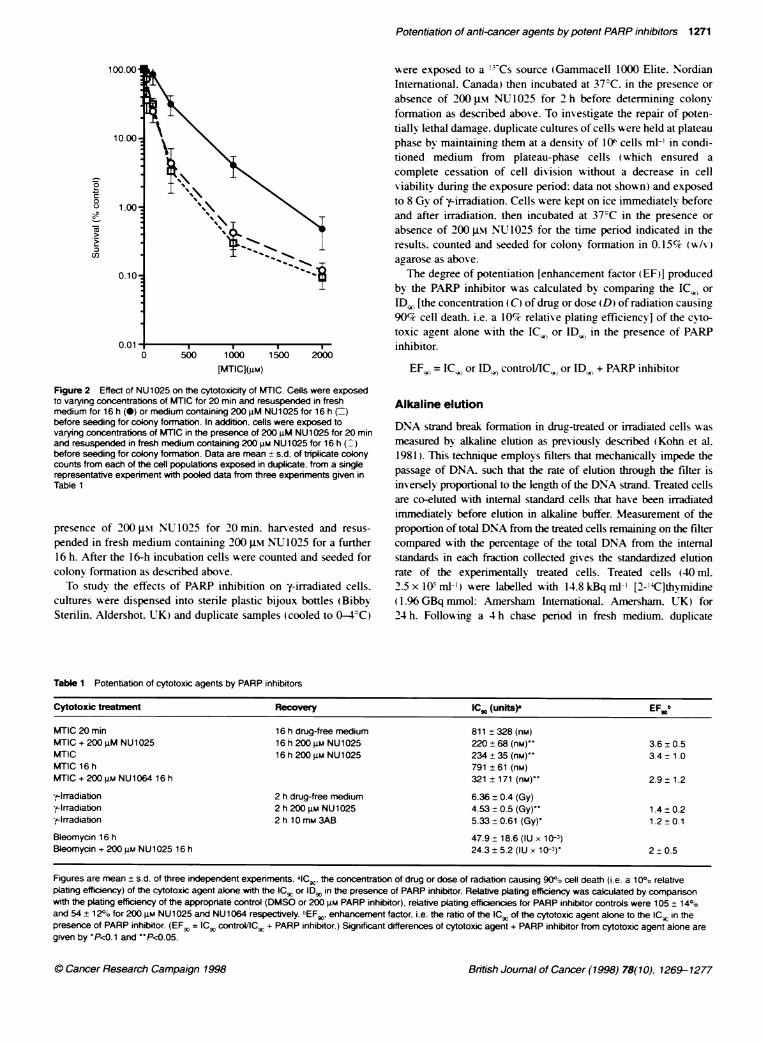
Figure 2 Effect of NU1025 on the cytotoxicity of MTIC. Cells were exposedto varying concentrations of MTIC for 20 min and resuspended in freshmedium for 16 h (-) or medium containing 200 JM NU1025 for 16 h (=)before seeding for colony formabon. In additon, cells were exposed tovarying concentrations of MT1C in the presence of 200 JM NU1025 for 20 minand resuspended in fresh medium containing 200 gm NU1025 for 16 h (:)before seeding for colony formation. Data are mean + s.d. of triplicate colonycounts from each of the cell populations exposed in duplicate. from a singlerepresentative experiment with pooled data from three experiments given inTable 1
presence of 200 gm NU 1025 for 20rmn. harvested and resus-
pended in fresh medium containing 200 im NU 1025 for a further16 h. After the 16-h incubation cells were counted and seeded forcolony formation as described above.
To study the effects of PARP inhibition on y-irradiated cells.cultures were dispensed into sterile plastic bijoux bottles (BibbySterilin. Aldershot. UK) and duplicate samples (cooled to 0-4C)
Alkaline elution
DNA strand break formation in druc-treated or irradiated cells wasmeasured by alkaline elution as previously described (Kohn et al.1981 ). This technique employs filters that mechanically impede thepassage of DNA. such that the rate of elution through the filter is
inversely proportional to the length of the DNA strand. Treated cellsare co-eluted with intemal standard cells that hase been irradiatedimmediately before elution in alkaline buffer. Measurement of theproportion of total DNA from the treated cells remaining on the filtercompared with the percentage of the total DNA from the intemalstandards in each fraction collected gives the standardized elutionrate of the experimentally treated cells. Treated cells (40 ml.
2.5 x I0W ml-' ) were labelled wvith 14.8 kBq ml' [2-'4C]thNmidine(1 96 GBq mmol: Amersham International. Amersham. TK) for24 h. Following a 4 h chase period in fresh medium. duplicate
Table 1 Potentiation of cytotoxic agents by PARP inhibitors
Cytotoxic treabTme Recovery IC,0 (units) EF b
MTIC 20 min 16 h drug-free medium 811 ± 328 (nM)MTIC + 200 iM NU1025 16 h 200 iLM NU1025 220 ± 68 (nM)" 3.6 0.5MTIC 16 h 200 gM NU1025 234 ± 35 (nm) 3.4-1.0MTIC 16 h 791 ± 61 (nM)MTIC + 200 gM NU1064 16 h 321 ± 171 (nM) 2.9 1.2
-y-Irradiation 2 h dnrg-free medium 6.36 ± 0.4 (Gy)-,.-Irradiation 2 h 200 g NU1025 4.53 ± 0.5 (Gy)' 1.4 0.2F-lrradiation 2 h 10 mM 3AB 5.33 ± 0.61 (Gy)' 1.2 0.1
Bleomycin 16 h 47.9 ± 18.6 (IU x 10-3)Bleomycin + 200 4M NU1025 16 h 24.3 ± 5.2 (IU x 10-3) 2 + 0.5
Figures are mean ± s.d. of three independent experiments. aIC., the concentration of drug or dose of radiation causing 90°o cell death (i.e. a 10%O relativeplating efficency) of the cytotoxic agent alone with the ICg or IDg in the presence of PARP inhibitor. Relative plating efficiency was calculated by comparisonwith the plating efficiency of the appropriate control (DMSO or 200 gM PARP inhibitor), relative plating efficienies for PARP inhibitor controls were 105 ± 14°oand 54± 120/o for 200 JM NU1025 and NU1064 respectively. tEFw. enhancement factor. i.e. the ratio of the ICw of the cytotoxic agent aklne to the ICg in thepresence of PARP inhibitor. (EF. = IC.C controVllC , + PARP inhibitor.) Significant differences of cytotoxic agent + PARP inhibitor from cytotoxic agent alone aregiven by 'P<z0.1 and 'P<c0.05.
