receptor sirp polymorphism in the innate immune
TRANSCRIPT

of April 6, 2018.This information is current as
MouseAutoimmunity in the Nonobese Diabetic
Controls CD47 Binding andαReceptor SIRPPolymorphism in the Innate Immune
S. DanskaJayneAngelo J. Canty, Omid Gulban, David R. Greaves and
Andrea Sut Ling Wong, Steven Mortin-Toth, Michael Sung,
ol.1401984http://www.jimmunol.org/content/early/2014/10/10/jimmun
published online 10 October 2014J Immunol
MaterialSupplementary
4.DCSupplementalhttp://www.jimmunol.org/content/suppl/2014/10/10/jimmunol.140198
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2014 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on April 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on April 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
Polymorphism in the Innate Immune Receptor SIRPaControls CD47 Binding and Autoimmunity in the NonobeseDiabetic Mouse
Andrea Sut Ling Wong,*,† Steven Mortin-Toth,† Michael Sung,† Angelo J. Canty,‡
Omid Gulban,† David R. Greaves,x and Jayne S. Danska*,†,{
The signal regulatory protein (SIRP) locus encodes a family of paired receptors that mediate both activating and inhibitory signals
and is associated with type 1 diabetes (T1D) risk. The NOD mouse model recapitulates multiple features of human T1D and
enables mechanistic analysis of the impact of genetic variations on disease. In this study, we identify Sirpa encoding an inhibitory
receptor on myeloid cells as a gene in the insulin-dependent diabetes locus 13.2 (Idd13.2) that drives islet inflammation and T1D.
Compared to T1D-resistant strains, the NOD variant of SIRPa displayed greater binding to its ligand CD47, as well as enhanced
T cell proliferation and diabetogenic potency. Myeloid cell–restricted expression of a Sirpa transgene accelerated disease in a dose-
dependent manner and displayed genetic and functional interaction with the Idd5 locus to potentiate insulitis progression. Our
study demonstrates that variations in both SIRPa sequence and expression level modulate T1D immunopathogenesis. Thus, we
identify Sirpa as a T1D risk gene and provide insight into the complex mechanisms by which disease-associated variants act in
concert to drive defined stages in disease progression. The Journal of Immunology, 2014, 193: 000–000.
Type I diabetes (T1D) is caused by autoreactive T cells thatinfiltrate and destroy pancreatic b cells, resulting in lossof insulin production and dependence on exogenous in-
sulin. The NOD mouse develops spontaneous autoimmune T1Dthat shares many features with the human disease (1). In humansand NOD mice, T1D is a complex disease, resulting from inter-action between multiple susceptibility genes and poorly definedenvironmental modifiers. The greatest genetic risk factor for T1Din humans and NOD mice is MHC class II (MHC II) haplotypes,in which the two species share remarkably similar amino acidresidues that influence peptide binding and Ag presentation toT cells. In addition to the MHC II, known causal variants includeregulator of immune homeostasis IL-2 and IL-2R and inhibitors ofT cell activation CTLA-4 and LYP (2). These disease-causingvariants are common functional alleles involved in maintaining
the balance between tolerance and immunity, suggesting that T1Ddevelops due to dysregulation of the immune response.Genome-wide association studies (GWAS) in humans have
identified .40 single nucleotide polymorphisms (SNPs) associ-ated with T1D susceptibility, including polymorphisms in thesignal regulatory protein (SIRP) locus that encodes a family ofactivating and inhibitory receptors (3). Members of the pairedreceptor family have similar extracellular regions but differentsignaling potentials that may have evolved to maintain the balancebetween tolerance and immunity in response to evolutionarypressure such as pathogens (4, 5). The majority of T1D-associatedhuman SNPs have small effect sizes in which each confers modestimpact on the phenotype: to date, few causal genetic variationshave been identified. Collectively, GWAS fail to fully explain theheritability of T1D likely because this approach did not capturethe contribution of gene–gene and gene–environment interactionsunderlying the complex disease phenotype (6). As shown in thisstudy, the NOD mouse model enables analysis of complex featuresof T1D heritability through enriched preclinical phenotypes, ac-cess to immune cell compartments, and germline manipulations totest the effects of both individual and combined genetic variationson disease.Diabetes onset in NOD mice is preceded by islet inflammation
(insulitis) by multiple leukocyte subsets. Insulitis begins withperi-islet accumulation of myeloid cells and T cells, which theninvades the b-cell–rich interior (invasive insulitis), leading toeventual loss of insulin secretion. In contrast, the nonobese T1D-resistant (NOR) strain displays only peri-islet infiltration by APCthat does not invade the islet interior or compromise b cellfunction (7). The NOR strain is 88% identical by descent toNOD, shares the high risk Mhc haplotype, but is protected fromT1D by genomic inheritance from C57BLKS/J mice (8). Previ-ously, we showed that progression from peri-islet to invasiveinsulitis in NOD mice was regulated by two Idd loci, Idd5 andIdd13 (7). In this study, we report high-resolution genomic,genetic, and functional analyses supporting Sirpa, encoding thesignal inhibitory receptor protein (SIRPa) as the candidate gene
*Department of Immunology, Faculty of Medicine, University of Toronto, Toronto,Ontario M5S1A8, Canada; †Program in Genetics and Genome Biology, TheHospital for Sick Children Research Institute, Toronto, Ontario M5G1X8, Can-ada; ‡Department of Mathematics and Statistics, McMaster University, Hamilton,Ontario L8S4L8, Canada; xSir William Dunn School of Pathology, University ofOxford, Oxford OX13RE, United Kingdom; and {Department of Medical Biophys-ics, Faculty of Medicine, University of Toronto, Toronto, Ontario M5S1A8, Canada
Received for publication August 12, 2014. Accepted for publication September 8,2014.
This work was supported by grants from the Juvenile Diabetes Research Foundation,Genome Canada (administered through the Ontario Genomics Institute), and theCanadian Institutes of Health Research (to J.S.D.).
Address correspondence and reprint requests to Dr. Jayne S. Danska, The Hospital forSick Children, Peter Gilgan Research Tower, 686 Bay Street, 15th Floor, Toronto, ONM5G 0A4, Canada. E-mail address: [email protected]
The online version of this article contains supplemental material.
Abbreviations used in this article: T1D, type 1 diabetes; DC, dendritic cell; EAE,experimental autoimmune encephalomyelitis; GWAS, genome-wide associationstudies; hCD47-Fc, human CD47-Fc protein; IgV, Ig variable; LN, lymph node;b2m, b2-microglobulin; Mf, macrophage; mCD47-Fc, mouse CD47-Ig variabledomain–human IgG-Fc fusion protein; MHC II, MHC class II; NOR, nonobese type 1diabetes–resistant; PDCA-1, plasmacytoid DC Ag-1; PLN, pancreatic LN; SIRP, signalregulatory protein; SNP, single nucleotide polymorphism.
Copyright� 2014 by The American Association of Immunologists, Inc. 0022-1767/14/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1401984
Published October 10, 2014, doi:10.4049/jimmunol.1401984 by guest on A
pril 6, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

at Idd13.2, which interacts with the Idd5 locus to drive insulitisprogression and T1D.
Materials and MethodsMice
All mice used in this study were maintained in specified pathogen-freeconditions at The Hospital for Sick Children (Toronto, Ontario, Canada)and followed the guidelines for the institutional animal care committee.
Generation of Idd13 congenic mice
All NOD.NOR-Idd13 congenic mice were generated by microsatellitemarker-directed selection of breeders (Supplemental Table I). NOR.NOD-Idd5, NOR.NOD-Idd13, and NOR.NOD-Idd5.Idd13 congenic mice weregenerated using similar strategies (Supplemental Table II). Microsatellitemarkers were developed using diNucleotide Tandem Repeat finder (http://www.gchelpdesk.ca/servers/servers.php), and genotyping was performedas previously described (9).
Generation of Sirpa transgenic mice
Sirpa cDNAwas generated by reverse transcription using RNA isolated fromNOD and NOR macrophages (Mf) and cloned into XbaI site in pCDNA3vector (10). Founders were identified by PCR with four pairs of primers: 1)59-CACGCTTTCCAGTCTTTCAGC-39 and 59- CTTAGAGTGCTGGTTC-CTTCCTG-39; 2) 59-GGGTTGCTAACCATCTCCTCTCT-39 and 59-GCT-TTGCCCTACTCCTCTGTACC-39; 3) 59-GGGTTGCTAACCATCTCCTC-TCT-39 and 59-CTCGCTGATCAGCTTCTGCTC-39; and 4) 59-CACATC-ACCTTGGATAGAAGCC-39 and 59-CAGCCAGATCAGCTGGAGATC-39.To generate NOR.NOD-SirpaTg.NOD-Idd5, NOR.NOD-SirpaTg wascrossed to NOR.NOD-Idd5, and pups were genotyped for Sirpa trans-gene and microsatellite markers (Supplemental Table II). To generateNOD.NOR-SirpaTg homo, hemizygous mice were intercrossed and screenedby real-time PCR with primers 59-CCTCACCTATGCTGACCTGG-39 and59-TCAGCTTCTGCTCCTTCCTC-39. Sirpa transgene copy number wasnormalized to Rag1, detected with primers 59- GACGGAATTCTGC-CATGACT-39 and 59-GTCTCTTCCTCTTGAGTCCC-39. Amplicon wasquantitated by SYBR Green on an ABI/PRIZM 7900 HT Cycler (LifeTechnologies).
Sequence analysis of Idd13.2
NOD-derived Chr.2 sequence from Next Generation Sequencing was down-loaded from the Sanger database (ftp://ftp.sanger.ac.uk/pub/NODmouse/) andformatted as a BLAST database. B6-derived sequences for Idd13.2 weredownloaded from the Ensembl Genome Browser and aligned with NODsequences. The resulting alignment was queried for exonic SNPs.
Insulitis assessment
Pancreata were fixed in formalin, paraffin embedded, and 5-mm sectionswere prepared at 200-mm spacing. Pancreatic sections were stained withMayer’s H&E Y (Sigma-Aldrich) to visualize cell infiltration. Each isletwas scored as: 0 indicates no infiltration, 1 for perivascular/periductalinfiltration touching islet perimeters but not penetrating, 2 for penetrationof up to 25% of islet mass, 3 for penetration up to 75% islet mass, and 4for ,20% of islet mass remaining. The number of islets scored were NOR-Tg neg (629 islets; n = 17), NOR.NOD-SirpaTg (1048 islets; n = 28),NOR-Tgneg.NOD-Idd5 (776 islets; n = 28), NOR.NOD-SirpaTg.NOD-Idd5(713 islets; n = 26), NOR (1908 islets; n = 53), NOR.NOD-Idd13 (1385islets; n = 26), NOR.NOD-Idd5 (1151 islets; n = 43), and NOR.NOD-Idd5.Idd13 (1778 islets; n = 38).
