radiographic and morphologic findings in a previously undescribed type of mesomelic dysplasia...
TRANSCRIPT

Radiographic and Morphologic Findings in aPreviously Undescribed Type of MesomelicDysplasia Resembling Atelosteogenesis Type II
Steven G. Brodie,1 Ralph S. Lachman,1,3 Barbara F. Crandall,2 Michelle A. Fox,2David L. Rimoin,1,2 Daniel H. Cohn,1,2 and William R. Wilcox1,2*1Ahmanson Department of Pediatrics, Steven Spielberg Pediatric Research Center, Cedars-Sinai Burns and AllenResearch Institute, UCLA School of Medicine, Los Angeles, California
2Department of Pediatrics, UCLA School of Medicine, Los Angeles, California3Department of Radiology, UCLA School of Medicine, Los Angeles, California
The mesomelic chondrodysplasias are a het-erogeneous group of dwarfing disorderscharacterized by shortness of the middlesegments of limbs. We report on a 25-weekfetus with disproportionate shortness oflimbs with an apparently distinct form ofmesomelic dysplasia. Radiographic findingsat necropsy included ulnar deviation ofhands, talipes equinovarus, distal taperingof the humeri, and hypoplastic fibulae, ra-dii, and ulnae. Chondro-osseous morphol-ogy showed mild shortness of the physealcolumns, overgrowth of perichondral bone,peripheral ingrowth of mesenchymal cellsinto the physis, and numerous areas of fi-brillar degeneration with rings of collagensurrounding the chondrocytes. Ultrastruc-tural findings included a degenerated terri-torial matrix, pericellular halos of collagen,and dilated loops of rough endoplasmic re-ticulum in chondrocytes. The radiographicappearance of the long bones is distinctfrom that of previously described meso-melic dysplasias. The chondro-osseous mor-phologic findings and the distal tapering ofthe humerus are somewhat reminiscent ofatelosteogenesis type II, but the pattern ofmatrix degeneration and the presence of in-clusion bodies in the chondrocytes distin-guish it from disorders of sulfate transport.Am. J. Med. Genet. 80:247–251, 1998.© 1998 Wiley-Liss, Inc.
KEY WORDS: lethal skeletal dysplasia; at-elosteogenesis; mesomelicdysplasia; dilated rough en-doplasmic reticulum; inclu-sion bodies
INTRODUCTION
The mesomelic dysplasias are a heterogeneous groupof disorders with disproportionate shortness in the ra-dioulnar and/or tibiofibular segments of the limbs,which range in severity from perinatal lethal to mildshort stature. Over 30 distinct entities have been de-scribed and are classified primarily by the radiographicappearance of the fibulae, radii, tibiae, and ulnae[Kaitila et al., 1976; Taybi and Lachman; 1996]. Addi-tional clinical and radiographic findings such as in-volvement of the proximal or distal limb segments, ver-tebral or craniofacial anomalies, or other skeletal mal-formations are used to distinguish further one disorderfrom another. The classification of the mesomelic dys-plasias is complicated further by disorders with meso-melic limb shortness as a secondary characteristic,such as the mesomelia found in the atelosteogenesissyndromes (AO) [Lohr and Wiedemann, 1981; Marote-aux et al., 1982; Sillence et al., 1982; Sillence et al.,1987; Stern et al., 1990; Taybi and Lachman; 1996]. Wepresent the clinical, radiographic, histologic, and ultra-structural findings in a fetus with a novel form of me-somelic dysplasia.
CLINICAL REPORT
[Registry #97-20] Routine prenatal ultrasound studyidentified a 25-week fetus with disproportionate short-ness of the long bones. Family histories of the 19-year-old G1 mothers and a 22-year-old nonconsanguineousfather were unremarkable. Physical examination of theaborted male fetus showed a normal face with over-folded ears and prominent superior helices. The chestwas narrow with Harrison grooves, and the limbs
Contract grant sponsor: NIH; Contract grant number: HD22657.
*Correspondence to: William R. Wilcox, M.D., Ph.D., MedicalGenetics, Cedars-Sinai Medical Center, 8700 Beverly Blvd., SSB-3, Los Angeles, CA 90048. E-mail: [email protected]
Received 27 March 1998; Accepted 29 June 1998
American Journal of Medical Genetics 80:247–251 (1998)
© 1998 Wiley-Liss, Inc.

