prediction of protein disorder zsuzsanna dosztányi institute of enzymology, budapest, hungary...
TRANSCRIPT

Prediction of protein Prediction of protein disorderdisorder
Zsuzsanna DosztányiInstitute of Enzymology, Budapest, [email protected]
Protein Structure/Function Paradigm
Dominant view: 3D structure is a prerequisite for protein function
Amino acid sequence Structure Function

But….
Heat stability Protease sensitivity Failed attempts to crystallize Lack of NMR signals “Weird” sequences …


IDPs
Intrinsically disordered proteins/regions (IDPs/IDRs)
Do not adopt a well-defined structure in isolation under native-like conditions
Highly flexible ensembles, little secondary structure, no folded structure
Functional proteins

Protein disorder is prevalent
20
40
60
0
LD
R (
40<
) p
rote
in, %
kingdom
B
A
E

Protein disorder is important
Prion protein Prion disease
CFTR Cystic fibrosis
Alzheimer’s
-synuclein Parkinson’s
p53, BRCA1 cancer

Protein disorder is functional
0
20
40
60
80
regulatorysignaling
biosynthetic
metabolic
pro
tein
(%
)
30< 40< 50< 60 <
length of disordered region
Iakoucheva et al. (2002) J. Mol. Biol. 323, 573

p53 tumor suppressor
TAD DBD TD RD
transactivation DNA-binding tetramerizationregulation
Wells et al. PNAS 2008; 105: 5762

Heterogeneity in protein disorder
Flexible loop
RC-like Compact
Transient structures

Modularity in proteins
Many proteins contains multiple domains
Composed of ordered and disordered segments
Average length of a PDB chain is < 300
Average length of a human proteins ~ 500
Average length of cancer-related proteins > 900
Structural properties of full length proteins …

Bioinformatics of protein disorder
Part 1 Databases Prediction of protein disorder
Part 2 Prediction of functional regions within IDPs

Datasets Ordered proteins in the PDB
over 94000 structures few 1000 folds
Some structures in the PDB classify as disordered! only adopt a well-defined structure in complex in crystals, with cofactors, proteins, …
Disorder in the PDB Missing electron density regions from the PDB
NMR structures with large structural variations
Less than 10% of all positions
Usually short (<10 residues), often at the termini

Disprot
www.disprot.org
Current release: 6.02Release date: 05/24/2013Number of proteins: 694Number of disordered regions: 1539
Experimentally verified disordered
proteins collected from literature
(X-ray, NMR, CD, proteolysis, SAXS,
heat stability, gel filtration, …)

Additional databases Combining experiments and predictions
Genome level annotations
MobiDB: http://mobidb.bio.unipd.it D2P2: http://d2p2.pro IDEAL: http://www.ideal.force.cs.is.nagoya-u.ac.jp/IDEAL
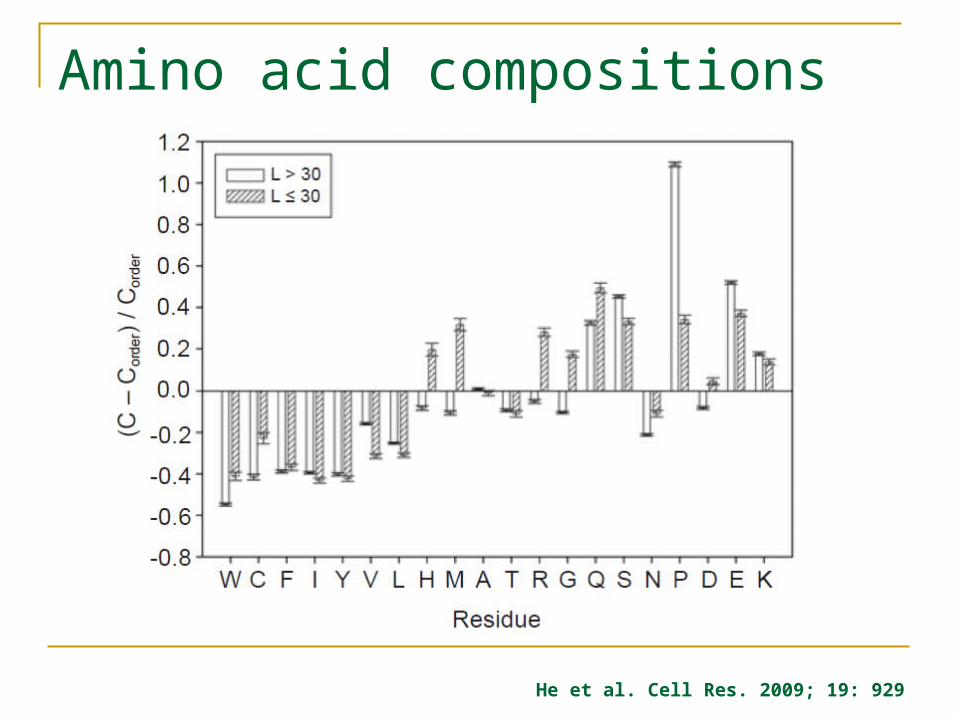
Amino acid compositions
He et al. Cell Res. 2009; 19: 929