Statistical analysis of insulitis scores
To increase the power to detect differences between mouse strains, thedistribution of insulitis scores, rather than a single mean insulitis score, wasexamined for each mouse. We used a Poisson model in which the log countat a given score for a mouse was modeled as a linear function of the totalnumber of islets scored for that mouse as well as indicators for whether themouse carried the NOD Sirpa transgene and/or the NOD Idd5 locus. Thepairwise analyses included one of these indicators (the other was fixed) andassessed the evidence of differences between strains by comparing theincrease in the log-likelihood caused by inclusion of the indicator, to theasymptotic x2 distribution with four df. To assess the evidence for inter-action, we analyzed all mice together with both indicators and also theproduct of the indicators. If this product was different from 0 for any count,there was evidence of an interaction between the transgenic Sirpa statusand the NOD Idd5 congenic status. Assessment of the defect of the in-
teraction term was done by comparing the increased in log-likelihoodcaused by its inclusion to the asymptotic x2 distribution with four df(11). All statistical test analyses were carried out using R 2.13.1 availableat http://www.R-project.org/ (12).
Generation of mouse CD47-IgV domain–human IgG-Fc fusionprotein and human CD47-Fc
The plasmid pIAP369 containing mouse CD47 was a gift from Dr. EricBrown (13). FreeStyle 293-F cells (Life Technologies) were transfectedaccording to the manufacturer’s guidelines. The mouse CD47-IgV do-main–human IgG-Fc fusion protein (mCD47-Fc) was purified on protein GSepharose (Pierce) and conjugated to biotin for flow cytometry–basedbinding assay. Human CD47-Fc protein (hCD47-Fc) was generated aspreviously described (14).
Flow cytometry
For analysis of SIRPa expression, cells were stained with Abs specific forCD19, CD4, CD8a, plasmacytoid dendritic cell (DC) Ag-1 (PDCA-1),CD11b, and CD11c from eBioscience; P84 and CD3 from BD Biosciences;and hCD47-biotin, detected with streptavidin–PE-Cy7 (Caltag Laboratories).Data were collected on the LSR II (BD Biosciences), and analysis was per-formed using FlowJo (Tree Star). The gating strategy used is detailed in thefollowing sections. In all flow cytometry analysis, live cells were identified bypropidium iodide exclusion. To exclude doublets, live single cells were furtheridentified based on their forward and side scatter profile.
mCD47-Fc binding assay
For flow-based mCD47-Fc binding assay, cells were blocked with anti-CD16/32 followed by preclustering of cell-surface SIRPa with unconju-gated P84. Cells were stained with Abs specific for CD11b, F4/80-FITC(Cedarlane Laboratories), and biotinylated mCD47-Fc detected withStreptavidin-PE (Molecular Probes). Cells were gated on propidium io-dide–negative, forward versus side scatter, and CD11b+F4/80+.
Plate-based mCD47-Fc binding assay was performed on bone marrow–derived Mf, cultured as previously described (14). Cells were blockedwith anti-CD16/32 (2.4G2) and stained with mCD47-Fc. Binding wasdetected with HRP-conjugated anti-human IgG (Bethyl Laboratories) andchromogenic peroxidase substrate tetramethylbenzidine (Mendell Scien-tific). Nonlinear regression analysis with one site binding equation wasperformed to generate a ligand-binding curve and calculate equilibriumconstant Kd using GraphPad Prism (GraphPad).
T cell proliferation assay
Splenic DC from 8–10-wk-old mice were isolated using anti-CD11cmagnetic beads (Miltenyi Biotec), and a purity of .85% was routinelyobtained. CD4+ cells from spleen and peripheral lymph nodes (LN) of 8–15-wk-old NOD.BDC2.5 transgenic mice (stock #004460; The JacksonLaboratory) were isolated using the CD4+ T cell isolation kit II (MiltenyiBiotec) and labeled with CFSE (Molecular Probes). A total of 2.5 3 104
DC and 5 3 104 BDC2.5 T cells was cocultured with BDC2.5 1040-63peptide (RTRPLWVRME; AnaSpec) and proliferation measured 3 d later.
Adoptive transfer
LN were harvested from 12-wk-old NOD mice. Total T cells were isolatedusing the Pan T cell isolation Kit II (Miltenyi Biotec), and a purity of .90%was routinely obtained. A total of 10 3 106 T cells was delivered via i.v.injection into 4- to 5-wk-old NOD.SCID and NOD.NOR-Idd13.SCIDrecipients. Recipients were aged up to 21–27 wk posttransfer and diabetesassessed by urine glucose test using Chemstrip from Roche. PancreaticLN (PLN) of disease-free NOD.NOR-Idd13.SCID recipients at the end ofthe study were analyzed for presence of T cells by staining for Absspecific for CD3, CD4, and CD8. NOD.NOR-Idd13.SCID recipients thatreceived T cells (+T cells) were compared with NOD.NOR-Idd13.SCIDmice that did not receive any donor T cells (2T cells).
ResultsRefined genomic mapping of the Idd13.2 locus
Idd13 was originally mapped to a 4-cM region of chromosome 2 inlinkage analysis of T1D in (NOD 3 NOR)F2 mice in which theNOR allele conferred T1D resistance (15). To refine the Idd13 locusto a tractable size for candidate gene analyses, we generated a seriesof congenic strains by introgression of NOR-derived Idd13 onto theNOD background (Fig. 1A). We developed an algorithm to identify
2 SIPRa-MEDIATED CONTROL OF AUTOIMMUNITY IN THE NOD MOUSE
by guest on April 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
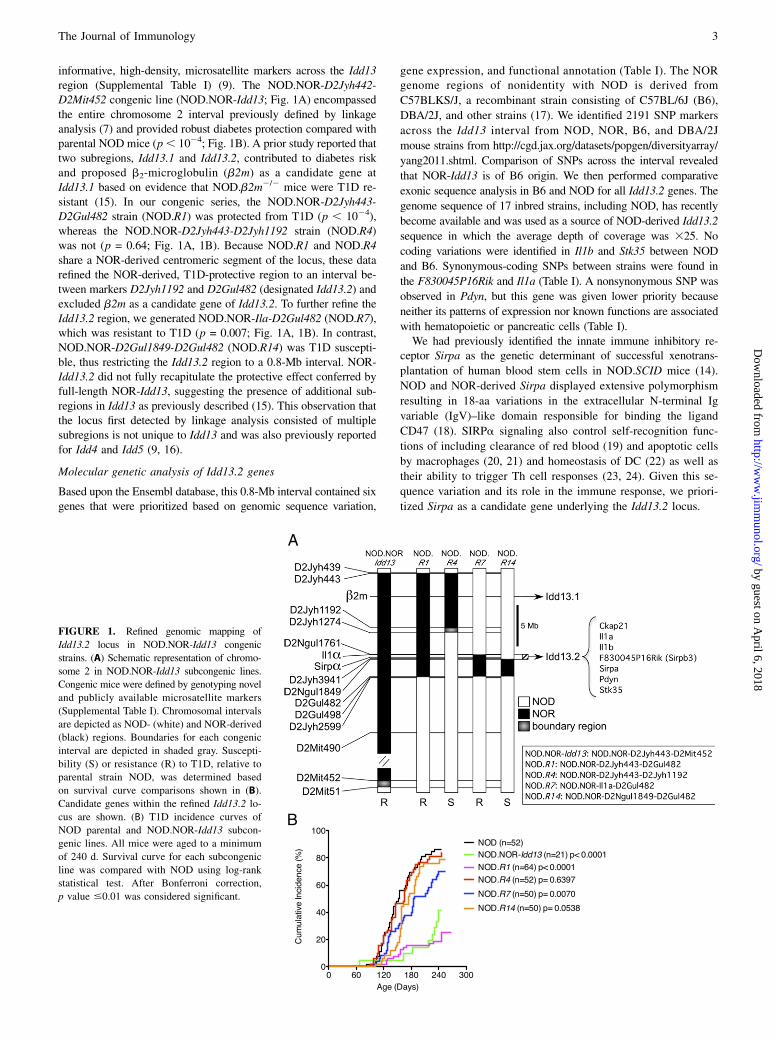
informative, high-density, microsatellite markers across the Idd13region (Supplemental Table I) (9). The NOD.NOR-D2Jyh442-D2Mit452 congenic line (NOD.NOR-Idd13; Fig. 1A) encompassedthe entire chromosome 2 interval previously defined by linkageanalysis (7) and provided robust diabetes protection compared withparental NOD mice (p, 1024; Fig. 1B). A prior study reported thattwo subregions, Idd13.1 and Idd13.2, contributed to diabetes riskand proposed b2-microglobulin (b2m) as a candidate gene atIdd13.1 based on evidence that NOD.b2m2/2 mice were T1D re-sistant (15). In our congenic series, the NOD.NOR-D2Jyh443-D2Gul482 strain (NOD.R1) was protected from T1D (p , 1024),whereas the NOD.NOR-D2Jyh443-D2Jyh1192 strain (NOD.R4)was not (p = 0.64; Fig. 1A, 1B). Because NOD.R1 and NOD.R4share a NOR-derived centromeric segment of the locus, these datarefined the NOR-derived, T1D-protective region to an interval be-tween markers D2Jyh1192 and D2Gul482 (designated Idd13.2) andexcluded b2m as a candidate gene of Idd13.2. To further refine theIdd13.2 region, we generated NOD.NOR-Ila-D2Gul482 (NOD.R7),which was resistant to T1D (p = 0.007; Fig. 1A, 1B). In contrast,NOD.NOR-D2Gul1849-D2Gul482 (NOD.R14) was T1D suscepti-ble, thus restricting the Idd13.2 region to a 0.8-Mb interval. NOR-Idd13.2 did not fully recapitulate the protective effect conferred byfull-length NOR-Idd13, suggesting the presence of additional sub-regions in Idd13 as previously described (15). This observation thatthe locus first detected by linkage analysis consisted of multiplesubregions is not unique to Idd13 and was also previously reportedfor Idd4 and Idd5 (9, 16).
