protocol representation model in the real worldprotocol representation model in the real world...
TRANSCRIPT

Optimizing Clinical Trials: Concept to Conclusion™ © 2012 Medidata Solutions, Inc. § 1
Optimizing Clinical Trials: Concept to Conclusion™
Protocol Representation Model in the Real World
Joshua Pines Senior Manager, Medidata Solutions

Optimizing Clinical Trials: Concept to Conclusion™ © 2012 Medidata Solutions, Inc. § 2
Agenda
• Introduction
• Challenges
• Opportunities
• Application

Optimizing Clinical Trials: Concept to Conclusion™ © 2012 Medidata Solutions, Inc. § 3
Brief Intro - The Theory Behind PRM!

Optimizing Clinical Trials: Concept to Conclusion™ © 2012 Medidata Solutions, Inc. § 4
The PRM and BRIDG Relationship
• PRM is a domain analysis model
• PRM is a collection of BRIDG Classes
• Protocol Representation Sub Domain View + Additional Classes
4

Optimizing Clinical Trials: Concept to Conclusion™ © 2012 Medidata Solutions, Inc. § 5
Why Use BRIDG?
• Protocol ID = Protocol ID = Protocol ID
• Whatever the implementation, we can be assured that “Protocol ID” is semantically the same across the entire study lifecycle
• BRIDG ensures it is technically the same as well
5

Optimizing Clinical Trials: Concept to Conclusion™ © 2012 Medidata Solutions, Inc. § 6
But People Think it’s Not That Simple

Optimizing Clinical Trials: Concept to Conclusion™ © 2012 Medidata Solutions, Inc. § 7
Challenges – A Reality Check!
7

Optimizing Clinical Trials: Concept to Conclusion™ © 2012 Medidata Solutions, Inc. § 8
PRM is a Scary-Sounding Acronym

Optimizing Clinical Trials: Concept to Conclusion™ © 2012 Medidata Solutions, Inc. § 9
Analysis Model vs. Implementation

Optimizing Clinical Trials: Concept to Conclusion™ © 2012 Medidata Solutions, Inc. § 10
Document-Centric Thinking Abounds
“My protocol standards will be different than
yours so how can there be an ‘industry
standard’?”

Optimizing Clinical Trials: Concept to Conclusion™ © 2012 Medidata Solutions, Inc. § 11
Opportunities for PRM
11

Optimizing Clinical Trials: Concept to Conclusion™ © 2012 Medidata Solutions, Inc. § 12
There is More Complexity and Data
• Studies are increasingly longer and more complex1
• 49% higher procedure frequency • 74% longer studies • 270% longer CRFs • 6.5% more unique procedures
• 15-30% of data not used in NDA2
• 24% of procedures are non-core3 • Contributing 18% of per patient procedure budget
1 Tufts Center for the Study of Drug Development: Assessing the impact of protocol design changes on clinical trial performance,
2 Tufts Center for Drug Development: Assessing the down stream impact of protocol design complexity
3 Tufts Center for Drug Development: 2012 Protocol Study

Optimizing Clinical Trials: Concept to Conclusion™ © 2012 Medidata Solutions, Inc. § 13
Which is Driving Up Protocol Amendments
Getz, Zuckerman, Cropp, Hindle , Krauss. Measuring the Incidence, Causes and Repercussions of Protocol Amendments. Drug information Journal. 2011 45(3); 265 - 275
13
*Analysis of those protocols with at least one amendment Note: All values are means
• 69% of all protocols have at least one amendment
• 46% of all amendments occur BEFORE first patient first dose
• 34% are considered “somewhat” or “completely’” avoidable
• Adds 61-days and cost $450,000+ to implement each amendment

Optimizing Clinical Trials: Concept to Conclusion™ © 2012 Medidata Solutions, Inc. § 14
Application of PRM
14

