protein purification lab c2 pages 115 to 168 lab c.2 four periods protocol page 135-160 benchtop...
TRANSCRIPT

Protein Purification Lab C2Pages 115 to 168
Lab C.2
Four Periods
Protocol Page 135-160
Benchtop Protocols begin on page 398
Be sure to read theory starting page 120

Exam
• Exam March 3,4
• Includes Carbohydrates, Enzyme kinetics, and all protein labs and material related there to.
• Pay attention to the powerpoints– Read theory sections in the lab manual
• Will be about one hour in length
• Example of exam with answers is posted on web

You Have:
• Become skilled at using micro pipetters• Have learned to use the spectrophotometer
– To determine concentration of an unknown• Beers Law
– To measure activity of an enzyme
• Have learned how to organize experimental protocols
• Have learned how to prepare a report.

In the next days
• You will use all of these skills to perform a fundamental exercise in Biochemistry/Molecular Biology
• Will learn basic protocols in protein purification and analysis

Protein Purification
• A black art (proteins have personality)• Requires knowledge of protein
– What kind of cell is it coming from– What part of cell– What does it do
• Particularly helpful– Size– Composition

Strategy
• Move from organism to pure protein in as few steps as possible with as little loss of activity (assayable quality) as possible– Time and temperature are factors

Protocols for Protein Purification
• Highly individualized
• Use a common approach– Fractionate crude extract in a way that protein
of interest always goes into the pellet or the supernatant.
– Follow progress with functional assay

Lactate Dehydrogenase
• NADH + H+ + Pyruvate =NAD+ + Lactate• Enzyme clears lactic acid from working muscles• The obvious source of enzyme is muscle tissue
(heart & skeletal muscle, H&M, isomers)• We will assay for the enzymes ability to convert
Pyruvate to Lactate

Begin with intact tissue• Disrupt (step4&5)
– Blender, homoginizer
• Remove debris (step7)– Centrifugation
• Precipitate/concentrate (step 14-16)– Ammonium sulfate
• Remove salt (step 22)– dialysis
• Purify (next Lab)– Chromatography
• Analyze (Part B and week 3 & 4)– Activity, molecular weight

Ammonium Sulfate pptpage 124
• Has a wide range of application• Relies on fact that proteins loose solubility as
concentration of salt is increased– Is characteristic of particular protein– Results in a partial purification of all proteins with
similar solubility characteristics– Must determine [amm sulf] to precipitate your protein
empirically.
• Produces “salt cuts”

Salting in / Salting out
• Salting IN• At low concentrations,
added salt usually increases the solubility of charged macromolecules because the salt screens out charge-charge interactions.
• So low [salt] prevents aggregation and therefore precipitation or “crashing.”
• Salting OUT• At high concentrations
added salt lowers the solubility of macromolecules because it competes for the solvent (H2O) needed to solvate the macromolecules.
• So high [salt] removes the solvation sphere from the protein molecules and they come out of solution.

Kosmotrope vs. ChaotropePage 125
• Ammonium Sulfate• Increasing conc
causes proteins to precipitate stably.
• Kosmotropic ion = stabilizing ion.
• Urea• Increasing conc
denatures proteins; when they finally do precipitate, it is random and aggregated.
• Chaotropic ion = denaturing ion.

