proteomics and mass spectroscopy. proteomics the dream of having genomes completely sequenced is now...
TRANSCRIPT

Proteomicsand
Mass Spectroscopy

Proteomics• The dream of having genomes completely sequenced is now
a reality. The complete sequence of many genomes including the human one is known.
• However, the understanding of probably half a million human proteins encoded by less than 30,000 genes is still a long way away and the hard work to unravel the complexity of biological systems is yet to come.
• A new fundamental concept called proteome (PROTEin complement to a genOME) has recently emerged.

Proteomics• should drastically help to unravel biochemical and
physiological mechanisms of complex multivariate diseases at the functional molecular level.
• The discipline of proteomics has been initiated to complement physical genomic research.
• The term “proteome” was coined in 1994 by an Australian graduate student (Mark Wilkins), it has come to be used and defined in a variety of different ways

Proteomics
• Definition-The identification, characterization and quantification of all proteins involved in a particular pathway, organelle, cell, tissue, organ or organism that can be studied in concert to provide accurate and comprehensive data about that system.
• Or - A complete description of proteins expressed in any given cell at any given time

Proteomics• A cellular proteome is the collection of proteins found in
a particular cell type under a particular set of environmental conditions such as exposure to hormone stimulation
• It can also be useful to consider an organism's complete proteome, which can be conceptualized as the complete set of proteins from all of the various cellular proteomes. This is very roughly the protein equivalent of the genome.
• The term "proteome" has also been used to refer to the collection of proteins in certain sub-cellular biological systems. For example, all of the proteins in a virus can be called a viral proteome.

Proteomics
• So where are we in our understanding of the cell?– 31-60 K total genes in the human genome with little
difference between the fruit fly and us!
– Where does the diversity come from?
Answer: It’s the proteins!!!!!!!!!!!!

Proteomics
• The proteome is larger than the genome, especially in eukaryotes, in the sense that there are more proteins than genes.
• This is due to:
• alternative splicing of genes
• post-translational modifications like glycosylation or Phosphorylation.

alternative splicing of genes
• A given piece of pre-mRNA which has been transcribed from one gene can be chopped and reconnected in different ways to yield various new mRNAs which then exit the nucleus to be translated in the cytoplasm.
• When the pre-mRNA has been transcribed from the DNA, it includes several introns and exons.
• The regulation and selection of splice sites is done by Serine/Arginine-residue proteins

alternative splicing of genes
• Four known modes • A - Alternative selection of promoters:
this is the only method of splicing which can produce an alternative N-terminus domain in proteins. In this case, different sets of promoters can be spliced with certain sets of other exons.
• B - Alternative selection of cleavage/polyadenylation sites:
• this is the only method of splicing which can produce an alternative C-terminus domain in proteins. In this case, different sets of polyadenylation sites can be spliced with the other exons.

alternative splicing of genes
• Four known modes • C - Intron retaining mode: Instead of
splicing out an intron, the intron is retained in the mRNA transcript.
– However, the intron must be properly encoding for amino acids. The intron's code must be properly expressible, otherwise a stop codon or a shift in the reading frame will cause the protein to
be non-functional.
• D -Exon cassette mode: Certain exons are spliced out to alter the sequence of amino acids in the expressed protein.

post-translational modifications
• PTMs involving addition include:• Acetylation - the addition of an acetyl group, usually at the
N-terminus of the protein • Alkylation - the addition of an alkyl group (e.g. methyl,
ethyl) • Methylation -the addition of a methyl group, usually at
lysine or arginine residues. (This is a type of alkylation.) • Biotinylation - acylation of conserved lysine residues with a
biotin appendage • Glutamylation - covalent linkage of glutamic acid residues
to tubulin and some other proteins.

post-translational modifications
• PTMs involving addition include:• Glycylation - covalent linkage of one to more than 40
glycine residues to the tubulin C-terminal tail
• Glycosylation - the addition of a glycosyl group to either asparagine, hydroxylysine, serine, or threonine, resulting in a glycoprotein
• Isoprenylation - the addition of an isoprenoid group (e.g. farnesol and geranylgeraniol)
• Lipoylation - attachment of a lipoate functionality

post-translational modifications
• PTMs involving addition include:• Phosphopantetheinylation - the addition of a 4'-
phosphopantetheinyl moiety from coenzyme A, as in fatty acid, polyketide, non-ribosomal peptide and leucine biosynthesis
• Phosphorylation - the addition of a phosphate group, usually to serine, tyrosine, threonine or histidine

post-translational modifications• For instance, the peptide
hormone insulin
• Cut twice after disulfide bonds are formed, and a propeptide is removed from the middle of the chain;
• The resulting protein consists of two polypeptide chains connected by disulfide bonds.