showed prominent severe mesomelic and mild rhizo-melic shortness. There was ulnar deviation of thehands, bilateral transverse palmar creases, and bilat-eral equinovarus deformities of the feet.
METHODSLight Microscopy
Formalin fixed chondro-osseous tissue was processedundecalcified and embedded in glycol and methylmethacrylate. Thin sections were stained with vonKossa trichrome, toluidine blue, alcian blue, silvermethanamine, and Goldner’s stains.
Electron Microscopy
For electron microscopy, fresh chondro-osseous tis-sue was fixed in 2.5% glutaraldehyde in 0.2 M phos-phate buffer (pH 7.3) overnight. The tissue was washedtwice in 0.2 M phosphate buffer, postfixed for 15 min-utes with 3% osmium tetraoxide, and dehydrated withan acetone gradient (50–100%) over 2 hours. After de-hydration the tissue was infiltrated with acetone/Spurrresin (1:1) overnight and then for an additional 4 hourswith four changes of resin. The resin was polymerizedat 60°C under vacuum for 24 hours, sectioned (500–600Å) using a Leica ultracut UCT ultramicrotome, andtransfered to Formvar-coated one-hole-grids. The sec-tions were stained with uranyl acetate for 1 hour,washed with distilled water, stained with lead citratefor 10 minutes, and examined with a Zeiss 902 trans-mission electron microscope.
Immunohistochemistry
Frozen sections (6 mm) of cartilage were fixed with4% paraformaldehyde and washed with phosphatebuffered saline (pH 7.4). Endogenous enzyme activitywas blocked using Endo/Blocker (Biømeda, Foster City,CA) and the tissue was partially digested with Auto/Zyme solution (Biømeda) for 5 minutes. The sectionswere blocked for 20 minutes with 0.1% bovine serumalbumin and 1% nonfat dry milk, incubated with theprimary antibody for 1 hour, followed by a peroxidase-tagged secondary antibody for 1 hour, and ABC solu-tion (Vector Labs, Burlingame, CA) for 30 minutes.Sections were developed with a peroxidase substrateDAB solution (Biømeda) and mounted.
Proteoglycan Sulfation
Proteoglycan sulfation assays were performed ac-cording to the method of Hastbacka et al. [1996].
RESULTSRadiographic Findings
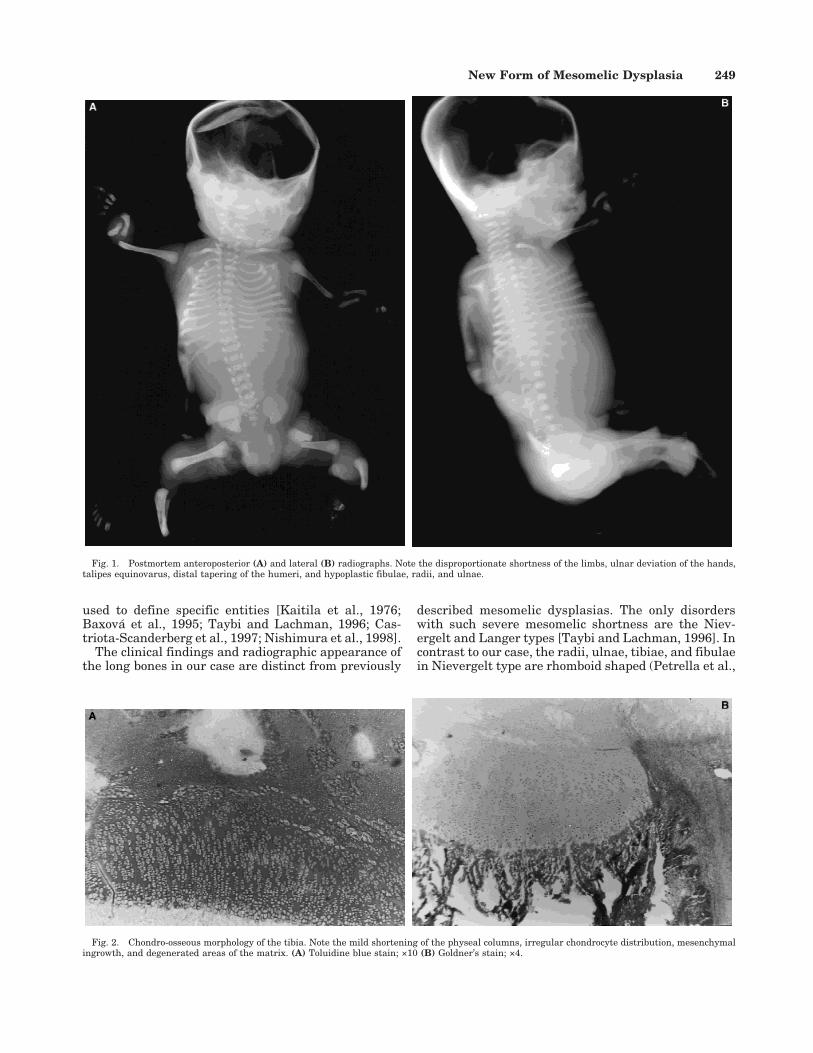
Postmortem radiographs (Fig. 1) showed a mild cer-vical kyphosis and decreased ossification in the C1-C2cervical region with coronal and sagittal clefts. Therewas marked glenoid hypoplasia, mild shortness of thehumeri with distal tapering, and frank dislocations atthe elbows. Both radii and ulnae were hypoplastic. Themetacarpals were bent and short, and the first meta-carpal had an ovoid shape. The pelvis showed an in-creased iliac flare. The femora were dumbbell-shaped
with widened proximal and distal metaphyses andshortened diaphyses. The fibulae were hypoplasticwhile the tibiae had widened proximal metaphyses andshortened diaphyses. There were equinovarus deformi-ties of both feet and decreased ossification of the tarsalbones.
