recent developments in textal phenix workshop berkeley sept. 2006 thomas r. ioerger texas a&m...
Post on 21-Dec-2015
218 views
TRANSCRIPT

Recent Developmentsin TEXTAL
Phenix WorkshopBerkeley
Sept. 2006
Thomas R. IoergerTexas A&M University

NCS Identification via Pattern Recognition
• Pai, R., Sacchettini, J.C. and Ioerger, T.R. (2006). Identifying non-crystallographic symmetry in protein electron-density maps: a feature-based approach. Acta Crystallographica, D62(9):1012-1021.
• The Problem:– Symmetry averaging can greatly improve phases.– Typical methods for finding NCS require ≥ 3 heavy
atoms, and are sensitive to errors in coordinates.– Despite noise and breaks from symmetry, similar
patterns of density exist over large regions of real space (even if imperfectly phased).
– How to efficiently identify these similarities and derive symmetry operators?

Our Approach to NCS• Step 1: calculate backbone using CAPRA
– Putative C-alpha atoms become centers of regions for initial matching
• Step 2: Calculate local features for each CA based on pattern of surround CA’s and density; select subset of candidates that are likely to be similar– Example features: #CAs, center of mass, moments of inertia,
std.dev., skewness, kurtosis…

• Step 3: Calculate local density correlation between each pair of CA’s (over 5A spheres), with rotation-optimization
• Step 4: Cluster pairs of matching regions with similar rotation matrices– How can you tell if two local transformations are
related (from same pair of domains)?– Each can transform the coordinates of the other.
Definition 1: similar rotation matrices. Given RUV and RPQ asrotation matrices that optimally superpose regions U and Vand regions P and Q, respectively, and u, v, p and q as thecoordinates of the centers of regions U, V, P and Q, respectively,then RUV is similar to RPQ if q RUV p ≤ 2 A° andu RPQ v ≤ 2 A°.
U VP
Q

• Step 5: Extend regions to molecular boundaries (excluding non-symmetric deviations)
• - caveat: doesn’t work for proper symmetry (can’t identify unique boundaries)
• Step 6: Organize and output N-1 operators
• (Step 7): Run DM to do symmetry-averaging

Protein Native reso (Å)
# NCS subunits
in ASU
Map corr.
# NCS subunits found
RMS of superposition
NCS-averaged map corr.
1a7a 2.8 2 0.845 2 0.670 0.859
1bkj 1.8 2 0.443 2 0.819 0.600
1l1e 2.8 2 0.505 2 0.739
2gmf 2.35 2 2 0.857
1f61 1.8 2 2 0.655
1nye 3 8 0.506 7 0.713, 0.757, 0.771, 0.819, 0.844, 0.917
1kwa 1.93 2 0.475 2 1.43 0.531
1l8w 2.3 4 0.454 4 0.82, 0.858, 1.09
1p32 2.25 3 3 0.801, 0.883
1nf2 3 3 0.313 3 0.954, 0.979
1ytt 1.8 2 0.667 2 0.780 0.692
Results on Experimental Maps
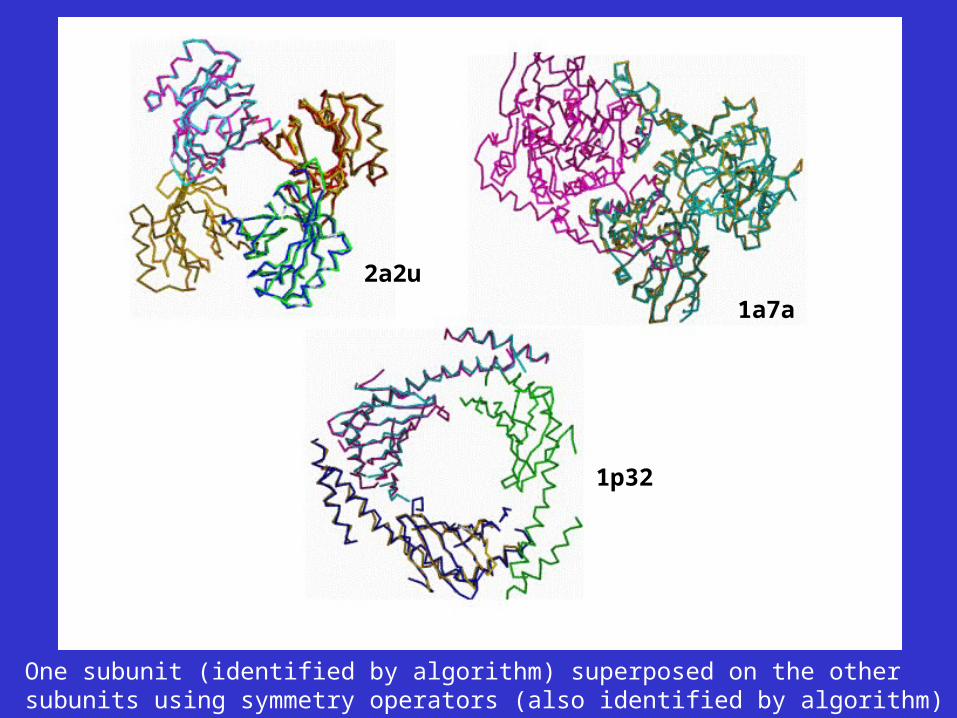
1p32
1a7a
2a2u
One subunit (identified by algorithm) superposed on the othersubunits using symmetry operators (also identified by algorithm)

