reactions of the class ii peroxidases, lignin peroxidase ... · compound i reduction steps have...
TRANSCRIPT

1
Reactions of the Class II Peroxidases, Lignin Peroxidase and Arthromyces
ramosus Peroxidase, with Hydrogen Peroxide: Catalase-Like Activity,
Compound III Formation and Enzyme Inactivation*
Alexander N. P. Hiner‡, Josefa Hernández Ruiz‡, José Neptuno Rodríguez López§,
Francisco García Cánovas§, Nigel C. Brisset||, Andrew T. Smith||, Marino B. Arnao‡
and Manuel Acosta‡**
From the ‡Departamento de Biología Vegetal (Fisiología Vegetal) and the §Departamento de
Bioquímica y Biología Molecular-A, Universidad de Murcia, E-30100 Espinardo, (Murcia)
Spain, and the ||School of Biological Sciences, University of Sussex, Brighton, Sussex. BN1
9QG, UK.
Copyright 2002 by The American Society for Biochemistry and Molecular Biology, Inc.
JBC Papers in Press. Published on April 30, 2002 as Manuscript M200002200 by guest on M
arch 31, 2020http://w
ww
.jbc.org/D
ownloaded from

2
Footnotes to the text.
*This work was supported in part by a grant from the EU Training and Mobility of
Researchers Programme-TMR Network: “Peroxidases in Agriculture, the Environment and
Industry” (Contract FMRX-CT98-0200), EU COST D21/0004/00, and Comisión
Interministerial de Ciencia y Tecnología (Spain): CICYT-ALI98-0524. A.N.P.H. is a TMR
network young researcher. J.H.R. has a grant from CajaMurcia (Spain). N.C.B. is supported
by an EU RTD Network grant (01LK3-1999-00590) to A.T.S. The costs of publication of this
article were defrayed in part by the payment of page charges. This article must therefore be
hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate
this fact.
**To whom correspondence should be addressed. Tel: +34 968 364940; Fax: +34 968
363963; E-mail: [email protected]
1The abbreviations used are: LiP, lignin peroxidase; ARP, Arthromyces ramosus
peroxidase; CiP, Coprinus cinereus peroxidase; MnP, manganese peroxidase; HRP,
horseradish peroxidase; APX, ascorbate peroxidase; CcP, cytochrome c peroxidase; H2O2,
hydrogen peroxide; m-CPBA, m-chloroperoxybenzoic acid; ABTS, 2,2′-azino-bis-(3-
ethylbenzthiazoline-6-sulfonic acid); O2· -, superoxide radical; SOD, superoxide dismutase;
TNM, tetranitromethane; Mn2+, manganous ion.
Running Title.
LiP and ARP reactions with H2O2
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

3
The reactions of the fungal enzymes Arthromyces ramosus peroxidase (ARP) and
Phanerochaete chrysosporium lignin peroxidase (LiP) with hydrogen peroxide (H2O2) have
been studied. Both enzymes exhibited catalase activity with hyperbolic H2O2 concentration
dependence (KM≈8-10mM, kcat≈1-3s-1). The catalase and peroxidase activities of LiP were
inhibited within 10 minutes and those of ARP in 1 hour. The inactivation constants were
calculated using two independent methods; LiP, ki≈19×10-3s-1; ARP, ki≈1.6×10-3s-1.
Compound III (oxyperoxidase) was detected as the majority species after addition of H2O2 to
LiP or ARP, its formation was accompanied by loss of enzyme activity. A reaction scheme is
presented that rationalizes the turnover and inactivation of LiP and ARP with H2O2. A similar
model is applicable to horseradish peroxidase. The scheme links catalase and compound III-
forming catalytic pathways, and inactivation at the level of the compound I-H2O2 complex.
Inactivation does not occur from compound III. All peroxidases studied to date are sensitive
to inactivation by H2O2 and it is suggested that the model will be generally applicable to
peroxidases of the plant, fungal and prokaryotic superfamily.
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

4
Peroxidases (donor: hydrogen peroxide oxidoreductases) are ubiquitous enzymes that
catalyze the oxidation of substrate at the expense of hydrogen peroxide (H2O2)1. The heme
peroxidases have been classified into two distinct groups, termed the animal (found only in
animals) and plant (found in plants, fungi and prokaryotes) superfamilies (1). The plant
peroxidases, which share similar overall protein folds and specific features, such as
catalytically essential histidine and arginine residues in their active sites, have been sub-
divided into three classes on the basis of sequence comparison (2, 3). Class I are intracellular
enzymes including yeast cytochrome c peroxidase (CcP), ascorbate peroxidase (APX) from
plants, and bacterial gene-duplicated catalase-peroxidases (4). Class III contains the secretory
plant peroxidases such as those from horseradish (HRP), barley or soybean. These
peroxidases seem to be biosynthetic enzymes involved in processes such as plant cell wall
formation and lignification. Class II consists of the secretory fungal peroxidases such as
lignin peroxidase (LiP) from Phanerochaete chrysosporium, manganese peroxidase (MnP)
from the same source, and Coprinus cinereus peroxidase (CiP) or Arthromyces ramosus
peroxidase (ARP), which have been shown to be essentially identical in both sequence and
properties (5). The main role of class II peroxidases appears to be the degradation of lignin in
wood.
All peroxidases so far studied share much the same catalytic cycle that proceeds in
three distinct and essentially irreversible steps (6), and is often referred to as the ‘peroxidase
ping-pong’. The resting ferric enzyme reacts with H2O2 in a two-electron process to generate
the intermediate known as compound I. Compound I is discharged in two sequential single-
electron reactions with reducing substrate yielding radical products, which are often highly
reactive, and water. The first reduction step results in the formation of another enzyme
intermediate, compound II. In the final step compound II is reduced back to ferric
peroxidase.
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