Chondro-osseous Morphology
Chondro-osseous specimens from the humerus, ra-dius, ulna, femur, tibia, fibula, vertebra, and rib wereexamined. The resting cartilage had an uneven chon-drocyte distribution and increased vascularity (Fig. 2).There were areas of fibrillar degeneration resulting incollagenous rings around the chondrocytes. The growthplate showed mild shortness of the physeal columns, adecrease in the width of the hypertrophic zone, over-growth of perichondral bone, and peripheral ingrowthof mesenchymal cells. The histologic abnormalitieswere present in all the sites studied, but were moresevere in the forearm and leg (the mesomelic areas).Areas of focal degeneration and the normal-appearingmatrix stained positively with alcian blue, whereas asection of cartilage from a representative case of atelo-steogenesis type II (AO II) did not. Alcian blue stainedmatrix indicates the presence of sulfated proteogly-cans. The rings stained darkly with silver methena-mine consistent with the presence of collagen.
Ultrastructural examination of the resting cartilagefrom the femur, vertebrae, and tibia showed a normalinterterritorial matrix. However, the territorial matrixwas abnormal consisting of rings of aggregated colla-gen fibrils. The chondrocytes contained numerous loopsof dilated rough endoplasmic reticulum (rER) (Fig. 3).
Immunohistochemistry
Immunohistochemical studies using antibodies spe-cific for collagens (types II, IX, and X), aggrecan, ver-sican, and cartilage oligomeric matrix protein (COMP)were performed on frozen tissues. There were no de-tectable differences in matrix staining compared tocontrols, and there was no staining of the rER inclu-sions. Control sections of achondrogenesis II/hypochondrogenesis material showed staining of inclu-sions only with type II collagen antibodies.
DISCUSSION
Mesomelic shortness is common in many skeletaldysplasias. In the international nomenclature, disor-ders with prominent mesomelia that have not beenplaced in other groups are grouped together as ‘‘meso-melic chondrodysplasias’’ [Rimoin et al., in press]. Con-sequently, the group is heterogenous and individualdisorders are inherited as autosomal dominant or re-cessive traits, usually identifiable at birth [Kaitila etal., 1976; Taybi and Lachman, 1996]. They range inseverity from perinatal lethal to mild short stature anddemonstrate varying degrees of limb shortness. Thisvariability in shape and involvement of the ulnae, ra-dii, tibiae, or fibulae, as well as additional clinical orradiographic findings such as involvement of the proxi-mal or distal limb segments, vertebral or craniofacialanomalies, or other skeletal malformations have been
248 Brodie et al.
used to define specific entities [Kaitila et al., 1976;Baxova et al., 1995; Taybi and Lachman, 1996; Cas-triota-Scanderberg et al., 1997; Nishimura et al., 1998].