Availability• Pattern Recognition Algorithm for NCS (by Reetal Pai,
PhD student in Ioerger lab)– Initial implementation in C and csh scripts– User input: structure factors (.mtz), expected # copies– Runs CAPRA, extracts features, matches regions…– Automatically runs DM to improve phases via averaging– Output:
• NCS operators • masks for each region• C-alpha chains for each region• NCS-averaged structure factors (.mtz)
• Web server: textal.tamu.edu/NCS– Users can upload reflection file; results emailed back

• Command line# first source phenix_setup and ccp4_setup
>textal.find_ncs prot.mtz <N> <FP> <PHIB> <FOM>
...
Outputs: prot_ncs_ops.dat, prot_ncs_avg.mtz
prot_mask_1.xplor, prot_mask_2.xplor...
prot_region_1.pdb, prog_region_2.pdb...
• Script-level APIfrom textal.find_ncs import find_ncs
from textal.io.reflection_file import reflection_file
ref = reflection_file("mbp.mtz")
obj = find_ncs(reflections=ref,copies=2,
amplitude='FP',phases='PHIB',FOM='FOM')
obj.find_ncs()
(rot_mat,trans_vec) = obj.get_operators(0)
model1 = obj.get_subunit(0) # type pdb_extended
mask1 = obj.get_mask(0) # type emap
Port to Python

Improving Sequence Alignment with Simplex
• Romo, T.R., Sacchettini, J.C. and Ioerger, T.R. (2006). Improving Amino Acid Identification, Fit, and C-alpha Prediction using the Simplex Method in Automated Model-Building. Acta Crystallographica, accepted.
• The Problem:– Most model-building programs build backbone first,
then try to recognize side-chains (using probabilities, free atoms, features…)
– Identification of amino acids is sensitive to errors in predicted Ca coordinates (often up to 1Å rms)
– Even if sequence alignment is used to correct mistakes, initial side-chains must be sufficiently accurate

