public assessment report decentralised procedure · par cefixime 100 mg/5 ml powder for oral...
TRANSCRIPT

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
1
Public Assessment Report
Decentralised Procedure
Cefixime 100 mg/5 mL Powder for Oral Suspension
UK/H/2828/001/DC
UK licence no: PL 04569/1118
Generics (UK) Limited

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
2
LAY SUMMARY On 6 Septemeber 2011, the Medicine and Healthcare products Regulatory Agency (MHRA) granted Generics (UK) Limited a Marketing Authorisation (licence) for the medicinal product Cefixime 100 mg/5 mL Powder for Oral Suspension (PL 04569/1118). This licence was granted via the decentralised procedure (UK/H/2828/001/DC). This is a prescription-only medicine. Cefixime 100 mg/5 mL Powder for Oral Suspension contains the active ingredient cefixime and belongs to a group of medicines called cephalsporins which are used for treating bacterial infections. Cefixime can be used to treat:
• Infection of the middle ear • Sinus infection • Throat infection • Infection causing sudden worsening of long standing bronchitis • Serious lung infections (such as pneumonia) acquired outside the hospital • Infections in the urinary tract • Kidney infection
No new or unexpected safety concerns arose from this application and it was therefore judged that the benefits of taking Cefixime 100 mg/5 mL Powder for Oral Suspension outweighs the risks and a Marketing Authorisation was granted.

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
3
TABLE OF CONTENTS
Module 1: Information about initial procedure Page 4 Module 2: Summary of Product Characteristics Page 5 Module 3: Product Information Leaflets Page 12 Module 4: Labelling Page 18 Module 5: Scientific Discussion Page 22 I Introduction Page 22 II About the product Page 24 III.1 Quality aspects Page 25 III.2 Non-clinical aspects Page 27 III.3 Clinical aspects Page 28 IV Overall Conclusion and Benefit:Risk Assessment Page 31 Module 6 Steps taken after initial procedure Page 32

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
4
Module 1
Product Name
Cefixime 100 mg/5 mL Powder for Oral Suspensin
Type of Application
Generic Application, Article 10(1)
Active Substance
Cefixime
Form
Powder for oral suspension
Strength
100mg/5ml
Marketing Authorisation Holder
Generics (UK) Limited Station Close Potters Bar, Hertsfordshire EN6 1TL UK
Reference Member State (RMS)
UK
Concerned Member State (CMS)
Italy
Procedure Number
UK/H/2828/001/DC
End of Procedure
3 August 2011

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
5
Module 2 SUMMARY OF PRODUCT CHARACTERISTICS
The UK Summary of Product Characteristics (SmPC) for Cefixime 100 mg/5 mL Powder for Oral Suspension (PL 04569/1118) is as follows: 1 NAME OF THE MEDICINAL PRODUCT
Cefixime 100 mg/5 ml Powder for Oral Suspension
2 QUALITATIVE AND QUANTITATIVE COMPOSITION Each 5 ml of reconstituted suspension contains 111.917 mg of cefixime trihydrate equivalent to 100 mg of cefixime (anhydrous). Excipient(s): Each 5 ml of reconstituted suspension contains 2.43 g of sucrose.
For a full list of excipients, see section 6.1.
3 PHARMACEUTICAL FORM Powder for oral suspension.
Off-white to pale yellow coloured powder with characteristic odour and gives a cream colour to pale yellow coloured viscous suspension after reconstitution with water.
4 CLINICAL PARTICULARS 4.1 Therapeutic indications
Cefixime is indicated for the treatment of the following infections when caused by susceptible organisms (see sections 4.4 and 5.1)
Acute exacerbations of chronic bronchitis Community-acquired Pneumonia Lower urinary tract infections Pyelonephritis
In the treatment of: Otitis media Sinusitis Pharyngitis
The use of Cefixime should be reserved for infections where the causative organism is known or suspected to be resistant to other commonly used antibiotics, or where treatment failure may carry significant risk.
Consideration should be given to official guidance on the appropriate use of antibacterial agents.
4.2 Posology and method of administration Adults and children over 10 years of age (body weight is greater than 50 kg) The recommended dose is 200-400 mg daily according to the severity of the infection, given either as a single dose or in two divided doses.
Elderly patients Elderly patients may be given the same dose as recommended for adults. Renal function should be assessed and dosage should be adjusted in severe impairment (See dosage for renal impairment and section 4.4).
Children younger than 10 years of age (body weight is lower than 50 kg) – Paediatric Oral Suspension The recommended dosage for children is 8 mg/kg/day administered as a single dose or in two divided doses. As a general guide for prescribing in children the following daily doses in terms of quantity and volume of paediatric oral suspension are suggested:

