proceedings of the 16th international students conference
TRANSCRIPT

ISBN 978-80-7444-079-3
Pro
ceedin
gs of th
e 16
th In
ternatio
nal Stu
den
ts Co
nferen
ce ldquoMo
dern
An
alytical Ch
emistryrdquo P
rague 2
02
0
788074 440793
Prague 17mdash18 September 2020
Edited by Karel Nesměraacutek
Prague 2020
Proceedings of the
16th International Students Conference
ldquoModern Analytical Chemistryrdquo
Proceedingsofthe
16thInternationalStudentsConference
ldquoModernAnalyticalChemistryrdquo
Prague 17mdash18 September 2020
Edited by Karel Nesměraacutek
Prague 2020
Proceedings of the
16th International Students Conference
ldquoModern Analytical Chemistryrdquo
CATALOGUING-IN-PUBLICATIONndashNATIONALLIBRARYOFTHECZECHREPUBLIC
KATALOGIZACEVKNIZEndashNA RODNIKNIHOVNACR
ModernAnalyticalChemistry(konference)(162020PrahaCesko)
Proceedingsofthe16thInternationalStudentsConferenceldquoModernAnalyticalChemistryrdquo
Prague17ndash18September2020editedbyKarelNesmerak--1stedition--PragueFacultyof
ScienceCharlesUniversity2020--vi154stran
Obsahujebibliografiearejstrıky
ISBN978-80-7444-079-3(brozovano)
543(062534)
analyticalchemistry
proceedingsofconferences
543ndashAnalyticalchemistry[10]
TheelectronicversionoftheProceedingsisavailableattheconferencewebpage
httpwwwnaturcuniczisc-mac
copyCharlesUniversityFacultyofScience2020
ISBN978-80-7444-079-3
Preface
Despitethefactthattheyear2020ismarkedbyCOVID-19morethan40young
analytical chemists gathered in Prague for the 16th annual international
conferenceldquoModernAnalyticalChemistryrdquoTheymeettopresenttheresultsof
theirresearchtomastertheirpresentationandlanguageskillsandtoexchange
anddiscussideasandexperiencesofanalyticalchemistry
Thisvolumeofconferenceproceedingsbringsyouatotalof25papersfrom
thisconferenceAsinpreviousyearsthecontributionspresentedareassortedby
the sequence of their delivery supplemented by indexes at the end of the
proceedingsallowingeasynavigationthroughthepagesYouwillseethattopics
of contributions cover all the aspects of modern analytical chemistry from
theoretical problems through development of new analytical methods and
improvementofanalyticaltechniquestotheapplicationsinvolvingthesolutionof
medicinaltechnicalorenvironmentalproblemsLetushopethatlikeprocee-
dingsofpreviousyearsofourconference thisonewillalsobean interesting
beneficialandenjoyablereading
Itseemstousthattheauthorsofthecontributionsareaguaranteeofthatanew
generationofanalyticalchemistswillprotectbrightandthrillingfutureofour
science
We are very grateful to the Division of
AnalyticalChemistryofEuChemSforitslong-
lasting auspices of our conference Also we
arethankfultooursponsorsnotonlyfortheir
kind sponsorship making the conference
possiblebutalsoforalltheircooperationand
supportinmanyofourotheractivities
Enjoyreadingtheseproceedings
docRNDrKarelNesmerakPhD
editor
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 iii
Sponsors
The organizersof16th International Students Conference ldquoModernAnalytical
Chemistryrdquo gratefully acknowledge the generous sponsorship of following
companies
wwwecomsrocom
wwwlach-nercom
iv Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
wwwthermofishercz
www2thetacz
wwwzentivacz wwwquintacz
wwwshimadzueucom
wwwwaterscom
Contents
AlikovaVChernovaAShormanovVKorotkovaEDeterminationof2-methoxyphenol inmodelsolutionsbyspectrophotometry 1
BohmDMatysikF-MTheeffectsoflinearlyassembledcapillarieswithvariousinnerdiametersoncapillaryelectrophoresis 6
PoradaRBasBVoltammetricdeterminationofvitamins 13Baluchova S Klouda J Barek J Schwarzova-Peckova K Wong DKY Dopamine detection at
antifoulingconical-tipcarbonelectrodes 19Tvorynska SBarek J JosypcukBA comparative study of covalentglucose oxidaseand laccase
immobilizationtechniquesatpowderedsupportsforbiosensorsfabrication 25Heigl N Matysik F-M Capillary flow injection analysis with electrochemical detection for
carbohydrateanalysis 31KravchenkoAVKolobovaEAKartsovaLAApplicationofcovalentcoatingsbasedonimidazolium
cations for separationandon-linepreconcentrationof basicandneutralanalytes in capillaryelectrophoresis35
Efremenko E Chernova A Bastrygina O Determination of vanillin in smoking mixtures byspectrophotometry41
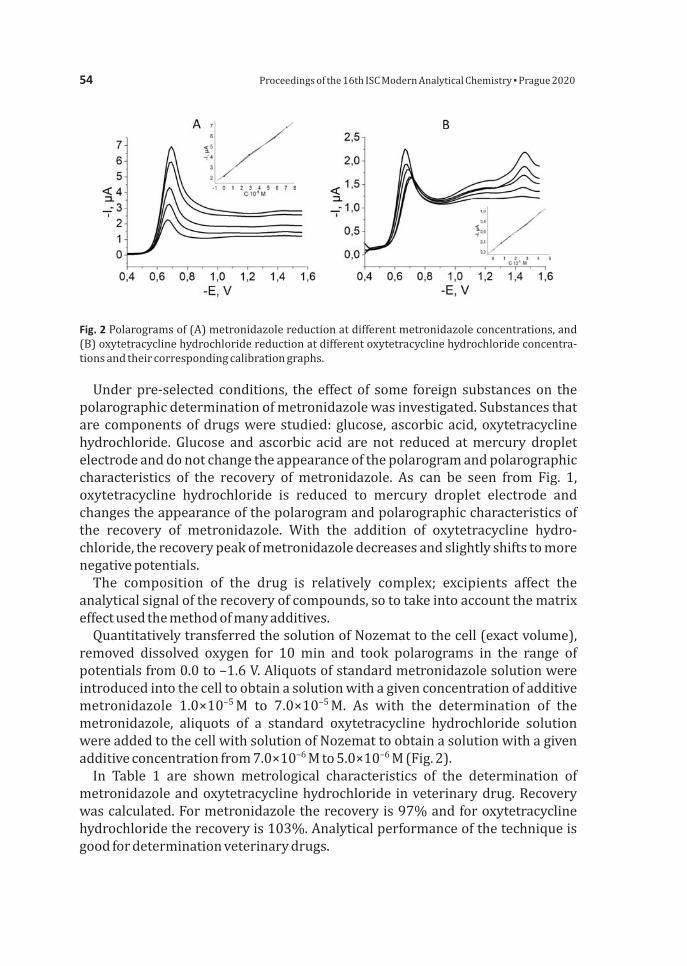
PietrzakKWardakCUranylion-selectiveelectrodewithsolidcontact 45Plotnikova K Dubenska L Zeleny I Polarographic determination of metronidazole and
oxytetracyclinehydrochlorideinveterinarydrugforhoneybees 51Deev V Bessonova E Kartsova L Application of microextraction techniques combined with
chromatographicmethodsfortheanalysisofcomplexobjects 57KralMDendisovaMMatejkaPThedevelopmentofreferenceprobesystemfortip-enhancedRaman
spectroscopy 63ChoinskaMHrdlickaVRedondoBRBarekJNavratilTDeterminationofheavymetalpoisoning
antidote23-dimercapto-1-propanesulfonicacidusingsilversolidamalgamelectrode70VymyslickyFKrızekTHert JCanagliflozinoxidation studyusingelectrochemical flowcelland
comparisonwithhydrogenperoxideoxidation 76AugustınMVyskocilVNovelhybridelectrochemicalDNAbiosensorformonitoringoxidativeDNA
damageviaoxidationreductionsignalsoflowmolecularweightdouble-strandedDNA 83SagapovaLKodrık ovaBSvobodaMMusilSKratzerJChemicalvaporgenerationofcadmiumfor
analyticalatomicspectrometry 90S tadlerova B Vyhnanovsky J Dedina J Musil S Photochemical vapour generation of bismuth
coupledwithatomicfluorescencespectrometry 97Cokrtova K Krızek T Separation of liquid crystals using non-aqueous capillary electrokinetic
chromatography104OndrackovaAStiborovaMHavranLSchwarzova-PeckovaKFojtaMElectrochemistryofSudanI
anditsderivatesinaqueousmedia 110BurkinKGalvidisIBurkinMGroupdetectionofaminoglycosidesusingELISAforcontroloffood
contamination 116Vyhnanovsky J Musil S Photochemical vapor generation of cobalt for detection by inductively
coupledplasmamassspectrometry 123LipinskaJMadejMBasBTyczkowskiJOptimizationofconditionforcoldplasmadepositionofthin
layersforsurfacemodificationofworkingelectrodes 129KorbanAAdvancedGC-MSmethodforqualityandsafetycontrolofalcoholicbeverages 135Baroch M Dejmkova H Sladkova S Utilization of a carbon felt as a material for working
electrodes 141Benesova L Zarybnicka A Klouda J Schwarzova-Peckova K Electroanalytical methods for
determinationof7-dehydrocholesterolinartificialserum146
Authorindex 151Keywordindex 152
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 v
1Introduction
2-Methoxyphenol(guaiacol)isusedinmedicineasanexpectorantThestructuralformulaisshowninFig1Itiswidelyusedinthepharmaceuticalindustry[1]forthesynthesisofantituberculosisexpectorantdrugs(Kas-nol Sudafed Ascoril Prothiazine Expectorant Guai-phenesinum) Moreover 2-methoxyphenol is often used as anaromaticsubstance [2] in the food industry Inparti-cular itwaswidelyusedintheproductionofsmokedfish and meat products using smokeless smokingtechnologyusingflavourings Ontheotherhand2-methoxyphenolhasthesymbolGHS07andhasahazardcodeXnTXi[2]accordingtotheGHSsystemItisverytoxicbyinhalationitcanirritatethemucousmembraneoftherespiratorytractandtheconjunctivaofthe
Determination of 2-methoxyphenol in model solutions by spectrophotometry
a a b aVALERIYAALIKOVA ANNACHERNOVA VLADIMIRSHORMANOV ELENAKOROTKOVA
a DepartmentofChemicalEngineeringEngineeringSchoolofNationalResourcesNationalResearchTomskPolytechnicUniversityLeninavenue30634050TomskRussiaalikovaleramailru
b DepartmentofPharmaceuticalToxicologicalandAnalyticalChemistryKurskStateMedicalUniversitystKarlaMarks3305000KurskRussia
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 1
AbstractA spectrophotometric approach for determination of 2-methoxy-phenol in model solutions has been developed The absorptionspectra of 2-methoxyphenol were determined in the wavelengthrangefrom200to400nminsolutionsof95ethanolacetonitrile01Msodiumhydroxideandethylacetatewithaconcentrationofthe
minus3analyte of 005mgdm For the quantitative determination of2-methoxyphenol a seriesof solutionswaspreparedwithvarious
minus3 minus3concentrationsfrom0001mgdm to005mgdm in95ethanolacetonitrile01MsodiumhydroxideTheopticaldensityof2-meth-oxyphenolinsolventswasmeasuredatawavelengthof276nmand289nmThedevelopedmethodwastestedusingthemethodanalysisofspikedsamples
Keywordsquantitation2-methoxyphenolUVVISspectrophoto-
metry
Fig 1Structuralformulaof2-methoxyphenol
eyeballinhighconcentrationswhenitpenetratestheskincanleadtoneurosiswhenadministeredorallycanstimulatetheesophagusandstomachresultinginheartfailurecollapseanddeathNowadaystherearepublisheddataoncasesofasystemicallergicreaction[3]causedby2-methoxyphenolderivativesandthereisa fatal case known [4] for oral administration of guaifenesin (3-(2-methoxy-phenoxy)propane-12-diol)oneofthecomponentsofcommonlyavailablecoughmedications Thedeterminationof2-methoxyphenolinenvironmentalobjectsaswellasinthefoodindustryiscarriedoutusinggaschromatographymethodswithsolid-phasemicroextraction[5]Inordertocontrol2-methoxyphenolinnaturaldrink-ing and treatedwastewater gas chromatography is used followed by opticaldetectionoftheeluate[6]Themaindisadvantagesofthismethodofanalysisarethelowselectivityanddurationofdetermination(about3hours)AlsoaccordingtoRussianStateStandartGOST33312-2015themethodofgaschromatographyisusedforthequalitativeandquantitativedeterminationof2-metoxyphenolinjuiceproducts Commonlyfortheanalysisoftoxicsubstancesinvariousbiologicalsamples(bloodplasmaurinesalivasweathair)bygaschromatographyitisnecessarytocarryoutmultistagesamplepreparationwhichcomplicatesandslowsdownthecourseofthestudy[7]Atthesametimeitisimportantthatduringtheprepa-rationofsamplesintheanalyzedcompoundstheirstructureisnotviolatedasthiswillleadtothedifficultyoftheiridentification Spectrophotometryintheultravioletregionhaslowersensitivitycomparedtothe abovemethods however thismethod does not require such complicatedpreparation of the analyzed samples it is a relatively affordable simple andinexpensive analysis method In addition its sensitivity can be significantlyimprovedbyapplyinganappropriateseparationprocedureandpreconcentrationbeforedetection[8]MethodUVspectrophotometryisusedtoassessthequalityof both medicinal substances and preparations made from them in terms ofauthenticitygoodqualityandquantitativecontentInadditionitisarelativelyaffordablesimpleandlow-costanalysismethod Ananalysisoftheliteraturedatashowedthattodaytherearefastandsensitivespectrophotometricmethodsforthedeterminationofpyrocatecholderivativesinmedicines[9]vanillininfoodproducts[10]andotherphenolsinwastewaterandwineproducts[1112]Howeverasfarasweknowinformationonthedetermi-nationof2-methoxyphenolfromtheabsorptionspectraintheultravioletregionisabsent Theaimofthisstudyistodevelopmethodsforthequalitativeandquantitativeof2-methoxyphenolinmodelsolutionsusingUVspectrophotometry
2 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
2Experimental
21Reagentsandchemicals
Asampleof2-methoxyphenolfromFlukawithabasicsubstancecontentofge98wastakenastheobjectofstudyAssolventsweusedacetonitrile(ChP)95ethanolethylacetateand01MsodiumhydroxidesolutionAllotherchemicalsusedwereofanalyticalreagentgrade
22Instrumentation
Theopticaldensitywasmeasuredincuvetteewithanabsorbinglayerthicknessof10mmusingaCary60spectrophotometer(AgilentUSA)Allmeasurementswerecarriedoutatroomtemperature
3Resultsanddiscussion
Thechangeinthebehavioroftheabsorptionspectrumwasinvestigatedinthewavelengthrangeof200ndash400nmFigure2isshowedthatwithanincreaseinthepolarityofthesolventtheabsorptionmaximumshiftstowardthevisiblepartofthe spectrum The wavelength of absorption maxima of 2-methoxyphenol ispresentedinTable1 A studyof thephotometricbehaviorof2-metoxyphenolinvarious solventsshowedthatacetonitrile95ethanoland01Msodiumhydroxidearethemostsuitablesolventsforthequalitativedeterminationofthetestsubstance
ndash3Fig 1Thespectraof2-methoxyphenolofconcentration005mgdm inthemediumofsolvents(anabsorbinglayerthicknessof10mm)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 3
Forthequantitativedeterminationof2-methoxyphenolaseriesofsolutionsndash3 ndash3withaconcentrationragefrom0001mgdm to005mgdm werepreparedin
acetonitrileand95ethanolTheopticaldensityof2-methoxyphenolinsolventswasmeasuredbywavelengthof276nmThedependenceoftheintensityoftheoptical density on the concentration of 2-methoxyphenol in 01 M sodium
ndash3hydroxide was plotted in the concentration range from 0005mgdm tondash3003mgdm Themeasurementwerecarriedoutbywavelengthof276nmThe
obtainedregressionequationsarepresentedinTable2Dataanalysisobtainedwasperformedusingleast-squaresmethodThedevelopedmethodwastestedusingthemethodanalysisofspikedsamplesTheresultsarepresentedinTable2
4Conclusions
Studieshaveshownthepossibilityofusingspectrophotometricanalysisforthequalitativeandquantitativedeterminationof2-methoxyphenolTheabsorptionmaximaofweredeterminedinsolutionsofethanolandacetonitrile(276nm)inasolutionofethylacetate(277nm)and01Msodiumhydroxide(289nm)Theconstructed calibration curves of thepure substance of 2-methoxyphenol hasshownagoodregressioncoefficient(Rgt099)andcanbeusedforquantitativedeterminationof2-metoxyphenolinbiologicalobjectsInthefutureitisplannedtoapplythistechniquetodetermine2-metoxyphenolinincadavericmaterial
Solvent Regressionequation Found S RSD Δх δ 2-metoxy- phenolg
ndash3 ndash6 ndash5Acetonitrile y=18294C+01130 499times10 50times10 028 2times10 044 Rsup2=09985
ndash3 ndash6 ndash595ethanol y=35131C+00269 503times10 03times10 021 7times10 137 Rsup2=09956
ndash3 ndash6 ndash501Msodium y=31196C+01101 495times10 01times10 018 1times10 028hydroxide Rsup2=09997
Table 2Results of the determination of 2-methoxyphenol (average of three measurements) in modelsolutionsbythemethodanalysisofspikedsamplestheconcentrationofintroduced2-methoxy-
ndash3phenolwas500times10 g(SndashstandarddeviationRSDndashrelativestandarddeviationΔхndashabsoluteerrorδndashrelativeerror)
Table 1Valuesofopticaldensityandwavelengthsinappropriatesolventswith2-methoxyphenol
minus3 minus1Solvent λnm εgdm cm
Acetonitrile 276 0082895ethanol 276 00853Ethylacetate 277 0083101Msodiumhydroxide 289 00744
4 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
References
[1] МельниковаИММизерницкии ЮЛКомбинированныеотхаркивающиепрепаратырастительного происхождения в педиатрическои практикеМедицинский совет 2(2018)93ndash97
[2] httpwwwthegoodscentscompanycomdatarw1032272html(accessed27stFebruary2020)
[3] RayMFaltayBHallerNACasereportanaphylacticreactiontoguaifenesinHospPract37(2009)60ndash63
[4] OkicMJohnsonTCrifasiJALongCMitchellEKSwiftonsetofcentralnervoussystemdepressionandasystolefollowinganoverdoseofguaifenesinJAnalToxicol37(2013)318ndash319
[5] ВолковCМЧерновецАНОпределениеконцентрациифеноловвгазовыхвыбросахпромышленных предприятии методом газовои хроматографии с твердофазнои микроэкстракциеи Сорбционныеихроматографическиепроцессы10(2010)723ndash728
[6] ШачневаЕЮОньковаДВСерековаСМСпособыопределенияфеноловвобъектахокружающеи среды Астраханский вестник экологического образования 4 (2013)138ndash142
[7] ГладиловичВДПодольскаяЕПВозможностипримененияметодаГХ-МС(Обзор)Научноеприборостроение4(2010)36ndash49
[8] Pena-PereiraFLavillaIBendichoCHeadspacesingle-dropmicroextractioncoupledtomicrovolumeUVndashVis spectrophotometry for iodine determinationAnal Chim Acta631(2009)223ndash228
[9] NagarajaPMurthyKCSRangappaKSGowdaNMMSpectrophotometricmethodsforthe determination of certain catecholamine derivatives in pharmaceutical preparationsTalanta46(1998)39ndash44
[10] Altunay N Development of vortex-assisted ionic liquid-dispersive microextractionmethodology for vanillin monitoring in food products using ultraviolet-visible spectro-photometryLWT93(2018)9ndash15
[11] Lupetti KO Rocha FRP Fatibello-Filho O An improved flow system for phenolsdetermination exploiting multicommutation and long pathlength spectrophotometryTalanta62(2004)463ndash467
[12] Figueiredo-Gonzalez M Cancho-Grande B Simal-Gandara J Garnacha tintorera-basedsweetwineschromaticpropertiesandglobalphenoliccompositionbymeansofUVndashVisspectrophotometryFoodChem140(2013)217ndash224
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 5
1Introduction
Thenumberofsamplesthesamplecomplexityandalsothenumberofsubstanceswhich need to be analysed simultaneously is increasing steadily ThereforepowerfulseparationanddetectionmethodsarerequiredOnewaytoachievethisisthecouplingofaseparationsystemwithmorethanonedetector[12] In recent years capillary electrophoresis (CE)was established as a potentseparation system due to its high separation efficiency and the low sampleconsumption [3] To generate more information numerous dual detectionconceptsforCEweredevelopedwhicharesummarisedelsewhere[12]Acom-binationofamperometricdetectionandmassspectrometry(MS)isaninterestingdual detection concept for CE because both detectors supply complementaryinformationForelectroactivespeciesamperometricdetectionisarobustandoneof the most sensitive detection method [4] Thus it is well suited for the
The effects of linearly assembled capillaries with various inner diameters on capillary electrophoresis
DANIELBO HMFRANK-MICHAELMATYSIK
InstituteofAnalyticalChemistryChemo-andBiosensorsFacultyofChemistryandPharmacyUniversityofRegensburgUniversitaumltsstraszlige3193053RegensburgGermanydanielboehmchemieuni-regensburgde
AbstractDuetotheincreasingneedofpowerfulanalyticalmethodsanewdualdetection concept for capillary electrophoresis (CE) with parallelamperometricdetectionandmassspectrometryshallbedevelopedFor this concept the CE flow has to be divided into two streamsutilizinga flowsplitter In thiswork theeffectsof combinedcapi-llarieswithvarious innerdiameterswerestudiedForpreliminaryinvestigationsthecapillarieswereconnectedinaserialconfigurationwithoutdeadvolumeUsingcapillaryflowinjectionanalysishyphe-natedtocontactlessconductivitydetectionitcouldbeshownthatthecouplingofidenticalcapillariesleadstoaslightdecreaseoftheflowratesWithCEhyphenatedtoUVdetection itcouldbeshownthatthecouplingofcapillarieswithdifferentinnerdiameterhasamuchstrongereffectontheelectroosmoticflowthanthecombinationwiththesameinnerdiameterFurthermorenosignificantchangeinpeakshapewasobserved
Keywordsassembledcapillariescapillaryelectrophoresiscapillaryflowinjection
analysisdualdetectionconceptnon-aqueoussystem
6 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
quantificationofsubstanceswhereasMSiswellsuitedfortheidentificationofunknown substances [3] In most dual detection concepts the detectors arearrangedinaserialconfigurationwhichisnotpossibleincaseofamperometricdetection-mass spectrometry [1] The instrumental implementation is morecomplicatedwithbothdetectorsbeingdestructiveFurthermoretheymustbedecoupledfromthehighvoltagefieldoftheCEThereforetheCEflowmustbedividedintotwostreamswithaflowsplitterAsimplifiedsketchofthepossiblenewdualdetectionconceptisshowninFig1 ForthedevelopmentofthenewdualdetectionconceptthreecapillarieswithpotentiallydifferentinnerdiametersmustbecoupledForthisreasonthedeadvolume-freecouplingofcapillarieswithdifferent innerdiameterswas investi-gatedinafirststepTokeepthesetupsimplewefocusedonthelinearcouplingofcapillariesandtheresultingeffectsNon-fragmentedcapillarieswerecomparedwithfragmentedcapillariesofthesameordifferentinnerdiametersEffectsonthe flow rate were investigated with capillary flow injection analysis (CFIA)
4hyphenatedtocontactlessconductivitydetection(C D)EffectslikechangesinthemigrationbehaviourorpeakshapesoccurringinCEwereinvestigatedwithCEhyphenatedtoUVdetection(CE-UV)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 7
Fig 1SchematicillustrationofthenewdualdetectionconceptwithparallelamperometricdetectionandmassspectrometricdetectionforCEAfterinjectionfrom(a)thesamplevialthecomponentsareseparatedbyCEnext(b)theflowsplitterdivides(c)thecapillaryintotwopartsandleadstheCEflowtowards(d)themassspectrometerand(e)theamperometricdetector
2Experimental
21Reagentsandchemicals
Thefollowingchemicalswereusedallofanalyticalgradeferrocenemethanoldecamethylferrocene(ABCRGermany)acetonitrileammoniumacetate01Msodiumhydroxidesolutionultra-purewaterprovidedbyaMilliQAdvantageA10system(MerckGermany)aceticacid(RothGermany)
22Instrumentation
221Capillaries
4Forbothexperiments(CFIA-C DandCE-UV)capillarieswithinnerdiametersof2550and75micromanouterdiameterof360micromandatotallengthof70cmwereusedTheywerepurchasedfromPolymicroTechnologies(USA)Measurementswere carried out with fragmented and non-fragmented capillaries For themeasurementswiththefragmentedcapillariestheoriginalcapillarieswerecutintotwopiecesyieldingatotalof9capillarycombinationswithlengthsof70cm(20cmfirstcapillarypieceand50cmsecondcapillarypiece)ThesecombinationsaresummarizedinTab1(section31)Atbothendsofthecapillariesabout02cmof thepolyimidecoatingwasremovedBothsidesof thecapillarypieceswerepolishedtoreceiveplanarcapillarytipsForthelinearassemblingofthecapillarypiecesMicroTightSleevesF185Xanda capillary connectorUnionAssemblyMicroTightP720fromIDEXHealthampScience(USA)wereusedPriortothefirstCEmeasurementsthecapillarieswereconditionedbyflushingthemfor10minwith01Msodiumhydroxidesolution5minwithultra-purewaterand30minwithseparationbuffer
222Capillaryflowinjectionanalysishyphenatedtocontactlessconductivitydetectionsetup
The flow rates for the fragmented and non-fragmented capillaries were4determinedwithaCFIA-C DsetupschematicallydepictedinFig2ATheflowin
thecapillarywasgravitationdrivenbyaheightdifferencebetweentheinletandoutletcarriersolutionvialTheconceptofCFIAwithgravitationdrivenflowwasfirstdescribedbyMatysiketal[5]AlaboratoryconstructedautosamplerofaCEdevicewasusedforthehydrodynamicinjectionThesamplesolutionconsistedof10mMdecamethylferrocene incarrier solution (10mMCH COONH and1M3 4
4CH COOH in acetonitrile) A high resolution C D was placed after 40 cm for3
detectionThedetectordescribedelsewhere[6]wasconstructedinthedoLagogroup(Brazil)Adoubledeterminationattwodifferentheightswasdoneforthedeterminationoftheflowrates
8 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
223Capillaryelectrophoresis-UVdetectionsetup
Fig2BshowsasketchoftheCE-UVsetupItconsistedofalab-builtCEdevicewhichwasconnectedtoahighvoltagepowersupplyfromISEG(Germany)Theseparationswerecarriedoutwithanon-fragmented50micromcapillaryandwithcapillary combinations implementing a 50micromdownstream capillary segment(25+5050+50and75+50microm)ALambda1010UV-VISdetectorfromBischoff(Germany)wasusedfordetectionat210nmThedetectorwasplacedafter40cm
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 9
Fig 2Schemeof(A)thecapillaryflowinjectionanalysis(CFIA)hyphenatedtocontactlessconduc-4tivity detection (C D) setup and (B) the capillary electrophoresis hyphenated to UV detection
4(CE-UV) setup Components of the CFIA-C D setup (a) sample (b) inlet and (c)outlet carrier4solution vial (d) fused silica capillary (e) linear capillary connector (f) C D and (g) stand
ComponentsoftheCE-UVsetup(h)sample(i)inletand(j)outletbuffervialand(k)UVdetector4therestofthecomponentswereidenticaltotheCFIA-C DsetupTheoutletbuffervialwaslowered
forthehydrodynamicinjection(j)Theenlargedview(k)depictsthecouplingoftwocapillarieswithdifferentinnerdiametersintheconnectionsidewithoutdeadvolume
Asamplesolutioncontaining1mMferrocenemethanolanddecamethylferroceneinseparationbuffer(10mMCH COONH and1MCH COOHinacetonitrile)was3 4 3
utilizedTheinjectionwasperformedhydrodynamicallybyloweringtheoutletbuffer vial by 20 cm A uniform sample plug was injected to compare bandbroadeningeffectsTheinjectionsegmenthadalengthof035cm(05ofthetotalcapillarylength)andtherespectiveinjectiontimewasdeterminedbasedontheflowratesofthecorrespondingcapillarycombinationFortheelectrophoreticseparationaseparationvoltageof25kVwasappliedandtheinletandtheoutletbuffervialswereplacedatthesameheightsothattherewasnogravityflowwhichaffectedthemigrationbehaviour
3Resultsanddiscussion
31Capillaryflowinjectionanalysishyphenatedtocontactlessconductivitydetectionexperiments
AsshowninTab1theflowratesforaheightdifferenceof20cmwerecalculated4basedontheCFIA-C DmeasurementsItwasobservablethattheflowratesare
slightlylowerforfragmentedcapillariesthanfornon-fragmentedcapillariesofthe same dimension This indicates that a flow resistance arises when twocapillaries are combined Furthermore it was observed that the flow ratedecreasesforupstreamcapillarieswithlowerinnerdiametersandviceversaTheflow rate for the combination 25+75microm could not be determineddue to theformationofairbubblesattheconnectionside
10 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Table 1Flowratesandthecorrespondingstandarddeviations(SD4measurements)ofdifferentcapillarycombinationsforaheightdifferencebetweeninletandoutletvialof20cmbymeansofcapillaryflowinjectionanalysishyphenatedtocontactlessconductivitydetection
ndash1capillary flowratenLs plusmnSDflowndash1combinationmicrom ratenLs
25 00577 0000525+25 0057 000150+25 00775 0000775+25 ndash ndash25+50 0137 000550 0865 000250+50 080 00575+50 105 00625+75 0187 000250+75 1941 000375 456 00275+75 42 01
32Capillaryelectrophoresis-UVdetectionexperiments
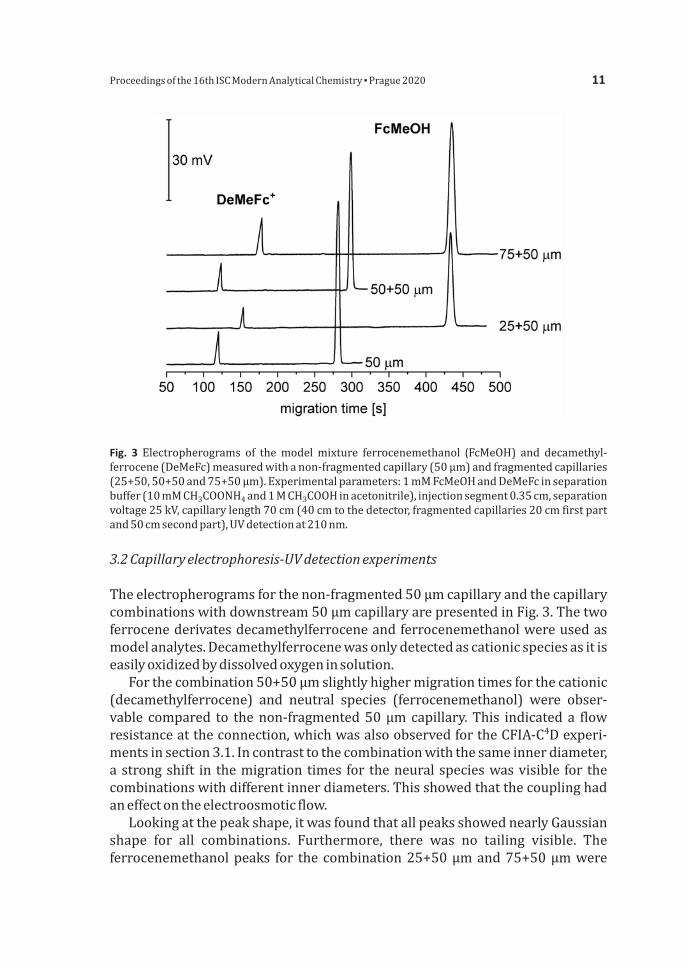
Theelectropherogramsforthenon-fragmented50micromcapillaryandthecapillarycombinationswithdownstream50micromcapillaryarepresentedinFig3ThetwoferrocenederivatesdecamethylferroceneandferrocenemethanolwereusedasmodelanalytesDecamethylferrocenewasonlydetectedascationicspeciesasitiseasilyoxidizedbydissolvedoxygeninsolution Forthecombination50+50micromslightlyhighermigrationtimesforthecationic(decamethylferrocene) and neutral species (ferrocenemethanol) were obser-vable compared to thenon-fragmented50micromcapillaryThis indicateda flow
4resistanceattheconnectionwhichwasalsoobservedfortheCFIA-C Dexperi-mentsinsection31Incontrasttothecombinationwiththesameinnerdiameterastrongshift in themigration times for theneuralspecieswasvisible for thecombinationswithdifferentinnerdiametersThisshowedthatthecouplinghadaneffectontheelectroosmoticflow LookingatthepeakshapeitwasfoundthatallpeaksshowednearlyGaussianshape for all combinations Furthermore there was no tailing visible Theferrocenemethanolpeaks for thecombination25+50micromand75+50micromwere
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 11
Fig 3Electropherograms of themodelmixture ferrocenemethanol (FcMeOH) and decamethyl-ferrocene(DeMeFc)measuredwithanon-fragmentedcapillary(50microm)andfragmentedcapillaries(25+5050+50and75+50microm)Experimentalparameters1mMFcMeOHandDeMeFcinseparationbuffer(10mMCH COONH and1MCH COOHinacetonitrile)injectionsegment035cmseparation3 4 3
voltage25kVcapillarylength70cm(40cmtothedetectorfragmentedcapillaries20cmfirstpartand50cmsecondpart)UVdetectionat210nm
slightlybroaderthanthepeaksforthenon-fragmented50micromcapillaryorforthe50+50 microm combination But this probably results from longitudinal diffusioneffectsduetothelongerresidencetimes
4Conclusions
FromtheCFIAmeasurementsitcanbeconcludedthattherewasamechanicaldisturbanceoftheflowduetothecouplingFurthermoreitcouldbeshownthatCEmeasurementswithlinearcoupledcapillariesofvariousinnerdiameterwerepossible Unlike to the capillary combinationwith the same inner diameter astrongshiftoftheelectroosmoticflowtowardshighermigrationtimeswasfoundfor capillary combinations with different inner diameters In this work thecapillaries were coupled with almost no dead volume which resulted in nosignificantchangesofthepeakshapeorpeaktailingContrarytoexpectationsthecouplingofcapillarieswithvariousinnerdiametershadnosignificantimpactonthepeakwidth TheknowledgegainedfromthelinearcouplingofcapillariesisagoodbasisforthedevelopmentofthenewdualdetectionconceptInanextstepthreecapillariesshouldbecoupledwitheachother
Acknowledgments
WethanktheGermanResearchFoundation(DFG)forfinancialsupport
References
[1] OpekarFS tulıkKSomeimportantcombinationsofdetectiontechniquesforelectrophoresisincapillariesandonchipswithemphasisonelectrochemicalprinciplesElectrophoresis32(2011)795ndash810
[2] BeutnerAHerlTMatysikF-MSelectivityenhancement incapillaryelectrophoresisbymeans of two-dimensional separation or dual detection conceptsAnal Chim Acta1057(2018)18ndash35
4[3] BeutnerACunhaRRRichterEMMatysikF-MCombiningC DandMSasadualdetectionapproachforcapillaryelectrophoresisElectrophoresis37(2016)931ndash935
[4] MatysikF-MEnd-columnelectrochemicaldetectionforcapillaryelectrophoresisElectro-analysis12(2000)1349ndash1355
[5] MatysikF-MWernerGTracemetaldeterminationintearsbyanodicstrippingvoltammetryinacapillaryflowinjectionsystemAnalyst118(1993)1523ndash1526
[6] FranciscoKJMdoLagoCLAcompactandhigh-resolutionversionofacapacitivelycoupledcontactlessconductivitydetectorElectrophoresis30(2009)3458ndash3464
12 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Thetermldquovitaminsrdquodescribestheheterogeneousgroupofchemicalcompoundswhich are important for the proper functioning of the human body [1 2] Bydefinitionvitaminsarenotsynthesizedbythehumanbodyorthesynthesizedamount is not sufficient to cover the demand That is why they have to besupplementedfromtheexternalsourceslikefoodproductsorpharmaceuticals[1ndash3]Basedontheirsolubilityvitaminsaredividedintowater-soluble(B-groupandvitaminC)andfat-solublevitamins(ADEandK)[3]VitaminC(ascorbicacid) is the most important antioxidant and participates in the activation ofenzymes[4]VitaminB1(thiamine)facilitateswoundhealingandiscrucialforthehumannervoussystem[15]VitaminB2(riboflavin)participatesintheenzy-maticreactionsandthebiotransformationofglucoseandaminoacids[6]Vitamin
+B3(niacin)isthemainconstituentoftheNAD andNADHcoenzymeswhichareresponsible for the transfer of electrons and hydrogen ions in the cellularrespiration [1ndash3] Vitamin B6 possesses six related structures (vitamers) thateasilyinterconvertThemostimportantoneispyridoxinewhichhelpstopreventtongue inflammation and microcytic anemia [2] For the production of well-functioningredbloodcellsandtheavoidanceofmegaloblasticanemiaandfetusdefectsvitaminB9(folicacid)hastobesupplementedintheproperamount[12]
Voltammetric determination of vitamins
RADOSŁAWPORADABOGUSŁAWBAS
DepartmentofAnalyticalChemistryFacultyofMaterialsScienceandCeramicsAGHUniversityofScienceandTechnologyMickiewicza3030-059KrakoacutewPolandrporadaaghedupl
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 13
AbstractVitaminsbelongtothegroupofchemicalcompoundsessentialfortheproperfunctioningofthebodySinceboththeirdeficiencyandexcessmay result in serious health problems the amount of vitaminssupplementedinthedietaswellasvitamincontentintheirsourceshavetobestrictlycontrolledInthisworkthepossibilityofsimulta-neous determination of vitamins by means of differential pulseadsorptive stripping voltammetry is discussed The research hasshownthatthedeterminationofsingularvitaminatthemicromolarlevel isrelativelyfastandstraightforwardandthemostimportanthindranceisrelatedtotheanalyteadsorptionattheelectrodesurfaceInthecaseofvitaminswithdifferentredoxpotentialstheycanbeanalyzedsimultaneouslywithouttheneedtoreachfortheadvancedmethodsforsignalprocessing
Keywordsmercuryelectrodevitaminsvoltammetry
VitaminK3(menadione)doesnotoccurnaturallybutitservesasaprecursorforthesynthesisofotherK-groupvitaminsandcanbeusedtotreathypoprothrom-binemiaVitaminK3ispartiallysolubleinwater[17] All of the vitamins are electrochemically active [3] therefore the electro-chemicalmethodscanbeappliedforthedeterminationofvitamincontentinfoodproductspharmaceuticalsandbodyfluidsVoltammetrictechniquesarecharac-terizedbyhighsensitivityandselectivityandtheydonotrequiretime-consumingsamplepreparationMoreovertheelectrochemicalinstrumentationisrelativelyinexpensiveandcanbeappliedintheon-siteconditionsfortheonlineanalyseseginqualitycontrolMostofthepapersreporttheconstructiondevelopmentand characterization of a novel modified working electrodes for quantitativeanalyses of a singular vitamin in the variety of matrices Unfortunately only alimitednumberofpapersdescribethesimultaneousdeterminationofmultiplevitaminsinasinglerun[23] The preliminary research devoted to the simultaneous determination ofB-groupCandK3vitaminswiththeuseofthecontrolledgrowthmercurydropworking electrode in aqueous solutions is presented in this work Particularattentionhasbeenpaidtotheredoxpotentialsofthestudiedcompoundstheshape of the calibration curves and adsorption phenomena As an attempt toovercomethelattertheneutralsurfactantTritonX-100hasbeenintroducedintothestudiedsystem
2Experimental
21Reagentsandchemicals
TheappliedreagentswereofanalyticalgradeandusedassuppliedPhosphateand McIlvaine buffers were obtained by mixing the appropriate amount of
ndash1 ndash1 ndash102molL Na HPO with 02molL NaH PO and 01molL citric acid2 4 2 4
respectively (all reagents purchased from Avantor Performance MaterialsPoland)ThestandardsolutionsofvitaminB1B2B3B9andCwerepreparedbydissolving the corresponding amount of the standard (all Sigma-Aldrich) in
ndash1distilledwater In thecaseofB2andB9 theadditionof02molL NaOHwasinevitabletoobtainaclearsolutionVitaminK3standard(Sigma-Aldrich)was
ndash1dissolvedinthemixtureofmethanoland1molL phosphatebuffer(pH=82)(vv=25)LaboratorygradeTritonX-100(Sigma-Aldrich)wasusedinthestudyoftheadsorptionprocesses22Instrumentation
All the electrochemical measurements were conducted in the three-electrodesystem composed of the Pt auxiliary electrode double-junction silversilverchloride reference electrode and controlled-growth mercury drop electrode
14 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
actingastheworkingelectrodeTheusedmeasurementequipmentinvolvedtheM164electrodestandandM161multipurposeelectrochemicalanalyzer(bothmtm-anko Krakow) To control the buffer pH-value the SevenCompact S210laboratorypH-meter(MettlerToledoSwitzerland)wasemployed
23Voltammetricmeasurements
Throughout the course of the study differential pulse adsorptive strippingvoltammetry has been used for recording the current-potential curves BothcathodicandanodicscanswererecordedinapotentialrangeadjustedforthestudiedvitaminsTheinfluenceofvariousmeasurementconditionsontheregis-teredsignalshasbeeninvestigatedFinallythepossibilityofthesimultaneousdeterminationofmultiplevitaminsinonescanhasbeenverified
3Resultsanddiscussion
Figure 1 depicts the redox potentials of the studied vitamins in the aqueoussolutions for the mercury electrode The only exception is vitamin B6 whoseredox potential is higher than the potential of mercury oxidation (ca +02 V)Therefore the given value refers to the glassy carbon electrode The redoxpotentialvalueofstudiedvitamins isnotasingularvaluebut it fallswithinacertainrangeThiscanbeascribedtothedependenceofredoxpotentialonthesolutionpHvaluewhichresultsfromtheparticipationofprotonsintheredoxreactions of vitamins Moreover the potentials for individual vitamins arerelatively well separated indicating that the simultaneous determination ofseveralvitaminsintheonerunmaybepossibleTheonlyencounteredproblems
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 15
Fig 1Redoxpotentialsofchosenvitamins
regardthevitaminsB1B3andB12whoseredoxpotentialsoscillatebetweenndash15andndash17VandtheK-groupvitamins(K1K2K3)inwhichonlythecommonstructuralmotifndashthequinoneringndashiselectrochemicallyactiveresultinginthevalueoftheredoxpotentialofcandash02V[8] AtypicalcalibrationcurveisdepictedinFig2Abasedonthedifferentialpulsevoltammograms of the vitamin B9 reduction in the concentration range from
ndash1blank to 01 mg L recorded in the McIlvaine buffer of pH = 52 using thecontrolledgrowthmercurydropelectrodeworkingelectrodeTherelationshipbetweenthepeakcurrentandtheconcentrationofVB9 is linear in thewholetestedrange(r=09999)Basedontheparametersoftheregressioncurvethelimit of detection and limit of quantitation were estimated to 42 and
ndash1142nmolL respectively Similar dependencies and figures of merit can beobtainedforothervitamins QuitedifferentbehaviorwasobservedinthecaseofthevitaminK3forwhichthe increase in current was not strictly proportional to the increase in theconcentrationandthecalibrationplotresemblesanS-shapecurve(Fig2B)ThelatterindicatesthatvitaminK3adsorbsatthesurfaceoftheworkingelectrodeHoweverasnopre-orpost-peakwereobservedwearedealingherewiththeweakadsorption[9]AdsorptionalsoplaysasignificantroleinthecaseofvitaminB2forwhichboththepre-andpost-peakswereobservedindicatingitsstrongaffinitytothemercuryelectrodes To overcome this issue attempts with Triton X-100 were performed TritonX-100isaneutralsurfactant thateasilyadsorbsat thesurfaceof themercury
16 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2Differentialpulseadsorptivestrippingvoltammogramsof(A)vitaminB9and(B)vitaminK3ndash1reduction recorded in the concentration range from blank to 01 and from blank to 04 mg L
respectivelyInsetcorrespondingcalibrationcurvesSupportingelectrolyte(A)McIlvainebufferndash1(pH=52)(B)04molL phosphatebuffer(pH=82)
electrode partially blocking its surface Doing so it prevents the undesiredadsorption of other molecules and thus allows to obtain a linear relationshipbetweenthepeakcurrentandthevitaminconcentration(Fig3)UnfortunatelyduetotheblockingoftheelectrodesurfacetheslopesofthecalibrationlinesaresmallerincomparisontotheonesobtainedintheabsenceofanysurfactantsThismeans that the sensitivity defined as the increase in current caused by a unitincrease in concentration and the resolution understood as the possibility todistinguishsmallvariationinconcentrationarecorrespondinglydecreased Figure3alsopresentsthepossibilitytodeterminemultiplevitaminsinasinglerunChosenvitaminshavewell-separatedpotentialsandtheydonotinterferewitheachotherthereforenoadvancedmultivariatecalibrationstrategiesareneededTheproblemsinthesimultaneousanalysisincludevarioussensitivitieswith respect to the studied analytes and differences in the influence of themeasurementconditionsontherecordedsignalsDuetothattheexperimentalconditionswillneverensurethehighestpossiblesignalvaluesforallanalyzedcompounds
4Conclusions
Differential pulse voltammetry in conjunction with the controlled growthmercurydropelectrodeisaperfecttoolforquantitativeanalysesofvitaminsTheadsorptionofvitaminB2andK3canbepreventedbytheadditionoftheneutralsurfactantTritonX-100whichselectivelyblockstheworkingelectrodesurface
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 17
Fig 3Cathodic voltammograms for the simulatanous determination of vitamin B2 B3 and K3Depictedintheinsetsarethevoltammogramsafterbackgroundsubtractionwiththecorresponding
ndash1callibrationplotsSupportingelectrolye04molL phosphatebuffer(pH=82)with40ppmTritonX-100AccumulationconditionsE =ndash005Vt =20sacc acc
The proposed methodology allows for the simultaneous determination ofmicromolaramountsofvitaminB2B3andK3Suchaprocedurewillhelptoreduce the time and costs of analyses of multivitamin formulations and foodproducts
Acknowledgments
RPhasbeenpartlysupportedbytheEUProjectPOWR0302-00-00-I00416
References
[1] Combs GF JrTheVitaminsFundamentalAspects inNutritionandHealth 3rd ed IthacaElsevierAcademicPress2008
[2] Lovander MD Lyon JD Parr DL Wang J Parke B Leddy J Review Electrochemicalpropertiesof13vitaminsAcriticalreviewandassessmentJElectrochmSoc165(2018)G18ndashG49
[3] Brunetti B Recent advances in electroanalysis of vitamins Electroanalysis 28 (2016)1930ndash1942
[4] BrubacherGMuller-MulotWSouthgateDATMethods forDeterminationofVitamins inFoodNewYorkElsevier1985
[5] Szpikowska-Sroka B A simple and sensitive analytical method for the determination ofthiamineinpharmaceuticalpreparationsJAnalChem68(2013)218ndash222
[6] PetteysBJFrankELRapiddeterminationofvitaminB (riboflavin)inplasmabyHPLCClin2
ChimActa412(2011)38ndash43[7] ZhangZXuJWenYZhangJDingWTheelectro-syntesizedimprintedPEDOTfilmasa
simple voltammetric sensor for highly sensitive and selective detection of vitamin K in3
poultrydrugsamplesSynthMet230(2017)79ndash88[8] JedlinskaKStrusMBasBAnewelectrochemicalsensorwiththeRefreshableSilverLiquid
Amalgam Film multi-Electrode for sensitive voltammetric determination of vitamin K2(menaquinone)ElectrochimActa265(2018)355ndash363
[9] SouthamptonElectrochemistryGroupInstrumentalMethodsinElectrochemistryChichesterHorwood1985
18 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Carbonelectrodesarecommonlyappliedtosensitiveelectrochemicaldetectionofneurotransmittersegdopamine (nor)epinephrineandserotonin in-vivoandin-vitro[12]Neverthelessadsorptionofhigh-molecularweightbiomoleculesinthe matrix on a sensing electrode which then hinders the electron transferreactionofneurotransmitterswillresultinbiofoulingofelectrodesThisremainsachallengingproblemasbiofoulingwillcompromiseelectrochemicalmeasure-mentsThusseveralstrategies foraddressingbiofoulinghavepreviouslybeenreported[34] Thisworkreportsonaneffectiveapproachforminimisingbiofoulingbasedonthehypothesisthatahydrophobicelectrodesurfacewillrepelagainstadsorptionof amphiphilic biomolecules Briefly structurally small conical-tip electrodes
Dopamine detection at antifouling conical-tip carbon electrodes
a a a aSIMONABALUCHOVA JANKLOUDA JIR IBAREK KAROLINASCHWARZOVA -PECKOVA bDANNYKYWONG
a UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova812800PragueCzechRepublicsimonabaluchovanaturcunicz
b DepartmentofMolecularSciencesMacquarieUniversitySydneyNSW2109Australia
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 19
AbstractA significant achievement in this work is the development ofantifoulingconical-tipcarbonelectrodes(~27micromtipdiameterand~165micromaxiallength)suitablefordetectionoftheneurotransmitterdopamine in-vivo These electrodes were hydrogenated using adiphenylsilanereductionmethodtoyieldahydrophobicsurfacetodeteradsorptionofamphiphilicbiomoleculesInitiallyhydrogenatedcarbonelectrodeswereelectrochemicallycharacterisedusingseveralredoxmarkersThedegreeofantifoulingwasthenassessedbythevoltammetricsignalchangeofdopamineattheseelectrodesbeforeand after being incubated in a fouling solution containing bovineserumalbumincytochromeC(bothareproteins)andcaproicacid(alipid) In our work we have obtained only a 69 (standarddeviation35N=40)decreaseindopaminesignalsatthehydro-genated carbon electrodes These results strongly support thediphenylsilanereductionstrategyforthedevelopmentofantifoulingbiosensorsfordopaminedetectioninbiologicalmatrices
Keywordsantifoulingelectrodesdiphenylsilanereduction
methodhydrogenatedconical-tip
carbonelectrodesvoltammetricdopamine
detection
(denoted as CTEs) are fabricated by thermally pyrolysing acetylene gas in anitrogenatmospheretodepositcarbonatthetipandontheshankofpulledquartzcapillaries [5] Spectroscopic studies confirmed that the electrode surface
2 3consistsofsp -likegraphiticcarbonandsp -hybridiseddiamond-likecarbon[6]In addition there is also a range of carbon-oxygen functionalities includingcarbonylquinonecarboxylphenolsalcoholsandethergroupsontheelectrodesurface[6]whichcaninteractwithspectatorbiomoleculesthroughdipole-dipoleorion-dipoleinteractionleadingtotheirirreversibleadsorptionontheelectrodesurface[7]HoweverbysubjectingthesecarbonelectrodestosilanereductionCndashObondsareconvertedtoCndashHbondsandphenolicgroupsaretransformedtosiloxane dendrimers [6] to yield a more hydrophobic carbon surface that isexpectedtobesimilarlylesssusceptibletobiofoulingcomparedtoboron-dopeddiamondelectrodes[78] In this work we will present a methodology involving diphenylsilanereduction to fabricate physically small hydrogenated conical-tip carbonelectrodes(denotedasHCTEs)withanti-foulingcapabilityBothCTEsandHCTEswere electrochemically characterised using several redox probes to elucidatetheirsurfacepropertiesbeforeevaluatingtheirresistancetobiofoulingduringdopaminedetectionin-vitro
2Experimental
21Reagentsandchemicals
Analyticalgradereagents(Sigma-AldrichAustralia)including4-methylcatecholhexaammineruthenium(III) chloride potassium hexacyanoferrate(III) dop-amine hydrochloride sodium phosphate dibasic citric acid perchloric acidpotassiumchloride sodiumhydroxide anhydrousdichloromethanediphenyl-silanetris-(pentafluorophenyl)boraneandgraphitepowderwereusedas-recei-ved Ultra-high purity gases acetylene and nitrogenwere obtained from BOCGases (Australia) All aqueous solutionswere preparedwith deionisedwater(MilliporeMiliplusQsystemUSA)witharesistivityof182MΩcm
22Instrumentation
Chronoamperometric and voltammetric experiments were carried out usingalow-currentpicostateDAQoperatedbyanEChemversion212softwareviaanE-corderinterface(eDAQPtyLtdAustralia)Athree-electrodeset-upinvolving
minus1eitheraCTEorHCTEasaworkingelectrodeaAgAgCl(3molL KCl)referenceelectrode (Bioanalytical SystemsUSA)andaplatinumwire counterelectrode
minus1(CypressSystemsUSA)wasusedCyclicvoltammetryatascanrateof100mVs anddifferential pulse voltammetry (pulse height +25mV pulsewidth 50ms
minus1sampling time 20 ms and scan rate 20mVs ) were used in this work All
20 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
electroanalyticalexperimentswereperformedinanaluminiumFaradaycageatanambienttemperature(23plusmn1degC)
23Preparationofhydrogenatedconical-tipcarbonelectrodes
Asreportedpreviously[5]structurallysmallCTEswerefabricatedbythermallypyrolysing C H (a pressure of 50 kPa) in a pulled quartz capillary (Sutter2 2
minus1InstrumentUSA)housedinaN atmosphere(counterflowof60mLmin )Prior2
to hydrogenation the catalyst tris(pentafluorophenyl) borane (100 mg) wasdissolvedinanhydrousCH Cl (50mL)bystirringfor5minbeforethehydroge-2 2
natingagentdiphenylsilane(25μL)wasaddedCTEswerethenplacedinthereagentmixturefor2hThepreparedHCTEsweredriedovernightbeforeuse
24Biofoulingexperiments
A laboratory synthetic fouling solution consisting of 4 (wv) bovine serumalbumin001(wv)cytochromeC(bothareproteins)and10(vv)caproicacid(alipid)waspreparedbyhomogenisingtheminapH=74citrate-phosphate
minus1buffer(01molL )AllfoulingcompoundswereacquiredfromSigmaAldrichAustralia
3Resultsanddiscussion
31Electrochemicalcharacterisation
minus1InthisworkallCTEswerecharacterisedbycyclicvoltammetryof10mmolL 3+ minus1[Ru(NH ) ] in10molL KClAsdisplayedinFig1(A)onlyCTEsthatshow3 6
asigmoidal-shapedvoltammogramwithasmallchargingcurrentwereemployedinfurtherexperimentsUsingchronoamperometry[5]ameantipdiameterof27μm(standarddeviation(SD)28μmN=142)ameanaxiallengthof165μm(SD=114μmN=142)wereestimatedfortheseCTEs TocomparesurfacecharacteristicsofbothCTEsandHCTEs cyclicvoltam-
minus1 3+2+ minus1 minus1metryof (1)10mmolL [Ru(NH ) ] in10molL KCl (2)10mmolL 3 63minus4minus minus1 minus1[Fe(CN) ] in 10 mol L KCl and (3) 10 mmol L 4-methylcatechol in6minus101molL HClO wasconductedatthesameelectrodesbeforeandafterhydroge-4
nation The results obtained are shown in Fig 1(A-C)We observed a ~20(SD=5N=10)decreaseinthelimitingcurrentofallthreeredoxmarkersafterdiphenylsilane reduction most likely attributable to the hindrance to theirelectron transfer reactions by the phenylsiloxane group formed on HCTEs
3minus4minusMoreover as an inner-sphere redox probe both [Fe(CN) ] and 4-methyl-6
catechol reactionsare sensitive to thepresenceofoxygen functionalitiesonacarbonsurface [7]Accordingly theconversionof these functionalities toCndashHbondsbydiphenylsilanereductionwasexpectedtoyieldmoresluggishelectron
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 21
transfer kinetics at HCTEs as supported by a negative potential shift (from3minus4minus+75mVtominus10mV)inthecyclicvoltammogramof[Fe(CN) ] andapositive6
potentialshift(from+580mVto+675mV)inthecorrespondingcyclicvoltammo-2gram of 4-methylcatechol In addition the conversion of sp -carbon to
3sp -diamond-likecarbon[6] isalsoexpectedtoreducetheconductivityof thecarbonelectrodesurface
32Dopaminedetectionduringbiofoulingexperiments
minus1Theelectrochemicalbehaviourof1mmolL dopamine inapH=74citrate-minus1phosphate buffer (01 mol L ) at CTEs and HCTEs was studied by cyclic
voltammetryTheresultsobtainedareshown inFig1(D)Acomparable12decrease(SD=6N=10)inthedopamineoxidationlimitingcurrenttothatof4-methylcatechol was observed A positive potential shift from +285 mV to+305mVinthevoltammogramsisalsoaccountedforasdescribedabove
22 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
minus1 3+ minus1 minus1Fig 1Cyclicvoltammetryof(A)10mmolL [Ru(NH ) ] in10molL KCl (B)10mmolL 3 63minus4minus minus1 minus1 minus1[Fe(CN) ] in10molL KCland(C)10mmolL 4-methylcatechol in01molL HClO at3 4
minus1(a)aCTEand(b)aHCTE(D)10mmolL dopamineinapH=74citrate-phosphatebufferrecordedminus1at(a)aCTEandaHCTE(b)beforeand(c)afterbiofoulingScanrate100mVs
Next HCTEs were incubated in a synthetic fouling solution containing4(wv)bovineserumalbumin001(wv)cytochromeC(bothareproteins)and 10 (vv) caproic acid (a lipid) for 30 min Cyclic voltammetry of
minus110mmolL dopamineattheseHCTEswasthenconductedinapH=74citrate-phosphatebuffertoevaluatetheirantifoulingpropertyNotablyaconsiderable515(SD=183N=6)decreaseindopaminesignalwasobservedatCTEsIncontrastonlyacorresponding69decrease(SD=35N=40)wasestimatedatHCTEsasshowninFig1(D)Clearlythisrepresentsamajorimprovementinthe antifouling capability of HCTEs obtained using diphenylsilane reductioncomparedtoCTEsandotherpreviouslytestedhydrogenatingagents[9]includingn-butylsilane (35decrease) triethylsilane (23decrease) andphenylsilane(18decrease)Thereforethisworkhassuccessfullydemonstratedtheeffecti-venessofdiphenylsilanereductionmethodindevelopingantifoulingelectrodesfordopaminedetection
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 23
Smallconical-tipelectrode
As-prepared Hydrogenated
minus1LinearrangemicromolL 1ndash10 1ndash20minus2 minus2Intercept10 pAmicrom 108plusmn004 273plusmn011
minus3 minus2 minus1Slope10 pALmicrom micromol 128plusmn007 443plusmn012R 0993 0998
minus1LimitofdetectionmicromolL 100 077
Table 1Analyticalparametersofconcentrationdependencesofdopamineobtainedbydifferentialpulse
minus1voltammetryinapH=74citrate-phosphtebuffer(01molL )Allquoteduncertaintiesrepresentthe95confidenceintervalandthecorrelationcoefficient(R)wasfoundtobestatisticallysigni-ficantatthe95usingStudentrsquost-test
Fig 2DifferentialpulsevoltammetryofdopamineataHCTEinapH=74citrate-phosphatebufferminus1 minus1(01molL )atconcentrations(a)1(b)2(c)4(d)6(e)8(f)10and(g)20μmolL
WehavealsostudiedtheconcentrationdependenceofdopamineinapH=74citrate-phosphatebufferbydifferentialpulsevoltammetryAtypicalcalibrationplotobtainedisshowninFig2Theanalyticalparametersandestimatedlimitsofdetection are summarised inTable 1 These results show thatHCTEs outper-formed CTEs because they exhibit a ~35times higher sensitivity a 23 lowerdetectionlimitandawiderlinearrange
4Conclusions
InthisstudyphysicallysmallHCTEswithanti-foulingcharacteristicsachievedbyhydrogenationusingdiphenylsilanereductionweresuccessfullyfabricatedandelectrochemically characterised using several redox probes Next dopaminedetectionwasperformedbeforeandafterincubationofelectrodesinasyntheticfoulingsolutioncontainingahighconcentrationofbiomoleculesOnlyalow69(SD=35)decreaseindopaminelimitingcurrentwasachievedatHCTEsobtain-edbydiphenylsilanereductionindicatingtheirsignificantlylesssusceptibilitytobiofoulingthanCTEsThesepromisingresultsindicatethatantifoulingHCTEswillpotentiallybenefitthedevelopmentofbiosensorsfordopaminedetectionin-vivoinbiologicalmedia
Acknowledgments
ThisresearchwasperformedwithintheframeworkofSpecificUniversityResearch(SVV260560)FinancialsupportsprovidedbytheGrantAgencyofCharlesUniversity(project390119)andbytheCzechScienceFoundation(project20-03187S)aregratefullyacknowledgedSBandJKalsothankthe Mobility Fund of Charles University and Hlavkova nadace for providing funding for theirresearchinternshipsatMacquarieUniversitySydneyAustralia
References[1] BaranwalAChandraPClinicalimplicationsandelectrochemicalbiosensingofmonoamine
neurotransmittersinbodyfluidsinvitroinvivoandexvivomodelsBiosensBioelectron121(2018)137ndash152
[2] CaoQPuthongkhamPJillVentonBReviewnewinsightsintooptimizingchemicaland3Dsurface structuresof carbonelectrodes forneurotransmitterdetectionAnalMethods11(2019)247ndash261
[3] LinP-HLinB-RAntifoulingstrategiesinadvancedelectrochemicalsensorsandbiosensorsAnalyst145(2020)1110ndash1120
[4] HanssenBLSirajSWongDKYRecentStrategiestoMinimiseFoulinginElectrochemicalDetectionSystemsRevAnalChem35(2016)1ndash28
[5] McNallyMWongDKYAnin-vivoprobebasedonmechanicallystrongbutstructurallysmallcarbonelectrodeswithanappreciablesurfaceareaAnalChem73(2001)4793ndash4800
[6] SirajSMcRaeCRWongDKYEffectiveactivationofphysicallysmallcarbonelectrodesbyn-butylsilanereductionElectrochemCommun64(2016)35ndash41
[7] ParkJShowYQuaiserovaVGalliganJFinkGDSwainGMDiamondmicroelectrodesforuseinbiologicalenvironmentsJElectroanalChem583(2005)56ndash68
[8] ShinDTrykDAFujishimaAMerkociAWang JResistance to surfactantandproteinfoulingeffectsatconductingdiamondelectrodesElectroanalysis17(2005)305ndash311
[9] Roshni RAnAntifouling Structurally Small Carbon Electrode forDetectionof theNeuro-transmitterDopaminePhDThesisMacquarieUniversitySydney2019
24 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Theanalyticalperformancesofenzymaticbiosensorsarestronglyaffectedbytheenzyme immobilization process There is no universal technique for enzymesattachmentThereforespecialattentionshouldbepaid to theselectionof theappropriatesupportandthedevelopmentoftheoptimalbindingstrategyinordertoensure thebestcharacteristicsof immobilizedenzymeDespiteavarietyofpreviouslyreportedcovalentimmobilizationmethodsfordifferentenzymesthepresentedprocedurescanbehardlycomparedtofindtheoptimalonesbecauseofdifferentanalyticalmethodsandexperimentalconditionsusedUptodatethere
A comparative study of covalent glucose oxidase and laccase immobilization techniques at powdered supports for biosensors fabrication
ab a bSOFIIATVORYNSKA JIR IBAREK BOHDANJOSYPCUK
a UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova2030812843Prague2CzechRepublicsofiiatvorynskajh-instcascz
b JHeyrovskyacuteInstituteofPhysicalChemistryoftheCzechAcademyofSciences Dolejškova318223Prague8CzechRepublic
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 25
AbstractInordertodeveloptheoptimalstrategyandtodeepentheknowledgeinthefieldofenzymeimmobilizationthreedifferenttechniquesofcovalentbindingfortwoenzymes(glucoseoxidaseandlaccase)atpowdered surfaces were compared Immobilization protocol wasoptimized by changing supports (twomesoporous silica powders(SBAminus15 MCMminus41) and a cellulose powder) the functionalizedgroupsintroducedatsupportsurfaces(minusNH andminusCOOH)andthe2
methodsofactivation(glutaraldehydeandcarbodiimide)Aminoandcarboxyl functionalized mesoporous silica and cellulose powderswerepreparedbysilanizationusing(3-aminopropyl)triethoxysilaneandcarboxyethylsilanetriolrespectivelyItwasfoundthatcouplingof both enzymes by their ndashNH groups through glutaraldehyde to2
ndashNH functionalized supports in particular SBA15minusNH and2 2
celluloseminusNH forglucoseoxidaseMCM41minusNH forlaccaseshowed2 2
thehighestactivityandthebeststability
Keywordsbiosensorcovalentimmobilizationenzymaticreactorglucoseoxidaselaccase
is still a lack of the comparative systematic studies focusing on the enzymesimmobilizationonthevarioussupportsusingdifferenttechniques The aim of this work is the systematic comparative study of the differenttechniques for covalent coupling of the enzymeswhich ensures not only thedevelopmentoftheoptimalimmobilizationstrategyfortheselectedenzymesbutalsoenablestofindoutsometendenciesinenzymeattachmentprocessgenerallyThusthisworkisfocusedonadetailedanalysisoftheeffectofthekindofsupportits anchor groups and the activation methods on activity and stability ofimmobilizedenzymesTwoenzymeswithdifferentnature(glucoseoxidase(GOx)andlaccase(Lac))werechosenasthetestingbioreceptors
2Experimental
21Reagentsandchemicals
AllchemicalswereofpaorbettergradeGlucoseoxidasefromAspergillusnigerminus1(GOxEC11341452Umg )laccasefromTrametesversicolor(LacEC11032
minus1129Umg )D-(+)-glucosedopamineglutaraldehyde(GAgradeII25aqueoussolution) N-(3-dimethylaminopropyl)-Nʹ-ethylcarbodiimide hydrochloride(EDC ge980) N-hydroxysuccinimide (NHS ge970) (3-aminopropyl)-triethoxysilane (APTES) mesoporous silica powder SBAminus15 (particle size
2 minus12ndash6μmporesizeasymp7nmsurfaceareaasymp600m g )mesoporoussilicapowder2 minus1MCMminus41 (pore size 21ndash27 nm surface area asymp 1000 m g ) cellulose (Cell
microcrystalline powder particle size 20 microm) were purchased from SigmaAldrichCarboxyethylsilanetriol(CEST25aqueoussolution)waspurchased
regfromabcr (Germany)
22Instrumentation
Amperometric measurements were carried out at room temperature usingcomputer-controlled electrochemical stand (Polaro-Sensors Czech Republic)withMultiElchemv31software(JHeyrovskyInstituteofPhysicalChemistryoftheCAS)Flowinjectionanalysis(FIA)withthethree-electrodelaboratory-madeflow-through cellwas usedworking electrode minus tubular detector of polishedsilversolidamalgam(TD-p-AgSAlaboratory-madeinnerdiameter05mmtheamalgamtube length60mm) referenceelectrodeminusaminiaturizedsaturatedcalomelelectrodebasedonsilverpasteamalgam[1](laboratory-madeithasthesamepotential as classical saturated calomel electrode) auxiliary electrodeminusplatinum wire (diameter 10 mm length 10 mm) The system for FIA withelectrochemicaldetectioncomprisedofalinearsyringepumpa2-position6-port
regsampleinjectorvalveaninjectionlooplaboratory-madeofTeflon (PTFE)tubing(100μL) a solenoid operatedmicro-pumpan enzymatic reactor and a flow-through cell for TD The enzymatic reactor consists of a tube filled by theenzymaticpowder
26 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
3Resultsanddiscussion
Basedonthedatareportedintheliterature[23]andontheresultsofourpreviousworks[4ndash8]forthisstudyCellandmesoporoussilicapowders(namelySBAminus15andMCMminus41)havebeenselectedas thepotentialpromisingsupports for thecovalent enzyme immobilizationBecauseof thehigh contentof surficial ndashOHgroupswhich are capable of chemical reactions these supports can be easilyfunctionalizedThewell-knownandfrequentlyutilizedtechniqueofsilanizationhasbeenusedtomodifythesurfacesofSBA15MCMminus41andCellbythedesiredfunctionalizedgroupsAminosilaneAPTESwasappliedtoformminusNH groupson2
thematrixsurfaceswhereascarboxylsilaneCESTwasusedtointroducendashCOOHgroups Generallytheprocedureofthecovalentimmobilizationofenzyme(eitherLacorGOx)onthefunctionalizedsupportconsistsofthreestepsI Synthesisofthefunctionalizedsupportwhichmeansthemodificationofthematrix(MCMminus41SBAminus15andCell)withsuitableanchoredgroups(minusNH or2
minusCOOH)II Activationstepofthefunctionalizedsupportwithspecificactivatingagents(glutaraldehydeorEDCNHS)tomakeitreactivetowardsenzyme
IIIEnzyme(LacorGOx)couplingtotheactivatedsupport
To investigate the effect of support its surface functionalizedgroups and themethodsofactivationontheefficiencyofthecovalentenzymeimmobilizationthreedifferentstrategies(ABandC)forLacandGOxattachmenthavebeenusedThedetailsoftheusedtechniquesandthedenotationsofthepreparedenzymaticpowdersaresummarizedinTable1(nextpage)ToexaminetheefficiencyofLacandGOximmobilizationtheenzymaticreactors(filledbytheenzymaticpowderspreparedwithdifferenttechniques)coupledwithTDwereusedforamperometricdetermination of dopamine and glucose respectively in flow systems Theprincipleofglucosedetection isbasedonamperometricmeasurementsof theenzymatically consumed oxygen whereas dopamine was detected by thereductionoftheenzymaticallyoxidiseddopamine Asdepicted inFig1 thebiosensors responsesare stronglyaffectedby thestrategyusedforLacorGOximmobilizationAsshowntheresponsesofLacandGOx biosensors decrease in the order strategy A gt strategy B gt strategy Cirrespectiveofthetypeofsupport It isclearlyseenthataminofunctionalizedsupports(SBA15minusNH MCM41minusNH andCellminusNH )providehigheractivitiesof2 2 2
the immobilized Lac andGOx than these supports functionalized by carboxylgroups(SBA15minusCOOHMCM41minusCOOHandCellminusCOOH)BycomparingactivitiesofimmobilizedenzymesusingstrategiesAandBtheinfluenceoftheactivationagenthasbeenevaluatedThebestresultsforbothenzymeswereobtainedforndashNH functionalizedsupportsactivatedbyGAItcouldbeexplainedbythefact2
thatGAcontrary tocarbodiimidewithnonemolecularspaceprovidesa long
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 27
spacerarmensuringminimalsterichindrancesforenzymesbindingItcanbeconcludedthatthecovalentimmobilizationofbothenzymesbytheirndashNH groups2
viaGAtondashNH functionalizedmesoporoussilicapowders(strategyA)provided2
the highest activities Interestingly in the similar comparative studies it isreportedthatamongndashOHminusCOOHandndashNH functionalizedsupportsactivatedby2
divinylsulfonecarbodiimideandGArespectivelythelastonewasfoundasthemostsuitabletechniqueforthecovalentbindingofLac[9]invertase[10]andpepsin[11]
28 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Tab
le 1
Theprinciplesofthecovalentimmobilizationmethodsofenzymesusedinthisstudy
StrategyA
StrategyB
StrategyC
Support
SBAminus15MCMminus41Cell
SBAminus15MCMminus41Cell
SBAminus15MCMminus41Cell
Supportfunctio-
minusNH
minusNH
minusCOOH
22
nalizedgroup
Activationagent
Glutaraldehyde(GA)
Carbodiimide
Carbodiimide
(EDCNHS)
(EDCNHS)
Enzymereactive
minusNH
minusCOOH
minusNH
22
group
Typeofbond
secondaryamine
amide
amide
Denotationsofthe
GOxminusNHminusCHminus(CH)minusCHminusNHminusSBA15
GOxminusC(=O)minusNHminusSBA15
GOxminusNHminusC(=O)minusSBA15
23
preparedenzy-
GOxminusNHminusCHminus(CH)minusCHminusNHminusMCM41
GOxminusC(=O)minusNHminusMCM41
GOxminusNHminusC(=O)minusMCM41
23
maticpowders
GOxminusNHminusCHminus(CH)minusCHminusNHminusCell
GOxminusC(=O)minusNHminusCell
GOxminusNHminusC(=O)minusCell
23
LacminusNHminusCHminus(CH)minusCHminusNHminusSBA15
LaxminusC(=O)minusNHminusSBA15
LacminusNHminusC(=O)minusSBA15
23
LacminusNHminusCHminus(CH)minusCHminusNHminusMCM41
LacminusC(=O)minusNHminusMCM41
LacminusNHminusC(=O)minusMCM41
23
LacminusNHminusCHminus(CH)minusCHminusNHminusCell
LacminusC(=O)minusNHminusCell
LacminusNHminusC(=O)minusCell
23
When the effect of the method of the covalent enzyme coupling on thebiosensorstabilitywasevaluateditwasfoundthatLacboundedtondashNH functio-2
nalizedsupportsviaGA(strategyA)hasshownthehigheststability(gt65oftheinitial responses after 1 month) compared to other strategies whereas GOximmobilizedwithtwostrategies(AandB)possessedapproximatelysimilarhighstability(gt80oftheinitialresponsesin1month)BothenzymesboundedviandashNH groupstondashCOOHfunctionalizedsupportsthroughEDCNHS(strategyC)2
showedquitelowstability
4Conclusions
Threedifferent strategies including the support selection the anchor surfacegroups and the activationmethod havebeen compared for efficient covalentimmobilization of Lac and GOx The results showed that ndashNH functionalized2
supports(SBA15minusNH CelluloseminusNH forGOxandMCMminusNH forLac)activatedby2 2 2
GAmaybeusedtoeffectivelybindenzymesintermsofhighactivityandstability
Acknowledgments
ThisworkwasfinanciallysupportedbytheGrantAgencyofCharlesUniversityinPrague(Project1356120)theGrantAgencyoftheCzechRepublic(Project20-07350S)anditwascarriedoutwithintheframeworkofSpecificCharlesUniversityResearch(SVV260440)
References
[1] YosypchukBBarekJYosypchukOPreparationandpropertiesofreferenceelectrodesbasedonsilverpasteamalgamElectroanalysis23(2011)2226minus2231
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 29
Fig 1Effectof the covalent attachment techniqueson (A) laccaseand (B) andglucoseoxidaseminus1biosensor responses Experimental conditions (A) c = 500 micromol L E = minus50 mVDOP det
minus1 minus1v = 01 mL min V = 40 microL carrier solution 01 mol L acetate buffer pH = 48flow DOPminus1 minus1(B)c =500micromol L E =minus1100mVv ==01mLmin V =40microL carrier solutionGlu det flow Glu
minus1 minus101molL acetatebuffer0001molL Na EDTApH=652
[2] LiuYChenJYEnzymeimmobilizationoncellulosematrixesJBioactCompactPolym31(2016)553ndash567
[3] Hartmann M Kostrov X Immobilization of enzymes on porous silicas ndash benefits andchallengesChemSocRev42(2013)6277minus6289
[4] JosypcukOBarekJJosypcukBElectrochemicalbiosensorsbasedonenzymaticreactorsfilledbyvarioustypesofsilicaandamalgampowdersformeasurements inflowsystemsElectroanalysis28(2016)3028minus3038
[5] Josypcuk O Barek J Josypcuk B Amperometric determination of catecholamines byenzymaticbiosensorsinflowsystemsElectroanalysis30(2018)1163minus1171
[6] TvorynskaSBarekJJosypcukBAmperometricbiosensorbasedonenzymaticreactorforcholinedeterminationinflowsystemsElectroanalysis31(2019)1901minus1912
[7] TvorynskaSBarekJJosypcukBFlowamperometricbiosensorbasedontwoenzymaticreactors (acetylcholinesterase-choline oxidase) for the detection of neurotransmitteracetylcholine In Proceedings of the 15th International Students Conference ldquoModernAnalyticalChemistryrdquoKNesmerak(ed)PragueFacultyofScienceCharlesUniversity2019p61minus66
[8] TvorynskaSBarekJJosypcukBAcetylcholinesterase-cholineoxidase-basedmini-reactorscoupledwithsilversolidamalgamelectrodeforamperometricdetectionofacetylcholineinflowinjectionanalysisJElectroanalChem860(2020)113883
[9] RekucAKruczkiewiczPJastrzembskaBLiesieneJPeczynska-CzochWBryjakJLaccaseimmobilizationonthetailoredcellulose-basedGranocelcarriersIntJBiolMacromol42(2008)208minus215
[10] Bryjak J Liesiene J S tefuca V Man-tailored cellulose-based carriers for invertaseimmobilizationCellulose15(2008)631minus640
[11] SzałapataKOsinska-JaroszukMBryjakJJaszekMJarosz-WilkołazkaANovelapplicationofporousandcellularmaterialsforcovalentimmobilizationofpepsinBrazJChemEng33(2016)251minus260
30 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Carbohydrates are crucial for energy structure and signaling in the humanbody[1]Thereisavarietyofcarbohydratesbutthemostimportantoneforlifeisglucoseasitisfundamentalinthemetabolismandphotosynthesis[2]GlucoseisclassifiedashexoseThesemonosaccharidesonlydifferinthepositionofhydroxylsubstituentsinsomecasesInadditiontothestructuralsimilaritiesthesemole-culeslackachromophoreandarenoteasilyionizable(pK ~12)Thusdetectiona
intheUVregionandseparationofanalytesbycapillaryelectrophoresis(CE)arechallenging [1 3] At the moment there are many different techniques for theanalysis of carbohydrates commonly including time-consuming derivatizationstepsoreluentswithhighpHvalues(pHgt12)inionchromatographyAwell-esta-blishedtechniquefortheanalysisofcarbohydratesishigh-performanceanion-exchangechromatographywithpulsedamperometricdetection(HPAE-PAD)[4]Electrochemical detection like AD is matching miniaturization simple instru-mentationlowcostandrobustnessandthusisoftenusedforflow-basedsystemssuchasCEandflowinjectionanalysis(FIA)[5]
Capillary flow injection analysis with electrochemical detection for carbohydrate analysis
NICOLEHEIGLFRANK-MICHAELMATYSIK
InstituteofAnalyticalChemistryChemo-andBiosensorsFacultyofChemistryandPharmacyUniversityofRegensburgUniversitaumltsstraszlige3193053RegensburgGermanynicoleheiglchemieuni-regensburgde
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 31
AbstractAsimplecapillaryflowinjectionanalysissystemwithamperometricdetection was arranged for the development of a method for fastoptimization of detection conditions in the context of thedetermination of carbohydrates by means of electrochemistry-capillary electrophoresis-mass spectrometry This setup is free ofelectricalinterferencebyhighvoltageandisperfectforstudyingtheoxidationofvariousanalytesFurthermoreitassureseasycouplingtoMS and thus is an useful tool to investigate the correspondingoxidationproductsofananalyte
Keywordscapillaryflowinjection
analysiscarbohydratesmassspectrometrydisposableelectrodespulsedamperometric
detection
Inthiscontributioncapillaryflowinjectionanalysis(CFIA)withADwillbepresentedasamethodtoapplyandtestADforthedetectionofmonosaccharidesondifferentdisposablethin-filmorscreen-printedelectrodesCFIAwaschosenoverconventionalFIAforthispurposeasthegravityflowinCFIAisstableforalongertimeandverylowsampleconsumptioncanbeachieved[6]TheCFIAsystemwasarrangedassimpleaspossibleandperformedhydrodynamicallytoavoidanyinterferencesFurthermoreitassuresthecouplingoftheflowsystemtoamassspectrometerThusthesamesetupasusedforCFIAcanbeutilizedforcapillaryelectrophoresis-massspectrometry(CE-MS)experimentsbychangingthe flow through the electrochemical flow cell in opposite direction In futureexperimentstheexperienceintermsofADonthoseelectrodeswillbeusedtodevelopelectrochemicalpretreatmentprotocolsforcarbohydratedeterminationbyCE-MS
2Experimental
21Reagentsandchemicals
The following chemicals were used for this study all of analytical gradeAmmoniumacetate(NH OAc)wasobtainedfromMerck(DarmstadtGermany)4
and ferrocene methanol (FcMeOH) from ABCR (Karlsruhe Germany) Milli-Qregwater(182MΩcm)wasgeneratedbyaMilli-QAdvantageA10 system(Merck
Millipore Darmstadt Germany) Carrier solution was prepared by dissolvingndash1NH OAc (50mmolL ) in Milli-Q water FcMeOH solution was prepared by4
dissolvingFcMeOHincarriersolution
22Instrumentation
Electrochemical measurements were performed using a microAutolab Type IIIpotentiostatgalvanostat (Metrohm Autolab B V Utrecht Netherlands)controlledbyNOVA20softwareforexperimentalcontrolanddataacquisitionCFIAwasperformedusingthesetupillustratedinFig1(A)consistingofacarrierreservoirsamplevialandtwofusedsilicacapillaries(PolymicroTechnologiesPhoenix AZ USA inner diameter 100 microm length inlet 40 cm length outlet10cm)connectedtoacommerciallyavailableflowcellfromMicruxTechnologies(model ED-FLOW-CELL Oviedo Spain) Inside of the flow cell the fused silicacapillary was placed in a so-called wall-jet configuration above the workingelectrode of a disposable thin-film gold electrode (model ED-SE1-Au MicruxTechnologiesOviedoSpain)ascanbeseeninFig1(B)Thethin-filmelectrodeswerebasedonathree-electrodesystemwithagoldworkingauxiliaryandquasi-referenceelectrodeThehydrostaticpressurewasachievedbyaheightdifferencebetweeninletandoutletreservoirof30cmresultinginagravityflowofthecarriersolutionthroughafusedsilicacapillaryandsubsequentlythroughtheflowcell
32 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Theinjectionwascarriedoutbyloweringthevialcontainingthecarriersolutiontotheleveloftheoutletexchangingthecarrierreservoirwiththesamplevialandliftingthesamplevialto20cmforadefinedperiodoftimeRe-establishingthecarrierreservoirtookplacethesameway
3Resultsanddiscussion
AsimpleCFIA-ADsystemwasarrangedwherehydrostaticpressurebyaheightdifferencebetweeninletandoutletreservoirresultedinagravitationalflowToobtain general information about the behavior of the assembled CFIA systempreliminaryexperimentswithFcMeOHwereperformedToassurecompatibilitywithMSlateronNH OAcwaschosenastheelectrolyteVariousheightdifferences4
andinjectiontimesweretestedandtheinjectionataheightdifferenceof20cmlasting for 10 s was found to be the optimum concerning feasibility and peakshapes Injections of several solutions of FcMeOH of different concentrationsshowed that the concentration dependence of FcMeOH was linear in theinvestigatedrange(Fig2)Furthermoreexperimentsrevealedthattheinjectionprocedure was established with reasonable precision When repeating the
ndash1injectionof05mmolL FcMeOHincarriersolutionfortentimestherelativestandarddeviationwasfoundtobe3forthemanualinjectionprotocol
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 33
Fig 1(A)Schemeoftheusedcapillaryflowinjectionanalysissetup(1)carrierreservoir(2)samplevial(3)inletcapillarywithalengthof40cmandaninnerdiameterof100microm(4)outletcapillarywithalengthof10cmandaninnerdiameterof100micrombothcapillariesconnectedto(5)acommerciallyavailableflowcellfromMicruxand(6)awastevial(B)Configurationinsidetheflowcell(7)thefusedsilicacapillarywasplacedinaso-calledwall-jetconfigurationabovetheworkingelectrodeof(8)adisposablethin-filmgoldelectrode
4Conclusions
The presented system for CFIA-AD was arranged as simple as possible andrepresents a useful approach for the development of a method for fastoptimization of detection conditions in the context of the determination ofcarbohydratesbymeansofelectrochemistry-CE-MSThesetupisfreeofelectricalinterferencebyhighvoltagecompatiblewithMSandthuspromisingforstudyingtheoxidationofvariousanalytes
References
[1] LuGCrihfieldCLGattuSVeltriLMHollandLACapillaryelectrophoresisseparationsofglycansChemRev118(2018)7867ndash7858
[2] GalantALKaufman RCWilson JDGlucoseDetectionandanalysisFoodChem188(2015)149ndash160
[3] Sarazin C Delaunay N Costanza C Eudes V Gareil P Application of a new capillaryelectrophoreticmethodforthedeterminationofcarbohydratesinforensicpharmaceuticalandbeveragesamplesTalanta99(2012)202ndash206
[4] Rohrer JS Basumallick L Hurum D High-performance anion-exchange chromatographywithpulsedamperometricdetectionforcarbohydrateanalysisofglycoproteinsBiochem78(2013)697ndash709
[5] IslamMAMahbubPNesterenkoPNPaullBMackaMProspectsofpulsedamperometricdetectioninflow-basedanalyticalsystemsndashAreviewAnalChimActa1052(2019)10ndash26
[6] MatysikF-MWernerGTracemetaldeterminationintearsbyanodicstrippingvoltammetryinacapillaryflowinjectionsystemAnalyst118(1993)1523ndash1526
34 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2ndash1 ndash1(A) CFIA-AD recordings of three consecutive injections of (1) 025mmolL (2) 05mmolL
ndash1 ndash1 ndash1(3) 075mmolL and (4) 1mmolL FcMeOH in 50 mmolL NH OAc detection at a Micrux4
thin-filmAuelectrodeataconstantpotentialof03VinaflowcellHydrodynamicinjectionlasted10sataheightdifferenceof20cm(B) Calibration dependence of FcMeOH for CFIA-AD determination and detection at a Micruxthin-filmAuelectrodeataconstantpotentialof03VinaflowcellThestandarddeviationsofpeakheights(n=3)areindicatedbyerrorbars
1Introduction
Topreventsorptiononthecapillarysurfaceandimproveseparationefficiencyand selectivity of determined analytes coatings are formed on the capillarysurface There are two types of ones namelydynamic and covalently bondedcoatings Despite the simplicity of creation dynamic coatings cannot providerequiredreproducibilitywhilecovalentcoatingscontributethestableelectro-osmoticflow(EOF)andhighreproducibilityoftheanalysisInmostcasesthe
Application of covalent coatings based on imidazolium cations for separation and on-line preconcentration of basic and neutral analytes in capillary electrophoresis
a ab aANASTASIAVKRAVCHENKO EKATERINAAKOLOBOVA LIUDMILAAKARTSOVA
a DepartmentofOrganicChemistryInstituteofChemistrySaintPetersburgStateUniversity 26Universitetskiiprospect198504StPetersburgPeterhofRussiakravchenko161216gmailcom
b TheFederalStateInstituteofPublicHealthldquoTheNikiforovRussianCenterofEmergencyandRadiationMedicinerdquoTheMinistryofRussianFederationforCivilDefenceEmergenciesandEliminationofConsequencesofNaturalDisasters54Optikovst197082StPetersburgRussia
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 35
AbstractThemethodofcapillaryelectrophoresis (CE) isactivelydevelopedandmoreandmoreattractsscientistsattentioneveryyearHoweverthesorptionofanalytesonsurfaceoffused-silicacapillarywallsisoneof thesignificantdisadvantagesof thisapproachThe formationofcoatings on the inner capillary surface is typical way to preventsorption and to increase separation efficiency and selectivity ofdetermined analytes Coatings that covalently bonded to capillarywalls is more suitable because they are stable and provides highreproducibilityofanalysisThepresentworkisfocusedonthedevelo-pmentofthemethodofelectrophoreticdeterminationofbiologicalactiveanalytesusingacovalentcoatingbasedonimidazoliumcationsTheeffectofsubstituentinimidazoliumringonmainelectrophoreticparameters was examined It was shown that alkylimidazoliumcoatingscontributetosignificantreducingofbiogenicamineslimitsof detection while β-cyclodextrinimidazolium covalent coatingallowstoseparatebothofhydrophobicandhydrophilicanalytesinonerun
Keywordsbiologicalactiveanalytescapillarycoatingcapillaryelectrophoresisimidazoliumionicliquids
analytes nature determines type of usedmodifiers because suitable ones canprovideaccessorial interactionbetweentheanalytesandthestationaryphaseimproving separation selectivity and efficiency [1 2] Ionic liquidshave beenwidelyusedinanalyticalchemistry[3]andseparationtechniquesparticularlyincapillaryelectrophoresis[4]Earlyresearches[5ndash9]haveshownopportunityofcovalently bonded imidazolium ionic liquids for electrophoretic separationHowever the effect of various substituents in imidazolium ring hasnot beendescribed previously Thus the purpose of this study was to create covalentcoatingsbasedon ionic liquidwithvarioussubstituentsand tocompare theiranalytical capabilities in the electrophoretic separation of biologically activecompounds
2Experimental
21Reagentsandchemicals
(3-Glycidyloxypropyl)trimethoxysilane (GPTMS) hydrochloric acid sodiumdodecyl sulfate (SDS) imidazole 22-diphenyl-1-picrylhydrazyl (DPPH)p-toluen-sulfonylchlorideβ-cyclodextrinhydrocortisone(F)11-deoxycortisol(S)Corticosterone(B)rac-ketoprofen(ndash)-adrenaline(A)L-(ndash)-norepinephrine(NE) DL-normetanephrine (NMN) dopamine (DA) DL-metanephrine hydro-chloride(Met)serotoninhydrochloride(Ser)homovanillicacid(HVA)24-di-hydroxy-benzoic acid (24-DHBA) 34-dihydroxy-L-phenylalanine (DOPA)L-tryptophan(Trp)L-tyrosine(Tyr)werepurchasedfromSigma-Aldrich(USA)1-Bromo-butane1-bromooctanewerepurchasedfromReagentPlus(Ukraine)Sodium dihydrogenphosphate dihydrate acetone NN-dimethylformamide(DMF)wereobtainedfromMerck(Germany)AllreagentsusedwereanalyticalgradeAllsolutionswerepreparedusingdeionizedwater
22Instrumentation
Capillary electrophoresis experiments were carried out using the system ofcapillaryelectrophoresisCAPEL-105M(LumexRussia)withUV-spectrophoto-metricdetector(wavelengthrange190ndash360nm)Separationswereperformedusing 58times49 cm (9 cm to the detector outside diameter 360 microm and innerdiameter50microm) coated silica capillaries (LumexRussia) Thebuffer pHwasmeasuredwithapH-meterHI2210ndash2216(Hanna)
23Capillarycoatingsynthesis
Earlier our research team has proposed the synthesis route for the covalentcoatingsbasedonimidazoliumcationfunctionalizedwithalkylgroup[10]andβ-cyclodextrin [11] All capillarieswere prepared according above-mentioned
36 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
manuscriptsandcharacterizedbytheEOFmobilitymeasurementandscanningelectronmicroscopyThesynthesisconsistedoffollowingstepspreparationofacapillarytocreateacovalentcoating(heatingcapillaryfilledwith2MNaOHat90degCfor1handdryingfollowed)silylationwithGPTMSandfunctionalizationwiththeimidazolesolutionfollowedmodificationbybutyl-andoctylbromideortosyl-β-cyclodextrin(seedetailsin[10]and[11])
24Solutions
A stock buffer solution in concentration 50mM was prepared by dissolvingappropriate amount of sodium dihydrogenphosphate dihydrate in deionizedwateradjustingpHto20with1MhydrochloricacidThisbuffersolutionwasthendilutedwithdeionizedwater
ndash1 Allthesamplestocksolutionswerepreparedwithconcentration10mgmL Thestocksolutionsoftheneurotransmittersandtheirmetabolites(adrenalinenoradrenaline dopamine normetanephrine metanephrine serotonin homo-vanillicacid)and24-dihydroxybenzoicacidasinnerstandardandaminoacids(tryptophan34-dihydroxy-L-phenylalanine tyrosine)wereprepared in01MhydrochloricacidThestocksolutionsofsteroids(hydrocortisone11-deoxycor-tisolandcorticosterone)wereprepared inacetonitrileThestocksolutionsofketoprofen racemate and S-ketoprofen were prepared in acetonitrilewatersolution(1090vv) Untilelectrophoreticanalysisthestocksolutionswerestoredatndash16degCTheworkingsolutionswerepreparedbydilutingtheinitialsolutionswithwaterjustbeforetheexperiments
3Resultsanddiscussion
CovalentcoatingsbasedonN-alkylimidazoliumcationwereespeciallysuitableforseparationofneurotransmittersandtheirmetabolites(Fig1)Inadditionthecombinationofcovalentcoatingwithon-linepreconcentrationtechniquesallowstothesignificantdecreaseoftheseanalyteslimitsofdetection(LOD)Accessorialinteractions positively charged analytes with positively charged imidazoleimproveseparationselectivity(viaπ-πinteraction)andefficiency(concentrationintightzonesviaelectrostaticrepulsion)Sodiumdodecylsulfate(SDS)addedintobackgroundelectrolyte(inconcentrationabovecriticalmicelleconcentra-tion) strongly interacts with hydrophobic alkyl groups in covalent coatingstructureThenegativelychargedSDSlayerisformedoninnercapillarysurfaceThedoublereversingEOFallowsustocarryoutelectrokineticinjectionofsampleandon-linepreconcentrationbysweepingsimultaneouslyLODweredeclineto
ndash108ndash20ngmL ThelengthofalkylsubstituentalsoaffectsthestackingefficiencyfactorandLODMorehydrophobicoctylgroupscomparetobutylprovidemoreeffectiveinteractionwithSDSandasresultlowerLOD
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 37
Covalentcoatingmodifiedβ-CDhasnotshownsharpreducingofLODbyon-linepreconcentrationStacking sweeping (SDSasmicelle reagent) field-enhancedsample injection were examined using different model mixtures of analytesNeverthelessthiscoatingallowssimultaneousseparationofbothofhydrophobicsteroidhormonesandhydrophilicbiogenicaminesinasinglerun(Fig2) The guest-host interaction hydrophobic cavity of β-cyclodextrin with thehydrophobicsteroids leadsto the formationofcomplexwhichaffectssteroidselectrophoretic mobility At the same time β-cyclodextrin can act as a chiralselector and baseline separation of ketoprofen enantiomers has also beenachieved
38 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Electropherogramofmixtureof neurotransmitters and theirmetabolitesadrenaline (A)norepinephrine (NE)normetanephrine (NMN)dopamine (DA)metanephrine (Met) serotonin(SER)homovanillicacid(HVA)andtheinnerstandard24-dihydroxybenzoicacid(24-DHBA)oncovalentlymodifiedwithN-buthylimidazoliumionicliquidscapillaryConditions10mMNaH PO 2 4
(adjusted to pH= 20 by 1MHCl) injection 50 stimes30mbar ndash20 kV 220 nmmodelmixurendash1 ndash1 ndash110microgmL (METADNMNNADA24-DHBA)5microgmL (SER)and20microgmL (HVA)
4Conclusions
ItwasshownthatstructurecovalentcoatingaffectsitsanalyticalcharacteristicsWecomparedtwotypesofcovalentcoatingdifferingsubstituentinimidazoliumringnamelyalkylgroupandβ-cyclodextrinThefirsttypeisgreatcoupledwithon-line preconcentration technic but it is limited to effectively determine ofbiogenicaminesonlywhilethesecondtype(withβ-cyclodextrin)showedthepossibilitiestoseparatevariousanalytesbutsuitableon-linemodehasnotbeenfoundThemainpointsaresummarizedinTable1
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 39
Covalentcoatingtype Electrophoreticseparationof On-lineprecon- centration biogenicamines amino steroid ketoprofen andtheirmeta- acids hormones enantiomers bolites N-β-cyclodextrinimida- yes yes yes yes thesuitableapproachzoliumcovalentcoatings wasnotfoundedN-alkylimidazolium yes yes nonsepa- nonsepa- thesignificantreducingcovalentcoatings rated rated forbiogenicaminesLOD
Table 1Thesummationofpossibilitiesofcovalentcoatingsbasedonimidazoliumcation
Fig 2Electropherogramofsimultaneousseparationofhydrophobic(steroidhormones)andhydro-philicanalytes (aminoacidsandbiogenicamines) insinglerunwithcovalentcoatingbasedonimidazoleandβ-CDConditions10mMNaH PO (adjusted topH = 20by1MHCl) injection2 4 20 stimes30mbarndash20kV254nm(1ndash8min)and220nm(8ndash15min)0mbar(1ndash10min)and40mbar(10ndash15 min) Model mixture corticosterone (B) hydrocortisone (F) 11-deoxycortisole (S)
ndash1 ndash15μgmL L-tryptophan(Trp)34-dihydroxy-L-phenylalanine(DOPA)10μgmL L-tyrosine(Tyr)ndash15 μg mL noradrenaline (NA) normetanephrine (NMN) adrenaline (AD) dopamine (DA)
ndash120μgmL
Acknowledgments
This work was supported by Russian Science Foundation (grant numbers 19-13-00370) Theauthors are also grateful to the Chemistry Education Centre and Nanothechnologies Centre ofResearchParkSaintPetersburgStateUniversityfortechnicalsupport
References
[1] HuLFYinSJZhangHYangFQRecentdevelopmentsofmonolithicandopen-tubularcapillaryelectrochromatography(2017ndash2019)JSepSci43(2020)1942ndash1966
[2] KartsovaLAKravchenkoAVKolobovaEACovalentcoatingsofquartzcapillariesfortheelectrophoretic determination of biologically active analytes J Anal Chem 74 (2019)729ndash737
[3] HoTDZhangCHantaoLWAndersonJLIonicliquidsinanalyticalchemistryFundamen-talsadvancesandperspectivesAnalChem86(2014)262minus285
[4] TangSLiuSGuoYLiuXJiangSRecentadvancesofionicliquidsandpolymericionicliquids incapillaryelectrophoresisandcapillaryelectrochromatography JChromatogrA1357(2014)147ndash157
[5] QinWLiSFYElectrophoresisofDNAinionicliquidcoatedcapillaryAnalyst128(2003)37ndash41
[6] QinWWeiH Li SFY 13-Dialkylimidazolium-based room-temperature ionic liquids asbackgroundelectrolyteand coatingmaterial in aqueous capillaryelectrophoresis JChro-matogrA985(2003)447ndash454
[7] QinW Fong S Li Y Determination of ammonium andmetal ions by capillary electro-phoresisndashpotential gradient detection using ionic liquid as background electrolyte andcovalentcoatingreagentJChromatogrA1048(2004)253ndash256
[8] QinWLiSFYAn ionic liquidcoating fordeterminationofsildenafilandUK-103320 inhumanserumbycapillaryzoneelectrophoresis-iontrapmassspectrometryElectrophoresis23(2002)4110ndash4116
[9] BorissovaMVaherMKoelMKaljurandMCapillaryzoneelectrophoresisonchemicallybondedimidazoliumbasedsaltsJChromatogrA1160(2007)320ndash332
[10] KolobovaEKartsovaLKravchenkoABessonovaEImidazoliumionicliquidsasdynamicand covalent modifiers of electrophoretic systems for determination of catecholaminesTalanta188(2018)183ndash191
[11] KravchenkoAKolobovaEKartsovaLMultifunctioncovalentcoatingsforseparationofaminoacidsbiogenicaminessteroidhormonesandketoprofenenantiomersbycapillaryelectrophoresisandcapillaryelectrochromatographySepSciplus3(2020)102ndash111
40 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Synthetic 4-hydroxy-3-methoxybenzaldehyde (vanillin) is used as a flavoringagent in foodsdrinksperfumesandpharmaceuticals [1]However at certainconcentrationsthesubstancemayaccumulateinthebodyhaveatoxiceffectand
ndash1at high concentrations may be fatal (lethal dose LD (oral rat) =2gkg 50ndash1 ndash1LD (oral guinea pig) = 14gkg LD (intravenous dog) = 132gkg lethal50 50 ndash1concentrationLC (inhalationmouse)=417gkg )[2]AccordingtoRussianState
StandartGOST121005-88thetoxiceffectsofvanillinintheworkplaceinthendash3formofvapoursoraerosolsareobservedatconcentrationsabove15mgm
Chromatography[3]spectrophotometry[4]capillaryelectrophoresisareusedforvanillindeterminationindifferentobjects CurrentlysmokingmixturesforhookahsandelectroniccigarettesarewidelyusedamongyoungpeopleThesemixturesarenotcontrolledforthecontentofsubstancesandarefreelyavailableconsideringthemmoreharmlesswithrespecttoordinarycigarettesThusthedevelopmentofamethodforthedeterminationof4-hydroxy-3-methoxybenzaldehydeinsmokingmixturesisrelevant
Determination of vanillin in smoking mixtures by spectrophotometry
ELIZAVETAEFREMENKOANNACHERNOVAOLGABASTRYGINA
DepartmentofChemicalEngineeringNationalResearchTomskPolytechnicUniversityLeninavenue30634050TomskRussiaeaetpuru
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 41
AbstractTheresearchdealswithdeterminationofvanillin insmokingmix-turesbyultraviolet-visiblespectrophotometryThemethodshowed
ndash1goodlinearityintherangeof005ndash012gL withalimitofdetectionndash1005gL After validation studies the method was successfully
applied to thedeterminationof vanillin in smokingmixtureswithsatisfactoryresultsItwasshownthattheerrorofthismethoddoesnot exceed 1 The developed spectrophotometric procedure fordeterminingvanillininsmokingmixturescanbeusedasacontrol
Keywordssmokingmixturesspectrophotometryvanillin
2Experimental
21Reagentsandchemicals
Asampleofvanillin(purity98)wastakenastheobjectofstudyAssolventsweused95ethanolAllchemicalsusedwereofanalyticalreagentgrade
22Instrumentation
Theopticaldensityofsampleswasmeasuredincuvettewithanabsorbinglayerthickness of 10 mm using a Cary 60 spectrophotometer (Agilent USA) Allmeasurementswerecarriedoutatroomtemperature
23Samplepreparation
Sample preparation of the investigated objects consisted of the preliminarydissolutionofthesamplein95ethanolThesample10mgoftobaccoldquoAdalyandashVanillardquo(Turkey)wasdiluted in10μLof95ethanol to theconcentrationof
ndash11gL Thesample10μLofldquoFlavoringTPAndashVanillaCustardrdquo(USA)wasdilutedin10 μL of 95 ethanol The resulting solution was diluted six times to the
ndash3concentrationof017μLcm
3Resultsanddiscussion
TodeterminevanillininthesamplestheopticalpropertiesofvanillininvarioussolventsweredeterminedAsaresultthe95ethanolwaschosenastheoptimalsolvent[4] IthasbeenestablishedthatintheUVspectraoftheanalyteabsorptionbondsareobservedwithmaximumvaluesat23002800and3100nmwhichcorres-pondstopublisheddata[45](Fig1) To quantify vanillin the calibration curve of the optical density on theconcentrationofvanillin in95ethanolwasobtainedatconcentrations005
ndash1006007008010and012gL Calibrationcurveofvanillinin95ethanolatawavelengthof280nmis
ndash1 A =81914c[gL ]+00357 (1)2802 R =1
Calibrationcurveofvanillinin95ethanolat310nmis
ndash1 A =73824c[gL ]+00301 (2)3102 R =1
42 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Inthespectraoftheanalyzedsamplesolutionsabsorptionmaxima(2800nmand3100nm)characteristicforvanillinwereobservedTheamountofvanillininthesamplewasdeterminedusingcalibrationcurvesat280and310nmWeightedleast square regressionwas applied to the calibration curves to improve theaccuracyespeciallyatinlowconcentrationlevelrangeGoodlinearitywasfound
ndash1 ndash1intherangeof005ndash012gL withadetectionlimitof005gL TheresultsarepresentedintheTable1
4Conclusions
ThedevelopedmethodcanbeusedasacontrolmethodTheerrorinthemethodfordeterminingvanillininthesampleldquoFlavoringTPAndashVanillaCustardrdquowithaknownconcentrationofvanillinwas0004Accordingtothedataobtainedwerecommendawavelengthof280nmforthedeterminationofvanillininsamples
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 43
λ nm
Absorban
ce
ndash1Fig 1 Absorption spectrum of vanilin solution in 95 ethanol at concentration 01 mol L (anabsorbinglayerthicknessof10mm)
Sample λnm Tookmg Foundmg S Sх Δх δ
FlavoringTRA 310 10850 104096 00024 00011 00006 00306ndashVanillaCustardAdalyandashVanilla 310 100000 10162 00019 00009 00002 00025
FlavoringTRA 280 100300 96062 00033 00015 00032 00042ndashVanillaCustardAdalyandashVanilla 280 100000 10122 00013 00006 00012 00017
Table 1Testingmethods introduced foundof vanillin in the samplesat310nmby spectrophotometricmethod(n=5p=099SndashstandarddeviationSxndashrelativestandarddeviationΔхndashabsoluteerrorδndashrelativeerror)
ThenthedevelopedmethodwastastedonthesampleldquoAdalyandashVanillardquosamplewithamorecomplexcompositionandanunknownconcentrationofvanillinwastaken The vanillin content in the sample was determined according to thedevelopedmethoditamountedto10ofthetotalmassStudieshaveshownthepossibility of using spectrophotometric analysis for the qualitative andquantitative determination of vanillin Also based on preliminary studies aspectrophotometricprocedurewasdevelopedforthequantitativedeterminationofvanillinbasedonabsorptioninethanolinthewavelengthrange200ndash400nm
References
[1] httpswwwrusnaukacom43_DWS_2015Chimia6_203179dochtm (accessed 25thFebruary2019)
[2] httpswwwcdcgovnioshrtecsdefaulthtm(accessed11stApril2020)[3] AliLPerfettiGDiachenkoGRapidmethodforthedeterminationof342coumarinvanillin
and ethyl vanillin in vanilla extract by reversed-phase liquid 343 chromatography withultravioletdetectionJAOACInt91(2008)383ndash386
[4] БастрыгинаОАЕфременкоЕАЧерноваАПВыделениеванилинаисследованиеегооптическихсвои ствопределениевбиологическомматериалеВХимияихимическаятехнология в XXI веке Материалы XX Международной научно-практическойконференции имени профессора ЛП Кулёва студентов и молодых ученых ТомскНациональныи исследовательскии Томскии политехническии университет 2019с301ndash302
[5] WeastRCHandbookofChemistryandPhysics60thedBocaRatonCRCPress1979p143
44 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
UraniumbelongstothegroupofhazardouselementsItisahighlyharmfulandradioactiveelementtoxictohumansandalllivingorganisms[12]Inhaledwithair it has a particularly destructive effect on the kidneys and as a result ofaccumulationinwhitebloodcellsitcanalsocauseimpairmentoftheimmunesystem[2]Uraniumoccursatseveraldegreesofoxidationhoweverinaqueoussolutionsthemoststableformisuranylion(UO (II))[12]Thepresenceofura-2
niumintheenvironmentiscausedbyamongothersnaturalsoilandrockerosionEnvironmentalpollutionwiththiselementisalsoconstantlyincreasingduetohumanactivitycoalcombustionuraniumoreminingandprocessingthearmsindustryandtheuseofuraniumasnuclearfuelinfissionreactors[3]Itisveryimportanttoconstantlymonitortheconcentrationofuraniumbothinthenaturalenvironment in order to assess its state and safety (especially in the case ofdrinkingwater)aswellasinallstagesofprocessingprocessesassociatedwiththenuclearindustrytoavoidtheoccurrenceofnuclearpollution[13] Scientists have made many attempts to develop research methods todeterminethecontentofuranylcompoundsinliquidsamplesEffortsweremadetousemanyanalyticalmethodsforthispurposeincludingspectrophotometry
Uranyl ion-selective electrode with solid contact
KAROLINAPIETRZAKCECYLIAWARDAK
DepartmentofAnalyticalChemistryInstituteofSciencesFacultyofChemistryMariaCurie-SklodowskaUniversityMariaCurie-SklodowskaSq320-031LublinPolandkarolinapietrzakpocztaumcslublinpl
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 45
AbstractNewallsolidstateuranylion-selectiveelectrodeswithlowdetection
ndash7 ndash1limits(71times10 molL )shortresponsetimegoodselectivityandstable and reproducible potential were developed Many types ofelectrodeswith different active ingredient content in ion-selectivemembrane (bis(244-trimethylpentyl)phosphonium acid Cyanex-272)were testedAs an additive an ionic liquid1-octyl-3-methyl-imidazole chloride was used The optimal composition of theion-selective membrane was chosen from all electrodes based onthedeterminationand comparisonof analyticalparametersof thesensors
Keywordsion-selectiveelectrodesolidcontacturanyl
plasma spectrometry luminescence spectroscopy voltammetry or chromato-graphymethods[2] Duetomanyadvantagesofpotentiometricmethods(amongthemlowercostseasieroperationofdevicesquickresponseandtheabilitytoperformmeasure-ments in flowmode) [3] a numberof potentiometric sensorshave alsobeendeveloped that could be successfully used in this type of research Themostpopularpotentiometricsensorsincludeion-selectiveelectrodes(ISEs)whicharecharacterized by low-energy consumption small size and portability and aresuccessfullywidelyusedforthedeterminationofbothinorganicandorganicionsinclinicalanalysisprocesstechnologyaswellasincontrolthestateofthenaturalenvironment[45]Removaloftheinternalsolutioncontainingthesameanalytetowhich theelectrode is sensitiveresulted in theso-calledsolidcontact ISEswhicharemuchsmallerinsizethantheirpredecessorsaremoreconvenienttouse and more mechanically resistant In this type of sensors however it isimportanttoachievesatisfactorypotentialstabilitywhichisnecessarytoobtainsatisfactoryresults[5]AveryimportantpartofISEsistheion-selectivemem-branewhosecompositiondeterminestheanalyticalparametersofthesensorsResearchers are currently focusing on the production and testing of newsubstancesthatcouldbesuccessfullyusedasmembranecomponentsandsolidcontacts thatwould allow to obtain new sensorswith lower detection limitslongerlifetimeandbetterpotentialstabilityandtodeterminenewpreviouslyunattainableanalytes[4] AstheactivecomponentsofthemembranesensitivetouranylionscientistshavealreadyusedKryptofix22DD(413-didecyl-171016-tetraoxa-413-diaza-cyclooctadecane)[2]Cyanexextractants(bis(244-trimethylpentyl)phosphinicacid bis(244-trimethylpentyl)monothiophosphinic acid and bis(244-tri-methylpentyl)dithiophosphinic)acid[3]DBBP(dibutylbutylphosphonate)andDOPP (di-n-octyl phenylphosphonate) [6] DMSO (dimethylsuphoxide) [7]TTPTP (5678-tetrahydro-8-thioxopyrido[4345]thieno[23-d]pyrimidine-4(3H)one)[8]orTEHP(tris(2-ethylhexyl)phosphate)andTPTU(O-(12-dihydro-2-oxo-1-pyridyl)-NNNN-bis(tetra-methylene)uronium hexafluorophos-phate)[9]
2Experimental
21Reagentsandchemicals
This paper presents research on the design and properties of ion-selectiveelectrodes with solid contact for the determination of uranyl ions Bis(244-trimethylpentyl)phosphonium acid (Cyanex-272) was used as the activecomponentof themembranewhichwasdescribed in the literatureasagooduranylextractant[10]Inordertoensureaconstantpotentialofthiselectrodeandreducetheelectroderesistancetheion-sensitivemembranewasenrichedwithafewpercentadditionof1-octyl-3-methylimidazolechlorideionicliquid
46 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Several types of ion-selective electrodes were prepared using an AgAgClelectrodeasaninternalelectrodewhichdifferinthequantitativeandqualitativecompositionofthemembranesAllcompositionsarelistedinTable1
22Instrumentation
Measurements were made at room temperature using a 16-channel datacollectionsystem(LawsonLabs IncUSA)coupled toa computer in solutionsmixedwithamechanicalstirrerAsilversilverchlorideelectrodewithdoublejunctionwasusedasthereferenceelectrode
3Resultsanddiscussion
The effect of ion-selective membrane composition on the properties of theobtained potentiometric sensors was examined by determining their basicanalyticalparametersincludingslopeoftheelectrodecharacteristicsdetectionlimitmeasuringrange(concentrationrangeinwhichthecourseoftheelectrodecharacteristics isrectilinear)pHrange(inwhich ithasnoeffect forelectrodepotential)andresponsetimeTheobtainedvaluesofthetestedparametersareshowninTable2 Figure1showsthecalibrationcurvesofthetestedelectrodesdeterminedin
ndash7 ndash1 ndash1UO (NO ) solutionsintheconcentrationrange1times10 ndash1times10 molL Asitcan2 3 2
beseeninFig1andTable2allelectrodesweresensitivetouranylionsbutindifferent extend The best response exhibited ISE-3 containing 1 (ww) ofionophore Increasing the ionophore content in themembrane shortened thelinearityrangeofthecalibrationcurveanditssupernenstianslope Theselectivityofthetestedelectrodeswasestimatedbydeterminingtheselec-tivitycoefficients inrelationto interfering ionsForthispurpose theseparate
ndash1solutionmethodwasused(extrapolatingresponsecurves toa =a =1molL )i j
ComparisonofISE-1andISE-3electrodeselectivityisshowninFig2
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 47
Table 1Quantitative and qualitative composition of electrode membranes Cyanex-272 (bis(244-tri-methylpentyl)phosphoric acid) TBP (tri-n-butyl phosphate) and OMImCl (1-octyl-3-methyl-imidazolechloride)
Abbreviation Membranecomposition(ww)ofelectrode Cyanex-272 PVC TBP OMImCL
ISE-1 00 33 620 5ISE-2 05 33 615 5ISE-3 10 33 610 5ISE-4 30 33 590 5ISE-5 50 33 570 5ISE-6 100 33 520 5
48 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Abbreviation Slope Detectionlimit Linearrange Response pHrange2+ ndash1 ndash1ofelectrode mVpa(UO ) molL molL times2
ndash5 ndash5 ndash1ISE-1 297 25times10 5times10 ndash1times10 5ndash8 28ndash42ndash6 ndash5 ndash1ISE-2 292 65times10 1times10 ndash1times10 5ndash8 25ndash60ndash7 ndash5 ndash1ISE-3 298 71times10 1times10 ndash1times10 5ndash8 24ndash60
ndash6 ndash4 ndash1ISE-4(I) 357 31times10 5times10 ndash1times10 5ndash8 ndndash6 ndash6 ndash4ISE-4(II) 242 31times10 5times10 ndash5times10 5ndash8 nd
ndash3 ndash1ISE-5(I) 638 nd 1times10 ndash1times10 5ndash10 ndndash5 ndash3ISE-5(II) 234 nd 5times10 ndash1times10 5ndash10 ndndash3 ndash1ISE-6(I) 733 nd 1times10 ndash1times10 5ndash10 ndndash5 ndash3ISE-6(II) 222 nd 5times10 ndash1times10 5ndash10 nd
Table 2Selectedparametersandtheirdeterminedvaluesoftestedionselectiveelectrodes
Fig 1 Calibration curves of the testedelectrodesobtainedinUO (NO ) solutionsin2 3 2 ndash7the concentration range from 1times10 to
ndash1 ndash11times10 molL
Fig 2Comparisonofselectivitycoefficientspot(log K (UO (II))M) for electrodes ISE-12
(1stcolumn)andISE-3(2ndcolumn)
Inordertoexaminethereversibilityofthepotentialofthetestedelectrodesndash4 ndash1potentialmeasurementsweremadealternatelyinsolutions1times10 molL and
ndash5 ndash11times10 molL ofUO (NO ) TherecordedpotentialreadingsareshowninFig32 3 2
Long-term potential stability and sensor reproducibility were evaluated byndash1determiningtheaveragevalueoftheelectrodepotentialina01molL UO (II)ion2
solutionovertimeforthreeidenticalISE-3Thesemeasurementsweremadetoobservechangesinthepotentialofelectrodeswiththesameconcentrationoveralongperiodoftime(30days)Figure3showsthelong-termpotentialstabilityandreproducibilitydeterminedforthreeidenticalsensors
4Conclusions
Asaresultofthetestsion-selectiveelectrodeforthedeterminationofuranylionswasobtainedwhich iseasy todesignanduseThebestanalyticalparametersexhibitedISE-3containing1ionophoreintheion-selectivemembraneForthis
ndash7 ndash1typeofelectrodesthedetectionlimitof71times10 molL linearityoftheelectrodendash6 ndash1 ndash1calibrationcurve in the range1times10 ndash1times10 molL andresponse time5ndash8s
were obtained In addition the manufactured sensors also showed stablereproducibleandreversiblepotentialandverygoodselectivityinrelationtothetestedinterferents
References
[1] AnsariRMosayebzadehZConstructionofanewsolid-stateU(VI)ion-selectiveelectrodebasedonpolypyrroleconductingpolymerJRadioanalNuclChem299(2014)1597ndash1605
[2] GhanbariMRounaghiGHAshrafNAnuranylsolidstatePVCmembranepotentiometricsensor based on 413-didecyl-171016-tetraoxa-413-diazacyclooctadecane and itsapplicationforenvironmentalsamplesIntJEnvironAnalChem97(2017)189ndash200
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 49
Fig 3 Stability () reproducibility andreversibility () of the potential of ISE-3Standard deviations given on the plot aredeterminedforthesamethreeISE-3
[3] Badr IHA Zidan WI Akl ZF Cyanex based uranyl sensitive polymeric membraneelectrodesTalanta118(2014)147ndash155
[4] BiegCFuchsbergerKStelzleMIntroductiontopolymer-basedsolid-contaction-selectiveelectrodes basic concepts practical considerations and current research topics AnalBioanalChem409(2017)45ndash61
[5] Bobacka J IvaskaA LewenstamA Potentiometric ion sensorsChem Rev108 (2008)329ndash351
[6] ZidanWI Badr IHA Akl ZF Development of potentiometric sensors for the selective2+determinationofUO ionsJRadioanalNuclChem303(2015)469ndash4772
[7] SalehMBSolimanEMGaberAAAAhmedSANovelPVCmembraneuranylion-selectivesensorSensActuatorsB114(2006)199ndash205
[8] SalehMBHassanSSMAbdelAAAbdelNAAnoveluranylion-selectivePVCmembranesensor based on 5678-tetrahydro-8-thioxopyrido[4345]thieno[23-d]pyrimidine-4(3H)oneSensActuatorsB94(2003)140ndash144
[9] HassanSSMAliMMAttawiyaAMYPVCmembranebasedpotentiometricsensorsforuraniumdeterminationTalanta54(2001)1153ndash1161
[10] Prabhu DR Ansari SA Raut DR Murali MS Mohapatra PK Extraction behaviour ofdioxouranium(VI) cation by two phosphorous-based liquid cation-exchangers in room-temperatureionicliquidsSepSciTechnol52(2017)2328ndash2337
50 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Metronidazole(2-methyl-5-nitroimidazole-1-ethanol)isoneofthemostwidelyused nitroimidazole antibiotics Metronidazole is used for the treatment ofinflammatorydiseasescausedbyanaerobicorganismsandsomeprotozoaandforpreventionofdysenterycolibacillosiseimeriosisbalantidiasissalmonellosisenteritissepticemiapost-surgicalcomplications[1ndash3]Oxytetracyclinehydro-chlorideisanantibioticofthetetracyclinefamilyItisoneofthemostcommonlyused antibiotics in poultry because of its low cost and effective [4] Thesecompounds are intensively used in poultry breeding and stockbreedingUnreasonableuseofthesedrugscancauseseriousfoodsafetyissues[5] The veterinary drug Nozemat which includemetronidazole and oxytetra-cyclinehydrochloridewaschosenfortheexperimentsNozematisusedtotreat
Polarographic determination of metronidazole and oxytetracycline hydrochloride in veterinary drug for honey bees
a a bKATERYNAPLOTNIKOVA LILIYADUBENSKA IVANZELENYI
a AnalyticalChemistryDepartmentIvanFrankoNationalUniversityofLvivKyrylaiMefodiaStr879005LvivUkrainekaterina27plgmailcom
b DrohobychPedagogicalLyceumIvanaFrankaStr3682100DrohobychUkraine
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 51
AbstractWehavedevelopedanewpolarographicmethodforthedetermin-ation of metronidazole and oxytetracycline hydrochloride in theveterinarydrugNozematforhoneybeesThetechniqueisbasedonthereductionofpolarographicallyactivecompoundsonamercurydropletelectrodeTheinfluenceofthecomponentsoftheveterinarydrugNozematonthepolarographicdeterminationofmetronidazolewasstudiedItwasfoundthatthereductionofmetronidazoleisnotaffected by glucose and ascorbic acid but is affected by oxytetra-cyclinehydrochloridewhichisreducedtomercurydropletelectrodeatapotentialofndash145VThedevelopedtechniqueischaracterizedbyeaseofsamplepreparationandcost-effectivenessThistechniquehastheabilitytoidentifysimultaneouslyanddeterminatemetronidazoleand oxytetracycline hydrochloride in solution without the use ofseparationandconcentrationmethods
Keywordselectrochemistrymetronidazoleoxytetracycline
hydrochloridepolarographyveterinarydrug
bees and it can be given in unregulated doses Because of this an unknownamountofmetronidazolecangetintothehoneyanditsometimescausessideeffectsofthehumanbodyanditcouldbeofgreatconcernforpublichealth[56]MedicinesforpeoplearemorestringentandbettertestedthanveterinarydrugsTheproblemofthecontroloftheveterinarydrugsisurgentnowadaysVeterinarymedicinescouldbeunauthorizedandtheuncontrolleduseofmedicinesexistsinretailpharmaciesofmedicineorimportedascontrabandfromothercountries The most widespreadof these classes in thequality controlare chromato-graphic [6ndash9] spectrophotometric [10ndash13] and electrochemical methods[14ndash17]Manyoftheknownmethodsforthedeterminationofmetronidazoleandoxytetracyclinehydrochloridehaveanumberofdisadvantagestime-consumingtheuseoforganicsolventsandexpensivereagentsthesideeffectsofexcipientsandotheractivesubstancesElectrochemicalmethodsarepromisingalternativefor the determination of the electroactive substances Their advantages aresimplicityminiaturizationhighsensitivityandrelativelylowcostThereforethesearch for simple express and affordable methods for the determination ofmetronidazoleremainsrelevantOneofthepromisingmethodsofdeterminationisvoltammetry
2Experimental
21Reagentsandchemicals
VeterinarydrugNozemat (manufacturerAPI-SANRussia) is a yellowpowderwithaslighttypicalodorAvailableinlaminatedbagsof25gCompositionper1gof the drug metronidazole 400 mg oxytetracycline hydrochloride 400 mgglucoseascorbicacid MetronidazoleandoxytetracyclinehydrochloridewerepurchasedfromSigmaAldrich(USA)Stockstandardsolutionofmetronidazolefordeterminationwaspreparedbydissolvingtheexactamountofstandardin7mLof2Mhydrochloricin 500 mL volumetric flask Stock standard solution of oxytetracyclinehydrochloride was prepared by dissolving the exact amount of standard indistilledwaterin500mLvolumetricflaskAfterthatthesolutionswereadjustedtothemarkwithdistilledwaterandmixedthoroughly The Britton-Robinson buffer preparationwas as follows 202 g of sodiumtetraboratedecahydrate287mLofglacialaceticacidand176mLofconcen-tratedorthophosphoricacidweredissolvedin10Lvolumetricflask Working solution preparation was as follows an aliquot of stock standardsolutionwasaddedintoa25mLvolumetricflasktoobtainasolutionwiththenecessaryconcentrationthen2mLofBritton-RobinsonbufferwithnecessarypHwasaddedtotheflaskanddistilledwaterwasaddedtothemark AqueoussolutionofNozematwaspreparedasfollowstheexactportionofthetestveterinarydrugwasdissolved ina250mlvolumetric flaskAnaliquotof
52 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Polarogramsof(A)metronidazoleand(B)metronidazolewithoxytetracyclinehydrochloridendash1solutionsat02MBritton-RobinsonbufferbackgroundatpH=96(υ=05Vs c(metronidazole)=
ndash5 ndash5=45times10 Мc(oxytetracyclinehydrochloride)=50times10 М
100mloftheresultingsolutionwasaddedtoa250mlvolumetricflaskandmadeuptothemarkwithwaterAnaliquotof100mloftheresultingsolutionwasaddedtoa250mlvolumetricflask2mlofBritton-RobinsonbufferwithapHof96wasaddedandthevolumewasadjustedtothemarkwithdistilledwater
22Instrumentation
ForpolarographicmeasurementsweuseddigitaldeviceMTechOVA-410 [18]temperature-controlledthree-electrodeamercurydropletindicatorelectrodeasaturatedcalomelreferenceelectrodeandplatinumwireauxiliaryelectrodeTheaccuracyofthepotentialmeasurementis1mVTheuncertaintyofcurrentmeasu-rement is 01 The employed mercury droplet electrode had the following
ndash4 ndash1characteristicsm=594times10 gs τ=10 min in 02 M NH Cl We used cyclic4
voltammetryforthestudyoftheelectrochemicalprocess WeusedMV870DIGITAL-pH-MESSERATpH-meterformeasuringpHofthesolutions Theobtainedworkingsolutionswereintroducedintothecellanddeoxyge-natedwithargonfor10minPolarogramswererecordedintherangeofpotentialsfrom00tondash16V
3Resultsanddiscussion
Previously it was found that using the Britton-Robinson buffer with pH = 96metronidazoleisreducedwiththeformationofacharacteristiconeirreversiblepeakatndash064V(Fig1A)UsingpolarographywithfastpotentialscanitwasfoundthatmetronidazoleisreducedonmercurydropletelectrodeintherangeofpH20to105ThepeakrecoverycurrentofthemetronidazolereachesthemaximumvalueatpH9ndash10againstthebackgroundofatheBritton-Robinsonbuffer
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 53
Underpre-selected conditions theeffectof some foreign substanceson thepolarographicdeterminationofmetronidazolewasinvestigatedSubstancesthatare componentsofdrugswere studiedglucose ascorbicacidoxytetracyclinehydrochlorideGlucose and ascorbic acid are not reduced atmercury dropletelectrodeanddonotchangetheappearanceofthepolarogramandpolarographiccharacteristics of the recovery of metronidazole As can be seen from Fig 1oxytetracycline hydrochloride is reduced to mercury droplet electrode andchangestheappearanceofthepolarogramandpolarographiccharacteristicsofthe recovery of metronidazole With the addition of oxytetracycline hydro-chloridetherecoverypeakofmetronidazoledecreasesandslightlyshiftstomorenegativepotentials The composition of the drug is relatively complex excipients affect theanalyticalsignaloftherecoveryofcompoundssototakeintoaccountthematrixeffectusedthemethodofmanyadditives QuantitativelytransferredthesolutionofNozemattothecell(exactvolume)removed dissolved oxygen for 10 min and took polarograms in the range ofpotentialsfrom00tondash16VAliquotsofstandardmetronidazolesolutionwereintroducedintothecelltoobtainasolutionwithagivenconcentrationofadditive
ndash5 ndash5metronidazole 10times10 M to 70times10 M As with the determination of themetronidazole aliquots of a standard oxytetracycline hydrochloride solutionwereaddedtothecellwithsolutionofNozemattoobtainasolutionwithagiven
ndash6 ndash6additiveconcentrationfrom70times10 Mto50times10 M(Fig2) In Table 1 are shown metrological characteristics of the determination ofmetronidazoleandoxytetracyclinehydrochloride inveterinarydrugRecoverywascalculatedFormetronidazoletherecoveryis97andforoxytetracyclinehydrochloridetherecoveryis103Analyticalperformanceofthetechniqueisgoodfordeterminationveterinarydrugs
54 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2Polarogramsof(A)metronidazolereductionatdifferentmetronidazoleconcentrationsand(B)oxytetracyclinehydrochloridereductionatdifferentoxytetracyclinehydrochlorideconcentra-tionsandtheircorrespondingcalibrationgraphs
Theaccuracywasverifiedbytheldquoadded-foundrdquomethodAliquotsofstandardsolutionofmetronidazoleweremadeina250mlvolumetricflasktoobtaina
ndash5solutionofagivenconcentrationof33times10 Mandthesolutionofoxytetracyclinendash5hydrochloridetoobtainasolutionofagivenconcentrationof15times10 M2mlof
Britton-RobinsonbufferwithpH96wasaddedtoflaskwithstirringandadjustedtothemarkwithwaterTheanalysisprocedureofmodelsolutionissimilartoanalysis procedure of the solution of Nozemat The calculated amount ofmetronidazolebythemethodofmultipleadditivesinthetestedmodelsolutionisinagreementwiththeamountthatwasintroducedintothesample
4Conclusions
The new polarographic method for the determination of metronidazole andoxytetracyclinehydrochlorideintheveterinarydrugNozematforhoneybeeswasdeveloped We conducted principal component analysis of veterinary drugNozemattoassesstheoveralleffectforthedeterminationofmetronidazoleWefoundthatoxytetracyclinehydrochlorideisreducedtomercurydropletelectrodeThismethodhastheabilitytoidentifysimultaneouslyanddeterminatemetro-nidazole and oxytetracycline hydrochloride in solution without the use ofseparationandconcentrationmethodsOnemoreofadvantagesoftechniquearefastprocedureofanalysissimplesamplepreparationlowcostthepossibilityofminiaturization
References
[1] Antibiotic and Chemotherapy Finch R Greenwood D Whitley R (edits) AmsterdamElsevier2006p292ndash299
[2] MitrowskaKPrzyczynyiskutkizakazustosowania5-nitroimidazoliuzwierzątktorychtkanki lub produkty przeznaczone są do spozycia przez ludziMed Weter 71 (2015)736ndash742
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 55
Metronidazole Oxytetracycline hydrochloride
PeakspotentialV ndash065 ndash144CorelationcoefficientR 099892 099922
ndash1 4 4SlopebmicroАM 656times10 176times10Δb 1763 400InterceptamicroА 2238 0219Δa 0075 0009
ndash1 ndash5 ndash5cmolL 342times10 125times10ndash1cmgg 389 413
Recovery 97 103
Table 1Validationparametersofthemethodofmetronidazoleandoxytetracyclinehydrochloridedetermi-nationinsolutionsofNozematbythemethodofmanyadditives
[3] VermaPNamboodiryVMishraSBhagwatABhoirSAstabilityindicatingHPLCmethodfor the determination of Metronidazole using Ecofriendly solvent as mobile phasecomponentIntJPharmPharmSci5(2013)496ndash501
[4] Cervini P Ambrozini B Machado LCM Ferreira Garcia AP Cavalheiro Gomes ETThermal behavior and decomposition of oxytetracycline hydrochloride J Therm AnalCalorim121(2015)347ndash352
[5] DangBNAnhNTKKyLXThaiPKAntibioticsintheaquaticenvironmentofVietnamsourcesconcentrationsriskandcontrolstrategyChemosphere197(2018)438ndash450
[6] QuintanillaPHettingaKABeltranMCEscricheIMolinaMPVolatileprofileofmaturedTronchon cheese affected by oxytetracycline in raw goat milk J Dairy Sci 103 (2020)6015ndash6021
[7] Chen F Yu L Jingdong P Xiang W Huanjun P Yu C Yan H Study on simultaneousdetermination of three nitroimidazole residues in honey by high performance liquidchromatographyndashresonanceRayleighscatteringspectraMicrochemJ141(2018)423ndash430
[8] Hernandez-MesaM Cruces-Blanco C Campana GA Simple and rapid determination of5-nitroimidazolesandmetabolitesinfishroesamplesbysalting-outassistedliquid-liquidextractionandUHPLC-MSMSFoodChem252(2018)294ndash302
[9] Xiu-ChunGZhao-YangXHai-HuiWWen-YiKLi-MingLWen-QingCHong-WeiZWen-HuiZMolecularlyimprintedsolidphaseextractionmethodforsimultaneousdeterminationofsevennitroimidazolesfromhoneybyHPLC-MSMSTalanta166(2017)101ndash108
[10] ТеплыхАНИлларионоваЕАКоличественноеопределениеметронидазоласпектро-фотометрическимметодомСибирскиймедицинскийжурнал5(2009)48ndash50
[11] ZheltvayOIZheltvayIISpinulVVAntonovichVPSpectrophotometricdeterminationofmetronidazoleandtinidazoleusingcopper(II)complexesJAnalChem68(2013)663ndash668
[12] Youssef AK Saleh MS Abdel-Kader DA Hashem Facile DY SpectrophotometricdeterminationofmetronidazoleandsecnidazoleinpharmaceuticalpreparationsbasedontheformatioonofdyesIntJPharmPharmSci6(2015)103ndash110
[13] Sversut RA Vieira JC Rosa AM Amaral MS Kassab NM Salgado H ValidatedspectrophotometricmethodsforsimultaneousdeterminationofoxytetracyclineassociatedwithdiclofenacsodiumorwithpiroxicaminveterinarypharmaceuticaldosageformArabianJChem13(2020)3159ndash3171
[14] Nikodimos Y Electrochemical determination of metronidazole in tablet samples usingcarbonpasteelectrodeJAnalMethodsChem(2016)361294
[15] Srivastava AK Upadhyay SS Rawool CR Punde NS Rajpurohit AS Voltammetrictechniques for the analysis of drugs using nanomaterials based chemically modifiedelectrodesCurrAnalChem15(2019)249ndash276
[16] Sahu G Voltammetric behaviour of metronidazole at a composite polymer membraneelectrodeOrienJChem26(2010)81ndash86
[17] Yang Y YanW Guo YWang X Zhang F Yu L Guo C Fang G Sensitive and selectiveelectrochemicalaptasensorviadiazonium-couplingreactionforlabel-freedeterminationofoxytetracyclineinmilksamplesSensorsandActuatorsReports2(2020)1ndash7
[18] httpchemlnueduuamtechdeviceshtml(accesed21stJune2020)
56 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
AnimportantpartofanyanalysisthatsignificantlyaffectsthefinalresultsisthesamplepreparationThelowconcentrationofbiologicallyactivecompoundsandthepresenceofaccompanyingcomponentspreventdirectanalysisofthesamplewithcomplexmatrixcompositionTraditionalmethodsofliquidandsolid-phaseextractionhaveaplentyoflimitationssuchashighlytime-consumingprocedureslarge volume of samples expensive cartridges toxic organic solvents andchallenges in automating the process Therefore the application of extractiontechniquesemployinglowamountofsolvents(microextractionmethods)andthelow toxicity extractantes has become the main research direction in recentyears[12] Solid-phasemicroextraction(SPME)wasproposedbyPavlishinin1989[3]Onevariantofthismethodistousethinrodswithvariouspolymercoatingssuch
Application of microextraction techniques combined with chromatographic methods for the analysis of complex objects
VLADISLAVDEEVELENABESSONOVALIUDMILAKARTSOVA
InstituteofChemistrySaint-PetersburgStateUniversityUniversitetskyprospect26198504PeterhofSaint-PetersburgRussiahitchervmailru
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 57
AbstractThelowconcentrationofanalytesandthepreventionofthematrixinfluence requires a stage for extraction and concentration of thestudiedcompoundsTheclassicalmethodsofliquidandsolid-phaseextractionhavemanylimitationsthatpreventtheiruseinsomecasesMicroextraction techniques are becoming more widespread WestudiedthepossibilityofusingionicliquidstoextractpesticidesfromwatersampleswiththeirsubsequentHPLS-MSdeterminationTheinfluenceonthedegreeofextractionofsuchparametersasanatureofionicliquidsanddispersersolventtheiramountssaltconcentrationvolumeratioofionicliquidsandwatersampledilutionoftheionicliquidsextractwithmethanolwasperformedBesidesconditionsofsolid-phase microextraction of volatile organic compounds fromurine samples obtained from healthy donors and donors withprostate cancer have been found The analysis of volatile organiccompoundsbyGC-MSfollowedbychemometricprocessingallowedachievingahighvalueofbinaryclassificationaccuracy(91)
Keywordschemometricsdispersiveliquid-liquid
microextractionsolid-phase
microextraction
asdivinylbenzenepolydimethylsiloxanepolyacrylateandpolyethyleneglycolwhichappliedtothesurface[4]Thepolymersorbentisplacedintheequilibriumheadspaceaboveacondensedphaseofthesampleandthevolatilecompoundsareextracted Liquidmicroextractionconsistsofusingsmallamountsofliquid(extractant)inequilibriumwiththegasorliquidphaseofthesampleDispersiveliquid-liquidmicroextraction(DLLME)isavariantofliquidmicroextractionTheessenceofthemethodisasfollowsextractantisdissolvedinthephaseofadispersingsolventandthemixtureisrapidlyinjectedintothesamplevolume[5]Inthiscasethedispersing solvent is dissolved and a ldquocloudrdquo of extractant is formed A largesurfaceareacontributestomasstransferprocessesThecombinationofDLLMEwiththeuseof ionic liquids(ionic liquids)asextractantsreducestheharmfulimpactontheenvironment[6] Sothegoalofthisstudywastheapplicationofmicroextractionmethodsfortheanalysisofrealsamples
2Experimental
21Reagents
Deionizedwaterwas obtained at the AQUILON D 301 deionizer (Russia) Allchemicals and reagents (the highest commercially available purity) werepurchasedfromReachimBakerAcrosorganicsandSigmaAldrich
22Instrumentation
HPLCanalysiswascarriedoutusinganHPLCLCMS-8030(Shimadzu)withatriplequadrupole mass-selective detector with electrospray ionization Analysis ofvolatileorganiccomponents(volatileorganiccompounds)ofurinesampleswasmade by GCMS-QP2010 SE (Shimadzu) Chemometric data processing wasperformedusingRStudio
23DeterminationofvolatileorganiccompoundsinurinesamplesbyGC-MSmethod
ForSPMEofvolatileorganiccompounds inurinesamplewasusedfibercoatedwith a polydimethylsiloxane (PDMS) The volatile organic compounds wereextractedontofibercoatingfor20minat50degCThentheanalytesweredesorbedinto the gas chromatography for 4 minutes at a temperature of 250degCChromatographic separation was carried out on a HP-5 capillary column(30mtimes250μmtimes025μm)usingtemperatureprogrammingmodeThetempera-
ndash1tureofovenwasincreasedfrom50degCupto250degCatarateof10degCmin Tionsourcewas200degCMassspectrometrywasusedinSIMmode(mz=35ndash900)
58 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
24ConditionsforLCMSMSdeterminationofpesticides
Separation of pesticides was performed by HPLCMSMS with positiveelectrosprayionizationoncolumnZorbaxBonusRP35μm(21times100mm)with40mMammoniumacetateandmethanolasmobilephaseAandBrespectivelyThefollowinggradientelutionwasapplied20ndash85B(8min)85B(8ndash15min)85ndash95B(150ndash155min)95B(155minus180min)95ndash20B(180minus185min)
ndash1Thevelocityof themobilephasewas03mlmin Thevolumeof the injectedsamplewas 20 microlMS detection capillary voltage +45 kV spray gas velocity
3 ndash1 3 ndash13dm min flow rate and drying gas temperature 15 dm min and 250 degCrespectively
25Selectionofconditionsfordispersiveliquid-liquidmicroextractionofpesticides
The influence of the natures of ionic liquids ([C MIM][PF ] [C MIM][NTf ]4 6 6 2
[C MIM][BF ])andthedispersingsolvent(methanolacetonitrileacetone) the6 4
weightoftheionicliquids(0060ndash0200g)thevolumeofthedispersingsolvent(02ndash10ml)onthedegreeofpesticidesextractionwerestudiedTheinfluenceofthepH(5422)theconcentrationofNaCl(0040ndash0200g)andextractiontime(1-6min)wereinvestigated The effects of different ionic liquids and disperser solvents on DLLMEprocedures were investigated and optimized by using standard solutions ofpesticidesIndetailasolutionofionicliquidsinadispersingsolventwaspreparedand rapidly injected into the aqueous sample solution (2ml) followed bytreatment for 2 min in an ultrasonic bath cooling at ndash4degC for 10 mincentrifugationfor10minat3500rpmandcollectionofionicliquidsThewaterphasewas separatedandanalyzedbyHPLC-MSThe ionic liquidsextractwasdilutedinmethanolandanalyzedbyHPLC-MS
3Resultsanddiscussion
31Microextractionofpesticides
One of the important tasks of environmental monitoring is to control traceconcentrationsofpesticidesinwatersamplesTechniqueofcombiningseveralpesticideshasbecomemorewidespread inagriculture It allowsreducing thetotalconcentrationoftheappliedcompoundsandtodecreasetheadaptabilityofpathogens and insects Therefore the analysis of real samples requires apreliminarystageofselectiveanalytesextractionandconcentration ExtractinganddispersingsolventsarebothimportantinDLLMEofanalytesTheinfluenceofthenatureofthedispersingsolvent(methanolacetonitrileandacetone) and the extractant (imidazolium ionic liquids [C MIM][PF ]4 6
[C MIM][BF ]and[C MIM][NTf ])onthedegreeofextractionofpesticideswas6 4 6 2
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 59
studied This parameter was controlled by the residual concentration ofpesticidesinthewaterphaseafterextractionThebestresultswereobtainedforionic liquids [C MIM][PF ] as an extractant and acetonitrile as a dispersing4 6
solvent Thenextstepwastoselecttheamountofionicliquids(0060ndash0200g)andthevolume of acetonitrile (02ndash10 ml) It was found that the highest degree ofextractionofanalyteswasachievedbyusing020gofionicliquidsand03mlofacetonitrile It was shown that the degree of extraction of selected pesticides does notdependonpHofthewatersamplewhichconfirmsthepartitionmechanismofextractionThedegreeofextractionofcarbofosincreasedslightlywithanincreaseinthesaltconcentrationandreachmaximumbyweightto4(008g) ItisknownthatthehighviscosityofionicliquidshavehinderedtheprocessesofelectrosprayionizationDilutionofthesamplewithmethanolby3timesgivesthebestresult(thesignalintensitywas373to845ofthesignalwithoutionicliquids) ThustheconditionsofDLLME-ionicliquidsextractionofpesticidesfromwatersampleswere found The limits of detection for pesticideswere from007 to
ndash1019ngml thereproducibilityofpeakareaswerefrom3to5theextractionrecoverywascloseto100
32SPMEofvolatileorganiccompoundsfromurinesamples
Oneof the importantdirections is the search for criteriaofearlydiagnosisofcancer Obtaining characteristic profiles of volatile organic compounds fromurinesamplescanhelpdevelopanon-invasivemethodforearlydiagnosisofthedisease ForthiswestudiedtheinfluenceofanumberoffactorsonthetotalnumberofpeaksandthetotalpeakareaTheywerethetemperatureatwhichthevaporandcondensedphasesofurinewerebalanced(30ndash60degC)thepreheatingtimeofthesample(10ndash40min)NaClconcentration(30ndash133wv)andthesorptiontimeonthePDMScoating(5ndash30min) Anincreaseinthepreheatingtemperatureofthesampleto500degCledtoanincreaseinthenumberofsignalswhichdidnotchangewithafurthertempera-tureincreasingNextparameterwasthetimeofachievementequilibriumofthevapor and condensed phases The largest number of peaks was observed at40minbutthisgreatlyincreasedthetimeofanalysisandsowechose20minAlsowestudiedthedesaltingeffectofsodiumchlorideonefficiencyofextractionThebestvolatileorganiccompounds sorptionwasachievedbyadding133saltingagent It is also shown that thenumberofpeaksdidnot changeafter20minutesofsorption Thus to obtain the characteristic profiles of urine samples the followingconditionswereselected1333NaClwasaddedtotheurinesample(3ml)
60 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
heatingfor20minat50degCthensorptionofvolatileorganiccompoundsonPDMSfibercoatingat50degCfor20min Undertheselectedconditionsweobtainedvapor-phaseprofilesof52urinesamples (32 normal and 20 pathology) Prior to performing chemometricprocessing of chromatographic profiles of urine samples preliminary datapreparation is necessary [7] The baseline was removed and the peaks werealignedusingdynamictimewarpingwithcontrollingbymassspectra ThePCAmodelwasbasedon52aligned characteristicprofilesThere is asatisfactoryseparationofdataintotwoclustersinthescoresplotrelativetothefirstandsecondprincipalcomponent(Fig1) The original data set (52 samples) was randomly divided into calibration(13pathology21normal)andtest(7pathology11normal)setsThenthePLS-DAmodelwasbuiltusingthecalibrationsetanditspredictiveabilitywasevaluatedusingthetestsetTheprocedurewasrepeated100timesTheaveragevaluesofsensitivityspecificityandaccuracyinthiscasewere9594and91
4Conclusions
Thepossibilityofusingimidazoliumionicliquids([C MIM][PF ])asextractants4 6
forquantitativeextractionandconcentrationofpesticidesundertheconditionsofDLLMEisshownThedegreeofconcentrationwas28ndash33whichallowedreaching
ndash1thedetectionlimits(006ndash019ngml )belowthemaximumpermissibleconcen-trationThepossibilityofnon-invasivediagnosisofprostatecancerbySPMEofvolatileorganiccompounds inurine isshownChemometricprocessingofgaschromatographic profiles using PLS-DA and PCA methods allowed achievingclassificationaccuracyvaluesmorethan90
Acknowledgments
ThisworkwassupportedbytheRussianFoundationforBasicResearchprojectno18-53-80010BRICS_t and the Russian Science Foundations (Projects 19-13-00370) We are grateful to
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 61
Fig 1Scoresplotrelativetothefirstandsecondprincipalcomponent
Resource Education Center in Chemistry of St Petersburg State University for the providedequipment
References
[1] Rutkowska M Płotka-Wasylka J Sajid M Andruch V Liquidndashphase microextractionAreviewofreviewsMicrochemJ149(2019)103989
[2] JaliliVBarkhordariAGhiasvandAAcomprehensivelookatsolid-phasemicroextractiontechniqueAreviewofreviewsMicrochemJ152(2020)104319
[3] ArthurCLPawliszynJSolidphasemicroextractionwiththermaldesorptionusingfusedsilicaopticalfibersAnalChem62(1990)2145ndash2148
[4] SchmidtKPodmoreISolidphasemicroextraction(SPME)methoddevelopmentinanalysisof volatile organic compounds (VOCs) as potential biomarkers of cancer JMol BiomarkDiagn6(2015)1000253
[5] Mousavi L Tamiji Z Khoshayand MR Applications and opportunities of experimentaldesign for the dispersive liquidndashliquidmicroextractionmethod ndash A review Talanta190(2018)225ndash356
[6] MarcinkowskaRKoniecznaKMarcinkowskiLNamiesnikJKloskowskiAApplicationofionic liquids inmicroextractiontechniquesCurrent trendsandfutureperspectivesTrACTrendsAnalChem119(2019)115614
[7] WehrensRChemometricswithRMultivariateDataAnalysisintheNaturalSciencesandLifeSciencesBerlinSpringer2011
62 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Theself-assembledtwo-dimensionalmonolayers(2DSAMs)ofvariousmolecules(eg graphene [1] MoS [2] rubrene [3]) offer beneficial properties for the2
constructionofnano-electronic andnano-opticaldevicesThe topological andchemicalcharacterizationof2DSAMsiscrucialtogatherinformationaboutthearrangementofdepositedmoleculesandtheirinteractionwiththesubstrateThistaskrequiresanalyticaltechniqueswith(sub)nanometerspatialresolutionandup to single-molecular detection sensitivity Only few techniques meet therequirements and one of them is tip-enhanced Raman spectroscopy (TERS)whichcombinestheexcellentspatialresolutionofscanningprobemicroscopy(SPM) and chemical sensitivity of surface-enhanced Raman scattering (SERS)spectroscopy[45] TheSERS spectroscopyutilizesplasmonicmetal nanostructures to cause ahighlocalenhancementoftheelectricfieldintheirclosevicinityviathesurfaceplasmonresonance(SPR)effectThelocalelectricfieldcausesanincreaseofthe
The development of reference probe system for tip-enhanced Raman spectroscopy
MARTINKRA LMARCELADENDISOVA PAVELMATE JKA
DepartmentofPhysicalChemistryFacultyofChemicalEngineeringUniversityofChemistryandTechnologyPragueTechnickaacute516628Prague6CzechRepublicMartinKralvschtcz
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 63
AbstractThetip-enhancedRamanspectroscopy(TERS)isamodernanalyticaltechniquewithanoutstandingspatialresolutionandchemicalsensi-tivityTheseparametersmainlydependon the structural integrityand chemical purity of employed plasmonic scanning probe tipsUsuallyeachtipistestedbeforeTERSmeasurementsusingcommer-ciallyavailablereferencesamplesHowevertheirpriceandrelativelyshortexpirationdatemustbeconsideredwhenplanningaresearchbudgetWedevelopedaproceduretoproduceself-madereferenceprobesamplesfortestingTERStipsusingcopper(II)phthalocyanineonaAunanolayerwhichispreparedbythermalvacuumevaporationofAuonaSiwaferOurresultsshowthatthepreparedsystemenablesrepeateddetectionofwell-resolvedTERSspectraThecollectedTERSspectraandspectralmapsexhibitsomedegreeofvariabilitywhichmaybeduetovariousphoto-inducedprocessesanditmustbeconsi-deredwhileperformingTERSmeasurements
Keywordscopper(II)
phthalocyaninescanningtunnelling
microscopyresonanceRaman
spectroscopysurface-enhancedRaman
spectroscopytip-enhancedRaman
spectroscopy
Ramanscatteringfrommoleculesboundtothemetalby6ndash8ordersofmagnitude[6]TheenhancementallowsSERSspectroscopytobeusedforsingle-moleculardetectionHoweverRamanmicroscopeshave limitedspatialresolutionbythelightdiffractionwiththeachievableresolutionbeingaroundhalfoftheexcitationwavelengthOntheotherhandthespatialresolutionofSPMtechniquesislimitedonlybythedimensionsoftheapexofthescanningprobetipwhichmayevenbeatomicallysharpByutilizingSPMtipandsubstratemadefromplasmonicmetalsan artificial ldquohotspotrdquomay be createdwith its position and dimensions beingdefinedbythetipItopensthepossibilitytocollectstronglyenhancedRamanspectrafromtheareapreciselylocalizedbellowthetipandthusovercometheopticaldiffraction limitTheartificialhotspotmayberelocatedbymovingthesamplebelowthetipwhichisthefoundationofTERSmapping[578] A successful TERS experiment requires an optimal combination of variousparameters the most important of which are tip sharpness and purity [7]Areferencesampleconsistingofaflatplasmonicnanolayerwithattachedprobemolecules is frequently used to check the state of the tip before using it forexperiments Unfortunately commercially available TERS standards areexpensiveandhaveanexpirationdateofseveralmonths Thegoalofthisstudywastofindapreparationprocedurewhichwouldbeableto produce cheap reference samples for repeated detection of intense TERSspectraAcombinationofaAunanolayeronaSisubstratepreparedbythermalvacuumevaporationwithadsorbedcopper(II)phthalocyanine(CuPc)whichisamoleculewithhighRamancross-sectionwastested[910]Copper(II)phthalo-cyanine known as phthalocyanine blue is a synthetic blue pigment and isfrequently used in paints It has been studied as a potentialmaterial for theconstruction of organic solar cells and other photoelectronic devices [11] AsindicatedbyitscolourCuPcexhibitsseveralabsorptionbandswithinthevisibleregion The effects of a transition to excited electronic states and subsequentluminescencemaybeobservable inRamanmeasurements[12]Au isahighlysuitablemetalforthesampleasitisboththermallyandelectricallyconductivewhichlimitsthelocalheatingofthesampleduringthemeasurementsandenablesthe use of scanning tunnelling microscopy (STM) for tip-surface interactionfeedback[13]
2Experimental
21Reagentsandchemicals
Thesubstrateforthesamplewaspreparedbythermalvacuumevaporationofgoldonsilicon(100)waferFirsta5nmthickCradhesionlayerwasdepositedon
ndash1thewaferfollowedby100nmofAuThedepositionratewas4nmmin forCrand ndash18nmmin for Au The base pressure of the evaporation system was below
ndash65times10 mbarFollowingthepreparationproceduredescribedbyJiangetal[14]
64 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
thecleansubstratewasimmersedintoasaturatedsolutionofCuPc(˃99SigmaAldrich USA) in dimethylformamide (˃98 Lach-ner CZ) for at least 12h atambienttemperatureSubsequentlythesamplewasremovedfromthesolutionrinsedwithMilli-Qwaterandmethanol(paPentaCZ)anddriedwithair
22Instrumentation
TheRamanSERSandTERSspectrawererecordedusingRamanspectrometerInVia Reflex (Renishaw UK) equipped with lasers emitting at two differentexcitation wavelengths 633nm (136mW max power output) and 785nm(204mWmaxpoweroutput)Thespectrometerhasathermoelectricallycooled
ndash1CCDdetectorwithaspectralresolutionof2cm and4microscopeobjectiveswith5times20times50timesand100timesmagnitudeForTERSexperiments the laserbeamwasredirectedtotheSPMplatformInnova-IRIS(BrukerUSA)viaasystemoflightguidesElectrochemicallyetchedAuTERS-STMtips(BrukerUSA)wereusedforallTERSmeasurements The spectra were processed using the Spectragryph software (F MengesldquoSpectragryph - optical spectroscopy softwarerdquo Version 1214 2020httpwwweffemm2despectragryph) Using this software all collectedspectra were treated by a Savitzky-Golay noise filter automatic baselinecorrectionspikeremovalandpeaknormalization
3Resultsanddiscussion
31Ramanmeasurementsofcrystallinecopper(II)phthalocyanine
AtfirsttheRamanspectraofpureCuPcwerecollectedtoprovidereferencedatawhileusingboth633and785-nmexcitationlasers(Fig1)Bothspectraexhibit
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 65
Fig 1RamanspectraofCu(II)phthalocyanineincrystallineformmeasuredat633(top)and785-nm(bottom)excitationThespectraareoffset
ndash1vibration bands in the 500ndash1600 cm region with slight differences in theirintensityratiosThe633-nmexcitationallowstheobservationofadditionalbands
ndash1inthe2000ndash3000cm region(onaluminescencebackground)whichoriginatefrom the resonance Raman effect as the excitation energy overlaps with theQ-bandofCuPc[12]MoreovertheprocessofelectronicexcitationmaylowertheD symmetry of CuPc during resonance Raman scattering and previously4h
forbiddenbandsmaybecomeobservable[15]Theluminescentbackgroundhasandash1maximumaround2200cm whichcorrespondstoamolecularemissionbandat
735nmEventhoughthespectrameasuredwiththe785-nmlaserlinedonotexhibitapparentresonanceenhancementapre-resonanceRamanenhancementmayoccur
32Surface-enhancedRamanmeasurementsofcopper(II)phthalocyaninelayeronagoldsubstrate
The prepared sample of CuPc on a Au layer was analysed using the Ramanmicroscope Both excitation laserswere used to obtain SERS spectra (Fig 2)whichwerecomparedtothespectraofapurecrystallineCuPc Thepositionsof bands in SERS spectra closelymatch their positions in thespectraofbulkCuPcHoweveraslightshiftofsomespectralbandsisobservable
ndash1(eg1528rarr1532cm )whichmaybeattributedtotheinteractionbetweenCuPcand the Au substrate The disappearance of luminescence background andresonance-enhancedbandsinthespectrumat633-nmexcitationalsosuggeststhemolecule-metalinteractionandthetransferofenergyfromCuPcmoleculestothesubstrateMoreovertherearevariationsintherelativeintensitiesofbands
ndash1whichdependontheexcitationenergyegthebandat1309cm isenhancedinSERSspectraatthe785-nmexcitationwhencomparedtothespectraofpureCuPcorevenSERSspectraat633-nmexcitation
66 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2SERS spectra of Cu(II) phthalocyanineon aAu layermeasured at 633 (top) and785-nm(bottom)excitationThespectraareoffset
33Tip-enhancedRamanmeasurementsofcopper(II)phthalocyaninelayeronagoldsubstrate
SERS microspectroscopy is a diffraction-limited technique as it provides anaveragedinformationaboutmoleculesintheilluminatedareaofseveralsquaremicrometers Meanwhile TERS spectra are collected from an area of tens ofnanometers and they contain specific information about the local moleculararrangement topography of the underlyingmetal and properties of the localelectric field between the tip and the substrate Therefore a higher spectralvariabilityshouldbeexpected Several TERS mapping experiments were carried out using both 633 and785-nmexcitationwithvaryingexperimentalparameterssuchasthenumberanddistance betweenmeasured points acquisition time number of acquisitionslaserpoweretc TheTERSspectrameasuredat633-nmexcitationexhibited lowersignal tonoiseratioandreproducibilityAsaconsequenceTERSmappingwasimpossibleandonlyafewone-pointTERSspectrawereobtained(Fig3) TheTERSspectracollectedat785-nmexcitationcontainedahighernumberofwell-resolvedbandsMoreoverthespectrawerestableintimeandsotheTERSmappingwaspossibleThe twopresentedTERSspectraareaveragesofTERSmaps which contained 16 and 80 points with 600 and 300-nm spacingrespectively(Fig4) Thespectracollectedusingbothexcitationwavelengthsexhibitavariabilityinrelative intensities and positions of bands between themeasured points ThevariabilitymaybeattributedtothelocalorientationofCuPcmoleculesbetweenthe tip and the Au surface and the properties of strongly enhanced andnon-homogeneous electromagnetic field which depend on the tip-surfacedistancetheirmorphologyandrelativepositionMoreovertheusedexcitation
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 67
Fig 3TwoexamplesofTERSspectraofCu(II)phthalocyaninemeasuredat633-nmexcitationThespectraareoffset
wavelengthsareclosetoabsorptionbandsofCuPcandthestrongelectricfieldmay give rise to photo-induced effects These effects include the electronicexcitationofCuPctohigherstateschargetransferbetweentheCuatomandthephthalocyaninering ionizationof themoleculeand formationofradicalsThephoto-inducedprocessesarelikelytoplayabiggerroleinTERSspectradetectedat633-nmexcitationduetotheoverlapwithQ-bandofCuPcwhichmaybethecauseoftheirlowersignaltonoiseratioandreproducibility
4Conclusions
Thedeveloped referenceprobe systemofCuPc adsorbedon aAu surfacehasprovedtobesuitablefortheintendeduseasitenabledthedetectionofintenseandwell-resolvedSERSandTERSspectraTheAulayerpreventsoverheatingofthesampleandallowsfortheuseofSTMTheSERSspectrawereinagoodmatchwith thespectraofpureCuPcAslight shiftof somebandsandchange in theluminescent background indicated the interaction between CuPc and the AusurfaceTheTERSexperimentsresultedinspectralmapswithhighintensitiesofindividualspectraIncreasedvariabilitybetweenmeasuredpointswasobservedPossiblesourcesofthevariabilityarephoto-inducedprocessesthatmayoccurinthestronglyenhancedelectricfieldTheseeffectsareaknownfeatureofTERSmeasurements and they offer valuable insight into the photophysics andphotochemistryofCuPcinteractingwiththeAusurfaceThedependenceofTERSspectra on experimental parameters and the preparation procedure of thereferencesampleshouldbefurtherstudied
Acknowledgments
ThisworkwassupportedfromthegrantofSpecificuniversityresearchndashA2_FCHI_2020_039
68 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 4TwoexamplesofaveragedTERSspectraofCu(II)phthalocyaninemeasuredwith785-nmexcitationThespectraareaveragesfromTERSmapsincluding16(top)and80(bottom)measuredpointsThespectraareoffset
References
[1] Mas-BallesteRGomez-NavarroCGomez-HerreroJZamoraF2DmaterialstographeneandbeyondNanoscale3(2011)20ndash30
[2] ZengHCuiXAnopticalspectroscopicstudyontwo-dimensionalgroup-VItransitionmetaldichalcogenidesChemSocRev44(2015)2629ndash2642
[3] SchultzJFLiLMahapatraSShawCZhangXJiangNDefiningmultipleconfigurationsofrubreneonaAg(100)surfacewith5A spatialresolutionviaultrahighvacuumtip-enhancedRamanspectroscopyJPhysChemC124(2020)2420ndash2426
[4] WhitemanPJSchultzJFPorachZDChenHNJiangNDualbindingconfigurationsofsubphthalocyanineonAg(100)substratecharacterizedbyscanningtunnelingmicroscopytip-enhanced Raman spectroscopy and density functional theory J Phys Chem C 122(2018)5489ndash5495
[5] ShaoFZenobiRTip-enhancedRamanspectroscopyprinciplespracticeandapplicationstonanospectroscopicimagingof2DmaterialsAnalBioanalChem411(2019)37ndash61
[6] ArocaRSurface-EnhancedVibrationalSpectroscopyHobokenWiley2006[7] KumarNMignuzziS SuWRoyDTip-enhancedRamanspectroscopyprinciplesand
applicationsEPJTechInstrum2(2015)9[8] BailoEDeckertVTip-enhancedRamanscatteringChemSocRev37(2008)921ndash930[9] BovillAJMcConnellAANimmoJASmithWEResonanceRamanspectraofα-copper
phthalocyanineJPhysChem90(1986)569ndash575[10] Shaibat MA Casabianca LB Siberio-Perez DY Matzger AJ Ishii Y Distinguishing
polymorphsofthesemiconductingpigmentcopperphthalocyaninebysolid-stateNMRandRamanspectroscopyJPhysChemB114(2010)4400ndash4406
[11] SzybowiczMRunkaTDrozdowskiMBałaWGrodzickiAPiszczekPBratkowskiAHightemperaturestudyofFT-IRandRamanscatteringspectraofvacuumdepositedCuPcthinfilmsJMolStruct704(2004)107ndash113
[12] CaplinsBWMullenbachTKHolmesRJBlankDAFemtosecondtonanosecondexcitedstatedynamicsofvapordepositedcopperphthalocyaninethinfilmsPhysChemChemPhys18(2016)11454ndash11459
[13] SaccoAImbraguglioDGiovannozziAndreaMPortesiCRossiAMDevelopmentofacandidatereferencesampleforthecharacterizationoftip-enhancedRamanspectroscopyspatialresolutionRSCAdv8(2018)27863ndash27869
[14] JiangSChenZChenXNguyenDMatteiMGoubertGVanDuyneRPInvestigationofcobaltphthalocyanineatthesolidliquidinterfacebyelectrochemicaltip-enhancedRamanspectroscopyJPhysChemC123(2019)9852ndash9859
[15] MelendresCAMaroniVARamanspectraandnormalcoordinateanalysisoftheplanarvibrationsofironphthalocyanineJRamanSpectrosc15(1984)319ndash326
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 69
1Introduction
The aim of this work has been develo-pmentofanewvoltammetricmethodforthe determination of 23-dimercapto-1-propane-sulfonic acid (DMPS) Fig 1Investigationwasdonetoobtainrelevantinformation about complexingbehaviorofDMPStowardsleadions Lead is one of heavy metals which can cause irreversible neurologicalproblems [1 2]DMPS is a synthetic antidotewith two thiol groups used fortreatmentofpoisoningbyheavymetals[3ndash6]StrongcomplexingpropertieshighwatersolubilityandnegligiblesideeffectsarethemostimportantadvantagesofDMPS[47]
Fig 1 Chemical structure of 23-dimercap-to-1-propane-sulfonicacid
Determination of heavy metal poisoning antidote 23-dimercapto-1-propanesulfonic acid using silver solid amalgam electrode
ab ab bc bMARTACHOIN SKA VOJTE CHHRDLICKA BEATRIZRUIZREDONDO JIR IBAREK aTOMA S NAVRATIL
a JHeyrovskyacuteInstituteofPhysicalChemistryoftheCzechAcademyofSciences Dolejškova21553182thinsp23Prague8CzechRepublicmartachoinskagmailcomb UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova20308128thinsp43Prague2CzechRepublic
c UniversityofValladolidPlazadeSantaCruz847002ValladolidSpain
Abstract23-Dimercapto-1-propane-sulfonic acid (DMPS) was investigatedusingdirectcurrentvoltammetry(DCV)differentialpulsecathodicstrippingvoltammetry(DPCSV)differentialpulseanodicstrippingvoltammetry(DPASV)andeliminationvoltammetrywithlinearscan(EVLS)atapolished(p-AgSAE)andatameniscusmodifiedsilversolidamalgam electrode (m-AgSAE) EVLS confirmed two consecutivereductions with coupled protonelectron transfer VoltammetrictitrationsofDMPSwithPb2+provedcomplexformationwithlimits
minus1ofquantification(LOQs)anddetection(LODs)03and01micromolL atminus1m-AgSAE and 08 and 03micromolL at p-AgSAE respectively
Determination of DMPS in commercial drug Dimaval and humanurine samples confirmed practical applicability of the developedmethod
Keywordscathodicstripping
voltammetry23-dimercapto-1-pro-
panesulfonicacideliminationvoltammetry
withlinearscansilversolidamalgam
electrodeunithiol
70 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Voltammetry was chosen as a determination technique due to its highsensitivity and selectivity speed low costs [8] Thiol groups in DMPS can beoxidatively chemisorbed on solid amalgam electrode It can be used as anaccumulation step for cathodic strippingvoltammetry [9ndash11]Moreover solidamalgamelectrode (SAE)was chosen as theworking electrodebecauseof itspropertiesashighsignaltonoiseratiowidepotentialwindowandabilitytoreachlowlimitsofdetection(LOD)[12ndash13]
2Experimental
21Reagentsandchemicals
Allsolutionswerepreparedusingdeionizedwater(Milli-Q-GradientMilliporendash1PragueCzechRepublic)withconductivitylt005microScm Britton-Robinsonbuffer
solutionspHrangefrom2to12werepreparedbymixingtheproperamountsof02MNaOH(alkalinesolution)andof004MH BO 004MH PO and004M3 3 3 4
CH COOH(allLachemaCzechRepublic)acidicsolutionTheacidicsolutionwas3
preparedbydissolutionof1235gofH BO pa088mLofH PO (85)paand3 3 3 4
1435mLofCH COOH(99)pa in500mLofdeionizedwaterThealkaline3
solution was prepared by dissolution of 3995 g of NaOH pa in 500mL ofdeionizedwater(allLachemaCzechRepublic) StocksolutionofDMPSwaspreparedbydissolving10mgofsolid23-dimer-capto-1-propanesulfonic acid monohydrate pa (Merck Czech Republic) in100mLofdeionizedwaterForthepreparationofthemodelsamplesolutiononecapsuleofdrugDimaval(HeylGermany)contains100mgofDMPSwasdissolved
ndash1in10LofdeionizedwatertotheDMPSconcentrationof0531mmolL Twomodel samplesofDimavalwerepreparedbydilutionof theabove-mentioned
ndash1solutionwithBritton-Robinsonbuffersolutiontoconcentrations10micromolL andndash1of 10micromolL respectively Urine model samples were prepared by mixing
Britton-Robinsonbuffer solutionwithurine samples obtained fromvolunteer(manhealthy30yearsold)inratio11SamplepHwasadjustedbyadditionof
ndash1proper amount of 02molL NaOH Before each measurement oxygen wasremoved for 5minbynitrogenbubbling (purity class 46MesserTechnogasPragueCzechRepublic)
22Instrumentation
Measurementswereperformedusingtwotypesofworkingelectrodesmeniscusmodified silver solid amalgam electrode (m-AgSAE working surface of
20382plusmn0025mm α lt 005) and polished silver solid amalgam electrode2(p-AgSAEworkingsurfaceof0196plusmn0015mm αlt005)Ag|AgCl|3MKClwas
used as the reference electrode andplatinumwire (Oslash1mm)wasused as theauxiliary electrode (both from Elektrochemicke detektory Czech Republic)Measurementswereperformedatlaboratorytemperature(25plusmn2degC)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 71
The pH was measured using pH-meter Jenway 3505 with combined glasselectrodetype924001(BibbyScientificLimitedUK)Voltammetricmeasure-ments were performed using the computer-controlled Eco-Tribo Polarograph(Polaro-Sensors Czech Republic) Software used for measurements wasMultiElChem 33 forWindows XP7810 (J Heyrovsky Institute of PhysicalChemistryoftheCzechAcademyofSciencesCzechRepublic)
3Resultsanddiscussion
OptimumconditionsformeasurementswereobtainedbyseriesofmeasurementinwiderangeofpHvaluesandtestingvariouscleaningproceduresOptimumpotentialof accumulation (E ) and timeof accumulation (t )ofDMPSwereacc acc
adjusted for differential pulse cathodic stripping voltammetry (DPCSV) atp-AgSAEandm-AgSAE ThedependencebetweenpeakheightandconcentrationofDMPSatp-AgSAEhasalogarithmicshapewhichcorrespondstotheaccumulationprocessattheelectrodesurfaceThelineardependencewasobservedintheDMPSconcentra-
ndash1 ndash1tionsfrom03micromolL to20micromolL Peakshifttowardsnegativepotentialwithincreasing concentration of DMPS corresponds to the metal-thiol bond andinfluenceofelectrodesurfacestructureonthisbondOnthecontrarysignalsonm-AgSAEweremorestableandthedependencebetweenconcentrationofDMPSandsignalwasalmostlinearinwholetestedrangeofconcentrations The developedmethodwas tested inmodel samples of Dimaval and urineFoundamountsofDMPSwereingoodagreementwithdeclaredcontentsusingbothelectrodesHowevertherepeatabilityofsignalsregisteredusingp-AgSAEinurinesamplesweresignificantlyworsethanthoseinDimavalsamplesItcanbecausedbycomplicatedbiologicalmatricesandfoulingeffectsofurine ELSV measurements confirmed two consecutive reductions of DMPS inadsorbed state At m-AgSAE signal were at about minus415 mV and minus440 mVrespectively and at p-AgSAE at about minus790 mV and minus830 mV respectively
ndash1 ndash1Reductionsatm-AgSAEatthescanratesfrom80mVs to640mVs havebeencontrolledbyakineticprocessatminus400mV Inanodicscansonm-AgSAEonlyonepeakwasvisibleatabout‒390mVItcorrespondswiththeoxidationofmercuryelectrodesurfaceontheelectrodeinthepresenceofDMPSandwithdiffusionfromthebulksolutionofproductsAtp-AgSAEnosignificantanodicsignalwasfound ThelastpartoftheresearchrevealedvoltammetricbehaviorofDMPSinthe
2+presence of Pb Voltammetric titration was investigated by DPCSV anddifferential pulse anodic stripping voltammetry (DPASV) during consecutive
ndash1 ndash1additionsof1mmolL ofPb(NO ) into100micromolL DMPSsolutioninacetate3 2
bufferofpH50InabsenceofPb2+ontheelectrodesurfaceHg(DMPS)complexisformedduringtheaccumulationstepDuringtheanodicscanthereisonlyonewelldevelopedreductionpeak(Fig2A)
72 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ndash1Fig 2DPCSandDPASvoltammogramsof10micromolL ofDMPSinacetatebufferpH=5correspond-2+ingto[Pb DMPS]ratiosof(A)01(B)11and(C)21Uppercurvecorrespondstothecathodic
scanE =0mVt =15sLowercurvecorrespondstoreverseanodicscanwitht =15satacc acc accndash1E =minus1000mVν=20mVs (Ref[16])acc
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 73
2+ WhenPb DMPSratioisequalto11twooxidationandthreereductionpeakswereregistered(Fig2B)PeakA ataboutndash300mVcorrespondstoformationofred
0Pb(DMPS)complexThiscomplexwasfurtherreducedtothePb (Hg)atabout2+ndash500mV(C )ReductionpeakoffreePb wasalsoregistered(B )Oxidationred red
peaksA andB correspondtoreverseprocessesandC isnotpresentbecauseox ox ox
nofreeDMPSispresentinthesolutionAtratio21thereisnofreeDMPSinthesolution however excess of lead ions As a consequence A and B peakred ox
2+increasedIncreaseofB correspondstothedepositionofPb duringtheaccu-ox
mulationstepPeaksBredandCredremainedpracticallyunchanged(Fig2C) Voltammetric titrationconfirmed themechanismof formationcomplexesofPb(DMPS)Hg(DMPS)andPb(Hg)Italsoconfirmedpossibilityofdetermination
2+Pb andDMPSinthesamesolution
4Conclusions
ValidationinmodelsampleofdrugDimavalandhumanurinespikedwithDMPSconfirmed that this method can be used for clinical purposes Voltammetric
2+titration of DMPS by Pb ions proved that it can be used for simultaneousdetermination of the drug and heavy metal ions in human urine Moreoverobtained LODs were two orders lower than those in the previously reportedvoltammetricmethod[14](Table1)
Acknowledgments
ResearchwascarriedoutwithintheframeworkofSpecificUniversityResearch(SVV260560)TheauthorsthanktheCzechScienceFoundation(GACRprojectNo20-01589S)
References
[1] AnHHLuchakMCopesRLeadtoxicityAsystematicreviewofrecentlypublishedcasesClinToxicol53(2015)757ndash758
[2] KimYLustMRKreimerbirnbaumM23-Dimercaptopropane-1-sulfonate(DMPS)inthetreatmentoflead-poisoningFasebJ2(1988)A1820ndashA1820
[3] AposhianHVDMSAandDMPS ndashwater-solubleantidotesforheavy-metalpoisoningAnnuRevPharmacol23(1983)193ndash215
74 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Table 1ComparisonofvoltammetricmethodsforDMPSdetermination(LDR
Method Workingelectrode Lineardynamic LOQ LOD Refminus1 minus1 minus1 rangemicromolL micromolL micromolL
LSV glassy-carbonelectrode 18ndash140 41 14 [14] modifiedwithmulti-walled 260ndash690 carbonnanotubes DPCSV p-AgSAE 03ndash20 08 03 thisworkDPCSV m-AgSAE 01ndash10 03 01 thiswork 10ndash100
[4] BjorklundG Crisponi G Nurchi VM Cappai R Djordjevic AB Aaseth J A review oncoordinationpropertiesof thiol-containingchelatingagents towardsmercury cadmiumandleadMolecules24(2019)3247
[5] DonnerAHrubyKDMPSinthetreatmentofacuteandchronicheavy-metalpoisoningActaMedAust14(1987)10ndash10
[6] DonnerAHrubyKPirichKKahlsPSchwarzacherKMeisingerVDimercaptopropan-sulfonate(DMPS) inthetreatmentofacute lead-poisoningVetHumToxicol29 (1987)37ndash37
[7] Blanusa M Varnai VM Piasek M Kostial K Chelators as antidotes of metal toxicityTherapeuticandexperimentalaspectsCurrMedChem12(2005)2771ndash2794
[8] BarekJMoreiraJCZimaJModernelectrochemicalmethodsformonitoringofchemicalcarcinogensSensors-Basel5(2005)148ndash158
[9] Josypcuk B FojtaM Yosypchuk O Thiolatemonolayers formed on different amalgamelectrodesPartIIPropertiesandapplicationJElectroanalChem694(2013)84ndash93
[10] YosypchukBMarecekVPropertiesofthiolatemonolayersformedondifferentamalgamelectrodesJElectroanalChem653(2011)7ndash13
[11] Alvarez JMF SmythMRCathodic strippingvoltammetryofpyridine-2-thiolandsomerelated-compoundsAnalyst114(1989)1603ndash1605
[12] DanhelABarekJAmalgamelectrodesinorganicelectrochemistryCurrOrgChem15(2011)2957ndash2969
[13] Fadrna R Polished silver solid amalgam electrode Further characterization and appli-cationsinvoltammetricmeasurementsAnalLett37(2004)3255ndash3270
[14] ZiyatdinovaGKGrigorevaLVBudnikovGKElectrochemicaldeterminationofunithioland lipoic acid at electrodesmodifiedwith carbonnanotubes J Anal Chem64 (2009)185ndash188
[15] HrdlickaVChoinskaMRedondoBRBarekJNavratilTDeterminationofheavymetalpoisoning antidote 23-dimercapto-1-propanesulfonic acid using silver solid amalgamelectrodeElectrochimActadoiorg101016jelectacta2020136623
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 75
Fig 1Structureofcanagliflozin
1Introduction
Canagliflozin is a selective sodium-glucosecotransportertype2inhibitorused for the treatment of type 2 dia-betes mellitus Canagliflozin inhibitssodium-glucose cotransporter type 2present in proximal tubules of the
Canagliflozin oxidation study using electrochemical flow cell and comparison with hydrogen peroxide oxidation
a a bFILIPVYMYSLICKY TOMA S KR IZ EK JAKUBHER T
a DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublicfvymyslickygmailcomb ZentivaGroupasUKabelovny13010237Prague10CzechRepublic
AbstractBystandardstheeffectonoxidationofanactivesubstanceistestedusinghydrogenperoxidesolutionatelevatedtemperatureinastresschamberfor1ndash7daysAnalternativewaytostudytheeffectofoxida-tion on an active substance is to use an electrochemical flow cellSolutionwith active substance flows at low flow rate into a smallreactorwheretheactivesubstanceisoxidizedonworkingelectrodesurfaceTheelectrolytestreamwiththeoxidizedactivesubstanceisthen directed to the sample collector Products of electrochemicaloxidationareanalyzedbyhighperformanceliquidchromatographywithultravioletndashvisiblespectrophotometrydetectionCanagliflozinhasbeenusedbecauseitsmaindegradationpathwayisoxidationThedesign of experiments approach was used to explore the experi-mentalspaceandoptimizeexperimentalconditionsofoxidationTheresultsoftheoxidationstudyperformedintheelectrochemicalflowcellwerestatisticallycomparedwiththeresultsofastandardstudyusinghydrogenperoxidesolutionThemostsuitableconditionsforelectrochemical oxidation were found Electrochemical oxidationproducedcomparableamountsofimpuritiesaschemicaloxidationwithhydrogenperoxide
KeywordscanagliflozindesignofexperimentselectrochemicalflowcellHPLCoxidation
76 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
kidneywhichrestrictsglucoseabsorptioninthekidneytherebyincreasingtheurinaryexcretionofglucoseandloweringthelevelofglucoseintheblood[1]TheformulaofcanagliflozinisC H FO SthestructureofcanagliflozinisinFig1The24 25 5
IUPAC name of canagliflozin is (2S3R4R5S6R)-2-[3-[5-(4-fluoro-phenyl)-thiophen-2-ylmethyl]-4-methyl-phenyl]-6-hydroxymethyltetrahydro-pyran-345-triol[2]CanagliflozinisawhitepowderinsolubleinwaterbutverysolubleinorganicsolventslikemethanolordimethylsulfoxideCanagliflozinissoldundertradenameINVOKANA Manyauthorshavestudiedtheelectrochemicalpropertiesofactivesubstancesin the literature One example is the study of electrochemical behaviour andoxidationofbromhexineThesepropertieswerestudiedusingdifferentialpulsevoltammetryandcyclicvoltammetryonacarbonelectrodeTheresultsofelectro-chemicalmethodswerecomparedwithhighperformanceliquidchromatography(HPLC)analysis[3]Anotherexampleisthestudyofelectrochemicalbehaviourand degradation study performed on the active substance atomoxetineDegradation was studied using differential pulse voltammetry and cyclicvoltammetryonacarbonelectrodeTheresultswerealsocomparedwithHPLCanalysis[4]Electrochemicalmethodsareusedmainlytostudythemechanismofoxidationbutinthisworktheelectrochemicalmethodwasusedtodegradetheactivepharmaceuticalingredient In the stability studies of active pharmaceutical ingredient properties theinfluence of temperature pH light and oxidation is studied [5] By TheInternationalCouncilforHarmonisationofTechnicalRequirementsforPharma-ceuticals forHumanUse (ICH) standards the influenceof oxidationon activepharmaceutical ingredient is studied using hydrogen peroxide at roomtemperatureorincreasedtemperatureinthestresschamberduring1ndash7days[6]Analternativewaytostudytheinfluenceofoxidationonactivepharmaceuticalingredient is using electrochemical flow cellwhere an electrolytewith activepharmaceuticalingredientisdrivenbylowflowrateintothesmallreactorInthesmallreactortheactivepharmaceuticalingredientisoxidizedonthesurfaceoftheworkingelectrodeThestreamofelectrolytewithoxidizedactivepharma-ceuticalingredientisdriventothesamplecollectorProductsofelectrochemicaloxidationareanalysedbyHPLCUVVISThedesignofexperimentsapproachwasusedfordevelopmentofanalternativemethodofoxidationofcanagliflozinusinganelectrochemical flowcellThedesignofexperimentsapproachwasusedtoexplore the experimental space of the method and to find the optimalexperimentalconditionsofelectrochemicaloxidationofcanagliflozin
2 Experimental
21Materialandreagents
Canagliflozin(ZentivaCzechRepublic)999methanol(HoneywellGermany)98ammoniumdihydrogenphosphate(Sigma-AldrichJapan)35ortho-phos-phoricacid(PentaCzechRepublic)25ammonia(LachnerCzechRepublic)30hydrogenperoxide (LachnerCzechRepublic)water forchromatography
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 77
Fig 2Schemeoftheelectrochemicalflowcell(1)input(2)workingelectrode(3)gasket(4)refe-renceelectrode(5)counterelectrode
wasobtainedbypurifyingdemineralisedwaterusingMilliporetypeSynergyUVpurificationinstrument
22Instruments
An Agilent 1290 HPLC system (Agilent Technologies Germany) with highpressure pump autosampler thermostat and DAD detector was used for allexperimentsThePinnacleDBbiphenylcolumn(100times21mm19micromRestekUSA)wasusedforseparationIntheHPLCmethod10mMammoniumdihydrogenphosphatebufferpH=25wasusedascomponentAandmethanolascomponentBofthemobilephaseThegradientprogramwassetasfollowst(min)B01555010551790229023152515Theflowrateofthemobilephasewas
ndash104mlmin and the injection volume was 2μl The detector operated at awavelengthof220nmTheautosamplertemperaturewassetat20degCandthecolumntemperatureat60degCTheEmpowersoftwarewasusedforevaluationForelectrochemicaloxidationelectrochemicalflowcellfromALS(Japan)wasusedGlassycarbonelectrode(=6mm)andsilversilverchlorideelectrodewereusedasworkingandreferenceelectroderespectivelyTheschemeofelectrochemicalflowcellisinFig2ElectrodeswereconnectedwithpotentiostatPalmSens3fromPalmsens (Netherlands) AnElmasonic S15Hultrasonic bath fromElma (Ger-many)wasusedforsamplepreparationForpHmeasurementspHmeterJenway3540fromJenway(UnitedKingdom)wasused
3Resultsanddiscussion
AtthedevelopmentofthemethodforthestudyofcanagliflozinoxidationusingelectrochemicalflowcellitwasfirstnecessarytofindtheoptimalconditionsofelectrochemicaloxidationThedesignofexperimentsapproachwasusedChosen
78 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ndash1Fig 3Cyclicvoltammogramofcanagliflozin(concentrationofcanagliflozin11mgml electrolyte300 mM ammonium dihydrogen phosphate pH = 40 and methanol (11 vv) and scan rate
ndash1001Vs )
independentvariablesandtheirlevelswereconcentrationofelectrolyte(100200300mM)pHofelectrolyte(406080)cellsize(50100200500microm)and
ndash1flowrate(0102504mlh )ThereducedcombinatorialdesignwasusedIntheModde12 software aworksheet containing 11 experimentswas created Theworkingpotentialof12Vwasselectedbasedoncyclicvoltammetryofcanagli-flozininFig3Fromthisfigureitcanbeseenthattheoxidationofcanagliflozinoccursintheregionfrom11Vto14VAllexperimentswereperformedwith
ndash111mgml canagliflozin samples The glassy carbon electrodewas used as aworkingelectrodeandthesilversilverchlorideelectrodewasusedasareferentelectrode The canagliflozin samples oxidized in the electrochemical flow cellunder theexperimentalconditionsgivenby theworksheetweremeasuredbyHPLCwithUVVISdetectionDependentvariablespeakareasofimpuritiesandpercentage of peak areas of impurities obtained from chromatograms wereevaluated by the partial least squaremethod in theModde 12 software Thevariableimportanceintheprojectionplottoolwasusedforinterpretationofthedata as a whole The significance values of the independent variables were
ndash1evaluatedbufferpH=137flowrateof125mlh bufferconcentration061mMandcell size04micromFromthis tool itwasconcluded that theelectrochemicaloxidationofcanagliflozinisthemostaffectedbythepHoftheelectrolyteandflowrateoftheelectrolyteUsingtheoptimizertoolthemostsuitableconditionsfor
ndash1the oxidation of canagliflozin were evaluated flow rate 01 ml h 300 mMammoniumdihydrogenphosphate pHof electrolyte40 and cell size500micromUsingonefactoratthetimeapproachthedependenceofthecellsizeonthetotalsumofimpuritieswastested(Fig4A) Itisvisiblefromthegraphthatthesmallerthecellweusethemoreoxidationproducts are formed Based on the graph the most suitable conditions were
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 79
Fig 4(A)Optimizationofelectrochemicaloxidationconditionsdependenceofsumofimpuritiesoncellsize(B)Chromatogramofasampleoxidizedundermostsuitableconditions
ndash1adjustedtoflowrate01mlh 300mMammoniumdihydrogenphosphatepHofelectrolyte40andcellsize12micromUnderthemostsuitableconditionsarepeat-ability test was performed by ten independent oxidation experiments Therelativestandarddeviationofthepercentageareaofcanagliflozinwas164atasignificant level of 095 The chromatogram of sample oxidized under mostsuitableconditionsisinFig4BThestandardstudyoftheeffectofoxidationoncanagliflozinusinghydrogenperoxideaccordingtoICHguidelineswasperfor-medThestudywasperformedundertwosetsofexperimentalconditionsInthefirstcaseasolutionof50methanolwiththeadditionof3H O wasusedIn2 2
thesecondcasetheconditionsintheelectrochemicalflowcellweresimulatedA300mMammoniumdihydrogenphosphate pH40 andmethanol in a ratio11(vv)withtheadditionof3H O wasusedSamplesfortheoxidationstudy2 2
werestressedinastabilitychamberfor13and7daysattheconstanttempe-ratureof50degC ThetotalsumsofimpuritiesformedduringchemicaloxidationusinghydrogenperoxideinbothmediawerecomparedasisshowninFig5AItisobviousthat
80 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 5(A)Acomparisonofastandardoxidationstudyusinghydrogenperoxidein50methanolwith added buffer andwithout them (B) Chromatogramof sample oxidized electrochemically(C)Chromatogramofsampleoxidizedchemically
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 81
ammoniumphosphatesuppressesoxidationofcanagliflozinThereasonofthisphenomenonisunknownFig5BandFig5CshowchromatogramsofsamplesoxidizedelectrochemicallyandchemicallyrespectivelyItcanbeseenthatfiveimpuritieswereformedbybothtypesofoxidationhoweverindifferentamounts
4Conclusion
AnalternativemethodfortheoxidativestudyofcanagliflozinwasdevelopedThedesign of experiments approach was used in the method development ThedevelopedmethodworkswithRSDof165(α=095)Oxidationofcanagliflozinbythedevelopedmethodproducedfiveimpuritiesthatareidenticalwiththoseproducedusingthestandardoxidationstudywithhydrogenperoxide
Acknowledgments
ThisworkhasbeensupportedbyCharlesUniversityResearchCentreprogramNoUNCESCI014SVV260560projectandpharmaceuticalappliedresearchcenter(TheParc)
References
[1] ChaoECCanagliflozinDrugsFuture36(2011)351ndash357[2] NislySAKolanczykDMWaltonAMCanagliflozinanewsodium-glucosecotransporter2
inhibitorinthetreatmentofdiabetesAmJHealthSystPharm70(2013)311ndash319[3] Turchan M Jara-Ulloa P Bollo S Nunez-Vergara LJ Squella JA Alvarez-Lueje A
VoltammetricbehaviourofbromhexineanditsdeterminationinpharmaceuticalsTalanta73(2007)913ndash919
[4] Perez-OrtizMMunoz C Zapata-Urzua C Alvarez-Lueje A Electrochemical behavior ofatomoxetineanditsvoltametricdeterminationincapsulesTalanta82(2010)398ndash403
[5] Baertschi SW Alsante KM Reed RA Pharmaceutical Stress Testing Predicting DrugDegradationLondonInformaHealthcare2011
[6] RignallA ICHQ1A(R2) stability testing of newdrug substance andproduct and ICHQ1CstabilitytestingofnewdosageformsInICH Quality Guidelines An Implementation GuideATeasdaleDElderRWNims(Eds)HobokenWiley2017p3ndash44
82 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
AlthoughDNArepresentsarelativelystablecomponentfromthechemicalpointofviewitremainsconstantlyexposedtoalargenumberofchemicalorphysicalagentscausingchemicalchangesinDNAmoleculesthatoccurintheenvironmentoraremajororminorproductsofcellularmetabolism[1] One-electronoxidationoftheDNArepresentsadamagingprocesswheretheloss of an electron (oxidation) fromduplexDNA results in the formationof anucleobase radical cation (electron ldquoholerdquo) that is subsequently consumed inchemicalreactionsthatoftenleadtomutationsAdefiningcharacteristicoftheone-electronoxidationofDNAisthepreferentialreactionattheguaninemoietythatisdetectedasstrandcleavagefollowingchemicalorenzymatictreatmentoftheoxidizedDNA[2ndash3]
Novel hybrid electrochemical DNA biosensor for monitoring oxidative DNA damage via oxidationreduction signals of low molecular weight double-stranded DNA
MICHALAUGUSTINVLASTIMILVYSKOCIL
UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova812843Prague2CzechRepublicmichalaugustinnaturcunicz
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 83
AbstractDeoxyribonucleicacid(DNA)representsamajortargetmoleculeformanydamagingagentscausingunfavorablechangesinastructureofDNAmoleculethatbindandinteractwithDNAThusahighdemandforreliabletoolsregardingabettercomprehensionofthenatureofDNAdamagingprocessesstillrepresentsoneofthemaingoalsinthisareaHereinwedescribeadevelopmentofanovelhybridelectro-chemicalDNAbiosensorbasedonanldquoedge-planerdquopyrolyticgraphiteelectrode (EPPGE) in connectionwith an elementaryoptimizationprocessprovidingacloserresolutionoftheredoxprocessesoflowmolecularweightdouble-strandedDNA(dsDNA)attheEPPGESub-sequentanalyticalapplicationincorporatinganemploymentofthemodel structure K [IrCl ] (representative of transition metal2 6
complexes)andevaluationofitsdamagingeffectinrelationtoDNAbymeansof linear sweepvoltammetry resp square-wavevoltam-metryarealsopresented
KeywordsbiosensordamageDNAgraphitevoltammetry
DNA-based electrochemical biosensors are successfully used in variousapplicationssuchasmonitoringandevaluatingthemechanismsof interactionbetweenDNAandvariousdrugsordamagingagentsrapidmonitoringoftracemetalsorpollutantspresent in theenvironmentordirectmonitoringofDNAhybridizationprocesses[4] Theelectrochemicalactivityofnucleicacids(boththenativehigh-molecularonesaswellasoligonucleotides)isingeneralreferredtotheelectroactivityofitscomponents ndash nucleobases and sugar residues At mercury-based electrodesadenine and cytosine residues undergo reduction processes close to ndash14 V(againstSCE)inneutralorweaklyacidicmedium(givingrisetothepeakCA)Ontheotherhandallbaseshavebeenreportedtobeelectrochemicallyoxidizedatcarbonelectrodesbutonlyadenineand(particularly)guanineoxidationsignalshavebeenwidelyutilizedinelectrochemicalDNAbiosensors[5] In2017theelectrochemistryofnucleicacidsachievedanimportantmilestoneasthereductionoftheDNAoligonucleotideswasperformedataldquobasal-planerdquopyrolytic graphite electrode which provided wide potential window allowingboththeelectrooxidationaswellastheeletroreductionofthenucleobasesatasingleelectrodefortheveryfirsttimeDespitethesefindingsutilizationoftheaforementionedbiosensorintermsofanalyticalapplicationshasyettobeverifiedandremainsunclearuptothisdate[6] TheaimoftheproposedcontributionisapresentationofthedevelopmentprocessandsubsequenttestingofanoveltypeofhybridelectrochemicalDNAbiosensoranditsverificationasareliableanalyticaltoolintermsofmonitoringDNAdamage
2Experimental
21Reagentsandchemicals
Low molecular weight double-stranded DNA (dsDNA) derived from salmonspermwasobtainedfromSigma-AldrichGermanyStocksolutions(01mgmL)
of dsDNA were prepared in a 01 molL phosphate buffer of pH=74 (PB)Dipotassium hexachloroiridate (K [IrCl ]) was purchased from Sigma-Aldrich2 6
GermanyStocksolutions(0001molL)ofK [IrCl ]werepreparedinthePB2 6
22Apparatus
Voltammetric measurements were performed using the μAutolab IIIFRA2potentiostatgalvanostat(EcoChemieTheNetherlands)drivenbyaNOVA111software(MetrohmAutolabSwitzerland)Allmeasurementswerecarriedoutina three-electrode system using an ldquoedge-planerdquo pyrolytic graphite workingelectrode(EPPGE)withanelectroactivesurfacediameterof3mm(BASJapan)asilver|silver chloride reference electrode (Ag|AgCl|sat KCl) and a platinum
84 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
counterelectrode(ElektrochemickeDetektoryCzechRepublic)ina20mLglassvoltammetriccellatambienttemperature
23Preparationofthebiosensor
PriortotheeverymeasurementsurfaceoftheEPPGEwasmechanicallycleanedbygentlewipingoftheelectrodeonthesoftpolishingpadrinsedwithdistilledwaterAfterwardstheelectrodewasrinsedwithdistilledwaterandplacedinthePBforthesubsequentelectrochemicalactivationElectrochemicalactivationwasperformed in thePBbyapplyingpotentialof15V for240swithoutstirringApotentialpulseinworkingrangeofpotentials(00ndash15V)wasthenapplied Additional electrochemical activationwasperformed in the solutionof the
3minus4minusredoxindicator([Fe(CN) ] )byconsecutivecyclingintherangeofpotentials6
from10tondash08V(15scans)andfrom055tondash015V(10scans) The electrochemical DNA biosensor based on the EPPGE (dsDNAEPPGEbiosensor)was prepared by the adsorption of dsDNAon the EPPGEOptimal
parametersofthedsDNAadsorptionwereaconcentrationof01mgmLinthePB
(c )adepositionpotentialof07V(E )andanadsorptiontimeof5ming(dsDNA) dep
(t )withoutstirringthesolutionads
Atlasttheelectrodewasimmersedinthesolutionoftheredoxindicatorandtheconsecutivecyclingintherangeofpotentialsfrom055tondash015V(20scans)wasperformedinordertosecurethestabilityoftheoxidationreductionsignalsofdsDNAattheEPPGE
24Procedures
Theexperimentalparameterswereasfollowssquarewavevoltammetry(SWV)inthePBwithapulseamplitudeof20mVafrequencyof50Hzascanrateof750mVsandapotentialstepof15mVlinearsweepvoltammetry(LSV)inthePBwithscanratesof02ndash10Vandapotentialstepof24mVAllcurveswere
recordedthreetimes(n=3)
3Resultsanddiscussion
Sincetheclosestresolutionoftheprocessesassociatedwiththeelectroreductionof singleDNAcomponents at pyrolytic graphitehasbeenperformedwith theldquobasal-planerdquopyrolyticgraphiteelectrode(BPPGE)wehavedecidedtotakeovercorrespondingexperimentaltechniqueandconditions(LSVscanrateof10Vssteppotentialof24mV)attheverybeginningofouroptimizationprocesswiththeEPPGE[6] Inthisparticularcaseitispossibletonoticetheoccurrenceofthetwomixedvoltammetricpeaksatthedefaultexperimentalconditions(greenlineFig1A)selectedforthereductionofdsDNAattheEPPGEBygraduallydecreasingthe
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 85
Fig 1Baseline-correctedLSVrecordingscorrespondingtothereductionofdsDNAattheEPPGEfordifferentvaluesofscanrate(02ndash10VsFig1A)respbaseline-correctedLSVrecordingscorres-pondingtothereductionofdsDNAattheEPPGEandthenegativetestperformedunderthesameexperimentalconditionswithintheblanksolution(phosphatebuffer)atthebareEPPGE(03VsFig1B)
scan rate the optimal conditions (νle03Vs) were found and the mutualseparationof the signalswas allowedndash characterizedby thepresenceof twosinglewell-developedvoltammetricpeaksatpotentialsofndash175Vrespndash190V(03Vs orange line Fig 1A) Taking into account previous work regardingprocessesassociatedwiththereductionofDNAatthemercuryelectrodesrespBPPGE we can assume that the peak appearing at the potential of ndash175 Vcorresponds to themixedpeak for the reductionof the cytosine and adenineresidueswithindsDNA(peakCA)[5ndash6] Closer resolution of the second voltammetric peak appears to be farmoreproblematic Regarding our previous study we have discovered that theutilizationofdifferentE fortheadsorptionofdsDNA(E lt07V)isconnecteddep dep
withanappearanceofthethirdoxidationsignal(besidestheoxidationsignalsofguanine resp adeninemoieties) at apotential of073V corresponding to theoxidationoffreeguaninebases(FGBs)presentwithinthesolutionofdsDNAInthiscasewecanassumethatthepeakappearingatapotentialofndash190VcanpossiblyrepresentthereductioncounterpartofFGBspresentwithinthesolutionofdsDNAThisassumptioncanalsobesupportedbytheaforementionedstudyandbythefactthatthereductionsignalatsuchahighnegativepotentialcanbeobservedfortheoligodeoxynucleotidescontainingguanineresidues[6] AdditionallyinordertoverifythetruenatureofthereductionsignalsdepictedatF ig1Bandtoexcludetheoptionthattherelatedsignalsdonotrepresenttheproductsofpriorelectrochemicalactivationof theEPPGE(variousCndashObasedchemicalspecies)wehavedecidedtoperformanegative(control)testwithinthe
86 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2Baseline-correctedLSVrecordingscorresponding to thereductionof thedsDNA(02Vs
Fig 2A)respbaseline-correctedSWVrecordingscorrespondingtotheoxidationoftheguanine(098V)respadenine(128V)moieties(075VsFig2B)attheEPPGEafteritsincubationinthephosphatebufferforadefinedtimeperiod(60ndash900s)
blanksolution(PB)employingthesameprotocolasforthedsDNAadsorptionattheEPPGEInthiscaseitispossibletoobservetheabsenceofanypronouncedvoltammetricpeakslinkedtothedsDNAadsorptionandonlythepresenceofoneirreversiblepeakatapotentialofndash153VcorrespondingtotheelectroreductionoftheCndashObasedmoietymoietieswhichdropsafterthedsDNAadsorptiontoonetenthofitsoriginalvalue(approximately) Perhaps the most important parameter regarding further optimizationprocessrepresentedthetime-dependentstabilityofthecorrespondingsignalsofdsDNAwhichcanbespecificallyimportantinrelationtothestudyofthetime-dependentoxidativedamageofdsDNA AsithasalreadybeenprovedasingleelectrochemicalactivationoftheEPPGEinthePBdoesnotrepresentasatisfyingtechniqueregardingstabilityofdsDNAoxidationsignalsat theEPPGEand theadditionalstabilization isachievedbyfurtherelectrochemicalactivationinthesolutionofaredoxindicator(Fig2B)[7]Basedonthisaverificationoftheproposedstabilizationprotocolintermsofthetime-dependent stabilityofdsDNAreduction signals in the solutionof thePBwithinthedefinedtimeperiod(60ndash900s)appearedasareasonablenextstep FromtheresultsdepictedinFig2Aitispossibletonoticethatwithinthefirst300sdsDNAreductionsignalsremainstableinrelationtothecurrentresponseaswellasintermsofthepotentialvalueWithanadditionalincubationtime(t )inc
(900sorangeline)thepeakcurrentofthevoltammetricsignalpresentatmorenegative potentials decreased which can probably be addressed as a slowprogressive elimination of the weak (electro)chemical forces related to theunspecificadsorptionoftheFGBsattheEPPGE
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 87
Fig 3Baseline-corrected SWV recordings corresponding to the oxidation of the guanine respadeninemoietiesattheEPPGEafteritsincubationinthesolutionofK [IrCl ](IR)foradefinedtime2 6
period(60ndash3600s)(075VsFig3A)andthecorrespondingrelativebiosensorresponses(ΔI )rel
evaluatedusingtheguanosine(turquoise)andadenosine(red)peaksplottedversustheincubationtime(Fig3C)Baseline-correctedLSVrecordingscorrespondingtothereductionofdsDNAattheEPPGEafteritsincubationinthesolutionofK [IrCl ](IR)fordifferenttimeperiods(60ndash3600s)2 6
(02 VsFig3B)andthecorrespondingrelativebiosensorresponses(ΔI )evaluatedusingtherelpeakCA(darkpink)plottedversustheincubationtime(Fig3-D)
AdditionallywehavedecidedtotesttheapplicabilityofthepresentedhybridbiosensorintermsofmonitoringdsDNAdamagecausedbyarepresentativeofone-electron oxidants ndash K [IrCl ] In this case the prepared dsDNAEPPGE2 6
biosensor was immersed into the solution of K [IrCl ] (0001molL) for the2 6
definedtimeperiod(60ndash3600s) In thecaseof theoxidationpath (SWVrecordingsdepicted inFig3A) it ispossible to observe a time-dependent decrease of the oxidation signal of theguaninemoietieswhereastheoxidationsignaloftheadeninemoietiesremainsunaffectedforthemostof the incubationperiodThisphenomenonis ingoodcorrelationwiththetheoreticalknowledgeregardingoxidativedamageofdsDNAcausedbyone-electronoxidants [3]Simultaneouslywith thisLSVrecordingsdepicted in Fig 3B followed the similar behavior (decrease in relation to the
88 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
currentresponseofthedsDNAreductionsignalndashpeakCA)asinthecaseofthesignal regarding oxidation of guanine moieties In addition according totheportionofthepreservedDNA(Fig3C3D)itispossibletoassumethatthepronouncedoxidativedamageofdsDNAcanbemonitoredquitepreciselynotonlydirectlyviathedsDNAoxidationsignaloftheguaninemoietiesbutevenindirectlythroughthedsDNAreductionsignalndashpeakCA
4Conclusions
Inthiscontributionwehavepresenteddevelopmentofanunorthodoxhybridelectrochemical DNA biosensor based on an EPPGE Optimization processconcerning some important parameters was performed as well as closerresolutionofthenatureofthereductionprocessesofdsDNAattheEPPGEwasachievedInordertoconfirmtheresultsoftheoptimizationprocessapplicabilityoftheproposedbiosensorhadbeenprobedintermsofmonitoringDNAdamagecausedbyK [IrCl ]Inthiscasethefinalresultshadprovedthattheprepared2 6
hybridbiosensorcanbeconsideredasaversatileanalyticaltoolformonitoringoxidativeDNAdamage(viaoxidationreductionsignals)andispresentedasafinealternative in comparisonwith conventional electrochemical DNA biosensorsprepared within the group of traditional transducer materials (mercury- orcarbon-based)
Acknowledgments
ThisresearchwassupportedbytheSpecificUniversityResearch(SVV260440)
References
[1] FojtaMDanhelAHavranLVyskocilVRecentprogressinelectrochemicalsensorsandassaysforDNAdamageandrepairTrACTrendsAnalChem79(2016)160ndash167
[2] GieseBSpichtyMWesselySLong-distancechargetransportthroughDNAAnextendedhoppingmodelPureApplChem73(2001)449ndash453
[3] Burrows CJ Muller JG Oxidative nucleobasemodifications leading to strand scissionChemRev98(1998)1109ndash1151
[4] DiculescuVC Chiorcea-PaquimAMOliveira-BrettAMApplications of aDNA-electro-chemicalbiosensorTrACTrendsAnalChem79(2016)23ndash36
[5] PalecekEJelenFElectrochemistryofnucleicacidsInElectrochemistryofNucleicAcidsandProteinsndashTowardsElectrochemicalSensorsforGenomicsandProteomicsPalecekESchellerFWangJ(edits)AmsterdamElsevier2005p74ndash174
[6] SpacekJDanhelAHasonSFojtaMLabel-freedetectionofcanonicalDNAbasesuraciland5-methylcytosineinDNAoligonucleotidesusinglinearsweepvoltammetryatapyrolyticgraphiteelectrodeElectrochemCommun82(2017)34ndash38
[7] AugustınMVyskocilVNovelelectrochemicalDNAbiosensorbasedonedge-planepyrolyticgraphite for DNA interaction studies In Proceedings of the 15th International StudentsConferenceldquoModernAnalyticalChemistryrdquoNesmerakK(edit)PragueFacultyofScienceCharlesUniversity2019p263ndash268
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 89
1Introduction
Cadmiumisoneofthemosttoxicmetalsanditswidespreadindustrialusesresultin increased environmental pollution Hence the development of sensitivemethodology for Cd determination is still highly desirable Chemical vaporgeneration(CVG)ofCdbythetetrahydroboratereductioninacidicmediumisasuitable alternative sample introduction technique compatible with atomicspectrometricdetectorsandofferingimproveddetectioncapabilityComparedtocommonliquidnebulizationCVGoffersseveraladvantagessuchassignificantlyhigheranalyteintroductionefficiencyandalsoanalyteseparationfromsamplematrix IncomparisontoCVGofcommonhydrideformingelementsthereisalackofliterature dealing with mechanistic aspects of CVG of Cd [1] as well as withstabilityandidentityofitsvolatilespecies(freeatomshydrideotherspecies)Very little information is also available on achieved generation efficiencyMoreover there are many discrepancies in the literature regarding optimum
Chemical vapor generation of cadmium for analytical atomic spectrometry
a b a b a aLINDASAGAPOVA BARBORAKODRIKOVA MILANSVOBODA STANISLAVMUSIL aJANKRATZER
a InstituteofAnalyticalChemistryoftheCzechAcademyofSciences Veveřiacute9760200BrnoCzechRepublicsagapovaiachczb DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublic
AbstractChemical vapor generation of cadmium volatile compounds wasoptimizedinordertodeterminetraceCdconcentrationsbyatomicabsorptionspectrometry(AAS)Severalreactionmodifiersbasedon
III+ II+ III+ IV+inorganicsaltsandcomplexesofCr Co Ti Ti weretestedtoimproveanalyticalperformanceandgenerationefficiencyTheuseofthese reaction modifiers resulted in 4ndash5 times enhancement insensitivity reflected also in corresponding increase of generationefficiency and better repeatability Generation efficiency wasdeterminedfromacomparisonbetweensensitivitiesobtainedwithchemicalvaporgenerationandconventionalsolutionnebulizationbothsimultaneouslycoupledwithinductivelycoupledplasmamassspectrometryTheidentityofthegeneratedcadmiumcompoundswillbediscussed
Keywordsatomicabsorption
spectrometryatomizationcadmiumchemicalvapor
generation
90 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
conditionsforCVGofCdAlthoughstrongacid(HClorHNO )isalwaysemployed3
asacarrierandNaBH asareductantsomeauthorsreportedvariousadditives4III+ III+ IV+(modifiers)basedontransitionmetalions(Cr Ti Ti )inthepresenceof
II+KCN[23]orCo inthepresenceofthioureaandascorbicacid[4]toimproveCdsignalssignificantly TheaimofthisworkwastoinvestigateCVGofCdinacomprehensivewayFirstlyCVGofCdwithoutandwithselectedmodifierswasoptimizedemployingatomicabsorptionspectrometry(AAS)asadetectorandexternallyheatedquartztube(QTA)astheatomizerSecondlytheeffectofatomizationtemperatureonCdsignalwasstudiedallowingthustodeducetheatomicormolecularstructureofgeneratedCd speciesThirdly generation efficiencyofCdvolatile specieswasquantified
2Experimental
21Reagentsandchemicals
minus1Boiled and bubled (Ar per 30min) deionizedwater (lt 01 μScm UltrapurWatrex USA) was used to prepare all solutionsWorking Cd standards were
minus1preparedfrom1000mgL Cdstocksolution(AstasolAnalytikaCzechRepublic)minus1bydilutionin01ndash048molL HCl(basedonthemodifieremployed)from37
HCl(paMerckGermany)Theoptimumgenerationconditionsbeingdifferentforeachmodifiertestedare listedinTable1ThereductantwasasolutionofNaBH (ge 97 Sigma-Aldrich Germany) in 04 (mv) KOH (pa Merck4
Germany) prepared fresh daily The solutions of modifiers were prepared as2+followsCo waspreparedfromCoCl 6H O(ge990PENTACzechRepublic)2 2
3+the solution of Cr from Cr(NO ) 9H O (ge 9999 tracemetal basis Sigma-3 3 23+AldrichGermany)thesolutionofTi fromTiCl solution(about15in10HCl3
4+Sigma-AldrichGermany)andthesolutionofTi fromTiOSO (ge999Sigma-4
AldrichGermany)Tostabilizethelattersolution1H SO wasusedprepared2 4
bydilutionof96H SO (paLach-NerSlovakia)SolutionofKCN(ge9702 43+ 3+FlukaSwitzerland)wasusedasasecondmodifierwhenworkingwithCr Ti or
4+ ndash3Ti asmodifiersitsconcentrationvariedfrom008to016moldm depending
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 91
Table 1OptimumconditionsforchemicalvaporgenerationofCdinthepresenceofmodifiersandtheirabsence
onthemetalionThiourea(CH N Sge980LachemaBrno)andascorbicacid4 2
(C H O ge997Riedel-deHaenGermany)wereusedasmodifierscombined6 8 62+withCo
22Instrumentation
221Chemicalvaporgenerationsystems
TwoCVG flow injection systemswere employed either a two channel systemwithoutadditionofamodifier(seeFig1A)orafourchannelsystemallowingadditionofmodifiers(seeFig1B)
ndash1 TheflowratesofHClandNaBH were42and10mLmin respectivelyinatwo4ndash1channelsystem(Fig1A)whiletheywerebothkeptat10mLmin inthefour
channelsystem(Fig1B)Theflowratesofmodifiersinthefourchannelsystemndash1were05mLmin Thevolumeofthesampleloopwas015mLinbothsystems
ndash1Carriergasflowrateof75mLmin Arwascontrolledbyamassflowcontroler(Cole-ParmerUSA)
92 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
(A)
(B)
Fig 1Schemesofthechemicalvaporgenerationflowinjectionsystemwith(A)twochannels(nomodifiers)and(B)fourchannels(modifiersemployed)
222Atomicabsorptionspectrometry
ThePerkin-Elmermodel503atomicabsorptionspectrometer(BodenseewerkGermany)wasequippedwithaCdelectrodelessdischargelamp(Perkin-ElmerUSA)operatedat228mAThemeasurementswereperformedat2288nmusinga07nmslitwidthTheShimadzumodelAA-7000atomicabsorptionspectrometer(ShimadzuJapan)wasalsousedACdhollowcathodelamp(PhotronAustralia)operatedat2288nmlinewith07nmspectralbandpassandalampcurrentof12mA Signals were recorded for 2 minutes and peak areas were taken forevaluation The QTA was heated electrically to the temperature required byfurnace(PerkinElmer)andanin-housemadefurnacecontrolledbytheREX-C100controller(SysconIndianaUSA)withtheK-typethermocouplesensor(OmegaEngineeringUSA)223QuantificationofCVGefficiencybyICP-MS
Overall CVG efficiency of Cd was quantified bymeans of inductively coupledplasmamassspectrometry(ICP-MS)fromcomparisonoftheslopesofcalibra-tionsobtainedwithnebulizationliquidCdstandardstothoseobtainedwithCVGThe efficiency of liquid nebulization was quantified using a modified wastecollection method (see reference [5] for details) The Agilent 7700x ICP-MSinstrument(AgilentUSA)wasoperatingat1600WofRFpowerThesignalwas
111 125monitoredat Cdisotopeandcorrectedforthesignalofinternalstandard( Tendash11000ngmL Tein2HNO )NebulizeranddilutionArgasflowrateswere11503
ndash1and0mLmin respectively
3Resultsanddiscussion
31Chemicalvaporgenerationconditions
UnivariateoptimizationswereperformedtofindoptimumconditionsforCVGofCdinpresenceandabsenceofmodifiersTheparameterstobeoptimizedwerecarrieracid(HCl)concentrationreductant(NaBH )concentrationmodifierIand4
modifier II concentrations carrier gas flow rate (Ar) length of reaction coilsRCI-III (see Fig 1B) The optimum conditions for individual modifiers aresummarizedinTable1
32IdentityofCdspecies
ThepeakareasofgeneratedCdspecieswereforagivenmodifiermeasuredintheQTAheatedto900degCandnon-heatedQTAsubsequentlyOptimumCVGconditionswere employed as summarized in Table 1 This simple experiment allowsdistinguishingbetweenatomic(freeatoms)andmolecularformsofgenerated
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 93
Fig 2RelativesignalofgeneratedCdspeciesmeasuredintheQTAheatedto900degC(blackbars)andnon-heatedQTA(whitebars)withoutorinthepresenceofmodifiers
speciesOnlyfreeatomscanbedetectedinnon-heatedQTAsimilarlyasincaseofmercurycoldvaporsOn thecontrarymolecularanalytespeciesareatomizedat900degCAsaconsequencethesignalregisteredintheheatedQTAcorrespondstobothatomicandmolecularspeciesgeneratedItmustbehighlightedthattheresidencetimeoffreeatomsintheatomizerisdependentonQTAtemperatureduetogasexpansionAsaconsequencethesignalinQTAheatedto900degCshouldreach25ofthesignalatambienttemperaturetakingintoaccountthatonlyfreeatomsaregeneratedSincethetemperaturealongtheopticalarmofQTAisnotdistributedhomogeneouslydecreasingtobothendtheeffectivetemperatureoftheatomizerislowerOurexperimentswithCVGofHgrevealedsignalinheatedQTAisaround40[6]TheresultsreachedforCVGofCdaredepictedinFig2ThesignalofCdinheatedQTAisaround50ofthesignaldetectedinnon-heatedQTA
4+whennomodifierisemployedorusingTi asthemodifierindicatingclearlyfreeCdatomsarethedominantvolatilespeciesgeneratedOnthecontraryalmostno
2+differenceinpeakareaswasobservedforCo asthemodifierwhilethesignalinheatedQTAwas even 5 times higher in heatedQTA compared to non-heated
3+atomizer with Cr as the modifier suggesting the dominant contribution of3+molecularstructurestoCdsignalespeciallyincaseofCr KCNreactionsystem
33Generationefficiency
TheoverallCVGefficiencywasestimatedfromacomparisonbetweensensitivitiesobtainedwithCVGsampleintroductionandconventionalsolutionnebulizationICP-MSunder the sameexperimental conditionsNebulization efficiency for aMicroMISTnebulizerwasdeterminedas79plusmn01ThegenerationefficiencyofCd was derived from the sensitivity enhancement between CVG and liquidnebulizationTheresultsaresummarizedinTable2indicatingthatCVGwithout
94 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
modifiersisonlycatwotimesmoresensitivecomparedtoliquidnebulization3+ 4+Generation efficiency of Cd increases to 60 in the presence of Ti and Ti
modifiers
4Conclusions
CVG of Cd was thoroughly optimized in the presence of selected modifiersreportedpreviouslyintheliteratureGenerationefficiencyofCdintheabsenceofanymodifierswasquantifiedto15whileitcanbeincreasedupto60inthe
3+ 4+presenceofTi KCNorTi KCNasmodifiersFreeCdatomsseemtobe the4+dominantCdformgeneratedintheabsenceofanymodifiersorusingTi KCN
3+modifierwhile rathermolecularCd structuresaregenerated inCr KCNand2+Co thioureaascorbicacidreactionsystems
ExperimentsareinprogresstofinishthiscomprehensivestudyOnlythebestmodifierwillbefurtherusedforCVGofCdtobecoupledwithotherspectrometricdetectorsandappliedtocertifiedreferencematerialsandrealsamples
Acknowledgments
ThisresearchhasbeensupportedbytheCzechScienceFoundationundercontract18-01116SandbytheInstituteofAnalyticalChemistryoftheCzechAcademyofSciences(InstitutionalResearchPlannoRVO68081715)andCharlesUniversity(ProjectnoSVV260440)
References
[1] PitzalisEAngeliniDMascherpaMCDacuteUlivoAInsightintothemechanismscontrollingthechemicalvaporgenerationofcadmiumJAnalAtSpectrom33(2018)2160ndash2171
[2] ArslanZYilmazVRoseLEfficientgenerationofvolatilecadmiumspeciesusingTi(III)andTi(IV)andapplicationtodeterminationofcadmiumbycoldvaporgenerationinductivelycoupledplasmamassspectrometryMicrochemJ123(2015)170ndash178
[3] YilmazVRoseLArslanZLittleMDOn-linechemicalvapourgenerationofcadmiuminthepresenceofhexacyanochromate(III)fordeterminationbyinductivelycoupledplasmamassspectrometryJAnalAtSpectrom27(2012)1895ndash1902
[4] Y Lu SunHW YuanCG YanXP Simultaneous determination of trace cadmiumandarsenic inbiologicalsamplesbyhydridegeneration-doublechannelAFSAnalChem74(2002)1525ndash1529
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 95
Table 2GenerationefficiencyofchemicalvaporgenerationofCdasquantifiedbyICP-MS
Modifiers Generationefficiency
Nomodifiers 15plusmn13+Cr KCN ndash2+Co thioureaascorbicacid ndash3+Ti KCN 58plusmn24+Ti KCN 61plusmn2
[5] VyhnanovskyJStrugeonREMusilSCadmiumassistedphotochemicalvaporgenerationoftungsten fordetectionby inductively coupledplasmamass spectrometryAnal Chem91(2019)13306ndash13312
[6] MigasovaMMatousekTSchrenkovaVZ ıdekRPetry-Podgorska IKratzer JMercuryvolatilespeciesgenerationfromHClandTRISbuffermediaAnalChimActa1119(2020)68ndash76
96 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Atomicfluorescencespectrometry(AFS)coupledwithvapourgenerationisanultrasensitive analytical method for determination of various elements ItsanalyticalperformancecanbecomparabletoICP-MSwithliquidnebulizationbutatsubstantiallylowercost[1]SampleintroductiontoAFSisacrucialstepoftheanalyticalproceduresincetheanalytehastobeintroducedtotheatomizerintheformofitsvolatilespecies Hydridegeneration(HG)isamaturetechniqueofsampleintroductionduringwhichvolatileanalytehydridesare formedbyreactionwithareducingagenttypicallysodiumborohydrideAnewemergingtechniquephotochemicalvapourgeneration(PVG)employsUV irradiationof theanalyte in liquidphase in thepresenceofaphotochemicalagent(usuallyalowmolarmassorganicacidformic
minusoraceticacid)Highlyreducingradicalspecies(HbullRbullandCOObull )andaquatedelectronsare formedduring irradiationandreactwith theanalyte to form its
Photochemical vapour generation of bismuth coupled with atomic fluorescence spectrometry
ab ab a aBARBORASTA DLEROVA JAROMIRVYHNANOVSKY JIR IDE DINA STANISLAVMUSIL
a InstituteofAnalyticalChemistryoftheCzechAcademyofSciences Veveřiacute9760200BrnoCzechRepublicstadlerovaiachczb DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublic
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 97
AbstractPhotochemical vapour generation of bismuth was successfullycoupledwithnon-dispersiveatomic fluorescencespectrometry forthefirsttimeVolatilespeciesofBiweregeneratedusingastandardmercurylow-pressuretubelampandacoiledreactorfromareaction
2+mediumwhichwas composedof acetic and formic acid Co ionswereusedasasensitizerOptimizationofatomizationconditionsinaflame-in-gas-shieldatomizerwasperformedThismethodologywascomparedtothecommonlyemployedhydridegenerationapproachAbsolutelimitofdetectionof68pgwasachievedwithphotochemicalvapour generation which is still about 7 times worse than withhydride generation The developed methodology was successfullyverifiedbyBideterminationinareferencematerialofwater
Keywordsatomicfluorescence
spectrometrybismuthhydridegenerationphotochemicalvapour
generation
volatilespeciesInbothcasesthegeneratedvolatilespeciesoftheanalytehavetobeseparatedfromtheliquidphaseinthegas-liquidseparatorandarecarriedtotheatomizerbyacarriergas[2] InthisworkanatomizerdesignedspecificallyforAFStheflame-in-gas-shieldatomizerwasused(Fig1) It consistsofaverticalquartz tubesuppliedwithargon and hydrogen together with the analyte volatile species Moreover acapillaryisinsertedintheverticalaxisoftheverticaltubethroughwhichoxygenisintroducedA hydrogen-oxygenmicroflameburns on top of the capillary Themicroflameisshieldedfromtheambientatmospherebyaflowofargonwhichisintroducedthroughashieldingunitfittedaroundtheverticaltube[34] Theaimofthisworkwastooptimizeatomizationconditionsintheflame-in-gas-shieldatomizerusingPVGasasampleintroductiontechniqueandtocomparetheanalyticalcharacteristicsofPVGandHGforultrasensitivedeterminationofbismuthbyAFS
2Experimental
21Reagentsandchemicals
Deionized water (Ultrapur Watrex USA) was used for preparation of all thesolutionsWorkingBisolutionswerepreparedfreshdailybyserialdilutionof
ndash1stock1000mgl BistandardforAAS(Sigma-AldrichGermany)RegardingHG05(mv)NaBH in04(mv)KOHwasusedasa reductantAsolutionof4 ndash11mol l HClwasusedasacarrierandblank
98 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Flame-in-gas-shieldatomizerOHndashobser-vationheight
RegardingPVGformicacid(98paLach-NerCzechRepublic)andaceticacid (998 pa Lach-Ner CzechRepublic)were used for preparation of thereaction medium they were purified in a Teflon BSB-939-IR sub-boilingdistillation apparatus (Berghof Germany) The composition of the reactionmedium(40(vv)acetic125(vv)formicacid)wasoptimizedearlier[5]
ndash1The 5000mgl Co stock solution was prepared from cobalt(II) acetatetetrahydrate (pa Lach-Ner Czech Republic) and used as a sensitizer ofphotochemicalreactionTheoptimalconcentrationofCointhestandardsamples
ndash1andblanksolutionscorrespondedto50mgl (ref[5]) Acertifiedreferencematerial(CRM)-1643fTraceElementsinWater(NationalInstituteofStandardsandTechnologyUSA)wasusedtochecktheprecisionofthedevelopedmethodology
22Instrumentation
221Atomicfluorescencespectrometer
An in-house assembled non-dispersive atomic fluorescence spectrometerconstructedatourlaboratorywasusedforBideterminationandisdescribedindetail elsewhere [3] The detector output provided signals in microV Peak areacorrectedtobaselineandmainlysignaltonoiseratioweretheparametersusedtoevaluatethedata
222Hydridegeneratorphotochemicalvapourgeneratorandatomizer
A flow injection hydride generator was employed (Fig 2a) The reductantndash1 ndash1(12mlmin )andthecarrier(4mlmin )werepumpedbyaperistalticpump
Thesamplewasinjectedthrougha1mlsampleloopintotheflowofcarrierAglassgas-liquid separator (5 ml) with forced waste removal was employed forseparatingthegasphasecontainingbismuthanewhichwasthencarriedtotheatomizerbyargon Thephotochemicalvapourgenerator(Fig2b)consistedofthephotoreactorconstructedwitha15Wlow-pressureHggermicidallamp(Cole-ParmerUSA)wrappedaroundwith6mofPTFEtubing(1mmidinternalvolume471ml)The
ndash1reactionmedium(3mlmin )waspumpedbyaperistalticpumpThesamplewasinjected througha056ml sample loopApolypropylenegas-liquid separator
(15ml)withforcedwasteremovalimmersedinanicebath[6]wasemployedforseparatingthegasphasecontainingBivolatilespeciessubsequentlycarriedtotheatomizerbyargon Theflame-in-gas-shieldatomizerisdepictedinFig1detaileddescriptionisgiveninRef[3]Theobservationheight(OH)isdefinedasthedistancefromthetopofthecapillarytothecentreoftheopticalbeam
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 99
23Samplepreparation
CRMNIST1643fwasdilutedwith1MHCl80-foldforBideterminationbyHG-AFSRegardingBideterminationbyPVG-AFSthesampleneededtobeevaporatedtodrynessinordertogetridofnitricacidthatseriouslyinterferesatmMlevel[5]Avolume of 3ml of CRMwere pipetted into a 40 ml quartz vial evaporated(temperature asymp100 degC two replicates) and subsequently diluted ca 33-foldAsamplepreparationblank3mlofdeionizedwaterwaspreparedaswell
3Resultsanddiscussion
TheatomizationconditionsforHG-AFSwereoptimizedinourpreviouswork[3]TheseconditionswereusedasinitialtofindtheoptimumconditionsforPVG-AFSwith the flame-in-gas-shieldatomizerwith respect to sensitivityandsignal tonoiseratioFirstlythehydrogenfractionintherange10ndash16wasoptimizedatconstant total gas flow rate (sum of total argon and total hydrogen) of
ndash1500mlmin the lower the hydrogen fraction the better However at 10hydrogenfractiontheflamewasnotstableenoughandwentoftenouthenceitwasoptedfor12hydrogenfraction
100 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2(a)Hydrideand(b)photochemicalvapourgenerator
The oxygen flow rate through the capillary was optimized in the rangendash15ndash30mlmin Thehighestsignaltonoiseratiowasachievedwiththeflowrateof
ndash120mlmin Thetotalgasflowratewasoptimizedatconstant12hydrogenfractioninthe
ndash1range500ndash800mlmin Theoptimumobservationheightvarieswithtotalgasflow rate so it had to be optimized as well The optimum conditions aresummarizedandcomparedtothoseachievedwithHGinTable1 TheanalyticalfiguresofmeritofPVG-AFSwiththeflame-in-gas-shieldatomi-zerweredeterminedThecalibrationfunctionconstructedwith010025050
ndash1 2100 and 200 microg l Bi standardswas linear (R = 09998) The repeatabilityndash1expressedastherelativestandarddeviation(n=10)was6at1microgl andthe
ndash1relativeandabsolutelimitsofdetection(3σn=10)achievedwere12ngl and68pgrespectively(Table2)TheabsolutelimitofdetectionachievedwithHGwas 76 times lower which can be attributed to several aspects Firstly thegenerationefficiencyforPVGapproachwasaround53while100isexpectedforHG[3]Secondlyafullwidthathalfmaximumofthemeasuredpeakswasca2-foldgreaterwhichnecessitatedlongerintegrationtimeandwasthusreflectedinhighernoiseofthesignalsFinallythelimitofdetectionforPVGapproachwas
ndash1affectedby seriouscontamination (around10ng l )mostprobably from thesensitizersolutionthatcontainedBiasimpurity Tovalidate theproposedmethodologyBi contentwasdetermined inCRMNIST1643f (Table3) and the resultswere compared to thosemeasuredwithHG-AFS[3]Duetosevereinterferencesfrominorganicacidsespeciallynitricacid[5]thesampleneededtobeevaporatedtodrynessandthenfilledupwiththe
2+reactionmediumcontainingCo as thesensitizer(NIST1643f isstabilized in
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 101
Parameter HG-AFS(ref[3]) PVG-AFS
ndash1Artotalmlmin 440 528ndash1H totalmlmin 60 722 ndash1O mlmin 7 202
OH mm 6 9ndash1Arshieldlmin 1515 1515
Table 1Atomizationconditionsforflame-in-gas-shieldatomizer
Parameter HG-AFS(ref[3]) PVG-AFS
LODpg 09 68ndash1LOQngl 09 12
Repeatability lt1 6
Table 2AnalyticalfiguresofmeritofHG-AFSandPVG-AFS
ndash1032moll nitricacid)Theresultsobtainedbybothmethodologiesareingoodagreementwiththecertifiedvalue
4Conclusion
Photochemical vapour generation of Bi was successfully coupled with non-dispersiveatomicfluorescencespectrometryforthefirsttimeanditsapplicabilitywas verified by determination of Bi in certified reference material of waterComparedtohydridegenerationconditionsofatomizationdifferinanoptimalobservationheightandsupplyofoxygenwhichmaybeneededtoldquoburnoutrdquotheorganicvapoursthatarereleasedfromthereactionmediumtothegasphasehowever this remains tobe verifiedAlthough there are still some limitationsregardingthelimitsofdetectionrepeatabilityandinterferencesthisnewsampleintroductionapproachseemstobepromising
Acknowledgments
The support of the Czech Science Foundation (19-17604Y) Czech Academy of Sciences(Institutional supportRVO68081715)andCharlesUniversity (ProjectSVV260560andProjectGAUK1048120)isgratefullyacknowledged
References
[1] Musil SMatousek T Currier JM StybloM Dedina J Speciation analysis of arsenic byselectivehydridegeneration-cryotrapping-atomicfluorescencespectrometrywithflame-in-gas-shield atomizer achieving extremely low detection limits with inexpensiveinstrumentationAnalChem86(2014)10422ndash10428
[2] SturgeonREPhotochemicalvaporgenerationaradicalapproachtoanalyteintroductionforatomicspectrometryJAnalAtSpectrom32(2017)2319ndash2340
[3] S tadlerova B Kolrosova M Dedina J Musil S Atomic fluorescence spectrometry forultrasensitivedeterminationofbismuthbasedonhydridegenerationndashtheroleofexcitationsourceinterferencefilterandflameatomizersJAnalAtSpectrom35(2020)993ndash1002
[4] DedinaJAtomizationofvolatilecompoundsforatomicabsorptionandatomicfluorescencespectrometryOnthewaytowardstheidealatomizerSpectrochimActaPartB62(2007)846ndash872
102 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Certified HG-AFS PVG-AFSndash1valuemicrogl
ndash1 a ndash1 a valueobtainedmicrogl recovery valueobtainedmicrogl recovery
1262plusmn011 128plusmn01 102plusmn1 121plusmn09 97plusmn5
a Spikedrecovery=slopeofstandardadditions(noadditionandtwospikedconcentrationstoasample)slopeofexternalcalibration
Table 3ThedeterminedcontentofBiinCRMNIST1643fpresentedasmedianvalueplusmncombineduncertainty(n=3)andrecoveries
[5] Vyhnanovsky J Yildiz D Musil S Effect of metal sensitizers on photochemical vaporgeneration of bismuth for analytical atomic spectrometry In Proceedings of the 15thInternationalStudentsConferenceModernAnalyticalChemistryKNesmerak(ed)PragueCharlesUniversity2019p257ndash262
[6] VyhnanovskyJSturgeonREMusilSCadmiumassistedphotochemicalvaporgenerationoftungstenfordetectionbyinductivelycoupledplasmamassspectrometryAnalChem91(2019)13306ndash13312
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 103
1Introduction
Liquidcrystalsareorganicsubstancesthatformamesomorphicphaseinsolu-tion[1]TheyareliquidlikeliquidsbuthaveaninternalconfigurationassolidsTheir light transmittance changes in the electric fieldwhich is used in liquidcrystaldisplays(LCDs)CholestericliquidcrystalsaretemperaturesensitiveThecolorofreflectedlightchangeswithsmalltemperaturechangeThisisusedinmedicineasasensitivetemperatureindicatorfordisease-infectedtissues High performance liquid chromatography or supercritical fluid chromato-graphy[23]areusedtoseparateanddetermineliquidcrystalcompoundsAnelectrophoretic method could be complementary to these commonly usedmethods To our best knowledge no study dealing with liquid crystal puritycontrolbyelectrokineticchromatographywaspublisheduntilnow Electroneutralsubstancesmoveincapillaryzoneelectrophoresisatthesamespeedcorrespondingtothespeedoftheelectroosmoticflow(EOF)andtherefore
Separation of liquid crystals using non-aqueous capillary electrokinetic chromatography
KATER INACOKRTOVATOMA S KR IZ EK
DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublickaterinacokrtovagmailcom
AbstractLiquid crystals arewidelyused in electronicsmedicine andotherfields Analytical separations are important in the development ofnewliquidcrystalstocontrolthepurityofsynthesizedsubstancesThesampleanalysisisimportantfordetectionofimpuritiesformedduring synthesis Liquid crystal-forming substances cannot beseparated by capillary zone electrophoresis due to the absence ofreadily ionizable groups Therefore electrokinetic chromatographywasused in thisworkAnotherproblemcomplicating theanalysiswastheverylowsolubilityofanalytesinwaterSeparationsinthisworkwere thereforecarriedoutundernon-aqueousconditions inacetonitrilewithaceticacidtoadjustthepHandhexadecyltrimethyl-ammonium chloride as a detergent to mobilize the non-ionizedanalytesUndertheseconditionsitwaspossibletoseparateimpu-ritiesfromsynthesizedanalytesinsamples
Keywordselectrokinetic
chromatographyliquidcrystalsnon-aqueouscapillary
electrophoresis
104 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
itisnotpossibletoseparatethemDuetothisanelectrokineticchromatographymethodwasdevelopedInthismethodasurfactantisaddedtothebackgroundelectrolyteMoleculesaggregateandformsphericalformationscalledmicelles[4]ifthesubstanceisaddedinsufficientconcentrationiehigherthanthecriticalmicellar concentration (CMC) Separation is possible due to interactions ofnonpolarmoleculepartswiththenonpolarmicelleinsideAlthoughwateristhemostusedsolventinelectrophoreticmethodsforseparationofwater-insolublesubstances organic solvents are selected However such solvent must meetcertaincriteriatobesuitableforuseincapillaryelectrophoresisAllcomponentsmustbesolubleinthesolvent[5]ItshouldnotbeflammabletoxicorreactiveforpracticalityitshouldbeliquidatroomtemperatureandalsoitspriceistakenintoaccountThevalueof its relativepermittivitywhichdescribes the strengthofinteractionsbetweenionsshouldbearound30Lowdynamicviscosityisalsopreferred to allow faster migration of analytes No organic solvent meets allparameters of the ideal solvent In practice methanol acetonitrile and theirmixturesarethemostusedTheseparationparameterscanbeinfluencedbyusingan organic solvent of the background electrolyte This topic has already beenwidelyexplored[6ndash8] Itwasgenerallyassumedthat inanhydrousconditionsmicellesarenotcreateddespitesufficientsurfactantconcentrationHoweveritwasfoundoutthatdodecylsulfatecanformstablemicelleswhenthebackgroundelectrolyteisdissolvedinformamide[9]Fortheanalysisofactivesubstancesinmedicinal plants Chen et al developed a method in which sodium cholatedissolvedinmethanolisusedasasurfactant[10]Theaddedpseudostationaryphase does not always form micelles but can still affect mobilization andseparationofanalytesiftheanalytesinteractdifferentlywithfreemoleculesofsurfactantInthisstudywater-insolubleliquidcrystalswereseparated(Fig1)Thereforenonaqueouselectrokineticchromatographymethodwasdeveloped
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 105
Fig 1 Structures of liquid crystals 4-([1-oxo-1-(pentyloxy)propan-2-yl]oxycarbonyl)phenyl4-(octyloxy)-[11-biphenyl]-4-carboxylate (ZL 85) and 4-([1-(decyloxy)-1-oxopropan-2-yl]oxycarbonyl)phenyl 4-(dodecyloxy)-[11- biphenyl]-4-carboxylate (ZL 1210) OpticalisomerismsitesaremarkedwithanasteriskStructurescreatedinMarvinSketch[11]
2Experimental
21Reagentsandchemicals
Acetonitrilege999fromSigma-Aldrich(Germany)aceticacid99fromLach-Ner Neratovice (Czech Republic) and hexadecyltrimethylammonium chloride25(ww)inwaterfromSigma-Aldrich(USA)wereusedforpreparationofback-groundelectrolyteMesyloxidepa(MO)suppliedbyLach-nerNeratovice(CzechRepublic)wasusedasareferencesubstance
22Instrumentation
ForexperimentsG7100ACapillaryElectrophoresisInstrument(AgilentTechno-logiesGermany)wasusedwithUV-VISdetectoroperatingat235nmand254nmwavelengthMeasurementswereconductedinafused-silicacapillaryof50microminner diameter with the total length 500cm and effective length 415cm(PolymicroTechnologiesUSA)
23Method
Capillarywas flushed for3minuteswith1MHCland for2minuteswith thebackgroundelectrolyteBackgroundelectrolytewaspreparedbymixingaceticacid (10mM) and hexadecyltrimethylammonium chloride (40mM) in aceto-nitrile Sampleswere introducedhydrodynamicallybyapressureof5kPa for1secondSampleswerefirstdissolvedinacetonitrileandthendilutedtwotimeswith the background electrolyte A voltage of 20 kV was applied during theseparation
3Resultsanddiscussion
Liquidcrystalsampleswerepractically insoluble inwater theirsolubilitywasndash3testedataconcentrationlevelof1mgcm inmethanolandacetonitrileWhile
samples were not sufficiently soluble in methanol they were successfullydissolvedinacetonitrile Becauseallanalytesaresubstancesthatdonothaveeasilyionizablefunctionalgroups theelectrokineticchromatographymethodwaschosen forseparationAsuitablesurfactantwassoughtCommonlyusedsodiumdodecylsulfate(SDS)isinsoluble in acetonitrile Therefore hexadecyltrimethylammonium chloride(CTAC) which had sufficient solubility for further experiments was chosenAlthoughasuitablebufferwassoughttoensureastablepHduetoproblemswithprecipitationofbuffercomponentsinthenon-aqueousenvironmentaceticacidwasusedtoadjustandmaintainpHofbackgroundelectrolytesolutionAstheadditionofcationicsurfactantsuchasCTACleadstoEOFreversalthedependence
106 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ofEOFmobilityontheconcentrationofCTACinthebackgroundelectrolytewasmeasuredContrarytowhatisobservedinaqueousbackgroundelectrolytesEOFwasnotreversedItsmobilitydecreasedwithincreasingCTACconcentrationbutnomajorchangesoccurredabove40mMconcentrationThecapillarywallwasprobablyalreadysaturatedbyCTACandthefurtherincreaseinconcentrationhadno signifficant effect on the conditionof the capillarywall Therefore a CTAC
ndash3concentrationof40mmoldm was chosenas sufficient for furthermeasure-mentswithrespecttotheincreasingcurrentwithincreasingionicstrengthofthebackgroundelectrolyte The optimized method was used for separation of several liquid crystalsamplesofdifferentpurityInthesampleoftheZL85liquidcrystalwith99purityonezoneoftheanalytewasdetectedImpuritieswereseparatedfromthisanalytewhen the samplewith lower puritywas introduced The peak of theanalytewasidentifiedbasedonrelativemigrationtimerelatedtomesityloxideSeparationoftheanalytefromanimpurityinthesampleZL8576isshowninFig 2The relativemigration timeof the firstpeak is 0834 therefore itwasidentifiedastheZL85analyteThestandarddeviationoftherelativemigrationtimesinfivemeasurementswas0002min(01) Using the available high purity sample it was possible to measure thecalibration line forquantificationof theanalyte in lesspuresamplesLimitof
ndash3detection was determined as 0009mgcm and limit of quantification as ndash30031mgcm FromthecalibrationlineconcentrationofZL85inthesample
with lower purity was calculated The concentration was determined as
48(ww)standarddeviation5(ww)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 107
Fig 2ElectropherogramobtainedwhenasampleofZL85liquidcrystalwithlowerpuritywasndash3introducedSamplewas introduced in01mgcm concentrationandwithaddedmesityloxide
ndash3(10mgcm )Capillarywithinnerdiameterof50micromtotallengthof500cm415cmeffectivelength The background electrolyte was acetonitrile with 10 mM acetic acid and 40 mMhexadecyltrimethylammonium chloride A voltage of 20 kVwith positive polaritywas appliedDetectionat254nm
ForsampleZL121099onlytheanalyteandmesityloxideweredetectedInthe sample ZL 1210 59 several impurities were separated and detected(Fig3)Accordingtotherelativemigrationtimetheanalyteofinterestcorres-pondstothefirstpeakPeakresolutionissufficientTheresolutionoftheanalytepeakandthesecondpeakis284andtheresolutionoftheothertwopeaksis230
4Conclusions
InthisstudyanewmethodforanalysisofnewlysynthesizedliquidcrystalswasdevelopedSomeparametersofthemethodwereoptimizedndashoptimumconcen-tration of hexadecyltrimethylammonium chloride was searched The identifi-cation of analyteswas based on a comparison of relativemigration times InsamplesZL85andZL1210withlowerpuritytheimpuritieswereseparatedfromthepeaksofliquidcrystalsthecontentofanalytewasdeterminedintheZL85sampleaccordingtothecalibrationline
Acknowledgments
IwouldliketothanktheInstituteofPhysicsoftheCzechAcademySciencesforprovidingnewlysynthesized liquid crystals This work has been supported by Specific University Research(SVV260560)andbyCharlesUniversityResearchCentreprogramNoUNCESCI014
References
[1] GennesPGProstJThePhysicsofLiquidCrystals2ndedNewYorkOxfordUniversityPress1993
108 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ndash3Fig 3ElectropherogramofsampleZL121059ataconcentrationof05mgcm withmesitylndash3oxideataconcentrationof10mgcm capillarywith innerdiameterof50microm total lengthof
500 cmeffectivelength415cmThebackgroundelectrolytewasacetonitrilewith10mMaceticacidand40mMCTACAppliedvoltage20kVpositivepolarityDetectionat235nm
[2] Vankatova P KalıkovaK KubıckovaA Ultra-performance supercritical fluid chromato-graphy A powerful tool for the enantioseparation of thermotropic fluorinated liquidcrystalsAnalChimActa1038(2018)191ndash197
[3] Vankatova P Kubıckova A Cigl M Kalıkova K Ultra-performance chromatographicmethodsforenantioseparationofliquidcrystalsbasedonlacticacidJSupercritFluids146(2019)217ndash125
[4] Terabe S Otsuka K Ichikawa K Tsuchiya A Ando T Electrokinetic separations withmicellarsolutionsandopen-tubularcapillariesAnalChem56(1984)111ndash113
[5] RiekkolaMLRecentadvancesinnonaqueouscapillaryelectrophoresisElectrophoresis23(2002)3865ndash3883
[6] Wright PB Lister AS Dorsey JG Behavior and use of nonaqueous media withoutsupporting electrolyte in capillary electrophoresis and capillary electrochromatographyAnalChem69(1997)3251ndash3259
[7] PorrasSPKenndlerECapillaryzoneelectrophoresisinnon-aqueoussolutionspHofthebackgroundelectrolyteJChromatogrA1037(2004)455ndash465
[8] PorrasSPRiekkolaMLKenndlerETheprinciplesofmigrationanddispersionincapillaryzoneelectrophoresisinnonaqueoussolventsElectrophoresis24(2003)1485ndash1498
[9] GuoXWangK ChenGH Shi JWuX Di L LWangY Determination of strobilurinfungicideresiduesinfruitsandvegetablesbynonaqueousmicellarelectrokineticcapillarychromatography with indirect laser-induced fluorescence Electrophoresis 38 (2017)2004ndash2010
[10] Chen AJ Li C Gao WH Hu ZD Chen XG Application of non-aqueous micellarelectrokinetic chromatography to the analysis of active components in radix SalviaemiltiorrhizaeanditsmedicinalpreparationsJPharmBiomedAnal37(2005)811ndash816
[11] MarvinSketch [computer program] version 1990 ChemAxon httpschemaxoncom-productsmarvin
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 109
1Introduction
Sudandyesaresyntheticazo-basedaromaticcompoundsTheyaretraditionallyusedinvariousindustriessuchaschemicaltextileandwoodworkingasdyestocolourwaxesplasticsoilspolishesandsoforthTheyhavebeencategorizedasclass3carcinogensbytheInternationalAgencyforResearchonCancerandtheiruseisthereforeforbiddeninthefoodindustryTheyareknownfortheirbrightcolours and easy and cost-effectivemanufactureThey arenearly insoluble inwater but soluble in various organic solvents such asmethanol or trichloro-methane[1] SudanI1-phenylazo-2-naphthol(Fig1A)isadyeusedasanorangecolouringagentItssometimesalsosoldundernamesSolventOrangeRorCISolventYellow14ItisformedasasecondaryproductinthemanufactureoftheSunsetYellowdye
Electrochemistry of Sudan I and its derivates in aqueous media
ad b aANNAONDRA CKOVA MARIESTIBOROVA LUDE KHAVRAN cd adKAROLINASCHWARZOVA -PECKOVA MIROSLAVFOJTA
a CentralEuropeanInstituteofTechnologyMasarykUniversity Kamenice753562500BrnoCzechRepublicannaondrackovaceitecmuniczb DepartmentofBiochemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublicc UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistry FacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublicd InstituteofBiophysicsCzechAcademyofSciences Kralovopolska13561265BrnoCzechRepublic
AbstractSudanIisanaromaticazo-compoundthathasbeenproventobeacar-cinogenDuringitsmetabolizationbycytochromeP450inliverafewmain derivates can be identified Thiswork sets out to assess themechanismofelectrochemicalreductionandoxidationofSudanIitshydroxylationderivativesfeaturingmetabolitesintheSudanIdetoxi-fication pathway and to introduce their selective voltammetricanalysis on boron-doped diamond electrode We show successfuldifferentiationamongthesecompoundsthankstothedifferencesintheelectrochemicaloxidationoftheirphenolicgroups
Keywordsborondopeddiamond
electrodecytochromeP450electrochemicalanalysisSudanI
110 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
InmammalianorganismsSudanIismetabolizedbythemicrosomaldetoxi-fying systemwitha central roleof cytochromeP450hydroxylationactivity inliver[2]DuringtheoxidativeprocessofmetabolizingSudanIseveralmetaboliteswereidentifiedbyprevioustestsThesearegt1-(phenylazo)-naphtalene-26-diol(further abbreviated SI-6OH) 1-(4-hydroxyphenylazo)-2-hydroxynaphtol(furtherabbreviatedSI-4OH)and1-(4-hydroxyphenylazo)-naphtalene-26-diol(furtherabbreviatedSI-46-diOH)thestructuresarepresentedinFig1 ThemainmethodcurrentlyusedtoidentifySudanIamongotherdyeswithsimilar structure is high-performance liquid chromatography (HPLC) It isrecommendedasthestandardmethodtoidentifythelevelofSudanIinfood[3] ComparedtoHPLCelectrochemicalmethodsareprovingtobefastercheaperandcomparablypreciseUnfortunatelyacomprehensiveelectrochemicalstudyofSudanIandparticularlyofitshydroxylatedmetaboliteshasnotbeencompletedyetThedyecanbedetectedthroughelectrochemistryeitherbytheoxidationofitsphenolicgrouporviareductionoftheazogrouppresentinitsmoleculeInbothcasesotherelectrochemicallyactivemoietiesareformedThederivatesofSudanIcanbedetectedandrecognizedfromSudanIthroughanalogousprocesses[4]InthisstudywefocusedoncomparisonofelectrochemicalbehaviourofSudanIanditshydroxylatedmetabolitesonborondopeddiamondelectrodetoaddressthepossibilitiesoftheirrecognitionsinmixturesbasedondifferencesinanodicandcathodicsignals
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 111
Fig 1 Chemical structure of (A) Sudan I (B) 1-(phenylazo)-naphtalene-26-diol (SI-6-OH)(C) 1-(4-hydroxyphenylazo)-2-hydroxynaphtol (SI-4-OH) and (D) 1-(4-hydroxyphenylazo)-naphtalene-26-diol(SI-46-diOH)
(A) (B)
(C) (D)
2Experimental
21Reagentsandchemicals
SudanI(Merckanalyticalstandardgrade)wasdissolvedinethanol(Merck)andkept at room temperatureChemicals forBritton-Robinsonbufferpreparation(acetic acid boric acid orthophosphoric acid sodium hydroxide) were fromMerckwithpurityge99pHofthebufferwasadjustedbymixingoftheacidsandsodium hydroxide solution at different ratios The Sudan I metabolites weresynthetized at the Department of Biochemistry Faculty of Science CharlesUniversityandkeptinmethanolattemperature3degC
22Instrumentation
Cyclicvoltammetric (CV)measurementswerecarriedout inBritton-RobinsonbufferofpH=70atroomtemperatureSudanIanditsderivativeswereaddedto
minus1thesolutionofBritton-Robinsonbuffertofinalconcentrationof5micromolL andstirred Before the measurement oxygen was removed from the solution bypurgingwithargonfor3minutesAutolabanalyzerPGSTAT20(EcochemieTheNetherlands)inconnectionwithVA-Stand663(MetrohmSwitzerland)GPES49(MetrohmSwitzerland)andathree-electrodesetup(withborondopeddiamond
2(WindsorScientificUKdiskdiameter3mmA=707mm )asworkingelectrodeminus1AgAgCl3molL KCl as reference electrode and platinum wire as auxiliary
electrode) Five cycles were performed for each measurement at scan rateminus1of1Vs
3Resultsanddiscussion
ForeachcompoundtwoseparateCVmeasurementswereperformedeachwithfive cycles performed in rapid succession For bothmeasurements the initialpotentialwassetat0VTheanodicscancontinuedto+1Vturnedtowardsndash1Vandfinishedat0VIntheothersetupcathodicscanwasperformedfirstfromthestartingpointtondash1Vturnedtowards+1Vandreturnedto0VThiswaywewereabletoobservethebehavioursofSudanIandcompareittothatofitsderivativeswhilefirstbeingreducedandthenoxidisedorviceversa In the anodic scan of Sudan I and its derivatives (Fig 2) differences in thepositionsoftheoxidationpeaksineachcompoundcanbeobservedWhileSudanIwithonlyonephenolgroupisoxidizedat+067VthederivativeswithtwophenolgroupsieSI-4OHandSI-6OHgiveoxidationpeaksatremarkablylesspositivepotentialofca+05VSI-46OHwiththepresenceofoverall3hydroxygroupsyieldstwooxidationpeaksInthereversecathodicscan(vertexpotential+1V)probablythankstothepresencereductionpeaksappearwhichcanbefurtherusedtodifferentiatebetweenSudanIandthederivativesSI-4OHandSI-6OH
112 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 113
Fig 3VoltammetricscanofSudanIanditsderivatesSI-4-OHSI-6-OHandSI-46-diOH1stscanincathodicdirectionfrom0Vvertexpotentialsndash1Vand+1VThemeasurementswereperformedinBritton-Robinsonbuffer(pH=7)withtheconcentrationofeachcompoundat5microMandatscan
ndash1rate1Vs
Fig 2 CyclicvoltammogramofSudanIanditsderivatesSI-4-OHSI-6-OHandSI-46-diOH1stscaninanodicdirectionfrom0Vvertexpotentials+1Vandndash1VThemeasurementswereperformedinBritton-Robinsonbuffer(pH=7)withtheconcentrationofeachcompoundat5microMandatscan
ndash1rate1Vs
possesstwoconjugatedhydroxylgroupswhichcanberegardedashydroquinonestructures and thus undergoing quasireversible redox process due to oxida-tionreductionofthehydroquinonetoquinonemoietyThisiswellvisibleattheCVsastheanodicsignalisfollowedbycathodiconeatthepotentialof+023VforSI-4OHand+03VforSI-6OHTheoxidationofSudanIproceedsbymechanism
minus +typicalforphenoliccompoundsatmorepositivepotentialsleading by1e 1H exchange to naphthoxy-type ndashO radical [5] This species undergoes furtherreactionsleadingtoformationofdimersandpolymersThecathodicpeakinthereversescanatndash02Varisesfromreductionofthesereactionproductsanditsoriginneeds tobe further investigatedSI-46OHwith thepresenceofoverallthree hydroxyl groups yields two oxidation peaks The first one is a result ofoxidationoftwoofthembeinginconjugationandthusbeingoxidizedtoquinonemoietyThesecondsignalatthesamepotentialastheoxidationsignalof2OHonnaphthaleneringofSudan I is consequenceofoxidationof the thirdhydroxylgroupofphenolictypeAsinglewidepeakat0Vinthereversescanispresumablyan overlap of signals arising from reduction of the quinonic moiety and by-products formed during oxidation processes Thanks to differences of theseprocessesspecificforindividualcompoundsitispossibletodifferentiateamongallfourofthemviapropersetting-upoftheinitialandvertexpotentialvalues The cathodic scan of Sudan I and its derivatives (Fig 3) shows a dominantreductionpeakaroundndash08Vwhichisduetoreductionoftheazogroupintheirstructuresaccompaniedbycleavageoftheirmoleculestoseparatethebenzeneandnaphthalene rings [4] The peaks in the subsequent anodic scans (vertexpotentialndash1V)arethereforetheresultoftheelectrochemicalreactionofmoietiesthatareproductsofthedivisionofthearomaticcirclesTheseproductsincludeaniline4-aminophenol1-amino-2-naphtholand1-amino-25-naphthalenediolwith irreversibly oxidizable amino moieties or (quasi)reversibly oxidizableamino-hydroxylsystemonthebenzeneornaphthaleneringandtispossibletodifferentiatebetweenthemTheobtainedoxidationpeaks(+02VforSI-4-OH+073VforSudanIandSI-6-OHand+025Vand+075VforSI-46-diOH)makeitpossibletodifferentiatebetweenallcompoundswiththeexceptionofSudanIandSI-6-OHForthereliablerecognitionofthesetwocompoundsanothermeasure-mentwithdifferentparametersisneeded
4Conclusions
ThestructuresofSudanIanditshydroxyderivatesthatarethemainproductsofthe metabolization of Sudan I by cytochrome P450 are similar and theirrecognitionwhenpresentinmixtureinsolutionisdemandingHereinwepresenta simple approach based on comparison of signals obtained in cathodic andanodic scan in CV measurements without the need of time-demandingchromatographicseparationstepFurtherworkwillbedevotedtoidentificationof observed redoxprocesses and applicationof themethod formonitoringofmetabolictransformationsofSudanIinvitro
114 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Acknowledgments
ThisresearchwassupportedbytheCzechScienceFoundation(projectNo18-01710S)
References
[1] ChailapakulOWonsawatW SiangprohW GrudpanK ZhaoYF Zhu ZW Analysis ofSudanISudanIISudanIIIandSudanIVinfoodbyHPLCwithelectrochemicaldetectionComparison of glassy carbon electrodewith carbon nanotube-ionic liquid gelmodifiedelectrodeFoodChem109(2008)876ndash882
[2] StiborovaMMartinekVRydlovaHHodekPFreiESudanIisapotentialcarcinogenforhumans Evidence for itsmetabolic activation and detoxication by human recombinantcytochromeP4501A1andlivermicrosomesCancerRes62(2002)5678ndash5684
[3] GomezMArancibiaVAliagaMNunezCRojas-RomoCDeterminationofSudan I indrinks containing Sunset yellow by adsorptive stripping voltammetry Food Chem 212(2016)807ndash813
[4] PrabakaranEPandianKAmperometricdetectionofSudanIinredchilipowdersamplesusingAgnanoparticlesdecoratedgrapheneoxidemodifiedglassycarbonelectrodeFoodChem166(2015)198ndash205
[5] Enache T A Oliveira-Brett A M Phenol and para-substituted phenols electrochemicaloxidationpathwaysJElectroanalChem655(2011)9ndash16
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 115
1Introduction
Aminoglycosidesarealargegroupofnaturalandsemi-syntheticantibioticswithawidespectrumofantimicrobialactivityagainstmostgram-positiveandgram-negativemicroorganismsCurrentlymultiplerepresentativesofaminoglycosidefamily ndash gentamicin (GM) neomycin B (NM) paromomycin (PM) kanamycin(KM)apramycin(AP) andstreptomycin(STM)Fig1ndashareapproved to treatinfectiousdiseasesinanimalsMaximumresiduelimitsfortheseaminoglycosidesinproductsandtissuesfromedibleanimalsareestablished[1]henceaneffectiveandrobustassayisnecessaryforcontrolofaminoglycosidescontamination Inthisstudyenzyme-linkedimmunosorbentassayforthedetectionofamino-glycosides in foodproductsandenvironmentalobjectswasdevelopedGroup-specificantibodieswereproducedowingtoimmunogenbasedonribostamycin(RS)whichexposedthecommonfragmentofmostaminoglycosides2-deoxy-streptamin (2-DOS) Fig 1 The developed assay was made suitable for thedetectionofresidualaminoglycosidesinhoney[2]
Group detection of aminoglycosides using ELISA for control of food contamination
ab a aKONSTANTINBURKIN INNAGALVIDIS MAXIMBURKIN
a DepartmentofImmunologyIMechnikovResearchInstituteofVaccinesandSera MalyjKazionnyjper5a105064MoscowRussianFederationburkin-kostyandexrub DepartmentofChemicalEnzymologyFacultyofChemistryLomonosovMoscowStateUniversityLeninskieGory1119991MoscowRussianFederation
AbstractThegrowingthreatofglobalantibioticresistanceisforcingtoreducenon-targetconsumptionofantibioticsandtomonitorcontaminationoffoodandenvironmentalobjectsInthisworkELISAwasdevelopedforgroupdetectionofaminoglycosidesToobtaingroup-specificanti-bodies a new immunogen based on ribostamycin was used Thedevelopedindirectcompetitiveformatofassayallowedtherecogni-tionof9aminoglycosidesnamelyneomycinribostamycinneaminparomomycin gentamicin sisomicin kanamycin tobramycin and
ndash1apramycinwithadetectionlimitrangedbetween002ndash020ngmL TheeffectivenessoftheproposedassaywasevaluatedinhoneyasafoodstuffmodelToneutralizea stronghoneymatrixeffect and toavoidalaborioussamplepre-treatmentanewmatriximitatorwassuggested 5 sucrose solution imitated the influence of 50-folddilutedhoneyTheproposedassayallowedustorevealanyofthe9
ndash1mentionedaminoglycosidesinhoneyata10microgkg level
KeywordsaminoglycosidesELISAhoney
116 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
2Experimental
21Reagentsandchemicals
NeomycinBribostamycinneamin(NA)paromomycinkanamycintobramycin(TM)amikacin(AM)gentamicinnetilmicin(NTM)sisomicin(SSM)geneticin(GC) apramycin and streptomycin were purchased from Chimmed (MoscowRussia) Bovine serumalbumin (BSA) complete Freund adjuvant 16-hexane-diamine1-ethyl-3-(3-dimethylaminopropyl)carbodiimide(EDC)sodiumperio-dateandsodiumborohydrideweretheproductsofSigma-Aldrich(USA)Gelatin(Gel)wasfromBio-Rad(USA)sucrosefromServa(Germany)two-componenttetramethylbenzidine (TMB) substrate solutionwas fromBioservice (Russia)andgoatanti-rabbitIgGantibodiesconjugatedtohorseradishperoxidase(anti-rIgG-HRP)werefromIMTEK(Russia)Honeysampleswerepurchasedfromlocaloutlets
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 117
Fig 1 Structuralformulasofstudiedaminoglycosides
22Preparationofconjugatedantigens
TwotypesofconjugateswerepreparedbasedonRSandBSAusingzero-lengthand C6 spacer arm between hapten and protein carrier RSwas treatedwithsodiumperiodatetooxidizehydroxylsofribosefragmenttoreactivealdehydegroupsandthencoupledtoBSAaminesthroughreductiveaminationToremoveuncoupledRSanexhaustingdialysiswascarriedoutusingdialysismembranetubes(MWCO14kDa)UsingthesameprocedureGel-RSconjugatewassynthe-sized ForpreparationofBSA-C6-RSwefirstlymodifiedBSAwith16-hexanediamineThemixtureofBSAandEDCinwaterwerestirredfor30minThen16-hexane-diaminewasaddedandstirredfor2hThemodifiedproteinwasdialyzedfromtheexcessive reagents and resultant BSA-C6-NH2was coupled to RS in reductiveaminationprocessasdescribedabove
23Immunizationandantibodypreparation
BSA-RSandBSA-C6-RSwereusedasimmunogensChinchillarabbits(20ndash25kg)weresubcutaneouslyinjectedat10ndash15pointsonthebackwith01mgofimmuno-gensemulsifiedinthecompleteFreundadjuvantThesamedosesofimmunogensinsalinewereadministeredmonthly forbooster immunizationsAweekaftereachinjectionabloodsamplefromearveinswastakenforthecontrolofimmuneresponseTheantiserainglycerol(11vv)werestoredatndash15degCuntiltestinginELISA
24TheELISAprocedure
AcompetitiveassaywasconductedaccordingtoclassicalprocedureGel-RSwascoatedovernightonpolystyrene96-wellCostarplatesNon-adsorbedconjugatewashedoutusingPBSwith005oftween20(PBS-T)Thenextcompetitivestep
ndash1includedtheadditionof01mLstandardaminoglycosidesolutions(1pgmL to ndash1 ndash11microgmL (B)and0microgmL (B ))inPBS-Tor01mLoftestedsampleand01mLof0
antibodiesinworkingdilution(1h25 degC)Afterwashingtheantibodiesboundtoimmobilized Gel-RS were detected using anti-rIgG-HRP (1h 37degC) Coloredproduct formedasaresultofenzymaticreactionwithTMBsubstratemixture(05h25degC)wasreadat450nmusingaStatFax2100platereader(AwarenessTechnologiesUSA) Relativeantibodybinding(BB )vstheanalyteconcentrationswasplottedas0
standardcurvesfittedtoafour-parameterlogisticfunctionThecross-reactivity(CR) for every aminoglycoside representative was calculated as ratio of half-inhibition concentrations IC NMIC aminoglycoside The dynamic range of50 50
assaywasacceptedasIC ndashIC andthelimitofdetection(LOD)wascalculatedas20 80
B ndash3timesSD0
118 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
3Resultsanddiscussion
31Immunogensynthesisandantibodypreparation
Inthemajorityofpublicationsdevotedtoimmunoassayofaminoglycosidestheimmunogenscoatingantigensenzymeconjugatesortracerswerepreparedbycarbodiimide or glutaraldehyde methods involving aminoglycosidesrsquo aminogroups [3ndash7] Due to several amino groups in aminoglycoside molecules theformationofconjugateswithavariableorientationofthehaptenoccurs InpresentstudyRSwaschosenasanimmunizinghaptenduetothefollowingadvantageous features Being a trisaccharide RS has the size of a moleculecomparable to themostof aminoglycosides Ithas three identical ringsA-B-CsimilartothoseinNMUsingaperiodateoxidationwecouldinvolvearibosesiteofRSincouplingtoproteinthatprovidedastrictorientationofhaptenonthecarrier with a favorable presentation of the 2-DOS fragment The resultantimmunogensBSA-RSandBSA-C6-RSwerecomparedtorevealwhichdesignisbetterforpresentationofacommonfragmentofaminoglycosidemoleculeandgenerationofgroup-specificantibody Antibodies to the BSA-RS demonstrated moderate sensitivity (NM
ndash1IC =10ngmL )andhighselectivitytowardsNMwithrelativelylowcross-reac-50
tivity(lt5)forGMKMandAPTheapplicationofthespacerintheimmunogenBSA-C6-RScontributedtoaprominentpresentationofthe2-DOSdeterminantandtheinductionofantibodieswithrecognitionofbroadspectrumofdifferentaminoglycosides In addition anti-BSA-C6-RS exhibited significantly better
ndash1sensitivity(NMIC =02ngmL )Thusallsubsequentstudieswereconducted50
usinganti-BSA-C6-RS
32Examinationofassayspecificityandselectionofimmunoreagents
TheindirectcompetitiveformatofassaywasdevelopedForevaluationofassayspecificity a panel of following aminoglycosideswas studied and their cross-reactivitywasdeterminedNA(625)RS(250)NM(100)KM(475)PM(173)GM(90)TM(78)AP(17)SSM(12)AM(lt01)GC(lt01)STM(lt01)andNTM(lt01)ThemostoftheseanalytesareusedinmedicalandveterinaryareashoweveronlyNMPMGMKMAPandSTMareappliedinanimalhusbandry[1]
33Determinationofaminoglycosidesinhoneyandselectionofthematriximitator
Honey is a complex product consisting of carbohydrates (75ndash80) vitaminsproteinsenzymesorganicacids traceelements inclusionsandothercompo-nentsThesecomponentsmightinterfereimmunochemicalreactionThereforetheisolationofaminoglycosidesfromhoneyisalaboriousandtime-consuming
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 119
120 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
procedureToavoidthisstephoneymatriximitatorswereappliedtomimictheinfluenceofhoneymatrixonantibodybindingSucrosewaschosenasthehoneyimitatorsinceitexposedastronghoney-matrix-likeeffectonantibodybindingTheadequacywasfoundbetweensolutionsofhoneyandsolutionsofsucroseexpressinganequalmatrixeffectTwopairswithequivalentmatrixeffectwere120honey=20sucroseand150honey=5sucroseThelatterconditionswere chosen asmore preferable due to inconvenience of operatingwith highviscous20sucrosesolution Thedeterminationofaminoglycosidesinhoneycouldbecarriedoutquantita-tivelyiftheanalytetobedetectedisknownForquantificationofaminoglycosideinhoneyasamplewasdiluted50timesinPBS-Tandaminoglycosidestandardcurvewas generated in 5 sucrose-PBST (Fig 2) If analyte is unknown thedevelopedgroup-specificELISAcanbeusedasascreeningtestInthiscasetheanalyzed sample can be considered as contaminated if it caused a relativeantibodybindingbelowthecut-off level (Fig3)Thus thedeveloped testwas
ndash1 ndash1 ndash1Analyte IC ngmL Dynamicrange LODngmL LODinhoneymicrogkg50 ndash1 IC ndashIC ngmL20 80
NM 02 003ndash21 002 10PM 07 008ndash71 005 25GM 15 015ndash133 011 55KM 035 005ndash39 004 20AP 68 05ndash968 023 115
Fig 2StandardcurvesandanalyticalparametersoftheELISA-systemforgroupdeterminationofaminoglycosidesinhoneyInteractionofanti-BSA-RSwithcoatingantigenGel-RSin5sucrosesolutionasthehoneyimitatorThedetectionlimitin5sucrosesolutionwasdeterminedaccordingtoLOD=B ndash3timesSD0
capabletorevealthecontaminationofhoneywith9aminoglycosides5amino-ndash1glycosidesapprovedforveterinary(NMPMGMKMandAP)ata10microgkg level
andalsoNARSSSMandTM
4Conclusions
A novel indirect competitive ELISA for the detection of aminoglycosides wasdevelopedRSwasusedasanewimmunizinghaptentoproducegroup-specificantibodiesagainst2-DOSacommonmoietyofalargenumberofaminoglycosideantibiotics A wide spectrum of aminoglycoside representatives could bedetectedincludingNMRSNAPMGMSSMKMTMandAPThedevelopedassay
ndash1was capable todetect theseanalyteswithaLOD up to002ndash020 ngmL ForanalysisofhoneyamatriximitatorwasdevelopedtoavoidhoneyinterferencesonimmunoassayTheanalysisofthehoneysampleallowedustorevealanyofthe
ndash1mentionedaminoglycosidesinhoneyata10microgkg level
References
[1] CouncilRegulation(EU)N372010OffJEurCommunitiesInfNotL15(2009)1ndash72[2] GalvidisIABurkinKMEreminSABurkinMA Group-specificdetectionof2-deoxy-
streptamineaminoglycosidesinhoneybasedonantibodiesagainstribostamycinAnalMeth11(2019)4620ndash4628
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 121
Fig 3Detectionofaminoglycosidesspikedinhoneysamplesata40ppblevelusinggroup-specificELISAEachsymbolcorrespondstotheaveragerelativebindingandtheerrorisSDobtainedforanindividualhoneysampleanalyzedintriplicateEmptycharactersrepresentindividualblankhoneysamples(limebuckwheatandflower)andfilledsymbolsrepresentthesamesamplesfortifiedwith
ndash1aminoglycosidesata40mgkg (level establishedonlyforSTMinseveralcountries)Thecut-offlevelcorrespondstothelimitofassaydetectionobtainedbythematriximitator(5sucrose-PBS-T)
[3] ThompsonSGBurdJFSubstrate-labeledfluorescentimmunoassayforamikacininhumanserumAntimicrobAgentsChemother18(1980)264ndash268
[4] LiCZhangYEreminSAYakupOYaoGZhangXDetectionofkanamycinandgentamicinresiduesinanimal-derivedfoodusingIgYantibodybasedic-ELISAandFPIAFoodChem227(2017)48ndash54
[5] GalvidisIABurkinMAMonoclonalantibody-basedenzyme-linkedimmunosorbentassayfortheaminoglycosideantibiotickanamycininfoodstuffsRussJBiorganChem36(2010)722ndash729
[6] HaasnootWStoutenPCazemierGLommenANouwsJFKeukensHJImmunochemicaldetectionofaminoglycosidesinmilkandkidneyAnalyst124(1999)301ndash305
[7] Peng JWangYLiuLKuangHLiAXuCMultiplex lateral flowimmunoassayfor fiveantibioticsdetectionbasedongoldnanoparticleaggregationsRSCAdv6(2016)7798ndash7805
122 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
PhotochemicalvaporgenerationisanalternativesampleintroductiontechniqueforanalyticalatomicspectrometryThistechniqueisbasedaroundasourceofUV-radiationthatirradiatesalowmolecularweightorganicacidmedium(mostcommonlyformicacidaceticacidortheircombinations)withananalyteHighlyreducingradicalsandaquatedelectronsareproducedandconverttheanalyteintoavolatilespecieswhichisthentransportedintoadetector[1]SofartheuseofPVGhasbeendescribedforhydride-formingelements(AsBiTeSbPbSeSnandTl)andmercury[12]transitionmetals(FeCoNiCuMoWCdAgAuIrPdPtRhandOs)[13ndash6]andevennon-metals(BrIClFandS)[16ndash9] AfirstsuccessfulphotochemicalvaporgenerationofcobaltwasdescribedbyGuoetalin2004[6]whichwasfollowedbymoresystematicstudiesbyGrinbergetalin2008[10]andDengetalin2010[11]LaterworksbydeQuadrosetal[12]anddeJesusetal[13]focusedontheanalysisofrealsamplesInthelatterwork
Photochemical vapor generation of cobalt for detection by inductively coupled plasma mass spectrometry
ab aJAROMIRVYHNANOVSKY STANISLAVMUSIL
a DepartmentofTraceElementAnalysisInstituteofAnalyticalChemistryoftheCzechAcademyofSciencesVeveřiacute9760200BrnoCzechRepublicjaromirvyhnanovskygmailcom
b DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublic
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 123
AbstractThisworkfocusedonthephotochemicalvaporgenerationofcobaltVolatilespeciesweregeneratedinaflow-injectionsystememployinga high-efficiency flow-through UV photoreactor and a formic acidbased medium and were introduced by an argon carrier into aninductivelycoupledplasmamassspectrometerfordetectionOptimalgeneration conditions were found as 10 (vv) formic acid and
ndash1 ndash14molL ammonium formate with a 4mLmin flow rate whichcorresponds to irradiation time of around 13 s The influence ofvariousmetalsensitizersofphotochemicalreactionwasinvestigated
2+andonlyCu ionsexhibitedapositiveeffectongenerationefficiencyndash ndash 2ndashInterferencesfromcommoninorganicanions(NO Cl SO ) were3 4
also examined Lastly the limit of detection and repeatability (atndash1 ndash1250ngL )weredeterminedtobe13ngL and41respectively
Keywordscobaltinductivelycoupled
plasmamassspectrometry
photochemicalvaporgeneration
the authors also presented a systematic study on generation conditions andachievedagenerationefficiencyofaround40 Themainaimofthisworkwastooptimizetheconditionsofgenerationwithinductivelycoupledplasmamassspectrometry(ICP-MS)detectionexaminetheeffectofvariousmetal sensitizers toachieve thehighestgenerationefficiencypossibleandreachthelowestlimitofdetectionpossible
2Experimental
21Reagentsandchemicals
minus1Deionizedwater (DIW lt 02μScm UltrapurWatrex USA)was used for thepreparationofallsolutionsFormicacid(98paLach-NerCzechRepublic)andammonium hydroxide (ge25 pa Sigma-Aldrich USA) were used for the
minus1preparationof the reactionmediumA1000mgL Co stock solution (Sigma-AldrichUSA)wasusedforthepreparationofallsamplesolutionsThefollowingcompounds were used as potential metal sensitizers cadmium(II) acetatedihydrate(paLach-NerCzechRepublic)zinc(II)acetatedihydrate(paSigma-AldrichUSA)copper(II)acetatemonohydrate(paMerckGermany)nickel(II)acetatetetrahydrate(paSigma-AldrichUSA)sodiumtungstatedihydrate(paCarlRothGermany)and iron(II) sulphateheptahydrate (pa LachemaCzechRepublic)Nitricacid(65semiconductorgradeSigma-AldrichUSA)hydro-chloricacid(37paMerckGermany)andsulfuricacid(98paLach-NerCzechRepublic)wereusedforaninterferencestudy
22Instrumentation
AschematicdiagramofthePVGsystemcoupledtoICP-MSisshowninFig1andamore detailed description can be found in reference [5] Briefly a singlequadrupole ICP-MS Agilent 7700x (Agilent Technologies USA)was used as a
minus1detector Deionized water was mixed with a 10μgL Rh internal standardsolutionin2HNO andwassubsequentlynebulizedbyaMicroMistnebulizer3
59 103during PVG Isotopes of Co and RhweremonitoredMeasurementswereperformed in time resolved analysis mode and in He collision mode
minus1(41mLmin )AlltubingusedwasmadefromPTFEwiththeexceptionoftygontubing in the peristaltic pump (Reglo ICC Ismatec Switzerland) The high-efficiency flow-through photoreactor was a 19 W low-pressure mercurydischarge lamp (Beijing Titan Instruments Co Beijing China) with a quartzcentralchannel(asymp720μL internalvolume)Samplesolutionswere introducedintoastreamofreactionmediumusinganinjectionvalve(V-451IDEXHealthandScienceUSAsampleloopvolume05ml)Effluentfromthephotoreactorwasmixedwithaflowofargonandcarriedtothechilledgas-liquidseparator(internalvolume15mL)wherethevolatilespecieswereseparatedfromtheliquidwaste
124 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
andcarried to the inletofaScott-typespraychamber (originally the inlet formakeupargon)oftheICP-MS
3Resultsanddiscussion
The starting conditionswere adopted fromour earlierwork [14]whichusedatomicabsorptionspectrometerasadetectorandminiaturediffusionflameasanatomizerThefirstparameteroptimizedwasthecompositionofreactionmedium(Fig2)Theadditionofammoniumformate(createdin-situbytheadditionofacalculatedamountofammoniumhydroxidetoformicacid)wasfoundcrucialto
ndash1effectivelygeneratevolatilespeciesofcobalt10(vv)formicacidand4molL ammonium formatewas chosen as the optimumandwasused for all further
experimentsAlthoughhigher concentrations of both components led to even
higher signalstheseconditionswerenotusedfurtherbecauseofthelaboriousprocessofpreparation(mixingofconcentratedacidwithconcentratedbase)andtolimittheconsumptionofchemicals Theinfluenceofirradiationtimewasalsoexaminedandthehighestpeakarea
-ndash1wasobtained for4mLmin corresponding to an irradiation timeof approximately13s ToenhancethegenerationefficiencyadditionofvariousmetalstothereactionmediumwastestedtoldquosensitizerdquothephotochemicalreactionThemetalswerechosenwithrespecttotheirsignificantenhancementeffectdescribedrecentlyforphotochemicalvaporgenerationofotheranalytes[158]Theonlymetalionthat
2+ledtoanenhancementofthesignalwasCu (Fig3a)buteveninthiscasetheeffect was rather negligible reaching only 12-fold enhancement in the range
ndash1 2+ 2+001to01mgL Cu FurtheradditionofmoreCu ledtoadecreaseinthesignal2+TheadditionofZn didnotexhibitanypositiveornegativeeffectacrossthetested ndash1range (01 to 1500mgL not shown in figure) and the addition of higher
ndash1 2+ 2+ 6+concentrations (tens to hundredths ofmg L ) of Cd Fe andW (Fig3b)causedsevereinterferences
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 125
ndash ndash 2ndash Interferencescausedbycommoninorganicanions(NO Cl SO addedas3 4ndashtheirrespectiveacids)werealso investigatedOutof theseNO was foundto3
ndash1causethemostsevereinterferencesevenatconcentrationsofsinglemmolL Thendash 2ndashmethodologywasmorerobusttowardstheinterferencesfromCl andSO but4
theystillcausedsignificantdropinsensitivityathigherconcentrationsConsi-deringthewideuseoftheseacidsinanalyticalchemistryforsamplepreparationthisposesabigchallengeintheapplicationofthismethodtorealsamples
ndash1 Usingoptimalconditions(10(vv)formicacid4molL ammoniumformateandirradiationtimeof13s)acalibrationcurvewasmeasuredandevaluatedThelimitofdetectionwasdeterminedas3timesthestandarddeviationof10blank
ndash1measurements and was calculated as 13 ng L The repeatability of 10ndash1consecutivemeasurementsof250ngL was41
126 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 3Effectofvariousmetalionsonthepeakarea(a)metalionswithapositiveeffect(b)metalionsndash1withoutapositiveeffectExperimentalconditions2microgL Coreactionmedium10(vv)formic
ndash1 ndash1acidand4molL ammoniumformateflowrate4mLmin
ndash1Fig 2Effectofthecompositionofreactionmediumonpeakareaexperimentalconditions2microgL ndash1Coreactionmediumflowrate4mLmin (blackdotscorrespondtomeasuredpoints)
4Conclusions
Theconditionsofthephotochemicalvaporgenerationofcobaltwereoptimizedand are in good agreement with previous works [11 13] Copper ions wereidentifiedasapotentialsensitizerincreasingthesignalbyabout12-foldbuttheirpotentialuseisseverelylimitedbythenarrowrangeofconcentrationsinwhichthepositiveeffectisexhibitedSevereinterferencesfrominorganicanionswereobservedwhichisinlinewithotherworksdealingwithphotochemicalgeneration[1358]Furtherexperimentswillfollownamely(i)furtherinvestigationsinnewpotentialsensitizerstoenhancegenerationefficiencyandthusdecreasethe
ndash1limit of detection to sub ng L levels (ii) determination of the generationefficiency(fromcomparisonwithnebulizationandorusingaradioactiveisotope58Co)(iii)verificationoftheaccuracyandpracticalfeasibilityofthismethodologybyanalysisofcertifiedreferencematerials
Acknowledgments
ThesupportofTheCzechScienceFoundation(ProjectNo19-17604Y)CzechAcademyofSciences(Institutional supportRVO68081715)andCharlesUniversity (project SVV260560andprojectGAUK60120)isgratefullyacknowledged
References
[1] SturgeonREPhotochemicalvaporgenerationaradicalapproachtoanalyteintroductionforatomicspectrometryJAnalAtomSpectrom32(2017)2319ndash2340
[2] XuTHuJChenHJTransitionmetalionCo(II)-assistedphotochemicalvaporgenerationofthalliumforitssensitivedeterminationbyinductivelycoupledplasmamassspectrometryMicrochemJ149(2019)103972
[3] SoukalJSturgeonREMusilSEfficientphotochemicalvaporgenerationofmolybdenumforICPMSdetectionAnalChem90(2018)11688ndash11695
[4] deOliveiraRMBorgesDLGUVphotochemicalvaporgenerationofnoblemetals(AuIrPdPtandRh)AfeasibilitystudyusinginductivelycoupledplasmamassspectrometryandseawaterasatestmatrixJAnalAtomSpectrom33(2018)1700ndash1706
[5] VyhnanovskyJSturgeonREMusilSCadmiumassistedphotochemicalvaporgenerationoftungstenfordetectionbyinductivelycoupledplasmamassspectrometryAnalChem91(2019)13306ndash13312
[6] GuoXSturgeonREMesterZGardnerGJVaporgenerationbyUVirradiationforsampleintroductionwithatomicspectrometryAnalChem76(2004)2401ndash2405
[7] HuJSturgeonRENadeauKHouXZhengCYangLCopperionassistedphotochemicalvapor generation of chlorine for its sensitive determination by sector field inductivelycoupledplasmamassspectrometryAnalChem90(2018)4112ndash4118
[8] LeonoriDSturgeonREAunifiedapproachtomechanisticaspectsofphotochemicalvaporgenerationJAnalAtomSpectrom34(2019)636ndash654
[9] SturgeonREPaglianoEEvidenceforphotochemicalsynthesisoffluoromethaneJAnalAtomSpectrom(2020)httpsdoiorg101039D0JA00108B
[10] GrinbergPMesterZSturgeonREFerrettiAGenerationofvolatilecobaltspeciesbyUVphotoreduction and their tentative identification J Anal Atom Spectrom 23 (2008)583ndash587
[11] DengHZhengCB LiuLWWuLHouXDLvYPhotochemicalvaporgenerationofcarbonyl for ultrasensitive atomic fluorescence spectrometric determination of cobaltMicrochemJ96(2010)277ndash282
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 127
[12] deQuadrosDPBorgesDLDirectanalysisofalcoholicbeveragesforthedeterminationofcobalt nickel and tellurium by inductively coupled plasmamass spectrometry followingphotochemicalvaporgenerationMicrochemJ116(2014)244ndash248
[13] deJesusHCGrinbergPSturgeonRESystemoptimizationfordeterminationofcobaltinbiologicalsamplesbyICP-OESusingphotochemicalvaporgenerationJAnalAtomSpectrom31(2016)1590ndash1604
[14] VyhnanovskyJFotochemickegenerovanıtekavychspeciı kobaltuproanalytickouatomovouspektrometriiMasterthesisFacultyofScienceCharlesUniversityPrague2018
128 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Oneof themethods thatallowobtainingmaterialswithnewproperties is theplasmaenhancedchemicalvapordepositionmethodInthismethodcompoundscalledprecursorsaresuppliedtotheplasmareactorasagasphaseThankstoplasmaenhancedchemicalvapordeposition it ispossible toobtainmaterialswithuniquepropertiesThisisduetothefactthattheplasmaaffectsthesurfaceinfourdifferentways etching cleaning chemicalmodification and crosslinking
Optimization of condition for cold plasma deposition of thin layers for surface modification of working electrodes
a b a cJUSTYNALIPIN SKA MARIAMADEJ BOGUSŁAWBAS JACEKTYCZKOWSKI
a DepartmentofAnalyticalChemistryFacultyofMaterialsScienceandCeramicsAGHUniversityofScienceandTechnologyAdamaMickiewicza3030-059KrakoacutewPolandjustynalipinskaaghedupl
b DepartmentofAnalyticalChemistryFacultyofChemistryJagiellonianUniversityinKrakoacutewGronostajowa230-387KrakoacutewPoland
c Departmentof MolecularEngineeringFacultyofProcessandEnvironmentalEngineeringLodzUniversityofTechnologyWolczanska21390-924ŁoacutedźPoland
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 129
AbstractCurrentlyresearchisfocusedonthesearchfornewphysicallyandchemicallystablematerialsaswellasvolumeorsurfacemodificationOneofthemethodsusedforsurfacemodificationistheapplicationofthin layers from inorganic and organic compounds The plasmaenhancedchemicalvapordepositionisamethodthatallowsmaterialmodificationandalsodepositionofthinlayersThisworkconcernsoptimizationofcoldplasmadepositionparametersandtoachievethebestelectrical conductivitywhilemaintaining thehighmechanicalstrength of the formed layers Preliminary tests were focused onoptimizing the layering parameters such as the deposition timedischargepowerpressureofmonomerandthe flowofargonTheobtainedsamplesweresubjectedtothermaltreatmentafterwhichtheywere coveredwitha layerof aluminumThe thicknessof theobtained layers was determined on the basis of interferencemicroscopymeasurementsAsaresultoftheexperimentslayerswithathicknessof20nmto600nmwereobtainedTheconductivityofthedeposited layers was also determined and values from 003 to
ndash1150Sm wereobtained
Keywordscoldplasmadepositionelectrochemical
applicationssurfacemodificationthinlayers
Thismethodisusedtoproducecatalyticstructuresortomodifythepropertiesofmaterials eg improve hydrophobicity The growing popularity of surfacemodificationmethodsusingcoldplasmaisassociatedwiththefactthatitisanenvironmentallyfriendlyandversatilemethod[12] Workingelectrodesusedinvoltammetryareasubgroupofchemicalsensorswhich are small devices that convert real-time chemical information into ameasurableandanalyticallyusefulmeasurementsignalChemicalinformationrangingfromtheconcentrationofaspecificcomponentofthetestedsampletotheoverallcompositionofthematrixcancomefromboththeinitiatedchemicalreactionandbetheresultofphysico-chemicaltransformationstakingplaceinthetested object Chemical sensors are equipped with two basic elements iereceptor and transducer The receptor is responsible for the conversion ofchemicalinformationfromthetestedobjectintoaspecificformofenergyintheconverterthisenergyistransformedintoausefulanalyticalsignal Parameters characterizing the electrochemical sensor include accuracyprecision selectivity accuracy presentation selectivity sensitivity dynamicrange limit of quantification limit of detection lifetime response time andreliability Themost numerous and the oldest group of chemical sensors areelectrochemicalsensorsCommonlyobservedinterest inthisgroupofsensorsresultsfromthefactthatwithrelativelylowproductionandoperatingcoststheyofferthebestmetrologicalandoperationalparameters[3ndash5]Oneofthemaintrendsofmodernanalyticsisthesearchfornewelectrodematerialsandvariousgeometries of working electrodes One way to improve the performance ofworkingelectrodesistomodifytheirsurfaceforexamplebyapplyingthinlayersIn this work were considered plasma enhanced chemical vapor depositionmethod as the method of surface modification designed to performworkingelectrodeforvoltammetricdeterminationsAspartoftheinitialresearchplasmaprocessing parameters such as discharge power time of treatment andcompositionofgasmixtureinwhichplasmawasgeneratedwereoptimizedThelayers obtained in different conditions have been tested for suitability forelectrochemical applications (layer thicknessmeasurement and themeasure-mentofconductivity)
2Experimental
21Reagentsandchemicals
Theprecursor solutions suchasacrylonitriledietoxydimethylsilane trietoxy-methylsilaneandtetramethyldisiloxanewhicharesuppliedbyABCRwereusedOtherreagentsofanalyticalpuritysuchasn-hexane(SigmaAldrich)andargonwereused
130 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
22Instrumentation
The thin layers were deposited in a parallel-plate plasma reactor (frequency1356MHz)ThesamplesobtainedwerecalcinedinatunnelfurnaceunderanargonatmosphereThethicknessofthedepositedlayerswasmeasuredafterthealuminum was sputtered using a Nikon microscope type ECLIPSE LV150NElectrometerhigh resistance system (KEITHLEY) was used to measureconductivity
3Resultsanddiscussion
Eachof themonomerswasdepositedonprepared1times1corningglasssamplesSamples prepared with n-hexane were placed in a plasma reactor andadditionallypartiallycoveredwithamicroscopecoverslipSchematiclayoutofsamplesinthereactorshowninFig1ThefirststepwastoetchthesystemusingargonplasmaThisstageallowedfortheeliminationofimpuritiesthatwerenotremovedbythehelpofn-hexaneandthepreparationofthesurfaceofthesamplesforthedepositionoftheproperlayerTheproperstageistheapplicationofathinlayerwiththeplasmainducedbytheselectedprecursoracrylonitriledietoxydi-methylsilanetrietoxymethylsilaneandtetramethyldisiloxaneThethicknessandpropertiesoftheobtainedlayersdependonthedepositionparameterssuchasdischarge power time of treatment and composition of gasmixture inwhichplasmawasgenerated Table1showsallcombinationsofparameterstestedforallfourprecursorsFour different discharge powers for acrylonitrile and two different dischargepowersfororganosiliconmonomersweretestedwithtwodifferenttreatmenttimesEachtimeandpowercombinationwasperformedinplasmainducedbypuremonomerandmonomerwithargonAfterapplyingthelayersthesampleswereplacedinaquartzboatandcalcinedinatunnelfurnaceAllsampleswerecalcinedat500degCfor2hoursunderargonflowSamplesaftercalcinationwerecoveredwithalayerofaluminum
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 131
Fig 1 Scheme of sample distribution in plasma reactor(a)reactorelectrode(b)corningglasssamples(c)micro-scopecoverslip
Thethicknessoftheobtainedlayerswasmeasuredusinganinterferencemicro-scopeMeasurements were carried out at amagnification 10times recording theimageinmonochromelightTheimagewassetsothattheinterferencefringeswere perpendicular to the arc on the sample In order to calculate the layerthicknessD[nm]thefollowingformulawasused
(1)
wheredandLweredeterminedonthebasisoftheregisteredimage(disfringeshiftduetorefractionoflightontheslopeLisdistancebetweenthefringes) Thelaststageofthestudywastodeterminethecurrent-voltagecharacteristicstodeterminetheconductivityoftheobtainedlayersThesamplewasplacedinameasuring cell and attachedwith silver paste to the electrometerwires ThechangeincurrentwasrecordedwiththeapplicationofalternatingvoltageintimeBasedontheresultsobtainedthegraphsofdependenceUndashIwereobtainedfromwhichthevalueofresistance(R)wasdeterminedAnexampleofcurrent-voltagecharacteristicsisshowninFig2
132 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Table 1Conditionsforlayersdeposition-parameterwhichweretested
Monomer DischargepowerW Timeoftreatmentmin
Acrylonitrile 10204080 24Dietoxydimethylsilane 2040 255Trietoxymethylsilane 2040 255Tetramethyldisiloxane 2040 255
Fig 2Current-voltagecharacteristicsdeterminedfortheacrylonitrilelayer(depositionparametersW=10Wt=25mingasmixtureonlyacrylonitrile)
Table 4Theresultsofthicknessmeasurementsandconductivityspecifictothetrietoxymethylsilanelayers
DischargepowerW 20 20 40Timeoftreatmentmin 25 5 5Argonflowsccm 10 10 10Thicknessnm 14686 24282 35311
ndash1SpecificconductivitySm 165 93 67
Knowing the value of the resistance and the geometry of the system thespecificresistancewasdeterminedfollowedbythespecificconductivityofthesamplethefollowingequationwasused
(2)
whereρisspecificresistance[Ωm]Risresistance[Ω]bissamplelength[m]Disdepositedlayerthickness[m]andldistancebetweenelectrodes(wires)[m] Theresultsofthicknessmeasurementsandspecificconductivityarepresentedin the Tables 2ndash5 For electrochemical applications it is important that theobtainedlayerhasthehighestspecificconductivityAnalyzingthedatapresentedin Tables 2ndash5 shows that the thinnest layers have the greatest applicationpotentialastheelectrodematerial
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 133
Table 2Theresultsofthicknessmeasurementsandconductivityspecifictotheacrylonitrilelayers
DischargepowerW 40 40 80 80 10 10 20 20Timeoftreatmentmin 2 4 2 4 2 4 2 4Thicknessnm 15845 31167 250 4644 247 67 7531 12236
ndash1SpecificconductivitySm 0002 156 191 0003 1512 454 311 185
Table 3The results of thickness measurements and conductivity specific to the dietoxydimethylsilanelayers
DischargepowerW 20 20 40 40Timeoftreatmentmin 25 5 25 5Argonflowsccm 10 10 10 10Thicknessnm 13998 37519 28431 49556
ndash1SpecificconductivitySm 1640 55 105 40