Our Approach: Simplex Optimization
• Simplex is a classic optimization algorithm – High radius of convergence– Does not require explicit computation of derivatives
• Simplex can be applied to refine individual residues as rigid bodies (translation+rotation)– Several programs do local real-space rigid-body
refinement of individual side-chains to improve fit.– Typically, applied after aa identity has been determined
• We apply Simplex in Textal (LOOKUP) during residue selection, to help pick the template from our database that matches the local density pattern best, allowing the Ca atom to shift up to 2Å
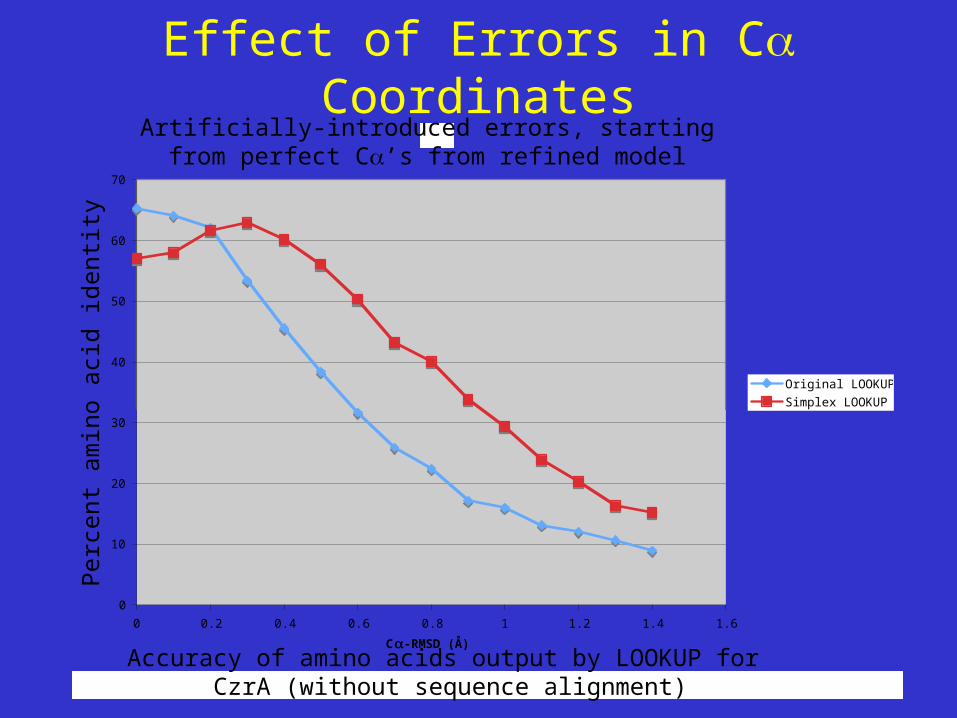
Effect of Errors in C CoordinatesC
0
10
20
30
40
50
60
70
0 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6
C-RMSD (Å)
Str
ict
Am
ino A
cid
Id
en
tity
Original LOOKUP
Simplex LOOKUP
Per
cent
am
ino
acid
iden
tity
Accuracy of amino acids output by LOOKUP for CzrA (without sequence alignment)
Artificially-introduced errors, startingfrom perfect C’s from refined model

Procedure• Step 1: Given a C, extract density-based
features and retrieve K=400 most similar regions from database
• Step 2: Re-rank by local density correlation (5Å)– Original method:
• try to find optimal rotation only– New method:
• Generate initial Simplex: N+1 perturbations of configuration vector (6-DOF)
• Evaluate density correlation coefficient of each• Pick the lowest, and ‘reflect’ over average of
remaining configuration vectors
Vector representing original position (3 coords) and orientation (3 angles) of side-chain
mean ofrest
worst score
new
6D config. space

Results on Experimental MapsProtein Reso. Mean phase
errorMap corr.
CzrA 2.3 Å 18.1 0.95
If5a 2.1 Å 36.8 0.91
MVK 2.4 Å 42.8 0.84
ICL 3.0 Å 44.1 0.81
PcaA 2.8 Å 54.2 0.73
without Simplex with Simplex
no alignment with alignment no alignment with alignment
CzrA 40.0 94.4 47.8 93.3
If5a 30.2 92.2 38.8 93.0
MVK 18.1 40.1 30.8 77.6
ICL 23.5 55.3 26.0 76.4
PcaA 15.6 38.7 19.3 47.4
average: 25.5 64.1 32.5 77.5
Percent identity of model compared to true (refined) structure:

Without Simplex With Simplex
Without SimplexWith SimplexTrue structure

TEXTAL for Molecular Replacement
• Motivation:– Why not exploit the MR
search model if available?– No excuse for mistakes in
connectivity or aa identities
• Steps toward larger goal of Model Completion
• Idea:– Rotate search model into
density (MR solution) – Replace amino acid identities
with new sequence– Run LOOKUP to build side-
chains into new density