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
6
Weight (kg)/Age Daily dose (mg) Daily dose (ml) 6 months upto 1 year 75 mg 3.75 ml Children aged 1 – 4 years 100 mg 5 ml
Children aged 5 – 10 years 200 mg 10 ml Children weighing more than 50 kg or older than 10 years should be treated with the recommended adult dose (200 -400 mg daily), depending on the severity of the infection.
Children younger than 6 months of age The safety and efficacy of cefixime has not been established in children less than 6 months.
Renal impairment Cefixime may be administered in the presence of impaired renal function. Normal dose and schedule may be given in patients with creatinine clearance of 20 ml/ min or greater. In patients whose creatinine clearance is less than 20 ml/min, it is recommended that a dose of 200 mg once daily should not be exceeded. The dose and regimen for patients who are maintained on chronic ambulatory dialysis or haemodialysis should follow the same recommendation as that for patients with creatinine clearance of less than 20 ml/min.
Method of administration Cefixime powder for oral suspension is for oral administration only. The absorption of cefixime is not significantly affected by the presence of food. Hence it can be administered with or without food.
For instructions on reconstitution of the medicinal product before administration, see section 6.6.
Duration of treatment The usual course of treatment is 7 days. In severe cases, this can be extended to 14 days.
4.3 Contraindications
Patients with known hypersensitivity to cefixime, other cephalosporin antibiotics or to any of the excipients.
Cefixime is also contraindicated in patients with previous, immediate and/or severe hypersensitivity to penicillin or any beta-lactam antibiotics and preterm and term newborn infants (0-27 days).
4.4 Special warnings and precautions for use
Cefixime should be given with caution to patients who have shown hypersensitivity to other drugs. Cephalsporins should be given with caution to penicillin-sensitive patients, as there is some evidence of partial cross-allerginicity between penicillin and cephalsporins.
Patients have had severe reactions (including anaphylaxis) to both classes of drugs. Special care is indicated in patients who have experienced any allergic reaction to penicillins or any beta-lactam antibiotics as cross-reactions may occur (see section 4.3).
If severe hypersensitivity reactions or anaphylactic reactions occur after administration of Cefixime, the medicine should be discontinued immediately and appropriate emergency measures should be initiated.
Prolonged use of cefixime may result in the overgrowth of non-susceptible organisms.
Treatment with a broad spectrum of antibiotics alters the normal flora of the colon and may permit the overgrowth of Clostridia. Studies indicate that a toxin produced by Clostridium difficile is a primary cause for antibiotic-associated diarrhoea. Pseudomembranous colitis is associated with the use of broad-spectrum antibiotics (including macrolides, semi-synthetic penicillins, lincosamides and cephalsporins). It is therefore important to consider its diagnosis in patients who develop diarrhoea in association with the use of antibiotics.
In patients who develop severe diarrhoea during or after use of cefixime, the risk of life threatening pseudo-membranous colitis should be taken into account (see section 4.8). The use of cefixime should

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
7
be discontinued and appropriate treatment measures should be established. Management of pseudomembranous colitis should include sigmoidoscopy, appropriate bacteriologic studies, fluids, electrolyte and protein supplementation. If the colitis does not improve after the drug has been discontinued or if the symptoms are severe, oral vancomycin is the drug of choice for antibiotic-associated pseudomembreanous colitis produced by C. Difficile. Other causes of colitis should be excluded. The use of medicinal products inhibiting the intestinal peristalsis is contra-indicated.
Cefixime contains sucrose. Patients with rare hereditary problems of galactose intolerance, the Lapp lactase deficiency or glucose-galactose malabsorption should not take this medicine.
Use of Nifedipine, a calcium channel blocker, may increase bioavailability of Cefixime upto 70%.
Renal insufficiency Cefixime should be administered with caution in adult patients with creatinine clearance <20ml/min (see sections 4.2 and 5.2). There are insufficient data regarding use of cefixime in the pediatric and adolescent age group in the presence of renal insufficiency: the use of cefixime in these patient-groups is not recommended.
4.5 Interaction with other medicinal products and other forms of interaction
The administration of cephalsporins may interfere with the results of some laboratory tests.
A false positive reaction for glucose in the urine may occur with the Benedict’s or Fehling’s solutions or with copper sulphate test tablets, but not with tests based on enzymatic glucose oxidase reactions.
A false positive direct Coombs’test has been reported during treatment with cephalosporin antibiotics, therefore it should be recognised that a positive Coombs’ test may be due to the drug.
In common with other cephalsporins, increases in prothrombin times have been noted in a few patients. Care should therefore be taken in patients receiving anticoagulation therapy.
In use with Nifedipine, a calcium channel blocker, may increase bioavailability of Cefixime upto 70%.
4.6 Fertility, pregnancy and lactation Pregnancy For cefixime, no clinical data on exposed pregnancies are available. Animal studies do not indicate direct or indirect harmful effects with respect to pregnancy, embryonal/foetal development, parturition or postnatal development (see section 5.3).
Caution should be exercised when prescribing to pregnant women. Cefixime should not be used in pregnant mothers unless considered essential by the physician.
Breast-feeding It is unknown whether cefixime is excreted in human milk and non-clinical studies have shown excretion of cefixime in animalmilk. A decision on whether to continue/discontinue breast-feeding or to continue/discontinue therapy with cefixime should be made taking into account the benefit of breast-feeding to the child and the benefit of cefixime therapy to the woman. However, until further clinical experience is available, cefixime should not be prescribed to breast-feeding mothers.
Fertility Animal studies do not indicate any harmful effects with respect to fertility, however, no clinical data are available
4.7 Effects on ability to drive and use machines
Cefixime has no known influence on the ability to drive and use machines. However, side effects may occur (see section 4.8), which may influence the ability to drive and use machines.
4.8 Undesirable effects
Cefixime, like other cephalsporin antibiotics, may be associated with adverse events.
Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness.
The following undesirable effects have been divided in the following categories:

