qcqa 3rd sessional lk
TRANSCRIPT

Drug Master File (DMF)-USA
Facilitator:
Dr.N.Vishal Kumar Guptha
Asst Professor,
Dept of Pharmaceutics,
J.S.S.C.P, Mysuru.
Submitted by:LakshmiKanth Reddy.P
1st M.PharmaDept of Pharmaceutical analysis
J.S.S.C.P, Mysuru
4/26/2015 1

What is drug master file ?
• Drug Master File (DMF) is a document prepared by a pharmaceutical manufacturer and submitted to the appropriate regulatory authority in the intended drug market.
• However, the document provides the regulatory authority with confidential, detailed information about facilities, processes, or articles used in the manufacturing, processing, packaging, and storing of one or more human drugs.
• Typically, a DMF is filed when two or more firms work in partnership on developing or manufacturing a drug product.4/26/2015 2

• The DMF filing allows a firm to protect its intellectual property from its partner while complying with regulatory requirements for disclosure of processing details.
• API manufacturers with a large number of DMFs are often considered more reliable in terms of quality, regulatory standing, and ability to meet Current Good Manufacturing Process (cGMP) requirements.
4/26/2015 3

REMEMBER
• There is no legal or regulatory requirement to file a DMF.
The information contained in DMF may be used to support an
- Investigational New Drug Application (IND),
- New Drug Application (NDA),
- Abbreviated New Drug Application (ANDA)
- An Export Application
• DMF is NOT a substitute for IND / NDA / ANDA or export application
4/26/2015 4

What does the DMF contains?
• Drug Master File (DMF) is a document
containing complete information on an
Active Pharmaceutical Ingredient (API) or
finished drug dosage form.
4/26/2015 5

• The DMF contains factual and complete information on a drug product's chemistry, manufacture, stability, purity, impurity profile, packaging, and the cGMP status of any human drug product.
DMF CONTAINS
Chemistry
Manufacture
Stability cGMP
Packaging
ImpurityProfile
Purity
4/26/2015 6

DRUG MASTER FILE - USA
• In the United States, DMFs are submitted to
the Food and Drug Administration (FDA).
• The Main Objective of the DMF is to support
regulatory requirements and to prove the
quality, safety and efficacy of the medicinal
product for obtaining an Investigational New
Drug Application (IND), New Drug Application
(NDA), An Abbreviated New Drug Application
(ANDA), another DMF, Or an Export
Application.
4/26/2015 7

Types of DRUG MASTER FILE:
Type I - Manufacturing Site, Facilities, Operating
Procedures, and Personnel
Type II -Drug Substance, Drug Substance Intermediate, and
Material Used in Their Preparation, or Drug
Product
Type III - Packaging Material
Type IV -Excipient, Colorant, Flavor, Essence, or Material Used in
Their Preparation
Type V- FDA Accepted Reference Information4/26/2015 8

Type I: Manufacturing Site, Facilities,
Operating Procedures, and Personnel
Type I drug master files are no longer used. Once they were used to describe facilities.
It is recommended for a person outside of the United States to assist FDA in conducting on site inspections of their manufacturing facilities.
The DMF should describe the-
Manufacturing site -operational lay out, area, map equipment capabilities - model, make, capacity operational layout - QA, QC, R&D
A diagram of major production and processing areas is helpful for understanding the operational layout.
4/26/2015 9

Type II: Drug Substance, Drug Substance
Intermediate, and Material Used in Their
Preparation, or Drug Product• Type II drug master files are required for drug
substance, also referred to as active ingredients.
• A separate DMF is filed for each active ingredient.
• The DMF should include the brief description of the manufacturing facilities, the address, a contact, phone number and fax number.
• The manufacturing facility must be registered and the registration number should be listed
• Summarize all significant steps in the manufacturing and controls of the drug intermediate or substance including the critical steps and in process tests performed at those steps.
4/26/2015 10

•The testing of all raw materials should be provided
along with the in process controls, packaging, release,
and stability testing.
•The impurity profile, particle size distribution, organic
volatile impurities, and the residual solvent test
results have become increasingly important.
•These tests should reference the current standards of
the International Conference on Harmonization and
The United States Pharmacopoeia, if not then it should
be validated
4/26/2015 11

TYPE III: Packaging Material
Contents:-
• Its components and composition.
• Packaging material intended for which use.
• Names of the suppliers or fabricators of the
components used in preparing the packaging
material.
• Acceptance specifications.
• Toxicological data on these materials.
4/26/2015 12

TYPE IV: Excipient, Colorant, Flavor, Essence, or
Material Used in Their Preparation
• Each additive should be identified and
characterized by its method of
manufacture, release specifications, and
testing methods.
• Toxicological data on these materials
would be included under this type of
DMF, if not otherwise available by cross
reference to another document.
4/26/2015 13

