radiosensitizing activity of a novel benzoxazine through the promotion of apoptosis and inhibition...
TRANSCRIPT

PRECLINICAL STUDIES
Radiosensitizing activity of a novel Benzoxazinethrough the promotion of apoptosis and inhibitionof DNA repair
Suraj Radhamani & Christopher Bradley &
Terri Meehan-Andrews & Saleh K. Ihmaid &
Jasim Al-Rawi
Received: 9 January 2014 /Accepted: 24 February 2014 /Published online: 14 March 2014# Springer Science+Business Media New York 2014
Summary The DNA dependant protein kinase (DNA-PK)enzyme plays a major part in the repair of double strandedbreaks induced by radiation and hence in the radio-resistanceof tumour cells. Inhibitors of DNA-PK have been testedsuccessfully in the past for their ability to sensitize cancercells to the effects of radiation. Here we present a novelbenzoxazine, 8-methyl-2-(morpholine-4yl)-7-(pyridine-3-methoxy)-4H-1,3-benzoxacine-4-one (LTU27) and analyseits ability to cause sensitization of lung cancer and coloncancer cells to radiation. There was a significant reduction insurvival rate, increase in apoptosis and inhibition in autophos-phorylation of DNA-PK and AKT1 after treating them con-comitantly with both radiation and LTU27. The mechanism ofaction appears to be through inhibition of DNA-PK leading todelayed DNA repair and promotion of apoptosis.
Keywords Radiosensitizing . Colon cancer . Lung cancer .
DNA-PK . Benzoxazines
Introduction
Radiotherapy is one of the most promising, effective andcommon methods for the treatment of cancer, despite poten-tially severe side effects [1, 2]. Irradiation of cancer cells
induces DNA double strand breaks (DNA-DSBs) which arecritical lesions for the success of radiotherapy [3–5]. However,cells are programmed with various DSB repair pathways tomaintain genomic integrity and to prevent misrepair and chro-mosomal rearrangements [6]. These DSB repair pathways areone of the main causes of radio-resistance and failure ofradiation treatment [7]. Specifically targeting proteins in-volved in this pathway, thereby inhibiting cellular repairmechanisms, could enhance radiation treatment, minimisingdosage and thus side effects, while maximising the results.
Mammalian cells primarily repair DSBs through variouspathways such as Non-Homologous End Joining (NHEJ) andHomologous Recombination (HR). The induction of DSBsfollowing ionizing radiation (IR) causes the free DNA ends tobind to heterodimeric Ku70 and Ku80 complex proteins thusinitiating NHEJ [8, 9]. Through binding of Ku proteins, theDNA-dependent protein kinase catalytic subunit (DNA-PKcs)is recruited to the DSBs. DNA-PKcs belongs to the phos-phatidylinositol 3-kinase-related kinase (PIKK) family and isa large kinase which requires DNA binding for its activity [10,11]. The kinase activity of DNA-PK is a crucial initiator ofDSB repair through NHEJ. Aside from its integral role inNHEJ, DNA-PK has also been shown to have importantdownstream targets such as the PIKK member AKT whichis involved in diverse cellular pathways including NHEJ[12–14]. AKT is a kinase which is activated after phosphory-lation on two key residues: serine 473 and threonine 308following which it has roles in cell survival, cell cycle andproliferation and has been associated with carcinogenesis andradiation/chemo resistance [15–17]. That DNA-PK catalysesthe phosphorylation of AKT at serine 473 is well established[18, 19].
Inhibition of these kinase enzymes through a targetedapproach not only sensitizes cancer cells to radiation, but alsoresults in a decrease in cancer cell multiplication rate [20, 21].
S. Radhamani : C. Bradley (*) : S. K. Ihmaid : J. Al-RawiSchool of Pharmacy and Applied Science, La Trobe Institute ofMolecular Sciences, La Trobe University, P.O. Box 199, Bendigo,VIC 3552, Australiae-mail: [email protected]
C. Bradley : T. Meehan-AndrewsLa Trobe Rural Health School, La Trobe University, P.O. Box 199,Bendigo, VIC 3552, Australia
Invest New Drugs (2014) 32:424–435DOI 10.1007/s10637-014-0079-4

In the absence of DNA repair cells will undergo apoptosis[22]. Early sensitizing compounds included LY294002 andWortmannin, two broad specificity PIKK family inhibitors.[23–25]. Various in vitro studies have shown thatWortmanninand LY294002 inhibit the re-joining of broken DSBs inducedby radiation [26, 27]. More recently, the pyrone NU7026 andthe chromone NU7441 have been developed as specificDNA-PK inhibitors [28] and are in pre-clinical trials [29,30]. In this study, we have examined the radio-sensitizingeffects of two novel benzoxazine derivatives: LTU27 andLTU11B . LTU27 has been shown to exhibit comparableinhibitory potency to LY294002 and NU7026 as a DNA-PKinhibitor whereas LTU11B has a DNA-PK inhibitory IC50 anorder of magnitude higher [31]. We have tested theradiosensitizing effects of these compounds in two cell lines:HT29 colorectal adenocarcinoma and A549 lung carcinoma.Here we report the potent radiosensitizing effects of thesecompounds and link their mechanism of action to theirDNA-PK inhibitory activity.
Materials and methods
Cell lines
Cell lines used in this study were A549 and HT29. The celllines were cultured in DMEM supplemented with 5.0 mMglutamine and 10 % foetal calf serum. Cells were incubated at37 °C under an atmosphere comprising 5 % CO2/95 % air.Cell lines were obtained from the American Type CultureCollection and were stored in liquid nitrogen until use. Cellswere used before 10 passages.
Compounds and stock Solutions
LTU27 and LTU11Bwere synthesized in the medicinal chem-istry laboratory, Latrobe University, Bendigo (Fig. 1) [31].
LY294002 was purchased from Cell Signalling Technology(Danvers, MA). NU7026 was obtained from Sigma AldrichPty. Ltd. (NSW, Australia). Stock solutions (1 mM) wereprepared for LTU27, LTU11B, LY294002 and NU7026 inDMSO and were stored at −20 °C. Compounds were dilutedin culture medium to obtain the required concentration. Thepercentage of DMSO was kept below 0.5 % to avoid toxicity[32]. LTU11b and LTU27 IC50s for PI3K isoforms and DNA-PK were performed by Reaction Biology Corporation USAand are presented below (Table 1) along with those of controlinhibitors LY294002 [33, 34] and NU7026 [35].
Cell culture
Cells from an actively growing stock were trypsinized, count-ed using haemocytometer, and seeded onto sterile six wellplates (Costar 3736) at 1×106 cells per well unless otherwisestated. Cells were then allowed to adhere overnight at 37 °Cbefore being cultured with test compounds for 40 min prior toirradiation.
Radiation treatment
To optimise the radiation dose and to ensure effective andeven delivery of radiation, a custom made Perspex phantomwas manufactured according to the method described earlier[36]. The significance of this phantom was to ensure fullscatter conditions, provide adequate backscatter and to mini-mize air gaps. The phantom was designed to position six wellplates, with cells attached to the bottom and growing as amonolayer at the maximum depth of 1.5 cm. An isodosecontrol virtual scan for checking percentage dose variationsaround the well plates was also conducted to ensure thedelivery of 100 % dose to the cells [37]. Plates were irradiatedat doses ranging from 1 to 8 Gray (Gy) using a Varian linearaccelerator (Peter MacCallum Cancer Centre, Bendigo, VIC).