Molecular genetic analysis of Idd13.2 genes
Based upon the Ensembl database, this 0.8-Mb interval contained sixgenes that were prioritized based on genomic sequence variation,
gene expression, and functional annotation (Table I). The NORgenome regions of nonidentity with NOD is derived from
C57BLKS/J, a recombinant strain consisting of C57BL/6J (B6),
DBA/2J, and other strains (17). We identified 2191 SNP markers
across the Idd13 interval from NOD, NOR, B6, and DBA/2J
mouse strains from http://cgd.jax.org/datasets/popgen/diversityarray/
yang2011.shtml. Comparison of SNPs across the interval revealed
that NOR-Idd13 is of B6 origin. We then performed comparative
exonic sequence analysis in B6 and NOD for all Idd13.2 genes. The
genome sequence of 17 inbred strains, including NOD, has recently
become available and was used as a source of NOD-derived Idd13.2
sequence in which the average depth of coverage was 325. No
coding variations were identified in Il1b and Stk35 between NOD
and B6. Synonymous-coding SNPs between strains were found in
the F830045P16Rik and Il1a (Table I). A nonsynonymous SNP was
observed in Pdyn, but this gene was given lower priority because
neither its patterns of expression nor known functions are associated
with hematopoietic or pancreatic cells (Table I).We had previously identified the innate immune inhibitory re-
ceptor Sirpa as the genetic determinant of successful xenotrans-
plantation of human blood stem cells in NOD.SCID mice (14).
NOD and NOR-derived Sirpa displayed extensive polymorphism
resulting in 18-aa variations in the extracellular N-terminal Ig
variable (IgV)–like domain responsible for binding the ligand
CD47 (18). SIRPa signaling also control self-recognition func-
tions of including clearance of red blood (19) and apoptotic cells
by macrophages (20, 21) and homeostasis of DC (22) as well as
their ability to trigger Th cell responses (23, 24). Given this se-
quence variation and its role in the immune response, we priori-
tized Sirpa as a candidate gene underlying the Idd13.2 locus.
FIGURE 1. Refined genomic mapping of
Idd13.2 locus in NOD.NOR-Idd13 congenic
strains. (A) Schematic representation of chromo-
some 2 in NOD.NOR-Idd13 subcongenic lines.
Congenic mice were defined by genotyping novel
and publicly available microsatellite markers
(Supplemental Table I). Chromosomal intervals
are depicted as NOD- (white) and NOR-derived
(black) regions. Boundaries for each congenic
interval are depicted in shaded gray. Suscepti-
bility (S) or resistance (R) to T1D, relative to
parental strain NOD, was determined based
on survival curve comparisons shown in (B).
Candidate genes within the refined Idd13.2 lo-
cus are shown. (B) T1D incidence curves of
NOD parental and NOD.NOR-Idd13 subcon-
genic lines. All mice were aged to a minimum
of 240 d. Survival curve for each subcongenic
line was compared with NOD using log-rank
statistical test. After Bonferroni correction,
p value #0.01 was considered significant.
The Journal of Immunology 3
by guest on April 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
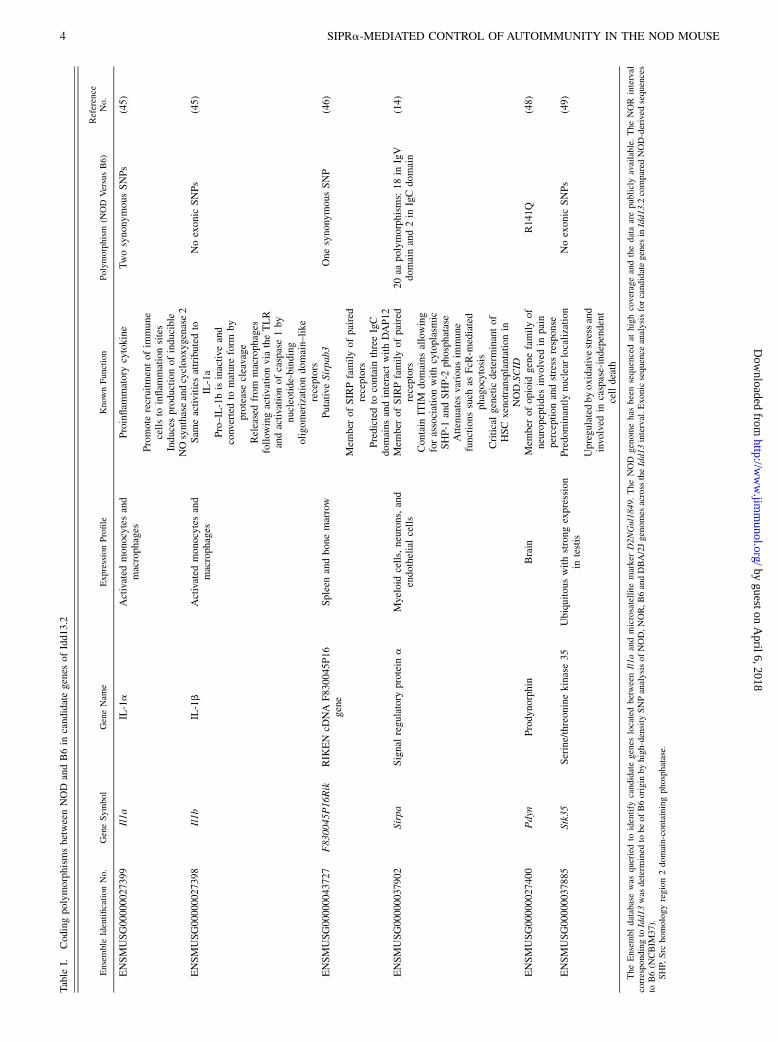
Table
I.CodingpolymorphismsbetweenNOD
andB6in
candidategenes
ofIdd13.2
Ensemble
IdentificationNo.
GeneSymbol
GeneNam
eExpressionProfile
Know
nFunction
Polymorphism
(NOD
VersusB6)
Reference
No.
ENSMUSG00000027399
Il1a
IL-1a
Activated
monocytesand
macrophages
Proinflam
matory
cytokine
TwosynonymousSNPs
(45)
Promote
recruitmentofim
mune
cellsto
inflam
mationsites
Inducesproductionofinducible
NOsynthaseandcyclooxygenase2
ENSMUSG00000027398
Il1b
IL-1b
Activated
monocytesand
macrophages
Sam
eactivitiesattributedto
IL-1a
Noexonic
SNPs
(45)
Pro–IL-1bisinactive
and
convertedto
mature
form
by
proteasecleavage
Releasedfrom
macrophages
follow
ingactivationvia
theTLR
andactivationofcaspase1by
nucleotide-binding
oligomerizationdomain–like
receptors
ENSMUSG00000043727
F830045P16Rik
RIK
EN
cDNA
F830045P16
gene
Spleen
andbonemarrow
Putative
Sirpab3
OnesynonymousSNP
(46)
Mem
ber
ofSIRPfamilyofpaired
receptors
Predictedto
contain
threeIgC
domains
andinteract
withDAP12
ENSMUSG00000037902
Sirpa
Signal
regulatory
protein
aMyeloid
cells,neurons,and
endothelialcells
Mem
ber
ofSIRPfamilyofpaired
receptors
20aa
polymorphisms:18in
IgV
domainand2in
IgCdomain
(14)
Contain
ITIM
domainsallowing
forassociationwithcytoplasmic
SHP-1
andSHP-2
phosphatase
Attenuates
variousim
mune
functions
such
asFcR
-mediated
phagocytosis
Criticalgenetic
determinantof
HSCxenotransplantationin
NOD.SCID
ENSMUSG00000027400
Pdyn
Prodynorphin
Brain
Mem
ber
ofopioid
genefamilyof
neuropeptides
involved
inpain
perceptionandstress
response
R141Q
(48)
ENSMUSG00000037885
Stk35
Serine/threoninekinase35
Ubiquitouswithstrong
expression
intestis
Predom
inantlynuclearlocalization
Noexonic
SNPs
(49)
Upregulatedbyoxidativestressand
involved
incaspase-independent
celldeath
TheEnsembldatabasewas
queriedto
identify
candidategenes
locatedbetweenIl1aandmicrosatellitemarker
D2NGul1849.TheNOD
genomehas
beensequencedat
highcoverageandthedataarepubliclyavailable.TheNOR
interval
correspondingto
Idd13was
determined
tobeofB6origin
byhigh-density
SNPanalysisofNOD,NOR,B6andDBA/2Jgenomes
across
theIdd13interval.Exonicsequence
analysisforcandidategenes
inIdd13.2
compared
NOD-derived
sequences
toB6(N
CBIM
37).
SHP,
Src
homologyregion2domain-containingphosphatase.
4 SIPRa-MEDIATED CONTROL OF AUTOIMMUNITY IN THE NOD MOUSE
by guest on April 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

Sirpa alleles confer differential binding to mouse CD47
We previously showed that in contrast to NOR and B6, the NODvariant of SIRPa IgV domain polymorphisms conferred bindingto hCD47 (14). Therefore, we questioned whether NOD and NORSIRPa would display differential binding to mouse CD47, whichis not polymorphic between these strains. We generated a mCD47-Fc for flow cytometric and chemiluminescent cell-based bindingassays. SIRPa is highly expressed on myeloid cells including Mf,neutrophils, and DC (25). Mf isolated from NOD and NOD.NOR-Idd13 congenic mice (Fig. 1) were stained with either mCD47-Fcor control human IgG-Fc and assessed by flow cytometry. NODand NOD.NOR-Idd13 Mf displayed similar surface expression ofSIRPa based upon staining with anti-mouse SIRPa Ab (Mab P84;Fig. 2A); however, NOD Mf displayed greater binding to mCD47-Fc at all concentrations tested compared with NOD.NOR-Idd13congenic Mf (Fig. 2B). To ensure that the observed differentialbinding of mCD47-Fc was not influenced by other NOD backgroundgenes, we also compared B6 and B6.NOD-Idd13 congenic Mf andobserved a similar dependence of mCD47-Fc binding on Idd13genotype (Fig. 2C, 2D).mCD47-Fc binding to Mf was also examined with a plate-
based assay using the conversion of chemiluminescent substratesby HRP-conjugated anti-human IgG Ab. Mf from NOD andNOD.R7 were incubated with a range of mCD47-Fc concentrationsto calculate a ligand-binding curve (Fig. 2E). Under these conditions,the dissociation constant (Kd) of NOD SIRPa expressed on Mf andmCD47-Fc was approximately three times lower than for NORSIRPa (171 6 12 versus 460 6 69 nM; Fig. 2E). Taken together,these data show that compared with the NOR protein, NOD variantof SIRPa displayed a greater affinity for mouse CD47, suggestinga potential functional impact of these sequence variations.
