protein s-nitrosylation: purview and parameters
TRANSCRIPT

150 | FEBRUARY 2005 | VOLUME 6 www.nature.com/reviews/molcellbio
R E V I E W S
of S-nitrosylation/de-nitrosylation, and the spatial regu-lation (targeting) of S-nitrosylation within and betweenproteins, confer specificity to NO-derived effects. Thespecific, regulated functions of S-nitrosylation contrastwith the exertion of global control that is ascribed to sys-tems that influence cellular redox state (BOX 1), and allowS-nitrosylation to function as a prototype of mechanismsthat convey redox-based cellular signals6.
Protein S-nitrosothiol: turnover and homeostasisAnalysis of the S–N-bond dissociation energies of manyRSNOs (where R is the organic moiety that bears theSNO) has shown that (unimolecular) homolyticdecomposition (that is, the scission of the S–N bond toyield the NO• and thiyl radicals) is typically precluded,and therefore that reductive mechanisms and NO-group transfer chemistry mainly control physiologicalRSNO decomposition15–17. It is important to note that,in diverse cell types, many SNO-proteins are detectedin situ7,14,18, and a portion of basal SNO content is stablein the presence of NOS inhibitors19,20, which reflects thelocalization of liganded NO to privileged sites withinproteins that are inaccessible to solvent and/or providestabilizing interactions with proximate residues (BOX 2).These findings emphasize that the regulation of proteinfunction by S-nitrosylation is subserved by mechanismsthat control both the addition and the removal of theNO group from a Cys thiol. As detailed below, thosemechanisms might be represented, in a significantpart, by changes in protein conformation that alter the
A function for nitric oxide (NO) in signal transductionwas established by the demonstration that NO that isgenerated by endothelial cells relaxes vascular smoothmuscle, in part, through the activation of guanylatecyclase1. The extensive range of NO-based signallingwas indicated by the discovery of constitutivelyexpressed NO SYNTHASES (NOSs) with pervasive phylogeneticand tissue distributions2. It was recognized early onthat, in addition to the well-characterized binding ofNO to the haem iron of guanylate cyclase, physiologi-cal NO-based protein modification might be effected byS-nitrosylation — that is, the coupling of an NO moietyto a reactive cysteine thiol (to form an S-nitrosothiol,SNO; FIG. 1)3–5. Over the past decade, the number ofreported substrates for S-nitrosylation has grown towell over a hundred6, which is consistent with theubiquity of regulatory and/or active-site thiols acrossprotein classes. Initial studies relied to a great extenton in vitro analysis using exogenous NO sources, whichdid not necessarily recapitulate the cellular milieu.However, a substantial body of recent work, which is described in this review, has directly implicated S-nitrosylation in the regulation of numerous signallingpathways in intact cellular systems, and recent geneticevidence supports a diversity of regulatory roles for thisprotein-modification reaction7–9.
The current zeitgeist reflects a growing awarenessthat S-nitrosylation is a post-translational protein mod-ification that is regulated precisely in time andspace6,10–14. The temporal (stimulus-coupled) regulation
PROTEIN S-NITROSYLATION:PURVIEW AND PARAMETERSDouglas T. Hess*, Akio Matsumoto‡, Sung-Oog Kim*, Harvey E. Marshall* andJonathan S. Stamler*‡
Abstract | S-nitrosylation, the covalent attachment of a nitrogen monoxide group to the thiol sidechain of cysteine, has emerged as an important mechanism for dynamic, post-translationalregulation of most or all main classes of protein. S-nitrosylation thereby conveys a large part of the ubiquitous influence of nitric oxide (NO) on cellular signal transduction, and provides amechanism for redox-based physiological regulation.
*Departments of Medicineand Biochemistry, Box 2612Duke University MedicalCenter, Durham, NorthCarolina 27710 USA.‡Howard Hughes MedicalInstitute, Duke UniversityMedical Center, Durham,North Carolina 27710, USA.Correspondence to J.S.S.e-mail:[email protected]:10.1038/nrm1569
NO SYNTHASE
(NOS). Mammals have three NO synthases that generate NOfrom Arg — NOS1 or nNOS,NOS2 or iNOS and NOS3 oreNOS — one or more of whichreside, or can be induced, inmost or all cell types. NOShomologues are distributedbroadly across the phylogeny.

NATURE REVIEWS | MOLECULAR CELL BIOLOGY VOLUME 6 | FEBRUARY 2005 | 151
R E V I E W S
S-NITROSO-GLUTATHIONE
(GSNO). The main non-protein S-nitrosothiol (SNO) in cells, which functions in anequilibrium with protein SNOs(the equilibrium is determinedin part by the enzyme GSNO-reductase, which metabolizesGSNO (see TABLE 1)).
OXIDOREDUCTASE
An enzyme that catalysesoxidation–reduction reactions,which entail the transfer ofelectrons from a substrate thatbecomes oxidized (electrondonor) to a substrate thatbecomes reduced (electronacceptor).
electrostatic environment, hydrophobicity, contiguityand orientation of aromatic side chains, and proximityof target thiols to transition metals or redox centres, aswell as by protein–protein interactions that might controlthose changes or otherwise promote S-nitrosylation/de-nitrosylation. In addition, S-nitrosylation can pro-mote or inhibit the formation of disulphide linkageswithin or between proteins, depending on thiol proximityand orientation15,21.
Promotion of S-nitrosylation/de-nitrosylation. In general,the NO moiety can be provided by NO itself, nitrite, otherNO
xspecies (higher NO oxides), metal–NO complexes or
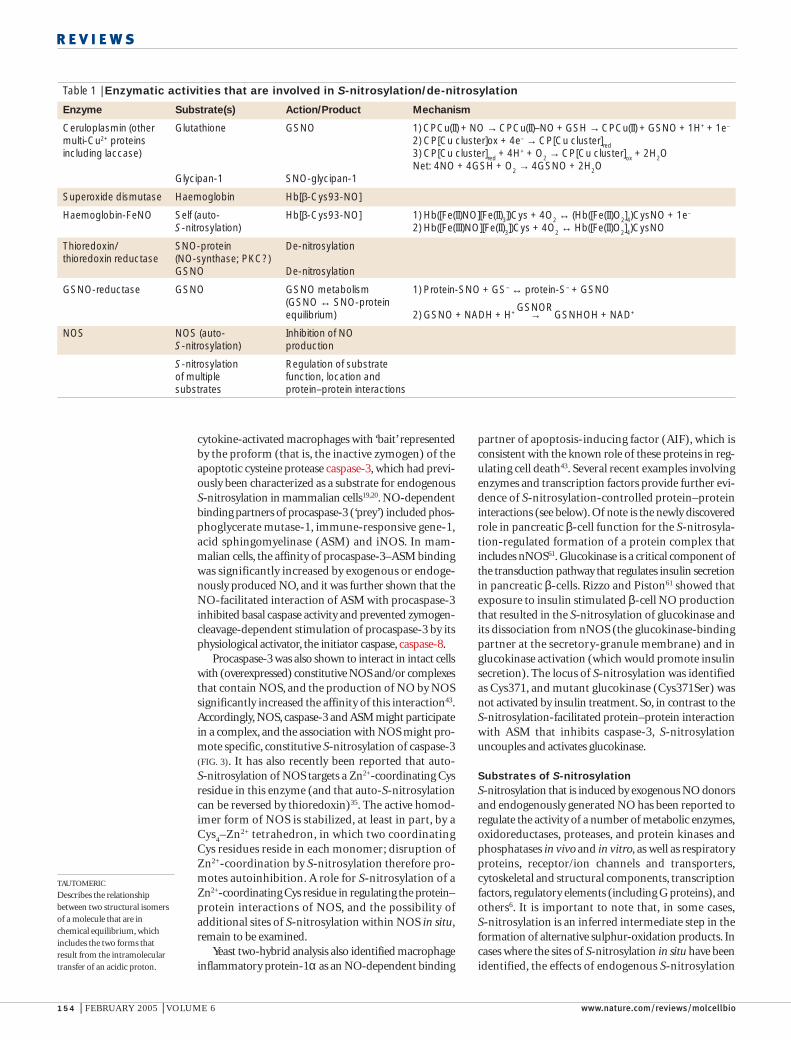
various SNOs10. Although specific enzymatic mecha-nisms that function primarily in protein S-nitrosylation(other than the NOSs) have not yet been described,several enzymes are known to promote S-nitrosylationor de-nitrosylation reactions. In particular (see TABLE 1),Cu,Zn superoxide dismutase (SOD) catalyses the S-nitrosylation of haemoglobin by NO in solution22 orNO that is derived from S-NITROSO-GLUTATHIONE (GSNO)23,and the Cu2+-containing protein ceruloplasmin catalysesS-nitrosylation of the heparan-sulphate proteoglycan,glypican, in situ24 and can catalyze the formation ofGSNO from NO25.
Protein-bound transition metals — in particularCu2+ or Fe2+ that are bound in close proximity to a tar-get thiol — can catalyse the transnitrosylation (that is,the NO-group transfer) from GSNO or nitrite of ‘reac-tive’ Cys residues within serum albumin, haemoglobin(HB) and calbindin, which represents a form of auto-S-nitrosylation26–29. Also flavins might support theauto-S-nitrosylation of mitochondrial proteins such assarcosine dehydrogenase30. A principal source of the NOgroup in mammalian S-nitroso-haemoglobin underphysiological conditions is the haem iron-nitrosylspecies (FeNO) that is formed initially from NO, whichoriginates from NOS, low-mass SNOs or nitrite29.Non-haem FeNOs, in particular dinitrosyl–iron com-plexes (DNICs), can also function as a source of NOgroups31,32, and DNICs and other Cu2+/Fe2+-containingproteins such as ceruloplasmin might account forsome examples of S-nitrosylation that are independentof combinatorial NO/O
2chemistry (that is, the O
2-
dependent generation of nitrosylating NOx
species)10,30.Transnitrosylative, thiol-to-thiol SNO formation isrepresented by the transfer of an NO group from athiol of S-nitrosohaemoglobin directly to an adjacentthiol within a haemoglobin-binding partner — theerythrocytic membrane protein Band-3 (REFS 17, 33).De-nitrosylation can be carried out by the OXIDOREDUCTASE
thioredoxin (TRX), which reverses S-nitrosylation-induced inhibition of NOS34,35 and NO-mediated inhi-bition of protein kinase C (REF. 36). Both TRX and SODcan also liberate NO from small-molecular-weightSNO in vivo23,37, as might protein disulphide isomerases.
GSNO is thought to be the main non-protein SNOin cells and in extracellular fluids38, and Liu et al.39 dis-covered that the glutathione-dependent formaldehydedehydrogenase functions across phylogeny and cell typeas a specific GSNO reductase (GSNOR) to metabolize
Figure 1 | Cellular sources and targets of nitric oxide. a |Nitric oxide (NO) is generated, in most cell types, from Arg andO2 by NO synthases (NOSs), and potentially by the reduction ofNOx species that are derived from exogenous and endogenoussources. Separate genes code for neuronal NOS (nNOS orNOS1) and for endothelial NOS (eNOS or NOS3), which arenamed after the tissues in which they were discovered, as wellas for inducible/Ca2+-independent NOS (iNOS or NOS2).Although nNOS and eNOS are expressed constitutively, theirlevels might be induced further, and iNOS can also beexpressed constitutively. Cells might possess more than oneNOS, in different subcellular compartments. Compart-mentalization of NOS and its targets is illustrated in part b. Thesources of nitrosylating groups might include NOx species thatoriginate exogenously (for example, NO2
– from food or frombacterial flora or infecting agents) or endogenously (for example,NO oxidation products, including N2O3). And NO might beexchanged between transition metals and thiols. Initial analysesemphasized the binding of NO to the haem Fe2+ of guanylatecyclase. Subsequent research indicates that a principal target of cellular NO is the thiol group of Cys in peptides and proteins,and that S-nitrosylation conveys a large part of the ubiquitousregulatory influence of NO on cellular signal transduction. b | In the neuronal signalling module that is illustrated, NO that is generated by NOS after NMDAR stimulation can activateguanylate cyclase, and S-nitrosylate and activate G proteins(green), whereas feedback S-nitrosylation of NMDAR and ofNOS is inhibitory (red; see main text).
Arginine+
O2
NO synthase(nNOS, eNOS, iNOS)
NO
Oxidation/reduction
NOx
Citrulline
Exogenous(microbial andfood sources)
Cys–S–NO
Metal–NO
a
NO
NONO
NO
S
S
S
Receptor(NMDA)
Scaffold(PSD95)
Cys
Cys
Cys
G-cyclase
HaemFe
NOS(type 1)
Adaptor(CAPON)
Dexras
NOSCysRas
b