• Issues:– Backbones sometimes diverge (e.g. in loops)– Phase improvement: How to identify and edit-out
incorrect parts of the model built?– Avoiding model bias
• Our Approach:– Use CAPRA to generate backbone for new density– Match up C’s with search model (core of protein)– Identify divergences (no nearby matches)– Fill in gaps with chains from new density
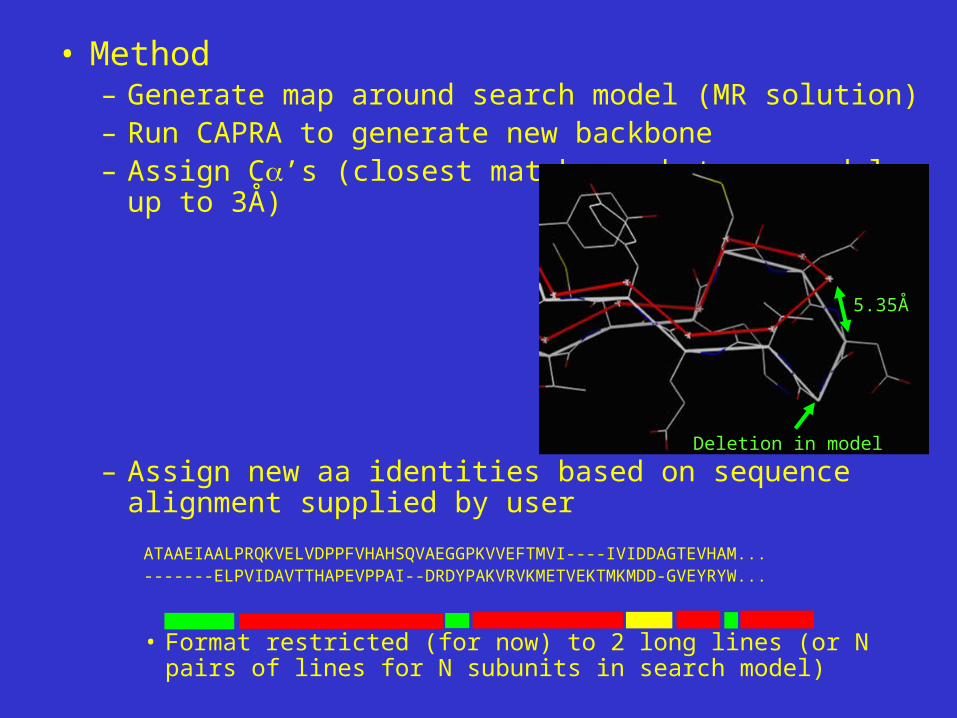
• Method– Generate map around search model (MR solution)– Run CAPRA to generate new backbone– Assign C’s (closest match
between models, up to 3Å)
– Assign new aa identities based on sequence alignment supplied by user
ATAAEIAALPRQKVELVDPPFVHAHSQVAEGGPKVVEFTMVI----IVIDDAGTEVHAM...-------ELPVIDAVTTHAPEVPPAI--DRDYPAKVRVKMETVEKTMKMDD-GVEYRYW...
• Format restricted (for now) to 2 long lines (or N pairs of lines for N subunits in search model)
5.35Å
Deletion in model

• Connect small gaps (len≤5)– Common (including due to alignment errors)– Method 1: Look for a bridge using existing C’s– Method 2: Use a fragment library
• 4188 9-mers extracted from 238 non-homologous proteins with min RMS of 1.25Å
• Superpose edges of each fragment on chain ends, with expected number of missing C’s in middle
• Select top 25 fragments by RMS (typically in range of 1-2Å)• Evaluate each fragment based on density measured every
0.5Å along fragment• Score(frag) = –exp(-(-1))
–exp(-(-1))

