proximate analysis: water, ash, minerals, proteins, fat ... · proximate analysis: water, ash,...
TRANSCRIPT

1|P a g e
Proximate Analysis: Water, Ash, Minerals, Proteins, Fat and Crude Fiber
Pre-lab Exercise: 1) Read about proximate analysis, water, ash, minerals, proteins, fat and the experiment and its background. 2) Prepare a flowchart(s) as appropriate for the lab exercises. 3) Prepare tables in anticipation of results for the various exercises. Introduction: Proximate analysis is a method for the quantification of different micronutrients in feed, also called the Weende analysis. This method divides all materials into six different categories, moisture or water, ash, crude protein (or Kjeldahl protein), crude lipid, crude fiber, and nitrogen free extracts (digestible carbohydrates). All nutrients can be placed into one of these categories whether they be amino acids, vitamins, mineral elements, enzymes, hormones, fats, carbohydrates etc. During this course we will be looking at some of the individual parts of this method of analysis. This is a three-week lab exercise: WEEK 1:
1. Take a representative sample – homogenize/grind sample (This sample will be used in subsequent exercises.)
2. Prepare sample for moisture content using vacuum oven. 3. Prepare sample for moisture analysis using freeze dryer. 4. Observe the apparatus for Dean Stark moisture determination 5. Prepare sample for ash analysis.
Week 2:
1. Prepare samples for fat determination by Soxhlet (must use the freeze dried samples for this analysis).
2. Take ashed samples and extract for mineral analysis. Use AA to determine mineral content.
3. Mass samples for protein analysis by Kjeldahl method. 4. Prepare and analyze sample for crude fiber analysis.

2|P a g e
Week 3: 1. Digest samples for protein analysis. Allow samples to cool and titrate to
determine protein content. 2. Mass samples from Soxhlet analysis to determine fat content. 3. Clean all glassware/equipment used.
Sampling: Raw materials and products must be sampled in order to conduct analysis. The size of the lot to be sampled can range from a few grams to thousands of kilos, and yet the sample used must be representative of the whole. Sampling methods will vary depending on the type of sample, i.e., gases, liquids or solids. Obtaining a representative sample from a nonhomogeneous solid sample requires that:
1. A gross sample must be taken 2. The gross sample be reduced to a representative laboratory – size sample. 3. The sample be prepared for analysis.
Two methods used for obtaining a solid representative sample are:
1. Coning and quartering: This method involves taking the gross sample mixing and pouring the sample into the shape of a cone. The gross sample may require grinding, blending, crushing etc. prior to coning and quartering. The cone is then flattened and then divided into four sections using a scoopula, stiff paper etc. Alternate quarters are combined to make a subsample, and one subsample is chosen at random for any additional mass reduction until the desired sample size is reached.

3|P a g e
Figure 1: Coning and quartering procedure (taken from: http://civilblog.org/2015/01/06/how-to-reduce-gross-aggregate-sample-to-test-sample-by-quartering-or-coning-method/)
.2. Rolling and quartering: In this method a representative sample obtained
from the coning and quartering method described above is placed in a conical pile on a piece of paper, wax paper, etc. The sample is then flattened and the material is mixed by rolling it back and forth. The material is then returned to the center by lifting the four corners of the paper and the sample is then split into half by using the quartering method. One half is selected by random for any further reduction or for analysis. The only difference between the two methods is that with the rolling and quartering method some mixing takes place when the sample is rolled. This method may allow for loosely bound clumps of particles to break apart, allowing for a more representative sample. Once the sample has been ground and representative samples have been obtained the remainder of the sample must be stored appropriately for future use.