152 | FEBRUARY 2005 | VOLUME 6 www.nature.com/reviews/molcellbio
R E V I E W S
most well-characterized determinants of specificityinclude: electrostatic interactions that control thiolpKa (NUCLEOPHILICITY), as initially described for HB45 ;hydrophobic compartmentalization41 (BOX 3); ALLOSTERIC
REGULATORS (for example, Ca2+, Mg2+ and O2/redox) that
can modulate thiol (solvent) accessibility or reactivity46,47;and, interactions between NOSs and proteins that aretargets for S-nitrosylation.
SNO motifs. The thiol of Cys93 within the β-subunitof HB is alternately apposed to basic or acidic sidechains as a function of the O
2-regulated conformation
of the HB tetramer45. And oxygenation and deoxy-genation of HB promotes the S-nitrosylation and de-nitrosylation of β-Cys93, which allows red blood cellsto deliver vasodilatory NO bioactivity along with O
2, in
accordance with local metabolic demand in the arterialperiphery (referred to as hypoxic vasodilation)33,45,48–51.
The analysis of NO transfer in HB provided the basisfor a proposed ‘acid–base’ motif for protein S-nitrosyla-tion (and de-nitrosylation)52, which has proved to bepredictive of sites of S-nitrosylation in a number of cases— especially when protein models are available thatallow deduction of the proximity of charged side groupsand thiol that emerges from tertiary or quaternarystructure41,52. As originally suggested52, the acid–basemotif comprises flanking acidic (Asp, Glu) and basic
GSNO39. Importantly, although this enzyme does notact on SNO-protein substrates, GSNOR deficiencyresults in increases in intracellular SNO-protein levelsunder basal conditions that are greatly enhanced in acti-vated (inducible (i)NOS-producing) cells. This demon-strates the existence of a cellular equilibrium that is basedon the transfer of NO groups between populations ofsmall-mass and protein thiols20,39. It should be noted thatGSNO might participate in protein S-nitrosylation notonly through NO+ chemistry (that is, thiol-to-thioltransnitrosylation), but also by functioning as a sourceof free-radical NO that is generated by homolytic cleav-age. NO release from GSNO is observed in the presenceof endogenous cellular reductants and transitionmetals23,26,40 and, as noted above, reductive cleavage ofGSNO might be catalysed by SOD and TRX23,37 (andperhaps other enzymes).
S-nitrosylation: determinants of specificityAlthough one or more isoforms of NOS reside in mostcell types, and most proteins possess Cys residues, sub-strate specificity is a salient characteristic of endoge-nous protein S-nitrosylation. In addition, in virtually allproteins in which the site (or sites) of S-nitrosylationhave been identified, the modulation of their functionunder physiological conditions results from the modi-fication of one, or only a few, Cys thiols12,41–44. The
NITROSATIVE AND OXIDATIVE
STRESS
The dysregulated productionand/or metabolism of reactivenitrogen and/or oxygen species,which generate nitrosativeand/or oxidative chemistries thatcan result in disrupted cellularsignalling, injury and death.
NUCLEOPHILICITY
Nucleophilic groups such as the sulphur group of Cys areelectron rich, and the degree ofnucleophilicity is a measure of their reactivity towardselectrophiles (electron-deficientspecies such as NO+), asexemplified by the increasedreactivity of cysteine thiolateversus sulfhydryl.
ALLOSTERIC REGULATOR
An ion or small molecule thatreacts with and therebymodulates the conformationand function of proteins.A number of such allostericregulators, including O
2and
Ca2+, have been implicated in the regulation of protein S-nitrosylation.
METALLOPROTEIN
A protein that contains a metalion (or ions; such as Fe2+, Cu2+
or Zn2+) as a prosthetic group,which is coordinated by amino-acid side chains.
Box 1 | Redox-based modification of protein cysteine thiols
Cysteine thiols (Cys-SH) can undergo a range of nitric oxide(NO)-dependent electrophilic and oxidative modifications aswell as NO-independent oxidation by reactive oxygen species(ROS). These modifications can be viewed as a continuum(see figure) that relates levels (amount, origin, spatio-temporal distribution) of reactive NO/higher NO oxides(NO
x)/SNO to the form and consequences of modification.
From this perspective, the progression from S-nitrosylationto sulphenic acid (SOH)/disulphide (SS), to sulphinic (SO
2–) and irreversible sulphonic (SO
3–) acids represents a graded
transition from signalling functions through adaptation to NITROSATIVE AND OXIDATIVE STRESS and, finally, to toxicity.(SS formation can be internal, or mixed between proteins or between a protein and a peptide; and other irreversiblemodifications such as tyrosine nitration can also occur.) It should be noted that the accumulation of protein SNOs is theonly mechanism of NO toxicity (nitrosative stress)8,116,117,144 that has been shown by strict genetic criteria8, althoughother mechanisms are likely.
Nitrosative or oxidative stress that results from increased or dysregulated production of reactive NO- or O2-derived
species is countered by several cellular mechanisms, which are triggered, in part, by nitrosative/oxidative thiolmodifications of proteins that directly, or through transcriptional activation, mobilize adaptive (protective) responses.However, sustained and/or extreme redox-related stress is associated with irreversible protein modification andpathophysiological sequelae. Therefore, reversible modifications — including the formation of protein sulphenic acid or disulphide linkages (intramolecular, or mixed disulphides including glutathionylation) — are, for the most part,associated with the homeostatic maintenance of the cellular redox state, whereas irreversible modifications, including the formation of some sulphinic acids and of sulphonic acids, are associated with irreversible alterations in cell fate.
Among the various possible nitrosative/oxidative modifications by SNO/NOx/NO, only S-nitrosylation has
unequivocally been shown to function not only in redox homeostasis (as exemplified by mammalian thioredoxin and bythe bacterial transcription factor OxyR) and toxicity (that is, apoptotic cell death and septic shock), but also in conveyingor regulating physiological cellular signals. These include signals that are transmitted along transduction cascadestriggered by ligand–receptor interactions (for example, the neuronal N-methyl-D-aspartate receptor (NMDAR) and thepancreatic β-cell insulin receptor), and the post-translational regulation that is exerted at several stages along signal-transduction pathways (see FIGS 4,5). In addition, S-nitrosylation functions uniquely to generate and deliver NO-derivedbioactivity (for example, the vasodilatory activity of erythrocytic haemoglobin). S-nitrosylation therefore functions asthe prototype of redox-based, thiol-dependent cellular-signalling mechanisms6.
NO/NOx/SNO
Signalling
SNOSOH
SSSO2H
SO3H
Adaptation (redox state)Pathophysiology

NATURE REVIEWS | MOLECULAR CELL BIOLOGY VOLUME 6 | FEBRUARY 2005 | 153
R E V I E W S
of ≤6 Å). In addition, the coordination of Zn2+ by Cysthiols in METALLOPROTEINS can result in significantdecreases in thiol pKa, and it has been shown that theinteraction between contiguous and appropriatelyorientated aromatic side chains and Cys thiols pro-motes formation of the thiolate anion53–55. Theacid–base catalysis of S-nitrosylation/de-nitrosylationis illustrated in the analysis by Pérez-Mato et al.56 ofGSNO-induced S-nitrosylation of hepatic methionineadenosyltransferase, which generates the methyl donorS-adenosylmethionine (FIG. 2). A novel, potential rolefor the coordination by acidic and basic side chains ofNO-group transfer from GSNO to protein is detailed inBOX 4 (‘GSNO motif ’). In addition, hydrophobic pock-ets in proteins might represent a ‘hydrophobic motif ’for S-nitrosylation (BOX 3).
Allostery. Ions (Ca2+, Mg2+, H+) and O2-based modifi-
cations, which can cause changes in protein structure,have been shown to promote S-nitrosylation and/orde-nitrosylation45–47,51. These findings indicate that the binding of NO to Cys residues might be linked to thebinding (or redox actions) of these regulators and that,more generally, protein allostery might be a significantfactor in specifying the sites of S-nitrosylation.
Compartmentalization of NO sources and targets. Animportant determinant of the selectivity of S-nitrosy-lation between proteins in situ is provided by the colocal-ization of NO sources and targets within subcellularcompartments on the basis of specific protein–proteininteractions of the NOSs. The most well-characterizedexample of such modularity is provided by the multipro-tein complex that incorporates neuronal (n)NOS, at thepostsynaptic specialization of mammalian neurons(FIG. 1b). nNOS in neurons is largely membrane bound, byvirtue of ‘post-synaptic density protein of 95 kDa, Discslarge and Zona occludens-1’ (PDZ)-domain interactionswith the postsynaptic scaffolding protein, PSD93/95(REF. 57). The N-methyl-D-aspartate receptor (NMDAR)also binds to PSD93/95 (REF. 58) and is thereby broughtinto proximity with nNOS. The NMDAR is both a prin-cipal source of Ca2+ flux that regulates nNOS activity anda substrate of nNOS-dependent S-nitrosylation7,59. Inaddition, nNOS is linked by the adaptor protein CAPONto the small G-protein Dexras1, which is S-nitrosylatedand activated by NMDAR-mediated nNOS stimulation60.
Some protein–protein interactions of the NOSs havebeen shown to be dynamic and regulated, and recentresults indicate that one important regulatory signal is,in fact, NOS-generated NO, which controls complexformation through S-nitrosylation of binding partners43.
S-nitrosylation and protein–protein interactionsCellular signal transduction relies essentially on induced,stimulus-coupled protein–protein interactions. The roleof S-nitrosylation in regulating the assembly of signallingcomplexes was examined by Matsumoto et al.43 whodeveloped a modified yeast two-hybrid system to iden-tify NO-facilitated protein–protein interactions (FIG. 3).They screened a cDNA library that was derived from
(Arg, His, Lys) residues, and it is well-established thatdeprotonation of thiol to the nucleophilic thiolate (RS–)is suppressed and enhanced, respectively, by neighbour-ing acidic and basic groups (generally, with a proximity
Box 2 | The nature of the S-nitrosothiol (SNO) linkage
Homolytic bond dissociation energies (BDEs) of the S–N bond in S-nitrosothiols (SNOs;shown in part a) can vary from ~22 to 32 kcal, which represents theoretical predictedlifetimes of seconds to years — in fact, protein SNOs will probably span that range15,16.The wide variation of homolytic and heterolytic BDEs results from the variable nature ofthe RS–NO linkage, which is influenced by the nature of the R-group, by the propensity toaccept an unpaired electron from elsewhere in the protein or from alternative e– donors,and by proximate ionizing and aromatic species. This variability also reflects themechanism of NO addition: NO+ transfer, versus NO-radical (NO•)–thyil-radical (S•)addition, versus the formation of an SNO radical anion intermediate (which is formed in adirect reaction of NO• and S–)10,145. That is, the SNO group in proteins and peptides mighthave more or less double-bond character (C–S–N=O ↔ C–S+=N–O–), might carry anunpaired electron (which creates radical species of varying predicted stability; RSNO•–,RSN•OH or RSNHO•), or might interact with a thiol or other nucleophile to form an SNO2(formally, a nitroxyl disulphide or N-hydroxy-nitrosamine)146. The nature of the SNOlinkage is reflected in the S–N bond length and in the CSNO dihedral angle.A 0° (syn) or180° (anti) planar CSNO dihedral angle is energetically preferred for S-nitrosothiols147
(see figure, part a). However, in the crystal structure of mammalian haemoglobin that is S-nitrosylated at Cys93 of the β-subunit, analysis indicates a CSNO dihedral angle of~70°–90° (see figure, part b), which might represent a Cys–NO radical148 that can exist inprotonated TAUTOMERIC forms (K. N. Houk, personal communication).An additionalprediction is that the CSNO dihedral in HB will vary depending on the proteinconformation51. The fact that the nature of the SNO linkage can vary substantially and beinfluenced by the presence of stabilizing and destabilizing groups, sources of protons andelectrons, and by other aspects of protein structure, has important methodologicalimplications for the analysis of protein (and small-molecular-weight) SNOs17. SNOstandards should replicate the BDEs and reactivities of the assayed SNOs, and deviationsfrom physiological pH, ionic strength and redox status that alter protein structure orelectronic state, and thereby profoundly alter SNO stability, should be avoided.
Dihedral 0°Syn (cis)
Dihedral 70°–90°
Dihedral 180°Anti (trans)
bβ-Cys93–NO β-Cys93–NO
S
NO
S
NO
a
Backbone
154 | FEBRUARY 2005 | VOLUME 6 www.nature.com/reviews/molcellbio
R E V I E W S
partner of apoptosis-inducing factor (AIF), which isconsistent with the known role of these proteins in reg-ulating cell death43. Several recent examples involvingenzymes and transcription factors provide further evi-dence of S-nitrosylation-controlled protein–proteininteractions (see below). Of note is the newly discoveredrole in pancreatic β-cell function for the S-nitrosyla-tion-regulated formation of a protein complex thatincludes nNOS61. Glucokinase is a critical component ofthe transduction pathway that regulates insulin secretionin pancreatic β-cells. Rizzo and Piston61 showed thatexposure to insulin stimulated β-cell NO productionthat resulted in the S-nitrosylation of glucokinase andits dissociation from nNOS (the glucokinase-bindingpartner at the secretory-granule membrane) and inglucokinase activation (which would promote insulinsecretion). The locus of S-nitrosylation was identifiedas Cys371, and mutant glucokinase (Cys371Ser) wasnot activated by insulin treatment. So, in contrast to theS-nitrosylation-facilitated protein–protein interactionwith ASM that inhibits caspase-3, S-nitrosylationuncouples and activates glucokinase.
Substrates of S-nitrosylationS-nitrosylation that is induced by exogenous NO donorsand endogenously generated NO has been reported toregulate the activity of a number of metabolic enzymes,oxidoreductases, proteases, and protein kinases andphosphatases in vivo and in vitro, as well as respiratoryproteins, receptor/ion channels and transporters,cytoskeletal and structural components, transcriptionfactors, regulatory elements (including G proteins), andothers6. It is important to note that, in some cases,S-nitrosylation is an inferred intermediate step in theformation of alternative sulphur-oxidation products. Incases where the sites of S-nitrosylation in situ have beenidentified, the effects of endogenous S-nitrosylation
cytokine-activated macrophages with ‘bait’ representedby the proform (that is, the inactive zymogen) of theapoptotic cysteine protease caspase-3, which had previ-ously been characterized as a substrate for endogenousS-nitrosylation in mammalian cells19,20. NO-dependentbinding partners of procaspase-3 (‘prey’) included phos-phoglycerate mutase-1, immune-responsive gene-1,acid sphingomyelinase (ASM) and iNOS. In mam-malian cells, the affinity of procaspase-3–ASM bindingwas significantly increased by exogenous or endoge-nously produced NO, and it was further shown that theNO-facilitated interaction of ASM with procaspase-3inhibited basal caspase activity and prevented zymogen-cleavage-dependent stimulation of procaspase-3 by itsphysiological activator, the initiator caspase, caspase-8.
Procaspase-3 was also shown to interact in intact cellswith (overexpressed) constitutive NOS and/or complexesthat contain NOS, and the production of NO by NOSsignificantly increased the affinity of this interaction43.Accordingly, NOS, caspase-3 and ASM might participatein a complex, and the association with NOS might pro-mote specific, constitutive S-nitrosylation of caspase-3(FIG. 3). It has also recently been reported that auto-S-nitrosylation of NOS targets a Zn2+-coordinating Cysresidue in this enzyme (and that auto-S-nitrosylationcan be reversed by thioredoxin)35. The active homod-imer form of NOS is stabilized, at least in part, by aCys
4–Zn2+ tetrahedron, in which two coordinating
Cys residues reside in each monomer; disruption ofZn2+-coordination by S-nitrosylation therefore pro-motes autoinhibition. A role for S-nitrosylation of aZn2+-coordinating Cys residue in regulating the protein–protein interactions of NOS, and the possibility ofadditional sites of S-nitrosylation within NOS in situ,remain to be examined.
Yeast two-hybrid analysis also identified macrophageinflammatory protein-1α as an NO-dependent binding
TAUTOMERIC
Describes the relationshipbetween two structural isomersof a molecule that are inchemical equilibrium, whichincludes the two forms thatresult from the intramoleculartransfer of an acidic proton.
Table 1 | Enzymatic activities that are involved in S-nitrosylation/de-nitrosylation
Enzyme Substrate(s) Action/Product Mechanism
Ceruloplasmin (other Glutathione GSNO 1) CPCu(II) + NO → CPCu(II)–NO + GSH → CPCu(II) + GSNO + 1H+ + 1e–
multi-Cu2+ proteins 2) CP[Cu cluster]ox + 4e– → CP[Cu cluster]redincluding laccase) 3) CP[Cu cluster]red + 4H+ + O2 → CP[Cu cluster]ox + 2H2O
Net: 4NO + 4GSH + O2 → 4GSNO + 2H2OGlycipan-1 SNO-glycipan-1
Superoxide dismutase Haemoglobin Hb[β-Cys93-NO]
Haemoglobin-FeNO Self (auto- Hb[β-Cys93-NO] 1) Hb([Fe(II)NO][Fe(II)3])Cys + 4O2 ↔ (Hb([Fe(II)O2]4)CysNO + 1e–
S-nitrosylation) 2) Hb([Fe(III)NO][Fe(II)3])Cys + 4O2 ↔ Hb([Fe(II)O2]4)CysNO
Thioredoxin/ SNO-protein De-nitrosylationthioredoxin reductase (NO-synthase; PKC?)
GSNO De-nitrosylation
GSNO-reductase GSNO GSNO metabolism 1) Protein-SNO + GS– ↔ protein-S– + GSNO(GSNO ↔ SNO-proteinequilibrium) 2) GSNO + NADH + H+ → GSNHOH + NAD+
NOS NOS (auto- Inhibition of NOS-nitrosylation) production
S-nitrosylation Regulation of substrateof multiple function, location and substrates protein–protein interactions
GSNOR