Sirpa variation on DC impacts T cell proliferation and transferof diabetes
SIRPa–CD47 interactions have been suggested to enhance T cellactivation in response to APC (24, 26). CD47 is a broadly ex-pressed protein that is often tightly associated with integrins (27).CD47 cell-surface expression on T cells is regulated during acti-vation (28, 29) and delivers a positive signal during encounterswith cognate Ag (30, 31). We asked whether the observed dif-ferential binding of NOD versus NOR SIRPa to mCD47 impactedT cell–APC interaction and T cell activation and proliferation.To test islet Ag-specific proliferation of T cells, we used theNOD.BDC2.5-Tcr (BDC2.5) TCR-transgenic mice, which expressesthe TCR from a diabetogenic CD4+ T cell clone specific forChromogranin A, a protein found in pancreatic b-cells and neu-roendocrine tissues (32). CFSE-labeled CD4+ BDC2.5 T cellswere cocultured with CD11c+ DC from NOD or NOD.R7 mice(Fig. 3A). A titration of the BDC2.5 mimotope peptide (1000–37ng/ml) was added to the T cell–DC culture (33) and T cell pro-liferation measured as CFSE dilution by flow cytometry. AlthoughDC from NOD and NOD.R7 mice had similar levels of costimu-latory molecules such as CD86 and MHC II (data not shown),a higher fraction of proliferating BDC2.5 T cells (CFSElo) cellswas observed in cultures with NOD-derived compared withNOD.R7-derived DC, particularly at low peptide concentrations(Fig. 3B). These data were consistent with prior evidence thatCD47 conveys costimulatory effects under conditions of subop-timal TCR ligation (31).Because BDC2.5 T cells cocultured with NOD compared
with DC resulted in greater proliferation, we asked whether thisdifference affected T cell pathogenicity in vivo. Previous workestablished that NOD-derived T cells can transfer T1D to immu-nodeficient NOD.SCID mice (34). To examine whether the ge-
notype of recipient myeloid cells affected the ability of NODT cells to transfer T1D, we generated NOD.NOR-Idd13.SCID
mice homozygous for NOR-derived Sirpa (14). NOD splenic
T cells were adoptively transferred into either NOD.SCID or
NOD.NOR-Idd13.SCID recipients. After 150 d posttransfer,
NOD.NOR-Idd13.SCID recipients of NOD T cells had significantly
lower T1D incidence than NOD.SCID recipients (p = 0.0013;
Fig. 3C). At the study end point, PLN of T1D-free NOD.NOR-
Idd13.SCID recipients were analyzed for T cell markers and found to
be successfully engrafted (data not shown). These results suggested
that, compared with the NOR variant, expression of NOD SIRPa on
myeloid cells enhanced T cell–mediated transfer of diabetes. Taken
together, these data demonstrated that NOD SIRPa expressed on
myeloid cells is associated with enhanced T cell proliferation and
diabetogenicity.
Generation of NOR.NOD-SirpaTg mice
We previously showed by genetic linkage analysis that Idd13 actsin concert with Idd5 (chromosome 1) to regulate progression of
peri-insulitis to invasive insulitis. We tested whether NOD Sirpa
was an Idd13 gene responsible for driving insulitis progression
using a gain of function approach. NOR.NOD-Idd13 congenic and
NOR.NOD-SirpaTg transgenic mice were generated to examine
the impact of the NOD-Sirpa allele on insulitis progression.NOR.NOD-Idd13 congenic mice were generated from (NOD 3
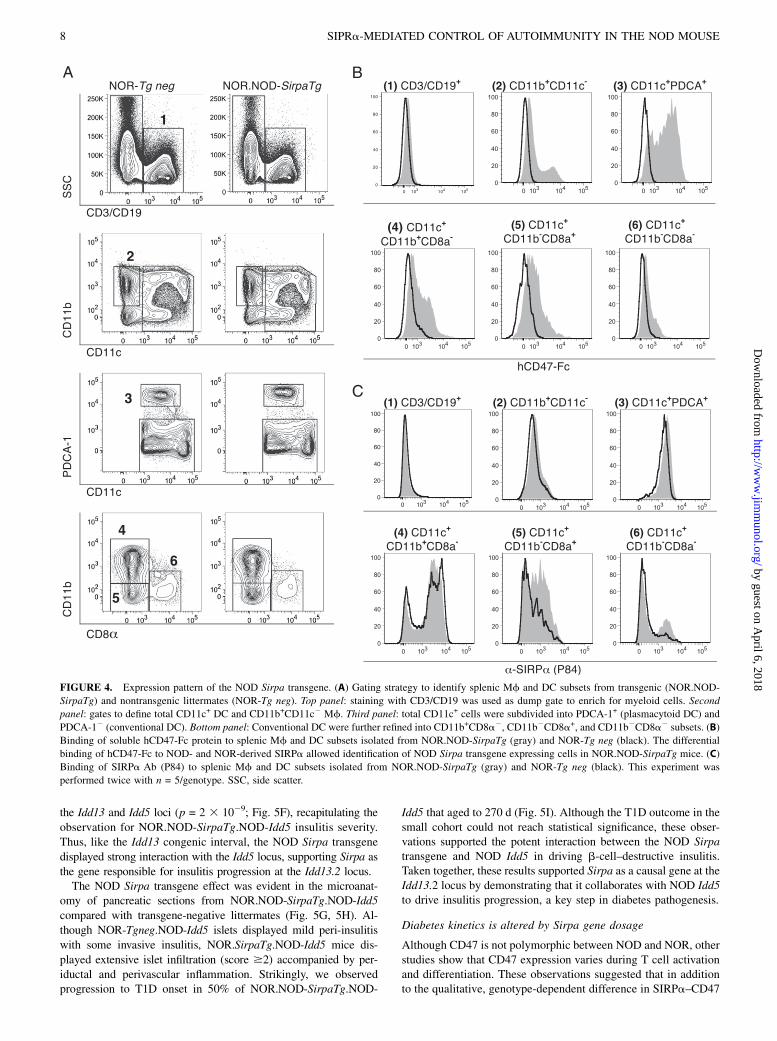
NOR)F1 mice followed by backcrossing to NOR with marker-directedselection of breeders at each generation (Supplemental Fig. 1,Supplemental Table II). To produce Sirpa transgenic mice witha myeloid-specific expression emulating the normal expressionof the gene, NOD-derived Sirpa cDNA was cloned downstreamof hCD68 previously reported to drive myeloid-lineage specificexpression in mice (10). Fluorescence in situ hybridization identifiedthe genomic insertion site of the NOD Sirpa transgene on chromo-some 10C1 (data not shown), where there are no known Idd loci.To characterize transgene expression, we compared the per-
centages of splenic lymphocytes (CD3+/CD19+), Mf (CD11b+
CD11c2), conventional DC (CD11c+PDCA-12), and plasmacy-
toid DC (CD11c+PDCA-1+) in transgenic (NOR.NOD-SirpaTg)
and transgene-negative littermates (NOR-Tg neg; Fig. 4A). The
conventional DC subset was further refined using CD11b and
CD8a markers (Fig. 4A, bottom panel). The frequency of these
leukocyte subsets was not altered by the presence of the transgene
(Fig. 4A). We previously showed that hCD47-Fc fusion protein
binds to the NOD but not to NOR SIRPa, so we used this reagent
to distinguish expression of the NOD Sirpa transgene from en-
dogenous NOR-derived SIRPa (Fig. 4B). Expression of both the
NOD and NOR variants of SIRPa was detected with mAb P84
(Fig. 4C). The NOD Sirpa transgene was not expressed on T or
B cells (Fig. 4B1), just as we observed for expression of endog-
enous SIRPa (Fig. 4C1). In contrast, the product of the NOD
Sirpa transgene was detected on Mf and all DC subsets
(Fig. 4B2–6). Although NOD Sirpa transgene expression was
robust in Mf and all DC subsets, the total SIRPa expression
levels in NOR.NOD-SirpaTg were mostly unaltered compared
with NOR-Tg neg mice (Fig. 4C2–4). Only a subset of the CD11b-
CD8a+ DC displayed a higher level of total SIRPa expression
in the NOR.NOD-SirpaTg compared with NOR-Tg neg mice
(Fig. 4C5). SIRPa expression on bone marrow Mf and neu-
trophils did not differ between NOR.NOD-SirpaTg and NOR-Tg
neg strains (data not shown). Thus, with the exception of greater
expression in CD11b2CD8a+ DC, the NOD Sirpa transgene was
expressed at similar levels to the endogenous locus and did not
affect the frequency of myeloid cell populations.
The Journal of Immunology 5
by guest on April 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
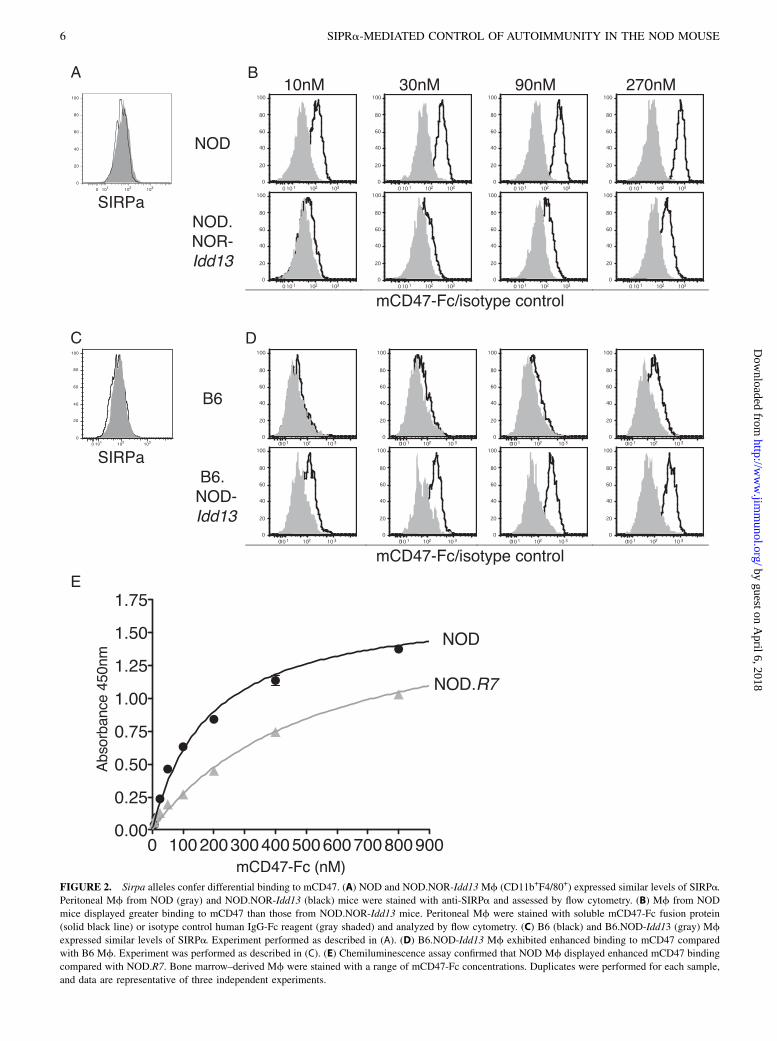
FIGURE 2. Sirpa alleles confer differential binding to mCD47. (A) NOD and NOD.NOR-Idd13 Mf (CD11b+F4/80+) expressed similar levels of SIRPa.