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
8
Very common: >1/10 Common: >1/100 to <1/10 Uncommon: >1/1,000 to <1/100 Rare: >1/10,000 to <1/1,000 Very rare: <1/10,000 Not known: cannot be estimated from the available data
MedDRA System Organ Class Adverse Reaction Frequency Superinfection bacterial, superinfection fungal
Rare
Infections and infestations Antibiotic-associated colitis (see section
Very rare
Eosinophilia Rare Blood and lymphatic system disorders
Leucopenia, agranulocytosis, pancytopenia, thrombocytopenia, haemolytic anaemia
Very rare
Hypersensitivity Rare Immune system disorders Anaphylactic shock, serum
sickness Very rare
Metabolism and nutrition disorders
Anorexia Rare
Headache Uncommon Vertigo, dizziness Rare Nervous system disorders Psychomotor hyperactivity Very rare Diarrhoea Common Abdominal pain, nausea, vomiting
Uncommon
Flatulence Rare Gastrointestinal disorders
Cases of pseudomembraneous colitis
Very rare
Hepatobiliary disorders Hepatitis, cholestatic jaundice Very rare Rash Uncommon Angioneurotic oedema, pruritus Rare Skin and subcutaneous
disorders Stevens-Johnson Syndrome, toxic epidermal necrolysis, Lyell syndrome
Very rare
Renal and urinary disorders Interstitial nephritis Very rare General disorders and administration site conditions
Mucosal inflammation, pyrexia Rare
Hepatic enzyme increased (transaminase, alkaline phosphatase)
Uncommon
Blood urea increased Rare Investigations
Blood creatinine increased Very rare
4.9 Overdose There is no experience with overdoses with Cefixime.
Adverse reactions seen at dose levels up to 2 g of cefixime in normal subjects did not differ from the profile seen in patients treated at the recommended doses. Gastric lavage may be indicated in overdosage. No specific antidote exists. Cefixime is not removed from the circulation in significant quantities by dialysis.
5 PHARMACOLOGICAL PROPERTIES 5.1 Pharmacodynamic properties
Pharmacotherapeutic group: antibacterial for systemic use, belonging to the class of cephalsporins, ATC code: J01DD08.
Mode of action Cefixime is an antibiotic belonging to the third generation cephalosporin group.

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
9
Like other cephalsporins, cefixime exerts antibacterial activity by binding to and inhibiting the action of penicillin-binding proteins involved in the synthesis of bacterial cell walls. This leads to bacterial cell lysis and cell death.
PK/PD Relationship The time that the plasma concentration of cefixime exceeds the MIC of the infecting organism has been shown to best correlate with efficacy in PK/PD studies.
Mechanism of resistance Bacterial resistance to cefixime may be due to one or more of the following mechanisms:
• Hydrolysis by extended-spectrum beta-lactamases and/or by chromosomally-encoded (AmpC)
enzymes that may be induced or de-repressed in certain aerobic gram- negative bacterial species • Reduced affinity of penicillin-binding proteins • Reduced permeability of the outer membrane of certain gram-negative organisms restricting access to
penicillin-binding proteins • Drug efflux pumps
More than one of these mechanisms of resistance may co-exist in a single bacterial cell. Depending on the mechanism(s) present, bacteria may express cross-resistance to several or all beta-lactams and/or antibacterial drugs of other classes.
Breakpoints Clinical minimum inhibitory concentration (MIC) breakpoints established by EUCAST (May 2009) for cefixime are: Breakpoints (MIC, mg/L)
Microorganism Susceptible (≤) Susceptible (≤) Resistant (>) Haemophilus influenzae 0.12 mg/L 0.12 mg/L Moraxella catarrhalis 0.5 mg/L 1.0 mg/L Neisseria gonorrhoeae 0.12 mg/L 0.12 mg/L Enterobacteriaceae 1.0 mg/L 1.0 mg/L Enterobacteriaceae: For uncomplicated urinary tract infections only. The breakpoints for Enterobacteriaceae will detect reduced susceptibility mediated by most clinically important beta-lactamases in Enterobacteriaceae. Occassional ESBL-producing strains will be reported susceptible. For purposes of infection control, epidemiology and surveillance, laboratories may wish to use specific tests to screen for and confirm ESBL-production. Non-species related breakpoints Insufficient data
Susceptibility The prevalence of resistance may vary geographically and over time for selected species and local information on resistance is desirable, particularly when treating severe infections. As necessary, expert advice should be sought when local prevalence if resistance is such that the utility of the agent in at least some types of infections is questionable.
Category 1: Commonly Susceptible organisms Aerobes, Gram-positive Aerobes, Gram-negative Streptococcus pneumoniae (penicillin-susceptible) Escherichia Coli% Streptococcus pyogenes Haemophilus influenzae Klebsiella species% Morexella catarrhalis Proteus mirabilis% Category 2: Organisms for which acquired resistance may be problematic Enterobacter species Category 3: Resistant organisms Clostridium difficile Bacteroides fragilis Enterococci