NATURE REVIEWS | MOLECULAR CELL BIOLOGY VOLUME 6 | FEBRUARY 2005 | 155
R E V I E W S
caspase-9 is de-nitrosylated following stimulation bytumour necrosis factor α (TNFα)65. The results ofMatsumoto et al.43 indicate that de-nitrosylation mightactivate caspase-3 not only by unblocking the active-siteCys, but also by relieving inhibitory protein–proteininteractions (FIG. 3). Inhibition of caspase activity by S-nitrosylation provides one mechanism throughwhich NO generated by constitutive NOSs exerts itswell-characterized anti-apoptotic function.
Phosphatases. Members of the protein tyrosinephosphatase (PTPase) superfamily are characterizedby a highly conserved active-site motif that includesa catalytic Cys residue flanked by a His residue — as isthe case for the cysteine proteases. Numerous studieshave shown the inhibition of PTPases by endogenouslyproduced REACTIVE OXYGEN SPECIES (ROS), and it has alsobeen shown that the exposure of purified PTPases toNO+ donors leads to reversible enzyme inhibition.Exposure of intact cells to exogenous NO/SNO orinduction of iNOS inhibits tyrosine-phosphatase activ-ity and results in increased levels of phosphotyrosyl
(activation versus inhibition, and the regulation ofprotein–protein interactions and/or subcellular localiza-tion) are correlated with targeting of catalytic and/orregulatory Cys residues.
Enzymes with active-site thiolsCaspases. Cys residues are present at the active site andare involved in catalysis in a wide range of enzymes62,63.In the original description of protein S-nitrosylation3,5,it was shown that the enzymatic activity of cathepsin Bwas inhibited by an NO-derived active-site thiol modifi-cation. It has been shown subsequently that most, or all,of the caspase cysteine proteases can be S-nitrosylated attheir active-site Cys with the consequent inhibition ofenzyme activity 64. Mannick et al.19 demonstrated thatprocaspase-3 is constitutively S-nitrosylated in situ at itsactive-site Cys, and that it is selectively de-nitrosylatedfollowing apoptotic stimulation by Fas. It was thenshown that constitutively S-nitrosylated procaspase-3was localized predominantly to mitochondria, andthat most mitochondrial caspase-9 (an initiator caspase)is constitutively S-nitrosylated as well64. Endogenous
REACTIVE OXYGEN SPECIES
Reduced derivatives ofmolecular oxygen (O
2),
including, in particular, thesuperoxide radical (O
2• –) and
hydrogen peroxide (H2O
2),
which can have significantreactivity towards biologicalmacromolecules and towardsother reactive small molecules.
Box 3 | Hydrophobicity and S-nitrosylation
Local hydrophobicity might promote specific S-nitrosylation.In support of this, a role has been shown for nitrosylation bynitric oxide (NO) itself10,145 or by other hydrophobic NOoxides (NO
x), including in particular NO
2/N
2O
3/N
2O
4
(which might be generated as NO/O2
or NO/superoxidereaction products). NO and O
2partition preferentially into
the hydrophobic phase of mixed (that is, hydrophilic andhydrophobic) systems — as shown by the large increase inthe reaction rate of the NO/O
2combination in biological
membranes149 — and hydrolysis of NOx
is inhibited in ahydrophobic milieu.Accordingly, S-nitrosylation that is basedon NO auto-oxidation or direct reaction of NO with thiolate(in the presence of electron acceptors or stabilizing groups)is favoured in hydrophobic compartments (see figure). Cysresidues that are subject to physiological S-nitrosylationhave been shown in a number of cases to reside in ajuxtamembrane zone90,99, which might expose them tobiologically significant fluxes of nitrosylating NO products.
Nedospasov et al.150 showed that regions of high localhydrophobicity within proteins promoted S-nitrosylation ofresident Cys residues (a form of micellar catalysis). Suchhydrophobic compartmentalization can arise from tertiaryprotein structure and protein–protein interactions. Theryanodine receptor/Ca2+-release channel of skeletal muscle(RyR1) is S-nitrosylated by endogenously generated NO at one of ~50 free thiols (Cys3635), which is intercalated within ahydrophobic region of an identified calmodulin (CaM)-binding domain41,46,91 (see figure).
Radical-based reaction mechanisms (reviewed in REF. 10) might generate protein S-nitrosothiols (SNOs)10,151, throughone of two mechanisms. First, the reaction of free-radical NO with the one-electron oxidation product of Cys, the thiylradical (as in S-nitroso-glutathione (GSNO)-mediated Cu,Zn superoxide dismutase (SOD)-catalysed S-nitrosylation ofCys93 in the β-subunit of haemoglobin23, and perhaps NO
2•-catalysed S-nitrosylation of Cys3635 by NO in RyR1 (REF. 10);
see figure, part b). Or, second, through the formation of a radical intermediate (RS– + NO → RSNO•–) that can yield SNOin the presence of an electron acceptor (for example, O
2reduced to superoxide, NO
2• reduced to NO
2– or NAD+ reduced to
NAD•)145. The concentration and stabilization of lipophilic NO2• might often promote the extraction of an electron from
thiol to enable thiyl-radical/NO-radical addition in a hydrophobic milieu10,151 (see figure, part b). In general, high localhydrophibicity (and/or protonation) would sequester (and stabilize) radical species and, in addition, impede hydrolysisof SNO to sulphenic acid (and further oxidation to sulphinic and sulphonic acids).
N2O3
NO
S
NO•
NO2•
NO•
O2
Cys3635
Hydrophobic
Hydrophillic
Calmodulin
KSKKAVWHKLLSKQRRRAVVACFRMTPLYNLPTHRACNMFLES
Ca2+ Ca 2+ Ca2
+Ca 2+
a
b

156 | FEBRUARY 2005 | VOLUME 6 www.nature.com/reviews/molcellbio
R E V I E W S
(ASK1). Dimmeler et al.75 showed that TRX is S-nitrosylated constitutively in endothelial cells at asingle allosteric Cys (Cys69) that lies outside theactive site (which contains Cys32 and Cys35), andthat S-nitrosylation of Cys69 is a principal, facilita-tory determinant of the oxidoreductase capacity ofendothelial cells (FIG. 4).
ASK1 functions as a mitogen-activated proteinkinase (MAPK) kinase kinase, which activates the JunN-terminal kinase (JNK) and p38 MAPK pathways andis required for TNFα-induced apoptosis. The analysisof Haendeler et al.75 indicated that S-nitrosylation ofTRX Cys69 is necessary for its basal anti-apoptoticfunction in endothelial cells. However, it has recentlybeen reported that treatment with GSNO can induceS-nitrosylation of TRX in intact cells and dissociateASK1 (REF. 76), possibly by targeting active-site Cys32and/or Cys35.Accordingly, whereas basal S-nitrosylationof Cys69 will facilitate oxidoreductase and anti-apoptoticfunctions, S-nitrosylation of active-site Cys (whichmight be induced by nitrosative stress8) might exert anapoptotic effect. This would occur through the releaseof ASK1 and the inhibition of oxidoreductase function(including ROS scavenging), which would result fromactive-site thiol modification (FIG. 4). Recently, Park et al.77 have reported that ASK1 is itself a substrate forinhibitory S-nitrosylation in situ (FIG. 4). S-nitrosylationof Cys869 inhibited the binding of ASK1 to the princi-pal downstream effector substrates, MAPK kinase(MKK)3 and 6.
The kinases JNK1–3 are additional members of theMAPK family. Park et al.78 showed that the inductionof iNOS, or treatment with an NO+ donor, inhibitedJNK-mediated phosphorylation and transactivation ofJun. JNK inhibition is mediated by S-nitrosylation of a single regulatory JNK thiol, Cys116, which is con-served in JNK1–3 (but, notably, not in other MAPKs).Control of JNK activity by NO emphasizes the evidentcomplexity of coordinate regulation by S-nitrosylationof several partners, the interactions of which promotesignal transduction that results in altered gene tran-scription. For example, JNK is activated downstreamof Ras and of ASK1, both of which are substrates forS-nitrosylation in situ77,79, as is the JNK substrateJun80,81 (FIG. 4).
Src proto-oncoproteinThe non-receptor protein tyrosine kinase p60Src (Src)is the prototypical proto-oncoprotein (FIG. 5). Src isphosphorylated constitutively at Tyr527, and dephos-phorylation of Tyr527 is the most well-characterizedmechanism of kinase activation. However, treatmentof fibroblasts82,83 (or of Src that was immunoprecipi-tated from those cells83) with NO donors activates Srcthrough autophosphorylation at Tyr416 (REF. 83).Autophosphorylation and activation by NO is associatedwith the formation of disulphide-linked Src multimers83,which indicates that NO/SNO targets neighbouringthiols within closely associated Src monomers21.Regulation of signal transduction through Src by S-nitrosylation is illustrated in FIG. 5.
proteins (the substrates of PTPases)66–68. Studies thatused both isolated proteins and intact cells indicatethat protein tyrosine phosphatase 1B (PTP1B), theprototypic tyrosine-specific PTPase, is a substrate forinhibitory S-nitrosylation68,69. Evidence for the physio-logical regulation of PTPase S-nitrosylation has beenprovided by Mikkelsen and Wardman70, who describedirradiation-induced transient S-nitrosylation of active-site Cys residues within the non-transmembrane, Srchomology-2 (SH2)-domain-containing PTPases, SHP1and SHP2 (SHP2 to a greater extent than SHP1).
Enzymes of arginine metabolism. All forms of thedimethylarginine dimethylaminohydrolases (DDAHs),which hydrolyse Arg-derived endogenous NOSinhibitors, are characterized by a Cys-His-(Glu/Asp) cat-alytic triad that conforms to a SNO motif. Leiper et al.71
demonstrated that the enzymatic activity of DDAHwas inhibited by NO, through S-nitrosylation of theactive-site Cys249. It is of note that the Cys-His-(Glu/Asp) catalytic triad that is found in DDAH definesa superfamily of Arg-handling enzymes that includesarginine glycine amidino transferase and arginine deimi-nase. Recently, Gross et al.72 reported that argininosucci-nate synthetase, which controls the regeneration of Argfrom citrulline, is inhibited by S-nitrosylation of non-cat-alytic Cys132. In addition, it has been shown thatornithine decarboxylase, which catalyses the conversionof ornithine (the product of arginase) to putrescine (aPOLYAMINE precursor), is inhibited by exposure toNO/SNO as a result of S-nitrosylation of the active-siteCys73. In combination with the recent description of theinhibition of S-adenosylmethionine decarboxylase by S-nitrosylation74 (and the inhibition of methionineadenosylytransferase56), these results emphasize that themetabolic fate of Arg (the substrate for NO productionby NOSs), which includes polyamine biosynthesis, mightbe controlled by S-nitrosylation of several functionallyrelated enzymes.
TRX–ASK1 and JNKThe oxidoreductase TRX is essential for the regula-tion of cellular redox state, and TRX also binds andinactivates the apoptosis signal-regulating kinase-1
POLYAMINE
A metabolic product of Arg (thesubstrate for NO synthases),which is generated through ahighly regulated sequence ofenzymatic reactions.
Figure 2 | Concerted acid–base catalysis of protein transnitrosylation. S-nitroso-glutathione (GSNO)-mediated S-nitrosylation of hepatic methionine adenosyltransferase(MAT) selectively targets Cys121, as does endogenous nitric oxide synthase (numbering isaccording to the rat MAT protein sequence). In the three-dimensional structure, the thiol ofCys121 is closely apposed (3.3–4.9 Å) to the basic guanidino groups of Arg357 and Arg363,and to the acidic γ-carboxylate of Asp355. Substitution of any of these residues with a residuethat bears an uncharged side chain inhibits S-nitrosylation of Cys121 (but has no effect on itsmodification by peroxynitrite). Pérez-Mato et al.56 concluded that electrostatic interaction withbasic side chains increases Cys121 nucleophilicity (lowers pKa), whereas the interaction withthe acidic Asp side chain reduces GSNO thiol nucleophilicity (increases pKa) and promotesNO+ donation. Accordingly, concerted acid–base catalysis provides a ‘push-pull’ mechanismfor transnitrosylation of MAT, thereby regulating cellular methylation. Adapted from REF. 56.
GSH+
Cys121 SNO
H O
OAsp355NO
GS
H
SCys121
NArg357 N Arg363H
NArg357 N Arg363+
O
OAsp355–