Proteomics - Bridging the genome to the functions of the
cellAreas of Proteomics• Protein Analysis/Chemistry - Look at PT modifications,
structure and function, enzyme behavior
• Expression - what why and when - 2D gels, MS, HPLC/protein chips,
• Cell Mapping - protein-protein interactions - affinity tags, two hybrid, antibody pull down

Proteomics• A surprising finding of the Human Genome Project is that
there are far fewer protein-coding genes in the human genome than proteins in the human proteome – 20,000 to 25,000 genes coding for proteins. – about 1,000,000 proteins.
• The human body may contain more than 2 million proteins, each having different functions.
• The discrepancy implies that protein diversity cannot be fully characterized by gene expression analysis, thus proteomics is useful for characterizing cells and tissues.

So how does it work?• Most proteins function in collaboration with other proteins,
and one goal of proteomics is to identify which proteins interact.
• This often gives important clues about the functions of newly discovered proteins

So how does it work?• Proteins are resolved, sometimes
on a massive scale. Protein separation can be performed using 2-D gel electrophoresis, – `usually separates proteins first by
isoelectric point and then by molecular weight.
• Once proteins are separated and quantified, they are identified
• Individual spots are cut out of the gel and cleaved into peptides with proteolytic enzymes

So how does it work?• These peptides can then be
identified by mass spectrometry,
• Specifically: matrix-assisted laser desorption-ionization time-of-flight (MALDI-TOF) mass spectrometry.
• In this procedure, a peptide is placed on a matrix, which causes the peptide to form crystals.

So how does it work?• Then the peptide on the matrix is
ionized with a laser beam and an increase in voltage at the matrix is used to shoot the ions toward a detector in which the time it takes an ion to reach the detector depends on its mass.
• The higher the mass, the longer the time of flight of the ion.

So how does it work?• In a MALDI-TOF mass
spectrometer, the ions can also be deflected with an electrostatic reflector that also focuses the ion beam.
• Thus, the masses of the ions reaching the second detector can be determined with high precision and these masses can reveal the exact chemical compositions of the peptides, and therefore their identities!

So how does it work?• Protein mixtures can also be analyzed without prior
separation.
• These procedures begin with proteolytic digestion of the proteins in a complex mixture
• The resulting peptides are often injected onto a high pressure liquid chromatography column (HPLC) that separates peptides based on hydrophobicity.
• HPLC can be coupled directly to a time-of-flight mass spectrometer using electrospray ionization

So how does it work?• electrospray ionization:
A technique used in mass spectrometry to produce ions.
• It is especially useful in producing ions from macromolecules because it overcomes the propensity of these molecules to fragment when ionized

So how does it work?• Peptides eluting from the column
can be identified by tandem mass spectrometry (MS/MS).
• The first stage of tandem MS/MS isolates individual peptide ions, and the second breaks the peptides into fragments and uses the fragmentation pattern to determine their amino acid sequences.
• Labeling with isotope tags can be used to quantitatively compare proteins concentration among two or more protein samples.

So how does it work?• Finally, use databases.
• Computer compares sequences to other sequences stored in an internationally accessible database.
• Determines the identity of the isolated protein
• As the entire human genome is known, computers are able to determine nearly every potential protein.
• New proteins are “discovered” when they match sequences predicted by the computer that have not previously been found.

Medical Applications Alzheimer’s disease: Elevations in beta secretase creates amyloid/beta-protein,
which causes plaque to build up in the patient's brain, which causes dementia.
Targeting this enzyme decreases the amyloid/beta-protein and so slows the progression of the disease.
A procedure to test for the increase in amyloid/beta-protein is immunohistochemical staining, in which antibodies bind to specific antigens or biological tissue of amyloid/beta-protein.