British Joumal of Cancer (1998) 78(10). 1269-1277
100.00
10.00
1.00o
0
0
._
c)
0.10
0.01
[MTIC]@m)
0 Cancer Research Campaign 1998
1272 KJ Bowman et al
samples were transferred to sterile Bijoux bottles and irradiated asdescribed above. Cells were either eluted immediately or were incu-bated at 37°C for 2 h in the presence or absence of 200 gM NU1025.At the end of the exposure period cells were harvested by centnfuga-tion and resuspended in ice-cold PBS (Gibco. Paisley. UK) beforeelution. Interal standard cells were labelled with 37 kBq ml-'[methyl-3H]thymidine (1.85 TBq mmol1: Amersham) for 24 h.chased for 4 h plus the exposure period of the experimental cells.irradiated with 3 Gy and resuspended in ice-cold PBS before elution.
Duplicate aliquots of treated cells were allowed to settle onto individual polycarbonate filters (pore size 0.8 gm. diameter25 mm. Whatman International. Maidstone. UK), replicatealiquots of intemal standard cells were also added to all filters andboth treated and internal standard cells were allowed to settle on tothe filters by gravity in ice-cold PBS. A solution of 2% (w/v)sodium dodecyl sulphate (SDS) in 25 mm EDTA pH 10 was addedto lyse the cells and the filters were then exposed to 0.5 mg ml'proteinase K in lysis solution for 1 h. The filters were washedthree times with 20 mm EDTA pH 10 and eluted with 20mmEDTA (acid form) + 1% (w/v) SDS adjusted to pH 12.1 withtetrapropylammonium hydroxide (Aldrich. Gillingham. UK). Theelution rate was 2 ml h-' and eight 90-min fractions were collectedinto scintillation vials containing 15 ml of scintillant (OptiphaseHisafe 2, Fisons. Loughborough. UK). The filters were transferredto scintillation vials. baked at 60°C in 0.4 ml of 1 M hydrochloricacid for 1 h then neutralized with 2.5 ml of 0.4M sodiumhydroxide at room temperature for 15 min before adding 15 ml ofscintillant and counting with the eluted samples. The total radio-activity collected in the fractions and hence that remaining on thefilters was determined. and the proportion of the total retained onthe filter for each of the 90 min intervals was calculated. Theproportion of the total '4C retained on the filter at each time pointwas plotted against the proportion of the total 3H retained to stan-dardize the assay against inter- and intra-assay variations in pumpefficiency.
Statistical analysis
Comparison of data sets was made by paired or unpaired Student'stwo-tailed t-test analyses as appropriate using Graphad Instat soft-ware (GraphPad Software. San Diego. CA. USA).
RESULTS
Monofunctional alkylating agents
Two monofunctional alkylating agents were used in these experi-ments: MTIC. a very reactive DNA-methylating agent with a half-life in aqueous medium of 8 min (Shealy and Krauth, 1966). andthe clinically used agent, temozolomide, which spontaneouslydecomposes to yield MTIC with a half-life of 1.24 h in phosphatebuffer and 0.42 h in human serum (Stevens et al, 1987). NU1025has previously been shown to potentiate the cytotoxicity of temo-zolomide when cells were exposed to both drugs simultaneously(Boulton et al. 1995). In order to investigate the mechanism ofNU1025 potentiation. experiments were performed with MTIC asits rapid half-life allows a pulse exposure to the methylatingspecies. Exposure to MTIC and concomitant or sequential expo-sure to NU1025 was used to determine if potentiation requiredboth drugs to be present at the same time. Cells were exposed for20 min to MTIC followed by a 16-h recovery period in fresh
000-
a-2:it
[MTWC]i)
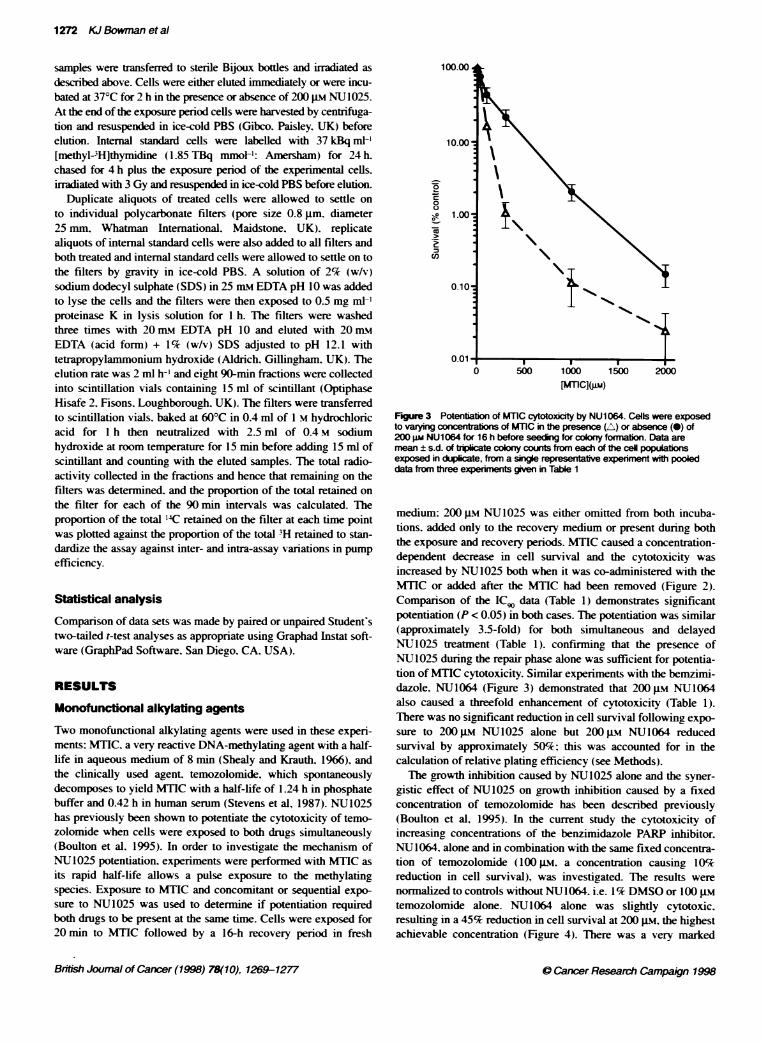
Figure 3 Potentiation of MTIC cytotoxicity by NU1064. Cells were exposedto varying c ations of MTIC in the presence (A) or absence (-) of200 gm NU1064 for 16 h before seeding for colony formation. Data aremean ± s.d. of triplicate coony counts from each of the ceU populabonsexposed in duplicate, from a single representative experiment with pooleddata from three experiments gven in Table 1
medium: 200 gM NU1025 was either omitted from both incuba-tions. added only to the recovery medium or present during boththe exposure and recovery periods. MTIC caused a concentration-dependent decrease in cell survival and the cytotoxicity wasincreased by NU1025 both when it was co-administered with theMTIC or added after the MTIC had been removed (Figure 2).Comparison of the IC, data (Table 1) demonstrates significantpotentiation (P < 0.05) in both cases. The potentiation was similar(approximately 3.5-fold) for both simultaneous and delayedNU1025 treatment (Table 1). confirming that the presence ofNU1025 during the repair phase alone was sufficient for potentia-tion of MTIC cytotoxicity. Similar experiments with the bemzimi-dazole. NU1064 (Figure 3) demonstrated that 200 gm NU1064also caused a threefold enhancement of cytotoxicity (Table 1).There was no significant reduction in cell survival following expo-sure to 200 gm NU1025 alone but 200 gM NU1064 reducedsurvival by approximately 50%; this was accounted for in thecalculation of relative plating efficiency (see Methods).