Table 5The results of thickness measurements and conductivity specific to the tetramethyldisiloxanelayers
DischargepowerW 20 40 40Timeoftreatmentmin 25 25 5Argonflowsccm 10 10 10Thicknessnm 16759 3159 61652
ndash1SpecificconductivitySm 213 95 31
4Conclusions
InthisworkplasmaenhancedchemicalvapordepositionmethodwasusedforapplyinglayersoffourdifferentmaterialsacrylonitriledietoxydimethylsilanetrietoxymethylsilaneandtetramethyldisiloxaneBychangingparameterssuchasdischargepowertimeoftreatmentandcompositionofgasmixtureanumberofsampleswereobtainedwithlayersofdifferentthicknessandwhatisassociatedwithotherelectricalproperties TestsperformedaspartofthisworkwereusedtoperforminnovativeworkingelectrodesforvoltammetricdeterminationsThesurfacewasmodifiedusingthecoldplasmaofthreesubstratesgraphiteglassycarbonandgold
Acknowledgments
JLandMMhavebeenpartlysupportedbytheEUProjectPOWR030200-00-I00416
References
[1] KapicaRTyczkowskiJBalcerzakJMakowskiMSielskiJWorwaEEnhancingadhesivejointsbetweencommercialrubber(SBS)andpolyurethanebylow-pressureplasmasurfacemodificationIntJAdhesAdhes95(2019)102415
[2] TyczkowskiJKapicaRŁojewskaJThincobaltoxidefilmsforcatalysisdepositedbyplasma-enhancedmetalndashorganicchemicalvapordepositionThinSolidFilms515(2007)6590ndash6595
[3] HulanickiAGłabSIngmanFChemicalsensorsDefinitionsandclassificationPureApplChem63(1991)1247ndash1250
[4] BrzozkaZWroblewskiWSensorychemiczneWarszawaOficynaWydawniczaPolitechnikiWarszawskiej1999(InPolish)
[5] SkoogDAWestDWHollerFJCrouchSRFundamentalsofAnalyticalChemistry9thEdBostonCengageLearning2013
134 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
ConcentrationandcompositionofvolatilecompoundsorcongenersisoneofthemostimportantparametersresponsibleforqualityofproducedalcoholicdrinksandhencefortheirsensorycharacteristicsandconsumeracceptanceTodaygaschromatography (GC) is conventionally used to determine qualitative andquantitativecompositionsofvolatilecompoundswithvariousexternalandorinternalstandardcalibrationprocedures Methodemployingethanol as an internal standard (IS) forGCquantitativedeterminationofvolatilecompoundsinalcoholicbeverageshasbeensuggestedquitelongago[1]andsincethattimegreatresearchworkhasbeencarriedoutRecentlyaninterlaboratorystudyofthemethodinvolving9testinglaboratoriesfrom4countrieswascarriedout[2]TheresultsdemonstratedgreatperspectivesofldquoEthanolasISrdquomethodandproveditsreferencecharacterandeaseofroutineimplementation
Advanced GC-MS method for quality and safety control of alcoholic beverages
abANTONKORBAN
a DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova2030812840Prague2CzechRepublickarbonat7gmailcomb DepartmentofAnalyticalChemistryChemistryFacultyBelarusianStateUniversityLeningradskaya14220050MinskBelarus
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 135
AbstractRecently developed and validated simple and reliable quantitativemethod employing ethanol as an internal standard for GC-MSquantification of volatile compounds in alcoholic products wasapplied to 36 samples including commercially available world-famousbrandspirits from18countriesandhomemadedistillatesThe GC-MS analyses were performed simultaneously by thesuggested approach and official internal standard method that isprescribedinthelegislationofEUandUSATheindependentsamplest-testwasemployedtoevaluatethestatisticaldifferenceofresultsofthesetwomethodsThetestrevealednodifferenceintheresultsandtheirrepeatabilityThemainbenefitsofthesuggestedmethodaretheeliminationofthenecessityofmanualinternalstandardadditionandsamplesdensitymeasurementthusmakingitmoreeconomicalandproductive
Keywordsalcoholicbeveragesgaschromatography-
massspectrometry(GC-MS)
internalstandardmethodvolatilecompounds
quantification
AllpreviousstudieswereutilizingflameionizationdetectorssinceGC-FIDisprescribedinthelegislation[3]wheremass-spectrometrydetectorsarenotyetofficially referred However GC-MS instruments are employed in practice toqualifyandorquantifyvolatilesincommercialspiritsintraditionalhomemadealcoholicdrinksinnewlydevelopedbeveragesinspiritwastesandindistillatesobtainedwithdifferentmanufacturingprocesses Our recent researchwasdirected towardsdevelopment of an algorithmofldquoEthanolasISrdquomethodapplicationonGC-MSinstruments[4]WehaveshowedthattopreventMSdetectorfromsaturationethanolshouldberegisteredinthecorresponding SIM timewindow at characteristicmz of low abundance forinstance bymz of 47 ions This ion corresponds to non-fragmented ethanol
13moleculescontaining1heavyisotope(mainly C)FinallytheresultsofmeasuredstandardsolutionsshowedthatthesuggestedapproachisvalidandldquoEthanolasISrdquomethodmaybesuccessfullyusedonGC-MSinstrumentstoo The objective of this studywas to test and further approve the suggestedapproachonalargersetof36realsamplesofalcoholicdrinkseithercommercialorhomemadeThesamplesweresimultaneouslyanalysedbytwoGC-MSmethodsndashaclassicalISmethodprescribedinthelegislationandthesuggestedldquoEthanolasISrdquomethod
2Experimental
21Reagentsandchemicals
Thefollowingvolatilecompoundsweredeterminedintestedsamplesofalcoholicbeverages 11-diethoxyethane (acetal) acetaldehyde methyl acetate ethylacetate methanol 2 propanol 1-propanol 2 methylpropan-1-ol (isobutanol)1-butanol2-butanoland3methylbutan-1-ol(isoamylalcohol)1-pentanolwasemployedasatraditionalIScompound
22Instrumentation
ShimadzuGCMS-QP2010Ultraequippedwithaquadrupolemassspectrometrydetector was employed for GC-MS measurements Rxi-1301Sil MS capillarycolumn(60mlength025mmid025micromfilmthicknessRestek)wasusedfortheseparationofcompoundsInjectionswereperformedinasplitmode(ratio175)Helium(99999purity)wasusedasacarriergasinjectortemperaturewas170degCTheoventemperaturewasheldat30degCfor5minthenraisedto210degC
ndash1at a rate of 30 degCmin and held isothermally for 4minMeasurementswereperformedinaSIMmodeFortheanalysedcompoundsand1-pentanol2ndash3mostabundantionsinthecorrespondingMSspectrumwereselectedethanolSIMtimewindowcontainedonly47mzionsAllGC-MSmeasurementswerecarriedoutintriplicateunderrepeatabilityconditions
136 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
AnalysisofeachalcoholicsamplewasperformedinafollowingwayAliquotof09mL of a tested sample was pipetted into a standard 2mL glass vial and
ndash1weighedAfterthat01mloftheISsolution(2355mgkg of1-pentanolinWES)wasaddedtothetestedsampleandthemasswasrecordedTheobtainedmixturewasmixedthoroughlyand05microlofitwasinjectedintotheGCsystem Theoriginoftestedalcoholicbeverageswaseithercommercialorhomemade33 world-famous spirits manufactured at different parts of the world werepurchasedfromcommerciallyavailablesourcesThelistoftypesofpurchasedandanalysedspiritsincludedbourboncalvadoscognacgingrappaliquormetaxaportwine rumsake tequilavodkawhiskeyandvarious fruitdistillatesThepurchased drinks were produced at the territory of the following countriesBelarus Bermuda Cuba Czech Republic Denmark France Germany GreeceGuatemala Jamaica Japan Mexico Moldova Portugal Slovakia Trinidad andTobago UK (England and Scotland) USA Three homemade fruit distillatesproducedbyfermentationofpulpyfruitsortheirmustswereobtainedfromlocalspiritmakersThedeclaredABVvaluesofalltestedsamplesvariedfrom15to81
3Resultsanddiscussion
To fulfil themaingoalof thiswork ie toevaluate thestatisticaldifferenceofresultsyieldedbythecomparedmethodswehaveemployedStudentst-testforindependentsamplestoverifystatisticaldifferencesonthesignificancelevelofp=005Theobtainedempiricalvaluesforallpairsofcongenersrsquoconcentrationswere lower than critical one in all cases demonstrating that concentrationsobtainedbythetwomethodshavenostatisticaldifferenceandleadtothesameresults InadditionrepeatabilityofthetwomethodswascomparedthereforeallRSDvalues obtained from triplicatemeasurements were split in two groupswith
ndash1respecttothecorrespondingconcentrations(lowerthan50mgL AAandhigherndash1than50mgL AA)Theobtainedresultsarepresentedintheformofboxplotin
Fig1AnalysisofthechartinFig1showedthatboththetestedmethodshaveyieldedstatisticallysimilarrepeatability AllofthetestedalcoholicdrinkssatisfiedtherequirementsofEURegulation(EC)no1102008[5]Theconcentrationsofundesirablecompoundssuchasmethanoldidnotexceedthelevelsspecifiedinthesameregulationforcorres-ponding beverages In Table 1 the description of the used SIM method andsummaryoftheexperimentalresultsarepresented Tocomparethetruenessofthemethodsoneofthespiritsampleswasspikedwithstandardsolutions(ABV40)containingallanalysedvolatilecompoundsat
ndash1concentrationsof50500and5000mgL AATheoriginalsamplewasusedasareferenceEachofthespikedsolutionswasmeasuredintriplicateSelectedspirit(cherry distillate) initially contained all 11 volatile compounds in various
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 137
138 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1BoxchartsofRSDsofusedISmethodsat2concentrationrangesMeanisequaltoarithmeticmeanoraverageInterquartileRange(IQR)meansisthedistancebetweentheupper(themedianoftheupperhalfofthedataset)andlower(themedianofthelowerhalfofthedataset)quartile
ndash1Compound Timemin Registeredmz Numberof ConcentrationmgL AA results Minimal Maximal
Acetaldehyde 0ndash42 314344 36 24 715Methanol 36 13 13600
Ethanol(IS) 42ndash48 47 mdash mdash mdash
2-Propanol 48ndash70 29314345 14 27 199Methylacetate 596174 10 34 3201-Propanol 26 361 12070Ethylacetate 27 166 107002-Butanol 11 18 2080
Isobutanol 70ndash200 3141ndash4345 28 19 20001-Butanol 5556 13 28 155Acetal 26 45 270Isoamylol 31 39 26501-Pentanol(IS) mdash mdash mdash
Table 1DescriptionoftheusedSIMmethodandsomestatisticsconcerningallmeasured36spiritsamplesbothpurchasedandhomemade
concentrationsTheobtainedrecoveriesboxchartsareshowninFig2Compa-risonoftherecoveriesobtainedwithtwomethodsindicatesthattheyhavenosignificantdifferenceintermsoftruenessAverageobtainedrecoverywas981whenusingsuggestedmethodand980whenusingtraditionalISmethod
4Conclusions
InthisworktheresultsoftestingtheadvantageousldquoEthanolasISrdquomethodfortheGC-MS quality control analysis of alcoholic beverages were presented33purchasedsamplesofworld-famousalcoholicbeveragesoriginatingfrom18countriesand3homemadefruitdistillateswereanalysedtomakeathoroughandcomprehensive studyof thedevelopedmethodTheconcentrationsofvolatile
ndash1compoundsinanalysedsamplesvariedfrom1to13500mgL AAtheABVvalueofanalysedsamplesvariedfrom15to81ThesuggestedmethodwascomparedwiththetraditionalISmethodthatiscurrentlystatedinlegislationTheindepen-dentsamplest-testrevealedthatwithaprobabilityof095resultsobtainedwithtwo methods do not differ significantly The results of within-run precision(repeatability)showedrelativestandarddeviationswithin3measurementstobelessthat6indicatingthatthetechniqueisreproducibleThetruenessofthemethodwasevaluatedbyrecoverycalculationAccordingtotheobtainedresultsrecoveryofthesuggestedmethod(981plusmn33)wasslightlybetterthanthatofthetraditionalone(980plusmn58) ThesefactsprovethatdevelopedldquoEthanolasISrdquomethodistruepreciseandreliable when employed on GC-MS instruments At the same time to obtain
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 139
Fig 2Boxchartsof recoveriesof thesuggested(dottedpattern)and traditional (brickpattern)ISmethodsatdifferentspikeconcentrationsSymbolsdefinitionsarethesameasinFigure1
concentrationsofvolatilecompoundsintheofficiallyrequiredunitsofmeasurendash1 ndash1(mgL AAgL AAetc)suggestedmethodrequiresnodensitometrymeasure-
mentsofthetestedsampleandnoadditionofIScompoundoranyothersamplepre-treatmentThismethodprovidesaninvaluableanalyticaltoolforthequalitycontrolofalcoholicproductsandshouldbeusedinroutineanalysis
Acknowledgments
ThisworkwasfinanciallysupportedbytheVisegradFund
References
[1] CherepitsaSVBychkovSMKovalenkoANMazanikALSeleminaNMSeredinskayaOBThe use of themajor component (solvent) as an internal standard in the gas-chromato-graphicdeterminationofimpuritiesJAnalChem58(2003)368ndash371
[2] CharapitsaSSytovaSKorbanASobolenkoLEgorovVLeschevSZakharovMCabalaRBusarovaRShestakovichITolstouhovaAOndrousekSVavraJYilmaztekinMCabarogluTInterlaboratorystudyofethanolusageasaninternalstandardindirectdeterminationofvolatile compounds in alcoholic products BIO Web Conf 15 (2019) 02030httpsdoiorg101051bioconf20191502030
[3] CommissionRegulation(EC)No28702000layingdownCommunityreferencemethodsfortheanalysisofspiritsdrinkshttpdataeuropaeuelireg20002870oj
[4] KorbanACharapitsaSCabalaRSobolenkoLSytovaSTheperspectivesofethanolusageasaninternalstandardforthequantificationofvolatilecompoundsinalcoholicproductsbyGC-MSJMassSpectr55(2020)e4493
[5] EuropeanUnion(2008)Regulation(EC)No1102008oftheEuropeanParliamentandoftheCouncilof15January2008ontheDefinitionDescriptionPresentationLabellingandtheProtectionofGeographical IndicationsofSpiritDrinksandRepealingCouncilRegulationhttpdataeuropaeuelireg2008110(1)oj
140 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Carbonorgraphitefeltsareusedaselectrodematerialsincethe1990sandtheirutilizationstillgrowsinanalyticalelectrochemistryaswellasinotherareasThisisduetotheirsuitablepropertiesfromwhichwecannamehighporosityhighspecificsurfaceareagoodelectricconductivityandhighphysicalandchemicalstabilityThefirsttwoparametersaregivenbystructureoffeltwhichconsistsoforderlesscarbonfibreswithabouttentotwentymicrometersindiameter[1]theothersby theadvantageouselectricalpropertiesof carbon fibreOn theotherhand porous flow-through electrodes including carbon felt electrode havedisadvantageinapotentialdropintheelectrodevolumewhichcausesdifficultcontrollingoftheexactpotentialappliedontheelectrodeandthereforeresultsindifferentcurrentefficienciesontheoppositesidesoftheelectrode[2] Carbon felt electrode can be utilized for detection of structurally differentcompoundsatvariousconditionsForexampleoperatingatreductionpotentialofndash08V[3]oxidationatrelativelyhighpotential+15V[4]ormeasuringatlowconcentrationsofelectrolyte[5]canbenamedDevelopedtechniquesalsoshowsthatcarbonfeltcanbeusedfordeterminationatsubmicromolarconcentrationsThis ismainly due to its ability to operate as a high-efficiency amperometricdetector Themainaimofthispaperistooverviewandcompareparametersofseveraldetermination methods of different analytes using carbon felt detector in
Utilization of a carbon felt as a material for working electrodes
MARTINBAROCHHANADEJMKOVA SA RKASLA DKOVA
DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublicmartinbarochnaturcunicz
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 141
AbstractWorkingelectrodemadeofcarbonfeltwasusedincombinationwithHPLC for verificationofpractical applicabilityof the electrodeAlldeveloped methods confirm advantageous physical and chemicalpropertiesofcarbonfeltForelectrochemicalutilizationitispossibleto operate at higher positive potentials and even in low concen-trationsofelectrolyteinmobilephaseObtainedlimitsofdetectionwere mostly in submicromolar range and standard deviations ofmeasurementrepeatabilitywereunder5
KeywordsamperometrycarbonfeltFIAHPLC
combinationwithHPLC(forexampletheirdetectionpotentialslimitsofdetectionorlinearrange)
2Experimental
21Reagentsandchemicals
Stock solutions of propyl gallate butylhroxyanisole tert-butylhydroquinonebutylhydroxytoluene chlortoluron 2-amino-4-nitrophenol and 4-amino-2-
ndash3 ndash1nitrophenol (all SigmandashAldrich) with concentration of 1times10 molL werepreparedbydissolving theappropriateamountof therespectivesubstance inmethanol(HPLCgradeLach-NerCzechRepublic)Stocksolutionsof indole-3-aceticacidandindole-3-butyricacidwerepreparedinthesamemannerbutindeionized water Mobile phase consisted of methanol and phosphate-acetatebufferpreparedfromphosphoricandaceticacid(bothLach-NerCzechRepublic)andsodiumhydroxide(Fluka)
22Instrumentation
Theelectrochemicalcellconsistedofcarbonfelt(KarbotechnikCzechRepublic)flow-throughelectrodewhichwasplacedincapwithplatinumwireelectricalcontactanddrilledoutletholeononesideandwithflatferrulawithcapillaryonthe other side Schematic picture of the assembly is shown in ref [5] Otherelectrodeswereauxiliaryplatinumwireelectrodeandreferencesilverchloride(3MKCl) electrode (bothMonokrystaly Turnov Czech Republic) Potentiostatused in combination with this cell was Amperometric Detector ADLC 2(LaboratonıprıstrojePrahaCzechRepublic) HPLCapparatusconsistedofBeta10gradientpump(ECOMCzechRepublic)degasser DG 4014 (ECOM Czech Republic) six-way valve with 20microl loop(Rheodyne USA) HPLC column used for separation of propyl gallatebutylhydroxytoluenetert-butylhydroquinoneandbutylhroxyanisolefromtheirmixtureandforindole-3-aceticacidandindole-3-butyricacidfromtheirmixture
regwasLichrospher RP-18(125times4mm5micromMerckGermany)ForseparationofregchlortoluroncolumnPurospher RP-18(125times4mm5micromMerckGermany)was
used In case of mixture 2-amino-4-nitrophenol and 4-amino-2-nitrophenolregcolumnGemini C18110A (150times46mm5micromPhenomenexUSA)wasusedfor
separation Measurements of pHwere carriedout at Conductivity andpH-meter3510usingcombinedglasselectrode(JenwayUK)
142 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
3Resultsanddiscussion
Performanceofcarbonfeltwastestedonseveraltypesofanalyteswhichneededdifferent separation conditions namely amount of organic solvent in mobilephase and buffer pH The lowest amount of methanol (30) was used forseparation of 2-amino-4-nitrophenol and 4-amino-2-nitrophenol [3] Higherconcentrations of methanol in mobile phase was used for determination ofchlortoluronandforseparationofindole-3-aceticacidandindole-3-butyricacid[4]namely40and60respectivelyThehighestconcentrationsofmethanoland therefore electrolyte with lowest conductivity was used in separation ofantioxidantsnamelypropylgallatebutylhroxyanisolebutylhydroxytolueneandtert-butylhydroquinonewhereamountofmethanolwasrampingfrom55to95[5]DetectionpotentialsofmentionedanalytesandtheirlimitsofdetectionsareshowninTable1 HPLCseparationofantioxidantswas the firstmethodchosen for testingofcarbonfeltelectrodeperformancewiththistechniqueDuetodifferencesintheirstructure when butylhydroxytoluene has a different oxidation mechanismdetectionwith two appliedpotentialswas necessary According to the hydro-dynamic voltammograms potentials 14V and 08Vwere chosen for determi-nationofbutylhydroxytolueneandfortheotherthreeanalytesrespectively[5]AsshowninTable1whenthehigherpotentialwasappliedlimitsofdetectionforpropyl gallate butylhroxyanisole and tert-butylhydroquinone had increasedContrary determination of butylhydroxytoluene had approximately six timeslowerdetectionlimitathigherpotential
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 143
ndash1Compound E VLODmicromolL Refdet
Propylgallate 080 088 [5] 140 186 [5]Butylhroxyanisole 080 144 [5] 140 348 [5]tert-Butylhydroquinone 080 121 [5] 140 266 [5]Butylhydroxytoluene 080 3128 [5] 140 463 [5]Chlortoluron 140 013Indole-3-aceticacid 150 033 [4]Indole-3-butyricacid 150 054 [4]4-Amino-2-nitrophenol 080 016 [3]2-Amino-4-nitrophenol 080 021 [3]4-Amino-2-nitrophenol ndash080 35 [3]2-Amino-4-nitrophenol ndash080 37 [3]
Table 1Parametersofdetectionpotentialsandlimitsofdetectionfordifferentcompoundsusingcarbonfeltdetector
Detectionofauxins(indole-3-aceticacidandindole-3-butyricacid)wascarriedout at potential 15VMeasurements at this potential gives repeatabilitywithstandarddeviation31forindole-3-aceticacidand25forindole-3-butyricacidevenwithexchangingoftheworkingelectrodematerialCalibrationcurves
ndash1forbothanalyteswereobservedfrom04to100μmolL withlinearityinwholeconcentrationrangeLimitsofdetectionforbothanalytesreachedsubmicromolarconcentrationsevenwithrelativelyhighpotential[4] Incaseofdeterminationof2-amino-4-nitrophenoland4-amino-2-nitrophenolcarbon felt electrodewasused inbothoxidationand reductionmodeHydro-dynamicvoltammogramsshowedthatoptimaldetectionpotentialinreductionmodewasndash08VThislowpotentialisclosetotheendofthepotentialwindowandthereforeinterferenceswithremnantsofdissolvedoxygeninmobilephasewereobservedTheseinterferencesresultedinapproximately20timeshigherlimitofdetection for 2-amino-4-nitrophenol or 4-amino-2-nitrophenol obtained inreductionthaninoxidationmodeOntheotherhandmaximumvaluesoflinearrangewerethesameforbothanalytesinbothdetectionmodes[3] ForHPLCofchlortolurontheoptimaldetectionpotentialof14VwasfoundIts
ndash1calibration dependence although observed from 025 to 1000μmolL was ndash1linearonly in therange from025to50μmolL Limitofdetectionbasedon
ndash1standardsolutionswas013μmolL andreproducibilityofmeasurementgivenbytwentyconsecutivemeasurementsgaverelativestandarddeviationof05 ForallthedeterminationmethodsattentionwaspaidtotheapplicabilityofthecarbonfeltindetectionofanalytesincomplexmatricesIncaseofantioxidantsedibleoilswerechosenasrealsamples[5]nitrophenolderivatesweredeter-minedinurinesamples[3]andauxinsinrootingpreparation[4]ChlortolurondeterminationwasperformedinsoilandsurfacewaterThefoundvaluesshowanegligiblematrixinfluenceondetection
4Conclusions
CarbonfeltworkingelectrodewassuccessfullyusedincombinationwithHPLCfordeterminationofdifferenttypesofelectroactivecompoundsegantioxidantsauxinsorpesticidesAllmentionedapplicationsshowgreatperformanceofthecarbonfeltasaflow-throughelectrodematerialinelectroanalyticalchemistryforoxidationandreductionwayofanalytesdeterminationLimitsofdetectionforanalytes are mostly in submicromolar concentrations the exceptions areoxidationof analytes athigherpotentials and their reductionwhere limitsofdetectionsareinmicromolarconcentrationsApplicabilityoftheelectrodeonrealmatriceswas proven on analysis of edible oil samples groundwater soil androotingpreparation
Acknowledgments
ThisworkhasbeensupportedbytheCzechScienceFoundation(projectGACR20-01589S)
144 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
References
[1] Gonzalez-Garcia J Bonete P Exposito E Montiel V Aldaz ATorregrosa-Macia RCharacterizationofacarbonfeltelectrodeStructuralandphysicalpropertiesJMaterChem9(1999)419ndash426
[2] NavaJLRecendizAGonzalezLGCarrenoGMartın ezFMassTransportandpotentialstudiesinaflow-throughporouselectrodereactorPortugalElectrochimActa27(2009)381ndash396
[3] Dejmkova H Knaf M Application of carbon felt detector for the determination ofdinitrophenolmetabolitesInXXXIXModernElectrochemicalMethodsFojtaMSchwarzovaKNavratilT(Eds)U stınadLabemBestServis2019p41ndash43
[4] DejmkovaHdeAraujoDanielMElectrochemicaldeterminationofindole-3-aceticacidandindole-3-butyric acid using hplc with carbon felt detectorMonatsh Chem150 (2019)439ndash442
[5] DejmkovaHBarochMKrejcovaMBarekJZimaJCoulometricdetectorbasedoncarbonfeltApplMaterToday9(2017)482ndash486
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 145
1Introduction
Smith-Lemli-Opitzsyndrome(SLOS)isan autosomal recessive genetic disor-der firstly described in 1964 [1] It iscaused by the inborn deficiency of7-dehydrocholesterol reductase Thisenzyme transforms 7-dehydrocholes-terol (7-DHC Fig 1) to cholesterolduringthefinalstepofbiosynthesisofcholesterolincells The clinical symptoms of SLOS aredecreasedbloodlevelofcholesteroland
Electroanalytical methods for determination of 7-dehydrocholesterol in artificial serum
LENKABENESOVAADE LAZA RYBNICKA JANKLOUDAKAROLINASCHWARZOVA -PECKOVA
UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublicbenesolenaturcunicz
Abstract7-DehydrocholesterolisabiomarkerofSmith-Lemli-Opitzsyndromeanautosomalrecessivegeneticdisordercausedbytheinborndefici-encyof7-dehydrocholesterolreductaseInthisstudyproceduresforitsdeterminationinartificialserumusingflowinjectionanalysiswithelectrochemical detection and voltammetric detection on borondoped diamond electrode were optimized The proteins wereprecipitatedbyacetonitrileandaftercentrifugationthesupernatantused for analysis For quantitation of 7-DHC by differential pulsevoltammetrytheoptimalratioacetonitrile-artificialserum91(vv)wasappliedInFIA-EDtheratio31(vv)runelectrolyteconsisting
minus1ofwater-acetonitrilecontaining001molL NaClO inthesameratio4minus1anddetectionpotentialof+13VvsAgAgCl(3molL KCl)were
usedQuantitationof7-DHCwaspossibleusingcalibrationdepen-minus1dencewithlimitdetectionof20micromolL inartificialserumNeverthe-
lessthemethodhaslowrecoveryandforsensitivedeterminationinreal matrices of human serum and amniotic fluid a liquid-liquidextraction needs to be applied to prevent presence of 7-dehydro-cholesterolinthephasewithprecipitatedproteins
Keywordsamperometricdetectionborondopeddiamond
electrode7-dehydrocholesteroldifferentialpulse
voltammetryflowinjectionanalysisSmith-Lemli-Opitz
syndrome
146 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Structureof7-dehydrocholesterol
increased concentrationof7-DHC inbloodandnervous system [2] SLOS is acomplexofmultipleanomaliesincludingmentalretardationItismanifestedbyholoprosencephaly(anomaliesinbraindevelopmentwithimprecisedivisionintotherightandlefthemispheres)milddysmorphismscardiacrenalandgastro-intestinalmalformations ThecharacteristicfacialanomaliesofSLOS[2]aremicrocephalybitemporalnarrowing ptosis short nasal root short nose with anteverted nares andmicrognathia epicanthal folds and capillary hemangioma over the nasal rootextendingontotheglabellatheearappearlow-setandareposteriorlyrotatedOral finding includes a high-arched and narrow hard plate broad and ridgealveoralridgesandredundancyofsublinqualtissuesCNSanomaliesareagenesisorhypoplasiaBilateralandunilateralpostaxialpolydactylycanbepresentedinthehandsorfeetorboth Concentrationof7-DHCinbloodiscrucialforclinicaldiagnosticofSLOSinpatients Concentration levels in amniotic fluid are used for fetal diagnosticsTable1 summarizes concentration of 7-DHC in plasma and amniotic fluid ofhealthypersonandSLOSpatientsAnalyticalmethodsusedfordeterminationofconcentrationof7-DHCinthesematricesincludecombinationofGCorHPLCwithMS[3]orGCwithflameionizationdetection(FID)orUVdetection[45] Thepossibilitiesofelectrochemicalmethodsfordetectionof7-DHCarelimitedasgenerallythesteroidcoreisratherredox-inactive(detailinreview[6])undervarietyofconditionsNevertheless7-DHCpossessesconjugateddoublebondsonsteroidcoreanditsoxidationwasreportedinseveralstudies[7ndash9]Itsvoltam-metricsignal+095VvsSCEonglassycarbonelectrodewasfirstlyobservedin
minus1non-aqueousmediaofmethanol-benzene7525(vv)using005molL LiClO as4
supporting electrolyte in a study dealing with electrochemical behaviour ofvitaminAandDandtheirprovitaminsD(7-DHCisprecursorofvitaminD3)[7]Determinationof7-DHCinhumanskinispossiblebyHPLCwithUV(λ=286nm)andamperometricdetection(E +17VvsAgAgCl)onglassycarbonelectrodedet
minus1usingmethanol-tetrahydrofuran175mmolL KH PO (9514vvv)asmobile2 4minus1phase7-DHCwasdetectedintherangefrom12to81microgg dryweightwith
minus1detectionlimitof39pmolL [8]Anotherstudy[9]isdevotedtodeterminationof
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 147
minus1Author(s)ref Matrix ConcentrationμmolL Healthy SLOS
Kelley[4] Plasma 03plusmn001 385plusmn309 Amnioticfluid lt02plusmn001 16plusmn9Rossiteretal[5] Plasma lt5 179ndash335 Amnioticfluid lt03 12ndash15
Table 1Concentrationof7-dehydrocholesterolinclinicalmatricesforhealthypersonsandforSmith-Lemli-Opitzsyndrome(SLOS)patientsdetectedbyGC-FID
7-DHCandvitaminD3 in fishusingHPLCwithelectrochemicaldetectionTheanalytical cell was a serial combination of two-flow-through porous graphiteworking electrodes The first standard coulometric electrode was used toeliminatepotentially interfering compounds using the second lineardynamic
minus1rangefrom0013to0312micromolL for7-DHCwasachieved Hereinwestudiedpossibilitiestodetect7-DHCbasedonitsoxidationonborondoped diamond (BDD) electrode using differential pulse voltammetry andelectrochemicaldetectioninflowinjectionanalysis(FIA-ED)inartificialserumandperipherallyinhumanserumandamnioticfluid
2Experimental
21Reagentsandchemicals
7-dehydrocholesterol (purity95)wasobtainedSigmaAldrich (USA)and itsstandard solution was prepared in acetonitrile (Honeywell Germany) Theartificial serum was prepared from KCl (Penta Czech Republic) CaCl 2H O2 2
(PentaCzechRepublic)NaCl(PentaPragueCzechRepublic)urineD-glucoseand01albuminfromSigma-Aldrich(USA)NaClO4(PentaCzechRepublic)wasusedassupportingelectrolyte
22Instrumentation
VoltammetricmeasurementweregovernedbythepotentiostatPalmSensusingworking BDD electrode (Windsor Scientific UK d = 31 mm) AgAgNO 3
minus1 minus1(01molL AgNO 1molL NaClO in acetonitrile) non-aqueous reference3 4electrodeandplatinumwirecounterelectrodeBDDsurfacewaspolishedbeforeeachscanusingsuspensionofAl O (ElektrochemickedetektoryTurnovCzech2 3
Republic) HPLC system (Hitachi Merck) consisting of control unit D-7000gradientpumpL-7100autosamplerL-7200andUVdetectorL-7400wasusedfor
minus1FIA-EDdetectionof7-DHCRunelectrolytewascomposedof001molL NaClO 4inacetonitrileanddeionisedwaterinratio31(vv)Flowrateofmobilephase
minus1was30mlmin injectionvolumewas40microLandλ=280nmwasusedforUVdetectionWall-jet detection cell was employedwith working BDD electrode
minus1AgAgCl (3molL KCl) reference electrode and platinum wire auxiliaryelectrode Optimal detection potential of +13 V was controlled using ADLC2potentiostat(Laboratornı prıstrojePragueCzechrepublic)
3Resultsanddiscussion
In this study electroanalytical methods were developed for determination of7-DHCinartificialserumnamelyFIA-EDandDPVBothmethodsarebasedondirectoxidationof7-DHConborondopeddiamondelectroderesultinginanodic
148 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
peak at ca +08 V (vs AgAgNO in acetonitrile) in non-aqueous medium of3
acetonitrile or mixedmedium acetonitrile-water using NaClO as supporting4
electrolyteTheoxidationispresumablyinitiatedbyoneelectronremovalfromtheconjugateddoublebondsonthesteroidcoreof7-DHC Fordeterminationof7-DHCinrealmatricesitisnecessarytoremovepresentproteinsArtificialserumcontainingalbuminwasusedasmodelmatrixtostudythe possibilities Firstly albumin was removed simply by precipitation withacetonitrile(serum-water13(vv))andthesupernatantwasanalysed Differentratioswateracetonitrileweretestedinrunelectrolyte(59510902080 2575 3070 4060 and 5050 (vv)) to evaluate the influence of itscompositionontheFIA-EDsignaloftheblankand7-DHCThesameratio13asusedforprecipitationofalbuminwaschosenasoptimalbecauseofminimalandstablesignaloftheblankinjectedinFIA-EDsystemFurtherdetectionpotentialE in the range from +10 V to +15 V was optimized by evaluation of thedet
hydrodynamicvoltammogramsresultinginE of+13Vsetasoptimalvaluedetminus1 Concentrationdependenceof7-DHCislinearintherangefrom25micromolL to
minus1 minus1300micromolL (concentrationinartificialserum)withdetectionlimitof20micromolL and this concentration dependence can be used for quantitation of 7-DHC inartificial serum Nevertheless determination of 7-DHC in human serum andamnioticserumfailedastheyrepresentmorecomplicatedmatricesand7-DHCispresumablypartiallyadsorbedinthepresentproteinsandcannotbequantifiedinthesupernatant Furtherdifferentialpulsevoltammetrywithoptimizedparameterswasusedfordeterminationof7-DHCInthepresenceofproteinsinartificialserum(human
minus1serum albumin) an unacceptably high detection limit of 178micromolL wasachieved When the proteins were precipitated using acetonitrile (aceto-nitrileartificialserumratio91(vv))thelimitofdetectionof7-DHCwaslowered
minus1to15micromolL inartificialserumNeverthelesstherecoveryofthemethodwasonly43to70dependingontheconcentrationof7-DHCagainreflectingthelossof7-DHCduetoproteinprecipitation Thereforeasecondapproachofsamplepretreatmentbasedonliquid-liquidextractionofalllipidsdescribedin[10]wastested(Bligh-Dyerextraction)TheprocedurehastwopartsFirstlymethanolchloroformandthesampleofartificialserum is mixed and shaken to form a monophasic system After addition ofchloroform and water a biphasic system is formed where chloroform phasecontainsalloflipidcompoundsandmethanol-waterphasecontainsallnon-lipidscompoundsChloroformphaseisthendriedunderN2atmosphereat50degCanddried extract dissolved in acetonitrile Preliminary experiments using DPVresultedinrecoveryof97forBligh-Dyerextractionof7-DHCfrominartificialserum
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 149
4Conclusions
FIA-EDandDPVwereoptimizedfordeterminationof7-DHCinartificialserumUsing precipitation of proteins by acetonitrile limit of detection of 7-DHC in
minus1artificialserumusingFIA-EDwas20micromolL andthismethodcanbeusedfortheirquantificationusingcalibrationdependenceNeverthelessdeterminationusingDPVisunreliableduetolowrecoveryoftheprocedureDevelopmentofamethodincludingliquid-liquidextractionstepisinprogresssothat7-DHCcouldbedeterminedinrealmatricesashumanserumandamnioticfluid
Acknowledgments
TheresearchwassupportedbytheCzechScienceFoundation(projectGACR19-11268S)andtheSpecificUniversityResearch(SVV260560)
References
[1] Smith DW Lemli L Opitz JM A newly recognized syndrome of multiple congenitalanomaliesJPediatr64(1964)210minus217
[2] Nowaczyk M Waye J The SmithndashLemlindashOpitz syndrome a novel metabolic way ofunderstandingdevelopmentalbiologyembryogenesisanddysmorphologyClinGenet59(2001)375minus386
[3] BeckerSRohnikeSEmptingSHaasDMohnikeKBebloSMutzeUHusainRAThieryJCeglarekULC-MSMS-basedquantificationofcholesterolandrelatedmetabolitesindriedblood for the screening of inborn errors of sterolmetabolismAnal Bioanal Chem407(2015)5227minus5233
[4] Kelley RI Diagnosis of Smith-Lemli-Opitz syndrome by gas-chromatography mass-spectrometryof7-dehydrocholesterolinplasmaamniotic-fluidandculturedskinfibroblastsClinChimActa236(1995)45minus58
[5] Rossiter JP Hofman KJ Kelley RI Smith-Lemli-Opitz SyndromePrenatal-diagnosis byquantification of cholesterol precursors in amniotic-fluid Am J Med Genet 56 (1995)272minus275
[6] KloudaJBarekJNesmerakKSchwarzova-PeckovaKNon-enzymaticelectrochemistryincharacterization and analysis of steroid compounds Crit Rev Anal Chem 47 (2017)384minus404
[7] AtumaSSLundstromKLindquistJTheelectrochemicaldeterminationofvitaminAPartIIFurthervoltammetricdeterminationofvitaminAandinitialworkonthedeterminationofvitaminDinthepresenceofvitaminAAnalyst100(1975)827minus834
[8] MoodyJPHumphriesCAAllanSMPatersonCRDeterminationof7-dehydrocholesterolinhumanskinbyhigh-performance liquid-chromatography JChromatogrB530 (1990)19minus27
[9] OstermeyerUSchmidtTVitaminDandprovitaminDinfishEurFoodResTechnol222(2005)403minus413
[10] BlighEGDyerWJArapidmethodoftotallipidextractionandpurificationCanJBiochemPhysiol37(1959)911minus917
150 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Author Index
AlikovaV1
AugustınM83
BaluchovaS19
BarekJ192570
BarochM141
BasB13129
BastryginaO41
BenesovaL146
BessonovaE57
BohmD6
BurkinK116
BurkinM116
ChernovaA141
ChoinskaM70
CokrtovaK104
DedinaJ97
DeevV57
DejmkovaH141
DendisovaM63
DubenskaL51
EfremenkoE41
FojtaM110
GalvidisI116
HavranL110
HeiglN31
HertJ76
HrdlickaV70
JosypcukB25
KartsovaLA3557
KloudaJ19146
Kodrık ovaB90
KolobovaEA35
KorbanA135
KorotkovaE1
KralM63
KratzerJ90
KravchenkoAV35
KrızekT76104
LipinskaJ129
MadejM129
MatejkaP63
MatysikF-M631
MusilS9097123
NavratilT70
OndrackovaA110
PietrzakK45
PlotnikovaK51
PoradaR13
RedondoBR70
SagapovaL90
ShormanovV1
Schwarzova-PeckovaK19110146
SladkovaS 141
S tadlerovaB97
StiborovaM110
SvobodaM90
TvorynskaS25
TyczkowskiJ129
VyhnanovskyJ97123
VymyslickyF76
VyskocilV83
WardakC45
WongDKY19
ZarybnickaA146
ZelenyI51
Proceedingsofthe16thISCModernAnalyticalChemistry Prague2020 151
152 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Keyword Index
alcoholicbeverages135
aminoglycosides116
amperometricdetection146
amperometry141
antifoulingelectrodes19
assembledcapillaries6
atomicabsorptionspectrometry90
atomicfluorescencespectro-
metry97
atomization90
biologicalactiveanalytes35
biosensor2583
bismuth97
borondopeddiamond
electrode110146
cadmium90
canagliflozin76
capillarycoating35
capillaryelectrophoresis635
capillaryflowinjectionanalysis631
carbohydrates31
carbonfelt141
cathodicstrippingvoltammetry70
chemicalvaporgeneration90
chemometrics57
cobalt123
coldplasmadeposition129
copper(II)phthalocyanine63
covalentimmobilization25
cytochromeP450110
damage83
7-dehydrocholesterol146
designofexperiments76
differentialpulsevoltammetry146
diphenylsilanereductionmethod19
dispersiveliquid-liquid
microextraction57
disposableelectrodes31
DNA83
dualdetectionconcept6
electrochemicalanalysis110
electrochemicalapplications129
electrochemicalflowcell76
electrochemistry51
electrokineticchromatography104
eliminationvoltammetrywith
linearscan70
ELISA116
enzymaticreactor25
FIA141146
gaschromatography-mass
spectrometry(GC-MS)135
glucoseoxidase25
graphite83
honey116
HPLC76141
hydridegeneration97
hydrogenatedconical-tipcarbon
electrodes19
imidazoliumionicliquids35
inductivelycoupledplasmamass
spectrometry123
internalstandardmethod135
ion-selectiveelectrode45
laccase25
liquidcrystals104
massspectrometry31
mercuryelectrode13
metronidazole51
non-aqueouscapillary
electrophoresis104
non-aqueoussystem6
oxidation76
oxytetracyclinehydrochloride51
phenol2-methoxy1
photochemicalvaporgene-
ration97123
polarography51
1-propanesulfonicacid23-
dimercapto-70
pulsedamperometricdetection31
quantitation1
resonanceRamanspectroscopy63
scanningtunnellingmicroscopy63
silversolidamalgamelectrode70
Smith-Lemli-Opitzsyndrome146
smokingmixtures41
solidcontact45
solid-phasemicroextraction57
spectrophotometry141
SudanI110
surfacemodification129
surface-enhancedRaman
spectroscopy63
thinlayers129
tip-enhancedRamanspectroscopy
63
unithiol70
uranyl45
vanillin41
veterinarydrug51
vitamins13
volatilecompoundsquantifi-
cation135
voltammetricdopaminedetec-
tion19
voltammetry1383
Proceedingsofthe16thISCModernAnalyticalChemistry Prague2020 153
Proceedings of the 16th International Students Conference ldquoModern Analytical Chemistryrdquo
EditedbyKarelNesmerak
PublishedbyCharlesUniversityFacultyofScience
Prague2020
1steditionndashvi154pages
ISBN978-80-7444-079-3
ISBN 978-80-7444-079-3
Pro
ceedin
gs of th
e 16
th In
ternatio
nal Stu
den
ts Co
nferen
ce ldquoMo
dern
An
alytical Ch
emistryrdquo P
rague 2
02
0
788074 440793
Prague 17mdash18 September 2020
Edited by Karel Nesměraacutek
Prague 2020
Proceedings of the
16th International Students Conference
ldquoModern Analytical Chemistryrdquo

Proceedingsofthe
16thInternationalStudentsConference
ldquoModernAnalyticalChemistryrdquo
Prague 17mdash18 September 2020
Edited by Karel Nesměraacutek
Prague 2020
Proceedings of the
16th International Students Conference
ldquoModern Analytical Chemistryrdquo
CATALOGUING-IN-PUBLICATIONndashNATIONALLIBRARYOFTHECZECHREPUBLIC
KATALOGIZACEVKNIZEndashNA RODNIKNIHOVNACR
ModernAnalyticalChemistry(konference)(162020PrahaCesko)
Proceedingsofthe16thInternationalStudentsConferenceldquoModernAnalyticalChemistryrdquo
Prague17ndash18September2020editedbyKarelNesmerak--1stedition--PragueFacultyof
ScienceCharlesUniversity2020--vi154stran
Obsahujebibliografiearejstrıky
ISBN978-80-7444-079-3(brozovano)
543(062534)
analyticalchemistry
proceedingsofconferences
543ndashAnalyticalchemistry[10]
TheelectronicversionoftheProceedingsisavailableattheconferencewebpage
httpwwwnaturcuniczisc-mac
copyCharlesUniversityFacultyofScience2020
ISBN978-80-7444-079-3
Preface
Despitethefactthattheyear2020ismarkedbyCOVID-19morethan40young
analytical chemists gathered in Prague for the 16th annual international
conferenceldquoModernAnalyticalChemistryrdquoTheymeettopresenttheresultsof
theirresearchtomastertheirpresentationandlanguageskillsandtoexchange
anddiscussideasandexperiencesofanalyticalchemistry
Thisvolumeofconferenceproceedingsbringsyouatotalof25papersfrom
thisconferenceAsinpreviousyearsthecontributionspresentedareassortedby
the sequence of their delivery supplemented by indexes at the end of the
proceedingsallowingeasynavigationthroughthepagesYouwillseethattopics
of contributions cover all the aspects of modern analytical chemistry from
theoretical problems through development of new analytical methods and
improvementofanalyticaltechniquestotheapplicationsinvolvingthesolutionof
medicinaltechnicalorenvironmentalproblemsLetushopethatlikeprocee-
dingsofpreviousyearsofourconference thisonewillalsobean interesting
beneficialandenjoyablereading
Itseemstousthattheauthorsofthecontributionsareaguaranteeofthatanew
generationofanalyticalchemistswillprotectbrightandthrillingfutureofour
science
We are very grateful to the Division of
AnalyticalChemistryofEuChemSforitslong-
lasting auspices of our conference Also we
arethankfultooursponsorsnotonlyfortheir
kind sponsorship making the conference
possiblebutalsoforalltheircooperationand
supportinmanyofourotheractivities
Enjoyreadingtheseproceedings
docRNDrKarelNesmerakPhD
editor
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 iii
Sponsors
The organizersof16th International Students Conference ldquoModernAnalytical
Chemistryrdquo gratefully acknowledge the generous sponsorship of following
companies
wwwecomsrocom
wwwlach-nercom
iv Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
wwwthermofishercz
www2thetacz
wwwzentivacz wwwquintacz
wwwshimadzueucom
wwwwaterscom
Contents
AlikovaVChernovaAShormanovVKorotkovaEDeterminationof2-methoxyphenol inmodelsolutionsbyspectrophotometry 1
BohmDMatysikF-MTheeffectsoflinearlyassembledcapillarieswithvariousinnerdiametersoncapillaryelectrophoresis 6
PoradaRBasBVoltammetricdeterminationofvitamins 13Baluchova S Klouda J Barek J Schwarzova-Peckova K Wong DKY Dopamine detection at
antifoulingconical-tipcarbonelectrodes 19Tvorynska SBarek J JosypcukBA comparative study of covalentglucose oxidaseand laccase
immobilizationtechniquesatpowderedsupportsforbiosensorsfabrication 25Heigl N Matysik F-M Capillary flow injection analysis with electrochemical detection for
carbohydrateanalysis 31KravchenkoAVKolobovaEAKartsovaLAApplicationofcovalentcoatingsbasedonimidazolium
cations for separationandon-linepreconcentrationof basicandneutralanalytes in capillaryelectrophoresis35
Efremenko E Chernova A Bastrygina O Determination of vanillin in smoking mixtures byspectrophotometry41
PietrzakKWardakCUranylion-selectiveelectrodewithsolidcontact 45Plotnikova K Dubenska L Zeleny I Polarographic determination of metronidazole and
oxytetracyclinehydrochlorideinveterinarydrugforhoneybees 51Deev V Bessonova E Kartsova L Application of microextraction techniques combined with
chromatographicmethodsfortheanalysisofcomplexobjects 57KralMDendisovaMMatejkaPThedevelopmentofreferenceprobesystemfortip-enhancedRaman
spectroscopy 63ChoinskaMHrdlickaVRedondoBRBarekJNavratilTDeterminationofheavymetalpoisoning
antidote23-dimercapto-1-propanesulfonicacidusingsilversolidamalgamelectrode70VymyslickyFKrızekTHert JCanagliflozinoxidation studyusingelectrochemical flowcelland
comparisonwithhydrogenperoxideoxidation 76AugustınMVyskocilVNovelhybridelectrochemicalDNAbiosensorformonitoringoxidativeDNA
damageviaoxidationreductionsignalsoflowmolecularweightdouble-strandedDNA 83SagapovaLKodrık ovaBSvobodaMMusilSKratzerJChemicalvaporgenerationofcadmiumfor
analyticalatomicspectrometry 90S tadlerova B Vyhnanovsky J Dedina J Musil S Photochemical vapour generation of bismuth
coupledwithatomicfluorescencespectrometry 97Cokrtova K Krızek T Separation of liquid crystals using non-aqueous capillary electrokinetic
chromatography104OndrackovaAStiborovaMHavranLSchwarzova-PeckovaKFojtaMElectrochemistryofSudanI
anditsderivatesinaqueousmedia 110BurkinKGalvidisIBurkinMGroupdetectionofaminoglycosidesusingELISAforcontroloffood
contamination 116Vyhnanovsky J Musil S Photochemical vapor generation of cobalt for detection by inductively
coupledplasmamassspectrometry 123LipinskaJMadejMBasBTyczkowskiJOptimizationofconditionforcoldplasmadepositionofthin
layersforsurfacemodificationofworkingelectrodes 129KorbanAAdvancedGC-MSmethodforqualityandsafetycontrolofalcoholicbeverages 135Baroch M Dejmkova H Sladkova S Utilization of a carbon felt as a material for working
electrodes 141Benesova L Zarybnicka A Klouda J Schwarzova-Peckova K Electroanalytical methods for
determinationof7-dehydrocholesterolinartificialserum146
Authorindex 151Keywordindex 152
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 v
1Introduction
2-Methoxyphenol(guaiacol)isusedinmedicineasanexpectorantThestructuralformulaisshowninFig1Itiswidelyusedinthepharmaceuticalindustry[1]forthesynthesisofantituberculosisexpectorantdrugs(Kas-nol Sudafed Ascoril Prothiazine Expectorant Guai-phenesinum) Moreover 2-methoxyphenol is often used as anaromaticsubstance [2] in the food industry Inparti-cular itwaswidelyusedintheproductionofsmokedfish and meat products using smokeless smokingtechnologyusingflavourings Ontheotherhand2-methoxyphenolhasthesymbolGHS07andhasahazardcodeXnTXi[2]accordingtotheGHSsystemItisverytoxicbyinhalationitcanirritatethemucousmembraneoftherespiratorytractandtheconjunctivaofthe
Determination of 2-methoxyphenol in model solutions by spectrophotometry
a a b aVALERIYAALIKOVA ANNACHERNOVA VLADIMIRSHORMANOV ELENAKOROTKOVA
a DepartmentofChemicalEngineeringEngineeringSchoolofNationalResourcesNationalResearchTomskPolytechnicUniversityLeninavenue30634050TomskRussiaalikovaleramailru
b DepartmentofPharmaceuticalToxicologicalandAnalyticalChemistryKurskStateMedicalUniversitystKarlaMarks3305000KurskRussia
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 1
AbstractA spectrophotometric approach for determination of 2-methoxy-phenol in model solutions has been developed The absorptionspectra of 2-methoxyphenol were determined in the wavelengthrangefrom200to400nminsolutionsof95ethanolacetonitrile01Msodiumhydroxideandethylacetatewithaconcentrationofthe
minus3analyte of 005mgdm For the quantitative determination of2-methoxyphenol a seriesof solutionswaspreparedwithvarious
minus3 minus3concentrationsfrom0001mgdm to005mgdm in95ethanolacetonitrile01MsodiumhydroxideTheopticaldensityof2-meth-oxyphenolinsolventswasmeasuredatawavelengthof276nmand289nmThedevelopedmethodwastestedusingthemethodanalysisofspikedsamples
Keywordsquantitation2-methoxyphenolUVVISspectrophoto-
metry
Fig 1Structuralformulaof2-methoxyphenol
eyeballinhighconcentrationswhenitpenetratestheskincanleadtoneurosiswhenadministeredorallycanstimulatetheesophagusandstomachresultinginheartfailurecollapseanddeathNowadaystherearepublisheddataoncasesofasystemicallergicreaction[3]causedby2-methoxyphenolderivativesandthereisa fatal case known [4] for oral administration of guaifenesin (3-(2-methoxy-phenoxy)propane-12-diol)oneofthecomponentsofcommonlyavailablecoughmedications Thedeterminationof2-methoxyphenolinenvironmentalobjectsaswellasinthefoodindustryiscarriedoutusinggaschromatographymethodswithsolid-phasemicroextraction[5]Inordertocontrol2-methoxyphenolinnaturaldrink-ing and treatedwastewater gas chromatography is used followed by opticaldetectionoftheeluate[6]Themaindisadvantagesofthismethodofanalysisarethelowselectivityanddurationofdetermination(about3hours)AlsoaccordingtoRussianStateStandartGOST33312-2015themethodofgaschromatographyisusedforthequalitativeandquantitativedeterminationof2-metoxyphenolinjuiceproducts Commonlyfortheanalysisoftoxicsubstancesinvariousbiologicalsamples(bloodplasmaurinesalivasweathair)bygaschromatographyitisnecessarytocarryoutmultistagesamplepreparationwhichcomplicatesandslowsdownthecourseofthestudy[7]Atthesametimeitisimportantthatduringtheprepa-rationofsamplesintheanalyzedcompoundstheirstructureisnotviolatedasthiswillleadtothedifficultyoftheiridentification Spectrophotometryintheultravioletregionhaslowersensitivitycomparedtothe abovemethods however thismethod does not require such complicatedpreparation of the analyzed samples it is a relatively affordable simple andinexpensive analysis method In addition its sensitivity can be significantlyimprovedbyapplyinganappropriateseparationprocedureandpreconcentrationbeforedetection[8]MethodUVspectrophotometryisusedtoassessthequalityof both medicinal substances and preparations made from them in terms ofauthenticitygoodqualityandquantitativecontentInadditionitisarelativelyaffordablesimpleandlow-costanalysismethod Ananalysisoftheliteraturedatashowedthattodaytherearefastandsensitivespectrophotometricmethodsforthedeterminationofpyrocatecholderivativesinmedicines[9]vanillininfoodproducts[10]andotherphenolsinwastewaterandwineproducts[1112]Howeverasfarasweknowinformationonthedetermi-nationof2-methoxyphenolfromtheabsorptionspectraintheultravioletregionisabsent Theaimofthisstudyistodevelopmethodsforthequalitativeandquantitativeof2-methoxyphenolinmodelsolutionsusingUVspectrophotometry
2 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
2Experimental
21Reagentsandchemicals
Asampleof2-methoxyphenolfromFlukawithabasicsubstancecontentofge98wastakenastheobjectofstudyAssolventsweusedacetonitrile(ChP)95ethanolethylacetateand01MsodiumhydroxidesolutionAllotherchemicalsusedwereofanalyticalreagentgrade
22Instrumentation
Theopticaldensitywasmeasuredincuvetteewithanabsorbinglayerthicknessof10mmusingaCary60spectrophotometer(AgilentUSA)Allmeasurementswerecarriedoutatroomtemperature
3Resultsanddiscussion
Thechangeinthebehavioroftheabsorptionspectrumwasinvestigatedinthewavelengthrangeof200ndash400nmFigure2isshowedthatwithanincreaseinthepolarityofthesolventtheabsorptionmaximumshiftstowardthevisiblepartofthe spectrum The wavelength of absorption maxima of 2-methoxyphenol ispresentedinTable1 A studyof thephotometricbehaviorof2-metoxyphenolinvarious solventsshowedthatacetonitrile95ethanoland01Msodiumhydroxidearethemostsuitablesolventsforthequalitativedeterminationofthetestsubstance
ndash3Fig 1Thespectraof2-methoxyphenolofconcentration005mgdm inthemediumofsolvents(anabsorbinglayerthicknessof10mm)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 3
Forthequantitativedeterminationof2-methoxyphenolaseriesofsolutionsndash3 ndash3withaconcentrationragefrom0001mgdm to005mgdm werepreparedin
acetonitrileand95ethanolTheopticaldensityof2-methoxyphenolinsolventswasmeasuredbywavelengthof276nmThedependenceoftheintensityoftheoptical density on the concentration of 2-methoxyphenol in 01 M sodium
ndash3hydroxide was plotted in the concentration range from 0005mgdm tondash3003mgdm Themeasurementwerecarriedoutbywavelengthof276nmThe
obtainedregressionequationsarepresentedinTable2Dataanalysisobtainedwasperformedusingleast-squaresmethodThedevelopedmethodwastestedusingthemethodanalysisofspikedsamplesTheresultsarepresentedinTable2
4Conclusions
Studieshaveshownthepossibilityofusingspectrophotometricanalysisforthequalitativeandquantitativedeterminationof2-methoxyphenolTheabsorptionmaximaofweredeterminedinsolutionsofethanolandacetonitrile(276nm)inasolutionofethylacetate(277nm)and01Msodiumhydroxide(289nm)Theconstructed calibration curves of thepure substance of 2-methoxyphenol hasshownagoodregressioncoefficient(Rgt099)andcanbeusedforquantitativedeterminationof2-metoxyphenolinbiologicalobjectsInthefutureitisplannedtoapplythistechniquetodetermine2-metoxyphenolinincadavericmaterial
Solvent Regressionequation Found S RSD Δх δ 2-metoxy- phenolg
ndash3 ndash6 ndash5Acetonitrile y=18294C+01130 499times10 50times10 028 2times10 044 Rsup2=09985
ndash3 ndash6 ndash595ethanol y=35131C+00269 503times10 03times10 021 7times10 137 Rsup2=09956
ndash3 ndash6 ndash501Msodium y=31196C+01101 495times10 01times10 018 1times10 028hydroxide Rsup2=09997
Table 2Results of the determination of 2-methoxyphenol (average of three measurements) in modelsolutionsbythemethodanalysisofspikedsamplestheconcentrationofintroduced2-methoxy-
ndash3phenolwas500times10 g(SndashstandarddeviationRSDndashrelativestandarddeviationΔхndashabsoluteerrorδndashrelativeerror)
Table 1Valuesofopticaldensityandwavelengthsinappropriatesolventswith2-methoxyphenol
minus3 minus1Solvent λnm εgdm cm
Acetonitrile 276 0082895ethanol 276 00853Ethylacetate 277 0083101Msodiumhydroxide 289 00744
4 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
References
[1] МельниковаИММизерницкии ЮЛКомбинированныеотхаркивающиепрепаратырастительного происхождения в педиатрическои практикеМедицинский совет 2(2018)93ndash97
[2] httpwwwthegoodscentscompanycomdatarw1032272html(accessed27stFebruary2020)
[3] RayMFaltayBHallerNACasereportanaphylacticreactiontoguaifenesinHospPract37(2009)60ndash63
[4] OkicMJohnsonTCrifasiJALongCMitchellEKSwiftonsetofcentralnervoussystemdepressionandasystolefollowinganoverdoseofguaifenesinJAnalToxicol37(2013)318ndash319
[5] ВолковCМЧерновецАНОпределениеконцентрациифеноловвгазовыхвыбросахпромышленных предприятии методом газовои хроматографии с твердофазнои микроэкстракциеи Сорбционныеихроматографическиепроцессы10(2010)723ndash728
[6] ШачневаЕЮОньковаДВСерековаСМСпособыопределенияфеноловвобъектахокружающеи среды Астраханский вестник экологического образования 4 (2013)138ndash142
[7] ГладиловичВДПодольскаяЕПВозможностипримененияметодаГХ-МС(Обзор)Научноеприборостроение4(2010)36ndash49
[8] Pena-PereiraFLavillaIBendichoCHeadspacesingle-dropmicroextractioncoupledtomicrovolumeUVndashVis spectrophotometry for iodine determinationAnal Chim Acta631(2009)223ndash228
[9] NagarajaPMurthyKCSRangappaKSGowdaNMMSpectrophotometricmethodsforthe determination of certain catecholamine derivatives in pharmaceutical preparationsTalanta46(1998)39ndash44
[10] Altunay N Development of vortex-assisted ionic liquid-dispersive microextractionmethodology for vanillin monitoring in food products using ultraviolet-visible spectro-photometryLWT93(2018)9ndash15
[11] Lupetti KO Rocha FRP Fatibello-Filho O An improved flow system for phenolsdetermination exploiting multicommutation and long pathlength spectrophotometryTalanta62(2004)463ndash467
[12] Figueiredo-Gonzalez M Cancho-Grande B Simal-Gandara J Garnacha tintorera-basedsweetwineschromaticpropertiesandglobalphenoliccompositionbymeansofUVndashVisspectrophotometryFoodChem140(2013)217ndash224
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 5
1Introduction
Thenumberofsamplesthesamplecomplexityandalsothenumberofsubstanceswhich need to be analysed simultaneously is increasing steadily ThereforepowerfulseparationanddetectionmethodsarerequiredOnewaytoachievethisisthecouplingofaseparationsystemwithmorethanonedetector[12] In recent years capillary electrophoresis (CE)was established as a potentseparation system due to its high separation efficiency and the low sampleconsumption [3] To generate more information numerous dual detectionconceptsforCEweredevelopedwhicharesummarisedelsewhere[12]Acom-binationofamperometricdetectionandmassspectrometry(MS)isaninterestingdual detection concept for CE because both detectors supply complementaryinformationForelectroactivespeciesamperometricdetectionisarobustandoneof the most sensitive detection method [4] Thus it is well suited for the
The effects of linearly assembled capillaries with various inner diameters on capillary electrophoresis
DANIELBO HMFRANK-MICHAELMATYSIK
InstituteofAnalyticalChemistryChemo-andBiosensorsFacultyofChemistryandPharmacyUniversityofRegensburgUniversitaumltsstraszlige3193053RegensburgGermanydanielboehmchemieuni-regensburgde
AbstractDuetotheincreasingneedofpowerfulanalyticalmethodsanewdualdetection concept for capillary electrophoresis (CE) with parallelamperometricdetectionandmassspectrometryshallbedevelopedFor this concept the CE flow has to be divided into two streamsutilizinga flowsplitter In thiswork theeffectsof combinedcapi-llarieswithvarious innerdiameterswerestudiedForpreliminaryinvestigationsthecapillarieswereconnectedinaserialconfigurationwithoutdeadvolumeUsingcapillaryflowinjectionanalysishyphe-natedtocontactlessconductivitydetectionitcouldbeshownthatthecouplingofidenticalcapillariesleadstoaslightdecreaseoftheflowratesWithCEhyphenatedtoUVdetection itcouldbeshownthatthecouplingofcapillarieswithdifferentinnerdiameterhasamuchstrongereffectontheelectroosmoticflowthanthecombinationwiththesameinnerdiameterFurthermorenosignificantchangeinpeakshapewasobserved
Keywordsassembledcapillariescapillaryelectrophoresiscapillaryflowinjection
analysisdualdetectionconceptnon-aqueoussystem
6 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
quantificationofsubstanceswhereasMSiswellsuitedfortheidentificationofunknown substances [3] In most dual detection concepts the detectors arearrangedinaserialconfigurationwhichisnotpossibleincaseofamperometricdetection-mass spectrometry [1] The instrumental implementation is morecomplicatedwithbothdetectorsbeingdestructiveFurthermoretheymustbedecoupledfromthehighvoltagefieldoftheCEThereforetheCEflowmustbedividedintotwostreamswithaflowsplitterAsimplifiedsketchofthepossiblenewdualdetectionconceptisshowninFig1 ForthedevelopmentofthenewdualdetectionconceptthreecapillarieswithpotentiallydifferentinnerdiametersmustbecoupledForthisreasonthedeadvolume-freecouplingofcapillarieswithdifferent innerdiameterswas investi-gatedinafirststepTokeepthesetupsimplewefocusedonthelinearcouplingofcapillariesandtheresultingeffectsNon-fragmentedcapillarieswerecomparedwithfragmentedcapillariesofthesameordifferentinnerdiametersEffectsonthe flow rate were investigated with capillary flow injection analysis (CFIA)
4hyphenatedtocontactlessconductivitydetection(C D)EffectslikechangesinthemigrationbehaviourorpeakshapesoccurringinCEwereinvestigatedwithCEhyphenatedtoUVdetection(CE-UV)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 7
Fig 1SchematicillustrationofthenewdualdetectionconceptwithparallelamperometricdetectionandmassspectrometricdetectionforCEAfterinjectionfrom(a)thesamplevialthecomponentsareseparatedbyCEnext(b)theflowsplitterdivides(c)thecapillaryintotwopartsandleadstheCEflowtowards(d)themassspectrometerand(e)theamperometricdetector
2Experimental
21Reagentsandchemicals
Thefollowingchemicalswereusedallofanalyticalgradeferrocenemethanoldecamethylferrocene(ABCRGermany)acetonitrileammoniumacetate01Msodiumhydroxidesolutionultra-purewaterprovidedbyaMilliQAdvantageA10system(MerckGermany)aceticacid(RothGermany)
22Instrumentation
221Capillaries
4Forbothexperiments(CFIA-C DandCE-UV)capillarieswithinnerdiametersof2550and75micromanouterdiameterof360micromandatotallengthof70cmwereusedTheywerepurchasedfromPolymicroTechnologies(USA)Measurementswere carried out with fragmented and non-fragmented capillaries For themeasurementswiththefragmentedcapillariestheoriginalcapillarieswerecutintotwopiecesyieldingatotalof9capillarycombinationswithlengthsof70cm(20cmfirstcapillarypieceand50cmsecondcapillarypiece)ThesecombinationsaresummarizedinTab1(section31)Atbothendsofthecapillariesabout02cmof thepolyimidecoatingwasremovedBothsidesof thecapillarypieceswerepolishedtoreceiveplanarcapillarytipsForthelinearassemblingofthecapillarypiecesMicroTightSleevesF185Xanda capillary connectorUnionAssemblyMicroTightP720fromIDEXHealthampScience(USA)wereusedPriortothefirstCEmeasurementsthecapillarieswereconditionedbyflushingthemfor10minwith01Msodiumhydroxidesolution5minwithultra-purewaterand30minwithseparationbuffer
222Capillaryflowinjectionanalysishyphenatedtocontactlessconductivitydetectionsetup
The flow rates for the fragmented and non-fragmented capillaries were4determinedwithaCFIA-C DsetupschematicallydepictedinFig2ATheflowin
thecapillarywasgravitationdrivenbyaheightdifferencebetweentheinletandoutletcarriersolutionvialTheconceptofCFIAwithgravitationdrivenflowwasfirstdescribedbyMatysiketal[5]AlaboratoryconstructedautosamplerofaCEdevicewasusedforthehydrodynamicinjectionThesamplesolutionconsistedof10mMdecamethylferrocene incarrier solution (10mMCH COONH and1M3 4
4CH COOH in acetonitrile) A high resolution C D was placed after 40 cm for3
detectionThedetectordescribedelsewhere[6]wasconstructedinthedoLagogroup(Brazil)Adoubledeterminationattwodifferentheightswasdoneforthedeterminationoftheflowrates
8 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
223Capillaryelectrophoresis-UVdetectionsetup
Fig2BshowsasketchoftheCE-UVsetupItconsistedofalab-builtCEdevicewhichwasconnectedtoahighvoltagepowersupplyfromISEG(Germany)Theseparationswerecarriedoutwithanon-fragmented50micromcapillaryandwithcapillary combinations implementing a 50micromdownstream capillary segment(25+5050+50and75+50microm)ALambda1010UV-VISdetectorfromBischoff(Germany)wasusedfordetectionat210nmThedetectorwasplacedafter40cm
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 9
Fig 2Schemeof(A)thecapillaryflowinjectionanalysis(CFIA)hyphenatedtocontactlessconduc-4tivity detection (C D) setup and (B) the capillary electrophoresis hyphenated to UV detection
4(CE-UV) setup Components of the CFIA-C D setup (a) sample (b) inlet and (c)outlet carrier4solution vial (d) fused silica capillary (e) linear capillary connector (f) C D and (g) stand
ComponentsoftheCE-UVsetup(h)sample(i)inletand(j)outletbuffervialand(k)UVdetector4therestofthecomponentswereidenticaltotheCFIA-C DsetupTheoutletbuffervialwaslowered
forthehydrodynamicinjection(j)Theenlargedview(k)depictsthecouplingoftwocapillarieswithdifferentinnerdiametersintheconnectionsidewithoutdeadvolume
Asamplesolutioncontaining1mMferrocenemethanolanddecamethylferroceneinseparationbuffer(10mMCH COONH and1MCH COOHinacetonitrile)was3 4 3
utilizedTheinjectionwasperformedhydrodynamicallybyloweringtheoutletbuffer vial by 20 cm A uniform sample plug was injected to compare bandbroadeningeffectsTheinjectionsegmenthadalengthof035cm(05ofthetotalcapillarylength)andtherespectiveinjectiontimewasdeterminedbasedontheflowratesofthecorrespondingcapillarycombinationFortheelectrophoreticseparationaseparationvoltageof25kVwasappliedandtheinletandtheoutletbuffervialswereplacedatthesameheightsothattherewasnogravityflowwhichaffectedthemigrationbehaviour
3Resultsanddiscussion
31Capillaryflowinjectionanalysishyphenatedtocontactlessconductivitydetectionexperiments
AsshowninTab1theflowratesforaheightdifferenceof20cmwerecalculated4basedontheCFIA-C DmeasurementsItwasobservablethattheflowratesare
slightlylowerforfragmentedcapillariesthanfornon-fragmentedcapillariesofthe same dimension This indicates that a flow resistance arises when twocapillaries are combined Furthermore it was observed that the flow ratedecreasesforupstreamcapillarieswithlowerinnerdiametersandviceversaTheflow rate for the combination 25+75microm could not be determineddue to theformationofairbubblesattheconnectionside
10 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Table 1Flowratesandthecorrespondingstandarddeviations(SD4measurements)ofdifferentcapillarycombinationsforaheightdifferencebetweeninletandoutletvialof20cmbymeansofcapillaryflowinjectionanalysishyphenatedtocontactlessconductivitydetection
ndash1capillary flowratenLs plusmnSDflowndash1combinationmicrom ratenLs
25 00577 0000525+25 0057 000150+25 00775 0000775+25 ndash ndash25+50 0137 000550 0865 000250+50 080 00575+50 105 00625+75 0187 000250+75 1941 000375 456 00275+75 42 01
32Capillaryelectrophoresis-UVdetectionexperiments
Theelectropherogramsforthenon-fragmented50micromcapillaryandthecapillarycombinationswithdownstream50micromcapillaryarepresentedinFig3ThetwoferrocenederivatesdecamethylferroceneandferrocenemethanolwereusedasmodelanalytesDecamethylferrocenewasonlydetectedascationicspeciesasitiseasilyoxidizedbydissolvedoxygeninsolution Forthecombination50+50micromslightlyhighermigrationtimesforthecationic(decamethylferrocene) and neutral species (ferrocenemethanol) were obser-vable compared to thenon-fragmented50micromcapillaryThis indicateda flow
4resistanceattheconnectionwhichwasalsoobservedfortheCFIA-C Dexperi-mentsinsection31Incontrasttothecombinationwiththesameinnerdiameterastrongshift in themigration times for theneuralspecieswasvisible for thecombinationswithdifferentinnerdiametersThisshowedthatthecouplinghadaneffectontheelectroosmoticflow LookingatthepeakshapeitwasfoundthatallpeaksshowednearlyGaussianshape for all combinations Furthermore there was no tailing visible Theferrocenemethanolpeaks for thecombination25+50micromand75+50micromwere
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 11
Fig 3Electropherograms of themodelmixture ferrocenemethanol (FcMeOH) and decamethyl-ferrocene(DeMeFc)measuredwithanon-fragmentedcapillary(50microm)andfragmentedcapillaries(25+5050+50and75+50microm)Experimentalparameters1mMFcMeOHandDeMeFcinseparationbuffer(10mMCH COONH and1MCH COOHinacetonitrile)injectionsegment035cmseparation3 4 3
voltage25kVcapillarylength70cm(40cmtothedetectorfragmentedcapillaries20cmfirstpartand50cmsecondpart)UVdetectionat210nm
slightlybroaderthanthepeaksforthenon-fragmented50micromcapillaryorforthe50+50 microm combination But this probably results from longitudinal diffusioneffectsduetothelongerresidencetimes
4Conclusions
FromtheCFIAmeasurementsitcanbeconcludedthattherewasamechanicaldisturbanceoftheflowduetothecouplingFurthermoreitcouldbeshownthatCEmeasurementswithlinearcoupledcapillariesofvariousinnerdiameterwerepossible Unlike to the capillary combinationwith the same inner diameter astrongshiftoftheelectroosmoticflowtowardshighermigrationtimeswasfoundfor capillary combinations with different inner diameters In this work thecapillaries were coupled with almost no dead volume which resulted in nosignificantchangesofthepeakshapeorpeaktailingContrarytoexpectationsthecouplingofcapillarieswithvariousinnerdiametershadnosignificantimpactonthepeakwidth TheknowledgegainedfromthelinearcouplingofcapillariesisagoodbasisforthedevelopmentofthenewdualdetectionconceptInanextstepthreecapillariesshouldbecoupledwitheachother
Acknowledgments
WethanktheGermanResearchFoundation(DFG)forfinancialsupport
References
[1] OpekarFS tulıkKSomeimportantcombinationsofdetectiontechniquesforelectrophoresisincapillariesandonchipswithemphasisonelectrochemicalprinciplesElectrophoresis32(2011)795ndash810
[2] BeutnerAHerlTMatysikF-MSelectivityenhancement incapillaryelectrophoresisbymeans of two-dimensional separation or dual detection conceptsAnal Chim Acta1057(2018)18ndash35
4[3] BeutnerACunhaRRRichterEMMatysikF-MCombiningC DandMSasadualdetectionapproachforcapillaryelectrophoresisElectrophoresis37(2016)931ndash935
[4] MatysikF-MEnd-columnelectrochemicaldetectionforcapillaryelectrophoresisElectro-analysis12(2000)1349ndash1355
[5] MatysikF-MWernerGTracemetaldeterminationintearsbyanodicstrippingvoltammetryinacapillaryflowinjectionsystemAnalyst118(1993)1523ndash1526
[6] FranciscoKJMdoLagoCLAcompactandhigh-resolutionversionofacapacitivelycoupledcontactlessconductivitydetectorElectrophoresis30(2009)3458ndash3464
12 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Thetermldquovitaminsrdquodescribestheheterogeneousgroupofchemicalcompoundswhich are important for the proper functioning of the human body [1 2] Bydefinitionvitaminsarenotsynthesizedbythehumanbodyorthesynthesizedamount is not sufficient to cover the demand That is why they have to besupplementedfromtheexternalsourceslikefoodproductsorpharmaceuticals[1ndash3]Basedontheirsolubilityvitaminsaredividedintowater-soluble(B-groupandvitaminC)andfat-solublevitamins(ADEandK)[3]VitaminC(ascorbicacid) is the most important antioxidant and participates in the activation ofenzymes[4]VitaminB1(thiamine)facilitateswoundhealingandiscrucialforthehumannervoussystem[15]VitaminB2(riboflavin)participatesintheenzy-maticreactionsandthebiotransformationofglucoseandaminoacids[6]Vitamin
+B3(niacin)isthemainconstituentoftheNAD andNADHcoenzymeswhichareresponsible for the transfer of electrons and hydrogen ions in the cellularrespiration [1ndash3] Vitamin B6 possesses six related structures (vitamers) thateasilyinterconvertThemostimportantoneispyridoxinewhichhelpstopreventtongue inflammation and microcytic anemia [2] For the production of well-functioningredbloodcellsandtheavoidanceofmegaloblasticanemiaandfetusdefectsvitaminB9(folicacid)hastobesupplementedintheproperamount[12]
Voltammetric determination of vitamins
RADOSŁAWPORADABOGUSŁAWBAS
DepartmentofAnalyticalChemistryFacultyofMaterialsScienceandCeramicsAGHUniversityofScienceandTechnologyMickiewicza3030-059KrakoacutewPolandrporadaaghedupl
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 13
AbstractVitaminsbelongtothegroupofchemicalcompoundsessentialfortheproperfunctioningofthebodySinceboththeirdeficiencyandexcessmay result in serious health problems the amount of vitaminssupplementedinthedietaswellasvitamincontentintheirsourceshavetobestrictlycontrolledInthisworkthepossibilityofsimulta-neous determination of vitamins by means of differential pulseadsorptive stripping voltammetry is discussed The research hasshownthatthedeterminationofsingularvitaminatthemicromolarlevel isrelativelyfastandstraightforwardandthemostimportanthindranceisrelatedtotheanalyteadsorptionattheelectrodesurfaceInthecaseofvitaminswithdifferentredoxpotentialstheycanbeanalyzedsimultaneouslywithouttheneedtoreachfortheadvancedmethodsforsignalprocessing
Keywordsmercuryelectrodevitaminsvoltammetry
VitaminK3(menadione)doesnotoccurnaturallybutitservesasaprecursorforthesynthesisofotherK-groupvitaminsandcanbeusedtotreathypoprothrom-binemiaVitaminK3ispartiallysolubleinwater[17] All of the vitamins are electrochemically active [3] therefore the electro-chemicalmethodscanbeappliedforthedeterminationofvitamincontentinfoodproductspharmaceuticalsandbodyfluidsVoltammetrictechniquesarecharac-terizedbyhighsensitivityandselectivityandtheydonotrequiretime-consumingsamplepreparationMoreovertheelectrochemicalinstrumentationisrelativelyinexpensiveandcanbeappliedintheon-siteconditionsfortheonlineanalyseseginqualitycontrolMostofthepapersreporttheconstructiondevelopmentand characterization of a novel modified working electrodes for quantitativeanalyses of a singular vitamin in the variety of matrices Unfortunately only alimitednumberofpapersdescribethesimultaneousdeterminationofmultiplevitaminsinasinglerun[23] The preliminary research devoted to the simultaneous determination ofB-groupCandK3vitaminswiththeuseofthecontrolledgrowthmercurydropworking electrode in aqueous solutions is presented in this work Particularattentionhasbeenpaidtotheredoxpotentialsofthestudiedcompoundstheshape of the calibration curves and adsorption phenomena As an attempt toovercomethelattertheneutralsurfactantTritonX-100hasbeenintroducedintothestudiedsystem
2Experimental
21Reagentsandchemicals
TheappliedreagentswereofanalyticalgradeandusedassuppliedPhosphateand McIlvaine buffers were obtained by mixing the appropriate amount of
ndash1 ndash1 ndash102molL Na HPO with 02molL NaH PO and 01molL citric acid2 4 2 4
respectively (all reagents purchased from Avantor Performance MaterialsPoland)ThestandardsolutionsofvitaminB1B2B3B9andCwerepreparedbydissolving the corresponding amount of the standard (all Sigma-Aldrich) in
ndash1distilledwater In thecaseofB2andB9 theadditionof02molL NaOHwasinevitabletoobtainaclearsolutionVitaminK3standard(Sigma-Aldrich)was
ndash1dissolvedinthemixtureofmethanoland1molL phosphatebuffer(pH=82)(vv=25)LaboratorygradeTritonX-100(Sigma-Aldrich)wasusedinthestudyoftheadsorptionprocesses22Instrumentation
All the electrochemical measurements were conducted in the three-electrodesystem composed of the Pt auxiliary electrode double-junction silversilverchloride reference electrode and controlled-growth mercury drop electrode
14 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
actingastheworkingelectrodeTheusedmeasurementequipmentinvolvedtheM164electrodestandandM161multipurposeelectrochemicalanalyzer(bothmtm-anko Krakow) To control the buffer pH-value the SevenCompact S210laboratorypH-meter(MettlerToledoSwitzerland)wasemployed
23Voltammetricmeasurements
Throughout the course of the study differential pulse adsorptive strippingvoltammetry has been used for recording the current-potential curves BothcathodicandanodicscanswererecordedinapotentialrangeadjustedforthestudiedvitaminsTheinfluenceofvariousmeasurementconditionsontheregis-teredsignalshasbeeninvestigatedFinallythepossibilityofthesimultaneousdeterminationofmultiplevitaminsinonescanhasbeenverified
3Resultsanddiscussion
Figure 1 depicts the redox potentials of the studied vitamins in the aqueoussolutions for the mercury electrode The only exception is vitamin B6 whoseredox potential is higher than the potential of mercury oxidation (ca +02 V)Therefore the given value refers to the glassy carbon electrode The redoxpotentialvalueofstudiedvitamins isnotasingularvaluebut it fallswithinacertainrangeThiscanbeascribedtothedependenceofredoxpotentialonthesolutionpHvaluewhichresultsfromtheparticipationofprotonsintheredoxreactions of vitamins Moreover the potentials for individual vitamins arerelatively well separated indicating that the simultaneous determination ofseveralvitaminsintheonerunmaybepossibleTheonlyencounteredproblems
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 15
Fig 1Redoxpotentialsofchosenvitamins
regardthevitaminsB1B3andB12whoseredoxpotentialsoscillatebetweenndash15andndash17VandtheK-groupvitamins(K1K2K3)inwhichonlythecommonstructuralmotifndashthequinoneringndashiselectrochemicallyactiveresultinginthevalueoftheredoxpotentialofcandash02V[8] AtypicalcalibrationcurveisdepictedinFig2Abasedonthedifferentialpulsevoltammograms of the vitamin B9 reduction in the concentration range from
ndash1blank to 01 mg L recorded in the McIlvaine buffer of pH = 52 using thecontrolledgrowthmercurydropelectrodeworkingelectrodeTherelationshipbetweenthepeakcurrentandtheconcentrationofVB9 is linear in thewholetestedrange(r=09999)Basedontheparametersoftheregressioncurvethelimit of detection and limit of quantitation were estimated to 42 and
ndash1142nmolL respectively Similar dependencies and figures of merit can beobtainedforothervitamins QuitedifferentbehaviorwasobservedinthecaseofthevitaminK3forwhichthe increase in current was not strictly proportional to the increase in theconcentrationandthecalibrationplotresemblesanS-shapecurve(Fig2B)ThelatterindicatesthatvitaminK3adsorbsatthesurfaceoftheworkingelectrodeHoweverasnopre-orpost-peakwereobservedwearedealingherewiththeweakadsorption[9]AdsorptionalsoplaysasignificantroleinthecaseofvitaminB2forwhichboththepre-andpost-peakswereobservedindicatingitsstrongaffinitytothemercuryelectrodes To overcome this issue attempts with Triton X-100 were performed TritonX-100isaneutralsurfactant thateasilyadsorbsat thesurfaceof themercury
16 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2Differentialpulseadsorptivestrippingvoltammogramsof(A)vitaminB9and(B)vitaminK3ndash1reduction recorded in the concentration range from blank to 01 and from blank to 04 mg L
respectivelyInsetcorrespondingcalibrationcurvesSupportingelectrolyte(A)McIlvainebufferndash1(pH=52)(B)04molL phosphatebuffer(pH=82)
electrode partially blocking its surface Doing so it prevents the undesiredadsorption of other molecules and thus allows to obtain a linear relationshipbetweenthepeakcurrentandthevitaminconcentration(Fig3)UnfortunatelyduetotheblockingoftheelectrodesurfacetheslopesofthecalibrationlinesaresmallerincomparisontotheonesobtainedintheabsenceofanysurfactantsThismeans that the sensitivity defined as the increase in current caused by a unitincrease in concentration and the resolution understood as the possibility todistinguishsmallvariationinconcentrationarecorrespondinglydecreased Figure3alsopresentsthepossibilitytodeterminemultiplevitaminsinasinglerunChosenvitaminshavewell-separatedpotentialsandtheydonotinterferewitheachotherthereforenoadvancedmultivariatecalibrationstrategiesareneededTheproblemsinthesimultaneousanalysisincludevarioussensitivitieswith respect to the studied analytes and differences in the influence of themeasurementconditionsontherecordedsignalsDuetothattheexperimentalconditionswillneverensurethehighestpossiblesignalvaluesforallanalyzedcompounds
4Conclusions
Differential pulse voltammetry in conjunction with the controlled growthmercurydropelectrodeisaperfecttoolforquantitativeanalysesofvitaminsTheadsorptionofvitaminB2andK3canbepreventedbytheadditionoftheneutralsurfactantTritonX-100whichselectivelyblockstheworkingelectrodesurface
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 17
Fig 3Cathodic voltammograms for the simulatanous determination of vitamin B2 B3 and K3Depictedintheinsetsarethevoltammogramsafterbackgroundsubtractionwiththecorresponding
ndash1callibrationplotsSupportingelectrolye04molL phosphatebuffer(pH=82)with40ppmTritonX-100AccumulationconditionsE =ndash005Vt =20sacc acc
The proposed methodology allows for the simultaneous determination ofmicromolaramountsofvitaminB2B3andK3Suchaprocedurewillhelptoreduce the time and costs of analyses of multivitamin formulations and foodproducts
Acknowledgments
RPhasbeenpartlysupportedbytheEUProjectPOWR0302-00-00-I00416
References
[1] Combs GF JrTheVitaminsFundamentalAspects inNutritionandHealth 3rd ed IthacaElsevierAcademicPress2008
[2] Lovander MD Lyon JD Parr DL Wang J Parke B Leddy J Review Electrochemicalpropertiesof13vitaminsAcriticalreviewandassessmentJElectrochmSoc165(2018)G18ndashG49
[3] Brunetti B Recent advances in electroanalysis of vitamins Electroanalysis 28 (2016)1930ndash1942
[4] BrubacherGMuller-MulotWSouthgateDATMethods forDeterminationofVitamins inFoodNewYorkElsevier1985
[5] Szpikowska-Sroka B A simple and sensitive analytical method for the determination ofthiamineinpharmaceuticalpreparationsJAnalChem68(2013)218ndash222
[6] PetteysBJFrankELRapiddeterminationofvitaminB (riboflavin)inplasmabyHPLCClin2
ChimActa412(2011)38ndash43[7] ZhangZXuJWenYZhangJDingWTheelectro-syntesizedimprintedPEDOTfilmasa
simple voltammetric sensor for highly sensitive and selective detection of vitamin K in3
poultrydrugsamplesSynthMet230(2017)79ndash88[8] JedlinskaKStrusMBasBAnewelectrochemicalsensorwiththeRefreshableSilverLiquid
Amalgam Film multi-Electrode for sensitive voltammetric determination of vitamin K2(menaquinone)ElectrochimActa265(2018)355ndash363
[9] SouthamptonElectrochemistryGroupInstrumentalMethodsinElectrochemistryChichesterHorwood1985
18 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Carbonelectrodesarecommonlyappliedtosensitiveelectrochemicaldetectionofneurotransmittersegdopamine (nor)epinephrineandserotonin in-vivoandin-vitro[12]Neverthelessadsorptionofhigh-molecularweightbiomoleculesinthe matrix on a sensing electrode which then hinders the electron transferreactionofneurotransmitterswillresultinbiofoulingofelectrodesThisremainsachallengingproblemasbiofoulingwillcompromiseelectrochemicalmeasure-mentsThusseveralstrategies foraddressingbiofoulinghavepreviouslybeenreported[34] Thisworkreportsonaneffectiveapproachforminimisingbiofoulingbasedonthehypothesisthatahydrophobicelectrodesurfacewillrepelagainstadsorptionof amphiphilic biomolecules Briefly structurally small conical-tip electrodes
Dopamine detection at antifouling conical-tip carbon electrodes
a a a aSIMONABALUCHOVA JANKLOUDA JIR IBAREK KAROLINASCHWARZOVA -PECKOVA bDANNYKYWONG
a UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova812800PragueCzechRepublicsimonabaluchovanaturcunicz
b DepartmentofMolecularSciencesMacquarieUniversitySydneyNSW2109Australia
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 19
AbstractA significant achievement in this work is the development ofantifoulingconical-tipcarbonelectrodes(~27micromtipdiameterand~165micromaxiallength)suitablefordetectionoftheneurotransmitterdopamine in-vivo These electrodes were hydrogenated using adiphenylsilanereductionmethodtoyieldahydrophobicsurfacetodeteradsorptionofamphiphilicbiomoleculesInitiallyhydrogenatedcarbonelectrodeswereelectrochemicallycharacterisedusingseveralredoxmarkersThedegreeofantifoulingwasthenassessedbythevoltammetricsignalchangeofdopamineattheseelectrodesbeforeand after being incubated in a fouling solution containing bovineserumalbumincytochromeC(bothareproteins)andcaproicacid(alipid) In our work we have obtained only a 69 (standarddeviation35N=40)decreaseindopaminesignalsatthehydro-genated carbon electrodes These results strongly support thediphenylsilanereductionstrategyforthedevelopmentofantifoulingbiosensorsfordopaminedetectioninbiologicalmatrices
Keywordsantifoulingelectrodesdiphenylsilanereduction
methodhydrogenatedconical-tip
carbonelectrodesvoltammetricdopamine
detection
(denoted as CTEs) are fabricated by thermally pyrolysing acetylene gas in anitrogenatmospheretodepositcarbonatthetipandontheshankofpulledquartzcapillaries [5] Spectroscopic studies confirmed that the electrode surface
2 3consistsofsp -likegraphiticcarbonandsp -hybridiseddiamond-likecarbon[6]In addition there is also a range of carbon-oxygen functionalities includingcarbonylquinonecarboxylphenolsalcoholsandethergroupsontheelectrodesurface[6]whichcaninteractwithspectatorbiomoleculesthroughdipole-dipoleorion-dipoleinteractionleadingtotheirirreversibleadsorptionontheelectrodesurface[7]HoweverbysubjectingthesecarbonelectrodestosilanereductionCndashObondsareconvertedtoCndashHbondsandphenolicgroupsaretransformedtosiloxane dendrimers [6] to yield a more hydrophobic carbon surface that isexpectedtobesimilarlylesssusceptibletobiofoulingcomparedtoboron-dopeddiamondelectrodes[78] In this work we will present a methodology involving diphenylsilanereduction to fabricate physically small hydrogenated conical-tip carbonelectrodes(denotedasHCTEs)withanti-foulingcapabilityBothCTEsandHCTEswere electrochemically characterised using several redox probes to elucidatetheirsurfacepropertiesbeforeevaluatingtheirresistancetobiofoulingduringdopaminedetectionin-vitro
2Experimental
21Reagentsandchemicals
Analyticalgradereagents(Sigma-AldrichAustralia)including4-methylcatecholhexaammineruthenium(III) chloride potassium hexacyanoferrate(III) dop-amine hydrochloride sodium phosphate dibasic citric acid perchloric acidpotassiumchloride sodiumhydroxide anhydrousdichloromethanediphenyl-silanetris-(pentafluorophenyl)boraneandgraphitepowderwereusedas-recei-ved Ultra-high purity gases acetylene and nitrogenwere obtained from BOCGases (Australia) All aqueous solutionswere preparedwith deionisedwater(MilliporeMiliplusQsystemUSA)witharesistivityof182MΩcm
22Instrumentation
Chronoamperometric and voltammetric experiments were carried out usingalow-currentpicostateDAQoperatedbyanEChemversion212softwareviaanE-corderinterface(eDAQPtyLtdAustralia)Athree-electrodeset-upinvolving
minus1eitheraCTEorHCTEasaworkingelectrodeaAgAgCl(3molL KCl)referenceelectrode (Bioanalytical SystemsUSA)andaplatinumwire counterelectrode
minus1(CypressSystemsUSA)wasusedCyclicvoltammetryatascanrateof100mVs anddifferential pulse voltammetry (pulse height +25mV pulsewidth 50ms
minus1sampling time 20 ms and scan rate 20mVs ) were used in this work All
20 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
electroanalyticalexperimentswereperformedinanaluminiumFaradaycageatanambienttemperature(23plusmn1degC)
23Preparationofhydrogenatedconical-tipcarbonelectrodes
Asreportedpreviously[5]structurallysmallCTEswerefabricatedbythermallypyrolysing C H (a pressure of 50 kPa) in a pulled quartz capillary (Sutter2 2
minus1InstrumentUSA)housedinaN atmosphere(counterflowof60mLmin )Prior2
to hydrogenation the catalyst tris(pentafluorophenyl) borane (100 mg) wasdissolvedinanhydrousCH Cl (50mL)bystirringfor5minbeforethehydroge-2 2
natingagentdiphenylsilane(25μL)wasaddedCTEswerethenplacedinthereagentmixturefor2hThepreparedHCTEsweredriedovernightbeforeuse
24Biofoulingexperiments
A laboratory synthetic fouling solution consisting of 4 (wv) bovine serumalbumin001(wv)cytochromeC(bothareproteins)and10(vv)caproicacid(alipid)waspreparedbyhomogenisingtheminapH=74citrate-phosphate
minus1buffer(01molL )AllfoulingcompoundswereacquiredfromSigmaAldrichAustralia
3Resultsanddiscussion
31Electrochemicalcharacterisation
minus1InthisworkallCTEswerecharacterisedbycyclicvoltammetryof10mmolL 3+ minus1[Ru(NH ) ] in10molL KClAsdisplayedinFig1(A)onlyCTEsthatshow3 6
asigmoidal-shapedvoltammogramwithasmallchargingcurrentwereemployedinfurtherexperimentsUsingchronoamperometry[5]ameantipdiameterof27μm(standarddeviation(SD)28μmN=142)ameanaxiallengthof165μm(SD=114μmN=142)wereestimatedfortheseCTEs TocomparesurfacecharacteristicsofbothCTEsandHCTEs cyclicvoltam-
minus1 3+2+ minus1 minus1metryof (1)10mmolL [Ru(NH ) ] in10molL KCl (2)10mmolL 3 63minus4minus minus1 minus1[Fe(CN) ] in 10 mol L KCl and (3) 10 mmol L 4-methylcatechol in6minus101molL HClO wasconductedatthesameelectrodesbeforeandafterhydroge-4
nation The results obtained are shown in Fig 1(A-C)We observed a ~20(SD=5N=10)decreaseinthelimitingcurrentofallthreeredoxmarkersafterdiphenylsilane reduction most likely attributable to the hindrance to theirelectron transfer reactions by the phenylsiloxane group formed on HCTEs
3minus4minusMoreover as an inner-sphere redox probe both [Fe(CN) ] and 4-methyl-6
catechol reactionsare sensitive to thepresenceofoxygen functionalitiesonacarbonsurface [7]Accordingly theconversionof these functionalities toCndashHbondsbydiphenylsilanereductionwasexpectedtoyieldmoresluggishelectron
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 21
transfer kinetics at HCTEs as supported by a negative potential shift (from3minus4minus+75mVtominus10mV)inthecyclicvoltammogramof[Fe(CN) ] andapositive6
potentialshift(from+580mVto+675mV)inthecorrespondingcyclicvoltammo-2gram of 4-methylcatechol In addition the conversion of sp -carbon to
3sp -diamond-likecarbon[6] isalsoexpectedtoreducetheconductivityof thecarbonelectrodesurface
32Dopaminedetectionduringbiofoulingexperiments
minus1Theelectrochemicalbehaviourof1mmolL dopamine inapH=74citrate-minus1phosphate buffer (01 mol L ) at CTEs and HCTEs was studied by cyclic
voltammetryTheresultsobtainedareshown inFig1(D)Acomparable12decrease(SD=6N=10)inthedopamineoxidationlimitingcurrenttothatof4-methylcatechol was observed A positive potential shift from +285 mV to+305mVinthevoltammogramsisalsoaccountedforasdescribedabove
22 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
minus1 3+ minus1 minus1Fig 1Cyclicvoltammetryof(A)10mmolL [Ru(NH ) ] in10molL KCl (B)10mmolL 3 63minus4minus minus1 minus1 minus1[Fe(CN) ] in10molL KCland(C)10mmolL 4-methylcatechol in01molL HClO at3 4
minus1(a)aCTEand(b)aHCTE(D)10mmolL dopamineinapH=74citrate-phosphatebufferrecordedminus1at(a)aCTEandaHCTE(b)beforeand(c)afterbiofoulingScanrate100mVs
Next HCTEs were incubated in a synthetic fouling solution containing4(wv)bovineserumalbumin001(wv)cytochromeC(bothareproteins)and 10 (vv) caproic acid (a lipid) for 30 min Cyclic voltammetry of
minus110mmolL dopamineattheseHCTEswasthenconductedinapH=74citrate-phosphatebuffertoevaluatetheirantifoulingpropertyNotablyaconsiderable515(SD=183N=6)decreaseindopaminesignalwasobservedatCTEsIncontrastonlyacorresponding69decrease(SD=35N=40)wasestimatedatHCTEsasshowninFig1(D)Clearlythisrepresentsamajorimprovementinthe antifouling capability of HCTEs obtained using diphenylsilane reductioncomparedtoCTEsandotherpreviouslytestedhydrogenatingagents[9]includingn-butylsilane (35decrease) triethylsilane (23decrease) andphenylsilane(18decrease)Thereforethisworkhassuccessfullydemonstratedtheeffecti-venessofdiphenylsilanereductionmethodindevelopingantifoulingelectrodesfordopaminedetection
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 23
Smallconical-tipelectrode
As-prepared Hydrogenated
minus1LinearrangemicromolL 1ndash10 1ndash20minus2 minus2Intercept10 pAmicrom 108plusmn004 273plusmn011
minus3 minus2 minus1Slope10 pALmicrom micromol 128plusmn007 443plusmn012R 0993 0998
minus1LimitofdetectionmicromolL 100 077
Table 1Analyticalparametersofconcentrationdependencesofdopamineobtainedbydifferentialpulse
minus1voltammetryinapH=74citrate-phosphtebuffer(01molL )Allquoteduncertaintiesrepresentthe95confidenceintervalandthecorrelationcoefficient(R)wasfoundtobestatisticallysigni-ficantatthe95usingStudentrsquost-test
Fig 2DifferentialpulsevoltammetryofdopamineataHCTEinapH=74citrate-phosphatebufferminus1 minus1(01molL )atconcentrations(a)1(b)2(c)4(d)6(e)8(f)10and(g)20μmolL
WehavealsostudiedtheconcentrationdependenceofdopamineinapH=74citrate-phosphatebufferbydifferentialpulsevoltammetryAtypicalcalibrationplotobtainedisshowninFig2Theanalyticalparametersandestimatedlimitsofdetection are summarised inTable 1 These results show thatHCTEs outper-formed CTEs because they exhibit a ~35times higher sensitivity a 23 lowerdetectionlimitandawiderlinearrange
4Conclusions
InthisstudyphysicallysmallHCTEswithanti-foulingcharacteristicsachievedbyhydrogenationusingdiphenylsilanereductionweresuccessfullyfabricatedandelectrochemically characterised using several redox probes Next dopaminedetectionwasperformedbeforeandafterincubationofelectrodesinasyntheticfoulingsolutioncontainingahighconcentrationofbiomoleculesOnlyalow69(SD=35)decreaseindopaminelimitingcurrentwasachievedatHCTEsobtain-edbydiphenylsilanereductionindicatingtheirsignificantlylesssusceptibilitytobiofoulingthanCTEsThesepromisingresultsindicatethatantifoulingHCTEswillpotentiallybenefitthedevelopmentofbiosensorsfordopaminedetectionin-vivoinbiologicalmedia
Acknowledgments
ThisresearchwasperformedwithintheframeworkofSpecificUniversityResearch(SVV260560)FinancialsupportsprovidedbytheGrantAgencyofCharlesUniversity(project390119)andbytheCzechScienceFoundation(project20-03187S)aregratefullyacknowledgedSBandJKalsothankthe Mobility Fund of Charles University and Hlavkova nadace for providing funding for theirresearchinternshipsatMacquarieUniversitySydneyAustralia
References[1] BaranwalAChandraPClinicalimplicationsandelectrochemicalbiosensingofmonoamine
neurotransmittersinbodyfluidsinvitroinvivoandexvivomodelsBiosensBioelectron121(2018)137ndash152
[2] CaoQPuthongkhamPJillVentonBReviewnewinsightsintooptimizingchemicaland3Dsurface structuresof carbonelectrodes forneurotransmitterdetectionAnalMethods11(2019)247ndash261
[3] LinP-HLinB-RAntifoulingstrategiesinadvancedelectrochemicalsensorsandbiosensorsAnalyst145(2020)1110ndash1120
[4] HanssenBLSirajSWongDKYRecentStrategiestoMinimiseFoulinginElectrochemicalDetectionSystemsRevAnalChem35(2016)1ndash28
[5] McNallyMWongDKYAnin-vivoprobebasedonmechanicallystrongbutstructurallysmallcarbonelectrodeswithanappreciablesurfaceareaAnalChem73(2001)4793ndash4800
[6] SirajSMcRaeCRWongDKYEffectiveactivationofphysicallysmallcarbonelectrodesbyn-butylsilanereductionElectrochemCommun64(2016)35ndash41
[7] ParkJShowYQuaiserovaVGalliganJFinkGDSwainGMDiamondmicroelectrodesforuseinbiologicalenvironmentsJElectroanalChem583(2005)56ndash68
[8] ShinDTrykDAFujishimaAMerkociAWang JResistance to surfactantandproteinfoulingeffectsatconductingdiamondelectrodesElectroanalysis17(2005)305ndash311
[9] Roshni RAnAntifouling Structurally Small Carbon Electrode forDetectionof theNeuro-transmitterDopaminePhDThesisMacquarieUniversitySydney2019
24 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Theanalyticalperformancesofenzymaticbiosensorsarestronglyaffectedbytheenzyme immobilization process There is no universal technique for enzymesattachmentThereforespecialattentionshouldbepaid to theselectionof theappropriatesupportandthedevelopmentoftheoptimalbindingstrategyinordertoensure thebestcharacteristicsof immobilizedenzymeDespiteavarietyofpreviouslyreportedcovalentimmobilizationmethodsfordifferentenzymesthepresentedprocedurescanbehardlycomparedtofindtheoptimalonesbecauseofdifferentanalyticalmethodsandexperimentalconditionsusedUptodatethere
A comparative study of covalent glucose oxidase and laccase immobilization techniques at powdered supports for biosensors fabrication
ab a bSOFIIATVORYNSKA JIR IBAREK BOHDANJOSYPCUK
a UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova2030812843Prague2CzechRepublicsofiiatvorynskajh-instcascz
b JHeyrovskyacuteInstituteofPhysicalChemistryoftheCzechAcademyofSciences Dolejškova318223Prague8CzechRepublic
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 25
AbstractInordertodeveloptheoptimalstrategyandtodeepentheknowledgeinthefieldofenzymeimmobilizationthreedifferenttechniquesofcovalentbindingfortwoenzymes(glucoseoxidaseandlaccase)atpowdered surfaces were compared Immobilization protocol wasoptimized by changing supports (twomesoporous silica powders(SBAminus15 MCMminus41) and a cellulose powder) the functionalizedgroupsintroducedatsupportsurfaces(minusNH andminusCOOH)andthe2
methodsofactivation(glutaraldehydeandcarbodiimide)Aminoandcarboxyl functionalized mesoporous silica and cellulose powderswerepreparedbysilanizationusing(3-aminopropyl)triethoxysilaneandcarboxyethylsilanetriolrespectivelyItwasfoundthatcouplingof both enzymes by their ndashNH groups through glutaraldehyde to2
ndashNH functionalized supports in particular SBA15minusNH and2 2
celluloseminusNH forglucoseoxidaseMCM41minusNH forlaccaseshowed2 2
thehighestactivityandthebeststability
Keywordsbiosensorcovalentimmobilizationenzymaticreactorglucoseoxidaselaccase
is still a lack of the comparative systematic studies focusing on the enzymesimmobilizationonthevarioussupportsusingdifferenttechniques The aim of this work is the systematic comparative study of the differenttechniques for covalent coupling of the enzymeswhich ensures not only thedevelopmentoftheoptimalimmobilizationstrategyfortheselectedenzymesbutalsoenablestofindoutsometendenciesinenzymeattachmentprocessgenerallyThusthisworkisfocusedonadetailedanalysisoftheeffectofthekindofsupportits anchor groups and the activation methods on activity and stability ofimmobilizedenzymesTwoenzymeswithdifferentnature(glucoseoxidase(GOx)andlaccase(Lac))werechosenasthetestingbioreceptors
2Experimental
21Reagentsandchemicals
AllchemicalswereofpaorbettergradeGlucoseoxidasefromAspergillusnigerminus1(GOxEC11341452Umg )laccasefromTrametesversicolor(LacEC11032
minus1129Umg )D-(+)-glucosedopamineglutaraldehyde(GAgradeII25aqueoussolution) N-(3-dimethylaminopropyl)-Nʹ-ethylcarbodiimide hydrochloride(EDC ge980) N-hydroxysuccinimide (NHS ge970) (3-aminopropyl)-triethoxysilane (APTES) mesoporous silica powder SBAminus15 (particle size
2 minus12ndash6μmporesizeasymp7nmsurfaceareaasymp600m g )mesoporoussilicapowder2 minus1MCMminus41 (pore size 21ndash27 nm surface area asymp 1000 m g ) cellulose (Cell
microcrystalline powder particle size 20 microm) were purchased from SigmaAldrichCarboxyethylsilanetriol(CEST25aqueoussolution)waspurchased
regfromabcr (Germany)
22Instrumentation
Amperometric measurements were carried out at room temperature usingcomputer-controlled electrochemical stand (Polaro-Sensors Czech Republic)withMultiElchemv31software(JHeyrovskyInstituteofPhysicalChemistryoftheCAS)Flowinjectionanalysis(FIA)withthethree-electrodelaboratory-madeflow-through cellwas usedworking electrode minus tubular detector of polishedsilversolidamalgam(TD-p-AgSAlaboratory-madeinnerdiameter05mmtheamalgamtube length60mm) referenceelectrodeminusaminiaturizedsaturatedcalomelelectrodebasedonsilverpasteamalgam[1](laboratory-madeithasthesamepotential as classical saturated calomel electrode) auxiliary electrodeminusplatinum wire (diameter 10 mm length 10 mm) The system for FIA withelectrochemicaldetectioncomprisedofalinearsyringepumpa2-position6-port
regsampleinjectorvalveaninjectionlooplaboratory-madeofTeflon (PTFE)tubing(100μL) a solenoid operatedmicro-pumpan enzymatic reactor and a flow-through cell for TD The enzymatic reactor consists of a tube filled by theenzymaticpowder
26 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
3Resultsanddiscussion
Basedonthedatareportedintheliterature[23]andontheresultsofourpreviousworks[4ndash8]forthisstudyCellandmesoporoussilicapowders(namelySBAminus15andMCMminus41)havebeenselectedas thepotentialpromisingsupports for thecovalent enzyme immobilizationBecauseof thehigh contentof surficial ndashOHgroupswhich are capable of chemical reactions these supports can be easilyfunctionalizedThewell-knownandfrequentlyutilizedtechniqueofsilanizationhasbeenusedtomodifythesurfacesofSBA15MCMminus41andCellbythedesiredfunctionalizedgroupsAminosilaneAPTESwasappliedtoformminusNH groupson2
thematrixsurfaceswhereascarboxylsilaneCESTwasusedtointroducendashCOOHgroups Generallytheprocedureofthecovalentimmobilizationofenzyme(eitherLacorGOx)onthefunctionalizedsupportconsistsofthreestepsI Synthesisofthefunctionalizedsupportwhichmeansthemodificationofthematrix(MCMminus41SBAminus15andCell)withsuitableanchoredgroups(minusNH or2
minusCOOH)II Activationstepofthefunctionalizedsupportwithspecificactivatingagents(glutaraldehydeorEDCNHS)tomakeitreactivetowardsenzyme
IIIEnzyme(LacorGOx)couplingtotheactivatedsupport
To investigate the effect of support its surface functionalizedgroups and themethodsofactivationontheefficiencyofthecovalentenzymeimmobilizationthreedifferentstrategies(ABandC)forLacandGOxattachmenthavebeenusedThedetailsoftheusedtechniquesandthedenotationsofthepreparedenzymaticpowdersaresummarizedinTable1(nextpage)ToexaminetheefficiencyofLacandGOximmobilizationtheenzymaticreactors(filledbytheenzymaticpowderspreparedwithdifferenttechniques)coupledwithTDwereusedforamperometricdetermination of dopamine and glucose respectively in flow systems Theprincipleofglucosedetection isbasedonamperometricmeasurementsof theenzymatically consumed oxygen whereas dopamine was detected by thereductionoftheenzymaticallyoxidiseddopamine Asdepicted inFig1 thebiosensors responsesare stronglyaffectedby thestrategyusedforLacorGOximmobilizationAsshowntheresponsesofLacandGOx biosensors decrease in the order strategy A gt strategy B gt strategy Cirrespectiveofthetypeofsupport It isclearlyseenthataminofunctionalizedsupports(SBA15minusNH MCM41minusNH andCellminusNH )providehigheractivitiesof2 2 2
the immobilized Lac andGOx than these supports functionalized by carboxylgroups(SBA15minusCOOHMCM41minusCOOHandCellminusCOOH)BycomparingactivitiesofimmobilizedenzymesusingstrategiesAandBtheinfluenceoftheactivationagenthasbeenevaluatedThebestresultsforbothenzymeswereobtainedforndashNH functionalizedsupportsactivatedbyGAItcouldbeexplainedbythefact2
thatGAcontrary tocarbodiimidewithnonemolecularspaceprovidesa long
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 27
spacerarmensuringminimalsterichindrancesforenzymesbindingItcanbeconcludedthatthecovalentimmobilizationofbothenzymesbytheirndashNH groups2
viaGAtondashNH functionalizedmesoporoussilicapowders(strategyA)provided2
the highest activities Interestingly in the similar comparative studies it isreportedthatamongndashOHminusCOOHandndashNH functionalizedsupportsactivatedby2
divinylsulfonecarbodiimideandGArespectivelythelastonewasfoundasthemostsuitabletechniqueforthecovalentbindingofLac[9]invertase[10]andpepsin[11]
28 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Tab
le 1
Theprinciplesofthecovalentimmobilizationmethodsofenzymesusedinthisstudy
StrategyA
StrategyB
StrategyC
Support
SBAminus15MCMminus41Cell
SBAminus15MCMminus41Cell
SBAminus15MCMminus41Cell
Supportfunctio-
minusNH
minusNH
minusCOOH
22
nalizedgroup
Activationagent
Glutaraldehyde(GA)
Carbodiimide
Carbodiimide
(EDCNHS)
(EDCNHS)
Enzymereactive
minusNH
minusCOOH
minusNH
22
group
Typeofbond
secondaryamine
amide
amide
Denotationsofthe
GOxminusNHminusCHminus(CH)minusCHminusNHminusSBA15
GOxminusC(=O)minusNHminusSBA15
GOxminusNHminusC(=O)minusSBA15
23
preparedenzy-
GOxminusNHminusCHminus(CH)minusCHminusNHminusMCM41
GOxminusC(=O)minusNHminusMCM41
GOxminusNHminusC(=O)minusMCM41
23
maticpowders
GOxminusNHminusCHminus(CH)minusCHminusNHminusCell
GOxminusC(=O)minusNHminusCell
GOxminusNHminusC(=O)minusCell
23
LacminusNHminusCHminus(CH)minusCHminusNHminusSBA15
LaxminusC(=O)minusNHminusSBA15
LacminusNHminusC(=O)minusSBA15
23
LacminusNHminusCHminus(CH)minusCHminusNHminusMCM41
LacminusC(=O)minusNHminusMCM41
LacminusNHminusC(=O)minusMCM41
23
LacminusNHminusCHminus(CH)minusCHminusNHminusCell
LacminusC(=O)minusNHminusCell
LacminusNHminusC(=O)minusCell
23
When the effect of the method of the covalent enzyme coupling on thebiosensorstabilitywasevaluateditwasfoundthatLacboundedtondashNH functio-2
nalizedsupportsviaGA(strategyA)hasshownthehigheststability(gt65oftheinitial responses after 1 month) compared to other strategies whereas GOximmobilizedwithtwostrategies(AandB)possessedapproximatelysimilarhighstability(gt80oftheinitialresponsesin1month)BothenzymesboundedviandashNH groupstondashCOOHfunctionalizedsupportsthroughEDCNHS(strategyC)2
showedquitelowstability
4Conclusions
Threedifferent strategies including the support selection the anchor surfacegroups and the activationmethod havebeen compared for efficient covalentimmobilization of Lac and GOx The results showed that ndashNH functionalized2
supports(SBA15minusNH CelluloseminusNH forGOxandMCMminusNH forLac)activatedby2 2 2
GAmaybeusedtoeffectivelybindenzymesintermsofhighactivityandstability
Acknowledgments
ThisworkwasfinanciallysupportedbytheGrantAgencyofCharlesUniversityinPrague(Project1356120)theGrantAgencyoftheCzechRepublic(Project20-07350S)anditwascarriedoutwithintheframeworkofSpecificCharlesUniversityResearch(SVV260440)
References
[1] YosypchukBBarekJYosypchukOPreparationandpropertiesofreferenceelectrodesbasedonsilverpasteamalgamElectroanalysis23(2011)2226minus2231
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 29
Fig 1Effectof the covalent attachment techniqueson (A) laccaseand (B) andglucoseoxidaseminus1biosensor responses Experimental conditions (A) c = 500 micromol L E = minus50 mVDOP det
minus1 minus1v = 01 mL min V = 40 microL carrier solution 01 mol L acetate buffer pH = 48flow DOPminus1 minus1(B)c =500micromol L E =minus1100mVv ==01mLmin V =40microL carrier solutionGlu det flow Glu
minus1 minus101molL acetatebuffer0001molL Na EDTApH=652
[2] LiuYChenJYEnzymeimmobilizationoncellulosematrixesJBioactCompactPolym31(2016)553ndash567
[3] Hartmann M Kostrov X Immobilization of enzymes on porous silicas ndash benefits andchallengesChemSocRev42(2013)6277minus6289
[4] JosypcukOBarekJJosypcukBElectrochemicalbiosensorsbasedonenzymaticreactorsfilledbyvarioustypesofsilicaandamalgampowdersformeasurements inflowsystemsElectroanalysis28(2016)3028minus3038
[5] Josypcuk O Barek J Josypcuk B Amperometric determination of catecholamines byenzymaticbiosensorsinflowsystemsElectroanalysis30(2018)1163minus1171
[6] TvorynskaSBarekJJosypcukBAmperometricbiosensorbasedonenzymaticreactorforcholinedeterminationinflowsystemsElectroanalysis31(2019)1901minus1912
[7] TvorynskaSBarekJJosypcukBFlowamperometricbiosensorbasedontwoenzymaticreactors (acetylcholinesterase-choline oxidase) for the detection of neurotransmitteracetylcholine In Proceedings of the 15th International Students Conference ldquoModernAnalyticalChemistryrdquoKNesmerak(ed)PragueFacultyofScienceCharlesUniversity2019p61minus66
[8] TvorynskaSBarekJJosypcukBAcetylcholinesterase-cholineoxidase-basedmini-reactorscoupledwithsilversolidamalgamelectrodeforamperometricdetectionofacetylcholineinflowinjectionanalysisJElectroanalChem860(2020)113883
[9] RekucAKruczkiewiczPJastrzembskaBLiesieneJPeczynska-CzochWBryjakJLaccaseimmobilizationonthetailoredcellulose-basedGranocelcarriersIntJBiolMacromol42(2008)208minus215
[10] Bryjak J Liesiene J S tefuca V Man-tailored cellulose-based carriers for invertaseimmobilizationCellulose15(2008)631minus640
[11] SzałapataKOsinska-JaroszukMBryjakJJaszekMJarosz-WilkołazkaANovelapplicationofporousandcellularmaterialsforcovalentimmobilizationofpepsinBrazJChemEng33(2016)251minus260
30 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Carbohydrates are crucial for energy structure and signaling in the humanbody[1]Thereisavarietyofcarbohydratesbutthemostimportantoneforlifeisglucoseasitisfundamentalinthemetabolismandphotosynthesis[2]GlucoseisclassifiedashexoseThesemonosaccharidesonlydifferinthepositionofhydroxylsubstituentsinsomecasesInadditiontothestructuralsimilaritiesthesemole-culeslackachromophoreandarenoteasilyionizable(pK ~12)Thusdetectiona
intheUVregionandseparationofanalytesbycapillaryelectrophoresis(CE)arechallenging [1 3] At the moment there are many different techniques for theanalysis of carbohydrates commonly including time-consuming derivatizationstepsoreluentswithhighpHvalues(pHgt12)inionchromatographyAwell-esta-blishedtechniquefortheanalysisofcarbohydratesishigh-performanceanion-exchangechromatographywithpulsedamperometricdetection(HPAE-PAD)[4]Electrochemical detection like AD is matching miniaturization simple instru-mentationlowcostandrobustnessandthusisoftenusedforflow-basedsystemssuchasCEandflowinjectionanalysis(FIA)[5]
Capillary flow injection analysis with electrochemical detection for carbohydrate analysis
NICOLEHEIGLFRANK-MICHAELMATYSIK
InstituteofAnalyticalChemistryChemo-andBiosensorsFacultyofChemistryandPharmacyUniversityofRegensburgUniversitaumltsstraszlige3193053RegensburgGermanynicoleheiglchemieuni-regensburgde
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 31
AbstractAsimplecapillaryflowinjectionanalysissystemwithamperometricdetection was arranged for the development of a method for fastoptimization of detection conditions in the context of thedetermination of carbohydrates by means of electrochemistry-capillary electrophoresis-mass spectrometry This setup is free ofelectricalinterferencebyhighvoltageandisperfectforstudyingtheoxidationofvariousanalytesFurthermoreitassureseasycouplingtoMS and thus is an useful tool to investigate the correspondingoxidationproductsofananalyte
Keywordscapillaryflowinjection
analysiscarbohydratesmassspectrometrydisposableelectrodespulsedamperometric
detection
Inthiscontributioncapillaryflowinjectionanalysis(CFIA)withADwillbepresentedasamethodtoapplyandtestADforthedetectionofmonosaccharidesondifferentdisposablethin-filmorscreen-printedelectrodesCFIAwaschosenoverconventionalFIAforthispurposeasthegravityflowinCFIAisstableforalongertimeandverylowsampleconsumptioncanbeachieved[6]TheCFIAsystemwasarrangedassimpleaspossibleandperformedhydrodynamicallytoavoidanyinterferencesFurthermoreitassuresthecouplingoftheflowsystemtoamassspectrometerThusthesamesetupasusedforCFIAcanbeutilizedforcapillaryelectrophoresis-massspectrometry(CE-MS)experimentsbychangingthe flow through the electrochemical flow cell in opposite direction In futureexperimentstheexperienceintermsofADonthoseelectrodeswillbeusedtodevelopelectrochemicalpretreatmentprotocolsforcarbohydratedeterminationbyCE-MS
2Experimental
21Reagentsandchemicals
The following chemicals were used for this study all of analytical gradeAmmoniumacetate(NH OAc)wasobtainedfromMerck(DarmstadtGermany)4
and ferrocene methanol (FcMeOH) from ABCR (Karlsruhe Germany) Milli-Qregwater(182MΩcm)wasgeneratedbyaMilli-QAdvantageA10 system(Merck
Millipore Darmstadt Germany) Carrier solution was prepared by dissolvingndash1NH OAc (50mmolL ) in Milli-Q water FcMeOH solution was prepared by4
dissolvingFcMeOHincarriersolution
22Instrumentation
Electrochemical measurements were performed using a microAutolab Type IIIpotentiostatgalvanostat (Metrohm Autolab B V Utrecht Netherlands)controlledbyNOVA20softwareforexperimentalcontrolanddataacquisitionCFIAwasperformedusingthesetupillustratedinFig1(A)consistingofacarrierreservoirsamplevialandtwofusedsilicacapillaries(PolymicroTechnologiesPhoenix AZ USA inner diameter 100 microm length inlet 40 cm length outlet10cm)connectedtoacommerciallyavailableflowcellfromMicruxTechnologies(model ED-FLOW-CELL Oviedo Spain) Inside of the flow cell the fused silicacapillary was placed in a so-called wall-jet configuration above the workingelectrode of a disposable thin-film gold electrode (model ED-SE1-Au MicruxTechnologiesOviedoSpain)ascanbeseeninFig1(B)Thethin-filmelectrodeswerebasedonathree-electrodesystemwithagoldworkingauxiliaryandquasi-referenceelectrodeThehydrostaticpressurewasachievedbyaheightdifferencebetweeninletandoutletreservoirof30cmresultinginagravityflowofthecarriersolutionthroughafusedsilicacapillaryandsubsequentlythroughtheflowcell
32 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Theinjectionwascarriedoutbyloweringthevialcontainingthecarriersolutiontotheleveloftheoutletexchangingthecarrierreservoirwiththesamplevialandliftingthesamplevialto20cmforadefinedperiodoftimeRe-establishingthecarrierreservoirtookplacethesameway
3Resultsanddiscussion
AsimpleCFIA-ADsystemwasarrangedwherehydrostaticpressurebyaheightdifferencebetweeninletandoutletreservoirresultedinagravitationalflowToobtain general information about the behavior of the assembled CFIA systempreliminaryexperimentswithFcMeOHwereperformedToassurecompatibilitywithMSlateronNH OAcwaschosenastheelectrolyteVariousheightdifferences4
andinjectiontimesweretestedandtheinjectionataheightdifferenceof20cmlasting for 10 s was found to be the optimum concerning feasibility and peakshapes Injections of several solutions of FcMeOH of different concentrationsshowed that the concentration dependence of FcMeOH was linear in theinvestigatedrange(Fig2)Furthermoreexperimentsrevealedthattheinjectionprocedure was established with reasonable precision When repeating the
ndash1injectionof05mmolL FcMeOHincarriersolutionfortentimestherelativestandarddeviationwasfoundtobe3forthemanualinjectionprotocol
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 33
Fig 1(A)Schemeoftheusedcapillaryflowinjectionanalysissetup(1)carrierreservoir(2)samplevial(3)inletcapillarywithalengthof40cmandaninnerdiameterof100microm(4)outletcapillarywithalengthof10cmandaninnerdiameterof100micrombothcapillariesconnectedto(5)acommerciallyavailableflowcellfromMicruxand(6)awastevial(B)Configurationinsidetheflowcell(7)thefusedsilicacapillarywasplacedinaso-calledwall-jetconfigurationabovetheworkingelectrodeof(8)adisposablethin-filmgoldelectrode
4Conclusions
The presented system for CFIA-AD was arranged as simple as possible andrepresents a useful approach for the development of a method for fastoptimization of detection conditions in the context of the determination ofcarbohydratesbymeansofelectrochemistry-CE-MSThesetupisfreeofelectricalinterferencebyhighvoltagecompatiblewithMSandthuspromisingforstudyingtheoxidationofvariousanalytes
References
[1] LuGCrihfieldCLGattuSVeltriLMHollandLACapillaryelectrophoresisseparationsofglycansChemRev118(2018)7867ndash7858
[2] GalantALKaufman RCWilson JDGlucoseDetectionandanalysisFoodChem188(2015)149ndash160
[3] Sarazin C Delaunay N Costanza C Eudes V Gareil P Application of a new capillaryelectrophoreticmethodforthedeterminationofcarbohydratesinforensicpharmaceuticalandbeveragesamplesTalanta99(2012)202ndash206
[4] Rohrer JS Basumallick L Hurum D High-performance anion-exchange chromatographywithpulsedamperometricdetectionforcarbohydrateanalysisofglycoproteinsBiochem78(2013)697ndash709
[5] IslamMAMahbubPNesterenkoPNPaullBMackaMProspectsofpulsedamperometricdetectioninflow-basedanalyticalsystemsndashAreviewAnalChimActa1052(2019)10ndash26
[6] MatysikF-MWernerGTracemetaldeterminationintearsbyanodicstrippingvoltammetryinacapillaryflowinjectionsystemAnalyst118(1993)1523ndash1526
34 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2ndash1 ndash1(A) CFIA-AD recordings of three consecutive injections of (1) 025mmolL (2) 05mmolL
ndash1 ndash1 ndash1(3) 075mmolL and (4) 1mmolL FcMeOH in 50 mmolL NH OAc detection at a Micrux4
thin-filmAuelectrodeataconstantpotentialof03VinaflowcellHydrodynamicinjectionlasted10sataheightdifferenceof20cm(B) Calibration dependence of FcMeOH for CFIA-AD determination and detection at a Micruxthin-filmAuelectrodeataconstantpotentialof03VinaflowcellThestandarddeviationsofpeakheights(n=3)areindicatedbyerrorbars
1Introduction
Topreventsorptiononthecapillarysurfaceandimproveseparationefficiencyand selectivity of determined analytes coatings are formed on the capillarysurface There are two types of ones namelydynamic and covalently bondedcoatings Despite the simplicity of creation dynamic coatings cannot providerequiredreproducibilitywhilecovalentcoatingscontributethestableelectro-osmoticflow(EOF)andhighreproducibilityoftheanalysisInmostcasesthe
Application of covalent coatings based on imidazolium cations for separation and on-line preconcentration of basic and neutral analytes in capillary electrophoresis
a ab aANASTASIAVKRAVCHENKO EKATERINAAKOLOBOVA LIUDMILAAKARTSOVA
a DepartmentofOrganicChemistryInstituteofChemistrySaintPetersburgStateUniversity 26Universitetskiiprospect198504StPetersburgPeterhofRussiakravchenko161216gmailcom
b TheFederalStateInstituteofPublicHealthldquoTheNikiforovRussianCenterofEmergencyandRadiationMedicinerdquoTheMinistryofRussianFederationforCivilDefenceEmergenciesandEliminationofConsequencesofNaturalDisasters54Optikovst197082StPetersburgRussia
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 35
AbstractThemethodofcapillaryelectrophoresis (CE) isactivelydevelopedandmoreandmoreattractsscientistsattentioneveryyearHoweverthesorptionofanalytesonsurfaceoffused-silicacapillarywallsisoneof thesignificantdisadvantagesof thisapproachThe formationofcoatings on the inner capillary surface is typical way to preventsorption and to increase separation efficiency and selectivity ofdetermined analytes Coatings that covalently bonded to capillarywalls is more suitable because they are stable and provides highreproducibilityofanalysisThepresentworkisfocusedonthedevelo-pmentofthemethodofelectrophoreticdeterminationofbiologicalactiveanalytesusingacovalentcoatingbasedonimidazoliumcationsTheeffectofsubstituentinimidazoliumringonmainelectrophoreticparameters was examined It was shown that alkylimidazoliumcoatingscontributetosignificantreducingofbiogenicamineslimitsof detection while β-cyclodextrinimidazolium covalent coatingallowstoseparatebothofhydrophobicandhydrophilicanalytesinonerun
Keywordsbiologicalactiveanalytescapillarycoatingcapillaryelectrophoresisimidazoliumionicliquids
analytes nature determines type of usedmodifiers because suitable ones canprovideaccessorial interactionbetweentheanalytesandthestationaryphaseimproving separation selectivity and efficiency [1 2] Ionic liquidshave beenwidelyusedinanalyticalchemistry[3]andseparationtechniquesparticularlyincapillaryelectrophoresis[4]Earlyresearches[5ndash9]haveshownopportunityofcovalently bonded imidazolium ionic liquids for electrophoretic separationHowever the effect of various substituents in imidazolium ring hasnot beendescribed previously Thus the purpose of this study was to create covalentcoatingsbasedon ionic liquidwithvarioussubstituentsand tocompare theiranalytical capabilities in the electrophoretic separation of biologically activecompounds
2Experimental
21Reagentsandchemicals
(3-Glycidyloxypropyl)trimethoxysilane (GPTMS) hydrochloric acid sodiumdodecyl sulfate (SDS) imidazole 22-diphenyl-1-picrylhydrazyl (DPPH)p-toluen-sulfonylchlorideβ-cyclodextrinhydrocortisone(F)11-deoxycortisol(S)Corticosterone(B)rac-ketoprofen(ndash)-adrenaline(A)L-(ndash)-norepinephrine(NE) DL-normetanephrine (NMN) dopamine (DA) DL-metanephrine hydro-chloride(Met)serotoninhydrochloride(Ser)homovanillicacid(HVA)24-di-hydroxy-benzoic acid (24-DHBA) 34-dihydroxy-L-phenylalanine (DOPA)L-tryptophan(Trp)L-tyrosine(Tyr)werepurchasedfromSigma-Aldrich(USA)1-Bromo-butane1-bromooctanewerepurchasedfromReagentPlus(Ukraine)Sodium dihydrogenphosphate dihydrate acetone NN-dimethylformamide(DMF)wereobtainedfromMerck(Germany)AllreagentsusedwereanalyticalgradeAllsolutionswerepreparedusingdeionizedwater
22Instrumentation
Capillary electrophoresis experiments were carried out using the system ofcapillaryelectrophoresisCAPEL-105M(LumexRussia)withUV-spectrophoto-metricdetector(wavelengthrange190ndash360nm)Separationswereperformedusing 58times49 cm (9 cm to the detector outside diameter 360 microm and innerdiameter50microm) coated silica capillaries (LumexRussia) Thebuffer pHwasmeasuredwithapH-meterHI2210ndash2216(Hanna)
23Capillarycoatingsynthesis
Earlier our research team has proposed the synthesis route for the covalentcoatingsbasedonimidazoliumcationfunctionalizedwithalkylgroup[10]andβ-cyclodextrin [11] All capillarieswere prepared according above-mentioned
36 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
manuscriptsandcharacterizedbytheEOFmobilitymeasurementandscanningelectronmicroscopyThesynthesisconsistedoffollowingstepspreparationofacapillarytocreateacovalentcoating(heatingcapillaryfilledwith2MNaOHat90degCfor1handdryingfollowed)silylationwithGPTMSandfunctionalizationwiththeimidazolesolutionfollowedmodificationbybutyl-andoctylbromideortosyl-β-cyclodextrin(seedetailsin[10]and[11])
24Solutions
A stock buffer solution in concentration 50mM was prepared by dissolvingappropriate amount of sodium dihydrogenphosphate dihydrate in deionizedwateradjustingpHto20with1MhydrochloricacidThisbuffersolutionwasthendilutedwithdeionizedwater
ndash1 Allthesamplestocksolutionswerepreparedwithconcentration10mgmL Thestocksolutionsoftheneurotransmittersandtheirmetabolites(adrenalinenoradrenaline dopamine normetanephrine metanephrine serotonin homo-vanillicacid)and24-dihydroxybenzoicacidasinnerstandardandaminoacids(tryptophan34-dihydroxy-L-phenylalanine tyrosine)wereprepared in01MhydrochloricacidThestocksolutionsofsteroids(hydrocortisone11-deoxycor-tisolandcorticosterone)wereprepared inacetonitrileThestocksolutionsofketoprofen racemate and S-ketoprofen were prepared in acetonitrilewatersolution(1090vv) Untilelectrophoreticanalysisthestocksolutionswerestoredatndash16degCTheworkingsolutionswerepreparedbydilutingtheinitialsolutionswithwaterjustbeforetheexperiments
3Resultsanddiscussion
CovalentcoatingsbasedonN-alkylimidazoliumcationwereespeciallysuitableforseparationofneurotransmittersandtheirmetabolites(Fig1)Inadditionthecombinationofcovalentcoatingwithon-linepreconcentrationtechniquesallowstothesignificantdecreaseoftheseanalyteslimitsofdetection(LOD)Accessorialinteractions positively charged analytes with positively charged imidazoleimproveseparationselectivity(viaπ-πinteraction)andefficiency(concentrationintightzonesviaelectrostaticrepulsion)Sodiumdodecylsulfate(SDS)addedintobackgroundelectrolyte(inconcentrationabovecriticalmicelleconcentra-tion) strongly interacts with hydrophobic alkyl groups in covalent coatingstructureThenegativelychargedSDSlayerisformedoninnercapillarysurfaceThedoublereversingEOFallowsustocarryoutelectrokineticinjectionofsampleandon-linepreconcentrationbysweepingsimultaneouslyLODweredeclineto
ndash108ndash20ngmL ThelengthofalkylsubstituentalsoaffectsthestackingefficiencyfactorandLODMorehydrophobicoctylgroupscomparetobutylprovidemoreeffectiveinteractionwithSDSandasresultlowerLOD
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 37
Covalentcoatingmodifiedβ-CDhasnotshownsharpreducingofLODbyon-linepreconcentrationStacking sweeping (SDSasmicelle reagent) field-enhancedsample injection were examined using different model mixtures of analytesNeverthelessthiscoatingallowssimultaneousseparationofbothofhydrophobicsteroidhormonesandhydrophilicbiogenicaminesinasinglerun(Fig2) The guest-host interaction hydrophobic cavity of β-cyclodextrin with thehydrophobicsteroids leadsto the formationofcomplexwhichaffectssteroidselectrophoretic mobility At the same time β-cyclodextrin can act as a chiralselector and baseline separation of ketoprofen enantiomers has also beenachieved
38 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Electropherogramofmixtureof neurotransmitters and theirmetabolitesadrenaline (A)norepinephrine (NE)normetanephrine (NMN)dopamine (DA)metanephrine (Met) serotonin(SER)homovanillicacid(HVA)andtheinnerstandard24-dihydroxybenzoicacid(24-DHBA)oncovalentlymodifiedwithN-buthylimidazoliumionicliquidscapillaryConditions10mMNaH PO 2 4
(adjusted to pH= 20 by 1MHCl) injection 50 stimes30mbar ndash20 kV 220 nmmodelmixurendash1 ndash1 ndash110microgmL (METADNMNNADA24-DHBA)5microgmL (SER)and20microgmL (HVA)
4Conclusions
ItwasshownthatstructurecovalentcoatingaffectsitsanalyticalcharacteristicsWecomparedtwotypesofcovalentcoatingdifferingsubstituentinimidazoliumringnamelyalkylgroupandβ-cyclodextrinThefirsttypeisgreatcoupledwithon-line preconcentration technic but it is limited to effectively determine ofbiogenicaminesonlywhilethesecondtype(withβ-cyclodextrin)showedthepossibilitiestoseparatevariousanalytesbutsuitableon-linemodehasnotbeenfoundThemainpointsaresummarizedinTable1
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 39
Covalentcoatingtype Electrophoreticseparationof On-lineprecon- centration biogenicamines amino steroid ketoprofen andtheirmeta- acids hormones enantiomers bolites N-β-cyclodextrinimida- yes yes yes yes thesuitableapproachzoliumcovalentcoatings wasnotfoundedN-alkylimidazolium yes yes nonsepa- nonsepa- thesignificantreducingcovalentcoatings rated rated forbiogenicaminesLOD
Table 1Thesummationofpossibilitiesofcovalentcoatingsbasedonimidazoliumcation
Fig 2Electropherogramofsimultaneousseparationofhydrophobic(steroidhormones)andhydro-philicanalytes (aminoacidsandbiogenicamines) insinglerunwithcovalentcoatingbasedonimidazoleandβ-CDConditions10mMNaH PO (adjusted topH = 20by1MHCl) injection2 4 20 stimes30mbarndash20kV254nm(1ndash8min)and220nm(8ndash15min)0mbar(1ndash10min)and40mbar(10ndash15 min) Model mixture corticosterone (B) hydrocortisone (F) 11-deoxycortisole (S)
ndash1 ndash15μgmL L-tryptophan(Trp)34-dihydroxy-L-phenylalanine(DOPA)10μgmL L-tyrosine(Tyr)ndash15 μg mL noradrenaline (NA) normetanephrine (NMN) adrenaline (AD) dopamine (DA)
ndash120μgmL
Acknowledgments
This work was supported by Russian Science Foundation (grant numbers 19-13-00370) Theauthors are also grateful to the Chemistry Education Centre and Nanothechnologies Centre ofResearchParkSaintPetersburgStateUniversityfortechnicalsupport
References
[1] HuLFYinSJZhangHYangFQRecentdevelopmentsofmonolithicandopen-tubularcapillaryelectrochromatography(2017ndash2019)JSepSci43(2020)1942ndash1966
[2] KartsovaLAKravchenkoAVKolobovaEACovalentcoatingsofquartzcapillariesfortheelectrophoretic determination of biologically active analytes J Anal Chem 74 (2019)729ndash737
[3] HoTDZhangCHantaoLWAndersonJLIonicliquidsinanalyticalchemistryFundamen-talsadvancesandperspectivesAnalChem86(2014)262minus285
[4] TangSLiuSGuoYLiuXJiangSRecentadvancesofionicliquidsandpolymericionicliquids incapillaryelectrophoresisandcapillaryelectrochromatography JChromatogrA1357(2014)147ndash157
[5] QinWLiSFYElectrophoresisofDNAinionicliquidcoatedcapillaryAnalyst128(2003)37ndash41
[6] QinWWeiH Li SFY 13-Dialkylimidazolium-based room-temperature ionic liquids asbackgroundelectrolyteand coatingmaterial in aqueous capillaryelectrophoresis JChro-matogrA985(2003)447ndash454
[7] QinW Fong S Li Y Determination of ammonium andmetal ions by capillary electro-phoresisndashpotential gradient detection using ionic liquid as background electrolyte andcovalentcoatingreagentJChromatogrA1048(2004)253ndash256
[8] QinWLiSFYAn ionic liquidcoating fordeterminationofsildenafilandUK-103320 inhumanserumbycapillaryzoneelectrophoresis-iontrapmassspectrometryElectrophoresis23(2002)4110ndash4116
[9] BorissovaMVaherMKoelMKaljurandMCapillaryzoneelectrophoresisonchemicallybondedimidazoliumbasedsaltsJChromatogrA1160(2007)320ndash332
[10] KolobovaEKartsovaLKravchenkoABessonovaEImidazoliumionicliquidsasdynamicand covalent modifiers of electrophoretic systems for determination of catecholaminesTalanta188(2018)183ndash191
[11] KravchenkoAKolobovaEKartsovaLMultifunctioncovalentcoatingsforseparationofaminoacidsbiogenicaminessteroidhormonesandketoprofenenantiomersbycapillaryelectrophoresisandcapillaryelectrochromatographySepSciplus3(2020)102ndash111
40 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Synthetic 4-hydroxy-3-methoxybenzaldehyde (vanillin) is used as a flavoringagent in foodsdrinksperfumesandpharmaceuticals [1]However at certainconcentrationsthesubstancemayaccumulateinthebodyhaveatoxiceffectand
ndash1at high concentrations may be fatal (lethal dose LD (oral rat) =2gkg 50ndash1 ndash1LD (oral guinea pig) = 14gkg LD (intravenous dog) = 132gkg lethal50 50 ndash1concentrationLC (inhalationmouse)=417gkg )[2]AccordingtoRussianState
StandartGOST121005-88thetoxiceffectsofvanillinintheworkplaceinthendash3formofvapoursoraerosolsareobservedatconcentrationsabove15mgm
Chromatography[3]spectrophotometry[4]capillaryelectrophoresisareusedforvanillindeterminationindifferentobjects CurrentlysmokingmixturesforhookahsandelectroniccigarettesarewidelyusedamongyoungpeopleThesemixturesarenotcontrolledforthecontentofsubstancesandarefreelyavailableconsideringthemmoreharmlesswithrespecttoordinarycigarettesThusthedevelopmentofamethodforthedeterminationof4-hydroxy-3-methoxybenzaldehydeinsmokingmixturesisrelevant
Determination of vanillin in smoking mixtures by spectrophotometry
ELIZAVETAEFREMENKOANNACHERNOVAOLGABASTRYGINA
DepartmentofChemicalEngineeringNationalResearchTomskPolytechnicUniversityLeninavenue30634050TomskRussiaeaetpuru
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 41
AbstractTheresearchdealswithdeterminationofvanillin insmokingmix-turesbyultraviolet-visiblespectrophotometryThemethodshowed
ndash1goodlinearityintherangeof005ndash012gL withalimitofdetectionndash1005gL After validation studies the method was successfully
applied to thedeterminationof vanillin in smokingmixtureswithsatisfactoryresultsItwasshownthattheerrorofthismethoddoesnot exceed 1 The developed spectrophotometric procedure fordeterminingvanillininsmokingmixturescanbeusedasacontrol
Keywordssmokingmixturesspectrophotometryvanillin
2Experimental
21Reagentsandchemicals
Asampleofvanillin(purity98)wastakenastheobjectofstudyAssolventsweused95ethanolAllchemicalsusedwereofanalyticalreagentgrade
22Instrumentation
Theopticaldensityofsampleswasmeasuredincuvettewithanabsorbinglayerthickness of 10 mm using a Cary 60 spectrophotometer (Agilent USA) Allmeasurementswerecarriedoutatroomtemperature
23Samplepreparation
Sample preparation of the investigated objects consisted of the preliminarydissolutionofthesamplein95ethanolThesample10mgoftobaccoldquoAdalyandashVanillardquo(Turkey)wasdiluted in10μLof95ethanol to theconcentrationof
ndash11gL Thesample10μLofldquoFlavoringTPAndashVanillaCustardrdquo(USA)wasdilutedin10 μL of 95 ethanol The resulting solution was diluted six times to the
ndash3concentrationof017μLcm
3Resultsanddiscussion
TodeterminevanillininthesamplestheopticalpropertiesofvanillininvarioussolventsweredeterminedAsaresultthe95ethanolwaschosenastheoptimalsolvent[4] IthasbeenestablishedthatintheUVspectraoftheanalyteabsorptionbondsareobservedwithmaximumvaluesat23002800and3100nmwhichcorres-pondstopublisheddata[45](Fig1) To quantify vanillin the calibration curve of the optical density on theconcentrationofvanillin in95ethanolwasobtainedatconcentrations005
ndash1006007008010and012gL Calibrationcurveofvanillinin95ethanolatawavelengthof280nmis
ndash1 A =81914c[gL ]+00357 (1)2802 R =1
Calibrationcurveofvanillinin95ethanolat310nmis
ndash1 A =73824c[gL ]+00301 (2)3102 R =1
42 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Inthespectraoftheanalyzedsamplesolutionsabsorptionmaxima(2800nmand3100nm)characteristicforvanillinwereobservedTheamountofvanillininthesamplewasdeterminedusingcalibrationcurvesat280and310nmWeightedleast square regressionwas applied to the calibration curves to improve theaccuracyespeciallyatinlowconcentrationlevelrangeGoodlinearitywasfound
ndash1 ndash1intherangeof005ndash012gL withadetectionlimitof005gL TheresultsarepresentedintheTable1
4Conclusions
ThedevelopedmethodcanbeusedasacontrolmethodTheerrorinthemethodfordeterminingvanillininthesampleldquoFlavoringTPAndashVanillaCustardrdquowithaknownconcentrationofvanillinwas0004Accordingtothedataobtainedwerecommendawavelengthof280nmforthedeterminationofvanillininsamples
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 43
λ nm
Absorban
ce
ndash1Fig 1 Absorption spectrum of vanilin solution in 95 ethanol at concentration 01 mol L (anabsorbinglayerthicknessof10mm)
Sample λnm Tookmg Foundmg S Sх Δх δ
FlavoringTRA 310 10850 104096 00024 00011 00006 00306ndashVanillaCustardAdalyandashVanilla 310 100000 10162 00019 00009 00002 00025
FlavoringTRA 280 100300 96062 00033 00015 00032 00042ndashVanillaCustardAdalyandashVanilla 280 100000 10122 00013 00006 00012 00017
Table 1Testingmethods introduced foundof vanillin in the samplesat310nmby spectrophotometricmethod(n=5p=099SndashstandarddeviationSxndashrelativestandarddeviationΔхndashabsoluteerrorδndashrelativeerror)
ThenthedevelopedmethodwastastedonthesampleldquoAdalyandashVanillardquosamplewithamorecomplexcompositionandanunknownconcentrationofvanillinwastaken The vanillin content in the sample was determined according to thedevelopedmethoditamountedto10ofthetotalmassStudieshaveshownthepossibility of using spectrophotometric analysis for the qualitative andquantitative determination of vanillin Also based on preliminary studies aspectrophotometricprocedurewasdevelopedforthequantitativedeterminationofvanillinbasedonabsorptioninethanolinthewavelengthrange200ndash400nm
References
[1] httpswwwrusnaukacom43_DWS_2015Chimia6_203179dochtm (accessed 25thFebruary2019)
[2] httpswwwcdcgovnioshrtecsdefaulthtm(accessed11stApril2020)[3] AliLPerfettiGDiachenkoGRapidmethodforthedeterminationof342coumarinvanillin
and ethyl vanillin in vanilla extract by reversed-phase liquid 343 chromatography withultravioletdetectionJAOACInt91(2008)383ndash386
[4] БастрыгинаОАЕфременкоЕАЧерноваАПВыделениеванилинаисследованиеегооптическихсвои ствопределениевбиологическомматериалеВХимияихимическаятехнология в XXI веке Материалы XX Международной научно-практическойконференции имени профессора ЛП Кулёва студентов и молодых ученых ТомскНациональныи исследовательскии Томскии политехническии университет 2019с301ndash302
[5] WeastRCHandbookofChemistryandPhysics60thedBocaRatonCRCPress1979p143
44 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
UraniumbelongstothegroupofhazardouselementsItisahighlyharmfulandradioactiveelementtoxictohumansandalllivingorganisms[12]Inhaledwithair it has a particularly destructive effect on the kidneys and as a result ofaccumulationinwhitebloodcellsitcanalsocauseimpairmentoftheimmunesystem[2]Uraniumoccursatseveraldegreesofoxidationhoweverinaqueoussolutionsthemoststableformisuranylion(UO (II))[12]Thepresenceofura-2
niumintheenvironmentiscausedbyamongothersnaturalsoilandrockerosionEnvironmentalpollutionwiththiselementisalsoconstantlyincreasingduetohumanactivitycoalcombustionuraniumoreminingandprocessingthearmsindustryandtheuseofuraniumasnuclearfuelinfissionreactors[3]Itisveryimportanttoconstantlymonitortheconcentrationofuraniumbothinthenaturalenvironment in order to assess its state and safety (especially in the case ofdrinkingwater)aswellasinallstagesofprocessingprocessesassociatedwiththenuclearindustrytoavoidtheoccurrenceofnuclearpollution[13] Scientists have made many attempts to develop research methods todeterminethecontentofuranylcompoundsinliquidsamplesEffortsweremadetousemanyanalyticalmethodsforthispurposeincludingspectrophotometry
Uranyl ion-selective electrode with solid contact
KAROLINAPIETRZAKCECYLIAWARDAK
DepartmentofAnalyticalChemistryInstituteofSciencesFacultyofChemistryMariaCurie-SklodowskaUniversityMariaCurie-SklodowskaSq320-031LublinPolandkarolinapietrzakpocztaumcslublinpl
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 45
AbstractNewallsolidstateuranylion-selectiveelectrodeswithlowdetection
ndash7 ndash1limits(71times10 molL )shortresponsetimegoodselectivityandstable and reproducible potential were developed Many types ofelectrodeswith different active ingredient content in ion-selectivemembrane (bis(244-trimethylpentyl)phosphonium acid Cyanex-272)were testedAs an additive an ionic liquid1-octyl-3-methyl-imidazole chloride was used The optimal composition of theion-selective membrane was chosen from all electrodes based onthedeterminationand comparisonof analyticalparametersof thesensors
Keywordsion-selectiveelectrodesolidcontacturanyl
plasma spectrometry luminescence spectroscopy voltammetry or chromato-graphymethods[2] Duetomanyadvantagesofpotentiometricmethods(amongthemlowercostseasieroperationofdevicesquickresponseandtheabilitytoperformmeasure-ments in flowmode) [3] a numberof potentiometric sensorshave alsobeendeveloped that could be successfully used in this type of research Themostpopularpotentiometricsensorsincludeion-selectiveelectrodes(ISEs)whicharecharacterized by low-energy consumption small size and portability and aresuccessfullywidelyusedforthedeterminationofbothinorganicandorganicionsinclinicalanalysisprocesstechnologyaswellasincontrolthestateofthenaturalenvironment[45]Removaloftheinternalsolutioncontainingthesameanalytetowhich theelectrode is sensitiveresulted in theso-calledsolidcontact ISEswhicharemuchsmallerinsizethantheirpredecessorsaremoreconvenienttouse and more mechanically resistant In this type of sensors however it isimportanttoachievesatisfactorypotentialstabilitywhichisnecessarytoobtainsatisfactoryresults[5]AveryimportantpartofISEsistheion-selectivemem-branewhosecompositiondeterminestheanalyticalparametersofthesensorsResearchers are currently focusing on the production and testing of newsubstancesthatcouldbesuccessfullyusedasmembranecomponentsandsolidcontacts thatwould allow to obtain new sensorswith lower detection limitslongerlifetimeandbetterpotentialstabilityandtodeterminenewpreviouslyunattainableanalytes[4] AstheactivecomponentsofthemembranesensitivetouranylionscientistshavealreadyusedKryptofix22DD(413-didecyl-171016-tetraoxa-413-diaza-cyclooctadecane)[2]Cyanexextractants(bis(244-trimethylpentyl)phosphinicacid bis(244-trimethylpentyl)monothiophosphinic acid and bis(244-tri-methylpentyl)dithiophosphinic)acid[3]DBBP(dibutylbutylphosphonate)andDOPP (di-n-octyl phenylphosphonate) [6] DMSO (dimethylsuphoxide) [7]TTPTP (5678-tetrahydro-8-thioxopyrido[4345]thieno[23-d]pyrimidine-4(3H)one)[8]orTEHP(tris(2-ethylhexyl)phosphate)andTPTU(O-(12-dihydro-2-oxo-1-pyridyl)-NNNN-bis(tetra-methylene)uronium hexafluorophos-phate)[9]
2Experimental
21Reagentsandchemicals
This paper presents research on the design and properties of ion-selectiveelectrodes with solid contact for the determination of uranyl ions Bis(244-trimethylpentyl)phosphonium acid (Cyanex-272) was used as the activecomponentof themembranewhichwasdescribed in the literatureasagooduranylextractant[10]Inordertoensureaconstantpotentialofthiselectrodeandreducetheelectroderesistancetheion-sensitivemembranewasenrichedwithafewpercentadditionof1-octyl-3-methylimidazolechlorideionicliquid
46 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Several types of ion-selective electrodes were prepared using an AgAgClelectrodeasaninternalelectrodewhichdifferinthequantitativeandqualitativecompositionofthemembranesAllcompositionsarelistedinTable1
22Instrumentation
Measurements were made at room temperature using a 16-channel datacollectionsystem(LawsonLabs IncUSA)coupled toa computer in solutionsmixedwithamechanicalstirrerAsilversilverchlorideelectrodewithdoublejunctionwasusedasthereferenceelectrode
3Resultsanddiscussion
The effect of ion-selective membrane composition on the properties of theobtained potentiometric sensors was examined by determining their basicanalyticalparametersincludingslopeoftheelectrodecharacteristicsdetectionlimitmeasuringrange(concentrationrangeinwhichthecourseoftheelectrodecharacteristics isrectilinear)pHrange(inwhich ithasnoeffect forelectrodepotential)andresponsetimeTheobtainedvaluesofthetestedparametersareshowninTable2 Figure1showsthecalibrationcurvesofthetestedelectrodesdeterminedin
ndash7 ndash1 ndash1UO (NO ) solutionsintheconcentrationrange1times10 ndash1times10 molL Asitcan2 3 2
beseeninFig1andTable2allelectrodesweresensitivetouranylionsbutindifferent extend The best response exhibited ISE-3 containing 1 (ww) ofionophore Increasing the ionophore content in themembrane shortened thelinearityrangeofthecalibrationcurveanditssupernenstianslope Theselectivityofthetestedelectrodeswasestimatedbydeterminingtheselec-tivitycoefficients inrelationto interfering ionsForthispurpose theseparate
ndash1solutionmethodwasused(extrapolatingresponsecurves toa =a =1molL )i j
ComparisonofISE-1andISE-3electrodeselectivityisshowninFig2
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 47
Table 1Quantitative and qualitative composition of electrode membranes Cyanex-272 (bis(244-tri-methylpentyl)phosphoric acid) TBP (tri-n-butyl phosphate) and OMImCl (1-octyl-3-methyl-imidazolechloride)
Abbreviation Membranecomposition(ww)ofelectrode Cyanex-272 PVC TBP OMImCL
ISE-1 00 33 620 5ISE-2 05 33 615 5ISE-3 10 33 610 5ISE-4 30 33 590 5ISE-5 50 33 570 5ISE-6 100 33 520 5
48 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Abbreviation Slope Detectionlimit Linearrange Response pHrange2+ ndash1 ndash1ofelectrode mVpa(UO ) molL molL times2
ndash5 ndash5 ndash1ISE-1 297 25times10 5times10 ndash1times10 5ndash8 28ndash42ndash6 ndash5 ndash1ISE-2 292 65times10 1times10 ndash1times10 5ndash8 25ndash60ndash7 ndash5 ndash1ISE-3 298 71times10 1times10 ndash1times10 5ndash8 24ndash60
ndash6 ndash4 ndash1ISE-4(I) 357 31times10 5times10 ndash1times10 5ndash8 ndndash6 ndash6 ndash4ISE-4(II) 242 31times10 5times10 ndash5times10 5ndash8 nd
ndash3 ndash1ISE-5(I) 638 nd 1times10 ndash1times10 5ndash10 ndndash5 ndash3ISE-5(II) 234 nd 5times10 ndash1times10 5ndash10 ndndash3 ndash1ISE-6(I) 733 nd 1times10 ndash1times10 5ndash10 ndndash5 ndash3ISE-6(II) 222 nd 5times10 ndash1times10 5ndash10 nd
Table 2Selectedparametersandtheirdeterminedvaluesoftestedionselectiveelectrodes
Fig 1 Calibration curves of the testedelectrodesobtainedinUO (NO ) solutionsin2 3 2 ndash7the concentration range from 1times10 to
ndash1 ndash11times10 molL
Fig 2Comparisonofselectivitycoefficientspot(log K (UO (II))M) for electrodes ISE-12
(1stcolumn)andISE-3(2ndcolumn)
Inordertoexaminethereversibilityofthepotentialofthetestedelectrodesndash4 ndash1potentialmeasurementsweremadealternatelyinsolutions1times10 molL and
ndash5 ndash11times10 molL ofUO (NO ) TherecordedpotentialreadingsareshowninFig32 3 2
Long-term potential stability and sensor reproducibility were evaluated byndash1determiningtheaveragevalueoftheelectrodepotentialina01molL UO (II)ion2
solutionovertimeforthreeidenticalISE-3Thesemeasurementsweremadetoobservechangesinthepotentialofelectrodeswiththesameconcentrationoveralongperiodoftime(30days)Figure3showsthelong-termpotentialstabilityandreproducibilitydeterminedforthreeidenticalsensors
4Conclusions
Asaresultofthetestsion-selectiveelectrodeforthedeterminationofuranylionswasobtainedwhich iseasy todesignanduseThebestanalyticalparametersexhibitedISE-3containing1ionophoreintheion-selectivemembraneForthis
ndash7 ndash1typeofelectrodesthedetectionlimitof71times10 molL linearityoftheelectrodendash6 ndash1 ndash1calibrationcurve in the range1times10 ndash1times10 molL andresponse time5ndash8s
were obtained In addition the manufactured sensors also showed stablereproducibleandreversiblepotentialandverygoodselectivityinrelationtothetestedinterferents
References
[1] AnsariRMosayebzadehZConstructionofanewsolid-stateU(VI)ion-selectiveelectrodebasedonpolypyrroleconductingpolymerJRadioanalNuclChem299(2014)1597ndash1605
[2] GhanbariMRounaghiGHAshrafNAnuranylsolidstatePVCmembranepotentiometricsensor based on 413-didecyl-171016-tetraoxa-413-diazacyclooctadecane and itsapplicationforenvironmentalsamplesIntJEnvironAnalChem97(2017)189ndash200
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 49
Fig 3 Stability () reproducibility andreversibility () of the potential of ISE-3Standard deviations given on the plot aredeterminedforthesamethreeISE-3
[3] Badr IHA Zidan WI Akl ZF Cyanex based uranyl sensitive polymeric membraneelectrodesTalanta118(2014)147ndash155
[4] BiegCFuchsbergerKStelzleMIntroductiontopolymer-basedsolid-contaction-selectiveelectrodes basic concepts practical considerations and current research topics AnalBioanalChem409(2017)45ndash61
[5] Bobacka J IvaskaA LewenstamA Potentiometric ion sensorsChem Rev108 (2008)329ndash351
[6] ZidanWI Badr IHA Akl ZF Development of potentiometric sensors for the selective2+determinationofUO ionsJRadioanalNuclChem303(2015)469ndash4772
[7] SalehMBSolimanEMGaberAAAAhmedSANovelPVCmembraneuranylion-selectivesensorSensActuatorsB114(2006)199ndash205
[8] SalehMBHassanSSMAbdelAAAbdelNAAnoveluranylion-selectivePVCmembranesensor based on 5678-tetrahydro-8-thioxopyrido[4345]thieno[23-d]pyrimidine-4(3H)oneSensActuatorsB94(2003)140ndash144
[9] HassanSSMAliMMAttawiyaAMYPVCmembranebasedpotentiometricsensorsforuraniumdeterminationTalanta54(2001)1153ndash1161
[10] Prabhu DR Ansari SA Raut DR Murali MS Mohapatra PK Extraction behaviour ofdioxouranium(VI) cation by two phosphorous-based liquid cation-exchangers in room-temperatureionicliquidsSepSciTechnol52(2017)2328ndash2337
50 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Metronidazole(2-methyl-5-nitroimidazole-1-ethanol)isoneofthemostwidelyused nitroimidazole antibiotics Metronidazole is used for the treatment ofinflammatorydiseasescausedbyanaerobicorganismsandsomeprotozoaandforpreventionofdysenterycolibacillosiseimeriosisbalantidiasissalmonellosisenteritissepticemiapost-surgicalcomplications[1ndash3]Oxytetracyclinehydro-chlorideisanantibioticofthetetracyclinefamilyItisoneofthemostcommonlyused antibiotics in poultry because of its low cost and effective [4] Thesecompounds are intensively used in poultry breeding and stockbreedingUnreasonableuseofthesedrugscancauseseriousfoodsafetyissues[5] The veterinary drug Nozemat which includemetronidazole and oxytetra-cyclinehydrochloridewaschosenfortheexperimentsNozematisusedtotreat
Polarographic determination of metronidazole and oxytetracycline hydrochloride in veterinary drug for honey bees
a a bKATERYNAPLOTNIKOVA LILIYADUBENSKA IVANZELENYI
a AnalyticalChemistryDepartmentIvanFrankoNationalUniversityofLvivKyrylaiMefodiaStr879005LvivUkrainekaterina27plgmailcom
b DrohobychPedagogicalLyceumIvanaFrankaStr3682100DrohobychUkraine
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 51
AbstractWehavedevelopedanewpolarographicmethodforthedetermin-ation of metronidazole and oxytetracycline hydrochloride in theveterinarydrugNozematforhoneybeesThetechniqueisbasedonthereductionofpolarographicallyactivecompoundsonamercurydropletelectrodeTheinfluenceofthecomponentsoftheveterinarydrugNozematonthepolarographicdeterminationofmetronidazolewasstudiedItwasfoundthatthereductionofmetronidazoleisnotaffected by glucose and ascorbic acid but is affected by oxytetra-cyclinehydrochloridewhichisreducedtomercurydropletelectrodeatapotentialofndash145VThedevelopedtechniqueischaracterizedbyeaseofsamplepreparationandcost-effectivenessThistechniquehastheabilitytoidentifysimultaneouslyanddeterminatemetronidazoleand oxytetracycline hydrochloride in solution without the use ofseparationandconcentrationmethods
Keywordselectrochemistrymetronidazoleoxytetracycline
hydrochloridepolarographyveterinarydrug
bees and it can be given in unregulated doses Because of this an unknownamountofmetronidazolecangetintothehoneyanditsometimescausessideeffectsofthehumanbodyanditcouldbeofgreatconcernforpublichealth[56]MedicinesforpeoplearemorestringentandbettertestedthanveterinarydrugsTheproblemofthecontroloftheveterinarydrugsisurgentnowadaysVeterinarymedicinescouldbeunauthorizedandtheuncontrolleduseofmedicinesexistsinretailpharmaciesofmedicineorimportedascontrabandfromothercountries The most widespreadof these classes in thequality controlare chromato-graphic [6ndash9] spectrophotometric [10ndash13] and electrochemical methods[14ndash17]Manyoftheknownmethodsforthedeterminationofmetronidazoleandoxytetracyclinehydrochloridehaveanumberofdisadvantagestime-consumingtheuseoforganicsolventsandexpensivereagentsthesideeffectsofexcipientsandotheractivesubstancesElectrochemicalmethodsarepromisingalternativefor the determination of the electroactive substances Their advantages aresimplicityminiaturizationhighsensitivityandrelativelylowcostThereforethesearch for simple express and affordable methods for the determination ofmetronidazoleremainsrelevantOneofthepromisingmethodsofdeterminationisvoltammetry
2Experimental
21Reagentsandchemicals
VeterinarydrugNozemat (manufacturerAPI-SANRussia) is a yellowpowderwithaslighttypicalodorAvailableinlaminatedbagsof25gCompositionper1gof the drug metronidazole 400 mg oxytetracycline hydrochloride 400 mgglucoseascorbicacid MetronidazoleandoxytetracyclinehydrochloridewerepurchasedfromSigmaAldrich(USA)Stockstandardsolutionofmetronidazolefordeterminationwaspreparedbydissolvingtheexactamountofstandardin7mLof2Mhydrochloricin 500 mL volumetric flask Stock standard solution of oxytetracyclinehydrochloride was prepared by dissolving the exact amount of standard indistilledwaterin500mLvolumetricflaskAfterthatthesolutionswereadjustedtothemarkwithdistilledwaterandmixedthoroughly The Britton-Robinson buffer preparationwas as follows 202 g of sodiumtetraboratedecahydrate287mLofglacialaceticacidand176mLofconcen-tratedorthophosphoricacidweredissolvedin10Lvolumetricflask Working solution preparation was as follows an aliquot of stock standardsolutionwasaddedintoa25mLvolumetricflasktoobtainasolutionwiththenecessaryconcentrationthen2mLofBritton-RobinsonbufferwithnecessarypHwasaddedtotheflaskanddistilledwaterwasaddedtothemark AqueoussolutionofNozematwaspreparedasfollowstheexactportionofthetestveterinarydrugwasdissolved ina250mlvolumetric flaskAnaliquotof
52 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Polarogramsof(A)metronidazoleand(B)metronidazolewithoxytetracyclinehydrochloridendash1solutionsat02MBritton-RobinsonbufferbackgroundatpH=96(υ=05Vs c(metronidazole)=
ndash5 ndash5=45times10 Мc(oxytetracyclinehydrochloride)=50times10 М
100mloftheresultingsolutionwasaddedtoa250mlvolumetricflaskandmadeuptothemarkwithwaterAnaliquotof100mloftheresultingsolutionwasaddedtoa250mlvolumetricflask2mlofBritton-RobinsonbufferwithapHof96wasaddedandthevolumewasadjustedtothemarkwithdistilledwater
22Instrumentation
ForpolarographicmeasurementsweuseddigitaldeviceMTechOVA-410 [18]temperature-controlledthree-electrodeamercurydropletindicatorelectrodeasaturatedcalomelreferenceelectrodeandplatinumwireauxiliaryelectrodeTheaccuracyofthepotentialmeasurementis1mVTheuncertaintyofcurrentmeasu-rement is 01 The employed mercury droplet electrode had the following
ndash4 ndash1characteristicsm=594times10 gs τ=10 min in 02 M NH Cl We used cyclic4
voltammetryforthestudyoftheelectrochemicalprocess WeusedMV870DIGITAL-pH-MESSERATpH-meterformeasuringpHofthesolutions Theobtainedworkingsolutionswereintroducedintothecellanddeoxyge-natedwithargonfor10minPolarogramswererecordedintherangeofpotentialsfrom00tondash16V
3Resultsanddiscussion
Previously it was found that using the Britton-Robinson buffer with pH = 96metronidazoleisreducedwiththeformationofacharacteristiconeirreversiblepeakatndash064V(Fig1A)UsingpolarographywithfastpotentialscanitwasfoundthatmetronidazoleisreducedonmercurydropletelectrodeintherangeofpH20to105ThepeakrecoverycurrentofthemetronidazolereachesthemaximumvalueatpH9ndash10againstthebackgroundofatheBritton-Robinsonbuffer
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 53
Underpre-selected conditions theeffectof some foreign substanceson thepolarographicdeterminationofmetronidazolewasinvestigatedSubstancesthatare componentsofdrugswere studiedglucose ascorbicacidoxytetracyclinehydrochlorideGlucose and ascorbic acid are not reduced atmercury dropletelectrodeanddonotchangetheappearanceofthepolarogramandpolarographiccharacteristics of the recovery of metronidazole As can be seen from Fig 1oxytetracycline hydrochloride is reduced to mercury droplet electrode andchangestheappearanceofthepolarogramandpolarographiccharacteristicsofthe recovery of metronidazole With the addition of oxytetracycline hydro-chloridetherecoverypeakofmetronidazoledecreasesandslightlyshiftstomorenegativepotentials The composition of the drug is relatively complex excipients affect theanalyticalsignaloftherecoveryofcompoundssototakeintoaccountthematrixeffectusedthemethodofmanyadditives QuantitativelytransferredthesolutionofNozemattothecell(exactvolume)removed dissolved oxygen for 10 min and took polarograms in the range ofpotentialsfrom00tondash16VAliquotsofstandardmetronidazolesolutionwereintroducedintothecelltoobtainasolutionwithagivenconcentrationofadditive
ndash5 ndash5metronidazole 10times10 M to 70times10 M As with the determination of themetronidazole aliquots of a standard oxytetracycline hydrochloride solutionwereaddedtothecellwithsolutionofNozemattoobtainasolutionwithagiven
ndash6 ndash6additiveconcentrationfrom70times10 Mto50times10 M(Fig2) In Table 1 are shown metrological characteristics of the determination ofmetronidazoleandoxytetracyclinehydrochloride inveterinarydrugRecoverywascalculatedFormetronidazoletherecoveryis97andforoxytetracyclinehydrochloridetherecoveryis103Analyticalperformanceofthetechniqueisgoodfordeterminationveterinarydrugs
54 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2Polarogramsof(A)metronidazolereductionatdifferentmetronidazoleconcentrationsand(B)oxytetracyclinehydrochloridereductionatdifferentoxytetracyclinehydrochlorideconcentra-tionsandtheircorrespondingcalibrationgraphs
Theaccuracywasverifiedbytheldquoadded-foundrdquomethodAliquotsofstandardsolutionofmetronidazoleweremadeina250mlvolumetricflasktoobtaina
ndash5solutionofagivenconcentrationof33times10 Mandthesolutionofoxytetracyclinendash5hydrochloridetoobtainasolutionofagivenconcentrationof15times10 M2mlof
Britton-RobinsonbufferwithpH96wasaddedtoflaskwithstirringandadjustedtothemarkwithwaterTheanalysisprocedureofmodelsolutionissimilartoanalysis procedure of the solution of Nozemat The calculated amount ofmetronidazolebythemethodofmultipleadditivesinthetestedmodelsolutionisinagreementwiththeamountthatwasintroducedintothesample
4Conclusions
The new polarographic method for the determination of metronidazole andoxytetracyclinehydrochlorideintheveterinarydrugNozematforhoneybeeswasdeveloped We conducted principal component analysis of veterinary drugNozemattoassesstheoveralleffectforthedeterminationofmetronidazoleWefoundthatoxytetracyclinehydrochlorideisreducedtomercurydropletelectrodeThismethodhastheabilitytoidentifysimultaneouslyanddeterminatemetro-nidazole and oxytetracycline hydrochloride in solution without the use ofseparationandconcentrationmethodsOnemoreofadvantagesoftechniquearefastprocedureofanalysissimplesamplepreparationlowcostthepossibilityofminiaturization
References
[1] Antibiotic and Chemotherapy Finch R Greenwood D Whitley R (edits) AmsterdamElsevier2006p292ndash299
[2] MitrowskaKPrzyczynyiskutkizakazustosowania5-nitroimidazoliuzwierzątktorychtkanki lub produkty przeznaczone są do spozycia przez ludziMed Weter 71 (2015)736ndash742
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 55
Metronidazole Oxytetracycline hydrochloride
PeakspotentialV ndash065 ndash144CorelationcoefficientR 099892 099922
ndash1 4 4SlopebmicroАM 656times10 176times10Δb 1763 400InterceptamicroА 2238 0219Δa 0075 0009
ndash1 ndash5 ndash5cmolL 342times10 125times10ndash1cmgg 389 413
Recovery 97 103
Table 1Validationparametersofthemethodofmetronidazoleandoxytetracyclinehydrochloridedetermi-nationinsolutionsofNozematbythemethodofmanyadditives
[3] VermaPNamboodiryVMishraSBhagwatABhoirSAstabilityindicatingHPLCmethodfor the determination of Metronidazole using Ecofriendly solvent as mobile phasecomponentIntJPharmPharmSci5(2013)496ndash501
[4] Cervini P Ambrozini B Machado LCM Ferreira Garcia AP Cavalheiro Gomes ETThermal behavior and decomposition of oxytetracycline hydrochloride J Therm AnalCalorim121(2015)347ndash352
[5] DangBNAnhNTKKyLXThaiPKAntibioticsintheaquaticenvironmentofVietnamsourcesconcentrationsriskandcontrolstrategyChemosphere197(2018)438ndash450
[6] QuintanillaPHettingaKABeltranMCEscricheIMolinaMPVolatileprofileofmaturedTronchon cheese affected by oxytetracycline in raw goat milk J Dairy Sci 103 (2020)6015ndash6021
[7] Chen F Yu L Jingdong P Xiang W Huanjun P Yu C Yan H Study on simultaneousdetermination of three nitroimidazole residues in honey by high performance liquidchromatographyndashresonanceRayleighscatteringspectraMicrochemJ141(2018)423ndash430
[8] Hernandez-MesaM Cruces-Blanco C Campana GA Simple and rapid determination of5-nitroimidazolesandmetabolitesinfishroesamplesbysalting-outassistedliquid-liquidextractionandUHPLC-MSMSFoodChem252(2018)294ndash302
[9] Xiu-ChunGZhao-YangXHai-HuiWWen-YiKLi-MingLWen-QingCHong-WeiZWen-HuiZMolecularlyimprintedsolidphaseextractionmethodforsimultaneousdeterminationofsevennitroimidazolesfromhoneybyHPLC-MSMSTalanta166(2017)101ndash108
[10] ТеплыхАНИлларионоваЕАКоличественноеопределениеметронидазоласпектро-фотометрическимметодомСибирскиймедицинскийжурнал5(2009)48ndash50
[11] ZheltvayOIZheltvayIISpinulVVAntonovichVPSpectrophotometricdeterminationofmetronidazoleandtinidazoleusingcopper(II)complexesJAnalChem68(2013)663ndash668
[12] Youssef AK Saleh MS Abdel-Kader DA Hashem Facile DY SpectrophotometricdeterminationofmetronidazoleandsecnidazoleinpharmaceuticalpreparationsbasedontheformatioonofdyesIntJPharmPharmSci6(2015)103ndash110
[13] Sversut RA Vieira JC Rosa AM Amaral MS Kassab NM Salgado H ValidatedspectrophotometricmethodsforsimultaneousdeterminationofoxytetracyclineassociatedwithdiclofenacsodiumorwithpiroxicaminveterinarypharmaceuticaldosageformArabianJChem13(2020)3159ndash3171
[14] Nikodimos Y Electrochemical determination of metronidazole in tablet samples usingcarbonpasteelectrodeJAnalMethodsChem(2016)361294
[15] Srivastava AK Upadhyay SS Rawool CR Punde NS Rajpurohit AS Voltammetrictechniques for the analysis of drugs using nanomaterials based chemically modifiedelectrodesCurrAnalChem15(2019)249ndash276
[16] Sahu G Voltammetric behaviour of metronidazole at a composite polymer membraneelectrodeOrienJChem26(2010)81ndash86
[17] Yang Y YanW Guo YWang X Zhang F Yu L Guo C Fang G Sensitive and selectiveelectrochemicalaptasensorviadiazonium-couplingreactionforlabel-freedeterminationofoxytetracyclineinmilksamplesSensorsandActuatorsReports2(2020)1ndash7
[18] httpchemlnueduuamtechdeviceshtml(accesed21stJune2020)
56 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
AnimportantpartofanyanalysisthatsignificantlyaffectsthefinalresultsisthesamplepreparationThelowconcentrationofbiologicallyactivecompoundsandthepresenceofaccompanyingcomponentspreventdirectanalysisofthesamplewithcomplexmatrixcompositionTraditionalmethodsofliquidandsolid-phaseextractionhaveaplentyoflimitationssuchashighlytime-consumingprocedureslarge volume of samples expensive cartridges toxic organic solvents andchallenges in automating the process Therefore the application of extractiontechniquesemployinglowamountofsolvents(microextractionmethods)andthelow toxicity extractantes has become the main research direction in recentyears[12] Solid-phasemicroextraction(SPME)wasproposedbyPavlishinin1989[3]Onevariantofthismethodistousethinrodswithvariouspolymercoatingssuch
Application of microextraction techniques combined with chromatographic methods for the analysis of complex objects
VLADISLAVDEEVELENABESSONOVALIUDMILAKARTSOVA
InstituteofChemistrySaint-PetersburgStateUniversityUniversitetskyprospect26198504PeterhofSaint-PetersburgRussiahitchervmailru
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 57
AbstractThelowconcentrationofanalytesandthepreventionofthematrixinfluence requires a stage for extraction and concentration of thestudiedcompoundsTheclassicalmethodsofliquidandsolid-phaseextractionhavemanylimitationsthatpreventtheiruseinsomecasesMicroextraction techniques are becoming more widespread WestudiedthepossibilityofusingionicliquidstoextractpesticidesfromwatersampleswiththeirsubsequentHPLS-MSdeterminationTheinfluenceonthedegreeofextractionofsuchparametersasanatureofionicliquidsanddispersersolventtheiramountssaltconcentrationvolumeratioofionicliquidsandwatersampledilutionoftheionicliquidsextractwithmethanolwasperformedBesidesconditionsofsolid-phase microextraction of volatile organic compounds fromurine samples obtained from healthy donors and donors withprostate cancer have been found The analysis of volatile organiccompoundsbyGC-MSfollowedbychemometricprocessingallowedachievingahighvalueofbinaryclassificationaccuracy(91)
Keywordschemometricsdispersiveliquid-liquid
microextractionsolid-phase
microextraction
asdivinylbenzenepolydimethylsiloxanepolyacrylateandpolyethyleneglycolwhichappliedtothesurface[4]Thepolymersorbentisplacedintheequilibriumheadspaceaboveacondensedphaseofthesampleandthevolatilecompoundsareextracted Liquidmicroextractionconsistsofusingsmallamountsofliquid(extractant)inequilibriumwiththegasorliquidphaseofthesampleDispersiveliquid-liquidmicroextraction(DLLME)isavariantofliquidmicroextractionTheessenceofthemethodisasfollowsextractantisdissolvedinthephaseofadispersingsolventandthemixtureisrapidlyinjectedintothesamplevolume[5]Inthiscasethedispersing solvent is dissolved and a ldquocloudrdquo of extractant is formed A largesurfaceareacontributestomasstransferprocessesThecombinationofDLLMEwiththeuseof ionic liquids(ionic liquids)asextractantsreducestheharmfulimpactontheenvironment[6] Sothegoalofthisstudywastheapplicationofmicroextractionmethodsfortheanalysisofrealsamples
2Experimental
21Reagents
Deionizedwaterwas obtained at the AQUILON D 301 deionizer (Russia) Allchemicals and reagents (the highest commercially available purity) werepurchasedfromReachimBakerAcrosorganicsandSigmaAldrich
22Instrumentation
HPLCanalysiswascarriedoutusinganHPLCLCMS-8030(Shimadzu)withatriplequadrupole mass-selective detector with electrospray ionization Analysis ofvolatileorganiccomponents(volatileorganiccompounds)ofurinesampleswasmade by GCMS-QP2010 SE (Shimadzu) Chemometric data processing wasperformedusingRStudio
23DeterminationofvolatileorganiccompoundsinurinesamplesbyGC-MSmethod
ForSPMEofvolatileorganiccompounds inurinesamplewasusedfibercoatedwith a polydimethylsiloxane (PDMS) The volatile organic compounds wereextractedontofibercoatingfor20minat50degCThentheanalytesweredesorbedinto the gas chromatography for 4 minutes at a temperature of 250degCChromatographic separation was carried out on a HP-5 capillary column(30mtimes250μmtimes025μm)usingtemperatureprogrammingmodeThetempera-
ndash1tureofovenwasincreasedfrom50degCupto250degCatarateof10degCmin Tionsourcewas200degCMassspectrometrywasusedinSIMmode(mz=35ndash900)
58 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
24ConditionsforLCMSMSdeterminationofpesticides
Separation of pesticides was performed by HPLCMSMS with positiveelectrosprayionizationoncolumnZorbaxBonusRP35μm(21times100mm)with40mMammoniumacetateandmethanolasmobilephaseAandBrespectivelyThefollowinggradientelutionwasapplied20ndash85B(8min)85B(8ndash15min)85ndash95B(150ndash155min)95B(155minus180min)95ndash20B(180minus185min)
ndash1Thevelocityof themobilephasewas03mlmin Thevolumeof the injectedsamplewas 20 microlMS detection capillary voltage +45 kV spray gas velocity
3 ndash1 3 ndash13dm min flow rate and drying gas temperature 15 dm min and 250 degCrespectively
25Selectionofconditionsfordispersiveliquid-liquidmicroextractionofpesticides
The influence of the natures of ionic liquids ([C MIM][PF ] [C MIM][NTf ]4 6 6 2
[C MIM][BF ])andthedispersingsolvent(methanolacetonitrileacetone) the6 4
weightoftheionicliquids(0060ndash0200g)thevolumeofthedispersingsolvent(02ndash10ml)onthedegreeofpesticidesextractionwerestudiedTheinfluenceofthepH(5422)theconcentrationofNaCl(0040ndash0200g)andextractiontime(1-6min)wereinvestigated The effects of different ionic liquids and disperser solvents on DLLMEprocedures were investigated and optimized by using standard solutions ofpesticidesIndetailasolutionofionicliquidsinadispersingsolventwaspreparedand rapidly injected into the aqueous sample solution (2ml) followed bytreatment for 2 min in an ultrasonic bath cooling at ndash4degC for 10 mincentrifugationfor10minat3500rpmandcollectionofionicliquidsThewaterphasewas separatedandanalyzedbyHPLC-MSThe ionic liquidsextractwasdilutedinmethanolandanalyzedbyHPLC-MS
3Resultsanddiscussion
31Microextractionofpesticides
One of the important tasks of environmental monitoring is to control traceconcentrationsofpesticidesinwatersamplesTechniqueofcombiningseveralpesticideshasbecomemorewidespread inagriculture It allowsreducing thetotalconcentrationoftheappliedcompoundsandtodecreasetheadaptabilityofpathogens and insects Therefore the analysis of real samples requires apreliminarystageofselectiveanalytesextractionandconcentration ExtractinganddispersingsolventsarebothimportantinDLLMEofanalytesTheinfluenceofthenatureofthedispersingsolvent(methanolacetonitrileandacetone) and the extractant (imidazolium ionic liquids [C MIM][PF ]4 6
[C MIM][BF ]and[C MIM][NTf ])onthedegreeofextractionofpesticideswas6 4 6 2
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 59
studied This parameter was controlled by the residual concentration ofpesticidesinthewaterphaseafterextractionThebestresultswereobtainedforionic liquids [C MIM][PF ] as an extractant and acetonitrile as a dispersing4 6
solvent Thenextstepwastoselecttheamountofionicliquids(0060ndash0200g)andthevolume of acetonitrile (02ndash10 ml) It was found that the highest degree ofextractionofanalyteswasachievedbyusing020gofionicliquidsand03mlofacetonitrile It was shown that the degree of extraction of selected pesticides does notdependonpHofthewatersamplewhichconfirmsthepartitionmechanismofextractionThedegreeofextractionofcarbofosincreasedslightlywithanincreaseinthesaltconcentrationandreachmaximumbyweightto4(008g) ItisknownthatthehighviscosityofionicliquidshavehinderedtheprocessesofelectrosprayionizationDilutionofthesamplewithmethanolby3timesgivesthebestresult(thesignalintensitywas373to845ofthesignalwithoutionicliquids) ThustheconditionsofDLLME-ionicliquidsextractionofpesticidesfromwatersampleswere found The limits of detection for pesticideswere from007 to
ndash1019ngml thereproducibilityofpeakareaswerefrom3to5theextractionrecoverywascloseto100
32SPMEofvolatileorganiccompoundsfromurinesamples
Oneof the importantdirections is the search for criteriaofearlydiagnosisofcancer Obtaining characteristic profiles of volatile organic compounds fromurinesamplescanhelpdevelopanon-invasivemethodforearlydiagnosisofthedisease ForthiswestudiedtheinfluenceofanumberoffactorsonthetotalnumberofpeaksandthetotalpeakareaTheywerethetemperatureatwhichthevaporandcondensedphasesofurinewerebalanced(30ndash60degC)thepreheatingtimeofthesample(10ndash40min)NaClconcentration(30ndash133wv)andthesorptiontimeonthePDMScoating(5ndash30min) Anincreaseinthepreheatingtemperatureofthesampleto500degCledtoanincreaseinthenumberofsignalswhichdidnotchangewithafurthertempera-tureincreasingNextparameterwasthetimeofachievementequilibriumofthevapor and condensed phases The largest number of peaks was observed at40minbutthisgreatlyincreasedthetimeofanalysisandsowechose20minAlsowestudiedthedesaltingeffectofsodiumchlorideonefficiencyofextractionThebestvolatileorganiccompounds sorptionwasachievedbyadding133saltingagent It is also shown that thenumberofpeaksdidnot changeafter20minutesofsorption Thus to obtain the characteristic profiles of urine samples the followingconditionswereselected1333NaClwasaddedtotheurinesample(3ml)
60 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
heatingfor20minat50degCthensorptionofvolatileorganiccompoundsonPDMSfibercoatingat50degCfor20min Undertheselectedconditionsweobtainedvapor-phaseprofilesof52urinesamples (32 normal and 20 pathology) Prior to performing chemometricprocessing of chromatographic profiles of urine samples preliminary datapreparation is necessary [7] The baseline was removed and the peaks werealignedusingdynamictimewarpingwithcontrollingbymassspectra ThePCAmodelwasbasedon52aligned characteristicprofilesThere is asatisfactoryseparationofdataintotwoclustersinthescoresplotrelativetothefirstandsecondprincipalcomponent(Fig1) The original data set (52 samples) was randomly divided into calibration(13pathology21normal)andtest(7pathology11normal)setsThenthePLS-DAmodelwasbuiltusingthecalibrationsetanditspredictiveabilitywasevaluatedusingthetestsetTheprocedurewasrepeated100timesTheaveragevaluesofsensitivityspecificityandaccuracyinthiscasewere9594and91
4Conclusions
Thepossibilityofusingimidazoliumionicliquids([C MIM][PF ])asextractants4 6
forquantitativeextractionandconcentrationofpesticidesundertheconditionsofDLLMEisshownThedegreeofconcentrationwas28ndash33whichallowedreaching
ndash1thedetectionlimits(006ndash019ngml )belowthemaximumpermissibleconcen-trationThepossibilityofnon-invasivediagnosisofprostatecancerbySPMEofvolatileorganiccompounds inurine isshownChemometricprocessingofgaschromatographic profiles using PLS-DA and PCA methods allowed achievingclassificationaccuracyvaluesmorethan90
Acknowledgments
ThisworkwassupportedbytheRussianFoundationforBasicResearchprojectno18-53-80010BRICS_t and the Russian Science Foundations (Projects 19-13-00370) We are grateful to
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 61
Fig 1Scoresplotrelativetothefirstandsecondprincipalcomponent
Resource Education Center in Chemistry of St Petersburg State University for the providedequipment
References
[1] Rutkowska M Płotka-Wasylka J Sajid M Andruch V Liquidndashphase microextractionAreviewofreviewsMicrochemJ149(2019)103989
[2] JaliliVBarkhordariAGhiasvandAAcomprehensivelookatsolid-phasemicroextractiontechniqueAreviewofreviewsMicrochemJ152(2020)104319
[3] ArthurCLPawliszynJSolidphasemicroextractionwiththermaldesorptionusingfusedsilicaopticalfibersAnalChem62(1990)2145ndash2148
[4] SchmidtKPodmoreISolidphasemicroextraction(SPME)methoddevelopmentinanalysisof volatile organic compounds (VOCs) as potential biomarkers of cancer JMol BiomarkDiagn6(2015)1000253
[5] Mousavi L Tamiji Z Khoshayand MR Applications and opportunities of experimentaldesign for the dispersive liquidndashliquidmicroextractionmethod ndash A review Talanta190(2018)225ndash356
[6] MarcinkowskaRKoniecznaKMarcinkowskiLNamiesnikJKloskowskiAApplicationofionic liquids inmicroextractiontechniquesCurrent trendsandfutureperspectivesTrACTrendsAnalChem119(2019)115614
[7] WehrensRChemometricswithRMultivariateDataAnalysisintheNaturalSciencesandLifeSciencesBerlinSpringer2011
62 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Theself-assembledtwo-dimensionalmonolayers(2DSAMs)ofvariousmolecules(eg graphene [1] MoS [2] rubrene [3]) offer beneficial properties for the2
constructionofnano-electronic andnano-opticaldevicesThe topological andchemicalcharacterizationof2DSAMsiscrucialtogatherinformationaboutthearrangementofdepositedmoleculesandtheirinteractionwiththesubstrateThistaskrequiresanalyticaltechniqueswith(sub)nanometerspatialresolutionandup to single-molecular detection sensitivity Only few techniques meet therequirements and one of them is tip-enhanced Raman spectroscopy (TERS)whichcombinestheexcellentspatialresolutionofscanningprobemicroscopy(SPM) and chemical sensitivity of surface-enhanced Raman scattering (SERS)spectroscopy[45] TheSERS spectroscopyutilizesplasmonicmetal nanostructures to cause ahighlocalenhancementoftheelectricfieldintheirclosevicinityviathesurfaceplasmonresonance(SPR)effectThelocalelectricfieldcausesanincreaseofthe
The development of reference probe system for tip-enhanced Raman spectroscopy
MARTINKRA LMARCELADENDISOVA PAVELMATE JKA
DepartmentofPhysicalChemistryFacultyofChemicalEngineeringUniversityofChemistryandTechnologyPragueTechnickaacute516628Prague6CzechRepublicMartinKralvschtcz
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 63
AbstractThetip-enhancedRamanspectroscopy(TERS)isamodernanalyticaltechniquewithanoutstandingspatialresolutionandchemicalsensi-tivityTheseparametersmainlydependon the structural integrityand chemical purity of employed plasmonic scanning probe tipsUsuallyeachtipistestedbeforeTERSmeasurementsusingcommer-ciallyavailablereferencesamplesHowevertheirpriceandrelativelyshortexpirationdatemustbeconsideredwhenplanningaresearchbudgetWedevelopedaproceduretoproduceself-madereferenceprobesamplesfortestingTERStipsusingcopper(II)phthalocyanineonaAunanolayerwhichispreparedbythermalvacuumevaporationofAuonaSiwaferOurresultsshowthatthepreparedsystemenablesrepeateddetectionofwell-resolvedTERSspectraThecollectedTERSspectraandspectralmapsexhibitsomedegreeofvariabilitywhichmaybeduetovariousphoto-inducedprocessesanditmustbeconsi-deredwhileperformingTERSmeasurements
Keywordscopper(II)
phthalocyaninescanningtunnelling
microscopyresonanceRaman
spectroscopysurface-enhancedRaman
spectroscopytip-enhancedRaman
spectroscopy
Ramanscatteringfrommoleculesboundtothemetalby6ndash8ordersofmagnitude[6]TheenhancementallowsSERSspectroscopytobeusedforsingle-moleculardetectionHoweverRamanmicroscopeshave limitedspatialresolutionbythelightdiffractionwiththeachievableresolutionbeingaroundhalfoftheexcitationwavelengthOntheotherhandthespatialresolutionofSPMtechniquesislimitedonlybythedimensionsoftheapexofthescanningprobetipwhichmayevenbeatomicallysharpByutilizingSPMtipandsubstratemadefromplasmonicmetalsan artificial ldquohotspotrdquomay be createdwith its position and dimensions beingdefinedbythetipItopensthepossibilitytocollectstronglyenhancedRamanspectrafromtheareapreciselylocalizedbellowthetipandthusovercometheopticaldiffraction limitTheartificialhotspotmayberelocatedbymovingthesamplebelowthetipwhichisthefoundationofTERSmapping[578] A successful TERS experiment requires an optimal combination of variousparameters the most important of which are tip sharpness and purity [7]Areferencesampleconsistingofaflatplasmonicnanolayerwithattachedprobemolecules is frequently used to check the state of the tip before using it forexperiments Unfortunately commercially available TERS standards areexpensiveandhaveanexpirationdateofseveralmonths Thegoalofthisstudywastofindapreparationprocedurewhichwouldbeableto produce cheap reference samples for repeated detection of intense TERSspectraAcombinationofaAunanolayeronaSisubstratepreparedbythermalvacuumevaporationwithadsorbedcopper(II)phthalocyanine(CuPc)whichisamoleculewithhighRamancross-sectionwastested[910]Copper(II)phthalo-cyanine known as phthalocyanine blue is a synthetic blue pigment and isfrequently used in paints It has been studied as a potentialmaterial for theconstruction of organic solar cells and other photoelectronic devices [11] AsindicatedbyitscolourCuPcexhibitsseveralabsorptionbandswithinthevisibleregion The effects of a transition to excited electronic states and subsequentluminescencemaybeobservable inRamanmeasurements[12]Au isahighlysuitablemetalforthesampleasitisboththermallyandelectricallyconductivewhichlimitsthelocalheatingofthesampleduringthemeasurementsandenablesthe use of scanning tunnelling microscopy (STM) for tip-surface interactionfeedback[13]
2Experimental
21Reagentsandchemicals
Thesubstrateforthesamplewaspreparedbythermalvacuumevaporationofgoldonsilicon(100)waferFirsta5nmthickCradhesionlayerwasdepositedon
ndash1thewaferfollowedby100nmofAuThedepositionratewas4nmmin forCrand ndash18nmmin for Au The base pressure of the evaporation system was below
ndash65times10 mbarFollowingthepreparationproceduredescribedbyJiangetal[14]
64 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
thecleansubstratewasimmersedintoasaturatedsolutionofCuPc(˃99SigmaAldrich USA) in dimethylformamide (˃98 Lach-ner CZ) for at least 12h atambienttemperatureSubsequentlythesamplewasremovedfromthesolutionrinsedwithMilli-Qwaterandmethanol(paPentaCZ)anddriedwithair
22Instrumentation
TheRamanSERSandTERSspectrawererecordedusingRamanspectrometerInVia Reflex (Renishaw UK) equipped with lasers emitting at two differentexcitation wavelengths 633nm (136mW max power output) and 785nm(204mWmaxpoweroutput)Thespectrometerhasathermoelectricallycooled
ndash1CCDdetectorwithaspectralresolutionof2cm and4microscopeobjectiveswith5times20times50timesand100timesmagnitudeForTERSexperiments the laserbeamwasredirectedtotheSPMplatformInnova-IRIS(BrukerUSA)viaasystemoflightguidesElectrochemicallyetchedAuTERS-STMtips(BrukerUSA)wereusedforallTERSmeasurements The spectra were processed using the Spectragryph software (F MengesldquoSpectragryph - optical spectroscopy softwarerdquo Version 1214 2020httpwwweffemm2despectragryph) Using this software all collectedspectra were treated by a Savitzky-Golay noise filter automatic baselinecorrectionspikeremovalandpeaknormalization
3Resultsanddiscussion
31Ramanmeasurementsofcrystallinecopper(II)phthalocyanine
AtfirsttheRamanspectraofpureCuPcwerecollectedtoprovidereferencedatawhileusingboth633and785-nmexcitationlasers(Fig1)Bothspectraexhibit
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 65
Fig 1RamanspectraofCu(II)phthalocyanineincrystallineformmeasuredat633(top)and785-nm(bottom)excitationThespectraareoffset
ndash1vibration bands in the 500ndash1600 cm region with slight differences in theirintensityratiosThe633-nmexcitationallowstheobservationofadditionalbands
ndash1inthe2000ndash3000cm region(onaluminescencebackground)whichoriginatefrom the resonance Raman effect as the excitation energy overlaps with theQ-bandofCuPc[12]MoreovertheprocessofelectronicexcitationmaylowertheD symmetry of CuPc during resonance Raman scattering and previously4h
forbiddenbandsmaybecomeobservable[15]Theluminescentbackgroundhasandash1maximumaround2200cm whichcorrespondstoamolecularemissionbandat
735nmEventhoughthespectrameasuredwiththe785-nmlaserlinedonotexhibitapparentresonanceenhancementapre-resonanceRamanenhancementmayoccur
32Surface-enhancedRamanmeasurementsofcopper(II)phthalocyaninelayeronagoldsubstrate
The prepared sample of CuPc on a Au layer was analysed using the Ramanmicroscope Both excitation laserswere used to obtain SERS spectra (Fig 2)whichwerecomparedtothespectraofapurecrystallineCuPc Thepositionsof bands in SERS spectra closelymatch their positions in thespectraofbulkCuPcHoweveraslightshiftofsomespectralbandsisobservable
ndash1(eg1528rarr1532cm )whichmaybeattributedtotheinteractionbetweenCuPcand the Au substrate The disappearance of luminescence background andresonance-enhancedbandsinthespectrumat633-nmexcitationalsosuggeststhemolecule-metalinteractionandthetransferofenergyfromCuPcmoleculestothesubstrateMoreovertherearevariationsintherelativeintensitiesofbands
ndash1whichdependontheexcitationenergyegthebandat1309cm isenhancedinSERSspectraatthe785-nmexcitationwhencomparedtothespectraofpureCuPcorevenSERSspectraat633-nmexcitation
66 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2SERS spectra of Cu(II) phthalocyanineon aAu layermeasured at 633 (top) and785-nm(bottom)excitationThespectraareoffset
33Tip-enhancedRamanmeasurementsofcopper(II)phthalocyaninelayeronagoldsubstrate
SERS microspectroscopy is a diffraction-limited technique as it provides anaveragedinformationaboutmoleculesintheilluminatedareaofseveralsquaremicrometers Meanwhile TERS spectra are collected from an area of tens ofnanometers and they contain specific information about the local moleculararrangement topography of the underlyingmetal and properties of the localelectric field between the tip and the substrate Therefore a higher spectralvariabilityshouldbeexpected Several TERS mapping experiments were carried out using both 633 and785-nmexcitationwithvaryingexperimentalparameterssuchasthenumberanddistance betweenmeasured points acquisition time number of acquisitionslaserpoweretc TheTERSspectrameasuredat633-nmexcitationexhibited lowersignal tonoiseratioandreproducibilityAsaconsequenceTERSmappingwasimpossibleandonlyafewone-pointTERSspectrawereobtained(Fig3) TheTERSspectracollectedat785-nmexcitationcontainedahighernumberofwell-resolvedbandsMoreoverthespectrawerestableintimeandsotheTERSmappingwaspossibleThe twopresentedTERSspectraareaveragesofTERSmaps which contained 16 and 80 points with 600 and 300-nm spacingrespectively(Fig4) Thespectracollectedusingbothexcitationwavelengthsexhibitavariabilityinrelative intensities and positions of bands between themeasured points ThevariabilitymaybeattributedtothelocalorientationofCuPcmoleculesbetweenthe tip and the Au surface and the properties of strongly enhanced andnon-homogeneous electromagnetic field which depend on the tip-surfacedistancetheirmorphologyandrelativepositionMoreovertheusedexcitation
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 67
Fig 3TwoexamplesofTERSspectraofCu(II)phthalocyaninemeasuredat633-nmexcitationThespectraareoffset
wavelengthsareclosetoabsorptionbandsofCuPcandthestrongelectricfieldmay give rise to photo-induced effects These effects include the electronicexcitationofCuPctohigherstateschargetransferbetweentheCuatomandthephthalocyaninering ionizationof themoleculeand formationofradicalsThephoto-inducedprocessesarelikelytoplayabiggerroleinTERSspectradetectedat633-nmexcitationduetotheoverlapwithQ-bandofCuPcwhichmaybethecauseoftheirlowersignaltonoiseratioandreproducibility
4Conclusions
Thedeveloped referenceprobe systemofCuPc adsorbedon aAu surfacehasprovedtobesuitablefortheintendeduseasitenabledthedetectionofintenseandwell-resolvedSERSandTERSspectraTheAulayerpreventsoverheatingofthesampleandallowsfortheuseofSTMTheSERSspectrawereinagoodmatchwith thespectraofpureCuPcAslight shiftof somebandsandchange in theluminescent background indicated the interaction between CuPc and the AusurfaceTheTERSexperimentsresultedinspectralmapswithhighintensitiesofindividualspectraIncreasedvariabilitybetweenmeasuredpointswasobservedPossiblesourcesofthevariabilityarephoto-inducedprocessesthatmayoccurinthestronglyenhancedelectricfieldTheseeffectsareaknownfeatureofTERSmeasurements and they offer valuable insight into the photophysics andphotochemistryofCuPcinteractingwiththeAusurfaceThedependenceofTERSspectra on experimental parameters and the preparation procedure of thereferencesampleshouldbefurtherstudied
Acknowledgments
ThisworkwassupportedfromthegrantofSpecificuniversityresearchndashA2_FCHI_2020_039
68 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 4TwoexamplesofaveragedTERSspectraofCu(II)phthalocyaninemeasuredwith785-nmexcitationThespectraareaveragesfromTERSmapsincluding16(top)and80(bottom)measuredpointsThespectraareoffset
References
[1] Mas-BallesteRGomez-NavarroCGomez-HerreroJZamoraF2DmaterialstographeneandbeyondNanoscale3(2011)20ndash30
[2] ZengHCuiXAnopticalspectroscopicstudyontwo-dimensionalgroup-VItransitionmetaldichalcogenidesChemSocRev44(2015)2629ndash2642
[3] SchultzJFLiLMahapatraSShawCZhangXJiangNDefiningmultipleconfigurationsofrubreneonaAg(100)surfacewith5A spatialresolutionviaultrahighvacuumtip-enhancedRamanspectroscopyJPhysChemC124(2020)2420ndash2426
[4] WhitemanPJSchultzJFPorachZDChenHNJiangNDualbindingconfigurationsofsubphthalocyanineonAg(100)substratecharacterizedbyscanningtunnelingmicroscopytip-enhanced Raman spectroscopy and density functional theory J Phys Chem C 122(2018)5489ndash5495
[5] ShaoFZenobiRTip-enhancedRamanspectroscopyprinciplespracticeandapplicationstonanospectroscopicimagingof2DmaterialsAnalBioanalChem411(2019)37ndash61
[6] ArocaRSurface-EnhancedVibrationalSpectroscopyHobokenWiley2006[7] KumarNMignuzziS SuWRoyDTip-enhancedRamanspectroscopyprinciplesand
applicationsEPJTechInstrum2(2015)9[8] BailoEDeckertVTip-enhancedRamanscatteringChemSocRev37(2008)921ndash930[9] BovillAJMcConnellAANimmoJASmithWEResonanceRamanspectraofα-copper
phthalocyanineJPhysChem90(1986)569ndash575[10] Shaibat MA Casabianca LB Siberio-Perez DY Matzger AJ Ishii Y Distinguishing
polymorphsofthesemiconductingpigmentcopperphthalocyaninebysolid-stateNMRandRamanspectroscopyJPhysChemB114(2010)4400ndash4406
[11] SzybowiczMRunkaTDrozdowskiMBałaWGrodzickiAPiszczekPBratkowskiAHightemperaturestudyofFT-IRandRamanscatteringspectraofvacuumdepositedCuPcthinfilmsJMolStruct704(2004)107ndash113
[12] CaplinsBWMullenbachTKHolmesRJBlankDAFemtosecondtonanosecondexcitedstatedynamicsofvapordepositedcopperphthalocyaninethinfilmsPhysChemChemPhys18(2016)11454ndash11459
[13] SaccoAImbraguglioDGiovannozziAndreaMPortesiCRossiAMDevelopmentofacandidatereferencesampleforthecharacterizationoftip-enhancedRamanspectroscopyspatialresolutionRSCAdv8(2018)27863ndash27869
[14] JiangSChenZChenXNguyenDMatteiMGoubertGVanDuyneRPInvestigationofcobaltphthalocyanineatthesolidliquidinterfacebyelectrochemicaltip-enhancedRamanspectroscopyJPhysChemC123(2019)9852ndash9859
[15] MelendresCAMaroniVARamanspectraandnormalcoordinateanalysisoftheplanarvibrationsofironphthalocyanineJRamanSpectrosc15(1984)319ndash326
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 69
1Introduction
The aim of this work has been develo-pmentofanewvoltammetricmethodforthe determination of 23-dimercapto-1-propane-sulfonic acid (DMPS) Fig 1Investigationwasdonetoobtainrelevantinformation about complexingbehaviorofDMPStowardsleadions Lead is one of heavy metals which can cause irreversible neurologicalproblems [1 2]DMPS is a synthetic antidotewith two thiol groups used fortreatmentofpoisoningbyheavymetals[3ndash6]StrongcomplexingpropertieshighwatersolubilityandnegligiblesideeffectsarethemostimportantadvantagesofDMPS[47]
Fig 1 Chemical structure of 23-dimercap-to-1-propane-sulfonicacid
Determination of heavy metal poisoning antidote 23-dimercapto-1-propanesulfonic acid using silver solid amalgam electrode
ab ab bc bMARTACHOIN SKA VOJTE CHHRDLICKA BEATRIZRUIZREDONDO JIR IBAREK aTOMA S NAVRATIL
a JHeyrovskyacuteInstituteofPhysicalChemistryoftheCzechAcademyofSciences Dolejškova21553182thinsp23Prague8CzechRepublicmartachoinskagmailcomb UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova20308128thinsp43Prague2CzechRepublic
c UniversityofValladolidPlazadeSantaCruz847002ValladolidSpain
Abstract23-Dimercapto-1-propane-sulfonic acid (DMPS) was investigatedusingdirectcurrentvoltammetry(DCV)differentialpulsecathodicstrippingvoltammetry(DPCSV)differentialpulseanodicstrippingvoltammetry(DPASV)andeliminationvoltammetrywithlinearscan(EVLS)atapolished(p-AgSAE)andatameniscusmodifiedsilversolidamalgam electrode (m-AgSAE) EVLS confirmed two consecutivereductions with coupled protonelectron transfer VoltammetrictitrationsofDMPSwithPb2+provedcomplexformationwithlimits
minus1ofquantification(LOQs)anddetection(LODs)03and01micromolL atminus1m-AgSAE and 08 and 03micromolL at p-AgSAE respectively
Determination of DMPS in commercial drug Dimaval and humanurine samples confirmed practical applicability of the developedmethod
Keywordscathodicstripping
voltammetry23-dimercapto-1-pro-
panesulfonicacideliminationvoltammetry
withlinearscansilversolidamalgam
electrodeunithiol
70 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Voltammetry was chosen as a determination technique due to its highsensitivity and selectivity speed low costs [8] Thiol groups in DMPS can beoxidatively chemisorbed on solid amalgam electrode It can be used as anaccumulation step for cathodic strippingvoltammetry [9ndash11]Moreover solidamalgamelectrode (SAE)was chosen as theworking electrodebecauseof itspropertiesashighsignaltonoiseratiowidepotentialwindowandabilitytoreachlowlimitsofdetection(LOD)[12ndash13]
2Experimental
21Reagentsandchemicals
Allsolutionswerepreparedusingdeionizedwater(Milli-Q-GradientMilliporendash1PragueCzechRepublic)withconductivitylt005microScm Britton-Robinsonbuffer
solutionspHrangefrom2to12werepreparedbymixingtheproperamountsof02MNaOH(alkalinesolution)andof004MH BO 004MH PO and004M3 3 3 4
CH COOH(allLachemaCzechRepublic)acidicsolutionTheacidicsolutionwas3
preparedbydissolutionof1235gofH BO pa088mLofH PO (85)paand3 3 3 4
1435mLofCH COOH(99)pa in500mLofdeionizedwaterThealkaline3
solution was prepared by dissolution of 3995 g of NaOH pa in 500mL ofdeionizedwater(allLachemaCzechRepublic) StocksolutionofDMPSwaspreparedbydissolving10mgofsolid23-dimer-capto-1-propanesulfonic acid monohydrate pa (Merck Czech Republic) in100mLofdeionizedwaterForthepreparationofthemodelsamplesolutiononecapsuleofdrugDimaval(HeylGermany)contains100mgofDMPSwasdissolved
ndash1in10LofdeionizedwatertotheDMPSconcentrationof0531mmolL Twomodel samplesofDimavalwerepreparedbydilutionof theabove-mentioned
ndash1solutionwithBritton-Robinsonbuffersolutiontoconcentrations10micromolL andndash1of 10micromolL respectively Urine model samples were prepared by mixing
Britton-Robinsonbuffer solutionwithurine samples obtained fromvolunteer(manhealthy30yearsold)inratio11SamplepHwasadjustedbyadditionof
ndash1proper amount of 02molL NaOH Before each measurement oxygen wasremoved for 5minbynitrogenbubbling (purity class 46MesserTechnogasPragueCzechRepublic)
22Instrumentation
Measurementswereperformedusingtwotypesofworkingelectrodesmeniscusmodified silver solid amalgam electrode (m-AgSAE working surface of
20382plusmn0025mm α lt 005) and polished silver solid amalgam electrode2(p-AgSAEworkingsurfaceof0196plusmn0015mm αlt005)Ag|AgCl|3MKClwas
used as the reference electrode andplatinumwire (Oslash1mm)wasused as theauxiliary electrode (both from Elektrochemicke detektory Czech Republic)Measurementswereperformedatlaboratorytemperature(25plusmn2degC)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 71
The pH was measured using pH-meter Jenway 3505 with combined glasselectrodetype924001(BibbyScientificLimitedUK)Voltammetricmeasure-ments were performed using the computer-controlled Eco-Tribo Polarograph(Polaro-Sensors Czech Republic) Software used for measurements wasMultiElChem 33 forWindows XP7810 (J Heyrovsky Institute of PhysicalChemistryoftheCzechAcademyofSciencesCzechRepublic)
3Resultsanddiscussion
OptimumconditionsformeasurementswereobtainedbyseriesofmeasurementinwiderangeofpHvaluesandtestingvariouscleaningproceduresOptimumpotentialof accumulation (E ) and timeof accumulation (t )ofDMPSwereacc acc
adjusted for differential pulse cathodic stripping voltammetry (DPCSV) atp-AgSAEandm-AgSAE ThedependencebetweenpeakheightandconcentrationofDMPSatp-AgSAEhasalogarithmicshapewhichcorrespondstotheaccumulationprocessattheelectrodesurfaceThelineardependencewasobservedintheDMPSconcentra-
ndash1 ndash1tionsfrom03micromolL to20micromolL Peakshifttowardsnegativepotentialwithincreasing concentration of DMPS corresponds to the metal-thiol bond andinfluenceofelectrodesurfacestructureonthisbondOnthecontrarysignalsonm-AgSAEweremorestableandthedependencebetweenconcentrationofDMPSandsignalwasalmostlinearinwholetestedrangeofconcentrations The developedmethodwas tested inmodel samples of Dimaval and urineFoundamountsofDMPSwereingoodagreementwithdeclaredcontentsusingbothelectrodesHowevertherepeatabilityofsignalsregisteredusingp-AgSAEinurinesamplesweresignificantlyworsethanthoseinDimavalsamplesItcanbecausedbycomplicatedbiologicalmatricesandfoulingeffectsofurine ELSV measurements confirmed two consecutive reductions of DMPS inadsorbed state At m-AgSAE signal were at about minus415 mV and minus440 mVrespectively and at p-AgSAE at about minus790 mV and minus830 mV respectively
ndash1 ndash1Reductionsatm-AgSAEatthescanratesfrom80mVs to640mVs havebeencontrolledbyakineticprocessatminus400mV Inanodicscansonm-AgSAEonlyonepeakwasvisibleatabout‒390mVItcorrespondswiththeoxidationofmercuryelectrodesurfaceontheelectrodeinthepresenceofDMPSandwithdiffusionfromthebulksolutionofproductsAtp-AgSAEnosignificantanodicsignalwasfound ThelastpartoftheresearchrevealedvoltammetricbehaviorofDMPSinthe
2+presence of Pb Voltammetric titration was investigated by DPCSV anddifferential pulse anodic stripping voltammetry (DPASV) during consecutive
ndash1 ndash1additionsof1mmolL ofPb(NO ) into100micromolL DMPSsolutioninacetate3 2
bufferofpH50InabsenceofPb2+ontheelectrodesurfaceHg(DMPS)complexisformedduringtheaccumulationstepDuringtheanodicscanthereisonlyonewelldevelopedreductionpeak(Fig2A)
72 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ndash1Fig 2DPCSandDPASvoltammogramsof10micromolL ofDMPSinacetatebufferpH=5correspond-2+ingto[Pb DMPS]ratiosof(A)01(B)11and(C)21Uppercurvecorrespondstothecathodic
scanE =0mVt =15sLowercurvecorrespondstoreverseanodicscanwitht =15satacc acc accndash1E =minus1000mVν=20mVs (Ref[16])acc
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 73
2+ WhenPb DMPSratioisequalto11twooxidationandthreereductionpeakswereregistered(Fig2B)PeakA ataboutndash300mVcorrespondstoformationofred
0Pb(DMPS)complexThiscomplexwasfurtherreducedtothePb (Hg)atabout2+ndash500mV(C )ReductionpeakoffreePb wasalsoregistered(B )Oxidationred red
peaksA andB correspondtoreverseprocessesandC isnotpresentbecauseox ox ox
nofreeDMPSispresentinthesolutionAtratio21thereisnofreeDMPSinthesolution however excess of lead ions As a consequence A and B peakred ox
2+increasedIncreaseofB correspondstothedepositionofPb duringtheaccu-ox
mulationstepPeaksBredandCredremainedpracticallyunchanged(Fig2C) Voltammetric titrationconfirmed themechanismof formationcomplexesofPb(DMPS)Hg(DMPS)andPb(Hg)Italsoconfirmedpossibilityofdetermination
2+Pb andDMPSinthesamesolution
4Conclusions
ValidationinmodelsampleofdrugDimavalandhumanurinespikedwithDMPSconfirmed that this method can be used for clinical purposes Voltammetric
2+titration of DMPS by Pb ions proved that it can be used for simultaneousdetermination of the drug and heavy metal ions in human urine Moreoverobtained LODs were two orders lower than those in the previously reportedvoltammetricmethod[14](Table1)
Acknowledgments
ResearchwascarriedoutwithintheframeworkofSpecificUniversityResearch(SVV260560)TheauthorsthanktheCzechScienceFoundation(GACRprojectNo20-01589S)
References
[1] AnHHLuchakMCopesRLeadtoxicityAsystematicreviewofrecentlypublishedcasesClinToxicol53(2015)757ndash758
[2] KimYLustMRKreimerbirnbaumM23-Dimercaptopropane-1-sulfonate(DMPS)inthetreatmentoflead-poisoningFasebJ2(1988)A1820ndashA1820
[3] AposhianHVDMSAandDMPS ndashwater-solubleantidotesforheavy-metalpoisoningAnnuRevPharmacol23(1983)193ndash215
74 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Table 1ComparisonofvoltammetricmethodsforDMPSdetermination(LDR
Method Workingelectrode Lineardynamic LOQ LOD Refminus1 minus1 minus1 rangemicromolL micromolL micromolL
LSV glassy-carbonelectrode 18ndash140 41 14 [14] modifiedwithmulti-walled 260ndash690 carbonnanotubes DPCSV p-AgSAE 03ndash20 08 03 thisworkDPCSV m-AgSAE 01ndash10 03 01 thiswork 10ndash100
[4] BjorklundG Crisponi G Nurchi VM Cappai R Djordjevic AB Aaseth J A review oncoordinationpropertiesof thiol-containingchelatingagents towardsmercury cadmiumandleadMolecules24(2019)3247
[5] DonnerAHrubyKDMPSinthetreatmentofacuteandchronicheavy-metalpoisoningActaMedAust14(1987)10ndash10
[6] DonnerAHrubyKPirichKKahlsPSchwarzacherKMeisingerVDimercaptopropan-sulfonate(DMPS) inthetreatmentofacute lead-poisoningVetHumToxicol29 (1987)37ndash37
[7] Blanusa M Varnai VM Piasek M Kostial K Chelators as antidotes of metal toxicityTherapeuticandexperimentalaspectsCurrMedChem12(2005)2771ndash2794
[8] BarekJMoreiraJCZimaJModernelectrochemicalmethodsformonitoringofchemicalcarcinogensSensors-Basel5(2005)148ndash158
[9] Josypcuk B FojtaM Yosypchuk O Thiolatemonolayers formed on different amalgamelectrodesPartIIPropertiesandapplicationJElectroanalChem694(2013)84ndash93
[10] YosypchukBMarecekVPropertiesofthiolatemonolayersformedondifferentamalgamelectrodesJElectroanalChem653(2011)7ndash13
[11] Alvarez JMF SmythMRCathodic strippingvoltammetryofpyridine-2-thiolandsomerelated-compoundsAnalyst114(1989)1603ndash1605
[12] DanhelABarekJAmalgamelectrodesinorganicelectrochemistryCurrOrgChem15(2011)2957ndash2969
[13] Fadrna R Polished silver solid amalgam electrode Further characterization and appli-cationsinvoltammetricmeasurementsAnalLett37(2004)3255ndash3270
[14] ZiyatdinovaGKGrigorevaLVBudnikovGKElectrochemicaldeterminationofunithioland lipoic acid at electrodesmodifiedwith carbonnanotubes J Anal Chem64 (2009)185ndash188
[15] HrdlickaVChoinskaMRedondoBRBarekJNavratilTDeterminationofheavymetalpoisoning antidote 23-dimercapto-1-propanesulfonic acid using silver solid amalgamelectrodeElectrochimActadoiorg101016jelectacta2020136623
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 75
Fig 1Structureofcanagliflozin
1Introduction
Canagliflozin is a selective sodium-glucosecotransportertype2inhibitorused for the treatment of type 2 dia-betes mellitus Canagliflozin inhibitssodium-glucose cotransporter type 2present in proximal tubules of the
Canagliflozin oxidation study using electrochemical flow cell and comparison with hydrogen peroxide oxidation
a a bFILIPVYMYSLICKY TOMA S KR IZ EK JAKUBHER T
a DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublicfvymyslickygmailcomb ZentivaGroupasUKabelovny13010237Prague10CzechRepublic
AbstractBystandardstheeffectonoxidationofanactivesubstanceistestedusinghydrogenperoxidesolutionatelevatedtemperatureinastresschamberfor1ndash7daysAnalternativewaytostudytheeffectofoxida-tion on an active substance is to use an electrochemical flow cellSolutionwith active substance flows at low flow rate into a smallreactorwheretheactivesubstanceisoxidizedonworkingelectrodesurfaceTheelectrolytestreamwiththeoxidizedactivesubstanceisthen directed to the sample collector Products of electrochemicaloxidationareanalyzedbyhighperformanceliquidchromatographywithultravioletndashvisiblespectrophotometrydetectionCanagliflozinhasbeenusedbecauseitsmaindegradationpathwayisoxidationThedesign of experiments approach was used to explore the experi-mentalspaceandoptimizeexperimentalconditionsofoxidationTheresultsoftheoxidationstudyperformedintheelectrochemicalflowcellwerestatisticallycomparedwiththeresultsofastandardstudyusinghydrogenperoxidesolutionThemostsuitableconditionsforelectrochemical oxidation were found Electrochemical oxidationproducedcomparableamountsofimpuritiesaschemicaloxidationwithhydrogenperoxide
KeywordscanagliflozindesignofexperimentselectrochemicalflowcellHPLCoxidation
76 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
kidneywhichrestrictsglucoseabsorptioninthekidneytherebyincreasingtheurinaryexcretionofglucoseandloweringthelevelofglucoseintheblood[1]TheformulaofcanagliflozinisC H FO SthestructureofcanagliflozinisinFig1The24 25 5
IUPAC name of canagliflozin is (2S3R4R5S6R)-2-[3-[5-(4-fluoro-phenyl)-thiophen-2-ylmethyl]-4-methyl-phenyl]-6-hydroxymethyltetrahydro-pyran-345-triol[2]CanagliflozinisawhitepowderinsolubleinwaterbutverysolubleinorganicsolventslikemethanolordimethylsulfoxideCanagliflozinissoldundertradenameINVOKANA Manyauthorshavestudiedtheelectrochemicalpropertiesofactivesubstancesin the literature One example is the study of electrochemical behaviour andoxidationofbromhexineThesepropertieswerestudiedusingdifferentialpulsevoltammetryandcyclicvoltammetryonacarbonelectrodeTheresultsofelectro-chemicalmethodswerecomparedwithhighperformanceliquidchromatography(HPLC)analysis[3]Anotherexampleisthestudyofelectrochemicalbehaviourand degradation study performed on the active substance atomoxetineDegradation was studied using differential pulse voltammetry and cyclicvoltammetryonacarbonelectrodeTheresultswerealsocomparedwithHPLCanalysis[4]Electrochemicalmethodsareusedmainlytostudythemechanismofoxidationbutinthisworktheelectrochemicalmethodwasusedtodegradetheactivepharmaceuticalingredient In the stability studies of active pharmaceutical ingredient properties theinfluence of temperature pH light and oxidation is studied [5] By TheInternationalCouncilforHarmonisationofTechnicalRequirementsforPharma-ceuticals forHumanUse (ICH) standards the influenceof oxidationon activepharmaceutical ingredient is studied using hydrogen peroxide at roomtemperatureorincreasedtemperatureinthestresschamberduring1ndash7days[6]Analternativewaytostudytheinfluenceofoxidationonactivepharmaceuticalingredient is using electrochemical flow cellwhere an electrolytewith activepharmaceuticalingredientisdrivenbylowflowrateintothesmallreactorInthesmallreactortheactivepharmaceuticalingredientisoxidizedonthesurfaceoftheworkingelectrodeThestreamofelectrolytewithoxidizedactivepharma-ceuticalingredientisdriventothesamplecollectorProductsofelectrochemicaloxidationareanalysedbyHPLCUVVISThedesignofexperimentsapproachwasusedfordevelopmentofanalternativemethodofoxidationofcanagliflozinusinganelectrochemical flowcellThedesignofexperimentsapproachwasusedtoexplore the experimental space of the method and to find the optimalexperimentalconditionsofelectrochemicaloxidationofcanagliflozin
2 Experimental
21Materialandreagents
Canagliflozin(ZentivaCzechRepublic)999methanol(HoneywellGermany)98ammoniumdihydrogenphosphate(Sigma-AldrichJapan)35ortho-phos-phoricacid(PentaCzechRepublic)25ammonia(LachnerCzechRepublic)30hydrogenperoxide (LachnerCzechRepublic)water forchromatography
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 77
Fig 2Schemeoftheelectrochemicalflowcell(1)input(2)workingelectrode(3)gasket(4)refe-renceelectrode(5)counterelectrode
wasobtainedbypurifyingdemineralisedwaterusingMilliporetypeSynergyUVpurificationinstrument
22Instruments
An Agilent 1290 HPLC system (Agilent Technologies Germany) with highpressure pump autosampler thermostat and DAD detector was used for allexperimentsThePinnacleDBbiphenylcolumn(100times21mm19micromRestekUSA)wasusedforseparationIntheHPLCmethod10mMammoniumdihydrogenphosphatebufferpH=25wasusedascomponentAandmethanolascomponentBofthemobilephaseThegradientprogramwassetasfollowst(min)B01555010551790229023152515Theflowrateofthemobilephasewas
ndash104mlmin and the injection volume was 2μl The detector operated at awavelengthof220nmTheautosamplertemperaturewassetat20degCandthecolumntemperatureat60degCTheEmpowersoftwarewasusedforevaluationForelectrochemicaloxidationelectrochemicalflowcellfromALS(Japan)wasusedGlassycarbonelectrode(=6mm)andsilversilverchlorideelectrodewereusedasworkingandreferenceelectroderespectivelyTheschemeofelectrochemicalflowcellisinFig2ElectrodeswereconnectedwithpotentiostatPalmSens3fromPalmsens (Netherlands) AnElmasonic S15Hultrasonic bath fromElma (Ger-many)wasusedforsamplepreparationForpHmeasurementspHmeterJenway3540fromJenway(UnitedKingdom)wasused
3Resultsanddiscussion
AtthedevelopmentofthemethodforthestudyofcanagliflozinoxidationusingelectrochemicalflowcellitwasfirstnecessarytofindtheoptimalconditionsofelectrochemicaloxidationThedesignofexperimentsapproachwasusedChosen
78 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ndash1Fig 3Cyclicvoltammogramofcanagliflozin(concentrationofcanagliflozin11mgml electrolyte300 mM ammonium dihydrogen phosphate pH = 40 and methanol (11 vv) and scan rate
ndash1001Vs )
independentvariablesandtheirlevelswereconcentrationofelectrolyte(100200300mM)pHofelectrolyte(406080)cellsize(50100200500microm)and
ndash1flowrate(0102504mlh )ThereducedcombinatorialdesignwasusedIntheModde12 software aworksheet containing 11 experimentswas created Theworkingpotentialof12Vwasselectedbasedoncyclicvoltammetryofcanagli-flozininFig3Fromthisfigureitcanbeseenthattheoxidationofcanagliflozinoccursintheregionfrom11Vto14VAllexperimentswereperformedwith
ndash111mgml canagliflozin samples The glassy carbon electrodewas used as aworkingelectrodeandthesilversilverchlorideelectrodewasusedasareferentelectrode The canagliflozin samples oxidized in the electrochemical flow cellunder theexperimentalconditionsgivenby theworksheetweremeasuredbyHPLCwithUVVISdetectionDependentvariablespeakareasofimpuritiesandpercentage of peak areas of impurities obtained from chromatograms wereevaluated by the partial least squaremethod in theModde 12 software Thevariableimportanceintheprojectionplottoolwasusedforinterpretationofthedata as a whole The significance values of the independent variables were
ndash1evaluatedbufferpH=137flowrateof125mlh bufferconcentration061mMandcell size04micromFromthis tool itwasconcluded that theelectrochemicaloxidationofcanagliflozinisthemostaffectedbythepHoftheelectrolyteandflowrateoftheelectrolyteUsingtheoptimizertoolthemostsuitableconditionsfor
ndash1the oxidation of canagliflozin were evaluated flow rate 01 ml h 300 mMammoniumdihydrogenphosphate pHof electrolyte40 and cell size500micromUsingonefactoratthetimeapproachthedependenceofthecellsizeonthetotalsumofimpuritieswastested(Fig4A) Itisvisiblefromthegraphthatthesmallerthecellweusethemoreoxidationproducts are formed Based on the graph the most suitable conditions were
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 79
Fig 4(A)Optimizationofelectrochemicaloxidationconditionsdependenceofsumofimpuritiesoncellsize(B)Chromatogramofasampleoxidizedundermostsuitableconditions
ndash1adjustedtoflowrate01mlh 300mMammoniumdihydrogenphosphatepHofelectrolyte40andcellsize12micromUnderthemostsuitableconditionsarepeat-ability test was performed by ten independent oxidation experiments Therelativestandarddeviationofthepercentageareaofcanagliflozinwas164atasignificant level of 095 The chromatogram of sample oxidized under mostsuitableconditionsisinFig4BThestandardstudyoftheeffectofoxidationoncanagliflozinusinghydrogenperoxideaccordingtoICHguidelineswasperfor-medThestudywasperformedundertwosetsofexperimentalconditionsInthefirstcaseasolutionof50methanolwiththeadditionof3H O wasusedIn2 2
thesecondcasetheconditionsintheelectrochemicalflowcellweresimulatedA300mMammoniumdihydrogenphosphate pH40 andmethanol in a ratio11(vv)withtheadditionof3H O wasusedSamplesfortheoxidationstudy2 2
werestressedinastabilitychamberfor13and7daysattheconstanttempe-ratureof50degC ThetotalsumsofimpuritiesformedduringchemicaloxidationusinghydrogenperoxideinbothmediawerecomparedasisshowninFig5AItisobviousthat
80 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 5(A)Acomparisonofastandardoxidationstudyusinghydrogenperoxidein50methanolwith added buffer andwithout them (B) Chromatogramof sample oxidized electrochemically(C)Chromatogramofsampleoxidizedchemically
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 81
ammoniumphosphatesuppressesoxidationofcanagliflozinThereasonofthisphenomenonisunknownFig5BandFig5CshowchromatogramsofsamplesoxidizedelectrochemicallyandchemicallyrespectivelyItcanbeseenthatfiveimpuritieswereformedbybothtypesofoxidationhoweverindifferentamounts
4Conclusion
AnalternativemethodfortheoxidativestudyofcanagliflozinwasdevelopedThedesign of experiments approach was used in the method development ThedevelopedmethodworkswithRSDof165(α=095)Oxidationofcanagliflozinbythedevelopedmethodproducedfiveimpuritiesthatareidenticalwiththoseproducedusingthestandardoxidationstudywithhydrogenperoxide
Acknowledgments
ThisworkhasbeensupportedbyCharlesUniversityResearchCentreprogramNoUNCESCI014SVV260560projectandpharmaceuticalappliedresearchcenter(TheParc)
References
[1] ChaoECCanagliflozinDrugsFuture36(2011)351ndash357[2] NislySAKolanczykDMWaltonAMCanagliflozinanewsodium-glucosecotransporter2
inhibitorinthetreatmentofdiabetesAmJHealthSystPharm70(2013)311ndash319[3] Turchan M Jara-Ulloa P Bollo S Nunez-Vergara LJ Squella JA Alvarez-Lueje A
VoltammetricbehaviourofbromhexineanditsdeterminationinpharmaceuticalsTalanta73(2007)913ndash919
[4] Perez-OrtizMMunoz C Zapata-Urzua C Alvarez-Lueje A Electrochemical behavior ofatomoxetineanditsvoltametricdeterminationincapsulesTalanta82(2010)398ndash403
[5] Baertschi SW Alsante KM Reed RA Pharmaceutical Stress Testing Predicting DrugDegradationLondonInformaHealthcare2011
[6] RignallA ICHQ1A(R2) stability testing of newdrug substance andproduct and ICHQ1CstabilitytestingofnewdosageformsInICH Quality Guidelines An Implementation GuideATeasdaleDElderRWNims(Eds)HobokenWiley2017p3ndash44
82 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
AlthoughDNArepresentsarelativelystablecomponentfromthechemicalpointofviewitremainsconstantlyexposedtoalargenumberofchemicalorphysicalagentscausingchemicalchangesinDNAmoleculesthatoccurintheenvironmentoraremajororminorproductsofcellularmetabolism[1] One-electronoxidationoftheDNArepresentsadamagingprocesswheretheloss of an electron (oxidation) fromduplexDNA results in the formationof anucleobase radical cation (electron ldquoholerdquo) that is subsequently consumed inchemicalreactionsthatoftenleadtomutationsAdefiningcharacteristicoftheone-electronoxidationofDNAisthepreferentialreactionattheguaninemoietythatisdetectedasstrandcleavagefollowingchemicalorenzymatictreatmentoftheoxidizedDNA[2ndash3]
Novel hybrid electrochemical DNA biosensor for monitoring oxidative DNA damage via oxidationreduction signals of low molecular weight double-stranded DNA
MICHALAUGUSTINVLASTIMILVYSKOCIL
UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova812843Prague2CzechRepublicmichalaugustinnaturcunicz
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 83
AbstractDeoxyribonucleicacid(DNA)representsamajortargetmoleculeformanydamagingagentscausingunfavorablechangesinastructureofDNAmoleculethatbindandinteractwithDNAThusahighdemandforreliabletoolsregardingabettercomprehensionofthenatureofDNAdamagingprocessesstillrepresentsoneofthemaingoalsinthisareaHereinwedescribeadevelopmentofanovelhybridelectro-chemicalDNAbiosensorbasedonanldquoedge-planerdquopyrolyticgraphiteelectrode (EPPGE) in connectionwith an elementaryoptimizationprocessprovidingacloserresolutionoftheredoxprocessesoflowmolecularweightdouble-strandedDNA(dsDNA)attheEPPGESub-sequentanalyticalapplicationincorporatinganemploymentofthemodel structure K [IrCl ] (representative of transition metal2 6
complexes)andevaluationofitsdamagingeffectinrelationtoDNAbymeansof linear sweepvoltammetry resp square-wavevoltam-metryarealsopresented
KeywordsbiosensordamageDNAgraphitevoltammetry
DNA-based electrochemical biosensors are successfully used in variousapplicationssuchasmonitoringandevaluatingthemechanismsof interactionbetweenDNAandvariousdrugsordamagingagentsrapidmonitoringoftracemetalsorpollutantspresent in theenvironmentordirectmonitoringofDNAhybridizationprocesses[4] Theelectrochemicalactivityofnucleicacids(boththenativehigh-molecularonesaswellasoligonucleotides)isingeneralreferredtotheelectroactivityofitscomponents ndash nucleobases and sugar residues At mercury-based electrodesadenine and cytosine residues undergo reduction processes close to ndash14 V(againstSCE)inneutralorweaklyacidicmedium(givingrisetothepeakCA)Ontheotherhandallbaseshavebeenreportedtobeelectrochemicallyoxidizedatcarbonelectrodesbutonlyadenineand(particularly)guanineoxidationsignalshavebeenwidelyutilizedinelectrochemicalDNAbiosensors[5] In2017theelectrochemistryofnucleicacidsachievedanimportantmilestoneasthereductionoftheDNAoligonucleotideswasperformedataldquobasal-planerdquopyrolytic graphite electrode which provided wide potential window allowingboththeelectrooxidationaswellastheeletroreductionofthenucleobasesatasingleelectrodefortheveryfirsttimeDespitethesefindingsutilizationoftheaforementionedbiosensorintermsofanalyticalapplicationshasyettobeverifiedandremainsunclearuptothisdate[6] TheaimoftheproposedcontributionisapresentationofthedevelopmentprocessandsubsequenttestingofanoveltypeofhybridelectrochemicalDNAbiosensoranditsverificationasareliableanalyticaltoolintermsofmonitoringDNAdamage
2Experimental
21Reagentsandchemicals
Low molecular weight double-stranded DNA (dsDNA) derived from salmonspermwasobtainedfromSigma-AldrichGermanyStocksolutions(01mgmL)
of dsDNA were prepared in a 01 molL phosphate buffer of pH=74 (PB)Dipotassium hexachloroiridate (K [IrCl ]) was purchased from Sigma-Aldrich2 6
GermanyStocksolutions(0001molL)ofK [IrCl ]werepreparedinthePB2 6
22Apparatus
Voltammetric measurements were performed using the μAutolab IIIFRA2potentiostatgalvanostat(EcoChemieTheNetherlands)drivenbyaNOVA111software(MetrohmAutolabSwitzerland)Allmeasurementswerecarriedoutina three-electrode system using an ldquoedge-planerdquo pyrolytic graphite workingelectrode(EPPGE)withanelectroactivesurfacediameterof3mm(BASJapan)asilver|silver chloride reference electrode (Ag|AgCl|sat KCl) and a platinum
84 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
counterelectrode(ElektrochemickeDetektoryCzechRepublic)ina20mLglassvoltammetriccellatambienttemperature
23Preparationofthebiosensor
PriortotheeverymeasurementsurfaceoftheEPPGEwasmechanicallycleanedbygentlewipingoftheelectrodeonthesoftpolishingpadrinsedwithdistilledwaterAfterwardstheelectrodewasrinsedwithdistilledwaterandplacedinthePBforthesubsequentelectrochemicalactivationElectrochemicalactivationwasperformed in thePBbyapplyingpotentialof15V for240swithoutstirringApotentialpulseinworkingrangeofpotentials(00ndash15V)wasthenapplied Additional electrochemical activationwasperformed in the solutionof the
3minus4minusredoxindicator([Fe(CN) ] )byconsecutivecyclingintherangeofpotentials6
from10tondash08V(15scans)andfrom055tondash015V(10scans) The electrochemical DNA biosensor based on the EPPGE (dsDNAEPPGEbiosensor)was prepared by the adsorption of dsDNAon the EPPGEOptimal
parametersofthedsDNAadsorptionwereaconcentrationof01mgmLinthePB
(c )adepositionpotentialof07V(E )andanadsorptiontimeof5ming(dsDNA) dep
(t )withoutstirringthesolutionads
Atlasttheelectrodewasimmersedinthesolutionoftheredoxindicatorandtheconsecutivecyclingintherangeofpotentialsfrom055tondash015V(20scans)wasperformedinordertosecurethestabilityoftheoxidationreductionsignalsofdsDNAattheEPPGE
24Procedures
Theexperimentalparameterswereasfollowssquarewavevoltammetry(SWV)inthePBwithapulseamplitudeof20mVafrequencyof50Hzascanrateof750mVsandapotentialstepof15mVlinearsweepvoltammetry(LSV)inthePBwithscanratesof02ndash10Vandapotentialstepof24mVAllcurveswere
recordedthreetimes(n=3)
3Resultsanddiscussion
Sincetheclosestresolutionoftheprocessesassociatedwiththeelectroreductionof singleDNAcomponents at pyrolytic graphitehasbeenperformedwith theldquobasal-planerdquopyrolyticgraphiteelectrode(BPPGE)wehavedecidedtotakeovercorrespondingexperimentaltechniqueandconditions(LSVscanrateof10Vssteppotentialof24mV)attheverybeginningofouroptimizationprocesswiththeEPPGE[6] Inthisparticularcaseitispossibletonoticetheoccurrenceofthetwomixedvoltammetricpeaksatthedefaultexperimentalconditions(greenlineFig1A)selectedforthereductionofdsDNAattheEPPGEBygraduallydecreasingthe
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 85
Fig 1Baseline-correctedLSVrecordingscorrespondingtothereductionofdsDNAattheEPPGEfordifferentvaluesofscanrate(02ndash10VsFig1A)respbaseline-correctedLSVrecordingscorres-pondingtothereductionofdsDNAattheEPPGEandthenegativetestperformedunderthesameexperimentalconditionswithintheblanksolution(phosphatebuffer)atthebareEPPGE(03VsFig1B)
scan rate the optimal conditions (νle03Vs) were found and the mutualseparationof the signalswas allowedndash characterizedby thepresenceof twosinglewell-developedvoltammetricpeaksatpotentialsofndash175Vrespndash190V(03Vs orange line Fig 1A) Taking into account previous work regardingprocessesassociatedwiththereductionofDNAatthemercuryelectrodesrespBPPGE we can assume that the peak appearing at the potential of ndash175 Vcorresponds to themixedpeak for the reductionof the cytosine and adenineresidueswithindsDNA(peakCA)[5ndash6] Closer resolution of the second voltammetric peak appears to be farmoreproblematic Regarding our previous study we have discovered that theutilizationofdifferentE fortheadsorptionofdsDNA(E lt07V)isconnecteddep dep
withanappearanceofthethirdoxidationsignal(besidestheoxidationsignalsofguanine resp adeninemoieties) at apotential of073V corresponding to theoxidationoffreeguaninebases(FGBs)presentwithinthesolutionofdsDNAInthiscasewecanassumethatthepeakappearingatapotentialofndash190VcanpossiblyrepresentthereductioncounterpartofFGBspresentwithinthesolutionofdsDNAThisassumptioncanalsobesupportedbytheaforementionedstudyandbythefactthatthereductionsignalatsuchahighnegativepotentialcanbeobservedfortheoligodeoxynucleotidescontainingguanineresidues[6] AdditionallyinordertoverifythetruenatureofthereductionsignalsdepictedatF ig1Bandtoexcludetheoptionthattherelatedsignalsdonotrepresenttheproductsofpriorelectrochemicalactivationof theEPPGE(variousCndashObasedchemicalspecies)wehavedecidedtoperformanegative(control)testwithinthe
86 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2Baseline-correctedLSVrecordingscorresponding to thereductionof thedsDNA(02Vs
Fig 2A)respbaseline-correctedSWVrecordingscorrespondingtotheoxidationoftheguanine(098V)respadenine(128V)moieties(075VsFig2B)attheEPPGEafteritsincubationinthephosphatebufferforadefinedtimeperiod(60ndash900s)
blanksolution(PB)employingthesameprotocolasforthedsDNAadsorptionattheEPPGEInthiscaseitispossibletoobservetheabsenceofanypronouncedvoltammetricpeakslinkedtothedsDNAadsorptionandonlythepresenceofoneirreversiblepeakatapotentialofndash153VcorrespondingtotheelectroreductionoftheCndashObasedmoietymoietieswhichdropsafterthedsDNAadsorptiontoonetenthofitsoriginalvalue(approximately) Perhaps the most important parameter regarding further optimizationprocessrepresentedthetime-dependentstabilityofthecorrespondingsignalsofdsDNAwhichcanbespecificallyimportantinrelationtothestudyofthetime-dependentoxidativedamageofdsDNA AsithasalreadybeenprovedasingleelectrochemicalactivationoftheEPPGEinthePBdoesnotrepresentasatisfyingtechniqueregardingstabilityofdsDNAoxidationsignalsat theEPPGEand theadditionalstabilization isachievedbyfurtherelectrochemicalactivationinthesolutionofaredoxindicator(Fig2B)[7]Basedonthisaverificationoftheproposedstabilizationprotocolintermsofthetime-dependent stabilityofdsDNAreduction signals in the solutionof thePBwithinthedefinedtimeperiod(60ndash900s)appearedasareasonablenextstep FromtheresultsdepictedinFig2Aitispossibletonoticethatwithinthefirst300sdsDNAreductionsignalsremainstableinrelationtothecurrentresponseaswellasintermsofthepotentialvalueWithanadditionalincubationtime(t )inc
(900sorangeline)thepeakcurrentofthevoltammetricsignalpresentatmorenegative potentials decreased which can probably be addressed as a slowprogressive elimination of the weak (electro)chemical forces related to theunspecificadsorptionoftheFGBsattheEPPGE
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 87
Fig 3Baseline-corrected SWV recordings corresponding to the oxidation of the guanine respadeninemoietiesattheEPPGEafteritsincubationinthesolutionofK [IrCl ](IR)foradefinedtime2 6
period(60ndash3600s)(075VsFig3A)andthecorrespondingrelativebiosensorresponses(ΔI )rel
evaluatedusingtheguanosine(turquoise)andadenosine(red)peaksplottedversustheincubationtime(Fig3C)Baseline-correctedLSVrecordingscorrespondingtothereductionofdsDNAattheEPPGEafteritsincubationinthesolutionofK [IrCl ](IR)fordifferenttimeperiods(60ndash3600s)2 6
(02 VsFig3B)andthecorrespondingrelativebiosensorresponses(ΔI )evaluatedusingtherelpeakCA(darkpink)plottedversustheincubationtime(Fig3-D)
AdditionallywehavedecidedtotesttheapplicabilityofthepresentedhybridbiosensorintermsofmonitoringdsDNAdamagecausedbyarepresentativeofone-electron oxidants ndash K [IrCl ] In this case the prepared dsDNAEPPGE2 6
biosensor was immersed into the solution of K [IrCl ] (0001molL) for the2 6
definedtimeperiod(60ndash3600s) In thecaseof theoxidationpath (SWVrecordingsdepicted inFig3A) it ispossible to observe a time-dependent decrease of the oxidation signal of theguaninemoietieswhereastheoxidationsignaloftheadeninemoietiesremainsunaffectedforthemostof the incubationperiodThisphenomenonis ingoodcorrelationwiththetheoreticalknowledgeregardingoxidativedamageofdsDNAcausedbyone-electronoxidants [3]Simultaneouslywith thisLSVrecordingsdepicted in Fig 3B followed the similar behavior (decrease in relation to the
88 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
currentresponseofthedsDNAreductionsignalndashpeakCA)asinthecaseofthesignal regarding oxidation of guanine moieties In addition according totheportionofthepreservedDNA(Fig3C3D)itispossibletoassumethatthepronouncedoxidativedamageofdsDNAcanbemonitoredquitepreciselynotonlydirectlyviathedsDNAoxidationsignaloftheguaninemoietiesbutevenindirectlythroughthedsDNAreductionsignalndashpeakCA
4Conclusions
Inthiscontributionwehavepresenteddevelopmentofanunorthodoxhybridelectrochemical DNA biosensor based on an EPPGE Optimization processconcerning some important parameters was performed as well as closerresolutionofthenatureofthereductionprocessesofdsDNAattheEPPGEwasachievedInordertoconfirmtheresultsoftheoptimizationprocessapplicabilityoftheproposedbiosensorhadbeenprobedintermsofmonitoringDNAdamagecausedbyK [IrCl ]Inthiscasethefinalresultshadprovedthattheprepared2 6
hybridbiosensorcanbeconsideredasaversatileanalyticaltoolformonitoringoxidativeDNAdamage(viaoxidationreductionsignals)andispresentedasafinealternative in comparisonwith conventional electrochemical DNA biosensorsprepared within the group of traditional transducer materials (mercury- orcarbon-based)
Acknowledgments
ThisresearchwassupportedbytheSpecificUniversityResearch(SVV260440)
References
[1] FojtaMDanhelAHavranLVyskocilVRecentprogressinelectrochemicalsensorsandassaysforDNAdamageandrepairTrACTrendsAnalChem79(2016)160ndash167
[2] GieseBSpichtyMWesselySLong-distancechargetransportthroughDNAAnextendedhoppingmodelPureApplChem73(2001)449ndash453
[3] Burrows CJ Muller JG Oxidative nucleobasemodifications leading to strand scissionChemRev98(1998)1109ndash1151
[4] DiculescuVC Chiorcea-PaquimAMOliveira-BrettAMApplications of aDNA-electro-chemicalbiosensorTrACTrendsAnalChem79(2016)23ndash36
[5] PalecekEJelenFElectrochemistryofnucleicacidsInElectrochemistryofNucleicAcidsandProteinsndashTowardsElectrochemicalSensorsforGenomicsandProteomicsPalecekESchellerFWangJ(edits)AmsterdamElsevier2005p74ndash174
[6] SpacekJDanhelAHasonSFojtaMLabel-freedetectionofcanonicalDNAbasesuraciland5-methylcytosineinDNAoligonucleotidesusinglinearsweepvoltammetryatapyrolyticgraphiteelectrodeElectrochemCommun82(2017)34ndash38
[7] AugustınMVyskocilVNovelelectrochemicalDNAbiosensorbasedonedge-planepyrolyticgraphite for DNA interaction studies In Proceedings of the 15th International StudentsConferenceldquoModernAnalyticalChemistryrdquoNesmerakK(edit)PragueFacultyofScienceCharlesUniversity2019p263ndash268
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 89
1Introduction
Cadmiumisoneofthemosttoxicmetalsanditswidespreadindustrialusesresultin increased environmental pollution Hence the development of sensitivemethodology for Cd determination is still highly desirable Chemical vaporgeneration(CVG)ofCdbythetetrahydroboratereductioninacidicmediumisasuitable alternative sample introduction technique compatible with atomicspectrometricdetectorsandofferingimproveddetectioncapabilityComparedtocommonliquidnebulizationCVGoffersseveraladvantagessuchassignificantlyhigheranalyteintroductionefficiencyandalsoanalyteseparationfromsamplematrix IncomparisontoCVGofcommonhydrideformingelementsthereisalackofliterature dealing with mechanistic aspects of CVG of Cd [1] as well as withstabilityandidentityofitsvolatilespecies(freeatomshydrideotherspecies)Very little information is also available on achieved generation efficiencyMoreover there are many discrepancies in the literature regarding optimum
Chemical vapor generation of cadmium for analytical atomic spectrometry
a b a b a aLINDASAGAPOVA BARBORAKODRIKOVA MILANSVOBODA STANISLAVMUSIL aJANKRATZER
a InstituteofAnalyticalChemistryoftheCzechAcademyofSciences Veveřiacute9760200BrnoCzechRepublicsagapovaiachczb DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublic
AbstractChemical vapor generation of cadmium volatile compounds wasoptimizedinordertodeterminetraceCdconcentrationsbyatomicabsorptionspectrometry(AAS)Severalreactionmodifiersbasedon
III+ II+ III+ IV+inorganicsaltsandcomplexesofCr Co Ti Ti weretestedtoimproveanalyticalperformanceandgenerationefficiencyTheuseofthese reaction modifiers resulted in 4ndash5 times enhancement insensitivity reflected also in corresponding increase of generationefficiency and better repeatability Generation efficiency wasdeterminedfromacomparisonbetweensensitivitiesobtainedwithchemicalvaporgenerationandconventionalsolutionnebulizationbothsimultaneouslycoupledwithinductivelycoupledplasmamassspectrometryTheidentityofthegeneratedcadmiumcompoundswillbediscussed
Keywordsatomicabsorption
spectrometryatomizationcadmiumchemicalvapor
generation
90 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
conditionsforCVGofCdAlthoughstrongacid(HClorHNO )isalwaysemployed3
asacarrierandNaBH asareductantsomeauthorsreportedvariousadditives4III+ III+ IV+(modifiers)basedontransitionmetalions(Cr Ti Ti )inthepresenceof
II+KCN[23]orCo inthepresenceofthioureaandascorbicacid[4]toimproveCdsignalssignificantly TheaimofthisworkwastoinvestigateCVGofCdinacomprehensivewayFirstlyCVGofCdwithoutandwithselectedmodifierswasoptimizedemployingatomicabsorptionspectrometry(AAS)asadetectorandexternallyheatedquartztube(QTA)astheatomizerSecondlytheeffectofatomizationtemperatureonCdsignalwasstudiedallowingthustodeducetheatomicormolecularstructureofgeneratedCd speciesThirdly generation efficiencyofCdvolatile specieswasquantified
2Experimental
21Reagentsandchemicals
minus1Boiled and bubled (Ar per 30min) deionizedwater (lt 01 μScm UltrapurWatrex USA) was used to prepare all solutionsWorking Cd standards were
minus1preparedfrom1000mgL Cdstocksolution(AstasolAnalytikaCzechRepublic)minus1bydilutionin01ndash048molL HCl(basedonthemodifieremployed)from37
HCl(paMerckGermany)Theoptimumgenerationconditionsbeingdifferentforeachmodifiertestedare listedinTable1ThereductantwasasolutionofNaBH (ge 97 Sigma-Aldrich Germany) in 04 (mv) KOH (pa Merck4
Germany) prepared fresh daily The solutions of modifiers were prepared as2+followsCo waspreparedfromCoCl 6H O(ge990PENTACzechRepublic)2 2
3+the solution of Cr from Cr(NO ) 9H O (ge 9999 tracemetal basis Sigma-3 3 23+AldrichGermany)thesolutionofTi fromTiCl solution(about15in10HCl3
4+Sigma-AldrichGermany)andthesolutionofTi fromTiOSO (ge999Sigma-4
AldrichGermany)Tostabilizethelattersolution1H SO wasusedprepared2 4
bydilutionof96H SO (paLach-NerSlovakia)SolutionofKCN(ge9702 43+ 3+FlukaSwitzerland)wasusedasasecondmodifierwhenworkingwithCr Ti or
4+ ndash3Ti asmodifiersitsconcentrationvariedfrom008to016moldm depending
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 91
Table 1OptimumconditionsforchemicalvaporgenerationofCdinthepresenceofmodifiersandtheirabsence
onthemetalionThiourea(CH N Sge980LachemaBrno)andascorbicacid4 2
(C H O ge997Riedel-deHaenGermany)wereusedasmodifierscombined6 8 62+withCo
22Instrumentation
221Chemicalvaporgenerationsystems
TwoCVG flow injection systemswere employed either a two channel systemwithoutadditionofamodifier(seeFig1A)orafourchannelsystemallowingadditionofmodifiers(seeFig1B)
ndash1 TheflowratesofHClandNaBH were42and10mLmin respectivelyinatwo4ndash1channelsystem(Fig1A)whiletheywerebothkeptat10mLmin inthefour
channelsystem(Fig1B)Theflowratesofmodifiersinthefourchannelsystemndash1were05mLmin Thevolumeofthesampleloopwas015mLinbothsystems
ndash1Carriergasflowrateof75mLmin Arwascontrolledbyamassflowcontroler(Cole-ParmerUSA)
92 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
(A)
(B)
Fig 1Schemesofthechemicalvaporgenerationflowinjectionsystemwith(A)twochannels(nomodifiers)and(B)fourchannels(modifiersemployed)
222Atomicabsorptionspectrometry
ThePerkin-Elmermodel503atomicabsorptionspectrometer(BodenseewerkGermany)wasequippedwithaCdelectrodelessdischargelamp(Perkin-ElmerUSA)operatedat228mAThemeasurementswereperformedat2288nmusinga07nmslitwidthTheShimadzumodelAA-7000atomicabsorptionspectrometer(ShimadzuJapan)wasalsousedACdhollowcathodelamp(PhotronAustralia)operatedat2288nmlinewith07nmspectralbandpassandalampcurrentof12mA Signals were recorded for 2 minutes and peak areas were taken forevaluation The QTA was heated electrically to the temperature required byfurnace(PerkinElmer)andanin-housemadefurnacecontrolledbytheREX-C100controller(SysconIndianaUSA)withtheK-typethermocouplesensor(OmegaEngineeringUSA)223QuantificationofCVGefficiencybyICP-MS
Overall CVG efficiency of Cd was quantified bymeans of inductively coupledplasmamassspectrometry(ICP-MS)fromcomparisonoftheslopesofcalibra-tionsobtainedwithnebulizationliquidCdstandardstothoseobtainedwithCVGThe efficiency of liquid nebulization was quantified using a modified wastecollection method (see reference [5] for details) The Agilent 7700x ICP-MSinstrument(AgilentUSA)wasoperatingat1600WofRFpowerThesignalwas
111 125monitoredat Cdisotopeandcorrectedforthesignalofinternalstandard( Tendash11000ngmL Tein2HNO )NebulizeranddilutionArgasflowrateswere11503
ndash1and0mLmin respectively
3Resultsanddiscussion
31Chemicalvaporgenerationconditions
UnivariateoptimizationswereperformedtofindoptimumconditionsforCVGofCdinpresenceandabsenceofmodifiersTheparameterstobeoptimizedwerecarrieracid(HCl)concentrationreductant(NaBH )concentrationmodifierIand4
modifier II concentrations carrier gas flow rate (Ar) length of reaction coilsRCI-III (see Fig 1B) The optimum conditions for individual modifiers aresummarizedinTable1
32IdentityofCdspecies
ThepeakareasofgeneratedCdspecieswereforagivenmodifiermeasuredintheQTAheatedto900degCandnon-heatedQTAsubsequentlyOptimumCVGconditionswere employed as summarized in Table 1 This simple experiment allowsdistinguishingbetweenatomic(freeatoms)andmolecularformsofgenerated
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 93
Fig 2RelativesignalofgeneratedCdspeciesmeasuredintheQTAheatedto900degC(blackbars)andnon-heatedQTA(whitebars)withoutorinthepresenceofmodifiers
speciesOnlyfreeatomscanbedetectedinnon-heatedQTAsimilarlyasincaseofmercurycoldvaporsOn thecontrarymolecularanalytespeciesareatomizedat900degCAsaconsequencethesignalregisteredintheheatedQTAcorrespondstobothatomicandmolecularspeciesgeneratedItmustbehighlightedthattheresidencetimeoffreeatomsintheatomizerisdependentonQTAtemperatureduetogasexpansionAsaconsequencethesignalinQTAheatedto900degCshouldreach25ofthesignalatambienttemperaturetakingintoaccountthatonlyfreeatomsaregeneratedSincethetemperaturealongtheopticalarmofQTAisnotdistributedhomogeneouslydecreasingtobothendtheeffectivetemperatureoftheatomizerislowerOurexperimentswithCVGofHgrevealedsignalinheatedQTAisaround40[6]TheresultsreachedforCVGofCdaredepictedinFig2ThesignalofCdinheatedQTAisaround50ofthesignaldetectedinnon-heatedQTA
4+whennomodifierisemployedorusingTi asthemodifierindicatingclearlyfreeCdatomsarethedominantvolatilespeciesgeneratedOnthecontraryalmostno
2+differenceinpeakareaswasobservedforCo asthemodifierwhilethesignalinheatedQTAwas even 5 times higher in heatedQTA compared to non-heated
3+atomizer with Cr as the modifier suggesting the dominant contribution of3+molecularstructurestoCdsignalespeciallyincaseofCr KCNreactionsystem
33Generationefficiency
TheoverallCVGefficiencywasestimatedfromacomparisonbetweensensitivitiesobtainedwithCVGsampleintroductionandconventionalsolutionnebulizationICP-MSunder the sameexperimental conditionsNebulization efficiency for aMicroMISTnebulizerwasdeterminedas79plusmn01ThegenerationefficiencyofCd was derived from the sensitivity enhancement between CVG and liquidnebulizationTheresultsaresummarizedinTable2indicatingthatCVGwithout
94 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
modifiersisonlycatwotimesmoresensitivecomparedtoliquidnebulization3+ 4+Generation efficiency of Cd increases to 60 in the presence of Ti and Ti
modifiers
4Conclusions
CVG of Cd was thoroughly optimized in the presence of selected modifiersreportedpreviouslyintheliteratureGenerationefficiencyofCdintheabsenceofanymodifierswasquantifiedto15whileitcanbeincreasedupto60inthe
3+ 4+presenceofTi KCNorTi KCNasmodifiersFreeCdatomsseemtobe the4+dominantCdformgeneratedintheabsenceofanymodifiersorusingTi KCN
3+modifierwhile rathermolecularCd structuresaregenerated inCr KCNand2+Co thioureaascorbicacidreactionsystems
ExperimentsareinprogresstofinishthiscomprehensivestudyOnlythebestmodifierwillbefurtherusedforCVGofCdtobecoupledwithotherspectrometricdetectorsandappliedtocertifiedreferencematerialsandrealsamples
Acknowledgments
ThisresearchhasbeensupportedbytheCzechScienceFoundationundercontract18-01116SandbytheInstituteofAnalyticalChemistryoftheCzechAcademyofSciences(InstitutionalResearchPlannoRVO68081715)andCharlesUniversity(ProjectnoSVV260440)
References
[1] PitzalisEAngeliniDMascherpaMCDacuteUlivoAInsightintothemechanismscontrollingthechemicalvaporgenerationofcadmiumJAnalAtSpectrom33(2018)2160ndash2171
[2] ArslanZYilmazVRoseLEfficientgenerationofvolatilecadmiumspeciesusingTi(III)andTi(IV)andapplicationtodeterminationofcadmiumbycoldvaporgenerationinductivelycoupledplasmamassspectrometryMicrochemJ123(2015)170ndash178
[3] YilmazVRoseLArslanZLittleMDOn-linechemicalvapourgenerationofcadmiuminthepresenceofhexacyanochromate(III)fordeterminationbyinductivelycoupledplasmamassspectrometryJAnalAtSpectrom27(2012)1895ndash1902
[4] Y Lu SunHW YuanCG YanXP Simultaneous determination of trace cadmiumandarsenic inbiologicalsamplesbyhydridegeneration-doublechannelAFSAnalChem74(2002)1525ndash1529
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 95
Table 2GenerationefficiencyofchemicalvaporgenerationofCdasquantifiedbyICP-MS
Modifiers Generationefficiency
Nomodifiers 15plusmn13+Cr KCN ndash2+Co thioureaascorbicacid ndash3+Ti KCN 58plusmn24+Ti KCN 61plusmn2
[5] VyhnanovskyJStrugeonREMusilSCadmiumassistedphotochemicalvaporgenerationoftungsten fordetectionby inductively coupledplasmamass spectrometryAnal Chem91(2019)13306ndash13312
[6] MigasovaMMatousekTSchrenkovaVZ ıdekRPetry-Podgorska IKratzer JMercuryvolatilespeciesgenerationfromHClandTRISbuffermediaAnalChimActa1119(2020)68ndash76
96 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Atomicfluorescencespectrometry(AFS)coupledwithvapourgenerationisanultrasensitive analytical method for determination of various elements ItsanalyticalperformancecanbecomparabletoICP-MSwithliquidnebulizationbutatsubstantiallylowercost[1]SampleintroductiontoAFSisacrucialstepoftheanalyticalproceduresincetheanalytehastobeintroducedtotheatomizerintheformofitsvolatilespecies Hydridegeneration(HG)isamaturetechniqueofsampleintroductionduringwhichvolatileanalytehydridesare formedbyreactionwithareducingagenttypicallysodiumborohydrideAnewemergingtechniquephotochemicalvapourgeneration(PVG)employsUV irradiationof theanalyte in liquidphase in thepresenceofaphotochemicalagent(usuallyalowmolarmassorganicacidformic
minusoraceticacid)Highlyreducingradicalspecies(HbullRbullandCOObull )andaquatedelectronsare formedduring irradiationandreactwith theanalyte to form its
Photochemical vapour generation of bismuth coupled with atomic fluorescence spectrometry
ab ab a aBARBORASTA DLEROVA JAROMIRVYHNANOVSKY JIR IDE DINA STANISLAVMUSIL
a InstituteofAnalyticalChemistryoftheCzechAcademyofSciences Veveřiacute9760200BrnoCzechRepublicstadlerovaiachczb DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublic
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 97
AbstractPhotochemical vapour generation of bismuth was successfullycoupledwithnon-dispersiveatomic fluorescencespectrometry forthefirsttimeVolatilespeciesofBiweregeneratedusingastandardmercurylow-pressuretubelampandacoiledreactorfromareaction
2+mediumwhichwas composedof acetic and formic acid Co ionswereusedasasensitizerOptimizationofatomizationconditionsinaflame-in-gas-shieldatomizerwasperformedThismethodologywascomparedtothecommonlyemployedhydridegenerationapproachAbsolutelimitofdetectionof68pgwasachievedwithphotochemicalvapour generation which is still about 7 times worse than withhydride generation The developed methodology was successfullyverifiedbyBideterminationinareferencematerialofwater
Keywordsatomicfluorescence
spectrometrybismuthhydridegenerationphotochemicalvapour
generation
volatilespeciesInbothcasesthegeneratedvolatilespeciesoftheanalytehavetobeseparatedfromtheliquidphaseinthegas-liquidseparatorandarecarriedtotheatomizerbyacarriergas[2] InthisworkanatomizerdesignedspecificallyforAFStheflame-in-gas-shieldatomizerwasused(Fig1) It consistsofaverticalquartz tubesuppliedwithargon and hydrogen together with the analyte volatile species Moreover acapillaryisinsertedintheverticalaxisoftheverticaltubethroughwhichoxygenisintroducedA hydrogen-oxygenmicroflameburns on top of the capillary Themicroflameisshieldedfromtheambientatmospherebyaflowofargonwhichisintroducedthroughashieldingunitfittedaroundtheverticaltube[34] Theaimofthisworkwastooptimizeatomizationconditionsintheflame-in-gas-shieldatomizerusingPVGasasampleintroductiontechniqueandtocomparetheanalyticalcharacteristicsofPVGandHGforultrasensitivedeterminationofbismuthbyAFS
2Experimental
21Reagentsandchemicals
Deionized water (Ultrapur Watrex USA) was used for preparation of all thesolutionsWorkingBisolutionswerepreparedfreshdailybyserialdilutionof
ndash1stock1000mgl BistandardforAAS(Sigma-AldrichGermany)RegardingHG05(mv)NaBH in04(mv)KOHwasusedasa reductantAsolutionof4 ndash11mol l HClwasusedasacarrierandblank
98 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Flame-in-gas-shieldatomizerOHndashobser-vationheight
RegardingPVGformicacid(98paLach-NerCzechRepublic)andaceticacid (998 pa Lach-Ner CzechRepublic)were used for preparation of thereaction medium they were purified in a Teflon BSB-939-IR sub-boilingdistillation apparatus (Berghof Germany) The composition of the reactionmedium(40(vv)acetic125(vv)formicacid)wasoptimizedearlier[5]
ndash1The 5000mgl Co stock solution was prepared from cobalt(II) acetatetetrahydrate (pa Lach-Ner Czech Republic) and used as a sensitizer ofphotochemicalreactionTheoptimalconcentrationofCointhestandardsamples
ndash1andblanksolutionscorrespondedto50mgl (ref[5]) Acertifiedreferencematerial(CRM)-1643fTraceElementsinWater(NationalInstituteofStandardsandTechnologyUSA)wasusedtochecktheprecisionofthedevelopedmethodology
22Instrumentation
221Atomicfluorescencespectrometer
An in-house assembled non-dispersive atomic fluorescence spectrometerconstructedatourlaboratorywasusedforBideterminationandisdescribedindetail elsewhere [3] The detector output provided signals in microV Peak areacorrectedtobaselineandmainlysignaltonoiseratioweretheparametersusedtoevaluatethedata
222Hydridegeneratorphotochemicalvapourgeneratorandatomizer
A flow injection hydride generator was employed (Fig 2a) The reductantndash1 ndash1(12mlmin )andthecarrier(4mlmin )werepumpedbyaperistalticpump
Thesamplewasinjectedthrougha1mlsampleloopintotheflowofcarrierAglassgas-liquid separator (5 ml) with forced waste removal was employed forseparatingthegasphasecontainingbismuthanewhichwasthencarriedtotheatomizerbyargon Thephotochemicalvapourgenerator(Fig2b)consistedofthephotoreactorconstructedwitha15Wlow-pressureHggermicidallamp(Cole-ParmerUSA)wrappedaroundwith6mofPTFEtubing(1mmidinternalvolume471ml)The
ndash1reactionmedium(3mlmin )waspumpedbyaperistalticpumpThesamplewasinjected througha056ml sample loopApolypropylenegas-liquid separator
(15ml)withforcedwasteremovalimmersedinanicebath[6]wasemployedforseparatingthegasphasecontainingBivolatilespeciessubsequentlycarriedtotheatomizerbyargon Theflame-in-gas-shieldatomizerisdepictedinFig1detaileddescriptionisgiveninRef[3]Theobservationheight(OH)isdefinedasthedistancefromthetopofthecapillarytothecentreoftheopticalbeam
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 99
23Samplepreparation
CRMNIST1643fwasdilutedwith1MHCl80-foldforBideterminationbyHG-AFSRegardingBideterminationbyPVG-AFSthesampleneededtobeevaporatedtodrynessinordertogetridofnitricacidthatseriouslyinterferesatmMlevel[5]Avolume of 3ml of CRMwere pipetted into a 40 ml quartz vial evaporated(temperature asymp100 degC two replicates) and subsequently diluted ca 33-foldAsamplepreparationblank3mlofdeionizedwaterwaspreparedaswell
3Resultsanddiscussion
TheatomizationconditionsforHG-AFSwereoptimizedinourpreviouswork[3]TheseconditionswereusedasinitialtofindtheoptimumconditionsforPVG-AFSwith the flame-in-gas-shieldatomizerwith respect to sensitivityandsignal tonoiseratioFirstlythehydrogenfractionintherange10ndash16wasoptimizedatconstant total gas flow rate (sum of total argon and total hydrogen) of
ndash1500mlmin the lower the hydrogen fraction the better However at 10hydrogenfractiontheflamewasnotstableenoughandwentoftenouthenceitwasoptedfor12hydrogenfraction
100 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2(a)Hydrideand(b)photochemicalvapourgenerator
The oxygen flow rate through the capillary was optimized in the rangendash15ndash30mlmin Thehighestsignaltonoiseratiowasachievedwiththeflowrateof
ndash120mlmin Thetotalgasflowratewasoptimizedatconstant12hydrogenfractioninthe
ndash1range500ndash800mlmin Theoptimumobservationheightvarieswithtotalgasflow rate so it had to be optimized as well The optimum conditions aresummarizedandcomparedtothoseachievedwithHGinTable1 TheanalyticalfiguresofmeritofPVG-AFSwiththeflame-in-gas-shieldatomi-zerweredeterminedThecalibrationfunctionconstructedwith010025050
ndash1 2100 and 200 microg l Bi standardswas linear (R = 09998) The repeatabilityndash1expressedastherelativestandarddeviation(n=10)was6at1microgl andthe
ndash1relativeandabsolutelimitsofdetection(3σn=10)achievedwere12ngl and68pgrespectively(Table2)TheabsolutelimitofdetectionachievedwithHGwas 76 times lower which can be attributed to several aspects Firstly thegenerationefficiencyforPVGapproachwasaround53while100isexpectedforHG[3]Secondlyafullwidthathalfmaximumofthemeasuredpeakswasca2-foldgreaterwhichnecessitatedlongerintegrationtimeandwasthusreflectedinhighernoiseofthesignalsFinallythelimitofdetectionforPVGapproachwas
ndash1affectedby seriouscontamination (around10ng l )mostprobably from thesensitizersolutionthatcontainedBiasimpurity Tovalidate theproposedmethodologyBi contentwasdetermined inCRMNIST1643f (Table3) and the resultswere compared to thosemeasuredwithHG-AFS[3]Duetosevereinterferencesfrominorganicacidsespeciallynitricacid[5]thesampleneededtobeevaporatedtodrynessandthenfilledupwiththe
2+reactionmediumcontainingCo as thesensitizer(NIST1643f isstabilized in
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 101
Parameter HG-AFS(ref[3]) PVG-AFS
ndash1Artotalmlmin 440 528ndash1H totalmlmin 60 722 ndash1O mlmin 7 202
OH mm 6 9ndash1Arshieldlmin 1515 1515
Table 1Atomizationconditionsforflame-in-gas-shieldatomizer
Parameter HG-AFS(ref[3]) PVG-AFS
LODpg 09 68ndash1LOQngl 09 12
Repeatability lt1 6
Table 2AnalyticalfiguresofmeritofHG-AFSandPVG-AFS
ndash1032moll nitricacid)Theresultsobtainedbybothmethodologiesareingoodagreementwiththecertifiedvalue
4Conclusion
Photochemical vapour generation of Bi was successfully coupled with non-dispersiveatomicfluorescencespectrometryforthefirsttimeanditsapplicabilitywas verified by determination of Bi in certified reference material of waterComparedtohydridegenerationconditionsofatomizationdifferinanoptimalobservationheightandsupplyofoxygenwhichmaybeneededtoldquoburnoutrdquotheorganicvapoursthatarereleasedfromthereactionmediumtothegasphasehowever this remains tobe verifiedAlthough there are still some limitationsregardingthelimitsofdetectionrepeatabilityandinterferencesthisnewsampleintroductionapproachseemstobepromising
Acknowledgments
The support of the Czech Science Foundation (19-17604Y) Czech Academy of Sciences(Institutional supportRVO68081715)andCharlesUniversity (ProjectSVV260560andProjectGAUK1048120)isgratefullyacknowledged
References
[1] Musil SMatousek T Currier JM StybloM Dedina J Speciation analysis of arsenic byselectivehydridegeneration-cryotrapping-atomicfluorescencespectrometrywithflame-in-gas-shield atomizer achieving extremely low detection limits with inexpensiveinstrumentationAnalChem86(2014)10422ndash10428
[2] SturgeonREPhotochemicalvaporgenerationaradicalapproachtoanalyteintroductionforatomicspectrometryJAnalAtSpectrom32(2017)2319ndash2340
[3] S tadlerova B Kolrosova M Dedina J Musil S Atomic fluorescence spectrometry forultrasensitivedeterminationofbismuthbasedonhydridegenerationndashtheroleofexcitationsourceinterferencefilterandflameatomizersJAnalAtSpectrom35(2020)993ndash1002
[4] DedinaJAtomizationofvolatilecompoundsforatomicabsorptionandatomicfluorescencespectrometryOnthewaytowardstheidealatomizerSpectrochimActaPartB62(2007)846ndash872
102 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Certified HG-AFS PVG-AFSndash1valuemicrogl
ndash1 a ndash1 a valueobtainedmicrogl recovery valueobtainedmicrogl recovery
1262plusmn011 128plusmn01 102plusmn1 121plusmn09 97plusmn5
a Spikedrecovery=slopeofstandardadditions(noadditionandtwospikedconcentrationstoasample)slopeofexternalcalibration
Table 3ThedeterminedcontentofBiinCRMNIST1643fpresentedasmedianvalueplusmncombineduncertainty(n=3)andrecoveries
[5] Vyhnanovsky J Yildiz D Musil S Effect of metal sensitizers on photochemical vaporgeneration of bismuth for analytical atomic spectrometry In Proceedings of the 15thInternationalStudentsConferenceModernAnalyticalChemistryKNesmerak(ed)PragueCharlesUniversity2019p257ndash262
[6] VyhnanovskyJSturgeonREMusilSCadmiumassistedphotochemicalvaporgenerationoftungstenfordetectionbyinductivelycoupledplasmamassspectrometryAnalChem91(2019)13306ndash13312
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 103
1Introduction
Liquidcrystalsareorganicsubstancesthatformamesomorphicphaseinsolu-tion[1]TheyareliquidlikeliquidsbuthaveaninternalconfigurationassolidsTheir light transmittance changes in the electric fieldwhich is used in liquidcrystaldisplays(LCDs)CholestericliquidcrystalsaretemperaturesensitiveThecolorofreflectedlightchangeswithsmalltemperaturechangeThisisusedinmedicineasasensitivetemperatureindicatorfordisease-infectedtissues High performance liquid chromatography or supercritical fluid chromato-graphy[23]areusedtoseparateanddetermineliquidcrystalcompoundsAnelectrophoretic method could be complementary to these commonly usedmethods To our best knowledge no study dealing with liquid crystal puritycontrolbyelectrokineticchromatographywaspublisheduntilnow Electroneutralsubstancesmoveincapillaryzoneelectrophoresisatthesamespeedcorrespondingtothespeedoftheelectroosmoticflow(EOF)andtherefore
Separation of liquid crystals using non-aqueous capillary electrokinetic chromatography
KATER INACOKRTOVATOMA S KR IZ EK
DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublickaterinacokrtovagmailcom
AbstractLiquid crystals arewidelyused in electronicsmedicine andotherfields Analytical separations are important in the development ofnewliquidcrystalstocontrolthepurityofsynthesizedsubstancesThesampleanalysisisimportantfordetectionofimpuritiesformedduring synthesis Liquid crystal-forming substances cannot beseparated by capillary zone electrophoresis due to the absence ofreadily ionizable groups Therefore electrokinetic chromatographywasused in thisworkAnotherproblemcomplicating theanalysiswastheverylowsolubilityofanalytesinwaterSeparationsinthisworkwere thereforecarriedoutundernon-aqueousconditions inacetonitrilewithaceticacidtoadjustthepHandhexadecyltrimethyl-ammonium chloride as a detergent to mobilize the non-ionizedanalytesUndertheseconditionsitwaspossibletoseparateimpu-ritiesfromsynthesizedanalytesinsamples
Keywordselectrokinetic
chromatographyliquidcrystalsnon-aqueouscapillary
electrophoresis
104 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
itisnotpossibletoseparatethemDuetothisanelectrokineticchromatographymethodwasdevelopedInthismethodasurfactantisaddedtothebackgroundelectrolyteMoleculesaggregateandformsphericalformationscalledmicelles[4]ifthesubstanceisaddedinsufficientconcentrationiehigherthanthecriticalmicellar concentration (CMC) Separation is possible due to interactions ofnonpolarmoleculepartswiththenonpolarmicelleinsideAlthoughwateristhemostusedsolventinelectrophoreticmethodsforseparationofwater-insolublesubstances organic solvents are selected However such solvent must meetcertaincriteriatobesuitableforuseincapillaryelectrophoresisAllcomponentsmustbesolubleinthesolvent[5]ItshouldnotbeflammabletoxicorreactiveforpracticalityitshouldbeliquidatroomtemperatureandalsoitspriceistakenintoaccountThevalueof its relativepermittivitywhichdescribes the strengthofinteractionsbetweenionsshouldbearound30Lowdynamicviscosityisalsopreferred to allow faster migration of analytes No organic solvent meets allparameters of the ideal solvent In practice methanol acetonitrile and theirmixturesarethemostusedTheseparationparameterscanbeinfluencedbyusingan organic solvent of the background electrolyte This topic has already beenwidelyexplored[6ndash8] Itwasgenerallyassumedthat inanhydrousconditionsmicellesarenotcreateddespitesufficientsurfactantconcentrationHoweveritwasfoundoutthatdodecylsulfatecanformstablemicelleswhenthebackgroundelectrolyteisdissolvedinformamide[9]Fortheanalysisofactivesubstancesinmedicinal plants Chen et al developed a method in which sodium cholatedissolvedinmethanolisusedasasurfactant[10]Theaddedpseudostationaryphase does not always form micelles but can still affect mobilization andseparationofanalytesiftheanalytesinteractdifferentlywithfreemoleculesofsurfactantInthisstudywater-insolubleliquidcrystalswereseparated(Fig1)Thereforenonaqueouselectrokineticchromatographymethodwasdeveloped
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 105
Fig 1 Structures of liquid crystals 4-([1-oxo-1-(pentyloxy)propan-2-yl]oxycarbonyl)phenyl4-(octyloxy)-[11-biphenyl]-4-carboxylate (ZL 85) and 4-([1-(decyloxy)-1-oxopropan-2-yl]oxycarbonyl)phenyl 4-(dodecyloxy)-[11- biphenyl]-4-carboxylate (ZL 1210) OpticalisomerismsitesaremarkedwithanasteriskStructurescreatedinMarvinSketch[11]
2Experimental
21Reagentsandchemicals
Acetonitrilege999fromSigma-Aldrich(Germany)aceticacid99fromLach-Ner Neratovice (Czech Republic) and hexadecyltrimethylammonium chloride25(ww)inwaterfromSigma-Aldrich(USA)wereusedforpreparationofback-groundelectrolyteMesyloxidepa(MO)suppliedbyLach-nerNeratovice(CzechRepublic)wasusedasareferencesubstance
22Instrumentation
ForexperimentsG7100ACapillaryElectrophoresisInstrument(AgilentTechno-logiesGermany)wasusedwithUV-VISdetectoroperatingat235nmand254nmwavelengthMeasurementswereconductedinafused-silicacapillaryof50microminner diameter with the total length 500cm and effective length 415cm(PolymicroTechnologiesUSA)
23Method
Capillarywas flushed for3minuteswith1MHCland for2minuteswith thebackgroundelectrolyteBackgroundelectrolytewaspreparedbymixingaceticacid (10mM) and hexadecyltrimethylammonium chloride (40mM) in aceto-nitrile Sampleswere introducedhydrodynamicallybyapressureof5kPa for1secondSampleswerefirstdissolvedinacetonitrileandthendilutedtwotimeswith the background electrolyte A voltage of 20 kV was applied during theseparation
3Resultsanddiscussion
Liquidcrystalsampleswerepractically insoluble inwater theirsolubilitywasndash3testedataconcentrationlevelof1mgcm inmethanolandacetonitrileWhile
samples were not sufficiently soluble in methanol they were successfullydissolvedinacetonitrile Becauseallanalytesaresubstancesthatdonothaveeasilyionizablefunctionalgroups theelectrokineticchromatographymethodwaschosen forseparationAsuitablesurfactantwassoughtCommonlyusedsodiumdodecylsulfate(SDS)isinsoluble in acetonitrile Therefore hexadecyltrimethylammonium chloride(CTAC) which had sufficient solubility for further experiments was chosenAlthoughasuitablebufferwassoughttoensureastablepHduetoproblemswithprecipitationofbuffercomponentsinthenon-aqueousenvironmentaceticacidwasusedtoadjustandmaintainpHofbackgroundelectrolytesolutionAstheadditionofcationicsurfactantsuchasCTACleadstoEOFreversalthedependence
106 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ofEOFmobilityontheconcentrationofCTACinthebackgroundelectrolytewasmeasuredContrarytowhatisobservedinaqueousbackgroundelectrolytesEOFwasnotreversedItsmobilitydecreasedwithincreasingCTACconcentrationbutnomajorchangesoccurredabove40mMconcentrationThecapillarywallwasprobablyalreadysaturatedbyCTACandthefurtherincreaseinconcentrationhadno signifficant effect on the conditionof the capillarywall Therefore a CTAC
ndash3concentrationof40mmoldm was chosenas sufficient for furthermeasure-mentswithrespecttotheincreasingcurrentwithincreasingionicstrengthofthebackgroundelectrolyte The optimized method was used for separation of several liquid crystalsamplesofdifferentpurityInthesampleoftheZL85liquidcrystalwith99purityonezoneoftheanalytewasdetectedImpuritieswereseparatedfromthisanalytewhen the samplewith lower puritywas introduced The peak of theanalytewasidentifiedbasedonrelativemigrationtimerelatedtomesityloxideSeparationoftheanalytefromanimpurityinthesampleZL8576isshowninFig 2The relativemigration timeof the firstpeak is 0834 therefore itwasidentifiedastheZL85analyteThestandarddeviationoftherelativemigrationtimesinfivemeasurementswas0002min(01) Using the available high purity sample it was possible to measure thecalibration line forquantificationof theanalyte in lesspuresamplesLimitof
ndash3detection was determined as 0009mgcm and limit of quantification as ndash30031mgcm FromthecalibrationlineconcentrationofZL85inthesample
with lower purity was calculated The concentration was determined as
48(ww)standarddeviation5(ww)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 107
Fig 2ElectropherogramobtainedwhenasampleofZL85liquidcrystalwithlowerpuritywasndash3introducedSamplewas introduced in01mgcm concentrationandwithaddedmesityloxide
ndash3(10mgcm )Capillarywithinnerdiameterof50micromtotallengthof500cm415cmeffectivelength The background electrolyte was acetonitrile with 10 mM acetic acid and 40 mMhexadecyltrimethylammonium chloride A voltage of 20 kVwith positive polaritywas appliedDetectionat254nm
ForsampleZL121099onlytheanalyteandmesityloxideweredetectedInthe sample ZL 1210 59 several impurities were separated and detected(Fig3)Accordingtotherelativemigrationtimetheanalyteofinterestcorres-pondstothefirstpeakPeakresolutionissufficientTheresolutionoftheanalytepeakandthesecondpeakis284andtheresolutionoftheothertwopeaksis230
4Conclusions
InthisstudyanewmethodforanalysisofnewlysynthesizedliquidcrystalswasdevelopedSomeparametersofthemethodwereoptimizedndashoptimumconcen-tration of hexadecyltrimethylammonium chloride was searched The identifi-cation of analyteswas based on a comparison of relativemigration times InsamplesZL85andZL1210withlowerpuritytheimpuritieswereseparatedfromthepeaksofliquidcrystalsthecontentofanalytewasdeterminedintheZL85sampleaccordingtothecalibrationline
Acknowledgments
IwouldliketothanktheInstituteofPhysicsoftheCzechAcademySciencesforprovidingnewlysynthesized liquid crystals This work has been supported by Specific University Research(SVV260560)andbyCharlesUniversityResearchCentreprogramNoUNCESCI014
References
[1] GennesPGProstJThePhysicsofLiquidCrystals2ndedNewYorkOxfordUniversityPress1993
108 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ndash3Fig 3ElectropherogramofsampleZL121059ataconcentrationof05mgcm withmesitylndash3oxideataconcentrationof10mgcm capillarywith innerdiameterof50microm total lengthof
500 cmeffectivelength415cmThebackgroundelectrolytewasacetonitrilewith10mMaceticacidand40mMCTACAppliedvoltage20kVpositivepolarityDetectionat235nm
[2] Vankatova P KalıkovaK KubıckovaA Ultra-performance supercritical fluid chromato-graphy A powerful tool for the enantioseparation of thermotropic fluorinated liquidcrystalsAnalChimActa1038(2018)191ndash197
[3] Vankatova P Kubıckova A Cigl M Kalıkova K Ultra-performance chromatographicmethodsforenantioseparationofliquidcrystalsbasedonlacticacidJSupercritFluids146(2019)217ndash125
[4] Terabe S Otsuka K Ichikawa K Tsuchiya A Ando T Electrokinetic separations withmicellarsolutionsandopen-tubularcapillariesAnalChem56(1984)111ndash113
[5] RiekkolaMLRecentadvancesinnonaqueouscapillaryelectrophoresisElectrophoresis23(2002)3865ndash3883
[6] Wright PB Lister AS Dorsey JG Behavior and use of nonaqueous media withoutsupporting electrolyte in capillary electrophoresis and capillary electrochromatographyAnalChem69(1997)3251ndash3259
[7] PorrasSPKenndlerECapillaryzoneelectrophoresisinnon-aqueoussolutionspHofthebackgroundelectrolyteJChromatogrA1037(2004)455ndash465
[8] PorrasSPRiekkolaMLKenndlerETheprinciplesofmigrationanddispersionincapillaryzoneelectrophoresisinnonaqueoussolventsElectrophoresis24(2003)1485ndash1498
[9] GuoXWangK ChenGH Shi JWuX Di L LWangY Determination of strobilurinfungicideresiduesinfruitsandvegetablesbynonaqueousmicellarelectrokineticcapillarychromatography with indirect laser-induced fluorescence Electrophoresis 38 (2017)2004ndash2010
[10] Chen AJ Li C Gao WH Hu ZD Chen XG Application of non-aqueous micellarelectrokinetic chromatography to the analysis of active components in radix SalviaemiltiorrhizaeanditsmedicinalpreparationsJPharmBiomedAnal37(2005)811ndash816
[11] MarvinSketch [computer program] version 1990 ChemAxon httpschemaxoncom-productsmarvin
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 109
1Introduction
Sudandyesaresyntheticazo-basedaromaticcompoundsTheyaretraditionallyusedinvariousindustriessuchaschemicaltextileandwoodworkingasdyestocolourwaxesplasticsoilspolishesandsoforthTheyhavebeencategorizedasclass3carcinogensbytheInternationalAgencyforResearchonCancerandtheiruseisthereforeforbiddeninthefoodindustryTheyareknownfortheirbrightcolours and easy and cost-effectivemanufactureThey arenearly insoluble inwater but soluble in various organic solvents such asmethanol or trichloro-methane[1] SudanI1-phenylazo-2-naphthol(Fig1A)isadyeusedasanorangecolouringagentItssometimesalsosoldundernamesSolventOrangeRorCISolventYellow14ItisformedasasecondaryproductinthemanufactureoftheSunsetYellowdye
Electrochemistry of Sudan I and its derivates in aqueous media
ad b aANNAONDRA CKOVA MARIESTIBOROVA LUDE KHAVRAN cd adKAROLINASCHWARZOVA -PECKOVA MIROSLAVFOJTA
a CentralEuropeanInstituteofTechnologyMasarykUniversity Kamenice753562500BrnoCzechRepublicannaondrackovaceitecmuniczb DepartmentofBiochemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublicc UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistry FacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublicd InstituteofBiophysicsCzechAcademyofSciences Kralovopolska13561265BrnoCzechRepublic
AbstractSudanIisanaromaticazo-compoundthathasbeenproventobeacar-cinogenDuringitsmetabolizationbycytochromeP450inliverafewmain derivates can be identified Thiswork sets out to assess themechanismofelectrochemicalreductionandoxidationofSudanIitshydroxylationderivativesfeaturingmetabolitesintheSudanIdetoxi-fication pathway and to introduce their selective voltammetricanalysis on boron-doped diamond electrode We show successfuldifferentiationamongthesecompoundsthankstothedifferencesintheelectrochemicaloxidationoftheirphenolicgroups
Keywordsborondopeddiamond
electrodecytochromeP450electrochemicalanalysisSudanI
110 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
InmammalianorganismsSudanIismetabolizedbythemicrosomaldetoxi-fying systemwitha central roleof cytochromeP450hydroxylationactivity inliver[2]DuringtheoxidativeprocessofmetabolizingSudanIseveralmetaboliteswereidentifiedbyprevioustestsThesearegt1-(phenylazo)-naphtalene-26-diol(further abbreviated SI-6OH) 1-(4-hydroxyphenylazo)-2-hydroxynaphtol(furtherabbreviatedSI-4OH)and1-(4-hydroxyphenylazo)-naphtalene-26-diol(furtherabbreviatedSI-46-diOH)thestructuresarepresentedinFig1 ThemainmethodcurrentlyusedtoidentifySudanIamongotherdyeswithsimilar structure is high-performance liquid chromatography (HPLC) It isrecommendedasthestandardmethodtoidentifythelevelofSudanIinfood[3] ComparedtoHPLCelectrochemicalmethodsareprovingtobefastercheaperandcomparablypreciseUnfortunatelyacomprehensiveelectrochemicalstudyofSudanIandparticularlyofitshydroxylatedmetaboliteshasnotbeencompletedyetThedyecanbedetectedthroughelectrochemistryeitherbytheoxidationofitsphenolicgrouporviareductionoftheazogrouppresentinitsmoleculeInbothcasesotherelectrochemicallyactivemoietiesareformedThederivatesofSudanIcanbedetectedandrecognizedfromSudanIthroughanalogousprocesses[4]InthisstudywefocusedoncomparisonofelectrochemicalbehaviourofSudanIanditshydroxylatedmetabolitesonborondopeddiamondelectrodetoaddressthepossibilitiesoftheirrecognitionsinmixturesbasedondifferencesinanodicandcathodicsignals
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 111
Fig 1 Chemical structure of (A) Sudan I (B) 1-(phenylazo)-naphtalene-26-diol (SI-6-OH)(C) 1-(4-hydroxyphenylazo)-2-hydroxynaphtol (SI-4-OH) and (D) 1-(4-hydroxyphenylazo)-naphtalene-26-diol(SI-46-diOH)
(A) (B)
(C) (D)
2Experimental
21Reagentsandchemicals
SudanI(Merckanalyticalstandardgrade)wasdissolvedinethanol(Merck)andkept at room temperatureChemicals forBritton-Robinsonbufferpreparation(acetic acid boric acid orthophosphoric acid sodium hydroxide) were fromMerckwithpurityge99pHofthebufferwasadjustedbymixingoftheacidsandsodium hydroxide solution at different ratios The Sudan I metabolites weresynthetized at the Department of Biochemistry Faculty of Science CharlesUniversityandkeptinmethanolattemperature3degC
22Instrumentation
Cyclicvoltammetric (CV)measurementswerecarriedout inBritton-RobinsonbufferofpH=70atroomtemperatureSudanIanditsderivativeswereaddedto
minus1thesolutionofBritton-Robinsonbuffertofinalconcentrationof5micromolL andstirred Before the measurement oxygen was removed from the solution bypurgingwithargonfor3minutesAutolabanalyzerPGSTAT20(EcochemieTheNetherlands)inconnectionwithVA-Stand663(MetrohmSwitzerland)GPES49(MetrohmSwitzerland)andathree-electrodesetup(withborondopeddiamond
2(WindsorScientificUKdiskdiameter3mmA=707mm )asworkingelectrodeminus1AgAgCl3molL KCl as reference electrode and platinum wire as auxiliary
electrode) Five cycles were performed for each measurement at scan rateminus1of1Vs
3Resultsanddiscussion