Medical Applications Heart disease: Commonly assessed using several key protein based biomarkers. Standard protein biomarkers for CVD include interleukin-6,
interleukin-8, serum amyloid A protein, fibrinogen, and troponins.
cTnI cardiac troponin I increases in concentration within 3 to 12 hours of initial cardiac injury and can be found elevated days after an acute myocardial infarction.
A number of commercial antibody based assays as well as other methods are used in hospitals as primary tests for acute myocardial infarction.

Medical Applications Renal cell carcinoma: Proteomic analysis of kidney cells and cancerous kidney
cells is producing promising leads for biomarkers and developing assays to test for this disease.
In kidney-related diseases, urine is a potential source for such biomarkers.
Recently, it has been shown that the identification of urinary polypeptides as biomarkers of kidney-related diseases allows to diagnose the severity of the disease several months before the appearance of the pathology.

Medical Applications . Phenylketonuria (PKU)
– Affects in in 5,000 newborns– Most common nervous system disorder
• Allele is on chromosome 12– Lack the enzyme needed for the metabolism of
the amino acid phenylalanine– A build up of abnormal breakdown pathway
• Phenylketone
• Accumulates in urine. If diet is not checked, can lead to severe mental retardation

Overview of proteomics
• Nucleus
• DNA – RNA
• Cytoplasm– Protein expression and
modification
• Protein isolation
• Mass spectroscopy
• Protein sequence
• Identification

Overview of proteomics• Proteomics research is
highly interdisciplinary, bringing together:
• biology
• chemistry
• instrumentation
• Statistics
• computer science

Overview of proteomics
Summary time!
• Spend 15- 20 mins going over this as a group
• MAKE NOTES!!!!!!!!!!!!
• http://www.childrenshospital.org/cfapps/research/data_admin/Site602/mainpageS602P0.html

Mass Spectroscopy

Mass Spectroscopy• An analytical technique used to measure the mass-to-charge
ratio of ions.
• It is most generally used to find the composition of a physical sample by generating a mass spectrum representing the masses of sample components.
• The technique has several applications, including:
• 1) identifying unknown compounds by the mass of the compound molecules or their fragments

Mass Spectroscopy• 2) determining the isotopic composition of elements in a
compound.
• 3) determining the structure of a compound by observing its
fragmentation
• 4) quantifying the amount of a compound in a sample using
carefully designed methods (mass spectrometry is not inherently quantitative)

Mass Spectroscopy• 5) studying the fundamentals of gas phase ion chemistry
(the chemistry of ions and neutrals in vacuum)
• 6) determining other physical, chemical, or even biological properties of compounds with a variety of other approaches.
• A mass spectrometer is a device that measures the mass-to-charge ratio of ions.
• This is achieved by ionizing the sample and separating ions of differing masses and recording their relative abundance by measuring intensities of ion flux.

Mass Spectroscopy• All Mass specs consist of:• A high vacuum system
– 10-6 torr
• A sample inlet– GC, HPLC, electron impact,
or direct chemical isolation
• An ion source– Converts molecules to gas-
phase ions– MALDI, fast atom
bombardment