Fig. 1 Chemical structure ofnovel Compounds. a 8-methyl-2-(morpholine-4yl)-7-(pyridine-3-methoxy)-4H-1, 3-benzoxacine-4-one (LTU27) and(b) 8-methyl-2[−(pyridine-3-yl-pyridine-2-yl methyl) amino]-4-H-1,3-benzoxacine-4-one(LTU11B)
Invest New Drugs (2014) 32:424–435 425

Clonogenic assay
Clonogenic assays were performed according to the methoddescribed earlier with slight modifications [38]. HT29 andA549 cell lines were selected for clonogenic assays becauseof their ability to form well defined colonies. Cells wereseeded at 3×102 cells per well and allowed to adhere over-night. Cells were treated with LTU27 and LTU11B at variousconcentrations (0.1 μM, 0.2 μM, 0.3 μM, 0.4 μM and0.8 μM) for 40 min prior to irradiation. These concentrationswere chosen based on previously published DNA-PK IC50’sfor these compounds [31] . After irradiation cells were furtherincubated for 10 days in the presence of test compounds toallow colony formation. Media and test compounds werechanged every 2nd day of the 10 day culture. After incubation,cells were washed with PBS and fixed with methanol andacetic acid (3:1). Fixed cells were stained with 0.5 % crystalviolet in ethanol and colonies were counted using an invertedmicroscope at 10x magnification. Only colonies containingmore than 50 cells were counted. The number of coloniesformed in treated and control groups were compared relativeto survival rate. Survival rate was calculated as (mean platingefficiency of treated cells/mean plating efficiency of controlcells) x 100 %. Plating efficiency was the number of coloniesdivided by the number of cells inoculated [39].
Analysis of apoptosis and cell cycle distribution by flowcytometry
Cell cycle analysis was conducted according to the methoddescribed earlier [40]. Following treatment, cells were fixedwith 70 % ethanol and stored at −20 °C. Samples were centri-fuged and washed with PBS to remove traces of ethanol andresuspended in 250 μl of PBS containing 25 μg/ml PI and100 μg/ml RNase A (Sigma Aldrich, R4642). All samples wereincubated for 30 min at 37 °C, protected from light. After30min, cells were stored on ice and analysed by flow cytometry.
Analysis of apoptosis by flow cytometry and Apostain
Apoptosis assay was performed as per the manufacturer’sinstruct ion using Anti-ssDNA/Apostain (BenderMedSystems, Austria). Following treatment with compoundsand radiation, cells were cultured for a further 24 h, thendetached and fixed with methanol, washed and heated with
formamide at 75 °C, washed with PBS and incubated with100 μl of monoclonal antibody F7-26 for 15 min (1 ml ofantibody in 9 ml of 5 % fetal bovine serum in PBS and storedin −20 °C). The cells were then washed again with PBS andincubated in 100 μl (1:500) of fluorescein conjugated goatanti-mouse IgM (Sigma, F-9259) in PBS containing 1 % non-fat milk powder, washed again with PBS and resuspended in300 μl of propidium iodide solution (1 μg/ml). Samples wereanalysed in flow cytometer and data were analysed using FCSexpress.
γ-H2AX and phosphorylated DNA-PK assay by flowcytometry
Cells were treated with 0.3μMof test compounds, NU7026 orLY294002 diluted in DMEM and incubated for 40 min. Cellswere then irradiated at 6 Gy and further incubated for 24 h(γ-H2AX assay) or 30 min (phosphorylated DNA-PK assay).Cells were then trypsinized and fixed with 1 % paraformalde-hyde at 4 °C for 15 min. Cells were washed with PBS andresuspended in 70 % ice cold ethanol and stored at −20 °C.Fixed cells were then washed with PBS and resuspended indilution buffer (PBS, 1 % bovine serum albumin and 0.1 %Triton X-100) and further incubated for 15 min at roomtemperature. After incubation, cells were pelleted again andresuspended in either 100 μl rabbit polyclonal anti-p-histoneH2AX (ser139) diluted 1:500 in dilution buffer (Santa CruzBiotechnology-101696) or 1:500 of 100 μl of rabbit Anti-DNA PKcs (phospho S2056) antibody (abcam, ab18192) indilution buffer and incubated for 2 h at 4 °C. After incubationcells were washed in PBS and resuspended in 100μl goat anti-rabbit IgG (H+L) fragment (Alexa fluor 488 conjugate, Cat-alogue no: A11008, Invitrogen) diluted 1:500 in dilutionbuffer. Samples were incubated at room temperature for 1 hunder constant agitation, protected from light. Cells were thenwashed twice in dilution buffer and resuspended in 5 μg/ml PI(Sigma Aldrich). Samples were analysed using a flowcytometer (BD Accuri C6, BD Biosciences, California,USA) and gated to omit doublets and debris. Data wasanalysed using FCS express 4 flow research edition.
Western blot analysis
After treatment with compounds and radiation, cells wereincubated for a further 30 min in the presence of compounds
Table. 1 DNA-PK and PI3KIC50 values for all inhibitors usedin this study. IC50 values greaterthan 100 μM were considered tobe inactive against the corre-sponding enzyme.
PI3K α PI3K β PI3K PI3K δ DNA-PKcs
LTU27 0.12 μM 1.42 μM 7.21 μM 2.00 μM 0.28 μM
LTU11B >100 >100 >100 86.70 μM 25.20 μM
LY294002 0.50 μM 0.97 μM NA 0.57 μM 6.00 μM
NU7026 13.00 μM NA NA NA 0.23 μM
426 Invest New Drugs (2014) 32:424–435

or control, harvested, and washed twice with ice-cold PBScontaining 1 mM PMSF (phenylmethylsulphonyl fluoride).Pelleted cells were resuspended in 100 μL of ice cold lysisbuffer (50 mM Tris–HCl pH 7.5, 150 mMNaCl, 1 % NonidetP-40, 1 x protease inhibitor cocktail (Roche), 1 mM PMSFand 10 μg/mL aprotinin, in distilled water) and incubated onice for 20 min. Lysates were centrifuged (14,000 rpm, 25 minat 4 °C) and the supernatant was used further. The proteinconcentration of each sample was determined using the Bio-Rad DC protein assay kit (Bio-Rad) according to the manu-facturer’s instructions. Typically 50μg protein per sample wasdenatured via addition of 1 x SDS loading buffer (abcam,ab119196) boiled for 10 min at 75 °C and loaded onto 4 % or10 % gels (abcam). Samples were run in the presence of SDSrunning buffer (abcam, ab119195), typically at 100 V forapproximately 90 min to separate proteins. Proteins weretransferred to PVDF membrane by using a Tank blottingapparatus (Biorad) according to the manufactures instructionwith 1 x Western transfer buffer (abcam). PVDF membraneswere blocked in 10 % (w/v) bovine serum albumin in TBS-T(Tris-buffered saline containing 0.1 % (v/v) Tween-20) for2 h. Membranes were incubated overnight at 4 °C with gentlerocking in anti-DNA PKcs, s2056 (abcam, ab18192), anti-AKT1, s473 (abcam, ab66138), anti-AKTI (abcam, ab91505),or anti-beta tubulin (abcam, ab6046). A 1 in 5,000 dilutionwas used for all the primary antibodies in TBS-T. Afterovernight incubation with primary antibodies membraneswere washed three times for 5 min each with TBS-T andincubated with HRP conjugated goat anti-rabbit IgG second-ary antibody (1 in 10,000) in TBS-T (abcam, ab97200)protected from light. Cells were washed with 1xTBS-T andincubated with ChemiFast Chemiluminescence substrate(Syngene, USA, CH-FAST/20) according to manufacturer’sinstructions. Chemiluminescent images were captured usingSyngene G-BOX (G: BOX-CHEMI-XL1.4, USA).