Dialysis Page 180
• Passage of solutes through a semi-permeable membrane.
• Pores in the dialysis membrane are of a certain size.
• Protein stays in; water, salts, protein fragments, and other molecules smaller than the pore size pass through.
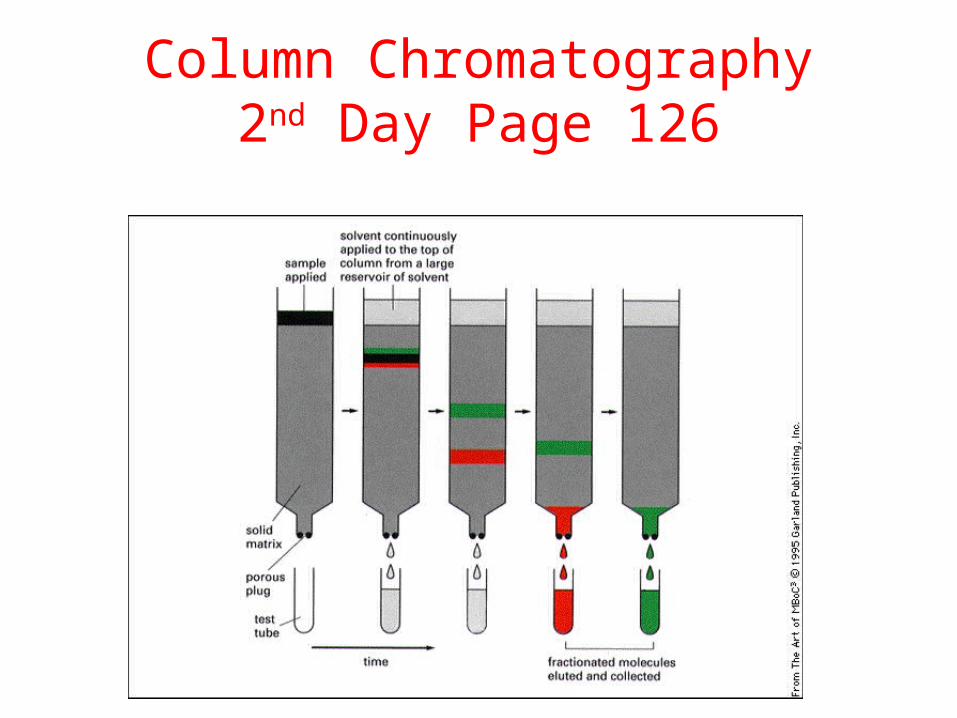
Column Chromatography2nd Day Page 126

Available in any volume

Gel Filtration
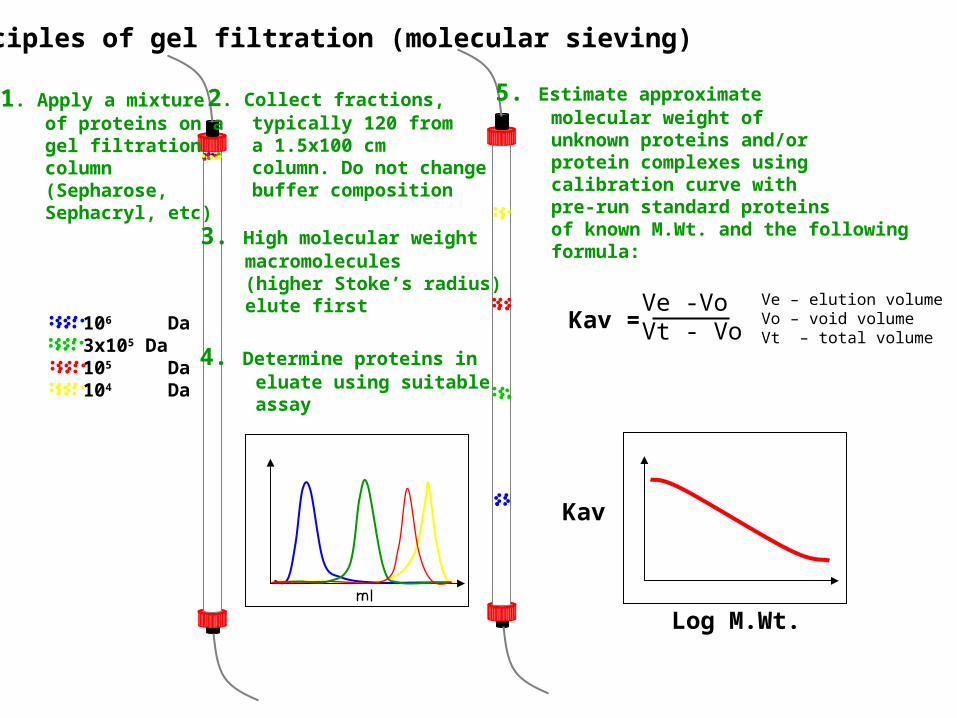
Principles of gel filtration (molecular sieving)
106 Da3x105 Da105 Da104 Da
1. Apply a mixture of proteins on a gel filtration column (Sepharose, Sephacryl, etc)
2. Collect fractions, typically 120 from a 1.5x100 cm column. Do not change buffer composition
3. High molecular weight macromolecules (higher Stoke’s radius) elute first
4. Determine proteins in eluate using suitable assay
5. Estimate approximate molecular weight of unknown proteins and/or protein complexes using calibration curve with pre-run standard proteins of known M.Wt. and the following formula:
Kav = Ve -VoVt - Vo
Ve – elution volumeVo – void volumeVt – total volume
Kav
Log M.Wt.