Sequence properties of disordered proteins Amino acid compositional bias High proportion of polar and charged amino acids
(Gln, Ser, Pro, Glu, Lys) Low proportion of bulky, hydrophobhic amino acids
(Val, Leu, Ile, Met, Phe, Trp, Tyr) Low sequence complexity Signature sequences identifying disordered proteins
Protein disorder is encoded in the amino acid sequence

Uversky plot: charge-hydrophobicity (two parameters)
Mean hydrophobicity
Mea
n n
et c
har
ge
Uversky (2002) Eur. J. Biochem. 269, 2

p53
Prilusky (2005) Bioinformatics 21, 3435
Making it position specific: FoldIndex http://bip.weizmann.ac.il/fldbin/findex

Disorder Prediction Methods
Amino acid propensity scales
GlobPlot
Compare the tendency of amino acids: to be in coil (irregular) structure. to be in regular secondary structure elements
Linding (2003) NAR 31, 3701

GlobPlot

GlobPlot
From position specific predictions
Where are the ordered domains?
Longer disordered segments?
Noise vs. real data

GlobPlot: http://globplot.embl.de/
downhill regions correspond to putative domains (GlobDom)
up-hill regions correspond to predicted protein disorder

Disorder Prediction Methods
Physical principles
IUPred
If a residue cannot form enough favorable interactions within its sequential environment, it will not adopt a well defined structure it will be disordered
Dosztanyi (2005) JMB 347, 827

Energy description of proteins
For example: L-I interaction is frequent (hydrophobic effect)
L-I interaction energy is low (favorable)
K-R interaction is rare (electrostatic repulsion)
K-R interaction energy is high (unfavorable)
Estimation of interaction energies based on statistical potentials:
Calculated from the frequency of amino acid interactions in globular proteins alone, based on the Boltzmann hypothesis.

Predicting protein disorder - IUPred The algorithm:
…PSVEPPLSQETFSDL WKLLPENNVLSPLPSQAMDDLMLSPDDIEQWFTEDPGPDEAPRMPEAAPRVA PAPAAPTPAA...
Based only on the composition of environment of D’swe try to predict if it is in a disordered region or not:
Amino acid composition of environ-ment:
A – 10%C – 0%D – 12 %E – 10 %F – 2 % etc…
Estimate the interaction energy between the residue and its environment
Decide the probability of the residue being disordered based on this

IUPred: http://iupred.enzim.hu/

Disorder Prediction Methods
Machine learning
DISOPRED2
Binary classification problem
Ward (2004) JMB 337, 635

DISOPRED2 …..AMDDLMLSPDDIEQWFTED…..
Assign label: D or O D O
F(inp)SVM with linear kernel

DISOPRED2
Cutoff value!

PONDR VSL2
Differences in short and long disorder amino acid composition methods trained on one type of dataset tested on
other dataset resulted in lower efficiencies
PONDR VSL2: separate predictors for short and long disorder combined
length independent predictions
Peng (2006) BMC Bioinformatics 7, 208

PONDR-FIT
Sequence
Disorder prediction methods
PONDR VLXT
PONDR VL3
PONDR VSL2
IUPred
FoldIndex
TopIDP
PredictionANN
Meta-predictor
Xue et al. Biochem Biophys Acta. 2010; 180: 996

Complexity of protein disorder

Prediction of protein disorder Disordered residues can be predicted from
the amino acid sequence ~ 80% at the residue level
Methods can be specific to certain type of disorder accordingly, accuracies vary depending on
datasets Predictions are based on binary
classification of disorder