ForeachcompoundtwoseparateCVmeasurementswereperformedeachwithfive cycles performed in rapid succession For bothmeasurements the initialpotentialwassetat0VTheanodicscancontinuedto+1Vturnedtowardsndash1Vandfinishedat0VIntheothersetupcathodicscanwasperformedfirstfromthestartingpointtondash1Vturnedtowards+1Vandreturnedto0VThiswaywewereabletoobservethebehavioursofSudanIandcompareittothatofitsderivativeswhilefirstbeingreducedandthenoxidisedorviceversa In the anodic scan of Sudan I and its derivatives (Fig 2) differences in thepositionsoftheoxidationpeaksineachcompoundcanbeobservedWhileSudanIwithonlyonephenolgroupisoxidizedat+067VthederivativeswithtwophenolgroupsieSI-4OHandSI-6OHgiveoxidationpeaksatremarkablylesspositivepotentialofca+05VSI-46OHwiththepresenceofoverall3hydroxygroupsyieldstwooxidationpeaksInthereversecathodicscan(vertexpotential+1V)probablythankstothepresencereductionpeaksappearwhichcanbefurtherusedtodifferentiatebetweenSudanIandthederivativesSI-4OHandSI-6OH
112 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 113
Fig 3VoltammetricscanofSudanIanditsderivatesSI-4-OHSI-6-OHandSI-46-diOH1stscanincathodicdirectionfrom0Vvertexpotentialsndash1Vand+1VThemeasurementswereperformedinBritton-Robinsonbuffer(pH=7)withtheconcentrationofeachcompoundat5microMandatscan
ndash1rate1Vs
Fig 2 CyclicvoltammogramofSudanIanditsderivatesSI-4-OHSI-6-OHandSI-46-diOH1stscaninanodicdirectionfrom0Vvertexpotentials+1Vandndash1VThemeasurementswereperformedinBritton-Robinsonbuffer(pH=7)withtheconcentrationofeachcompoundat5microMandatscan
ndash1rate1Vs
possesstwoconjugatedhydroxylgroupswhichcanberegardedashydroquinonestructures and thus undergoing quasireversible redox process due to oxida-tionreductionofthehydroquinonetoquinonemoietyThisiswellvisibleattheCVsastheanodicsignalisfollowedbycathodiconeatthepotentialof+023VforSI-4OHand+03VforSI-6OHTheoxidationofSudanIproceedsbymechanism
minus +typicalforphenoliccompoundsatmorepositivepotentialsleading by1e 1H exchange to naphthoxy-type ndashO radical [5] This species undergoes furtherreactionsleadingtoformationofdimersandpolymersThecathodicpeakinthereversescanatndash02Varisesfromreductionofthesereactionproductsanditsoriginneeds tobe further investigatedSI-46OHwith thepresenceofoverallthree hydroxyl groups yields two oxidation peaks The first one is a result ofoxidationoftwoofthembeinginconjugationandthusbeingoxidizedtoquinonemoietyThesecondsignalatthesamepotentialastheoxidationsignalof2OHonnaphthaleneringofSudan I is consequenceofoxidationof the thirdhydroxylgroupofphenolictypeAsinglewidepeakat0Vinthereversescanispresumablyan overlap of signals arising from reduction of the quinonic moiety and by-products formed during oxidation processes Thanks to differences of theseprocessesspecificforindividualcompoundsitispossibletodifferentiateamongallfourofthemviapropersetting-upoftheinitialandvertexpotentialvalues The cathodic scan of Sudan I and its derivatives (Fig 3) shows a dominantreductionpeakaroundndash08Vwhichisduetoreductionoftheazogroupintheirstructuresaccompaniedbycleavageoftheirmoleculestoseparatethebenzeneandnaphthalene rings [4] The peaks in the subsequent anodic scans (vertexpotentialndash1V)arethereforetheresultoftheelectrochemicalreactionofmoietiesthatareproductsofthedivisionofthearomaticcirclesTheseproductsincludeaniline4-aminophenol1-amino-2-naphtholand1-amino-25-naphthalenediolwith irreversibly oxidizable amino moieties or (quasi)reversibly oxidizableamino-hydroxylsystemonthebenzeneornaphthaleneringandtispossibletodifferentiatebetweenthemTheobtainedoxidationpeaks(+02VforSI-4-OH+073VforSudanIandSI-6-OHand+025Vand+075VforSI-46-diOH)makeitpossibletodifferentiatebetweenallcompoundswiththeexceptionofSudanIandSI-6-OHForthereliablerecognitionofthesetwocompoundsanothermeasure-mentwithdifferentparametersisneeded
4Conclusions
ThestructuresofSudanIanditshydroxyderivatesthatarethemainproductsofthe metabolization of Sudan I by cytochrome P450 are similar and theirrecognitionwhenpresentinmixtureinsolutionisdemandingHereinwepresenta simple approach based on comparison of signals obtained in cathodic andanodic scan in CV measurements without the need of time-demandingchromatographicseparationstepFurtherworkwillbedevotedtoidentificationof observed redoxprocesses and applicationof themethod formonitoringofmetabolictransformationsofSudanIinvitro
114 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Acknowledgments
ThisresearchwassupportedbytheCzechScienceFoundation(projectNo18-01710S)
References
[1] ChailapakulOWonsawatW SiangprohW GrudpanK ZhaoYF Zhu ZW Analysis ofSudanISudanIISudanIIIandSudanIVinfoodbyHPLCwithelectrochemicaldetectionComparison of glassy carbon electrodewith carbon nanotube-ionic liquid gelmodifiedelectrodeFoodChem109(2008)876ndash882
[2] StiborovaMMartinekVRydlovaHHodekPFreiESudanIisapotentialcarcinogenforhumans Evidence for itsmetabolic activation and detoxication by human recombinantcytochromeP4501A1andlivermicrosomesCancerRes62(2002)5678ndash5684
[3] GomezMArancibiaVAliagaMNunezCRojas-RomoCDeterminationofSudan I indrinks containing Sunset yellow by adsorptive stripping voltammetry Food Chem 212(2016)807ndash813
[4] PrabakaranEPandianKAmperometricdetectionofSudanIinredchilipowdersamplesusingAgnanoparticlesdecoratedgrapheneoxidemodifiedglassycarbonelectrodeFoodChem166(2015)198ndash205
[5] Enache T A Oliveira-Brett A M Phenol and para-substituted phenols electrochemicaloxidationpathwaysJElectroanalChem655(2011)9ndash16
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 115
1Introduction
Aminoglycosidesarealargegroupofnaturalandsemi-syntheticantibioticswithawidespectrumofantimicrobialactivityagainstmostgram-positiveandgram-negativemicroorganismsCurrentlymultiplerepresentativesofaminoglycosidefamily ndash gentamicin (GM) neomycin B (NM) paromomycin (PM) kanamycin(KM)apramycin(AP) andstreptomycin(STM)Fig1ndashareapproved to treatinfectiousdiseasesinanimalsMaximumresiduelimitsfortheseaminoglycosidesinproductsandtissuesfromedibleanimalsareestablished[1]henceaneffectiveandrobustassayisnecessaryforcontrolofaminoglycosidescontamination Inthisstudyenzyme-linkedimmunosorbentassayforthedetectionofamino-glycosides in foodproductsandenvironmentalobjectswasdevelopedGroup-specificantibodieswereproducedowingtoimmunogenbasedonribostamycin(RS)whichexposedthecommonfragmentofmostaminoglycosides2-deoxy-streptamin (2-DOS) Fig 1 The developed assay was made suitable for thedetectionofresidualaminoglycosidesinhoney[2]
Group detection of aminoglycosides using ELISA for control of food contamination
ab a aKONSTANTINBURKIN INNAGALVIDIS MAXIMBURKIN
a DepartmentofImmunologyIMechnikovResearchInstituteofVaccinesandSera MalyjKazionnyjper5a105064MoscowRussianFederationburkin-kostyandexrub DepartmentofChemicalEnzymologyFacultyofChemistryLomonosovMoscowStateUniversityLeninskieGory1119991MoscowRussianFederation
AbstractThegrowingthreatofglobalantibioticresistanceisforcingtoreducenon-targetconsumptionofantibioticsandtomonitorcontaminationoffoodandenvironmentalobjectsInthisworkELISAwasdevelopedforgroupdetectionofaminoglycosidesToobtaingroup-specificanti-bodies a new immunogen based on ribostamycin was used Thedevelopedindirectcompetitiveformatofassayallowedtherecogni-tionof9aminoglycosidesnamelyneomycinribostamycinneaminparomomycin gentamicin sisomicin kanamycin tobramycin and
ndash1apramycinwithadetectionlimitrangedbetween002ndash020ngmL TheeffectivenessoftheproposedassaywasevaluatedinhoneyasafoodstuffmodelToneutralizea stronghoneymatrixeffect and toavoidalaborioussamplepre-treatmentanewmatriximitatorwassuggested 5 sucrose solution imitated the influence of 50-folddilutedhoneyTheproposedassayallowedustorevealanyofthe9
ndash1mentionedaminoglycosidesinhoneyata10microgkg level
KeywordsaminoglycosidesELISAhoney
116 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
2Experimental
21Reagentsandchemicals
NeomycinBribostamycinneamin(NA)paromomycinkanamycintobramycin(TM)amikacin(AM)gentamicinnetilmicin(NTM)sisomicin(SSM)geneticin(GC) apramycin and streptomycin were purchased from Chimmed (MoscowRussia) Bovine serumalbumin (BSA) complete Freund adjuvant 16-hexane-diamine1-ethyl-3-(3-dimethylaminopropyl)carbodiimide(EDC)sodiumperio-dateandsodiumborohydrideweretheproductsofSigma-Aldrich(USA)Gelatin(Gel)wasfromBio-Rad(USA)sucrosefromServa(Germany)two-componenttetramethylbenzidine (TMB) substrate solutionwas fromBioservice (Russia)andgoatanti-rabbitIgGantibodiesconjugatedtohorseradishperoxidase(anti-rIgG-HRP)werefromIMTEK(Russia)Honeysampleswerepurchasedfromlocaloutlets
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 117
Fig 1 Structuralformulasofstudiedaminoglycosides
22Preparationofconjugatedantigens
TwotypesofconjugateswerepreparedbasedonRSandBSAusingzero-lengthand C6 spacer arm between hapten and protein carrier RSwas treatedwithsodiumperiodatetooxidizehydroxylsofribosefragmenttoreactivealdehydegroupsandthencoupledtoBSAaminesthroughreductiveaminationToremoveuncoupledRSanexhaustingdialysiswascarriedoutusingdialysismembranetubes(MWCO14kDa)UsingthesameprocedureGel-RSconjugatewassynthe-sized ForpreparationofBSA-C6-RSwefirstlymodifiedBSAwith16-hexanediamineThemixtureofBSAandEDCinwaterwerestirredfor30minThen16-hexane-diaminewasaddedandstirredfor2hThemodifiedproteinwasdialyzedfromtheexcessive reagents and resultant BSA-C6-NH2was coupled to RS in reductiveaminationprocessasdescribedabove
23Immunizationandantibodypreparation
BSA-RSandBSA-C6-RSwereusedasimmunogensChinchillarabbits(20ndash25kg)weresubcutaneouslyinjectedat10ndash15pointsonthebackwith01mgofimmuno-gensemulsifiedinthecompleteFreundadjuvantThesamedosesofimmunogensinsalinewereadministeredmonthly forbooster immunizationsAweekaftereachinjectionabloodsamplefromearveinswastakenforthecontrolofimmuneresponseTheantiserainglycerol(11vv)werestoredatndash15degCuntiltestinginELISA
24TheELISAprocedure
AcompetitiveassaywasconductedaccordingtoclassicalprocedureGel-RSwascoatedovernightonpolystyrene96-wellCostarplatesNon-adsorbedconjugatewashedoutusingPBSwith005oftween20(PBS-T)Thenextcompetitivestep
ndash1includedtheadditionof01mLstandardaminoglycosidesolutions(1pgmL to ndash1 ndash11microgmL (B)and0microgmL (B ))inPBS-Tor01mLoftestedsampleand01mLof0
antibodiesinworkingdilution(1h25 degC)Afterwashingtheantibodiesboundtoimmobilized Gel-RS were detected using anti-rIgG-HRP (1h 37degC) Coloredproduct formedasaresultofenzymaticreactionwithTMBsubstratemixture(05h25degC)wasreadat450nmusingaStatFax2100platereader(AwarenessTechnologiesUSA) Relativeantibodybinding(BB )vstheanalyteconcentrationswasplottedas0
standardcurvesfittedtoafour-parameterlogisticfunctionThecross-reactivity(CR) for every aminoglycoside representative was calculated as ratio of half-inhibition concentrations IC NMIC aminoglycoside The dynamic range of50 50
assaywasacceptedasIC ndashIC andthelimitofdetection(LOD)wascalculatedas20 80
B ndash3timesSD0
118 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
3Resultsanddiscussion
31Immunogensynthesisandantibodypreparation
Inthemajorityofpublicationsdevotedtoimmunoassayofaminoglycosidestheimmunogenscoatingantigensenzymeconjugatesortracerswerepreparedbycarbodiimide or glutaraldehyde methods involving aminoglycosidesrsquo aminogroups [3ndash7] Due to several amino groups in aminoglycoside molecules theformationofconjugateswithavariableorientationofthehaptenoccurs InpresentstudyRSwaschosenasanimmunizinghaptenduetothefollowingadvantageous features Being a trisaccharide RS has the size of a moleculecomparable to themostof aminoglycosides Ithas three identical ringsA-B-CsimilartothoseinNMUsingaperiodateoxidationwecouldinvolvearibosesiteofRSincouplingtoproteinthatprovidedastrictorientationofhaptenonthecarrier with a favorable presentation of the 2-DOS fragment The resultantimmunogensBSA-RSandBSA-C6-RSwerecomparedtorevealwhichdesignisbetterforpresentationofacommonfragmentofaminoglycosidemoleculeandgenerationofgroup-specificantibody Antibodies to the BSA-RS demonstrated moderate sensitivity (NM
ndash1IC =10ngmL )andhighselectivitytowardsNMwithrelativelylowcross-reac-50
tivity(lt5)forGMKMandAPTheapplicationofthespacerintheimmunogenBSA-C6-RScontributedtoaprominentpresentationofthe2-DOSdeterminantandtheinductionofantibodieswithrecognitionofbroadspectrumofdifferentaminoglycosides In addition anti-BSA-C6-RS exhibited significantly better
ndash1sensitivity(NMIC =02ngmL )Thusallsubsequentstudieswereconducted50
usinganti-BSA-C6-RS
32Examinationofassayspecificityandselectionofimmunoreagents
TheindirectcompetitiveformatofassaywasdevelopedForevaluationofassayspecificity a panel of following aminoglycosideswas studied and their cross-reactivitywasdeterminedNA(625)RS(250)NM(100)KM(475)PM(173)GM(90)TM(78)AP(17)SSM(12)AM(lt01)GC(lt01)STM(lt01)andNTM(lt01)ThemostoftheseanalytesareusedinmedicalandveterinaryareashoweveronlyNMPMGMKMAPandSTMareappliedinanimalhusbandry[1]
33Determinationofaminoglycosidesinhoneyandselectionofthematriximitator
Honey is a complex product consisting of carbohydrates (75ndash80) vitaminsproteinsenzymesorganicacids traceelements inclusionsandothercompo-nentsThesecomponentsmightinterfereimmunochemicalreactionThereforetheisolationofaminoglycosidesfromhoneyisalaboriousandtime-consuming
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 119
120 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
procedureToavoidthisstephoneymatriximitatorswereappliedtomimictheinfluenceofhoneymatrixonantibodybindingSucrosewaschosenasthehoneyimitatorsinceitexposedastronghoney-matrix-likeeffectonantibodybindingTheadequacywasfoundbetweensolutionsofhoneyandsolutionsofsucroseexpressinganequalmatrixeffectTwopairswithequivalentmatrixeffectwere120honey=20sucroseand150honey=5sucroseThelatterconditionswere chosen asmore preferable due to inconvenience of operatingwith highviscous20sucrosesolution Thedeterminationofaminoglycosidesinhoneycouldbecarriedoutquantita-tivelyiftheanalytetobedetectedisknownForquantificationofaminoglycosideinhoneyasamplewasdiluted50timesinPBS-Tandaminoglycosidestandardcurvewas generated in 5 sucrose-PBST (Fig 2) If analyte is unknown thedevelopedgroup-specificELISAcanbeusedasascreeningtestInthiscasetheanalyzed sample can be considered as contaminated if it caused a relativeantibodybindingbelowthecut-off level (Fig3)Thus thedeveloped testwas
ndash1 ndash1 ndash1Analyte IC ngmL Dynamicrange LODngmL LODinhoneymicrogkg50 ndash1 IC ndashIC ngmL20 80
NM 02 003ndash21 002 10PM 07 008ndash71 005 25GM 15 015ndash133 011 55KM 035 005ndash39 004 20AP 68 05ndash968 023 115
Fig 2StandardcurvesandanalyticalparametersoftheELISA-systemforgroupdeterminationofaminoglycosidesinhoneyInteractionofanti-BSA-RSwithcoatingantigenGel-RSin5sucrosesolutionasthehoneyimitatorThedetectionlimitin5sucrosesolutionwasdeterminedaccordingtoLOD=B ndash3timesSD0
capabletorevealthecontaminationofhoneywith9aminoglycosides5amino-ndash1glycosidesapprovedforveterinary(NMPMGMKMandAP)ata10microgkg level
andalsoNARSSSMandTM
4Conclusions
A novel indirect competitive ELISA for the detection of aminoglycosides wasdevelopedRSwasusedasanewimmunizinghaptentoproducegroup-specificantibodiesagainst2-DOSacommonmoietyofalargenumberofaminoglycosideantibiotics A wide spectrum of aminoglycoside representatives could bedetectedincludingNMRSNAPMGMSSMKMTMandAPThedevelopedassay
ndash1was capable todetect theseanalyteswithaLOD up to002ndash020 ngmL ForanalysisofhoneyamatriximitatorwasdevelopedtoavoidhoneyinterferencesonimmunoassayTheanalysisofthehoneysampleallowedustorevealanyofthe
ndash1mentionedaminoglycosidesinhoneyata10microgkg level
References
[1] CouncilRegulation(EU)N372010OffJEurCommunitiesInfNotL15(2009)1ndash72[2] GalvidisIABurkinKMEreminSABurkinMA Group-specificdetectionof2-deoxy-
streptamineaminoglycosidesinhoneybasedonantibodiesagainstribostamycinAnalMeth11(2019)4620ndash4628
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 121
Fig 3Detectionofaminoglycosidesspikedinhoneysamplesata40ppblevelusinggroup-specificELISAEachsymbolcorrespondstotheaveragerelativebindingandtheerrorisSDobtainedforanindividualhoneysampleanalyzedintriplicateEmptycharactersrepresentindividualblankhoneysamples(limebuckwheatandflower)andfilledsymbolsrepresentthesamesamplesfortifiedwith
ndash1aminoglycosidesata40mgkg (level establishedonlyforSTMinseveralcountries)Thecut-offlevelcorrespondstothelimitofassaydetectionobtainedbythematriximitator(5sucrose-PBS-T)
[3] ThompsonSGBurdJFSubstrate-labeledfluorescentimmunoassayforamikacininhumanserumAntimicrobAgentsChemother18(1980)264ndash268
[4] LiCZhangYEreminSAYakupOYaoGZhangXDetectionofkanamycinandgentamicinresiduesinanimal-derivedfoodusingIgYantibodybasedic-ELISAandFPIAFoodChem227(2017)48ndash54
[5] GalvidisIABurkinMAMonoclonalantibody-basedenzyme-linkedimmunosorbentassayfortheaminoglycosideantibiotickanamycininfoodstuffsRussJBiorganChem36(2010)722ndash729
[6] HaasnootWStoutenPCazemierGLommenANouwsJFKeukensHJImmunochemicaldetectionofaminoglycosidesinmilkandkidneyAnalyst124(1999)301ndash305
[7] Peng JWangYLiuLKuangHLiAXuCMultiplex lateral flowimmunoassayfor fiveantibioticsdetectionbasedongoldnanoparticleaggregationsRSCAdv6(2016)7798ndash7805
122 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
PhotochemicalvaporgenerationisanalternativesampleintroductiontechniqueforanalyticalatomicspectrometryThistechniqueisbasedaroundasourceofUV-radiationthatirradiatesalowmolecularweightorganicacidmedium(mostcommonlyformicacidaceticacidortheircombinations)withananalyteHighlyreducingradicalsandaquatedelectronsareproducedandconverttheanalyteintoavolatilespecieswhichisthentransportedintoadetector[1]SofartheuseofPVGhasbeendescribedforhydride-formingelements(AsBiTeSbPbSeSnandTl)andmercury[12]transitionmetals(FeCoNiCuMoWCdAgAuIrPdPtRhandOs)[13ndash6]andevennon-metals(BrIClFandS)[16ndash9] AfirstsuccessfulphotochemicalvaporgenerationofcobaltwasdescribedbyGuoetalin2004[6]whichwasfollowedbymoresystematicstudiesbyGrinbergetalin2008[10]andDengetalin2010[11]LaterworksbydeQuadrosetal[12]anddeJesusetal[13]focusedontheanalysisofrealsamplesInthelatterwork
Photochemical vapor generation of cobalt for detection by inductively coupled plasma mass spectrometry
ab aJAROMIRVYHNANOVSKY STANISLAVMUSIL
a DepartmentofTraceElementAnalysisInstituteofAnalyticalChemistryoftheCzechAcademyofSciencesVeveřiacute9760200BrnoCzechRepublicjaromirvyhnanovskygmailcom
b DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublic
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 123
AbstractThisworkfocusedonthephotochemicalvaporgenerationofcobaltVolatilespeciesweregeneratedinaflow-injectionsystememployinga high-efficiency flow-through UV photoreactor and a formic acidbased medium and were introduced by an argon carrier into aninductivelycoupledplasmamassspectrometerfordetectionOptimalgeneration conditions were found as 10 (vv) formic acid and
ndash1 ndash14molL ammonium formate with a 4mLmin flow rate whichcorresponds to irradiation time of around 13 s The influence ofvariousmetalsensitizersofphotochemicalreactionwasinvestigated
2+andonlyCu ionsexhibitedapositiveeffectongenerationefficiencyndash ndash 2ndashInterferencesfromcommoninorganicanions(NO Cl SO ) were3 4
also examined Lastly the limit of detection and repeatability (atndash1 ndash1250ngL )weredeterminedtobe13ngL and41respectively
Keywordscobaltinductivelycoupled
plasmamassspectrometry
photochemicalvaporgeneration
the authors also presented a systematic study on generation conditions andachievedagenerationefficiencyofaround40 Themainaimofthisworkwastooptimizetheconditionsofgenerationwithinductivelycoupledplasmamassspectrometry(ICP-MS)detectionexaminetheeffectofvariousmetal sensitizers toachieve thehighestgenerationefficiencypossibleandreachthelowestlimitofdetectionpossible
2Experimental
21Reagentsandchemicals
minus1Deionizedwater (DIW lt 02μScm UltrapurWatrex USA)was used for thepreparationofallsolutionsFormicacid(98paLach-NerCzechRepublic)andammonium hydroxide (ge25 pa Sigma-Aldrich USA) were used for the
minus1preparationof the reactionmediumA1000mgL Co stock solution (Sigma-AldrichUSA)wasusedforthepreparationofallsamplesolutionsThefollowingcompounds were used as potential metal sensitizers cadmium(II) acetatedihydrate(paLach-NerCzechRepublic)zinc(II)acetatedihydrate(paSigma-AldrichUSA)copper(II)acetatemonohydrate(paMerckGermany)nickel(II)acetatetetrahydrate(paSigma-AldrichUSA)sodiumtungstatedihydrate(paCarlRothGermany)and iron(II) sulphateheptahydrate (pa LachemaCzechRepublic)Nitricacid(65semiconductorgradeSigma-AldrichUSA)hydro-chloricacid(37paMerckGermany)andsulfuricacid(98paLach-NerCzechRepublic)wereusedforaninterferencestudy
22Instrumentation
AschematicdiagramofthePVGsystemcoupledtoICP-MSisshowninFig1andamore detailed description can be found in reference [5] Briefly a singlequadrupole ICP-MS Agilent 7700x (Agilent Technologies USA)was used as a
minus1detector Deionized water was mixed with a 10μgL Rh internal standardsolutionin2HNO andwassubsequentlynebulizedbyaMicroMistnebulizer3
59 103during PVG Isotopes of Co and RhweremonitoredMeasurementswereperformed in time resolved analysis mode and in He collision mode
minus1(41mLmin )AlltubingusedwasmadefromPTFEwiththeexceptionoftygontubing in the peristaltic pump (Reglo ICC Ismatec Switzerland) The high-efficiency flow-through photoreactor was a 19 W low-pressure mercurydischarge lamp (Beijing Titan Instruments Co Beijing China) with a quartzcentralchannel(asymp720μL internalvolume)Samplesolutionswere introducedintoastreamofreactionmediumusinganinjectionvalve(V-451IDEXHealthandScienceUSAsampleloopvolume05ml)Effluentfromthephotoreactorwasmixedwithaflowofargonandcarriedtothechilledgas-liquidseparator(internalvolume15mL)wherethevolatilespecieswereseparatedfromtheliquidwaste
124 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
andcarried to the inletofaScott-typespraychamber (originally the inlet formakeupargon)oftheICP-MS
3Resultsanddiscussion
The starting conditionswere adopted fromour earlierwork [14]whichusedatomicabsorptionspectrometerasadetectorandminiaturediffusionflameasanatomizerThefirstparameteroptimizedwasthecompositionofreactionmedium(Fig2)Theadditionofammoniumformate(createdin-situbytheadditionofacalculatedamountofammoniumhydroxidetoformicacid)wasfoundcrucialto
ndash1effectivelygeneratevolatilespeciesofcobalt10(vv)formicacidand4molL ammonium formatewas chosen as the optimumandwasused for all further
experimentsAlthoughhigher concentrations of both components led to even
higher signalstheseconditionswerenotusedfurtherbecauseofthelaboriousprocessofpreparation(mixingofconcentratedacidwithconcentratedbase)andtolimittheconsumptionofchemicals Theinfluenceofirradiationtimewasalsoexaminedandthehighestpeakarea
-ndash1wasobtained for4mLmin corresponding to an irradiation timeof approximately13s ToenhancethegenerationefficiencyadditionofvariousmetalstothereactionmediumwastestedtoldquosensitizerdquothephotochemicalreactionThemetalswerechosenwithrespecttotheirsignificantenhancementeffectdescribedrecentlyforphotochemicalvaporgenerationofotheranalytes[158]Theonlymetalionthat
2+ledtoanenhancementofthesignalwasCu (Fig3a)buteveninthiscasetheeffect was rather negligible reaching only 12-fold enhancement in the range
ndash1 2+ 2+001to01mgL Cu FurtheradditionofmoreCu ledtoadecreaseinthesignal2+TheadditionofZn didnotexhibitanypositiveornegativeeffectacrossthetested ndash1range (01 to 1500mgL not shown in figure) and the addition of higher
ndash1 2+ 2+ 6+concentrations (tens to hundredths ofmg L ) of Cd Fe andW (Fig3b)causedsevereinterferences
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 125
ndash ndash 2ndash Interferencescausedbycommoninorganicanions(NO Cl SO addedas3 4ndashtheirrespectiveacids)werealso investigatedOutof theseNO was foundto3
ndash1causethemostsevereinterferencesevenatconcentrationsofsinglemmolL Thendash 2ndashmethodologywasmorerobusttowardstheinterferencesfromCl andSO but4
theystillcausedsignificantdropinsensitivityathigherconcentrationsConsi-deringthewideuseoftheseacidsinanalyticalchemistryforsamplepreparationthisposesabigchallengeintheapplicationofthismethodtorealsamples
ndash1 Usingoptimalconditions(10(vv)formicacid4molL ammoniumformateandirradiationtimeof13s)acalibrationcurvewasmeasuredandevaluatedThelimitofdetectionwasdeterminedas3timesthestandarddeviationof10blank
ndash1measurements and was calculated as 13 ng L The repeatability of 10ndash1consecutivemeasurementsof250ngL was41
126 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 3Effectofvariousmetalionsonthepeakarea(a)metalionswithapositiveeffect(b)metalionsndash1withoutapositiveeffectExperimentalconditions2microgL Coreactionmedium10(vv)formic
ndash1 ndash1acidand4molL ammoniumformateflowrate4mLmin
ndash1Fig 2Effectofthecompositionofreactionmediumonpeakareaexperimentalconditions2microgL ndash1Coreactionmediumflowrate4mLmin (blackdotscorrespondtomeasuredpoints)
4Conclusions
Theconditionsofthephotochemicalvaporgenerationofcobaltwereoptimizedand are in good agreement with previous works [11 13] Copper ions wereidentifiedasapotentialsensitizerincreasingthesignalbyabout12-foldbuttheirpotentialuseisseverelylimitedbythenarrowrangeofconcentrationsinwhichthepositiveeffectisexhibitedSevereinterferencesfrominorganicanionswereobservedwhichisinlinewithotherworksdealingwithphotochemicalgeneration[1358]Furtherexperimentswillfollownamely(i)furtherinvestigationsinnewpotentialsensitizerstoenhancegenerationefficiencyandthusdecreasethe
ndash1limit of detection to sub ng L levels (ii) determination of the generationefficiency(fromcomparisonwithnebulizationandorusingaradioactiveisotope58Co)(iii)verificationoftheaccuracyandpracticalfeasibilityofthismethodologybyanalysisofcertifiedreferencematerials
Acknowledgments
ThesupportofTheCzechScienceFoundation(ProjectNo19-17604Y)CzechAcademyofSciences(Institutional supportRVO68081715)andCharlesUniversity (project SVV260560andprojectGAUK60120)isgratefullyacknowledged
References
[1] SturgeonREPhotochemicalvaporgenerationaradicalapproachtoanalyteintroductionforatomicspectrometryJAnalAtomSpectrom32(2017)2319ndash2340
[2] XuTHuJChenHJTransitionmetalionCo(II)-assistedphotochemicalvaporgenerationofthalliumforitssensitivedeterminationbyinductivelycoupledplasmamassspectrometryMicrochemJ149(2019)103972
[3] SoukalJSturgeonREMusilSEfficientphotochemicalvaporgenerationofmolybdenumforICPMSdetectionAnalChem90(2018)11688ndash11695
[4] deOliveiraRMBorgesDLGUVphotochemicalvaporgenerationofnoblemetals(AuIrPdPtandRh)AfeasibilitystudyusinginductivelycoupledplasmamassspectrometryandseawaterasatestmatrixJAnalAtomSpectrom33(2018)1700ndash1706
[5] VyhnanovskyJSturgeonREMusilSCadmiumassistedphotochemicalvaporgenerationoftungstenfordetectionbyinductivelycoupledplasmamassspectrometryAnalChem91(2019)13306ndash13312
[6] GuoXSturgeonREMesterZGardnerGJVaporgenerationbyUVirradiationforsampleintroductionwithatomicspectrometryAnalChem76(2004)2401ndash2405
[7] HuJSturgeonRENadeauKHouXZhengCYangLCopperionassistedphotochemicalvapor generation of chlorine for its sensitive determination by sector field inductivelycoupledplasmamassspectrometryAnalChem90(2018)4112ndash4118
[8] LeonoriDSturgeonREAunifiedapproachtomechanisticaspectsofphotochemicalvaporgenerationJAnalAtomSpectrom34(2019)636ndash654
[9] SturgeonREPaglianoEEvidenceforphotochemicalsynthesisoffluoromethaneJAnalAtomSpectrom(2020)httpsdoiorg101039D0JA00108B
[10] GrinbergPMesterZSturgeonREFerrettiAGenerationofvolatilecobaltspeciesbyUVphotoreduction and their tentative identification J Anal Atom Spectrom 23 (2008)583ndash587
[11] DengHZhengCB LiuLWWuLHouXDLvYPhotochemicalvaporgenerationofcarbonyl for ultrasensitive atomic fluorescence spectrometric determination of cobaltMicrochemJ96(2010)277ndash282
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 127
[12] deQuadrosDPBorgesDLDirectanalysisofalcoholicbeveragesforthedeterminationofcobalt nickel and tellurium by inductively coupled plasmamass spectrometry followingphotochemicalvaporgenerationMicrochemJ116(2014)244ndash248
[13] deJesusHCGrinbergPSturgeonRESystemoptimizationfordeterminationofcobaltinbiologicalsamplesbyICP-OESusingphotochemicalvaporgenerationJAnalAtomSpectrom31(2016)1590ndash1604
[14] VyhnanovskyJFotochemickegenerovanıtekavychspeciı kobaltuproanalytickouatomovouspektrometriiMasterthesisFacultyofScienceCharlesUniversityPrague2018
128 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Oneof themethods thatallowobtainingmaterialswithnewproperties is theplasmaenhancedchemicalvapordepositionmethodInthismethodcompoundscalledprecursorsaresuppliedtotheplasmareactorasagasphaseThankstoplasmaenhancedchemicalvapordeposition it ispossible toobtainmaterialswithuniquepropertiesThisisduetothefactthattheplasmaaffectsthesurfaceinfourdifferentways etching cleaning chemicalmodification and crosslinking
Optimization of condition for cold plasma deposition of thin layers for surface modification of working electrodes
a b a cJUSTYNALIPIN SKA MARIAMADEJ BOGUSŁAWBAS JACEKTYCZKOWSKI
a DepartmentofAnalyticalChemistryFacultyofMaterialsScienceandCeramicsAGHUniversityofScienceandTechnologyAdamaMickiewicza3030-059KrakoacutewPolandjustynalipinskaaghedupl
b DepartmentofAnalyticalChemistryFacultyofChemistryJagiellonianUniversityinKrakoacutewGronostajowa230-387KrakoacutewPoland
c Departmentof MolecularEngineeringFacultyofProcessandEnvironmentalEngineeringLodzUniversityofTechnologyWolczanska21390-924ŁoacutedźPoland
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 129
AbstractCurrentlyresearchisfocusedonthesearchfornewphysicallyandchemicallystablematerialsaswellasvolumeorsurfacemodificationOneofthemethodsusedforsurfacemodificationistheapplicationofthin layers from inorganic and organic compounds The plasmaenhancedchemicalvapordepositionisamethodthatallowsmaterialmodificationandalsodepositionofthinlayersThisworkconcernsoptimizationofcoldplasmadepositionparametersandtoachievethebestelectrical conductivitywhilemaintaining thehighmechanicalstrength of the formed layers Preliminary tests were focused onoptimizing the layering parameters such as the deposition timedischargepowerpressureofmonomerandthe flowofargonTheobtainedsamplesweresubjectedtothermaltreatmentafterwhichtheywere coveredwitha layerof aluminumThe thicknessof theobtained layers was determined on the basis of interferencemicroscopymeasurementsAsaresultoftheexperimentslayerswithathicknessof20nmto600nmwereobtainedTheconductivityofthedeposited layers was also determined and values from 003 to
ndash1150Sm wereobtained
Keywordscoldplasmadepositionelectrochemical
applicationssurfacemodificationthinlayers
Thismethodisusedtoproducecatalyticstructuresortomodifythepropertiesofmaterials eg improve hydrophobicity The growing popularity of surfacemodificationmethodsusingcoldplasmaisassociatedwiththefactthatitisanenvironmentallyfriendlyandversatilemethod[12] Workingelectrodesusedinvoltammetryareasubgroupofchemicalsensorswhich are small devices that convert real-time chemical information into ameasurableandanalyticallyusefulmeasurementsignalChemicalinformationrangingfromtheconcentrationofaspecificcomponentofthetestedsampletotheoverallcompositionofthematrixcancomefromboththeinitiatedchemicalreactionandbetheresultofphysico-chemicaltransformationstakingplaceinthetested object Chemical sensors are equipped with two basic elements iereceptor and transducer The receptor is responsible for the conversion ofchemicalinformationfromthetestedobjectintoaspecificformofenergyintheconverterthisenergyistransformedintoausefulanalyticalsignal Parameters characterizing the electrochemical sensor include accuracyprecision selectivity accuracy presentation selectivity sensitivity dynamicrange limit of quantification limit of detection lifetime response time andreliability Themost numerous and the oldest group of chemical sensors areelectrochemicalsensorsCommonlyobservedinterest inthisgroupofsensorsresultsfromthefactthatwithrelativelylowproductionandoperatingcoststheyofferthebestmetrologicalandoperationalparameters[3ndash5]Oneofthemaintrendsofmodernanalyticsisthesearchfornewelectrodematerialsandvariousgeometries of working electrodes One way to improve the performance ofworkingelectrodesistomodifytheirsurfaceforexamplebyapplyingthinlayersIn this work were considered plasma enhanced chemical vapor depositionmethod as the method of surface modification designed to performworkingelectrodeforvoltammetricdeterminationsAspartoftheinitialresearchplasmaprocessing parameters such as discharge power time of treatment andcompositionofgasmixtureinwhichplasmawasgeneratedwereoptimizedThelayers obtained in different conditions have been tested for suitability forelectrochemical applications (layer thicknessmeasurement and themeasure-mentofconductivity)
2Experimental
21Reagentsandchemicals
Theprecursor solutions suchasacrylonitriledietoxydimethylsilane trietoxy-methylsilaneandtetramethyldisiloxanewhicharesuppliedbyABCRwereusedOtherreagentsofanalyticalpuritysuchasn-hexane(SigmaAldrich)andargonwereused
130 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
22Instrumentation
The thin layers were deposited in a parallel-plate plasma reactor (frequency1356MHz)ThesamplesobtainedwerecalcinedinatunnelfurnaceunderanargonatmosphereThethicknessofthedepositedlayerswasmeasuredafterthealuminum was sputtered using a Nikon microscope type ECLIPSE LV150NElectrometerhigh resistance system (KEITHLEY) was used to measureconductivity
3Resultsanddiscussion
Eachof themonomerswasdepositedonprepared1times1corningglasssamplesSamples prepared with n-hexane were placed in a plasma reactor andadditionallypartiallycoveredwithamicroscopecoverslipSchematiclayoutofsamplesinthereactorshowninFig1ThefirststepwastoetchthesystemusingargonplasmaThisstageallowedfortheeliminationofimpuritiesthatwerenotremovedbythehelpofn-hexaneandthepreparationofthesurfaceofthesamplesforthedepositionoftheproperlayerTheproperstageistheapplicationofathinlayerwiththeplasmainducedbytheselectedprecursoracrylonitriledietoxydi-methylsilanetrietoxymethylsilaneandtetramethyldisiloxaneThethicknessandpropertiesoftheobtainedlayersdependonthedepositionparameterssuchasdischarge power time of treatment and composition of gasmixture inwhichplasmawasgenerated Table1showsallcombinationsofparameterstestedforallfourprecursorsFour different discharge powers for acrylonitrile and two different dischargepowersfororganosiliconmonomersweretestedwithtwodifferenttreatmenttimesEachtimeandpowercombinationwasperformedinplasmainducedbypuremonomerandmonomerwithargonAfterapplyingthelayersthesampleswereplacedinaquartzboatandcalcinedinatunnelfurnaceAllsampleswerecalcinedat500degCfor2hoursunderargonflowSamplesaftercalcinationwerecoveredwithalayerofaluminum
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 131
Fig 1 Scheme of sample distribution in plasma reactor(a)reactorelectrode(b)corningglasssamples(c)micro-scopecoverslip
Thethicknessoftheobtainedlayerswasmeasuredusinganinterferencemicro-scopeMeasurements were carried out at amagnification 10times recording theimageinmonochromelightTheimagewassetsothattheinterferencefringeswere perpendicular to the arc on the sample In order to calculate the layerthicknessD[nm]thefollowingformulawasused
(1)
wheredandLweredeterminedonthebasisoftheregisteredimage(disfringeshiftduetorefractionoflightontheslopeLisdistancebetweenthefringes) Thelaststageofthestudywastodeterminethecurrent-voltagecharacteristicstodeterminetheconductivityoftheobtainedlayersThesamplewasplacedinameasuring cell and attachedwith silver paste to the electrometerwires ThechangeincurrentwasrecordedwiththeapplicationofalternatingvoltageintimeBasedontheresultsobtainedthegraphsofdependenceUndashIwereobtainedfromwhichthevalueofresistance(R)wasdeterminedAnexampleofcurrent-voltagecharacteristicsisshowninFig2
132 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Table 1Conditionsforlayersdeposition-parameterwhichweretested
Monomer DischargepowerW Timeoftreatmentmin
Acrylonitrile 10204080 24Dietoxydimethylsilane 2040 255Trietoxymethylsilane 2040 255Tetramethyldisiloxane 2040 255
Fig 2Current-voltagecharacteristicsdeterminedfortheacrylonitrilelayer(depositionparametersW=10Wt=25mingasmixtureonlyacrylonitrile)
Table 4Theresultsofthicknessmeasurementsandconductivityspecifictothetrietoxymethylsilanelayers
DischargepowerW 20 20 40Timeoftreatmentmin 25 5 5Argonflowsccm 10 10 10Thicknessnm 14686 24282 35311
ndash1SpecificconductivitySm 165 93 67
Knowing the value of the resistance and the geometry of the system thespecificresistancewasdeterminedfollowedbythespecificconductivityofthesamplethefollowingequationwasused
(2)
whereρisspecificresistance[Ωm]Risresistance[Ω]bissamplelength[m]Disdepositedlayerthickness[m]andldistancebetweenelectrodes(wires)[m] Theresultsofthicknessmeasurementsandspecificconductivityarepresentedin the Tables 2ndash5 For electrochemical applications it is important that theobtainedlayerhasthehighestspecificconductivityAnalyzingthedatapresentedin Tables 2ndash5 shows that the thinnest layers have the greatest applicationpotentialastheelectrodematerial
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 133
Table 2Theresultsofthicknessmeasurementsandconductivityspecifictotheacrylonitrilelayers
DischargepowerW 40 40 80 80 10 10 20 20Timeoftreatmentmin 2 4 2 4 2 4 2 4Thicknessnm 15845 31167 250 4644 247 67 7531 12236
ndash1SpecificconductivitySm 0002 156 191 0003 1512 454 311 185
Table 3The results of thickness measurements and conductivity specific to the dietoxydimethylsilanelayers
DischargepowerW 20 20 40 40Timeoftreatmentmin 25 5 25 5Argonflowsccm 10 10 10 10Thicknessnm 13998 37519 28431 49556
ndash1SpecificconductivitySm 1640 55 105 40
Table 5The results of thickness measurements and conductivity specific to the tetramethyldisiloxanelayers
DischargepowerW 20 40 40Timeoftreatmentmin 25 25 5Argonflowsccm 10 10 10Thicknessnm 16759 3159 61652
ndash1SpecificconductivitySm 213 95 31
4Conclusions
InthisworkplasmaenhancedchemicalvapordepositionmethodwasusedforapplyinglayersoffourdifferentmaterialsacrylonitriledietoxydimethylsilanetrietoxymethylsilaneandtetramethyldisiloxaneBychangingparameterssuchasdischargepowertimeoftreatmentandcompositionofgasmixtureanumberofsampleswereobtainedwithlayersofdifferentthicknessandwhatisassociatedwithotherelectricalproperties TestsperformedaspartofthisworkwereusedtoperforminnovativeworkingelectrodesforvoltammetricdeterminationsThesurfacewasmodifiedusingthecoldplasmaofthreesubstratesgraphiteglassycarbonandgold
Acknowledgments
JLandMMhavebeenpartlysupportedbytheEUProjectPOWR030200-00-I00416
References
[1] KapicaRTyczkowskiJBalcerzakJMakowskiMSielskiJWorwaEEnhancingadhesivejointsbetweencommercialrubber(SBS)andpolyurethanebylow-pressureplasmasurfacemodificationIntJAdhesAdhes95(2019)102415
[2] TyczkowskiJKapicaRŁojewskaJThincobaltoxidefilmsforcatalysisdepositedbyplasma-enhancedmetalndashorganicchemicalvapordepositionThinSolidFilms515(2007)6590ndash6595
[3] HulanickiAGłabSIngmanFChemicalsensorsDefinitionsandclassificationPureApplChem63(1991)1247ndash1250
[4] BrzozkaZWroblewskiWSensorychemiczneWarszawaOficynaWydawniczaPolitechnikiWarszawskiej1999(InPolish)
[5] SkoogDAWestDWHollerFJCrouchSRFundamentalsofAnalyticalChemistry9thEdBostonCengageLearning2013
134 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
ConcentrationandcompositionofvolatilecompoundsorcongenersisoneofthemostimportantparametersresponsibleforqualityofproducedalcoholicdrinksandhencefortheirsensorycharacteristicsandconsumeracceptanceTodaygaschromatography (GC) is conventionally used to determine qualitative andquantitativecompositionsofvolatilecompoundswithvariousexternalandorinternalstandardcalibrationprocedures Methodemployingethanol as an internal standard (IS) forGCquantitativedeterminationofvolatilecompoundsinalcoholicbeverageshasbeensuggestedquitelongago[1]andsincethattimegreatresearchworkhasbeencarriedoutRecentlyaninterlaboratorystudyofthemethodinvolving9testinglaboratoriesfrom4countrieswascarriedout[2]TheresultsdemonstratedgreatperspectivesofldquoEthanolasISrdquomethodandproveditsreferencecharacterandeaseofroutineimplementation
Advanced GC-MS method for quality and safety control of alcoholic beverages
abANTONKORBAN
a DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova2030812840Prague2CzechRepublickarbonat7gmailcomb DepartmentofAnalyticalChemistryChemistryFacultyBelarusianStateUniversityLeningradskaya14220050MinskBelarus
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 135
AbstractRecently developed and validated simple and reliable quantitativemethod employing ethanol as an internal standard for GC-MSquantification of volatile compounds in alcoholic products wasapplied to 36 samples including commercially available world-famousbrandspirits from18countriesandhomemadedistillatesThe GC-MS analyses were performed simultaneously by thesuggested approach and official internal standard method that isprescribedinthelegislationofEUandUSATheindependentsamplest-testwasemployedtoevaluatethestatisticaldifferenceofresultsofthesetwomethodsThetestrevealednodifferenceintheresultsandtheirrepeatabilityThemainbenefitsofthesuggestedmethodaretheeliminationofthenecessityofmanualinternalstandardadditionandsamplesdensitymeasurementthusmakingitmoreeconomicalandproductive
Keywordsalcoholicbeveragesgaschromatography-
massspectrometry(GC-MS)
internalstandardmethodvolatilecompounds
quantification
AllpreviousstudieswereutilizingflameionizationdetectorssinceGC-FIDisprescribedinthelegislation[3]wheremass-spectrometrydetectorsarenotyetofficially referred However GC-MS instruments are employed in practice toqualifyandorquantifyvolatilesincommercialspiritsintraditionalhomemadealcoholicdrinksinnewlydevelopedbeveragesinspiritwastesandindistillatesobtainedwithdifferentmanufacturingprocesses Our recent researchwasdirected towardsdevelopment of an algorithmofldquoEthanolasISrdquomethodapplicationonGC-MSinstruments[4]WehaveshowedthattopreventMSdetectorfromsaturationethanolshouldberegisteredinthecorresponding SIM timewindow at characteristicmz of low abundance forinstance bymz of 47 ions This ion corresponds to non-fragmented ethanol
13moleculescontaining1heavyisotope(mainly C)FinallytheresultsofmeasuredstandardsolutionsshowedthatthesuggestedapproachisvalidandldquoEthanolasISrdquomethodmaybesuccessfullyusedonGC-MSinstrumentstoo The objective of this studywas to test and further approve the suggestedapproachonalargersetof36realsamplesofalcoholicdrinkseithercommercialorhomemadeThesamplesweresimultaneouslyanalysedbytwoGC-MSmethodsndashaclassicalISmethodprescribedinthelegislationandthesuggestedldquoEthanolasISrdquomethod
2Experimental
21Reagentsandchemicals
Thefollowingvolatilecompoundsweredeterminedintestedsamplesofalcoholicbeverages 11-diethoxyethane (acetal) acetaldehyde methyl acetate ethylacetate methanol 2 propanol 1-propanol 2 methylpropan-1-ol (isobutanol)1-butanol2-butanoland3methylbutan-1-ol(isoamylalcohol)1-pentanolwasemployedasatraditionalIScompound
22Instrumentation
ShimadzuGCMS-QP2010Ultraequippedwithaquadrupolemassspectrometrydetector was employed for GC-MS measurements Rxi-1301Sil MS capillarycolumn(60mlength025mmid025micromfilmthicknessRestek)wasusedfortheseparationofcompoundsInjectionswereperformedinasplitmode(ratio175)Helium(99999purity)wasusedasacarriergasinjectortemperaturewas170degCTheoventemperaturewasheldat30degCfor5minthenraisedto210degC
ndash1at a rate of 30 degCmin and held isothermally for 4minMeasurementswereperformedinaSIMmodeFortheanalysedcompoundsand1-pentanol2ndash3mostabundantionsinthecorrespondingMSspectrumwereselectedethanolSIMtimewindowcontainedonly47mzionsAllGC-MSmeasurementswerecarriedoutintriplicateunderrepeatabilityconditions
136 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
AnalysisofeachalcoholicsamplewasperformedinafollowingwayAliquotof09mL of a tested sample was pipetted into a standard 2mL glass vial and
ndash1weighedAfterthat01mloftheISsolution(2355mgkg of1-pentanolinWES)wasaddedtothetestedsampleandthemasswasrecordedTheobtainedmixturewasmixedthoroughlyand05microlofitwasinjectedintotheGCsystem Theoriginoftestedalcoholicbeverageswaseithercommercialorhomemade33 world-famous spirits manufactured at different parts of the world werepurchasedfromcommerciallyavailablesourcesThelistoftypesofpurchasedandanalysedspiritsincludedbourboncalvadoscognacgingrappaliquormetaxaportwine rumsake tequilavodkawhiskeyandvarious fruitdistillatesThepurchased drinks were produced at the territory of the following countriesBelarus Bermuda Cuba Czech Republic Denmark France Germany GreeceGuatemala Jamaica Japan Mexico Moldova Portugal Slovakia Trinidad andTobago UK (England and Scotland) USA Three homemade fruit distillatesproducedbyfermentationofpulpyfruitsortheirmustswereobtainedfromlocalspiritmakersThedeclaredABVvaluesofalltestedsamplesvariedfrom15to81
3Resultsanddiscussion
To fulfil themaingoalof thiswork ie toevaluate thestatisticaldifferenceofresultsyieldedbythecomparedmethodswehaveemployedStudentst-testforindependentsamplestoverifystatisticaldifferencesonthesignificancelevelofp=005Theobtainedempiricalvaluesforallpairsofcongenersrsquoconcentrationswere lower than critical one in all cases demonstrating that concentrationsobtainedbythetwomethodshavenostatisticaldifferenceandleadtothesameresults InadditionrepeatabilityofthetwomethodswascomparedthereforeallRSDvalues obtained from triplicatemeasurements were split in two groupswith
ndash1respecttothecorrespondingconcentrations(lowerthan50mgL AAandhigherndash1than50mgL AA)Theobtainedresultsarepresentedintheformofboxplotin
Fig1AnalysisofthechartinFig1showedthatboththetestedmethodshaveyieldedstatisticallysimilarrepeatability AllofthetestedalcoholicdrinkssatisfiedtherequirementsofEURegulation(EC)no1102008[5]Theconcentrationsofundesirablecompoundssuchasmethanoldidnotexceedthelevelsspecifiedinthesameregulationforcorres-ponding beverages In Table 1 the description of the used SIM method andsummaryoftheexperimentalresultsarepresented Tocomparethetruenessofthemethodsoneofthespiritsampleswasspikedwithstandardsolutions(ABV40)containingallanalysedvolatilecompoundsat
ndash1concentrationsof50500and5000mgL AATheoriginalsamplewasusedasareferenceEachofthespikedsolutionswasmeasuredintriplicateSelectedspirit(cherry distillate) initially contained all 11 volatile compounds in various
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 137
138 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1BoxchartsofRSDsofusedISmethodsat2concentrationrangesMeanisequaltoarithmeticmeanoraverageInterquartileRange(IQR)meansisthedistancebetweentheupper(themedianoftheupperhalfofthedataset)andlower(themedianofthelowerhalfofthedataset)quartile
ndash1Compound Timemin Registeredmz Numberof ConcentrationmgL AA results Minimal Maximal
Acetaldehyde 0ndash42 314344 36 24 715Methanol 36 13 13600
Ethanol(IS) 42ndash48 47 mdash mdash mdash
2-Propanol 48ndash70 29314345 14 27 199Methylacetate 596174 10 34 3201-Propanol 26 361 12070Ethylacetate 27 166 107002-Butanol 11 18 2080
Isobutanol 70ndash200 3141ndash4345 28 19 20001-Butanol 5556 13 28 155Acetal 26 45 270Isoamylol 31 39 26501-Pentanol(IS) mdash mdash mdash
Table 1DescriptionoftheusedSIMmethodandsomestatisticsconcerningallmeasured36spiritsamplesbothpurchasedandhomemade
concentrationsTheobtainedrecoveriesboxchartsareshowninFig2Compa-risonoftherecoveriesobtainedwithtwomethodsindicatesthattheyhavenosignificantdifferenceintermsoftruenessAverageobtainedrecoverywas981whenusingsuggestedmethodand980whenusingtraditionalISmethod
4Conclusions
InthisworktheresultsoftestingtheadvantageousldquoEthanolasISrdquomethodfortheGC-MS quality control analysis of alcoholic beverages were presented33purchasedsamplesofworld-famousalcoholicbeveragesoriginatingfrom18countriesand3homemadefruitdistillateswereanalysedtomakeathoroughandcomprehensive studyof thedevelopedmethodTheconcentrationsofvolatile
ndash1compoundsinanalysedsamplesvariedfrom1to13500mgL AAtheABVvalueofanalysedsamplesvariedfrom15to81ThesuggestedmethodwascomparedwiththetraditionalISmethodthatiscurrentlystatedinlegislationTheindepen-dentsamplest-testrevealedthatwithaprobabilityof095resultsobtainedwithtwo methods do not differ significantly The results of within-run precision(repeatability)showedrelativestandarddeviationswithin3measurementstobelessthat6indicatingthatthetechniqueisreproducibleThetruenessofthemethodwasevaluatedbyrecoverycalculationAccordingtotheobtainedresultsrecoveryofthesuggestedmethod(981plusmn33)wasslightlybetterthanthatofthetraditionalone(980plusmn58) ThesefactsprovethatdevelopedldquoEthanolasISrdquomethodistruepreciseandreliable when employed on GC-MS instruments At the same time to obtain
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 139
Fig 2Boxchartsof recoveriesof thesuggested(dottedpattern)and traditional (brickpattern)ISmethodsatdifferentspikeconcentrationsSymbolsdefinitionsarethesameasinFigure1
concentrationsofvolatilecompoundsintheofficiallyrequiredunitsofmeasurendash1 ndash1(mgL AAgL AAetc)suggestedmethodrequiresnodensitometrymeasure-
mentsofthetestedsampleandnoadditionofIScompoundoranyothersamplepre-treatmentThismethodprovidesaninvaluableanalyticaltoolforthequalitycontrolofalcoholicproductsandshouldbeusedinroutineanalysis
Acknowledgments
ThisworkwasfinanciallysupportedbytheVisegradFund
References
[1] CherepitsaSVBychkovSMKovalenkoANMazanikALSeleminaNMSeredinskayaOBThe use of themajor component (solvent) as an internal standard in the gas-chromato-graphicdeterminationofimpuritiesJAnalChem58(2003)368ndash371
[2] CharapitsaSSytovaSKorbanASobolenkoLEgorovVLeschevSZakharovMCabalaRBusarovaRShestakovichITolstouhovaAOndrousekSVavraJYilmaztekinMCabarogluTInterlaboratorystudyofethanolusageasaninternalstandardindirectdeterminationofvolatile compounds in alcoholic products BIO Web Conf 15 (2019) 02030httpsdoiorg101051bioconf20191502030
[3] CommissionRegulation(EC)No28702000layingdownCommunityreferencemethodsfortheanalysisofspiritsdrinkshttpdataeuropaeuelireg20002870oj
[4] KorbanACharapitsaSCabalaRSobolenkoLSytovaSTheperspectivesofethanolusageasaninternalstandardforthequantificationofvolatilecompoundsinalcoholicproductsbyGC-MSJMassSpectr55(2020)e4493
[5] EuropeanUnion(2008)Regulation(EC)No1102008oftheEuropeanParliamentandoftheCouncilof15January2008ontheDefinitionDescriptionPresentationLabellingandtheProtectionofGeographical IndicationsofSpiritDrinksandRepealingCouncilRegulationhttpdataeuropaeuelireg2008110(1)oj
140 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Carbonorgraphitefeltsareusedaselectrodematerialsincethe1990sandtheirutilizationstillgrowsinanalyticalelectrochemistryaswellasinotherareasThisisduetotheirsuitablepropertiesfromwhichwecannamehighporosityhighspecificsurfaceareagoodelectricconductivityandhighphysicalandchemicalstabilityThefirsttwoparametersaregivenbystructureoffeltwhichconsistsoforderlesscarbonfibreswithabouttentotwentymicrometersindiameter[1]theothersby theadvantageouselectricalpropertiesof carbon fibreOn theotherhand porous flow-through electrodes including carbon felt electrode havedisadvantageinapotentialdropintheelectrodevolumewhichcausesdifficultcontrollingoftheexactpotentialappliedontheelectrodeandthereforeresultsindifferentcurrentefficienciesontheoppositesidesoftheelectrode[2] Carbon felt electrode can be utilized for detection of structurally differentcompoundsatvariousconditionsForexampleoperatingatreductionpotentialofndash08V[3]oxidationatrelativelyhighpotential+15V[4]ormeasuringatlowconcentrationsofelectrolyte[5]canbenamedDevelopedtechniquesalsoshowsthatcarbonfeltcanbeusedfordeterminationatsubmicromolarconcentrationsThis ismainly due to its ability to operate as a high-efficiency amperometricdetector Themainaimofthispaperistooverviewandcompareparametersofseveraldetermination methods of different analytes using carbon felt detector in
Utilization of a carbon felt as a material for working electrodes
MARTINBAROCHHANADEJMKOVA SA RKASLA DKOVA
DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublicmartinbarochnaturcunicz
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 141
AbstractWorkingelectrodemadeofcarbonfeltwasusedincombinationwithHPLC for verificationofpractical applicabilityof the electrodeAlldeveloped methods confirm advantageous physical and chemicalpropertiesofcarbonfeltForelectrochemicalutilizationitispossibleto operate at higher positive potentials and even in low concen-trationsofelectrolyteinmobilephaseObtainedlimitsofdetectionwere mostly in submicromolar range and standard deviations ofmeasurementrepeatabilitywereunder5
KeywordsamperometrycarbonfeltFIAHPLC
combinationwithHPLC(forexampletheirdetectionpotentialslimitsofdetectionorlinearrange)
2Experimental
21Reagentsandchemicals
Stock solutions of propyl gallate butylhroxyanisole tert-butylhydroquinonebutylhydroxytoluene chlortoluron 2-amino-4-nitrophenol and 4-amino-2-
ndash3 ndash1nitrophenol (all SigmandashAldrich) with concentration of 1times10 molL werepreparedbydissolving theappropriateamountof therespectivesubstance inmethanol(HPLCgradeLach-NerCzechRepublic)Stocksolutionsof indole-3-aceticacidandindole-3-butyricacidwerepreparedinthesamemannerbutindeionized water Mobile phase consisted of methanol and phosphate-acetatebufferpreparedfromphosphoricandaceticacid(bothLach-NerCzechRepublic)andsodiumhydroxide(Fluka)
22Instrumentation
Theelectrochemicalcellconsistedofcarbonfelt(KarbotechnikCzechRepublic)flow-throughelectrodewhichwasplacedincapwithplatinumwireelectricalcontactanddrilledoutletholeononesideandwithflatferrulawithcapillaryonthe other side Schematic picture of the assembly is shown in ref [5] Otherelectrodeswereauxiliaryplatinumwireelectrodeandreferencesilverchloride(3MKCl) electrode (bothMonokrystaly Turnov Czech Republic) Potentiostatused in combination with this cell was Amperometric Detector ADLC 2(LaboratonıprıstrojePrahaCzechRepublic) HPLCapparatusconsistedofBeta10gradientpump(ECOMCzechRepublic)degasser DG 4014 (ECOM Czech Republic) six-way valve with 20microl loop(Rheodyne USA) HPLC column used for separation of propyl gallatebutylhydroxytoluenetert-butylhydroquinoneandbutylhroxyanisolefromtheirmixtureandforindole-3-aceticacidandindole-3-butyricacidfromtheirmixture
regwasLichrospher RP-18(125times4mm5micromMerckGermany)ForseparationofregchlortoluroncolumnPurospher RP-18(125times4mm5micromMerckGermany)was
used In case of mixture 2-amino-4-nitrophenol and 4-amino-2-nitrophenolregcolumnGemini C18110A (150times46mm5micromPhenomenexUSA)wasusedfor
separation Measurements of pHwere carriedout at Conductivity andpH-meter3510usingcombinedglasselectrode(JenwayUK)
142 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
3Resultsanddiscussion
Performanceofcarbonfeltwastestedonseveraltypesofanalyteswhichneededdifferent separation conditions namely amount of organic solvent in mobilephase and buffer pH The lowest amount of methanol (30) was used forseparation of 2-amino-4-nitrophenol and 4-amino-2-nitrophenol [3] Higherconcentrations of methanol in mobile phase was used for determination ofchlortoluronandforseparationofindole-3-aceticacidandindole-3-butyricacid[4]namely40and60respectivelyThehighestconcentrationsofmethanoland therefore electrolyte with lowest conductivity was used in separation ofantioxidantsnamelypropylgallatebutylhroxyanisolebutylhydroxytolueneandtert-butylhydroquinonewhereamountofmethanolwasrampingfrom55to95[5]DetectionpotentialsofmentionedanalytesandtheirlimitsofdetectionsareshowninTable1 HPLCseparationofantioxidantswas the firstmethodchosen for testingofcarbonfeltelectrodeperformancewiththistechniqueDuetodifferencesintheirstructure when butylhydroxytoluene has a different oxidation mechanismdetectionwith two appliedpotentialswas necessary According to the hydro-dynamic voltammograms potentials 14V and 08Vwere chosen for determi-nationofbutylhydroxytolueneandfortheotherthreeanalytesrespectively[5]AsshowninTable1whenthehigherpotentialwasappliedlimitsofdetectionforpropyl gallate butylhroxyanisole and tert-butylhydroquinone had increasedContrary determination of butylhydroxytoluene had approximately six timeslowerdetectionlimitathigherpotential
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 143
ndash1Compound E VLODmicromolL Refdet
Propylgallate 080 088 [5] 140 186 [5]Butylhroxyanisole 080 144 [5] 140 348 [5]tert-Butylhydroquinone 080 121 [5] 140 266 [5]Butylhydroxytoluene 080 3128 [5] 140 463 [5]Chlortoluron 140 013Indole-3-aceticacid 150 033 [4]Indole-3-butyricacid 150 054 [4]4-Amino-2-nitrophenol 080 016 [3]2-Amino-4-nitrophenol 080 021 [3]4-Amino-2-nitrophenol ndash080 35 [3]2-Amino-4-nitrophenol ndash080 37 [3]
Table 1Parametersofdetectionpotentialsandlimitsofdetectionfordifferentcompoundsusingcarbonfeltdetector
Detectionofauxins(indole-3-aceticacidandindole-3-butyricacid)wascarriedout at potential 15VMeasurements at this potential gives repeatabilitywithstandarddeviation31forindole-3-aceticacidand25forindole-3-butyricacidevenwithexchangingoftheworkingelectrodematerialCalibrationcurves
ndash1forbothanalyteswereobservedfrom04to100μmolL withlinearityinwholeconcentrationrangeLimitsofdetectionforbothanalytesreachedsubmicromolarconcentrationsevenwithrelativelyhighpotential[4] Incaseofdeterminationof2-amino-4-nitrophenoland4-amino-2-nitrophenolcarbon felt electrodewasused inbothoxidationand reductionmodeHydro-dynamicvoltammogramsshowedthatoptimaldetectionpotentialinreductionmodewasndash08VThislowpotentialisclosetotheendofthepotentialwindowandthereforeinterferenceswithremnantsofdissolvedoxygeninmobilephasewereobservedTheseinterferencesresultedinapproximately20timeshigherlimitofdetection for 2-amino-4-nitrophenol or 4-amino-2-nitrophenol obtained inreductionthaninoxidationmodeOntheotherhandmaximumvaluesoflinearrangewerethesameforbothanalytesinbothdetectionmodes[3] ForHPLCofchlortolurontheoptimaldetectionpotentialof14VwasfoundIts
ndash1calibration dependence although observed from 025 to 1000μmolL was ndash1linearonly in therange from025to50μmolL Limitofdetectionbasedon
ndash1standardsolutionswas013μmolL andreproducibilityofmeasurementgivenbytwentyconsecutivemeasurementsgaverelativestandarddeviationof05 ForallthedeterminationmethodsattentionwaspaidtotheapplicabilityofthecarbonfeltindetectionofanalytesincomplexmatricesIncaseofantioxidantsedibleoilswerechosenasrealsamples[5]nitrophenolderivatesweredeter-minedinurinesamples[3]andauxinsinrootingpreparation[4]ChlortolurondeterminationwasperformedinsoilandsurfacewaterThefoundvaluesshowanegligiblematrixinfluenceondetection
4Conclusions
CarbonfeltworkingelectrodewassuccessfullyusedincombinationwithHPLCfordeterminationofdifferenttypesofelectroactivecompoundsegantioxidantsauxinsorpesticidesAllmentionedapplicationsshowgreatperformanceofthecarbonfeltasaflow-throughelectrodematerialinelectroanalyticalchemistryforoxidationandreductionwayofanalytesdeterminationLimitsofdetectionforanalytes are mostly in submicromolar concentrations the exceptions areoxidationof analytes athigherpotentials and their reductionwhere limitsofdetectionsareinmicromolarconcentrationsApplicabilityoftheelectrodeonrealmatriceswas proven on analysis of edible oil samples groundwater soil androotingpreparation
Acknowledgments
ThisworkhasbeensupportedbytheCzechScienceFoundation(projectGACR20-01589S)
144 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
References
[1] Gonzalez-Garcia J Bonete P Exposito E Montiel V Aldaz ATorregrosa-Macia RCharacterizationofacarbonfeltelectrodeStructuralandphysicalpropertiesJMaterChem9(1999)419ndash426
[2] NavaJLRecendizAGonzalezLGCarrenoGMartın ezFMassTransportandpotentialstudiesinaflow-throughporouselectrodereactorPortugalElectrochimActa27(2009)381ndash396
[3] Dejmkova H Knaf M Application of carbon felt detector for the determination ofdinitrophenolmetabolitesInXXXIXModernElectrochemicalMethodsFojtaMSchwarzovaKNavratilT(Eds)U stınadLabemBestServis2019p41ndash43
[4] DejmkovaHdeAraujoDanielMElectrochemicaldeterminationofindole-3-aceticacidandindole-3-butyric acid using hplc with carbon felt detectorMonatsh Chem150 (2019)439ndash442
[5] DejmkovaHBarochMKrejcovaMBarekJZimaJCoulometricdetectorbasedoncarbonfeltApplMaterToday9(2017)482ndash486
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 145
1Introduction
Smith-Lemli-Opitzsyndrome(SLOS)isan autosomal recessive genetic disor-der firstly described in 1964 [1] It iscaused by the inborn deficiency of7-dehydrocholesterol reductase Thisenzyme transforms 7-dehydrocholes-terol (7-DHC Fig 1) to cholesterolduringthefinalstepofbiosynthesisofcholesterolincells The clinical symptoms of SLOS aredecreasedbloodlevelofcholesteroland
Electroanalytical methods for determination of 7-dehydrocholesterol in artificial serum
LENKABENESOVAADE LAZA RYBNICKA JANKLOUDAKAROLINASCHWARZOVA -PECKOVA
UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublicbenesolenaturcunicz
Abstract7-DehydrocholesterolisabiomarkerofSmith-Lemli-Opitzsyndromeanautosomalrecessivegeneticdisordercausedbytheinborndefici-encyof7-dehydrocholesterolreductaseInthisstudyproceduresforitsdeterminationinartificialserumusingflowinjectionanalysiswithelectrochemical detection and voltammetric detection on borondoped diamond electrode were optimized The proteins wereprecipitatedbyacetonitrileandaftercentrifugationthesupernatantused for analysis For quantitation of 7-DHC by differential pulsevoltammetrytheoptimalratioacetonitrile-artificialserum91(vv)wasappliedInFIA-EDtheratio31(vv)runelectrolyteconsisting
minus1ofwater-acetonitrilecontaining001molL NaClO inthesameratio4minus1anddetectionpotentialof+13VvsAgAgCl(3molL KCl)were
usedQuantitationof7-DHCwaspossibleusingcalibrationdepen-minus1dencewithlimitdetectionof20micromolL inartificialserumNeverthe-
lessthemethodhaslowrecoveryandforsensitivedeterminationinreal matrices of human serum and amniotic fluid a liquid-liquidextraction needs to be applied to prevent presence of 7-dehydro-cholesterolinthephasewithprecipitatedproteins
Keywordsamperometricdetectionborondopeddiamond
electrode7-dehydrocholesteroldifferentialpulse
voltammetryflowinjectionanalysisSmith-Lemli-Opitz
syndrome
146 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Structureof7-dehydrocholesterol
increased concentrationof7-DHC inbloodandnervous system [2] SLOS is acomplexofmultipleanomaliesincludingmentalretardationItismanifestedbyholoprosencephaly(anomaliesinbraindevelopmentwithimprecisedivisionintotherightandlefthemispheres)milddysmorphismscardiacrenalandgastro-intestinalmalformations ThecharacteristicfacialanomaliesofSLOS[2]aremicrocephalybitemporalnarrowing ptosis short nasal root short nose with anteverted nares andmicrognathia epicanthal folds and capillary hemangioma over the nasal rootextendingontotheglabellatheearappearlow-setandareposteriorlyrotatedOral finding includes a high-arched and narrow hard plate broad and ridgealveoralridgesandredundancyofsublinqualtissuesCNSanomaliesareagenesisorhypoplasiaBilateralandunilateralpostaxialpolydactylycanbepresentedinthehandsorfeetorboth Concentrationof7-DHCinbloodiscrucialforclinicaldiagnosticofSLOSinpatients Concentration levels in amniotic fluid are used for fetal diagnosticsTable1 summarizes concentration of 7-DHC in plasma and amniotic fluid ofhealthypersonandSLOSpatientsAnalyticalmethodsusedfordeterminationofconcentrationof7-DHCinthesematricesincludecombinationofGCorHPLCwithMS[3]orGCwithflameionizationdetection(FID)orUVdetection[45] Thepossibilitiesofelectrochemicalmethodsfordetectionof7-DHCarelimitedasgenerallythesteroidcoreisratherredox-inactive(detailinreview[6])undervarietyofconditionsNevertheless7-DHCpossessesconjugateddoublebondsonsteroidcoreanditsoxidationwasreportedinseveralstudies[7ndash9]Itsvoltam-metricsignal+095VvsSCEonglassycarbonelectrodewasfirstlyobservedin
minus1non-aqueousmediaofmethanol-benzene7525(vv)using005molL LiClO as4
supporting electrolyte in a study dealing with electrochemical behaviour ofvitaminAandDandtheirprovitaminsD(7-DHCisprecursorofvitaminD3)[7]Determinationof7-DHCinhumanskinispossiblebyHPLCwithUV(λ=286nm)andamperometricdetection(E +17VvsAgAgCl)onglassycarbonelectrodedet
minus1usingmethanol-tetrahydrofuran175mmolL KH PO (9514vvv)asmobile2 4minus1phase7-DHCwasdetectedintherangefrom12to81microgg dryweightwith
minus1detectionlimitof39pmolL [8]Anotherstudy[9]isdevotedtodeterminationof
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 147
minus1Author(s)ref Matrix ConcentrationμmolL Healthy SLOS
Kelley[4] Plasma 03plusmn001 385plusmn309 Amnioticfluid lt02plusmn001 16plusmn9Rossiteretal[5] Plasma lt5 179ndash335 Amnioticfluid lt03 12ndash15
Table 1Concentrationof7-dehydrocholesterolinclinicalmatricesforhealthypersonsandforSmith-Lemli-Opitzsyndrome(SLOS)patientsdetectedbyGC-FID
7-DHCandvitaminD3 in fishusingHPLCwithelectrochemicaldetectionTheanalytical cell was a serial combination of two-flow-through porous graphiteworking electrodes The first standard coulometric electrode was used toeliminatepotentially interfering compounds using the second lineardynamic
minus1rangefrom0013to0312micromolL for7-DHCwasachieved Hereinwestudiedpossibilitiestodetect7-DHCbasedonitsoxidationonborondoped diamond (BDD) electrode using differential pulse voltammetry andelectrochemicaldetectioninflowinjectionanalysis(FIA-ED)inartificialserumandperipherallyinhumanserumandamnioticfluid
2Experimental
21Reagentsandchemicals
7-dehydrocholesterol (purity95)wasobtainedSigmaAldrich (USA)and itsstandard solution was prepared in acetonitrile (Honeywell Germany) Theartificial serum was prepared from KCl (Penta Czech Republic) CaCl 2H O2 2
(PentaCzechRepublic)NaCl(PentaPragueCzechRepublic)urineD-glucoseand01albuminfromSigma-Aldrich(USA)NaClO4(PentaCzechRepublic)wasusedassupportingelectrolyte
22Instrumentation
VoltammetricmeasurementweregovernedbythepotentiostatPalmSensusingworking BDD electrode (Windsor Scientific UK d = 31 mm) AgAgNO 3
minus1 minus1(01molL AgNO 1molL NaClO in acetonitrile) non-aqueous reference3 4electrodeandplatinumwirecounterelectrodeBDDsurfacewaspolishedbeforeeachscanusingsuspensionofAl O (ElektrochemickedetektoryTurnovCzech2 3
Republic) HPLC system (Hitachi Merck) consisting of control unit D-7000gradientpumpL-7100autosamplerL-7200andUVdetectorL-7400wasusedfor
minus1FIA-EDdetectionof7-DHCRunelectrolytewascomposedof001molL NaClO 4inacetonitrileanddeionisedwaterinratio31(vv)Flowrateofmobilephase
minus1was30mlmin injectionvolumewas40microLandλ=280nmwasusedforUVdetectionWall-jet detection cell was employedwith working BDD electrode
minus1AgAgCl (3molL KCl) reference electrode and platinum wire auxiliaryelectrode Optimal detection potential of +13 V was controlled using ADLC2potentiostat(Laboratornı prıstrojePragueCzechrepublic)
3Resultsanddiscussion
In this study electroanalytical methods were developed for determination of7-DHCinartificialserumnamelyFIA-EDandDPVBothmethodsarebasedondirectoxidationof7-DHConborondopeddiamondelectroderesultinginanodic
148 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
peak at ca +08 V (vs AgAgNO in acetonitrile) in non-aqueous medium of3
acetonitrile or mixedmedium acetonitrile-water using NaClO as supporting4
electrolyteTheoxidationispresumablyinitiatedbyoneelectronremovalfromtheconjugateddoublebondsonthesteroidcoreof7-DHC Fordeterminationof7-DHCinrealmatricesitisnecessarytoremovepresentproteinsArtificialserumcontainingalbuminwasusedasmodelmatrixtostudythe possibilities Firstly albumin was removed simply by precipitation withacetonitrile(serum-water13(vv))andthesupernatantwasanalysed Differentratioswateracetonitrileweretestedinrunelectrolyte(59510902080 2575 3070 4060 and 5050 (vv)) to evaluate the influence of itscompositionontheFIA-EDsignaloftheblankand7-DHCThesameratio13asusedforprecipitationofalbuminwaschosenasoptimalbecauseofminimalandstablesignaloftheblankinjectedinFIA-EDsystemFurtherdetectionpotentialE in the range from +10 V to +15 V was optimized by evaluation of thedet
hydrodynamicvoltammogramsresultinginE of+13Vsetasoptimalvaluedetminus1 Concentrationdependenceof7-DHCislinearintherangefrom25micromolL to
minus1 minus1300micromolL (concentrationinartificialserum)withdetectionlimitof20micromolL and this concentration dependence can be used for quantitation of 7-DHC inartificial serum Nevertheless determination of 7-DHC in human serum andamnioticserumfailedastheyrepresentmorecomplicatedmatricesand7-DHCispresumablypartiallyadsorbedinthepresentproteinsandcannotbequantifiedinthesupernatant Furtherdifferentialpulsevoltammetrywithoptimizedparameterswasusedfordeterminationof7-DHCInthepresenceofproteinsinartificialserum(human
minus1serum albumin) an unacceptably high detection limit of 178micromolL wasachieved When the proteins were precipitated using acetonitrile (aceto-nitrileartificialserumratio91(vv))thelimitofdetectionof7-DHCwaslowered
minus1to15micromolL inartificialserumNeverthelesstherecoveryofthemethodwasonly43to70dependingontheconcentrationof7-DHCagainreflectingthelossof7-DHCduetoproteinprecipitation Thereforeasecondapproachofsamplepretreatmentbasedonliquid-liquidextractionofalllipidsdescribedin[10]wastested(Bligh-Dyerextraction)TheprocedurehastwopartsFirstlymethanolchloroformandthesampleofartificialserum is mixed and shaken to form a monophasic system After addition ofchloroform and water a biphasic system is formed where chloroform phasecontainsalloflipidcompoundsandmethanol-waterphasecontainsallnon-lipidscompoundsChloroformphaseisthendriedunderN2atmosphereat50degCanddried extract dissolved in acetonitrile Preliminary experiments using DPVresultedinrecoveryof97forBligh-Dyerextractionof7-DHCfrominartificialserum
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 149
4Conclusions
FIA-EDandDPVwereoptimizedfordeterminationof7-DHCinartificialserumUsing precipitation of proteins by acetonitrile limit of detection of 7-DHC in
minus1artificialserumusingFIA-EDwas20micromolL andthismethodcanbeusedfortheirquantificationusingcalibrationdependenceNeverthelessdeterminationusingDPVisunreliableduetolowrecoveryoftheprocedureDevelopmentofamethodincludingliquid-liquidextractionstepisinprogresssothat7-DHCcouldbedeterminedinrealmatricesashumanserumandamnioticfluid
Acknowledgments
TheresearchwassupportedbytheCzechScienceFoundation(projectGACR19-11268S)andtheSpecificUniversityResearch(SVV260560)
References
[1] Smith DW Lemli L Opitz JM A newly recognized syndrome of multiple congenitalanomaliesJPediatr64(1964)210minus217
[2] Nowaczyk M Waye J The SmithndashLemlindashOpitz syndrome a novel metabolic way ofunderstandingdevelopmentalbiologyembryogenesisanddysmorphologyClinGenet59(2001)375minus386
[3] BeckerSRohnikeSEmptingSHaasDMohnikeKBebloSMutzeUHusainRAThieryJCeglarekULC-MSMS-basedquantificationofcholesterolandrelatedmetabolitesindriedblood for the screening of inborn errors of sterolmetabolismAnal Bioanal Chem407(2015)5227minus5233
[4] Kelley RI Diagnosis of Smith-Lemli-Opitz syndrome by gas-chromatography mass-spectrometryof7-dehydrocholesterolinplasmaamniotic-fluidandculturedskinfibroblastsClinChimActa236(1995)45minus58
[5] Rossiter JP Hofman KJ Kelley RI Smith-Lemli-Opitz SyndromePrenatal-diagnosis byquantification of cholesterol precursors in amniotic-fluid Am J Med Genet 56 (1995)272minus275
[6] KloudaJBarekJNesmerakKSchwarzova-PeckovaKNon-enzymaticelectrochemistryincharacterization and analysis of steroid compounds Crit Rev Anal Chem 47 (2017)384minus404
[7] AtumaSSLundstromKLindquistJTheelectrochemicaldeterminationofvitaminAPartIIFurthervoltammetricdeterminationofvitaminAandinitialworkonthedeterminationofvitaminDinthepresenceofvitaminAAnalyst100(1975)827minus834
[8] MoodyJPHumphriesCAAllanSMPatersonCRDeterminationof7-dehydrocholesterolinhumanskinbyhigh-performance liquid-chromatography JChromatogrB530 (1990)19minus27
[9] OstermeyerUSchmidtTVitaminDandprovitaminDinfishEurFoodResTechnol222(2005)403minus413
[10] BlighEGDyerWJArapidmethodoftotallipidextractionandpurificationCanJBiochemPhysiol37(1959)911minus917
150 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Author Index
AlikovaV1
AugustınM83
BaluchovaS19
BarekJ192570
BarochM141
BasB13129
BastryginaO41
BenesovaL146
BessonovaE57
BohmD6
BurkinK116
BurkinM116
ChernovaA141
ChoinskaM70
CokrtovaK104
DedinaJ97
DeevV57
DejmkovaH141
DendisovaM63
DubenskaL51
EfremenkoE41
FojtaM110
GalvidisI116
HavranL110
HeiglN31
HertJ76
HrdlickaV70
JosypcukB25
KartsovaLA3557
KloudaJ19146
Kodrık ovaB90
KolobovaEA35
KorbanA135
KorotkovaE1
KralM63
KratzerJ90
KravchenkoAV35
KrızekT76104
LipinskaJ129
MadejM129
MatejkaP63
MatysikF-M631
MusilS9097123
NavratilT70
OndrackovaA110
PietrzakK45
PlotnikovaK51
PoradaR13
RedondoBR70
SagapovaL90
ShormanovV1
Schwarzova-PeckovaK19110146
SladkovaS 141
S tadlerovaB97
StiborovaM110
SvobodaM90
TvorynskaS25
TyczkowskiJ129
VyhnanovskyJ97123
VymyslickyF76
VyskocilV83
WardakC45
WongDKY19
ZarybnickaA146
ZelenyI51
Proceedingsofthe16thISCModernAnalyticalChemistry Prague2020 151
152 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Keyword Index
alcoholicbeverages135
aminoglycosides116
amperometricdetection146
amperometry141
antifoulingelectrodes19
assembledcapillaries6
atomicabsorptionspectrometry90
atomicfluorescencespectro-
metry97
atomization90
biologicalactiveanalytes35
biosensor2583
bismuth97
borondopeddiamond
electrode110146
cadmium90
canagliflozin76
capillarycoating35
capillaryelectrophoresis635
capillaryflowinjectionanalysis631
carbohydrates31
carbonfelt141
cathodicstrippingvoltammetry70
chemicalvaporgeneration90
chemometrics57
cobalt123
coldplasmadeposition129
copper(II)phthalocyanine63
covalentimmobilization25
cytochromeP450110
damage83
7-dehydrocholesterol146
designofexperiments76
differentialpulsevoltammetry146
diphenylsilanereductionmethod19
dispersiveliquid-liquid
microextraction57
disposableelectrodes31
DNA83
dualdetectionconcept6
electrochemicalanalysis110
electrochemicalapplications129
electrochemicalflowcell76
electrochemistry51
electrokineticchromatography104
eliminationvoltammetrywith
linearscan70
ELISA116
enzymaticreactor25
FIA141146
gaschromatography-mass
spectrometry(GC-MS)135
glucoseoxidase25
graphite83
honey116
HPLC76141
hydridegeneration97
hydrogenatedconical-tipcarbon
electrodes19
imidazoliumionicliquids35
inductivelycoupledplasmamass
spectrometry123
internalstandardmethod135
ion-selectiveelectrode45
laccase25
liquidcrystals104
massspectrometry31
mercuryelectrode13
metronidazole51
non-aqueouscapillary
electrophoresis104
non-aqueoussystem6
oxidation76
oxytetracyclinehydrochloride51
phenol2-methoxy1
photochemicalvaporgene-
ration97123
polarography51
1-propanesulfonicacid23-
dimercapto-70
pulsedamperometricdetection31
quantitation1
resonanceRamanspectroscopy63
scanningtunnellingmicroscopy63
silversolidamalgamelectrode70
Smith-Lemli-Opitzsyndrome146
smokingmixtures41
solidcontact45
solid-phasemicroextraction57
spectrophotometry141
SudanI110
surfacemodification129
surface-enhancedRaman
spectroscopy63
thinlayers129
tip-enhancedRamanspectroscopy
63
unithiol70
uranyl45
vanillin41
veterinarydrug51
vitamins13
volatilecompoundsquantifi-
cation135
voltammetricdopaminedetec-
tion19
voltammetry1383
Proceedingsofthe16thISCModernAnalyticalChemistry Prague2020 153
Proceedings of the 16th International Students Conference ldquoModern Analytical Chemistryrdquo
EditedbyKarelNesmerak
PublishedbyCharlesUniversityFacultyofScience
Prague2020
1steditionndashvi154pages
ISBN978-80-7444-079-3
ISBN 978-80-7444-079-3
Pro
ceedin
gs of th
e 16
th In
ternatio
nal Stu
den
ts Co
nferen
ce ldquoMo
dern
An
alytical Ch
emistryrdquo P
rague 2
02
0
788074 440793
Prague 17mdash18 September 2020
Edited by Karel Nesměraacutek
Prague 2020
Proceedings of the
16th International Students Conference
ldquoModern Analytical Chemistryrdquo

Prague 17mdash18 September 2020
Edited by Karel Nesměraacutek
Prague 2020
Proceedings of the
16th International Students Conference
ldquoModern Analytical Chemistryrdquo
CATALOGUING-IN-PUBLICATIONndashNATIONALLIBRARYOFTHECZECHREPUBLIC
KATALOGIZACEVKNIZEndashNA RODNIKNIHOVNACR
ModernAnalyticalChemistry(konference)(162020PrahaCesko)
Proceedingsofthe16thInternationalStudentsConferenceldquoModernAnalyticalChemistryrdquo
Prague17ndash18September2020editedbyKarelNesmerak--1stedition--PragueFacultyof
ScienceCharlesUniversity2020--vi154stran
Obsahujebibliografiearejstrıky
ISBN978-80-7444-079-3(brozovano)
543(062534)
analyticalchemistry
proceedingsofconferences
543ndashAnalyticalchemistry[10]
TheelectronicversionoftheProceedingsisavailableattheconferencewebpage
httpwwwnaturcuniczisc-mac
copyCharlesUniversityFacultyofScience2020
ISBN978-80-7444-079-3
Preface
Despitethefactthattheyear2020ismarkedbyCOVID-19morethan40young
analytical chemists gathered in Prague for the 16th annual international
conferenceldquoModernAnalyticalChemistryrdquoTheymeettopresenttheresultsof
theirresearchtomastertheirpresentationandlanguageskillsandtoexchange
anddiscussideasandexperiencesofanalyticalchemistry
Thisvolumeofconferenceproceedingsbringsyouatotalof25papersfrom
thisconferenceAsinpreviousyearsthecontributionspresentedareassortedby
the sequence of their delivery supplemented by indexes at the end of the
proceedingsallowingeasynavigationthroughthepagesYouwillseethattopics
of contributions cover all the aspects of modern analytical chemistry from
theoretical problems through development of new analytical methods and
improvementofanalyticaltechniquestotheapplicationsinvolvingthesolutionof
medicinaltechnicalorenvironmentalproblemsLetushopethatlikeprocee-
dingsofpreviousyearsofourconference thisonewillalsobean interesting
beneficialandenjoyablereading
Itseemstousthattheauthorsofthecontributionsareaguaranteeofthatanew
generationofanalyticalchemistswillprotectbrightandthrillingfutureofour
science
We are very grateful to the Division of
AnalyticalChemistryofEuChemSforitslong-
lasting auspices of our conference Also we
arethankfultooursponsorsnotonlyfortheir
kind sponsorship making the conference
possiblebutalsoforalltheircooperationand
supportinmanyofourotheractivities
Enjoyreadingtheseproceedings
docRNDrKarelNesmerakPhD
editor
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 iii
Sponsors
The organizersof16th International Students Conference ldquoModernAnalytical
Chemistryrdquo gratefully acknowledge the generous sponsorship of following
companies
wwwecomsrocom
wwwlach-nercom
iv Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
wwwthermofishercz
www2thetacz
wwwzentivacz wwwquintacz
wwwshimadzueucom
wwwwaterscom
Contents
AlikovaVChernovaAShormanovVKorotkovaEDeterminationof2-methoxyphenol inmodelsolutionsbyspectrophotometry 1
BohmDMatysikF-MTheeffectsoflinearlyassembledcapillarieswithvariousinnerdiametersoncapillaryelectrophoresis 6
PoradaRBasBVoltammetricdeterminationofvitamins 13Baluchova S Klouda J Barek J Schwarzova-Peckova K Wong DKY Dopamine detection at
antifoulingconical-tipcarbonelectrodes 19Tvorynska SBarek J JosypcukBA comparative study of covalentglucose oxidaseand laccase
immobilizationtechniquesatpowderedsupportsforbiosensorsfabrication 25Heigl N Matysik F-M Capillary flow injection analysis with electrochemical detection for
carbohydrateanalysis 31KravchenkoAVKolobovaEAKartsovaLAApplicationofcovalentcoatingsbasedonimidazolium
cations for separationandon-linepreconcentrationof basicandneutralanalytes in capillaryelectrophoresis35
Efremenko E Chernova A Bastrygina O Determination of vanillin in smoking mixtures byspectrophotometry41
PietrzakKWardakCUranylion-selectiveelectrodewithsolidcontact 45Plotnikova K Dubenska L Zeleny I Polarographic determination of metronidazole and
oxytetracyclinehydrochlorideinveterinarydrugforhoneybees 51Deev V Bessonova E Kartsova L Application of microextraction techniques combined with
chromatographicmethodsfortheanalysisofcomplexobjects 57KralMDendisovaMMatejkaPThedevelopmentofreferenceprobesystemfortip-enhancedRaman
spectroscopy 63ChoinskaMHrdlickaVRedondoBRBarekJNavratilTDeterminationofheavymetalpoisoning
antidote23-dimercapto-1-propanesulfonicacidusingsilversolidamalgamelectrode70VymyslickyFKrızekTHert JCanagliflozinoxidation studyusingelectrochemical flowcelland
comparisonwithhydrogenperoxideoxidation 76AugustınMVyskocilVNovelhybridelectrochemicalDNAbiosensorformonitoringoxidativeDNA
damageviaoxidationreductionsignalsoflowmolecularweightdouble-strandedDNA 83SagapovaLKodrık ovaBSvobodaMMusilSKratzerJChemicalvaporgenerationofcadmiumfor
analyticalatomicspectrometry 90S tadlerova B Vyhnanovsky J Dedina J Musil S Photochemical vapour generation of bismuth
coupledwithatomicfluorescencespectrometry 97Cokrtova K Krızek T Separation of liquid crystals using non-aqueous capillary electrokinetic
chromatography104OndrackovaAStiborovaMHavranLSchwarzova-PeckovaKFojtaMElectrochemistryofSudanI
anditsderivatesinaqueousmedia 110BurkinKGalvidisIBurkinMGroupdetectionofaminoglycosidesusingELISAforcontroloffood
contamination 116Vyhnanovsky J Musil S Photochemical vapor generation of cobalt for detection by inductively
coupledplasmamassspectrometry 123LipinskaJMadejMBasBTyczkowskiJOptimizationofconditionforcoldplasmadepositionofthin
layersforsurfacemodificationofworkingelectrodes 129KorbanAAdvancedGC-MSmethodforqualityandsafetycontrolofalcoholicbeverages 135Baroch M Dejmkova H Sladkova S Utilization of a carbon felt as a material for working
electrodes 141Benesova L Zarybnicka A Klouda J Schwarzova-Peckova K Electroanalytical methods for
determinationof7-dehydrocholesterolinartificialserum146
Authorindex 151Keywordindex 152
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 v
1Introduction
2-Methoxyphenol(guaiacol)isusedinmedicineasanexpectorantThestructuralformulaisshowninFig1Itiswidelyusedinthepharmaceuticalindustry[1]forthesynthesisofantituberculosisexpectorantdrugs(Kas-nol Sudafed Ascoril Prothiazine Expectorant Guai-phenesinum) Moreover 2-methoxyphenol is often used as anaromaticsubstance [2] in the food industry Inparti-cular itwaswidelyusedintheproductionofsmokedfish and meat products using smokeless smokingtechnologyusingflavourings Ontheotherhand2-methoxyphenolhasthesymbolGHS07andhasahazardcodeXnTXi[2]accordingtotheGHSsystemItisverytoxicbyinhalationitcanirritatethemucousmembraneoftherespiratorytractandtheconjunctivaofthe
Determination of 2-methoxyphenol in model solutions by spectrophotometry
a a b aVALERIYAALIKOVA ANNACHERNOVA VLADIMIRSHORMANOV ELENAKOROTKOVA
a DepartmentofChemicalEngineeringEngineeringSchoolofNationalResourcesNationalResearchTomskPolytechnicUniversityLeninavenue30634050TomskRussiaalikovaleramailru
b DepartmentofPharmaceuticalToxicologicalandAnalyticalChemistryKurskStateMedicalUniversitystKarlaMarks3305000KurskRussia
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 1
AbstractA spectrophotometric approach for determination of 2-methoxy-phenol in model solutions has been developed The absorptionspectra of 2-methoxyphenol were determined in the wavelengthrangefrom200to400nminsolutionsof95ethanolacetonitrile01Msodiumhydroxideandethylacetatewithaconcentrationofthe
minus3analyte of 005mgdm For the quantitative determination of2-methoxyphenol a seriesof solutionswaspreparedwithvarious
minus3 minus3concentrationsfrom0001mgdm to005mgdm in95ethanolacetonitrile01MsodiumhydroxideTheopticaldensityof2-meth-oxyphenolinsolventswasmeasuredatawavelengthof276nmand289nmThedevelopedmethodwastestedusingthemethodanalysisofspikedsamples
Keywordsquantitation2-methoxyphenolUVVISspectrophoto-
metry
Fig 1Structuralformulaof2-methoxyphenol
eyeballinhighconcentrationswhenitpenetratestheskincanleadtoneurosiswhenadministeredorallycanstimulatetheesophagusandstomachresultinginheartfailurecollapseanddeathNowadaystherearepublisheddataoncasesofasystemicallergicreaction[3]causedby2-methoxyphenolderivativesandthereisa fatal case known [4] for oral administration of guaifenesin (3-(2-methoxy-phenoxy)propane-12-diol)oneofthecomponentsofcommonlyavailablecoughmedications Thedeterminationof2-methoxyphenolinenvironmentalobjectsaswellasinthefoodindustryiscarriedoutusinggaschromatographymethodswithsolid-phasemicroextraction[5]Inordertocontrol2-methoxyphenolinnaturaldrink-ing and treatedwastewater gas chromatography is used followed by opticaldetectionoftheeluate[6]Themaindisadvantagesofthismethodofanalysisarethelowselectivityanddurationofdetermination(about3hours)AlsoaccordingtoRussianStateStandartGOST33312-2015themethodofgaschromatographyisusedforthequalitativeandquantitativedeterminationof2-metoxyphenolinjuiceproducts Commonlyfortheanalysisoftoxicsubstancesinvariousbiologicalsamples(bloodplasmaurinesalivasweathair)bygaschromatographyitisnecessarytocarryoutmultistagesamplepreparationwhichcomplicatesandslowsdownthecourseofthestudy[7]Atthesametimeitisimportantthatduringtheprepa-rationofsamplesintheanalyzedcompoundstheirstructureisnotviolatedasthiswillleadtothedifficultyoftheiridentification Spectrophotometryintheultravioletregionhaslowersensitivitycomparedtothe abovemethods however thismethod does not require such complicatedpreparation of the analyzed samples it is a relatively affordable simple andinexpensive analysis method In addition its sensitivity can be significantlyimprovedbyapplyinganappropriateseparationprocedureandpreconcentrationbeforedetection[8]MethodUVspectrophotometryisusedtoassessthequalityof both medicinal substances and preparations made from them in terms ofauthenticitygoodqualityandquantitativecontentInadditionitisarelativelyaffordablesimpleandlow-costanalysismethod Ananalysisoftheliteraturedatashowedthattodaytherearefastandsensitivespectrophotometricmethodsforthedeterminationofpyrocatecholderivativesinmedicines[9]vanillininfoodproducts[10]andotherphenolsinwastewaterandwineproducts[1112]Howeverasfarasweknowinformationonthedetermi-nationof2-methoxyphenolfromtheabsorptionspectraintheultravioletregionisabsent Theaimofthisstudyistodevelopmethodsforthequalitativeandquantitativeof2-methoxyphenolinmodelsolutionsusingUVspectrophotometry
2 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
2Experimental
21Reagentsandchemicals
Asampleof2-methoxyphenolfromFlukawithabasicsubstancecontentofge98wastakenastheobjectofstudyAssolventsweusedacetonitrile(ChP)95ethanolethylacetateand01MsodiumhydroxidesolutionAllotherchemicalsusedwereofanalyticalreagentgrade
22Instrumentation
Theopticaldensitywasmeasuredincuvetteewithanabsorbinglayerthicknessof10mmusingaCary60spectrophotometer(AgilentUSA)Allmeasurementswerecarriedoutatroomtemperature
3Resultsanddiscussion
Thechangeinthebehavioroftheabsorptionspectrumwasinvestigatedinthewavelengthrangeof200ndash400nmFigure2isshowedthatwithanincreaseinthepolarityofthesolventtheabsorptionmaximumshiftstowardthevisiblepartofthe spectrum The wavelength of absorption maxima of 2-methoxyphenol ispresentedinTable1 A studyof thephotometricbehaviorof2-metoxyphenolinvarious solventsshowedthatacetonitrile95ethanoland01Msodiumhydroxidearethemostsuitablesolventsforthequalitativedeterminationofthetestsubstance
ndash3Fig 1Thespectraof2-methoxyphenolofconcentration005mgdm inthemediumofsolvents(anabsorbinglayerthicknessof10mm)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 3
Forthequantitativedeterminationof2-methoxyphenolaseriesofsolutionsndash3 ndash3withaconcentrationragefrom0001mgdm to005mgdm werepreparedin
acetonitrileand95ethanolTheopticaldensityof2-methoxyphenolinsolventswasmeasuredbywavelengthof276nmThedependenceoftheintensityoftheoptical density on the concentration of 2-methoxyphenol in 01 M sodium
ndash3hydroxide was plotted in the concentration range from 0005mgdm tondash3003mgdm Themeasurementwerecarriedoutbywavelengthof276nmThe
obtainedregressionequationsarepresentedinTable2Dataanalysisobtainedwasperformedusingleast-squaresmethodThedevelopedmethodwastestedusingthemethodanalysisofspikedsamplesTheresultsarepresentedinTable2
4Conclusions
Studieshaveshownthepossibilityofusingspectrophotometricanalysisforthequalitativeandquantitativedeterminationof2-methoxyphenolTheabsorptionmaximaofweredeterminedinsolutionsofethanolandacetonitrile(276nm)inasolutionofethylacetate(277nm)and01Msodiumhydroxide(289nm)Theconstructed calibration curves of thepure substance of 2-methoxyphenol hasshownagoodregressioncoefficient(Rgt099)andcanbeusedforquantitativedeterminationof2-metoxyphenolinbiologicalobjectsInthefutureitisplannedtoapplythistechniquetodetermine2-metoxyphenolinincadavericmaterial
Solvent Regressionequation Found S RSD Δх δ 2-metoxy- phenolg
ndash3 ndash6 ndash5Acetonitrile y=18294C+01130 499times10 50times10 028 2times10 044 Rsup2=09985
ndash3 ndash6 ndash595ethanol y=35131C+00269 503times10 03times10 021 7times10 137 Rsup2=09956
ndash3 ndash6 ndash501Msodium y=31196C+01101 495times10 01times10 018 1times10 028hydroxide Rsup2=09997
Table 2Results of the determination of 2-methoxyphenol (average of three measurements) in modelsolutionsbythemethodanalysisofspikedsamplestheconcentrationofintroduced2-methoxy-
ndash3phenolwas500times10 g(SndashstandarddeviationRSDndashrelativestandarddeviationΔхndashabsoluteerrorδndashrelativeerror)
Table 1Valuesofopticaldensityandwavelengthsinappropriatesolventswith2-methoxyphenol
minus3 minus1Solvent λnm εgdm cm
Acetonitrile 276 0082895ethanol 276 00853Ethylacetate 277 0083101Msodiumhydroxide 289 00744
4 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
References
[1] МельниковаИММизерницкии ЮЛКомбинированныеотхаркивающиепрепаратырастительного происхождения в педиатрическои практикеМедицинский совет 2(2018)93ndash97
[2] httpwwwthegoodscentscompanycomdatarw1032272html(accessed27stFebruary2020)
[3] RayMFaltayBHallerNACasereportanaphylacticreactiontoguaifenesinHospPract37(2009)60ndash63
[4] OkicMJohnsonTCrifasiJALongCMitchellEKSwiftonsetofcentralnervoussystemdepressionandasystolefollowinganoverdoseofguaifenesinJAnalToxicol37(2013)318ndash319
[5] ВолковCМЧерновецАНОпределениеконцентрациифеноловвгазовыхвыбросахпромышленных предприятии методом газовои хроматографии с твердофазнои микроэкстракциеи Сорбционныеихроматографическиепроцессы10(2010)723ndash728
[6] ШачневаЕЮОньковаДВСерековаСМСпособыопределенияфеноловвобъектахокружающеи среды Астраханский вестник экологического образования 4 (2013)138ndash142
[7] ГладиловичВДПодольскаяЕПВозможностипримененияметодаГХ-МС(Обзор)Научноеприборостроение4(2010)36ndash49
[8] Pena-PereiraFLavillaIBendichoCHeadspacesingle-dropmicroextractioncoupledtomicrovolumeUVndashVis spectrophotometry for iodine determinationAnal Chim Acta631(2009)223ndash228
[9] NagarajaPMurthyKCSRangappaKSGowdaNMMSpectrophotometricmethodsforthe determination of certain catecholamine derivatives in pharmaceutical preparationsTalanta46(1998)39ndash44
[10] Altunay N Development of vortex-assisted ionic liquid-dispersive microextractionmethodology for vanillin monitoring in food products using ultraviolet-visible spectro-photometryLWT93(2018)9ndash15
[11] Lupetti KO Rocha FRP Fatibello-Filho O An improved flow system for phenolsdetermination exploiting multicommutation and long pathlength spectrophotometryTalanta62(2004)463ndash467
[12] Figueiredo-Gonzalez M Cancho-Grande B Simal-Gandara J Garnacha tintorera-basedsweetwineschromaticpropertiesandglobalphenoliccompositionbymeansofUVndashVisspectrophotometryFoodChem140(2013)217ndash224
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 5
1Introduction
Thenumberofsamplesthesamplecomplexityandalsothenumberofsubstanceswhich need to be analysed simultaneously is increasing steadily ThereforepowerfulseparationanddetectionmethodsarerequiredOnewaytoachievethisisthecouplingofaseparationsystemwithmorethanonedetector[12] In recent years capillary electrophoresis (CE)was established as a potentseparation system due to its high separation efficiency and the low sampleconsumption [3] To generate more information numerous dual detectionconceptsforCEweredevelopedwhicharesummarisedelsewhere[12]Acom-binationofamperometricdetectionandmassspectrometry(MS)isaninterestingdual detection concept for CE because both detectors supply complementaryinformationForelectroactivespeciesamperometricdetectionisarobustandoneof the most sensitive detection method [4] Thus it is well suited for the
The effects of linearly assembled capillaries with various inner diameters on capillary electrophoresis
DANIELBO HMFRANK-MICHAELMATYSIK
InstituteofAnalyticalChemistryChemo-andBiosensorsFacultyofChemistryandPharmacyUniversityofRegensburgUniversitaumltsstraszlige3193053RegensburgGermanydanielboehmchemieuni-regensburgde
AbstractDuetotheincreasingneedofpowerfulanalyticalmethodsanewdualdetection concept for capillary electrophoresis (CE) with parallelamperometricdetectionandmassspectrometryshallbedevelopedFor this concept the CE flow has to be divided into two streamsutilizinga flowsplitter In thiswork theeffectsof combinedcapi-llarieswithvarious innerdiameterswerestudiedForpreliminaryinvestigationsthecapillarieswereconnectedinaserialconfigurationwithoutdeadvolumeUsingcapillaryflowinjectionanalysishyphe-natedtocontactlessconductivitydetectionitcouldbeshownthatthecouplingofidenticalcapillariesleadstoaslightdecreaseoftheflowratesWithCEhyphenatedtoUVdetection itcouldbeshownthatthecouplingofcapillarieswithdifferentinnerdiameterhasamuchstrongereffectontheelectroosmoticflowthanthecombinationwiththesameinnerdiameterFurthermorenosignificantchangeinpeakshapewasobserved
Keywordsassembledcapillariescapillaryelectrophoresiscapillaryflowinjection
analysisdualdetectionconceptnon-aqueoussystem
6 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
quantificationofsubstanceswhereasMSiswellsuitedfortheidentificationofunknown substances [3] In most dual detection concepts the detectors arearrangedinaserialconfigurationwhichisnotpossibleincaseofamperometricdetection-mass spectrometry [1] The instrumental implementation is morecomplicatedwithbothdetectorsbeingdestructiveFurthermoretheymustbedecoupledfromthehighvoltagefieldoftheCEThereforetheCEflowmustbedividedintotwostreamswithaflowsplitterAsimplifiedsketchofthepossiblenewdualdetectionconceptisshowninFig1 ForthedevelopmentofthenewdualdetectionconceptthreecapillarieswithpotentiallydifferentinnerdiametersmustbecoupledForthisreasonthedeadvolume-freecouplingofcapillarieswithdifferent innerdiameterswas investi-gatedinafirststepTokeepthesetupsimplewefocusedonthelinearcouplingofcapillariesandtheresultingeffectsNon-fragmentedcapillarieswerecomparedwithfragmentedcapillariesofthesameordifferentinnerdiametersEffectsonthe flow rate were investigated with capillary flow injection analysis (CFIA)
4hyphenatedtocontactlessconductivitydetection(C D)EffectslikechangesinthemigrationbehaviourorpeakshapesoccurringinCEwereinvestigatedwithCEhyphenatedtoUVdetection(CE-UV)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 7
Fig 1SchematicillustrationofthenewdualdetectionconceptwithparallelamperometricdetectionandmassspectrometricdetectionforCEAfterinjectionfrom(a)thesamplevialthecomponentsareseparatedbyCEnext(b)theflowsplitterdivides(c)thecapillaryintotwopartsandleadstheCEflowtowards(d)themassspectrometerand(e)theamperometricdetector
2Experimental
21Reagentsandchemicals
Thefollowingchemicalswereusedallofanalyticalgradeferrocenemethanoldecamethylferrocene(ABCRGermany)acetonitrileammoniumacetate01Msodiumhydroxidesolutionultra-purewaterprovidedbyaMilliQAdvantageA10system(MerckGermany)aceticacid(RothGermany)
22Instrumentation
221Capillaries
4Forbothexperiments(CFIA-C DandCE-UV)capillarieswithinnerdiametersof2550and75micromanouterdiameterof360micromandatotallengthof70cmwereusedTheywerepurchasedfromPolymicroTechnologies(USA)Measurementswere carried out with fragmented and non-fragmented capillaries For themeasurementswiththefragmentedcapillariestheoriginalcapillarieswerecutintotwopiecesyieldingatotalof9capillarycombinationswithlengthsof70cm(20cmfirstcapillarypieceand50cmsecondcapillarypiece)ThesecombinationsaresummarizedinTab1(section31)Atbothendsofthecapillariesabout02cmof thepolyimidecoatingwasremovedBothsidesof thecapillarypieceswerepolishedtoreceiveplanarcapillarytipsForthelinearassemblingofthecapillarypiecesMicroTightSleevesF185Xanda capillary connectorUnionAssemblyMicroTightP720fromIDEXHealthampScience(USA)wereusedPriortothefirstCEmeasurementsthecapillarieswereconditionedbyflushingthemfor10minwith01Msodiumhydroxidesolution5minwithultra-purewaterand30minwithseparationbuffer
222Capillaryflowinjectionanalysishyphenatedtocontactlessconductivitydetectionsetup
The flow rates for the fragmented and non-fragmented capillaries were4determinedwithaCFIA-C DsetupschematicallydepictedinFig2ATheflowin
thecapillarywasgravitationdrivenbyaheightdifferencebetweentheinletandoutletcarriersolutionvialTheconceptofCFIAwithgravitationdrivenflowwasfirstdescribedbyMatysiketal[5]AlaboratoryconstructedautosamplerofaCEdevicewasusedforthehydrodynamicinjectionThesamplesolutionconsistedof10mMdecamethylferrocene incarrier solution (10mMCH COONH and1M3 4
4CH COOH in acetonitrile) A high resolution C D was placed after 40 cm for3
detectionThedetectordescribedelsewhere[6]wasconstructedinthedoLagogroup(Brazil)Adoubledeterminationattwodifferentheightswasdoneforthedeterminationoftheflowrates
8 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
223Capillaryelectrophoresis-UVdetectionsetup
Fig2BshowsasketchoftheCE-UVsetupItconsistedofalab-builtCEdevicewhichwasconnectedtoahighvoltagepowersupplyfromISEG(Germany)Theseparationswerecarriedoutwithanon-fragmented50micromcapillaryandwithcapillary combinations implementing a 50micromdownstream capillary segment(25+5050+50and75+50microm)ALambda1010UV-VISdetectorfromBischoff(Germany)wasusedfordetectionat210nmThedetectorwasplacedafter40cm
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 9
Fig 2Schemeof(A)thecapillaryflowinjectionanalysis(CFIA)hyphenatedtocontactlessconduc-4tivity detection (C D) setup and (B) the capillary electrophoresis hyphenated to UV detection
4(CE-UV) setup Components of the CFIA-C D setup (a) sample (b) inlet and (c)outlet carrier4solution vial (d) fused silica capillary (e) linear capillary connector (f) C D and (g) stand
ComponentsoftheCE-UVsetup(h)sample(i)inletand(j)outletbuffervialand(k)UVdetector4therestofthecomponentswereidenticaltotheCFIA-C DsetupTheoutletbuffervialwaslowered
forthehydrodynamicinjection(j)Theenlargedview(k)depictsthecouplingoftwocapillarieswithdifferentinnerdiametersintheconnectionsidewithoutdeadvolume
Asamplesolutioncontaining1mMferrocenemethanolanddecamethylferroceneinseparationbuffer(10mMCH COONH and1MCH COOHinacetonitrile)was3 4 3
utilizedTheinjectionwasperformedhydrodynamicallybyloweringtheoutletbuffer vial by 20 cm A uniform sample plug was injected to compare bandbroadeningeffectsTheinjectionsegmenthadalengthof035cm(05ofthetotalcapillarylength)andtherespectiveinjectiontimewasdeterminedbasedontheflowratesofthecorrespondingcapillarycombinationFortheelectrophoreticseparationaseparationvoltageof25kVwasappliedandtheinletandtheoutletbuffervialswereplacedatthesameheightsothattherewasnogravityflowwhichaffectedthemigrationbehaviour
3Resultsanddiscussion
31Capillaryflowinjectionanalysishyphenatedtocontactlessconductivitydetectionexperiments
AsshowninTab1theflowratesforaheightdifferenceof20cmwerecalculated4basedontheCFIA-C DmeasurementsItwasobservablethattheflowratesare
slightlylowerforfragmentedcapillariesthanfornon-fragmentedcapillariesofthe same dimension This indicates that a flow resistance arises when twocapillaries are combined Furthermore it was observed that the flow ratedecreasesforupstreamcapillarieswithlowerinnerdiametersandviceversaTheflow rate for the combination 25+75microm could not be determineddue to theformationofairbubblesattheconnectionside
10 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Table 1Flowratesandthecorrespondingstandarddeviations(SD4measurements)ofdifferentcapillarycombinationsforaheightdifferencebetweeninletandoutletvialof20cmbymeansofcapillaryflowinjectionanalysishyphenatedtocontactlessconductivitydetection
ndash1capillary flowratenLs plusmnSDflowndash1combinationmicrom ratenLs
25 00577 0000525+25 0057 000150+25 00775 0000775+25 ndash ndash25+50 0137 000550 0865 000250+50 080 00575+50 105 00625+75 0187 000250+75 1941 000375 456 00275+75 42 01
32Capillaryelectrophoresis-UVdetectionexperiments
Theelectropherogramsforthenon-fragmented50micromcapillaryandthecapillarycombinationswithdownstream50micromcapillaryarepresentedinFig3ThetwoferrocenederivatesdecamethylferroceneandferrocenemethanolwereusedasmodelanalytesDecamethylferrocenewasonlydetectedascationicspeciesasitiseasilyoxidizedbydissolvedoxygeninsolution Forthecombination50+50micromslightlyhighermigrationtimesforthecationic(decamethylferrocene) and neutral species (ferrocenemethanol) were obser-vable compared to thenon-fragmented50micromcapillaryThis indicateda flow
4resistanceattheconnectionwhichwasalsoobservedfortheCFIA-C Dexperi-mentsinsection31Incontrasttothecombinationwiththesameinnerdiameterastrongshift in themigration times for theneuralspecieswasvisible for thecombinationswithdifferentinnerdiametersThisshowedthatthecouplinghadaneffectontheelectroosmoticflow LookingatthepeakshapeitwasfoundthatallpeaksshowednearlyGaussianshape for all combinations Furthermore there was no tailing visible Theferrocenemethanolpeaks for thecombination25+50micromand75+50micromwere
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 11
Fig 3Electropherograms of themodelmixture ferrocenemethanol (FcMeOH) and decamethyl-ferrocene(DeMeFc)measuredwithanon-fragmentedcapillary(50microm)andfragmentedcapillaries(25+5050+50and75+50microm)Experimentalparameters1mMFcMeOHandDeMeFcinseparationbuffer(10mMCH COONH and1MCH COOHinacetonitrile)injectionsegment035cmseparation3 4 3
voltage25kVcapillarylength70cm(40cmtothedetectorfragmentedcapillaries20cmfirstpartand50cmsecondpart)UVdetectionat210nm
slightlybroaderthanthepeaksforthenon-fragmented50micromcapillaryorforthe50+50 microm combination But this probably results from longitudinal diffusioneffectsduetothelongerresidencetimes
4Conclusions
FromtheCFIAmeasurementsitcanbeconcludedthattherewasamechanicaldisturbanceoftheflowduetothecouplingFurthermoreitcouldbeshownthatCEmeasurementswithlinearcoupledcapillariesofvariousinnerdiameterwerepossible Unlike to the capillary combinationwith the same inner diameter astrongshiftoftheelectroosmoticflowtowardshighermigrationtimeswasfoundfor capillary combinations with different inner diameters In this work thecapillaries were coupled with almost no dead volume which resulted in nosignificantchangesofthepeakshapeorpeaktailingContrarytoexpectationsthecouplingofcapillarieswithvariousinnerdiametershadnosignificantimpactonthepeakwidth TheknowledgegainedfromthelinearcouplingofcapillariesisagoodbasisforthedevelopmentofthenewdualdetectionconceptInanextstepthreecapillariesshouldbecoupledwitheachother
Acknowledgments
WethanktheGermanResearchFoundation(DFG)forfinancialsupport
References
[1] OpekarFS tulıkKSomeimportantcombinationsofdetectiontechniquesforelectrophoresisincapillariesandonchipswithemphasisonelectrochemicalprinciplesElectrophoresis32(2011)795ndash810
[2] BeutnerAHerlTMatysikF-MSelectivityenhancement incapillaryelectrophoresisbymeans of two-dimensional separation or dual detection conceptsAnal Chim Acta1057(2018)18ndash35
4[3] BeutnerACunhaRRRichterEMMatysikF-MCombiningC DandMSasadualdetectionapproachforcapillaryelectrophoresisElectrophoresis37(2016)931ndash935
[4] MatysikF-MEnd-columnelectrochemicaldetectionforcapillaryelectrophoresisElectro-analysis12(2000)1349ndash1355
[5] MatysikF-MWernerGTracemetaldeterminationintearsbyanodicstrippingvoltammetryinacapillaryflowinjectionsystemAnalyst118(1993)1523ndash1526
[6] FranciscoKJMdoLagoCLAcompactandhigh-resolutionversionofacapacitivelycoupledcontactlessconductivitydetectorElectrophoresis30(2009)3458ndash3464
12 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Thetermldquovitaminsrdquodescribestheheterogeneousgroupofchemicalcompoundswhich are important for the proper functioning of the human body [1 2] Bydefinitionvitaminsarenotsynthesizedbythehumanbodyorthesynthesizedamount is not sufficient to cover the demand That is why they have to besupplementedfromtheexternalsourceslikefoodproductsorpharmaceuticals[1ndash3]Basedontheirsolubilityvitaminsaredividedintowater-soluble(B-groupandvitaminC)andfat-solublevitamins(ADEandK)[3]VitaminC(ascorbicacid) is the most important antioxidant and participates in the activation ofenzymes[4]VitaminB1(thiamine)facilitateswoundhealingandiscrucialforthehumannervoussystem[15]VitaminB2(riboflavin)participatesintheenzy-maticreactionsandthebiotransformationofglucoseandaminoacids[6]Vitamin
+B3(niacin)isthemainconstituentoftheNAD andNADHcoenzymeswhichareresponsible for the transfer of electrons and hydrogen ions in the cellularrespiration [1ndash3] Vitamin B6 possesses six related structures (vitamers) thateasilyinterconvertThemostimportantoneispyridoxinewhichhelpstopreventtongue inflammation and microcytic anemia [2] For the production of well-functioningredbloodcellsandtheavoidanceofmegaloblasticanemiaandfetusdefectsvitaminB9(folicacid)hastobesupplementedintheproperamount[12]
Voltammetric determination of vitamins
RADOSŁAWPORADABOGUSŁAWBAS
DepartmentofAnalyticalChemistryFacultyofMaterialsScienceandCeramicsAGHUniversityofScienceandTechnologyMickiewicza3030-059KrakoacutewPolandrporadaaghedupl
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 13
AbstractVitaminsbelongtothegroupofchemicalcompoundsessentialfortheproperfunctioningofthebodySinceboththeirdeficiencyandexcessmay result in serious health problems the amount of vitaminssupplementedinthedietaswellasvitamincontentintheirsourceshavetobestrictlycontrolledInthisworkthepossibilityofsimulta-neous determination of vitamins by means of differential pulseadsorptive stripping voltammetry is discussed The research hasshownthatthedeterminationofsingularvitaminatthemicromolarlevel isrelativelyfastandstraightforwardandthemostimportanthindranceisrelatedtotheanalyteadsorptionattheelectrodesurfaceInthecaseofvitaminswithdifferentredoxpotentialstheycanbeanalyzedsimultaneouslywithouttheneedtoreachfortheadvancedmethodsforsignalprocessing
Keywordsmercuryelectrodevitaminsvoltammetry
VitaminK3(menadione)doesnotoccurnaturallybutitservesasaprecursorforthesynthesisofotherK-groupvitaminsandcanbeusedtotreathypoprothrom-binemiaVitaminK3ispartiallysolubleinwater[17] All of the vitamins are electrochemically active [3] therefore the electro-chemicalmethodscanbeappliedforthedeterminationofvitamincontentinfoodproductspharmaceuticalsandbodyfluidsVoltammetrictechniquesarecharac-terizedbyhighsensitivityandselectivityandtheydonotrequiretime-consumingsamplepreparationMoreovertheelectrochemicalinstrumentationisrelativelyinexpensiveandcanbeappliedintheon-siteconditionsfortheonlineanalyseseginqualitycontrolMostofthepapersreporttheconstructiondevelopmentand characterization of a novel modified working electrodes for quantitativeanalyses of a singular vitamin in the variety of matrices Unfortunately only alimitednumberofpapersdescribethesimultaneousdeterminationofmultiplevitaminsinasinglerun[23] The preliminary research devoted to the simultaneous determination ofB-groupCandK3vitaminswiththeuseofthecontrolledgrowthmercurydropworking electrode in aqueous solutions is presented in this work Particularattentionhasbeenpaidtotheredoxpotentialsofthestudiedcompoundstheshape of the calibration curves and adsorption phenomena As an attempt toovercomethelattertheneutralsurfactantTritonX-100hasbeenintroducedintothestudiedsystem
2Experimental
21Reagentsandchemicals
TheappliedreagentswereofanalyticalgradeandusedassuppliedPhosphateand McIlvaine buffers were obtained by mixing the appropriate amount of
ndash1 ndash1 ndash102molL Na HPO with 02molL NaH PO and 01molL citric acid2 4 2 4
respectively (all reagents purchased from Avantor Performance MaterialsPoland)ThestandardsolutionsofvitaminB1B2B3B9andCwerepreparedbydissolving the corresponding amount of the standard (all Sigma-Aldrich) in
ndash1distilledwater In thecaseofB2andB9 theadditionof02molL NaOHwasinevitabletoobtainaclearsolutionVitaminK3standard(Sigma-Aldrich)was
ndash1dissolvedinthemixtureofmethanoland1molL phosphatebuffer(pH=82)(vv=25)LaboratorygradeTritonX-100(Sigma-Aldrich)wasusedinthestudyoftheadsorptionprocesses22Instrumentation
All the electrochemical measurements were conducted in the three-electrodesystem composed of the Pt auxiliary electrode double-junction silversilverchloride reference electrode and controlled-growth mercury drop electrode
14 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
actingastheworkingelectrodeTheusedmeasurementequipmentinvolvedtheM164electrodestandandM161multipurposeelectrochemicalanalyzer(bothmtm-anko Krakow) To control the buffer pH-value the SevenCompact S210laboratorypH-meter(MettlerToledoSwitzerland)wasemployed
23Voltammetricmeasurements
Throughout the course of the study differential pulse adsorptive strippingvoltammetry has been used for recording the current-potential curves BothcathodicandanodicscanswererecordedinapotentialrangeadjustedforthestudiedvitaminsTheinfluenceofvariousmeasurementconditionsontheregis-teredsignalshasbeeninvestigatedFinallythepossibilityofthesimultaneousdeterminationofmultiplevitaminsinonescanhasbeenverified
3Resultsanddiscussion
Figure 1 depicts the redox potentials of the studied vitamins in the aqueoussolutions for the mercury electrode The only exception is vitamin B6 whoseredox potential is higher than the potential of mercury oxidation (ca +02 V)Therefore the given value refers to the glassy carbon electrode The redoxpotentialvalueofstudiedvitamins isnotasingularvaluebut it fallswithinacertainrangeThiscanbeascribedtothedependenceofredoxpotentialonthesolutionpHvaluewhichresultsfromtheparticipationofprotonsintheredoxreactions of vitamins Moreover the potentials for individual vitamins arerelatively well separated indicating that the simultaneous determination ofseveralvitaminsintheonerunmaybepossibleTheonlyencounteredproblems
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 15
Fig 1Redoxpotentialsofchosenvitamins
regardthevitaminsB1B3andB12whoseredoxpotentialsoscillatebetweenndash15andndash17VandtheK-groupvitamins(K1K2K3)inwhichonlythecommonstructuralmotifndashthequinoneringndashiselectrochemicallyactiveresultinginthevalueoftheredoxpotentialofcandash02V[8] AtypicalcalibrationcurveisdepictedinFig2Abasedonthedifferentialpulsevoltammograms of the vitamin B9 reduction in the concentration range from
ndash1blank to 01 mg L recorded in the McIlvaine buffer of pH = 52 using thecontrolledgrowthmercurydropelectrodeworkingelectrodeTherelationshipbetweenthepeakcurrentandtheconcentrationofVB9 is linear in thewholetestedrange(r=09999)Basedontheparametersoftheregressioncurvethelimit of detection and limit of quantitation were estimated to 42 and
ndash1142nmolL respectively Similar dependencies and figures of merit can beobtainedforothervitamins QuitedifferentbehaviorwasobservedinthecaseofthevitaminK3forwhichthe increase in current was not strictly proportional to the increase in theconcentrationandthecalibrationplotresemblesanS-shapecurve(Fig2B)ThelatterindicatesthatvitaminK3adsorbsatthesurfaceoftheworkingelectrodeHoweverasnopre-orpost-peakwereobservedwearedealingherewiththeweakadsorption[9]AdsorptionalsoplaysasignificantroleinthecaseofvitaminB2forwhichboththepre-andpost-peakswereobservedindicatingitsstrongaffinitytothemercuryelectrodes To overcome this issue attempts with Triton X-100 were performed TritonX-100isaneutralsurfactant thateasilyadsorbsat thesurfaceof themercury
16 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2Differentialpulseadsorptivestrippingvoltammogramsof(A)vitaminB9and(B)vitaminK3ndash1reduction recorded in the concentration range from blank to 01 and from blank to 04 mg L
respectivelyInsetcorrespondingcalibrationcurvesSupportingelectrolyte(A)McIlvainebufferndash1(pH=52)(B)04molL phosphatebuffer(pH=82)
electrode partially blocking its surface Doing so it prevents the undesiredadsorption of other molecules and thus allows to obtain a linear relationshipbetweenthepeakcurrentandthevitaminconcentration(Fig3)UnfortunatelyduetotheblockingoftheelectrodesurfacetheslopesofthecalibrationlinesaresmallerincomparisontotheonesobtainedintheabsenceofanysurfactantsThismeans that the sensitivity defined as the increase in current caused by a unitincrease in concentration and the resolution understood as the possibility todistinguishsmallvariationinconcentrationarecorrespondinglydecreased Figure3alsopresentsthepossibilitytodeterminemultiplevitaminsinasinglerunChosenvitaminshavewell-separatedpotentialsandtheydonotinterferewitheachotherthereforenoadvancedmultivariatecalibrationstrategiesareneededTheproblemsinthesimultaneousanalysisincludevarioussensitivitieswith respect to the studied analytes and differences in the influence of themeasurementconditionsontherecordedsignalsDuetothattheexperimentalconditionswillneverensurethehighestpossiblesignalvaluesforallanalyzedcompounds
4Conclusions
Differential pulse voltammetry in conjunction with the controlled growthmercurydropelectrodeisaperfecttoolforquantitativeanalysesofvitaminsTheadsorptionofvitaminB2andK3canbepreventedbytheadditionoftheneutralsurfactantTritonX-100whichselectivelyblockstheworkingelectrodesurface
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 17
Fig 3Cathodic voltammograms for the simulatanous determination of vitamin B2 B3 and K3Depictedintheinsetsarethevoltammogramsafterbackgroundsubtractionwiththecorresponding
ndash1callibrationplotsSupportingelectrolye04molL phosphatebuffer(pH=82)with40ppmTritonX-100AccumulationconditionsE =ndash005Vt =20sacc acc
The proposed methodology allows for the simultaneous determination ofmicromolaramountsofvitaminB2B3andK3Suchaprocedurewillhelptoreduce the time and costs of analyses of multivitamin formulations and foodproducts
Acknowledgments
RPhasbeenpartlysupportedbytheEUProjectPOWR0302-00-00-I00416
References
[1] Combs GF JrTheVitaminsFundamentalAspects inNutritionandHealth 3rd ed IthacaElsevierAcademicPress2008
[2] Lovander MD Lyon JD Parr DL Wang J Parke B Leddy J Review Electrochemicalpropertiesof13vitaminsAcriticalreviewandassessmentJElectrochmSoc165(2018)G18ndashG49
[3] Brunetti B Recent advances in electroanalysis of vitamins Electroanalysis 28 (2016)1930ndash1942
[4] BrubacherGMuller-MulotWSouthgateDATMethods forDeterminationofVitamins inFoodNewYorkElsevier1985
[5] Szpikowska-Sroka B A simple and sensitive analytical method for the determination ofthiamineinpharmaceuticalpreparationsJAnalChem68(2013)218ndash222
[6] PetteysBJFrankELRapiddeterminationofvitaminB (riboflavin)inplasmabyHPLCClin2
ChimActa412(2011)38ndash43[7] ZhangZXuJWenYZhangJDingWTheelectro-syntesizedimprintedPEDOTfilmasa
simple voltammetric sensor for highly sensitive and selective detection of vitamin K in3
poultrydrugsamplesSynthMet230(2017)79ndash88[8] JedlinskaKStrusMBasBAnewelectrochemicalsensorwiththeRefreshableSilverLiquid
Amalgam Film multi-Electrode for sensitive voltammetric determination of vitamin K2(menaquinone)ElectrochimActa265(2018)355ndash363
[9] SouthamptonElectrochemistryGroupInstrumentalMethodsinElectrochemistryChichesterHorwood1985
18 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Carbonelectrodesarecommonlyappliedtosensitiveelectrochemicaldetectionofneurotransmittersegdopamine (nor)epinephrineandserotonin in-vivoandin-vitro[12]Neverthelessadsorptionofhigh-molecularweightbiomoleculesinthe matrix on a sensing electrode which then hinders the electron transferreactionofneurotransmitterswillresultinbiofoulingofelectrodesThisremainsachallengingproblemasbiofoulingwillcompromiseelectrochemicalmeasure-mentsThusseveralstrategies foraddressingbiofoulinghavepreviouslybeenreported[34] Thisworkreportsonaneffectiveapproachforminimisingbiofoulingbasedonthehypothesisthatahydrophobicelectrodesurfacewillrepelagainstadsorptionof amphiphilic biomolecules Briefly structurally small conical-tip electrodes
Dopamine detection at antifouling conical-tip carbon electrodes
a a a aSIMONABALUCHOVA JANKLOUDA JIR IBAREK KAROLINASCHWARZOVA -PECKOVA bDANNYKYWONG
a UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova812800PragueCzechRepublicsimonabaluchovanaturcunicz
b DepartmentofMolecularSciencesMacquarieUniversitySydneyNSW2109Australia
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 19
AbstractA significant achievement in this work is the development ofantifoulingconical-tipcarbonelectrodes(~27micromtipdiameterand~165micromaxiallength)suitablefordetectionoftheneurotransmitterdopamine in-vivo These electrodes were hydrogenated using adiphenylsilanereductionmethodtoyieldahydrophobicsurfacetodeteradsorptionofamphiphilicbiomoleculesInitiallyhydrogenatedcarbonelectrodeswereelectrochemicallycharacterisedusingseveralredoxmarkersThedegreeofantifoulingwasthenassessedbythevoltammetricsignalchangeofdopamineattheseelectrodesbeforeand after being incubated in a fouling solution containing bovineserumalbumincytochromeC(bothareproteins)andcaproicacid(alipid) In our work we have obtained only a 69 (standarddeviation35N=40)decreaseindopaminesignalsatthehydro-genated carbon electrodes These results strongly support thediphenylsilanereductionstrategyforthedevelopmentofantifoulingbiosensorsfordopaminedetectioninbiologicalmatrices
Keywordsantifoulingelectrodesdiphenylsilanereduction
methodhydrogenatedconical-tip
carbonelectrodesvoltammetricdopamine
detection
(denoted as CTEs) are fabricated by thermally pyrolysing acetylene gas in anitrogenatmospheretodepositcarbonatthetipandontheshankofpulledquartzcapillaries [5] Spectroscopic studies confirmed that the electrode surface
2 3consistsofsp -likegraphiticcarbonandsp -hybridiseddiamond-likecarbon[6]In addition there is also a range of carbon-oxygen functionalities includingcarbonylquinonecarboxylphenolsalcoholsandethergroupsontheelectrodesurface[6]whichcaninteractwithspectatorbiomoleculesthroughdipole-dipoleorion-dipoleinteractionleadingtotheirirreversibleadsorptionontheelectrodesurface[7]HoweverbysubjectingthesecarbonelectrodestosilanereductionCndashObondsareconvertedtoCndashHbondsandphenolicgroupsaretransformedtosiloxane dendrimers [6] to yield a more hydrophobic carbon surface that isexpectedtobesimilarlylesssusceptibletobiofoulingcomparedtoboron-dopeddiamondelectrodes[78] In this work we will present a methodology involving diphenylsilanereduction to fabricate physically small hydrogenated conical-tip carbonelectrodes(denotedasHCTEs)withanti-foulingcapabilityBothCTEsandHCTEswere electrochemically characterised using several redox probes to elucidatetheirsurfacepropertiesbeforeevaluatingtheirresistancetobiofoulingduringdopaminedetectionin-vitro
2Experimental
21Reagentsandchemicals
Analyticalgradereagents(Sigma-AldrichAustralia)including4-methylcatecholhexaammineruthenium(III) chloride potassium hexacyanoferrate(III) dop-amine hydrochloride sodium phosphate dibasic citric acid perchloric acidpotassiumchloride sodiumhydroxide anhydrousdichloromethanediphenyl-silanetris-(pentafluorophenyl)boraneandgraphitepowderwereusedas-recei-ved Ultra-high purity gases acetylene and nitrogenwere obtained from BOCGases (Australia) All aqueous solutionswere preparedwith deionisedwater(MilliporeMiliplusQsystemUSA)witharesistivityof182MΩcm
22Instrumentation
Chronoamperometric and voltammetric experiments were carried out usingalow-currentpicostateDAQoperatedbyanEChemversion212softwareviaanE-corderinterface(eDAQPtyLtdAustralia)Athree-electrodeset-upinvolving
minus1eitheraCTEorHCTEasaworkingelectrodeaAgAgCl(3molL KCl)referenceelectrode (Bioanalytical SystemsUSA)andaplatinumwire counterelectrode
minus1(CypressSystemsUSA)wasusedCyclicvoltammetryatascanrateof100mVs anddifferential pulse voltammetry (pulse height +25mV pulsewidth 50ms
minus1sampling time 20 ms and scan rate 20mVs ) were used in this work All
20 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
electroanalyticalexperimentswereperformedinanaluminiumFaradaycageatanambienttemperature(23plusmn1degC)
23Preparationofhydrogenatedconical-tipcarbonelectrodes
Asreportedpreviously[5]structurallysmallCTEswerefabricatedbythermallypyrolysing C H (a pressure of 50 kPa) in a pulled quartz capillary (Sutter2 2
minus1InstrumentUSA)housedinaN atmosphere(counterflowof60mLmin )Prior2
to hydrogenation the catalyst tris(pentafluorophenyl) borane (100 mg) wasdissolvedinanhydrousCH Cl (50mL)bystirringfor5minbeforethehydroge-2 2
natingagentdiphenylsilane(25μL)wasaddedCTEswerethenplacedinthereagentmixturefor2hThepreparedHCTEsweredriedovernightbeforeuse
24Biofoulingexperiments
A laboratory synthetic fouling solution consisting of 4 (wv) bovine serumalbumin001(wv)cytochromeC(bothareproteins)and10(vv)caproicacid(alipid)waspreparedbyhomogenisingtheminapH=74citrate-phosphate
minus1buffer(01molL )AllfoulingcompoundswereacquiredfromSigmaAldrichAustralia
3Resultsanddiscussion
31Electrochemicalcharacterisation
minus1InthisworkallCTEswerecharacterisedbycyclicvoltammetryof10mmolL 3+ minus1[Ru(NH ) ] in10molL KClAsdisplayedinFig1(A)onlyCTEsthatshow3 6
asigmoidal-shapedvoltammogramwithasmallchargingcurrentwereemployedinfurtherexperimentsUsingchronoamperometry[5]ameantipdiameterof27μm(standarddeviation(SD)28μmN=142)ameanaxiallengthof165μm(SD=114μmN=142)wereestimatedfortheseCTEs TocomparesurfacecharacteristicsofbothCTEsandHCTEs cyclicvoltam-
minus1 3+2+ minus1 minus1metryof (1)10mmolL [Ru(NH ) ] in10molL KCl (2)10mmolL 3 63minus4minus minus1 minus1[Fe(CN) ] in 10 mol L KCl and (3) 10 mmol L 4-methylcatechol in6minus101molL HClO wasconductedatthesameelectrodesbeforeandafterhydroge-4
nation The results obtained are shown in Fig 1(A-C)We observed a ~20(SD=5N=10)decreaseinthelimitingcurrentofallthreeredoxmarkersafterdiphenylsilane reduction most likely attributable to the hindrance to theirelectron transfer reactions by the phenylsiloxane group formed on HCTEs
3minus4minusMoreover as an inner-sphere redox probe both [Fe(CN) ] and 4-methyl-6
catechol reactionsare sensitive to thepresenceofoxygen functionalitiesonacarbonsurface [7]Accordingly theconversionof these functionalities toCndashHbondsbydiphenylsilanereductionwasexpectedtoyieldmoresluggishelectron
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 21
transfer kinetics at HCTEs as supported by a negative potential shift (from3minus4minus+75mVtominus10mV)inthecyclicvoltammogramof[Fe(CN) ] andapositive6
potentialshift(from+580mVto+675mV)inthecorrespondingcyclicvoltammo-2gram of 4-methylcatechol In addition the conversion of sp -carbon to
3sp -diamond-likecarbon[6] isalsoexpectedtoreducetheconductivityof thecarbonelectrodesurface
32Dopaminedetectionduringbiofoulingexperiments
minus1Theelectrochemicalbehaviourof1mmolL dopamine inapH=74citrate-minus1phosphate buffer (01 mol L ) at CTEs and HCTEs was studied by cyclic
voltammetryTheresultsobtainedareshown inFig1(D)Acomparable12decrease(SD=6N=10)inthedopamineoxidationlimitingcurrenttothatof4-methylcatechol was observed A positive potential shift from +285 mV to+305mVinthevoltammogramsisalsoaccountedforasdescribedabove
22 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
minus1 3+ minus1 minus1Fig 1Cyclicvoltammetryof(A)10mmolL [Ru(NH ) ] in10molL KCl (B)10mmolL 3 63minus4minus minus1 minus1 minus1[Fe(CN) ] in10molL KCland(C)10mmolL 4-methylcatechol in01molL HClO at3 4
minus1(a)aCTEand(b)aHCTE(D)10mmolL dopamineinapH=74citrate-phosphatebufferrecordedminus1at(a)aCTEandaHCTE(b)beforeand(c)afterbiofoulingScanrate100mVs
Next HCTEs were incubated in a synthetic fouling solution containing4(wv)bovineserumalbumin001(wv)cytochromeC(bothareproteins)and 10 (vv) caproic acid (a lipid) for 30 min Cyclic voltammetry of
minus110mmolL dopamineattheseHCTEswasthenconductedinapH=74citrate-phosphatebuffertoevaluatetheirantifoulingpropertyNotablyaconsiderable515(SD=183N=6)decreaseindopaminesignalwasobservedatCTEsIncontrastonlyacorresponding69decrease(SD=35N=40)wasestimatedatHCTEsasshowninFig1(D)Clearlythisrepresentsamajorimprovementinthe antifouling capability of HCTEs obtained using diphenylsilane reductioncomparedtoCTEsandotherpreviouslytestedhydrogenatingagents[9]includingn-butylsilane (35decrease) triethylsilane (23decrease) andphenylsilane(18decrease)Thereforethisworkhassuccessfullydemonstratedtheeffecti-venessofdiphenylsilanereductionmethodindevelopingantifoulingelectrodesfordopaminedetection
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 23
Smallconical-tipelectrode
As-prepared Hydrogenated
minus1LinearrangemicromolL 1ndash10 1ndash20minus2 minus2Intercept10 pAmicrom 108plusmn004 273plusmn011
minus3 minus2 minus1Slope10 pALmicrom micromol 128plusmn007 443plusmn012R 0993 0998
minus1LimitofdetectionmicromolL 100 077
Table 1Analyticalparametersofconcentrationdependencesofdopamineobtainedbydifferentialpulse
minus1voltammetryinapH=74citrate-phosphtebuffer(01molL )Allquoteduncertaintiesrepresentthe95confidenceintervalandthecorrelationcoefficient(R)wasfoundtobestatisticallysigni-ficantatthe95usingStudentrsquost-test
Fig 2DifferentialpulsevoltammetryofdopamineataHCTEinapH=74citrate-phosphatebufferminus1 minus1(01molL )atconcentrations(a)1(b)2(c)4(d)6(e)8(f)10and(g)20μmolL
WehavealsostudiedtheconcentrationdependenceofdopamineinapH=74citrate-phosphatebufferbydifferentialpulsevoltammetryAtypicalcalibrationplotobtainedisshowninFig2Theanalyticalparametersandestimatedlimitsofdetection are summarised inTable 1 These results show thatHCTEs outper-formed CTEs because they exhibit a ~35times higher sensitivity a 23 lowerdetectionlimitandawiderlinearrange
4Conclusions
InthisstudyphysicallysmallHCTEswithanti-foulingcharacteristicsachievedbyhydrogenationusingdiphenylsilanereductionweresuccessfullyfabricatedandelectrochemically characterised using several redox probes Next dopaminedetectionwasperformedbeforeandafterincubationofelectrodesinasyntheticfoulingsolutioncontainingahighconcentrationofbiomoleculesOnlyalow69(SD=35)decreaseindopaminelimitingcurrentwasachievedatHCTEsobtain-edbydiphenylsilanereductionindicatingtheirsignificantlylesssusceptibilitytobiofoulingthanCTEsThesepromisingresultsindicatethatantifoulingHCTEswillpotentiallybenefitthedevelopmentofbiosensorsfordopaminedetectionin-vivoinbiologicalmedia
Acknowledgments
ThisresearchwasperformedwithintheframeworkofSpecificUniversityResearch(SVV260560)FinancialsupportsprovidedbytheGrantAgencyofCharlesUniversity(project390119)andbytheCzechScienceFoundation(project20-03187S)aregratefullyacknowledgedSBandJKalsothankthe Mobility Fund of Charles University and Hlavkova nadace for providing funding for theirresearchinternshipsatMacquarieUniversitySydneyAustralia
References[1] BaranwalAChandraPClinicalimplicationsandelectrochemicalbiosensingofmonoamine
neurotransmittersinbodyfluidsinvitroinvivoandexvivomodelsBiosensBioelectron121(2018)137ndash152
[2] CaoQPuthongkhamPJillVentonBReviewnewinsightsintooptimizingchemicaland3Dsurface structuresof carbonelectrodes forneurotransmitterdetectionAnalMethods11(2019)247ndash261
[3] LinP-HLinB-RAntifoulingstrategiesinadvancedelectrochemicalsensorsandbiosensorsAnalyst145(2020)1110ndash1120
[4] HanssenBLSirajSWongDKYRecentStrategiestoMinimiseFoulinginElectrochemicalDetectionSystemsRevAnalChem35(2016)1ndash28
[5] McNallyMWongDKYAnin-vivoprobebasedonmechanicallystrongbutstructurallysmallcarbonelectrodeswithanappreciablesurfaceareaAnalChem73(2001)4793ndash4800
[6] SirajSMcRaeCRWongDKYEffectiveactivationofphysicallysmallcarbonelectrodesbyn-butylsilanereductionElectrochemCommun64(2016)35ndash41
[7] ParkJShowYQuaiserovaVGalliganJFinkGDSwainGMDiamondmicroelectrodesforuseinbiologicalenvironmentsJElectroanalChem583(2005)56ndash68
[8] ShinDTrykDAFujishimaAMerkociAWang JResistance to surfactantandproteinfoulingeffectsatconductingdiamondelectrodesElectroanalysis17(2005)305ndash311
[9] Roshni RAnAntifouling Structurally Small Carbon Electrode forDetectionof theNeuro-transmitterDopaminePhDThesisMacquarieUniversitySydney2019
24 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Theanalyticalperformancesofenzymaticbiosensorsarestronglyaffectedbytheenzyme immobilization process There is no universal technique for enzymesattachmentThereforespecialattentionshouldbepaid to theselectionof theappropriatesupportandthedevelopmentoftheoptimalbindingstrategyinordertoensure thebestcharacteristicsof immobilizedenzymeDespiteavarietyofpreviouslyreportedcovalentimmobilizationmethodsfordifferentenzymesthepresentedprocedurescanbehardlycomparedtofindtheoptimalonesbecauseofdifferentanalyticalmethodsandexperimentalconditionsusedUptodatethere
A comparative study of covalent glucose oxidase and laccase immobilization techniques at powdered supports for biosensors fabrication
ab a bSOFIIATVORYNSKA JIR IBAREK BOHDANJOSYPCUK
a UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova2030812843Prague2CzechRepublicsofiiatvorynskajh-instcascz
b JHeyrovskyacuteInstituteofPhysicalChemistryoftheCzechAcademyofSciences Dolejškova318223Prague8CzechRepublic
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 25
AbstractInordertodeveloptheoptimalstrategyandtodeepentheknowledgeinthefieldofenzymeimmobilizationthreedifferenttechniquesofcovalentbindingfortwoenzymes(glucoseoxidaseandlaccase)atpowdered surfaces were compared Immobilization protocol wasoptimized by changing supports (twomesoporous silica powders(SBAminus15 MCMminus41) and a cellulose powder) the functionalizedgroupsintroducedatsupportsurfaces(minusNH andminusCOOH)andthe2
methodsofactivation(glutaraldehydeandcarbodiimide)Aminoandcarboxyl functionalized mesoporous silica and cellulose powderswerepreparedbysilanizationusing(3-aminopropyl)triethoxysilaneandcarboxyethylsilanetriolrespectivelyItwasfoundthatcouplingof both enzymes by their ndashNH groups through glutaraldehyde to2
ndashNH functionalized supports in particular SBA15minusNH and2 2
celluloseminusNH forglucoseoxidaseMCM41minusNH forlaccaseshowed2 2
thehighestactivityandthebeststability
Keywordsbiosensorcovalentimmobilizationenzymaticreactorglucoseoxidaselaccase
is still a lack of the comparative systematic studies focusing on the enzymesimmobilizationonthevarioussupportsusingdifferenttechniques The aim of this work is the systematic comparative study of the differenttechniques for covalent coupling of the enzymeswhich ensures not only thedevelopmentoftheoptimalimmobilizationstrategyfortheselectedenzymesbutalsoenablestofindoutsometendenciesinenzymeattachmentprocessgenerallyThusthisworkisfocusedonadetailedanalysisoftheeffectofthekindofsupportits anchor groups and the activation methods on activity and stability ofimmobilizedenzymesTwoenzymeswithdifferentnature(glucoseoxidase(GOx)andlaccase(Lac))werechosenasthetestingbioreceptors
2Experimental
21Reagentsandchemicals
AllchemicalswereofpaorbettergradeGlucoseoxidasefromAspergillusnigerminus1(GOxEC11341452Umg )laccasefromTrametesversicolor(LacEC11032
minus1129Umg )D-(+)-glucosedopamineglutaraldehyde(GAgradeII25aqueoussolution) N-(3-dimethylaminopropyl)-Nʹ-ethylcarbodiimide hydrochloride(EDC ge980) N-hydroxysuccinimide (NHS ge970) (3-aminopropyl)-triethoxysilane (APTES) mesoporous silica powder SBAminus15 (particle size
2 minus12ndash6μmporesizeasymp7nmsurfaceareaasymp600m g )mesoporoussilicapowder2 minus1MCMminus41 (pore size 21ndash27 nm surface area asymp 1000 m g ) cellulose (Cell
microcrystalline powder particle size 20 microm) were purchased from SigmaAldrichCarboxyethylsilanetriol(CEST25aqueoussolution)waspurchased
regfromabcr (Germany)
22Instrumentation
Amperometric measurements were carried out at room temperature usingcomputer-controlled electrochemical stand (Polaro-Sensors Czech Republic)withMultiElchemv31software(JHeyrovskyInstituteofPhysicalChemistryoftheCAS)Flowinjectionanalysis(FIA)withthethree-electrodelaboratory-madeflow-through cellwas usedworking electrode minus tubular detector of polishedsilversolidamalgam(TD-p-AgSAlaboratory-madeinnerdiameter05mmtheamalgamtube length60mm) referenceelectrodeminusaminiaturizedsaturatedcalomelelectrodebasedonsilverpasteamalgam[1](laboratory-madeithasthesamepotential as classical saturated calomel electrode) auxiliary electrodeminusplatinum wire (diameter 10 mm length 10 mm) The system for FIA withelectrochemicaldetectioncomprisedofalinearsyringepumpa2-position6-port
regsampleinjectorvalveaninjectionlooplaboratory-madeofTeflon (PTFE)tubing(100μL) a solenoid operatedmicro-pumpan enzymatic reactor and a flow-through cell for TD The enzymatic reactor consists of a tube filled by theenzymaticpowder
26 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
3Resultsanddiscussion
Basedonthedatareportedintheliterature[23]andontheresultsofourpreviousworks[4ndash8]forthisstudyCellandmesoporoussilicapowders(namelySBAminus15andMCMminus41)havebeenselectedas thepotentialpromisingsupports for thecovalent enzyme immobilizationBecauseof thehigh contentof surficial ndashOHgroupswhich are capable of chemical reactions these supports can be easilyfunctionalizedThewell-knownandfrequentlyutilizedtechniqueofsilanizationhasbeenusedtomodifythesurfacesofSBA15MCMminus41andCellbythedesiredfunctionalizedgroupsAminosilaneAPTESwasappliedtoformminusNH groupson2
thematrixsurfaceswhereascarboxylsilaneCESTwasusedtointroducendashCOOHgroups Generallytheprocedureofthecovalentimmobilizationofenzyme(eitherLacorGOx)onthefunctionalizedsupportconsistsofthreestepsI Synthesisofthefunctionalizedsupportwhichmeansthemodificationofthematrix(MCMminus41SBAminus15andCell)withsuitableanchoredgroups(minusNH or2
minusCOOH)II Activationstepofthefunctionalizedsupportwithspecificactivatingagents(glutaraldehydeorEDCNHS)tomakeitreactivetowardsenzyme
IIIEnzyme(LacorGOx)couplingtotheactivatedsupport
To investigate the effect of support its surface functionalizedgroups and themethodsofactivationontheefficiencyofthecovalentenzymeimmobilizationthreedifferentstrategies(ABandC)forLacandGOxattachmenthavebeenusedThedetailsoftheusedtechniquesandthedenotationsofthepreparedenzymaticpowdersaresummarizedinTable1(nextpage)ToexaminetheefficiencyofLacandGOximmobilizationtheenzymaticreactors(filledbytheenzymaticpowderspreparedwithdifferenttechniques)coupledwithTDwereusedforamperometricdetermination of dopamine and glucose respectively in flow systems Theprincipleofglucosedetection isbasedonamperometricmeasurementsof theenzymatically consumed oxygen whereas dopamine was detected by thereductionoftheenzymaticallyoxidiseddopamine Asdepicted inFig1 thebiosensors responsesare stronglyaffectedby thestrategyusedforLacorGOximmobilizationAsshowntheresponsesofLacandGOx biosensors decrease in the order strategy A gt strategy B gt strategy Cirrespectiveofthetypeofsupport It isclearlyseenthataminofunctionalizedsupports(SBA15minusNH MCM41minusNH andCellminusNH )providehigheractivitiesof2 2 2
the immobilized Lac andGOx than these supports functionalized by carboxylgroups(SBA15minusCOOHMCM41minusCOOHandCellminusCOOH)BycomparingactivitiesofimmobilizedenzymesusingstrategiesAandBtheinfluenceoftheactivationagenthasbeenevaluatedThebestresultsforbothenzymeswereobtainedforndashNH functionalizedsupportsactivatedbyGAItcouldbeexplainedbythefact2
thatGAcontrary tocarbodiimidewithnonemolecularspaceprovidesa long
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 27
spacerarmensuringminimalsterichindrancesforenzymesbindingItcanbeconcludedthatthecovalentimmobilizationofbothenzymesbytheirndashNH groups2
viaGAtondashNH functionalizedmesoporoussilicapowders(strategyA)provided2
the highest activities Interestingly in the similar comparative studies it isreportedthatamongndashOHminusCOOHandndashNH functionalizedsupportsactivatedby2
divinylsulfonecarbodiimideandGArespectivelythelastonewasfoundasthemostsuitabletechniqueforthecovalentbindingofLac[9]invertase[10]andpepsin[11]
28 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Tab
le 1
Theprinciplesofthecovalentimmobilizationmethodsofenzymesusedinthisstudy
StrategyA
StrategyB
StrategyC
Support
SBAminus15MCMminus41Cell
SBAminus15MCMminus41Cell
SBAminus15MCMminus41Cell
Supportfunctio-
minusNH
minusNH
minusCOOH
22
nalizedgroup
Activationagent
Glutaraldehyde(GA)
Carbodiimide
Carbodiimide
(EDCNHS)
(EDCNHS)
Enzymereactive
minusNH
minusCOOH
minusNH
22
group
Typeofbond
secondaryamine
amide
amide
Denotationsofthe
GOxminusNHminusCHminus(CH)minusCHminusNHminusSBA15
GOxminusC(=O)minusNHminusSBA15
GOxminusNHminusC(=O)minusSBA15
23
preparedenzy-
GOxminusNHminusCHminus(CH)minusCHminusNHminusMCM41
GOxminusC(=O)minusNHminusMCM41
GOxminusNHminusC(=O)minusMCM41
23
maticpowders
GOxminusNHminusCHminus(CH)minusCHminusNHminusCell
GOxminusC(=O)minusNHminusCell
GOxminusNHminusC(=O)minusCell
23
LacminusNHminusCHminus(CH)minusCHminusNHminusSBA15
LaxminusC(=O)minusNHminusSBA15
LacminusNHminusC(=O)minusSBA15
23
LacminusNHminusCHminus(CH)minusCHminusNHminusMCM41
LacminusC(=O)minusNHminusMCM41
LacminusNHminusC(=O)minusMCM41
23
LacminusNHminusCHminus(CH)minusCHminusNHminusCell
LacminusC(=O)minusNHminusCell
LacminusNHminusC(=O)minusCell
23
When the effect of the method of the covalent enzyme coupling on thebiosensorstabilitywasevaluateditwasfoundthatLacboundedtondashNH functio-2
nalizedsupportsviaGA(strategyA)hasshownthehigheststability(gt65oftheinitial responses after 1 month) compared to other strategies whereas GOximmobilizedwithtwostrategies(AandB)possessedapproximatelysimilarhighstability(gt80oftheinitialresponsesin1month)BothenzymesboundedviandashNH groupstondashCOOHfunctionalizedsupportsthroughEDCNHS(strategyC)2
showedquitelowstability
4Conclusions
Threedifferent strategies including the support selection the anchor surfacegroups and the activationmethod havebeen compared for efficient covalentimmobilization of Lac and GOx The results showed that ndashNH functionalized2
supports(SBA15minusNH CelluloseminusNH forGOxandMCMminusNH forLac)activatedby2 2 2
GAmaybeusedtoeffectivelybindenzymesintermsofhighactivityandstability
Acknowledgments
ThisworkwasfinanciallysupportedbytheGrantAgencyofCharlesUniversityinPrague(Project1356120)theGrantAgencyoftheCzechRepublic(Project20-07350S)anditwascarriedoutwithintheframeworkofSpecificCharlesUniversityResearch(SVV260440)
References
[1] YosypchukBBarekJYosypchukOPreparationandpropertiesofreferenceelectrodesbasedonsilverpasteamalgamElectroanalysis23(2011)2226minus2231
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 29
Fig 1Effectof the covalent attachment techniqueson (A) laccaseand (B) andglucoseoxidaseminus1biosensor responses Experimental conditions (A) c = 500 micromol L E = minus50 mVDOP det
minus1 minus1v = 01 mL min V = 40 microL carrier solution 01 mol L acetate buffer pH = 48flow DOPminus1 minus1(B)c =500micromol L E =minus1100mVv ==01mLmin V =40microL carrier solutionGlu det flow Glu
minus1 minus101molL acetatebuffer0001molL Na EDTApH=652
[2] LiuYChenJYEnzymeimmobilizationoncellulosematrixesJBioactCompactPolym31(2016)553ndash567
[3] Hartmann M Kostrov X Immobilization of enzymes on porous silicas ndash benefits andchallengesChemSocRev42(2013)6277minus6289
[4] JosypcukOBarekJJosypcukBElectrochemicalbiosensorsbasedonenzymaticreactorsfilledbyvarioustypesofsilicaandamalgampowdersformeasurements inflowsystemsElectroanalysis28(2016)3028minus3038
[5] Josypcuk O Barek J Josypcuk B Amperometric determination of catecholamines byenzymaticbiosensorsinflowsystemsElectroanalysis30(2018)1163minus1171
[6] TvorynskaSBarekJJosypcukBAmperometricbiosensorbasedonenzymaticreactorforcholinedeterminationinflowsystemsElectroanalysis31(2019)1901minus1912
[7] TvorynskaSBarekJJosypcukBFlowamperometricbiosensorbasedontwoenzymaticreactors (acetylcholinesterase-choline oxidase) for the detection of neurotransmitteracetylcholine In Proceedings of the 15th International Students Conference ldquoModernAnalyticalChemistryrdquoKNesmerak(ed)PragueFacultyofScienceCharlesUniversity2019p61minus66
[8] TvorynskaSBarekJJosypcukBAcetylcholinesterase-cholineoxidase-basedmini-reactorscoupledwithsilversolidamalgamelectrodeforamperometricdetectionofacetylcholineinflowinjectionanalysisJElectroanalChem860(2020)113883
[9] RekucAKruczkiewiczPJastrzembskaBLiesieneJPeczynska-CzochWBryjakJLaccaseimmobilizationonthetailoredcellulose-basedGranocelcarriersIntJBiolMacromol42(2008)208minus215
[10] Bryjak J Liesiene J S tefuca V Man-tailored cellulose-based carriers for invertaseimmobilizationCellulose15(2008)631minus640
[11] SzałapataKOsinska-JaroszukMBryjakJJaszekMJarosz-WilkołazkaANovelapplicationofporousandcellularmaterialsforcovalentimmobilizationofpepsinBrazJChemEng33(2016)251minus260
30 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Carbohydrates are crucial for energy structure and signaling in the humanbody[1]Thereisavarietyofcarbohydratesbutthemostimportantoneforlifeisglucoseasitisfundamentalinthemetabolismandphotosynthesis[2]GlucoseisclassifiedashexoseThesemonosaccharidesonlydifferinthepositionofhydroxylsubstituentsinsomecasesInadditiontothestructuralsimilaritiesthesemole-culeslackachromophoreandarenoteasilyionizable(pK ~12)Thusdetectiona
intheUVregionandseparationofanalytesbycapillaryelectrophoresis(CE)arechallenging [1 3] At the moment there are many different techniques for theanalysis of carbohydrates commonly including time-consuming derivatizationstepsoreluentswithhighpHvalues(pHgt12)inionchromatographyAwell-esta-blishedtechniquefortheanalysisofcarbohydratesishigh-performanceanion-exchangechromatographywithpulsedamperometricdetection(HPAE-PAD)[4]Electrochemical detection like AD is matching miniaturization simple instru-mentationlowcostandrobustnessandthusisoftenusedforflow-basedsystemssuchasCEandflowinjectionanalysis(FIA)[5]
Capillary flow injection analysis with electrochemical detection for carbohydrate analysis
NICOLEHEIGLFRANK-MICHAELMATYSIK
InstituteofAnalyticalChemistryChemo-andBiosensorsFacultyofChemistryandPharmacyUniversityofRegensburgUniversitaumltsstraszlige3193053RegensburgGermanynicoleheiglchemieuni-regensburgde
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 31
AbstractAsimplecapillaryflowinjectionanalysissystemwithamperometricdetection was arranged for the development of a method for fastoptimization of detection conditions in the context of thedetermination of carbohydrates by means of electrochemistry-capillary electrophoresis-mass spectrometry This setup is free ofelectricalinterferencebyhighvoltageandisperfectforstudyingtheoxidationofvariousanalytesFurthermoreitassureseasycouplingtoMS and thus is an useful tool to investigate the correspondingoxidationproductsofananalyte
Keywordscapillaryflowinjection
analysiscarbohydratesmassspectrometrydisposableelectrodespulsedamperometric
detection
Inthiscontributioncapillaryflowinjectionanalysis(CFIA)withADwillbepresentedasamethodtoapplyandtestADforthedetectionofmonosaccharidesondifferentdisposablethin-filmorscreen-printedelectrodesCFIAwaschosenoverconventionalFIAforthispurposeasthegravityflowinCFIAisstableforalongertimeandverylowsampleconsumptioncanbeachieved[6]TheCFIAsystemwasarrangedassimpleaspossibleandperformedhydrodynamicallytoavoidanyinterferencesFurthermoreitassuresthecouplingoftheflowsystemtoamassspectrometerThusthesamesetupasusedforCFIAcanbeutilizedforcapillaryelectrophoresis-massspectrometry(CE-MS)experimentsbychangingthe flow through the electrochemical flow cell in opposite direction In futureexperimentstheexperienceintermsofADonthoseelectrodeswillbeusedtodevelopelectrochemicalpretreatmentprotocolsforcarbohydratedeterminationbyCE-MS
2Experimental
21Reagentsandchemicals
The following chemicals were used for this study all of analytical gradeAmmoniumacetate(NH OAc)wasobtainedfromMerck(DarmstadtGermany)4
and ferrocene methanol (FcMeOH) from ABCR (Karlsruhe Germany) Milli-Qregwater(182MΩcm)wasgeneratedbyaMilli-QAdvantageA10 system(Merck
Millipore Darmstadt Germany) Carrier solution was prepared by dissolvingndash1NH OAc (50mmolL ) in Milli-Q water FcMeOH solution was prepared by4
dissolvingFcMeOHincarriersolution
22Instrumentation
Electrochemical measurements were performed using a microAutolab Type IIIpotentiostatgalvanostat (Metrohm Autolab B V Utrecht Netherlands)controlledbyNOVA20softwareforexperimentalcontrolanddataacquisitionCFIAwasperformedusingthesetupillustratedinFig1(A)consistingofacarrierreservoirsamplevialandtwofusedsilicacapillaries(PolymicroTechnologiesPhoenix AZ USA inner diameter 100 microm length inlet 40 cm length outlet10cm)connectedtoacommerciallyavailableflowcellfromMicruxTechnologies(model ED-FLOW-CELL Oviedo Spain) Inside of the flow cell the fused silicacapillary was placed in a so-called wall-jet configuration above the workingelectrode of a disposable thin-film gold electrode (model ED-SE1-Au MicruxTechnologiesOviedoSpain)ascanbeseeninFig1(B)Thethin-filmelectrodeswerebasedonathree-electrodesystemwithagoldworkingauxiliaryandquasi-referenceelectrodeThehydrostaticpressurewasachievedbyaheightdifferencebetweeninletandoutletreservoirof30cmresultinginagravityflowofthecarriersolutionthroughafusedsilicacapillaryandsubsequentlythroughtheflowcell
32 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Theinjectionwascarriedoutbyloweringthevialcontainingthecarriersolutiontotheleveloftheoutletexchangingthecarrierreservoirwiththesamplevialandliftingthesamplevialto20cmforadefinedperiodoftimeRe-establishingthecarrierreservoirtookplacethesameway
3Resultsanddiscussion
AsimpleCFIA-ADsystemwasarrangedwherehydrostaticpressurebyaheightdifferencebetweeninletandoutletreservoirresultedinagravitationalflowToobtain general information about the behavior of the assembled CFIA systempreliminaryexperimentswithFcMeOHwereperformedToassurecompatibilitywithMSlateronNH OAcwaschosenastheelectrolyteVariousheightdifferences4
andinjectiontimesweretestedandtheinjectionataheightdifferenceof20cmlasting for 10 s was found to be the optimum concerning feasibility and peakshapes Injections of several solutions of FcMeOH of different concentrationsshowed that the concentration dependence of FcMeOH was linear in theinvestigatedrange(Fig2)Furthermoreexperimentsrevealedthattheinjectionprocedure was established with reasonable precision When repeating the
ndash1injectionof05mmolL FcMeOHincarriersolutionfortentimestherelativestandarddeviationwasfoundtobe3forthemanualinjectionprotocol
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 33
Fig 1(A)Schemeoftheusedcapillaryflowinjectionanalysissetup(1)carrierreservoir(2)samplevial(3)inletcapillarywithalengthof40cmandaninnerdiameterof100microm(4)outletcapillarywithalengthof10cmandaninnerdiameterof100micrombothcapillariesconnectedto(5)acommerciallyavailableflowcellfromMicruxand(6)awastevial(B)Configurationinsidetheflowcell(7)thefusedsilicacapillarywasplacedinaso-calledwall-jetconfigurationabovetheworkingelectrodeof(8)adisposablethin-filmgoldelectrode
4Conclusions
The presented system for CFIA-AD was arranged as simple as possible andrepresents a useful approach for the development of a method for fastoptimization of detection conditions in the context of the determination ofcarbohydratesbymeansofelectrochemistry-CE-MSThesetupisfreeofelectricalinterferencebyhighvoltagecompatiblewithMSandthuspromisingforstudyingtheoxidationofvariousanalytes
References
[1] LuGCrihfieldCLGattuSVeltriLMHollandLACapillaryelectrophoresisseparationsofglycansChemRev118(2018)7867ndash7858
[2] GalantALKaufman RCWilson JDGlucoseDetectionandanalysisFoodChem188(2015)149ndash160
[3] Sarazin C Delaunay N Costanza C Eudes V Gareil P Application of a new capillaryelectrophoreticmethodforthedeterminationofcarbohydratesinforensicpharmaceuticalandbeveragesamplesTalanta99(2012)202ndash206
[4] Rohrer JS Basumallick L Hurum D High-performance anion-exchange chromatographywithpulsedamperometricdetectionforcarbohydrateanalysisofglycoproteinsBiochem78(2013)697ndash709
[5] IslamMAMahbubPNesterenkoPNPaullBMackaMProspectsofpulsedamperometricdetectioninflow-basedanalyticalsystemsndashAreviewAnalChimActa1052(2019)10ndash26
[6] MatysikF-MWernerGTracemetaldeterminationintearsbyanodicstrippingvoltammetryinacapillaryflowinjectionsystemAnalyst118(1993)1523ndash1526
34 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2ndash1 ndash1(A) CFIA-AD recordings of three consecutive injections of (1) 025mmolL (2) 05mmolL
ndash1 ndash1 ndash1(3) 075mmolL and (4) 1mmolL FcMeOH in 50 mmolL NH OAc detection at a Micrux4
thin-filmAuelectrodeataconstantpotentialof03VinaflowcellHydrodynamicinjectionlasted10sataheightdifferenceof20cm(B) Calibration dependence of FcMeOH for CFIA-AD determination and detection at a Micruxthin-filmAuelectrodeataconstantpotentialof03VinaflowcellThestandarddeviationsofpeakheights(n=3)areindicatedbyerrorbars
1Introduction
Topreventsorptiononthecapillarysurfaceandimproveseparationefficiencyand selectivity of determined analytes coatings are formed on the capillarysurface There are two types of ones namelydynamic and covalently bondedcoatings Despite the simplicity of creation dynamic coatings cannot providerequiredreproducibilitywhilecovalentcoatingscontributethestableelectro-osmoticflow(EOF)andhighreproducibilityoftheanalysisInmostcasesthe
Application of covalent coatings based on imidazolium cations for separation and on-line preconcentration of basic and neutral analytes in capillary electrophoresis
a ab aANASTASIAVKRAVCHENKO EKATERINAAKOLOBOVA LIUDMILAAKARTSOVA
a DepartmentofOrganicChemistryInstituteofChemistrySaintPetersburgStateUniversity 26Universitetskiiprospect198504StPetersburgPeterhofRussiakravchenko161216gmailcom
b TheFederalStateInstituteofPublicHealthldquoTheNikiforovRussianCenterofEmergencyandRadiationMedicinerdquoTheMinistryofRussianFederationforCivilDefenceEmergenciesandEliminationofConsequencesofNaturalDisasters54Optikovst197082StPetersburgRussia
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 35
AbstractThemethodofcapillaryelectrophoresis (CE) isactivelydevelopedandmoreandmoreattractsscientistsattentioneveryyearHoweverthesorptionofanalytesonsurfaceoffused-silicacapillarywallsisoneof thesignificantdisadvantagesof thisapproachThe formationofcoatings on the inner capillary surface is typical way to preventsorption and to increase separation efficiency and selectivity ofdetermined analytes Coatings that covalently bonded to capillarywalls is more suitable because they are stable and provides highreproducibilityofanalysisThepresentworkisfocusedonthedevelo-pmentofthemethodofelectrophoreticdeterminationofbiologicalactiveanalytesusingacovalentcoatingbasedonimidazoliumcationsTheeffectofsubstituentinimidazoliumringonmainelectrophoreticparameters was examined It was shown that alkylimidazoliumcoatingscontributetosignificantreducingofbiogenicamineslimitsof detection while β-cyclodextrinimidazolium covalent coatingallowstoseparatebothofhydrophobicandhydrophilicanalytesinonerun
Keywordsbiologicalactiveanalytescapillarycoatingcapillaryelectrophoresisimidazoliumionicliquids
analytes nature determines type of usedmodifiers because suitable ones canprovideaccessorial interactionbetweentheanalytesandthestationaryphaseimproving separation selectivity and efficiency [1 2] Ionic liquidshave beenwidelyusedinanalyticalchemistry[3]andseparationtechniquesparticularlyincapillaryelectrophoresis[4]Earlyresearches[5ndash9]haveshownopportunityofcovalently bonded imidazolium ionic liquids for electrophoretic separationHowever the effect of various substituents in imidazolium ring hasnot beendescribed previously Thus the purpose of this study was to create covalentcoatingsbasedon ionic liquidwithvarioussubstituentsand tocompare theiranalytical capabilities in the electrophoretic separation of biologically activecompounds
2Experimental
21Reagentsandchemicals
(3-Glycidyloxypropyl)trimethoxysilane (GPTMS) hydrochloric acid sodiumdodecyl sulfate (SDS) imidazole 22-diphenyl-1-picrylhydrazyl (DPPH)p-toluen-sulfonylchlorideβ-cyclodextrinhydrocortisone(F)11-deoxycortisol(S)Corticosterone(B)rac-ketoprofen(ndash)-adrenaline(A)L-(ndash)-norepinephrine(NE) DL-normetanephrine (NMN) dopamine (DA) DL-metanephrine hydro-chloride(Met)serotoninhydrochloride(Ser)homovanillicacid(HVA)24-di-hydroxy-benzoic acid (24-DHBA) 34-dihydroxy-L-phenylalanine (DOPA)L-tryptophan(Trp)L-tyrosine(Tyr)werepurchasedfromSigma-Aldrich(USA)1-Bromo-butane1-bromooctanewerepurchasedfromReagentPlus(Ukraine)Sodium dihydrogenphosphate dihydrate acetone NN-dimethylformamide(DMF)wereobtainedfromMerck(Germany)AllreagentsusedwereanalyticalgradeAllsolutionswerepreparedusingdeionizedwater
22Instrumentation
Capillary electrophoresis experiments were carried out using the system ofcapillaryelectrophoresisCAPEL-105M(LumexRussia)withUV-spectrophoto-metricdetector(wavelengthrange190ndash360nm)Separationswereperformedusing 58times49 cm (9 cm to the detector outside diameter 360 microm and innerdiameter50microm) coated silica capillaries (LumexRussia) Thebuffer pHwasmeasuredwithapH-meterHI2210ndash2216(Hanna)
23Capillarycoatingsynthesis
Earlier our research team has proposed the synthesis route for the covalentcoatingsbasedonimidazoliumcationfunctionalizedwithalkylgroup[10]andβ-cyclodextrin [11] All capillarieswere prepared according above-mentioned
36 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
manuscriptsandcharacterizedbytheEOFmobilitymeasurementandscanningelectronmicroscopyThesynthesisconsistedoffollowingstepspreparationofacapillarytocreateacovalentcoating(heatingcapillaryfilledwith2MNaOHat90degCfor1handdryingfollowed)silylationwithGPTMSandfunctionalizationwiththeimidazolesolutionfollowedmodificationbybutyl-andoctylbromideortosyl-β-cyclodextrin(seedetailsin[10]and[11])
24Solutions
A stock buffer solution in concentration 50mM was prepared by dissolvingappropriate amount of sodium dihydrogenphosphate dihydrate in deionizedwateradjustingpHto20with1MhydrochloricacidThisbuffersolutionwasthendilutedwithdeionizedwater
ndash1 Allthesamplestocksolutionswerepreparedwithconcentration10mgmL Thestocksolutionsoftheneurotransmittersandtheirmetabolites(adrenalinenoradrenaline dopamine normetanephrine metanephrine serotonin homo-vanillicacid)and24-dihydroxybenzoicacidasinnerstandardandaminoacids(tryptophan34-dihydroxy-L-phenylalanine tyrosine)wereprepared in01MhydrochloricacidThestocksolutionsofsteroids(hydrocortisone11-deoxycor-tisolandcorticosterone)wereprepared inacetonitrileThestocksolutionsofketoprofen racemate and S-ketoprofen were prepared in acetonitrilewatersolution(1090vv) Untilelectrophoreticanalysisthestocksolutionswerestoredatndash16degCTheworkingsolutionswerepreparedbydilutingtheinitialsolutionswithwaterjustbeforetheexperiments
3Resultsanddiscussion
CovalentcoatingsbasedonN-alkylimidazoliumcationwereespeciallysuitableforseparationofneurotransmittersandtheirmetabolites(Fig1)Inadditionthecombinationofcovalentcoatingwithon-linepreconcentrationtechniquesallowstothesignificantdecreaseoftheseanalyteslimitsofdetection(LOD)Accessorialinteractions positively charged analytes with positively charged imidazoleimproveseparationselectivity(viaπ-πinteraction)andefficiency(concentrationintightzonesviaelectrostaticrepulsion)Sodiumdodecylsulfate(SDS)addedintobackgroundelectrolyte(inconcentrationabovecriticalmicelleconcentra-tion) strongly interacts with hydrophobic alkyl groups in covalent coatingstructureThenegativelychargedSDSlayerisformedoninnercapillarysurfaceThedoublereversingEOFallowsustocarryoutelectrokineticinjectionofsampleandon-linepreconcentrationbysweepingsimultaneouslyLODweredeclineto
ndash108ndash20ngmL ThelengthofalkylsubstituentalsoaffectsthestackingefficiencyfactorandLODMorehydrophobicoctylgroupscomparetobutylprovidemoreeffectiveinteractionwithSDSandasresultlowerLOD
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 37
Covalentcoatingmodifiedβ-CDhasnotshownsharpreducingofLODbyon-linepreconcentrationStacking sweeping (SDSasmicelle reagent) field-enhancedsample injection were examined using different model mixtures of analytesNeverthelessthiscoatingallowssimultaneousseparationofbothofhydrophobicsteroidhormonesandhydrophilicbiogenicaminesinasinglerun(Fig2) The guest-host interaction hydrophobic cavity of β-cyclodextrin with thehydrophobicsteroids leadsto the formationofcomplexwhichaffectssteroidselectrophoretic mobility At the same time β-cyclodextrin can act as a chiralselector and baseline separation of ketoprofen enantiomers has also beenachieved
38 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Electropherogramofmixtureof neurotransmitters and theirmetabolitesadrenaline (A)norepinephrine (NE)normetanephrine (NMN)dopamine (DA)metanephrine (Met) serotonin(SER)homovanillicacid(HVA)andtheinnerstandard24-dihydroxybenzoicacid(24-DHBA)oncovalentlymodifiedwithN-buthylimidazoliumionicliquidscapillaryConditions10mMNaH PO 2 4
(adjusted to pH= 20 by 1MHCl) injection 50 stimes30mbar ndash20 kV 220 nmmodelmixurendash1 ndash1 ndash110microgmL (METADNMNNADA24-DHBA)5microgmL (SER)and20microgmL (HVA)
4Conclusions
ItwasshownthatstructurecovalentcoatingaffectsitsanalyticalcharacteristicsWecomparedtwotypesofcovalentcoatingdifferingsubstituentinimidazoliumringnamelyalkylgroupandβ-cyclodextrinThefirsttypeisgreatcoupledwithon-line preconcentration technic but it is limited to effectively determine ofbiogenicaminesonlywhilethesecondtype(withβ-cyclodextrin)showedthepossibilitiestoseparatevariousanalytesbutsuitableon-linemodehasnotbeenfoundThemainpointsaresummarizedinTable1
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 39
Covalentcoatingtype Electrophoreticseparationof On-lineprecon- centration biogenicamines amino steroid ketoprofen andtheirmeta- acids hormones enantiomers bolites N-β-cyclodextrinimida- yes yes yes yes thesuitableapproachzoliumcovalentcoatings wasnotfoundedN-alkylimidazolium yes yes nonsepa- nonsepa- thesignificantreducingcovalentcoatings rated rated forbiogenicaminesLOD
Table 1Thesummationofpossibilitiesofcovalentcoatingsbasedonimidazoliumcation
Fig 2Electropherogramofsimultaneousseparationofhydrophobic(steroidhormones)andhydro-philicanalytes (aminoacidsandbiogenicamines) insinglerunwithcovalentcoatingbasedonimidazoleandβ-CDConditions10mMNaH PO (adjusted topH = 20by1MHCl) injection2 4 20 stimes30mbarndash20kV254nm(1ndash8min)and220nm(8ndash15min)0mbar(1ndash10min)and40mbar(10ndash15 min) Model mixture corticosterone (B) hydrocortisone (F) 11-deoxycortisole (S)
ndash1 ndash15μgmL L-tryptophan(Trp)34-dihydroxy-L-phenylalanine(DOPA)10μgmL L-tyrosine(Tyr)ndash15 μg mL noradrenaline (NA) normetanephrine (NMN) adrenaline (AD) dopamine (DA)
ndash120μgmL
Acknowledgments
This work was supported by Russian Science Foundation (grant numbers 19-13-00370) Theauthors are also grateful to the Chemistry Education Centre and Nanothechnologies Centre ofResearchParkSaintPetersburgStateUniversityfortechnicalsupport
References
[1] HuLFYinSJZhangHYangFQRecentdevelopmentsofmonolithicandopen-tubularcapillaryelectrochromatography(2017ndash2019)JSepSci43(2020)1942ndash1966
[2] KartsovaLAKravchenkoAVKolobovaEACovalentcoatingsofquartzcapillariesfortheelectrophoretic determination of biologically active analytes J Anal Chem 74 (2019)729ndash737
[3] HoTDZhangCHantaoLWAndersonJLIonicliquidsinanalyticalchemistryFundamen-talsadvancesandperspectivesAnalChem86(2014)262minus285
[4] TangSLiuSGuoYLiuXJiangSRecentadvancesofionicliquidsandpolymericionicliquids incapillaryelectrophoresisandcapillaryelectrochromatography JChromatogrA1357(2014)147ndash157
[5] QinWLiSFYElectrophoresisofDNAinionicliquidcoatedcapillaryAnalyst128(2003)37ndash41
[6] QinWWeiH Li SFY 13-Dialkylimidazolium-based room-temperature ionic liquids asbackgroundelectrolyteand coatingmaterial in aqueous capillaryelectrophoresis JChro-matogrA985(2003)447ndash454
[7] QinW Fong S Li Y Determination of ammonium andmetal ions by capillary electro-phoresisndashpotential gradient detection using ionic liquid as background electrolyte andcovalentcoatingreagentJChromatogrA1048(2004)253ndash256
[8] QinWLiSFYAn ionic liquidcoating fordeterminationofsildenafilandUK-103320 inhumanserumbycapillaryzoneelectrophoresis-iontrapmassspectrometryElectrophoresis23(2002)4110ndash4116
[9] BorissovaMVaherMKoelMKaljurandMCapillaryzoneelectrophoresisonchemicallybondedimidazoliumbasedsaltsJChromatogrA1160(2007)320ndash332
[10] KolobovaEKartsovaLKravchenkoABessonovaEImidazoliumionicliquidsasdynamicand covalent modifiers of electrophoretic systems for determination of catecholaminesTalanta188(2018)183ndash191
[11] KravchenkoAKolobovaEKartsovaLMultifunctioncovalentcoatingsforseparationofaminoacidsbiogenicaminessteroidhormonesandketoprofenenantiomersbycapillaryelectrophoresisandcapillaryelectrochromatographySepSciplus3(2020)102ndash111
40 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Synthetic 4-hydroxy-3-methoxybenzaldehyde (vanillin) is used as a flavoringagent in foodsdrinksperfumesandpharmaceuticals [1]However at certainconcentrationsthesubstancemayaccumulateinthebodyhaveatoxiceffectand
ndash1at high concentrations may be fatal (lethal dose LD (oral rat) =2gkg 50ndash1 ndash1LD (oral guinea pig) = 14gkg LD (intravenous dog) = 132gkg lethal50 50 ndash1concentrationLC (inhalationmouse)=417gkg )[2]AccordingtoRussianState
StandartGOST121005-88thetoxiceffectsofvanillinintheworkplaceinthendash3formofvapoursoraerosolsareobservedatconcentrationsabove15mgm
Chromatography[3]spectrophotometry[4]capillaryelectrophoresisareusedforvanillindeterminationindifferentobjects CurrentlysmokingmixturesforhookahsandelectroniccigarettesarewidelyusedamongyoungpeopleThesemixturesarenotcontrolledforthecontentofsubstancesandarefreelyavailableconsideringthemmoreharmlesswithrespecttoordinarycigarettesThusthedevelopmentofamethodforthedeterminationof4-hydroxy-3-methoxybenzaldehydeinsmokingmixturesisrelevant
Determination of vanillin in smoking mixtures by spectrophotometry
ELIZAVETAEFREMENKOANNACHERNOVAOLGABASTRYGINA
DepartmentofChemicalEngineeringNationalResearchTomskPolytechnicUniversityLeninavenue30634050TomskRussiaeaetpuru
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 41
AbstractTheresearchdealswithdeterminationofvanillin insmokingmix-turesbyultraviolet-visiblespectrophotometryThemethodshowed
ndash1goodlinearityintherangeof005ndash012gL withalimitofdetectionndash1005gL After validation studies the method was successfully
applied to thedeterminationof vanillin in smokingmixtureswithsatisfactoryresultsItwasshownthattheerrorofthismethoddoesnot exceed 1 The developed spectrophotometric procedure fordeterminingvanillininsmokingmixturescanbeusedasacontrol
Keywordssmokingmixturesspectrophotometryvanillin
2Experimental
21Reagentsandchemicals
Asampleofvanillin(purity98)wastakenastheobjectofstudyAssolventsweused95ethanolAllchemicalsusedwereofanalyticalreagentgrade
22Instrumentation
Theopticaldensityofsampleswasmeasuredincuvettewithanabsorbinglayerthickness of 10 mm using a Cary 60 spectrophotometer (Agilent USA) Allmeasurementswerecarriedoutatroomtemperature
23Samplepreparation
Sample preparation of the investigated objects consisted of the preliminarydissolutionofthesamplein95ethanolThesample10mgoftobaccoldquoAdalyandashVanillardquo(Turkey)wasdiluted in10μLof95ethanol to theconcentrationof
ndash11gL Thesample10μLofldquoFlavoringTPAndashVanillaCustardrdquo(USA)wasdilutedin10 μL of 95 ethanol The resulting solution was diluted six times to the
ndash3concentrationof017μLcm
3Resultsanddiscussion
TodeterminevanillininthesamplestheopticalpropertiesofvanillininvarioussolventsweredeterminedAsaresultthe95ethanolwaschosenastheoptimalsolvent[4] IthasbeenestablishedthatintheUVspectraoftheanalyteabsorptionbondsareobservedwithmaximumvaluesat23002800and3100nmwhichcorres-pondstopublisheddata[45](Fig1) To quantify vanillin the calibration curve of the optical density on theconcentrationofvanillin in95ethanolwasobtainedatconcentrations005
ndash1006007008010and012gL Calibrationcurveofvanillinin95ethanolatawavelengthof280nmis
ndash1 A =81914c[gL ]+00357 (1)2802 R =1
Calibrationcurveofvanillinin95ethanolat310nmis
ndash1 A =73824c[gL ]+00301 (2)3102 R =1
42 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Inthespectraoftheanalyzedsamplesolutionsabsorptionmaxima(2800nmand3100nm)characteristicforvanillinwereobservedTheamountofvanillininthesamplewasdeterminedusingcalibrationcurvesat280and310nmWeightedleast square regressionwas applied to the calibration curves to improve theaccuracyespeciallyatinlowconcentrationlevelrangeGoodlinearitywasfound
ndash1 ndash1intherangeof005ndash012gL withadetectionlimitof005gL TheresultsarepresentedintheTable1
4Conclusions
ThedevelopedmethodcanbeusedasacontrolmethodTheerrorinthemethodfordeterminingvanillininthesampleldquoFlavoringTPAndashVanillaCustardrdquowithaknownconcentrationofvanillinwas0004Accordingtothedataobtainedwerecommendawavelengthof280nmforthedeterminationofvanillininsamples
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 43
λ nm
Absorban
ce
ndash1Fig 1 Absorption spectrum of vanilin solution in 95 ethanol at concentration 01 mol L (anabsorbinglayerthicknessof10mm)
Sample λnm Tookmg Foundmg S Sх Δх δ
FlavoringTRA 310 10850 104096 00024 00011 00006 00306ndashVanillaCustardAdalyandashVanilla 310 100000 10162 00019 00009 00002 00025
FlavoringTRA 280 100300 96062 00033 00015 00032 00042ndashVanillaCustardAdalyandashVanilla 280 100000 10122 00013 00006 00012 00017
Table 1Testingmethods introduced foundof vanillin in the samplesat310nmby spectrophotometricmethod(n=5p=099SndashstandarddeviationSxndashrelativestandarddeviationΔхndashabsoluteerrorδndashrelativeerror)
ThenthedevelopedmethodwastastedonthesampleldquoAdalyandashVanillardquosamplewithamorecomplexcompositionandanunknownconcentrationofvanillinwastaken The vanillin content in the sample was determined according to thedevelopedmethoditamountedto10ofthetotalmassStudieshaveshownthepossibility of using spectrophotometric analysis for the qualitative andquantitative determination of vanillin Also based on preliminary studies aspectrophotometricprocedurewasdevelopedforthequantitativedeterminationofvanillinbasedonabsorptioninethanolinthewavelengthrange200ndash400nm
References
[1] httpswwwrusnaukacom43_DWS_2015Chimia6_203179dochtm (accessed 25thFebruary2019)
[2] httpswwwcdcgovnioshrtecsdefaulthtm(accessed11stApril2020)[3] AliLPerfettiGDiachenkoGRapidmethodforthedeterminationof342coumarinvanillin
and ethyl vanillin in vanilla extract by reversed-phase liquid 343 chromatography withultravioletdetectionJAOACInt91(2008)383ndash386
[4] БастрыгинаОАЕфременкоЕАЧерноваАПВыделениеванилинаисследованиеегооптическихсвои ствопределениевбиологическомматериалеВХимияихимическаятехнология в XXI веке Материалы XX Международной научно-практическойконференции имени профессора ЛП Кулёва студентов и молодых ученых ТомскНациональныи исследовательскии Томскии политехническии университет 2019с301ndash302
[5] WeastRCHandbookofChemistryandPhysics60thedBocaRatonCRCPress1979p143
44 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
UraniumbelongstothegroupofhazardouselementsItisahighlyharmfulandradioactiveelementtoxictohumansandalllivingorganisms[12]Inhaledwithair it has a particularly destructive effect on the kidneys and as a result ofaccumulationinwhitebloodcellsitcanalsocauseimpairmentoftheimmunesystem[2]Uraniumoccursatseveraldegreesofoxidationhoweverinaqueoussolutionsthemoststableformisuranylion(UO (II))[12]Thepresenceofura-2
niumintheenvironmentiscausedbyamongothersnaturalsoilandrockerosionEnvironmentalpollutionwiththiselementisalsoconstantlyincreasingduetohumanactivitycoalcombustionuraniumoreminingandprocessingthearmsindustryandtheuseofuraniumasnuclearfuelinfissionreactors[3]Itisveryimportanttoconstantlymonitortheconcentrationofuraniumbothinthenaturalenvironment in order to assess its state and safety (especially in the case ofdrinkingwater)aswellasinallstagesofprocessingprocessesassociatedwiththenuclearindustrytoavoidtheoccurrenceofnuclearpollution[13] Scientists have made many attempts to develop research methods todeterminethecontentofuranylcompoundsinliquidsamplesEffortsweremadetousemanyanalyticalmethodsforthispurposeincludingspectrophotometry
Uranyl ion-selective electrode with solid contact
KAROLINAPIETRZAKCECYLIAWARDAK
DepartmentofAnalyticalChemistryInstituteofSciencesFacultyofChemistryMariaCurie-SklodowskaUniversityMariaCurie-SklodowskaSq320-031LublinPolandkarolinapietrzakpocztaumcslublinpl
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 45
AbstractNewallsolidstateuranylion-selectiveelectrodeswithlowdetection
ndash7 ndash1limits(71times10 molL )shortresponsetimegoodselectivityandstable and reproducible potential were developed Many types ofelectrodeswith different active ingredient content in ion-selectivemembrane (bis(244-trimethylpentyl)phosphonium acid Cyanex-272)were testedAs an additive an ionic liquid1-octyl-3-methyl-imidazole chloride was used The optimal composition of theion-selective membrane was chosen from all electrodes based onthedeterminationand comparisonof analyticalparametersof thesensors
Keywordsion-selectiveelectrodesolidcontacturanyl
plasma spectrometry luminescence spectroscopy voltammetry or chromato-graphymethods[2] Duetomanyadvantagesofpotentiometricmethods(amongthemlowercostseasieroperationofdevicesquickresponseandtheabilitytoperformmeasure-ments in flowmode) [3] a numberof potentiometric sensorshave alsobeendeveloped that could be successfully used in this type of research Themostpopularpotentiometricsensorsincludeion-selectiveelectrodes(ISEs)whicharecharacterized by low-energy consumption small size and portability and aresuccessfullywidelyusedforthedeterminationofbothinorganicandorganicionsinclinicalanalysisprocesstechnologyaswellasincontrolthestateofthenaturalenvironment[45]Removaloftheinternalsolutioncontainingthesameanalytetowhich theelectrode is sensitiveresulted in theso-calledsolidcontact ISEswhicharemuchsmallerinsizethantheirpredecessorsaremoreconvenienttouse and more mechanically resistant In this type of sensors however it isimportanttoachievesatisfactorypotentialstabilitywhichisnecessarytoobtainsatisfactoryresults[5]AveryimportantpartofISEsistheion-selectivemem-branewhosecompositiondeterminestheanalyticalparametersofthesensorsResearchers are currently focusing on the production and testing of newsubstancesthatcouldbesuccessfullyusedasmembranecomponentsandsolidcontacts thatwould allow to obtain new sensorswith lower detection limitslongerlifetimeandbetterpotentialstabilityandtodeterminenewpreviouslyunattainableanalytes[4] AstheactivecomponentsofthemembranesensitivetouranylionscientistshavealreadyusedKryptofix22DD(413-didecyl-171016-tetraoxa-413-diaza-cyclooctadecane)[2]Cyanexextractants(bis(244-trimethylpentyl)phosphinicacid bis(244-trimethylpentyl)monothiophosphinic acid and bis(244-tri-methylpentyl)dithiophosphinic)acid[3]DBBP(dibutylbutylphosphonate)andDOPP (di-n-octyl phenylphosphonate) [6] DMSO (dimethylsuphoxide) [7]TTPTP (5678-tetrahydro-8-thioxopyrido[4345]thieno[23-d]pyrimidine-4(3H)one)[8]orTEHP(tris(2-ethylhexyl)phosphate)andTPTU(O-(12-dihydro-2-oxo-1-pyridyl)-NNNN-bis(tetra-methylene)uronium hexafluorophos-phate)[9]
2Experimental
21Reagentsandchemicals
This paper presents research on the design and properties of ion-selectiveelectrodes with solid contact for the determination of uranyl ions Bis(244-trimethylpentyl)phosphonium acid (Cyanex-272) was used as the activecomponentof themembranewhichwasdescribed in the literatureasagooduranylextractant[10]Inordertoensureaconstantpotentialofthiselectrodeandreducetheelectroderesistancetheion-sensitivemembranewasenrichedwithafewpercentadditionof1-octyl-3-methylimidazolechlorideionicliquid
46 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Several types of ion-selective electrodes were prepared using an AgAgClelectrodeasaninternalelectrodewhichdifferinthequantitativeandqualitativecompositionofthemembranesAllcompositionsarelistedinTable1
22Instrumentation
Measurements were made at room temperature using a 16-channel datacollectionsystem(LawsonLabs IncUSA)coupled toa computer in solutionsmixedwithamechanicalstirrerAsilversilverchlorideelectrodewithdoublejunctionwasusedasthereferenceelectrode
3Resultsanddiscussion
The effect of ion-selective membrane composition on the properties of theobtained potentiometric sensors was examined by determining their basicanalyticalparametersincludingslopeoftheelectrodecharacteristicsdetectionlimitmeasuringrange(concentrationrangeinwhichthecourseoftheelectrodecharacteristics isrectilinear)pHrange(inwhich ithasnoeffect forelectrodepotential)andresponsetimeTheobtainedvaluesofthetestedparametersareshowninTable2 Figure1showsthecalibrationcurvesofthetestedelectrodesdeterminedin
ndash7 ndash1 ndash1UO (NO ) solutionsintheconcentrationrange1times10 ndash1times10 molL Asitcan2 3 2
beseeninFig1andTable2allelectrodesweresensitivetouranylionsbutindifferent extend The best response exhibited ISE-3 containing 1 (ww) ofionophore Increasing the ionophore content in themembrane shortened thelinearityrangeofthecalibrationcurveanditssupernenstianslope Theselectivityofthetestedelectrodeswasestimatedbydeterminingtheselec-tivitycoefficients inrelationto interfering ionsForthispurpose theseparate
ndash1solutionmethodwasused(extrapolatingresponsecurves toa =a =1molL )i j
ComparisonofISE-1andISE-3electrodeselectivityisshowninFig2
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 47
Table 1Quantitative and qualitative composition of electrode membranes Cyanex-272 (bis(244-tri-methylpentyl)phosphoric acid) TBP (tri-n-butyl phosphate) and OMImCl (1-octyl-3-methyl-imidazolechloride)
Abbreviation Membranecomposition(ww)ofelectrode Cyanex-272 PVC TBP OMImCL
ISE-1 00 33 620 5ISE-2 05 33 615 5ISE-3 10 33 610 5ISE-4 30 33 590 5ISE-5 50 33 570 5ISE-6 100 33 520 5
48 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Abbreviation Slope Detectionlimit Linearrange Response pHrange2+ ndash1 ndash1ofelectrode mVpa(UO ) molL molL times2
ndash5 ndash5 ndash1ISE-1 297 25times10 5times10 ndash1times10 5ndash8 28ndash42ndash6 ndash5 ndash1ISE-2 292 65times10 1times10 ndash1times10 5ndash8 25ndash60ndash7 ndash5 ndash1ISE-3 298 71times10 1times10 ndash1times10 5ndash8 24ndash60
ndash6 ndash4 ndash1ISE-4(I) 357 31times10 5times10 ndash1times10 5ndash8 ndndash6 ndash6 ndash4ISE-4(II) 242 31times10 5times10 ndash5times10 5ndash8 nd
ndash3 ndash1ISE-5(I) 638 nd 1times10 ndash1times10 5ndash10 ndndash5 ndash3ISE-5(II) 234 nd 5times10 ndash1times10 5ndash10 ndndash3 ndash1ISE-6(I) 733 nd 1times10 ndash1times10 5ndash10 ndndash5 ndash3ISE-6(II) 222 nd 5times10 ndash1times10 5ndash10 nd
Table 2Selectedparametersandtheirdeterminedvaluesoftestedionselectiveelectrodes
Fig 1 Calibration curves of the testedelectrodesobtainedinUO (NO ) solutionsin2 3 2 ndash7the concentration range from 1times10 to
ndash1 ndash11times10 molL
Fig 2Comparisonofselectivitycoefficientspot(log K (UO (II))M) for electrodes ISE-12
(1stcolumn)andISE-3(2ndcolumn)
Inordertoexaminethereversibilityofthepotentialofthetestedelectrodesndash4 ndash1potentialmeasurementsweremadealternatelyinsolutions1times10 molL and
ndash5 ndash11times10 molL ofUO (NO ) TherecordedpotentialreadingsareshowninFig32 3 2
Long-term potential stability and sensor reproducibility were evaluated byndash1determiningtheaveragevalueoftheelectrodepotentialina01molL UO (II)ion2
solutionovertimeforthreeidenticalISE-3Thesemeasurementsweremadetoobservechangesinthepotentialofelectrodeswiththesameconcentrationoveralongperiodoftime(30days)Figure3showsthelong-termpotentialstabilityandreproducibilitydeterminedforthreeidenticalsensors
4Conclusions
Asaresultofthetestsion-selectiveelectrodeforthedeterminationofuranylionswasobtainedwhich iseasy todesignanduseThebestanalyticalparametersexhibitedISE-3containing1ionophoreintheion-selectivemembraneForthis
ndash7 ndash1typeofelectrodesthedetectionlimitof71times10 molL linearityoftheelectrodendash6 ndash1 ndash1calibrationcurve in the range1times10 ndash1times10 molL andresponse time5ndash8s
were obtained In addition the manufactured sensors also showed stablereproducibleandreversiblepotentialandverygoodselectivityinrelationtothetestedinterferents
References
[1] AnsariRMosayebzadehZConstructionofanewsolid-stateU(VI)ion-selectiveelectrodebasedonpolypyrroleconductingpolymerJRadioanalNuclChem299(2014)1597ndash1605
[2] GhanbariMRounaghiGHAshrafNAnuranylsolidstatePVCmembranepotentiometricsensor based on 413-didecyl-171016-tetraoxa-413-diazacyclooctadecane and itsapplicationforenvironmentalsamplesIntJEnvironAnalChem97(2017)189ndash200
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 49
Fig 3 Stability () reproducibility andreversibility () of the potential of ISE-3Standard deviations given on the plot aredeterminedforthesamethreeISE-3
[3] Badr IHA Zidan WI Akl ZF Cyanex based uranyl sensitive polymeric membraneelectrodesTalanta118(2014)147ndash155
[4] BiegCFuchsbergerKStelzleMIntroductiontopolymer-basedsolid-contaction-selectiveelectrodes basic concepts practical considerations and current research topics AnalBioanalChem409(2017)45ndash61
[5] Bobacka J IvaskaA LewenstamA Potentiometric ion sensorsChem Rev108 (2008)329ndash351
[6] ZidanWI Badr IHA Akl ZF Development of potentiometric sensors for the selective2+determinationofUO ionsJRadioanalNuclChem303(2015)469ndash4772
[7] SalehMBSolimanEMGaberAAAAhmedSANovelPVCmembraneuranylion-selectivesensorSensActuatorsB114(2006)199ndash205
[8] SalehMBHassanSSMAbdelAAAbdelNAAnoveluranylion-selectivePVCmembranesensor based on 5678-tetrahydro-8-thioxopyrido[4345]thieno[23-d]pyrimidine-4(3H)oneSensActuatorsB94(2003)140ndash144
[9] HassanSSMAliMMAttawiyaAMYPVCmembranebasedpotentiometricsensorsforuraniumdeterminationTalanta54(2001)1153ndash1161
[10] Prabhu DR Ansari SA Raut DR Murali MS Mohapatra PK Extraction behaviour ofdioxouranium(VI) cation by two phosphorous-based liquid cation-exchangers in room-temperatureionicliquidsSepSciTechnol52(2017)2328ndash2337
50 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Metronidazole(2-methyl-5-nitroimidazole-1-ethanol)isoneofthemostwidelyused nitroimidazole antibiotics Metronidazole is used for the treatment ofinflammatorydiseasescausedbyanaerobicorganismsandsomeprotozoaandforpreventionofdysenterycolibacillosiseimeriosisbalantidiasissalmonellosisenteritissepticemiapost-surgicalcomplications[1ndash3]Oxytetracyclinehydro-chlorideisanantibioticofthetetracyclinefamilyItisoneofthemostcommonlyused antibiotics in poultry because of its low cost and effective [4] Thesecompounds are intensively used in poultry breeding and stockbreedingUnreasonableuseofthesedrugscancauseseriousfoodsafetyissues[5] The veterinary drug Nozemat which includemetronidazole and oxytetra-cyclinehydrochloridewaschosenfortheexperimentsNozematisusedtotreat
Polarographic determination of metronidazole and oxytetracycline hydrochloride in veterinary drug for honey bees
a a bKATERYNAPLOTNIKOVA LILIYADUBENSKA IVANZELENYI
a AnalyticalChemistryDepartmentIvanFrankoNationalUniversityofLvivKyrylaiMefodiaStr879005LvivUkrainekaterina27plgmailcom
b DrohobychPedagogicalLyceumIvanaFrankaStr3682100DrohobychUkraine
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 51
AbstractWehavedevelopedanewpolarographicmethodforthedetermin-ation of metronidazole and oxytetracycline hydrochloride in theveterinarydrugNozematforhoneybeesThetechniqueisbasedonthereductionofpolarographicallyactivecompoundsonamercurydropletelectrodeTheinfluenceofthecomponentsoftheveterinarydrugNozematonthepolarographicdeterminationofmetronidazolewasstudiedItwasfoundthatthereductionofmetronidazoleisnotaffected by glucose and ascorbic acid but is affected by oxytetra-cyclinehydrochloridewhichisreducedtomercurydropletelectrodeatapotentialofndash145VThedevelopedtechniqueischaracterizedbyeaseofsamplepreparationandcost-effectivenessThistechniquehastheabilitytoidentifysimultaneouslyanddeterminatemetronidazoleand oxytetracycline hydrochloride in solution without the use ofseparationandconcentrationmethods
Keywordselectrochemistrymetronidazoleoxytetracycline
hydrochloridepolarographyveterinarydrug
bees and it can be given in unregulated doses Because of this an unknownamountofmetronidazolecangetintothehoneyanditsometimescausessideeffectsofthehumanbodyanditcouldbeofgreatconcernforpublichealth[56]MedicinesforpeoplearemorestringentandbettertestedthanveterinarydrugsTheproblemofthecontroloftheveterinarydrugsisurgentnowadaysVeterinarymedicinescouldbeunauthorizedandtheuncontrolleduseofmedicinesexistsinretailpharmaciesofmedicineorimportedascontrabandfromothercountries The most widespreadof these classes in thequality controlare chromato-graphic [6ndash9] spectrophotometric [10ndash13] and electrochemical methods[14ndash17]Manyoftheknownmethodsforthedeterminationofmetronidazoleandoxytetracyclinehydrochloridehaveanumberofdisadvantagestime-consumingtheuseoforganicsolventsandexpensivereagentsthesideeffectsofexcipientsandotheractivesubstancesElectrochemicalmethodsarepromisingalternativefor the determination of the electroactive substances Their advantages aresimplicityminiaturizationhighsensitivityandrelativelylowcostThereforethesearch for simple express and affordable methods for the determination ofmetronidazoleremainsrelevantOneofthepromisingmethodsofdeterminationisvoltammetry
2Experimental
21Reagentsandchemicals
VeterinarydrugNozemat (manufacturerAPI-SANRussia) is a yellowpowderwithaslighttypicalodorAvailableinlaminatedbagsof25gCompositionper1gof the drug metronidazole 400 mg oxytetracycline hydrochloride 400 mgglucoseascorbicacid MetronidazoleandoxytetracyclinehydrochloridewerepurchasedfromSigmaAldrich(USA)Stockstandardsolutionofmetronidazolefordeterminationwaspreparedbydissolvingtheexactamountofstandardin7mLof2Mhydrochloricin 500 mL volumetric flask Stock standard solution of oxytetracyclinehydrochloride was prepared by dissolving the exact amount of standard indistilledwaterin500mLvolumetricflaskAfterthatthesolutionswereadjustedtothemarkwithdistilledwaterandmixedthoroughly The Britton-Robinson buffer preparationwas as follows 202 g of sodiumtetraboratedecahydrate287mLofglacialaceticacidand176mLofconcen-tratedorthophosphoricacidweredissolvedin10Lvolumetricflask Working solution preparation was as follows an aliquot of stock standardsolutionwasaddedintoa25mLvolumetricflasktoobtainasolutionwiththenecessaryconcentrationthen2mLofBritton-RobinsonbufferwithnecessarypHwasaddedtotheflaskanddistilledwaterwasaddedtothemark AqueoussolutionofNozematwaspreparedasfollowstheexactportionofthetestveterinarydrugwasdissolved ina250mlvolumetric flaskAnaliquotof
52 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Polarogramsof(A)metronidazoleand(B)metronidazolewithoxytetracyclinehydrochloridendash1solutionsat02MBritton-RobinsonbufferbackgroundatpH=96(υ=05Vs c(metronidazole)=
ndash5 ndash5=45times10 Мc(oxytetracyclinehydrochloride)=50times10 М
100mloftheresultingsolutionwasaddedtoa250mlvolumetricflaskandmadeuptothemarkwithwaterAnaliquotof100mloftheresultingsolutionwasaddedtoa250mlvolumetricflask2mlofBritton-RobinsonbufferwithapHof96wasaddedandthevolumewasadjustedtothemarkwithdistilledwater
22Instrumentation
ForpolarographicmeasurementsweuseddigitaldeviceMTechOVA-410 [18]temperature-controlledthree-electrodeamercurydropletindicatorelectrodeasaturatedcalomelreferenceelectrodeandplatinumwireauxiliaryelectrodeTheaccuracyofthepotentialmeasurementis1mVTheuncertaintyofcurrentmeasu-rement is 01 The employed mercury droplet electrode had the following
ndash4 ndash1characteristicsm=594times10 gs τ=10 min in 02 M NH Cl We used cyclic4
voltammetryforthestudyoftheelectrochemicalprocess WeusedMV870DIGITAL-pH-MESSERATpH-meterformeasuringpHofthesolutions Theobtainedworkingsolutionswereintroducedintothecellanddeoxyge-natedwithargonfor10minPolarogramswererecordedintherangeofpotentialsfrom00tondash16V
3Resultsanddiscussion
Previously it was found that using the Britton-Robinson buffer with pH = 96metronidazoleisreducedwiththeformationofacharacteristiconeirreversiblepeakatndash064V(Fig1A)UsingpolarographywithfastpotentialscanitwasfoundthatmetronidazoleisreducedonmercurydropletelectrodeintherangeofpH20to105ThepeakrecoverycurrentofthemetronidazolereachesthemaximumvalueatpH9ndash10againstthebackgroundofatheBritton-Robinsonbuffer
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 53
Underpre-selected conditions theeffectof some foreign substanceson thepolarographicdeterminationofmetronidazolewasinvestigatedSubstancesthatare componentsofdrugswere studiedglucose ascorbicacidoxytetracyclinehydrochlorideGlucose and ascorbic acid are not reduced atmercury dropletelectrodeanddonotchangetheappearanceofthepolarogramandpolarographiccharacteristics of the recovery of metronidazole As can be seen from Fig 1oxytetracycline hydrochloride is reduced to mercury droplet electrode andchangestheappearanceofthepolarogramandpolarographiccharacteristicsofthe recovery of metronidazole With the addition of oxytetracycline hydro-chloridetherecoverypeakofmetronidazoledecreasesandslightlyshiftstomorenegativepotentials The composition of the drug is relatively complex excipients affect theanalyticalsignaloftherecoveryofcompoundssototakeintoaccountthematrixeffectusedthemethodofmanyadditives QuantitativelytransferredthesolutionofNozemattothecell(exactvolume)removed dissolved oxygen for 10 min and took polarograms in the range ofpotentialsfrom00tondash16VAliquotsofstandardmetronidazolesolutionwereintroducedintothecelltoobtainasolutionwithagivenconcentrationofadditive
ndash5 ndash5metronidazole 10times10 M to 70times10 M As with the determination of themetronidazole aliquots of a standard oxytetracycline hydrochloride solutionwereaddedtothecellwithsolutionofNozemattoobtainasolutionwithagiven
ndash6 ndash6additiveconcentrationfrom70times10 Mto50times10 M(Fig2) In Table 1 are shown metrological characteristics of the determination ofmetronidazoleandoxytetracyclinehydrochloride inveterinarydrugRecoverywascalculatedFormetronidazoletherecoveryis97andforoxytetracyclinehydrochloridetherecoveryis103Analyticalperformanceofthetechniqueisgoodfordeterminationveterinarydrugs
54 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2Polarogramsof(A)metronidazolereductionatdifferentmetronidazoleconcentrationsand(B)oxytetracyclinehydrochloridereductionatdifferentoxytetracyclinehydrochlorideconcentra-tionsandtheircorrespondingcalibrationgraphs
Theaccuracywasverifiedbytheldquoadded-foundrdquomethodAliquotsofstandardsolutionofmetronidazoleweremadeina250mlvolumetricflasktoobtaina
ndash5solutionofagivenconcentrationof33times10 Mandthesolutionofoxytetracyclinendash5hydrochloridetoobtainasolutionofagivenconcentrationof15times10 M2mlof
Britton-RobinsonbufferwithpH96wasaddedtoflaskwithstirringandadjustedtothemarkwithwaterTheanalysisprocedureofmodelsolutionissimilartoanalysis procedure of the solution of Nozemat The calculated amount ofmetronidazolebythemethodofmultipleadditivesinthetestedmodelsolutionisinagreementwiththeamountthatwasintroducedintothesample
4Conclusions
The new polarographic method for the determination of metronidazole andoxytetracyclinehydrochlorideintheveterinarydrugNozematforhoneybeeswasdeveloped We conducted principal component analysis of veterinary drugNozemattoassesstheoveralleffectforthedeterminationofmetronidazoleWefoundthatoxytetracyclinehydrochlorideisreducedtomercurydropletelectrodeThismethodhastheabilitytoidentifysimultaneouslyanddeterminatemetro-nidazole and oxytetracycline hydrochloride in solution without the use ofseparationandconcentrationmethodsOnemoreofadvantagesoftechniquearefastprocedureofanalysissimplesamplepreparationlowcostthepossibilityofminiaturization
References
[1] Antibiotic and Chemotherapy Finch R Greenwood D Whitley R (edits) AmsterdamElsevier2006p292ndash299
[2] MitrowskaKPrzyczynyiskutkizakazustosowania5-nitroimidazoliuzwierzątktorychtkanki lub produkty przeznaczone są do spozycia przez ludziMed Weter 71 (2015)736ndash742
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 55
Metronidazole Oxytetracycline hydrochloride
PeakspotentialV ndash065 ndash144CorelationcoefficientR 099892 099922
ndash1 4 4SlopebmicroАM 656times10 176times10Δb 1763 400InterceptamicroА 2238 0219Δa 0075 0009
ndash1 ndash5 ndash5cmolL 342times10 125times10ndash1cmgg 389 413
Recovery 97 103
Table 1Validationparametersofthemethodofmetronidazoleandoxytetracyclinehydrochloridedetermi-nationinsolutionsofNozematbythemethodofmanyadditives
[3] VermaPNamboodiryVMishraSBhagwatABhoirSAstabilityindicatingHPLCmethodfor the determination of Metronidazole using Ecofriendly solvent as mobile phasecomponentIntJPharmPharmSci5(2013)496ndash501
[4] Cervini P Ambrozini B Machado LCM Ferreira Garcia AP Cavalheiro Gomes ETThermal behavior and decomposition of oxytetracycline hydrochloride J Therm AnalCalorim121(2015)347ndash352
[5] DangBNAnhNTKKyLXThaiPKAntibioticsintheaquaticenvironmentofVietnamsourcesconcentrationsriskandcontrolstrategyChemosphere197(2018)438ndash450
[6] QuintanillaPHettingaKABeltranMCEscricheIMolinaMPVolatileprofileofmaturedTronchon cheese affected by oxytetracycline in raw goat milk J Dairy Sci 103 (2020)6015ndash6021
[7] Chen F Yu L Jingdong P Xiang W Huanjun P Yu C Yan H Study on simultaneousdetermination of three nitroimidazole residues in honey by high performance liquidchromatographyndashresonanceRayleighscatteringspectraMicrochemJ141(2018)423ndash430
[8] Hernandez-MesaM Cruces-Blanco C Campana GA Simple and rapid determination of5-nitroimidazolesandmetabolitesinfishroesamplesbysalting-outassistedliquid-liquidextractionandUHPLC-MSMSFoodChem252(2018)294ndash302
[9] Xiu-ChunGZhao-YangXHai-HuiWWen-YiKLi-MingLWen-QingCHong-WeiZWen-HuiZMolecularlyimprintedsolidphaseextractionmethodforsimultaneousdeterminationofsevennitroimidazolesfromhoneybyHPLC-MSMSTalanta166(2017)101ndash108
[10] ТеплыхАНИлларионоваЕАКоличественноеопределениеметронидазоласпектро-фотометрическимметодомСибирскиймедицинскийжурнал5(2009)48ndash50
[11] ZheltvayOIZheltvayIISpinulVVAntonovichVPSpectrophotometricdeterminationofmetronidazoleandtinidazoleusingcopper(II)complexesJAnalChem68(2013)663ndash668
[12] Youssef AK Saleh MS Abdel-Kader DA Hashem Facile DY SpectrophotometricdeterminationofmetronidazoleandsecnidazoleinpharmaceuticalpreparationsbasedontheformatioonofdyesIntJPharmPharmSci6(2015)103ndash110
[13] Sversut RA Vieira JC Rosa AM Amaral MS Kassab NM Salgado H ValidatedspectrophotometricmethodsforsimultaneousdeterminationofoxytetracyclineassociatedwithdiclofenacsodiumorwithpiroxicaminveterinarypharmaceuticaldosageformArabianJChem13(2020)3159ndash3171
[14] Nikodimos Y Electrochemical determination of metronidazole in tablet samples usingcarbonpasteelectrodeJAnalMethodsChem(2016)361294
[15] Srivastava AK Upadhyay SS Rawool CR Punde NS Rajpurohit AS Voltammetrictechniques for the analysis of drugs using nanomaterials based chemically modifiedelectrodesCurrAnalChem15(2019)249ndash276
[16] Sahu G Voltammetric behaviour of metronidazole at a composite polymer membraneelectrodeOrienJChem26(2010)81ndash86
[17] Yang Y YanW Guo YWang X Zhang F Yu L Guo C Fang G Sensitive and selectiveelectrochemicalaptasensorviadiazonium-couplingreactionforlabel-freedeterminationofoxytetracyclineinmilksamplesSensorsandActuatorsReports2(2020)1ndash7
[18] httpchemlnueduuamtechdeviceshtml(accesed21stJune2020)
56 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
AnimportantpartofanyanalysisthatsignificantlyaffectsthefinalresultsisthesamplepreparationThelowconcentrationofbiologicallyactivecompoundsandthepresenceofaccompanyingcomponentspreventdirectanalysisofthesamplewithcomplexmatrixcompositionTraditionalmethodsofliquidandsolid-phaseextractionhaveaplentyoflimitationssuchashighlytime-consumingprocedureslarge volume of samples expensive cartridges toxic organic solvents andchallenges in automating the process Therefore the application of extractiontechniquesemployinglowamountofsolvents(microextractionmethods)andthelow toxicity extractantes has become the main research direction in recentyears[12] Solid-phasemicroextraction(SPME)wasproposedbyPavlishinin1989[3]Onevariantofthismethodistousethinrodswithvariouspolymercoatingssuch
Application of microextraction techniques combined with chromatographic methods for the analysis of complex objects
VLADISLAVDEEVELENABESSONOVALIUDMILAKARTSOVA
InstituteofChemistrySaint-PetersburgStateUniversityUniversitetskyprospect26198504PeterhofSaint-PetersburgRussiahitchervmailru
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 57
AbstractThelowconcentrationofanalytesandthepreventionofthematrixinfluence requires a stage for extraction and concentration of thestudiedcompoundsTheclassicalmethodsofliquidandsolid-phaseextractionhavemanylimitationsthatpreventtheiruseinsomecasesMicroextraction techniques are becoming more widespread WestudiedthepossibilityofusingionicliquidstoextractpesticidesfromwatersampleswiththeirsubsequentHPLS-MSdeterminationTheinfluenceonthedegreeofextractionofsuchparametersasanatureofionicliquidsanddispersersolventtheiramountssaltconcentrationvolumeratioofionicliquidsandwatersampledilutionoftheionicliquidsextractwithmethanolwasperformedBesidesconditionsofsolid-phase microextraction of volatile organic compounds fromurine samples obtained from healthy donors and donors withprostate cancer have been found The analysis of volatile organiccompoundsbyGC-MSfollowedbychemometricprocessingallowedachievingahighvalueofbinaryclassificationaccuracy(91)
Keywordschemometricsdispersiveliquid-liquid
microextractionsolid-phase
microextraction
asdivinylbenzenepolydimethylsiloxanepolyacrylateandpolyethyleneglycolwhichappliedtothesurface[4]Thepolymersorbentisplacedintheequilibriumheadspaceaboveacondensedphaseofthesampleandthevolatilecompoundsareextracted Liquidmicroextractionconsistsofusingsmallamountsofliquid(extractant)inequilibriumwiththegasorliquidphaseofthesampleDispersiveliquid-liquidmicroextraction(DLLME)isavariantofliquidmicroextractionTheessenceofthemethodisasfollowsextractantisdissolvedinthephaseofadispersingsolventandthemixtureisrapidlyinjectedintothesamplevolume[5]Inthiscasethedispersing solvent is dissolved and a ldquocloudrdquo of extractant is formed A largesurfaceareacontributestomasstransferprocessesThecombinationofDLLMEwiththeuseof ionic liquids(ionic liquids)asextractantsreducestheharmfulimpactontheenvironment[6] Sothegoalofthisstudywastheapplicationofmicroextractionmethodsfortheanalysisofrealsamples
2Experimental
21Reagents
Deionizedwaterwas obtained at the AQUILON D 301 deionizer (Russia) Allchemicals and reagents (the highest commercially available purity) werepurchasedfromReachimBakerAcrosorganicsandSigmaAldrich
22Instrumentation
HPLCanalysiswascarriedoutusinganHPLCLCMS-8030(Shimadzu)withatriplequadrupole mass-selective detector with electrospray ionization Analysis ofvolatileorganiccomponents(volatileorganiccompounds)ofurinesampleswasmade by GCMS-QP2010 SE (Shimadzu) Chemometric data processing wasperformedusingRStudio
23DeterminationofvolatileorganiccompoundsinurinesamplesbyGC-MSmethod
ForSPMEofvolatileorganiccompounds inurinesamplewasusedfibercoatedwith a polydimethylsiloxane (PDMS) The volatile organic compounds wereextractedontofibercoatingfor20minat50degCThentheanalytesweredesorbedinto the gas chromatography for 4 minutes at a temperature of 250degCChromatographic separation was carried out on a HP-5 capillary column(30mtimes250μmtimes025μm)usingtemperatureprogrammingmodeThetempera-
ndash1tureofovenwasincreasedfrom50degCupto250degCatarateof10degCmin Tionsourcewas200degCMassspectrometrywasusedinSIMmode(mz=35ndash900)
58 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
24ConditionsforLCMSMSdeterminationofpesticides
Separation of pesticides was performed by HPLCMSMS with positiveelectrosprayionizationoncolumnZorbaxBonusRP35μm(21times100mm)with40mMammoniumacetateandmethanolasmobilephaseAandBrespectivelyThefollowinggradientelutionwasapplied20ndash85B(8min)85B(8ndash15min)85ndash95B(150ndash155min)95B(155minus180min)95ndash20B(180minus185min)
ndash1Thevelocityof themobilephasewas03mlmin Thevolumeof the injectedsamplewas 20 microlMS detection capillary voltage +45 kV spray gas velocity
3 ndash1 3 ndash13dm min flow rate and drying gas temperature 15 dm min and 250 degCrespectively
25Selectionofconditionsfordispersiveliquid-liquidmicroextractionofpesticides
The influence of the natures of ionic liquids ([C MIM][PF ] [C MIM][NTf ]4 6 6 2
[C MIM][BF ])andthedispersingsolvent(methanolacetonitrileacetone) the6 4
weightoftheionicliquids(0060ndash0200g)thevolumeofthedispersingsolvent(02ndash10ml)onthedegreeofpesticidesextractionwerestudiedTheinfluenceofthepH(5422)theconcentrationofNaCl(0040ndash0200g)andextractiontime(1-6min)wereinvestigated The effects of different ionic liquids and disperser solvents on DLLMEprocedures were investigated and optimized by using standard solutions ofpesticidesIndetailasolutionofionicliquidsinadispersingsolventwaspreparedand rapidly injected into the aqueous sample solution (2ml) followed bytreatment for 2 min in an ultrasonic bath cooling at ndash4degC for 10 mincentrifugationfor10minat3500rpmandcollectionofionicliquidsThewaterphasewas separatedandanalyzedbyHPLC-MSThe ionic liquidsextractwasdilutedinmethanolandanalyzedbyHPLC-MS
3Resultsanddiscussion
31Microextractionofpesticides
One of the important tasks of environmental monitoring is to control traceconcentrationsofpesticidesinwatersamplesTechniqueofcombiningseveralpesticideshasbecomemorewidespread inagriculture It allowsreducing thetotalconcentrationoftheappliedcompoundsandtodecreasetheadaptabilityofpathogens and insects Therefore the analysis of real samples requires apreliminarystageofselectiveanalytesextractionandconcentration ExtractinganddispersingsolventsarebothimportantinDLLMEofanalytesTheinfluenceofthenatureofthedispersingsolvent(methanolacetonitrileandacetone) and the extractant (imidazolium ionic liquids [C MIM][PF ]4 6
[C MIM][BF ]and[C MIM][NTf ])onthedegreeofextractionofpesticideswas6 4 6 2
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 59
studied This parameter was controlled by the residual concentration ofpesticidesinthewaterphaseafterextractionThebestresultswereobtainedforionic liquids [C MIM][PF ] as an extractant and acetonitrile as a dispersing4 6
solvent Thenextstepwastoselecttheamountofionicliquids(0060ndash0200g)andthevolume of acetonitrile (02ndash10 ml) It was found that the highest degree ofextractionofanalyteswasachievedbyusing020gofionicliquidsand03mlofacetonitrile It was shown that the degree of extraction of selected pesticides does notdependonpHofthewatersamplewhichconfirmsthepartitionmechanismofextractionThedegreeofextractionofcarbofosincreasedslightlywithanincreaseinthesaltconcentrationandreachmaximumbyweightto4(008g) ItisknownthatthehighviscosityofionicliquidshavehinderedtheprocessesofelectrosprayionizationDilutionofthesamplewithmethanolby3timesgivesthebestresult(thesignalintensitywas373to845ofthesignalwithoutionicliquids) ThustheconditionsofDLLME-ionicliquidsextractionofpesticidesfromwatersampleswere found The limits of detection for pesticideswere from007 to
ndash1019ngml thereproducibilityofpeakareaswerefrom3to5theextractionrecoverywascloseto100
32SPMEofvolatileorganiccompoundsfromurinesamples
Oneof the importantdirections is the search for criteriaofearlydiagnosisofcancer Obtaining characteristic profiles of volatile organic compounds fromurinesamplescanhelpdevelopanon-invasivemethodforearlydiagnosisofthedisease ForthiswestudiedtheinfluenceofanumberoffactorsonthetotalnumberofpeaksandthetotalpeakareaTheywerethetemperatureatwhichthevaporandcondensedphasesofurinewerebalanced(30ndash60degC)thepreheatingtimeofthesample(10ndash40min)NaClconcentration(30ndash133wv)andthesorptiontimeonthePDMScoating(5ndash30min) Anincreaseinthepreheatingtemperatureofthesampleto500degCledtoanincreaseinthenumberofsignalswhichdidnotchangewithafurthertempera-tureincreasingNextparameterwasthetimeofachievementequilibriumofthevapor and condensed phases The largest number of peaks was observed at40minbutthisgreatlyincreasedthetimeofanalysisandsowechose20minAlsowestudiedthedesaltingeffectofsodiumchlorideonefficiencyofextractionThebestvolatileorganiccompounds sorptionwasachievedbyadding133saltingagent It is also shown that thenumberofpeaksdidnot changeafter20minutesofsorption Thus to obtain the characteristic profiles of urine samples the followingconditionswereselected1333NaClwasaddedtotheurinesample(3ml)
60 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
heatingfor20minat50degCthensorptionofvolatileorganiccompoundsonPDMSfibercoatingat50degCfor20min Undertheselectedconditionsweobtainedvapor-phaseprofilesof52urinesamples (32 normal and 20 pathology) Prior to performing chemometricprocessing of chromatographic profiles of urine samples preliminary datapreparation is necessary [7] The baseline was removed and the peaks werealignedusingdynamictimewarpingwithcontrollingbymassspectra ThePCAmodelwasbasedon52aligned characteristicprofilesThere is asatisfactoryseparationofdataintotwoclustersinthescoresplotrelativetothefirstandsecondprincipalcomponent(Fig1) The original data set (52 samples) was randomly divided into calibration(13pathology21normal)andtest(7pathology11normal)setsThenthePLS-DAmodelwasbuiltusingthecalibrationsetanditspredictiveabilitywasevaluatedusingthetestsetTheprocedurewasrepeated100timesTheaveragevaluesofsensitivityspecificityandaccuracyinthiscasewere9594and91
4Conclusions
Thepossibilityofusingimidazoliumionicliquids([C MIM][PF ])asextractants4 6
forquantitativeextractionandconcentrationofpesticidesundertheconditionsofDLLMEisshownThedegreeofconcentrationwas28ndash33whichallowedreaching
ndash1thedetectionlimits(006ndash019ngml )belowthemaximumpermissibleconcen-trationThepossibilityofnon-invasivediagnosisofprostatecancerbySPMEofvolatileorganiccompounds inurine isshownChemometricprocessingofgaschromatographic profiles using PLS-DA and PCA methods allowed achievingclassificationaccuracyvaluesmorethan90
Acknowledgments
ThisworkwassupportedbytheRussianFoundationforBasicResearchprojectno18-53-80010BRICS_t and the Russian Science Foundations (Projects 19-13-00370) We are grateful to
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 61
Fig 1Scoresplotrelativetothefirstandsecondprincipalcomponent
Resource Education Center in Chemistry of St Petersburg State University for the providedequipment
References
[1] Rutkowska M Płotka-Wasylka J Sajid M Andruch V Liquidndashphase microextractionAreviewofreviewsMicrochemJ149(2019)103989
[2] JaliliVBarkhordariAGhiasvandAAcomprehensivelookatsolid-phasemicroextractiontechniqueAreviewofreviewsMicrochemJ152(2020)104319
[3] ArthurCLPawliszynJSolidphasemicroextractionwiththermaldesorptionusingfusedsilicaopticalfibersAnalChem62(1990)2145ndash2148
[4] SchmidtKPodmoreISolidphasemicroextraction(SPME)methoddevelopmentinanalysisof volatile organic compounds (VOCs) as potential biomarkers of cancer JMol BiomarkDiagn6(2015)1000253
[5] Mousavi L Tamiji Z Khoshayand MR Applications and opportunities of experimentaldesign for the dispersive liquidndashliquidmicroextractionmethod ndash A review Talanta190(2018)225ndash356
[6] MarcinkowskaRKoniecznaKMarcinkowskiLNamiesnikJKloskowskiAApplicationofionic liquids inmicroextractiontechniquesCurrent trendsandfutureperspectivesTrACTrendsAnalChem119(2019)115614
[7] WehrensRChemometricswithRMultivariateDataAnalysisintheNaturalSciencesandLifeSciencesBerlinSpringer2011
62 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Theself-assembledtwo-dimensionalmonolayers(2DSAMs)ofvariousmolecules(eg graphene [1] MoS [2] rubrene [3]) offer beneficial properties for the2
constructionofnano-electronic andnano-opticaldevicesThe topological andchemicalcharacterizationof2DSAMsiscrucialtogatherinformationaboutthearrangementofdepositedmoleculesandtheirinteractionwiththesubstrateThistaskrequiresanalyticaltechniqueswith(sub)nanometerspatialresolutionandup to single-molecular detection sensitivity Only few techniques meet therequirements and one of them is tip-enhanced Raman spectroscopy (TERS)whichcombinestheexcellentspatialresolutionofscanningprobemicroscopy(SPM) and chemical sensitivity of surface-enhanced Raman scattering (SERS)spectroscopy[45] TheSERS spectroscopyutilizesplasmonicmetal nanostructures to cause ahighlocalenhancementoftheelectricfieldintheirclosevicinityviathesurfaceplasmonresonance(SPR)effectThelocalelectricfieldcausesanincreaseofthe
The development of reference probe system for tip-enhanced Raman spectroscopy
MARTINKRA LMARCELADENDISOVA PAVELMATE JKA
DepartmentofPhysicalChemistryFacultyofChemicalEngineeringUniversityofChemistryandTechnologyPragueTechnickaacute516628Prague6CzechRepublicMartinKralvschtcz
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 63
AbstractThetip-enhancedRamanspectroscopy(TERS)isamodernanalyticaltechniquewithanoutstandingspatialresolutionandchemicalsensi-tivityTheseparametersmainlydependon the structural integrityand chemical purity of employed plasmonic scanning probe tipsUsuallyeachtipistestedbeforeTERSmeasurementsusingcommer-ciallyavailablereferencesamplesHowevertheirpriceandrelativelyshortexpirationdatemustbeconsideredwhenplanningaresearchbudgetWedevelopedaproceduretoproduceself-madereferenceprobesamplesfortestingTERStipsusingcopper(II)phthalocyanineonaAunanolayerwhichispreparedbythermalvacuumevaporationofAuonaSiwaferOurresultsshowthatthepreparedsystemenablesrepeateddetectionofwell-resolvedTERSspectraThecollectedTERSspectraandspectralmapsexhibitsomedegreeofvariabilitywhichmaybeduetovariousphoto-inducedprocessesanditmustbeconsi-deredwhileperformingTERSmeasurements
Keywordscopper(II)
phthalocyaninescanningtunnelling
microscopyresonanceRaman
spectroscopysurface-enhancedRaman
spectroscopytip-enhancedRaman
spectroscopy
Ramanscatteringfrommoleculesboundtothemetalby6ndash8ordersofmagnitude[6]TheenhancementallowsSERSspectroscopytobeusedforsingle-moleculardetectionHoweverRamanmicroscopeshave limitedspatialresolutionbythelightdiffractionwiththeachievableresolutionbeingaroundhalfoftheexcitationwavelengthOntheotherhandthespatialresolutionofSPMtechniquesislimitedonlybythedimensionsoftheapexofthescanningprobetipwhichmayevenbeatomicallysharpByutilizingSPMtipandsubstratemadefromplasmonicmetalsan artificial ldquohotspotrdquomay be createdwith its position and dimensions beingdefinedbythetipItopensthepossibilitytocollectstronglyenhancedRamanspectrafromtheareapreciselylocalizedbellowthetipandthusovercometheopticaldiffraction limitTheartificialhotspotmayberelocatedbymovingthesamplebelowthetipwhichisthefoundationofTERSmapping[578] A successful TERS experiment requires an optimal combination of variousparameters the most important of which are tip sharpness and purity [7]Areferencesampleconsistingofaflatplasmonicnanolayerwithattachedprobemolecules is frequently used to check the state of the tip before using it forexperiments Unfortunately commercially available TERS standards areexpensiveandhaveanexpirationdateofseveralmonths Thegoalofthisstudywastofindapreparationprocedurewhichwouldbeableto produce cheap reference samples for repeated detection of intense TERSspectraAcombinationofaAunanolayeronaSisubstratepreparedbythermalvacuumevaporationwithadsorbedcopper(II)phthalocyanine(CuPc)whichisamoleculewithhighRamancross-sectionwastested[910]Copper(II)phthalo-cyanine known as phthalocyanine blue is a synthetic blue pigment and isfrequently used in paints It has been studied as a potentialmaterial for theconstruction of organic solar cells and other photoelectronic devices [11] AsindicatedbyitscolourCuPcexhibitsseveralabsorptionbandswithinthevisibleregion The effects of a transition to excited electronic states and subsequentluminescencemaybeobservable inRamanmeasurements[12]Au isahighlysuitablemetalforthesampleasitisboththermallyandelectricallyconductivewhichlimitsthelocalheatingofthesampleduringthemeasurementsandenablesthe use of scanning tunnelling microscopy (STM) for tip-surface interactionfeedback[13]
2Experimental
21Reagentsandchemicals
Thesubstrateforthesamplewaspreparedbythermalvacuumevaporationofgoldonsilicon(100)waferFirsta5nmthickCradhesionlayerwasdepositedon
ndash1thewaferfollowedby100nmofAuThedepositionratewas4nmmin forCrand ndash18nmmin for Au The base pressure of the evaporation system was below
ndash65times10 mbarFollowingthepreparationproceduredescribedbyJiangetal[14]
64 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
thecleansubstratewasimmersedintoasaturatedsolutionofCuPc(˃99SigmaAldrich USA) in dimethylformamide (˃98 Lach-ner CZ) for at least 12h atambienttemperatureSubsequentlythesamplewasremovedfromthesolutionrinsedwithMilli-Qwaterandmethanol(paPentaCZ)anddriedwithair
22Instrumentation
TheRamanSERSandTERSspectrawererecordedusingRamanspectrometerInVia Reflex (Renishaw UK) equipped with lasers emitting at two differentexcitation wavelengths 633nm (136mW max power output) and 785nm(204mWmaxpoweroutput)Thespectrometerhasathermoelectricallycooled
ndash1CCDdetectorwithaspectralresolutionof2cm and4microscopeobjectiveswith5times20times50timesand100timesmagnitudeForTERSexperiments the laserbeamwasredirectedtotheSPMplatformInnova-IRIS(BrukerUSA)viaasystemoflightguidesElectrochemicallyetchedAuTERS-STMtips(BrukerUSA)wereusedforallTERSmeasurements The spectra were processed using the Spectragryph software (F MengesldquoSpectragryph - optical spectroscopy softwarerdquo Version 1214 2020httpwwweffemm2despectragryph) Using this software all collectedspectra were treated by a Savitzky-Golay noise filter automatic baselinecorrectionspikeremovalandpeaknormalization
3Resultsanddiscussion
31Ramanmeasurementsofcrystallinecopper(II)phthalocyanine
AtfirsttheRamanspectraofpureCuPcwerecollectedtoprovidereferencedatawhileusingboth633and785-nmexcitationlasers(Fig1)Bothspectraexhibit
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 65
Fig 1RamanspectraofCu(II)phthalocyanineincrystallineformmeasuredat633(top)and785-nm(bottom)excitationThespectraareoffset
ndash1vibration bands in the 500ndash1600 cm region with slight differences in theirintensityratiosThe633-nmexcitationallowstheobservationofadditionalbands
ndash1inthe2000ndash3000cm region(onaluminescencebackground)whichoriginatefrom the resonance Raman effect as the excitation energy overlaps with theQ-bandofCuPc[12]MoreovertheprocessofelectronicexcitationmaylowertheD symmetry of CuPc during resonance Raman scattering and previously4h
forbiddenbandsmaybecomeobservable[15]Theluminescentbackgroundhasandash1maximumaround2200cm whichcorrespondstoamolecularemissionbandat
735nmEventhoughthespectrameasuredwiththe785-nmlaserlinedonotexhibitapparentresonanceenhancementapre-resonanceRamanenhancementmayoccur
32Surface-enhancedRamanmeasurementsofcopper(II)phthalocyaninelayeronagoldsubstrate
The prepared sample of CuPc on a Au layer was analysed using the Ramanmicroscope Both excitation laserswere used to obtain SERS spectra (Fig 2)whichwerecomparedtothespectraofapurecrystallineCuPc Thepositionsof bands in SERS spectra closelymatch their positions in thespectraofbulkCuPcHoweveraslightshiftofsomespectralbandsisobservable
ndash1(eg1528rarr1532cm )whichmaybeattributedtotheinteractionbetweenCuPcand the Au substrate The disappearance of luminescence background andresonance-enhancedbandsinthespectrumat633-nmexcitationalsosuggeststhemolecule-metalinteractionandthetransferofenergyfromCuPcmoleculestothesubstrateMoreovertherearevariationsintherelativeintensitiesofbands
ndash1whichdependontheexcitationenergyegthebandat1309cm isenhancedinSERSspectraatthe785-nmexcitationwhencomparedtothespectraofpureCuPcorevenSERSspectraat633-nmexcitation
66 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2SERS spectra of Cu(II) phthalocyanineon aAu layermeasured at 633 (top) and785-nm(bottom)excitationThespectraareoffset
33Tip-enhancedRamanmeasurementsofcopper(II)phthalocyaninelayeronagoldsubstrate
SERS microspectroscopy is a diffraction-limited technique as it provides anaveragedinformationaboutmoleculesintheilluminatedareaofseveralsquaremicrometers Meanwhile TERS spectra are collected from an area of tens ofnanometers and they contain specific information about the local moleculararrangement topography of the underlyingmetal and properties of the localelectric field between the tip and the substrate Therefore a higher spectralvariabilityshouldbeexpected Several TERS mapping experiments were carried out using both 633 and785-nmexcitationwithvaryingexperimentalparameterssuchasthenumberanddistance betweenmeasured points acquisition time number of acquisitionslaserpoweretc TheTERSspectrameasuredat633-nmexcitationexhibited lowersignal tonoiseratioandreproducibilityAsaconsequenceTERSmappingwasimpossibleandonlyafewone-pointTERSspectrawereobtained(Fig3) TheTERSspectracollectedat785-nmexcitationcontainedahighernumberofwell-resolvedbandsMoreoverthespectrawerestableintimeandsotheTERSmappingwaspossibleThe twopresentedTERSspectraareaveragesofTERSmaps which contained 16 and 80 points with 600 and 300-nm spacingrespectively(Fig4) Thespectracollectedusingbothexcitationwavelengthsexhibitavariabilityinrelative intensities and positions of bands between themeasured points ThevariabilitymaybeattributedtothelocalorientationofCuPcmoleculesbetweenthe tip and the Au surface and the properties of strongly enhanced andnon-homogeneous electromagnetic field which depend on the tip-surfacedistancetheirmorphologyandrelativepositionMoreovertheusedexcitation
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 67
Fig 3TwoexamplesofTERSspectraofCu(II)phthalocyaninemeasuredat633-nmexcitationThespectraareoffset
wavelengthsareclosetoabsorptionbandsofCuPcandthestrongelectricfieldmay give rise to photo-induced effects These effects include the electronicexcitationofCuPctohigherstateschargetransferbetweentheCuatomandthephthalocyaninering ionizationof themoleculeand formationofradicalsThephoto-inducedprocessesarelikelytoplayabiggerroleinTERSspectradetectedat633-nmexcitationduetotheoverlapwithQ-bandofCuPcwhichmaybethecauseoftheirlowersignaltonoiseratioandreproducibility
4Conclusions
Thedeveloped referenceprobe systemofCuPc adsorbedon aAu surfacehasprovedtobesuitablefortheintendeduseasitenabledthedetectionofintenseandwell-resolvedSERSandTERSspectraTheAulayerpreventsoverheatingofthesampleandallowsfortheuseofSTMTheSERSspectrawereinagoodmatchwith thespectraofpureCuPcAslight shiftof somebandsandchange in theluminescent background indicated the interaction between CuPc and the AusurfaceTheTERSexperimentsresultedinspectralmapswithhighintensitiesofindividualspectraIncreasedvariabilitybetweenmeasuredpointswasobservedPossiblesourcesofthevariabilityarephoto-inducedprocessesthatmayoccurinthestronglyenhancedelectricfieldTheseeffectsareaknownfeatureofTERSmeasurements and they offer valuable insight into the photophysics andphotochemistryofCuPcinteractingwiththeAusurfaceThedependenceofTERSspectra on experimental parameters and the preparation procedure of thereferencesampleshouldbefurtherstudied
Acknowledgments
ThisworkwassupportedfromthegrantofSpecificuniversityresearchndashA2_FCHI_2020_039
68 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 4TwoexamplesofaveragedTERSspectraofCu(II)phthalocyaninemeasuredwith785-nmexcitationThespectraareaveragesfromTERSmapsincluding16(top)and80(bottom)measuredpointsThespectraareoffset
References
[1] Mas-BallesteRGomez-NavarroCGomez-HerreroJZamoraF2DmaterialstographeneandbeyondNanoscale3(2011)20ndash30
[2] ZengHCuiXAnopticalspectroscopicstudyontwo-dimensionalgroup-VItransitionmetaldichalcogenidesChemSocRev44(2015)2629ndash2642
[3] SchultzJFLiLMahapatraSShawCZhangXJiangNDefiningmultipleconfigurationsofrubreneonaAg(100)surfacewith5A spatialresolutionviaultrahighvacuumtip-enhancedRamanspectroscopyJPhysChemC124(2020)2420ndash2426
[4] WhitemanPJSchultzJFPorachZDChenHNJiangNDualbindingconfigurationsofsubphthalocyanineonAg(100)substratecharacterizedbyscanningtunnelingmicroscopytip-enhanced Raman spectroscopy and density functional theory J Phys Chem C 122(2018)5489ndash5495
[5] ShaoFZenobiRTip-enhancedRamanspectroscopyprinciplespracticeandapplicationstonanospectroscopicimagingof2DmaterialsAnalBioanalChem411(2019)37ndash61
[6] ArocaRSurface-EnhancedVibrationalSpectroscopyHobokenWiley2006[7] KumarNMignuzziS SuWRoyDTip-enhancedRamanspectroscopyprinciplesand
applicationsEPJTechInstrum2(2015)9[8] BailoEDeckertVTip-enhancedRamanscatteringChemSocRev37(2008)921ndash930[9] BovillAJMcConnellAANimmoJASmithWEResonanceRamanspectraofα-copper
phthalocyanineJPhysChem90(1986)569ndash575[10] Shaibat MA Casabianca LB Siberio-Perez DY Matzger AJ Ishii Y Distinguishing
polymorphsofthesemiconductingpigmentcopperphthalocyaninebysolid-stateNMRandRamanspectroscopyJPhysChemB114(2010)4400ndash4406
[11] SzybowiczMRunkaTDrozdowskiMBałaWGrodzickiAPiszczekPBratkowskiAHightemperaturestudyofFT-IRandRamanscatteringspectraofvacuumdepositedCuPcthinfilmsJMolStruct704(2004)107ndash113
[12] CaplinsBWMullenbachTKHolmesRJBlankDAFemtosecondtonanosecondexcitedstatedynamicsofvapordepositedcopperphthalocyaninethinfilmsPhysChemChemPhys18(2016)11454ndash11459
[13] SaccoAImbraguglioDGiovannozziAndreaMPortesiCRossiAMDevelopmentofacandidatereferencesampleforthecharacterizationoftip-enhancedRamanspectroscopyspatialresolutionRSCAdv8(2018)27863ndash27869
[14] JiangSChenZChenXNguyenDMatteiMGoubertGVanDuyneRPInvestigationofcobaltphthalocyanineatthesolidliquidinterfacebyelectrochemicaltip-enhancedRamanspectroscopyJPhysChemC123(2019)9852ndash9859
[15] MelendresCAMaroniVARamanspectraandnormalcoordinateanalysisoftheplanarvibrationsofironphthalocyanineJRamanSpectrosc15(1984)319ndash326
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 69
1Introduction
The aim of this work has been develo-pmentofanewvoltammetricmethodforthe determination of 23-dimercapto-1-propane-sulfonic acid (DMPS) Fig 1Investigationwasdonetoobtainrelevantinformation about complexingbehaviorofDMPStowardsleadions Lead is one of heavy metals which can cause irreversible neurologicalproblems [1 2]DMPS is a synthetic antidotewith two thiol groups used fortreatmentofpoisoningbyheavymetals[3ndash6]StrongcomplexingpropertieshighwatersolubilityandnegligiblesideeffectsarethemostimportantadvantagesofDMPS[47]
Fig 1 Chemical structure of 23-dimercap-to-1-propane-sulfonicacid
Determination of heavy metal poisoning antidote 23-dimercapto-1-propanesulfonic acid using silver solid amalgam electrode
ab ab bc bMARTACHOIN SKA VOJTE CHHRDLICKA BEATRIZRUIZREDONDO JIR IBAREK aTOMA S NAVRATIL
a JHeyrovskyacuteInstituteofPhysicalChemistryoftheCzechAcademyofSciences Dolejškova21553182thinsp23Prague8CzechRepublicmartachoinskagmailcomb UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova20308128thinsp43Prague2CzechRepublic
c UniversityofValladolidPlazadeSantaCruz847002ValladolidSpain
Abstract23-Dimercapto-1-propane-sulfonic acid (DMPS) was investigatedusingdirectcurrentvoltammetry(DCV)differentialpulsecathodicstrippingvoltammetry(DPCSV)differentialpulseanodicstrippingvoltammetry(DPASV)andeliminationvoltammetrywithlinearscan(EVLS)atapolished(p-AgSAE)andatameniscusmodifiedsilversolidamalgam electrode (m-AgSAE) EVLS confirmed two consecutivereductions with coupled protonelectron transfer VoltammetrictitrationsofDMPSwithPb2+provedcomplexformationwithlimits
minus1ofquantification(LOQs)anddetection(LODs)03and01micromolL atminus1m-AgSAE and 08 and 03micromolL at p-AgSAE respectively
Determination of DMPS in commercial drug Dimaval and humanurine samples confirmed practical applicability of the developedmethod
Keywordscathodicstripping
voltammetry23-dimercapto-1-pro-
panesulfonicacideliminationvoltammetry
withlinearscansilversolidamalgam
electrodeunithiol
70 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Voltammetry was chosen as a determination technique due to its highsensitivity and selectivity speed low costs [8] Thiol groups in DMPS can beoxidatively chemisorbed on solid amalgam electrode It can be used as anaccumulation step for cathodic strippingvoltammetry [9ndash11]Moreover solidamalgamelectrode (SAE)was chosen as theworking electrodebecauseof itspropertiesashighsignaltonoiseratiowidepotentialwindowandabilitytoreachlowlimitsofdetection(LOD)[12ndash13]
2Experimental
21Reagentsandchemicals
Allsolutionswerepreparedusingdeionizedwater(Milli-Q-GradientMilliporendash1PragueCzechRepublic)withconductivitylt005microScm Britton-Robinsonbuffer
solutionspHrangefrom2to12werepreparedbymixingtheproperamountsof02MNaOH(alkalinesolution)andof004MH BO 004MH PO and004M3 3 3 4
CH COOH(allLachemaCzechRepublic)acidicsolutionTheacidicsolutionwas3
preparedbydissolutionof1235gofH BO pa088mLofH PO (85)paand3 3 3 4
1435mLofCH COOH(99)pa in500mLofdeionizedwaterThealkaline3
solution was prepared by dissolution of 3995 g of NaOH pa in 500mL ofdeionizedwater(allLachemaCzechRepublic) StocksolutionofDMPSwaspreparedbydissolving10mgofsolid23-dimer-capto-1-propanesulfonic acid monohydrate pa (Merck Czech Republic) in100mLofdeionizedwaterForthepreparationofthemodelsamplesolutiononecapsuleofdrugDimaval(HeylGermany)contains100mgofDMPSwasdissolved
ndash1in10LofdeionizedwatertotheDMPSconcentrationof0531mmolL Twomodel samplesofDimavalwerepreparedbydilutionof theabove-mentioned
ndash1solutionwithBritton-Robinsonbuffersolutiontoconcentrations10micromolL andndash1of 10micromolL respectively Urine model samples were prepared by mixing
Britton-Robinsonbuffer solutionwithurine samples obtained fromvolunteer(manhealthy30yearsold)inratio11SamplepHwasadjustedbyadditionof
ndash1proper amount of 02molL NaOH Before each measurement oxygen wasremoved for 5minbynitrogenbubbling (purity class 46MesserTechnogasPragueCzechRepublic)
22Instrumentation
Measurementswereperformedusingtwotypesofworkingelectrodesmeniscusmodified silver solid amalgam electrode (m-AgSAE working surface of
20382plusmn0025mm α lt 005) and polished silver solid amalgam electrode2(p-AgSAEworkingsurfaceof0196plusmn0015mm αlt005)Ag|AgCl|3MKClwas
used as the reference electrode andplatinumwire (Oslash1mm)wasused as theauxiliary electrode (both from Elektrochemicke detektory Czech Republic)Measurementswereperformedatlaboratorytemperature(25plusmn2degC)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 71
The pH was measured using pH-meter Jenway 3505 with combined glasselectrodetype924001(BibbyScientificLimitedUK)Voltammetricmeasure-ments were performed using the computer-controlled Eco-Tribo Polarograph(Polaro-Sensors Czech Republic) Software used for measurements wasMultiElChem 33 forWindows XP7810 (J Heyrovsky Institute of PhysicalChemistryoftheCzechAcademyofSciencesCzechRepublic)
3Resultsanddiscussion
OptimumconditionsformeasurementswereobtainedbyseriesofmeasurementinwiderangeofpHvaluesandtestingvariouscleaningproceduresOptimumpotentialof accumulation (E ) and timeof accumulation (t )ofDMPSwereacc acc
adjusted for differential pulse cathodic stripping voltammetry (DPCSV) atp-AgSAEandm-AgSAE ThedependencebetweenpeakheightandconcentrationofDMPSatp-AgSAEhasalogarithmicshapewhichcorrespondstotheaccumulationprocessattheelectrodesurfaceThelineardependencewasobservedintheDMPSconcentra-
ndash1 ndash1tionsfrom03micromolL to20micromolL Peakshifttowardsnegativepotentialwithincreasing concentration of DMPS corresponds to the metal-thiol bond andinfluenceofelectrodesurfacestructureonthisbondOnthecontrarysignalsonm-AgSAEweremorestableandthedependencebetweenconcentrationofDMPSandsignalwasalmostlinearinwholetestedrangeofconcentrations The developedmethodwas tested inmodel samples of Dimaval and urineFoundamountsofDMPSwereingoodagreementwithdeclaredcontentsusingbothelectrodesHowevertherepeatabilityofsignalsregisteredusingp-AgSAEinurinesamplesweresignificantlyworsethanthoseinDimavalsamplesItcanbecausedbycomplicatedbiologicalmatricesandfoulingeffectsofurine ELSV measurements confirmed two consecutive reductions of DMPS inadsorbed state At m-AgSAE signal were at about minus415 mV and minus440 mVrespectively and at p-AgSAE at about minus790 mV and minus830 mV respectively
ndash1 ndash1Reductionsatm-AgSAEatthescanratesfrom80mVs to640mVs havebeencontrolledbyakineticprocessatminus400mV Inanodicscansonm-AgSAEonlyonepeakwasvisibleatabout‒390mVItcorrespondswiththeoxidationofmercuryelectrodesurfaceontheelectrodeinthepresenceofDMPSandwithdiffusionfromthebulksolutionofproductsAtp-AgSAEnosignificantanodicsignalwasfound ThelastpartoftheresearchrevealedvoltammetricbehaviorofDMPSinthe
2+presence of Pb Voltammetric titration was investigated by DPCSV anddifferential pulse anodic stripping voltammetry (DPASV) during consecutive
ndash1 ndash1additionsof1mmolL ofPb(NO ) into100micromolL DMPSsolutioninacetate3 2
bufferofpH50InabsenceofPb2+ontheelectrodesurfaceHg(DMPS)complexisformedduringtheaccumulationstepDuringtheanodicscanthereisonlyonewelldevelopedreductionpeak(Fig2A)
72 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ndash1Fig 2DPCSandDPASvoltammogramsof10micromolL ofDMPSinacetatebufferpH=5correspond-2+ingto[Pb DMPS]ratiosof(A)01(B)11and(C)21Uppercurvecorrespondstothecathodic
scanE =0mVt =15sLowercurvecorrespondstoreverseanodicscanwitht =15satacc acc accndash1E =minus1000mVν=20mVs (Ref[16])acc
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 73
2+ WhenPb DMPSratioisequalto11twooxidationandthreereductionpeakswereregistered(Fig2B)PeakA ataboutndash300mVcorrespondstoformationofred
0Pb(DMPS)complexThiscomplexwasfurtherreducedtothePb (Hg)atabout2+ndash500mV(C )ReductionpeakoffreePb wasalsoregistered(B )Oxidationred red
peaksA andB correspondtoreverseprocessesandC isnotpresentbecauseox ox ox
nofreeDMPSispresentinthesolutionAtratio21thereisnofreeDMPSinthesolution however excess of lead ions As a consequence A and B peakred ox
2+increasedIncreaseofB correspondstothedepositionofPb duringtheaccu-ox
mulationstepPeaksBredandCredremainedpracticallyunchanged(Fig2C) Voltammetric titrationconfirmed themechanismof formationcomplexesofPb(DMPS)Hg(DMPS)andPb(Hg)Italsoconfirmedpossibilityofdetermination
2+Pb andDMPSinthesamesolution
4Conclusions
ValidationinmodelsampleofdrugDimavalandhumanurinespikedwithDMPSconfirmed that this method can be used for clinical purposes Voltammetric
2+titration of DMPS by Pb ions proved that it can be used for simultaneousdetermination of the drug and heavy metal ions in human urine Moreoverobtained LODs were two orders lower than those in the previously reportedvoltammetricmethod[14](Table1)
Acknowledgments
ResearchwascarriedoutwithintheframeworkofSpecificUniversityResearch(SVV260560)TheauthorsthanktheCzechScienceFoundation(GACRprojectNo20-01589S)
References
[1] AnHHLuchakMCopesRLeadtoxicityAsystematicreviewofrecentlypublishedcasesClinToxicol53(2015)757ndash758
[2] KimYLustMRKreimerbirnbaumM23-Dimercaptopropane-1-sulfonate(DMPS)inthetreatmentoflead-poisoningFasebJ2(1988)A1820ndashA1820
[3] AposhianHVDMSAandDMPS ndashwater-solubleantidotesforheavy-metalpoisoningAnnuRevPharmacol23(1983)193ndash215
74 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Table 1ComparisonofvoltammetricmethodsforDMPSdetermination(LDR
Method Workingelectrode Lineardynamic LOQ LOD Refminus1 minus1 minus1 rangemicromolL micromolL micromolL
LSV glassy-carbonelectrode 18ndash140 41 14 [14] modifiedwithmulti-walled 260ndash690 carbonnanotubes DPCSV p-AgSAE 03ndash20 08 03 thisworkDPCSV m-AgSAE 01ndash10 03 01 thiswork 10ndash100
[4] BjorklundG Crisponi G Nurchi VM Cappai R Djordjevic AB Aaseth J A review oncoordinationpropertiesof thiol-containingchelatingagents towardsmercury cadmiumandleadMolecules24(2019)3247
[5] DonnerAHrubyKDMPSinthetreatmentofacuteandchronicheavy-metalpoisoningActaMedAust14(1987)10ndash10
[6] DonnerAHrubyKPirichKKahlsPSchwarzacherKMeisingerVDimercaptopropan-sulfonate(DMPS) inthetreatmentofacute lead-poisoningVetHumToxicol29 (1987)37ndash37
[7] Blanusa M Varnai VM Piasek M Kostial K Chelators as antidotes of metal toxicityTherapeuticandexperimentalaspectsCurrMedChem12(2005)2771ndash2794
[8] BarekJMoreiraJCZimaJModernelectrochemicalmethodsformonitoringofchemicalcarcinogensSensors-Basel5(2005)148ndash158
[9] Josypcuk B FojtaM Yosypchuk O Thiolatemonolayers formed on different amalgamelectrodesPartIIPropertiesandapplicationJElectroanalChem694(2013)84ndash93
[10] YosypchukBMarecekVPropertiesofthiolatemonolayersformedondifferentamalgamelectrodesJElectroanalChem653(2011)7ndash13
[11] Alvarez JMF SmythMRCathodic strippingvoltammetryofpyridine-2-thiolandsomerelated-compoundsAnalyst114(1989)1603ndash1605
[12] DanhelABarekJAmalgamelectrodesinorganicelectrochemistryCurrOrgChem15(2011)2957ndash2969
[13] Fadrna R Polished silver solid amalgam electrode Further characterization and appli-cationsinvoltammetricmeasurementsAnalLett37(2004)3255ndash3270
[14] ZiyatdinovaGKGrigorevaLVBudnikovGKElectrochemicaldeterminationofunithioland lipoic acid at electrodesmodifiedwith carbonnanotubes J Anal Chem64 (2009)185ndash188
[15] HrdlickaVChoinskaMRedondoBRBarekJNavratilTDeterminationofheavymetalpoisoning antidote 23-dimercapto-1-propanesulfonic acid using silver solid amalgamelectrodeElectrochimActadoiorg101016jelectacta2020136623
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 75
Fig 1Structureofcanagliflozin
1Introduction
Canagliflozin is a selective sodium-glucosecotransportertype2inhibitorused for the treatment of type 2 dia-betes mellitus Canagliflozin inhibitssodium-glucose cotransporter type 2present in proximal tubules of the
Canagliflozin oxidation study using electrochemical flow cell and comparison with hydrogen peroxide oxidation
a a bFILIPVYMYSLICKY TOMA S KR IZ EK JAKUBHER T
a DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublicfvymyslickygmailcomb ZentivaGroupasUKabelovny13010237Prague10CzechRepublic
AbstractBystandardstheeffectonoxidationofanactivesubstanceistestedusinghydrogenperoxidesolutionatelevatedtemperatureinastresschamberfor1ndash7daysAnalternativewaytostudytheeffectofoxida-tion on an active substance is to use an electrochemical flow cellSolutionwith active substance flows at low flow rate into a smallreactorwheretheactivesubstanceisoxidizedonworkingelectrodesurfaceTheelectrolytestreamwiththeoxidizedactivesubstanceisthen directed to the sample collector Products of electrochemicaloxidationareanalyzedbyhighperformanceliquidchromatographywithultravioletndashvisiblespectrophotometrydetectionCanagliflozinhasbeenusedbecauseitsmaindegradationpathwayisoxidationThedesign of experiments approach was used to explore the experi-mentalspaceandoptimizeexperimentalconditionsofoxidationTheresultsoftheoxidationstudyperformedintheelectrochemicalflowcellwerestatisticallycomparedwiththeresultsofastandardstudyusinghydrogenperoxidesolutionThemostsuitableconditionsforelectrochemical oxidation were found Electrochemical oxidationproducedcomparableamountsofimpuritiesaschemicaloxidationwithhydrogenperoxide
KeywordscanagliflozindesignofexperimentselectrochemicalflowcellHPLCoxidation
76 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
kidneywhichrestrictsglucoseabsorptioninthekidneytherebyincreasingtheurinaryexcretionofglucoseandloweringthelevelofglucoseintheblood[1]TheformulaofcanagliflozinisC H FO SthestructureofcanagliflozinisinFig1The24 25 5
IUPAC name of canagliflozin is (2S3R4R5S6R)-2-[3-[5-(4-fluoro-phenyl)-thiophen-2-ylmethyl]-4-methyl-phenyl]-6-hydroxymethyltetrahydro-pyran-345-triol[2]CanagliflozinisawhitepowderinsolubleinwaterbutverysolubleinorganicsolventslikemethanolordimethylsulfoxideCanagliflozinissoldundertradenameINVOKANA Manyauthorshavestudiedtheelectrochemicalpropertiesofactivesubstancesin the literature One example is the study of electrochemical behaviour andoxidationofbromhexineThesepropertieswerestudiedusingdifferentialpulsevoltammetryandcyclicvoltammetryonacarbonelectrodeTheresultsofelectro-chemicalmethodswerecomparedwithhighperformanceliquidchromatography(HPLC)analysis[3]Anotherexampleisthestudyofelectrochemicalbehaviourand degradation study performed on the active substance atomoxetineDegradation was studied using differential pulse voltammetry and cyclicvoltammetryonacarbonelectrodeTheresultswerealsocomparedwithHPLCanalysis[4]Electrochemicalmethodsareusedmainlytostudythemechanismofoxidationbutinthisworktheelectrochemicalmethodwasusedtodegradetheactivepharmaceuticalingredient In the stability studies of active pharmaceutical ingredient properties theinfluence of temperature pH light and oxidation is studied [5] By TheInternationalCouncilforHarmonisationofTechnicalRequirementsforPharma-ceuticals forHumanUse (ICH) standards the influenceof oxidationon activepharmaceutical ingredient is studied using hydrogen peroxide at roomtemperatureorincreasedtemperatureinthestresschamberduring1ndash7days[6]Analternativewaytostudytheinfluenceofoxidationonactivepharmaceuticalingredient is using electrochemical flow cellwhere an electrolytewith activepharmaceuticalingredientisdrivenbylowflowrateintothesmallreactorInthesmallreactortheactivepharmaceuticalingredientisoxidizedonthesurfaceoftheworkingelectrodeThestreamofelectrolytewithoxidizedactivepharma-ceuticalingredientisdriventothesamplecollectorProductsofelectrochemicaloxidationareanalysedbyHPLCUVVISThedesignofexperimentsapproachwasusedfordevelopmentofanalternativemethodofoxidationofcanagliflozinusinganelectrochemical flowcellThedesignofexperimentsapproachwasusedtoexplore the experimental space of the method and to find the optimalexperimentalconditionsofelectrochemicaloxidationofcanagliflozin
2 Experimental
21Materialandreagents
Canagliflozin(ZentivaCzechRepublic)999methanol(HoneywellGermany)98ammoniumdihydrogenphosphate(Sigma-AldrichJapan)35ortho-phos-phoricacid(PentaCzechRepublic)25ammonia(LachnerCzechRepublic)30hydrogenperoxide (LachnerCzechRepublic)water forchromatography
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 77
Fig 2Schemeoftheelectrochemicalflowcell(1)input(2)workingelectrode(3)gasket(4)refe-renceelectrode(5)counterelectrode
wasobtainedbypurifyingdemineralisedwaterusingMilliporetypeSynergyUVpurificationinstrument
22Instruments
An Agilent 1290 HPLC system (Agilent Technologies Germany) with highpressure pump autosampler thermostat and DAD detector was used for allexperimentsThePinnacleDBbiphenylcolumn(100times21mm19micromRestekUSA)wasusedforseparationIntheHPLCmethod10mMammoniumdihydrogenphosphatebufferpH=25wasusedascomponentAandmethanolascomponentBofthemobilephaseThegradientprogramwassetasfollowst(min)B01555010551790229023152515Theflowrateofthemobilephasewas
ndash104mlmin and the injection volume was 2μl The detector operated at awavelengthof220nmTheautosamplertemperaturewassetat20degCandthecolumntemperatureat60degCTheEmpowersoftwarewasusedforevaluationForelectrochemicaloxidationelectrochemicalflowcellfromALS(Japan)wasusedGlassycarbonelectrode(=6mm)andsilversilverchlorideelectrodewereusedasworkingandreferenceelectroderespectivelyTheschemeofelectrochemicalflowcellisinFig2ElectrodeswereconnectedwithpotentiostatPalmSens3fromPalmsens (Netherlands) AnElmasonic S15Hultrasonic bath fromElma (Ger-many)wasusedforsamplepreparationForpHmeasurementspHmeterJenway3540fromJenway(UnitedKingdom)wasused
3Resultsanddiscussion
AtthedevelopmentofthemethodforthestudyofcanagliflozinoxidationusingelectrochemicalflowcellitwasfirstnecessarytofindtheoptimalconditionsofelectrochemicaloxidationThedesignofexperimentsapproachwasusedChosen
78 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ndash1Fig 3Cyclicvoltammogramofcanagliflozin(concentrationofcanagliflozin11mgml electrolyte300 mM ammonium dihydrogen phosphate pH = 40 and methanol (11 vv) and scan rate
ndash1001Vs )
independentvariablesandtheirlevelswereconcentrationofelectrolyte(100200300mM)pHofelectrolyte(406080)cellsize(50100200500microm)and
ndash1flowrate(0102504mlh )ThereducedcombinatorialdesignwasusedIntheModde12 software aworksheet containing 11 experimentswas created Theworkingpotentialof12Vwasselectedbasedoncyclicvoltammetryofcanagli-flozininFig3Fromthisfigureitcanbeseenthattheoxidationofcanagliflozinoccursintheregionfrom11Vto14VAllexperimentswereperformedwith
ndash111mgml canagliflozin samples The glassy carbon electrodewas used as aworkingelectrodeandthesilversilverchlorideelectrodewasusedasareferentelectrode The canagliflozin samples oxidized in the electrochemical flow cellunder theexperimentalconditionsgivenby theworksheetweremeasuredbyHPLCwithUVVISdetectionDependentvariablespeakareasofimpuritiesandpercentage of peak areas of impurities obtained from chromatograms wereevaluated by the partial least squaremethod in theModde 12 software Thevariableimportanceintheprojectionplottoolwasusedforinterpretationofthedata as a whole The significance values of the independent variables were
ndash1evaluatedbufferpH=137flowrateof125mlh bufferconcentration061mMandcell size04micromFromthis tool itwasconcluded that theelectrochemicaloxidationofcanagliflozinisthemostaffectedbythepHoftheelectrolyteandflowrateoftheelectrolyteUsingtheoptimizertoolthemostsuitableconditionsfor
ndash1the oxidation of canagliflozin were evaluated flow rate 01 ml h 300 mMammoniumdihydrogenphosphate pHof electrolyte40 and cell size500micromUsingonefactoratthetimeapproachthedependenceofthecellsizeonthetotalsumofimpuritieswastested(Fig4A) Itisvisiblefromthegraphthatthesmallerthecellweusethemoreoxidationproducts are formed Based on the graph the most suitable conditions were
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 79
Fig 4(A)Optimizationofelectrochemicaloxidationconditionsdependenceofsumofimpuritiesoncellsize(B)Chromatogramofasampleoxidizedundermostsuitableconditions
ndash1adjustedtoflowrate01mlh 300mMammoniumdihydrogenphosphatepHofelectrolyte40andcellsize12micromUnderthemostsuitableconditionsarepeat-ability test was performed by ten independent oxidation experiments Therelativestandarddeviationofthepercentageareaofcanagliflozinwas164atasignificant level of 095 The chromatogram of sample oxidized under mostsuitableconditionsisinFig4BThestandardstudyoftheeffectofoxidationoncanagliflozinusinghydrogenperoxideaccordingtoICHguidelineswasperfor-medThestudywasperformedundertwosetsofexperimentalconditionsInthefirstcaseasolutionof50methanolwiththeadditionof3H O wasusedIn2 2
thesecondcasetheconditionsintheelectrochemicalflowcellweresimulatedA300mMammoniumdihydrogenphosphate pH40 andmethanol in a ratio11(vv)withtheadditionof3H O wasusedSamplesfortheoxidationstudy2 2
werestressedinastabilitychamberfor13and7daysattheconstanttempe-ratureof50degC ThetotalsumsofimpuritiesformedduringchemicaloxidationusinghydrogenperoxideinbothmediawerecomparedasisshowninFig5AItisobviousthat
80 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 5(A)Acomparisonofastandardoxidationstudyusinghydrogenperoxidein50methanolwith added buffer andwithout them (B) Chromatogramof sample oxidized electrochemically(C)Chromatogramofsampleoxidizedchemically
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 81
ammoniumphosphatesuppressesoxidationofcanagliflozinThereasonofthisphenomenonisunknownFig5BandFig5CshowchromatogramsofsamplesoxidizedelectrochemicallyandchemicallyrespectivelyItcanbeseenthatfiveimpuritieswereformedbybothtypesofoxidationhoweverindifferentamounts
4Conclusion
AnalternativemethodfortheoxidativestudyofcanagliflozinwasdevelopedThedesign of experiments approach was used in the method development ThedevelopedmethodworkswithRSDof165(α=095)Oxidationofcanagliflozinbythedevelopedmethodproducedfiveimpuritiesthatareidenticalwiththoseproducedusingthestandardoxidationstudywithhydrogenperoxide
Acknowledgments
ThisworkhasbeensupportedbyCharlesUniversityResearchCentreprogramNoUNCESCI014SVV260560projectandpharmaceuticalappliedresearchcenter(TheParc)
References
[1] ChaoECCanagliflozinDrugsFuture36(2011)351ndash357[2] NislySAKolanczykDMWaltonAMCanagliflozinanewsodium-glucosecotransporter2
inhibitorinthetreatmentofdiabetesAmJHealthSystPharm70(2013)311ndash319[3] Turchan M Jara-Ulloa P Bollo S Nunez-Vergara LJ Squella JA Alvarez-Lueje A
VoltammetricbehaviourofbromhexineanditsdeterminationinpharmaceuticalsTalanta73(2007)913ndash919
[4] Perez-OrtizMMunoz C Zapata-Urzua C Alvarez-Lueje A Electrochemical behavior ofatomoxetineanditsvoltametricdeterminationincapsulesTalanta82(2010)398ndash403
[5] Baertschi SW Alsante KM Reed RA Pharmaceutical Stress Testing Predicting DrugDegradationLondonInformaHealthcare2011
[6] RignallA ICHQ1A(R2) stability testing of newdrug substance andproduct and ICHQ1CstabilitytestingofnewdosageformsInICH Quality Guidelines An Implementation GuideATeasdaleDElderRWNims(Eds)HobokenWiley2017p3ndash44
82 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
AlthoughDNArepresentsarelativelystablecomponentfromthechemicalpointofviewitremainsconstantlyexposedtoalargenumberofchemicalorphysicalagentscausingchemicalchangesinDNAmoleculesthatoccurintheenvironmentoraremajororminorproductsofcellularmetabolism[1] One-electronoxidationoftheDNArepresentsadamagingprocesswheretheloss of an electron (oxidation) fromduplexDNA results in the formationof anucleobase radical cation (electron ldquoholerdquo) that is subsequently consumed inchemicalreactionsthatoftenleadtomutationsAdefiningcharacteristicoftheone-electronoxidationofDNAisthepreferentialreactionattheguaninemoietythatisdetectedasstrandcleavagefollowingchemicalorenzymatictreatmentoftheoxidizedDNA[2ndash3]
Novel hybrid electrochemical DNA biosensor for monitoring oxidative DNA damage via oxidationreduction signals of low molecular weight double-stranded DNA
MICHALAUGUSTINVLASTIMILVYSKOCIL
UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova812843Prague2CzechRepublicmichalaugustinnaturcunicz
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 83
AbstractDeoxyribonucleicacid(DNA)representsamajortargetmoleculeformanydamagingagentscausingunfavorablechangesinastructureofDNAmoleculethatbindandinteractwithDNAThusahighdemandforreliabletoolsregardingabettercomprehensionofthenatureofDNAdamagingprocessesstillrepresentsoneofthemaingoalsinthisareaHereinwedescribeadevelopmentofanovelhybridelectro-chemicalDNAbiosensorbasedonanldquoedge-planerdquopyrolyticgraphiteelectrode (EPPGE) in connectionwith an elementaryoptimizationprocessprovidingacloserresolutionoftheredoxprocessesoflowmolecularweightdouble-strandedDNA(dsDNA)attheEPPGESub-sequentanalyticalapplicationincorporatinganemploymentofthemodel structure K [IrCl ] (representative of transition metal2 6
complexes)andevaluationofitsdamagingeffectinrelationtoDNAbymeansof linear sweepvoltammetry resp square-wavevoltam-metryarealsopresented
KeywordsbiosensordamageDNAgraphitevoltammetry
DNA-based electrochemical biosensors are successfully used in variousapplicationssuchasmonitoringandevaluatingthemechanismsof interactionbetweenDNAandvariousdrugsordamagingagentsrapidmonitoringoftracemetalsorpollutantspresent in theenvironmentordirectmonitoringofDNAhybridizationprocesses[4] Theelectrochemicalactivityofnucleicacids(boththenativehigh-molecularonesaswellasoligonucleotides)isingeneralreferredtotheelectroactivityofitscomponents ndash nucleobases and sugar residues At mercury-based electrodesadenine and cytosine residues undergo reduction processes close to ndash14 V(againstSCE)inneutralorweaklyacidicmedium(givingrisetothepeakCA)Ontheotherhandallbaseshavebeenreportedtobeelectrochemicallyoxidizedatcarbonelectrodesbutonlyadenineand(particularly)guanineoxidationsignalshavebeenwidelyutilizedinelectrochemicalDNAbiosensors[5] In2017theelectrochemistryofnucleicacidsachievedanimportantmilestoneasthereductionoftheDNAoligonucleotideswasperformedataldquobasal-planerdquopyrolytic graphite electrode which provided wide potential window allowingboththeelectrooxidationaswellastheeletroreductionofthenucleobasesatasingleelectrodefortheveryfirsttimeDespitethesefindingsutilizationoftheaforementionedbiosensorintermsofanalyticalapplicationshasyettobeverifiedandremainsunclearuptothisdate[6] TheaimoftheproposedcontributionisapresentationofthedevelopmentprocessandsubsequenttestingofanoveltypeofhybridelectrochemicalDNAbiosensoranditsverificationasareliableanalyticaltoolintermsofmonitoringDNAdamage
2Experimental
21Reagentsandchemicals
Low molecular weight double-stranded DNA (dsDNA) derived from salmonspermwasobtainedfromSigma-AldrichGermanyStocksolutions(01mgmL)
of dsDNA were prepared in a 01 molL phosphate buffer of pH=74 (PB)Dipotassium hexachloroiridate (K [IrCl ]) was purchased from Sigma-Aldrich2 6
GermanyStocksolutions(0001molL)ofK [IrCl ]werepreparedinthePB2 6
22Apparatus
Voltammetric measurements were performed using the μAutolab IIIFRA2potentiostatgalvanostat(EcoChemieTheNetherlands)drivenbyaNOVA111software(MetrohmAutolabSwitzerland)Allmeasurementswerecarriedoutina three-electrode system using an ldquoedge-planerdquo pyrolytic graphite workingelectrode(EPPGE)withanelectroactivesurfacediameterof3mm(BASJapan)asilver|silver chloride reference electrode (Ag|AgCl|sat KCl) and a platinum
84 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
counterelectrode(ElektrochemickeDetektoryCzechRepublic)ina20mLglassvoltammetriccellatambienttemperature
23Preparationofthebiosensor
PriortotheeverymeasurementsurfaceoftheEPPGEwasmechanicallycleanedbygentlewipingoftheelectrodeonthesoftpolishingpadrinsedwithdistilledwaterAfterwardstheelectrodewasrinsedwithdistilledwaterandplacedinthePBforthesubsequentelectrochemicalactivationElectrochemicalactivationwasperformed in thePBbyapplyingpotentialof15V for240swithoutstirringApotentialpulseinworkingrangeofpotentials(00ndash15V)wasthenapplied Additional electrochemical activationwasperformed in the solutionof the
3minus4minusredoxindicator([Fe(CN) ] )byconsecutivecyclingintherangeofpotentials6
from10tondash08V(15scans)andfrom055tondash015V(10scans) The electrochemical DNA biosensor based on the EPPGE (dsDNAEPPGEbiosensor)was prepared by the adsorption of dsDNAon the EPPGEOptimal
parametersofthedsDNAadsorptionwereaconcentrationof01mgmLinthePB
(c )adepositionpotentialof07V(E )andanadsorptiontimeof5ming(dsDNA) dep
(t )withoutstirringthesolutionads
Atlasttheelectrodewasimmersedinthesolutionoftheredoxindicatorandtheconsecutivecyclingintherangeofpotentialsfrom055tondash015V(20scans)wasperformedinordertosecurethestabilityoftheoxidationreductionsignalsofdsDNAattheEPPGE
24Procedures
Theexperimentalparameterswereasfollowssquarewavevoltammetry(SWV)inthePBwithapulseamplitudeof20mVafrequencyof50Hzascanrateof750mVsandapotentialstepof15mVlinearsweepvoltammetry(LSV)inthePBwithscanratesof02ndash10Vandapotentialstepof24mVAllcurveswere
recordedthreetimes(n=3)
3Resultsanddiscussion
Sincetheclosestresolutionoftheprocessesassociatedwiththeelectroreductionof singleDNAcomponents at pyrolytic graphitehasbeenperformedwith theldquobasal-planerdquopyrolyticgraphiteelectrode(BPPGE)wehavedecidedtotakeovercorrespondingexperimentaltechniqueandconditions(LSVscanrateof10Vssteppotentialof24mV)attheverybeginningofouroptimizationprocesswiththeEPPGE[6] Inthisparticularcaseitispossibletonoticetheoccurrenceofthetwomixedvoltammetricpeaksatthedefaultexperimentalconditions(greenlineFig1A)selectedforthereductionofdsDNAattheEPPGEBygraduallydecreasingthe
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 85
Fig 1Baseline-correctedLSVrecordingscorrespondingtothereductionofdsDNAattheEPPGEfordifferentvaluesofscanrate(02ndash10VsFig1A)respbaseline-correctedLSVrecordingscorres-pondingtothereductionofdsDNAattheEPPGEandthenegativetestperformedunderthesameexperimentalconditionswithintheblanksolution(phosphatebuffer)atthebareEPPGE(03VsFig1B)
scan rate the optimal conditions (νle03Vs) were found and the mutualseparationof the signalswas allowedndash characterizedby thepresenceof twosinglewell-developedvoltammetricpeaksatpotentialsofndash175Vrespndash190V(03Vs orange line Fig 1A) Taking into account previous work regardingprocessesassociatedwiththereductionofDNAatthemercuryelectrodesrespBPPGE we can assume that the peak appearing at the potential of ndash175 Vcorresponds to themixedpeak for the reductionof the cytosine and adenineresidueswithindsDNA(peakCA)[5ndash6] Closer resolution of the second voltammetric peak appears to be farmoreproblematic Regarding our previous study we have discovered that theutilizationofdifferentE fortheadsorptionofdsDNA(E lt07V)isconnecteddep dep
withanappearanceofthethirdoxidationsignal(besidestheoxidationsignalsofguanine resp adeninemoieties) at apotential of073V corresponding to theoxidationoffreeguaninebases(FGBs)presentwithinthesolutionofdsDNAInthiscasewecanassumethatthepeakappearingatapotentialofndash190VcanpossiblyrepresentthereductioncounterpartofFGBspresentwithinthesolutionofdsDNAThisassumptioncanalsobesupportedbytheaforementionedstudyandbythefactthatthereductionsignalatsuchahighnegativepotentialcanbeobservedfortheoligodeoxynucleotidescontainingguanineresidues[6] AdditionallyinordertoverifythetruenatureofthereductionsignalsdepictedatF ig1Bandtoexcludetheoptionthattherelatedsignalsdonotrepresenttheproductsofpriorelectrochemicalactivationof theEPPGE(variousCndashObasedchemicalspecies)wehavedecidedtoperformanegative(control)testwithinthe
86 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2Baseline-correctedLSVrecordingscorresponding to thereductionof thedsDNA(02Vs
Fig 2A)respbaseline-correctedSWVrecordingscorrespondingtotheoxidationoftheguanine(098V)respadenine(128V)moieties(075VsFig2B)attheEPPGEafteritsincubationinthephosphatebufferforadefinedtimeperiod(60ndash900s)
blanksolution(PB)employingthesameprotocolasforthedsDNAadsorptionattheEPPGEInthiscaseitispossibletoobservetheabsenceofanypronouncedvoltammetricpeakslinkedtothedsDNAadsorptionandonlythepresenceofoneirreversiblepeakatapotentialofndash153VcorrespondingtotheelectroreductionoftheCndashObasedmoietymoietieswhichdropsafterthedsDNAadsorptiontoonetenthofitsoriginalvalue(approximately) Perhaps the most important parameter regarding further optimizationprocessrepresentedthetime-dependentstabilityofthecorrespondingsignalsofdsDNAwhichcanbespecificallyimportantinrelationtothestudyofthetime-dependentoxidativedamageofdsDNA AsithasalreadybeenprovedasingleelectrochemicalactivationoftheEPPGEinthePBdoesnotrepresentasatisfyingtechniqueregardingstabilityofdsDNAoxidationsignalsat theEPPGEand theadditionalstabilization isachievedbyfurtherelectrochemicalactivationinthesolutionofaredoxindicator(Fig2B)[7]Basedonthisaverificationoftheproposedstabilizationprotocolintermsofthetime-dependent stabilityofdsDNAreduction signals in the solutionof thePBwithinthedefinedtimeperiod(60ndash900s)appearedasareasonablenextstep FromtheresultsdepictedinFig2Aitispossibletonoticethatwithinthefirst300sdsDNAreductionsignalsremainstableinrelationtothecurrentresponseaswellasintermsofthepotentialvalueWithanadditionalincubationtime(t )inc
(900sorangeline)thepeakcurrentofthevoltammetricsignalpresentatmorenegative potentials decreased which can probably be addressed as a slowprogressive elimination of the weak (electro)chemical forces related to theunspecificadsorptionoftheFGBsattheEPPGE
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 87
Fig 3Baseline-corrected SWV recordings corresponding to the oxidation of the guanine respadeninemoietiesattheEPPGEafteritsincubationinthesolutionofK [IrCl ](IR)foradefinedtime2 6
period(60ndash3600s)(075VsFig3A)andthecorrespondingrelativebiosensorresponses(ΔI )rel
evaluatedusingtheguanosine(turquoise)andadenosine(red)peaksplottedversustheincubationtime(Fig3C)Baseline-correctedLSVrecordingscorrespondingtothereductionofdsDNAattheEPPGEafteritsincubationinthesolutionofK [IrCl ](IR)fordifferenttimeperiods(60ndash3600s)2 6
(02 VsFig3B)andthecorrespondingrelativebiosensorresponses(ΔI )evaluatedusingtherelpeakCA(darkpink)plottedversustheincubationtime(Fig3-D)
AdditionallywehavedecidedtotesttheapplicabilityofthepresentedhybridbiosensorintermsofmonitoringdsDNAdamagecausedbyarepresentativeofone-electron oxidants ndash K [IrCl ] In this case the prepared dsDNAEPPGE2 6
biosensor was immersed into the solution of K [IrCl ] (0001molL) for the2 6
definedtimeperiod(60ndash3600s) In thecaseof theoxidationpath (SWVrecordingsdepicted inFig3A) it ispossible to observe a time-dependent decrease of the oxidation signal of theguaninemoietieswhereastheoxidationsignaloftheadeninemoietiesremainsunaffectedforthemostof the incubationperiodThisphenomenonis ingoodcorrelationwiththetheoreticalknowledgeregardingoxidativedamageofdsDNAcausedbyone-electronoxidants [3]Simultaneouslywith thisLSVrecordingsdepicted in Fig 3B followed the similar behavior (decrease in relation to the
88 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
currentresponseofthedsDNAreductionsignalndashpeakCA)asinthecaseofthesignal regarding oxidation of guanine moieties In addition according totheportionofthepreservedDNA(Fig3C3D)itispossibletoassumethatthepronouncedoxidativedamageofdsDNAcanbemonitoredquitepreciselynotonlydirectlyviathedsDNAoxidationsignaloftheguaninemoietiesbutevenindirectlythroughthedsDNAreductionsignalndashpeakCA
4Conclusions
Inthiscontributionwehavepresenteddevelopmentofanunorthodoxhybridelectrochemical DNA biosensor based on an EPPGE Optimization processconcerning some important parameters was performed as well as closerresolutionofthenatureofthereductionprocessesofdsDNAattheEPPGEwasachievedInordertoconfirmtheresultsoftheoptimizationprocessapplicabilityoftheproposedbiosensorhadbeenprobedintermsofmonitoringDNAdamagecausedbyK [IrCl ]Inthiscasethefinalresultshadprovedthattheprepared2 6
hybridbiosensorcanbeconsideredasaversatileanalyticaltoolformonitoringoxidativeDNAdamage(viaoxidationreductionsignals)andispresentedasafinealternative in comparisonwith conventional electrochemical DNA biosensorsprepared within the group of traditional transducer materials (mercury- orcarbon-based)
Acknowledgments
ThisresearchwassupportedbytheSpecificUniversityResearch(SVV260440)
References
[1] FojtaMDanhelAHavranLVyskocilVRecentprogressinelectrochemicalsensorsandassaysforDNAdamageandrepairTrACTrendsAnalChem79(2016)160ndash167
[2] GieseBSpichtyMWesselySLong-distancechargetransportthroughDNAAnextendedhoppingmodelPureApplChem73(2001)449ndash453
[3] Burrows CJ Muller JG Oxidative nucleobasemodifications leading to strand scissionChemRev98(1998)1109ndash1151
[4] DiculescuVC Chiorcea-PaquimAMOliveira-BrettAMApplications of aDNA-electro-chemicalbiosensorTrACTrendsAnalChem79(2016)23ndash36
[5] PalecekEJelenFElectrochemistryofnucleicacidsInElectrochemistryofNucleicAcidsandProteinsndashTowardsElectrochemicalSensorsforGenomicsandProteomicsPalecekESchellerFWangJ(edits)AmsterdamElsevier2005p74ndash174
[6] SpacekJDanhelAHasonSFojtaMLabel-freedetectionofcanonicalDNAbasesuraciland5-methylcytosineinDNAoligonucleotidesusinglinearsweepvoltammetryatapyrolyticgraphiteelectrodeElectrochemCommun82(2017)34ndash38
[7] AugustınMVyskocilVNovelelectrochemicalDNAbiosensorbasedonedge-planepyrolyticgraphite for DNA interaction studies In Proceedings of the 15th International StudentsConferenceldquoModernAnalyticalChemistryrdquoNesmerakK(edit)PragueFacultyofScienceCharlesUniversity2019p263ndash268
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 89
1Introduction
Cadmiumisoneofthemosttoxicmetalsanditswidespreadindustrialusesresultin increased environmental pollution Hence the development of sensitivemethodology for Cd determination is still highly desirable Chemical vaporgeneration(CVG)ofCdbythetetrahydroboratereductioninacidicmediumisasuitable alternative sample introduction technique compatible with atomicspectrometricdetectorsandofferingimproveddetectioncapabilityComparedtocommonliquidnebulizationCVGoffersseveraladvantagessuchassignificantlyhigheranalyteintroductionefficiencyandalsoanalyteseparationfromsamplematrix IncomparisontoCVGofcommonhydrideformingelementsthereisalackofliterature dealing with mechanistic aspects of CVG of Cd [1] as well as withstabilityandidentityofitsvolatilespecies(freeatomshydrideotherspecies)Very little information is also available on achieved generation efficiencyMoreover there are many discrepancies in the literature regarding optimum
Chemical vapor generation of cadmium for analytical atomic spectrometry
a b a b a aLINDASAGAPOVA BARBORAKODRIKOVA MILANSVOBODA STANISLAVMUSIL aJANKRATZER
a InstituteofAnalyticalChemistryoftheCzechAcademyofSciences Veveřiacute9760200BrnoCzechRepublicsagapovaiachczb DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublic
AbstractChemical vapor generation of cadmium volatile compounds wasoptimizedinordertodeterminetraceCdconcentrationsbyatomicabsorptionspectrometry(AAS)Severalreactionmodifiersbasedon
III+ II+ III+ IV+inorganicsaltsandcomplexesofCr Co Ti Ti weretestedtoimproveanalyticalperformanceandgenerationefficiencyTheuseofthese reaction modifiers resulted in 4ndash5 times enhancement insensitivity reflected also in corresponding increase of generationefficiency and better repeatability Generation efficiency wasdeterminedfromacomparisonbetweensensitivitiesobtainedwithchemicalvaporgenerationandconventionalsolutionnebulizationbothsimultaneouslycoupledwithinductivelycoupledplasmamassspectrometryTheidentityofthegeneratedcadmiumcompoundswillbediscussed
Keywordsatomicabsorption
spectrometryatomizationcadmiumchemicalvapor
generation
90 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