The growth inhibition caused by NU1025 alone and the syner-gistic effect of NU1025 on growth inhibition caused by a fixedconcentration of temozolomide has been described previously(Boulton et al, 1995). In the current study the cytotoxicity ofincreasing concentrations of the benzimidazole PARP inhibitor,NU1064. alone and in combination with the same fixed concentra-tion of temozolomide (100 gM, a concentration causing 10%reduction in cell survival), was investigated. The results werenormalized to controls without NUI1064, i.e. 1% DMSO or 100 gIMtemozolomide alone. NU1064 alone was slightly cytotoxic.resulting in a 45% reduction in cell survival at 200 gs, the highestachievable concentration (Figure 4). There was a very marked
Brinsh Joumral of Cancer (1998) 78(10), 1269-1277
1
0 Cancer Research Campaign 1996
Potentiation of anti-cancer agents by potent PARP inhibitors 1273
A
-*Irradiation (Gy)
[NU 1 064](Jim)
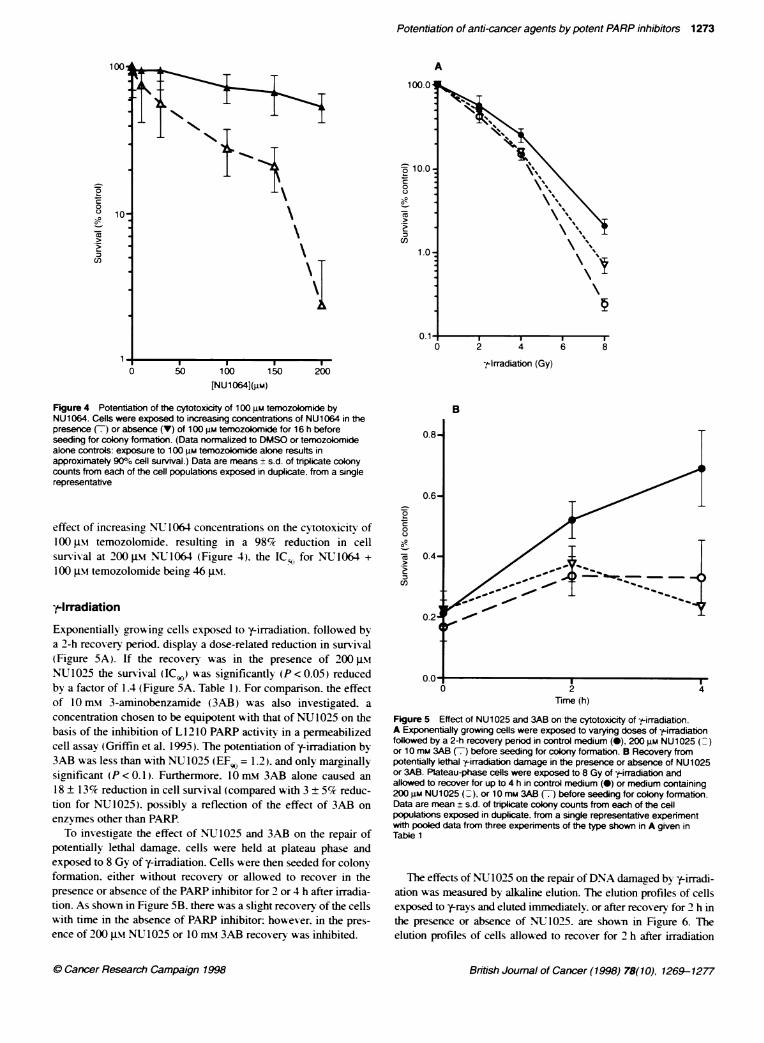
Figure 4 PotentiatKon of the cytotoxicity of 100 uM temozolomide byNU1064. Cells were exposed to increasing concentrations of NU1064 in thepresence (7) or absence (V) of 100 gM temozokomide for 16 h beforeseeding for colony formation. (Data norrnalized to DMSO or temozolomidealone controls: exposure to 100 gm temozolomide alone results inapproximatety 900% cell survival.) Data are means + s.d. of triplicate colonycounts from each of the cell populations exposed in duplicate, from a singlerepresentative
effect of increasing NU 1064 concentrations on the cytotoxicitv of100 JIM temozolomide. resulting in a 98% reduction in cellsurvival at 200 -iM NU1064 (Figure 4). the IC-o for NLT1064 +