– Run patch to make any remaining connections
• More indiscriminant; may skip residues or insert extra atoms not consistent with alignment
• Can turn off via --connectivity=conservative
– Run ca_refine • reduces variance in inter-C distances
– Run LOOKUP to build side-chains– Run simulated annealing
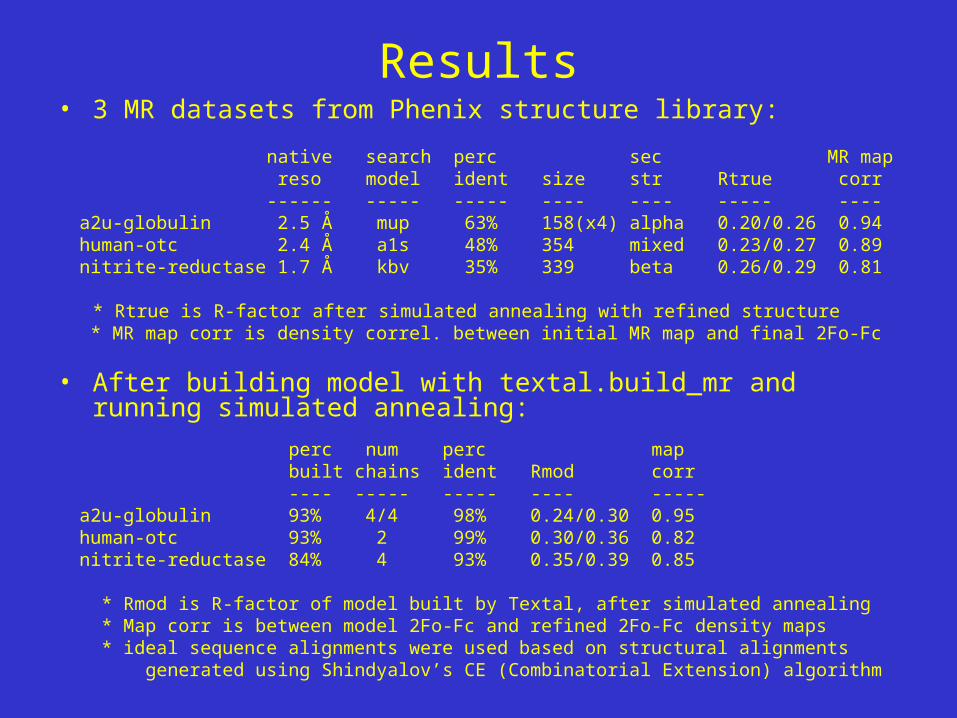
Results• 3 MR datasets from Phenix structure library:
native search perc sec MR map reso model ident size str Rtrue corr ------ ----- ----- ---- ---- ----- ---- a2u-globulin 2.5 Å mup 63% 158(x4) alpha 0.20/0.26 0.94 human-otc 2.4 Å a1s 48% 354 mixed 0.23/0.27 0.89 nitrite-reductase 1.7 Å kbv 35% 339 beta 0.26/0.29 0.81
* Rtrue is R-factor after simulated annealing with refined structure * MR map corr is density correl. between initial MR map and final 2Fo-Fc
• After building model with textal.build_mr and running simulated annealing:
perc num perc map built chains ident Rmod corr ---- ----- ----- ---- ----- a2u-globulin 93% 4/4 98% 0.24/0.30 0.95 human-otc 93% 2 99% 0.30/0.36 0.82 nitrite-reductase 84% 4 93% 0.35/0.39 0.85
* Rmod is R-factor of model built by Textal, after simulated annealing * Map corr is between model 2Fo-Fc and refined 2Fo-Fc density maps * ideal sequence alignments were used based on structural alignments generated using Shindyalov’s CE (Combinatorial Extension) algorithm

11 resN-term tailnot built
a2u-globulin (white)Textal model (green)
disorderedloop, res 60-64

human-otc (white)Textal (red, green)
loop not built,res 266-275
C-term not built,res 345-352

human-otc (white)Textal (red, green)

nitrite-reductase (white)Textal model (colors)
missing loop: res 186-205
missing loop: res 29-36
missing loop: res 159-170
missing term: res 334-342
missing term: res 5-10

nitrite-reductase (white)kbv (MR solution, purple)
largedivergent
loop
loop insertion
smalldifferences
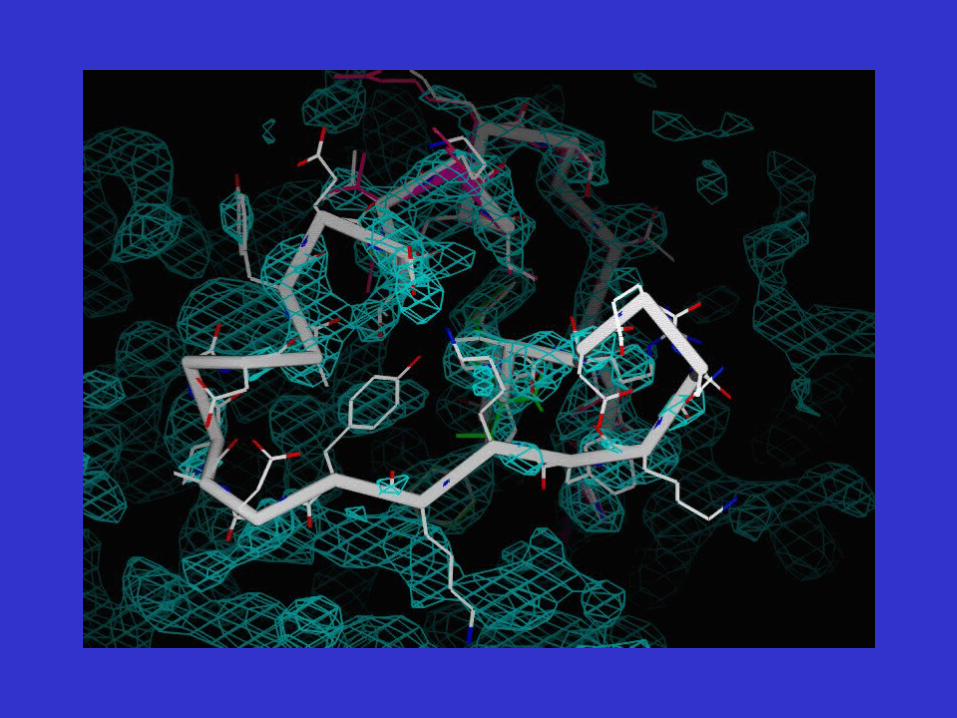