conditionsforCVGofCdAlthoughstrongacid(HClorHNO )isalwaysemployed3
asacarrierandNaBH asareductantsomeauthorsreportedvariousadditives4III+ III+ IV+(modifiers)basedontransitionmetalions(Cr Ti Ti )inthepresenceof
II+KCN[23]orCo inthepresenceofthioureaandascorbicacid[4]toimproveCdsignalssignificantly TheaimofthisworkwastoinvestigateCVGofCdinacomprehensivewayFirstlyCVGofCdwithoutandwithselectedmodifierswasoptimizedemployingatomicabsorptionspectrometry(AAS)asadetectorandexternallyheatedquartztube(QTA)astheatomizerSecondlytheeffectofatomizationtemperatureonCdsignalwasstudiedallowingthustodeducetheatomicormolecularstructureofgeneratedCd speciesThirdly generation efficiencyofCdvolatile specieswasquantified
2Experimental
21Reagentsandchemicals
minus1Boiled and bubled (Ar per 30min) deionizedwater (lt 01 μScm UltrapurWatrex USA) was used to prepare all solutionsWorking Cd standards were
minus1preparedfrom1000mgL Cdstocksolution(AstasolAnalytikaCzechRepublic)minus1bydilutionin01ndash048molL HCl(basedonthemodifieremployed)from37
HCl(paMerckGermany)Theoptimumgenerationconditionsbeingdifferentforeachmodifiertestedare listedinTable1ThereductantwasasolutionofNaBH (ge 97 Sigma-Aldrich Germany) in 04 (mv) KOH (pa Merck4
Germany) prepared fresh daily The solutions of modifiers were prepared as2+followsCo waspreparedfromCoCl 6H O(ge990PENTACzechRepublic)2 2
3+the solution of Cr from Cr(NO ) 9H O (ge 9999 tracemetal basis Sigma-3 3 23+AldrichGermany)thesolutionofTi fromTiCl solution(about15in10HCl3
4+Sigma-AldrichGermany)andthesolutionofTi fromTiOSO (ge999Sigma-4
AldrichGermany)Tostabilizethelattersolution1H SO wasusedprepared2 4
bydilutionof96H SO (paLach-NerSlovakia)SolutionofKCN(ge9702 43+ 3+FlukaSwitzerland)wasusedasasecondmodifierwhenworkingwithCr Ti or
4+ ndash3Ti asmodifiersitsconcentrationvariedfrom008to016moldm depending
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 91
Table 1OptimumconditionsforchemicalvaporgenerationofCdinthepresenceofmodifiersandtheirabsence
onthemetalionThiourea(CH N Sge980LachemaBrno)andascorbicacid4 2
(C H O ge997Riedel-deHaenGermany)wereusedasmodifierscombined6 8 62+withCo
22Instrumentation
221Chemicalvaporgenerationsystems
TwoCVG flow injection systemswere employed either a two channel systemwithoutadditionofamodifier(seeFig1A)orafourchannelsystemallowingadditionofmodifiers(seeFig1B)
ndash1 TheflowratesofHClandNaBH were42and10mLmin respectivelyinatwo4ndash1channelsystem(Fig1A)whiletheywerebothkeptat10mLmin inthefour
channelsystem(Fig1B)Theflowratesofmodifiersinthefourchannelsystemndash1were05mLmin Thevolumeofthesampleloopwas015mLinbothsystems
ndash1Carriergasflowrateof75mLmin Arwascontrolledbyamassflowcontroler(Cole-ParmerUSA)
92 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
(A)
(B)
Fig 1Schemesofthechemicalvaporgenerationflowinjectionsystemwith(A)twochannels(nomodifiers)and(B)fourchannels(modifiersemployed)
222Atomicabsorptionspectrometry
ThePerkin-Elmermodel503atomicabsorptionspectrometer(BodenseewerkGermany)wasequippedwithaCdelectrodelessdischargelamp(Perkin-ElmerUSA)operatedat228mAThemeasurementswereperformedat2288nmusinga07nmslitwidthTheShimadzumodelAA-7000atomicabsorptionspectrometer(ShimadzuJapan)wasalsousedACdhollowcathodelamp(PhotronAustralia)operatedat2288nmlinewith07nmspectralbandpassandalampcurrentof12mA Signals were recorded for 2 minutes and peak areas were taken forevaluation The QTA was heated electrically to the temperature required byfurnace(PerkinElmer)andanin-housemadefurnacecontrolledbytheREX-C100controller(SysconIndianaUSA)withtheK-typethermocouplesensor(OmegaEngineeringUSA)223QuantificationofCVGefficiencybyICP-MS
Overall CVG efficiency of Cd was quantified bymeans of inductively coupledplasmamassspectrometry(ICP-MS)fromcomparisonoftheslopesofcalibra-tionsobtainedwithnebulizationliquidCdstandardstothoseobtainedwithCVGThe efficiency of liquid nebulization was quantified using a modified wastecollection method (see reference [5] for details) The Agilent 7700x ICP-MSinstrument(AgilentUSA)wasoperatingat1600WofRFpowerThesignalwas
111 125monitoredat Cdisotopeandcorrectedforthesignalofinternalstandard( Tendash11000ngmL Tein2HNO )NebulizeranddilutionArgasflowrateswere11503
ndash1and0mLmin respectively
3Resultsanddiscussion
31Chemicalvaporgenerationconditions
UnivariateoptimizationswereperformedtofindoptimumconditionsforCVGofCdinpresenceandabsenceofmodifiersTheparameterstobeoptimizedwerecarrieracid(HCl)concentrationreductant(NaBH )concentrationmodifierIand4
modifier II concentrations carrier gas flow rate (Ar) length of reaction coilsRCI-III (see Fig 1B) The optimum conditions for individual modifiers aresummarizedinTable1
32IdentityofCdspecies
ThepeakareasofgeneratedCdspecieswereforagivenmodifiermeasuredintheQTAheatedto900degCandnon-heatedQTAsubsequentlyOptimumCVGconditionswere employed as summarized in Table 1 This simple experiment allowsdistinguishingbetweenatomic(freeatoms)andmolecularformsofgenerated
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 93
Fig 2RelativesignalofgeneratedCdspeciesmeasuredintheQTAheatedto900degC(blackbars)andnon-heatedQTA(whitebars)withoutorinthepresenceofmodifiers
speciesOnlyfreeatomscanbedetectedinnon-heatedQTAsimilarlyasincaseofmercurycoldvaporsOn thecontrarymolecularanalytespeciesareatomizedat900degCAsaconsequencethesignalregisteredintheheatedQTAcorrespondstobothatomicandmolecularspeciesgeneratedItmustbehighlightedthattheresidencetimeoffreeatomsintheatomizerisdependentonQTAtemperatureduetogasexpansionAsaconsequencethesignalinQTAheatedto900degCshouldreach25ofthesignalatambienttemperaturetakingintoaccountthatonlyfreeatomsaregeneratedSincethetemperaturealongtheopticalarmofQTAisnotdistributedhomogeneouslydecreasingtobothendtheeffectivetemperatureoftheatomizerislowerOurexperimentswithCVGofHgrevealedsignalinheatedQTAisaround40[6]TheresultsreachedforCVGofCdaredepictedinFig2ThesignalofCdinheatedQTAisaround50ofthesignaldetectedinnon-heatedQTA
4+whennomodifierisemployedorusingTi asthemodifierindicatingclearlyfreeCdatomsarethedominantvolatilespeciesgeneratedOnthecontraryalmostno
2+differenceinpeakareaswasobservedforCo asthemodifierwhilethesignalinheatedQTAwas even 5 times higher in heatedQTA compared to non-heated
3+atomizer with Cr as the modifier suggesting the dominant contribution of3+molecularstructurestoCdsignalespeciallyincaseofCr KCNreactionsystem
33Generationefficiency
TheoverallCVGefficiencywasestimatedfromacomparisonbetweensensitivitiesobtainedwithCVGsampleintroductionandconventionalsolutionnebulizationICP-MSunder the sameexperimental conditionsNebulization efficiency for aMicroMISTnebulizerwasdeterminedas79plusmn01ThegenerationefficiencyofCd was derived from the sensitivity enhancement between CVG and liquidnebulizationTheresultsaresummarizedinTable2indicatingthatCVGwithout
94 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
modifiersisonlycatwotimesmoresensitivecomparedtoliquidnebulization3+ 4+Generation efficiency of Cd increases to 60 in the presence of Ti and Ti
modifiers
4Conclusions
CVG of Cd was thoroughly optimized in the presence of selected modifiersreportedpreviouslyintheliteratureGenerationefficiencyofCdintheabsenceofanymodifierswasquantifiedto15whileitcanbeincreasedupto60inthe
3+ 4+presenceofTi KCNorTi KCNasmodifiersFreeCdatomsseemtobe the4+dominantCdformgeneratedintheabsenceofanymodifiersorusingTi KCN
3+modifierwhile rathermolecularCd structuresaregenerated inCr KCNand2+Co thioureaascorbicacidreactionsystems
ExperimentsareinprogresstofinishthiscomprehensivestudyOnlythebestmodifierwillbefurtherusedforCVGofCdtobecoupledwithotherspectrometricdetectorsandappliedtocertifiedreferencematerialsandrealsamples
Acknowledgments
ThisresearchhasbeensupportedbytheCzechScienceFoundationundercontract18-01116SandbytheInstituteofAnalyticalChemistryoftheCzechAcademyofSciences(InstitutionalResearchPlannoRVO68081715)andCharlesUniversity(ProjectnoSVV260440)
References
[1] PitzalisEAngeliniDMascherpaMCDacuteUlivoAInsightintothemechanismscontrollingthechemicalvaporgenerationofcadmiumJAnalAtSpectrom33(2018)2160ndash2171
[2] ArslanZYilmazVRoseLEfficientgenerationofvolatilecadmiumspeciesusingTi(III)andTi(IV)andapplicationtodeterminationofcadmiumbycoldvaporgenerationinductivelycoupledplasmamassspectrometryMicrochemJ123(2015)170ndash178
[3] YilmazVRoseLArslanZLittleMDOn-linechemicalvapourgenerationofcadmiuminthepresenceofhexacyanochromate(III)fordeterminationbyinductivelycoupledplasmamassspectrometryJAnalAtSpectrom27(2012)1895ndash1902
[4] Y Lu SunHW YuanCG YanXP Simultaneous determination of trace cadmiumandarsenic inbiologicalsamplesbyhydridegeneration-doublechannelAFSAnalChem74(2002)1525ndash1529
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 95
Table 2GenerationefficiencyofchemicalvaporgenerationofCdasquantifiedbyICP-MS
Modifiers Generationefficiency
Nomodifiers 15plusmn13+Cr KCN ndash2+Co thioureaascorbicacid ndash3+Ti KCN 58plusmn24+Ti KCN 61plusmn2
[5] VyhnanovskyJStrugeonREMusilSCadmiumassistedphotochemicalvaporgenerationoftungsten fordetectionby inductively coupledplasmamass spectrometryAnal Chem91(2019)13306ndash13312
[6] MigasovaMMatousekTSchrenkovaVZ ıdekRPetry-Podgorska IKratzer JMercuryvolatilespeciesgenerationfromHClandTRISbuffermediaAnalChimActa1119(2020)68ndash76
96 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Atomicfluorescencespectrometry(AFS)coupledwithvapourgenerationisanultrasensitive analytical method for determination of various elements ItsanalyticalperformancecanbecomparabletoICP-MSwithliquidnebulizationbutatsubstantiallylowercost[1]SampleintroductiontoAFSisacrucialstepoftheanalyticalproceduresincetheanalytehastobeintroducedtotheatomizerintheformofitsvolatilespecies Hydridegeneration(HG)isamaturetechniqueofsampleintroductionduringwhichvolatileanalytehydridesare formedbyreactionwithareducingagenttypicallysodiumborohydrideAnewemergingtechniquephotochemicalvapourgeneration(PVG)employsUV irradiationof theanalyte in liquidphase in thepresenceofaphotochemicalagent(usuallyalowmolarmassorganicacidformic
minusoraceticacid)Highlyreducingradicalspecies(HbullRbullandCOObull )andaquatedelectronsare formedduring irradiationandreactwith theanalyte to form its
Photochemical vapour generation of bismuth coupled with atomic fluorescence spectrometry
ab ab a aBARBORASTA DLEROVA JAROMIRVYHNANOVSKY JIR IDE DINA STANISLAVMUSIL
a InstituteofAnalyticalChemistryoftheCzechAcademyofSciences Veveřiacute9760200BrnoCzechRepublicstadlerovaiachczb DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublic
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 97
AbstractPhotochemical vapour generation of bismuth was successfullycoupledwithnon-dispersiveatomic fluorescencespectrometry forthefirsttimeVolatilespeciesofBiweregeneratedusingastandardmercurylow-pressuretubelampandacoiledreactorfromareaction
2+mediumwhichwas composedof acetic and formic acid Co ionswereusedasasensitizerOptimizationofatomizationconditionsinaflame-in-gas-shieldatomizerwasperformedThismethodologywascomparedtothecommonlyemployedhydridegenerationapproachAbsolutelimitofdetectionof68pgwasachievedwithphotochemicalvapour generation which is still about 7 times worse than withhydride generation The developed methodology was successfullyverifiedbyBideterminationinareferencematerialofwater
Keywordsatomicfluorescence
spectrometrybismuthhydridegenerationphotochemicalvapour
generation
volatilespeciesInbothcasesthegeneratedvolatilespeciesoftheanalytehavetobeseparatedfromtheliquidphaseinthegas-liquidseparatorandarecarriedtotheatomizerbyacarriergas[2] InthisworkanatomizerdesignedspecificallyforAFStheflame-in-gas-shieldatomizerwasused(Fig1) It consistsofaverticalquartz tubesuppliedwithargon and hydrogen together with the analyte volatile species Moreover acapillaryisinsertedintheverticalaxisoftheverticaltubethroughwhichoxygenisintroducedA hydrogen-oxygenmicroflameburns on top of the capillary Themicroflameisshieldedfromtheambientatmospherebyaflowofargonwhichisintroducedthroughashieldingunitfittedaroundtheverticaltube[34] Theaimofthisworkwastooptimizeatomizationconditionsintheflame-in-gas-shieldatomizerusingPVGasasampleintroductiontechniqueandtocomparetheanalyticalcharacteristicsofPVGandHGforultrasensitivedeterminationofbismuthbyAFS
2Experimental
21Reagentsandchemicals
Deionized water (Ultrapur Watrex USA) was used for preparation of all thesolutionsWorkingBisolutionswerepreparedfreshdailybyserialdilutionof
ndash1stock1000mgl BistandardforAAS(Sigma-AldrichGermany)RegardingHG05(mv)NaBH in04(mv)KOHwasusedasa reductantAsolutionof4 ndash11mol l HClwasusedasacarrierandblank
98 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Flame-in-gas-shieldatomizerOHndashobser-vationheight
RegardingPVGformicacid(98paLach-NerCzechRepublic)andaceticacid (998 pa Lach-Ner CzechRepublic)were used for preparation of thereaction medium they were purified in a Teflon BSB-939-IR sub-boilingdistillation apparatus (Berghof Germany) The composition of the reactionmedium(40(vv)acetic125(vv)formicacid)wasoptimizedearlier[5]
ndash1The 5000mgl Co stock solution was prepared from cobalt(II) acetatetetrahydrate (pa Lach-Ner Czech Republic) and used as a sensitizer ofphotochemicalreactionTheoptimalconcentrationofCointhestandardsamples
ndash1andblanksolutionscorrespondedto50mgl (ref[5]) Acertifiedreferencematerial(CRM)-1643fTraceElementsinWater(NationalInstituteofStandardsandTechnologyUSA)wasusedtochecktheprecisionofthedevelopedmethodology
22Instrumentation
221Atomicfluorescencespectrometer
An in-house assembled non-dispersive atomic fluorescence spectrometerconstructedatourlaboratorywasusedforBideterminationandisdescribedindetail elsewhere [3] The detector output provided signals in microV Peak areacorrectedtobaselineandmainlysignaltonoiseratioweretheparametersusedtoevaluatethedata
222Hydridegeneratorphotochemicalvapourgeneratorandatomizer
A flow injection hydride generator was employed (Fig 2a) The reductantndash1 ndash1(12mlmin )andthecarrier(4mlmin )werepumpedbyaperistalticpump
Thesamplewasinjectedthrougha1mlsampleloopintotheflowofcarrierAglassgas-liquid separator (5 ml) with forced waste removal was employed forseparatingthegasphasecontainingbismuthanewhichwasthencarriedtotheatomizerbyargon Thephotochemicalvapourgenerator(Fig2b)consistedofthephotoreactorconstructedwitha15Wlow-pressureHggermicidallamp(Cole-ParmerUSA)wrappedaroundwith6mofPTFEtubing(1mmidinternalvolume471ml)The
ndash1reactionmedium(3mlmin )waspumpedbyaperistalticpumpThesamplewasinjected througha056ml sample loopApolypropylenegas-liquid separator
(15ml)withforcedwasteremovalimmersedinanicebath[6]wasemployedforseparatingthegasphasecontainingBivolatilespeciessubsequentlycarriedtotheatomizerbyargon Theflame-in-gas-shieldatomizerisdepictedinFig1detaileddescriptionisgiveninRef[3]Theobservationheight(OH)isdefinedasthedistancefromthetopofthecapillarytothecentreoftheopticalbeam
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 99
23Samplepreparation
CRMNIST1643fwasdilutedwith1MHCl80-foldforBideterminationbyHG-AFSRegardingBideterminationbyPVG-AFSthesampleneededtobeevaporatedtodrynessinordertogetridofnitricacidthatseriouslyinterferesatmMlevel[5]Avolume of 3ml of CRMwere pipetted into a 40 ml quartz vial evaporated(temperature asymp100 degC two replicates) and subsequently diluted ca 33-foldAsamplepreparationblank3mlofdeionizedwaterwaspreparedaswell
3Resultsanddiscussion
TheatomizationconditionsforHG-AFSwereoptimizedinourpreviouswork[3]TheseconditionswereusedasinitialtofindtheoptimumconditionsforPVG-AFSwith the flame-in-gas-shieldatomizerwith respect to sensitivityandsignal tonoiseratioFirstlythehydrogenfractionintherange10ndash16wasoptimizedatconstant total gas flow rate (sum of total argon and total hydrogen) of
ndash1500mlmin the lower the hydrogen fraction the better However at 10hydrogenfractiontheflamewasnotstableenoughandwentoftenouthenceitwasoptedfor12hydrogenfraction
100 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2(a)Hydrideand(b)photochemicalvapourgenerator
The oxygen flow rate through the capillary was optimized in the rangendash15ndash30mlmin Thehighestsignaltonoiseratiowasachievedwiththeflowrateof
ndash120mlmin Thetotalgasflowratewasoptimizedatconstant12hydrogenfractioninthe
ndash1range500ndash800mlmin Theoptimumobservationheightvarieswithtotalgasflow rate so it had to be optimized as well The optimum conditions aresummarizedandcomparedtothoseachievedwithHGinTable1 TheanalyticalfiguresofmeritofPVG-AFSwiththeflame-in-gas-shieldatomi-zerweredeterminedThecalibrationfunctionconstructedwith010025050
ndash1 2100 and 200 microg l Bi standardswas linear (R = 09998) The repeatabilityndash1expressedastherelativestandarddeviation(n=10)was6at1microgl andthe
ndash1relativeandabsolutelimitsofdetection(3σn=10)achievedwere12ngl and68pgrespectively(Table2)TheabsolutelimitofdetectionachievedwithHGwas 76 times lower which can be attributed to several aspects Firstly thegenerationefficiencyforPVGapproachwasaround53while100isexpectedforHG[3]Secondlyafullwidthathalfmaximumofthemeasuredpeakswasca2-foldgreaterwhichnecessitatedlongerintegrationtimeandwasthusreflectedinhighernoiseofthesignalsFinallythelimitofdetectionforPVGapproachwas
ndash1affectedby seriouscontamination (around10ng l )mostprobably from thesensitizersolutionthatcontainedBiasimpurity Tovalidate theproposedmethodologyBi contentwasdetermined inCRMNIST1643f (Table3) and the resultswere compared to thosemeasuredwithHG-AFS[3]Duetosevereinterferencesfrominorganicacidsespeciallynitricacid[5]thesampleneededtobeevaporatedtodrynessandthenfilledupwiththe
2+reactionmediumcontainingCo as thesensitizer(NIST1643f isstabilized in
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 101
Parameter HG-AFS(ref[3]) PVG-AFS
ndash1Artotalmlmin 440 528ndash1H totalmlmin 60 722 ndash1O mlmin 7 202
OH mm 6 9ndash1Arshieldlmin 1515 1515
Table 1Atomizationconditionsforflame-in-gas-shieldatomizer
Parameter HG-AFS(ref[3]) PVG-AFS
LODpg 09 68ndash1LOQngl 09 12
Repeatability lt1 6
Table 2AnalyticalfiguresofmeritofHG-AFSandPVG-AFS
ndash1032moll nitricacid)Theresultsobtainedbybothmethodologiesareingoodagreementwiththecertifiedvalue
4Conclusion
Photochemical vapour generation of Bi was successfully coupled with non-dispersiveatomicfluorescencespectrometryforthefirsttimeanditsapplicabilitywas verified by determination of Bi in certified reference material of waterComparedtohydridegenerationconditionsofatomizationdifferinanoptimalobservationheightandsupplyofoxygenwhichmaybeneededtoldquoburnoutrdquotheorganicvapoursthatarereleasedfromthereactionmediumtothegasphasehowever this remains tobe verifiedAlthough there are still some limitationsregardingthelimitsofdetectionrepeatabilityandinterferencesthisnewsampleintroductionapproachseemstobepromising
Acknowledgments
The support of the Czech Science Foundation (19-17604Y) Czech Academy of Sciences(Institutional supportRVO68081715)andCharlesUniversity (ProjectSVV260560andProjectGAUK1048120)isgratefullyacknowledged
References
[1] Musil SMatousek T Currier JM StybloM Dedina J Speciation analysis of arsenic byselectivehydridegeneration-cryotrapping-atomicfluorescencespectrometrywithflame-in-gas-shield atomizer achieving extremely low detection limits with inexpensiveinstrumentationAnalChem86(2014)10422ndash10428
[2] SturgeonREPhotochemicalvaporgenerationaradicalapproachtoanalyteintroductionforatomicspectrometryJAnalAtSpectrom32(2017)2319ndash2340
[3] S tadlerova B Kolrosova M Dedina J Musil S Atomic fluorescence spectrometry forultrasensitivedeterminationofbismuthbasedonhydridegenerationndashtheroleofexcitationsourceinterferencefilterandflameatomizersJAnalAtSpectrom35(2020)993ndash1002
[4] DedinaJAtomizationofvolatilecompoundsforatomicabsorptionandatomicfluorescencespectrometryOnthewaytowardstheidealatomizerSpectrochimActaPartB62(2007)846ndash872
102 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Certified HG-AFS PVG-AFSndash1valuemicrogl
ndash1 a ndash1 a valueobtainedmicrogl recovery valueobtainedmicrogl recovery
1262plusmn011 128plusmn01 102plusmn1 121plusmn09 97plusmn5
a Spikedrecovery=slopeofstandardadditions(noadditionandtwospikedconcentrationstoasample)slopeofexternalcalibration
Table 3ThedeterminedcontentofBiinCRMNIST1643fpresentedasmedianvalueplusmncombineduncertainty(n=3)andrecoveries
[5] Vyhnanovsky J Yildiz D Musil S Effect of metal sensitizers on photochemical vaporgeneration of bismuth for analytical atomic spectrometry In Proceedings of the 15thInternationalStudentsConferenceModernAnalyticalChemistryKNesmerak(ed)PragueCharlesUniversity2019p257ndash262
[6] VyhnanovskyJSturgeonREMusilSCadmiumassistedphotochemicalvaporgenerationoftungstenfordetectionbyinductivelycoupledplasmamassspectrometryAnalChem91(2019)13306ndash13312
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 103
1Introduction
Liquidcrystalsareorganicsubstancesthatformamesomorphicphaseinsolu-tion[1]TheyareliquidlikeliquidsbuthaveaninternalconfigurationassolidsTheir light transmittance changes in the electric fieldwhich is used in liquidcrystaldisplays(LCDs)CholestericliquidcrystalsaretemperaturesensitiveThecolorofreflectedlightchangeswithsmalltemperaturechangeThisisusedinmedicineasasensitivetemperatureindicatorfordisease-infectedtissues High performance liquid chromatography or supercritical fluid chromato-graphy[23]areusedtoseparateanddetermineliquidcrystalcompoundsAnelectrophoretic method could be complementary to these commonly usedmethods To our best knowledge no study dealing with liquid crystal puritycontrolbyelectrokineticchromatographywaspublisheduntilnow Electroneutralsubstancesmoveincapillaryzoneelectrophoresisatthesamespeedcorrespondingtothespeedoftheelectroosmoticflow(EOF)andtherefore
Separation of liquid crystals using non-aqueous capillary electrokinetic chromatography
KATER INACOKRTOVATOMA S KR IZ EK
DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublickaterinacokrtovagmailcom
AbstractLiquid crystals arewidelyused in electronicsmedicine andotherfields Analytical separations are important in the development ofnewliquidcrystalstocontrolthepurityofsynthesizedsubstancesThesampleanalysisisimportantfordetectionofimpuritiesformedduring synthesis Liquid crystal-forming substances cannot beseparated by capillary zone electrophoresis due to the absence ofreadily ionizable groups Therefore electrokinetic chromatographywasused in thisworkAnotherproblemcomplicating theanalysiswastheverylowsolubilityofanalytesinwaterSeparationsinthisworkwere thereforecarriedoutundernon-aqueousconditions inacetonitrilewithaceticacidtoadjustthepHandhexadecyltrimethyl-ammonium chloride as a detergent to mobilize the non-ionizedanalytesUndertheseconditionsitwaspossibletoseparateimpu-ritiesfromsynthesizedanalytesinsamples
Keywordselectrokinetic
chromatographyliquidcrystalsnon-aqueouscapillary
electrophoresis
104 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
itisnotpossibletoseparatethemDuetothisanelectrokineticchromatographymethodwasdevelopedInthismethodasurfactantisaddedtothebackgroundelectrolyteMoleculesaggregateandformsphericalformationscalledmicelles[4]ifthesubstanceisaddedinsufficientconcentrationiehigherthanthecriticalmicellar concentration (CMC) Separation is possible due to interactions ofnonpolarmoleculepartswiththenonpolarmicelleinsideAlthoughwateristhemostusedsolventinelectrophoreticmethodsforseparationofwater-insolublesubstances organic solvents are selected However such solvent must meetcertaincriteriatobesuitableforuseincapillaryelectrophoresisAllcomponentsmustbesolubleinthesolvent[5]ItshouldnotbeflammabletoxicorreactiveforpracticalityitshouldbeliquidatroomtemperatureandalsoitspriceistakenintoaccountThevalueof its relativepermittivitywhichdescribes the strengthofinteractionsbetweenionsshouldbearound30Lowdynamicviscosityisalsopreferred to allow faster migration of analytes No organic solvent meets allparameters of the ideal solvent In practice methanol acetonitrile and theirmixturesarethemostusedTheseparationparameterscanbeinfluencedbyusingan organic solvent of the background electrolyte This topic has already beenwidelyexplored[6ndash8] Itwasgenerallyassumedthat inanhydrousconditionsmicellesarenotcreateddespitesufficientsurfactantconcentrationHoweveritwasfoundoutthatdodecylsulfatecanformstablemicelleswhenthebackgroundelectrolyteisdissolvedinformamide[9]Fortheanalysisofactivesubstancesinmedicinal plants Chen et al developed a method in which sodium cholatedissolvedinmethanolisusedasasurfactant[10]Theaddedpseudostationaryphase does not always form micelles but can still affect mobilization andseparationofanalytesiftheanalytesinteractdifferentlywithfreemoleculesofsurfactantInthisstudywater-insolubleliquidcrystalswereseparated(Fig1)Thereforenonaqueouselectrokineticchromatographymethodwasdeveloped
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 105
Fig 1 Structures of liquid crystals 4-([1-oxo-1-(pentyloxy)propan-2-yl]oxycarbonyl)phenyl4-(octyloxy)-[11-biphenyl]-4-carboxylate (ZL 85) and 4-([1-(decyloxy)-1-oxopropan-2-yl]oxycarbonyl)phenyl 4-(dodecyloxy)-[11- biphenyl]-4-carboxylate (ZL 1210) OpticalisomerismsitesaremarkedwithanasteriskStructurescreatedinMarvinSketch[11]
2Experimental
21Reagentsandchemicals
Acetonitrilege999fromSigma-Aldrich(Germany)aceticacid99fromLach-Ner Neratovice (Czech Republic) and hexadecyltrimethylammonium chloride25(ww)inwaterfromSigma-Aldrich(USA)wereusedforpreparationofback-groundelectrolyteMesyloxidepa(MO)suppliedbyLach-nerNeratovice(CzechRepublic)wasusedasareferencesubstance
22Instrumentation
ForexperimentsG7100ACapillaryElectrophoresisInstrument(AgilentTechno-logiesGermany)wasusedwithUV-VISdetectoroperatingat235nmand254nmwavelengthMeasurementswereconductedinafused-silicacapillaryof50microminner diameter with the total length 500cm and effective length 415cm(PolymicroTechnologiesUSA)
23Method
Capillarywas flushed for3minuteswith1MHCland for2minuteswith thebackgroundelectrolyteBackgroundelectrolytewaspreparedbymixingaceticacid (10mM) and hexadecyltrimethylammonium chloride (40mM) in aceto-nitrile Sampleswere introducedhydrodynamicallybyapressureof5kPa for1secondSampleswerefirstdissolvedinacetonitrileandthendilutedtwotimeswith the background electrolyte A voltage of 20 kV was applied during theseparation
3Resultsanddiscussion
Liquidcrystalsampleswerepractically insoluble inwater theirsolubilitywasndash3testedataconcentrationlevelof1mgcm inmethanolandacetonitrileWhile
samples were not sufficiently soluble in methanol they were successfullydissolvedinacetonitrile Becauseallanalytesaresubstancesthatdonothaveeasilyionizablefunctionalgroups theelectrokineticchromatographymethodwaschosen forseparationAsuitablesurfactantwassoughtCommonlyusedsodiumdodecylsulfate(SDS)isinsoluble in acetonitrile Therefore hexadecyltrimethylammonium chloride(CTAC) which had sufficient solubility for further experiments was chosenAlthoughasuitablebufferwassoughttoensureastablepHduetoproblemswithprecipitationofbuffercomponentsinthenon-aqueousenvironmentaceticacidwasusedtoadjustandmaintainpHofbackgroundelectrolytesolutionAstheadditionofcationicsurfactantsuchasCTACleadstoEOFreversalthedependence
106 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ofEOFmobilityontheconcentrationofCTACinthebackgroundelectrolytewasmeasuredContrarytowhatisobservedinaqueousbackgroundelectrolytesEOFwasnotreversedItsmobilitydecreasedwithincreasingCTACconcentrationbutnomajorchangesoccurredabove40mMconcentrationThecapillarywallwasprobablyalreadysaturatedbyCTACandthefurtherincreaseinconcentrationhadno signifficant effect on the conditionof the capillarywall Therefore a CTAC
ndash3concentrationof40mmoldm was chosenas sufficient for furthermeasure-mentswithrespecttotheincreasingcurrentwithincreasingionicstrengthofthebackgroundelectrolyte The optimized method was used for separation of several liquid crystalsamplesofdifferentpurityInthesampleoftheZL85liquidcrystalwith99purityonezoneoftheanalytewasdetectedImpuritieswereseparatedfromthisanalytewhen the samplewith lower puritywas introduced The peak of theanalytewasidentifiedbasedonrelativemigrationtimerelatedtomesityloxideSeparationoftheanalytefromanimpurityinthesampleZL8576isshowninFig 2The relativemigration timeof the firstpeak is 0834 therefore itwasidentifiedastheZL85analyteThestandarddeviationoftherelativemigrationtimesinfivemeasurementswas0002min(01) Using the available high purity sample it was possible to measure thecalibration line forquantificationof theanalyte in lesspuresamplesLimitof
ndash3detection was determined as 0009mgcm and limit of quantification as ndash30031mgcm FromthecalibrationlineconcentrationofZL85inthesample
with lower purity was calculated The concentration was determined as
48(ww)standarddeviation5(ww)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 107
Fig 2ElectropherogramobtainedwhenasampleofZL85liquidcrystalwithlowerpuritywasndash3introducedSamplewas introduced in01mgcm concentrationandwithaddedmesityloxide
ndash3(10mgcm )Capillarywithinnerdiameterof50micromtotallengthof500cm415cmeffectivelength The background electrolyte was acetonitrile with 10 mM acetic acid and 40 mMhexadecyltrimethylammonium chloride A voltage of 20 kVwith positive polaritywas appliedDetectionat254nm
ForsampleZL121099onlytheanalyteandmesityloxideweredetectedInthe sample ZL 1210 59 several impurities were separated and detected(Fig3)Accordingtotherelativemigrationtimetheanalyteofinterestcorres-pondstothefirstpeakPeakresolutionissufficientTheresolutionoftheanalytepeakandthesecondpeakis284andtheresolutionoftheothertwopeaksis230
4Conclusions
InthisstudyanewmethodforanalysisofnewlysynthesizedliquidcrystalswasdevelopedSomeparametersofthemethodwereoptimizedndashoptimumconcen-tration of hexadecyltrimethylammonium chloride was searched The identifi-cation of analyteswas based on a comparison of relativemigration times InsamplesZL85andZL1210withlowerpuritytheimpuritieswereseparatedfromthepeaksofliquidcrystalsthecontentofanalytewasdeterminedintheZL85sampleaccordingtothecalibrationline
Acknowledgments
IwouldliketothanktheInstituteofPhysicsoftheCzechAcademySciencesforprovidingnewlysynthesized liquid crystals This work has been supported by Specific University Research(SVV260560)andbyCharlesUniversityResearchCentreprogramNoUNCESCI014
References
[1] GennesPGProstJThePhysicsofLiquidCrystals2ndedNewYorkOxfordUniversityPress1993
108 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ndash3Fig 3ElectropherogramofsampleZL121059ataconcentrationof05mgcm withmesitylndash3oxideataconcentrationof10mgcm capillarywith innerdiameterof50microm total lengthof
500 cmeffectivelength415cmThebackgroundelectrolytewasacetonitrilewith10mMaceticacidand40mMCTACAppliedvoltage20kVpositivepolarityDetectionat235nm
[2] Vankatova P KalıkovaK KubıckovaA Ultra-performance supercritical fluid chromato-graphy A powerful tool for the enantioseparation of thermotropic fluorinated liquidcrystalsAnalChimActa1038(2018)191ndash197
[3] Vankatova P Kubıckova A Cigl M Kalıkova K Ultra-performance chromatographicmethodsforenantioseparationofliquidcrystalsbasedonlacticacidJSupercritFluids146(2019)217ndash125
[4] Terabe S Otsuka K Ichikawa K Tsuchiya A Ando T Electrokinetic separations withmicellarsolutionsandopen-tubularcapillariesAnalChem56(1984)111ndash113
[5] RiekkolaMLRecentadvancesinnonaqueouscapillaryelectrophoresisElectrophoresis23(2002)3865ndash3883
[6] Wright PB Lister AS Dorsey JG Behavior and use of nonaqueous media withoutsupporting electrolyte in capillary electrophoresis and capillary electrochromatographyAnalChem69(1997)3251ndash3259
[7] PorrasSPKenndlerECapillaryzoneelectrophoresisinnon-aqueoussolutionspHofthebackgroundelectrolyteJChromatogrA1037(2004)455ndash465
[8] PorrasSPRiekkolaMLKenndlerETheprinciplesofmigrationanddispersionincapillaryzoneelectrophoresisinnonaqueoussolventsElectrophoresis24(2003)1485ndash1498
[9] GuoXWangK ChenGH Shi JWuX Di L LWangY Determination of strobilurinfungicideresiduesinfruitsandvegetablesbynonaqueousmicellarelectrokineticcapillarychromatography with indirect laser-induced fluorescence Electrophoresis 38 (2017)2004ndash2010
[10] Chen AJ Li C Gao WH Hu ZD Chen XG Application of non-aqueous micellarelectrokinetic chromatography to the analysis of active components in radix SalviaemiltiorrhizaeanditsmedicinalpreparationsJPharmBiomedAnal37(2005)811ndash816
[11] MarvinSketch [computer program] version 1990 ChemAxon httpschemaxoncom-productsmarvin
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 109
1Introduction
Sudandyesaresyntheticazo-basedaromaticcompoundsTheyaretraditionallyusedinvariousindustriessuchaschemicaltextileandwoodworkingasdyestocolourwaxesplasticsoilspolishesandsoforthTheyhavebeencategorizedasclass3carcinogensbytheInternationalAgencyforResearchonCancerandtheiruseisthereforeforbiddeninthefoodindustryTheyareknownfortheirbrightcolours and easy and cost-effectivemanufactureThey arenearly insoluble inwater but soluble in various organic solvents such asmethanol or trichloro-methane[1] SudanI1-phenylazo-2-naphthol(Fig1A)isadyeusedasanorangecolouringagentItssometimesalsosoldundernamesSolventOrangeRorCISolventYellow14ItisformedasasecondaryproductinthemanufactureoftheSunsetYellowdye
Electrochemistry of Sudan I and its derivates in aqueous media
ad b aANNAONDRA CKOVA MARIESTIBOROVA LUDE KHAVRAN cd adKAROLINASCHWARZOVA -PECKOVA MIROSLAVFOJTA
a CentralEuropeanInstituteofTechnologyMasarykUniversity Kamenice753562500BrnoCzechRepublicannaondrackovaceitecmuniczb DepartmentofBiochemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublicc UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistry FacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublicd InstituteofBiophysicsCzechAcademyofSciences Kralovopolska13561265BrnoCzechRepublic
AbstractSudanIisanaromaticazo-compoundthathasbeenproventobeacar-cinogenDuringitsmetabolizationbycytochromeP450inliverafewmain derivates can be identified Thiswork sets out to assess themechanismofelectrochemicalreductionandoxidationofSudanIitshydroxylationderivativesfeaturingmetabolitesintheSudanIdetoxi-fication pathway and to introduce their selective voltammetricanalysis on boron-doped diamond electrode We show successfuldifferentiationamongthesecompoundsthankstothedifferencesintheelectrochemicaloxidationoftheirphenolicgroups
Keywordsborondopeddiamond
electrodecytochromeP450electrochemicalanalysisSudanI
110 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
InmammalianorganismsSudanIismetabolizedbythemicrosomaldetoxi-fying systemwitha central roleof cytochromeP450hydroxylationactivity inliver[2]DuringtheoxidativeprocessofmetabolizingSudanIseveralmetaboliteswereidentifiedbyprevioustestsThesearegt1-(phenylazo)-naphtalene-26-diol(further abbreviated SI-6OH) 1-(4-hydroxyphenylazo)-2-hydroxynaphtol(furtherabbreviatedSI-4OH)and1-(4-hydroxyphenylazo)-naphtalene-26-diol(furtherabbreviatedSI-46-diOH)thestructuresarepresentedinFig1 ThemainmethodcurrentlyusedtoidentifySudanIamongotherdyeswithsimilar structure is high-performance liquid chromatography (HPLC) It isrecommendedasthestandardmethodtoidentifythelevelofSudanIinfood[3] ComparedtoHPLCelectrochemicalmethodsareprovingtobefastercheaperandcomparablypreciseUnfortunatelyacomprehensiveelectrochemicalstudyofSudanIandparticularlyofitshydroxylatedmetaboliteshasnotbeencompletedyetThedyecanbedetectedthroughelectrochemistryeitherbytheoxidationofitsphenolicgrouporviareductionoftheazogrouppresentinitsmoleculeInbothcasesotherelectrochemicallyactivemoietiesareformedThederivatesofSudanIcanbedetectedandrecognizedfromSudanIthroughanalogousprocesses[4]InthisstudywefocusedoncomparisonofelectrochemicalbehaviourofSudanIanditshydroxylatedmetabolitesonborondopeddiamondelectrodetoaddressthepossibilitiesoftheirrecognitionsinmixturesbasedondifferencesinanodicandcathodicsignals
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 111
Fig 1 Chemical structure of (A) Sudan I (B) 1-(phenylazo)-naphtalene-26-diol (SI-6-OH)(C) 1-(4-hydroxyphenylazo)-2-hydroxynaphtol (SI-4-OH) and (D) 1-(4-hydroxyphenylazo)-naphtalene-26-diol(SI-46-diOH)
(A) (B)
(C) (D)
2Experimental
21Reagentsandchemicals
SudanI(Merckanalyticalstandardgrade)wasdissolvedinethanol(Merck)andkept at room temperatureChemicals forBritton-Robinsonbufferpreparation(acetic acid boric acid orthophosphoric acid sodium hydroxide) were fromMerckwithpurityge99pHofthebufferwasadjustedbymixingoftheacidsandsodium hydroxide solution at different ratios The Sudan I metabolites weresynthetized at the Department of Biochemistry Faculty of Science CharlesUniversityandkeptinmethanolattemperature3degC
22Instrumentation
Cyclicvoltammetric (CV)measurementswerecarriedout inBritton-RobinsonbufferofpH=70atroomtemperatureSudanIanditsderivativeswereaddedto
minus1thesolutionofBritton-Robinsonbuffertofinalconcentrationof5micromolL andstirred Before the measurement oxygen was removed from the solution bypurgingwithargonfor3minutesAutolabanalyzerPGSTAT20(EcochemieTheNetherlands)inconnectionwithVA-Stand663(MetrohmSwitzerland)GPES49(MetrohmSwitzerland)andathree-electrodesetup(withborondopeddiamond
2(WindsorScientificUKdiskdiameter3mmA=707mm )asworkingelectrodeminus1AgAgCl3molL KCl as reference electrode and platinum wire as auxiliary
electrode) Five cycles were performed for each measurement at scan rateminus1of1Vs
3Resultsanddiscussion
ForeachcompoundtwoseparateCVmeasurementswereperformedeachwithfive cycles performed in rapid succession For bothmeasurements the initialpotentialwassetat0VTheanodicscancontinuedto+1Vturnedtowardsndash1Vandfinishedat0VIntheothersetupcathodicscanwasperformedfirstfromthestartingpointtondash1Vturnedtowards+1Vandreturnedto0VThiswaywewereabletoobservethebehavioursofSudanIandcompareittothatofitsderivativeswhilefirstbeingreducedandthenoxidisedorviceversa In the anodic scan of Sudan I and its derivatives (Fig 2) differences in thepositionsoftheoxidationpeaksineachcompoundcanbeobservedWhileSudanIwithonlyonephenolgroupisoxidizedat+067VthederivativeswithtwophenolgroupsieSI-4OHandSI-6OHgiveoxidationpeaksatremarkablylesspositivepotentialofca+05VSI-46OHwiththepresenceofoverall3hydroxygroupsyieldstwooxidationpeaksInthereversecathodicscan(vertexpotential+1V)probablythankstothepresencereductionpeaksappearwhichcanbefurtherusedtodifferentiatebetweenSudanIandthederivativesSI-4OHandSI-6OH
112 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 113
Fig 3VoltammetricscanofSudanIanditsderivatesSI-4-OHSI-6-OHandSI-46-diOH1stscanincathodicdirectionfrom0Vvertexpotentialsndash1Vand+1VThemeasurementswereperformedinBritton-Robinsonbuffer(pH=7)withtheconcentrationofeachcompoundat5microMandatscan
ndash1rate1Vs
Fig 2 CyclicvoltammogramofSudanIanditsderivatesSI-4-OHSI-6-OHandSI-46-diOH1stscaninanodicdirectionfrom0Vvertexpotentials+1Vandndash1VThemeasurementswereperformedinBritton-Robinsonbuffer(pH=7)withtheconcentrationofeachcompoundat5microMandatscan
ndash1rate1Vs
possesstwoconjugatedhydroxylgroupswhichcanberegardedashydroquinonestructures and thus undergoing quasireversible redox process due to oxida-tionreductionofthehydroquinonetoquinonemoietyThisiswellvisibleattheCVsastheanodicsignalisfollowedbycathodiconeatthepotentialof+023VforSI-4OHand+03VforSI-6OHTheoxidationofSudanIproceedsbymechanism
minus +typicalforphenoliccompoundsatmorepositivepotentialsleading by1e 1H exchange to naphthoxy-type ndashO radical [5] This species undergoes furtherreactionsleadingtoformationofdimersandpolymersThecathodicpeakinthereversescanatndash02Varisesfromreductionofthesereactionproductsanditsoriginneeds tobe further investigatedSI-46OHwith thepresenceofoverallthree hydroxyl groups yields two oxidation peaks The first one is a result ofoxidationoftwoofthembeinginconjugationandthusbeingoxidizedtoquinonemoietyThesecondsignalatthesamepotentialastheoxidationsignalof2OHonnaphthaleneringofSudan I is consequenceofoxidationof the thirdhydroxylgroupofphenolictypeAsinglewidepeakat0Vinthereversescanispresumablyan overlap of signals arising from reduction of the quinonic moiety and by-products formed during oxidation processes Thanks to differences of theseprocessesspecificforindividualcompoundsitispossibletodifferentiateamongallfourofthemviapropersetting-upoftheinitialandvertexpotentialvalues The cathodic scan of Sudan I and its derivatives (Fig 3) shows a dominantreductionpeakaroundndash08Vwhichisduetoreductionoftheazogroupintheirstructuresaccompaniedbycleavageoftheirmoleculestoseparatethebenzeneandnaphthalene rings [4] The peaks in the subsequent anodic scans (vertexpotentialndash1V)arethereforetheresultoftheelectrochemicalreactionofmoietiesthatareproductsofthedivisionofthearomaticcirclesTheseproductsincludeaniline4-aminophenol1-amino-2-naphtholand1-amino-25-naphthalenediolwith irreversibly oxidizable amino moieties or (quasi)reversibly oxidizableamino-hydroxylsystemonthebenzeneornaphthaleneringandtispossibletodifferentiatebetweenthemTheobtainedoxidationpeaks(+02VforSI-4-OH+073VforSudanIandSI-6-OHand+025Vand+075VforSI-46-diOH)makeitpossibletodifferentiatebetweenallcompoundswiththeexceptionofSudanIandSI-6-OHForthereliablerecognitionofthesetwocompoundsanothermeasure-mentwithdifferentparametersisneeded
4Conclusions
ThestructuresofSudanIanditshydroxyderivatesthatarethemainproductsofthe metabolization of Sudan I by cytochrome P450 are similar and theirrecognitionwhenpresentinmixtureinsolutionisdemandingHereinwepresenta simple approach based on comparison of signals obtained in cathodic andanodic scan in CV measurements without the need of time-demandingchromatographicseparationstepFurtherworkwillbedevotedtoidentificationof observed redoxprocesses and applicationof themethod formonitoringofmetabolictransformationsofSudanIinvitro
114 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Acknowledgments
ThisresearchwassupportedbytheCzechScienceFoundation(projectNo18-01710S)
References
[1] ChailapakulOWonsawatW SiangprohW GrudpanK ZhaoYF Zhu ZW Analysis ofSudanISudanIISudanIIIandSudanIVinfoodbyHPLCwithelectrochemicaldetectionComparison of glassy carbon electrodewith carbon nanotube-ionic liquid gelmodifiedelectrodeFoodChem109(2008)876ndash882
[2] StiborovaMMartinekVRydlovaHHodekPFreiESudanIisapotentialcarcinogenforhumans Evidence for itsmetabolic activation and detoxication by human recombinantcytochromeP4501A1andlivermicrosomesCancerRes62(2002)5678ndash5684
[3] GomezMArancibiaVAliagaMNunezCRojas-RomoCDeterminationofSudan I indrinks containing Sunset yellow by adsorptive stripping voltammetry Food Chem 212(2016)807ndash813
[4] PrabakaranEPandianKAmperometricdetectionofSudanIinredchilipowdersamplesusingAgnanoparticlesdecoratedgrapheneoxidemodifiedglassycarbonelectrodeFoodChem166(2015)198ndash205
[5] Enache T A Oliveira-Brett A M Phenol and para-substituted phenols electrochemicaloxidationpathwaysJElectroanalChem655(2011)9ndash16
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 115
1Introduction
Aminoglycosidesarealargegroupofnaturalandsemi-syntheticantibioticswithawidespectrumofantimicrobialactivityagainstmostgram-positiveandgram-negativemicroorganismsCurrentlymultiplerepresentativesofaminoglycosidefamily ndash gentamicin (GM) neomycin B (NM) paromomycin (PM) kanamycin(KM)apramycin(AP) andstreptomycin(STM)Fig1ndashareapproved to treatinfectiousdiseasesinanimalsMaximumresiduelimitsfortheseaminoglycosidesinproductsandtissuesfromedibleanimalsareestablished[1]henceaneffectiveandrobustassayisnecessaryforcontrolofaminoglycosidescontamination Inthisstudyenzyme-linkedimmunosorbentassayforthedetectionofamino-glycosides in foodproductsandenvironmentalobjectswasdevelopedGroup-specificantibodieswereproducedowingtoimmunogenbasedonribostamycin(RS)whichexposedthecommonfragmentofmostaminoglycosides2-deoxy-streptamin (2-DOS) Fig 1 The developed assay was made suitable for thedetectionofresidualaminoglycosidesinhoney[2]
Group detection of aminoglycosides using ELISA for control of food contamination
ab a aKONSTANTINBURKIN INNAGALVIDIS MAXIMBURKIN
a DepartmentofImmunologyIMechnikovResearchInstituteofVaccinesandSera MalyjKazionnyjper5a105064MoscowRussianFederationburkin-kostyandexrub DepartmentofChemicalEnzymologyFacultyofChemistryLomonosovMoscowStateUniversityLeninskieGory1119991MoscowRussianFederation
AbstractThegrowingthreatofglobalantibioticresistanceisforcingtoreducenon-targetconsumptionofantibioticsandtomonitorcontaminationoffoodandenvironmentalobjectsInthisworkELISAwasdevelopedforgroupdetectionofaminoglycosidesToobtaingroup-specificanti-bodies a new immunogen based on ribostamycin was used Thedevelopedindirectcompetitiveformatofassayallowedtherecogni-tionof9aminoglycosidesnamelyneomycinribostamycinneaminparomomycin gentamicin sisomicin kanamycin tobramycin and
ndash1apramycinwithadetectionlimitrangedbetween002ndash020ngmL TheeffectivenessoftheproposedassaywasevaluatedinhoneyasafoodstuffmodelToneutralizea stronghoneymatrixeffect and toavoidalaborioussamplepre-treatmentanewmatriximitatorwassuggested 5 sucrose solution imitated the influence of 50-folddilutedhoneyTheproposedassayallowedustorevealanyofthe9
ndash1mentionedaminoglycosidesinhoneyata10microgkg level
KeywordsaminoglycosidesELISAhoney
116 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
2Experimental
21Reagentsandchemicals
NeomycinBribostamycinneamin(NA)paromomycinkanamycintobramycin(TM)amikacin(AM)gentamicinnetilmicin(NTM)sisomicin(SSM)geneticin(GC) apramycin and streptomycin were purchased from Chimmed (MoscowRussia) Bovine serumalbumin (BSA) complete Freund adjuvant 16-hexane-diamine1-ethyl-3-(3-dimethylaminopropyl)carbodiimide(EDC)sodiumperio-dateandsodiumborohydrideweretheproductsofSigma-Aldrich(USA)Gelatin(Gel)wasfromBio-Rad(USA)sucrosefromServa(Germany)two-componenttetramethylbenzidine (TMB) substrate solutionwas fromBioservice (Russia)andgoatanti-rabbitIgGantibodiesconjugatedtohorseradishperoxidase(anti-rIgG-HRP)werefromIMTEK(Russia)Honeysampleswerepurchasedfromlocaloutlets
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 117
Fig 1 Structuralformulasofstudiedaminoglycosides
22Preparationofconjugatedantigens
TwotypesofconjugateswerepreparedbasedonRSandBSAusingzero-lengthand C6 spacer arm between hapten and protein carrier RSwas treatedwithsodiumperiodatetooxidizehydroxylsofribosefragmenttoreactivealdehydegroupsandthencoupledtoBSAaminesthroughreductiveaminationToremoveuncoupledRSanexhaustingdialysiswascarriedoutusingdialysismembranetubes(MWCO14kDa)UsingthesameprocedureGel-RSconjugatewassynthe-sized ForpreparationofBSA-C6-RSwefirstlymodifiedBSAwith16-hexanediamineThemixtureofBSAandEDCinwaterwerestirredfor30minThen16-hexane-diaminewasaddedandstirredfor2hThemodifiedproteinwasdialyzedfromtheexcessive reagents and resultant BSA-C6-NH2was coupled to RS in reductiveaminationprocessasdescribedabove
23Immunizationandantibodypreparation
BSA-RSandBSA-C6-RSwereusedasimmunogensChinchillarabbits(20ndash25kg)weresubcutaneouslyinjectedat10ndash15pointsonthebackwith01mgofimmuno-gensemulsifiedinthecompleteFreundadjuvantThesamedosesofimmunogensinsalinewereadministeredmonthly forbooster immunizationsAweekaftereachinjectionabloodsamplefromearveinswastakenforthecontrolofimmuneresponseTheantiserainglycerol(11vv)werestoredatndash15degCuntiltestinginELISA
24TheELISAprocedure
AcompetitiveassaywasconductedaccordingtoclassicalprocedureGel-RSwascoatedovernightonpolystyrene96-wellCostarplatesNon-adsorbedconjugatewashedoutusingPBSwith005oftween20(PBS-T)Thenextcompetitivestep
ndash1includedtheadditionof01mLstandardaminoglycosidesolutions(1pgmL to ndash1 ndash11microgmL (B)and0microgmL (B ))inPBS-Tor01mLoftestedsampleand01mLof0
antibodiesinworkingdilution(1h25 degC)Afterwashingtheantibodiesboundtoimmobilized Gel-RS were detected using anti-rIgG-HRP (1h 37degC) Coloredproduct formedasaresultofenzymaticreactionwithTMBsubstratemixture(05h25degC)wasreadat450nmusingaStatFax2100platereader(AwarenessTechnologiesUSA) Relativeantibodybinding(BB )vstheanalyteconcentrationswasplottedas0
standardcurvesfittedtoafour-parameterlogisticfunctionThecross-reactivity(CR) for every aminoglycoside representative was calculated as ratio of half-inhibition concentrations IC NMIC aminoglycoside The dynamic range of50 50
assaywasacceptedasIC ndashIC andthelimitofdetection(LOD)wascalculatedas20 80
B ndash3timesSD0
118 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
3Resultsanddiscussion
31Immunogensynthesisandantibodypreparation
Inthemajorityofpublicationsdevotedtoimmunoassayofaminoglycosidestheimmunogenscoatingantigensenzymeconjugatesortracerswerepreparedbycarbodiimide or glutaraldehyde methods involving aminoglycosidesrsquo aminogroups [3ndash7] Due to several amino groups in aminoglycoside molecules theformationofconjugateswithavariableorientationofthehaptenoccurs InpresentstudyRSwaschosenasanimmunizinghaptenduetothefollowingadvantageous features Being a trisaccharide RS has the size of a moleculecomparable to themostof aminoglycosides Ithas three identical ringsA-B-CsimilartothoseinNMUsingaperiodateoxidationwecouldinvolvearibosesiteofRSincouplingtoproteinthatprovidedastrictorientationofhaptenonthecarrier with a favorable presentation of the 2-DOS fragment The resultantimmunogensBSA-RSandBSA-C6-RSwerecomparedtorevealwhichdesignisbetterforpresentationofacommonfragmentofaminoglycosidemoleculeandgenerationofgroup-specificantibody Antibodies to the BSA-RS demonstrated moderate sensitivity (NM
ndash1IC =10ngmL )andhighselectivitytowardsNMwithrelativelylowcross-reac-50
tivity(lt5)forGMKMandAPTheapplicationofthespacerintheimmunogenBSA-C6-RScontributedtoaprominentpresentationofthe2-DOSdeterminantandtheinductionofantibodieswithrecognitionofbroadspectrumofdifferentaminoglycosides In addition anti-BSA-C6-RS exhibited significantly better
ndash1sensitivity(NMIC =02ngmL )Thusallsubsequentstudieswereconducted50
usinganti-BSA-C6-RS
32Examinationofassayspecificityandselectionofimmunoreagents
TheindirectcompetitiveformatofassaywasdevelopedForevaluationofassayspecificity a panel of following aminoglycosideswas studied and their cross-reactivitywasdeterminedNA(625)RS(250)NM(100)KM(475)PM(173)GM(90)TM(78)AP(17)SSM(12)AM(lt01)GC(lt01)STM(lt01)andNTM(lt01)ThemostoftheseanalytesareusedinmedicalandveterinaryareashoweveronlyNMPMGMKMAPandSTMareappliedinanimalhusbandry[1]
33Determinationofaminoglycosidesinhoneyandselectionofthematriximitator
Honey is a complex product consisting of carbohydrates (75ndash80) vitaminsproteinsenzymesorganicacids traceelements inclusionsandothercompo-nentsThesecomponentsmightinterfereimmunochemicalreactionThereforetheisolationofaminoglycosidesfromhoneyisalaboriousandtime-consuming
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 119
120 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
procedureToavoidthisstephoneymatriximitatorswereappliedtomimictheinfluenceofhoneymatrixonantibodybindingSucrosewaschosenasthehoneyimitatorsinceitexposedastronghoney-matrix-likeeffectonantibodybindingTheadequacywasfoundbetweensolutionsofhoneyandsolutionsofsucroseexpressinganequalmatrixeffectTwopairswithequivalentmatrixeffectwere120honey=20sucroseand150honey=5sucroseThelatterconditionswere chosen asmore preferable due to inconvenience of operatingwith highviscous20sucrosesolution Thedeterminationofaminoglycosidesinhoneycouldbecarriedoutquantita-tivelyiftheanalytetobedetectedisknownForquantificationofaminoglycosideinhoneyasamplewasdiluted50timesinPBS-Tandaminoglycosidestandardcurvewas generated in 5 sucrose-PBST (Fig 2) If analyte is unknown thedevelopedgroup-specificELISAcanbeusedasascreeningtestInthiscasetheanalyzed sample can be considered as contaminated if it caused a relativeantibodybindingbelowthecut-off level (Fig3)Thus thedeveloped testwas
ndash1 ndash1 ndash1Analyte IC ngmL Dynamicrange LODngmL LODinhoneymicrogkg50 ndash1 IC ndashIC ngmL20 80
NM 02 003ndash21 002 10PM 07 008ndash71 005 25GM 15 015ndash133 011 55KM 035 005ndash39 004 20AP 68 05ndash968 023 115
Fig 2StandardcurvesandanalyticalparametersoftheELISA-systemforgroupdeterminationofaminoglycosidesinhoneyInteractionofanti-BSA-RSwithcoatingantigenGel-RSin5sucrosesolutionasthehoneyimitatorThedetectionlimitin5sucrosesolutionwasdeterminedaccordingtoLOD=B ndash3timesSD0
capabletorevealthecontaminationofhoneywith9aminoglycosides5amino-ndash1glycosidesapprovedforveterinary(NMPMGMKMandAP)ata10microgkg level
andalsoNARSSSMandTM
4Conclusions
A novel indirect competitive ELISA for the detection of aminoglycosides wasdevelopedRSwasusedasanewimmunizinghaptentoproducegroup-specificantibodiesagainst2-DOSacommonmoietyofalargenumberofaminoglycosideantibiotics A wide spectrum of aminoglycoside representatives could bedetectedincludingNMRSNAPMGMSSMKMTMandAPThedevelopedassay
ndash1was capable todetect theseanalyteswithaLOD up to002ndash020 ngmL ForanalysisofhoneyamatriximitatorwasdevelopedtoavoidhoneyinterferencesonimmunoassayTheanalysisofthehoneysampleallowedustorevealanyofthe
ndash1mentionedaminoglycosidesinhoneyata10microgkg level
References
[1] CouncilRegulation(EU)N372010OffJEurCommunitiesInfNotL15(2009)1ndash72[2] GalvidisIABurkinKMEreminSABurkinMA Group-specificdetectionof2-deoxy-
streptamineaminoglycosidesinhoneybasedonantibodiesagainstribostamycinAnalMeth11(2019)4620ndash4628
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 121
Fig 3Detectionofaminoglycosidesspikedinhoneysamplesata40ppblevelusinggroup-specificELISAEachsymbolcorrespondstotheaveragerelativebindingandtheerrorisSDobtainedforanindividualhoneysampleanalyzedintriplicateEmptycharactersrepresentindividualblankhoneysamples(limebuckwheatandflower)andfilledsymbolsrepresentthesamesamplesfortifiedwith
ndash1aminoglycosidesata40mgkg (level establishedonlyforSTMinseveralcountries)Thecut-offlevelcorrespondstothelimitofassaydetectionobtainedbythematriximitator(5sucrose-PBS-T)
[3] ThompsonSGBurdJFSubstrate-labeledfluorescentimmunoassayforamikacininhumanserumAntimicrobAgentsChemother18(1980)264ndash268
[4] LiCZhangYEreminSAYakupOYaoGZhangXDetectionofkanamycinandgentamicinresiduesinanimal-derivedfoodusingIgYantibodybasedic-ELISAandFPIAFoodChem227(2017)48ndash54
[5] GalvidisIABurkinMAMonoclonalantibody-basedenzyme-linkedimmunosorbentassayfortheaminoglycosideantibiotickanamycininfoodstuffsRussJBiorganChem36(2010)722ndash729
[6] HaasnootWStoutenPCazemierGLommenANouwsJFKeukensHJImmunochemicaldetectionofaminoglycosidesinmilkandkidneyAnalyst124(1999)301ndash305
[7] Peng JWangYLiuLKuangHLiAXuCMultiplex lateral flowimmunoassayfor fiveantibioticsdetectionbasedongoldnanoparticleaggregationsRSCAdv6(2016)7798ndash7805
122 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
PhotochemicalvaporgenerationisanalternativesampleintroductiontechniqueforanalyticalatomicspectrometryThistechniqueisbasedaroundasourceofUV-radiationthatirradiatesalowmolecularweightorganicacidmedium(mostcommonlyformicacidaceticacidortheircombinations)withananalyteHighlyreducingradicalsandaquatedelectronsareproducedandconverttheanalyteintoavolatilespecieswhichisthentransportedintoadetector[1]SofartheuseofPVGhasbeendescribedforhydride-formingelements(AsBiTeSbPbSeSnandTl)andmercury[12]transitionmetals(FeCoNiCuMoWCdAgAuIrPdPtRhandOs)[13ndash6]andevennon-metals(BrIClFandS)[16ndash9] AfirstsuccessfulphotochemicalvaporgenerationofcobaltwasdescribedbyGuoetalin2004[6]whichwasfollowedbymoresystematicstudiesbyGrinbergetalin2008[10]andDengetalin2010[11]LaterworksbydeQuadrosetal[12]anddeJesusetal[13]focusedontheanalysisofrealsamplesInthelatterwork
Photochemical vapor generation of cobalt for detection by inductively coupled plasma mass spectrometry
ab aJAROMIRVYHNANOVSKY STANISLAVMUSIL
a DepartmentofTraceElementAnalysisInstituteofAnalyticalChemistryoftheCzechAcademyofSciencesVeveřiacute9760200BrnoCzechRepublicjaromirvyhnanovskygmailcom
b DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublic
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 123
AbstractThisworkfocusedonthephotochemicalvaporgenerationofcobaltVolatilespeciesweregeneratedinaflow-injectionsystememployinga high-efficiency flow-through UV photoreactor and a formic acidbased medium and were introduced by an argon carrier into aninductivelycoupledplasmamassspectrometerfordetectionOptimalgeneration conditions were found as 10 (vv) formic acid and
ndash1 ndash14molL ammonium formate with a 4mLmin flow rate whichcorresponds to irradiation time of around 13 s The influence ofvariousmetalsensitizersofphotochemicalreactionwasinvestigated
2+andonlyCu ionsexhibitedapositiveeffectongenerationefficiencyndash ndash 2ndashInterferencesfromcommoninorganicanions(NO Cl SO ) were3 4
also examined Lastly the limit of detection and repeatability (atndash1 ndash1250ngL )weredeterminedtobe13ngL and41respectively
Keywordscobaltinductivelycoupled
plasmamassspectrometry
photochemicalvaporgeneration
the authors also presented a systematic study on generation conditions andachievedagenerationefficiencyofaround40 Themainaimofthisworkwastooptimizetheconditionsofgenerationwithinductivelycoupledplasmamassspectrometry(ICP-MS)detectionexaminetheeffectofvariousmetal sensitizers toachieve thehighestgenerationefficiencypossibleandreachthelowestlimitofdetectionpossible
2Experimental
21Reagentsandchemicals
minus1Deionizedwater (DIW lt 02μScm UltrapurWatrex USA)was used for thepreparationofallsolutionsFormicacid(98paLach-NerCzechRepublic)andammonium hydroxide (ge25 pa Sigma-Aldrich USA) were used for the
minus1preparationof the reactionmediumA1000mgL Co stock solution (Sigma-AldrichUSA)wasusedforthepreparationofallsamplesolutionsThefollowingcompounds were used as potential metal sensitizers cadmium(II) acetatedihydrate(paLach-NerCzechRepublic)zinc(II)acetatedihydrate(paSigma-AldrichUSA)copper(II)acetatemonohydrate(paMerckGermany)nickel(II)acetatetetrahydrate(paSigma-AldrichUSA)sodiumtungstatedihydrate(paCarlRothGermany)and iron(II) sulphateheptahydrate (pa LachemaCzechRepublic)Nitricacid(65semiconductorgradeSigma-AldrichUSA)hydro-chloricacid(37paMerckGermany)andsulfuricacid(98paLach-NerCzechRepublic)wereusedforaninterferencestudy
22Instrumentation
AschematicdiagramofthePVGsystemcoupledtoICP-MSisshowninFig1andamore detailed description can be found in reference [5] Briefly a singlequadrupole ICP-MS Agilent 7700x (Agilent Technologies USA)was used as a
minus1detector Deionized water was mixed with a 10μgL Rh internal standardsolutionin2HNO andwassubsequentlynebulizedbyaMicroMistnebulizer3
59 103during PVG Isotopes of Co and RhweremonitoredMeasurementswereperformed in time resolved analysis mode and in He collision mode
minus1(41mLmin )AlltubingusedwasmadefromPTFEwiththeexceptionoftygontubing in the peristaltic pump (Reglo ICC Ismatec Switzerland) The high-efficiency flow-through photoreactor was a 19 W low-pressure mercurydischarge lamp (Beijing Titan Instruments Co Beijing China) with a quartzcentralchannel(asymp720μL internalvolume)Samplesolutionswere introducedintoastreamofreactionmediumusinganinjectionvalve(V-451IDEXHealthandScienceUSAsampleloopvolume05ml)Effluentfromthephotoreactorwasmixedwithaflowofargonandcarriedtothechilledgas-liquidseparator(internalvolume15mL)wherethevolatilespecieswereseparatedfromtheliquidwaste
124 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
andcarried to the inletofaScott-typespraychamber (originally the inlet formakeupargon)oftheICP-MS
3Resultsanddiscussion
The starting conditionswere adopted fromour earlierwork [14]whichusedatomicabsorptionspectrometerasadetectorandminiaturediffusionflameasanatomizerThefirstparameteroptimizedwasthecompositionofreactionmedium(Fig2)Theadditionofammoniumformate(createdin-situbytheadditionofacalculatedamountofammoniumhydroxidetoformicacid)wasfoundcrucialto
ndash1effectivelygeneratevolatilespeciesofcobalt10(vv)formicacidand4molL ammonium formatewas chosen as the optimumandwasused for all further
experimentsAlthoughhigher concentrations of both components led to even
higher signalstheseconditionswerenotusedfurtherbecauseofthelaboriousprocessofpreparation(mixingofconcentratedacidwithconcentratedbase)andtolimittheconsumptionofchemicals Theinfluenceofirradiationtimewasalsoexaminedandthehighestpeakarea
-ndash1wasobtained for4mLmin corresponding to an irradiation timeof approximately13s ToenhancethegenerationefficiencyadditionofvariousmetalstothereactionmediumwastestedtoldquosensitizerdquothephotochemicalreactionThemetalswerechosenwithrespecttotheirsignificantenhancementeffectdescribedrecentlyforphotochemicalvaporgenerationofotheranalytes[158]Theonlymetalionthat
2+ledtoanenhancementofthesignalwasCu (Fig3a)buteveninthiscasetheeffect was rather negligible reaching only 12-fold enhancement in the range
ndash1 2+ 2+001to01mgL Cu FurtheradditionofmoreCu ledtoadecreaseinthesignal2+TheadditionofZn didnotexhibitanypositiveornegativeeffectacrossthetested ndash1range (01 to 1500mgL not shown in figure) and the addition of higher
ndash1 2+ 2+ 6+concentrations (tens to hundredths ofmg L ) of Cd Fe andW (Fig3b)causedsevereinterferences
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 125
ndash ndash 2ndash Interferencescausedbycommoninorganicanions(NO Cl SO addedas3 4ndashtheirrespectiveacids)werealso investigatedOutof theseNO was foundto3
ndash1causethemostsevereinterferencesevenatconcentrationsofsinglemmolL Thendash 2ndashmethodologywasmorerobusttowardstheinterferencesfromCl andSO but4
theystillcausedsignificantdropinsensitivityathigherconcentrationsConsi-deringthewideuseoftheseacidsinanalyticalchemistryforsamplepreparationthisposesabigchallengeintheapplicationofthismethodtorealsamples
ndash1 Usingoptimalconditions(10(vv)formicacid4molL ammoniumformateandirradiationtimeof13s)acalibrationcurvewasmeasuredandevaluatedThelimitofdetectionwasdeterminedas3timesthestandarddeviationof10blank
ndash1measurements and was calculated as 13 ng L The repeatability of 10ndash1consecutivemeasurementsof250ngL was41
126 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 3Effectofvariousmetalionsonthepeakarea(a)metalionswithapositiveeffect(b)metalionsndash1withoutapositiveeffectExperimentalconditions2microgL Coreactionmedium10(vv)formic
ndash1 ndash1acidand4molL ammoniumformateflowrate4mLmin
ndash1Fig 2Effectofthecompositionofreactionmediumonpeakareaexperimentalconditions2microgL ndash1Coreactionmediumflowrate4mLmin (blackdotscorrespondtomeasuredpoints)
4Conclusions
Theconditionsofthephotochemicalvaporgenerationofcobaltwereoptimizedand are in good agreement with previous works [11 13] Copper ions wereidentifiedasapotentialsensitizerincreasingthesignalbyabout12-foldbuttheirpotentialuseisseverelylimitedbythenarrowrangeofconcentrationsinwhichthepositiveeffectisexhibitedSevereinterferencesfrominorganicanionswereobservedwhichisinlinewithotherworksdealingwithphotochemicalgeneration[1358]Furtherexperimentswillfollownamely(i)furtherinvestigationsinnewpotentialsensitizerstoenhancegenerationefficiencyandthusdecreasethe
ndash1limit of detection to sub ng L levels (ii) determination of the generationefficiency(fromcomparisonwithnebulizationandorusingaradioactiveisotope58Co)(iii)verificationoftheaccuracyandpracticalfeasibilityofthismethodologybyanalysisofcertifiedreferencematerials
Acknowledgments
ThesupportofTheCzechScienceFoundation(ProjectNo19-17604Y)CzechAcademyofSciences(Institutional supportRVO68081715)andCharlesUniversity (project SVV260560andprojectGAUK60120)isgratefullyacknowledged
References
[1] SturgeonREPhotochemicalvaporgenerationaradicalapproachtoanalyteintroductionforatomicspectrometryJAnalAtomSpectrom32(2017)2319ndash2340
[2] XuTHuJChenHJTransitionmetalionCo(II)-assistedphotochemicalvaporgenerationofthalliumforitssensitivedeterminationbyinductivelycoupledplasmamassspectrometryMicrochemJ149(2019)103972
[3] SoukalJSturgeonREMusilSEfficientphotochemicalvaporgenerationofmolybdenumforICPMSdetectionAnalChem90(2018)11688ndash11695
[4] deOliveiraRMBorgesDLGUVphotochemicalvaporgenerationofnoblemetals(AuIrPdPtandRh)AfeasibilitystudyusinginductivelycoupledplasmamassspectrometryandseawaterasatestmatrixJAnalAtomSpectrom33(2018)1700ndash1706
[5] VyhnanovskyJSturgeonREMusilSCadmiumassistedphotochemicalvaporgenerationoftungstenfordetectionbyinductivelycoupledplasmamassspectrometryAnalChem91(2019)13306ndash13312
[6] GuoXSturgeonREMesterZGardnerGJVaporgenerationbyUVirradiationforsampleintroductionwithatomicspectrometryAnalChem76(2004)2401ndash2405
[7] HuJSturgeonRENadeauKHouXZhengCYangLCopperionassistedphotochemicalvapor generation of chlorine for its sensitive determination by sector field inductivelycoupledplasmamassspectrometryAnalChem90(2018)4112ndash4118
[8] LeonoriDSturgeonREAunifiedapproachtomechanisticaspectsofphotochemicalvaporgenerationJAnalAtomSpectrom34(2019)636ndash654
[9] SturgeonREPaglianoEEvidenceforphotochemicalsynthesisoffluoromethaneJAnalAtomSpectrom(2020)httpsdoiorg101039D0JA00108B
[10] GrinbergPMesterZSturgeonREFerrettiAGenerationofvolatilecobaltspeciesbyUVphotoreduction and their tentative identification J Anal Atom Spectrom 23 (2008)583ndash587
[11] DengHZhengCB LiuLWWuLHouXDLvYPhotochemicalvaporgenerationofcarbonyl for ultrasensitive atomic fluorescence spectrometric determination of cobaltMicrochemJ96(2010)277ndash282
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 127
[12] deQuadrosDPBorgesDLDirectanalysisofalcoholicbeveragesforthedeterminationofcobalt nickel and tellurium by inductively coupled plasmamass spectrometry followingphotochemicalvaporgenerationMicrochemJ116(2014)244ndash248
[13] deJesusHCGrinbergPSturgeonRESystemoptimizationfordeterminationofcobaltinbiologicalsamplesbyICP-OESusingphotochemicalvaporgenerationJAnalAtomSpectrom31(2016)1590ndash1604
[14] VyhnanovskyJFotochemickegenerovanıtekavychspeciı kobaltuproanalytickouatomovouspektrometriiMasterthesisFacultyofScienceCharlesUniversityPrague2018
128 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Oneof themethods thatallowobtainingmaterialswithnewproperties is theplasmaenhancedchemicalvapordepositionmethodInthismethodcompoundscalledprecursorsaresuppliedtotheplasmareactorasagasphaseThankstoplasmaenhancedchemicalvapordeposition it ispossible toobtainmaterialswithuniquepropertiesThisisduetothefactthattheplasmaaffectsthesurfaceinfourdifferentways etching cleaning chemicalmodification and crosslinking
Optimization of condition for cold plasma deposition of thin layers for surface modification of working electrodes
a b a cJUSTYNALIPIN SKA MARIAMADEJ BOGUSŁAWBAS JACEKTYCZKOWSKI
a DepartmentofAnalyticalChemistryFacultyofMaterialsScienceandCeramicsAGHUniversityofScienceandTechnologyAdamaMickiewicza3030-059KrakoacutewPolandjustynalipinskaaghedupl
b DepartmentofAnalyticalChemistryFacultyofChemistryJagiellonianUniversityinKrakoacutewGronostajowa230-387KrakoacutewPoland
c Departmentof MolecularEngineeringFacultyofProcessandEnvironmentalEngineeringLodzUniversityofTechnologyWolczanska21390-924ŁoacutedźPoland
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 129
AbstractCurrentlyresearchisfocusedonthesearchfornewphysicallyandchemicallystablematerialsaswellasvolumeorsurfacemodificationOneofthemethodsusedforsurfacemodificationistheapplicationofthin layers from inorganic and organic compounds The plasmaenhancedchemicalvapordepositionisamethodthatallowsmaterialmodificationandalsodepositionofthinlayersThisworkconcernsoptimizationofcoldplasmadepositionparametersandtoachievethebestelectrical conductivitywhilemaintaining thehighmechanicalstrength of the formed layers Preliminary tests were focused onoptimizing the layering parameters such as the deposition timedischargepowerpressureofmonomerandthe flowofargonTheobtainedsamplesweresubjectedtothermaltreatmentafterwhichtheywere coveredwitha layerof aluminumThe thicknessof theobtained layers was determined on the basis of interferencemicroscopymeasurementsAsaresultoftheexperimentslayerswithathicknessof20nmto600nmwereobtainedTheconductivityofthedeposited layers was also determined and values from 003 to
ndash1150Sm wereobtained
Keywordscoldplasmadepositionelectrochemical
applicationssurfacemodificationthinlayers
Thismethodisusedtoproducecatalyticstructuresortomodifythepropertiesofmaterials eg improve hydrophobicity The growing popularity of surfacemodificationmethodsusingcoldplasmaisassociatedwiththefactthatitisanenvironmentallyfriendlyandversatilemethod[12] Workingelectrodesusedinvoltammetryareasubgroupofchemicalsensorswhich are small devices that convert real-time chemical information into ameasurableandanalyticallyusefulmeasurementsignalChemicalinformationrangingfromtheconcentrationofaspecificcomponentofthetestedsampletotheoverallcompositionofthematrixcancomefromboththeinitiatedchemicalreactionandbetheresultofphysico-chemicaltransformationstakingplaceinthetested object Chemical sensors are equipped with two basic elements iereceptor and transducer The receptor is responsible for the conversion ofchemicalinformationfromthetestedobjectintoaspecificformofenergyintheconverterthisenergyistransformedintoausefulanalyticalsignal Parameters characterizing the electrochemical sensor include accuracyprecision selectivity accuracy presentation selectivity sensitivity dynamicrange limit of quantification limit of detection lifetime response time andreliability Themost numerous and the oldest group of chemical sensors areelectrochemicalsensorsCommonlyobservedinterest inthisgroupofsensorsresultsfromthefactthatwithrelativelylowproductionandoperatingcoststheyofferthebestmetrologicalandoperationalparameters[3ndash5]Oneofthemaintrendsofmodernanalyticsisthesearchfornewelectrodematerialsandvariousgeometries of working electrodes One way to improve the performance ofworkingelectrodesistomodifytheirsurfaceforexamplebyapplyingthinlayersIn this work were considered plasma enhanced chemical vapor depositionmethod as the method of surface modification designed to performworkingelectrodeforvoltammetricdeterminationsAspartoftheinitialresearchplasmaprocessing parameters such as discharge power time of treatment andcompositionofgasmixtureinwhichplasmawasgeneratedwereoptimizedThelayers obtained in different conditions have been tested for suitability forelectrochemical applications (layer thicknessmeasurement and themeasure-mentofconductivity)
2Experimental
21Reagentsandchemicals
Theprecursor solutions suchasacrylonitriledietoxydimethylsilane trietoxy-methylsilaneandtetramethyldisiloxanewhicharesuppliedbyABCRwereusedOtherreagentsofanalyticalpuritysuchasn-hexane(SigmaAldrich)andargonwereused
130 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
22Instrumentation
The thin layers were deposited in a parallel-plate plasma reactor (frequency1356MHz)ThesamplesobtainedwerecalcinedinatunnelfurnaceunderanargonatmosphereThethicknessofthedepositedlayerswasmeasuredafterthealuminum was sputtered using a Nikon microscope type ECLIPSE LV150NElectrometerhigh resistance system (KEITHLEY) was used to measureconductivity
3Resultsanddiscussion
Eachof themonomerswasdepositedonprepared1times1corningglasssamplesSamples prepared with n-hexane were placed in a plasma reactor andadditionallypartiallycoveredwithamicroscopecoverslipSchematiclayoutofsamplesinthereactorshowninFig1ThefirststepwastoetchthesystemusingargonplasmaThisstageallowedfortheeliminationofimpuritiesthatwerenotremovedbythehelpofn-hexaneandthepreparationofthesurfaceofthesamplesforthedepositionoftheproperlayerTheproperstageistheapplicationofathinlayerwiththeplasmainducedbytheselectedprecursoracrylonitriledietoxydi-methylsilanetrietoxymethylsilaneandtetramethyldisiloxaneThethicknessandpropertiesoftheobtainedlayersdependonthedepositionparameterssuchasdischarge power time of treatment and composition of gasmixture inwhichplasmawasgenerated Table1showsallcombinationsofparameterstestedforallfourprecursorsFour different discharge powers for acrylonitrile and two different dischargepowersfororganosiliconmonomersweretestedwithtwodifferenttreatmenttimesEachtimeandpowercombinationwasperformedinplasmainducedbypuremonomerandmonomerwithargonAfterapplyingthelayersthesampleswereplacedinaquartzboatandcalcinedinatunnelfurnaceAllsampleswerecalcinedat500degCfor2hoursunderargonflowSamplesaftercalcinationwerecoveredwithalayerofaluminum
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 131
Fig 1 Scheme of sample distribution in plasma reactor(a)reactorelectrode(b)corningglasssamples(c)micro-scopecoverslip
Thethicknessoftheobtainedlayerswasmeasuredusinganinterferencemicro-scopeMeasurements were carried out at amagnification 10times recording theimageinmonochromelightTheimagewassetsothattheinterferencefringeswere perpendicular to the arc on the sample In order to calculate the layerthicknessD[nm]thefollowingformulawasused
(1)
wheredandLweredeterminedonthebasisoftheregisteredimage(disfringeshiftduetorefractionoflightontheslopeLisdistancebetweenthefringes) Thelaststageofthestudywastodeterminethecurrent-voltagecharacteristicstodeterminetheconductivityoftheobtainedlayersThesamplewasplacedinameasuring cell and attachedwith silver paste to the electrometerwires ThechangeincurrentwasrecordedwiththeapplicationofalternatingvoltageintimeBasedontheresultsobtainedthegraphsofdependenceUndashIwereobtainedfromwhichthevalueofresistance(R)wasdeterminedAnexampleofcurrent-voltagecharacteristicsisshowninFig2
132 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Table 1Conditionsforlayersdeposition-parameterwhichweretested
Monomer DischargepowerW Timeoftreatmentmin
Acrylonitrile 10204080 24Dietoxydimethylsilane 2040 255Trietoxymethylsilane 2040 255Tetramethyldisiloxane 2040 255
Fig 2Current-voltagecharacteristicsdeterminedfortheacrylonitrilelayer(depositionparametersW=10Wt=25mingasmixtureonlyacrylonitrile)
Table 4Theresultsofthicknessmeasurementsandconductivityspecifictothetrietoxymethylsilanelayers
DischargepowerW 20 20 40Timeoftreatmentmin 25 5 5Argonflowsccm 10 10 10Thicknessnm 14686 24282 35311
ndash1SpecificconductivitySm 165 93 67
Knowing the value of the resistance and the geometry of the system thespecificresistancewasdeterminedfollowedbythespecificconductivityofthesamplethefollowingequationwasused
(2)
whereρisspecificresistance[Ωm]Risresistance[Ω]bissamplelength[m]Disdepositedlayerthickness[m]andldistancebetweenelectrodes(wires)[m] Theresultsofthicknessmeasurementsandspecificconductivityarepresentedin the Tables 2ndash5 For electrochemical applications it is important that theobtainedlayerhasthehighestspecificconductivityAnalyzingthedatapresentedin Tables 2ndash5 shows that the thinnest layers have the greatest applicationpotentialastheelectrodematerial
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 133
Table 2Theresultsofthicknessmeasurementsandconductivityspecifictotheacrylonitrilelayers
DischargepowerW 40 40 80 80 10 10 20 20Timeoftreatmentmin 2 4 2 4 2 4 2 4Thicknessnm 15845 31167 250 4644 247 67 7531 12236
ndash1SpecificconductivitySm 0002 156 191 0003 1512 454 311 185
Table 3The results of thickness measurements and conductivity specific to the dietoxydimethylsilanelayers
DischargepowerW 20 20 40 40Timeoftreatmentmin 25 5 25 5Argonflowsccm 10 10 10 10Thicknessnm 13998 37519 28431 49556
ndash1SpecificconductivitySm 1640 55 105 40
Table 5The results of thickness measurements and conductivity specific to the tetramethyldisiloxanelayers
DischargepowerW 20 40 40Timeoftreatmentmin 25 25 5Argonflowsccm 10 10 10Thicknessnm 16759 3159 61652
ndash1SpecificconductivitySm 213 95 31
4Conclusions
InthisworkplasmaenhancedchemicalvapordepositionmethodwasusedforapplyinglayersoffourdifferentmaterialsacrylonitriledietoxydimethylsilanetrietoxymethylsilaneandtetramethyldisiloxaneBychangingparameterssuchasdischargepowertimeoftreatmentandcompositionofgasmixtureanumberofsampleswereobtainedwithlayersofdifferentthicknessandwhatisassociatedwithotherelectricalproperties TestsperformedaspartofthisworkwereusedtoperforminnovativeworkingelectrodesforvoltammetricdeterminationsThesurfacewasmodifiedusingthecoldplasmaofthreesubstratesgraphiteglassycarbonandgold
Acknowledgments
JLandMMhavebeenpartlysupportedbytheEUProjectPOWR030200-00-I00416
References
[1] KapicaRTyczkowskiJBalcerzakJMakowskiMSielskiJWorwaEEnhancingadhesivejointsbetweencommercialrubber(SBS)andpolyurethanebylow-pressureplasmasurfacemodificationIntJAdhesAdhes95(2019)102415
[2] TyczkowskiJKapicaRŁojewskaJThincobaltoxidefilmsforcatalysisdepositedbyplasma-enhancedmetalndashorganicchemicalvapordepositionThinSolidFilms515(2007)6590ndash6595
[3] HulanickiAGłabSIngmanFChemicalsensorsDefinitionsandclassificationPureApplChem63(1991)1247ndash1250
[4] BrzozkaZWroblewskiWSensorychemiczneWarszawaOficynaWydawniczaPolitechnikiWarszawskiej1999(InPolish)
[5] SkoogDAWestDWHollerFJCrouchSRFundamentalsofAnalyticalChemistry9thEdBostonCengageLearning2013
134 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
ConcentrationandcompositionofvolatilecompoundsorcongenersisoneofthemostimportantparametersresponsibleforqualityofproducedalcoholicdrinksandhencefortheirsensorycharacteristicsandconsumeracceptanceTodaygaschromatography (GC) is conventionally used to determine qualitative andquantitativecompositionsofvolatilecompoundswithvariousexternalandorinternalstandardcalibrationprocedures Methodemployingethanol as an internal standard (IS) forGCquantitativedeterminationofvolatilecompoundsinalcoholicbeverageshasbeensuggestedquitelongago[1]andsincethattimegreatresearchworkhasbeencarriedoutRecentlyaninterlaboratorystudyofthemethodinvolving9testinglaboratoriesfrom4countrieswascarriedout[2]TheresultsdemonstratedgreatperspectivesofldquoEthanolasISrdquomethodandproveditsreferencecharacterandeaseofroutineimplementation
Advanced GC-MS method for quality and safety control of alcoholic beverages
abANTONKORBAN
a DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova2030812840Prague2CzechRepublickarbonat7gmailcomb DepartmentofAnalyticalChemistryChemistryFacultyBelarusianStateUniversityLeningradskaya14220050MinskBelarus
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 135
AbstractRecently developed and validated simple and reliable quantitativemethod employing ethanol as an internal standard for GC-MSquantification of volatile compounds in alcoholic products wasapplied to 36 samples including commercially available world-famousbrandspirits from18countriesandhomemadedistillatesThe GC-MS analyses were performed simultaneously by thesuggested approach and official internal standard method that isprescribedinthelegislationofEUandUSATheindependentsamplest-testwasemployedtoevaluatethestatisticaldifferenceofresultsofthesetwomethodsThetestrevealednodifferenceintheresultsandtheirrepeatabilityThemainbenefitsofthesuggestedmethodaretheeliminationofthenecessityofmanualinternalstandardadditionandsamplesdensitymeasurementthusmakingitmoreeconomicalandproductive
Keywordsalcoholicbeveragesgaschromatography-
massspectrometry(GC-MS)
internalstandardmethodvolatilecompounds
quantification
AllpreviousstudieswereutilizingflameionizationdetectorssinceGC-FIDisprescribedinthelegislation[3]wheremass-spectrometrydetectorsarenotyetofficially referred However GC-MS instruments are employed in practice toqualifyandorquantifyvolatilesincommercialspiritsintraditionalhomemadealcoholicdrinksinnewlydevelopedbeveragesinspiritwastesandindistillatesobtainedwithdifferentmanufacturingprocesses Our recent researchwasdirected towardsdevelopment of an algorithmofldquoEthanolasISrdquomethodapplicationonGC-MSinstruments[4]WehaveshowedthattopreventMSdetectorfromsaturationethanolshouldberegisteredinthecorresponding SIM timewindow at characteristicmz of low abundance forinstance bymz of 47 ions This ion corresponds to non-fragmented ethanol
13moleculescontaining1heavyisotope(mainly C)FinallytheresultsofmeasuredstandardsolutionsshowedthatthesuggestedapproachisvalidandldquoEthanolasISrdquomethodmaybesuccessfullyusedonGC-MSinstrumentstoo The objective of this studywas to test and further approve the suggestedapproachonalargersetof36realsamplesofalcoholicdrinkseithercommercialorhomemadeThesamplesweresimultaneouslyanalysedbytwoGC-MSmethodsndashaclassicalISmethodprescribedinthelegislationandthesuggestedldquoEthanolasISrdquomethod
2Experimental
21Reagentsandchemicals
Thefollowingvolatilecompoundsweredeterminedintestedsamplesofalcoholicbeverages 11-diethoxyethane (acetal) acetaldehyde methyl acetate ethylacetate methanol 2 propanol 1-propanol 2 methylpropan-1-ol (isobutanol)1-butanol2-butanoland3methylbutan-1-ol(isoamylalcohol)1-pentanolwasemployedasatraditionalIScompound
22Instrumentation
ShimadzuGCMS-QP2010Ultraequippedwithaquadrupolemassspectrometrydetector was employed for GC-MS measurements Rxi-1301Sil MS capillarycolumn(60mlength025mmid025micromfilmthicknessRestek)wasusedfortheseparationofcompoundsInjectionswereperformedinasplitmode(ratio175)Helium(99999purity)wasusedasacarriergasinjectortemperaturewas170degCTheoventemperaturewasheldat30degCfor5minthenraisedto210degC
ndash1at a rate of 30 degCmin and held isothermally for 4minMeasurementswereperformedinaSIMmodeFortheanalysedcompoundsand1-pentanol2ndash3mostabundantionsinthecorrespondingMSspectrumwereselectedethanolSIMtimewindowcontainedonly47mzionsAllGC-MSmeasurementswerecarriedoutintriplicateunderrepeatabilityconditions
136 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
AnalysisofeachalcoholicsamplewasperformedinafollowingwayAliquotof09mL of a tested sample was pipetted into a standard 2mL glass vial and
ndash1weighedAfterthat01mloftheISsolution(2355mgkg of1-pentanolinWES)wasaddedtothetestedsampleandthemasswasrecordedTheobtainedmixturewasmixedthoroughlyand05microlofitwasinjectedintotheGCsystem Theoriginoftestedalcoholicbeverageswaseithercommercialorhomemade33 world-famous spirits manufactured at different parts of the world werepurchasedfromcommerciallyavailablesourcesThelistoftypesofpurchasedandanalysedspiritsincludedbourboncalvadoscognacgingrappaliquormetaxaportwine rumsake tequilavodkawhiskeyandvarious fruitdistillatesThepurchased drinks were produced at the territory of the following countriesBelarus Bermuda Cuba Czech Republic Denmark France Germany GreeceGuatemala Jamaica Japan Mexico Moldova Portugal Slovakia Trinidad andTobago UK (England and Scotland) USA Three homemade fruit distillatesproducedbyfermentationofpulpyfruitsortheirmustswereobtainedfromlocalspiritmakersThedeclaredABVvaluesofalltestedsamplesvariedfrom15to81
3Resultsanddiscussion
To fulfil themaingoalof thiswork ie toevaluate thestatisticaldifferenceofresultsyieldedbythecomparedmethodswehaveemployedStudentst-testforindependentsamplestoverifystatisticaldifferencesonthesignificancelevelofp=005Theobtainedempiricalvaluesforallpairsofcongenersrsquoconcentrationswere lower than critical one in all cases demonstrating that concentrationsobtainedbythetwomethodshavenostatisticaldifferenceandleadtothesameresults InadditionrepeatabilityofthetwomethodswascomparedthereforeallRSDvalues obtained from triplicatemeasurements were split in two groupswith
ndash1respecttothecorrespondingconcentrations(lowerthan50mgL AAandhigherndash1than50mgL AA)Theobtainedresultsarepresentedintheformofboxplotin
Fig1AnalysisofthechartinFig1showedthatboththetestedmethodshaveyieldedstatisticallysimilarrepeatability AllofthetestedalcoholicdrinkssatisfiedtherequirementsofEURegulation(EC)no1102008[5]Theconcentrationsofundesirablecompoundssuchasmethanoldidnotexceedthelevelsspecifiedinthesameregulationforcorres-ponding beverages In Table 1 the description of the used SIM method andsummaryoftheexperimentalresultsarepresented Tocomparethetruenessofthemethodsoneofthespiritsampleswasspikedwithstandardsolutions(ABV40)containingallanalysedvolatilecompoundsat
ndash1concentrationsof50500and5000mgL AATheoriginalsamplewasusedasareferenceEachofthespikedsolutionswasmeasuredintriplicateSelectedspirit(cherry distillate) initially contained all 11 volatile compounds in various
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 137
138 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1BoxchartsofRSDsofusedISmethodsat2concentrationrangesMeanisequaltoarithmeticmeanoraverageInterquartileRange(IQR)meansisthedistancebetweentheupper(themedianoftheupperhalfofthedataset)andlower(themedianofthelowerhalfofthedataset)quartile
ndash1Compound Timemin Registeredmz Numberof ConcentrationmgL AA results Minimal Maximal
Acetaldehyde 0ndash42 314344 36 24 715Methanol 36 13 13600
Ethanol(IS) 42ndash48 47 mdash mdash mdash
2-Propanol 48ndash70 29314345 14 27 199Methylacetate 596174 10 34 3201-Propanol 26 361 12070Ethylacetate 27 166 107002-Butanol 11 18 2080
Isobutanol 70ndash200 3141ndash4345 28 19 20001-Butanol 5556 13 28 155Acetal 26 45 270Isoamylol 31 39 26501-Pentanol(IS) mdash mdash mdash
Table 1DescriptionoftheusedSIMmethodandsomestatisticsconcerningallmeasured36spiritsamplesbothpurchasedandhomemade
concentrationsTheobtainedrecoveriesboxchartsareshowninFig2Compa-risonoftherecoveriesobtainedwithtwomethodsindicatesthattheyhavenosignificantdifferenceintermsoftruenessAverageobtainedrecoverywas981whenusingsuggestedmethodand980whenusingtraditionalISmethod
4Conclusions
InthisworktheresultsoftestingtheadvantageousldquoEthanolasISrdquomethodfortheGC-MS quality control analysis of alcoholic beverages were presented33purchasedsamplesofworld-famousalcoholicbeveragesoriginatingfrom18countriesand3homemadefruitdistillateswereanalysedtomakeathoroughandcomprehensive studyof thedevelopedmethodTheconcentrationsofvolatile
ndash1compoundsinanalysedsamplesvariedfrom1to13500mgL AAtheABVvalueofanalysedsamplesvariedfrom15to81ThesuggestedmethodwascomparedwiththetraditionalISmethodthatiscurrentlystatedinlegislationTheindepen-dentsamplest-testrevealedthatwithaprobabilityof095resultsobtainedwithtwo methods do not differ significantly The results of within-run precision(repeatability)showedrelativestandarddeviationswithin3measurementstobelessthat6indicatingthatthetechniqueisreproducibleThetruenessofthemethodwasevaluatedbyrecoverycalculationAccordingtotheobtainedresultsrecoveryofthesuggestedmethod(981plusmn33)wasslightlybetterthanthatofthetraditionalone(980plusmn58) ThesefactsprovethatdevelopedldquoEthanolasISrdquomethodistruepreciseandreliable when employed on GC-MS instruments At the same time to obtain
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 139
Fig 2Boxchartsof recoveriesof thesuggested(dottedpattern)and traditional (brickpattern)ISmethodsatdifferentspikeconcentrationsSymbolsdefinitionsarethesameasinFigure1
concentrationsofvolatilecompoundsintheofficiallyrequiredunitsofmeasurendash1 ndash1(mgL AAgL AAetc)suggestedmethodrequiresnodensitometrymeasure-
mentsofthetestedsampleandnoadditionofIScompoundoranyothersamplepre-treatmentThismethodprovidesaninvaluableanalyticaltoolforthequalitycontrolofalcoholicproductsandshouldbeusedinroutineanalysis
Acknowledgments
ThisworkwasfinanciallysupportedbytheVisegradFund
References
[1] CherepitsaSVBychkovSMKovalenkoANMazanikALSeleminaNMSeredinskayaOBThe use of themajor component (solvent) as an internal standard in the gas-chromato-graphicdeterminationofimpuritiesJAnalChem58(2003)368ndash371
[2] CharapitsaSSytovaSKorbanASobolenkoLEgorovVLeschevSZakharovMCabalaRBusarovaRShestakovichITolstouhovaAOndrousekSVavraJYilmaztekinMCabarogluTInterlaboratorystudyofethanolusageasaninternalstandardindirectdeterminationofvolatile compounds in alcoholic products BIO Web Conf 15 (2019) 02030httpsdoiorg101051bioconf20191502030
[3] CommissionRegulation(EC)No28702000layingdownCommunityreferencemethodsfortheanalysisofspiritsdrinkshttpdataeuropaeuelireg20002870oj
[4] KorbanACharapitsaSCabalaRSobolenkoLSytovaSTheperspectivesofethanolusageasaninternalstandardforthequantificationofvolatilecompoundsinalcoholicproductsbyGC-MSJMassSpectr55(2020)e4493
[5] EuropeanUnion(2008)Regulation(EC)No1102008oftheEuropeanParliamentandoftheCouncilof15January2008ontheDefinitionDescriptionPresentationLabellingandtheProtectionofGeographical IndicationsofSpiritDrinksandRepealingCouncilRegulationhttpdataeuropaeuelireg2008110(1)oj
140 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Carbonorgraphitefeltsareusedaselectrodematerialsincethe1990sandtheirutilizationstillgrowsinanalyticalelectrochemistryaswellasinotherareasThisisduetotheirsuitablepropertiesfromwhichwecannamehighporosityhighspecificsurfaceareagoodelectricconductivityandhighphysicalandchemicalstabilityThefirsttwoparametersaregivenbystructureoffeltwhichconsistsoforderlesscarbonfibreswithabouttentotwentymicrometersindiameter[1]theothersby theadvantageouselectricalpropertiesof carbon fibreOn theotherhand porous flow-through electrodes including carbon felt electrode havedisadvantageinapotentialdropintheelectrodevolumewhichcausesdifficultcontrollingoftheexactpotentialappliedontheelectrodeandthereforeresultsindifferentcurrentefficienciesontheoppositesidesoftheelectrode[2] Carbon felt electrode can be utilized for detection of structurally differentcompoundsatvariousconditionsForexampleoperatingatreductionpotentialofndash08V[3]oxidationatrelativelyhighpotential+15V[4]ormeasuringatlowconcentrationsofelectrolyte[5]canbenamedDevelopedtechniquesalsoshowsthatcarbonfeltcanbeusedfordeterminationatsubmicromolarconcentrationsThis ismainly due to its ability to operate as a high-efficiency amperometricdetector Themainaimofthispaperistooverviewandcompareparametersofseveraldetermination methods of different analytes using carbon felt detector in
Utilization of a carbon felt as a material for working electrodes
MARTINBAROCHHANADEJMKOVA SA RKASLA DKOVA
DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublicmartinbarochnaturcunicz
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 141
AbstractWorkingelectrodemadeofcarbonfeltwasusedincombinationwithHPLC for verificationofpractical applicabilityof the electrodeAlldeveloped methods confirm advantageous physical and chemicalpropertiesofcarbonfeltForelectrochemicalutilizationitispossibleto operate at higher positive potentials and even in low concen-trationsofelectrolyteinmobilephaseObtainedlimitsofdetectionwere mostly in submicromolar range and standard deviations ofmeasurementrepeatabilitywereunder5
KeywordsamperometrycarbonfeltFIAHPLC
combinationwithHPLC(forexampletheirdetectionpotentialslimitsofdetectionorlinearrange)
2Experimental
21Reagentsandchemicals
Stock solutions of propyl gallate butylhroxyanisole tert-butylhydroquinonebutylhydroxytoluene chlortoluron 2-amino-4-nitrophenol and 4-amino-2-
ndash3 ndash1nitrophenol (all SigmandashAldrich) with concentration of 1times10 molL werepreparedbydissolving theappropriateamountof therespectivesubstance inmethanol(HPLCgradeLach-NerCzechRepublic)Stocksolutionsof indole-3-aceticacidandindole-3-butyricacidwerepreparedinthesamemannerbutindeionized water Mobile phase consisted of methanol and phosphate-acetatebufferpreparedfromphosphoricandaceticacid(bothLach-NerCzechRepublic)andsodiumhydroxide(Fluka)
22Instrumentation
Theelectrochemicalcellconsistedofcarbonfelt(KarbotechnikCzechRepublic)flow-throughelectrodewhichwasplacedincapwithplatinumwireelectricalcontactanddrilledoutletholeononesideandwithflatferrulawithcapillaryonthe other side Schematic picture of the assembly is shown in ref [5] Otherelectrodeswereauxiliaryplatinumwireelectrodeandreferencesilverchloride(3MKCl) electrode (bothMonokrystaly Turnov Czech Republic) Potentiostatused in combination with this cell was Amperometric Detector ADLC 2(LaboratonıprıstrojePrahaCzechRepublic) HPLCapparatusconsistedofBeta10gradientpump(ECOMCzechRepublic)degasser DG 4014 (ECOM Czech Republic) six-way valve with 20microl loop(Rheodyne USA) HPLC column used for separation of propyl gallatebutylhydroxytoluenetert-butylhydroquinoneandbutylhroxyanisolefromtheirmixtureandforindole-3-aceticacidandindole-3-butyricacidfromtheirmixture
regwasLichrospher RP-18(125times4mm5micromMerckGermany)ForseparationofregchlortoluroncolumnPurospher RP-18(125times4mm5micromMerckGermany)was
used In case of mixture 2-amino-4-nitrophenol and 4-amino-2-nitrophenolregcolumnGemini C18110A (150times46mm5micromPhenomenexUSA)wasusedfor
separation Measurements of pHwere carriedout at Conductivity andpH-meter3510usingcombinedglasselectrode(JenwayUK)
142 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
3Resultsanddiscussion
Performanceofcarbonfeltwastestedonseveraltypesofanalyteswhichneededdifferent separation conditions namely amount of organic solvent in mobilephase and buffer pH The lowest amount of methanol (30) was used forseparation of 2-amino-4-nitrophenol and 4-amino-2-nitrophenol [3] Higherconcentrations of methanol in mobile phase was used for determination ofchlortoluronandforseparationofindole-3-aceticacidandindole-3-butyricacid[4]namely40and60respectivelyThehighestconcentrationsofmethanoland therefore electrolyte with lowest conductivity was used in separation ofantioxidantsnamelypropylgallatebutylhroxyanisolebutylhydroxytolueneandtert-butylhydroquinonewhereamountofmethanolwasrampingfrom55to95[5]DetectionpotentialsofmentionedanalytesandtheirlimitsofdetectionsareshowninTable1 HPLCseparationofantioxidantswas the firstmethodchosen for testingofcarbonfeltelectrodeperformancewiththistechniqueDuetodifferencesintheirstructure when butylhydroxytoluene has a different oxidation mechanismdetectionwith two appliedpotentialswas necessary According to the hydro-dynamic voltammograms potentials 14V and 08Vwere chosen for determi-nationofbutylhydroxytolueneandfortheotherthreeanalytesrespectively[5]AsshowninTable1whenthehigherpotentialwasappliedlimitsofdetectionforpropyl gallate butylhroxyanisole and tert-butylhydroquinone had increasedContrary determination of butylhydroxytoluene had approximately six timeslowerdetectionlimitathigherpotential
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 143
ndash1Compound E VLODmicromolL Refdet
Propylgallate 080 088 [5] 140 186 [5]Butylhroxyanisole 080 144 [5] 140 348 [5]tert-Butylhydroquinone 080 121 [5] 140 266 [5]Butylhydroxytoluene 080 3128 [5] 140 463 [5]Chlortoluron 140 013Indole-3-aceticacid 150 033 [4]Indole-3-butyricacid 150 054 [4]4-Amino-2-nitrophenol 080 016 [3]2-Amino-4-nitrophenol 080 021 [3]4-Amino-2-nitrophenol ndash080 35 [3]2-Amino-4-nitrophenol ndash080 37 [3]
Table 1Parametersofdetectionpotentialsandlimitsofdetectionfordifferentcompoundsusingcarbonfeltdetector
Detectionofauxins(indole-3-aceticacidandindole-3-butyricacid)wascarriedout at potential 15VMeasurements at this potential gives repeatabilitywithstandarddeviation31forindole-3-aceticacidand25forindole-3-butyricacidevenwithexchangingoftheworkingelectrodematerialCalibrationcurves
ndash1forbothanalyteswereobservedfrom04to100μmolL withlinearityinwholeconcentrationrangeLimitsofdetectionforbothanalytesreachedsubmicromolarconcentrationsevenwithrelativelyhighpotential[4] Incaseofdeterminationof2-amino-4-nitrophenoland4-amino-2-nitrophenolcarbon felt electrodewasused inbothoxidationand reductionmodeHydro-dynamicvoltammogramsshowedthatoptimaldetectionpotentialinreductionmodewasndash08VThislowpotentialisclosetotheendofthepotentialwindowandthereforeinterferenceswithremnantsofdissolvedoxygeninmobilephasewereobservedTheseinterferencesresultedinapproximately20timeshigherlimitofdetection for 2-amino-4-nitrophenol or 4-amino-2-nitrophenol obtained inreductionthaninoxidationmodeOntheotherhandmaximumvaluesoflinearrangewerethesameforbothanalytesinbothdetectionmodes[3] ForHPLCofchlortolurontheoptimaldetectionpotentialof14VwasfoundIts
ndash1calibration dependence although observed from 025 to 1000μmolL was ndash1linearonly in therange from025to50μmolL Limitofdetectionbasedon
ndash1standardsolutionswas013μmolL andreproducibilityofmeasurementgivenbytwentyconsecutivemeasurementsgaverelativestandarddeviationof05 ForallthedeterminationmethodsattentionwaspaidtotheapplicabilityofthecarbonfeltindetectionofanalytesincomplexmatricesIncaseofantioxidantsedibleoilswerechosenasrealsamples[5]nitrophenolderivatesweredeter-minedinurinesamples[3]andauxinsinrootingpreparation[4]ChlortolurondeterminationwasperformedinsoilandsurfacewaterThefoundvaluesshowanegligiblematrixinfluenceondetection
4Conclusions
CarbonfeltworkingelectrodewassuccessfullyusedincombinationwithHPLCfordeterminationofdifferenttypesofelectroactivecompoundsegantioxidantsauxinsorpesticidesAllmentionedapplicationsshowgreatperformanceofthecarbonfeltasaflow-throughelectrodematerialinelectroanalyticalchemistryforoxidationandreductionwayofanalytesdeterminationLimitsofdetectionforanalytes are mostly in submicromolar concentrations the exceptions areoxidationof analytes athigherpotentials and their reductionwhere limitsofdetectionsareinmicromolarconcentrationsApplicabilityoftheelectrodeonrealmatriceswas proven on analysis of edible oil samples groundwater soil androotingpreparation
Acknowledgments
ThisworkhasbeensupportedbytheCzechScienceFoundation(projectGACR20-01589S)
144 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
References
[1] Gonzalez-Garcia J Bonete P Exposito E Montiel V Aldaz ATorregrosa-Macia RCharacterizationofacarbonfeltelectrodeStructuralandphysicalpropertiesJMaterChem9(1999)419ndash426
[2] NavaJLRecendizAGonzalezLGCarrenoGMartın ezFMassTransportandpotentialstudiesinaflow-throughporouselectrodereactorPortugalElectrochimActa27(2009)381ndash396
[3] Dejmkova H Knaf M Application of carbon felt detector for the determination ofdinitrophenolmetabolitesInXXXIXModernElectrochemicalMethodsFojtaMSchwarzovaKNavratilT(Eds)U stınadLabemBestServis2019p41ndash43
[4] DejmkovaHdeAraujoDanielMElectrochemicaldeterminationofindole-3-aceticacidandindole-3-butyric acid using hplc with carbon felt detectorMonatsh Chem150 (2019)439ndash442
[5] DejmkovaHBarochMKrejcovaMBarekJZimaJCoulometricdetectorbasedoncarbonfeltApplMaterToday9(2017)482ndash486
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 145
1Introduction
Smith-Lemli-Opitzsyndrome(SLOS)isan autosomal recessive genetic disor-der firstly described in 1964 [1] It iscaused by the inborn deficiency of7-dehydrocholesterol reductase Thisenzyme transforms 7-dehydrocholes-terol (7-DHC Fig 1) to cholesterolduringthefinalstepofbiosynthesisofcholesterolincells The clinical symptoms of SLOS aredecreasedbloodlevelofcholesteroland
Electroanalytical methods for determination of 7-dehydrocholesterol in artificial serum
LENKABENESOVAADE LAZA RYBNICKA JANKLOUDAKAROLINASCHWARZOVA -PECKOVA
UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublicbenesolenaturcunicz
Abstract7-DehydrocholesterolisabiomarkerofSmith-Lemli-Opitzsyndromeanautosomalrecessivegeneticdisordercausedbytheinborndefici-encyof7-dehydrocholesterolreductaseInthisstudyproceduresforitsdeterminationinartificialserumusingflowinjectionanalysiswithelectrochemical detection and voltammetric detection on borondoped diamond electrode were optimized The proteins wereprecipitatedbyacetonitrileandaftercentrifugationthesupernatantused for analysis For quantitation of 7-DHC by differential pulsevoltammetrytheoptimalratioacetonitrile-artificialserum91(vv)wasappliedInFIA-EDtheratio31(vv)runelectrolyteconsisting
minus1ofwater-acetonitrilecontaining001molL NaClO inthesameratio4minus1anddetectionpotentialof+13VvsAgAgCl(3molL KCl)were
usedQuantitationof7-DHCwaspossibleusingcalibrationdepen-minus1dencewithlimitdetectionof20micromolL inartificialserumNeverthe-
lessthemethodhaslowrecoveryandforsensitivedeterminationinreal matrices of human serum and amniotic fluid a liquid-liquidextraction needs to be applied to prevent presence of 7-dehydro-cholesterolinthephasewithprecipitatedproteins
Keywordsamperometricdetectionborondopeddiamond
electrode7-dehydrocholesteroldifferentialpulse
voltammetryflowinjectionanalysisSmith-Lemli-Opitz
syndrome
146 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Structureof7-dehydrocholesterol
increased concentrationof7-DHC inbloodandnervous system [2] SLOS is acomplexofmultipleanomaliesincludingmentalretardationItismanifestedbyholoprosencephaly(anomaliesinbraindevelopmentwithimprecisedivisionintotherightandlefthemispheres)milddysmorphismscardiacrenalandgastro-intestinalmalformations ThecharacteristicfacialanomaliesofSLOS[2]aremicrocephalybitemporalnarrowing ptosis short nasal root short nose with anteverted nares andmicrognathia epicanthal folds and capillary hemangioma over the nasal rootextendingontotheglabellatheearappearlow-setandareposteriorlyrotatedOral finding includes a high-arched and narrow hard plate broad and ridgealveoralridgesandredundancyofsublinqualtissuesCNSanomaliesareagenesisorhypoplasiaBilateralandunilateralpostaxialpolydactylycanbepresentedinthehandsorfeetorboth Concentrationof7-DHCinbloodiscrucialforclinicaldiagnosticofSLOSinpatients Concentration levels in amniotic fluid are used for fetal diagnosticsTable1 summarizes concentration of 7-DHC in plasma and amniotic fluid ofhealthypersonandSLOSpatientsAnalyticalmethodsusedfordeterminationofconcentrationof7-DHCinthesematricesincludecombinationofGCorHPLCwithMS[3]orGCwithflameionizationdetection(FID)orUVdetection[45] Thepossibilitiesofelectrochemicalmethodsfordetectionof7-DHCarelimitedasgenerallythesteroidcoreisratherredox-inactive(detailinreview[6])undervarietyofconditionsNevertheless7-DHCpossessesconjugateddoublebondsonsteroidcoreanditsoxidationwasreportedinseveralstudies[7ndash9]Itsvoltam-metricsignal+095VvsSCEonglassycarbonelectrodewasfirstlyobservedin
minus1non-aqueousmediaofmethanol-benzene7525(vv)using005molL LiClO as4
supporting electrolyte in a study dealing with electrochemical behaviour ofvitaminAandDandtheirprovitaminsD(7-DHCisprecursorofvitaminD3)[7]Determinationof7-DHCinhumanskinispossiblebyHPLCwithUV(λ=286nm)andamperometricdetection(E +17VvsAgAgCl)onglassycarbonelectrodedet
minus1usingmethanol-tetrahydrofuran175mmolL KH PO (9514vvv)asmobile2 4minus1phase7-DHCwasdetectedintherangefrom12to81microgg dryweightwith
minus1detectionlimitof39pmolL [8]Anotherstudy[9]isdevotedtodeterminationof
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 147
minus1Author(s)ref Matrix ConcentrationμmolL Healthy SLOS
Kelley[4] Plasma 03plusmn001 385plusmn309 Amnioticfluid lt02plusmn001 16plusmn9Rossiteretal[5] Plasma lt5 179ndash335 Amnioticfluid lt03 12ndash15
Table 1Concentrationof7-dehydrocholesterolinclinicalmatricesforhealthypersonsandforSmith-Lemli-Opitzsyndrome(SLOS)patientsdetectedbyGC-FID
7-DHCandvitaminD3 in fishusingHPLCwithelectrochemicaldetectionTheanalytical cell was a serial combination of two-flow-through porous graphiteworking electrodes The first standard coulometric electrode was used toeliminatepotentially interfering compounds using the second lineardynamic
minus1rangefrom0013to0312micromolL for7-DHCwasachieved Hereinwestudiedpossibilitiestodetect7-DHCbasedonitsoxidationonborondoped diamond (BDD) electrode using differential pulse voltammetry andelectrochemicaldetectioninflowinjectionanalysis(FIA-ED)inartificialserumandperipherallyinhumanserumandamnioticfluid
2Experimental
21Reagentsandchemicals
7-dehydrocholesterol (purity95)wasobtainedSigmaAldrich (USA)and itsstandard solution was prepared in acetonitrile (Honeywell Germany) Theartificial serum was prepared from KCl (Penta Czech Republic) CaCl 2H O2 2
(PentaCzechRepublic)NaCl(PentaPragueCzechRepublic)urineD-glucoseand01albuminfromSigma-Aldrich(USA)NaClO4(PentaCzechRepublic)wasusedassupportingelectrolyte
22Instrumentation
VoltammetricmeasurementweregovernedbythepotentiostatPalmSensusingworking BDD electrode (Windsor Scientific UK d = 31 mm) AgAgNO 3
minus1 minus1(01molL AgNO 1molL NaClO in acetonitrile) non-aqueous reference3 4electrodeandplatinumwirecounterelectrodeBDDsurfacewaspolishedbeforeeachscanusingsuspensionofAl O (ElektrochemickedetektoryTurnovCzech2 3
Republic) HPLC system (Hitachi Merck) consisting of control unit D-7000gradientpumpL-7100autosamplerL-7200andUVdetectorL-7400wasusedfor
minus1FIA-EDdetectionof7-DHCRunelectrolytewascomposedof001molL NaClO 4inacetonitrileanddeionisedwaterinratio31(vv)Flowrateofmobilephase
minus1was30mlmin injectionvolumewas40microLandλ=280nmwasusedforUVdetectionWall-jet detection cell was employedwith working BDD electrode
minus1AgAgCl (3molL KCl) reference electrode and platinum wire auxiliaryelectrode Optimal detection potential of +13 V was controlled using ADLC2potentiostat(Laboratornı prıstrojePragueCzechrepublic)
3Resultsanddiscussion
In this study electroanalytical methods were developed for determination of7-DHCinartificialserumnamelyFIA-EDandDPVBothmethodsarebasedondirectoxidationof7-DHConborondopeddiamondelectroderesultinginanodic
148 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
peak at ca +08 V (vs AgAgNO in acetonitrile) in non-aqueous medium of3
acetonitrile or mixedmedium acetonitrile-water using NaClO as supporting4
electrolyteTheoxidationispresumablyinitiatedbyoneelectronremovalfromtheconjugateddoublebondsonthesteroidcoreof7-DHC Fordeterminationof7-DHCinrealmatricesitisnecessarytoremovepresentproteinsArtificialserumcontainingalbuminwasusedasmodelmatrixtostudythe possibilities Firstly albumin was removed simply by precipitation withacetonitrile(serum-water13(vv))andthesupernatantwasanalysed Differentratioswateracetonitrileweretestedinrunelectrolyte(59510902080 2575 3070 4060 and 5050 (vv)) to evaluate the influence of itscompositionontheFIA-EDsignaloftheblankand7-DHCThesameratio13asusedforprecipitationofalbuminwaschosenasoptimalbecauseofminimalandstablesignaloftheblankinjectedinFIA-EDsystemFurtherdetectionpotentialE in the range from +10 V to +15 V was optimized by evaluation of thedet
hydrodynamicvoltammogramsresultinginE of+13Vsetasoptimalvaluedetminus1 Concentrationdependenceof7-DHCislinearintherangefrom25micromolL to
minus1 minus1300micromolL (concentrationinartificialserum)withdetectionlimitof20micromolL and this concentration dependence can be used for quantitation of 7-DHC inartificial serum Nevertheless determination of 7-DHC in human serum andamnioticserumfailedastheyrepresentmorecomplicatedmatricesand7-DHCispresumablypartiallyadsorbedinthepresentproteinsandcannotbequantifiedinthesupernatant Furtherdifferentialpulsevoltammetrywithoptimizedparameterswasusedfordeterminationof7-DHCInthepresenceofproteinsinartificialserum(human
minus1serum albumin) an unacceptably high detection limit of 178micromolL wasachieved When the proteins were precipitated using acetonitrile (aceto-nitrileartificialserumratio91(vv))thelimitofdetectionof7-DHCwaslowered
minus1to15micromolL inartificialserumNeverthelesstherecoveryofthemethodwasonly43to70dependingontheconcentrationof7-DHCagainreflectingthelossof7-DHCduetoproteinprecipitation Thereforeasecondapproachofsamplepretreatmentbasedonliquid-liquidextractionofalllipidsdescribedin[10]wastested(Bligh-Dyerextraction)TheprocedurehastwopartsFirstlymethanolchloroformandthesampleofartificialserum is mixed and shaken to form a monophasic system After addition ofchloroform and water a biphasic system is formed where chloroform phasecontainsalloflipidcompoundsandmethanol-waterphasecontainsallnon-lipidscompoundsChloroformphaseisthendriedunderN2atmosphereat50degCanddried extract dissolved in acetonitrile Preliminary experiments using DPVresultedinrecoveryof97forBligh-Dyerextractionof7-DHCfrominartificialserum
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 149
4Conclusions
FIA-EDandDPVwereoptimizedfordeterminationof7-DHCinartificialserumUsing precipitation of proteins by acetonitrile limit of detection of 7-DHC in
minus1artificialserumusingFIA-EDwas20micromolL andthismethodcanbeusedfortheirquantificationusingcalibrationdependenceNeverthelessdeterminationusingDPVisunreliableduetolowrecoveryoftheprocedureDevelopmentofamethodincludingliquid-liquidextractionstepisinprogresssothat7-DHCcouldbedeterminedinrealmatricesashumanserumandamnioticfluid
Acknowledgments
TheresearchwassupportedbytheCzechScienceFoundation(projectGACR19-11268S)andtheSpecificUniversityResearch(SVV260560)
References
[1] Smith DW Lemli L Opitz JM A newly recognized syndrome of multiple congenitalanomaliesJPediatr64(1964)210minus217
[2] Nowaczyk M Waye J The SmithndashLemlindashOpitz syndrome a novel metabolic way ofunderstandingdevelopmentalbiologyembryogenesisanddysmorphologyClinGenet59(2001)375minus386
[3] BeckerSRohnikeSEmptingSHaasDMohnikeKBebloSMutzeUHusainRAThieryJCeglarekULC-MSMS-basedquantificationofcholesterolandrelatedmetabolitesindriedblood for the screening of inborn errors of sterolmetabolismAnal Bioanal Chem407(2015)5227minus5233
[4] Kelley RI Diagnosis of Smith-Lemli-Opitz syndrome by gas-chromatography mass-spectrometryof7-dehydrocholesterolinplasmaamniotic-fluidandculturedskinfibroblastsClinChimActa236(1995)45minus58
[5] Rossiter JP Hofman KJ Kelley RI Smith-Lemli-Opitz SyndromePrenatal-diagnosis byquantification of cholesterol precursors in amniotic-fluid Am J Med Genet 56 (1995)272minus275
[6] KloudaJBarekJNesmerakKSchwarzova-PeckovaKNon-enzymaticelectrochemistryincharacterization and analysis of steroid compounds Crit Rev Anal Chem 47 (2017)384minus404
[7] AtumaSSLundstromKLindquistJTheelectrochemicaldeterminationofvitaminAPartIIFurthervoltammetricdeterminationofvitaminAandinitialworkonthedeterminationofvitaminDinthepresenceofvitaminAAnalyst100(1975)827minus834
[8] MoodyJPHumphriesCAAllanSMPatersonCRDeterminationof7-dehydrocholesterolinhumanskinbyhigh-performance liquid-chromatography JChromatogrB530 (1990)19minus27
[9] OstermeyerUSchmidtTVitaminDandprovitaminDinfishEurFoodResTechnol222(2005)403minus413
[10] BlighEGDyerWJArapidmethodoftotallipidextractionandpurificationCanJBiochemPhysiol37(1959)911minus917
150 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Author Index
AlikovaV1
AugustınM83
BaluchovaS19
BarekJ192570
BarochM141
BasB13129
BastryginaO41
BenesovaL146
BessonovaE57
BohmD6
BurkinK116
BurkinM116
ChernovaA141
ChoinskaM70
CokrtovaK104
DedinaJ97
DeevV57
DejmkovaH141
DendisovaM63
DubenskaL51
EfremenkoE41
FojtaM110
GalvidisI116
HavranL110
HeiglN31
HertJ76
HrdlickaV70
JosypcukB25
KartsovaLA3557
KloudaJ19146
Kodrık ovaB90
KolobovaEA35
KorbanA135
KorotkovaE1
KralM63
KratzerJ90
KravchenkoAV35
KrızekT76104
LipinskaJ129
MadejM129
MatejkaP63
MatysikF-M631
MusilS9097123
NavratilT70
OndrackovaA110
PietrzakK45
PlotnikovaK51
PoradaR13
RedondoBR70
SagapovaL90
ShormanovV1
Schwarzova-PeckovaK19110146
SladkovaS 141
S tadlerovaB97
StiborovaM110
SvobodaM90
TvorynskaS25
TyczkowskiJ129
VyhnanovskyJ97123
VymyslickyF76
VyskocilV83
WardakC45
WongDKY19
ZarybnickaA146
ZelenyI51
Proceedingsofthe16thISCModernAnalyticalChemistry Prague2020 151
152 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Keyword Index
alcoholicbeverages135
aminoglycosides116
amperometricdetection146
amperometry141
antifoulingelectrodes19
assembledcapillaries6
atomicabsorptionspectrometry90
atomicfluorescencespectro-
metry97
atomization90
biologicalactiveanalytes35
biosensor2583
bismuth97
borondopeddiamond
electrode110146
cadmium90
canagliflozin76
capillarycoating35
capillaryelectrophoresis635
capillaryflowinjectionanalysis631
carbohydrates31
carbonfelt141
cathodicstrippingvoltammetry70
chemicalvaporgeneration90
chemometrics57
cobalt123
coldplasmadeposition129
copper(II)phthalocyanine63
covalentimmobilization25
cytochromeP450110
damage83
7-dehydrocholesterol146
designofexperiments76
differentialpulsevoltammetry146
diphenylsilanereductionmethod19
dispersiveliquid-liquid
microextraction57
disposableelectrodes31
DNA83
dualdetectionconcept6
electrochemicalanalysis110
electrochemicalapplications129
electrochemicalflowcell76
electrochemistry51
electrokineticchromatography104
eliminationvoltammetrywith
linearscan70
ELISA116
enzymaticreactor25
FIA141146
gaschromatography-mass
spectrometry(GC-MS)135
glucoseoxidase25
graphite83
honey116
HPLC76141
hydridegeneration97
hydrogenatedconical-tipcarbon
electrodes19
imidazoliumionicliquids35
inductivelycoupledplasmamass
spectrometry123
internalstandardmethod135
ion-selectiveelectrode45
laccase25
liquidcrystals104
massspectrometry31
mercuryelectrode13
metronidazole51
non-aqueouscapillary
electrophoresis104
non-aqueoussystem6
oxidation76
oxytetracyclinehydrochloride51
phenol2-methoxy1
photochemicalvaporgene-
ration97123
polarography51
1-propanesulfonicacid23-
dimercapto-70
pulsedamperometricdetection31
quantitation1
resonanceRamanspectroscopy63
scanningtunnellingmicroscopy63
silversolidamalgamelectrode70
Smith-Lemli-Opitzsyndrome146
smokingmixtures41
solidcontact45
solid-phasemicroextraction57
spectrophotometry141
SudanI110
surfacemodification129
surface-enhancedRaman
spectroscopy63
thinlayers129
tip-enhancedRamanspectroscopy
63
unithiol70
uranyl45
vanillin41
veterinarydrug51
vitamins13
volatilecompoundsquantifi-
cation135
voltammetricdopaminedetec-
tion19
voltammetry1383
Proceedingsofthe16thISCModernAnalyticalChemistry Prague2020 153
Proceedings of the 16th International Students Conference ldquoModern Analytical Chemistryrdquo
EditedbyKarelNesmerak
PublishedbyCharlesUniversityFacultyofScience
Prague2020
1steditionndashvi154pages
ISBN978-80-7444-079-3
ISBN 978-80-7444-079-3
Pro
ceedin
gs of th
e 16
th In
ternatio
nal Stu
den
ts Co
nferen
ce ldquoMo
dern
An
alytical Ch
emistryrdquo P
rague 2
02
0
788074 440793
Prague 17mdash18 September 2020
Edited by Karel Nesměraacutek
Prague 2020
Proceedings of the
16th International Students Conference
ldquoModern Analytical Chemistryrdquo

CATALOGUING-IN-PUBLICATIONndashNATIONALLIBRARYOFTHECZECHREPUBLIC
KATALOGIZACEVKNIZEndashNA RODNIKNIHOVNACR
ModernAnalyticalChemistry(konference)(162020PrahaCesko)
Proceedingsofthe16thInternationalStudentsConferenceldquoModernAnalyticalChemistryrdquo
Prague17ndash18September2020editedbyKarelNesmerak--1stedition--PragueFacultyof
ScienceCharlesUniversity2020--vi154stran
Obsahujebibliografiearejstrıky
ISBN978-80-7444-079-3(brozovano)
543(062534)
analyticalchemistry
proceedingsofconferences
543ndashAnalyticalchemistry[10]
TheelectronicversionoftheProceedingsisavailableattheconferencewebpage
httpwwwnaturcuniczisc-mac
copyCharlesUniversityFacultyofScience2020
ISBN978-80-7444-079-3
Preface
Despitethefactthattheyear2020ismarkedbyCOVID-19morethan40young
analytical chemists gathered in Prague for the 16th annual international
conferenceldquoModernAnalyticalChemistryrdquoTheymeettopresenttheresultsof
theirresearchtomastertheirpresentationandlanguageskillsandtoexchange
anddiscussideasandexperiencesofanalyticalchemistry
Thisvolumeofconferenceproceedingsbringsyouatotalof25papersfrom
thisconferenceAsinpreviousyearsthecontributionspresentedareassortedby
the sequence of their delivery supplemented by indexes at the end of the
proceedingsallowingeasynavigationthroughthepagesYouwillseethattopics
of contributions cover all the aspects of modern analytical chemistry from
theoretical problems through development of new analytical methods and
improvementofanalyticaltechniquestotheapplicationsinvolvingthesolutionof
medicinaltechnicalorenvironmentalproblemsLetushopethatlikeprocee-
dingsofpreviousyearsofourconference thisonewillalsobean interesting
beneficialandenjoyablereading
Itseemstousthattheauthorsofthecontributionsareaguaranteeofthatanew
generationofanalyticalchemistswillprotectbrightandthrillingfutureofour
science
We are very grateful to the Division of
AnalyticalChemistryofEuChemSforitslong-
lasting auspices of our conference Also we
arethankfultooursponsorsnotonlyfortheir
kind sponsorship making the conference
possiblebutalsoforalltheircooperationand
supportinmanyofourotheractivities
Enjoyreadingtheseproceedings
docRNDrKarelNesmerakPhD
editor
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 iii
Sponsors
The organizersof16th International Students Conference ldquoModernAnalytical
Chemistryrdquo gratefully acknowledge the generous sponsorship of following
companies
wwwecomsrocom
wwwlach-nercom
iv Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
wwwthermofishercz
www2thetacz
wwwzentivacz wwwquintacz
wwwshimadzueucom
wwwwaterscom
Contents
AlikovaVChernovaAShormanovVKorotkovaEDeterminationof2-methoxyphenol inmodelsolutionsbyspectrophotometry 1
BohmDMatysikF-MTheeffectsoflinearlyassembledcapillarieswithvariousinnerdiametersoncapillaryelectrophoresis 6
PoradaRBasBVoltammetricdeterminationofvitamins 13Baluchova S Klouda J Barek J Schwarzova-Peckova K Wong DKY Dopamine detection at
antifoulingconical-tipcarbonelectrodes 19Tvorynska SBarek J JosypcukBA comparative study of covalentglucose oxidaseand laccase
immobilizationtechniquesatpowderedsupportsforbiosensorsfabrication 25Heigl N Matysik F-M Capillary flow injection analysis with electrochemical detection for
carbohydrateanalysis 31KravchenkoAVKolobovaEAKartsovaLAApplicationofcovalentcoatingsbasedonimidazolium
cations for separationandon-linepreconcentrationof basicandneutralanalytes in capillaryelectrophoresis35
Efremenko E Chernova A Bastrygina O Determination of vanillin in smoking mixtures byspectrophotometry41
PietrzakKWardakCUranylion-selectiveelectrodewithsolidcontact 45Plotnikova K Dubenska L Zeleny I Polarographic determination of metronidazole and
oxytetracyclinehydrochlorideinveterinarydrugforhoneybees 51Deev V Bessonova E Kartsova L Application of microextraction techniques combined with
chromatographicmethodsfortheanalysisofcomplexobjects 57KralMDendisovaMMatejkaPThedevelopmentofreferenceprobesystemfortip-enhancedRaman
spectroscopy 63ChoinskaMHrdlickaVRedondoBRBarekJNavratilTDeterminationofheavymetalpoisoning
antidote23-dimercapto-1-propanesulfonicacidusingsilversolidamalgamelectrode70VymyslickyFKrızekTHert JCanagliflozinoxidation studyusingelectrochemical flowcelland
comparisonwithhydrogenperoxideoxidation 76AugustınMVyskocilVNovelhybridelectrochemicalDNAbiosensorformonitoringoxidativeDNA
damageviaoxidationreductionsignalsoflowmolecularweightdouble-strandedDNA 83SagapovaLKodrık ovaBSvobodaMMusilSKratzerJChemicalvaporgenerationofcadmiumfor
analyticalatomicspectrometry 90S tadlerova B Vyhnanovsky J Dedina J Musil S Photochemical vapour generation of bismuth
coupledwithatomicfluorescencespectrometry 97Cokrtova K Krızek T Separation of liquid crystals using non-aqueous capillary electrokinetic
chromatography104OndrackovaAStiborovaMHavranLSchwarzova-PeckovaKFojtaMElectrochemistryofSudanI
anditsderivatesinaqueousmedia 110BurkinKGalvidisIBurkinMGroupdetectionofaminoglycosidesusingELISAforcontroloffood
contamination 116Vyhnanovsky J Musil S Photochemical vapor generation of cobalt for detection by inductively
coupledplasmamassspectrometry 123LipinskaJMadejMBasBTyczkowskiJOptimizationofconditionforcoldplasmadepositionofthin
layersforsurfacemodificationofworkingelectrodes 129KorbanAAdvancedGC-MSmethodforqualityandsafetycontrolofalcoholicbeverages 135Baroch M Dejmkova H Sladkova S Utilization of a carbon felt as a material for working
electrodes 141Benesova L Zarybnicka A Klouda J Schwarzova-Peckova K Electroanalytical methods for
determinationof7-dehydrocholesterolinartificialserum146
Authorindex 151Keywordindex 152
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 v
1Introduction
2-Methoxyphenol(guaiacol)isusedinmedicineasanexpectorantThestructuralformulaisshowninFig1Itiswidelyusedinthepharmaceuticalindustry[1]forthesynthesisofantituberculosisexpectorantdrugs(Kas-nol Sudafed Ascoril Prothiazine Expectorant Guai-phenesinum) Moreover 2-methoxyphenol is often used as anaromaticsubstance [2] in the food industry Inparti-cular itwaswidelyusedintheproductionofsmokedfish and meat products using smokeless smokingtechnologyusingflavourings Ontheotherhand2-methoxyphenolhasthesymbolGHS07andhasahazardcodeXnTXi[2]accordingtotheGHSsystemItisverytoxicbyinhalationitcanirritatethemucousmembraneoftherespiratorytractandtheconjunctivaofthe
Determination of 2-methoxyphenol in model solutions by spectrophotometry
a a b aVALERIYAALIKOVA ANNACHERNOVA VLADIMIRSHORMANOV ELENAKOROTKOVA
a DepartmentofChemicalEngineeringEngineeringSchoolofNationalResourcesNationalResearchTomskPolytechnicUniversityLeninavenue30634050TomskRussiaalikovaleramailru
b DepartmentofPharmaceuticalToxicologicalandAnalyticalChemistryKurskStateMedicalUniversitystKarlaMarks3305000KurskRussia
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 1
AbstractA spectrophotometric approach for determination of 2-methoxy-phenol in model solutions has been developed The absorptionspectra of 2-methoxyphenol were determined in the wavelengthrangefrom200to400nminsolutionsof95ethanolacetonitrile01Msodiumhydroxideandethylacetatewithaconcentrationofthe
minus3analyte of 005mgdm For the quantitative determination of2-methoxyphenol a seriesof solutionswaspreparedwithvarious
minus3 minus3concentrationsfrom0001mgdm to005mgdm in95ethanolacetonitrile01MsodiumhydroxideTheopticaldensityof2-meth-oxyphenolinsolventswasmeasuredatawavelengthof276nmand289nmThedevelopedmethodwastestedusingthemethodanalysisofspikedsamples
Keywordsquantitation2-methoxyphenolUVVISspectrophoto-
metry
Fig 1Structuralformulaof2-methoxyphenol
eyeballinhighconcentrationswhenitpenetratestheskincanleadtoneurosiswhenadministeredorallycanstimulatetheesophagusandstomachresultinginheartfailurecollapseanddeathNowadaystherearepublisheddataoncasesofasystemicallergicreaction[3]causedby2-methoxyphenolderivativesandthereisa fatal case known [4] for oral administration of guaifenesin (3-(2-methoxy-phenoxy)propane-12-diol)oneofthecomponentsofcommonlyavailablecoughmedications Thedeterminationof2-methoxyphenolinenvironmentalobjectsaswellasinthefoodindustryiscarriedoutusinggaschromatographymethodswithsolid-phasemicroextraction[5]Inordertocontrol2-methoxyphenolinnaturaldrink-ing and treatedwastewater gas chromatography is used followed by opticaldetectionoftheeluate[6]Themaindisadvantagesofthismethodofanalysisarethelowselectivityanddurationofdetermination(about3hours)AlsoaccordingtoRussianStateStandartGOST33312-2015themethodofgaschromatographyisusedforthequalitativeandquantitativedeterminationof2-metoxyphenolinjuiceproducts Commonlyfortheanalysisoftoxicsubstancesinvariousbiologicalsamples(bloodplasmaurinesalivasweathair)bygaschromatographyitisnecessarytocarryoutmultistagesamplepreparationwhichcomplicatesandslowsdownthecourseofthestudy[7]Atthesametimeitisimportantthatduringtheprepa-rationofsamplesintheanalyzedcompoundstheirstructureisnotviolatedasthiswillleadtothedifficultyoftheiridentification Spectrophotometryintheultravioletregionhaslowersensitivitycomparedtothe abovemethods however thismethod does not require such complicatedpreparation of the analyzed samples it is a relatively affordable simple andinexpensive analysis method In addition its sensitivity can be significantlyimprovedbyapplyinganappropriateseparationprocedureandpreconcentrationbeforedetection[8]MethodUVspectrophotometryisusedtoassessthequalityof both medicinal substances and preparations made from them in terms ofauthenticitygoodqualityandquantitativecontentInadditionitisarelativelyaffordablesimpleandlow-costanalysismethod Ananalysisoftheliteraturedatashowedthattodaytherearefastandsensitivespectrophotometricmethodsforthedeterminationofpyrocatecholderivativesinmedicines[9]vanillininfoodproducts[10]andotherphenolsinwastewaterandwineproducts[1112]Howeverasfarasweknowinformationonthedetermi-nationof2-methoxyphenolfromtheabsorptionspectraintheultravioletregionisabsent Theaimofthisstudyistodevelopmethodsforthequalitativeandquantitativeof2-methoxyphenolinmodelsolutionsusingUVspectrophotometry
2 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
2Experimental
21Reagentsandchemicals
Asampleof2-methoxyphenolfromFlukawithabasicsubstancecontentofge98wastakenastheobjectofstudyAssolventsweusedacetonitrile(ChP)95ethanolethylacetateand01MsodiumhydroxidesolutionAllotherchemicalsusedwereofanalyticalreagentgrade
22Instrumentation
Theopticaldensitywasmeasuredincuvetteewithanabsorbinglayerthicknessof10mmusingaCary60spectrophotometer(AgilentUSA)Allmeasurementswerecarriedoutatroomtemperature
3Resultsanddiscussion
Thechangeinthebehavioroftheabsorptionspectrumwasinvestigatedinthewavelengthrangeof200ndash400nmFigure2isshowedthatwithanincreaseinthepolarityofthesolventtheabsorptionmaximumshiftstowardthevisiblepartofthe spectrum The wavelength of absorption maxima of 2-methoxyphenol ispresentedinTable1 A studyof thephotometricbehaviorof2-metoxyphenolinvarious solventsshowedthatacetonitrile95ethanoland01Msodiumhydroxidearethemostsuitablesolventsforthequalitativedeterminationofthetestsubstance
ndash3Fig 1Thespectraof2-methoxyphenolofconcentration005mgdm inthemediumofsolvents(anabsorbinglayerthicknessof10mm)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 3
Forthequantitativedeterminationof2-methoxyphenolaseriesofsolutionsndash3 ndash3withaconcentrationragefrom0001mgdm to005mgdm werepreparedin
acetonitrileand95ethanolTheopticaldensityof2-methoxyphenolinsolventswasmeasuredbywavelengthof276nmThedependenceoftheintensityoftheoptical density on the concentration of 2-methoxyphenol in 01 M sodium
ndash3hydroxide was plotted in the concentration range from 0005mgdm tondash3003mgdm Themeasurementwerecarriedoutbywavelengthof276nmThe
obtainedregressionequationsarepresentedinTable2Dataanalysisobtainedwasperformedusingleast-squaresmethodThedevelopedmethodwastestedusingthemethodanalysisofspikedsamplesTheresultsarepresentedinTable2
4Conclusions
Studieshaveshownthepossibilityofusingspectrophotometricanalysisforthequalitativeandquantitativedeterminationof2-methoxyphenolTheabsorptionmaximaofweredeterminedinsolutionsofethanolandacetonitrile(276nm)inasolutionofethylacetate(277nm)and01Msodiumhydroxide(289nm)Theconstructed calibration curves of thepure substance of 2-methoxyphenol hasshownagoodregressioncoefficient(Rgt099)andcanbeusedforquantitativedeterminationof2-metoxyphenolinbiologicalobjectsInthefutureitisplannedtoapplythistechniquetodetermine2-metoxyphenolinincadavericmaterial
Solvent Regressionequation Found S RSD Δх δ 2-metoxy- phenolg
ndash3 ndash6 ndash5Acetonitrile y=18294C+01130 499times10 50times10 028 2times10 044 Rsup2=09985
ndash3 ndash6 ndash595ethanol y=35131C+00269 503times10 03times10 021 7times10 137 Rsup2=09956
ndash3 ndash6 ndash501Msodium y=31196C+01101 495times10 01times10 018 1times10 028hydroxide Rsup2=09997
Table 2Results of the determination of 2-methoxyphenol (average of three measurements) in modelsolutionsbythemethodanalysisofspikedsamplestheconcentrationofintroduced2-methoxy-
ndash3phenolwas500times10 g(SndashstandarddeviationRSDndashrelativestandarddeviationΔхndashabsoluteerrorδndashrelativeerror)
Table 1Valuesofopticaldensityandwavelengthsinappropriatesolventswith2-methoxyphenol
minus3 minus1Solvent λnm εgdm cm
Acetonitrile 276 0082895ethanol 276 00853Ethylacetate 277 0083101Msodiumhydroxide 289 00744
4 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
References
[1] МельниковаИММизерницкии ЮЛКомбинированныеотхаркивающиепрепаратырастительного происхождения в педиатрическои практикеМедицинский совет 2(2018)93ndash97
[2] httpwwwthegoodscentscompanycomdatarw1032272html(accessed27stFebruary2020)
[3] RayMFaltayBHallerNACasereportanaphylacticreactiontoguaifenesinHospPract37(2009)60ndash63
[4] OkicMJohnsonTCrifasiJALongCMitchellEKSwiftonsetofcentralnervoussystemdepressionandasystolefollowinganoverdoseofguaifenesinJAnalToxicol37(2013)318ndash319
[5] ВолковCМЧерновецАНОпределениеконцентрациифеноловвгазовыхвыбросахпромышленных предприятии методом газовои хроматографии с твердофазнои микроэкстракциеи Сорбционныеихроматографическиепроцессы10(2010)723ndash728
[6] ШачневаЕЮОньковаДВСерековаСМСпособыопределенияфеноловвобъектахокружающеи среды Астраханский вестник экологического образования 4 (2013)138ndash142
[7] ГладиловичВДПодольскаяЕПВозможностипримененияметодаГХ-МС(Обзор)Научноеприборостроение4(2010)36ndash49
[8] Pena-PereiraFLavillaIBendichoCHeadspacesingle-dropmicroextractioncoupledtomicrovolumeUVndashVis spectrophotometry for iodine determinationAnal Chim Acta631(2009)223ndash228
[9] NagarajaPMurthyKCSRangappaKSGowdaNMMSpectrophotometricmethodsforthe determination of certain catecholamine derivatives in pharmaceutical preparationsTalanta46(1998)39ndash44
[10] Altunay N Development of vortex-assisted ionic liquid-dispersive microextractionmethodology for vanillin monitoring in food products using ultraviolet-visible spectro-photometryLWT93(2018)9ndash15
[11] Lupetti KO Rocha FRP Fatibello-Filho O An improved flow system for phenolsdetermination exploiting multicommutation and long pathlength spectrophotometryTalanta62(2004)463ndash467
[12] Figueiredo-Gonzalez M Cancho-Grande B Simal-Gandara J Garnacha tintorera-basedsweetwineschromaticpropertiesandglobalphenoliccompositionbymeansofUVndashVisspectrophotometryFoodChem140(2013)217ndash224
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 5
1Introduction
Thenumberofsamplesthesamplecomplexityandalsothenumberofsubstanceswhich need to be analysed simultaneously is increasing steadily ThereforepowerfulseparationanddetectionmethodsarerequiredOnewaytoachievethisisthecouplingofaseparationsystemwithmorethanonedetector[12] In recent years capillary electrophoresis (CE)was established as a potentseparation system due to its high separation efficiency and the low sampleconsumption [3] To generate more information numerous dual detectionconceptsforCEweredevelopedwhicharesummarisedelsewhere[12]Acom-binationofamperometricdetectionandmassspectrometry(MS)isaninterestingdual detection concept for CE because both detectors supply complementaryinformationForelectroactivespeciesamperometricdetectionisarobustandoneof the most sensitive detection method [4] Thus it is well suited for the
The effects of linearly assembled capillaries with various inner diameters on capillary electrophoresis
DANIELBO HMFRANK-MICHAELMATYSIK
InstituteofAnalyticalChemistryChemo-andBiosensorsFacultyofChemistryandPharmacyUniversityofRegensburgUniversitaumltsstraszlige3193053RegensburgGermanydanielboehmchemieuni-regensburgde
AbstractDuetotheincreasingneedofpowerfulanalyticalmethodsanewdualdetection concept for capillary electrophoresis (CE) with parallelamperometricdetectionandmassspectrometryshallbedevelopedFor this concept the CE flow has to be divided into two streamsutilizinga flowsplitter In thiswork theeffectsof combinedcapi-llarieswithvarious innerdiameterswerestudiedForpreliminaryinvestigationsthecapillarieswereconnectedinaserialconfigurationwithoutdeadvolumeUsingcapillaryflowinjectionanalysishyphe-natedtocontactlessconductivitydetectionitcouldbeshownthatthecouplingofidenticalcapillariesleadstoaslightdecreaseoftheflowratesWithCEhyphenatedtoUVdetection itcouldbeshownthatthecouplingofcapillarieswithdifferentinnerdiameterhasamuchstrongereffectontheelectroosmoticflowthanthecombinationwiththesameinnerdiameterFurthermorenosignificantchangeinpeakshapewasobserved
Keywordsassembledcapillariescapillaryelectrophoresiscapillaryflowinjection
analysisdualdetectionconceptnon-aqueoussystem
6 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
quantificationofsubstanceswhereasMSiswellsuitedfortheidentificationofunknown substances [3] In most dual detection concepts the detectors arearrangedinaserialconfigurationwhichisnotpossibleincaseofamperometricdetection-mass spectrometry [1] The instrumental implementation is morecomplicatedwithbothdetectorsbeingdestructiveFurthermoretheymustbedecoupledfromthehighvoltagefieldoftheCEThereforetheCEflowmustbedividedintotwostreamswithaflowsplitterAsimplifiedsketchofthepossiblenewdualdetectionconceptisshowninFig1 ForthedevelopmentofthenewdualdetectionconceptthreecapillarieswithpotentiallydifferentinnerdiametersmustbecoupledForthisreasonthedeadvolume-freecouplingofcapillarieswithdifferent innerdiameterswas investi-gatedinafirststepTokeepthesetupsimplewefocusedonthelinearcouplingofcapillariesandtheresultingeffectsNon-fragmentedcapillarieswerecomparedwithfragmentedcapillariesofthesameordifferentinnerdiametersEffectsonthe flow rate were investigated with capillary flow injection analysis (CFIA)
4hyphenatedtocontactlessconductivitydetection(C D)EffectslikechangesinthemigrationbehaviourorpeakshapesoccurringinCEwereinvestigatedwithCEhyphenatedtoUVdetection(CE-UV)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 7
Fig 1SchematicillustrationofthenewdualdetectionconceptwithparallelamperometricdetectionandmassspectrometricdetectionforCEAfterinjectionfrom(a)thesamplevialthecomponentsareseparatedbyCEnext(b)theflowsplitterdivides(c)thecapillaryintotwopartsandleadstheCEflowtowards(d)themassspectrometerand(e)theamperometricdetector
2Experimental
21Reagentsandchemicals
Thefollowingchemicalswereusedallofanalyticalgradeferrocenemethanoldecamethylferrocene(ABCRGermany)acetonitrileammoniumacetate01Msodiumhydroxidesolutionultra-purewaterprovidedbyaMilliQAdvantageA10system(MerckGermany)aceticacid(RothGermany)
22Instrumentation
221Capillaries
4Forbothexperiments(CFIA-C DandCE-UV)capillarieswithinnerdiametersof2550and75micromanouterdiameterof360micromandatotallengthof70cmwereusedTheywerepurchasedfromPolymicroTechnologies(USA)Measurementswere carried out with fragmented and non-fragmented capillaries For themeasurementswiththefragmentedcapillariestheoriginalcapillarieswerecutintotwopiecesyieldingatotalof9capillarycombinationswithlengthsof70cm(20cmfirstcapillarypieceand50cmsecondcapillarypiece)ThesecombinationsaresummarizedinTab1(section31)Atbothendsofthecapillariesabout02cmof thepolyimidecoatingwasremovedBothsidesof thecapillarypieceswerepolishedtoreceiveplanarcapillarytipsForthelinearassemblingofthecapillarypiecesMicroTightSleevesF185Xanda capillary connectorUnionAssemblyMicroTightP720fromIDEXHealthampScience(USA)wereusedPriortothefirstCEmeasurementsthecapillarieswereconditionedbyflushingthemfor10minwith01Msodiumhydroxidesolution5minwithultra-purewaterand30minwithseparationbuffer
222Capillaryflowinjectionanalysishyphenatedtocontactlessconductivitydetectionsetup
The flow rates for the fragmented and non-fragmented capillaries were4determinedwithaCFIA-C DsetupschematicallydepictedinFig2ATheflowin
thecapillarywasgravitationdrivenbyaheightdifferencebetweentheinletandoutletcarriersolutionvialTheconceptofCFIAwithgravitationdrivenflowwasfirstdescribedbyMatysiketal[5]AlaboratoryconstructedautosamplerofaCEdevicewasusedforthehydrodynamicinjectionThesamplesolutionconsistedof10mMdecamethylferrocene incarrier solution (10mMCH COONH and1M3 4
4CH COOH in acetonitrile) A high resolution C D was placed after 40 cm for3
detectionThedetectordescribedelsewhere[6]wasconstructedinthedoLagogroup(Brazil)Adoubledeterminationattwodifferentheightswasdoneforthedeterminationoftheflowrates
8 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
223Capillaryelectrophoresis-UVdetectionsetup
Fig2BshowsasketchoftheCE-UVsetupItconsistedofalab-builtCEdevicewhichwasconnectedtoahighvoltagepowersupplyfromISEG(Germany)Theseparationswerecarriedoutwithanon-fragmented50micromcapillaryandwithcapillary combinations implementing a 50micromdownstream capillary segment(25+5050+50and75+50microm)ALambda1010UV-VISdetectorfromBischoff(Germany)wasusedfordetectionat210nmThedetectorwasplacedafter40cm
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 9
Fig 2Schemeof(A)thecapillaryflowinjectionanalysis(CFIA)hyphenatedtocontactlessconduc-4tivity detection (C D) setup and (B) the capillary electrophoresis hyphenated to UV detection
4(CE-UV) setup Components of the CFIA-C D setup (a) sample (b) inlet and (c)outlet carrier4solution vial (d) fused silica capillary (e) linear capillary connector (f) C D and (g) stand
ComponentsoftheCE-UVsetup(h)sample(i)inletand(j)outletbuffervialand(k)UVdetector4therestofthecomponentswereidenticaltotheCFIA-C DsetupTheoutletbuffervialwaslowered
forthehydrodynamicinjection(j)Theenlargedview(k)depictsthecouplingoftwocapillarieswithdifferentinnerdiametersintheconnectionsidewithoutdeadvolume
Asamplesolutioncontaining1mMferrocenemethanolanddecamethylferroceneinseparationbuffer(10mMCH COONH and1MCH COOHinacetonitrile)was3 4 3
utilizedTheinjectionwasperformedhydrodynamicallybyloweringtheoutletbuffer vial by 20 cm A uniform sample plug was injected to compare bandbroadeningeffectsTheinjectionsegmenthadalengthof035cm(05ofthetotalcapillarylength)andtherespectiveinjectiontimewasdeterminedbasedontheflowratesofthecorrespondingcapillarycombinationFortheelectrophoreticseparationaseparationvoltageof25kVwasappliedandtheinletandtheoutletbuffervialswereplacedatthesameheightsothattherewasnogravityflowwhichaffectedthemigrationbehaviour
3Resultsanddiscussion
31Capillaryflowinjectionanalysishyphenatedtocontactlessconductivitydetectionexperiments
AsshowninTab1theflowratesforaheightdifferenceof20cmwerecalculated4basedontheCFIA-C DmeasurementsItwasobservablethattheflowratesare
slightlylowerforfragmentedcapillariesthanfornon-fragmentedcapillariesofthe same dimension This indicates that a flow resistance arises when twocapillaries are combined Furthermore it was observed that the flow ratedecreasesforupstreamcapillarieswithlowerinnerdiametersandviceversaTheflow rate for the combination 25+75microm could not be determineddue to theformationofairbubblesattheconnectionside
10 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Table 1Flowratesandthecorrespondingstandarddeviations(SD4measurements)ofdifferentcapillarycombinationsforaheightdifferencebetweeninletandoutletvialof20cmbymeansofcapillaryflowinjectionanalysishyphenatedtocontactlessconductivitydetection
ndash1capillary flowratenLs plusmnSDflowndash1combinationmicrom ratenLs
25 00577 0000525+25 0057 000150+25 00775 0000775+25 ndash ndash25+50 0137 000550 0865 000250+50 080 00575+50 105 00625+75 0187 000250+75 1941 000375 456 00275+75 42 01
32Capillaryelectrophoresis-UVdetectionexperiments
Theelectropherogramsforthenon-fragmented50micromcapillaryandthecapillarycombinationswithdownstream50micromcapillaryarepresentedinFig3ThetwoferrocenederivatesdecamethylferroceneandferrocenemethanolwereusedasmodelanalytesDecamethylferrocenewasonlydetectedascationicspeciesasitiseasilyoxidizedbydissolvedoxygeninsolution Forthecombination50+50micromslightlyhighermigrationtimesforthecationic(decamethylferrocene) and neutral species (ferrocenemethanol) were obser-vable compared to thenon-fragmented50micromcapillaryThis indicateda flow
4resistanceattheconnectionwhichwasalsoobservedfortheCFIA-C Dexperi-mentsinsection31Incontrasttothecombinationwiththesameinnerdiameterastrongshift in themigration times for theneuralspecieswasvisible for thecombinationswithdifferentinnerdiametersThisshowedthatthecouplinghadaneffectontheelectroosmoticflow LookingatthepeakshapeitwasfoundthatallpeaksshowednearlyGaussianshape for all combinations Furthermore there was no tailing visible Theferrocenemethanolpeaks for thecombination25+50micromand75+50micromwere
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 11
Fig 3Electropherograms of themodelmixture ferrocenemethanol (FcMeOH) and decamethyl-ferrocene(DeMeFc)measuredwithanon-fragmentedcapillary(50microm)andfragmentedcapillaries(25+5050+50and75+50microm)Experimentalparameters1mMFcMeOHandDeMeFcinseparationbuffer(10mMCH COONH and1MCH COOHinacetonitrile)injectionsegment035cmseparation3 4 3
voltage25kVcapillarylength70cm(40cmtothedetectorfragmentedcapillaries20cmfirstpartand50cmsecondpart)UVdetectionat210nm
slightlybroaderthanthepeaksforthenon-fragmented50micromcapillaryorforthe50+50 microm combination But this probably results from longitudinal diffusioneffectsduetothelongerresidencetimes
4Conclusions
FromtheCFIAmeasurementsitcanbeconcludedthattherewasamechanicaldisturbanceoftheflowduetothecouplingFurthermoreitcouldbeshownthatCEmeasurementswithlinearcoupledcapillariesofvariousinnerdiameterwerepossible Unlike to the capillary combinationwith the same inner diameter astrongshiftoftheelectroosmoticflowtowardshighermigrationtimeswasfoundfor capillary combinations with different inner diameters In this work thecapillaries were coupled with almost no dead volume which resulted in nosignificantchangesofthepeakshapeorpeaktailingContrarytoexpectationsthecouplingofcapillarieswithvariousinnerdiametershadnosignificantimpactonthepeakwidth TheknowledgegainedfromthelinearcouplingofcapillariesisagoodbasisforthedevelopmentofthenewdualdetectionconceptInanextstepthreecapillariesshouldbecoupledwitheachother
Acknowledgments
WethanktheGermanResearchFoundation(DFG)forfinancialsupport
References
[1] OpekarFS tulıkKSomeimportantcombinationsofdetectiontechniquesforelectrophoresisincapillariesandonchipswithemphasisonelectrochemicalprinciplesElectrophoresis32(2011)795ndash810
[2] BeutnerAHerlTMatysikF-MSelectivityenhancement incapillaryelectrophoresisbymeans of two-dimensional separation or dual detection conceptsAnal Chim Acta1057(2018)18ndash35
4[3] BeutnerACunhaRRRichterEMMatysikF-MCombiningC DandMSasadualdetectionapproachforcapillaryelectrophoresisElectrophoresis37(2016)931ndash935
[4] MatysikF-MEnd-columnelectrochemicaldetectionforcapillaryelectrophoresisElectro-analysis12(2000)1349ndash1355
[5] MatysikF-MWernerGTracemetaldeterminationintearsbyanodicstrippingvoltammetryinacapillaryflowinjectionsystemAnalyst118(1993)1523ndash1526
[6] FranciscoKJMdoLagoCLAcompactandhigh-resolutionversionofacapacitivelycoupledcontactlessconductivitydetectorElectrophoresis30(2009)3458ndash3464
12 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Thetermldquovitaminsrdquodescribestheheterogeneousgroupofchemicalcompoundswhich are important for the proper functioning of the human body [1 2] Bydefinitionvitaminsarenotsynthesizedbythehumanbodyorthesynthesizedamount is not sufficient to cover the demand That is why they have to besupplementedfromtheexternalsourceslikefoodproductsorpharmaceuticals[1ndash3]Basedontheirsolubilityvitaminsaredividedintowater-soluble(B-groupandvitaminC)andfat-solublevitamins(ADEandK)[3]VitaminC(ascorbicacid) is the most important antioxidant and participates in the activation ofenzymes[4]VitaminB1(thiamine)facilitateswoundhealingandiscrucialforthehumannervoussystem[15]VitaminB2(riboflavin)participatesintheenzy-maticreactionsandthebiotransformationofglucoseandaminoacids[6]Vitamin
+B3(niacin)isthemainconstituentoftheNAD andNADHcoenzymeswhichareresponsible for the transfer of electrons and hydrogen ions in the cellularrespiration [1ndash3] Vitamin B6 possesses six related structures (vitamers) thateasilyinterconvertThemostimportantoneispyridoxinewhichhelpstopreventtongue inflammation and microcytic anemia [2] For the production of well-functioningredbloodcellsandtheavoidanceofmegaloblasticanemiaandfetusdefectsvitaminB9(folicacid)hastobesupplementedintheproperamount[12]
Voltammetric determination of vitamins
RADOSŁAWPORADABOGUSŁAWBAS
DepartmentofAnalyticalChemistryFacultyofMaterialsScienceandCeramicsAGHUniversityofScienceandTechnologyMickiewicza3030-059KrakoacutewPolandrporadaaghedupl
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 13
AbstractVitaminsbelongtothegroupofchemicalcompoundsessentialfortheproperfunctioningofthebodySinceboththeirdeficiencyandexcessmay result in serious health problems the amount of vitaminssupplementedinthedietaswellasvitamincontentintheirsourceshavetobestrictlycontrolledInthisworkthepossibilityofsimulta-neous determination of vitamins by means of differential pulseadsorptive stripping voltammetry is discussed The research hasshownthatthedeterminationofsingularvitaminatthemicromolarlevel isrelativelyfastandstraightforwardandthemostimportanthindranceisrelatedtotheanalyteadsorptionattheelectrodesurfaceInthecaseofvitaminswithdifferentredoxpotentialstheycanbeanalyzedsimultaneouslywithouttheneedtoreachfortheadvancedmethodsforsignalprocessing
Keywordsmercuryelectrodevitaminsvoltammetry
VitaminK3(menadione)doesnotoccurnaturallybutitservesasaprecursorforthesynthesisofotherK-groupvitaminsandcanbeusedtotreathypoprothrom-binemiaVitaminK3ispartiallysolubleinwater[17] All of the vitamins are electrochemically active [3] therefore the electro-chemicalmethodscanbeappliedforthedeterminationofvitamincontentinfoodproductspharmaceuticalsandbodyfluidsVoltammetrictechniquesarecharac-terizedbyhighsensitivityandselectivityandtheydonotrequiretime-consumingsamplepreparationMoreovertheelectrochemicalinstrumentationisrelativelyinexpensiveandcanbeappliedintheon-siteconditionsfortheonlineanalyseseginqualitycontrolMostofthepapersreporttheconstructiondevelopmentand characterization of a novel modified working electrodes for quantitativeanalyses of a singular vitamin in the variety of matrices Unfortunately only alimitednumberofpapersdescribethesimultaneousdeterminationofmultiplevitaminsinasinglerun[23] The preliminary research devoted to the simultaneous determination ofB-groupCandK3vitaminswiththeuseofthecontrolledgrowthmercurydropworking electrode in aqueous solutions is presented in this work Particularattentionhasbeenpaidtotheredoxpotentialsofthestudiedcompoundstheshape of the calibration curves and adsorption phenomena As an attempt toovercomethelattertheneutralsurfactantTritonX-100hasbeenintroducedintothestudiedsystem
2Experimental
21Reagentsandchemicals
TheappliedreagentswereofanalyticalgradeandusedassuppliedPhosphateand McIlvaine buffers were obtained by mixing the appropriate amount of
ndash1 ndash1 ndash102molL Na HPO with 02molL NaH PO and 01molL citric acid2 4 2 4
respectively (all reagents purchased from Avantor Performance MaterialsPoland)ThestandardsolutionsofvitaminB1B2B3B9andCwerepreparedbydissolving the corresponding amount of the standard (all Sigma-Aldrich) in
ndash1distilledwater In thecaseofB2andB9 theadditionof02molL NaOHwasinevitabletoobtainaclearsolutionVitaminK3standard(Sigma-Aldrich)was
ndash1dissolvedinthemixtureofmethanoland1molL phosphatebuffer(pH=82)(vv=25)LaboratorygradeTritonX-100(Sigma-Aldrich)wasusedinthestudyoftheadsorptionprocesses22Instrumentation
All the electrochemical measurements were conducted in the three-electrodesystem composed of the Pt auxiliary electrode double-junction silversilverchloride reference electrode and controlled-growth mercury drop electrode
14 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
actingastheworkingelectrodeTheusedmeasurementequipmentinvolvedtheM164electrodestandandM161multipurposeelectrochemicalanalyzer(bothmtm-anko Krakow) To control the buffer pH-value the SevenCompact S210laboratorypH-meter(MettlerToledoSwitzerland)wasemployed
23Voltammetricmeasurements
Throughout the course of the study differential pulse adsorptive strippingvoltammetry has been used for recording the current-potential curves BothcathodicandanodicscanswererecordedinapotentialrangeadjustedforthestudiedvitaminsTheinfluenceofvariousmeasurementconditionsontheregis-teredsignalshasbeeninvestigatedFinallythepossibilityofthesimultaneousdeterminationofmultiplevitaminsinonescanhasbeenverified
3Resultsanddiscussion
Figure 1 depicts the redox potentials of the studied vitamins in the aqueoussolutions for the mercury electrode The only exception is vitamin B6 whoseredox potential is higher than the potential of mercury oxidation (ca +02 V)Therefore the given value refers to the glassy carbon electrode The redoxpotentialvalueofstudiedvitamins isnotasingularvaluebut it fallswithinacertainrangeThiscanbeascribedtothedependenceofredoxpotentialonthesolutionpHvaluewhichresultsfromtheparticipationofprotonsintheredoxreactions of vitamins Moreover the potentials for individual vitamins arerelatively well separated indicating that the simultaneous determination ofseveralvitaminsintheonerunmaybepossibleTheonlyencounteredproblems
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 15
Fig 1Redoxpotentialsofchosenvitamins
regardthevitaminsB1B3andB12whoseredoxpotentialsoscillatebetweenndash15andndash17VandtheK-groupvitamins(K1K2K3)inwhichonlythecommonstructuralmotifndashthequinoneringndashiselectrochemicallyactiveresultinginthevalueoftheredoxpotentialofcandash02V[8] AtypicalcalibrationcurveisdepictedinFig2Abasedonthedifferentialpulsevoltammograms of the vitamin B9 reduction in the concentration range from
ndash1blank to 01 mg L recorded in the McIlvaine buffer of pH = 52 using thecontrolledgrowthmercurydropelectrodeworkingelectrodeTherelationshipbetweenthepeakcurrentandtheconcentrationofVB9 is linear in thewholetestedrange(r=09999)Basedontheparametersoftheregressioncurvethelimit of detection and limit of quantitation were estimated to 42 and
ndash1142nmolL respectively Similar dependencies and figures of merit can beobtainedforothervitamins QuitedifferentbehaviorwasobservedinthecaseofthevitaminK3forwhichthe increase in current was not strictly proportional to the increase in theconcentrationandthecalibrationplotresemblesanS-shapecurve(Fig2B)ThelatterindicatesthatvitaminK3adsorbsatthesurfaceoftheworkingelectrodeHoweverasnopre-orpost-peakwereobservedwearedealingherewiththeweakadsorption[9]AdsorptionalsoplaysasignificantroleinthecaseofvitaminB2forwhichboththepre-andpost-peakswereobservedindicatingitsstrongaffinitytothemercuryelectrodes To overcome this issue attempts with Triton X-100 were performed TritonX-100isaneutralsurfactant thateasilyadsorbsat thesurfaceof themercury
16 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2Differentialpulseadsorptivestrippingvoltammogramsof(A)vitaminB9and(B)vitaminK3ndash1reduction recorded in the concentration range from blank to 01 and from blank to 04 mg L
respectivelyInsetcorrespondingcalibrationcurvesSupportingelectrolyte(A)McIlvainebufferndash1(pH=52)(B)04molL phosphatebuffer(pH=82)
electrode partially blocking its surface Doing so it prevents the undesiredadsorption of other molecules and thus allows to obtain a linear relationshipbetweenthepeakcurrentandthevitaminconcentration(Fig3)UnfortunatelyduetotheblockingoftheelectrodesurfacetheslopesofthecalibrationlinesaresmallerincomparisontotheonesobtainedintheabsenceofanysurfactantsThismeans that the sensitivity defined as the increase in current caused by a unitincrease in concentration and the resolution understood as the possibility todistinguishsmallvariationinconcentrationarecorrespondinglydecreased Figure3alsopresentsthepossibilitytodeterminemultiplevitaminsinasinglerunChosenvitaminshavewell-separatedpotentialsandtheydonotinterferewitheachotherthereforenoadvancedmultivariatecalibrationstrategiesareneededTheproblemsinthesimultaneousanalysisincludevarioussensitivitieswith respect to the studied analytes and differences in the influence of themeasurementconditionsontherecordedsignalsDuetothattheexperimentalconditionswillneverensurethehighestpossiblesignalvaluesforallanalyzedcompounds
4Conclusions
Differential pulse voltammetry in conjunction with the controlled growthmercurydropelectrodeisaperfecttoolforquantitativeanalysesofvitaminsTheadsorptionofvitaminB2andK3canbepreventedbytheadditionoftheneutralsurfactantTritonX-100whichselectivelyblockstheworkingelectrodesurface
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 17
Fig 3Cathodic voltammograms for the simulatanous determination of vitamin B2 B3 and K3Depictedintheinsetsarethevoltammogramsafterbackgroundsubtractionwiththecorresponding
ndash1callibrationplotsSupportingelectrolye04molL phosphatebuffer(pH=82)with40ppmTritonX-100AccumulationconditionsE =ndash005Vt =20sacc acc
The proposed methodology allows for the simultaneous determination ofmicromolaramountsofvitaminB2B3andK3Suchaprocedurewillhelptoreduce the time and costs of analyses of multivitamin formulations and foodproducts
Acknowledgments
RPhasbeenpartlysupportedbytheEUProjectPOWR0302-00-00-I00416
References
[1] Combs GF JrTheVitaminsFundamentalAspects inNutritionandHealth 3rd ed IthacaElsevierAcademicPress2008
[2] Lovander MD Lyon JD Parr DL Wang J Parke B Leddy J Review Electrochemicalpropertiesof13vitaminsAcriticalreviewandassessmentJElectrochmSoc165(2018)G18ndashG49
[3] Brunetti B Recent advances in electroanalysis of vitamins Electroanalysis 28 (2016)1930ndash1942
[4] BrubacherGMuller-MulotWSouthgateDATMethods forDeterminationofVitamins inFoodNewYorkElsevier1985
[5] Szpikowska-Sroka B A simple and sensitive analytical method for the determination ofthiamineinpharmaceuticalpreparationsJAnalChem68(2013)218ndash222
[6] PetteysBJFrankELRapiddeterminationofvitaminB (riboflavin)inplasmabyHPLCClin2
ChimActa412(2011)38ndash43[7] ZhangZXuJWenYZhangJDingWTheelectro-syntesizedimprintedPEDOTfilmasa
simple voltammetric sensor for highly sensitive and selective detection of vitamin K in3
poultrydrugsamplesSynthMet230(2017)79ndash88[8] JedlinskaKStrusMBasBAnewelectrochemicalsensorwiththeRefreshableSilverLiquid
Amalgam Film multi-Electrode for sensitive voltammetric determination of vitamin K2(menaquinone)ElectrochimActa265(2018)355ndash363
[9] SouthamptonElectrochemistryGroupInstrumentalMethodsinElectrochemistryChichesterHorwood1985
18 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Carbonelectrodesarecommonlyappliedtosensitiveelectrochemicaldetectionofneurotransmittersegdopamine (nor)epinephrineandserotonin in-vivoandin-vitro[12]Neverthelessadsorptionofhigh-molecularweightbiomoleculesinthe matrix on a sensing electrode which then hinders the electron transferreactionofneurotransmitterswillresultinbiofoulingofelectrodesThisremainsachallengingproblemasbiofoulingwillcompromiseelectrochemicalmeasure-mentsThusseveralstrategies foraddressingbiofoulinghavepreviouslybeenreported[34] Thisworkreportsonaneffectiveapproachforminimisingbiofoulingbasedonthehypothesisthatahydrophobicelectrodesurfacewillrepelagainstadsorptionof amphiphilic biomolecules Briefly structurally small conical-tip electrodes
Dopamine detection at antifouling conical-tip carbon electrodes
a a a aSIMONABALUCHOVA JANKLOUDA JIR IBAREK KAROLINASCHWARZOVA -PECKOVA bDANNYKYWONG
a UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova812800PragueCzechRepublicsimonabaluchovanaturcunicz
b DepartmentofMolecularSciencesMacquarieUniversitySydneyNSW2109Australia
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 19
AbstractA significant achievement in this work is the development ofantifoulingconical-tipcarbonelectrodes(~27micromtipdiameterand~165micromaxiallength)suitablefordetectionoftheneurotransmitterdopamine in-vivo These electrodes were hydrogenated using adiphenylsilanereductionmethodtoyieldahydrophobicsurfacetodeteradsorptionofamphiphilicbiomoleculesInitiallyhydrogenatedcarbonelectrodeswereelectrochemicallycharacterisedusingseveralredoxmarkersThedegreeofantifoulingwasthenassessedbythevoltammetricsignalchangeofdopamineattheseelectrodesbeforeand after being incubated in a fouling solution containing bovineserumalbumincytochromeC(bothareproteins)andcaproicacid(alipid) In our work we have obtained only a 69 (standarddeviation35N=40)decreaseindopaminesignalsatthehydro-genated carbon electrodes These results strongly support thediphenylsilanereductionstrategyforthedevelopmentofantifoulingbiosensorsfordopaminedetectioninbiologicalmatrices
Keywordsantifoulingelectrodesdiphenylsilanereduction
methodhydrogenatedconical-tip
carbonelectrodesvoltammetricdopamine
detection
(denoted as CTEs) are fabricated by thermally pyrolysing acetylene gas in anitrogenatmospheretodepositcarbonatthetipandontheshankofpulledquartzcapillaries [5] Spectroscopic studies confirmed that the electrode surface
2 3consistsofsp -likegraphiticcarbonandsp -hybridiseddiamond-likecarbon[6]In addition there is also a range of carbon-oxygen functionalities includingcarbonylquinonecarboxylphenolsalcoholsandethergroupsontheelectrodesurface[6]whichcaninteractwithspectatorbiomoleculesthroughdipole-dipoleorion-dipoleinteractionleadingtotheirirreversibleadsorptionontheelectrodesurface[7]HoweverbysubjectingthesecarbonelectrodestosilanereductionCndashObondsareconvertedtoCndashHbondsandphenolicgroupsaretransformedtosiloxane dendrimers [6] to yield a more hydrophobic carbon surface that isexpectedtobesimilarlylesssusceptibletobiofoulingcomparedtoboron-dopeddiamondelectrodes[78] In this work we will present a methodology involving diphenylsilanereduction to fabricate physically small hydrogenated conical-tip carbonelectrodes(denotedasHCTEs)withanti-foulingcapabilityBothCTEsandHCTEswere electrochemically characterised using several redox probes to elucidatetheirsurfacepropertiesbeforeevaluatingtheirresistancetobiofoulingduringdopaminedetectionin-vitro
2Experimental
21Reagentsandchemicals
Analyticalgradereagents(Sigma-AldrichAustralia)including4-methylcatecholhexaammineruthenium(III) chloride potassium hexacyanoferrate(III) dop-amine hydrochloride sodium phosphate dibasic citric acid perchloric acidpotassiumchloride sodiumhydroxide anhydrousdichloromethanediphenyl-silanetris-(pentafluorophenyl)boraneandgraphitepowderwereusedas-recei-ved Ultra-high purity gases acetylene and nitrogenwere obtained from BOCGases (Australia) All aqueous solutionswere preparedwith deionisedwater(MilliporeMiliplusQsystemUSA)witharesistivityof182MΩcm
22Instrumentation
Chronoamperometric and voltammetric experiments were carried out usingalow-currentpicostateDAQoperatedbyanEChemversion212softwareviaanE-corderinterface(eDAQPtyLtdAustralia)Athree-electrodeset-upinvolving
minus1eitheraCTEorHCTEasaworkingelectrodeaAgAgCl(3molL KCl)referenceelectrode (Bioanalytical SystemsUSA)andaplatinumwire counterelectrode
minus1(CypressSystemsUSA)wasusedCyclicvoltammetryatascanrateof100mVs anddifferential pulse voltammetry (pulse height +25mV pulsewidth 50ms
minus1sampling time 20 ms and scan rate 20mVs ) were used in this work All
20 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
electroanalyticalexperimentswereperformedinanaluminiumFaradaycageatanambienttemperature(23plusmn1degC)
23Preparationofhydrogenatedconical-tipcarbonelectrodes
Asreportedpreviously[5]structurallysmallCTEswerefabricatedbythermallypyrolysing C H (a pressure of 50 kPa) in a pulled quartz capillary (Sutter2 2
minus1InstrumentUSA)housedinaN atmosphere(counterflowof60mLmin )Prior2
to hydrogenation the catalyst tris(pentafluorophenyl) borane (100 mg) wasdissolvedinanhydrousCH Cl (50mL)bystirringfor5minbeforethehydroge-2 2
natingagentdiphenylsilane(25μL)wasaddedCTEswerethenplacedinthereagentmixturefor2hThepreparedHCTEsweredriedovernightbeforeuse
24Biofoulingexperiments
A laboratory synthetic fouling solution consisting of 4 (wv) bovine serumalbumin001(wv)cytochromeC(bothareproteins)and10(vv)caproicacid(alipid)waspreparedbyhomogenisingtheminapH=74citrate-phosphate
minus1buffer(01molL )AllfoulingcompoundswereacquiredfromSigmaAldrichAustralia
3Resultsanddiscussion
31Electrochemicalcharacterisation
minus1InthisworkallCTEswerecharacterisedbycyclicvoltammetryof10mmolL 3+ minus1[Ru(NH ) ] in10molL KClAsdisplayedinFig1(A)onlyCTEsthatshow3 6
asigmoidal-shapedvoltammogramwithasmallchargingcurrentwereemployedinfurtherexperimentsUsingchronoamperometry[5]ameantipdiameterof27μm(standarddeviation(SD)28μmN=142)ameanaxiallengthof165μm(SD=114μmN=142)wereestimatedfortheseCTEs TocomparesurfacecharacteristicsofbothCTEsandHCTEs cyclicvoltam-
minus1 3+2+ minus1 minus1metryof (1)10mmolL [Ru(NH ) ] in10molL KCl (2)10mmolL 3 63minus4minus minus1 minus1[Fe(CN) ] in 10 mol L KCl and (3) 10 mmol L 4-methylcatechol in6minus101molL HClO wasconductedatthesameelectrodesbeforeandafterhydroge-4
nation The results obtained are shown in Fig 1(A-C)We observed a ~20(SD=5N=10)decreaseinthelimitingcurrentofallthreeredoxmarkersafterdiphenylsilane reduction most likely attributable to the hindrance to theirelectron transfer reactions by the phenylsiloxane group formed on HCTEs
3minus4minusMoreover as an inner-sphere redox probe both [Fe(CN) ] and 4-methyl-6
catechol reactionsare sensitive to thepresenceofoxygen functionalitiesonacarbonsurface [7]Accordingly theconversionof these functionalities toCndashHbondsbydiphenylsilanereductionwasexpectedtoyieldmoresluggishelectron
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 21
transfer kinetics at HCTEs as supported by a negative potential shift (from3minus4minus+75mVtominus10mV)inthecyclicvoltammogramof[Fe(CN) ] andapositive6
potentialshift(from+580mVto+675mV)inthecorrespondingcyclicvoltammo-2gram of 4-methylcatechol In addition the conversion of sp -carbon to
3sp -diamond-likecarbon[6] isalsoexpectedtoreducetheconductivityof thecarbonelectrodesurface
32Dopaminedetectionduringbiofoulingexperiments
minus1Theelectrochemicalbehaviourof1mmolL dopamine inapH=74citrate-minus1phosphate buffer (01 mol L ) at CTEs and HCTEs was studied by cyclic
voltammetryTheresultsobtainedareshown inFig1(D)Acomparable12decrease(SD=6N=10)inthedopamineoxidationlimitingcurrenttothatof4-methylcatechol was observed A positive potential shift from +285 mV to+305mVinthevoltammogramsisalsoaccountedforasdescribedabove
22 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
minus1 3+ minus1 minus1Fig 1Cyclicvoltammetryof(A)10mmolL [Ru(NH ) ] in10molL KCl (B)10mmolL 3 63minus4minus minus1 minus1 minus1[Fe(CN) ] in10molL KCland(C)10mmolL 4-methylcatechol in01molL HClO at3 4
minus1(a)aCTEand(b)aHCTE(D)10mmolL dopamineinapH=74citrate-phosphatebufferrecordedminus1at(a)aCTEandaHCTE(b)beforeand(c)afterbiofoulingScanrate100mVs
Next HCTEs were incubated in a synthetic fouling solution containing4(wv)bovineserumalbumin001(wv)cytochromeC(bothareproteins)and 10 (vv) caproic acid (a lipid) for 30 min Cyclic voltammetry of
minus110mmolL dopamineattheseHCTEswasthenconductedinapH=74citrate-phosphatebuffertoevaluatetheirantifoulingpropertyNotablyaconsiderable515(SD=183N=6)decreaseindopaminesignalwasobservedatCTEsIncontrastonlyacorresponding69decrease(SD=35N=40)wasestimatedatHCTEsasshowninFig1(D)Clearlythisrepresentsamajorimprovementinthe antifouling capability of HCTEs obtained using diphenylsilane reductioncomparedtoCTEsandotherpreviouslytestedhydrogenatingagents[9]includingn-butylsilane (35decrease) triethylsilane (23decrease) andphenylsilane(18decrease)Thereforethisworkhassuccessfullydemonstratedtheeffecti-venessofdiphenylsilanereductionmethodindevelopingantifoulingelectrodesfordopaminedetection
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 23
Smallconical-tipelectrode
As-prepared Hydrogenated
minus1LinearrangemicromolL 1ndash10 1ndash20minus2 minus2Intercept10 pAmicrom 108plusmn004 273plusmn011
minus3 minus2 minus1Slope10 pALmicrom micromol 128plusmn007 443plusmn012R 0993 0998
minus1LimitofdetectionmicromolL 100 077
Table 1Analyticalparametersofconcentrationdependencesofdopamineobtainedbydifferentialpulse
minus1voltammetryinapH=74citrate-phosphtebuffer(01molL )Allquoteduncertaintiesrepresentthe95confidenceintervalandthecorrelationcoefficient(R)wasfoundtobestatisticallysigni-ficantatthe95usingStudentrsquost-test
Fig 2DifferentialpulsevoltammetryofdopamineataHCTEinapH=74citrate-phosphatebufferminus1 minus1(01molL )atconcentrations(a)1(b)2(c)4(d)6(e)8(f)10and(g)20μmolL
WehavealsostudiedtheconcentrationdependenceofdopamineinapH=74citrate-phosphatebufferbydifferentialpulsevoltammetryAtypicalcalibrationplotobtainedisshowninFig2Theanalyticalparametersandestimatedlimitsofdetection are summarised inTable 1 These results show thatHCTEs outper-formed CTEs because they exhibit a ~35times higher sensitivity a 23 lowerdetectionlimitandawiderlinearrange
4Conclusions
InthisstudyphysicallysmallHCTEswithanti-foulingcharacteristicsachievedbyhydrogenationusingdiphenylsilanereductionweresuccessfullyfabricatedandelectrochemically characterised using several redox probes Next dopaminedetectionwasperformedbeforeandafterincubationofelectrodesinasyntheticfoulingsolutioncontainingahighconcentrationofbiomoleculesOnlyalow69(SD=35)decreaseindopaminelimitingcurrentwasachievedatHCTEsobtain-edbydiphenylsilanereductionindicatingtheirsignificantlylesssusceptibilitytobiofoulingthanCTEsThesepromisingresultsindicatethatantifoulingHCTEswillpotentiallybenefitthedevelopmentofbiosensorsfordopaminedetectionin-vivoinbiologicalmedia
Acknowledgments
ThisresearchwasperformedwithintheframeworkofSpecificUniversityResearch(SVV260560)FinancialsupportsprovidedbytheGrantAgencyofCharlesUniversity(project390119)andbytheCzechScienceFoundation(project20-03187S)aregratefullyacknowledgedSBandJKalsothankthe Mobility Fund of Charles University and Hlavkova nadace for providing funding for theirresearchinternshipsatMacquarieUniversitySydneyAustralia
References[1] BaranwalAChandraPClinicalimplicationsandelectrochemicalbiosensingofmonoamine
neurotransmittersinbodyfluidsinvitroinvivoandexvivomodelsBiosensBioelectron121(2018)137ndash152
[2] CaoQPuthongkhamPJillVentonBReviewnewinsightsintooptimizingchemicaland3Dsurface structuresof carbonelectrodes forneurotransmitterdetectionAnalMethods11(2019)247ndash261
[3] LinP-HLinB-RAntifoulingstrategiesinadvancedelectrochemicalsensorsandbiosensorsAnalyst145(2020)1110ndash1120
[4] HanssenBLSirajSWongDKYRecentStrategiestoMinimiseFoulinginElectrochemicalDetectionSystemsRevAnalChem35(2016)1ndash28
[5] McNallyMWongDKYAnin-vivoprobebasedonmechanicallystrongbutstructurallysmallcarbonelectrodeswithanappreciablesurfaceareaAnalChem73(2001)4793ndash4800
[6] SirajSMcRaeCRWongDKYEffectiveactivationofphysicallysmallcarbonelectrodesbyn-butylsilanereductionElectrochemCommun64(2016)35ndash41
[7] ParkJShowYQuaiserovaVGalliganJFinkGDSwainGMDiamondmicroelectrodesforuseinbiologicalenvironmentsJElectroanalChem583(2005)56ndash68
[8] ShinDTrykDAFujishimaAMerkociAWang JResistance to surfactantandproteinfoulingeffectsatconductingdiamondelectrodesElectroanalysis17(2005)305ndash311
[9] Roshni RAnAntifouling Structurally Small Carbon Electrode forDetectionof theNeuro-transmitterDopaminePhDThesisMacquarieUniversitySydney2019
24 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Theanalyticalperformancesofenzymaticbiosensorsarestronglyaffectedbytheenzyme immobilization process There is no universal technique for enzymesattachmentThereforespecialattentionshouldbepaid to theselectionof theappropriatesupportandthedevelopmentoftheoptimalbindingstrategyinordertoensure thebestcharacteristicsof immobilizedenzymeDespiteavarietyofpreviouslyreportedcovalentimmobilizationmethodsfordifferentenzymesthepresentedprocedurescanbehardlycomparedtofindtheoptimalonesbecauseofdifferentanalyticalmethodsandexperimentalconditionsusedUptodatethere
A comparative study of covalent glucose oxidase and laccase immobilization techniques at powdered supports for biosensors fabrication
ab a bSOFIIATVORYNSKA JIR IBAREK BOHDANJOSYPCUK
a UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova2030812843Prague2CzechRepublicsofiiatvorynskajh-instcascz
b JHeyrovskyacuteInstituteofPhysicalChemistryoftheCzechAcademyofSciences Dolejškova318223Prague8CzechRepublic
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 25
AbstractInordertodeveloptheoptimalstrategyandtodeepentheknowledgeinthefieldofenzymeimmobilizationthreedifferenttechniquesofcovalentbindingfortwoenzymes(glucoseoxidaseandlaccase)atpowdered surfaces were compared Immobilization protocol wasoptimized by changing supports (twomesoporous silica powders(SBAminus15 MCMminus41) and a cellulose powder) the functionalizedgroupsintroducedatsupportsurfaces(minusNH andminusCOOH)andthe2
methodsofactivation(glutaraldehydeandcarbodiimide)Aminoandcarboxyl functionalized mesoporous silica and cellulose powderswerepreparedbysilanizationusing(3-aminopropyl)triethoxysilaneandcarboxyethylsilanetriolrespectivelyItwasfoundthatcouplingof both enzymes by their ndashNH groups through glutaraldehyde to2
ndashNH functionalized supports in particular SBA15minusNH and2 2
celluloseminusNH forglucoseoxidaseMCM41minusNH forlaccaseshowed2 2
thehighestactivityandthebeststability
Keywordsbiosensorcovalentimmobilizationenzymaticreactorglucoseoxidaselaccase
is still a lack of the comparative systematic studies focusing on the enzymesimmobilizationonthevarioussupportsusingdifferenttechniques The aim of this work is the systematic comparative study of the differenttechniques for covalent coupling of the enzymeswhich ensures not only thedevelopmentoftheoptimalimmobilizationstrategyfortheselectedenzymesbutalsoenablestofindoutsometendenciesinenzymeattachmentprocessgenerallyThusthisworkisfocusedonadetailedanalysisoftheeffectofthekindofsupportits anchor groups and the activation methods on activity and stability ofimmobilizedenzymesTwoenzymeswithdifferentnature(glucoseoxidase(GOx)andlaccase(Lac))werechosenasthetestingbioreceptors
2Experimental
21Reagentsandchemicals
AllchemicalswereofpaorbettergradeGlucoseoxidasefromAspergillusnigerminus1(GOxEC11341452Umg )laccasefromTrametesversicolor(LacEC11032
minus1129Umg )D-(+)-glucosedopamineglutaraldehyde(GAgradeII25aqueoussolution) N-(3-dimethylaminopropyl)-Nʹ-ethylcarbodiimide hydrochloride(EDC ge980) N-hydroxysuccinimide (NHS ge970) (3-aminopropyl)-triethoxysilane (APTES) mesoporous silica powder SBAminus15 (particle size
2 minus12ndash6μmporesizeasymp7nmsurfaceareaasymp600m g )mesoporoussilicapowder2 minus1MCMminus41 (pore size 21ndash27 nm surface area asymp 1000 m g ) cellulose (Cell
microcrystalline powder particle size 20 microm) were purchased from SigmaAldrichCarboxyethylsilanetriol(CEST25aqueoussolution)waspurchased
regfromabcr (Germany)
22Instrumentation
Amperometric measurements were carried out at room temperature usingcomputer-controlled electrochemical stand (Polaro-Sensors Czech Republic)withMultiElchemv31software(JHeyrovskyInstituteofPhysicalChemistryoftheCAS)Flowinjectionanalysis(FIA)withthethree-electrodelaboratory-madeflow-through cellwas usedworking electrode minus tubular detector of polishedsilversolidamalgam(TD-p-AgSAlaboratory-madeinnerdiameter05mmtheamalgamtube length60mm) referenceelectrodeminusaminiaturizedsaturatedcalomelelectrodebasedonsilverpasteamalgam[1](laboratory-madeithasthesamepotential as classical saturated calomel electrode) auxiliary electrodeminusplatinum wire (diameter 10 mm length 10 mm) The system for FIA withelectrochemicaldetectioncomprisedofalinearsyringepumpa2-position6-port
regsampleinjectorvalveaninjectionlooplaboratory-madeofTeflon (PTFE)tubing(100μL) a solenoid operatedmicro-pumpan enzymatic reactor and a flow-through cell for TD The enzymatic reactor consists of a tube filled by theenzymaticpowder
26 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
3Resultsanddiscussion
Basedonthedatareportedintheliterature[23]andontheresultsofourpreviousworks[4ndash8]forthisstudyCellandmesoporoussilicapowders(namelySBAminus15andMCMminus41)havebeenselectedas thepotentialpromisingsupports for thecovalent enzyme immobilizationBecauseof thehigh contentof surficial ndashOHgroupswhich are capable of chemical reactions these supports can be easilyfunctionalizedThewell-knownandfrequentlyutilizedtechniqueofsilanizationhasbeenusedtomodifythesurfacesofSBA15MCMminus41andCellbythedesiredfunctionalizedgroupsAminosilaneAPTESwasappliedtoformminusNH groupson2
thematrixsurfaceswhereascarboxylsilaneCESTwasusedtointroducendashCOOHgroups Generallytheprocedureofthecovalentimmobilizationofenzyme(eitherLacorGOx)onthefunctionalizedsupportconsistsofthreestepsI Synthesisofthefunctionalizedsupportwhichmeansthemodificationofthematrix(MCMminus41SBAminus15andCell)withsuitableanchoredgroups(minusNH or2
minusCOOH)II Activationstepofthefunctionalizedsupportwithspecificactivatingagents(glutaraldehydeorEDCNHS)tomakeitreactivetowardsenzyme
IIIEnzyme(LacorGOx)couplingtotheactivatedsupport
To investigate the effect of support its surface functionalizedgroups and themethodsofactivationontheefficiencyofthecovalentenzymeimmobilizationthreedifferentstrategies(ABandC)forLacandGOxattachmenthavebeenusedThedetailsoftheusedtechniquesandthedenotationsofthepreparedenzymaticpowdersaresummarizedinTable1(nextpage)ToexaminetheefficiencyofLacandGOximmobilizationtheenzymaticreactors(filledbytheenzymaticpowderspreparedwithdifferenttechniques)coupledwithTDwereusedforamperometricdetermination of dopamine and glucose respectively in flow systems Theprincipleofglucosedetection isbasedonamperometricmeasurementsof theenzymatically consumed oxygen whereas dopamine was detected by thereductionoftheenzymaticallyoxidiseddopamine Asdepicted inFig1 thebiosensors responsesare stronglyaffectedby thestrategyusedforLacorGOximmobilizationAsshowntheresponsesofLacandGOx biosensors decrease in the order strategy A gt strategy B gt strategy Cirrespectiveofthetypeofsupport It isclearlyseenthataminofunctionalizedsupports(SBA15minusNH MCM41minusNH andCellminusNH )providehigheractivitiesof2 2 2
the immobilized Lac andGOx than these supports functionalized by carboxylgroups(SBA15minusCOOHMCM41minusCOOHandCellminusCOOH)BycomparingactivitiesofimmobilizedenzymesusingstrategiesAandBtheinfluenceoftheactivationagenthasbeenevaluatedThebestresultsforbothenzymeswereobtainedforndashNH functionalizedsupportsactivatedbyGAItcouldbeexplainedbythefact2
thatGAcontrary tocarbodiimidewithnonemolecularspaceprovidesa long
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 27
spacerarmensuringminimalsterichindrancesforenzymesbindingItcanbeconcludedthatthecovalentimmobilizationofbothenzymesbytheirndashNH groups2
viaGAtondashNH functionalizedmesoporoussilicapowders(strategyA)provided2
the highest activities Interestingly in the similar comparative studies it isreportedthatamongndashOHminusCOOHandndashNH functionalizedsupportsactivatedby2
divinylsulfonecarbodiimideandGArespectivelythelastonewasfoundasthemostsuitabletechniqueforthecovalentbindingofLac[9]invertase[10]andpepsin[11]
28 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Tab
le 1
Theprinciplesofthecovalentimmobilizationmethodsofenzymesusedinthisstudy
StrategyA
StrategyB
StrategyC
Support
SBAminus15MCMminus41Cell
SBAminus15MCMminus41Cell
SBAminus15MCMminus41Cell
Supportfunctio-
minusNH
minusNH
minusCOOH
22
nalizedgroup
Activationagent
Glutaraldehyde(GA)
Carbodiimide
Carbodiimide
(EDCNHS)
(EDCNHS)
Enzymereactive
minusNH
minusCOOH
minusNH
22
group
Typeofbond
secondaryamine
amide
amide
Denotationsofthe
GOxminusNHminusCHminus(CH)minusCHminusNHminusSBA15
GOxminusC(=O)minusNHminusSBA15
GOxminusNHminusC(=O)minusSBA15
23
preparedenzy-
GOxminusNHminusCHminus(CH)minusCHminusNHminusMCM41
GOxminusC(=O)minusNHminusMCM41
GOxminusNHminusC(=O)minusMCM41
23
maticpowders
GOxminusNHminusCHminus(CH)minusCHminusNHminusCell
GOxminusC(=O)minusNHminusCell
GOxminusNHminusC(=O)minusCell
23
LacminusNHminusCHminus(CH)minusCHminusNHminusSBA15
LaxminusC(=O)minusNHminusSBA15
LacminusNHminusC(=O)minusSBA15
23
LacminusNHminusCHminus(CH)minusCHminusNHminusMCM41
LacminusC(=O)minusNHminusMCM41
LacminusNHminusC(=O)minusMCM41
23
LacminusNHminusCHminus(CH)minusCHminusNHminusCell
LacminusC(=O)minusNHminusCell
LacminusNHminusC(=O)minusCell
23
When the effect of the method of the covalent enzyme coupling on thebiosensorstabilitywasevaluateditwasfoundthatLacboundedtondashNH functio-2
nalizedsupportsviaGA(strategyA)hasshownthehigheststability(gt65oftheinitial responses after 1 month) compared to other strategies whereas GOximmobilizedwithtwostrategies(AandB)possessedapproximatelysimilarhighstability(gt80oftheinitialresponsesin1month)BothenzymesboundedviandashNH groupstondashCOOHfunctionalizedsupportsthroughEDCNHS(strategyC)2
showedquitelowstability
4Conclusions
Threedifferent strategies including the support selection the anchor surfacegroups and the activationmethod havebeen compared for efficient covalentimmobilization of Lac and GOx The results showed that ndashNH functionalized2
supports(SBA15minusNH CelluloseminusNH forGOxandMCMminusNH forLac)activatedby2 2 2
GAmaybeusedtoeffectivelybindenzymesintermsofhighactivityandstability
Acknowledgments
ThisworkwasfinanciallysupportedbytheGrantAgencyofCharlesUniversityinPrague(Project1356120)theGrantAgencyoftheCzechRepublic(Project20-07350S)anditwascarriedoutwithintheframeworkofSpecificCharlesUniversityResearch(SVV260440)
References
[1] YosypchukBBarekJYosypchukOPreparationandpropertiesofreferenceelectrodesbasedonsilverpasteamalgamElectroanalysis23(2011)2226minus2231
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 29
Fig 1Effectof the covalent attachment techniqueson (A) laccaseand (B) andglucoseoxidaseminus1biosensor responses Experimental conditions (A) c = 500 micromol L E = minus50 mVDOP det
minus1 minus1v = 01 mL min V = 40 microL carrier solution 01 mol L acetate buffer pH = 48flow DOPminus1 minus1(B)c =500micromol L E =minus1100mVv ==01mLmin V =40microL carrier solutionGlu det flow Glu
minus1 minus101molL acetatebuffer0001molL Na EDTApH=652
[2] LiuYChenJYEnzymeimmobilizationoncellulosematrixesJBioactCompactPolym31(2016)553ndash567
[3] Hartmann M Kostrov X Immobilization of enzymes on porous silicas ndash benefits andchallengesChemSocRev42(2013)6277minus6289
[4] JosypcukOBarekJJosypcukBElectrochemicalbiosensorsbasedonenzymaticreactorsfilledbyvarioustypesofsilicaandamalgampowdersformeasurements inflowsystemsElectroanalysis28(2016)3028minus3038
[5] Josypcuk O Barek J Josypcuk B Amperometric determination of catecholamines byenzymaticbiosensorsinflowsystemsElectroanalysis30(2018)1163minus1171
[6] TvorynskaSBarekJJosypcukBAmperometricbiosensorbasedonenzymaticreactorforcholinedeterminationinflowsystemsElectroanalysis31(2019)1901minus1912
[7] TvorynskaSBarekJJosypcukBFlowamperometricbiosensorbasedontwoenzymaticreactors (acetylcholinesterase-choline oxidase) for the detection of neurotransmitteracetylcholine In Proceedings of the 15th International Students Conference ldquoModernAnalyticalChemistryrdquoKNesmerak(ed)PragueFacultyofScienceCharlesUniversity2019p61minus66
[8] TvorynskaSBarekJJosypcukBAcetylcholinesterase-cholineoxidase-basedmini-reactorscoupledwithsilversolidamalgamelectrodeforamperometricdetectionofacetylcholineinflowinjectionanalysisJElectroanalChem860(2020)113883
[9] RekucAKruczkiewiczPJastrzembskaBLiesieneJPeczynska-CzochWBryjakJLaccaseimmobilizationonthetailoredcellulose-basedGranocelcarriersIntJBiolMacromol42(2008)208minus215
[10] Bryjak J Liesiene J S tefuca V Man-tailored cellulose-based carriers for invertaseimmobilizationCellulose15(2008)631minus640
[11] SzałapataKOsinska-JaroszukMBryjakJJaszekMJarosz-WilkołazkaANovelapplicationofporousandcellularmaterialsforcovalentimmobilizationofpepsinBrazJChemEng33(2016)251minus260
30 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Carbohydrates are crucial for energy structure and signaling in the humanbody[1]Thereisavarietyofcarbohydratesbutthemostimportantoneforlifeisglucoseasitisfundamentalinthemetabolismandphotosynthesis[2]GlucoseisclassifiedashexoseThesemonosaccharidesonlydifferinthepositionofhydroxylsubstituentsinsomecasesInadditiontothestructuralsimilaritiesthesemole-culeslackachromophoreandarenoteasilyionizable(pK ~12)Thusdetectiona
intheUVregionandseparationofanalytesbycapillaryelectrophoresis(CE)arechallenging [1 3] At the moment there are many different techniques for theanalysis of carbohydrates commonly including time-consuming derivatizationstepsoreluentswithhighpHvalues(pHgt12)inionchromatographyAwell-esta-blishedtechniquefortheanalysisofcarbohydratesishigh-performanceanion-exchangechromatographywithpulsedamperometricdetection(HPAE-PAD)[4]Electrochemical detection like AD is matching miniaturization simple instru-mentationlowcostandrobustnessandthusisoftenusedforflow-basedsystemssuchasCEandflowinjectionanalysis(FIA)[5]
Capillary flow injection analysis with electrochemical detection for carbohydrate analysis
NICOLEHEIGLFRANK-MICHAELMATYSIK
InstituteofAnalyticalChemistryChemo-andBiosensorsFacultyofChemistryandPharmacyUniversityofRegensburgUniversitaumltsstraszlige3193053RegensburgGermanynicoleheiglchemieuni-regensburgde
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 31
AbstractAsimplecapillaryflowinjectionanalysissystemwithamperometricdetection was arranged for the development of a method for fastoptimization of detection conditions in the context of thedetermination of carbohydrates by means of electrochemistry-capillary electrophoresis-mass spectrometry This setup is free ofelectricalinterferencebyhighvoltageandisperfectforstudyingtheoxidationofvariousanalytesFurthermoreitassureseasycouplingtoMS and thus is an useful tool to investigate the correspondingoxidationproductsofananalyte
Keywordscapillaryflowinjection
analysiscarbohydratesmassspectrometrydisposableelectrodespulsedamperometric
detection
Inthiscontributioncapillaryflowinjectionanalysis(CFIA)withADwillbepresentedasamethodtoapplyandtestADforthedetectionofmonosaccharidesondifferentdisposablethin-filmorscreen-printedelectrodesCFIAwaschosenoverconventionalFIAforthispurposeasthegravityflowinCFIAisstableforalongertimeandverylowsampleconsumptioncanbeachieved[6]TheCFIAsystemwasarrangedassimpleaspossibleandperformedhydrodynamicallytoavoidanyinterferencesFurthermoreitassuresthecouplingoftheflowsystemtoamassspectrometerThusthesamesetupasusedforCFIAcanbeutilizedforcapillaryelectrophoresis-massspectrometry(CE-MS)experimentsbychangingthe flow through the electrochemical flow cell in opposite direction In futureexperimentstheexperienceintermsofADonthoseelectrodeswillbeusedtodevelopelectrochemicalpretreatmentprotocolsforcarbohydratedeterminationbyCE-MS
2Experimental
21Reagentsandchemicals
The following chemicals were used for this study all of analytical gradeAmmoniumacetate(NH OAc)wasobtainedfromMerck(DarmstadtGermany)4
and ferrocene methanol (FcMeOH) from ABCR (Karlsruhe Germany) Milli-Qregwater(182MΩcm)wasgeneratedbyaMilli-QAdvantageA10 system(Merck
Millipore Darmstadt Germany) Carrier solution was prepared by dissolvingndash1NH OAc (50mmolL ) in Milli-Q water FcMeOH solution was prepared by4
dissolvingFcMeOHincarriersolution
22Instrumentation
Electrochemical measurements were performed using a microAutolab Type IIIpotentiostatgalvanostat (Metrohm Autolab B V Utrecht Netherlands)controlledbyNOVA20softwareforexperimentalcontrolanddataacquisitionCFIAwasperformedusingthesetupillustratedinFig1(A)consistingofacarrierreservoirsamplevialandtwofusedsilicacapillaries(PolymicroTechnologiesPhoenix AZ USA inner diameter 100 microm length inlet 40 cm length outlet10cm)connectedtoacommerciallyavailableflowcellfromMicruxTechnologies(model ED-FLOW-CELL Oviedo Spain) Inside of the flow cell the fused silicacapillary was placed in a so-called wall-jet configuration above the workingelectrode of a disposable thin-film gold electrode (model ED-SE1-Au MicruxTechnologiesOviedoSpain)ascanbeseeninFig1(B)Thethin-filmelectrodeswerebasedonathree-electrodesystemwithagoldworkingauxiliaryandquasi-referenceelectrodeThehydrostaticpressurewasachievedbyaheightdifferencebetweeninletandoutletreservoirof30cmresultinginagravityflowofthecarriersolutionthroughafusedsilicacapillaryandsubsequentlythroughtheflowcell
32 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Theinjectionwascarriedoutbyloweringthevialcontainingthecarriersolutiontotheleveloftheoutletexchangingthecarrierreservoirwiththesamplevialandliftingthesamplevialto20cmforadefinedperiodoftimeRe-establishingthecarrierreservoirtookplacethesameway
3Resultsanddiscussion
AsimpleCFIA-ADsystemwasarrangedwherehydrostaticpressurebyaheightdifferencebetweeninletandoutletreservoirresultedinagravitationalflowToobtain general information about the behavior of the assembled CFIA systempreliminaryexperimentswithFcMeOHwereperformedToassurecompatibilitywithMSlateronNH OAcwaschosenastheelectrolyteVariousheightdifferences4
andinjectiontimesweretestedandtheinjectionataheightdifferenceof20cmlasting for 10 s was found to be the optimum concerning feasibility and peakshapes Injections of several solutions of FcMeOH of different concentrationsshowed that the concentration dependence of FcMeOH was linear in theinvestigatedrange(Fig2)Furthermoreexperimentsrevealedthattheinjectionprocedure was established with reasonable precision When repeating the
ndash1injectionof05mmolL FcMeOHincarriersolutionfortentimestherelativestandarddeviationwasfoundtobe3forthemanualinjectionprotocol
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 33
Fig 1(A)Schemeoftheusedcapillaryflowinjectionanalysissetup(1)carrierreservoir(2)samplevial(3)inletcapillarywithalengthof40cmandaninnerdiameterof100microm(4)outletcapillarywithalengthof10cmandaninnerdiameterof100micrombothcapillariesconnectedto(5)acommerciallyavailableflowcellfromMicruxand(6)awastevial(B)Configurationinsidetheflowcell(7)thefusedsilicacapillarywasplacedinaso-calledwall-jetconfigurationabovetheworkingelectrodeof(8)adisposablethin-filmgoldelectrode
4Conclusions
The presented system for CFIA-AD was arranged as simple as possible andrepresents a useful approach for the development of a method for fastoptimization of detection conditions in the context of the determination ofcarbohydratesbymeansofelectrochemistry-CE-MSThesetupisfreeofelectricalinterferencebyhighvoltagecompatiblewithMSandthuspromisingforstudyingtheoxidationofvariousanalytes
References
[1] LuGCrihfieldCLGattuSVeltriLMHollandLACapillaryelectrophoresisseparationsofglycansChemRev118(2018)7867ndash7858
[2] GalantALKaufman RCWilson JDGlucoseDetectionandanalysisFoodChem188(2015)149ndash160
[3] Sarazin C Delaunay N Costanza C Eudes V Gareil P Application of a new capillaryelectrophoreticmethodforthedeterminationofcarbohydratesinforensicpharmaceuticalandbeveragesamplesTalanta99(2012)202ndash206
[4] Rohrer JS Basumallick L Hurum D High-performance anion-exchange chromatographywithpulsedamperometricdetectionforcarbohydrateanalysisofglycoproteinsBiochem78(2013)697ndash709
[5] IslamMAMahbubPNesterenkoPNPaullBMackaMProspectsofpulsedamperometricdetectioninflow-basedanalyticalsystemsndashAreviewAnalChimActa1052(2019)10ndash26
[6] MatysikF-MWernerGTracemetaldeterminationintearsbyanodicstrippingvoltammetryinacapillaryflowinjectionsystemAnalyst118(1993)1523ndash1526
34 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2ndash1 ndash1(A) CFIA-AD recordings of three consecutive injections of (1) 025mmolL (2) 05mmolL
ndash1 ndash1 ndash1(3) 075mmolL and (4) 1mmolL FcMeOH in 50 mmolL NH OAc detection at a Micrux4
thin-filmAuelectrodeataconstantpotentialof03VinaflowcellHydrodynamicinjectionlasted10sataheightdifferenceof20cm(B) Calibration dependence of FcMeOH for CFIA-AD determination and detection at a Micruxthin-filmAuelectrodeataconstantpotentialof03VinaflowcellThestandarddeviationsofpeakheights(n=3)areindicatedbyerrorbars
1Introduction
Topreventsorptiononthecapillarysurfaceandimproveseparationefficiencyand selectivity of determined analytes coatings are formed on the capillarysurface There are two types of ones namelydynamic and covalently bondedcoatings Despite the simplicity of creation dynamic coatings cannot providerequiredreproducibilitywhilecovalentcoatingscontributethestableelectro-osmoticflow(EOF)andhighreproducibilityoftheanalysisInmostcasesthe
Application of covalent coatings based on imidazolium cations for separation and on-line preconcentration of basic and neutral analytes in capillary electrophoresis
a ab aANASTASIAVKRAVCHENKO EKATERINAAKOLOBOVA LIUDMILAAKARTSOVA
a DepartmentofOrganicChemistryInstituteofChemistrySaintPetersburgStateUniversity 26Universitetskiiprospect198504StPetersburgPeterhofRussiakravchenko161216gmailcom
b TheFederalStateInstituteofPublicHealthldquoTheNikiforovRussianCenterofEmergencyandRadiationMedicinerdquoTheMinistryofRussianFederationforCivilDefenceEmergenciesandEliminationofConsequencesofNaturalDisasters54Optikovst197082StPetersburgRussia
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 35
AbstractThemethodofcapillaryelectrophoresis (CE) isactivelydevelopedandmoreandmoreattractsscientistsattentioneveryyearHoweverthesorptionofanalytesonsurfaceoffused-silicacapillarywallsisoneof thesignificantdisadvantagesof thisapproachThe formationofcoatings on the inner capillary surface is typical way to preventsorption and to increase separation efficiency and selectivity ofdetermined analytes Coatings that covalently bonded to capillarywalls is more suitable because they are stable and provides highreproducibilityofanalysisThepresentworkisfocusedonthedevelo-pmentofthemethodofelectrophoreticdeterminationofbiologicalactiveanalytesusingacovalentcoatingbasedonimidazoliumcationsTheeffectofsubstituentinimidazoliumringonmainelectrophoreticparameters was examined It was shown that alkylimidazoliumcoatingscontributetosignificantreducingofbiogenicamineslimitsof detection while β-cyclodextrinimidazolium covalent coatingallowstoseparatebothofhydrophobicandhydrophilicanalytesinonerun
Keywordsbiologicalactiveanalytescapillarycoatingcapillaryelectrophoresisimidazoliumionicliquids
analytes nature determines type of usedmodifiers because suitable ones canprovideaccessorial interactionbetweentheanalytesandthestationaryphaseimproving separation selectivity and efficiency [1 2] Ionic liquidshave beenwidelyusedinanalyticalchemistry[3]andseparationtechniquesparticularlyincapillaryelectrophoresis[4]Earlyresearches[5ndash9]haveshownopportunityofcovalently bonded imidazolium ionic liquids for electrophoretic separationHowever the effect of various substituents in imidazolium ring hasnot beendescribed previously Thus the purpose of this study was to create covalentcoatingsbasedon ionic liquidwithvarioussubstituentsand tocompare theiranalytical capabilities in the electrophoretic separation of biologically activecompounds
2Experimental
21Reagentsandchemicals
(3-Glycidyloxypropyl)trimethoxysilane (GPTMS) hydrochloric acid sodiumdodecyl sulfate (SDS) imidazole 22-diphenyl-1-picrylhydrazyl (DPPH)p-toluen-sulfonylchlorideβ-cyclodextrinhydrocortisone(F)11-deoxycortisol(S)Corticosterone(B)rac-ketoprofen(ndash)-adrenaline(A)L-(ndash)-norepinephrine(NE) DL-normetanephrine (NMN) dopamine (DA) DL-metanephrine hydro-chloride(Met)serotoninhydrochloride(Ser)homovanillicacid(HVA)24-di-hydroxy-benzoic acid (24-DHBA) 34-dihydroxy-L-phenylalanine (DOPA)L-tryptophan(Trp)L-tyrosine(Tyr)werepurchasedfromSigma-Aldrich(USA)1-Bromo-butane1-bromooctanewerepurchasedfromReagentPlus(Ukraine)Sodium dihydrogenphosphate dihydrate acetone NN-dimethylformamide(DMF)wereobtainedfromMerck(Germany)AllreagentsusedwereanalyticalgradeAllsolutionswerepreparedusingdeionizedwater
22Instrumentation
Capillary electrophoresis experiments were carried out using the system ofcapillaryelectrophoresisCAPEL-105M(LumexRussia)withUV-spectrophoto-metricdetector(wavelengthrange190ndash360nm)Separationswereperformedusing 58times49 cm (9 cm to the detector outside diameter 360 microm and innerdiameter50microm) coated silica capillaries (LumexRussia) Thebuffer pHwasmeasuredwithapH-meterHI2210ndash2216(Hanna)
23Capillarycoatingsynthesis
Earlier our research team has proposed the synthesis route for the covalentcoatingsbasedonimidazoliumcationfunctionalizedwithalkylgroup[10]andβ-cyclodextrin [11] All capillarieswere prepared according above-mentioned
36 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
manuscriptsandcharacterizedbytheEOFmobilitymeasurementandscanningelectronmicroscopyThesynthesisconsistedoffollowingstepspreparationofacapillarytocreateacovalentcoating(heatingcapillaryfilledwith2MNaOHat90degCfor1handdryingfollowed)silylationwithGPTMSandfunctionalizationwiththeimidazolesolutionfollowedmodificationbybutyl-andoctylbromideortosyl-β-cyclodextrin(seedetailsin[10]and[11])
24Solutions
A stock buffer solution in concentration 50mM was prepared by dissolvingappropriate amount of sodium dihydrogenphosphate dihydrate in deionizedwateradjustingpHto20with1MhydrochloricacidThisbuffersolutionwasthendilutedwithdeionizedwater
ndash1 Allthesamplestocksolutionswerepreparedwithconcentration10mgmL Thestocksolutionsoftheneurotransmittersandtheirmetabolites(adrenalinenoradrenaline dopamine normetanephrine metanephrine serotonin homo-vanillicacid)and24-dihydroxybenzoicacidasinnerstandardandaminoacids(tryptophan34-dihydroxy-L-phenylalanine tyrosine)wereprepared in01MhydrochloricacidThestocksolutionsofsteroids(hydrocortisone11-deoxycor-tisolandcorticosterone)wereprepared inacetonitrileThestocksolutionsofketoprofen racemate and S-ketoprofen were prepared in acetonitrilewatersolution(1090vv) Untilelectrophoreticanalysisthestocksolutionswerestoredatndash16degCTheworkingsolutionswerepreparedbydilutingtheinitialsolutionswithwaterjustbeforetheexperiments
3Resultsanddiscussion
CovalentcoatingsbasedonN-alkylimidazoliumcationwereespeciallysuitableforseparationofneurotransmittersandtheirmetabolites(Fig1)Inadditionthecombinationofcovalentcoatingwithon-linepreconcentrationtechniquesallowstothesignificantdecreaseoftheseanalyteslimitsofdetection(LOD)Accessorialinteractions positively charged analytes with positively charged imidazoleimproveseparationselectivity(viaπ-πinteraction)andefficiency(concentrationintightzonesviaelectrostaticrepulsion)Sodiumdodecylsulfate(SDS)addedintobackgroundelectrolyte(inconcentrationabovecriticalmicelleconcentra-tion) strongly interacts with hydrophobic alkyl groups in covalent coatingstructureThenegativelychargedSDSlayerisformedoninnercapillarysurfaceThedoublereversingEOFallowsustocarryoutelectrokineticinjectionofsampleandon-linepreconcentrationbysweepingsimultaneouslyLODweredeclineto
ndash108ndash20ngmL ThelengthofalkylsubstituentalsoaffectsthestackingefficiencyfactorandLODMorehydrophobicoctylgroupscomparetobutylprovidemoreeffectiveinteractionwithSDSandasresultlowerLOD
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 37
Covalentcoatingmodifiedβ-CDhasnotshownsharpreducingofLODbyon-linepreconcentrationStacking sweeping (SDSasmicelle reagent) field-enhancedsample injection were examined using different model mixtures of analytesNeverthelessthiscoatingallowssimultaneousseparationofbothofhydrophobicsteroidhormonesandhydrophilicbiogenicaminesinasinglerun(Fig2) The guest-host interaction hydrophobic cavity of β-cyclodextrin with thehydrophobicsteroids leadsto the formationofcomplexwhichaffectssteroidselectrophoretic mobility At the same time β-cyclodextrin can act as a chiralselector and baseline separation of ketoprofen enantiomers has also beenachieved
38 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Electropherogramofmixtureof neurotransmitters and theirmetabolitesadrenaline (A)norepinephrine (NE)normetanephrine (NMN)dopamine (DA)metanephrine (Met) serotonin(SER)homovanillicacid(HVA)andtheinnerstandard24-dihydroxybenzoicacid(24-DHBA)oncovalentlymodifiedwithN-buthylimidazoliumionicliquidscapillaryConditions10mMNaH PO 2 4
(adjusted to pH= 20 by 1MHCl) injection 50 stimes30mbar ndash20 kV 220 nmmodelmixurendash1 ndash1 ndash110microgmL (METADNMNNADA24-DHBA)5microgmL (SER)and20microgmL (HVA)
4Conclusions
ItwasshownthatstructurecovalentcoatingaffectsitsanalyticalcharacteristicsWecomparedtwotypesofcovalentcoatingdifferingsubstituentinimidazoliumringnamelyalkylgroupandβ-cyclodextrinThefirsttypeisgreatcoupledwithon-line preconcentration technic but it is limited to effectively determine ofbiogenicaminesonlywhilethesecondtype(withβ-cyclodextrin)showedthepossibilitiestoseparatevariousanalytesbutsuitableon-linemodehasnotbeenfoundThemainpointsaresummarizedinTable1
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 39
Covalentcoatingtype Electrophoreticseparationof On-lineprecon- centration biogenicamines amino steroid ketoprofen andtheirmeta- acids hormones enantiomers bolites N-β-cyclodextrinimida- yes yes yes yes thesuitableapproachzoliumcovalentcoatings wasnotfoundedN-alkylimidazolium yes yes nonsepa- nonsepa- thesignificantreducingcovalentcoatings rated rated forbiogenicaminesLOD
Table 1Thesummationofpossibilitiesofcovalentcoatingsbasedonimidazoliumcation
Fig 2Electropherogramofsimultaneousseparationofhydrophobic(steroidhormones)andhydro-philicanalytes (aminoacidsandbiogenicamines) insinglerunwithcovalentcoatingbasedonimidazoleandβ-CDConditions10mMNaH PO (adjusted topH = 20by1MHCl) injection2 4 20 stimes30mbarndash20kV254nm(1ndash8min)and220nm(8ndash15min)0mbar(1ndash10min)and40mbar(10ndash15 min) Model mixture corticosterone (B) hydrocortisone (F) 11-deoxycortisole (S)
ndash1 ndash15μgmL L-tryptophan(Trp)34-dihydroxy-L-phenylalanine(DOPA)10μgmL L-tyrosine(Tyr)ndash15 μg mL noradrenaline (NA) normetanephrine (NMN) adrenaline (AD) dopamine (DA)
ndash120μgmL
Acknowledgments
This work was supported by Russian Science Foundation (grant numbers 19-13-00370) Theauthors are also grateful to the Chemistry Education Centre and Nanothechnologies Centre ofResearchParkSaintPetersburgStateUniversityfortechnicalsupport
References
[1] HuLFYinSJZhangHYangFQRecentdevelopmentsofmonolithicandopen-tubularcapillaryelectrochromatography(2017ndash2019)JSepSci43(2020)1942ndash1966
[2] KartsovaLAKravchenkoAVKolobovaEACovalentcoatingsofquartzcapillariesfortheelectrophoretic determination of biologically active analytes J Anal Chem 74 (2019)729ndash737
[3] HoTDZhangCHantaoLWAndersonJLIonicliquidsinanalyticalchemistryFundamen-talsadvancesandperspectivesAnalChem86(2014)262minus285
[4] TangSLiuSGuoYLiuXJiangSRecentadvancesofionicliquidsandpolymericionicliquids incapillaryelectrophoresisandcapillaryelectrochromatography JChromatogrA1357(2014)147ndash157
[5] QinWLiSFYElectrophoresisofDNAinionicliquidcoatedcapillaryAnalyst128(2003)37ndash41
[6] QinWWeiH Li SFY 13-Dialkylimidazolium-based room-temperature ionic liquids asbackgroundelectrolyteand coatingmaterial in aqueous capillaryelectrophoresis JChro-matogrA985(2003)447ndash454
[7] QinW Fong S Li Y Determination of ammonium andmetal ions by capillary electro-phoresisndashpotential gradient detection using ionic liquid as background electrolyte andcovalentcoatingreagentJChromatogrA1048(2004)253ndash256
[8] QinWLiSFYAn ionic liquidcoating fordeterminationofsildenafilandUK-103320 inhumanserumbycapillaryzoneelectrophoresis-iontrapmassspectrometryElectrophoresis23(2002)4110ndash4116
[9] BorissovaMVaherMKoelMKaljurandMCapillaryzoneelectrophoresisonchemicallybondedimidazoliumbasedsaltsJChromatogrA1160(2007)320ndash332
[10] KolobovaEKartsovaLKravchenkoABessonovaEImidazoliumionicliquidsasdynamicand covalent modifiers of electrophoretic systems for determination of catecholaminesTalanta188(2018)183ndash191
[11] KravchenkoAKolobovaEKartsovaLMultifunctioncovalentcoatingsforseparationofaminoacidsbiogenicaminessteroidhormonesandketoprofenenantiomersbycapillaryelectrophoresisandcapillaryelectrochromatographySepSciplus3(2020)102ndash111
40 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Synthetic 4-hydroxy-3-methoxybenzaldehyde (vanillin) is used as a flavoringagent in foodsdrinksperfumesandpharmaceuticals [1]However at certainconcentrationsthesubstancemayaccumulateinthebodyhaveatoxiceffectand
ndash1at high concentrations may be fatal (lethal dose LD (oral rat) =2gkg 50ndash1 ndash1LD (oral guinea pig) = 14gkg LD (intravenous dog) = 132gkg lethal50 50 ndash1concentrationLC (inhalationmouse)=417gkg )[2]AccordingtoRussianState
StandartGOST121005-88thetoxiceffectsofvanillinintheworkplaceinthendash3formofvapoursoraerosolsareobservedatconcentrationsabove15mgm
Chromatography[3]spectrophotometry[4]capillaryelectrophoresisareusedforvanillindeterminationindifferentobjects CurrentlysmokingmixturesforhookahsandelectroniccigarettesarewidelyusedamongyoungpeopleThesemixturesarenotcontrolledforthecontentofsubstancesandarefreelyavailableconsideringthemmoreharmlesswithrespecttoordinarycigarettesThusthedevelopmentofamethodforthedeterminationof4-hydroxy-3-methoxybenzaldehydeinsmokingmixturesisrelevant
Determination of vanillin in smoking mixtures by spectrophotometry
ELIZAVETAEFREMENKOANNACHERNOVAOLGABASTRYGINA
DepartmentofChemicalEngineeringNationalResearchTomskPolytechnicUniversityLeninavenue30634050TomskRussiaeaetpuru
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 41
AbstractTheresearchdealswithdeterminationofvanillin insmokingmix-turesbyultraviolet-visiblespectrophotometryThemethodshowed
ndash1goodlinearityintherangeof005ndash012gL withalimitofdetectionndash1005gL After validation studies the method was successfully
applied to thedeterminationof vanillin in smokingmixtureswithsatisfactoryresultsItwasshownthattheerrorofthismethoddoesnot exceed 1 The developed spectrophotometric procedure fordeterminingvanillininsmokingmixturescanbeusedasacontrol
Keywordssmokingmixturesspectrophotometryvanillin
2Experimental
21Reagentsandchemicals
Asampleofvanillin(purity98)wastakenastheobjectofstudyAssolventsweused95ethanolAllchemicalsusedwereofanalyticalreagentgrade
22Instrumentation
Theopticaldensityofsampleswasmeasuredincuvettewithanabsorbinglayerthickness of 10 mm using a Cary 60 spectrophotometer (Agilent USA) Allmeasurementswerecarriedoutatroomtemperature
23Samplepreparation
Sample preparation of the investigated objects consisted of the preliminarydissolutionofthesamplein95ethanolThesample10mgoftobaccoldquoAdalyandashVanillardquo(Turkey)wasdiluted in10μLof95ethanol to theconcentrationof
ndash11gL Thesample10μLofldquoFlavoringTPAndashVanillaCustardrdquo(USA)wasdilutedin10 μL of 95 ethanol The resulting solution was diluted six times to the
ndash3concentrationof017μLcm
3Resultsanddiscussion
TodeterminevanillininthesamplestheopticalpropertiesofvanillininvarioussolventsweredeterminedAsaresultthe95ethanolwaschosenastheoptimalsolvent[4] IthasbeenestablishedthatintheUVspectraoftheanalyteabsorptionbondsareobservedwithmaximumvaluesat23002800and3100nmwhichcorres-pondstopublisheddata[45](Fig1) To quantify vanillin the calibration curve of the optical density on theconcentrationofvanillin in95ethanolwasobtainedatconcentrations005
ndash1006007008010and012gL Calibrationcurveofvanillinin95ethanolatawavelengthof280nmis
ndash1 A =81914c[gL ]+00357 (1)2802 R =1
Calibrationcurveofvanillinin95ethanolat310nmis
ndash1 A =73824c[gL ]+00301 (2)3102 R =1
42 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Inthespectraoftheanalyzedsamplesolutionsabsorptionmaxima(2800nmand3100nm)characteristicforvanillinwereobservedTheamountofvanillininthesamplewasdeterminedusingcalibrationcurvesat280and310nmWeightedleast square regressionwas applied to the calibration curves to improve theaccuracyespeciallyatinlowconcentrationlevelrangeGoodlinearitywasfound
ndash1 ndash1intherangeof005ndash012gL withadetectionlimitof005gL TheresultsarepresentedintheTable1
4Conclusions
ThedevelopedmethodcanbeusedasacontrolmethodTheerrorinthemethodfordeterminingvanillininthesampleldquoFlavoringTPAndashVanillaCustardrdquowithaknownconcentrationofvanillinwas0004Accordingtothedataobtainedwerecommendawavelengthof280nmforthedeterminationofvanillininsamples
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 43
λ nm
Absorban
ce
ndash1Fig 1 Absorption spectrum of vanilin solution in 95 ethanol at concentration 01 mol L (anabsorbinglayerthicknessof10mm)
Sample λnm Tookmg Foundmg S Sх Δх δ
FlavoringTRA 310 10850 104096 00024 00011 00006 00306ndashVanillaCustardAdalyandashVanilla 310 100000 10162 00019 00009 00002 00025
FlavoringTRA 280 100300 96062 00033 00015 00032 00042ndashVanillaCustardAdalyandashVanilla 280 100000 10122 00013 00006 00012 00017
Table 1Testingmethods introduced foundof vanillin in the samplesat310nmby spectrophotometricmethod(n=5p=099SndashstandarddeviationSxndashrelativestandarddeviationΔхndashabsoluteerrorδndashrelativeerror)
ThenthedevelopedmethodwastastedonthesampleldquoAdalyandashVanillardquosamplewithamorecomplexcompositionandanunknownconcentrationofvanillinwastaken The vanillin content in the sample was determined according to thedevelopedmethoditamountedto10ofthetotalmassStudieshaveshownthepossibility of using spectrophotometric analysis for the qualitative andquantitative determination of vanillin Also based on preliminary studies aspectrophotometricprocedurewasdevelopedforthequantitativedeterminationofvanillinbasedonabsorptioninethanolinthewavelengthrange200ndash400nm
References
[1] httpswwwrusnaukacom43_DWS_2015Chimia6_203179dochtm (accessed 25thFebruary2019)
[2] httpswwwcdcgovnioshrtecsdefaulthtm(accessed11stApril2020)[3] AliLPerfettiGDiachenkoGRapidmethodforthedeterminationof342coumarinvanillin
and ethyl vanillin in vanilla extract by reversed-phase liquid 343 chromatography withultravioletdetectionJAOACInt91(2008)383ndash386
[4] БастрыгинаОАЕфременкоЕАЧерноваАПВыделениеванилинаисследованиеегооптическихсвои ствопределениевбиологическомматериалеВХимияихимическаятехнология в XXI веке Материалы XX Международной научно-практическойконференции имени профессора ЛП Кулёва студентов и молодых ученых ТомскНациональныи исследовательскии Томскии политехническии университет 2019с301ndash302
[5] WeastRCHandbookofChemistryandPhysics60thedBocaRatonCRCPress1979p143
44 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
UraniumbelongstothegroupofhazardouselementsItisahighlyharmfulandradioactiveelementtoxictohumansandalllivingorganisms[12]Inhaledwithair it has a particularly destructive effect on the kidneys and as a result ofaccumulationinwhitebloodcellsitcanalsocauseimpairmentoftheimmunesystem[2]Uraniumoccursatseveraldegreesofoxidationhoweverinaqueoussolutionsthemoststableformisuranylion(UO (II))[12]Thepresenceofura-2
niumintheenvironmentiscausedbyamongothersnaturalsoilandrockerosionEnvironmentalpollutionwiththiselementisalsoconstantlyincreasingduetohumanactivitycoalcombustionuraniumoreminingandprocessingthearmsindustryandtheuseofuraniumasnuclearfuelinfissionreactors[3]Itisveryimportanttoconstantlymonitortheconcentrationofuraniumbothinthenaturalenvironment in order to assess its state and safety (especially in the case ofdrinkingwater)aswellasinallstagesofprocessingprocessesassociatedwiththenuclearindustrytoavoidtheoccurrenceofnuclearpollution[13] Scientists have made many attempts to develop research methods todeterminethecontentofuranylcompoundsinliquidsamplesEffortsweremadetousemanyanalyticalmethodsforthispurposeincludingspectrophotometry
Uranyl ion-selective electrode with solid contact
KAROLINAPIETRZAKCECYLIAWARDAK
DepartmentofAnalyticalChemistryInstituteofSciencesFacultyofChemistryMariaCurie-SklodowskaUniversityMariaCurie-SklodowskaSq320-031LublinPolandkarolinapietrzakpocztaumcslublinpl
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 45
AbstractNewallsolidstateuranylion-selectiveelectrodeswithlowdetection
ndash7 ndash1limits(71times10 molL )shortresponsetimegoodselectivityandstable and reproducible potential were developed Many types ofelectrodeswith different active ingredient content in ion-selectivemembrane (bis(244-trimethylpentyl)phosphonium acid Cyanex-272)were testedAs an additive an ionic liquid1-octyl-3-methyl-imidazole chloride was used The optimal composition of theion-selective membrane was chosen from all electrodes based onthedeterminationand comparisonof analyticalparametersof thesensors
Keywordsion-selectiveelectrodesolidcontacturanyl
plasma spectrometry luminescence spectroscopy voltammetry or chromato-graphymethods[2] Duetomanyadvantagesofpotentiometricmethods(amongthemlowercostseasieroperationofdevicesquickresponseandtheabilitytoperformmeasure-ments in flowmode) [3] a numberof potentiometric sensorshave alsobeendeveloped that could be successfully used in this type of research Themostpopularpotentiometricsensorsincludeion-selectiveelectrodes(ISEs)whicharecharacterized by low-energy consumption small size and portability and aresuccessfullywidelyusedforthedeterminationofbothinorganicandorganicionsinclinicalanalysisprocesstechnologyaswellasincontrolthestateofthenaturalenvironment[45]Removaloftheinternalsolutioncontainingthesameanalytetowhich theelectrode is sensitiveresulted in theso-calledsolidcontact ISEswhicharemuchsmallerinsizethantheirpredecessorsaremoreconvenienttouse and more mechanically resistant In this type of sensors however it isimportanttoachievesatisfactorypotentialstabilitywhichisnecessarytoobtainsatisfactoryresults[5]AveryimportantpartofISEsistheion-selectivemem-branewhosecompositiondeterminestheanalyticalparametersofthesensorsResearchers are currently focusing on the production and testing of newsubstancesthatcouldbesuccessfullyusedasmembranecomponentsandsolidcontacts thatwould allow to obtain new sensorswith lower detection limitslongerlifetimeandbetterpotentialstabilityandtodeterminenewpreviouslyunattainableanalytes[4] AstheactivecomponentsofthemembranesensitivetouranylionscientistshavealreadyusedKryptofix22DD(413-didecyl-171016-tetraoxa-413-diaza-cyclooctadecane)[2]Cyanexextractants(bis(244-trimethylpentyl)phosphinicacid bis(244-trimethylpentyl)monothiophosphinic acid and bis(244-tri-methylpentyl)dithiophosphinic)acid[3]DBBP(dibutylbutylphosphonate)andDOPP (di-n-octyl phenylphosphonate) [6] DMSO (dimethylsuphoxide) [7]TTPTP (5678-tetrahydro-8-thioxopyrido[4345]thieno[23-d]pyrimidine-4(3H)one)[8]orTEHP(tris(2-ethylhexyl)phosphate)andTPTU(O-(12-dihydro-2-oxo-1-pyridyl)-NNNN-bis(tetra-methylene)uronium hexafluorophos-phate)[9]
2Experimental
21Reagentsandchemicals
This paper presents research on the design and properties of ion-selectiveelectrodes with solid contact for the determination of uranyl ions Bis(244-trimethylpentyl)phosphonium acid (Cyanex-272) was used as the activecomponentof themembranewhichwasdescribed in the literatureasagooduranylextractant[10]Inordertoensureaconstantpotentialofthiselectrodeandreducetheelectroderesistancetheion-sensitivemembranewasenrichedwithafewpercentadditionof1-octyl-3-methylimidazolechlorideionicliquid
46 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Several types of ion-selective electrodes were prepared using an AgAgClelectrodeasaninternalelectrodewhichdifferinthequantitativeandqualitativecompositionofthemembranesAllcompositionsarelistedinTable1
22Instrumentation
Measurements were made at room temperature using a 16-channel datacollectionsystem(LawsonLabs IncUSA)coupled toa computer in solutionsmixedwithamechanicalstirrerAsilversilverchlorideelectrodewithdoublejunctionwasusedasthereferenceelectrode
3Resultsanddiscussion
The effect of ion-selective membrane composition on the properties of theobtained potentiometric sensors was examined by determining their basicanalyticalparametersincludingslopeoftheelectrodecharacteristicsdetectionlimitmeasuringrange(concentrationrangeinwhichthecourseoftheelectrodecharacteristics isrectilinear)pHrange(inwhich ithasnoeffect forelectrodepotential)andresponsetimeTheobtainedvaluesofthetestedparametersareshowninTable2 Figure1showsthecalibrationcurvesofthetestedelectrodesdeterminedin
ndash7 ndash1 ndash1UO (NO ) solutionsintheconcentrationrange1times10 ndash1times10 molL Asitcan2 3 2
beseeninFig1andTable2allelectrodesweresensitivetouranylionsbutindifferent extend The best response exhibited ISE-3 containing 1 (ww) ofionophore Increasing the ionophore content in themembrane shortened thelinearityrangeofthecalibrationcurveanditssupernenstianslope Theselectivityofthetestedelectrodeswasestimatedbydeterminingtheselec-tivitycoefficients inrelationto interfering ionsForthispurpose theseparate
ndash1solutionmethodwasused(extrapolatingresponsecurves toa =a =1molL )i j
ComparisonofISE-1andISE-3electrodeselectivityisshowninFig2
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 47
Table 1Quantitative and qualitative composition of electrode membranes Cyanex-272 (bis(244-tri-methylpentyl)phosphoric acid) TBP (tri-n-butyl phosphate) and OMImCl (1-octyl-3-methyl-imidazolechloride)
Abbreviation Membranecomposition(ww)ofelectrode Cyanex-272 PVC TBP OMImCL
ISE-1 00 33 620 5ISE-2 05 33 615 5ISE-3 10 33 610 5ISE-4 30 33 590 5ISE-5 50 33 570 5ISE-6 100 33 520 5
48 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Abbreviation Slope Detectionlimit Linearrange Response pHrange2+ ndash1 ndash1ofelectrode mVpa(UO ) molL molL times2
ndash5 ndash5 ndash1ISE-1 297 25times10 5times10 ndash1times10 5ndash8 28ndash42ndash6 ndash5 ndash1ISE-2 292 65times10 1times10 ndash1times10 5ndash8 25ndash60ndash7 ndash5 ndash1ISE-3 298 71times10 1times10 ndash1times10 5ndash8 24ndash60
ndash6 ndash4 ndash1ISE-4(I) 357 31times10 5times10 ndash1times10 5ndash8 ndndash6 ndash6 ndash4ISE-4(II) 242 31times10 5times10 ndash5times10 5ndash8 nd
ndash3 ndash1ISE-5(I) 638 nd 1times10 ndash1times10 5ndash10 ndndash5 ndash3ISE-5(II) 234 nd 5times10 ndash1times10 5ndash10 ndndash3 ndash1ISE-6(I) 733 nd 1times10 ndash1times10 5ndash10 ndndash5 ndash3ISE-6(II) 222 nd 5times10 ndash1times10 5ndash10 nd
Table 2Selectedparametersandtheirdeterminedvaluesoftestedionselectiveelectrodes
Fig 1 Calibration curves of the testedelectrodesobtainedinUO (NO ) solutionsin2 3 2 ndash7the concentration range from 1times10 to
ndash1 ndash11times10 molL
Fig 2Comparisonofselectivitycoefficientspot(log K (UO (II))M) for electrodes ISE-12
(1stcolumn)andISE-3(2ndcolumn)
Inordertoexaminethereversibilityofthepotentialofthetestedelectrodesndash4 ndash1potentialmeasurementsweremadealternatelyinsolutions1times10 molL and
ndash5 ndash11times10 molL ofUO (NO ) TherecordedpotentialreadingsareshowninFig32 3 2
Long-term potential stability and sensor reproducibility were evaluated byndash1determiningtheaveragevalueoftheelectrodepotentialina01molL UO (II)ion2
solutionovertimeforthreeidenticalISE-3Thesemeasurementsweremadetoobservechangesinthepotentialofelectrodeswiththesameconcentrationoveralongperiodoftime(30days)Figure3showsthelong-termpotentialstabilityandreproducibilitydeterminedforthreeidenticalsensors
4Conclusions
Asaresultofthetestsion-selectiveelectrodeforthedeterminationofuranylionswasobtainedwhich iseasy todesignanduseThebestanalyticalparametersexhibitedISE-3containing1ionophoreintheion-selectivemembraneForthis
ndash7 ndash1typeofelectrodesthedetectionlimitof71times10 molL linearityoftheelectrodendash6 ndash1 ndash1calibrationcurve in the range1times10 ndash1times10 molL andresponse time5ndash8s
were obtained In addition the manufactured sensors also showed stablereproducibleandreversiblepotentialandverygoodselectivityinrelationtothetestedinterferents
References
[1] AnsariRMosayebzadehZConstructionofanewsolid-stateU(VI)ion-selectiveelectrodebasedonpolypyrroleconductingpolymerJRadioanalNuclChem299(2014)1597ndash1605
[2] GhanbariMRounaghiGHAshrafNAnuranylsolidstatePVCmembranepotentiometricsensor based on 413-didecyl-171016-tetraoxa-413-diazacyclooctadecane and itsapplicationforenvironmentalsamplesIntJEnvironAnalChem97(2017)189ndash200
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 49
Fig 3 Stability () reproducibility andreversibility () of the potential of ISE-3Standard deviations given on the plot aredeterminedforthesamethreeISE-3
[3] Badr IHA Zidan WI Akl ZF Cyanex based uranyl sensitive polymeric membraneelectrodesTalanta118(2014)147ndash155
[4] BiegCFuchsbergerKStelzleMIntroductiontopolymer-basedsolid-contaction-selectiveelectrodes basic concepts practical considerations and current research topics AnalBioanalChem409(2017)45ndash61
[5] Bobacka J IvaskaA LewenstamA Potentiometric ion sensorsChem Rev108 (2008)329ndash351
[6] ZidanWI Badr IHA Akl ZF Development of potentiometric sensors for the selective2+determinationofUO ionsJRadioanalNuclChem303(2015)469ndash4772
[7] SalehMBSolimanEMGaberAAAAhmedSANovelPVCmembraneuranylion-selectivesensorSensActuatorsB114(2006)199ndash205
[8] SalehMBHassanSSMAbdelAAAbdelNAAnoveluranylion-selectivePVCmembranesensor based on 5678-tetrahydro-8-thioxopyrido[4345]thieno[23-d]pyrimidine-4(3H)oneSensActuatorsB94(2003)140ndash144
[9] HassanSSMAliMMAttawiyaAMYPVCmembranebasedpotentiometricsensorsforuraniumdeterminationTalanta54(2001)1153ndash1161
[10] Prabhu DR Ansari SA Raut DR Murali MS Mohapatra PK Extraction behaviour ofdioxouranium(VI) cation by two phosphorous-based liquid cation-exchangers in room-temperatureionicliquidsSepSciTechnol52(2017)2328ndash2337
50 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Metronidazole(2-methyl-5-nitroimidazole-1-ethanol)isoneofthemostwidelyused nitroimidazole antibiotics Metronidazole is used for the treatment ofinflammatorydiseasescausedbyanaerobicorganismsandsomeprotozoaandforpreventionofdysenterycolibacillosiseimeriosisbalantidiasissalmonellosisenteritissepticemiapost-surgicalcomplications[1ndash3]Oxytetracyclinehydro-chlorideisanantibioticofthetetracyclinefamilyItisoneofthemostcommonlyused antibiotics in poultry because of its low cost and effective [4] Thesecompounds are intensively used in poultry breeding and stockbreedingUnreasonableuseofthesedrugscancauseseriousfoodsafetyissues[5] The veterinary drug Nozemat which includemetronidazole and oxytetra-cyclinehydrochloridewaschosenfortheexperimentsNozematisusedtotreat
Polarographic determination of metronidazole and oxytetracycline hydrochloride in veterinary drug for honey bees
a a bKATERYNAPLOTNIKOVA LILIYADUBENSKA IVANZELENYI
a AnalyticalChemistryDepartmentIvanFrankoNationalUniversityofLvivKyrylaiMefodiaStr879005LvivUkrainekaterina27plgmailcom
b DrohobychPedagogicalLyceumIvanaFrankaStr3682100DrohobychUkraine
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 51
AbstractWehavedevelopedanewpolarographicmethodforthedetermin-ation of metronidazole and oxytetracycline hydrochloride in theveterinarydrugNozematforhoneybeesThetechniqueisbasedonthereductionofpolarographicallyactivecompoundsonamercurydropletelectrodeTheinfluenceofthecomponentsoftheveterinarydrugNozematonthepolarographicdeterminationofmetronidazolewasstudiedItwasfoundthatthereductionofmetronidazoleisnotaffected by glucose and ascorbic acid but is affected by oxytetra-cyclinehydrochloridewhichisreducedtomercurydropletelectrodeatapotentialofndash145VThedevelopedtechniqueischaracterizedbyeaseofsamplepreparationandcost-effectivenessThistechniquehastheabilitytoidentifysimultaneouslyanddeterminatemetronidazoleand oxytetracycline hydrochloride in solution without the use ofseparationandconcentrationmethods
Keywordselectrochemistrymetronidazoleoxytetracycline
hydrochloridepolarographyveterinarydrug
bees and it can be given in unregulated doses Because of this an unknownamountofmetronidazolecangetintothehoneyanditsometimescausessideeffectsofthehumanbodyanditcouldbeofgreatconcernforpublichealth[56]MedicinesforpeoplearemorestringentandbettertestedthanveterinarydrugsTheproblemofthecontroloftheveterinarydrugsisurgentnowadaysVeterinarymedicinescouldbeunauthorizedandtheuncontrolleduseofmedicinesexistsinretailpharmaciesofmedicineorimportedascontrabandfromothercountries The most widespreadof these classes in thequality controlare chromato-graphic [6ndash9] spectrophotometric [10ndash13] and electrochemical methods[14ndash17]Manyoftheknownmethodsforthedeterminationofmetronidazoleandoxytetracyclinehydrochloridehaveanumberofdisadvantagestime-consumingtheuseoforganicsolventsandexpensivereagentsthesideeffectsofexcipientsandotheractivesubstancesElectrochemicalmethodsarepromisingalternativefor the determination of the electroactive substances Their advantages aresimplicityminiaturizationhighsensitivityandrelativelylowcostThereforethesearch for simple express and affordable methods for the determination ofmetronidazoleremainsrelevantOneofthepromisingmethodsofdeterminationisvoltammetry
2Experimental
21Reagentsandchemicals
VeterinarydrugNozemat (manufacturerAPI-SANRussia) is a yellowpowderwithaslighttypicalodorAvailableinlaminatedbagsof25gCompositionper1gof the drug metronidazole 400 mg oxytetracycline hydrochloride 400 mgglucoseascorbicacid MetronidazoleandoxytetracyclinehydrochloridewerepurchasedfromSigmaAldrich(USA)Stockstandardsolutionofmetronidazolefordeterminationwaspreparedbydissolvingtheexactamountofstandardin7mLof2Mhydrochloricin 500 mL volumetric flask Stock standard solution of oxytetracyclinehydrochloride was prepared by dissolving the exact amount of standard indistilledwaterin500mLvolumetricflaskAfterthatthesolutionswereadjustedtothemarkwithdistilledwaterandmixedthoroughly The Britton-Robinson buffer preparationwas as follows 202 g of sodiumtetraboratedecahydrate287mLofglacialaceticacidand176mLofconcen-tratedorthophosphoricacidweredissolvedin10Lvolumetricflask Working solution preparation was as follows an aliquot of stock standardsolutionwasaddedintoa25mLvolumetricflasktoobtainasolutionwiththenecessaryconcentrationthen2mLofBritton-RobinsonbufferwithnecessarypHwasaddedtotheflaskanddistilledwaterwasaddedtothemark AqueoussolutionofNozematwaspreparedasfollowstheexactportionofthetestveterinarydrugwasdissolved ina250mlvolumetric flaskAnaliquotof
52 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Polarogramsof(A)metronidazoleand(B)metronidazolewithoxytetracyclinehydrochloridendash1solutionsat02MBritton-RobinsonbufferbackgroundatpH=96(υ=05Vs c(metronidazole)=
ndash5 ndash5=45times10 Мc(oxytetracyclinehydrochloride)=50times10 М
100mloftheresultingsolutionwasaddedtoa250mlvolumetricflaskandmadeuptothemarkwithwaterAnaliquotof100mloftheresultingsolutionwasaddedtoa250mlvolumetricflask2mlofBritton-RobinsonbufferwithapHof96wasaddedandthevolumewasadjustedtothemarkwithdistilledwater
22Instrumentation
ForpolarographicmeasurementsweuseddigitaldeviceMTechOVA-410 [18]temperature-controlledthree-electrodeamercurydropletindicatorelectrodeasaturatedcalomelreferenceelectrodeandplatinumwireauxiliaryelectrodeTheaccuracyofthepotentialmeasurementis1mVTheuncertaintyofcurrentmeasu-rement is 01 The employed mercury droplet electrode had the following
ndash4 ndash1characteristicsm=594times10 gs τ=10 min in 02 M NH Cl We used cyclic4
voltammetryforthestudyoftheelectrochemicalprocess WeusedMV870DIGITAL-pH-MESSERATpH-meterformeasuringpHofthesolutions Theobtainedworkingsolutionswereintroducedintothecellanddeoxyge-natedwithargonfor10minPolarogramswererecordedintherangeofpotentialsfrom00tondash16V
3Resultsanddiscussion
Previously it was found that using the Britton-Robinson buffer with pH = 96metronidazoleisreducedwiththeformationofacharacteristiconeirreversiblepeakatndash064V(Fig1A)UsingpolarographywithfastpotentialscanitwasfoundthatmetronidazoleisreducedonmercurydropletelectrodeintherangeofpH20to105ThepeakrecoverycurrentofthemetronidazolereachesthemaximumvalueatpH9ndash10againstthebackgroundofatheBritton-Robinsonbuffer
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 53
Underpre-selected conditions theeffectof some foreign substanceson thepolarographicdeterminationofmetronidazolewasinvestigatedSubstancesthatare componentsofdrugswere studiedglucose ascorbicacidoxytetracyclinehydrochlorideGlucose and ascorbic acid are not reduced atmercury dropletelectrodeanddonotchangetheappearanceofthepolarogramandpolarographiccharacteristics of the recovery of metronidazole As can be seen from Fig 1oxytetracycline hydrochloride is reduced to mercury droplet electrode andchangestheappearanceofthepolarogramandpolarographiccharacteristicsofthe recovery of metronidazole With the addition of oxytetracycline hydro-chloridetherecoverypeakofmetronidazoledecreasesandslightlyshiftstomorenegativepotentials The composition of the drug is relatively complex excipients affect theanalyticalsignaloftherecoveryofcompoundssototakeintoaccountthematrixeffectusedthemethodofmanyadditives QuantitativelytransferredthesolutionofNozemattothecell(exactvolume)removed dissolved oxygen for 10 min and took polarograms in the range ofpotentialsfrom00tondash16VAliquotsofstandardmetronidazolesolutionwereintroducedintothecelltoobtainasolutionwithagivenconcentrationofadditive
ndash5 ndash5metronidazole 10times10 M to 70times10 M As with the determination of themetronidazole aliquots of a standard oxytetracycline hydrochloride solutionwereaddedtothecellwithsolutionofNozemattoobtainasolutionwithagiven
ndash6 ndash6additiveconcentrationfrom70times10 Mto50times10 M(Fig2) In Table 1 are shown metrological characteristics of the determination ofmetronidazoleandoxytetracyclinehydrochloride inveterinarydrugRecoverywascalculatedFormetronidazoletherecoveryis97andforoxytetracyclinehydrochloridetherecoveryis103Analyticalperformanceofthetechniqueisgoodfordeterminationveterinarydrugs
54 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2Polarogramsof(A)metronidazolereductionatdifferentmetronidazoleconcentrationsand(B)oxytetracyclinehydrochloridereductionatdifferentoxytetracyclinehydrochlorideconcentra-tionsandtheircorrespondingcalibrationgraphs
Theaccuracywasverifiedbytheldquoadded-foundrdquomethodAliquotsofstandardsolutionofmetronidazoleweremadeina250mlvolumetricflasktoobtaina
ndash5solutionofagivenconcentrationof33times10 Mandthesolutionofoxytetracyclinendash5hydrochloridetoobtainasolutionofagivenconcentrationof15times10 M2mlof
Britton-RobinsonbufferwithpH96wasaddedtoflaskwithstirringandadjustedtothemarkwithwaterTheanalysisprocedureofmodelsolutionissimilartoanalysis procedure of the solution of Nozemat The calculated amount ofmetronidazolebythemethodofmultipleadditivesinthetestedmodelsolutionisinagreementwiththeamountthatwasintroducedintothesample
4Conclusions
The new polarographic method for the determination of metronidazole andoxytetracyclinehydrochlorideintheveterinarydrugNozematforhoneybeeswasdeveloped We conducted principal component analysis of veterinary drugNozemattoassesstheoveralleffectforthedeterminationofmetronidazoleWefoundthatoxytetracyclinehydrochlorideisreducedtomercurydropletelectrodeThismethodhastheabilitytoidentifysimultaneouslyanddeterminatemetro-nidazole and oxytetracycline hydrochloride in solution without the use ofseparationandconcentrationmethodsOnemoreofadvantagesoftechniquearefastprocedureofanalysissimplesamplepreparationlowcostthepossibilityofminiaturization
References
[1] Antibiotic and Chemotherapy Finch R Greenwood D Whitley R (edits) AmsterdamElsevier2006p292ndash299
[2] MitrowskaKPrzyczynyiskutkizakazustosowania5-nitroimidazoliuzwierzątktorychtkanki lub produkty przeznaczone są do spozycia przez ludziMed Weter 71 (2015)736ndash742
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 55
Metronidazole Oxytetracycline hydrochloride
PeakspotentialV ndash065 ndash144CorelationcoefficientR 099892 099922
ndash1 4 4SlopebmicroАM 656times10 176times10Δb 1763 400InterceptamicroА 2238 0219Δa 0075 0009
ndash1 ndash5 ndash5cmolL 342times10 125times10ndash1cmgg 389 413
Recovery 97 103
Table 1Validationparametersofthemethodofmetronidazoleandoxytetracyclinehydrochloridedetermi-nationinsolutionsofNozematbythemethodofmanyadditives
[3] VermaPNamboodiryVMishraSBhagwatABhoirSAstabilityindicatingHPLCmethodfor the determination of Metronidazole using Ecofriendly solvent as mobile phasecomponentIntJPharmPharmSci5(2013)496ndash501
[4] Cervini P Ambrozini B Machado LCM Ferreira Garcia AP Cavalheiro Gomes ETThermal behavior and decomposition of oxytetracycline hydrochloride J Therm AnalCalorim121(2015)347ndash352
[5] DangBNAnhNTKKyLXThaiPKAntibioticsintheaquaticenvironmentofVietnamsourcesconcentrationsriskandcontrolstrategyChemosphere197(2018)438ndash450
[6] QuintanillaPHettingaKABeltranMCEscricheIMolinaMPVolatileprofileofmaturedTronchon cheese affected by oxytetracycline in raw goat milk J Dairy Sci 103 (2020)6015ndash6021
[7] Chen F Yu L Jingdong P Xiang W Huanjun P Yu C Yan H Study on simultaneousdetermination of three nitroimidazole residues in honey by high performance liquidchromatographyndashresonanceRayleighscatteringspectraMicrochemJ141(2018)423ndash430
[8] Hernandez-MesaM Cruces-Blanco C Campana GA Simple and rapid determination of5-nitroimidazolesandmetabolitesinfishroesamplesbysalting-outassistedliquid-liquidextractionandUHPLC-MSMSFoodChem252(2018)294ndash302
[9] Xiu-ChunGZhao-YangXHai-HuiWWen-YiKLi-MingLWen-QingCHong-WeiZWen-HuiZMolecularlyimprintedsolidphaseextractionmethodforsimultaneousdeterminationofsevennitroimidazolesfromhoneybyHPLC-MSMSTalanta166(2017)101ndash108
[10] ТеплыхАНИлларионоваЕАКоличественноеопределениеметронидазоласпектро-фотометрическимметодомСибирскиймедицинскийжурнал5(2009)48ndash50
[11] ZheltvayOIZheltvayIISpinulVVAntonovichVPSpectrophotometricdeterminationofmetronidazoleandtinidazoleusingcopper(II)complexesJAnalChem68(2013)663ndash668
[12] Youssef AK Saleh MS Abdel-Kader DA Hashem Facile DY SpectrophotometricdeterminationofmetronidazoleandsecnidazoleinpharmaceuticalpreparationsbasedontheformatioonofdyesIntJPharmPharmSci6(2015)103ndash110
[13] Sversut RA Vieira JC Rosa AM Amaral MS Kassab NM Salgado H ValidatedspectrophotometricmethodsforsimultaneousdeterminationofoxytetracyclineassociatedwithdiclofenacsodiumorwithpiroxicaminveterinarypharmaceuticaldosageformArabianJChem13(2020)3159ndash3171
[14] Nikodimos Y Electrochemical determination of metronidazole in tablet samples usingcarbonpasteelectrodeJAnalMethodsChem(2016)361294
[15] Srivastava AK Upadhyay SS Rawool CR Punde NS Rajpurohit AS Voltammetrictechniques for the analysis of drugs using nanomaterials based chemically modifiedelectrodesCurrAnalChem15(2019)249ndash276
[16] Sahu G Voltammetric behaviour of metronidazole at a composite polymer membraneelectrodeOrienJChem26(2010)81ndash86
[17] Yang Y YanW Guo YWang X Zhang F Yu L Guo C Fang G Sensitive and selectiveelectrochemicalaptasensorviadiazonium-couplingreactionforlabel-freedeterminationofoxytetracyclineinmilksamplesSensorsandActuatorsReports2(2020)1ndash7
[18] httpchemlnueduuamtechdeviceshtml(accesed21stJune2020)
56 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
AnimportantpartofanyanalysisthatsignificantlyaffectsthefinalresultsisthesamplepreparationThelowconcentrationofbiologicallyactivecompoundsandthepresenceofaccompanyingcomponentspreventdirectanalysisofthesamplewithcomplexmatrixcompositionTraditionalmethodsofliquidandsolid-phaseextractionhaveaplentyoflimitationssuchashighlytime-consumingprocedureslarge volume of samples expensive cartridges toxic organic solvents andchallenges in automating the process Therefore the application of extractiontechniquesemployinglowamountofsolvents(microextractionmethods)andthelow toxicity extractantes has become the main research direction in recentyears[12] Solid-phasemicroextraction(SPME)wasproposedbyPavlishinin1989[3]Onevariantofthismethodistousethinrodswithvariouspolymercoatingssuch
Application of microextraction techniques combined with chromatographic methods for the analysis of complex objects
VLADISLAVDEEVELENABESSONOVALIUDMILAKARTSOVA
InstituteofChemistrySaint-PetersburgStateUniversityUniversitetskyprospect26198504PeterhofSaint-PetersburgRussiahitchervmailru
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 57
AbstractThelowconcentrationofanalytesandthepreventionofthematrixinfluence requires a stage for extraction and concentration of thestudiedcompoundsTheclassicalmethodsofliquidandsolid-phaseextractionhavemanylimitationsthatpreventtheiruseinsomecasesMicroextraction techniques are becoming more widespread WestudiedthepossibilityofusingionicliquidstoextractpesticidesfromwatersampleswiththeirsubsequentHPLS-MSdeterminationTheinfluenceonthedegreeofextractionofsuchparametersasanatureofionicliquidsanddispersersolventtheiramountssaltconcentrationvolumeratioofionicliquidsandwatersampledilutionoftheionicliquidsextractwithmethanolwasperformedBesidesconditionsofsolid-phase microextraction of volatile organic compounds fromurine samples obtained from healthy donors and donors withprostate cancer have been found The analysis of volatile organiccompoundsbyGC-MSfollowedbychemometricprocessingallowedachievingahighvalueofbinaryclassificationaccuracy(91)
Keywordschemometricsdispersiveliquid-liquid
microextractionsolid-phase
microextraction
asdivinylbenzenepolydimethylsiloxanepolyacrylateandpolyethyleneglycolwhichappliedtothesurface[4]Thepolymersorbentisplacedintheequilibriumheadspaceaboveacondensedphaseofthesampleandthevolatilecompoundsareextracted Liquidmicroextractionconsistsofusingsmallamountsofliquid(extractant)inequilibriumwiththegasorliquidphaseofthesampleDispersiveliquid-liquidmicroextraction(DLLME)isavariantofliquidmicroextractionTheessenceofthemethodisasfollowsextractantisdissolvedinthephaseofadispersingsolventandthemixtureisrapidlyinjectedintothesamplevolume[5]Inthiscasethedispersing solvent is dissolved and a ldquocloudrdquo of extractant is formed A largesurfaceareacontributestomasstransferprocessesThecombinationofDLLMEwiththeuseof ionic liquids(ionic liquids)asextractantsreducestheharmfulimpactontheenvironment[6] Sothegoalofthisstudywastheapplicationofmicroextractionmethodsfortheanalysisofrealsamples
2Experimental
21Reagents
Deionizedwaterwas obtained at the AQUILON D 301 deionizer (Russia) Allchemicals and reagents (the highest commercially available purity) werepurchasedfromReachimBakerAcrosorganicsandSigmaAldrich
22Instrumentation
HPLCanalysiswascarriedoutusinganHPLCLCMS-8030(Shimadzu)withatriplequadrupole mass-selective detector with electrospray ionization Analysis ofvolatileorganiccomponents(volatileorganiccompounds)ofurinesampleswasmade by GCMS-QP2010 SE (Shimadzu) Chemometric data processing wasperformedusingRStudio
23DeterminationofvolatileorganiccompoundsinurinesamplesbyGC-MSmethod
ForSPMEofvolatileorganiccompounds inurinesamplewasusedfibercoatedwith a polydimethylsiloxane (PDMS) The volatile organic compounds wereextractedontofibercoatingfor20minat50degCThentheanalytesweredesorbedinto the gas chromatography for 4 minutes at a temperature of 250degCChromatographic separation was carried out on a HP-5 capillary column(30mtimes250μmtimes025μm)usingtemperatureprogrammingmodeThetempera-
ndash1tureofovenwasincreasedfrom50degCupto250degCatarateof10degCmin Tionsourcewas200degCMassspectrometrywasusedinSIMmode(mz=35ndash900)
58 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
24ConditionsforLCMSMSdeterminationofpesticides
Separation of pesticides was performed by HPLCMSMS with positiveelectrosprayionizationoncolumnZorbaxBonusRP35μm(21times100mm)with40mMammoniumacetateandmethanolasmobilephaseAandBrespectivelyThefollowinggradientelutionwasapplied20ndash85B(8min)85B(8ndash15min)85ndash95B(150ndash155min)95B(155minus180min)95ndash20B(180minus185min)
ndash1Thevelocityof themobilephasewas03mlmin Thevolumeof the injectedsamplewas 20 microlMS detection capillary voltage +45 kV spray gas velocity
3 ndash1 3 ndash13dm min flow rate and drying gas temperature 15 dm min and 250 degCrespectively
25Selectionofconditionsfordispersiveliquid-liquidmicroextractionofpesticides
The influence of the natures of ionic liquids ([C MIM][PF ] [C MIM][NTf ]4 6 6 2
[C MIM][BF ])andthedispersingsolvent(methanolacetonitrileacetone) the6 4
weightoftheionicliquids(0060ndash0200g)thevolumeofthedispersingsolvent(02ndash10ml)onthedegreeofpesticidesextractionwerestudiedTheinfluenceofthepH(5422)theconcentrationofNaCl(0040ndash0200g)andextractiontime(1-6min)wereinvestigated The effects of different ionic liquids and disperser solvents on DLLMEprocedures were investigated and optimized by using standard solutions ofpesticidesIndetailasolutionofionicliquidsinadispersingsolventwaspreparedand rapidly injected into the aqueous sample solution (2ml) followed bytreatment for 2 min in an ultrasonic bath cooling at ndash4degC for 10 mincentrifugationfor10minat3500rpmandcollectionofionicliquidsThewaterphasewas separatedandanalyzedbyHPLC-MSThe ionic liquidsextractwasdilutedinmethanolandanalyzedbyHPLC-MS
3Resultsanddiscussion
31Microextractionofpesticides
One of the important tasks of environmental monitoring is to control traceconcentrationsofpesticidesinwatersamplesTechniqueofcombiningseveralpesticideshasbecomemorewidespread inagriculture It allowsreducing thetotalconcentrationoftheappliedcompoundsandtodecreasetheadaptabilityofpathogens and insects Therefore the analysis of real samples requires apreliminarystageofselectiveanalytesextractionandconcentration ExtractinganddispersingsolventsarebothimportantinDLLMEofanalytesTheinfluenceofthenatureofthedispersingsolvent(methanolacetonitrileandacetone) and the extractant (imidazolium ionic liquids [C MIM][PF ]4 6
[C MIM][BF ]and[C MIM][NTf ])onthedegreeofextractionofpesticideswas6 4 6 2
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 59
studied This parameter was controlled by the residual concentration ofpesticidesinthewaterphaseafterextractionThebestresultswereobtainedforionic liquids [C MIM][PF ] as an extractant and acetonitrile as a dispersing4 6
solvent Thenextstepwastoselecttheamountofionicliquids(0060ndash0200g)andthevolume of acetonitrile (02ndash10 ml) It was found that the highest degree ofextractionofanalyteswasachievedbyusing020gofionicliquidsand03mlofacetonitrile It was shown that the degree of extraction of selected pesticides does notdependonpHofthewatersamplewhichconfirmsthepartitionmechanismofextractionThedegreeofextractionofcarbofosincreasedslightlywithanincreaseinthesaltconcentrationandreachmaximumbyweightto4(008g) ItisknownthatthehighviscosityofionicliquidshavehinderedtheprocessesofelectrosprayionizationDilutionofthesamplewithmethanolby3timesgivesthebestresult(thesignalintensitywas373to845ofthesignalwithoutionicliquids) ThustheconditionsofDLLME-ionicliquidsextractionofpesticidesfromwatersampleswere found The limits of detection for pesticideswere from007 to
ndash1019ngml thereproducibilityofpeakareaswerefrom3to5theextractionrecoverywascloseto100
32SPMEofvolatileorganiccompoundsfromurinesamples
Oneof the importantdirections is the search for criteriaofearlydiagnosisofcancer Obtaining characteristic profiles of volatile organic compounds fromurinesamplescanhelpdevelopanon-invasivemethodforearlydiagnosisofthedisease ForthiswestudiedtheinfluenceofanumberoffactorsonthetotalnumberofpeaksandthetotalpeakareaTheywerethetemperatureatwhichthevaporandcondensedphasesofurinewerebalanced(30ndash60degC)thepreheatingtimeofthesample(10ndash40min)NaClconcentration(30ndash133wv)andthesorptiontimeonthePDMScoating(5ndash30min) Anincreaseinthepreheatingtemperatureofthesampleto500degCledtoanincreaseinthenumberofsignalswhichdidnotchangewithafurthertempera-tureincreasingNextparameterwasthetimeofachievementequilibriumofthevapor and condensed phases The largest number of peaks was observed at40minbutthisgreatlyincreasedthetimeofanalysisandsowechose20minAlsowestudiedthedesaltingeffectofsodiumchlorideonefficiencyofextractionThebestvolatileorganiccompounds sorptionwasachievedbyadding133saltingagent It is also shown that thenumberofpeaksdidnot changeafter20minutesofsorption Thus to obtain the characteristic profiles of urine samples the followingconditionswereselected1333NaClwasaddedtotheurinesample(3ml)
60 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
heatingfor20minat50degCthensorptionofvolatileorganiccompoundsonPDMSfibercoatingat50degCfor20min Undertheselectedconditionsweobtainedvapor-phaseprofilesof52urinesamples (32 normal and 20 pathology) Prior to performing chemometricprocessing of chromatographic profiles of urine samples preliminary datapreparation is necessary [7] The baseline was removed and the peaks werealignedusingdynamictimewarpingwithcontrollingbymassspectra ThePCAmodelwasbasedon52aligned characteristicprofilesThere is asatisfactoryseparationofdataintotwoclustersinthescoresplotrelativetothefirstandsecondprincipalcomponent(Fig1) The original data set (52 samples) was randomly divided into calibration(13pathology21normal)andtest(7pathology11normal)setsThenthePLS-DAmodelwasbuiltusingthecalibrationsetanditspredictiveabilitywasevaluatedusingthetestsetTheprocedurewasrepeated100timesTheaveragevaluesofsensitivityspecificityandaccuracyinthiscasewere9594and91
4Conclusions
Thepossibilityofusingimidazoliumionicliquids([C MIM][PF ])asextractants4 6
forquantitativeextractionandconcentrationofpesticidesundertheconditionsofDLLMEisshownThedegreeofconcentrationwas28ndash33whichallowedreaching
ndash1thedetectionlimits(006ndash019ngml )belowthemaximumpermissibleconcen-trationThepossibilityofnon-invasivediagnosisofprostatecancerbySPMEofvolatileorganiccompounds inurine isshownChemometricprocessingofgaschromatographic profiles using PLS-DA and PCA methods allowed achievingclassificationaccuracyvaluesmorethan90
Acknowledgments
ThisworkwassupportedbytheRussianFoundationforBasicResearchprojectno18-53-80010BRICS_t and the Russian Science Foundations (Projects 19-13-00370) We are grateful to
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 61
Fig 1Scoresplotrelativetothefirstandsecondprincipalcomponent
Resource Education Center in Chemistry of St Petersburg State University for the providedequipment
References
[1] Rutkowska M Płotka-Wasylka J Sajid M Andruch V Liquidndashphase microextractionAreviewofreviewsMicrochemJ149(2019)103989
[2] JaliliVBarkhordariAGhiasvandAAcomprehensivelookatsolid-phasemicroextractiontechniqueAreviewofreviewsMicrochemJ152(2020)104319
[3] ArthurCLPawliszynJSolidphasemicroextractionwiththermaldesorptionusingfusedsilicaopticalfibersAnalChem62(1990)2145ndash2148
[4] SchmidtKPodmoreISolidphasemicroextraction(SPME)methoddevelopmentinanalysisof volatile organic compounds (VOCs) as potential biomarkers of cancer JMol BiomarkDiagn6(2015)1000253
[5] Mousavi L Tamiji Z Khoshayand MR Applications and opportunities of experimentaldesign for the dispersive liquidndashliquidmicroextractionmethod ndash A review Talanta190(2018)225ndash356
[6] MarcinkowskaRKoniecznaKMarcinkowskiLNamiesnikJKloskowskiAApplicationofionic liquids inmicroextractiontechniquesCurrent trendsandfutureperspectivesTrACTrendsAnalChem119(2019)115614
[7] WehrensRChemometricswithRMultivariateDataAnalysisintheNaturalSciencesandLifeSciencesBerlinSpringer2011
62 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Theself-assembledtwo-dimensionalmonolayers(2DSAMs)ofvariousmolecules(eg graphene [1] MoS [2] rubrene [3]) offer beneficial properties for the2
constructionofnano-electronic andnano-opticaldevicesThe topological andchemicalcharacterizationof2DSAMsiscrucialtogatherinformationaboutthearrangementofdepositedmoleculesandtheirinteractionwiththesubstrateThistaskrequiresanalyticaltechniqueswith(sub)nanometerspatialresolutionandup to single-molecular detection sensitivity Only few techniques meet therequirements and one of them is tip-enhanced Raman spectroscopy (TERS)whichcombinestheexcellentspatialresolutionofscanningprobemicroscopy(SPM) and chemical sensitivity of surface-enhanced Raman scattering (SERS)spectroscopy[45] TheSERS spectroscopyutilizesplasmonicmetal nanostructures to cause ahighlocalenhancementoftheelectricfieldintheirclosevicinityviathesurfaceplasmonresonance(SPR)effectThelocalelectricfieldcausesanincreaseofthe
The development of reference probe system for tip-enhanced Raman spectroscopy
MARTINKRA LMARCELADENDISOVA PAVELMATE JKA
DepartmentofPhysicalChemistryFacultyofChemicalEngineeringUniversityofChemistryandTechnologyPragueTechnickaacute516628Prague6CzechRepublicMartinKralvschtcz
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 63
AbstractThetip-enhancedRamanspectroscopy(TERS)isamodernanalyticaltechniquewithanoutstandingspatialresolutionandchemicalsensi-tivityTheseparametersmainlydependon the structural integrityand chemical purity of employed plasmonic scanning probe tipsUsuallyeachtipistestedbeforeTERSmeasurementsusingcommer-ciallyavailablereferencesamplesHowevertheirpriceandrelativelyshortexpirationdatemustbeconsideredwhenplanningaresearchbudgetWedevelopedaproceduretoproduceself-madereferenceprobesamplesfortestingTERStipsusingcopper(II)phthalocyanineonaAunanolayerwhichispreparedbythermalvacuumevaporationofAuonaSiwaferOurresultsshowthatthepreparedsystemenablesrepeateddetectionofwell-resolvedTERSspectraThecollectedTERSspectraandspectralmapsexhibitsomedegreeofvariabilitywhichmaybeduetovariousphoto-inducedprocessesanditmustbeconsi-deredwhileperformingTERSmeasurements
Keywordscopper(II)
phthalocyaninescanningtunnelling
microscopyresonanceRaman
spectroscopysurface-enhancedRaman
spectroscopytip-enhancedRaman
spectroscopy
Ramanscatteringfrommoleculesboundtothemetalby6ndash8ordersofmagnitude[6]TheenhancementallowsSERSspectroscopytobeusedforsingle-moleculardetectionHoweverRamanmicroscopeshave limitedspatialresolutionbythelightdiffractionwiththeachievableresolutionbeingaroundhalfoftheexcitationwavelengthOntheotherhandthespatialresolutionofSPMtechniquesislimitedonlybythedimensionsoftheapexofthescanningprobetipwhichmayevenbeatomicallysharpByutilizingSPMtipandsubstratemadefromplasmonicmetalsan artificial ldquohotspotrdquomay be createdwith its position and dimensions beingdefinedbythetipItopensthepossibilitytocollectstronglyenhancedRamanspectrafromtheareapreciselylocalizedbellowthetipandthusovercometheopticaldiffraction limitTheartificialhotspotmayberelocatedbymovingthesamplebelowthetipwhichisthefoundationofTERSmapping[578] A successful TERS experiment requires an optimal combination of variousparameters the most important of which are tip sharpness and purity [7]Areferencesampleconsistingofaflatplasmonicnanolayerwithattachedprobemolecules is frequently used to check the state of the tip before using it forexperiments Unfortunately commercially available TERS standards areexpensiveandhaveanexpirationdateofseveralmonths Thegoalofthisstudywastofindapreparationprocedurewhichwouldbeableto produce cheap reference samples for repeated detection of intense TERSspectraAcombinationofaAunanolayeronaSisubstratepreparedbythermalvacuumevaporationwithadsorbedcopper(II)phthalocyanine(CuPc)whichisamoleculewithhighRamancross-sectionwastested[910]Copper(II)phthalo-cyanine known as phthalocyanine blue is a synthetic blue pigment and isfrequently used in paints It has been studied as a potentialmaterial for theconstruction of organic solar cells and other photoelectronic devices [11] AsindicatedbyitscolourCuPcexhibitsseveralabsorptionbandswithinthevisibleregion The effects of a transition to excited electronic states and subsequentluminescencemaybeobservable inRamanmeasurements[12]Au isahighlysuitablemetalforthesampleasitisboththermallyandelectricallyconductivewhichlimitsthelocalheatingofthesampleduringthemeasurementsandenablesthe use of scanning tunnelling microscopy (STM) for tip-surface interactionfeedback[13]
2Experimental
21Reagentsandchemicals
Thesubstrateforthesamplewaspreparedbythermalvacuumevaporationofgoldonsilicon(100)waferFirsta5nmthickCradhesionlayerwasdepositedon
ndash1thewaferfollowedby100nmofAuThedepositionratewas4nmmin forCrand ndash18nmmin for Au The base pressure of the evaporation system was below
ndash65times10 mbarFollowingthepreparationproceduredescribedbyJiangetal[14]
64 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
thecleansubstratewasimmersedintoasaturatedsolutionofCuPc(˃99SigmaAldrich USA) in dimethylformamide (˃98 Lach-ner CZ) for at least 12h atambienttemperatureSubsequentlythesamplewasremovedfromthesolutionrinsedwithMilli-Qwaterandmethanol(paPentaCZ)anddriedwithair
22Instrumentation
TheRamanSERSandTERSspectrawererecordedusingRamanspectrometerInVia Reflex (Renishaw UK) equipped with lasers emitting at two differentexcitation wavelengths 633nm (136mW max power output) and 785nm(204mWmaxpoweroutput)Thespectrometerhasathermoelectricallycooled
ndash1CCDdetectorwithaspectralresolutionof2cm and4microscopeobjectiveswith5times20times50timesand100timesmagnitudeForTERSexperiments the laserbeamwasredirectedtotheSPMplatformInnova-IRIS(BrukerUSA)viaasystemoflightguidesElectrochemicallyetchedAuTERS-STMtips(BrukerUSA)wereusedforallTERSmeasurements The spectra were processed using the Spectragryph software (F MengesldquoSpectragryph - optical spectroscopy softwarerdquo Version 1214 2020httpwwweffemm2despectragryph) Using this software all collectedspectra were treated by a Savitzky-Golay noise filter automatic baselinecorrectionspikeremovalandpeaknormalization
3Resultsanddiscussion
31Ramanmeasurementsofcrystallinecopper(II)phthalocyanine
AtfirsttheRamanspectraofpureCuPcwerecollectedtoprovidereferencedatawhileusingboth633and785-nmexcitationlasers(Fig1)Bothspectraexhibit
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 65
Fig 1RamanspectraofCu(II)phthalocyanineincrystallineformmeasuredat633(top)and785-nm(bottom)excitationThespectraareoffset
ndash1vibration bands in the 500ndash1600 cm region with slight differences in theirintensityratiosThe633-nmexcitationallowstheobservationofadditionalbands
ndash1inthe2000ndash3000cm region(onaluminescencebackground)whichoriginatefrom the resonance Raman effect as the excitation energy overlaps with theQ-bandofCuPc[12]MoreovertheprocessofelectronicexcitationmaylowertheD symmetry of CuPc during resonance Raman scattering and previously4h
forbiddenbandsmaybecomeobservable[15]Theluminescentbackgroundhasandash1maximumaround2200cm whichcorrespondstoamolecularemissionbandat
735nmEventhoughthespectrameasuredwiththe785-nmlaserlinedonotexhibitapparentresonanceenhancementapre-resonanceRamanenhancementmayoccur
32Surface-enhancedRamanmeasurementsofcopper(II)phthalocyaninelayeronagoldsubstrate
The prepared sample of CuPc on a Au layer was analysed using the Ramanmicroscope Both excitation laserswere used to obtain SERS spectra (Fig 2)whichwerecomparedtothespectraofapurecrystallineCuPc Thepositionsof bands in SERS spectra closelymatch their positions in thespectraofbulkCuPcHoweveraslightshiftofsomespectralbandsisobservable
ndash1(eg1528rarr1532cm )whichmaybeattributedtotheinteractionbetweenCuPcand the Au substrate The disappearance of luminescence background andresonance-enhancedbandsinthespectrumat633-nmexcitationalsosuggeststhemolecule-metalinteractionandthetransferofenergyfromCuPcmoleculestothesubstrateMoreovertherearevariationsintherelativeintensitiesofbands
ndash1whichdependontheexcitationenergyegthebandat1309cm isenhancedinSERSspectraatthe785-nmexcitationwhencomparedtothespectraofpureCuPcorevenSERSspectraat633-nmexcitation
66 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2SERS spectra of Cu(II) phthalocyanineon aAu layermeasured at 633 (top) and785-nm(bottom)excitationThespectraareoffset
33Tip-enhancedRamanmeasurementsofcopper(II)phthalocyaninelayeronagoldsubstrate
SERS microspectroscopy is a diffraction-limited technique as it provides anaveragedinformationaboutmoleculesintheilluminatedareaofseveralsquaremicrometers Meanwhile TERS spectra are collected from an area of tens ofnanometers and they contain specific information about the local moleculararrangement topography of the underlyingmetal and properties of the localelectric field between the tip and the substrate Therefore a higher spectralvariabilityshouldbeexpected Several TERS mapping experiments were carried out using both 633 and785-nmexcitationwithvaryingexperimentalparameterssuchasthenumberanddistance betweenmeasured points acquisition time number of acquisitionslaserpoweretc TheTERSspectrameasuredat633-nmexcitationexhibited lowersignal tonoiseratioandreproducibilityAsaconsequenceTERSmappingwasimpossibleandonlyafewone-pointTERSspectrawereobtained(Fig3) TheTERSspectracollectedat785-nmexcitationcontainedahighernumberofwell-resolvedbandsMoreoverthespectrawerestableintimeandsotheTERSmappingwaspossibleThe twopresentedTERSspectraareaveragesofTERSmaps which contained 16 and 80 points with 600 and 300-nm spacingrespectively(Fig4) Thespectracollectedusingbothexcitationwavelengthsexhibitavariabilityinrelative intensities and positions of bands between themeasured points ThevariabilitymaybeattributedtothelocalorientationofCuPcmoleculesbetweenthe tip and the Au surface and the properties of strongly enhanced andnon-homogeneous electromagnetic field which depend on the tip-surfacedistancetheirmorphologyandrelativepositionMoreovertheusedexcitation
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 67
Fig 3TwoexamplesofTERSspectraofCu(II)phthalocyaninemeasuredat633-nmexcitationThespectraareoffset
wavelengthsareclosetoabsorptionbandsofCuPcandthestrongelectricfieldmay give rise to photo-induced effects These effects include the electronicexcitationofCuPctohigherstateschargetransferbetweentheCuatomandthephthalocyaninering ionizationof themoleculeand formationofradicalsThephoto-inducedprocessesarelikelytoplayabiggerroleinTERSspectradetectedat633-nmexcitationduetotheoverlapwithQ-bandofCuPcwhichmaybethecauseoftheirlowersignaltonoiseratioandreproducibility
4Conclusions
Thedeveloped referenceprobe systemofCuPc adsorbedon aAu surfacehasprovedtobesuitablefortheintendeduseasitenabledthedetectionofintenseandwell-resolvedSERSandTERSspectraTheAulayerpreventsoverheatingofthesampleandallowsfortheuseofSTMTheSERSspectrawereinagoodmatchwith thespectraofpureCuPcAslight shiftof somebandsandchange in theluminescent background indicated the interaction between CuPc and the AusurfaceTheTERSexperimentsresultedinspectralmapswithhighintensitiesofindividualspectraIncreasedvariabilitybetweenmeasuredpointswasobservedPossiblesourcesofthevariabilityarephoto-inducedprocessesthatmayoccurinthestronglyenhancedelectricfieldTheseeffectsareaknownfeatureofTERSmeasurements and they offer valuable insight into the photophysics andphotochemistryofCuPcinteractingwiththeAusurfaceThedependenceofTERSspectra on experimental parameters and the preparation procedure of thereferencesampleshouldbefurtherstudied
Acknowledgments
ThisworkwassupportedfromthegrantofSpecificuniversityresearchndashA2_FCHI_2020_039
68 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 4TwoexamplesofaveragedTERSspectraofCu(II)phthalocyaninemeasuredwith785-nmexcitationThespectraareaveragesfromTERSmapsincluding16(top)and80(bottom)measuredpointsThespectraareoffset
References
[1] Mas-BallesteRGomez-NavarroCGomez-HerreroJZamoraF2DmaterialstographeneandbeyondNanoscale3(2011)20ndash30
[2] ZengHCuiXAnopticalspectroscopicstudyontwo-dimensionalgroup-VItransitionmetaldichalcogenidesChemSocRev44(2015)2629ndash2642
[3] SchultzJFLiLMahapatraSShawCZhangXJiangNDefiningmultipleconfigurationsofrubreneonaAg(100)surfacewith5A spatialresolutionviaultrahighvacuumtip-enhancedRamanspectroscopyJPhysChemC124(2020)2420ndash2426
[4] WhitemanPJSchultzJFPorachZDChenHNJiangNDualbindingconfigurationsofsubphthalocyanineonAg(100)substratecharacterizedbyscanningtunnelingmicroscopytip-enhanced Raman spectroscopy and density functional theory J Phys Chem C 122(2018)5489ndash5495
[5] ShaoFZenobiRTip-enhancedRamanspectroscopyprinciplespracticeandapplicationstonanospectroscopicimagingof2DmaterialsAnalBioanalChem411(2019)37ndash61
[6] ArocaRSurface-EnhancedVibrationalSpectroscopyHobokenWiley2006[7] KumarNMignuzziS SuWRoyDTip-enhancedRamanspectroscopyprinciplesand
applicationsEPJTechInstrum2(2015)9[8] BailoEDeckertVTip-enhancedRamanscatteringChemSocRev37(2008)921ndash930[9] BovillAJMcConnellAANimmoJASmithWEResonanceRamanspectraofα-copper
phthalocyanineJPhysChem90(1986)569ndash575[10] Shaibat MA Casabianca LB Siberio-Perez DY Matzger AJ Ishii Y Distinguishing
polymorphsofthesemiconductingpigmentcopperphthalocyaninebysolid-stateNMRandRamanspectroscopyJPhysChemB114(2010)4400ndash4406
[11] SzybowiczMRunkaTDrozdowskiMBałaWGrodzickiAPiszczekPBratkowskiAHightemperaturestudyofFT-IRandRamanscatteringspectraofvacuumdepositedCuPcthinfilmsJMolStruct704(2004)107ndash113
[12] CaplinsBWMullenbachTKHolmesRJBlankDAFemtosecondtonanosecondexcitedstatedynamicsofvapordepositedcopperphthalocyaninethinfilmsPhysChemChemPhys18(2016)11454ndash11459
[13] SaccoAImbraguglioDGiovannozziAndreaMPortesiCRossiAMDevelopmentofacandidatereferencesampleforthecharacterizationoftip-enhancedRamanspectroscopyspatialresolutionRSCAdv8(2018)27863ndash27869
[14] JiangSChenZChenXNguyenDMatteiMGoubertGVanDuyneRPInvestigationofcobaltphthalocyanineatthesolidliquidinterfacebyelectrochemicaltip-enhancedRamanspectroscopyJPhysChemC123(2019)9852ndash9859
[15] MelendresCAMaroniVARamanspectraandnormalcoordinateanalysisoftheplanarvibrationsofironphthalocyanineJRamanSpectrosc15(1984)319ndash326
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 69
1Introduction
The aim of this work has been develo-pmentofanewvoltammetricmethodforthe determination of 23-dimercapto-1-propane-sulfonic acid (DMPS) Fig 1Investigationwasdonetoobtainrelevantinformation about complexingbehaviorofDMPStowardsleadions Lead is one of heavy metals which can cause irreversible neurologicalproblems [1 2]DMPS is a synthetic antidotewith two thiol groups used fortreatmentofpoisoningbyheavymetals[3ndash6]StrongcomplexingpropertieshighwatersolubilityandnegligiblesideeffectsarethemostimportantadvantagesofDMPS[47]
Fig 1 Chemical structure of 23-dimercap-to-1-propane-sulfonicacid
Determination of heavy metal poisoning antidote 23-dimercapto-1-propanesulfonic acid using silver solid amalgam electrode
ab ab bc bMARTACHOIN SKA VOJTE CHHRDLICKA BEATRIZRUIZREDONDO JIR IBAREK aTOMA S NAVRATIL
a JHeyrovskyacuteInstituteofPhysicalChemistryoftheCzechAcademyofSciences Dolejškova21553182thinsp23Prague8CzechRepublicmartachoinskagmailcomb UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova20308128thinsp43Prague2CzechRepublic
c UniversityofValladolidPlazadeSantaCruz847002ValladolidSpain
Abstract23-Dimercapto-1-propane-sulfonic acid (DMPS) was investigatedusingdirectcurrentvoltammetry(DCV)differentialpulsecathodicstrippingvoltammetry(DPCSV)differentialpulseanodicstrippingvoltammetry(DPASV)andeliminationvoltammetrywithlinearscan(EVLS)atapolished(p-AgSAE)andatameniscusmodifiedsilversolidamalgam electrode (m-AgSAE) EVLS confirmed two consecutivereductions with coupled protonelectron transfer VoltammetrictitrationsofDMPSwithPb2+provedcomplexformationwithlimits
minus1ofquantification(LOQs)anddetection(LODs)03and01micromolL atminus1m-AgSAE and 08 and 03micromolL at p-AgSAE respectively
Determination of DMPS in commercial drug Dimaval and humanurine samples confirmed practical applicability of the developedmethod
Keywordscathodicstripping
voltammetry23-dimercapto-1-pro-
panesulfonicacideliminationvoltammetry
withlinearscansilversolidamalgam
electrodeunithiol