100 JIM temozolomide being 46 gtm.
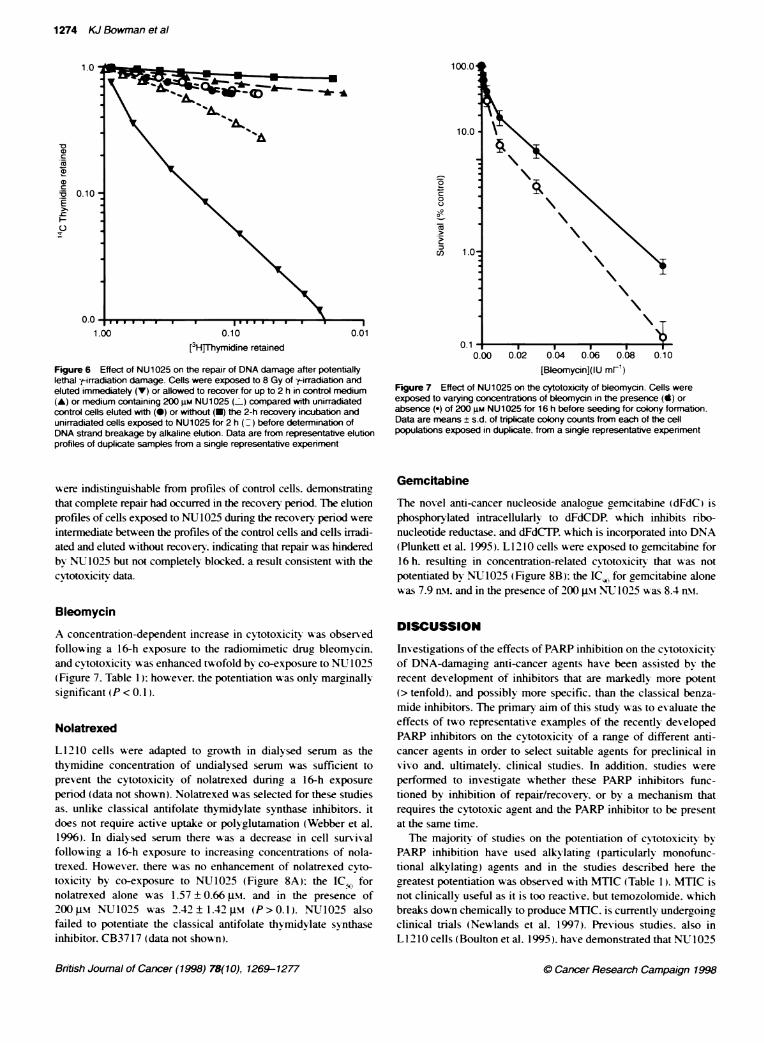
f-IrradiationExponentially grow-ing cells exposed to y-irradiation. followed bya 2-h recovery period. display a dose-related reduction in survival(Figrure 5A). If the recovery was in the presence of 200 j.si
NU1025 the survival (ICG) was significantly (P<0.05) reducedby a factor of 1.4 (Figure 5A. Table 1). For companrson, the effectof 10 m-M 3-aminobenzamide (3AB) was also investigated. a
concentration chosen to be equipotent with that of NU 1025 on thebasis of the inhibition of L 1210 PARP activitv in a permeabilizedcell assay (Griffin et al. 1995). The potentiation of y-irradiation by3AB was less than with NU1025 (EF9 = 1.2). and only marginallysignificant (P < 0.1). Furthermore. 10 mm 3AB alone caused an
18 ± 13% reduction in cell survival (compared with 3 ± 5% reduc-tion for NU1025). possibly a reflection of the effect of 3AB on
enzymes other than PARP.To investigate the effect of NUJ1025 and 3AB on the repair of
potentially lethal damage. cells were held at plateau phase andexposed to 8 Gy of y-irradiation. Cells were then seeded for colonyformation. either without recovery or allowed to recover in thepresence or absence of the PARP inhibitor for 2 or 4 h after irradia-tion. As shown in Figure SB. there was a slight recovery of the cellswith time in the absence of PARP inhibitor: however. in the pres-
ence of 200 ptM NV 1025 or 10 Xm1 3AB recovery was inhibited.
-a
-a00
-ac:2)
B
lime (h)
Figure 5 Effect of NU1025 and 3AB on the cytotoxicity of --irradiation.A Exponentially growing cells were exposed to varying doses of -tLirradiationfollowed by a 2-h recovery period in control medium (0), 200 gM NU1025or 10 mm 3AB (7) before seeding for colony formation. B Recovery frompotentially lethalk-irradiation damage in the presence or absence of NU1025or 3AB. Plateau-phase cells were exposed to 8 Gy of y-irradiation andallowed to recover for up to 4 h in control medium (0) or medium containing200 gm NU1025 (.), or 10 mm 3AB (7) before seeding for colony formation.Data are mean ± s.d. of triplicate colony counts from each of the cellpopulations exposed in duplicate, from a single representative experimentwith pooled data from three expenments of the type shown in A given inTable 1
The effects ofNU 1025 on the repair ofDNA damaged by y-irradi-ation was measured by alkaline elution. The elution profiles of cellsexposed to y-rays and eluted immediatelv. or after recovery for 2 h inthe presence or absence of NU1025. are shown in Figure 6. Theelution profiles of cells allowed to recover for 2 h after inradiation
British Joumal of Cancer (1998) 78(10), 1269-1277
N*1_%
0
0
-a
cn
0 Cancer Research Campaign 1998
1274 KJ Bowman et al
'a-aaac
E
I-U1
0
0
-n
C/)
[3H]Thymidine retained
Figure 6 Effect of NU1025 on the repair of DNA damage after potentialtylethal y-irradiation damage. Cells were exposed to 8 Gy of y-irradiaton andeluted immediatety (V) or allowed to recover for up to 2 h in control medium(A) or medium containing 200 gM NU1025 ( ) compared with unirradiatedcontrol cells eluted with (-) or wrthout (U) the 2-h recovery incubation andunirradiated cells exposed to NU1025 for 2 h (_ ) before determination ofDNA strand breakage by alkaline elution. Data are from representative elubonprofiles of duplicate samples from a single representative experiment
were indistinguishable from profiles of control cells. demonstratin,that complete repair had occurred in the recovery period- The elutionprofiles of cells exposed to NU1025 dungn the recovery period wereintermediate between the profiles of the control cells and cells irradi-ated and eluted without recovery. indicating that repair A-as hinderedby NU1025 but not completely blocked a result consistent with thecvtotoxicitx data.
Bleomycin
A concentration-dependent increase in cytotoxicity A as observedfollowing a 16-h exposure to the radiomimetic drug, bleomvcin.and cytotoxicio-swas enhanced twofold by co-exposure to NU 1025(Figure 7. Table 1): however. the potentiation was only marginallvsignificant (P < 0.1 ).