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
10
Pseudomonas species Staphylococcus aureus+ Streptococcus pneumoniae (Penicillin resistant) % Extended spectrum beta-lactamase (ESBL) producing isolates are always resistant + Cefixime has poor activity against staphylococci (regardless of susceptibility to methicillin)
5.2 Pharmacokinetic properties
Absorption The absolute bioavailability of cefixime is in the range of 22-54%. Absorption is not significantly modified by the presence of food. Cefixime may therefore be given without regard for meals.
Distribution Serum protein binding is well characterised for human and animal sera; cefixime is almost exclusively bound to the albumin fraction, the mean free fraction being approximately 30%. Protein binding of cefixime is only concentration dependent in human serum at very high concentrations which are not seen following clinical dosing.
From in vitro studies, serum or urine concentrations of 1 mg/L or greater were considered to be adequate for most common pathogens against which cefixime is active. Typically, the peak serum levels following the recommended adult or paediatric doses are between 1.5 and 3 mg/L. Little or no accumulation of cefixime occurs following multiple dosing.
Metabolism and elimination Cefixime is predominantly eliminated as unchanged drug in the urine. Glomerular filtration is considered the predominant mechanism. Metabolites of cefixime have not been isolated from human serum or urine.
Transfer of 14C-labelled cefixime from lactating rats to their nursing offspring through breast milk was quantitatively small (approximately 1.5% of the mothers' body content of cefixime in the pup). No data are available on secretion of cefixime in human breast milk. Placental transfer of cefixime was small in pregnant rats dosed with labelled cefixime.
Special age groups The pharmacokinetics of cefixime in healthy elderly (aged > 64 years) and young volunteers (11-35) compared the administration of 400 mg doses once daily for 5 days. Mean Cmax and AUC values were slightly greater in the elderly. Elderly patients may be given the same dose as the general population (see section 4.2).
5.3 Preclinical safety data
There are no findings from chronic toxicity investigations suggesting that any side effects unknown to date could occur in humans. Furthermore, in vivo and in vitro studies did not yield any indication of a potential to cause mutagenicity. Long-term studies on carcinogenicity have not been conducted. Reproduction studies have been performed in mice and rats at does up to 400 times the human dose and have revealed no evidence of impaired fertility or harm to the foetus due to cefixime. In the rabbit, at doses up to 4 times the human dose, there was no evidence of a teratogenic effect; there was a high incidence of abortion and maternal death, which is an expected consequence of the known hypersensitivity of rabbits to antibiotic-induced changes in the population of the microflora of the intestine.
6 PHARMACEUTICAL PARTICULARS 6.1 List of excipients
Xanthum gum Sodium benzoate Silica colloidal, anhydrous Sucrose Flavour strawberry (maltodextrin, triethyl acetate –E1505, propylene glycol – E1520)
6.2 Incompatibilities Not applicable.

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
11
6.3 Shelf life
Unopened: 2 years. Do not store above 25oC. After reconstitution: The reconstituted suspension may be stored for 14 days below 25oC.
6.4 Special precautions for storage Unopened: This medicinal product does not require any special storage conditions.
For storage conditions of the reconstituted medicinal product, see section 6.3.
After reconstitution, the suspension can be stored below 25º C for 14 days without significant loss of potency. Do not freeze. Keep bottles tightly closed and shake well before use. Discard any unused portion after 14 days. Dilution of the suspension is not recommended.
6.5 Nature and contents of container Nature of container: Type III molded, amber coloured, round glass bottle with 100ml ring mark and with a plastic child resistant cap with induction seal liner.
Contents of container: Bottle of size 100 ml.
Bottles are supplied with a single 5 ml plastic dosing pipette (plunger, barrel and piston).
6.6 Special precautions for disposal Instructions for the preparation of the oral suspension:
Cefixime 100 mg/5 ml Powder for Oral Suspension Bottle size Directions for Reconstitution
100 mg/ 5 ml 100 ml Add 68 ml of water in two portions to the dry mixture in the bottle. Shake well after each addition.
Any unused product or waste material should be disposed of in accordance with local requirements.
7 MARKETING AUTHORISATION HOLDER Generics [UK] Ltd t/a Mylan Station Close, Potters Bar, EN6 1AG United Kingdom
8 MARKETING AUTHORISATION NUMBER(S)
PL 04569/0118 9 DATE OF FIRST AUTHORISATION/RENEWAL OF THE AUTHORISATION 06/09/2011 10 DATE OF REVISION OF THE TEXT
06/09/2011

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
12
Module 3 Product Information Leaflet
The text version of the PIL was provided and approved as part of this application. In accordance with medicines legislation, this product shall not be marketed in the UK until the PIL mock-up has been submitted to and approved by the competent authority.

