principi di chimica degli equilibri ionici in … · algebra avanzata e un po' di sensibilità...
TRANSCRIPT

1
CAP. I
PRINCIPI DI CHIMICA DEGLI EQUILIBRI IONICI IN SOLUZIONE
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-1

2
1.0.0.- Premesse L’Ecotossicologia e’ fondamentalmente basata sulle reazioni chimiche che un composto inquinante subisce nel suo percorso ambientale. Nella stragrande maggioranza dei casi queste reazioni avvengono in mezzo acquoso o mediate da una condizione acquosa. La conoscenza, quindi, degli equilibri ionici in soluzione diventa importante in particolare per cio’ che riguarda alcuni specifici ambiti: gli equilibri acido-base, la solubilita’ e la precipitazione, i complessi e le reazioni ossido-riduttive. In questo capitolo non tratteremo dei principi basici della Termodinamica che determinano l'andamento delle reazioni. Tale argomento sara' ampiamente sviluppato nel Capitolo II per quello che riguarda i processi ambientali. Va qui, peraltro, ricordato che lo stato di equilibrio e lo svolgersi di una reazione acido-base, di precipitazione, di complessazione e di ossido-riduzione, sono sempre condizionate dal principio fondamentale di raggiungimento della minima energia finale. Cio' vuol dire che, in assenza di apporti energetici esterni, le reazioni chimiche si sposteranno nella direzione della minima energia ed i prodotti di reazione saranno quelli con il contenuto energetico piu' basso in rispetto al II° Principio della Termodinamica.. 1.1.0 - Lo stato di equilibrio Quando alcuni composti vengono mescolati e subiscono una variazione chimica, diciamo che è avvenuta una reazione chimica. In realtà poche reazioni procedono in modo tale che i reagenti siano consumati completamente e rimanga soltanto il prodotto: cio’ che risulta e’ un equilibrio tra reazioni dirette ed inverse. Il concetto di equilibrio è abbastanza intuitivo: se un sistema è stabile nel tempo, nel senso che conserva inalterate le sue proprietà fisiche e la sua composizione chimica, si dice che è in stato di equilibrio; in esso non risultano reazioni chimiche in atto, né si manifestano trasformazioni di alcun genere. In Ecotossicologia gli equilibri di reazioni chimiche che avvengono tra ioni in soluzione acquosa, (ambito in cui avvengono la maggior parte delle reazioni della Chimica Ambientale) esistono come ioni in soluzione; le reazioni tra ioni sono spesso molto rapide e possono raggiungere lo stato di equilibrio quasi con la stessa velocità con cui i reagenti vengono mescolati. Le reazioni ioniche all'equilibrio sono state studiate a fondo e sono disponibili semplici ma fondamentali principi teorici che governano le reazioni all'equilibrio. Applicando tali principi possiamo prevedere teoricamente i risultati delle reazioni chimiche, compresa la valutazione numerica di molte quantità che sarebbe estremamente difficile misurare direttamente. Sebbene il gran numero di specie presenti in soluzione possa portare alla formulazione di equazioni di grado elevato, in pratica è necessaria soltanto un poco di algebra avanzata e un po' di sensibilità chimica, poiché spesso è possibile semplificare il grado delle equazioni trascurando in modo logico alcuni termini. Una descrizione immediata di un equilibrio chimico è quella di un bilanciamento di reazioni apposte. All'avvicinarsi all'equilibrio i reagenti si combinano per dare i prodotti, ma i prodotti stessi possono combinarsi tra loro per dare i reagenti originari. Un bilanciamento di equilibrio è raggiunto quando la velocità con cui procede la reazione diretta è esattamente bilanciata dalla velocità della reazione inversa. L'equilibrio è cioè un bilanciamento dinamico, e variazioni nelle concentrazioni delle sostanze e della temperatura della soluzione sposteranno la posizione dell'equilibrio, alterando le velocità relative delle reazioni dirette e inverse. 1.1.1.0 - La costante di equilibrio
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-2

3
Il principio fondamentale su cui si basa lo studio quantitativo dei sistemi di equilibrio è l'esistenza della costante di equilibrio. Consideriamo la dissociazione dell'acido acetico: la costante di equilibrio delle concentrazioni per la dissociazione dell'acido acetico è definita dall'equazione:
Ka =H+⎡
⎣ ⎢ ⎤ ⎦ ⎥ + Ac−[ ]HAc[ ] (1.1)
in cui le parentesi quadre indicano concentrazioni in grammo-ioni. litro-1 o in mole.litro-1. La costante Ka varia soltanto leggermente con la concentrazione analitica, dipende dalla temperatura della soluzione, ma non dalla pressione atmosferica, almeno in entità rilevabile. Notiamo che nell'espressione della costante non interviene il volume totale della soluzione, ma è soltanto il rapporto tra il numero di ioni di un dato tipo ed il numero totale di molecole di solvente, cioè la concentrazione degli ioni, che determina la composizione dell'equilibrio della soluzione. Se si richiede una relazione più accurata o si devono considerare soluzioni più concentrate, le concentrazioni indicate in (1.1) devono essere sostituite con le "attività", che contengono correzioni per le forze interioniche, che, in questo caso, non sono più trascurabili. Per una reazione della forma :
aA + bB = cC + dD dove A,B,C e D sono specie chimiche e a, b, c e d sono i coefficienti numerici richiesti per bilanciare l'equazione, all'equilibrio sarà rispettata la seguente relazione:
K0 =C{ }c D{ }d
A{ }a B{ }b (1.2)
dove le graffe indicano l'attività della specie entro le graffe stesse. L'attività è interpretata come segue: - ioni e molecole in soluzione diluita: l'attività è approssimativamente eguale alla concentrazione in mole/litro; - il solvente in una soluzione diluita: l'attività è eguale alla frazione molare del solvente ed è approssimativamente unitaria; - solidi o liquidi puri in equilibrio con la soluzione: l'attività è esattamente unitaria; - gas in equilibrio con la soluzione: l'attività è la pressione parziale del gas in atmosfere; - miscele di liquidi: l'attività di un dato componente è approssimativamente
eguale alla sua frazione molare.
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-3

4
La costante K° dipende soltanto dalla temperatura, e non dalla pressione o dalla composizione del sistema. Consideriamo di seguito alcuni importanti esempi della legge di equilibrio applicata a soluzioni ioniche. 1.1.1.1.- Il prodotto ionico dell'acqua Un equilibrio fondamentale che sempre esiste in soluzioni acquose è e’ quello di autoprotolisi dell’acqua che fornisce ioni idrogeno e ossidrili:
H2O + H2O ⇔ H3O+ + OH−
(1.3) che è scritta convenzionalmente:
H2O ⇔ H+ + OH −
In termini di attività la relazione della costante di equilibrio è:
K 0 =H +{ } OH −{ }
H2O{ } (1.4)
Le attività degli ioni sono approssimativamente eguali alle loro concentrazioni e l'attività del solvente acqua è approssimativamente unitaria; per questi motivi è d'uso comune la relazione approssimata
Kw = H +[ ]OH−[ ]= 10−14 (1.5)
dove Kw è chiamata "prodotto ionico dell'acqua". 1.2.0.0.- Bilanci 1.2.1.0.- Il bilancio di massa Un bilancio di massa stabilisce che il numero di atomi di un certo tipo deve rimanere costante nelle reazioni chimiche ordinarie. Consideriamo, per esempio, una soluzione satura si solfato di bario (completamente dissociato) in acqua. Se S è la sua solubilità molare, cioé il numero di moli di BaSO4 che si scioglie in 1 litro di soluzione, allora: S = [Ba++] (1.6) è il bilancio di massa del bario nella soluzione. La relazione indica che il bario è presente nella soluzione soltanto come ioni Ba++. Un bilancio di massa per il solfato è: S = [SO4
=] + [HSO4-] (1.7)
poiché il gruppo solfato esiste in sole due forme: ioni solfato e ioni solfato acido. Nella soluzione satura di cloruro mercurico in acqua si formano molti complessi. Un bilancio di massa per il mercurio in tale soluzione è: S = [Hg++] + [HgCl+] + [HgCl2] + [HgCl3-] + [HgCl4=]+[Hg(OH)2] ed indica che le 7 specie riportate costituiscono tutte le varie forme in cui il mercurio esiste in soluzione quando è presente lo ione cloro.
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-4