Mass Spectroscopy• All Mass specs consist of:• A mass filter/ analyzer
– Time of flight, magnetic sector, MALDI, or ion trap
• A detector– Array detector, conversion
dynode, or electron multiplyer
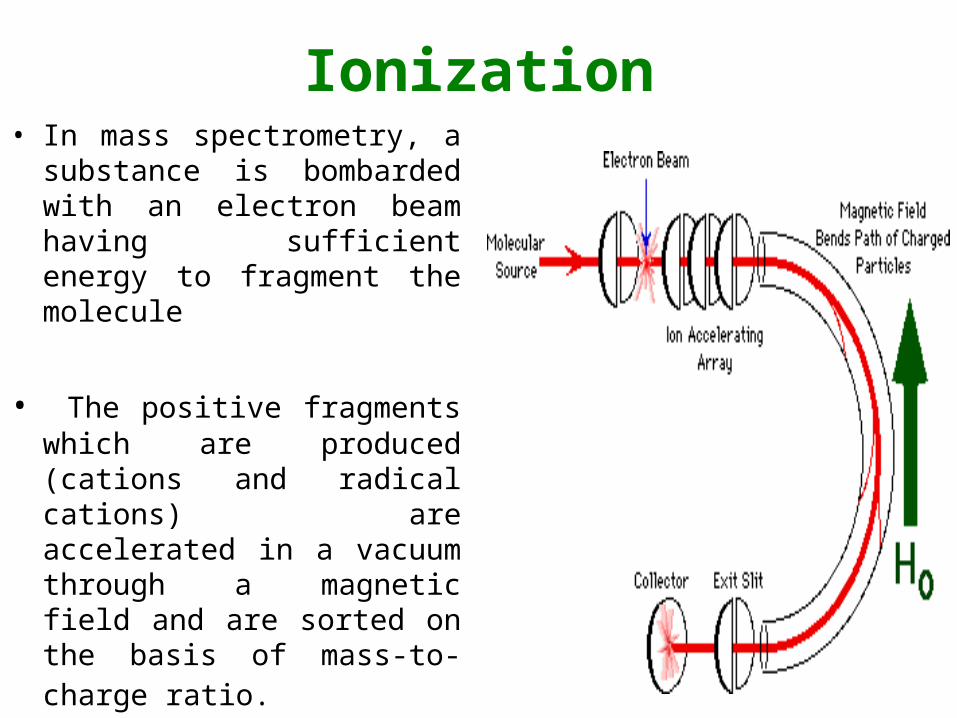
Ionization• In mass spectrometry, a
substance is bombarded with an electron beam having sufficient energy to fragment the molecule
• The positive fragments which are produced (cations and radical cations) are accelerated in a vacuum through a magnetic field and are sorted on the basis of mass-to-charge ratio.

Ionization• Since the bulk of the ions
produced in the mass spectrometer carry a unit positive charge, the value m/e is equivalent to the molecular weight of the fragment.
• The analysis of mass spectroscopy information involves the re-assembling of fragments, working backwards to generate the original molecule.

Ionization• Electron Impact ionization (EI):• It comprises an electron gun, a time-of-flight mass spectrometer with
position-sensitive detector (PSD). • Analyte must be in a vapor state, limiting biological material below
400Da
• Useful for metabolites, pollutants, and pharmaceutical compounds

Ionization• Chemical ionization:• Especially useful technique for organic chemists when no
molecular ion is observed in EI mass spectrum
• Ionization of sample (analyte) is achieved by interaction of its molecules with reagents such as CH4 or NH3
• Very good for determining molecular mass– as high intensity molecular ions are produced owing to less
fragmentation.

Ionization• electrospray ionization: A
technique used in mass spectrometry to produce ions.
• It is especially useful in producing ions from macromolecules because it overcomes the propensity of these molecules to fragment when ionized
• Is the primary ion source used in liquid chromatography-mass spec because it's a liquid-gas interface that is capable of coupling liquid chomatography with mass spectrometry

Mass Analyzers • Once ions are created and leave the ion source they pass
into a mass analyzer
• This separates the ions and measures their masses– What is actually measured is the mass to charge ratio (m/z) of each
ion
• At any given time, ions of a particular mass pass through and are counted by the detector– In this way, the analyzer scans through a large range of masses

Mass Analyzers• Quadrupole mass spectrometry:
• Essentially a mass filter that is capable of transmitting only the ion of choice. A mass spectrum is obtained by scanning through the mass range of interest over time.
• Two opposite rods have an opposite applied potential and affect the trajectory of ions traveling down the flight path centered between the four rods

Mass Analyzers• Quadrupole mass spectrometry:
• The mass range of the oscillating ions is scanned by changing the DC voltage and the frequency.
• The resolution of the spectrometer can be increased by either employing eight poles or by connecting two or three qaudupoles in series
• Excel at applications where particular ions of interest are being studied because they can stay tuned on a single ion for extended periods

Mass Analyzers• Ion trap mass spectrometry:
• The ion trap consists of three electrodes with hyperbolic surfaces, the central ring electrode, and two adjacent endcap electrodes
• The device is radially symmetrical the electrodes are aligned and isolated using ceramic spacers and posts

Mass Analyzers• Ion trap mass spectrometry:
• Advantages:
• (i) high sensitivity
• (ii) compactness and mechanical simplicity in a device which is nevertheless capable of high performance
• (iii) tandem mass spectrometry experiments are available by performing sequential mass analysis measurements
• (iv) high resolution

Mass Analyzers• Magnetic sector analyzer:
• Generated ions are accelerated and are passed around a curved track (the sector) leading to a detector.
• By increasing the magnetic field applied to the ions, heavier ions with higher momentum can be induced to follow the curved track.