5
The ‘peroxidase ping-pong’ provides an adequate description of the peroxidase
reaction; however, continuing work has revealed some limitations of the basic model. The
compound I reduction steps have been shown to consist of reversible substrate binding
followed by substrate oxidation (7). The formation and nature of compound I has been
intensively studied. Both a neutral peroxidase-peroxide complex and a charged complex
(known as compound 0) have been observed (8), and variations have been identified in the
electronic structures of the compound Is of different peroxidases (9-14). Furthermore, certain
peroxidases have been found to utilize H2O2 to reduce compound I when no other substrate is
available. Foremost amongst the enzymes capable of this are the catalase-peroxidases, which
act as highly efficient catalases (kcat/KM ≈ 106 M-1 s-1) (15, 16). Unexpectedly, APX appears
incapable of turnover in the presence of only H2O2 (17) despite also being classified in class
I. HRP, from class III, has been shown to possess catalase activity, albeit with much lower
efficiency (kcat/KM ≈ 102-103 M-1 s-1) than the catalase-peroxidases (18), but to our
knowledge, no data are available on catalase activity in class II peroxidases. HRP compound
I can also carry out a single-electron reduction of H2O2 to generate compound II and
superoxide radical (O2· -). Reaction of compound II with more H2O2 yields compound III (a
complex between ferric peroxidase and O2· - also known as oxyperoxidase).
Catalase activity and compound III formation result in enzyme turnover in the
absence of normal reducing substrates, however, a further reaction with H2O2 has been
identified that leads to progressive irreversible enzyme inactivation. In the case of HRP, the
complex between compound I and peroxide ([compound I•H2O2]) has been unambiguously
identified as the pivotal point connecting the three simultaneous pathways (19, 20). Thus, the
‘peroxidase ping-pong’ mechanism can be replaced by an alternative catalytic cycle (Scheme
I) when no substrate other than H2O2 is present.
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

6
Studies on the susceptibility of different peroxidases to H2O2 suggest that all will be
sensitive to inactivation to a greater or lesser extent. Even catalase-peroxidase loses activity
after repeated exposure to H2O2 (21) with inactivation suggested to occur at the level of the
[compound I•H2O2] complex. Previous studies on LiP compound III found comparatively
facile degradation of the heme by H2O2 (that resulted in loss of activity), but aspects of this
process proved rather contentious (22-27). A reaction model was developed that suggested
that the inactivation of LiP might occur from compound III or a modified compound III
species. (24).
Following on from our work with class I (12, 17) and class III peroxidases (18-20, 28-
38), we have now examined the class II enzymes, LiP and ARP. Apart from observing
catalase activity, compound III formation and inactivation in fungal peroxidases another
important aim of this work was to ascertain if a model for turn over in the presence of only
H2O2 that essentially unifies the behavior of all or most peroxidases may likely exist, just as
the ‘peroxidase ping-pong’ describes activity with reducing substrates even though these
substrates can be so varied. We have, therefore, systematically examined the enzymes using
a set of experimental procedures that have proved very successful for the determination of
the controlling kinetic parameters under such conditions.
Further impetus for the present study was provided by the important physiological
implications of the reactions of LiP under highly oxidizing conditions and because ARP is a
potentially important commercial alternative to HRP. Directed evolution techniques have
previously been applied to ARP (39) to produce an enzyme that is hyper resistant to
conditions of high pH, temperature and H2O2 concentrations. A better understanding of H2O2
dependent turnover and inactivation may point to new strategies to further enhance resistance
during applications.
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

7
EXPERIMENTAL PROCEDURES
Materials
Enzymes – Non-glycosylated recombinant lignin peroxidase isoenzyme H8 (LiP) from
Phanerochaete chrysosporium was expressed in Escherichia coli, the polypeptide was
recovered from inclusion bodies and refolded in vitro in the presence of heme, calcium ions
and glutathione (40). After purification the preparation had an RZ (A409 nm / A280 nm) = 1.8 and
was homogeneous by SDS/PAGE. Recombinant LiP has so far has exhibited identical
behavior to the natural fungal enzyme (40, 41). Arthromyces ramosus peroxidase (ARP, lot
48H0555, RZ (A405 nm / A275 nm) = 2.5) and horseradish peroxidase (type IX, RZ (A403 nm / A275
nm) = 3.2) were obtained from Sigma as lyophilized powders. The HRP preparation has been
characterized previously (33) by isoelectric focussing as a single band with pI 8.5 and
confirmed to be isoenzyme C. ARP was similarly characterized as a single peroxidase band
with pI 3.5. Enzyme concentrations were determined spectrophotometrically using ε403 nm =
100 mM-1 cm-1 (for HRP-C), ε409 nm = 168 mM-1 cm-1 (for LiP) and ε405 nm = 109 mM-1 cm-1
(for ARP). Bovine erythrocyte superoxide dismutase (SOD) (product code S-2515) was
purchased from Sigma as a lyophilized powder containing 4200 U mg-1.
Chemicals - Hydrogen peroxide (30 % by vol.) and buffer substances (analytical
reagent grade) were obtained from Merck. The concentration of H2O2 was determined using
ε240 nm = 43.6 M-1 cm-1. 2,2′-azino-bis-(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS) in the
form of the crystallized diammonium salt was supplied by Boehringer-Mannheim, its
concentration was measured spectrophotometrically using ε340 nm = 36 mM-1 cm-1.
Tetranitromethane (TNM) and manganese (II) chloride were from Aldrich. All solutions were
prepared using de-ionized water drawn from a Milli-Q system (Millipore).
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