4|P a g e
Determination of Moisture: Reading and preparation assignment: Chapter 1, pages 1-33 in Principles of food chemistry, Second Edition by John M. deMan. Water, although not considered a nutrient, is an integral part of all animals and their diets. Moisture content of foods varies widely. Fluid dairy products range from 87-91% water, while some fresh fruits contain 86-89% moisture and some breakfast cereals have below 4% moisture. The moisture content of foods is one of the most important and widely used measurements in the processing and testing of foods. Moisture content has a great effect on the stability and quality of foods. For example, if a grain contains too much water it is subject to rapid deterioration from mold growth, heating, insect damage, and sprouting. Moisture content is used in determining the nutritive value of foods, in expressing results of analytical determinations on a uniform basis, in meeting compositional standards or laws and is desirable to weigh samples for analytical determinations on a given moisture basis. Water may occur in foods in three forms. Some may be present as free water. This is found in the intergranular spaces and within pores of the material. Some water may also be absorbed on the surface of macromolecular colloids and finally there is some water that is in the bound form. This type of water is in combination with various other substances. The determination of moisture can be divided into 4 different methods; drying methods, distillation methods, chemical assays and physical procedures. In proximate analysis the moisture is determined by heating the sample until a constant mass is obtained. This treatment will drive off any volatile substances present. The mass of such compounds is generally insignificant and is included in the moisture content. Drying methods:
A. Vacuum Oven Method: Moisture determination can be accomplished by the means of a vacuum oven. This method takes advantage of the fact that the evaporation of water occurs more readily at lower pressures than at higher ones. This method allows for the removal

5|P a g e
of most of the water with relative ease, however the last 1% or so is quite difficult to remove. Procedure:
1. Weigh 2- 5 ± 0.001g of prepared sample into a predried, cooled in a desiccator, and weighed metal dish. (Remember not to handle the dish with your hands as they contain moisture, dirt, etc. Use tongs or a kimwipe)
2. Dry partially on a steam bath if necessary (only necessary if sample is liquid)
3. Place loosely covered dish on metal shelf in the vacuum oven kept at 70oC. 4. Dry to a constant weight (4-5 hours or overnight) at a pressure not to
exceed 200mm Hg. During drying admit into oven slow current of air (about 2 bubbles/sec) dried by passing through H2SO4.
5. Shut off the vacuum line. Carefully admit dried air; open oven; press cover onto dish; remove dish from oven; cool in desiccator for approximately 30 and weigh.
Calculations: Sample weight (g) – dry weight (g) *100% Sample weight (g) Table # 1A: Information for Moisture Determination by Vacuum Oven Replicate 1 Replicate 2 Replicate 3 ID of tin Mass of tin + lid Mass of tin + lid + sample Mass of sample Mass of tin + lid + dried sample Mass of dried sample Mass of moisture lost % moisture

6|P a g e
B. Freeze Drying Method: Freeze drying is a means of preserving wet substances, or solutions in water, that are usually of biological origin. Substances treated are living micro-organisms for preventative or curative medicine or for “banking” in culture collections, starters for fermentation processes, blood fractions, biological standards, hormones, enzymes, antibiotics, vitamins and unstable fine chemicals and pieces of tissue. After pre-freezing, the frozen product is exposed to vacuum and the contained ice allowed to sublime – that is, to change directly from the solid to the vapor state without melting. During freeze drying the ice does not evaporate simultaneously from all parts of the material, but a frozen interface continuously moves away from a dry outer boundary. The resulting “cake” has substantially the same size and shape as the original frozen mass and it may usually be stored at room temperature until required for use. When reconstitution is necessary this can be done by the addition of a suitable quantity of water, when the properties which survived the initial freezing are restored. The apparatus for freeze drying consists essentially of a vacuum vessel, a water trap and a mechanical vacuum pump. The water vapor which evolves in the process is trapped before entering the pump, which cannot deal with large volumes of vapor generated in the process. Methods in common used to for trap water are desiccants (drying agents), or refrigerated condensers as in the Supermodulyo. Containers of the prefrozen material are either attached externally to a vacuum manifold or placed in a chamber which in turn is evacuated. Vacuum creates conditions favorable to freeze drying and sublimation only occurs if heat is supplied to the frozen material. Room heat is usually sufficient for the manifold or small chamber freeze dryers. Procedure:
1. Weigh 2- 5 ± 0.001g of prepared sample into a preweighed large weigh boat. You should have 2 weights – mass of the empty boat and mass of the boat and sample.
2. Cover with saran wrap – poke holes to allow moisture to escape. 3. Samples are then placed in the freezer until frozen. 4. Place frozen samples on freeze dryer. 5. Turn on freeze dryer. Ensure that a good vacuum is obtained. 6. Samples are removed when the temperature of the shelf reached room
temperature (the time required depends on the amount of sample).