The clinical findings and radiographic appearance ofthe long bones in our case are distinct from previously
described mesomelic dysplasias. The only disorderswith such severe mesomelic shortness are the Niev-ergelt and Langer types [Taybi and Lachman, 1996]. Incontrast to our case, the radii, ulnae, tibiae, and fibulaein Nievergelt type are rhomboid shaped (Petrella et al.,
Fig. 2. Chondro-osseous morphology of the tibia. Note the mild shortening of the physeal columns, irregular chondrocyte distribution, mesenchymalingrowth, and degenerated areas of the matrix. (A) Toluidine blue stain; ×10 (B) Goldner’s stain; ×4.
Fig. 1. Postmortem anteroposterior (A) and lateral (B) radiographs. Note the disproportionate shortness of the limbs, ulnar deviation of the hands,talipes equinovarus, distal tapering of the humeri, and hypoplastic fibulae, radii, and ulnae.
New Form of Mesomelic Dysplasia 249

1990]. The pattern of shortness was also different fromthe Langer type, and our case had abnormalities of thespine [Langer, 1967].
Our case also has some similarity to skeletal dyspla-sias known as atelosteogenesis (AO) [Table I]. Thethree recognized forms of AO share similar clinical andradiographic manifestations, such as generalized limbshortness, cleft palate, midface hypoplasia, microgna-thia, depressed nasal root, equinovarus deformities,vertebral and hand anomalies, aplastic or hypoplasticfibula, and joint dislocations [Sillence et al., 1987;Stern et al., 1990]. In the present case, the ulnar de-viation of the hands, talipes equinovarus, distal taper-ing of the humerus, and hypoplastic fibulae, radii, andulnae are manifestations shared with AO [Maroteauxet al., 1982; Sillence et al., 1982, 1987; Stern et al.,1990; Fallon et al., 1994]. However, our case has radio-logic changes in the long bones not observed in the AOsyndromes. The morphologic findings of degenerationand rings of collagen around the chondrocytes superfi-cially resemble those in AO II, which has been reclas-sified and grouped with the diastrophic dysplasia sul-fate transporter (DTDST) disorders [Hastbacka et al.,1996; Rimoin et al., in press]. However, the appearanceof the degeneration, the positive staining of the matrixwith alcian blue, and the presence of rER inclusionsbodies distinguish this case from cases with DTDST
defects. Furthermore, in vitro sulfation of proteogly-cans by cultured fibroblasts from our case were notdifferent from age-matched controls and were muchgreater than those from cases of diastrophic dysplasiaand achondrogenesis IB (data not shown).
Several chondrodysplasias demonstrate the presenceof rER inclusion bodies within chondrocytes. For ex-ample, in the type II collagenopathies (achondrogen-esis, II/hypochondrogenesis, Kniest dysplasia, andspondyloepi(meta)physeal dysplasia) and COMP disor-ders (pseudoachondroplasia and some cases of multipleepiphyseal dysplasia) immunohistochemical studieshave shown the defective extracellular matrix proteinis retained within the inclusions [Godfrey et al., 1988].The rER inclusion bodies in our case suggest this dis-order may also be due to a defect in a protein traffickedthrough the rER. Negative immuno-staining of the in-clusions in this case with antibodies to collagens typeII, IX, X, aggrecan, versican, and COMP indicates thatthis disorder is unlikely to result from defects in theseproteins.
In summary, we have described a new form of meso-melic dysplasia with chondro-osseous morphology re-sembling AO II but with normal in vitro proteoglycansulfation and intracellular inclusion bodies suggestinga new and separate disorder.
Fig. 3. Transmission electron micrographs of resting cartilage from the tibia. Note the rings of aggregated collagen fibrils in the territorial matrix (A)8,400×, and chondrocytes containing loops of dilated rough endoplasmic reticulum (B) 12,960×.
250 Brodie et al.

ACKNOWLEDGMENTS
We thank Betty Mekikian, Christine Kim, and LoydaNolasco for technical assistance, and Maryann Prioreand Roberta Bonacquisti for administering the Inter-national Skeletal Dysplasia Registry.
REFERENCES
Baxova A, Kozlowski K, Netriova I (1995): A new form of rhizo-mesomelicbone dysplasia. Pediatr. Radiol 25:300–302.