Ion Exchange
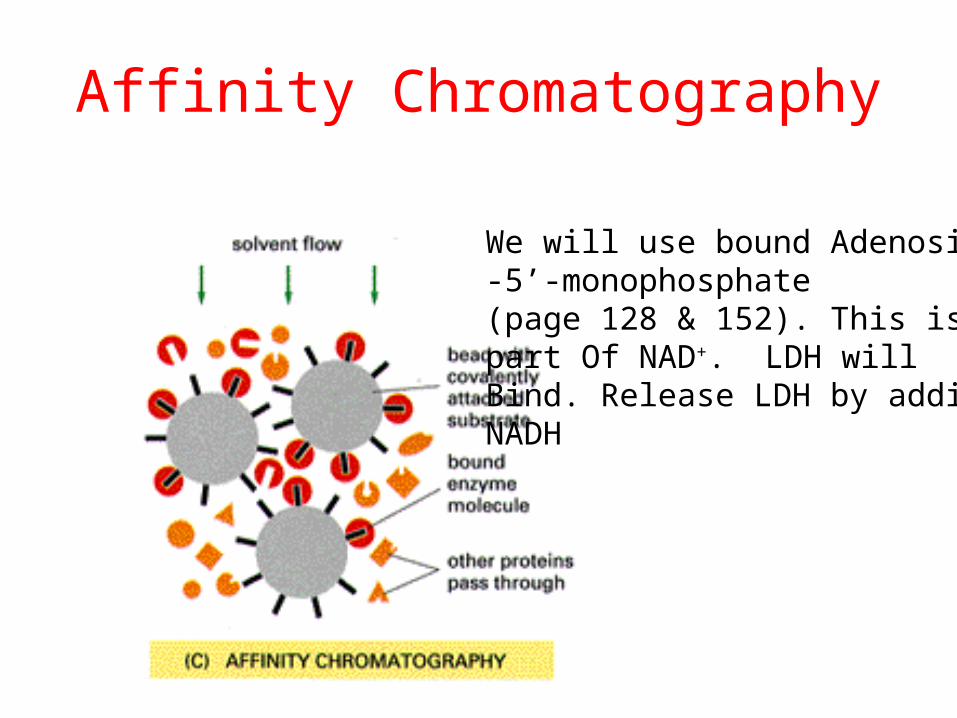
Affinity Chromatography
We will use bound Adenosine-5’-monophosphate (page 128 & 152). This is part Of NAD+. LDH will Bind. Release LDH by adding NADH