Statistical analysis
Experiments were done in triplicate; data was analysed usingGraph Pad prism 5.0 and presented as mean±SEM. Statisticalsignificance was determined using two-tailed unpaired Stu-dents t -test, p <0.05 were considered as statistically significant.
Results
LTU27 is a potent radiosensitizer in both HT-29 and A549cells
The survival rates for HT29 and A549 by clonogenic assay areshown in Fig. 2. Neither cell line showed a significant differ-ence between control and cells treated with 0.1 μM and0.2 μM LTU27 at any radiation dose. However, the survival
rate for HT29 and A549 was significantly decreased in groupstreated with 0.3 μM, 0.4 μMor 0.8 μMLTU27when exposedto radiation of 2 Gy or above (p<0.05) (Fig.2a and Fig.2b,respectively). When both cell lines were treated with LTU11Bthere was no significant reduction in survival compared to thecontrol at any concentration or radiation dose (Fig.2c andFig.2d). Since there was a significant decrease in survival at0.3 μM LTU27 and 6 Gy for both cell lines, these conditionswere used for further studies. To compare the efficacy ofLTU27 compared with two other known radiosensitizingagents, LY294002 [41] and NU7026 [35] the same concen-trations and radiation doses were applied to each cell line(Fig. 2). When cells were treated with LTU27 followingradiation exposure, there was a significant decrease in survivalin HT29 and A549 cell lines (p<0.05) when compared withother treatment groups (Fig.2e and Fig.2f respectively). At theconcentration tested, 0.3uM for all inhibitors, LTU27 wasfound to have the greatest radiosensitizing effect, followedby LY294002. NU7026 did not show radiosensitizing effect atthe tested concentration in either cell line.
LTU27 delays repair of DSB in both HT-29 and A549 cells
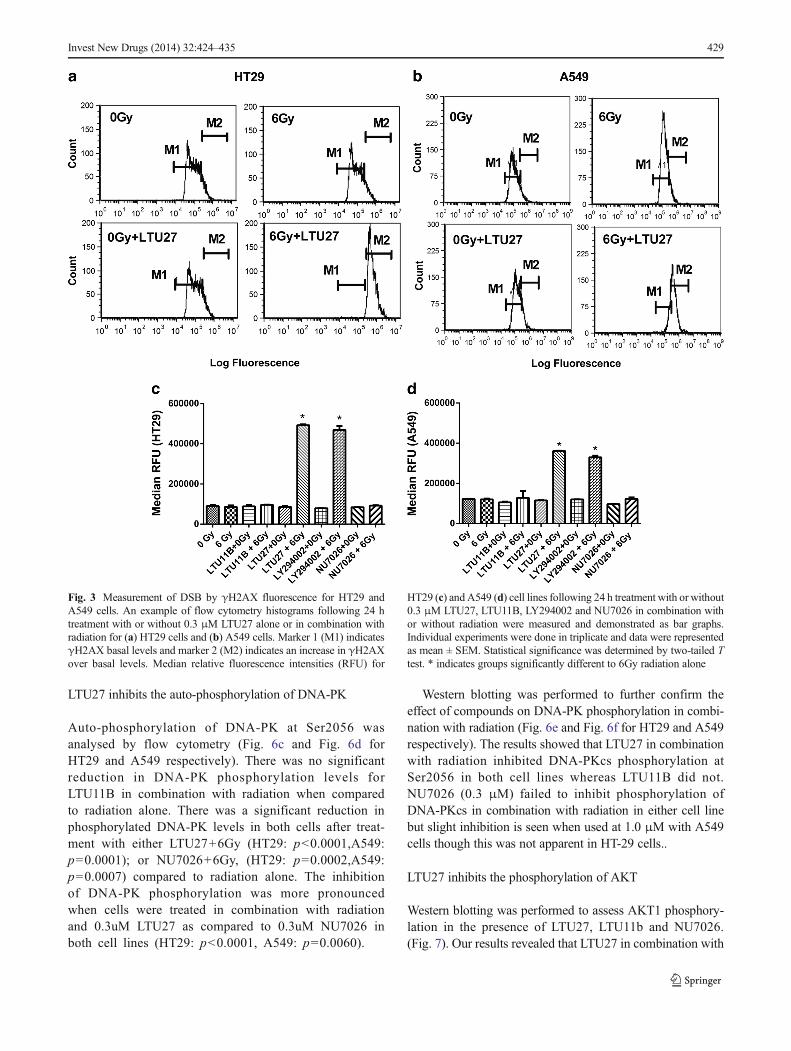
γ-H2AXwas assayed as a measure of DSBs and their repair inHT29 (Fig. 3c) and A549 (Fig. 3d) cells 24 h after treatmentwith compounds alone or in combination with 6 Gy radiation.When cells were treated with either LTU27 or LY294002 incombination with 6Gy radiation there was a significant re-sponse in both cell lines. HT29 cells treated with LTU27showed a more than 4 fold increase in γ-H2AX levels after24 h compared to 6Gy radiation alone (p<0.0001). LY294002in combination with 6Gy also increased γ-H2AX expressioncompared to 6Gy alone (p<0.0001), but was significantly lessthan treatment group LTU27+6Gy (p=0.0002). γ-H2AX levelsin A549 cells treated with LTU27+6Gy (p<0.0001) andLY294002+6Gy (p<0.0001) showed lesser response thanthose similarly treated HT29 cells. In both the cell lines,LTU27+6Gy exhibited highest γ-H2AX levels after 24 hand showed a slightly increased level when comparedwith LY294002+6Gy. When cells were treated with NU7026following radiation exposure, there was no significantincrease of γ-H2AX levels in either cell line.