8
Methods
Oxygen production - Oxygen production was measured using a Clark-type electrode
coupled to a Hansatech (Kings Lynn, Cambs., UK) CB1D oxygraph unit. The equipment was
calibrated using the tyrosinase / 4-tert-butylcatechol method (42). The temperature of the
reaction chamber was controlled at 25 ± 0.1ºC using a Haake circulating water bath. Nitrogen
was bubbled through the reaction medium to remove dissolved oxygen. A baseline rise in
oxygen concentration of less than 0.5 µM min-1 without enzyme was obtained. The reaction
medium (2 ml total volume) contained H2O2 at the appropriate concentrations (see results) in
the following buffers: 50 mM Na acetate (pH 3.0), 50 mM Na citrate, (pH: 4.5, 5.5) and 50
mM Na phosphate (pH: 6.5, 7.0, 7.5). The reactions were started by addition of peroxidase.
Additional reagents (Mn2+, TNM or SOD) were added as required. Two types of experiments
were performed using the system described.
Determination of the initial rate of H2O2-decomposing activity (V0) - Values of V0
were determined at short reaction times in the initial linear phase of oxygen production. The
initial linear phase lasted approximately 10 to 30 seconds, departure from linearity at later
times indicated that enzyme inactivation was being observed, and such data were therefore
not used in calculations. Values of Vmax and KM were obtained by non-linear regression of a
hyperbolic function to a plot of V0 against [H2O2] using the program SigmaPlot for Windows
(version 2, Jandel Scientific Software, San Rafael, CA, USA). These data could be fitted
using the Michaelis-Menten equation, which in this case takes the following form (18):
[ ] [ ] [ ][ ]222
22T32
OH
OHE
d
Od
+=
K
k
t (Eq. 1)
where 2M KK = and [ ]T3max EkV = (thus cat3 kk = ), the steps to which k3 and K2 refer being
shown in Scheme I.
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

9
Determination of the total oxygen produced in the reaction ([O2]∞) - The reaction end-
point was reached when no further O2 production was observed. In all experiments, the final
concentration of oxygen produced ([O2]∞) was much less than 0.24 mM (i.e. the oxygen
concentration in air-saturated medium at 25ºC).
Inactivation and the determination of residual activity - Peroxidase was inactivated at
25ºC in 100 µl incubations in similar buffers to those used in the oxygraph. Each incubation
contained enzyme (1 µM) and H2O2 at different concentrations (giving the desired
[H2O2]/[peroxidase] ratios). When the reaction was complete, the peroxidase activity was
measured spectrophotometrically by the increase in the absorbance at 414 nm (ε = 31.1 mM-1
cm-1) in an assay system comprising 10 mM ABTS and 5 mM H2O2 in 50 mM Na phosphate
buffer (pH 6.5). The residual enzyme activity (AR) (expressed as %) was taken as the activity
remaining at the end of the reaction (At) compared to the initial activity (A0). Assays were
recorded on a Perkin-Elmer Lambda-2S UV-vis spectrophotometer. The temperature was
controlled at 25 ± 0.1ºC using a Haake circulating water bath. The partition ratio (r, the
number of turnovers with H2O2 before the enzyme was inactivated; ( ) i3i43 kkkkkr ≈+= ,
since k3 >> k4 (18)) was calculated by inserting the value of [H2O2]/[peroxidase] at which %
AR = 0 in graphical representations of % AR against [H2O2]/[peroxidase] into the equation
(18):
[ ][ ]0
22R E
OH
22
11
+−=
rA (Eq. 2)
Kinetics of inactivation - The inactivation of ARP was followed against time at pH
7.5. ARP (1 µM) was incubated with H2O2 (0, 1, 2, 5, 10, 20 and 50 mM). At appropriate
times after addition of the enzyme, aliquots were removed and the activity with ABTS was
determined. The conditions used were such that the [H2O2] added with the enzyme aliquot,
which varied with time as inactivation proceeded, did not affect the assayed activity. The AR
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

10
(%) was calculated for each time point and H2O2 concentration. The data were plotted and
fits to a first-order exponential decay to obtain values of kobs (observed rate constants of
inactivation) were made using SigmaPlot. The rate constant of inactivation (ki) and
dissocation constant (Ki) were calculated from a hyperbolic secondary plot of kobs against
[H2O2] using the Michaelis-Menten equation in the form (17):
[ ][ ]22I
22iobs OH
OH
+=
K
kk (Eq. 3)
Spectral changes on addition of H2O2 - The UV-vis spectral changes that occurred
over time to peroxidase (LiP or ARP) upon addition of H2O2 (100 or 500 equivalents
respectively) were observed, at pH 3.0 and pH 7.0, using a Perkin-Elmer Lambda-2S
spectrophotometer. The reaction of LiP (0.8 µM) with H2O2 (5 µM) was measured at pH 4.0
and pH 6.0 in stopped-flow experiments done on an Applied Photophysics (Leatherhead,
Surrey, UK) SX18.MV stopped-flow instrument fitted with a PDA.1 photodiode array
detector. The temperature was controlled at 25 ± 0.1ºC using a Neslab circulating water bath.
The multivariate data sets from the stopped-flow were fitted globally using the program
Pro/K (Applied Photophysics).
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