NAD+
AMP

Affinity chromatography
• Remember: NADH is a co-substrate for lactate dehydrogenase.
• We use AMP-Sepharose: AMP is covalently bound to the affinity gel, which will not pass through the filter.
• LDH binds to the AMP b/c it looks like half an NADH.
• Thus LDH remains immobilized in the column until we add NADH which binds tighter to the LDH.
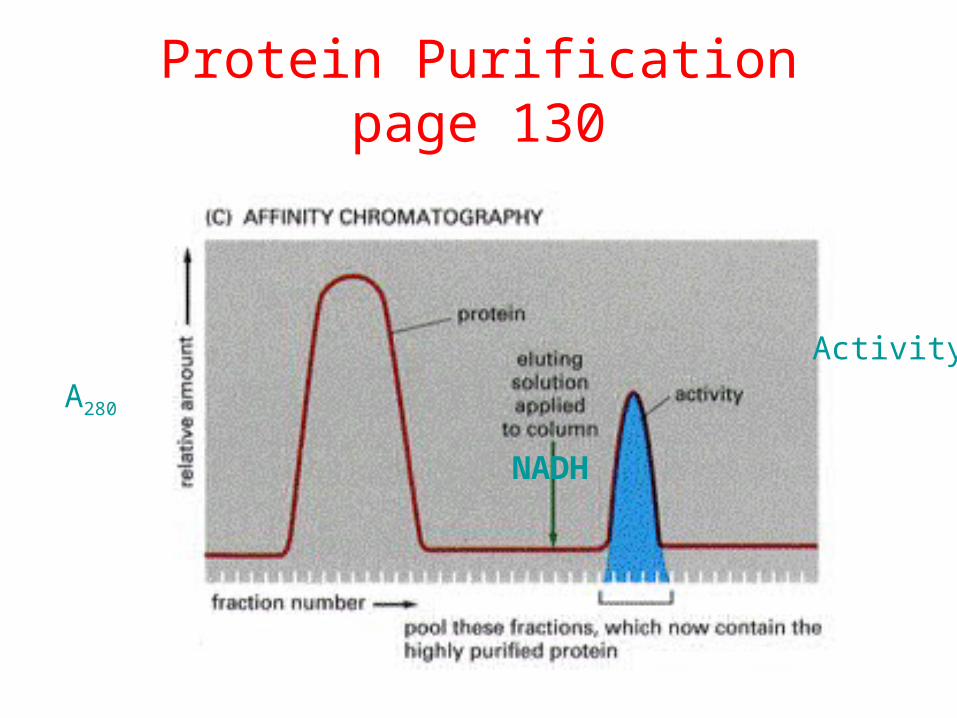
Protein Purificationpage 130
A280
Activity
NADH

Protein Concentration
• Lowry ( most cited reference in biology)– Color assay
• A280
– Intrinsic absorbance Page 132– Relies on aromatic amino acids
• BCA page 133– Modification of Lowry: increased sensitivity and
consistency
• Bradford– Shifts Amax of dye from 465nm to 595nm

A280 Page 132
• Uses intrinsic absorbance
• Detects aromatic residues – Resonating bonds
• Depends on protein structure, native state and AA composition
• Retains protein function
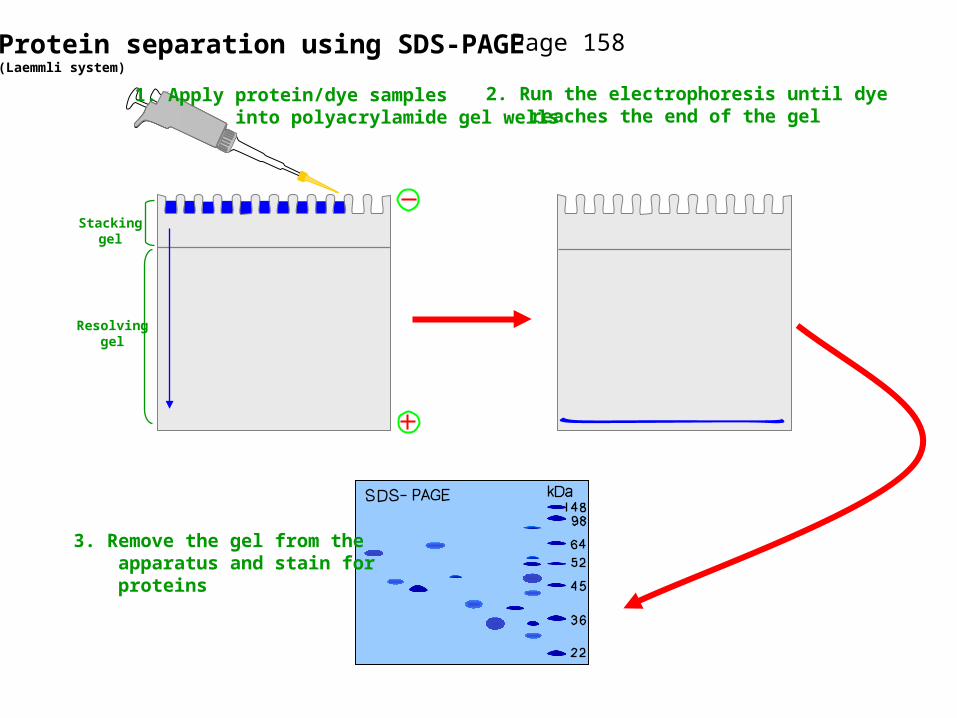
Protein separation using SDS-PAGE(Laemmli system)
Stackinggel
Resolvinggel
1. Apply protein/dye samples into polyacrylamide gel wells
2. Run the electrophoresis until dye reaches the end of the gel
3. Remove the gel from the apparatus and stain for proteins
Page 158