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
13

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
14

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
15

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
16

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
17
Module 4 Labelling
The text version of the labelling has been provided and approved as part of this application. No label mock-ups have been provided. In accordance with medicines legislation, this product shall not be marketed in the UK until approval of the label mock-ups has been obtained. Carton

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
18

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
19

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
20

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
21
Module 5 Scientific discussion during initial procedure
I INTRODUCTION On 3 August 2011, Italy and the UK agreed to grant a Marketing Authorisation (MA) to Generics (UK) Limited for the medicinal product Cefixime 100 mg/5 mL Powder for Oral Suspension. The MA was granted via a Decentralised Procedure (DCP), with the UK as Reference Member State (UK/H/2828/001/DC). After the national phase, a licence was granted in the UK on 6 September 2011 (PL 04569/1118). This is a generic application for Cefixime 100 mg/5 mL Powder for Oral Suspension, submitted under Article 10.1 of Directive 2001/83/EC, as amended. The application refers to Suprax Powder Paediatric 100 mg/5 mL Powder for Oral Suspension, first authorised to Sanofi-Aventis Ireland Ltd in Ireland on 5 January 1989. The period of exclusivity has expired. Cefixime is an oral third-generation cephalosporin. It exerts its bactericidal effect by attaching to penicillin-binding proteins and inhibiting peptidoglycan synthesis, thus causing damage to the bacterial cell wall. Cefixime has marked in-vitro bactericidal activity against a wide variety of gram-positive and gram-negative organisms. It exhibits a broad spectrum of antibacterial activity with minimum inhibitory concentrations similar to or less than those for other oral cephalosporins against many Gram-negative and Gram-positive microorganisms. It is approved for the treatment of upper and lower respiratory tract infections and urinary tract infections. No new non-clinical or clinical efficacy studies were conducted for this application, which is acceptable given that the application was for a generic version of a product that have been licensed for over 10 years. The application is supported by a bioequivalence study comparing the pharmacokinetic profile of the test product, Cefixime 100 mg/5 mL Powder for Oral Suspension, to that of the reference product, Suprax powder for Paediatric Oral Suspension (Sanofi-Aventis). The bioequivalence study was carried out in accordance with Good Clinical Prcatice (GCP). The RMS has been assured that acceptable standards of Good Manufacturing Practice (GMP) are in place for these product types at all sites responsible for the manufacture and assembly of these products. Evidence of compliance with GMP has been provided for the named manufacturing and assembly sites. For manufacturing sites within the Community, the RMS has accepted copies of current manufacturer authorisations issued by inspection services of the competent authorities as certification that acceptable standards of GMP are in place at those sites. For manufacturing sites outside the community, the RMS has accepted copies of current GMP certificates or satisfactory inspection summary reports, ‘close-out letters’ or ‘exchange of information’ issued by the inspection services of the competent authorities (or those countries with which the EEA has a Mutual Recognition Agreement for their own territories) as certification that acceptable standards of GMP are in place at those non-Community sites. The RMS considers that the pharmacovigilance system, as described by the MAH, fulfils the requirements and provides adequate evidence that the MAH has the services of a qualified

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
22
person responsible for pharmacovigilance and has the necessary means for the notification of any adverse reaction suspected of occurring either in the Community or in a third country. The Marketing Authorisation Holder has provided adequate justification for not submitting a Risk Management Plan (RMP). As the application is for a generic version of an already authorised reference product, for which safety concerns requiring additional risk minimisation have not been identified, a risk minimisation system is not considered necessary. The reference product has been in use for many years and the safety profile of the active substances is well established. The Marketing Authorisation Holder has provided adequate justification for not submitting an Environmental Risk Assessment (ERA). This was an application for a generic product and there is no reason to conclude that the marketing of this product will change the overall use pattern of the existing market.

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
23
II. ABOUT THE PRODUCT
Name of the product in the Reference Member State
Cefixime 100mg/5ml Powder for oral suspension
Name(s) of the active substance(s) (INN) Cefixime Pharmacotherapeutic classification (ATC code)
J01DD08 (Third generation Cephalosporin)
Pharmaceutical form and strength(s) Powder for oral suspension 100mg/5ml
Reference numbers for the Decentralised Procedure
UK/H/2828/001/DC
Reference Member State United Kingdom Member States concerned Italy Marketing Authorisation Number(s) PL 04569/1118 Name and address of the authorisation holder
Generics (UK) Limited Station Close Potters Bar, Hertsfordshire EN6 1TL UK