70 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Voltammetry was chosen as a determination technique due to its highsensitivity and selectivity speed low costs [8] Thiol groups in DMPS can beoxidatively chemisorbed on solid amalgam electrode It can be used as anaccumulation step for cathodic strippingvoltammetry [9ndash11]Moreover solidamalgamelectrode (SAE)was chosen as theworking electrodebecauseof itspropertiesashighsignaltonoiseratiowidepotentialwindowandabilitytoreachlowlimitsofdetection(LOD)[12ndash13]
2Experimental
21Reagentsandchemicals
Allsolutionswerepreparedusingdeionizedwater(Milli-Q-GradientMilliporendash1PragueCzechRepublic)withconductivitylt005microScm Britton-Robinsonbuffer
solutionspHrangefrom2to12werepreparedbymixingtheproperamountsof02MNaOH(alkalinesolution)andof004MH BO 004MH PO and004M3 3 3 4
CH COOH(allLachemaCzechRepublic)acidicsolutionTheacidicsolutionwas3
preparedbydissolutionof1235gofH BO pa088mLofH PO (85)paand3 3 3 4
1435mLofCH COOH(99)pa in500mLofdeionizedwaterThealkaline3
solution was prepared by dissolution of 3995 g of NaOH pa in 500mL ofdeionizedwater(allLachemaCzechRepublic) StocksolutionofDMPSwaspreparedbydissolving10mgofsolid23-dimer-capto-1-propanesulfonic acid monohydrate pa (Merck Czech Republic) in100mLofdeionizedwaterForthepreparationofthemodelsamplesolutiononecapsuleofdrugDimaval(HeylGermany)contains100mgofDMPSwasdissolved
ndash1in10LofdeionizedwatertotheDMPSconcentrationof0531mmolL Twomodel samplesofDimavalwerepreparedbydilutionof theabove-mentioned
ndash1solutionwithBritton-Robinsonbuffersolutiontoconcentrations10micromolL andndash1of 10micromolL respectively Urine model samples were prepared by mixing
Britton-Robinsonbuffer solutionwithurine samples obtained fromvolunteer(manhealthy30yearsold)inratio11SamplepHwasadjustedbyadditionof
ndash1proper amount of 02molL NaOH Before each measurement oxygen wasremoved for 5minbynitrogenbubbling (purity class 46MesserTechnogasPragueCzechRepublic)
22Instrumentation
Measurementswereperformedusingtwotypesofworkingelectrodesmeniscusmodified silver solid amalgam electrode (m-AgSAE working surface of
20382plusmn0025mm α lt 005) and polished silver solid amalgam electrode2(p-AgSAEworkingsurfaceof0196plusmn0015mm αlt005)Ag|AgCl|3MKClwas
used as the reference electrode andplatinumwire (Oslash1mm)wasused as theauxiliary electrode (both from Elektrochemicke detektory Czech Republic)Measurementswereperformedatlaboratorytemperature(25plusmn2degC)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 71
The pH was measured using pH-meter Jenway 3505 with combined glasselectrodetype924001(BibbyScientificLimitedUK)Voltammetricmeasure-ments were performed using the computer-controlled Eco-Tribo Polarograph(Polaro-Sensors Czech Republic) Software used for measurements wasMultiElChem 33 forWindows XP7810 (J Heyrovsky Institute of PhysicalChemistryoftheCzechAcademyofSciencesCzechRepublic)
3Resultsanddiscussion
OptimumconditionsformeasurementswereobtainedbyseriesofmeasurementinwiderangeofpHvaluesandtestingvariouscleaningproceduresOptimumpotentialof accumulation (E ) and timeof accumulation (t )ofDMPSwereacc acc
adjusted for differential pulse cathodic stripping voltammetry (DPCSV) atp-AgSAEandm-AgSAE ThedependencebetweenpeakheightandconcentrationofDMPSatp-AgSAEhasalogarithmicshapewhichcorrespondstotheaccumulationprocessattheelectrodesurfaceThelineardependencewasobservedintheDMPSconcentra-
ndash1 ndash1tionsfrom03micromolL to20micromolL Peakshifttowardsnegativepotentialwithincreasing concentration of DMPS corresponds to the metal-thiol bond andinfluenceofelectrodesurfacestructureonthisbondOnthecontrarysignalsonm-AgSAEweremorestableandthedependencebetweenconcentrationofDMPSandsignalwasalmostlinearinwholetestedrangeofconcentrations The developedmethodwas tested inmodel samples of Dimaval and urineFoundamountsofDMPSwereingoodagreementwithdeclaredcontentsusingbothelectrodesHowevertherepeatabilityofsignalsregisteredusingp-AgSAEinurinesamplesweresignificantlyworsethanthoseinDimavalsamplesItcanbecausedbycomplicatedbiologicalmatricesandfoulingeffectsofurine ELSV measurements confirmed two consecutive reductions of DMPS inadsorbed state At m-AgSAE signal were at about minus415 mV and minus440 mVrespectively and at p-AgSAE at about minus790 mV and minus830 mV respectively
ndash1 ndash1Reductionsatm-AgSAEatthescanratesfrom80mVs to640mVs havebeencontrolledbyakineticprocessatminus400mV Inanodicscansonm-AgSAEonlyonepeakwasvisibleatabout‒390mVItcorrespondswiththeoxidationofmercuryelectrodesurfaceontheelectrodeinthepresenceofDMPSandwithdiffusionfromthebulksolutionofproductsAtp-AgSAEnosignificantanodicsignalwasfound ThelastpartoftheresearchrevealedvoltammetricbehaviorofDMPSinthe
2+presence of Pb Voltammetric titration was investigated by DPCSV anddifferential pulse anodic stripping voltammetry (DPASV) during consecutive
ndash1 ndash1additionsof1mmolL ofPb(NO ) into100micromolL DMPSsolutioninacetate3 2
bufferofpH50InabsenceofPb2+ontheelectrodesurfaceHg(DMPS)complexisformedduringtheaccumulationstepDuringtheanodicscanthereisonlyonewelldevelopedreductionpeak(Fig2A)
72 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ndash1Fig 2DPCSandDPASvoltammogramsof10micromolL ofDMPSinacetatebufferpH=5correspond-2+ingto[Pb DMPS]ratiosof(A)01(B)11and(C)21Uppercurvecorrespondstothecathodic
scanE =0mVt =15sLowercurvecorrespondstoreverseanodicscanwitht =15satacc acc accndash1E =minus1000mVν=20mVs (Ref[16])acc
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 73
2+ WhenPb DMPSratioisequalto11twooxidationandthreereductionpeakswereregistered(Fig2B)PeakA ataboutndash300mVcorrespondstoformationofred
0Pb(DMPS)complexThiscomplexwasfurtherreducedtothePb (Hg)atabout2+ndash500mV(C )ReductionpeakoffreePb wasalsoregistered(B )Oxidationred red
peaksA andB correspondtoreverseprocessesandC isnotpresentbecauseox ox ox
nofreeDMPSispresentinthesolutionAtratio21thereisnofreeDMPSinthesolution however excess of lead ions As a consequence A and B peakred ox
2+increasedIncreaseofB correspondstothedepositionofPb duringtheaccu-ox
mulationstepPeaksBredandCredremainedpracticallyunchanged(Fig2C) Voltammetric titrationconfirmed themechanismof formationcomplexesofPb(DMPS)Hg(DMPS)andPb(Hg)Italsoconfirmedpossibilityofdetermination
2+Pb andDMPSinthesamesolution
4Conclusions
ValidationinmodelsampleofdrugDimavalandhumanurinespikedwithDMPSconfirmed that this method can be used for clinical purposes Voltammetric
2+titration of DMPS by Pb ions proved that it can be used for simultaneousdetermination of the drug and heavy metal ions in human urine Moreoverobtained LODs were two orders lower than those in the previously reportedvoltammetricmethod[14](Table1)
Acknowledgments
ResearchwascarriedoutwithintheframeworkofSpecificUniversityResearch(SVV260560)TheauthorsthanktheCzechScienceFoundation(GACRprojectNo20-01589S)
References
[1] AnHHLuchakMCopesRLeadtoxicityAsystematicreviewofrecentlypublishedcasesClinToxicol53(2015)757ndash758
[2] KimYLustMRKreimerbirnbaumM23-Dimercaptopropane-1-sulfonate(DMPS)inthetreatmentoflead-poisoningFasebJ2(1988)A1820ndashA1820
[3] AposhianHVDMSAandDMPS ndashwater-solubleantidotesforheavy-metalpoisoningAnnuRevPharmacol23(1983)193ndash215
74 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Table 1ComparisonofvoltammetricmethodsforDMPSdetermination(LDR
Method Workingelectrode Lineardynamic LOQ LOD Refminus1 minus1 minus1 rangemicromolL micromolL micromolL
LSV glassy-carbonelectrode 18ndash140 41 14 [14] modifiedwithmulti-walled 260ndash690 carbonnanotubes DPCSV p-AgSAE 03ndash20 08 03 thisworkDPCSV m-AgSAE 01ndash10 03 01 thiswork 10ndash100
[4] BjorklundG Crisponi G Nurchi VM Cappai R Djordjevic AB Aaseth J A review oncoordinationpropertiesof thiol-containingchelatingagents towardsmercury cadmiumandleadMolecules24(2019)3247
[5] DonnerAHrubyKDMPSinthetreatmentofacuteandchronicheavy-metalpoisoningActaMedAust14(1987)10ndash10
[6] DonnerAHrubyKPirichKKahlsPSchwarzacherKMeisingerVDimercaptopropan-sulfonate(DMPS) inthetreatmentofacute lead-poisoningVetHumToxicol29 (1987)37ndash37
[7] Blanusa M Varnai VM Piasek M Kostial K Chelators as antidotes of metal toxicityTherapeuticandexperimentalaspectsCurrMedChem12(2005)2771ndash2794
[8] BarekJMoreiraJCZimaJModernelectrochemicalmethodsformonitoringofchemicalcarcinogensSensors-Basel5(2005)148ndash158
[9] Josypcuk B FojtaM Yosypchuk O Thiolatemonolayers formed on different amalgamelectrodesPartIIPropertiesandapplicationJElectroanalChem694(2013)84ndash93
[10] YosypchukBMarecekVPropertiesofthiolatemonolayersformedondifferentamalgamelectrodesJElectroanalChem653(2011)7ndash13
[11] Alvarez JMF SmythMRCathodic strippingvoltammetryofpyridine-2-thiolandsomerelated-compoundsAnalyst114(1989)1603ndash1605
[12] DanhelABarekJAmalgamelectrodesinorganicelectrochemistryCurrOrgChem15(2011)2957ndash2969
[13] Fadrna R Polished silver solid amalgam electrode Further characterization and appli-cationsinvoltammetricmeasurementsAnalLett37(2004)3255ndash3270
[14] ZiyatdinovaGKGrigorevaLVBudnikovGKElectrochemicaldeterminationofunithioland lipoic acid at electrodesmodifiedwith carbonnanotubes J Anal Chem64 (2009)185ndash188
[15] HrdlickaVChoinskaMRedondoBRBarekJNavratilTDeterminationofheavymetalpoisoning antidote 23-dimercapto-1-propanesulfonic acid using silver solid amalgamelectrodeElectrochimActadoiorg101016jelectacta2020136623
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 75
Fig 1Structureofcanagliflozin
1Introduction
Canagliflozin is a selective sodium-glucosecotransportertype2inhibitorused for the treatment of type 2 dia-betes mellitus Canagliflozin inhibitssodium-glucose cotransporter type 2present in proximal tubules of the
Canagliflozin oxidation study using electrochemical flow cell and comparison with hydrogen peroxide oxidation
a a bFILIPVYMYSLICKY TOMA S KR IZ EK JAKUBHER T
a DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublicfvymyslickygmailcomb ZentivaGroupasUKabelovny13010237Prague10CzechRepublic
AbstractBystandardstheeffectonoxidationofanactivesubstanceistestedusinghydrogenperoxidesolutionatelevatedtemperatureinastresschamberfor1ndash7daysAnalternativewaytostudytheeffectofoxida-tion on an active substance is to use an electrochemical flow cellSolutionwith active substance flows at low flow rate into a smallreactorwheretheactivesubstanceisoxidizedonworkingelectrodesurfaceTheelectrolytestreamwiththeoxidizedactivesubstanceisthen directed to the sample collector Products of electrochemicaloxidationareanalyzedbyhighperformanceliquidchromatographywithultravioletndashvisiblespectrophotometrydetectionCanagliflozinhasbeenusedbecauseitsmaindegradationpathwayisoxidationThedesign of experiments approach was used to explore the experi-mentalspaceandoptimizeexperimentalconditionsofoxidationTheresultsoftheoxidationstudyperformedintheelectrochemicalflowcellwerestatisticallycomparedwiththeresultsofastandardstudyusinghydrogenperoxidesolutionThemostsuitableconditionsforelectrochemical oxidation were found Electrochemical oxidationproducedcomparableamountsofimpuritiesaschemicaloxidationwithhydrogenperoxide
KeywordscanagliflozindesignofexperimentselectrochemicalflowcellHPLCoxidation
76 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
kidneywhichrestrictsglucoseabsorptioninthekidneytherebyincreasingtheurinaryexcretionofglucoseandloweringthelevelofglucoseintheblood[1]TheformulaofcanagliflozinisC H FO SthestructureofcanagliflozinisinFig1The24 25 5
IUPAC name of canagliflozin is (2S3R4R5S6R)-2-[3-[5-(4-fluoro-phenyl)-thiophen-2-ylmethyl]-4-methyl-phenyl]-6-hydroxymethyltetrahydro-pyran-345-triol[2]CanagliflozinisawhitepowderinsolubleinwaterbutverysolubleinorganicsolventslikemethanolordimethylsulfoxideCanagliflozinissoldundertradenameINVOKANA Manyauthorshavestudiedtheelectrochemicalpropertiesofactivesubstancesin the literature One example is the study of electrochemical behaviour andoxidationofbromhexineThesepropertieswerestudiedusingdifferentialpulsevoltammetryandcyclicvoltammetryonacarbonelectrodeTheresultsofelectro-chemicalmethodswerecomparedwithhighperformanceliquidchromatography(HPLC)analysis[3]Anotherexampleisthestudyofelectrochemicalbehaviourand degradation study performed on the active substance atomoxetineDegradation was studied using differential pulse voltammetry and cyclicvoltammetryonacarbonelectrodeTheresultswerealsocomparedwithHPLCanalysis[4]Electrochemicalmethodsareusedmainlytostudythemechanismofoxidationbutinthisworktheelectrochemicalmethodwasusedtodegradetheactivepharmaceuticalingredient In the stability studies of active pharmaceutical ingredient properties theinfluence of temperature pH light and oxidation is studied [5] By TheInternationalCouncilforHarmonisationofTechnicalRequirementsforPharma-ceuticals forHumanUse (ICH) standards the influenceof oxidationon activepharmaceutical ingredient is studied using hydrogen peroxide at roomtemperatureorincreasedtemperatureinthestresschamberduring1ndash7days[6]Analternativewaytostudytheinfluenceofoxidationonactivepharmaceuticalingredient is using electrochemical flow cellwhere an electrolytewith activepharmaceuticalingredientisdrivenbylowflowrateintothesmallreactorInthesmallreactortheactivepharmaceuticalingredientisoxidizedonthesurfaceoftheworkingelectrodeThestreamofelectrolytewithoxidizedactivepharma-ceuticalingredientisdriventothesamplecollectorProductsofelectrochemicaloxidationareanalysedbyHPLCUVVISThedesignofexperimentsapproachwasusedfordevelopmentofanalternativemethodofoxidationofcanagliflozinusinganelectrochemical flowcellThedesignofexperimentsapproachwasusedtoexplore the experimental space of the method and to find the optimalexperimentalconditionsofelectrochemicaloxidationofcanagliflozin
2 Experimental
21Materialandreagents
Canagliflozin(ZentivaCzechRepublic)999methanol(HoneywellGermany)98ammoniumdihydrogenphosphate(Sigma-AldrichJapan)35ortho-phos-phoricacid(PentaCzechRepublic)25ammonia(LachnerCzechRepublic)30hydrogenperoxide (LachnerCzechRepublic)water forchromatography
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 77
Fig 2Schemeoftheelectrochemicalflowcell(1)input(2)workingelectrode(3)gasket(4)refe-renceelectrode(5)counterelectrode
wasobtainedbypurifyingdemineralisedwaterusingMilliporetypeSynergyUVpurificationinstrument
22Instruments
An Agilent 1290 HPLC system (Agilent Technologies Germany) with highpressure pump autosampler thermostat and DAD detector was used for allexperimentsThePinnacleDBbiphenylcolumn(100times21mm19micromRestekUSA)wasusedforseparationIntheHPLCmethod10mMammoniumdihydrogenphosphatebufferpH=25wasusedascomponentAandmethanolascomponentBofthemobilephaseThegradientprogramwassetasfollowst(min)B01555010551790229023152515Theflowrateofthemobilephasewas
ndash104mlmin and the injection volume was 2μl The detector operated at awavelengthof220nmTheautosamplertemperaturewassetat20degCandthecolumntemperatureat60degCTheEmpowersoftwarewasusedforevaluationForelectrochemicaloxidationelectrochemicalflowcellfromALS(Japan)wasusedGlassycarbonelectrode(=6mm)andsilversilverchlorideelectrodewereusedasworkingandreferenceelectroderespectivelyTheschemeofelectrochemicalflowcellisinFig2ElectrodeswereconnectedwithpotentiostatPalmSens3fromPalmsens (Netherlands) AnElmasonic S15Hultrasonic bath fromElma (Ger-many)wasusedforsamplepreparationForpHmeasurementspHmeterJenway3540fromJenway(UnitedKingdom)wasused
3Resultsanddiscussion
AtthedevelopmentofthemethodforthestudyofcanagliflozinoxidationusingelectrochemicalflowcellitwasfirstnecessarytofindtheoptimalconditionsofelectrochemicaloxidationThedesignofexperimentsapproachwasusedChosen
78 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ndash1Fig 3Cyclicvoltammogramofcanagliflozin(concentrationofcanagliflozin11mgml electrolyte300 mM ammonium dihydrogen phosphate pH = 40 and methanol (11 vv) and scan rate
ndash1001Vs )
independentvariablesandtheirlevelswereconcentrationofelectrolyte(100200300mM)pHofelectrolyte(406080)cellsize(50100200500microm)and
ndash1flowrate(0102504mlh )ThereducedcombinatorialdesignwasusedIntheModde12 software aworksheet containing 11 experimentswas created Theworkingpotentialof12Vwasselectedbasedoncyclicvoltammetryofcanagli-flozininFig3Fromthisfigureitcanbeseenthattheoxidationofcanagliflozinoccursintheregionfrom11Vto14VAllexperimentswereperformedwith
ndash111mgml canagliflozin samples The glassy carbon electrodewas used as aworkingelectrodeandthesilversilverchlorideelectrodewasusedasareferentelectrode The canagliflozin samples oxidized in the electrochemical flow cellunder theexperimentalconditionsgivenby theworksheetweremeasuredbyHPLCwithUVVISdetectionDependentvariablespeakareasofimpuritiesandpercentage of peak areas of impurities obtained from chromatograms wereevaluated by the partial least squaremethod in theModde 12 software Thevariableimportanceintheprojectionplottoolwasusedforinterpretationofthedata as a whole The significance values of the independent variables were
ndash1evaluatedbufferpH=137flowrateof125mlh bufferconcentration061mMandcell size04micromFromthis tool itwasconcluded that theelectrochemicaloxidationofcanagliflozinisthemostaffectedbythepHoftheelectrolyteandflowrateoftheelectrolyteUsingtheoptimizertoolthemostsuitableconditionsfor
ndash1the oxidation of canagliflozin were evaluated flow rate 01 ml h 300 mMammoniumdihydrogenphosphate pHof electrolyte40 and cell size500micromUsingonefactoratthetimeapproachthedependenceofthecellsizeonthetotalsumofimpuritieswastested(Fig4A) Itisvisiblefromthegraphthatthesmallerthecellweusethemoreoxidationproducts are formed Based on the graph the most suitable conditions were
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 79
Fig 4(A)Optimizationofelectrochemicaloxidationconditionsdependenceofsumofimpuritiesoncellsize(B)Chromatogramofasampleoxidizedundermostsuitableconditions
ndash1adjustedtoflowrate01mlh 300mMammoniumdihydrogenphosphatepHofelectrolyte40andcellsize12micromUnderthemostsuitableconditionsarepeat-ability test was performed by ten independent oxidation experiments Therelativestandarddeviationofthepercentageareaofcanagliflozinwas164atasignificant level of 095 The chromatogram of sample oxidized under mostsuitableconditionsisinFig4BThestandardstudyoftheeffectofoxidationoncanagliflozinusinghydrogenperoxideaccordingtoICHguidelineswasperfor-medThestudywasperformedundertwosetsofexperimentalconditionsInthefirstcaseasolutionof50methanolwiththeadditionof3H O wasusedIn2 2
thesecondcasetheconditionsintheelectrochemicalflowcellweresimulatedA300mMammoniumdihydrogenphosphate pH40 andmethanol in a ratio11(vv)withtheadditionof3H O wasusedSamplesfortheoxidationstudy2 2
werestressedinastabilitychamberfor13and7daysattheconstanttempe-ratureof50degC ThetotalsumsofimpuritiesformedduringchemicaloxidationusinghydrogenperoxideinbothmediawerecomparedasisshowninFig5AItisobviousthat
80 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 5(A)Acomparisonofastandardoxidationstudyusinghydrogenperoxidein50methanolwith added buffer andwithout them (B) Chromatogramof sample oxidized electrochemically(C)Chromatogramofsampleoxidizedchemically
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 81
ammoniumphosphatesuppressesoxidationofcanagliflozinThereasonofthisphenomenonisunknownFig5BandFig5CshowchromatogramsofsamplesoxidizedelectrochemicallyandchemicallyrespectivelyItcanbeseenthatfiveimpuritieswereformedbybothtypesofoxidationhoweverindifferentamounts
4Conclusion
AnalternativemethodfortheoxidativestudyofcanagliflozinwasdevelopedThedesign of experiments approach was used in the method development ThedevelopedmethodworkswithRSDof165(α=095)Oxidationofcanagliflozinbythedevelopedmethodproducedfiveimpuritiesthatareidenticalwiththoseproducedusingthestandardoxidationstudywithhydrogenperoxide
Acknowledgments
ThisworkhasbeensupportedbyCharlesUniversityResearchCentreprogramNoUNCESCI014SVV260560projectandpharmaceuticalappliedresearchcenter(TheParc)
References
[1] ChaoECCanagliflozinDrugsFuture36(2011)351ndash357[2] NislySAKolanczykDMWaltonAMCanagliflozinanewsodium-glucosecotransporter2
inhibitorinthetreatmentofdiabetesAmJHealthSystPharm70(2013)311ndash319[3] Turchan M Jara-Ulloa P Bollo S Nunez-Vergara LJ Squella JA Alvarez-Lueje A
VoltammetricbehaviourofbromhexineanditsdeterminationinpharmaceuticalsTalanta73(2007)913ndash919
[4] Perez-OrtizMMunoz C Zapata-Urzua C Alvarez-Lueje A Electrochemical behavior ofatomoxetineanditsvoltametricdeterminationincapsulesTalanta82(2010)398ndash403
[5] Baertschi SW Alsante KM Reed RA Pharmaceutical Stress Testing Predicting DrugDegradationLondonInformaHealthcare2011
[6] RignallA ICHQ1A(R2) stability testing of newdrug substance andproduct and ICHQ1CstabilitytestingofnewdosageformsInICH Quality Guidelines An Implementation GuideATeasdaleDElderRWNims(Eds)HobokenWiley2017p3ndash44
82 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
AlthoughDNArepresentsarelativelystablecomponentfromthechemicalpointofviewitremainsconstantlyexposedtoalargenumberofchemicalorphysicalagentscausingchemicalchangesinDNAmoleculesthatoccurintheenvironmentoraremajororminorproductsofcellularmetabolism[1] One-electronoxidationoftheDNArepresentsadamagingprocesswheretheloss of an electron (oxidation) fromduplexDNA results in the formationof anucleobase radical cation (electron ldquoholerdquo) that is subsequently consumed inchemicalreactionsthatoftenleadtomutationsAdefiningcharacteristicoftheone-electronoxidationofDNAisthepreferentialreactionattheguaninemoietythatisdetectedasstrandcleavagefollowingchemicalorenzymatictreatmentoftheoxidizedDNA[2ndash3]
Novel hybrid electrochemical DNA biosensor for monitoring oxidative DNA damage via oxidationreduction signals of low molecular weight double-stranded DNA
MICHALAUGUSTINVLASTIMILVYSKOCIL
UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova812843Prague2CzechRepublicmichalaugustinnaturcunicz
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 83
AbstractDeoxyribonucleicacid(DNA)representsamajortargetmoleculeformanydamagingagentscausingunfavorablechangesinastructureofDNAmoleculethatbindandinteractwithDNAThusahighdemandforreliabletoolsregardingabettercomprehensionofthenatureofDNAdamagingprocessesstillrepresentsoneofthemaingoalsinthisareaHereinwedescribeadevelopmentofanovelhybridelectro-chemicalDNAbiosensorbasedonanldquoedge-planerdquopyrolyticgraphiteelectrode (EPPGE) in connectionwith an elementaryoptimizationprocessprovidingacloserresolutionoftheredoxprocessesoflowmolecularweightdouble-strandedDNA(dsDNA)attheEPPGESub-sequentanalyticalapplicationincorporatinganemploymentofthemodel structure K [IrCl ] (representative of transition metal2 6
complexes)andevaluationofitsdamagingeffectinrelationtoDNAbymeansof linear sweepvoltammetry resp square-wavevoltam-metryarealsopresented
KeywordsbiosensordamageDNAgraphitevoltammetry
DNA-based electrochemical biosensors are successfully used in variousapplicationssuchasmonitoringandevaluatingthemechanismsof interactionbetweenDNAandvariousdrugsordamagingagentsrapidmonitoringoftracemetalsorpollutantspresent in theenvironmentordirectmonitoringofDNAhybridizationprocesses[4] Theelectrochemicalactivityofnucleicacids(boththenativehigh-molecularonesaswellasoligonucleotides)isingeneralreferredtotheelectroactivityofitscomponents ndash nucleobases and sugar residues At mercury-based electrodesadenine and cytosine residues undergo reduction processes close to ndash14 V(againstSCE)inneutralorweaklyacidicmedium(givingrisetothepeakCA)Ontheotherhandallbaseshavebeenreportedtobeelectrochemicallyoxidizedatcarbonelectrodesbutonlyadenineand(particularly)guanineoxidationsignalshavebeenwidelyutilizedinelectrochemicalDNAbiosensors[5] In2017theelectrochemistryofnucleicacidsachievedanimportantmilestoneasthereductionoftheDNAoligonucleotideswasperformedataldquobasal-planerdquopyrolytic graphite electrode which provided wide potential window allowingboththeelectrooxidationaswellastheeletroreductionofthenucleobasesatasingleelectrodefortheveryfirsttimeDespitethesefindingsutilizationoftheaforementionedbiosensorintermsofanalyticalapplicationshasyettobeverifiedandremainsunclearuptothisdate[6] TheaimoftheproposedcontributionisapresentationofthedevelopmentprocessandsubsequenttestingofanoveltypeofhybridelectrochemicalDNAbiosensoranditsverificationasareliableanalyticaltoolintermsofmonitoringDNAdamage
2Experimental
21Reagentsandchemicals
Low molecular weight double-stranded DNA (dsDNA) derived from salmonspermwasobtainedfromSigma-AldrichGermanyStocksolutions(01mgmL)
of dsDNA were prepared in a 01 molL phosphate buffer of pH=74 (PB)Dipotassium hexachloroiridate (K [IrCl ]) was purchased from Sigma-Aldrich2 6
GermanyStocksolutions(0001molL)ofK [IrCl ]werepreparedinthePB2 6
22Apparatus
Voltammetric measurements were performed using the μAutolab IIIFRA2potentiostatgalvanostat(EcoChemieTheNetherlands)drivenbyaNOVA111software(MetrohmAutolabSwitzerland)Allmeasurementswerecarriedoutina three-electrode system using an ldquoedge-planerdquo pyrolytic graphite workingelectrode(EPPGE)withanelectroactivesurfacediameterof3mm(BASJapan)asilver|silver chloride reference electrode (Ag|AgCl|sat KCl) and a platinum
84 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
counterelectrode(ElektrochemickeDetektoryCzechRepublic)ina20mLglassvoltammetriccellatambienttemperature
23Preparationofthebiosensor
PriortotheeverymeasurementsurfaceoftheEPPGEwasmechanicallycleanedbygentlewipingoftheelectrodeonthesoftpolishingpadrinsedwithdistilledwaterAfterwardstheelectrodewasrinsedwithdistilledwaterandplacedinthePBforthesubsequentelectrochemicalactivationElectrochemicalactivationwasperformed in thePBbyapplyingpotentialof15V for240swithoutstirringApotentialpulseinworkingrangeofpotentials(00ndash15V)wasthenapplied Additional electrochemical activationwasperformed in the solutionof the
3minus4minusredoxindicator([Fe(CN) ] )byconsecutivecyclingintherangeofpotentials6
from10tondash08V(15scans)andfrom055tondash015V(10scans) The electrochemical DNA biosensor based on the EPPGE (dsDNAEPPGEbiosensor)was prepared by the adsorption of dsDNAon the EPPGEOptimal
parametersofthedsDNAadsorptionwereaconcentrationof01mgmLinthePB
(c )adepositionpotentialof07V(E )andanadsorptiontimeof5ming(dsDNA) dep
(t )withoutstirringthesolutionads
Atlasttheelectrodewasimmersedinthesolutionoftheredoxindicatorandtheconsecutivecyclingintherangeofpotentialsfrom055tondash015V(20scans)wasperformedinordertosecurethestabilityoftheoxidationreductionsignalsofdsDNAattheEPPGE
24Procedures
Theexperimentalparameterswereasfollowssquarewavevoltammetry(SWV)inthePBwithapulseamplitudeof20mVafrequencyof50Hzascanrateof750mVsandapotentialstepof15mVlinearsweepvoltammetry(LSV)inthePBwithscanratesof02ndash10Vandapotentialstepof24mVAllcurveswere
recordedthreetimes(n=3)
3Resultsanddiscussion
Sincetheclosestresolutionoftheprocessesassociatedwiththeelectroreductionof singleDNAcomponents at pyrolytic graphitehasbeenperformedwith theldquobasal-planerdquopyrolyticgraphiteelectrode(BPPGE)wehavedecidedtotakeovercorrespondingexperimentaltechniqueandconditions(LSVscanrateof10Vssteppotentialof24mV)attheverybeginningofouroptimizationprocesswiththeEPPGE[6] Inthisparticularcaseitispossibletonoticetheoccurrenceofthetwomixedvoltammetricpeaksatthedefaultexperimentalconditions(greenlineFig1A)selectedforthereductionofdsDNAattheEPPGEBygraduallydecreasingthe
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 85
Fig 1Baseline-correctedLSVrecordingscorrespondingtothereductionofdsDNAattheEPPGEfordifferentvaluesofscanrate(02ndash10VsFig1A)respbaseline-correctedLSVrecordingscorres-pondingtothereductionofdsDNAattheEPPGEandthenegativetestperformedunderthesameexperimentalconditionswithintheblanksolution(phosphatebuffer)atthebareEPPGE(03VsFig1B)
scan rate the optimal conditions (νle03Vs) were found and the mutualseparationof the signalswas allowedndash characterizedby thepresenceof twosinglewell-developedvoltammetricpeaksatpotentialsofndash175Vrespndash190V(03Vs orange line Fig 1A) Taking into account previous work regardingprocessesassociatedwiththereductionofDNAatthemercuryelectrodesrespBPPGE we can assume that the peak appearing at the potential of ndash175 Vcorresponds to themixedpeak for the reductionof the cytosine and adenineresidueswithindsDNA(peakCA)[5ndash6] Closer resolution of the second voltammetric peak appears to be farmoreproblematic Regarding our previous study we have discovered that theutilizationofdifferentE fortheadsorptionofdsDNA(E lt07V)isconnecteddep dep
withanappearanceofthethirdoxidationsignal(besidestheoxidationsignalsofguanine resp adeninemoieties) at apotential of073V corresponding to theoxidationoffreeguaninebases(FGBs)presentwithinthesolutionofdsDNAInthiscasewecanassumethatthepeakappearingatapotentialofndash190VcanpossiblyrepresentthereductioncounterpartofFGBspresentwithinthesolutionofdsDNAThisassumptioncanalsobesupportedbytheaforementionedstudyandbythefactthatthereductionsignalatsuchahighnegativepotentialcanbeobservedfortheoligodeoxynucleotidescontainingguanineresidues[6] AdditionallyinordertoverifythetruenatureofthereductionsignalsdepictedatF ig1Bandtoexcludetheoptionthattherelatedsignalsdonotrepresenttheproductsofpriorelectrochemicalactivationof theEPPGE(variousCndashObasedchemicalspecies)wehavedecidedtoperformanegative(control)testwithinthe
86 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2Baseline-correctedLSVrecordingscorresponding to thereductionof thedsDNA(02Vs
Fig 2A)respbaseline-correctedSWVrecordingscorrespondingtotheoxidationoftheguanine(098V)respadenine(128V)moieties(075VsFig2B)attheEPPGEafteritsincubationinthephosphatebufferforadefinedtimeperiod(60ndash900s)
blanksolution(PB)employingthesameprotocolasforthedsDNAadsorptionattheEPPGEInthiscaseitispossibletoobservetheabsenceofanypronouncedvoltammetricpeakslinkedtothedsDNAadsorptionandonlythepresenceofoneirreversiblepeakatapotentialofndash153VcorrespondingtotheelectroreductionoftheCndashObasedmoietymoietieswhichdropsafterthedsDNAadsorptiontoonetenthofitsoriginalvalue(approximately) Perhaps the most important parameter regarding further optimizationprocessrepresentedthetime-dependentstabilityofthecorrespondingsignalsofdsDNAwhichcanbespecificallyimportantinrelationtothestudyofthetime-dependentoxidativedamageofdsDNA AsithasalreadybeenprovedasingleelectrochemicalactivationoftheEPPGEinthePBdoesnotrepresentasatisfyingtechniqueregardingstabilityofdsDNAoxidationsignalsat theEPPGEand theadditionalstabilization isachievedbyfurtherelectrochemicalactivationinthesolutionofaredoxindicator(Fig2B)[7]Basedonthisaverificationoftheproposedstabilizationprotocolintermsofthetime-dependent stabilityofdsDNAreduction signals in the solutionof thePBwithinthedefinedtimeperiod(60ndash900s)appearedasareasonablenextstep FromtheresultsdepictedinFig2Aitispossibletonoticethatwithinthefirst300sdsDNAreductionsignalsremainstableinrelationtothecurrentresponseaswellasintermsofthepotentialvalueWithanadditionalincubationtime(t )inc
(900sorangeline)thepeakcurrentofthevoltammetricsignalpresentatmorenegative potentials decreased which can probably be addressed as a slowprogressive elimination of the weak (electro)chemical forces related to theunspecificadsorptionoftheFGBsattheEPPGE
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 87
Fig 3Baseline-corrected SWV recordings corresponding to the oxidation of the guanine respadeninemoietiesattheEPPGEafteritsincubationinthesolutionofK [IrCl ](IR)foradefinedtime2 6
period(60ndash3600s)(075VsFig3A)andthecorrespondingrelativebiosensorresponses(ΔI )rel
evaluatedusingtheguanosine(turquoise)andadenosine(red)peaksplottedversustheincubationtime(Fig3C)Baseline-correctedLSVrecordingscorrespondingtothereductionofdsDNAattheEPPGEafteritsincubationinthesolutionofK [IrCl ](IR)fordifferenttimeperiods(60ndash3600s)2 6
(02 VsFig3B)andthecorrespondingrelativebiosensorresponses(ΔI )evaluatedusingtherelpeakCA(darkpink)plottedversustheincubationtime(Fig3-D)
AdditionallywehavedecidedtotesttheapplicabilityofthepresentedhybridbiosensorintermsofmonitoringdsDNAdamagecausedbyarepresentativeofone-electron oxidants ndash K [IrCl ] In this case the prepared dsDNAEPPGE2 6
biosensor was immersed into the solution of K [IrCl ] (0001molL) for the2 6
definedtimeperiod(60ndash3600s) In thecaseof theoxidationpath (SWVrecordingsdepicted inFig3A) it ispossible to observe a time-dependent decrease of the oxidation signal of theguaninemoietieswhereastheoxidationsignaloftheadeninemoietiesremainsunaffectedforthemostof the incubationperiodThisphenomenonis ingoodcorrelationwiththetheoreticalknowledgeregardingoxidativedamageofdsDNAcausedbyone-electronoxidants [3]Simultaneouslywith thisLSVrecordingsdepicted in Fig 3B followed the similar behavior (decrease in relation to the
88 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
currentresponseofthedsDNAreductionsignalndashpeakCA)asinthecaseofthesignal regarding oxidation of guanine moieties In addition according totheportionofthepreservedDNA(Fig3C3D)itispossibletoassumethatthepronouncedoxidativedamageofdsDNAcanbemonitoredquitepreciselynotonlydirectlyviathedsDNAoxidationsignaloftheguaninemoietiesbutevenindirectlythroughthedsDNAreductionsignalndashpeakCA
4Conclusions
Inthiscontributionwehavepresenteddevelopmentofanunorthodoxhybridelectrochemical DNA biosensor based on an EPPGE Optimization processconcerning some important parameters was performed as well as closerresolutionofthenatureofthereductionprocessesofdsDNAattheEPPGEwasachievedInordertoconfirmtheresultsoftheoptimizationprocessapplicabilityoftheproposedbiosensorhadbeenprobedintermsofmonitoringDNAdamagecausedbyK [IrCl ]Inthiscasethefinalresultshadprovedthattheprepared2 6
hybridbiosensorcanbeconsideredasaversatileanalyticaltoolformonitoringoxidativeDNAdamage(viaoxidationreductionsignals)andispresentedasafinealternative in comparisonwith conventional electrochemical DNA biosensorsprepared within the group of traditional transducer materials (mercury- orcarbon-based)
Acknowledgments
ThisresearchwassupportedbytheSpecificUniversityResearch(SVV260440)
References
[1] FojtaMDanhelAHavranLVyskocilVRecentprogressinelectrochemicalsensorsandassaysforDNAdamageandrepairTrACTrendsAnalChem79(2016)160ndash167
[2] GieseBSpichtyMWesselySLong-distancechargetransportthroughDNAAnextendedhoppingmodelPureApplChem73(2001)449ndash453
[3] Burrows CJ Muller JG Oxidative nucleobasemodifications leading to strand scissionChemRev98(1998)1109ndash1151
[4] DiculescuVC Chiorcea-PaquimAMOliveira-BrettAMApplications of aDNA-electro-chemicalbiosensorTrACTrendsAnalChem79(2016)23ndash36
[5] PalecekEJelenFElectrochemistryofnucleicacidsInElectrochemistryofNucleicAcidsandProteinsndashTowardsElectrochemicalSensorsforGenomicsandProteomicsPalecekESchellerFWangJ(edits)AmsterdamElsevier2005p74ndash174
[6] SpacekJDanhelAHasonSFojtaMLabel-freedetectionofcanonicalDNAbasesuraciland5-methylcytosineinDNAoligonucleotidesusinglinearsweepvoltammetryatapyrolyticgraphiteelectrodeElectrochemCommun82(2017)34ndash38
[7] AugustınMVyskocilVNovelelectrochemicalDNAbiosensorbasedonedge-planepyrolyticgraphite for DNA interaction studies In Proceedings of the 15th International StudentsConferenceldquoModernAnalyticalChemistryrdquoNesmerakK(edit)PragueFacultyofScienceCharlesUniversity2019p263ndash268
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 89
1Introduction
Cadmiumisoneofthemosttoxicmetalsanditswidespreadindustrialusesresultin increased environmental pollution Hence the development of sensitivemethodology for Cd determination is still highly desirable Chemical vaporgeneration(CVG)ofCdbythetetrahydroboratereductioninacidicmediumisasuitable alternative sample introduction technique compatible with atomicspectrometricdetectorsandofferingimproveddetectioncapabilityComparedtocommonliquidnebulizationCVGoffersseveraladvantagessuchassignificantlyhigheranalyteintroductionefficiencyandalsoanalyteseparationfromsamplematrix IncomparisontoCVGofcommonhydrideformingelementsthereisalackofliterature dealing with mechanistic aspects of CVG of Cd [1] as well as withstabilityandidentityofitsvolatilespecies(freeatomshydrideotherspecies)Very little information is also available on achieved generation efficiencyMoreover there are many discrepancies in the literature regarding optimum
Chemical vapor generation of cadmium for analytical atomic spectrometry
a b a b a aLINDASAGAPOVA BARBORAKODRIKOVA MILANSVOBODA STANISLAVMUSIL aJANKRATZER
a InstituteofAnalyticalChemistryoftheCzechAcademyofSciences Veveřiacute9760200BrnoCzechRepublicsagapovaiachczb DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublic
AbstractChemical vapor generation of cadmium volatile compounds wasoptimizedinordertodeterminetraceCdconcentrationsbyatomicabsorptionspectrometry(AAS)Severalreactionmodifiersbasedon
III+ II+ III+ IV+inorganicsaltsandcomplexesofCr Co Ti Ti weretestedtoimproveanalyticalperformanceandgenerationefficiencyTheuseofthese reaction modifiers resulted in 4ndash5 times enhancement insensitivity reflected also in corresponding increase of generationefficiency and better repeatability Generation efficiency wasdeterminedfromacomparisonbetweensensitivitiesobtainedwithchemicalvaporgenerationandconventionalsolutionnebulizationbothsimultaneouslycoupledwithinductivelycoupledplasmamassspectrometryTheidentityofthegeneratedcadmiumcompoundswillbediscussed
Keywordsatomicabsorption
spectrometryatomizationcadmiumchemicalvapor
generation
90 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
conditionsforCVGofCdAlthoughstrongacid(HClorHNO )isalwaysemployed3
asacarrierandNaBH asareductantsomeauthorsreportedvariousadditives4III+ III+ IV+(modifiers)basedontransitionmetalions(Cr Ti Ti )inthepresenceof
II+KCN[23]orCo inthepresenceofthioureaandascorbicacid[4]toimproveCdsignalssignificantly TheaimofthisworkwastoinvestigateCVGofCdinacomprehensivewayFirstlyCVGofCdwithoutandwithselectedmodifierswasoptimizedemployingatomicabsorptionspectrometry(AAS)asadetectorandexternallyheatedquartztube(QTA)astheatomizerSecondlytheeffectofatomizationtemperatureonCdsignalwasstudiedallowingthustodeducetheatomicormolecularstructureofgeneratedCd speciesThirdly generation efficiencyofCdvolatile specieswasquantified
2Experimental
21Reagentsandchemicals
minus1Boiled and bubled (Ar per 30min) deionizedwater (lt 01 μScm UltrapurWatrex USA) was used to prepare all solutionsWorking Cd standards were
minus1preparedfrom1000mgL Cdstocksolution(AstasolAnalytikaCzechRepublic)minus1bydilutionin01ndash048molL HCl(basedonthemodifieremployed)from37
HCl(paMerckGermany)Theoptimumgenerationconditionsbeingdifferentforeachmodifiertestedare listedinTable1ThereductantwasasolutionofNaBH (ge 97 Sigma-Aldrich Germany) in 04 (mv) KOH (pa Merck4
Germany) prepared fresh daily The solutions of modifiers were prepared as2+followsCo waspreparedfromCoCl 6H O(ge990PENTACzechRepublic)2 2
3+the solution of Cr from Cr(NO ) 9H O (ge 9999 tracemetal basis Sigma-3 3 23+AldrichGermany)thesolutionofTi fromTiCl solution(about15in10HCl3
4+Sigma-AldrichGermany)andthesolutionofTi fromTiOSO (ge999Sigma-4
AldrichGermany)Tostabilizethelattersolution1H SO wasusedprepared2 4
bydilutionof96H SO (paLach-NerSlovakia)SolutionofKCN(ge9702 43+ 3+FlukaSwitzerland)wasusedasasecondmodifierwhenworkingwithCr Ti or
4+ ndash3Ti asmodifiersitsconcentrationvariedfrom008to016moldm depending
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 91
Table 1OptimumconditionsforchemicalvaporgenerationofCdinthepresenceofmodifiersandtheirabsence
onthemetalionThiourea(CH N Sge980LachemaBrno)andascorbicacid4 2
(C H O ge997Riedel-deHaenGermany)wereusedasmodifierscombined6 8 62+withCo
22Instrumentation
221Chemicalvaporgenerationsystems
TwoCVG flow injection systemswere employed either a two channel systemwithoutadditionofamodifier(seeFig1A)orafourchannelsystemallowingadditionofmodifiers(seeFig1B)
ndash1 TheflowratesofHClandNaBH were42and10mLmin respectivelyinatwo4ndash1channelsystem(Fig1A)whiletheywerebothkeptat10mLmin inthefour
channelsystem(Fig1B)Theflowratesofmodifiersinthefourchannelsystemndash1were05mLmin Thevolumeofthesampleloopwas015mLinbothsystems
ndash1Carriergasflowrateof75mLmin Arwascontrolledbyamassflowcontroler(Cole-ParmerUSA)
92 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
(A)
(B)
Fig 1Schemesofthechemicalvaporgenerationflowinjectionsystemwith(A)twochannels(nomodifiers)and(B)fourchannels(modifiersemployed)
222Atomicabsorptionspectrometry
ThePerkin-Elmermodel503atomicabsorptionspectrometer(BodenseewerkGermany)wasequippedwithaCdelectrodelessdischargelamp(Perkin-ElmerUSA)operatedat228mAThemeasurementswereperformedat2288nmusinga07nmslitwidthTheShimadzumodelAA-7000atomicabsorptionspectrometer(ShimadzuJapan)wasalsousedACdhollowcathodelamp(PhotronAustralia)operatedat2288nmlinewith07nmspectralbandpassandalampcurrentof12mA Signals were recorded for 2 minutes and peak areas were taken forevaluation The QTA was heated electrically to the temperature required byfurnace(PerkinElmer)andanin-housemadefurnacecontrolledbytheREX-C100controller(SysconIndianaUSA)withtheK-typethermocouplesensor(OmegaEngineeringUSA)223QuantificationofCVGefficiencybyICP-MS
Overall CVG efficiency of Cd was quantified bymeans of inductively coupledplasmamassspectrometry(ICP-MS)fromcomparisonoftheslopesofcalibra-tionsobtainedwithnebulizationliquidCdstandardstothoseobtainedwithCVGThe efficiency of liquid nebulization was quantified using a modified wastecollection method (see reference [5] for details) The Agilent 7700x ICP-MSinstrument(AgilentUSA)wasoperatingat1600WofRFpowerThesignalwas
111 125monitoredat Cdisotopeandcorrectedforthesignalofinternalstandard( Tendash11000ngmL Tein2HNO )NebulizeranddilutionArgasflowrateswere11503
ndash1and0mLmin respectively
3Resultsanddiscussion
31Chemicalvaporgenerationconditions
UnivariateoptimizationswereperformedtofindoptimumconditionsforCVGofCdinpresenceandabsenceofmodifiersTheparameterstobeoptimizedwerecarrieracid(HCl)concentrationreductant(NaBH )concentrationmodifierIand4
modifier II concentrations carrier gas flow rate (Ar) length of reaction coilsRCI-III (see Fig 1B) The optimum conditions for individual modifiers aresummarizedinTable1
32IdentityofCdspecies
ThepeakareasofgeneratedCdspecieswereforagivenmodifiermeasuredintheQTAheatedto900degCandnon-heatedQTAsubsequentlyOptimumCVGconditionswere employed as summarized in Table 1 This simple experiment allowsdistinguishingbetweenatomic(freeatoms)andmolecularformsofgenerated
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 93
Fig 2RelativesignalofgeneratedCdspeciesmeasuredintheQTAheatedto900degC(blackbars)andnon-heatedQTA(whitebars)withoutorinthepresenceofmodifiers
speciesOnlyfreeatomscanbedetectedinnon-heatedQTAsimilarlyasincaseofmercurycoldvaporsOn thecontrarymolecularanalytespeciesareatomizedat900degCAsaconsequencethesignalregisteredintheheatedQTAcorrespondstobothatomicandmolecularspeciesgeneratedItmustbehighlightedthattheresidencetimeoffreeatomsintheatomizerisdependentonQTAtemperatureduetogasexpansionAsaconsequencethesignalinQTAheatedto900degCshouldreach25ofthesignalatambienttemperaturetakingintoaccountthatonlyfreeatomsaregeneratedSincethetemperaturealongtheopticalarmofQTAisnotdistributedhomogeneouslydecreasingtobothendtheeffectivetemperatureoftheatomizerislowerOurexperimentswithCVGofHgrevealedsignalinheatedQTAisaround40[6]TheresultsreachedforCVGofCdaredepictedinFig2ThesignalofCdinheatedQTAisaround50ofthesignaldetectedinnon-heatedQTA
4+whennomodifierisemployedorusingTi asthemodifierindicatingclearlyfreeCdatomsarethedominantvolatilespeciesgeneratedOnthecontraryalmostno
2+differenceinpeakareaswasobservedforCo asthemodifierwhilethesignalinheatedQTAwas even 5 times higher in heatedQTA compared to non-heated
3+atomizer with Cr as the modifier suggesting the dominant contribution of3+molecularstructurestoCdsignalespeciallyincaseofCr KCNreactionsystem
33Generationefficiency
TheoverallCVGefficiencywasestimatedfromacomparisonbetweensensitivitiesobtainedwithCVGsampleintroductionandconventionalsolutionnebulizationICP-MSunder the sameexperimental conditionsNebulization efficiency for aMicroMISTnebulizerwasdeterminedas79plusmn01ThegenerationefficiencyofCd was derived from the sensitivity enhancement between CVG and liquidnebulizationTheresultsaresummarizedinTable2indicatingthatCVGwithout
94 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
modifiersisonlycatwotimesmoresensitivecomparedtoliquidnebulization3+ 4+Generation efficiency of Cd increases to 60 in the presence of Ti and Ti
modifiers
4Conclusions
CVG of Cd was thoroughly optimized in the presence of selected modifiersreportedpreviouslyintheliteratureGenerationefficiencyofCdintheabsenceofanymodifierswasquantifiedto15whileitcanbeincreasedupto60inthe
3+ 4+presenceofTi KCNorTi KCNasmodifiersFreeCdatomsseemtobe the4+dominantCdformgeneratedintheabsenceofanymodifiersorusingTi KCN
3+modifierwhile rathermolecularCd structuresaregenerated inCr KCNand2+Co thioureaascorbicacidreactionsystems
ExperimentsareinprogresstofinishthiscomprehensivestudyOnlythebestmodifierwillbefurtherusedforCVGofCdtobecoupledwithotherspectrometricdetectorsandappliedtocertifiedreferencematerialsandrealsamples
Acknowledgments
ThisresearchhasbeensupportedbytheCzechScienceFoundationundercontract18-01116SandbytheInstituteofAnalyticalChemistryoftheCzechAcademyofSciences(InstitutionalResearchPlannoRVO68081715)andCharlesUniversity(ProjectnoSVV260440)
References
[1] PitzalisEAngeliniDMascherpaMCDacuteUlivoAInsightintothemechanismscontrollingthechemicalvaporgenerationofcadmiumJAnalAtSpectrom33(2018)2160ndash2171
[2] ArslanZYilmazVRoseLEfficientgenerationofvolatilecadmiumspeciesusingTi(III)andTi(IV)andapplicationtodeterminationofcadmiumbycoldvaporgenerationinductivelycoupledplasmamassspectrometryMicrochemJ123(2015)170ndash178
[3] YilmazVRoseLArslanZLittleMDOn-linechemicalvapourgenerationofcadmiuminthepresenceofhexacyanochromate(III)fordeterminationbyinductivelycoupledplasmamassspectrometryJAnalAtSpectrom27(2012)1895ndash1902
[4] Y Lu SunHW YuanCG YanXP Simultaneous determination of trace cadmiumandarsenic inbiologicalsamplesbyhydridegeneration-doublechannelAFSAnalChem74(2002)1525ndash1529
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 95
Table 2GenerationefficiencyofchemicalvaporgenerationofCdasquantifiedbyICP-MS
Modifiers Generationefficiency
Nomodifiers 15plusmn13+Cr KCN ndash2+Co thioureaascorbicacid ndash3+Ti KCN 58plusmn24+Ti KCN 61plusmn2
[5] VyhnanovskyJStrugeonREMusilSCadmiumassistedphotochemicalvaporgenerationoftungsten fordetectionby inductively coupledplasmamass spectrometryAnal Chem91(2019)13306ndash13312
[6] MigasovaMMatousekTSchrenkovaVZ ıdekRPetry-Podgorska IKratzer JMercuryvolatilespeciesgenerationfromHClandTRISbuffermediaAnalChimActa1119(2020)68ndash76
96 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Atomicfluorescencespectrometry(AFS)coupledwithvapourgenerationisanultrasensitive analytical method for determination of various elements ItsanalyticalperformancecanbecomparabletoICP-MSwithliquidnebulizationbutatsubstantiallylowercost[1]SampleintroductiontoAFSisacrucialstepoftheanalyticalproceduresincetheanalytehastobeintroducedtotheatomizerintheformofitsvolatilespecies Hydridegeneration(HG)isamaturetechniqueofsampleintroductionduringwhichvolatileanalytehydridesare formedbyreactionwithareducingagenttypicallysodiumborohydrideAnewemergingtechniquephotochemicalvapourgeneration(PVG)employsUV irradiationof theanalyte in liquidphase in thepresenceofaphotochemicalagent(usuallyalowmolarmassorganicacidformic
minusoraceticacid)Highlyreducingradicalspecies(HbullRbullandCOObull )andaquatedelectronsare formedduring irradiationandreactwith theanalyte to form its
Photochemical vapour generation of bismuth coupled with atomic fluorescence spectrometry
ab ab a aBARBORASTA DLEROVA JAROMIRVYHNANOVSKY JIR IDE DINA STANISLAVMUSIL
a InstituteofAnalyticalChemistryoftheCzechAcademyofSciences Veveřiacute9760200BrnoCzechRepublicstadlerovaiachczb DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublic
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 97
AbstractPhotochemical vapour generation of bismuth was successfullycoupledwithnon-dispersiveatomic fluorescencespectrometry forthefirsttimeVolatilespeciesofBiweregeneratedusingastandardmercurylow-pressuretubelampandacoiledreactorfromareaction
2+mediumwhichwas composedof acetic and formic acid Co ionswereusedasasensitizerOptimizationofatomizationconditionsinaflame-in-gas-shieldatomizerwasperformedThismethodologywascomparedtothecommonlyemployedhydridegenerationapproachAbsolutelimitofdetectionof68pgwasachievedwithphotochemicalvapour generation which is still about 7 times worse than withhydride generation The developed methodology was successfullyverifiedbyBideterminationinareferencematerialofwater
Keywordsatomicfluorescence
spectrometrybismuthhydridegenerationphotochemicalvapour
generation
volatilespeciesInbothcasesthegeneratedvolatilespeciesoftheanalytehavetobeseparatedfromtheliquidphaseinthegas-liquidseparatorandarecarriedtotheatomizerbyacarriergas[2] InthisworkanatomizerdesignedspecificallyforAFStheflame-in-gas-shieldatomizerwasused(Fig1) It consistsofaverticalquartz tubesuppliedwithargon and hydrogen together with the analyte volatile species Moreover acapillaryisinsertedintheverticalaxisoftheverticaltubethroughwhichoxygenisintroducedA hydrogen-oxygenmicroflameburns on top of the capillary Themicroflameisshieldedfromtheambientatmospherebyaflowofargonwhichisintroducedthroughashieldingunitfittedaroundtheverticaltube[34] Theaimofthisworkwastooptimizeatomizationconditionsintheflame-in-gas-shieldatomizerusingPVGasasampleintroductiontechniqueandtocomparetheanalyticalcharacteristicsofPVGandHGforultrasensitivedeterminationofbismuthbyAFS
2Experimental
21Reagentsandchemicals
Deionized water (Ultrapur Watrex USA) was used for preparation of all thesolutionsWorkingBisolutionswerepreparedfreshdailybyserialdilutionof
ndash1stock1000mgl BistandardforAAS(Sigma-AldrichGermany)RegardingHG05(mv)NaBH in04(mv)KOHwasusedasa reductantAsolutionof4 ndash11mol l HClwasusedasacarrierandblank
98 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Flame-in-gas-shieldatomizerOHndashobser-vationheight
RegardingPVGformicacid(98paLach-NerCzechRepublic)andaceticacid (998 pa Lach-Ner CzechRepublic)were used for preparation of thereaction medium they were purified in a Teflon BSB-939-IR sub-boilingdistillation apparatus (Berghof Germany) The composition of the reactionmedium(40(vv)acetic125(vv)formicacid)wasoptimizedearlier[5]
ndash1The 5000mgl Co stock solution was prepared from cobalt(II) acetatetetrahydrate (pa Lach-Ner Czech Republic) and used as a sensitizer ofphotochemicalreactionTheoptimalconcentrationofCointhestandardsamples
ndash1andblanksolutionscorrespondedto50mgl (ref[5]) Acertifiedreferencematerial(CRM)-1643fTraceElementsinWater(NationalInstituteofStandardsandTechnologyUSA)wasusedtochecktheprecisionofthedevelopedmethodology
22Instrumentation
221Atomicfluorescencespectrometer
An in-house assembled non-dispersive atomic fluorescence spectrometerconstructedatourlaboratorywasusedforBideterminationandisdescribedindetail elsewhere [3] The detector output provided signals in microV Peak areacorrectedtobaselineandmainlysignaltonoiseratioweretheparametersusedtoevaluatethedata
222Hydridegeneratorphotochemicalvapourgeneratorandatomizer
A flow injection hydride generator was employed (Fig 2a) The reductantndash1 ndash1(12mlmin )andthecarrier(4mlmin )werepumpedbyaperistalticpump
Thesamplewasinjectedthrougha1mlsampleloopintotheflowofcarrierAglassgas-liquid separator (5 ml) with forced waste removal was employed forseparatingthegasphasecontainingbismuthanewhichwasthencarriedtotheatomizerbyargon Thephotochemicalvapourgenerator(Fig2b)consistedofthephotoreactorconstructedwitha15Wlow-pressureHggermicidallamp(Cole-ParmerUSA)wrappedaroundwith6mofPTFEtubing(1mmidinternalvolume471ml)The
ndash1reactionmedium(3mlmin )waspumpedbyaperistalticpumpThesamplewasinjected througha056ml sample loopApolypropylenegas-liquid separator
(15ml)withforcedwasteremovalimmersedinanicebath[6]wasemployedforseparatingthegasphasecontainingBivolatilespeciessubsequentlycarriedtotheatomizerbyargon Theflame-in-gas-shieldatomizerisdepictedinFig1detaileddescriptionisgiveninRef[3]Theobservationheight(OH)isdefinedasthedistancefromthetopofthecapillarytothecentreoftheopticalbeam
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 99
23Samplepreparation
CRMNIST1643fwasdilutedwith1MHCl80-foldforBideterminationbyHG-AFSRegardingBideterminationbyPVG-AFSthesampleneededtobeevaporatedtodrynessinordertogetridofnitricacidthatseriouslyinterferesatmMlevel[5]Avolume of 3ml of CRMwere pipetted into a 40 ml quartz vial evaporated(temperature asymp100 degC two replicates) and subsequently diluted ca 33-foldAsamplepreparationblank3mlofdeionizedwaterwaspreparedaswell
3Resultsanddiscussion
TheatomizationconditionsforHG-AFSwereoptimizedinourpreviouswork[3]TheseconditionswereusedasinitialtofindtheoptimumconditionsforPVG-AFSwith the flame-in-gas-shieldatomizerwith respect to sensitivityandsignal tonoiseratioFirstlythehydrogenfractionintherange10ndash16wasoptimizedatconstant total gas flow rate (sum of total argon and total hydrogen) of
ndash1500mlmin the lower the hydrogen fraction the better However at 10hydrogenfractiontheflamewasnotstableenoughandwentoftenouthenceitwasoptedfor12hydrogenfraction
100 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2(a)Hydrideand(b)photochemicalvapourgenerator
The oxygen flow rate through the capillary was optimized in the rangendash15ndash30mlmin Thehighestsignaltonoiseratiowasachievedwiththeflowrateof
ndash120mlmin Thetotalgasflowratewasoptimizedatconstant12hydrogenfractioninthe
ndash1range500ndash800mlmin Theoptimumobservationheightvarieswithtotalgasflow rate so it had to be optimized as well The optimum conditions aresummarizedandcomparedtothoseachievedwithHGinTable1 TheanalyticalfiguresofmeritofPVG-AFSwiththeflame-in-gas-shieldatomi-zerweredeterminedThecalibrationfunctionconstructedwith010025050
ndash1 2100 and 200 microg l Bi standardswas linear (R = 09998) The repeatabilityndash1expressedastherelativestandarddeviation(n=10)was6at1microgl andthe
ndash1relativeandabsolutelimitsofdetection(3σn=10)achievedwere12ngl and68pgrespectively(Table2)TheabsolutelimitofdetectionachievedwithHGwas 76 times lower which can be attributed to several aspects Firstly thegenerationefficiencyforPVGapproachwasaround53while100isexpectedforHG[3]Secondlyafullwidthathalfmaximumofthemeasuredpeakswasca2-foldgreaterwhichnecessitatedlongerintegrationtimeandwasthusreflectedinhighernoiseofthesignalsFinallythelimitofdetectionforPVGapproachwas
ndash1affectedby seriouscontamination (around10ng l )mostprobably from thesensitizersolutionthatcontainedBiasimpurity Tovalidate theproposedmethodologyBi contentwasdetermined inCRMNIST1643f (Table3) and the resultswere compared to thosemeasuredwithHG-AFS[3]Duetosevereinterferencesfrominorganicacidsespeciallynitricacid[5]thesampleneededtobeevaporatedtodrynessandthenfilledupwiththe
2+reactionmediumcontainingCo as thesensitizer(NIST1643f isstabilized in
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 101
Parameter HG-AFS(ref[3]) PVG-AFS
ndash1Artotalmlmin 440 528ndash1H totalmlmin 60 722 ndash1O mlmin 7 202
OH mm 6 9ndash1Arshieldlmin 1515 1515
Table 1Atomizationconditionsforflame-in-gas-shieldatomizer
Parameter HG-AFS(ref[3]) PVG-AFS
LODpg 09 68ndash1LOQngl 09 12
Repeatability lt1 6
Table 2AnalyticalfiguresofmeritofHG-AFSandPVG-AFS
ndash1032moll nitricacid)Theresultsobtainedbybothmethodologiesareingoodagreementwiththecertifiedvalue
4Conclusion
Photochemical vapour generation of Bi was successfully coupled with non-dispersiveatomicfluorescencespectrometryforthefirsttimeanditsapplicabilitywas verified by determination of Bi in certified reference material of waterComparedtohydridegenerationconditionsofatomizationdifferinanoptimalobservationheightandsupplyofoxygenwhichmaybeneededtoldquoburnoutrdquotheorganicvapoursthatarereleasedfromthereactionmediumtothegasphasehowever this remains tobe verifiedAlthough there are still some limitationsregardingthelimitsofdetectionrepeatabilityandinterferencesthisnewsampleintroductionapproachseemstobepromising
Acknowledgments
The support of the Czech Science Foundation (19-17604Y) Czech Academy of Sciences(Institutional supportRVO68081715)andCharlesUniversity (ProjectSVV260560andProjectGAUK1048120)isgratefullyacknowledged
References
[1] Musil SMatousek T Currier JM StybloM Dedina J Speciation analysis of arsenic byselectivehydridegeneration-cryotrapping-atomicfluorescencespectrometrywithflame-in-gas-shield atomizer achieving extremely low detection limits with inexpensiveinstrumentationAnalChem86(2014)10422ndash10428
[2] SturgeonREPhotochemicalvaporgenerationaradicalapproachtoanalyteintroductionforatomicspectrometryJAnalAtSpectrom32(2017)2319ndash2340
[3] S tadlerova B Kolrosova M Dedina J Musil S Atomic fluorescence spectrometry forultrasensitivedeterminationofbismuthbasedonhydridegenerationndashtheroleofexcitationsourceinterferencefilterandflameatomizersJAnalAtSpectrom35(2020)993ndash1002
[4] DedinaJAtomizationofvolatilecompoundsforatomicabsorptionandatomicfluorescencespectrometryOnthewaytowardstheidealatomizerSpectrochimActaPartB62(2007)846ndash872
102 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Certified HG-AFS PVG-AFSndash1valuemicrogl
ndash1 a ndash1 a valueobtainedmicrogl recovery valueobtainedmicrogl recovery
1262plusmn011 128plusmn01 102plusmn1 121plusmn09 97plusmn5
a Spikedrecovery=slopeofstandardadditions(noadditionandtwospikedconcentrationstoasample)slopeofexternalcalibration
Table 3ThedeterminedcontentofBiinCRMNIST1643fpresentedasmedianvalueplusmncombineduncertainty(n=3)andrecoveries
[5] Vyhnanovsky J Yildiz D Musil S Effect of metal sensitizers on photochemical vaporgeneration of bismuth for analytical atomic spectrometry In Proceedings of the 15thInternationalStudentsConferenceModernAnalyticalChemistryKNesmerak(ed)PragueCharlesUniversity2019p257ndash262
[6] VyhnanovskyJSturgeonREMusilSCadmiumassistedphotochemicalvaporgenerationoftungstenfordetectionbyinductivelycoupledplasmamassspectrometryAnalChem91(2019)13306ndash13312
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 103
1Introduction
Liquidcrystalsareorganicsubstancesthatformamesomorphicphaseinsolu-tion[1]TheyareliquidlikeliquidsbuthaveaninternalconfigurationassolidsTheir light transmittance changes in the electric fieldwhich is used in liquidcrystaldisplays(LCDs)CholestericliquidcrystalsaretemperaturesensitiveThecolorofreflectedlightchangeswithsmalltemperaturechangeThisisusedinmedicineasasensitivetemperatureindicatorfordisease-infectedtissues High performance liquid chromatography or supercritical fluid chromato-graphy[23]areusedtoseparateanddetermineliquidcrystalcompoundsAnelectrophoretic method could be complementary to these commonly usedmethods To our best knowledge no study dealing with liquid crystal puritycontrolbyelectrokineticchromatographywaspublisheduntilnow Electroneutralsubstancesmoveincapillaryzoneelectrophoresisatthesamespeedcorrespondingtothespeedoftheelectroosmoticflow(EOF)andtherefore
Separation of liquid crystals using non-aqueous capillary electrokinetic chromatography
KATER INACOKRTOVATOMA S KR IZ EK
DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublickaterinacokrtovagmailcom
AbstractLiquid crystals arewidelyused in electronicsmedicine andotherfields Analytical separations are important in the development ofnewliquidcrystalstocontrolthepurityofsynthesizedsubstancesThesampleanalysisisimportantfordetectionofimpuritiesformedduring synthesis Liquid crystal-forming substances cannot beseparated by capillary zone electrophoresis due to the absence ofreadily ionizable groups Therefore electrokinetic chromatographywasused in thisworkAnotherproblemcomplicating theanalysiswastheverylowsolubilityofanalytesinwaterSeparationsinthisworkwere thereforecarriedoutundernon-aqueousconditions inacetonitrilewithaceticacidtoadjustthepHandhexadecyltrimethyl-ammonium chloride as a detergent to mobilize the non-ionizedanalytesUndertheseconditionsitwaspossibletoseparateimpu-ritiesfromsynthesizedanalytesinsamples
Keywordselectrokinetic
chromatographyliquidcrystalsnon-aqueouscapillary
electrophoresis
104 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
itisnotpossibletoseparatethemDuetothisanelectrokineticchromatographymethodwasdevelopedInthismethodasurfactantisaddedtothebackgroundelectrolyteMoleculesaggregateandformsphericalformationscalledmicelles[4]ifthesubstanceisaddedinsufficientconcentrationiehigherthanthecriticalmicellar concentration (CMC) Separation is possible due to interactions ofnonpolarmoleculepartswiththenonpolarmicelleinsideAlthoughwateristhemostusedsolventinelectrophoreticmethodsforseparationofwater-insolublesubstances organic solvents are selected However such solvent must meetcertaincriteriatobesuitableforuseincapillaryelectrophoresisAllcomponentsmustbesolubleinthesolvent[5]ItshouldnotbeflammabletoxicorreactiveforpracticalityitshouldbeliquidatroomtemperatureandalsoitspriceistakenintoaccountThevalueof its relativepermittivitywhichdescribes the strengthofinteractionsbetweenionsshouldbearound30Lowdynamicviscosityisalsopreferred to allow faster migration of analytes No organic solvent meets allparameters of the ideal solvent In practice methanol acetonitrile and theirmixturesarethemostusedTheseparationparameterscanbeinfluencedbyusingan organic solvent of the background electrolyte This topic has already beenwidelyexplored[6ndash8] Itwasgenerallyassumedthat inanhydrousconditionsmicellesarenotcreateddespitesufficientsurfactantconcentrationHoweveritwasfoundoutthatdodecylsulfatecanformstablemicelleswhenthebackgroundelectrolyteisdissolvedinformamide[9]Fortheanalysisofactivesubstancesinmedicinal plants Chen et al developed a method in which sodium cholatedissolvedinmethanolisusedasasurfactant[10]Theaddedpseudostationaryphase does not always form micelles but can still affect mobilization andseparationofanalytesiftheanalytesinteractdifferentlywithfreemoleculesofsurfactantInthisstudywater-insolubleliquidcrystalswereseparated(Fig1)Thereforenonaqueouselectrokineticchromatographymethodwasdeveloped
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 105
Fig 1 Structures of liquid crystals 4-([1-oxo-1-(pentyloxy)propan-2-yl]oxycarbonyl)phenyl4-(octyloxy)-[11-biphenyl]-4-carboxylate (ZL 85) and 4-([1-(decyloxy)-1-oxopropan-2-yl]oxycarbonyl)phenyl 4-(dodecyloxy)-[11- biphenyl]-4-carboxylate (ZL 1210) OpticalisomerismsitesaremarkedwithanasteriskStructurescreatedinMarvinSketch[11]
2Experimental
21Reagentsandchemicals
Acetonitrilege999fromSigma-Aldrich(Germany)aceticacid99fromLach-Ner Neratovice (Czech Republic) and hexadecyltrimethylammonium chloride25(ww)inwaterfromSigma-Aldrich(USA)wereusedforpreparationofback-groundelectrolyteMesyloxidepa(MO)suppliedbyLach-nerNeratovice(CzechRepublic)wasusedasareferencesubstance
22Instrumentation
ForexperimentsG7100ACapillaryElectrophoresisInstrument(AgilentTechno-logiesGermany)wasusedwithUV-VISdetectoroperatingat235nmand254nmwavelengthMeasurementswereconductedinafused-silicacapillaryof50microminner diameter with the total length 500cm and effective length 415cm(PolymicroTechnologiesUSA)
23Method
Capillarywas flushed for3minuteswith1MHCland for2minuteswith thebackgroundelectrolyteBackgroundelectrolytewaspreparedbymixingaceticacid (10mM) and hexadecyltrimethylammonium chloride (40mM) in aceto-nitrile Sampleswere introducedhydrodynamicallybyapressureof5kPa for1secondSampleswerefirstdissolvedinacetonitrileandthendilutedtwotimeswith the background electrolyte A voltage of 20 kV was applied during theseparation
3Resultsanddiscussion
Liquidcrystalsampleswerepractically insoluble inwater theirsolubilitywasndash3testedataconcentrationlevelof1mgcm inmethanolandacetonitrileWhile
samples were not sufficiently soluble in methanol they were successfullydissolvedinacetonitrile Becauseallanalytesaresubstancesthatdonothaveeasilyionizablefunctionalgroups theelectrokineticchromatographymethodwaschosen forseparationAsuitablesurfactantwassoughtCommonlyusedsodiumdodecylsulfate(SDS)isinsoluble in acetonitrile Therefore hexadecyltrimethylammonium chloride(CTAC) which had sufficient solubility for further experiments was chosenAlthoughasuitablebufferwassoughttoensureastablepHduetoproblemswithprecipitationofbuffercomponentsinthenon-aqueousenvironmentaceticacidwasusedtoadjustandmaintainpHofbackgroundelectrolytesolutionAstheadditionofcationicsurfactantsuchasCTACleadstoEOFreversalthedependence
106 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ofEOFmobilityontheconcentrationofCTACinthebackgroundelectrolytewasmeasuredContrarytowhatisobservedinaqueousbackgroundelectrolytesEOFwasnotreversedItsmobilitydecreasedwithincreasingCTACconcentrationbutnomajorchangesoccurredabove40mMconcentrationThecapillarywallwasprobablyalreadysaturatedbyCTACandthefurtherincreaseinconcentrationhadno signifficant effect on the conditionof the capillarywall Therefore a CTAC
ndash3concentrationof40mmoldm was chosenas sufficient for furthermeasure-mentswithrespecttotheincreasingcurrentwithincreasingionicstrengthofthebackgroundelectrolyte The optimized method was used for separation of several liquid crystalsamplesofdifferentpurityInthesampleoftheZL85liquidcrystalwith99purityonezoneoftheanalytewasdetectedImpuritieswereseparatedfromthisanalytewhen the samplewith lower puritywas introduced The peak of theanalytewasidentifiedbasedonrelativemigrationtimerelatedtomesityloxideSeparationoftheanalytefromanimpurityinthesampleZL8576isshowninFig 2The relativemigration timeof the firstpeak is 0834 therefore itwasidentifiedastheZL85analyteThestandarddeviationoftherelativemigrationtimesinfivemeasurementswas0002min(01) Using the available high purity sample it was possible to measure thecalibration line forquantificationof theanalyte in lesspuresamplesLimitof
ndash3detection was determined as 0009mgcm and limit of quantification as ndash30031mgcm FromthecalibrationlineconcentrationofZL85inthesample
with lower purity was calculated The concentration was determined as
48(ww)standarddeviation5(ww)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 107
Fig 2ElectropherogramobtainedwhenasampleofZL85liquidcrystalwithlowerpuritywasndash3introducedSamplewas introduced in01mgcm concentrationandwithaddedmesityloxide
ndash3(10mgcm )Capillarywithinnerdiameterof50micromtotallengthof500cm415cmeffectivelength The background electrolyte was acetonitrile with 10 mM acetic acid and 40 mMhexadecyltrimethylammonium chloride A voltage of 20 kVwith positive polaritywas appliedDetectionat254nm
ForsampleZL121099onlytheanalyteandmesityloxideweredetectedInthe sample ZL 1210 59 several impurities were separated and detected(Fig3)Accordingtotherelativemigrationtimetheanalyteofinterestcorres-pondstothefirstpeakPeakresolutionissufficientTheresolutionoftheanalytepeakandthesecondpeakis284andtheresolutionoftheothertwopeaksis230
4Conclusions
InthisstudyanewmethodforanalysisofnewlysynthesizedliquidcrystalswasdevelopedSomeparametersofthemethodwereoptimizedndashoptimumconcen-tration of hexadecyltrimethylammonium chloride was searched The identifi-cation of analyteswas based on a comparison of relativemigration times InsamplesZL85andZL1210withlowerpuritytheimpuritieswereseparatedfromthepeaksofliquidcrystalsthecontentofanalytewasdeterminedintheZL85sampleaccordingtothecalibrationline
Acknowledgments
IwouldliketothanktheInstituteofPhysicsoftheCzechAcademySciencesforprovidingnewlysynthesized liquid crystals This work has been supported by Specific University Research(SVV260560)andbyCharlesUniversityResearchCentreprogramNoUNCESCI014
References
[1] GennesPGProstJThePhysicsofLiquidCrystals2ndedNewYorkOxfordUniversityPress1993
108 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ndash3Fig 3ElectropherogramofsampleZL121059ataconcentrationof05mgcm withmesitylndash3oxideataconcentrationof10mgcm capillarywith innerdiameterof50microm total lengthof
500 cmeffectivelength415cmThebackgroundelectrolytewasacetonitrilewith10mMaceticacidand40mMCTACAppliedvoltage20kVpositivepolarityDetectionat235nm
[2] Vankatova P KalıkovaK KubıckovaA Ultra-performance supercritical fluid chromato-graphy A powerful tool for the enantioseparation of thermotropic fluorinated liquidcrystalsAnalChimActa1038(2018)191ndash197
[3] Vankatova P Kubıckova A Cigl M Kalıkova K Ultra-performance chromatographicmethodsforenantioseparationofliquidcrystalsbasedonlacticacidJSupercritFluids146(2019)217ndash125
[4] Terabe S Otsuka K Ichikawa K Tsuchiya A Ando T Electrokinetic separations withmicellarsolutionsandopen-tubularcapillariesAnalChem56(1984)111ndash113
[5] RiekkolaMLRecentadvancesinnonaqueouscapillaryelectrophoresisElectrophoresis23(2002)3865ndash3883
[6] Wright PB Lister AS Dorsey JG Behavior and use of nonaqueous media withoutsupporting electrolyte in capillary electrophoresis and capillary electrochromatographyAnalChem69(1997)3251ndash3259
[7] PorrasSPKenndlerECapillaryzoneelectrophoresisinnon-aqueoussolutionspHofthebackgroundelectrolyteJChromatogrA1037(2004)455ndash465
[8] PorrasSPRiekkolaMLKenndlerETheprinciplesofmigrationanddispersionincapillaryzoneelectrophoresisinnonaqueoussolventsElectrophoresis24(2003)1485ndash1498
[9] GuoXWangK ChenGH Shi JWuX Di L LWangY Determination of strobilurinfungicideresiduesinfruitsandvegetablesbynonaqueousmicellarelectrokineticcapillarychromatography with indirect laser-induced fluorescence Electrophoresis 38 (2017)2004ndash2010
[10] Chen AJ Li C Gao WH Hu ZD Chen XG Application of non-aqueous micellarelectrokinetic chromatography to the analysis of active components in radix SalviaemiltiorrhizaeanditsmedicinalpreparationsJPharmBiomedAnal37(2005)811ndash816
[11] MarvinSketch [computer program] version 1990 ChemAxon httpschemaxoncom-productsmarvin
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 109
1Introduction
Sudandyesaresyntheticazo-basedaromaticcompoundsTheyaretraditionallyusedinvariousindustriessuchaschemicaltextileandwoodworkingasdyestocolourwaxesplasticsoilspolishesandsoforthTheyhavebeencategorizedasclass3carcinogensbytheInternationalAgencyforResearchonCancerandtheiruseisthereforeforbiddeninthefoodindustryTheyareknownfortheirbrightcolours and easy and cost-effectivemanufactureThey arenearly insoluble inwater but soluble in various organic solvents such asmethanol or trichloro-methane[1] SudanI1-phenylazo-2-naphthol(Fig1A)isadyeusedasanorangecolouringagentItssometimesalsosoldundernamesSolventOrangeRorCISolventYellow14ItisformedasasecondaryproductinthemanufactureoftheSunsetYellowdye
Electrochemistry of Sudan I and its derivates in aqueous media
ad b aANNAONDRA CKOVA MARIESTIBOROVA LUDE KHAVRAN cd adKAROLINASCHWARZOVA -PECKOVA MIROSLAVFOJTA
a CentralEuropeanInstituteofTechnologyMasarykUniversity Kamenice753562500BrnoCzechRepublicannaondrackovaceitecmuniczb DepartmentofBiochemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublicc UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistry FacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublicd InstituteofBiophysicsCzechAcademyofSciences Kralovopolska13561265BrnoCzechRepublic
AbstractSudanIisanaromaticazo-compoundthathasbeenproventobeacar-cinogenDuringitsmetabolizationbycytochromeP450inliverafewmain derivates can be identified Thiswork sets out to assess themechanismofelectrochemicalreductionandoxidationofSudanIitshydroxylationderivativesfeaturingmetabolitesintheSudanIdetoxi-fication pathway and to introduce their selective voltammetricanalysis on boron-doped diamond electrode We show successfuldifferentiationamongthesecompoundsthankstothedifferencesintheelectrochemicaloxidationoftheirphenolicgroups
Keywordsborondopeddiamond
electrodecytochromeP450electrochemicalanalysisSudanI
110 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
InmammalianorganismsSudanIismetabolizedbythemicrosomaldetoxi-fying systemwitha central roleof cytochromeP450hydroxylationactivity inliver[2]DuringtheoxidativeprocessofmetabolizingSudanIseveralmetaboliteswereidentifiedbyprevioustestsThesearegt1-(phenylazo)-naphtalene-26-diol(further abbreviated SI-6OH) 1-(4-hydroxyphenylazo)-2-hydroxynaphtol(furtherabbreviatedSI-4OH)and1-(4-hydroxyphenylazo)-naphtalene-26-diol(furtherabbreviatedSI-46-diOH)thestructuresarepresentedinFig1 ThemainmethodcurrentlyusedtoidentifySudanIamongotherdyeswithsimilar structure is high-performance liquid chromatography (HPLC) It isrecommendedasthestandardmethodtoidentifythelevelofSudanIinfood[3] ComparedtoHPLCelectrochemicalmethodsareprovingtobefastercheaperandcomparablypreciseUnfortunatelyacomprehensiveelectrochemicalstudyofSudanIandparticularlyofitshydroxylatedmetaboliteshasnotbeencompletedyetThedyecanbedetectedthroughelectrochemistryeitherbytheoxidationofitsphenolicgrouporviareductionoftheazogrouppresentinitsmoleculeInbothcasesotherelectrochemicallyactivemoietiesareformedThederivatesofSudanIcanbedetectedandrecognizedfromSudanIthroughanalogousprocesses[4]InthisstudywefocusedoncomparisonofelectrochemicalbehaviourofSudanIanditshydroxylatedmetabolitesonborondopeddiamondelectrodetoaddressthepossibilitiesoftheirrecognitionsinmixturesbasedondifferencesinanodicandcathodicsignals
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 111
Fig 1 Chemical structure of (A) Sudan I (B) 1-(phenylazo)-naphtalene-26-diol (SI-6-OH)(C) 1-(4-hydroxyphenylazo)-2-hydroxynaphtol (SI-4-OH) and (D) 1-(4-hydroxyphenylazo)-naphtalene-26-diol(SI-46-diOH)
(A) (B)
(C) (D)
2Experimental
21Reagentsandchemicals
SudanI(Merckanalyticalstandardgrade)wasdissolvedinethanol(Merck)andkept at room temperatureChemicals forBritton-Robinsonbufferpreparation(acetic acid boric acid orthophosphoric acid sodium hydroxide) were fromMerckwithpurityge99pHofthebufferwasadjustedbymixingoftheacidsandsodium hydroxide solution at different ratios The Sudan I metabolites weresynthetized at the Department of Biochemistry Faculty of Science CharlesUniversityandkeptinmethanolattemperature3degC
22Instrumentation
Cyclicvoltammetric (CV)measurementswerecarriedout inBritton-RobinsonbufferofpH=70atroomtemperatureSudanIanditsderivativeswereaddedto
minus1thesolutionofBritton-Robinsonbuffertofinalconcentrationof5micromolL andstirred Before the measurement oxygen was removed from the solution bypurgingwithargonfor3minutesAutolabanalyzerPGSTAT20(EcochemieTheNetherlands)inconnectionwithVA-Stand663(MetrohmSwitzerland)GPES49(MetrohmSwitzerland)andathree-electrodesetup(withborondopeddiamond
2(WindsorScientificUKdiskdiameter3mmA=707mm )asworkingelectrodeminus1AgAgCl3molL KCl as reference electrode and platinum wire as auxiliary
electrode) Five cycles were performed for each measurement at scan rateminus1of1Vs
3Resultsanddiscussion
ForeachcompoundtwoseparateCVmeasurementswereperformedeachwithfive cycles performed in rapid succession For bothmeasurements the initialpotentialwassetat0VTheanodicscancontinuedto+1Vturnedtowardsndash1Vandfinishedat0VIntheothersetupcathodicscanwasperformedfirstfromthestartingpointtondash1Vturnedtowards+1Vandreturnedto0VThiswaywewereabletoobservethebehavioursofSudanIandcompareittothatofitsderivativeswhilefirstbeingreducedandthenoxidisedorviceversa In the anodic scan of Sudan I and its derivatives (Fig 2) differences in thepositionsoftheoxidationpeaksineachcompoundcanbeobservedWhileSudanIwithonlyonephenolgroupisoxidizedat+067VthederivativeswithtwophenolgroupsieSI-4OHandSI-6OHgiveoxidationpeaksatremarkablylesspositivepotentialofca+05VSI-46OHwiththepresenceofoverall3hydroxygroupsyieldstwooxidationpeaksInthereversecathodicscan(vertexpotential+1V)probablythankstothepresencereductionpeaksappearwhichcanbefurtherusedtodifferentiatebetweenSudanIandthederivativesSI-4OHandSI-6OH
112 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 113
Fig 3VoltammetricscanofSudanIanditsderivatesSI-4-OHSI-6-OHandSI-46-diOH1stscanincathodicdirectionfrom0Vvertexpotentialsndash1Vand+1VThemeasurementswereperformedinBritton-Robinsonbuffer(pH=7)withtheconcentrationofeachcompoundat5microMandatscan
ndash1rate1Vs
Fig 2 CyclicvoltammogramofSudanIanditsderivatesSI-4-OHSI-6-OHandSI-46-diOH1stscaninanodicdirectionfrom0Vvertexpotentials+1Vandndash1VThemeasurementswereperformedinBritton-Robinsonbuffer(pH=7)withtheconcentrationofeachcompoundat5microMandatscan
ndash1rate1Vs
possesstwoconjugatedhydroxylgroupswhichcanberegardedashydroquinonestructures and thus undergoing quasireversible redox process due to oxida-tionreductionofthehydroquinonetoquinonemoietyThisiswellvisibleattheCVsastheanodicsignalisfollowedbycathodiconeatthepotentialof+023VforSI-4OHand+03VforSI-6OHTheoxidationofSudanIproceedsbymechanism
minus +typicalforphenoliccompoundsatmorepositivepotentialsleading by1e 1H exchange to naphthoxy-type ndashO radical [5] This species undergoes furtherreactionsleadingtoformationofdimersandpolymersThecathodicpeakinthereversescanatndash02Varisesfromreductionofthesereactionproductsanditsoriginneeds tobe further investigatedSI-46OHwith thepresenceofoverallthree hydroxyl groups yields two oxidation peaks The first one is a result ofoxidationoftwoofthembeinginconjugationandthusbeingoxidizedtoquinonemoietyThesecondsignalatthesamepotentialastheoxidationsignalof2OHonnaphthaleneringofSudan I is consequenceofoxidationof the thirdhydroxylgroupofphenolictypeAsinglewidepeakat0Vinthereversescanispresumablyan overlap of signals arising from reduction of the quinonic moiety and by-products formed during oxidation processes Thanks to differences of theseprocessesspecificforindividualcompoundsitispossibletodifferentiateamongallfourofthemviapropersetting-upoftheinitialandvertexpotentialvalues The cathodic scan of Sudan I and its derivatives (Fig 3) shows a dominantreductionpeakaroundndash08Vwhichisduetoreductionoftheazogroupintheirstructuresaccompaniedbycleavageoftheirmoleculestoseparatethebenzeneandnaphthalene rings [4] The peaks in the subsequent anodic scans (vertexpotentialndash1V)arethereforetheresultoftheelectrochemicalreactionofmoietiesthatareproductsofthedivisionofthearomaticcirclesTheseproductsincludeaniline4-aminophenol1-amino-2-naphtholand1-amino-25-naphthalenediolwith irreversibly oxidizable amino moieties or (quasi)reversibly oxidizableamino-hydroxylsystemonthebenzeneornaphthaleneringandtispossibletodifferentiatebetweenthemTheobtainedoxidationpeaks(+02VforSI-4-OH+073VforSudanIandSI-6-OHand+025Vand+075VforSI-46-diOH)makeitpossibletodifferentiatebetweenallcompoundswiththeexceptionofSudanIandSI-6-OHForthereliablerecognitionofthesetwocompoundsanothermeasure-mentwithdifferentparametersisneeded
4Conclusions
ThestructuresofSudanIanditshydroxyderivatesthatarethemainproductsofthe metabolization of Sudan I by cytochrome P450 are similar and theirrecognitionwhenpresentinmixtureinsolutionisdemandingHereinwepresenta simple approach based on comparison of signals obtained in cathodic andanodic scan in CV measurements without the need of time-demandingchromatographicseparationstepFurtherworkwillbedevotedtoidentificationof observed redoxprocesses and applicationof themethod formonitoringofmetabolictransformationsofSudanIinvitro
114 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Acknowledgments
ThisresearchwassupportedbytheCzechScienceFoundation(projectNo18-01710S)
References
[1] ChailapakulOWonsawatW SiangprohW GrudpanK ZhaoYF Zhu ZW Analysis ofSudanISudanIISudanIIIandSudanIVinfoodbyHPLCwithelectrochemicaldetectionComparison of glassy carbon electrodewith carbon nanotube-ionic liquid gelmodifiedelectrodeFoodChem109(2008)876ndash882
[2] StiborovaMMartinekVRydlovaHHodekPFreiESudanIisapotentialcarcinogenforhumans Evidence for itsmetabolic activation and detoxication by human recombinantcytochromeP4501A1andlivermicrosomesCancerRes62(2002)5678ndash5684
[3] GomezMArancibiaVAliagaMNunezCRojas-RomoCDeterminationofSudan I indrinks containing Sunset yellow by adsorptive stripping voltammetry Food Chem 212(2016)807ndash813
[4] PrabakaranEPandianKAmperometricdetectionofSudanIinredchilipowdersamplesusingAgnanoparticlesdecoratedgrapheneoxidemodifiedglassycarbonelectrodeFoodChem166(2015)198ndash205
[5] Enache T A Oliveira-Brett A M Phenol and para-substituted phenols electrochemicaloxidationpathwaysJElectroanalChem655(2011)9ndash16
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 115
1Introduction
Aminoglycosidesarealargegroupofnaturalandsemi-syntheticantibioticswithawidespectrumofantimicrobialactivityagainstmostgram-positiveandgram-negativemicroorganismsCurrentlymultiplerepresentativesofaminoglycosidefamily ndash gentamicin (GM) neomycin B (NM) paromomycin (PM) kanamycin(KM)apramycin(AP) andstreptomycin(STM)Fig1ndashareapproved to treatinfectiousdiseasesinanimalsMaximumresiduelimitsfortheseaminoglycosidesinproductsandtissuesfromedibleanimalsareestablished[1]henceaneffectiveandrobustassayisnecessaryforcontrolofaminoglycosidescontamination Inthisstudyenzyme-linkedimmunosorbentassayforthedetectionofamino-glycosides in foodproductsandenvironmentalobjectswasdevelopedGroup-specificantibodieswereproducedowingtoimmunogenbasedonribostamycin(RS)whichexposedthecommonfragmentofmostaminoglycosides2-deoxy-streptamin (2-DOS) Fig 1 The developed assay was made suitable for thedetectionofresidualaminoglycosidesinhoney[2]
Group detection of aminoglycosides using ELISA for control of food contamination
ab a aKONSTANTINBURKIN INNAGALVIDIS MAXIMBURKIN
a DepartmentofImmunologyIMechnikovResearchInstituteofVaccinesandSera MalyjKazionnyjper5a105064MoscowRussianFederationburkin-kostyandexrub DepartmentofChemicalEnzymologyFacultyofChemistryLomonosovMoscowStateUniversityLeninskieGory1119991MoscowRussianFederation
AbstractThegrowingthreatofglobalantibioticresistanceisforcingtoreducenon-targetconsumptionofantibioticsandtomonitorcontaminationoffoodandenvironmentalobjectsInthisworkELISAwasdevelopedforgroupdetectionofaminoglycosidesToobtaingroup-specificanti-bodies a new immunogen based on ribostamycin was used Thedevelopedindirectcompetitiveformatofassayallowedtherecogni-tionof9aminoglycosidesnamelyneomycinribostamycinneaminparomomycin gentamicin sisomicin kanamycin tobramycin and
ndash1apramycinwithadetectionlimitrangedbetween002ndash020ngmL TheeffectivenessoftheproposedassaywasevaluatedinhoneyasafoodstuffmodelToneutralizea stronghoneymatrixeffect and toavoidalaborioussamplepre-treatmentanewmatriximitatorwassuggested 5 sucrose solution imitated the influence of 50-folddilutedhoneyTheproposedassayallowedustorevealanyofthe9
ndash1mentionedaminoglycosidesinhoneyata10microgkg level
KeywordsaminoglycosidesELISAhoney
116 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
2Experimental
21Reagentsandchemicals
NeomycinBribostamycinneamin(NA)paromomycinkanamycintobramycin(TM)amikacin(AM)gentamicinnetilmicin(NTM)sisomicin(SSM)geneticin(GC) apramycin and streptomycin were purchased from Chimmed (MoscowRussia) Bovine serumalbumin (BSA) complete Freund adjuvant 16-hexane-diamine1-ethyl-3-(3-dimethylaminopropyl)carbodiimide(EDC)sodiumperio-dateandsodiumborohydrideweretheproductsofSigma-Aldrich(USA)Gelatin(Gel)wasfromBio-Rad(USA)sucrosefromServa(Germany)two-componenttetramethylbenzidine (TMB) substrate solutionwas fromBioservice (Russia)andgoatanti-rabbitIgGantibodiesconjugatedtohorseradishperoxidase(anti-rIgG-HRP)werefromIMTEK(Russia)Honeysampleswerepurchasedfromlocaloutlets
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 117
Fig 1 Structuralformulasofstudiedaminoglycosides
22Preparationofconjugatedantigens
TwotypesofconjugateswerepreparedbasedonRSandBSAusingzero-lengthand C6 spacer arm between hapten and protein carrier RSwas treatedwithsodiumperiodatetooxidizehydroxylsofribosefragmenttoreactivealdehydegroupsandthencoupledtoBSAaminesthroughreductiveaminationToremoveuncoupledRSanexhaustingdialysiswascarriedoutusingdialysismembranetubes(MWCO14kDa)UsingthesameprocedureGel-RSconjugatewassynthe-sized ForpreparationofBSA-C6-RSwefirstlymodifiedBSAwith16-hexanediamineThemixtureofBSAandEDCinwaterwerestirredfor30minThen16-hexane-diaminewasaddedandstirredfor2hThemodifiedproteinwasdialyzedfromtheexcessive reagents and resultant BSA-C6-NH2was coupled to RS in reductiveaminationprocessasdescribedabove
23Immunizationandantibodypreparation
BSA-RSandBSA-C6-RSwereusedasimmunogensChinchillarabbits(20ndash25kg)weresubcutaneouslyinjectedat10ndash15pointsonthebackwith01mgofimmuno-gensemulsifiedinthecompleteFreundadjuvantThesamedosesofimmunogensinsalinewereadministeredmonthly forbooster immunizationsAweekaftereachinjectionabloodsamplefromearveinswastakenforthecontrolofimmuneresponseTheantiserainglycerol(11vv)werestoredatndash15degCuntiltestinginELISA
24TheELISAprocedure
AcompetitiveassaywasconductedaccordingtoclassicalprocedureGel-RSwascoatedovernightonpolystyrene96-wellCostarplatesNon-adsorbedconjugatewashedoutusingPBSwith005oftween20(PBS-T)Thenextcompetitivestep
ndash1includedtheadditionof01mLstandardaminoglycosidesolutions(1pgmL to ndash1 ndash11microgmL (B)and0microgmL (B ))inPBS-Tor01mLoftestedsampleand01mLof0
antibodiesinworkingdilution(1h25 degC)Afterwashingtheantibodiesboundtoimmobilized Gel-RS were detected using anti-rIgG-HRP (1h 37degC) Coloredproduct formedasaresultofenzymaticreactionwithTMBsubstratemixture(05h25degC)wasreadat450nmusingaStatFax2100platereader(AwarenessTechnologiesUSA) Relativeantibodybinding(BB )vstheanalyteconcentrationswasplottedas0
standardcurvesfittedtoafour-parameterlogisticfunctionThecross-reactivity(CR) for every aminoglycoside representative was calculated as ratio of half-inhibition concentrations IC NMIC aminoglycoside The dynamic range of50 50
assaywasacceptedasIC ndashIC andthelimitofdetection(LOD)wascalculatedas20 80
B ndash3timesSD0
118 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
3Resultsanddiscussion
31Immunogensynthesisandantibodypreparation
Inthemajorityofpublicationsdevotedtoimmunoassayofaminoglycosidestheimmunogenscoatingantigensenzymeconjugatesortracerswerepreparedbycarbodiimide or glutaraldehyde methods involving aminoglycosidesrsquo aminogroups [3ndash7] Due to several amino groups in aminoglycoside molecules theformationofconjugateswithavariableorientationofthehaptenoccurs InpresentstudyRSwaschosenasanimmunizinghaptenduetothefollowingadvantageous features Being a trisaccharide RS has the size of a moleculecomparable to themostof aminoglycosides Ithas three identical ringsA-B-CsimilartothoseinNMUsingaperiodateoxidationwecouldinvolvearibosesiteofRSincouplingtoproteinthatprovidedastrictorientationofhaptenonthecarrier with a favorable presentation of the 2-DOS fragment The resultantimmunogensBSA-RSandBSA-C6-RSwerecomparedtorevealwhichdesignisbetterforpresentationofacommonfragmentofaminoglycosidemoleculeandgenerationofgroup-specificantibody Antibodies to the BSA-RS demonstrated moderate sensitivity (NM
ndash1IC =10ngmL )andhighselectivitytowardsNMwithrelativelylowcross-reac-50
tivity(lt5)forGMKMandAPTheapplicationofthespacerintheimmunogenBSA-C6-RScontributedtoaprominentpresentationofthe2-DOSdeterminantandtheinductionofantibodieswithrecognitionofbroadspectrumofdifferentaminoglycosides In addition anti-BSA-C6-RS exhibited significantly better
ndash1sensitivity(NMIC =02ngmL )Thusallsubsequentstudieswereconducted50
usinganti-BSA-C6-RS
32Examinationofassayspecificityandselectionofimmunoreagents
TheindirectcompetitiveformatofassaywasdevelopedForevaluationofassayspecificity a panel of following aminoglycosideswas studied and their cross-reactivitywasdeterminedNA(625)RS(250)NM(100)KM(475)PM(173)GM(90)TM(78)AP(17)SSM(12)AM(lt01)GC(lt01)STM(lt01)andNTM(lt01)ThemostoftheseanalytesareusedinmedicalandveterinaryareashoweveronlyNMPMGMKMAPandSTMareappliedinanimalhusbandry[1]
33Determinationofaminoglycosidesinhoneyandselectionofthematriximitator
Honey is a complex product consisting of carbohydrates (75ndash80) vitaminsproteinsenzymesorganicacids traceelements inclusionsandothercompo-nentsThesecomponentsmightinterfereimmunochemicalreactionThereforetheisolationofaminoglycosidesfromhoneyisalaboriousandtime-consuming
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 119
120 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
procedureToavoidthisstephoneymatriximitatorswereappliedtomimictheinfluenceofhoneymatrixonantibodybindingSucrosewaschosenasthehoneyimitatorsinceitexposedastronghoney-matrix-likeeffectonantibodybindingTheadequacywasfoundbetweensolutionsofhoneyandsolutionsofsucroseexpressinganequalmatrixeffectTwopairswithequivalentmatrixeffectwere120honey=20sucroseand150honey=5sucroseThelatterconditionswere chosen asmore preferable due to inconvenience of operatingwith highviscous20sucrosesolution Thedeterminationofaminoglycosidesinhoneycouldbecarriedoutquantita-tivelyiftheanalytetobedetectedisknownForquantificationofaminoglycosideinhoneyasamplewasdiluted50timesinPBS-Tandaminoglycosidestandardcurvewas generated in 5 sucrose-PBST (Fig 2) If analyte is unknown thedevelopedgroup-specificELISAcanbeusedasascreeningtestInthiscasetheanalyzed sample can be considered as contaminated if it caused a relativeantibodybindingbelowthecut-off level (Fig3)Thus thedeveloped testwas
ndash1 ndash1 ndash1Analyte IC ngmL Dynamicrange LODngmL LODinhoneymicrogkg50 ndash1 IC ndashIC ngmL20 80
NM 02 003ndash21 002 10PM 07 008ndash71 005 25GM 15 015ndash133 011 55KM 035 005ndash39 004 20AP 68 05ndash968 023 115
Fig 2StandardcurvesandanalyticalparametersoftheELISA-systemforgroupdeterminationofaminoglycosidesinhoneyInteractionofanti-BSA-RSwithcoatingantigenGel-RSin5sucrosesolutionasthehoneyimitatorThedetectionlimitin5sucrosesolutionwasdeterminedaccordingtoLOD=B ndash3timesSD0
capabletorevealthecontaminationofhoneywith9aminoglycosides5amino-ndash1glycosidesapprovedforveterinary(NMPMGMKMandAP)ata10microgkg level
andalsoNARSSSMandTM
4Conclusions
A novel indirect competitive ELISA for the detection of aminoglycosides wasdevelopedRSwasusedasanewimmunizinghaptentoproducegroup-specificantibodiesagainst2-DOSacommonmoietyofalargenumberofaminoglycosideantibiotics A wide spectrum of aminoglycoside representatives could bedetectedincludingNMRSNAPMGMSSMKMTMandAPThedevelopedassay
ndash1was capable todetect theseanalyteswithaLOD up to002ndash020 ngmL ForanalysisofhoneyamatriximitatorwasdevelopedtoavoidhoneyinterferencesonimmunoassayTheanalysisofthehoneysampleallowedustorevealanyofthe
ndash1mentionedaminoglycosidesinhoneyata10microgkg level
References
[1] CouncilRegulation(EU)N372010OffJEurCommunitiesInfNotL15(2009)1ndash72[2] GalvidisIABurkinKMEreminSABurkinMA Group-specificdetectionof2-deoxy-
streptamineaminoglycosidesinhoneybasedonantibodiesagainstribostamycinAnalMeth11(2019)4620ndash4628
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 121
Fig 3Detectionofaminoglycosidesspikedinhoneysamplesata40ppblevelusinggroup-specificELISAEachsymbolcorrespondstotheaveragerelativebindingandtheerrorisSDobtainedforanindividualhoneysampleanalyzedintriplicateEmptycharactersrepresentindividualblankhoneysamples(limebuckwheatandflower)andfilledsymbolsrepresentthesamesamplesfortifiedwith
ndash1aminoglycosidesata40mgkg (level establishedonlyforSTMinseveralcountries)Thecut-offlevelcorrespondstothelimitofassaydetectionobtainedbythematriximitator(5sucrose-PBS-T)
[3] ThompsonSGBurdJFSubstrate-labeledfluorescentimmunoassayforamikacininhumanserumAntimicrobAgentsChemother18(1980)264ndash268
[4] LiCZhangYEreminSAYakupOYaoGZhangXDetectionofkanamycinandgentamicinresiduesinanimal-derivedfoodusingIgYantibodybasedic-ELISAandFPIAFoodChem227(2017)48ndash54
[5] GalvidisIABurkinMAMonoclonalantibody-basedenzyme-linkedimmunosorbentassayfortheaminoglycosideantibiotickanamycininfoodstuffsRussJBiorganChem36(2010)722ndash729
[6] HaasnootWStoutenPCazemierGLommenANouwsJFKeukensHJImmunochemicaldetectionofaminoglycosidesinmilkandkidneyAnalyst124(1999)301ndash305
[7] Peng JWangYLiuLKuangHLiAXuCMultiplex lateral flowimmunoassayfor fiveantibioticsdetectionbasedongoldnanoparticleaggregationsRSCAdv6(2016)7798ndash7805
122 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
PhotochemicalvaporgenerationisanalternativesampleintroductiontechniqueforanalyticalatomicspectrometryThistechniqueisbasedaroundasourceofUV-radiationthatirradiatesalowmolecularweightorganicacidmedium(mostcommonlyformicacidaceticacidortheircombinations)withananalyteHighlyreducingradicalsandaquatedelectronsareproducedandconverttheanalyteintoavolatilespecieswhichisthentransportedintoadetector[1]SofartheuseofPVGhasbeendescribedforhydride-formingelements(AsBiTeSbPbSeSnandTl)andmercury[12]transitionmetals(FeCoNiCuMoWCdAgAuIrPdPtRhandOs)[13ndash6]andevennon-metals(BrIClFandS)[16ndash9] AfirstsuccessfulphotochemicalvaporgenerationofcobaltwasdescribedbyGuoetalin2004[6]whichwasfollowedbymoresystematicstudiesbyGrinbergetalin2008[10]andDengetalin2010[11]LaterworksbydeQuadrosetal[12]anddeJesusetal[13]focusedontheanalysisofrealsamplesInthelatterwork
Photochemical vapor generation of cobalt for detection by inductively coupled plasma mass spectrometry
ab aJAROMIRVYHNANOVSKY STANISLAVMUSIL
a DepartmentofTraceElementAnalysisInstituteofAnalyticalChemistryoftheCzechAcademyofSciencesVeveřiacute9760200BrnoCzechRepublicjaromirvyhnanovskygmailcom
b DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublic
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 123
AbstractThisworkfocusedonthephotochemicalvaporgenerationofcobaltVolatilespeciesweregeneratedinaflow-injectionsystememployinga high-efficiency flow-through UV photoreactor and a formic acidbased medium and were introduced by an argon carrier into aninductivelycoupledplasmamassspectrometerfordetectionOptimalgeneration conditions were found as 10 (vv) formic acid and
ndash1 ndash14molL ammonium formate with a 4mLmin flow rate whichcorresponds to irradiation time of around 13 s The influence ofvariousmetalsensitizersofphotochemicalreactionwasinvestigated
2+andonlyCu ionsexhibitedapositiveeffectongenerationefficiencyndash ndash 2ndashInterferencesfromcommoninorganicanions(NO Cl SO ) were3 4
also examined Lastly the limit of detection and repeatability (atndash1 ndash1250ngL )weredeterminedtobe13ngL and41respectively
Keywordscobaltinductivelycoupled
plasmamassspectrometry
photochemicalvaporgeneration
the authors also presented a systematic study on generation conditions andachievedagenerationefficiencyofaround40 Themainaimofthisworkwastooptimizetheconditionsofgenerationwithinductivelycoupledplasmamassspectrometry(ICP-MS)detectionexaminetheeffectofvariousmetal sensitizers toachieve thehighestgenerationefficiencypossibleandreachthelowestlimitofdetectionpossible
2Experimental
21Reagentsandchemicals
minus1Deionizedwater (DIW lt 02μScm UltrapurWatrex USA)was used for thepreparationofallsolutionsFormicacid(98paLach-NerCzechRepublic)andammonium hydroxide (ge25 pa Sigma-Aldrich USA) were used for the
minus1preparationof the reactionmediumA1000mgL Co stock solution (Sigma-AldrichUSA)wasusedforthepreparationofallsamplesolutionsThefollowingcompounds were used as potential metal sensitizers cadmium(II) acetatedihydrate(paLach-NerCzechRepublic)zinc(II)acetatedihydrate(paSigma-AldrichUSA)copper(II)acetatemonohydrate(paMerckGermany)nickel(II)acetatetetrahydrate(paSigma-AldrichUSA)sodiumtungstatedihydrate(paCarlRothGermany)and iron(II) sulphateheptahydrate (pa LachemaCzechRepublic)Nitricacid(65semiconductorgradeSigma-AldrichUSA)hydro-chloricacid(37paMerckGermany)andsulfuricacid(98paLach-NerCzechRepublic)wereusedforaninterferencestudy
22Instrumentation
AschematicdiagramofthePVGsystemcoupledtoICP-MSisshowninFig1andamore detailed description can be found in reference [5] Briefly a singlequadrupole ICP-MS Agilent 7700x (Agilent Technologies USA)was used as a
minus1detector Deionized water was mixed with a 10μgL Rh internal standardsolutionin2HNO andwassubsequentlynebulizedbyaMicroMistnebulizer3
59 103during PVG Isotopes of Co and RhweremonitoredMeasurementswereperformed in time resolved analysis mode and in He collision mode
minus1(41mLmin )AlltubingusedwasmadefromPTFEwiththeexceptionoftygontubing in the peristaltic pump (Reglo ICC Ismatec Switzerland) The high-efficiency flow-through photoreactor was a 19 W low-pressure mercurydischarge lamp (Beijing Titan Instruments Co Beijing China) with a quartzcentralchannel(asymp720μL internalvolume)Samplesolutionswere introducedintoastreamofreactionmediumusinganinjectionvalve(V-451IDEXHealthandScienceUSAsampleloopvolume05ml)Effluentfromthephotoreactorwasmixedwithaflowofargonandcarriedtothechilledgas-liquidseparator(internalvolume15mL)wherethevolatilespecieswereseparatedfromtheliquidwaste
124 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
andcarried to the inletofaScott-typespraychamber (originally the inlet formakeupargon)oftheICP-MS
3Resultsanddiscussion
The starting conditionswere adopted fromour earlierwork [14]whichusedatomicabsorptionspectrometerasadetectorandminiaturediffusionflameasanatomizerThefirstparameteroptimizedwasthecompositionofreactionmedium(Fig2)Theadditionofammoniumformate(createdin-situbytheadditionofacalculatedamountofammoniumhydroxidetoformicacid)wasfoundcrucialto
ndash1effectivelygeneratevolatilespeciesofcobalt10(vv)formicacidand4molL ammonium formatewas chosen as the optimumandwasused for all further
experimentsAlthoughhigher concentrations of both components led to even
higher signalstheseconditionswerenotusedfurtherbecauseofthelaboriousprocessofpreparation(mixingofconcentratedacidwithconcentratedbase)andtolimittheconsumptionofchemicals Theinfluenceofirradiationtimewasalsoexaminedandthehighestpeakarea
-ndash1wasobtained for4mLmin corresponding to an irradiation timeof approximately13s ToenhancethegenerationefficiencyadditionofvariousmetalstothereactionmediumwastestedtoldquosensitizerdquothephotochemicalreactionThemetalswerechosenwithrespecttotheirsignificantenhancementeffectdescribedrecentlyforphotochemicalvaporgenerationofotheranalytes[158]Theonlymetalionthat
2+ledtoanenhancementofthesignalwasCu (Fig3a)buteveninthiscasetheeffect was rather negligible reaching only 12-fold enhancement in the range
ndash1 2+ 2+001to01mgL Cu FurtheradditionofmoreCu ledtoadecreaseinthesignal2+TheadditionofZn didnotexhibitanypositiveornegativeeffectacrossthetested ndash1range (01 to 1500mgL not shown in figure) and the addition of higher
ndash1 2+ 2+ 6+concentrations (tens to hundredths ofmg L ) of Cd Fe andW (Fig3b)causedsevereinterferences
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 125
ndash ndash 2ndash Interferencescausedbycommoninorganicanions(NO Cl SO addedas3 4ndashtheirrespectiveacids)werealso investigatedOutof theseNO was foundto3
ndash1causethemostsevereinterferencesevenatconcentrationsofsinglemmolL Thendash 2ndashmethodologywasmorerobusttowardstheinterferencesfromCl andSO but4
theystillcausedsignificantdropinsensitivityathigherconcentrationsConsi-deringthewideuseoftheseacidsinanalyticalchemistryforsamplepreparationthisposesabigchallengeintheapplicationofthismethodtorealsamples
ndash1 Usingoptimalconditions(10(vv)formicacid4molL ammoniumformateandirradiationtimeof13s)acalibrationcurvewasmeasuredandevaluatedThelimitofdetectionwasdeterminedas3timesthestandarddeviationof10blank
ndash1measurements and was calculated as 13 ng L The repeatability of 10ndash1consecutivemeasurementsof250ngL was41
126 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 3Effectofvariousmetalionsonthepeakarea(a)metalionswithapositiveeffect(b)metalionsndash1withoutapositiveeffectExperimentalconditions2microgL Coreactionmedium10(vv)formic
ndash1 ndash1acidand4molL ammoniumformateflowrate4mLmin
ndash1Fig 2Effectofthecompositionofreactionmediumonpeakareaexperimentalconditions2microgL ndash1Coreactionmediumflowrate4mLmin (blackdotscorrespondtomeasuredpoints)
4Conclusions
Theconditionsofthephotochemicalvaporgenerationofcobaltwereoptimizedand are in good agreement with previous works [11 13] Copper ions wereidentifiedasapotentialsensitizerincreasingthesignalbyabout12-foldbuttheirpotentialuseisseverelylimitedbythenarrowrangeofconcentrationsinwhichthepositiveeffectisexhibitedSevereinterferencesfrominorganicanionswereobservedwhichisinlinewithotherworksdealingwithphotochemicalgeneration[1358]Furtherexperimentswillfollownamely(i)furtherinvestigationsinnewpotentialsensitizerstoenhancegenerationefficiencyandthusdecreasethe
ndash1limit of detection to sub ng L levels (ii) determination of the generationefficiency(fromcomparisonwithnebulizationandorusingaradioactiveisotope58Co)(iii)verificationoftheaccuracyandpracticalfeasibilityofthismethodologybyanalysisofcertifiedreferencematerials
Acknowledgments
ThesupportofTheCzechScienceFoundation(ProjectNo19-17604Y)CzechAcademyofSciences(Institutional supportRVO68081715)andCharlesUniversity (project SVV260560andprojectGAUK60120)isgratefullyacknowledged
References
[1] SturgeonREPhotochemicalvaporgenerationaradicalapproachtoanalyteintroductionforatomicspectrometryJAnalAtomSpectrom32(2017)2319ndash2340
[2] XuTHuJChenHJTransitionmetalionCo(II)-assistedphotochemicalvaporgenerationofthalliumforitssensitivedeterminationbyinductivelycoupledplasmamassspectrometryMicrochemJ149(2019)103972
[3] SoukalJSturgeonREMusilSEfficientphotochemicalvaporgenerationofmolybdenumforICPMSdetectionAnalChem90(2018)11688ndash11695
[4] deOliveiraRMBorgesDLGUVphotochemicalvaporgenerationofnoblemetals(AuIrPdPtandRh)AfeasibilitystudyusinginductivelycoupledplasmamassspectrometryandseawaterasatestmatrixJAnalAtomSpectrom33(2018)1700ndash1706
[5] VyhnanovskyJSturgeonREMusilSCadmiumassistedphotochemicalvaporgenerationoftungstenfordetectionbyinductivelycoupledplasmamassspectrometryAnalChem91(2019)13306ndash13312
[6] GuoXSturgeonREMesterZGardnerGJVaporgenerationbyUVirradiationforsampleintroductionwithatomicspectrometryAnalChem76(2004)2401ndash2405
[7] HuJSturgeonRENadeauKHouXZhengCYangLCopperionassistedphotochemicalvapor generation of chlorine for its sensitive determination by sector field inductivelycoupledplasmamassspectrometryAnalChem90(2018)4112ndash4118
[8] LeonoriDSturgeonREAunifiedapproachtomechanisticaspectsofphotochemicalvaporgenerationJAnalAtomSpectrom34(2019)636ndash654
[9] SturgeonREPaglianoEEvidenceforphotochemicalsynthesisoffluoromethaneJAnalAtomSpectrom(2020)httpsdoiorg101039D0JA00108B
[10] GrinbergPMesterZSturgeonREFerrettiAGenerationofvolatilecobaltspeciesbyUVphotoreduction and their tentative identification J Anal Atom Spectrom 23 (2008)583ndash587
[11] DengHZhengCB LiuLWWuLHouXDLvYPhotochemicalvaporgenerationofcarbonyl for ultrasensitive atomic fluorescence spectrometric determination of cobaltMicrochemJ96(2010)277ndash282
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 127
[12] deQuadrosDPBorgesDLDirectanalysisofalcoholicbeveragesforthedeterminationofcobalt nickel and tellurium by inductively coupled plasmamass spectrometry followingphotochemicalvaporgenerationMicrochemJ116(2014)244ndash248
[13] deJesusHCGrinbergPSturgeonRESystemoptimizationfordeterminationofcobaltinbiologicalsamplesbyICP-OESusingphotochemicalvaporgenerationJAnalAtomSpectrom31(2016)1590ndash1604
[14] VyhnanovskyJFotochemickegenerovanıtekavychspeciı kobaltuproanalytickouatomovouspektrometriiMasterthesisFacultyofScienceCharlesUniversityPrague2018
128 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Oneof themethods thatallowobtainingmaterialswithnewproperties is theplasmaenhancedchemicalvapordepositionmethodInthismethodcompoundscalledprecursorsaresuppliedtotheplasmareactorasagasphaseThankstoplasmaenhancedchemicalvapordeposition it ispossible toobtainmaterialswithuniquepropertiesThisisduetothefactthattheplasmaaffectsthesurfaceinfourdifferentways etching cleaning chemicalmodification and crosslinking
Optimization of condition for cold plasma deposition of thin layers for surface modification of working electrodes
a b a cJUSTYNALIPIN SKA MARIAMADEJ BOGUSŁAWBAS JACEKTYCZKOWSKI
a DepartmentofAnalyticalChemistryFacultyofMaterialsScienceandCeramicsAGHUniversityofScienceandTechnologyAdamaMickiewicza3030-059KrakoacutewPolandjustynalipinskaaghedupl
b DepartmentofAnalyticalChemistryFacultyofChemistryJagiellonianUniversityinKrakoacutewGronostajowa230-387KrakoacutewPoland
c Departmentof MolecularEngineeringFacultyofProcessandEnvironmentalEngineeringLodzUniversityofTechnologyWolczanska21390-924ŁoacutedźPoland
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 129
AbstractCurrentlyresearchisfocusedonthesearchfornewphysicallyandchemicallystablematerialsaswellasvolumeorsurfacemodificationOneofthemethodsusedforsurfacemodificationistheapplicationofthin layers from inorganic and organic compounds The plasmaenhancedchemicalvapordepositionisamethodthatallowsmaterialmodificationandalsodepositionofthinlayersThisworkconcernsoptimizationofcoldplasmadepositionparametersandtoachievethebestelectrical conductivitywhilemaintaining thehighmechanicalstrength of the formed layers Preliminary tests were focused onoptimizing the layering parameters such as the deposition timedischargepowerpressureofmonomerandthe flowofargonTheobtainedsamplesweresubjectedtothermaltreatmentafterwhichtheywere coveredwitha layerof aluminumThe thicknessof theobtained layers was determined on the basis of interferencemicroscopymeasurementsAsaresultoftheexperimentslayerswithathicknessof20nmto600nmwereobtainedTheconductivityofthedeposited layers was also determined and values from 003 to
ndash1150Sm wereobtained
Keywordscoldplasmadepositionelectrochemical
applicationssurfacemodificationthinlayers
Thismethodisusedtoproducecatalyticstructuresortomodifythepropertiesofmaterials eg improve hydrophobicity The growing popularity of surfacemodificationmethodsusingcoldplasmaisassociatedwiththefactthatitisanenvironmentallyfriendlyandversatilemethod[12] Workingelectrodesusedinvoltammetryareasubgroupofchemicalsensorswhich are small devices that convert real-time chemical information into ameasurableandanalyticallyusefulmeasurementsignalChemicalinformationrangingfromtheconcentrationofaspecificcomponentofthetestedsampletotheoverallcompositionofthematrixcancomefromboththeinitiatedchemicalreactionandbetheresultofphysico-chemicaltransformationstakingplaceinthetested object Chemical sensors are equipped with two basic elements iereceptor and transducer The receptor is responsible for the conversion ofchemicalinformationfromthetestedobjectintoaspecificformofenergyintheconverterthisenergyistransformedintoausefulanalyticalsignal Parameters characterizing the electrochemical sensor include accuracyprecision selectivity accuracy presentation selectivity sensitivity dynamicrange limit of quantification limit of detection lifetime response time andreliability Themost numerous and the oldest group of chemical sensors areelectrochemicalsensorsCommonlyobservedinterest inthisgroupofsensorsresultsfromthefactthatwithrelativelylowproductionandoperatingcoststheyofferthebestmetrologicalandoperationalparameters[3ndash5]Oneofthemaintrendsofmodernanalyticsisthesearchfornewelectrodematerialsandvariousgeometries of working electrodes One way to improve the performance ofworkingelectrodesistomodifytheirsurfaceforexamplebyapplyingthinlayersIn this work were considered plasma enhanced chemical vapor depositionmethod as the method of surface modification designed to performworkingelectrodeforvoltammetricdeterminationsAspartoftheinitialresearchplasmaprocessing parameters such as discharge power time of treatment andcompositionofgasmixtureinwhichplasmawasgeneratedwereoptimizedThelayers obtained in different conditions have been tested for suitability forelectrochemical applications (layer thicknessmeasurement and themeasure-mentofconductivity)
2Experimental
21Reagentsandchemicals
Theprecursor solutions suchasacrylonitriledietoxydimethylsilane trietoxy-methylsilaneandtetramethyldisiloxanewhicharesuppliedbyABCRwereusedOtherreagentsofanalyticalpuritysuchasn-hexane(SigmaAldrich)andargonwereused
130 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
22Instrumentation
The thin layers were deposited in a parallel-plate plasma reactor (frequency1356MHz)ThesamplesobtainedwerecalcinedinatunnelfurnaceunderanargonatmosphereThethicknessofthedepositedlayerswasmeasuredafterthealuminum was sputtered using a Nikon microscope type ECLIPSE LV150NElectrometerhigh resistance system (KEITHLEY) was used to measureconductivity
3Resultsanddiscussion
Eachof themonomerswasdepositedonprepared1times1corningglasssamplesSamples prepared with n-hexane were placed in a plasma reactor andadditionallypartiallycoveredwithamicroscopecoverslipSchematiclayoutofsamplesinthereactorshowninFig1ThefirststepwastoetchthesystemusingargonplasmaThisstageallowedfortheeliminationofimpuritiesthatwerenotremovedbythehelpofn-hexaneandthepreparationofthesurfaceofthesamplesforthedepositionoftheproperlayerTheproperstageistheapplicationofathinlayerwiththeplasmainducedbytheselectedprecursoracrylonitriledietoxydi-methylsilanetrietoxymethylsilaneandtetramethyldisiloxaneThethicknessandpropertiesoftheobtainedlayersdependonthedepositionparameterssuchasdischarge power time of treatment and composition of gasmixture inwhichplasmawasgenerated Table1showsallcombinationsofparameterstestedforallfourprecursorsFour different discharge powers for acrylonitrile and two different dischargepowersfororganosiliconmonomersweretestedwithtwodifferenttreatmenttimesEachtimeandpowercombinationwasperformedinplasmainducedbypuremonomerandmonomerwithargonAfterapplyingthelayersthesampleswereplacedinaquartzboatandcalcinedinatunnelfurnaceAllsampleswerecalcinedat500degCfor2hoursunderargonflowSamplesaftercalcinationwerecoveredwithalayerofaluminum
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 131
Fig 1 Scheme of sample distribution in plasma reactor(a)reactorelectrode(b)corningglasssamples(c)micro-scopecoverslip
Thethicknessoftheobtainedlayerswasmeasuredusinganinterferencemicro-scopeMeasurements were carried out at amagnification 10times recording theimageinmonochromelightTheimagewassetsothattheinterferencefringeswere perpendicular to the arc on the sample In order to calculate the layerthicknessD[nm]thefollowingformulawasused
(1)
wheredandLweredeterminedonthebasisoftheregisteredimage(disfringeshiftduetorefractionoflightontheslopeLisdistancebetweenthefringes) Thelaststageofthestudywastodeterminethecurrent-voltagecharacteristicstodeterminetheconductivityoftheobtainedlayersThesamplewasplacedinameasuring cell and attachedwith silver paste to the electrometerwires ThechangeincurrentwasrecordedwiththeapplicationofalternatingvoltageintimeBasedontheresultsobtainedthegraphsofdependenceUndashIwereobtainedfromwhichthevalueofresistance(R)wasdeterminedAnexampleofcurrent-voltagecharacteristicsisshowninFig2
132 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Table 1Conditionsforlayersdeposition-parameterwhichweretested
Monomer DischargepowerW Timeoftreatmentmin
Acrylonitrile 10204080 24Dietoxydimethylsilane 2040 255Trietoxymethylsilane 2040 255Tetramethyldisiloxane 2040 255
Fig 2Current-voltagecharacteristicsdeterminedfortheacrylonitrilelayer(depositionparametersW=10Wt=25mingasmixtureonlyacrylonitrile)
Table 4Theresultsofthicknessmeasurementsandconductivityspecifictothetrietoxymethylsilanelayers
DischargepowerW 20 20 40Timeoftreatmentmin 25 5 5Argonflowsccm 10 10 10Thicknessnm 14686 24282 35311
ndash1SpecificconductivitySm 165 93 67
Knowing the value of the resistance and the geometry of the system thespecificresistancewasdeterminedfollowedbythespecificconductivityofthesamplethefollowingequationwasused
(2)
whereρisspecificresistance[Ωm]Risresistance[Ω]bissamplelength[m]Disdepositedlayerthickness[m]andldistancebetweenelectrodes(wires)[m] Theresultsofthicknessmeasurementsandspecificconductivityarepresentedin the Tables 2ndash5 For electrochemical applications it is important that theobtainedlayerhasthehighestspecificconductivityAnalyzingthedatapresentedin Tables 2ndash5 shows that the thinnest layers have the greatest applicationpotentialastheelectrodematerial
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 133
Table 2Theresultsofthicknessmeasurementsandconductivityspecifictotheacrylonitrilelayers
DischargepowerW 40 40 80 80 10 10 20 20Timeoftreatmentmin 2 4 2 4 2 4 2 4Thicknessnm 15845 31167 250 4644 247 67 7531 12236
ndash1SpecificconductivitySm 0002 156 191 0003 1512 454 311 185
Table 3The results of thickness measurements and conductivity specific to the dietoxydimethylsilanelayers
DischargepowerW 20 20 40 40Timeoftreatmentmin 25 5 25 5Argonflowsccm 10 10 10 10Thicknessnm 13998 37519 28431 49556
ndash1SpecificconductivitySm 1640 55 105 40
Table 5The results of thickness measurements and conductivity specific to the tetramethyldisiloxanelayers
DischargepowerW 20 40 40Timeoftreatmentmin 25 25 5Argonflowsccm 10 10 10Thicknessnm 16759 3159 61652
ndash1SpecificconductivitySm 213 95 31
4Conclusions
InthisworkplasmaenhancedchemicalvapordepositionmethodwasusedforapplyinglayersoffourdifferentmaterialsacrylonitriledietoxydimethylsilanetrietoxymethylsilaneandtetramethyldisiloxaneBychangingparameterssuchasdischargepowertimeoftreatmentandcompositionofgasmixtureanumberofsampleswereobtainedwithlayersofdifferentthicknessandwhatisassociatedwithotherelectricalproperties TestsperformedaspartofthisworkwereusedtoperforminnovativeworkingelectrodesforvoltammetricdeterminationsThesurfacewasmodifiedusingthecoldplasmaofthreesubstratesgraphiteglassycarbonandgold
Acknowledgments
JLandMMhavebeenpartlysupportedbytheEUProjectPOWR030200-00-I00416
References
[1] KapicaRTyczkowskiJBalcerzakJMakowskiMSielskiJWorwaEEnhancingadhesivejointsbetweencommercialrubber(SBS)andpolyurethanebylow-pressureplasmasurfacemodificationIntJAdhesAdhes95(2019)102415
[2] TyczkowskiJKapicaRŁojewskaJThincobaltoxidefilmsforcatalysisdepositedbyplasma-enhancedmetalndashorganicchemicalvapordepositionThinSolidFilms515(2007)6590ndash6595
[3] HulanickiAGłabSIngmanFChemicalsensorsDefinitionsandclassificationPureApplChem63(1991)1247ndash1250
[4] BrzozkaZWroblewskiWSensorychemiczneWarszawaOficynaWydawniczaPolitechnikiWarszawskiej1999(InPolish)
[5] SkoogDAWestDWHollerFJCrouchSRFundamentalsofAnalyticalChemistry9thEdBostonCengageLearning2013
134 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
ConcentrationandcompositionofvolatilecompoundsorcongenersisoneofthemostimportantparametersresponsibleforqualityofproducedalcoholicdrinksandhencefortheirsensorycharacteristicsandconsumeracceptanceTodaygaschromatography (GC) is conventionally used to determine qualitative andquantitativecompositionsofvolatilecompoundswithvariousexternalandorinternalstandardcalibrationprocedures Methodemployingethanol as an internal standard (IS) forGCquantitativedeterminationofvolatilecompoundsinalcoholicbeverageshasbeensuggestedquitelongago[1]andsincethattimegreatresearchworkhasbeencarriedoutRecentlyaninterlaboratorystudyofthemethodinvolving9testinglaboratoriesfrom4countrieswascarriedout[2]TheresultsdemonstratedgreatperspectivesofldquoEthanolasISrdquomethodandproveditsreferencecharacterandeaseofroutineimplementation
Advanced GC-MS method for quality and safety control of alcoholic beverages
abANTONKORBAN
a DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova2030812840Prague2CzechRepublickarbonat7gmailcomb DepartmentofAnalyticalChemistryChemistryFacultyBelarusianStateUniversityLeningradskaya14220050MinskBelarus
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 135
AbstractRecently developed and validated simple and reliable quantitativemethod employing ethanol as an internal standard for GC-MSquantification of volatile compounds in alcoholic products wasapplied to 36 samples including commercially available world-famousbrandspirits from18countriesandhomemadedistillatesThe GC-MS analyses were performed simultaneously by thesuggested approach and official internal standard method that isprescribedinthelegislationofEUandUSATheindependentsamplest-testwasemployedtoevaluatethestatisticaldifferenceofresultsofthesetwomethodsThetestrevealednodifferenceintheresultsandtheirrepeatabilityThemainbenefitsofthesuggestedmethodaretheeliminationofthenecessityofmanualinternalstandardadditionandsamplesdensitymeasurementthusmakingitmoreeconomicalandproductive
Keywordsalcoholicbeveragesgaschromatography-
massspectrometry(GC-MS)
internalstandardmethodvolatilecompounds
quantification
AllpreviousstudieswereutilizingflameionizationdetectorssinceGC-FIDisprescribedinthelegislation[3]wheremass-spectrometrydetectorsarenotyetofficially referred However GC-MS instruments are employed in practice toqualifyandorquantifyvolatilesincommercialspiritsintraditionalhomemadealcoholicdrinksinnewlydevelopedbeveragesinspiritwastesandindistillatesobtainedwithdifferentmanufacturingprocesses Our recent researchwasdirected towardsdevelopment of an algorithmofldquoEthanolasISrdquomethodapplicationonGC-MSinstruments[4]WehaveshowedthattopreventMSdetectorfromsaturationethanolshouldberegisteredinthecorresponding SIM timewindow at characteristicmz of low abundance forinstance bymz of 47 ions This ion corresponds to non-fragmented ethanol
13moleculescontaining1heavyisotope(mainly C)FinallytheresultsofmeasuredstandardsolutionsshowedthatthesuggestedapproachisvalidandldquoEthanolasISrdquomethodmaybesuccessfullyusedonGC-MSinstrumentstoo The objective of this studywas to test and further approve the suggestedapproachonalargersetof36realsamplesofalcoholicdrinkseithercommercialorhomemadeThesamplesweresimultaneouslyanalysedbytwoGC-MSmethodsndashaclassicalISmethodprescribedinthelegislationandthesuggestedldquoEthanolasISrdquomethod
2Experimental
21Reagentsandchemicals
Thefollowingvolatilecompoundsweredeterminedintestedsamplesofalcoholicbeverages 11-diethoxyethane (acetal) acetaldehyde methyl acetate ethylacetate methanol 2 propanol 1-propanol 2 methylpropan-1-ol (isobutanol)1-butanol2-butanoland3methylbutan-1-ol(isoamylalcohol)1-pentanolwasemployedasatraditionalIScompound
22Instrumentation
ShimadzuGCMS-QP2010Ultraequippedwithaquadrupolemassspectrometrydetector was employed for GC-MS measurements Rxi-1301Sil MS capillarycolumn(60mlength025mmid025micromfilmthicknessRestek)wasusedfortheseparationofcompoundsInjectionswereperformedinasplitmode(ratio175)Helium(99999purity)wasusedasacarriergasinjectortemperaturewas170degCTheoventemperaturewasheldat30degCfor5minthenraisedto210degC
ndash1at a rate of 30 degCmin and held isothermally for 4minMeasurementswereperformedinaSIMmodeFortheanalysedcompoundsand1-pentanol2ndash3mostabundantionsinthecorrespondingMSspectrumwereselectedethanolSIMtimewindowcontainedonly47mzionsAllGC-MSmeasurementswerecarriedoutintriplicateunderrepeatabilityconditions
136 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
AnalysisofeachalcoholicsamplewasperformedinafollowingwayAliquotof09mL of a tested sample was pipetted into a standard 2mL glass vial and
ndash1weighedAfterthat01mloftheISsolution(2355mgkg of1-pentanolinWES)wasaddedtothetestedsampleandthemasswasrecordedTheobtainedmixturewasmixedthoroughlyand05microlofitwasinjectedintotheGCsystem Theoriginoftestedalcoholicbeverageswaseithercommercialorhomemade33 world-famous spirits manufactured at different parts of the world werepurchasedfromcommerciallyavailablesourcesThelistoftypesofpurchasedandanalysedspiritsincludedbourboncalvadoscognacgingrappaliquormetaxaportwine rumsake tequilavodkawhiskeyandvarious fruitdistillatesThepurchased drinks were produced at the territory of the following countriesBelarus Bermuda Cuba Czech Republic Denmark France Germany GreeceGuatemala Jamaica Japan Mexico Moldova Portugal Slovakia Trinidad andTobago UK (England and Scotland) USA Three homemade fruit distillatesproducedbyfermentationofpulpyfruitsortheirmustswereobtainedfromlocalspiritmakersThedeclaredABVvaluesofalltestedsamplesvariedfrom15to81
3Resultsanddiscussion
To fulfil themaingoalof thiswork ie toevaluate thestatisticaldifferenceofresultsyieldedbythecomparedmethodswehaveemployedStudentst-testforindependentsamplestoverifystatisticaldifferencesonthesignificancelevelofp=005Theobtainedempiricalvaluesforallpairsofcongenersrsquoconcentrationswere lower than critical one in all cases demonstrating that concentrationsobtainedbythetwomethodshavenostatisticaldifferenceandleadtothesameresults InadditionrepeatabilityofthetwomethodswascomparedthereforeallRSDvalues obtained from triplicatemeasurements were split in two groupswith
ndash1respecttothecorrespondingconcentrations(lowerthan50mgL AAandhigherndash1than50mgL AA)Theobtainedresultsarepresentedintheformofboxplotin
Fig1AnalysisofthechartinFig1showedthatboththetestedmethodshaveyieldedstatisticallysimilarrepeatability AllofthetestedalcoholicdrinkssatisfiedtherequirementsofEURegulation(EC)no1102008[5]Theconcentrationsofundesirablecompoundssuchasmethanoldidnotexceedthelevelsspecifiedinthesameregulationforcorres-ponding beverages In Table 1 the description of the used SIM method andsummaryoftheexperimentalresultsarepresented Tocomparethetruenessofthemethodsoneofthespiritsampleswasspikedwithstandardsolutions(ABV40)containingallanalysedvolatilecompoundsat
ndash1concentrationsof50500and5000mgL AATheoriginalsamplewasusedasareferenceEachofthespikedsolutionswasmeasuredintriplicateSelectedspirit(cherry distillate) initially contained all 11 volatile compounds in various
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 137
138 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1BoxchartsofRSDsofusedISmethodsat2concentrationrangesMeanisequaltoarithmeticmeanoraverageInterquartileRange(IQR)meansisthedistancebetweentheupper(themedianoftheupperhalfofthedataset)andlower(themedianofthelowerhalfofthedataset)quartile
ndash1Compound Timemin Registeredmz Numberof ConcentrationmgL AA results Minimal Maximal
Acetaldehyde 0ndash42 314344 36 24 715Methanol 36 13 13600
Ethanol(IS) 42ndash48 47 mdash mdash mdash
2-Propanol 48ndash70 29314345 14 27 199Methylacetate 596174 10 34 3201-Propanol 26 361 12070Ethylacetate 27 166 107002-Butanol 11 18 2080
Isobutanol 70ndash200 3141ndash4345 28 19 20001-Butanol 5556 13 28 155Acetal 26 45 270Isoamylol 31 39 26501-Pentanol(IS) mdash mdash mdash
Table 1DescriptionoftheusedSIMmethodandsomestatisticsconcerningallmeasured36spiritsamplesbothpurchasedandhomemade
concentrationsTheobtainedrecoveriesboxchartsareshowninFig2Compa-risonoftherecoveriesobtainedwithtwomethodsindicatesthattheyhavenosignificantdifferenceintermsoftruenessAverageobtainedrecoverywas981whenusingsuggestedmethodand980whenusingtraditionalISmethod
4Conclusions
InthisworktheresultsoftestingtheadvantageousldquoEthanolasISrdquomethodfortheGC-MS quality control analysis of alcoholic beverages were presented33purchasedsamplesofworld-famousalcoholicbeveragesoriginatingfrom18countriesand3homemadefruitdistillateswereanalysedtomakeathoroughandcomprehensive studyof thedevelopedmethodTheconcentrationsofvolatile
ndash1compoundsinanalysedsamplesvariedfrom1to13500mgL AAtheABVvalueofanalysedsamplesvariedfrom15to81ThesuggestedmethodwascomparedwiththetraditionalISmethodthatiscurrentlystatedinlegislationTheindepen-dentsamplest-testrevealedthatwithaprobabilityof095resultsobtainedwithtwo methods do not differ significantly The results of within-run precision(repeatability)showedrelativestandarddeviationswithin3measurementstobelessthat6indicatingthatthetechniqueisreproducibleThetruenessofthemethodwasevaluatedbyrecoverycalculationAccordingtotheobtainedresultsrecoveryofthesuggestedmethod(981plusmn33)wasslightlybetterthanthatofthetraditionalone(980plusmn58) ThesefactsprovethatdevelopedldquoEthanolasISrdquomethodistruepreciseandreliable when employed on GC-MS instruments At the same time to obtain
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 139
Fig 2Boxchartsof recoveriesof thesuggested(dottedpattern)and traditional (brickpattern)ISmethodsatdifferentspikeconcentrationsSymbolsdefinitionsarethesameasinFigure1
concentrationsofvolatilecompoundsintheofficiallyrequiredunitsofmeasurendash1 ndash1(mgL AAgL AAetc)suggestedmethodrequiresnodensitometrymeasure-
mentsofthetestedsampleandnoadditionofIScompoundoranyothersamplepre-treatmentThismethodprovidesaninvaluableanalyticaltoolforthequalitycontrolofalcoholicproductsandshouldbeusedinroutineanalysis
Acknowledgments
ThisworkwasfinanciallysupportedbytheVisegradFund
References
[1] CherepitsaSVBychkovSMKovalenkoANMazanikALSeleminaNMSeredinskayaOBThe use of themajor component (solvent) as an internal standard in the gas-chromato-graphicdeterminationofimpuritiesJAnalChem58(2003)368ndash371
[2] CharapitsaSSytovaSKorbanASobolenkoLEgorovVLeschevSZakharovMCabalaRBusarovaRShestakovichITolstouhovaAOndrousekSVavraJYilmaztekinMCabarogluTInterlaboratorystudyofethanolusageasaninternalstandardindirectdeterminationofvolatile compounds in alcoholic products BIO Web Conf 15 (2019) 02030httpsdoiorg101051bioconf20191502030
[3] CommissionRegulation(EC)No28702000layingdownCommunityreferencemethodsfortheanalysisofspiritsdrinkshttpdataeuropaeuelireg20002870oj
[4] KorbanACharapitsaSCabalaRSobolenkoLSytovaSTheperspectivesofethanolusageasaninternalstandardforthequantificationofvolatilecompoundsinalcoholicproductsbyGC-MSJMassSpectr55(2020)e4493
[5] EuropeanUnion(2008)Regulation(EC)No1102008oftheEuropeanParliamentandoftheCouncilof15January2008ontheDefinitionDescriptionPresentationLabellingandtheProtectionofGeographical IndicationsofSpiritDrinksandRepealingCouncilRegulationhttpdataeuropaeuelireg2008110(1)oj
140 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Carbonorgraphitefeltsareusedaselectrodematerialsincethe1990sandtheirutilizationstillgrowsinanalyticalelectrochemistryaswellasinotherareasThisisduetotheirsuitablepropertiesfromwhichwecannamehighporosityhighspecificsurfaceareagoodelectricconductivityandhighphysicalandchemicalstabilityThefirsttwoparametersaregivenbystructureoffeltwhichconsistsoforderlesscarbonfibreswithabouttentotwentymicrometersindiameter[1]theothersby theadvantageouselectricalpropertiesof carbon fibreOn theotherhand porous flow-through electrodes including carbon felt electrode havedisadvantageinapotentialdropintheelectrodevolumewhichcausesdifficultcontrollingoftheexactpotentialappliedontheelectrodeandthereforeresultsindifferentcurrentefficienciesontheoppositesidesoftheelectrode[2] Carbon felt electrode can be utilized for detection of structurally differentcompoundsatvariousconditionsForexampleoperatingatreductionpotentialofndash08V[3]oxidationatrelativelyhighpotential+15V[4]ormeasuringatlowconcentrationsofelectrolyte[5]canbenamedDevelopedtechniquesalsoshowsthatcarbonfeltcanbeusedfordeterminationatsubmicromolarconcentrationsThis ismainly due to its ability to operate as a high-efficiency amperometricdetector Themainaimofthispaperistooverviewandcompareparametersofseveraldetermination methods of different analytes using carbon felt detector in
Utilization of a carbon felt as a material for working electrodes
MARTINBAROCHHANADEJMKOVA SA RKASLA DKOVA
DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublicmartinbarochnaturcunicz
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 141
AbstractWorkingelectrodemadeofcarbonfeltwasusedincombinationwithHPLC for verificationofpractical applicabilityof the electrodeAlldeveloped methods confirm advantageous physical and chemicalpropertiesofcarbonfeltForelectrochemicalutilizationitispossibleto operate at higher positive potentials and even in low concen-trationsofelectrolyteinmobilephaseObtainedlimitsofdetectionwere mostly in submicromolar range and standard deviations ofmeasurementrepeatabilitywereunder5
KeywordsamperometrycarbonfeltFIAHPLC
combinationwithHPLC(forexampletheirdetectionpotentialslimitsofdetectionorlinearrange)
2Experimental
21Reagentsandchemicals
Stock solutions of propyl gallate butylhroxyanisole tert-butylhydroquinonebutylhydroxytoluene chlortoluron 2-amino-4-nitrophenol and 4-amino-2-
ndash3 ndash1nitrophenol (all SigmandashAldrich) with concentration of 1times10 molL werepreparedbydissolving theappropriateamountof therespectivesubstance inmethanol(HPLCgradeLach-NerCzechRepublic)Stocksolutionsof indole-3-aceticacidandindole-3-butyricacidwerepreparedinthesamemannerbutindeionized water Mobile phase consisted of methanol and phosphate-acetatebufferpreparedfromphosphoricandaceticacid(bothLach-NerCzechRepublic)andsodiumhydroxide(Fluka)
22Instrumentation
Theelectrochemicalcellconsistedofcarbonfelt(KarbotechnikCzechRepublic)flow-throughelectrodewhichwasplacedincapwithplatinumwireelectricalcontactanddrilledoutletholeononesideandwithflatferrulawithcapillaryonthe other side Schematic picture of the assembly is shown in ref [5] Otherelectrodeswereauxiliaryplatinumwireelectrodeandreferencesilverchloride(3MKCl) electrode (bothMonokrystaly Turnov Czech Republic) Potentiostatused in combination with this cell was Amperometric Detector ADLC 2(LaboratonıprıstrojePrahaCzechRepublic) HPLCapparatusconsistedofBeta10gradientpump(ECOMCzechRepublic)degasser DG 4014 (ECOM Czech Republic) six-way valve with 20microl loop(Rheodyne USA) HPLC column used for separation of propyl gallatebutylhydroxytoluenetert-butylhydroquinoneandbutylhroxyanisolefromtheirmixtureandforindole-3-aceticacidandindole-3-butyricacidfromtheirmixture
regwasLichrospher RP-18(125times4mm5micromMerckGermany)ForseparationofregchlortoluroncolumnPurospher RP-18(125times4mm5micromMerckGermany)was
used In case of mixture 2-amino-4-nitrophenol and 4-amino-2-nitrophenolregcolumnGemini C18110A (150times46mm5micromPhenomenexUSA)wasusedfor
separation Measurements of pHwere carriedout at Conductivity andpH-meter3510usingcombinedglasselectrode(JenwayUK)
142 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
3Resultsanddiscussion
Performanceofcarbonfeltwastestedonseveraltypesofanalyteswhichneededdifferent separation conditions namely amount of organic solvent in mobilephase and buffer pH The lowest amount of methanol (30) was used forseparation of 2-amino-4-nitrophenol and 4-amino-2-nitrophenol [3] Higherconcentrations of methanol in mobile phase was used for determination ofchlortoluronandforseparationofindole-3-aceticacidandindole-3-butyricacid[4]namely40and60respectivelyThehighestconcentrationsofmethanoland therefore electrolyte with lowest conductivity was used in separation ofantioxidantsnamelypropylgallatebutylhroxyanisolebutylhydroxytolueneandtert-butylhydroquinonewhereamountofmethanolwasrampingfrom55to95[5]DetectionpotentialsofmentionedanalytesandtheirlimitsofdetectionsareshowninTable1 HPLCseparationofantioxidantswas the firstmethodchosen for testingofcarbonfeltelectrodeperformancewiththistechniqueDuetodifferencesintheirstructure when butylhydroxytoluene has a different oxidation mechanismdetectionwith two appliedpotentialswas necessary According to the hydro-dynamic voltammograms potentials 14V and 08Vwere chosen for determi-nationofbutylhydroxytolueneandfortheotherthreeanalytesrespectively[5]AsshowninTable1whenthehigherpotentialwasappliedlimitsofdetectionforpropyl gallate butylhroxyanisole and tert-butylhydroquinone had increasedContrary determination of butylhydroxytoluene had approximately six timeslowerdetectionlimitathigherpotential
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 143
ndash1Compound E VLODmicromolL Refdet
Propylgallate 080 088 [5] 140 186 [5]Butylhroxyanisole 080 144 [5] 140 348 [5]tert-Butylhydroquinone 080 121 [5] 140 266 [5]Butylhydroxytoluene 080 3128 [5] 140 463 [5]Chlortoluron 140 013Indole-3-aceticacid 150 033 [4]Indole-3-butyricacid 150 054 [4]4-Amino-2-nitrophenol 080 016 [3]2-Amino-4-nitrophenol 080 021 [3]4-Amino-2-nitrophenol ndash080 35 [3]2-Amino-4-nitrophenol ndash080 37 [3]
Table 1Parametersofdetectionpotentialsandlimitsofdetectionfordifferentcompoundsusingcarbonfeltdetector
Detectionofauxins(indole-3-aceticacidandindole-3-butyricacid)wascarriedout at potential 15VMeasurements at this potential gives repeatabilitywithstandarddeviation31forindole-3-aceticacidand25forindole-3-butyricacidevenwithexchangingoftheworkingelectrodematerialCalibrationcurves
ndash1forbothanalyteswereobservedfrom04to100μmolL withlinearityinwholeconcentrationrangeLimitsofdetectionforbothanalytesreachedsubmicromolarconcentrationsevenwithrelativelyhighpotential[4] Incaseofdeterminationof2-amino-4-nitrophenoland4-amino-2-nitrophenolcarbon felt electrodewasused inbothoxidationand reductionmodeHydro-dynamicvoltammogramsshowedthatoptimaldetectionpotentialinreductionmodewasndash08VThislowpotentialisclosetotheendofthepotentialwindowandthereforeinterferenceswithremnantsofdissolvedoxygeninmobilephasewereobservedTheseinterferencesresultedinapproximately20timeshigherlimitofdetection for 2-amino-4-nitrophenol or 4-amino-2-nitrophenol obtained inreductionthaninoxidationmodeOntheotherhandmaximumvaluesoflinearrangewerethesameforbothanalytesinbothdetectionmodes[3] ForHPLCofchlortolurontheoptimaldetectionpotentialof14VwasfoundIts
ndash1calibration dependence although observed from 025 to 1000μmolL was ndash1linearonly in therange from025to50μmolL Limitofdetectionbasedon
ndash1standardsolutionswas013μmolL andreproducibilityofmeasurementgivenbytwentyconsecutivemeasurementsgaverelativestandarddeviationof05 ForallthedeterminationmethodsattentionwaspaidtotheapplicabilityofthecarbonfeltindetectionofanalytesincomplexmatricesIncaseofantioxidantsedibleoilswerechosenasrealsamples[5]nitrophenolderivatesweredeter-minedinurinesamples[3]andauxinsinrootingpreparation[4]ChlortolurondeterminationwasperformedinsoilandsurfacewaterThefoundvaluesshowanegligiblematrixinfluenceondetection
4Conclusions
CarbonfeltworkingelectrodewassuccessfullyusedincombinationwithHPLCfordeterminationofdifferenttypesofelectroactivecompoundsegantioxidantsauxinsorpesticidesAllmentionedapplicationsshowgreatperformanceofthecarbonfeltasaflow-throughelectrodematerialinelectroanalyticalchemistryforoxidationandreductionwayofanalytesdeterminationLimitsofdetectionforanalytes are mostly in submicromolar concentrations the exceptions areoxidationof analytes athigherpotentials and their reductionwhere limitsofdetectionsareinmicromolarconcentrationsApplicabilityoftheelectrodeonrealmatriceswas proven on analysis of edible oil samples groundwater soil androotingpreparation
Acknowledgments
ThisworkhasbeensupportedbytheCzechScienceFoundation(projectGACR20-01589S)
144 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
References
[1] Gonzalez-Garcia J Bonete P Exposito E Montiel V Aldaz ATorregrosa-Macia RCharacterizationofacarbonfeltelectrodeStructuralandphysicalpropertiesJMaterChem9(1999)419ndash426
[2] NavaJLRecendizAGonzalezLGCarrenoGMartın ezFMassTransportandpotentialstudiesinaflow-throughporouselectrodereactorPortugalElectrochimActa27(2009)381ndash396
[3] Dejmkova H Knaf M Application of carbon felt detector for the determination ofdinitrophenolmetabolitesInXXXIXModernElectrochemicalMethodsFojtaMSchwarzovaKNavratilT(Eds)U stınadLabemBestServis2019p41ndash43
[4] DejmkovaHdeAraujoDanielMElectrochemicaldeterminationofindole-3-aceticacidandindole-3-butyric acid using hplc with carbon felt detectorMonatsh Chem150 (2019)439ndash442
[5] DejmkovaHBarochMKrejcovaMBarekJZimaJCoulometricdetectorbasedoncarbonfeltApplMaterToday9(2017)482ndash486
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 145
1Introduction
Smith-Lemli-Opitzsyndrome(SLOS)isan autosomal recessive genetic disor-der firstly described in 1964 [1] It iscaused by the inborn deficiency of7-dehydrocholesterol reductase Thisenzyme transforms 7-dehydrocholes-terol (7-DHC Fig 1) to cholesterolduringthefinalstepofbiosynthesisofcholesterolincells The clinical symptoms of SLOS aredecreasedbloodlevelofcholesteroland
Electroanalytical methods for determination of 7-dehydrocholesterol in artificial serum
LENKABENESOVAADE LAZA RYBNICKA JANKLOUDAKAROLINASCHWARZOVA -PECKOVA
UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublicbenesolenaturcunicz
Abstract7-DehydrocholesterolisabiomarkerofSmith-Lemli-Opitzsyndromeanautosomalrecessivegeneticdisordercausedbytheinborndefici-encyof7-dehydrocholesterolreductaseInthisstudyproceduresforitsdeterminationinartificialserumusingflowinjectionanalysiswithelectrochemical detection and voltammetric detection on borondoped diamond electrode were optimized The proteins wereprecipitatedbyacetonitrileandaftercentrifugationthesupernatantused for analysis For quantitation of 7-DHC by differential pulsevoltammetrytheoptimalratioacetonitrile-artificialserum91(vv)wasappliedInFIA-EDtheratio31(vv)runelectrolyteconsisting
minus1ofwater-acetonitrilecontaining001molL NaClO inthesameratio4minus1anddetectionpotentialof+13VvsAgAgCl(3molL KCl)were
usedQuantitationof7-DHCwaspossibleusingcalibrationdepen-minus1dencewithlimitdetectionof20micromolL inartificialserumNeverthe-
lessthemethodhaslowrecoveryandforsensitivedeterminationinreal matrices of human serum and amniotic fluid a liquid-liquidextraction needs to be applied to prevent presence of 7-dehydro-cholesterolinthephasewithprecipitatedproteins
Keywordsamperometricdetectionborondopeddiamond
electrode7-dehydrocholesteroldifferentialpulse
voltammetryflowinjectionanalysisSmith-Lemli-Opitz
syndrome
146 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Structureof7-dehydrocholesterol
increased concentrationof7-DHC inbloodandnervous system [2] SLOS is acomplexofmultipleanomaliesincludingmentalretardationItismanifestedbyholoprosencephaly(anomaliesinbraindevelopmentwithimprecisedivisionintotherightandlefthemispheres)milddysmorphismscardiacrenalandgastro-intestinalmalformations ThecharacteristicfacialanomaliesofSLOS[2]aremicrocephalybitemporalnarrowing ptosis short nasal root short nose with anteverted nares andmicrognathia epicanthal folds and capillary hemangioma over the nasal rootextendingontotheglabellatheearappearlow-setandareposteriorlyrotatedOral finding includes a high-arched and narrow hard plate broad and ridgealveoralridgesandredundancyofsublinqualtissuesCNSanomaliesareagenesisorhypoplasiaBilateralandunilateralpostaxialpolydactylycanbepresentedinthehandsorfeetorboth Concentrationof7-DHCinbloodiscrucialforclinicaldiagnosticofSLOSinpatients Concentration levels in amniotic fluid are used for fetal diagnosticsTable1 summarizes concentration of 7-DHC in plasma and amniotic fluid ofhealthypersonandSLOSpatientsAnalyticalmethodsusedfordeterminationofconcentrationof7-DHCinthesematricesincludecombinationofGCorHPLCwithMS[3]orGCwithflameionizationdetection(FID)orUVdetection[45] Thepossibilitiesofelectrochemicalmethodsfordetectionof7-DHCarelimitedasgenerallythesteroidcoreisratherredox-inactive(detailinreview[6])undervarietyofconditionsNevertheless7-DHCpossessesconjugateddoublebondsonsteroidcoreanditsoxidationwasreportedinseveralstudies[7ndash9]Itsvoltam-metricsignal+095VvsSCEonglassycarbonelectrodewasfirstlyobservedin
minus1non-aqueousmediaofmethanol-benzene7525(vv)using005molL LiClO as4
supporting electrolyte in a study dealing with electrochemical behaviour ofvitaminAandDandtheirprovitaminsD(7-DHCisprecursorofvitaminD3)[7]Determinationof7-DHCinhumanskinispossiblebyHPLCwithUV(λ=286nm)andamperometricdetection(E +17VvsAgAgCl)onglassycarbonelectrodedet
minus1usingmethanol-tetrahydrofuran175mmolL KH PO (9514vvv)asmobile2 4minus1phase7-DHCwasdetectedintherangefrom12to81microgg dryweightwith
minus1detectionlimitof39pmolL [8]Anotherstudy[9]isdevotedtodeterminationof
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 147
minus1Author(s)ref Matrix ConcentrationμmolL Healthy SLOS
Kelley[4] Plasma 03plusmn001 385plusmn309 Amnioticfluid lt02plusmn001 16plusmn9Rossiteretal[5] Plasma lt5 179ndash335 Amnioticfluid lt03 12ndash15
Table 1Concentrationof7-dehydrocholesterolinclinicalmatricesforhealthypersonsandforSmith-Lemli-Opitzsyndrome(SLOS)patientsdetectedbyGC-FID
7-DHCandvitaminD3 in fishusingHPLCwithelectrochemicaldetectionTheanalytical cell was a serial combination of two-flow-through porous graphiteworking electrodes The first standard coulometric electrode was used toeliminatepotentially interfering compounds using the second lineardynamic
minus1rangefrom0013to0312micromolL for7-DHCwasachieved Hereinwestudiedpossibilitiestodetect7-DHCbasedonitsoxidationonborondoped diamond (BDD) electrode using differential pulse voltammetry andelectrochemicaldetectioninflowinjectionanalysis(FIA-ED)inartificialserumandperipherallyinhumanserumandamnioticfluid
2Experimental
21Reagentsandchemicals
7-dehydrocholesterol (purity95)wasobtainedSigmaAldrich (USA)and itsstandard solution was prepared in acetonitrile (Honeywell Germany) Theartificial serum was prepared from KCl (Penta Czech Republic) CaCl 2H O2 2
(PentaCzechRepublic)NaCl(PentaPragueCzechRepublic)urineD-glucoseand01albuminfromSigma-Aldrich(USA)NaClO4(PentaCzechRepublic)wasusedassupportingelectrolyte
22Instrumentation
VoltammetricmeasurementweregovernedbythepotentiostatPalmSensusingworking BDD electrode (Windsor Scientific UK d = 31 mm) AgAgNO 3
minus1 minus1(01molL AgNO 1molL NaClO in acetonitrile) non-aqueous reference3 4electrodeandplatinumwirecounterelectrodeBDDsurfacewaspolishedbeforeeachscanusingsuspensionofAl O (ElektrochemickedetektoryTurnovCzech2 3
Republic) HPLC system (Hitachi Merck) consisting of control unit D-7000gradientpumpL-7100autosamplerL-7200andUVdetectorL-7400wasusedfor
minus1FIA-EDdetectionof7-DHCRunelectrolytewascomposedof001molL NaClO 4inacetonitrileanddeionisedwaterinratio31(vv)Flowrateofmobilephase
minus1was30mlmin injectionvolumewas40microLandλ=280nmwasusedforUVdetectionWall-jet detection cell was employedwith working BDD electrode
minus1AgAgCl (3molL KCl) reference electrode and platinum wire auxiliaryelectrode Optimal detection potential of +13 V was controlled using ADLC2potentiostat(Laboratornı prıstrojePragueCzechrepublic)
3Resultsanddiscussion
In this study electroanalytical methods were developed for determination of7-DHCinartificialserumnamelyFIA-EDandDPVBothmethodsarebasedondirectoxidationof7-DHConborondopeddiamondelectroderesultinginanodic
148 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
peak at ca +08 V (vs AgAgNO in acetonitrile) in non-aqueous medium of3
acetonitrile or mixedmedium acetonitrile-water using NaClO as supporting4
electrolyteTheoxidationispresumablyinitiatedbyoneelectronremovalfromtheconjugateddoublebondsonthesteroidcoreof7-DHC Fordeterminationof7-DHCinrealmatricesitisnecessarytoremovepresentproteinsArtificialserumcontainingalbuminwasusedasmodelmatrixtostudythe possibilities Firstly albumin was removed simply by precipitation withacetonitrile(serum-water13(vv))andthesupernatantwasanalysed Differentratioswateracetonitrileweretestedinrunelectrolyte(59510902080 2575 3070 4060 and 5050 (vv)) to evaluate the influence of itscompositionontheFIA-EDsignaloftheblankand7-DHCThesameratio13asusedforprecipitationofalbuminwaschosenasoptimalbecauseofminimalandstablesignaloftheblankinjectedinFIA-EDsystemFurtherdetectionpotentialE in the range from +10 V to +15 V was optimized by evaluation of thedet
hydrodynamicvoltammogramsresultinginE of+13Vsetasoptimalvaluedetminus1 Concentrationdependenceof7-DHCislinearintherangefrom25micromolL to
minus1 minus1300micromolL (concentrationinartificialserum)withdetectionlimitof20micromolL and this concentration dependence can be used for quantitation of 7-DHC inartificial serum Nevertheless determination of 7-DHC in human serum andamnioticserumfailedastheyrepresentmorecomplicatedmatricesand7-DHCispresumablypartiallyadsorbedinthepresentproteinsandcannotbequantifiedinthesupernatant Furtherdifferentialpulsevoltammetrywithoptimizedparameterswasusedfordeterminationof7-DHCInthepresenceofproteinsinartificialserum(human
minus1serum albumin) an unacceptably high detection limit of 178micromolL wasachieved When the proteins were precipitated using acetonitrile (aceto-nitrileartificialserumratio91(vv))thelimitofdetectionof7-DHCwaslowered
minus1to15micromolL inartificialserumNeverthelesstherecoveryofthemethodwasonly43to70dependingontheconcentrationof7-DHCagainreflectingthelossof7-DHCduetoproteinprecipitation Thereforeasecondapproachofsamplepretreatmentbasedonliquid-liquidextractionofalllipidsdescribedin[10]wastested(Bligh-Dyerextraction)TheprocedurehastwopartsFirstlymethanolchloroformandthesampleofartificialserum is mixed and shaken to form a monophasic system After addition ofchloroform and water a biphasic system is formed where chloroform phasecontainsalloflipidcompoundsandmethanol-waterphasecontainsallnon-lipidscompoundsChloroformphaseisthendriedunderN2atmosphereat50degCanddried extract dissolved in acetonitrile Preliminary experiments using DPVresultedinrecoveryof97forBligh-Dyerextractionof7-DHCfrominartificialserum
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 149
4Conclusions
FIA-EDandDPVwereoptimizedfordeterminationof7-DHCinartificialserumUsing precipitation of proteins by acetonitrile limit of detection of 7-DHC in
minus1artificialserumusingFIA-EDwas20micromolL andthismethodcanbeusedfortheirquantificationusingcalibrationdependenceNeverthelessdeterminationusingDPVisunreliableduetolowrecoveryoftheprocedureDevelopmentofamethodincludingliquid-liquidextractionstepisinprogresssothat7-DHCcouldbedeterminedinrealmatricesashumanserumandamnioticfluid
Acknowledgments
TheresearchwassupportedbytheCzechScienceFoundation(projectGACR19-11268S)andtheSpecificUniversityResearch(SVV260560)
References
[1] Smith DW Lemli L Opitz JM A newly recognized syndrome of multiple congenitalanomaliesJPediatr64(1964)210minus217
[2] Nowaczyk M Waye J The SmithndashLemlindashOpitz syndrome a novel metabolic way ofunderstandingdevelopmentalbiologyembryogenesisanddysmorphologyClinGenet59(2001)375minus386
[3] BeckerSRohnikeSEmptingSHaasDMohnikeKBebloSMutzeUHusainRAThieryJCeglarekULC-MSMS-basedquantificationofcholesterolandrelatedmetabolitesindriedblood for the screening of inborn errors of sterolmetabolismAnal Bioanal Chem407(2015)5227minus5233
[4] Kelley RI Diagnosis of Smith-Lemli-Opitz syndrome by gas-chromatography mass-spectrometryof7-dehydrocholesterolinplasmaamniotic-fluidandculturedskinfibroblastsClinChimActa236(1995)45minus58
[5] Rossiter JP Hofman KJ Kelley RI Smith-Lemli-Opitz SyndromePrenatal-diagnosis byquantification of cholesterol precursors in amniotic-fluid Am J Med Genet 56 (1995)272minus275
[6] KloudaJBarekJNesmerakKSchwarzova-PeckovaKNon-enzymaticelectrochemistryincharacterization and analysis of steroid compounds Crit Rev Anal Chem 47 (2017)384minus404
[7] AtumaSSLundstromKLindquistJTheelectrochemicaldeterminationofvitaminAPartIIFurthervoltammetricdeterminationofvitaminAandinitialworkonthedeterminationofvitaminDinthepresenceofvitaminAAnalyst100(1975)827minus834
[8] MoodyJPHumphriesCAAllanSMPatersonCRDeterminationof7-dehydrocholesterolinhumanskinbyhigh-performance liquid-chromatography JChromatogrB530 (1990)19minus27
[9] OstermeyerUSchmidtTVitaminDandprovitaminDinfishEurFoodResTechnol222(2005)403minus413
[10] BlighEGDyerWJArapidmethodoftotallipidextractionandpurificationCanJBiochemPhysiol37(1959)911minus917
150 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Author Index
AlikovaV1
AugustınM83
BaluchovaS19
BarekJ192570
BarochM141
BasB13129
BastryginaO41
BenesovaL146
BessonovaE57
BohmD6
BurkinK116
BurkinM116
ChernovaA141
ChoinskaM70
CokrtovaK104
DedinaJ97
DeevV57
DejmkovaH141
DendisovaM63
DubenskaL51
EfremenkoE41
FojtaM110
GalvidisI116
HavranL110
HeiglN31
HertJ76
HrdlickaV70
JosypcukB25
KartsovaLA3557
KloudaJ19146
Kodrık ovaB90
KolobovaEA35
KorbanA135
KorotkovaE1
KralM63
KratzerJ90
KravchenkoAV35
KrızekT76104
LipinskaJ129
MadejM129
MatejkaP63
MatysikF-M631
MusilS9097123
NavratilT70
OndrackovaA110
PietrzakK45
PlotnikovaK51
PoradaR13
RedondoBR70
SagapovaL90
ShormanovV1
Schwarzova-PeckovaK19110146
SladkovaS 141
S tadlerovaB97
StiborovaM110
SvobodaM90
TvorynskaS25
TyczkowskiJ129
VyhnanovskyJ97123
VymyslickyF76
VyskocilV83
WardakC45
WongDKY19
ZarybnickaA146
ZelenyI51
Proceedingsofthe16thISCModernAnalyticalChemistry Prague2020 151
152 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Keyword Index
alcoholicbeverages135
aminoglycosides116
amperometricdetection146
amperometry141
antifoulingelectrodes19
assembledcapillaries6
atomicabsorptionspectrometry90
atomicfluorescencespectro-
metry97
atomization90
biologicalactiveanalytes35
biosensor2583
bismuth97
borondopeddiamond
electrode110146
cadmium90
canagliflozin76
capillarycoating35
capillaryelectrophoresis635
capillaryflowinjectionanalysis631
carbohydrates31
carbonfelt141
cathodicstrippingvoltammetry70
chemicalvaporgeneration90
chemometrics57
cobalt123
coldplasmadeposition129
copper(II)phthalocyanine63
covalentimmobilization25
cytochromeP450110
damage83
7-dehydrocholesterol146
designofexperiments76
differentialpulsevoltammetry146
diphenylsilanereductionmethod19
dispersiveliquid-liquid
microextraction57
disposableelectrodes31
DNA83
dualdetectionconcept6
electrochemicalanalysis110
electrochemicalapplications129
electrochemicalflowcell76
electrochemistry51
electrokineticchromatography104
eliminationvoltammetrywith
linearscan70
ELISA116
enzymaticreactor25
FIA141146
gaschromatography-mass
spectrometry(GC-MS)135
glucoseoxidase25
graphite83
honey116
HPLC76141
hydridegeneration97
hydrogenatedconical-tipcarbon
electrodes19
imidazoliumionicliquids35
inductivelycoupledplasmamass
spectrometry123
internalstandardmethod135
ion-selectiveelectrode45
laccase25
liquidcrystals104
massspectrometry31
mercuryelectrode13
metronidazole51
non-aqueouscapillary
electrophoresis104
non-aqueoussystem6
oxidation76
oxytetracyclinehydrochloride51
phenol2-methoxy1
photochemicalvaporgene-
ration97123
polarography51
1-propanesulfonicacid23-
dimercapto-70
pulsedamperometricdetection31
quantitation1
resonanceRamanspectroscopy63
scanningtunnellingmicroscopy63
silversolidamalgamelectrode70
Smith-Lemli-Opitzsyndrome146
smokingmixtures41
solidcontact45
solid-phasemicroextraction57
spectrophotometry141
SudanI110
surfacemodification129
surface-enhancedRaman
spectroscopy63
thinlayers129
tip-enhancedRamanspectroscopy
63
unithiol70
uranyl45
vanillin41
veterinarydrug51
vitamins13
volatilecompoundsquantifi-
cation135
voltammetricdopaminedetec-
tion19
voltammetry1383
Proceedingsofthe16thISCModernAnalyticalChemistry Prague2020 153
Proceedings of the 16th International Students Conference ldquoModern Analytical Chemistryrdquo
EditedbyKarelNesmerak
PublishedbyCharlesUniversityFacultyofScience
Prague2020
1steditionndashvi154pages
ISBN978-80-7444-079-3
ISBN 978-80-7444-079-3
Pro
ceedin
gs of th
e 16
th In
ternatio
nal Stu
den
ts Co
nferen
ce ldquoMo
dern
An
alytical Ch
emistryrdquo P
rague 2
02
0
788074 440793
Prague 17mdash18 September 2020
Edited by Karel Nesměraacutek
Prague 2020
Proceedings of the
16th International Students Conference
ldquoModern Analytical Chemistryrdquo

Preface
Despitethefactthattheyear2020ismarkedbyCOVID-19morethan40young
analytical chemists gathered in Prague for the 16th annual international
conferenceldquoModernAnalyticalChemistryrdquoTheymeettopresenttheresultsof
theirresearchtomastertheirpresentationandlanguageskillsandtoexchange
anddiscussideasandexperiencesofanalyticalchemistry
Thisvolumeofconferenceproceedingsbringsyouatotalof25papersfrom
thisconferenceAsinpreviousyearsthecontributionspresentedareassortedby
the sequence of their delivery supplemented by indexes at the end of the
proceedingsallowingeasynavigationthroughthepagesYouwillseethattopics
of contributions cover all the aspects of modern analytical chemistry from
theoretical problems through development of new analytical methods and
improvementofanalyticaltechniquestotheapplicationsinvolvingthesolutionof
medicinaltechnicalorenvironmentalproblemsLetushopethatlikeprocee-
dingsofpreviousyearsofourconference thisonewillalsobean interesting
beneficialandenjoyablereading
Itseemstousthattheauthorsofthecontributionsareaguaranteeofthatanew
generationofanalyticalchemistswillprotectbrightandthrillingfutureofour
science
We are very grateful to the Division of
AnalyticalChemistryofEuChemSforitslong-
lasting auspices of our conference Also we
arethankfultooursponsorsnotonlyfortheir
kind sponsorship making the conference
possiblebutalsoforalltheircooperationand
supportinmanyofourotheractivities
Enjoyreadingtheseproceedings
docRNDrKarelNesmerakPhD
editor
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 iii
Sponsors
The organizersof16th International Students Conference ldquoModernAnalytical
Chemistryrdquo gratefully acknowledge the generous sponsorship of following
companies
wwwecomsrocom
wwwlach-nercom
iv Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
wwwthermofishercz
www2thetacz
wwwzentivacz wwwquintacz
wwwshimadzueucom
wwwwaterscom
Contents
AlikovaVChernovaAShormanovVKorotkovaEDeterminationof2-methoxyphenol inmodelsolutionsbyspectrophotometry 1
BohmDMatysikF-MTheeffectsoflinearlyassembledcapillarieswithvariousinnerdiametersoncapillaryelectrophoresis 6
PoradaRBasBVoltammetricdeterminationofvitamins 13Baluchova S Klouda J Barek J Schwarzova-Peckova K Wong DKY Dopamine detection at
antifoulingconical-tipcarbonelectrodes 19Tvorynska SBarek J JosypcukBA comparative study of covalentglucose oxidaseand laccase
immobilizationtechniquesatpowderedsupportsforbiosensorsfabrication 25Heigl N Matysik F-M Capillary flow injection analysis with electrochemical detection for
carbohydrateanalysis 31KravchenkoAVKolobovaEAKartsovaLAApplicationofcovalentcoatingsbasedonimidazolium
cations for separationandon-linepreconcentrationof basicandneutralanalytes in capillaryelectrophoresis35
Efremenko E Chernova A Bastrygina O Determination of vanillin in smoking mixtures byspectrophotometry41
PietrzakKWardakCUranylion-selectiveelectrodewithsolidcontact 45Plotnikova K Dubenska L Zeleny I Polarographic determination of metronidazole and
oxytetracyclinehydrochlorideinveterinarydrugforhoneybees 51Deev V Bessonova E Kartsova L Application of microextraction techniques combined with
chromatographicmethodsfortheanalysisofcomplexobjects 57KralMDendisovaMMatejkaPThedevelopmentofreferenceprobesystemfortip-enhancedRaman
spectroscopy 63ChoinskaMHrdlickaVRedondoBRBarekJNavratilTDeterminationofheavymetalpoisoning
antidote23-dimercapto-1-propanesulfonicacidusingsilversolidamalgamelectrode70VymyslickyFKrızekTHert JCanagliflozinoxidation studyusingelectrochemical flowcelland
comparisonwithhydrogenperoxideoxidation 76AugustınMVyskocilVNovelhybridelectrochemicalDNAbiosensorformonitoringoxidativeDNA
damageviaoxidationreductionsignalsoflowmolecularweightdouble-strandedDNA 83SagapovaLKodrık ovaBSvobodaMMusilSKratzerJChemicalvaporgenerationofcadmiumfor
analyticalatomicspectrometry 90S tadlerova B Vyhnanovsky J Dedina J Musil S Photochemical vapour generation of bismuth
coupledwithatomicfluorescencespectrometry 97Cokrtova K Krızek T Separation of liquid crystals using non-aqueous capillary electrokinetic
chromatography104OndrackovaAStiborovaMHavranLSchwarzova-PeckovaKFojtaMElectrochemistryofSudanI
anditsderivatesinaqueousmedia 110BurkinKGalvidisIBurkinMGroupdetectionofaminoglycosidesusingELISAforcontroloffood
contamination 116Vyhnanovsky J Musil S Photochemical vapor generation of cobalt for detection by inductively
coupledplasmamassspectrometry 123LipinskaJMadejMBasBTyczkowskiJOptimizationofconditionforcoldplasmadepositionofthin
layersforsurfacemodificationofworkingelectrodes 129KorbanAAdvancedGC-MSmethodforqualityandsafetycontrolofalcoholicbeverages 135Baroch M Dejmkova H Sladkova S Utilization of a carbon felt as a material for working
electrodes 141Benesova L Zarybnicka A Klouda J Schwarzova-Peckova K Electroanalytical methods for
determinationof7-dehydrocholesterolinartificialserum146
Authorindex 151Keywordindex 152
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 v
1Introduction
2-Methoxyphenol(guaiacol)isusedinmedicineasanexpectorantThestructuralformulaisshowninFig1Itiswidelyusedinthepharmaceuticalindustry[1]forthesynthesisofantituberculosisexpectorantdrugs(Kas-nol Sudafed Ascoril Prothiazine Expectorant Guai-phenesinum) Moreover 2-methoxyphenol is often used as anaromaticsubstance [2] in the food industry Inparti-cular itwaswidelyusedintheproductionofsmokedfish and meat products using smokeless smokingtechnologyusingflavourings Ontheotherhand2-methoxyphenolhasthesymbolGHS07andhasahazardcodeXnTXi[2]accordingtotheGHSsystemItisverytoxicbyinhalationitcanirritatethemucousmembraneoftherespiratorytractandtheconjunctivaofthe
Determination of 2-methoxyphenol in model solutions by spectrophotometry
a a b aVALERIYAALIKOVA ANNACHERNOVA VLADIMIRSHORMANOV ELENAKOROTKOVA
a DepartmentofChemicalEngineeringEngineeringSchoolofNationalResourcesNationalResearchTomskPolytechnicUniversityLeninavenue30634050TomskRussiaalikovaleramailru
b DepartmentofPharmaceuticalToxicologicalandAnalyticalChemistryKurskStateMedicalUniversitystKarlaMarks3305000KurskRussia
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 1
AbstractA spectrophotometric approach for determination of 2-methoxy-phenol in model solutions has been developed The absorptionspectra of 2-methoxyphenol were determined in the wavelengthrangefrom200to400nminsolutionsof95ethanolacetonitrile01Msodiumhydroxideandethylacetatewithaconcentrationofthe
minus3analyte of 005mgdm For the quantitative determination of2-methoxyphenol a seriesof solutionswaspreparedwithvarious
minus3 minus3concentrationsfrom0001mgdm to005mgdm in95ethanolacetonitrile01MsodiumhydroxideTheopticaldensityof2-meth-oxyphenolinsolventswasmeasuredatawavelengthof276nmand289nmThedevelopedmethodwastestedusingthemethodanalysisofspikedsamples
Keywordsquantitation2-methoxyphenolUVVISspectrophoto-
metry
Fig 1Structuralformulaof2-methoxyphenol
eyeballinhighconcentrationswhenitpenetratestheskincanleadtoneurosiswhenadministeredorallycanstimulatetheesophagusandstomachresultinginheartfailurecollapseanddeathNowadaystherearepublisheddataoncasesofasystemicallergicreaction[3]causedby2-methoxyphenolderivativesandthereisa fatal case known [4] for oral administration of guaifenesin (3-(2-methoxy-phenoxy)propane-12-diol)oneofthecomponentsofcommonlyavailablecoughmedications Thedeterminationof2-methoxyphenolinenvironmentalobjectsaswellasinthefoodindustryiscarriedoutusinggaschromatographymethodswithsolid-phasemicroextraction[5]Inordertocontrol2-methoxyphenolinnaturaldrink-ing and treatedwastewater gas chromatography is used followed by opticaldetectionoftheeluate[6]Themaindisadvantagesofthismethodofanalysisarethelowselectivityanddurationofdetermination(about3hours)AlsoaccordingtoRussianStateStandartGOST33312-2015themethodofgaschromatographyisusedforthequalitativeandquantitativedeterminationof2-metoxyphenolinjuiceproducts Commonlyfortheanalysisoftoxicsubstancesinvariousbiologicalsamples(bloodplasmaurinesalivasweathair)bygaschromatographyitisnecessarytocarryoutmultistagesamplepreparationwhichcomplicatesandslowsdownthecourseofthestudy[7]Atthesametimeitisimportantthatduringtheprepa-rationofsamplesintheanalyzedcompoundstheirstructureisnotviolatedasthiswillleadtothedifficultyoftheiridentification Spectrophotometryintheultravioletregionhaslowersensitivitycomparedtothe abovemethods however thismethod does not require such complicatedpreparation of the analyzed samples it is a relatively affordable simple andinexpensive analysis method In addition its sensitivity can be significantlyimprovedbyapplyinganappropriateseparationprocedureandpreconcentrationbeforedetection[8]MethodUVspectrophotometryisusedtoassessthequalityof both medicinal substances and preparations made from them in terms ofauthenticitygoodqualityandquantitativecontentInadditionitisarelativelyaffordablesimpleandlow-costanalysismethod Ananalysisoftheliteraturedatashowedthattodaytherearefastandsensitivespectrophotometricmethodsforthedeterminationofpyrocatecholderivativesinmedicines[9]vanillininfoodproducts[10]andotherphenolsinwastewaterandwineproducts[1112]Howeverasfarasweknowinformationonthedetermi-nationof2-methoxyphenolfromtheabsorptionspectraintheultravioletregionisabsent Theaimofthisstudyistodevelopmethodsforthequalitativeandquantitativeof2-methoxyphenolinmodelsolutionsusingUVspectrophotometry
2 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
2Experimental
21Reagentsandchemicals
Asampleof2-methoxyphenolfromFlukawithabasicsubstancecontentofge98wastakenastheobjectofstudyAssolventsweusedacetonitrile(ChP)95ethanolethylacetateand01MsodiumhydroxidesolutionAllotherchemicalsusedwereofanalyticalreagentgrade
22Instrumentation
Theopticaldensitywasmeasuredincuvetteewithanabsorbinglayerthicknessof10mmusingaCary60spectrophotometer(AgilentUSA)Allmeasurementswerecarriedoutatroomtemperature
3Resultsanddiscussion
Thechangeinthebehavioroftheabsorptionspectrumwasinvestigatedinthewavelengthrangeof200ndash400nmFigure2isshowedthatwithanincreaseinthepolarityofthesolventtheabsorptionmaximumshiftstowardthevisiblepartofthe spectrum The wavelength of absorption maxima of 2-methoxyphenol ispresentedinTable1 A studyof thephotometricbehaviorof2-metoxyphenolinvarious solventsshowedthatacetonitrile95ethanoland01Msodiumhydroxidearethemostsuitablesolventsforthequalitativedeterminationofthetestsubstance
ndash3Fig 1Thespectraof2-methoxyphenolofconcentration005mgdm inthemediumofsolvents(anabsorbinglayerthicknessof10mm)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 3
Forthequantitativedeterminationof2-methoxyphenolaseriesofsolutionsndash3 ndash3withaconcentrationragefrom0001mgdm to005mgdm werepreparedin
acetonitrileand95ethanolTheopticaldensityof2-methoxyphenolinsolventswasmeasuredbywavelengthof276nmThedependenceoftheintensityoftheoptical density on the concentration of 2-methoxyphenol in 01 M sodium
ndash3hydroxide was plotted in the concentration range from 0005mgdm tondash3003mgdm Themeasurementwerecarriedoutbywavelengthof276nmThe
obtainedregressionequationsarepresentedinTable2Dataanalysisobtainedwasperformedusingleast-squaresmethodThedevelopedmethodwastestedusingthemethodanalysisofspikedsamplesTheresultsarepresentedinTable2
4Conclusions
Studieshaveshownthepossibilityofusingspectrophotometricanalysisforthequalitativeandquantitativedeterminationof2-methoxyphenolTheabsorptionmaximaofweredeterminedinsolutionsofethanolandacetonitrile(276nm)inasolutionofethylacetate(277nm)and01Msodiumhydroxide(289nm)Theconstructed calibration curves of thepure substance of 2-methoxyphenol hasshownagoodregressioncoefficient(Rgt099)andcanbeusedforquantitativedeterminationof2-metoxyphenolinbiologicalobjectsInthefutureitisplannedtoapplythistechniquetodetermine2-metoxyphenolinincadavericmaterial
Solvent Regressionequation Found S RSD Δх δ 2-metoxy- phenolg
ndash3 ndash6 ndash5Acetonitrile y=18294C+01130 499times10 50times10 028 2times10 044 Rsup2=09985
ndash3 ndash6 ndash595ethanol y=35131C+00269 503times10 03times10 021 7times10 137 Rsup2=09956
ndash3 ndash6 ndash501Msodium y=31196C+01101 495times10 01times10 018 1times10 028hydroxide Rsup2=09997
Table 2Results of the determination of 2-methoxyphenol (average of three measurements) in modelsolutionsbythemethodanalysisofspikedsamplestheconcentrationofintroduced2-methoxy-
ndash3phenolwas500times10 g(SndashstandarddeviationRSDndashrelativestandarddeviationΔхndashabsoluteerrorδndashrelativeerror)
Table 1Valuesofopticaldensityandwavelengthsinappropriatesolventswith2-methoxyphenol
minus3 minus1Solvent λnm εgdm cm
Acetonitrile 276 0082895ethanol 276 00853Ethylacetate 277 0083101Msodiumhydroxide 289 00744
4 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
References
[1] МельниковаИММизерницкии ЮЛКомбинированныеотхаркивающиепрепаратырастительного происхождения в педиатрическои практикеМедицинский совет 2(2018)93ndash97
[2] httpwwwthegoodscentscompanycomdatarw1032272html(accessed27stFebruary2020)
[3] RayMFaltayBHallerNACasereportanaphylacticreactiontoguaifenesinHospPract37(2009)60ndash63
[4] OkicMJohnsonTCrifasiJALongCMitchellEKSwiftonsetofcentralnervoussystemdepressionandasystolefollowinganoverdoseofguaifenesinJAnalToxicol37(2013)318ndash319
[5] ВолковCМЧерновецАНОпределениеконцентрациифеноловвгазовыхвыбросахпромышленных предприятии методом газовои хроматографии с твердофазнои микроэкстракциеи Сорбционныеихроматографическиепроцессы10(2010)723ndash728
[6] ШачневаЕЮОньковаДВСерековаСМСпособыопределенияфеноловвобъектахокружающеи среды Астраханский вестник экологического образования 4 (2013)138ndash142
[7] ГладиловичВДПодольскаяЕПВозможностипримененияметодаГХ-МС(Обзор)Научноеприборостроение4(2010)36ndash49
[8] Pena-PereiraFLavillaIBendichoCHeadspacesingle-dropmicroextractioncoupledtomicrovolumeUVndashVis spectrophotometry for iodine determinationAnal Chim Acta631(2009)223ndash228
[9] NagarajaPMurthyKCSRangappaKSGowdaNMMSpectrophotometricmethodsforthe determination of certain catecholamine derivatives in pharmaceutical preparationsTalanta46(1998)39ndash44
[10] Altunay N Development of vortex-assisted ionic liquid-dispersive microextractionmethodology for vanillin monitoring in food products using ultraviolet-visible spectro-photometryLWT93(2018)9ndash15
[11] Lupetti KO Rocha FRP Fatibello-Filho O An improved flow system for phenolsdetermination exploiting multicommutation and long pathlength spectrophotometryTalanta62(2004)463ndash467
[12] Figueiredo-Gonzalez M Cancho-Grande B Simal-Gandara J Garnacha tintorera-basedsweetwineschromaticpropertiesandglobalphenoliccompositionbymeansofUVndashVisspectrophotometryFoodChem140(2013)217ndash224
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 5
1Introduction
Thenumberofsamplesthesamplecomplexityandalsothenumberofsubstanceswhich need to be analysed simultaneously is increasing steadily ThereforepowerfulseparationanddetectionmethodsarerequiredOnewaytoachievethisisthecouplingofaseparationsystemwithmorethanonedetector[12] In recent years capillary electrophoresis (CE)was established as a potentseparation system due to its high separation efficiency and the low sampleconsumption [3] To generate more information numerous dual detectionconceptsforCEweredevelopedwhicharesummarisedelsewhere[12]Acom-binationofamperometricdetectionandmassspectrometry(MS)isaninterestingdual detection concept for CE because both detectors supply complementaryinformationForelectroactivespeciesamperometricdetectionisarobustandoneof the most sensitive detection method [4] Thus it is well suited for the
The effects of linearly assembled capillaries with various inner diameters on capillary electrophoresis
DANIELBO HMFRANK-MICHAELMATYSIK
InstituteofAnalyticalChemistryChemo-andBiosensorsFacultyofChemistryandPharmacyUniversityofRegensburgUniversitaumltsstraszlige3193053RegensburgGermanydanielboehmchemieuni-regensburgde
AbstractDuetotheincreasingneedofpowerfulanalyticalmethodsanewdualdetection concept for capillary electrophoresis (CE) with parallelamperometricdetectionandmassspectrometryshallbedevelopedFor this concept the CE flow has to be divided into two streamsutilizinga flowsplitter In thiswork theeffectsof combinedcapi-llarieswithvarious innerdiameterswerestudiedForpreliminaryinvestigationsthecapillarieswereconnectedinaserialconfigurationwithoutdeadvolumeUsingcapillaryflowinjectionanalysishyphe-natedtocontactlessconductivitydetectionitcouldbeshownthatthecouplingofidenticalcapillariesleadstoaslightdecreaseoftheflowratesWithCEhyphenatedtoUVdetection itcouldbeshownthatthecouplingofcapillarieswithdifferentinnerdiameterhasamuchstrongereffectontheelectroosmoticflowthanthecombinationwiththesameinnerdiameterFurthermorenosignificantchangeinpeakshapewasobserved
Keywordsassembledcapillariescapillaryelectrophoresiscapillaryflowinjection
analysisdualdetectionconceptnon-aqueoussystem
6 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
quantificationofsubstanceswhereasMSiswellsuitedfortheidentificationofunknown substances [3] In most dual detection concepts the detectors arearrangedinaserialconfigurationwhichisnotpossibleincaseofamperometricdetection-mass spectrometry [1] The instrumental implementation is morecomplicatedwithbothdetectorsbeingdestructiveFurthermoretheymustbedecoupledfromthehighvoltagefieldoftheCEThereforetheCEflowmustbedividedintotwostreamswithaflowsplitterAsimplifiedsketchofthepossiblenewdualdetectionconceptisshowninFig1 ForthedevelopmentofthenewdualdetectionconceptthreecapillarieswithpotentiallydifferentinnerdiametersmustbecoupledForthisreasonthedeadvolume-freecouplingofcapillarieswithdifferent innerdiameterswas investi-gatedinafirststepTokeepthesetupsimplewefocusedonthelinearcouplingofcapillariesandtheresultingeffectsNon-fragmentedcapillarieswerecomparedwithfragmentedcapillariesofthesameordifferentinnerdiametersEffectsonthe flow rate were investigated with capillary flow injection analysis (CFIA)
4hyphenatedtocontactlessconductivitydetection(C D)EffectslikechangesinthemigrationbehaviourorpeakshapesoccurringinCEwereinvestigatedwithCEhyphenatedtoUVdetection(CE-UV)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 7
Fig 1SchematicillustrationofthenewdualdetectionconceptwithparallelamperometricdetectionandmassspectrometricdetectionforCEAfterinjectionfrom(a)thesamplevialthecomponentsareseparatedbyCEnext(b)theflowsplitterdivides(c)thecapillaryintotwopartsandleadstheCEflowtowards(d)themassspectrometerand(e)theamperometricdetector
2Experimental
21Reagentsandchemicals
Thefollowingchemicalswereusedallofanalyticalgradeferrocenemethanoldecamethylferrocene(ABCRGermany)acetonitrileammoniumacetate01Msodiumhydroxidesolutionultra-purewaterprovidedbyaMilliQAdvantageA10system(MerckGermany)aceticacid(RothGermany)
22Instrumentation
221Capillaries
4Forbothexperiments(CFIA-C DandCE-UV)capillarieswithinnerdiametersof2550and75micromanouterdiameterof360micromandatotallengthof70cmwereusedTheywerepurchasedfromPolymicroTechnologies(USA)Measurementswere carried out with fragmented and non-fragmented capillaries For themeasurementswiththefragmentedcapillariestheoriginalcapillarieswerecutintotwopiecesyieldingatotalof9capillarycombinationswithlengthsof70cm(20cmfirstcapillarypieceand50cmsecondcapillarypiece)ThesecombinationsaresummarizedinTab1(section31)Atbothendsofthecapillariesabout02cmof thepolyimidecoatingwasremovedBothsidesof thecapillarypieceswerepolishedtoreceiveplanarcapillarytipsForthelinearassemblingofthecapillarypiecesMicroTightSleevesF185Xanda capillary connectorUnionAssemblyMicroTightP720fromIDEXHealthampScience(USA)wereusedPriortothefirstCEmeasurementsthecapillarieswereconditionedbyflushingthemfor10minwith01Msodiumhydroxidesolution5minwithultra-purewaterand30minwithseparationbuffer
222Capillaryflowinjectionanalysishyphenatedtocontactlessconductivitydetectionsetup
The flow rates for the fragmented and non-fragmented capillaries were4determinedwithaCFIA-C DsetupschematicallydepictedinFig2ATheflowin
thecapillarywasgravitationdrivenbyaheightdifferencebetweentheinletandoutletcarriersolutionvialTheconceptofCFIAwithgravitationdrivenflowwasfirstdescribedbyMatysiketal[5]AlaboratoryconstructedautosamplerofaCEdevicewasusedforthehydrodynamicinjectionThesamplesolutionconsistedof10mMdecamethylferrocene incarrier solution (10mMCH COONH and1M3 4
4CH COOH in acetonitrile) A high resolution C D was placed after 40 cm for3
detectionThedetectordescribedelsewhere[6]wasconstructedinthedoLagogroup(Brazil)Adoubledeterminationattwodifferentheightswasdoneforthedeterminationoftheflowrates
8 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
223Capillaryelectrophoresis-UVdetectionsetup
Fig2BshowsasketchoftheCE-UVsetupItconsistedofalab-builtCEdevicewhichwasconnectedtoahighvoltagepowersupplyfromISEG(Germany)Theseparationswerecarriedoutwithanon-fragmented50micromcapillaryandwithcapillary combinations implementing a 50micromdownstream capillary segment(25+5050+50and75+50microm)ALambda1010UV-VISdetectorfromBischoff(Germany)wasusedfordetectionat210nmThedetectorwasplacedafter40cm
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 9
Fig 2Schemeof(A)thecapillaryflowinjectionanalysis(CFIA)hyphenatedtocontactlessconduc-4tivity detection (C D) setup and (B) the capillary electrophoresis hyphenated to UV detection
4(CE-UV) setup Components of the CFIA-C D setup (a) sample (b) inlet and (c)outlet carrier4solution vial (d) fused silica capillary (e) linear capillary connector (f) C D and (g) stand
ComponentsoftheCE-UVsetup(h)sample(i)inletand(j)outletbuffervialand(k)UVdetector4therestofthecomponentswereidenticaltotheCFIA-C DsetupTheoutletbuffervialwaslowered
forthehydrodynamicinjection(j)Theenlargedview(k)depictsthecouplingoftwocapillarieswithdifferentinnerdiametersintheconnectionsidewithoutdeadvolume
Asamplesolutioncontaining1mMferrocenemethanolanddecamethylferroceneinseparationbuffer(10mMCH COONH and1MCH COOHinacetonitrile)was3 4 3
utilizedTheinjectionwasperformedhydrodynamicallybyloweringtheoutletbuffer vial by 20 cm A uniform sample plug was injected to compare bandbroadeningeffectsTheinjectionsegmenthadalengthof035cm(05ofthetotalcapillarylength)andtherespectiveinjectiontimewasdeterminedbasedontheflowratesofthecorrespondingcapillarycombinationFortheelectrophoreticseparationaseparationvoltageof25kVwasappliedandtheinletandtheoutletbuffervialswereplacedatthesameheightsothattherewasnogravityflowwhichaffectedthemigrationbehaviour
3Resultsanddiscussion
31Capillaryflowinjectionanalysishyphenatedtocontactlessconductivitydetectionexperiments
AsshowninTab1theflowratesforaheightdifferenceof20cmwerecalculated4basedontheCFIA-C DmeasurementsItwasobservablethattheflowratesare
slightlylowerforfragmentedcapillariesthanfornon-fragmentedcapillariesofthe same dimension This indicates that a flow resistance arises when twocapillaries are combined Furthermore it was observed that the flow ratedecreasesforupstreamcapillarieswithlowerinnerdiametersandviceversaTheflow rate for the combination 25+75microm could not be determineddue to theformationofairbubblesattheconnectionside
10 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Table 1Flowratesandthecorrespondingstandarddeviations(SD4measurements)ofdifferentcapillarycombinationsforaheightdifferencebetweeninletandoutletvialof20cmbymeansofcapillaryflowinjectionanalysishyphenatedtocontactlessconductivitydetection
ndash1capillary flowratenLs plusmnSDflowndash1combinationmicrom ratenLs
25 00577 0000525+25 0057 000150+25 00775 0000775+25 ndash ndash25+50 0137 000550 0865 000250+50 080 00575+50 105 00625+75 0187 000250+75 1941 000375 456 00275+75 42 01
32Capillaryelectrophoresis-UVdetectionexperiments
Theelectropherogramsforthenon-fragmented50micromcapillaryandthecapillarycombinationswithdownstream50micromcapillaryarepresentedinFig3ThetwoferrocenederivatesdecamethylferroceneandferrocenemethanolwereusedasmodelanalytesDecamethylferrocenewasonlydetectedascationicspeciesasitiseasilyoxidizedbydissolvedoxygeninsolution Forthecombination50+50micromslightlyhighermigrationtimesforthecationic(decamethylferrocene) and neutral species (ferrocenemethanol) were obser-vable compared to thenon-fragmented50micromcapillaryThis indicateda flow
4resistanceattheconnectionwhichwasalsoobservedfortheCFIA-C Dexperi-mentsinsection31Incontrasttothecombinationwiththesameinnerdiameterastrongshift in themigration times for theneuralspecieswasvisible for thecombinationswithdifferentinnerdiametersThisshowedthatthecouplinghadaneffectontheelectroosmoticflow LookingatthepeakshapeitwasfoundthatallpeaksshowednearlyGaussianshape for all combinations Furthermore there was no tailing visible Theferrocenemethanolpeaks for thecombination25+50micromand75+50micromwere
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 11
Fig 3Electropherograms of themodelmixture ferrocenemethanol (FcMeOH) and decamethyl-ferrocene(DeMeFc)measuredwithanon-fragmentedcapillary(50microm)andfragmentedcapillaries(25+5050+50and75+50microm)Experimentalparameters1mMFcMeOHandDeMeFcinseparationbuffer(10mMCH COONH and1MCH COOHinacetonitrile)injectionsegment035cmseparation3 4 3
voltage25kVcapillarylength70cm(40cmtothedetectorfragmentedcapillaries20cmfirstpartand50cmsecondpart)UVdetectionat210nm
slightlybroaderthanthepeaksforthenon-fragmented50micromcapillaryorforthe50+50 microm combination But this probably results from longitudinal diffusioneffectsduetothelongerresidencetimes
4Conclusions
FromtheCFIAmeasurementsitcanbeconcludedthattherewasamechanicaldisturbanceoftheflowduetothecouplingFurthermoreitcouldbeshownthatCEmeasurementswithlinearcoupledcapillariesofvariousinnerdiameterwerepossible Unlike to the capillary combinationwith the same inner diameter astrongshiftoftheelectroosmoticflowtowardshighermigrationtimeswasfoundfor capillary combinations with different inner diameters In this work thecapillaries were coupled with almost no dead volume which resulted in nosignificantchangesofthepeakshapeorpeaktailingContrarytoexpectationsthecouplingofcapillarieswithvariousinnerdiametershadnosignificantimpactonthepeakwidth TheknowledgegainedfromthelinearcouplingofcapillariesisagoodbasisforthedevelopmentofthenewdualdetectionconceptInanextstepthreecapillariesshouldbecoupledwitheachother
Acknowledgments
WethanktheGermanResearchFoundation(DFG)forfinancialsupport
References
[1] OpekarFS tulıkKSomeimportantcombinationsofdetectiontechniquesforelectrophoresisincapillariesandonchipswithemphasisonelectrochemicalprinciplesElectrophoresis32(2011)795ndash810
[2] BeutnerAHerlTMatysikF-MSelectivityenhancement incapillaryelectrophoresisbymeans of two-dimensional separation or dual detection conceptsAnal Chim Acta1057(2018)18ndash35
4[3] BeutnerACunhaRRRichterEMMatysikF-MCombiningC DandMSasadualdetectionapproachforcapillaryelectrophoresisElectrophoresis37(2016)931ndash935
[4] MatysikF-MEnd-columnelectrochemicaldetectionforcapillaryelectrophoresisElectro-analysis12(2000)1349ndash1355
[5] MatysikF-MWernerGTracemetaldeterminationintearsbyanodicstrippingvoltammetryinacapillaryflowinjectionsystemAnalyst118(1993)1523ndash1526
[6] FranciscoKJMdoLagoCLAcompactandhigh-resolutionversionofacapacitivelycoupledcontactlessconductivitydetectorElectrophoresis30(2009)3458ndash3464
12 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Thetermldquovitaminsrdquodescribestheheterogeneousgroupofchemicalcompoundswhich are important for the proper functioning of the human body [1 2] Bydefinitionvitaminsarenotsynthesizedbythehumanbodyorthesynthesizedamount is not sufficient to cover the demand That is why they have to besupplementedfromtheexternalsourceslikefoodproductsorpharmaceuticals[1ndash3]Basedontheirsolubilityvitaminsaredividedintowater-soluble(B-groupandvitaminC)andfat-solublevitamins(ADEandK)[3]VitaminC(ascorbicacid) is the most important antioxidant and participates in the activation ofenzymes[4]VitaminB1(thiamine)facilitateswoundhealingandiscrucialforthehumannervoussystem[15]VitaminB2(riboflavin)participatesintheenzy-maticreactionsandthebiotransformationofglucoseandaminoacids[6]Vitamin
+B3(niacin)isthemainconstituentoftheNAD andNADHcoenzymeswhichareresponsible for the transfer of electrons and hydrogen ions in the cellularrespiration [1ndash3] Vitamin B6 possesses six related structures (vitamers) thateasilyinterconvertThemostimportantoneispyridoxinewhichhelpstopreventtongue inflammation and microcytic anemia [2] For the production of well-functioningredbloodcellsandtheavoidanceofmegaloblasticanemiaandfetusdefectsvitaminB9(folicacid)hastobesupplementedintheproperamount[12]
Voltammetric determination of vitamins
RADOSŁAWPORADABOGUSŁAWBAS
DepartmentofAnalyticalChemistryFacultyofMaterialsScienceandCeramicsAGHUniversityofScienceandTechnologyMickiewicza3030-059KrakoacutewPolandrporadaaghedupl
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 13
AbstractVitaminsbelongtothegroupofchemicalcompoundsessentialfortheproperfunctioningofthebodySinceboththeirdeficiencyandexcessmay result in serious health problems the amount of vitaminssupplementedinthedietaswellasvitamincontentintheirsourceshavetobestrictlycontrolledInthisworkthepossibilityofsimulta-neous determination of vitamins by means of differential pulseadsorptive stripping voltammetry is discussed The research hasshownthatthedeterminationofsingularvitaminatthemicromolarlevel isrelativelyfastandstraightforwardandthemostimportanthindranceisrelatedtotheanalyteadsorptionattheelectrodesurfaceInthecaseofvitaminswithdifferentredoxpotentialstheycanbeanalyzedsimultaneouslywithouttheneedtoreachfortheadvancedmethodsforsignalprocessing
Keywordsmercuryelectrodevitaminsvoltammetry
VitaminK3(menadione)doesnotoccurnaturallybutitservesasaprecursorforthesynthesisofotherK-groupvitaminsandcanbeusedtotreathypoprothrom-binemiaVitaminK3ispartiallysolubleinwater[17] All of the vitamins are electrochemically active [3] therefore the electro-chemicalmethodscanbeappliedforthedeterminationofvitamincontentinfoodproductspharmaceuticalsandbodyfluidsVoltammetrictechniquesarecharac-terizedbyhighsensitivityandselectivityandtheydonotrequiretime-consumingsamplepreparationMoreovertheelectrochemicalinstrumentationisrelativelyinexpensiveandcanbeappliedintheon-siteconditionsfortheonlineanalyseseginqualitycontrolMostofthepapersreporttheconstructiondevelopmentand characterization of a novel modified working electrodes for quantitativeanalyses of a singular vitamin in the variety of matrices Unfortunately only alimitednumberofpapersdescribethesimultaneousdeterminationofmultiplevitaminsinasinglerun[23] The preliminary research devoted to the simultaneous determination ofB-groupCandK3vitaminswiththeuseofthecontrolledgrowthmercurydropworking electrode in aqueous solutions is presented in this work Particularattentionhasbeenpaidtotheredoxpotentialsofthestudiedcompoundstheshape of the calibration curves and adsorption phenomena As an attempt toovercomethelattertheneutralsurfactantTritonX-100hasbeenintroducedintothestudiedsystem
2Experimental
21Reagentsandchemicals
TheappliedreagentswereofanalyticalgradeandusedassuppliedPhosphateand McIlvaine buffers were obtained by mixing the appropriate amount of
ndash1 ndash1 ndash102molL Na HPO with 02molL NaH PO and 01molL citric acid2 4 2 4
respectively (all reagents purchased from Avantor Performance MaterialsPoland)ThestandardsolutionsofvitaminB1B2B3B9andCwerepreparedbydissolving the corresponding amount of the standard (all Sigma-Aldrich) in
ndash1distilledwater In thecaseofB2andB9 theadditionof02molL NaOHwasinevitabletoobtainaclearsolutionVitaminK3standard(Sigma-Aldrich)was
ndash1dissolvedinthemixtureofmethanoland1molL phosphatebuffer(pH=82)(vv=25)LaboratorygradeTritonX-100(Sigma-Aldrich)wasusedinthestudyoftheadsorptionprocesses22Instrumentation
All the electrochemical measurements were conducted in the three-electrodesystem composed of the Pt auxiliary electrode double-junction silversilverchloride reference electrode and controlled-growth mercury drop electrode
14 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
actingastheworkingelectrodeTheusedmeasurementequipmentinvolvedtheM164electrodestandandM161multipurposeelectrochemicalanalyzer(bothmtm-anko Krakow) To control the buffer pH-value the SevenCompact S210laboratorypH-meter(MettlerToledoSwitzerland)wasemployed
23Voltammetricmeasurements
Throughout the course of the study differential pulse adsorptive strippingvoltammetry has been used for recording the current-potential curves BothcathodicandanodicscanswererecordedinapotentialrangeadjustedforthestudiedvitaminsTheinfluenceofvariousmeasurementconditionsontheregis-teredsignalshasbeeninvestigatedFinallythepossibilityofthesimultaneousdeterminationofmultiplevitaminsinonescanhasbeenverified
3Resultsanddiscussion
Figure 1 depicts the redox potentials of the studied vitamins in the aqueoussolutions for the mercury electrode The only exception is vitamin B6 whoseredox potential is higher than the potential of mercury oxidation (ca +02 V)Therefore the given value refers to the glassy carbon electrode The redoxpotentialvalueofstudiedvitamins isnotasingularvaluebut it fallswithinacertainrangeThiscanbeascribedtothedependenceofredoxpotentialonthesolutionpHvaluewhichresultsfromtheparticipationofprotonsintheredoxreactions of vitamins Moreover the potentials for individual vitamins arerelatively well separated indicating that the simultaneous determination ofseveralvitaminsintheonerunmaybepossibleTheonlyencounteredproblems
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 15
Fig 1Redoxpotentialsofchosenvitamins
regardthevitaminsB1B3andB12whoseredoxpotentialsoscillatebetweenndash15andndash17VandtheK-groupvitamins(K1K2K3)inwhichonlythecommonstructuralmotifndashthequinoneringndashiselectrochemicallyactiveresultinginthevalueoftheredoxpotentialofcandash02V[8] AtypicalcalibrationcurveisdepictedinFig2Abasedonthedifferentialpulsevoltammograms of the vitamin B9 reduction in the concentration range from
ndash1blank to 01 mg L recorded in the McIlvaine buffer of pH = 52 using thecontrolledgrowthmercurydropelectrodeworkingelectrodeTherelationshipbetweenthepeakcurrentandtheconcentrationofVB9 is linear in thewholetestedrange(r=09999)Basedontheparametersoftheregressioncurvethelimit of detection and limit of quantitation were estimated to 42 and
ndash1142nmolL respectively Similar dependencies and figures of merit can beobtainedforothervitamins QuitedifferentbehaviorwasobservedinthecaseofthevitaminK3forwhichthe increase in current was not strictly proportional to the increase in theconcentrationandthecalibrationplotresemblesanS-shapecurve(Fig2B)ThelatterindicatesthatvitaminK3adsorbsatthesurfaceoftheworkingelectrodeHoweverasnopre-orpost-peakwereobservedwearedealingherewiththeweakadsorption[9]AdsorptionalsoplaysasignificantroleinthecaseofvitaminB2forwhichboththepre-andpost-peakswereobservedindicatingitsstrongaffinitytothemercuryelectrodes To overcome this issue attempts with Triton X-100 were performed TritonX-100isaneutralsurfactant thateasilyadsorbsat thesurfaceof themercury
16 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2Differentialpulseadsorptivestrippingvoltammogramsof(A)vitaminB9and(B)vitaminK3ndash1reduction recorded in the concentration range from blank to 01 and from blank to 04 mg L
respectivelyInsetcorrespondingcalibrationcurvesSupportingelectrolyte(A)McIlvainebufferndash1(pH=52)(B)04molL phosphatebuffer(pH=82)
electrode partially blocking its surface Doing so it prevents the undesiredadsorption of other molecules and thus allows to obtain a linear relationshipbetweenthepeakcurrentandthevitaminconcentration(Fig3)UnfortunatelyduetotheblockingoftheelectrodesurfacetheslopesofthecalibrationlinesaresmallerincomparisontotheonesobtainedintheabsenceofanysurfactantsThismeans that the sensitivity defined as the increase in current caused by a unitincrease in concentration and the resolution understood as the possibility todistinguishsmallvariationinconcentrationarecorrespondinglydecreased Figure3alsopresentsthepossibilitytodeterminemultiplevitaminsinasinglerunChosenvitaminshavewell-separatedpotentialsandtheydonotinterferewitheachotherthereforenoadvancedmultivariatecalibrationstrategiesareneededTheproblemsinthesimultaneousanalysisincludevarioussensitivitieswith respect to the studied analytes and differences in the influence of themeasurementconditionsontherecordedsignalsDuetothattheexperimentalconditionswillneverensurethehighestpossiblesignalvaluesforallanalyzedcompounds
4Conclusions
Differential pulse voltammetry in conjunction with the controlled growthmercurydropelectrodeisaperfecttoolforquantitativeanalysesofvitaminsTheadsorptionofvitaminB2andK3canbepreventedbytheadditionoftheneutralsurfactantTritonX-100whichselectivelyblockstheworkingelectrodesurface
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 17
Fig 3Cathodic voltammograms for the simulatanous determination of vitamin B2 B3 and K3Depictedintheinsetsarethevoltammogramsafterbackgroundsubtractionwiththecorresponding
ndash1callibrationplotsSupportingelectrolye04molL phosphatebuffer(pH=82)with40ppmTritonX-100AccumulationconditionsE =ndash005Vt =20sacc acc
The proposed methodology allows for the simultaneous determination ofmicromolaramountsofvitaminB2B3andK3Suchaprocedurewillhelptoreduce the time and costs of analyses of multivitamin formulations and foodproducts
Acknowledgments
RPhasbeenpartlysupportedbytheEUProjectPOWR0302-00-00-I00416
References
[1] Combs GF JrTheVitaminsFundamentalAspects inNutritionandHealth 3rd ed IthacaElsevierAcademicPress2008
[2] Lovander MD Lyon JD Parr DL Wang J Parke B Leddy J Review Electrochemicalpropertiesof13vitaminsAcriticalreviewandassessmentJElectrochmSoc165(2018)G18ndashG49
[3] Brunetti B Recent advances in electroanalysis of vitamins Electroanalysis 28 (2016)1930ndash1942
[4] BrubacherGMuller-MulotWSouthgateDATMethods forDeterminationofVitamins inFoodNewYorkElsevier1985
[5] Szpikowska-Sroka B A simple and sensitive analytical method for the determination ofthiamineinpharmaceuticalpreparationsJAnalChem68(2013)218ndash222
[6] PetteysBJFrankELRapiddeterminationofvitaminB (riboflavin)inplasmabyHPLCClin2
ChimActa412(2011)38ndash43[7] ZhangZXuJWenYZhangJDingWTheelectro-syntesizedimprintedPEDOTfilmasa
simple voltammetric sensor for highly sensitive and selective detection of vitamin K in3
poultrydrugsamplesSynthMet230(2017)79ndash88[8] JedlinskaKStrusMBasBAnewelectrochemicalsensorwiththeRefreshableSilverLiquid
Amalgam Film multi-Electrode for sensitive voltammetric determination of vitamin K2(menaquinone)ElectrochimActa265(2018)355ndash363
[9] SouthamptonElectrochemistryGroupInstrumentalMethodsinElectrochemistryChichesterHorwood1985
18 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Carbonelectrodesarecommonlyappliedtosensitiveelectrochemicaldetectionofneurotransmittersegdopamine (nor)epinephrineandserotonin in-vivoandin-vitro[12]Neverthelessadsorptionofhigh-molecularweightbiomoleculesinthe matrix on a sensing electrode which then hinders the electron transferreactionofneurotransmitterswillresultinbiofoulingofelectrodesThisremainsachallengingproblemasbiofoulingwillcompromiseelectrochemicalmeasure-mentsThusseveralstrategies foraddressingbiofoulinghavepreviouslybeenreported[34] Thisworkreportsonaneffectiveapproachforminimisingbiofoulingbasedonthehypothesisthatahydrophobicelectrodesurfacewillrepelagainstadsorptionof amphiphilic biomolecules Briefly structurally small conical-tip electrodes
Dopamine detection at antifouling conical-tip carbon electrodes
a a a aSIMONABALUCHOVA JANKLOUDA JIR IBAREK KAROLINASCHWARZOVA -PECKOVA bDANNYKYWONG
a UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova812800PragueCzechRepublicsimonabaluchovanaturcunicz
b DepartmentofMolecularSciencesMacquarieUniversitySydneyNSW2109Australia
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 19
AbstractA significant achievement in this work is the development ofantifoulingconical-tipcarbonelectrodes(~27micromtipdiameterand~165micromaxiallength)suitablefordetectionoftheneurotransmitterdopamine in-vivo These electrodes were hydrogenated using adiphenylsilanereductionmethodtoyieldahydrophobicsurfacetodeteradsorptionofamphiphilicbiomoleculesInitiallyhydrogenatedcarbonelectrodeswereelectrochemicallycharacterisedusingseveralredoxmarkersThedegreeofantifoulingwasthenassessedbythevoltammetricsignalchangeofdopamineattheseelectrodesbeforeand after being incubated in a fouling solution containing bovineserumalbumincytochromeC(bothareproteins)andcaproicacid(alipid) In our work we have obtained only a 69 (standarddeviation35N=40)decreaseindopaminesignalsatthehydro-genated carbon electrodes These results strongly support thediphenylsilanereductionstrategyforthedevelopmentofantifoulingbiosensorsfordopaminedetectioninbiologicalmatrices
Keywordsantifoulingelectrodesdiphenylsilanereduction
methodhydrogenatedconical-tip
carbonelectrodesvoltammetricdopamine
detection
(denoted as CTEs) are fabricated by thermally pyrolysing acetylene gas in anitrogenatmospheretodepositcarbonatthetipandontheshankofpulledquartzcapillaries [5] Spectroscopic studies confirmed that the electrode surface
2 3consistsofsp -likegraphiticcarbonandsp -hybridiseddiamond-likecarbon[6]In addition there is also a range of carbon-oxygen functionalities includingcarbonylquinonecarboxylphenolsalcoholsandethergroupsontheelectrodesurface[6]whichcaninteractwithspectatorbiomoleculesthroughdipole-dipoleorion-dipoleinteractionleadingtotheirirreversibleadsorptionontheelectrodesurface[7]HoweverbysubjectingthesecarbonelectrodestosilanereductionCndashObondsareconvertedtoCndashHbondsandphenolicgroupsaretransformedtosiloxane dendrimers [6] to yield a more hydrophobic carbon surface that isexpectedtobesimilarlylesssusceptibletobiofoulingcomparedtoboron-dopeddiamondelectrodes[78] In this work we will present a methodology involving diphenylsilanereduction to fabricate physically small hydrogenated conical-tip carbonelectrodes(denotedasHCTEs)withanti-foulingcapabilityBothCTEsandHCTEswere electrochemically characterised using several redox probes to elucidatetheirsurfacepropertiesbeforeevaluatingtheirresistancetobiofoulingduringdopaminedetectionin-vitro
2Experimental
21Reagentsandchemicals
Analyticalgradereagents(Sigma-AldrichAustralia)including4-methylcatecholhexaammineruthenium(III) chloride potassium hexacyanoferrate(III) dop-amine hydrochloride sodium phosphate dibasic citric acid perchloric acidpotassiumchloride sodiumhydroxide anhydrousdichloromethanediphenyl-silanetris-(pentafluorophenyl)boraneandgraphitepowderwereusedas-recei-ved Ultra-high purity gases acetylene and nitrogenwere obtained from BOCGases (Australia) All aqueous solutionswere preparedwith deionisedwater(MilliporeMiliplusQsystemUSA)witharesistivityof182MΩcm
22Instrumentation
Chronoamperometric and voltammetric experiments were carried out usingalow-currentpicostateDAQoperatedbyanEChemversion212softwareviaanE-corderinterface(eDAQPtyLtdAustralia)Athree-electrodeset-upinvolving
minus1eitheraCTEorHCTEasaworkingelectrodeaAgAgCl(3molL KCl)referenceelectrode (Bioanalytical SystemsUSA)andaplatinumwire counterelectrode
minus1(CypressSystemsUSA)wasusedCyclicvoltammetryatascanrateof100mVs anddifferential pulse voltammetry (pulse height +25mV pulsewidth 50ms
minus1sampling time 20 ms and scan rate 20mVs ) were used in this work All
20 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
electroanalyticalexperimentswereperformedinanaluminiumFaradaycageatanambienttemperature(23plusmn1degC)
23Preparationofhydrogenatedconical-tipcarbonelectrodes
Asreportedpreviously[5]structurallysmallCTEswerefabricatedbythermallypyrolysing C H (a pressure of 50 kPa) in a pulled quartz capillary (Sutter2 2
minus1InstrumentUSA)housedinaN atmosphere(counterflowof60mLmin )Prior2
to hydrogenation the catalyst tris(pentafluorophenyl) borane (100 mg) wasdissolvedinanhydrousCH Cl (50mL)bystirringfor5minbeforethehydroge-2 2
natingagentdiphenylsilane(25μL)wasaddedCTEswerethenplacedinthereagentmixturefor2hThepreparedHCTEsweredriedovernightbeforeuse
24Biofoulingexperiments
A laboratory synthetic fouling solution consisting of 4 (wv) bovine serumalbumin001(wv)cytochromeC(bothareproteins)and10(vv)caproicacid(alipid)waspreparedbyhomogenisingtheminapH=74citrate-phosphate
minus1buffer(01molL )AllfoulingcompoundswereacquiredfromSigmaAldrichAustralia
3Resultsanddiscussion
31Electrochemicalcharacterisation
minus1InthisworkallCTEswerecharacterisedbycyclicvoltammetryof10mmolL 3+ minus1[Ru(NH ) ] in10molL KClAsdisplayedinFig1(A)onlyCTEsthatshow3 6
asigmoidal-shapedvoltammogramwithasmallchargingcurrentwereemployedinfurtherexperimentsUsingchronoamperometry[5]ameantipdiameterof27μm(standarddeviation(SD)28μmN=142)ameanaxiallengthof165μm(SD=114μmN=142)wereestimatedfortheseCTEs TocomparesurfacecharacteristicsofbothCTEsandHCTEs cyclicvoltam-
minus1 3+2+ minus1 minus1metryof (1)10mmolL [Ru(NH ) ] in10molL KCl (2)10mmolL 3 63minus4minus minus1 minus1[Fe(CN) ] in 10 mol L KCl and (3) 10 mmol L 4-methylcatechol in6minus101molL HClO wasconductedatthesameelectrodesbeforeandafterhydroge-4
nation The results obtained are shown in Fig 1(A-C)We observed a ~20(SD=5N=10)decreaseinthelimitingcurrentofallthreeredoxmarkersafterdiphenylsilane reduction most likely attributable to the hindrance to theirelectron transfer reactions by the phenylsiloxane group formed on HCTEs
3minus4minusMoreover as an inner-sphere redox probe both [Fe(CN) ] and 4-methyl-6
catechol reactionsare sensitive to thepresenceofoxygen functionalitiesonacarbonsurface [7]Accordingly theconversionof these functionalities toCndashHbondsbydiphenylsilanereductionwasexpectedtoyieldmoresluggishelectron
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 21
transfer kinetics at HCTEs as supported by a negative potential shift (from3minus4minus+75mVtominus10mV)inthecyclicvoltammogramof[Fe(CN) ] andapositive6
potentialshift(from+580mVto+675mV)inthecorrespondingcyclicvoltammo-2gram of 4-methylcatechol In addition the conversion of sp -carbon to
3sp -diamond-likecarbon[6] isalsoexpectedtoreducetheconductivityof thecarbonelectrodesurface
32Dopaminedetectionduringbiofoulingexperiments
minus1Theelectrochemicalbehaviourof1mmolL dopamine inapH=74citrate-minus1phosphate buffer (01 mol L ) at CTEs and HCTEs was studied by cyclic
voltammetryTheresultsobtainedareshown inFig1(D)Acomparable12decrease(SD=6N=10)inthedopamineoxidationlimitingcurrenttothatof4-methylcatechol was observed A positive potential shift from +285 mV to+305mVinthevoltammogramsisalsoaccountedforasdescribedabove
22 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
minus1 3+ minus1 minus1Fig 1Cyclicvoltammetryof(A)10mmolL [Ru(NH ) ] in10molL KCl (B)10mmolL 3 63minus4minus minus1 minus1 minus1[Fe(CN) ] in10molL KCland(C)10mmolL 4-methylcatechol in01molL HClO at3 4
minus1(a)aCTEand(b)aHCTE(D)10mmolL dopamineinapH=74citrate-phosphatebufferrecordedminus1at(a)aCTEandaHCTE(b)beforeand(c)afterbiofoulingScanrate100mVs
Next HCTEs were incubated in a synthetic fouling solution containing4(wv)bovineserumalbumin001(wv)cytochromeC(bothareproteins)and 10 (vv) caproic acid (a lipid) for 30 min Cyclic voltammetry of
minus110mmolL dopamineattheseHCTEswasthenconductedinapH=74citrate-phosphatebuffertoevaluatetheirantifoulingpropertyNotablyaconsiderable515(SD=183N=6)decreaseindopaminesignalwasobservedatCTEsIncontrastonlyacorresponding69decrease(SD=35N=40)wasestimatedatHCTEsasshowninFig1(D)Clearlythisrepresentsamajorimprovementinthe antifouling capability of HCTEs obtained using diphenylsilane reductioncomparedtoCTEsandotherpreviouslytestedhydrogenatingagents[9]includingn-butylsilane (35decrease) triethylsilane (23decrease) andphenylsilane(18decrease)Thereforethisworkhassuccessfullydemonstratedtheeffecti-venessofdiphenylsilanereductionmethodindevelopingantifoulingelectrodesfordopaminedetection
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 23
Smallconical-tipelectrode
As-prepared Hydrogenated
minus1LinearrangemicromolL 1ndash10 1ndash20minus2 minus2Intercept10 pAmicrom 108plusmn004 273plusmn011
minus3 minus2 minus1Slope10 pALmicrom micromol 128plusmn007 443plusmn012R 0993 0998
minus1LimitofdetectionmicromolL 100 077
Table 1Analyticalparametersofconcentrationdependencesofdopamineobtainedbydifferentialpulse
minus1voltammetryinapH=74citrate-phosphtebuffer(01molL )Allquoteduncertaintiesrepresentthe95confidenceintervalandthecorrelationcoefficient(R)wasfoundtobestatisticallysigni-ficantatthe95usingStudentrsquost-test
Fig 2DifferentialpulsevoltammetryofdopamineataHCTEinapH=74citrate-phosphatebufferminus1 minus1(01molL )atconcentrations(a)1(b)2(c)4(d)6(e)8(f)10and(g)20μmolL
WehavealsostudiedtheconcentrationdependenceofdopamineinapH=74citrate-phosphatebufferbydifferentialpulsevoltammetryAtypicalcalibrationplotobtainedisshowninFig2Theanalyticalparametersandestimatedlimitsofdetection are summarised inTable 1 These results show thatHCTEs outper-formed CTEs because they exhibit a ~35times higher sensitivity a 23 lowerdetectionlimitandawiderlinearrange
4Conclusions
InthisstudyphysicallysmallHCTEswithanti-foulingcharacteristicsachievedbyhydrogenationusingdiphenylsilanereductionweresuccessfullyfabricatedandelectrochemically characterised using several redox probes Next dopaminedetectionwasperformedbeforeandafterincubationofelectrodesinasyntheticfoulingsolutioncontainingahighconcentrationofbiomoleculesOnlyalow69(SD=35)decreaseindopaminelimitingcurrentwasachievedatHCTEsobtain-edbydiphenylsilanereductionindicatingtheirsignificantlylesssusceptibilitytobiofoulingthanCTEsThesepromisingresultsindicatethatantifoulingHCTEswillpotentiallybenefitthedevelopmentofbiosensorsfordopaminedetectionin-vivoinbiologicalmedia
Acknowledgments
ThisresearchwasperformedwithintheframeworkofSpecificUniversityResearch(SVV260560)FinancialsupportsprovidedbytheGrantAgencyofCharlesUniversity(project390119)andbytheCzechScienceFoundation(project20-03187S)aregratefullyacknowledgedSBandJKalsothankthe Mobility Fund of Charles University and Hlavkova nadace for providing funding for theirresearchinternshipsatMacquarieUniversitySydneyAustralia
References[1] BaranwalAChandraPClinicalimplicationsandelectrochemicalbiosensingofmonoamine
neurotransmittersinbodyfluidsinvitroinvivoandexvivomodelsBiosensBioelectron121(2018)137ndash152
[2] CaoQPuthongkhamPJillVentonBReviewnewinsightsintooptimizingchemicaland3Dsurface structuresof carbonelectrodes forneurotransmitterdetectionAnalMethods11(2019)247ndash261
[3] LinP-HLinB-RAntifoulingstrategiesinadvancedelectrochemicalsensorsandbiosensorsAnalyst145(2020)1110ndash1120
[4] HanssenBLSirajSWongDKYRecentStrategiestoMinimiseFoulinginElectrochemicalDetectionSystemsRevAnalChem35(2016)1ndash28
[5] McNallyMWongDKYAnin-vivoprobebasedonmechanicallystrongbutstructurallysmallcarbonelectrodeswithanappreciablesurfaceareaAnalChem73(2001)4793ndash4800
[6] SirajSMcRaeCRWongDKYEffectiveactivationofphysicallysmallcarbonelectrodesbyn-butylsilanereductionElectrochemCommun64(2016)35ndash41
[7] ParkJShowYQuaiserovaVGalliganJFinkGDSwainGMDiamondmicroelectrodesforuseinbiologicalenvironmentsJElectroanalChem583(2005)56ndash68
[8] ShinDTrykDAFujishimaAMerkociAWang JResistance to surfactantandproteinfoulingeffectsatconductingdiamondelectrodesElectroanalysis17(2005)305ndash311
[9] Roshni RAnAntifouling Structurally Small Carbon Electrode forDetectionof theNeuro-transmitterDopaminePhDThesisMacquarieUniversitySydney2019
24 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Theanalyticalperformancesofenzymaticbiosensorsarestronglyaffectedbytheenzyme immobilization process There is no universal technique for enzymesattachmentThereforespecialattentionshouldbepaid to theselectionof theappropriatesupportandthedevelopmentoftheoptimalbindingstrategyinordertoensure thebestcharacteristicsof immobilizedenzymeDespiteavarietyofpreviouslyreportedcovalentimmobilizationmethodsfordifferentenzymesthepresentedprocedurescanbehardlycomparedtofindtheoptimalonesbecauseofdifferentanalyticalmethodsandexperimentalconditionsusedUptodatethere
A comparative study of covalent glucose oxidase and laccase immobilization techniques at powdered supports for biosensors fabrication
ab a bSOFIIATVORYNSKA JIR IBAREK BOHDANJOSYPCUK
a UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova2030812843Prague2CzechRepublicsofiiatvorynskajh-instcascz
b JHeyrovskyacuteInstituteofPhysicalChemistryoftheCzechAcademyofSciences Dolejškova318223Prague8CzechRepublic
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 25
AbstractInordertodeveloptheoptimalstrategyandtodeepentheknowledgeinthefieldofenzymeimmobilizationthreedifferenttechniquesofcovalentbindingfortwoenzymes(glucoseoxidaseandlaccase)atpowdered surfaces were compared Immobilization protocol wasoptimized by changing supports (twomesoporous silica powders(SBAminus15 MCMminus41) and a cellulose powder) the functionalizedgroupsintroducedatsupportsurfaces(minusNH andminusCOOH)andthe2
methodsofactivation(glutaraldehydeandcarbodiimide)Aminoandcarboxyl functionalized mesoporous silica and cellulose powderswerepreparedbysilanizationusing(3-aminopropyl)triethoxysilaneandcarboxyethylsilanetriolrespectivelyItwasfoundthatcouplingof both enzymes by their ndashNH groups through glutaraldehyde to2
ndashNH functionalized supports in particular SBA15minusNH and2 2
celluloseminusNH forglucoseoxidaseMCM41minusNH forlaccaseshowed2 2
thehighestactivityandthebeststability
Keywordsbiosensorcovalentimmobilizationenzymaticreactorglucoseoxidaselaccase
is still a lack of the comparative systematic studies focusing on the enzymesimmobilizationonthevarioussupportsusingdifferenttechniques The aim of this work is the systematic comparative study of the differenttechniques for covalent coupling of the enzymeswhich ensures not only thedevelopmentoftheoptimalimmobilizationstrategyfortheselectedenzymesbutalsoenablestofindoutsometendenciesinenzymeattachmentprocessgenerallyThusthisworkisfocusedonadetailedanalysisoftheeffectofthekindofsupportits anchor groups and the activation methods on activity and stability ofimmobilizedenzymesTwoenzymeswithdifferentnature(glucoseoxidase(GOx)andlaccase(Lac))werechosenasthetestingbioreceptors
2Experimental
21Reagentsandchemicals
AllchemicalswereofpaorbettergradeGlucoseoxidasefromAspergillusnigerminus1(GOxEC11341452Umg )laccasefromTrametesversicolor(LacEC11032
minus1129Umg )D-(+)-glucosedopamineglutaraldehyde(GAgradeII25aqueoussolution) N-(3-dimethylaminopropyl)-Nʹ-ethylcarbodiimide hydrochloride(EDC ge980) N-hydroxysuccinimide (NHS ge970) (3-aminopropyl)-triethoxysilane (APTES) mesoporous silica powder SBAminus15 (particle size
2 minus12ndash6μmporesizeasymp7nmsurfaceareaasymp600m g )mesoporoussilicapowder2 minus1MCMminus41 (pore size 21ndash27 nm surface area asymp 1000 m g ) cellulose (Cell
microcrystalline powder particle size 20 microm) were purchased from SigmaAldrichCarboxyethylsilanetriol(CEST25aqueoussolution)waspurchased
regfromabcr (Germany)
22Instrumentation
Amperometric measurements were carried out at room temperature usingcomputer-controlled electrochemical stand (Polaro-Sensors Czech Republic)withMultiElchemv31software(JHeyrovskyInstituteofPhysicalChemistryoftheCAS)Flowinjectionanalysis(FIA)withthethree-electrodelaboratory-madeflow-through cellwas usedworking electrode minus tubular detector of polishedsilversolidamalgam(TD-p-AgSAlaboratory-madeinnerdiameter05mmtheamalgamtube length60mm) referenceelectrodeminusaminiaturizedsaturatedcalomelelectrodebasedonsilverpasteamalgam[1](laboratory-madeithasthesamepotential as classical saturated calomel electrode) auxiliary electrodeminusplatinum wire (diameter 10 mm length 10 mm) The system for FIA withelectrochemicaldetectioncomprisedofalinearsyringepumpa2-position6-port
regsampleinjectorvalveaninjectionlooplaboratory-madeofTeflon (PTFE)tubing(100μL) a solenoid operatedmicro-pumpan enzymatic reactor and a flow-through cell for TD The enzymatic reactor consists of a tube filled by theenzymaticpowder
26 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
3Resultsanddiscussion
Basedonthedatareportedintheliterature[23]andontheresultsofourpreviousworks[4ndash8]forthisstudyCellandmesoporoussilicapowders(namelySBAminus15andMCMminus41)havebeenselectedas thepotentialpromisingsupports for thecovalent enzyme immobilizationBecauseof thehigh contentof surficial ndashOHgroupswhich are capable of chemical reactions these supports can be easilyfunctionalizedThewell-knownandfrequentlyutilizedtechniqueofsilanizationhasbeenusedtomodifythesurfacesofSBA15MCMminus41andCellbythedesiredfunctionalizedgroupsAminosilaneAPTESwasappliedtoformminusNH groupson2
thematrixsurfaceswhereascarboxylsilaneCESTwasusedtointroducendashCOOHgroups Generallytheprocedureofthecovalentimmobilizationofenzyme(eitherLacorGOx)onthefunctionalizedsupportconsistsofthreestepsI Synthesisofthefunctionalizedsupportwhichmeansthemodificationofthematrix(MCMminus41SBAminus15andCell)withsuitableanchoredgroups(minusNH or2
minusCOOH)II Activationstepofthefunctionalizedsupportwithspecificactivatingagents(glutaraldehydeorEDCNHS)tomakeitreactivetowardsenzyme
IIIEnzyme(LacorGOx)couplingtotheactivatedsupport
To investigate the effect of support its surface functionalizedgroups and themethodsofactivationontheefficiencyofthecovalentenzymeimmobilizationthreedifferentstrategies(ABandC)forLacandGOxattachmenthavebeenusedThedetailsoftheusedtechniquesandthedenotationsofthepreparedenzymaticpowdersaresummarizedinTable1(nextpage)ToexaminetheefficiencyofLacandGOximmobilizationtheenzymaticreactors(filledbytheenzymaticpowderspreparedwithdifferenttechniques)coupledwithTDwereusedforamperometricdetermination of dopamine and glucose respectively in flow systems Theprincipleofglucosedetection isbasedonamperometricmeasurementsof theenzymatically consumed oxygen whereas dopamine was detected by thereductionoftheenzymaticallyoxidiseddopamine Asdepicted inFig1 thebiosensors responsesare stronglyaffectedby thestrategyusedforLacorGOximmobilizationAsshowntheresponsesofLacandGOx biosensors decrease in the order strategy A gt strategy B gt strategy Cirrespectiveofthetypeofsupport It isclearlyseenthataminofunctionalizedsupports(SBA15minusNH MCM41minusNH andCellminusNH )providehigheractivitiesof2 2 2
the immobilized Lac andGOx than these supports functionalized by carboxylgroups(SBA15minusCOOHMCM41minusCOOHandCellminusCOOH)BycomparingactivitiesofimmobilizedenzymesusingstrategiesAandBtheinfluenceoftheactivationagenthasbeenevaluatedThebestresultsforbothenzymeswereobtainedforndashNH functionalizedsupportsactivatedbyGAItcouldbeexplainedbythefact2
thatGAcontrary tocarbodiimidewithnonemolecularspaceprovidesa long
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 27
spacerarmensuringminimalsterichindrancesforenzymesbindingItcanbeconcludedthatthecovalentimmobilizationofbothenzymesbytheirndashNH groups2
viaGAtondashNH functionalizedmesoporoussilicapowders(strategyA)provided2
the highest activities Interestingly in the similar comparative studies it isreportedthatamongndashOHminusCOOHandndashNH functionalizedsupportsactivatedby2
divinylsulfonecarbodiimideandGArespectivelythelastonewasfoundasthemostsuitabletechniqueforthecovalentbindingofLac[9]invertase[10]andpepsin[11]
28 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Tab
le 1
Theprinciplesofthecovalentimmobilizationmethodsofenzymesusedinthisstudy
StrategyA
StrategyB
StrategyC
Support
SBAminus15MCMminus41Cell
SBAminus15MCMminus41Cell
SBAminus15MCMminus41Cell
Supportfunctio-
minusNH
minusNH
minusCOOH
22
nalizedgroup
Activationagent
Glutaraldehyde(GA)
Carbodiimide
Carbodiimide
(EDCNHS)
(EDCNHS)
Enzymereactive
minusNH
minusCOOH
minusNH
22
group
Typeofbond
secondaryamine
amide
amide
Denotationsofthe
GOxminusNHminusCHminus(CH)minusCHminusNHminusSBA15
GOxminusC(=O)minusNHminusSBA15
GOxminusNHminusC(=O)minusSBA15
23
preparedenzy-
GOxminusNHminusCHminus(CH)minusCHminusNHminusMCM41
GOxminusC(=O)minusNHminusMCM41
GOxminusNHminusC(=O)minusMCM41
23
maticpowders
GOxminusNHminusCHminus(CH)minusCHminusNHminusCell
GOxminusC(=O)minusNHminusCell
GOxminusNHminusC(=O)minusCell
23
LacminusNHminusCHminus(CH)minusCHminusNHminusSBA15
LaxminusC(=O)minusNHminusSBA15
LacminusNHminusC(=O)minusSBA15
23
LacminusNHminusCHminus(CH)minusCHminusNHminusMCM41
LacminusC(=O)minusNHminusMCM41
LacminusNHminusC(=O)minusMCM41
23
LacminusNHminusCHminus(CH)minusCHminusNHminusCell
LacminusC(=O)minusNHminusCell
LacminusNHminusC(=O)minusCell
23
When the effect of the method of the covalent enzyme coupling on thebiosensorstabilitywasevaluateditwasfoundthatLacboundedtondashNH functio-2
nalizedsupportsviaGA(strategyA)hasshownthehigheststability(gt65oftheinitial responses after 1 month) compared to other strategies whereas GOximmobilizedwithtwostrategies(AandB)possessedapproximatelysimilarhighstability(gt80oftheinitialresponsesin1month)BothenzymesboundedviandashNH groupstondashCOOHfunctionalizedsupportsthroughEDCNHS(strategyC)2
showedquitelowstability
4Conclusions
Threedifferent strategies including the support selection the anchor surfacegroups and the activationmethod havebeen compared for efficient covalentimmobilization of Lac and GOx The results showed that ndashNH functionalized2
supports(SBA15minusNH CelluloseminusNH forGOxandMCMminusNH forLac)activatedby2 2 2
GAmaybeusedtoeffectivelybindenzymesintermsofhighactivityandstability
Acknowledgments
ThisworkwasfinanciallysupportedbytheGrantAgencyofCharlesUniversityinPrague(Project1356120)theGrantAgencyoftheCzechRepublic(Project20-07350S)anditwascarriedoutwithintheframeworkofSpecificCharlesUniversityResearch(SVV260440)
References
[1] YosypchukBBarekJYosypchukOPreparationandpropertiesofreferenceelectrodesbasedonsilverpasteamalgamElectroanalysis23(2011)2226minus2231
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 29
Fig 1Effectof the covalent attachment techniqueson (A) laccaseand (B) andglucoseoxidaseminus1biosensor responses Experimental conditions (A) c = 500 micromol L E = minus50 mVDOP det
minus1 minus1v = 01 mL min V = 40 microL carrier solution 01 mol L acetate buffer pH = 48flow DOPminus1 minus1(B)c =500micromol L E =minus1100mVv ==01mLmin V =40microL carrier solutionGlu det flow Glu
minus1 minus101molL acetatebuffer0001molL Na EDTApH=652
[2] LiuYChenJYEnzymeimmobilizationoncellulosematrixesJBioactCompactPolym31(2016)553ndash567
[3] Hartmann M Kostrov X Immobilization of enzymes on porous silicas ndash benefits andchallengesChemSocRev42(2013)6277minus6289
[4] JosypcukOBarekJJosypcukBElectrochemicalbiosensorsbasedonenzymaticreactorsfilledbyvarioustypesofsilicaandamalgampowdersformeasurements inflowsystemsElectroanalysis28(2016)3028minus3038
[5] Josypcuk O Barek J Josypcuk B Amperometric determination of catecholamines byenzymaticbiosensorsinflowsystemsElectroanalysis30(2018)1163minus1171
[6] TvorynskaSBarekJJosypcukBAmperometricbiosensorbasedonenzymaticreactorforcholinedeterminationinflowsystemsElectroanalysis31(2019)1901minus1912
[7] TvorynskaSBarekJJosypcukBFlowamperometricbiosensorbasedontwoenzymaticreactors (acetylcholinesterase-choline oxidase) for the detection of neurotransmitteracetylcholine In Proceedings of the 15th International Students Conference ldquoModernAnalyticalChemistryrdquoKNesmerak(ed)PragueFacultyofScienceCharlesUniversity2019p61minus66
[8] TvorynskaSBarekJJosypcukBAcetylcholinesterase-cholineoxidase-basedmini-reactorscoupledwithsilversolidamalgamelectrodeforamperometricdetectionofacetylcholineinflowinjectionanalysisJElectroanalChem860(2020)113883
[9] RekucAKruczkiewiczPJastrzembskaBLiesieneJPeczynska-CzochWBryjakJLaccaseimmobilizationonthetailoredcellulose-basedGranocelcarriersIntJBiolMacromol42(2008)208minus215
[10] Bryjak J Liesiene J S tefuca V Man-tailored cellulose-based carriers for invertaseimmobilizationCellulose15(2008)631minus640
[11] SzałapataKOsinska-JaroszukMBryjakJJaszekMJarosz-WilkołazkaANovelapplicationofporousandcellularmaterialsforcovalentimmobilizationofpepsinBrazJChemEng33(2016)251minus260
30 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Carbohydrates are crucial for energy structure and signaling in the humanbody[1]Thereisavarietyofcarbohydratesbutthemostimportantoneforlifeisglucoseasitisfundamentalinthemetabolismandphotosynthesis[2]GlucoseisclassifiedashexoseThesemonosaccharidesonlydifferinthepositionofhydroxylsubstituentsinsomecasesInadditiontothestructuralsimilaritiesthesemole-culeslackachromophoreandarenoteasilyionizable(pK ~12)Thusdetectiona
intheUVregionandseparationofanalytesbycapillaryelectrophoresis(CE)arechallenging [1 3] At the moment there are many different techniques for theanalysis of carbohydrates commonly including time-consuming derivatizationstepsoreluentswithhighpHvalues(pHgt12)inionchromatographyAwell-esta-blishedtechniquefortheanalysisofcarbohydratesishigh-performanceanion-exchangechromatographywithpulsedamperometricdetection(HPAE-PAD)[4]Electrochemical detection like AD is matching miniaturization simple instru-mentationlowcostandrobustnessandthusisoftenusedforflow-basedsystemssuchasCEandflowinjectionanalysis(FIA)[5]
Capillary flow injection analysis with electrochemical detection for carbohydrate analysis
NICOLEHEIGLFRANK-MICHAELMATYSIK
InstituteofAnalyticalChemistryChemo-andBiosensorsFacultyofChemistryandPharmacyUniversityofRegensburgUniversitaumltsstraszlige3193053RegensburgGermanynicoleheiglchemieuni-regensburgde
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 31
AbstractAsimplecapillaryflowinjectionanalysissystemwithamperometricdetection was arranged for the development of a method for fastoptimization of detection conditions in the context of thedetermination of carbohydrates by means of electrochemistry-capillary electrophoresis-mass spectrometry This setup is free ofelectricalinterferencebyhighvoltageandisperfectforstudyingtheoxidationofvariousanalytesFurthermoreitassureseasycouplingtoMS and thus is an useful tool to investigate the correspondingoxidationproductsofananalyte
Keywordscapillaryflowinjection
analysiscarbohydratesmassspectrometrydisposableelectrodespulsedamperometric
detection
Inthiscontributioncapillaryflowinjectionanalysis(CFIA)withADwillbepresentedasamethodtoapplyandtestADforthedetectionofmonosaccharidesondifferentdisposablethin-filmorscreen-printedelectrodesCFIAwaschosenoverconventionalFIAforthispurposeasthegravityflowinCFIAisstableforalongertimeandverylowsampleconsumptioncanbeachieved[6]TheCFIAsystemwasarrangedassimpleaspossibleandperformedhydrodynamicallytoavoidanyinterferencesFurthermoreitassuresthecouplingoftheflowsystemtoamassspectrometerThusthesamesetupasusedforCFIAcanbeutilizedforcapillaryelectrophoresis-massspectrometry(CE-MS)experimentsbychangingthe flow through the electrochemical flow cell in opposite direction In futureexperimentstheexperienceintermsofADonthoseelectrodeswillbeusedtodevelopelectrochemicalpretreatmentprotocolsforcarbohydratedeterminationbyCE-MS
2Experimental
21Reagentsandchemicals
The following chemicals were used for this study all of analytical gradeAmmoniumacetate(NH OAc)wasobtainedfromMerck(DarmstadtGermany)4
and ferrocene methanol (FcMeOH) from ABCR (Karlsruhe Germany) Milli-Qregwater(182MΩcm)wasgeneratedbyaMilli-QAdvantageA10 system(Merck
Millipore Darmstadt Germany) Carrier solution was prepared by dissolvingndash1NH OAc (50mmolL ) in Milli-Q water FcMeOH solution was prepared by4
dissolvingFcMeOHincarriersolution
22Instrumentation
Electrochemical measurements were performed using a microAutolab Type IIIpotentiostatgalvanostat (Metrohm Autolab B V Utrecht Netherlands)controlledbyNOVA20softwareforexperimentalcontrolanddataacquisitionCFIAwasperformedusingthesetupillustratedinFig1(A)consistingofacarrierreservoirsamplevialandtwofusedsilicacapillaries(PolymicroTechnologiesPhoenix AZ USA inner diameter 100 microm length inlet 40 cm length outlet10cm)connectedtoacommerciallyavailableflowcellfromMicruxTechnologies(model ED-FLOW-CELL Oviedo Spain) Inside of the flow cell the fused silicacapillary was placed in a so-called wall-jet configuration above the workingelectrode of a disposable thin-film gold electrode (model ED-SE1-Au MicruxTechnologiesOviedoSpain)ascanbeseeninFig1(B)Thethin-filmelectrodeswerebasedonathree-electrodesystemwithagoldworkingauxiliaryandquasi-referenceelectrodeThehydrostaticpressurewasachievedbyaheightdifferencebetweeninletandoutletreservoirof30cmresultinginagravityflowofthecarriersolutionthroughafusedsilicacapillaryandsubsequentlythroughtheflowcell
32 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Theinjectionwascarriedoutbyloweringthevialcontainingthecarriersolutiontotheleveloftheoutletexchangingthecarrierreservoirwiththesamplevialandliftingthesamplevialto20cmforadefinedperiodoftimeRe-establishingthecarrierreservoirtookplacethesameway
3Resultsanddiscussion
AsimpleCFIA-ADsystemwasarrangedwherehydrostaticpressurebyaheightdifferencebetweeninletandoutletreservoirresultedinagravitationalflowToobtain general information about the behavior of the assembled CFIA systempreliminaryexperimentswithFcMeOHwereperformedToassurecompatibilitywithMSlateronNH OAcwaschosenastheelectrolyteVariousheightdifferences4
andinjectiontimesweretestedandtheinjectionataheightdifferenceof20cmlasting for 10 s was found to be the optimum concerning feasibility and peakshapes Injections of several solutions of FcMeOH of different concentrationsshowed that the concentration dependence of FcMeOH was linear in theinvestigatedrange(Fig2)Furthermoreexperimentsrevealedthattheinjectionprocedure was established with reasonable precision When repeating the
ndash1injectionof05mmolL FcMeOHincarriersolutionfortentimestherelativestandarddeviationwasfoundtobe3forthemanualinjectionprotocol
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 33
Fig 1(A)Schemeoftheusedcapillaryflowinjectionanalysissetup(1)carrierreservoir(2)samplevial(3)inletcapillarywithalengthof40cmandaninnerdiameterof100microm(4)outletcapillarywithalengthof10cmandaninnerdiameterof100micrombothcapillariesconnectedto(5)acommerciallyavailableflowcellfromMicruxand(6)awastevial(B)Configurationinsidetheflowcell(7)thefusedsilicacapillarywasplacedinaso-calledwall-jetconfigurationabovetheworkingelectrodeof(8)adisposablethin-filmgoldelectrode
4Conclusions
The presented system for CFIA-AD was arranged as simple as possible andrepresents a useful approach for the development of a method for fastoptimization of detection conditions in the context of the determination ofcarbohydratesbymeansofelectrochemistry-CE-MSThesetupisfreeofelectricalinterferencebyhighvoltagecompatiblewithMSandthuspromisingforstudyingtheoxidationofvariousanalytes
References
[1] LuGCrihfieldCLGattuSVeltriLMHollandLACapillaryelectrophoresisseparationsofglycansChemRev118(2018)7867ndash7858
[2] GalantALKaufman RCWilson JDGlucoseDetectionandanalysisFoodChem188(2015)149ndash160
[3] Sarazin C Delaunay N Costanza C Eudes V Gareil P Application of a new capillaryelectrophoreticmethodforthedeterminationofcarbohydratesinforensicpharmaceuticalandbeveragesamplesTalanta99(2012)202ndash206
[4] Rohrer JS Basumallick L Hurum D High-performance anion-exchange chromatographywithpulsedamperometricdetectionforcarbohydrateanalysisofglycoproteinsBiochem78(2013)697ndash709
[5] IslamMAMahbubPNesterenkoPNPaullBMackaMProspectsofpulsedamperometricdetectioninflow-basedanalyticalsystemsndashAreviewAnalChimActa1052(2019)10ndash26
[6] MatysikF-MWernerGTracemetaldeterminationintearsbyanodicstrippingvoltammetryinacapillaryflowinjectionsystemAnalyst118(1993)1523ndash1526
34 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2ndash1 ndash1(A) CFIA-AD recordings of three consecutive injections of (1) 025mmolL (2) 05mmolL
ndash1 ndash1 ndash1(3) 075mmolL and (4) 1mmolL FcMeOH in 50 mmolL NH OAc detection at a Micrux4
thin-filmAuelectrodeataconstantpotentialof03VinaflowcellHydrodynamicinjectionlasted10sataheightdifferenceof20cm(B) Calibration dependence of FcMeOH for CFIA-AD determination and detection at a Micruxthin-filmAuelectrodeataconstantpotentialof03VinaflowcellThestandarddeviationsofpeakheights(n=3)areindicatedbyerrorbars
1Introduction
Topreventsorptiononthecapillarysurfaceandimproveseparationefficiencyand selectivity of determined analytes coatings are formed on the capillarysurface There are two types of ones namelydynamic and covalently bondedcoatings Despite the simplicity of creation dynamic coatings cannot providerequiredreproducibilitywhilecovalentcoatingscontributethestableelectro-osmoticflow(EOF)andhighreproducibilityoftheanalysisInmostcasesthe
Application of covalent coatings based on imidazolium cations for separation and on-line preconcentration of basic and neutral analytes in capillary electrophoresis
a ab aANASTASIAVKRAVCHENKO EKATERINAAKOLOBOVA LIUDMILAAKARTSOVA
a DepartmentofOrganicChemistryInstituteofChemistrySaintPetersburgStateUniversity 26Universitetskiiprospect198504StPetersburgPeterhofRussiakravchenko161216gmailcom
b TheFederalStateInstituteofPublicHealthldquoTheNikiforovRussianCenterofEmergencyandRadiationMedicinerdquoTheMinistryofRussianFederationforCivilDefenceEmergenciesandEliminationofConsequencesofNaturalDisasters54Optikovst197082StPetersburgRussia
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 35
AbstractThemethodofcapillaryelectrophoresis (CE) isactivelydevelopedandmoreandmoreattractsscientistsattentioneveryyearHoweverthesorptionofanalytesonsurfaceoffused-silicacapillarywallsisoneof thesignificantdisadvantagesof thisapproachThe formationofcoatings on the inner capillary surface is typical way to preventsorption and to increase separation efficiency and selectivity ofdetermined analytes Coatings that covalently bonded to capillarywalls is more suitable because they are stable and provides highreproducibilityofanalysisThepresentworkisfocusedonthedevelo-pmentofthemethodofelectrophoreticdeterminationofbiologicalactiveanalytesusingacovalentcoatingbasedonimidazoliumcationsTheeffectofsubstituentinimidazoliumringonmainelectrophoreticparameters was examined It was shown that alkylimidazoliumcoatingscontributetosignificantreducingofbiogenicamineslimitsof detection while β-cyclodextrinimidazolium covalent coatingallowstoseparatebothofhydrophobicandhydrophilicanalytesinonerun
Keywordsbiologicalactiveanalytescapillarycoatingcapillaryelectrophoresisimidazoliumionicliquids
analytes nature determines type of usedmodifiers because suitable ones canprovideaccessorial interactionbetweentheanalytesandthestationaryphaseimproving separation selectivity and efficiency [1 2] Ionic liquidshave beenwidelyusedinanalyticalchemistry[3]andseparationtechniquesparticularlyincapillaryelectrophoresis[4]Earlyresearches[5ndash9]haveshownopportunityofcovalently bonded imidazolium ionic liquids for electrophoretic separationHowever the effect of various substituents in imidazolium ring hasnot beendescribed previously Thus the purpose of this study was to create covalentcoatingsbasedon ionic liquidwithvarioussubstituentsand tocompare theiranalytical capabilities in the electrophoretic separation of biologically activecompounds
2Experimental
21Reagentsandchemicals
(3-Glycidyloxypropyl)trimethoxysilane (GPTMS) hydrochloric acid sodiumdodecyl sulfate (SDS) imidazole 22-diphenyl-1-picrylhydrazyl (DPPH)p-toluen-sulfonylchlorideβ-cyclodextrinhydrocortisone(F)11-deoxycortisol(S)Corticosterone(B)rac-ketoprofen(ndash)-adrenaline(A)L-(ndash)-norepinephrine(NE) DL-normetanephrine (NMN) dopamine (DA) DL-metanephrine hydro-chloride(Met)serotoninhydrochloride(Ser)homovanillicacid(HVA)24-di-hydroxy-benzoic acid (24-DHBA) 34-dihydroxy-L-phenylalanine (DOPA)L-tryptophan(Trp)L-tyrosine(Tyr)werepurchasedfromSigma-Aldrich(USA)1-Bromo-butane1-bromooctanewerepurchasedfromReagentPlus(Ukraine)Sodium dihydrogenphosphate dihydrate acetone NN-dimethylformamide(DMF)wereobtainedfromMerck(Germany)AllreagentsusedwereanalyticalgradeAllsolutionswerepreparedusingdeionizedwater
22Instrumentation
Capillary electrophoresis experiments were carried out using the system ofcapillaryelectrophoresisCAPEL-105M(LumexRussia)withUV-spectrophoto-metricdetector(wavelengthrange190ndash360nm)Separationswereperformedusing 58times49 cm (9 cm to the detector outside diameter 360 microm and innerdiameter50microm) coated silica capillaries (LumexRussia) Thebuffer pHwasmeasuredwithapH-meterHI2210ndash2216(Hanna)
23Capillarycoatingsynthesis
Earlier our research team has proposed the synthesis route for the covalentcoatingsbasedonimidazoliumcationfunctionalizedwithalkylgroup[10]andβ-cyclodextrin [11] All capillarieswere prepared according above-mentioned
36 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
manuscriptsandcharacterizedbytheEOFmobilitymeasurementandscanningelectronmicroscopyThesynthesisconsistedoffollowingstepspreparationofacapillarytocreateacovalentcoating(heatingcapillaryfilledwith2MNaOHat90degCfor1handdryingfollowed)silylationwithGPTMSandfunctionalizationwiththeimidazolesolutionfollowedmodificationbybutyl-andoctylbromideortosyl-β-cyclodextrin(seedetailsin[10]and[11])
24Solutions
A stock buffer solution in concentration 50mM was prepared by dissolvingappropriate amount of sodium dihydrogenphosphate dihydrate in deionizedwateradjustingpHto20with1MhydrochloricacidThisbuffersolutionwasthendilutedwithdeionizedwater
ndash1 Allthesamplestocksolutionswerepreparedwithconcentration10mgmL Thestocksolutionsoftheneurotransmittersandtheirmetabolites(adrenalinenoradrenaline dopamine normetanephrine metanephrine serotonin homo-vanillicacid)and24-dihydroxybenzoicacidasinnerstandardandaminoacids(tryptophan34-dihydroxy-L-phenylalanine tyrosine)wereprepared in01MhydrochloricacidThestocksolutionsofsteroids(hydrocortisone11-deoxycor-tisolandcorticosterone)wereprepared inacetonitrileThestocksolutionsofketoprofen racemate and S-ketoprofen were prepared in acetonitrilewatersolution(1090vv) Untilelectrophoreticanalysisthestocksolutionswerestoredatndash16degCTheworkingsolutionswerepreparedbydilutingtheinitialsolutionswithwaterjustbeforetheexperiments
3Resultsanddiscussion
CovalentcoatingsbasedonN-alkylimidazoliumcationwereespeciallysuitableforseparationofneurotransmittersandtheirmetabolites(Fig1)Inadditionthecombinationofcovalentcoatingwithon-linepreconcentrationtechniquesallowstothesignificantdecreaseoftheseanalyteslimitsofdetection(LOD)Accessorialinteractions positively charged analytes with positively charged imidazoleimproveseparationselectivity(viaπ-πinteraction)andefficiency(concentrationintightzonesviaelectrostaticrepulsion)Sodiumdodecylsulfate(SDS)addedintobackgroundelectrolyte(inconcentrationabovecriticalmicelleconcentra-tion) strongly interacts with hydrophobic alkyl groups in covalent coatingstructureThenegativelychargedSDSlayerisformedoninnercapillarysurfaceThedoublereversingEOFallowsustocarryoutelectrokineticinjectionofsampleandon-linepreconcentrationbysweepingsimultaneouslyLODweredeclineto
ndash108ndash20ngmL ThelengthofalkylsubstituentalsoaffectsthestackingefficiencyfactorandLODMorehydrophobicoctylgroupscomparetobutylprovidemoreeffectiveinteractionwithSDSandasresultlowerLOD
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 37
Covalentcoatingmodifiedβ-CDhasnotshownsharpreducingofLODbyon-linepreconcentrationStacking sweeping (SDSasmicelle reagent) field-enhancedsample injection were examined using different model mixtures of analytesNeverthelessthiscoatingallowssimultaneousseparationofbothofhydrophobicsteroidhormonesandhydrophilicbiogenicaminesinasinglerun(Fig2) The guest-host interaction hydrophobic cavity of β-cyclodextrin with thehydrophobicsteroids leadsto the formationofcomplexwhichaffectssteroidselectrophoretic mobility At the same time β-cyclodextrin can act as a chiralselector and baseline separation of ketoprofen enantiomers has also beenachieved
38 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Electropherogramofmixtureof neurotransmitters and theirmetabolitesadrenaline (A)norepinephrine (NE)normetanephrine (NMN)dopamine (DA)metanephrine (Met) serotonin(SER)homovanillicacid(HVA)andtheinnerstandard24-dihydroxybenzoicacid(24-DHBA)oncovalentlymodifiedwithN-buthylimidazoliumionicliquidscapillaryConditions10mMNaH PO 2 4
(adjusted to pH= 20 by 1MHCl) injection 50 stimes30mbar ndash20 kV 220 nmmodelmixurendash1 ndash1 ndash110microgmL (METADNMNNADA24-DHBA)5microgmL (SER)and20microgmL (HVA)
4Conclusions
ItwasshownthatstructurecovalentcoatingaffectsitsanalyticalcharacteristicsWecomparedtwotypesofcovalentcoatingdifferingsubstituentinimidazoliumringnamelyalkylgroupandβ-cyclodextrinThefirsttypeisgreatcoupledwithon-line preconcentration technic but it is limited to effectively determine ofbiogenicaminesonlywhilethesecondtype(withβ-cyclodextrin)showedthepossibilitiestoseparatevariousanalytesbutsuitableon-linemodehasnotbeenfoundThemainpointsaresummarizedinTable1
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 39
Covalentcoatingtype Electrophoreticseparationof On-lineprecon- centration biogenicamines amino steroid ketoprofen andtheirmeta- acids hormones enantiomers bolites N-β-cyclodextrinimida- yes yes yes yes thesuitableapproachzoliumcovalentcoatings wasnotfoundedN-alkylimidazolium yes yes nonsepa- nonsepa- thesignificantreducingcovalentcoatings rated rated forbiogenicaminesLOD
Table 1Thesummationofpossibilitiesofcovalentcoatingsbasedonimidazoliumcation
Fig 2Electropherogramofsimultaneousseparationofhydrophobic(steroidhormones)andhydro-philicanalytes (aminoacidsandbiogenicamines) insinglerunwithcovalentcoatingbasedonimidazoleandβ-CDConditions10mMNaH PO (adjusted topH = 20by1MHCl) injection2 4 20 stimes30mbarndash20kV254nm(1ndash8min)and220nm(8ndash15min)0mbar(1ndash10min)and40mbar(10ndash15 min) Model mixture corticosterone (B) hydrocortisone (F) 11-deoxycortisole (S)
ndash1 ndash15μgmL L-tryptophan(Trp)34-dihydroxy-L-phenylalanine(DOPA)10μgmL L-tyrosine(Tyr)ndash15 μg mL noradrenaline (NA) normetanephrine (NMN) adrenaline (AD) dopamine (DA)
ndash120μgmL
Acknowledgments
This work was supported by Russian Science Foundation (grant numbers 19-13-00370) Theauthors are also grateful to the Chemistry Education Centre and Nanothechnologies Centre ofResearchParkSaintPetersburgStateUniversityfortechnicalsupport
References
[1] HuLFYinSJZhangHYangFQRecentdevelopmentsofmonolithicandopen-tubularcapillaryelectrochromatography(2017ndash2019)JSepSci43(2020)1942ndash1966
[2] KartsovaLAKravchenkoAVKolobovaEACovalentcoatingsofquartzcapillariesfortheelectrophoretic determination of biologically active analytes J Anal Chem 74 (2019)729ndash737
[3] HoTDZhangCHantaoLWAndersonJLIonicliquidsinanalyticalchemistryFundamen-talsadvancesandperspectivesAnalChem86(2014)262minus285
[4] TangSLiuSGuoYLiuXJiangSRecentadvancesofionicliquidsandpolymericionicliquids incapillaryelectrophoresisandcapillaryelectrochromatography JChromatogrA1357(2014)147ndash157
[5] QinWLiSFYElectrophoresisofDNAinionicliquidcoatedcapillaryAnalyst128(2003)37ndash41
[6] QinWWeiH Li SFY 13-Dialkylimidazolium-based room-temperature ionic liquids asbackgroundelectrolyteand coatingmaterial in aqueous capillaryelectrophoresis JChro-matogrA985(2003)447ndash454
[7] QinW Fong S Li Y Determination of ammonium andmetal ions by capillary electro-phoresisndashpotential gradient detection using ionic liquid as background electrolyte andcovalentcoatingreagentJChromatogrA1048(2004)253ndash256
[8] QinWLiSFYAn ionic liquidcoating fordeterminationofsildenafilandUK-103320 inhumanserumbycapillaryzoneelectrophoresis-iontrapmassspectrometryElectrophoresis23(2002)4110ndash4116
[9] BorissovaMVaherMKoelMKaljurandMCapillaryzoneelectrophoresisonchemicallybondedimidazoliumbasedsaltsJChromatogrA1160(2007)320ndash332
[10] KolobovaEKartsovaLKravchenkoABessonovaEImidazoliumionicliquidsasdynamicand covalent modifiers of electrophoretic systems for determination of catecholaminesTalanta188(2018)183ndash191
[11] KravchenkoAKolobovaEKartsovaLMultifunctioncovalentcoatingsforseparationofaminoacidsbiogenicaminessteroidhormonesandketoprofenenantiomersbycapillaryelectrophoresisandcapillaryelectrochromatographySepSciplus3(2020)102ndash111
40 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Synthetic 4-hydroxy-3-methoxybenzaldehyde (vanillin) is used as a flavoringagent in foodsdrinksperfumesandpharmaceuticals [1]However at certainconcentrationsthesubstancemayaccumulateinthebodyhaveatoxiceffectand
ndash1at high concentrations may be fatal (lethal dose LD (oral rat) =2gkg 50ndash1 ndash1LD (oral guinea pig) = 14gkg LD (intravenous dog) = 132gkg lethal50 50 ndash1concentrationLC (inhalationmouse)=417gkg )[2]AccordingtoRussianState
StandartGOST121005-88thetoxiceffectsofvanillinintheworkplaceinthendash3formofvapoursoraerosolsareobservedatconcentrationsabove15mgm
Chromatography[3]spectrophotometry[4]capillaryelectrophoresisareusedforvanillindeterminationindifferentobjects CurrentlysmokingmixturesforhookahsandelectroniccigarettesarewidelyusedamongyoungpeopleThesemixturesarenotcontrolledforthecontentofsubstancesandarefreelyavailableconsideringthemmoreharmlesswithrespecttoordinarycigarettesThusthedevelopmentofamethodforthedeterminationof4-hydroxy-3-methoxybenzaldehydeinsmokingmixturesisrelevant
Determination of vanillin in smoking mixtures by spectrophotometry
ELIZAVETAEFREMENKOANNACHERNOVAOLGABASTRYGINA
DepartmentofChemicalEngineeringNationalResearchTomskPolytechnicUniversityLeninavenue30634050TomskRussiaeaetpuru
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 41
AbstractTheresearchdealswithdeterminationofvanillin insmokingmix-turesbyultraviolet-visiblespectrophotometryThemethodshowed
ndash1goodlinearityintherangeof005ndash012gL withalimitofdetectionndash1005gL After validation studies the method was successfully
applied to thedeterminationof vanillin in smokingmixtureswithsatisfactoryresultsItwasshownthattheerrorofthismethoddoesnot exceed 1 The developed spectrophotometric procedure fordeterminingvanillininsmokingmixturescanbeusedasacontrol
Keywordssmokingmixturesspectrophotometryvanillin
2Experimental
21Reagentsandchemicals
Asampleofvanillin(purity98)wastakenastheobjectofstudyAssolventsweused95ethanolAllchemicalsusedwereofanalyticalreagentgrade
22Instrumentation
Theopticaldensityofsampleswasmeasuredincuvettewithanabsorbinglayerthickness of 10 mm using a Cary 60 spectrophotometer (Agilent USA) Allmeasurementswerecarriedoutatroomtemperature
23Samplepreparation
Sample preparation of the investigated objects consisted of the preliminarydissolutionofthesamplein95ethanolThesample10mgoftobaccoldquoAdalyandashVanillardquo(Turkey)wasdiluted in10μLof95ethanol to theconcentrationof
ndash11gL Thesample10μLofldquoFlavoringTPAndashVanillaCustardrdquo(USA)wasdilutedin10 μL of 95 ethanol The resulting solution was diluted six times to the
ndash3concentrationof017μLcm
3Resultsanddiscussion
TodeterminevanillininthesamplestheopticalpropertiesofvanillininvarioussolventsweredeterminedAsaresultthe95ethanolwaschosenastheoptimalsolvent[4] IthasbeenestablishedthatintheUVspectraoftheanalyteabsorptionbondsareobservedwithmaximumvaluesat23002800and3100nmwhichcorres-pondstopublisheddata[45](Fig1) To quantify vanillin the calibration curve of the optical density on theconcentrationofvanillin in95ethanolwasobtainedatconcentrations005
ndash1006007008010and012gL Calibrationcurveofvanillinin95ethanolatawavelengthof280nmis
ndash1 A =81914c[gL ]+00357 (1)2802 R =1
Calibrationcurveofvanillinin95ethanolat310nmis
ndash1 A =73824c[gL ]+00301 (2)3102 R =1
42 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Inthespectraoftheanalyzedsamplesolutionsabsorptionmaxima(2800nmand3100nm)characteristicforvanillinwereobservedTheamountofvanillininthesamplewasdeterminedusingcalibrationcurvesat280and310nmWeightedleast square regressionwas applied to the calibration curves to improve theaccuracyespeciallyatinlowconcentrationlevelrangeGoodlinearitywasfound
ndash1 ndash1intherangeof005ndash012gL withadetectionlimitof005gL TheresultsarepresentedintheTable1
4Conclusions
ThedevelopedmethodcanbeusedasacontrolmethodTheerrorinthemethodfordeterminingvanillininthesampleldquoFlavoringTPAndashVanillaCustardrdquowithaknownconcentrationofvanillinwas0004Accordingtothedataobtainedwerecommendawavelengthof280nmforthedeterminationofvanillininsamples
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 43
λ nm
Absorban
ce
ndash1Fig 1 Absorption spectrum of vanilin solution in 95 ethanol at concentration 01 mol L (anabsorbinglayerthicknessof10mm)
Sample λnm Tookmg Foundmg S Sх Δх δ
FlavoringTRA 310 10850 104096 00024 00011 00006 00306ndashVanillaCustardAdalyandashVanilla 310 100000 10162 00019 00009 00002 00025
FlavoringTRA 280 100300 96062 00033 00015 00032 00042ndashVanillaCustardAdalyandashVanilla 280 100000 10122 00013 00006 00012 00017
Table 1Testingmethods introduced foundof vanillin in the samplesat310nmby spectrophotometricmethod(n=5p=099SndashstandarddeviationSxndashrelativestandarddeviationΔхndashabsoluteerrorδndashrelativeerror)
ThenthedevelopedmethodwastastedonthesampleldquoAdalyandashVanillardquosamplewithamorecomplexcompositionandanunknownconcentrationofvanillinwastaken The vanillin content in the sample was determined according to thedevelopedmethoditamountedto10ofthetotalmassStudieshaveshownthepossibility of using spectrophotometric analysis for the qualitative andquantitative determination of vanillin Also based on preliminary studies aspectrophotometricprocedurewasdevelopedforthequantitativedeterminationofvanillinbasedonabsorptioninethanolinthewavelengthrange200ndash400nm
References
[1] httpswwwrusnaukacom43_DWS_2015Chimia6_203179dochtm (accessed 25thFebruary2019)
[2] httpswwwcdcgovnioshrtecsdefaulthtm(accessed11stApril2020)[3] AliLPerfettiGDiachenkoGRapidmethodforthedeterminationof342coumarinvanillin
and ethyl vanillin in vanilla extract by reversed-phase liquid 343 chromatography withultravioletdetectionJAOACInt91(2008)383ndash386
[4] БастрыгинаОАЕфременкоЕАЧерноваАПВыделениеванилинаисследованиеегооптическихсвои ствопределениевбиологическомматериалеВХимияихимическаятехнология в XXI веке Материалы XX Международной научно-практическойконференции имени профессора ЛП Кулёва студентов и молодых ученых ТомскНациональныи исследовательскии Томскии политехническии университет 2019с301ndash302
[5] WeastRCHandbookofChemistryandPhysics60thedBocaRatonCRCPress1979p143
44 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
UraniumbelongstothegroupofhazardouselementsItisahighlyharmfulandradioactiveelementtoxictohumansandalllivingorganisms[12]Inhaledwithair it has a particularly destructive effect on the kidneys and as a result ofaccumulationinwhitebloodcellsitcanalsocauseimpairmentoftheimmunesystem[2]Uraniumoccursatseveraldegreesofoxidationhoweverinaqueoussolutionsthemoststableformisuranylion(UO (II))[12]Thepresenceofura-2
niumintheenvironmentiscausedbyamongothersnaturalsoilandrockerosionEnvironmentalpollutionwiththiselementisalsoconstantlyincreasingduetohumanactivitycoalcombustionuraniumoreminingandprocessingthearmsindustryandtheuseofuraniumasnuclearfuelinfissionreactors[3]Itisveryimportanttoconstantlymonitortheconcentrationofuraniumbothinthenaturalenvironment in order to assess its state and safety (especially in the case ofdrinkingwater)aswellasinallstagesofprocessingprocessesassociatedwiththenuclearindustrytoavoidtheoccurrenceofnuclearpollution[13] Scientists have made many attempts to develop research methods todeterminethecontentofuranylcompoundsinliquidsamplesEffortsweremadetousemanyanalyticalmethodsforthispurposeincludingspectrophotometry
Uranyl ion-selective electrode with solid contact
KAROLINAPIETRZAKCECYLIAWARDAK
DepartmentofAnalyticalChemistryInstituteofSciencesFacultyofChemistryMariaCurie-SklodowskaUniversityMariaCurie-SklodowskaSq320-031LublinPolandkarolinapietrzakpocztaumcslublinpl
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 45
AbstractNewallsolidstateuranylion-selectiveelectrodeswithlowdetection
ndash7 ndash1limits(71times10 molL )shortresponsetimegoodselectivityandstable and reproducible potential were developed Many types ofelectrodeswith different active ingredient content in ion-selectivemembrane (bis(244-trimethylpentyl)phosphonium acid Cyanex-272)were testedAs an additive an ionic liquid1-octyl-3-methyl-imidazole chloride was used The optimal composition of theion-selective membrane was chosen from all electrodes based onthedeterminationand comparisonof analyticalparametersof thesensors
Keywordsion-selectiveelectrodesolidcontacturanyl
plasma spectrometry luminescence spectroscopy voltammetry or chromato-graphymethods[2] Duetomanyadvantagesofpotentiometricmethods(amongthemlowercostseasieroperationofdevicesquickresponseandtheabilitytoperformmeasure-ments in flowmode) [3] a numberof potentiometric sensorshave alsobeendeveloped that could be successfully used in this type of research Themostpopularpotentiometricsensorsincludeion-selectiveelectrodes(ISEs)whicharecharacterized by low-energy consumption small size and portability and aresuccessfullywidelyusedforthedeterminationofbothinorganicandorganicionsinclinicalanalysisprocesstechnologyaswellasincontrolthestateofthenaturalenvironment[45]Removaloftheinternalsolutioncontainingthesameanalytetowhich theelectrode is sensitiveresulted in theso-calledsolidcontact ISEswhicharemuchsmallerinsizethantheirpredecessorsaremoreconvenienttouse and more mechanically resistant In this type of sensors however it isimportanttoachievesatisfactorypotentialstabilitywhichisnecessarytoobtainsatisfactoryresults[5]AveryimportantpartofISEsistheion-selectivemem-branewhosecompositiondeterminestheanalyticalparametersofthesensorsResearchers are currently focusing on the production and testing of newsubstancesthatcouldbesuccessfullyusedasmembranecomponentsandsolidcontacts thatwould allow to obtain new sensorswith lower detection limitslongerlifetimeandbetterpotentialstabilityandtodeterminenewpreviouslyunattainableanalytes[4] AstheactivecomponentsofthemembranesensitivetouranylionscientistshavealreadyusedKryptofix22DD(413-didecyl-171016-tetraoxa-413-diaza-cyclooctadecane)[2]Cyanexextractants(bis(244-trimethylpentyl)phosphinicacid bis(244-trimethylpentyl)monothiophosphinic acid and bis(244-tri-methylpentyl)dithiophosphinic)acid[3]DBBP(dibutylbutylphosphonate)andDOPP (di-n-octyl phenylphosphonate) [6] DMSO (dimethylsuphoxide) [7]TTPTP (5678-tetrahydro-8-thioxopyrido[4345]thieno[23-d]pyrimidine-4(3H)one)[8]orTEHP(tris(2-ethylhexyl)phosphate)andTPTU(O-(12-dihydro-2-oxo-1-pyridyl)-NNNN-bis(tetra-methylene)uronium hexafluorophos-phate)[9]
2Experimental
21Reagentsandchemicals
This paper presents research on the design and properties of ion-selectiveelectrodes with solid contact for the determination of uranyl ions Bis(244-trimethylpentyl)phosphonium acid (Cyanex-272) was used as the activecomponentof themembranewhichwasdescribed in the literatureasagooduranylextractant[10]Inordertoensureaconstantpotentialofthiselectrodeandreducetheelectroderesistancetheion-sensitivemembranewasenrichedwithafewpercentadditionof1-octyl-3-methylimidazolechlorideionicliquid
46 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Several types of ion-selective electrodes were prepared using an AgAgClelectrodeasaninternalelectrodewhichdifferinthequantitativeandqualitativecompositionofthemembranesAllcompositionsarelistedinTable1
22Instrumentation
Measurements were made at room temperature using a 16-channel datacollectionsystem(LawsonLabs IncUSA)coupled toa computer in solutionsmixedwithamechanicalstirrerAsilversilverchlorideelectrodewithdoublejunctionwasusedasthereferenceelectrode
3Resultsanddiscussion
The effect of ion-selective membrane composition on the properties of theobtained potentiometric sensors was examined by determining their basicanalyticalparametersincludingslopeoftheelectrodecharacteristicsdetectionlimitmeasuringrange(concentrationrangeinwhichthecourseoftheelectrodecharacteristics isrectilinear)pHrange(inwhich ithasnoeffect forelectrodepotential)andresponsetimeTheobtainedvaluesofthetestedparametersareshowninTable2 Figure1showsthecalibrationcurvesofthetestedelectrodesdeterminedin
ndash7 ndash1 ndash1UO (NO ) solutionsintheconcentrationrange1times10 ndash1times10 molL Asitcan2 3 2
beseeninFig1andTable2allelectrodesweresensitivetouranylionsbutindifferent extend The best response exhibited ISE-3 containing 1 (ww) ofionophore Increasing the ionophore content in themembrane shortened thelinearityrangeofthecalibrationcurveanditssupernenstianslope Theselectivityofthetestedelectrodeswasestimatedbydeterminingtheselec-tivitycoefficients inrelationto interfering ionsForthispurpose theseparate
ndash1solutionmethodwasused(extrapolatingresponsecurves toa =a =1molL )i j
ComparisonofISE-1andISE-3electrodeselectivityisshowninFig2
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 47
Table 1Quantitative and qualitative composition of electrode membranes Cyanex-272 (bis(244-tri-methylpentyl)phosphoric acid) TBP (tri-n-butyl phosphate) and OMImCl (1-octyl-3-methyl-imidazolechloride)
Abbreviation Membranecomposition(ww)ofelectrode Cyanex-272 PVC TBP OMImCL
ISE-1 00 33 620 5ISE-2 05 33 615 5ISE-3 10 33 610 5ISE-4 30 33 590 5ISE-5 50 33 570 5ISE-6 100 33 520 5
48 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Abbreviation Slope Detectionlimit Linearrange Response pHrange2+ ndash1 ndash1ofelectrode mVpa(UO ) molL molL times2
ndash5 ndash5 ndash1ISE-1 297 25times10 5times10 ndash1times10 5ndash8 28ndash42ndash6 ndash5 ndash1ISE-2 292 65times10 1times10 ndash1times10 5ndash8 25ndash60ndash7 ndash5 ndash1ISE-3 298 71times10 1times10 ndash1times10 5ndash8 24ndash60
ndash6 ndash4 ndash1ISE-4(I) 357 31times10 5times10 ndash1times10 5ndash8 ndndash6 ndash6 ndash4ISE-4(II) 242 31times10 5times10 ndash5times10 5ndash8 nd
ndash3 ndash1ISE-5(I) 638 nd 1times10 ndash1times10 5ndash10 ndndash5 ndash3ISE-5(II) 234 nd 5times10 ndash1times10 5ndash10 ndndash3 ndash1ISE-6(I) 733 nd 1times10 ndash1times10 5ndash10 ndndash5 ndash3ISE-6(II) 222 nd 5times10 ndash1times10 5ndash10 nd
Table 2Selectedparametersandtheirdeterminedvaluesoftestedionselectiveelectrodes
Fig 1 Calibration curves of the testedelectrodesobtainedinUO (NO ) solutionsin2 3 2 ndash7the concentration range from 1times10 to
ndash1 ndash11times10 molL
Fig 2Comparisonofselectivitycoefficientspot(log K (UO (II))M) for electrodes ISE-12
(1stcolumn)andISE-3(2ndcolumn)
Inordertoexaminethereversibilityofthepotentialofthetestedelectrodesndash4 ndash1potentialmeasurementsweremadealternatelyinsolutions1times10 molL and
ndash5 ndash11times10 molL ofUO (NO ) TherecordedpotentialreadingsareshowninFig32 3 2
Long-term potential stability and sensor reproducibility were evaluated byndash1determiningtheaveragevalueoftheelectrodepotentialina01molL UO (II)ion2
solutionovertimeforthreeidenticalISE-3Thesemeasurementsweremadetoobservechangesinthepotentialofelectrodeswiththesameconcentrationoveralongperiodoftime(30days)Figure3showsthelong-termpotentialstabilityandreproducibilitydeterminedforthreeidenticalsensors
4Conclusions
Asaresultofthetestsion-selectiveelectrodeforthedeterminationofuranylionswasobtainedwhich iseasy todesignanduseThebestanalyticalparametersexhibitedISE-3containing1ionophoreintheion-selectivemembraneForthis
ndash7 ndash1typeofelectrodesthedetectionlimitof71times10 molL linearityoftheelectrodendash6 ndash1 ndash1calibrationcurve in the range1times10 ndash1times10 molL andresponse time5ndash8s
were obtained In addition the manufactured sensors also showed stablereproducibleandreversiblepotentialandverygoodselectivityinrelationtothetestedinterferents
References
[1] AnsariRMosayebzadehZConstructionofanewsolid-stateU(VI)ion-selectiveelectrodebasedonpolypyrroleconductingpolymerJRadioanalNuclChem299(2014)1597ndash1605
[2] GhanbariMRounaghiGHAshrafNAnuranylsolidstatePVCmembranepotentiometricsensor based on 413-didecyl-171016-tetraoxa-413-diazacyclooctadecane and itsapplicationforenvironmentalsamplesIntJEnvironAnalChem97(2017)189ndash200
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 49
Fig 3 Stability () reproducibility andreversibility () of the potential of ISE-3Standard deviations given on the plot aredeterminedforthesamethreeISE-3
[3] Badr IHA Zidan WI Akl ZF Cyanex based uranyl sensitive polymeric membraneelectrodesTalanta118(2014)147ndash155
[4] BiegCFuchsbergerKStelzleMIntroductiontopolymer-basedsolid-contaction-selectiveelectrodes basic concepts practical considerations and current research topics AnalBioanalChem409(2017)45ndash61
[5] Bobacka J IvaskaA LewenstamA Potentiometric ion sensorsChem Rev108 (2008)329ndash351
[6] ZidanWI Badr IHA Akl ZF Development of potentiometric sensors for the selective2+determinationofUO ionsJRadioanalNuclChem303(2015)469ndash4772
[7] SalehMBSolimanEMGaberAAAAhmedSANovelPVCmembraneuranylion-selectivesensorSensActuatorsB114(2006)199ndash205
[8] SalehMBHassanSSMAbdelAAAbdelNAAnoveluranylion-selectivePVCmembranesensor based on 5678-tetrahydro-8-thioxopyrido[4345]thieno[23-d]pyrimidine-4(3H)oneSensActuatorsB94(2003)140ndash144
[9] HassanSSMAliMMAttawiyaAMYPVCmembranebasedpotentiometricsensorsforuraniumdeterminationTalanta54(2001)1153ndash1161
[10] Prabhu DR Ansari SA Raut DR Murali MS Mohapatra PK Extraction behaviour ofdioxouranium(VI) cation by two phosphorous-based liquid cation-exchangers in room-temperatureionicliquidsSepSciTechnol52(2017)2328ndash2337
50 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Metronidazole(2-methyl-5-nitroimidazole-1-ethanol)isoneofthemostwidelyused nitroimidazole antibiotics Metronidazole is used for the treatment ofinflammatorydiseasescausedbyanaerobicorganismsandsomeprotozoaandforpreventionofdysenterycolibacillosiseimeriosisbalantidiasissalmonellosisenteritissepticemiapost-surgicalcomplications[1ndash3]Oxytetracyclinehydro-chlorideisanantibioticofthetetracyclinefamilyItisoneofthemostcommonlyused antibiotics in poultry because of its low cost and effective [4] Thesecompounds are intensively used in poultry breeding and stockbreedingUnreasonableuseofthesedrugscancauseseriousfoodsafetyissues[5] The veterinary drug Nozemat which includemetronidazole and oxytetra-cyclinehydrochloridewaschosenfortheexperimentsNozematisusedtotreat
Polarographic determination of metronidazole and oxytetracycline hydrochloride in veterinary drug for honey bees
a a bKATERYNAPLOTNIKOVA LILIYADUBENSKA IVANZELENYI
a AnalyticalChemistryDepartmentIvanFrankoNationalUniversityofLvivKyrylaiMefodiaStr879005LvivUkrainekaterina27plgmailcom
b DrohobychPedagogicalLyceumIvanaFrankaStr3682100DrohobychUkraine
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 51
AbstractWehavedevelopedanewpolarographicmethodforthedetermin-ation of metronidazole and oxytetracycline hydrochloride in theveterinarydrugNozematforhoneybeesThetechniqueisbasedonthereductionofpolarographicallyactivecompoundsonamercurydropletelectrodeTheinfluenceofthecomponentsoftheveterinarydrugNozematonthepolarographicdeterminationofmetronidazolewasstudiedItwasfoundthatthereductionofmetronidazoleisnotaffected by glucose and ascorbic acid but is affected by oxytetra-cyclinehydrochloridewhichisreducedtomercurydropletelectrodeatapotentialofndash145VThedevelopedtechniqueischaracterizedbyeaseofsamplepreparationandcost-effectivenessThistechniquehastheabilitytoidentifysimultaneouslyanddeterminatemetronidazoleand oxytetracycline hydrochloride in solution without the use ofseparationandconcentrationmethods
Keywordselectrochemistrymetronidazoleoxytetracycline
hydrochloridepolarographyveterinarydrug
bees and it can be given in unregulated doses Because of this an unknownamountofmetronidazolecangetintothehoneyanditsometimescausessideeffectsofthehumanbodyanditcouldbeofgreatconcernforpublichealth[56]MedicinesforpeoplearemorestringentandbettertestedthanveterinarydrugsTheproblemofthecontroloftheveterinarydrugsisurgentnowadaysVeterinarymedicinescouldbeunauthorizedandtheuncontrolleduseofmedicinesexistsinretailpharmaciesofmedicineorimportedascontrabandfromothercountries The most widespreadof these classes in thequality controlare chromato-graphic [6ndash9] spectrophotometric [10ndash13] and electrochemical methods[14ndash17]Manyoftheknownmethodsforthedeterminationofmetronidazoleandoxytetracyclinehydrochloridehaveanumberofdisadvantagestime-consumingtheuseoforganicsolventsandexpensivereagentsthesideeffectsofexcipientsandotheractivesubstancesElectrochemicalmethodsarepromisingalternativefor the determination of the electroactive substances Their advantages aresimplicityminiaturizationhighsensitivityandrelativelylowcostThereforethesearch for simple express and affordable methods for the determination ofmetronidazoleremainsrelevantOneofthepromisingmethodsofdeterminationisvoltammetry
2Experimental
21Reagentsandchemicals
VeterinarydrugNozemat (manufacturerAPI-SANRussia) is a yellowpowderwithaslighttypicalodorAvailableinlaminatedbagsof25gCompositionper1gof the drug metronidazole 400 mg oxytetracycline hydrochloride 400 mgglucoseascorbicacid MetronidazoleandoxytetracyclinehydrochloridewerepurchasedfromSigmaAldrich(USA)Stockstandardsolutionofmetronidazolefordeterminationwaspreparedbydissolvingtheexactamountofstandardin7mLof2Mhydrochloricin 500 mL volumetric flask Stock standard solution of oxytetracyclinehydrochloride was prepared by dissolving the exact amount of standard indistilledwaterin500mLvolumetricflaskAfterthatthesolutionswereadjustedtothemarkwithdistilledwaterandmixedthoroughly The Britton-Robinson buffer preparationwas as follows 202 g of sodiumtetraboratedecahydrate287mLofglacialaceticacidand176mLofconcen-tratedorthophosphoricacidweredissolvedin10Lvolumetricflask Working solution preparation was as follows an aliquot of stock standardsolutionwasaddedintoa25mLvolumetricflasktoobtainasolutionwiththenecessaryconcentrationthen2mLofBritton-RobinsonbufferwithnecessarypHwasaddedtotheflaskanddistilledwaterwasaddedtothemark AqueoussolutionofNozematwaspreparedasfollowstheexactportionofthetestveterinarydrugwasdissolved ina250mlvolumetric flaskAnaliquotof
52 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Polarogramsof(A)metronidazoleand(B)metronidazolewithoxytetracyclinehydrochloridendash1solutionsat02MBritton-RobinsonbufferbackgroundatpH=96(υ=05Vs c(metronidazole)=
ndash5 ndash5=45times10 Мc(oxytetracyclinehydrochloride)=50times10 М
100mloftheresultingsolutionwasaddedtoa250mlvolumetricflaskandmadeuptothemarkwithwaterAnaliquotof100mloftheresultingsolutionwasaddedtoa250mlvolumetricflask2mlofBritton-RobinsonbufferwithapHof96wasaddedandthevolumewasadjustedtothemarkwithdistilledwater
22Instrumentation
ForpolarographicmeasurementsweuseddigitaldeviceMTechOVA-410 [18]temperature-controlledthree-electrodeamercurydropletindicatorelectrodeasaturatedcalomelreferenceelectrodeandplatinumwireauxiliaryelectrodeTheaccuracyofthepotentialmeasurementis1mVTheuncertaintyofcurrentmeasu-rement is 01 The employed mercury droplet electrode had the following