SDS PAGE of Purification Process
1. Complete mix of proteins2. High Salt3. Ion exchange4. Gel-filtratio5. Affinity
10micrograms loaded in each lane

IMPORTANT
• Do not throw away anything until you are certain you no longer need it– Biggest source of problem in this lab
• Label everything clearly copy labels into lab book
• Throwing out wrong fraction results in starting over– 3 days into experiment huge problem
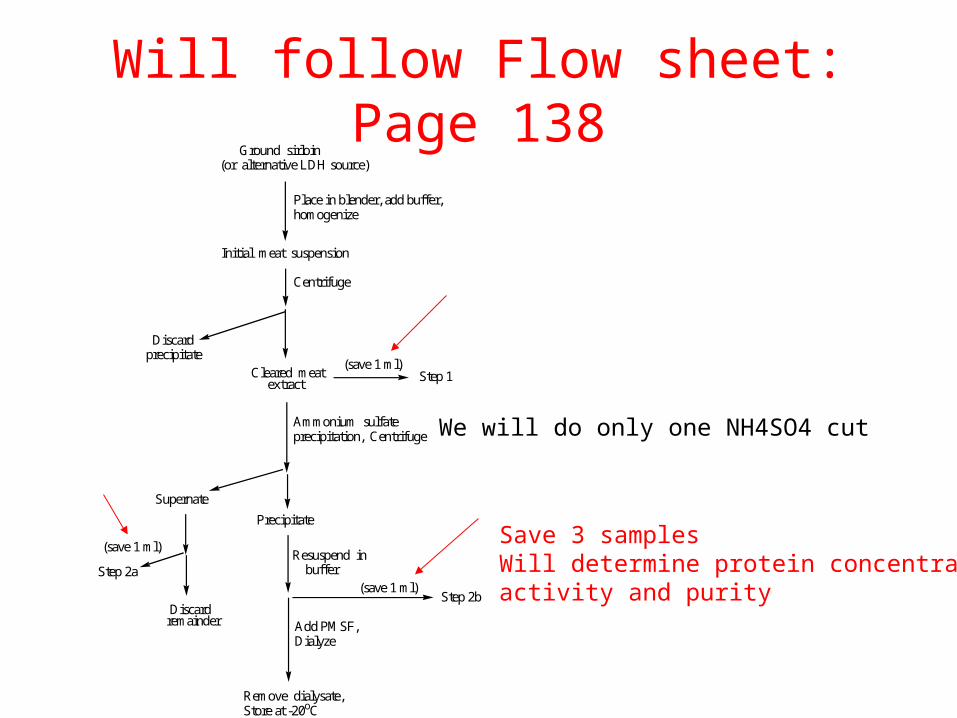
Will follow Flow sheet: Page 138
We will do only one NH4SO4 cutAmmonium sulfate precipitation, Centrifuge
Precipitate
Supernate
Step 2b
Remove dialysate,Store at -20oC
Ground sirloin(or alternative LDH source)
Place in blender, add buffer,homogenize
Initial meat suspension
Centrifuge
Cleared meat extract
(save 1 ml)
Discard precipitate
(save 1 ml)
Step 1
Discard remainder
Resuspend in buffer
Add PMSF,Dialyze
Step 2a(save 1 ml)
Save 3 samplesWill determine protein concentrationactivity and purity