Nolatrexed
L1210 cells were adapted to growth in dialysed serum as thethvmidine concentration of undialysed serum was sufficient toprevent the cytotoxicity of nolatrexed during a 16-h exposureperiod (data not shown). Nolatrexed wvas selected for these studiesas. unlike classical antifolate thymidylate synthase inhibitors. itdoes not require active uptak-e or polyglutamation (Webber et al.1996). In dialysed serum there was a decrease in cell survivalfollowinc a 16-h exposure to increasing concentrations of nola-trexed. However. there was no enhancement of nolatrexed cyto-toxicity by co-exposure to NU1025 (Figure 8A}: the ICSo fornolatrexed alone was 1.57 ± 0.66 Jm. and in the presence of200JM NU1025 was 2.42±1.42 -i (P>0.1). NU1025 alsofailed to potentiate the classical antifolate thymidylate synthaseinhibitor. CB3717 (data not shown).
[Bleomycin](IU ml')Fgure 7 Effect of NU1025 on the cytotoxicity of bleomycin. Cells wereexposed to varying concentrations of bleomycin in the presence (X) orabsence (e) of 200 gm NUl 025 for 16 h before seeding for colony formation.Data are means ± s.d. of triplicate colony counts from each of te cellpopulations exposed in duplicate, from a single representative experiment
Gemcitabine
The novel anti-cancer nucleoside analogue gremcitabine WdFdCH isphosphorylated intracellularlv to dFdCDP. vAhich inhibits nrbo-nucleotide reductase. and dFdCTPC which is incorporated into DNA(Plunkett et al. 1995). L 1210 cells were exposed to gremcitabine for16 h. resulting, in concentration-related cvtotoxicity that wvas notpotentiated by NUJ1025 (Figure 8B): the IC, for gaemcitabine alonewas 7.9 n-Nw and in the presence of 200 JIm NU 1025 was 8.4 nmt.
DISCUSSION
Investigations of the effects of PARP inhibition on the cvtotoxicitV
of DNA-damaging anti-cancer agents have been assisted by therecent development of inhibitors that are markedly more potent(> tenfold). and possibly more specific. than the classical benza-mide inhibitors. The primary aim of this study as to evaluate theeffects of two representative examples of the recently developedPARP inhibitors on the cv-totoxicitv of a range of different anti-cancer aaents in order to select suitable agents for preclinical in
vivo and. ultimately. clinical studies. In addition. studies were
performed to investigate whether these PARP inhibitors func-tioned by inhibition of repair/recovery. or by a mechanism thatrequires the cytotoxic agyent and the PARP inhibitor to be presentat the same time.The majonrtx of studies on the potentiation of cvtotoxicity by
PARP inhibition have used alky lating (particularly monofunc-tional alkylating) agents and in the studies described here thegreatest potentiation was observed with MTIC (Table 1). MTIC is
not clinically useful as it is too reactive. but temozolomide. whichbreaks down chemically to produce MTIC. is currently undergoingclinical trials (Newlands et al. 1997). Previous studies. also inL12 10 cells (Boulton et al. 1995). have demonstrated that NIT 1025
British Joumal of Cancer (1998) 78(10), 1269-1277 0 Cancer Research Campaign 1998
Potentiation of anti-cancer agents by potent PARP inhibitors 1275
A
1)c
00
C/)
[Nolatrexed] (gM)
B10
0
--
0001
-Ea
Cl)
[Gemcitabine] (nm)
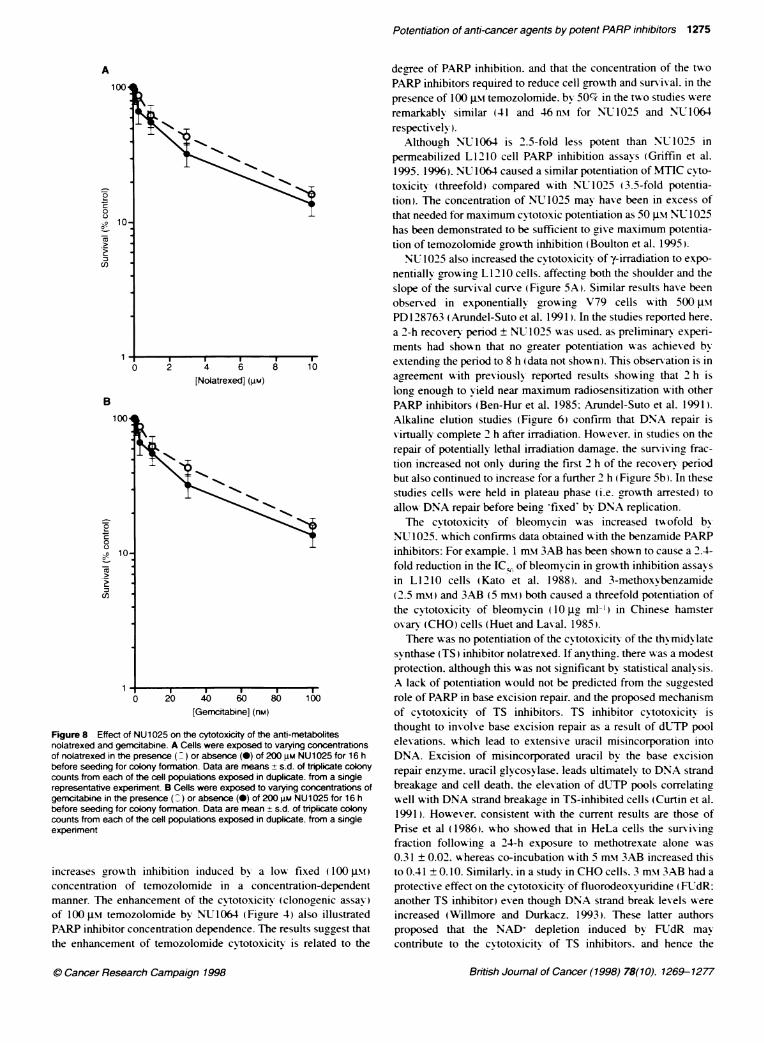
Figure 8 Effect of NU1025 on the cytotoxicity of the anti-metabolitesnolatrexed and gemcitabine. A Cells were exposed to varying concentrationsof nolatrexed in the presence ( ) or absence (-) of 200 gm NU1025 for 16 hbefore seeding for colony formation. Data are means + s.d. of tripicate colonycounts from each of the cell populations exposed in duplicate. from a singlerepresentative experiment. B Cells were exposed to varying concentratons ofgemcitabine in the presence ( ) or absence (0) of 200 gm NU1025 for 16 hbefore seeding for colony formation. Data are mean ± s.d. of triplicate colonycounts from each of the cell populations exposed in duplicate. from a singleexperiment
increases growth inhibition induced by a low fixed ( 100 JIM)concentration of temozolomide in a concentration-dependentmanner. The enhancement of the cytotoxicitx (clonogenic assay)of 100 Im temozolomide by NU1064 (Figure 4) also illustratedPARP inhibitor concentration dependence. The results suggest thatthe enhancement of temozolomide cvtotoxicitv is related to the
degree of PARP inhibition. and that the concentration of the tw-oPARP inhibitors required to reduce cell growth and survival. in thepresence of 100 gm temozolomide. by 50%c in the two studies wereremarkablx similar (41 and 46nm for NU1025 and NU1064respectivelx).