5
Se una specie contiene più atomi o gruppi dello stesso tipo, dobbiamo contare ciascuna specie tante volte quante appaiono gli atomi o i gruppi nelle specie. Un bilancio di massa per il cloro è: 2 S = [Cl-] + [HgCl+]+ 2[HgCl2] + 3[HgCl3-] + 4[HgCl4=] Poiché ciascuna mole di HgCl2 dissolvendosi dà 2 atomi di cloro, al lato sinistro dell'equazione compare il termine 2 S. Quando ci sono due sorgenti dello stesso atomo o gruppo nel bilancio di massa, si deve tenere conto di entrambi. Per esempio, consideriamo una soluzione contenente CHA mole.litro-1 di acido acetico e CA mole.litro-1 di acetato. Un bilancio di massa per l'acetato è: CHA + CA = [HAc] + [Ac-] che stabilisce che tutti i gruppi acetati introdotti dai due composti esistono in soluzione o come ioni acetato o come molecole di acido acetico indissociato. Posto che tutte le possibili sorgenti dell'atomo o del gruppo e tutte le possibili specie che contengono l'atomo o il gruppo siano state prese in considerazione, il bilancio di massa è una relazione "esatta". 1.2.2.0.- Il bilancio protonico o la condizione protonica In sistemi acido-base, dove gli ioni dell'acqua prendono parte all'equilibrio, è necessario avere una relazione, chiamata "condizione protonica", che implichi almeno uno degli ioni solvente. Ci sono 4 metodi per ottenere questa relazione: si può fare un bilancio di massa sugli ioni idrogeno o sugli ioni ossidrile; oppure si può far uso del bilancio protonico o del bilancio di carica . Per fare un bilancio di massa sugli ioni idrogeno occorre tener conto di quanti di essi provengono dalla ionizzazione dell'acqua e quanti invece dalla ionizzazione dell'acido. Se non vi è alcuna sorgente esterna di ioni ossidrile (tali come NH3 o NaOH) possiamo assumere che ciascuna molecola di acqua che si ionizza produca uno ione H+ e uno ione OH-. Per esempio, in una soluzione di acido cloridrico di concentrazione C moleL-1, l'acido è completamente dissociato in ioni idrogeno e ioni cloro. Un bilancio di massa per gli ioni idrogeno è: [H+] = C + [OH-] Esso stabilisce che tutti gli ioni idrogeno in soluzione provengono dall'acido (C) e dalla dissociazione dell'acqua (un H+ per ciascun OH-). Similmente, se non vi è alcuna sorgente esterna di ioni idrogeno (come HCl o acido acetico), possiamo fare un bilancio di massa sugli ioni ossidrile. In sistemi più complicati, come una soluzione contenente NaH2PO4 o (NH4)2HPO4, il metodo del bilancio protonico per ottenere la condizione protonica diviene più facile, anche se è un po' più sofisticato di un semplice bilancio di massa. 1.2.3.0.- Il bilancio di carica Anche in sistemi abbastanza semplici, come quelli descritti, è necessario essere accurati nel formulare la condizione protonica dai bilanci di massa che implicano ioni solvente, perché è facile trascurare una possibile reazione con il solvente.
Perciò è spesso preferibile usare un "bilancio di carica", a volte chiamato "relazione di elettroneutralità", che esprime il fatto che una soluzione contenente ioni debba essere elettricamente neutra. Tale relazione è ottenuta considerando "il numero totale di cariche positive per unità di volume" e ponendolo uguale al "numero totale di cariche negative per unità di volume".
Ciò significa naturalmente che tutti gli ioni in soluzione devono essere presi in considerazione, compresi quelli che sono irrilevanti per gli equilibri in considerazione. Il
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-5

6
bilancio di carica non dà una relazione indipendente, ma può essere sempre combinato con i bilanci di massa per dare la "condizione protonica". Consideriamo di nuovo una soluzione contenente C moli di acido cloridrico per litro. Un bilancio di carica è: [H+] = [Cl-] + [OH-] Poiché tutti gli ioni cloro provengono dall'acido cloridrico, un bilancio di massa per il cloro è semplicemente [Cl-] = C e combinando le due equazioni otteniamo la "condizione protonica", la stessa relazione ottenuta precedentemente da un bilancio di massa sugli ioni idrogeno:
[H+] = C + [OH-]
Per una soluzione contenente C mole di cianuro sodico, il bilancio di carica è:
[H+] + [Na+] = [OH-] + [CN-] La concentrazione di HCN indissociato non è inclusa perché la molecola non è caricata. Un bilancio di massa per il sodio è semplicemente
[Na+] = C e per il cianuro:
[HCN] + [CN-] = C Sostituendo Na+ e CN-, gli ioni del sale, nel bilancio di carica otteniamo la "condizione protonica" [H+] + [HCN] = [OH-] che è identica alla relazione ottenuta da un bilancio del protone e da un bilancio di massa sugli ioni idrogeno.
Per sistemi più complicati può essere necessaria una considerevole manipolazione algebrica per ridurre il bilancio di carica alla "condizione protonica", ma è probabilmente meno facile ottenere la relazione sbagliata se viene impiegato il metodo del bilancio di carica. In problemi che implicano il prodotto di solubilità o la formazione di ioni complessi, si possono ottenere un sufficiente numero di relazioni per risolvere il problema dagli equilibri e dai bilanci di massa.
Nei sistemi acido-base, dove gli ioni solvente entrano negli equilibri, è richiesta una relazione addizionale che può essere o la "condizione protonica" o il "bilancio di carica", ma non entrambe. Un'altra relazione ancora è richiesta per gli equilibri di ossido-riduzione: "il bilancio elettronico", che collega le concentrazioni delle specie ossidate e ridotte. 1.3.0.0.- Acidi e Basi 1.3.1.0.- Definizione di acido e base
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-6

7
Gli equilibri acido-base sono importanti perche’ il sistema ambientale e’, di solito, temponato dalla presenza di elevate quantita’di composti ad elevato potere tampone (ad es.: il sistema carbonati/bicarbonati).
Se tale potere tamponente viene a mancare possiamo avere la diminuzione rapida del pH di un comparto ambientale con drastiche alterazioni biocenotiche e, al limite, pesanti danni anche di tipo economico. Un esempio tipico di cio’ e’ dato dalle piogge acide in Scandinavia che hanno creato addirittura un caso di conflittualita’ internazionale.
Come si ricordera’ l’emissioni di anidride solforosa e solforica dei bacini della Ruhr e della Centrali termoelettriche inglesi inviavano in Scandinavia ingenti masse di SO2/SO3 che, tramite le piogge, alteravano il pH dei fiumi in cui venivano cresciuti i salmoni. Il pH raggiungeva valori tali da provocare la totale distruzione degli allevamenti poiche’ l’acqua, povera di carbonati, non neutralizzava l’acidita’ atmosferica come invece succede, ad esempio, nelle regioni Mediterranee.
In tali regioni, infatti, la grande quantita’ di polveri che si trovano in atmosfera sono dovute a dolomite (carbonati di calcio e magnesio) in grado di reagire con l’acidita’, neutralizzandola e creando, inoltre, un buon buffer sistematico.
Grandi variazioni di pH possono ancora verificarsi in casi particolari, quali incidenti di trasporto od esplosioni di serbatoi.
Fig.1.1 - Interazione tra i diversi equilibri del sistema anidride carbonica-
acido carbonico- bicarbonati - carbonati
____________________________________________________________________
Arrehnius introdusse la teoria della dissociazione elettronica e spiegò le caratteristiche comuni a tutti gli acidi (sapore acido, capacità di reagire con certi metalli
G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-7

8
con sviluppo di idrogeno gassoso, proprietà di arrossare il tornasole ecc.), ammettendo che tali composti siano in grado di dissociarsi liberando ioni H+:
HA <====> H+ + A- (1.8)
In pratica si poteva constatare che tutti gli acidi noti erano sostanze idrogenate: HCl, H2SO4, HCN, H2S, ecc. Sostanze non idrogenate ma che in soluzione acquosa mostrano caratteristiche acide, per es. CO2, SO2, SO3, NO2, si trasformano in composti idrogenati in seguito alla reazione con l'acqua: CO2 + H2O <====> H2CO3. Le basi secondo Arrhenius sarebbero le sostanze in grado di dissociarsi mettendo in libertà ioni OH-
BOH <====> B+ + OH- (1.9) Tipiche basi sono infatti gli idrossidi: NaOH, KOH, Ca(OH)2 ecc. Altre sostanze che, pur non avendo nella loro molecola gruppi OH-, per es. NH3, C6H5NH2 ecc. si comportano da basi, sarebbero in grado di reagire con l’acqua dando origine a sostanze contenenti l'ossidrile: es.: NH3 + H2O <====> NH4OH Secondo una teoria più recente (Bronsted e Lowry, 1923) un acido è una sostanza (molecola o ione) che ha la tendenza a cedere un protone, e una base è una sostanza che ha la tendenza a ricevere un protone. Perdendo un protone (p) un acido si trasforma nella sua base coniugata: A1 <====> B1 + p (1.10) Una base, assumendo un protone, si trasforma nel suo acido coniugato. La (1.10) rappresenta una semireazione acido-base e come tale non ha nessuna possibilità di realizzarsi. Un acido A1 può agire come tale solo se può cedere il suo protone alla base B2 di un altro sistema coniugato: A1 + B2 <====> A2 + B1 (1.11) Quindi nell'equilibrio agiscono contemporaneamente i due sistemi :
A1 <====> B1 + p e B2 + p <====> A2 Uno dei due sistemi acido-base può essere costituito dallo stesso solvente. Secondo la teoria di Bronsted, gli acidi possono essere non solo molecole neutre, ma anche cationi ed anioni. A titolo di esempio si riportano alcuni sistemi acido-base nella tabella che segue. Si dice che un acido è tanto più forte quanto maggiore è la sua tendenza a cedere il protone, e una base è tanto più forte quanto maggiore é la tendenza a fissare il protone.
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-8