11
RESULTS
Catalase activities of LiP and ARP - Both LiP and ARP showed clear catalase
activity. The H2O2 concentration dependence of the initial rates (V0) of O2 production by the
two fungal enzymes, shown in Fig. 1, demonstrated that both exhibited saturation kinetics.
From the curves, values of K2 (KM) and k3 (kcat) (see Scheme I) could be obtained using Eq.
1. Panel A shows the saturation curve for LiP with K2 = 8.6 ± 0.4 mM and k3 = 2.87 ± 0.21 s-
1, in panel B the corresponding values for ARP were K2 = 10.2 ± 2.3 mM and k3 = 1.15 ±
0.09 s-1. Thus both LiP and ARP had kinetic parameters for O2 generation of a similar order
of magnitude to those previously determined for HRP-C (18) and HRP-A2 (35). However,
during experiments it was noticed that the values of V0 for LiP were maintained for only a
very short time and that O2 production essentially ceased in approximately 10 mins, whereas
for ARP it took about 1 hour for activity to be lost. This suggested that LiP was being
inactivated more rapidly than ARP.
Determinations of the total O2 gas produced by a given amount of enzyme before its
inactivation (i.e. the infinite product of the catalase reaction) confirmed that LiP lost activity
much more rapidly than ARP. The values of the partition ratios (r), obtained from the slopes
of the straight line fits to plots of [O2]∞ against [peroxidase] (not shown) (18), indicated that
LiP (r = 155 ± 25) was more sensitive to inactivation by H2O2 than ARP (r = 620 ± 60).
Additionally, using the relationship i3 kkr ≈ (18) (see Scheme I), it was calculated that the
inactivation constant of LiP (ki = (18.5 ± 2.3) × 10-3 s-1) was 10-fold higher than for ARP (ki
= (1.85 ± 0.18) × 10-3 s-1). Thus the ki of ARP was approximately equivalent to the value for
HRP-C (18) but LiP was inactivated much more rapidly (Table I).
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

12
Both ARP and LiP were more effective catalases at neutral than at acidic pH (data not
shown). The apparent pKA ≈ 6 observed for ARP was quite similar to the value with HRP-C
(18), suggesting the involvement of the conserved distal histidine residue, and that catalase
activity is an enzymatic reaction.
It has recently been shown that O2· - scavengers (SOD, TNM and Mn2+) have only
limited effects on HRP-C catalase activity (38, 43). Fig. 2 shows the effects of O2· -
scavengers on ARP. SOD and TNM, which generate O2 and H2O2 from O2· -, slightly
increased O2 production. In contrast Mn2+, which regenerates H2O2 but does not yield O2,
slightly reduced the level of O2 released. Thus it appears that ARP, like HRP-C, produces O2
in an enzymatic reaction and not, except for a very minor component, as the result of the
chemical disproportionation of O2· - in solution.
Inactivation of LiP and ARP - Incubation experiments over a range of H2O2
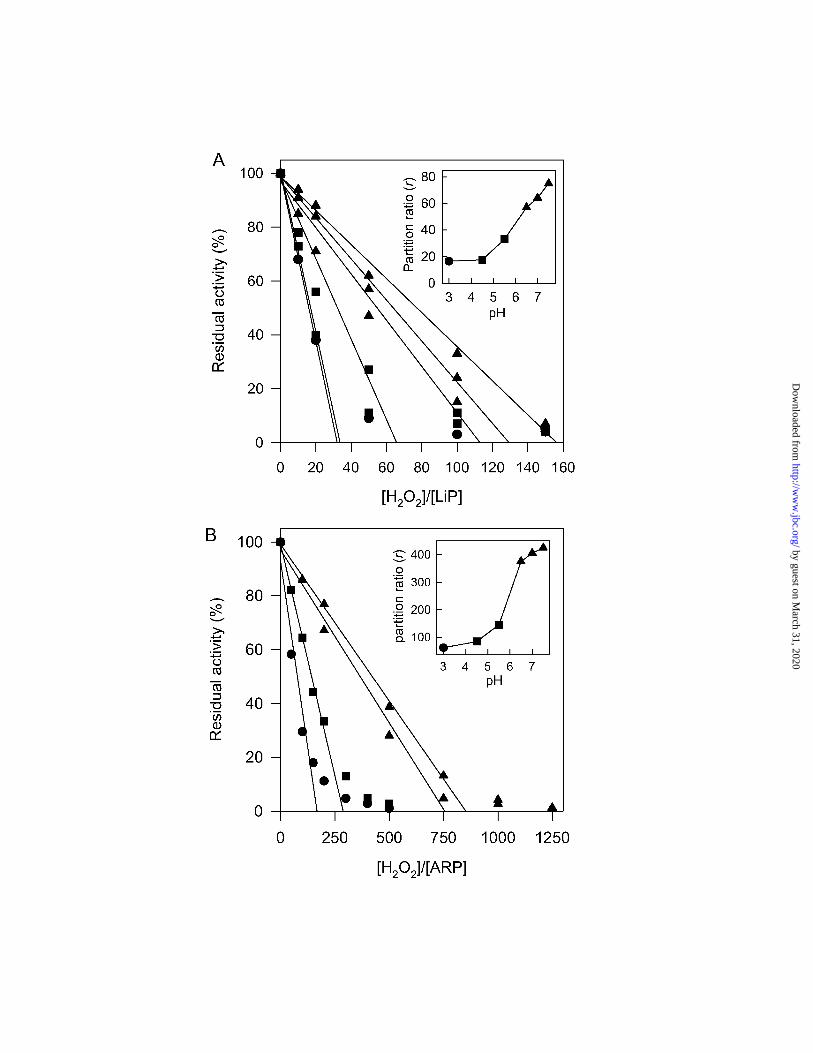
concentrations and pH values were used to further probe the inactivation of LiP and ARP. In
Fig. 3 it can be seen that LiP (panel A) was more sensitive to H2O2 than was ARP (panel B),
as expected from the O2 measurements above. The insets show that both enzymes were more
sensitive at acidic than at neutral pH. The values of r obtained here, calculated using Eq. 2,
were in reasonable agreement with those from oxygen measurements at the same pH (Table
I). This suggests that, as for HRP-C (18), the level of turnover through the catalase reaction is
strongly correlated with protection of the peroxidase against inactivation by H2O2.
The kinetics of inactivation were directly measured by following the time-dependent
fall in peroxidase activity, determined using ABTS, as a function of H2O2 concentration. In
Fig. 4 panel A the inactivation curves for ARP are shown, and in panel B the H2O2
concentration-dependent saturation curve from which values of ki = (1.37 ± 0.27) × 10-3 s-1
and KI = 3.83 ± 0.35 mM were calculated (see Eq. 3). The equivalent experiments performed
for LiP (data not shown) yielded values of ki = (20.0 ± 3.0) × 10-3 s-1 and KI = 6.1 ± 0.45
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