NATURE REVIEWS | MOLECULAR CELL BIOLOGY VOLUME 6 | FEBRUARY 2005 | 157
R E V I E W S
NSF ATPaseThe homohexameric ATPase that is designated N-ethylmaleimide (NEM)-sensitive factor (NSF) is acritical component in vesicular membrane trafficking,and therefore in both exocytosis and transport betweenmembrane-bound intracellular compartments. Fusionof vesicular and target membranes is mediated by thebinding of soluble NSF-attachment protein (SNAP)receptors (SNAREs) that are associated, respectively,with vesicular (v-SNARE) or target (t-SNARE) mem-branes. Disassembly of SNARE complexes to allowrecycling of SNAREs and subsequent rounds of fusionrequires ATP hydrolysis by NSF, which associates withthe SNARE complex through SNAPs. Matsushita et al.44
showed that NSF in vitro, and in intact endothelialcells, is S-nitrosylated by exogenous or endogenousNO. S-nitrosylation had no effect on basal NSF ATPaseactivity, but greatly inhibited disassembly of SNAREcomplexes by NSF. The inhibition of SNARE disassem-bly might reflect an effect on the protein–protein interac-tions of NSF (specifically, SNARE-complex binding) aswell as the uncoupling of ATP hydrolysis from the con-formational change that drives complex disassembly. Infunctional assays, S-nitrosylation inhibited the stimu-lus-induced exocytosis of WEIBEL–PALADE BODIES and theconsequent release of von Willebrand factor fromendothelial cells. And the introduction of native, but notS-nitrosylated, NSF restored exocytosis from NO-treatedcells, which is powerful evidence that S-nitrosylationof NSF is a principal mechanism for NO-dependentinhibition of SNARE-mediated membrane fusion44.
Monomeric GTPasesLander and colleagues79 showed initially that the small G protein p21Ras was S-nitrosylated at a single site(Cys118), and that S-nitrosylation increased the propor-tion of GTP-bound (active) p21Ras by promotingGDP–GTP exchange. Within Ras proteins, Cys118 isphylogenetically conserved. And within the Ras super-family, Cys is the variable X within the NKXD (where Xis any amino acid) consensus in Ha-, Ki- and N-Ras aswell as in Rap1A, Rap1B, Rab1 and Rab3, whereas otherresidues substitute in this position in Ral, Tc21, R-Ras,Rap2 and Rho. A physiological role for Ras S-nitrosyla-tion was confirmed in intact mammalian neurons byYun et al.84. They demonstrated that the stimulation ofNMDARs results in the activation of p21Ras, which isblocked by NOS inhibition, is absent in mutant micethat lacked nNOS (nNOS–/–), and is independent ofcGMP (FIG. 1b). In addition, Lin et al.85 showed that theactivation of ATP-sensitive K+ channels by NO resultsfrom S-nitrosylation of Ras and the subsequent stimula-tion of MKK. More recently, it has been reported thatthe G protein Ran, which regulates nucleocytoplasmictransport, is S-nitrosylated under basal conditions18.
The dexamethasone-inducible G protein Dexras,which is enriched in brain, is activated by NO donorsas well as by NMDAR-stimulated NO synthesis inneurons, and the basal activity of Dexras is decreasedin the brains of nNOS–/– mice60 (FIG. 1b). Cys11, whichis located in an N-terminal domain that is unique
Box 4 | A potential motif for GSNO-mediated protein S-nitrosylation
Exposure of the bacterial transcription factor OxyR to S-nitroso-glutathione (GSNO)results in S-nitrosylation of a single, regulatory Cys residue (Cys199)109. Molecularmodelling shows that GSNO docks in the binding pocket that was previously definedfor GSSG (oxidized glutathione dimer)109, through hydrogen bonding between the γ-glutamyl amine of GSNO and the γ-carboxylate of Asp202 (translucent purplecircle). This positions the NO group within ~4 Å of the Cys199 sulphur moiety (see figure, part a). A potential motif for trans-S-nitrosylation (NO+ transfer andperhaps NO-radical addition), which is derived from the sequence around Cys199,includes flanking basic and hydrophobic residues and an acidic, GSNO-coordinatingresidue: (H,K,R)(C)(hydrophobic)X(D,E) where X is any amino acid. This motif wasidentified by database analysis (see eMOTIF in the online links box), and was shownto be generally present in one or very few copies, within ~630 vertebrate proteins, anumber of which have been described as substrates for S-nitrosylation. Thefrequencies of residues with aliphatic or aromatic R-groups at the hydrophobicposition closely match their frequencies within the vertebrate proteome.
A portion of the crystal structures of four motif-bearing proteins, including OxyR, isshown in part b of the figure. This portion is defined by a sphere of 40-Å diameter aroundthe target thiol. Motif residues are shown using conventional colouring for Cys, basic andacidic residues (S, yellow; O, red; N, blue; C, white); the hydrophobic and X residues areshown in magenta and brown, respectively. Note that motif residues form a pocket at theprotein surface (which has been defined by Connolly rendering of solvent accessibility),which is created, in part, by the buried location of the hydrophobic residue.Also note thatthe target thiol is exposed. In annexin-6, the pocket is situated around Cys113 (numberingis according to the human protein sequence); the motif is present in several annexins,some of which are modified by S-nitroso-glutathione (GSNO; REF. 152). In glutathione-S-transferase (GST)-mu the pocket is situated around Cys178, and GST might be regulated byS-nitrosylation153. For semaphorin-4D the pocket is found at Cys156, which is conservedacross multiple semaphorins that have been implicated in redox regulation of axonguidance154. Other motif-bearing proteins include metallothionein-3, the NSF ATPase,the von Willebrand factor and the ryanodine receptor/Ca2+-release channels RyR1–3 (see main text). Transposition of the hydrophobic and X residues yields a second, partiallyoverlapping set of proteins, which includes creatine kinase (which is characterized by facileGSNO-mediated S-nitrosylation).
Asp202
GSNO
4.4 Å
Cys199
His198
OxyR Annexin-6 GST-mu Semaphorin-4D
a
b

158 | FEBRUARY 2005 | VOLUME 6 www.nature.com/reviews/molcellbio
R E V I E W S
single, critical Cys residue, Cys399 of NR2A89,90. NR2Awas subsequently identified as an endogenous substratefor nNOS-dependent S-nitrosylation7. Zn2+ is aninhibitory modulator of NMDAR that functions toenhance glutamate affinity and thereby promote recep-tor desensitization, and it has been reported59 that S-nitrosylation of Cys399 within NR2A enhancesNMDAR sensitivity to Zn2+-mediated inhibition.Cys399 resides in a linker region that connects Zn2+-binding and agonist-binding domains, and it has there-fore been suggested that S-nitrosylation of Cys399might allosterically regulate the interaction of ligand-binding, regulatory and linker regions.
The ryanodine receptor. The type 1 ryanodinereceptor/Ca2+ ionophore of skeletal muscle (RyR1) func-tions as the principal ion channel that controls the releaseof Ca2+ from the SARCOPLASMIC RETICULUM (SR) and, thereby,muscle contraction. Eu et al.46 showed in isolated SRvesicles that RyR1 is activated by S-nitrosylation. NOthat is derived from endogenous nNOS yields only asingle S-nitrosylated Cys from among ~50 free thiols ineach homotetrameric RyR1 monomer46, and the targetof S-nitrosylation has been identified as Cys3635, whichis intercalated within the hydrophobic, calmodulin(CaM)-binding domain of RyR141,91 (BOX 3). In addition,RyR1 is S-nitrosylated by physiological concentrations ofNO at physiological but not at ambient (or high) tissueO
2concentrations (pO
2)46. The molecular mechanism of
regulation by pO2
was shown to entail dynamic control
among Ras proteins, was identified as the single site ofS-nitrosylation in vitro, and mutation of Cys11 preventedthe activation of Dexras by an NO+ donor86.As for p21Ras,S-nitrosylation activates Dexras by increasing the rateof GDP dissociation.
Membrane receptors/ion channelsExposure of cells and tissues to S-nitrosylating agents hasbeen shown to inhibit the kinase activity of the epidermalgrowth factor receptor (EGFR) tyrosine kinase (RTK)87.Similarly, S-nitrosylation might regulate the signallingthrough several G-protein-coupled receptors (GPCRs)including the serotonin (5-hydroxytryptamine, 5-HT)5-HT
2receptor 88. However, the site (or sites) of pre-
sumed S-nitrosylation within RTK and GPCR systems(receptor and/or associated regulatory proteins) hasnot yet been determined. In addition, the activity ofa number of receptor-coupled, cation-activated orvoltage-regulated ion channels can be modulated byendogenous S-nitrosylation (examples provided inREFS 6,12,85).
The NMDA receptor. The neuronal NMDAR mediatesthe postsynaptic Ca2+ flux that functions as a principalregulator of nNOS activity, and the NMDAR is, in turn,a substrate for S-nitrosylation59 (FIG. 1b). Studies in vitroof heteromers that comprise the NR1 and NR2A sub-units of the NMDAR showed that S-nitrosylation inhib-ited NMDAR function (that is, Ca2+ flux), and that thiseffect was exerted primarily through modification of a
WEIBEL–PALADE BODY
A secretory vesicle containingagents, including the vonWillebrand factor, which areexocytosed from endothelialcells following inflammatorystimulation.
SARCOPLASMIC RETICULUM
(SR). A specialized form ofendoplasmic reticulum inmuscle cells that sequesters andreleases Ca2+ to control musclecontractility.
Figure 3 | Regulation by S-nitrosylation of protein–protein interactions of caspase-3. (a) At the mitochondrion, (b) S-nitrosylation of a regulatory Cys residue within the proform (that is, the inactive zymogen) of the apoptotic cysteine proteasecaspase-3 (c) promotes assembly of a complex that contains procaspase-3 and acid sphingomyelinase (ASM), and possibly nitric-oxide synthase (NOS)43. (d) Complex formation and S-nitrosylation of the active-site Cys within procaspase-3 (which would befacilitated by proximity with NOS) inhibit the activation of caspase-3 that is dependent on zymogen cleavage by an initiator caspase— for example, caspase-8 (REF. 43). (e) De-nitrosylation, which can be induced by Fas stimulation of apoptosis19, would promotecomplex disassembly and (f) caspase-3 activation. Note that, first, the cytosolic release of active caspase-3 and the interaction in situ with caspase-8 remain speculative; second, the exact location of the protein complex (at or in the mitochondrion) isundetermined; and third, the participation of endogenous (as opposed to overexpressed) NOS in the complex has not been shown.
Cytosol
Mitochondrion
ASMProcaspase-3
NOS
NO
NOCaspase-8
Caspase-3
a b c
d e f
NO
NO
NO
NO
NO

NATURE REVIEWS | MOLECULAR CELL BIOLOGY VOLUME 6 | FEBRUARY 2005 | 159
R E V I E W S
additional two Cys thiols per RyR1 monomer undergoglutathionylation (that is, the formation of a mixeddisulphide with glutathione) on exposure to GSNO93.Treatment with GSNO both enhances the activation ofrelease kinetics by Ca2+, and diminishes the inhibitoryeffect of Mg2+ on the Ca2+-release mechanism, whichrepresents a combination of results that are seen in isola-tion with glutathione-disulphide-derived oxidation andwith NO-donor-derived S-nitrosylation93.An additionalclass of thiols, which might overlap with the class that isreactive towards GSNO, is responsive to the cellular (orSR) redox potential, which is reflected in the ratio ofreduced and oxidized glutathione (GSH/GSSG) and possiblyother oxidants94,95.
of the redox state of a small set of RyR1-associatedthiols, which therefore functions as a novel O
2sensor.
Cys3635 is not a member of that set, and therefore theoxidation/reduction of pO
2-sensitive thiols regulates
Cys3635 S-nitrosylation allosterically 40. Contractilityand Ca2+ flux, which was assessed in intact skeletalmuscle and myocytes, is enhanced by endogenous NOat physiological but not at supra-normal pO
2 (REF. 92).
The distinction between classes of reactive Cys thiolswithin RyR1 encompasses differential effects on RyR1that are exerted by different sources of modifying NO reagents40. Exposure to GSNO resulted in theS-nitrosylation of two Cys residues and the activation ofRyR1, and these effects did not involve Cys3635. An
GSH/GSSG
The ratio of reduced to oxidized(disulphide-linked) glutathione.This ratio is a principal indicatorof the redox state of a cell orsubcellular compartment.
Figure 4 | Regulation of apoptosis through TRX–ASK1 and preservation of the redox equilibrium. The oxidoreductasethioredoxin (TRX) exerts an anti-apoptotic influence (shown in red) by binding and inactivating apoptosis signalling kinase-1 (ASK1),and by maintaining the cellular redox/S-nitrosothiol (SNO) equilibrium. In addition, TRX can de-nitrosylate and thereby activate auto-S-nitrosylated nitric-oxide synthase (NOS), which is consistent with the finding that active TRX helps maintain cellular SNO content,as indicated by the red interrupted line. NOS, in turn, maintains the activity of TRX by S-nitrosylation of Cys69 (REF. 75). In addition,S-nitrosylation inhibits the kinase activity of both ASK1 and Jun N-terminal kinase (JNK), and inhibits transcriptional activation byJun. NOS activity can therefore propagate an anti-apoptotic influence through S-nitrosylation of several elements in transductionpathways that are initiated by ASK1 and, in addition, exerts an inhibitory influence on other pro-apoptotic proteins, including, inparticular, the caspases. TRX also exerts an anti-apoptotic influence that is independent of ASK1 by scavenging reactive oxygenspecies (ROS; not shown). An oxidative stress, which is represented by ROS-induced modification of active-site Cys residues thatinactivates TRX, exerts a pro-apoptotic effect (shown in green). This is promoted, in part, by the release and activation of ASK1 andthe subsequent transduction through the mitogen-activated protein (MAP) kinase (MAPK) cascade that involves the sequentialphosphorylation of MAPK kinase (MKK), JNK and AP-1 (Jun–Fos). The release of ASK1 and apoptotic signalling can also beinitiated by S-nitrosylation of a site (or sites) within TRX other than Cys69 (REF. 76), probably active-site Cys32 and/or Cys35, whichis associated with nitrosative stress. By maintaining physiological levels of SNO and preventing accumulation of ROS, TRX exertsan anti-apoptotic influence that can be interrupted by significant nitrosative or oxidative stress.
Thioredoxin
S S
Cys32 Cys35
S
NO
Cys69
ASK1
S Cys869
SON
ON
SON
S Cys869
ASK1
Thioredoxin
S S
Cys32 Cys35
JNK
Jun
MKK
Cys116
NONO
MAP kinasecascade
Transcription
Fos
Apoptosis
NOS
Cys69
Thioredoxin
S S
Cys32 Cys35
S
NO
Cys69
O2–/ H2O2
NOS
S
NO
Caspase
S
NO