9
BOX 1.1 Alcune coppie coniugate acido-base
________________Acido_______________Base___________ Più forte HClO ClO4 4
- Più debole H2SO4 HSO4
-
HCl Cl-
H O3 2+ H O
HSO4- SO4
=
HF F-
CH COOH CH COO-3 3
Al3+,H2O AlOH +2
CO2 acq. HCO3-
H2S HS-
NH4 3+ NH
HCO3- CO3
=
HS- S=
H O OH-2
Più debole OH- O= Più forte
Nel BOX 1 gli acidi sono incolonnati in ordine di forza decrescente. E' ovvio che tanto più forte è l'acido tanto più debole è la sua base coniugata. Lo schema di reazione di cui sopra ha un significato del tutto generale; esso comprende reazioni che nello schema di Arrhenius venivano contraddistinte con nomi diversi: dissociazione, neutralizzazione o idrolisi, e considerate concettualmente diverse. La dissociazione è caratteristica di un acido o di una base disciolti in acqua:
HF <====> H+ + F-
NH3 + H2O <====> NH4OH <====> NH4+ + OH-
La neutralizzazione é la reazione di una acido con una base e dà origine ad acqua e ad un sale:
HF + NaOH <====> NaF + H2O L'idrolisi viene considerata da Arrhenius una decomposizione più o meno spinta, subita da sali provenienti da acidi e da basi di forza diversa o da acidi e basi deboli per azione dell’acqua:
CH3OONa + H2O <====> CH3COOH + NaOH Nella teoria di Bronsted, tutti questi tipi di reazione rientrano nell’unico schema di reazione
A1 + B2 <====> A2 + B1 quando si comprende anche il solvente nella classe degli acidi o delle basi. Le precedenti reazioni divengono infatti:
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-9

10
HF + H2O <====> H3O+ + F-
NH3 + H2O <====> NH4+ + OH-
HF + OH- <====> F- + H2O CH3COO- + H2O <====> CH3COOH + OH-
1.3.1.1.- Le costanti di ionizzazione a stadi di acidi poliprotici Quando un acido può perdere più di un atomo di idrogeno, sono interessati parecchi equilibri ed esistono diverse espressioni indipendenti dalle costanti di equilibrio. Per l'acido fosforico hanno luogo tre successive dissociazioni e sono richieste tre indipendenti costanti di equilibrio per descrivere i dati sperimentali H3PO4 <===> H2PO4
- + H+ [H+][H2PO4-] = Ka1 [H3PO4]
H2PO4- <===> HPO4
= + H+ [H+][HPO4=] = Ka2 [H2PO4
-] HPO4
= <===> HPO4≡ + H+ [H+][PO4≡] = Ka3 [HPO4
=] 1.3.2.0.- Il Potere Tamponante In Ecotossicologia il potere tamponante, ossia la capacita’ di una miscela di un sistema composto da un acido debole e dalla sua base coniugata di mantenere il valore di pH praticamente costante anche in presenza di relativamente elevate quantita’ di un acido forte, e’ un fattore assai importante.
Basti pensare all’importanza del sistema carbonati/bicarbonati non solo nlle acque marine e non ma anche negli stessi liquidi organici. Il sangue stesso, per scendere alla struttura biologica, e’ tamponato grazie a questi sistemi.
Se non vi fosse il potere tamponante, gli organismi dovrebbero sviluppare sistemi di gran lunga piu’ costosi, in termini energetici, di quello che la natura ha loro fornito. E' conveniente definire matematicamente il potere tamponante in termini differenziali: β = dCb/dpH (1.12) L'aggiunta di Cb moli di una base forte ad un litro di tampone aumenta la concentrazione analitica del componente basico (acetato di sodio) di una quantità dCb a spese del componente acido (acido acetico)
Poiché 1'aggiunta di un acido forte produce l'effetto inverso, possiamo scrivere β in termini di concentrazione di acido forte Ca aggiunto: β = -dCa/dpH (1.13) In ambedue i casi β è positivo. Il potere tamponante di un "acido monoprotico" e della sua base coniugata ha una espressione matematica semplice che può essere ottenuta facilmente differenziando il bilancio di carica rispetto al pH. Consideriamo una soluzione contenente Cb moli di NaOH, Ca moli di HCl e C moli di un acido debole sotto forma sia di HA che di A-. Gli equilibri in gioco sono:
[H+][A-] = Ka[HA] (1)
[H+][OH-] = Kw (2) I bilanci di massa sono:
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-10

11
[Na+] = Cb (1.14)
[Cl-] = Ca (1.15) [HA] + [A-] = C (1.16) Il bilancio di carica è:
[H+] + [Na+] = [OH-] + [Cl-] + [A-] (1.17) Combinando la (1) e la (1.16) si ottiene:
[A-] = C Ka / (Ka + [H+]) (1.18) Sostituendo la (2), la (1.14), la (1.15) e la (1.18) nella (1.17) otteniamo:
Cb = Ca + Kw - [H+] + C Ka (1.19)
[H+] Ka + [H+] Il potere tamponante è ottenuto differenziando la (1.19) rispetto al pH, ma prima è necessario esprimere β come derivata rispetto ad [H+]. Poiché
pH = -log10[H+] = - ln[H+]/2,303 l’equazione (1.12) può essere scritta come segue:
β =dCb
dpH=
dCb
d H +[ ]d H +[ ]dpH
= −2,303 H +[ ]dCb
dpH (1.20)
Differenziano la (1.19) rispetto ad [H+], mantenendo costante Ca ed usando la (1.20), otterremo l’espressione del “potere tamponante”:
β = 2,303Kw
H +[ ]+ H +[ ]+CKa H +[ ]
Ka + H +[ ]( )2
⎧ ⎨ ⎩
⎫ ⎬ (1.21)
⎭ Così, se si conosce il pH del tampone, è possibile calcolare il suo potere tamponante sostituendo direttamente nella (1.21), nella quale i primi due termini sono legati all’effetto tamponante dell’acqua, mentre il terzo alla coppia coniugata acido-base utilizzata. 1.3.2.1.- Sistemi di tamponi multipli Tali sono soluzioni contenenti sali di acidi poliprotici o miscele di acidi e basi monoprotici. Il calcolo del pH e’, comunque, semplice. In natura sono noti moltissimi sistemi multipli di temponi.
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-11

12
Basta pensare ai gia’ citati carbonati/bicarbonati, ai borati, ai silicati ecc. contamporaneamente presenti nelle acque superficiali; od ai sistemi carbonato/bicarbonato, acetati, proprionati, solfuri/solfuri acidi ecc. propri delle fermentazioni biologiche aerobie od anaerobie 1.3.2.2.- Potere tamponante di tamponi multipli Se un sistema è formato da più acidi (HA, HA’, ecc.) il potere tamponante risulta additivamente dal contributo delle singole coppie coniugate. Si ha cioè: β = βH2O + BHA + βHA’ + ..... (1.22) dove βH2O = 2,303 ([H+] - [OH-]) (1.23) βHA = 2,303 (C Ka [H+]) / (Ka + [H+])2 (1.24) con espressioni simili anche per HA’ ecc. Gli acidi possono essere neutri (come l’acido acetico), cationi (come NH4
+) o anioni (come HPO4-).
1.4.0.0.- Rappresentazione grafica del sistema acido/base 1.4.1.0.- Il diagramma di concentrazione logaritmico
Rappresentare funzioni mediante diagrammi logaritmici e' cosa molto comune nella scienza poiche', con pochi tratti, si possono ottenere moltissime informazioni sull'andamento di un fenomeno ed in tempi immediati.
La chimica ambientale mutua dalla chimica tradizionale molte di queste descrizioni diagrammatiche del tipo: concentrazione di una specie in funzione di altre. La variabile indipendente e', di solito, rappresentata dal fattore agente (il pH o -log della concentrazione degli ioni Idrogeno - in g ioni L-1, per l'acidita', lo ione precipitante per i composti poco solubili ed ancora il pH, la concentrazione del complessante per gli equilibri complessi ecc.).
Un diagramma logaritmico assai importante e' rappresentato dal sistema acido dissociato-acido indissociato (o meglio, dall'acido e dalla sua base coniugata) che consente di conoscere le "specie" chimiche derivate dalla sequenza di equilibri ad un certo valore del pH.
Poiche' sono derivate da equilibri termodinamicamente corretti le concentrazioni (o attivita' ) delle specie in gioco non sartanno mai zero ma esistera' sempre una infinitesima quantita' di qualunque specie del sistema considerato anche quando diagrammaticamente il suo valore e' trascurabile.
Infatti in chimica non esiste la concentrazione nulla quando siamo di fronte a sistemi in equilibrio.
L'equilibrio puo' essere totalmente spostato in una direzione per cui il 99.999999% ha reagito ma vi sara' sempre almeno un 0.0000001% del composto che rimane inalterato e che dinamicamente si sposta nel sistema in equilibrio.
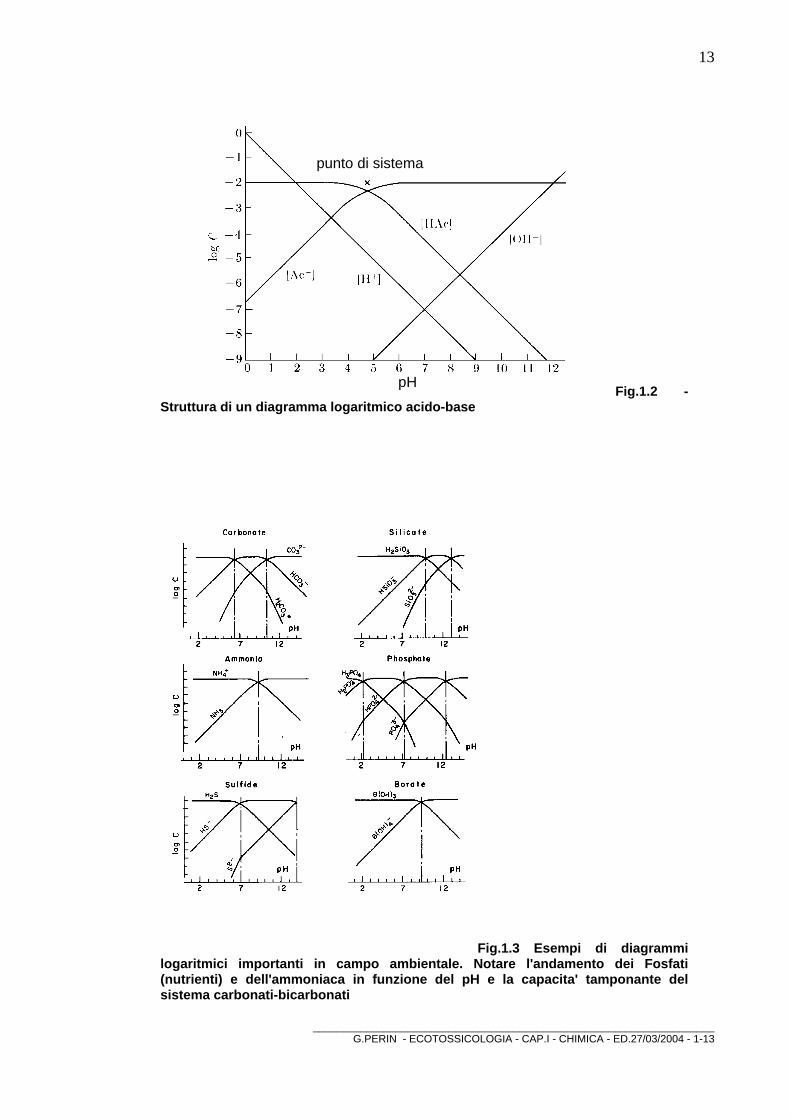
In figura è riportato il diagramma ottenuto riportando i logaritmi delle concentrazioni delle varie specie in funzione del pH per una soluzione 0,02 M di acido acetico.
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-12
13
punto di sistema
pH Fig.1.2 - Struttura di un diagramma logaritmico acido-base
Fig.1.3 Esempi di diagrammi logaritmici importanti in campo ambientale. Notare l'andamento dei Fosfati (nutrienti) e dell'ammoniaca in funzione del pH e la capacita' tamponante del sistema carbonati-bicarbonati
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-13