LTU27 increases the proportion of cells in sub-G1 followingradiation
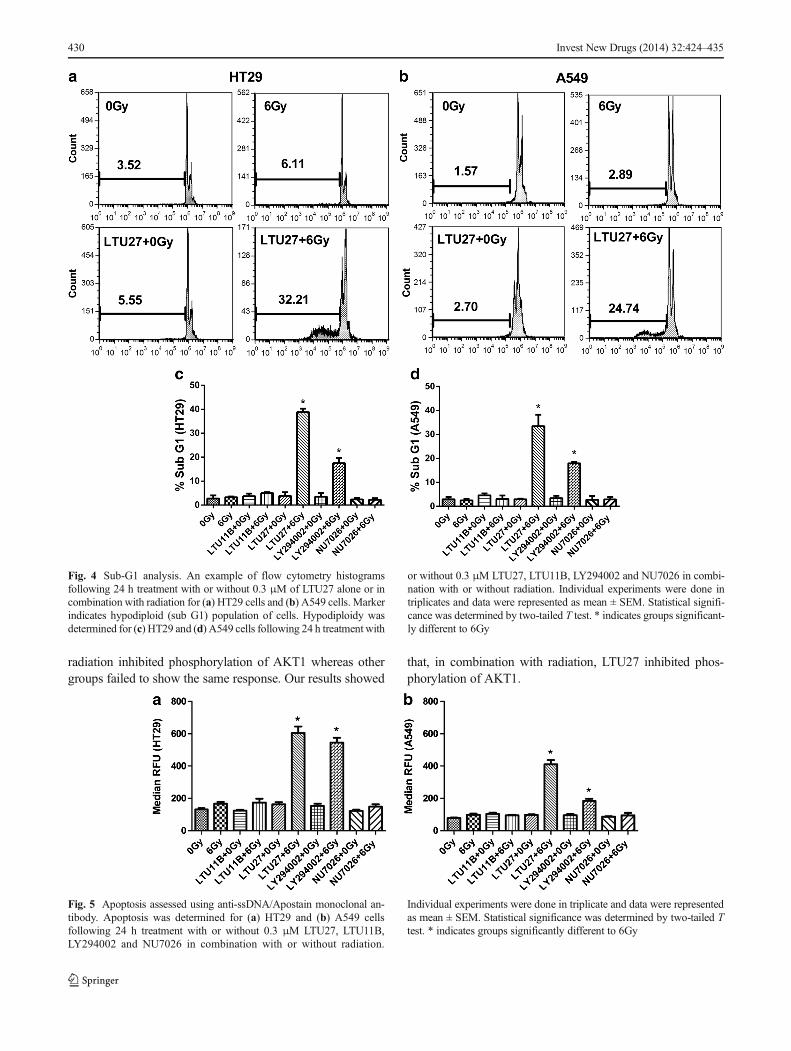
Analysis of cell death was determined initially by analysingDNA fragmentation and hypodiploidy expressed as the subG1fraction in cell cycle analysis [42, 43] (Fig. 4). In the HT29 cellline, cells treated with LTU27 in combination with radiationshowed a significantly higher percentage of sub G1 populationcompared with radiation alone and the non-irradiated groupafter 24 h post radiation (p<0.0001) (Fig. 4c). There was a
Invest New Drugs (2014) 32:424–435 427

significant increase in percentage of sub G1 for LY294002+6Gy (p=0.004) in combination with radiation when comparedto 6Gy radiated group, but this was significantly less than cellstreated with LTU27+6Gy (p=0.0015). The pattern was similarin A549 cells with a significant increase in the proportion ofcells in sub G1 for LTU27+6Gy when compared toradiation alone (p=0.0004) (Fig. 4d). There was a sig-nificant increase in the percentage of sub G1 cellsfollowing treatment with LY294002 in combination withradiation (p<0.0001) when compared to radiation alone,but this was significantly less than in the LTU27+6Gytreatment group (p=0.0046). LTU11B and NU7026 incombination with radiation did not induce a significantincrease or decrease in the percentage of cells withinsub G1 in either cell line.
LTU27 promotes apoptosis following radiation
To confirm that the hypodiploid cells in sub-G1 representapoptotic cells, the Apostain assay for ssDNAwas employed.This assay has proven specificity for apoptotic cells [44].
Fluorescence intensities obtained were plotted as bar graphsfor HT29 (Fig. 5a) and A549 (Fig. 5b). Cells exposed toradiation treatment alone showed a slight increase in apopto-sis. HT-29 and A549 cells treated with LTU11B in combina-tion with radiation showed no significant difference in theamount of apoptosis when compared to the radiation alonegroup. Cells treated with LTU27+6Gy, displayed cell linespecific apoptotic responses with HT29 being more sensitiveto the induction of apoptosis than A549. HT29 cells treatedwith LTU27 in combination with radiation had a significantincrease in apoptosis when compared to radiation alone(p<0.0001). A549 cells were less responsive when comparedto HT29 cells, but still had a significant increase in apoptosiswhen compared to radiation alone (p<0.0001). When cellswere treated with LY294002+6Gy, a similar trend was ob-served. HT29 cells were again more sensitive to the initiationof apoptosis, when compared to the non-radiated group(p<0.0001), and were comparable to LTU27+6Gy treatedcells. Cells treated with NU7026 in combination with radia-tion showed no significant difference in apoptosis to the non-radiated group.
Fig. 2 Radiosensitizing effect onHT29 and A549 cells wasdetermined by clonogenic assay.a Survival curves after treatmentwith LTU27 and radiation forHT29 and (b) for A549. cSurvival curves after treatmentwith LTU11B and radiation forHT29 and (d) for A549.Radiosensitizing effect on HT29(e) and A549 (f) cells alone(0Gy), or exposed to 6 Gyradiation alone, or treated with0.3 μM of LTU27, LTU11B,LY294002 and NU7026 alone orin combination with radiation(6Gy). Individual experimentswere done in triplicate and datawere represented as mean ± SEM.* indicates significant differenceversus 6Gy
428 Invest New Drugs (2014) 32:424–435
LTU27 inhibits the auto-phosphorylation of DNA-PK
Auto-phosphorylation of DNA-PK at Ser2056 wasanalysed by flow cytometry (Fig. 6c and Fig. 6d forHT29 and A549 respectively). There was no significantreduction in DNA-PK phosphorylation levels forLTU11B in combination with radiation when comparedto radiation alone. There was a significant reduction inphosphorylated DNA-PK levels in both cells after treat-ment with either LTU27+6Gy (HT29: p<0.0001,A549:p=0.0001); or NU7026+6Gy, (HT29: p=0.0002,A549:p=0.0007) compared to radiation alone. The inhibitionof DNA-PK phosphorylation was more pronouncedwhen cells were treated in combination with radiationand 0.3uM LTU27 as compared to 0.3uM NU7026 inboth cell lines (HT29: p<0.0001, A549: p=0.0060).
Western blotting was performed to further confirm theeffect of compounds on DNA-PK phosphorylation in combi-nation with radiation (Fig. 6e and Fig. 6f for HT29 and A549respectively). The results showed that LTU27 in combinationwith radiation inhibited DNA-PKcs phosphorylation atSer2056 in both cell lines whereas LTU11B did not.NU7026 (0.3 μM) failed to inhibit phosphorylation ofDNA-PKcs in combination with radiation in either cell linebut slight inhibition is seen when used at 1.0 μM with A549cells though this was not apparent in HT-29 cells..
LTU27 inhibits the phosphorylation of AKT
Western blotting was performed to assess AKT1 phosphory-lation in the presence of LTU27, LTU11b and NU7026.(Fig. 7). Our results revealed that LTU27 in combination with
Fig. 3 Measurement of DSB by γH2AX fluorescence for HT29 andA549 cells. An example of flow cytometry histograms following 24 htreatment with or without 0.3 μM LTU27 alone or in combination withradiation for (a) HT29 cells and (b) A549 cells. Marker 1 (M1) indicatesγH2AX basal levels and marker 2 (M2) indicates an increase in γH2AXover basal levels. Median relative fluorescence intensities (RFU) for
HT29 (c) and A549 (d) cell lines following 24 h treatment with or without0.3 μM LTU27, LTU11B, LY294002 and NU7026 in combination withor without radiation were measured and demonstrated as bar graphs.Individual experiments were done in triplicate and data were representedas mean ± SEM. Statistical significance was determined by two-tailed Ttest. * indicates groups significantly different to 6Gy radiation alone
Invest New Drugs (2014) 32:424–435 429
radiation inhibited phosphorylation of AKT1 whereas othergroups failed to show the same response. Our results showed
that, in combination with radiation, LTU27 inhibited phos-phorylation of AKT1.
Fig. 5 Apoptosis assessed using anti-ssDNA/Apostain monoclonal an-tibody. Apoptosis was determined for (a) HT29 and (b) A549 cellsfollowing 24 h treatment with or without 0.3 μM LTU27, LTU11B,LY294002 and NU7026 in combination with or without radiation.