TYPE V: FDA Accepted Reference
Information
• FDA discourages the use of Type V DMFs
for miscellaneous information, duplicate
information, or information that should be
included in one of the other types of
DMFs.
4/26/2015 14
4/26/2015 15
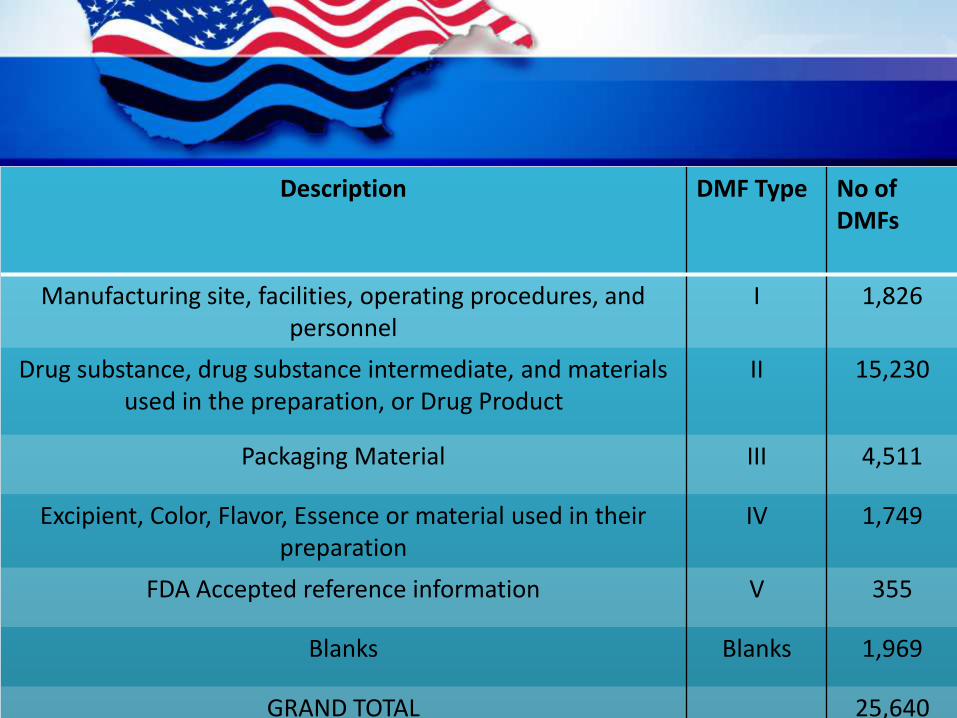
Description DMF Type No of DMFs
Manufacturing site, facilities, operating procedures, and personnel
I 1,826
Drug substance, drug substance intermediate, and materialsused in the preparation, or Drug Product
II 15,230
Packaging Material III 4,511
Excipient, Color, Flavor, Essence or material used in theirpreparation
IV 1,749
FDA Accepted reference information V 355
Blanks Blanks 1,969
GRAND TOTAL 25,640
4/26/2015 16
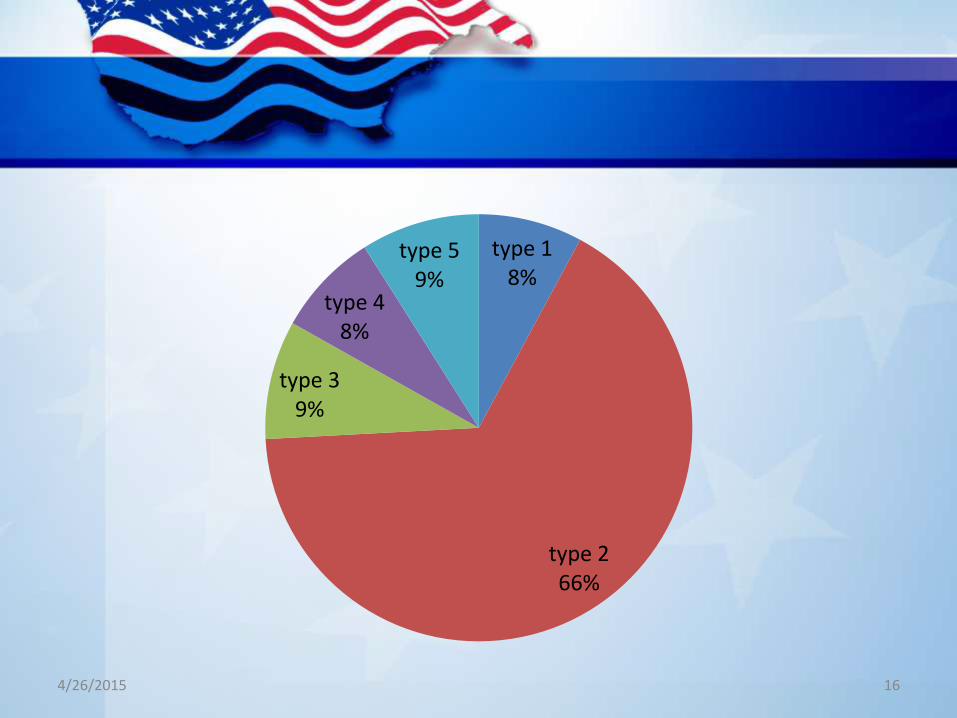
type 18%
type 266%
type 39%
type 48%
type 59%