160 | FEBRUARY 2005 | VOLUME 6 www.nature.com/reviews/molcellbio
R E V I E W S
that deletion of OxyR rendered Escherichia coli hyper-sensitive to SNOs,as well as to hydrogen peroxide (H
2O
2).
Cys199 within OxyR is essential for sensing redox dyse-quilibrium108, and Kim et al.109 showed subsequently thatCys199 is modified, both in vitro and in situ, under differ-ent conditions of oxidative or nitrosative stress (whichwas represented by exposure to GSNO, ambient O
2, H
2O
2
or GSSG), to yield selectively the S–NO (from GSNO),S–OH (from O
2or low concentrations of H
2O
2) or S–SG
(from GSSG or higher concentrations of H2O
2or
GSNO) derivatives. Functional analysis revealed thateach form activated transcription to a greater extent thanunmodified OxyR, but that they differed in structure,
The cardiac form of RyR (RyR2), which is S-nitro-sylated under basal conditions96, colocalizes with themuscle-specific form of nNOS (nNOSµ) at the SR97
and co-immunoprecipitates with nNOS from muscleextracts13. S-nitrosylation activates RyR2 (REF. 96) andmight also inhibit the Ca2+-ATPase-mediated uptakeof Ca2+ into the SR97. Accordingly, nNOSµ activity incardiomyocytes enhances contractility, as shown bythe suppressed INOTROPIC response to β-adrenergicstimulation in nNOS–/– mice13.
Ion channels gated/modulated by cyclic nucleotides.Cyclic-nucleotide-gated, Ca2+-permeable channels(CNGcs) constitute the critical, final elements for visualand olfactory transduction in sensory-receptor cells,and NO donors directly activate native olfactoryCNGcs98. Mutational analysis of homomeric CNGαchannels showed that substitution of Cys460 completelyeliminated NO sensitivity99. Cys460 is located immedi-ately adjacent to the cyclic-nucleotide-binding domain(CNBd) within the unordered stretch of residues thatlink the final, C-terminal transmembrane domain to theCNBd. These findings indicate that, as is the case forNMDAR, S-nitrosylation of a critical Cys residue withina linker region that is coupled to the ligand-bindingdomain might allosterically regulate channel activitythrough changes in tertiary or quaternary structure. Inaddition, members of the closely related family of hyper-polarization-activated (cyclic-nucleotide-modulated)cation channels have been identified as prominentneural substrates for endogenous S-nitrosylation7.
Transcription factorsRecently, S-nitrosylated proteins have been localized tothe nuclear compartment under basal conditions, and itwas shown that S-nitrosylation was enhanced by iNOSinduction or blockade of nuclear export18. These findingsare consistent with the extensive evidence for modulationof transcription-factor function by S-nitrosylation. Theeffect of S-nitrosylation on the activity of several tran-scription factors depends, at least in part, on whethermodified Cys residues reside in DNA-binding (‘activesite’) regions or allosteric regulatory regions6. In particu-lar, S-nitrosylation of single, critical Cys residues inDNA-binding regions is consistently inhibitory (whichmight activate transcription if the target is a transcrip-tional repressor100). As illustrated in FIG. 6, regulationby S-nitrosylation of gene transcription (or mRNAtranslation/stability) is often exerted at the level ofubiquitylation-dependent proteasomal targeting, andthereby degradation/stability of signalling elements. Inaddition, it is important to note that, as in the well-characterized cases discussed below (HIF, p53 andnuclear factor (NF)-κB; see FIGS 5,6), transcription factorsthat are regulated by S-nitrosylation have an importantrole in controlling the expression of one or more formsof NOS101–106.
Bacterial OxyR. Hausladen et al.107 showed that exposureto SNOs in vivo resulted in the S-nitrosylation of OxyRand the activation of OxyR-dependent transcription, and
INOTROPIC
Influencing muscle contractility.
Figure 5 | Regulation of signal transduction through Srcby S-nitrosylation at multiple loci. Src, the product of theproto-oncogene p60Src, transduces signals that regulate theenzymatic activity of endothelial nitric-oxide synthase (eNOS orNOS3) and transcription of the eNOS gene (as well as theinducible NOS2 (iNOS) gene). In endothelial cells, shear stressfunctions through Src to initiate sequential phosphorylationthrough a mitogen-activated protein kinase (MAPK) pathwaythat includes Ras, Raf/MEK (where MEK stands for MAPK andextracellular signal-regulated kinase (ERK) kinase) and ERK1/2.This leads to activation of the transcription factor nuclear factor (NF)-κB and initiation of eNOS transcription68,106. S-nitrosylation facilitates the kinase activity of Src (throughdisulphide formation) and activates Ras directly, but inhibits theproteosomal translocation of inhibitor of NF-κB (IκB) (FIG. 6)
and DNA-binding and transcriptional activation by NF-κB. So,S-nitrosylation regulates signal transduction that is initiated bySrc at several steps, each of which represents a point ofconvergence for a progressively larger set of signals (manysignal-transduction pathways share NF-κB as a final stage).In addition, Src/Ras mediate the activation of eNOS bymembrane receptors — including the receptors for high-density lipoprotein (HDL) and oestrogen — through thephosphoinositide 3-kinase (PI3K)–AKT/protein kinase B (PKB)pathway that promotes the activating phosphorylation ofeNOS. The activation of Src by S-nitrosylation thereforefunctions as part of a positive-feedback loop that controlseNOS enzymatic activity. Finally, as indicated by dashedarrows, Src might activate NF-κB through the PI3K–AKT/PKBpathway. The coordination of signalling through theseconverging pathways allows for both short- and long-termregulation of eNOS activity.
Src
Shear stress
Ras
NO
NO Ras
AKT/PKB
PI3K
Src
HDLR Oestrogen R
eNOS
eNOS (iNOS)abundance
eNOSactivity
Raf/MEK
ERK1/2
NF-κB (p50/p65)
NF-κB
ActivationInhibition
eNOS

NATURE REVIEWS | MOLECULAR CELL BIOLOGY VOLUME 6 | FEBRUARY 2005 | 161
R E V I E W S
Hypoxia-inducible factor. The coordinated transcrip-tional control of several genes through binding tohypoxia-responsive elements (HREs) by the α,β-het-erodimeric proteins hypoxia-inducible factor (HIF)1and 2 has an important role in regulating cellular O
2
cooperativity and affinity of DNA binding, as well as inpromoter activity. Therefore, differential modificationof a regulatory Cys thiol confers on OxyR the ability toprocess different redox-related signals into distincttranscriptional responses.
Figure 6 | Regulation by S-nitrosylation of protein ubiquitylation and proteasomal targeting. a | The nitric oxide (NO)-dependent increase in gene transcription through hypoxia-inducible factor α (HIFα) is mediated, in part, by S-nitrosylation of a singleCys residue within HIFα (purple), which facilitates binding of the transactivator cyclic-AMP-responsive-element-binding protein(CREB)/p300. In addition, an NO-dependent modification, presumably S-nitrosylation of an as-yet-unidentified protein (or proteins),inhibits HIFα proline hydroxylation, which is required for binding of the von Hippel–Lindau protein that couples a ubiquitin ligase toHIFα. As a result, NO also prevents the ubiquitylation-dependent routing of HIFα to the proteasome (red). b | Ubiquitylation andproteasomal targeting of the tumour-suppressor protein p53 is mediated by its interaction with human homologue of mousedouble-minute-2 (HDM2), which is disrupted by S-nitrosylation of a single, critical Cys within HDM2 (red), which results in increasedp53 stability. c | Phosphorylation by inhibitor of nuclear factor (NF)-κB (IκB) kinase (IKK) promotes the ubiquitylation and proteasomaltargeting of IκB , which allows NF-κB to translocate to the nucleus and initiate transcription. S-nitrosylation of a critical Cys residuewithin IKKβ inhibits IκB phosphorylation (red). In addition, S-nitrosylation of a single Cys within the p50 subunit of NF-κB inhibits DNAbinding (purple). d | Ubiquitylation and proteasomal targeting of iron-regulatory protein-2 (IRP2) can be triggered not only by cellulariron, but also by S-nitrosylation of a single critical Cys residue within the region that mediates iron-dependent degradation (green). It is possible that iron might promote the S-nitrosylation. See main text for more details.
IRP2
Cys
Fe2+-dependentdegradationsignal
IRE
Transferrin receptor mRNAIRE
Ferritin mRNA
Fe2+
SON
Proteasome
Ubiquitylation
Ubiquitylation
b
NF-κB
p50p65Cys S
IKKβ
Ubiquitylation
Proteasome
Nucleus
NF-κB
p50p65
IKKγ IKKα
p53
Proteasome
HDM2
S
NO
Cys
Ubiquitylation
d
von Hippel–Lindauprotein
Pro Pro
OH
Pro
OH
Pro
E3 ubiquitin ligase
Proteasome
NO
HIFα
HIFα
Prolyl-hydroxylase
CREB/p300
NO
NO
Cys
Ubiquitylation
a c
OH
Pro
OH
Pro
HIFα
von Hippel–Lindauprotein
OH
Pro
OH
Pro
HIFα
IκBα
IκBα
NOCys S
Ser
Ser
Ser
IRP2
CysS
Fe2+-dependentdegradationsignal
IRE
Transferrin receptor mRNA
P
P
P
IREFerritin mRNA
S

162 | FEBRUARY 2005 | VOLUME 6 www.nature.com/reviews/molcellbio
R E V I E W S
the case of HIF1α/p300, S-nitrosylation of a singlecritical thiol controls the (hydrophobic) binding ofthe transcription factor and its regulator. In the caseof p53–HDM2, however, that interaction is inhibitory,and S-nitrosylation would therefore activate theDNA-binding and transcriptional activity of p53.Disruption of p53–HDM2 binding would alsoreduce p53 ubiquitylation and degradation throughHDM2 (FIG. 6b).
Nuclear factor κB. NF-κB is complexed with andsequestered in the cytoplasm by inhibitor of NF-κB(IκB) proteins, and many activating stimuli promotethe phosphorylation of IκB by the IκB-kinase complex(which contains the catalytic IKKα and IKKβ subunitsand a non-catalytic IKKγsubunit). This induces IκBubiquitylation and degradation by the 26S proteasome,to allow translocation of NF-κB to the nucleus andDNA binding122.
The prototype of the NF-κB family is the p50–p65heterodimer, which is expressed constitutively in mostmammalian cells. S-nitrosylation of NF-κB with exoge-nous NO, or NO that is dependent on iNOS induction,inhibits NF-κB-dependent DNA binding, promoteractivity and gene transcription80,123–127. In vitro analysisindicated that p50 is S-nitrosylated at Cys62 (REF. 126),which is conserved in other Rel-homology-domain-containing proteins that can function as NF-κB sub-units. In addition, it was shown that the inhibition byNO of TNFα-induced apoptosis can involve decreasedIκB degradation128,129. Recently, Reynaert et al.130 showedthat endogenous S-nitrosylation of Cys179 withinIKKβ (this residue is conserved in IKKα) inhibits IκBphosphorylation, and that the activation of IKKβ byTNFα is coordinated with de-nitrosylation. Therefore,S-nitrosylation regulates the proteasomal targeting ofIκB, similar to the regulation by NO of p53 and HIFhydroxylases, except that in those cases stabilization ofthe transcription factor rather than the regulatorypartner is influenced by S-nitrosylation (FIG. 6c).
Iron-regulatory proteinsThe iron-regulatory proteins IRP1 and IRP2 have a central role in cellular Fe2+ homeostasis, principallythrough their regulated binding to iron-responsive ele-ments (IREs) within transferrin receptor and ferritinmRNAs. Binding of IRPs to IREs results in increasedFe2+ bioavailability. It is well established that exposure toexogenous or endogenous NO can disassemble the Fe–Scluster of IRP1 and so promote IRE binding. By con-trast, exogenous or endogenously produced NO reducesthe levels of IRE-bound IRP2, and the levels of transfer-rin receptor mRNA and protein are, in fact, decreased inintact cells by iNOS induction or by exposure to NOdonors131–133.
Kim et al.134 have shown recently that one of threeCys residues within the Fe2+-dependent degradationsequence of IRP2 is specifically S-nitrosylated in situ,which results in increased ubiquitylation and proteaso-mal degradation (FIG. 6d). These findings indicate that,similar to the case of the transcription factors HIF1α
homeostasis110. HIFα function is controlled by pO2
through changes in both abundance and transactivation,which are mediated by O
2-regulated hydroxylation that
controls both the proteasomal targeting of HIFα and theinteraction of HIFα with transcriptional coactivators,in particular cyclic-AMP-responsive-element-bindingprotein (CREB)/p300 proteins110.
Hydroxylation of Asn803 within the C-terminaltransactivation domain inhibits the interaction ofHIF1α with CREB/p300, and this inhibition is relievedwhen hypoxia reduces the activity of O
2-requiring HIF
hydroxylases, thereby allowing transcriptional activationthrough HRE binding. In intact cells, exposure to GSNOor induction of iNOS results in the S-nitrosylation ofHIF1α and an increase in transcriptional activity111,112.The NO-dependent increase in transactivation is elimi-nated by mutation of Cys800, and Cys800 (which isconserved in HIF2α) has been identified as a site ofS-nitrosylation112 (FIG. 6a). Furthermore, it has beenshown that S-nitrosylation of Cys800 facilitates the bind-ing of p300 to the C-terminal transactivation domain112.Therefore, targeted S-nitrosylation of a single regulatoryCys thiol controls protein–protein interactions of HIFαand p300, and thereby their transcriptional activity,independently of the O
2-regulated de-hydroxylation
of Asn803.NO also regulates HIFα stability (FIG. 6a).
Hydroxylation of Pro402 and Pro564 facilitates theinteraction of HIFα with the von Hippel–Lindau(VHL) protein, which couples the E3 ubiquitin ligasecomplex that targets HIFα through polyubiquitylationto proteasomal degradation110. HIFα is subject to rapidproteolysis at NORMOXIA, and accumulates when O
2-
dependent prolyl-hydroxylase activity is inhibited byhypoxia. However, it was observed113,114 that NO/SNOs,which were generated by induced NOS in situ, stabilizedHIFα at normoxia. Subsequently, it was reported thatGSNO decreased the binding of HIFα and VHL, andtherefore HIFα ubiquitylation, and that this effect wascorrelated with the ability of GSNO to inhibit the activ-ity of prolyl hydoxylases in vitro115. Therefore, alongwith the regulation of transactivation by S-nitrosylationof HIFα itself, NO might also target elements that con-trol HIFα ubiquitylation to regulate HIFα stability. Itshould be noted that the ubiquitylating activity of atleast one E3 ubiquitin ligase, parkin, is regulated directlyby S-nitrosylation116,117.
Tumour suppressor p53. p53 is rapidly turned over byproteasomal degradation and is targeted for ubiquity-lation through interaction with the RING finger E3ubiquitin ligase, human homologue of mouse doubleminute-2 (HDM2)118. In addition, the binding of HDM2to p53 directly inhibits transcriptional activity. Inductionof iNOS or exposure to exogenous NO results in p53accumulation and the activation of p53-dependenttranscription119,120.
Exposure of HDM2 to NO inhibits the subsequentbinding to p53 (FIG. 6b), and the site of S-nitrosylationthat controls p53 binding has been localized to thehighly conserved Cys77 residue121. Therefore, similar to
NORMOXIA
Ambient O2
(~21%) or, inreference to tissue, O
2
concentrations that representthe normal physiological state(~2–3%).