14
1.5.0. Equilibri di formazione dei complessi Per [H+] si ottiene una linea retta di pendenza -1 dalla definizione del pH: log [H+] = - pH. Per [OH-], il prodotto ionico dell'acqua dà una linea retta di pendenza +1 dalla definizione: log[OH-] = pH - pKw Dalla combinazione del bilancio di massa: [HAc] + [Ac-] = C e dell'equilibrio di dissociazione: [H+][Ac-]= Ka[HAc] si ottengono: [Ac-] = C Ka/(Ka + [H+]) e [HAc] = C [H+]/(Ka + [H+]) i cui logaritmi appaiono in figura come due segmenti rettilinei congiunti da una breve curva. Viene detto "system point" il punto per cui pH= pKa e la concentrazione è uguale a C (pH = 4,75 e log C = -2). A sinistra di tale punto, [H+] è grande rispetto a Ka e le relazioni si riducono a: log [Ac-] = log C - pKa + pH log [HAc] = - log C che sono rappresentate da una linea di pendenza -1 e da una linea orizzontale. A destra del punto, Ka è grande rispetto a [H+] e le relazioni si riducono a: log [Ac-] = log C log [HAc] = log C + pK - pH che sono rappresentate da una linea orizzontale e da una linea di pendenza -1, entrambe passanti per il punto. Vicino al punto, sia [H+] che Ka hanno circa lo stesso valore e le variazioni di [HAc] e [Ac-] in funzione del pH sono date dalle relazioni originarie. Le curve si intersecano dove [HAc] = [Ac-] ad una concentrazione 1/2C e a pH = pKa, cioè a 0,30 unità logaritmiche al di sotto del punto sistema.
BOX 1.2 Costruzione di un diagramma di concentrazione logaritmico Per costruire un diagramma come quello in figura, si segna per prima cosa il "system point" a pH = pKa e concentrazione C, si tracciano poi una linea orizzontale e le linee di pendenza +1 e -1 attraverso tale punto. Si congiungono infine le linee appropriate con brevi curve che si incrociano in un punto a 0,3 unità logaritmiche al di sotto del "system point". Le linee per [OH-] e [H+] hanno pendenze +1 e -1 e si intersecano a pH = 7 alla concentrazione di 10-7. Notiamo che variando la concentrazione della soluzione, le curve per [Ac-] e [HAc] si spostano assieme su o giù, ma non si spostano le curve per [H+] e [OH-].
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-14

15
Una qualsiasi specie in soluzione, formata dalla combinazione di due o più specie più semplici che possono anche esistere indipendentemente nella soluzione, è chiamata "complesso". Un complesso è usualmente uno ione positivo o negativo, ma può anche essere una molecola neutra.
La stabilità che viene dimostrata dai complessi metallici in soluzione varia notevolmente: i metalli alcalini ed alcalino-terrosi formano complessi molto deboli che sono meglio descritti come "paia di ioni", poiché il catione e l'anione mostrano un'affinità l'uno per l'altro soltanto leggermente più elevata di quella che può essere spiegata dalla semplice interazioneelettrostatica.
Dall'altro estremo vi sono i complessi molto stabili dell’ammoniaca con il cobalto III: ad es. Co(NH3)3+ non reagisce nemmeno con l'HCl bollente o con 1'H2SO4 concentrato caldo.
La formazione di complessi e' basilare ai fini della comprensione della tossicita' che un composto chimico (prevalentemente inorganico ma il concetto puo' essere applicato a qualunque sostanza) esplica su un substrato biologico.
Infatti la tossicita' di un composto - come sara' discusso nei prossimi capitoli - e' condizionata dalla speciazione del composto medesimo.
Vale a dire dalla "forma" o specie con la quale il composto si avvicina alla membrana cellulare.
Come tossicita' si intende appunto l'azione "interna" alla cellula del composto chimico (anche se biologicamente mediato ossia se perviene al citoplasma per grazia o causa dell'azione di microorganismi - esempio.: tossine batteriche ecc.); per esplicare tale azione il composto deve "passare" la membrana cellulare e penetrare il citoplasma.
E' allora intuitivo che i composti coordinati con altre strutture leganti (i complessi, appunto) troveranno una maggior o minor resistenza nell'attraversare la membrana a seconda dei "gruppi" chimici ad essi legati o complessati.
Alle volte la speciazione (complessazione) limita o impedisce il passaggio intramembrana; alle volte, invece, lo facilita poiche' la complessazione puo' ridurre la polarita' del composto (o esaltarla) facilitando legami tra gruppi a polarita' opposta. In altri casi la polarita' si inverte con corrispondenti interazioni laddove la membrana ha siti polarizzati nello stesso verso o nel verso opposto. Un classico esempio e' dato dai complessi del Cadmio e del Mercurio in aree estuarine ove la salinita' eccede 0.1 moli L-1.
Con un semplice calcolo si puo' dimostrare che, in quelle condizioni il Cadmio libero (Cd++) non esiste se non a livello di ultratracce (in funzione necessariamente dell'equilibrio termodinamico v. Vedi BOX 1.3 e Fig.1.7) ma che i complessi prevalenti sono il CdCl2 (neutro) e i complessi CdCl3- e CdCl-- (con una e due cariche negative).
E' ovvio che il complesso neutro verra' facilitato nella sua entrata in aree della membrana cellulare polarizzate poiche' non interagira' con il campo elettrico provocato dalle molecole terminali lipidiche e/o proteiche caricate mentre il complesso a forte carica negativa incontrera' difficolta' ad entrare il citoplasma per la repulsione coulombica descritta in Fig.1.4.
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-15

16
Fig. 1.4 Interazione e repulsioni tra cariche elettriche nel caso dei colloidi. Il principio e' comunque applicabile a tutti i sistemi ove si incontrano strati polarizzati
Ne consegue che la tossicita' del Cadmio in acqua di mare o salmastra non e' dovuta al Cadmio ione (Cd++) ma al complesso neutro od ai complessi carcati negativamente (che in parte possono penetrare la membrana). In maniera anloga si comporta il mercurio.
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-16

17
Fig.1.5 Modalita' di "entrata" del metallo complessato (ML) nella cellula attraverso i vari "canali" della membrana cellulare. Notare il processo di endocitosi (4).
I complessi "mononucleari' consistono di uno ione metallico centrale a cui è legato un certo numero di gruppi neutri anionici chiamati "leganti". Il numero di leganti attaccato allo ione centrale è chiamato numero di coordinazione.
Molti leganti, di struttura relativamente semplice, come gli ioni Cl-, OH-, CN-,o le molecole H2O, NH3, R-NH2, NO, occupano un solo posto nella sfera di coordinazione. Tali leganti si dicono monodentati. Leganti che avendo nelle loro molecole più di un gruppo atto a coordinarsi, atto cioè a cedere una coppia di elettroni, sono detti polidentati: esempi importanti sono le diammine gli amminoacidi, gli ossiacidi, le poliammine, i polialcoli e l'EDTA. Un legante polidentato nel formare un complesso con un catione blocca due o più posti di coordinazione, dando origine a strutture cicliche dotate di straordinaria stabilità: i complessi di tale tipo sono detti composti chelati e i leganti polidentati in grado di formarli si dicono agenti chelanti.
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-17