Castriota-Scanderberg A, Mingarelli R, Caramia G, Osimani P, LachmanRS, Rimoin DL, Wilcox WR, Dallapiccola B (1997): Spondylo-mesomelic-acrodysplasia with joint dislocations and severe combinedimmunodeficiency: A newly recognised immuno-osseous dysplasia. JMed Genet 34:854–856.
Fallon MJ, Hockey A, Hallam LA (1994): Atelosteogenesis type III: A casereport. Pediatr Radiol 24:47–49.
Godfrey M, Keene DR, Blank E, Hori H, Sakai LY, Sherwin LA, HollisterDW (1988): Type II achondrogenesis-hypochondrogenesis: Morpho-logic and immunohistopathologic studies. Am J Hum Genet 43:894–903.
Hastbacka J, Superti-Furga A, Wilcox WR, Rimoin DL, Cohn DH, LanderES (1996): Atelosteogenesis type II is caused by mutations in the dias-trophic dysplasia sulfate-transporter gene (DTDST): Evidence for aphenotypic series involving three chondrodysplasias. Am J Hum Genet58:255–262.
The International Working Group on Constitutional Diseases of Bone.(1998): International nomenclature and classification of the osteochon-drodysplasias. Am J Med Genet 79:376–382.
Kaitila II, Leisti JT, Rimoin DL (1976): Mesomelic skeletal dysplasias. ClinOrthop 114:94–106.
Langer LO (1967): Mesomelic dwarfism of hypoplastic unla, fibulae, man-dible type. Radiology 89:654–660.
Löhr H, Wiedemann HR (1981): Mesomelic dysplasia—associated withother abnormalities. Eur J Pediatr 137:313–316.
Maroteaux P, Spranger J, Stanescu V, Le Marec B, Pfeiffer RA, BeightonP, Mattei JF (1982): Atelosteogenesis. Am J Med Genet 13:15–25.
Nishimura G, Nakayama M, Fuke Y, Suehara N (1998): A lethal osteo-chondrodysplasia with mesomelic brachymelia, round pelvis, and con-genital hepatic fibrosis: Two siblings born to consanguineous parents.Pediatr Radiol 28:43–47.
Petrella R, Ludman MD, Rabinowitz JG, Gilbert F, Hirschhorn K (1990):Mesomelic dysplasia with absence of fibulae and hexadactyly: Niev-ergelt syndrome or new syndrome? Am J Med Genet 37:10–14.
Sillence D, Kozlowski K, Rogers J, Sprague P, Cullity G, Osborn R (1987):Atelosteogenesis: Evidence for heterogeneity. Pediatr Radiol 17:112–118.
Sillence DO, Lachman RS, Jenkins T, Riccardi VM, Rimoin DL (1982):Spondylohumerofemoral hypoplasia (giant cell chondrodysplasia): Aneonatally lethal short-limb skeletal dysplasia. Am J Med Genet 13:7–14.
Stern HJ, Graham JM Jr, Lachman RS, Horton W, Bernini PM, SpiegelPK, Bodurtha J, Ives EJ, Bocian M, Rimoin DL (1990): Atelosteogen-esis type III: A distinct skeletal dysplasia with features overlappingatelosteogenesis and oto-palato-digital syndrome type II. Am J MedGenet 36:183–195.
Taybi H, Lachman RS (1996): ‘‘Radiology of Syndromes, Metabolic Disor-ders, and Skeletal Dysplasias, Fourth Edition.’’ St. Louis: Mosby-YearBook, Inc.
TABLE I. Radiographic and Morphologic Comparison of thePresent Case With the Atelosteogenesis Group of Skeletal
Dysplasias
Radiographicfindings
Presentcase
Atelosteogenesis
TypeI
TypeII
TypeIII
UlnaeHypoplastic + − + −Shortened diaphysis + − − −
RadiiHypoplastic + − + −Short diaphysis + − − −Widened metaphysis + − − −
TibiaeHypoplastic − − + −Short diaphysis + − − −Widened metaphysis + − − −
FibulaeHypoplastic + − + −Short diaphysis + − − −
Metacarpals/tarsalsDecreased ossification + − − −Absent calcaneous + − − −Ovoid 1st metacarpal + − − −
PhalangesClinodactyly + − − −
Chondro-osseous morphologyrER inclusions + + − −Degenerated matrix + − + −
Abnormal chondrocytecolumns + + + −
New Form of Mesomelic Dysplasia 251