7|P a g e
7. Cool to room temperature in a desiccator. 8. Weigh the dried sample + weigh boat (don’t forget to remove the plastic
saran wrap before weighing).
Calculations: Sample weight (g) – dry weight (g) *100% Sample weight (g) Table # 1B: Information for Moisture Determination by Freeze Dryer Replicate 1 Replicate 2 Replicate 3 ID of sample Mass of weigh boat Mass of weigh boat + sample Mass of sample Mass of weigh boat + dried sample Mass of dried sample Mass of moisture lost % moisture
C. Dean – Stark Method:
This method involves the use of distillation in the determination of the moisture content of a sample. A sample is distilled using an immiscible organic solvent such as toluene. The mixture of water and the immiscible solvent (toluene) is distilled off and collected in a suitable measuring apparatus in which the water separates and its volume can be measured. Procedure:
1. Weigh an appropriate amount of prepared sample (amount will depend on the type of sample used) into a 250mL round-bottomed flask.
2. Add approximately 120mL of toluene to the flask. 3. Attach the moisture trap and condenser and connect the condenser up to
the water and drain into the sink. 4. Distill the toluene, using a heating mantle until the volume of water in the
trap is constant. 5. Allow the apparatus to cool.

8|P a g e
6. Carefully decant (remove the liquid while leaving the solid behind) the toluene into a pre-weighed 400mL beaker (ensure that your beaker is properly labelled).
7. Add 35mL of toluene to the sample, swirl and decant into same beaker. 8. Repeat two more times with new 35mL volumes of toluene. 9. Place labelled beaker into the fume hood. Allow the toluene to evaporate.
Reweigh the beaker to determine the fat content by Dean Stark. 10. Calculate the percentage of water in the sample by using the volume
measured and the weight of the initial sample used.
Determination of Ash: Ash is the inorganic fraction which does not give any specific information on any one nutrient. This value is used mostly to calculate the soluble carbohydrate by difference. The amount and composition of ash in a food product will depend upon the nature of the food used and on the method of ashing. The ashed sample can be further treated to analyze for 21-26 mineral elements. Procedure:
1. Acid wash 4 evaporating dishes, rinse well with deionized water and place them in the oven to dry. Once dry cool to room temperature in a desiccator. Remember to use tongs or a kimwipe when handling the evaporating dishes.
2. Weigh 2- 5 ± 0.001g of prepared sample into the preweighed acid washed evaporating dish. You should have 2 weights – mass of the empty evaporating dish and mass of the dish and sample. One of the dishes should be your blank (remember that a blank does not contain sample).
3. Preash the samples on the electric burners that are set up in the fume hood. When the samples have been preashed place them in a muffle furnace preheated to 550oC. Hold this temperature for at least 3hours.
4. Transfer samples to a desiccator to cool. Mass sample and calculate % ash.
These samples are used for Mineral Extraction. Do not discard!
9|P a g e
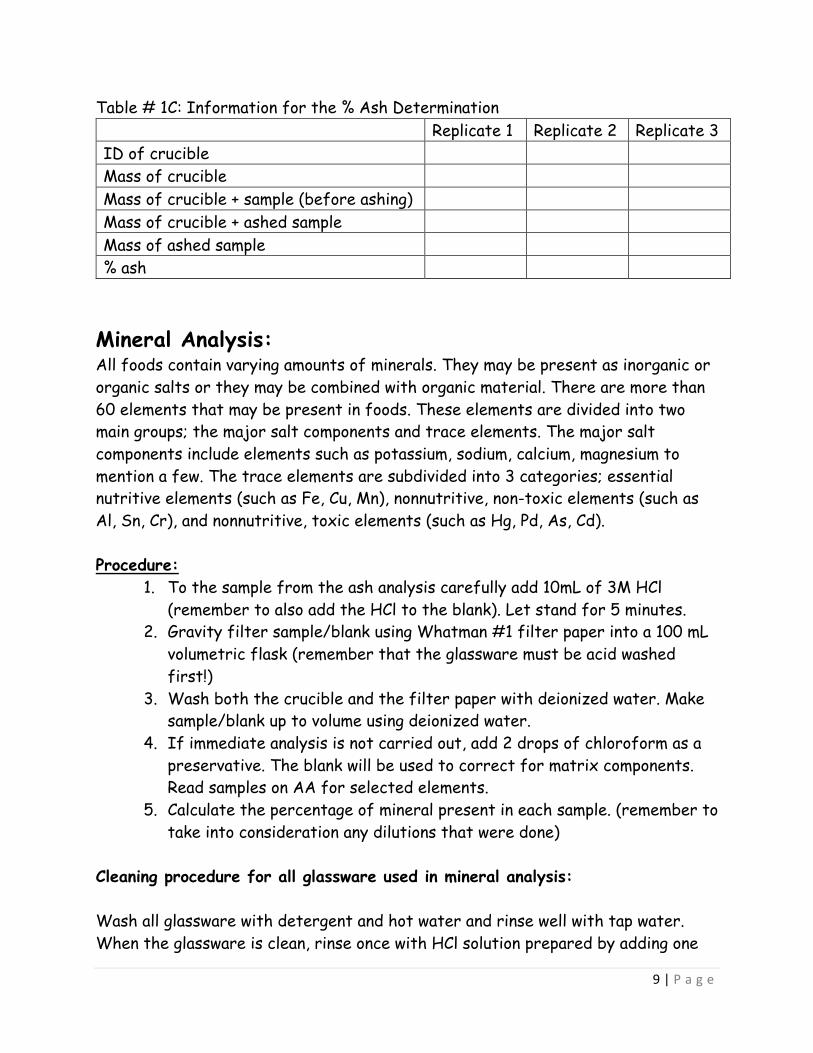
Table # 1C: Information for the % Ash Determination Replicate 1 Replicate 2 Replicate 3 ID of crucible Mass of crucible Mass of crucible + sample (before ashing) Mass of crucible + ashed sample Mass of ashed sample % ash