18
Fig.1.6 Agenti complessanti comunemente riscontrati nelle acque superficiali e
profonde
Come negli equilibri acido-base si ha lo scambio o il trasferimento del protone (e negli equilibri di ossido-riduzione di elettroni), cosi negli equilibri di complessazione la particella scambiata è il legante.
La stabilità di un complesso viene definita quantitativamente dalla costante di stabilità o dalla costante di formazione (Kf), relativa all'equilibrio scritto nel verso che porta alla formazione del complesso
M + nL <====> Mln (1.25) da cui Kf = [MLn]/[M][L]n (1.26)
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-18

19
1.5.1.- Le costanti di formazione a stadi per i complessi Gli equilibri di formazione dei complessi sono usualmente scritti nella direzione inversa degli acidi poliprotici. Per esempio, la formazione dei 4 complessi del cloruro di cadmio possono essere rappresentati dai 4 equilibri: Cd++ + Cl- <===> CdCl+ [CdCl+] = K1 [Cd++][Cl-] CdCl+ + Cl- <===> CdCl2 [CdCl2] = K2 [CdCl+][Cl-] CdCl2 + Cl- <===> CdCl3- [CdCl3-] = K3 [CdCl2][Cl-] CdCl3- + Cl- <===> CdCl4= [CdCl4=] = K4 [CdCl3-][Cl-] oppure: Cd++ + Cl- <===> CdCl+ [CdCl+] = β1 [Cd++][Cl-] Cd++ + 2Cl- <===> CdCl2 [CdCl2]= β2 [Cd++][Cl-]2
Cd++ + 3Cl- <===> CdCl3- [CdCl3-]= β3 [Cd++][Cl-]3
Cd++ + 4Cl- <===> CdCl4= [CdCl4=]= β4 [Cd++][Cl-]4
Occasionalmente il reciproco della costante di formazione complessiva per il complesso più elevato è indicato come "costante di instabilità" del complesso (notare che β1=K1; β2=K1K2; β3=K1K2K3; β4=K1K2K3K4 ). Per il complesso cloruro di cadmio questa dovrebbe essere CdCl4= <====> Cd++ + 4Cl- [Cd++][Cl-]4 =Kinst. [CdCl4=] Tale costante non può essere usata da sola a meno che le concentrazioni di tutti gli altri complessi siano trascurabili: il che è raramente il caso. 1.5.2. - Formazione di complessi da solidi
Chiarita l'importanza della complessazione - che noi ambientalmente chiameremo speciazione - e ricordando i principi dell'equilibrio chimico, possiamo affermare che anche un composto classificato come insolubile mette "a disposizione" un certo numero di ioni per confortare i principi termodinamici dell'equilibrio.
Se ci si trova in presenza di agenti complessanti in grado di appropriarsi (per maggiore capacita' competitiva = forza chimica = potenziale chimico = attivita' maggiore), la formazione dei complessi solubilizza sempre piu' un composto "insolubile". E' il caso, gia' accennato degli estuari ove un composto che arriva sotto forma insolubile, viene "sciolto" dalla formazione dei complessi con i cloruri presenti nell'acqua salata.
Cosi' quando un metallo forma un sale poco solubile, come pure una serie di complessi con un dato anione, gli equilibri sono spesso dati in termini delle reazioni che formano i vari complessi del sale.
Per esempio, nel sistema cloruro di argento alcuni degli equilibri sono: AgCl(s) = Ag+ + Cl- [Ag+][Cl-] = Ks0AgCl(s) = AgCl(acq) [AgCl] = Ks1AgCl(s) + Cl- = AgCl2- [AgCl2-] = Ks2[Cl-]
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-19

20
AgCl(s) + 2Cl- = AgCl3= [AgCl3=] = Ks3[Cl-]2
AgCl(s) + 3Cl- = AgCl4≡ [AgCl4≡ ] = Ks4[Cl-]3
AgCl(acq) indica le molecole di sale non dissociato disciolte in soluzione, in modo distinto dal sale solido e dagli ioni dissociati.
Le costanti di formazione a stadi (K) o le costanti di formazione globali (β) per i complessi del cloruro di argento possono essere ottenute dalle appropriate combinazioni delle costanti Ks sopradescritte. Per esempio:
K3 = Ks2/Ks3; β3=Ks3/Ks0; K1 = Ks1/Ks0 1.5.3 - Complessi mononucleari
Consideriamo le seguenti serie di equilibri reversibili tra uno ione centrale metallico M ed un legante L, in grado di formare con il metallo diversi complessi mononucleari ML, ML2, ML3 ... MLn, dove n può assumere i valori 1, 2, 3, ... fino ad un massimo corrispondente al numero di coordinazione. Si avrà allora: M + L <====> ML e [ML] = K1 [M][L] ML + L <====> ML2 e [ML2]= K2 ML][L] ML2 + L <====> ML3 e [ML3]=K3[ML2][L] ................................................................................................... MLn-1 + L <====> MLn e [MLn] = Kn [MLn-1][L] Le costanti K1, K2, ... Kn sono dette costanti di stabilità parziali. E' più conveniente tuttavia usare le costanti di formazione, o stabilità, globali, che corrispondono agli equilibri: M + L <====> ML e [ML] = β1 [M][L] M + 2L <====> ML2 e [ML2] = β2 [M][L]2
.......................................................................................... M + nL <====> MLn e [MLn]= βn [M][L]n
con βl = K1. β2 = K1K2, .... βn = K1K2 .... Kn Non vengono indicate le molecole coordinate del solvente: quest’ultimo si trova generalmente in eccesso tanto forte, rispetto ai componenti disciolti, da potersi considerare costante la sua attività. Nell’esempio riportato qui di seguito viene analizzato un interessante caso di contaminazione da Cadmio in presenza di cloruri. Teniamo presente che i cloruri in acqua di mare sono presenti a livello di circa 0.5 - 0.8 moli.L-1 e che i diversi complessi del cadmio con i cloruri hanno tossicita’ assai diversa (come verra’ analizzato piu’ propriamente nel capitolo relativo alla tossicita’ in funzione della speciazione) .
Nel primo esempio esamineremo le concentrazioni dei vari complessi in presenza di rilevanti quantita’ di cloruri (esempio: estuari) o in presenza di bassa quantita’ di cloruri quali quelli ad esempio prodotti dalla dissociazione di cloruro di Cadmio.
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-20

21
Un modo elegante e molto usato per descrivere la distribuzione dei vari complessi (specie) in funzione della quantita’ di legante (nel nostro caso i cloruri) e’ quello dei diagrammi di distribuzione.
Rimanendo nel caso del Cadmio (e non e’ casuale la scelta poiche il cadmio e’ uno dei piu’ importanti metalli sotto il profilo tossicologico e cancerogenico) il diagramma di distribuzione e’ calcolabile secondo quanto riportato nell’esempio che segue.
BOX 1.3 Calcolo della frazione di cadmio presente nelle varie specie di complessi-cadmio cloruro in funzione di [Cl-]. Il metodo matematico è una generalizzazione di quanto descritto nell'es.1: [CdCl+] = β1 [Cd2+][Cl-] β1 = K1 = 21 [CdCl2] = β2 [Cd2+][Cl-]2 β2 = K1K2 = 166 [CdCl3-] = β3 [Cd2+][Cl-]3 β3 = K1K2K3 = 204 [CdCl4=] = β4 [Cd2+][Cl-]4 β4 = K1K2K3 K4= 71,5 Se C è la concentrazione analitica del cadmio, allora il bilancio di massa per il cadmio è: C = [Cd2+] + [CdCl+] + [CdCl2] + [CdCl3-] + [CdCl4=] = [Cd2+] (1 + β1[Cl-] + β2[Cl-]2 + β3[Cl-]3 + β4[Cl-]4) La frazione di cadmio nelle varie specie può essere calcolata in funzione di [C1-]: α0 = [Cd2+] = 1/(1+ β1[Cl-] + β2[Cl-]2 + β3[Cl-]3 + β4[Cl-]4) C α1 = [CdCl+] = β1 [Cl-]α0 C α2 = [CdCl2] = β2 [Cl-]2α0 C α3 = [CdCl3-] = β3 [Cl-]3α0 C α4 = [CdCl4-] = β4 [Cl-]4α0 C La frazione di cadmio presente in ciascuna specie è indipendente dalla quantità totale di cadmio in soluzione, e dipende soltanto dalla concentrazione di cloruri. Notiamo che[Cl-] è la concentrazione effettiva e non quella analitica di cloruro ed è data dal secondo bilancio di massa: C1 = [Cl-] + [CdCl+] + 2[CdCl2] + 3[CdCl3-] + 4[CdCl4=] E' possibile rappresentare gli equilibri di formazione degli ioni complessi mediante gli stessi metodi grafici usati per gli acidi poliprotici, salvo il fatto che la variabile fondamentale è la concentrazione del legante: nel nostro caso [Cl-], invece del pH. Dalla figura si può ottenere la risposta all'es.l, in cui [Cl-] è 1 M ----> pCl = 0; le frazioni delle varie specie sono lette in corrispondenza dell'incontro di tale ordinata con le varie curve e moltiplicate per la concentrazione analitica totale del cadmio, che nell'es. 1 è 1,0.10-2 mole/l: La curva più a sinistra è α4, la frazione di Cd presente come CdCl4=; la curva successiva a destra è α4
+ α3, la frazione di Cd presente come CdCl4= e CdCl3-; quella successiva è α4 + α3 + α2 e l’ultima curva a destra è α4 + α3 + α2 + α1. Tali curve divideranno una linea verticale tracciata ad un dato valore di [Cl-] in parecchi segmenti. Per esempio, a [Cl-] = 1,00 (pCl= 0) una linea verticale è tagliata a 15,4% dalla Ia curva, indicando che 15,4% del cadmio è presente come CdCl4=. La distanza dalla curva successiva è α3 = 44,0%, la distanza dalla curva seguente è α2 = 35,8%, la distanza tra la IIIa e IVa curva è α1 = 4,6% e la distanza tra la IVa curva e la linea α = 1,00 è α0 = 0,2%.
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-21