ndash4 ndash1characteristicsm=594times10 gs τ=10 min in 02 M NH Cl We used cyclic4
voltammetryforthestudyoftheelectrochemicalprocess WeusedMV870DIGITAL-pH-MESSERATpH-meterformeasuringpHofthesolutions Theobtainedworkingsolutionswereintroducedintothecellanddeoxyge-natedwithargonfor10minPolarogramswererecordedintherangeofpotentialsfrom00tondash16V
3Resultsanddiscussion
Previously it was found that using the Britton-Robinson buffer with pH = 96metronidazoleisreducedwiththeformationofacharacteristiconeirreversiblepeakatndash064V(Fig1A)UsingpolarographywithfastpotentialscanitwasfoundthatmetronidazoleisreducedonmercurydropletelectrodeintherangeofpH20to105ThepeakrecoverycurrentofthemetronidazolereachesthemaximumvalueatpH9ndash10againstthebackgroundofatheBritton-Robinsonbuffer
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 53
Underpre-selected conditions theeffectof some foreign substanceson thepolarographicdeterminationofmetronidazolewasinvestigatedSubstancesthatare componentsofdrugswere studiedglucose ascorbicacidoxytetracyclinehydrochlorideGlucose and ascorbic acid are not reduced atmercury dropletelectrodeanddonotchangetheappearanceofthepolarogramandpolarographiccharacteristics of the recovery of metronidazole As can be seen from Fig 1oxytetracycline hydrochloride is reduced to mercury droplet electrode andchangestheappearanceofthepolarogramandpolarographiccharacteristicsofthe recovery of metronidazole With the addition of oxytetracycline hydro-chloridetherecoverypeakofmetronidazoledecreasesandslightlyshiftstomorenegativepotentials The composition of the drug is relatively complex excipients affect theanalyticalsignaloftherecoveryofcompoundssototakeintoaccountthematrixeffectusedthemethodofmanyadditives QuantitativelytransferredthesolutionofNozemattothecell(exactvolume)removed dissolved oxygen for 10 min and took polarograms in the range ofpotentialsfrom00tondash16VAliquotsofstandardmetronidazolesolutionwereintroducedintothecelltoobtainasolutionwithagivenconcentrationofadditive
ndash5 ndash5metronidazole 10times10 M to 70times10 M As with the determination of themetronidazole aliquots of a standard oxytetracycline hydrochloride solutionwereaddedtothecellwithsolutionofNozemattoobtainasolutionwithagiven
ndash6 ndash6additiveconcentrationfrom70times10 Mto50times10 M(Fig2) In Table 1 are shown metrological characteristics of the determination ofmetronidazoleandoxytetracyclinehydrochloride inveterinarydrugRecoverywascalculatedFormetronidazoletherecoveryis97andforoxytetracyclinehydrochloridetherecoveryis103Analyticalperformanceofthetechniqueisgoodfordeterminationveterinarydrugs
54 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2Polarogramsof(A)metronidazolereductionatdifferentmetronidazoleconcentrationsand(B)oxytetracyclinehydrochloridereductionatdifferentoxytetracyclinehydrochlorideconcentra-tionsandtheircorrespondingcalibrationgraphs
Theaccuracywasverifiedbytheldquoadded-foundrdquomethodAliquotsofstandardsolutionofmetronidazoleweremadeina250mlvolumetricflasktoobtaina
ndash5solutionofagivenconcentrationof33times10 Mandthesolutionofoxytetracyclinendash5hydrochloridetoobtainasolutionofagivenconcentrationof15times10 M2mlof
Britton-RobinsonbufferwithpH96wasaddedtoflaskwithstirringandadjustedtothemarkwithwaterTheanalysisprocedureofmodelsolutionissimilartoanalysis procedure of the solution of Nozemat The calculated amount ofmetronidazolebythemethodofmultipleadditivesinthetestedmodelsolutionisinagreementwiththeamountthatwasintroducedintothesample
4Conclusions
The new polarographic method for the determination of metronidazole andoxytetracyclinehydrochlorideintheveterinarydrugNozematforhoneybeeswasdeveloped We conducted principal component analysis of veterinary drugNozemattoassesstheoveralleffectforthedeterminationofmetronidazoleWefoundthatoxytetracyclinehydrochlorideisreducedtomercurydropletelectrodeThismethodhastheabilitytoidentifysimultaneouslyanddeterminatemetro-nidazole and oxytetracycline hydrochloride in solution without the use ofseparationandconcentrationmethodsOnemoreofadvantagesoftechniquearefastprocedureofanalysissimplesamplepreparationlowcostthepossibilityofminiaturization
References
[1] Antibiotic and Chemotherapy Finch R Greenwood D Whitley R (edits) AmsterdamElsevier2006p292ndash299
[2] MitrowskaKPrzyczynyiskutkizakazustosowania5-nitroimidazoliuzwierzątktorychtkanki lub produkty przeznaczone są do spozycia przez ludziMed Weter 71 (2015)736ndash742
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 55
Metronidazole Oxytetracycline hydrochloride
PeakspotentialV ndash065 ndash144CorelationcoefficientR 099892 099922
ndash1 4 4SlopebmicroАM 656times10 176times10Δb 1763 400InterceptamicroА 2238 0219Δa 0075 0009
ndash1 ndash5 ndash5cmolL 342times10 125times10ndash1cmgg 389 413
Recovery 97 103
Table 1Validationparametersofthemethodofmetronidazoleandoxytetracyclinehydrochloridedetermi-nationinsolutionsofNozematbythemethodofmanyadditives
[3] VermaPNamboodiryVMishraSBhagwatABhoirSAstabilityindicatingHPLCmethodfor the determination of Metronidazole using Ecofriendly solvent as mobile phasecomponentIntJPharmPharmSci5(2013)496ndash501
[4] Cervini P Ambrozini B Machado LCM Ferreira Garcia AP Cavalheiro Gomes ETThermal behavior and decomposition of oxytetracycline hydrochloride J Therm AnalCalorim121(2015)347ndash352
[5] DangBNAnhNTKKyLXThaiPKAntibioticsintheaquaticenvironmentofVietnamsourcesconcentrationsriskandcontrolstrategyChemosphere197(2018)438ndash450
[6] QuintanillaPHettingaKABeltranMCEscricheIMolinaMPVolatileprofileofmaturedTronchon cheese affected by oxytetracycline in raw goat milk J Dairy Sci 103 (2020)6015ndash6021
[7] Chen F Yu L Jingdong P Xiang W Huanjun P Yu C Yan H Study on simultaneousdetermination of three nitroimidazole residues in honey by high performance liquidchromatographyndashresonanceRayleighscatteringspectraMicrochemJ141(2018)423ndash430
[8] Hernandez-MesaM Cruces-Blanco C Campana GA Simple and rapid determination of5-nitroimidazolesandmetabolitesinfishroesamplesbysalting-outassistedliquid-liquidextractionandUHPLC-MSMSFoodChem252(2018)294ndash302
[9] Xiu-ChunGZhao-YangXHai-HuiWWen-YiKLi-MingLWen-QingCHong-WeiZWen-HuiZMolecularlyimprintedsolidphaseextractionmethodforsimultaneousdeterminationofsevennitroimidazolesfromhoneybyHPLC-MSMSTalanta166(2017)101ndash108
[10] ТеплыхАНИлларионоваЕАКоличественноеопределениеметронидазоласпектро-фотометрическимметодомСибирскиймедицинскийжурнал5(2009)48ndash50
[11] ZheltvayOIZheltvayIISpinulVVAntonovichVPSpectrophotometricdeterminationofmetronidazoleandtinidazoleusingcopper(II)complexesJAnalChem68(2013)663ndash668
[12] Youssef AK Saleh MS Abdel-Kader DA Hashem Facile DY SpectrophotometricdeterminationofmetronidazoleandsecnidazoleinpharmaceuticalpreparationsbasedontheformatioonofdyesIntJPharmPharmSci6(2015)103ndash110
[13] Sversut RA Vieira JC Rosa AM Amaral MS Kassab NM Salgado H ValidatedspectrophotometricmethodsforsimultaneousdeterminationofoxytetracyclineassociatedwithdiclofenacsodiumorwithpiroxicaminveterinarypharmaceuticaldosageformArabianJChem13(2020)3159ndash3171
[14] Nikodimos Y Electrochemical determination of metronidazole in tablet samples usingcarbonpasteelectrodeJAnalMethodsChem(2016)361294
[15] Srivastava AK Upadhyay SS Rawool CR Punde NS Rajpurohit AS Voltammetrictechniques for the analysis of drugs using nanomaterials based chemically modifiedelectrodesCurrAnalChem15(2019)249ndash276
[16] Sahu G Voltammetric behaviour of metronidazole at a composite polymer membraneelectrodeOrienJChem26(2010)81ndash86
[17] Yang Y YanW Guo YWang X Zhang F Yu L Guo C Fang G Sensitive and selectiveelectrochemicalaptasensorviadiazonium-couplingreactionforlabel-freedeterminationofoxytetracyclineinmilksamplesSensorsandActuatorsReports2(2020)1ndash7
[18] httpchemlnueduuamtechdeviceshtml(accesed21stJune2020)
56 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
AnimportantpartofanyanalysisthatsignificantlyaffectsthefinalresultsisthesamplepreparationThelowconcentrationofbiologicallyactivecompoundsandthepresenceofaccompanyingcomponentspreventdirectanalysisofthesamplewithcomplexmatrixcompositionTraditionalmethodsofliquidandsolid-phaseextractionhaveaplentyoflimitationssuchashighlytime-consumingprocedureslarge volume of samples expensive cartridges toxic organic solvents andchallenges in automating the process Therefore the application of extractiontechniquesemployinglowamountofsolvents(microextractionmethods)andthelow toxicity extractantes has become the main research direction in recentyears[12] Solid-phasemicroextraction(SPME)wasproposedbyPavlishinin1989[3]Onevariantofthismethodistousethinrodswithvariouspolymercoatingssuch
Application of microextraction techniques combined with chromatographic methods for the analysis of complex objects
VLADISLAVDEEVELENABESSONOVALIUDMILAKARTSOVA
InstituteofChemistrySaint-PetersburgStateUniversityUniversitetskyprospect26198504PeterhofSaint-PetersburgRussiahitchervmailru
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 57
AbstractThelowconcentrationofanalytesandthepreventionofthematrixinfluence requires a stage for extraction and concentration of thestudiedcompoundsTheclassicalmethodsofliquidandsolid-phaseextractionhavemanylimitationsthatpreventtheiruseinsomecasesMicroextraction techniques are becoming more widespread WestudiedthepossibilityofusingionicliquidstoextractpesticidesfromwatersampleswiththeirsubsequentHPLS-MSdeterminationTheinfluenceonthedegreeofextractionofsuchparametersasanatureofionicliquidsanddispersersolventtheiramountssaltconcentrationvolumeratioofionicliquidsandwatersampledilutionoftheionicliquidsextractwithmethanolwasperformedBesidesconditionsofsolid-phase microextraction of volatile organic compounds fromurine samples obtained from healthy donors and donors withprostate cancer have been found The analysis of volatile organiccompoundsbyGC-MSfollowedbychemometricprocessingallowedachievingahighvalueofbinaryclassificationaccuracy(91)
Keywordschemometricsdispersiveliquid-liquid
microextractionsolid-phase
microextraction
asdivinylbenzenepolydimethylsiloxanepolyacrylateandpolyethyleneglycolwhichappliedtothesurface[4]Thepolymersorbentisplacedintheequilibriumheadspaceaboveacondensedphaseofthesampleandthevolatilecompoundsareextracted Liquidmicroextractionconsistsofusingsmallamountsofliquid(extractant)inequilibriumwiththegasorliquidphaseofthesampleDispersiveliquid-liquidmicroextraction(DLLME)isavariantofliquidmicroextractionTheessenceofthemethodisasfollowsextractantisdissolvedinthephaseofadispersingsolventandthemixtureisrapidlyinjectedintothesamplevolume[5]Inthiscasethedispersing solvent is dissolved and a ldquocloudrdquo of extractant is formed A largesurfaceareacontributestomasstransferprocessesThecombinationofDLLMEwiththeuseof ionic liquids(ionic liquids)asextractantsreducestheharmfulimpactontheenvironment[6] Sothegoalofthisstudywastheapplicationofmicroextractionmethodsfortheanalysisofrealsamples
2Experimental
21Reagents
Deionizedwaterwas obtained at the AQUILON D 301 deionizer (Russia) Allchemicals and reagents (the highest commercially available purity) werepurchasedfromReachimBakerAcrosorganicsandSigmaAldrich
22Instrumentation
HPLCanalysiswascarriedoutusinganHPLCLCMS-8030(Shimadzu)withatriplequadrupole mass-selective detector with electrospray ionization Analysis ofvolatileorganiccomponents(volatileorganiccompounds)ofurinesampleswasmade by GCMS-QP2010 SE (Shimadzu) Chemometric data processing wasperformedusingRStudio
23DeterminationofvolatileorganiccompoundsinurinesamplesbyGC-MSmethod
ForSPMEofvolatileorganiccompounds inurinesamplewasusedfibercoatedwith a polydimethylsiloxane (PDMS) The volatile organic compounds wereextractedontofibercoatingfor20minat50degCThentheanalytesweredesorbedinto the gas chromatography for 4 minutes at a temperature of 250degCChromatographic separation was carried out on a HP-5 capillary column(30mtimes250μmtimes025μm)usingtemperatureprogrammingmodeThetempera-
ndash1tureofovenwasincreasedfrom50degCupto250degCatarateof10degCmin Tionsourcewas200degCMassspectrometrywasusedinSIMmode(mz=35ndash900)
58 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
24ConditionsforLCMSMSdeterminationofpesticides
Separation of pesticides was performed by HPLCMSMS with positiveelectrosprayionizationoncolumnZorbaxBonusRP35μm(21times100mm)with40mMammoniumacetateandmethanolasmobilephaseAandBrespectivelyThefollowinggradientelutionwasapplied20ndash85B(8min)85B(8ndash15min)85ndash95B(150ndash155min)95B(155minus180min)95ndash20B(180minus185min)
ndash1Thevelocityof themobilephasewas03mlmin Thevolumeof the injectedsamplewas 20 microlMS detection capillary voltage +45 kV spray gas velocity
3 ndash1 3 ndash13dm min flow rate and drying gas temperature 15 dm min and 250 degCrespectively
25Selectionofconditionsfordispersiveliquid-liquidmicroextractionofpesticides
The influence of the natures of ionic liquids ([C MIM][PF ] [C MIM][NTf ]4 6 6 2
[C MIM][BF ])andthedispersingsolvent(methanolacetonitrileacetone) the6 4
weightoftheionicliquids(0060ndash0200g)thevolumeofthedispersingsolvent(02ndash10ml)onthedegreeofpesticidesextractionwerestudiedTheinfluenceofthepH(5422)theconcentrationofNaCl(0040ndash0200g)andextractiontime(1-6min)wereinvestigated The effects of different ionic liquids and disperser solvents on DLLMEprocedures were investigated and optimized by using standard solutions ofpesticidesIndetailasolutionofionicliquidsinadispersingsolventwaspreparedand rapidly injected into the aqueous sample solution (2ml) followed bytreatment for 2 min in an ultrasonic bath cooling at ndash4degC for 10 mincentrifugationfor10minat3500rpmandcollectionofionicliquidsThewaterphasewas separatedandanalyzedbyHPLC-MSThe ionic liquidsextractwasdilutedinmethanolandanalyzedbyHPLC-MS
3Resultsanddiscussion
31Microextractionofpesticides
One of the important tasks of environmental monitoring is to control traceconcentrationsofpesticidesinwatersamplesTechniqueofcombiningseveralpesticideshasbecomemorewidespread inagriculture It allowsreducing thetotalconcentrationoftheappliedcompoundsandtodecreasetheadaptabilityofpathogens and insects Therefore the analysis of real samples requires apreliminarystageofselectiveanalytesextractionandconcentration ExtractinganddispersingsolventsarebothimportantinDLLMEofanalytesTheinfluenceofthenatureofthedispersingsolvent(methanolacetonitrileandacetone) and the extractant (imidazolium ionic liquids [C MIM][PF ]4 6
[C MIM][BF ]and[C MIM][NTf ])onthedegreeofextractionofpesticideswas6 4 6 2
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 59
studied This parameter was controlled by the residual concentration ofpesticidesinthewaterphaseafterextractionThebestresultswereobtainedforionic liquids [C MIM][PF ] as an extractant and acetonitrile as a dispersing4 6
solvent Thenextstepwastoselecttheamountofionicliquids(0060ndash0200g)andthevolume of acetonitrile (02ndash10 ml) It was found that the highest degree ofextractionofanalyteswasachievedbyusing020gofionicliquidsand03mlofacetonitrile It was shown that the degree of extraction of selected pesticides does notdependonpHofthewatersamplewhichconfirmsthepartitionmechanismofextractionThedegreeofextractionofcarbofosincreasedslightlywithanincreaseinthesaltconcentrationandreachmaximumbyweightto4(008g) ItisknownthatthehighviscosityofionicliquidshavehinderedtheprocessesofelectrosprayionizationDilutionofthesamplewithmethanolby3timesgivesthebestresult(thesignalintensitywas373to845ofthesignalwithoutionicliquids) ThustheconditionsofDLLME-ionicliquidsextractionofpesticidesfromwatersampleswere found The limits of detection for pesticideswere from007 to
ndash1019ngml thereproducibilityofpeakareaswerefrom3to5theextractionrecoverywascloseto100
32SPMEofvolatileorganiccompoundsfromurinesamples
Oneof the importantdirections is the search for criteriaofearlydiagnosisofcancer Obtaining characteristic profiles of volatile organic compounds fromurinesamplescanhelpdevelopanon-invasivemethodforearlydiagnosisofthedisease ForthiswestudiedtheinfluenceofanumberoffactorsonthetotalnumberofpeaksandthetotalpeakareaTheywerethetemperatureatwhichthevaporandcondensedphasesofurinewerebalanced(30ndash60degC)thepreheatingtimeofthesample(10ndash40min)NaClconcentration(30ndash133wv)andthesorptiontimeonthePDMScoating(5ndash30min) Anincreaseinthepreheatingtemperatureofthesampleto500degCledtoanincreaseinthenumberofsignalswhichdidnotchangewithafurthertempera-tureincreasingNextparameterwasthetimeofachievementequilibriumofthevapor and condensed phases The largest number of peaks was observed at40minbutthisgreatlyincreasedthetimeofanalysisandsowechose20minAlsowestudiedthedesaltingeffectofsodiumchlorideonefficiencyofextractionThebestvolatileorganiccompounds sorptionwasachievedbyadding133saltingagent It is also shown that thenumberofpeaksdidnot changeafter20minutesofsorption Thus to obtain the characteristic profiles of urine samples the followingconditionswereselected1333NaClwasaddedtotheurinesample(3ml)
60 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
heatingfor20minat50degCthensorptionofvolatileorganiccompoundsonPDMSfibercoatingat50degCfor20min Undertheselectedconditionsweobtainedvapor-phaseprofilesof52urinesamples (32 normal and 20 pathology) Prior to performing chemometricprocessing of chromatographic profiles of urine samples preliminary datapreparation is necessary [7] The baseline was removed and the peaks werealignedusingdynamictimewarpingwithcontrollingbymassspectra ThePCAmodelwasbasedon52aligned characteristicprofilesThere is asatisfactoryseparationofdataintotwoclustersinthescoresplotrelativetothefirstandsecondprincipalcomponent(Fig1) The original data set (52 samples) was randomly divided into calibration(13pathology21normal)andtest(7pathology11normal)setsThenthePLS-DAmodelwasbuiltusingthecalibrationsetanditspredictiveabilitywasevaluatedusingthetestsetTheprocedurewasrepeated100timesTheaveragevaluesofsensitivityspecificityandaccuracyinthiscasewere9594and91
4Conclusions
Thepossibilityofusingimidazoliumionicliquids([C MIM][PF ])asextractants4 6
forquantitativeextractionandconcentrationofpesticidesundertheconditionsofDLLMEisshownThedegreeofconcentrationwas28ndash33whichallowedreaching
ndash1thedetectionlimits(006ndash019ngml )belowthemaximumpermissibleconcen-trationThepossibilityofnon-invasivediagnosisofprostatecancerbySPMEofvolatileorganiccompounds inurine isshownChemometricprocessingofgaschromatographic profiles using PLS-DA and PCA methods allowed achievingclassificationaccuracyvaluesmorethan90
Acknowledgments
ThisworkwassupportedbytheRussianFoundationforBasicResearchprojectno18-53-80010BRICS_t and the Russian Science Foundations (Projects 19-13-00370) We are grateful to
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 61
Fig 1Scoresplotrelativetothefirstandsecondprincipalcomponent
Resource Education Center in Chemistry of St Petersburg State University for the providedequipment
References
[1] Rutkowska M Płotka-Wasylka J Sajid M Andruch V Liquidndashphase microextractionAreviewofreviewsMicrochemJ149(2019)103989
[2] JaliliVBarkhordariAGhiasvandAAcomprehensivelookatsolid-phasemicroextractiontechniqueAreviewofreviewsMicrochemJ152(2020)104319
[3] ArthurCLPawliszynJSolidphasemicroextractionwiththermaldesorptionusingfusedsilicaopticalfibersAnalChem62(1990)2145ndash2148
[4] SchmidtKPodmoreISolidphasemicroextraction(SPME)methoddevelopmentinanalysisof volatile organic compounds (VOCs) as potential biomarkers of cancer JMol BiomarkDiagn6(2015)1000253
[5] Mousavi L Tamiji Z Khoshayand MR Applications and opportunities of experimentaldesign for the dispersive liquidndashliquidmicroextractionmethod ndash A review Talanta190(2018)225ndash356
[6] MarcinkowskaRKoniecznaKMarcinkowskiLNamiesnikJKloskowskiAApplicationofionic liquids inmicroextractiontechniquesCurrent trendsandfutureperspectivesTrACTrendsAnalChem119(2019)115614
[7] WehrensRChemometricswithRMultivariateDataAnalysisintheNaturalSciencesandLifeSciencesBerlinSpringer2011
62 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Theself-assembledtwo-dimensionalmonolayers(2DSAMs)ofvariousmolecules(eg graphene [1] MoS [2] rubrene [3]) offer beneficial properties for the2
constructionofnano-electronic andnano-opticaldevicesThe topological andchemicalcharacterizationof2DSAMsiscrucialtogatherinformationaboutthearrangementofdepositedmoleculesandtheirinteractionwiththesubstrateThistaskrequiresanalyticaltechniqueswith(sub)nanometerspatialresolutionandup to single-molecular detection sensitivity Only few techniques meet therequirements and one of them is tip-enhanced Raman spectroscopy (TERS)whichcombinestheexcellentspatialresolutionofscanningprobemicroscopy(SPM) and chemical sensitivity of surface-enhanced Raman scattering (SERS)spectroscopy[45] TheSERS spectroscopyutilizesplasmonicmetal nanostructures to cause ahighlocalenhancementoftheelectricfieldintheirclosevicinityviathesurfaceplasmonresonance(SPR)effectThelocalelectricfieldcausesanincreaseofthe
The development of reference probe system for tip-enhanced Raman spectroscopy
MARTINKRA LMARCELADENDISOVA PAVELMATE JKA
DepartmentofPhysicalChemistryFacultyofChemicalEngineeringUniversityofChemistryandTechnologyPragueTechnickaacute516628Prague6CzechRepublicMartinKralvschtcz
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 63
AbstractThetip-enhancedRamanspectroscopy(TERS)isamodernanalyticaltechniquewithanoutstandingspatialresolutionandchemicalsensi-tivityTheseparametersmainlydependon the structural integrityand chemical purity of employed plasmonic scanning probe tipsUsuallyeachtipistestedbeforeTERSmeasurementsusingcommer-ciallyavailablereferencesamplesHowevertheirpriceandrelativelyshortexpirationdatemustbeconsideredwhenplanningaresearchbudgetWedevelopedaproceduretoproduceself-madereferenceprobesamplesfortestingTERStipsusingcopper(II)phthalocyanineonaAunanolayerwhichispreparedbythermalvacuumevaporationofAuonaSiwaferOurresultsshowthatthepreparedsystemenablesrepeateddetectionofwell-resolvedTERSspectraThecollectedTERSspectraandspectralmapsexhibitsomedegreeofvariabilitywhichmaybeduetovariousphoto-inducedprocessesanditmustbeconsi-deredwhileperformingTERSmeasurements
Keywordscopper(II)
phthalocyaninescanningtunnelling
microscopyresonanceRaman
spectroscopysurface-enhancedRaman
spectroscopytip-enhancedRaman
spectroscopy
Ramanscatteringfrommoleculesboundtothemetalby6ndash8ordersofmagnitude[6]TheenhancementallowsSERSspectroscopytobeusedforsingle-moleculardetectionHoweverRamanmicroscopeshave limitedspatialresolutionbythelightdiffractionwiththeachievableresolutionbeingaroundhalfoftheexcitationwavelengthOntheotherhandthespatialresolutionofSPMtechniquesislimitedonlybythedimensionsoftheapexofthescanningprobetipwhichmayevenbeatomicallysharpByutilizingSPMtipandsubstratemadefromplasmonicmetalsan artificial ldquohotspotrdquomay be createdwith its position and dimensions beingdefinedbythetipItopensthepossibilitytocollectstronglyenhancedRamanspectrafromtheareapreciselylocalizedbellowthetipandthusovercometheopticaldiffraction limitTheartificialhotspotmayberelocatedbymovingthesamplebelowthetipwhichisthefoundationofTERSmapping[578] A successful TERS experiment requires an optimal combination of variousparameters the most important of which are tip sharpness and purity [7]Areferencesampleconsistingofaflatplasmonicnanolayerwithattachedprobemolecules is frequently used to check the state of the tip before using it forexperiments Unfortunately commercially available TERS standards areexpensiveandhaveanexpirationdateofseveralmonths Thegoalofthisstudywastofindapreparationprocedurewhichwouldbeableto produce cheap reference samples for repeated detection of intense TERSspectraAcombinationofaAunanolayeronaSisubstratepreparedbythermalvacuumevaporationwithadsorbedcopper(II)phthalocyanine(CuPc)whichisamoleculewithhighRamancross-sectionwastested[910]Copper(II)phthalo-cyanine known as phthalocyanine blue is a synthetic blue pigment and isfrequently used in paints It has been studied as a potentialmaterial for theconstruction of organic solar cells and other photoelectronic devices [11] AsindicatedbyitscolourCuPcexhibitsseveralabsorptionbandswithinthevisibleregion The effects of a transition to excited electronic states and subsequentluminescencemaybeobservable inRamanmeasurements[12]Au isahighlysuitablemetalforthesampleasitisboththermallyandelectricallyconductivewhichlimitsthelocalheatingofthesampleduringthemeasurementsandenablesthe use of scanning tunnelling microscopy (STM) for tip-surface interactionfeedback[13]
2Experimental
21Reagentsandchemicals
Thesubstrateforthesamplewaspreparedbythermalvacuumevaporationofgoldonsilicon(100)waferFirsta5nmthickCradhesionlayerwasdepositedon
ndash1thewaferfollowedby100nmofAuThedepositionratewas4nmmin forCrand ndash18nmmin for Au The base pressure of the evaporation system was below
ndash65times10 mbarFollowingthepreparationproceduredescribedbyJiangetal[14]
64 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
thecleansubstratewasimmersedintoasaturatedsolutionofCuPc(˃99SigmaAldrich USA) in dimethylformamide (˃98 Lach-ner CZ) for at least 12h atambienttemperatureSubsequentlythesamplewasremovedfromthesolutionrinsedwithMilli-Qwaterandmethanol(paPentaCZ)anddriedwithair
22Instrumentation
TheRamanSERSandTERSspectrawererecordedusingRamanspectrometerInVia Reflex (Renishaw UK) equipped with lasers emitting at two differentexcitation wavelengths 633nm (136mW max power output) and 785nm(204mWmaxpoweroutput)Thespectrometerhasathermoelectricallycooled
ndash1CCDdetectorwithaspectralresolutionof2cm and4microscopeobjectiveswith5times20times50timesand100timesmagnitudeForTERSexperiments the laserbeamwasredirectedtotheSPMplatformInnova-IRIS(BrukerUSA)viaasystemoflightguidesElectrochemicallyetchedAuTERS-STMtips(BrukerUSA)wereusedforallTERSmeasurements The spectra were processed using the Spectragryph software (F MengesldquoSpectragryph - optical spectroscopy softwarerdquo Version 1214 2020httpwwweffemm2despectragryph) Using this software all collectedspectra were treated by a Savitzky-Golay noise filter automatic baselinecorrectionspikeremovalandpeaknormalization
3Resultsanddiscussion
31Ramanmeasurementsofcrystallinecopper(II)phthalocyanine
AtfirsttheRamanspectraofpureCuPcwerecollectedtoprovidereferencedatawhileusingboth633and785-nmexcitationlasers(Fig1)Bothspectraexhibit
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 65
Fig 1RamanspectraofCu(II)phthalocyanineincrystallineformmeasuredat633(top)and785-nm(bottom)excitationThespectraareoffset
ndash1vibration bands in the 500ndash1600 cm region with slight differences in theirintensityratiosThe633-nmexcitationallowstheobservationofadditionalbands
ndash1inthe2000ndash3000cm region(onaluminescencebackground)whichoriginatefrom the resonance Raman effect as the excitation energy overlaps with theQ-bandofCuPc[12]MoreovertheprocessofelectronicexcitationmaylowertheD symmetry of CuPc during resonance Raman scattering and previously4h
forbiddenbandsmaybecomeobservable[15]Theluminescentbackgroundhasandash1maximumaround2200cm whichcorrespondstoamolecularemissionbandat
735nmEventhoughthespectrameasuredwiththe785-nmlaserlinedonotexhibitapparentresonanceenhancementapre-resonanceRamanenhancementmayoccur
32Surface-enhancedRamanmeasurementsofcopper(II)phthalocyaninelayeronagoldsubstrate
The prepared sample of CuPc on a Au layer was analysed using the Ramanmicroscope Both excitation laserswere used to obtain SERS spectra (Fig 2)whichwerecomparedtothespectraofapurecrystallineCuPc Thepositionsof bands in SERS spectra closelymatch their positions in thespectraofbulkCuPcHoweveraslightshiftofsomespectralbandsisobservable
ndash1(eg1528rarr1532cm )whichmaybeattributedtotheinteractionbetweenCuPcand the Au substrate The disappearance of luminescence background andresonance-enhancedbandsinthespectrumat633-nmexcitationalsosuggeststhemolecule-metalinteractionandthetransferofenergyfromCuPcmoleculestothesubstrateMoreovertherearevariationsintherelativeintensitiesofbands
ndash1whichdependontheexcitationenergyegthebandat1309cm isenhancedinSERSspectraatthe785-nmexcitationwhencomparedtothespectraofpureCuPcorevenSERSspectraat633-nmexcitation
66 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2SERS spectra of Cu(II) phthalocyanineon aAu layermeasured at 633 (top) and785-nm(bottom)excitationThespectraareoffset
33Tip-enhancedRamanmeasurementsofcopper(II)phthalocyaninelayeronagoldsubstrate
SERS microspectroscopy is a diffraction-limited technique as it provides anaveragedinformationaboutmoleculesintheilluminatedareaofseveralsquaremicrometers Meanwhile TERS spectra are collected from an area of tens ofnanometers and they contain specific information about the local moleculararrangement topography of the underlyingmetal and properties of the localelectric field between the tip and the substrate Therefore a higher spectralvariabilityshouldbeexpected Several TERS mapping experiments were carried out using both 633 and785-nmexcitationwithvaryingexperimentalparameterssuchasthenumberanddistance betweenmeasured points acquisition time number of acquisitionslaserpoweretc TheTERSspectrameasuredat633-nmexcitationexhibited lowersignal tonoiseratioandreproducibilityAsaconsequenceTERSmappingwasimpossibleandonlyafewone-pointTERSspectrawereobtained(Fig3) TheTERSspectracollectedat785-nmexcitationcontainedahighernumberofwell-resolvedbandsMoreoverthespectrawerestableintimeandsotheTERSmappingwaspossibleThe twopresentedTERSspectraareaveragesofTERSmaps which contained 16 and 80 points with 600 and 300-nm spacingrespectively(Fig4) Thespectracollectedusingbothexcitationwavelengthsexhibitavariabilityinrelative intensities and positions of bands between themeasured points ThevariabilitymaybeattributedtothelocalorientationofCuPcmoleculesbetweenthe tip and the Au surface and the properties of strongly enhanced andnon-homogeneous electromagnetic field which depend on the tip-surfacedistancetheirmorphologyandrelativepositionMoreovertheusedexcitation
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 67
Fig 3TwoexamplesofTERSspectraofCu(II)phthalocyaninemeasuredat633-nmexcitationThespectraareoffset
wavelengthsareclosetoabsorptionbandsofCuPcandthestrongelectricfieldmay give rise to photo-induced effects These effects include the electronicexcitationofCuPctohigherstateschargetransferbetweentheCuatomandthephthalocyaninering ionizationof themoleculeand formationofradicalsThephoto-inducedprocessesarelikelytoplayabiggerroleinTERSspectradetectedat633-nmexcitationduetotheoverlapwithQ-bandofCuPcwhichmaybethecauseoftheirlowersignaltonoiseratioandreproducibility
4Conclusions
Thedeveloped referenceprobe systemofCuPc adsorbedon aAu surfacehasprovedtobesuitablefortheintendeduseasitenabledthedetectionofintenseandwell-resolvedSERSandTERSspectraTheAulayerpreventsoverheatingofthesampleandallowsfortheuseofSTMTheSERSspectrawereinagoodmatchwith thespectraofpureCuPcAslight shiftof somebandsandchange in theluminescent background indicated the interaction between CuPc and the AusurfaceTheTERSexperimentsresultedinspectralmapswithhighintensitiesofindividualspectraIncreasedvariabilitybetweenmeasuredpointswasobservedPossiblesourcesofthevariabilityarephoto-inducedprocessesthatmayoccurinthestronglyenhancedelectricfieldTheseeffectsareaknownfeatureofTERSmeasurements and they offer valuable insight into the photophysics andphotochemistryofCuPcinteractingwiththeAusurfaceThedependenceofTERSspectra on experimental parameters and the preparation procedure of thereferencesampleshouldbefurtherstudied
Acknowledgments
ThisworkwassupportedfromthegrantofSpecificuniversityresearchndashA2_FCHI_2020_039
68 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 4TwoexamplesofaveragedTERSspectraofCu(II)phthalocyaninemeasuredwith785-nmexcitationThespectraareaveragesfromTERSmapsincluding16(top)and80(bottom)measuredpointsThespectraareoffset
References
[1] Mas-BallesteRGomez-NavarroCGomez-HerreroJZamoraF2DmaterialstographeneandbeyondNanoscale3(2011)20ndash30
[2] ZengHCuiXAnopticalspectroscopicstudyontwo-dimensionalgroup-VItransitionmetaldichalcogenidesChemSocRev44(2015)2629ndash2642
[3] SchultzJFLiLMahapatraSShawCZhangXJiangNDefiningmultipleconfigurationsofrubreneonaAg(100)surfacewith5A spatialresolutionviaultrahighvacuumtip-enhancedRamanspectroscopyJPhysChemC124(2020)2420ndash2426
[4] WhitemanPJSchultzJFPorachZDChenHNJiangNDualbindingconfigurationsofsubphthalocyanineonAg(100)substratecharacterizedbyscanningtunnelingmicroscopytip-enhanced Raman spectroscopy and density functional theory J Phys Chem C 122(2018)5489ndash5495
[5] ShaoFZenobiRTip-enhancedRamanspectroscopyprinciplespracticeandapplicationstonanospectroscopicimagingof2DmaterialsAnalBioanalChem411(2019)37ndash61
[6] ArocaRSurface-EnhancedVibrationalSpectroscopyHobokenWiley2006[7] KumarNMignuzziS SuWRoyDTip-enhancedRamanspectroscopyprinciplesand
applicationsEPJTechInstrum2(2015)9[8] BailoEDeckertVTip-enhancedRamanscatteringChemSocRev37(2008)921ndash930[9] BovillAJMcConnellAANimmoJASmithWEResonanceRamanspectraofα-copper
phthalocyanineJPhysChem90(1986)569ndash575[10] Shaibat MA Casabianca LB Siberio-Perez DY Matzger AJ Ishii Y Distinguishing
polymorphsofthesemiconductingpigmentcopperphthalocyaninebysolid-stateNMRandRamanspectroscopyJPhysChemB114(2010)4400ndash4406
[11] SzybowiczMRunkaTDrozdowskiMBałaWGrodzickiAPiszczekPBratkowskiAHightemperaturestudyofFT-IRandRamanscatteringspectraofvacuumdepositedCuPcthinfilmsJMolStruct704(2004)107ndash113
[12] CaplinsBWMullenbachTKHolmesRJBlankDAFemtosecondtonanosecondexcitedstatedynamicsofvapordepositedcopperphthalocyaninethinfilmsPhysChemChemPhys18(2016)11454ndash11459
[13] SaccoAImbraguglioDGiovannozziAndreaMPortesiCRossiAMDevelopmentofacandidatereferencesampleforthecharacterizationoftip-enhancedRamanspectroscopyspatialresolutionRSCAdv8(2018)27863ndash27869
[14] JiangSChenZChenXNguyenDMatteiMGoubertGVanDuyneRPInvestigationofcobaltphthalocyanineatthesolidliquidinterfacebyelectrochemicaltip-enhancedRamanspectroscopyJPhysChemC123(2019)9852ndash9859
[15] MelendresCAMaroniVARamanspectraandnormalcoordinateanalysisoftheplanarvibrationsofironphthalocyanineJRamanSpectrosc15(1984)319ndash326
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 69
1Introduction
The aim of this work has been develo-pmentofanewvoltammetricmethodforthe determination of 23-dimercapto-1-propane-sulfonic acid (DMPS) Fig 1Investigationwasdonetoobtainrelevantinformation about complexingbehaviorofDMPStowardsleadions Lead is one of heavy metals which can cause irreversible neurologicalproblems [1 2]DMPS is a synthetic antidotewith two thiol groups used fortreatmentofpoisoningbyheavymetals[3ndash6]StrongcomplexingpropertieshighwatersolubilityandnegligiblesideeffectsarethemostimportantadvantagesofDMPS[47]
Fig 1 Chemical structure of 23-dimercap-to-1-propane-sulfonicacid
Determination of heavy metal poisoning antidote 23-dimercapto-1-propanesulfonic acid using silver solid amalgam electrode
ab ab bc bMARTACHOIN SKA VOJTE CHHRDLICKA BEATRIZRUIZREDONDO JIR IBAREK aTOMA S NAVRATIL
a JHeyrovskyacuteInstituteofPhysicalChemistryoftheCzechAcademyofSciences Dolejškova21553182thinsp23Prague8CzechRepublicmartachoinskagmailcomb UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova20308128thinsp43Prague2CzechRepublic
c UniversityofValladolidPlazadeSantaCruz847002ValladolidSpain
Abstract23-Dimercapto-1-propane-sulfonic acid (DMPS) was investigatedusingdirectcurrentvoltammetry(DCV)differentialpulsecathodicstrippingvoltammetry(DPCSV)differentialpulseanodicstrippingvoltammetry(DPASV)andeliminationvoltammetrywithlinearscan(EVLS)atapolished(p-AgSAE)andatameniscusmodifiedsilversolidamalgam electrode (m-AgSAE) EVLS confirmed two consecutivereductions with coupled protonelectron transfer VoltammetrictitrationsofDMPSwithPb2+provedcomplexformationwithlimits
minus1ofquantification(LOQs)anddetection(LODs)03and01micromolL atminus1m-AgSAE and 08 and 03micromolL at p-AgSAE respectively
Determination of DMPS in commercial drug Dimaval and humanurine samples confirmed practical applicability of the developedmethod
Keywordscathodicstripping
voltammetry23-dimercapto-1-pro-
panesulfonicacideliminationvoltammetry
withlinearscansilversolidamalgam
electrodeunithiol
70 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Voltammetry was chosen as a determination technique due to its highsensitivity and selectivity speed low costs [8] Thiol groups in DMPS can beoxidatively chemisorbed on solid amalgam electrode It can be used as anaccumulation step for cathodic strippingvoltammetry [9ndash11]Moreover solidamalgamelectrode (SAE)was chosen as theworking electrodebecauseof itspropertiesashighsignaltonoiseratiowidepotentialwindowandabilitytoreachlowlimitsofdetection(LOD)[12ndash13]
2Experimental
21Reagentsandchemicals
Allsolutionswerepreparedusingdeionizedwater(Milli-Q-GradientMilliporendash1PragueCzechRepublic)withconductivitylt005microScm Britton-Robinsonbuffer
solutionspHrangefrom2to12werepreparedbymixingtheproperamountsof02MNaOH(alkalinesolution)andof004MH BO 004MH PO and004M3 3 3 4
CH COOH(allLachemaCzechRepublic)acidicsolutionTheacidicsolutionwas3
preparedbydissolutionof1235gofH BO pa088mLofH PO (85)paand3 3 3 4
1435mLofCH COOH(99)pa in500mLofdeionizedwaterThealkaline3
solution was prepared by dissolution of 3995 g of NaOH pa in 500mL ofdeionizedwater(allLachemaCzechRepublic) StocksolutionofDMPSwaspreparedbydissolving10mgofsolid23-dimer-capto-1-propanesulfonic acid monohydrate pa (Merck Czech Republic) in100mLofdeionizedwaterForthepreparationofthemodelsamplesolutiononecapsuleofdrugDimaval(HeylGermany)contains100mgofDMPSwasdissolved
ndash1in10LofdeionizedwatertotheDMPSconcentrationof0531mmolL Twomodel samplesofDimavalwerepreparedbydilutionof theabove-mentioned
ndash1solutionwithBritton-Robinsonbuffersolutiontoconcentrations10micromolL andndash1of 10micromolL respectively Urine model samples were prepared by mixing
Britton-Robinsonbuffer solutionwithurine samples obtained fromvolunteer(manhealthy30yearsold)inratio11SamplepHwasadjustedbyadditionof
ndash1proper amount of 02molL NaOH Before each measurement oxygen wasremoved for 5minbynitrogenbubbling (purity class 46MesserTechnogasPragueCzechRepublic)
22Instrumentation
Measurementswereperformedusingtwotypesofworkingelectrodesmeniscusmodified silver solid amalgam electrode (m-AgSAE working surface of
20382plusmn0025mm α lt 005) and polished silver solid amalgam electrode2(p-AgSAEworkingsurfaceof0196plusmn0015mm αlt005)Ag|AgCl|3MKClwas
used as the reference electrode andplatinumwire (Oslash1mm)wasused as theauxiliary electrode (both from Elektrochemicke detektory Czech Republic)Measurementswereperformedatlaboratorytemperature(25plusmn2degC)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 71
The pH was measured using pH-meter Jenway 3505 with combined glasselectrodetype924001(BibbyScientificLimitedUK)Voltammetricmeasure-ments were performed using the computer-controlled Eco-Tribo Polarograph(Polaro-Sensors Czech Republic) Software used for measurements wasMultiElChem 33 forWindows XP7810 (J Heyrovsky Institute of PhysicalChemistryoftheCzechAcademyofSciencesCzechRepublic)
3Resultsanddiscussion
OptimumconditionsformeasurementswereobtainedbyseriesofmeasurementinwiderangeofpHvaluesandtestingvariouscleaningproceduresOptimumpotentialof accumulation (E ) and timeof accumulation (t )ofDMPSwereacc acc
adjusted for differential pulse cathodic stripping voltammetry (DPCSV) atp-AgSAEandm-AgSAE ThedependencebetweenpeakheightandconcentrationofDMPSatp-AgSAEhasalogarithmicshapewhichcorrespondstotheaccumulationprocessattheelectrodesurfaceThelineardependencewasobservedintheDMPSconcentra-
ndash1 ndash1tionsfrom03micromolL to20micromolL Peakshifttowardsnegativepotentialwithincreasing concentration of DMPS corresponds to the metal-thiol bond andinfluenceofelectrodesurfacestructureonthisbondOnthecontrarysignalsonm-AgSAEweremorestableandthedependencebetweenconcentrationofDMPSandsignalwasalmostlinearinwholetestedrangeofconcentrations The developedmethodwas tested inmodel samples of Dimaval and urineFoundamountsofDMPSwereingoodagreementwithdeclaredcontentsusingbothelectrodesHowevertherepeatabilityofsignalsregisteredusingp-AgSAEinurinesamplesweresignificantlyworsethanthoseinDimavalsamplesItcanbecausedbycomplicatedbiologicalmatricesandfoulingeffectsofurine ELSV measurements confirmed two consecutive reductions of DMPS inadsorbed state At m-AgSAE signal were at about minus415 mV and minus440 mVrespectively and at p-AgSAE at about minus790 mV and minus830 mV respectively
ndash1 ndash1Reductionsatm-AgSAEatthescanratesfrom80mVs to640mVs havebeencontrolledbyakineticprocessatminus400mV Inanodicscansonm-AgSAEonlyonepeakwasvisibleatabout‒390mVItcorrespondswiththeoxidationofmercuryelectrodesurfaceontheelectrodeinthepresenceofDMPSandwithdiffusionfromthebulksolutionofproductsAtp-AgSAEnosignificantanodicsignalwasfound ThelastpartoftheresearchrevealedvoltammetricbehaviorofDMPSinthe
2+presence of Pb Voltammetric titration was investigated by DPCSV anddifferential pulse anodic stripping voltammetry (DPASV) during consecutive
ndash1 ndash1additionsof1mmolL ofPb(NO ) into100micromolL DMPSsolutioninacetate3 2
bufferofpH50InabsenceofPb2+ontheelectrodesurfaceHg(DMPS)complexisformedduringtheaccumulationstepDuringtheanodicscanthereisonlyonewelldevelopedreductionpeak(Fig2A)
72 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ndash1Fig 2DPCSandDPASvoltammogramsof10micromolL ofDMPSinacetatebufferpH=5correspond-2+ingto[Pb DMPS]ratiosof(A)01(B)11and(C)21Uppercurvecorrespondstothecathodic
scanE =0mVt =15sLowercurvecorrespondstoreverseanodicscanwitht =15satacc acc accndash1E =minus1000mVν=20mVs (Ref[16])acc
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 73
2+ WhenPb DMPSratioisequalto11twooxidationandthreereductionpeakswereregistered(Fig2B)PeakA ataboutndash300mVcorrespondstoformationofred
0Pb(DMPS)complexThiscomplexwasfurtherreducedtothePb (Hg)atabout2+ndash500mV(C )ReductionpeakoffreePb wasalsoregistered(B )Oxidationred red
peaksA andB correspondtoreverseprocessesandC isnotpresentbecauseox ox ox
nofreeDMPSispresentinthesolutionAtratio21thereisnofreeDMPSinthesolution however excess of lead ions As a consequence A and B peakred ox
2+increasedIncreaseofB correspondstothedepositionofPb duringtheaccu-ox
mulationstepPeaksBredandCredremainedpracticallyunchanged(Fig2C) Voltammetric titrationconfirmed themechanismof formationcomplexesofPb(DMPS)Hg(DMPS)andPb(Hg)Italsoconfirmedpossibilityofdetermination
2+Pb andDMPSinthesamesolution
4Conclusions
ValidationinmodelsampleofdrugDimavalandhumanurinespikedwithDMPSconfirmed that this method can be used for clinical purposes Voltammetric
2+titration of DMPS by Pb ions proved that it can be used for simultaneousdetermination of the drug and heavy metal ions in human urine Moreoverobtained LODs were two orders lower than those in the previously reportedvoltammetricmethod[14](Table1)
Acknowledgments
ResearchwascarriedoutwithintheframeworkofSpecificUniversityResearch(SVV260560)TheauthorsthanktheCzechScienceFoundation(GACRprojectNo20-01589S)
References
[1] AnHHLuchakMCopesRLeadtoxicityAsystematicreviewofrecentlypublishedcasesClinToxicol53(2015)757ndash758
[2] KimYLustMRKreimerbirnbaumM23-Dimercaptopropane-1-sulfonate(DMPS)inthetreatmentoflead-poisoningFasebJ2(1988)A1820ndashA1820
[3] AposhianHVDMSAandDMPS ndashwater-solubleantidotesforheavy-metalpoisoningAnnuRevPharmacol23(1983)193ndash215
74 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Table 1ComparisonofvoltammetricmethodsforDMPSdetermination(LDR
Method Workingelectrode Lineardynamic LOQ LOD Refminus1 minus1 minus1 rangemicromolL micromolL micromolL
LSV glassy-carbonelectrode 18ndash140 41 14 [14] modifiedwithmulti-walled 260ndash690 carbonnanotubes DPCSV p-AgSAE 03ndash20 08 03 thisworkDPCSV m-AgSAE 01ndash10 03 01 thiswork 10ndash100
[4] BjorklundG Crisponi G Nurchi VM Cappai R Djordjevic AB Aaseth J A review oncoordinationpropertiesof thiol-containingchelatingagents towardsmercury cadmiumandleadMolecules24(2019)3247
[5] DonnerAHrubyKDMPSinthetreatmentofacuteandchronicheavy-metalpoisoningActaMedAust14(1987)10ndash10
[6] DonnerAHrubyKPirichKKahlsPSchwarzacherKMeisingerVDimercaptopropan-sulfonate(DMPS) inthetreatmentofacute lead-poisoningVetHumToxicol29 (1987)37ndash37
[7] Blanusa M Varnai VM Piasek M Kostial K Chelators as antidotes of metal toxicityTherapeuticandexperimentalaspectsCurrMedChem12(2005)2771ndash2794
[8] BarekJMoreiraJCZimaJModernelectrochemicalmethodsformonitoringofchemicalcarcinogensSensors-Basel5(2005)148ndash158
[9] Josypcuk B FojtaM Yosypchuk O Thiolatemonolayers formed on different amalgamelectrodesPartIIPropertiesandapplicationJElectroanalChem694(2013)84ndash93
[10] YosypchukBMarecekVPropertiesofthiolatemonolayersformedondifferentamalgamelectrodesJElectroanalChem653(2011)7ndash13
[11] Alvarez JMF SmythMRCathodic strippingvoltammetryofpyridine-2-thiolandsomerelated-compoundsAnalyst114(1989)1603ndash1605
[12] DanhelABarekJAmalgamelectrodesinorganicelectrochemistryCurrOrgChem15(2011)2957ndash2969
[13] Fadrna R Polished silver solid amalgam electrode Further characterization and appli-cationsinvoltammetricmeasurementsAnalLett37(2004)3255ndash3270
[14] ZiyatdinovaGKGrigorevaLVBudnikovGKElectrochemicaldeterminationofunithioland lipoic acid at electrodesmodifiedwith carbonnanotubes J Anal Chem64 (2009)185ndash188
[15] HrdlickaVChoinskaMRedondoBRBarekJNavratilTDeterminationofheavymetalpoisoning antidote 23-dimercapto-1-propanesulfonic acid using silver solid amalgamelectrodeElectrochimActadoiorg101016jelectacta2020136623
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 75
Fig 1Structureofcanagliflozin
1Introduction
Canagliflozin is a selective sodium-glucosecotransportertype2inhibitorused for the treatment of type 2 dia-betes mellitus Canagliflozin inhibitssodium-glucose cotransporter type 2present in proximal tubules of the
Canagliflozin oxidation study using electrochemical flow cell and comparison with hydrogen peroxide oxidation
a a bFILIPVYMYSLICKY TOMA S KR IZ EK JAKUBHER T
a DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublicfvymyslickygmailcomb ZentivaGroupasUKabelovny13010237Prague10CzechRepublic
AbstractBystandardstheeffectonoxidationofanactivesubstanceistestedusinghydrogenperoxidesolutionatelevatedtemperatureinastresschamberfor1ndash7daysAnalternativewaytostudytheeffectofoxida-tion on an active substance is to use an electrochemical flow cellSolutionwith active substance flows at low flow rate into a smallreactorwheretheactivesubstanceisoxidizedonworkingelectrodesurfaceTheelectrolytestreamwiththeoxidizedactivesubstanceisthen directed to the sample collector Products of electrochemicaloxidationareanalyzedbyhighperformanceliquidchromatographywithultravioletndashvisiblespectrophotometrydetectionCanagliflozinhasbeenusedbecauseitsmaindegradationpathwayisoxidationThedesign of experiments approach was used to explore the experi-mentalspaceandoptimizeexperimentalconditionsofoxidationTheresultsoftheoxidationstudyperformedintheelectrochemicalflowcellwerestatisticallycomparedwiththeresultsofastandardstudyusinghydrogenperoxidesolutionThemostsuitableconditionsforelectrochemical oxidation were found Electrochemical oxidationproducedcomparableamountsofimpuritiesaschemicaloxidationwithhydrogenperoxide
KeywordscanagliflozindesignofexperimentselectrochemicalflowcellHPLCoxidation
76 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
kidneywhichrestrictsglucoseabsorptioninthekidneytherebyincreasingtheurinaryexcretionofglucoseandloweringthelevelofglucoseintheblood[1]TheformulaofcanagliflozinisC H FO SthestructureofcanagliflozinisinFig1The24 25 5
IUPAC name of canagliflozin is (2S3R4R5S6R)-2-[3-[5-(4-fluoro-phenyl)-thiophen-2-ylmethyl]-4-methyl-phenyl]-6-hydroxymethyltetrahydro-pyran-345-triol[2]CanagliflozinisawhitepowderinsolubleinwaterbutverysolubleinorganicsolventslikemethanolordimethylsulfoxideCanagliflozinissoldundertradenameINVOKANA Manyauthorshavestudiedtheelectrochemicalpropertiesofactivesubstancesin the literature One example is the study of electrochemical behaviour andoxidationofbromhexineThesepropertieswerestudiedusingdifferentialpulsevoltammetryandcyclicvoltammetryonacarbonelectrodeTheresultsofelectro-chemicalmethodswerecomparedwithhighperformanceliquidchromatography(HPLC)analysis[3]Anotherexampleisthestudyofelectrochemicalbehaviourand degradation study performed on the active substance atomoxetineDegradation was studied using differential pulse voltammetry and cyclicvoltammetryonacarbonelectrodeTheresultswerealsocomparedwithHPLCanalysis[4]Electrochemicalmethodsareusedmainlytostudythemechanismofoxidationbutinthisworktheelectrochemicalmethodwasusedtodegradetheactivepharmaceuticalingredient In the stability studies of active pharmaceutical ingredient properties theinfluence of temperature pH light and oxidation is studied [5] By TheInternationalCouncilforHarmonisationofTechnicalRequirementsforPharma-ceuticals forHumanUse (ICH) standards the influenceof oxidationon activepharmaceutical ingredient is studied using hydrogen peroxide at roomtemperatureorincreasedtemperatureinthestresschamberduring1ndash7days[6]Analternativewaytostudytheinfluenceofoxidationonactivepharmaceuticalingredient is using electrochemical flow cellwhere an electrolytewith activepharmaceuticalingredientisdrivenbylowflowrateintothesmallreactorInthesmallreactortheactivepharmaceuticalingredientisoxidizedonthesurfaceoftheworkingelectrodeThestreamofelectrolytewithoxidizedactivepharma-ceuticalingredientisdriventothesamplecollectorProductsofelectrochemicaloxidationareanalysedbyHPLCUVVISThedesignofexperimentsapproachwasusedfordevelopmentofanalternativemethodofoxidationofcanagliflozinusinganelectrochemical flowcellThedesignofexperimentsapproachwasusedtoexplore the experimental space of the method and to find the optimalexperimentalconditionsofelectrochemicaloxidationofcanagliflozin
2 Experimental
21Materialandreagents
Canagliflozin(ZentivaCzechRepublic)999methanol(HoneywellGermany)98ammoniumdihydrogenphosphate(Sigma-AldrichJapan)35ortho-phos-phoricacid(PentaCzechRepublic)25ammonia(LachnerCzechRepublic)30hydrogenperoxide (LachnerCzechRepublic)water forchromatography
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 77
Fig 2Schemeoftheelectrochemicalflowcell(1)input(2)workingelectrode(3)gasket(4)refe-renceelectrode(5)counterelectrode
wasobtainedbypurifyingdemineralisedwaterusingMilliporetypeSynergyUVpurificationinstrument
22Instruments
An Agilent 1290 HPLC system (Agilent Technologies Germany) with highpressure pump autosampler thermostat and DAD detector was used for allexperimentsThePinnacleDBbiphenylcolumn(100times21mm19micromRestekUSA)wasusedforseparationIntheHPLCmethod10mMammoniumdihydrogenphosphatebufferpH=25wasusedascomponentAandmethanolascomponentBofthemobilephaseThegradientprogramwassetasfollowst(min)B01555010551790229023152515Theflowrateofthemobilephasewas
ndash104mlmin and the injection volume was 2μl The detector operated at awavelengthof220nmTheautosamplertemperaturewassetat20degCandthecolumntemperatureat60degCTheEmpowersoftwarewasusedforevaluationForelectrochemicaloxidationelectrochemicalflowcellfromALS(Japan)wasusedGlassycarbonelectrode(=6mm)andsilversilverchlorideelectrodewereusedasworkingandreferenceelectroderespectivelyTheschemeofelectrochemicalflowcellisinFig2ElectrodeswereconnectedwithpotentiostatPalmSens3fromPalmsens (Netherlands) AnElmasonic S15Hultrasonic bath fromElma (Ger-many)wasusedforsamplepreparationForpHmeasurementspHmeterJenway3540fromJenway(UnitedKingdom)wasused
3Resultsanddiscussion
AtthedevelopmentofthemethodforthestudyofcanagliflozinoxidationusingelectrochemicalflowcellitwasfirstnecessarytofindtheoptimalconditionsofelectrochemicaloxidationThedesignofexperimentsapproachwasusedChosen
78 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ndash1Fig 3Cyclicvoltammogramofcanagliflozin(concentrationofcanagliflozin11mgml electrolyte300 mM ammonium dihydrogen phosphate pH = 40 and methanol (11 vv) and scan rate
ndash1001Vs )
independentvariablesandtheirlevelswereconcentrationofelectrolyte(100200300mM)pHofelectrolyte(406080)cellsize(50100200500microm)and
ndash1flowrate(0102504mlh )ThereducedcombinatorialdesignwasusedIntheModde12 software aworksheet containing 11 experimentswas created Theworkingpotentialof12Vwasselectedbasedoncyclicvoltammetryofcanagli-flozininFig3Fromthisfigureitcanbeseenthattheoxidationofcanagliflozinoccursintheregionfrom11Vto14VAllexperimentswereperformedwith
ndash111mgml canagliflozin samples The glassy carbon electrodewas used as aworkingelectrodeandthesilversilverchlorideelectrodewasusedasareferentelectrode The canagliflozin samples oxidized in the electrochemical flow cellunder theexperimentalconditionsgivenby theworksheetweremeasuredbyHPLCwithUVVISdetectionDependentvariablespeakareasofimpuritiesandpercentage of peak areas of impurities obtained from chromatograms wereevaluated by the partial least squaremethod in theModde 12 software Thevariableimportanceintheprojectionplottoolwasusedforinterpretationofthedata as a whole The significance values of the independent variables were
ndash1evaluatedbufferpH=137flowrateof125mlh bufferconcentration061mMandcell size04micromFromthis tool itwasconcluded that theelectrochemicaloxidationofcanagliflozinisthemostaffectedbythepHoftheelectrolyteandflowrateoftheelectrolyteUsingtheoptimizertoolthemostsuitableconditionsfor
ndash1the oxidation of canagliflozin were evaluated flow rate 01 ml h 300 mMammoniumdihydrogenphosphate pHof electrolyte40 and cell size500micromUsingonefactoratthetimeapproachthedependenceofthecellsizeonthetotalsumofimpuritieswastested(Fig4A) Itisvisiblefromthegraphthatthesmallerthecellweusethemoreoxidationproducts are formed Based on the graph the most suitable conditions were
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 79
Fig 4(A)Optimizationofelectrochemicaloxidationconditionsdependenceofsumofimpuritiesoncellsize(B)Chromatogramofasampleoxidizedundermostsuitableconditions
ndash1adjustedtoflowrate01mlh 300mMammoniumdihydrogenphosphatepHofelectrolyte40andcellsize12micromUnderthemostsuitableconditionsarepeat-ability test was performed by ten independent oxidation experiments Therelativestandarddeviationofthepercentageareaofcanagliflozinwas164atasignificant level of 095 The chromatogram of sample oxidized under mostsuitableconditionsisinFig4BThestandardstudyoftheeffectofoxidationoncanagliflozinusinghydrogenperoxideaccordingtoICHguidelineswasperfor-medThestudywasperformedundertwosetsofexperimentalconditionsInthefirstcaseasolutionof50methanolwiththeadditionof3H O wasusedIn2 2
thesecondcasetheconditionsintheelectrochemicalflowcellweresimulatedA300mMammoniumdihydrogenphosphate pH40 andmethanol in a ratio11(vv)withtheadditionof3H O wasusedSamplesfortheoxidationstudy2 2
werestressedinastabilitychamberfor13and7daysattheconstanttempe-ratureof50degC ThetotalsumsofimpuritiesformedduringchemicaloxidationusinghydrogenperoxideinbothmediawerecomparedasisshowninFig5AItisobviousthat
80 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 5(A)Acomparisonofastandardoxidationstudyusinghydrogenperoxidein50methanolwith added buffer andwithout them (B) Chromatogramof sample oxidized electrochemically(C)Chromatogramofsampleoxidizedchemically
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 81
ammoniumphosphatesuppressesoxidationofcanagliflozinThereasonofthisphenomenonisunknownFig5BandFig5CshowchromatogramsofsamplesoxidizedelectrochemicallyandchemicallyrespectivelyItcanbeseenthatfiveimpuritieswereformedbybothtypesofoxidationhoweverindifferentamounts
4Conclusion
AnalternativemethodfortheoxidativestudyofcanagliflozinwasdevelopedThedesign of experiments approach was used in the method development ThedevelopedmethodworkswithRSDof165(α=095)Oxidationofcanagliflozinbythedevelopedmethodproducedfiveimpuritiesthatareidenticalwiththoseproducedusingthestandardoxidationstudywithhydrogenperoxide
Acknowledgments
ThisworkhasbeensupportedbyCharlesUniversityResearchCentreprogramNoUNCESCI014SVV260560projectandpharmaceuticalappliedresearchcenter(TheParc)
References
[1] ChaoECCanagliflozinDrugsFuture36(2011)351ndash357[2] NislySAKolanczykDMWaltonAMCanagliflozinanewsodium-glucosecotransporter2
inhibitorinthetreatmentofdiabetesAmJHealthSystPharm70(2013)311ndash319[3] Turchan M Jara-Ulloa P Bollo S Nunez-Vergara LJ Squella JA Alvarez-Lueje A
VoltammetricbehaviourofbromhexineanditsdeterminationinpharmaceuticalsTalanta73(2007)913ndash919
[4] Perez-OrtizMMunoz C Zapata-Urzua C Alvarez-Lueje A Electrochemical behavior ofatomoxetineanditsvoltametricdeterminationincapsulesTalanta82(2010)398ndash403
[5] Baertschi SW Alsante KM Reed RA Pharmaceutical Stress Testing Predicting DrugDegradationLondonInformaHealthcare2011
[6] RignallA ICHQ1A(R2) stability testing of newdrug substance andproduct and ICHQ1CstabilitytestingofnewdosageformsInICH Quality Guidelines An Implementation GuideATeasdaleDElderRWNims(Eds)HobokenWiley2017p3ndash44
82 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
AlthoughDNArepresentsarelativelystablecomponentfromthechemicalpointofviewitremainsconstantlyexposedtoalargenumberofchemicalorphysicalagentscausingchemicalchangesinDNAmoleculesthatoccurintheenvironmentoraremajororminorproductsofcellularmetabolism[1] One-electronoxidationoftheDNArepresentsadamagingprocesswheretheloss of an electron (oxidation) fromduplexDNA results in the formationof anucleobase radical cation (electron ldquoholerdquo) that is subsequently consumed inchemicalreactionsthatoftenleadtomutationsAdefiningcharacteristicoftheone-electronoxidationofDNAisthepreferentialreactionattheguaninemoietythatisdetectedasstrandcleavagefollowingchemicalorenzymatictreatmentoftheoxidizedDNA[2ndash3]
Novel hybrid electrochemical DNA biosensor for monitoring oxidative DNA damage via oxidationreduction signals of low molecular weight double-stranded DNA
MICHALAUGUSTINVLASTIMILVYSKOCIL
UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova812843Prague2CzechRepublicmichalaugustinnaturcunicz
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 83
AbstractDeoxyribonucleicacid(DNA)representsamajortargetmoleculeformanydamagingagentscausingunfavorablechangesinastructureofDNAmoleculethatbindandinteractwithDNAThusahighdemandforreliabletoolsregardingabettercomprehensionofthenatureofDNAdamagingprocessesstillrepresentsoneofthemaingoalsinthisareaHereinwedescribeadevelopmentofanovelhybridelectro-chemicalDNAbiosensorbasedonanldquoedge-planerdquopyrolyticgraphiteelectrode (EPPGE) in connectionwith an elementaryoptimizationprocessprovidingacloserresolutionoftheredoxprocessesoflowmolecularweightdouble-strandedDNA(dsDNA)attheEPPGESub-sequentanalyticalapplicationincorporatinganemploymentofthemodel structure K [IrCl ] (representative of transition metal2 6
complexes)andevaluationofitsdamagingeffectinrelationtoDNAbymeansof linear sweepvoltammetry resp square-wavevoltam-metryarealsopresented
KeywordsbiosensordamageDNAgraphitevoltammetry
DNA-based electrochemical biosensors are successfully used in variousapplicationssuchasmonitoringandevaluatingthemechanismsof interactionbetweenDNAandvariousdrugsordamagingagentsrapidmonitoringoftracemetalsorpollutantspresent in theenvironmentordirectmonitoringofDNAhybridizationprocesses[4] Theelectrochemicalactivityofnucleicacids(boththenativehigh-molecularonesaswellasoligonucleotides)isingeneralreferredtotheelectroactivityofitscomponents ndash nucleobases and sugar residues At mercury-based electrodesadenine and cytosine residues undergo reduction processes close to ndash14 V(againstSCE)inneutralorweaklyacidicmedium(givingrisetothepeakCA)Ontheotherhandallbaseshavebeenreportedtobeelectrochemicallyoxidizedatcarbonelectrodesbutonlyadenineand(particularly)guanineoxidationsignalshavebeenwidelyutilizedinelectrochemicalDNAbiosensors[5] In2017theelectrochemistryofnucleicacidsachievedanimportantmilestoneasthereductionoftheDNAoligonucleotideswasperformedataldquobasal-planerdquopyrolytic graphite electrode which provided wide potential window allowingboththeelectrooxidationaswellastheeletroreductionofthenucleobasesatasingleelectrodefortheveryfirsttimeDespitethesefindingsutilizationoftheaforementionedbiosensorintermsofanalyticalapplicationshasyettobeverifiedandremainsunclearuptothisdate[6] TheaimoftheproposedcontributionisapresentationofthedevelopmentprocessandsubsequenttestingofanoveltypeofhybridelectrochemicalDNAbiosensoranditsverificationasareliableanalyticaltoolintermsofmonitoringDNAdamage
2Experimental
21Reagentsandchemicals
Low molecular weight double-stranded DNA (dsDNA) derived from salmonspermwasobtainedfromSigma-AldrichGermanyStocksolutions(01mgmL)
of dsDNA were prepared in a 01 molL phosphate buffer of pH=74 (PB)Dipotassium hexachloroiridate (K [IrCl ]) was purchased from Sigma-Aldrich2 6
GermanyStocksolutions(0001molL)ofK [IrCl ]werepreparedinthePB2 6
22Apparatus
Voltammetric measurements were performed using the μAutolab IIIFRA2potentiostatgalvanostat(EcoChemieTheNetherlands)drivenbyaNOVA111software(MetrohmAutolabSwitzerland)Allmeasurementswerecarriedoutina three-electrode system using an ldquoedge-planerdquo pyrolytic graphite workingelectrode(EPPGE)withanelectroactivesurfacediameterof3mm(BASJapan)asilver|silver chloride reference electrode (Ag|AgCl|sat KCl) and a platinum
84 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
counterelectrode(ElektrochemickeDetektoryCzechRepublic)ina20mLglassvoltammetriccellatambienttemperature
23Preparationofthebiosensor
PriortotheeverymeasurementsurfaceoftheEPPGEwasmechanicallycleanedbygentlewipingoftheelectrodeonthesoftpolishingpadrinsedwithdistilledwaterAfterwardstheelectrodewasrinsedwithdistilledwaterandplacedinthePBforthesubsequentelectrochemicalactivationElectrochemicalactivationwasperformed in thePBbyapplyingpotentialof15V for240swithoutstirringApotentialpulseinworkingrangeofpotentials(00ndash15V)wasthenapplied Additional electrochemical activationwasperformed in the solutionof the
3minus4minusredoxindicator([Fe(CN) ] )byconsecutivecyclingintherangeofpotentials6
from10tondash08V(15scans)andfrom055tondash015V(10scans) The electrochemical DNA biosensor based on the EPPGE (dsDNAEPPGEbiosensor)was prepared by the adsorption of dsDNAon the EPPGEOptimal
parametersofthedsDNAadsorptionwereaconcentrationof01mgmLinthePB
(c )adepositionpotentialof07V(E )andanadsorptiontimeof5ming(dsDNA) dep
(t )withoutstirringthesolutionads
Atlasttheelectrodewasimmersedinthesolutionoftheredoxindicatorandtheconsecutivecyclingintherangeofpotentialsfrom055tondash015V(20scans)wasperformedinordertosecurethestabilityoftheoxidationreductionsignalsofdsDNAattheEPPGE
24Procedures
Theexperimentalparameterswereasfollowssquarewavevoltammetry(SWV)inthePBwithapulseamplitudeof20mVafrequencyof50Hzascanrateof750mVsandapotentialstepof15mVlinearsweepvoltammetry(LSV)inthePBwithscanratesof02ndash10Vandapotentialstepof24mVAllcurveswere
recordedthreetimes(n=3)
3Resultsanddiscussion
Sincetheclosestresolutionoftheprocessesassociatedwiththeelectroreductionof singleDNAcomponents at pyrolytic graphitehasbeenperformedwith theldquobasal-planerdquopyrolyticgraphiteelectrode(BPPGE)wehavedecidedtotakeovercorrespondingexperimentaltechniqueandconditions(LSVscanrateof10Vssteppotentialof24mV)attheverybeginningofouroptimizationprocesswiththeEPPGE[6] Inthisparticularcaseitispossibletonoticetheoccurrenceofthetwomixedvoltammetricpeaksatthedefaultexperimentalconditions(greenlineFig1A)selectedforthereductionofdsDNAattheEPPGEBygraduallydecreasingthe
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 85
Fig 1Baseline-correctedLSVrecordingscorrespondingtothereductionofdsDNAattheEPPGEfordifferentvaluesofscanrate(02ndash10VsFig1A)respbaseline-correctedLSVrecordingscorres-pondingtothereductionofdsDNAattheEPPGEandthenegativetestperformedunderthesameexperimentalconditionswithintheblanksolution(phosphatebuffer)atthebareEPPGE(03VsFig1B)
scan rate the optimal conditions (νle03Vs) were found and the mutualseparationof the signalswas allowedndash characterizedby thepresenceof twosinglewell-developedvoltammetricpeaksatpotentialsofndash175Vrespndash190V(03Vs orange line Fig 1A) Taking into account previous work regardingprocessesassociatedwiththereductionofDNAatthemercuryelectrodesrespBPPGE we can assume that the peak appearing at the potential of ndash175 Vcorresponds to themixedpeak for the reductionof the cytosine and adenineresidueswithindsDNA(peakCA)[5ndash6] Closer resolution of the second voltammetric peak appears to be farmoreproblematic Regarding our previous study we have discovered that theutilizationofdifferentE fortheadsorptionofdsDNA(E lt07V)isconnecteddep dep
withanappearanceofthethirdoxidationsignal(besidestheoxidationsignalsofguanine resp adeninemoieties) at apotential of073V corresponding to theoxidationoffreeguaninebases(FGBs)presentwithinthesolutionofdsDNAInthiscasewecanassumethatthepeakappearingatapotentialofndash190VcanpossiblyrepresentthereductioncounterpartofFGBspresentwithinthesolutionofdsDNAThisassumptioncanalsobesupportedbytheaforementionedstudyandbythefactthatthereductionsignalatsuchahighnegativepotentialcanbeobservedfortheoligodeoxynucleotidescontainingguanineresidues[6] AdditionallyinordertoverifythetruenatureofthereductionsignalsdepictedatF ig1Bandtoexcludetheoptionthattherelatedsignalsdonotrepresenttheproductsofpriorelectrochemicalactivationof theEPPGE(variousCndashObasedchemicalspecies)wehavedecidedtoperformanegative(control)testwithinthe
86 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2Baseline-correctedLSVrecordingscorresponding to thereductionof thedsDNA(02Vs
Fig 2A)respbaseline-correctedSWVrecordingscorrespondingtotheoxidationoftheguanine(098V)respadenine(128V)moieties(075VsFig2B)attheEPPGEafteritsincubationinthephosphatebufferforadefinedtimeperiod(60ndash900s)
blanksolution(PB)employingthesameprotocolasforthedsDNAadsorptionattheEPPGEInthiscaseitispossibletoobservetheabsenceofanypronouncedvoltammetricpeakslinkedtothedsDNAadsorptionandonlythepresenceofoneirreversiblepeakatapotentialofndash153VcorrespondingtotheelectroreductionoftheCndashObasedmoietymoietieswhichdropsafterthedsDNAadsorptiontoonetenthofitsoriginalvalue(approximately) Perhaps the most important parameter regarding further optimizationprocessrepresentedthetime-dependentstabilityofthecorrespondingsignalsofdsDNAwhichcanbespecificallyimportantinrelationtothestudyofthetime-dependentoxidativedamageofdsDNA AsithasalreadybeenprovedasingleelectrochemicalactivationoftheEPPGEinthePBdoesnotrepresentasatisfyingtechniqueregardingstabilityofdsDNAoxidationsignalsat theEPPGEand theadditionalstabilization isachievedbyfurtherelectrochemicalactivationinthesolutionofaredoxindicator(Fig2B)[7]Basedonthisaverificationoftheproposedstabilizationprotocolintermsofthetime-dependent stabilityofdsDNAreduction signals in the solutionof thePBwithinthedefinedtimeperiod(60ndash900s)appearedasareasonablenextstep FromtheresultsdepictedinFig2Aitispossibletonoticethatwithinthefirst300sdsDNAreductionsignalsremainstableinrelationtothecurrentresponseaswellasintermsofthepotentialvalueWithanadditionalincubationtime(t )inc
(900sorangeline)thepeakcurrentofthevoltammetricsignalpresentatmorenegative potentials decreased which can probably be addressed as a slowprogressive elimination of the weak (electro)chemical forces related to theunspecificadsorptionoftheFGBsattheEPPGE
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 87
Fig 3Baseline-corrected SWV recordings corresponding to the oxidation of the guanine respadeninemoietiesattheEPPGEafteritsincubationinthesolutionofK [IrCl ](IR)foradefinedtime2 6
period(60ndash3600s)(075VsFig3A)andthecorrespondingrelativebiosensorresponses(ΔI )rel
evaluatedusingtheguanosine(turquoise)andadenosine(red)peaksplottedversustheincubationtime(Fig3C)Baseline-correctedLSVrecordingscorrespondingtothereductionofdsDNAattheEPPGEafteritsincubationinthesolutionofK [IrCl ](IR)fordifferenttimeperiods(60ndash3600s)2 6
(02 VsFig3B)andthecorrespondingrelativebiosensorresponses(ΔI )evaluatedusingtherelpeakCA(darkpink)plottedversustheincubationtime(Fig3-D)
AdditionallywehavedecidedtotesttheapplicabilityofthepresentedhybridbiosensorintermsofmonitoringdsDNAdamagecausedbyarepresentativeofone-electron oxidants ndash K [IrCl ] In this case the prepared dsDNAEPPGE2 6
biosensor was immersed into the solution of K [IrCl ] (0001molL) for the2 6
definedtimeperiod(60ndash3600s) In thecaseof theoxidationpath (SWVrecordingsdepicted inFig3A) it ispossible to observe a time-dependent decrease of the oxidation signal of theguaninemoietieswhereastheoxidationsignaloftheadeninemoietiesremainsunaffectedforthemostof the incubationperiodThisphenomenonis ingoodcorrelationwiththetheoreticalknowledgeregardingoxidativedamageofdsDNAcausedbyone-electronoxidants [3]Simultaneouslywith thisLSVrecordingsdepicted in Fig 3B followed the similar behavior (decrease in relation to the
88 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
currentresponseofthedsDNAreductionsignalndashpeakCA)asinthecaseofthesignal regarding oxidation of guanine moieties In addition according totheportionofthepreservedDNA(Fig3C3D)itispossibletoassumethatthepronouncedoxidativedamageofdsDNAcanbemonitoredquitepreciselynotonlydirectlyviathedsDNAoxidationsignaloftheguaninemoietiesbutevenindirectlythroughthedsDNAreductionsignalndashpeakCA
4Conclusions
Inthiscontributionwehavepresenteddevelopmentofanunorthodoxhybridelectrochemical DNA biosensor based on an EPPGE Optimization processconcerning some important parameters was performed as well as closerresolutionofthenatureofthereductionprocessesofdsDNAattheEPPGEwasachievedInordertoconfirmtheresultsoftheoptimizationprocessapplicabilityoftheproposedbiosensorhadbeenprobedintermsofmonitoringDNAdamagecausedbyK [IrCl ]Inthiscasethefinalresultshadprovedthattheprepared2 6
hybridbiosensorcanbeconsideredasaversatileanalyticaltoolformonitoringoxidativeDNAdamage(viaoxidationreductionsignals)andispresentedasafinealternative in comparisonwith conventional electrochemical DNA biosensorsprepared within the group of traditional transducer materials (mercury- orcarbon-based)
Acknowledgments
ThisresearchwassupportedbytheSpecificUniversityResearch(SVV260440)
References
[1] FojtaMDanhelAHavranLVyskocilVRecentprogressinelectrochemicalsensorsandassaysforDNAdamageandrepairTrACTrendsAnalChem79(2016)160ndash167
[2] GieseBSpichtyMWesselySLong-distancechargetransportthroughDNAAnextendedhoppingmodelPureApplChem73(2001)449ndash453
[3] Burrows CJ Muller JG Oxidative nucleobasemodifications leading to strand scissionChemRev98(1998)1109ndash1151
[4] DiculescuVC Chiorcea-PaquimAMOliveira-BrettAMApplications of aDNA-electro-chemicalbiosensorTrACTrendsAnalChem79(2016)23ndash36
[5] PalecekEJelenFElectrochemistryofnucleicacidsInElectrochemistryofNucleicAcidsandProteinsndashTowardsElectrochemicalSensorsforGenomicsandProteomicsPalecekESchellerFWangJ(edits)AmsterdamElsevier2005p74ndash174
[6] SpacekJDanhelAHasonSFojtaMLabel-freedetectionofcanonicalDNAbasesuraciland5-methylcytosineinDNAoligonucleotidesusinglinearsweepvoltammetryatapyrolyticgraphiteelectrodeElectrochemCommun82(2017)34ndash38
[7] AugustınMVyskocilVNovelelectrochemicalDNAbiosensorbasedonedge-planepyrolyticgraphite for DNA interaction studies In Proceedings of the 15th International StudentsConferenceldquoModernAnalyticalChemistryrdquoNesmerakK(edit)PragueFacultyofScienceCharlesUniversity2019p263ndash268
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 89
1Introduction
Cadmiumisoneofthemosttoxicmetalsanditswidespreadindustrialusesresultin increased environmental pollution Hence the development of sensitivemethodology for Cd determination is still highly desirable Chemical vaporgeneration(CVG)ofCdbythetetrahydroboratereductioninacidicmediumisasuitable alternative sample introduction technique compatible with atomicspectrometricdetectorsandofferingimproveddetectioncapabilityComparedtocommonliquidnebulizationCVGoffersseveraladvantagessuchassignificantlyhigheranalyteintroductionefficiencyandalsoanalyteseparationfromsamplematrix IncomparisontoCVGofcommonhydrideformingelementsthereisalackofliterature dealing with mechanistic aspects of CVG of Cd [1] as well as withstabilityandidentityofitsvolatilespecies(freeatomshydrideotherspecies)Very little information is also available on achieved generation efficiencyMoreover there are many discrepancies in the literature regarding optimum
Chemical vapor generation of cadmium for analytical atomic spectrometry
a b a b a aLINDASAGAPOVA BARBORAKODRIKOVA MILANSVOBODA STANISLAVMUSIL aJANKRATZER
a InstituteofAnalyticalChemistryoftheCzechAcademyofSciences Veveřiacute9760200BrnoCzechRepublicsagapovaiachczb DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublic
AbstractChemical vapor generation of cadmium volatile compounds wasoptimizedinordertodeterminetraceCdconcentrationsbyatomicabsorptionspectrometry(AAS)Severalreactionmodifiersbasedon
III+ II+ III+ IV+inorganicsaltsandcomplexesofCr Co Ti Ti weretestedtoimproveanalyticalperformanceandgenerationefficiencyTheuseofthese reaction modifiers resulted in 4ndash5 times enhancement insensitivity reflected also in corresponding increase of generationefficiency and better repeatability Generation efficiency wasdeterminedfromacomparisonbetweensensitivitiesobtainedwithchemicalvaporgenerationandconventionalsolutionnebulizationbothsimultaneouslycoupledwithinductivelycoupledplasmamassspectrometryTheidentityofthegeneratedcadmiumcompoundswillbediscussed
Keywordsatomicabsorption
spectrometryatomizationcadmiumchemicalvapor
generation
90 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
conditionsforCVGofCdAlthoughstrongacid(HClorHNO )isalwaysemployed3
asacarrierandNaBH asareductantsomeauthorsreportedvariousadditives4III+ III+ IV+(modifiers)basedontransitionmetalions(Cr Ti Ti )inthepresenceof
II+KCN[23]orCo inthepresenceofthioureaandascorbicacid[4]toimproveCdsignalssignificantly TheaimofthisworkwastoinvestigateCVGofCdinacomprehensivewayFirstlyCVGofCdwithoutandwithselectedmodifierswasoptimizedemployingatomicabsorptionspectrometry(AAS)asadetectorandexternallyheatedquartztube(QTA)astheatomizerSecondlytheeffectofatomizationtemperatureonCdsignalwasstudiedallowingthustodeducetheatomicormolecularstructureofgeneratedCd speciesThirdly generation efficiencyofCdvolatile specieswasquantified
2Experimental
21Reagentsandchemicals
minus1Boiled and bubled (Ar per 30min) deionizedwater (lt 01 μScm UltrapurWatrex USA) was used to prepare all solutionsWorking Cd standards were
minus1preparedfrom1000mgL Cdstocksolution(AstasolAnalytikaCzechRepublic)minus1bydilutionin01ndash048molL HCl(basedonthemodifieremployed)from37
HCl(paMerckGermany)Theoptimumgenerationconditionsbeingdifferentforeachmodifiertestedare listedinTable1ThereductantwasasolutionofNaBH (ge 97 Sigma-Aldrich Germany) in 04 (mv) KOH (pa Merck4
Germany) prepared fresh daily The solutions of modifiers were prepared as2+followsCo waspreparedfromCoCl 6H O(ge990PENTACzechRepublic)2 2
3+the solution of Cr from Cr(NO ) 9H O (ge 9999 tracemetal basis Sigma-3 3 23+AldrichGermany)thesolutionofTi fromTiCl solution(about15in10HCl3
4+Sigma-AldrichGermany)andthesolutionofTi fromTiOSO (ge999Sigma-4
AldrichGermany)Tostabilizethelattersolution1H SO wasusedprepared2 4
bydilutionof96H SO (paLach-NerSlovakia)SolutionofKCN(ge9702 43+ 3+FlukaSwitzerland)wasusedasasecondmodifierwhenworkingwithCr Ti or
4+ ndash3Ti asmodifiersitsconcentrationvariedfrom008to016moldm depending
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 91
Table 1OptimumconditionsforchemicalvaporgenerationofCdinthepresenceofmodifiersandtheirabsence
onthemetalionThiourea(CH N Sge980LachemaBrno)andascorbicacid4 2
(C H O ge997Riedel-deHaenGermany)wereusedasmodifierscombined6 8 62+withCo
22Instrumentation
221Chemicalvaporgenerationsystems
TwoCVG flow injection systemswere employed either a two channel systemwithoutadditionofamodifier(seeFig1A)orafourchannelsystemallowingadditionofmodifiers(seeFig1B)
ndash1 TheflowratesofHClandNaBH were42and10mLmin respectivelyinatwo4ndash1channelsystem(Fig1A)whiletheywerebothkeptat10mLmin inthefour
channelsystem(Fig1B)Theflowratesofmodifiersinthefourchannelsystemndash1were05mLmin Thevolumeofthesampleloopwas015mLinbothsystems
ndash1Carriergasflowrateof75mLmin Arwascontrolledbyamassflowcontroler(Cole-ParmerUSA)
92 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
(A)
(B)
Fig 1Schemesofthechemicalvaporgenerationflowinjectionsystemwith(A)twochannels(nomodifiers)and(B)fourchannels(modifiersemployed)
222Atomicabsorptionspectrometry
ThePerkin-Elmermodel503atomicabsorptionspectrometer(BodenseewerkGermany)wasequippedwithaCdelectrodelessdischargelamp(Perkin-ElmerUSA)operatedat228mAThemeasurementswereperformedat2288nmusinga07nmslitwidthTheShimadzumodelAA-7000atomicabsorptionspectrometer(ShimadzuJapan)wasalsousedACdhollowcathodelamp(PhotronAustralia)operatedat2288nmlinewith07nmspectralbandpassandalampcurrentof12mA Signals were recorded for 2 minutes and peak areas were taken forevaluation The QTA was heated electrically to the temperature required byfurnace(PerkinElmer)andanin-housemadefurnacecontrolledbytheREX-C100controller(SysconIndianaUSA)withtheK-typethermocouplesensor(OmegaEngineeringUSA)223QuantificationofCVGefficiencybyICP-MS
Overall CVG efficiency of Cd was quantified bymeans of inductively coupledplasmamassspectrometry(ICP-MS)fromcomparisonoftheslopesofcalibra-tionsobtainedwithnebulizationliquidCdstandardstothoseobtainedwithCVGThe efficiency of liquid nebulization was quantified using a modified wastecollection method (see reference [5] for details) The Agilent 7700x ICP-MSinstrument(AgilentUSA)wasoperatingat1600WofRFpowerThesignalwas
111 125monitoredat Cdisotopeandcorrectedforthesignalofinternalstandard( Tendash11000ngmL Tein2HNO )NebulizeranddilutionArgasflowrateswere11503
ndash1and0mLmin respectively
3Resultsanddiscussion
31Chemicalvaporgenerationconditions
UnivariateoptimizationswereperformedtofindoptimumconditionsforCVGofCdinpresenceandabsenceofmodifiersTheparameterstobeoptimizedwerecarrieracid(HCl)concentrationreductant(NaBH )concentrationmodifierIand4
modifier II concentrations carrier gas flow rate (Ar) length of reaction coilsRCI-III (see Fig 1B) The optimum conditions for individual modifiers aresummarizedinTable1
32IdentityofCdspecies
ThepeakareasofgeneratedCdspecieswereforagivenmodifiermeasuredintheQTAheatedto900degCandnon-heatedQTAsubsequentlyOptimumCVGconditionswere employed as summarized in Table 1 This simple experiment allowsdistinguishingbetweenatomic(freeatoms)andmolecularformsofgenerated
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 93
Fig 2RelativesignalofgeneratedCdspeciesmeasuredintheQTAheatedto900degC(blackbars)andnon-heatedQTA(whitebars)withoutorinthepresenceofmodifiers
speciesOnlyfreeatomscanbedetectedinnon-heatedQTAsimilarlyasincaseofmercurycoldvaporsOn thecontrarymolecularanalytespeciesareatomizedat900degCAsaconsequencethesignalregisteredintheheatedQTAcorrespondstobothatomicandmolecularspeciesgeneratedItmustbehighlightedthattheresidencetimeoffreeatomsintheatomizerisdependentonQTAtemperatureduetogasexpansionAsaconsequencethesignalinQTAheatedto900degCshouldreach25ofthesignalatambienttemperaturetakingintoaccountthatonlyfreeatomsaregeneratedSincethetemperaturealongtheopticalarmofQTAisnotdistributedhomogeneouslydecreasingtobothendtheeffectivetemperatureoftheatomizerislowerOurexperimentswithCVGofHgrevealedsignalinheatedQTAisaround40[6]TheresultsreachedforCVGofCdaredepictedinFig2ThesignalofCdinheatedQTAisaround50ofthesignaldetectedinnon-heatedQTA
4+whennomodifierisemployedorusingTi asthemodifierindicatingclearlyfreeCdatomsarethedominantvolatilespeciesgeneratedOnthecontraryalmostno
2+differenceinpeakareaswasobservedforCo asthemodifierwhilethesignalinheatedQTAwas even 5 times higher in heatedQTA compared to non-heated
3+atomizer with Cr as the modifier suggesting the dominant contribution of3+molecularstructurestoCdsignalespeciallyincaseofCr KCNreactionsystem
33Generationefficiency
TheoverallCVGefficiencywasestimatedfromacomparisonbetweensensitivitiesobtainedwithCVGsampleintroductionandconventionalsolutionnebulizationICP-MSunder the sameexperimental conditionsNebulization efficiency for aMicroMISTnebulizerwasdeterminedas79plusmn01ThegenerationefficiencyofCd was derived from the sensitivity enhancement between CVG and liquidnebulizationTheresultsaresummarizedinTable2indicatingthatCVGwithout
94 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
modifiersisonlycatwotimesmoresensitivecomparedtoliquidnebulization3+ 4+Generation efficiency of Cd increases to 60 in the presence of Ti and Ti
modifiers
4Conclusions
CVG of Cd was thoroughly optimized in the presence of selected modifiersreportedpreviouslyintheliteratureGenerationefficiencyofCdintheabsenceofanymodifierswasquantifiedto15whileitcanbeincreasedupto60inthe
3+ 4+presenceofTi KCNorTi KCNasmodifiersFreeCdatomsseemtobe the4+dominantCdformgeneratedintheabsenceofanymodifiersorusingTi KCN
3+modifierwhile rathermolecularCd structuresaregenerated inCr KCNand2+Co thioureaascorbicacidreactionsystems
ExperimentsareinprogresstofinishthiscomprehensivestudyOnlythebestmodifierwillbefurtherusedforCVGofCdtobecoupledwithotherspectrometricdetectorsandappliedtocertifiedreferencematerialsandrealsamples
Acknowledgments
ThisresearchhasbeensupportedbytheCzechScienceFoundationundercontract18-01116SandbytheInstituteofAnalyticalChemistryoftheCzechAcademyofSciences(InstitutionalResearchPlannoRVO68081715)andCharlesUniversity(ProjectnoSVV260440)
References
[1] PitzalisEAngeliniDMascherpaMCDacuteUlivoAInsightintothemechanismscontrollingthechemicalvaporgenerationofcadmiumJAnalAtSpectrom33(2018)2160ndash2171
[2] ArslanZYilmazVRoseLEfficientgenerationofvolatilecadmiumspeciesusingTi(III)andTi(IV)andapplicationtodeterminationofcadmiumbycoldvaporgenerationinductivelycoupledplasmamassspectrometryMicrochemJ123(2015)170ndash178
[3] YilmazVRoseLArslanZLittleMDOn-linechemicalvapourgenerationofcadmiuminthepresenceofhexacyanochromate(III)fordeterminationbyinductivelycoupledplasmamassspectrometryJAnalAtSpectrom27(2012)1895ndash1902
[4] Y Lu SunHW YuanCG YanXP Simultaneous determination of trace cadmiumandarsenic inbiologicalsamplesbyhydridegeneration-doublechannelAFSAnalChem74(2002)1525ndash1529
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 95
Table 2GenerationefficiencyofchemicalvaporgenerationofCdasquantifiedbyICP-MS
Modifiers Generationefficiency
Nomodifiers 15plusmn13+Cr KCN ndash2+Co thioureaascorbicacid ndash3+Ti KCN 58plusmn24+Ti KCN 61plusmn2
[5] VyhnanovskyJStrugeonREMusilSCadmiumassistedphotochemicalvaporgenerationoftungsten fordetectionby inductively coupledplasmamass spectrometryAnal Chem91(2019)13306ndash13312
[6] MigasovaMMatousekTSchrenkovaVZ ıdekRPetry-Podgorska IKratzer JMercuryvolatilespeciesgenerationfromHClandTRISbuffermediaAnalChimActa1119(2020)68ndash76
96 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Atomicfluorescencespectrometry(AFS)coupledwithvapourgenerationisanultrasensitive analytical method for determination of various elements ItsanalyticalperformancecanbecomparabletoICP-MSwithliquidnebulizationbutatsubstantiallylowercost[1]SampleintroductiontoAFSisacrucialstepoftheanalyticalproceduresincetheanalytehastobeintroducedtotheatomizerintheformofitsvolatilespecies Hydridegeneration(HG)isamaturetechniqueofsampleintroductionduringwhichvolatileanalytehydridesare formedbyreactionwithareducingagenttypicallysodiumborohydrideAnewemergingtechniquephotochemicalvapourgeneration(PVG)employsUV irradiationof theanalyte in liquidphase in thepresenceofaphotochemicalagent(usuallyalowmolarmassorganicacidformic
minusoraceticacid)Highlyreducingradicalspecies(HbullRbullandCOObull )andaquatedelectronsare formedduring irradiationandreactwith theanalyte to form its
Photochemical vapour generation of bismuth coupled with atomic fluorescence spectrometry
ab ab a aBARBORASTA DLEROVA JAROMIRVYHNANOVSKY JIR IDE DINA STANISLAVMUSIL
a InstituteofAnalyticalChemistryoftheCzechAcademyofSciences Veveřiacute9760200BrnoCzechRepublicstadlerovaiachczb DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublic
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 97
AbstractPhotochemical vapour generation of bismuth was successfullycoupledwithnon-dispersiveatomic fluorescencespectrometry forthefirsttimeVolatilespeciesofBiweregeneratedusingastandardmercurylow-pressuretubelampandacoiledreactorfromareaction
2+mediumwhichwas composedof acetic and formic acid Co ionswereusedasasensitizerOptimizationofatomizationconditionsinaflame-in-gas-shieldatomizerwasperformedThismethodologywascomparedtothecommonlyemployedhydridegenerationapproachAbsolutelimitofdetectionof68pgwasachievedwithphotochemicalvapour generation which is still about 7 times worse than withhydride generation The developed methodology was successfullyverifiedbyBideterminationinareferencematerialofwater
Keywordsatomicfluorescence
spectrometrybismuthhydridegenerationphotochemicalvapour
generation
volatilespeciesInbothcasesthegeneratedvolatilespeciesoftheanalytehavetobeseparatedfromtheliquidphaseinthegas-liquidseparatorandarecarriedtotheatomizerbyacarriergas[2] InthisworkanatomizerdesignedspecificallyforAFStheflame-in-gas-shieldatomizerwasused(Fig1) It consistsofaverticalquartz tubesuppliedwithargon and hydrogen together with the analyte volatile species Moreover acapillaryisinsertedintheverticalaxisoftheverticaltubethroughwhichoxygenisintroducedA hydrogen-oxygenmicroflameburns on top of the capillary Themicroflameisshieldedfromtheambientatmospherebyaflowofargonwhichisintroducedthroughashieldingunitfittedaroundtheverticaltube[34] Theaimofthisworkwastooptimizeatomizationconditionsintheflame-in-gas-shieldatomizerusingPVGasasampleintroductiontechniqueandtocomparetheanalyticalcharacteristicsofPVGandHGforultrasensitivedeterminationofbismuthbyAFS
2Experimental
21Reagentsandchemicals
Deionized water (Ultrapur Watrex USA) was used for preparation of all thesolutionsWorkingBisolutionswerepreparedfreshdailybyserialdilutionof
ndash1stock1000mgl BistandardforAAS(Sigma-AldrichGermany)RegardingHG05(mv)NaBH in04(mv)KOHwasusedasa reductantAsolutionof4 ndash11mol l HClwasusedasacarrierandblank
98 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Flame-in-gas-shieldatomizerOHndashobser-vationheight
RegardingPVGformicacid(98paLach-NerCzechRepublic)andaceticacid (998 pa Lach-Ner CzechRepublic)were used for preparation of thereaction medium they were purified in a Teflon BSB-939-IR sub-boilingdistillation apparatus (Berghof Germany) The composition of the reactionmedium(40(vv)acetic125(vv)formicacid)wasoptimizedearlier[5]
ndash1The 5000mgl Co stock solution was prepared from cobalt(II) acetatetetrahydrate (pa Lach-Ner Czech Republic) and used as a sensitizer ofphotochemicalreactionTheoptimalconcentrationofCointhestandardsamples
ndash1andblanksolutionscorrespondedto50mgl (ref[5]) Acertifiedreferencematerial(CRM)-1643fTraceElementsinWater(NationalInstituteofStandardsandTechnologyUSA)wasusedtochecktheprecisionofthedevelopedmethodology
22Instrumentation
221Atomicfluorescencespectrometer
An in-house assembled non-dispersive atomic fluorescence spectrometerconstructedatourlaboratorywasusedforBideterminationandisdescribedindetail elsewhere [3] The detector output provided signals in microV Peak areacorrectedtobaselineandmainlysignaltonoiseratioweretheparametersusedtoevaluatethedata
222Hydridegeneratorphotochemicalvapourgeneratorandatomizer
A flow injection hydride generator was employed (Fig 2a) The reductantndash1 ndash1(12mlmin )andthecarrier(4mlmin )werepumpedbyaperistalticpump
Thesamplewasinjectedthrougha1mlsampleloopintotheflowofcarrierAglassgas-liquid separator (5 ml) with forced waste removal was employed forseparatingthegasphasecontainingbismuthanewhichwasthencarriedtotheatomizerbyargon Thephotochemicalvapourgenerator(Fig2b)consistedofthephotoreactorconstructedwitha15Wlow-pressureHggermicidallamp(Cole-ParmerUSA)wrappedaroundwith6mofPTFEtubing(1mmidinternalvolume471ml)The
ndash1reactionmedium(3mlmin )waspumpedbyaperistalticpumpThesamplewasinjected througha056ml sample loopApolypropylenegas-liquid separator
(15ml)withforcedwasteremovalimmersedinanicebath[6]wasemployedforseparatingthegasphasecontainingBivolatilespeciessubsequentlycarriedtotheatomizerbyargon Theflame-in-gas-shieldatomizerisdepictedinFig1detaileddescriptionisgiveninRef[3]Theobservationheight(OH)isdefinedasthedistancefromthetopofthecapillarytothecentreoftheopticalbeam
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 99
23Samplepreparation
CRMNIST1643fwasdilutedwith1MHCl80-foldforBideterminationbyHG-AFSRegardingBideterminationbyPVG-AFSthesampleneededtobeevaporatedtodrynessinordertogetridofnitricacidthatseriouslyinterferesatmMlevel[5]Avolume of 3ml of CRMwere pipetted into a 40 ml quartz vial evaporated(temperature asymp100 degC two replicates) and subsequently diluted ca 33-foldAsamplepreparationblank3mlofdeionizedwaterwaspreparedaswell
3Resultsanddiscussion
TheatomizationconditionsforHG-AFSwereoptimizedinourpreviouswork[3]TheseconditionswereusedasinitialtofindtheoptimumconditionsforPVG-AFSwith the flame-in-gas-shieldatomizerwith respect to sensitivityandsignal tonoiseratioFirstlythehydrogenfractionintherange10ndash16wasoptimizedatconstant total gas flow rate (sum of total argon and total hydrogen) of
ndash1500mlmin the lower the hydrogen fraction the better However at 10hydrogenfractiontheflamewasnotstableenoughandwentoftenouthenceitwasoptedfor12hydrogenfraction
100 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2(a)Hydrideand(b)photochemicalvapourgenerator
The oxygen flow rate through the capillary was optimized in the rangendash15ndash30mlmin Thehighestsignaltonoiseratiowasachievedwiththeflowrateof
ndash120mlmin Thetotalgasflowratewasoptimizedatconstant12hydrogenfractioninthe
ndash1range500ndash800mlmin Theoptimumobservationheightvarieswithtotalgasflow rate so it had to be optimized as well The optimum conditions aresummarizedandcomparedtothoseachievedwithHGinTable1 TheanalyticalfiguresofmeritofPVG-AFSwiththeflame-in-gas-shieldatomi-zerweredeterminedThecalibrationfunctionconstructedwith010025050
ndash1 2100 and 200 microg l Bi standardswas linear (R = 09998) The repeatabilityndash1expressedastherelativestandarddeviation(n=10)was6at1microgl andthe
ndash1relativeandabsolutelimitsofdetection(3σn=10)achievedwere12ngl and68pgrespectively(Table2)TheabsolutelimitofdetectionachievedwithHGwas 76 times lower which can be attributed to several aspects Firstly thegenerationefficiencyforPVGapproachwasaround53while100isexpectedforHG[3]Secondlyafullwidthathalfmaximumofthemeasuredpeakswasca2-foldgreaterwhichnecessitatedlongerintegrationtimeandwasthusreflectedinhighernoiseofthesignalsFinallythelimitofdetectionforPVGapproachwas
ndash1affectedby seriouscontamination (around10ng l )mostprobably from thesensitizersolutionthatcontainedBiasimpurity Tovalidate theproposedmethodologyBi contentwasdetermined inCRMNIST1643f (Table3) and the resultswere compared to thosemeasuredwithHG-AFS[3]Duetosevereinterferencesfrominorganicacidsespeciallynitricacid[5]thesampleneededtobeevaporatedtodrynessandthenfilledupwiththe
2+reactionmediumcontainingCo as thesensitizer(NIST1643f isstabilized in
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 101
Parameter HG-AFS(ref[3]) PVG-AFS
ndash1Artotalmlmin 440 528ndash1H totalmlmin 60 722 ndash1O mlmin 7 202
OH mm 6 9ndash1Arshieldlmin 1515 1515
Table 1Atomizationconditionsforflame-in-gas-shieldatomizer
Parameter HG-AFS(ref[3]) PVG-AFS
LODpg 09 68ndash1LOQngl 09 12
Repeatability lt1 6
Table 2AnalyticalfiguresofmeritofHG-AFSandPVG-AFS
ndash1032moll nitricacid)Theresultsobtainedbybothmethodologiesareingoodagreementwiththecertifiedvalue
4Conclusion
Photochemical vapour generation of Bi was successfully coupled with non-dispersiveatomicfluorescencespectrometryforthefirsttimeanditsapplicabilitywas verified by determination of Bi in certified reference material of waterComparedtohydridegenerationconditionsofatomizationdifferinanoptimalobservationheightandsupplyofoxygenwhichmaybeneededtoldquoburnoutrdquotheorganicvapoursthatarereleasedfromthereactionmediumtothegasphasehowever this remains tobe verifiedAlthough there are still some limitationsregardingthelimitsofdetectionrepeatabilityandinterferencesthisnewsampleintroductionapproachseemstobepromising
Acknowledgments
The support of the Czech Science Foundation (19-17604Y) Czech Academy of Sciences(Institutional supportRVO68081715)andCharlesUniversity (ProjectSVV260560andProjectGAUK1048120)isgratefullyacknowledged
References
[1] Musil SMatousek T Currier JM StybloM Dedina J Speciation analysis of arsenic byselectivehydridegeneration-cryotrapping-atomicfluorescencespectrometrywithflame-in-gas-shield atomizer achieving extremely low detection limits with inexpensiveinstrumentationAnalChem86(2014)10422ndash10428
[2] SturgeonREPhotochemicalvaporgenerationaradicalapproachtoanalyteintroductionforatomicspectrometryJAnalAtSpectrom32(2017)2319ndash2340
[3] S tadlerova B Kolrosova M Dedina J Musil S Atomic fluorescence spectrometry forultrasensitivedeterminationofbismuthbasedonhydridegenerationndashtheroleofexcitationsourceinterferencefilterandflameatomizersJAnalAtSpectrom35(2020)993ndash1002
[4] DedinaJAtomizationofvolatilecompoundsforatomicabsorptionandatomicfluorescencespectrometryOnthewaytowardstheidealatomizerSpectrochimActaPartB62(2007)846ndash872
102 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Certified HG-AFS PVG-AFSndash1valuemicrogl
ndash1 a ndash1 a valueobtainedmicrogl recovery valueobtainedmicrogl recovery
1262plusmn011 128plusmn01 102plusmn1 121plusmn09 97plusmn5
a Spikedrecovery=slopeofstandardadditions(noadditionandtwospikedconcentrationstoasample)slopeofexternalcalibration
Table 3ThedeterminedcontentofBiinCRMNIST1643fpresentedasmedianvalueplusmncombineduncertainty(n=3)andrecoveries
[5] Vyhnanovsky J Yildiz D Musil S Effect of metal sensitizers on photochemical vaporgeneration of bismuth for analytical atomic spectrometry In Proceedings of the 15thInternationalStudentsConferenceModernAnalyticalChemistryKNesmerak(ed)PragueCharlesUniversity2019p257ndash262
[6] VyhnanovskyJSturgeonREMusilSCadmiumassistedphotochemicalvaporgenerationoftungstenfordetectionbyinductivelycoupledplasmamassspectrometryAnalChem91(2019)13306ndash13312
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 103
1Introduction
Liquidcrystalsareorganicsubstancesthatformamesomorphicphaseinsolu-tion[1]TheyareliquidlikeliquidsbuthaveaninternalconfigurationassolidsTheir light transmittance changes in the electric fieldwhich is used in liquidcrystaldisplays(LCDs)CholestericliquidcrystalsaretemperaturesensitiveThecolorofreflectedlightchangeswithsmalltemperaturechangeThisisusedinmedicineasasensitivetemperatureindicatorfordisease-infectedtissues High performance liquid chromatography or supercritical fluid chromato-graphy[23]areusedtoseparateanddetermineliquidcrystalcompoundsAnelectrophoretic method could be complementary to these commonly usedmethods To our best knowledge no study dealing with liquid crystal puritycontrolbyelectrokineticchromatographywaspublisheduntilnow Electroneutralsubstancesmoveincapillaryzoneelectrophoresisatthesamespeedcorrespondingtothespeedoftheelectroosmoticflow(EOF)andtherefore
Separation of liquid crystals using non-aqueous capillary electrokinetic chromatography
KATER INACOKRTOVATOMA S KR IZ EK
DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublickaterinacokrtovagmailcom
AbstractLiquid crystals arewidelyused in electronicsmedicine andotherfields Analytical separations are important in the development ofnewliquidcrystalstocontrolthepurityofsynthesizedsubstancesThesampleanalysisisimportantfordetectionofimpuritiesformedduring synthesis Liquid crystal-forming substances cannot beseparated by capillary zone electrophoresis due to the absence ofreadily ionizable groups Therefore electrokinetic chromatographywasused in thisworkAnotherproblemcomplicating theanalysiswastheverylowsolubilityofanalytesinwaterSeparationsinthisworkwere thereforecarriedoutundernon-aqueousconditions inacetonitrilewithaceticacidtoadjustthepHandhexadecyltrimethyl-ammonium chloride as a detergent to mobilize the non-ionizedanalytesUndertheseconditionsitwaspossibletoseparateimpu-ritiesfromsynthesizedanalytesinsamples
Keywordselectrokinetic
chromatographyliquidcrystalsnon-aqueouscapillary
electrophoresis
104 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
itisnotpossibletoseparatethemDuetothisanelectrokineticchromatographymethodwasdevelopedInthismethodasurfactantisaddedtothebackgroundelectrolyteMoleculesaggregateandformsphericalformationscalledmicelles[4]ifthesubstanceisaddedinsufficientconcentrationiehigherthanthecriticalmicellar concentration (CMC) Separation is possible due to interactions ofnonpolarmoleculepartswiththenonpolarmicelleinsideAlthoughwateristhemostusedsolventinelectrophoreticmethodsforseparationofwater-insolublesubstances organic solvents are selected However such solvent must meetcertaincriteriatobesuitableforuseincapillaryelectrophoresisAllcomponentsmustbesolubleinthesolvent[5]ItshouldnotbeflammabletoxicorreactiveforpracticalityitshouldbeliquidatroomtemperatureandalsoitspriceistakenintoaccountThevalueof its relativepermittivitywhichdescribes the strengthofinteractionsbetweenionsshouldbearound30Lowdynamicviscosityisalsopreferred to allow faster migration of analytes No organic solvent meets allparameters of the ideal solvent In practice methanol acetonitrile and theirmixturesarethemostusedTheseparationparameterscanbeinfluencedbyusingan organic solvent of the background electrolyte This topic has already beenwidelyexplored[6ndash8] Itwasgenerallyassumedthat inanhydrousconditionsmicellesarenotcreateddespitesufficientsurfactantconcentrationHoweveritwasfoundoutthatdodecylsulfatecanformstablemicelleswhenthebackgroundelectrolyteisdissolvedinformamide[9]Fortheanalysisofactivesubstancesinmedicinal plants Chen et al developed a method in which sodium cholatedissolvedinmethanolisusedasasurfactant[10]Theaddedpseudostationaryphase does not always form micelles but can still affect mobilization andseparationofanalytesiftheanalytesinteractdifferentlywithfreemoleculesofsurfactantInthisstudywater-insolubleliquidcrystalswereseparated(Fig1)Thereforenonaqueouselectrokineticchromatographymethodwasdeveloped
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 105
Fig 1 Structures of liquid crystals 4-([1-oxo-1-(pentyloxy)propan-2-yl]oxycarbonyl)phenyl4-(octyloxy)-[11-biphenyl]-4-carboxylate (ZL 85) and 4-([1-(decyloxy)-1-oxopropan-2-yl]oxycarbonyl)phenyl 4-(dodecyloxy)-[11- biphenyl]-4-carboxylate (ZL 1210) OpticalisomerismsitesaremarkedwithanasteriskStructurescreatedinMarvinSketch[11]
2Experimental
21Reagentsandchemicals
Acetonitrilege999fromSigma-Aldrich(Germany)aceticacid99fromLach-Ner Neratovice (Czech Republic) and hexadecyltrimethylammonium chloride25(ww)inwaterfromSigma-Aldrich(USA)wereusedforpreparationofback-groundelectrolyteMesyloxidepa(MO)suppliedbyLach-nerNeratovice(CzechRepublic)wasusedasareferencesubstance
22Instrumentation
ForexperimentsG7100ACapillaryElectrophoresisInstrument(AgilentTechno-logiesGermany)wasusedwithUV-VISdetectoroperatingat235nmand254nmwavelengthMeasurementswereconductedinafused-silicacapillaryof50microminner diameter with the total length 500cm and effective length 415cm(PolymicroTechnologiesUSA)
23Method
Capillarywas flushed for3minuteswith1MHCland for2minuteswith thebackgroundelectrolyteBackgroundelectrolytewaspreparedbymixingaceticacid (10mM) and hexadecyltrimethylammonium chloride (40mM) in aceto-nitrile Sampleswere introducedhydrodynamicallybyapressureof5kPa for1secondSampleswerefirstdissolvedinacetonitrileandthendilutedtwotimeswith the background electrolyte A voltage of 20 kV was applied during theseparation
3Resultsanddiscussion
Liquidcrystalsampleswerepractically insoluble inwater theirsolubilitywasndash3testedataconcentrationlevelof1mgcm inmethanolandacetonitrileWhile
samples were not sufficiently soluble in methanol they were successfullydissolvedinacetonitrile Becauseallanalytesaresubstancesthatdonothaveeasilyionizablefunctionalgroups theelectrokineticchromatographymethodwaschosen forseparationAsuitablesurfactantwassoughtCommonlyusedsodiumdodecylsulfate(SDS)isinsoluble in acetonitrile Therefore hexadecyltrimethylammonium chloride(CTAC) which had sufficient solubility for further experiments was chosenAlthoughasuitablebufferwassoughttoensureastablepHduetoproblemswithprecipitationofbuffercomponentsinthenon-aqueousenvironmentaceticacidwasusedtoadjustandmaintainpHofbackgroundelectrolytesolutionAstheadditionofcationicsurfactantsuchasCTACleadstoEOFreversalthedependence
106 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ofEOFmobilityontheconcentrationofCTACinthebackgroundelectrolytewasmeasuredContrarytowhatisobservedinaqueousbackgroundelectrolytesEOFwasnotreversedItsmobilitydecreasedwithincreasingCTACconcentrationbutnomajorchangesoccurredabove40mMconcentrationThecapillarywallwasprobablyalreadysaturatedbyCTACandthefurtherincreaseinconcentrationhadno signifficant effect on the conditionof the capillarywall Therefore a CTAC
ndash3concentrationof40mmoldm was chosenas sufficient for furthermeasure-mentswithrespecttotheincreasingcurrentwithincreasingionicstrengthofthebackgroundelectrolyte The optimized method was used for separation of several liquid crystalsamplesofdifferentpurityInthesampleoftheZL85liquidcrystalwith99purityonezoneoftheanalytewasdetectedImpuritieswereseparatedfromthisanalytewhen the samplewith lower puritywas introduced The peak of theanalytewasidentifiedbasedonrelativemigrationtimerelatedtomesityloxideSeparationoftheanalytefromanimpurityinthesampleZL8576isshowninFig 2The relativemigration timeof the firstpeak is 0834 therefore itwasidentifiedastheZL85analyteThestandarddeviationoftherelativemigrationtimesinfivemeasurementswas0002min(01) Using the available high purity sample it was possible to measure thecalibration line forquantificationof theanalyte in lesspuresamplesLimitof
ndash3detection was determined as 0009mgcm and limit of quantification as ndash30031mgcm FromthecalibrationlineconcentrationofZL85inthesample
with lower purity was calculated The concentration was determined as
48(ww)standarddeviation5(ww)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 107
Fig 2ElectropherogramobtainedwhenasampleofZL85liquidcrystalwithlowerpuritywasndash3introducedSamplewas introduced in01mgcm concentrationandwithaddedmesityloxide
ndash3(10mgcm )Capillarywithinnerdiameterof50micromtotallengthof500cm415cmeffectivelength The background electrolyte was acetonitrile with 10 mM acetic acid and 40 mMhexadecyltrimethylammonium chloride A voltage of 20 kVwith positive polaritywas appliedDetectionat254nm
ForsampleZL121099onlytheanalyteandmesityloxideweredetectedInthe sample ZL 1210 59 several impurities were separated and detected(Fig3)Accordingtotherelativemigrationtimetheanalyteofinterestcorres-pondstothefirstpeakPeakresolutionissufficientTheresolutionoftheanalytepeakandthesecondpeakis284andtheresolutionoftheothertwopeaksis230
4Conclusions
InthisstudyanewmethodforanalysisofnewlysynthesizedliquidcrystalswasdevelopedSomeparametersofthemethodwereoptimizedndashoptimumconcen-tration of hexadecyltrimethylammonium chloride was searched The identifi-cation of analyteswas based on a comparison of relativemigration times InsamplesZL85andZL1210withlowerpuritytheimpuritieswereseparatedfromthepeaksofliquidcrystalsthecontentofanalytewasdeterminedintheZL85sampleaccordingtothecalibrationline
Acknowledgments
IwouldliketothanktheInstituteofPhysicsoftheCzechAcademySciencesforprovidingnewlysynthesized liquid crystals This work has been supported by Specific University Research(SVV260560)andbyCharlesUniversityResearchCentreprogramNoUNCESCI014
References
[1] GennesPGProstJThePhysicsofLiquidCrystals2ndedNewYorkOxfordUniversityPress1993
108 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ndash3Fig 3ElectropherogramofsampleZL121059ataconcentrationof05mgcm withmesitylndash3oxideataconcentrationof10mgcm capillarywith innerdiameterof50microm total lengthof
500 cmeffectivelength415cmThebackgroundelectrolytewasacetonitrilewith10mMaceticacidand40mMCTACAppliedvoltage20kVpositivepolarityDetectionat235nm
[2] Vankatova P KalıkovaK KubıckovaA Ultra-performance supercritical fluid chromato-graphy A powerful tool for the enantioseparation of thermotropic fluorinated liquidcrystalsAnalChimActa1038(2018)191ndash197
[3] Vankatova P Kubıckova A Cigl M Kalıkova K Ultra-performance chromatographicmethodsforenantioseparationofliquidcrystalsbasedonlacticacidJSupercritFluids146(2019)217ndash125
[4] Terabe S Otsuka K Ichikawa K Tsuchiya A Ando T Electrokinetic separations withmicellarsolutionsandopen-tubularcapillariesAnalChem56(1984)111ndash113
[5] RiekkolaMLRecentadvancesinnonaqueouscapillaryelectrophoresisElectrophoresis23(2002)3865ndash3883
[6] Wright PB Lister AS Dorsey JG Behavior and use of nonaqueous media withoutsupporting electrolyte in capillary electrophoresis and capillary electrochromatographyAnalChem69(1997)3251ndash3259
[7] PorrasSPKenndlerECapillaryzoneelectrophoresisinnon-aqueoussolutionspHofthebackgroundelectrolyteJChromatogrA1037(2004)455ndash465
[8] PorrasSPRiekkolaMLKenndlerETheprinciplesofmigrationanddispersionincapillaryzoneelectrophoresisinnonaqueoussolventsElectrophoresis24(2003)1485ndash1498
[9] GuoXWangK ChenGH Shi JWuX Di L LWangY Determination of strobilurinfungicideresiduesinfruitsandvegetablesbynonaqueousmicellarelectrokineticcapillarychromatography with indirect laser-induced fluorescence Electrophoresis 38 (2017)2004ndash2010
[10] Chen AJ Li C Gao WH Hu ZD Chen XG Application of non-aqueous micellarelectrokinetic chromatography to the analysis of active components in radix SalviaemiltiorrhizaeanditsmedicinalpreparationsJPharmBiomedAnal37(2005)811ndash816
[11] MarvinSketch [computer program] version 1990 ChemAxon httpschemaxoncom-productsmarvin
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 109
1Introduction
Sudandyesaresyntheticazo-basedaromaticcompoundsTheyaretraditionallyusedinvariousindustriessuchaschemicaltextileandwoodworkingasdyestocolourwaxesplasticsoilspolishesandsoforthTheyhavebeencategorizedasclass3carcinogensbytheInternationalAgencyforResearchonCancerandtheiruseisthereforeforbiddeninthefoodindustryTheyareknownfortheirbrightcolours and easy and cost-effectivemanufactureThey arenearly insoluble inwater but soluble in various organic solvents such asmethanol or trichloro-methane[1] SudanI1-phenylazo-2-naphthol(Fig1A)isadyeusedasanorangecolouringagentItssometimesalsosoldundernamesSolventOrangeRorCISolventYellow14ItisformedasasecondaryproductinthemanufactureoftheSunsetYellowdye
Electrochemistry of Sudan I and its derivates in aqueous media
ad b aANNAONDRA CKOVA MARIESTIBOROVA LUDE KHAVRAN cd adKAROLINASCHWARZOVA -PECKOVA MIROSLAVFOJTA
a CentralEuropeanInstituteofTechnologyMasarykUniversity Kamenice753562500BrnoCzechRepublicannaondrackovaceitecmuniczb DepartmentofBiochemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublicc UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistry FacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublicd InstituteofBiophysicsCzechAcademyofSciences Kralovopolska13561265BrnoCzechRepublic
AbstractSudanIisanaromaticazo-compoundthathasbeenproventobeacar-cinogenDuringitsmetabolizationbycytochromeP450inliverafewmain derivates can be identified Thiswork sets out to assess themechanismofelectrochemicalreductionandoxidationofSudanIitshydroxylationderivativesfeaturingmetabolitesintheSudanIdetoxi-fication pathway and to introduce their selective voltammetricanalysis on boron-doped diamond electrode We show successfuldifferentiationamongthesecompoundsthankstothedifferencesintheelectrochemicaloxidationoftheirphenolicgroups
Keywordsborondopeddiamond
electrodecytochromeP450electrochemicalanalysisSudanI
110 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
InmammalianorganismsSudanIismetabolizedbythemicrosomaldetoxi-fying systemwitha central roleof cytochromeP450hydroxylationactivity inliver[2]DuringtheoxidativeprocessofmetabolizingSudanIseveralmetaboliteswereidentifiedbyprevioustestsThesearegt1-(phenylazo)-naphtalene-26-diol(further abbreviated SI-6OH) 1-(4-hydroxyphenylazo)-2-hydroxynaphtol(furtherabbreviatedSI-4OH)and1-(4-hydroxyphenylazo)-naphtalene-26-diol(furtherabbreviatedSI-46-diOH)thestructuresarepresentedinFig1 ThemainmethodcurrentlyusedtoidentifySudanIamongotherdyeswithsimilar structure is high-performance liquid chromatography (HPLC) It isrecommendedasthestandardmethodtoidentifythelevelofSudanIinfood[3] ComparedtoHPLCelectrochemicalmethodsareprovingtobefastercheaperandcomparablypreciseUnfortunatelyacomprehensiveelectrochemicalstudyofSudanIandparticularlyofitshydroxylatedmetaboliteshasnotbeencompletedyetThedyecanbedetectedthroughelectrochemistryeitherbytheoxidationofitsphenolicgrouporviareductionoftheazogrouppresentinitsmoleculeInbothcasesotherelectrochemicallyactivemoietiesareformedThederivatesofSudanIcanbedetectedandrecognizedfromSudanIthroughanalogousprocesses[4]InthisstudywefocusedoncomparisonofelectrochemicalbehaviourofSudanIanditshydroxylatedmetabolitesonborondopeddiamondelectrodetoaddressthepossibilitiesoftheirrecognitionsinmixturesbasedondifferencesinanodicandcathodicsignals
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 111
Fig 1 Chemical structure of (A) Sudan I (B) 1-(phenylazo)-naphtalene-26-diol (SI-6-OH)(C) 1-(4-hydroxyphenylazo)-2-hydroxynaphtol (SI-4-OH) and (D) 1-(4-hydroxyphenylazo)-naphtalene-26-diol(SI-46-diOH)
(A) (B)
(C) (D)
2Experimental
21Reagentsandchemicals
SudanI(Merckanalyticalstandardgrade)wasdissolvedinethanol(Merck)andkept at room temperatureChemicals forBritton-Robinsonbufferpreparation(acetic acid boric acid orthophosphoric acid sodium hydroxide) were fromMerckwithpurityge99pHofthebufferwasadjustedbymixingoftheacidsandsodium hydroxide solution at different ratios The Sudan I metabolites weresynthetized at the Department of Biochemistry Faculty of Science CharlesUniversityandkeptinmethanolattemperature3degC
22Instrumentation
Cyclicvoltammetric (CV)measurementswerecarriedout inBritton-RobinsonbufferofpH=70atroomtemperatureSudanIanditsderivativeswereaddedto
minus1thesolutionofBritton-Robinsonbuffertofinalconcentrationof5micromolL andstirred Before the measurement oxygen was removed from the solution bypurgingwithargonfor3minutesAutolabanalyzerPGSTAT20(EcochemieTheNetherlands)inconnectionwithVA-Stand663(MetrohmSwitzerland)GPES49(MetrohmSwitzerland)andathree-electrodesetup(withborondopeddiamond
2(WindsorScientificUKdiskdiameter3mmA=707mm )asworkingelectrodeminus1AgAgCl3molL KCl as reference electrode and platinum wire as auxiliary
electrode) Five cycles were performed for each measurement at scan rateminus1of1Vs
3Resultsanddiscussion
ForeachcompoundtwoseparateCVmeasurementswereperformedeachwithfive cycles performed in rapid succession For bothmeasurements the initialpotentialwassetat0VTheanodicscancontinuedto+1Vturnedtowardsndash1Vandfinishedat0VIntheothersetupcathodicscanwasperformedfirstfromthestartingpointtondash1Vturnedtowards+1Vandreturnedto0VThiswaywewereabletoobservethebehavioursofSudanIandcompareittothatofitsderivativeswhilefirstbeingreducedandthenoxidisedorviceversa In the anodic scan of Sudan I and its derivatives (Fig 2) differences in thepositionsoftheoxidationpeaksineachcompoundcanbeobservedWhileSudanIwithonlyonephenolgroupisoxidizedat+067VthederivativeswithtwophenolgroupsieSI-4OHandSI-6OHgiveoxidationpeaksatremarkablylesspositivepotentialofca+05VSI-46OHwiththepresenceofoverall3hydroxygroupsyieldstwooxidationpeaksInthereversecathodicscan(vertexpotential+1V)probablythankstothepresencereductionpeaksappearwhichcanbefurtherusedtodifferentiatebetweenSudanIandthederivativesSI-4OHandSI-6OH
112 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 113
Fig 3VoltammetricscanofSudanIanditsderivatesSI-4-OHSI-6-OHandSI-46-diOH1stscanincathodicdirectionfrom0Vvertexpotentialsndash1Vand+1VThemeasurementswereperformedinBritton-Robinsonbuffer(pH=7)withtheconcentrationofeachcompoundat5microMandatscan
ndash1rate1Vs
Fig 2 CyclicvoltammogramofSudanIanditsderivatesSI-4-OHSI-6-OHandSI-46-diOH1stscaninanodicdirectionfrom0Vvertexpotentials+1Vandndash1VThemeasurementswereperformedinBritton-Robinsonbuffer(pH=7)withtheconcentrationofeachcompoundat5microMandatscan
ndash1rate1Vs
possesstwoconjugatedhydroxylgroupswhichcanberegardedashydroquinonestructures and thus undergoing quasireversible redox process due to oxida-tionreductionofthehydroquinonetoquinonemoietyThisiswellvisibleattheCVsastheanodicsignalisfollowedbycathodiconeatthepotentialof+023VforSI-4OHand+03VforSI-6OHTheoxidationofSudanIproceedsbymechanism
minus +typicalforphenoliccompoundsatmorepositivepotentialsleading by1e 1H exchange to naphthoxy-type ndashO radical [5] This species undergoes furtherreactionsleadingtoformationofdimersandpolymersThecathodicpeakinthereversescanatndash02Varisesfromreductionofthesereactionproductsanditsoriginneeds tobe further investigatedSI-46OHwith thepresenceofoverallthree hydroxyl groups yields two oxidation peaks The first one is a result ofoxidationoftwoofthembeinginconjugationandthusbeingoxidizedtoquinonemoietyThesecondsignalatthesamepotentialastheoxidationsignalof2OHonnaphthaleneringofSudan I is consequenceofoxidationof the thirdhydroxylgroupofphenolictypeAsinglewidepeakat0Vinthereversescanispresumablyan overlap of signals arising from reduction of the quinonic moiety and by-products formed during oxidation processes Thanks to differences of theseprocessesspecificforindividualcompoundsitispossibletodifferentiateamongallfourofthemviapropersetting-upoftheinitialandvertexpotentialvalues The cathodic scan of Sudan I and its derivatives (Fig 3) shows a dominantreductionpeakaroundndash08Vwhichisduetoreductionoftheazogroupintheirstructuresaccompaniedbycleavageoftheirmoleculestoseparatethebenzeneandnaphthalene rings [4] The peaks in the subsequent anodic scans (vertexpotentialndash1V)arethereforetheresultoftheelectrochemicalreactionofmoietiesthatareproductsofthedivisionofthearomaticcirclesTheseproductsincludeaniline4-aminophenol1-amino-2-naphtholand1-amino-25-naphthalenediolwith irreversibly oxidizable amino moieties or (quasi)reversibly oxidizableamino-hydroxylsystemonthebenzeneornaphthaleneringandtispossibletodifferentiatebetweenthemTheobtainedoxidationpeaks(+02VforSI-4-OH+073VforSudanIandSI-6-OHand+025Vand+075VforSI-46-diOH)makeitpossibletodifferentiatebetweenallcompoundswiththeexceptionofSudanIandSI-6-OHForthereliablerecognitionofthesetwocompoundsanothermeasure-mentwithdifferentparametersisneeded
4Conclusions
ThestructuresofSudanIanditshydroxyderivatesthatarethemainproductsofthe metabolization of Sudan I by cytochrome P450 are similar and theirrecognitionwhenpresentinmixtureinsolutionisdemandingHereinwepresenta simple approach based on comparison of signals obtained in cathodic andanodic scan in CV measurements without the need of time-demandingchromatographicseparationstepFurtherworkwillbedevotedtoidentificationof observed redoxprocesses and applicationof themethod formonitoringofmetabolictransformationsofSudanIinvitro
114 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Acknowledgments
ThisresearchwassupportedbytheCzechScienceFoundation(projectNo18-01710S)
References
[1] ChailapakulOWonsawatW SiangprohW GrudpanK ZhaoYF Zhu ZW Analysis ofSudanISudanIISudanIIIandSudanIVinfoodbyHPLCwithelectrochemicaldetectionComparison of glassy carbon electrodewith carbon nanotube-ionic liquid gelmodifiedelectrodeFoodChem109(2008)876ndash882
[2] StiborovaMMartinekVRydlovaHHodekPFreiESudanIisapotentialcarcinogenforhumans Evidence for itsmetabolic activation and detoxication by human recombinantcytochromeP4501A1andlivermicrosomesCancerRes62(2002)5678ndash5684
[3] GomezMArancibiaVAliagaMNunezCRojas-RomoCDeterminationofSudan I indrinks containing Sunset yellow by adsorptive stripping voltammetry Food Chem 212(2016)807ndash813
[4] PrabakaranEPandianKAmperometricdetectionofSudanIinredchilipowdersamplesusingAgnanoparticlesdecoratedgrapheneoxidemodifiedglassycarbonelectrodeFoodChem166(2015)198ndash205
[5] Enache T A Oliveira-Brett A M Phenol and para-substituted phenols electrochemicaloxidationpathwaysJElectroanalChem655(2011)9ndash16
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 115
1Introduction
Aminoglycosidesarealargegroupofnaturalandsemi-syntheticantibioticswithawidespectrumofantimicrobialactivityagainstmostgram-positiveandgram-negativemicroorganismsCurrentlymultiplerepresentativesofaminoglycosidefamily ndash gentamicin (GM) neomycin B (NM) paromomycin (PM) kanamycin(KM)apramycin(AP) andstreptomycin(STM)Fig1ndashareapproved to treatinfectiousdiseasesinanimalsMaximumresiduelimitsfortheseaminoglycosidesinproductsandtissuesfromedibleanimalsareestablished[1]henceaneffectiveandrobustassayisnecessaryforcontrolofaminoglycosidescontamination Inthisstudyenzyme-linkedimmunosorbentassayforthedetectionofamino-glycosides in foodproductsandenvironmentalobjectswasdevelopedGroup-specificantibodieswereproducedowingtoimmunogenbasedonribostamycin(RS)whichexposedthecommonfragmentofmostaminoglycosides2-deoxy-streptamin (2-DOS) Fig 1 The developed assay was made suitable for thedetectionofresidualaminoglycosidesinhoney[2]
Group detection of aminoglycosides using ELISA for control of food contamination
ab a aKONSTANTINBURKIN INNAGALVIDIS MAXIMBURKIN
a DepartmentofImmunologyIMechnikovResearchInstituteofVaccinesandSera MalyjKazionnyjper5a105064MoscowRussianFederationburkin-kostyandexrub DepartmentofChemicalEnzymologyFacultyofChemistryLomonosovMoscowStateUniversityLeninskieGory1119991MoscowRussianFederation
AbstractThegrowingthreatofglobalantibioticresistanceisforcingtoreducenon-targetconsumptionofantibioticsandtomonitorcontaminationoffoodandenvironmentalobjectsInthisworkELISAwasdevelopedforgroupdetectionofaminoglycosidesToobtaingroup-specificanti-bodies a new immunogen based on ribostamycin was used Thedevelopedindirectcompetitiveformatofassayallowedtherecogni-tionof9aminoglycosidesnamelyneomycinribostamycinneaminparomomycin gentamicin sisomicin kanamycin tobramycin and
ndash1apramycinwithadetectionlimitrangedbetween002ndash020ngmL TheeffectivenessoftheproposedassaywasevaluatedinhoneyasafoodstuffmodelToneutralizea stronghoneymatrixeffect and toavoidalaborioussamplepre-treatmentanewmatriximitatorwassuggested 5 sucrose solution imitated the influence of 50-folddilutedhoneyTheproposedassayallowedustorevealanyofthe9
ndash1mentionedaminoglycosidesinhoneyata10microgkg level
KeywordsaminoglycosidesELISAhoney
116 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
2Experimental
21Reagentsandchemicals
NeomycinBribostamycinneamin(NA)paromomycinkanamycintobramycin(TM)amikacin(AM)gentamicinnetilmicin(NTM)sisomicin(SSM)geneticin(GC) apramycin and streptomycin were purchased from Chimmed (MoscowRussia) Bovine serumalbumin (BSA) complete Freund adjuvant 16-hexane-diamine1-ethyl-3-(3-dimethylaminopropyl)carbodiimide(EDC)sodiumperio-dateandsodiumborohydrideweretheproductsofSigma-Aldrich(USA)Gelatin(Gel)wasfromBio-Rad(USA)sucrosefromServa(Germany)two-componenttetramethylbenzidine (TMB) substrate solutionwas fromBioservice (Russia)andgoatanti-rabbitIgGantibodiesconjugatedtohorseradishperoxidase(anti-rIgG-HRP)werefromIMTEK(Russia)Honeysampleswerepurchasedfromlocaloutlets
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 117
Fig 1 Structuralformulasofstudiedaminoglycosides
22Preparationofconjugatedantigens
TwotypesofconjugateswerepreparedbasedonRSandBSAusingzero-lengthand C6 spacer arm between hapten and protein carrier RSwas treatedwithsodiumperiodatetooxidizehydroxylsofribosefragmenttoreactivealdehydegroupsandthencoupledtoBSAaminesthroughreductiveaminationToremoveuncoupledRSanexhaustingdialysiswascarriedoutusingdialysismembranetubes(MWCO14kDa)UsingthesameprocedureGel-RSconjugatewassynthe-sized ForpreparationofBSA-C6-RSwefirstlymodifiedBSAwith16-hexanediamineThemixtureofBSAandEDCinwaterwerestirredfor30minThen16-hexane-diaminewasaddedandstirredfor2hThemodifiedproteinwasdialyzedfromtheexcessive reagents and resultant BSA-C6-NH2was coupled to RS in reductiveaminationprocessasdescribedabove
23Immunizationandantibodypreparation
BSA-RSandBSA-C6-RSwereusedasimmunogensChinchillarabbits(20ndash25kg)weresubcutaneouslyinjectedat10ndash15pointsonthebackwith01mgofimmuno-gensemulsifiedinthecompleteFreundadjuvantThesamedosesofimmunogensinsalinewereadministeredmonthly forbooster immunizationsAweekaftereachinjectionabloodsamplefromearveinswastakenforthecontrolofimmuneresponseTheantiserainglycerol(11vv)werestoredatndash15degCuntiltestinginELISA
24TheELISAprocedure
AcompetitiveassaywasconductedaccordingtoclassicalprocedureGel-RSwascoatedovernightonpolystyrene96-wellCostarplatesNon-adsorbedconjugatewashedoutusingPBSwith005oftween20(PBS-T)Thenextcompetitivestep
ndash1includedtheadditionof01mLstandardaminoglycosidesolutions(1pgmL to ndash1 ndash11microgmL (B)and0microgmL (B ))inPBS-Tor01mLoftestedsampleand01mLof0
antibodiesinworkingdilution(1h25 degC)Afterwashingtheantibodiesboundtoimmobilized Gel-RS were detected using anti-rIgG-HRP (1h 37degC) Coloredproduct formedasaresultofenzymaticreactionwithTMBsubstratemixture(05h25degC)wasreadat450nmusingaStatFax2100platereader(AwarenessTechnologiesUSA) Relativeantibodybinding(BB )vstheanalyteconcentrationswasplottedas0
standardcurvesfittedtoafour-parameterlogisticfunctionThecross-reactivity(CR) for every aminoglycoside representative was calculated as ratio of half-inhibition concentrations IC NMIC aminoglycoside The dynamic range of50 50
assaywasacceptedasIC ndashIC andthelimitofdetection(LOD)wascalculatedas20 80
B ndash3timesSD0
118 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
3Resultsanddiscussion
31Immunogensynthesisandantibodypreparation
Inthemajorityofpublicationsdevotedtoimmunoassayofaminoglycosidestheimmunogenscoatingantigensenzymeconjugatesortracerswerepreparedbycarbodiimide or glutaraldehyde methods involving aminoglycosidesrsquo aminogroups [3ndash7] Due to several amino groups in aminoglycoside molecules theformationofconjugateswithavariableorientationofthehaptenoccurs InpresentstudyRSwaschosenasanimmunizinghaptenduetothefollowingadvantageous features Being a trisaccharide RS has the size of a moleculecomparable to themostof aminoglycosides Ithas three identical ringsA-B-CsimilartothoseinNMUsingaperiodateoxidationwecouldinvolvearibosesiteofRSincouplingtoproteinthatprovidedastrictorientationofhaptenonthecarrier with a favorable presentation of the 2-DOS fragment The resultantimmunogensBSA-RSandBSA-C6-RSwerecomparedtorevealwhichdesignisbetterforpresentationofacommonfragmentofaminoglycosidemoleculeandgenerationofgroup-specificantibody Antibodies to the BSA-RS demonstrated moderate sensitivity (NM
ndash1IC =10ngmL )andhighselectivitytowardsNMwithrelativelylowcross-reac-50
tivity(lt5)forGMKMandAPTheapplicationofthespacerintheimmunogenBSA-C6-RScontributedtoaprominentpresentationofthe2-DOSdeterminantandtheinductionofantibodieswithrecognitionofbroadspectrumofdifferentaminoglycosides In addition anti-BSA-C6-RS exhibited significantly better
ndash1sensitivity(NMIC =02ngmL )Thusallsubsequentstudieswereconducted50
usinganti-BSA-C6-RS
32Examinationofassayspecificityandselectionofimmunoreagents
TheindirectcompetitiveformatofassaywasdevelopedForevaluationofassayspecificity a panel of following aminoglycosideswas studied and their cross-reactivitywasdeterminedNA(625)RS(250)NM(100)KM(475)PM(173)GM(90)TM(78)AP(17)SSM(12)AM(lt01)GC(lt01)STM(lt01)andNTM(lt01)ThemostoftheseanalytesareusedinmedicalandveterinaryareashoweveronlyNMPMGMKMAPandSTMareappliedinanimalhusbandry[1]
33Determinationofaminoglycosidesinhoneyandselectionofthematriximitator
Honey is a complex product consisting of carbohydrates (75ndash80) vitaminsproteinsenzymesorganicacids traceelements inclusionsandothercompo-nentsThesecomponentsmightinterfereimmunochemicalreactionThereforetheisolationofaminoglycosidesfromhoneyisalaboriousandtime-consuming
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 119
120 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
procedureToavoidthisstephoneymatriximitatorswereappliedtomimictheinfluenceofhoneymatrixonantibodybindingSucrosewaschosenasthehoneyimitatorsinceitexposedastronghoney-matrix-likeeffectonantibodybindingTheadequacywasfoundbetweensolutionsofhoneyandsolutionsofsucroseexpressinganequalmatrixeffectTwopairswithequivalentmatrixeffectwere120honey=20sucroseand150honey=5sucroseThelatterconditionswere chosen asmore preferable due to inconvenience of operatingwith highviscous20sucrosesolution Thedeterminationofaminoglycosidesinhoneycouldbecarriedoutquantita-tivelyiftheanalytetobedetectedisknownForquantificationofaminoglycosideinhoneyasamplewasdiluted50timesinPBS-Tandaminoglycosidestandardcurvewas generated in 5 sucrose-PBST (Fig 2) If analyte is unknown thedevelopedgroup-specificELISAcanbeusedasascreeningtestInthiscasetheanalyzed sample can be considered as contaminated if it caused a relativeantibodybindingbelowthecut-off level (Fig3)Thus thedeveloped testwas
ndash1 ndash1 ndash1Analyte IC ngmL Dynamicrange LODngmL LODinhoneymicrogkg50 ndash1 IC ndashIC ngmL20 80
NM 02 003ndash21 002 10PM 07 008ndash71 005 25GM 15 015ndash133 011 55KM 035 005ndash39 004 20AP 68 05ndash968 023 115
Fig 2StandardcurvesandanalyticalparametersoftheELISA-systemforgroupdeterminationofaminoglycosidesinhoneyInteractionofanti-BSA-RSwithcoatingantigenGel-RSin5sucrosesolutionasthehoneyimitatorThedetectionlimitin5sucrosesolutionwasdeterminedaccordingtoLOD=B ndash3timesSD0
capabletorevealthecontaminationofhoneywith9aminoglycosides5amino-ndash1glycosidesapprovedforveterinary(NMPMGMKMandAP)ata10microgkg level
andalsoNARSSSMandTM
4Conclusions
A novel indirect competitive ELISA for the detection of aminoglycosides wasdevelopedRSwasusedasanewimmunizinghaptentoproducegroup-specificantibodiesagainst2-DOSacommonmoietyofalargenumberofaminoglycosideantibiotics A wide spectrum of aminoglycoside representatives could bedetectedincludingNMRSNAPMGMSSMKMTMandAPThedevelopedassay
ndash1was capable todetect theseanalyteswithaLOD up to002ndash020 ngmL ForanalysisofhoneyamatriximitatorwasdevelopedtoavoidhoneyinterferencesonimmunoassayTheanalysisofthehoneysampleallowedustorevealanyofthe
ndash1mentionedaminoglycosidesinhoneyata10microgkg level
References
[1] CouncilRegulation(EU)N372010OffJEurCommunitiesInfNotL15(2009)1ndash72[2] GalvidisIABurkinKMEreminSABurkinMA Group-specificdetectionof2-deoxy-
streptamineaminoglycosidesinhoneybasedonantibodiesagainstribostamycinAnalMeth11(2019)4620ndash4628
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 121
Fig 3Detectionofaminoglycosidesspikedinhoneysamplesata40ppblevelusinggroup-specificELISAEachsymbolcorrespondstotheaveragerelativebindingandtheerrorisSDobtainedforanindividualhoneysampleanalyzedintriplicateEmptycharactersrepresentindividualblankhoneysamples(limebuckwheatandflower)andfilledsymbolsrepresentthesamesamplesfortifiedwith
ndash1aminoglycosidesata40mgkg (level establishedonlyforSTMinseveralcountries)Thecut-offlevelcorrespondstothelimitofassaydetectionobtainedbythematriximitator(5sucrose-PBS-T)
[3] ThompsonSGBurdJFSubstrate-labeledfluorescentimmunoassayforamikacininhumanserumAntimicrobAgentsChemother18(1980)264ndash268
[4] LiCZhangYEreminSAYakupOYaoGZhangXDetectionofkanamycinandgentamicinresiduesinanimal-derivedfoodusingIgYantibodybasedic-ELISAandFPIAFoodChem227(2017)48ndash54
[5] GalvidisIABurkinMAMonoclonalantibody-basedenzyme-linkedimmunosorbentassayfortheaminoglycosideantibiotickanamycininfoodstuffsRussJBiorganChem36(2010)722ndash729
[6] HaasnootWStoutenPCazemierGLommenANouwsJFKeukensHJImmunochemicaldetectionofaminoglycosidesinmilkandkidneyAnalyst124(1999)301ndash305
[7] Peng JWangYLiuLKuangHLiAXuCMultiplex lateral flowimmunoassayfor fiveantibioticsdetectionbasedongoldnanoparticleaggregationsRSCAdv6(2016)7798ndash7805
122 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
PhotochemicalvaporgenerationisanalternativesampleintroductiontechniqueforanalyticalatomicspectrometryThistechniqueisbasedaroundasourceofUV-radiationthatirradiatesalowmolecularweightorganicacidmedium(mostcommonlyformicacidaceticacidortheircombinations)withananalyteHighlyreducingradicalsandaquatedelectronsareproducedandconverttheanalyteintoavolatilespecieswhichisthentransportedintoadetector[1]SofartheuseofPVGhasbeendescribedforhydride-formingelements(AsBiTeSbPbSeSnandTl)andmercury[12]transitionmetals(FeCoNiCuMoWCdAgAuIrPdPtRhandOs)[13ndash6]andevennon-metals(BrIClFandS)[16ndash9] AfirstsuccessfulphotochemicalvaporgenerationofcobaltwasdescribedbyGuoetalin2004[6]whichwasfollowedbymoresystematicstudiesbyGrinbergetalin2008[10]andDengetalin2010[11]LaterworksbydeQuadrosetal[12]anddeJesusetal[13]focusedontheanalysisofrealsamplesInthelatterwork
Photochemical vapor generation of cobalt for detection by inductively coupled plasma mass spectrometry
ab aJAROMIRVYHNANOVSKY STANISLAVMUSIL
a DepartmentofTraceElementAnalysisInstituteofAnalyticalChemistryoftheCzechAcademyofSciencesVeveřiacute9760200BrnoCzechRepublicjaromirvyhnanovskygmailcom
b DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublic
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 123
AbstractThisworkfocusedonthephotochemicalvaporgenerationofcobaltVolatilespeciesweregeneratedinaflow-injectionsystememployinga high-efficiency flow-through UV photoreactor and a formic acidbased medium and were introduced by an argon carrier into aninductivelycoupledplasmamassspectrometerfordetectionOptimalgeneration conditions were found as 10 (vv) formic acid and
ndash1 ndash14molL ammonium formate with a 4mLmin flow rate whichcorresponds to irradiation time of around 13 s The influence ofvariousmetalsensitizersofphotochemicalreactionwasinvestigated
2+andonlyCu ionsexhibitedapositiveeffectongenerationefficiencyndash ndash 2ndashInterferencesfromcommoninorganicanions(NO Cl SO ) were3 4
also examined Lastly the limit of detection and repeatability (atndash1 ndash1250ngL )weredeterminedtobe13ngL and41respectively
Keywordscobaltinductivelycoupled
plasmamassspectrometry
photochemicalvaporgeneration
the authors also presented a systematic study on generation conditions andachievedagenerationefficiencyofaround40 Themainaimofthisworkwastooptimizetheconditionsofgenerationwithinductivelycoupledplasmamassspectrometry(ICP-MS)detectionexaminetheeffectofvariousmetal sensitizers toachieve thehighestgenerationefficiencypossibleandreachthelowestlimitofdetectionpossible
2Experimental
21Reagentsandchemicals
minus1Deionizedwater (DIW lt 02μScm UltrapurWatrex USA)was used for thepreparationofallsolutionsFormicacid(98paLach-NerCzechRepublic)andammonium hydroxide (ge25 pa Sigma-Aldrich USA) were used for the
minus1preparationof the reactionmediumA1000mgL Co stock solution (Sigma-AldrichUSA)wasusedforthepreparationofallsamplesolutionsThefollowingcompounds were used as potential metal sensitizers cadmium(II) acetatedihydrate(paLach-NerCzechRepublic)zinc(II)acetatedihydrate(paSigma-AldrichUSA)copper(II)acetatemonohydrate(paMerckGermany)nickel(II)acetatetetrahydrate(paSigma-AldrichUSA)sodiumtungstatedihydrate(paCarlRothGermany)and iron(II) sulphateheptahydrate (pa LachemaCzechRepublic)Nitricacid(65semiconductorgradeSigma-AldrichUSA)hydro-chloricacid(37paMerckGermany)andsulfuricacid(98paLach-NerCzechRepublic)wereusedforaninterferencestudy
22Instrumentation
AschematicdiagramofthePVGsystemcoupledtoICP-MSisshowninFig1andamore detailed description can be found in reference [5] Briefly a singlequadrupole ICP-MS Agilent 7700x (Agilent Technologies USA)was used as a
minus1detector Deionized water was mixed with a 10μgL Rh internal standardsolutionin2HNO andwassubsequentlynebulizedbyaMicroMistnebulizer3
59 103during PVG Isotopes of Co and RhweremonitoredMeasurementswereperformed in time resolved analysis mode and in He collision mode
minus1(41mLmin )AlltubingusedwasmadefromPTFEwiththeexceptionoftygontubing in the peristaltic pump (Reglo ICC Ismatec Switzerland) The high-efficiency flow-through photoreactor was a 19 W low-pressure mercurydischarge lamp (Beijing Titan Instruments Co Beijing China) with a quartzcentralchannel(asymp720μL internalvolume)Samplesolutionswere introducedintoastreamofreactionmediumusinganinjectionvalve(V-451IDEXHealthandScienceUSAsampleloopvolume05ml)Effluentfromthephotoreactorwasmixedwithaflowofargonandcarriedtothechilledgas-liquidseparator(internalvolume15mL)wherethevolatilespecieswereseparatedfromtheliquidwaste
124 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
andcarried to the inletofaScott-typespraychamber (originally the inlet formakeupargon)oftheICP-MS
3Resultsanddiscussion
The starting conditionswere adopted fromour earlierwork [14]whichusedatomicabsorptionspectrometerasadetectorandminiaturediffusionflameasanatomizerThefirstparameteroptimizedwasthecompositionofreactionmedium(Fig2)Theadditionofammoniumformate(createdin-situbytheadditionofacalculatedamountofammoniumhydroxidetoformicacid)wasfoundcrucialto
ndash1effectivelygeneratevolatilespeciesofcobalt10(vv)formicacidand4molL ammonium formatewas chosen as the optimumandwasused for all further
experimentsAlthoughhigher concentrations of both components led to even
higher signalstheseconditionswerenotusedfurtherbecauseofthelaboriousprocessofpreparation(mixingofconcentratedacidwithconcentratedbase)andtolimittheconsumptionofchemicals Theinfluenceofirradiationtimewasalsoexaminedandthehighestpeakarea
-ndash1wasobtained for4mLmin corresponding to an irradiation timeof approximately13s ToenhancethegenerationefficiencyadditionofvariousmetalstothereactionmediumwastestedtoldquosensitizerdquothephotochemicalreactionThemetalswerechosenwithrespecttotheirsignificantenhancementeffectdescribedrecentlyforphotochemicalvaporgenerationofotheranalytes[158]Theonlymetalionthat
2+ledtoanenhancementofthesignalwasCu (Fig3a)buteveninthiscasetheeffect was rather negligible reaching only 12-fold enhancement in the range
ndash1 2+ 2+001to01mgL Cu FurtheradditionofmoreCu ledtoadecreaseinthesignal2+TheadditionofZn didnotexhibitanypositiveornegativeeffectacrossthetested ndash1range (01 to 1500mgL not shown in figure) and the addition of higher
ndash1 2+ 2+ 6+concentrations (tens to hundredths ofmg L ) of Cd Fe andW (Fig3b)causedsevereinterferences
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 125
ndash ndash 2ndash Interferencescausedbycommoninorganicanions(NO Cl SO addedas3 4ndashtheirrespectiveacids)werealso investigatedOutof theseNO was foundto3
ndash1causethemostsevereinterferencesevenatconcentrationsofsinglemmolL Thendash 2ndashmethodologywasmorerobusttowardstheinterferencesfromCl andSO but4
theystillcausedsignificantdropinsensitivityathigherconcentrationsConsi-deringthewideuseoftheseacidsinanalyticalchemistryforsamplepreparationthisposesabigchallengeintheapplicationofthismethodtorealsamples
ndash1 Usingoptimalconditions(10(vv)formicacid4molL ammoniumformateandirradiationtimeof13s)acalibrationcurvewasmeasuredandevaluatedThelimitofdetectionwasdeterminedas3timesthestandarddeviationof10blank
ndash1measurements and was calculated as 13 ng L The repeatability of 10ndash1consecutivemeasurementsof250ngL was41
126 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 3Effectofvariousmetalionsonthepeakarea(a)metalionswithapositiveeffect(b)metalionsndash1withoutapositiveeffectExperimentalconditions2microgL Coreactionmedium10(vv)formic
ndash1 ndash1acidand4molL ammoniumformateflowrate4mLmin
ndash1Fig 2Effectofthecompositionofreactionmediumonpeakareaexperimentalconditions2microgL ndash1Coreactionmediumflowrate4mLmin (blackdotscorrespondtomeasuredpoints)
4Conclusions
Theconditionsofthephotochemicalvaporgenerationofcobaltwereoptimizedand are in good agreement with previous works [11 13] Copper ions wereidentifiedasapotentialsensitizerincreasingthesignalbyabout12-foldbuttheirpotentialuseisseverelylimitedbythenarrowrangeofconcentrationsinwhichthepositiveeffectisexhibitedSevereinterferencesfrominorganicanionswereobservedwhichisinlinewithotherworksdealingwithphotochemicalgeneration[1358]Furtherexperimentswillfollownamely(i)furtherinvestigationsinnewpotentialsensitizerstoenhancegenerationefficiencyandthusdecreasethe
ndash1limit of detection to sub ng L levels (ii) determination of the generationefficiency(fromcomparisonwithnebulizationandorusingaradioactiveisotope58Co)(iii)verificationoftheaccuracyandpracticalfeasibilityofthismethodologybyanalysisofcertifiedreferencematerials
Acknowledgments
ThesupportofTheCzechScienceFoundation(ProjectNo19-17604Y)CzechAcademyofSciences(Institutional supportRVO68081715)andCharlesUniversity (project SVV260560andprojectGAUK60120)isgratefullyacknowledged
References
[1] SturgeonREPhotochemicalvaporgenerationaradicalapproachtoanalyteintroductionforatomicspectrometryJAnalAtomSpectrom32(2017)2319ndash2340
[2] XuTHuJChenHJTransitionmetalionCo(II)-assistedphotochemicalvaporgenerationofthalliumforitssensitivedeterminationbyinductivelycoupledplasmamassspectrometryMicrochemJ149(2019)103972
[3] SoukalJSturgeonREMusilSEfficientphotochemicalvaporgenerationofmolybdenumforICPMSdetectionAnalChem90(2018)11688ndash11695
[4] deOliveiraRMBorgesDLGUVphotochemicalvaporgenerationofnoblemetals(AuIrPdPtandRh)AfeasibilitystudyusinginductivelycoupledplasmamassspectrometryandseawaterasatestmatrixJAnalAtomSpectrom33(2018)1700ndash1706
[5] VyhnanovskyJSturgeonREMusilSCadmiumassistedphotochemicalvaporgenerationoftungstenfordetectionbyinductivelycoupledplasmamassspectrometryAnalChem91(2019)13306ndash13312
[6] GuoXSturgeonREMesterZGardnerGJVaporgenerationbyUVirradiationforsampleintroductionwithatomicspectrometryAnalChem76(2004)2401ndash2405
[7] HuJSturgeonRENadeauKHouXZhengCYangLCopperionassistedphotochemicalvapor generation of chlorine for its sensitive determination by sector field inductivelycoupledplasmamassspectrometryAnalChem90(2018)4112ndash4118
[8] LeonoriDSturgeonREAunifiedapproachtomechanisticaspectsofphotochemicalvaporgenerationJAnalAtomSpectrom34(2019)636ndash654
[9] SturgeonREPaglianoEEvidenceforphotochemicalsynthesisoffluoromethaneJAnalAtomSpectrom(2020)httpsdoiorg101039D0JA00108B
[10] GrinbergPMesterZSturgeonREFerrettiAGenerationofvolatilecobaltspeciesbyUVphotoreduction and their tentative identification J Anal Atom Spectrom 23 (2008)583ndash587
[11] DengHZhengCB LiuLWWuLHouXDLvYPhotochemicalvaporgenerationofcarbonyl for ultrasensitive atomic fluorescence spectrometric determination of cobaltMicrochemJ96(2010)277ndash282
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 127
[12] deQuadrosDPBorgesDLDirectanalysisofalcoholicbeveragesforthedeterminationofcobalt nickel and tellurium by inductively coupled plasmamass spectrometry followingphotochemicalvaporgenerationMicrochemJ116(2014)244ndash248
[13] deJesusHCGrinbergPSturgeonRESystemoptimizationfordeterminationofcobaltinbiologicalsamplesbyICP-OESusingphotochemicalvaporgenerationJAnalAtomSpectrom31(2016)1590ndash1604
[14] VyhnanovskyJFotochemickegenerovanıtekavychspeciı kobaltuproanalytickouatomovouspektrometriiMasterthesisFacultyofScienceCharlesUniversityPrague2018
128 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Oneof themethods thatallowobtainingmaterialswithnewproperties is theplasmaenhancedchemicalvapordepositionmethodInthismethodcompoundscalledprecursorsaresuppliedtotheplasmareactorasagasphaseThankstoplasmaenhancedchemicalvapordeposition it ispossible toobtainmaterialswithuniquepropertiesThisisduetothefactthattheplasmaaffectsthesurfaceinfourdifferentways etching cleaning chemicalmodification and crosslinking
Optimization of condition for cold plasma deposition of thin layers for surface modification of working electrodes
a b a cJUSTYNALIPIN SKA MARIAMADEJ BOGUSŁAWBAS JACEKTYCZKOWSKI
a DepartmentofAnalyticalChemistryFacultyofMaterialsScienceandCeramicsAGHUniversityofScienceandTechnologyAdamaMickiewicza3030-059KrakoacutewPolandjustynalipinskaaghedupl
b DepartmentofAnalyticalChemistryFacultyofChemistryJagiellonianUniversityinKrakoacutewGronostajowa230-387KrakoacutewPoland
c Departmentof MolecularEngineeringFacultyofProcessandEnvironmentalEngineeringLodzUniversityofTechnologyWolczanska21390-924ŁoacutedźPoland
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 129
AbstractCurrentlyresearchisfocusedonthesearchfornewphysicallyandchemicallystablematerialsaswellasvolumeorsurfacemodificationOneofthemethodsusedforsurfacemodificationistheapplicationofthin layers from inorganic and organic compounds The plasmaenhancedchemicalvapordepositionisamethodthatallowsmaterialmodificationandalsodepositionofthinlayersThisworkconcernsoptimizationofcoldplasmadepositionparametersandtoachievethebestelectrical conductivitywhilemaintaining thehighmechanicalstrength of the formed layers Preliminary tests were focused onoptimizing the layering parameters such as the deposition timedischargepowerpressureofmonomerandthe flowofargonTheobtainedsamplesweresubjectedtothermaltreatmentafterwhichtheywere coveredwitha layerof aluminumThe thicknessof theobtained layers was determined on the basis of interferencemicroscopymeasurementsAsaresultoftheexperimentslayerswithathicknessof20nmto600nmwereobtainedTheconductivityofthedeposited layers was also determined and values from 003 to
ndash1150Sm wereobtained
Keywordscoldplasmadepositionelectrochemical
applicationssurfacemodificationthinlayers
Thismethodisusedtoproducecatalyticstructuresortomodifythepropertiesofmaterials eg improve hydrophobicity The growing popularity of surfacemodificationmethodsusingcoldplasmaisassociatedwiththefactthatitisanenvironmentallyfriendlyandversatilemethod[12] Workingelectrodesusedinvoltammetryareasubgroupofchemicalsensorswhich are small devices that convert real-time chemical information into ameasurableandanalyticallyusefulmeasurementsignalChemicalinformationrangingfromtheconcentrationofaspecificcomponentofthetestedsampletotheoverallcompositionofthematrixcancomefromboththeinitiatedchemicalreactionandbetheresultofphysico-chemicaltransformationstakingplaceinthetested object Chemical sensors are equipped with two basic elements iereceptor and transducer The receptor is responsible for the conversion ofchemicalinformationfromthetestedobjectintoaspecificformofenergyintheconverterthisenergyistransformedintoausefulanalyticalsignal Parameters characterizing the electrochemical sensor include accuracyprecision selectivity accuracy presentation selectivity sensitivity dynamicrange limit of quantification limit of detection lifetime response time andreliability Themost numerous and the oldest group of chemical sensors areelectrochemicalsensorsCommonlyobservedinterest inthisgroupofsensorsresultsfromthefactthatwithrelativelylowproductionandoperatingcoststheyofferthebestmetrologicalandoperationalparameters[3ndash5]Oneofthemaintrendsofmodernanalyticsisthesearchfornewelectrodematerialsandvariousgeometries of working electrodes One way to improve the performance ofworkingelectrodesistomodifytheirsurfaceforexamplebyapplyingthinlayersIn this work were considered plasma enhanced chemical vapor depositionmethod as the method of surface modification designed to performworkingelectrodeforvoltammetricdeterminationsAspartoftheinitialresearchplasmaprocessing parameters such as discharge power time of treatment andcompositionofgasmixtureinwhichplasmawasgeneratedwereoptimizedThelayers obtained in different conditions have been tested for suitability forelectrochemical applications (layer thicknessmeasurement and themeasure-mentofconductivity)
2Experimental
21Reagentsandchemicals
Theprecursor solutions suchasacrylonitriledietoxydimethylsilane trietoxy-methylsilaneandtetramethyldisiloxanewhicharesuppliedbyABCRwereusedOtherreagentsofanalyticalpuritysuchasn-hexane(SigmaAldrich)andargonwereused
130 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
22Instrumentation
The thin layers were deposited in a parallel-plate plasma reactor (frequency1356MHz)ThesamplesobtainedwerecalcinedinatunnelfurnaceunderanargonatmosphereThethicknessofthedepositedlayerswasmeasuredafterthealuminum was sputtered using a Nikon microscope type ECLIPSE LV150NElectrometerhigh resistance system (KEITHLEY) was used to measureconductivity
3Resultsanddiscussion
Eachof themonomerswasdepositedonprepared1times1corningglasssamplesSamples prepared with n-hexane were placed in a plasma reactor andadditionallypartiallycoveredwithamicroscopecoverslipSchematiclayoutofsamplesinthereactorshowninFig1ThefirststepwastoetchthesystemusingargonplasmaThisstageallowedfortheeliminationofimpuritiesthatwerenotremovedbythehelpofn-hexaneandthepreparationofthesurfaceofthesamplesforthedepositionoftheproperlayerTheproperstageistheapplicationofathinlayerwiththeplasmainducedbytheselectedprecursoracrylonitriledietoxydi-methylsilanetrietoxymethylsilaneandtetramethyldisiloxaneThethicknessandpropertiesoftheobtainedlayersdependonthedepositionparameterssuchasdischarge power time of treatment and composition of gasmixture inwhichplasmawasgenerated Table1showsallcombinationsofparameterstestedforallfourprecursorsFour different discharge powers for acrylonitrile and two different dischargepowersfororganosiliconmonomersweretestedwithtwodifferenttreatmenttimesEachtimeandpowercombinationwasperformedinplasmainducedbypuremonomerandmonomerwithargonAfterapplyingthelayersthesampleswereplacedinaquartzboatandcalcinedinatunnelfurnaceAllsampleswerecalcinedat500degCfor2hoursunderargonflowSamplesaftercalcinationwerecoveredwithalayerofaluminum
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 131
Fig 1 Scheme of sample distribution in plasma reactor(a)reactorelectrode(b)corningglasssamples(c)micro-scopecoverslip
Thethicknessoftheobtainedlayerswasmeasuredusinganinterferencemicro-scopeMeasurements were carried out at amagnification 10times recording theimageinmonochromelightTheimagewassetsothattheinterferencefringeswere perpendicular to the arc on the sample In order to calculate the layerthicknessD[nm]thefollowingformulawasused
(1)
wheredandLweredeterminedonthebasisoftheregisteredimage(disfringeshiftduetorefractionoflightontheslopeLisdistancebetweenthefringes) Thelaststageofthestudywastodeterminethecurrent-voltagecharacteristicstodeterminetheconductivityoftheobtainedlayersThesamplewasplacedinameasuring cell and attachedwith silver paste to the electrometerwires ThechangeincurrentwasrecordedwiththeapplicationofalternatingvoltageintimeBasedontheresultsobtainedthegraphsofdependenceUndashIwereobtainedfromwhichthevalueofresistance(R)wasdeterminedAnexampleofcurrent-voltagecharacteristicsisshowninFig2
132 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Table 1Conditionsforlayersdeposition-parameterwhichweretested
Monomer DischargepowerW Timeoftreatmentmin
Acrylonitrile 10204080 24Dietoxydimethylsilane 2040 255Trietoxymethylsilane 2040 255Tetramethyldisiloxane 2040 255
Fig 2Current-voltagecharacteristicsdeterminedfortheacrylonitrilelayer(depositionparametersW=10Wt=25mingasmixtureonlyacrylonitrile)
Table 4Theresultsofthicknessmeasurementsandconductivityspecifictothetrietoxymethylsilanelayers
DischargepowerW 20 20 40Timeoftreatmentmin 25 5 5Argonflowsccm 10 10 10Thicknessnm 14686 24282 35311
ndash1SpecificconductivitySm 165 93 67
Knowing the value of the resistance and the geometry of the system thespecificresistancewasdeterminedfollowedbythespecificconductivityofthesamplethefollowingequationwasused
(2)
whereρisspecificresistance[Ωm]Risresistance[Ω]bissamplelength[m]Disdepositedlayerthickness[m]andldistancebetweenelectrodes(wires)[m] Theresultsofthicknessmeasurementsandspecificconductivityarepresentedin the Tables 2ndash5 For electrochemical applications it is important that theobtainedlayerhasthehighestspecificconductivityAnalyzingthedatapresentedin Tables 2ndash5 shows that the thinnest layers have the greatest applicationpotentialastheelectrodematerial
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 133
Table 2Theresultsofthicknessmeasurementsandconductivityspecifictotheacrylonitrilelayers
DischargepowerW 40 40 80 80 10 10 20 20Timeoftreatmentmin 2 4 2 4 2 4 2 4Thicknessnm 15845 31167 250 4644 247 67 7531 12236
ndash1SpecificconductivitySm 0002 156 191 0003 1512 454 311 185
Table 3The results of thickness measurements and conductivity specific to the dietoxydimethylsilanelayers
DischargepowerW 20 20 40 40Timeoftreatmentmin 25 5 25 5Argonflowsccm 10 10 10 10Thicknessnm 13998 37519 28431 49556
ndash1SpecificconductivitySm 1640 55 105 40
Table 5The results of thickness measurements and conductivity specific to the tetramethyldisiloxanelayers
DischargepowerW 20 40 40Timeoftreatmentmin 25 25 5Argonflowsccm 10 10 10Thicknessnm 16759 3159 61652
ndash1SpecificconductivitySm 213 95 31
4Conclusions
InthisworkplasmaenhancedchemicalvapordepositionmethodwasusedforapplyinglayersoffourdifferentmaterialsacrylonitriledietoxydimethylsilanetrietoxymethylsilaneandtetramethyldisiloxaneBychangingparameterssuchasdischargepowertimeoftreatmentandcompositionofgasmixtureanumberofsampleswereobtainedwithlayersofdifferentthicknessandwhatisassociatedwithotherelectricalproperties TestsperformedaspartofthisworkwereusedtoperforminnovativeworkingelectrodesforvoltammetricdeterminationsThesurfacewasmodifiedusingthecoldplasmaofthreesubstratesgraphiteglassycarbonandgold
Acknowledgments
JLandMMhavebeenpartlysupportedbytheEUProjectPOWR030200-00-I00416
References
[1] KapicaRTyczkowskiJBalcerzakJMakowskiMSielskiJWorwaEEnhancingadhesivejointsbetweencommercialrubber(SBS)andpolyurethanebylow-pressureplasmasurfacemodificationIntJAdhesAdhes95(2019)102415
[2] TyczkowskiJKapicaRŁojewskaJThincobaltoxidefilmsforcatalysisdepositedbyplasma-enhancedmetalndashorganicchemicalvapordepositionThinSolidFilms515(2007)6590ndash6595
[3] HulanickiAGłabSIngmanFChemicalsensorsDefinitionsandclassificationPureApplChem63(1991)1247ndash1250
[4] BrzozkaZWroblewskiWSensorychemiczneWarszawaOficynaWydawniczaPolitechnikiWarszawskiej1999(InPolish)
[5] SkoogDAWestDWHollerFJCrouchSRFundamentalsofAnalyticalChemistry9thEdBostonCengageLearning2013
134 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
ConcentrationandcompositionofvolatilecompoundsorcongenersisoneofthemostimportantparametersresponsibleforqualityofproducedalcoholicdrinksandhencefortheirsensorycharacteristicsandconsumeracceptanceTodaygaschromatography (GC) is conventionally used to determine qualitative andquantitativecompositionsofvolatilecompoundswithvariousexternalandorinternalstandardcalibrationprocedures Methodemployingethanol as an internal standard (IS) forGCquantitativedeterminationofvolatilecompoundsinalcoholicbeverageshasbeensuggestedquitelongago[1]andsincethattimegreatresearchworkhasbeencarriedoutRecentlyaninterlaboratorystudyofthemethodinvolving9testinglaboratoriesfrom4countrieswascarriedout[2]TheresultsdemonstratedgreatperspectivesofldquoEthanolasISrdquomethodandproveditsreferencecharacterandeaseofroutineimplementation
Advanced GC-MS method for quality and safety control of alcoholic beverages
abANTONKORBAN
a DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova2030812840Prague2CzechRepublickarbonat7gmailcomb DepartmentofAnalyticalChemistryChemistryFacultyBelarusianStateUniversityLeningradskaya14220050MinskBelarus
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 135
AbstractRecently developed and validated simple and reliable quantitativemethod employing ethanol as an internal standard for GC-MSquantification of volatile compounds in alcoholic products wasapplied to 36 samples including commercially available world-famousbrandspirits from18countriesandhomemadedistillatesThe GC-MS analyses were performed simultaneously by thesuggested approach and official internal standard method that isprescribedinthelegislationofEUandUSATheindependentsamplest-testwasemployedtoevaluatethestatisticaldifferenceofresultsofthesetwomethodsThetestrevealednodifferenceintheresultsandtheirrepeatabilityThemainbenefitsofthesuggestedmethodaretheeliminationofthenecessityofmanualinternalstandardadditionandsamplesdensitymeasurementthusmakingitmoreeconomicalandproductive
Keywordsalcoholicbeveragesgaschromatography-
massspectrometry(GC-MS)
internalstandardmethodvolatilecompounds
quantification
AllpreviousstudieswereutilizingflameionizationdetectorssinceGC-FIDisprescribedinthelegislation[3]wheremass-spectrometrydetectorsarenotyetofficially referred However GC-MS instruments are employed in practice toqualifyandorquantifyvolatilesincommercialspiritsintraditionalhomemadealcoholicdrinksinnewlydevelopedbeveragesinspiritwastesandindistillatesobtainedwithdifferentmanufacturingprocesses Our recent researchwasdirected towardsdevelopment of an algorithmofldquoEthanolasISrdquomethodapplicationonGC-MSinstruments[4]WehaveshowedthattopreventMSdetectorfromsaturationethanolshouldberegisteredinthecorresponding SIM timewindow at characteristicmz of low abundance forinstance bymz of 47 ions This ion corresponds to non-fragmented ethanol
13moleculescontaining1heavyisotope(mainly C)FinallytheresultsofmeasuredstandardsolutionsshowedthatthesuggestedapproachisvalidandldquoEthanolasISrdquomethodmaybesuccessfullyusedonGC-MSinstrumentstoo The objective of this studywas to test and further approve the suggestedapproachonalargersetof36realsamplesofalcoholicdrinkseithercommercialorhomemadeThesamplesweresimultaneouslyanalysedbytwoGC-MSmethodsndashaclassicalISmethodprescribedinthelegislationandthesuggestedldquoEthanolasISrdquomethod
2Experimental
21Reagentsandchemicals
Thefollowingvolatilecompoundsweredeterminedintestedsamplesofalcoholicbeverages 11-diethoxyethane (acetal) acetaldehyde methyl acetate ethylacetate methanol 2 propanol 1-propanol 2 methylpropan-1-ol (isobutanol)1-butanol2-butanoland3methylbutan-1-ol(isoamylalcohol)1-pentanolwasemployedasatraditionalIScompound
22Instrumentation
ShimadzuGCMS-QP2010Ultraequippedwithaquadrupolemassspectrometrydetector was employed for GC-MS measurements Rxi-1301Sil MS capillarycolumn(60mlength025mmid025micromfilmthicknessRestek)wasusedfortheseparationofcompoundsInjectionswereperformedinasplitmode(ratio175)Helium(99999purity)wasusedasacarriergasinjectortemperaturewas170degCTheoventemperaturewasheldat30degCfor5minthenraisedto210degC
ndash1at a rate of 30 degCmin and held isothermally for 4minMeasurementswereperformedinaSIMmodeFortheanalysedcompoundsand1-pentanol2ndash3mostabundantionsinthecorrespondingMSspectrumwereselectedethanolSIMtimewindowcontainedonly47mzionsAllGC-MSmeasurementswerecarriedoutintriplicateunderrepeatabilityconditions
136 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
AnalysisofeachalcoholicsamplewasperformedinafollowingwayAliquotof09mL of a tested sample was pipetted into a standard 2mL glass vial and
ndash1weighedAfterthat01mloftheISsolution(2355mgkg of1-pentanolinWES)wasaddedtothetestedsampleandthemasswasrecordedTheobtainedmixturewasmixedthoroughlyand05microlofitwasinjectedintotheGCsystem Theoriginoftestedalcoholicbeverageswaseithercommercialorhomemade33 world-famous spirits manufactured at different parts of the world werepurchasedfromcommerciallyavailablesourcesThelistoftypesofpurchasedandanalysedspiritsincludedbourboncalvadoscognacgingrappaliquormetaxaportwine rumsake tequilavodkawhiskeyandvarious fruitdistillatesThepurchased drinks were produced at the territory of the following countriesBelarus Bermuda Cuba Czech Republic Denmark France Germany GreeceGuatemala Jamaica Japan Mexico Moldova Portugal Slovakia Trinidad andTobago UK (England and Scotland) USA Three homemade fruit distillatesproducedbyfermentationofpulpyfruitsortheirmustswereobtainedfromlocalspiritmakersThedeclaredABVvaluesofalltestedsamplesvariedfrom15to81
3Resultsanddiscussion
To fulfil themaingoalof thiswork ie toevaluate thestatisticaldifferenceofresultsyieldedbythecomparedmethodswehaveemployedStudentst-testforindependentsamplestoverifystatisticaldifferencesonthesignificancelevelofp=005Theobtainedempiricalvaluesforallpairsofcongenersrsquoconcentrationswere lower than critical one in all cases demonstrating that concentrationsobtainedbythetwomethodshavenostatisticaldifferenceandleadtothesameresults InadditionrepeatabilityofthetwomethodswascomparedthereforeallRSDvalues obtained from triplicatemeasurements were split in two groupswith
ndash1respecttothecorrespondingconcentrations(lowerthan50mgL AAandhigherndash1than50mgL AA)Theobtainedresultsarepresentedintheformofboxplotin
Fig1AnalysisofthechartinFig1showedthatboththetestedmethodshaveyieldedstatisticallysimilarrepeatability AllofthetestedalcoholicdrinkssatisfiedtherequirementsofEURegulation(EC)no1102008[5]Theconcentrationsofundesirablecompoundssuchasmethanoldidnotexceedthelevelsspecifiedinthesameregulationforcorres-ponding beverages In Table 1 the description of the used SIM method andsummaryoftheexperimentalresultsarepresented Tocomparethetruenessofthemethodsoneofthespiritsampleswasspikedwithstandardsolutions(ABV40)containingallanalysedvolatilecompoundsat
ndash1concentrationsof50500and5000mgL AATheoriginalsamplewasusedasareferenceEachofthespikedsolutionswasmeasuredintriplicateSelectedspirit(cherry distillate) initially contained all 11 volatile compounds in various
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 137
138 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1BoxchartsofRSDsofusedISmethodsat2concentrationrangesMeanisequaltoarithmeticmeanoraverageInterquartileRange(IQR)meansisthedistancebetweentheupper(themedianoftheupperhalfofthedataset)andlower(themedianofthelowerhalfofthedataset)quartile
ndash1Compound Timemin Registeredmz Numberof ConcentrationmgL AA results Minimal Maximal
Acetaldehyde 0ndash42 314344 36 24 715Methanol 36 13 13600
Ethanol(IS) 42ndash48 47 mdash mdash mdash
2-Propanol 48ndash70 29314345 14 27 199Methylacetate 596174 10 34 3201-Propanol 26 361 12070Ethylacetate 27 166 107002-Butanol 11 18 2080
Isobutanol 70ndash200 3141ndash4345 28 19 20001-Butanol 5556 13 28 155Acetal 26 45 270Isoamylol 31 39 26501-Pentanol(IS) mdash mdash mdash
Table 1DescriptionoftheusedSIMmethodandsomestatisticsconcerningallmeasured36spiritsamplesbothpurchasedandhomemade
concentrationsTheobtainedrecoveriesboxchartsareshowninFig2Compa-risonoftherecoveriesobtainedwithtwomethodsindicatesthattheyhavenosignificantdifferenceintermsoftruenessAverageobtainedrecoverywas981whenusingsuggestedmethodand980whenusingtraditionalISmethod
4Conclusions
InthisworktheresultsoftestingtheadvantageousldquoEthanolasISrdquomethodfortheGC-MS quality control analysis of alcoholic beverages were presented33purchasedsamplesofworld-famousalcoholicbeveragesoriginatingfrom18countriesand3homemadefruitdistillateswereanalysedtomakeathoroughandcomprehensive studyof thedevelopedmethodTheconcentrationsofvolatile
ndash1compoundsinanalysedsamplesvariedfrom1to13500mgL AAtheABVvalueofanalysedsamplesvariedfrom15to81ThesuggestedmethodwascomparedwiththetraditionalISmethodthatiscurrentlystatedinlegislationTheindepen-dentsamplest-testrevealedthatwithaprobabilityof095resultsobtainedwithtwo methods do not differ significantly The results of within-run precision(repeatability)showedrelativestandarddeviationswithin3measurementstobelessthat6indicatingthatthetechniqueisreproducibleThetruenessofthemethodwasevaluatedbyrecoverycalculationAccordingtotheobtainedresultsrecoveryofthesuggestedmethod(981plusmn33)wasslightlybetterthanthatofthetraditionalone(980plusmn58) ThesefactsprovethatdevelopedldquoEthanolasISrdquomethodistruepreciseandreliable when employed on GC-MS instruments At the same time to obtain
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 139
Fig 2Boxchartsof recoveriesof thesuggested(dottedpattern)and traditional (brickpattern)ISmethodsatdifferentspikeconcentrationsSymbolsdefinitionsarethesameasinFigure1
concentrationsofvolatilecompoundsintheofficiallyrequiredunitsofmeasurendash1 ndash1(mgL AAgL AAetc)suggestedmethodrequiresnodensitometrymeasure-
mentsofthetestedsampleandnoadditionofIScompoundoranyothersamplepre-treatmentThismethodprovidesaninvaluableanalyticaltoolforthequalitycontrolofalcoholicproductsandshouldbeusedinroutineanalysis
Acknowledgments
ThisworkwasfinanciallysupportedbytheVisegradFund
References
[1] CherepitsaSVBychkovSMKovalenkoANMazanikALSeleminaNMSeredinskayaOBThe use of themajor component (solvent) as an internal standard in the gas-chromato-graphicdeterminationofimpuritiesJAnalChem58(2003)368ndash371
[2] CharapitsaSSytovaSKorbanASobolenkoLEgorovVLeschevSZakharovMCabalaRBusarovaRShestakovichITolstouhovaAOndrousekSVavraJYilmaztekinMCabarogluTInterlaboratorystudyofethanolusageasaninternalstandardindirectdeterminationofvolatile compounds in alcoholic products BIO Web Conf 15 (2019) 02030httpsdoiorg101051bioconf20191502030
[3] CommissionRegulation(EC)No28702000layingdownCommunityreferencemethodsfortheanalysisofspiritsdrinkshttpdataeuropaeuelireg20002870oj
[4] KorbanACharapitsaSCabalaRSobolenkoLSytovaSTheperspectivesofethanolusageasaninternalstandardforthequantificationofvolatilecompoundsinalcoholicproductsbyGC-MSJMassSpectr55(2020)e4493
[5] EuropeanUnion(2008)Regulation(EC)No1102008oftheEuropeanParliamentandoftheCouncilof15January2008ontheDefinitionDescriptionPresentationLabellingandtheProtectionofGeographical IndicationsofSpiritDrinksandRepealingCouncilRegulationhttpdataeuropaeuelireg2008110(1)oj
140 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Carbonorgraphitefeltsareusedaselectrodematerialsincethe1990sandtheirutilizationstillgrowsinanalyticalelectrochemistryaswellasinotherareasThisisduetotheirsuitablepropertiesfromwhichwecannamehighporosityhighspecificsurfaceareagoodelectricconductivityandhighphysicalandchemicalstabilityThefirsttwoparametersaregivenbystructureoffeltwhichconsistsoforderlesscarbonfibreswithabouttentotwentymicrometersindiameter[1]theothersby theadvantageouselectricalpropertiesof carbon fibreOn theotherhand porous flow-through electrodes including carbon felt electrode havedisadvantageinapotentialdropintheelectrodevolumewhichcausesdifficultcontrollingoftheexactpotentialappliedontheelectrodeandthereforeresultsindifferentcurrentefficienciesontheoppositesidesoftheelectrode[2] Carbon felt electrode can be utilized for detection of structurally differentcompoundsatvariousconditionsForexampleoperatingatreductionpotentialofndash08V[3]oxidationatrelativelyhighpotential+15V[4]ormeasuringatlowconcentrationsofelectrolyte[5]canbenamedDevelopedtechniquesalsoshowsthatcarbonfeltcanbeusedfordeterminationatsubmicromolarconcentrationsThis ismainly due to its ability to operate as a high-efficiency amperometricdetector Themainaimofthispaperistooverviewandcompareparametersofseveraldetermination methods of different analytes using carbon felt detector in
Utilization of a carbon felt as a material for working electrodes
MARTINBAROCHHANADEJMKOVA SA RKASLA DKOVA
DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublicmartinbarochnaturcunicz
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 141
AbstractWorkingelectrodemadeofcarbonfeltwasusedincombinationwithHPLC for verificationofpractical applicabilityof the electrodeAlldeveloped methods confirm advantageous physical and chemicalpropertiesofcarbonfeltForelectrochemicalutilizationitispossibleto operate at higher positive potentials and even in low concen-trationsofelectrolyteinmobilephaseObtainedlimitsofdetectionwere mostly in submicromolar range and standard deviations ofmeasurementrepeatabilitywereunder5
KeywordsamperometrycarbonfeltFIAHPLC
combinationwithHPLC(forexampletheirdetectionpotentialslimitsofdetectionorlinearrange)
2Experimental
21Reagentsandchemicals
Stock solutions of propyl gallate butylhroxyanisole tert-butylhydroquinonebutylhydroxytoluene chlortoluron 2-amino-4-nitrophenol and 4-amino-2-
ndash3 ndash1nitrophenol (all SigmandashAldrich) with concentration of 1times10 molL werepreparedbydissolving theappropriateamountof therespectivesubstance inmethanol(HPLCgradeLach-NerCzechRepublic)Stocksolutionsof indole-3-aceticacidandindole-3-butyricacidwerepreparedinthesamemannerbutindeionized water Mobile phase consisted of methanol and phosphate-acetatebufferpreparedfromphosphoricandaceticacid(bothLach-NerCzechRepublic)andsodiumhydroxide(Fluka)
22Instrumentation
Theelectrochemicalcellconsistedofcarbonfelt(KarbotechnikCzechRepublic)flow-throughelectrodewhichwasplacedincapwithplatinumwireelectricalcontactanddrilledoutletholeononesideandwithflatferrulawithcapillaryonthe other side Schematic picture of the assembly is shown in ref [5] Otherelectrodeswereauxiliaryplatinumwireelectrodeandreferencesilverchloride(3MKCl) electrode (bothMonokrystaly Turnov Czech Republic) Potentiostatused in combination with this cell was Amperometric Detector ADLC 2(LaboratonıprıstrojePrahaCzechRepublic) HPLCapparatusconsistedofBeta10gradientpump(ECOMCzechRepublic)degasser DG 4014 (ECOM Czech Republic) six-way valve with 20microl loop(Rheodyne USA) HPLC column used for separation of propyl gallatebutylhydroxytoluenetert-butylhydroquinoneandbutylhroxyanisolefromtheirmixtureandforindole-3-aceticacidandindole-3-butyricacidfromtheirmixture
regwasLichrospher RP-18(125times4mm5micromMerckGermany)ForseparationofregchlortoluroncolumnPurospher RP-18(125times4mm5micromMerckGermany)was
used In case of mixture 2-amino-4-nitrophenol and 4-amino-2-nitrophenolregcolumnGemini C18110A (150times46mm5micromPhenomenexUSA)wasusedfor
separation Measurements of pHwere carriedout at Conductivity andpH-meter3510usingcombinedglasselectrode(JenwayUK)
142 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
3Resultsanddiscussion
Performanceofcarbonfeltwastestedonseveraltypesofanalyteswhichneededdifferent separation conditions namely amount of organic solvent in mobilephase and buffer pH The lowest amount of methanol (30) was used forseparation of 2-amino-4-nitrophenol and 4-amino-2-nitrophenol [3] Higherconcentrations of methanol in mobile phase was used for determination ofchlortoluronandforseparationofindole-3-aceticacidandindole-3-butyricacid[4]namely40and60respectivelyThehighestconcentrationsofmethanoland therefore electrolyte with lowest conductivity was used in separation ofantioxidantsnamelypropylgallatebutylhroxyanisolebutylhydroxytolueneandtert-butylhydroquinonewhereamountofmethanolwasrampingfrom55to95[5]DetectionpotentialsofmentionedanalytesandtheirlimitsofdetectionsareshowninTable1 HPLCseparationofantioxidantswas the firstmethodchosen for testingofcarbonfeltelectrodeperformancewiththistechniqueDuetodifferencesintheirstructure when butylhydroxytoluene has a different oxidation mechanismdetectionwith two appliedpotentialswas necessary According to the hydro-dynamic voltammograms potentials 14V and 08Vwere chosen for determi-nationofbutylhydroxytolueneandfortheotherthreeanalytesrespectively[5]AsshowninTable1whenthehigherpotentialwasappliedlimitsofdetectionforpropyl gallate butylhroxyanisole and tert-butylhydroquinone had increasedContrary determination of butylhydroxytoluene had approximately six timeslowerdetectionlimitathigherpotential
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 143
ndash1Compound E VLODmicromolL Refdet
Propylgallate 080 088 [5] 140 186 [5]Butylhroxyanisole 080 144 [5] 140 348 [5]tert-Butylhydroquinone 080 121 [5] 140 266 [5]Butylhydroxytoluene 080 3128 [5] 140 463 [5]Chlortoluron 140 013Indole-3-aceticacid 150 033 [4]Indole-3-butyricacid 150 054 [4]4-Amino-2-nitrophenol 080 016 [3]2-Amino-4-nitrophenol 080 021 [3]4-Amino-2-nitrophenol ndash080 35 [3]2-Amino-4-nitrophenol ndash080 37 [3]
Table 1Parametersofdetectionpotentialsandlimitsofdetectionfordifferentcompoundsusingcarbonfeltdetector
Detectionofauxins(indole-3-aceticacidandindole-3-butyricacid)wascarriedout at potential 15VMeasurements at this potential gives repeatabilitywithstandarddeviation31forindole-3-aceticacidand25forindole-3-butyricacidevenwithexchangingoftheworkingelectrodematerialCalibrationcurves
ndash1forbothanalyteswereobservedfrom04to100μmolL withlinearityinwholeconcentrationrangeLimitsofdetectionforbothanalytesreachedsubmicromolarconcentrationsevenwithrelativelyhighpotential[4] Incaseofdeterminationof2-amino-4-nitrophenoland4-amino-2-nitrophenolcarbon felt electrodewasused inbothoxidationand reductionmodeHydro-dynamicvoltammogramsshowedthatoptimaldetectionpotentialinreductionmodewasndash08VThislowpotentialisclosetotheendofthepotentialwindowandthereforeinterferenceswithremnantsofdissolvedoxygeninmobilephasewereobservedTheseinterferencesresultedinapproximately20timeshigherlimitofdetection for 2-amino-4-nitrophenol or 4-amino-2-nitrophenol obtained inreductionthaninoxidationmodeOntheotherhandmaximumvaluesoflinearrangewerethesameforbothanalytesinbothdetectionmodes[3] ForHPLCofchlortolurontheoptimaldetectionpotentialof14VwasfoundIts
ndash1calibration dependence although observed from 025 to 1000μmolL was ndash1linearonly in therange from025to50μmolL Limitofdetectionbasedon
ndash1standardsolutionswas013μmolL andreproducibilityofmeasurementgivenbytwentyconsecutivemeasurementsgaverelativestandarddeviationof05 ForallthedeterminationmethodsattentionwaspaidtotheapplicabilityofthecarbonfeltindetectionofanalytesincomplexmatricesIncaseofantioxidantsedibleoilswerechosenasrealsamples[5]nitrophenolderivatesweredeter-minedinurinesamples[3]andauxinsinrootingpreparation[4]ChlortolurondeterminationwasperformedinsoilandsurfacewaterThefoundvaluesshowanegligiblematrixinfluenceondetection
4Conclusions
CarbonfeltworkingelectrodewassuccessfullyusedincombinationwithHPLCfordeterminationofdifferenttypesofelectroactivecompoundsegantioxidantsauxinsorpesticidesAllmentionedapplicationsshowgreatperformanceofthecarbonfeltasaflow-throughelectrodematerialinelectroanalyticalchemistryforoxidationandreductionwayofanalytesdeterminationLimitsofdetectionforanalytes are mostly in submicromolar concentrations the exceptions areoxidationof analytes athigherpotentials and their reductionwhere limitsofdetectionsareinmicromolarconcentrationsApplicabilityoftheelectrodeonrealmatriceswas proven on analysis of edible oil samples groundwater soil androotingpreparation
Acknowledgments
ThisworkhasbeensupportedbytheCzechScienceFoundation(projectGACR20-01589S)
144 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
References
[1] Gonzalez-Garcia J Bonete P Exposito E Montiel V Aldaz ATorregrosa-Macia RCharacterizationofacarbonfeltelectrodeStructuralandphysicalpropertiesJMaterChem9(1999)419ndash426
[2] NavaJLRecendizAGonzalezLGCarrenoGMartın ezFMassTransportandpotentialstudiesinaflow-throughporouselectrodereactorPortugalElectrochimActa27(2009)381ndash396
[3] Dejmkova H Knaf M Application of carbon felt detector for the determination ofdinitrophenolmetabolitesInXXXIXModernElectrochemicalMethodsFojtaMSchwarzovaKNavratilT(Eds)U stınadLabemBestServis2019p41ndash43
[4] DejmkovaHdeAraujoDanielMElectrochemicaldeterminationofindole-3-aceticacidandindole-3-butyric acid using hplc with carbon felt detectorMonatsh Chem150 (2019)439ndash442
[5] DejmkovaHBarochMKrejcovaMBarekJZimaJCoulometricdetectorbasedoncarbonfeltApplMaterToday9(2017)482ndash486
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 145
1Introduction
Smith-Lemli-Opitzsyndrome(SLOS)isan autosomal recessive genetic disor-der firstly described in 1964 [1] It iscaused by the inborn deficiency of7-dehydrocholesterol reductase Thisenzyme transforms 7-dehydrocholes-terol (7-DHC Fig 1) to cholesterolduringthefinalstepofbiosynthesisofcholesterolincells The clinical symptoms of SLOS aredecreasedbloodlevelofcholesteroland
Electroanalytical methods for determination of 7-dehydrocholesterol in artificial serum
LENKABENESOVAADE LAZA RYBNICKA JANKLOUDAKAROLINASCHWARZOVA -PECKOVA
UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublicbenesolenaturcunicz
Abstract7-DehydrocholesterolisabiomarkerofSmith-Lemli-Opitzsyndromeanautosomalrecessivegeneticdisordercausedbytheinborndefici-encyof7-dehydrocholesterolreductaseInthisstudyproceduresforitsdeterminationinartificialserumusingflowinjectionanalysiswithelectrochemical detection and voltammetric detection on borondoped diamond electrode were optimized The proteins wereprecipitatedbyacetonitrileandaftercentrifugationthesupernatantused for analysis For quantitation of 7-DHC by differential pulsevoltammetrytheoptimalratioacetonitrile-artificialserum91(vv)wasappliedInFIA-EDtheratio31(vv)runelectrolyteconsisting
minus1ofwater-acetonitrilecontaining001molL NaClO inthesameratio4minus1anddetectionpotentialof+13VvsAgAgCl(3molL KCl)were
usedQuantitationof7-DHCwaspossibleusingcalibrationdepen-minus1dencewithlimitdetectionof20micromolL inartificialserumNeverthe-
lessthemethodhaslowrecoveryandforsensitivedeterminationinreal matrices of human serum and amniotic fluid a liquid-liquidextraction needs to be applied to prevent presence of 7-dehydro-cholesterolinthephasewithprecipitatedproteins
Keywordsamperometricdetectionborondopeddiamond
electrode7-dehydrocholesteroldifferentialpulse
voltammetryflowinjectionanalysisSmith-Lemli-Opitz
syndrome
146 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Structureof7-dehydrocholesterol
increased concentrationof7-DHC inbloodandnervous system [2] SLOS is acomplexofmultipleanomaliesincludingmentalretardationItismanifestedbyholoprosencephaly(anomaliesinbraindevelopmentwithimprecisedivisionintotherightandlefthemispheres)milddysmorphismscardiacrenalandgastro-intestinalmalformations ThecharacteristicfacialanomaliesofSLOS[2]aremicrocephalybitemporalnarrowing ptosis short nasal root short nose with anteverted nares andmicrognathia epicanthal folds and capillary hemangioma over the nasal rootextendingontotheglabellatheearappearlow-setandareposteriorlyrotatedOral finding includes a high-arched and narrow hard plate broad and ridgealveoralridgesandredundancyofsublinqualtissuesCNSanomaliesareagenesisorhypoplasiaBilateralandunilateralpostaxialpolydactylycanbepresentedinthehandsorfeetorboth Concentrationof7-DHCinbloodiscrucialforclinicaldiagnosticofSLOSinpatients Concentration levels in amniotic fluid are used for fetal diagnosticsTable1 summarizes concentration of 7-DHC in plasma and amniotic fluid ofhealthypersonandSLOSpatientsAnalyticalmethodsusedfordeterminationofconcentrationof7-DHCinthesematricesincludecombinationofGCorHPLCwithMS[3]orGCwithflameionizationdetection(FID)orUVdetection[45] Thepossibilitiesofelectrochemicalmethodsfordetectionof7-DHCarelimitedasgenerallythesteroidcoreisratherredox-inactive(detailinreview[6])undervarietyofconditionsNevertheless7-DHCpossessesconjugateddoublebondsonsteroidcoreanditsoxidationwasreportedinseveralstudies[7ndash9]Itsvoltam-metricsignal+095VvsSCEonglassycarbonelectrodewasfirstlyobservedin
minus1non-aqueousmediaofmethanol-benzene7525(vv)using005molL LiClO as4
supporting electrolyte in a study dealing with electrochemical behaviour ofvitaminAandDandtheirprovitaminsD(7-DHCisprecursorofvitaminD3)[7]Determinationof7-DHCinhumanskinispossiblebyHPLCwithUV(λ=286nm)andamperometricdetection(E +17VvsAgAgCl)onglassycarbonelectrodedet
minus1usingmethanol-tetrahydrofuran175mmolL KH PO (9514vvv)asmobile2 4minus1phase7-DHCwasdetectedintherangefrom12to81microgg dryweightwith
minus1detectionlimitof39pmolL [8]Anotherstudy[9]isdevotedtodeterminationof
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 147
minus1Author(s)ref Matrix ConcentrationμmolL Healthy SLOS
Kelley[4] Plasma 03plusmn001 385plusmn309 Amnioticfluid lt02plusmn001 16plusmn9Rossiteretal[5] Plasma lt5 179ndash335 Amnioticfluid lt03 12ndash15
Table 1Concentrationof7-dehydrocholesterolinclinicalmatricesforhealthypersonsandforSmith-Lemli-Opitzsyndrome(SLOS)patientsdetectedbyGC-FID
7-DHCandvitaminD3 in fishusingHPLCwithelectrochemicaldetectionTheanalytical cell was a serial combination of two-flow-through porous graphiteworking electrodes The first standard coulometric electrode was used toeliminatepotentially interfering compounds using the second lineardynamic
minus1rangefrom0013to0312micromolL for7-DHCwasachieved Hereinwestudiedpossibilitiestodetect7-DHCbasedonitsoxidationonborondoped diamond (BDD) electrode using differential pulse voltammetry andelectrochemicaldetectioninflowinjectionanalysis(FIA-ED)inartificialserumandperipherallyinhumanserumandamnioticfluid
2Experimental
21Reagentsandchemicals
7-dehydrocholesterol (purity95)wasobtainedSigmaAldrich (USA)and itsstandard solution was prepared in acetonitrile (Honeywell Germany) Theartificial serum was prepared from KCl (Penta Czech Republic) CaCl 2H O2 2
(PentaCzechRepublic)NaCl(PentaPragueCzechRepublic)urineD-glucoseand01albuminfromSigma-Aldrich(USA)NaClO4(PentaCzechRepublic)wasusedassupportingelectrolyte
22Instrumentation
VoltammetricmeasurementweregovernedbythepotentiostatPalmSensusingworking BDD electrode (Windsor Scientific UK d = 31 mm) AgAgNO 3
minus1 minus1(01molL AgNO 1molL NaClO in acetonitrile) non-aqueous reference3 4electrodeandplatinumwirecounterelectrodeBDDsurfacewaspolishedbeforeeachscanusingsuspensionofAl O (ElektrochemickedetektoryTurnovCzech2 3
Republic) HPLC system (Hitachi Merck) consisting of control unit D-7000gradientpumpL-7100autosamplerL-7200andUVdetectorL-7400wasusedfor
minus1FIA-EDdetectionof7-DHCRunelectrolytewascomposedof001molL NaClO 4inacetonitrileanddeionisedwaterinratio31(vv)Flowrateofmobilephase
minus1was30mlmin injectionvolumewas40microLandλ=280nmwasusedforUVdetectionWall-jet detection cell was employedwith working BDD electrode
minus1AgAgCl (3molL KCl) reference electrode and platinum wire auxiliaryelectrode Optimal detection potential of +13 V was controlled using ADLC2potentiostat(Laboratornı prıstrojePragueCzechrepublic)
3Resultsanddiscussion
In this study electroanalytical methods were developed for determination of7-DHCinartificialserumnamelyFIA-EDandDPVBothmethodsarebasedondirectoxidationof7-DHConborondopeddiamondelectroderesultinginanodic
148 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
peak at ca +08 V (vs AgAgNO in acetonitrile) in non-aqueous medium of3
acetonitrile or mixedmedium acetonitrile-water using NaClO as supporting4
electrolyteTheoxidationispresumablyinitiatedbyoneelectronremovalfromtheconjugateddoublebondsonthesteroidcoreof7-DHC Fordeterminationof7-DHCinrealmatricesitisnecessarytoremovepresentproteinsArtificialserumcontainingalbuminwasusedasmodelmatrixtostudythe possibilities Firstly albumin was removed simply by precipitation withacetonitrile(serum-water13(vv))andthesupernatantwasanalysed Differentratioswateracetonitrileweretestedinrunelectrolyte(59510902080 2575 3070 4060 and 5050 (vv)) to evaluate the influence of itscompositionontheFIA-EDsignaloftheblankand7-DHCThesameratio13asusedforprecipitationofalbuminwaschosenasoptimalbecauseofminimalandstablesignaloftheblankinjectedinFIA-EDsystemFurtherdetectionpotentialE in the range from +10 V to +15 V was optimized by evaluation of thedet
hydrodynamicvoltammogramsresultinginE of+13Vsetasoptimalvaluedetminus1 Concentrationdependenceof7-DHCislinearintherangefrom25micromolL to
minus1 minus1300micromolL (concentrationinartificialserum)withdetectionlimitof20micromolL and this concentration dependence can be used for quantitation of 7-DHC inartificial serum Nevertheless determination of 7-DHC in human serum andamnioticserumfailedastheyrepresentmorecomplicatedmatricesand7-DHCispresumablypartiallyadsorbedinthepresentproteinsandcannotbequantifiedinthesupernatant Furtherdifferentialpulsevoltammetrywithoptimizedparameterswasusedfordeterminationof7-DHCInthepresenceofproteinsinartificialserum(human
minus1serum albumin) an unacceptably high detection limit of 178micromolL wasachieved When the proteins were precipitated using acetonitrile (aceto-nitrileartificialserumratio91(vv))thelimitofdetectionof7-DHCwaslowered
minus1to15micromolL inartificialserumNeverthelesstherecoveryofthemethodwasonly43to70dependingontheconcentrationof7-DHCagainreflectingthelossof7-DHCduetoproteinprecipitation Thereforeasecondapproachofsamplepretreatmentbasedonliquid-liquidextractionofalllipidsdescribedin[10]wastested(Bligh-Dyerextraction)TheprocedurehastwopartsFirstlymethanolchloroformandthesampleofartificialserum is mixed and shaken to form a monophasic system After addition ofchloroform and water a biphasic system is formed where chloroform phasecontainsalloflipidcompoundsandmethanol-waterphasecontainsallnon-lipidscompoundsChloroformphaseisthendriedunderN2atmosphereat50degCanddried extract dissolved in acetonitrile Preliminary experiments using DPVresultedinrecoveryof97forBligh-Dyerextractionof7-DHCfrominartificialserum
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 149
4Conclusions
FIA-EDandDPVwereoptimizedfordeterminationof7-DHCinartificialserumUsing precipitation of proteins by acetonitrile limit of detection of 7-DHC in
minus1artificialserumusingFIA-EDwas20micromolL andthismethodcanbeusedfortheirquantificationusingcalibrationdependenceNeverthelessdeterminationusingDPVisunreliableduetolowrecoveryoftheprocedureDevelopmentofamethodincludingliquid-liquidextractionstepisinprogresssothat7-DHCcouldbedeterminedinrealmatricesashumanserumandamnioticfluid
Acknowledgments
TheresearchwassupportedbytheCzechScienceFoundation(projectGACR19-11268S)andtheSpecificUniversityResearch(SVV260560)
References
[1] Smith DW Lemli L Opitz JM A newly recognized syndrome of multiple congenitalanomaliesJPediatr64(1964)210minus217
[2] Nowaczyk M Waye J The SmithndashLemlindashOpitz syndrome a novel metabolic way ofunderstandingdevelopmentalbiologyembryogenesisanddysmorphologyClinGenet59(2001)375minus386
[3] BeckerSRohnikeSEmptingSHaasDMohnikeKBebloSMutzeUHusainRAThieryJCeglarekULC-MSMS-basedquantificationofcholesterolandrelatedmetabolitesindriedblood for the screening of inborn errors of sterolmetabolismAnal Bioanal Chem407(2015)5227minus5233
[4] Kelley RI Diagnosis of Smith-Lemli-Opitz syndrome by gas-chromatography mass-spectrometryof7-dehydrocholesterolinplasmaamniotic-fluidandculturedskinfibroblastsClinChimActa236(1995)45minus58
[5] Rossiter JP Hofman KJ Kelley RI Smith-Lemli-Opitz SyndromePrenatal-diagnosis byquantification of cholesterol precursors in amniotic-fluid Am J Med Genet 56 (1995)272minus275
[6] KloudaJBarekJNesmerakKSchwarzova-PeckovaKNon-enzymaticelectrochemistryincharacterization and analysis of steroid compounds Crit Rev Anal Chem 47 (2017)384minus404
[7] AtumaSSLundstromKLindquistJTheelectrochemicaldeterminationofvitaminAPartIIFurthervoltammetricdeterminationofvitaminAandinitialworkonthedeterminationofvitaminDinthepresenceofvitaminAAnalyst100(1975)827minus834
[8] MoodyJPHumphriesCAAllanSMPatersonCRDeterminationof7-dehydrocholesterolinhumanskinbyhigh-performance liquid-chromatography JChromatogrB530 (1990)19minus27
[9] OstermeyerUSchmidtTVitaminDandprovitaminDinfishEurFoodResTechnol222(2005)403minus413
[10] BlighEGDyerWJArapidmethodoftotallipidextractionandpurificationCanJBiochemPhysiol37(1959)911minus917
150 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Author Index
AlikovaV1
AugustınM83
BaluchovaS19
BarekJ192570
BarochM141
BasB13129
BastryginaO41
BenesovaL146
BessonovaE57
BohmD6
BurkinK116
BurkinM116
ChernovaA141
ChoinskaM70
CokrtovaK104
DedinaJ97
DeevV57
DejmkovaH141
DendisovaM63
DubenskaL51
EfremenkoE41
FojtaM110
GalvidisI116
HavranL110
HeiglN31
HertJ76
HrdlickaV70
JosypcukB25
KartsovaLA3557
KloudaJ19146
Kodrık ovaB90
KolobovaEA35
KorbanA135
KorotkovaE1
KralM63
KratzerJ90
KravchenkoAV35
KrızekT76104
LipinskaJ129
MadejM129
MatejkaP63
MatysikF-M631
MusilS9097123
NavratilT70
OndrackovaA110
PietrzakK45
PlotnikovaK51
PoradaR13
RedondoBR70
SagapovaL90
ShormanovV1
Schwarzova-PeckovaK19110146
SladkovaS 141
S tadlerovaB97
StiborovaM110
SvobodaM90
TvorynskaS25
TyczkowskiJ129
VyhnanovskyJ97123
VymyslickyF76
VyskocilV83
WardakC45
WongDKY19
ZarybnickaA146
ZelenyI51
Proceedingsofthe16thISCModernAnalyticalChemistry Prague2020 151
152 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Keyword Index
alcoholicbeverages135
aminoglycosides116
amperometricdetection146
amperometry141
antifoulingelectrodes19
assembledcapillaries6
atomicabsorptionspectrometry90
atomicfluorescencespectro-
metry97
atomization90
biologicalactiveanalytes35
biosensor2583
bismuth97
borondopeddiamond
electrode110146
cadmium90
canagliflozin76
capillarycoating35
capillaryelectrophoresis635
capillaryflowinjectionanalysis631
carbohydrates31
carbonfelt141
cathodicstrippingvoltammetry70
chemicalvaporgeneration90
chemometrics57
cobalt123
coldplasmadeposition129
copper(II)phthalocyanine63
covalentimmobilization25
cytochromeP450110
damage83
7-dehydrocholesterol146
designofexperiments76
differentialpulsevoltammetry146
diphenylsilanereductionmethod19
dispersiveliquid-liquid
microextraction57
disposableelectrodes31
DNA83
dualdetectionconcept6
electrochemicalanalysis110
electrochemicalapplications129
electrochemicalflowcell76
electrochemistry51
electrokineticchromatography104
eliminationvoltammetrywith
linearscan70
ELISA116
enzymaticreactor25
FIA141146
gaschromatography-mass
spectrometry(GC-MS)135
glucoseoxidase25
graphite83
honey116
HPLC76141
hydridegeneration97
hydrogenatedconical-tipcarbon
electrodes19
imidazoliumionicliquids35
inductivelycoupledplasmamass
spectrometry123
internalstandardmethod135
ion-selectiveelectrode45
laccase25
liquidcrystals104
massspectrometry31
mercuryelectrode13
metronidazole51
non-aqueouscapillary
electrophoresis104
non-aqueoussystem6
oxidation76
oxytetracyclinehydrochloride51
phenol2-methoxy1
photochemicalvaporgene-
ration97123
polarography51
1-propanesulfonicacid23-
dimercapto-70
pulsedamperometricdetection31
quantitation1
resonanceRamanspectroscopy63
scanningtunnellingmicroscopy63
silversolidamalgamelectrode70
Smith-Lemli-Opitzsyndrome146
smokingmixtures41
solidcontact45
solid-phasemicroextraction57
spectrophotometry141
SudanI110
surfacemodification129
surface-enhancedRaman
spectroscopy63
thinlayers129
tip-enhancedRamanspectroscopy
63
unithiol70
uranyl45
vanillin41
veterinarydrug51
vitamins13
volatilecompoundsquantifi-
cation135
voltammetricdopaminedetec-
tion19
voltammetry1383
Proceedingsofthe16thISCModernAnalyticalChemistry Prague2020 153
Proceedings of the 16th International Students Conference ldquoModern Analytical Chemistryrdquo
EditedbyKarelNesmerak
PublishedbyCharlesUniversityFacultyofScience
Prague2020
1steditionndashvi154pages
ISBN978-80-7444-079-3
ISBN 978-80-7444-079-3
Pro
ceedin
gs of th
e 16
th In
ternatio
nal Stu
den
ts Co
nferen
ce ldquoMo
dern
An
alytical Ch
emistryrdquo P
rague 2
02
0
788074 440793
Prague 17mdash18 September 2020
Edited by Karel Nesměraacutek
Prague 2020
Proceedings of the
16th International Students Conference
ldquoModern Analytical Chemistryrdquo

Sponsors
The organizersof16th International Students Conference ldquoModernAnalytical
Chemistryrdquo gratefully acknowledge the generous sponsorship of following
companies
wwwecomsrocom
wwwlach-nercom
iv Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
wwwthermofishercz
www2thetacz
wwwzentivacz wwwquintacz
wwwshimadzueucom
wwwwaterscom
Contents
AlikovaVChernovaAShormanovVKorotkovaEDeterminationof2-methoxyphenol inmodelsolutionsbyspectrophotometry 1
BohmDMatysikF-MTheeffectsoflinearlyassembledcapillarieswithvariousinnerdiametersoncapillaryelectrophoresis 6
PoradaRBasBVoltammetricdeterminationofvitamins 13Baluchova S Klouda J Barek J Schwarzova-Peckova K Wong DKY Dopamine detection at
antifoulingconical-tipcarbonelectrodes 19Tvorynska SBarek J JosypcukBA comparative study of covalentglucose oxidaseand laccase
immobilizationtechniquesatpowderedsupportsforbiosensorsfabrication 25Heigl N Matysik F-M Capillary flow injection analysis with electrochemical detection for
carbohydrateanalysis 31KravchenkoAVKolobovaEAKartsovaLAApplicationofcovalentcoatingsbasedonimidazolium
cations for separationandon-linepreconcentrationof basicandneutralanalytes in capillaryelectrophoresis35
Efremenko E Chernova A Bastrygina O Determination of vanillin in smoking mixtures byspectrophotometry41
PietrzakKWardakCUranylion-selectiveelectrodewithsolidcontact 45Plotnikova K Dubenska L Zeleny I Polarographic determination of metronidazole and
oxytetracyclinehydrochlorideinveterinarydrugforhoneybees 51Deev V Bessonova E Kartsova L Application of microextraction techniques combined with
chromatographicmethodsfortheanalysisofcomplexobjects 57KralMDendisovaMMatejkaPThedevelopmentofreferenceprobesystemfortip-enhancedRaman
spectroscopy 63ChoinskaMHrdlickaVRedondoBRBarekJNavratilTDeterminationofheavymetalpoisoning
antidote23-dimercapto-1-propanesulfonicacidusingsilversolidamalgamelectrode70VymyslickyFKrızekTHert JCanagliflozinoxidation studyusingelectrochemical flowcelland
comparisonwithhydrogenperoxideoxidation 76AugustınMVyskocilVNovelhybridelectrochemicalDNAbiosensorformonitoringoxidativeDNA
damageviaoxidationreductionsignalsoflowmolecularweightdouble-strandedDNA 83SagapovaLKodrık ovaBSvobodaMMusilSKratzerJChemicalvaporgenerationofcadmiumfor
analyticalatomicspectrometry 90S tadlerova B Vyhnanovsky J Dedina J Musil S Photochemical vapour generation of bismuth
coupledwithatomicfluorescencespectrometry 97Cokrtova K Krızek T Separation of liquid crystals using non-aqueous capillary electrokinetic
chromatography104OndrackovaAStiborovaMHavranLSchwarzova-PeckovaKFojtaMElectrochemistryofSudanI
anditsderivatesinaqueousmedia 110BurkinKGalvidisIBurkinMGroupdetectionofaminoglycosidesusingELISAforcontroloffood
contamination 116Vyhnanovsky J Musil S Photochemical vapor generation of cobalt for detection by inductively
coupledplasmamassspectrometry 123LipinskaJMadejMBasBTyczkowskiJOptimizationofconditionforcoldplasmadepositionofthin
layersforsurfacemodificationofworkingelectrodes 129KorbanAAdvancedGC-MSmethodforqualityandsafetycontrolofalcoholicbeverages 135Baroch M Dejmkova H Sladkova S Utilization of a carbon felt as a material for working
electrodes 141Benesova L Zarybnicka A Klouda J Schwarzova-Peckova K Electroanalytical methods for
determinationof7-dehydrocholesterolinartificialserum146
Authorindex 151Keywordindex 152
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 v
1Introduction
2-Methoxyphenol(guaiacol)isusedinmedicineasanexpectorantThestructuralformulaisshowninFig1Itiswidelyusedinthepharmaceuticalindustry[1]forthesynthesisofantituberculosisexpectorantdrugs(Kas-nol Sudafed Ascoril Prothiazine Expectorant Guai-phenesinum) Moreover 2-methoxyphenol is often used as anaromaticsubstance [2] in the food industry Inparti-cular itwaswidelyusedintheproductionofsmokedfish and meat products using smokeless smokingtechnologyusingflavourings Ontheotherhand2-methoxyphenolhasthesymbolGHS07andhasahazardcodeXnTXi[2]accordingtotheGHSsystemItisverytoxicbyinhalationitcanirritatethemucousmembraneoftherespiratorytractandtheconjunctivaofthe
Determination of 2-methoxyphenol in model solutions by spectrophotometry
a a b aVALERIYAALIKOVA ANNACHERNOVA VLADIMIRSHORMANOV ELENAKOROTKOVA
a DepartmentofChemicalEngineeringEngineeringSchoolofNationalResourcesNationalResearchTomskPolytechnicUniversityLeninavenue30634050TomskRussiaalikovaleramailru
b DepartmentofPharmaceuticalToxicologicalandAnalyticalChemistryKurskStateMedicalUniversitystKarlaMarks3305000KurskRussia
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 1
AbstractA spectrophotometric approach for determination of 2-methoxy-phenol in model solutions has been developed The absorptionspectra of 2-methoxyphenol were determined in the wavelengthrangefrom200to400nminsolutionsof95ethanolacetonitrile01Msodiumhydroxideandethylacetatewithaconcentrationofthe
minus3analyte of 005mgdm For the quantitative determination of2-methoxyphenol a seriesof solutionswaspreparedwithvarious
minus3 minus3concentrationsfrom0001mgdm to005mgdm in95ethanolacetonitrile01MsodiumhydroxideTheopticaldensityof2-meth-oxyphenolinsolventswasmeasuredatawavelengthof276nmand289nmThedevelopedmethodwastestedusingthemethodanalysisofspikedsamples
Keywordsquantitation2-methoxyphenolUVVISspectrophoto-
metry
Fig 1Structuralformulaof2-methoxyphenol
eyeballinhighconcentrationswhenitpenetratestheskincanleadtoneurosiswhenadministeredorallycanstimulatetheesophagusandstomachresultinginheartfailurecollapseanddeathNowadaystherearepublisheddataoncasesofasystemicallergicreaction[3]causedby2-methoxyphenolderivativesandthereisa fatal case known [4] for oral administration of guaifenesin (3-(2-methoxy-phenoxy)propane-12-diol)oneofthecomponentsofcommonlyavailablecoughmedications Thedeterminationof2-methoxyphenolinenvironmentalobjectsaswellasinthefoodindustryiscarriedoutusinggaschromatographymethodswithsolid-phasemicroextraction[5]Inordertocontrol2-methoxyphenolinnaturaldrink-ing and treatedwastewater gas chromatography is used followed by opticaldetectionoftheeluate[6]Themaindisadvantagesofthismethodofanalysisarethelowselectivityanddurationofdetermination(about3hours)AlsoaccordingtoRussianStateStandartGOST33312-2015themethodofgaschromatographyisusedforthequalitativeandquantitativedeterminationof2-metoxyphenolinjuiceproducts Commonlyfortheanalysisoftoxicsubstancesinvariousbiologicalsamples(bloodplasmaurinesalivasweathair)bygaschromatographyitisnecessarytocarryoutmultistagesamplepreparationwhichcomplicatesandslowsdownthecourseofthestudy[7]Atthesametimeitisimportantthatduringtheprepa-rationofsamplesintheanalyzedcompoundstheirstructureisnotviolatedasthiswillleadtothedifficultyoftheiridentification Spectrophotometryintheultravioletregionhaslowersensitivitycomparedtothe abovemethods however thismethod does not require such complicatedpreparation of the analyzed samples it is a relatively affordable simple andinexpensive analysis method In addition its sensitivity can be significantlyimprovedbyapplyinganappropriateseparationprocedureandpreconcentrationbeforedetection[8]MethodUVspectrophotometryisusedtoassessthequalityof both medicinal substances and preparations made from them in terms ofauthenticitygoodqualityandquantitativecontentInadditionitisarelativelyaffordablesimpleandlow-costanalysismethod Ananalysisoftheliteraturedatashowedthattodaytherearefastandsensitivespectrophotometricmethodsforthedeterminationofpyrocatecholderivativesinmedicines[9]vanillininfoodproducts[10]andotherphenolsinwastewaterandwineproducts[1112]Howeverasfarasweknowinformationonthedetermi-nationof2-methoxyphenolfromtheabsorptionspectraintheultravioletregionisabsent Theaimofthisstudyistodevelopmethodsforthequalitativeandquantitativeof2-methoxyphenolinmodelsolutionsusingUVspectrophotometry
2 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
2Experimental
21Reagentsandchemicals
Asampleof2-methoxyphenolfromFlukawithabasicsubstancecontentofge98wastakenastheobjectofstudyAssolventsweusedacetonitrile(ChP)95ethanolethylacetateand01MsodiumhydroxidesolutionAllotherchemicalsusedwereofanalyticalreagentgrade
22Instrumentation
Theopticaldensitywasmeasuredincuvetteewithanabsorbinglayerthicknessof10mmusingaCary60spectrophotometer(AgilentUSA)Allmeasurementswerecarriedoutatroomtemperature
3Resultsanddiscussion
Thechangeinthebehavioroftheabsorptionspectrumwasinvestigatedinthewavelengthrangeof200ndash400nmFigure2isshowedthatwithanincreaseinthepolarityofthesolventtheabsorptionmaximumshiftstowardthevisiblepartofthe spectrum The wavelength of absorption maxima of 2-methoxyphenol ispresentedinTable1 A studyof thephotometricbehaviorof2-metoxyphenolinvarious solventsshowedthatacetonitrile95ethanoland01Msodiumhydroxidearethemostsuitablesolventsforthequalitativedeterminationofthetestsubstance
ndash3Fig 1Thespectraof2-methoxyphenolofconcentration005mgdm inthemediumofsolvents(anabsorbinglayerthicknessof10mm)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 3
Forthequantitativedeterminationof2-methoxyphenolaseriesofsolutionsndash3 ndash3withaconcentrationragefrom0001mgdm to005mgdm werepreparedin
acetonitrileand95ethanolTheopticaldensityof2-methoxyphenolinsolventswasmeasuredbywavelengthof276nmThedependenceoftheintensityoftheoptical density on the concentration of 2-methoxyphenol in 01 M sodium
ndash3hydroxide was plotted in the concentration range from 0005mgdm tondash3003mgdm Themeasurementwerecarriedoutbywavelengthof276nmThe
obtainedregressionequationsarepresentedinTable2Dataanalysisobtainedwasperformedusingleast-squaresmethodThedevelopedmethodwastestedusingthemethodanalysisofspikedsamplesTheresultsarepresentedinTable2
4Conclusions
Studieshaveshownthepossibilityofusingspectrophotometricanalysisforthequalitativeandquantitativedeterminationof2-methoxyphenolTheabsorptionmaximaofweredeterminedinsolutionsofethanolandacetonitrile(276nm)inasolutionofethylacetate(277nm)and01Msodiumhydroxide(289nm)Theconstructed calibration curves of thepure substance of 2-methoxyphenol hasshownagoodregressioncoefficient(Rgt099)andcanbeusedforquantitativedeterminationof2-metoxyphenolinbiologicalobjectsInthefutureitisplannedtoapplythistechniquetodetermine2-metoxyphenolinincadavericmaterial
Solvent Regressionequation Found S RSD Δх δ 2-metoxy- phenolg
ndash3 ndash6 ndash5Acetonitrile y=18294C+01130 499times10 50times10 028 2times10 044 Rsup2=09985
ndash3 ndash6 ndash595ethanol y=35131C+00269 503times10 03times10 021 7times10 137 Rsup2=09956
ndash3 ndash6 ndash501Msodium y=31196C+01101 495times10 01times10 018 1times10 028hydroxide Rsup2=09997
Table 2Results of the determination of 2-methoxyphenol (average of three measurements) in modelsolutionsbythemethodanalysisofspikedsamplestheconcentrationofintroduced2-methoxy-
ndash3phenolwas500times10 g(SndashstandarddeviationRSDndashrelativestandarddeviationΔхndashabsoluteerrorδndashrelativeerror)
Table 1Valuesofopticaldensityandwavelengthsinappropriatesolventswith2-methoxyphenol
minus3 minus1Solvent λnm εgdm cm
Acetonitrile 276 0082895ethanol 276 00853Ethylacetate 277 0083101Msodiumhydroxide 289 00744
4 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
References
[1] МельниковаИММизерницкии ЮЛКомбинированныеотхаркивающиепрепаратырастительного происхождения в педиатрическои практикеМедицинский совет 2(2018)93ndash97
[2] httpwwwthegoodscentscompanycomdatarw1032272html(accessed27stFebruary2020)
[3] RayMFaltayBHallerNACasereportanaphylacticreactiontoguaifenesinHospPract37(2009)60ndash63
[4] OkicMJohnsonTCrifasiJALongCMitchellEKSwiftonsetofcentralnervoussystemdepressionandasystolefollowinganoverdoseofguaifenesinJAnalToxicol37(2013)318ndash319
[5] ВолковCМЧерновецАНОпределениеконцентрациифеноловвгазовыхвыбросахпромышленных предприятии методом газовои хроматографии с твердофазнои микроэкстракциеи Сорбционныеихроматографическиепроцессы10(2010)723ndash728
[6] ШачневаЕЮОньковаДВСерековаСМСпособыопределенияфеноловвобъектахокружающеи среды Астраханский вестник экологического образования 4 (2013)138ndash142
[7] ГладиловичВДПодольскаяЕПВозможностипримененияметодаГХ-МС(Обзор)Научноеприборостроение4(2010)36ndash49
[8] Pena-PereiraFLavillaIBendichoCHeadspacesingle-dropmicroextractioncoupledtomicrovolumeUVndashVis spectrophotometry for iodine determinationAnal Chim Acta631(2009)223ndash228
[9] NagarajaPMurthyKCSRangappaKSGowdaNMMSpectrophotometricmethodsforthe determination of certain catecholamine derivatives in pharmaceutical preparationsTalanta46(1998)39ndash44
[10] Altunay N Development of vortex-assisted ionic liquid-dispersive microextractionmethodology for vanillin monitoring in food products using ultraviolet-visible spectro-photometryLWT93(2018)9ndash15
[11] Lupetti KO Rocha FRP Fatibello-Filho O An improved flow system for phenolsdetermination exploiting multicommutation and long pathlength spectrophotometryTalanta62(2004)463ndash467
[12] Figueiredo-Gonzalez M Cancho-Grande B Simal-Gandara J Garnacha tintorera-basedsweetwineschromaticpropertiesandglobalphenoliccompositionbymeansofUVndashVisspectrophotometryFoodChem140(2013)217ndash224
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 5
1Introduction
Thenumberofsamplesthesamplecomplexityandalsothenumberofsubstanceswhich need to be analysed simultaneously is increasing steadily ThereforepowerfulseparationanddetectionmethodsarerequiredOnewaytoachievethisisthecouplingofaseparationsystemwithmorethanonedetector[12] In recent years capillary electrophoresis (CE)was established as a potentseparation system due to its high separation efficiency and the low sampleconsumption [3] To generate more information numerous dual detectionconceptsforCEweredevelopedwhicharesummarisedelsewhere[12]Acom-binationofamperometricdetectionandmassspectrometry(MS)isaninterestingdual detection concept for CE because both detectors supply complementaryinformationForelectroactivespeciesamperometricdetectionisarobustandoneof the most sensitive detection method [4] Thus it is well suited for the
The effects of linearly assembled capillaries with various inner diameters on capillary electrophoresis
DANIELBO HMFRANK-MICHAELMATYSIK
InstituteofAnalyticalChemistryChemo-andBiosensorsFacultyofChemistryandPharmacyUniversityofRegensburgUniversitaumltsstraszlige3193053RegensburgGermanydanielboehmchemieuni-regensburgde
AbstractDuetotheincreasingneedofpowerfulanalyticalmethodsanewdualdetection concept for capillary electrophoresis (CE) with parallelamperometricdetectionandmassspectrometryshallbedevelopedFor this concept the CE flow has to be divided into two streamsutilizinga flowsplitter In thiswork theeffectsof combinedcapi-llarieswithvarious innerdiameterswerestudiedForpreliminaryinvestigationsthecapillarieswereconnectedinaserialconfigurationwithoutdeadvolumeUsingcapillaryflowinjectionanalysishyphe-natedtocontactlessconductivitydetectionitcouldbeshownthatthecouplingofidenticalcapillariesleadstoaslightdecreaseoftheflowratesWithCEhyphenatedtoUVdetection itcouldbeshownthatthecouplingofcapillarieswithdifferentinnerdiameterhasamuchstrongereffectontheelectroosmoticflowthanthecombinationwiththesameinnerdiameterFurthermorenosignificantchangeinpeakshapewasobserved
Keywordsassembledcapillariescapillaryelectrophoresiscapillaryflowinjection
analysisdualdetectionconceptnon-aqueoussystem
6 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
quantificationofsubstanceswhereasMSiswellsuitedfortheidentificationofunknown substances [3] In most dual detection concepts the detectors arearrangedinaserialconfigurationwhichisnotpossibleincaseofamperometricdetection-mass spectrometry [1] The instrumental implementation is morecomplicatedwithbothdetectorsbeingdestructiveFurthermoretheymustbedecoupledfromthehighvoltagefieldoftheCEThereforetheCEflowmustbedividedintotwostreamswithaflowsplitterAsimplifiedsketchofthepossiblenewdualdetectionconceptisshowninFig1 ForthedevelopmentofthenewdualdetectionconceptthreecapillarieswithpotentiallydifferentinnerdiametersmustbecoupledForthisreasonthedeadvolume-freecouplingofcapillarieswithdifferent innerdiameterswas investi-gatedinafirststepTokeepthesetupsimplewefocusedonthelinearcouplingofcapillariesandtheresultingeffectsNon-fragmentedcapillarieswerecomparedwithfragmentedcapillariesofthesameordifferentinnerdiametersEffectsonthe flow rate were investigated with capillary flow injection analysis (CFIA)
4hyphenatedtocontactlessconductivitydetection(C D)EffectslikechangesinthemigrationbehaviourorpeakshapesoccurringinCEwereinvestigatedwithCEhyphenatedtoUVdetection(CE-UV)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 7
Fig 1SchematicillustrationofthenewdualdetectionconceptwithparallelamperometricdetectionandmassspectrometricdetectionforCEAfterinjectionfrom(a)thesamplevialthecomponentsareseparatedbyCEnext(b)theflowsplitterdivides(c)thecapillaryintotwopartsandleadstheCEflowtowards(d)themassspectrometerand(e)theamperometricdetector
2Experimental
21Reagentsandchemicals
Thefollowingchemicalswereusedallofanalyticalgradeferrocenemethanoldecamethylferrocene(ABCRGermany)acetonitrileammoniumacetate01Msodiumhydroxidesolutionultra-purewaterprovidedbyaMilliQAdvantageA10system(MerckGermany)aceticacid(RothGermany)
22Instrumentation
221Capillaries
4Forbothexperiments(CFIA-C DandCE-UV)capillarieswithinnerdiametersof2550and75micromanouterdiameterof360micromandatotallengthof70cmwereusedTheywerepurchasedfromPolymicroTechnologies(USA)Measurementswere carried out with fragmented and non-fragmented capillaries For themeasurementswiththefragmentedcapillariestheoriginalcapillarieswerecutintotwopiecesyieldingatotalof9capillarycombinationswithlengthsof70cm(20cmfirstcapillarypieceand50cmsecondcapillarypiece)ThesecombinationsaresummarizedinTab1(section31)Atbothendsofthecapillariesabout02cmof thepolyimidecoatingwasremovedBothsidesof thecapillarypieceswerepolishedtoreceiveplanarcapillarytipsForthelinearassemblingofthecapillarypiecesMicroTightSleevesF185Xanda capillary connectorUnionAssemblyMicroTightP720fromIDEXHealthampScience(USA)wereusedPriortothefirstCEmeasurementsthecapillarieswereconditionedbyflushingthemfor10minwith01Msodiumhydroxidesolution5minwithultra-purewaterand30minwithseparationbuffer
222Capillaryflowinjectionanalysishyphenatedtocontactlessconductivitydetectionsetup
The flow rates for the fragmented and non-fragmented capillaries were4determinedwithaCFIA-C DsetupschematicallydepictedinFig2ATheflowin
thecapillarywasgravitationdrivenbyaheightdifferencebetweentheinletandoutletcarriersolutionvialTheconceptofCFIAwithgravitationdrivenflowwasfirstdescribedbyMatysiketal[5]AlaboratoryconstructedautosamplerofaCEdevicewasusedforthehydrodynamicinjectionThesamplesolutionconsistedof10mMdecamethylferrocene incarrier solution (10mMCH COONH and1M3 4
4CH COOH in acetonitrile) A high resolution C D was placed after 40 cm for3
detectionThedetectordescribedelsewhere[6]wasconstructedinthedoLagogroup(Brazil)Adoubledeterminationattwodifferentheightswasdoneforthedeterminationoftheflowrates
8 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
223Capillaryelectrophoresis-UVdetectionsetup
Fig2BshowsasketchoftheCE-UVsetupItconsistedofalab-builtCEdevicewhichwasconnectedtoahighvoltagepowersupplyfromISEG(Germany)Theseparationswerecarriedoutwithanon-fragmented50micromcapillaryandwithcapillary combinations implementing a 50micromdownstream capillary segment(25+5050+50and75+50microm)ALambda1010UV-VISdetectorfromBischoff(Germany)wasusedfordetectionat210nmThedetectorwasplacedafter40cm
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 9
Fig 2Schemeof(A)thecapillaryflowinjectionanalysis(CFIA)hyphenatedtocontactlessconduc-4tivity detection (C D) setup and (B) the capillary electrophoresis hyphenated to UV detection
4(CE-UV) setup Components of the CFIA-C D setup (a) sample (b) inlet and (c)outlet carrier4solution vial (d) fused silica capillary (e) linear capillary connector (f) C D and (g) stand
ComponentsoftheCE-UVsetup(h)sample(i)inletand(j)outletbuffervialand(k)UVdetector4therestofthecomponentswereidenticaltotheCFIA-C DsetupTheoutletbuffervialwaslowered
forthehydrodynamicinjection(j)Theenlargedview(k)depictsthecouplingoftwocapillarieswithdifferentinnerdiametersintheconnectionsidewithoutdeadvolume
Asamplesolutioncontaining1mMferrocenemethanolanddecamethylferroceneinseparationbuffer(10mMCH COONH and1MCH COOHinacetonitrile)was3 4 3
utilizedTheinjectionwasperformedhydrodynamicallybyloweringtheoutletbuffer vial by 20 cm A uniform sample plug was injected to compare bandbroadeningeffectsTheinjectionsegmenthadalengthof035cm(05ofthetotalcapillarylength)andtherespectiveinjectiontimewasdeterminedbasedontheflowratesofthecorrespondingcapillarycombinationFortheelectrophoreticseparationaseparationvoltageof25kVwasappliedandtheinletandtheoutletbuffervialswereplacedatthesameheightsothattherewasnogravityflowwhichaffectedthemigrationbehaviour
3Resultsanddiscussion
31Capillaryflowinjectionanalysishyphenatedtocontactlessconductivitydetectionexperiments
AsshowninTab1theflowratesforaheightdifferenceof20cmwerecalculated4basedontheCFIA-C DmeasurementsItwasobservablethattheflowratesare
slightlylowerforfragmentedcapillariesthanfornon-fragmentedcapillariesofthe same dimension This indicates that a flow resistance arises when twocapillaries are combined Furthermore it was observed that the flow ratedecreasesforupstreamcapillarieswithlowerinnerdiametersandviceversaTheflow rate for the combination 25+75microm could not be determineddue to theformationofairbubblesattheconnectionside
10 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Table 1Flowratesandthecorrespondingstandarddeviations(SD4measurements)ofdifferentcapillarycombinationsforaheightdifferencebetweeninletandoutletvialof20cmbymeansofcapillaryflowinjectionanalysishyphenatedtocontactlessconductivitydetection
ndash1capillary flowratenLs plusmnSDflowndash1combinationmicrom ratenLs
25 00577 0000525+25 0057 000150+25 00775 0000775+25 ndash ndash25+50 0137 000550 0865 000250+50 080 00575+50 105 00625+75 0187 000250+75 1941 000375 456 00275+75 42 01
32Capillaryelectrophoresis-UVdetectionexperiments
Theelectropherogramsforthenon-fragmented50micromcapillaryandthecapillarycombinationswithdownstream50micromcapillaryarepresentedinFig3ThetwoferrocenederivatesdecamethylferroceneandferrocenemethanolwereusedasmodelanalytesDecamethylferrocenewasonlydetectedascationicspeciesasitiseasilyoxidizedbydissolvedoxygeninsolution Forthecombination50+50micromslightlyhighermigrationtimesforthecationic(decamethylferrocene) and neutral species (ferrocenemethanol) were obser-vable compared to thenon-fragmented50micromcapillaryThis indicateda flow
4resistanceattheconnectionwhichwasalsoobservedfortheCFIA-C Dexperi-mentsinsection31Incontrasttothecombinationwiththesameinnerdiameterastrongshift in themigration times for theneuralspecieswasvisible for thecombinationswithdifferentinnerdiametersThisshowedthatthecouplinghadaneffectontheelectroosmoticflow LookingatthepeakshapeitwasfoundthatallpeaksshowednearlyGaussianshape for all combinations Furthermore there was no tailing visible Theferrocenemethanolpeaks for thecombination25+50micromand75+50micromwere
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 11
Fig 3Electropherograms of themodelmixture ferrocenemethanol (FcMeOH) and decamethyl-ferrocene(DeMeFc)measuredwithanon-fragmentedcapillary(50microm)andfragmentedcapillaries(25+5050+50and75+50microm)Experimentalparameters1mMFcMeOHandDeMeFcinseparationbuffer(10mMCH COONH and1MCH COOHinacetonitrile)injectionsegment035cmseparation3 4 3
voltage25kVcapillarylength70cm(40cmtothedetectorfragmentedcapillaries20cmfirstpartand50cmsecondpart)UVdetectionat210nm
slightlybroaderthanthepeaksforthenon-fragmented50micromcapillaryorforthe50+50 microm combination But this probably results from longitudinal diffusioneffectsduetothelongerresidencetimes
4Conclusions
FromtheCFIAmeasurementsitcanbeconcludedthattherewasamechanicaldisturbanceoftheflowduetothecouplingFurthermoreitcouldbeshownthatCEmeasurementswithlinearcoupledcapillariesofvariousinnerdiameterwerepossible Unlike to the capillary combinationwith the same inner diameter astrongshiftoftheelectroosmoticflowtowardshighermigrationtimeswasfoundfor capillary combinations with different inner diameters In this work thecapillaries were coupled with almost no dead volume which resulted in nosignificantchangesofthepeakshapeorpeaktailingContrarytoexpectationsthecouplingofcapillarieswithvariousinnerdiametershadnosignificantimpactonthepeakwidth TheknowledgegainedfromthelinearcouplingofcapillariesisagoodbasisforthedevelopmentofthenewdualdetectionconceptInanextstepthreecapillariesshouldbecoupledwitheachother
Acknowledgments
WethanktheGermanResearchFoundation(DFG)forfinancialsupport
References
[1] OpekarFS tulıkKSomeimportantcombinationsofdetectiontechniquesforelectrophoresisincapillariesandonchipswithemphasisonelectrochemicalprinciplesElectrophoresis32(2011)795ndash810
[2] BeutnerAHerlTMatysikF-MSelectivityenhancement incapillaryelectrophoresisbymeans of two-dimensional separation or dual detection conceptsAnal Chim Acta1057(2018)18ndash35
4[3] BeutnerACunhaRRRichterEMMatysikF-MCombiningC DandMSasadualdetectionapproachforcapillaryelectrophoresisElectrophoresis37(2016)931ndash935
[4] MatysikF-MEnd-columnelectrochemicaldetectionforcapillaryelectrophoresisElectro-analysis12(2000)1349ndash1355
[5] MatysikF-MWernerGTracemetaldeterminationintearsbyanodicstrippingvoltammetryinacapillaryflowinjectionsystemAnalyst118(1993)1523ndash1526
[6] FranciscoKJMdoLagoCLAcompactandhigh-resolutionversionofacapacitivelycoupledcontactlessconductivitydetectorElectrophoresis30(2009)3458ndash3464
12 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Thetermldquovitaminsrdquodescribestheheterogeneousgroupofchemicalcompoundswhich are important for the proper functioning of the human body [1 2] Bydefinitionvitaminsarenotsynthesizedbythehumanbodyorthesynthesizedamount is not sufficient to cover the demand That is why they have to besupplementedfromtheexternalsourceslikefoodproductsorpharmaceuticals[1ndash3]Basedontheirsolubilityvitaminsaredividedintowater-soluble(B-groupandvitaminC)andfat-solublevitamins(ADEandK)[3]VitaminC(ascorbicacid) is the most important antioxidant and participates in the activation ofenzymes[4]VitaminB1(thiamine)facilitateswoundhealingandiscrucialforthehumannervoussystem[15]VitaminB2(riboflavin)participatesintheenzy-maticreactionsandthebiotransformationofglucoseandaminoacids[6]Vitamin
+B3(niacin)isthemainconstituentoftheNAD andNADHcoenzymeswhichareresponsible for the transfer of electrons and hydrogen ions in the cellularrespiration [1ndash3] Vitamin B6 possesses six related structures (vitamers) thateasilyinterconvertThemostimportantoneispyridoxinewhichhelpstopreventtongue inflammation and microcytic anemia [2] For the production of well-functioningredbloodcellsandtheavoidanceofmegaloblasticanemiaandfetusdefectsvitaminB9(folicacid)hastobesupplementedintheproperamount[12]
Voltammetric determination of vitamins
RADOSŁAWPORADABOGUSŁAWBAS
DepartmentofAnalyticalChemistryFacultyofMaterialsScienceandCeramicsAGHUniversityofScienceandTechnologyMickiewicza3030-059KrakoacutewPolandrporadaaghedupl
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 13
AbstractVitaminsbelongtothegroupofchemicalcompoundsessentialfortheproperfunctioningofthebodySinceboththeirdeficiencyandexcessmay result in serious health problems the amount of vitaminssupplementedinthedietaswellasvitamincontentintheirsourceshavetobestrictlycontrolledInthisworkthepossibilityofsimulta-neous determination of vitamins by means of differential pulseadsorptive stripping voltammetry is discussed The research hasshownthatthedeterminationofsingularvitaminatthemicromolarlevel isrelativelyfastandstraightforwardandthemostimportanthindranceisrelatedtotheanalyteadsorptionattheelectrodesurfaceInthecaseofvitaminswithdifferentredoxpotentialstheycanbeanalyzedsimultaneouslywithouttheneedtoreachfortheadvancedmethodsforsignalprocessing
Keywordsmercuryelectrodevitaminsvoltammetry
VitaminK3(menadione)doesnotoccurnaturallybutitservesasaprecursorforthesynthesisofotherK-groupvitaminsandcanbeusedtotreathypoprothrom-binemiaVitaminK3ispartiallysolubleinwater[17] All of the vitamins are electrochemically active [3] therefore the electro-chemicalmethodscanbeappliedforthedeterminationofvitamincontentinfoodproductspharmaceuticalsandbodyfluidsVoltammetrictechniquesarecharac-terizedbyhighsensitivityandselectivityandtheydonotrequiretime-consumingsamplepreparationMoreovertheelectrochemicalinstrumentationisrelativelyinexpensiveandcanbeappliedintheon-siteconditionsfortheonlineanalyseseginqualitycontrolMostofthepapersreporttheconstructiondevelopmentand characterization of a novel modified working electrodes for quantitativeanalyses of a singular vitamin in the variety of matrices Unfortunately only alimitednumberofpapersdescribethesimultaneousdeterminationofmultiplevitaminsinasinglerun[23] The preliminary research devoted to the simultaneous determination ofB-groupCandK3vitaminswiththeuseofthecontrolledgrowthmercurydropworking electrode in aqueous solutions is presented in this work Particularattentionhasbeenpaidtotheredoxpotentialsofthestudiedcompoundstheshape of the calibration curves and adsorption phenomena As an attempt toovercomethelattertheneutralsurfactantTritonX-100hasbeenintroducedintothestudiedsystem
2Experimental
21Reagentsandchemicals
TheappliedreagentswereofanalyticalgradeandusedassuppliedPhosphateand McIlvaine buffers were obtained by mixing the appropriate amount of
ndash1 ndash1 ndash102molL Na HPO with 02molL NaH PO and 01molL citric acid2 4 2 4
respectively (all reagents purchased from Avantor Performance MaterialsPoland)ThestandardsolutionsofvitaminB1B2B3B9andCwerepreparedbydissolving the corresponding amount of the standard (all Sigma-Aldrich) in
ndash1distilledwater In thecaseofB2andB9 theadditionof02molL NaOHwasinevitabletoobtainaclearsolutionVitaminK3standard(Sigma-Aldrich)was
ndash1dissolvedinthemixtureofmethanoland1molL phosphatebuffer(pH=82)(vv=25)LaboratorygradeTritonX-100(Sigma-Aldrich)wasusedinthestudyoftheadsorptionprocesses22Instrumentation
All the electrochemical measurements were conducted in the three-electrodesystem composed of the Pt auxiliary electrode double-junction silversilverchloride reference electrode and controlled-growth mercury drop electrode
14 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
actingastheworkingelectrodeTheusedmeasurementequipmentinvolvedtheM164electrodestandandM161multipurposeelectrochemicalanalyzer(bothmtm-anko Krakow) To control the buffer pH-value the SevenCompact S210laboratorypH-meter(MettlerToledoSwitzerland)wasemployed
23Voltammetricmeasurements
Throughout the course of the study differential pulse adsorptive strippingvoltammetry has been used for recording the current-potential curves BothcathodicandanodicscanswererecordedinapotentialrangeadjustedforthestudiedvitaminsTheinfluenceofvariousmeasurementconditionsontheregis-teredsignalshasbeeninvestigatedFinallythepossibilityofthesimultaneousdeterminationofmultiplevitaminsinonescanhasbeenverified
3Resultsanddiscussion
Figure 1 depicts the redox potentials of the studied vitamins in the aqueoussolutions for the mercury electrode The only exception is vitamin B6 whoseredox potential is higher than the potential of mercury oxidation (ca +02 V)Therefore the given value refers to the glassy carbon electrode The redoxpotentialvalueofstudiedvitamins isnotasingularvaluebut it fallswithinacertainrangeThiscanbeascribedtothedependenceofredoxpotentialonthesolutionpHvaluewhichresultsfromtheparticipationofprotonsintheredoxreactions of vitamins Moreover the potentials for individual vitamins arerelatively well separated indicating that the simultaneous determination ofseveralvitaminsintheonerunmaybepossibleTheonlyencounteredproblems
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 15
Fig 1Redoxpotentialsofchosenvitamins
regardthevitaminsB1B3andB12whoseredoxpotentialsoscillatebetweenndash15andndash17VandtheK-groupvitamins(K1K2K3)inwhichonlythecommonstructuralmotifndashthequinoneringndashiselectrochemicallyactiveresultinginthevalueoftheredoxpotentialofcandash02V[8] AtypicalcalibrationcurveisdepictedinFig2Abasedonthedifferentialpulsevoltammograms of the vitamin B9 reduction in the concentration range from
ndash1blank to 01 mg L recorded in the McIlvaine buffer of pH = 52 using thecontrolledgrowthmercurydropelectrodeworkingelectrodeTherelationshipbetweenthepeakcurrentandtheconcentrationofVB9 is linear in thewholetestedrange(r=09999)Basedontheparametersoftheregressioncurvethelimit of detection and limit of quantitation were estimated to 42 and
ndash1142nmolL respectively Similar dependencies and figures of merit can beobtainedforothervitamins QuitedifferentbehaviorwasobservedinthecaseofthevitaminK3forwhichthe increase in current was not strictly proportional to the increase in theconcentrationandthecalibrationplotresemblesanS-shapecurve(Fig2B)ThelatterindicatesthatvitaminK3adsorbsatthesurfaceoftheworkingelectrodeHoweverasnopre-orpost-peakwereobservedwearedealingherewiththeweakadsorption[9]AdsorptionalsoplaysasignificantroleinthecaseofvitaminB2forwhichboththepre-andpost-peakswereobservedindicatingitsstrongaffinitytothemercuryelectrodes To overcome this issue attempts with Triton X-100 were performed TritonX-100isaneutralsurfactant thateasilyadsorbsat thesurfaceof themercury
16 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2Differentialpulseadsorptivestrippingvoltammogramsof(A)vitaminB9and(B)vitaminK3ndash1reduction recorded in the concentration range from blank to 01 and from blank to 04 mg L
respectivelyInsetcorrespondingcalibrationcurvesSupportingelectrolyte(A)McIlvainebufferndash1(pH=52)(B)04molL phosphatebuffer(pH=82)
electrode partially blocking its surface Doing so it prevents the undesiredadsorption of other molecules and thus allows to obtain a linear relationshipbetweenthepeakcurrentandthevitaminconcentration(Fig3)UnfortunatelyduetotheblockingoftheelectrodesurfacetheslopesofthecalibrationlinesaresmallerincomparisontotheonesobtainedintheabsenceofanysurfactantsThismeans that the sensitivity defined as the increase in current caused by a unitincrease in concentration and the resolution understood as the possibility todistinguishsmallvariationinconcentrationarecorrespondinglydecreased Figure3alsopresentsthepossibilitytodeterminemultiplevitaminsinasinglerunChosenvitaminshavewell-separatedpotentialsandtheydonotinterferewitheachotherthereforenoadvancedmultivariatecalibrationstrategiesareneededTheproblemsinthesimultaneousanalysisincludevarioussensitivitieswith respect to the studied analytes and differences in the influence of themeasurementconditionsontherecordedsignalsDuetothattheexperimentalconditionswillneverensurethehighestpossiblesignalvaluesforallanalyzedcompounds
4Conclusions
Differential pulse voltammetry in conjunction with the controlled growthmercurydropelectrodeisaperfecttoolforquantitativeanalysesofvitaminsTheadsorptionofvitaminB2andK3canbepreventedbytheadditionoftheneutralsurfactantTritonX-100whichselectivelyblockstheworkingelectrodesurface
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 17
Fig 3Cathodic voltammograms for the simulatanous determination of vitamin B2 B3 and K3Depictedintheinsetsarethevoltammogramsafterbackgroundsubtractionwiththecorresponding
ndash1callibrationplotsSupportingelectrolye04molL phosphatebuffer(pH=82)with40ppmTritonX-100AccumulationconditionsE =ndash005Vt =20sacc acc
The proposed methodology allows for the simultaneous determination ofmicromolaramountsofvitaminB2B3andK3Suchaprocedurewillhelptoreduce the time and costs of analyses of multivitamin formulations and foodproducts
Acknowledgments
RPhasbeenpartlysupportedbytheEUProjectPOWR0302-00-00-I00416
References
[1] Combs GF JrTheVitaminsFundamentalAspects inNutritionandHealth 3rd ed IthacaElsevierAcademicPress2008
[2] Lovander MD Lyon JD Parr DL Wang J Parke B Leddy J Review Electrochemicalpropertiesof13vitaminsAcriticalreviewandassessmentJElectrochmSoc165(2018)G18ndashG49
[3] Brunetti B Recent advances in electroanalysis of vitamins Electroanalysis 28 (2016)1930ndash1942
[4] BrubacherGMuller-MulotWSouthgateDATMethods forDeterminationofVitamins inFoodNewYorkElsevier1985
[5] Szpikowska-Sroka B A simple and sensitive analytical method for the determination ofthiamineinpharmaceuticalpreparationsJAnalChem68(2013)218ndash222
[6] PetteysBJFrankELRapiddeterminationofvitaminB (riboflavin)inplasmabyHPLCClin2
ChimActa412(2011)38ndash43[7] ZhangZXuJWenYZhangJDingWTheelectro-syntesizedimprintedPEDOTfilmasa
simple voltammetric sensor for highly sensitive and selective detection of vitamin K in3
poultrydrugsamplesSynthMet230(2017)79ndash88[8] JedlinskaKStrusMBasBAnewelectrochemicalsensorwiththeRefreshableSilverLiquid
Amalgam Film multi-Electrode for sensitive voltammetric determination of vitamin K2(menaquinone)ElectrochimActa265(2018)355ndash363
[9] SouthamptonElectrochemistryGroupInstrumentalMethodsinElectrochemistryChichesterHorwood1985
18 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Carbonelectrodesarecommonlyappliedtosensitiveelectrochemicaldetectionofneurotransmittersegdopamine (nor)epinephrineandserotonin in-vivoandin-vitro[12]Neverthelessadsorptionofhigh-molecularweightbiomoleculesinthe matrix on a sensing electrode which then hinders the electron transferreactionofneurotransmitterswillresultinbiofoulingofelectrodesThisremainsachallengingproblemasbiofoulingwillcompromiseelectrochemicalmeasure-mentsThusseveralstrategies foraddressingbiofoulinghavepreviouslybeenreported[34] Thisworkreportsonaneffectiveapproachforminimisingbiofoulingbasedonthehypothesisthatahydrophobicelectrodesurfacewillrepelagainstadsorptionof amphiphilic biomolecules Briefly structurally small conical-tip electrodes
Dopamine detection at antifouling conical-tip carbon electrodes
a a a aSIMONABALUCHOVA JANKLOUDA JIR IBAREK KAROLINASCHWARZOVA -PECKOVA bDANNYKYWONG
a UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova812800PragueCzechRepublicsimonabaluchovanaturcunicz
b DepartmentofMolecularSciencesMacquarieUniversitySydneyNSW2109Australia
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 19
AbstractA significant achievement in this work is the development ofantifoulingconical-tipcarbonelectrodes(~27micromtipdiameterand~165micromaxiallength)suitablefordetectionoftheneurotransmitterdopamine in-vivo These electrodes were hydrogenated using adiphenylsilanereductionmethodtoyieldahydrophobicsurfacetodeteradsorptionofamphiphilicbiomoleculesInitiallyhydrogenatedcarbonelectrodeswereelectrochemicallycharacterisedusingseveralredoxmarkersThedegreeofantifoulingwasthenassessedbythevoltammetricsignalchangeofdopamineattheseelectrodesbeforeand after being incubated in a fouling solution containing bovineserumalbumincytochromeC(bothareproteins)andcaproicacid(alipid) In our work we have obtained only a 69 (standarddeviation35N=40)decreaseindopaminesignalsatthehydro-genated carbon electrodes These results strongly support thediphenylsilanereductionstrategyforthedevelopmentofantifoulingbiosensorsfordopaminedetectioninbiologicalmatrices
Keywordsantifoulingelectrodesdiphenylsilanereduction
methodhydrogenatedconical-tip
carbonelectrodesvoltammetricdopamine
detection
(denoted as CTEs) are fabricated by thermally pyrolysing acetylene gas in anitrogenatmospheretodepositcarbonatthetipandontheshankofpulledquartzcapillaries [5] Spectroscopic studies confirmed that the electrode surface
2 3consistsofsp -likegraphiticcarbonandsp -hybridiseddiamond-likecarbon[6]In addition there is also a range of carbon-oxygen functionalities includingcarbonylquinonecarboxylphenolsalcoholsandethergroupsontheelectrodesurface[6]whichcaninteractwithspectatorbiomoleculesthroughdipole-dipoleorion-dipoleinteractionleadingtotheirirreversibleadsorptionontheelectrodesurface[7]HoweverbysubjectingthesecarbonelectrodestosilanereductionCndashObondsareconvertedtoCndashHbondsandphenolicgroupsaretransformedtosiloxane dendrimers [6] to yield a more hydrophobic carbon surface that isexpectedtobesimilarlylesssusceptibletobiofoulingcomparedtoboron-dopeddiamondelectrodes[78] In this work we will present a methodology involving diphenylsilanereduction to fabricate physically small hydrogenated conical-tip carbonelectrodes(denotedasHCTEs)withanti-foulingcapabilityBothCTEsandHCTEswere electrochemically characterised using several redox probes to elucidatetheirsurfacepropertiesbeforeevaluatingtheirresistancetobiofoulingduringdopaminedetectionin-vitro
2Experimental
21Reagentsandchemicals
Analyticalgradereagents(Sigma-AldrichAustralia)including4-methylcatecholhexaammineruthenium(III) chloride potassium hexacyanoferrate(III) dop-amine hydrochloride sodium phosphate dibasic citric acid perchloric acidpotassiumchloride sodiumhydroxide anhydrousdichloromethanediphenyl-silanetris-(pentafluorophenyl)boraneandgraphitepowderwereusedas-recei-ved Ultra-high purity gases acetylene and nitrogenwere obtained from BOCGases (Australia) All aqueous solutionswere preparedwith deionisedwater(MilliporeMiliplusQsystemUSA)witharesistivityof182MΩcm
22Instrumentation
Chronoamperometric and voltammetric experiments were carried out usingalow-currentpicostateDAQoperatedbyanEChemversion212softwareviaanE-corderinterface(eDAQPtyLtdAustralia)Athree-electrodeset-upinvolving
minus1eitheraCTEorHCTEasaworkingelectrodeaAgAgCl(3molL KCl)referenceelectrode (Bioanalytical SystemsUSA)andaplatinumwire counterelectrode
minus1(CypressSystemsUSA)wasusedCyclicvoltammetryatascanrateof100mVs anddifferential pulse voltammetry (pulse height +25mV pulsewidth 50ms
minus1sampling time 20 ms and scan rate 20mVs ) were used in this work All
20 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
electroanalyticalexperimentswereperformedinanaluminiumFaradaycageatanambienttemperature(23plusmn1degC)
23Preparationofhydrogenatedconical-tipcarbonelectrodes
Asreportedpreviously[5]structurallysmallCTEswerefabricatedbythermallypyrolysing C H (a pressure of 50 kPa) in a pulled quartz capillary (Sutter2 2
minus1InstrumentUSA)housedinaN atmosphere(counterflowof60mLmin )Prior2
to hydrogenation the catalyst tris(pentafluorophenyl) borane (100 mg) wasdissolvedinanhydrousCH Cl (50mL)bystirringfor5minbeforethehydroge-2 2
natingagentdiphenylsilane(25μL)wasaddedCTEswerethenplacedinthereagentmixturefor2hThepreparedHCTEsweredriedovernightbeforeuse
24Biofoulingexperiments
A laboratory synthetic fouling solution consisting of 4 (wv) bovine serumalbumin001(wv)cytochromeC(bothareproteins)and10(vv)caproicacid(alipid)waspreparedbyhomogenisingtheminapH=74citrate-phosphate
minus1buffer(01molL )AllfoulingcompoundswereacquiredfromSigmaAldrichAustralia
3Resultsanddiscussion
31Electrochemicalcharacterisation
minus1InthisworkallCTEswerecharacterisedbycyclicvoltammetryof10mmolL 3+ minus1[Ru(NH ) ] in10molL KClAsdisplayedinFig1(A)onlyCTEsthatshow3 6
asigmoidal-shapedvoltammogramwithasmallchargingcurrentwereemployedinfurtherexperimentsUsingchronoamperometry[5]ameantipdiameterof27μm(standarddeviation(SD)28μmN=142)ameanaxiallengthof165μm(SD=114μmN=142)wereestimatedfortheseCTEs TocomparesurfacecharacteristicsofbothCTEsandHCTEs cyclicvoltam-
minus1 3+2+ minus1 minus1metryof (1)10mmolL [Ru(NH ) ] in10molL KCl (2)10mmolL 3 63minus4minus minus1 minus1[Fe(CN) ] in 10 mol L KCl and (3) 10 mmol L 4-methylcatechol in6minus101molL HClO wasconductedatthesameelectrodesbeforeandafterhydroge-4
nation The results obtained are shown in Fig 1(A-C)We observed a ~20(SD=5N=10)decreaseinthelimitingcurrentofallthreeredoxmarkersafterdiphenylsilane reduction most likely attributable to the hindrance to theirelectron transfer reactions by the phenylsiloxane group formed on HCTEs
3minus4minusMoreover as an inner-sphere redox probe both [Fe(CN) ] and 4-methyl-6
catechol reactionsare sensitive to thepresenceofoxygen functionalitiesonacarbonsurface [7]Accordingly theconversionof these functionalities toCndashHbondsbydiphenylsilanereductionwasexpectedtoyieldmoresluggishelectron
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 21
transfer kinetics at HCTEs as supported by a negative potential shift (from3minus4minus+75mVtominus10mV)inthecyclicvoltammogramof[Fe(CN) ] andapositive6
potentialshift(from+580mVto+675mV)inthecorrespondingcyclicvoltammo-2gram of 4-methylcatechol In addition the conversion of sp -carbon to
3sp -diamond-likecarbon[6] isalsoexpectedtoreducetheconductivityof thecarbonelectrodesurface
32Dopaminedetectionduringbiofoulingexperiments
minus1Theelectrochemicalbehaviourof1mmolL dopamine inapH=74citrate-minus1phosphate buffer (01 mol L ) at CTEs and HCTEs was studied by cyclic
voltammetryTheresultsobtainedareshown inFig1(D)Acomparable12decrease(SD=6N=10)inthedopamineoxidationlimitingcurrenttothatof4-methylcatechol was observed A positive potential shift from +285 mV to+305mVinthevoltammogramsisalsoaccountedforasdescribedabove
22 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
minus1 3+ minus1 minus1Fig 1Cyclicvoltammetryof(A)10mmolL [Ru(NH ) ] in10molL KCl (B)10mmolL 3 63minus4minus minus1 minus1 minus1[Fe(CN) ] in10molL KCland(C)10mmolL 4-methylcatechol in01molL HClO at3 4
minus1(a)aCTEand(b)aHCTE(D)10mmolL dopamineinapH=74citrate-phosphatebufferrecordedminus1at(a)aCTEandaHCTE(b)beforeand(c)afterbiofoulingScanrate100mVs
Next HCTEs were incubated in a synthetic fouling solution containing4(wv)bovineserumalbumin001(wv)cytochromeC(bothareproteins)and 10 (vv) caproic acid (a lipid) for 30 min Cyclic voltammetry of
minus110mmolL dopamineattheseHCTEswasthenconductedinapH=74citrate-phosphatebuffertoevaluatetheirantifoulingpropertyNotablyaconsiderable515(SD=183N=6)decreaseindopaminesignalwasobservedatCTEsIncontrastonlyacorresponding69decrease(SD=35N=40)wasestimatedatHCTEsasshowninFig1(D)Clearlythisrepresentsamajorimprovementinthe antifouling capability of HCTEs obtained using diphenylsilane reductioncomparedtoCTEsandotherpreviouslytestedhydrogenatingagents[9]includingn-butylsilane (35decrease) triethylsilane (23decrease) andphenylsilane(18decrease)Thereforethisworkhassuccessfullydemonstratedtheeffecti-venessofdiphenylsilanereductionmethodindevelopingantifoulingelectrodesfordopaminedetection
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 23
Smallconical-tipelectrode
As-prepared Hydrogenated
minus1LinearrangemicromolL 1ndash10 1ndash20minus2 minus2Intercept10 pAmicrom 108plusmn004 273plusmn011
minus3 minus2 minus1Slope10 pALmicrom micromol 128plusmn007 443plusmn012R 0993 0998
minus1LimitofdetectionmicromolL 100 077
Table 1Analyticalparametersofconcentrationdependencesofdopamineobtainedbydifferentialpulse
minus1voltammetryinapH=74citrate-phosphtebuffer(01molL )Allquoteduncertaintiesrepresentthe95confidenceintervalandthecorrelationcoefficient(R)wasfoundtobestatisticallysigni-ficantatthe95usingStudentrsquost-test
Fig 2DifferentialpulsevoltammetryofdopamineataHCTEinapH=74citrate-phosphatebufferminus1 minus1(01molL )atconcentrations(a)1(b)2(c)4(d)6(e)8(f)10and(g)20μmolL
WehavealsostudiedtheconcentrationdependenceofdopamineinapH=74citrate-phosphatebufferbydifferentialpulsevoltammetryAtypicalcalibrationplotobtainedisshowninFig2Theanalyticalparametersandestimatedlimitsofdetection are summarised inTable 1 These results show thatHCTEs outper-formed CTEs because they exhibit a ~35times higher sensitivity a 23 lowerdetectionlimitandawiderlinearrange
4Conclusions
InthisstudyphysicallysmallHCTEswithanti-foulingcharacteristicsachievedbyhydrogenationusingdiphenylsilanereductionweresuccessfullyfabricatedandelectrochemically characterised using several redox probes Next dopaminedetectionwasperformedbeforeandafterincubationofelectrodesinasyntheticfoulingsolutioncontainingahighconcentrationofbiomoleculesOnlyalow69(SD=35)decreaseindopaminelimitingcurrentwasachievedatHCTEsobtain-edbydiphenylsilanereductionindicatingtheirsignificantlylesssusceptibilitytobiofoulingthanCTEsThesepromisingresultsindicatethatantifoulingHCTEswillpotentiallybenefitthedevelopmentofbiosensorsfordopaminedetectionin-vivoinbiologicalmedia
Acknowledgments
ThisresearchwasperformedwithintheframeworkofSpecificUniversityResearch(SVV260560)FinancialsupportsprovidedbytheGrantAgencyofCharlesUniversity(project390119)andbytheCzechScienceFoundation(project20-03187S)aregratefullyacknowledgedSBandJKalsothankthe Mobility Fund of Charles University and Hlavkova nadace for providing funding for theirresearchinternshipsatMacquarieUniversitySydneyAustralia
References[1] BaranwalAChandraPClinicalimplicationsandelectrochemicalbiosensingofmonoamine
neurotransmittersinbodyfluidsinvitroinvivoandexvivomodelsBiosensBioelectron121(2018)137ndash152
[2] CaoQPuthongkhamPJillVentonBReviewnewinsightsintooptimizingchemicaland3Dsurface structuresof carbonelectrodes forneurotransmitterdetectionAnalMethods11(2019)247ndash261
[3] LinP-HLinB-RAntifoulingstrategiesinadvancedelectrochemicalsensorsandbiosensorsAnalyst145(2020)1110ndash1120
[4] HanssenBLSirajSWongDKYRecentStrategiestoMinimiseFoulinginElectrochemicalDetectionSystemsRevAnalChem35(2016)1ndash28
[5] McNallyMWongDKYAnin-vivoprobebasedonmechanicallystrongbutstructurallysmallcarbonelectrodeswithanappreciablesurfaceareaAnalChem73(2001)4793ndash4800
[6] SirajSMcRaeCRWongDKYEffectiveactivationofphysicallysmallcarbonelectrodesbyn-butylsilanereductionElectrochemCommun64(2016)35ndash41
[7] ParkJShowYQuaiserovaVGalliganJFinkGDSwainGMDiamondmicroelectrodesforuseinbiologicalenvironmentsJElectroanalChem583(2005)56ndash68
[8] ShinDTrykDAFujishimaAMerkociAWang JResistance to surfactantandproteinfoulingeffectsatconductingdiamondelectrodesElectroanalysis17(2005)305ndash311
[9] Roshni RAnAntifouling Structurally Small Carbon Electrode forDetectionof theNeuro-transmitterDopaminePhDThesisMacquarieUniversitySydney2019
24 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Theanalyticalperformancesofenzymaticbiosensorsarestronglyaffectedbytheenzyme immobilization process There is no universal technique for enzymesattachmentThereforespecialattentionshouldbepaid to theselectionof theappropriatesupportandthedevelopmentoftheoptimalbindingstrategyinordertoensure thebestcharacteristicsof immobilizedenzymeDespiteavarietyofpreviouslyreportedcovalentimmobilizationmethodsfordifferentenzymesthepresentedprocedurescanbehardlycomparedtofindtheoptimalonesbecauseofdifferentanalyticalmethodsandexperimentalconditionsusedUptodatethere
A comparative study of covalent glucose oxidase and laccase immobilization techniques at powdered supports for biosensors fabrication
ab a bSOFIIATVORYNSKA JIR IBAREK BOHDANJOSYPCUK
a UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova2030812843Prague2CzechRepublicsofiiatvorynskajh-instcascz
b JHeyrovskyacuteInstituteofPhysicalChemistryoftheCzechAcademyofSciences Dolejškova318223Prague8CzechRepublic
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 25
AbstractInordertodeveloptheoptimalstrategyandtodeepentheknowledgeinthefieldofenzymeimmobilizationthreedifferenttechniquesofcovalentbindingfortwoenzymes(glucoseoxidaseandlaccase)atpowdered surfaces were compared Immobilization protocol wasoptimized by changing supports (twomesoporous silica powders(SBAminus15 MCMminus41) and a cellulose powder) the functionalizedgroupsintroducedatsupportsurfaces(minusNH andminusCOOH)andthe2
methodsofactivation(glutaraldehydeandcarbodiimide)Aminoandcarboxyl functionalized mesoporous silica and cellulose powderswerepreparedbysilanizationusing(3-aminopropyl)triethoxysilaneandcarboxyethylsilanetriolrespectivelyItwasfoundthatcouplingof both enzymes by their ndashNH groups through glutaraldehyde to2
ndashNH functionalized supports in particular SBA15minusNH and2 2
celluloseminusNH forglucoseoxidaseMCM41minusNH forlaccaseshowed2 2
thehighestactivityandthebeststability
Keywordsbiosensorcovalentimmobilizationenzymaticreactorglucoseoxidaselaccase
is still a lack of the comparative systematic studies focusing on the enzymesimmobilizationonthevarioussupportsusingdifferenttechniques The aim of this work is the systematic comparative study of the differenttechniques for covalent coupling of the enzymeswhich ensures not only thedevelopmentoftheoptimalimmobilizationstrategyfortheselectedenzymesbutalsoenablestofindoutsometendenciesinenzymeattachmentprocessgenerallyThusthisworkisfocusedonadetailedanalysisoftheeffectofthekindofsupportits anchor groups and the activation methods on activity and stability ofimmobilizedenzymesTwoenzymeswithdifferentnature(glucoseoxidase(GOx)andlaccase(Lac))werechosenasthetestingbioreceptors
2Experimental
21Reagentsandchemicals
AllchemicalswereofpaorbettergradeGlucoseoxidasefromAspergillusnigerminus1(GOxEC11341452Umg )laccasefromTrametesversicolor(LacEC11032
minus1129Umg )D-(+)-glucosedopamineglutaraldehyde(GAgradeII25aqueoussolution) N-(3-dimethylaminopropyl)-Nʹ-ethylcarbodiimide hydrochloride(EDC ge980) N-hydroxysuccinimide (NHS ge970) (3-aminopropyl)-triethoxysilane (APTES) mesoporous silica powder SBAminus15 (particle size
2 minus12ndash6μmporesizeasymp7nmsurfaceareaasymp600m g )mesoporoussilicapowder2 minus1MCMminus41 (pore size 21ndash27 nm surface area asymp 1000 m g ) cellulose (Cell
microcrystalline powder particle size 20 microm) were purchased from SigmaAldrichCarboxyethylsilanetriol(CEST25aqueoussolution)waspurchased
regfromabcr (Germany)
22Instrumentation
Amperometric measurements were carried out at room temperature usingcomputer-controlled electrochemical stand (Polaro-Sensors Czech Republic)withMultiElchemv31software(JHeyrovskyInstituteofPhysicalChemistryoftheCAS)Flowinjectionanalysis(FIA)withthethree-electrodelaboratory-madeflow-through cellwas usedworking electrode minus tubular detector of polishedsilversolidamalgam(TD-p-AgSAlaboratory-madeinnerdiameter05mmtheamalgamtube length60mm) referenceelectrodeminusaminiaturizedsaturatedcalomelelectrodebasedonsilverpasteamalgam[1](laboratory-madeithasthesamepotential as classical saturated calomel electrode) auxiliary electrodeminusplatinum wire (diameter 10 mm length 10 mm) The system for FIA withelectrochemicaldetectioncomprisedofalinearsyringepumpa2-position6-port
regsampleinjectorvalveaninjectionlooplaboratory-madeofTeflon (PTFE)tubing(100μL) a solenoid operatedmicro-pumpan enzymatic reactor and a flow-through cell for TD The enzymatic reactor consists of a tube filled by theenzymaticpowder
26 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
3Resultsanddiscussion
Basedonthedatareportedintheliterature[23]andontheresultsofourpreviousworks[4ndash8]forthisstudyCellandmesoporoussilicapowders(namelySBAminus15andMCMminus41)havebeenselectedas thepotentialpromisingsupports for thecovalent enzyme immobilizationBecauseof thehigh contentof surficial ndashOHgroupswhich are capable of chemical reactions these supports can be easilyfunctionalizedThewell-knownandfrequentlyutilizedtechniqueofsilanizationhasbeenusedtomodifythesurfacesofSBA15MCMminus41andCellbythedesiredfunctionalizedgroupsAminosilaneAPTESwasappliedtoformminusNH groupson2
thematrixsurfaceswhereascarboxylsilaneCESTwasusedtointroducendashCOOHgroups Generallytheprocedureofthecovalentimmobilizationofenzyme(eitherLacorGOx)onthefunctionalizedsupportconsistsofthreestepsI Synthesisofthefunctionalizedsupportwhichmeansthemodificationofthematrix(MCMminus41SBAminus15andCell)withsuitableanchoredgroups(minusNH or2
minusCOOH)II Activationstepofthefunctionalizedsupportwithspecificactivatingagents(glutaraldehydeorEDCNHS)tomakeitreactivetowardsenzyme
IIIEnzyme(LacorGOx)couplingtotheactivatedsupport
To investigate the effect of support its surface functionalizedgroups and themethodsofactivationontheefficiencyofthecovalentenzymeimmobilizationthreedifferentstrategies(ABandC)forLacandGOxattachmenthavebeenusedThedetailsoftheusedtechniquesandthedenotationsofthepreparedenzymaticpowdersaresummarizedinTable1(nextpage)ToexaminetheefficiencyofLacandGOximmobilizationtheenzymaticreactors(filledbytheenzymaticpowderspreparedwithdifferenttechniques)coupledwithTDwereusedforamperometricdetermination of dopamine and glucose respectively in flow systems Theprincipleofglucosedetection isbasedonamperometricmeasurementsof theenzymatically consumed oxygen whereas dopamine was detected by thereductionoftheenzymaticallyoxidiseddopamine Asdepicted inFig1 thebiosensors responsesare stronglyaffectedby thestrategyusedforLacorGOximmobilizationAsshowntheresponsesofLacandGOx biosensors decrease in the order strategy A gt strategy B gt strategy Cirrespectiveofthetypeofsupport It isclearlyseenthataminofunctionalizedsupports(SBA15minusNH MCM41minusNH andCellminusNH )providehigheractivitiesof2 2 2
the immobilized Lac andGOx than these supports functionalized by carboxylgroups(SBA15minusCOOHMCM41minusCOOHandCellminusCOOH)BycomparingactivitiesofimmobilizedenzymesusingstrategiesAandBtheinfluenceoftheactivationagenthasbeenevaluatedThebestresultsforbothenzymeswereobtainedforndashNH functionalizedsupportsactivatedbyGAItcouldbeexplainedbythefact2
thatGAcontrary tocarbodiimidewithnonemolecularspaceprovidesa long
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 27
spacerarmensuringminimalsterichindrancesforenzymesbindingItcanbeconcludedthatthecovalentimmobilizationofbothenzymesbytheirndashNH groups2
viaGAtondashNH functionalizedmesoporoussilicapowders(strategyA)provided2
the highest activities Interestingly in the similar comparative studies it isreportedthatamongndashOHminusCOOHandndashNH functionalizedsupportsactivatedby2
divinylsulfonecarbodiimideandGArespectivelythelastonewasfoundasthemostsuitabletechniqueforthecovalentbindingofLac[9]invertase[10]andpepsin[11]
28 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Tab
le 1
Theprinciplesofthecovalentimmobilizationmethodsofenzymesusedinthisstudy
StrategyA
StrategyB
StrategyC
Support
SBAminus15MCMminus41Cell
SBAminus15MCMminus41Cell
SBAminus15MCMminus41Cell
Supportfunctio-
minusNH
minusNH
minusCOOH
22
nalizedgroup
Activationagent
Glutaraldehyde(GA)
Carbodiimide
Carbodiimide
(EDCNHS)
(EDCNHS)
Enzymereactive
minusNH
minusCOOH
minusNH
22
group
Typeofbond
secondaryamine
amide
amide
Denotationsofthe
GOxminusNHminusCHminus(CH)minusCHminusNHminusSBA15
GOxminusC(=O)minusNHminusSBA15
GOxminusNHminusC(=O)minusSBA15
23
preparedenzy-
GOxminusNHminusCHminus(CH)minusCHminusNHminusMCM41
GOxminusC(=O)minusNHminusMCM41
GOxminusNHminusC(=O)minusMCM41
23
maticpowders
GOxminusNHminusCHminus(CH)minusCHminusNHminusCell
GOxminusC(=O)minusNHminusCell
GOxminusNHminusC(=O)minusCell
23
LacminusNHminusCHminus(CH)minusCHminusNHminusSBA15
LaxminusC(=O)minusNHminusSBA15
LacminusNHminusC(=O)minusSBA15
23
LacminusNHminusCHminus(CH)minusCHminusNHminusMCM41
LacminusC(=O)minusNHminusMCM41
LacminusNHminusC(=O)minusMCM41
23
LacminusNHminusCHminus(CH)minusCHminusNHminusCell
LacminusC(=O)minusNHminusCell
LacminusNHminusC(=O)minusCell
23
When the effect of the method of the covalent enzyme coupling on thebiosensorstabilitywasevaluateditwasfoundthatLacboundedtondashNH functio-2
nalizedsupportsviaGA(strategyA)hasshownthehigheststability(gt65oftheinitial responses after 1 month) compared to other strategies whereas GOximmobilizedwithtwostrategies(AandB)possessedapproximatelysimilarhighstability(gt80oftheinitialresponsesin1month)BothenzymesboundedviandashNH groupstondashCOOHfunctionalizedsupportsthroughEDCNHS(strategyC)2
showedquitelowstability
4Conclusions
Threedifferent strategies including the support selection the anchor surfacegroups and the activationmethod havebeen compared for efficient covalentimmobilization of Lac and GOx The results showed that ndashNH functionalized2
supports(SBA15minusNH CelluloseminusNH forGOxandMCMminusNH forLac)activatedby2 2 2
GAmaybeusedtoeffectivelybindenzymesintermsofhighactivityandstability
Acknowledgments
ThisworkwasfinanciallysupportedbytheGrantAgencyofCharlesUniversityinPrague(Project1356120)theGrantAgencyoftheCzechRepublic(Project20-07350S)anditwascarriedoutwithintheframeworkofSpecificCharlesUniversityResearch(SVV260440)
References
[1] YosypchukBBarekJYosypchukOPreparationandpropertiesofreferenceelectrodesbasedonsilverpasteamalgamElectroanalysis23(2011)2226minus2231
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 29
Fig 1Effectof the covalent attachment techniqueson (A) laccaseand (B) andglucoseoxidaseminus1biosensor responses Experimental conditions (A) c = 500 micromol L E = minus50 mVDOP det
minus1 minus1v = 01 mL min V = 40 microL carrier solution 01 mol L acetate buffer pH = 48flow DOPminus1 minus1(B)c =500micromol L E =minus1100mVv ==01mLmin V =40microL carrier solutionGlu det flow Glu
minus1 minus101molL acetatebuffer0001molL Na EDTApH=652
[2] LiuYChenJYEnzymeimmobilizationoncellulosematrixesJBioactCompactPolym31(2016)553ndash567
[3] Hartmann M Kostrov X Immobilization of enzymes on porous silicas ndash benefits andchallengesChemSocRev42(2013)6277minus6289
[4] JosypcukOBarekJJosypcukBElectrochemicalbiosensorsbasedonenzymaticreactorsfilledbyvarioustypesofsilicaandamalgampowdersformeasurements inflowsystemsElectroanalysis28(2016)3028minus3038
[5] Josypcuk O Barek J Josypcuk B Amperometric determination of catecholamines byenzymaticbiosensorsinflowsystemsElectroanalysis30(2018)1163minus1171
[6] TvorynskaSBarekJJosypcukBAmperometricbiosensorbasedonenzymaticreactorforcholinedeterminationinflowsystemsElectroanalysis31(2019)1901minus1912
[7] TvorynskaSBarekJJosypcukBFlowamperometricbiosensorbasedontwoenzymaticreactors (acetylcholinesterase-choline oxidase) for the detection of neurotransmitteracetylcholine In Proceedings of the 15th International Students Conference ldquoModernAnalyticalChemistryrdquoKNesmerak(ed)PragueFacultyofScienceCharlesUniversity2019p61minus66
[8] TvorynskaSBarekJJosypcukBAcetylcholinesterase-cholineoxidase-basedmini-reactorscoupledwithsilversolidamalgamelectrodeforamperometricdetectionofacetylcholineinflowinjectionanalysisJElectroanalChem860(2020)113883
[9] RekucAKruczkiewiczPJastrzembskaBLiesieneJPeczynska-CzochWBryjakJLaccaseimmobilizationonthetailoredcellulose-basedGranocelcarriersIntJBiolMacromol42(2008)208minus215
[10] Bryjak J Liesiene J S tefuca V Man-tailored cellulose-based carriers for invertaseimmobilizationCellulose15(2008)631minus640
[11] SzałapataKOsinska-JaroszukMBryjakJJaszekMJarosz-WilkołazkaANovelapplicationofporousandcellularmaterialsforcovalentimmobilizationofpepsinBrazJChemEng33(2016)251minus260
30 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Carbohydrates are crucial for energy structure and signaling in the humanbody[1]Thereisavarietyofcarbohydratesbutthemostimportantoneforlifeisglucoseasitisfundamentalinthemetabolismandphotosynthesis[2]GlucoseisclassifiedashexoseThesemonosaccharidesonlydifferinthepositionofhydroxylsubstituentsinsomecasesInadditiontothestructuralsimilaritiesthesemole-culeslackachromophoreandarenoteasilyionizable(pK ~12)Thusdetectiona
intheUVregionandseparationofanalytesbycapillaryelectrophoresis(CE)arechallenging [1 3] At the moment there are many different techniques for theanalysis of carbohydrates commonly including time-consuming derivatizationstepsoreluentswithhighpHvalues(pHgt12)inionchromatographyAwell-esta-blishedtechniquefortheanalysisofcarbohydratesishigh-performanceanion-exchangechromatographywithpulsedamperometricdetection(HPAE-PAD)[4]Electrochemical detection like AD is matching miniaturization simple instru-mentationlowcostandrobustnessandthusisoftenusedforflow-basedsystemssuchasCEandflowinjectionanalysis(FIA)[5]
Capillary flow injection analysis with electrochemical detection for carbohydrate analysis
NICOLEHEIGLFRANK-MICHAELMATYSIK
InstituteofAnalyticalChemistryChemo-andBiosensorsFacultyofChemistryandPharmacyUniversityofRegensburgUniversitaumltsstraszlige3193053RegensburgGermanynicoleheiglchemieuni-regensburgde
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 31
AbstractAsimplecapillaryflowinjectionanalysissystemwithamperometricdetection was arranged for the development of a method for fastoptimization of detection conditions in the context of thedetermination of carbohydrates by means of electrochemistry-capillary electrophoresis-mass spectrometry This setup is free ofelectricalinterferencebyhighvoltageandisperfectforstudyingtheoxidationofvariousanalytesFurthermoreitassureseasycouplingtoMS and thus is an useful tool to investigate the correspondingoxidationproductsofananalyte
Keywordscapillaryflowinjection
analysiscarbohydratesmassspectrometrydisposableelectrodespulsedamperometric
detection
Inthiscontributioncapillaryflowinjectionanalysis(CFIA)withADwillbepresentedasamethodtoapplyandtestADforthedetectionofmonosaccharidesondifferentdisposablethin-filmorscreen-printedelectrodesCFIAwaschosenoverconventionalFIAforthispurposeasthegravityflowinCFIAisstableforalongertimeandverylowsampleconsumptioncanbeachieved[6]TheCFIAsystemwasarrangedassimpleaspossibleandperformedhydrodynamicallytoavoidanyinterferencesFurthermoreitassuresthecouplingoftheflowsystemtoamassspectrometerThusthesamesetupasusedforCFIAcanbeutilizedforcapillaryelectrophoresis-massspectrometry(CE-MS)experimentsbychangingthe flow through the electrochemical flow cell in opposite direction In futureexperimentstheexperienceintermsofADonthoseelectrodeswillbeusedtodevelopelectrochemicalpretreatmentprotocolsforcarbohydratedeterminationbyCE-MS
2Experimental
21Reagentsandchemicals
The following chemicals were used for this study all of analytical gradeAmmoniumacetate(NH OAc)wasobtainedfromMerck(DarmstadtGermany)4
and ferrocene methanol (FcMeOH) from ABCR (Karlsruhe Germany) Milli-Qregwater(182MΩcm)wasgeneratedbyaMilli-QAdvantageA10 system(Merck
Millipore Darmstadt Germany) Carrier solution was prepared by dissolvingndash1NH OAc (50mmolL ) in Milli-Q water FcMeOH solution was prepared by4
dissolvingFcMeOHincarriersolution
22Instrumentation
Electrochemical measurements were performed using a microAutolab Type IIIpotentiostatgalvanostat (Metrohm Autolab B V Utrecht Netherlands)controlledbyNOVA20softwareforexperimentalcontrolanddataacquisitionCFIAwasperformedusingthesetupillustratedinFig1(A)consistingofacarrierreservoirsamplevialandtwofusedsilicacapillaries(PolymicroTechnologiesPhoenix AZ USA inner diameter 100 microm length inlet 40 cm length outlet10cm)connectedtoacommerciallyavailableflowcellfromMicruxTechnologies(model ED-FLOW-CELL Oviedo Spain) Inside of the flow cell the fused silicacapillary was placed in a so-called wall-jet configuration above the workingelectrode of a disposable thin-film gold electrode (model ED-SE1-Au MicruxTechnologiesOviedoSpain)ascanbeseeninFig1(B)Thethin-filmelectrodeswerebasedonathree-electrodesystemwithagoldworkingauxiliaryandquasi-referenceelectrodeThehydrostaticpressurewasachievedbyaheightdifferencebetweeninletandoutletreservoirof30cmresultinginagravityflowofthecarriersolutionthroughafusedsilicacapillaryandsubsequentlythroughtheflowcell
32 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Theinjectionwascarriedoutbyloweringthevialcontainingthecarriersolutiontotheleveloftheoutletexchangingthecarrierreservoirwiththesamplevialandliftingthesamplevialto20cmforadefinedperiodoftimeRe-establishingthecarrierreservoirtookplacethesameway
3Resultsanddiscussion
AsimpleCFIA-ADsystemwasarrangedwherehydrostaticpressurebyaheightdifferencebetweeninletandoutletreservoirresultedinagravitationalflowToobtain general information about the behavior of the assembled CFIA systempreliminaryexperimentswithFcMeOHwereperformedToassurecompatibilitywithMSlateronNH OAcwaschosenastheelectrolyteVariousheightdifferences4
andinjectiontimesweretestedandtheinjectionataheightdifferenceof20cmlasting for 10 s was found to be the optimum concerning feasibility and peakshapes Injections of several solutions of FcMeOH of different concentrationsshowed that the concentration dependence of FcMeOH was linear in theinvestigatedrange(Fig2)Furthermoreexperimentsrevealedthattheinjectionprocedure was established with reasonable precision When repeating the
ndash1injectionof05mmolL FcMeOHincarriersolutionfortentimestherelativestandarddeviationwasfoundtobe3forthemanualinjectionprotocol
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 33
Fig 1(A)Schemeoftheusedcapillaryflowinjectionanalysissetup(1)carrierreservoir(2)samplevial(3)inletcapillarywithalengthof40cmandaninnerdiameterof100microm(4)outletcapillarywithalengthof10cmandaninnerdiameterof100micrombothcapillariesconnectedto(5)acommerciallyavailableflowcellfromMicruxand(6)awastevial(B)Configurationinsidetheflowcell(7)thefusedsilicacapillarywasplacedinaso-calledwall-jetconfigurationabovetheworkingelectrodeof(8)adisposablethin-filmgoldelectrode
4Conclusions
The presented system for CFIA-AD was arranged as simple as possible andrepresents a useful approach for the development of a method for fastoptimization of detection conditions in the context of the determination ofcarbohydratesbymeansofelectrochemistry-CE-MSThesetupisfreeofelectricalinterferencebyhighvoltagecompatiblewithMSandthuspromisingforstudyingtheoxidationofvariousanalytes
References
[1] LuGCrihfieldCLGattuSVeltriLMHollandLACapillaryelectrophoresisseparationsofglycansChemRev118(2018)7867ndash7858
[2] GalantALKaufman RCWilson JDGlucoseDetectionandanalysisFoodChem188(2015)149ndash160
[3] Sarazin C Delaunay N Costanza C Eudes V Gareil P Application of a new capillaryelectrophoreticmethodforthedeterminationofcarbohydratesinforensicpharmaceuticalandbeveragesamplesTalanta99(2012)202ndash206
[4] Rohrer JS Basumallick L Hurum D High-performance anion-exchange chromatographywithpulsedamperometricdetectionforcarbohydrateanalysisofglycoproteinsBiochem78(2013)697ndash709
[5] IslamMAMahbubPNesterenkoPNPaullBMackaMProspectsofpulsedamperometricdetectioninflow-basedanalyticalsystemsndashAreviewAnalChimActa1052(2019)10ndash26
[6] MatysikF-MWernerGTracemetaldeterminationintearsbyanodicstrippingvoltammetryinacapillaryflowinjectionsystemAnalyst118(1993)1523ndash1526
34 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2ndash1 ndash1(A) CFIA-AD recordings of three consecutive injections of (1) 025mmolL (2) 05mmolL