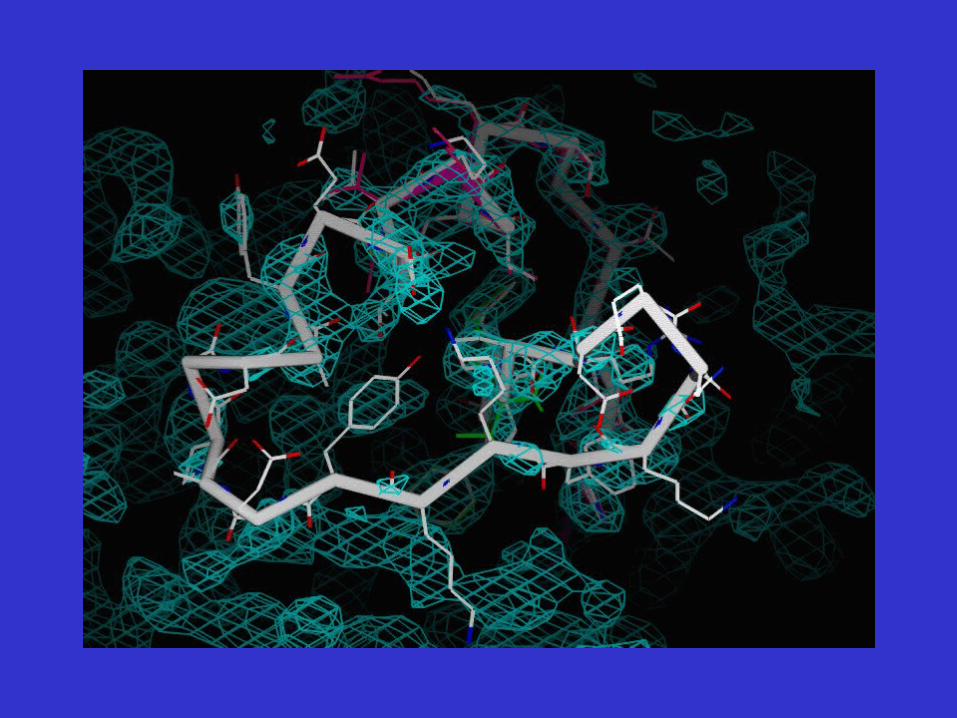

Initial Steps Toward Model EvaluationRun SFCHECK on model built…

Identifying errors with SFCHECK
• Which combination of values correlates best with errors in model?
• Use backbone_density_index from SFCHECK as residue quality score
Thr-203 0.092Gly-226 0.297Glu-236 0.306Thr-269 0.354...
residues (sorted)
qu
alit
y sc
ore
(S
fch
eck)

Residues in purple (50/284) are those with low backbonedensity index scores(<0.92)

Re-running SA on editted modelsHypothesis: impact of completeness versus accuracy of model on R-factor
Issues: • B-factors• side-chains• lack of HETATMs (2 Cu, 3Cd, 244 HOH in refined structure)• avoid model bias (use omit maps?)
Num residues deleted
Rwork Rfree Rwork Rfree
0 35.0 38.5 35.0 38.5
10 35.2 37.9 36.4 39.3
20 35.9 38.8 37.2 39.0
30 35.1 37.4 37.7 39.8
40 35.2 37.5 38.3 42.6
50 35.9 38.6 39.0 41.6
random deletions

Availability
• Phenix command line:
textal.build_mr [-c] [--symmetry] [--amplitudes] [--phases]
<reflections> <search_model> <alignment_file>
textal.build_mr --symmetry=nitrite-redct.inp –amplitudes=FULL_MOD nitrite-reduce.hkl kbv_mr_solution.pdb NR-KBV-align.txt
• Python API:
from textal.users.tom.textal_mr import MR_build
MR_build(reflections=rx,model=mod,alignment=algn,capra_only=True)

• Phenix GUI task: (textal/MR_Build):

• TEXTAL can build highly accurate models for Molecular Replacement (completely automatically), with almost perfect coordinates for backbone and side-chains atoms (with the help of simulated annealing), at least in the core (80-90%)
• Handle missing domains in the search model• Incorporate better model evaluation methods• Automate the whole improvement cycle
Future Work
Conclusion