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
24
III SCIENTIFIC OVERVIEW AND DISCUSSION III.1 QUALITY ASPECTS
DRUG SUBSTANCE Cefixime INN: Cefixime Chemical name: (6R, 7R0-7-[[(2Z)-(2-amino-4-thioazolyl)[(carboxymethoxy)imino]acetyl]amino]-3-ethenyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboylic acid.
Structure:
Molecular formula: C16H15N5O7S2.3H20 Molecular weight: 507.5 General Properties Description: A white or almost white powder, slightly hygroscopic. Solubility: Soluble in methanol, slightly soluble in water and ethanol and practically insoluble in ethyl acetate. Cefixime is the subject of a European Pharmacopoeia (Eur Ph.) monograph. Manufacture All aspects of the manufacture and control of the active substance cefixime are covered by a European Directorate for the Quality of Medicines (EDQM) Certificate of Suitability.
DRUG PRODUCT Description and Composition Cefixime 100 mg/5 mL Powder for Oral Suspension is presented as off-white to pale yellow coloured powder with characteristic odour and gives a cream colour to pale yellow coloured viscous suspension after reconstitution with water. Each 5 mL of reconstituted suspension contains 111.917 mg of cefixime trihydrate equivalent to 100 mg of the active ingredient, cefixime (anhydrous). Other ingredients consist of the pharmaceutical excipients, xanthum gum, sodium benzoate, silica colloidal, anhydrous, sucrose and flavour strawberry (maltodextrin, triethyl acetate-E1505, propylene glycol-E1520). All the ingredients in the tablets comply with their relevant Ph.Eur monographs with the exception of the strawberry flavour which complies with an in-house specification which is satisfactory.

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
25
Appropriate justification for the inclusion of each of the excipients has been provided. Satisfactory Certificates of Analysis for each of the excipients have been presented. The applicant has provided a declaration confirming that there are no materials of human or animal origin contained in the product, or used in the manufacturing process. Furthermore, no genetically modified organisms are used in the manufacture of the excipients. Pharmaceutical Development Details of the pharmaceutical development of the medicinal product have been supplied and are satisfactory. The aim was to develop a stable, robust, generic dosage form of Cefixime 100 mg/5 mL Powder for Oral Suspension, which is pharmaceutically equivalent to the reference product Suprax 100 mg/5 mL Powder for Oral Suspension (Sanofi-Aventis). Comparative dissolution data were provided for bio-batches of the test and reference products. The dissolution profiles were satisfactory. Manufacture A description and flow-chart of the manufacturing method has been provided. In-process controls are appropriate considering the nature of the product and the method of manufacture. Process validation studies have been conducted on two pilot-scale batches and the results were satisfactory. The validation data demonstrated consistency of the manufacturing process. A commitment has been made by the MAH that full process validation will be conducted on commercial scale batches in accordance with the process validation protocol.
Finished Product Specification Finished product specifications are provided for both release and shelf–life, and are satisfactory. These provide an assurance of the quality and consistency of the finished product. Acceptance limits have been justified with respect to conventional pharmaceutical requirements and, where appropriate, safety. Test methods have been described and adequately validated, as appropriate. Batch data are provided, which comply with the release specifications. Certificates of Analysis have been provided for all working standards used. Container Closure System The finished product is licensed for marketing in Type III moulded, amber coloured, round glass bottles with 100 mL ring mark and with a plastic child resistant cap with induction seal liner. Each bottle is supplied with a single 5 mL plastic dosing pipette (plunger, barrel and piston) and packed with the Patient Information Leaflet (PIL) into a cardboard outer carton. Satisfactory specifications and Certificates of Analysis for all packaging components used have been provided. . All primary product packaging complies with EU legislation, Directive 2002/72/EC (as amended), and is suitable for contact with foodstuffs. The glass components used have been provided. The glass bottles comply with Ph Eur requirements and are suitable for contact with oral solution products; the caps comply with child resistant packaging legislation. Stability Finished product stability studies have been conducted in accordance with current guidelines and results were within the proposed specification limits. Based on the results, a shelf-life of 2 years has been set, when the bottle is unopened, which is satisfactory. The storage conditions are ‘Do not store above 25oC’.

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
26
An ‘in-use’ product stability study was carried out on Cefixime 100 mg/5 mL Powder for Oral Suspension to establish the time period over which the product could be used after the bottle has been opened. Please refer to Section 6.3 and 6.4 for shelf-life and storage conditions of the product after reconstitution. Quality Overall Summary A satisfactory quality overview is provided and has been prepared by an appropriately qualified expert. The curriculum vitae of the expert has been provided. Summary of Product Characteristics (SmPC), Patient Information Leaflet (PIL), Labels The SmPC, PIL and labelling are acceptable from a pharmaceutical perspective. The labelling is satisfactory and fulfils the statutory requirements for Braille. Text versions of the PILs and labels have been provided and are satisfactory. In accordance with medicines legislation, the Marketing Authorisation Holder (MAH) has provided a commitment that the product shall not be marketed in the UK until approval of the PIL and label mock-ups has been obtained. The applicant has submitted results of PIL user testing. The results indicate that the PIL is well-structured and organised, easy to understand and written in a comprehensive manner. The test shows that the patients/users are able to act upon the information that it contains. MAA Form The MAA forms are satisfactory from a pharmaceutical perspective. Conclusion The test product has been demonstrated to be equivalent pharmaceutically to the reference product, which has been licensed in the UK for over 10 years. There are no objections to the approval of Cefixime 100 mg/5 mL Powder for Oral Solution from a pharmaceutical point of view. III.2 NON-CLINICAL ASPECTS Pharmacodynamic, pharmacokinetic and toxicological properties of cefixime are well known. As this is a widely used, well-known active substance, the applicant has not provided additional studies and further studies are not required. Overview based on literature review is, thus, appropriate. Environmental risk assessment No formal Environmental Risk Assessment has been provided. The applicant has justified the absence adequately. As a generic product, the use of this product is not expected to increase the overall use of cefixime and so no additional increase in environmental risk has been identified. Non-Clinical Overview The non-clinical overview was written by a suitably qualified person and is satisfactory. The curriculum vitae of the expert has been provided.