ndash1 ndash1 ndash1(3) 075mmolL and (4) 1mmolL FcMeOH in 50 mmolL NH OAc detection at a Micrux4
thin-filmAuelectrodeataconstantpotentialof03VinaflowcellHydrodynamicinjectionlasted10sataheightdifferenceof20cm(B) Calibration dependence of FcMeOH for CFIA-AD determination and detection at a Micruxthin-filmAuelectrodeataconstantpotentialof03VinaflowcellThestandarddeviationsofpeakheights(n=3)areindicatedbyerrorbars
1Introduction
Topreventsorptiononthecapillarysurfaceandimproveseparationefficiencyand selectivity of determined analytes coatings are formed on the capillarysurface There are two types of ones namelydynamic and covalently bondedcoatings Despite the simplicity of creation dynamic coatings cannot providerequiredreproducibilitywhilecovalentcoatingscontributethestableelectro-osmoticflow(EOF)andhighreproducibilityoftheanalysisInmostcasesthe
Application of covalent coatings based on imidazolium cations for separation and on-line preconcentration of basic and neutral analytes in capillary electrophoresis
a ab aANASTASIAVKRAVCHENKO EKATERINAAKOLOBOVA LIUDMILAAKARTSOVA
a DepartmentofOrganicChemistryInstituteofChemistrySaintPetersburgStateUniversity 26Universitetskiiprospect198504StPetersburgPeterhofRussiakravchenko161216gmailcom
b TheFederalStateInstituteofPublicHealthldquoTheNikiforovRussianCenterofEmergencyandRadiationMedicinerdquoTheMinistryofRussianFederationforCivilDefenceEmergenciesandEliminationofConsequencesofNaturalDisasters54Optikovst197082StPetersburgRussia
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 35
AbstractThemethodofcapillaryelectrophoresis (CE) isactivelydevelopedandmoreandmoreattractsscientistsattentioneveryyearHoweverthesorptionofanalytesonsurfaceoffused-silicacapillarywallsisoneof thesignificantdisadvantagesof thisapproachThe formationofcoatings on the inner capillary surface is typical way to preventsorption and to increase separation efficiency and selectivity ofdetermined analytes Coatings that covalently bonded to capillarywalls is more suitable because they are stable and provides highreproducibilityofanalysisThepresentworkisfocusedonthedevelo-pmentofthemethodofelectrophoreticdeterminationofbiologicalactiveanalytesusingacovalentcoatingbasedonimidazoliumcationsTheeffectofsubstituentinimidazoliumringonmainelectrophoreticparameters was examined It was shown that alkylimidazoliumcoatingscontributetosignificantreducingofbiogenicamineslimitsof detection while β-cyclodextrinimidazolium covalent coatingallowstoseparatebothofhydrophobicandhydrophilicanalytesinonerun
Keywordsbiologicalactiveanalytescapillarycoatingcapillaryelectrophoresisimidazoliumionicliquids
analytes nature determines type of usedmodifiers because suitable ones canprovideaccessorial interactionbetweentheanalytesandthestationaryphaseimproving separation selectivity and efficiency [1 2] Ionic liquidshave beenwidelyusedinanalyticalchemistry[3]andseparationtechniquesparticularlyincapillaryelectrophoresis[4]Earlyresearches[5ndash9]haveshownopportunityofcovalently bonded imidazolium ionic liquids for electrophoretic separationHowever the effect of various substituents in imidazolium ring hasnot beendescribed previously Thus the purpose of this study was to create covalentcoatingsbasedon ionic liquidwithvarioussubstituentsand tocompare theiranalytical capabilities in the electrophoretic separation of biologically activecompounds
2Experimental
21Reagentsandchemicals
(3-Glycidyloxypropyl)trimethoxysilane (GPTMS) hydrochloric acid sodiumdodecyl sulfate (SDS) imidazole 22-diphenyl-1-picrylhydrazyl (DPPH)p-toluen-sulfonylchlorideβ-cyclodextrinhydrocortisone(F)11-deoxycortisol(S)Corticosterone(B)rac-ketoprofen(ndash)-adrenaline(A)L-(ndash)-norepinephrine(NE) DL-normetanephrine (NMN) dopamine (DA) DL-metanephrine hydro-chloride(Met)serotoninhydrochloride(Ser)homovanillicacid(HVA)24-di-hydroxy-benzoic acid (24-DHBA) 34-dihydroxy-L-phenylalanine (DOPA)L-tryptophan(Trp)L-tyrosine(Tyr)werepurchasedfromSigma-Aldrich(USA)1-Bromo-butane1-bromooctanewerepurchasedfromReagentPlus(Ukraine)Sodium dihydrogenphosphate dihydrate acetone NN-dimethylformamide(DMF)wereobtainedfromMerck(Germany)AllreagentsusedwereanalyticalgradeAllsolutionswerepreparedusingdeionizedwater
22Instrumentation
Capillary electrophoresis experiments were carried out using the system ofcapillaryelectrophoresisCAPEL-105M(LumexRussia)withUV-spectrophoto-metricdetector(wavelengthrange190ndash360nm)Separationswereperformedusing 58times49 cm (9 cm to the detector outside diameter 360 microm and innerdiameter50microm) coated silica capillaries (LumexRussia) Thebuffer pHwasmeasuredwithapH-meterHI2210ndash2216(Hanna)
23Capillarycoatingsynthesis
Earlier our research team has proposed the synthesis route for the covalentcoatingsbasedonimidazoliumcationfunctionalizedwithalkylgroup[10]andβ-cyclodextrin [11] All capillarieswere prepared according above-mentioned
36 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
manuscriptsandcharacterizedbytheEOFmobilitymeasurementandscanningelectronmicroscopyThesynthesisconsistedoffollowingstepspreparationofacapillarytocreateacovalentcoating(heatingcapillaryfilledwith2MNaOHat90degCfor1handdryingfollowed)silylationwithGPTMSandfunctionalizationwiththeimidazolesolutionfollowedmodificationbybutyl-andoctylbromideortosyl-β-cyclodextrin(seedetailsin[10]and[11])
24Solutions
A stock buffer solution in concentration 50mM was prepared by dissolvingappropriate amount of sodium dihydrogenphosphate dihydrate in deionizedwateradjustingpHto20with1MhydrochloricacidThisbuffersolutionwasthendilutedwithdeionizedwater
ndash1 Allthesamplestocksolutionswerepreparedwithconcentration10mgmL Thestocksolutionsoftheneurotransmittersandtheirmetabolites(adrenalinenoradrenaline dopamine normetanephrine metanephrine serotonin homo-vanillicacid)and24-dihydroxybenzoicacidasinnerstandardandaminoacids(tryptophan34-dihydroxy-L-phenylalanine tyrosine)wereprepared in01MhydrochloricacidThestocksolutionsofsteroids(hydrocortisone11-deoxycor-tisolandcorticosterone)wereprepared inacetonitrileThestocksolutionsofketoprofen racemate and S-ketoprofen were prepared in acetonitrilewatersolution(1090vv) Untilelectrophoreticanalysisthestocksolutionswerestoredatndash16degCTheworkingsolutionswerepreparedbydilutingtheinitialsolutionswithwaterjustbeforetheexperiments
3Resultsanddiscussion
CovalentcoatingsbasedonN-alkylimidazoliumcationwereespeciallysuitableforseparationofneurotransmittersandtheirmetabolites(Fig1)Inadditionthecombinationofcovalentcoatingwithon-linepreconcentrationtechniquesallowstothesignificantdecreaseoftheseanalyteslimitsofdetection(LOD)Accessorialinteractions positively charged analytes with positively charged imidazoleimproveseparationselectivity(viaπ-πinteraction)andefficiency(concentrationintightzonesviaelectrostaticrepulsion)Sodiumdodecylsulfate(SDS)addedintobackgroundelectrolyte(inconcentrationabovecriticalmicelleconcentra-tion) strongly interacts with hydrophobic alkyl groups in covalent coatingstructureThenegativelychargedSDSlayerisformedoninnercapillarysurfaceThedoublereversingEOFallowsustocarryoutelectrokineticinjectionofsampleandon-linepreconcentrationbysweepingsimultaneouslyLODweredeclineto
ndash108ndash20ngmL ThelengthofalkylsubstituentalsoaffectsthestackingefficiencyfactorandLODMorehydrophobicoctylgroupscomparetobutylprovidemoreeffectiveinteractionwithSDSandasresultlowerLOD
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 37
Covalentcoatingmodifiedβ-CDhasnotshownsharpreducingofLODbyon-linepreconcentrationStacking sweeping (SDSasmicelle reagent) field-enhancedsample injection were examined using different model mixtures of analytesNeverthelessthiscoatingallowssimultaneousseparationofbothofhydrophobicsteroidhormonesandhydrophilicbiogenicaminesinasinglerun(Fig2) The guest-host interaction hydrophobic cavity of β-cyclodextrin with thehydrophobicsteroids leadsto the formationofcomplexwhichaffectssteroidselectrophoretic mobility At the same time β-cyclodextrin can act as a chiralselector and baseline separation of ketoprofen enantiomers has also beenachieved
38 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Electropherogramofmixtureof neurotransmitters and theirmetabolitesadrenaline (A)norepinephrine (NE)normetanephrine (NMN)dopamine (DA)metanephrine (Met) serotonin(SER)homovanillicacid(HVA)andtheinnerstandard24-dihydroxybenzoicacid(24-DHBA)oncovalentlymodifiedwithN-buthylimidazoliumionicliquidscapillaryConditions10mMNaH PO 2 4
(adjusted to pH= 20 by 1MHCl) injection 50 stimes30mbar ndash20 kV 220 nmmodelmixurendash1 ndash1 ndash110microgmL (METADNMNNADA24-DHBA)5microgmL (SER)and20microgmL (HVA)
4Conclusions
ItwasshownthatstructurecovalentcoatingaffectsitsanalyticalcharacteristicsWecomparedtwotypesofcovalentcoatingdifferingsubstituentinimidazoliumringnamelyalkylgroupandβ-cyclodextrinThefirsttypeisgreatcoupledwithon-line preconcentration technic but it is limited to effectively determine ofbiogenicaminesonlywhilethesecondtype(withβ-cyclodextrin)showedthepossibilitiestoseparatevariousanalytesbutsuitableon-linemodehasnotbeenfoundThemainpointsaresummarizedinTable1
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 39
Covalentcoatingtype Electrophoreticseparationof On-lineprecon- centration biogenicamines amino steroid ketoprofen andtheirmeta- acids hormones enantiomers bolites N-β-cyclodextrinimida- yes yes yes yes thesuitableapproachzoliumcovalentcoatings wasnotfoundedN-alkylimidazolium yes yes nonsepa- nonsepa- thesignificantreducingcovalentcoatings rated rated forbiogenicaminesLOD
Table 1Thesummationofpossibilitiesofcovalentcoatingsbasedonimidazoliumcation
Fig 2Electropherogramofsimultaneousseparationofhydrophobic(steroidhormones)andhydro-philicanalytes (aminoacidsandbiogenicamines) insinglerunwithcovalentcoatingbasedonimidazoleandβ-CDConditions10mMNaH PO (adjusted topH = 20by1MHCl) injection2 4 20 stimes30mbarndash20kV254nm(1ndash8min)and220nm(8ndash15min)0mbar(1ndash10min)and40mbar(10ndash15 min) Model mixture corticosterone (B) hydrocortisone (F) 11-deoxycortisole (S)
ndash1 ndash15μgmL L-tryptophan(Trp)34-dihydroxy-L-phenylalanine(DOPA)10μgmL L-tyrosine(Tyr)ndash15 μg mL noradrenaline (NA) normetanephrine (NMN) adrenaline (AD) dopamine (DA)
ndash120μgmL
Acknowledgments
This work was supported by Russian Science Foundation (grant numbers 19-13-00370) Theauthors are also grateful to the Chemistry Education Centre and Nanothechnologies Centre ofResearchParkSaintPetersburgStateUniversityfortechnicalsupport
References
[1] HuLFYinSJZhangHYangFQRecentdevelopmentsofmonolithicandopen-tubularcapillaryelectrochromatography(2017ndash2019)JSepSci43(2020)1942ndash1966
[2] KartsovaLAKravchenkoAVKolobovaEACovalentcoatingsofquartzcapillariesfortheelectrophoretic determination of biologically active analytes J Anal Chem 74 (2019)729ndash737
[3] HoTDZhangCHantaoLWAndersonJLIonicliquidsinanalyticalchemistryFundamen-talsadvancesandperspectivesAnalChem86(2014)262minus285
[4] TangSLiuSGuoYLiuXJiangSRecentadvancesofionicliquidsandpolymericionicliquids incapillaryelectrophoresisandcapillaryelectrochromatography JChromatogrA1357(2014)147ndash157
[5] QinWLiSFYElectrophoresisofDNAinionicliquidcoatedcapillaryAnalyst128(2003)37ndash41
[6] QinWWeiH Li SFY 13-Dialkylimidazolium-based room-temperature ionic liquids asbackgroundelectrolyteand coatingmaterial in aqueous capillaryelectrophoresis JChro-matogrA985(2003)447ndash454
[7] QinW Fong S Li Y Determination of ammonium andmetal ions by capillary electro-phoresisndashpotential gradient detection using ionic liquid as background electrolyte andcovalentcoatingreagentJChromatogrA1048(2004)253ndash256
[8] QinWLiSFYAn ionic liquidcoating fordeterminationofsildenafilandUK-103320 inhumanserumbycapillaryzoneelectrophoresis-iontrapmassspectrometryElectrophoresis23(2002)4110ndash4116
[9] BorissovaMVaherMKoelMKaljurandMCapillaryzoneelectrophoresisonchemicallybondedimidazoliumbasedsaltsJChromatogrA1160(2007)320ndash332
[10] KolobovaEKartsovaLKravchenkoABessonovaEImidazoliumionicliquidsasdynamicand covalent modifiers of electrophoretic systems for determination of catecholaminesTalanta188(2018)183ndash191
[11] KravchenkoAKolobovaEKartsovaLMultifunctioncovalentcoatingsforseparationofaminoacidsbiogenicaminessteroidhormonesandketoprofenenantiomersbycapillaryelectrophoresisandcapillaryelectrochromatographySepSciplus3(2020)102ndash111
40 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Synthetic 4-hydroxy-3-methoxybenzaldehyde (vanillin) is used as a flavoringagent in foodsdrinksperfumesandpharmaceuticals [1]However at certainconcentrationsthesubstancemayaccumulateinthebodyhaveatoxiceffectand
ndash1at high concentrations may be fatal (lethal dose LD (oral rat) =2gkg 50ndash1 ndash1LD (oral guinea pig) = 14gkg LD (intravenous dog) = 132gkg lethal50 50 ndash1concentrationLC (inhalationmouse)=417gkg )[2]AccordingtoRussianState
StandartGOST121005-88thetoxiceffectsofvanillinintheworkplaceinthendash3formofvapoursoraerosolsareobservedatconcentrationsabove15mgm
Chromatography[3]spectrophotometry[4]capillaryelectrophoresisareusedforvanillindeterminationindifferentobjects CurrentlysmokingmixturesforhookahsandelectroniccigarettesarewidelyusedamongyoungpeopleThesemixturesarenotcontrolledforthecontentofsubstancesandarefreelyavailableconsideringthemmoreharmlesswithrespecttoordinarycigarettesThusthedevelopmentofamethodforthedeterminationof4-hydroxy-3-methoxybenzaldehydeinsmokingmixturesisrelevant
Determination of vanillin in smoking mixtures by spectrophotometry
ELIZAVETAEFREMENKOANNACHERNOVAOLGABASTRYGINA
DepartmentofChemicalEngineeringNationalResearchTomskPolytechnicUniversityLeninavenue30634050TomskRussiaeaetpuru
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 41
AbstractTheresearchdealswithdeterminationofvanillin insmokingmix-turesbyultraviolet-visiblespectrophotometryThemethodshowed
ndash1goodlinearityintherangeof005ndash012gL withalimitofdetectionndash1005gL After validation studies the method was successfully
applied to thedeterminationof vanillin in smokingmixtureswithsatisfactoryresultsItwasshownthattheerrorofthismethoddoesnot exceed 1 The developed spectrophotometric procedure fordeterminingvanillininsmokingmixturescanbeusedasacontrol
Keywordssmokingmixturesspectrophotometryvanillin
2Experimental
21Reagentsandchemicals
Asampleofvanillin(purity98)wastakenastheobjectofstudyAssolventsweused95ethanolAllchemicalsusedwereofanalyticalreagentgrade
22Instrumentation
Theopticaldensityofsampleswasmeasuredincuvettewithanabsorbinglayerthickness of 10 mm using a Cary 60 spectrophotometer (Agilent USA) Allmeasurementswerecarriedoutatroomtemperature
23Samplepreparation
Sample preparation of the investigated objects consisted of the preliminarydissolutionofthesamplein95ethanolThesample10mgoftobaccoldquoAdalyandashVanillardquo(Turkey)wasdiluted in10μLof95ethanol to theconcentrationof
ndash11gL Thesample10μLofldquoFlavoringTPAndashVanillaCustardrdquo(USA)wasdilutedin10 μL of 95 ethanol The resulting solution was diluted six times to the
ndash3concentrationof017μLcm
3Resultsanddiscussion
TodeterminevanillininthesamplestheopticalpropertiesofvanillininvarioussolventsweredeterminedAsaresultthe95ethanolwaschosenastheoptimalsolvent[4] IthasbeenestablishedthatintheUVspectraoftheanalyteabsorptionbondsareobservedwithmaximumvaluesat23002800and3100nmwhichcorres-pondstopublisheddata[45](Fig1) To quantify vanillin the calibration curve of the optical density on theconcentrationofvanillin in95ethanolwasobtainedatconcentrations005
ndash1006007008010and012gL Calibrationcurveofvanillinin95ethanolatawavelengthof280nmis
ndash1 A =81914c[gL ]+00357 (1)2802 R =1
Calibrationcurveofvanillinin95ethanolat310nmis
ndash1 A =73824c[gL ]+00301 (2)3102 R =1
42 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Inthespectraoftheanalyzedsamplesolutionsabsorptionmaxima(2800nmand3100nm)characteristicforvanillinwereobservedTheamountofvanillininthesamplewasdeterminedusingcalibrationcurvesat280and310nmWeightedleast square regressionwas applied to the calibration curves to improve theaccuracyespeciallyatinlowconcentrationlevelrangeGoodlinearitywasfound
ndash1 ndash1intherangeof005ndash012gL withadetectionlimitof005gL TheresultsarepresentedintheTable1
4Conclusions
ThedevelopedmethodcanbeusedasacontrolmethodTheerrorinthemethodfordeterminingvanillininthesampleldquoFlavoringTPAndashVanillaCustardrdquowithaknownconcentrationofvanillinwas0004Accordingtothedataobtainedwerecommendawavelengthof280nmforthedeterminationofvanillininsamples
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 43
λ nm
Absorban
ce
ndash1Fig 1 Absorption spectrum of vanilin solution in 95 ethanol at concentration 01 mol L (anabsorbinglayerthicknessof10mm)
Sample λnm Tookmg Foundmg S Sх Δх δ
FlavoringTRA 310 10850 104096 00024 00011 00006 00306ndashVanillaCustardAdalyandashVanilla 310 100000 10162 00019 00009 00002 00025
FlavoringTRA 280 100300 96062 00033 00015 00032 00042ndashVanillaCustardAdalyandashVanilla 280 100000 10122 00013 00006 00012 00017
Table 1Testingmethods introduced foundof vanillin in the samplesat310nmby spectrophotometricmethod(n=5p=099SndashstandarddeviationSxndashrelativestandarddeviationΔхndashabsoluteerrorδndashrelativeerror)
ThenthedevelopedmethodwastastedonthesampleldquoAdalyandashVanillardquosamplewithamorecomplexcompositionandanunknownconcentrationofvanillinwastaken The vanillin content in the sample was determined according to thedevelopedmethoditamountedto10ofthetotalmassStudieshaveshownthepossibility of using spectrophotometric analysis for the qualitative andquantitative determination of vanillin Also based on preliminary studies aspectrophotometricprocedurewasdevelopedforthequantitativedeterminationofvanillinbasedonabsorptioninethanolinthewavelengthrange200ndash400nm
References
[1] httpswwwrusnaukacom43_DWS_2015Chimia6_203179dochtm (accessed 25thFebruary2019)
[2] httpswwwcdcgovnioshrtecsdefaulthtm(accessed11stApril2020)[3] AliLPerfettiGDiachenkoGRapidmethodforthedeterminationof342coumarinvanillin
and ethyl vanillin in vanilla extract by reversed-phase liquid 343 chromatography withultravioletdetectionJAOACInt91(2008)383ndash386
[4] БастрыгинаОАЕфременкоЕАЧерноваАПВыделениеванилинаисследованиеегооптическихсвои ствопределениевбиологическомматериалеВХимияихимическаятехнология в XXI веке Материалы XX Международной научно-практическойконференции имени профессора ЛП Кулёва студентов и молодых ученых ТомскНациональныи исследовательскии Томскии политехническии университет 2019с301ndash302
[5] WeastRCHandbookofChemistryandPhysics60thedBocaRatonCRCPress1979p143
44 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
UraniumbelongstothegroupofhazardouselementsItisahighlyharmfulandradioactiveelementtoxictohumansandalllivingorganisms[12]Inhaledwithair it has a particularly destructive effect on the kidneys and as a result ofaccumulationinwhitebloodcellsitcanalsocauseimpairmentoftheimmunesystem[2]Uraniumoccursatseveraldegreesofoxidationhoweverinaqueoussolutionsthemoststableformisuranylion(UO (II))[12]Thepresenceofura-2
niumintheenvironmentiscausedbyamongothersnaturalsoilandrockerosionEnvironmentalpollutionwiththiselementisalsoconstantlyincreasingduetohumanactivitycoalcombustionuraniumoreminingandprocessingthearmsindustryandtheuseofuraniumasnuclearfuelinfissionreactors[3]Itisveryimportanttoconstantlymonitortheconcentrationofuraniumbothinthenaturalenvironment in order to assess its state and safety (especially in the case ofdrinkingwater)aswellasinallstagesofprocessingprocessesassociatedwiththenuclearindustrytoavoidtheoccurrenceofnuclearpollution[13] Scientists have made many attempts to develop research methods todeterminethecontentofuranylcompoundsinliquidsamplesEffortsweremadetousemanyanalyticalmethodsforthispurposeincludingspectrophotometry
Uranyl ion-selective electrode with solid contact
KAROLINAPIETRZAKCECYLIAWARDAK
DepartmentofAnalyticalChemistryInstituteofSciencesFacultyofChemistryMariaCurie-SklodowskaUniversityMariaCurie-SklodowskaSq320-031LublinPolandkarolinapietrzakpocztaumcslublinpl
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 45
AbstractNewallsolidstateuranylion-selectiveelectrodeswithlowdetection
ndash7 ndash1limits(71times10 molL )shortresponsetimegoodselectivityandstable and reproducible potential were developed Many types ofelectrodeswith different active ingredient content in ion-selectivemembrane (bis(244-trimethylpentyl)phosphonium acid Cyanex-272)were testedAs an additive an ionic liquid1-octyl-3-methyl-imidazole chloride was used The optimal composition of theion-selective membrane was chosen from all electrodes based onthedeterminationand comparisonof analyticalparametersof thesensors
Keywordsion-selectiveelectrodesolidcontacturanyl
plasma spectrometry luminescence spectroscopy voltammetry or chromato-graphymethods[2] Duetomanyadvantagesofpotentiometricmethods(amongthemlowercostseasieroperationofdevicesquickresponseandtheabilitytoperformmeasure-ments in flowmode) [3] a numberof potentiometric sensorshave alsobeendeveloped that could be successfully used in this type of research Themostpopularpotentiometricsensorsincludeion-selectiveelectrodes(ISEs)whicharecharacterized by low-energy consumption small size and portability and aresuccessfullywidelyusedforthedeterminationofbothinorganicandorganicionsinclinicalanalysisprocesstechnologyaswellasincontrolthestateofthenaturalenvironment[45]Removaloftheinternalsolutioncontainingthesameanalytetowhich theelectrode is sensitiveresulted in theso-calledsolidcontact ISEswhicharemuchsmallerinsizethantheirpredecessorsaremoreconvenienttouse and more mechanically resistant In this type of sensors however it isimportanttoachievesatisfactorypotentialstabilitywhichisnecessarytoobtainsatisfactoryresults[5]AveryimportantpartofISEsistheion-selectivemem-branewhosecompositiondeterminestheanalyticalparametersofthesensorsResearchers are currently focusing on the production and testing of newsubstancesthatcouldbesuccessfullyusedasmembranecomponentsandsolidcontacts thatwould allow to obtain new sensorswith lower detection limitslongerlifetimeandbetterpotentialstabilityandtodeterminenewpreviouslyunattainableanalytes[4] AstheactivecomponentsofthemembranesensitivetouranylionscientistshavealreadyusedKryptofix22DD(413-didecyl-171016-tetraoxa-413-diaza-cyclooctadecane)[2]Cyanexextractants(bis(244-trimethylpentyl)phosphinicacid bis(244-trimethylpentyl)monothiophosphinic acid and bis(244-tri-methylpentyl)dithiophosphinic)acid[3]DBBP(dibutylbutylphosphonate)andDOPP (di-n-octyl phenylphosphonate) [6] DMSO (dimethylsuphoxide) [7]TTPTP (5678-tetrahydro-8-thioxopyrido[4345]thieno[23-d]pyrimidine-4(3H)one)[8]orTEHP(tris(2-ethylhexyl)phosphate)andTPTU(O-(12-dihydro-2-oxo-1-pyridyl)-NNNN-bis(tetra-methylene)uronium hexafluorophos-phate)[9]
2Experimental
21Reagentsandchemicals
This paper presents research on the design and properties of ion-selectiveelectrodes with solid contact for the determination of uranyl ions Bis(244-trimethylpentyl)phosphonium acid (Cyanex-272) was used as the activecomponentof themembranewhichwasdescribed in the literatureasagooduranylextractant[10]Inordertoensureaconstantpotentialofthiselectrodeandreducetheelectroderesistancetheion-sensitivemembranewasenrichedwithafewpercentadditionof1-octyl-3-methylimidazolechlorideionicliquid
46 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Several types of ion-selective electrodes were prepared using an AgAgClelectrodeasaninternalelectrodewhichdifferinthequantitativeandqualitativecompositionofthemembranesAllcompositionsarelistedinTable1
22Instrumentation
Measurements were made at room temperature using a 16-channel datacollectionsystem(LawsonLabs IncUSA)coupled toa computer in solutionsmixedwithamechanicalstirrerAsilversilverchlorideelectrodewithdoublejunctionwasusedasthereferenceelectrode
3Resultsanddiscussion
The effect of ion-selective membrane composition on the properties of theobtained potentiometric sensors was examined by determining their basicanalyticalparametersincludingslopeoftheelectrodecharacteristicsdetectionlimitmeasuringrange(concentrationrangeinwhichthecourseoftheelectrodecharacteristics isrectilinear)pHrange(inwhich ithasnoeffect forelectrodepotential)andresponsetimeTheobtainedvaluesofthetestedparametersareshowninTable2 Figure1showsthecalibrationcurvesofthetestedelectrodesdeterminedin
ndash7 ndash1 ndash1UO (NO ) solutionsintheconcentrationrange1times10 ndash1times10 molL Asitcan2 3 2
beseeninFig1andTable2allelectrodesweresensitivetouranylionsbutindifferent extend The best response exhibited ISE-3 containing 1 (ww) ofionophore Increasing the ionophore content in themembrane shortened thelinearityrangeofthecalibrationcurveanditssupernenstianslope Theselectivityofthetestedelectrodeswasestimatedbydeterminingtheselec-tivitycoefficients inrelationto interfering ionsForthispurpose theseparate
ndash1solutionmethodwasused(extrapolatingresponsecurves toa =a =1molL )i j
ComparisonofISE-1andISE-3electrodeselectivityisshowninFig2
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 47
Table 1Quantitative and qualitative composition of electrode membranes Cyanex-272 (bis(244-tri-methylpentyl)phosphoric acid) TBP (tri-n-butyl phosphate) and OMImCl (1-octyl-3-methyl-imidazolechloride)
Abbreviation Membranecomposition(ww)ofelectrode Cyanex-272 PVC TBP OMImCL
ISE-1 00 33 620 5ISE-2 05 33 615 5ISE-3 10 33 610 5ISE-4 30 33 590 5ISE-5 50 33 570 5ISE-6 100 33 520 5
48 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Abbreviation Slope Detectionlimit Linearrange Response pHrange2+ ndash1 ndash1ofelectrode mVpa(UO ) molL molL times2
ndash5 ndash5 ndash1ISE-1 297 25times10 5times10 ndash1times10 5ndash8 28ndash42ndash6 ndash5 ndash1ISE-2 292 65times10 1times10 ndash1times10 5ndash8 25ndash60ndash7 ndash5 ndash1ISE-3 298 71times10 1times10 ndash1times10 5ndash8 24ndash60
ndash6 ndash4 ndash1ISE-4(I) 357 31times10 5times10 ndash1times10 5ndash8 ndndash6 ndash6 ndash4ISE-4(II) 242 31times10 5times10 ndash5times10 5ndash8 nd
ndash3 ndash1ISE-5(I) 638 nd 1times10 ndash1times10 5ndash10 ndndash5 ndash3ISE-5(II) 234 nd 5times10 ndash1times10 5ndash10 ndndash3 ndash1ISE-6(I) 733 nd 1times10 ndash1times10 5ndash10 ndndash5 ndash3ISE-6(II) 222 nd 5times10 ndash1times10 5ndash10 nd
Table 2Selectedparametersandtheirdeterminedvaluesoftestedionselectiveelectrodes
Fig 1 Calibration curves of the testedelectrodesobtainedinUO (NO ) solutionsin2 3 2 ndash7the concentration range from 1times10 to
ndash1 ndash11times10 molL
Fig 2Comparisonofselectivitycoefficientspot(log K (UO (II))M) for electrodes ISE-12
(1stcolumn)andISE-3(2ndcolumn)
Inordertoexaminethereversibilityofthepotentialofthetestedelectrodesndash4 ndash1potentialmeasurementsweremadealternatelyinsolutions1times10 molL and
ndash5 ndash11times10 molL ofUO (NO ) TherecordedpotentialreadingsareshowninFig32 3 2
Long-term potential stability and sensor reproducibility were evaluated byndash1determiningtheaveragevalueoftheelectrodepotentialina01molL UO (II)ion2
solutionovertimeforthreeidenticalISE-3Thesemeasurementsweremadetoobservechangesinthepotentialofelectrodeswiththesameconcentrationoveralongperiodoftime(30days)Figure3showsthelong-termpotentialstabilityandreproducibilitydeterminedforthreeidenticalsensors
4Conclusions
Asaresultofthetestsion-selectiveelectrodeforthedeterminationofuranylionswasobtainedwhich iseasy todesignanduseThebestanalyticalparametersexhibitedISE-3containing1ionophoreintheion-selectivemembraneForthis
ndash7 ndash1typeofelectrodesthedetectionlimitof71times10 molL linearityoftheelectrodendash6 ndash1 ndash1calibrationcurve in the range1times10 ndash1times10 molL andresponse time5ndash8s
were obtained In addition the manufactured sensors also showed stablereproducibleandreversiblepotentialandverygoodselectivityinrelationtothetestedinterferents
References
[1] AnsariRMosayebzadehZConstructionofanewsolid-stateU(VI)ion-selectiveelectrodebasedonpolypyrroleconductingpolymerJRadioanalNuclChem299(2014)1597ndash1605
[2] GhanbariMRounaghiGHAshrafNAnuranylsolidstatePVCmembranepotentiometricsensor based on 413-didecyl-171016-tetraoxa-413-diazacyclooctadecane and itsapplicationforenvironmentalsamplesIntJEnvironAnalChem97(2017)189ndash200
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 49
Fig 3 Stability () reproducibility andreversibility () of the potential of ISE-3Standard deviations given on the plot aredeterminedforthesamethreeISE-3
[3] Badr IHA Zidan WI Akl ZF Cyanex based uranyl sensitive polymeric membraneelectrodesTalanta118(2014)147ndash155
[4] BiegCFuchsbergerKStelzleMIntroductiontopolymer-basedsolid-contaction-selectiveelectrodes basic concepts practical considerations and current research topics AnalBioanalChem409(2017)45ndash61
[5] Bobacka J IvaskaA LewenstamA Potentiometric ion sensorsChem Rev108 (2008)329ndash351
[6] ZidanWI Badr IHA Akl ZF Development of potentiometric sensors for the selective2+determinationofUO ionsJRadioanalNuclChem303(2015)469ndash4772
[7] SalehMBSolimanEMGaberAAAAhmedSANovelPVCmembraneuranylion-selectivesensorSensActuatorsB114(2006)199ndash205
[8] SalehMBHassanSSMAbdelAAAbdelNAAnoveluranylion-selectivePVCmembranesensor based on 5678-tetrahydro-8-thioxopyrido[4345]thieno[23-d]pyrimidine-4(3H)oneSensActuatorsB94(2003)140ndash144
[9] HassanSSMAliMMAttawiyaAMYPVCmembranebasedpotentiometricsensorsforuraniumdeterminationTalanta54(2001)1153ndash1161
[10] Prabhu DR Ansari SA Raut DR Murali MS Mohapatra PK Extraction behaviour ofdioxouranium(VI) cation by two phosphorous-based liquid cation-exchangers in room-temperatureionicliquidsSepSciTechnol52(2017)2328ndash2337
50 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Metronidazole(2-methyl-5-nitroimidazole-1-ethanol)isoneofthemostwidelyused nitroimidazole antibiotics Metronidazole is used for the treatment ofinflammatorydiseasescausedbyanaerobicorganismsandsomeprotozoaandforpreventionofdysenterycolibacillosiseimeriosisbalantidiasissalmonellosisenteritissepticemiapost-surgicalcomplications[1ndash3]Oxytetracyclinehydro-chlorideisanantibioticofthetetracyclinefamilyItisoneofthemostcommonlyused antibiotics in poultry because of its low cost and effective [4] Thesecompounds are intensively used in poultry breeding and stockbreedingUnreasonableuseofthesedrugscancauseseriousfoodsafetyissues[5] The veterinary drug Nozemat which includemetronidazole and oxytetra-cyclinehydrochloridewaschosenfortheexperimentsNozematisusedtotreat
Polarographic determination of metronidazole and oxytetracycline hydrochloride in veterinary drug for honey bees
a a bKATERYNAPLOTNIKOVA LILIYADUBENSKA IVANZELENYI
a AnalyticalChemistryDepartmentIvanFrankoNationalUniversityofLvivKyrylaiMefodiaStr879005LvivUkrainekaterina27plgmailcom
b DrohobychPedagogicalLyceumIvanaFrankaStr3682100DrohobychUkraine
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 51
AbstractWehavedevelopedanewpolarographicmethodforthedetermin-ation of metronidazole and oxytetracycline hydrochloride in theveterinarydrugNozematforhoneybeesThetechniqueisbasedonthereductionofpolarographicallyactivecompoundsonamercurydropletelectrodeTheinfluenceofthecomponentsoftheveterinarydrugNozematonthepolarographicdeterminationofmetronidazolewasstudiedItwasfoundthatthereductionofmetronidazoleisnotaffected by glucose and ascorbic acid but is affected by oxytetra-cyclinehydrochloridewhichisreducedtomercurydropletelectrodeatapotentialofndash145VThedevelopedtechniqueischaracterizedbyeaseofsamplepreparationandcost-effectivenessThistechniquehastheabilitytoidentifysimultaneouslyanddeterminatemetronidazoleand oxytetracycline hydrochloride in solution without the use ofseparationandconcentrationmethods
Keywordselectrochemistrymetronidazoleoxytetracycline
hydrochloridepolarographyveterinarydrug
bees and it can be given in unregulated doses Because of this an unknownamountofmetronidazolecangetintothehoneyanditsometimescausessideeffectsofthehumanbodyanditcouldbeofgreatconcernforpublichealth[56]MedicinesforpeoplearemorestringentandbettertestedthanveterinarydrugsTheproblemofthecontroloftheveterinarydrugsisurgentnowadaysVeterinarymedicinescouldbeunauthorizedandtheuncontrolleduseofmedicinesexistsinretailpharmaciesofmedicineorimportedascontrabandfromothercountries The most widespreadof these classes in thequality controlare chromato-graphic [6ndash9] spectrophotometric [10ndash13] and electrochemical methods[14ndash17]Manyoftheknownmethodsforthedeterminationofmetronidazoleandoxytetracyclinehydrochloridehaveanumberofdisadvantagestime-consumingtheuseoforganicsolventsandexpensivereagentsthesideeffectsofexcipientsandotheractivesubstancesElectrochemicalmethodsarepromisingalternativefor the determination of the electroactive substances Their advantages aresimplicityminiaturizationhighsensitivityandrelativelylowcostThereforethesearch for simple express and affordable methods for the determination ofmetronidazoleremainsrelevantOneofthepromisingmethodsofdeterminationisvoltammetry
2Experimental
21Reagentsandchemicals
VeterinarydrugNozemat (manufacturerAPI-SANRussia) is a yellowpowderwithaslighttypicalodorAvailableinlaminatedbagsof25gCompositionper1gof the drug metronidazole 400 mg oxytetracycline hydrochloride 400 mgglucoseascorbicacid MetronidazoleandoxytetracyclinehydrochloridewerepurchasedfromSigmaAldrich(USA)Stockstandardsolutionofmetronidazolefordeterminationwaspreparedbydissolvingtheexactamountofstandardin7mLof2Mhydrochloricin 500 mL volumetric flask Stock standard solution of oxytetracyclinehydrochloride was prepared by dissolving the exact amount of standard indistilledwaterin500mLvolumetricflaskAfterthatthesolutionswereadjustedtothemarkwithdistilledwaterandmixedthoroughly The Britton-Robinson buffer preparationwas as follows 202 g of sodiumtetraboratedecahydrate287mLofglacialaceticacidand176mLofconcen-tratedorthophosphoricacidweredissolvedin10Lvolumetricflask Working solution preparation was as follows an aliquot of stock standardsolutionwasaddedintoa25mLvolumetricflasktoobtainasolutionwiththenecessaryconcentrationthen2mLofBritton-RobinsonbufferwithnecessarypHwasaddedtotheflaskanddistilledwaterwasaddedtothemark AqueoussolutionofNozematwaspreparedasfollowstheexactportionofthetestveterinarydrugwasdissolved ina250mlvolumetric flaskAnaliquotof
52 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Polarogramsof(A)metronidazoleand(B)metronidazolewithoxytetracyclinehydrochloridendash1solutionsat02MBritton-RobinsonbufferbackgroundatpH=96(υ=05Vs c(metronidazole)=
ndash5 ndash5=45times10 Мc(oxytetracyclinehydrochloride)=50times10 М
100mloftheresultingsolutionwasaddedtoa250mlvolumetricflaskandmadeuptothemarkwithwaterAnaliquotof100mloftheresultingsolutionwasaddedtoa250mlvolumetricflask2mlofBritton-RobinsonbufferwithapHof96wasaddedandthevolumewasadjustedtothemarkwithdistilledwater
22Instrumentation
ForpolarographicmeasurementsweuseddigitaldeviceMTechOVA-410 [18]temperature-controlledthree-electrodeamercurydropletindicatorelectrodeasaturatedcalomelreferenceelectrodeandplatinumwireauxiliaryelectrodeTheaccuracyofthepotentialmeasurementis1mVTheuncertaintyofcurrentmeasu-rement is 01 The employed mercury droplet electrode had the following
ndash4 ndash1characteristicsm=594times10 gs τ=10 min in 02 M NH Cl We used cyclic4
voltammetryforthestudyoftheelectrochemicalprocess WeusedMV870DIGITAL-pH-MESSERATpH-meterformeasuringpHofthesolutions Theobtainedworkingsolutionswereintroducedintothecellanddeoxyge-natedwithargonfor10minPolarogramswererecordedintherangeofpotentialsfrom00tondash16V
3Resultsanddiscussion
Previously it was found that using the Britton-Robinson buffer with pH = 96metronidazoleisreducedwiththeformationofacharacteristiconeirreversiblepeakatndash064V(Fig1A)UsingpolarographywithfastpotentialscanitwasfoundthatmetronidazoleisreducedonmercurydropletelectrodeintherangeofpH20to105ThepeakrecoverycurrentofthemetronidazolereachesthemaximumvalueatpH9ndash10againstthebackgroundofatheBritton-Robinsonbuffer
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 53
Underpre-selected conditions theeffectof some foreign substanceson thepolarographicdeterminationofmetronidazolewasinvestigatedSubstancesthatare componentsofdrugswere studiedglucose ascorbicacidoxytetracyclinehydrochlorideGlucose and ascorbic acid are not reduced atmercury dropletelectrodeanddonotchangetheappearanceofthepolarogramandpolarographiccharacteristics of the recovery of metronidazole As can be seen from Fig 1oxytetracycline hydrochloride is reduced to mercury droplet electrode andchangestheappearanceofthepolarogramandpolarographiccharacteristicsofthe recovery of metronidazole With the addition of oxytetracycline hydro-chloridetherecoverypeakofmetronidazoledecreasesandslightlyshiftstomorenegativepotentials The composition of the drug is relatively complex excipients affect theanalyticalsignaloftherecoveryofcompoundssototakeintoaccountthematrixeffectusedthemethodofmanyadditives QuantitativelytransferredthesolutionofNozemattothecell(exactvolume)removed dissolved oxygen for 10 min and took polarograms in the range ofpotentialsfrom00tondash16VAliquotsofstandardmetronidazolesolutionwereintroducedintothecelltoobtainasolutionwithagivenconcentrationofadditive
ndash5 ndash5metronidazole 10times10 M to 70times10 M As with the determination of themetronidazole aliquots of a standard oxytetracycline hydrochloride solutionwereaddedtothecellwithsolutionofNozemattoobtainasolutionwithagiven
ndash6 ndash6additiveconcentrationfrom70times10 Mto50times10 M(Fig2) In Table 1 are shown metrological characteristics of the determination ofmetronidazoleandoxytetracyclinehydrochloride inveterinarydrugRecoverywascalculatedFormetronidazoletherecoveryis97andforoxytetracyclinehydrochloridetherecoveryis103Analyticalperformanceofthetechniqueisgoodfordeterminationveterinarydrugs
54 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2Polarogramsof(A)metronidazolereductionatdifferentmetronidazoleconcentrationsand(B)oxytetracyclinehydrochloridereductionatdifferentoxytetracyclinehydrochlorideconcentra-tionsandtheircorrespondingcalibrationgraphs
Theaccuracywasverifiedbytheldquoadded-foundrdquomethodAliquotsofstandardsolutionofmetronidazoleweremadeina250mlvolumetricflasktoobtaina
ndash5solutionofagivenconcentrationof33times10 Mandthesolutionofoxytetracyclinendash5hydrochloridetoobtainasolutionofagivenconcentrationof15times10 M2mlof
Britton-RobinsonbufferwithpH96wasaddedtoflaskwithstirringandadjustedtothemarkwithwaterTheanalysisprocedureofmodelsolutionissimilartoanalysis procedure of the solution of Nozemat The calculated amount ofmetronidazolebythemethodofmultipleadditivesinthetestedmodelsolutionisinagreementwiththeamountthatwasintroducedintothesample
4Conclusions
The new polarographic method for the determination of metronidazole andoxytetracyclinehydrochlorideintheveterinarydrugNozematforhoneybeeswasdeveloped We conducted principal component analysis of veterinary drugNozemattoassesstheoveralleffectforthedeterminationofmetronidazoleWefoundthatoxytetracyclinehydrochlorideisreducedtomercurydropletelectrodeThismethodhastheabilitytoidentifysimultaneouslyanddeterminatemetro-nidazole and oxytetracycline hydrochloride in solution without the use ofseparationandconcentrationmethodsOnemoreofadvantagesoftechniquearefastprocedureofanalysissimplesamplepreparationlowcostthepossibilityofminiaturization
References
[1] Antibiotic and Chemotherapy Finch R Greenwood D Whitley R (edits) AmsterdamElsevier2006p292ndash299
[2] MitrowskaKPrzyczynyiskutkizakazustosowania5-nitroimidazoliuzwierzątktorychtkanki lub produkty przeznaczone są do spozycia przez ludziMed Weter 71 (2015)736ndash742
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 55
Metronidazole Oxytetracycline hydrochloride
PeakspotentialV ndash065 ndash144CorelationcoefficientR 099892 099922
ndash1 4 4SlopebmicroАM 656times10 176times10Δb 1763 400InterceptamicroА 2238 0219Δa 0075 0009
ndash1 ndash5 ndash5cmolL 342times10 125times10ndash1cmgg 389 413
Recovery 97 103
Table 1Validationparametersofthemethodofmetronidazoleandoxytetracyclinehydrochloridedetermi-nationinsolutionsofNozematbythemethodofmanyadditives
[3] VermaPNamboodiryVMishraSBhagwatABhoirSAstabilityindicatingHPLCmethodfor the determination of Metronidazole using Ecofriendly solvent as mobile phasecomponentIntJPharmPharmSci5(2013)496ndash501
[4] Cervini P Ambrozini B Machado LCM Ferreira Garcia AP Cavalheiro Gomes ETThermal behavior and decomposition of oxytetracycline hydrochloride J Therm AnalCalorim121(2015)347ndash352
[5] DangBNAnhNTKKyLXThaiPKAntibioticsintheaquaticenvironmentofVietnamsourcesconcentrationsriskandcontrolstrategyChemosphere197(2018)438ndash450
[6] QuintanillaPHettingaKABeltranMCEscricheIMolinaMPVolatileprofileofmaturedTronchon cheese affected by oxytetracycline in raw goat milk J Dairy Sci 103 (2020)6015ndash6021
[7] Chen F Yu L Jingdong P Xiang W Huanjun P Yu C Yan H Study on simultaneousdetermination of three nitroimidazole residues in honey by high performance liquidchromatographyndashresonanceRayleighscatteringspectraMicrochemJ141(2018)423ndash430
[8] Hernandez-MesaM Cruces-Blanco C Campana GA Simple and rapid determination of5-nitroimidazolesandmetabolitesinfishroesamplesbysalting-outassistedliquid-liquidextractionandUHPLC-MSMSFoodChem252(2018)294ndash302
[9] Xiu-ChunGZhao-YangXHai-HuiWWen-YiKLi-MingLWen-QingCHong-WeiZWen-HuiZMolecularlyimprintedsolidphaseextractionmethodforsimultaneousdeterminationofsevennitroimidazolesfromhoneybyHPLC-MSMSTalanta166(2017)101ndash108
[10] ТеплыхАНИлларионоваЕАКоличественноеопределениеметронидазоласпектро-фотометрическимметодомСибирскиймедицинскийжурнал5(2009)48ndash50
[11] ZheltvayOIZheltvayIISpinulVVAntonovichVPSpectrophotometricdeterminationofmetronidazoleandtinidazoleusingcopper(II)complexesJAnalChem68(2013)663ndash668
[12] Youssef AK Saleh MS Abdel-Kader DA Hashem Facile DY SpectrophotometricdeterminationofmetronidazoleandsecnidazoleinpharmaceuticalpreparationsbasedontheformatioonofdyesIntJPharmPharmSci6(2015)103ndash110
[13] Sversut RA Vieira JC Rosa AM Amaral MS Kassab NM Salgado H ValidatedspectrophotometricmethodsforsimultaneousdeterminationofoxytetracyclineassociatedwithdiclofenacsodiumorwithpiroxicaminveterinarypharmaceuticaldosageformArabianJChem13(2020)3159ndash3171
[14] Nikodimos Y Electrochemical determination of metronidazole in tablet samples usingcarbonpasteelectrodeJAnalMethodsChem(2016)361294
[15] Srivastava AK Upadhyay SS Rawool CR Punde NS Rajpurohit AS Voltammetrictechniques for the analysis of drugs using nanomaterials based chemically modifiedelectrodesCurrAnalChem15(2019)249ndash276
[16] Sahu G Voltammetric behaviour of metronidazole at a composite polymer membraneelectrodeOrienJChem26(2010)81ndash86
[17] Yang Y YanW Guo YWang X Zhang F Yu L Guo C Fang G Sensitive and selectiveelectrochemicalaptasensorviadiazonium-couplingreactionforlabel-freedeterminationofoxytetracyclineinmilksamplesSensorsandActuatorsReports2(2020)1ndash7
[18] httpchemlnueduuamtechdeviceshtml(accesed21stJune2020)
56 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
AnimportantpartofanyanalysisthatsignificantlyaffectsthefinalresultsisthesamplepreparationThelowconcentrationofbiologicallyactivecompoundsandthepresenceofaccompanyingcomponentspreventdirectanalysisofthesamplewithcomplexmatrixcompositionTraditionalmethodsofliquidandsolid-phaseextractionhaveaplentyoflimitationssuchashighlytime-consumingprocedureslarge volume of samples expensive cartridges toxic organic solvents andchallenges in automating the process Therefore the application of extractiontechniquesemployinglowamountofsolvents(microextractionmethods)andthelow toxicity extractantes has become the main research direction in recentyears[12] Solid-phasemicroextraction(SPME)wasproposedbyPavlishinin1989[3]Onevariantofthismethodistousethinrodswithvariouspolymercoatingssuch
Application of microextraction techniques combined with chromatographic methods for the analysis of complex objects
VLADISLAVDEEVELENABESSONOVALIUDMILAKARTSOVA
InstituteofChemistrySaint-PetersburgStateUniversityUniversitetskyprospect26198504PeterhofSaint-PetersburgRussiahitchervmailru
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 57
AbstractThelowconcentrationofanalytesandthepreventionofthematrixinfluence requires a stage for extraction and concentration of thestudiedcompoundsTheclassicalmethodsofliquidandsolid-phaseextractionhavemanylimitationsthatpreventtheiruseinsomecasesMicroextraction techniques are becoming more widespread WestudiedthepossibilityofusingionicliquidstoextractpesticidesfromwatersampleswiththeirsubsequentHPLS-MSdeterminationTheinfluenceonthedegreeofextractionofsuchparametersasanatureofionicliquidsanddispersersolventtheiramountssaltconcentrationvolumeratioofionicliquidsandwatersampledilutionoftheionicliquidsextractwithmethanolwasperformedBesidesconditionsofsolid-phase microextraction of volatile organic compounds fromurine samples obtained from healthy donors and donors withprostate cancer have been found The analysis of volatile organiccompoundsbyGC-MSfollowedbychemometricprocessingallowedachievingahighvalueofbinaryclassificationaccuracy(91)
Keywordschemometricsdispersiveliquid-liquid
microextractionsolid-phase
microextraction
asdivinylbenzenepolydimethylsiloxanepolyacrylateandpolyethyleneglycolwhichappliedtothesurface[4]Thepolymersorbentisplacedintheequilibriumheadspaceaboveacondensedphaseofthesampleandthevolatilecompoundsareextracted Liquidmicroextractionconsistsofusingsmallamountsofliquid(extractant)inequilibriumwiththegasorliquidphaseofthesampleDispersiveliquid-liquidmicroextraction(DLLME)isavariantofliquidmicroextractionTheessenceofthemethodisasfollowsextractantisdissolvedinthephaseofadispersingsolventandthemixtureisrapidlyinjectedintothesamplevolume[5]Inthiscasethedispersing solvent is dissolved and a ldquocloudrdquo of extractant is formed A largesurfaceareacontributestomasstransferprocessesThecombinationofDLLMEwiththeuseof ionic liquids(ionic liquids)asextractantsreducestheharmfulimpactontheenvironment[6] Sothegoalofthisstudywastheapplicationofmicroextractionmethodsfortheanalysisofrealsamples
2Experimental
21Reagents
Deionizedwaterwas obtained at the AQUILON D 301 deionizer (Russia) Allchemicals and reagents (the highest commercially available purity) werepurchasedfromReachimBakerAcrosorganicsandSigmaAldrich
22Instrumentation
HPLCanalysiswascarriedoutusinganHPLCLCMS-8030(Shimadzu)withatriplequadrupole mass-selective detector with electrospray ionization Analysis ofvolatileorganiccomponents(volatileorganiccompounds)ofurinesampleswasmade by GCMS-QP2010 SE (Shimadzu) Chemometric data processing wasperformedusingRStudio
23DeterminationofvolatileorganiccompoundsinurinesamplesbyGC-MSmethod
ForSPMEofvolatileorganiccompounds inurinesamplewasusedfibercoatedwith a polydimethylsiloxane (PDMS) The volatile organic compounds wereextractedontofibercoatingfor20minat50degCThentheanalytesweredesorbedinto the gas chromatography for 4 minutes at a temperature of 250degCChromatographic separation was carried out on a HP-5 capillary column(30mtimes250μmtimes025μm)usingtemperatureprogrammingmodeThetempera-
ndash1tureofovenwasincreasedfrom50degCupto250degCatarateof10degCmin Tionsourcewas200degCMassspectrometrywasusedinSIMmode(mz=35ndash900)
58 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
24ConditionsforLCMSMSdeterminationofpesticides
Separation of pesticides was performed by HPLCMSMS with positiveelectrosprayionizationoncolumnZorbaxBonusRP35μm(21times100mm)with40mMammoniumacetateandmethanolasmobilephaseAandBrespectivelyThefollowinggradientelutionwasapplied20ndash85B(8min)85B(8ndash15min)85ndash95B(150ndash155min)95B(155minus180min)95ndash20B(180minus185min)
ndash1Thevelocityof themobilephasewas03mlmin Thevolumeof the injectedsamplewas 20 microlMS detection capillary voltage +45 kV spray gas velocity
3 ndash1 3 ndash13dm min flow rate and drying gas temperature 15 dm min and 250 degCrespectively
25Selectionofconditionsfordispersiveliquid-liquidmicroextractionofpesticides
The influence of the natures of ionic liquids ([C MIM][PF ] [C MIM][NTf ]4 6 6 2
[C MIM][BF ])andthedispersingsolvent(methanolacetonitrileacetone) the6 4
weightoftheionicliquids(0060ndash0200g)thevolumeofthedispersingsolvent(02ndash10ml)onthedegreeofpesticidesextractionwerestudiedTheinfluenceofthepH(5422)theconcentrationofNaCl(0040ndash0200g)andextractiontime(1-6min)wereinvestigated The effects of different ionic liquids and disperser solvents on DLLMEprocedures were investigated and optimized by using standard solutions ofpesticidesIndetailasolutionofionicliquidsinadispersingsolventwaspreparedand rapidly injected into the aqueous sample solution (2ml) followed bytreatment for 2 min in an ultrasonic bath cooling at ndash4degC for 10 mincentrifugationfor10minat3500rpmandcollectionofionicliquidsThewaterphasewas separatedandanalyzedbyHPLC-MSThe ionic liquidsextractwasdilutedinmethanolandanalyzedbyHPLC-MS
3Resultsanddiscussion
31Microextractionofpesticides
One of the important tasks of environmental monitoring is to control traceconcentrationsofpesticidesinwatersamplesTechniqueofcombiningseveralpesticideshasbecomemorewidespread inagriculture It allowsreducing thetotalconcentrationoftheappliedcompoundsandtodecreasetheadaptabilityofpathogens and insects Therefore the analysis of real samples requires apreliminarystageofselectiveanalytesextractionandconcentration ExtractinganddispersingsolventsarebothimportantinDLLMEofanalytesTheinfluenceofthenatureofthedispersingsolvent(methanolacetonitrileandacetone) and the extractant (imidazolium ionic liquids [C MIM][PF ]4 6
[C MIM][BF ]and[C MIM][NTf ])onthedegreeofextractionofpesticideswas6 4 6 2
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 59
studied This parameter was controlled by the residual concentration ofpesticidesinthewaterphaseafterextractionThebestresultswereobtainedforionic liquids [C MIM][PF ] as an extractant and acetonitrile as a dispersing4 6
solvent Thenextstepwastoselecttheamountofionicliquids(0060ndash0200g)andthevolume of acetonitrile (02ndash10 ml) It was found that the highest degree ofextractionofanalyteswasachievedbyusing020gofionicliquidsand03mlofacetonitrile It was shown that the degree of extraction of selected pesticides does notdependonpHofthewatersamplewhichconfirmsthepartitionmechanismofextractionThedegreeofextractionofcarbofosincreasedslightlywithanincreaseinthesaltconcentrationandreachmaximumbyweightto4(008g) ItisknownthatthehighviscosityofionicliquidshavehinderedtheprocessesofelectrosprayionizationDilutionofthesamplewithmethanolby3timesgivesthebestresult(thesignalintensitywas373to845ofthesignalwithoutionicliquids) ThustheconditionsofDLLME-ionicliquidsextractionofpesticidesfromwatersampleswere found The limits of detection for pesticideswere from007 to
ndash1019ngml thereproducibilityofpeakareaswerefrom3to5theextractionrecoverywascloseto100
32SPMEofvolatileorganiccompoundsfromurinesamples
Oneof the importantdirections is the search for criteriaofearlydiagnosisofcancer Obtaining characteristic profiles of volatile organic compounds fromurinesamplescanhelpdevelopanon-invasivemethodforearlydiagnosisofthedisease ForthiswestudiedtheinfluenceofanumberoffactorsonthetotalnumberofpeaksandthetotalpeakareaTheywerethetemperatureatwhichthevaporandcondensedphasesofurinewerebalanced(30ndash60degC)thepreheatingtimeofthesample(10ndash40min)NaClconcentration(30ndash133wv)andthesorptiontimeonthePDMScoating(5ndash30min) Anincreaseinthepreheatingtemperatureofthesampleto500degCledtoanincreaseinthenumberofsignalswhichdidnotchangewithafurthertempera-tureincreasingNextparameterwasthetimeofachievementequilibriumofthevapor and condensed phases The largest number of peaks was observed at40minbutthisgreatlyincreasedthetimeofanalysisandsowechose20minAlsowestudiedthedesaltingeffectofsodiumchlorideonefficiencyofextractionThebestvolatileorganiccompounds sorptionwasachievedbyadding133saltingagent It is also shown that thenumberofpeaksdidnot changeafter20minutesofsorption Thus to obtain the characteristic profiles of urine samples the followingconditionswereselected1333NaClwasaddedtotheurinesample(3ml)
60 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
heatingfor20minat50degCthensorptionofvolatileorganiccompoundsonPDMSfibercoatingat50degCfor20min Undertheselectedconditionsweobtainedvapor-phaseprofilesof52urinesamples (32 normal and 20 pathology) Prior to performing chemometricprocessing of chromatographic profiles of urine samples preliminary datapreparation is necessary [7] The baseline was removed and the peaks werealignedusingdynamictimewarpingwithcontrollingbymassspectra ThePCAmodelwasbasedon52aligned characteristicprofilesThere is asatisfactoryseparationofdataintotwoclustersinthescoresplotrelativetothefirstandsecondprincipalcomponent(Fig1) The original data set (52 samples) was randomly divided into calibration(13pathology21normal)andtest(7pathology11normal)setsThenthePLS-DAmodelwasbuiltusingthecalibrationsetanditspredictiveabilitywasevaluatedusingthetestsetTheprocedurewasrepeated100timesTheaveragevaluesofsensitivityspecificityandaccuracyinthiscasewere9594and91
4Conclusions
Thepossibilityofusingimidazoliumionicliquids([C MIM][PF ])asextractants4 6
forquantitativeextractionandconcentrationofpesticidesundertheconditionsofDLLMEisshownThedegreeofconcentrationwas28ndash33whichallowedreaching
ndash1thedetectionlimits(006ndash019ngml )belowthemaximumpermissibleconcen-trationThepossibilityofnon-invasivediagnosisofprostatecancerbySPMEofvolatileorganiccompounds inurine isshownChemometricprocessingofgaschromatographic profiles using PLS-DA and PCA methods allowed achievingclassificationaccuracyvaluesmorethan90
Acknowledgments
ThisworkwassupportedbytheRussianFoundationforBasicResearchprojectno18-53-80010BRICS_t and the Russian Science Foundations (Projects 19-13-00370) We are grateful to
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 61
Fig 1Scoresplotrelativetothefirstandsecondprincipalcomponent
Resource Education Center in Chemistry of St Petersburg State University for the providedequipment
References
[1] Rutkowska M Płotka-Wasylka J Sajid M Andruch V Liquidndashphase microextractionAreviewofreviewsMicrochemJ149(2019)103989
[2] JaliliVBarkhordariAGhiasvandAAcomprehensivelookatsolid-phasemicroextractiontechniqueAreviewofreviewsMicrochemJ152(2020)104319
[3] ArthurCLPawliszynJSolidphasemicroextractionwiththermaldesorptionusingfusedsilicaopticalfibersAnalChem62(1990)2145ndash2148
[4] SchmidtKPodmoreISolidphasemicroextraction(SPME)methoddevelopmentinanalysisof volatile organic compounds (VOCs) as potential biomarkers of cancer JMol BiomarkDiagn6(2015)1000253
[5] Mousavi L Tamiji Z Khoshayand MR Applications and opportunities of experimentaldesign for the dispersive liquidndashliquidmicroextractionmethod ndash A review Talanta190(2018)225ndash356
[6] MarcinkowskaRKoniecznaKMarcinkowskiLNamiesnikJKloskowskiAApplicationofionic liquids inmicroextractiontechniquesCurrent trendsandfutureperspectivesTrACTrendsAnalChem119(2019)115614
[7] WehrensRChemometricswithRMultivariateDataAnalysisintheNaturalSciencesandLifeSciencesBerlinSpringer2011
62 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Theself-assembledtwo-dimensionalmonolayers(2DSAMs)ofvariousmolecules(eg graphene [1] MoS [2] rubrene [3]) offer beneficial properties for the2
constructionofnano-electronic andnano-opticaldevicesThe topological andchemicalcharacterizationof2DSAMsiscrucialtogatherinformationaboutthearrangementofdepositedmoleculesandtheirinteractionwiththesubstrateThistaskrequiresanalyticaltechniqueswith(sub)nanometerspatialresolutionandup to single-molecular detection sensitivity Only few techniques meet therequirements and one of them is tip-enhanced Raman spectroscopy (TERS)whichcombinestheexcellentspatialresolutionofscanningprobemicroscopy(SPM) and chemical sensitivity of surface-enhanced Raman scattering (SERS)spectroscopy[45] TheSERS spectroscopyutilizesplasmonicmetal nanostructures to cause ahighlocalenhancementoftheelectricfieldintheirclosevicinityviathesurfaceplasmonresonance(SPR)effectThelocalelectricfieldcausesanincreaseofthe
The development of reference probe system for tip-enhanced Raman spectroscopy
MARTINKRA LMARCELADENDISOVA PAVELMATE JKA
DepartmentofPhysicalChemistryFacultyofChemicalEngineeringUniversityofChemistryandTechnologyPragueTechnickaacute516628Prague6CzechRepublicMartinKralvschtcz
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 63
AbstractThetip-enhancedRamanspectroscopy(TERS)isamodernanalyticaltechniquewithanoutstandingspatialresolutionandchemicalsensi-tivityTheseparametersmainlydependon the structural integrityand chemical purity of employed plasmonic scanning probe tipsUsuallyeachtipistestedbeforeTERSmeasurementsusingcommer-ciallyavailablereferencesamplesHowevertheirpriceandrelativelyshortexpirationdatemustbeconsideredwhenplanningaresearchbudgetWedevelopedaproceduretoproduceself-madereferenceprobesamplesfortestingTERStipsusingcopper(II)phthalocyanineonaAunanolayerwhichispreparedbythermalvacuumevaporationofAuonaSiwaferOurresultsshowthatthepreparedsystemenablesrepeateddetectionofwell-resolvedTERSspectraThecollectedTERSspectraandspectralmapsexhibitsomedegreeofvariabilitywhichmaybeduetovariousphoto-inducedprocessesanditmustbeconsi-deredwhileperformingTERSmeasurements
Keywordscopper(II)
phthalocyaninescanningtunnelling
microscopyresonanceRaman
spectroscopysurface-enhancedRaman
spectroscopytip-enhancedRaman
spectroscopy
Ramanscatteringfrommoleculesboundtothemetalby6ndash8ordersofmagnitude[6]TheenhancementallowsSERSspectroscopytobeusedforsingle-moleculardetectionHoweverRamanmicroscopeshave limitedspatialresolutionbythelightdiffractionwiththeachievableresolutionbeingaroundhalfoftheexcitationwavelengthOntheotherhandthespatialresolutionofSPMtechniquesislimitedonlybythedimensionsoftheapexofthescanningprobetipwhichmayevenbeatomicallysharpByutilizingSPMtipandsubstratemadefromplasmonicmetalsan artificial ldquohotspotrdquomay be createdwith its position and dimensions beingdefinedbythetipItopensthepossibilitytocollectstronglyenhancedRamanspectrafromtheareapreciselylocalizedbellowthetipandthusovercometheopticaldiffraction limitTheartificialhotspotmayberelocatedbymovingthesamplebelowthetipwhichisthefoundationofTERSmapping[578] A successful TERS experiment requires an optimal combination of variousparameters the most important of which are tip sharpness and purity [7]Areferencesampleconsistingofaflatplasmonicnanolayerwithattachedprobemolecules is frequently used to check the state of the tip before using it forexperiments Unfortunately commercially available TERS standards areexpensiveandhaveanexpirationdateofseveralmonths Thegoalofthisstudywastofindapreparationprocedurewhichwouldbeableto produce cheap reference samples for repeated detection of intense TERSspectraAcombinationofaAunanolayeronaSisubstratepreparedbythermalvacuumevaporationwithadsorbedcopper(II)phthalocyanine(CuPc)whichisamoleculewithhighRamancross-sectionwastested[910]Copper(II)phthalo-cyanine known as phthalocyanine blue is a synthetic blue pigment and isfrequently used in paints It has been studied as a potentialmaterial for theconstruction of organic solar cells and other photoelectronic devices [11] AsindicatedbyitscolourCuPcexhibitsseveralabsorptionbandswithinthevisibleregion The effects of a transition to excited electronic states and subsequentluminescencemaybeobservable inRamanmeasurements[12]Au isahighlysuitablemetalforthesampleasitisboththermallyandelectricallyconductivewhichlimitsthelocalheatingofthesampleduringthemeasurementsandenablesthe use of scanning tunnelling microscopy (STM) for tip-surface interactionfeedback[13]
2Experimental
21Reagentsandchemicals
Thesubstrateforthesamplewaspreparedbythermalvacuumevaporationofgoldonsilicon(100)waferFirsta5nmthickCradhesionlayerwasdepositedon
ndash1thewaferfollowedby100nmofAuThedepositionratewas4nmmin forCrand ndash18nmmin for Au The base pressure of the evaporation system was below
ndash65times10 mbarFollowingthepreparationproceduredescribedbyJiangetal[14]
64 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
thecleansubstratewasimmersedintoasaturatedsolutionofCuPc(˃99SigmaAldrich USA) in dimethylformamide (˃98 Lach-ner CZ) for at least 12h atambienttemperatureSubsequentlythesamplewasremovedfromthesolutionrinsedwithMilli-Qwaterandmethanol(paPentaCZ)anddriedwithair
22Instrumentation
TheRamanSERSandTERSspectrawererecordedusingRamanspectrometerInVia Reflex (Renishaw UK) equipped with lasers emitting at two differentexcitation wavelengths 633nm (136mW max power output) and 785nm(204mWmaxpoweroutput)Thespectrometerhasathermoelectricallycooled
ndash1CCDdetectorwithaspectralresolutionof2cm and4microscopeobjectiveswith5times20times50timesand100timesmagnitudeForTERSexperiments the laserbeamwasredirectedtotheSPMplatformInnova-IRIS(BrukerUSA)viaasystemoflightguidesElectrochemicallyetchedAuTERS-STMtips(BrukerUSA)wereusedforallTERSmeasurements The spectra were processed using the Spectragryph software (F MengesldquoSpectragryph - optical spectroscopy softwarerdquo Version 1214 2020httpwwweffemm2despectragryph) Using this software all collectedspectra were treated by a Savitzky-Golay noise filter automatic baselinecorrectionspikeremovalandpeaknormalization
3Resultsanddiscussion
31Ramanmeasurementsofcrystallinecopper(II)phthalocyanine
AtfirsttheRamanspectraofpureCuPcwerecollectedtoprovidereferencedatawhileusingboth633and785-nmexcitationlasers(Fig1)Bothspectraexhibit
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 65
Fig 1RamanspectraofCu(II)phthalocyanineincrystallineformmeasuredat633(top)and785-nm(bottom)excitationThespectraareoffset
ndash1vibration bands in the 500ndash1600 cm region with slight differences in theirintensityratiosThe633-nmexcitationallowstheobservationofadditionalbands
ndash1inthe2000ndash3000cm region(onaluminescencebackground)whichoriginatefrom the resonance Raman effect as the excitation energy overlaps with theQ-bandofCuPc[12]MoreovertheprocessofelectronicexcitationmaylowertheD symmetry of CuPc during resonance Raman scattering and previously4h
forbiddenbandsmaybecomeobservable[15]Theluminescentbackgroundhasandash1maximumaround2200cm whichcorrespondstoamolecularemissionbandat
735nmEventhoughthespectrameasuredwiththe785-nmlaserlinedonotexhibitapparentresonanceenhancementapre-resonanceRamanenhancementmayoccur
32Surface-enhancedRamanmeasurementsofcopper(II)phthalocyaninelayeronagoldsubstrate
The prepared sample of CuPc on a Au layer was analysed using the Ramanmicroscope Both excitation laserswere used to obtain SERS spectra (Fig 2)whichwerecomparedtothespectraofapurecrystallineCuPc Thepositionsof bands in SERS spectra closelymatch their positions in thespectraofbulkCuPcHoweveraslightshiftofsomespectralbandsisobservable
ndash1(eg1528rarr1532cm )whichmaybeattributedtotheinteractionbetweenCuPcand the Au substrate The disappearance of luminescence background andresonance-enhancedbandsinthespectrumat633-nmexcitationalsosuggeststhemolecule-metalinteractionandthetransferofenergyfromCuPcmoleculestothesubstrateMoreovertherearevariationsintherelativeintensitiesofbands
ndash1whichdependontheexcitationenergyegthebandat1309cm isenhancedinSERSspectraatthe785-nmexcitationwhencomparedtothespectraofpureCuPcorevenSERSspectraat633-nmexcitation
66 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2SERS spectra of Cu(II) phthalocyanineon aAu layermeasured at 633 (top) and785-nm(bottom)excitationThespectraareoffset
33Tip-enhancedRamanmeasurementsofcopper(II)phthalocyaninelayeronagoldsubstrate
SERS microspectroscopy is a diffraction-limited technique as it provides anaveragedinformationaboutmoleculesintheilluminatedareaofseveralsquaremicrometers Meanwhile TERS spectra are collected from an area of tens ofnanometers and they contain specific information about the local moleculararrangement topography of the underlyingmetal and properties of the localelectric field between the tip and the substrate Therefore a higher spectralvariabilityshouldbeexpected Several TERS mapping experiments were carried out using both 633 and785-nmexcitationwithvaryingexperimentalparameterssuchasthenumberanddistance betweenmeasured points acquisition time number of acquisitionslaserpoweretc TheTERSspectrameasuredat633-nmexcitationexhibited lowersignal tonoiseratioandreproducibilityAsaconsequenceTERSmappingwasimpossibleandonlyafewone-pointTERSspectrawereobtained(Fig3) TheTERSspectracollectedat785-nmexcitationcontainedahighernumberofwell-resolvedbandsMoreoverthespectrawerestableintimeandsotheTERSmappingwaspossibleThe twopresentedTERSspectraareaveragesofTERSmaps which contained 16 and 80 points with 600 and 300-nm spacingrespectively(Fig4) Thespectracollectedusingbothexcitationwavelengthsexhibitavariabilityinrelative intensities and positions of bands between themeasured points ThevariabilitymaybeattributedtothelocalorientationofCuPcmoleculesbetweenthe tip and the Au surface and the properties of strongly enhanced andnon-homogeneous electromagnetic field which depend on the tip-surfacedistancetheirmorphologyandrelativepositionMoreovertheusedexcitation
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 67
Fig 3TwoexamplesofTERSspectraofCu(II)phthalocyaninemeasuredat633-nmexcitationThespectraareoffset
wavelengthsareclosetoabsorptionbandsofCuPcandthestrongelectricfieldmay give rise to photo-induced effects These effects include the electronicexcitationofCuPctohigherstateschargetransferbetweentheCuatomandthephthalocyaninering ionizationof themoleculeand formationofradicalsThephoto-inducedprocessesarelikelytoplayabiggerroleinTERSspectradetectedat633-nmexcitationduetotheoverlapwithQ-bandofCuPcwhichmaybethecauseoftheirlowersignaltonoiseratioandreproducibility
4Conclusions
Thedeveloped referenceprobe systemofCuPc adsorbedon aAu surfacehasprovedtobesuitablefortheintendeduseasitenabledthedetectionofintenseandwell-resolvedSERSandTERSspectraTheAulayerpreventsoverheatingofthesampleandallowsfortheuseofSTMTheSERSspectrawereinagoodmatchwith thespectraofpureCuPcAslight shiftof somebandsandchange in theluminescent background indicated the interaction between CuPc and the AusurfaceTheTERSexperimentsresultedinspectralmapswithhighintensitiesofindividualspectraIncreasedvariabilitybetweenmeasuredpointswasobservedPossiblesourcesofthevariabilityarephoto-inducedprocessesthatmayoccurinthestronglyenhancedelectricfieldTheseeffectsareaknownfeatureofTERSmeasurements and they offer valuable insight into the photophysics andphotochemistryofCuPcinteractingwiththeAusurfaceThedependenceofTERSspectra on experimental parameters and the preparation procedure of thereferencesampleshouldbefurtherstudied
Acknowledgments
ThisworkwassupportedfromthegrantofSpecificuniversityresearchndashA2_FCHI_2020_039
68 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 4TwoexamplesofaveragedTERSspectraofCu(II)phthalocyaninemeasuredwith785-nmexcitationThespectraareaveragesfromTERSmapsincluding16(top)and80(bottom)measuredpointsThespectraareoffset
References
[1] Mas-BallesteRGomez-NavarroCGomez-HerreroJZamoraF2DmaterialstographeneandbeyondNanoscale3(2011)20ndash30
[2] ZengHCuiXAnopticalspectroscopicstudyontwo-dimensionalgroup-VItransitionmetaldichalcogenidesChemSocRev44(2015)2629ndash2642
[3] SchultzJFLiLMahapatraSShawCZhangXJiangNDefiningmultipleconfigurationsofrubreneonaAg(100)surfacewith5A spatialresolutionviaultrahighvacuumtip-enhancedRamanspectroscopyJPhysChemC124(2020)2420ndash2426
[4] WhitemanPJSchultzJFPorachZDChenHNJiangNDualbindingconfigurationsofsubphthalocyanineonAg(100)substratecharacterizedbyscanningtunnelingmicroscopytip-enhanced Raman spectroscopy and density functional theory J Phys Chem C 122(2018)5489ndash5495
[5] ShaoFZenobiRTip-enhancedRamanspectroscopyprinciplespracticeandapplicationstonanospectroscopicimagingof2DmaterialsAnalBioanalChem411(2019)37ndash61
[6] ArocaRSurface-EnhancedVibrationalSpectroscopyHobokenWiley2006[7] KumarNMignuzziS SuWRoyDTip-enhancedRamanspectroscopyprinciplesand
applicationsEPJTechInstrum2(2015)9[8] BailoEDeckertVTip-enhancedRamanscatteringChemSocRev37(2008)921ndash930[9] BovillAJMcConnellAANimmoJASmithWEResonanceRamanspectraofα-copper
phthalocyanineJPhysChem90(1986)569ndash575[10] Shaibat MA Casabianca LB Siberio-Perez DY Matzger AJ Ishii Y Distinguishing
polymorphsofthesemiconductingpigmentcopperphthalocyaninebysolid-stateNMRandRamanspectroscopyJPhysChemB114(2010)4400ndash4406
[11] SzybowiczMRunkaTDrozdowskiMBałaWGrodzickiAPiszczekPBratkowskiAHightemperaturestudyofFT-IRandRamanscatteringspectraofvacuumdepositedCuPcthinfilmsJMolStruct704(2004)107ndash113
[12] CaplinsBWMullenbachTKHolmesRJBlankDAFemtosecondtonanosecondexcitedstatedynamicsofvapordepositedcopperphthalocyaninethinfilmsPhysChemChemPhys18(2016)11454ndash11459
[13] SaccoAImbraguglioDGiovannozziAndreaMPortesiCRossiAMDevelopmentofacandidatereferencesampleforthecharacterizationoftip-enhancedRamanspectroscopyspatialresolutionRSCAdv8(2018)27863ndash27869
[14] JiangSChenZChenXNguyenDMatteiMGoubertGVanDuyneRPInvestigationofcobaltphthalocyanineatthesolidliquidinterfacebyelectrochemicaltip-enhancedRamanspectroscopyJPhysChemC123(2019)9852ndash9859
[15] MelendresCAMaroniVARamanspectraandnormalcoordinateanalysisoftheplanarvibrationsofironphthalocyanineJRamanSpectrosc15(1984)319ndash326
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 69
1Introduction
The aim of this work has been develo-pmentofanewvoltammetricmethodforthe determination of 23-dimercapto-1-propane-sulfonic acid (DMPS) Fig 1Investigationwasdonetoobtainrelevantinformation about complexingbehaviorofDMPStowardsleadions Lead is one of heavy metals which can cause irreversible neurologicalproblems [1 2]DMPS is a synthetic antidotewith two thiol groups used fortreatmentofpoisoningbyheavymetals[3ndash6]StrongcomplexingpropertieshighwatersolubilityandnegligiblesideeffectsarethemostimportantadvantagesofDMPS[47]
Fig 1 Chemical structure of 23-dimercap-to-1-propane-sulfonicacid
Determination of heavy metal poisoning antidote 23-dimercapto-1-propanesulfonic acid using silver solid amalgam electrode
ab ab bc bMARTACHOIN SKA VOJTE CHHRDLICKA BEATRIZRUIZREDONDO JIR IBAREK aTOMA S NAVRATIL
a JHeyrovskyacuteInstituteofPhysicalChemistryoftheCzechAcademyofSciences Dolejškova21553182thinsp23Prague8CzechRepublicmartachoinskagmailcomb UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova20308128thinsp43Prague2CzechRepublic
c UniversityofValladolidPlazadeSantaCruz847002ValladolidSpain
Abstract23-Dimercapto-1-propane-sulfonic acid (DMPS) was investigatedusingdirectcurrentvoltammetry(DCV)differentialpulsecathodicstrippingvoltammetry(DPCSV)differentialpulseanodicstrippingvoltammetry(DPASV)andeliminationvoltammetrywithlinearscan(EVLS)atapolished(p-AgSAE)andatameniscusmodifiedsilversolidamalgam electrode (m-AgSAE) EVLS confirmed two consecutivereductions with coupled protonelectron transfer VoltammetrictitrationsofDMPSwithPb2+provedcomplexformationwithlimits
minus1ofquantification(LOQs)anddetection(LODs)03and01micromolL atminus1m-AgSAE and 08 and 03micromolL at p-AgSAE respectively
Determination of DMPS in commercial drug Dimaval and humanurine samples confirmed practical applicability of the developedmethod
Keywordscathodicstripping
voltammetry23-dimercapto-1-pro-
panesulfonicacideliminationvoltammetry
withlinearscansilversolidamalgam
electrodeunithiol
70 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Voltammetry was chosen as a determination technique due to its highsensitivity and selectivity speed low costs [8] Thiol groups in DMPS can beoxidatively chemisorbed on solid amalgam electrode It can be used as anaccumulation step for cathodic strippingvoltammetry [9ndash11]Moreover solidamalgamelectrode (SAE)was chosen as theworking electrodebecauseof itspropertiesashighsignaltonoiseratiowidepotentialwindowandabilitytoreachlowlimitsofdetection(LOD)[12ndash13]
2Experimental
21Reagentsandchemicals
Allsolutionswerepreparedusingdeionizedwater(Milli-Q-GradientMilliporendash1PragueCzechRepublic)withconductivitylt005microScm Britton-Robinsonbuffer
solutionspHrangefrom2to12werepreparedbymixingtheproperamountsof02MNaOH(alkalinesolution)andof004MH BO 004MH PO and004M3 3 3 4
CH COOH(allLachemaCzechRepublic)acidicsolutionTheacidicsolutionwas3
preparedbydissolutionof1235gofH BO pa088mLofH PO (85)paand3 3 3 4
1435mLofCH COOH(99)pa in500mLofdeionizedwaterThealkaline3
solution was prepared by dissolution of 3995 g of NaOH pa in 500mL ofdeionizedwater(allLachemaCzechRepublic) StocksolutionofDMPSwaspreparedbydissolving10mgofsolid23-dimer-capto-1-propanesulfonic acid monohydrate pa (Merck Czech Republic) in100mLofdeionizedwaterForthepreparationofthemodelsamplesolutiononecapsuleofdrugDimaval(HeylGermany)contains100mgofDMPSwasdissolved
ndash1in10LofdeionizedwatertotheDMPSconcentrationof0531mmolL Twomodel samplesofDimavalwerepreparedbydilutionof theabove-mentioned
ndash1solutionwithBritton-Robinsonbuffersolutiontoconcentrations10micromolL andndash1of 10micromolL respectively Urine model samples were prepared by mixing
Britton-Robinsonbuffer solutionwithurine samples obtained fromvolunteer(manhealthy30yearsold)inratio11SamplepHwasadjustedbyadditionof
ndash1proper amount of 02molL NaOH Before each measurement oxygen wasremoved for 5minbynitrogenbubbling (purity class 46MesserTechnogasPragueCzechRepublic)
22Instrumentation
Measurementswereperformedusingtwotypesofworkingelectrodesmeniscusmodified silver solid amalgam electrode (m-AgSAE working surface of
20382plusmn0025mm α lt 005) and polished silver solid amalgam electrode2(p-AgSAEworkingsurfaceof0196plusmn0015mm αlt005)Ag|AgCl|3MKClwas
used as the reference electrode andplatinumwire (Oslash1mm)wasused as theauxiliary electrode (both from Elektrochemicke detektory Czech Republic)Measurementswereperformedatlaboratorytemperature(25plusmn2degC)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 71
The pH was measured using pH-meter Jenway 3505 with combined glasselectrodetype924001(BibbyScientificLimitedUK)Voltammetricmeasure-ments were performed using the computer-controlled Eco-Tribo Polarograph(Polaro-Sensors Czech Republic) Software used for measurements wasMultiElChem 33 forWindows XP7810 (J Heyrovsky Institute of PhysicalChemistryoftheCzechAcademyofSciencesCzechRepublic)
3Resultsanddiscussion
OptimumconditionsformeasurementswereobtainedbyseriesofmeasurementinwiderangeofpHvaluesandtestingvariouscleaningproceduresOptimumpotentialof accumulation (E ) and timeof accumulation (t )ofDMPSwereacc acc
adjusted for differential pulse cathodic stripping voltammetry (DPCSV) atp-AgSAEandm-AgSAE ThedependencebetweenpeakheightandconcentrationofDMPSatp-AgSAEhasalogarithmicshapewhichcorrespondstotheaccumulationprocessattheelectrodesurfaceThelineardependencewasobservedintheDMPSconcentra-
ndash1 ndash1tionsfrom03micromolL to20micromolL Peakshifttowardsnegativepotentialwithincreasing concentration of DMPS corresponds to the metal-thiol bond andinfluenceofelectrodesurfacestructureonthisbondOnthecontrarysignalsonm-AgSAEweremorestableandthedependencebetweenconcentrationofDMPSandsignalwasalmostlinearinwholetestedrangeofconcentrations The developedmethodwas tested inmodel samples of Dimaval and urineFoundamountsofDMPSwereingoodagreementwithdeclaredcontentsusingbothelectrodesHowevertherepeatabilityofsignalsregisteredusingp-AgSAEinurinesamplesweresignificantlyworsethanthoseinDimavalsamplesItcanbecausedbycomplicatedbiologicalmatricesandfoulingeffectsofurine ELSV measurements confirmed two consecutive reductions of DMPS inadsorbed state At m-AgSAE signal were at about minus415 mV and minus440 mVrespectively and at p-AgSAE at about minus790 mV and minus830 mV respectively
ndash1 ndash1Reductionsatm-AgSAEatthescanratesfrom80mVs to640mVs havebeencontrolledbyakineticprocessatminus400mV Inanodicscansonm-AgSAEonlyonepeakwasvisibleatabout‒390mVItcorrespondswiththeoxidationofmercuryelectrodesurfaceontheelectrodeinthepresenceofDMPSandwithdiffusionfromthebulksolutionofproductsAtp-AgSAEnosignificantanodicsignalwasfound ThelastpartoftheresearchrevealedvoltammetricbehaviorofDMPSinthe
2+presence of Pb Voltammetric titration was investigated by DPCSV anddifferential pulse anodic stripping voltammetry (DPASV) during consecutive
ndash1 ndash1additionsof1mmolL ofPb(NO ) into100micromolL DMPSsolutioninacetate3 2
bufferofpH50InabsenceofPb2+ontheelectrodesurfaceHg(DMPS)complexisformedduringtheaccumulationstepDuringtheanodicscanthereisonlyonewelldevelopedreductionpeak(Fig2A)
72 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ndash1Fig 2DPCSandDPASvoltammogramsof10micromolL ofDMPSinacetatebufferpH=5correspond-2+ingto[Pb DMPS]ratiosof(A)01(B)11and(C)21Uppercurvecorrespondstothecathodic
scanE =0mVt =15sLowercurvecorrespondstoreverseanodicscanwitht =15satacc acc accndash1E =minus1000mVν=20mVs (Ref[16])acc
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 73
2+ WhenPb DMPSratioisequalto11twooxidationandthreereductionpeakswereregistered(Fig2B)PeakA ataboutndash300mVcorrespondstoformationofred
0Pb(DMPS)complexThiscomplexwasfurtherreducedtothePb (Hg)atabout2+ndash500mV(C )ReductionpeakoffreePb wasalsoregistered(B )Oxidationred red
peaksA andB correspondtoreverseprocessesandC isnotpresentbecauseox ox ox
nofreeDMPSispresentinthesolutionAtratio21thereisnofreeDMPSinthesolution however excess of lead ions As a consequence A and B peakred ox
2+increasedIncreaseofB correspondstothedepositionofPb duringtheaccu-ox
mulationstepPeaksBredandCredremainedpracticallyunchanged(Fig2C) Voltammetric titrationconfirmed themechanismof formationcomplexesofPb(DMPS)Hg(DMPS)andPb(Hg)Italsoconfirmedpossibilityofdetermination
2+Pb andDMPSinthesamesolution
4Conclusions
ValidationinmodelsampleofdrugDimavalandhumanurinespikedwithDMPSconfirmed that this method can be used for clinical purposes Voltammetric
2+titration of DMPS by Pb ions proved that it can be used for simultaneousdetermination of the drug and heavy metal ions in human urine Moreoverobtained LODs were two orders lower than those in the previously reportedvoltammetricmethod[14](Table1)
Acknowledgments
ResearchwascarriedoutwithintheframeworkofSpecificUniversityResearch(SVV260560)TheauthorsthanktheCzechScienceFoundation(GACRprojectNo20-01589S)
References
[1] AnHHLuchakMCopesRLeadtoxicityAsystematicreviewofrecentlypublishedcasesClinToxicol53(2015)757ndash758
[2] KimYLustMRKreimerbirnbaumM23-Dimercaptopropane-1-sulfonate(DMPS)inthetreatmentoflead-poisoningFasebJ2(1988)A1820ndashA1820
[3] AposhianHVDMSAandDMPS ndashwater-solubleantidotesforheavy-metalpoisoningAnnuRevPharmacol23(1983)193ndash215
74 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Table 1ComparisonofvoltammetricmethodsforDMPSdetermination(LDR
Method Workingelectrode Lineardynamic LOQ LOD Refminus1 minus1 minus1 rangemicromolL micromolL micromolL
LSV glassy-carbonelectrode 18ndash140 41 14 [14] modifiedwithmulti-walled 260ndash690 carbonnanotubes DPCSV p-AgSAE 03ndash20 08 03 thisworkDPCSV m-AgSAE 01ndash10 03 01 thiswork 10ndash100
[4] BjorklundG Crisponi G Nurchi VM Cappai R Djordjevic AB Aaseth J A review oncoordinationpropertiesof thiol-containingchelatingagents towardsmercury cadmiumandleadMolecules24(2019)3247
[5] DonnerAHrubyKDMPSinthetreatmentofacuteandchronicheavy-metalpoisoningActaMedAust14(1987)10ndash10
[6] DonnerAHrubyKPirichKKahlsPSchwarzacherKMeisingerVDimercaptopropan-sulfonate(DMPS) inthetreatmentofacute lead-poisoningVetHumToxicol29 (1987)37ndash37
[7] Blanusa M Varnai VM Piasek M Kostial K Chelators as antidotes of metal toxicityTherapeuticandexperimentalaspectsCurrMedChem12(2005)2771ndash2794
[8] BarekJMoreiraJCZimaJModernelectrochemicalmethodsformonitoringofchemicalcarcinogensSensors-Basel5(2005)148ndash158
[9] Josypcuk B FojtaM Yosypchuk O Thiolatemonolayers formed on different amalgamelectrodesPartIIPropertiesandapplicationJElectroanalChem694(2013)84ndash93
[10] YosypchukBMarecekVPropertiesofthiolatemonolayersformedondifferentamalgamelectrodesJElectroanalChem653(2011)7ndash13
[11] Alvarez JMF SmythMRCathodic strippingvoltammetryofpyridine-2-thiolandsomerelated-compoundsAnalyst114(1989)1603ndash1605
[12] DanhelABarekJAmalgamelectrodesinorganicelectrochemistryCurrOrgChem15(2011)2957ndash2969
[13] Fadrna R Polished silver solid amalgam electrode Further characterization and appli-cationsinvoltammetricmeasurementsAnalLett37(2004)3255ndash3270
[14] ZiyatdinovaGKGrigorevaLVBudnikovGKElectrochemicaldeterminationofunithioland lipoic acid at electrodesmodifiedwith carbonnanotubes J Anal Chem64 (2009)185ndash188
[15] HrdlickaVChoinskaMRedondoBRBarekJNavratilTDeterminationofheavymetalpoisoning antidote 23-dimercapto-1-propanesulfonic acid using silver solid amalgamelectrodeElectrochimActadoiorg101016jelectacta2020136623
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 75
Fig 1Structureofcanagliflozin
1Introduction
Canagliflozin is a selective sodium-glucosecotransportertype2inhibitorused for the treatment of type 2 dia-betes mellitus Canagliflozin inhibitssodium-glucose cotransporter type 2present in proximal tubules of the
Canagliflozin oxidation study using electrochemical flow cell and comparison with hydrogen peroxide oxidation
a a bFILIPVYMYSLICKY TOMA S KR IZ EK JAKUBHER T
a DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublicfvymyslickygmailcomb ZentivaGroupasUKabelovny13010237Prague10CzechRepublic
AbstractBystandardstheeffectonoxidationofanactivesubstanceistestedusinghydrogenperoxidesolutionatelevatedtemperatureinastresschamberfor1ndash7daysAnalternativewaytostudytheeffectofoxida-tion on an active substance is to use an electrochemical flow cellSolutionwith active substance flows at low flow rate into a smallreactorwheretheactivesubstanceisoxidizedonworkingelectrodesurfaceTheelectrolytestreamwiththeoxidizedactivesubstanceisthen directed to the sample collector Products of electrochemicaloxidationareanalyzedbyhighperformanceliquidchromatographywithultravioletndashvisiblespectrophotometrydetectionCanagliflozinhasbeenusedbecauseitsmaindegradationpathwayisoxidationThedesign of experiments approach was used to explore the experi-mentalspaceandoptimizeexperimentalconditionsofoxidationTheresultsoftheoxidationstudyperformedintheelectrochemicalflowcellwerestatisticallycomparedwiththeresultsofastandardstudyusinghydrogenperoxidesolutionThemostsuitableconditionsforelectrochemical oxidation were found Electrochemical oxidationproducedcomparableamountsofimpuritiesaschemicaloxidationwithhydrogenperoxide
KeywordscanagliflozindesignofexperimentselectrochemicalflowcellHPLCoxidation
76 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
kidneywhichrestrictsglucoseabsorptioninthekidneytherebyincreasingtheurinaryexcretionofglucoseandloweringthelevelofglucoseintheblood[1]TheformulaofcanagliflozinisC H FO SthestructureofcanagliflozinisinFig1The24 25 5
IUPAC name of canagliflozin is (2S3R4R5S6R)-2-[3-[5-(4-fluoro-phenyl)-thiophen-2-ylmethyl]-4-methyl-phenyl]-6-hydroxymethyltetrahydro-pyran-345-triol[2]CanagliflozinisawhitepowderinsolubleinwaterbutverysolubleinorganicsolventslikemethanolordimethylsulfoxideCanagliflozinissoldundertradenameINVOKANA Manyauthorshavestudiedtheelectrochemicalpropertiesofactivesubstancesin the literature One example is the study of electrochemical behaviour andoxidationofbromhexineThesepropertieswerestudiedusingdifferentialpulsevoltammetryandcyclicvoltammetryonacarbonelectrodeTheresultsofelectro-chemicalmethodswerecomparedwithhighperformanceliquidchromatography(HPLC)analysis[3]Anotherexampleisthestudyofelectrochemicalbehaviourand degradation study performed on the active substance atomoxetineDegradation was studied using differential pulse voltammetry and cyclicvoltammetryonacarbonelectrodeTheresultswerealsocomparedwithHPLCanalysis[4]Electrochemicalmethodsareusedmainlytostudythemechanismofoxidationbutinthisworktheelectrochemicalmethodwasusedtodegradetheactivepharmaceuticalingredient In the stability studies of active pharmaceutical ingredient properties theinfluence of temperature pH light and oxidation is studied [5] By TheInternationalCouncilforHarmonisationofTechnicalRequirementsforPharma-ceuticals forHumanUse (ICH) standards the influenceof oxidationon activepharmaceutical ingredient is studied using hydrogen peroxide at roomtemperatureorincreasedtemperatureinthestresschamberduring1ndash7days[6]Analternativewaytostudytheinfluenceofoxidationonactivepharmaceuticalingredient is using electrochemical flow cellwhere an electrolytewith activepharmaceuticalingredientisdrivenbylowflowrateintothesmallreactorInthesmallreactortheactivepharmaceuticalingredientisoxidizedonthesurfaceoftheworkingelectrodeThestreamofelectrolytewithoxidizedactivepharma-ceuticalingredientisdriventothesamplecollectorProductsofelectrochemicaloxidationareanalysedbyHPLCUVVISThedesignofexperimentsapproachwasusedfordevelopmentofanalternativemethodofoxidationofcanagliflozinusinganelectrochemical flowcellThedesignofexperimentsapproachwasusedtoexplore the experimental space of the method and to find the optimalexperimentalconditionsofelectrochemicaloxidationofcanagliflozin
2 Experimental
21Materialandreagents
Canagliflozin(ZentivaCzechRepublic)999methanol(HoneywellGermany)98ammoniumdihydrogenphosphate(Sigma-AldrichJapan)35ortho-phos-phoricacid(PentaCzechRepublic)25ammonia(LachnerCzechRepublic)30hydrogenperoxide (LachnerCzechRepublic)water forchromatography
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 77
Fig 2Schemeoftheelectrochemicalflowcell(1)input(2)workingelectrode(3)gasket(4)refe-renceelectrode(5)counterelectrode
wasobtainedbypurifyingdemineralisedwaterusingMilliporetypeSynergyUVpurificationinstrument
22Instruments
An Agilent 1290 HPLC system (Agilent Technologies Germany) with highpressure pump autosampler thermostat and DAD detector was used for allexperimentsThePinnacleDBbiphenylcolumn(100times21mm19micromRestekUSA)wasusedforseparationIntheHPLCmethod10mMammoniumdihydrogenphosphatebufferpH=25wasusedascomponentAandmethanolascomponentBofthemobilephaseThegradientprogramwassetasfollowst(min)B01555010551790229023152515Theflowrateofthemobilephasewas
ndash104mlmin and the injection volume was 2μl The detector operated at awavelengthof220nmTheautosamplertemperaturewassetat20degCandthecolumntemperatureat60degCTheEmpowersoftwarewasusedforevaluationForelectrochemicaloxidationelectrochemicalflowcellfromALS(Japan)wasusedGlassycarbonelectrode(=6mm)andsilversilverchlorideelectrodewereusedasworkingandreferenceelectroderespectivelyTheschemeofelectrochemicalflowcellisinFig2ElectrodeswereconnectedwithpotentiostatPalmSens3fromPalmsens (Netherlands) AnElmasonic S15Hultrasonic bath fromElma (Ger-many)wasusedforsamplepreparationForpHmeasurementspHmeterJenway3540fromJenway(UnitedKingdom)wasused
3Resultsanddiscussion
AtthedevelopmentofthemethodforthestudyofcanagliflozinoxidationusingelectrochemicalflowcellitwasfirstnecessarytofindtheoptimalconditionsofelectrochemicaloxidationThedesignofexperimentsapproachwasusedChosen
78 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ndash1Fig 3Cyclicvoltammogramofcanagliflozin(concentrationofcanagliflozin11mgml electrolyte300 mM ammonium dihydrogen phosphate pH = 40 and methanol (11 vv) and scan rate
ndash1001Vs )
independentvariablesandtheirlevelswereconcentrationofelectrolyte(100200300mM)pHofelectrolyte(406080)cellsize(50100200500microm)and
ndash1flowrate(0102504mlh )ThereducedcombinatorialdesignwasusedIntheModde12 software aworksheet containing 11 experimentswas created Theworkingpotentialof12Vwasselectedbasedoncyclicvoltammetryofcanagli-flozininFig3Fromthisfigureitcanbeseenthattheoxidationofcanagliflozinoccursintheregionfrom11Vto14VAllexperimentswereperformedwith
ndash111mgml canagliflozin samples The glassy carbon electrodewas used as aworkingelectrodeandthesilversilverchlorideelectrodewasusedasareferentelectrode The canagliflozin samples oxidized in the electrochemical flow cellunder theexperimentalconditionsgivenby theworksheetweremeasuredbyHPLCwithUVVISdetectionDependentvariablespeakareasofimpuritiesandpercentage of peak areas of impurities obtained from chromatograms wereevaluated by the partial least squaremethod in theModde 12 software Thevariableimportanceintheprojectionplottoolwasusedforinterpretationofthedata as a whole The significance values of the independent variables were
ndash1evaluatedbufferpH=137flowrateof125mlh bufferconcentration061mMandcell size04micromFromthis tool itwasconcluded that theelectrochemicaloxidationofcanagliflozinisthemostaffectedbythepHoftheelectrolyteandflowrateoftheelectrolyteUsingtheoptimizertoolthemostsuitableconditionsfor
ndash1the oxidation of canagliflozin were evaluated flow rate 01 ml h 300 mMammoniumdihydrogenphosphate pHof electrolyte40 and cell size500micromUsingonefactoratthetimeapproachthedependenceofthecellsizeonthetotalsumofimpuritieswastested(Fig4A) Itisvisiblefromthegraphthatthesmallerthecellweusethemoreoxidationproducts are formed Based on the graph the most suitable conditions were
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 79
Fig 4(A)Optimizationofelectrochemicaloxidationconditionsdependenceofsumofimpuritiesoncellsize(B)Chromatogramofasampleoxidizedundermostsuitableconditions
ndash1adjustedtoflowrate01mlh 300mMammoniumdihydrogenphosphatepHofelectrolyte40andcellsize12micromUnderthemostsuitableconditionsarepeat-ability test was performed by ten independent oxidation experiments Therelativestandarddeviationofthepercentageareaofcanagliflozinwas164atasignificant level of 095 The chromatogram of sample oxidized under mostsuitableconditionsisinFig4BThestandardstudyoftheeffectofoxidationoncanagliflozinusinghydrogenperoxideaccordingtoICHguidelineswasperfor-medThestudywasperformedundertwosetsofexperimentalconditionsInthefirstcaseasolutionof50methanolwiththeadditionof3H O wasusedIn2 2
thesecondcasetheconditionsintheelectrochemicalflowcellweresimulatedA300mMammoniumdihydrogenphosphate pH40 andmethanol in a ratio11(vv)withtheadditionof3H O wasusedSamplesfortheoxidationstudy2 2
werestressedinastabilitychamberfor13and7daysattheconstanttempe-ratureof50degC ThetotalsumsofimpuritiesformedduringchemicaloxidationusinghydrogenperoxideinbothmediawerecomparedasisshowninFig5AItisobviousthat
80 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 5(A)Acomparisonofastandardoxidationstudyusinghydrogenperoxidein50methanolwith added buffer andwithout them (B) Chromatogramof sample oxidized electrochemically(C)Chromatogramofsampleoxidizedchemically
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 81
ammoniumphosphatesuppressesoxidationofcanagliflozinThereasonofthisphenomenonisunknownFig5BandFig5CshowchromatogramsofsamplesoxidizedelectrochemicallyandchemicallyrespectivelyItcanbeseenthatfiveimpuritieswereformedbybothtypesofoxidationhoweverindifferentamounts
4Conclusion
AnalternativemethodfortheoxidativestudyofcanagliflozinwasdevelopedThedesign of experiments approach was used in the method development ThedevelopedmethodworkswithRSDof165(α=095)Oxidationofcanagliflozinbythedevelopedmethodproducedfiveimpuritiesthatareidenticalwiththoseproducedusingthestandardoxidationstudywithhydrogenperoxide
Acknowledgments
ThisworkhasbeensupportedbyCharlesUniversityResearchCentreprogramNoUNCESCI014SVV260560projectandpharmaceuticalappliedresearchcenter(TheParc)
References
[1] ChaoECCanagliflozinDrugsFuture36(2011)351ndash357[2] NislySAKolanczykDMWaltonAMCanagliflozinanewsodium-glucosecotransporter2
inhibitorinthetreatmentofdiabetesAmJHealthSystPharm70(2013)311ndash319[3] Turchan M Jara-Ulloa P Bollo S Nunez-Vergara LJ Squella JA Alvarez-Lueje A
VoltammetricbehaviourofbromhexineanditsdeterminationinpharmaceuticalsTalanta73(2007)913ndash919
[4] Perez-OrtizMMunoz C Zapata-Urzua C Alvarez-Lueje A Electrochemical behavior ofatomoxetineanditsvoltametricdeterminationincapsulesTalanta82(2010)398ndash403
[5] Baertschi SW Alsante KM Reed RA Pharmaceutical Stress Testing Predicting DrugDegradationLondonInformaHealthcare2011
[6] RignallA ICHQ1A(R2) stability testing of newdrug substance andproduct and ICHQ1CstabilitytestingofnewdosageformsInICH Quality Guidelines An Implementation GuideATeasdaleDElderRWNims(Eds)HobokenWiley2017p3ndash44
82 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
AlthoughDNArepresentsarelativelystablecomponentfromthechemicalpointofviewitremainsconstantlyexposedtoalargenumberofchemicalorphysicalagentscausingchemicalchangesinDNAmoleculesthatoccurintheenvironmentoraremajororminorproductsofcellularmetabolism[1] One-electronoxidationoftheDNArepresentsadamagingprocesswheretheloss of an electron (oxidation) fromduplexDNA results in the formationof anucleobase radical cation (electron ldquoholerdquo) that is subsequently consumed inchemicalreactionsthatoftenleadtomutationsAdefiningcharacteristicoftheone-electronoxidationofDNAisthepreferentialreactionattheguaninemoietythatisdetectedasstrandcleavagefollowingchemicalorenzymatictreatmentoftheoxidizedDNA[2ndash3]
Novel hybrid electrochemical DNA biosensor for monitoring oxidative DNA damage via oxidationreduction signals of low molecular weight double-stranded DNA
MICHALAUGUSTINVLASTIMILVYSKOCIL
UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova812843Prague2CzechRepublicmichalaugustinnaturcunicz
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 83
AbstractDeoxyribonucleicacid(DNA)representsamajortargetmoleculeformanydamagingagentscausingunfavorablechangesinastructureofDNAmoleculethatbindandinteractwithDNAThusahighdemandforreliabletoolsregardingabettercomprehensionofthenatureofDNAdamagingprocessesstillrepresentsoneofthemaingoalsinthisareaHereinwedescribeadevelopmentofanovelhybridelectro-chemicalDNAbiosensorbasedonanldquoedge-planerdquopyrolyticgraphiteelectrode (EPPGE) in connectionwith an elementaryoptimizationprocessprovidingacloserresolutionoftheredoxprocessesoflowmolecularweightdouble-strandedDNA(dsDNA)attheEPPGESub-sequentanalyticalapplicationincorporatinganemploymentofthemodel structure K [IrCl ] (representative of transition metal2 6
complexes)andevaluationofitsdamagingeffectinrelationtoDNAbymeansof linear sweepvoltammetry resp square-wavevoltam-metryarealsopresented
KeywordsbiosensordamageDNAgraphitevoltammetry
DNA-based electrochemical biosensors are successfully used in variousapplicationssuchasmonitoringandevaluatingthemechanismsof interactionbetweenDNAandvariousdrugsordamagingagentsrapidmonitoringoftracemetalsorpollutantspresent in theenvironmentordirectmonitoringofDNAhybridizationprocesses[4] Theelectrochemicalactivityofnucleicacids(boththenativehigh-molecularonesaswellasoligonucleotides)isingeneralreferredtotheelectroactivityofitscomponents ndash nucleobases and sugar residues At mercury-based electrodesadenine and cytosine residues undergo reduction processes close to ndash14 V(againstSCE)inneutralorweaklyacidicmedium(givingrisetothepeakCA)Ontheotherhandallbaseshavebeenreportedtobeelectrochemicallyoxidizedatcarbonelectrodesbutonlyadenineand(particularly)guanineoxidationsignalshavebeenwidelyutilizedinelectrochemicalDNAbiosensors[5] In2017theelectrochemistryofnucleicacidsachievedanimportantmilestoneasthereductionoftheDNAoligonucleotideswasperformedataldquobasal-planerdquopyrolytic graphite electrode which provided wide potential window allowingboththeelectrooxidationaswellastheeletroreductionofthenucleobasesatasingleelectrodefortheveryfirsttimeDespitethesefindingsutilizationoftheaforementionedbiosensorintermsofanalyticalapplicationshasyettobeverifiedandremainsunclearuptothisdate[6] TheaimoftheproposedcontributionisapresentationofthedevelopmentprocessandsubsequenttestingofanoveltypeofhybridelectrochemicalDNAbiosensoranditsverificationasareliableanalyticaltoolintermsofmonitoringDNAdamage
2Experimental
21Reagentsandchemicals
Low molecular weight double-stranded DNA (dsDNA) derived from salmonspermwasobtainedfromSigma-AldrichGermanyStocksolutions(01mgmL)
of dsDNA were prepared in a 01 molL phosphate buffer of pH=74 (PB)Dipotassium hexachloroiridate (K [IrCl ]) was purchased from Sigma-Aldrich2 6
GermanyStocksolutions(0001molL)ofK [IrCl ]werepreparedinthePB2 6
22Apparatus
Voltammetric measurements were performed using the μAutolab IIIFRA2potentiostatgalvanostat(EcoChemieTheNetherlands)drivenbyaNOVA111software(MetrohmAutolabSwitzerland)Allmeasurementswerecarriedoutina three-electrode system using an ldquoedge-planerdquo pyrolytic graphite workingelectrode(EPPGE)withanelectroactivesurfacediameterof3mm(BASJapan)asilver|silver chloride reference electrode (Ag|AgCl|sat KCl) and a platinum
84 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
counterelectrode(ElektrochemickeDetektoryCzechRepublic)ina20mLglassvoltammetriccellatambienttemperature
23Preparationofthebiosensor
PriortotheeverymeasurementsurfaceoftheEPPGEwasmechanicallycleanedbygentlewipingoftheelectrodeonthesoftpolishingpadrinsedwithdistilledwaterAfterwardstheelectrodewasrinsedwithdistilledwaterandplacedinthePBforthesubsequentelectrochemicalactivationElectrochemicalactivationwasperformed in thePBbyapplyingpotentialof15V for240swithoutstirringApotentialpulseinworkingrangeofpotentials(00ndash15V)wasthenapplied Additional electrochemical activationwasperformed in the solutionof the
3minus4minusredoxindicator([Fe(CN) ] )byconsecutivecyclingintherangeofpotentials6
from10tondash08V(15scans)andfrom055tondash015V(10scans) The electrochemical DNA biosensor based on the EPPGE (dsDNAEPPGEbiosensor)was prepared by the adsorption of dsDNAon the EPPGEOptimal
parametersofthedsDNAadsorptionwereaconcentrationof01mgmLinthePB
(c )adepositionpotentialof07V(E )andanadsorptiontimeof5ming(dsDNA) dep
(t )withoutstirringthesolutionads
Atlasttheelectrodewasimmersedinthesolutionoftheredoxindicatorandtheconsecutivecyclingintherangeofpotentialsfrom055tondash015V(20scans)wasperformedinordertosecurethestabilityoftheoxidationreductionsignalsofdsDNAattheEPPGE
24Procedures
Theexperimentalparameterswereasfollowssquarewavevoltammetry(SWV)inthePBwithapulseamplitudeof20mVafrequencyof50Hzascanrateof750mVsandapotentialstepof15mVlinearsweepvoltammetry(LSV)inthePBwithscanratesof02ndash10Vandapotentialstepof24mVAllcurveswere
recordedthreetimes(n=3)
3Resultsanddiscussion
Sincetheclosestresolutionoftheprocessesassociatedwiththeelectroreductionof singleDNAcomponents at pyrolytic graphitehasbeenperformedwith theldquobasal-planerdquopyrolyticgraphiteelectrode(BPPGE)wehavedecidedtotakeovercorrespondingexperimentaltechniqueandconditions(LSVscanrateof10Vssteppotentialof24mV)attheverybeginningofouroptimizationprocesswiththeEPPGE[6] Inthisparticularcaseitispossibletonoticetheoccurrenceofthetwomixedvoltammetricpeaksatthedefaultexperimentalconditions(greenlineFig1A)selectedforthereductionofdsDNAattheEPPGEBygraduallydecreasingthe
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 85
Fig 1Baseline-correctedLSVrecordingscorrespondingtothereductionofdsDNAattheEPPGEfordifferentvaluesofscanrate(02ndash10VsFig1A)respbaseline-correctedLSVrecordingscorres-pondingtothereductionofdsDNAattheEPPGEandthenegativetestperformedunderthesameexperimentalconditionswithintheblanksolution(phosphatebuffer)atthebareEPPGE(03VsFig1B)
scan rate the optimal conditions (νle03Vs) were found and the mutualseparationof the signalswas allowedndash characterizedby thepresenceof twosinglewell-developedvoltammetricpeaksatpotentialsofndash175Vrespndash190V(03Vs orange line Fig 1A) Taking into account previous work regardingprocessesassociatedwiththereductionofDNAatthemercuryelectrodesrespBPPGE we can assume that the peak appearing at the potential of ndash175 Vcorresponds to themixedpeak for the reductionof the cytosine and adenineresidueswithindsDNA(peakCA)[5ndash6] Closer resolution of the second voltammetric peak appears to be farmoreproblematic Regarding our previous study we have discovered that theutilizationofdifferentE fortheadsorptionofdsDNA(E lt07V)isconnecteddep dep
withanappearanceofthethirdoxidationsignal(besidestheoxidationsignalsofguanine resp adeninemoieties) at apotential of073V corresponding to theoxidationoffreeguaninebases(FGBs)presentwithinthesolutionofdsDNAInthiscasewecanassumethatthepeakappearingatapotentialofndash190VcanpossiblyrepresentthereductioncounterpartofFGBspresentwithinthesolutionofdsDNAThisassumptioncanalsobesupportedbytheaforementionedstudyandbythefactthatthereductionsignalatsuchahighnegativepotentialcanbeobservedfortheoligodeoxynucleotidescontainingguanineresidues[6] AdditionallyinordertoverifythetruenatureofthereductionsignalsdepictedatF ig1Bandtoexcludetheoptionthattherelatedsignalsdonotrepresenttheproductsofpriorelectrochemicalactivationof theEPPGE(variousCndashObasedchemicalspecies)wehavedecidedtoperformanegative(control)testwithinthe
86 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2Baseline-correctedLSVrecordingscorresponding to thereductionof thedsDNA(02Vs
Fig 2A)respbaseline-correctedSWVrecordingscorrespondingtotheoxidationoftheguanine(098V)respadenine(128V)moieties(075VsFig2B)attheEPPGEafteritsincubationinthephosphatebufferforadefinedtimeperiod(60ndash900s)
blanksolution(PB)employingthesameprotocolasforthedsDNAadsorptionattheEPPGEInthiscaseitispossibletoobservetheabsenceofanypronouncedvoltammetricpeakslinkedtothedsDNAadsorptionandonlythepresenceofoneirreversiblepeakatapotentialofndash153VcorrespondingtotheelectroreductionoftheCndashObasedmoietymoietieswhichdropsafterthedsDNAadsorptiontoonetenthofitsoriginalvalue(approximately) Perhaps the most important parameter regarding further optimizationprocessrepresentedthetime-dependentstabilityofthecorrespondingsignalsofdsDNAwhichcanbespecificallyimportantinrelationtothestudyofthetime-dependentoxidativedamageofdsDNA AsithasalreadybeenprovedasingleelectrochemicalactivationoftheEPPGEinthePBdoesnotrepresentasatisfyingtechniqueregardingstabilityofdsDNAoxidationsignalsat theEPPGEand theadditionalstabilization isachievedbyfurtherelectrochemicalactivationinthesolutionofaredoxindicator(Fig2B)[7]Basedonthisaverificationoftheproposedstabilizationprotocolintermsofthetime-dependent stabilityofdsDNAreduction signals in the solutionof thePBwithinthedefinedtimeperiod(60ndash900s)appearedasareasonablenextstep FromtheresultsdepictedinFig2Aitispossibletonoticethatwithinthefirst300sdsDNAreductionsignalsremainstableinrelationtothecurrentresponseaswellasintermsofthepotentialvalueWithanadditionalincubationtime(t )inc
(900sorangeline)thepeakcurrentofthevoltammetricsignalpresentatmorenegative potentials decreased which can probably be addressed as a slowprogressive elimination of the weak (electro)chemical forces related to theunspecificadsorptionoftheFGBsattheEPPGE
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 87
Fig 3Baseline-corrected SWV recordings corresponding to the oxidation of the guanine respadeninemoietiesattheEPPGEafteritsincubationinthesolutionofK [IrCl ](IR)foradefinedtime2 6
period(60ndash3600s)(075VsFig3A)andthecorrespondingrelativebiosensorresponses(ΔI )rel
evaluatedusingtheguanosine(turquoise)andadenosine(red)peaksplottedversustheincubationtime(Fig3C)Baseline-correctedLSVrecordingscorrespondingtothereductionofdsDNAattheEPPGEafteritsincubationinthesolutionofK [IrCl ](IR)fordifferenttimeperiods(60ndash3600s)2 6
(02 VsFig3B)andthecorrespondingrelativebiosensorresponses(ΔI )evaluatedusingtherelpeakCA(darkpink)plottedversustheincubationtime(Fig3-D)
AdditionallywehavedecidedtotesttheapplicabilityofthepresentedhybridbiosensorintermsofmonitoringdsDNAdamagecausedbyarepresentativeofone-electron oxidants ndash K [IrCl ] In this case the prepared dsDNAEPPGE2 6
biosensor was immersed into the solution of K [IrCl ] (0001molL) for the2 6
definedtimeperiod(60ndash3600s) In thecaseof theoxidationpath (SWVrecordingsdepicted inFig3A) it ispossible to observe a time-dependent decrease of the oxidation signal of theguaninemoietieswhereastheoxidationsignaloftheadeninemoietiesremainsunaffectedforthemostof the incubationperiodThisphenomenonis ingoodcorrelationwiththetheoreticalknowledgeregardingoxidativedamageofdsDNAcausedbyone-electronoxidants [3]Simultaneouslywith thisLSVrecordingsdepicted in Fig 3B followed the similar behavior (decrease in relation to the
88 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
currentresponseofthedsDNAreductionsignalndashpeakCA)asinthecaseofthesignal regarding oxidation of guanine moieties In addition according totheportionofthepreservedDNA(Fig3C3D)itispossibletoassumethatthepronouncedoxidativedamageofdsDNAcanbemonitoredquitepreciselynotonlydirectlyviathedsDNAoxidationsignaloftheguaninemoietiesbutevenindirectlythroughthedsDNAreductionsignalndashpeakCA
4Conclusions
Inthiscontributionwehavepresenteddevelopmentofanunorthodoxhybridelectrochemical DNA biosensor based on an EPPGE Optimization processconcerning some important parameters was performed as well as closerresolutionofthenatureofthereductionprocessesofdsDNAattheEPPGEwasachievedInordertoconfirmtheresultsoftheoptimizationprocessapplicabilityoftheproposedbiosensorhadbeenprobedintermsofmonitoringDNAdamagecausedbyK [IrCl ]Inthiscasethefinalresultshadprovedthattheprepared2 6
hybridbiosensorcanbeconsideredasaversatileanalyticaltoolformonitoringoxidativeDNAdamage(viaoxidationreductionsignals)andispresentedasafinealternative in comparisonwith conventional electrochemical DNA biosensorsprepared within the group of traditional transducer materials (mercury- orcarbon-based)
Acknowledgments
ThisresearchwassupportedbytheSpecificUniversityResearch(SVV260440)
References
[1] FojtaMDanhelAHavranLVyskocilVRecentprogressinelectrochemicalsensorsandassaysforDNAdamageandrepairTrACTrendsAnalChem79(2016)160ndash167
[2] GieseBSpichtyMWesselySLong-distancechargetransportthroughDNAAnextendedhoppingmodelPureApplChem73(2001)449ndash453
[3] Burrows CJ Muller JG Oxidative nucleobasemodifications leading to strand scissionChemRev98(1998)1109ndash1151
[4] DiculescuVC Chiorcea-PaquimAMOliveira-BrettAMApplications of aDNA-electro-chemicalbiosensorTrACTrendsAnalChem79(2016)23ndash36
[5] PalecekEJelenFElectrochemistryofnucleicacidsInElectrochemistryofNucleicAcidsandProteinsndashTowardsElectrochemicalSensorsforGenomicsandProteomicsPalecekESchellerFWangJ(edits)AmsterdamElsevier2005p74ndash174
[6] SpacekJDanhelAHasonSFojtaMLabel-freedetectionofcanonicalDNAbasesuraciland5-methylcytosineinDNAoligonucleotidesusinglinearsweepvoltammetryatapyrolyticgraphiteelectrodeElectrochemCommun82(2017)34ndash38
[7] AugustınMVyskocilVNovelelectrochemicalDNAbiosensorbasedonedge-planepyrolyticgraphite for DNA interaction studies In Proceedings of the 15th International StudentsConferenceldquoModernAnalyticalChemistryrdquoNesmerakK(edit)PragueFacultyofScienceCharlesUniversity2019p263ndash268
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 89
1Introduction
Cadmiumisoneofthemosttoxicmetalsanditswidespreadindustrialusesresultin increased environmental pollution Hence the development of sensitivemethodology for Cd determination is still highly desirable Chemical vaporgeneration(CVG)ofCdbythetetrahydroboratereductioninacidicmediumisasuitable alternative sample introduction technique compatible with atomicspectrometricdetectorsandofferingimproveddetectioncapabilityComparedtocommonliquidnebulizationCVGoffersseveraladvantagessuchassignificantlyhigheranalyteintroductionefficiencyandalsoanalyteseparationfromsamplematrix IncomparisontoCVGofcommonhydrideformingelementsthereisalackofliterature dealing with mechanistic aspects of CVG of Cd [1] as well as withstabilityandidentityofitsvolatilespecies(freeatomshydrideotherspecies)Very little information is also available on achieved generation efficiencyMoreover there are many discrepancies in the literature regarding optimum
Chemical vapor generation of cadmium for analytical atomic spectrometry
a b a b a aLINDASAGAPOVA BARBORAKODRIKOVA MILANSVOBODA STANISLAVMUSIL aJANKRATZER
a InstituteofAnalyticalChemistryoftheCzechAcademyofSciences Veveřiacute9760200BrnoCzechRepublicsagapovaiachczb DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublic
AbstractChemical vapor generation of cadmium volatile compounds wasoptimizedinordertodeterminetraceCdconcentrationsbyatomicabsorptionspectrometry(AAS)Severalreactionmodifiersbasedon
III+ II+ III+ IV+inorganicsaltsandcomplexesofCr Co Ti Ti weretestedtoimproveanalyticalperformanceandgenerationefficiencyTheuseofthese reaction modifiers resulted in 4ndash5 times enhancement insensitivity reflected also in corresponding increase of generationefficiency and better repeatability Generation efficiency wasdeterminedfromacomparisonbetweensensitivitiesobtainedwithchemicalvaporgenerationandconventionalsolutionnebulizationbothsimultaneouslycoupledwithinductivelycoupledplasmamassspectrometryTheidentityofthegeneratedcadmiumcompoundswillbediscussed
Keywordsatomicabsorption
spectrometryatomizationcadmiumchemicalvapor
generation
90 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
conditionsforCVGofCdAlthoughstrongacid(HClorHNO )isalwaysemployed3
asacarrierandNaBH asareductantsomeauthorsreportedvariousadditives4III+ III+ IV+(modifiers)basedontransitionmetalions(Cr Ti Ti )inthepresenceof
II+KCN[23]orCo inthepresenceofthioureaandascorbicacid[4]toimproveCdsignalssignificantly TheaimofthisworkwastoinvestigateCVGofCdinacomprehensivewayFirstlyCVGofCdwithoutandwithselectedmodifierswasoptimizedemployingatomicabsorptionspectrometry(AAS)asadetectorandexternallyheatedquartztube(QTA)astheatomizerSecondlytheeffectofatomizationtemperatureonCdsignalwasstudiedallowingthustodeducetheatomicormolecularstructureofgeneratedCd speciesThirdly generation efficiencyofCdvolatile specieswasquantified
2Experimental
21Reagentsandchemicals
minus1Boiled and bubled (Ar per 30min) deionizedwater (lt 01 μScm UltrapurWatrex USA) was used to prepare all solutionsWorking Cd standards were
minus1preparedfrom1000mgL Cdstocksolution(AstasolAnalytikaCzechRepublic)minus1bydilutionin01ndash048molL HCl(basedonthemodifieremployed)from37
HCl(paMerckGermany)Theoptimumgenerationconditionsbeingdifferentforeachmodifiertestedare listedinTable1ThereductantwasasolutionofNaBH (ge 97 Sigma-Aldrich Germany) in 04 (mv) KOH (pa Merck4
Germany) prepared fresh daily The solutions of modifiers were prepared as2+followsCo waspreparedfromCoCl 6H O(ge990PENTACzechRepublic)2 2
3+the solution of Cr from Cr(NO ) 9H O (ge 9999 tracemetal basis Sigma-3 3 23+AldrichGermany)thesolutionofTi fromTiCl solution(about15in10HCl3
4+Sigma-AldrichGermany)andthesolutionofTi fromTiOSO (ge999Sigma-4
AldrichGermany)Tostabilizethelattersolution1H SO wasusedprepared2 4
bydilutionof96H SO (paLach-NerSlovakia)SolutionofKCN(ge9702 43+ 3+FlukaSwitzerland)wasusedasasecondmodifierwhenworkingwithCr Ti or
4+ ndash3Ti asmodifiersitsconcentrationvariedfrom008to016moldm depending
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 91
Table 1OptimumconditionsforchemicalvaporgenerationofCdinthepresenceofmodifiersandtheirabsence
onthemetalionThiourea(CH N Sge980LachemaBrno)andascorbicacid4 2
(C H O ge997Riedel-deHaenGermany)wereusedasmodifierscombined6 8 62+withCo
22Instrumentation
221Chemicalvaporgenerationsystems
TwoCVG flow injection systemswere employed either a two channel systemwithoutadditionofamodifier(seeFig1A)orafourchannelsystemallowingadditionofmodifiers(seeFig1B)
ndash1 TheflowratesofHClandNaBH were42and10mLmin respectivelyinatwo4ndash1channelsystem(Fig1A)whiletheywerebothkeptat10mLmin inthefour
channelsystem(Fig1B)Theflowratesofmodifiersinthefourchannelsystemndash1were05mLmin Thevolumeofthesampleloopwas015mLinbothsystems
ndash1Carriergasflowrateof75mLmin Arwascontrolledbyamassflowcontroler(Cole-ParmerUSA)
92 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
(A)
(B)
Fig 1Schemesofthechemicalvaporgenerationflowinjectionsystemwith(A)twochannels(nomodifiers)and(B)fourchannels(modifiersemployed)
222Atomicabsorptionspectrometry
ThePerkin-Elmermodel503atomicabsorptionspectrometer(BodenseewerkGermany)wasequippedwithaCdelectrodelessdischargelamp(Perkin-ElmerUSA)operatedat228mAThemeasurementswereperformedat2288nmusinga07nmslitwidthTheShimadzumodelAA-7000atomicabsorptionspectrometer(ShimadzuJapan)wasalsousedACdhollowcathodelamp(PhotronAustralia)operatedat2288nmlinewith07nmspectralbandpassandalampcurrentof12mA Signals were recorded for 2 minutes and peak areas were taken forevaluation The QTA was heated electrically to the temperature required byfurnace(PerkinElmer)andanin-housemadefurnacecontrolledbytheREX-C100controller(SysconIndianaUSA)withtheK-typethermocouplesensor(OmegaEngineeringUSA)223QuantificationofCVGefficiencybyICP-MS
Overall CVG efficiency of Cd was quantified bymeans of inductively coupledplasmamassspectrometry(ICP-MS)fromcomparisonoftheslopesofcalibra-tionsobtainedwithnebulizationliquidCdstandardstothoseobtainedwithCVGThe efficiency of liquid nebulization was quantified using a modified wastecollection method (see reference [5] for details) The Agilent 7700x ICP-MSinstrument(AgilentUSA)wasoperatingat1600WofRFpowerThesignalwas
111 125monitoredat Cdisotopeandcorrectedforthesignalofinternalstandard( Tendash11000ngmL Tein2HNO )NebulizeranddilutionArgasflowrateswere11503
ndash1and0mLmin respectively
3Resultsanddiscussion
31Chemicalvaporgenerationconditions
UnivariateoptimizationswereperformedtofindoptimumconditionsforCVGofCdinpresenceandabsenceofmodifiersTheparameterstobeoptimizedwerecarrieracid(HCl)concentrationreductant(NaBH )concentrationmodifierIand4
modifier II concentrations carrier gas flow rate (Ar) length of reaction coilsRCI-III (see Fig 1B) The optimum conditions for individual modifiers aresummarizedinTable1
32IdentityofCdspecies
ThepeakareasofgeneratedCdspecieswereforagivenmodifiermeasuredintheQTAheatedto900degCandnon-heatedQTAsubsequentlyOptimumCVGconditionswere employed as summarized in Table 1 This simple experiment allowsdistinguishingbetweenatomic(freeatoms)andmolecularformsofgenerated
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 93
Fig 2RelativesignalofgeneratedCdspeciesmeasuredintheQTAheatedto900degC(blackbars)andnon-heatedQTA(whitebars)withoutorinthepresenceofmodifiers
speciesOnlyfreeatomscanbedetectedinnon-heatedQTAsimilarlyasincaseofmercurycoldvaporsOn thecontrarymolecularanalytespeciesareatomizedat900degCAsaconsequencethesignalregisteredintheheatedQTAcorrespondstobothatomicandmolecularspeciesgeneratedItmustbehighlightedthattheresidencetimeoffreeatomsintheatomizerisdependentonQTAtemperatureduetogasexpansionAsaconsequencethesignalinQTAheatedto900degCshouldreach25ofthesignalatambienttemperaturetakingintoaccountthatonlyfreeatomsaregeneratedSincethetemperaturealongtheopticalarmofQTAisnotdistributedhomogeneouslydecreasingtobothendtheeffectivetemperatureoftheatomizerislowerOurexperimentswithCVGofHgrevealedsignalinheatedQTAisaround40[6]TheresultsreachedforCVGofCdaredepictedinFig2ThesignalofCdinheatedQTAisaround50ofthesignaldetectedinnon-heatedQTA
4+whennomodifierisemployedorusingTi asthemodifierindicatingclearlyfreeCdatomsarethedominantvolatilespeciesgeneratedOnthecontraryalmostno
2+differenceinpeakareaswasobservedforCo asthemodifierwhilethesignalinheatedQTAwas even 5 times higher in heatedQTA compared to non-heated
3+atomizer with Cr as the modifier suggesting the dominant contribution of3+molecularstructurestoCdsignalespeciallyincaseofCr KCNreactionsystem
33Generationefficiency
TheoverallCVGefficiencywasestimatedfromacomparisonbetweensensitivitiesobtainedwithCVGsampleintroductionandconventionalsolutionnebulizationICP-MSunder the sameexperimental conditionsNebulization efficiency for aMicroMISTnebulizerwasdeterminedas79plusmn01ThegenerationefficiencyofCd was derived from the sensitivity enhancement between CVG and liquidnebulizationTheresultsaresummarizedinTable2indicatingthatCVGwithout
94 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
modifiersisonlycatwotimesmoresensitivecomparedtoliquidnebulization3+ 4+Generation efficiency of Cd increases to 60 in the presence of Ti and Ti
modifiers
4Conclusions
CVG of Cd was thoroughly optimized in the presence of selected modifiersreportedpreviouslyintheliteratureGenerationefficiencyofCdintheabsenceofanymodifierswasquantifiedto15whileitcanbeincreasedupto60inthe
3+ 4+presenceofTi KCNorTi KCNasmodifiersFreeCdatomsseemtobe the4+dominantCdformgeneratedintheabsenceofanymodifiersorusingTi KCN
3+modifierwhile rathermolecularCd structuresaregenerated inCr KCNand2+Co thioureaascorbicacidreactionsystems
ExperimentsareinprogresstofinishthiscomprehensivestudyOnlythebestmodifierwillbefurtherusedforCVGofCdtobecoupledwithotherspectrometricdetectorsandappliedtocertifiedreferencematerialsandrealsamples
Acknowledgments
ThisresearchhasbeensupportedbytheCzechScienceFoundationundercontract18-01116SandbytheInstituteofAnalyticalChemistryoftheCzechAcademyofSciences(InstitutionalResearchPlannoRVO68081715)andCharlesUniversity(ProjectnoSVV260440)
References
[1] PitzalisEAngeliniDMascherpaMCDacuteUlivoAInsightintothemechanismscontrollingthechemicalvaporgenerationofcadmiumJAnalAtSpectrom33(2018)2160ndash2171
[2] ArslanZYilmazVRoseLEfficientgenerationofvolatilecadmiumspeciesusingTi(III)andTi(IV)andapplicationtodeterminationofcadmiumbycoldvaporgenerationinductivelycoupledplasmamassspectrometryMicrochemJ123(2015)170ndash178
[3] YilmazVRoseLArslanZLittleMDOn-linechemicalvapourgenerationofcadmiuminthepresenceofhexacyanochromate(III)fordeterminationbyinductivelycoupledplasmamassspectrometryJAnalAtSpectrom27(2012)1895ndash1902
[4] Y Lu SunHW YuanCG YanXP Simultaneous determination of trace cadmiumandarsenic inbiologicalsamplesbyhydridegeneration-doublechannelAFSAnalChem74(2002)1525ndash1529
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 95
Table 2GenerationefficiencyofchemicalvaporgenerationofCdasquantifiedbyICP-MS
Modifiers Generationefficiency
Nomodifiers 15plusmn13+Cr KCN ndash2+Co thioureaascorbicacid ndash3+Ti KCN 58plusmn24+Ti KCN 61plusmn2
[5] VyhnanovskyJStrugeonREMusilSCadmiumassistedphotochemicalvaporgenerationoftungsten fordetectionby inductively coupledplasmamass spectrometryAnal Chem91(2019)13306ndash13312
[6] MigasovaMMatousekTSchrenkovaVZ ıdekRPetry-Podgorska IKratzer JMercuryvolatilespeciesgenerationfromHClandTRISbuffermediaAnalChimActa1119(2020)68ndash76
96 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Atomicfluorescencespectrometry(AFS)coupledwithvapourgenerationisanultrasensitive analytical method for determination of various elements ItsanalyticalperformancecanbecomparabletoICP-MSwithliquidnebulizationbutatsubstantiallylowercost[1]SampleintroductiontoAFSisacrucialstepoftheanalyticalproceduresincetheanalytehastobeintroducedtotheatomizerintheformofitsvolatilespecies Hydridegeneration(HG)isamaturetechniqueofsampleintroductionduringwhichvolatileanalytehydridesare formedbyreactionwithareducingagenttypicallysodiumborohydrideAnewemergingtechniquephotochemicalvapourgeneration(PVG)employsUV irradiationof theanalyte in liquidphase in thepresenceofaphotochemicalagent(usuallyalowmolarmassorganicacidformic
minusoraceticacid)Highlyreducingradicalspecies(HbullRbullandCOObull )andaquatedelectronsare formedduring irradiationandreactwith theanalyte to form its
Photochemical vapour generation of bismuth coupled with atomic fluorescence spectrometry
ab ab a aBARBORASTA DLEROVA JAROMIRVYHNANOVSKY JIR IDE DINA STANISLAVMUSIL
a InstituteofAnalyticalChemistryoftheCzechAcademyofSciences Veveřiacute9760200BrnoCzechRepublicstadlerovaiachczb DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublic
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 97
AbstractPhotochemical vapour generation of bismuth was successfullycoupledwithnon-dispersiveatomic fluorescencespectrometry forthefirsttimeVolatilespeciesofBiweregeneratedusingastandardmercurylow-pressuretubelampandacoiledreactorfromareaction
2+mediumwhichwas composedof acetic and formic acid Co ionswereusedasasensitizerOptimizationofatomizationconditionsinaflame-in-gas-shieldatomizerwasperformedThismethodologywascomparedtothecommonlyemployedhydridegenerationapproachAbsolutelimitofdetectionof68pgwasachievedwithphotochemicalvapour generation which is still about 7 times worse than withhydride generation The developed methodology was successfullyverifiedbyBideterminationinareferencematerialofwater
Keywordsatomicfluorescence
spectrometrybismuthhydridegenerationphotochemicalvapour
generation
volatilespeciesInbothcasesthegeneratedvolatilespeciesoftheanalytehavetobeseparatedfromtheliquidphaseinthegas-liquidseparatorandarecarriedtotheatomizerbyacarriergas[2] InthisworkanatomizerdesignedspecificallyforAFStheflame-in-gas-shieldatomizerwasused(Fig1) It consistsofaverticalquartz tubesuppliedwithargon and hydrogen together with the analyte volatile species Moreover acapillaryisinsertedintheverticalaxisoftheverticaltubethroughwhichoxygenisintroducedA hydrogen-oxygenmicroflameburns on top of the capillary Themicroflameisshieldedfromtheambientatmospherebyaflowofargonwhichisintroducedthroughashieldingunitfittedaroundtheverticaltube[34] Theaimofthisworkwastooptimizeatomizationconditionsintheflame-in-gas-shieldatomizerusingPVGasasampleintroductiontechniqueandtocomparetheanalyticalcharacteristicsofPVGandHGforultrasensitivedeterminationofbismuthbyAFS
2Experimental
21Reagentsandchemicals
Deionized water (Ultrapur Watrex USA) was used for preparation of all thesolutionsWorkingBisolutionswerepreparedfreshdailybyserialdilutionof
ndash1stock1000mgl BistandardforAAS(Sigma-AldrichGermany)RegardingHG05(mv)NaBH in04(mv)KOHwasusedasa reductantAsolutionof4 ndash11mol l HClwasusedasacarrierandblank
98 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Flame-in-gas-shieldatomizerOHndashobser-vationheight
RegardingPVGformicacid(98paLach-NerCzechRepublic)andaceticacid (998 pa Lach-Ner CzechRepublic)were used for preparation of thereaction medium they were purified in a Teflon BSB-939-IR sub-boilingdistillation apparatus (Berghof Germany) The composition of the reactionmedium(40(vv)acetic125(vv)formicacid)wasoptimizedearlier[5]
ndash1The 5000mgl Co stock solution was prepared from cobalt(II) acetatetetrahydrate (pa Lach-Ner Czech Republic) and used as a sensitizer ofphotochemicalreactionTheoptimalconcentrationofCointhestandardsamples
ndash1andblanksolutionscorrespondedto50mgl (ref[5]) Acertifiedreferencematerial(CRM)-1643fTraceElementsinWater(NationalInstituteofStandardsandTechnologyUSA)wasusedtochecktheprecisionofthedevelopedmethodology
22Instrumentation
221Atomicfluorescencespectrometer
An in-house assembled non-dispersive atomic fluorescence spectrometerconstructedatourlaboratorywasusedforBideterminationandisdescribedindetail elsewhere [3] The detector output provided signals in microV Peak areacorrectedtobaselineandmainlysignaltonoiseratioweretheparametersusedtoevaluatethedata
222Hydridegeneratorphotochemicalvapourgeneratorandatomizer
A flow injection hydride generator was employed (Fig 2a) The reductantndash1 ndash1(12mlmin )andthecarrier(4mlmin )werepumpedbyaperistalticpump
Thesamplewasinjectedthrougha1mlsampleloopintotheflowofcarrierAglassgas-liquid separator (5 ml) with forced waste removal was employed forseparatingthegasphasecontainingbismuthanewhichwasthencarriedtotheatomizerbyargon Thephotochemicalvapourgenerator(Fig2b)consistedofthephotoreactorconstructedwitha15Wlow-pressureHggermicidallamp(Cole-ParmerUSA)wrappedaroundwith6mofPTFEtubing(1mmidinternalvolume471ml)The
ndash1reactionmedium(3mlmin )waspumpedbyaperistalticpumpThesamplewasinjected througha056ml sample loopApolypropylenegas-liquid separator
(15ml)withforcedwasteremovalimmersedinanicebath[6]wasemployedforseparatingthegasphasecontainingBivolatilespeciessubsequentlycarriedtotheatomizerbyargon Theflame-in-gas-shieldatomizerisdepictedinFig1detaileddescriptionisgiveninRef[3]Theobservationheight(OH)isdefinedasthedistancefromthetopofthecapillarytothecentreoftheopticalbeam
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 99
23Samplepreparation
CRMNIST1643fwasdilutedwith1MHCl80-foldforBideterminationbyHG-AFSRegardingBideterminationbyPVG-AFSthesampleneededtobeevaporatedtodrynessinordertogetridofnitricacidthatseriouslyinterferesatmMlevel[5]Avolume of 3ml of CRMwere pipetted into a 40 ml quartz vial evaporated(temperature asymp100 degC two replicates) and subsequently diluted ca 33-foldAsamplepreparationblank3mlofdeionizedwaterwaspreparedaswell
3Resultsanddiscussion
TheatomizationconditionsforHG-AFSwereoptimizedinourpreviouswork[3]TheseconditionswereusedasinitialtofindtheoptimumconditionsforPVG-AFSwith the flame-in-gas-shieldatomizerwith respect to sensitivityandsignal tonoiseratioFirstlythehydrogenfractionintherange10ndash16wasoptimizedatconstant total gas flow rate (sum of total argon and total hydrogen) of
ndash1500mlmin the lower the hydrogen fraction the better However at 10hydrogenfractiontheflamewasnotstableenoughandwentoftenouthenceitwasoptedfor12hydrogenfraction
100 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2(a)Hydrideand(b)photochemicalvapourgenerator
The oxygen flow rate through the capillary was optimized in the rangendash15ndash30mlmin Thehighestsignaltonoiseratiowasachievedwiththeflowrateof
ndash120mlmin Thetotalgasflowratewasoptimizedatconstant12hydrogenfractioninthe
ndash1range500ndash800mlmin Theoptimumobservationheightvarieswithtotalgasflow rate so it had to be optimized as well The optimum conditions aresummarizedandcomparedtothoseachievedwithHGinTable1 TheanalyticalfiguresofmeritofPVG-AFSwiththeflame-in-gas-shieldatomi-zerweredeterminedThecalibrationfunctionconstructedwith010025050
ndash1 2100 and 200 microg l Bi standardswas linear (R = 09998) The repeatabilityndash1expressedastherelativestandarddeviation(n=10)was6at1microgl andthe
ndash1relativeandabsolutelimitsofdetection(3σn=10)achievedwere12ngl and68pgrespectively(Table2)TheabsolutelimitofdetectionachievedwithHGwas 76 times lower which can be attributed to several aspects Firstly thegenerationefficiencyforPVGapproachwasaround53while100isexpectedforHG[3]Secondlyafullwidthathalfmaximumofthemeasuredpeakswasca2-foldgreaterwhichnecessitatedlongerintegrationtimeandwasthusreflectedinhighernoiseofthesignalsFinallythelimitofdetectionforPVGapproachwas
ndash1affectedby seriouscontamination (around10ng l )mostprobably from thesensitizersolutionthatcontainedBiasimpurity Tovalidate theproposedmethodologyBi contentwasdetermined inCRMNIST1643f (Table3) and the resultswere compared to thosemeasuredwithHG-AFS[3]Duetosevereinterferencesfrominorganicacidsespeciallynitricacid[5]thesampleneededtobeevaporatedtodrynessandthenfilledupwiththe
2+reactionmediumcontainingCo as thesensitizer(NIST1643f isstabilized in
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 101
Parameter HG-AFS(ref[3]) PVG-AFS
ndash1Artotalmlmin 440 528ndash1H totalmlmin 60 722 ndash1O mlmin 7 202
OH mm 6 9ndash1Arshieldlmin 1515 1515
Table 1Atomizationconditionsforflame-in-gas-shieldatomizer
Parameter HG-AFS(ref[3]) PVG-AFS
LODpg 09 68ndash1LOQngl 09 12
Repeatability lt1 6
Table 2AnalyticalfiguresofmeritofHG-AFSandPVG-AFS
ndash1032moll nitricacid)Theresultsobtainedbybothmethodologiesareingoodagreementwiththecertifiedvalue
4Conclusion
Photochemical vapour generation of Bi was successfully coupled with non-dispersiveatomicfluorescencespectrometryforthefirsttimeanditsapplicabilitywas verified by determination of Bi in certified reference material of waterComparedtohydridegenerationconditionsofatomizationdifferinanoptimalobservationheightandsupplyofoxygenwhichmaybeneededtoldquoburnoutrdquotheorganicvapoursthatarereleasedfromthereactionmediumtothegasphasehowever this remains tobe verifiedAlthough there are still some limitationsregardingthelimitsofdetectionrepeatabilityandinterferencesthisnewsampleintroductionapproachseemstobepromising
Acknowledgments
The support of the Czech Science Foundation (19-17604Y) Czech Academy of Sciences(Institutional supportRVO68081715)andCharlesUniversity (ProjectSVV260560andProjectGAUK1048120)isgratefullyacknowledged
References
[1] Musil SMatousek T Currier JM StybloM Dedina J Speciation analysis of arsenic byselectivehydridegeneration-cryotrapping-atomicfluorescencespectrometrywithflame-in-gas-shield atomizer achieving extremely low detection limits with inexpensiveinstrumentationAnalChem86(2014)10422ndash10428
[2] SturgeonREPhotochemicalvaporgenerationaradicalapproachtoanalyteintroductionforatomicspectrometryJAnalAtSpectrom32(2017)2319ndash2340
[3] S tadlerova B Kolrosova M Dedina J Musil S Atomic fluorescence spectrometry forultrasensitivedeterminationofbismuthbasedonhydridegenerationndashtheroleofexcitationsourceinterferencefilterandflameatomizersJAnalAtSpectrom35(2020)993ndash1002
[4] DedinaJAtomizationofvolatilecompoundsforatomicabsorptionandatomicfluorescencespectrometryOnthewaytowardstheidealatomizerSpectrochimActaPartB62(2007)846ndash872
102 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Certified HG-AFS PVG-AFSndash1valuemicrogl
ndash1 a ndash1 a valueobtainedmicrogl recovery valueobtainedmicrogl recovery
1262plusmn011 128plusmn01 102plusmn1 121plusmn09 97plusmn5
a Spikedrecovery=slopeofstandardadditions(noadditionandtwospikedconcentrationstoasample)slopeofexternalcalibration
Table 3ThedeterminedcontentofBiinCRMNIST1643fpresentedasmedianvalueplusmncombineduncertainty(n=3)andrecoveries
[5] Vyhnanovsky J Yildiz D Musil S Effect of metal sensitizers on photochemical vaporgeneration of bismuth for analytical atomic spectrometry In Proceedings of the 15thInternationalStudentsConferenceModernAnalyticalChemistryKNesmerak(ed)PragueCharlesUniversity2019p257ndash262
[6] VyhnanovskyJSturgeonREMusilSCadmiumassistedphotochemicalvaporgenerationoftungstenfordetectionbyinductivelycoupledplasmamassspectrometryAnalChem91(2019)13306ndash13312
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 103
1Introduction
Liquidcrystalsareorganicsubstancesthatformamesomorphicphaseinsolu-tion[1]TheyareliquidlikeliquidsbuthaveaninternalconfigurationassolidsTheir light transmittance changes in the electric fieldwhich is used in liquidcrystaldisplays(LCDs)CholestericliquidcrystalsaretemperaturesensitiveThecolorofreflectedlightchangeswithsmalltemperaturechangeThisisusedinmedicineasasensitivetemperatureindicatorfordisease-infectedtissues High performance liquid chromatography or supercritical fluid chromato-graphy[23]areusedtoseparateanddetermineliquidcrystalcompoundsAnelectrophoretic method could be complementary to these commonly usedmethods To our best knowledge no study dealing with liquid crystal puritycontrolbyelectrokineticchromatographywaspublisheduntilnow Electroneutralsubstancesmoveincapillaryzoneelectrophoresisatthesamespeedcorrespondingtothespeedoftheelectroosmoticflow(EOF)andtherefore
Separation of liquid crystals using non-aqueous capillary electrokinetic chromatography
KATER INACOKRTOVATOMA S KR IZ EK
DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublickaterinacokrtovagmailcom
AbstractLiquid crystals arewidelyused in electronicsmedicine andotherfields Analytical separations are important in the development ofnewliquidcrystalstocontrolthepurityofsynthesizedsubstancesThesampleanalysisisimportantfordetectionofimpuritiesformedduring synthesis Liquid crystal-forming substances cannot beseparated by capillary zone electrophoresis due to the absence ofreadily ionizable groups Therefore electrokinetic chromatographywasused in thisworkAnotherproblemcomplicating theanalysiswastheverylowsolubilityofanalytesinwaterSeparationsinthisworkwere thereforecarriedoutundernon-aqueousconditions inacetonitrilewithaceticacidtoadjustthepHandhexadecyltrimethyl-ammonium chloride as a detergent to mobilize the non-ionizedanalytesUndertheseconditionsitwaspossibletoseparateimpu-ritiesfromsynthesizedanalytesinsamples
Keywordselectrokinetic
chromatographyliquidcrystalsnon-aqueouscapillary
electrophoresis
104 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
itisnotpossibletoseparatethemDuetothisanelectrokineticchromatographymethodwasdevelopedInthismethodasurfactantisaddedtothebackgroundelectrolyteMoleculesaggregateandformsphericalformationscalledmicelles[4]ifthesubstanceisaddedinsufficientconcentrationiehigherthanthecriticalmicellar concentration (CMC) Separation is possible due to interactions ofnonpolarmoleculepartswiththenonpolarmicelleinsideAlthoughwateristhemostusedsolventinelectrophoreticmethodsforseparationofwater-insolublesubstances organic solvents are selected However such solvent must meetcertaincriteriatobesuitableforuseincapillaryelectrophoresisAllcomponentsmustbesolubleinthesolvent[5]ItshouldnotbeflammabletoxicorreactiveforpracticalityitshouldbeliquidatroomtemperatureandalsoitspriceistakenintoaccountThevalueof its relativepermittivitywhichdescribes the strengthofinteractionsbetweenionsshouldbearound30Lowdynamicviscosityisalsopreferred to allow faster migration of analytes No organic solvent meets allparameters of the ideal solvent In practice methanol acetonitrile and theirmixturesarethemostusedTheseparationparameterscanbeinfluencedbyusingan organic solvent of the background electrolyte This topic has already beenwidelyexplored[6ndash8] Itwasgenerallyassumedthat inanhydrousconditionsmicellesarenotcreateddespitesufficientsurfactantconcentrationHoweveritwasfoundoutthatdodecylsulfatecanformstablemicelleswhenthebackgroundelectrolyteisdissolvedinformamide[9]Fortheanalysisofactivesubstancesinmedicinal plants Chen et al developed a method in which sodium cholatedissolvedinmethanolisusedasasurfactant[10]Theaddedpseudostationaryphase does not always form micelles but can still affect mobilization andseparationofanalytesiftheanalytesinteractdifferentlywithfreemoleculesofsurfactantInthisstudywater-insolubleliquidcrystalswereseparated(Fig1)Thereforenonaqueouselectrokineticchromatographymethodwasdeveloped
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 105
Fig 1 Structures of liquid crystals 4-([1-oxo-1-(pentyloxy)propan-2-yl]oxycarbonyl)phenyl4-(octyloxy)-[11-biphenyl]-4-carboxylate (ZL 85) and 4-([1-(decyloxy)-1-oxopropan-2-yl]oxycarbonyl)phenyl 4-(dodecyloxy)-[11- biphenyl]-4-carboxylate (ZL 1210) OpticalisomerismsitesaremarkedwithanasteriskStructurescreatedinMarvinSketch[11]
2Experimental
21Reagentsandchemicals
Acetonitrilege999fromSigma-Aldrich(Germany)aceticacid99fromLach-Ner Neratovice (Czech Republic) and hexadecyltrimethylammonium chloride25(ww)inwaterfromSigma-Aldrich(USA)wereusedforpreparationofback-groundelectrolyteMesyloxidepa(MO)suppliedbyLach-nerNeratovice(CzechRepublic)wasusedasareferencesubstance
22Instrumentation
ForexperimentsG7100ACapillaryElectrophoresisInstrument(AgilentTechno-logiesGermany)wasusedwithUV-VISdetectoroperatingat235nmand254nmwavelengthMeasurementswereconductedinafused-silicacapillaryof50microminner diameter with the total length 500cm and effective length 415cm(PolymicroTechnologiesUSA)
23Method
Capillarywas flushed for3minuteswith1MHCland for2minuteswith thebackgroundelectrolyteBackgroundelectrolytewaspreparedbymixingaceticacid (10mM) and hexadecyltrimethylammonium chloride (40mM) in aceto-nitrile Sampleswere introducedhydrodynamicallybyapressureof5kPa for1secondSampleswerefirstdissolvedinacetonitrileandthendilutedtwotimeswith the background electrolyte A voltage of 20 kV was applied during theseparation
3Resultsanddiscussion
Liquidcrystalsampleswerepractically insoluble inwater theirsolubilitywasndash3testedataconcentrationlevelof1mgcm inmethanolandacetonitrileWhile
samples were not sufficiently soluble in methanol they were successfullydissolvedinacetonitrile Becauseallanalytesaresubstancesthatdonothaveeasilyionizablefunctionalgroups theelectrokineticchromatographymethodwaschosen forseparationAsuitablesurfactantwassoughtCommonlyusedsodiumdodecylsulfate(SDS)isinsoluble in acetonitrile Therefore hexadecyltrimethylammonium chloride(CTAC) which had sufficient solubility for further experiments was chosenAlthoughasuitablebufferwassoughttoensureastablepHduetoproblemswithprecipitationofbuffercomponentsinthenon-aqueousenvironmentaceticacidwasusedtoadjustandmaintainpHofbackgroundelectrolytesolutionAstheadditionofcationicsurfactantsuchasCTACleadstoEOFreversalthedependence
106 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ofEOFmobilityontheconcentrationofCTACinthebackgroundelectrolytewasmeasuredContrarytowhatisobservedinaqueousbackgroundelectrolytesEOFwasnotreversedItsmobilitydecreasedwithincreasingCTACconcentrationbutnomajorchangesoccurredabove40mMconcentrationThecapillarywallwasprobablyalreadysaturatedbyCTACandthefurtherincreaseinconcentrationhadno signifficant effect on the conditionof the capillarywall Therefore a CTAC
ndash3concentrationof40mmoldm was chosenas sufficient for furthermeasure-mentswithrespecttotheincreasingcurrentwithincreasingionicstrengthofthebackgroundelectrolyte The optimized method was used for separation of several liquid crystalsamplesofdifferentpurityInthesampleoftheZL85liquidcrystalwith99purityonezoneoftheanalytewasdetectedImpuritieswereseparatedfromthisanalytewhen the samplewith lower puritywas introduced The peak of theanalytewasidentifiedbasedonrelativemigrationtimerelatedtomesityloxideSeparationoftheanalytefromanimpurityinthesampleZL8576isshowninFig 2The relativemigration timeof the firstpeak is 0834 therefore itwasidentifiedastheZL85analyteThestandarddeviationoftherelativemigrationtimesinfivemeasurementswas0002min(01) Using the available high purity sample it was possible to measure thecalibration line forquantificationof theanalyte in lesspuresamplesLimitof
ndash3detection was determined as 0009mgcm and limit of quantification as ndash30031mgcm FromthecalibrationlineconcentrationofZL85inthesample
with lower purity was calculated The concentration was determined as
48(ww)standarddeviation5(ww)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 107
Fig 2ElectropherogramobtainedwhenasampleofZL85liquidcrystalwithlowerpuritywasndash3introducedSamplewas introduced in01mgcm concentrationandwithaddedmesityloxide
ndash3(10mgcm )Capillarywithinnerdiameterof50micromtotallengthof500cm415cmeffectivelength The background electrolyte was acetonitrile with 10 mM acetic acid and 40 mMhexadecyltrimethylammonium chloride A voltage of 20 kVwith positive polaritywas appliedDetectionat254nm
ForsampleZL121099onlytheanalyteandmesityloxideweredetectedInthe sample ZL 1210 59 several impurities were separated and detected(Fig3)Accordingtotherelativemigrationtimetheanalyteofinterestcorres-pondstothefirstpeakPeakresolutionissufficientTheresolutionoftheanalytepeakandthesecondpeakis284andtheresolutionoftheothertwopeaksis230
4Conclusions
InthisstudyanewmethodforanalysisofnewlysynthesizedliquidcrystalswasdevelopedSomeparametersofthemethodwereoptimizedndashoptimumconcen-tration of hexadecyltrimethylammonium chloride was searched The identifi-cation of analyteswas based on a comparison of relativemigration times InsamplesZL85andZL1210withlowerpuritytheimpuritieswereseparatedfromthepeaksofliquidcrystalsthecontentofanalytewasdeterminedintheZL85sampleaccordingtothecalibrationline
Acknowledgments
IwouldliketothanktheInstituteofPhysicsoftheCzechAcademySciencesforprovidingnewlysynthesized liquid crystals This work has been supported by Specific University Research(SVV260560)andbyCharlesUniversityResearchCentreprogramNoUNCESCI014
References
[1] GennesPGProstJThePhysicsofLiquidCrystals2ndedNewYorkOxfordUniversityPress1993
108 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ndash3Fig 3ElectropherogramofsampleZL121059ataconcentrationof05mgcm withmesitylndash3oxideataconcentrationof10mgcm capillarywith innerdiameterof50microm total lengthof
500 cmeffectivelength415cmThebackgroundelectrolytewasacetonitrilewith10mMaceticacidand40mMCTACAppliedvoltage20kVpositivepolarityDetectionat235nm
[2] Vankatova P KalıkovaK KubıckovaA Ultra-performance supercritical fluid chromato-graphy A powerful tool for the enantioseparation of thermotropic fluorinated liquidcrystalsAnalChimActa1038(2018)191ndash197
[3] Vankatova P Kubıckova A Cigl M Kalıkova K Ultra-performance chromatographicmethodsforenantioseparationofliquidcrystalsbasedonlacticacidJSupercritFluids146(2019)217ndash125
[4] Terabe S Otsuka K Ichikawa K Tsuchiya A Ando T Electrokinetic separations withmicellarsolutionsandopen-tubularcapillariesAnalChem56(1984)111ndash113
[5] RiekkolaMLRecentadvancesinnonaqueouscapillaryelectrophoresisElectrophoresis23(2002)3865ndash3883
[6] Wright PB Lister AS Dorsey JG Behavior and use of nonaqueous media withoutsupporting electrolyte in capillary electrophoresis and capillary electrochromatographyAnalChem69(1997)3251ndash3259
[7] PorrasSPKenndlerECapillaryzoneelectrophoresisinnon-aqueoussolutionspHofthebackgroundelectrolyteJChromatogrA1037(2004)455ndash465
[8] PorrasSPRiekkolaMLKenndlerETheprinciplesofmigrationanddispersionincapillaryzoneelectrophoresisinnonaqueoussolventsElectrophoresis24(2003)1485ndash1498
[9] GuoXWangK ChenGH Shi JWuX Di L LWangY Determination of strobilurinfungicideresiduesinfruitsandvegetablesbynonaqueousmicellarelectrokineticcapillarychromatography with indirect laser-induced fluorescence Electrophoresis 38 (2017)2004ndash2010
[10] Chen AJ Li C Gao WH Hu ZD Chen XG Application of non-aqueous micellarelectrokinetic chromatography to the analysis of active components in radix SalviaemiltiorrhizaeanditsmedicinalpreparationsJPharmBiomedAnal37(2005)811ndash816
[11] MarvinSketch [computer program] version 1990 ChemAxon httpschemaxoncom-productsmarvin
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 109
1Introduction
Sudandyesaresyntheticazo-basedaromaticcompoundsTheyaretraditionallyusedinvariousindustriessuchaschemicaltextileandwoodworkingasdyestocolourwaxesplasticsoilspolishesandsoforthTheyhavebeencategorizedasclass3carcinogensbytheInternationalAgencyforResearchonCancerandtheiruseisthereforeforbiddeninthefoodindustryTheyareknownfortheirbrightcolours and easy and cost-effectivemanufactureThey arenearly insoluble inwater but soluble in various organic solvents such asmethanol or trichloro-methane[1] SudanI1-phenylazo-2-naphthol(Fig1A)isadyeusedasanorangecolouringagentItssometimesalsosoldundernamesSolventOrangeRorCISolventYellow14ItisformedasasecondaryproductinthemanufactureoftheSunsetYellowdye
Electrochemistry of Sudan I and its derivates in aqueous media
ad b aANNAONDRA CKOVA MARIESTIBOROVA LUDE KHAVRAN cd adKAROLINASCHWARZOVA -PECKOVA MIROSLAVFOJTA
a CentralEuropeanInstituteofTechnologyMasarykUniversity Kamenice753562500BrnoCzechRepublicannaondrackovaceitecmuniczb DepartmentofBiochemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublicc UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistry FacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublicd InstituteofBiophysicsCzechAcademyofSciences Kralovopolska13561265BrnoCzechRepublic
AbstractSudanIisanaromaticazo-compoundthathasbeenproventobeacar-cinogenDuringitsmetabolizationbycytochromeP450inliverafewmain derivates can be identified Thiswork sets out to assess themechanismofelectrochemicalreductionandoxidationofSudanIitshydroxylationderivativesfeaturingmetabolitesintheSudanIdetoxi-fication pathway and to introduce their selective voltammetricanalysis on boron-doped diamond electrode We show successfuldifferentiationamongthesecompoundsthankstothedifferencesintheelectrochemicaloxidationoftheirphenolicgroups
Keywordsborondopeddiamond
electrodecytochromeP450electrochemicalanalysisSudanI
110 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
InmammalianorganismsSudanIismetabolizedbythemicrosomaldetoxi-fying systemwitha central roleof cytochromeP450hydroxylationactivity inliver[2]DuringtheoxidativeprocessofmetabolizingSudanIseveralmetaboliteswereidentifiedbyprevioustestsThesearegt1-(phenylazo)-naphtalene-26-diol(further abbreviated SI-6OH) 1-(4-hydroxyphenylazo)-2-hydroxynaphtol(furtherabbreviatedSI-4OH)and1-(4-hydroxyphenylazo)-naphtalene-26-diol(furtherabbreviatedSI-46-diOH)thestructuresarepresentedinFig1 ThemainmethodcurrentlyusedtoidentifySudanIamongotherdyeswithsimilar structure is high-performance liquid chromatography (HPLC) It isrecommendedasthestandardmethodtoidentifythelevelofSudanIinfood[3] ComparedtoHPLCelectrochemicalmethodsareprovingtobefastercheaperandcomparablypreciseUnfortunatelyacomprehensiveelectrochemicalstudyofSudanIandparticularlyofitshydroxylatedmetaboliteshasnotbeencompletedyetThedyecanbedetectedthroughelectrochemistryeitherbytheoxidationofitsphenolicgrouporviareductionoftheazogrouppresentinitsmoleculeInbothcasesotherelectrochemicallyactivemoietiesareformedThederivatesofSudanIcanbedetectedandrecognizedfromSudanIthroughanalogousprocesses[4]InthisstudywefocusedoncomparisonofelectrochemicalbehaviourofSudanIanditshydroxylatedmetabolitesonborondopeddiamondelectrodetoaddressthepossibilitiesoftheirrecognitionsinmixturesbasedondifferencesinanodicandcathodicsignals
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 111
Fig 1 Chemical structure of (A) Sudan I (B) 1-(phenylazo)-naphtalene-26-diol (SI-6-OH)(C) 1-(4-hydroxyphenylazo)-2-hydroxynaphtol (SI-4-OH) and (D) 1-(4-hydroxyphenylazo)-naphtalene-26-diol(SI-46-diOH)
(A) (B)
(C) (D)
2Experimental
21Reagentsandchemicals
SudanI(Merckanalyticalstandardgrade)wasdissolvedinethanol(Merck)andkept at room temperatureChemicals forBritton-Robinsonbufferpreparation(acetic acid boric acid orthophosphoric acid sodium hydroxide) were fromMerckwithpurityge99pHofthebufferwasadjustedbymixingoftheacidsandsodium hydroxide solution at different ratios The Sudan I metabolites weresynthetized at the Department of Biochemistry Faculty of Science CharlesUniversityandkeptinmethanolattemperature3degC
22Instrumentation
Cyclicvoltammetric (CV)measurementswerecarriedout inBritton-RobinsonbufferofpH=70atroomtemperatureSudanIanditsderivativeswereaddedto
minus1thesolutionofBritton-Robinsonbuffertofinalconcentrationof5micromolL andstirred Before the measurement oxygen was removed from the solution bypurgingwithargonfor3minutesAutolabanalyzerPGSTAT20(EcochemieTheNetherlands)inconnectionwithVA-Stand663(MetrohmSwitzerland)GPES49(MetrohmSwitzerland)andathree-electrodesetup(withborondopeddiamond
2(WindsorScientificUKdiskdiameter3mmA=707mm )asworkingelectrodeminus1AgAgCl3molL KCl as reference electrode and platinum wire as auxiliary
electrode) Five cycles were performed for each measurement at scan rateminus1of1Vs
3Resultsanddiscussion
ForeachcompoundtwoseparateCVmeasurementswereperformedeachwithfive cycles performed in rapid succession For bothmeasurements the initialpotentialwassetat0VTheanodicscancontinuedto+1Vturnedtowardsndash1Vandfinishedat0VIntheothersetupcathodicscanwasperformedfirstfromthestartingpointtondash1Vturnedtowards+1Vandreturnedto0VThiswaywewereabletoobservethebehavioursofSudanIandcompareittothatofitsderivativeswhilefirstbeingreducedandthenoxidisedorviceversa In the anodic scan of Sudan I and its derivatives (Fig 2) differences in thepositionsoftheoxidationpeaksineachcompoundcanbeobservedWhileSudanIwithonlyonephenolgroupisoxidizedat+067VthederivativeswithtwophenolgroupsieSI-4OHandSI-6OHgiveoxidationpeaksatremarkablylesspositivepotentialofca+05VSI-46OHwiththepresenceofoverall3hydroxygroupsyieldstwooxidationpeaksInthereversecathodicscan(vertexpotential+1V)probablythankstothepresencereductionpeaksappearwhichcanbefurtherusedtodifferentiatebetweenSudanIandthederivativesSI-4OHandSI-6OH
112 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 113
Fig 3VoltammetricscanofSudanIanditsderivatesSI-4-OHSI-6-OHandSI-46-diOH1stscanincathodicdirectionfrom0Vvertexpotentialsndash1Vand+1VThemeasurementswereperformedinBritton-Robinsonbuffer(pH=7)withtheconcentrationofeachcompoundat5microMandatscan
ndash1rate1Vs
Fig 2 CyclicvoltammogramofSudanIanditsderivatesSI-4-OHSI-6-OHandSI-46-diOH1stscaninanodicdirectionfrom0Vvertexpotentials+1Vandndash1VThemeasurementswereperformedinBritton-Robinsonbuffer(pH=7)withtheconcentrationofeachcompoundat5microMandatscan
ndash1rate1Vs
possesstwoconjugatedhydroxylgroupswhichcanberegardedashydroquinonestructures and thus undergoing quasireversible redox process due to oxida-tionreductionofthehydroquinonetoquinonemoietyThisiswellvisibleattheCVsastheanodicsignalisfollowedbycathodiconeatthepotentialof+023VforSI-4OHand+03VforSI-6OHTheoxidationofSudanIproceedsbymechanism
minus +typicalforphenoliccompoundsatmorepositivepotentialsleading by1e 1H exchange to naphthoxy-type ndashO radical [5] This species undergoes furtherreactionsleadingtoformationofdimersandpolymersThecathodicpeakinthereversescanatndash02Varisesfromreductionofthesereactionproductsanditsoriginneeds tobe further investigatedSI-46OHwith thepresenceofoverallthree hydroxyl groups yields two oxidation peaks The first one is a result ofoxidationoftwoofthembeinginconjugationandthusbeingoxidizedtoquinonemoietyThesecondsignalatthesamepotentialastheoxidationsignalof2OHonnaphthaleneringofSudan I is consequenceofoxidationof the thirdhydroxylgroupofphenolictypeAsinglewidepeakat0Vinthereversescanispresumablyan overlap of signals arising from reduction of the quinonic moiety and by-products formed during oxidation processes Thanks to differences of theseprocessesspecificforindividualcompoundsitispossibletodifferentiateamongallfourofthemviapropersetting-upoftheinitialandvertexpotentialvalues The cathodic scan of Sudan I and its derivatives (Fig 3) shows a dominantreductionpeakaroundndash08Vwhichisduetoreductionoftheazogroupintheirstructuresaccompaniedbycleavageoftheirmoleculestoseparatethebenzeneandnaphthalene rings [4] The peaks in the subsequent anodic scans (vertexpotentialndash1V)arethereforetheresultoftheelectrochemicalreactionofmoietiesthatareproductsofthedivisionofthearomaticcirclesTheseproductsincludeaniline4-aminophenol1-amino-2-naphtholand1-amino-25-naphthalenediolwith irreversibly oxidizable amino moieties or (quasi)reversibly oxidizableamino-hydroxylsystemonthebenzeneornaphthaleneringandtispossibletodifferentiatebetweenthemTheobtainedoxidationpeaks(+02VforSI-4-OH+073VforSudanIandSI-6-OHand+025Vand+075VforSI-46-diOH)makeitpossibletodifferentiatebetweenallcompoundswiththeexceptionofSudanIandSI-6-OHForthereliablerecognitionofthesetwocompoundsanothermeasure-mentwithdifferentparametersisneeded
4Conclusions
ThestructuresofSudanIanditshydroxyderivatesthatarethemainproductsofthe metabolization of Sudan I by cytochrome P450 are similar and theirrecognitionwhenpresentinmixtureinsolutionisdemandingHereinwepresenta simple approach based on comparison of signals obtained in cathodic andanodic scan in CV measurements without the need of time-demandingchromatographicseparationstepFurtherworkwillbedevotedtoidentificationof observed redoxprocesses and applicationof themethod formonitoringofmetabolictransformationsofSudanIinvitro
114 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Acknowledgments
ThisresearchwassupportedbytheCzechScienceFoundation(projectNo18-01710S)
References
[1] ChailapakulOWonsawatW SiangprohW GrudpanK ZhaoYF Zhu ZW Analysis ofSudanISudanIISudanIIIandSudanIVinfoodbyHPLCwithelectrochemicaldetectionComparison of glassy carbon electrodewith carbon nanotube-ionic liquid gelmodifiedelectrodeFoodChem109(2008)876ndash882
[2] StiborovaMMartinekVRydlovaHHodekPFreiESudanIisapotentialcarcinogenforhumans Evidence for itsmetabolic activation and detoxication by human recombinantcytochromeP4501A1andlivermicrosomesCancerRes62(2002)5678ndash5684
[3] GomezMArancibiaVAliagaMNunezCRojas-RomoCDeterminationofSudan I indrinks containing Sunset yellow by adsorptive stripping voltammetry Food Chem 212(2016)807ndash813
[4] PrabakaranEPandianKAmperometricdetectionofSudanIinredchilipowdersamplesusingAgnanoparticlesdecoratedgrapheneoxidemodifiedglassycarbonelectrodeFoodChem166(2015)198ndash205
[5] Enache T A Oliveira-Brett A M Phenol and para-substituted phenols electrochemicaloxidationpathwaysJElectroanalChem655(2011)9ndash16
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 115
1Introduction
Aminoglycosidesarealargegroupofnaturalandsemi-syntheticantibioticswithawidespectrumofantimicrobialactivityagainstmostgram-positiveandgram-negativemicroorganismsCurrentlymultiplerepresentativesofaminoglycosidefamily ndash gentamicin (GM) neomycin B (NM) paromomycin (PM) kanamycin(KM)apramycin(AP) andstreptomycin(STM)Fig1ndashareapproved to treatinfectiousdiseasesinanimalsMaximumresiduelimitsfortheseaminoglycosidesinproductsandtissuesfromedibleanimalsareestablished[1]henceaneffectiveandrobustassayisnecessaryforcontrolofaminoglycosidescontamination Inthisstudyenzyme-linkedimmunosorbentassayforthedetectionofamino-glycosides in foodproductsandenvironmentalobjectswasdevelopedGroup-specificantibodieswereproducedowingtoimmunogenbasedonribostamycin(RS)whichexposedthecommonfragmentofmostaminoglycosides2-deoxy-streptamin (2-DOS) Fig 1 The developed assay was made suitable for thedetectionofresidualaminoglycosidesinhoney[2]
Group detection of aminoglycosides using ELISA for control of food contamination
ab a aKONSTANTINBURKIN INNAGALVIDIS MAXIMBURKIN
a DepartmentofImmunologyIMechnikovResearchInstituteofVaccinesandSera MalyjKazionnyjper5a105064MoscowRussianFederationburkin-kostyandexrub DepartmentofChemicalEnzymologyFacultyofChemistryLomonosovMoscowStateUniversityLeninskieGory1119991MoscowRussianFederation
AbstractThegrowingthreatofglobalantibioticresistanceisforcingtoreducenon-targetconsumptionofantibioticsandtomonitorcontaminationoffoodandenvironmentalobjectsInthisworkELISAwasdevelopedforgroupdetectionofaminoglycosidesToobtaingroup-specificanti-bodies a new immunogen based on ribostamycin was used Thedevelopedindirectcompetitiveformatofassayallowedtherecogni-tionof9aminoglycosidesnamelyneomycinribostamycinneaminparomomycin gentamicin sisomicin kanamycin tobramycin and
ndash1apramycinwithadetectionlimitrangedbetween002ndash020ngmL TheeffectivenessoftheproposedassaywasevaluatedinhoneyasafoodstuffmodelToneutralizea stronghoneymatrixeffect and toavoidalaborioussamplepre-treatmentanewmatriximitatorwassuggested 5 sucrose solution imitated the influence of 50-folddilutedhoneyTheproposedassayallowedustorevealanyofthe9
ndash1mentionedaminoglycosidesinhoneyata10microgkg level
KeywordsaminoglycosidesELISAhoney
116 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
2Experimental
21Reagentsandchemicals
NeomycinBribostamycinneamin(NA)paromomycinkanamycintobramycin(TM)amikacin(AM)gentamicinnetilmicin(NTM)sisomicin(SSM)geneticin(GC) apramycin and streptomycin were purchased from Chimmed (MoscowRussia) Bovine serumalbumin (BSA) complete Freund adjuvant 16-hexane-diamine1-ethyl-3-(3-dimethylaminopropyl)carbodiimide(EDC)sodiumperio-dateandsodiumborohydrideweretheproductsofSigma-Aldrich(USA)Gelatin(Gel)wasfromBio-Rad(USA)sucrosefromServa(Germany)two-componenttetramethylbenzidine (TMB) substrate solutionwas fromBioservice (Russia)andgoatanti-rabbitIgGantibodiesconjugatedtohorseradishperoxidase(anti-rIgG-HRP)werefromIMTEK(Russia)Honeysampleswerepurchasedfromlocaloutlets
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 117
Fig 1 Structuralformulasofstudiedaminoglycosides
22Preparationofconjugatedantigens
TwotypesofconjugateswerepreparedbasedonRSandBSAusingzero-lengthand C6 spacer arm between hapten and protein carrier RSwas treatedwithsodiumperiodatetooxidizehydroxylsofribosefragmenttoreactivealdehydegroupsandthencoupledtoBSAaminesthroughreductiveaminationToremoveuncoupledRSanexhaustingdialysiswascarriedoutusingdialysismembranetubes(MWCO14kDa)UsingthesameprocedureGel-RSconjugatewassynthe-sized ForpreparationofBSA-C6-RSwefirstlymodifiedBSAwith16-hexanediamineThemixtureofBSAandEDCinwaterwerestirredfor30minThen16-hexane-diaminewasaddedandstirredfor2hThemodifiedproteinwasdialyzedfromtheexcessive reagents and resultant BSA-C6-NH2was coupled to RS in reductiveaminationprocessasdescribedabove
23Immunizationandantibodypreparation
BSA-RSandBSA-C6-RSwereusedasimmunogensChinchillarabbits(20ndash25kg)weresubcutaneouslyinjectedat10ndash15pointsonthebackwith01mgofimmuno-gensemulsifiedinthecompleteFreundadjuvantThesamedosesofimmunogensinsalinewereadministeredmonthly forbooster immunizationsAweekaftereachinjectionabloodsamplefromearveinswastakenforthecontrolofimmuneresponseTheantiserainglycerol(11vv)werestoredatndash15degCuntiltestinginELISA
24TheELISAprocedure
AcompetitiveassaywasconductedaccordingtoclassicalprocedureGel-RSwascoatedovernightonpolystyrene96-wellCostarplatesNon-adsorbedconjugatewashedoutusingPBSwith005oftween20(PBS-T)Thenextcompetitivestep
ndash1includedtheadditionof01mLstandardaminoglycosidesolutions(1pgmL to ndash1 ndash11microgmL (B)and0microgmL (B ))inPBS-Tor01mLoftestedsampleand01mLof0
antibodiesinworkingdilution(1h25 degC)Afterwashingtheantibodiesboundtoimmobilized Gel-RS were detected using anti-rIgG-HRP (1h 37degC) Coloredproduct formedasaresultofenzymaticreactionwithTMBsubstratemixture(05h25degC)wasreadat450nmusingaStatFax2100platereader(AwarenessTechnologiesUSA) Relativeantibodybinding(BB )vstheanalyteconcentrationswasplottedas0
standardcurvesfittedtoafour-parameterlogisticfunctionThecross-reactivity(CR) for every aminoglycoside representative was calculated as ratio of half-inhibition concentrations IC NMIC aminoglycoside The dynamic range of50 50
assaywasacceptedasIC ndashIC andthelimitofdetection(LOD)wascalculatedas20 80
B ndash3timesSD0
118 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
3Resultsanddiscussion
31Immunogensynthesisandantibodypreparation
Inthemajorityofpublicationsdevotedtoimmunoassayofaminoglycosidestheimmunogenscoatingantigensenzymeconjugatesortracerswerepreparedbycarbodiimide or glutaraldehyde methods involving aminoglycosidesrsquo aminogroups [3ndash7] Due to several amino groups in aminoglycoside molecules theformationofconjugateswithavariableorientationofthehaptenoccurs InpresentstudyRSwaschosenasanimmunizinghaptenduetothefollowingadvantageous features Being a trisaccharide RS has the size of a moleculecomparable to themostof aminoglycosides Ithas three identical ringsA-B-CsimilartothoseinNMUsingaperiodateoxidationwecouldinvolvearibosesiteofRSincouplingtoproteinthatprovidedastrictorientationofhaptenonthecarrier with a favorable presentation of the 2-DOS fragment The resultantimmunogensBSA-RSandBSA-C6-RSwerecomparedtorevealwhichdesignisbetterforpresentationofacommonfragmentofaminoglycosidemoleculeandgenerationofgroup-specificantibody Antibodies to the BSA-RS demonstrated moderate sensitivity (NM
ndash1IC =10ngmL )andhighselectivitytowardsNMwithrelativelylowcross-reac-50
tivity(lt5)forGMKMandAPTheapplicationofthespacerintheimmunogenBSA-C6-RScontributedtoaprominentpresentationofthe2-DOSdeterminantandtheinductionofantibodieswithrecognitionofbroadspectrumofdifferentaminoglycosides In addition anti-BSA-C6-RS exhibited significantly better
ndash1sensitivity(NMIC =02ngmL )Thusallsubsequentstudieswereconducted50
usinganti-BSA-C6-RS
32Examinationofassayspecificityandselectionofimmunoreagents
TheindirectcompetitiveformatofassaywasdevelopedForevaluationofassayspecificity a panel of following aminoglycosideswas studied and their cross-reactivitywasdeterminedNA(625)RS(250)NM(100)KM(475)PM(173)GM(90)TM(78)AP(17)SSM(12)AM(lt01)GC(lt01)STM(lt01)andNTM(lt01)ThemostoftheseanalytesareusedinmedicalandveterinaryareashoweveronlyNMPMGMKMAPandSTMareappliedinanimalhusbandry[1]
33Determinationofaminoglycosidesinhoneyandselectionofthematriximitator
Honey is a complex product consisting of carbohydrates (75ndash80) vitaminsproteinsenzymesorganicacids traceelements inclusionsandothercompo-nentsThesecomponentsmightinterfereimmunochemicalreactionThereforetheisolationofaminoglycosidesfromhoneyisalaboriousandtime-consuming
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 119
120 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
procedureToavoidthisstephoneymatriximitatorswereappliedtomimictheinfluenceofhoneymatrixonantibodybindingSucrosewaschosenasthehoneyimitatorsinceitexposedastronghoney-matrix-likeeffectonantibodybindingTheadequacywasfoundbetweensolutionsofhoneyandsolutionsofsucroseexpressinganequalmatrixeffectTwopairswithequivalentmatrixeffectwere120honey=20sucroseand150honey=5sucroseThelatterconditionswere chosen asmore preferable due to inconvenience of operatingwith highviscous20sucrosesolution Thedeterminationofaminoglycosidesinhoneycouldbecarriedoutquantita-tivelyiftheanalytetobedetectedisknownForquantificationofaminoglycosideinhoneyasamplewasdiluted50timesinPBS-Tandaminoglycosidestandardcurvewas generated in 5 sucrose-PBST (Fig 2) If analyte is unknown thedevelopedgroup-specificELISAcanbeusedasascreeningtestInthiscasetheanalyzed sample can be considered as contaminated if it caused a relativeantibodybindingbelowthecut-off level (Fig3)Thus thedeveloped testwas
ndash1 ndash1 ndash1Analyte IC ngmL Dynamicrange LODngmL LODinhoneymicrogkg50 ndash1 IC ndashIC ngmL20 80
NM 02 003ndash21 002 10PM 07 008ndash71 005 25GM 15 015ndash133 011 55KM 035 005ndash39 004 20AP 68 05ndash968 023 115
Fig 2StandardcurvesandanalyticalparametersoftheELISA-systemforgroupdeterminationofaminoglycosidesinhoneyInteractionofanti-BSA-RSwithcoatingantigenGel-RSin5sucrosesolutionasthehoneyimitatorThedetectionlimitin5sucrosesolutionwasdeterminedaccordingtoLOD=B ndash3timesSD0
capabletorevealthecontaminationofhoneywith9aminoglycosides5amino-ndash1glycosidesapprovedforveterinary(NMPMGMKMandAP)ata10microgkg level
andalsoNARSSSMandTM
4Conclusions
A novel indirect competitive ELISA for the detection of aminoglycosides wasdevelopedRSwasusedasanewimmunizinghaptentoproducegroup-specificantibodiesagainst2-DOSacommonmoietyofalargenumberofaminoglycosideantibiotics A wide spectrum of aminoglycoside representatives could bedetectedincludingNMRSNAPMGMSSMKMTMandAPThedevelopedassay
ndash1was capable todetect theseanalyteswithaLOD up to002ndash020 ngmL ForanalysisofhoneyamatriximitatorwasdevelopedtoavoidhoneyinterferencesonimmunoassayTheanalysisofthehoneysampleallowedustorevealanyofthe
ndash1mentionedaminoglycosidesinhoneyata10microgkg level
References
[1] CouncilRegulation(EU)N372010OffJEurCommunitiesInfNotL15(2009)1ndash72[2] GalvidisIABurkinKMEreminSABurkinMA Group-specificdetectionof2-deoxy-
streptamineaminoglycosidesinhoneybasedonantibodiesagainstribostamycinAnalMeth11(2019)4620ndash4628
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 121
Fig 3Detectionofaminoglycosidesspikedinhoneysamplesata40ppblevelusinggroup-specificELISAEachsymbolcorrespondstotheaveragerelativebindingandtheerrorisSDobtainedforanindividualhoneysampleanalyzedintriplicateEmptycharactersrepresentindividualblankhoneysamples(limebuckwheatandflower)andfilledsymbolsrepresentthesamesamplesfortifiedwith
ndash1aminoglycosidesata40mgkg (level establishedonlyforSTMinseveralcountries)Thecut-offlevelcorrespondstothelimitofassaydetectionobtainedbythematriximitator(5sucrose-PBS-T)
[3] ThompsonSGBurdJFSubstrate-labeledfluorescentimmunoassayforamikacininhumanserumAntimicrobAgentsChemother18(1980)264ndash268
[4] LiCZhangYEreminSAYakupOYaoGZhangXDetectionofkanamycinandgentamicinresiduesinanimal-derivedfoodusingIgYantibodybasedic-ELISAandFPIAFoodChem227(2017)48ndash54
[5] GalvidisIABurkinMAMonoclonalantibody-basedenzyme-linkedimmunosorbentassayfortheaminoglycosideantibiotickanamycininfoodstuffsRussJBiorganChem36(2010)722ndash729
[6] HaasnootWStoutenPCazemierGLommenANouwsJFKeukensHJImmunochemicaldetectionofaminoglycosidesinmilkandkidneyAnalyst124(1999)301ndash305
[7] Peng JWangYLiuLKuangHLiAXuCMultiplex lateral flowimmunoassayfor fiveantibioticsdetectionbasedongoldnanoparticleaggregationsRSCAdv6(2016)7798ndash7805
122 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
PhotochemicalvaporgenerationisanalternativesampleintroductiontechniqueforanalyticalatomicspectrometryThistechniqueisbasedaroundasourceofUV-radiationthatirradiatesalowmolecularweightorganicacidmedium(mostcommonlyformicacidaceticacidortheircombinations)withananalyteHighlyreducingradicalsandaquatedelectronsareproducedandconverttheanalyteintoavolatilespecieswhichisthentransportedintoadetector[1]SofartheuseofPVGhasbeendescribedforhydride-formingelements(AsBiTeSbPbSeSnandTl)andmercury[12]transitionmetals(FeCoNiCuMoWCdAgAuIrPdPtRhandOs)[13ndash6]andevennon-metals(BrIClFandS)[16ndash9] AfirstsuccessfulphotochemicalvaporgenerationofcobaltwasdescribedbyGuoetalin2004[6]whichwasfollowedbymoresystematicstudiesbyGrinbergetalin2008[10]andDengetalin2010[11]LaterworksbydeQuadrosetal[12]anddeJesusetal[13]focusedontheanalysisofrealsamplesInthelatterwork
Photochemical vapor generation of cobalt for detection by inductively coupled plasma mass spectrometry
ab aJAROMIRVYHNANOVSKY STANISLAVMUSIL
a DepartmentofTraceElementAnalysisInstituteofAnalyticalChemistryoftheCzechAcademyofSciencesVeveřiacute9760200BrnoCzechRepublicjaromirvyhnanovskygmailcom
b DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublic
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 123
AbstractThisworkfocusedonthephotochemicalvaporgenerationofcobaltVolatilespeciesweregeneratedinaflow-injectionsystememployinga high-efficiency flow-through UV photoreactor and a formic acidbased medium and were introduced by an argon carrier into aninductivelycoupledplasmamassspectrometerfordetectionOptimalgeneration conditions were found as 10 (vv) formic acid and
ndash1 ndash14molL ammonium formate with a 4mLmin flow rate whichcorresponds to irradiation time of around 13 s The influence ofvariousmetalsensitizersofphotochemicalreactionwasinvestigated
2+andonlyCu ionsexhibitedapositiveeffectongenerationefficiencyndash ndash 2ndashInterferencesfromcommoninorganicanions(NO Cl SO ) were3 4
also examined Lastly the limit of detection and repeatability (atndash1 ndash1250ngL )weredeterminedtobe13ngL and41respectively
Keywordscobaltinductivelycoupled
plasmamassspectrometry
photochemicalvaporgeneration
the authors also presented a systematic study on generation conditions andachievedagenerationefficiencyofaround40 Themainaimofthisworkwastooptimizetheconditionsofgenerationwithinductivelycoupledplasmamassspectrometry(ICP-MS)detectionexaminetheeffectofvariousmetal sensitizers toachieve thehighestgenerationefficiencypossibleandreachthelowestlimitofdetectionpossible
2Experimental
21Reagentsandchemicals
minus1Deionizedwater (DIW lt 02μScm UltrapurWatrex USA)was used for thepreparationofallsolutionsFormicacid(98paLach-NerCzechRepublic)andammonium hydroxide (ge25 pa Sigma-Aldrich USA) were used for the
minus1preparationof the reactionmediumA1000mgL Co stock solution (Sigma-AldrichUSA)wasusedforthepreparationofallsamplesolutionsThefollowingcompounds were used as potential metal sensitizers cadmium(II) acetatedihydrate(paLach-NerCzechRepublic)zinc(II)acetatedihydrate(paSigma-AldrichUSA)copper(II)acetatemonohydrate(paMerckGermany)nickel(II)acetatetetrahydrate(paSigma-AldrichUSA)sodiumtungstatedihydrate(paCarlRothGermany)and iron(II) sulphateheptahydrate (pa LachemaCzechRepublic)Nitricacid(65semiconductorgradeSigma-AldrichUSA)hydro-chloricacid(37paMerckGermany)andsulfuricacid(98paLach-NerCzechRepublic)wereusedforaninterferencestudy
22Instrumentation
AschematicdiagramofthePVGsystemcoupledtoICP-MSisshowninFig1andamore detailed description can be found in reference [5] Briefly a singlequadrupole ICP-MS Agilent 7700x (Agilent Technologies USA)was used as a
minus1detector Deionized water was mixed with a 10μgL Rh internal standardsolutionin2HNO andwassubsequentlynebulizedbyaMicroMistnebulizer3
59 103during PVG Isotopes of Co and RhweremonitoredMeasurementswereperformed in time resolved analysis mode and in He collision mode
minus1(41mLmin )AlltubingusedwasmadefromPTFEwiththeexceptionoftygontubing in the peristaltic pump (Reglo ICC Ismatec Switzerland) The high-efficiency flow-through photoreactor was a 19 W low-pressure mercurydischarge lamp (Beijing Titan Instruments Co Beijing China) with a quartzcentralchannel(asymp720μL internalvolume)Samplesolutionswere introducedintoastreamofreactionmediumusinganinjectionvalve(V-451IDEXHealthandScienceUSAsampleloopvolume05ml)Effluentfromthephotoreactorwasmixedwithaflowofargonandcarriedtothechilledgas-liquidseparator(internalvolume15mL)wherethevolatilespecieswereseparatedfromtheliquidwaste
124 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
andcarried to the inletofaScott-typespraychamber (originally the inlet formakeupargon)oftheICP-MS
3Resultsanddiscussion
The starting conditionswere adopted fromour earlierwork [14]whichusedatomicabsorptionspectrometerasadetectorandminiaturediffusionflameasanatomizerThefirstparameteroptimizedwasthecompositionofreactionmedium(Fig2)Theadditionofammoniumformate(createdin-situbytheadditionofacalculatedamountofammoniumhydroxidetoformicacid)wasfoundcrucialto
ndash1effectivelygeneratevolatilespeciesofcobalt10(vv)formicacidand4molL ammonium formatewas chosen as the optimumandwasused for all further
experimentsAlthoughhigher concentrations of both components led to even
higher signalstheseconditionswerenotusedfurtherbecauseofthelaboriousprocessofpreparation(mixingofconcentratedacidwithconcentratedbase)andtolimittheconsumptionofchemicals Theinfluenceofirradiationtimewasalsoexaminedandthehighestpeakarea
-ndash1wasobtained for4mLmin corresponding to an irradiation timeof approximately13s ToenhancethegenerationefficiencyadditionofvariousmetalstothereactionmediumwastestedtoldquosensitizerdquothephotochemicalreactionThemetalswerechosenwithrespecttotheirsignificantenhancementeffectdescribedrecentlyforphotochemicalvaporgenerationofotheranalytes[158]Theonlymetalionthat
2+ledtoanenhancementofthesignalwasCu (Fig3a)buteveninthiscasetheeffect was rather negligible reaching only 12-fold enhancement in the range
ndash1 2+ 2+001to01mgL Cu FurtheradditionofmoreCu ledtoadecreaseinthesignal2+TheadditionofZn didnotexhibitanypositiveornegativeeffectacrossthetested ndash1range (01 to 1500mgL not shown in figure) and the addition of higher
ndash1 2+ 2+ 6+concentrations (tens to hundredths ofmg L ) of Cd Fe andW (Fig3b)causedsevereinterferences
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 125
ndash ndash 2ndash Interferencescausedbycommoninorganicanions(NO Cl SO addedas3 4ndashtheirrespectiveacids)werealso investigatedOutof theseNO was foundto3
ndash1causethemostsevereinterferencesevenatconcentrationsofsinglemmolL Thendash 2ndashmethodologywasmorerobusttowardstheinterferencesfromCl andSO but4
theystillcausedsignificantdropinsensitivityathigherconcentrationsConsi-deringthewideuseoftheseacidsinanalyticalchemistryforsamplepreparationthisposesabigchallengeintheapplicationofthismethodtorealsamples
ndash1 Usingoptimalconditions(10(vv)formicacid4molL ammoniumformateandirradiationtimeof13s)acalibrationcurvewasmeasuredandevaluatedThelimitofdetectionwasdeterminedas3timesthestandarddeviationof10blank
ndash1measurements and was calculated as 13 ng L The repeatability of 10ndash1consecutivemeasurementsof250ngL was41
126 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 3Effectofvariousmetalionsonthepeakarea(a)metalionswithapositiveeffect(b)metalionsndash1withoutapositiveeffectExperimentalconditions2microgL Coreactionmedium10(vv)formic
ndash1 ndash1acidand4molL ammoniumformateflowrate4mLmin
ndash1Fig 2Effectofthecompositionofreactionmediumonpeakareaexperimentalconditions2microgL ndash1Coreactionmediumflowrate4mLmin (blackdotscorrespondtomeasuredpoints)
4Conclusions
Theconditionsofthephotochemicalvaporgenerationofcobaltwereoptimizedand are in good agreement with previous works [11 13] Copper ions wereidentifiedasapotentialsensitizerincreasingthesignalbyabout12-foldbuttheirpotentialuseisseverelylimitedbythenarrowrangeofconcentrationsinwhichthepositiveeffectisexhibitedSevereinterferencesfrominorganicanionswereobservedwhichisinlinewithotherworksdealingwithphotochemicalgeneration[1358]Furtherexperimentswillfollownamely(i)furtherinvestigationsinnewpotentialsensitizerstoenhancegenerationefficiencyandthusdecreasethe
ndash1limit of detection to sub ng L levels (ii) determination of the generationefficiency(fromcomparisonwithnebulizationandorusingaradioactiveisotope58Co)(iii)verificationoftheaccuracyandpracticalfeasibilityofthismethodologybyanalysisofcertifiedreferencematerials
Acknowledgments
ThesupportofTheCzechScienceFoundation(ProjectNo19-17604Y)CzechAcademyofSciences(Institutional supportRVO68081715)andCharlesUniversity (project SVV260560andprojectGAUK60120)isgratefullyacknowledged
References
[1] SturgeonREPhotochemicalvaporgenerationaradicalapproachtoanalyteintroductionforatomicspectrometryJAnalAtomSpectrom32(2017)2319ndash2340
[2] XuTHuJChenHJTransitionmetalionCo(II)-assistedphotochemicalvaporgenerationofthalliumforitssensitivedeterminationbyinductivelycoupledplasmamassspectrometryMicrochemJ149(2019)103972
[3] SoukalJSturgeonREMusilSEfficientphotochemicalvaporgenerationofmolybdenumforICPMSdetectionAnalChem90(2018)11688ndash11695
[4] deOliveiraRMBorgesDLGUVphotochemicalvaporgenerationofnoblemetals(AuIrPdPtandRh)AfeasibilitystudyusinginductivelycoupledplasmamassspectrometryandseawaterasatestmatrixJAnalAtomSpectrom33(2018)1700ndash1706
[5] VyhnanovskyJSturgeonREMusilSCadmiumassistedphotochemicalvaporgenerationoftungstenfordetectionbyinductivelycoupledplasmamassspectrometryAnalChem91(2019)13306ndash13312
[6] GuoXSturgeonREMesterZGardnerGJVaporgenerationbyUVirradiationforsampleintroductionwithatomicspectrometryAnalChem76(2004)2401ndash2405
[7] HuJSturgeonRENadeauKHouXZhengCYangLCopperionassistedphotochemicalvapor generation of chlorine for its sensitive determination by sector field inductivelycoupledplasmamassspectrometryAnalChem90(2018)4112ndash4118
[8] LeonoriDSturgeonREAunifiedapproachtomechanisticaspectsofphotochemicalvaporgenerationJAnalAtomSpectrom34(2019)636ndash654
[9] SturgeonREPaglianoEEvidenceforphotochemicalsynthesisoffluoromethaneJAnalAtomSpectrom(2020)httpsdoiorg101039D0JA00108B
[10] GrinbergPMesterZSturgeonREFerrettiAGenerationofvolatilecobaltspeciesbyUVphotoreduction and their tentative identification J Anal Atom Spectrom 23 (2008)583ndash587
[11] DengHZhengCB LiuLWWuLHouXDLvYPhotochemicalvaporgenerationofcarbonyl for ultrasensitive atomic fluorescence spectrometric determination of cobaltMicrochemJ96(2010)277ndash282
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 127
[12] deQuadrosDPBorgesDLDirectanalysisofalcoholicbeveragesforthedeterminationofcobalt nickel and tellurium by inductively coupled plasmamass spectrometry followingphotochemicalvaporgenerationMicrochemJ116(2014)244ndash248
[13] deJesusHCGrinbergPSturgeonRESystemoptimizationfordeterminationofcobaltinbiologicalsamplesbyICP-OESusingphotochemicalvaporgenerationJAnalAtomSpectrom31(2016)1590ndash1604
[14] VyhnanovskyJFotochemickegenerovanıtekavychspeciı kobaltuproanalytickouatomovouspektrometriiMasterthesisFacultyofScienceCharlesUniversityPrague2018
128 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Oneof themethods thatallowobtainingmaterialswithnewproperties is theplasmaenhancedchemicalvapordepositionmethodInthismethodcompoundscalledprecursorsaresuppliedtotheplasmareactorasagasphaseThankstoplasmaenhancedchemicalvapordeposition it ispossible toobtainmaterialswithuniquepropertiesThisisduetothefactthattheplasmaaffectsthesurfaceinfourdifferentways etching cleaning chemicalmodification and crosslinking
Optimization of condition for cold plasma deposition of thin layers for surface modification of working electrodes
a b a cJUSTYNALIPIN SKA MARIAMADEJ BOGUSŁAWBAS JACEKTYCZKOWSKI
a DepartmentofAnalyticalChemistryFacultyofMaterialsScienceandCeramicsAGHUniversityofScienceandTechnologyAdamaMickiewicza3030-059KrakoacutewPolandjustynalipinskaaghedupl
b DepartmentofAnalyticalChemistryFacultyofChemistryJagiellonianUniversityinKrakoacutewGronostajowa230-387KrakoacutewPoland
c Departmentof MolecularEngineeringFacultyofProcessandEnvironmentalEngineeringLodzUniversityofTechnologyWolczanska21390-924ŁoacutedźPoland
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 129
AbstractCurrentlyresearchisfocusedonthesearchfornewphysicallyandchemicallystablematerialsaswellasvolumeorsurfacemodificationOneofthemethodsusedforsurfacemodificationistheapplicationofthin layers from inorganic and organic compounds The plasmaenhancedchemicalvapordepositionisamethodthatallowsmaterialmodificationandalsodepositionofthinlayersThisworkconcernsoptimizationofcoldplasmadepositionparametersandtoachievethebestelectrical conductivitywhilemaintaining thehighmechanicalstrength of the formed layers Preliminary tests were focused onoptimizing the layering parameters such as the deposition timedischargepowerpressureofmonomerandthe flowofargonTheobtainedsamplesweresubjectedtothermaltreatmentafterwhichtheywere coveredwitha layerof aluminumThe thicknessof theobtained layers was determined on the basis of interferencemicroscopymeasurementsAsaresultoftheexperimentslayerswithathicknessof20nmto600nmwereobtainedTheconductivityofthedeposited layers was also determined and values from 003 to
ndash1150Sm wereobtained
Keywordscoldplasmadepositionelectrochemical
applicationssurfacemodificationthinlayers
Thismethodisusedtoproducecatalyticstructuresortomodifythepropertiesofmaterials eg improve hydrophobicity The growing popularity of surfacemodificationmethodsusingcoldplasmaisassociatedwiththefactthatitisanenvironmentallyfriendlyandversatilemethod[12] Workingelectrodesusedinvoltammetryareasubgroupofchemicalsensorswhich are small devices that convert real-time chemical information into ameasurableandanalyticallyusefulmeasurementsignalChemicalinformationrangingfromtheconcentrationofaspecificcomponentofthetestedsampletotheoverallcompositionofthematrixcancomefromboththeinitiatedchemicalreactionandbetheresultofphysico-chemicaltransformationstakingplaceinthetested object Chemical sensors are equipped with two basic elements iereceptor and transducer The receptor is responsible for the conversion ofchemicalinformationfromthetestedobjectintoaspecificformofenergyintheconverterthisenergyistransformedintoausefulanalyticalsignal Parameters characterizing the electrochemical sensor include accuracyprecision selectivity accuracy presentation selectivity sensitivity dynamicrange limit of quantification limit of detection lifetime response time andreliability Themost numerous and the oldest group of chemical sensors areelectrochemicalsensorsCommonlyobservedinterest inthisgroupofsensorsresultsfromthefactthatwithrelativelylowproductionandoperatingcoststheyofferthebestmetrologicalandoperationalparameters[3ndash5]Oneofthemaintrendsofmodernanalyticsisthesearchfornewelectrodematerialsandvariousgeometries of working electrodes One way to improve the performance ofworkingelectrodesistomodifytheirsurfaceforexamplebyapplyingthinlayersIn this work were considered plasma enhanced chemical vapor depositionmethod as the method of surface modification designed to performworkingelectrodeforvoltammetricdeterminationsAspartoftheinitialresearchplasmaprocessing parameters such as discharge power time of treatment andcompositionofgasmixtureinwhichplasmawasgeneratedwereoptimizedThelayers obtained in different conditions have been tested for suitability forelectrochemical applications (layer thicknessmeasurement and themeasure-mentofconductivity)
2Experimental
21Reagentsandchemicals
Theprecursor solutions suchasacrylonitriledietoxydimethylsilane trietoxy-methylsilaneandtetramethyldisiloxanewhicharesuppliedbyABCRwereusedOtherreagentsofanalyticalpuritysuchasn-hexane(SigmaAldrich)andargonwereused
130 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
22Instrumentation
The thin layers were deposited in a parallel-plate plasma reactor (frequency1356MHz)ThesamplesobtainedwerecalcinedinatunnelfurnaceunderanargonatmosphereThethicknessofthedepositedlayerswasmeasuredafterthealuminum was sputtered using a Nikon microscope type ECLIPSE LV150NElectrometerhigh resistance system (KEITHLEY) was used to measureconductivity
3Resultsanddiscussion
Eachof themonomerswasdepositedonprepared1times1corningglasssamplesSamples prepared with n-hexane were placed in a plasma reactor andadditionallypartiallycoveredwithamicroscopecoverslipSchematiclayoutofsamplesinthereactorshowninFig1ThefirststepwastoetchthesystemusingargonplasmaThisstageallowedfortheeliminationofimpuritiesthatwerenotremovedbythehelpofn-hexaneandthepreparationofthesurfaceofthesamplesforthedepositionoftheproperlayerTheproperstageistheapplicationofathinlayerwiththeplasmainducedbytheselectedprecursoracrylonitriledietoxydi-methylsilanetrietoxymethylsilaneandtetramethyldisiloxaneThethicknessandpropertiesoftheobtainedlayersdependonthedepositionparameterssuchasdischarge power time of treatment and composition of gasmixture inwhichplasmawasgenerated Table1showsallcombinationsofparameterstestedforallfourprecursorsFour different discharge powers for acrylonitrile and two different dischargepowersfororganosiliconmonomersweretestedwithtwodifferenttreatmenttimesEachtimeandpowercombinationwasperformedinplasmainducedbypuremonomerandmonomerwithargonAfterapplyingthelayersthesampleswereplacedinaquartzboatandcalcinedinatunnelfurnaceAllsampleswerecalcinedat500degCfor2hoursunderargonflowSamplesaftercalcinationwerecoveredwithalayerofaluminum
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 131
Fig 1 Scheme of sample distribution in plasma reactor(a)reactorelectrode(b)corningglasssamples(c)micro-scopecoverslip
Thethicknessoftheobtainedlayerswasmeasuredusinganinterferencemicro-scopeMeasurements were carried out at amagnification 10times recording theimageinmonochromelightTheimagewassetsothattheinterferencefringeswere perpendicular to the arc on the sample In order to calculate the layerthicknessD[nm]thefollowingformulawasused
(1)
wheredandLweredeterminedonthebasisoftheregisteredimage(disfringeshiftduetorefractionoflightontheslopeLisdistancebetweenthefringes) Thelaststageofthestudywastodeterminethecurrent-voltagecharacteristicstodeterminetheconductivityoftheobtainedlayersThesamplewasplacedinameasuring cell and attachedwith silver paste to the electrometerwires ThechangeincurrentwasrecordedwiththeapplicationofalternatingvoltageintimeBasedontheresultsobtainedthegraphsofdependenceUndashIwereobtainedfromwhichthevalueofresistance(R)wasdeterminedAnexampleofcurrent-voltagecharacteristicsisshowninFig2
132 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Table 1Conditionsforlayersdeposition-parameterwhichweretested
Monomer DischargepowerW Timeoftreatmentmin
Acrylonitrile 10204080 24Dietoxydimethylsilane 2040 255Trietoxymethylsilane 2040 255Tetramethyldisiloxane 2040 255
Fig 2Current-voltagecharacteristicsdeterminedfortheacrylonitrilelayer(depositionparametersW=10Wt=25mingasmixtureonlyacrylonitrile)
Table 4Theresultsofthicknessmeasurementsandconductivityspecifictothetrietoxymethylsilanelayers
DischargepowerW 20 20 40Timeoftreatmentmin 25 5 5Argonflowsccm 10 10 10Thicknessnm 14686 24282 35311
ndash1SpecificconductivitySm 165 93 67
Knowing the value of the resistance and the geometry of the system thespecificresistancewasdeterminedfollowedbythespecificconductivityofthesamplethefollowingequationwasused
(2)
whereρisspecificresistance[Ωm]Risresistance[Ω]bissamplelength[m]Disdepositedlayerthickness[m]andldistancebetweenelectrodes(wires)[m] Theresultsofthicknessmeasurementsandspecificconductivityarepresentedin the Tables 2ndash5 For electrochemical applications it is important that theobtainedlayerhasthehighestspecificconductivityAnalyzingthedatapresentedin Tables 2ndash5 shows that the thinnest layers have the greatest applicationpotentialastheelectrodematerial
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 133
Table 2Theresultsofthicknessmeasurementsandconductivityspecifictotheacrylonitrilelayers
DischargepowerW 40 40 80 80 10 10 20 20Timeoftreatmentmin 2 4 2 4 2 4 2 4Thicknessnm 15845 31167 250 4644 247 67 7531 12236
ndash1SpecificconductivitySm 0002 156 191 0003 1512 454 311 185
Table 3The results of thickness measurements and conductivity specific to the dietoxydimethylsilanelayers
DischargepowerW 20 20 40 40Timeoftreatmentmin 25 5 25 5Argonflowsccm 10 10 10 10Thicknessnm 13998 37519 28431 49556
ndash1SpecificconductivitySm 1640 55 105 40
Table 5The results of thickness measurements and conductivity specific to the tetramethyldisiloxanelayers
DischargepowerW 20 40 40Timeoftreatmentmin 25 25 5Argonflowsccm 10 10 10Thicknessnm 16759 3159 61652
ndash1SpecificconductivitySm 213 95 31
4Conclusions
InthisworkplasmaenhancedchemicalvapordepositionmethodwasusedforapplyinglayersoffourdifferentmaterialsacrylonitriledietoxydimethylsilanetrietoxymethylsilaneandtetramethyldisiloxaneBychangingparameterssuchasdischargepowertimeoftreatmentandcompositionofgasmixtureanumberofsampleswereobtainedwithlayersofdifferentthicknessandwhatisassociatedwithotherelectricalproperties TestsperformedaspartofthisworkwereusedtoperforminnovativeworkingelectrodesforvoltammetricdeterminationsThesurfacewasmodifiedusingthecoldplasmaofthreesubstratesgraphiteglassycarbonandgold
Acknowledgments
JLandMMhavebeenpartlysupportedbytheEUProjectPOWR030200-00-I00416
References
[1] KapicaRTyczkowskiJBalcerzakJMakowskiMSielskiJWorwaEEnhancingadhesivejointsbetweencommercialrubber(SBS)andpolyurethanebylow-pressureplasmasurfacemodificationIntJAdhesAdhes95(2019)102415
[2] TyczkowskiJKapicaRŁojewskaJThincobaltoxidefilmsforcatalysisdepositedbyplasma-enhancedmetalndashorganicchemicalvapordepositionThinSolidFilms515(2007)6590ndash6595
[3] HulanickiAGłabSIngmanFChemicalsensorsDefinitionsandclassificationPureApplChem63(1991)1247ndash1250
[4] BrzozkaZWroblewskiWSensorychemiczneWarszawaOficynaWydawniczaPolitechnikiWarszawskiej1999(InPolish)
[5] SkoogDAWestDWHollerFJCrouchSRFundamentalsofAnalyticalChemistry9thEdBostonCengageLearning2013
134 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
ConcentrationandcompositionofvolatilecompoundsorcongenersisoneofthemostimportantparametersresponsibleforqualityofproducedalcoholicdrinksandhencefortheirsensorycharacteristicsandconsumeracceptanceTodaygaschromatography (GC) is conventionally used to determine qualitative andquantitativecompositionsofvolatilecompoundswithvariousexternalandorinternalstandardcalibrationprocedures Methodemployingethanol as an internal standard (IS) forGCquantitativedeterminationofvolatilecompoundsinalcoholicbeverageshasbeensuggestedquitelongago[1]andsincethattimegreatresearchworkhasbeencarriedoutRecentlyaninterlaboratorystudyofthemethodinvolving9testinglaboratoriesfrom4countrieswascarriedout[2]TheresultsdemonstratedgreatperspectivesofldquoEthanolasISrdquomethodandproveditsreferencecharacterandeaseofroutineimplementation
Advanced GC-MS method for quality and safety control of alcoholic beverages
abANTONKORBAN
a DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova2030812840Prague2CzechRepublickarbonat7gmailcomb DepartmentofAnalyticalChemistryChemistryFacultyBelarusianStateUniversityLeningradskaya14220050MinskBelarus
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 135
AbstractRecently developed and validated simple and reliable quantitativemethod employing ethanol as an internal standard for GC-MSquantification of volatile compounds in alcoholic products wasapplied to 36 samples including commercially available world-famousbrandspirits from18countriesandhomemadedistillatesThe GC-MS analyses were performed simultaneously by thesuggested approach and official internal standard method that isprescribedinthelegislationofEUandUSATheindependentsamplest-testwasemployedtoevaluatethestatisticaldifferenceofresultsofthesetwomethodsThetestrevealednodifferenceintheresultsandtheirrepeatabilityThemainbenefitsofthesuggestedmethodaretheeliminationofthenecessityofmanualinternalstandardadditionandsamplesdensitymeasurementthusmakingitmoreeconomicalandproductive
Keywordsalcoholicbeveragesgaschromatography-
massspectrometry(GC-MS)
internalstandardmethodvolatilecompounds
quantification
AllpreviousstudieswereutilizingflameionizationdetectorssinceGC-FIDisprescribedinthelegislation[3]wheremass-spectrometrydetectorsarenotyetofficially referred However GC-MS instruments are employed in practice toqualifyandorquantifyvolatilesincommercialspiritsintraditionalhomemadealcoholicdrinksinnewlydevelopedbeveragesinspiritwastesandindistillatesobtainedwithdifferentmanufacturingprocesses Our recent researchwasdirected towardsdevelopment of an algorithmofldquoEthanolasISrdquomethodapplicationonGC-MSinstruments[4]WehaveshowedthattopreventMSdetectorfromsaturationethanolshouldberegisteredinthecorresponding SIM timewindow at characteristicmz of low abundance forinstance bymz of 47 ions This ion corresponds to non-fragmented ethanol
13moleculescontaining1heavyisotope(mainly C)FinallytheresultsofmeasuredstandardsolutionsshowedthatthesuggestedapproachisvalidandldquoEthanolasISrdquomethodmaybesuccessfullyusedonGC-MSinstrumentstoo The objective of this studywas to test and further approve the suggestedapproachonalargersetof36realsamplesofalcoholicdrinkseithercommercialorhomemadeThesamplesweresimultaneouslyanalysedbytwoGC-MSmethodsndashaclassicalISmethodprescribedinthelegislationandthesuggestedldquoEthanolasISrdquomethod
2Experimental
21Reagentsandchemicals
Thefollowingvolatilecompoundsweredeterminedintestedsamplesofalcoholicbeverages 11-diethoxyethane (acetal) acetaldehyde methyl acetate ethylacetate methanol 2 propanol 1-propanol 2 methylpropan-1-ol (isobutanol)1-butanol2-butanoland3methylbutan-1-ol(isoamylalcohol)1-pentanolwasemployedasatraditionalIScompound
22Instrumentation
ShimadzuGCMS-QP2010Ultraequippedwithaquadrupolemassspectrometrydetector was employed for GC-MS measurements Rxi-1301Sil MS capillarycolumn(60mlength025mmid025micromfilmthicknessRestek)wasusedfortheseparationofcompoundsInjectionswereperformedinasplitmode(ratio175)Helium(99999purity)wasusedasacarriergasinjectortemperaturewas170degCTheoventemperaturewasheldat30degCfor5minthenraisedto210degC
ndash1at a rate of 30 degCmin and held isothermally for 4minMeasurementswereperformedinaSIMmodeFortheanalysedcompoundsand1-pentanol2ndash3mostabundantionsinthecorrespondingMSspectrumwereselectedethanolSIMtimewindowcontainedonly47mzionsAllGC-MSmeasurementswerecarriedoutintriplicateunderrepeatabilityconditions
136 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
AnalysisofeachalcoholicsamplewasperformedinafollowingwayAliquotof09mL of a tested sample was pipetted into a standard 2mL glass vial and
ndash1weighedAfterthat01mloftheISsolution(2355mgkg of1-pentanolinWES)wasaddedtothetestedsampleandthemasswasrecordedTheobtainedmixturewasmixedthoroughlyand05microlofitwasinjectedintotheGCsystem Theoriginoftestedalcoholicbeverageswaseithercommercialorhomemade33 world-famous spirits manufactured at different parts of the world werepurchasedfromcommerciallyavailablesourcesThelistoftypesofpurchasedandanalysedspiritsincludedbourboncalvadoscognacgingrappaliquormetaxaportwine rumsake tequilavodkawhiskeyandvarious fruitdistillatesThepurchased drinks were produced at the territory of the following countriesBelarus Bermuda Cuba Czech Republic Denmark France Germany GreeceGuatemala Jamaica Japan Mexico Moldova Portugal Slovakia Trinidad andTobago UK (England and Scotland) USA Three homemade fruit distillatesproducedbyfermentationofpulpyfruitsortheirmustswereobtainedfromlocalspiritmakersThedeclaredABVvaluesofalltestedsamplesvariedfrom15to81
3Resultsanddiscussion
To fulfil themaingoalof thiswork ie toevaluate thestatisticaldifferenceofresultsyieldedbythecomparedmethodswehaveemployedStudentst-testforindependentsamplestoverifystatisticaldifferencesonthesignificancelevelofp=005Theobtainedempiricalvaluesforallpairsofcongenersrsquoconcentrationswere lower than critical one in all cases demonstrating that concentrationsobtainedbythetwomethodshavenostatisticaldifferenceandleadtothesameresults InadditionrepeatabilityofthetwomethodswascomparedthereforeallRSDvalues obtained from triplicatemeasurements were split in two groupswith
ndash1respecttothecorrespondingconcentrations(lowerthan50mgL AAandhigherndash1than50mgL AA)Theobtainedresultsarepresentedintheformofboxplotin
Fig1AnalysisofthechartinFig1showedthatboththetestedmethodshaveyieldedstatisticallysimilarrepeatability AllofthetestedalcoholicdrinkssatisfiedtherequirementsofEURegulation(EC)no1102008[5]Theconcentrationsofundesirablecompoundssuchasmethanoldidnotexceedthelevelsspecifiedinthesameregulationforcorres-ponding beverages In Table 1 the description of the used SIM method andsummaryoftheexperimentalresultsarepresented Tocomparethetruenessofthemethodsoneofthespiritsampleswasspikedwithstandardsolutions(ABV40)containingallanalysedvolatilecompoundsat
ndash1concentrationsof50500and5000mgL AATheoriginalsamplewasusedasareferenceEachofthespikedsolutionswasmeasuredintriplicateSelectedspirit(cherry distillate) initially contained all 11 volatile compounds in various
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 137
138 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1BoxchartsofRSDsofusedISmethodsat2concentrationrangesMeanisequaltoarithmeticmeanoraverageInterquartileRange(IQR)meansisthedistancebetweentheupper(themedianoftheupperhalfofthedataset)andlower(themedianofthelowerhalfofthedataset)quartile
ndash1Compound Timemin Registeredmz Numberof ConcentrationmgL AA results Minimal Maximal
Acetaldehyde 0ndash42 314344 36 24 715Methanol 36 13 13600
Ethanol(IS) 42ndash48 47 mdash mdash mdash
2-Propanol 48ndash70 29314345 14 27 199Methylacetate 596174 10 34 3201-Propanol 26 361 12070Ethylacetate 27 166 107002-Butanol 11 18 2080
Isobutanol 70ndash200 3141ndash4345 28 19 20001-Butanol 5556 13 28 155Acetal 26 45 270Isoamylol 31 39 26501-Pentanol(IS) mdash mdash mdash
Table 1DescriptionoftheusedSIMmethodandsomestatisticsconcerningallmeasured36spiritsamplesbothpurchasedandhomemade
concentrationsTheobtainedrecoveriesboxchartsareshowninFig2Compa-risonoftherecoveriesobtainedwithtwomethodsindicatesthattheyhavenosignificantdifferenceintermsoftruenessAverageobtainedrecoverywas981whenusingsuggestedmethodand980whenusingtraditionalISmethod
4Conclusions
InthisworktheresultsoftestingtheadvantageousldquoEthanolasISrdquomethodfortheGC-MS quality control analysis of alcoholic beverages were presented33purchasedsamplesofworld-famousalcoholicbeveragesoriginatingfrom18countriesand3homemadefruitdistillateswereanalysedtomakeathoroughandcomprehensive studyof thedevelopedmethodTheconcentrationsofvolatile
ndash1compoundsinanalysedsamplesvariedfrom1to13500mgL AAtheABVvalueofanalysedsamplesvariedfrom15to81ThesuggestedmethodwascomparedwiththetraditionalISmethodthatiscurrentlystatedinlegislationTheindepen-dentsamplest-testrevealedthatwithaprobabilityof095resultsobtainedwithtwo methods do not differ significantly The results of within-run precision(repeatability)showedrelativestandarddeviationswithin3measurementstobelessthat6indicatingthatthetechniqueisreproducibleThetruenessofthemethodwasevaluatedbyrecoverycalculationAccordingtotheobtainedresultsrecoveryofthesuggestedmethod(981plusmn33)wasslightlybetterthanthatofthetraditionalone(980plusmn58) ThesefactsprovethatdevelopedldquoEthanolasISrdquomethodistruepreciseandreliable when employed on GC-MS instruments At the same time to obtain
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 139
Fig 2Boxchartsof recoveriesof thesuggested(dottedpattern)and traditional (brickpattern)ISmethodsatdifferentspikeconcentrationsSymbolsdefinitionsarethesameasinFigure1
concentrationsofvolatilecompoundsintheofficiallyrequiredunitsofmeasurendash1 ndash1(mgL AAgL AAetc)suggestedmethodrequiresnodensitometrymeasure-
mentsofthetestedsampleandnoadditionofIScompoundoranyothersamplepre-treatmentThismethodprovidesaninvaluableanalyticaltoolforthequalitycontrolofalcoholicproductsandshouldbeusedinroutineanalysis
Acknowledgments
ThisworkwasfinanciallysupportedbytheVisegradFund
References
[1] CherepitsaSVBychkovSMKovalenkoANMazanikALSeleminaNMSeredinskayaOBThe use of themajor component (solvent) as an internal standard in the gas-chromato-graphicdeterminationofimpuritiesJAnalChem58(2003)368ndash371
[2] CharapitsaSSytovaSKorbanASobolenkoLEgorovVLeschevSZakharovMCabalaRBusarovaRShestakovichITolstouhovaAOndrousekSVavraJYilmaztekinMCabarogluTInterlaboratorystudyofethanolusageasaninternalstandardindirectdeterminationofvolatile compounds in alcoholic products BIO Web Conf 15 (2019) 02030httpsdoiorg101051bioconf20191502030
[3] CommissionRegulation(EC)No28702000layingdownCommunityreferencemethodsfortheanalysisofspiritsdrinkshttpdataeuropaeuelireg20002870oj
[4] KorbanACharapitsaSCabalaRSobolenkoLSytovaSTheperspectivesofethanolusageasaninternalstandardforthequantificationofvolatilecompoundsinalcoholicproductsbyGC-MSJMassSpectr55(2020)e4493
[5] EuropeanUnion(2008)Regulation(EC)No1102008oftheEuropeanParliamentandoftheCouncilof15January2008ontheDefinitionDescriptionPresentationLabellingandtheProtectionofGeographical IndicationsofSpiritDrinksandRepealingCouncilRegulationhttpdataeuropaeuelireg2008110(1)oj
140 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Carbonorgraphitefeltsareusedaselectrodematerialsincethe1990sandtheirutilizationstillgrowsinanalyticalelectrochemistryaswellasinotherareasThisisduetotheirsuitablepropertiesfromwhichwecannamehighporosityhighspecificsurfaceareagoodelectricconductivityandhighphysicalandchemicalstabilityThefirsttwoparametersaregivenbystructureoffeltwhichconsistsoforderlesscarbonfibreswithabouttentotwentymicrometersindiameter[1]theothersby theadvantageouselectricalpropertiesof carbon fibreOn theotherhand porous flow-through electrodes including carbon felt electrode havedisadvantageinapotentialdropintheelectrodevolumewhichcausesdifficultcontrollingoftheexactpotentialappliedontheelectrodeandthereforeresultsindifferentcurrentefficienciesontheoppositesidesoftheelectrode[2] Carbon felt electrode can be utilized for detection of structurally differentcompoundsatvariousconditionsForexampleoperatingatreductionpotentialofndash08V[3]oxidationatrelativelyhighpotential+15V[4]ormeasuringatlowconcentrationsofelectrolyte[5]canbenamedDevelopedtechniquesalsoshowsthatcarbonfeltcanbeusedfordeterminationatsubmicromolarconcentrationsThis ismainly due to its ability to operate as a high-efficiency amperometricdetector Themainaimofthispaperistooverviewandcompareparametersofseveraldetermination methods of different analytes using carbon felt detector in
Utilization of a carbon felt as a material for working electrodes
MARTINBAROCHHANADEJMKOVA SA RKASLA DKOVA
DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublicmartinbarochnaturcunicz
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 141
AbstractWorkingelectrodemadeofcarbonfeltwasusedincombinationwithHPLC for verificationofpractical applicabilityof the electrodeAlldeveloped methods confirm advantageous physical and chemicalpropertiesofcarbonfeltForelectrochemicalutilizationitispossibleto operate at higher positive potentials and even in low concen-trationsofelectrolyteinmobilephaseObtainedlimitsofdetectionwere mostly in submicromolar range and standard deviations ofmeasurementrepeatabilitywereunder5
KeywordsamperometrycarbonfeltFIAHPLC
combinationwithHPLC(forexampletheirdetectionpotentialslimitsofdetectionorlinearrange)
2Experimental
21Reagentsandchemicals
Stock solutions of propyl gallate butylhroxyanisole tert-butylhydroquinonebutylhydroxytoluene chlortoluron 2-amino-4-nitrophenol and 4-amino-2-
ndash3 ndash1nitrophenol (all SigmandashAldrich) with concentration of 1times10 molL werepreparedbydissolving theappropriateamountof therespectivesubstance inmethanol(HPLCgradeLach-NerCzechRepublic)Stocksolutionsof indole-3-aceticacidandindole-3-butyricacidwerepreparedinthesamemannerbutindeionized water Mobile phase consisted of methanol and phosphate-acetatebufferpreparedfromphosphoricandaceticacid(bothLach-NerCzechRepublic)andsodiumhydroxide(Fluka)
22Instrumentation
Theelectrochemicalcellconsistedofcarbonfelt(KarbotechnikCzechRepublic)flow-throughelectrodewhichwasplacedincapwithplatinumwireelectricalcontactanddrilledoutletholeononesideandwithflatferrulawithcapillaryonthe other side Schematic picture of the assembly is shown in ref [5] Otherelectrodeswereauxiliaryplatinumwireelectrodeandreferencesilverchloride(3MKCl) electrode (bothMonokrystaly Turnov Czech Republic) Potentiostatused in combination with this cell was Amperometric Detector ADLC 2(LaboratonıprıstrojePrahaCzechRepublic) HPLCapparatusconsistedofBeta10gradientpump(ECOMCzechRepublic)degasser DG 4014 (ECOM Czech Republic) six-way valve with 20microl loop(Rheodyne USA) HPLC column used for separation of propyl gallatebutylhydroxytoluenetert-butylhydroquinoneandbutylhroxyanisolefromtheirmixtureandforindole-3-aceticacidandindole-3-butyricacidfromtheirmixture
regwasLichrospher RP-18(125times4mm5micromMerckGermany)ForseparationofregchlortoluroncolumnPurospher RP-18(125times4mm5micromMerckGermany)was
used In case of mixture 2-amino-4-nitrophenol and 4-amino-2-nitrophenolregcolumnGemini C18110A (150times46mm5micromPhenomenexUSA)wasusedfor
separation Measurements of pHwere carriedout at Conductivity andpH-meter3510usingcombinedglasselectrode(JenwayUK)
142 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
3Resultsanddiscussion
Performanceofcarbonfeltwastestedonseveraltypesofanalyteswhichneededdifferent separation conditions namely amount of organic solvent in mobilephase and buffer pH The lowest amount of methanol (30) was used forseparation of 2-amino-4-nitrophenol and 4-amino-2-nitrophenol [3] Higherconcentrations of methanol in mobile phase was used for determination ofchlortoluronandforseparationofindole-3-aceticacidandindole-3-butyricacid[4]namely40and60respectivelyThehighestconcentrationsofmethanoland therefore electrolyte with lowest conductivity was used in separation ofantioxidantsnamelypropylgallatebutylhroxyanisolebutylhydroxytolueneandtert-butylhydroquinonewhereamountofmethanolwasrampingfrom55to95[5]DetectionpotentialsofmentionedanalytesandtheirlimitsofdetectionsareshowninTable1 HPLCseparationofantioxidantswas the firstmethodchosen for testingofcarbonfeltelectrodeperformancewiththistechniqueDuetodifferencesintheirstructure when butylhydroxytoluene has a different oxidation mechanismdetectionwith two appliedpotentialswas necessary According to the hydro-dynamic voltammograms potentials 14V and 08Vwere chosen for determi-nationofbutylhydroxytolueneandfortheotherthreeanalytesrespectively[5]AsshowninTable1whenthehigherpotentialwasappliedlimitsofdetectionforpropyl gallate butylhroxyanisole and tert-butylhydroquinone had increasedContrary determination of butylhydroxytoluene had approximately six timeslowerdetectionlimitathigherpotential
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 143
ndash1Compound E VLODmicromolL Refdet
Propylgallate 080 088 [5] 140 186 [5]Butylhroxyanisole 080 144 [5] 140 348 [5]tert-Butylhydroquinone 080 121 [5] 140 266 [5]Butylhydroxytoluene 080 3128 [5] 140 463 [5]Chlortoluron 140 013Indole-3-aceticacid 150 033 [4]Indole-3-butyricacid 150 054 [4]4-Amino-2-nitrophenol 080 016 [3]2-Amino-4-nitrophenol 080 021 [3]4-Amino-2-nitrophenol ndash080 35 [3]2-Amino-4-nitrophenol ndash080 37 [3]
Table 1Parametersofdetectionpotentialsandlimitsofdetectionfordifferentcompoundsusingcarbonfeltdetector
Detectionofauxins(indole-3-aceticacidandindole-3-butyricacid)wascarriedout at potential 15VMeasurements at this potential gives repeatabilitywithstandarddeviation31forindole-3-aceticacidand25forindole-3-butyricacidevenwithexchangingoftheworkingelectrodematerialCalibrationcurves
ndash1forbothanalyteswereobservedfrom04to100μmolL withlinearityinwholeconcentrationrangeLimitsofdetectionforbothanalytesreachedsubmicromolarconcentrationsevenwithrelativelyhighpotential[4] Incaseofdeterminationof2-amino-4-nitrophenoland4-amino-2-nitrophenolcarbon felt electrodewasused inbothoxidationand reductionmodeHydro-dynamicvoltammogramsshowedthatoptimaldetectionpotentialinreductionmodewasndash08VThislowpotentialisclosetotheendofthepotentialwindowandthereforeinterferenceswithremnantsofdissolvedoxygeninmobilephasewereobservedTheseinterferencesresultedinapproximately20timeshigherlimitofdetection for 2-amino-4-nitrophenol or 4-amino-2-nitrophenol obtained inreductionthaninoxidationmodeOntheotherhandmaximumvaluesoflinearrangewerethesameforbothanalytesinbothdetectionmodes[3] ForHPLCofchlortolurontheoptimaldetectionpotentialof14VwasfoundIts
ndash1calibration dependence although observed from 025 to 1000μmolL was ndash1linearonly in therange from025to50μmolL Limitofdetectionbasedon
ndash1standardsolutionswas013μmolL andreproducibilityofmeasurementgivenbytwentyconsecutivemeasurementsgaverelativestandarddeviationof05 ForallthedeterminationmethodsattentionwaspaidtotheapplicabilityofthecarbonfeltindetectionofanalytesincomplexmatricesIncaseofantioxidantsedibleoilswerechosenasrealsamples[5]nitrophenolderivatesweredeter-minedinurinesamples[3]andauxinsinrootingpreparation[4]ChlortolurondeterminationwasperformedinsoilandsurfacewaterThefoundvaluesshowanegligiblematrixinfluenceondetection
4Conclusions
CarbonfeltworkingelectrodewassuccessfullyusedincombinationwithHPLCfordeterminationofdifferenttypesofelectroactivecompoundsegantioxidantsauxinsorpesticidesAllmentionedapplicationsshowgreatperformanceofthecarbonfeltasaflow-throughelectrodematerialinelectroanalyticalchemistryforoxidationandreductionwayofanalytesdeterminationLimitsofdetectionforanalytes are mostly in submicromolar concentrations the exceptions areoxidationof analytes athigherpotentials and their reductionwhere limitsofdetectionsareinmicromolarconcentrationsApplicabilityoftheelectrodeonrealmatriceswas proven on analysis of edible oil samples groundwater soil androotingpreparation
Acknowledgments
ThisworkhasbeensupportedbytheCzechScienceFoundation(projectGACR20-01589S)
144 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
References
[1] Gonzalez-Garcia J Bonete P Exposito E Montiel V Aldaz ATorregrosa-Macia RCharacterizationofacarbonfeltelectrodeStructuralandphysicalpropertiesJMaterChem9(1999)419ndash426
[2] NavaJLRecendizAGonzalezLGCarrenoGMartın ezFMassTransportandpotentialstudiesinaflow-throughporouselectrodereactorPortugalElectrochimActa27(2009)381ndash396
[3] Dejmkova H Knaf M Application of carbon felt detector for the determination ofdinitrophenolmetabolitesInXXXIXModernElectrochemicalMethodsFojtaMSchwarzovaKNavratilT(Eds)U stınadLabemBestServis2019p41ndash43
[4] DejmkovaHdeAraujoDanielMElectrochemicaldeterminationofindole-3-aceticacidandindole-3-butyric acid using hplc with carbon felt detectorMonatsh Chem150 (2019)439ndash442
[5] DejmkovaHBarochMKrejcovaMBarekJZimaJCoulometricdetectorbasedoncarbonfeltApplMaterToday9(2017)482ndash486
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 145
1Introduction
Smith-Lemli-Opitzsyndrome(SLOS)isan autosomal recessive genetic disor-der firstly described in 1964 [1] It iscaused by the inborn deficiency of7-dehydrocholesterol reductase Thisenzyme transforms 7-dehydrocholes-terol (7-DHC Fig 1) to cholesterolduringthefinalstepofbiosynthesisofcholesterolincells The clinical symptoms of SLOS aredecreasedbloodlevelofcholesteroland
Electroanalytical methods for determination of 7-dehydrocholesterol in artificial serum
LENKABENESOVAADE LAZA RYBNICKA JANKLOUDAKAROLINASCHWARZOVA -PECKOVA
UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublicbenesolenaturcunicz
Abstract7-DehydrocholesterolisabiomarkerofSmith-Lemli-Opitzsyndromeanautosomalrecessivegeneticdisordercausedbytheinborndefici-encyof7-dehydrocholesterolreductaseInthisstudyproceduresforitsdeterminationinartificialserumusingflowinjectionanalysiswithelectrochemical detection and voltammetric detection on borondoped diamond electrode were optimized The proteins wereprecipitatedbyacetonitrileandaftercentrifugationthesupernatantused for analysis For quantitation of 7-DHC by differential pulsevoltammetrytheoptimalratioacetonitrile-artificialserum91(vv)wasappliedInFIA-EDtheratio31(vv)runelectrolyteconsisting
minus1ofwater-acetonitrilecontaining001molL NaClO inthesameratio4minus1anddetectionpotentialof+13VvsAgAgCl(3molL KCl)were
usedQuantitationof7-DHCwaspossibleusingcalibrationdepen-minus1dencewithlimitdetectionof20micromolL inartificialserumNeverthe-
lessthemethodhaslowrecoveryandforsensitivedeterminationinreal matrices of human serum and amniotic fluid a liquid-liquidextraction needs to be applied to prevent presence of 7-dehydro-cholesterolinthephasewithprecipitatedproteins
Keywordsamperometricdetectionborondopeddiamond
electrode7-dehydrocholesteroldifferentialpulse
voltammetryflowinjectionanalysisSmith-Lemli-Opitz
syndrome
146 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Structureof7-dehydrocholesterol
increased concentrationof7-DHC inbloodandnervous system [2] SLOS is acomplexofmultipleanomaliesincludingmentalretardationItismanifestedbyholoprosencephaly(anomaliesinbraindevelopmentwithimprecisedivisionintotherightandlefthemispheres)milddysmorphismscardiacrenalandgastro-intestinalmalformations ThecharacteristicfacialanomaliesofSLOS[2]aremicrocephalybitemporalnarrowing ptosis short nasal root short nose with anteverted nares andmicrognathia epicanthal folds and capillary hemangioma over the nasal rootextendingontotheglabellatheearappearlow-setandareposteriorlyrotatedOral finding includes a high-arched and narrow hard plate broad and ridgealveoralridgesandredundancyofsublinqualtissuesCNSanomaliesareagenesisorhypoplasiaBilateralandunilateralpostaxialpolydactylycanbepresentedinthehandsorfeetorboth Concentrationof7-DHCinbloodiscrucialforclinicaldiagnosticofSLOSinpatients Concentration levels in amniotic fluid are used for fetal diagnosticsTable1 summarizes concentration of 7-DHC in plasma and amniotic fluid ofhealthypersonandSLOSpatientsAnalyticalmethodsusedfordeterminationofconcentrationof7-DHCinthesematricesincludecombinationofGCorHPLCwithMS[3]orGCwithflameionizationdetection(FID)orUVdetection[45] Thepossibilitiesofelectrochemicalmethodsfordetectionof7-DHCarelimitedasgenerallythesteroidcoreisratherredox-inactive(detailinreview[6])undervarietyofconditionsNevertheless7-DHCpossessesconjugateddoublebondsonsteroidcoreanditsoxidationwasreportedinseveralstudies[7ndash9]Itsvoltam-metricsignal+095VvsSCEonglassycarbonelectrodewasfirstlyobservedin
minus1non-aqueousmediaofmethanol-benzene7525(vv)using005molL LiClO as4
supporting electrolyte in a study dealing with electrochemical behaviour ofvitaminAandDandtheirprovitaminsD(7-DHCisprecursorofvitaminD3)[7]Determinationof7-DHCinhumanskinispossiblebyHPLCwithUV(λ=286nm)andamperometricdetection(E +17VvsAgAgCl)onglassycarbonelectrodedet
minus1usingmethanol-tetrahydrofuran175mmolL KH PO (9514vvv)asmobile2 4minus1phase7-DHCwasdetectedintherangefrom12to81microgg dryweightwith
minus1detectionlimitof39pmolL [8]Anotherstudy[9]isdevotedtodeterminationof
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 147
minus1Author(s)ref Matrix ConcentrationμmolL Healthy SLOS
Kelley[4] Plasma 03plusmn001 385plusmn309 Amnioticfluid lt02plusmn001 16plusmn9Rossiteretal[5] Plasma lt5 179ndash335 Amnioticfluid lt03 12ndash15
Table 1Concentrationof7-dehydrocholesterolinclinicalmatricesforhealthypersonsandforSmith-Lemli-Opitzsyndrome(SLOS)patientsdetectedbyGC-FID
7-DHCandvitaminD3 in fishusingHPLCwithelectrochemicaldetectionTheanalytical cell was a serial combination of two-flow-through porous graphiteworking electrodes The first standard coulometric electrode was used toeliminatepotentially interfering compounds using the second lineardynamic
minus1rangefrom0013to0312micromolL for7-DHCwasachieved Hereinwestudiedpossibilitiestodetect7-DHCbasedonitsoxidationonborondoped diamond (BDD) electrode using differential pulse voltammetry andelectrochemicaldetectioninflowinjectionanalysis(FIA-ED)inartificialserumandperipherallyinhumanserumandamnioticfluid
2Experimental
21Reagentsandchemicals
7-dehydrocholesterol (purity95)wasobtainedSigmaAldrich (USA)and itsstandard solution was prepared in acetonitrile (Honeywell Germany) Theartificial serum was prepared from KCl (Penta Czech Republic) CaCl 2H O2 2
(PentaCzechRepublic)NaCl(PentaPragueCzechRepublic)urineD-glucoseand01albuminfromSigma-Aldrich(USA)NaClO4(PentaCzechRepublic)wasusedassupportingelectrolyte
22Instrumentation
VoltammetricmeasurementweregovernedbythepotentiostatPalmSensusingworking BDD electrode (Windsor Scientific UK d = 31 mm) AgAgNO 3
minus1 minus1(01molL AgNO 1molL NaClO in acetonitrile) non-aqueous reference3 4electrodeandplatinumwirecounterelectrodeBDDsurfacewaspolishedbeforeeachscanusingsuspensionofAl O (ElektrochemickedetektoryTurnovCzech2 3
Republic) HPLC system (Hitachi Merck) consisting of control unit D-7000gradientpumpL-7100autosamplerL-7200andUVdetectorL-7400wasusedfor
minus1FIA-EDdetectionof7-DHCRunelectrolytewascomposedof001molL NaClO 4inacetonitrileanddeionisedwaterinratio31(vv)Flowrateofmobilephase
minus1was30mlmin injectionvolumewas40microLandλ=280nmwasusedforUVdetectionWall-jet detection cell was employedwith working BDD electrode
minus1AgAgCl (3molL KCl) reference electrode and platinum wire auxiliaryelectrode Optimal detection potential of +13 V was controlled using ADLC2potentiostat(Laboratornı prıstrojePragueCzechrepublic)
3Resultsanddiscussion
In this study electroanalytical methods were developed for determination of7-DHCinartificialserumnamelyFIA-EDandDPVBothmethodsarebasedondirectoxidationof7-DHConborondopeddiamondelectroderesultinginanodic
148 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
peak at ca +08 V (vs AgAgNO in acetonitrile) in non-aqueous medium of3
acetonitrile or mixedmedium acetonitrile-water using NaClO as supporting4
electrolyteTheoxidationispresumablyinitiatedbyoneelectronremovalfromtheconjugateddoublebondsonthesteroidcoreof7-DHC Fordeterminationof7-DHCinrealmatricesitisnecessarytoremovepresentproteinsArtificialserumcontainingalbuminwasusedasmodelmatrixtostudythe possibilities Firstly albumin was removed simply by precipitation withacetonitrile(serum-water13(vv))andthesupernatantwasanalysed Differentratioswateracetonitrileweretestedinrunelectrolyte(59510902080 2575 3070 4060 and 5050 (vv)) to evaluate the influence of itscompositionontheFIA-EDsignaloftheblankand7-DHCThesameratio13asusedforprecipitationofalbuminwaschosenasoptimalbecauseofminimalandstablesignaloftheblankinjectedinFIA-EDsystemFurtherdetectionpotentialE in the range from +10 V to +15 V was optimized by evaluation of thedet
hydrodynamicvoltammogramsresultinginE of+13Vsetasoptimalvaluedetminus1 Concentrationdependenceof7-DHCislinearintherangefrom25micromolL to
minus1 minus1300micromolL (concentrationinartificialserum)withdetectionlimitof20micromolL and this concentration dependence can be used for quantitation of 7-DHC inartificial serum Nevertheless determination of 7-DHC in human serum andamnioticserumfailedastheyrepresentmorecomplicatedmatricesand7-DHCispresumablypartiallyadsorbedinthepresentproteinsandcannotbequantifiedinthesupernatant Furtherdifferentialpulsevoltammetrywithoptimizedparameterswasusedfordeterminationof7-DHCInthepresenceofproteinsinartificialserum(human
minus1serum albumin) an unacceptably high detection limit of 178micromolL wasachieved When the proteins were precipitated using acetonitrile (aceto-nitrileartificialserumratio91(vv))thelimitofdetectionof7-DHCwaslowered
minus1to15micromolL inartificialserumNeverthelesstherecoveryofthemethodwasonly43to70dependingontheconcentrationof7-DHCagainreflectingthelossof7-DHCduetoproteinprecipitation Thereforeasecondapproachofsamplepretreatmentbasedonliquid-liquidextractionofalllipidsdescribedin[10]wastested(Bligh-Dyerextraction)TheprocedurehastwopartsFirstlymethanolchloroformandthesampleofartificialserum is mixed and shaken to form a monophasic system After addition ofchloroform and water a biphasic system is formed where chloroform phasecontainsalloflipidcompoundsandmethanol-waterphasecontainsallnon-lipidscompoundsChloroformphaseisthendriedunderN2atmosphereat50degCanddried extract dissolved in acetonitrile Preliminary experiments using DPVresultedinrecoveryof97forBligh-Dyerextractionof7-DHCfrominartificialserum
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 149
4Conclusions
FIA-EDandDPVwereoptimizedfordeterminationof7-DHCinartificialserumUsing precipitation of proteins by acetonitrile limit of detection of 7-DHC in
minus1artificialserumusingFIA-EDwas20micromolL andthismethodcanbeusedfortheirquantificationusingcalibrationdependenceNeverthelessdeterminationusingDPVisunreliableduetolowrecoveryoftheprocedureDevelopmentofamethodincludingliquid-liquidextractionstepisinprogresssothat7-DHCcouldbedeterminedinrealmatricesashumanserumandamnioticfluid
Acknowledgments
TheresearchwassupportedbytheCzechScienceFoundation(projectGACR19-11268S)andtheSpecificUniversityResearch(SVV260560)
References
[1] Smith DW Lemli L Opitz JM A newly recognized syndrome of multiple congenitalanomaliesJPediatr64(1964)210minus217
[2] Nowaczyk M Waye J The SmithndashLemlindashOpitz syndrome a novel metabolic way ofunderstandingdevelopmentalbiologyembryogenesisanddysmorphologyClinGenet59(2001)375minus386
[3] BeckerSRohnikeSEmptingSHaasDMohnikeKBebloSMutzeUHusainRAThieryJCeglarekULC-MSMS-basedquantificationofcholesterolandrelatedmetabolitesindriedblood for the screening of inborn errors of sterolmetabolismAnal Bioanal Chem407(2015)5227minus5233
[4] Kelley RI Diagnosis of Smith-Lemli-Opitz syndrome by gas-chromatography mass-spectrometryof7-dehydrocholesterolinplasmaamniotic-fluidandculturedskinfibroblastsClinChimActa236(1995)45minus58
[5] Rossiter JP Hofman KJ Kelley RI Smith-Lemli-Opitz SyndromePrenatal-diagnosis byquantification of cholesterol precursors in amniotic-fluid Am J Med Genet 56 (1995)272minus275
[6] KloudaJBarekJNesmerakKSchwarzova-PeckovaKNon-enzymaticelectrochemistryincharacterization and analysis of steroid compounds Crit Rev Anal Chem 47 (2017)384minus404
[7] AtumaSSLundstromKLindquistJTheelectrochemicaldeterminationofvitaminAPartIIFurthervoltammetricdeterminationofvitaminAandinitialworkonthedeterminationofvitaminDinthepresenceofvitaminAAnalyst100(1975)827minus834
[8] MoodyJPHumphriesCAAllanSMPatersonCRDeterminationof7-dehydrocholesterolinhumanskinbyhigh-performance liquid-chromatography JChromatogrB530 (1990)19minus27
[9] OstermeyerUSchmidtTVitaminDandprovitaminDinfishEurFoodResTechnol222(2005)403minus413
[10] BlighEGDyerWJArapidmethodoftotallipidextractionandpurificationCanJBiochemPhysiol37(1959)911minus917
150 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Author Index
AlikovaV1
AugustınM83
BaluchovaS19
BarekJ192570
BarochM141
BasB13129
BastryginaO41
BenesovaL146
BessonovaE57
BohmD6
BurkinK116
BurkinM116
ChernovaA141
ChoinskaM70
CokrtovaK104
DedinaJ97
DeevV57
DejmkovaH141
DendisovaM63
DubenskaL51
EfremenkoE41
FojtaM110
GalvidisI116
HavranL110
HeiglN31
HertJ76
HrdlickaV70
JosypcukB25
KartsovaLA3557
KloudaJ19146
Kodrık ovaB90
KolobovaEA35
KorbanA135
KorotkovaE1
KralM63
KratzerJ90
KravchenkoAV35
KrızekT76104
LipinskaJ129
MadejM129
MatejkaP63
MatysikF-M631
MusilS9097123
NavratilT70
OndrackovaA110
PietrzakK45
PlotnikovaK51
PoradaR13
RedondoBR70
SagapovaL90
ShormanovV1
Schwarzova-PeckovaK19110146
SladkovaS 141
S tadlerovaB97
StiborovaM110
SvobodaM90
TvorynskaS25
TyczkowskiJ129
VyhnanovskyJ97123
VymyslickyF76
VyskocilV83
WardakC45
WongDKY19
ZarybnickaA146
ZelenyI51
Proceedingsofthe16thISCModernAnalyticalChemistry Prague2020 151
152 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Keyword Index
alcoholicbeverages135
aminoglycosides116
amperometricdetection146
amperometry141
antifoulingelectrodes19
assembledcapillaries6
atomicabsorptionspectrometry90
atomicfluorescencespectro-
metry97
atomization90
biologicalactiveanalytes35
biosensor2583
bismuth97
borondopeddiamond
electrode110146
cadmium90
canagliflozin76
capillarycoating35
capillaryelectrophoresis635
capillaryflowinjectionanalysis631
carbohydrates31
carbonfelt141
cathodicstrippingvoltammetry70
chemicalvaporgeneration90
chemometrics57
cobalt123
coldplasmadeposition129
copper(II)phthalocyanine63
covalentimmobilization25
cytochromeP450110
damage83
7-dehydrocholesterol146
designofexperiments76
differentialpulsevoltammetry146
diphenylsilanereductionmethod19
dispersiveliquid-liquid
microextraction57
disposableelectrodes31
DNA83
dualdetectionconcept6
electrochemicalanalysis110
electrochemicalapplications129
electrochemicalflowcell76
electrochemistry51
electrokineticchromatography104
eliminationvoltammetrywith
linearscan70
ELISA116
enzymaticreactor25
FIA141146
gaschromatography-mass
spectrometry(GC-MS)135
glucoseoxidase25
graphite83
honey116
HPLC76141
hydridegeneration97
hydrogenatedconical-tipcarbon
electrodes19
imidazoliumionicliquids35
inductivelycoupledplasmamass
spectrometry123
internalstandardmethod135
ion-selectiveelectrode45
laccase25
liquidcrystals104
massspectrometry31
mercuryelectrode13
metronidazole51
non-aqueouscapillary
electrophoresis104
non-aqueoussystem6
oxidation76
oxytetracyclinehydrochloride51
phenol2-methoxy1
photochemicalvaporgene-
ration97123
polarography51
1-propanesulfonicacid23-
dimercapto-70
pulsedamperometricdetection31
quantitation1
resonanceRamanspectroscopy63
scanningtunnellingmicroscopy63
silversolidamalgamelectrode70
Smith-Lemli-Opitzsyndrome146
smokingmixtures41
solidcontact45
solid-phasemicroextraction57
spectrophotometry141
SudanI110
surfacemodification129
surface-enhancedRaman
spectroscopy63
thinlayers129
tip-enhancedRamanspectroscopy
63
unithiol70
uranyl45
vanillin41
veterinarydrug51
vitamins13
volatilecompoundsquantifi-
cation135
voltammetricdopaminedetec-
tion19
voltammetry1383
Proceedingsofthe16thISCModernAnalyticalChemistry Prague2020 153
Proceedings of the 16th International Students Conference ldquoModern Analytical Chemistryrdquo
EditedbyKarelNesmerak
PublishedbyCharlesUniversityFacultyofScience
Prague2020
1steditionndashvi154pages
ISBN978-80-7444-079-3
ISBN 978-80-7444-079-3
Pro
ceedin
gs of th
e 16
th In
ternatio
nal Stu
den
ts Co
nferen
ce ldquoMo
dern
An
alytical Ch
emistryrdquo P
rague 2
02
0
788074 440793
Prague 17mdash18 September 2020
Edited by Karel Nesměraacutek
Prague 2020
Proceedings of the
16th International Students Conference
ldquoModern Analytical Chemistryrdquo

Contents
AlikovaVChernovaAShormanovVKorotkovaEDeterminationof2-methoxyphenol inmodelsolutionsbyspectrophotometry 1
BohmDMatysikF-MTheeffectsoflinearlyassembledcapillarieswithvariousinnerdiametersoncapillaryelectrophoresis 6
PoradaRBasBVoltammetricdeterminationofvitamins 13Baluchova S Klouda J Barek J Schwarzova-Peckova K Wong DKY Dopamine detection at
antifoulingconical-tipcarbonelectrodes 19Tvorynska SBarek J JosypcukBA comparative study of covalentglucose oxidaseand laccase
immobilizationtechniquesatpowderedsupportsforbiosensorsfabrication 25Heigl N Matysik F-M Capillary flow injection analysis with electrochemical detection for
carbohydrateanalysis 31KravchenkoAVKolobovaEAKartsovaLAApplicationofcovalentcoatingsbasedonimidazolium
cations for separationandon-linepreconcentrationof basicandneutralanalytes in capillaryelectrophoresis35
Efremenko E Chernova A Bastrygina O Determination of vanillin in smoking mixtures byspectrophotometry41
PietrzakKWardakCUranylion-selectiveelectrodewithsolidcontact 45Plotnikova K Dubenska L Zeleny I Polarographic determination of metronidazole and
oxytetracyclinehydrochlorideinveterinarydrugforhoneybees 51Deev V Bessonova E Kartsova L Application of microextraction techniques combined with
chromatographicmethodsfortheanalysisofcomplexobjects 57KralMDendisovaMMatejkaPThedevelopmentofreferenceprobesystemfortip-enhancedRaman
spectroscopy 63ChoinskaMHrdlickaVRedondoBRBarekJNavratilTDeterminationofheavymetalpoisoning
antidote23-dimercapto-1-propanesulfonicacidusingsilversolidamalgamelectrode70VymyslickyFKrızekTHert JCanagliflozinoxidation studyusingelectrochemical flowcelland
comparisonwithhydrogenperoxideoxidation 76AugustınMVyskocilVNovelhybridelectrochemicalDNAbiosensorformonitoringoxidativeDNA
damageviaoxidationreductionsignalsoflowmolecularweightdouble-strandedDNA 83SagapovaLKodrık ovaBSvobodaMMusilSKratzerJChemicalvaporgenerationofcadmiumfor
analyticalatomicspectrometry 90S tadlerova B Vyhnanovsky J Dedina J Musil S Photochemical vapour generation of bismuth
coupledwithatomicfluorescencespectrometry 97Cokrtova K Krızek T Separation of liquid crystals using non-aqueous capillary electrokinetic
chromatography104OndrackovaAStiborovaMHavranLSchwarzova-PeckovaKFojtaMElectrochemistryofSudanI
anditsderivatesinaqueousmedia 110BurkinKGalvidisIBurkinMGroupdetectionofaminoglycosidesusingELISAforcontroloffood
contamination 116Vyhnanovsky J Musil S Photochemical vapor generation of cobalt for detection by inductively
coupledplasmamassspectrometry 123LipinskaJMadejMBasBTyczkowskiJOptimizationofconditionforcoldplasmadepositionofthin
layersforsurfacemodificationofworkingelectrodes 129KorbanAAdvancedGC-MSmethodforqualityandsafetycontrolofalcoholicbeverages 135Baroch M Dejmkova H Sladkova S Utilization of a carbon felt as a material for working
electrodes 141Benesova L Zarybnicka A Klouda J Schwarzova-Peckova K Electroanalytical methods for
determinationof7-dehydrocholesterolinartificialserum146
Authorindex 151Keywordindex 152
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 v
1Introduction
2-Methoxyphenol(guaiacol)isusedinmedicineasanexpectorantThestructuralformulaisshowninFig1Itiswidelyusedinthepharmaceuticalindustry[1]forthesynthesisofantituberculosisexpectorantdrugs(Kas-nol Sudafed Ascoril Prothiazine Expectorant Guai-phenesinum) Moreover 2-methoxyphenol is often used as anaromaticsubstance [2] in the food industry Inparti-cular itwaswidelyusedintheproductionofsmokedfish and meat products using smokeless smokingtechnologyusingflavourings Ontheotherhand2-methoxyphenolhasthesymbolGHS07andhasahazardcodeXnTXi[2]accordingtotheGHSsystemItisverytoxicbyinhalationitcanirritatethemucousmembraneoftherespiratorytractandtheconjunctivaofthe
Determination of 2-methoxyphenol in model solutions by spectrophotometry
a a b aVALERIYAALIKOVA ANNACHERNOVA VLADIMIRSHORMANOV ELENAKOROTKOVA
a DepartmentofChemicalEngineeringEngineeringSchoolofNationalResourcesNationalResearchTomskPolytechnicUniversityLeninavenue30634050TomskRussiaalikovaleramailru
b DepartmentofPharmaceuticalToxicologicalandAnalyticalChemistryKurskStateMedicalUniversitystKarlaMarks3305000KurskRussia
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 1
AbstractA spectrophotometric approach for determination of 2-methoxy-phenol in model solutions has been developed The absorptionspectra of 2-methoxyphenol were determined in the wavelengthrangefrom200to400nminsolutionsof95ethanolacetonitrile01Msodiumhydroxideandethylacetatewithaconcentrationofthe
minus3analyte of 005mgdm For the quantitative determination of2-methoxyphenol a seriesof solutionswaspreparedwithvarious
minus3 minus3concentrationsfrom0001mgdm to005mgdm in95ethanolacetonitrile01MsodiumhydroxideTheopticaldensityof2-meth-oxyphenolinsolventswasmeasuredatawavelengthof276nmand289nmThedevelopedmethodwastestedusingthemethodanalysisofspikedsamples
Keywordsquantitation2-methoxyphenolUVVISspectrophoto-
metry
Fig 1Structuralformulaof2-methoxyphenol
eyeballinhighconcentrationswhenitpenetratestheskincanleadtoneurosiswhenadministeredorallycanstimulatetheesophagusandstomachresultinginheartfailurecollapseanddeathNowadaystherearepublisheddataoncasesofasystemicallergicreaction[3]causedby2-methoxyphenolderivativesandthereisa fatal case known [4] for oral administration of guaifenesin (3-(2-methoxy-phenoxy)propane-12-diol)oneofthecomponentsofcommonlyavailablecoughmedications Thedeterminationof2-methoxyphenolinenvironmentalobjectsaswellasinthefoodindustryiscarriedoutusinggaschromatographymethodswithsolid-phasemicroextraction[5]Inordertocontrol2-methoxyphenolinnaturaldrink-ing and treatedwastewater gas chromatography is used followed by opticaldetectionoftheeluate[6]Themaindisadvantagesofthismethodofanalysisarethelowselectivityanddurationofdetermination(about3hours)AlsoaccordingtoRussianStateStandartGOST33312-2015themethodofgaschromatographyisusedforthequalitativeandquantitativedeterminationof2-metoxyphenolinjuiceproducts Commonlyfortheanalysisoftoxicsubstancesinvariousbiologicalsamples(bloodplasmaurinesalivasweathair)bygaschromatographyitisnecessarytocarryoutmultistagesamplepreparationwhichcomplicatesandslowsdownthecourseofthestudy[7]Atthesametimeitisimportantthatduringtheprepa-rationofsamplesintheanalyzedcompoundstheirstructureisnotviolatedasthiswillleadtothedifficultyoftheiridentification Spectrophotometryintheultravioletregionhaslowersensitivitycomparedtothe abovemethods however thismethod does not require such complicatedpreparation of the analyzed samples it is a relatively affordable simple andinexpensive analysis method In addition its sensitivity can be significantlyimprovedbyapplyinganappropriateseparationprocedureandpreconcentrationbeforedetection[8]MethodUVspectrophotometryisusedtoassessthequalityof both medicinal substances and preparations made from them in terms ofauthenticitygoodqualityandquantitativecontentInadditionitisarelativelyaffordablesimpleandlow-costanalysismethod Ananalysisoftheliteraturedatashowedthattodaytherearefastandsensitivespectrophotometricmethodsforthedeterminationofpyrocatecholderivativesinmedicines[9]vanillininfoodproducts[10]andotherphenolsinwastewaterandwineproducts[1112]Howeverasfarasweknowinformationonthedetermi-nationof2-methoxyphenolfromtheabsorptionspectraintheultravioletregionisabsent Theaimofthisstudyistodevelopmethodsforthequalitativeandquantitativeof2-methoxyphenolinmodelsolutionsusingUVspectrophotometry
2 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
2Experimental
21Reagentsandchemicals
Asampleof2-methoxyphenolfromFlukawithabasicsubstancecontentofge98wastakenastheobjectofstudyAssolventsweusedacetonitrile(ChP)95ethanolethylacetateand01MsodiumhydroxidesolutionAllotherchemicalsusedwereofanalyticalreagentgrade
22Instrumentation
Theopticaldensitywasmeasuredincuvetteewithanabsorbinglayerthicknessof10mmusingaCary60spectrophotometer(AgilentUSA)Allmeasurementswerecarriedoutatroomtemperature
3Resultsanddiscussion
Thechangeinthebehavioroftheabsorptionspectrumwasinvestigatedinthewavelengthrangeof200ndash400nmFigure2isshowedthatwithanincreaseinthepolarityofthesolventtheabsorptionmaximumshiftstowardthevisiblepartofthe spectrum The wavelength of absorption maxima of 2-methoxyphenol ispresentedinTable1 A studyof thephotometricbehaviorof2-metoxyphenolinvarious solventsshowedthatacetonitrile95ethanoland01Msodiumhydroxidearethemostsuitablesolventsforthequalitativedeterminationofthetestsubstance
ndash3Fig 1Thespectraof2-methoxyphenolofconcentration005mgdm inthemediumofsolvents(anabsorbinglayerthicknessof10mm)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 3
Forthequantitativedeterminationof2-methoxyphenolaseriesofsolutionsndash3 ndash3withaconcentrationragefrom0001mgdm to005mgdm werepreparedin
acetonitrileand95ethanolTheopticaldensityof2-methoxyphenolinsolventswasmeasuredbywavelengthof276nmThedependenceoftheintensityoftheoptical density on the concentration of 2-methoxyphenol in 01 M sodium
ndash3hydroxide was plotted in the concentration range from 0005mgdm tondash3003mgdm Themeasurementwerecarriedoutbywavelengthof276nmThe
obtainedregressionequationsarepresentedinTable2Dataanalysisobtainedwasperformedusingleast-squaresmethodThedevelopedmethodwastestedusingthemethodanalysisofspikedsamplesTheresultsarepresentedinTable2
4Conclusions
Studieshaveshownthepossibilityofusingspectrophotometricanalysisforthequalitativeandquantitativedeterminationof2-methoxyphenolTheabsorptionmaximaofweredeterminedinsolutionsofethanolandacetonitrile(276nm)inasolutionofethylacetate(277nm)and01Msodiumhydroxide(289nm)Theconstructed calibration curves of thepure substance of 2-methoxyphenol hasshownagoodregressioncoefficient(Rgt099)andcanbeusedforquantitativedeterminationof2-metoxyphenolinbiologicalobjectsInthefutureitisplannedtoapplythistechniquetodetermine2-metoxyphenolinincadavericmaterial
Solvent Regressionequation Found S RSD Δх δ 2-metoxy- phenolg
ndash3 ndash6 ndash5Acetonitrile y=18294C+01130 499times10 50times10 028 2times10 044 Rsup2=09985
ndash3 ndash6 ndash595ethanol y=35131C+00269 503times10 03times10 021 7times10 137 Rsup2=09956
ndash3 ndash6 ndash501Msodium y=31196C+01101 495times10 01times10 018 1times10 028hydroxide Rsup2=09997
Table 2Results of the determination of 2-methoxyphenol (average of three measurements) in modelsolutionsbythemethodanalysisofspikedsamplestheconcentrationofintroduced2-methoxy-
ndash3phenolwas500times10 g(SndashstandarddeviationRSDndashrelativestandarddeviationΔхndashabsoluteerrorδndashrelativeerror)
Table 1Valuesofopticaldensityandwavelengthsinappropriatesolventswith2-methoxyphenol
minus3 minus1Solvent λnm εgdm cm
Acetonitrile 276 0082895ethanol 276 00853Ethylacetate 277 0083101Msodiumhydroxide 289 00744
4 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
References
[1] МельниковаИММизерницкии ЮЛКомбинированныеотхаркивающиепрепаратырастительного происхождения в педиатрическои практикеМедицинский совет 2(2018)93ndash97
[2] httpwwwthegoodscentscompanycomdatarw1032272html(accessed27stFebruary2020)
[3] RayMFaltayBHallerNACasereportanaphylacticreactiontoguaifenesinHospPract37(2009)60ndash63
[4] OkicMJohnsonTCrifasiJALongCMitchellEKSwiftonsetofcentralnervoussystemdepressionandasystolefollowinganoverdoseofguaifenesinJAnalToxicol37(2013)318ndash319
[5] ВолковCМЧерновецАНОпределениеконцентрациифеноловвгазовыхвыбросахпромышленных предприятии методом газовои хроматографии с твердофазнои микроэкстракциеи Сорбционныеихроматографическиепроцессы10(2010)723ndash728
[6] ШачневаЕЮОньковаДВСерековаСМСпособыопределенияфеноловвобъектахокружающеи среды Астраханский вестник экологического образования 4 (2013)138ndash142
[7] ГладиловичВДПодольскаяЕПВозможностипримененияметодаГХ-МС(Обзор)Научноеприборостроение4(2010)36ndash49
[8] Pena-PereiraFLavillaIBendichoCHeadspacesingle-dropmicroextractioncoupledtomicrovolumeUVndashVis spectrophotometry for iodine determinationAnal Chim Acta631(2009)223ndash228
[9] NagarajaPMurthyKCSRangappaKSGowdaNMMSpectrophotometricmethodsforthe determination of certain catecholamine derivatives in pharmaceutical preparationsTalanta46(1998)39ndash44
[10] Altunay N Development of vortex-assisted ionic liquid-dispersive microextractionmethodology for vanillin monitoring in food products using ultraviolet-visible spectro-photometryLWT93(2018)9ndash15
[11] Lupetti KO Rocha FRP Fatibello-Filho O An improved flow system for phenolsdetermination exploiting multicommutation and long pathlength spectrophotometryTalanta62(2004)463ndash467
[12] Figueiredo-Gonzalez M Cancho-Grande B Simal-Gandara J Garnacha tintorera-basedsweetwineschromaticpropertiesandglobalphenoliccompositionbymeansofUVndashVisspectrophotometryFoodChem140(2013)217ndash224
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 5
1Introduction
Thenumberofsamplesthesamplecomplexityandalsothenumberofsubstanceswhich need to be analysed simultaneously is increasing steadily ThereforepowerfulseparationanddetectionmethodsarerequiredOnewaytoachievethisisthecouplingofaseparationsystemwithmorethanonedetector[12] In recent years capillary electrophoresis (CE)was established as a potentseparation system due to its high separation efficiency and the low sampleconsumption [3] To generate more information numerous dual detectionconceptsforCEweredevelopedwhicharesummarisedelsewhere[12]Acom-binationofamperometricdetectionandmassspectrometry(MS)isaninterestingdual detection concept for CE because both detectors supply complementaryinformationForelectroactivespeciesamperometricdetectionisarobustandoneof the most sensitive detection method [4] Thus it is well suited for the
The effects of linearly assembled capillaries with various inner diameters on capillary electrophoresis
DANIELBO HMFRANK-MICHAELMATYSIK
InstituteofAnalyticalChemistryChemo-andBiosensorsFacultyofChemistryandPharmacyUniversityofRegensburgUniversitaumltsstraszlige3193053RegensburgGermanydanielboehmchemieuni-regensburgde
AbstractDuetotheincreasingneedofpowerfulanalyticalmethodsanewdualdetection concept for capillary electrophoresis (CE) with parallelamperometricdetectionandmassspectrometryshallbedevelopedFor this concept the CE flow has to be divided into two streamsutilizinga flowsplitter In thiswork theeffectsof combinedcapi-llarieswithvarious innerdiameterswerestudiedForpreliminaryinvestigationsthecapillarieswereconnectedinaserialconfigurationwithoutdeadvolumeUsingcapillaryflowinjectionanalysishyphe-natedtocontactlessconductivitydetectionitcouldbeshownthatthecouplingofidenticalcapillariesleadstoaslightdecreaseoftheflowratesWithCEhyphenatedtoUVdetection itcouldbeshownthatthecouplingofcapillarieswithdifferentinnerdiameterhasamuchstrongereffectontheelectroosmoticflowthanthecombinationwiththesameinnerdiameterFurthermorenosignificantchangeinpeakshapewasobserved
Keywordsassembledcapillariescapillaryelectrophoresiscapillaryflowinjection
analysisdualdetectionconceptnon-aqueoussystem
6 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
quantificationofsubstanceswhereasMSiswellsuitedfortheidentificationofunknown substances [3] In most dual detection concepts the detectors arearrangedinaserialconfigurationwhichisnotpossibleincaseofamperometricdetection-mass spectrometry [1] The instrumental implementation is morecomplicatedwithbothdetectorsbeingdestructiveFurthermoretheymustbedecoupledfromthehighvoltagefieldoftheCEThereforetheCEflowmustbedividedintotwostreamswithaflowsplitterAsimplifiedsketchofthepossiblenewdualdetectionconceptisshowninFig1 ForthedevelopmentofthenewdualdetectionconceptthreecapillarieswithpotentiallydifferentinnerdiametersmustbecoupledForthisreasonthedeadvolume-freecouplingofcapillarieswithdifferent innerdiameterswas investi-gatedinafirststepTokeepthesetupsimplewefocusedonthelinearcouplingofcapillariesandtheresultingeffectsNon-fragmentedcapillarieswerecomparedwithfragmentedcapillariesofthesameordifferentinnerdiametersEffectsonthe flow rate were investigated with capillary flow injection analysis (CFIA)
4hyphenatedtocontactlessconductivitydetection(C D)EffectslikechangesinthemigrationbehaviourorpeakshapesoccurringinCEwereinvestigatedwithCEhyphenatedtoUVdetection(CE-UV)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 7
Fig 1SchematicillustrationofthenewdualdetectionconceptwithparallelamperometricdetectionandmassspectrometricdetectionforCEAfterinjectionfrom(a)thesamplevialthecomponentsareseparatedbyCEnext(b)theflowsplitterdivides(c)thecapillaryintotwopartsandleadstheCEflowtowards(d)themassspectrometerand(e)theamperometricdetector
2Experimental
21Reagentsandchemicals
Thefollowingchemicalswereusedallofanalyticalgradeferrocenemethanoldecamethylferrocene(ABCRGermany)acetonitrileammoniumacetate01Msodiumhydroxidesolutionultra-purewaterprovidedbyaMilliQAdvantageA10system(MerckGermany)aceticacid(RothGermany)
22Instrumentation
221Capillaries
4Forbothexperiments(CFIA-C DandCE-UV)capillarieswithinnerdiametersof2550and75micromanouterdiameterof360micromandatotallengthof70cmwereusedTheywerepurchasedfromPolymicroTechnologies(USA)Measurementswere carried out with fragmented and non-fragmented capillaries For themeasurementswiththefragmentedcapillariestheoriginalcapillarieswerecutintotwopiecesyieldingatotalof9capillarycombinationswithlengthsof70cm(20cmfirstcapillarypieceand50cmsecondcapillarypiece)ThesecombinationsaresummarizedinTab1(section31)Atbothendsofthecapillariesabout02cmof thepolyimidecoatingwasremovedBothsidesof thecapillarypieceswerepolishedtoreceiveplanarcapillarytipsForthelinearassemblingofthecapillarypiecesMicroTightSleevesF185Xanda capillary connectorUnionAssemblyMicroTightP720fromIDEXHealthampScience(USA)wereusedPriortothefirstCEmeasurementsthecapillarieswereconditionedbyflushingthemfor10minwith01Msodiumhydroxidesolution5minwithultra-purewaterand30minwithseparationbuffer
222Capillaryflowinjectionanalysishyphenatedtocontactlessconductivitydetectionsetup
The flow rates for the fragmented and non-fragmented capillaries were4determinedwithaCFIA-C DsetupschematicallydepictedinFig2ATheflowin
thecapillarywasgravitationdrivenbyaheightdifferencebetweentheinletandoutletcarriersolutionvialTheconceptofCFIAwithgravitationdrivenflowwasfirstdescribedbyMatysiketal[5]AlaboratoryconstructedautosamplerofaCEdevicewasusedforthehydrodynamicinjectionThesamplesolutionconsistedof10mMdecamethylferrocene incarrier solution (10mMCH COONH and1M3 4
4CH COOH in acetonitrile) A high resolution C D was placed after 40 cm for3
detectionThedetectordescribedelsewhere[6]wasconstructedinthedoLagogroup(Brazil)Adoubledeterminationattwodifferentheightswasdoneforthedeterminationoftheflowrates
8 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
223Capillaryelectrophoresis-UVdetectionsetup
Fig2BshowsasketchoftheCE-UVsetupItconsistedofalab-builtCEdevicewhichwasconnectedtoahighvoltagepowersupplyfromISEG(Germany)Theseparationswerecarriedoutwithanon-fragmented50micromcapillaryandwithcapillary combinations implementing a 50micromdownstream capillary segment(25+5050+50and75+50microm)ALambda1010UV-VISdetectorfromBischoff(Germany)wasusedfordetectionat210nmThedetectorwasplacedafter40cm
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 9
Fig 2Schemeof(A)thecapillaryflowinjectionanalysis(CFIA)hyphenatedtocontactlessconduc-4tivity detection (C D) setup and (B) the capillary electrophoresis hyphenated to UV detection
4(CE-UV) setup Components of the CFIA-C D setup (a) sample (b) inlet and (c)outlet carrier4solution vial (d) fused silica capillary (e) linear capillary connector (f) C D and (g) stand
ComponentsoftheCE-UVsetup(h)sample(i)inletand(j)outletbuffervialand(k)UVdetector4therestofthecomponentswereidenticaltotheCFIA-C DsetupTheoutletbuffervialwaslowered
forthehydrodynamicinjection(j)Theenlargedview(k)depictsthecouplingoftwocapillarieswithdifferentinnerdiametersintheconnectionsidewithoutdeadvolume
Asamplesolutioncontaining1mMferrocenemethanolanddecamethylferroceneinseparationbuffer(10mMCH COONH and1MCH COOHinacetonitrile)was3 4 3
utilizedTheinjectionwasperformedhydrodynamicallybyloweringtheoutletbuffer vial by 20 cm A uniform sample plug was injected to compare bandbroadeningeffectsTheinjectionsegmenthadalengthof035cm(05ofthetotalcapillarylength)andtherespectiveinjectiontimewasdeterminedbasedontheflowratesofthecorrespondingcapillarycombinationFortheelectrophoreticseparationaseparationvoltageof25kVwasappliedandtheinletandtheoutletbuffervialswereplacedatthesameheightsothattherewasnogravityflowwhichaffectedthemigrationbehaviour
3Resultsanddiscussion
31Capillaryflowinjectionanalysishyphenatedtocontactlessconductivitydetectionexperiments
AsshowninTab1theflowratesforaheightdifferenceof20cmwerecalculated4basedontheCFIA-C DmeasurementsItwasobservablethattheflowratesare
slightlylowerforfragmentedcapillariesthanfornon-fragmentedcapillariesofthe same dimension This indicates that a flow resistance arises when twocapillaries are combined Furthermore it was observed that the flow ratedecreasesforupstreamcapillarieswithlowerinnerdiametersandviceversaTheflow rate for the combination 25+75microm could not be determineddue to theformationofairbubblesattheconnectionside
10 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Table 1Flowratesandthecorrespondingstandarddeviations(SD4measurements)ofdifferentcapillarycombinationsforaheightdifferencebetweeninletandoutletvialof20cmbymeansofcapillaryflowinjectionanalysishyphenatedtocontactlessconductivitydetection
ndash1capillary flowratenLs plusmnSDflowndash1combinationmicrom ratenLs
25 00577 0000525+25 0057 000150+25 00775 0000775+25 ndash ndash25+50 0137 000550 0865 000250+50 080 00575+50 105 00625+75 0187 000250+75 1941 000375 456 00275+75 42 01
32Capillaryelectrophoresis-UVdetectionexperiments
Theelectropherogramsforthenon-fragmented50micromcapillaryandthecapillarycombinationswithdownstream50micromcapillaryarepresentedinFig3ThetwoferrocenederivatesdecamethylferroceneandferrocenemethanolwereusedasmodelanalytesDecamethylferrocenewasonlydetectedascationicspeciesasitiseasilyoxidizedbydissolvedoxygeninsolution Forthecombination50+50micromslightlyhighermigrationtimesforthecationic(decamethylferrocene) and neutral species (ferrocenemethanol) were obser-vable compared to thenon-fragmented50micromcapillaryThis indicateda flow
4resistanceattheconnectionwhichwasalsoobservedfortheCFIA-C Dexperi-mentsinsection31Incontrasttothecombinationwiththesameinnerdiameterastrongshift in themigration times for theneuralspecieswasvisible for thecombinationswithdifferentinnerdiametersThisshowedthatthecouplinghadaneffectontheelectroosmoticflow LookingatthepeakshapeitwasfoundthatallpeaksshowednearlyGaussianshape for all combinations Furthermore there was no tailing visible Theferrocenemethanolpeaks for thecombination25+50micromand75+50micromwere
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 11
Fig 3Electropherograms of themodelmixture ferrocenemethanol (FcMeOH) and decamethyl-ferrocene(DeMeFc)measuredwithanon-fragmentedcapillary(50microm)andfragmentedcapillaries(25+5050+50and75+50microm)Experimentalparameters1mMFcMeOHandDeMeFcinseparationbuffer(10mMCH COONH and1MCH COOHinacetonitrile)injectionsegment035cmseparation3 4 3
voltage25kVcapillarylength70cm(40cmtothedetectorfragmentedcapillaries20cmfirstpartand50cmsecondpart)UVdetectionat210nm
slightlybroaderthanthepeaksforthenon-fragmented50micromcapillaryorforthe50+50 microm combination But this probably results from longitudinal diffusioneffectsduetothelongerresidencetimes
4Conclusions
FromtheCFIAmeasurementsitcanbeconcludedthattherewasamechanicaldisturbanceoftheflowduetothecouplingFurthermoreitcouldbeshownthatCEmeasurementswithlinearcoupledcapillariesofvariousinnerdiameterwerepossible Unlike to the capillary combinationwith the same inner diameter astrongshiftoftheelectroosmoticflowtowardshighermigrationtimeswasfoundfor capillary combinations with different inner diameters In this work thecapillaries were coupled with almost no dead volume which resulted in nosignificantchangesofthepeakshapeorpeaktailingContrarytoexpectationsthecouplingofcapillarieswithvariousinnerdiametershadnosignificantimpactonthepeakwidth TheknowledgegainedfromthelinearcouplingofcapillariesisagoodbasisforthedevelopmentofthenewdualdetectionconceptInanextstepthreecapillariesshouldbecoupledwitheachother
Acknowledgments
WethanktheGermanResearchFoundation(DFG)forfinancialsupport
References
[1] OpekarFS tulıkKSomeimportantcombinationsofdetectiontechniquesforelectrophoresisincapillariesandonchipswithemphasisonelectrochemicalprinciplesElectrophoresis32(2011)795ndash810
[2] BeutnerAHerlTMatysikF-MSelectivityenhancement incapillaryelectrophoresisbymeans of two-dimensional separation or dual detection conceptsAnal Chim Acta1057(2018)18ndash35
4[3] BeutnerACunhaRRRichterEMMatysikF-MCombiningC DandMSasadualdetectionapproachforcapillaryelectrophoresisElectrophoresis37(2016)931ndash935
[4] MatysikF-MEnd-columnelectrochemicaldetectionforcapillaryelectrophoresisElectro-analysis12(2000)1349ndash1355
[5] MatysikF-MWernerGTracemetaldeterminationintearsbyanodicstrippingvoltammetryinacapillaryflowinjectionsystemAnalyst118(1993)1523ndash1526
[6] FranciscoKJMdoLagoCLAcompactandhigh-resolutionversionofacapacitivelycoupledcontactlessconductivitydetectorElectrophoresis30(2009)3458ndash3464
12 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Thetermldquovitaminsrdquodescribestheheterogeneousgroupofchemicalcompoundswhich are important for the proper functioning of the human body [1 2] Bydefinitionvitaminsarenotsynthesizedbythehumanbodyorthesynthesizedamount is not sufficient to cover the demand That is why they have to besupplementedfromtheexternalsourceslikefoodproductsorpharmaceuticals[1ndash3]Basedontheirsolubilityvitaminsaredividedintowater-soluble(B-groupandvitaminC)andfat-solublevitamins(ADEandK)[3]VitaminC(ascorbicacid) is the most important antioxidant and participates in the activation ofenzymes[4]VitaminB1(thiamine)facilitateswoundhealingandiscrucialforthehumannervoussystem[15]VitaminB2(riboflavin)participatesintheenzy-maticreactionsandthebiotransformationofglucoseandaminoacids[6]Vitamin
+B3(niacin)isthemainconstituentoftheNAD andNADHcoenzymeswhichareresponsible for the transfer of electrons and hydrogen ions in the cellularrespiration [1ndash3] Vitamin B6 possesses six related structures (vitamers) thateasilyinterconvertThemostimportantoneispyridoxinewhichhelpstopreventtongue inflammation and microcytic anemia [2] For the production of well-functioningredbloodcellsandtheavoidanceofmegaloblasticanemiaandfetusdefectsvitaminB9(folicacid)hastobesupplementedintheproperamount[12]
Voltammetric determination of vitamins
RADOSŁAWPORADABOGUSŁAWBAS
DepartmentofAnalyticalChemistryFacultyofMaterialsScienceandCeramicsAGHUniversityofScienceandTechnologyMickiewicza3030-059KrakoacutewPolandrporadaaghedupl
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 13
AbstractVitaminsbelongtothegroupofchemicalcompoundsessentialfortheproperfunctioningofthebodySinceboththeirdeficiencyandexcessmay result in serious health problems the amount of vitaminssupplementedinthedietaswellasvitamincontentintheirsourceshavetobestrictlycontrolledInthisworkthepossibilityofsimulta-neous determination of vitamins by means of differential pulseadsorptive stripping voltammetry is discussed The research hasshownthatthedeterminationofsingularvitaminatthemicromolarlevel isrelativelyfastandstraightforwardandthemostimportanthindranceisrelatedtotheanalyteadsorptionattheelectrodesurfaceInthecaseofvitaminswithdifferentredoxpotentialstheycanbeanalyzedsimultaneouslywithouttheneedtoreachfortheadvancedmethodsforsignalprocessing
Keywordsmercuryelectrodevitaminsvoltammetry
VitaminK3(menadione)doesnotoccurnaturallybutitservesasaprecursorforthesynthesisofotherK-groupvitaminsandcanbeusedtotreathypoprothrom-binemiaVitaminK3ispartiallysolubleinwater[17] All of the vitamins are electrochemically active [3] therefore the electro-chemicalmethodscanbeappliedforthedeterminationofvitamincontentinfoodproductspharmaceuticalsandbodyfluidsVoltammetrictechniquesarecharac-terizedbyhighsensitivityandselectivityandtheydonotrequiretime-consumingsamplepreparationMoreovertheelectrochemicalinstrumentationisrelativelyinexpensiveandcanbeappliedintheon-siteconditionsfortheonlineanalyseseginqualitycontrolMostofthepapersreporttheconstructiondevelopmentand characterization of a novel modified working electrodes for quantitativeanalyses of a singular vitamin in the variety of matrices Unfortunately only alimitednumberofpapersdescribethesimultaneousdeterminationofmultiplevitaminsinasinglerun[23] The preliminary research devoted to the simultaneous determination ofB-groupCandK3vitaminswiththeuseofthecontrolledgrowthmercurydropworking electrode in aqueous solutions is presented in this work Particularattentionhasbeenpaidtotheredoxpotentialsofthestudiedcompoundstheshape of the calibration curves and adsorption phenomena As an attempt toovercomethelattertheneutralsurfactantTritonX-100hasbeenintroducedintothestudiedsystem
2Experimental
21Reagentsandchemicals
TheappliedreagentswereofanalyticalgradeandusedassuppliedPhosphateand McIlvaine buffers were obtained by mixing the appropriate amount of
ndash1 ndash1 ndash102molL Na HPO with 02molL NaH PO and 01molL citric acid2 4 2 4
respectively (all reagents purchased from Avantor Performance MaterialsPoland)ThestandardsolutionsofvitaminB1B2B3B9andCwerepreparedbydissolving the corresponding amount of the standard (all Sigma-Aldrich) in
ndash1distilledwater In thecaseofB2andB9 theadditionof02molL NaOHwasinevitabletoobtainaclearsolutionVitaminK3standard(Sigma-Aldrich)was
ndash1dissolvedinthemixtureofmethanoland1molL phosphatebuffer(pH=82)(vv=25)LaboratorygradeTritonX-100(Sigma-Aldrich)wasusedinthestudyoftheadsorptionprocesses22Instrumentation
All the electrochemical measurements were conducted in the three-electrodesystem composed of the Pt auxiliary electrode double-junction silversilverchloride reference electrode and controlled-growth mercury drop electrode
14 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
actingastheworkingelectrodeTheusedmeasurementequipmentinvolvedtheM164electrodestandandM161multipurposeelectrochemicalanalyzer(bothmtm-anko Krakow) To control the buffer pH-value the SevenCompact S210laboratorypH-meter(MettlerToledoSwitzerland)wasemployed
23Voltammetricmeasurements
Throughout the course of the study differential pulse adsorptive strippingvoltammetry has been used for recording the current-potential curves BothcathodicandanodicscanswererecordedinapotentialrangeadjustedforthestudiedvitaminsTheinfluenceofvariousmeasurementconditionsontheregis-teredsignalshasbeeninvestigatedFinallythepossibilityofthesimultaneousdeterminationofmultiplevitaminsinonescanhasbeenverified
3Resultsanddiscussion
Figure 1 depicts the redox potentials of the studied vitamins in the aqueoussolutions for the mercury electrode The only exception is vitamin B6 whoseredox potential is higher than the potential of mercury oxidation (ca +02 V)Therefore the given value refers to the glassy carbon electrode The redoxpotentialvalueofstudiedvitamins isnotasingularvaluebut it fallswithinacertainrangeThiscanbeascribedtothedependenceofredoxpotentialonthesolutionpHvaluewhichresultsfromtheparticipationofprotonsintheredoxreactions of vitamins Moreover the potentials for individual vitamins arerelatively well separated indicating that the simultaneous determination ofseveralvitaminsintheonerunmaybepossibleTheonlyencounteredproblems
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 15
Fig 1Redoxpotentialsofchosenvitamins
regardthevitaminsB1B3andB12whoseredoxpotentialsoscillatebetweenndash15andndash17VandtheK-groupvitamins(K1K2K3)inwhichonlythecommonstructuralmotifndashthequinoneringndashiselectrochemicallyactiveresultinginthevalueoftheredoxpotentialofcandash02V[8] AtypicalcalibrationcurveisdepictedinFig2Abasedonthedifferentialpulsevoltammograms of the vitamin B9 reduction in the concentration range from
ndash1blank to 01 mg L recorded in the McIlvaine buffer of pH = 52 using thecontrolledgrowthmercurydropelectrodeworkingelectrodeTherelationshipbetweenthepeakcurrentandtheconcentrationofVB9 is linear in thewholetestedrange(r=09999)Basedontheparametersoftheregressioncurvethelimit of detection and limit of quantitation were estimated to 42 and
ndash1142nmolL respectively Similar dependencies and figures of merit can beobtainedforothervitamins QuitedifferentbehaviorwasobservedinthecaseofthevitaminK3forwhichthe increase in current was not strictly proportional to the increase in theconcentrationandthecalibrationplotresemblesanS-shapecurve(Fig2B)ThelatterindicatesthatvitaminK3adsorbsatthesurfaceoftheworkingelectrodeHoweverasnopre-orpost-peakwereobservedwearedealingherewiththeweakadsorption[9]AdsorptionalsoplaysasignificantroleinthecaseofvitaminB2forwhichboththepre-andpost-peakswereobservedindicatingitsstrongaffinitytothemercuryelectrodes To overcome this issue attempts with Triton X-100 were performed TritonX-100isaneutralsurfactant thateasilyadsorbsat thesurfaceof themercury
16 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2Differentialpulseadsorptivestrippingvoltammogramsof(A)vitaminB9and(B)vitaminK3ndash1reduction recorded in the concentration range from blank to 01 and from blank to 04 mg L
respectivelyInsetcorrespondingcalibrationcurvesSupportingelectrolyte(A)McIlvainebufferndash1(pH=52)(B)04molL phosphatebuffer(pH=82)
electrode partially blocking its surface Doing so it prevents the undesiredadsorption of other molecules and thus allows to obtain a linear relationshipbetweenthepeakcurrentandthevitaminconcentration(Fig3)UnfortunatelyduetotheblockingoftheelectrodesurfacetheslopesofthecalibrationlinesaresmallerincomparisontotheonesobtainedintheabsenceofanysurfactantsThismeans that the sensitivity defined as the increase in current caused by a unitincrease in concentration and the resolution understood as the possibility todistinguishsmallvariationinconcentrationarecorrespondinglydecreased Figure3alsopresentsthepossibilitytodeterminemultiplevitaminsinasinglerunChosenvitaminshavewell-separatedpotentialsandtheydonotinterferewitheachotherthereforenoadvancedmultivariatecalibrationstrategiesareneededTheproblemsinthesimultaneousanalysisincludevarioussensitivitieswith respect to the studied analytes and differences in the influence of themeasurementconditionsontherecordedsignalsDuetothattheexperimentalconditionswillneverensurethehighestpossiblesignalvaluesforallanalyzedcompounds
4Conclusions
Differential pulse voltammetry in conjunction with the controlled growthmercurydropelectrodeisaperfecttoolforquantitativeanalysesofvitaminsTheadsorptionofvitaminB2andK3canbepreventedbytheadditionoftheneutralsurfactantTritonX-100whichselectivelyblockstheworkingelectrodesurface
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 17