Optimizing Clinical Trials: Concept to Conclusion™ © 2012 Medidata Solutions, Inc. § 15
Results
• Simplified protocols • Reduced number of procedures and CPP • Reduced effort on downstream systems • Minimized cleaning and analysis of excess data
• Avoid amendments/rework • Reductions in cycle times • Minimized duplicate entry
• Shift from CDM to information management • Increased quality, consistency of clinical info • Align objectives, endpoints and procedures
A New Approach to Study Design Strategy/Goal
• Line of sight from strategy to plan to study design • Parallel development of Protocol, RAP, eCRF, CSR • Leverage industry benchmarks
Modular Authoring
Tem
plat
es a
nd R
euse
Feas
ibilit
y As
sess
men
tRe
view/
App
rova
l/ Di
strib
utio
nAu
thor
ing
Plan
ning
& s
tudy
des
ign
CIL
Medidata Designer
Enter study id
1
Trigger: CIL ‘wants to’
1
Study Information Skeleton Created
Automatic
Medidata Designer2, 3
Grow study information in Design environment
8Medidata Designer
CIL / EST2,3,4,5,6,7
Medidata Designer
Select delivery template(s)
9CIL / EST
9
8,9 10Automatic
Medidata Designer
Populate study information from Design env
Medidata Designer
CIL / EST11
Develop WORD content within Design env
9
Templates include:- CSP- Protocol- RAP- eT&E- CSR focus document / shell- Disclosure summary (tbd)
12CIL / EST
12
Upload deliverables to eDX for review
eDX
eDX
Approval of deliverables
Governance
eDX
13
15
12
Review of deliverables
Internal / External
14
17
IMMS
CIL / EST
PIER
16CIL / EST
Upload approved deliverables to PIER
16
15
Upload approved deliverables to IMMS
Conduct Protocol Specific Feasibility
??
Regional Medical Director
Or does this input to WORD content?
Do we need Inc/Exc flow into eDX in order to perform
feasibility analysis (ref SB)? Or is this just manual
reference?
Draft
Final
Internal functionality not mapped: review, report, ppt
Internal references utilised in growing the information but not mapped:
- Reference content- Standard procedures
- Indicative costs
What is captured here?- new content or
- content that cannot go into design environment
IR interface for tables/listing & graphs – where?
Would details for adaptive trials have to be added here?
Trigger:1, Want wider input2, Develop WORD
content
Trigger??
Comments / Edits
Comments / Edits
‘Approved’ / FPAData Input and Output
1.Study id’s 2. Medium term study design (OneCDP)3. Study Master Data (MDM)4. Library of approved standard content (eProtocol) {All studies, Asset, study level}5. Legacy information (IMMS)6. Standard procedures (within system from MDR?)7. Indicative costs (eProtocol)8. Study information 9. Document Template Library10. Study information from design environment in template11. Additional information captured in WORD in design environment12. Draft deliverable13. Review deliverable14. Final ‘Approvable’ Deliverable15. Approved Deliverable16. Published Deliverable17 Archived Deliverable18. eDocument Metadata
2
15,16Technical Authoring Team
Store & Manage Deliverable Metadata
Medidata Designer9
Technical Authoring Team
6
??
eProtocol, IMMS
Technical Authoring Team
Metadata Search of Ref Content for Reuse
9
eProtocol
18
2
Define Deliverables Templates
Technical Authoring Team
Copy Existing Deliverables Content: ??
??
??
Scope:Start = medium term study design onwardsEnd = approval, publishing & archiving of all study deliverables; deliverable metadata to facilitate re-use
eSignature or workflow transfer
to IMMS

Optimizing Clinical Trials: Concept to Conclusion™ © 2012 Medidata Solutions, Inc. § 16
Objectives
Endpoints
Schedule
What We Mean by Structured Design
80% protocol standard, design is buried as unstructured text
Optimizing Clinical Trials: Concept to Conclusion™ © 2012 Medidata Solutions, Inc. § 17
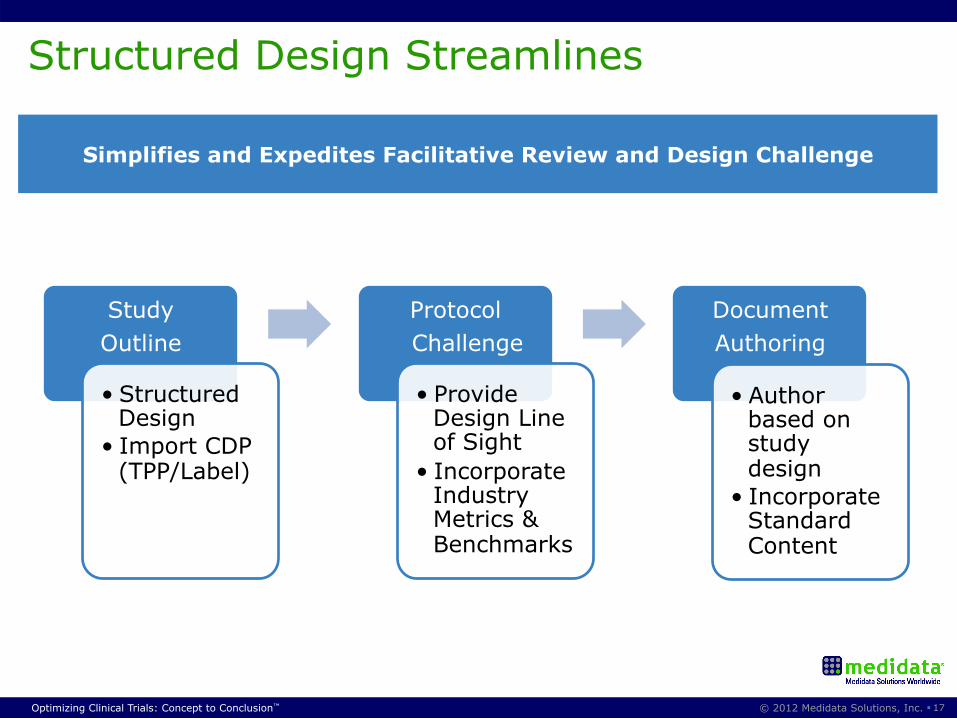
Structured Design Streamlines
Simplifies and Expedites Facilitative Review and Design Challenge
Study Outline
• Structured Design
• Import CDP (TPP/Label)
Protocol Challenge
• Provide Design Line of Sight
• Incorporate Industry Metrics & Benchmarks
Document Authoring
• Author based on study design
• Incorporate Standard Content