Mineral Analysis: All foods contain varying amounts of minerals. They may be present as inorganic or organic salts or they may be combined with organic material. There are more than 60 elements that may be present in foods. These elements are divided into two main groups; the major salt components and trace elements. The major salt components include elements such as potassium, sodium, calcium, magnesium to mention a few. The trace elements are subdivided into 3 categories; essential nutritive elements (such as Fe, Cu, Mn), nonnutritive, non-toxic elements (such as Al, Sn, Cr), and nonnutritive, toxic elements (such as Hg, Pd, As, Cd). Procedure:
1. To the sample from the ash analysis carefully add 10mL of 3M HCl (remember to also add the HCl to the blank). Let stand for 5 minutes.
2. Gravity filter sample/blank using Whatman #1 filter paper into a 100 mL volumetric flask (remember that the glassware must be acid washed first!)
3. Wash both the crucible and the filter paper with deionized water. Make sample/blank up to volume using deionized water.
4. If immediate analysis is not carried out, add 2 drops of chloroform as a preservative. The blank will be used to correct for matrix components. Read samples on AA for selected elements.
5. Calculate the percentage of mineral present in each sample. (remember to take into consideration any dilutions that were done)
Cleaning procedure for all glassware used in mineral analysis: Wash all glassware with detergent and hot water and rinse well with tap water. When the glassware is clean, rinse once with HCl solution prepared by adding one
10|P a g e
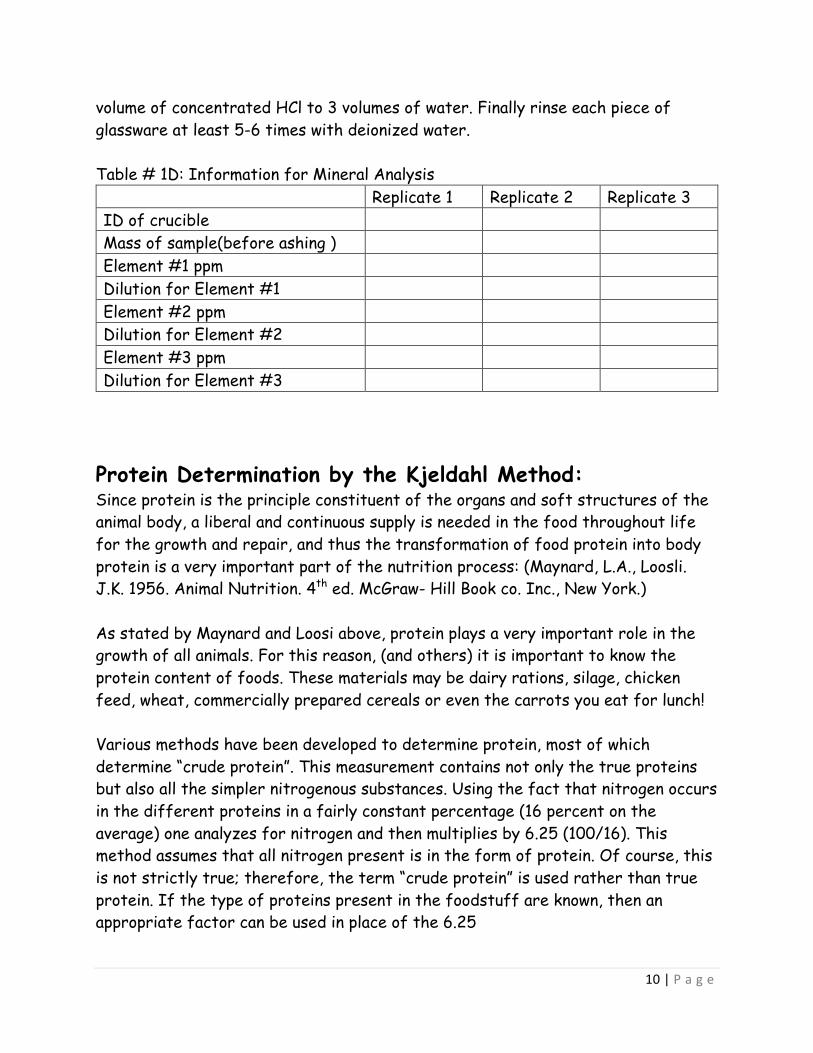
volume of concentrated HCl to 3 volumes of water. Finally rinse each piece of glassware at least 5-6 times with deionized water. Table # 1D: Information for Mineral Analysis Replicate 1 Replicate 2 Replicate 3 ID of crucible Mass of sample(before ashing ) Element #1 ppm Dilution for Element #1 Element #2 ppm Dilution for Element #2 Element #3 ppm Dilution for Element #3
Protein Determination by the Kjeldahl Method: Since protein is the principle constituent of the organs and soft structures of the animal body, a liberal and continuous supply is needed in the food throughout life for the growth and repair, and thus the transformation of food protein into body protein is a very important part of the nutrition process: (Maynard, L.A., Loosli. J.K. 1956. Animal Nutrition. 4th ed. McGraw- Hill Book co. Inc., New York.) As stated by Maynard and Loosi above, protein plays a very important role in the growth of all animals. For this reason, (and others) it is important to know the protein content of foods. These materials may be dairy rations, silage, chicken feed, wheat, commercially prepared cereals or even the carrots you eat for lunch! Various methods have been developed to determine protein, most of which determine “crude protein”. This measurement contains not only the true proteins but also all the simpler nitrogenous substances. Using the fact that nitrogen occurs in the different proteins in a fairly constant percentage (16 percent on the average) one analyzes for nitrogen and then multiplies by 6.25 (100/16). This method assumes that all nitrogen present is in the form of protein. Of course, this is not strictly true; therefore, the term “crude protein” is used rather than true protein. If the type of proteins present in the foodstuff are known, then an appropriate factor can be used in place of the 6.25