Mass Analyzers• Magnetic sector analyzer:
• Only ions of mass-to-charge ratio that have equal centripetal and centrifugal forces pass through the flight tube
• The ions that reach the detector can be varied by changing either the magnetic field or the applied voltage of the ion optics.
• So the individual ion beams are separated spatially and each has a unique radius of curvature according to its mass/charge ratio

Mass Analyzers• Plasma desorption ionization:
• First available to analyze proteins and other large biomolecules (well, less than 35,000 Mr).
• The technology is now relatively obsolete
• Used radioactive californium (252Cf)
• Required a time of flight (TOF) mass detector

Mass Analyzers• matrix-assisted laser desorption-ionization time-of-flight
(MALDI-TOF) mass spectrometry. • Relatively novel technique in which a co-precipitate of an
UV-light absorbing matrix and a biomolecule is irradiated by a nanosecond laser pulse,which causes the peptide to form crystals.
• Most of the laser energy is absorbed by the matrix, which prevents unwanted fragmentation of the biomolecule.
• The ionized biomolecules are accelerated in an elctric field and enter the flight tube

Mass Analyzers• matrix-assisted laser desorption-ionization time-of-flight
(MALDI-TOF) mass spectrometry.
• During the flight in this tube, different molecules are separated according to their mass to charge ratio and reach the detector at different times.
• In this way each molecule yields a distinct signal.
• It is a very sensitive method, which allows the detection of low (10-15 to 10-18 mole) quantities of sample with an accuracy of 0.1 - 0.01 %.
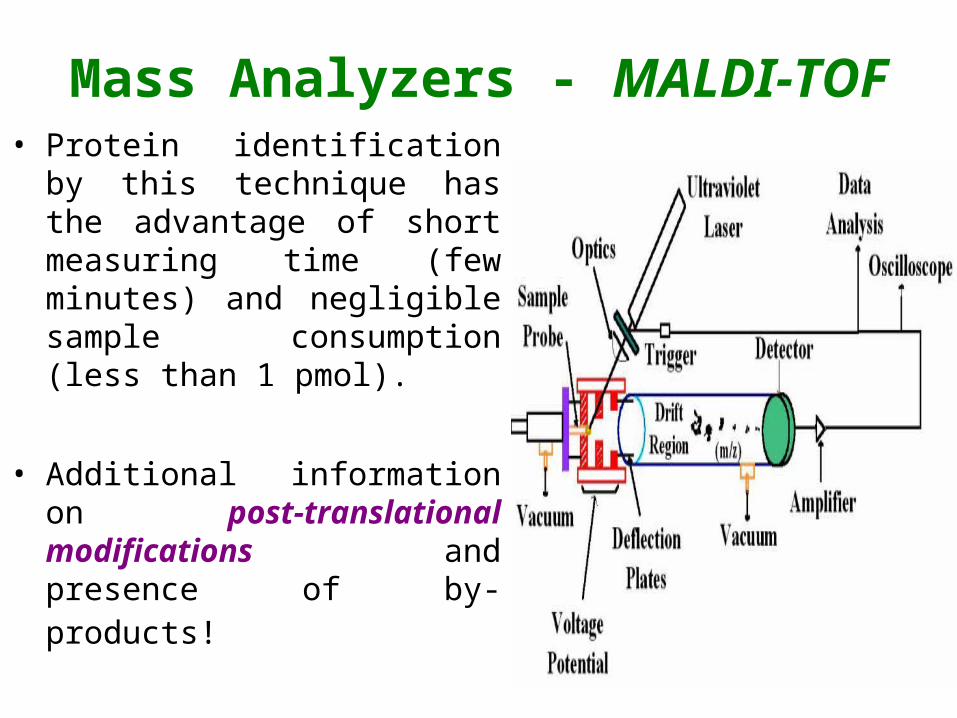
Mass Analyzers - MALDI-TOF• Protein identification by this
technique has the advantage of short measuring time (few minutes) and negligible sample consumption (less than 1 pmol).
• Additional information on post-translational modifications and presence of by-products!