Fig 3Cathodic voltammograms for the simulatanous determination of vitamin B2 B3 and K3Depictedintheinsetsarethevoltammogramsafterbackgroundsubtractionwiththecorresponding
ndash1callibrationplotsSupportingelectrolye04molL phosphatebuffer(pH=82)with40ppmTritonX-100AccumulationconditionsE =ndash005Vt =20sacc acc
The proposed methodology allows for the simultaneous determination ofmicromolaramountsofvitaminB2B3andK3Suchaprocedurewillhelptoreduce the time and costs of analyses of multivitamin formulations and foodproducts
Acknowledgments
RPhasbeenpartlysupportedbytheEUProjectPOWR0302-00-00-I00416
References
[1] Combs GF JrTheVitaminsFundamentalAspects inNutritionandHealth 3rd ed IthacaElsevierAcademicPress2008
[2] Lovander MD Lyon JD Parr DL Wang J Parke B Leddy J Review Electrochemicalpropertiesof13vitaminsAcriticalreviewandassessmentJElectrochmSoc165(2018)G18ndashG49
[3] Brunetti B Recent advances in electroanalysis of vitamins Electroanalysis 28 (2016)1930ndash1942
[4] BrubacherGMuller-MulotWSouthgateDATMethods forDeterminationofVitamins inFoodNewYorkElsevier1985
[5] Szpikowska-Sroka B A simple and sensitive analytical method for the determination ofthiamineinpharmaceuticalpreparationsJAnalChem68(2013)218ndash222
[6] PetteysBJFrankELRapiddeterminationofvitaminB (riboflavin)inplasmabyHPLCClin2
ChimActa412(2011)38ndash43[7] ZhangZXuJWenYZhangJDingWTheelectro-syntesizedimprintedPEDOTfilmasa
simple voltammetric sensor for highly sensitive and selective detection of vitamin K in3
poultrydrugsamplesSynthMet230(2017)79ndash88[8] JedlinskaKStrusMBasBAnewelectrochemicalsensorwiththeRefreshableSilverLiquid
Amalgam Film multi-Electrode for sensitive voltammetric determination of vitamin K2(menaquinone)ElectrochimActa265(2018)355ndash363
[9] SouthamptonElectrochemistryGroupInstrumentalMethodsinElectrochemistryChichesterHorwood1985
18 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Carbonelectrodesarecommonlyappliedtosensitiveelectrochemicaldetectionofneurotransmittersegdopamine (nor)epinephrineandserotonin in-vivoandin-vitro[12]Neverthelessadsorptionofhigh-molecularweightbiomoleculesinthe matrix on a sensing electrode which then hinders the electron transferreactionofneurotransmitterswillresultinbiofoulingofelectrodesThisremainsachallengingproblemasbiofoulingwillcompromiseelectrochemicalmeasure-mentsThusseveralstrategies foraddressingbiofoulinghavepreviouslybeenreported[34] Thisworkreportsonaneffectiveapproachforminimisingbiofoulingbasedonthehypothesisthatahydrophobicelectrodesurfacewillrepelagainstadsorptionof amphiphilic biomolecules Briefly structurally small conical-tip electrodes
Dopamine detection at antifouling conical-tip carbon electrodes
a a a aSIMONABALUCHOVA JANKLOUDA JIR IBAREK KAROLINASCHWARZOVA -PECKOVA bDANNYKYWONG
a UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova812800PragueCzechRepublicsimonabaluchovanaturcunicz
b DepartmentofMolecularSciencesMacquarieUniversitySydneyNSW2109Australia
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 19
AbstractA significant achievement in this work is the development ofantifoulingconical-tipcarbonelectrodes(~27micromtipdiameterand~165micromaxiallength)suitablefordetectionoftheneurotransmitterdopamine in-vivo These electrodes were hydrogenated using adiphenylsilanereductionmethodtoyieldahydrophobicsurfacetodeteradsorptionofamphiphilicbiomoleculesInitiallyhydrogenatedcarbonelectrodeswereelectrochemicallycharacterisedusingseveralredoxmarkersThedegreeofantifoulingwasthenassessedbythevoltammetricsignalchangeofdopamineattheseelectrodesbeforeand after being incubated in a fouling solution containing bovineserumalbumincytochromeC(bothareproteins)andcaproicacid(alipid) In our work we have obtained only a 69 (standarddeviation35N=40)decreaseindopaminesignalsatthehydro-genated carbon electrodes These results strongly support thediphenylsilanereductionstrategyforthedevelopmentofantifoulingbiosensorsfordopaminedetectioninbiologicalmatrices
Keywordsantifoulingelectrodesdiphenylsilanereduction
methodhydrogenatedconical-tip
carbonelectrodesvoltammetricdopamine
detection
(denoted as CTEs) are fabricated by thermally pyrolysing acetylene gas in anitrogenatmospheretodepositcarbonatthetipandontheshankofpulledquartzcapillaries [5] Spectroscopic studies confirmed that the electrode surface
2 3consistsofsp -likegraphiticcarbonandsp -hybridiseddiamond-likecarbon[6]In addition there is also a range of carbon-oxygen functionalities includingcarbonylquinonecarboxylphenolsalcoholsandethergroupsontheelectrodesurface[6]whichcaninteractwithspectatorbiomoleculesthroughdipole-dipoleorion-dipoleinteractionleadingtotheirirreversibleadsorptionontheelectrodesurface[7]HoweverbysubjectingthesecarbonelectrodestosilanereductionCndashObondsareconvertedtoCndashHbondsandphenolicgroupsaretransformedtosiloxane dendrimers [6] to yield a more hydrophobic carbon surface that isexpectedtobesimilarlylesssusceptibletobiofoulingcomparedtoboron-dopeddiamondelectrodes[78] In this work we will present a methodology involving diphenylsilanereduction to fabricate physically small hydrogenated conical-tip carbonelectrodes(denotedasHCTEs)withanti-foulingcapabilityBothCTEsandHCTEswere electrochemically characterised using several redox probes to elucidatetheirsurfacepropertiesbeforeevaluatingtheirresistancetobiofoulingduringdopaminedetectionin-vitro
2Experimental
21Reagentsandchemicals
Analyticalgradereagents(Sigma-AldrichAustralia)including4-methylcatecholhexaammineruthenium(III) chloride potassium hexacyanoferrate(III) dop-amine hydrochloride sodium phosphate dibasic citric acid perchloric acidpotassiumchloride sodiumhydroxide anhydrousdichloromethanediphenyl-silanetris-(pentafluorophenyl)boraneandgraphitepowderwereusedas-recei-ved Ultra-high purity gases acetylene and nitrogenwere obtained from BOCGases (Australia) All aqueous solutionswere preparedwith deionisedwater(MilliporeMiliplusQsystemUSA)witharesistivityof182MΩcm
22Instrumentation
Chronoamperometric and voltammetric experiments were carried out usingalow-currentpicostateDAQoperatedbyanEChemversion212softwareviaanE-corderinterface(eDAQPtyLtdAustralia)Athree-electrodeset-upinvolving
minus1eitheraCTEorHCTEasaworkingelectrodeaAgAgCl(3molL KCl)referenceelectrode (Bioanalytical SystemsUSA)andaplatinumwire counterelectrode
minus1(CypressSystemsUSA)wasusedCyclicvoltammetryatascanrateof100mVs anddifferential pulse voltammetry (pulse height +25mV pulsewidth 50ms
minus1sampling time 20 ms and scan rate 20mVs ) were used in this work All
20 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
electroanalyticalexperimentswereperformedinanaluminiumFaradaycageatanambienttemperature(23plusmn1degC)
23Preparationofhydrogenatedconical-tipcarbonelectrodes
Asreportedpreviously[5]structurallysmallCTEswerefabricatedbythermallypyrolysing C H (a pressure of 50 kPa) in a pulled quartz capillary (Sutter2 2
minus1InstrumentUSA)housedinaN atmosphere(counterflowof60mLmin )Prior2
to hydrogenation the catalyst tris(pentafluorophenyl) borane (100 mg) wasdissolvedinanhydrousCH Cl (50mL)bystirringfor5minbeforethehydroge-2 2
natingagentdiphenylsilane(25μL)wasaddedCTEswerethenplacedinthereagentmixturefor2hThepreparedHCTEsweredriedovernightbeforeuse
24Biofoulingexperiments
A laboratory synthetic fouling solution consisting of 4 (wv) bovine serumalbumin001(wv)cytochromeC(bothareproteins)and10(vv)caproicacid(alipid)waspreparedbyhomogenisingtheminapH=74citrate-phosphate
minus1buffer(01molL )AllfoulingcompoundswereacquiredfromSigmaAldrichAustralia
3Resultsanddiscussion
31Electrochemicalcharacterisation
minus1InthisworkallCTEswerecharacterisedbycyclicvoltammetryof10mmolL 3+ minus1[Ru(NH ) ] in10molL KClAsdisplayedinFig1(A)onlyCTEsthatshow3 6
asigmoidal-shapedvoltammogramwithasmallchargingcurrentwereemployedinfurtherexperimentsUsingchronoamperometry[5]ameantipdiameterof27μm(standarddeviation(SD)28μmN=142)ameanaxiallengthof165μm(SD=114μmN=142)wereestimatedfortheseCTEs TocomparesurfacecharacteristicsofbothCTEsandHCTEs cyclicvoltam-
minus1 3+2+ minus1 minus1metryof (1)10mmolL [Ru(NH ) ] in10molL KCl (2)10mmolL 3 63minus4minus minus1 minus1[Fe(CN) ] in 10 mol L KCl and (3) 10 mmol L 4-methylcatechol in6minus101molL HClO wasconductedatthesameelectrodesbeforeandafterhydroge-4
nation The results obtained are shown in Fig 1(A-C)We observed a ~20(SD=5N=10)decreaseinthelimitingcurrentofallthreeredoxmarkersafterdiphenylsilane reduction most likely attributable to the hindrance to theirelectron transfer reactions by the phenylsiloxane group formed on HCTEs
3minus4minusMoreover as an inner-sphere redox probe both [Fe(CN) ] and 4-methyl-6
catechol reactionsare sensitive to thepresenceofoxygen functionalitiesonacarbonsurface [7]Accordingly theconversionof these functionalities toCndashHbondsbydiphenylsilanereductionwasexpectedtoyieldmoresluggishelectron
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 21
transfer kinetics at HCTEs as supported by a negative potential shift (from3minus4minus+75mVtominus10mV)inthecyclicvoltammogramof[Fe(CN) ] andapositive6
potentialshift(from+580mVto+675mV)inthecorrespondingcyclicvoltammo-2gram of 4-methylcatechol In addition the conversion of sp -carbon to
3sp -diamond-likecarbon[6] isalsoexpectedtoreducetheconductivityof thecarbonelectrodesurface
32Dopaminedetectionduringbiofoulingexperiments
minus1Theelectrochemicalbehaviourof1mmolL dopamine inapH=74citrate-minus1phosphate buffer (01 mol L ) at CTEs and HCTEs was studied by cyclic
voltammetryTheresultsobtainedareshown inFig1(D)Acomparable12decrease(SD=6N=10)inthedopamineoxidationlimitingcurrenttothatof4-methylcatechol was observed A positive potential shift from +285 mV to+305mVinthevoltammogramsisalsoaccountedforasdescribedabove
22 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
minus1 3+ minus1 minus1Fig 1Cyclicvoltammetryof(A)10mmolL [Ru(NH ) ] in10molL KCl (B)10mmolL 3 63minus4minus minus1 minus1 minus1[Fe(CN) ] in10molL KCland(C)10mmolL 4-methylcatechol in01molL HClO at3 4
minus1(a)aCTEand(b)aHCTE(D)10mmolL dopamineinapH=74citrate-phosphatebufferrecordedminus1at(a)aCTEandaHCTE(b)beforeand(c)afterbiofoulingScanrate100mVs
Next HCTEs were incubated in a synthetic fouling solution containing4(wv)bovineserumalbumin001(wv)cytochromeC(bothareproteins)and 10 (vv) caproic acid (a lipid) for 30 min Cyclic voltammetry of
minus110mmolL dopamineattheseHCTEswasthenconductedinapH=74citrate-phosphatebuffertoevaluatetheirantifoulingpropertyNotablyaconsiderable515(SD=183N=6)decreaseindopaminesignalwasobservedatCTEsIncontrastonlyacorresponding69decrease(SD=35N=40)wasestimatedatHCTEsasshowninFig1(D)Clearlythisrepresentsamajorimprovementinthe antifouling capability of HCTEs obtained using diphenylsilane reductioncomparedtoCTEsandotherpreviouslytestedhydrogenatingagents[9]includingn-butylsilane (35decrease) triethylsilane (23decrease) andphenylsilane(18decrease)Thereforethisworkhassuccessfullydemonstratedtheeffecti-venessofdiphenylsilanereductionmethodindevelopingantifoulingelectrodesfordopaminedetection
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 23
Smallconical-tipelectrode
As-prepared Hydrogenated
minus1LinearrangemicromolL 1ndash10 1ndash20minus2 minus2Intercept10 pAmicrom 108plusmn004 273plusmn011
minus3 minus2 minus1Slope10 pALmicrom micromol 128plusmn007 443plusmn012R 0993 0998
minus1LimitofdetectionmicromolL 100 077
Table 1Analyticalparametersofconcentrationdependencesofdopamineobtainedbydifferentialpulse
minus1voltammetryinapH=74citrate-phosphtebuffer(01molL )Allquoteduncertaintiesrepresentthe95confidenceintervalandthecorrelationcoefficient(R)wasfoundtobestatisticallysigni-ficantatthe95usingStudentrsquost-test
Fig 2DifferentialpulsevoltammetryofdopamineataHCTEinapH=74citrate-phosphatebufferminus1 minus1(01molL )atconcentrations(a)1(b)2(c)4(d)6(e)8(f)10and(g)20μmolL
WehavealsostudiedtheconcentrationdependenceofdopamineinapH=74citrate-phosphatebufferbydifferentialpulsevoltammetryAtypicalcalibrationplotobtainedisshowninFig2Theanalyticalparametersandestimatedlimitsofdetection are summarised inTable 1 These results show thatHCTEs outper-formed CTEs because they exhibit a ~35times higher sensitivity a 23 lowerdetectionlimitandawiderlinearrange
4Conclusions
InthisstudyphysicallysmallHCTEswithanti-foulingcharacteristicsachievedbyhydrogenationusingdiphenylsilanereductionweresuccessfullyfabricatedandelectrochemically characterised using several redox probes Next dopaminedetectionwasperformedbeforeandafterincubationofelectrodesinasyntheticfoulingsolutioncontainingahighconcentrationofbiomoleculesOnlyalow69(SD=35)decreaseindopaminelimitingcurrentwasachievedatHCTEsobtain-edbydiphenylsilanereductionindicatingtheirsignificantlylesssusceptibilitytobiofoulingthanCTEsThesepromisingresultsindicatethatantifoulingHCTEswillpotentiallybenefitthedevelopmentofbiosensorsfordopaminedetectionin-vivoinbiologicalmedia
Acknowledgments
ThisresearchwasperformedwithintheframeworkofSpecificUniversityResearch(SVV260560)FinancialsupportsprovidedbytheGrantAgencyofCharlesUniversity(project390119)andbytheCzechScienceFoundation(project20-03187S)aregratefullyacknowledgedSBandJKalsothankthe Mobility Fund of Charles University and Hlavkova nadace for providing funding for theirresearchinternshipsatMacquarieUniversitySydneyAustralia
References[1] BaranwalAChandraPClinicalimplicationsandelectrochemicalbiosensingofmonoamine
neurotransmittersinbodyfluidsinvitroinvivoandexvivomodelsBiosensBioelectron121(2018)137ndash152
[2] CaoQPuthongkhamPJillVentonBReviewnewinsightsintooptimizingchemicaland3Dsurface structuresof carbonelectrodes forneurotransmitterdetectionAnalMethods11(2019)247ndash261
[3] LinP-HLinB-RAntifoulingstrategiesinadvancedelectrochemicalsensorsandbiosensorsAnalyst145(2020)1110ndash1120
[4] HanssenBLSirajSWongDKYRecentStrategiestoMinimiseFoulinginElectrochemicalDetectionSystemsRevAnalChem35(2016)1ndash28
[5] McNallyMWongDKYAnin-vivoprobebasedonmechanicallystrongbutstructurallysmallcarbonelectrodeswithanappreciablesurfaceareaAnalChem73(2001)4793ndash4800
[6] SirajSMcRaeCRWongDKYEffectiveactivationofphysicallysmallcarbonelectrodesbyn-butylsilanereductionElectrochemCommun64(2016)35ndash41
[7] ParkJShowYQuaiserovaVGalliganJFinkGDSwainGMDiamondmicroelectrodesforuseinbiologicalenvironmentsJElectroanalChem583(2005)56ndash68
[8] ShinDTrykDAFujishimaAMerkociAWang JResistance to surfactantandproteinfoulingeffectsatconductingdiamondelectrodesElectroanalysis17(2005)305ndash311
[9] Roshni RAnAntifouling Structurally Small Carbon Electrode forDetectionof theNeuro-transmitterDopaminePhDThesisMacquarieUniversitySydney2019
24 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Theanalyticalperformancesofenzymaticbiosensorsarestronglyaffectedbytheenzyme immobilization process There is no universal technique for enzymesattachmentThereforespecialattentionshouldbepaid to theselectionof theappropriatesupportandthedevelopmentoftheoptimalbindingstrategyinordertoensure thebestcharacteristicsof immobilizedenzymeDespiteavarietyofpreviouslyreportedcovalentimmobilizationmethodsfordifferentenzymesthepresentedprocedurescanbehardlycomparedtofindtheoptimalonesbecauseofdifferentanalyticalmethodsandexperimentalconditionsusedUptodatethere
A comparative study of covalent glucose oxidase and laccase immobilization techniques at powdered supports for biosensors fabrication
ab a bSOFIIATVORYNSKA JIR IBAREK BOHDANJOSYPCUK
a UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova2030812843Prague2CzechRepublicsofiiatvorynskajh-instcascz
b JHeyrovskyacuteInstituteofPhysicalChemistryoftheCzechAcademyofSciences Dolejškova318223Prague8CzechRepublic
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 25
AbstractInordertodeveloptheoptimalstrategyandtodeepentheknowledgeinthefieldofenzymeimmobilizationthreedifferenttechniquesofcovalentbindingfortwoenzymes(glucoseoxidaseandlaccase)atpowdered surfaces were compared Immobilization protocol wasoptimized by changing supports (twomesoporous silica powders(SBAminus15 MCMminus41) and a cellulose powder) the functionalizedgroupsintroducedatsupportsurfaces(minusNH andminusCOOH)andthe2
methodsofactivation(glutaraldehydeandcarbodiimide)Aminoandcarboxyl functionalized mesoporous silica and cellulose powderswerepreparedbysilanizationusing(3-aminopropyl)triethoxysilaneandcarboxyethylsilanetriolrespectivelyItwasfoundthatcouplingof both enzymes by their ndashNH groups through glutaraldehyde to2
ndashNH functionalized supports in particular SBA15minusNH and2 2
celluloseminusNH forglucoseoxidaseMCM41minusNH forlaccaseshowed2 2
thehighestactivityandthebeststability
Keywordsbiosensorcovalentimmobilizationenzymaticreactorglucoseoxidaselaccase
is still a lack of the comparative systematic studies focusing on the enzymesimmobilizationonthevarioussupportsusingdifferenttechniques The aim of this work is the systematic comparative study of the differenttechniques for covalent coupling of the enzymeswhich ensures not only thedevelopmentoftheoptimalimmobilizationstrategyfortheselectedenzymesbutalsoenablestofindoutsometendenciesinenzymeattachmentprocessgenerallyThusthisworkisfocusedonadetailedanalysisoftheeffectofthekindofsupportits anchor groups and the activation methods on activity and stability ofimmobilizedenzymesTwoenzymeswithdifferentnature(glucoseoxidase(GOx)andlaccase(Lac))werechosenasthetestingbioreceptors
2Experimental
21Reagentsandchemicals
AllchemicalswereofpaorbettergradeGlucoseoxidasefromAspergillusnigerminus1(GOxEC11341452Umg )laccasefromTrametesversicolor(LacEC11032
minus1129Umg )D-(+)-glucosedopamineglutaraldehyde(GAgradeII25aqueoussolution) N-(3-dimethylaminopropyl)-Nʹ-ethylcarbodiimide hydrochloride(EDC ge980) N-hydroxysuccinimide (NHS ge970) (3-aminopropyl)-triethoxysilane (APTES) mesoporous silica powder SBAminus15 (particle size
2 minus12ndash6μmporesizeasymp7nmsurfaceareaasymp600m g )mesoporoussilicapowder2 minus1MCMminus41 (pore size 21ndash27 nm surface area asymp 1000 m g ) cellulose (Cell
microcrystalline powder particle size 20 microm) were purchased from SigmaAldrichCarboxyethylsilanetriol(CEST25aqueoussolution)waspurchased
regfromabcr (Germany)
22Instrumentation
Amperometric measurements were carried out at room temperature usingcomputer-controlled electrochemical stand (Polaro-Sensors Czech Republic)withMultiElchemv31software(JHeyrovskyInstituteofPhysicalChemistryoftheCAS)Flowinjectionanalysis(FIA)withthethree-electrodelaboratory-madeflow-through cellwas usedworking electrode minus tubular detector of polishedsilversolidamalgam(TD-p-AgSAlaboratory-madeinnerdiameter05mmtheamalgamtube length60mm) referenceelectrodeminusaminiaturizedsaturatedcalomelelectrodebasedonsilverpasteamalgam[1](laboratory-madeithasthesamepotential as classical saturated calomel electrode) auxiliary electrodeminusplatinum wire (diameter 10 mm length 10 mm) The system for FIA withelectrochemicaldetectioncomprisedofalinearsyringepumpa2-position6-port
regsampleinjectorvalveaninjectionlooplaboratory-madeofTeflon (PTFE)tubing(100μL) a solenoid operatedmicro-pumpan enzymatic reactor and a flow-through cell for TD The enzymatic reactor consists of a tube filled by theenzymaticpowder
26 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
3Resultsanddiscussion
Basedonthedatareportedintheliterature[23]andontheresultsofourpreviousworks[4ndash8]forthisstudyCellandmesoporoussilicapowders(namelySBAminus15andMCMminus41)havebeenselectedas thepotentialpromisingsupports for thecovalent enzyme immobilizationBecauseof thehigh contentof surficial ndashOHgroupswhich are capable of chemical reactions these supports can be easilyfunctionalizedThewell-knownandfrequentlyutilizedtechniqueofsilanizationhasbeenusedtomodifythesurfacesofSBA15MCMminus41andCellbythedesiredfunctionalizedgroupsAminosilaneAPTESwasappliedtoformminusNH groupson2
thematrixsurfaceswhereascarboxylsilaneCESTwasusedtointroducendashCOOHgroups Generallytheprocedureofthecovalentimmobilizationofenzyme(eitherLacorGOx)onthefunctionalizedsupportconsistsofthreestepsI Synthesisofthefunctionalizedsupportwhichmeansthemodificationofthematrix(MCMminus41SBAminus15andCell)withsuitableanchoredgroups(minusNH or2
minusCOOH)II Activationstepofthefunctionalizedsupportwithspecificactivatingagents(glutaraldehydeorEDCNHS)tomakeitreactivetowardsenzyme
IIIEnzyme(LacorGOx)couplingtotheactivatedsupport
To investigate the effect of support its surface functionalizedgroups and themethodsofactivationontheefficiencyofthecovalentenzymeimmobilizationthreedifferentstrategies(ABandC)forLacandGOxattachmenthavebeenusedThedetailsoftheusedtechniquesandthedenotationsofthepreparedenzymaticpowdersaresummarizedinTable1(nextpage)ToexaminetheefficiencyofLacandGOximmobilizationtheenzymaticreactors(filledbytheenzymaticpowderspreparedwithdifferenttechniques)coupledwithTDwereusedforamperometricdetermination of dopamine and glucose respectively in flow systems Theprincipleofglucosedetection isbasedonamperometricmeasurementsof theenzymatically consumed oxygen whereas dopamine was detected by thereductionoftheenzymaticallyoxidiseddopamine Asdepicted inFig1 thebiosensors responsesare stronglyaffectedby thestrategyusedforLacorGOximmobilizationAsshowntheresponsesofLacandGOx biosensors decrease in the order strategy A gt strategy B gt strategy Cirrespectiveofthetypeofsupport It isclearlyseenthataminofunctionalizedsupports(SBA15minusNH MCM41minusNH andCellminusNH )providehigheractivitiesof2 2 2
the immobilized Lac andGOx than these supports functionalized by carboxylgroups(SBA15minusCOOHMCM41minusCOOHandCellminusCOOH)BycomparingactivitiesofimmobilizedenzymesusingstrategiesAandBtheinfluenceoftheactivationagenthasbeenevaluatedThebestresultsforbothenzymeswereobtainedforndashNH functionalizedsupportsactivatedbyGAItcouldbeexplainedbythefact2
thatGAcontrary tocarbodiimidewithnonemolecularspaceprovidesa long
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 27
spacerarmensuringminimalsterichindrancesforenzymesbindingItcanbeconcludedthatthecovalentimmobilizationofbothenzymesbytheirndashNH groups2
viaGAtondashNH functionalizedmesoporoussilicapowders(strategyA)provided2
the highest activities Interestingly in the similar comparative studies it isreportedthatamongndashOHminusCOOHandndashNH functionalizedsupportsactivatedby2
divinylsulfonecarbodiimideandGArespectivelythelastonewasfoundasthemostsuitabletechniqueforthecovalentbindingofLac[9]invertase[10]andpepsin[11]
28 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Tab
le 1
Theprinciplesofthecovalentimmobilizationmethodsofenzymesusedinthisstudy
StrategyA
StrategyB
StrategyC
Support
SBAminus15MCMminus41Cell
SBAminus15MCMminus41Cell
SBAminus15MCMminus41Cell
Supportfunctio-
minusNH
minusNH
minusCOOH
22
nalizedgroup
Activationagent
Glutaraldehyde(GA)
Carbodiimide
Carbodiimide
(EDCNHS)
(EDCNHS)
Enzymereactive
minusNH
minusCOOH
minusNH
22
group
Typeofbond
secondaryamine
amide
amide
Denotationsofthe
GOxminusNHminusCHminus(CH)minusCHminusNHminusSBA15
GOxminusC(=O)minusNHminusSBA15
GOxminusNHminusC(=O)minusSBA15
23
preparedenzy-
GOxminusNHminusCHminus(CH)minusCHminusNHminusMCM41
GOxminusC(=O)minusNHminusMCM41
GOxminusNHminusC(=O)minusMCM41
23
maticpowders
GOxminusNHminusCHminus(CH)minusCHminusNHminusCell
GOxminusC(=O)minusNHminusCell
GOxminusNHminusC(=O)minusCell
23
LacminusNHminusCHminus(CH)minusCHminusNHminusSBA15
LaxminusC(=O)minusNHminusSBA15
LacminusNHminusC(=O)minusSBA15
23
LacminusNHminusCHminus(CH)minusCHminusNHminusMCM41
LacminusC(=O)minusNHminusMCM41
LacminusNHminusC(=O)minusMCM41
23
LacminusNHminusCHminus(CH)minusCHminusNHminusCell
LacminusC(=O)minusNHminusCell
LacminusNHminusC(=O)minusCell
23
When the effect of the method of the covalent enzyme coupling on thebiosensorstabilitywasevaluateditwasfoundthatLacboundedtondashNH functio-2
nalizedsupportsviaGA(strategyA)hasshownthehigheststability(gt65oftheinitial responses after 1 month) compared to other strategies whereas GOximmobilizedwithtwostrategies(AandB)possessedapproximatelysimilarhighstability(gt80oftheinitialresponsesin1month)BothenzymesboundedviandashNH groupstondashCOOHfunctionalizedsupportsthroughEDCNHS(strategyC)2
showedquitelowstability
4Conclusions
Threedifferent strategies including the support selection the anchor surfacegroups and the activationmethod havebeen compared for efficient covalentimmobilization of Lac and GOx The results showed that ndashNH functionalized2
supports(SBA15minusNH CelluloseminusNH forGOxandMCMminusNH forLac)activatedby2 2 2
GAmaybeusedtoeffectivelybindenzymesintermsofhighactivityandstability
Acknowledgments
ThisworkwasfinanciallysupportedbytheGrantAgencyofCharlesUniversityinPrague(Project1356120)theGrantAgencyoftheCzechRepublic(Project20-07350S)anditwascarriedoutwithintheframeworkofSpecificCharlesUniversityResearch(SVV260440)
References
[1] YosypchukBBarekJYosypchukOPreparationandpropertiesofreferenceelectrodesbasedonsilverpasteamalgamElectroanalysis23(2011)2226minus2231
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 29
Fig 1Effectof the covalent attachment techniqueson (A) laccaseand (B) andglucoseoxidaseminus1biosensor responses Experimental conditions (A) c = 500 micromol L E = minus50 mVDOP det
minus1 minus1v = 01 mL min V = 40 microL carrier solution 01 mol L acetate buffer pH = 48flow DOPminus1 minus1(B)c =500micromol L E =minus1100mVv ==01mLmin V =40microL carrier solutionGlu det flow Glu
minus1 minus101molL acetatebuffer0001molL Na EDTApH=652
[2] LiuYChenJYEnzymeimmobilizationoncellulosematrixesJBioactCompactPolym31(2016)553ndash567
[3] Hartmann M Kostrov X Immobilization of enzymes on porous silicas ndash benefits andchallengesChemSocRev42(2013)6277minus6289
[4] JosypcukOBarekJJosypcukBElectrochemicalbiosensorsbasedonenzymaticreactorsfilledbyvarioustypesofsilicaandamalgampowdersformeasurements inflowsystemsElectroanalysis28(2016)3028minus3038
[5] Josypcuk O Barek J Josypcuk B Amperometric determination of catecholamines byenzymaticbiosensorsinflowsystemsElectroanalysis30(2018)1163minus1171
[6] TvorynskaSBarekJJosypcukBAmperometricbiosensorbasedonenzymaticreactorforcholinedeterminationinflowsystemsElectroanalysis31(2019)1901minus1912
[7] TvorynskaSBarekJJosypcukBFlowamperometricbiosensorbasedontwoenzymaticreactors (acetylcholinesterase-choline oxidase) for the detection of neurotransmitteracetylcholine In Proceedings of the 15th International Students Conference ldquoModernAnalyticalChemistryrdquoKNesmerak(ed)PragueFacultyofScienceCharlesUniversity2019p61minus66
[8] TvorynskaSBarekJJosypcukBAcetylcholinesterase-cholineoxidase-basedmini-reactorscoupledwithsilversolidamalgamelectrodeforamperometricdetectionofacetylcholineinflowinjectionanalysisJElectroanalChem860(2020)113883
[9] RekucAKruczkiewiczPJastrzembskaBLiesieneJPeczynska-CzochWBryjakJLaccaseimmobilizationonthetailoredcellulose-basedGranocelcarriersIntJBiolMacromol42(2008)208minus215
[10] Bryjak J Liesiene J S tefuca V Man-tailored cellulose-based carriers for invertaseimmobilizationCellulose15(2008)631minus640
[11] SzałapataKOsinska-JaroszukMBryjakJJaszekMJarosz-WilkołazkaANovelapplicationofporousandcellularmaterialsforcovalentimmobilizationofpepsinBrazJChemEng33(2016)251minus260
30 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Carbohydrates are crucial for energy structure and signaling in the humanbody[1]Thereisavarietyofcarbohydratesbutthemostimportantoneforlifeisglucoseasitisfundamentalinthemetabolismandphotosynthesis[2]GlucoseisclassifiedashexoseThesemonosaccharidesonlydifferinthepositionofhydroxylsubstituentsinsomecasesInadditiontothestructuralsimilaritiesthesemole-culeslackachromophoreandarenoteasilyionizable(pK ~12)Thusdetectiona
intheUVregionandseparationofanalytesbycapillaryelectrophoresis(CE)arechallenging [1 3] At the moment there are many different techniques for theanalysis of carbohydrates commonly including time-consuming derivatizationstepsoreluentswithhighpHvalues(pHgt12)inionchromatographyAwell-esta-blishedtechniquefortheanalysisofcarbohydratesishigh-performanceanion-exchangechromatographywithpulsedamperometricdetection(HPAE-PAD)[4]Electrochemical detection like AD is matching miniaturization simple instru-mentationlowcostandrobustnessandthusisoftenusedforflow-basedsystemssuchasCEandflowinjectionanalysis(FIA)[5]
Capillary flow injection analysis with electrochemical detection for carbohydrate analysis
NICOLEHEIGLFRANK-MICHAELMATYSIK
InstituteofAnalyticalChemistryChemo-andBiosensorsFacultyofChemistryandPharmacyUniversityofRegensburgUniversitaumltsstraszlige3193053RegensburgGermanynicoleheiglchemieuni-regensburgde
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 31
AbstractAsimplecapillaryflowinjectionanalysissystemwithamperometricdetection was arranged for the development of a method for fastoptimization of detection conditions in the context of thedetermination of carbohydrates by means of electrochemistry-capillary electrophoresis-mass spectrometry This setup is free ofelectricalinterferencebyhighvoltageandisperfectforstudyingtheoxidationofvariousanalytesFurthermoreitassureseasycouplingtoMS and thus is an useful tool to investigate the correspondingoxidationproductsofananalyte
Keywordscapillaryflowinjection
analysiscarbohydratesmassspectrometrydisposableelectrodespulsedamperometric
detection
Inthiscontributioncapillaryflowinjectionanalysis(CFIA)withADwillbepresentedasamethodtoapplyandtestADforthedetectionofmonosaccharidesondifferentdisposablethin-filmorscreen-printedelectrodesCFIAwaschosenoverconventionalFIAforthispurposeasthegravityflowinCFIAisstableforalongertimeandverylowsampleconsumptioncanbeachieved[6]TheCFIAsystemwasarrangedassimpleaspossibleandperformedhydrodynamicallytoavoidanyinterferencesFurthermoreitassuresthecouplingoftheflowsystemtoamassspectrometerThusthesamesetupasusedforCFIAcanbeutilizedforcapillaryelectrophoresis-massspectrometry(CE-MS)experimentsbychangingthe flow through the electrochemical flow cell in opposite direction In futureexperimentstheexperienceintermsofADonthoseelectrodeswillbeusedtodevelopelectrochemicalpretreatmentprotocolsforcarbohydratedeterminationbyCE-MS
2Experimental
21Reagentsandchemicals
The following chemicals were used for this study all of analytical gradeAmmoniumacetate(NH OAc)wasobtainedfromMerck(DarmstadtGermany)4
and ferrocene methanol (FcMeOH) from ABCR (Karlsruhe Germany) Milli-Qregwater(182MΩcm)wasgeneratedbyaMilli-QAdvantageA10 system(Merck
Millipore Darmstadt Germany) Carrier solution was prepared by dissolvingndash1NH OAc (50mmolL ) in Milli-Q water FcMeOH solution was prepared by4
dissolvingFcMeOHincarriersolution
22Instrumentation
Electrochemical measurements were performed using a microAutolab Type IIIpotentiostatgalvanostat (Metrohm Autolab B V Utrecht Netherlands)controlledbyNOVA20softwareforexperimentalcontrolanddataacquisitionCFIAwasperformedusingthesetupillustratedinFig1(A)consistingofacarrierreservoirsamplevialandtwofusedsilicacapillaries(PolymicroTechnologiesPhoenix AZ USA inner diameter 100 microm length inlet 40 cm length outlet10cm)connectedtoacommerciallyavailableflowcellfromMicruxTechnologies(model ED-FLOW-CELL Oviedo Spain) Inside of the flow cell the fused silicacapillary was placed in a so-called wall-jet configuration above the workingelectrode of a disposable thin-film gold electrode (model ED-SE1-Au MicruxTechnologiesOviedoSpain)ascanbeseeninFig1(B)Thethin-filmelectrodeswerebasedonathree-electrodesystemwithagoldworkingauxiliaryandquasi-referenceelectrodeThehydrostaticpressurewasachievedbyaheightdifferencebetweeninletandoutletreservoirof30cmresultinginagravityflowofthecarriersolutionthroughafusedsilicacapillaryandsubsequentlythroughtheflowcell
32 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Theinjectionwascarriedoutbyloweringthevialcontainingthecarriersolutiontotheleveloftheoutletexchangingthecarrierreservoirwiththesamplevialandliftingthesamplevialto20cmforadefinedperiodoftimeRe-establishingthecarrierreservoirtookplacethesameway
3Resultsanddiscussion
AsimpleCFIA-ADsystemwasarrangedwherehydrostaticpressurebyaheightdifferencebetweeninletandoutletreservoirresultedinagravitationalflowToobtain general information about the behavior of the assembled CFIA systempreliminaryexperimentswithFcMeOHwereperformedToassurecompatibilitywithMSlateronNH OAcwaschosenastheelectrolyteVariousheightdifferences4
andinjectiontimesweretestedandtheinjectionataheightdifferenceof20cmlasting for 10 s was found to be the optimum concerning feasibility and peakshapes Injections of several solutions of FcMeOH of different concentrationsshowed that the concentration dependence of FcMeOH was linear in theinvestigatedrange(Fig2)Furthermoreexperimentsrevealedthattheinjectionprocedure was established with reasonable precision When repeating the
ndash1injectionof05mmolL FcMeOHincarriersolutionfortentimestherelativestandarddeviationwasfoundtobe3forthemanualinjectionprotocol
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 33
Fig 1(A)Schemeoftheusedcapillaryflowinjectionanalysissetup(1)carrierreservoir(2)samplevial(3)inletcapillarywithalengthof40cmandaninnerdiameterof100microm(4)outletcapillarywithalengthof10cmandaninnerdiameterof100micrombothcapillariesconnectedto(5)acommerciallyavailableflowcellfromMicruxand(6)awastevial(B)Configurationinsidetheflowcell(7)thefusedsilicacapillarywasplacedinaso-calledwall-jetconfigurationabovetheworkingelectrodeof(8)adisposablethin-filmgoldelectrode
4Conclusions
The presented system for CFIA-AD was arranged as simple as possible andrepresents a useful approach for the development of a method for fastoptimization of detection conditions in the context of the determination ofcarbohydratesbymeansofelectrochemistry-CE-MSThesetupisfreeofelectricalinterferencebyhighvoltagecompatiblewithMSandthuspromisingforstudyingtheoxidationofvariousanalytes
References
[1] LuGCrihfieldCLGattuSVeltriLMHollandLACapillaryelectrophoresisseparationsofglycansChemRev118(2018)7867ndash7858
[2] GalantALKaufman RCWilson JDGlucoseDetectionandanalysisFoodChem188(2015)149ndash160
[3] Sarazin C Delaunay N Costanza C Eudes V Gareil P Application of a new capillaryelectrophoreticmethodforthedeterminationofcarbohydratesinforensicpharmaceuticalandbeveragesamplesTalanta99(2012)202ndash206
[4] Rohrer JS Basumallick L Hurum D High-performance anion-exchange chromatographywithpulsedamperometricdetectionforcarbohydrateanalysisofglycoproteinsBiochem78(2013)697ndash709
[5] IslamMAMahbubPNesterenkoPNPaullBMackaMProspectsofpulsedamperometricdetectioninflow-basedanalyticalsystemsndashAreviewAnalChimActa1052(2019)10ndash26
[6] MatysikF-MWernerGTracemetaldeterminationintearsbyanodicstrippingvoltammetryinacapillaryflowinjectionsystemAnalyst118(1993)1523ndash1526
34 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2ndash1 ndash1(A) CFIA-AD recordings of three consecutive injections of (1) 025mmolL (2) 05mmolL
ndash1 ndash1 ndash1(3) 075mmolL and (4) 1mmolL FcMeOH in 50 mmolL NH OAc detection at a Micrux4
thin-filmAuelectrodeataconstantpotentialof03VinaflowcellHydrodynamicinjectionlasted10sataheightdifferenceof20cm(B) Calibration dependence of FcMeOH for CFIA-AD determination and detection at a Micruxthin-filmAuelectrodeataconstantpotentialof03VinaflowcellThestandarddeviationsofpeakheights(n=3)areindicatedbyerrorbars
1Introduction
Topreventsorptiononthecapillarysurfaceandimproveseparationefficiencyand selectivity of determined analytes coatings are formed on the capillarysurface There are two types of ones namelydynamic and covalently bondedcoatings Despite the simplicity of creation dynamic coatings cannot providerequiredreproducibilitywhilecovalentcoatingscontributethestableelectro-osmoticflow(EOF)andhighreproducibilityoftheanalysisInmostcasesthe
Application of covalent coatings based on imidazolium cations for separation and on-line preconcentration of basic and neutral analytes in capillary electrophoresis
a ab aANASTASIAVKRAVCHENKO EKATERINAAKOLOBOVA LIUDMILAAKARTSOVA
a DepartmentofOrganicChemistryInstituteofChemistrySaintPetersburgStateUniversity 26Universitetskiiprospect198504StPetersburgPeterhofRussiakravchenko161216gmailcom
b TheFederalStateInstituteofPublicHealthldquoTheNikiforovRussianCenterofEmergencyandRadiationMedicinerdquoTheMinistryofRussianFederationforCivilDefenceEmergenciesandEliminationofConsequencesofNaturalDisasters54Optikovst197082StPetersburgRussia
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 35
AbstractThemethodofcapillaryelectrophoresis (CE) isactivelydevelopedandmoreandmoreattractsscientistsattentioneveryyearHoweverthesorptionofanalytesonsurfaceoffused-silicacapillarywallsisoneof thesignificantdisadvantagesof thisapproachThe formationofcoatings on the inner capillary surface is typical way to preventsorption and to increase separation efficiency and selectivity ofdetermined analytes Coatings that covalently bonded to capillarywalls is more suitable because they are stable and provides highreproducibilityofanalysisThepresentworkisfocusedonthedevelo-pmentofthemethodofelectrophoreticdeterminationofbiologicalactiveanalytesusingacovalentcoatingbasedonimidazoliumcationsTheeffectofsubstituentinimidazoliumringonmainelectrophoreticparameters was examined It was shown that alkylimidazoliumcoatingscontributetosignificantreducingofbiogenicamineslimitsof detection while β-cyclodextrinimidazolium covalent coatingallowstoseparatebothofhydrophobicandhydrophilicanalytesinonerun
Keywordsbiologicalactiveanalytescapillarycoatingcapillaryelectrophoresisimidazoliumionicliquids
analytes nature determines type of usedmodifiers because suitable ones canprovideaccessorial interactionbetweentheanalytesandthestationaryphaseimproving separation selectivity and efficiency [1 2] Ionic liquidshave beenwidelyusedinanalyticalchemistry[3]andseparationtechniquesparticularlyincapillaryelectrophoresis[4]Earlyresearches[5ndash9]haveshownopportunityofcovalently bonded imidazolium ionic liquids for electrophoretic separationHowever the effect of various substituents in imidazolium ring hasnot beendescribed previously Thus the purpose of this study was to create covalentcoatingsbasedon ionic liquidwithvarioussubstituentsand tocompare theiranalytical capabilities in the electrophoretic separation of biologically activecompounds
2Experimental
21Reagentsandchemicals
(3-Glycidyloxypropyl)trimethoxysilane (GPTMS) hydrochloric acid sodiumdodecyl sulfate (SDS) imidazole 22-diphenyl-1-picrylhydrazyl (DPPH)p-toluen-sulfonylchlorideβ-cyclodextrinhydrocortisone(F)11-deoxycortisol(S)Corticosterone(B)rac-ketoprofen(ndash)-adrenaline(A)L-(ndash)-norepinephrine(NE) DL-normetanephrine (NMN) dopamine (DA) DL-metanephrine hydro-chloride(Met)serotoninhydrochloride(Ser)homovanillicacid(HVA)24-di-hydroxy-benzoic acid (24-DHBA) 34-dihydroxy-L-phenylalanine (DOPA)L-tryptophan(Trp)L-tyrosine(Tyr)werepurchasedfromSigma-Aldrich(USA)1-Bromo-butane1-bromooctanewerepurchasedfromReagentPlus(Ukraine)Sodium dihydrogenphosphate dihydrate acetone NN-dimethylformamide(DMF)wereobtainedfromMerck(Germany)AllreagentsusedwereanalyticalgradeAllsolutionswerepreparedusingdeionizedwater
22Instrumentation
Capillary electrophoresis experiments were carried out using the system ofcapillaryelectrophoresisCAPEL-105M(LumexRussia)withUV-spectrophoto-metricdetector(wavelengthrange190ndash360nm)Separationswereperformedusing 58times49 cm (9 cm to the detector outside diameter 360 microm and innerdiameter50microm) coated silica capillaries (LumexRussia) Thebuffer pHwasmeasuredwithapH-meterHI2210ndash2216(Hanna)
23Capillarycoatingsynthesis
Earlier our research team has proposed the synthesis route for the covalentcoatingsbasedonimidazoliumcationfunctionalizedwithalkylgroup[10]andβ-cyclodextrin [11] All capillarieswere prepared according above-mentioned
36 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
manuscriptsandcharacterizedbytheEOFmobilitymeasurementandscanningelectronmicroscopyThesynthesisconsistedoffollowingstepspreparationofacapillarytocreateacovalentcoating(heatingcapillaryfilledwith2MNaOHat90degCfor1handdryingfollowed)silylationwithGPTMSandfunctionalizationwiththeimidazolesolutionfollowedmodificationbybutyl-andoctylbromideortosyl-β-cyclodextrin(seedetailsin[10]and[11])
24Solutions
A stock buffer solution in concentration 50mM was prepared by dissolvingappropriate amount of sodium dihydrogenphosphate dihydrate in deionizedwateradjustingpHto20with1MhydrochloricacidThisbuffersolutionwasthendilutedwithdeionizedwater
ndash1 Allthesamplestocksolutionswerepreparedwithconcentration10mgmL Thestocksolutionsoftheneurotransmittersandtheirmetabolites(adrenalinenoradrenaline dopamine normetanephrine metanephrine serotonin homo-vanillicacid)and24-dihydroxybenzoicacidasinnerstandardandaminoacids(tryptophan34-dihydroxy-L-phenylalanine tyrosine)wereprepared in01MhydrochloricacidThestocksolutionsofsteroids(hydrocortisone11-deoxycor-tisolandcorticosterone)wereprepared inacetonitrileThestocksolutionsofketoprofen racemate and S-ketoprofen were prepared in acetonitrilewatersolution(1090vv) Untilelectrophoreticanalysisthestocksolutionswerestoredatndash16degCTheworkingsolutionswerepreparedbydilutingtheinitialsolutionswithwaterjustbeforetheexperiments
3Resultsanddiscussion
CovalentcoatingsbasedonN-alkylimidazoliumcationwereespeciallysuitableforseparationofneurotransmittersandtheirmetabolites(Fig1)Inadditionthecombinationofcovalentcoatingwithon-linepreconcentrationtechniquesallowstothesignificantdecreaseoftheseanalyteslimitsofdetection(LOD)Accessorialinteractions positively charged analytes with positively charged imidazoleimproveseparationselectivity(viaπ-πinteraction)andefficiency(concentrationintightzonesviaelectrostaticrepulsion)Sodiumdodecylsulfate(SDS)addedintobackgroundelectrolyte(inconcentrationabovecriticalmicelleconcentra-tion) strongly interacts with hydrophobic alkyl groups in covalent coatingstructureThenegativelychargedSDSlayerisformedoninnercapillarysurfaceThedoublereversingEOFallowsustocarryoutelectrokineticinjectionofsampleandon-linepreconcentrationbysweepingsimultaneouslyLODweredeclineto
ndash108ndash20ngmL ThelengthofalkylsubstituentalsoaffectsthestackingefficiencyfactorandLODMorehydrophobicoctylgroupscomparetobutylprovidemoreeffectiveinteractionwithSDSandasresultlowerLOD
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 37
Covalentcoatingmodifiedβ-CDhasnotshownsharpreducingofLODbyon-linepreconcentrationStacking sweeping (SDSasmicelle reagent) field-enhancedsample injection were examined using different model mixtures of analytesNeverthelessthiscoatingallowssimultaneousseparationofbothofhydrophobicsteroidhormonesandhydrophilicbiogenicaminesinasinglerun(Fig2) The guest-host interaction hydrophobic cavity of β-cyclodextrin with thehydrophobicsteroids leadsto the formationofcomplexwhichaffectssteroidselectrophoretic mobility At the same time β-cyclodextrin can act as a chiralselector and baseline separation of ketoprofen enantiomers has also beenachieved
38 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Electropherogramofmixtureof neurotransmitters and theirmetabolitesadrenaline (A)norepinephrine (NE)normetanephrine (NMN)dopamine (DA)metanephrine (Met) serotonin(SER)homovanillicacid(HVA)andtheinnerstandard24-dihydroxybenzoicacid(24-DHBA)oncovalentlymodifiedwithN-buthylimidazoliumionicliquidscapillaryConditions10mMNaH PO 2 4
(adjusted to pH= 20 by 1MHCl) injection 50 stimes30mbar ndash20 kV 220 nmmodelmixurendash1 ndash1 ndash110microgmL (METADNMNNADA24-DHBA)5microgmL (SER)and20microgmL (HVA)
4Conclusions
ItwasshownthatstructurecovalentcoatingaffectsitsanalyticalcharacteristicsWecomparedtwotypesofcovalentcoatingdifferingsubstituentinimidazoliumringnamelyalkylgroupandβ-cyclodextrinThefirsttypeisgreatcoupledwithon-line preconcentration technic but it is limited to effectively determine ofbiogenicaminesonlywhilethesecondtype(withβ-cyclodextrin)showedthepossibilitiestoseparatevariousanalytesbutsuitableon-linemodehasnotbeenfoundThemainpointsaresummarizedinTable1
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 39
Covalentcoatingtype Electrophoreticseparationof On-lineprecon- centration biogenicamines amino steroid ketoprofen andtheirmeta- acids hormones enantiomers bolites N-β-cyclodextrinimida- yes yes yes yes thesuitableapproachzoliumcovalentcoatings wasnotfoundedN-alkylimidazolium yes yes nonsepa- nonsepa- thesignificantreducingcovalentcoatings rated rated forbiogenicaminesLOD
Table 1Thesummationofpossibilitiesofcovalentcoatingsbasedonimidazoliumcation
Fig 2Electropherogramofsimultaneousseparationofhydrophobic(steroidhormones)andhydro-philicanalytes (aminoacidsandbiogenicamines) insinglerunwithcovalentcoatingbasedonimidazoleandβ-CDConditions10mMNaH PO (adjusted topH = 20by1MHCl) injection2 4 20 stimes30mbarndash20kV254nm(1ndash8min)and220nm(8ndash15min)0mbar(1ndash10min)and40mbar(10ndash15 min) Model mixture corticosterone (B) hydrocortisone (F) 11-deoxycortisole (S)
ndash1 ndash15μgmL L-tryptophan(Trp)34-dihydroxy-L-phenylalanine(DOPA)10μgmL L-tyrosine(Tyr)ndash15 μg mL noradrenaline (NA) normetanephrine (NMN) adrenaline (AD) dopamine (DA)
ndash120μgmL
Acknowledgments
This work was supported by Russian Science Foundation (grant numbers 19-13-00370) Theauthors are also grateful to the Chemistry Education Centre and Nanothechnologies Centre ofResearchParkSaintPetersburgStateUniversityfortechnicalsupport
References
[1] HuLFYinSJZhangHYangFQRecentdevelopmentsofmonolithicandopen-tubularcapillaryelectrochromatography(2017ndash2019)JSepSci43(2020)1942ndash1966
[2] KartsovaLAKravchenkoAVKolobovaEACovalentcoatingsofquartzcapillariesfortheelectrophoretic determination of biologically active analytes J Anal Chem 74 (2019)729ndash737
[3] HoTDZhangCHantaoLWAndersonJLIonicliquidsinanalyticalchemistryFundamen-talsadvancesandperspectivesAnalChem86(2014)262minus285
[4] TangSLiuSGuoYLiuXJiangSRecentadvancesofionicliquidsandpolymericionicliquids incapillaryelectrophoresisandcapillaryelectrochromatography JChromatogrA1357(2014)147ndash157
[5] QinWLiSFYElectrophoresisofDNAinionicliquidcoatedcapillaryAnalyst128(2003)37ndash41
[6] QinWWeiH Li SFY 13-Dialkylimidazolium-based room-temperature ionic liquids asbackgroundelectrolyteand coatingmaterial in aqueous capillaryelectrophoresis JChro-matogrA985(2003)447ndash454
[7] QinW Fong S Li Y Determination of ammonium andmetal ions by capillary electro-phoresisndashpotential gradient detection using ionic liquid as background electrolyte andcovalentcoatingreagentJChromatogrA1048(2004)253ndash256
[8] QinWLiSFYAn ionic liquidcoating fordeterminationofsildenafilandUK-103320 inhumanserumbycapillaryzoneelectrophoresis-iontrapmassspectrometryElectrophoresis23(2002)4110ndash4116
[9] BorissovaMVaherMKoelMKaljurandMCapillaryzoneelectrophoresisonchemicallybondedimidazoliumbasedsaltsJChromatogrA1160(2007)320ndash332
[10] KolobovaEKartsovaLKravchenkoABessonovaEImidazoliumionicliquidsasdynamicand covalent modifiers of electrophoretic systems for determination of catecholaminesTalanta188(2018)183ndash191
[11] KravchenkoAKolobovaEKartsovaLMultifunctioncovalentcoatingsforseparationofaminoacidsbiogenicaminessteroidhormonesandketoprofenenantiomersbycapillaryelectrophoresisandcapillaryelectrochromatographySepSciplus3(2020)102ndash111
40 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Synthetic 4-hydroxy-3-methoxybenzaldehyde (vanillin) is used as a flavoringagent in foodsdrinksperfumesandpharmaceuticals [1]However at certainconcentrationsthesubstancemayaccumulateinthebodyhaveatoxiceffectand
ndash1at high concentrations may be fatal (lethal dose LD (oral rat) =2gkg 50ndash1 ndash1LD (oral guinea pig) = 14gkg LD (intravenous dog) = 132gkg lethal50 50 ndash1concentrationLC (inhalationmouse)=417gkg )[2]AccordingtoRussianState
StandartGOST121005-88thetoxiceffectsofvanillinintheworkplaceinthendash3formofvapoursoraerosolsareobservedatconcentrationsabove15mgm
Chromatography[3]spectrophotometry[4]capillaryelectrophoresisareusedforvanillindeterminationindifferentobjects CurrentlysmokingmixturesforhookahsandelectroniccigarettesarewidelyusedamongyoungpeopleThesemixturesarenotcontrolledforthecontentofsubstancesandarefreelyavailableconsideringthemmoreharmlesswithrespecttoordinarycigarettesThusthedevelopmentofamethodforthedeterminationof4-hydroxy-3-methoxybenzaldehydeinsmokingmixturesisrelevant
Determination of vanillin in smoking mixtures by spectrophotometry
ELIZAVETAEFREMENKOANNACHERNOVAOLGABASTRYGINA
DepartmentofChemicalEngineeringNationalResearchTomskPolytechnicUniversityLeninavenue30634050TomskRussiaeaetpuru
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 41
AbstractTheresearchdealswithdeterminationofvanillin insmokingmix-turesbyultraviolet-visiblespectrophotometryThemethodshowed
ndash1goodlinearityintherangeof005ndash012gL withalimitofdetectionndash1005gL After validation studies the method was successfully
applied to thedeterminationof vanillin in smokingmixtureswithsatisfactoryresultsItwasshownthattheerrorofthismethoddoesnot exceed 1 The developed spectrophotometric procedure fordeterminingvanillininsmokingmixturescanbeusedasacontrol
Keywordssmokingmixturesspectrophotometryvanillin
2Experimental
21Reagentsandchemicals
Asampleofvanillin(purity98)wastakenastheobjectofstudyAssolventsweused95ethanolAllchemicalsusedwereofanalyticalreagentgrade
22Instrumentation
Theopticaldensityofsampleswasmeasuredincuvettewithanabsorbinglayerthickness of 10 mm using a Cary 60 spectrophotometer (Agilent USA) Allmeasurementswerecarriedoutatroomtemperature
23Samplepreparation
Sample preparation of the investigated objects consisted of the preliminarydissolutionofthesamplein95ethanolThesample10mgoftobaccoldquoAdalyandashVanillardquo(Turkey)wasdiluted in10μLof95ethanol to theconcentrationof
ndash11gL Thesample10μLofldquoFlavoringTPAndashVanillaCustardrdquo(USA)wasdilutedin10 μL of 95 ethanol The resulting solution was diluted six times to the
ndash3concentrationof017μLcm
3Resultsanddiscussion
TodeterminevanillininthesamplestheopticalpropertiesofvanillininvarioussolventsweredeterminedAsaresultthe95ethanolwaschosenastheoptimalsolvent[4] IthasbeenestablishedthatintheUVspectraoftheanalyteabsorptionbondsareobservedwithmaximumvaluesat23002800and3100nmwhichcorres-pondstopublisheddata[45](Fig1) To quantify vanillin the calibration curve of the optical density on theconcentrationofvanillin in95ethanolwasobtainedatconcentrations005
ndash1006007008010and012gL Calibrationcurveofvanillinin95ethanolatawavelengthof280nmis
ndash1 A =81914c[gL ]+00357 (1)2802 R =1
Calibrationcurveofvanillinin95ethanolat310nmis
ndash1 A =73824c[gL ]+00301 (2)3102 R =1
42 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Inthespectraoftheanalyzedsamplesolutionsabsorptionmaxima(2800nmand3100nm)characteristicforvanillinwereobservedTheamountofvanillininthesamplewasdeterminedusingcalibrationcurvesat280and310nmWeightedleast square regressionwas applied to the calibration curves to improve theaccuracyespeciallyatinlowconcentrationlevelrangeGoodlinearitywasfound
ndash1 ndash1intherangeof005ndash012gL withadetectionlimitof005gL TheresultsarepresentedintheTable1
4Conclusions
ThedevelopedmethodcanbeusedasacontrolmethodTheerrorinthemethodfordeterminingvanillininthesampleldquoFlavoringTPAndashVanillaCustardrdquowithaknownconcentrationofvanillinwas0004Accordingtothedataobtainedwerecommendawavelengthof280nmforthedeterminationofvanillininsamples
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 43
λ nm
Absorban
ce
ndash1Fig 1 Absorption spectrum of vanilin solution in 95 ethanol at concentration 01 mol L (anabsorbinglayerthicknessof10mm)
Sample λnm Tookmg Foundmg S Sх Δх δ
FlavoringTRA 310 10850 104096 00024 00011 00006 00306ndashVanillaCustardAdalyandashVanilla 310 100000 10162 00019 00009 00002 00025
FlavoringTRA 280 100300 96062 00033 00015 00032 00042ndashVanillaCustardAdalyandashVanilla 280 100000 10122 00013 00006 00012 00017
Table 1Testingmethods introduced foundof vanillin in the samplesat310nmby spectrophotometricmethod(n=5p=099SndashstandarddeviationSxndashrelativestandarddeviationΔхndashabsoluteerrorδndashrelativeerror)
ThenthedevelopedmethodwastastedonthesampleldquoAdalyandashVanillardquosamplewithamorecomplexcompositionandanunknownconcentrationofvanillinwastaken The vanillin content in the sample was determined according to thedevelopedmethoditamountedto10ofthetotalmassStudieshaveshownthepossibility of using spectrophotometric analysis for the qualitative andquantitative determination of vanillin Also based on preliminary studies aspectrophotometricprocedurewasdevelopedforthequantitativedeterminationofvanillinbasedonabsorptioninethanolinthewavelengthrange200ndash400nm
References
[1] httpswwwrusnaukacom43_DWS_2015Chimia6_203179dochtm (accessed 25thFebruary2019)
[2] httpswwwcdcgovnioshrtecsdefaulthtm(accessed11stApril2020)[3] AliLPerfettiGDiachenkoGRapidmethodforthedeterminationof342coumarinvanillin
and ethyl vanillin in vanilla extract by reversed-phase liquid 343 chromatography withultravioletdetectionJAOACInt91(2008)383ndash386
[4] БастрыгинаОАЕфременкоЕАЧерноваАПВыделениеванилинаисследованиеегооптическихсвои ствопределениевбиологическомматериалеВХимияихимическаятехнология в XXI веке Материалы XX Международной научно-практическойконференции имени профессора ЛП Кулёва студентов и молодых ученых ТомскНациональныи исследовательскии Томскии политехническии университет 2019с301ndash302
[5] WeastRCHandbookofChemistryandPhysics60thedBocaRatonCRCPress1979p143
44 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
UraniumbelongstothegroupofhazardouselementsItisahighlyharmfulandradioactiveelementtoxictohumansandalllivingorganisms[12]Inhaledwithair it has a particularly destructive effect on the kidneys and as a result ofaccumulationinwhitebloodcellsitcanalsocauseimpairmentoftheimmunesystem[2]Uraniumoccursatseveraldegreesofoxidationhoweverinaqueoussolutionsthemoststableformisuranylion(UO (II))[12]Thepresenceofura-2
niumintheenvironmentiscausedbyamongothersnaturalsoilandrockerosionEnvironmentalpollutionwiththiselementisalsoconstantlyincreasingduetohumanactivitycoalcombustionuraniumoreminingandprocessingthearmsindustryandtheuseofuraniumasnuclearfuelinfissionreactors[3]Itisveryimportanttoconstantlymonitortheconcentrationofuraniumbothinthenaturalenvironment in order to assess its state and safety (especially in the case ofdrinkingwater)aswellasinallstagesofprocessingprocessesassociatedwiththenuclearindustrytoavoidtheoccurrenceofnuclearpollution[13] Scientists have made many attempts to develop research methods todeterminethecontentofuranylcompoundsinliquidsamplesEffortsweremadetousemanyanalyticalmethodsforthispurposeincludingspectrophotometry
Uranyl ion-selective electrode with solid contact
KAROLINAPIETRZAKCECYLIAWARDAK
DepartmentofAnalyticalChemistryInstituteofSciencesFacultyofChemistryMariaCurie-SklodowskaUniversityMariaCurie-SklodowskaSq320-031LublinPolandkarolinapietrzakpocztaumcslublinpl
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 45
AbstractNewallsolidstateuranylion-selectiveelectrodeswithlowdetection
ndash7 ndash1limits(71times10 molL )shortresponsetimegoodselectivityandstable and reproducible potential were developed Many types ofelectrodeswith different active ingredient content in ion-selectivemembrane (bis(244-trimethylpentyl)phosphonium acid Cyanex-272)were testedAs an additive an ionic liquid1-octyl-3-methyl-imidazole chloride was used The optimal composition of theion-selective membrane was chosen from all electrodes based onthedeterminationand comparisonof analyticalparametersof thesensors
Keywordsion-selectiveelectrodesolidcontacturanyl
plasma spectrometry luminescence spectroscopy voltammetry or chromato-graphymethods[2] Duetomanyadvantagesofpotentiometricmethods(amongthemlowercostseasieroperationofdevicesquickresponseandtheabilitytoperformmeasure-ments in flowmode) [3] a numberof potentiometric sensorshave alsobeendeveloped that could be successfully used in this type of research Themostpopularpotentiometricsensorsincludeion-selectiveelectrodes(ISEs)whicharecharacterized by low-energy consumption small size and portability and aresuccessfullywidelyusedforthedeterminationofbothinorganicandorganicionsinclinicalanalysisprocesstechnologyaswellasincontrolthestateofthenaturalenvironment[45]Removaloftheinternalsolutioncontainingthesameanalytetowhich theelectrode is sensitiveresulted in theso-calledsolidcontact ISEswhicharemuchsmallerinsizethantheirpredecessorsaremoreconvenienttouse and more mechanically resistant In this type of sensors however it isimportanttoachievesatisfactorypotentialstabilitywhichisnecessarytoobtainsatisfactoryresults[5]AveryimportantpartofISEsistheion-selectivemem-branewhosecompositiondeterminestheanalyticalparametersofthesensorsResearchers are currently focusing on the production and testing of newsubstancesthatcouldbesuccessfullyusedasmembranecomponentsandsolidcontacts thatwould allow to obtain new sensorswith lower detection limitslongerlifetimeandbetterpotentialstabilityandtodeterminenewpreviouslyunattainableanalytes[4] AstheactivecomponentsofthemembranesensitivetouranylionscientistshavealreadyusedKryptofix22DD(413-didecyl-171016-tetraoxa-413-diaza-cyclooctadecane)[2]Cyanexextractants(bis(244-trimethylpentyl)phosphinicacid bis(244-trimethylpentyl)monothiophosphinic acid and bis(244-tri-methylpentyl)dithiophosphinic)acid[3]DBBP(dibutylbutylphosphonate)andDOPP (di-n-octyl phenylphosphonate) [6] DMSO (dimethylsuphoxide) [7]TTPTP (5678-tetrahydro-8-thioxopyrido[4345]thieno[23-d]pyrimidine-4(3H)one)[8]orTEHP(tris(2-ethylhexyl)phosphate)andTPTU(O-(12-dihydro-2-oxo-1-pyridyl)-NNNN-bis(tetra-methylene)uronium hexafluorophos-phate)[9]
2Experimental
21Reagentsandchemicals
This paper presents research on the design and properties of ion-selectiveelectrodes with solid contact for the determination of uranyl ions Bis(244-trimethylpentyl)phosphonium acid (Cyanex-272) was used as the activecomponentof themembranewhichwasdescribed in the literatureasagooduranylextractant[10]Inordertoensureaconstantpotentialofthiselectrodeandreducetheelectroderesistancetheion-sensitivemembranewasenrichedwithafewpercentadditionof1-octyl-3-methylimidazolechlorideionicliquid
46 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Several types of ion-selective electrodes were prepared using an AgAgClelectrodeasaninternalelectrodewhichdifferinthequantitativeandqualitativecompositionofthemembranesAllcompositionsarelistedinTable1
22Instrumentation
Measurements were made at room temperature using a 16-channel datacollectionsystem(LawsonLabs IncUSA)coupled toa computer in solutionsmixedwithamechanicalstirrerAsilversilverchlorideelectrodewithdoublejunctionwasusedasthereferenceelectrode
3Resultsanddiscussion
The effect of ion-selective membrane composition on the properties of theobtained potentiometric sensors was examined by determining their basicanalyticalparametersincludingslopeoftheelectrodecharacteristicsdetectionlimitmeasuringrange(concentrationrangeinwhichthecourseoftheelectrodecharacteristics isrectilinear)pHrange(inwhich ithasnoeffect forelectrodepotential)andresponsetimeTheobtainedvaluesofthetestedparametersareshowninTable2 Figure1showsthecalibrationcurvesofthetestedelectrodesdeterminedin
ndash7 ndash1 ndash1UO (NO ) solutionsintheconcentrationrange1times10 ndash1times10 molL Asitcan2 3 2
beseeninFig1andTable2allelectrodesweresensitivetouranylionsbutindifferent extend The best response exhibited ISE-3 containing 1 (ww) ofionophore Increasing the ionophore content in themembrane shortened thelinearityrangeofthecalibrationcurveanditssupernenstianslope Theselectivityofthetestedelectrodeswasestimatedbydeterminingtheselec-tivitycoefficients inrelationto interfering ionsForthispurpose theseparate
ndash1solutionmethodwasused(extrapolatingresponsecurves toa =a =1molL )i j
ComparisonofISE-1andISE-3electrodeselectivityisshowninFig2
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 47
Table 1Quantitative and qualitative composition of electrode membranes Cyanex-272 (bis(244-tri-methylpentyl)phosphoric acid) TBP (tri-n-butyl phosphate) and OMImCl (1-octyl-3-methyl-imidazolechloride)
Abbreviation Membranecomposition(ww)ofelectrode Cyanex-272 PVC TBP OMImCL
ISE-1 00 33 620 5ISE-2 05 33 615 5ISE-3 10 33 610 5ISE-4 30 33 590 5ISE-5 50 33 570 5ISE-6 100 33 520 5
48 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Abbreviation Slope Detectionlimit Linearrange Response pHrange2+ ndash1 ndash1ofelectrode mVpa(UO ) molL molL times2
ndash5 ndash5 ndash1ISE-1 297 25times10 5times10 ndash1times10 5ndash8 28ndash42ndash6 ndash5 ndash1ISE-2 292 65times10 1times10 ndash1times10 5ndash8 25ndash60ndash7 ndash5 ndash1ISE-3 298 71times10 1times10 ndash1times10 5ndash8 24ndash60
ndash6 ndash4 ndash1ISE-4(I) 357 31times10 5times10 ndash1times10 5ndash8 ndndash6 ndash6 ndash4ISE-4(II) 242 31times10 5times10 ndash5times10 5ndash8 nd
ndash3 ndash1ISE-5(I) 638 nd 1times10 ndash1times10 5ndash10 ndndash5 ndash3ISE-5(II) 234 nd 5times10 ndash1times10 5ndash10 ndndash3 ndash1ISE-6(I) 733 nd 1times10 ndash1times10 5ndash10 ndndash5 ndash3ISE-6(II) 222 nd 5times10 ndash1times10 5ndash10 nd
Table 2Selectedparametersandtheirdeterminedvaluesoftestedionselectiveelectrodes
Fig 1 Calibration curves of the testedelectrodesobtainedinUO (NO ) solutionsin2 3 2 ndash7the concentration range from 1times10 to
ndash1 ndash11times10 molL
Fig 2Comparisonofselectivitycoefficientspot(log K (UO (II))M) for electrodes ISE-12
(1stcolumn)andISE-3(2ndcolumn)
Inordertoexaminethereversibilityofthepotentialofthetestedelectrodesndash4 ndash1potentialmeasurementsweremadealternatelyinsolutions1times10 molL and
ndash5 ndash11times10 molL ofUO (NO ) TherecordedpotentialreadingsareshowninFig32 3 2
Long-term potential stability and sensor reproducibility were evaluated byndash1determiningtheaveragevalueoftheelectrodepotentialina01molL UO (II)ion2
solutionovertimeforthreeidenticalISE-3Thesemeasurementsweremadetoobservechangesinthepotentialofelectrodeswiththesameconcentrationoveralongperiodoftime(30days)Figure3showsthelong-termpotentialstabilityandreproducibilitydeterminedforthreeidenticalsensors
4Conclusions
Asaresultofthetestsion-selectiveelectrodeforthedeterminationofuranylionswasobtainedwhich iseasy todesignanduseThebestanalyticalparametersexhibitedISE-3containing1ionophoreintheion-selectivemembraneForthis
ndash7 ndash1typeofelectrodesthedetectionlimitof71times10 molL linearityoftheelectrodendash6 ndash1 ndash1calibrationcurve in the range1times10 ndash1times10 molL andresponse time5ndash8s
were obtained In addition the manufactured sensors also showed stablereproducibleandreversiblepotentialandverygoodselectivityinrelationtothetestedinterferents
References
[1] AnsariRMosayebzadehZConstructionofanewsolid-stateU(VI)ion-selectiveelectrodebasedonpolypyrroleconductingpolymerJRadioanalNuclChem299(2014)1597ndash1605
[2] GhanbariMRounaghiGHAshrafNAnuranylsolidstatePVCmembranepotentiometricsensor based on 413-didecyl-171016-tetraoxa-413-diazacyclooctadecane and itsapplicationforenvironmentalsamplesIntJEnvironAnalChem97(2017)189ndash200
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 49
Fig 3 Stability () reproducibility andreversibility () of the potential of ISE-3Standard deviations given on the plot aredeterminedforthesamethreeISE-3
[3] Badr IHA Zidan WI Akl ZF Cyanex based uranyl sensitive polymeric membraneelectrodesTalanta118(2014)147ndash155
[4] BiegCFuchsbergerKStelzleMIntroductiontopolymer-basedsolid-contaction-selectiveelectrodes basic concepts practical considerations and current research topics AnalBioanalChem409(2017)45ndash61
[5] Bobacka J IvaskaA LewenstamA Potentiometric ion sensorsChem Rev108 (2008)329ndash351
[6] ZidanWI Badr IHA Akl ZF Development of potentiometric sensors for the selective2+determinationofUO ionsJRadioanalNuclChem303(2015)469ndash4772
[7] SalehMBSolimanEMGaberAAAAhmedSANovelPVCmembraneuranylion-selectivesensorSensActuatorsB114(2006)199ndash205
[8] SalehMBHassanSSMAbdelAAAbdelNAAnoveluranylion-selectivePVCmembranesensor based on 5678-tetrahydro-8-thioxopyrido[4345]thieno[23-d]pyrimidine-4(3H)oneSensActuatorsB94(2003)140ndash144
[9] HassanSSMAliMMAttawiyaAMYPVCmembranebasedpotentiometricsensorsforuraniumdeterminationTalanta54(2001)1153ndash1161
[10] Prabhu DR Ansari SA Raut DR Murali MS Mohapatra PK Extraction behaviour ofdioxouranium(VI) cation by two phosphorous-based liquid cation-exchangers in room-temperatureionicliquidsSepSciTechnol52(2017)2328ndash2337
50 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Metronidazole(2-methyl-5-nitroimidazole-1-ethanol)isoneofthemostwidelyused nitroimidazole antibiotics Metronidazole is used for the treatment ofinflammatorydiseasescausedbyanaerobicorganismsandsomeprotozoaandforpreventionofdysenterycolibacillosiseimeriosisbalantidiasissalmonellosisenteritissepticemiapost-surgicalcomplications[1ndash3]Oxytetracyclinehydro-chlorideisanantibioticofthetetracyclinefamilyItisoneofthemostcommonlyused antibiotics in poultry because of its low cost and effective [4] Thesecompounds are intensively used in poultry breeding and stockbreedingUnreasonableuseofthesedrugscancauseseriousfoodsafetyissues[5] The veterinary drug Nozemat which includemetronidazole and oxytetra-cyclinehydrochloridewaschosenfortheexperimentsNozematisusedtotreat
Polarographic determination of metronidazole and oxytetracycline hydrochloride in veterinary drug for honey bees
a a bKATERYNAPLOTNIKOVA LILIYADUBENSKA IVANZELENYI
a AnalyticalChemistryDepartmentIvanFrankoNationalUniversityofLvivKyrylaiMefodiaStr879005LvivUkrainekaterina27plgmailcom
b DrohobychPedagogicalLyceumIvanaFrankaStr3682100DrohobychUkraine
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 51
AbstractWehavedevelopedanewpolarographicmethodforthedetermin-ation of metronidazole and oxytetracycline hydrochloride in theveterinarydrugNozematforhoneybeesThetechniqueisbasedonthereductionofpolarographicallyactivecompoundsonamercurydropletelectrodeTheinfluenceofthecomponentsoftheveterinarydrugNozematonthepolarographicdeterminationofmetronidazolewasstudiedItwasfoundthatthereductionofmetronidazoleisnotaffected by glucose and ascorbic acid but is affected by oxytetra-cyclinehydrochloridewhichisreducedtomercurydropletelectrodeatapotentialofndash145VThedevelopedtechniqueischaracterizedbyeaseofsamplepreparationandcost-effectivenessThistechniquehastheabilitytoidentifysimultaneouslyanddeterminatemetronidazoleand oxytetracycline hydrochloride in solution without the use ofseparationandconcentrationmethods
Keywordselectrochemistrymetronidazoleoxytetracycline
hydrochloridepolarographyveterinarydrug
bees and it can be given in unregulated doses Because of this an unknownamountofmetronidazolecangetintothehoneyanditsometimescausessideeffectsofthehumanbodyanditcouldbeofgreatconcernforpublichealth[56]MedicinesforpeoplearemorestringentandbettertestedthanveterinarydrugsTheproblemofthecontroloftheveterinarydrugsisurgentnowadaysVeterinarymedicinescouldbeunauthorizedandtheuncontrolleduseofmedicinesexistsinretailpharmaciesofmedicineorimportedascontrabandfromothercountries The most widespreadof these classes in thequality controlare chromato-graphic [6ndash9] spectrophotometric [10ndash13] and electrochemical methods[14ndash17]Manyoftheknownmethodsforthedeterminationofmetronidazoleandoxytetracyclinehydrochloridehaveanumberofdisadvantagestime-consumingtheuseoforganicsolventsandexpensivereagentsthesideeffectsofexcipientsandotheractivesubstancesElectrochemicalmethodsarepromisingalternativefor the determination of the electroactive substances Their advantages aresimplicityminiaturizationhighsensitivityandrelativelylowcostThereforethesearch for simple express and affordable methods for the determination ofmetronidazoleremainsrelevantOneofthepromisingmethodsofdeterminationisvoltammetry
2Experimental
21Reagentsandchemicals
VeterinarydrugNozemat (manufacturerAPI-SANRussia) is a yellowpowderwithaslighttypicalodorAvailableinlaminatedbagsof25gCompositionper1gof the drug metronidazole 400 mg oxytetracycline hydrochloride 400 mgglucoseascorbicacid MetronidazoleandoxytetracyclinehydrochloridewerepurchasedfromSigmaAldrich(USA)Stockstandardsolutionofmetronidazolefordeterminationwaspreparedbydissolvingtheexactamountofstandardin7mLof2Mhydrochloricin 500 mL volumetric flask Stock standard solution of oxytetracyclinehydrochloride was prepared by dissolving the exact amount of standard indistilledwaterin500mLvolumetricflaskAfterthatthesolutionswereadjustedtothemarkwithdistilledwaterandmixedthoroughly The Britton-Robinson buffer preparationwas as follows 202 g of sodiumtetraboratedecahydrate287mLofglacialaceticacidand176mLofconcen-tratedorthophosphoricacidweredissolvedin10Lvolumetricflask Working solution preparation was as follows an aliquot of stock standardsolutionwasaddedintoa25mLvolumetricflasktoobtainasolutionwiththenecessaryconcentrationthen2mLofBritton-RobinsonbufferwithnecessarypHwasaddedtotheflaskanddistilledwaterwasaddedtothemark AqueoussolutionofNozematwaspreparedasfollowstheexactportionofthetestveterinarydrugwasdissolved ina250mlvolumetric flaskAnaliquotof
52 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Polarogramsof(A)metronidazoleand(B)metronidazolewithoxytetracyclinehydrochloridendash1solutionsat02MBritton-RobinsonbufferbackgroundatpH=96(υ=05Vs c(metronidazole)=
ndash5 ndash5=45times10 Мc(oxytetracyclinehydrochloride)=50times10 М
100mloftheresultingsolutionwasaddedtoa250mlvolumetricflaskandmadeuptothemarkwithwaterAnaliquotof100mloftheresultingsolutionwasaddedtoa250mlvolumetricflask2mlofBritton-RobinsonbufferwithapHof96wasaddedandthevolumewasadjustedtothemarkwithdistilledwater
22Instrumentation
ForpolarographicmeasurementsweuseddigitaldeviceMTechOVA-410 [18]temperature-controlledthree-electrodeamercurydropletindicatorelectrodeasaturatedcalomelreferenceelectrodeandplatinumwireauxiliaryelectrodeTheaccuracyofthepotentialmeasurementis1mVTheuncertaintyofcurrentmeasu-rement is 01 The employed mercury droplet electrode had the following
ndash4 ndash1characteristicsm=594times10 gs τ=10 min in 02 M NH Cl We used cyclic4
voltammetryforthestudyoftheelectrochemicalprocess WeusedMV870DIGITAL-pH-MESSERATpH-meterformeasuringpHofthesolutions Theobtainedworkingsolutionswereintroducedintothecellanddeoxyge-natedwithargonfor10minPolarogramswererecordedintherangeofpotentialsfrom00tondash16V
3Resultsanddiscussion
Previously it was found that using the Britton-Robinson buffer with pH = 96metronidazoleisreducedwiththeformationofacharacteristiconeirreversiblepeakatndash064V(Fig1A)UsingpolarographywithfastpotentialscanitwasfoundthatmetronidazoleisreducedonmercurydropletelectrodeintherangeofpH20to105ThepeakrecoverycurrentofthemetronidazolereachesthemaximumvalueatpH9ndash10againstthebackgroundofatheBritton-Robinsonbuffer
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 53
Underpre-selected conditions theeffectof some foreign substanceson thepolarographicdeterminationofmetronidazolewasinvestigatedSubstancesthatare componentsofdrugswere studiedglucose ascorbicacidoxytetracyclinehydrochlorideGlucose and ascorbic acid are not reduced atmercury dropletelectrodeanddonotchangetheappearanceofthepolarogramandpolarographiccharacteristics of the recovery of metronidazole As can be seen from Fig 1oxytetracycline hydrochloride is reduced to mercury droplet electrode andchangestheappearanceofthepolarogramandpolarographiccharacteristicsofthe recovery of metronidazole With the addition of oxytetracycline hydro-chloridetherecoverypeakofmetronidazoledecreasesandslightlyshiftstomorenegativepotentials The composition of the drug is relatively complex excipients affect theanalyticalsignaloftherecoveryofcompoundssototakeintoaccountthematrixeffectusedthemethodofmanyadditives QuantitativelytransferredthesolutionofNozemattothecell(exactvolume)removed dissolved oxygen for 10 min and took polarograms in the range ofpotentialsfrom00tondash16VAliquotsofstandardmetronidazolesolutionwereintroducedintothecelltoobtainasolutionwithagivenconcentrationofadditive
ndash5 ndash5metronidazole 10times10 M to 70times10 M As with the determination of themetronidazole aliquots of a standard oxytetracycline hydrochloride solutionwereaddedtothecellwithsolutionofNozemattoobtainasolutionwithagiven
ndash6 ndash6additiveconcentrationfrom70times10 Mto50times10 M(Fig2) In Table 1 are shown metrological characteristics of the determination ofmetronidazoleandoxytetracyclinehydrochloride inveterinarydrugRecoverywascalculatedFormetronidazoletherecoveryis97andforoxytetracyclinehydrochloridetherecoveryis103Analyticalperformanceofthetechniqueisgoodfordeterminationveterinarydrugs
54 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2Polarogramsof(A)metronidazolereductionatdifferentmetronidazoleconcentrationsand(B)oxytetracyclinehydrochloridereductionatdifferentoxytetracyclinehydrochlorideconcentra-tionsandtheircorrespondingcalibrationgraphs
Theaccuracywasverifiedbytheldquoadded-foundrdquomethodAliquotsofstandardsolutionofmetronidazoleweremadeina250mlvolumetricflasktoobtaina
ndash5solutionofagivenconcentrationof33times10 Mandthesolutionofoxytetracyclinendash5hydrochloridetoobtainasolutionofagivenconcentrationof15times10 M2mlof
Britton-RobinsonbufferwithpH96wasaddedtoflaskwithstirringandadjustedtothemarkwithwaterTheanalysisprocedureofmodelsolutionissimilartoanalysis procedure of the solution of Nozemat The calculated amount ofmetronidazolebythemethodofmultipleadditivesinthetestedmodelsolutionisinagreementwiththeamountthatwasintroducedintothesample
4Conclusions
The new polarographic method for the determination of metronidazole andoxytetracyclinehydrochlorideintheveterinarydrugNozematforhoneybeeswasdeveloped We conducted principal component analysis of veterinary drugNozemattoassesstheoveralleffectforthedeterminationofmetronidazoleWefoundthatoxytetracyclinehydrochlorideisreducedtomercurydropletelectrodeThismethodhastheabilitytoidentifysimultaneouslyanddeterminatemetro-nidazole and oxytetracycline hydrochloride in solution without the use ofseparationandconcentrationmethodsOnemoreofadvantagesoftechniquearefastprocedureofanalysissimplesamplepreparationlowcostthepossibilityofminiaturization
References
[1] Antibiotic and Chemotherapy Finch R Greenwood D Whitley R (edits) AmsterdamElsevier2006p292ndash299
[2] MitrowskaKPrzyczynyiskutkizakazustosowania5-nitroimidazoliuzwierzątktorychtkanki lub produkty przeznaczone są do spozycia przez ludziMed Weter 71 (2015)736ndash742
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 55
Metronidazole Oxytetracycline hydrochloride
PeakspotentialV ndash065 ndash144CorelationcoefficientR 099892 099922
ndash1 4 4SlopebmicroАM 656times10 176times10Δb 1763 400InterceptamicroА 2238 0219Δa 0075 0009
ndash1 ndash5 ndash5cmolL 342times10 125times10ndash1cmgg 389 413
Recovery 97 103
Table 1Validationparametersofthemethodofmetronidazoleandoxytetracyclinehydrochloridedetermi-nationinsolutionsofNozematbythemethodofmanyadditives
[3] VermaPNamboodiryVMishraSBhagwatABhoirSAstabilityindicatingHPLCmethodfor the determination of Metronidazole using Ecofriendly solvent as mobile phasecomponentIntJPharmPharmSci5(2013)496ndash501
[4] Cervini P Ambrozini B Machado LCM Ferreira Garcia AP Cavalheiro Gomes ETThermal behavior and decomposition of oxytetracycline hydrochloride J Therm AnalCalorim121(2015)347ndash352
[5] DangBNAnhNTKKyLXThaiPKAntibioticsintheaquaticenvironmentofVietnamsourcesconcentrationsriskandcontrolstrategyChemosphere197(2018)438ndash450
[6] QuintanillaPHettingaKABeltranMCEscricheIMolinaMPVolatileprofileofmaturedTronchon cheese affected by oxytetracycline in raw goat milk J Dairy Sci 103 (2020)6015ndash6021
[7] Chen F Yu L Jingdong P Xiang W Huanjun P Yu C Yan H Study on simultaneousdetermination of three nitroimidazole residues in honey by high performance liquidchromatographyndashresonanceRayleighscatteringspectraMicrochemJ141(2018)423ndash430
[8] Hernandez-MesaM Cruces-Blanco C Campana GA Simple and rapid determination of5-nitroimidazolesandmetabolitesinfishroesamplesbysalting-outassistedliquid-liquidextractionandUHPLC-MSMSFoodChem252(2018)294ndash302
[9] Xiu-ChunGZhao-YangXHai-HuiWWen-YiKLi-MingLWen-QingCHong-WeiZWen-HuiZMolecularlyimprintedsolidphaseextractionmethodforsimultaneousdeterminationofsevennitroimidazolesfromhoneybyHPLC-MSMSTalanta166(2017)101ndash108
[10] ТеплыхАНИлларионоваЕАКоличественноеопределениеметронидазоласпектро-фотометрическимметодомСибирскиймедицинскийжурнал5(2009)48ndash50
[11] ZheltvayOIZheltvayIISpinulVVAntonovichVPSpectrophotometricdeterminationofmetronidazoleandtinidazoleusingcopper(II)complexesJAnalChem68(2013)663ndash668
[12] Youssef AK Saleh MS Abdel-Kader DA Hashem Facile DY SpectrophotometricdeterminationofmetronidazoleandsecnidazoleinpharmaceuticalpreparationsbasedontheformatioonofdyesIntJPharmPharmSci6(2015)103ndash110
[13] Sversut RA Vieira JC Rosa AM Amaral MS Kassab NM Salgado H ValidatedspectrophotometricmethodsforsimultaneousdeterminationofoxytetracyclineassociatedwithdiclofenacsodiumorwithpiroxicaminveterinarypharmaceuticaldosageformArabianJChem13(2020)3159ndash3171
[14] Nikodimos Y Electrochemical determination of metronidazole in tablet samples usingcarbonpasteelectrodeJAnalMethodsChem(2016)361294
[15] Srivastava AK Upadhyay SS Rawool CR Punde NS Rajpurohit AS Voltammetrictechniques for the analysis of drugs using nanomaterials based chemically modifiedelectrodesCurrAnalChem15(2019)249ndash276
[16] Sahu G Voltammetric behaviour of metronidazole at a composite polymer membraneelectrodeOrienJChem26(2010)81ndash86
[17] Yang Y YanW Guo YWang X Zhang F Yu L Guo C Fang G Sensitive and selectiveelectrochemicalaptasensorviadiazonium-couplingreactionforlabel-freedeterminationofoxytetracyclineinmilksamplesSensorsandActuatorsReports2(2020)1ndash7
[18] httpchemlnueduuamtechdeviceshtml(accesed21stJune2020)
56 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
AnimportantpartofanyanalysisthatsignificantlyaffectsthefinalresultsisthesamplepreparationThelowconcentrationofbiologicallyactivecompoundsandthepresenceofaccompanyingcomponentspreventdirectanalysisofthesamplewithcomplexmatrixcompositionTraditionalmethodsofliquidandsolid-phaseextractionhaveaplentyoflimitationssuchashighlytime-consumingprocedureslarge volume of samples expensive cartridges toxic organic solvents andchallenges in automating the process Therefore the application of extractiontechniquesemployinglowamountofsolvents(microextractionmethods)andthelow toxicity extractantes has become the main research direction in recentyears[12] Solid-phasemicroextraction(SPME)wasproposedbyPavlishinin1989[3]Onevariantofthismethodistousethinrodswithvariouspolymercoatingssuch
Application of microextraction techniques combined with chromatographic methods for the analysis of complex objects
VLADISLAVDEEVELENABESSONOVALIUDMILAKARTSOVA
InstituteofChemistrySaint-PetersburgStateUniversityUniversitetskyprospect26198504PeterhofSaint-PetersburgRussiahitchervmailru
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 57
AbstractThelowconcentrationofanalytesandthepreventionofthematrixinfluence requires a stage for extraction and concentration of thestudiedcompoundsTheclassicalmethodsofliquidandsolid-phaseextractionhavemanylimitationsthatpreventtheiruseinsomecasesMicroextraction techniques are becoming more widespread WestudiedthepossibilityofusingionicliquidstoextractpesticidesfromwatersampleswiththeirsubsequentHPLS-MSdeterminationTheinfluenceonthedegreeofextractionofsuchparametersasanatureofionicliquidsanddispersersolventtheiramountssaltconcentrationvolumeratioofionicliquidsandwatersampledilutionoftheionicliquidsextractwithmethanolwasperformedBesidesconditionsofsolid-phase microextraction of volatile organic compounds fromurine samples obtained from healthy donors and donors withprostate cancer have been found The analysis of volatile organiccompoundsbyGC-MSfollowedbychemometricprocessingallowedachievingahighvalueofbinaryclassificationaccuracy(91)
Keywordschemometricsdispersiveliquid-liquid
microextractionsolid-phase
microextraction
asdivinylbenzenepolydimethylsiloxanepolyacrylateandpolyethyleneglycolwhichappliedtothesurface[4]Thepolymersorbentisplacedintheequilibriumheadspaceaboveacondensedphaseofthesampleandthevolatilecompoundsareextracted Liquidmicroextractionconsistsofusingsmallamountsofliquid(extractant)inequilibriumwiththegasorliquidphaseofthesampleDispersiveliquid-liquidmicroextraction(DLLME)isavariantofliquidmicroextractionTheessenceofthemethodisasfollowsextractantisdissolvedinthephaseofadispersingsolventandthemixtureisrapidlyinjectedintothesamplevolume[5]Inthiscasethedispersing solvent is dissolved and a ldquocloudrdquo of extractant is formed A largesurfaceareacontributestomasstransferprocessesThecombinationofDLLMEwiththeuseof ionic liquids(ionic liquids)asextractantsreducestheharmfulimpactontheenvironment[6] Sothegoalofthisstudywastheapplicationofmicroextractionmethodsfortheanalysisofrealsamples
2Experimental
21Reagents
Deionizedwaterwas obtained at the AQUILON D 301 deionizer (Russia) Allchemicals and reagents (the highest commercially available purity) werepurchasedfromReachimBakerAcrosorganicsandSigmaAldrich
22Instrumentation
HPLCanalysiswascarriedoutusinganHPLCLCMS-8030(Shimadzu)withatriplequadrupole mass-selective detector with electrospray ionization Analysis ofvolatileorganiccomponents(volatileorganiccompounds)ofurinesampleswasmade by GCMS-QP2010 SE (Shimadzu) Chemometric data processing wasperformedusingRStudio
23DeterminationofvolatileorganiccompoundsinurinesamplesbyGC-MSmethod
ForSPMEofvolatileorganiccompounds inurinesamplewasusedfibercoatedwith a polydimethylsiloxane (PDMS) The volatile organic compounds wereextractedontofibercoatingfor20minat50degCThentheanalytesweredesorbedinto the gas chromatography for 4 minutes at a temperature of 250degCChromatographic separation was carried out on a HP-5 capillary column(30mtimes250μmtimes025μm)usingtemperatureprogrammingmodeThetempera-
ndash1tureofovenwasincreasedfrom50degCupto250degCatarateof10degCmin Tionsourcewas200degCMassspectrometrywasusedinSIMmode(mz=35ndash900)
58 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
24ConditionsforLCMSMSdeterminationofpesticides
Separation of pesticides was performed by HPLCMSMS with positiveelectrosprayionizationoncolumnZorbaxBonusRP35μm(21times100mm)with40mMammoniumacetateandmethanolasmobilephaseAandBrespectivelyThefollowinggradientelutionwasapplied20ndash85B(8min)85B(8ndash15min)85ndash95B(150ndash155min)95B(155minus180min)95ndash20B(180minus185min)
ndash1Thevelocityof themobilephasewas03mlmin Thevolumeof the injectedsamplewas 20 microlMS detection capillary voltage +45 kV spray gas velocity
3 ndash1 3 ndash13dm min flow rate and drying gas temperature 15 dm min and 250 degCrespectively
25Selectionofconditionsfordispersiveliquid-liquidmicroextractionofpesticides
The influence of the natures of ionic liquids ([C MIM][PF ] [C MIM][NTf ]4 6 6 2
[C MIM][BF ])andthedispersingsolvent(methanolacetonitrileacetone) the6 4
weightoftheionicliquids(0060ndash0200g)thevolumeofthedispersingsolvent(02ndash10ml)onthedegreeofpesticidesextractionwerestudiedTheinfluenceofthepH(5422)theconcentrationofNaCl(0040ndash0200g)andextractiontime(1-6min)wereinvestigated The effects of different ionic liquids and disperser solvents on DLLMEprocedures were investigated and optimized by using standard solutions ofpesticidesIndetailasolutionofionicliquidsinadispersingsolventwaspreparedand rapidly injected into the aqueous sample solution (2ml) followed bytreatment for 2 min in an ultrasonic bath cooling at ndash4degC for 10 mincentrifugationfor10minat3500rpmandcollectionofionicliquidsThewaterphasewas separatedandanalyzedbyHPLC-MSThe ionic liquidsextractwasdilutedinmethanolandanalyzedbyHPLC-MS
3Resultsanddiscussion
31Microextractionofpesticides
One of the important tasks of environmental monitoring is to control traceconcentrationsofpesticidesinwatersamplesTechniqueofcombiningseveralpesticideshasbecomemorewidespread inagriculture It allowsreducing thetotalconcentrationoftheappliedcompoundsandtodecreasetheadaptabilityofpathogens and insects Therefore the analysis of real samples requires apreliminarystageofselectiveanalytesextractionandconcentration ExtractinganddispersingsolventsarebothimportantinDLLMEofanalytesTheinfluenceofthenatureofthedispersingsolvent(methanolacetonitrileandacetone) and the extractant (imidazolium ionic liquids [C MIM][PF ]4 6
[C MIM][BF ]and[C MIM][NTf ])onthedegreeofextractionofpesticideswas6 4 6 2
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 59
studied This parameter was controlled by the residual concentration ofpesticidesinthewaterphaseafterextractionThebestresultswereobtainedforionic liquids [C MIM][PF ] as an extractant and acetonitrile as a dispersing4 6
solvent Thenextstepwastoselecttheamountofionicliquids(0060ndash0200g)andthevolume of acetonitrile (02ndash10 ml) It was found that the highest degree ofextractionofanalyteswasachievedbyusing020gofionicliquidsand03mlofacetonitrile It was shown that the degree of extraction of selected pesticides does notdependonpHofthewatersamplewhichconfirmsthepartitionmechanismofextractionThedegreeofextractionofcarbofosincreasedslightlywithanincreaseinthesaltconcentrationandreachmaximumbyweightto4(008g) ItisknownthatthehighviscosityofionicliquidshavehinderedtheprocessesofelectrosprayionizationDilutionofthesamplewithmethanolby3timesgivesthebestresult(thesignalintensitywas373to845ofthesignalwithoutionicliquids) ThustheconditionsofDLLME-ionicliquidsextractionofpesticidesfromwatersampleswere found The limits of detection for pesticideswere from007 to
ndash1019ngml thereproducibilityofpeakareaswerefrom3to5theextractionrecoverywascloseto100
32SPMEofvolatileorganiccompoundsfromurinesamples
Oneof the importantdirections is the search for criteriaofearlydiagnosisofcancer Obtaining characteristic profiles of volatile organic compounds fromurinesamplescanhelpdevelopanon-invasivemethodforearlydiagnosisofthedisease ForthiswestudiedtheinfluenceofanumberoffactorsonthetotalnumberofpeaksandthetotalpeakareaTheywerethetemperatureatwhichthevaporandcondensedphasesofurinewerebalanced(30ndash60degC)thepreheatingtimeofthesample(10ndash40min)NaClconcentration(30ndash133wv)andthesorptiontimeonthePDMScoating(5ndash30min) Anincreaseinthepreheatingtemperatureofthesampleto500degCledtoanincreaseinthenumberofsignalswhichdidnotchangewithafurthertempera-tureincreasingNextparameterwasthetimeofachievementequilibriumofthevapor and condensed phases The largest number of peaks was observed at40minbutthisgreatlyincreasedthetimeofanalysisandsowechose20minAlsowestudiedthedesaltingeffectofsodiumchlorideonefficiencyofextractionThebestvolatileorganiccompounds sorptionwasachievedbyadding133saltingagent It is also shown that thenumberofpeaksdidnot changeafter20minutesofsorption Thus to obtain the characteristic profiles of urine samples the followingconditionswereselected1333NaClwasaddedtotheurinesample(3ml)
60 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
heatingfor20minat50degCthensorptionofvolatileorganiccompoundsonPDMSfibercoatingat50degCfor20min Undertheselectedconditionsweobtainedvapor-phaseprofilesof52urinesamples (32 normal and 20 pathology) Prior to performing chemometricprocessing of chromatographic profiles of urine samples preliminary datapreparation is necessary [7] The baseline was removed and the peaks werealignedusingdynamictimewarpingwithcontrollingbymassspectra ThePCAmodelwasbasedon52aligned characteristicprofilesThere is asatisfactoryseparationofdataintotwoclustersinthescoresplotrelativetothefirstandsecondprincipalcomponent(Fig1) The original data set (52 samples) was randomly divided into calibration(13pathology21normal)andtest(7pathology11normal)setsThenthePLS-DAmodelwasbuiltusingthecalibrationsetanditspredictiveabilitywasevaluatedusingthetestsetTheprocedurewasrepeated100timesTheaveragevaluesofsensitivityspecificityandaccuracyinthiscasewere9594and91
4Conclusions
Thepossibilityofusingimidazoliumionicliquids([C MIM][PF ])asextractants4 6
forquantitativeextractionandconcentrationofpesticidesundertheconditionsofDLLMEisshownThedegreeofconcentrationwas28ndash33whichallowedreaching
ndash1thedetectionlimits(006ndash019ngml )belowthemaximumpermissibleconcen-trationThepossibilityofnon-invasivediagnosisofprostatecancerbySPMEofvolatileorganiccompounds inurine isshownChemometricprocessingofgaschromatographic profiles using PLS-DA and PCA methods allowed achievingclassificationaccuracyvaluesmorethan90
Acknowledgments
ThisworkwassupportedbytheRussianFoundationforBasicResearchprojectno18-53-80010BRICS_t and the Russian Science Foundations (Projects 19-13-00370) We are grateful to
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 61
Fig 1Scoresplotrelativetothefirstandsecondprincipalcomponent
Resource Education Center in Chemistry of St Petersburg State University for the providedequipment
References
[1] Rutkowska M Płotka-Wasylka J Sajid M Andruch V Liquidndashphase microextractionAreviewofreviewsMicrochemJ149(2019)103989
[2] JaliliVBarkhordariAGhiasvandAAcomprehensivelookatsolid-phasemicroextractiontechniqueAreviewofreviewsMicrochemJ152(2020)104319
[3] ArthurCLPawliszynJSolidphasemicroextractionwiththermaldesorptionusingfusedsilicaopticalfibersAnalChem62(1990)2145ndash2148
[4] SchmidtKPodmoreISolidphasemicroextraction(SPME)methoddevelopmentinanalysisof volatile organic compounds (VOCs) as potential biomarkers of cancer JMol BiomarkDiagn6(2015)1000253
[5] Mousavi L Tamiji Z Khoshayand MR Applications and opportunities of experimentaldesign for the dispersive liquidndashliquidmicroextractionmethod ndash A review Talanta190(2018)225ndash356
[6] MarcinkowskaRKoniecznaKMarcinkowskiLNamiesnikJKloskowskiAApplicationofionic liquids inmicroextractiontechniquesCurrent trendsandfutureperspectivesTrACTrendsAnalChem119(2019)115614
[7] WehrensRChemometricswithRMultivariateDataAnalysisintheNaturalSciencesandLifeSciencesBerlinSpringer2011
62 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Theself-assembledtwo-dimensionalmonolayers(2DSAMs)ofvariousmolecules(eg graphene [1] MoS [2] rubrene [3]) offer beneficial properties for the2
constructionofnano-electronic andnano-opticaldevicesThe topological andchemicalcharacterizationof2DSAMsiscrucialtogatherinformationaboutthearrangementofdepositedmoleculesandtheirinteractionwiththesubstrateThistaskrequiresanalyticaltechniqueswith(sub)nanometerspatialresolutionandup to single-molecular detection sensitivity Only few techniques meet therequirements and one of them is tip-enhanced Raman spectroscopy (TERS)whichcombinestheexcellentspatialresolutionofscanningprobemicroscopy(SPM) and chemical sensitivity of surface-enhanced Raman scattering (SERS)spectroscopy[45] TheSERS spectroscopyutilizesplasmonicmetal nanostructures to cause ahighlocalenhancementoftheelectricfieldintheirclosevicinityviathesurfaceplasmonresonance(SPR)effectThelocalelectricfieldcausesanincreaseofthe
The development of reference probe system for tip-enhanced Raman spectroscopy
MARTINKRA LMARCELADENDISOVA PAVELMATE JKA
DepartmentofPhysicalChemistryFacultyofChemicalEngineeringUniversityofChemistryandTechnologyPragueTechnickaacute516628Prague6CzechRepublicMartinKralvschtcz
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 63
AbstractThetip-enhancedRamanspectroscopy(TERS)isamodernanalyticaltechniquewithanoutstandingspatialresolutionandchemicalsensi-tivityTheseparametersmainlydependon the structural integrityand chemical purity of employed plasmonic scanning probe tipsUsuallyeachtipistestedbeforeTERSmeasurementsusingcommer-ciallyavailablereferencesamplesHowevertheirpriceandrelativelyshortexpirationdatemustbeconsideredwhenplanningaresearchbudgetWedevelopedaproceduretoproduceself-madereferenceprobesamplesfortestingTERStipsusingcopper(II)phthalocyanineonaAunanolayerwhichispreparedbythermalvacuumevaporationofAuonaSiwaferOurresultsshowthatthepreparedsystemenablesrepeateddetectionofwell-resolvedTERSspectraThecollectedTERSspectraandspectralmapsexhibitsomedegreeofvariabilitywhichmaybeduetovariousphoto-inducedprocessesanditmustbeconsi-deredwhileperformingTERSmeasurements
Keywordscopper(II)
phthalocyaninescanningtunnelling
microscopyresonanceRaman
spectroscopysurface-enhancedRaman
spectroscopytip-enhancedRaman
spectroscopy
Ramanscatteringfrommoleculesboundtothemetalby6ndash8ordersofmagnitude[6]TheenhancementallowsSERSspectroscopytobeusedforsingle-moleculardetectionHoweverRamanmicroscopeshave limitedspatialresolutionbythelightdiffractionwiththeachievableresolutionbeingaroundhalfoftheexcitationwavelengthOntheotherhandthespatialresolutionofSPMtechniquesislimitedonlybythedimensionsoftheapexofthescanningprobetipwhichmayevenbeatomicallysharpByutilizingSPMtipandsubstratemadefromplasmonicmetalsan artificial ldquohotspotrdquomay be createdwith its position and dimensions beingdefinedbythetipItopensthepossibilitytocollectstronglyenhancedRamanspectrafromtheareapreciselylocalizedbellowthetipandthusovercometheopticaldiffraction limitTheartificialhotspotmayberelocatedbymovingthesamplebelowthetipwhichisthefoundationofTERSmapping[578] A successful TERS experiment requires an optimal combination of variousparameters the most important of which are tip sharpness and purity [7]Areferencesampleconsistingofaflatplasmonicnanolayerwithattachedprobemolecules is frequently used to check the state of the tip before using it forexperiments Unfortunately commercially available TERS standards areexpensiveandhaveanexpirationdateofseveralmonths Thegoalofthisstudywastofindapreparationprocedurewhichwouldbeableto produce cheap reference samples for repeated detection of intense TERSspectraAcombinationofaAunanolayeronaSisubstratepreparedbythermalvacuumevaporationwithadsorbedcopper(II)phthalocyanine(CuPc)whichisamoleculewithhighRamancross-sectionwastested[910]Copper(II)phthalo-cyanine known as phthalocyanine blue is a synthetic blue pigment and isfrequently used in paints It has been studied as a potentialmaterial for theconstruction of organic solar cells and other photoelectronic devices [11] AsindicatedbyitscolourCuPcexhibitsseveralabsorptionbandswithinthevisibleregion The effects of a transition to excited electronic states and subsequentluminescencemaybeobservable inRamanmeasurements[12]Au isahighlysuitablemetalforthesampleasitisboththermallyandelectricallyconductivewhichlimitsthelocalheatingofthesampleduringthemeasurementsandenablesthe use of scanning tunnelling microscopy (STM) for tip-surface interactionfeedback[13]
2Experimental
21Reagentsandchemicals
Thesubstrateforthesamplewaspreparedbythermalvacuumevaporationofgoldonsilicon(100)waferFirsta5nmthickCradhesionlayerwasdepositedon
ndash1thewaferfollowedby100nmofAuThedepositionratewas4nmmin forCrand ndash18nmmin for Au The base pressure of the evaporation system was below
ndash65times10 mbarFollowingthepreparationproceduredescribedbyJiangetal[14]
64 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
thecleansubstratewasimmersedintoasaturatedsolutionofCuPc(˃99SigmaAldrich USA) in dimethylformamide (˃98 Lach-ner CZ) for at least 12h atambienttemperatureSubsequentlythesamplewasremovedfromthesolutionrinsedwithMilli-Qwaterandmethanol(paPentaCZ)anddriedwithair
22Instrumentation
TheRamanSERSandTERSspectrawererecordedusingRamanspectrometerInVia Reflex (Renishaw UK) equipped with lasers emitting at two differentexcitation wavelengths 633nm (136mW max power output) and 785nm(204mWmaxpoweroutput)Thespectrometerhasathermoelectricallycooled
ndash1CCDdetectorwithaspectralresolutionof2cm and4microscopeobjectiveswith5times20times50timesand100timesmagnitudeForTERSexperiments the laserbeamwasredirectedtotheSPMplatformInnova-IRIS(BrukerUSA)viaasystemoflightguidesElectrochemicallyetchedAuTERS-STMtips(BrukerUSA)wereusedforallTERSmeasurements The spectra were processed using the Spectragryph software (F MengesldquoSpectragryph - optical spectroscopy softwarerdquo Version 1214 2020httpwwweffemm2despectragryph) Using this software all collectedspectra were treated by a Savitzky-Golay noise filter automatic baselinecorrectionspikeremovalandpeaknormalization
3Resultsanddiscussion
31Ramanmeasurementsofcrystallinecopper(II)phthalocyanine
AtfirsttheRamanspectraofpureCuPcwerecollectedtoprovidereferencedatawhileusingboth633and785-nmexcitationlasers(Fig1)Bothspectraexhibit
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 65
Fig 1RamanspectraofCu(II)phthalocyanineincrystallineformmeasuredat633(top)and785-nm(bottom)excitationThespectraareoffset
ndash1vibration bands in the 500ndash1600 cm region with slight differences in theirintensityratiosThe633-nmexcitationallowstheobservationofadditionalbands
ndash1inthe2000ndash3000cm region(onaluminescencebackground)whichoriginatefrom the resonance Raman effect as the excitation energy overlaps with theQ-bandofCuPc[12]MoreovertheprocessofelectronicexcitationmaylowertheD symmetry of CuPc during resonance Raman scattering and previously4h
forbiddenbandsmaybecomeobservable[15]Theluminescentbackgroundhasandash1maximumaround2200cm whichcorrespondstoamolecularemissionbandat
735nmEventhoughthespectrameasuredwiththe785-nmlaserlinedonotexhibitapparentresonanceenhancementapre-resonanceRamanenhancementmayoccur
32Surface-enhancedRamanmeasurementsofcopper(II)phthalocyaninelayeronagoldsubstrate
The prepared sample of CuPc on a Au layer was analysed using the Ramanmicroscope Both excitation laserswere used to obtain SERS spectra (Fig 2)whichwerecomparedtothespectraofapurecrystallineCuPc Thepositionsof bands in SERS spectra closelymatch their positions in thespectraofbulkCuPcHoweveraslightshiftofsomespectralbandsisobservable
ndash1(eg1528rarr1532cm )whichmaybeattributedtotheinteractionbetweenCuPcand the Au substrate The disappearance of luminescence background andresonance-enhancedbandsinthespectrumat633-nmexcitationalsosuggeststhemolecule-metalinteractionandthetransferofenergyfromCuPcmoleculestothesubstrateMoreovertherearevariationsintherelativeintensitiesofbands
ndash1whichdependontheexcitationenergyegthebandat1309cm isenhancedinSERSspectraatthe785-nmexcitationwhencomparedtothespectraofpureCuPcorevenSERSspectraat633-nmexcitation
66 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2SERS spectra of Cu(II) phthalocyanineon aAu layermeasured at 633 (top) and785-nm(bottom)excitationThespectraareoffset
33Tip-enhancedRamanmeasurementsofcopper(II)phthalocyaninelayeronagoldsubstrate
SERS microspectroscopy is a diffraction-limited technique as it provides anaveragedinformationaboutmoleculesintheilluminatedareaofseveralsquaremicrometers Meanwhile TERS spectra are collected from an area of tens ofnanometers and they contain specific information about the local moleculararrangement topography of the underlyingmetal and properties of the localelectric field between the tip and the substrate Therefore a higher spectralvariabilityshouldbeexpected Several TERS mapping experiments were carried out using both 633 and785-nmexcitationwithvaryingexperimentalparameterssuchasthenumberanddistance betweenmeasured points acquisition time number of acquisitionslaserpoweretc TheTERSspectrameasuredat633-nmexcitationexhibited lowersignal tonoiseratioandreproducibilityAsaconsequenceTERSmappingwasimpossibleandonlyafewone-pointTERSspectrawereobtained(Fig3) TheTERSspectracollectedat785-nmexcitationcontainedahighernumberofwell-resolvedbandsMoreoverthespectrawerestableintimeandsotheTERSmappingwaspossibleThe twopresentedTERSspectraareaveragesofTERSmaps which contained 16 and 80 points with 600 and 300-nm spacingrespectively(Fig4) Thespectracollectedusingbothexcitationwavelengthsexhibitavariabilityinrelative intensities and positions of bands between themeasured points ThevariabilitymaybeattributedtothelocalorientationofCuPcmoleculesbetweenthe tip and the Au surface and the properties of strongly enhanced andnon-homogeneous electromagnetic field which depend on the tip-surfacedistancetheirmorphologyandrelativepositionMoreovertheusedexcitation
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 67
Fig 3TwoexamplesofTERSspectraofCu(II)phthalocyaninemeasuredat633-nmexcitationThespectraareoffset
wavelengthsareclosetoabsorptionbandsofCuPcandthestrongelectricfieldmay give rise to photo-induced effects These effects include the electronicexcitationofCuPctohigherstateschargetransferbetweentheCuatomandthephthalocyaninering ionizationof themoleculeand formationofradicalsThephoto-inducedprocessesarelikelytoplayabiggerroleinTERSspectradetectedat633-nmexcitationduetotheoverlapwithQ-bandofCuPcwhichmaybethecauseoftheirlowersignaltonoiseratioandreproducibility
4Conclusions
Thedeveloped referenceprobe systemofCuPc adsorbedon aAu surfacehasprovedtobesuitablefortheintendeduseasitenabledthedetectionofintenseandwell-resolvedSERSandTERSspectraTheAulayerpreventsoverheatingofthesampleandallowsfortheuseofSTMTheSERSspectrawereinagoodmatchwith thespectraofpureCuPcAslight shiftof somebandsandchange in theluminescent background indicated the interaction between CuPc and the AusurfaceTheTERSexperimentsresultedinspectralmapswithhighintensitiesofindividualspectraIncreasedvariabilitybetweenmeasuredpointswasobservedPossiblesourcesofthevariabilityarephoto-inducedprocessesthatmayoccurinthestronglyenhancedelectricfieldTheseeffectsareaknownfeatureofTERSmeasurements and they offer valuable insight into the photophysics andphotochemistryofCuPcinteractingwiththeAusurfaceThedependenceofTERSspectra on experimental parameters and the preparation procedure of thereferencesampleshouldbefurtherstudied
Acknowledgments
ThisworkwassupportedfromthegrantofSpecificuniversityresearchndashA2_FCHI_2020_039
68 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 4TwoexamplesofaveragedTERSspectraofCu(II)phthalocyaninemeasuredwith785-nmexcitationThespectraareaveragesfromTERSmapsincluding16(top)and80(bottom)measuredpointsThespectraareoffset
References
[1] Mas-BallesteRGomez-NavarroCGomez-HerreroJZamoraF2DmaterialstographeneandbeyondNanoscale3(2011)20ndash30
[2] ZengHCuiXAnopticalspectroscopicstudyontwo-dimensionalgroup-VItransitionmetaldichalcogenidesChemSocRev44(2015)2629ndash2642
[3] SchultzJFLiLMahapatraSShawCZhangXJiangNDefiningmultipleconfigurationsofrubreneonaAg(100)surfacewith5A spatialresolutionviaultrahighvacuumtip-enhancedRamanspectroscopyJPhysChemC124(2020)2420ndash2426
[4] WhitemanPJSchultzJFPorachZDChenHNJiangNDualbindingconfigurationsofsubphthalocyanineonAg(100)substratecharacterizedbyscanningtunnelingmicroscopytip-enhanced Raman spectroscopy and density functional theory J Phys Chem C 122(2018)5489ndash5495
[5] ShaoFZenobiRTip-enhancedRamanspectroscopyprinciplespracticeandapplicationstonanospectroscopicimagingof2DmaterialsAnalBioanalChem411(2019)37ndash61
[6] ArocaRSurface-EnhancedVibrationalSpectroscopyHobokenWiley2006[7] KumarNMignuzziS SuWRoyDTip-enhancedRamanspectroscopyprinciplesand
applicationsEPJTechInstrum2(2015)9[8] BailoEDeckertVTip-enhancedRamanscatteringChemSocRev37(2008)921ndash930[9] BovillAJMcConnellAANimmoJASmithWEResonanceRamanspectraofα-copper
phthalocyanineJPhysChem90(1986)569ndash575[10] Shaibat MA Casabianca LB Siberio-Perez DY Matzger AJ Ishii Y Distinguishing
polymorphsofthesemiconductingpigmentcopperphthalocyaninebysolid-stateNMRandRamanspectroscopyJPhysChemB114(2010)4400ndash4406
[11] SzybowiczMRunkaTDrozdowskiMBałaWGrodzickiAPiszczekPBratkowskiAHightemperaturestudyofFT-IRandRamanscatteringspectraofvacuumdepositedCuPcthinfilmsJMolStruct704(2004)107ndash113
[12] CaplinsBWMullenbachTKHolmesRJBlankDAFemtosecondtonanosecondexcitedstatedynamicsofvapordepositedcopperphthalocyaninethinfilmsPhysChemChemPhys18(2016)11454ndash11459
[13] SaccoAImbraguglioDGiovannozziAndreaMPortesiCRossiAMDevelopmentofacandidatereferencesampleforthecharacterizationoftip-enhancedRamanspectroscopyspatialresolutionRSCAdv8(2018)27863ndash27869
[14] JiangSChenZChenXNguyenDMatteiMGoubertGVanDuyneRPInvestigationofcobaltphthalocyanineatthesolidliquidinterfacebyelectrochemicaltip-enhancedRamanspectroscopyJPhysChemC123(2019)9852ndash9859
[15] MelendresCAMaroniVARamanspectraandnormalcoordinateanalysisoftheplanarvibrationsofironphthalocyanineJRamanSpectrosc15(1984)319ndash326
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 69
1Introduction
The aim of this work has been develo-pmentofanewvoltammetricmethodforthe determination of 23-dimercapto-1-propane-sulfonic acid (DMPS) Fig 1Investigationwasdonetoobtainrelevantinformation about complexingbehaviorofDMPStowardsleadions Lead is one of heavy metals which can cause irreversible neurologicalproblems [1 2]DMPS is a synthetic antidotewith two thiol groups used fortreatmentofpoisoningbyheavymetals[3ndash6]StrongcomplexingpropertieshighwatersolubilityandnegligiblesideeffectsarethemostimportantadvantagesofDMPS[47]
Fig 1 Chemical structure of 23-dimercap-to-1-propane-sulfonicacid
Determination of heavy metal poisoning antidote 23-dimercapto-1-propanesulfonic acid using silver solid amalgam electrode
ab ab bc bMARTACHOIN SKA VOJTE CHHRDLICKA BEATRIZRUIZREDONDO JIR IBAREK aTOMA S NAVRATIL
a JHeyrovskyacuteInstituteofPhysicalChemistryoftheCzechAcademyofSciences Dolejškova21553182thinsp23Prague8CzechRepublicmartachoinskagmailcomb UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova20308128thinsp43Prague2CzechRepublic
c UniversityofValladolidPlazadeSantaCruz847002ValladolidSpain
Abstract23-Dimercapto-1-propane-sulfonic acid (DMPS) was investigatedusingdirectcurrentvoltammetry(DCV)differentialpulsecathodicstrippingvoltammetry(DPCSV)differentialpulseanodicstrippingvoltammetry(DPASV)andeliminationvoltammetrywithlinearscan(EVLS)atapolished(p-AgSAE)andatameniscusmodifiedsilversolidamalgam electrode (m-AgSAE) EVLS confirmed two consecutivereductions with coupled protonelectron transfer VoltammetrictitrationsofDMPSwithPb2+provedcomplexformationwithlimits
minus1ofquantification(LOQs)anddetection(LODs)03and01micromolL atminus1m-AgSAE and 08 and 03micromolL at p-AgSAE respectively
Determination of DMPS in commercial drug Dimaval and humanurine samples confirmed practical applicability of the developedmethod
Keywordscathodicstripping
voltammetry23-dimercapto-1-pro-
panesulfonicacideliminationvoltammetry
withlinearscansilversolidamalgam
electrodeunithiol
70 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Voltammetry was chosen as a determination technique due to its highsensitivity and selectivity speed low costs [8] Thiol groups in DMPS can beoxidatively chemisorbed on solid amalgam electrode It can be used as anaccumulation step for cathodic strippingvoltammetry [9ndash11]Moreover solidamalgamelectrode (SAE)was chosen as theworking electrodebecauseof itspropertiesashighsignaltonoiseratiowidepotentialwindowandabilitytoreachlowlimitsofdetection(LOD)[12ndash13]
2Experimental
21Reagentsandchemicals
Allsolutionswerepreparedusingdeionizedwater(Milli-Q-GradientMilliporendash1PragueCzechRepublic)withconductivitylt005microScm Britton-Robinsonbuffer
solutionspHrangefrom2to12werepreparedbymixingtheproperamountsof02MNaOH(alkalinesolution)andof004MH BO 004MH PO and004M3 3 3 4
CH COOH(allLachemaCzechRepublic)acidicsolutionTheacidicsolutionwas3
preparedbydissolutionof1235gofH BO pa088mLofH PO (85)paand3 3 3 4
1435mLofCH COOH(99)pa in500mLofdeionizedwaterThealkaline3
solution was prepared by dissolution of 3995 g of NaOH pa in 500mL ofdeionizedwater(allLachemaCzechRepublic) StocksolutionofDMPSwaspreparedbydissolving10mgofsolid23-dimer-capto-1-propanesulfonic acid monohydrate pa (Merck Czech Republic) in100mLofdeionizedwaterForthepreparationofthemodelsamplesolutiononecapsuleofdrugDimaval(HeylGermany)contains100mgofDMPSwasdissolved
ndash1in10LofdeionizedwatertotheDMPSconcentrationof0531mmolL Twomodel samplesofDimavalwerepreparedbydilutionof theabove-mentioned
ndash1solutionwithBritton-Robinsonbuffersolutiontoconcentrations10micromolL andndash1of 10micromolL respectively Urine model samples were prepared by mixing
Britton-Robinsonbuffer solutionwithurine samples obtained fromvolunteer(manhealthy30yearsold)inratio11SamplepHwasadjustedbyadditionof
ndash1proper amount of 02molL NaOH Before each measurement oxygen wasremoved for 5minbynitrogenbubbling (purity class 46MesserTechnogasPragueCzechRepublic)
22Instrumentation
Measurementswereperformedusingtwotypesofworkingelectrodesmeniscusmodified silver solid amalgam electrode (m-AgSAE working surface of
20382plusmn0025mm α lt 005) and polished silver solid amalgam electrode2(p-AgSAEworkingsurfaceof0196plusmn0015mm αlt005)Ag|AgCl|3MKClwas
used as the reference electrode andplatinumwire (Oslash1mm)wasused as theauxiliary electrode (both from Elektrochemicke detektory Czech Republic)Measurementswereperformedatlaboratorytemperature(25plusmn2degC)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 71
The pH was measured using pH-meter Jenway 3505 with combined glasselectrodetype924001(BibbyScientificLimitedUK)Voltammetricmeasure-ments were performed using the computer-controlled Eco-Tribo Polarograph(Polaro-Sensors Czech Republic) Software used for measurements wasMultiElChem 33 forWindows XP7810 (J Heyrovsky Institute of PhysicalChemistryoftheCzechAcademyofSciencesCzechRepublic)
3Resultsanddiscussion
OptimumconditionsformeasurementswereobtainedbyseriesofmeasurementinwiderangeofpHvaluesandtestingvariouscleaningproceduresOptimumpotentialof accumulation (E ) and timeof accumulation (t )ofDMPSwereacc acc
adjusted for differential pulse cathodic stripping voltammetry (DPCSV) atp-AgSAEandm-AgSAE ThedependencebetweenpeakheightandconcentrationofDMPSatp-AgSAEhasalogarithmicshapewhichcorrespondstotheaccumulationprocessattheelectrodesurfaceThelineardependencewasobservedintheDMPSconcentra-
ndash1 ndash1tionsfrom03micromolL to20micromolL Peakshifttowardsnegativepotentialwithincreasing concentration of DMPS corresponds to the metal-thiol bond andinfluenceofelectrodesurfacestructureonthisbondOnthecontrarysignalsonm-AgSAEweremorestableandthedependencebetweenconcentrationofDMPSandsignalwasalmostlinearinwholetestedrangeofconcentrations The developedmethodwas tested inmodel samples of Dimaval and urineFoundamountsofDMPSwereingoodagreementwithdeclaredcontentsusingbothelectrodesHowevertherepeatabilityofsignalsregisteredusingp-AgSAEinurinesamplesweresignificantlyworsethanthoseinDimavalsamplesItcanbecausedbycomplicatedbiologicalmatricesandfoulingeffectsofurine ELSV measurements confirmed two consecutive reductions of DMPS inadsorbed state At m-AgSAE signal were at about minus415 mV and minus440 mVrespectively and at p-AgSAE at about minus790 mV and minus830 mV respectively
ndash1 ndash1Reductionsatm-AgSAEatthescanratesfrom80mVs to640mVs havebeencontrolledbyakineticprocessatminus400mV Inanodicscansonm-AgSAEonlyonepeakwasvisibleatabout‒390mVItcorrespondswiththeoxidationofmercuryelectrodesurfaceontheelectrodeinthepresenceofDMPSandwithdiffusionfromthebulksolutionofproductsAtp-AgSAEnosignificantanodicsignalwasfound ThelastpartoftheresearchrevealedvoltammetricbehaviorofDMPSinthe
2+presence of Pb Voltammetric titration was investigated by DPCSV anddifferential pulse anodic stripping voltammetry (DPASV) during consecutive
ndash1 ndash1additionsof1mmolL ofPb(NO ) into100micromolL DMPSsolutioninacetate3 2
bufferofpH50InabsenceofPb2+ontheelectrodesurfaceHg(DMPS)complexisformedduringtheaccumulationstepDuringtheanodicscanthereisonlyonewelldevelopedreductionpeak(Fig2A)
72 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ndash1Fig 2DPCSandDPASvoltammogramsof10micromolL ofDMPSinacetatebufferpH=5correspond-2+ingto[Pb DMPS]ratiosof(A)01(B)11and(C)21Uppercurvecorrespondstothecathodic
scanE =0mVt =15sLowercurvecorrespondstoreverseanodicscanwitht =15satacc acc accndash1E =minus1000mVν=20mVs (Ref[16])acc
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 73
2+ WhenPb DMPSratioisequalto11twooxidationandthreereductionpeakswereregistered(Fig2B)PeakA ataboutndash300mVcorrespondstoformationofred
0Pb(DMPS)complexThiscomplexwasfurtherreducedtothePb (Hg)atabout2+ndash500mV(C )ReductionpeakoffreePb wasalsoregistered(B )Oxidationred red
peaksA andB correspondtoreverseprocessesandC isnotpresentbecauseox ox ox
nofreeDMPSispresentinthesolutionAtratio21thereisnofreeDMPSinthesolution however excess of lead ions As a consequence A and B peakred ox
2+increasedIncreaseofB correspondstothedepositionofPb duringtheaccu-ox
mulationstepPeaksBredandCredremainedpracticallyunchanged(Fig2C) Voltammetric titrationconfirmed themechanismof formationcomplexesofPb(DMPS)Hg(DMPS)andPb(Hg)Italsoconfirmedpossibilityofdetermination
2+Pb andDMPSinthesamesolution
4Conclusions
ValidationinmodelsampleofdrugDimavalandhumanurinespikedwithDMPSconfirmed that this method can be used for clinical purposes Voltammetric
2+titration of DMPS by Pb ions proved that it can be used for simultaneousdetermination of the drug and heavy metal ions in human urine Moreoverobtained LODs were two orders lower than those in the previously reportedvoltammetricmethod[14](Table1)
Acknowledgments
ResearchwascarriedoutwithintheframeworkofSpecificUniversityResearch(SVV260560)TheauthorsthanktheCzechScienceFoundation(GACRprojectNo20-01589S)
References
[1] AnHHLuchakMCopesRLeadtoxicityAsystematicreviewofrecentlypublishedcasesClinToxicol53(2015)757ndash758
[2] KimYLustMRKreimerbirnbaumM23-Dimercaptopropane-1-sulfonate(DMPS)inthetreatmentoflead-poisoningFasebJ2(1988)A1820ndashA1820
[3] AposhianHVDMSAandDMPS ndashwater-solubleantidotesforheavy-metalpoisoningAnnuRevPharmacol23(1983)193ndash215
74 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Table 1ComparisonofvoltammetricmethodsforDMPSdetermination(LDR
Method Workingelectrode Lineardynamic LOQ LOD Refminus1 minus1 minus1 rangemicromolL micromolL micromolL
LSV glassy-carbonelectrode 18ndash140 41 14 [14] modifiedwithmulti-walled 260ndash690 carbonnanotubes DPCSV p-AgSAE 03ndash20 08 03 thisworkDPCSV m-AgSAE 01ndash10 03 01 thiswork 10ndash100
[4] BjorklundG Crisponi G Nurchi VM Cappai R Djordjevic AB Aaseth J A review oncoordinationpropertiesof thiol-containingchelatingagents towardsmercury cadmiumandleadMolecules24(2019)3247
[5] DonnerAHrubyKDMPSinthetreatmentofacuteandchronicheavy-metalpoisoningActaMedAust14(1987)10ndash10
[6] DonnerAHrubyKPirichKKahlsPSchwarzacherKMeisingerVDimercaptopropan-sulfonate(DMPS) inthetreatmentofacute lead-poisoningVetHumToxicol29 (1987)37ndash37
[7] Blanusa M Varnai VM Piasek M Kostial K Chelators as antidotes of metal toxicityTherapeuticandexperimentalaspectsCurrMedChem12(2005)2771ndash2794
[8] BarekJMoreiraJCZimaJModernelectrochemicalmethodsformonitoringofchemicalcarcinogensSensors-Basel5(2005)148ndash158
[9] Josypcuk B FojtaM Yosypchuk O Thiolatemonolayers formed on different amalgamelectrodesPartIIPropertiesandapplicationJElectroanalChem694(2013)84ndash93
[10] YosypchukBMarecekVPropertiesofthiolatemonolayersformedondifferentamalgamelectrodesJElectroanalChem653(2011)7ndash13
[11] Alvarez JMF SmythMRCathodic strippingvoltammetryofpyridine-2-thiolandsomerelated-compoundsAnalyst114(1989)1603ndash1605
[12] DanhelABarekJAmalgamelectrodesinorganicelectrochemistryCurrOrgChem15(2011)2957ndash2969
[13] Fadrna R Polished silver solid amalgam electrode Further characterization and appli-cationsinvoltammetricmeasurementsAnalLett37(2004)3255ndash3270
[14] ZiyatdinovaGKGrigorevaLVBudnikovGKElectrochemicaldeterminationofunithioland lipoic acid at electrodesmodifiedwith carbonnanotubes J Anal Chem64 (2009)185ndash188
[15] HrdlickaVChoinskaMRedondoBRBarekJNavratilTDeterminationofheavymetalpoisoning antidote 23-dimercapto-1-propanesulfonic acid using silver solid amalgamelectrodeElectrochimActadoiorg101016jelectacta2020136623
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 75
Fig 1Structureofcanagliflozin
1Introduction
Canagliflozin is a selective sodium-glucosecotransportertype2inhibitorused for the treatment of type 2 dia-betes mellitus Canagliflozin inhibitssodium-glucose cotransporter type 2present in proximal tubules of the
Canagliflozin oxidation study using electrochemical flow cell and comparison with hydrogen peroxide oxidation
a a bFILIPVYMYSLICKY TOMA S KR IZ EK JAKUBHER T
a DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublicfvymyslickygmailcomb ZentivaGroupasUKabelovny13010237Prague10CzechRepublic
AbstractBystandardstheeffectonoxidationofanactivesubstanceistestedusinghydrogenperoxidesolutionatelevatedtemperatureinastresschamberfor1ndash7daysAnalternativewaytostudytheeffectofoxida-tion on an active substance is to use an electrochemical flow cellSolutionwith active substance flows at low flow rate into a smallreactorwheretheactivesubstanceisoxidizedonworkingelectrodesurfaceTheelectrolytestreamwiththeoxidizedactivesubstanceisthen directed to the sample collector Products of electrochemicaloxidationareanalyzedbyhighperformanceliquidchromatographywithultravioletndashvisiblespectrophotometrydetectionCanagliflozinhasbeenusedbecauseitsmaindegradationpathwayisoxidationThedesign of experiments approach was used to explore the experi-mentalspaceandoptimizeexperimentalconditionsofoxidationTheresultsoftheoxidationstudyperformedintheelectrochemicalflowcellwerestatisticallycomparedwiththeresultsofastandardstudyusinghydrogenperoxidesolutionThemostsuitableconditionsforelectrochemical oxidation were found Electrochemical oxidationproducedcomparableamountsofimpuritiesaschemicaloxidationwithhydrogenperoxide
KeywordscanagliflozindesignofexperimentselectrochemicalflowcellHPLCoxidation
76 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
kidneywhichrestrictsglucoseabsorptioninthekidneytherebyincreasingtheurinaryexcretionofglucoseandloweringthelevelofglucoseintheblood[1]TheformulaofcanagliflozinisC H FO SthestructureofcanagliflozinisinFig1The24 25 5
IUPAC name of canagliflozin is (2S3R4R5S6R)-2-[3-[5-(4-fluoro-phenyl)-thiophen-2-ylmethyl]-4-methyl-phenyl]-6-hydroxymethyltetrahydro-pyran-345-triol[2]CanagliflozinisawhitepowderinsolubleinwaterbutverysolubleinorganicsolventslikemethanolordimethylsulfoxideCanagliflozinissoldundertradenameINVOKANA Manyauthorshavestudiedtheelectrochemicalpropertiesofactivesubstancesin the literature One example is the study of electrochemical behaviour andoxidationofbromhexineThesepropertieswerestudiedusingdifferentialpulsevoltammetryandcyclicvoltammetryonacarbonelectrodeTheresultsofelectro-chemicalmethodswerecomparedwithhighperformanceliquidchromatography(HPLC)analysis[3]Anotherexampleisthestudyofelectrochemicalbehaviourand degradation study performed on the active substance atomoxetineDegradation was studied using differential pulse voltammetry and cyclicvoltammetryonacarbonelectrodeTheresultswerealsocomparedwithHPLCanalysis[4]Electrochemicalmethodsareusedmainlytostudythemechanismofoxidationbutinthisworktheelectrochemicalmethodwasusedtodegradetheactivepharmaceuticalingredient In the stability studies of active pharmaceutical ingredient properties theinfluence of temperature pH light and oxidation is studied [5] By TheInternationalCouncilforHarmonisationofTechnicalRequirementsforPharma-ceuticals forHumanUse (ICH) standards the influenceof oxidationon activepharmaceutical ingredient is studied using hydrogen peroxide at roomtemperatureorincreasedtemperatureinthestresschamberduring1ndash7days[6]Analternativewaytostudytheinfluenceofoxidationonactivepharmaceuticalingredient is using electrochemical flow cellwhere an electrolytewith activepharmaceuticalingredientisdrivenbylowflowrateintothesmallreactorInthesmallreactortheactivepharmaceuticalingredientisoxidizedonthesurfaceoftheworkingelectrodeThestreamofelectrolytewithoxidizedactivepharma-ceuticalingredientisdriventothesamplecollectorProductsofelectrochemicaloxidationareanalysedbyHPLCUVVISThedesignofexperimentsapproachwasusedfordevelopmentofanalternativemethodofoxidationofcanagliflozinusinganelectrochemical flowcellThedesignofexperimentsapproachwasusedtoexplore the experimental space of the method and to find the optimalexperimentalconditionsofelectrochemicaloxidationofcanagliflozin
2 Experimental
21Materialandreagents
Canagliflozin(ZentivaCzechRepublic)999methanol(HoneywellGermany)98ammoniumdihydrogenphosphate(Sigma-AldrichJapan)35ortho-phos-phoricacid(PentaCzechRepublic)25ammonia(LachnerCzechRepublic)30hydrogenperoxide (LachnerCzechRepublic)water forchromatography
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 77
Fig 2Schemeoftheelectrochemicalflowcell(1)input(2)workingelectrode(3)gasket(4)refe-renceelectrode(5)counterelectrode
wasobtainedbypurifyingdemineralisedwaterusingMilliporetypeSynergyUVpurificationinstrument
22Instruments
An Agilent 1290 HPLC system (Agilent Technologies Germany) with highpressure pump autosampler thermostat and DAD detector was used for allexperimentsThePinnacleDBbiphenylcolumn(100times21mm19micromRestekUSA)wasusedforseparationIntheHPLCmethod10mMammoniumdihydrogenphosphatebufferpH=25wasusedascomponentAandmethanolascomponentBofthemobilephaseThegradientprogramwassetasfollowst(min)B01555010551790229023152515Theflowrateofthemobilephasewas
ndash104mlmin and the injection volume was 2μl The detector operated at awavelengthof220nmTheautosamplertemperaturewassetat20degCandthecolumntemperatureat60degCTheEmpowersoftwarewasusedforevaluationForelectrochemicaloxidationelectrochemicalflowcellfromALS(Japan)wasusedGlassycarbonelectrode(=6mm)andsilversilverchlorideelectrodewereusedasworkingandreferenceelectroderespectivelyTheschemeofelectrochemicalflowcellisinFig2ElectrodeswereconnectedwithpotentiostatPalmSens3fromPalmsens (Netherlands) AnElmasonic S15Hultrasonic bath fromElma (Ger-many)wasusedforsamplepreparationForpHmeasurementspHmeterJenway3540fromJenway(UnitedKingdom)wasused
3Resultsanddiscussion
AtthedevelopmentofthemethodforthestudyofcanagliflozinoxidationusingelectrochemicalflowcellitwasfirstnecessarytofindtheoptimalconditionsofelectrochemicaloxidationThedesignofexperimentsapproachwasusedChosen
78 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ndash1Fig 3Cyclicvoltammogramofcanagliflozin(concentrationofcanagliflozin11mgml electrolyte300 mM ammonium dihydrogen phosphate pH = 40 and methanol (11 vv) and scan rate
ndash1001Vs )
independentvariablesandtheirlevelswereconcentrationofelectrolyte(100200300mM)pHofelectrolyte(406080)cellsize(50100200500microm)and
ndash1flowrate(0102504mlh )ThereducedcombinatorialdesignwasusedIntheModde12 software aworksheet containing 11 experimentswas created Theworkingpotentialof12Vwasselectedbasedoncyclicvoltammetryofcanagli-flozininFig3Fromthisfigureitcanbeseenthattheoxidationofcanagliflozinoccursintheregionfrom11Vto14VAllexperimentswereperformedwith
ndash111mgml canagliflozin samples The glassy carbon electrodewas used as aworkingelectrodeandthesilversilverchlorideelectrodewasusedasareferentelectrode The canagliflozin samples oxidized in the electrochemical flow cellunder theexperimentalconditionsgivenby theworksheetweremeasuredbyHPLCwithUVVISdetectionDependentvariablespeakareasofimpuritiesandpercentage of peak areas of impurities obtained from chromatograms wereevaluated by the partial least squaremethod in theModde 12 software Thevariableimportanceintheprojectionplottoolwasusedforinterpretationofthedata as a whole The significance values of the independent variables were
ndash1evaluatedbufferpH=137flowrateof125mlh bufferconcentration061mMandcell size04micromFromthis tool itwasconcluded that theelectrochemicaloxidationofcanagliflozinisthemostaffectedbythepHoftheelectrolyteandflowrateoftheelectrolyteUsingtheoptimizertoolthemostsuitableconditionsfor
ndash1the oxidation of canagliflozin were evaluated flow rate 01 ml h 300 mMammoniumdihydrogenphosphate pHof electrolyte40 and cell size500micromUsingonefactoratthetimeapproachthedependenceofthecellsizeonthetotalsumofimpuritieswastested(Fig4A) Itisvisiblefromthegraphthatthesmallerthecellweusethemoreoxidationproducts are formed Based on the graph the most suitable conditions were
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 79
Fig 4(A)Optimizationofelectrochemicaloxidationconditionsdependenceofsumofimpuritiesoncellsize(B)Chromatogramofasampleoxidizedundermostsuitableconditions
ndash1adjustedtoflowrate01mlh 300mMammoniumdihydrogenphosphatepHofelectrolyte40andcellsize12micromUnderthemostsuitableconditionsarepeat-ability test was performed by ten independent oxidation experiments Therelativestandarddeviationofthepercentageareaofcanagliflozinwas164atasignificant level of 095 The chromatogram of sample oxidized under mostsuitableconditionsisinFig4BThestandardstudyoftheeffectofoxidationoncanagliflozinusinghydrogenperoxideaccordingtoICHguidelineswasperfor-medThestudywasperformedundertwosetsofexperimentalconditionsInthefirstcaseasolutionof50methanolwiththeadditionof3H O wasusedIn2 2
thesecondcasetheconditionsintheelectrochemicalflowcellweresimulatedA300mMammoniumdihydrogenphosphate pH40 andmethanol in a ratio11(vv)withtheadditionof3H O wasusedSamplesfortheoxidationstudy2 2
werestressedinastabilitychamberfor13and7daysattheconstanttempe-ratureof50degC ThetotalsumsofimpuritiesformedduringchemicaloxidationusinghydrogenperoxideinbothmediawerecomparedasisshowninFig5AItisobviousthat
80 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 5(A)Acomparisonofastandardoxidationstudyusinghydrogenperoxidein50methanolwith added buffer andwithout them (B) Chromatogramof sample oxidized electrochemically(C)Chromatogramofsampleoxidizedchemically
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 81
ammoniumphosphatesuppressesoxidationofcanagliflozinThereasonofthisphenomenonisunknownFig5BandFig5CshowchromatogramsofsamplesoxidizedelectrochemicallyandchemicallyrespectivelyItcanbeseenthatfiveimpuritieswereformedbybothtypesofoxidationhoweverindifferentamounts
4Conclusion
AnalternativemethodfortheoxidativestudyofcanagliflozinwasdevelopedThedesign of experiments approach was used in the method development ThedevelopedmethodworkswithRSDof165(α=095)Oxidationofcanagliflozinbythedevelopedmethodproducedfiveimpuritiesthatareidenticalwiththoseproducedusingthestandardoxidationstudywithhydrogenperoxide
Acknowledgments
ThisworkhasbeensupportedbyCharlesUniversityResearchCentreprogramNoUNCESCI014SVV260560projectandpharmaceuticalappliedresearchcenter(TheParc)
References
[1] ChaoECCanagliflozinDrugsFuture36(2011)351ndash357[2] NislySAKolanczykDMWaltonAMCanagliflozinanewsodium-glucosecotransporter2
inhibitorinthetreatmentofdiabetesAmJHealthSystPharm70(2013)311ndash319[3] Turchan M Jara-Ulloa P Bollo S Nunez-Vergara LJ Squella JA Alvarez-Lueje A
VoltammetricbehaviourofbromhexineanditsdeterminationinpharmaceuticalsTalanta73(2007)913ndash919
[4] Perez-OrtizMMunoz C Zapata-Urzua C Alvarez-Lueje A Electrochemical behavior ofatomoxetineanditsvoltametricdeterminationincapsulesTalanta82(2010)398ndash403
[5] Baertschi SW Alsante KM Reed RA Pharmaceutical Stress Testing Predicting DrugDegradationLondonInformaHealthcare2011
[6] RignallA ICHQ1A(R2) stability testing of newdrug substance andproduct and ICHQ1CstabilitytestingofnewdosageformsInICH Quality Guidelines An Implementation GuideATeasdaleDElderRWNims(Eds)HobokenWiley2017p3ndash44
82 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
AlthoughDNArepresentsarelativelystablecomponentfromthechemicalpointofviewitremainsconstantlyexposedtoalargenumberofchemicalorphysicalagentscausingchemicalchangesinDNAmoleculesthatoccurintheenvironmentoraremajororminorproductsofcellularmetabolism[1] One-electronoxidationoftheDNArepresentsadamagingprocesswheretheloss of an electron (oxidation) fromduplexDNA results in the formationof anucleobase radical cation (electron ldquoholerdquo) that is subsequently consumed inchemicalreactionsthatoftenleadtomutationsAdefiningcharacteristicoftheone-electronoxidationofDNAisthepreferentialreactionattheguaninemoietythatisdetectedasstrandcleavagefollowingchemicalorenzymatictreatmentoftheoxidizedDNA[2ndash3]
Novel hybrid electrochemical DNA biosensor for monitoring oxidative DNA damage via oxidationreduction signals of low molecular weight double-stranded DNA
MICHALAUGUSTINVLASTIMILVYSKOCIL
UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova812843Prague2CzechRepublicmichalaugustinnaturcunicz
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 83
AbstractDeoxyribonucleicacid(DNA)representsamajortargetmoleculeformanydamagingagentscausingunfavorablechangesinastructureofDNAmoleculethatbindandinteractwithDNAThusahighdemandforreliabletoolsregardingabettercomprehensionofthenatureofDNAdamagingprocessesstillrepresentsoneofthemaingoalsinthisareaHereinwedescribeadevelopmentofanovelhybridelectro-chemicalDNAbiosensorbasedonanldquoedge-planerdquopyrolyticgraphiteelectrode (EPPGE) in connectionwith an elementaryoptimizationprocessprovidingacloserresolutionoftheredoxprocessesoflowmolecularweightdouble-strandedDNA(dsDNA)attheEPPGESub-sequentanalyticalapplicationincorporatinganemploymentofthemodel structure K [IrCl ] (representative of transition metal2 6
complexes)andevaluationofitsdamagingeffectinrelationtoDNAbymeansof linear sweepvoltammetry resp square-wavevoltam-metryarealsopresented
KeywordsbiosensordamageDNAgraphitevoltammetry
DNA-based electrochemical biosensors are successfully used in variousapplicationssuchasmonitoringandevaluatingthemechanismsof interactionbetweenDNAandvariousdrugsordamagingagentsrapidmonitoringoftracemetalsorpollutantspresent in theenvironmentordirectmonitoringofDNAhybridizationprocesses[4] Theelectrochemicalactivityofnucleicacids(boththenativehigh-molecularonesaswellasoligonucleotides)isingeneralreferredtotheelectroactivityofitscomponents ndash nucleobases and sugar residues At mercury-based electrodesadenine and cytosine residues undergo reduction processes close to ndash14 V(againstSCE)inneutralorweaklyacidicmedium(givingrisetothepeakCA)Ontheotherhandallbaseshavebeenreportedtobeelectrochemicallyoxidizedatcarbonelectrodesbutonlyadenineand(particularly)guanineoxidationsignalshavebeenwidelyutilizedinelectrochemicalDNAbiosensors[5] In2017theelectrochemistryofnucleicacidsachievedanimportantmilestoneasthereductionoftheDNAoligonucleotideswasperformedataldquobasal-planerdquopyrolytic graphite electrode which provided wide potential window allowingboththeelectrooxidationaswellastheeletroreductionofthenucleobasesatasingleelectrodefortheveryfirsttimeDespitethesefindingsutilizationoftheaforementionedbiosensorintermsofanalyticalapplicationshasyettobeverifiedandremainsunclearuptothisdate[6] TheaimoftheproposedcontributionisapresentationofthedevelopmentprocessandsubsequenttestingofanoveltypeofhybridelectrochemicalDNAbiosensoranditsverificationasareliableanalyticaltoolintermsofmonitoringDNAdamage
2Experimental
21Reagentsandchemicals
Low molecular weight double-stranded DNA (dsDNA) derived from salmonspermwasobtainedfromSigma-AldrichGermanyStocksolutions(01mgmL)
of dsDNA were prepared in a 01 molL phosphate buffer of pH=74 (PB)Dipotassium hexachloroiridate (K [IrCl ]) was purchased from Sigma-Aldrich2 6
GermanyStocksolutions(0001molL)ofK [IrCl ]werepreparedinthePB2 6
22Apparatus
Voltammetric measurements were performed using the μAutolab IIIFRA2potentiostatgalvanostat(EcoChemieTheNetherlands)drivenbyaNOVA111software(MetrohmAutolabSwitzerland)Allmeasurementswerecarriedoutina three-electrode system using an ldquoedge-planerdquo pyrolytic graphite workingelectrode(EPPGE)withanelectroactivesurfacediameterof3mm(BASJapan)asilver|silver chloride reference electrode (Ag|AgCl|sat KCl) and a platinum
84 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
counterelectrode(ElektrochemickeDetektoryCzechRepublic)ina20mLglassvoltammetriccellatambienttemperature
23Preparationofthebiosensor
PriortotheeverymeasurementsurfaceoftheEPPGEwasmechanicallycleanedbygentlewipingoftheelectrodeonthesoftpolishingpadrinsedwithdistilledwaterAfterwardstheelectrodewasrinsedwithdistilledwaterandplacedinthePBforthesubsequentelectrochemicalactivationElectrochemicalactivationwasperformed in thePBbyapplyingpotentialof15V for240swithoutstirringApotentialpulseinworkingrangeofpotentials(00ndash15V)wasthenapplied Additional electrochemical activationwasperformed in the solutionof the
3minus4minusredoxindicator([Fe(CN) ] )byconsecutivecyclingintherangeofpotentials6
from10tondash08V(15scans)andfrom055tondash015V(10scans) The electrochemical DNA biosensor based on the EPPGE (dsDNAEPPGEbiosensor)was prepared by the adsorption of dsDNAon the EPPGEOptimal
parametersofthedsDNAadsorptionwereaconcentrationof01mgmLinthePB
(c )adepositionpotentialof07V(E )andanadsorptiontimeof5ming(dsDNA) dep
(t )withoutstirringthesolutionads
Atlasttheelectrodewasimmersedinthesolutionoftheredoxindicatorandtheconsecutivecyclingintherangeofpotentialsfrom055tondash015V(20scans)wasperformedinordertosecurethestabilityoftheoxidationreductionsignalsofdsDNAattheEPPGE
24Procedures
Theexperimentalparameterswereasfollowssquarewavevoltammetry(SWV)inthePBwithapulseamplitudeof20mVafrequencyof50Hzascanrateof750mVsandapotentialstepof15mVlinearsweepvoltammetry(LSV)inthePBwithscanratesof02ndash10Vandapotentialstepof24mVAllcurveswere
recordedthreetimes(n=3)
3Resultsanddiscussion
Sincetheclosestresolutionoftheprocessesassociatedwiththeelectroreductionof singleDNAcomponents at pyrolytic graphitehasbeenperformedwith theldquobasal-planerdquopyrolyticgraphiteelectrode(BPPGE)wehavedecidedtotakeovercorrespondingexperimentaltechniqueandconditions(LSVscanrateof10Vssteppotentialof24mV)attheverybeginningofouroptimizationprocesswiththeEPPGE[6] Inthisparticularcaseitispossibletonoticetheoccurrenceofthetwomixedvoltammetricpeaksatthedefaultexperimentalconditions(greenlineFig1A)selectedforthereductionofdsDNAattheEPPGEBygraduallydecreasingthe
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 85
Fig 1Baseline-correctedLSVrecordingscorrespondingtothereductionofdsDNAattheEPPGEfordifferentvaluesofscanrate(02ndash10VsFig1A)respbaseline-correctedLSVrecordingscorres-pondingtothereductionofdsDNAattheEPPGEandthenegativetestperformedunderthesameexperimentalconditionswithintheblanksolution(phosphatebuffer)atthebareEPPGE(03VsFig1B)
scan rate the optimal conditions (νle03Vs) were found and the mutualseparationof the signalswas allowedndash characterizedby thepresenceof twosinglewell-developedvoltammetricpeaksatpotentialsofndash175Vrespndash190V(03Vs orange line Fig 1A) Taking into account previous work regardingprocessesassociatedwiththereductionofDNAatthemercuryelectrodesrespBPPGE we can assume that the peak appearing at the potential of ndash175 Vcorresponds to themixedpeak for the reductionof the cytosine and adenineresidueswithindsDNA(peakCA)[5ndash6] Closer resolution of the second voltammetric peak appears to be farmoreproblematic Regarding our previous study we have discovered that theutilizationofdifferentE fortheadsorptionofdsDNA(E lt07V)isconnecteddep dep
withanappearanceofthethirdoxidationsignal(besidestheoxidationsignalsofguanine resp adeninemoieties) at apotential of073V corresponding to theoxidationoffreeguaninebases(FGBs)presentwithinthesolutionofdsDNAInthiscasewecanassumethatthepeakappearingatapotentialofndash190VcanpossiblyrepresentthereductioncounterpartofFGBspresentwithinthesolutionofdsDNAThisassumptioncanalsobesupportedbytheaforementionedstudyandbythefactthatthereductionsignalatsuchahighnegativepotentialcanbeobservedfortheoligodeoxynucleotidescontainingguanineresidues[6] AdditionallyinordertoverifythetruenatureofthereductionsignalsdepictedatF ig1Bandtoexcludetheoptionthattherelatedsignalsdonotrepresenttheproductsofpriorelectrochemicalactivationof theEPPGE(variousCndashObasedchemicalspecies)wehavedecidedtoperformanegative(control)testwithinthe
86 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2Baseline-correctedLSVrecordingscorresponding to thereductionof thedsDNA(02Vs
Fig 2A)respbaseline-correctedSWVrecordingscorrespondingtotheoxidationoftheguanine(098V)respadenine(128V)moieties(075VsFig2B)attheEPPGEafteritsincubationinthephosphatebufferforadefinedtimeperiod(60ndash900s)
blanksolution(PB)employingthesameprotocolasforthedsDNAadsorptionattheEPPGEInthiscaseitispossibletoobservetheabsenceofanypronouncedvoltammetricpeakslinkedtothedsDNAadsorptionandonlythepresenceofoneirreversiblepeakatapotentialofndash153VcorrespondingtotheelectroreductionoftheCndashObasedmoietymoietieswhichdropsafterthedsDNAadsorptiontoonetenthofitsoriginalvalue(approximately) Perhaps the most important parameter regarding further optimizationprocessrepresentedthetime-dependentstabilityofthecorrespondingsignalsofdsDNAwhichcanbespecificallyimportantinrelationtothestudyofthetime-dependentoxidativedamageofdsDNA AsithasalreadybeenprovedasingleelectrochemicalactivationoftheEPPGEinthePBdoesnotrepresentasatisfyingtechniqueregardingstabilityofdsDNAoxidationsignalsat theEPPGEand theadditionalstabilization isachievedbyfurtherelectrochemicalactivationinthesolutionofaredoxindicator(Fig2B)[7]Basedonthisaverificationoftheproposedstabilizationprotocolintermsofthetime-dependent stabilityofdsDNAreduction signals in the solutionof thePBwithinthedefinedtimeperiod(60ndash900s)appearedasareasonablenextstep FromtheresultsdepictedinFig2Aitispossibletonoticethatwithinthefirst300sdsDNAreductionsignalsremainstableinrelationtothecurrentresponseaswellasintermsofthepotentialvalueWithanadditionalincubationtime(t )inc
(900sorangeline)thepeakcurrentofthevoltammetricsignalpresentatmorenegative potentials decreased which can probably be addressed as a slowprogressive elimination of the weak (electro)chemical forces related to theunspecificadsorptionoftheFGBsattheEPPGE
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 87
Fig 3Baseline-corrected SWV recordings corresponding to the oxidation of the guanine respadeninemoietiesattheEPPGEafteritsincubationinthesolutionofK [IrCl ](IR)foradefinedtime2 6
period(60ndash3600s)(075VsFig3A)andthecorrespondingrelativebiosensorresponses(ΔI )rel
evaluatedusingtheguanosine(turquoise)andadenosine(red)peaksplottedversustheincubationtime(Fig3C)Baseline-correctedLSVrecordingscorrespondingtothereductionofdsDNAattheEPPGEafteritsincubationinthesolutionofK [IrCl ](IR)fordifferenttimeperiods(60ndash3600s)2 6
(02 VsFig3B)andthecorrespondingrelativebiosensorresponses(ΔI )evaluatedusingtherelpeakCA(darkpink)plottedversustheincubationtime(Fig3-D)
AdditionallywehavedecidedtotesttheapplicabilityofthepresentedhybridbiosensorintermsofmonitoringdsDNAdamagecausedbyarepresentativeofone-electron oxidants ndash K [IrCl ] In this case the prepared dsDNAEPPGE2 6
biosensor was immersed into the solution of K [IrCl ] (0001molL) for the2 6
definedtimeperiod(60ndash3600s) In thecaseof theoxidationpath (SWVrecordingsdepicted inFig3A) it ispossible to observe a time-dependent decrease of the oxidation signal of theguaninemoietieswhereastheoxidationsignaloftheadeninemoietiesremainsunaffectedforthemostof the incubationperiodThisphenomenonis ingoodcorrelationwiththetheoreticalknowledgeregardingoxidativedamageofdsDNAcausedbyone-electronoxidants [3]Simultaneouslywith thisLSVrecordingsdepicted in Fig 3B followed the similar behavior (decrease in relation to the
88 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
currentresponseofthedsDNAreductionsignalndashpeakCA)asinthecaseofthesignal regarding oxidation of guanine moieties In addition according totheportionofthepreservedDNA(Fig3C3D)itispossibletoassumethatthepronouncedoxidativedamageofdsDNAcanbemonitoredquitepreciselynotonlydirectlyviathedsDNAoxidationsignaloftheguaninemoietiesbutevenindirectlythroughthedsDNAreductionsignalndashpeakCA
4Conclusions
Inthiscontributionwehavepresenteddevelopmentofanunorthodoxhybridelectrochemical DNA biosensor based on an EPPGE Optimization processconcerning some important parameters was performed as well as closerresolutionofthenatureofthereductionprocessesofdsDNAattheEPPGEwasachievedInordertoconfirmtheresultsoftheoptimizationprocessapplicabilityoftheproposedbiosensorhadbeenprobedintermsofmonitoringDNAdamagecausedbyK [IrCl ]Inthiscasethefinalresultshadprovedthattheprepared2 6
hybridbiosensorcanbeconsideredasaversatileanalyticaltoolformonitoringoxidativeDNAdamage(viaoxidationreductionsignals)andispresentedasafinealternative in comparisonwith conventional electrochemical DNA biosensorsprepared within the group of traditional transducer materials (mercury- orcarbon-based)
Acknowledgments
ThisresearchwassupportedbytheSpecificUniversityResearch(SVV260440)
References
[1] FojtaMDanhelAHavranLVyskocilVRecentprogressinelectrochemicalsensorsandassaysforDNAdamageandrepairTrACTrendsAnalChem79(2016)160ndash167
[2] GieseBSpichtyMWesselySLong-distancechargetransportthroughDNAAnextendedhoppingmodelPureApplChem73(2001)449ndash453
[3] Burrows CJ Muller JG Oxidative nucleobasemodifications leading to strand scissionChemRev98(1998)1109ndash1151
[4] DiculescuVC Chiorcea-PaquimAMOliveira-BrettAMApplications of aDNA-electro-chemicalbiosensorTrACTrendsAnalChem79(2016)23ndash36
[5] PalecekEJelenFElectrochemistryofnucleicacidsInElectrochemistryofNucleicAcidsandProteinsndashTowardsElectrochemicalSensorsforGenomicsandProteomicsPalecekESchellerFWangJ(edits)AmsterdamElsevier2005p74ndash174
[6] SpacekJDanhelAHasonSFojtaMLabel-freedetectionofcanonicalDNAbasesuraciland5-methylcytosineinDNAoligonucleotidesusinglinearsweepvoltammetryatapyrolyticgraphiteelectrodeElectrochemCommun82(2017)34ndash38
[7] AugustınMVyskocilVNovelelectrochemicalDNAbiosensorbasedonedge-planepyrolyticgraphite for DNA interaction studies In Proceedings of the 15th International StudentsConferenceldquoModernAnalyticalChemistryrdquoNesmerakK(edit)PragueFacultyofScienceCharlesUniversity2019p263ndash268
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 89
1Introduction
Cadmiumisoneofthemosttoxicmetalsanditswidespreadindustrialusesresultin increased environmental pollution Hence the development of sensitivemethodology for Cd determination is still highly desirable Chemical vaporgeneration(CVG)ofCdbythetetrahydroboratereductioninacidicmediumisasuitable alternative sample introduction technique compatible with atomicspectrometricdetectorsandofferingimproveddetectioncapabilityComparedtocommonliquidnebulizationCVGoffersseveraladvantagessuchassignificantlyhigheranalyteintroductionefficiencyandalsoanalyteseparationfromsamplematrix IncomparisontoCVGofcommonhydrideformingelementsthereisalackofliterature dealing with mechanistic aspects of CVG of Cd [1] as well as withstabilityandidentityofitsvolatilespecies(freeatomshydrideotherspecies)Very little information is also available on achieved generation efficiencyMoreover there are many discrepancies in the literature regarding optimum
Chemical vapor generation of cadmium for analytical atomic spectrometry
a b a b a aLINDASAGAPOVA BARBORAKODRIKOVA MILANSVOBODA STANISLAVMUSIL aJANKRATZER
a InstituteofAnalyticalChemistryoftheCzechAcademyofSciences Veveřiacute9760200BrnoCzechRepublicsagapovaiachczb DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublic
AbstractChemical vapor generation of cadmium volatile compounds wasoptimizedinordertodeterminetraceCdconcentrationsbyatomicabsorptionspectrometry(AAS)Severalreactionmodifiersbasedon
III+ II+ III+ IV+inorganicsaltsandcomplexesofCr Co Ti Ti weretestedtoimproveanalyticalperformanceandgenerationefficiencyTheuseofthese reaction modifiers resulted in 4ndash5 times enhancement insensitivity reflected also in corresponding increase of generationefficiency and better repeatability Generation efficiency wasdeterminedfromacomparisonbetweensensitivitiesobtainedwithchemicalvaporgenerationandconventionalsolutionnebulizationbothsimultaneouslycoupledwithinductivelycoupledplasmamassspectrometryTheidentityofthegeneratedcadmiumcompoundswillbediscussed
Keywordsatomicabsorption
spectrometryatomizationcadmiumchemicalvapor
generation
90 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
conditionsforCVGofCdAlthoughstrongacid(HClorHNO )isalwaysemployed3
asacarrierandNaBH asareductantsomeauthorsreportedvariousadditives4III+ III+ IV+(modifiers)basedontransitionmetalions(Cr Ti Ti )inthepresenceof
II+KCN[23]orCo inthepresenceofthioureaandascorbicacid[4]toimproveCdsignalssignificantly TheaimofthisworkwastoinvestigateCVGofCdinacomprehensivewayFirstlyCVGofCdwithoutandwithselectedmodifierswasoptimizedemployingatomicabsorptionspectrometry(AAS)asadetectorandexternallyheatedquartztube(QTA)astheatomizerSecondlytheeffectofatomizationtemperatureonCdsignalwasstudiedallowingthustodeducetheatomicormolecularstructureofgeneratedCd speciesThirdly generation efficiencyofCdvolatile specieswasquantified
2Experimental
21Reagentsandchemicals
minus1Boiled and bubled (Ar per 30min) deionizedwater (lt 01 μScm UltrapurWatrex USA) was used to prepare all solutionsWorking Cd standards were
minus1preparedfrom1000mgL Cdstocksolution(AstasolAnalytikaCzechRepublic)minus1bydilutionin01ndash048molL HCl(basedonthemodifieremployed)from37
HCl(paMerckGermany)Theoptimumgenerationconditionsbeingdifferentforeachmodifiertestedare listedinTable1ThereductantwasasolutionofNaBH (ge 97 Sigma-Aldrich Germany) in 04 (mv) KOH (pa Merck4
Germany) prepared fresh daily The solutions of modifiers were prepared as2+followsCo waspreparedfromCoCl 6H O(ge990PENTACzechRepublic)2 2
3+the solution of Cr from Cr(NO ) 9H O (ge 9999 tracemetal basis Sigma-3 3 23+AldrichGermany)thesolutionofTi fromTiCl solution(about15in10HCl3
4+Sigma-AldrichGermany)andthesolutionofTi fromTiOSO (ge999Sigma-4
AldrichGermany)Tostabilizethelattersolution1H SO wasusedprepared2 4
bydilutionof96H SO (paLach-NerSlovakia)SolutionofKCN(ge9702 43+ 3+FlukaSwitzerland)wasusedasasecondmodifierwhenworkingwithCr Ti or
4+ ndash3Ti asmodifiersitsconcentrationvariedfrom008to016moldm depending
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 91
Table 1OptimumconditionsforchemicalvaporgenerationofCdinthepresenceofmodifiersandtheirabsence
onthemetalionThiourea(CH N Sge980LachemaBrno)andascorbicacid4 2
(C H O ge997Riedel-deHaenGermany)wereusedasmodifierscombined6 8 62+withCo
22Instrumentation
221Chemicalvaporgenerationsystems
TwoCVG flow injection systemswere employed either a two channel systemwithoutadditionofamodifier(seeFig1A)orafourchannelsystemallowingadditionofmodifiers(seeFig1B)
ndash1 TheflowratesofHClandNaBH were42and10mLmin respectivelyinatwo4ndash1channelsystem(Fig1A)whiletheywerebothkeptat10mLmin inthefour
channelsystem(Fig1B)Theflowratesofmodifiersinthefourchannelsystemndash1were05mLmin Thevolumeofthesampleloopwas015mLinbothsystems
ndash1Carriergasflowrateof75mLmin Arwascontrolledbyamassflowcontroler(Cole-ParmerUSA)
92 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
(A)
(B)
Fig 1Schemesofthechemicalvaporgenerationflowinjectionsystemwith(A)twochannels(nomodifiers)and(B)fourchannels(modifiersemployed)
222Atomicabsorptionspectrometry
ThePerkin-Elmermodel503atomicabsorptionspectrometer(BodenseewerkGermany)wasequippedwithaCdelectrodelessdischargelamp(Perkin-ElmerUSA)operatedat228mAThemeasurementswereperformedat2288nmusinga07nmslitwidthTheShimadzumodelAA-7000atomicabsorptionspectrometer(ShimadzuJapan)wasalsousedACdhollowcathodelamp(PhotronAustralia)operatedat2288nmlinewith07nmspectralbandpassandalampcurrentof12mA Signals were recorded for 2 minutes and peak areas were taken forevaluation The QTA was heated electrically to the temperature required byfurnace(PerkinElmer)andanin-housemadefurnacecontrolledbytheREX-C100controller(SysconIndianaUSA)withtheK-typethermocouplesensor(OmegaEngineeringUSA)223QuantificationofCVGefficiencybyICP-MS
Overall CVG efficiency of Cd was quantified bymeans of inductively coupledplasmamassspectrometry(ICP-MS)fromcomparisonoftheslopesofcalibra-tionsobtainedwithnebulizationliquidCdstandardstothoseobtainedwithCVGThe efficiency of liquid nebulization was quantified using a modified wastecollection method (see reference [5] for details) The Agilent 7700x ICP-MSinstrument(AgilentUSA)wasoperatingat1600WofRFpowerThesignalwas
111 125monitoredat Cdisotopeandcorrectedforthesignalofinternalstandard( Tendash11000ngmL Tein2HNO )NebulizeranddilutionArgasflowrateswere11503
ndash1and0mLmin respectively
3Resultsanddiscussion
31Chemicalvaporgenerationconditions
UnivariateoptimizationswereperformedtofindoptimumconditionsforCVGofCdinpresenceandabsenceofmodifiersTheparameterstobeoptimizedwerecarrieracid(HCl)concentrationreductant(NaBH )concentrationmodifierIand4
modifier II concentrations carrier gas flow rate (Ar) length of reaction coilsRCI-III (see Fig 1B) The optimum conditions for individual modifiers aresummarizedinTable1
32IdentityofCdspecies
ThepeakareasofgeneratedCdspecieswereforagivenmodifiermeasuredintheQTAheatedto900degCandnon-heatedQTAsubsequentlyOptimumCVGconditionswere employed as summarized in Table 1 This simple experiment allowsdistinguishingbetweenatomic(freeatoms)andmolecularformsofgenerated
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 93
Fig 2RelativesignalofgeneratedCdspeciesmeasuredintheQTAheatedto900degC(blackbars)andnon-heatedQTA(whitebars)withoutorinthepresenceofmodifiers
speciesOnlyfreeatomscanbedetectedinnon-heatedQTAsimilarlyasincaseofmercurycoldvaporsOn thecontrarymolecularanalytespeciesareatomizedat900degCAsaconsequencethesignalregisteredintheheatedQTAcorrespondstobothatomicandmolecularspeciesgeneratedItmustbehighlightedthattheresidencetimeoffreeatomsintheatomizerisdependentonQTAtemperatureduetogasexpansionAsaconsequencethesignalinQTAheatedto900degCshouldreach25ofthesignalatambienttemperaturetakingintoaccountthatonlyfreeatomsaregeneratedSincethetemperaturealongtheopticalarmofQTAisnotdistributedhomogeneouslydecreasingtobothendtheeffectivetemperatureoftheatomizerislowerOurexperimentswithCVGofHgrevealedsignalinheatedQTAisaround40[6]TheresultsreachedforCVGofCdaredepictedinFig2ThesignalofCdinheatedQTAisaround50ofthesignaldetectedinnon-heatedQTA
4+whennomodifierisemployedorusingTi asthemodifierindicatingclearlyfreeCdatomsarethedominantvolatilespeciesgeneratedOnthecontraryalmostno
2+differenceinpeakareaswasobservedforCo asthemodifierwhilethesignalinheatedQTAwas even 5 times higher in heatedQTA compared to non-heated
3+atomizer with Cr as the modifier suggesting the dominant contribution of3+molecularstructurestoCdsignalespeciallyincaseofCr KCNreactionsystem
33Generationefficiency
TheoverallCVGefficiencywasestimatedfromacomparisonbetweensensitivitiesobtainedwithCVGsampleintroductionandconventionalsolutionnebulizationICP-MSunder the sameexperimental conditionsNebulization efficiency for aMicroMISTnebulizerwasdeterminedas79plusmn01ThegenerationefficiencyofCd was derived from the sensitivity enhancement between CVG and liquidnebulizationTheresultsaresummarizedinTable2indicatingthatCVGwithout
94 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
modifiersisonlycatwotimesmoresensitivecomparedtoliquidnebulization3+ 4+Generation efficiency of Cd increases to 60 in the presence of Ti and Ti
modifiers
4Conclusions
CVG of Cd was thoroughly optimized in the presence of selected modifiersreportedpreviouslyintheliteratureGenerationefficiencyofCdintheabsenceofanymodifierswasquantifiedto15whileitcanbeincreasedupto60inthe
3+ 4+presenceofTi KCNorTi KCNasmodifiersFreeCdatomsseemtobe the4+dominantCdformgeneratedintheabsenceofanymodifiersorusingTi KCN
3+modifierwhile rathermolecularCd structuresaregenerated inCr KCNand2+Co thioureaascorbicacidreactionsystems
ExperimentsareinprogresstofinishthiscomprehensivestudyOnlythebestmodifierwillbefurtherusedforCVGofCdtobecoupledwithotherspectrometricdetectorsandappliedtocertifiedreferencematerialsandrealsamples
Acknowledgments
ThisresearchhasbeensupportedbytheCzechScienceFoundationundercontract18-01116SandbytheInstituteofAnalyticalChemistryoftheCzechAcademyofSciences(InstitutionalResearchPlannoRVO68081715)andCharlesUniversity(ProjectnoSVV260440)
References
[1] PitzalisEAngeliniDMascherpaMCDacuteUlivoAInsightintothemechanismscontrollingthechemicalvaporgenerationofcadmiumJAnalAtSpectrom33(2018)2160ndash2171
[2] ArslanZYilmazVRoseLEfficientgenerationofvolatilecadmiumspeciesusingTi(III)andTi(IV)andapplicationtodeterminationofcadmiumbycoldvaporgenerationinductivelycoupledplasmamassspectrometryMicrochemJ123(2015)170ndash178
[3] YilmazVRoseLArslanZLittleMDOn-linechemicalvapourgenerationofcadmiuminthepresenceofhexacyanochromate(III)fordeterminationbyinductivelycoupledplasmamassspectrometryJAnalAtSpectrom27(2012)1895ndash1902
[4] Y Lu SunHW YuanCG YanXP Simultaneous determination of trace cadmiumandarsenic inbiologicalsamplesbyhydridegeneration-doublechannelAFSAnalChem74(2002)1525ndash1529
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 95
Table 2GenerationefficiencyofchemicalvaporgenerationofCdasquantifiedbyICP-MS
Modifiers Generationefficiency
Nomodifiers 15plusmn13+Cr KCN ndash2+Co thioureaascorbicacid ndash3+Ti KCN 58plusmn24+Ti KCN 61plusmn2
[5] VyhnanovskyJStrugeonREMusilSCadmiumassistedphotochemicalvaporgenerationoftungsten fordetectionby inductively coupledplasmamass spectrometryAnal Chem91(2019)13306ndash13312
[6] MigasovaMMatousekTSchrenkovaVZ ıdekRPetry-Podgorska IKratzer JMercuryvolatilespeciesgenerationfromHClandTRISbuffermediaAnalChimActa1119(2020)68ndash76
96 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Atomicfluorescencespectrometry(AFS)coupledwithvapourgenerationisanultrasensitive analytical method for determination of various elements ItsanalyticalperformancecanbecomparabletoICP-MSwithliquidnebulizationbutatsubstantiallylowercost[1]SampleintroductiontoAFSisacrucialstepoftheanalyticalproceduresincetheanalytehastobeintroducedtotheatomizerintheformofitsvolatilespecies Hydridegeneration(HG)isamaturetechniqueofsampleintroductionduringwhichvolatileanalytehydridesare formedbyreactionwithareducingagenttypicallysodiumborohydrideAnewemergingtechniquephotochemicalvapourgeneration(PVG)employsUV irradiationof theanalyte in liquidphase in thepresenceofaphotochemicalagent(usuallyalowmolarmassorganicacidformic
minusoraceticacid)Highlyreducingradicalspecies(HbullRbullandCOObull )andaquatedelectronsare formedduring irradiationandreactwith theanalyte to form its
Photochemical vapour generation of bismuth coupled with atomic fluorescence spectrometry
ab ab a aBARBORASTA DLEROVA JAROMIRVYHNANOVSKY JIR IDE DINA STANISLAVMUSIL
a InstituteofAnalyticalChemistryoftheCzechAcademyofSciences Veveřiacute9760200BrnoCzechRepublicstadlerovaiachczb DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublic
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 97
AbstractPhotochemical vapour generation of bismuth was successfullycoupledwithnon-dispersiveatomic fluorescencespectrometry forthefirsttimeVolatilespeciesofBiweregeneratedusingastandardmercurylow-pressuretubelampandacoiledreactorfromareaction
2+mediumwhichwas composedof acetic and formic acid Co ionswereusedasasensitizerOptimizationofatomizationconditionsinaflame-in-gas-shieldatomizerwasperformedThismethodologywascomparedtothecommonlyemployedhydridegenerationapproachAbsolutelimitofdetectionof68pgwasachievedwithphotochemicalvapour generation which is still about 7 times worse than withhydride generation The developed methodology was successfullyverifiedbyBideterminationinareferencematerialofwater
Keywordsatomicfluorescence
spectrometrybismuthhydridegenerationphotochemicalvapour
generation
volatilespeciesInbothcasesthegeneratedvolatilespeciesoftheanalytehavetobeseparatedfromtheliquidphaseinthegas-liquidseparatorandarecarriedtotheatomizerbyacarriergas[2] InthisworkanatomizerdesignedspecificallyforAFStheflame-in-gas-shieldatomizerwasused(Fig1) It consistsofaverticalquartz tubesuppliedwithargon and hydrogen together with the analyte volatile species Moreover acapillaryisinsertedintheverticalaxisoftheverticaltubethroughwhichoxygenisintroducedA hydrogen-oxygenmicroflameburns on top of the capillary Themicroflameisshieldedfromtheambientatmospherebyaflowofargonwhichisintroducedthroughashieldingunitfittedaroundtheverticaltube[34] Theaimofthisworkwastooptimizeatomizationconditionsintheflame-in-gas-shieldatomizerusingPVGasasampleintroductiontechniqueandtocomparetheanalyticalcharacteristicsofPVGandHGforultrasensitivedeterminationofbismuthbyAFS
2Experimental
21Reagentsandchemicals
Deionized water (Ultrapur Watrex USA) was used for preparation of all thesolutionsWorkingBisolutionswerepreparedfreshdailybyserialdilutionof
ndash1stock1000mgl BistandardforAAS(Sigma-AldrichGermany)RegardingHG05(mv)NaBH in04(mv)KOHwasusedasa reductantAsolutionof4 ndash11mol l HClwasusedasacarrierandblank
98 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Flame-in-gas-shieldatomizerOHndashobser-vationheight
RegardingPVGformicacid(98paLach-NerCzechRepublic)andaceticacid (998 pa Lach-Ner CzechRepublic)were used for preparation of thereaction medium they were purified in a Teflon BSB-939-IR sub-boilingdistillation apparatus (Berghof Germany) The composition of the reactionmedium(40(vv)acetic125(vv)formicacid)wasoptimizedearlier[5]
ndash1The 5000mgl Co stock solution was prepared from cobalt(II) acetatetetrahydrate (pa Lach-Ner Czech Republic) and used as a sensitizer ofphotochemicalreactionTheoptimalconcentrationofCointhestandardsamples
ndash1andblanksolutionscorrespondedto50mgl (ref[5]) Acertifiedreferencematerial(CRM)-1643fTraceElementsinWater(NationalInstituteofStandardsandTechnologyUSA)wasusedtochecktheprecisionofthedevelopedmethodology
22Instrumentation
221Atomicfluorescencespectrometer
An in-house assembled non-dispersive atomic fluorescence spectrometerconstructedatourlaboratorywasusedforBideterminationandisdescribedindetail elsewhere [3] The detector output provided signals in microV Peak areacorrectedtobaselineandmainlysignaltonoiseratioweretheparametersusedtoevaluatethedata
222Hydridegeneratorphotochemicalvapourgeneratorandatomizer
A flow injection hydride generator was employed (Fig 2a) The reductantndash1 ndash1(12mlmin )andthecarrier(4mlmin )werepumpedbyaperistalticpump
Thesamplewasinjectedthrougha1mlsampleloopintotheflowofcarrierAglassgas-liquid separator (5 ml) with forced waste removal was employed forseparatingthegasphasecontainingbismuthanewhichwasthencarriedtotheatomizerbyargon Thephotochemicalvapourgenerator(Fig2b)consistedofthephotoreactorconstructedwitha15Wlow-pressureHggermicidallamp(Cole-ParmerUSA)wrappedaroundwith6mofPTFEtubing(1mmidinternalvolume471ml)The
ndash1reactionmedium(3mlmin )waspumpedbyaperistalticpumpThesamplewasinjected througha056ml sample loopApolypropylenegas-liquid separator
(15ml)withforcedwasteremovalimmersedinanicebath[6]wasemployedforseparatingthegasphasecontainingBivolatilespeciessubsequentlycarriedtotheatomizerbyargon Theflame-in-gas-shieldatomizerisdepictedinFig1detaileddescriptionisgiveninRef[3]Theobservationheight(OH)isdefinedasthedistancefromthetopofthecapillarytothecentreoftheopticalbeam
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 99
23Samplepreparation
CRMNIST1643fwasdilutedwith1MHCl80-foldforBideterminationbyHG-AFSRegardingBideterminationbyPVG-AFSthesampleneededtobeevaporatedtodrynessinordertogetridofnitricacidthatseriouslyinterferesatmMlevel[5]Avolume of 3ml of CRMwere pipetted into a 40 ml quartz vial evaporated(temperature asymp100 degC two replicates) and subsequently diluted ca 33-foldAsamplepreparationblank3mlofdeionizedwaterwaspreparedaswell
3Resultsanddiscussion
TheatomizationconditionsforHG-AFSwereoptimizedinourpreviouswork[3]TheseconditionswereusedasinitialtofindtheoptimumconditionsforPVG-AFSwith the flame-in-gas-shieldatomizerwith respect to sensitivityandsignal tonoiseratioFirstlythehydrogenfractionintherange10ndash16wasoptimizedatconstant total gas flow rate (sum of total argon and total hydrogen) of
ndash1500mlmin the lower the hydrogen fraction the better However at 10hydrogenfractiontheflamewasnotstableenoughandwentoftenouthenceitwasoptedfor12hydrogenfraction
100 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 2(a)Hydrideand(b)photochemicalvapourgenerator
The oxygen flow rate through the capillary was optimized in the rangendash15ndash30mlmin Thehighestsignaltonoiseratiowasachievedwiththeflowrateof
ndash120mlmin Thetotalgasflowratewasoptimizedatconstant12hydrogenfractioninthe
ndash1range500ndash800mlmin Theoptimumobservationheightvarieswithtotalgasflow rate so it had to be optimized as well The optimum conditions aresummarizedandcomparedtothoseachievedwithHGinTable1 TheanalyticalfiguresofmeritofPVG-AFSwiththeflame-in-gas-shieldatomi-zerweredeterminedThecalibrationfunctionconstructedwith010025050
ndash1 2100 and 200 microg l Bi standardswas linear (R = 09998) The repeatabilityndash1expressedastherelativestandarddeviation(n=10)was6at1microgl andthe
ndash1relativeandabsolutelimitsofdetection(3σn=10)achievedwere12ngl and68pgrespectively(Table2)TheabsolutelimitofdetectionachievedwithHGwas 76 times lower which can be attributed to several aspects Firstly thegenerationefficiencyforPVGapproachwasaround53while100isexpectedforHG[3]Secondlyafullwidthathalfmaximumofthemeasuredpeakswasca2-foldgreaterwhichnecessitatedlongerintegrationtimeandwasthusreflectedinhighernoiseofthesignalsFinallythelimitofdetectionforPVGapproachwas
ndash1affectedby seriouscontamination (around10ng l )mostprobably from thesensitizersolutionthatcontainedBiasimpurity Tovalidate theproposedmethodologyBi contentwasdetermined inCRMNIST1643f (Table3) and the resultswere compared to thosemeasuredwithHG-AFS[3]Duetosevereinterferencesfrominorganicacidsespeciallynitricacid[5]thesampleneededtobeevaporatedtodrynessandthenfilledupwiththe
2+reactionmediumcontainingCo as thesensitizer(NIST1643f isstabilized in
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 101
Parameter HG-AFS(ref[3]) PVG-AFS
ndash1Artotalmlmin 440 528ndash1H totalmlmin 60 722 ndash1O mlmin 7 202
OH mm 6 9ndash1Arshieldlmin 1515 1515
Table 1Atomizationconditionsforflame-in-gas-shieldatomizer
Parameter HG-AFS(ref[3]) PVG-AFS
LODpg 09 68ndash1LOQngl 09 12
Repeatability lt1 6
Table 2AnalyticalfiguresofmeritofHG-AFSandPVG-AFS
ndash1032moll nitricacid)Theresultsobtainedbybothmethodologiesareingoodagreementwiththecertifiedvalue
4Conclusion
Photochemical vapour generation of Bi was successfully coupled with non-dispersiveatomicfluorescencespectrometryforthefirsttimeanditsapplicabilitywas verified by determination of Bi in certified reference material of waterComparedtohydridegenerationconditionsofatomizationdifferinanoptimalobservationheightandsupplyofoxygenwhichmaybeneededtoldquoburnoutrdquotheorganicvapoursthatarereleasedfromthereactionmediumtothegasphasehowever this remains tobe verifiedAlthough there are still some limitationsregardingthelimitsofdetectionrepeatabilityandinterferencesthisnewsampleintroductionapproachseemstobepromising
Acknowledgments
The support of the Czech Science Foundation (19-17604Y) Czech Academy of Sciences(Institutional supportRVO68081715)andCharlesUniversity (ProjectSVV260560andProjectGAUK1048120)isgratefullyacknowledged
References
[1] Musil SMatousek T Currier JM StybloM Dedina J Speciation analysis of arsenic byselectivehydridegeneration-cryotrapping-atomicfluorescencespectrometrywithflame-in-gas-shield atomizer achieving extremely low detection limits with inexpensiveinstrumentationAnalChem86(2014)10422ndash10428
[2] SturgeonREPhotochemicalvaporgenerationaradicalapproachtoanalyteintroductionforatomicspectrometryJAnalAtSpectrom32(2017)2319ndash2340
[3] S tadlerova B Kolrosova M Dedina J Musil S Atomic fluorescence spectrometry forultrasensitivedeterminationofbismuthbasedonhydridegenerationndashtheroleofexcitationsourceinterferencefilterandflameatomizersJAnalAtSpectrom35(2020)993ndash1002
[4] DedinaJAtomizationofvolatilecompoundsforatomicabsorptionandatomicfluorescencespectrometryOnthewaytowardstheidealatomizerSpectrochimActaPartB62(2007)846ndash872
102 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Certified HG-AFS PVG-AFSndash1valuemicrogl
ndash1 a ndash1 a valueobtainedmicrogl recovery valueobtainedmicrogl recovery
1262plusmn011 128plusmn01 102plusmn1 121plusmn09 97plusmn5
a Spikedrecovery=slopeofstandardadditions(noadditionandtwospikedconcentrationstoasample)slopeofexternalcalibration
Table 3ThedeterminedcontentofBiinCRMNIST1643fpresentedasmedianvalueplusmncombineduncertainty(n=3)andrecoveries
[5] Vyhnanovsky J Yildiz D Musil S Effect of metal sensitizers on photochemical vaporgeneration of bismuth for analytical atomic spectrometry In Proceedings of the 15thInternationalStudentsConferenceModernAnalyticalChemistryKNesmerak(ed)PragueCharlesUniversity2019p257ndash262
[6] VyhnanovskyJSturgeonREMusilSCadmiumassistedphotochemicalvaporgenerationoftungstenfordetectionbyinductivelycoupledplasmamassspectrometryAnalChem91(2019)13306ndash13312
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 103
1Introduction
Liquidcrystalsareorganicsubstancesthatformamesomorphicphaseinsolu-tion[1]TheyareliquidlikeliquidsbuthaveaninternalconfigurationassolidsTheir light transmittance changes in the electric fieldwhich is used in liquidcrystaldisplays(LCDs)CholestericliquidcrystalsaretemperaturesensitiveThecolorofreflectedlightchangeswithsmalltemperaturechangeThisisusedinmedicineasasensitivetemperatureindicatorfordisease-infectedtissues High performance liquid chromatography or supercritical fluid chromato-graphy[23]areusedtoseparateanddetermineliquidcrystalcompoundsAnelectrophoretic method could be complementary to these commonly usedmethods To our best knowledge no study dealing with liquid crystal puritycontrolbyelectrokineticchromatographywaspublisheduntilnow Electroneutralsubstancesmoveincapillaryzoneelectrophoresisatthesamespeedcorrespondingtothespeedoftheelectroosmoticflow(EOF)andtherefore
Separation of liquid crystals using non-aqueous capillary electrokinetic chromatography
KATER INACOKRTOVATOMA S KR IZ EK
DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublickaterinacokrtovagmailcom
AbstractLiquid crystals arewidelyused in electronicsmedicine andotherfields Analytical separations are important in the development ofnewliquidcrystalstocontrolthepurityofsynthesizedsubstancesThesampleanalysisisimportantfordetectionofimpuritiesformedduring synthesis Liquid crystal-forming substances cannot beseparated by capillary zone electrophoresis due to the absence ofreadily ionizable groups Therefore electrokinetic chromatographywasused in thisworkAnotherproblemcomplicating theanalysiswastheverylowsolubilityofanalytesinwaterSeparationsinthisworkwere thereforecarriedoutundernon-aqueousconditions inacetonitrilewithaceticacidtoadjustthepHandhexadecyltrimethyl-ammonium chloride as a detergent to mobilize the non-ionizedanalytesUndertheseconditionsitwaspossibletoseparateimpu-ritiesfromsynthesizedanalytesinsamples
Keywordselectrokinetic
chromatographyliquidcrystalsnon-aqueouscapillary
electrophoresis
104 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
itisnotpossibletoseparatethemDuetothisanelectrokineticchromatographymethodwasdevelopedInthismethodasurfactantisaddedtothebackgroundelectrolyteMoleculesaggregateandformsphericalformationscalledmicelles[4]ifthesubstanceisaddedinsufficientconcentrationiehigherthanthecriticalmicellar concentration (CMC) Separation is possible due to interactions ofnonpolarmoleculepartswiththenonpolarmicelleinsideAlthoughwateristhemostusedsolventinelectrophoreticmethodsforseparationofwater-insolublesubstances organic solvents are selected However such solvent must meetcertaincriteriatobesuitableforuseincapillaryelectrophoresisAllcomponentsmustbesolubleinthesolvent[5]ItshouldnotbeflammabletoxicorreactiveforpracticalityitshouldbeliquidatroomtemperatureandalsoitspriceistakenintoaccountThevalueof its relativepermittivitywhichdescribes the strengthofinteractionsbetweenionsshouldbearound30Lowdynamicviscosityisalsopreferred to allow faster migration of analytes No organic solvent meets allparameters of the ideal solvent In practice methanol acetonitrile and theirmixturesarethemostusedTheseparationparameterscanbeinfluencedbyusingan organic solvent of the background electrolyte This topic has already beenwidelyexplored[6ndash8] Itwasgenerallyassumedthat inanhydrousconditionsmicellesarenotcreateddespitesufficientsurfactantconcentrationHoweveritwasfoundoutthatdodecylsulfatecanformstablemicelleswhenthebackgroundelectrolyteisdissolvedinformamide[9]Fortheanalysisofactivesubstancesinmedicinal plants Chen et al developed a method in which sodium cholatedissolvedinmethanolisusedasasurfactant[10]Theaddedpseudostationaryphase does not always form micelles but can still affect mobilization andseparationofanalytesiftheanalytesinteractdifferentlywithfreemoleculesofsurfactantInthisstudywater-insolubleliquidcrystalswereseparated(Fig1)Thereforenonaqueouselectrokineticchromatographymethodwasdeveloped
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 105
Fig 1 Structures of liquid crystals 4-([1-oxo-1-(pentyloxy)propan-2-yl]oxycarbonyl)phenyl4-(octyloxy)-[11-biphenyl]-4-carboxylate (ZL 85) and 4-([1-(decyloxy)-1-oxopropan-2-yl]oxycarbonyl)phenyl 4-(dodecyloxy)-[11- biphenyl]-4-carboxylate (ZL 1210) OpticalisomerismsitesaremarkedwithanasteriskStructurescreatedinMarvinSketch[11]
2Experimental
21Reagentsandchemicals
Acetonitrilege999fromSigma-Aldrich(Germany)aceticacid99fromLach-Ner Neratovice (Czech Republic) and hexadecyltrimethylammonium chloride25(ww)inwaterfromSigma-Aldrich(USA)wereusedforpreparationofback-groundelectrolyteMesyloxidepa(MO)suppliedbyLach-nerNeratovice(CzechRepublic)wasusedasareferencesubstance
22Instrumentation
ForexperimentsG7100ACapillaryElectrophoresisInstrument(AgilentTechno-logiesGermany)wasusedwithUV-VISdetectoroperatingat235nmand254nmwavelengthMeasurementswereconductedinafused-silicacapillaryof50microminner diameter with the total length 500cm and effective length 415cm(PolymicroTechnologiesUSA)
23Method
Capillarywas flushed for3minuteswith1MHCland for2minuteswith thebackgroundelectrolyteBackgroundelectrolytewaspreparedbymixingaceticacid (10mM) and hexadecyltrimethylammonium chloride (40mM) in aceto-nitrile Sampleswere introducedhydrodynamicallybyapressureof5kPa for1secondSampleswerefirstdissolvedinacetonitrileandthendilutedtwotimeswith the background electrolyte A voltage of 20 kV was applied during theseparation
3Resultsanddiscussion
Liquidcrystalsampleswerepractically insoluble inwater theirsolubilitywasndash3testedataconcentrationlevelof1mgcm inmethanolandacetonitrileWhile
samples were not sufficiently soluble in methanol they were successfullydissolvedinacetonitrile Becauseallanalytesaresubstancesthatdonothaveeasilyionizablefunctionalgroups theelectrokineticchromatographymethodwaschosen forseparationAsuitablesurfactantwassoughtCommonlyusedsodiumdodecylsulfate(SDS)isinsoluble in acetonitrile Therefore hexadecyltrimethylammonium chloride(CTAC) which had sufficient solubility for further experiments was chosenAlthoughasuitablebufferwassoughttoensureastablepHduetoproblemswithprecipitationofbuffercomponentsinthenon-aqueousenvironmentaceticacidwasusedtoadjustandmaintainpHofbackgroundelectrolytesolutionAstheadditionofcationicsurfactantsuchasCTACleadstoEOFreversalthedependence
106 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ofEOFmobilityontheconcentrationofCTACinthebackgroundelectrolytewasmeasuredContrarytowhatisobservedinaqueousbackgroundelectrolytesEOFwasnotreversedItsmobilitydecreasedwithincreasingCTACconcentrationbutnomajorchangesoccurredabove40mMconcentrationThecapillarywallwasprobablyalreadysaturatedbyCTACandthefurtherincreaseinconcentrationhadno signifficant effect on the conditionof the capillarywall Therefore a CTAC
ndash3concentrationof40mmoldm was chosenas sufficient for furthermeasure-mentswithrespecttotheincreasingcurrentwithincreasingionicstrengthofthebackgroundelectrolyte The optimized method was used for separation of several liquid crystalsamplesofdifferentpurityInthesampleoftheZL85liquidcrystalwith99purityonezoneoftheanalytewasdetectedImpuritieswereseparatedfromthisanalytewhen the samplewith lower puritywas introduced The peak of theanalytewasidentifiedbasedonrelativemigrationtimerelatedtomesityloxideSeparationoftheanalytefromanimpurityinthesampleZL8576isshowninFig 2The relativemigration timeof the firstpeak is 0834 therefore itwasidentifiedastheZL85analyteThestandarddeviationoftherelativemigrationtimesinfivemeasurementswas0002min(01) Using the available high purity sample it was possible to measure thecalibration line forquantificationof theanalyte in lesspuresamplesLimitof
ndash3detection was determined as 0009mgcm and limit of quantification as ndash30031mgcm FromthecalibrationlineconcentrationofZL85inthesample
with lower purity was calculated The concentration was determined as
48(ww)standarddeviation5(ww)
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 107
Fig 2ElectropherogramobtainedwhenasampleofZL85liquidcrystalwithlowerpuritywasndash3introducedSamplewas introduced in01mgcm concentrationandwithaddedmesityloxide
ndash3(10mgcm )Capillarywithinnerdiameterof50micromtotallengthof500cm415cmeffectivelength The background electrolyte was acetonitrile with 10 mM acetic acid and 40 mMhexadecyltrimethylammonium chloride A voltage of 20 kVwith positive polaritywas appliedDetectionat254nm
ForsampleZL121099onlytheanalyteandmesityloxideweredetectedInthe sample ZL 1210 59 several impurities were separated and detected(Fig3)Accordingtotherelativemigrationtimetheanalyteofinterestcorres-pondstothefirstpeakPeakresolutionissufficientTheresolutionoftheanalytepeakandthesecondpeakis284andtheresolutionoftheothertwopeaksis230
4Conclusions
InthisstudyanewmethodforanalysisofnewlysynthesizedliquidcrystalswasdevelopedSomeparametersofthemethodwereoptimizedndashoptimumconcen-tration of hexadecyltrimethylammonium chloride was searched The identifi-cation of analyteswas based on a comparison of relativemigration times InsamplesZL85andZL1210withlowerpuritytheimpuritieswereseparatedfromthepeaksofliquidcrystalsthecontentofanalytewasdeterminedintheZL85sampleaccordingtothecalibrationline
Acknowledgments
IwouldliketothanktheInstituteofPhysicsoftheCzechAcademySciencesforprovidingnewlysynthesized liquid crystals This work has been supported by Specific University Research(SVV260560)andbyCharlesUniversityResearchCentreprogramNoUNCESCI014
References
[1] GennesPGProstJThePhysicsofLiquidCrystals2ndedNewYorkOxfordUniversityPress1993
108 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
ndash3Fig 3ElectropherogramofsampleZL121059ataconcentrationof05mgcm withmesitylndash3oxideataconcentrationof10mgcm capillarywith innerdiameterof50microm total lengthof
500 cmeffectivelength415cmThebackgroundelectrolytewasacetonitrilewith10mMaceticacidand40mMCTACAppliedvoltage20kVpositivepolarityDetectionat235nm
[2] Vankatova P KalıkovaK KubıckovaA Ultra-performance supercritical fluid chromato-graphy A powerful tool for the enantioseparation of thermotropic fluorinated liquidcrystalsAnalChimActa1038(2018)191ndash197
[3] Vankatova P Kubıckova A Cigl M Kalıkova K Ultra-performance chromatographicmethodsforenantioseparationofliquidcrystalsbasedonlacticacidJSupercritFluids146(2019)217ndash125
[4] Terabe S Otsuka K Ichikawa K Tsuchiya A Ando T Electrokinetic separations withmicellarsolutionsandopen-tubularcapillariesAnalChem56(1984)111ndash113
[5] RiekkolaMLRecentadvancesinnonaqueouscapillaryelectrophoresisElectrophoresis23(2002)3865ndash3883
[6] Wright PB Lister AS Dorsey JG Behavior and use of nonaqueous media withoutsupporting electrolyte in capillary electrophoresis and capillary electrochromatographyAnalChem69(1997)3251ndash3259
[7] PorrasSPKenndlerECapillaryzoneelectrophoresisinnon-aqueoussolutionspHofthebackgroundelectrolyteJChromatogrA1037(2004)455ndash465
[8] PorrasSPRiekkolaMLKenndlerETheprinciplesofmigrationanddispersionincapillaryzoneelectrophoresisinnonaqueoussolventsElectrophoresis24(2003)1485ndash1498
[9] GuoXWangK ChenGH Shi JWuX Di L LWangY Determination of strobilurinfungicideresiduesinfruitsandvegetablesbynonaqueousmicellarelectrokineticcapillarychromatography with indirect laser-induced fluorescence Electrophoresis 38 (2017)2004ndash2010
[10] Chen AJ Li C Gao WH Hu ZD Chen XG Application of non-aqueous micellarelectrokinetic chromatography to the analysis of active components in radix SalviaemiltiorrhizaeanditsmedicinalpreparationsJPharmBiomedAnal37(2005)811ndash816
[11] MarvinSketch [computer program] version 1990 ChemAxon httpschemaxoncom-productsmarvin
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 109
1Introduction
Sudandyesaresyntheticazo-basedaromaticcompoundsTheyaretraditionallyusedinvariousindustriessuchaschemicaltextileandwoodworkingasdyestocolourwaxesplasticsoilspolishesandsoforthTheyhavebeencategorizedasclass3carcinogensbytheInternationalAgencyforResearchonCancerandtheiruseisthereforeforbiddeninthefoodindustryTheyareknownfortheirbrightcolours and easy and cost-effectivemanufactureThey arenearly insoluble inwater but soluble in various organic solvents such asmethanol or trichloro-methane[1] SudanI1-phenylazo-2-naphthol(Fig1A)isadyeusedasanorangecolouringagentItssometimesalsosoldundernamesSolventOrangeRorCISolventYellow14ItisformedasasecondaryproductinthemanufactureoftheSunsetYellowdye
Electrochemistry of Sudan I and its derivates in aqueous media
ad b aANNAONDRA CKOVA MARIESTIBOROVA LUDE KHAVRAN cd adKAROLINASCHWARZOVA -PECKOVA MIROSLAVFOJTA
a CentralEuropeanInstituteofTechnologyMasarykUniversity Kamenice753562500BrnoCzechRepublicannaondrackovaceitecmuniczb DepartmentofBiochemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublicc UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistry FacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublicd InstituteofBiophysicsCzechAcademyofSciences Kralovopolska13561265BrnoCzechRepublic
AbstractSudanIisanaromaticazo-compoundthathasbeenproventobeacar-cinogenDuringitsmetabolizationbycytochromeP450inliverafewmain derivates can be identified Thiswork sets out to assess themechanismofelectrochemicalreductionandoxidationofSudanIitshydroxylationderivativesfeaturingmetabolitesintheSudanIdetoxi-fication pathway and to introduce their selective voltammetricanalysis on boron-doped diamond electrode We show successfuldifferentiationamongthesecompoundsthankstothedifferencesintheelectrochemicaloxidationoftheirphenolicgroups
Keywordsborondopeddiamond
electrodecytochromeP450electrochemicalanalysisSudanI
110 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
InmammalianorganismsSudanIismetabolizedbythemicrosomaldetoxi-fying systemwitha central roleof cytochromeP450hydroxylationactivity inliver[2]DuringtheoxidativeprocessofmetabolizingSudanIseveralmetaboliteswereidentifiedbyprevioustestsThesearegt1-(phenylazo)-naphtalene-26-diol(further abbreviated SI-6OH) 1-(4-hydroxyphenylazo)-2-hydroxynaphtol(furtherabbreviatedSI-4OH)and1-(4-hydroxyphenylazo)-naphtalene-26-diol(furtherabbreviatedSI-46-diOH)thestructuresarepresentedinFig1 ThemainmethodcurrentlyusedtoidentifySudanIamongotherdyeswithsimilar structure is high-performance liquid chromatography (HPLC) It isrecommendedasthestandardmethodtoidentifythelevelofSudanIinfood[3] ComparedtoHPLCelectrochemicalmethodsareprovingtobefastercheaperandcomparablypreciseUnfortunatelyacomprehensiveelectrochemicalstudyofSudanIandparticularlyofitshydroxylatedmetaboliteshasnotbeencompletedyetThedyecanbedetectedthroughelectrochemistryeitherbytheoxidationofitsphenolicgrouporviareductionoftheazogrouppresentinitsmoleculeInbothcasesotherelectrochemicallyactivemoietiesareformedThederivatesofSudanIcanbedetectedandrecognizedfromSudanIthroughanalogousprocesses[4]InthisstudywefocusedoncomparisonofelectrochemicalbehaviourofSudanIanditshydroxylatedmetabolitesonborondopeddiamondelectrodetoaddressthepossibilitiesoftheirrecognitionsinmixturesbasedondifferencesinanodicandcathodicsignals
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 111
Fig 1 Chemical structure of (A) Sudan I (B) 1-(phenylazo)-naphtalene-26-diol (SI-6-OH)(C) 1-(4-hydroxyphenylazo)-2-hydroxynaphtol (SI-4-OH) and (D) 1-(4-hydroxyphenylazo)-naphtalene-26-diol(SI-46-diOH)
(A) (B)
(C) (D)
2Experimental
21Reagentsandchemicals
SudanI(Merckanalyticalstandardgrade)wasdissolvedinethanol(Merck)andkept at room temperatureChemicals forBritton-Robinsonbufferpreparation(acetic acid boric acid orthophosphoric acid sodium hydroxide) were fromMerckwithpurityge99pHofthebufferwasadjustedbymixingoftheacidsandsodium hydroxide solution at different ratios The Sudan I metabolites weresynthetized at the Department of Biochemistry Faculty of Science CharlesUniversityandkeptinmethanolattemperature3degC
22Instrumentation
Cyclicvoltammetric (CV)measurementswerecarriedout inBritton-RobinsonbufferofpH=70atroomtemperatureSudanIanditsderivativeswereaddedto
minus1thesolutionofBritton-Robinsonbuffertofinalconcentrationof5micromolL andstirred Before the measurement oxygen was removed from the solution bypurgingwithargonfor3minutesAutolabanalyzerPGSTAT20(EcochemieTheNetherlands)inconnectionwithVA-Stand663(MetrohmSwitzerland)GPES49(MetrohmSwitzerland)andathree-electrodesetup(withborondopeddiamond
2(WindsorScientificUKdiskdiameter3mmA=707mm )asworkingelectrodeminus1AgAgCl3molL KCl as reference electrode and platinum wire as auxiliary
electrode) Five cycles were performed for each measurement at scan rateminus1of1Vs
3Resultsanddiscussion
ForeachcompoundtwoseparateCVmeasurementswereperformedeachwithfive cycles performed in rapid succession For bothmeasurements the initialpotentialwassetat0VTheanodicscancontinuedto+1Vturnedtowardsndash1Vandfinishedat0VIntheothersetupcathodicscanwasperformedfirstfromthestartingpointtondash1Vturnedtowards+1Vandreturnedto0VThiswaywewereabletoobservethebehavioursofSudanIandcompareittothatofitsderivativeswhilefirstbeingreducedandthenoxidisedorviceversa In the anodic scan of Sudan I and its derivatives (Fig 2) differences in thepositionsoftheoxidationpeaksineachcompoundcanbeobservedWhileSudanIwithonlyonephenolgroupisoxidizedat+067VthederivativeswithtwophenolgroupsieSI-4OHandSI-6OHgiveoxidationpeaksatremarkablylesspositivepotentialofca+05VSI-46OHwiththepresenceofoverall3hydroxygroupsyieldstwooxidationpeaksInthereversecathodicscan(vertexpotential+1V)probablythankstothepresencereductionpeaksappearwhichcanbefurtherusedtodifferentiatebetweenSudanIandthederivativesSI-4OHandSI-6OH
112 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 113
Fig 3VoltammetricscanofSudanIanditsderivatesSI-4-OHSI-6-OHandSI-46-diOH1stscanincathodicdirectionfrom0Vvertexpotentialsndash1Vand+1VThemeasurementswereperformedinBritton-Robinsonbuffer(pH=7)withtheconcentrationofeachcompoundat5microMandatscan
ndash1rate1Vs
Fig 2 CyclicvoltammogramofSudanIanditsderivatesSI-4-OHSI-6-OHandSI-46-diOH1stscaninanodicdirectionfrom0Vvertexpotentials+1Vandndash1VThemeasurementswereperformedinBritton-Robinsonbuffer(pH=7)withtheconcentrationofeachcompoundat5microMandatscan
ndash1rate1Vs
possesstwoconjugatedhydroxylgroupswhichcanberegardedashydroquinonestructures and thus undergoing quasireversible redox process due to oxida-tionreductionofthehydroquinonetoquinonemoietyThisiswellvisibleattheCVsastheanodicsignalisfollowedbycathodiconeatthepotentialof+023VforSI-4OHand+03VforSI-6OHTheoxidationofSudanIproceedsbymechanism
minus +typicalforphenoliccompoundsatmorepositivepotentialsleading by1e 1H exchange to naphthoxy-type ndashO radical [5] This species undergoes furtherreactionsleadingtoformationofdimersandpolymersThecathodicpeakinthereversescanatndash02Varisesfromreductionofthesereactionproductsanditsoriginneeds tobe further investigatedSI-46OHwith thepresenceofoverallthree hydroxyl groups yields two oxidation peaks The first one is a result ofoxidationoftwoofthembeinginconjugationandthusbeingoxidizedtoquinonemoietyThesecondsignalatthesamepotentialastheoxidationsignalof2OHonnaphthaleneringofSudan I is consequenceofoxidationof the thirdhydroxylgroupofphenolictypeAsinglewidepeakat0Vinthereversescanispresumablyan overlap of signals arising from reduction of the quinonic moiety and by-products formed during oxidation processes Thanks to differences of theseprocessesspecificforindividualcompoundsitispossibletodifferentiateamongallfourofthemviapropersetting-upoftheinitialandvertexpotentialvalues The cathodic scan of Sudan I and its derivatives (Fig 3) shows a dominantreductionpeakaroundndash08Vwhichisduetoreductionoftheazogroupintheirstructuresaccompaniedbycleavageoftheirmoleculestoseparatethebenzeneandnaphthalene rings [4] The peaks in the subsequent anodic scans (vertexpotentialndash1V)arethereforetheresultoftheelectrochemicalreactionofmoietiesthatareproductsofthedivisionofthearomaticcirclesTheseproductsincludeaniline4-aminophenol1-amino-2-naphtholand1-amino-25-naphthalenediolwith irreversibly oxidizable amino moieties or (quasi)reversibly oxidizableamino-hydroxylsystemonthebenzeneornaphthaleneringandtispossibletodifferentiatebetweenthemTheobtainedoxidationpeaks(+02VforSI-4-OH+073VforSudanIandSI-6-OHand+025Vand+075VforSI-46-diOH)makeitpossibletodifferentiatebetweenallcompoundswiththeexceptionofSudanIandSI-6-OHForthereliablerecognitionofthesetwocompoundsanothermeasure-mentwithdifferentparametersisneeded
4Conclusions
ThestructuresofSudanIanditshydroxyderivatesthatarethemainproductsofthe metabolization of Sudan I by cytochrome P450 are similar and theirrecognitionwhenpresentinmixtureinsolutionisdemandingHereinwepresenta simple approach based on comparison of signals obtained in cathodic andanodic scan in CV measurements without the need of time-demandingchromatographicseparationstepFurtherworkwillbedevotedtoidentificationof observed redoxprocesses and applicationof themethod formonitoringofmetabolictransformationsofSudanIinvitro
114 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Acknowledgments
ThisresearchwassupportedbytheCzechScienceFoundation(projectNo18-01710S)
References
[1] ChailapakulOWonsawatW SiangprohW GrudpanK ZhaoYF Zhu ZW Analysis ofSudanISudanIISudanIIIandSudanIVinfoodbyHPLCwithelectrochemicaldetectionComparison of glassy carbon electrodewith carbon nanotube-ionic liquid gelmodifiedelectrodeFoodChem109(2008)876ndash882
[2] StiborovaMMartinekVRydlovaHHodekPFreiESudanIisapotentialcarcinogenforhumans Evidence for itsmetabolic activation and detoxication by human recombinantcytochromeP4501A1andlivermicrosomesCancerRes62(2002)5678ndash5684
[3] GomezMArancibiaVAliagaMNunezCRojas-RomoCDeterminationofSudan I indrinks containing Sunset yellow by adsorptive stripping voltammetry Food Chem 212(2016)807ndash813
[4] PrabakaranEPandianKAmperometricdetectionofSudanIinredchilipowdersamplesusingAgnanoparticlesdecoratedgrapheneoxidemodifiedglassycarbonelectrodeFoodChem166(2015)198ndash205
[5] Enache T A Oliveira-Brett A M Phenol and para-substituted phenols electrochemicaloxidationpathwaysJElectroanalChem655(2011)9ndash16
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 115
1Introduction
Aminoglycosidesarealargegroupofnaturalandsemi-syntheticantibioticswithawidespectrumofantimicrobialactivityagainstmostgram-positiveandgram-negativemicroorganismsCurrentlymultiplerepresentativesofaminoglycosidefamily ndash gentamicin (GM) neomycin B (NM) paromomycin (PM) kanamycin(KM)apramycin(AP) andstreptomycin(STM)Fig1ndashareapproved to treatinfectiousdiseasesinanimalsMaximumresiduelimitsfortheseaminoglycosidesinproductsandtissuesfromedibleanimalsareestablished[1]henceaneffectiveandrobustassayisnecessaryforcontrolofaminoglycosidescontamination Inthisstudyenzyme-linkedimmunosorbentassayforthedetectionofamino-glycosides in foodproductsandenvironmentalobjectswasdevelopedGroup-specificantibodieswereproducedowingtoimmunogenbasedonribostamycin(RS)whichexposedthecommonfragmentofmostaminoglycosides2-deoxy-streptamin (2-DOS) Fig 1 The developed assay was made suitable for thedetectionofresidualaminoglycosidesinhoney[2]
Group detection of aminoglycosides using ELISA for control of food contamination
ab a aKONSTANTINBURKIN INNAGALVIDIS MAXIMBURKIN
a DepartmentofImmunologyIMechnikovResearchInstituteofVaccinesandSera MalyjKazionnyjper5a105064MoscowRussianFederationburkin-kostyandexrub DepartmentofChemicalEnzymologyFacultyofChemistryLomonosovMoscowStateUniversityLeninskieGory1119991MoscowRussianFederation
AbstractThegrowingthreatofglobalantibioticresistanceisforcingtoreducenon-targetconsumptionofantibioticsandtomonitorcontaminationoffoodandenvironmentalobjectsInthisworkELISAwasdevelopedforgroupdetectionofaminoglycosidesToobtaingroup-specificanti-bodies a new immunogen based on ribostamycin was used Thedevelopedindirectcompetitiveformatofassayallowedtherecogni-tionof9aminoglycosidesnamelyneomycinribostamycinneaminparomomycin gentamicin sisomicin kanamycin tobramycin and
ndash1apramycinwithadetectionlimitrangedbetween002ndash020ngmL TheeffectivenessoftheproposedassaywasevaluatedinhoneyasafoodstuffmodelToneutralizea stronghoneymatrixeffect and toavoidalaborioussamplepre-treatmentanewmatriximitatorwassuggested 5 sucrose solution imitated the influence of 50-folddilutedhoneyTheproposedassayallowedustorevealanyofthe9
ndash1mentionedaminoglycosidesinhoneyata10microgkg level
KeywordsaminoglycosidesELISAhoney
116 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
2Experimental
21Reagentsandchemicals
NeomycinBribostamycinneamin(NA)paromomycinkanamycintobramycin(TM)amikacin(AM)gentamicinnetilmicin(NTM)sisomicin(SSM)geneticin(GC) apramycin and streptomycin were purchased from Chimmed (MoscowRussia) Bovine serumalbumin (BSA) complete Freund adjuvant 16-hexane-diamine1-ethyl-3-(3-dimethylaminopropyl)carbodiimide(EDC)sodiumperio-dateandsodiumborohydrideweretheproductsofSigma-Aldrich(USA)Gelatin(Gel)wasfromBio-Rad(USA)sucrosefromServa(Germany)two-componenttetramethylbenzidine (TMB) substrate solutionwas fromBioservice (Russia)andgoatanti-rabbitIgGantibodiesconjugatedtohorseradishperoxidase(anti-rIgG-HRP)werefromIMTEK(Russia)Honeysampleswerepurchasedfromlocaloutlets
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 117
Fig 1 Structuralformulasofstudiedaminoglycosides
22Preparationofconjugatedantigens
TwotypesofconjugateswerepreparedbasedonRSandBSAusingzero-lengthand C6 spacer arm between hapten and protein carrier RSwas treatedwithsodiumperiodatetooxidizehydroxylsofribosefragmenttoreactivealdehydegroupsandthencoupledtoBSAaminesthroughreductiveaminationToremoveuncoupledRSanexhaustingdialysiswascarriedoutusingdialysismembranetubes(MWCO14kDa)UsingthesameprocedureGel-RSconjugatewassynthe-sized ForpreparationofBSA-C6-RSwefirstlymodifiedBSAwith16-hexanediamineThemixtureofBSAandEDCinwaterwerestirredfor30minThen16-hexane-diaminewasaddedandstirredfor2hThemodifiedproteinwasdialyzedfromtheexcessive reagents and resultant BSA-C6-NH2was coupled to RS in reductiveaminationprocessasdescribedabove
23Immunizationandantibodypreparation
BSA-RSandBSA-C6-RSwereusedasimmunogensChinchillarabbits(20ndash25kg)weresubcutaneouslyinjectedat10ndash15pointsonthebackwith01mgofimmuno-gensemulsifiedinthecompleteFreundadjuvantThesamedosesofimmunogensinsalinewereadministeredmonthly forbooster immunizationsAweekaftereachinjectionabloodsamplefromearveinswastakenforthecontrolofimmuneresponseTheantiserainglycerol(11vv)werestoredatndash15degCuntiltestinginELISA
24TheELISAprocedure
AcompetitiveassaywasconductedaccordingtoclassicalprocedureGel-RSwascoatedovernightonpolystyrene96-wellCostarplatesNon-adsorbedconjugatewashedoutusingPBSwith005oftween20(PBS-T)Thenextcompetitivestep
ndash1includedtheadditionof01mLstandardaminoglycosidesolutions(1pgmL to ndash1 ndash11microgmL (B)and0microgmL (B ))inPBS-Tor01mLoftestedsampleand01mLof0
antibodiesinworkingdilution(1h25 degC)Afterwashingtheantibodiesboundtoimmobilized Gel-RS were detected using anti-rIgG-HRP (1h 37degC) Coloredproduct formedasaresultofenzymaticreactionwithTMBsubstratemixture(05h25degC)wasreadat450nmusingaStatFax2100platereader(AwarenessTechnologiesUSA) Relativeantibodybinding(BB )vstheanalyteconcentrationswasplottedas0
standardcurvesfittedtoafour-parameterlogisticfunctionThecross-reactivity(CR) for every aminoglycoside representative was calculated as ratio of half-inhibition concentrations IC NMIC aminoglycoside The dynamic range of50 50
assaywasacceptedasIC ndashIC andthelimitofdetection(LOD)wascalculatedas20 80
B ndash3timesSD0
118 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
3Resultsanddiscussion
31Immunogensynthesisandantibodypreparation
Inthemajorityofpublicationsdevotedtoimmunoassayofaminoglycosidestheimmunogenscoatingantigensenzymeconjugatesortracerswerepreparedbycarbodiimide or glutaraldehyde methods involving aminoglycosidesrsquo aminogroups [3ndash7] Due to several amino groups in aminoglycoside molecules theformationofconjugateswithavariableorientationofthehaptenoccurs InpresentstudyRSwaschosenasanimmunizinghaptenduetothefollowingadvantageous features Being a trisaccharide RS has the size of a moleculecomparable to themostof aminoglycosides Ithas three identical ringsA-B-CsimilartothoseinNMUsingaperiodateoxidationwecouldinvolvearibosesiteofRSincouplingtoproteinthatprovidedastrictorientationofhaptenonthecarrier with a favorable presentation of the 2-DOS fragment The resultantimmunogensBSA-RSandBSA-C6-RSwerecomparedtorevealwhichdesignisbetterforpresentationofacommonfragmentofaminoglycosidemoleculeandgenerationofgroup-specificantibody Antibodies to the BSA-RS demonstrated moderate sensitivity (NM
ndash1IC =10ngmL )andhighselectivitytowardsNMwithrelativelylowcross-reac-50
tivity(lt5)forGMKMandAPTheapplicationofthespacerintheimmunogenBSA-C6-RScontributedtoaprominentpresentationofthe2-DOSdeterminantandtheinductionofantibodieswithrecognitionofbroadspectrumofdifferentaminoglycosides In addition anti-BSA-C6-RS exhibited significantly better
ndash1sensitivity(NMIC =02ngmL )Thusallsubsequentstudieswereconducted50
usinganti-BSA-C6-RS
32Examinationofassayspecificityandselectionofimmunoreagents
TheindirectcompetitiveformatofassaywasdevelopedForevaluationofassayspecificity a panel of following aminoglycosideswas studied and their cross-reactivitywasdeterminedNA(625)RS(250)NM(100)KM(475)PM(173)GM(90)TM(78)AP(17)SSM(12)AM(lt01)GC(lt01)STM(lt01)andNTM(lt01)ThemostoftheseanalytesareusedinmedicalandveterinaryareashoweveronlyNMPMGMKMAPandSTMareappliedinanimalhusbandry[1]
33Determinationofaminoglycosidesinhoneyandselectionofthematriximitator
Honey is a complex product consisting of carbohydrates (75ndash80) vitaminsproteinsenzymesorganicacids traceelements inclusionsandothercompo-nentsThesecomponentsmightinterfereimmunochemicalreactionThereforetheisolationofaminoglycosidesfromhoneyisalaboriousandtime-consuming
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 119
120 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
procedureToavoidthisstephoneymatriximitatorswereappliedtomimictheinfluenceofhoneymatrixonantibodybindingSucrosewaschosenasthehoneyimitatorsinceitexposedastronghoney-matrix-likeeffectonantibodybindingTheadequacywasfoundbetweensolutionsofhoneyandsolutionsofsucroseexpressinganequalmatrixeffectTwopairswithequivalentmatrixeffectwere120honey=20sucroseand150honey=5sucroseThelatterconditionswere chosen asmore preferable due to inconvenience of operatingwith highviscous20sucrosesolution Thedeterminationofaminoglycosidesinhoneycouldbecarriedoutquantita-tivelyiftheanalytetobedetectedisknownForquantificationofaminoglycosideinhoneyasamplewasdiluted50timesinPBS-Tandaminoglycosidestandardcurvewas generated in 5 sucrose-PBST (Fig 2) If analyte is unknown thedevelopedgroup-specificELISAcanbeusedasascreeningtestInthiscasetheanalyzed sample can be considered as contaminated if it caused a relativeantibodybindingbelowthecut-off level (Fig3)Thus thedeveloped testwas
ndash1 ndash1 ndash1Analyte IC ngmL Dynamicrange LODngmL LODinhoneymicrogkg50 ndash1 IC ndashIC ngmL20 80
NM 02 003ndash21 002 10PM 07 008ndash71 005 25GM 15 015ndash133 011 55KM 035 005ndash39 004 20AP 68 05ndash968 023 115
Fig 2StandardcurvesandanalyticalparametersoftheELISA-systemforgroupdeterminationofaminoglycosidesinhoneyInteractionofanti-BSA-RSwithcoatingantigenGel-RSin5sucrosesolutionasthehoneyimitatorThedetectionlimitin5sucrosesolutionwasdeterminedaccordingtoLOD=B ndash3timesSD0
capabletorevealthecontaminationofhoneywith9aminoglycosides5amino-ndash1glycosidesapprovedforveterinary(NMPMGMKMandAP)ata10microgkg level
andalsoNARSSSMandTM
4Conclusions
A novel indirect competitive ELISA for the detection of aminoglycosides wasdevelopedRSwasusedasanewimmunizinghaptentoproducegroup-specificantibodiesagainst2-DOSacommonmoietyofalargenumberofaminoglycosideantibiotics A wide spectrum of aminoglycoside representatives could bedetectedincludingNMRSNAPMGMSSMKMTMandAPThedevelopedassay
ndash1was capable todetect theseanalyteswithaLOD up to002ndash020 ngmL ForanalysisofhoneyamatriximitatorwasdevelopedtoavoidhoneyinterferencesonimmunoassayTheanalysisofthehoneysampleallowedustorevealanyofthe
ndash1mentionedaminoglycosidesinhoneyata10microgkg level
References
[1] CouncilRegulation(EU)N372010OffJEurCommunitiesInfNotL15(2009)1ndash72[2] GalvidisIABurkinKMEreminSABurkinMA Group-specificdetectionof2-deoxy-
streptamineaminoglycosidesinhoneybasedonantibodiesagainstribostamycinAnalMeth11(2019)4620ndash4628
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 121
Fig 3Detectionofaminoglycosidesspikedinhoneysamplesata40ppblevelusinggroup-specificELISAEachsymbolcorrespondstotheaveragerelativebindingandtheerrorisSDobtainedforanindividualhoneysampleanalyzedintriplicateEmptycharactersrepresentindividualblankhoneysamples(limebuckwheatandflower)andfilledsymbolsrepresentthesamesamplesfortifiedwith
ndash1aminoglycosidesata40mgkg (level establishedonlyforSTMinseveralcountries)Thecut-offlevelcorrespondstothelimitofassaydetectionobtainedbythematriximitator(5sucrose-PBS-T)
[3] ThompsonSGBurdJFSubstrate-labeledfluorescentimmunoassayforamikacininhumanserumAntimicrobAgentsChemother18(1980)264ndash268
[4] LiCZhangYEreminSAYakupOYaoGZhangXDetectionofkanamycinandgentamicinresiduesinanimal-derivedfoodusingIgYantibodybasedic-ELISAandFPIAFoodChem227(2017)48ndash54
[5] GalvidisIABurkinMAMonoclonalantibody-basedenzyme-linkedimmunosorbentassayfortheaminoglycosideantibiotickanamycininfoodstuffsRussJBiorganChem36(2010)722ndash729
[6] HaasnootWStoutenPCazemierGLommenANouwsJFKeukensHJImmunochemicaldetectionofaminoglycosidesinmilkandkidneyAnalyst124(1999)301ndash305
[7] Peng JWangYLiuLKuangHLiAXuCMultiplex lateral flowimmunoassayfor fiveantibioticsdetectionbasedongoldnanoparticleaggregationsRSCAdv6(2016)7798ndash7805
122 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
PhotochemicalvaporgenerationisanalternativesampleintroductiontechniqueforanalyticalatomicspectrometryThistechniqueisbasedaroundasourceofUV-radiationthatirradiatesalowmolecularweightorganicacidmedium(mostcommonlyformicacidaceticacidortheircombinations)withananalyteHighlyreducingradicalsandaquatedelectronsareproducedandconverttheanalyteintoavolatilespecieswhichisthentransportedintoadetector[1]SofartheuseofPVGhasbeendescribedforhydride-formingelements(AsBiTeSbPbSeSnandTl)andmercury[12]transitionmetals(FeCoNiCuMoWCdAgAuIrPdPtRhandOs)[13ndash6]andevennon-metals(BrIClFandS)[16ndash9] AfirstsuccessfulphotochemicalvaporgenerationofcobaltwasdescribedbyGuoetalin2004[6]whichwasfollowedbymoresystematicstudiesbyGrinbergetalin2008[10]andDengetalin2010[11]LaterworksbydeQuadrosetal[12]anddeJesusetal[13]focusedontheanalysisofrealsamplesInthelatterwork
Photochemical vapor generation of cobalt for detection by inductively coupled plasma mass spectrometry
ab aJAROMIRVYHNANOVSKY STANISLAVMUSIL
a DepartmentofTraceElementAnalysisInstituteofAnalyticalChemistryoftheCzechAcademyofSciencesVeveřiacute9760200BrnoCzechRepublicjaromirvyhnanovskygmailcom
b DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova8203012843Prague2CzechRepublic
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 123
AbstractThisworkfocusedonthephotochemicalvaporgenerationofcobaltVolatilespeciesweregeneratedinaflow-injectionsystememployinga high-efficiency flow-through UV photoreactor and a formic acidbased medium and were introduced by an argon carrier into aninductivelycoupledplasmamassspectrometerfordetectionOptimalgeneration conditions were found as 10 (vv) formic acid and
ndash1 ndash14molL ammonium formate with a 4mLmin flow rate whichcorresponds to irradiation time of around 13 s The influence ofvariousmetalsensitizersofphotochemicalreactionwasinvestigated
2+andonlyCu ionsexhibitedapositiveeffectongenerationefficiencyndash ndash 2ndashInterferencesfromcommoninorganicanions(NO Cl SO ) were3 4
also examined Lastly the limit of detection and repeatability (atndash1 ndash1250ngL )weredeterminedtobe13ngL and41respectively
Keywordscobaltinductivelycoupled
plasmamassspectrometry
photochemicalvaporgeneration
the authors also presented a systematic study on generation conditions andachievedagenerationefficiencyofaround40 Themainaimofthisworkwastooptimizetheconditionsofgenerationwithinductivelycoupledplasmamassspectrometry(ICP-MS)detectionexaminetheeffectofvariousmetal sensitizers toachieve thehighestgenerationefficiencypossibleandreachthelowestlimitofdetectionpossible
2Experimental
21Reagentsandchemicals
minus1Deionizedwater (DIW lt 02μScm UltrapurWatrex USA)was used for thepreparationofallsolutionsFormicacid(98paLach-NerCzechRepublic)andammonium hydroxide (ge25 pa Sigma-Aldrich USA) were used for the
minus1preparationof the reactionmediumA1000mgL Co stock solution (Sigma-AldrichUSA)wasusedforthepreparationofallsamplesolutionsThefollowingcompounds were used as potential metal sensitizers cadmium(II) acetatedihydrate(paLach-NerCzechRepublic)zinc(II)acetatedihydrate(paSigma-AldrichUSA)copper(II)acetatemonohydrate(paMerckGermany)nickel(II)acetatetetrahydrate(paSigma-AldrichUSA)sodiumtungstatedihydrate(paCarlRothGermany)and iron(II) sulphateheptahydrate (pa LachemaCzechRepublic)Nitricacid(65semiconductorgradeSigma-AldrichUSA)hydro-chloricacid(37paMerckGermany)andsulfuricacid(98paLach-NerCzechRepublic)wereusedforaninterferencestudy
22Instrumentation
AschematicdiagramofthePVGsystemcoupledtoICP-MSisshowninFig1andamore detailed description can be found in reference [5] Briefly a singlequadrupole ICP-MS Agilent 7700x (Agilent Technologies USA)was used as a
minus1detector Deionized water was mixed with a 10μgL Rh internal standardsolutionin2HNO andwassubsequentlynebulizedbyaMicroMistnebulizer3
59 103during PVG Isotopes of Co and RhweremonitoredMeasurementswereperformed in time resolved analysis mode and in He collision mode
minus1(41mLmin )AlltubingusedwasmadefromPTFEwiththeexceptionoftygontubing in the peristaltic pump (Reglo ICC Ismatec Switzerland) The high-efficiency flow-through photoreactor was a 19 W low-pressure mercurydischarge lamp (Beijing Titan Instruments Co Beijing China) with a quartzcentralchannel(asymp720μL internalvolume)Samplesolutionswere introducedintoastreamofreactionmediumusinganinjectionvalve(V-451IDEXHealthandScienceUSAsampleloopvolume05ml)Effluentfromthephotoreactorwasmixedwithaflowofargonandcarriedtothechilledgas-liquidseparator(internalvolume15mL)wherethevolatilespecieswereseparatedfromtheliquidwaste
124 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
andcarried to the inletofaScott-typespraychamber (originally the inlet formakeupargon)oftheICP-MS
3Resultsanddiscussion
The starting conditionswere adopted fromour earlierwork [14]whichusedatomicabsorptionspectrometerasadetectorandminiaturediffusionflameasanatomizerThefirstparameteroptimizedwasthecompositionofreactionmedium(Fig2)Theadditionofammoniumformate(createdin-situbytheadditionofacalculatedamountofammoniumhydroxidetoformicacid)wasfoundcrucialto
ndash1effectivelygeneratevolatilespeciesofcobalt10(vv)formicacidand4molL ammonium formatewas chosen as the optimumandwasused for all further
experimentsAlthoughhigher concentrations of both components led to even
higher signalstheseconditionswerenotusedfurtherbecauseofthelaboriousprocessofpreparation(mixingofconcentratedacidwithconcentratedbase)andtolimittheconsumptionofchemicals Theinfluenceofirradiationtimewasalsoexaminedandthehighestpeakarea
-ndash1wasobtained for4mLmin corresponding to an irradiation timeof approximately13s ToenhancethegenerationefficiencyadditionofvariousmetalstothereactionmediumwastestedtoldquosensitizerdquothephotochemicalreactionThemetalswerechosenwithrespecttotheirsignificantenhancementeffectdescribedrecentlyforphotochemicalvaporgenerationofotheranalytes[158]Theonlymetalionthat
2+ledtoanenhancementofthesignalwasCu (Fig3a)buteveninthiscasetheeffect was rather negligible reaching only 12-fold enhancement in the range
ndash1 2+ 2+001to01mgL Cu FurtheradditionofmoreCu ledtoadecreaseinthesignal2+TheadditionofZn didnotexhibitanypositiveornegativeeffectacrossthetested ndash1range (01 to 1500mgL not shown in figure) and the addition of higher
ndash1 2+ 2+ 6+concentrations (tens to hundredths ofmg L ) of Cd Fe andW (Fig3b)causedsevereinterferences
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 125
ndash ndash 2ndash Interferencescausedbycommoninorganicanions(NO Cl SO addedas3 4ndashtheirrespectiveacids)werealso investigatedOutof theseNO was foundto3
ndash1causethemostsevereinterferencesevenatconcentrationsofsinglemmolL Thendash 2ndashmethodologywasmorerobusttowardstheinterferencesfromCl andSO but4
theystillcausedsignificantdropinsensitivityathigherconcentrationsConsi-deringthewideuseoftheseacidsinanalyticalchemistryforsamplepreparationthisposesabigchallengeintheapplicationofthismethodtorealsamples
ndash1 Usingoptimalconditions(10(vv)formicacid4molL ammoniumformateandirradiationtimeof13s)acalibrationcurvewasmeasuredandevaluatedThelimitofdetectionwasdeterminedas3timesthestandarddeviationof10blank
ndash1measurements and was calculated as 13 ng L The repeatability of 10ndash1consecutivemeasurementsof250ngL was41
126 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 3Effectofvariousmetalionsonthepeakarea(a)metalionswithapositiveeffect(b)metalionsndash1withoutapositiveeffectExperimentalconditions2microgL Coreactionmedium10(vv)formic
ndash1 ndash1acidand4molL ammoniumformateflowrate4mLmin
ndash1Fig 2Effectofthecompositionofreactionmediumonpeakareaexperimentalconditions2microgL ndash1Coreactionmediumflowrate4mLmin (blackdotscorrespondtomeasuredpoints)
4Conclusions
Theconditionsofthephotochemicalvaporgenerationofcobaltwereoptimizedand are in good agreement with previous works [11 13] Copper ions wereidentifiedasapotentialsensitizerincreasingthesignalbyabout12-foldbuttheirpotentialuseisseverelylimitedbythenarrowrangeofconcentrationsinwhichthepositiveeffectisexhibitedSevereinterferencesfrominorganicanionswereobservedwhichisinlinewithotherworksdealingwithphotochemicalgeneration[1358]Furtherexperimentswillfollownamely(i)furtherinvestigationsinnewpotentialsensitizerstoenhancegenerationefficiencyandthusdecreasethe
ndash1limit of detection to sub ng L levels (ii) determination of the generationefficiency(fromcomparisonwithnebulizationandorusingaradioactiveisotope58Co)(iii)verificationoftheaccuracyandpracticalfeasibilityofthismethodologybyanalysisofcertifiedreferencematerials
Acknowledgments
ThesupportofTheCzechScienceFoundation(ProjectNo19-17604Y)CzechAcademyofSciences(Institutional supportRVO68081715)andCharlesUniversity (project SVV260560andprojectGAUK60120)isgratefullyacknowledged
References
[1] SturgeonREPhotochemicalvaporgenerationaradicalapproachtoanalyteintroductionforatomicspectrometryJAnalAtomSpectrom32(2017)2319ndash2340
[2] XuTHuJChenHJTransitionmetalionCo(II)-assistedphotochemicalvaporgenerationofthalliumforitssensitivedeterminationbyinductivelycoupledplasmamassspectrometryMicrochemJ149(2019)103972
[3] SoukalJSturgeonREMusilSEfficientphotochemicalvaporgenerationofmolybdenumforICPMSdetectionAnalChem90(2018)11688ndash11695
[4] deOliveiraRMBorgesDLGUVphotochemicalvaporgenerationofnoblemetals(AuIrPdPtandRh)AfeasibilitystudyusinginductivelycoupledplasmamassspectrometryandseawaterasatestmatrixJAnalAtomSpectrom33(2018)1700ndash1706
[5] VyhnanovskyJSturgeonREMusilSCadmiumassistedphotochemicalvaporgenerationoftungstenfordetectionbyinductivelycoupledplasmamassspectrometryAnalChem91(2019)13306ndash13312
[6] GuoXSturgeonREMesterZGardnerGJVaporgenerationbyUVirradiationforsampleintroductionwithatomicspectrometryAnalChem76(2004)2401ndash2405
[7] HuJSturgeonRENadeauKHouXZhengCYangLCopperionassistedphotochemicalvapor generation of chlorine for its sensitive determination by sector field inductivelycoupledplasmamassspectrometryAnalChem90(2018)4112ndash4118
[8] LeonoriDSturgeonREAunifiedapproachtomechanisticaspectsofphotochemicalvaporgenerationJAnalAtomSpectrom34(2019)636ndash654
[9] SturgeonREPaglianoEEvidenceforphotochemicalsynthesisoffluoromethaneJAnalAtomSpectrom(2020)httpsdoiorg101039D0JA00108B
[10] GrinbergPMesterZSturgeonREFerrettiAGenerationofvolatilecobaltspeciesbyUVphotoreduction and their tentative identification J Anal Atom Spectrom 23 (2008)583ndash587
[11] DengHZhengCB LiuLWWuLHouXDLvYPhotochemicalvaporgenerationofcarbonyl for ultrasensitive atomic fluorescence spectrometric determination of cobaltMicrochemJ96(2010)277ndash282
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 127
[12] deQuadrosDPBorgesDLDirectanalysisofalcoholicbeveragesforthedeterminationofcobalt nickel and tellurium by inductively coupled plasmamass spectrometry followingphotochemicalvaporgenerationMicrochemJ116(2014)244ndash248
[13] deJesusHCGrinbergPSturgeonRESystemoptimizationfordeterminationofcobaltinbiologicalsamplesbyICP-OESusingphotochemicalvaporgenerationJAnalAtomSpectrom31(2016)1590ndash1604
[14] VyhnanovskyJFotochemickegenerovanıtekavychspeciı kobaltuproanalytickouatomovouspektrometriiMasterthesisFacultyofScienceCharlesUniversityPrague2018
128 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Oneof themethods thatallowobtainingmaterialswithnewproperties is theplasmaenhancedchemicalvapordepositionmethodInthismethodcompoundscalledprecursorsaresuppliedtotheplasmareactorasagasphaseThankstoplasmaenhancedchemicalvapordeposition it ispossible toobtainmaterialswithuniquepropertiesThisisduetothefactthattheplasmaaffectsthesurfaceinfourdifferentways etching cleaning chemicalmodification and crosslinking
Optimization of condition for cold plasma deposition of thin layers for surface modification of working electrodes
a b a cJUSTYNALIPIN SKA MARIAMADEJ BOGUSŁAWBAS JACEKTYCZKOWSKI
a DepartmentofAnalyticalChemistryFacultyofMaterialsScienceandCeramicsAGHUniversityofScienceandTechnologyAdamaMickiewicza3030-059KrakoacutewPolandjustynalipinskaaghedupl
b DepartmentofAnalyticalChemistryFacultyofChemistryJagiellonianUniversityinKrakoacutewGronostajowa230-387KrakoacutewPoland
c Departmentof MolecularEngineeringFacultyofProcessandEnvironmentalEngineeringLodzUniversityofTechnologyWolczanska21390-924ŁoacutedźPoland
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 129
AbstractCurrentlyresearchisfocusedonthesearchfornewphysicallyandchemicallystablematerialsaswellasvolumeorsurfacemodificationOneofthemethodsusedforsurfacemodificationistheapplicationofthin layers from inorganic and organic compounds The plasmaenhancedchemicalvapordepositionisamethodthatallowsmaterialmodificationandalsodepositionofthinlayersThisworkconcernsoptimizationofcoldplasmadepositionparametersandtoachievethebestelectrical conductivitywhilemaintaining thehighmechanicalstrength of the formed layers Preliminary tests were focused onoptimizing the layering parameters such as the deposition timedischargepowerpressureofmonomerandthe flowofargonTheobtainedsamplesweresubjectedtothermaltreatmentafterwhichtheywere coveredwitha layerof aluminumThe thicknessof theobtained layers was determined on the basis of interferencemicroscopymeasurementsAsaresultoftheexperimentslayerswithathicknessof20nmto600nmwereobtainedTheconductivityofthedeposited layers was also determined and values from 003 to
ndash1150Sm wereobtained
Keywordscoldplasmadepositionelectrochemical
applicationssurfacemodificationthinlayers
Thismethodisusedtoproducecatalyticstructuresortomodifythepropertiesofmaterials eg improve hydrophobicity The growing popularity of surfacemodificationmethodsusingcoldplasmaisassociatedwiththefactthatitisanenvironmentallyfriendlyandversatilemethod[12] Workingelectrodesusedinvoltammetryareasubgroupofchemicalsensorswhich are small devices that convert real-time chemical information into ameasurableandanalyticallyusefulmeasurementsignalChemicalinformationrangingfromtheconcentrationofaspecificcomponentofthetestedsampletotheoverallcompositionofthematrixcancomefromboththeinitiatedchemicalreactionandbetheresultofphysico-chemicaltransformationstakingplaceinthetested object Chemical sensors are equipped with two basic elements iereceptor and transducer The receptor is responsible for the conversion ofchemicalinformationfromthetestedobjectintoaspecificformofenergyintheconverterthisenergyistransformedintoausefulanalyticalsignal Parameters characterizing the electrochemical sensor include accuracyprecision selectivity accuracy presentation selectivity sensitivity dynamicrange limit of quantification limit of detection lifetime response time andreliability Themost numerous and the oldest group of chemical sensors areelectrochemicalsensorsCommonlyobservedinterest inthisgroupofsensorsresultsfromthefactthatwithrelativelylowproductionandoperatingcoststheyofferthebestmetrologicalandoperationalparameters[3ndash5]Oneofthemaintrendsofmodernanalyticsisthesearchfornewelectrodematerialsandvariousgeometries of working electrodes One way to improve the performance ofworkingelectrodesistomodifytheirsurfaceforexamplebyapplyingthinlayersIn this work were considered plasma enhanced chemical vapor depositionmethod as the method of surface modification designed to performworkingelectrodeforvoltammetricdeterminationsAspartoftheinitialresearchplasmaprocessing parameters such as discharge power time of treatment andcompositionofgasmixtureinwhichplasmawasgeneratedwereoptimizedThelayers obtained in different conditions have been tested for suitability forelectrochemical applications (layer thicknessmeasurement and themeasure-mentofconductivity)
2Experimental
21Reagentsandchemicals
Theprecursor solutions suchasacrylonitriledietoxydimethylsilane trietoxy-methylsilaneandtetramethyldisiloxanewhicharesuppliedbyABCRwereusedOtherreagentsofanalyticalpuritysuchasn-hexane(SigmaAldrich)andargonwereused
130 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
22Instrumentation
The thin layers were deposited in a parallel-plate plasma reactor (frequency1356MHz)ThesamplesobtainedwerecalcinedinatunnelfurnaceunderanargonatmosphereThethicknessofthedepositedlayerswasmeasuredafterthealuminum was sputtered using a Nikon microscope type ECLIPSE LV150NElectrometerhigh resistance system (KEITHLEY) was used to measureconductivity
3Resultsanddiscussion
Eachof themonomerswasdepositedonprepared1times1corningglasssamplesSamples prepared with n-hexane were placed in a plasma reactor andadditionallypartiallycoveredwithamicroscopecoverslipSchematiclayoutofsamplesinthereactorshowninFig1ThefirststepwastoetchthesystemusingargonplasmaThisstageallowedfortheeliminationofimpuritiesthatwerenotremovedbythehelpofn-hexaneandthepreparationofthesurfaceofthesamplesforthedepositionoftheproperlayerTheproperstageistheapplicationofathinlayerwiththeplasmainducedbytheselectedprecursoracrylonitriledietoxydi-methylsilanetrietoxymethylsilaneandtetramethyldisiloxaneThethicknessandpropertiesoftheobtainedlayersdependonthedepositionparameterssuchasdischarge power time of treatment and composition of gasmixture inwhichplasmawasgenerated Table1showsallcombinationsofparameterstestedforallfourprecursorsFour different discharge powers for acrylonitrile and two different dischargepowersfororganosiliconmonomersweretestedwithtwodifferenttreatmenttimesEachtimeandpowercombinationwasperformedinplasmainducedbypuremonomerandmonomerwithargonAfterapplyingthelayersthesampleswereplacedinaquartzboatandcalcinedinatunnelfurnaceAllsampleswerecalcinedat500degCfor2hoursunderargonflowSamplesaftercalcinationwerecoveredwithalayerofaluminum
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 131
Fig 1 Scheme of sample distribution in plasma reactor(a)reactorelectrode(b)corningglasssamples(c)micro-scopecoverslip
Thethicknessoftheobtainedlayerswasmeasuredusinganinterferencemicro-scopeMeasurements were carried out at amagnification 10times recording theimageinmonochromelightTheimagewassetsothattheinterferencefringeswere perpendicular to the arc on the sample In order to calculate the layerthicknessD[nm]thefollowingformulawasused
(1)
wheredandLweredeterminedonthebasisoftheregisteredimage(disfringeshiftduetorefractionoflightontheslopeLisdistancebetweenthefringes) Thelaststageofthestudywastodeterminethecurrent-voltagecharacteristicstodeterminetheconductivityoftheobtainedlayersThesamplewasplacedinameasuring cell and attachedwith silver paste to the electrometerwires ThechangeincurrentwasrecordedwiththeapplicationofalternatingvoltageintimeBasedontheresultsobtainedthegraphsofdependenceUndashIwereobtainedfromwhichthevalueofresistance(R)wasdeterminedAnexampleofcurrent-voltagecharacteristicsisshowninFig2
132 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Table 1Conditionsforlayersdeposition-parameterwhichweretested
Monomer DischargepowerW Timeoftreatmentmin
Acrylonitrile 10204080 24Dietoxydimethylsilane 2040 255Trietoxymethylsilane 2040 255Tetramethyldisiloxane 2040 255
Fig 2Current-voltagecharacteristicsdeterminedfortheacrylonitrilelayer(depositionparametersW=10Wt=25mingasmixtureonlyacrylonitrile)
Table 4Theresultsofthicknessmeasurementsandconductivityspecifictothetrietoxymethylsilanelayers
DischargepowerW 20 20 40Timeoftreatmentmin 25 5 5Argonflowsccm 10 10 10Thicknessnm 14686 24282 35311
ndash1SpecificconductivitySm 165 93 67
Knowing the value of the resistance and the geometry of the system thespecificresistancewasdeterminedfollowedbythespecificconductivityofthesamplethefollowingequationwasused
(2)
whereρisspecificresistance[Ωm]Risresistance[Ω]bissamplelength[m]Disdepositedlayerthickness[m]andldistancebetweenelectrodes(wires)[m] Theresultsofthicknessmeasurementsandspecificconductivityarepresentedin the Tables 2ndash5 For electrochemical applications it is important that theobtainedlayerhasthehighestspecificconductivityAnalyzingthedatapresentedin Tables 2ndash5 shows that the thinnest layers have the greatest applicationpotentialastheelectrodematerial
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 133
Table 2Theresultsofthicknessmeasurementsandconductivityspecifictotheacrylonitrilelayers
DischargepowerW 40 40 80 80 10 10 20 20Timeoftreatmentmin 2 4 2 4 2 4 2 4Thicknessnm 15845 31167 250 4644 247 67 7531 12236
ndash1SpecificconductivitySm 0002 156 191 0003 1512 454 311 185
Table 3The results of thickness measurements and conductivity specific to the dietoxydimethylsilanelayers
DischargepowerW 20 20 40 40Timeoftreatmentmin 25 5 25 5Argonflowsccm 10 10 10 10Thicknessnm 13998 37519 28431 49556
ndash1SpecificconductivitySm 1640 55 105 40
Table 5The results of thickness measurements and conductivity specific to the tetramethyldisiloxanelayers
DischargepowerW 20 40 40Timeoftreatmentmin 25 25 5Argonflowsccm 10 10 10Thicknessnm 16759 3159 61652
ndash1SpecificconductivitySm 213 95 31
4Conclusions
InthisworkplasmaenhancedchemicalvapordepositionmethodwasusedforapplyinglayersoffourdifferentmaterialsacrylonitriledietoxydimethylsilanetrietoxymethylsilaneandtetramethyldisiloxaneBychangingparameterssuchasdischargepowertimeoftreatmentandcompositionofgasmixtureanumberofsampleswereobtainedwithlayersofdifferentthicknessandwhatisassociatedwithotherelectricalproperties TestsperformedaspartofthisworkwereusedtoperforminnovativeworkingelectrodesforvoltammetricdeterminationsThesurfacewasmodifiedusingthecoldplasmaofthreesubstratesgraphiteglassycarbonandgold
Acknowledgments
JLandMMhavebeenpartlysupportedbytheEUProjectPOWR030200-00-I00416
References
[1] KapicaRTyczkowskiJBalcerzakJMakowskiMSielskiJWorwaEEnhancingadhesivejointsbetweencommercialrubber(SBS)andpolyurethanebylow-pressureplasmasurfacemodificationIntJAdhesAdhes95(2019)102415
[2] TyczkowskiJKapicaRŁojewskaJThincobaltoxidefilmsforcatalysisdepositedbyplasma-enhancedmetalndashorganicchemicalvapordepositionThinSolidFilms515(2007)6590ndash6595
[3] HulanickiAGłabSIngmanFChemicalsensorsDefinitionsandclassificationPureApplChem63(1991)1247ndash1250
[4] BrzozkaZWroblewskiWSensorychemiczneWarszawaOficynaWydawniczaPolitechnikiWarszawskiej1999(InPolish)
[5] SkoogDAWestDWHollerFJCrouchSRFundamentalsofAnalyticalChemistry9thEdBostonCengageLearning2013
134 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
ConcentrationandcompositionofvolatilecompoundsorcongenersisoneofthemostimportantparametersresponsibleforqualityofproducedalcoholicdrinksandhencefortheirsensorycharacteristicsandconsumeracceptanceTodaygaschromatography (GC) is conventionally used to determine qualitative andquantitativecompositionsofvolatilecompoundswithvariousexternalandorinternalstandardcalibrationprocedures Methodemployingethanol as an internal standard (IS) forGCquantitativedeterminationofvolatilecompoundsinalcoholicbeverageshasbeensuggestedquitelongago[1]andsincethattimegreatresearchworkhasbeencarriedoutRecentlyaninterlaboratorystudyofthemethodinvolving9testinglaboratoriesfrom4countrieswascarriedout[2]TheresultsdemonstratedgreatperspectivesofldquoEthanolasISrdquomethodandproveditsreferencecharacterandeaseofroutineimplementation
Advanced GC-MS method for quality and safety control of alcoholic beverages
abANTONKORBAN
a DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversity Hlavova2030812840Prague2CzechRepublickarbonat7gmailcomb DepartmentofAnalyticalChemistryChemistryFacultyBelarusianStateUniversityLeningradskaya14220050MinskBelarus
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 135
AbstractRecently developed and validated simple and reliable quantitativemethod employing ethanol as an internal standard for GC-MSquantification of volatile compounds in alcoholic products wasapplied to 36 samples including commercially available world-famousbrandspirits from18countriesandhomemadedistillatesThe GC-MS analyses were performed simultaneously by thesuggested approach and official internal standard method that isprescribedinthelegislationofEUandUSATheindependentsamplest-testwasemployedtoevaluatethestatisticaldifferenceofresultsofthesetwomethodsThetestrevealednodifferenceintheresultsandtheirrepeatabilityThemainbenefitsofthesuggestedmethodaretheeliminationofthenecessityofmanualinternalstandardadditionandsamplesdensitymeasurementthusmakingitmoreeconomicalandproductive
Keywordsalcoholicbeveragesgaschromatography-
massspectrometry(GC-MS)
internalstandardmethodvolatilecompounds
quantification
AllpreviousstudieswereutilizingflameionizationdetectorssinceGC-FIDisprescribedinthelegislation[3]wheremass-spectrometrydetectorsarenotyetofficially referred However GC-MS instruments are employed in practice toqualifyandorquantifyvolatilesincommercialspiritsintraditionalhomemadealcoholicdrinksinnewlydevelopedbeveragesinspiritwastesandindistillatesobtainedwithdifferentmanufacturingprocesses Our recent researchwasdirected towardsdevelopment of an algorithmofldquoEthanolasISrdquomethodapplicationonGC-MSinstruments[4]WehaveshowedthattopreventMSdetectorfromsaturationethanolshouldberegisteredinthecorresponding SIM timewindow at characteristicmz of low abundance forinstance bymz of 47 ions This ion corresponds to non-fragmented ethanol
13moleculescontaining1heavyisotope(mainly C)FinallytheresultsofmeasuredstandardsolutionsshowedthatthesuggestedapproachisvalidandldquoEthanolasISrdquomethodmaybesuccessfullyusedonGC-MSinstrumentstoo The objective of this studywas to test and further approve the suggestedapproachonalargersetof36realsamplesofalcoholicdrinkseithercommercialorhomemadeThesamplesweresimultaneouslyanalysedbytwoGC-MSmethodsndashaclassicalISmethodprescribedinthelegislationandthesuggestedldquoEthanolasISrdquomethod
2Experimental
21Reagentsandchemicals
Thefollowingvolatilecompoundsweredeterminedintestedsamplesofalcoholicbeverages 11-diethoxyethane (acetal) acetaldehyde methyl acetate ethylacetate methanol 2 propanol 1-propanol 2 methylpropan-1-ol (isobutanol)1-butanol2-butanoland3methylbutan-1-ol(isoamylalcohol)1-pentanolwasemployedasatraditionalIScompound
22Instrumentation
ShimadzuGCMS-QP2010Ultraequippedwithaquadrupolemassspectrometrydetector was employed for GC-MS measurements Rxi-1301Sil MS capillarycolumn(60mlength025mmid025micromfilmthicknessRestek)wasusedfortheseparationofcompoundsInjectionswereperformedinasplitmode(ratio175)Helium(99999purity)wasusedasacarriergasinjectortemperaturewas170degCTheoventemperaturewasheldat30degCfor5minthenraisedto210degC
ndash1at a rate of 30 degCmin and held isothermally for 4minMeasurementswereperformedinaSIMmodeFortheanalysedcompoundsand1-pentanol2ndash3mostabundantionsinthecorrespondingMSspectrumwereselectedethanolSIMtimewindowcontainedonly47mzionsAllGC-MSmeasurementswerecarriedoutintriplicateunderrepeatabilityconditions
136 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
AnalysisofeachalcoholicsamplewasperformedinafollowingwayAliquotof09mL of a tested sample was pipetted into a standard 2mL glass vial and
ndash1weighedAfterthat01mloftheISsolution(2355mgkg of1-pentanolinWES)wasaddedtothetestedsampleandthemasswasrecordedTheobtainedmixturewasmixedthoroughlyand05microlofitwasinjectedintotheGCsystem Theoriginoftestedalcoholicbeverageswaseithercommercialorhomemade33 world-famous spirits manufactured at different parts of the world werepurchasedfromcommerciallyavailablesourcesThelistoftypesofpurchasedandanalysedspiritsincludedbourboncalvadoscognacgingrappaliquormetaxaportwine rumsake tequilavodkawhiskeyandvarious fruitdistillatesThepurchased drinks were produced at the territory of the following countriesBelarus Bermuda Cuba Czech Republic Denmark France Germany GreeceGuatemala Jamaica Japan Mexico Moldova Portugal Slovakia Trinidad andTobago UK (England and Scotland) USA Three homemade fruit distillatesproducedbyfermentationofpulpyfruitsortheirmustswereobtainedfromlocalspiritmakersThedeclaredABVvaluesofalltestedsamplesvariedfrom15to81
3Resultsanddiscussion
To fulfil themaingoalof thiswork ie toevaluate thestatisticaldifferenceofresultsyieldedbythecomparedmethodswehaveemployedStudentst-testforindependentsamplestoverifystatisticaldifferencesonthesignificancelevelofp=005Theobtainedempiricalvaluesforallpairsofcongenersrsquoconcentrationswere lower than critical one in all cases demonstrating that concentrationsobtainedbythetwomethodshavenostatisticaldifferenceandleadtothesameresults InadditionrepeatabilityofthetwomethodswascomparedthereforeallRSDvalues obtained from triplicatemeasurements were split in two groupswith
ndash1respecttothecorrespondingconcentrations(lowerthan50mgL AAandhigherndash1than50mgL AA)Theobtainedresultsarepresentedintheformofboxplotin
Fig1AnalysisofthechartinFig1showedthatboththetestedmethodshaveyieldedstatisticallysimilarrepeatability AllofthetestedalcoholicdrinkssatisfiedtherequirementsofEURegulation(EC)no1102008[5]Theconcentrationsofundesirablecompoundssuchasmethanoldidnotexceedthelevelsspecifiedinthesameregulationforcorres-ponding beverages In Table 1 the description of the used SIM method andsummaryoftheexperimentalresultsarepresented Tocomparethetruenessofthemethodsoneofthespiritsampleswasspikedwithstandardsolutions(ABV40)containingallanalysedvolatilecompoundsat
ndash1concentrationsof50500and5000mgL AATheoriginalsamplewasusedasareferenceEachofthespikedsolutionswasmeasuredintriplicateSelectedspirit(cherry distillate) initially contained all 11 volatile compounds in various
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 137
138 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1BoxchartsofRSDsofusedISmethodsat2concentrationrangesMeanisequaltoarithmeticmeanoraverageInterquartileRange(IQR)meansisthedistancebetweentheupper(themedianoftheupperhalfofthedataset)andlower(themedianofthelowerhalfofthedataset)quartile
ndash1Compound Timemin Registeredmz Numberof ConcentrationmgL AA results Minimal Maximal
Acetaldehyde 0ndash42 314344 36 24 715Methanol 36 13 13600
Ethanol(IS) 42ndash48 47 mdash mdash mdash
2-Propanol 48ndash70 29314345 14 27 199Methylacetate 596174 10 34 3201-Propanol 26 361 12070Ethylacetate 27 166 107002-Butanol 11 18 2080
Isobutanol 70ndash200 3141ndash4345 28 19 20001-Butanol 5556 13 28 155Acetal 26 45 270Isoamylol 31 39 26501-Pentanol(IS) mdash mdash mdash
Table 1DescriptionoftheusedSIMmethodandsomestatisticsconcerningallmeasured36spiritsamplesbothpurchasedandhomemade
concentrationsTheobtainedrecoveriesboxchartsareshowninFig2Compa-risonoftherecoveriesobtainedwithtwomethodsindicatesthattheyhavenosignificantdifferenceintermsoftruenessAverageobtainedrecoverywas981whenusingsuggestedmethodand980whenusingtraditionalISmethod
4Conclusions
InthisworktheresultsoftestingtheadvantageousldquoEthanolasISrdquomethodfortheGC-MS quality control analysis of alcoholic beverages were presented33purchasedsamplesofworld-famousalcoholicbeveragesoriginatingfrom18countriesand3homemadefruitdistillateswereanalysedtomakeathoroughandcomprehensive studyof thedevelopedmethodTheconcentrationsofvolatile
ndash1compoundsinanalysedsamplesvariedfrom1to13500mgL AAtheABVvalueofanalysedsamplesvariedfrom15to81ThesuggestedmethodwascomparedwiththetraditionalISmethodthatiscurrentlystatedinlegislationTheindepen-dentsamplest-testrevealedthatwithaprobabilityof095resultsobtainedwithtwo methods do not differ significantly The results of within-run precision(repeatability)showedrelativestandarddeviationswithin3measurementstobelessthat6indicatingthatthetechniqueisreproducibleThetruenessofthemethodwasevaluatedbyrecoverycalculationAccordingtotheobtainedresultsrecoveryofthesuggestedmethod(981plusmn33)wasslightlybetterthanthatofthetraditionalone(980plusmn58) ThesefactsprovethatdevelopedldquoEthanolasISrdquomethodistruepreciseandreliable when employed on GC-MS instruments At the same time to obtain
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 139
Fig 2Boxchartsof recoveriesof thesuggested(dottedpattern)and traditional (brickpattern)ISmethodsatdifferentspikeconcentrationsSymbolsdefinitionsarethesameasinFigure1
concentrationsofvolatilecompoundsintheofficiallyrequiredunitsofmeasurendash1 ndash1(mgL AAgL AAetc)suggestedmethodrequiresnodensitometrymeasure-
mentsofthetestedsampleandnoadditionofIScompoundoranyothersamplepre-treatmentThismethodprovidesaninvaluableanalyticaltoolforthequalitycontrolofalcoholicproductsandshouldbeusedinroutineanalysis
Acknowledgments
ThisworkwasfinanciallysupportedbytheVisegradFund
References
[1] CherepitsaSVBychkovSMKovalenkoANMazanikALSeleminaNMSeredinskayaOBThe use of themajor component (solvent) as an internal standard in the gas-chromato-graphicdeterminationofimpuritiesJAnalChem58(2003)368ndash371
[2] CharapitsaSSytovaSKorbanASobolenkoLEgorovVLeschevSZakharovMCabalaRBusarovaRShestakovichITolstouhovaAOndrousekSVavraJYilmaztekinMCabarogluTInterlaboratorystudyofethanolusageasaninternalstandardindirectdeterminationofvolatile compounds in alcoholic products BIO Web Conf 15 (2019) 02030httpsdoiorg101051bioconf20191502030
[3] CommissionRegulation(EC)No28702000layingdownCommunityreferencemethodsfortheanalysisofspiritsdrinkshttpdataeuropaeuelireg20002870oj
[4] KorbanACharapitsaSCabalaRSobolenkoLSytovaSTheperspectivesofethanolusageasaninternalstandardforthequantificationofvolatilecompoundsinalcoholicproductsbyGC-MSJMassSpectr55(2020)e4493
[5] EuropeanUnion(2008)Regulation(EC)No1102008oftheEuropeanParliamentandoftheCouncilof15January2008ontheDefinitionDescriptionPresentationLabellingandtheProtectionofGeographical IndicationsofSpiritDrinksandRepealingCouncilRegulationhttpdataeuropaeuelireg2008110(1)oj
140 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
1Introduction
Carbonorgraphitefeltsareusedaselectrodematerialsincethe1990sandtheirutilizationstillgrowsinanalyticalelectrochemistryaswellasinotherareasThisisduetotheirsuitablepropertiesfromwhichwecannamehighporosityhighspecificsurfaceareagoodelectricconductivityandhighphysicalandchemicalstabilityThefirsttwoparametersaregivenbystructureoffeltwhichconsistsoforderlesscarbonfibreswithabouttentotwentymicrometersindiameter[1]theothersby theadvantageouselectricalpropertiesof carbon fibreOn theotherhand porous flow-through electrodes including carbon felt electrode havedisadvantageinapotentialdropintheelectrodevolumewhichcausesdifficultcontrollingoftheexactpotentialappliedontheelectrodeandthereforeresultsindifferentcurrentefficienciesontheoppositesidesoftheelectrode[2] Carbon felt electrode can be utilized for detection of structurally differentcompoundsatvariousconditionsForexampleoperatingatreductionpotentialofndash08V[3]oxidationatrelativelyhighpotential+15V[4]ormeasuringatlowconcentrationsofelectrolyte[5]canbenamedDevelopedtechniquesalsoshowsthatcarbonfeltcanbeusedfordeterminationatsubmicromolarconcentrationsThis ismainly due to its ability to operate as a high-efficiency amperometricdetector Themainaimofthispaperistooverviewandcompareparametersofseveraldetermination methods of different analytes using carbon felt detector in
Utilization of a carbon felt as a material for working electrodes
MARTINBAROCHHANADEJMKOVA SA RKASLA DKOVA
DepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublicmartinbarochnaturcunicz
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 141
AbstractWorkingelectrodemadeofcarbonfeltwasusedincombinationwithHPLC for verificationofpractical applicabilityof the electrodeAlldeveloped methods confirm advantageous physical and chemicalpropertiesofcarbonfeltForelectrochemicalutilizationitispossibleto operate at higher positive potentials and even in low concen-trationsofelectrolyteinmobilephaseObtainedlimitsofdetectionwere mostly in submicromolar range and standard deviations ofmeasurementrepeatabilitywereunder5
KeywordsamperometrycarbonfeltFIAHPLC
combinationwithHPLC(forexampletheirdetectionpotentialslimitsofdetectionorlinearrange)
2Experimental
21Reagentsandchemicals
Stock solutions of propyl gallate butylhroxyanisole tert-butylhydroquinonebutylhydroxytoluene chlortoluron 2-amino-4-nitrophenol and 4-amino-2-
ndash3 ndash1nitrophenol (all SigmandashAldrich) with concentration of 1times10 molL werepreparedbydissolving theappropriateamountof therespectivesubstance inmethanol(HPLCgradeLach-NerCzechRepublic)Stocksolutionsof indole-3-aceticacidandindole-3-butyricacidwerepreparedinthesamemannerbutindeionized water Mobile phase consisted of methanol and phosphate-acetatebufferpreparedfromphosphoricandaceticacid(bothLach-NerCzechRepublic)andsodiumhydroxide(Fluka)
22Instrumentation
Theelectrochemicalcellconsistedofcarbonfelt(KarbotechnikCzechRepublic)flow-throughelectrodewhichwasplacedincapwithplatinumwireelectricalcontactanddrilledoutletholeononesideandwithflatferrulawithcapillaryonthe other side Schematic picture of the assembly is shown in ref [5] Otherelectrodeswereauxiliaryplatinumwireelectrodeandreferencesilverchloride(3MKCl) electrode (bothMonokrystaly Turnov Czech Republic) Potentiostatused in combination with this cell was Amperometric Detector ADLC 2(LaboratonıprıstrojePrahaCzechRepublic) HPLCapparatusconsistedofBeta10gradientpump(ECOMCzechRepublic)degasser DG 4014 (ECOM Czech Republic) six-way valve with 20microl loop(Rheodyne USA) HPLC column used for separation of propyl gallatebutylhydroxytoluenetert-butylhydroquinoneandbutylhroxyanisolefromtheirmixtureandforindole-3-aceticacidandindole-3-butyricacidfromtheirmixture
regwasLichrospher RP-18(125times4mm5micromMerckGermany)ForseparationofregchlortoluroncolumnPurospher RP-18(125times4mm5micromMerckGermany)was
used In case of mixture 2-amino-4-nitrophenol and 4-amino-2-nitrophenolregcolumnGemini C18110A (150times46mm5micromPhenomenexUSA)wasusedfor
separation Measurements of pHwere carriedout at Conductivity andpH-meter3510usingcombinedglasselectrode(JenwayUK)
142 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
3Resultsanddiscussion
Performanceofcarbonfeltwastestedonseveraltypesofanalyteswhichneededdifferent separation conditions namely amount of organic solvent in mobilephase and buffer pH The lowest amount of methanol (30) was used forseparation of 2-amino-4-nitrophenol and 4-amino-2-nitrophenol [3] Higherconcentrations of methanol in mobile phase was used for determination ofchlortoluronandforseparationofindole-3-aceticacidandindole-3-butyricacid[4]namely40and60respectivelyThehighestconcentrationsofmethanoland therefore electrolyte with lowest conductivity was used in separation ofantioxidantsnamelypropylgallatebutylhroxyanisolebutylhydroxytolueneandtert-butylhydroquinonewhereamountofmethanolwasrampingfrom55to95[5]DetectionpotentialsofmentionedanalytesandtheirlimitsofdetectionsareshowninTable1 HPLCseparationofantioxidantswas the firstmethodchosen for testingofcarbonfeltelectrodeperformancewiththistechniqueDuetodifferencesintheirstructure when butylhydroxytoluene has a different oxidation mechanismdetectionwith two appliedpotentialswas necessary According to the hydro-dynamic voltammograms potentials 14V and 08Vwere chosen for determi-nationofbutylhydroxytolueneandfortheotherthreeanalytesrespectively[5]AsshowninTable1whenthehigherpotentialwasappliedlimitsofdetectionforpropyl gallate butylhroxyanisole and tert-butylhydroquinone had increasedContrary determination of butylhydroxytoluene had approximately six timeslowerdetectionlimitathigherpotential
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 143
ndash1Compound E VLODmicromolL Refdet
Propylgallate 080 088 [5] 140 186 [5]Butylhroxyanisole 080 144 [5] 140 348 [5]tert-Butylhydroquinone 080 121 [5] 140 266 [5]Butylhydroxytoluene 080 3128 [5] 140 463 [5]Chlortoluron 140 013Indole-3-aceticacid 150 033 [4]Indole-3-butyricacid 150 054 [4]4-Amino-2-nitrophenol 080 016 [3]2-Amino-4-nitrophenol 080 021 [3]4-Amino-2-nitrophenol ndash080 35 [3]2-Amino-4-nitrophenol ndash080 37 [3]
Table 1Parametersofdetectionpotentialsandlimitsofdetectionfordifferentcompoundsusingcarbonfeltdetector
Detectionofauxins(indole-3-aceticacidandindole-3-butyricacid)wascarriedout at potential 15VMeasurements at this potential gives repeatabilitywithstandarddeviation31forindole-3-aceticacidand25forindole-3-butyricacidevenwithexchangingoftheworkingelectrodematerialCalibrationcurves
ndash1forbothanalyteswereobservedfrom04to100μmolL withlinearityinwholeconcentrationrangeLimitsofdetectionforbothanalytesreachedsubmicromolarconcentrationsevenwithrelativelyhighpotential[4] Incaseofdeterminationof2-amino-4-nitrophenoland4-amino-2-nitrophenolcarbon felt electrodewasused inbothoxidationand reductionmodeHydro-dynamicvoltammogramsshowedthatoptimaldetectionpotentialinreductionmodewasndash08VThislowpotentialisclosetotheendofthepotentialwindowandthereforeinterferenceswithremnantsofdissolvedoxygeninmobilephasewereobservedTheseinterferencesresultedinapproximately20timeshigherlimitofdetection for 2-amino-4-nitrophenol or 4-amino-2-nitrophenol obtained inreductionthaninoxidationmodeOntheotherhandmaximumvaluesoflinearrangewerethesameforbothanalytesinbothdetectionmodes[3] ForHPLCofchlortolurontheoptimaldetectionpotentialof14VwasfoundIts
ndash1calibration dependence although observed from 025 to 1000μmolL was ndash1linearonly in therange from025to50μmolL Limitofdetectionbasedon
ndash1standardsolutionswas013μmolL andreproducibilityofmeasurementgivenbytwentyconsecutivemeasurementsgaverelativestandarddeviationof05 ForallthedeterminationmethodsattentionwaspaidtotheapplicabilityofthecarbonfeltindetectionofanalytesincomplexmatricesIncaseofantioxidantsedibleoilswerechosenasrealsamples[5]nitrophenolderivatesweredeter-minedinurinesamples[3]andauxinsinrootingpreparation[4]ChlortolurondeterminationwasperformedinsoilandsurfacewaterThefoundvaluesshowanegligiblematrixinfluenceondetection
4Conclusions
CarbonfeltworkingelectrodewassuccessfullyusedincombinationwithHPLCfordeterminationofdifferenttypesofelectroactivecompoundsegantioxidantsauxinsorpesticidesAllmentionedapplicationsshowgreatperformanceofthecarbonfeltasaflow-throughelectrodematerialinelectroanalyticalchemistryforoxidationandreductionwayofanalytesdeterminationLimitsofdetectionforanalytes are mostly in submicromolar concentrations the exceptions areoxidationof analytes athigherpotentials and their reductionwhere limitsofdetectionsareinmicromolarconcentrationsApplicabilityoftheelectrodeonrealmatriceswas proven on analysis of edible oil samples groundwater soil androotingpreparation
Acknowledgments
ThisworkhasbeensupportedbytheCzechScienceFoundation(projectGACR20-01589S)
144 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
References
[1] Gonzalez-Garcia J Bonete P Exposito E Montiel V Aldaz ATorregrosa-Macia RCharacterizationofacarbonfeltelectrodeStructuralandphysicalpropertiesJMaterChem9(1999)419ndash426
[2] NavaJLRecendizAGonzalezLGCarrenoGMartın ezFMassTransportandpotentialstudiesinaflow-throughporouselectrodereactorPortugalElectrochimActa27(2009)381ndash396
[3] Dejmkova H Knaf M Application of carbon felt detector for the determination ofdinitrophenolmetabolitesInXXXIXModernElectrochemicalMethodsFojtaMSchwarzovaKNavratilT(Eds)U stınadLabemBestServis2019p41ndash43
[4] DejmkovaHdeAraujoDanielMElectrochemicaldeterminationofindole-3-aceticacidandindole-3-butyric acid using hplc with carbon felt detectorMonatsh Chem150 (2019)439ndash442
[5] DejmkovaHBarochMKrejcovaMBarekJZimaJCoulometricdetectorbasedoncarbonfeltApplMaterToday9(2017)482ndash486
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 145
1Introduction
Smith-Lemli-Opitzsyndrome(SLOS)isan autosomal recessive genetic disor-der firstly described in 1964 [1] It iscaused by the inborn deficiency of7-dehydrocholesterol reductase Thisenzyme transforms 7-dehydrocholes-terol (7-DHC Fig 1) to cholesterolduringthefinalstepofbiosynthesisofcholesterolincells The clinical symptoms of SLOS aredecreasedbloodlevelofcholesteroland
Electroanalytical methods for determination of 7-dehydrocholesterol in artificial serum
LENKABENESOVAADE LAZA RYBNICKA JANKLOUDAKAROLINASCHWARZOVA -PECKOVA
UNESCOLaboratoryofEnvironmentalElectrochemistryDepartmentofAnalyticalChemistryFacultyofScienceCharlesUniversityHlavova8203012843Prague2CzechRepublicbenesolenaturcunicz
Abstract7-DehydrocholesterolisabiomarkerofSmith-Lemli-Opitzsyndromeanautosomalrecessivegeneticdisordercausedbytheinborndefici-encyof7-dehydrocholesterolreductaseInthisstudyproceduresforitsdeterminationinartificialserumusingflowinjectionanalysiswithelectrochemical detection and voltammetric detection on borondoped diamond electrode were optimized The proteins wereprecipitatedbyacetonitrileandaftercentrifugationthesupernatantused for analysis For quantitation of 7-DHC by differential pulsevoltammetrytheoptimalratioacetonitrile-artificialserum91(vv)wasappliedInFIA-EDtheratio31(vv)runelectrolyteconsisting
minus1ofwater-acetonitrilecontaining001molL NaClO inthesameratio4minus1anddetectionpotentialof+13VvsAgAgCl(3molL KCl)were
usedQuantitationof7-DHCwaspossibleusingcalibrationdepen-minus1dencewithlimitdetectionof20micromolL inartificialserumNeverthe-
lessthemethodhaslowrecoveryandforsensitivedeterminationinreal matrices of human serum and amniotic fluid a liquid-liquidextraction needs to be applied to prevent presence of 7-dehydro-cholesterolinthephasewithprecipitatedproteins
Keywordsamperometricdetectionborondopeddiamond
electrode7-dehydrocholesteroldifferentialpulse
voltammetryflowinjectionanalysisSmith-Lemli-Opitz
syndrome
146 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Fig 1Structureof7-dehydrocholesterol
increased concentrationof7-DHC inbloodandnervous system [2] SLOS is acomplexofmultipleanomaliesincludingmentalretardationItismanifestedbyholoprosencephaly(anomaliesinbraindevelopmentwithimprecisedivisionintotherightandlefthemispheres)milddysmorphismscardiacrenalandgastro-intestinalmalformations ThecharacteristicfacialanomaliesofSLOS[2]aremicrocephalybitemporalnarrowing ptosis short nasal root short nose with anteverted nares andmicrognathia epicanthal folds and capillary hemangioma over the nasal rootextendingontotheglabellatheearappearlow-setandareposteriorlyrotatedOral finding includes a high-arched and narrow hard plate broad and ridgealveoralridgesandredundancyofsublinqualtissuesCNSanomaliesareagenesisorhypoplasiaBilateralandunilateralpostaxialpolydactylycanbepresentedinthehandsorfeetorboth Concentrationof7-DHCinbloodiscrucialforclinicaldiagnosticofSLOSinpatients Concentration levels in amniotic fluid are used for fetal diagnosticsTable1 summarizes concentration of 7-DHC in plasma and amniotic fluid ofhealthypersonandSLOSpatientsAnalyticalmethodsusedfordeterminationofconcentrationof7-DHCinthesematricesincludecombinationofGCorHPLCwithMS[3]orGCwithflameionizationdetection(FID)orUVdetection[45] Thepossibilitiesofelectrochemicalmethodsfordetectionof7-DHCarelimitedasgenerallythesteroidcoreisratherredox-inactive(detailinreview[6])undervarietyofconditionsNevertheless7-DHCpossessesconjugateddoublebondsonsteroidcoreanditsoxidationwasreportedinseveralstudies[7ndash9]Itsvoltam-metricsignal+095VvsSCEonglassycarbonelectrodewasfirstlyobservedin
minus1non-aqueousmediaofmethanol-benzene7525(vv)using005molL LiClO as4
supporting electrolyte in a study dealing with electrochemical behaviour ofvitaminAandDandtheirprovitaminsD(7-DHCisprecursorofvitaminD3)[7]Determinationof7-DHCinhumanskinispossiblebyHPLCwithUV(λ=286nm)andamperometricdetection(E +17VvsAgAgCl)onglassycarbonelectrodedet
minus1usingmethanol-tetrahydrofuran175mmolL KH PO (9514vvv)asmobile2 4minus1phase7-DHCwasdetectedintherangefrom12to81microgg dryweightwith
minus1detectionlimitof39pmolL [8]Anotherstudy[9]isdevotedtodeterminationof
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 147
minus1Author(s)ref Matrix ConcentrationμmolL Healthy SLOS
Kelley[4] Plasma 03plusmn001 385plusmn309 Amnioticfluid lt02plusmn001 16plusmn9Rossiteretal[5] Plasma lt5 179ndash335 Amnioticfluid lt03 12ndash15
Table 1Concentrationof7-dehydrocholesterolinclinicalmatricesforhealthypersonsandforSmith-Lemli-Opitzsyndrome(SLOS)patientsdetectedbyGC-FID
7-DHCandvitaminD3 in fishusingHPLCwithelectrochemicaldetectionTheanalytical cell was a serial combination of two-flow-through porous graphiteworking electrodes The first standard coulometric electrode was used toeliminatepotentially interfering compounds using the second lineardynamic
minus1rangefrom0013to0312micromolL for7-DHCwasachieved Hereinwestudiedpossibilitiestodetect7-DHCbasedonitsoxidationonborondoped diamond (BDD) electrode using differential pulse voltammetry andelectrochemicaldetectioninflowinjectionanalysis(FIA-ED)inartificialserumandperipherallyinhumanserumandamnioticfluid
2Experimental
21Reagentsandchemicals
7-dehydrocholesterol (purity95)wasobtainedSigmaAldrich (USA)and itsstandard solution was prepared in acetonitrile (Honeywell Germany) Theartificial serum was prepared from KCl (Penta Czech Republic) CaCl 2H O2 2
(PentaCzechRepublic)NaCl(PentaPragueCzechRepublic)urineD-glucoseand01albuminfromSigma-Aldrich(USA)NaClO4(PentaCzechRepublic)wasusedassupportingelectrolyte
22Instrumentation
VoltammetricmeasurementweregovernedbythepotentiostatPalmSensusingworking BDD electrode (Windsor Scientific UK d = 31 mm) AgAgNO 3
minus1 minus1(01molL AgNO 1molL NaClO in acetonitrile) non-aqueous reference3 4electrodeandplatinumwirecounterelectrodeBDDsurfacewaspolishedbeforeeachscanusingsuspensionofAl O (ElektrochemickedetektoryTurnovCzech2 3
Republic) HPLC system (Hitachi Merck) consisting of control unit D-7000gradientpumpL-7100autosamplerL-7200andUVdetectorL-7400wasusedfor
minus1FIA-EDdetectionof7-DHCRunelectrolytewascomposedof001molL NaClO 4inacetonitrileanddeionisedwaterinratio31(vv)Flowrateofmobilephase
minus1was30mlmin injectionvolumewas40microLandλ=280nmwasusedforUVdetectionWall-jet detection cell was employedwith working BDD electrode
minus1AgAgCl (3molL KCl) reference electrode and platinum wire auxiliaryelectrode Optimal detection potential of +13 V was controlled using ADLC2potentiostat(Laboratornı prıstrojePragueCzechrepublic)
3Resultsanddiscussion
In this study electroanalytical methods were developed for determination of7-DHCinartificialserumnamelyFIA-EDandDPVBothmethodsarebasedondirectoxidationof7-DHConborondopeddiamondelectroderesultinginanodic
148 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
peak at ca +08 V (vs AgAgNO in acetonitrile) in non-aqueous medium of3
acetonitrile or mixedmedium acetonitrile-water using NaClO as supporting4
electrolyteTheoxidationispresumablyinitiatedbyoneelectronremovalfromtheconjugateddoublebondsonthesteroidcoreof7-DHC Fordeterminationof7-DHCinrealmatricesitisnecessarytoremovepresentproteinsArtificialserumcontainingalbuminwasusedasmodelmatrixtostudythe possibilities Firstly albumin was removed simply by precipitation withacetonitrile(serum-water13(vv))andthesupernatantwasanalysed Differentratioswateracetonitrileweretestedinrunelectrolyte(59510902080 2575 3070 4060 and 5050 (vv)) to evaluate the influence of itscompositionontheFIA-EDsignaloftheblankand7-DHCThesameratio13asusedforprecipitationofalbuminwaschosenasoptimalbecauseofminimalandstablesignaloftheblankinjectedinFIA-EDsystemFurtherdetectionpotentialE in the range from +10 V to +15 V was optimized by evaluation of thedet
hydrodynamicvoltammogramsresultinginE of+13Vsetasoptimalvaluedetminus1 Concentrationdependenceof7-DHCislinearintherangefrom25micromolL to
minus1 minus1300micromolL (concentrationinartificialserum)withdetectionlimitof20micromolL and this concentration dependence can be used for quantitation of 7-DHC inartificial serum Nevertheless determination of 7-DHC in human serum andamnioticserumfailedastheyrepresentmorecomplicatedmatricesand7-DHCispresumablypartiallyadsorbedinthepresentproteinsandcannotbequantifiedinthesupernatant Furtherdifferentialpulsevoltammetrywithoptimizedparameterswasusedfordeterminationof7-DHCInthepresenceofproteinsinartificialserum(human
minus1serum albumin) an unacceptably high detection limit of 178micromolL wasachieved When the proteins were precipitated using acetonitrile (aceto-nitrileartificialserumratio91(vv))thelimitofdetectionof7-DHCwaslowered
minus1to15micromolL inartificialserumNeverthelesstherecoveryofthemethodwasonly43to70dependingontheconcentrationof7-DHCagainreflectingthelossof7-DHCduetoproteinprecipitation Thereforeasecondapproachofsamplepretreatmentbasedonliquid-liquidextractionofalllipidsdescribedin[10]wastested(Bligh-Dyerextraction)TheprocedurehastwopartsFirstlymethanolchloroformandthesampleofartificialserum is mixed and shaken to form a monophasic system After addition ofchloroform and water a biphasic system is formed where chloroform phasecontainsalloflipidcompoundsandmethanol-waterphasecontainsallnon-lipidscompoundsChloroformphaseisthendriedunderN2atmosphereat50degCanddried extract dissolved in acetonitrile Preliminary experiments using DPVresultedinrecoveryof97forBligh-Dyerextractionof7-DHCfrominartificialserum
Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020 149
4Conclusions
FIA-EDandDPVwereoptimizedfordeterminationof7-DHCinartificialserumUsing precipitation of proteins by acetonitrile limit of detection of 7-DHC in
minus1artificialserumusingFIA-EDwas20micromolL andthismethodcanbeusedfortheirquantificationusingcalibrationdependenceNeverthelessdeterminationusingDPVisunreliableduetolowrecoveryoftheprocedureDevelopmentofamethodincludingliquid-liquidextractionstepisinprogresssothat7-DHCcouldbedeterminedinrealmatricesashumanserumandamnioticfluid
Acknowledgments
TheresearchwassupportedbytheCzechScienceFoundation(projectGACR19-11268S)andtheSpecificUniversityResearch(SVV260560)
References
[1] Smith DW Lemli L Opitz JM A newly recognized syndrome of multiple congenitalanomaliesJPediatr64(1964)210minus217
[2] Nowaczyk M Waye J The SmithndashLemlindashOpitz syndrome a novel metabolic way ofunderstandingdevelopmentalbiologyembryogenesisanddysmorphologyClinGenet59(2001)375minus386
[3] BeckerSRohnikeSEmptingSHaasDMohnikeKBebloSMutzeUHusainRAThieryJCeglarekULC-MSMS-basedquantificationofcholesterolandrelatedmetabolitesindriedblood for the screening of inborn errors of sterolmetabolismAnal Bioanal Chem407(2015)5227minus5233
[4] Kelley RI Diagnosis of Smith-Lemli-Opitz syndrome by gas-chromatography mass-spectrometryof7-dehydrocholesterolinplasmaamniotic-fluidandculturedskinfibroblastsClinChimActa236(1995)45minus58
[5] Rossiter JP Hofman KJ Kelley RI Smith-Lemli-Opitz SyndromePrenatal-diagnosis byquantification of cholesterol precursors in amniotic-fluid Am J Med Genet 56 (1995)272minus275
[6] KloudaJBarekJNesmerakKSchwarzova-PeckovaKNon-enzymaticelectrochemistryincharacterization and analysis of steroid compounds Crit Rev Anal Chem 47 (2017)384minus404
[7] AtumaSSLundstromKLindquistJTheelectrochemicaldeterminationofvitaminAPartIIFurthervoltammetricdeterminationofvitaminAandinitialworkonthedeterminationofvitaminDinthepresenceofvitaminAAnalyst100(1975)827minus834
[8] MoodyJPHumphriesCAAllanSMPatersonCRDeterminationof7-dehydrocholesterolinhumanskinbyhigh-performance liquid-chromatography JChromatogrB530 (1990)19minus27
[9] OstermeyerUSchmidtTVitaminDandprovitaminDinfishEurFoodResTechnol222(2005)403minus413
[10] BlighEGDyerWJArapidmethodoftotallipidextractionandpurificationCanJBiochemPhysiol37(1959)911minus917
150 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Author Index
AlikovaV1
AugustınM83
BaluchovaS19
BarekJ192570
BarochM141
BasB13129
BastryginaO41
BenesovaL146
BessonovaE57
BohmD6
BurkinK116
BurkinM116
ChernovaA141
ChoinskaM70
CokrtovaK104
DedinaJ97
DeevV57
DejmkovaH141
DendisovaM63
DubenskaL51
EfremenkoE41
FojtaM110
GalvidisI116
HavranL110
HeiglN31
HertJ76
HrdlickaV70
JosypcukB25
KartsovaLA3557
KloudaJ19146
Kodrık ovaB90
KolobovaEA35
KorbanA135
KorotkovaE1
KralM63
KratzerJ90
KravchenkoAV35
KrızekT76104
LipinskaJ129
MadejM129
MatejkaP63
MatysikF-M631
MusilS9097123
NavratilT70
OndrackovaA110
PietrzakK45
PlotnikovaK51
PoradaR13
RedondoBR70
SagapovaL90
ShormanovV1
Schwarzova-PeckovaK19110146
SladkovaS 141
S tadlerovaB97
StiborovaM110
SvobodaM90
TvorynskaS25
TyczkowskiJ129
VyhnanovskyJ97123
VymyslickyF76
VyskocilV83
WardakC45
WongDKY19
ZarybnickaA146
ZelenyI51
Proceedingsofthe16thISCModernAnalyticalChemistry Prague2020 151
152 Proceedingsofthe16thISCModernAnalyticalChemistryPrague2020
Keyword Index
alcoholicbeverages135
aminoglycosides116
amperometricdetection146
amperometry141
antifoulingelectrodes19
assembledcapillaries6
atomicabsorptionspectrometry90
atomicfluorescencespectro-
metry97
atomization90
biologicalactiveanalytes35
biosensor2583
bismuth97
borondopeddiamond
electrode110146
cadmium90
canagliflozin76
capillarycoating35
capillaryelectrophoresis635
capillaryflowinjectionanalysis631
carbohydrates31
carbonfelt141
cathodicstrippingvoltammetry70
chemicalvaporgeneration90
chemometrics57
cobalt123
coldplasmadeposition129
copper(II)phthalocyanine63
covalentimmobilization25
cytochromeP450110
damage83
7-dehydrocholesterol146
designofexperiments76
differentialpulsevoltammetry146
diphenylsilanereductionmethod19
dispersiveliquid-liquid
microextraction57
disposableelectrodes31
DNA83
dualdetectionconcept6
electrochemicalanalysis110
electrochemicalapplications129
electrochemicalflowcell76
electrochemistry51
electrokineticchromatography104
eliminationvoltammetrywith
linearscan70
ELISA116
enzymaticreactor25
FIA141146
gaschromatography-mass
spectrometry(GC-MS)135
glucoseoxidase25
graphite83
honey116
HPLC76141
hydridegeneration97
hydrogenatedconical-tipcarbon
electrodes19
imidazoliumionicliquids35
inductivelycoupledplasmamass
spectrometry123
internalstandardmethod135
ion-selectiveelectrode45
laccase25
liquidcrystals104
massspectrometry31
mercuryelectrode13
metronidazole51
non-aqueouscapillary
electrophoresis104
non-aqueoussystem6
oxidation76
oxytetracyclinehydrochloride51
phenol2-methoxy1
photochemicalvaporgene-
ration97123
polarography51
1-propanesulfonicacid23-
dimercapto-70
pulsedamperometricdetection31
quantitation1
resonanceRamanspectroscopy63
scanningtunnellingmicroscopy63
silversolidamalgamelectrode70
Smith-Lemli-Opitzsyndrome146
smokingmixtures41
solidcontact45
solid-phasemicroextraction57
spectrophotometry141
SudanI110
surfacemodification129
surface-enhancedRaman
spectroscopy63
thinlayers129
tip-enhancedRamanspectroscopy
63
unithiol70
uranyl45
vanillin41
veterinarydrug51
vitamins13
volatilecompoundsquantifi-
cation135
voltammetricdopaminedetec-
tion19
voltammetry1383
Proceedingsofthe16thISCModernAnalyticalChemistry Prague2020 153
Proceedings of the 16th International Students Conference ldquoModern Analytical Chemistryrdquo
EditedbyKarelNesmerak
PublishedbyCharlesUniversityFacultyofScience
Prague2020
1steditionndashvi154pages
ISBN978-80-7444-079-3
ISBN 978-80-7444-079-3
Pro
ceedin
gs of th
e 16
th In
ternatio
nal Stu
den
ts Co
nferen
ce ldquoMo
dern
An
alytical Ch
emistryrdquo P
rague 2
02
0
788074 440793
Prague 17mdash18 September 2020
Edited by Karel Nesměraacutek
Prague 2020
Proceedings of the
16th International Students Conference
ldquoModern Analytical Chemistryrdquo