13
mM. In both cases the values of ki were in good agreement with those independently
calculated from the oxygraph data. The value of KI is related to the K2 obtained from oxygen
measurements. However, the longer periods needed for inactivation studies mean that the
constants controlling the compound III and inactivation pathways (Scheme I) have time to
affect the determination of KI whereas with the more rapid oxygraph method to obtain K2
these additional factors are insignificant. Thus, for this reason, KI < K2.
Spectral changes observed during the reaction of peroxidase with H2O2 - Upon
addition of 100 molar equivalents of H2O2 to a sample of LiP or 500 equivalents to ARP the
enzyme intermediate compound III was formed (Fig. 5 panel A, LiP; panel B, ARP)
accounting for a large proportion of the total enzyme present. The formation of compound III
was observed at both pH 7 (Fig. 5) and pH 3 (not shown). Repeat scans over time showed
that the concentration of compound III fell concomitant with a heme bleaching process. Both
compound IIIs were considerably less stable at acid pH. Heme bleaching proceeded without
the detectable accumulation of verdohemochrome P670 for either LiP or ARP.
Stopped-flow experiments (data not shown) using LiP (0.8 µM) and H2O2 (5 µM)
indicated the generation of compound I followed by the reduction of compound I to a species
with a compound II-like spectrum in a biphasic process with kfast = 2.3 s-1 (41). The heme in
this species then bleached in an H2O2 concentration-independent process with a rate k = 2.1 ×
10-3 s-1 at pH 4.0, rising to 9 × 10-3 s-1 at pH 6.0. Compound III was not observed.
The kinetic constants determined in the present study for LiP and ARP are compared
in Table I to those previously obtained for HRP-C.
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

14
DISCUSSION
The aim of this study was to examine in some detail the reactions of the class II
fungal peroxidases, LiP and ARP, with H2O2 in the absence of additional substrates. This has
involved the kinetic analysis of their catalase activity, inactivation reactions and compound
III formation using methods previously applied by us to the class I peroxidase APX (12, 17)
and the class III peroxidase HRP (18-20, 28-36, 38). In both these cases it was found that
H2O2 acted as a mechanism-based (suicide) inactivator (44, 45), however, important
differences were detected between these enzymes. HRP showed considerable catalase
activity (18, 35, 38) and a large stoichiometric excess of H2O2 was required for inactivation
(19). From the data that were amassed we formulated a reaction model for HRP plus H2O2
(Scheme I). In contrast, APX was extremely sensitive to inactivation (17), only 2-3
equivalents of H2O2 being required. To compound matters, APX did not exhibit a catalase
reaction. An alternative model was therefore developed, which in essence did not involve the
turnover of H2O2 but, significantly, retained a partition of the enzyme from [compound
I•H2O2] (17). In light of these distinct results for HRP and APX, we wished to establish
whether either (or even both) of these models was applicable to the class II peroxidases.
The data tend to indicate that Scheme I describes of the reactions of both LiP and
ARP better than the APX model (17). LiP and ARP showed catalase activity of a similar
magnitude to that of HRP (18, 35, 38), which has been shown to be the main pathway with
H2O2 protecting the enzyme from inactivation. One unexpected observation was that LiP
appeared to exhibit a higher initial velocity of O2 production than either ARP or HRP but that
this rate was sustained for only a short period resulting in a lower overall turnover of H2O2
and a correspondingly greater sensitivity to inactivation. The inactivation constant (ki),
determined from the catalase reaction and as the fall in peroxidase activity, of LiP was an
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

15
order of magnitude higher than those of ARP or HRP-C. Thus, the partition ratio between
turnover and inactivation ( ( ) i43 kkkr += - see Scheme I) was much lower for LiP. The pH
dependencies of inactivation (more sensitive at acid pH) and the catalase activity (lower at
acid pH), of LiP and ARP and the effects of O2· - scavengers on the catalase reaction were
essentially similar to HRP (38) suggesting the involvement of common active site residues
and a similar overall mechanism for all three enzymes. An additional point to note is that
LiP, during reaction with H2O2, undergoes an autocatalytic covalent modification of Trp 171
to a β-hydroxy adduct (13). This modification process may be connected to the spontaneous
reduction of LiP compound I to a compound II-like species (41) followed by heme bleaching.
It is therefore possible that at very low H2O2 concentrations and higher pH values, when
heme bleaching from the compound II-type material is more rapid (in contrast to the general
sensitivity to inactivation, suggesting that the mechanism is not the same), a proportion of
LiP activity loss occurs in this way. There may be parallels between this reaction of LiP and
the inactivation of APX (17) since it is unlikely that the catalase cycle of LiP will be active
with so little H2O2 and in neither example was compound III detected.
Upon addition of large stoichiometric excesses of H2O2 to LiP or ARP the majority of
the peroxidase was converted to compound III. A similar effect has been seen with HRP (19,
35). The formation of compound III under conditions where inactivation occurs has been
used in some studies with LiP and HRP as evidence that inactivation results from
modification of this intermediate (24, 43). However, computer simulations of Scheme I (done
using the rate constants available for HRP) showed that compound III could be present at
high concentrations even though inactivation proceeded from the [compound I•H2O2]
complex (38). This was because of the overwhelming proportion of catalytic turnover via the
catalase cycle (independent of compound III) and the low rate of reversion of compound III
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