Peritoneal Mf from NOD (gray) and NOD.NOR-Idd13 (black) mice were stained with anti-SIRPa and assessed by flow cytometry. (B) Mf from NOD
mice displayed greater binding to mCD47 than those from NOD.NOR-Idd13 mice. Peritoneal Mf were stained with soluble mCD47-Fc fusion protein
(solid black line) or isotype control human IgG-Fc reagent (gray shaded) and analyzed by flow cytometry. (C) B6 (black) and B6.NOD-Idd13 (gray) Mf
expressed similar levels of SIRPa. Experiment performed as described in (A). (D) B6.NOD-Idd13 Mf exhibited enhanced binding to mCD47 compared
with B6 Mf. Experiment was performed as described in (C). (E) Chemiluminescence assay confirmed that NOD Mf displayed enhanced mCD47 binding
compared with NOD.R7. Bone marrow–derived Mf were stained with a range of mCD47-Fc concentrations. Duplicates were performed for each sample,
and data are representative of three independent experiments.
6 SIPRa-MEDIATED CONTROL OF AUTOIMMUNITY IN THE NOD MOUSE
by guest on April 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
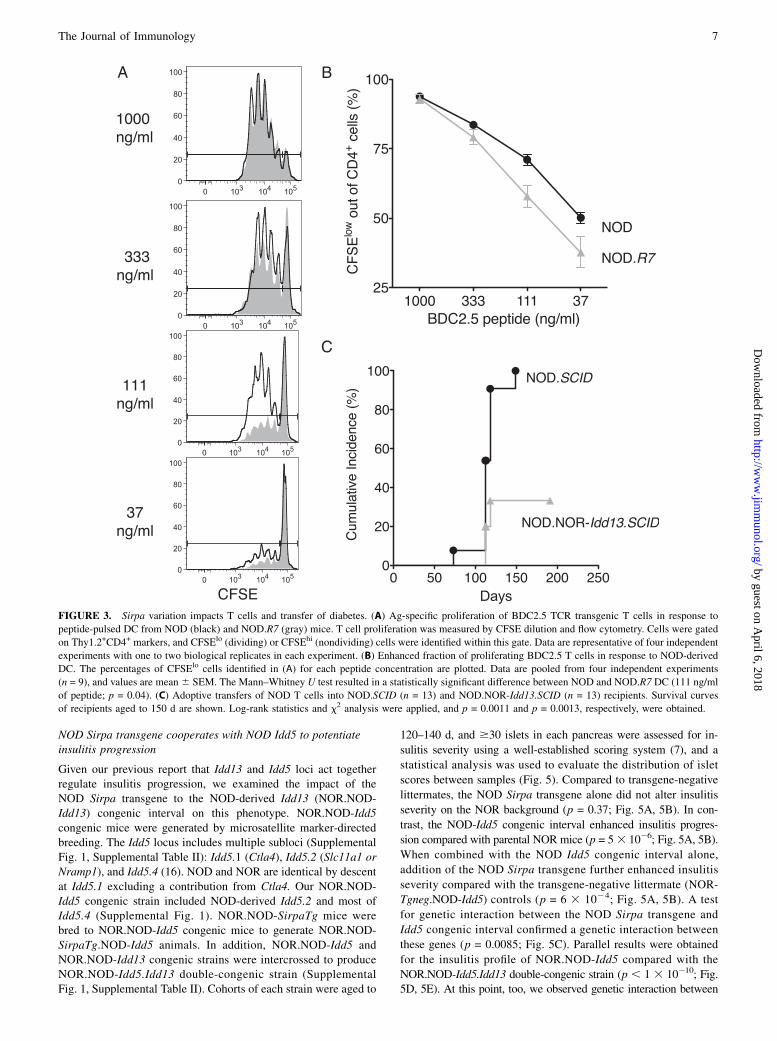
NOD Sirpa transgene cooperates with NOD Idd5 to potentiateinsulitis progression
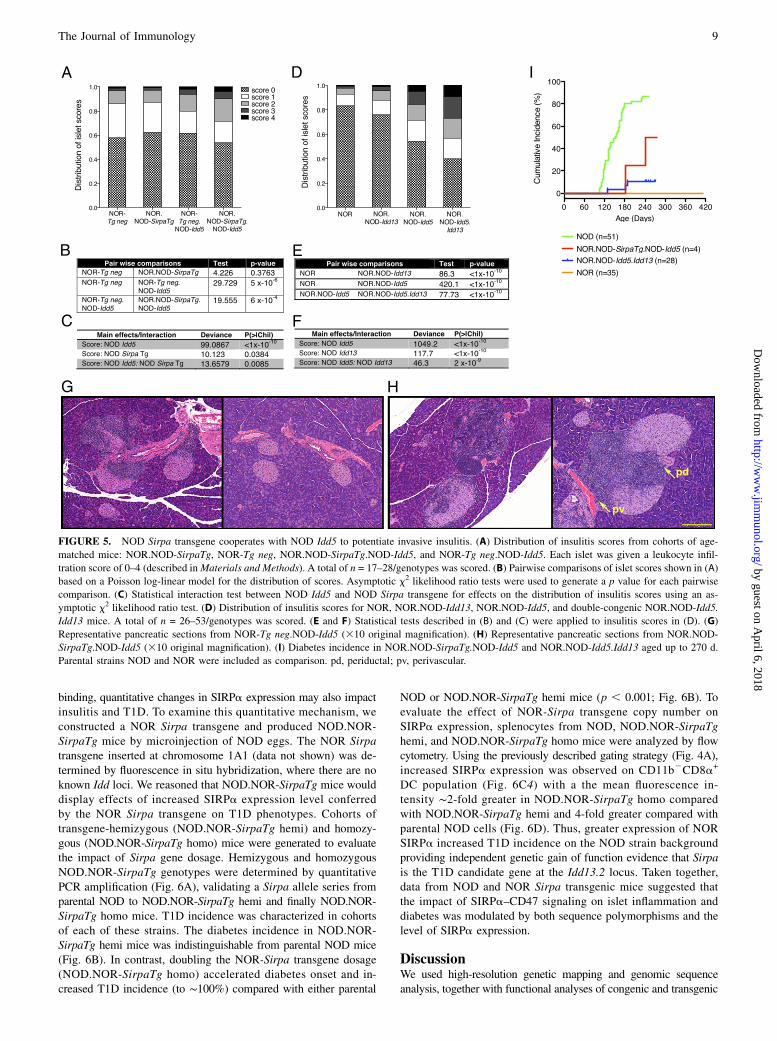
Given our previous report that Idd13 and Idd5 loci act togetherregulate insulitis progression, we examined the impact of theNOD Sirpa transgene to the NOD-derived Idd13 (NOR.NOD-Idd13) congenic interval on this phenotype. NOR.NOD-Idd5congenic mice were generated by microsatellite marker-directedbreeding. The Idd5 locus includes multiple subloci (SupplementalFig. 1, Supplemental Table II): Idd5.1 (Ctla4), Idd5.2 (Slc11a1 orNramp1), and Idd5.4 (16). NOD and NOR are identical by descentat Idd5.1 excluding a contribution from Ctla4. Our NOR.NOD-Idd5 congenic strain included NOD-derived Idd5.2 and most ofIdd5.4 (Supplemental Fig. 1). NOR.NOD-SirpaTg mice werebred to NOR.NOD-Idd5 congenic mice to generate NOR.NOD-SirpaTg.NOD-Idd5 animals. In addition, NOR.NOD-Idd5 andNOR.NOD-Idd13 congenic strains were intercrossed to produceNOR.NOD-Idd5.Idd13 double-congenic strain (SupplementalFig. 1, Supplemental Table II). Cohorts of each strain were aged to
120–140 d, and $30 islets in each pancreas were assessed for in-
sulitis severity using a well-established scoring system (7), and a
statistical analysis was used to evaluate the distribution of islet
scores between samples (Fig. 5). Compared to transgene-negative
littermates, the NOD Sirpa transgene alone did not alter insulitis
severity on the NOR background (p = 0.37; Fig. 5A, 5B). In con-
trast, the NOD-Idd5 congenic interval enhanced insulitis progres-
sion compared with parental NOR mice (p = 53 1026; Fig. 5A, 5B).
When combined with the NOD Idd5 congenic interval alone,
addition of the NOD Sirpa transgene further enhanced insulitis
severity compared with the transgene-negative littermate (NOR-
Tgneg.NOD-Idd5) controls (p = 6 3 1024; Fig. 5A, 5B). A test
for genetic interaction between the NOD Sirpa transgene and
Idd5 congenic interval confirmed a genetic interaction between
these genes (p = 0.0085; Fig. 5C). Parallel results were obtained
for the insulitis profile of NOR.NOD-Idd5 compared with the
NOR.NOD-Idd5.Idd13 double-congenic strain (p , 1 3 10210; Fig.5D, 5E). At this point, too, we observed genetic interaction between
FIGURE 3. Sirpa variation impacts T cells and transfer of diabetes. (A) Ag-specific proliferation of BDC2.5 TCR transgenic T cells in response to
peptide-pulsed DC from NOD (black) and NOD.R7 (gray) mice. T cell proliferation was measured by CFSE dilution and flow cytometry. Cells were gated
on Thy1.2+CD4+ markers, and CFSElo (dividing) or CFSEhi (nondividing) cells were identified within this gate. Data are representative of four independent
experiments with one to two biological replicates in each experiment. (B) Enhanced fraction of proliferating BDC2.5 T cells in response to NOD-derived
DC. The percentages of CFSElo cells identified in (A) for each peptide concentration are plotted. Data are pooled from four independent experiments
(n = 9), and values are mean6 SEM. The Mann–Whitney U test resulted in a statistically significant difference between NOD and NOD.R7 DC (111 ng/ml
of peptide; p = 0.04). (C) Adoptive transfers of NOD T cells into NOD.SCID (n = 13) and NOD.NOR-Idd13.SCID (n = 13) recipients. Survival curves
of recipients aged to 150 d are shown. Log-rank statistics and x2 analysis were applied, and p = 0.0011 and p = 0.0013, respectively, were obtained.