Optimizing Clinical Trials: Concept to Conclusion™ © 2012 Medidata Solutions, Inc. § 18
And Facilitates Reuse
PRM

Optimizing Clinical Trials: Concept to Conclusion™ © 2012 Medidata Solutions, Inc. § 19
"Within Study" Re-Use Instances
% Re-Use
Study 1 As-Is Transformed Total
Stats plan 13 1.5% 1.6% 3.1%
CSR Introduction & Methodology sections 37 4% 39% 43%
CSR Synopsis 22 17% 82% 99%
CTR Summary 22 1% 91% 92%
Ct.gov ----- 1% 99% 100%
CSR Study Results in text tables2 ----- 0% 100% 100%
"Within Study" Re-Use Instances
% Re-Use
Study 2 As-Is Transformed Total
CSR Introduction & Methodology sections 41 2% 66% 67%
CSR Synopsis 13 5% 93% 98%
CTR Summary 16 19% 76% 95%
"Across Program" Re-Use Instances
% Re-Use
Study x to Study y As-Is Transformed Total
Protocol to Protocol 73 23%3 24% 47%3
Stats plan to stats plan 29 3% 27% 30%
Which Can Enable Better Analysis

© 2011 Medidata Solutions, Inc. – Proprietary and Confidential © 2012 Medidata Solutions, Inc. § 20 Optimizing Clinical Trials: Concept to Conclusion TM
And Comparisons Across Designs
Complexity - Study Work Effort Sponsor 80.57 Industry 36.05
Total Procedures 254.27 Industry 117.25
Unique Procedures 31.69 Industry 21.27
Total Study Visits 10.94 Industry 8.58
Patients per site 15.40 Industry 9.63
Cost Per Patient $13,810 Industry $8,083
Benefit Per Study (Potential) Sponsor spend compared to Industry Avg/study $69,205 Avg of individual study details
Clinical Study Analysis (ANTI-INFECTIVE) Sponsor vs Industry Median (indications, phase and year)
Analysis includes 32 sponsor studies
Sponsor
Sponsor
Sponsor
Sponsor
Sponsor
224% higher
217% higher
149% higher
128% higher
160% higher
171% higher

Optimizing Clinical Trials: Concept to Conclusion™ © 2012 Medidata Solutions, Inc. § 21
Some Real-Life PRM Benefits
*24 studies across phases II – IV
Prior to data-centric process improvements
After data-centric process improvements
§ Avg. Protocol Amendments: 1.94 § Sites w/zero enrollment: 14% § Median recruitment cycle: 68 weeks
§ Avg. Protocol Amendments: 1.54 § Sites w/zero enrollment: 10% § Median recruitment cycle: 38 weeks
§ Handoffs in review process: 312 § Authoring & Review Cycle Time: 268 Days
§ Authoring & Review Effort: 57.5 Days
§ Process value added time: 48.7%
§ Handoffs in review process: 78 § Authoring & Review Cycle Time: 99 Days
§ Authoring & Review Effort: 39.5 Days
§ Process value added time: 84%

Optimizing Clinical Trials: Concept to Conclusion™ © 2012 Medidata Solutions, Inc. § 22
Summary

Optimizing Clinical Trials: Concept to Conclusion™ © 2012 Medidata Solutions, Inc. § 23
PRM Can Drastically Influence Study Design
• Significant process change involved in adoption
• Real business value can be demonstrated
• Opportunities • Influence trial cost and complexity • Drive reuse and efficiency • Interface with other standards
• Analytics and business intelligence

Optimizing Clinical Trials: Concept to Conclusion™ © 2012 Medidata Solutions, Inc. § 24
Learn More
• CDISC.org
• Forthcoming PRM Toolkit Wizard
• November 1 webinar with Jeff Abolafia, Rho
• MDSOL.com

Optimizing Clinical Trials: Concept to Conclusion™ © 2012 Medidata Solutions, Inc. § 25
Questions Thank you! Joshua Pines [email protected] Twitter: @JoshPines LinkedIn: JoshPines