22
Fig. 1.7 Distribuzione dei metalli nei complessi dell'Acido Etilendiamino Tetraacetico in funzione della concentrazione dell'agente complessante
Fig. 1.8 Distribiuzione delle specie del Mercurio in ambiente estuarino in funzione della salinità
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-22

23
BOX 1.4
1.6.0. - Precipitazione e prodotto di solubilita’ 1.6.1. - Solubilita’ di sali semplici in acqua 1.6.1.1.- Prodotto di solubilità Un'altra costante di equilibrio, che è matematicamente simile al prodotto ionico dell'acqua, è il prodotto di solubilità di un sale. Se un sale ionico è presente in eccesso all'equilibrio con la sua soluzione satura (cioè è presente anche come corpo di fondo), le concentrazioni degli ioni saranno governate dalla legge di equilibrio. Consideriamo ad esempio il sale cloruro di argento:
AgCls ⇔ Ag+ + Cl−
Il termine (s) che segue la formula del composto indica che esso è presente come solido puro e quindi la sua attività è esattamente unitaria. L'espressione dell'equilibrio è:
KS00 = Ag +{ } Cl −{ } (1.27)
Sostituendo le attività con le concentrazioni:
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-23

24
KS00 = Ag+[ ]Cl −[ ] (1.28)
K°s0 è detta costante prodotto di solubilità del sale. Le concentrazioni degli ioni possono essere usate per calcoli approssimati per valori di concentrazione inferiori a 10-3 mole/l. L'equilibrio tra un sale ionico solido e la sua soluzione in acqua è governata dall’espressione del prodotto di solubilità. Il cloruro di argento si scioglie debolmente in acqua per dare il catione Ag+ e l’anione Cl-. La reazione: AgCl(s) <====> Ag+ + Cl- è rappresentata dall'espressione di equilibrio: [Ag+][Cl-] = Ks0 (1.29) dove Ks0 è la costante prodotto di solubilità dell'AgCl (l’attività del solido puro è unitaria; la concentrazione del solido puro è costante ed è inclusa nella costante di equilibrio). Il prodotto di solubilità si applica soltanto alle soluzioni sature. Se S moli di AgCl si disciolgono in acqua per formare 1 litro di soluzione, un bilancio di massa del catione e dell'anione dà [Ag+]= S; [Cl-] = S e dal prodotto di solubilità: [Ag+][Cl-] = Ks0 = S2 (1.30) La solubilità di un sale ionico in acqua pura dipende soltanto dal prodotto di solubilità, purché solo questi due ioni siano in soluzione. Se le cariche dei due ioni non sono eguali, la solubilità è ancora determinata solo dal P.S.. ma le equazioni sono un po' più complicate. un sale MzXy , che dà soltanto gli ioni M+y e X-z in soluzione acquosa, la reazione MzXy (s) <====> zM+y +y X-z (1.31) ha un'espressione per il prodotto di solubilità:
[M+y]z[X-z]y = Ks0
I bilanci di massa sono:
[M+y] = zS [X-z] = yS da cui:
(zS)z(yS)y = Ks0 (1.32)
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-24

25
BOX 1.5 Prodotti di solubilità a 25°C per sali che formano soltantodue specie ioniche principali in soluzioni acquose diluite Ioni di carica uguale Ioni di carica diversa Sale pKs0 Sale pKs0* TlCl 3,72 Ag2SO4 4,80 AgBrO3 4,28 Ag2C2O4 11,30 Hg2SO4 6,17 BaF2 5,76 SrSO4 6,55 Cu(IO3)2 7,13 AgIO3 7,52 MgF2 8,18 PbSO4 7,80 SrF2 8,54 AgCl 9,75 CaF2 10,40 BaSO4 9,96 Mg(OH)2 10,74 AgSCN 12,00 Pb(IO3)2 12,59 AgBr 12,28 Hg2Cl2 17,88 AgI 16,08 Ce(IO3)3 9,50 La(IO3)3 11,21 * Ks0 è stato estrapolato a forza ionica zero ** Hg2
2+ è uno ione singolo
BOX 1.6 Prodotto di solubilità del solfato di bario La sua solubilità in acqua pura (determinata da misure di conducibilità a 10°C) 1,0.10-5M. I bilanci di massa danno: [Ba2+] = 1,0.10-5
[SO42-] = 1,0.10-5
La sostituzione nel prodotto di solubilità dà: Ks0 = [Ba2+][SO4
2-] = 1,0.10-10
BOX 1.7 Solubilità del fluoruro di calcio in acqua Il prodotto di solubilità di CaF2 è 4,0.10-11. La reazione che avviene è: CaF2 (s) <====> Ca2+ + 2F-
e l'espressione del prodotto di solubilità è [Ca2+][F-]2 = 4,0.10-11
Usando i bilanci di massa: [Ca2+] = S [F-] = 2S otteniamo: S = 2,15.10-4 M pari a 1,68.10-2 g/l
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-25

26
1.6.2.- Effetto dello ione in comune In una soluzione satura di un sale ionico, come BaSO4 la concentrazione degli ioni è governata dal prodotto di solubilità: Ks0 = [Ba2+][SO4
2-] anche se altri sali sono disciolti in soluzione. Se la concentrazione di uno degli ioni costituenti il sale ionico in questione è aumentata per aggiunta di BaCl2 o Na2SO4, la concentrazione dell’altro deve essere diminuita per mantenere valido l'equilibrio. In altre parole, l'aggiunta di uno ione in comune porta ad una diminuzione della solubilità del sale, purché non si formino ioni complessi. 1.6.3 –Solubilità dei solfuri I solfuri metallici occupano un posto centrale nello schema tradizionale dell'analisi qualitativa. Per tale ragione in quasi tutti i testi elementari di chimica analitica viene discusso L'effetto dell'acidità sulla loro solubilità: sfortunatamente tali discussioni non sono molto realistiche per molte ragioni.La più importante è che raramente nelle condizioni in cui precipitano i solfuri nell'analisi qualitativa si raggiunge l'equilibrio. A causa dell'elevata concentrazione di acido cloridrico presente nelle soluzioni, si formano cloruri complessi, che portano la concentrazione di ione metallo libero a valori molto più bassi della sua concentrazione analitica.
La formazione di solfuri complessi e l'idrolisi degli ioni metallici possono avvenire estesamente specie in soluzioni alcaline. Tutti questi fattori tendono ad aumentare la solubilità dei solfuri di un fattore a volte di 105 o più al di sopra del valore calcolato trascurando la formazione dei complessi.
La formazione dei solfuri complessi è stata studiata solo per pochi sistemi ed esiste qualche dubbio sulla validità delle costanti riportate. Anche gli equilibri di ionizzazione dell'acido solfidrico sono difficili da misurare ed i valori di Ka2 trovati in letteratura variano di un fattore 100. Anche se i calcoli di equilibrio della solubilità dei solfuri non possono rappresentare la realtà delle condizioni incontrate nell'analisi qualitativa, essi forniscono esempi interessanti che considerano parecchi tipi di equilibri: dissociazione dell'acido diprotico H2S e formazione di complessi cloruri, idrossidi e solfuri degli ioni metallici.
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-26