The Journal of Immunology 7
by guest on April 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
the Idd13 and Idd5 loci (p = 2 3 1029; Fig. 5F), recapitulating the
observation for NOR.NOD-SirpaTg.NOD-Idd5 insulitis severity.Thus, like the Idd13 congenic interval, the NOD Sirpa transgene
displayed strong interaction with the Idd5 locus, supporting Sirpa as
the gene responsible for insulitis progression at the Idd13.2 locus.The NOD Sirpa transgene effect was evident in the microanat-
omy of pancreatic sections from NOR.NOD-SirpaTg.NOD-Idd5
compared with transgene-negative littermates (Fig. 5G, 5H). Al-though NOR-Tgneg.NOD-Idd5 islets displayed mild peri-insulitis
with some invasive insulitis, NOR.SirpaTg.NOD-Idd5 mice dis-
played extensive islet infiltration (score $2) accompanied by per-iductal and perivascular inflammation. Strikingly, we observedprogression to T1D onset in 50% of NOR.NOD-SirpaTg.NOD-
Idd5 that aged to 270 d (Fig. 5I). Although the T1D outcome in thesmall cohort could not reach statistical significance, these obser-vations supported the potent interaction between the NOD Sirpatransgene and NOD Idd5 in driving b-cell–destructive insulitis.Taken together, these results supported Sirpa as a causal gene at theIdd13.2 locus by demonstrating that it collaborates with NOD Idd5to drive insulitis progression, a key step in diabetes pathogenesis.
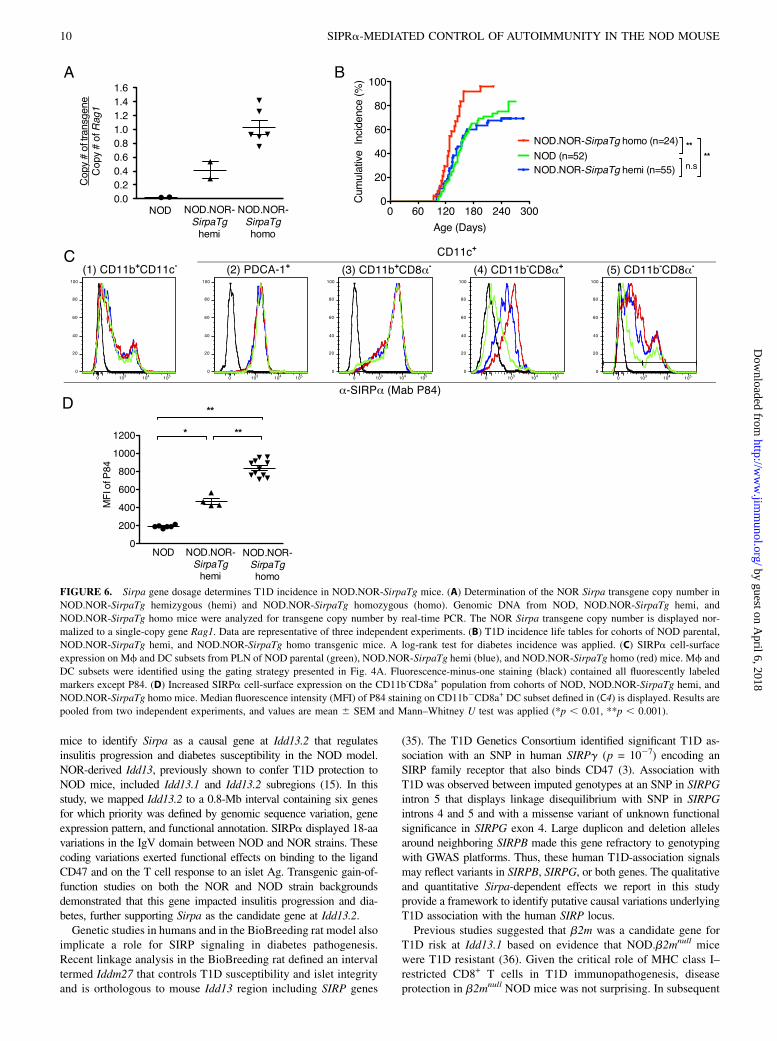
Diabetes kinetics is altered by Sirpa gene dosage
Although CD47 is not polymorphic between NOD and NOR, otherstudies show that CD47 expression varies during T cell activationand differentiation. These observations suggested that in additionto the qualitative, genotype-dependent difference in SIRPa–CD47
FIGURE 4. Expression pattern of the NOD Sirpa transgene. (A) Gating strategy to identify splenic Mf and DC subsets from transgenic (NOR.NOD-
SirpaTg) and nontransgenic littermates (NOR-Tg neg). Top panel: staining with CD3/CD19 was used as dump gate to enrich for myeloid cells. Second
panel: gates to define total CD11c+ DC and CD11b+CD11c2 Mf. Third panel: total CD11c+ cells were subdivided into PDCA-1+ (plasmacytoid DC) and
PDCA-12 (conventional DC). Bottom panel: Conventional DC were further refined into CD11b+CD8a2, CD11b2CD8a+, and CD11b2CD8a2 subsets. (B)
Binding of soluble hCD47-Fc protein to splenic Mf and DC subsets isolated from NOR.NOD-SirpaTg (gray) and NOR-Tg neg (black). The differential
binding of hCD47-Fc to NOD- and NOR-derived SIRPa allowed identification of NOD Sirpa transgene expressing cells in NOR.NOD-SirpaTg mice. (C)
Binding of SIRPa Ab (P84) to splenic Mf and DC subsets isolated from NOR.NOD-SirpaTg (gray) and NOR-Tg neg (black). This experiment was
performed twice with n = 5/genotype. SSC, side scatter.
8 SIPRa-MEDIATED CONTROL OF AUTOIMMUNITY IN THE NOD MOUSE
by guest on April 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
binding, quantitative changes in SIRPa expression may also impactinsulitis and T1D. To examine this quantitative mechanism, weconstructed a NOR Sirpa transgene and produced NOD.NOR-SirpaTg mice by microinjection of NOD eggs. The NOR Sirpatransgene inserted at chromosome 1A1 (data not shown) was de-termined by fluorescence in situ hybridization, where there are noknown Idd loci. We reasoned that NOD.NOR-SirpaTg mice woulddisplay effects of increased SIRPa expression level conferredby the NOR Sirpa transgene on T1D phenotypes. Cohorts oftransgene-hemizygous (NOD.NOR-SirpaTg hemi) and homozy-gous (NOD.NOR-SirpaTg homo) mice were generated to evaluatethe impact of Sirpa gene dosage. Hemizygous and homozygousNOD.NOR-SirpaTg genotypes were determined by quantitativePCR amplification (Fig. 6A), validating a Sirpa allele series fromparental NOD to NOD.NOR-SirpaTg hemi and finally NOD.NOR-SirpaTg homo mice. T1D incidence was characterized in cohortsof each of these strains. The diabetes incidence in NOD.NOR-SirpaTg hemi mice was indistinguishable from parental NOD mice(Fig. 6B). In contrast, doubling the NOR-Sirpa transgene dosage(NOD.NOR-SirpaTg homo) accelerated diabetes onset and in-creased T1D incidence (to ∼100%) compared with either parental
NOD or NOD.NOR-SirpaTg hemi mice (p , 0.001; Fig. 6B). Toevaluate the effect of NOR-Sirpa transgene copy number onSIRPa expression, splenocytes from NOD, NOD.NOR-SirpaTghemi, and NOD.NOR-SirpaTg homo mice were analyzed by flowcytometry. Using the previously described gating strategy (Fig. 4A),increased SIRPa expression was observed on CD11b2CD8a+
DC population (Fig. 6C4) with a the mean fluorescence in-tensity ∼2-fold greater in NOD.NOR-SirpaTg homo comparedwith NOD.NOR-SirpaTg hemi and 4-fold greater compared withparental NOD cells (Fig. 6D). Thus, greater expression of NORSIRPa increased T1D incidence on the NOD strain backgroundproviding independent genetic gain of function evidence that Sirpais the T1D candidate gene at the Idd13.2 locus. Taken together,data from NOD and NOR Sirpa transgenic mice suggested thatthe impact of SIRPa–CD47 signaling on islet inflammation anddiabetes was modulated by both sequence polymorphisms and thelevel of SIRPa expression.
DiscussionWe used high-resolution genetic mapping and genomic sequenceanalysis, together with functional analyses of congenic and transgenic
FIGURE 5. NOD Sirpa transgene cooperates with NOD Idd5 to potentiate invasive insulitis. (A) Distribution of insulitis scores from cohorts of age-
matched mice: NOR.NOD-SirpaTg, NOR-Tg neg, NOR.NOD-SirpaTg.NOD-Idd5, and NOR-Tg neg.NOD-Idd5. Each islet was given a leukocyte infil-
tration score of 0–4 (described inMaterials and Methods). A total of n = 17–28/genotypes was scored. (B) Pairwise comparisons of islet scores shown in (A)
based on a Poisson log-linear model for the distribution of scores. Asymptotic x2 likelihood ratio tests were used to generate a p value for each pairwise
comparison. (C) Statistical interaction test between NOD Idd5 and NOD Sirpa transgene for effects on the distribution of insulitis scores using an as-
ymptotic x2 likelihood ratio test. (D) Distribution of insulitis scores for NOR, NOR.NOD-Idd13, NOR.NOD-Idd5, and double-congenic NOR.NOD-Idd5.
Idd13 mice. A total of n = 26–53/genotypes was scored. (E and F) Statistical tests described in (B) and (C) were applied to insulitis scores in (D). (G)
Representative pancreatic sections from NOR-Tg neg.NOD-Idd5 (310 original magnification). (H) Representative pancreatic sections from NOR.NOD-
SirpaTg.NOD-Idd5 (310 original magnification). (I) Diabetes incidence in NOR.NOD-SirpaTg.NOD-Idd5 and NOR.NOD-Idd5.Idd13 aged up to 270 d.
Parental strains NOD and NOR were included as comparison. pd, periductal; pv, perivascular.