16
to ferric peroxidase by loss of O2· -. The involvement of O2
· - in the degeneration of LiP
compound III to the ferric form has been demonstrated (26). The [compound I•H2O2]
complex was not seen in the spectra obtained with LiP or ARP in this study, however, the
absorption peak of this species is around 940-965 nm making its observation difficult.
Additionally, the amount of complex that accumulates towards the start of the reaction is
related to the affinity of compound I for peroxide. Nor were the inactive forms of the fungal
enzymes, compound P670, detected. This was probably due to the inherently unstable nature
of this material that easily loses iron to leave colorless products. The P670 species of APX
(17) and HRP-A2 (35) proved equally hard to detect and that of HRP-C was most stable at
10ºC (20).
The role of compound III in the activity of LiP has in the past been the subject of
some discussion (22-27). A particularly contentious issue has been the presence or absence
of a second compound III species, termed LiP III*. Two distinct compound IIIs have also
been postulated to exist in HRP (46) with the superoxide ligand either loosely or tightly
bound to the iron. In HRP-A2 (35) the more stable form (observed after 5 days in the
presence of excess H2O2) has been suggested to account for the presence of a highly H2O2
resistant fraction (≈ 20 %) of the peroxidase seen in plots of residual activity (% AR) against
[H2O2]/[HRP-A2], similar to those shown in Fig. 3. The data in this paper do not demonstrate
the existence of a particularly long lasting form of LiP compound III or of a resistant enzyme
fraction. The presence of LiP III* would not necessarily affect the inactivation profile unless
it was either stable and unreactive towards H2O2 or directly involved in the inactivation
pathway. The first of these possibilities was not supported by the spectroscopic data and the
second has been suggested previously (24) but can be discarded for the reasons discussed
previously. No spectral or kinetic evidence of a second form of ARP compound III was
found.
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

17
Summing up, a number of conclusions can be drawn from the data presented here.
LiP has generally been considered to be a fragile enzyme in the presence of H2O2, however,
it will, in fact, turn over H2O2 quite successfully when compared to APX. LiP was actually a
better catalase than either ARP or HRP, but, the higher rate constant of LiP inactivation led
to the greater sensitivity of this enzyme. Many studies (1 and references therein) with LiP
have tended to focus on its behavior at acid pH with veratryl alcohol (a secondary metabolite
of P. chrysosporium which, when oxidized by LiP, is capable of attacking lignin (1)). The pH
dependence of inactivation observed in this study suggests that LiP may be especially
vulnerable under such conditions implying that the fungus must maintain strict control over
the oxidant / reductant ratio in order to avoid inactivation of the peroxidase. The inactivation
profile of ARP, as well as its catalase activity, were quite similar to HRP-C suggesting that
the fungal enzyme could indeed be suitable for many applications normally performed by the
plant peroxidase, at least from the stand-point of its stability under oxidizing conditions. The
firm understanding of the effects of H2O2 on ARP, presented here, should allow continued
development of peroxide resistant mutants.
Finally, the general mechanistic model shown in Scheme I is applicable to
peroxidases from class II. Since class III and class II peroxidases essentially catalyze the
same reaction, but in opposite directions, this may justify the apparently closer mechanistic
similarities observed here compared to the class I enzymes. Differences in detail may exist in
the behavior of specific enzymes but the central motif of the scheme, the partition between
turnover and inactivation from [compound I•H2O2], appears in the reactions of peroxidases
from each of the three classes of the plant superfamily. Therefore, just as the ‘peroxidase
ping-pong’ represents the turnover of peroxidases with both an oxidizing and a reducing
substrates, the model shown in Scheme I provides a framework in which to understand the
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

18
reactivity of peroxidases with peroxides that we fully expect to be applicable to enzymes that
have not yet been studied in detail or that remain to be identified.
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

19
REFERENCES
1. Dunford, H. B. (1999) Heme Peroxidases, pp 281-308, Wiley-VCH, New York,
Chichester, Weinheim, Brisbane, Singapore, Toronto.
2. Welinder, K. G. (1985) Eur. J. Biochem. 151, 497-450.
3. Welinder, K. G. (1992) Curr. Opin. Struct. Biol. 2, 388-393.
4. Welinder, K. G. (1991) Biochim. Biophys. Acta 1080, 215-220.
5. Kjalke, M., Andersen, M. B., Schneider, P., Christensen, B., Schülein, M., and
Welinder, K. G. (1992) Biochim. Biophys. Acta 1120, 248-256.
6. Dunford, H. B. (1990) in Peroxidases in Chemistry and Biology (Everse, J., Everse,
K. E., and Grisham, M. B., eds.) pp 1-24, CRC Press, Boca Raton, FL.
7. Rodríguez-López, J. N., Gilabert, M. A., Tudela, J., Thorneley, R. N. F., and García-
Cánovas, F. (2000) Biochemistry 39, 13201-13209.
8. Rodríguez-López, J. N., Lowe, D. J., Hernández-Ruiz, J., Hiner, A. N. P., García-
Cánovas, F., and Thorneley, R. N. F. (2001) J. Amer. Chem. Soc. 123, 11838-11847.
9. Dolphin, D., Forman, A., Borg, D. C., Fajer, J., and Felton, R. H. (1971) Rec. Res.
Dev. Agric. Food Chem. 68, 614-618.
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

20
10. Yonetani, T., Schleyer, H., and Ehrenberg, A. (1966) J. Biol. Chem. 259, 13027-
13036.
11. Sivaraja, M., Goodin, D. B., Smith, M., and Hoffman, B. M. (1989) Science 245, 738-
740.
12. Hiner, A. N. P., Martínez, J. I., Arnao, M. B., Acosta, M., Turner, D. D., Raven, E.
L., and Rodríguez-López, J. N. (2001) Eur. J. Biochem. 268, 3091-3098.
13. Blodig, W., Doyle, W. A., Smith, A. T., Winterhalter, K., Choinowski, T., and
Piontek, K. (1998) Biochemistry 37, 8832-8838.
14. Converso, D. A., and Fernández, M. E. (1998) Arch. Biochem. Biophys. 357, 22-26.
15. Claiborne, A., and Fridovich, I. (1979) J. Biol. Chem. 254, 4245-4252.
16. Obinger, C., Regelsberger, G., Strasser, G., Burner, U., and Peschek, G. A. (1997)
Biochem. Biophys. Res. Commun. 235, 545-552.
17. Hiner, A. N. P., Rodriguez-Lopez, J. N., Arnao, M. B., Lloyd Raven, E., García-
Cánovas, F., and Acosta, M. (2000) Biochem. J. 348, 321-328.
18. Hernández-Ruiz, J., Arnao, M. B., Hiner, A. N. P., García-Cánovas, F., and Acosta,
M. (2001) Biochem. J. 354, 107-114.
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