Individual experiments were done in triplicate and data were representedas mean ± SEM. Statistical significance was determined by two-tailed Ttest. * indicates groups significantly different to 6Gy
Fig. 4 Sub-G1 analysis. An example of flow cytometry histogramsfollowing 24 h treatment with or without 0.3 μM of LTU27 alone or incombination with radiation for (a) HT29 cells and (b) A549 cells. Markerindicates hypodiploid (sub G1) population of cells. Hypodiploidy wasdetermined for (c) HT29 and (d) A549 cells following 24 h treatment with
or without 0.3 μM LTU27, LTU11B, LY294002 and NU7026 in combi-nation with or without radiation. Individual experiments were done intriplicates and data were represented as mean ± SEM. Statistical signifi-cance was determined by two-tailed T test. * indicates groups significant-ly different to 6Gy
430 Invest New Drugs (2014) 32:424–435

Discussion
The substituted benzoxazine, LTU27 (2-morpholino, 1,3-ben-zoxazine), is a novel compound that has been developed to
specifically target and inhibit DNA-PK activity and sensitizecells to the effects of radiation. Both cell lines, HT29 and A549,showed highest radiosensitivity when treated with LTU27 ascompared to the known broad spectrum PI3K inhibitor
Fig. 7 Phosphorylation of AKT1 in HT29 and A549 cells (a and b respectively) was analysed by western blotting. Cells were irradiated in the presenceof 0.3 μM LTU27, 0.3 μM LTU11B, 0.3 and 1 μM of NU7026
Fig. 6 Measurement of DNA-PK(Ser2056) phosphorylation inHT29 and A549 cells by flowcytometry. An example of flowcytometry histograms following1 h treatment with or without0.3 μM of LTU27 alone or incombination with or withoutradiation for (a) HT29 cells and(b) A549 cells. Marker 1 (M1)indicates negative and marker 2(M2) indicates positive for DNA-PK phosphorylation. (c) Medianfluorescence intensities for HT29and (d) and A549 cell linesfollowing 1 h treatment with orwithout 0.3 μM LTU27, LTU11Band NU7026 in combination withor without radiation weremeasured and demonstrated asbar graphs. Individualexperiments were done intriplicate and data are representedas mean±SEM. Statisticalsignificance was determined bytwo-tailed T test. * indicatesgroups significantly different to6Gy. Expression ofPhosphorylated DNA-PK(Ser2056) in HT29 and A549cells (e and f respectively) wasanalysed by western blotting.Cells were irradiated in thepresence of 0.3 μM LTU27,0.3 μM LTU11B, 0.3 and 1 μMof NU7026
Invest New Drugs (2014) 32:424–435 431

LY294002 and the more specific DNA-PK inhibitor NU7026with the increase in sensitization occurring from 2Gy. NU7026did not show any effect at 0.3 μM. These results are somewhatunexpected since the DNA-PK IC50 for LY294002 is 6 μM[33]while the DNA-PK IC50’s for LTU27 and NU7026 havebeen reported to be very similar at 0.28μMfor LTU27[31], and0.23 μM for NU7026[35]. However, LY294002 has beenshown to cause radiosentization at 5 μM [45] while NU7026has only been shown to cause radiosensitization at 10 μM [46].The IC50 value of NU7026 was determined in purified enzymemodels and a lack of radiosensitization at the low dose of0.3 μMmight be due to the more complex environment of cellbased assays. This has been shown previously in chemo sensi-tivity studies employing the inhibitor KU-0060648 where a 10fold reduction in potency against DNA-PK inMCF7 cells and a250 fold reduction in potency against PI3K in SW620 cells wasfound as compared to the IC50 values derived from purifiedenzyme models [47]. Another study on radiotherapy of ovariancancer has shown radiosensitization byNU7026 at 10μM [46].Another possibility has to do with the broader specificity ofLTU27 when compared to NU7026. It is possible that activitiesother than against DNA-PK are responsible for the greaterpotency of LTU27 as a sensitizing agent to radiation althoughthe evidence presented in this study strongly supports a role forDNA-PK inhibition. Inhibition to other members of the PIKKfamily was not determined although ATM activation was un-affected by LTU11b and LTU27 when examined by Westernblotting (results not shown).
To confirm our hypothesis that LTU27 is sensitizing thecells to the effects of radiation through the inhibition of DNA-PK, we first investigated the inhibition of DSB repair byassaying γ-H2AX. The histone protein, H2AX, is an importantconstituent in DNA repair and is rapidly phosphorylated onfour serine residues to γH2AX at sites of DSB [48]. Althoughother factors such as hypoxia can cause the phosphorylation ofH2AX it is considered to be a reliable indicator of the presenceof DSBs [49]. Various studies have reported the disappearanceof γ-H2AX as a result of DSB repair thus enabling us to assessthe kinetics of repair [50–52]. If LTU27 is causing sensitizationthrough inhibition of DNA-PK we would expect to see aprolongation of DSB repair. LTU27 was most potent ininhibiting the repair of DSBs 24 h post-irradiation followedby LY294002. At the 0.3 μM concentration used for all inhib-itors NU7026 showed no significant DSBs remaining after24 h. DSBs were most frequent in HT29 cells when treatedwith LTU27 and LY294002 combined with radiation butmarkedly fewer in A549. A few studies have reported previ-ously on varying levels of phosphorylated H2AX expressionfollowing irradiation [53, 54]. A previous study profiling theradiosensitivity of various human ovarian cancer cell linesreported detectable difference in DNA-PK levels and varia-tions in DSB repair patterns among them [55]. Another studyreported that a low level of DNA-PK activity causes decreased
H2AX phosphorylation [56]. A previous study on KU-0060648 also shows varying degrees of inhibition of DNA-PK and PI3K in various cancer cell lines during chemo sensi-tization [47] . Consequently the cell line dependent variationsin DNA-PK expression could account for the varying degreeof DSB repair inhibition by these compounds.
The delay in DNA repair seen when LTU27 and LY294002are present following irradiation should result in an increase inapoptotic cell death. To first assess cell death in general wemeasured DNA fragmentation seen as hypodiploidy repre-sented as the sub-G1 section in the cell cycle [57, 58]. Fromthe results we can see similar patterns to those seen whenassaying sensitization and DSBs. Our results showed thatLTU27 increased the percentage of sub G1 cells more thanLY294002 when treated in combination with radiation. Thissupports the hypothesis that these compounds are sensitizingthe cell lines to radiation by inhibiting the repair of DSBs,which then leads to cell death through processes such asapoptosis.
To confirm that this cell death is occurring through apopto-tic mechanisms, an apoptosis assay using a monoclonal anti-body to single stranded DNA (anti-ssDNA MAb) was con-ducted [59, 60]. Our results showed that the cell death mea-sured by subG1 analysis is mostly apoptosis, with slight cellline differences observed. Irradiated HT29 cells revealedhigher levels of apoptosis than those seen in A549 cellsfollowing treatment with LTU27.
To confirm that the mechanism of action of LTU27 isthrough DNA-PK inhibition, we assessed DNA-PK autophos-phorylation by flow cytometry [61, 62] and western blotting.The phosphorylation at Ser2056 is primarily mediated byautophosphorylation and is considered to be a reliable markerof DNA-PK activation in vivo [63]. Previous studies haveshown that irradiation induced DNA-PKcs autophosphoryl-ation occurs at Ser2056 and that this plays a direct role in DSBrepair through NHEJ [64, 65]. Our results revealed the inhi-bition of DNA-PK phosphorylation at Ser2056 in irradiatedcells treated with 0.3uM LTU27. At this concentration LTU27was more potent than NU7026, which only showed inhibitionof phosphorylation at 10 μM (data not shown). As AKT hasbeen shown to be an important downstream target of DNA-PKwe expected that inhibition of DNA-PK would result in inhi-bition of AKT phosphorylation. LTU27 inhibited the phos-phorylation of AKT in irradiated A549 cells though notcompletely. This is to be expected as AKT activation mayalso be due to the involvement of other signalling pathwayssuch as mammalian target of rapamycin (mTOR) [66], insulinreceptor substrate (IRS) [67] and pro apoptotic FOXO pro-teins [68]. It is interesting to note that, in HT-29 cells the basallevels of AKT phosphorylation were much higher than thosefound in A549 cells and this could explain why LTU27 wasless effective in inhibiting AKT phosphorylation in this cellline.