The Journal of Immunology 9
by guest on April 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
mice to identify Sirpa as a causal gene at Idd13.2 that regulatesinsulitis progression and diabetes susceptibility in the NOD model.NOR-derived Idd13, previously shown to confer T1D protection toNOD mice, included Idd13.1 and Idd13.2 subregions (15). In thisstudy, we mapped Idd13.2 to a 0.8-Mb interval containing six genesfor which priority was defined by genomic sequence variation, geneexpression pattern, and functional annotation. SIRPa displayed 18-aavariations in the IgV domain between NOD and NOR strains. Thesecoding variations exerted functional effects on binding to the ligandCD47 and on the T cell response to an islet Ag. Transgenic gain-of-function studies on both the NOR and NOD strain backgroundsdemonstrated that this gene impacted insulitis progression and dia-betes, further supporting Sirpa as the candidate gene at Idd13.2.Genetic studies in humans and in the BioBreeding rat model also
implicate a role for SIRP signaling in diabetes pathogenesis.Recent linkage analysis in the BioBreeding rat defined an intervaltermed Iddm27 that controls T1D susceptibility and islet integrityand is orthologous to mouse Idd13 region including SIRP genes
(35). The T1D Genetics Consortium identified significant T1D as-sociation with an SNP in human SIRPg (p = 1027) encoding anSIRP family receptor that also binds CD47 (3). Association withT1D was observed between imputed genotypes at an SNP in SIRPGintron 5 that displays linkage disequilibrium with SNP in SIRPGintrons 4 and 5 and with a missense variant of unknown functionalsignificance in SIRPG exon 4. Large duplicon and deletion allelesaround neighboring SIRPB made this gene refractory to genotypingwith GWAS platforms. Thus, these human T1D-association signalsmay reflect variants in SIRPB, SIRPG, or both genes. The qualitativeand quantitative Sirpa-dependent effects we report in this studyprovide a framework to identify putative causal variations underlyingT1D association with the human SIRP locus.Previous studies suggested that b2m was a candidate gene for
T1D risk at Idd13.1 based on evidence that NOD.b2mnull micewere T1D resistant (36). Given the critical role of MHC class I–restricted CD8+ T cells in T1D immunopathogenesis, diseaseprotection in b2mnull NOD mice was not surprising. In subsequent
FIGURE 6. Sirpa gene dosage determines T1D incidence in NOD.NOR-SirpaTg mice. (A) Determination of the NOR Sirpa transgene copy number in
NOD.NOR-SirpaTg hemizygous (hemi) and NOD.NOR-SirpaTg homozygous (homo). Genomic DNA from NOD, NOD.NOR-SirpaTg hemi, and
NOD.NOR-SirpaTg homo mice were analyzed for transgene copy number by real-time PCR. The NOR Sirpa transgene copy number is displayed nor-
malized to a single-copy gene Rag1. Data are representative of three independent experiments. (B) T1D incidence life tables for cohorts of NOD parental,
NOD.NOR-SirpaTg hemi, and NOD.NOR-SirpaTg homo transgenic mice. A log-rank test for diabetes incidence was applied. (C) SIRPa cell-surface
expression on Mf and DC subsets from PLN of NOD parental (green), NOD.NOR-SirpaTg hemi (blue), and NOD.NOR-SirpaTg homo (red) mice. Mf and
DC subsets were identified using the gating strategy presented in Fig. 4A. Fluorescence-minus-one staining (black) contained all fluorescently labeled
markers except P84. (D) Increased SIRPa cell-surface expression on the CD11b-CD8a+ population from cohorts of NOD, NOD.NOR-SirpaTg hemi, and
NOD.NOR-SirpaTg homo mice. Median fluorescence intensity (MFI) of P84 staining on CD11b2CD8a+ DC subset defined in (C4) is displayed. Results are
pooled from two independent experiments, and values are mean 6 SEM and Mann–Whitney U test was applied (*p , 0.01, **p , 0.001).
10 SIPRa-MEDIATED CONTROL OF AUTOIMMUNITY IN THE NOD MOUSE
by guest on April 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

analyses, it was demonstrated that transgenic expression of NOD-b2m restored T1D incidence on the NOD.b2mnull background, butexpression of a NOR-b2m transgene resulted in lower T1D incidence(37). In our hands, NOD mice congenic for a NOR-derived b2m-containing interval (NOD.R4; Fig. 1A), but not carrying NOD-derived alleles at Idd13.2, were diabetes susceptible, suggesting thatB2m allelic variation alone was insufficient to protect the mice. Alikely explanation for these disparate findings is that the NOD.b2mnull
strain reported previously harbored non-NOD–derived genomicregions extending well beyond b2m. To address this possibility,we performed high-resolution genotyping of the Idd13 region ofthe NOD.b2mnull genome and found that the 129/SV-derivedb2mnull gene was flanked by $1.96 Mb of B6 and 129/SV-derived segments that included all the genes in Idd13.2 interval(Supplemental Table III). The likely origins of this region is fromthe 129/SV-derived embryonic stem cells used to generate theb2mnull mutation (38) and subsequent crosses to the B6 strain. Thus,the discrepancy between the T1D susceptibility of the NOD.R4 con-genic strain reported in this study and the T1D resistance previouslyreported in NOD.b2mnull.NOR-b2m transgenic mice is that the pro-tection conferred by NOR-b2m also reflected the protective 129/SV-and B6 derived-Idd13.2 locus. This predicted b2m-Idd13.2 “gene 3gene” interaction highlights the complex genetic architecture of T1D.The T1D phenotype reflects interactions between multiple
susceptibility genes and environmental factors that collectivelyregulate the immune response. Of the human T1D genes in whichplausible causal variations have been reported (e.g., INS andPTPN22), they are common, functional variants (2). Such func-tional variations are distinct from loss of function mutationsobserved in monogenic forms of autoimmunity such as the tran-scription factor autoimmune regulator responsible for the rareautoimmune polyendocrine syndrome type 1 (39). The odds ratioscomputed for most T1D-associated SNPs identified by GWAS are,2 (3). Variants with low genetic effect sizes likely confer theireffects on the complex disease phenotype through gene–gene (andgene–environment) interactions. As we show in this study, theNOD model enables mechanistic analysis of gene–gene inter-actions under controlled environmental conditions that are farmore difficult to demonstrate in genetically heterogeneous humansubjects. In this study, we show that the effect of Sirpa is greatlypotentiated by functional interaction with the Idd5 locus. Geneticinteraction was observed between the NOD Idd13 locus and NODSirpa transgene with NOD Idd5, supporting Sirpa as the causalgene of Idd13.2 that drives insulitis progression together witha gene(s) at Idd5. Other examples of interlocus effects havebeen reported in the NOD model for Idd3+Idd10 and also Idd5.1/Idd5.3+Idd3 (40). In the context of the current study, the relevantregions of the Idd5 locus in this study include Idd5.2 (Slc11a1) andpart of Idd5.4 for which a strong candidate gene is not yet defined(Supplemental Fig. 1). Further mapping efforts to identify the Idd5sub loci interacting with Sirpa will provide a useful framework foridentification and mechanistic analysis of causal variants in humanT1D. Prioritizing T1D associated variants that act in convergentpathways may provide more effective therapeutic strategiesSIRPa–CD47 interaction is also required in other T cell–me-
diated autoimmune syndromes including colitis (23, 41) and ex-perimental autoimmune encephalomyelitis (EAE) (24). Mutantmice lacking the SIRPa cytoplasmic signaling domain were re-sistant to myelin oligodendrocyte glycoprotein–induced EAEand displayed reduced DC stimulation of myelin oligodendrocyteglycoprotein–dependent T cell proliferation (24). Similarly, CD472/2
mice were also EAE resistant due to impaired T cell responses (42).These observations highlight role of SIRPa–CD47 signaling in reg-ulating DC function and T cell activation (43).
We report in this paper that NOD variant of SIRPa displayedenhanced CD47 binding and was associated with greater isletantigen–specific T cell proliferation compared with the NORprotein. Moreover, NOD SIRPa expressed on a recipient’s mye-loid cells enabled greater diabetogenic potential of adoptivelytransferred NOD T cells. Collectively, these results suggest thatpolymorphisms affecting SIRPa–CD47 binding exerts controlover spontaneous NOD diabetes through a T cell–mediatedmechanism. Our study describes mechanisms through which Sirpaimpacts islet-autoimmunity and is one of multiple T1D-associatedgenes that alter the balance between lymphocyte activation andattenuation.GWAS reveal that many common human variants associated
with complex diseases reflect regulatory rather than coding var-iations (44). This study of Sirpa in the NOD model provides anexample of effects on autoimmunity conferred by coding poly-morphisms impacting receptor–ligand binding and also by geneticvariations that alter protein expression. Despite the lower level ofCD47 binding by the NOR compared with NOD SIRPa, an in-crease in NOR-Sirpa transgene dosage accelerated diabetes ki-netics and increased susceptibility, suggesting that SIRPa–CD47signaling outcomes are also subjected to quantitative influences.Identification of T1D-associated variants in immune receptors andfunctional analysis of their effects may provide targets to attenuateanti-islet immunity and improve outcomes of b-cell replacementstrategies in persons with diabetes.
AcknowledgmentsWe thank S. Rajakumar and R. Sampson for establishing the Sirpa trans-
genic mice genotyping assay, T. Prasolava for assistance with SIRPa
binding assay, and E. Brown (University of California, San Francisco,
San Francisco, CA) for the mCD47-Fc plasmid.
DisclosuresThe authors have no financial conflicts of interest.
References1. Driver, J. P., D. V. Serreze, and Y. G. Chen. 2011. Mouse models for the study of
autoimmune type 1 diabetes: a NOD to similarities and differences to humandisease. Semin. Immunopathol. 33: 67–87.
2. Polychronakos, C., and Q. Li. 2011. Understanding type 1 diabetes throughgenetics: advances and prospects. Nat. Rev. Genet. 12: 781–792.
3. Barrett, J. C., D. G. Clayton, P. Concannon, B. Akolkar, J. D. Cooper,H. A. Erlich, C. Julier, G. Morahan, J. Nerup, C. Nierras, et al; Type 1 DiabetesGenetics Consortium. 2009. Genome-wide association study and meta-analysisfind that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 41: 703–707.
4. Akkaya, M., and A. N. Barclay. 2013. How do pathogens drive the evolution ofpaired receptors? Eur. J. Immunol. 43: 303–313.
5. Barclay, A. N., and D. Hatherley. 2008. The counterbalance theory for evolutionand function of paired receptors. Immunity 29: 675–678.
6. Pociot, F., B. Akolkar, P. Concannon, H. A. Erlich, C. Julier, G. Morahan,C. R. Nierras, J. A. Todd, S. S. Rich, and J. Nerup. 2010. Genetics of type 1diabetes: what’s next? Diabetes 59: 1561–1571.
7. Fox, C. J., A. D. Paterson, S. M. Mortin-Toth, and J. S. Danska. 2000. Twogenetic loci regulate T cell-dependent islet inflammation and drive autoimmunediabetes pathogenesis. Am. J. Hum. Genet. 67: 67–81.
8. Prochazka, M., D. V. Serreze, W. N. Frankel, and E. H. Leiter. 1992. NOR/Ltmice: MHC-matched diabetes-resistant control strain for NOD mice. Diabetes41: 98–106.
9. Ivakine, E. A., O. M. Gulban, S. M. Mortin-Toth, E. Wankiewicz, C. Scott,D. Spurrell, A. Canty, and J. S. Danska. 2006. Molecular genetic analysis of theIdd4 locus implicates the IFN response in type 1 diabetes susceptibility innonobese diabetic mice. J. Immunol. 176: 2976–2990.
10. Gough, P. J., S. Gordon, and D. R. Greaves. 2001. The use of human CD68transcriptional regulatory sequences to direct high-level expression of class Ascavenger receptor in macrophages in vitro and in vivo. Immunology 103: 351–361.
11. Venables, W. N., and B. D. Ripley. 2002. Modern applied statistics withS. Springer, New York.
12. R Development Core Team. 2011. R: A Language and Environment for Statis-tical Computing. R Foundation for Statistical Computing, Vienna, Austria.