21
19. Arnao, M. B., Acosta, M., del Rio, J. A., Varón, R., and García-Cánovas, F. (1990)
Biochim. Biophys. Acta 1041, 43-47.
20. Rodriguez-Lopez, J. N., Hernández-Ruiz, J., García-Cánovas, F., Thorneley, R. N. F.,
Acosta, M., and Arnao, M. B. (1997) J. Biol. Chem. 272, 5469-5476.
21. Obinger, C., Regelsberger, G., Pircher, A., Sevcik-Klöckler, A., Strasser, G., and
Peschek, G. A. (1999) in The Phototrophic Prokaryotes (Peschek, G. A., ed.) pp 719-
731, Kluwer Academic / Plenum Publishers, New York.
22. Wariishi, H., and Gold, M. H. (1989) FEBS Lett. 243, 165-168.
23. Cai, D., and Tien, M. (1989) Biochem. Biophys. Res. Commun. 162, 464-469.
24. Wariishi, H., and Gold, M. H. (1990) J. Biol. Chem. 265, 2070-2077.
25. Wariishi, H., Marquez, L. A., Dunford, H. B., and Gold, M. H. (1990) J. Biol. Chem.
265, 11137-11142.
26. Cai, D., and Tien, M. (1992) J. Biol. Chem. 267, 11149-11155.
27. Cai, D., and Tien, M. (1990) Biochemistry 29, 2085-2091.
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

22
28. Arnao, M. B., Acosta, M., del Rio, J. A., and García-Canovas, F. (1990) Biochim.
Biophys. Acta 1038, 85-89.
29. Acosta, M., Arnao, M. B., Hernández-Ruiz, J., and García-Cánovas, F. (1993) in
Plant Peroxidases: Biochemistry and Physiology. (Welinder, K. G., Rasmussen, S. K.,
Penel, C., and Greppin, H., eds.) pp 201-205, University of Geneva, Geneva.
30. Acosta, M., Arnao, M. B., del Rio, J. A., and García-Cánovas, F. (1991) in
Biochemical, Molecular and Physiological Aspects of Plant Peroxidases.
(Lobarzewski, H., Greppin, H., Penel, C., and Gaspar, T., eds.) pp 175-184,
University of Geneva, Geneva.
31. Acosta, M., Hernández-Ruiz, J., García-Cánovas, F., Rodríguez-López, J. N., and
Arnao, M. B. (1996) in Plant Peroxidases: Biochemistry and Physiology (Obinger, C.,
Burner, U., Ebermann, R., Penel, C., and Greppin, H., eds.) pp 76-81, University of
Geneva, Geneva.
32. Hiner, A. N. P., Hernández-Ruiz, J., García-Cánovas, F., Smith, A. T., Arnao, M. B.,
and Acosta, M. (1995) Eur. J. Biochem. 234, 506-512.
33. Hiner, A. N. P., Hernández-Ruiz, J., Arnao, M. B., García-Cánovas, F., and Acosta,
M. (1996) Biotechnol. Bioeng. 50, 655-662.
34. Arnao, M. B., Hernández-Ruiz, J., Varón, R., García-Cánovas, F., and Acosta, M.
(1995) J. Mol. Catal. A: Chem. 104, 179-191.
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

23
35. Hiner, A. N. P., Hernández-Ruiz, J., Rodríguez-López, J. N., Arnao, M. B., Varón,
R., García-Cánovas, F., and Acosta, M. (2001) J. Biol. Inorg. Chem. 6, 504-516.
36. Arnao, M. B., García-Cánovas, F., and Acosta, M. (1996) Biochem. Mol. Biol.
Internat. 39, 97-107.
37. Hernández-Ruiz, J., Rodríguez-López, J. N., García-Cánovas, F., Acosta, M., and
Arnao, M. B. (2000) Biochim. Biophys. Acta 1478, 78-88.
38. Hiner, A. N. P., Hernández-Ruiz, J., Williams, G. A., Arnao, M. B., García-Cánovas,
F., and Acosta, M. (2001) Arch. Biochem. Biophys. 392, 295-302.
39. Cherry, J. R., Lamsa, M. H., Schneider, P., Vind, J., Svendsen, A., Jones, A., and
Pedersen, A. H. (1999) Nat. Biotechnol. 17, 379-384.
40. Doyle, W. A., and Smith, A. T. (1996) Biochem. J. 315, 15-19.
41. Doyle, W. A., Blodig, W., Veitch, N. C., Piontek, K., and Smith, A. T. (1998)
Biochemistry 37, 15097-15105.
42. Rodríguez-López, J. N., Ros-Martínez, J. R., Varón, R., and García-Cánovas, F.
(1992) Anal. Biochem. 202, 356-360.
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

24
43. Baker, C. J., Deahl, K., Domek, J., and Orlandi, E. W. (2000) Arch. Biochem.
Biophys 382, 232-237.
44. Silverman, R. B. (1995) in Methods Enzymol. 249, 240-283.
45. García-Cánovas, F., Tudela, J., Varón, R., and Vazquez, A. M. (1989) J. Enz. Inhib.
3, 81-90.
46. Nakajima, R., and Yamazaki, I. (1987) J. Biol. Chem. 262, 2576-2581.
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