432 Invest New Drugs (2014) 32:424–435

These studies confirm that LTU27 is a new radiosensitizingagent that inhibits DSB repair and promotes apoptosis inirradiated cells. We propose that its mechanism of action isthrough the inhibition of DNA-PK. Evidence for this is thatLTU27 inhibited the autophosphorylation of DNA-PK andthis inhibition of activation is reflected in the decreased phos-phorylation of AKT, a known downstream target of DNA-PK.LTU27 has shown favourable potency as a radiosensitizingagent when compared to both the broad range PI3K inhibitorLY294002 and the more specific DNA-PK inhibitor NU7026.These results make it a promising candidate for furtherstudies.
Acknowledgements The authors would like to thank the staff of PeterMacCallum Bendigo Radiotherapy Centre for their support and technicalassistance for irradiation of cell lines. We also would like to thankProfessor Roger Martin, head of the molecular radiation biology andresearch division and Andrea Smith, Peter MacCallum cancer centre,Melbourne for their valuable advice and support. Suraj Radhamani wasa recipient of Latrobe University Postgraduate Research scholarship(LTUPS) and Full fee Research scholarship (LTUFFRS), with additionalresearch funding by Latrobe University.
Conflict of Interest The authors declare that they have no conflict ofinterest.
References
1. De Schutter H, Nuyts S (2009) Radiosensitizing Potential ofEpigenetic Anticancer Drugs. Anti-Cancer Agent Me 9(1):99–108
2. Willner J, Flentje M (1999) Radiochemotherapy with Taxol forlocally advanced non-small-cell lung cancer. Strahlenther Onkol175:14–19
3. Bogdanov KV, Chukhlovin AB, Zaritskey AY, Frolova OI,Afanasiev BV (1997) Ultraviolet irradiation induces multiple DNAdouble-strand breaks and apoptosis in normal granulocytes andchronic myeloid leukaemia blasts. Brit J Haematol 98(4):869–872
4. Dextraze ME, Gantchev T, Girouard S, Hunting D (2010) DNAinterstrand cross-links induced by ionizing radiation: An unsunglesion. Mutat Res-Rev Mutat 704(1–3):101–107. doi:10.1016/j.mrrev.2009.12.007
5. Quanz M, Chassoux D, Berthault N, Agrario C, Sun JS, Dutreix M(2009) Hyperactivation of DNA-PK by Double-Strand BreakMimicking Molecules Disorganizes DNA Damage Response. PLoSOne 4 (7). doi:Artn E6298 Doi 10.1371/Journal.Pone.0006298
6. Jankovic M, Nussenzweig A, Nussenzweig MC (2007) Antigenreceptor diversification and chromosome translocations. NatImmunol 8(8):801–808. doi:10.1038/Ni1498
7. Shimura T, Kakuda S, Ochiai Y, Nakagawa H, Kuwahara Y, Takai Y,Kobayashi J, Komatsu K, Fukumoto M (2010) Acquiredradioresistance of human tumor cells by DNA-PK/AKT/GSK3beta-mediated cyclin D1 overexpression. Oncogene 29(34):4826–4837.doi:10.1038/onc.2010.238
8. Olsen BB, Wang SY, Svenstrup TH, Chen BP, Guerra B (2012)Protein kinase CK2 localizes to sites of DNA double-strand breakregulating the cellular response to DNA damage. BmcMol Biol 13:7.doi:10.1186/1471-2199-13-7
9. Costantini S, Woodbine L, Andreoli L, Jeggo PA, Vindigni A (2007)Interaction of the Ku heterodimer with the DNA ligase IV/Xrcc4
complex and its regulation by DNA-PK. DNA Repair (Amst) 6(6):712–722. doi:10.1016/j.dnarep.2006.12.007
10. Hartley KO, Gell D, Smith GCM, Zhang H, Divecha N,Connelly MA, Admon A, Leesmiller SP, Anderson CW,Jackson SP (1995) DNA-Dependent Protein-Kinase CatalyticSubunit - a Relative of Phosphatidylinositol 3-Kinase and theAtaxia-Telangiectasia Gene-Product. Cell 82(5):849–856. doi:10.1016/0092-8674(95)90482-4
11. Durocher D, Jackson SP (2001) DNA-PK, ATM and ATR as sensorsof DNA damage: variations on a theme? Curr Opin Cell Biol 13(2):225–231. doi:10.1016/S0955-0674(00)00201-5
12. Dragoi AM, Fu XY, Ivanov S, Zhang P, Sheng LB, Wu DQ, Li GC,ChuWM (2005) DNA-PKcs, but not TLR9, is required for activationof Akt by CpG-DNA. Embo J 24(4):779–789. doi:10.1038/sj.emboj.7600539
13. Stronach EA, ChenM,Maginn EN, Agarwal R, Mills GB, Wasan H,Gabra H (2011) DNA-PK Mediates AKT Activation and ApoptosisInhibition in Clinically Acquired Platinum Resistance. Neoplasia13(11):1069–U1114. doi:10.1593/Neo.111032
14. Begg AC, Stewart FA, Vens C (2011) GENOMIC INSTABILITY INCANCER Strategies to improve radiotherapy with targeted drugs.Nat Rev Cancer 11(4):239–253. doi:10.1038/Nrc3007
15. Fraser M, Leung BM, Yan XJ, Dan HC, Cheng JQ, Tsang BK (2003)p53 is a determinant of X-linked inhibitor of apoptosis protein/Akt-mediated chemoresistance in human ovarian cancer cells. Cancer Res63(21):7081–7088
16. Molina JR, Hayashi Y, Stephens C, Georgescu MM (2010) InvasiveGlioblastoma Cells Acquire Stemness and Increased Akt Activation.Neoplasia 12(6):453–U437. doi:10.1593/Neo.10126
17. Vivanco I, Sawyers CL (2002) The phosphatidylinositol 3-kinase-AKT pathway in human cancer. Nat Rev Cancer 2(7):489–501. doi:10.1038/Mrc839
18. Bozulic L, Surucu B, Hynx D, Hemmings BA (2008) PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand breakresponse and promotes survival. Mol Cell 30(2):203–213. doi:10.1016/j.molcel.2008.02.024
19. Feng JH, Park J, Cron P, Hess D, Hemmings BA (2004)Identification of a PKB/Akt hydrophobic motif Ser-473 kinase asDNA-dependent protein kinase. J Biol Chem 279(39):41189–41196.doi:10.1074/jbc.M406731200
20. Kim CH, Park SJ, Lee SH (2002) A targeted inhibition of DNA-dependent protein kinase sensitizes breast cancer cells followingionizing radiation. Journal of Pharmacology and ExperimentalTherapeutics 303(2):753–759. doi:10.1124/jpet.102.038505
21. Pajonk F, van Ophoven A, Weissenberger C, McBride WH (2005)The proteasome inhibitor MG-132 sensitizes PC-3 prostate cancercells to ionizing radiation by a DNA-PK-independent mechanism.BMC Cancer 5:76. doi:10.1186/1471-2407-5-76
22. Martin NMB (2001) DNA repair inhibition and cancer therapy. JPhotoch Photobio B 63(1–3):162–170