Will fill out this critical table as we proceed page 162
Table C.2-4. Enzyme Purification Table
Net volume(ml)
V0 units per
ml
V0 units
Total(an “amount”)
Protein content
(% of total)
Proteinconcentration
(mg/ml)
Net amount
of protein(mg)
Specific Activity(V0/mg
protein)
Step A B C D E F G
1. Cleared
2. (NH4)2SO4
Supernatant
3. diluted dialyzed sample/ solution placed on column
4. pooled peak tubes from column
Column C = (Column A)(Column B)Column F = (Column A)(Column E)Column G = Column C/Column F = Column B / Column EColumn D = Column C/first value in Column C

Today. Page 138 (part of group)
• Steps 1-5: Weigh muscle sample place in blender with 50ml ice cold buffer homogenize for 2 minutes.
• Steps 6&7: remove large debris by centrifugation Save Supernatant (remove 1ml (Microfuge tube) for later analysis).
• Steps 9-13: Measure the volume of the supernatant determine amount of ammonium sulfate required for precipitation, weigh out 0.4 grams per/ml (NH4)2SO4

Today group 1 continued
• Step14-16: Slowly add salt to gently stirred supernatant . Keep Cold!!See step 12
• Step 17: Centrifuge precipitate to a pellet• Step 18-21: Save supernatant (1ml in microfuge
tube). Suspend pellets in 5ml cold buffer• Step 22, 23: Add PMSF and place suspended
pellet in dialysis tubing and give to TA

Today group 2
• Set up standard assay as on page 142– Measure loss of absorbance as NADH is converted to
NAD+• Step 4 is similar to Kinetic curve you did for ADH
(page 124) only reversed as measure loss of absorbance
• Steps 8-12: You will determine the velocity of LDH catalyzed reaction by varying the concentration of LDH with constant substrate and cofactor. Be sure to adjust the amount of reaction buffer to give 3.2 ml final volume in each assay

Very Important: Page 145
0
0.2
0.4
time (sec)60 120 1800
0
0.2
0.4
time (sec)60 120 1800
A B
observed
observed
extrapolated timecourse
Blank without NADH Blank with NADH

Today group 2 continued
• You are establishing the assay conditions you will use next week to follow the purification of LDH. You must become proficient at this assay.