11|P a g e
The nitrogenous substance is oxidized by boiling with concentrated sulfuric acid. The nitrogen is converted to ammonia and is fixed as ammonium sulfate. Copper sulfate, mercury and selenium are some of the catalysts used in the reaction. Furthermore, potassium sulfate is frequently added to raise the boiling point of the sulfuric acid. All reagents used must be nitrogen- free and should be checked by blank determinations. During the digestion carbon is converted to carbon dioxide, hydrogen to water and nitrogen to ammonia. The CO2 and H2O boil off from the hot H2SO4. The NH3 formed reacts further with the H2SO4 to form ammonium hydrogen sulfate which remains in the sulfuric acid. After digestion is complete the excess H2SO4 is neutralized with NaOH. The NaOH also reacts in a 2:1 with the NH4HSO4 to release ammonia. The ammonia is then removed by distillation and trapped in boric acid solution. The NH3-boric acid complex is then titrated with standard HCL to determine the number of moles present. For the titration an indicator with a low pH range must be chosen to avoid interference by the buffering of the ammonium chloride in solution. Methyl red (pH 5.6) is a widely used indicator. One may also use alizarin or the methyl red-methylene blue indicators. Procedure:
1. Weight about 1 ± 0.001g of ground and well mixed sample onto a piece of lens paper (remember to record the exact weight of the sample used). Fold the lens paper so as not to lose any of the sample and place it in a digestion tube. (Remember to include a blank in your analysis. A blank should include all components except the sample itself).
2. Add 2 pellets of Kjeltabs (use tongs) and 10 mL of digestion acid (100parts H2SO4 + 5 parts Concentrated H3PO4)
3. Carefully add 5mL of 30% hydrogen peroxide. (Do this in the fume hood and use gloves or hot hands to hold the tube as it will become very hot). Caution, too fast addition of peroxide may cause losses of sample due to the violent reaction.
4. Place the tubes in the preheated digester at 420oC. Connect the aspirator to the tubes and digest the samples for 20 minutes and then place the side