25
TABLE I
Rate constants for the catalase-like activity and inactivation of LiP and ARP compared
to those of HRP-C.
Enzyme LiP ARP HRP-C
Constants from oxygen
measurements
K2 (mM)a 8.6 ± 0.4 10.2 ± 2.3 4.0 ± 0.6d
k3 (s-1)a 2.87 ± 0.21 1.15 ± 0.09 1.78 ± 0.12d
rb 155 ± 25 620 ± 60 700 ± 100d
ki (× 10-3 s-1) 18.5 ± 2.3 1.85 ± 0.18 2.5 ± 0.2d
Constants from
inactivation studies
ki (× 10-3 s-1)c 20.0 ± 3.0 1.37 ± 0.27 3.92 ± 0.06e
KI (mM)c 6.1 ± 0.45 3.83 ± 0.35 1.3 ± 0.2f
rb 65 ± 15 425 ± 35 624 ± 40f
a see Eq. 1
b the partition ratio, ( ) i43 kkkr += ; where 43 kk >> , i3 kkr ≈
c see Eq. 3
d from ref. (18)
e from ref. (19)
f from ref. (32)
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

26
Legend to Scheme I.
SCHEME I. Mechanistic model of the reaction of peroxidase with H2O2 in the absence
of other substrates. E is native ferric peroxidase; S is H2O2; E′, E′′ and E′′′ are the enzyme
intermediates, compound I, II, III, respectively; E′S and E′′S are complexes between the
respective enzyme intermediates and H2O2, [compound I•H2O2] and [compound II•H2O2]; Ei
is inactive peroxidase. In the scheme, peroxidase exhibits two catalytic cycles (catalase and
compound III pathways) with distinct rate constants and stoichiometries and the inactivation
route to Ei, with the [compound I•H2O2] complex playing a central role. The reaction of
peroxidase with H2O2 proceeds in the transition phase until all the enzyme has been
inactivated or the substrate exhausted. The distribution of enzyme between the catalytic and
inactivating pathways is described by two partition ratios: i3cat kkr = (catalase activity /
inactivation) and i4coIII kkr = (compound III route / inactivation). A global partition ratio
may be defined: ( ) i43coIIIcat kkkrrr +=+= . In the case of HRP-C it has previously been
shown that coIIIcat rr >> (18, 30) and thus catrr ≈ . The data obtained in the present study
suggest that this relationship also applies to LiP and ARP. The ki for LiP was 10-fold higher
than that of ARP (or HRP-C) resulting in faster inactivation and a lower value of r for this
enzyme despite a somewhat higher value of k3. An additional factor that may contribute to
inactivation at low [H2O2] in the case of LiP is the spontaneous conversion of LiP compound
I to a compound II-like species followed by [H2O2]-independent heme bleaching.
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

27
Figure Legends
FIG. 1. H2O2 concentration dependence of catalase-like oxygen production by fungal
peroxidases. A, LiP (0.25 µM) and B, ARP (0.25 µM) on H2O2 concentration at pH 7.0 (50
mM Na phosphate buffer). Values of K2 and k3 were calculated from the data shown using
Eq. 1 in methods.
FIG. 2. Effect of superoxide radical scavengers on catalase-like oxygen production by
Arthromyces ramosus peroxidase. Superoxide dismutase (SOD, 50 nM), tetranitromethane
(TNM, 100 µM) or manganous ion (Mn2+, 100 µM) were added to the reaction medium in
the oxygraph. The concentration of ARP was 0.25 µM with 5 mM H2O2 in 50 mM Na
phosphate buffer, pH 7.0 at 25ºC.
FIG. 3. Sensitivity of the fungal peroxidases to inactivation by H2O2 in incubation
experiments. A, LiP and B, ARP at various pH values. Insets, Plots of the partition ratio (r)
against pH. The heme concentration was 1 µM with the appropriate number of equivalents of
H2O2 in 50 mM Na acetate (½), Na citrate (�) or Na phosphate (�). Incubations were
allowed to proceed to the end of reaction. Residual activity (AR) was measured using the
ABTS assay system. Values of r were calculated using Eq. 2 in methods.
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

28
FIG. 4. Inactivation kinetics of Arthromyces ramosus peroxidase. A, Time courses
determined with ARP (1 µM) and H2O2 (1 mM, »; 2 mM, ¹; 5 mM, i; 10 mM, �; 20
mM, �; 50 mM, ½) in 50 mM Na phosphate buffer, pH 7.5, and used to obtain values of
kobs. B, Plot of kobs against H2O2 concentration from which ki and KI were calculated using
Eq. 3 in methods.
FIG. 5. Spectral changes upon addition of H2O2 to the fungal peroxidases. A, 100
equivalents of H2O2 to LiP (2.4 µM) or B, 500 equivalents to ARP (4.1 µM) in 50 mM Na
phosphate buffer, pH 7.0. Repeat scans were obtained for 60 minutes. The expanded sections
show the visible spectra of ferric peroxidase and of compound III (C-III) detected soon after
H2O2 addition. The concentration of compound III fell with time due to the inactivation
process.
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from

E
S
E' E'S
O2
Ei
E''
S
E''SE'''
H2O
O2
S
k1 K2
k3
ki
k4
K5k6
H2O
H2O
-·
·-O2
k7
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from





García Cánovas, Nigel C. Brisset, Andrew T. Smith, Marino B. Arnao and Manuel AcostaAlexander N.P. Hiner, Josefa Hernández Ruiz, José Neptuno Rodríguez López, Francisco
and enzyme inactivationperoxidase, with hydrogen peroxide: Catalase-like activity, compound III formation
Reactions of the class II peroxidases, lignin peroxidase and arthromyces ramosus
published online April 30, 2002J. Biol. Chem.
10.1074/jbc.M200002200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on March 31, 2020
http://ww
w.jbc.org/
Dow
nloaded from