NATURE REVIEWS | MOLECULAR CELL BIOLOGY VOLUME 6 | FEBRUARY 2005 | 163
R E V I E W S
greatly reduced by pharmacological nNOS inhibitionand by nNOS knockout in mice. Furthermore, MMP9was S-nitrosylated and activated by exposure toCys–NO in vitro, and treatment of neurons with S-nitrosylated MMP9 induced apoptosis. In addition,Gu et al.141 reported that the Zn2+-coordinating Cysresidue in MMP9 was oxidized further to sulphinic orsulphonic acid after S-nitrosylation, which mightcause prolonged activation of the enzyme underpathophysiological conditions (BOX 1).
Activation by S-nitrosylation has also been describedin TNFα-converting enzyme (TACE), a member of aMETALLOPROTEINASE/DISINTEGRIN (ADAM) FAMILY, and the TACEactive-site domain contains a His
3–Cys-coordinated
catalytic Zn2+, similar to MMPs142. Zhang et al.142
showed in intact cells that exogenous or endogenouslyproduced NO augmented TACE-dependent cleavageand shedding (secretion) of TNFα, and that the Zn2+-coordinating Cys residue within the inhibitoryprodomain was a substrate for S-nitrosylation in vitro.Accordingly, S-nitrosylation of ADAM-family metallo-proteinases might regulate ectodomain shedding, whichis involved centrally in several processes that entail therelease of membrane-bound molecules, includinginflammation and haematopoiesis.
The futureProtein S-nitrosylation is established as a significantroute through which NO transmits its ubiquitous cellu-lar influence, and as a broad-based mechanism for post-translational regulation of protein function. Among theplethora of post-translational modifications, S-nitrosy-lation has in common with phosphorylation the controlof most or all classes of protein. S-nitrosylation is themost well-established example of specific, redox-basedsignalling (BOX 1) and provides a mechanism for physio-logical regulation through the modification of proteinCys residues. An expanded perspective on physiologicalredox-based signalling, in which S-nitrosylation mighthave a primary effector role, while O
2-derived and other
redox-based modifications influence the occurrence of,or responsiveness to, S-nitrosylation (much as oxidativemodifications can influence the responsiveness to phos-phorylation through inhibition of phosphatases), is out-lined in several examples. The ryanodine receptor/Ca2+
channel of skeletal muscle presents classes of allostericCys residues that are differentially susceptible tonitrosative and oxidative modifications. These modifica-tions exert distinct but interacting effects on channelfunction, and O
2-mediated oxidative modification
decreases channel responsiveness to NO. Also, reactiveCys residues within erythrocytic haemoglobin are thesites of S-nitrosylation, but O
2(and haem oxidation)
function as allosteric regulators of S-nitrosylation/de-nitrosylation. In addition, thioredoxin activity is regu-lated directly by S-nitrosylation,which maintains a cellularredox milieu that is conducive to S-nitrosylation. And,the transcription factor OxyR can differentiate betweenmutually exclusive oxidative and nitrosative modifica-tions of a single regulatory Cys (oxidation would pre-clude S-nitrosylation). The mechanisms of regulated
and p53–HDM2, S-nitrosylation inhibits IRP2 activity,at least in part, by regulating IRP2 stability. It should benoted that, whereas IRP1 ‘knockout’ mice have noapparent phenotype, IRP2–/– mice have pronouncedneurodegenerative effects that are correlated with ele-vated ferritin expression and Fe2+ deposition in brainregions that are enriched in both NOS and IRPs135,136.
MetalloproteinsMetalloproteins comprise about a third of all struc-turally characterized proteins, and a significant portionof metalloproteins contain a Zn2+ ion that is coordi-nated in a tetrahedral configuration by protein-specificcombinations of Cys thiols and/or His imidazole nitro-gens. This coordination forms zinc-finger domainswithin transcription factors that are essential inprotein–nucleic-acid interactions, and other relateddomains (for example, RING finger or LIM domains)that mediate protein–protein interactions. In addition,Cys thiols participate in coordinating active-site Zn2+
within metalloproteinases and in metal-thiolate pros-thetic complexes in a range of other proteins. BoundZn2+ often functions as a LEWIS ACID to effectively reducethe pKa of Cys (REF. 137).
Zinc-finger proteins and metallothioneins. Treatmentwith exogenous NO has been reported to disrupt DNAbinding of, and transcriptional regulation by, a widerange of eukaryotic zinc-finger transcription factors(ZFTFs). Inhibition of ZFTFs by NO is associated withthe loss of the Zn2+ moiety. However, disruption of thezinc-finger structure by NO (which involves disulphideformation that is dependent on S-nitrosylation) isreversible by reduction, whereas cellular repair of ZFTFsafter disruption by ROS might be precluded in manycases138. In this fashion, ZFTFs potentially mediate dif-ferential transcriptional responsiveness to ROS versusnitrosylating stimuli. Similarly, METALLOTHIONEINS are pre-sent in vivo mainly in Zn2+-bound or mixed-metal (forexample, Zn2+/Cu2+) form. Pearce et al.139 showed thatZn2+ is released from metallothioneins in endothelialcells following physiological activation of eNOS, andBossy-Wetzel et al.140 have shown that intraneuronalZn2+ release is mediated by NMDAR-coupled nNOSactivation in situ.
Metalloproteinases. Metalloproteinases comprise athird class of Zn2+-coordinating proteins that are sus-ceptible to regulation by S-nitrosylation. The active siteof EXTRACELLULAR MATRIX METALLOPROTEINASES (MMPs) containsa catalytic Zn2+ that is coordinated in a His
3–Cys tetra-
hedron, and the coordinate complex blocks the activesite in the proenzyme and excludes the obligate fourthcatalytic ligand, H
2O. MMPs are activated following dis-
ruption of Zn2+–Cys coordination (Zn2+ is retained byvirtue of the remaining interaction with His), but thenature of the physiological trigger (or triggers) for this‘cysteine switch’ has remained enigmatic. Gu et al.141
showed that focal ischaemia/reperfusion caused activa-tion of MMP9 and induced neuronal apoptosis inrodent brain, and that activation and apoptosis were
LEWIS ACID
An electron-pair acceptor thatcan participate as one memberof a conjugate acid–base pair inacid–base reactions.
METALLOTHIONEIN
A small Cys-rich protein thatbinds metal ions — in particular,Zn2+ and/or Cu2+ — at least inpart, through complexformation with cysteine thiolate.
EXTRACELLULAR MATRIX
METALLOPROTEINASE
(MMP). A Zn2+-dependentproteolytic enzyme that has anextracellular active site, which iscapable of breaking downextracellular-matrixcomponents.
METALLOPROTEINASE/
DISINTEGRIN (ADAM) FAMILY
A family of extracellularmetalloproteinases that areimplicated in the proteolyticprocessing of membrane-boundsubstrates, which results inectodomain shedding, and arenamed after their characteristicADAM-domain structure.

164 | FEBRUARY 2005 | VOLUME 6 www.nature.com/reviews/molcellbio
R E V I E W S
of pathophysiological conditions, including endo-toxic shock, multiple sclerosis, Parkinson’s disease,sickle cell disease, pulmonary hypertension, cysticfibrosis and asthma8,10,38,117. And altered blood levelsof SNO proteins have been associated with impairedclinical outcome in patients with cardiovascular dis-ease49,143,144. The therapeutic opportunities, presentedby the emerging recognition that S-nitrosylation has apanoply of roles in normal and disturbed cell function,remain largely untapped.
S-nitrosylation/de-nitrosylation remain outstandingissues in many cases.
The extensive functional purview of S-nitrosyla-tion, which encompasses the regulation of activity,turnover, subcellular localization and molecularinteractions (protein–protein or protein–nucleotide)of diverse proteins, indicates a role as a principaleffector mechanism in redox-based regulation ofcellular function. Accordingly, dysregulation of pro-tein S-nitrosylation is associated with a growing list
1. Murad, F. Cyclic guanosine monophosphate as a mediatorof vasodilation. J. Clin. Invest. 78, 1–5 (1986).
2. Bredt, D. S. Endogenous nitric oxide synthesis: biologicalfunctions and pathophysiology. Free Radic. Res. 31,577–596 (1999).
3. Stamler, J. S. et al. S-nitrosylation of proteins with nitricoxide: synthesis and characterization of biologically activecompounds. Proc. Natl Acad. Sci. USA 89, 444–448(1992).The first demonstration of protein S-nitrosylation,which showed that proteins in several classes couldbe modified at active-site or allosteric Cys residues byendogenous and exogenous NO.
4. Stamler, J. S. et al. S-nitrosylation of tissue-typeplasminogen activator confers vasodilatory and antiplateletproperties on the enzyme. Proc. Natl Acad. Sci. USA 89,8087–8091 (1992).
5. Stamler, J. S. et al. in Biology of Nitric Oxide (eds, Moncada, S.,Marletta, M. A. & Hibbs, J. B. J.) 20–23 (Portland Press,London, UK, 1992).
6. Stamler, J. S., Lamas, S. & Fang, F. C. Nitrosylation: theprototypic redox-based signaling mechanism. Cell 106,675–683 (2001).
7. Jaffrey, S. R., Erdjument-Bromage, H., Ferris, C. D., Tempst, P.& Snyder, S. H. Protein S-nitrosylation: a physiological signalfor neuronal nitric oxide. Nature Cell Biol. 3, 193–197 (2001).Introduced a method to selectively biotinylate sites ofS-nitrosylation within proteins, and applied thismethod to reveal several endogenous substrates inneural tissue of nNOS-dependent S-nitrosylation.
8. Liu, L. et al. Essential roles of S-nitrosothiols in vascularhomeostasis and endotoxic shock. Cell 116, 617–628(2004).Found that mice with a targeted gene deletion ofGSNO reductase have large increases in tissuedamage and mortality following endotoxic challenge,which indicated an important role for S-nitrosothiolmetabolism in innate immunity, as well as effects onbasal SNO levels and vascular tone.
9. de Jesus-Berrios, M. et al. Enzymes that counteractnitrosative stress promote fungal virulence. Curr. Biol. 13,1963–1968 (2003).
10. Foster, M. W., McMahon, T. J. & Stamler, J. S. S-nitrosylation in health and disease. Trends Mol. Med. 9,160–168 (2003).
11. Lane, P., Hao, G. & Gross, S. S. S-nitrosylation is emergingas a specific and fundamental posttranslational proteinmodification: head-to-head comparison with O-phosphorylation. Sci. STKE, RE1 (2001).
12. Boehning, D. & Snyder, S. H. Novel neural modulators.Annu. Rev. Neurosci. 26, 105–131 (2003).
13. Barouch, L. A. et al. Nitric oxide regulates the heart byspatial confinement of nitric oxide synthase isoforms. Nature416, 337–339 (2002).
14. Gow, A. J. et al. Basal and stimulated protein S-nitrosylationin multiple cell types and tissues. J. Biol. Chem. 277,9637–9640 (2002).
15. Stamler, J. S. & Toone, E. J. The decomposition ofthionitrites. Curr. Opin. Chem. Biol. 6, 779–785 (2002).
16. Bartberger, M. D. et al. S–N dissociation energies of S-nitrosothiols: on the origins of nitrosothiol decompositionrates. J. Am. Chem. Soc. 123, 8868–8869 (2001).
17. Stamler, J. S. S-nitrosothiols in the blood: roles, amounts,and methods of analysis. Circ. Res. 94, 414–417 (2004).
18. Ckless, K. et al. In situ detection and visualization of S-nitrosylated proteins following chemical derivatization:identification of Ran GTPase as a target for S-nitrosylation.Nitric Oxide 11, 216–217 (2004).
19. Mannick, J. B. et al. Fas-induced caspase denitrosylation.Science 284, 651–654 (1999).Provided the first demonstration of stimulus-coupledprotein de-nitrosylation, which was triggered by
activation of a membrane receptor that subservesapoptotic stimulation.
20. Hoffmann, J., Haendeler, J., Zeiher, A. M. & Dimmeler, S.TNFα and oxLDL reduce protein S-nitrosylation inendothelial cells. J. Biol. Chem. 276, 41383–41387(2001).
21. Arnelle, D. R. & Stamler, J. S. NO+, NO, and NO– donationby S-nitrosothiols: implications for regulation of physiologicalfunctions by S-nitrosylation and acceleration of disulfideformation. Arch. Biochem. Biophys. 318, 279–285 (1995).
22. Gow, A. J., Luchsinger, B. P., Pawloski, J. R., Singel, D. J. &Stamler, J. S. The oxyhemoglobin reaction of nitric oxide.Proc. Natl Acad. Sci. USA 96, 9027–9032 (1999).
23. Romeo, A. A., Capobianco, J. A. & English, A. M.Superoxide dismutase targets NO from GSNO to Cysβ93 ofoxyhemoglobin in concentrated but not dilute solutions ofthe protein. J. Am. Chem. Soc. 125, 14370–14378 (2003).
24. Mani, K., Cheng, F., Havsmark, B., David, S. & Fransson, L. A.Involvement of glycosylphosphatidylinositol-linkedceruloplasmin in the copper/zinc-nitric oxide-dependentdegradation of glypican-1 heparan sulfate in rat C6 gliomacells. J. Biol. Chem. 279, 12918–12923 (2004).
25. Inoue, K. et al. Nitrosothiol formation catalyzed byceruloplasmin. Implication for cytoprotective mechanism in vivo. J. Biol. Chem. 274, 27069–27075 (1999).
26. Stubauer, G., Giuffre, A. & Sarti, P. Mechanism of S-nitrosothiol formation and degradation mediated bycopper ions. J. Biol. Chem. 274, 28128–28133 (1999).
27. Tao, L. & English, A. M. Mechanism of S-nitrosation ofrecombinant human brain calbindin D28K. Biochemistry 42,3326–3334 (2003).
28. Romeo, A. A., Capobianco, J. A. & English, A. M. Hemenitrosylation of deoxyhemoglobin by S-nitrosoglutathionerequires copper. J. Biol. Chem. 277, 24135–24141 (2002).
29. Luchsinger, B. P. et al. Routes to S-nitroso-hemoglobinformation with heme redox and preferential reactivity in the β subunits. Proc. Natl Acad. Sci. USA 100, 461–466 (2003).
30. Foster, M. W. & Stamler, J. S. New insights into protein S-nitrosylation: mitochondria as a model system. J. Biol.Chem. 279, 25891–25897 (2004).
31. Mulsch, A., Mordvintcev, P. I., Vanin, A. F. & Busse, R.Formation and release of dinitrosyl iron complexes byendothelial cells. Biochem. Biophys. Res. Commun. 196,1303–1308 (1993).
32. Vanin, A. F., Mordvintcev, P. I., Hauschildt, S. & Mulsch, A.The relationship between L-arginine-dependent nitric oxidesynthesis, nitrite release and dinitrosyl–iron complexformation by activated macrophages. Biochim. Biophys.Acta 1177, 37–42 (1993).
33. Pawloski, J. R., Hess, D. T. & Stamler, J. S. Export by redblood cells of nitric oxide bioactivity. Nature 409, 622–626(2001).
34. Patel, J. M., Zhang, J. & Block, E. R. Nitric oxide-inducedinhibition of lung endothelial cell nitric oxide synthase viainteraction with allosteric thiols: role of thioredoxin inregulation of catalytic activity. Am. J. Respir. Cell Mol. Biol.15, 410–419 (1996).
35. Ravi, K., Brennan, L. A., Levic, S., Ross, P. A. & Black, S. M.S-nitrosylation of endothelial nitric oxide synthase isassociated with monomerization and decreased enzymeactivity. Proc. Natl Acad. Sci. USA 101, 2619–2624 (2004).
36. Kahlos, K., Zhang, J., Block, E. R. & Patel, J. M. Thioredoxinrestores nitric oxide-induced inhibition of protein kinase Cactivity in lung endothelial cells. Mol. Cell. Biochem. 254,47–54 (2003).
37. Nikitovic, D. & Holmgren, A. S-nitrosoglutathione is cleavedby the thioredoxin system with liberation of glutathione andredox regulating nitric oxide. J. Biol. Chem. 271,19180–19185 (1996).
38. Gaston, B. et al. Endogenous nitrogen oxides andbronchodilator S-nitrosothiols in human airways. Proc. NatlAcad. Sci. USA 90, 10957–10961 (1993).
39. Liu, L. et al. A metabolic enzyme for S-nitrosothiol conservedfrom bacteria to humans. Nature 410, 490–494 (2001).
40. Sun, J., Xu, L., Eu, J. P., Stamler, J. S. & Meissner, G. Nitricoxide, NOC-12, and S-nitrosoglutathione modulate theskeletal muscle calcium release channel/ryanodine receptorby different mechanisms. An allosteric function for O2 in S-nitrosylation of the channel. J. Biol. Chem. 278,8184–8189 (2003).
41. Hess, D. T., Matsumoto, A., Nudelman, R. & Stamler, J. S.S-nitrosylation: spectrum and specificity. Nature Cell Biol. 3,E46–E49 (2001).
42. Campbell, D. L., Stamler, J. S. & Strauss, H. C. Redoxmodulation of L-type calcium channels in ferret ventricularmyocytes. Dual mechanism regulation by nitric oxide and S-nitrosothiols. J. Gen. Physiol. 108, 277–293 (1996).
43. Matsumoto, A., Comatas, K. E., Liu, L. & Stamler, J. S.Screening for nitric oxide-dependent protein–proteininteractions. Science 301, 657–661 (2003).Used yeast two-hybrid screening to show S-nitrosylation-facilitated protein–proteininteractions, including interactions of NOS withsubstrates for S-nitrosylation, which were verifiedin mammalian cells.
44. Matsushita, K. et al. Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell115, 139–150 (2003).Showed that the activity of NSF, an essentialcomponent of most or all membrane trafficking, isregulated in situ by endogenous S-nitrosylation.
45. Stamler, J. S. et al. Blood flow regulation by S-nitrosohemoglobin in the physiological oxygen gradient. Science 276, 2034–2037 (1997).
46. Eu, J. P., Sun, J., Xu, L., Stamler, J. S. & Meissner, G. The skeletal muscle calcium release channel: coupled O2
sensor and NO signaling functions. Cell 102, 499–509(2000).Showed that Ca2+ flux through the ryanodinereceptor/Ca2+-release channel of skeletal muscle(RyR1) was activated in situ by S-nitrosylation of asingle regulatory Cys, and that S-nitrosylation wasgated by oxygen-dependent alteration in the redoxstatus of a small additional set of Cys residues.
47. Lai, T. S. et al. Calcium regulates S-nitrosylation,denitrosylation, and activity of tissue transglutaminase.Biochemistry 40, 4904–4910 (2001).
48. Jia, L., Bonaventura, C., Bonaventura, J. & Stamler, J. S. S-nitrosohaemoglobin: a dynamic activity of blood involvedin vascular control. Nature 380, 221–226 (1996).Showed that haemoglobin is S-nitrosylatedendogenously (rather than eliminating nitric oxide)and thereby functions as a source of vasodilatoryactivity conveyed by red blood cells (which subserveshypoxic vasodilation).
49. James, P. E., Lang, D., Tufnell-Barret, T., Milsom, A. B. &Frenneaux, M. P. Vasorelaxation by red blood cells andimpairment in diabetes. Reduced nitric oxide and oxygendelivery by glycated hemoglobin. Circ. Res. 94, 976–983(2004).
50. Funai, E. F., Davidson, A., Seligman, S. P. & Finlay, T. H. S-nitrosohemoglobin in the fetal circulation may represent acycle for blood pressure regulation. Biochem. Biophys. Res.Commun. 239, 875–877 (1997).
51. Singel, D. J. & Stamler, J. S. Chemical physiology of bloodflow regulation by red blood cells: role of nitric oxide and S-nitrosohemoglobin. Annu. Rev. Physiol. Oct 19 2004(doi:10.1146/annurev.physiol.67.060603.090918).
52. Stamler, J. S., Toone, E. J., Lipton, S. A. & Sucher, N. J.(S)NO signals: translocation, regulation, and a consensusmotif. Neuron 18, 691–696 (1997).
53. Britto, P. J., Knipling, L. & Wolff, J. The local electrostaticenvironment determines cysteine reactivity of tubulin. J. Biol. Chem. 277, 29018–29027 (2002).