12|P a g e
shields on the holder and continue to digest until no more black particles are on the sides of the tubes.
5. Remove the aspirator and the tubes from the digestion block and allow to cool for 5-10 minutes.
6. Dilute the samples with 75mL of water using a graduated cylinder.
7. Add 10mL of sodium thiosulfate solution (300grams of Na2S2O3. 5H2O/L)
just before distillation. (This should be done in the fume hood). 8. Fix the tube to the distilling unit. Place a 250mL Erlenmeyer flask containing
25mL boric acid solution (4%) and a couple of drops of indicator under the condenser outlet.
9. Dispense the alkali (50mL of 40% NaOH) slowly and then start the steam distillation. Collect the distillate until it reaches the 125mL mark on the flask. (This will take about 4-5 minutes).
10. Lower the flask and then turn off the steam. Pour the material in the distillation tube down the sink with lots of cold water.
11. Titrate the distillation of ammonia (material in the 250mL Erlenmeyer flask) with ~0.2M hydrochloric acid using methyl red: bromocresol indicator provided (0.07g methyl red in 70 mL ethanol +0.10g bromocresol green in ethanol).
12. Calculate results. Calculation:
% protein = (mL HCl – mL blank) * M of HCl *14.007 *f * 100 (mg of sample) (Typically f = 6.25 as mentioned above).
Table # 1D: Information for the determination of Crude Protein Replicate #1 Replicate #2 Replicate #3 Mass of Sample (g) mL of HCl for blank mL of HCl for sample Molarity of HCl % protein

13|P a g e
Fat Determination (Soxhlet Analysis): The fat content of foods can range from very low to very high in both vegetable and animal products. Lipids (fats and fat-like substances) have three important functions in foods; culinary, physiological and nutritional. Fats are insoluble in water and soluble in organic solvents. All substance soluble in ether are included in the crude fat fraction. True fats, as well as fat soluble vitamins, waxes, pigments, etc. will all be in this portion which is sometimes called the ether fraction, lipid fraction of fat fraction. There are many methods for lipid extraction. The method used depends on the type of material to be analyzed. Diethyl ether is the solvent most commonly used in the Soxhlet extraction method. The percentage of fat present is found by extracting a moisture free sample with ether continuously then evaporating the ether from the extraction and then measuring the weight of the residue. Procedure:
1. Mass out 3-5 ± 0.001g of freeze dried sample into extraction thimbles. 2. Cover with a small amount of defatted cotton wool. This is to prevent
sample from floating out with the ether. 3. Weight a 250mL beaker and ensure that the beaker is labelled the same
as the thimble. 4. Place the thimble in the center part of the Soxhlet apparatus. The lower
flask is filled with about 150mL of solvent (diethyl ether) and a few glass boiling beads. This apparatus is then assembled and attached to the condenser which is in turn attached to a tap with running water. As the sample is heated the condensing vapors will fill the middle part containing the sample and carry the dissolved lipid into the flask by a siphoning action each time the height of the siphon is attained.
5. After 4-5 hours turn off heat, disconnect the apparatus and remove the solvent to the appropriately pre-weighed labelled beaker.
6. Place labelled beakers in the fume hood and allow the solvent to evaporate.
7. Mass the beaker and the residue and calculate the percent crude fat.

14|P a g e
Table #1E: Information for the Determination of Crude Fat by Soxhlet Extraction Replicate 1 Replicate 2 Replicate 3 Mass of thimble Mass of thimble + sample Mass of sample Mass of beaker Mass of beaker + fatty residue Mass of fatty residue % crude fat
Simple Extraction: Procedure:
1. Beakers from the Dean Stark analysis are placed in the fume hood and allowed to evaporate.
2. Once solvent has evaporated determine the weight of the residue in the beaker and then calculate the % fat found in the sample
3. Compare this results with the results obtained by the Soxhlet method.
Last updated May, 2017