SUBMISSIONS REQUIRMENTS TO
DRUG MASTER FILES
• Each DMF submission should contain a transmittal letter, administrative information about the submission, and the specific information to be included in the DMF.
• The DMF must be in the English language. Whenever a submission contains information in another language, an accurate certified English translation must also be included.
• Each page of each copy of the DMF should be dated and consecutively numbered. An updated table of contents should be included with each submission.
4/26/2015 17

A. Transmittal Letters
• Original Submissions
• Amendments
B. Administrative Information
• Original Submissions
• Amendments
Cont…!!!
4/26/2015 18

A. Transmittal Letters (The following should be included)
• Original Submissions
a. Identification of submission: Original, the type of DMF and its subject.
b. Identification of the applications, if known, that the DMF is intended to support, including the name and address of each sponsor, applicant, or holder, and all relevant document numbers.
c. Signature of the holder or the authorized representative.
d. Typewritten name and title of the signer.
4/26/2015 19

• Amendments
a. Identification of submission: Amendment, the DMF number, type of DMF, and the subject of the amendment.
b. A description of the purpose of submission, e.g., update, revised formula, or revised process.
c. Signature of the holder or the authorized representative.
d. Typewritten name and title of the signer.
Transmittal Letters (The following should be included)
Cont…
4/26/2015 20

B. Administrative Information (The following should be included)
• Original Submissions
A. Names and addresses of the following:
(1) DMF holder.
(2) Corporate headquarters.
(3) Manufacturing/processing facility.
(4) Contact for FDA correspondence.
(5) Agent(s), if any.
B. The specific responsibilities of each person listed in any of the categories in Section a.
C. Statement of commitment.
A signed statement by the holder certifying that the DMF is current and that the DMF holder will comply with the statements made in it.
4/26/2015 21

Administrative Information (The following should be included)
• Amendments
a. Name of DMF holder.
b. DMF number.
c. Name and address for correspondence.
d. Affected section and/or page numbers of the DMF.
e. The name and address of each person whose IND, NDA, ANDA, DMF, or Export Application relies on the subject of the amendment for support.
f. The number of each IND, NDA, ANDA, DMF, and Export Application that relies on the subject of the amendment for support, if known.
g. Particular items within the IND, NDA, ANDA, DMF, and Export Application that are affected, if known.
Cont..!!
4/26/2015 22

Steps for Filing A DMF:
1. Set the document margins at 3/4 inch for the left (at least) and 1/2 inch for the right.
2. Print the transmittal page, administrative information and DMF information on standard letter-size paper. If a larger sheet of paper is required for a diagram or schematic, fold the sheet and attach it to a letter-sized page in a manner that will allow for the page to be opened and refolded. At a maximum, each volume of a DMF should be no more than 2 inches thick.
3. Number multiple volumes for one submission according to the total number of volumes (if more than one). (For example, 1 of 3, 2 of 3, etc.)
4. Sign all documents requiring signature (only if you are the DMF holder or authorized representative).
4/26/2015 23

Cont…
5. Copy and collate the document; FDA requires you submit both.
6. Punch documents with a standard hole-punch.
7. Cover each original and copy of each volume with a document jacket. Prepare the submission for shipping and mail to:
Drug Master File Staff
Food and Drug Administration
5901-B Ammendale Rd.
Beltsville, MD 20705-1266
4/26/2015 24

In General:
To submit the data
First select type of DMF I.e. type I to IV DMF
A holder must first submit A LETTER OF
INTENT to the drug master file staff
FDA then will contact the holder to discuss
the proposed subbmission.4/26/2015 25

Transfer of Ownership
To transfer ownership of a DMF to another party, the holder should so notify FDA and authorized persons in writing. The letter should include the following:
– Name of transferee
– Address of transferee
– Name of responsible official of transferee
– Effective date of transfer
– Signature of the transferring official
– Typewritten name and title of the transferring official.
The new holder should submit a letter of acceptance of the transfer and an update of the information contained in the DMF, where appropriate. Any change relating to the new ownership (e.g., plant location and methods) should be included.
4/26/2015 26

Closure of a drug master file
• A holder who wishes to close a DMF should submit a request to the Drug Master File Staff stating the reason for the closure.
• The request should include a statement of holder's obligations.
• The Agency may close a DMF that does not contain an annual update of persons authorized to incorporate information in the DMF by reference and a list of changes made since the previous annual report. The holder will be notified of FDA's intent to close the DMF.
4/26/2015 27

Reference:
1. Guideline for Drug Master Files, Centre for Drug Evaluation and Research, Food and Drug Administration, Department of Health and Human Services, Office of Drug Evaluation I (HFD-100), 5600 Fishers Lane, Rockville, Maryland 20857, September 1989.
2. Guidelines for drug master files, United States, Food and Drug Administration, Retrieved 2009-09-19. DMF search engine. FDA: Guideline for drug master file
4/26/2015 28

THANK YOU…!!!!4/26/2015 29

http://www.registrarcorp.com/fda-drugs/master-files/registration.jsp?lang=en
4/26/2015 30