Although NU1064 is 2.5-fold less potent than NU1025 inpermeabilized L1210 cell PARP inhibition assavs (Gnrffin et al.1995. 1996). NU1064 caused a similar potentiation of MTIC cyto-toxicitv (threefold) compared with NU,1025 (3.5-fold potentia-tion). The concentration of NUT1025 mav has-e been in excess ofthat needed for maximum cy totoxic potentiation as 50 msi N- 1025has been demonstrated to be sufficient to give maximum potentia-tion of temozolomide growth inhibition (Boulton et al. 1995).NU 1025 also increased the cytotoxicity of y-irradiation to expo-
nentially arowing L 1210 cells. affecting both the shoulder and theslope of the survival curve (Figure 5A). Similar results have beenobserved in exponentially growing V79 cells with 500 JimPD 128763 (Arundel-Suto et al. 1991). In the studies reported here.a 2-h recovery period ± NU 1025 was used. as preliminary experi-ments had shown that no greater potentiation Awas achieved bvextending the period to 8 h (data not shown). This obserxation is inagreement with previously reported results showing that 2 h islong, enough to vield near maximum radiosensitization with otherPARP inhibitors (Ben-Hur et al. 1985: Arundel-Suto et al. 1991).Alkaline elution studies (Figure 6) confirm that DNA repair isXvirtuallx complete 2 h after irradiation. Howuever. in studies on therepair of potentially lethal irradiation damage. the sunriving frac-tion increased not only during the first 2 h of the recovery periodbut also continued to increase for a further 2 h (Fiaure Sb). In thesestudies cells were held in plateau phase (i.e. growth arrested) toallow DNA repair before being 'fixed' by DNA replication.The cvtotoxicity of bleom cin w as increased tw ofold by
NU 1025. which confirms data obtained w-ith the benzamide PARPinhibitors: For example. 1 mrm 3AB has been shown to cause a 2.4-fold reduction in the IC; of bleomycin in growth inhibition assaysin L1210 cells (Kato et al. 1988). and 3-methoxNbenzamide(2.5 mxt) and 3AB (5 mm) both caused a threefold potentiation ofthe cytotoxicity of bleomycmn (10 pzg ml') in Chinese hamsteroxarv (CHO) cells (Huet and Laval. 1985).
There w as no potentiation of the cvtotoxicity of the thymidy latesvynthase (TS) inhibitor nolatrexed. If anvthing. there w-as a modestprotection. although this was not significant by statistical analysis.A lack of potentiation would not be predicted from the suggestedrole of PARP in base excision repair. and the proposed mechanismof cvtotoxicity of TS inhibitors. TS inhibitor cvtotoxicitx isthought to involve base excision repair as a result of dUTP poolelexations. which lead to extensive uracil misincorporation intoDNA. Excision of misincorporated uracil by the base excisionrepair enzyme. uracil glvcosvlase. leads ultimately to DNA strandbreakaae and cell death. the elevation of dUTP pools correlatincwell with DNA strand breakaae in TS-inhibited cells (Curtin et al.1991). However. consistent with the current results are those ofPrise et al (1986). who showed that in HeLa cells the survixingfraction follow ing a 24-h exposure to methotrexate alone w as0.31 ± 0.02. whereas co-incubation with 5 mM\ 3AB increased thisto 0.41 ± 0.10. Similarlv. in a studx in CHO cells. 3 mM\ 3AB had aprotective effect on the cytotoxicitv of fluorodeoxvuridine (FUTdR:another TS inhibitor) even though DNA strand break levels wereincreased (Willmore and Durkacz. 1993). These latter authorsproposed that the NAD+ depletion induced by FLTdR maycontribute to the cvtotoxicitv of TS inhibitors, and hence the
British Joumal of Cancer (1998) 78(10). 1269-12770 Cancer Research Campaign 1998

1276 KJ Bowman et al
prevention of this depletion by 3AB may exert a protective effect.In the studies described here. a modest protection from gem-
citabine-induced cytotoxicity by NU 1025 wvas also observed. S-phase cells that most actively incorporate the drug are the mostsensitive to gemcitabine (Huang and Plunkett. 1995). and protec-tion against a v-ariety of S-phase acting drugs (hydroxyurea.fluorodeoxvuridine and thioguanine) by 3AB has been reportedand attributed to the cytostatic effects of the benzamides (Moses etal. 1988). The growth-inhibitory effects of NU1025 alone inL1210 cells has been described previously: IC for a 48 h exposure is 410 Im (Boulton et al. 1995). It is possible that the mildlycvtostatic effect of NUL1025 at 200 -iM may hinder entry into S-phase and hence protect cells from agents acting in this phase.such as the anti-metabolites gemcitabine and nolatrexed.PARP inhibitors are considered to potentiate cytotoxic arents bv
inhibiting DNA repair and hence the recovery from genotoxicinsult. In order to verify that NU 1025 was acting by repair inhibi-tion. the survival of cells exposed to an alkylating agent orionizing radiation. with a recovery period in the presence orabsence of NU1025. were compared. The highly reactive methyl-ating, agent MTIC was selected in order to achieve rapid inductionof DNA damage for a limited period. NUT1025 caused a similarpotentiation of MTIC cytotoxicity w-hether it was present from thebeginningr of the experiment or added only after the MTIC hadbeen removed. This result indicates that NU1025 does not interactwith MTIC to influence the initial level of DNA damage. butrather that it suppresses recovery. In the case of y-iradiation. both200 g.m NJ 1025 and 10 mM 3AB inhibited recovery from poten-tially lethal damage. DNA repair following y-irradiation byNU 1025 was investigated by alkaline elution. and 2 h in drur-freemedium at 37°C was sufficient to pernit complete repair of DNAstrand breaks. NU1025 significantly retarded the repair of DNAstrand breaks but did not block it completely. Taken together thesedata indicate that NU 1025 is indeed inhibiting the repair of alkyl-ating agent- and y-irradiation-induced DNA damage. and that thiseffect underlies the potentiation produced by PARP inhibition.The normal growth and development of PARP knockout mice
(Wang et al. 1997). which exhibit normal DNA repair capacityafter MNNG exposure. suggests that PARP activity is not neces-sary for cell survival. However. the use of PARP inhibitors toretard DNA repair and hence increase the cytotoxicitv of certainclasses of anti-cancer agrents remains a viable option as the effectsof inhibited PARP are arguably different from those of PARP defi-ciency. This difference has been most clearly illustrated bv thework of Satoh and Lindahl (1992). who demonstrated that cellextracts depleted of PARP (analooous to PARP -I- mice) couldrepair nicked DNA with similar efficiency to those containingPARP. As expected. in whole-cell extracts containing PARP. theabsence of NAD+ or the presence of 3-aminobenzamide inhibitedDNA repair. Thus. the inhibition of PARP has a deleterious effecton DNA repair. whereas its removal is neutral. A similar situationto inhibited PARP is found when the DNA binding domain (DBD)is transfected into cells. resulting in the dominant negative inhibi-tion of PARP activity (Mohnete et al. 1993). In this system theDBD binds to the DNA nick and. as there is no catal-tic or auto-modification domain. no automodification and dissociation of theDBD occurs. Lack of dissociation prevents access of full-lengthPARP and DNA repair (Molinete et al. 1993). On the basis of thismodel it may be no coincidence that during apoptosis PARP iscleaved into two fragments. one comprising the DBD and the otherthe automodification and catalytic domains (Kaufmann et al.