Flow chart 1B (page 142)Prepare the reaction mixtures
Each reaction will contain 3.200 ml:
3.00 ml 50 mM buffer, pH 7.5
50 l NADH
100 l Enzyme solution, column fraction or diluted Step 1, 2, 3 or 4
Zero the spectrophotometer:Add buffer and pyruvate to the cuvette then set the zero.
Add NADH and check the A340 value.
Determine A340 at 15 sec and 45 sec after adding the enzyme sample.
Note: You may have to adjust the time frame of the rate measurement or the amount of added enzyme to achieve a non-spurious V0 value.
50 l pyruvate
Calculate V0.
Divide the raw answer by the product of 340 (for NADH) times the cuvette path length to convert the units to mole/liter per sec units.
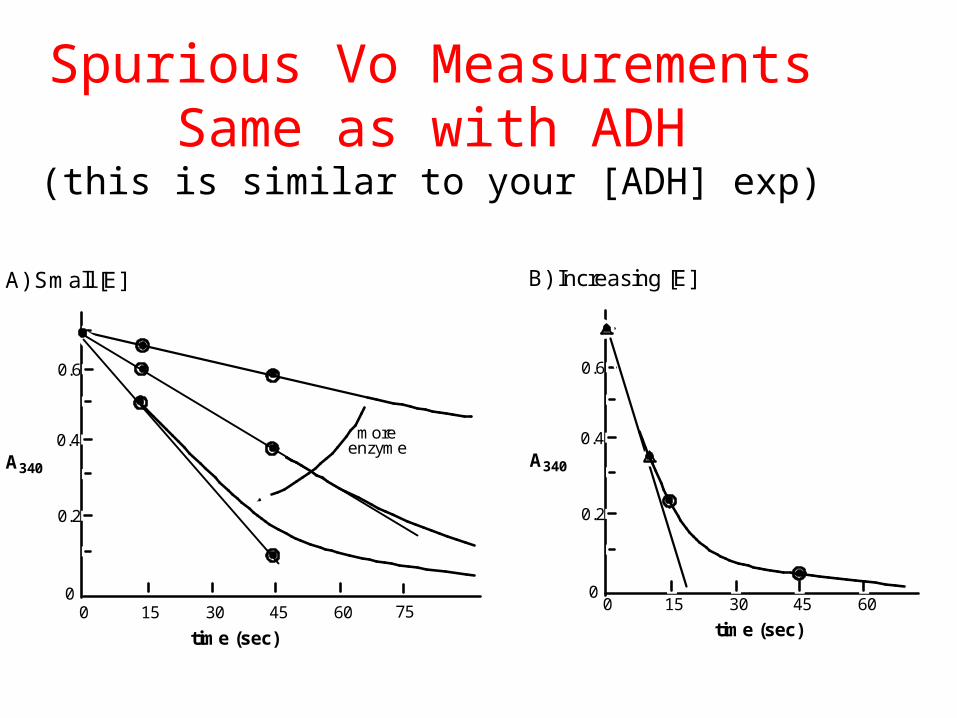
Spurious Vo MeasurementsSame as with ADH
(this is similar to your [ADH] exp)
0
0.2
0.4
0.6
0 15 30 45 60
more enzyme
time (sec)
A340
0
0.2
0.4
0.6
0 15 30 45 60
time (sec)
A340
A) Small [E] B) Increasing [E]
75

Procedure (Page 143)
• 1 Step 1-6. Will create a kinetic curve for LDH (adjust volume of buffer to make 3.2ml)– Similar to ADH
• 2. Repeat kinetic curve with different concentrations of enzyme– This is protocol you will use as you purify LDH
• Do this assay on the unknown samples from step one and 2a from group 1.

C2-3. Page 144Table C.2-3. Lactate Dehydrogenase Reaction Time Courses
Reading number
time(seconds)
A340 readings
50 ml sample
100 ml sample
200 ml sample
300 ml sample
400 ml sample
1 0
2 15
3 30
4 45
5 60
6 75
7 90
8 105
9 120

Next Week Column Chromatography
• Due next time: Prelab assignment for period 2 of ‘LDH Purification’
• You really should write up or otherwise arrange what you did today as soon as possible. Do Not Trust Your Memory

Next lab
• Need member of group to be here at 1:30 to begin washing column
• Will need to measure absorbance at 280 to determine that contaminating protein is lost from column. Wash and measure until A280 is constant.

Strategy
• For samples generated determine amount of protein (A280 ) and activity
• Activity per microgram of protein =s specific activity
• You strive for maximal activity per unit of protein. (table C2-4 Column G, Page 162)

Will generate this elution profilePage 153
A280 V0
fraction (tube) number (approximate only)
NADH added
0
0 10 20 30 40 50 60 70 80
contaminant protein
LDH

Will fill out this critical table as we proceed page 162 (day 4)
Table C.2-4. Enzyme Purification Table
Net volume(ml)
V0 units per
ml
V0 units
Total(an “amount”)
Protein content
(% of total)
Proteinconcentration
(mg/ml)
Net amount
of protein(mg)
Specific Activity(V0/mg
protein)
Step A B C D E F G
1. Cleared
2. (NH4)2SO4
Supernatant
3. diluted dialyzed sample/ solution placed on column
4. pooled peak tubes from column
Column C = (Column A)(Column B)Column F = (Column A)(Column E)Column G = Column C/Column F = Column B / Column EColumn D = Column C/first value in Column C

This Lab
• 4 lab periods
• Prelab= 12 points
• Lab Report= 50 points
• First exam in period 4