27
BOX 1.8 Calcolo della solubilita’ dell’ HgS in una soluzione di cloruri 0,10 M Gli equilibri di dissociazione dell’H2S sono: [H+][HS-] = 1,0.10-7 [H2S] (1) [H+][S=] = 1,2.1013 [HS-] (2) In soluzione satura di solfuro valgono i seguenti equilibri: [Hg2+][S=] = 1,6.10-52 = Ks0 logKs0 = 51,8 (3) [HgS2
=] = 0,58[S=] logK = 0,58 (4) [Hg(OH)2] = 2,5.1011[HgOH+][OH-] logK = 7,0 (5) Le costanti per la formazione dei complessi di idrossido di mercurio sono: [HgOH+] = 2,0.1010[Hg2+][OH-] logK1 = 10,30 (6) [Hg(OH)2] = 2,5.1011[HgOH+][OH-] logK1 = 11,40 (7) Il diagramma di distribuzione per questi complessi di cloruro mercurico sono: [HgCl+] = 5,5.106[Hg2+][Cl-] logK1 = 6,74 (8) [HgCl2] = 3,0.106[HgCl+][Cl-] logK2 = 6,48 (9) [HgCl3-] = 7,8[HgCl2][Cl-] logK3 = 0,85 (10) [HgCl4=] = 10,0[HgCl3-][Cl-] logK4 = 1,00 (11) Il bilancio di massa per il mercurio è: S = [Hg2+] + [HgS2
=] + [Hg(SH)2] + [HgOH+] + [Hg(OH)2] + [HgCl+] + [HgCl2] + [HgCl3-] + [HgCl4=] (12) Il bilancio di massa per il solfuro è: S = [S=] + [HS-] + [H2S] + [2HgS2
=] + 2[Hg(SH)2] (13) Il bilancio di massa per il cloruro è: 0,10 = [Cl-] + [HgCl+] + 2[HgCl2] + 3[HgCl3-] + 4[HgCl4=] (14) e la condizione protonica, che può essere ottenuta combinando il bilancio di carica con i tre bilanci di massa, è: 0,10 + [HgOH+] + 2[Hg(OH)2] + [OH-] = [H+] +[HS-] + 2[H2S] + 2[Hg(SH)2] (15) La prima approssimazione che possiamo fare è che la solubilità, e quindi la concentrazione, di tutte le specie che contengono Hg e S sia piccola in confronto con la concentrazione analitica di HCl; ne deriva che: [Cl-] = 0,10 (14a) [H+] = 0,10 (15a Come già detto, [Hg2+] e [HgCl+] sono trascurabili in confronto ai complessi predominanti, per cui: S = [HgCl2] + [HgCl3-] + [HgCl4=] (12a) Poiché la soluzione è fortemente acida, [HS-] e [S=] saranno trascurabili in confronto a [H2S], Dalla (4), [HgS2
=] è circa la metà di [S=] e quindi trascurabile.. Combinando la (4), la (5), la (1) e la (2) otteniamo: [Hg(SH)2] = 1,0.10-14[H2S] che mostra che anche questo termine è trascurabile nella (13), dando così la nuova equazione approssimata: S = [H2S] (13a) Sostituendo (14a) in (10) e (11) otteniamo: S = [HgCl2] + 0,708[HgCl2] + 0,708[HgCl2] = 2,416[HgCl2] (16) Le equazioni (13a) e (16) forniscono 2 delle 3 equazioni che correlano S, [H2S] e [HgCl2]. La relazione finale è ottenuta combinando (1), (2), (3), (8) e (9): [H2S][HgCl2] = 2,2.10-19[H+]2[Cl-]2 (17) Se sostituiamo (14a), (15a), (13a) e (16) in (17) otteniamo: S = 7,15.10-12
Questa solubilità è estremamente piccola, troppo piccola per essesere misurata direttamente. Il solfuro di mercurio può essere disciolto in acqua regia poiché questa miscela di acido nitrico e cloridrico ossida l'H2S in soluzione liberando zolfo e quindi spostando l'equilibrio. Il prodotto di solubilità di HgS è misurato potenziometricamente usando un elettrodo a mercurio. In una soluzione di nota concentrazione di H2S (normalmente satura) e nota acidità, la concentrazione di solfuro può essere calcolata dagli equilibri (1) e (2). Il potenziale della cella dà la concentrazione dello ione mercurio libero. Il prodotto di solubilità può essere calcolato da questi dati. Un calcolo in cui siano trascurati tutti i complessi e siano considerati soltanto gli equilibri (1), (2) e (3) non rappresenta la realtà di questo problema, giacché la solubilità calcolata è: S = 1,15.10-17 cioè
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-27

28
105 volte più piccola del valore ottenuto sopra (7,15.10-12). Una volta nota la solubilità del solfuro di mercurio. possono essere facilmente calcolate le concentrazioni di tutte le altre specie: da (14a) [Cl-] = 0,10 da (15a) [H+] = 0,10 da (13a) [H2S] = 7,15.10-12
da (1) [HS-] = 7,15.10-18
da (2) [S=] = 8,6.10-30
da (3) [Hg2+] = 1,86.10-23
da (4) [HgS=] = 5,0.10-30
da (5) [Hg(SH)2] = 5.0.10-25
da Kw [OH-] = 1,00.10-13
da (6) [HgOH+] = 3,7.10-26
da (7) [Hg(OH)2] 0 9,3.10-28
da (8) [HgCl+] = 1,0.10-17
da (9) e (16) [HgCl2] = 3.0.10-12
da (10) [HgCl3-] = 2,1.10-12
da (11) [HgCl4=] = 2,1.10-12
Si noti che le concentrazioni di molte specie sono "impossibilmente" piccole, meno di uno ione in 106 litri. Il bilancio (12) è verificato entro l'1%, (13) ad 1 parte su 105, (14) e (15) ad una parte su 1010. 1.7.0.- Equilibri di ossido - riduzione Mentre nelle reazioni acido-base si assiste al trasferimento di un protone da una specie ionica all'altra, nelle reazioni di ossido-riduzione si verifica il trasferimento di uno o più elettroni. La specie che cede elettroni è la specie riducente, mentre quella che accetta elettroni è la specie ossidante. In modo del tutto generale si può quindi indicare tale comportamento con la reazione: Oxi + ne <====> Ridi (1.33) La (1.25) non ha, tuttavia, un significato fisico ben definito, in quanto non è sperimentalmente realizzabile come tale. Si sa infatti, che una sostanza può manifestare le sue caratteristiche riducenti solo se viene posta in condizioni di poter cedere le sue cariche negative a qualche altra specie in grado di accettarle.
Analogamente un ossidante può manifestarsi tale solo in presenza di un riducente. Le specie Ridi/Oxi della (1.25) costituiscono una coppia coniugata di ossido-riduzione. In ogni reazione di ossido-riduzione sono impiegate due coppie coniugate.
La (1.25) assume perciò il nome di semireazione. Una reazione di ossido-riduzione completa risulta dalla somma delle due semireazioni: Rid1 <====> Ox1 + n e (1.34) Ox2 + n e <====> Rid2 (1.35) Rid1 + Ox2 <====> Ox1 + Rid2 (1.36) La (1.36) può essere generalizzata nell'espressione a Rid1 + b Ox2 <====> c Ox1 + d Rid2 (1.37) L’equilibrio (5) viene definito quantitativamente da una costante di equilibrio:
K =Ox1{ }c Rid2{ }d
Rid1{ }a Ox2{ }b (1.38)
Il valore numerico di K dipende dalla forza relativa delle due coppie coniugate interessate, cioè dalla tendenza che hanno le diversa specie a cedere o acquistare
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-28

29
elettroni. Ad esempio, il cloro elementare è in grado di ossidare gli ioni Br presenti in soluzioni acquose di bromuri, mentre lo iodio non è in grado di provocare la stessa reazione. Cioè, mentre è possibile il processo:
0,5 Cl2 + Br- ====> Cl- + 0,5 Br2 non avviene la reazione:
0,5 I2 + Br- ====> I- + 0,5 Br2 Si può quindi concludere che l'ossidante Cl2 è più forte dell'ossidante I2. Quanto è più forte un ossidante, tanto più debole è il riducente ad esso coniugato. La forza ossidante o riducente può essere misurata con esattezza ed espressa in termini quantitativi
La misura si basa sull’osservazione che un elettrodo di platino, immerso in una soluzione contenente sostanze ossidanti o riducenti, assume un potenziale elettrico tanto più positivo quanto più la soluzione manifesta un carattere ossidante, e tanto più negativo quanto più la soluzione ha carattere riducente. Il potenziale assunto dall’elettrodo di platino (potenziale di ossido-riduzione), misurato contro un elettrodo di riferimento, è una grandezza del tutto soddisfacente per esprimere le caratteristiche ossido-riduttive del sistema. Il potenziale di ossido-riduzione può essere considerato una misura della tendenza al trasferimento di elettroni.
Un potenziale molto negativo, ad esempio, denota una notevole attività di elettroni nel sistema in cui è immerso l’elettrodo di Pt e quindi un elevato potere riducente. Se si immerge un elettrodo di Pt in una soluzione contenente la coppia coniugata Ox,Rid, atta a stabilire un equilibrio Ox + n e <====> Rid (1.39) il metallo inerte assume un potenziale di equilibrio E. Il valore assoluto di tale potenziale non può essere misurato. Si può tuttavia misurarne un valore relativo rispetto ad un opportuno elettrodo di riferimento formando una cella galvanica del tipo Pt : Ox,Rid :: elettrodo di riferimento (1.40) II potenziale risulta esprimibile mediante la ben nota equazione di Nernst:
E =RTnF
logOx[ ]Rid[ ]
(1.41)
dove n è il numero degli elettroni in gioco nel processo e·il valore della costante dipende dalla natura dell'elettrodo di riferimento e dalla temperatura. Assumendo come elettrodo di riferimento l’elettrodo standard ad idrogeno, la costante assume un determinato valore, corrispondente al potenziale standard della coppia Ox,Rid e viene indicato col simbolo E°Ox,Rid. Se si ammette un comportamento ideale, per cui si possano sostituire alle attività delle specie Ox e Rid le rispettive concentrazioni molari, e passando ai logaritmi in base 10, la (9) si può scrivere
E = EOx ,Rid0 +
2,303RTnF
logOx[ ]Rid[ ]
(1.42)
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-29