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
27
Summary of Product Characteristics (SmPC) Section 4.6 and 5.3 are satisfactory from a non-clinical viewpoint. There are no objections to approval of Cefixime 100 mg/5 mL Powder for Oral Suspensiont from a non-clinical point of view. III.3 CLINICAL ASPECTS Indications Cefixime 100 mg/5 mL Powder for Oral Suspension is indicated in the treatment of:
• infection of the middle ear • sinus infection • throat infection • infection causing sudden worsening of long-standing bronchitis • serious lung infections (such as pneumonia) acquired outside hospital • urinary tract infections • kidney infection
The indications are in-line with thos for the reference product and are satisfactory. Posology and Method of Administration The recommended dose in adults and children over 10 years of age (body weight greater than 50kg) is 200-400 mg daily according to the severity of the infection, given either as a single dose or in two divided doses. For elderly patients and children younger than 10 years the dose may vary and details these patients can be found in the SmPC. Full details concerning the posology are provided in the SmPC. The posology is consistent with that for the reference product and is satisfactory. Pharmacokinetics- bioequivalence study The application is supported by the bioequivalence study presented by the applicant comparing the pharmacokinetic profile of the test product, Cefixime 100 mg/5 mL Powder for Oral Suspension, to that of the reference product, Suprax Powder for Paediatric Oral Suspension (Sanofi-Aventis). The tudy was of an appropriate design and was conducted to principles of Good Clinical Practice (GCP). Certificates of Analysis were provided for both the test and reference products. The UK reference product, Suprax Powder for Paediatric Oral Suspension (Sanofi-Aventis) is considered to be equivalent to the originator and clinical reference product Suprax Powder for Paediatric 100mg/5 mL (Sanofi-Aventis Ireland Ltd). This was a randomised open label, two treatment, two period, two sequence, single dose crossover bioequivalence study conducted in adult male subjects under fasting conditions. A single dose of the investigational products was administered orally to each subject in each period. A satisfactory washout period of 7 days was maintained between the dosing days in each group. Blood samples were taken pre-dose (0.0) and at specified time points up to 24 hours after administration of test or reference product. Plasma levels of cefixime were quanitifed by a validated LC-MS/MS method.
PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
28
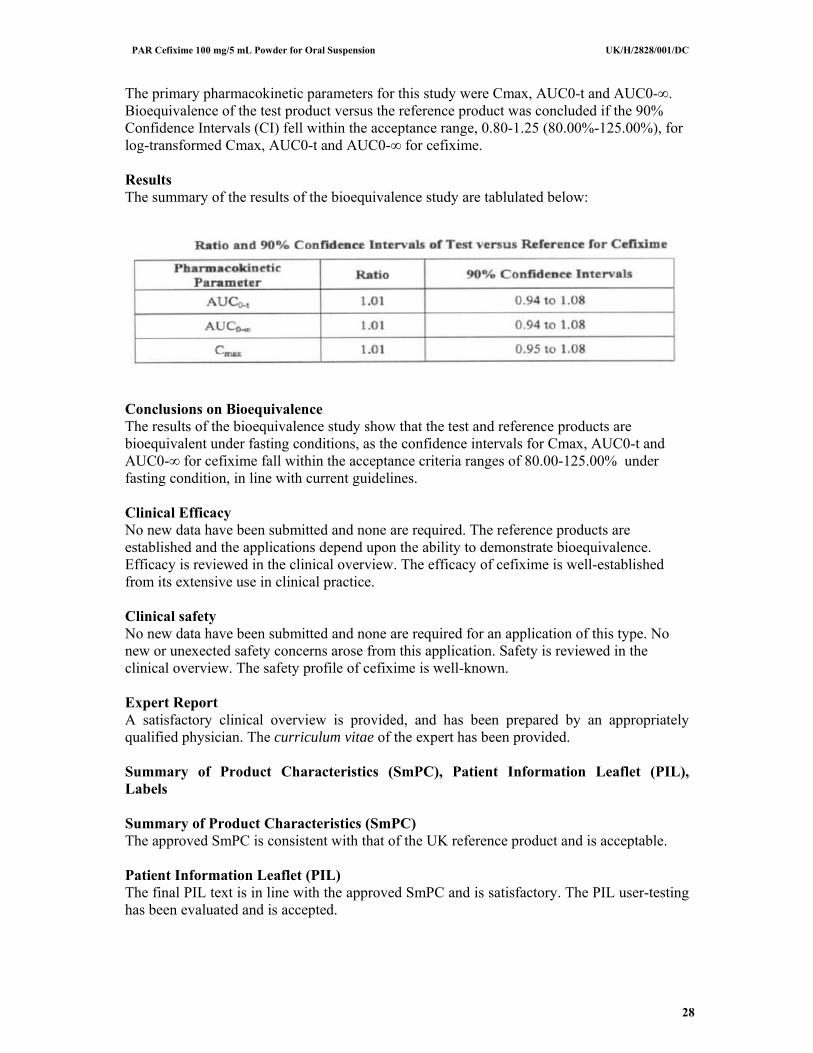
The primary pharmacokinetic parameters for this study were Cmax, AUC0-t and AUC0-∞. Bioequivalence of the test product versus the reference product was concluded if the 90% Confidence Intervals (CI) fell within the acceptance range, 0.80-1.25 (80.00%-125.00%), for log-transformed Cmax, AUC0-t and AUC0-∞ for cefixime. Results The summary of the results of the bioequivalence study are tablulated below:
Conclusions on Bioequivalence The results of the bioequivalence study show that the test and reference products are bioequivalent under fasting conditions, as the confidence intervals for Cmax, AUC0-t and AUC0-∞ for cefixime fall within the acceptance criteria ranges of 80.00-125.00% under fasting condition, in line with current guidelines. Clinical Efficacy No new data have been submitted and none are required. The reference products are established and the applications depend upon the ability to demonstrate bioequivalence. Efficacy is reviewed in the clinical overview. The efficacy of cefixime is well-established from its extensive use in clinical practice. Clinical safety No new data have been submitted and none are required for an application of this type. No new or unexected safety concerns arose from this application. Safety is reviewed in the clinical overview. The safety profile of cefixime is well-known. Expert Report A satisfactory clinical overview is provided, and has been prepared by an appropriately qualified physician. The curriculum vitae of the expert has been provided. Summary of Product Characteristics (SmPC), Patient Information Leaflet (PIL), Labels Summary of Product Characteristics (SmPC) The approved SmPC is consistent with that of the UK reference product and is acceptable. Patient Information Leaflet (PIL) The final PIL text is in line with the approved SmPC and is satisfactory. The PIL user-testing has been evaluated and is accepted.