Mass Analyzers - MALDI-TOF
• Drawbacks of Maldi-tof• The sample preparation for MALDI is important for the
result.
• Inorganic salts which are also part of protein extracts interfere with the ionization process.
• The matrix protein mixture is not homogenous because the polarity difference leads to a separation of the two substances during crystallization

Mass Analyzers - MALDI-TOF
• The spot diameter of the target is much larger than that of the laser, which makes it necessary to do several laser shots at different places of the target, to get the statistical average of the substance concentration within the target spot.
• Delay between laser pulses, delay time of the acceleration power, laser wavelength, energy density of the laser and the impact angle of the laser on the target are among others the critical values for the quality and reproducibility of the method.

Detector• The electron multiplier is a
highly sensitive device to detect individual energetic particles such as electron, photons, or ions
• Multipliers are based on two principles:
• (1) the particle(s) to be detected have to be converted to electrons before the amplification can take place (using a so called conversion dynode)

Detector• (2) the amplification is caused by
a cascade of acceleration electrodes (called dynodes) which accelerate the electrons to speeds which allow them to generate more than one new electron when hitting the next dynode.

Protein identification• Peptide sequencing – a quick
example.• A peptide and a protein digest of
it were studied by Mass Spec
• MALDI-TOF detected a peek at 3840.2
• Following HPLC-MS of the 3480.2 Da peak showed signals at m/z = 176, 624, 1129, 1508.
• Looked at the 624 component
• The ions appeared at m/z = 521, 406, 293, 130, and 43

Protein identification• Peptide sequencing – a
quick example.• What is the molecular mass of
the peptide?
• 176+624+1229+1508 = 3538 Da
• But the peak was 3840.2!
• Why the difference?


What is the molecular mass of the peptide?
• Why the difference?
• 3538 – 3480.2 = 57.8 (58 Da) why a difference of 58 Da
• There were four peaks following enzyme digest– Take into account enzymatic hydrolysis
– Three cleavage points give four parts
– Each requires the imput of one water molecule (H2O = 18)
– 18 X 3 =58
– So peak is heavier than the actual peptide

What is the sequence of the 624 component?

The Mass (DA) of the amino acids
Symbol Structure Mass (Da)Ala A -NH.CH.(CH3).CO- 71.0
Arg R -NH.CH.[(CH2)3.NH.C(NH).NH2].CO- 156.1
Asn N -NH.CH.(CH2CONH2).CO- 114.0
Asp D -NH.CH.(CH2COOH).CO- 115.0
Cys C -NH.CH.(CH2SH).CO- 103.0
Gln Q -NH.CH.(CH2CH2CONH2).CO- 128.1
Glu E -NH.CH.(CH2CH2COOH).CO- 129.0
Gly G -NH.CH2.CO- 57.0
His H -NH.CH.(CH2C3H3N2).CO- 137.1
Ile I -NH.CH.[CH.(CH3)CH2.CH3].CO- 113.1
Leu -NH.CH.[CH2CH(CH3)2].CO- 113.1
Lys K -NH.CH.[(CH2)4NH2].CO- 128.1
Met M -NH.CH.[(CH2)2.SCH3].CO- 131.0
Phe F -NH.CH.(CH2Ph).CO- 147.1
Pro P -NH.(CH2)3.CH.CO- 97.1
Ser S -NH.CH.(CH2OH).CO- 87.0
Thr T -NH.CH.[CH(OH)CH3).CO- 101.0
Trp W -NH.CH.[CH2.C8H6N].CO- 186.1
Tyr Y -NH.CH.[(CH2).C6H4.OH].CO- 163.1
Val V -NH.CH.[CH(CH3)2].CO- 99.1

Protein identification• What is the sequence of the 624 component?
• m/z 624 521 406 293 130 43
• ▲ 103 115 115 163 87
• aa Cys Asp Asp Tyr Ser
• The difference in the m/z valve gives you the identity of the corresponding amino acid
• So the peptide sequence for the 625 component is:– Cys-Asp-Asp-Tyr-Ser

THE END!