NATURE REVIEWS | MOLECULAR CELL BIOLOGY VOLUME 6 | FEBRUARY 2005 | 165
R E V I E W S
54. Bizzozero, O. A., Bixler, H. A. & Pastuszyn, A. Structuraldeterminants influencing the reaction of cysteine-containingpeptides with palmitoyl-coenzyme A and other thioesters.Biochim. Biophys. Acta 1545, 278–288 (2001).
55. Atkins, W. M., Wang, R. W., Bird, A. W., Newton, D. J. & Lu, A. Y. The catalytic mechanism of glutathione S-transferase (GST). Spectroscopic determination of thepKa of Tyr-9 in rat α1-1 GST. J. Biol. Chem. 268,19188–19191 (1993).
56. Pérez-Mato, I., Castro, C., Ruiz, F. A., Corrales, F. J. & Mato, J. M. Methionine adenosyltransferase S-nitrosylationis regulated by the basic and acidic amino acids surroundingthe target thiol. J. Biol. Chem. 274, 17075–17079 (1999).Showed with site-specific mutation the important roleof acidic and basic side chains, proximate to Cys thiol,in targeting S-nitrosylation within protein substrates(transnitrosylation by GSNO).
57. Brenman, J. E. et al. Interaction of nitric oxide synthase withthe postsynaptic density protein PSD-95 and α1-syntrophinmediated by PDZ domains. Cell 84, 757–767 (1996).
58. Kornau, H. C., Schenker, L. T., Kennedy, M. B. &Seeburg, P. H. Domain interaction between NMDA receptorsubunits and the postsynaptic density protein PSD-95.Science 269, 1737–1740 (1995).
59. Lipton, S. A. et al. Cysteine regulation of protein function —as exemplified by NMDA-receptor modulation. TrendsNeurosci. 25, 474–480 (2002).
60. Fang, M. et al. Dexras1: a G protein specifically coupled toneuronal nitric oxide synthase via CAPON. Neuron 28,183–193 (2000).
61. Rizzo, M. A. & Piston, D. W. Regulation of β cell glucokinaseby S-nitrosylation and association with nitric oxide synthase.J. Cell Biol. 161, 243–248 (2003).Showed that glucokinase is bound to nNOS inpancreatic β-cells, and that physiological activation ofnNOS by insulin results in S-nitrosylation ofglucokinase, its release from nNOS and activation ofkinase activity.
62. Giles, N. M., Giles, G. I. & Jacob, C. Multiple roles ofcysteine in biocatalysis. Biochem. Biophys. Res. Commun.300, 1–4 (2003).
63. Stamler, J. S., Singel, D. J. & Loscalzo, J. Biochemistry ofnitric oxide and its redox-activated forms. Science 258,1898–1902 (1992).
64. Mannick, J. B. et al. S-Nitrosylation of mitochondrialcaspases. J. Cell Biol. 154, 1111–1116 (2001).
65. Kim, J. E. & Tannenbaum, S. R. S-nitrosation regulates theactivation of endogenous procaspase-9 in HT-29 humancolon carcinoma cells. J. Biol. Chem. 279, 9758–9764(2004).
66. Caselli, A., Chiarugi, P., Camici, G., Manao, G. & Ramponi, G.In vivo inactivation of phosphotyrosine protein phosphatasesby nitric oxide. FEBS Lett. 374, 249–252 (1995).
67. Callsen, D., Sandau, K. B. & Brune, B. Nitric oxide andsuperoxide inhibit platelet-derived growth factor receptorphosphotyrosine phosphatases. Free Radic. Biol. Med. 26,1544–1553 (1999).
68. Xian, M. et al. Inhibition of protein tyrosine phosphatases bylow-molecular-weight S-nitrosothiols and S-nitrosylatedhuman serum albumin. Biochem. Biophys. Res. Commun.268, 310–314 (2000).
69. Li, S. & Whorton, A. R. Regulation of protein tyrosinephosphatase 1B in intact cells by S-nitrosothiols. Arch.Biochem. Biophys. 410, 269–279 (2003).
70. Mikkelsen, R. B. & Wardman, P. Biological chemistry ofreactive oxygen and nitrogen and radiation-induced signaltransduction mechanisms. Oncogene 22, 5734–5754(2003).
71. Leiper, J., Murray-Rust, J., McDonald, N. & Vallance, P. S-nitrosylation of dimethylarginine dimethylaminohydrolaseregulates enzyme activity: further interactions between nitricoxide synthase and dimethylargininedimethylaminohydrolase. Proc. Natl Acad. Sci. USA 99,13527–13532 (2002).
72. Hao, G., Xie, L. & Gross, S. S. Argininosuccinate synthetaseis reversibly inactivated by S-nitrosylation in vitro and in vivo.J. Biol. Chem. 279, 36192–36200 (2004).
73. Bauer, P. M., Buga, G. M., Fukuto, J. M., Pegg, A. E. &Ignarro, L. J. Nitric oxide inhibits ornithine decarboxylase viaS-nitrosylation of cysteine 360 in the active site of theenzyme. J. Biol. Chem. 276, 34458–34464 (2001).
74. Hillary, R. A. & Pegg, A. E. Decarboxylases involved inpolyamine biosynthesis and their inactivation by nitric oxide.Biochim. Biophys. Acta 1647, 161–166 (2003).
75. Haendeler, J. et al. Redox regulatory and anti-apoptoticfunctions of thioredoxin depend on S-nitrosylation atcysteine 69. Nature Cell Biol. 4, 743–749 (2002).
76. Sumbayev, V. V. S-nitrosylation of thioredoxin mediatesactivation of apoptosis signal-regulating kinase 1. Arch. Biochem. Biophys. 415, 133–136 (2003).
77. Park, H. S. et al. Inhibition of apoptosis signal-regulatingkinase 1 (ASK1) by nitric oxide through a thiol-redoxmechanism. J. Biol. Chem. 279, 7584–7590 (2003).
78. Park, H. S., Huh, S. H., Kim, M. S., Lee, S. H. & Choi, E. J.Nitric oxide negatively regulates c-Jun N-terminalkinase/stress-activated protein kinase by means of S-nitrosylation. Proc. Natl Acad. Sci. USA 97, 14382–14387(2000).Showed in intact cells that endogenously producedNO can suppress activity of a protein kinase, JNK,which could be ascribed to S-nitrosylation of a singleregulatory cysteine.
79. Lander, H. M., Ogiste, J. S., Pearce, S. F., Levi, R. &Novogrodsky, A. Nitric oxide-stimulated guanine nucleotideexchange on p21ras. J. Biol. Chem. 270, 7017–7020(1995).
80. dela Torre, A., Schroeder, R. A., Bartlett, S. T. & Kuo, P. C.Differential effects of nitric oxide-mediated S-nitrosylation onp50 and c-jun DNA binding. Surgery 124, 137–141 (1998).
81. Nikitovic, D., Holmgren, A. & Spyrou, G. Inhibition of AP-1DNA binding by nitric oxide involving conserved cysteineresidues in Jun and Fos. Biochem. Biophys. Res. Commun.242, 109–112 (1998).
82. Monteiro, H. P., Gruia-Gray, J., Peranovich, T. M.,de Oliveira, L. C. & Stern, A. Nitric oxide stimulates tyrosinephosphorylation of focal adhesion kinase, Src kinase, andmitogen-activated protein kinases in murine fibroblasts. Free Radic. Biol. Med. 28, 174–182 (2000).
83. Akhand, A. A. et al. Nitric oxide controls src kinase activitythrough a sulfhydryl group modification-mediated Tyr-527-independent and Tyr-416-linked mechanism. J. Biol. Chem.274, 25821–25826 (1999).
84. Yun, H. Y., Gonzalez-Zulueta, M., Dawson, V. L. &Dawson, T. M. Nitric oxide mediates N-methyl-D-aspartatereceptor-induced activation of p21ras. Proc. Natl Acad. Sci.USA 95, 5773–5778 (1998).
85. Lin, Y. F., Raab-Graham, K., Jan, Y. N. & Jan, L. Y. NO stimulation of ATP-sensitive potassium channels:involvement of Ras/mitogen-activated protein kinasepathway and contribution to neuroprotection. Proc. NatlAcad. Sci. USA 101, 7799–7804 (2004).
86. Jaffrey, S. R., Fang, M. & Snyder, S. H. Nitrosopeptidemapping: a novel methodology reveals S-nitrosylation ofdexras1 on a single cysteine residue. Chem. Biol. 9,1329–1335 (2002).
87. Estrada, C. et al. Nitric oxide reversibly inhibits the epidermalgrowth factor receptor tyrosine kinase. Biochem. J. 326,369–376 (1997).
88. Nozik-Grayck, E. et al. Pulmonary vasoconstriction byserotonin is inhibited by S-nitrosoglutathione. Am. J. Physiol.Lung Cell. Mol. Physiol. 282, L1057–L1065 (2002).
89. Lipton, S. A. et al. A redox-based mechanism for theneuroprotective and neurodestructive effects of nitric oxideand related nitroso-compounds. Nature 364, 626–632(1993).
90. Choi, Y. B. et al. Molecular basis of NMDA receptor-coupledion channel modulation by S-nitrosylation. Nature Neurosci.3, 15–21 (2000).
91. Sun, J., Xin, C., Eu, J. P., Stamler, J. S. & Meissner, G.Cysteine-3635 is responsible for skeletal muscle ryanodinereceptor modulation by NO. Proc. Natl Acad. Sci. USA 98,11158–11162 (2001).
92. Eu, J. P. et al. Concerted regulation of skeletal musclecontractility by oxygen tension and endogenous nitric oxide.Proc. Natl Acad. Sci. USA 100, 15229–15234 (2003).
93. Aracena, P., Sanchez, G., Donoso, P., Hamilton, S. L. &Hidalgo, C. S-glutathionylation decreases Mg2+ inhibitionand S-nitrosylation enhances Ca2+ activation of RyR1channels. J. Biol. Chem. 278, 42927–42935 (2003).
94. Sun, J., Xu, L., Eu, J. P., Stamler, J. S. & Meissner, G.Classes of thiols that influence the activity of the skeletalmuscle calcium release channel. J. Biol. Chem. 276,15625–15630 (2001).
95. Zable, A. C., Favero, T. G. & Abramson, J. J. Glutathionemodulates ryanodine receptor from skeletal musclesarcoplasmic reticulum. Evidence for redox regulation of theCa2+ release mechanism. J. Biol. Chem. 272, 7069–7077(1997).
96. Xu, L., Eu, J. P., Meissner, G. & Stamler, J. S. Activation ofthe cardiac calcium release channel (ryanodine receptor) bypoly-S-nitrosylation. Science 279, 234–237 (1998).
97. Xu, K. Y., Huso, D. L., Dawson, T. M., Bredt, D. S. & Becker, L. C. Nitric oxide synthase in cardiac sarcoplasmicreticulum. Proc. Natl Acad. Sci. USA 96, 657–662 (1999).
98. Broillet, M. C. & Firestein, S. Direct activation of the olfactorycyclic nucleotide-gated channel through modification ofsulfhydryl groups by NO compounds. Neuron 16, 377–385(1996).
99. Broillet, M. C. A single intracellular cysteine residue isresponsible for the activation of the olfactory cyclic
nucleotide-gated channel by NO. J. Biol. Chem. 275,15135–15141 (2000).
100. Gao, C. et al. S-nitrosylation of heterogeneous nuclearribonucleoprotein A/B regulates osteopontin transcription inendotoxin-stimulated murine macrophages. J. Biol. Chem.279, 11236–11243 (2004).
101. Melillo, G. et al. A hypoxia-responsive element mediates anovel pathway of activation of the inducible nitric oxidesynthase promoter. J. Exp. Med. 182, 1683–1693 (1995).
102. Palmer, L. A., Semenza, G. L., Stoler, M. H. & Johns, R. A.Hypoxia induces type II NOS gene expression in pulmonaryartery endothelial cells via HIF-1. Am. J. Physiol. 274,L212–L219 (1998).
103. Ambs, S., Hussain, S. P. & Harris, C. C. Interactive effects ofnitric oxide and the p53 tumor suppressor gene incarcinogenesis and tumor progression. FASEB J. 11,443–448 (1997).
104. Davis, M. E., Grumbach, I. M., Fukai, T., Cutchins, A. &Harrison, D. G. Shear stress regulates endothelial nitric-oxide synthase promoter activity through nuclear factor κBbinding. J. Biol. Chem. 279, 163–168 (2004).
105. Lowenstein, C. J. et al. Macrophage nitric oxide synthasegene: two upstream regions mediate induction by interferon γand lipopolysaccharide. Proc. Natl Acad. Sci. USA 90,9730–9734 (1993).
106. Xie, Q. W., Kashiwabara, Y. & Nathan, C. Role oftranscription factor NF-κB/Rel in induction of nitric oxidesynthase. J. Biol. Chem. 269, 4705–4708 (1994).
107. Hausladen, A., Privalle, C. T., Keng, T., DeAngelo, J. &Stamler, J. S. Nitrosative stress: activation of thetranscription factor OxyR. Cell 86, 719–729 (1996).
108. Kullik, I., Toledano, M. B., Tartaglia, L. A. & Storz, G.Mutational analysis of the redox-sensitive transcriptionalregulator OxyR: regions important for oxidation andtranscriptional activation. J. Bacteriol. 177. 1275–1284 (1995)
109. Kim, S. O. et al. OxyR: a molecular code for redox-relatedsignaling. Cell 109, 383–396 (2002).Showed that a single regulatory Cys within thebacterial transcription factor OxyR is subject not onlyto S-nitrosylation but also to additional NO-dependentand NO-independent oxidative modifications, withdifferential effects on DNA binding and transcriptionalresponse.
110. Pugh, C. W. & Ratcliffe, P. J. Regulation of angiogenesis byhypoxia: role of the HIF system. Nature Med. 9, 677–684(2003).
111. Sumbayev, V. V., Budde, A., Zhou, J. & Brune, B. HIF-1αprotein as a target for S-nitrosation. FEBS Lett. 535,106–112 (2003).
112. Yasinska, I. M. & Sumbayev, V. V. S-nitrosation of Cys-800 ofHIF-1α protein activates its interaction with p300 andstimulates its transcriptional activity. FEBS Lett. 549,105–109 (2003).
113. Palmer, L. A., Gaston, B. & Johns, R. A. Normoxicstabilization of hypoxia-inducible factor-1 expression andactivity: redox-dependent effect of nitrogen oxides. Mol. Pharmacol. 58, 1197–1203 (2000).Provided an initial demonstration, with HIF1, that NOcould control the activity of transcription factors byregulating their stabiltity (ubiquitylation andproteasomal degradation).
114. Sandau, K. B., Fandrey, J. & Brune, B. Accumulation of HIF-1α under the influence of nitric oxide. Blood 97,1009–1015 (2001).
115. Metzen, E., Zhou, J., Jelkmann, W., Fandrey, J. & Brune, B.Nitric oxide impairs normoxic degradation of HIF-1α byinhibition of prolyl hydroxylases. Mol. Biol. Cell 14,3470–3481 (2003).
116. Yao, D. et al. Nitrosative stress linked to sporadicParkinson’s disease: S-nitrosylation of parkin regulates its E3ubiquitin ligase activity. Proc. Natl Acad. Sci. USA 101,10810–10814 (2004).Showed, together with reference 117, that the activityof the neuronal E3 ubiquitin ligase parkin is regulatedby S-nitrosylation in situ as a result of nitrosativestress induced experimentally or occuringendogenously in the brains of Parkinson’s diseasepatients.
117. Chung, K. K. K. et al. S-nitrosylation of parkin regulatesubiquitination and compromises parkin’s protective function.Science 304, 1328–1331 (2004).
118. Michael, D. & Oren, M. The p53–Mdm2 module and theubiquitin system. Semin. Cancer Biol. 13, 49–58 (2003).
119. Calmels, S., Hainaut, P. & Ohshima, H. Nitric oxide inducesconformational and functional modifications of wild-type p53tumor suppressor protein. Cancer Res. 57, 3365–3369(1997).
120. Brune, B., von Knethen, A. & Sandau, K. B. Transcriptionfactors p53 and HIF-1α as targets of nitric oxide. Cell. Signal. 13, 525–533 (2001).