23. Murr MDE, Cano C, Golding BT, Hardcastle IR, Hummersome M,FrigerioM, Curtin NJ,Menear K, RichardsonC, Smith GCM,GriffinRJ (2008) 8-biarylchromen-4-one inhibitors of the DNA-dependentprotein kinase (DNA-PK). Bioorg Med Chem Lett 18(17):4885–4890. doi:10.1016/j.bmcl.2008.07.066
24. Yano H, Agatsuma T, Nakanishi S, Saitoh Y, Fukui Y, Nonomura Y,Matsuda Y (1995) Biochemical and Pharmacological Studies withKt7692 and Ly294002 on the Rare of Phosphatidylinositol 3-Kinasein Fc-Epsilon-Ri-Mediated Signal-Transduction. Biochem J 312:145–150
25. Poh TW, Pervaiz S (2005) LY294002 and LY303511 sensitize tumorcells to drug-induced apoptosis via intracellular hydrogen peroxideproduction independent of the phosphoinositide 3-kinase-akt pathway.Cancer Res 65(14):6264–6274. doi:10.1158/0008-5472.Can-05-0152
26. Dibiase SJ, Zeng ZC, Chen R, Hyslop T, CurranWJ, Iliakis G (2000)DNA-dependent protein kinase stimulates an independently active,
Invest New Drugs (2014) 32:424–435 433

nonhomologous, end-joining apparatus. Cancer Res 60(5):1245–1253
27. Rosenzweig KE, Youmell MB, Palayoor ST, Price BD (1997)Radiosensitization of human tumor cells by the phosphatidylinositol3-kinase inhibitors Wortmannin and LY294002 correlates with inhi-bition of DNA-dependent protein kinase and prolonged G(2)-Mdelay. Clinical Cancer Research 3(7):1149–1156
28. Hollick JJ, Golding BT, Hardcastle IR, Martin N, Richardson C,Rigoreau LJ, Smith GC, Griffin RJ (2003) 2,6-disubstitutedpyran-4-one and thiopyran-4-one inhibitors of DNA-Dependentprotein kinase (DNA-PK). Bioorg Med Chem Lett 13(18):3083–3086
29. Zhao Y, Thomas HD, Batey MA, Cowell IG, Richardson CJ, GriffinRJ, Calvert AH, Newell DR, Smith GCM, Curtin NJ (2006)Preclinical evaluation of a potent novel DNA-dependent proteinkinase inhibitor NU7441. Cancer Res 66(10):5354–5362. doi:10.1158/0008-5472.Can-05-4275
30. Mukherjee B, Tomimatsu N, Amancherla K, Camacho CV,Pichamoorthy N, Burma S (2012) The Dual PI3K/mTOR InhibitorNVP-BEZ235 Is a Potent Inhibitor of ATM- and DNA-PKCs-Mediated DNA Damage Responses. Neoplasia 14(1):34–U53. doi:10.1593/Neo.111512
31. Ihmaid SK, Al-Rawi JMA, Bradley CJ, Angove MJ, Robertson MN(2012) Synthesis, DNA-PK inhibition, anti-platelet activity studies of2-(N-substituted-3-aminopyridine)-substituted-1,3-benzoxazines andDNA-PK and PI3K inhibition, homology modelling studies of 2-morpholino-(7,8-di and 8-substituted)-1,3-benzoxazines. Eur J MedChem 57:85–101. doi:10.1016/j.ejmech.2012.08.035
32. Lee CM, Fuhrman CB, Planelles V, Peltier MR, Gaffney DK,Soisson AP, Dodson MK, Tolley HD, Green CL, Zempolich KA(2006) Phosphatidylinositol 3-kinase inhibition by LY294002radiosensitizes human cervical cancer cell lines. Clin Cancer Res12(1):250–256. doi:10.1158/1078-0432.Ccr-05-1084
33. Izzard RA, Jackson SP, Smith GCM (1999) Competitive and non-competitive inhibition of the DNA-dependent protein kinase. CancerRes 59(11):2581–2586
34. Chaussade C, Rewcastle GW, Kendall JD, Denny WA, Cho K,Gronning LM, Chong ML, Anagnostou SH, Jackson SP, DanieleN, Shepherd PR (2007) Evidence for functional redundancy of classIA PI3K isoforms in insulin signalling. Biochem J 404:449–458. doi:10.1042/Bj20070003
35. Veuger SJ, Curtin NJ, Richardson CJ, Smith GCM, Durkacz BW(2003) Radiosensitization and DNA repair inhibition by the com-bined use of novel inhibitors of DNA-dependent protein ki-nase and poly(ADP-ribose) polymerase-1. Cancer Res 63(18):6008–6015
36. Bromley R, Oliver L, Davey R, Harvie R, Baldock C (2009)Predicting the clonogenic survival of A549 cells after modulated x-ray irradiation using the linear quadratic model. Phys Med Biol54(2):187–206. doi:10.1088/0031-9155/54/2/002
37. Bromley R, Davey R, Oliver L, Harvie R, Baldock C (2006) Apreliminary investigation of cell growth after irradiation using amodulated x-ray intensity pattern. Phys Med Biol 51(15):3639–3651. doi:10.1088/0031-9155/51/15/003
38. Buch K, Peters T, Nawroth T, Sanger M, Schmidberger H, LangguthP (2012) Determination of cell survival after irradiation viaclonogenic assay versus multiple MTTAssay - A comparative study.Radiat Oncol 7. doi:Artn 1 Doi 10.1186/1748-717x-7-1
39. Pauwels B, Korst AEC, de Pooter CMJ, Pattyn GGO, LambrechtsHAJ, BaayMFD, Lardon F, Vermorken JB (2003) Comparison of thesulforhodamine B assay and the clonogenic assay for in vitro che-moradiation studies. Cancer Chemoth Pharm 51(3):221–226. doi:10.1007/s00280-002-0557-9
40. Zhou Y, Gwadry FG, Reinhold WC,Miller LD, Smith LH, Scherf U,Liu ET, Kohn KW, Pommier Y, Weinstein JN (2002) Transcriptionalregulation of mitotic genes by camptothecin-induced DNA damage:
Microarray analysis of dose- and time-dependent effects. Cancer Res62(6):1688–1695
41. Vlahos CJ, Matter WF, Hui KY, Brown RF (1994) A specific inhib-itor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). The Journal of biologicalchemistry 269(7):5241–5248
42. Wang TS, Kuo CF, Jan KY, Huang HM (1996) Arsenite inducesapoptosis in Chinese hamster ovary cells by generation of reactiveoxygen species. J Cell Physiol 169(2):256–268. doi:10.1002/(Sici)1097-4652(199611)169:2<256::Aid-Jcp5>3.0.Co;2-N
43. Yuan XL, Shan YJ, Zhao ZH, Chen JP, Cong YW (2005) G0/G1arrest and apoptosis induced by SARS-CoV 3b protein in transfectedcells. Virol J 2. doi:Artn 66 Doi 10.1186/1743-422x-2-66
44. Frankfurt O, Krishan A (2008) Application of Anti-ssDNAMonoclonal Antibody to Study Exogenous and Apoptosis-Associated DNA Damage. Cytom Part A 73A(12):1114–1115. doi:10.1002/Cyto.A.20629
45. Liang K, Lu Y, Jin WD, Ang KK, Milas L, Fan Z (2003)Sensitization of breast cancer cells to radiation by trastuzumab.Mol Cancer Ther 2(11):1113–1120
46. Nutley BP, Smith NF, Hayes A, Kelland LR, Brunton L, Golding BT,Smith GCM, Martin NMB, Workman P, Raynaud FI (2005)Preclinical pharmacokinetics and metabolism of a novel prototypeDNA-PK inhibitor NU7026. Brit J Cancer 93(9):1011–1018. doi:10.1038/sj.bjc.6602823