30
Il potenziale E corrisponde quindi, per convenzione, alla f.e.m. della cella: Pt : Ox,Rid :: H+ (aH
+ = 1) : H2 (1 atm), Pt (1.43) Per un generico sistema a Ox + ne <====> b Rid si avrà:
E = EOx ,Rid0 +
2,303RTnF
logOx[ ]a
Rid[ ]b (1.44)
Generalmente i sistemi di ossido-riduzione, implicando spesso specie ioniche a
carica elettrica elevata, sono notevolmente sensibili all'effetto della forza ionica del mezzo. In pratica, in chimica analitica, si fa largo uso dei potenziali formali, quali risultano dalla legge di Nernst quando si usino le concentrazioni analitiche al posto delle attività.
Il potenziale formale varia con i coefficienti di attività e quindi con la forza ionica della soluzione e risente in modo particolare della presenza in soluzione di specie chimiche a carattere più o meno complessante. I potenziali formali relativi ad una semireazione vanno per tanto espressi indicando la composizione della soluzione cui si riferiscono. Per esempio il potenziale formale della coppia Fe2+, Fe3+ è diverso per diverse concentrazioni di HCl, H2SO4 o HClO4 (vedi tabella 1).
BOX 1.9 Potenziali formali del Fe in diversi ambienti (il potenziale standard E°= 0,771 V) Composizione E°Fe2+, Fe3+___________________________________________________________ HCl 0,1 M 0,73 V HCl 1,0 M 0,70 V H2SO4 0,1 M 0,68 V H2SO4 1,0 M 0,68 V HClO4 0,1 M 0,735 V HClO4 1,0 M 0,735 V ___________________________________________________________
Per quanto riguarda la segnatura e la rappresentazione schematica degli elettrodi e delle celle, vengono qui adottate le convenzioni fissate dallo I.U.P.A.C. nel 1953. La pila viene rappresentata scrivendo a sinistra l’elettrodo negativo.
In tal caso alla f.e.m. della cella si attribuisce un segno positivo, per indicare che la reazione decorre spontaneamente nel senso indicato dalla cella: nel semielemento di sinistra procede l'ossidazione e in quello di destra la riduzione.
Il potenziale della cella è una misura diretta della variazione di energia libera per la reazione di cella secondo la:
∆G = - nFE (1.45) dove n è il numero di elettroni trasferiti nella reazione ed F è il faraday, 9,65.104 coulomb, la carica di una mole di elettroni. Se la f.e.m. della cella è positiva, la reazione associata procede spontaneamente e la variazione di energia libera è negativa (∆G < 0). 1.7.1. - Equilibri di ossido-riduzione e velocita’ di reazione
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-30

31
Sebbene vi siano molte reazioni di ossido-riduzione che procedono rapidamente in soluzione, ve ne sono di più numerose che procedono lentamente o non procedono affatto: Per esempio, lo ione perclorato è un agente ossidante molto forte e se esso potesse ottenere l’equilibrio molto rapidamente, potrebbe non esistere nella stessa soluzione con ioni Fe2+ , Cr2+, Mn2+. Tuttavia esso reagisce così lentamente che soluzioni contenenti ioni cromoso e ioni perclorato sono stabili per mesi. E' possibile usare permanganato come agente ossidante in soluzioni acquose soltanto a causa della sua bassa velocità di reazione con l'acqua; se si potesse raggiungere l’equilibrio nel processo, il permanganato ossiderebbe l’acqua ad ossigeno e sarebbe ridotto a MnO2. La costante di equilibrio per la reazione: 4 MnO4
- + 4H+ <====> 4 MnO2 (s) + 3 O2 (g) + 2H2O dà: [H+]4[MnO4
-]4 = [H+]4[MnO4-]4 = 10-68.1
pO23 aMnO2
4 aH2O2 pO2
3
In una soluzione di permanganato di potassio in equilibrio con l’atmosfera, pO2 è circa 0,2 atm., [H+] è 10-6M e quindi la concentrazione di ione permanganato all’equilibrio è [MnO4
-] = 10-11,6 che è notevolmente piccola. Tuttavia le soluzioni di permanganato 0,1 M sono stabili per parecchie settimane e quindi si decompongono lentamente.
La decomposizione è catalizzata dalla presenza di MnO4, cosicché, se è presente
inizialmente una traccia di sostanze organiche, che reagisca MnO4- con per dare MnO2,
la velocità di decomposizione sarà molto più elevata. I calcoli di equilibrio non ci diranno quindi mai quanto sia veloce una reazione, che invero potrà avvenire cosi lentamente da essere completamente inutilizzabile. 1.7.2.- I diagrammi E/(pH)
Numerosi processi di ossidoriduzione sono legati alla produzione o al consumo di ioni H+ o OH-. In questi casi tali specie, comparendo nelle semireazioni, entrano anche nelle espressioni di E, per cui il potere ossidante o riducente delle coppie in gioco diventa funzione del pH. Per esempio, una generica semireazione Ox + mH+ + ne <====> Rid (1.46) stabilirà un potenziale, funzione di aH+ secondo l'equazione: E = E° + RT ln aOx aH+
m (1.47) nF arid cioè, a 25°C: E = E° + 0,059 log aOx - m 0059 pH (1.48) n aRid n
In altri casi le variazioni di pH possono influenzare indirettamente il valore del potenziale redox. Ciò succede quando gli ioni H+ o OH-, pur non essendo implicati direttamente nel processo redox,, condizionano la concentrazione degli ioni partecipanti al processo, provocando associazioni o dissociazioni (qualora le specie ossidanti o riducenti abbiano carattere acido o basico) o la precipitazione di idrossidi o la formazione di complessi, ecc.
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-31

32
Di uso assai generale sono i diagrammi E/pH, detti diagrammi di predominanza o diagrammi Pourbaix. Essi sono particolarmente efficaci per rappresentare le condizioni di stabilità delle specie costituenti i sistemi redox. In tali diagrammi si riporta generalmente il cosiddetto potenziale normale apparente E°', che è il potenziale del sistema a concentrazioni analitiche totali unitarie di ossidante e di riducente al valore dato di pH. Questo potenziale è quindi soggetto a variare in funzione del pH o perché gli ioni H+ intervengono direttamente nel processo redox, come si è detto, per azione indiretta in seguito a fenomeni acido-basici o di formazione di composti poco solubili interessanti le specie ossidanti o riducenti. Questi potenziali, di valore essenzialmente pratico, si esprimono di solito in funzione delle concentrazioni molari e non delle attività delle specie implicate, e si rivelano particolarmente utili per prevedere reazioni redox, di dismutazione, ecc. nelle condizioni date. I diagrammi si costruiscono facilmente. Si consideri per esempio, il generico sistema (1). Dalla (3), ponendo [Ox] = [Rid] = 1, si ottiene: E°' = E° - m 0,059 pH (1.49) n direttamente trasferibile in un diagramma E/pH.
Fig. 1.8 Diagramma pe/pH del cadmio in presenza di altri ioni
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-32

33
Nella (1.49) m ed n rappresentano rispettivamente il numero di protoni e di elettroni in gioco nel processo considerato.
0
Diagramma potenziale/pH per lo zolfo
Fig. 1.9 Diagramma pe/pH per lo zolfo
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-33

34
BOX 1.10 Diagramma E(pH) per il sistema S° + 2e <====> S= (E° = -0,50 V) Questo sistema e’ assai importante in Ecotossicologia perche’ e’ il tipico sistema che si realizza in
un sedimento anossico in presenza (ma cio’ e’ quasi una costante in un sedimento inquinato) di sostanza organica contenente zolfo.
In questo caso la variazione di acidità ha un effetto indiretto per azione degli ioni H+ sulla specie S. Dalla reazione:
S° + 2e <====> S= considerando unitaria l'attività dello zolfo solido, arrotondando il termine 2,303 RT/nF, si ottiene: E = - 0,50 - 0,03 log[S=] In soluzione valgono gli equilibri acido-base: [H+][HS-] = K1[H2S] K1 = 10-7 [H+][S=] = K2[HS-] K2 = 10-13 [H+][S=] = K1K2[H2S] K1K2 = 10-20 Se la concentrazione totale è: [S=] + [HS-] + [H2S] = 1 usando [HS-] dalla (4) e [H2S] dalla (5) in (6) si ha: CS = [S=] { 1 + [H+] + [H+]2 } = 1 K2 K1K2e si può ricavare E in funzione di [H+]: E = - 0,50 - 0,03 log { 1 + [H+] + [H+]2 }-1 K2 K1K2Per valori di pH<pK1 (<7), la forma prevalente è H2S; nella si può semplificare: E = 0,50 - 0,03 log K1K2 [H+] da cui: E°' = 0,10 - 0.06 pH Questa rappresenta l'equazione di una retta, di pendenza dE/dpH = -0.06 e passante per il punto (0;
0.10) (vedi Figura, retta a). I tre processi redox sono quindi: S + 2H+ + 2e <====> H2S pH < 7 S + H+ + 2e <====> HS- 7 < pH < 13 S + 2e <====> S= pH > 13 Nella figura è riportato (I) il diagramma completo delle tre rette a, b e c raccordate, (II) le specie stabili e presenti quindi in gran prevalenza nei diversi campi di pH e potenziale. Lungo le linee verticali. in corrispondenza dei due valori di pK, le specie che formano le due
coppie coniugate sono presenti in eguale concentrazione. Come in tutti i diagrammi di questo tipo, attraversare una retta verticale comporta uno.
scambio di protoni; attraversare una retta orizzontale implica uno scambio di elettroni; passare attraverso rette inclinate comporta processi che mettono in gioco nello stesso tempo protoni ed elettroni.
____________________________________________________________________ G.PERIN - ECOTOSSICOLOGIA - CAP.I - CHIMICA - ED.27/03/2004 - 1-34