1993). Cleavage would presumably be analogous in effect to theoverproduction of the DBD and the use of PARP inhibitors. i.e. aninhibition of DNA repair. which is presumably counter-productiVeduring programmed cell death.
In recent years considerable effort has been devoted to theinvestigation of the role of PARP and its inhibition in the cvto-toxicitv of anti-cancer acents using a wide variety of molecularbiological approaches. as w-ell as inhibitor studies. The precise roleof the enzyme still remains to be fully elucidated. but the use ofthe new generation of potent and structurally different PARPinhibitors mav facilitate the probing of its function. The datapresented here. using two novel potent inhibitors that are struc-turallv different from the classical benzamides. supports many ofthe studies w-ith the benzamides and conclusions drawn from them.Furthermore. these data suggest that for the preclinical evaluationof PARP inhibitors it would be appropriate to use combinationswith alkvlating agents. bleomvcin and ionizinc radiation. but notantimetabolites.
ABBREVIATIONS
NlT1025. 8-hvdroxv-2-methylquinazolin4-[3H]one: N'U1064. 2-methvlbenzimidazole-4-carboxamide: 3AB. 3-aminobenzamide:DBD. DNA bindingy domain: IC. the concentration of druccausing 90% cell death: ID,. dose of radiation causing 90% celldeath: EF90 enhancement factor (ICg,- or ID90 controUIC, or ID, +PARP inhibitor): MNNG. N-methvl-M-nitro-N-nitrosoguanidine:MTIC. 5-( 3-methyltriazen- 1 -vl )imidazole-4-carboxamide: MTT.3-[4.5-dimethylthiazol-2-vl]-2.5-diphenvltetrazolium bromide:PBS. phosphate-buffered saline: SDS. sodium dodecyl sulphate:TS. thvmidylate synthase.
REFERENCES
.Athaus FR. Hofferer L. KIeczkow ska HE. Malana2a MI. Naeeeli H. Panzeter P andRealini C 1993) Histone shuttle driven bs the automodification c\cle ofpolv ADP-ribose) polymerase. Environ Mkol Muragen 2: 278-282
Arundel-Suto CM. Scavone S\: Turner WR. Suto mI and Sebolt-Leopold JS ( 1991Effects of PD 1 28763. a ness potent inhibitor of pol\ A.ADP-ribose pol-rnerase.on X-rav induced cellular recovers processes in Chinese hamster V79 cells.RadiatRes 126: 367-371
Ben-Hur E. Chen C-C and Elkind WMM 1985 (Inhibitors of pol\ adenosinediphosphonrbose synthetase. examination of metabolic perturbations andenhancement of radiation response in Chinese hamster cells. Cancer Res 45:2123-2127
Boulikas T ( 1991 Relation betseen carcinogenesis. chromatin structure andpol%<(.ADP-n'bos, lation ..Anticancer Res 11: 489-528
Boulton S. Pemberton LC. Porteous JK. Curtin NJ. Griffin RJ. Golding BT andDurkacz BW (1995) Potentiation of temozolomide c\totoxicity: a comparativestud%- of the biological effects of polv ADP-ribose )pol rmerase inhibitors. Br JCancer 72: 849-856
Bowman KJ. Cal\-ert .AH. Curtin NJ. Golding BT. Griffin RJ. Nesell DR.Srinis asan S and White A (1996) Effect of novel pol\ ADP-ribose pol meraseinhibitors on the cvtotoxicitx of anticancer agents. Br J C 73 suppl. 26: 1 3
Chen TR (1977 In situ detection of mycoplasma contamination in cell cultures bsfluorescent Hoechst 33258 stain. Erp Cell Res 104: 255-262
Cleaver JE (1984) Differential toxocitv of 3-amiinobenzamide to s ild-type and 6-thio2uanine resistant CHO cells b\ interference w-ith pathways of purinebiosvrnthesis .Uutation Res 131: 123-127
Cleaver JE and NMorgan WFT (199 1) Polv ADP-ribose ipolymerase: a perplexingparticipant in cellular responses to DN-A breakage. Mutation Res 257: 1-18
Curtin NJ. Harris AL and Aherne W (1991 Nlechanism of cell death followingth\mid, late s -nthase inhibition: 2'-deox\uridine-5'-triphosphate accumulation.DNA damage. and growth inhibition follow-ing exposure to CB37 17 anddipvridamole. Cancer Res 51: '346-25'
Gnrffin RJ. Pemberton LC. Rhkodes D. Bleasdale C. Bowsman K. Calenrt AH. CurtinNJ. Durkacz BU:~NesselI DR. Porteus IK and Golding BT (1995)> Nos el
British Joumal of Cancer (1998) 78(10), 1269-1277 C Cancer Research Campaign 1998

Potentiation of anti-cancer agents by potent PARP inhibitors 1277
potent inhibitors of the DNA repair enzyme pol A.ADP-ribose polrmerasePARP Anticancer Drug Design 10: 507-5 14
Gnrffin RJ. Srinivasan S. A-hite AW Bowman K. Calvert AH. Curtin NI. Neswell DRand Goldine BT 1996). Novel benzimidazole and quinazolinone inhibitors ofthe DNA repair enzyme. pol^-lADP-ribosepolN7erase. Pharm Sci 2: 43-47