166 | FEBRUARY 2005 | VOLUME 6 www.nature.com/reviews/molcellbio
R E V I E W S
121. Schonhoff, C. M., Daou, M. C., Jones, S. N., Schiffer, C. A.& Ross, A. H. Nitric oxide-mediated inhibition of Hdm2–p53binding. Biochemistry 41, 13570–13574 (2002).
122. Karin, M. & Ben-Neriah, Y. Phosphorylation meetsubiquitination: the control of NFκB activity. Annu. Rev.Immunol. 18, 621–663 (2000).
123. dela Torre, A., Schroeder, R. A., Punzalan, C. & Kuo, P. C.Endotoxin-mediated S-nitrosylation of p50 alters NF-κB-dependent gene transcription in ANA-1 murinemacrophages. J. Immunol. 162, 4101–4108 (1999).
124. Park, S. K., Lin, H. L. & Murphy, S. Nitric oxide regulatesnitric oxide synthase-2 gene expression by inhibiting NF-κBbinding to DNA. Biochem. J. 322, 609–613 (1997).
125. dela Torre, A., Schroeder, R. A. & Kuo, P. C. Alteration of NF-κB p50 DNA binding kinetics by S-nitrosylation.Biochem. Biophys. Res. Commun. 238, 703–706 (1997).
126. Matthews, J. R., Botting, C. H., Panico, M., Morris, H. R. &Hay, R. T. Inhibition of NF-κB DNA binding by nitric oxide.Nucleic Acids Res. 24, 2236–2242 (1996).
127. Marshall, H. E. & Stamler, J. S. Inhibition of NF-κB by S-nitrosylation. Biochemistry 40, 1688–1693 (2001).
128. Peng, H. B., Libby, P. & Liao, J. K. Induction and stabilizationof IκBα by nitric oxide mediates inhibition of NF-κB. J. Biol.Chem. 270, 14214–14219 (1995).
129. Marshall, H. E. & Stamler, J. S. Nitrosative stress-inducedapoptosis through inhibition of NF-κB. J. Biol. Chem. 277,34223–34228 (2002).
130. Reynaert, N. L. et al. Nitric oxide represses inhibitory κBkinase through S-nitrosylation. Proc. Natl Acad. Sci. USA101, 8945–8950 (2004).
131. Pantopoulos, K. & Hentze, M. W. Nitric oxide signaling toiron-regulatory protein: direct control of ferritin mRNAtranslation and transferrin receptor mRNA stability intransfected fibroblasts. Proc. Natl Acad. Sci. USA 92,1267–1271 (1995).
132. Kim, S. & Ponka, P. Effects of interferon-γandlipopolysaccharide on macrophage iron metabolism aremediated by nitric oxide-induced degradation of ironregulatory protein 2. J. Biol. Chem. 275, 6220–6226 (2000).
133. Bouton, C., Oliveira, L. & Drapier, J. C. Converse modulationof IRP1 and IRP2 by immunological stimuli in murine RAW264.7 macrophages. J. Biol. Chem. 273, 9403–9408 (1998).
134. Kim, S., Wing, S. S. & Ponka, P. S-nitrosylation of IRP2regulates its stability via the ubiquitin-proteasome pathway.Mol. Cell. Biol. 24, 330–337 (2004).
135. LaVaute, T. et al. Targeted deletion of the gene encoding ironregulatory protein-2 causes misregulation of iron metabolismand neurodegenerative disease in mice. Nature Genet. 27,209–214 (2001).
136. Jaffrey, S. R., Cohen, N. A., Rouault, T. A., Klausner, R. D. &Snyder, S. H. The iron-responsive element binding protein: atarget for synaptic actions of nitric oxide. Proc. Natl Acad.Sci. USA 91, 12994–12998 (1994).
137. Dudev, T. & Lim, C. Factors governing the protonation stateof cysteines in proteins: an Ab initio/CDM study. J. Am.Chem. Soc. 124, 6759–6766 (2002).
138. Kroncke, K. D., Klotz, L. O., Suschek, C. V. & Sies, H.Comparing nitrosative versus oxidative stress toward zincfinger-dependent transcription. Unique role for NO. J. Biol.Chem. 277, 13294–13301 (2002).
139. Pearce, L. L. et al. Role of metallothionein in nitric oxidesignaling as revealed by a green fluorescent fusion protein.Proc. Natl Acad. Sci. USA 97, 477–482 (2000).
140. Bossy-Wetzel, E. et al. Crosstalk between nitric oxide andzinc pathways to neuronal cell death involving mitochondrialdysfunction and p38-activated K+ channels. Neuron 41,351–365 (2004).
141. Gu, Z. et al. S-nitrosylation of matrix metalloproteinases:signaling pathway to neuronal cell death. Science 297,1186–1190 (2002).
142. Zhang, Z. et al. Activation of tumor necrosis factor-α-converting enzyme-mediated ectodomain shedding by nitricoxide. J. Biol. Chem. 275, 15839–15844 (2000).
143. Datta, B. et al. Red blood cell nitric oxide as an endocrinevasoregulator: a potential role in congestive heart failure.Circulation 109, 1339–1342 (2004).
144. Massy, Z. A. et al. Increased plasma S-nitrosothiolconcentrations predict cardiovascular outcomes amongpatients with end-stage renal disease: a prospective study.J. Am. Soc. Nephrol. 15, 470–476 (2004).
145. Gow, A. J., Buerk, D. G. & Ischiropoulos, H. A novel reactionmechanism for the formation of S-nitrosothiol in vivo. J. Biol.Chem. 272, 2841–2845 (1997).
146. Houk, K. N. et al. Nitroxyl disulfides, novel intermediates intransnitrosation reactions. J. Am. Chem. Soc. 125,6972–6976 (2003).
147. Bartberger, M. D. et al. Theory, spectroscopy, andcrystallographic analysis of S-nitrosothiols: conformationaldistribution dictates spectroscopic behavior. J. Am. Chem.Soc. 122, 5889–5890 (2000).
148. Chan, N. L., Kavanaugh, J. S., Rogers, P. H. & Arnone, A.Crystallographic analysis of the interaction of nitric oxide withquaternary-T human hemoglobin. Biochemistry 43,118–132 (2004).
149. Liu, X., Miller, M. J., Joshi, M. S., Thomas, D. D. & Lancaster, J. R. Jr. Accelerated reaction of nitric oxide withO2 within the hydrophobic interior of biological membranes.Proc. Natl Acad. Sci. USA 95, 2175–2179 (1998).
150. Nedospasov, A., Rafikov, R., Beda, N. & Nudler, E. Anautocatalytic mechanism of protein nitrosylation. Proc. NatlAcad. Sci. USA 97, 13543–13548 (2000).
151. Jourd’heuil, D., Jourd’heuil, F. L. & Feelisch, M. Oxidationand nitrosation of thiols at low micromolar exposure to nitricoxide. Evidence for a free radical mechanism. J. Biol. Chem.278, 15720–15726 (2003).
152. Liu, L. et al. Inactivation of annexin II tetramer by S-nitrosoglutathione. Eur. J. Biochem. 269, 4277–4286(2002).
153. Ji, Y., Toader, V. & Bennett, B. M. Regulation of microsomaland cytosolic glutathione S-transferase activities by S-nitrosylation. Biochem. Pharmacol. 63, 1397–1404 (2002).
154. Ventura, A. & Pelicci, P. G. Semaphorins: green light forredox signaling? Sci. STKE 2002, PE44 (2002).
Competing interests statementThe author declares competing financial interests: see Web versionfor details.
Online links
DATABASESThe following terms in this article are linked online to:Swiss-Prot: http://us.expasy.org/sprot/ASK1 | CAPON | caspase-3 | caspase-8 | caspase-9 | Dexras1 |eNOS | HDM2 | HIF1α | HIF2α | IKKα | IKKβ | IKKγ | iNOS | IRP1 |IRP2 | JNK1 | JNK3 | MKK3 | MKK6 | MMP9 | nNOS | OxyR |p21Ras | p53 | p60Src | PSD95 | RyR1 | RyR2 | SOD | TACE | TNFα |TRX
FURTHER INFORMATIONeMOTIF: http://motif.stanford.edu/emotif/emotif-scan.htmlAccess to this links box is available online.