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
29
Labelling The labelling text is satisfactory. Clinical overview A satisfactory clinical overview is provided and has been prepared by an appropriately qualified expert. The CV of the clinical expert has been supplied. MAA form The MAA form is satisfactory. Conclusion There are no objections to the approval of Cefixime 100 mg/5 mL powder for Oral Suspension from a clinical point of view.

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
30
IV OVERALL CONCLUSION AND BENEFIT/RISK ASSESSMENT QUALITY The important quality characteristics of Cefixime 100 mg/5 mL Powder for Oral Suspension are well-defined and controlled. The specifications and batch analytical results indicate consistency from batch to batch. There are no outstanding quality issues that would have a negative impact on the benefit/risk balance. NON-CLINICAL No new non-clinical data were submitted and none are required for an application of this type. EFFICACY Bioequivalence has been demonstrated between the applicant’s Cefixime 100 mg/5 mL Powder for Oral Suspension and the reference product Suprax Powder Paediatric 100 mg/5 mL Powder for Oral Suspension (Sanofi-Aventis Ireland Limited). The UK reference product, Suprax Powder Paediatric 100 mg/5 mL Powder for Oral Suspension, is considered to be equivalent tot eh originator and clionical reference product, Suprax Powder Paediatric 100 mg/5 mL Powder for Oral Suspension (Sanofi-Aventis- PL 00012/0318). No new or unexpected safety concerns arose from this application. PRODUCT LITERATURE
The SmPC and PIL are acceptable, and consistent with those for the reference product. The labelling is acceptable and in-line with current requirements. A package leaflet has been evaluated via a user consultation study in accordance with the requirements of Articles 59(3) and 61(1) of Directive 2001/83/EC. The results show that the package leaflet meets the criteria for readability as set out in the Guideline on the readability of the label and package leaflet of medicinal products for human use. The approved labeling text is satiactory and fulfils the stautory requirements for Braille. The MAH has submitted text versions only for the PIL and labeling and has committed to submitteing mock-up livery to the reevant regulatory authorities for approval before packs are marketed. BENEFIT/RISK ASSESSMENT The quality of the product is acceptable, and no new non-clinical or clinical safety concerns have been identified. The bioequivalence study and its conclusions support the claim that the applicant’s Cefixime 100 mg/5 mL Powder for Oral Suspension is a generic version of the reference product uprax Powder Paediatric 100 mg/5 mL Powder for Oral Suspension (Sanofi-Aventis Ireland Limited). Extensive clinical experience with cefixime is considered to have demonstrated the therapeutic value of the active substances. The benefit/risk ratio is therefore considered to be positive.

PAR Cefixime 100 mg/5 mL Powder for Oral Suspension UK/H/2828/001/DC
31
Module 6
STEPS TAKEN AFTER INITIAL PROCEDURE - SUMMARY
Date submitted
Application type
Scope Outcome