Huang P and Plunkett W 1995) Fludarabine- and gemcitabine-induced apoptosis:incorporation of analogues into DNA is a critical event Cancer ChemotherPharmaco 36: 181-188
Huet J and Laval F) 1985) Potentiation of cell killtnc bv inhibitors of polsyadenosinediphosphate-ribose) synthesis in bleomrncin-treated Chinese hamster ovaocells. Cancer Res 45: 987-991
Hunting, DJ. Gowans BJ and Henderson IF ( 1985) Specificity of inhibitors ofpolyl ADP-ribose) synthesis. Effects on nucleocide metabolism in cultured cells.Mol Pharnacol 28: 200-206
Kato T. Suzumura Y and Fukushima M 11988) Enhancement of bleornvm'c activitsby 3-aminobenzamide. a pol- ADP-ribose) synthesis inhibitor. in *iirro andin vivo. Anticancer Res 8: 239-244
Kaufmann SH. Desnovers S. Ottasiano Y. Dasidson NE and Poirier GG (1993)Specific proteolytic cleasvage of pols) ADP-nbose) polyrnerase: an earfl markerof chemotherapy-induced apoptosis. Cancer Res 53: 3976-3985
Kohn KW Ew.i2 RA. Erickson LC and Zwelling LA ( 1981 Measurement of strandbreaks and cross-links by alkaline elution. In DNA Repair: a LaboratoroManual ofResearch Procedures. Vol. I B. Friberg EC and Hanaswalt PC eds).pp. 379-401. Marcel Dekk-er Nesw York-
Kupper JH. van Gool L and Burkle A ( 1 995) Molecular genetic systems to study therole of poly(ADP-ribose ation in the cellular response to DNA dama-e.Biochimie 77: 450-455
Lautier D. Lagueux J. Thibodeau J. Menard L and Poirier GGO 1993) Molecular andbiochemical features of polv)ADP-ribose) metabolism. Mol Cell Biochem 122:171-193
Lindahl T. Satoh MIS. Poirier GG and Kluneland A (1995) Post-translationalmodification of polpADP-ribose "po1>merase induced bs DNA strand breaks.Trends Biol Sci 20: 405-411
Milam KM. Thomas GH and Cleaser IE (1986) Disturbances in DNA precursormetabolism associated with exposure to an inhibitor of polv l ADP-ribose)synthetase. ELp Cell Res 165: 260-268
Molinete M. Vermeulen W Burile A. Menissier-de Murcia J. Kupper IH.Hoejimakers JHJ and de Murcia G (1993) Overproduction of poly(ADP-ribose)
polvrmerase DNA-bindinu domain blocks alk-vlation-induced DNA repairsrnthesis in mamualian cells. EMBO J 12: 2109-2117
Moses K. Harris AL and Durkacz BW (1988 Adenosine-diphosphoribosyltransferase inhibitors can protect against or potentiate thecytoto.xicitr of S-phase acting drugs. Biochem Pharmacol 37: 2155-2160
de Murcia G and Menissier de Murcia J ( 1994) Polv(ADP-ribose)po1vmerase: amolecular nick-sensor. Trends Biol Sci 19: 172-176
Newlands ES. Stevens MFG. Wedge SR. Wheelhouse RT and Brock C (1997)Temozolomide: a revies of its discovers. chemical properties. pre-clinicaldevelopment and clinical tnials. Cancer Treat Rev 23: 35-61
Plunkett W. Huang P. Xu Y-Z. Heinemann Xl Gruneswald R and Gandi V (1995)Gemcitabine: metabolism, mechanisms of action and self-potentiation. SeminOncol22(suppl.) 1: 3-10
Prise K.M. Gaal JC and Pearson CK (1986) Increased protein ADPribosvlation inHeLa cells exposed to the anti-cancer drug methotrexate. Bichim Bioph s Acra887: 13-22
Satoh M and Lindahl T ( 1992) Role of pol%-(ADP-nrbose (formation in DNA repair.,Vature 356: 356-358
Shall S (1984) ADP-ribose in DNA repair a new concept of excision repair. AdvRadiar Biol 11: 1-69
Shealv FY and Krauth DA ( 1 966) Imidazoles 1. 5 (or 4 -Carboxarnides. J.MedChem 9 34-38
Stevens MFG. Hickman JA. Langdon SP. Chubb D. Vickers L Stone R. Baig G.Goddard C. Gibson NXA Slack JA. Newton C. Lunt E. Fizames C and LavelleF ( 1987) Antitumour activity and pharmacokinetics in mice of 8-carbamov1-3-meth% l-imi'dazo[5.1-d-1.2.35-tetrazin-4)3H-one (CCRG 81045: M&B39831). a novel drugo with potential as an altemative to dacarbazine. CancerRes 47: 5846-5852
Webber S. Barlett C.- Boritzki TJ. Hilliard J.- Howland EF. Johnston AJ. Kosa MI.Margosiak SA. Morde CA and Shetty BV (1996) AG337. a novel lipophilicthvmidWvlate svnthase inhibitor in * itro and in vii o precliical studies. CancerChemother Pharmacol 37: 509-517
Wang Z-Q. Auer B. Stingl L BerJhammer H. Haidacher D. Schuweiger M andWagner EF ( 1995) Mice lacking ADPRT and poly ADP-ribosyI ation developnormally but are susceptible to skin disease. Genes Dev 9: 509-520
Aillmnore E and Durkacz BW (1993) Cvtotoxic mechanisms of 5-fluoropynrmidines.Relationships with polv(ADP-nboseipolvmerase activity. DNA stand breakageand incorporation into nucleic acids. Biochem Pharmacol 46: 205-211
C Cancer Research Campaign 1998 British Joumal of Cancer (1998) 78(10), 1269-1277