13. Johansen, M. L., and E. J. Brown. 2007. Dual regulation of SIRPalpha phos-phorylation by integrins and CD47. J. Biol. Chem. 282: 24219–24230.
The Journal of Immunology 11
by guest on April 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

14. Takenaka, K., T. K. Prasolava, J. C. Wang, S. M. Mortin-Toth, S. Khalouei,O. I. Gan, J. E. Dick, and J. S. Danska. 2007. Polymorphism in Sirpa modulatesengraftment of human hematopoietic stem cells. Nat. Immunol. 8: 1313–1323.
15. Serreze, D. V., M. Bridgett, H. D. Chapman, E. Chen, S. D. Richard, andE. H. Leiter. 1998. Subcongenic analysis of the Idd13 locus in NOD/Lt mice:evidence for several susceptibility genes including a possible diabetogenic rolefor beta 2-microglobulin. J. Immunol. 160: 1472–1478.
16. Hunter, K., D. Rainbow, V. Plagnol, J. A. Todd, L. B. Peterson, and L. S. Wicker.2007. Interactions between Idd5.1/Ctla4 and other type 1 diabetes genes. J.Immunol. 179: 8341–8349.
17. Mao, H. Z., E. T. Roussos, and M. Peterfy. 2006. Genetic analysis of thediabetes-prone C57BLKS/J mouse strain reveals genetic contribution frommultiple strains. Biochim. Biophys. Acta 1762: 440–446.
18. Hatherley, D., S. C. Graham, K. Harlos, D. I. Stuart, and A. N. Barclay. 2009.Structure of signal-regulatory protein alpha: a link to antigen receptor evolution.J. Biol. Chem. 284: 26613–26619.
19. Oldenborg, P. A., A. Zheleznyak, Y. F. Fang, C. F. Lagenaur, H. D. Gresham, andF. P. Lindberg. 2000. Role of CD47 as a marker of self on red blood cells.Science 288: 2051–2054.
20. Gardai, S. J., K. A. McPhillips, S. C. Frasch, W. J. Janssen, A. Starefeldt,J. E. Murphy-Ullrich, D. L. Bratton, P. A. Oldenborg, M. Michalak, andP. M. Henson. 2005. Cell-surface calreticulin initiates clearance of viable orapoptotic cells through trans-activation of LRP on the phagocyte. Cell 123: 321–334.
21. Nuvolone, M., V. Kana, G. Hutter, D. Sakata, S. M. Mortin-Toth, G. Russo,J. S. Danska, and A. Aguzzi. 2013. SIRPa polymorphisms, but not the prionprotein, control phagocytosis of apoptotic cells. J. Exp. Med. 210: 2539–2552.
22. Saito, Y., H. Iwamura, T. Kaneko, H. Ohnishi, Y. Murata, H. Okazawa,Y. Kanazawa, M. Sato-Hashimoto, H. Kobayashi, P. A. Oldenborg, et al. 2010.Regulation by SIRPa of dendritic cell homeostasis in lymphoid tissues. Blood116: 3517–3525.
23. Fortin, G., M. Raymond, V. Q. Van, M. Rubio, P. Gautier, M. Sarfati, andD. Franchimont. 2009. A role for CD47 in the development of experimentalcolitis mediated by SIRPalpha+CD103- dendritic cells. J. Exp. Med. 206: 1995–2011.
24. Tomizawa, T., Y. Kaneko, Y. Kaneko, Y. Saito, H. Ohnishi, J. Okajo,C. Okuzawa, T. Ishikawa-Sekigami, Y. Murata, H. Okazawa, et al. 2007. Re-sistance to experimental autoimmune encephalomyelitis and impaired T cellpriming by dendritic cells in Src homology 2 domain-containing protein tyro-sine phosphatase substrate-1 mutant mice. J. Immunol. 179: 869–877.
25. Veillette, A., E. Thibaudeau, and S. Latour. 1998. High expression of inhibitoryreceptor SHPS-1 and its association with protein-tyrosine phosphatase SHP-1 inmacrophages. J. Biol. Chem. 273: 22719–22728.
26. Seiffert, M., P. Brossart, C. Cant, M. Cella, M. Colonna, W. Brugger, L. Kanz,A. Ullrich, and H. J. B€uhring. 2001. Signal-regulatory protein alpha (SIRPalpha)but not SIRPbeta is involved in T-cell activation, binds to CD47 with high af-finity, and is expressed on immature CD34(+)CD38(-) hematopoietic cells.Blood 97: 2741–2749.
27. Brown, E. J., and W. A. Frazier. 2001. Integrin-associated protein (CD47) and itsligands. Trends Cell Biol. 11: 130–135.
28. Brooke, G., J. D. Holbrook, M. H. Brown, and A. N. Barclay. 2004.Human lymphocytes interact directly with CD47 through a novel member of thesignal regulatory protein (SIRP) family. J. Immunol. 173: 2562–2570.
29. Van, V. Q., M. Raymond, N. Baba, M. Rubio, K. Wakahara, S. A. Susin, andM. Sarfati. 2012. CD47(high) expression on CD4 effectors identifies functionallong-lived memory T cell progenitors. J. Immunol. 188: 4249–4255.
30. Rebres, R. A., J. M. Green, M. I. Reinhold, M. Ticchioni, and E. J. Brown. 2001.Membrane raft association of CD47 is necessary for actin polymerization andprotein kinase C theta translocation in its synergistic activation of T cells. J. Biol.Chem. 276: 7672–7680.
31. Reinhold, M. I., F. P. Lindberg, G. J. Kersh, P. M. Allen, and E. J. Brown. 1997.Costimulation of T cell activation by integrin-associated protein (CD47) is anadhesion-dependent, CD28-independent signaling pathway. J. Exp. Med. 185: 1–11.
32. Stadinski, B. D., T. Delong, N. Reisdorph, R. Reisdorph, R. L. Powell,M. Armstrong, J. D. Piganelli, G. Barbour, B. Bradley, F. Crawford, et al. 2010.Chromogranin A is an autoantigen in type 1 diabetes. Nat. Immunol. 11: 225–231.
33. Judkowski, V., C. Pinilla, K. Schroder, L. Tucker, N. Sarvetnick, andD. B. Wilson. 2001. Identification of MHC class II-restricted peptide ligands,including a glutamic acid decarboxylase 65 sequence, that stimulate diabeto-genic T cells from transgenic BDC2.5 nonobese diabetic mice. J. Immunol. 166:908–917.
34. Rohane, P. W., A. Shimada, D. T. Kim, C. T. Edwards, B. Charlton, L. D. Shultz,and C. G. Fathman. 1995. Islet-infiltrating lymphocytes from prediabetic NODmice rapidly transfer diabetes to NOD-scid/scid mice. Diabetes 44: 550–554.
35. Wallis, R. H., K. Wang, L. Marandi, E. Hsieh, T. Ning, G. Y. Chao, J. Sarmiento,A. D. Paterson, and P. Poussier. 2009. Type 1 diabetes in the BB rat: a polygenicdisease. Diabetes 58: 1007–1017.
36. Serreze, D. V., E. H. Leiter, G. J. Christianson, D. Greiner, and D. C. Roopenian.1994. Major histocompatibility complex class I-deficient NOD-B2mnull miceare diabetes and insulitis resistant. Diabetes 43: 505–509.
37. Hamilton-Williams, E. E., D. V. Serreze, B. Charlton, E. A. Johnson,M. P. Marron, A. Mullbacher, and R. M. Slattery. 2001. Transgenic rescueimplicates beta2-microglobulin as a diabetes susceptibility gene in nonobesediabetic (NOD) mice. Proc. Natl. Acad. Sci. USA 98: 11533–11538.
38. Koller, B. H., and O. Smithies. 1989. Inactivating the beta 2-microglobulin locusin mouse embryonic stem cells by homologous recombination. Proc. Natl. Acad.Sci. USA 86: 8932–8935.
39. Finnish-German APECED Consortium. 1997. An autoimmune disease,APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat. Genet. 17: 399–403.
40. Cordell, H. J., J. A. Todd, N. J. Hill, C. J. Lord, P. A. Lyons, L. B. Peterson,L. S. Wicker, and D. G. Clayton. 2001. Statistical modeling of interlocusinteractions in a complex disease: rejection of the multiplicative model ofepistasis in type 1 diabetes. Genetics 158: 357–367.
41. Bian, Z., Y. Guo, B. Ha, K. Zen, and Y. Liu. 2012. Regulation of the inflam-matory response: enhancing neutrophil infiltration under chronic inflammatoryconditions. J. Immunol. 188: 844–853.
42. Han, M. H., D. H. Lundgren, S. Jaiswal, M. Chao, K. L. Graham, C. S. Garris,R. C. Axtell, P. P. Ho, C. B. Lock, J. I. Woodard, et al. 2012. Janus-like opposingroles of CD47 in autoimmune brain inflammation in humans and mice. J. Exp.Med. 209: 1325–1334.
43. Barclay, A. N., and T. K. Van den Berg. 2014. The interaction between signalregulatory protein alpha (SIRPa) and CD47: structure, function, and therapeutictarget. Annu. Rev. Immunol. 32: 25–50.
44. Maurano, M. T., R. Humbert, E. Rynes, R. E. Thurman, E. Haugen, H. Wang,A. P. Reynolds, R. Sandstrom, H. Qu, J. Brody, et al. 2012. Systematic locali-zation of common disease-associated variation in regulatory DNA. Science 337:1190–1195.
45. Dinarello, C. A. 2009. Immunological and inflammatory functions of theinterleukin-1 family. Annu. Rev. Immunol. 27: 519–550.
46. van Beek, E. M., F. Cochrane, A. N. Barclay, and T. K. van den Berg. 2005.Signal regulatory proteins in the immune system. J. Immunol. 175: 7781–7787.
47. Takenaka, K., T. K. Prasolava, J. C. Wang, S. M. Mortin-Toth, S. Khalouei,O. I. Gan, J. E. Dick, and J. S. Danska. 2007. Polymorphism in Sirpa modulatesengraftment of human hematopoietic stem cells. Nat. Immunol. 8: 1311–1323.
48. Kieffer, B. L., and C. Gaveriaux-Ruff. 2002. Exploring the opioid system bygene knockout. Prog. Neurobiol. 66: 285–306.
49. Yasuda, Y., Y. Miyamoto, T. Yamashiro, M. Asally, A. Masui, C. Wong,K. L. Loveland, and Y. Yoneda. 2012. Nuclear retention of importin alphacoordinates cell fate through changes in gene expression. EMBO J. 31: 83–94.
12 SIPRa-MEDIATED CONTROL OF AUTOIMMUNITY IN THE NOD MOUSE
by guest on April 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from