47. Munck JM, Batey MA, Zhao Y, Jenkins H, Richardson CJ, Cano C,Tavecchio M, Barbeau J, Bardos J, Cornell L, Griffin RJ, Menear K,Slade A, Thommes P, Martin NMB, Newell DR, Smith GCM, CurtinNJ (2012) Chemosensitization of Cancer Cells by KU-0060648, aDual Inhibitor of DNA-PK and PI-3K.Mol Cancer Ther 11(8):1789–1798. doi:10.1158/1535-7163.Mct-11-0535
48. Redon C, Boon C, Johnson K, Bonner WM, Rogakou EP (1999)Megabase chromatin domains involved in DNA double-strandbreaks in vivo. Mol Biol Cell 10:282a–282a
49. Wrann S, Kaufmann MR, Wirthner R, Stiehl DP, Wenger RH (2013)HIF mediated and DNA damage independent histone H2AX phos-phorylation in chronic hypoxia. Biol Chem 394(4):519–528. doi:10.1515/hsz-2012-0311
50. Ismail IH, Wadhra TI, Hammarsten O (2007) An optimized methodfor detecting gamma-H2AX in blood cells reveals a significantinterindividual variation in the gamma-H2AX response amonghumans. Nucleic Acids Res 35 (5). doi:Artn E36 Doi 10.1093/Nar/Gkl1169
51. Prendergast AM, Cruet-Hennequart S, Shaw G, Barry FP, Carty MP(2011) Activation of DNA damage response pathways in humanmesenchymal stem cells exposed to cisplatin or gamma-irradiation.Cell Cycle 10(21):3768–3777. doi:10.4161/cc.10.21.17972
52. Wang JH, Pluth JM, Cooper PK, Cowan MJ, Chen DJ, Yannone SM(2005) Artemis deficiency confers a DNA double-strand break repairdefect and Artemis phosphorylation status is altered byDNA damageand cell cycle progression. DNA Repair (Amst) 4(5):556–570. doi:10.1016/j.dnarep.2005.02.001
53. Francisco DC, Peddi P, Hair JM, Flood BA, Cecil AM, KalogerinisPT, Sigounas G, Georgakilas AG (2008) Induction and processing ofcomplex DNA damage in human breast cancer cells MCF-7 andnonmalignant MCF-10A cells. Free Radical Bio Med 44(4):558–569. doi:10.1016/j.freeradbiomed.2007.10.045
54. MacPhail SH, Banath JP, Yu TY, Chu EHM, Lambur H, Olive PL(2003) Expression of phosphorylated histone H2AX in cultured celllines following exposure to X-rays. Int J Radiat Biol 79(5):351–358.doi:10.1080/0955300032000093128
55. Langland GT, Yannone SM, Langland RA, Nakao A, Guan Y, LongSB, Vonguyen L, Chen DJ, Gray JW, Chen F (2010) Radiosensitivityprofiles from a panel of ovarian cancer cell lines exhibiting geneticalterations in p53 and disparate DNA-dependent protein kinase ac-tivities. Oncol Rep 23(4):1021–1026
434 Invest New Drugs (2014) 32:424–435

56. Urushihara Y, Kobayashi J, Matsumoto Y, Komatsu K, Oda S,MitaniH (2012) DNA-PK inhibition causes a low level of H2AX phosphor-ylation and homologous recombination repair in Medaka (Oryziaslatipes) cells. Biochem Biophys Res Commun 429(3–4):131–136.doi:10.1016/j.bbrc.2012.10.128
57. Yu M, Dai J, Huang WW, Jiao Y, Liu L, Wu M, Tan DY (2011)hMTERF4 knockdown in HeLa cells results in sub-G1 cell accumu-lation and cell death. Acta Bioch Bioph Sin 43(5):372–379. doi:10.1093/Abbs/Gmr020
58. Sun PC, Tzao C, Chen BH, Liu CW, Yu CP, Jin JS (2010)Suberoylanilide hydroxamic acid induces apoptosis and sub-G1arrest of 320 HSR colon cancer cells. J Biomed Sci 17. doi:Artn 76Doi 10.1186/1423-0127-17-76
59. Kunkl A, Terranova MP, Ferlini C, Astegiano G, Mazzarello G,Scambia G, Fattorossi A (2000) Detection of apoptotic T lympho-cytes in peripheral blood of human immunodeficiency virus (HIV)-infected subjects by Apostain. Cytometry 42(1):67–73. doi:10.1002/(Sici)1097-0320(20000215)42:1<67::Aid-Cyto10>3.0.Co;2–1
60. Bressenot A, Pooya S, Bossenmeyer-Pourie C, Gauchotte G,Germain A, Chevaux JB, Coste F, Vignaud JM, Gueant JL, Peyrin-Biroulet L (2013) Methyl donor deficiency affects small-intestinaldifferentiation and barrier function in rats. Brit J Nutr 109(4):667–677. doi:10.1017/S0007114512001869
61. Shawi M, Chu TW, Martinez-Marignac V, Yu Y, Gryaznov SM,Johnston JB, Lees-Miller SP, Assouline SE, Autexier C, Aloyz R(2013) Telomerase Contributes to Fludarabine Resistance in PrimaryHuman Leukemic Lymphocytes. Plos One 8 (7). doi:ARTN e70428DOI 10.1371/journal.pone.0070428
62. Amrein L, Loignon M, Goulet AC, Dunn M, Jean-Claude B,Aloyz R, Panasci L (2007) Chlorambucil cytotoxicity in malig-nant B lymphocytes is synergistically increased by 2-(morpholin-
4-yl)benzo-[h]chomen-4-one (NU7026)-mediated inhibition ofDNA double-strand break repair via inhibition of DNA-dependent protein kinase. J Pharmacol Exp Ther 321(3):848–855. doi:10.1124/jpet.106.118356
63. Hsu FM, Zhang S, Chen BP (2012) Role of DNA-dependentprotein kinase catalytic subunit in cancer development and treatment.Transl Cancer Res 1(1):22–34. doi:10.3978/j.issn.2218-676X.2012.04.01
64. Chen BPC, Chan DW, Kobayashi J, Burma S, Asaithamby A,Morotomi-Yano K, Botvinick E, Qin J, Chen DJ (2005) Cell cycledependence of DNA-dependent protein kinase phosphorylation inresponse to DNA double strand breaks. J Biol Chem 280(15):14709–14715. doi:10.1074/jbc.M408827200
65. Chen BPC, Uematsu N, Kobayashi J, Lerenthal Y, Krempler A,Yajima H, Lobrich M, Shiloh Y, Chen DJ (2007) Ataxia telangiecta-sia mutated (ATM) is essential for DNA-PKcs phosphorylations atthe Thr-2609 cluster upon DNA double strand break. J Biol Chem282(9):6582–6587. doi:10.1074/jbc.M611605200
66. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM (2005)Phosphorylation and regulation of Akt/PKB by the rictor-mTORcomplex. Science 307(5712):1098–1101. doi:10.1126/science.1106148
67. Brazil DP, Hemmings BA (2001) Ten years of protein kinase Bsignalling: a hard Akt to follow. Trends Biochem Sci 26(11):657–664
68. Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY,Moffat J, Brown M, Fitzgerald KJ, Sabatini DM (2006) Ablation inmice of themTORC components raptor, rictor, ormLST8 reveals thatmTORC2 is required for signaling to Akt-FOXO and PKC alpha butnot S6K1. Dev Cell 11(6):859–871. doi:10.1016/j.devcel.2006.10.007
Invest New Drugs (2014) 32:424–435 435