regulated bythe malt product - journal of bacteriology - american
TRANSCRIPT

JOURNAL OF BACTERIOLOGY, Nov. 1985, p. 639-645 Vol. 164, No. 20021-9193/85/110639-07$02.00/0Copyright C) 1985, American Society for Microbiology
Structure of Two Divergent Promoters Located in Front of the GeneEncoding Pullulanase in Klebsiella pneumoniae and Positively
Regulated by the malT ProductCHRISTINE CHAPON AND OLIVIER RAIBAUD*
Unite de Genetique Moleculaire, Institut Pasteur, 75724 Paris Cedex 15, France
Received 29 April 1985/Accepted 26 July 1985
Pullulanase ig ph extraceliular starch-debranching enzyme produced by Klebsiella pneumoniae. When itsstructural gene, pulA, is introduced into Escherichia coli, it is controlled by malT, the positive reguilator geneof the maltose regulon. Characterization of the region 5' to pulA and of the beginning of the gene describedherein demonstrate that (i) pullulanase is probably a lipoprotein; (ii) an additional malT-controlled promoter(the malX promoter) lies adjacent to the pulA promoter and is oriented in the opposite direction; (iii) in commonwith the three previously described malT-controlled promoters~the puUl and maiX promoters have a conservedhexanucleotide (consensus sequence, 5'-GGAGGA) 35 base pairs upstream from the transcription initiationsite; and (iv) upstream from this conserved hexanucleotide the pulA and maiX promoters differ from the othermal promoters in that they lack any detectable binding site for the cyclic AMP-binding protein.
Kiebsiella pneumoniae secretes an enzyme, pullulanase,which cleaves a(1-6) bonds in branched a(1-4), oa(1-6)glucans such as amylopectin and glycogen, producing linearox(1-4) polymers (3). Pullulanase also converts pullulan,which is a linear polymer composed of maltotriose unitslinked by a(1-6) bonds, into maltotriose (3). The synthesis ofpullulanase is induced when cells are grown in the presenceof maltose, maltodextrin, or pullulan and is sensitive tocatabolite repression (17). As an extension of our studies onthe maltose regulon of Escherichia coli, we recently startedanalyzing the regulation of pullulanase expression in K.pneumoniae. We have cloned pulA, the structural gene forpullulanase, and introduced it into E. coli, which does notnormally produce this enzyme (10, 18). In E. coli, pulA wasstill maltose inducible and was controlled by the product otmalT, the positive regulator gene of the maltose regulon (9,18). This result provided a strong indication that (i) a genehomologous to malT exists in K. pneumoniae, (ii) theproduct of this gene controls the expression ofpulA, and (iii)the malT products of the two species are at least partlyinterchangeable. In the present work, we analyzed thestructure of the promoter which controls pulA expression,with the aim of comparing it with that of the three previouslydescribed malT-controlled promoters in E. coli, i.e., thepromoters of the malPQ, malEFG, and malK-lamB operons(1, 23). An unexpected outcome of this work has been thefinding that an additional malT-controlled promoter liesadjacent to the pulA promoter and is oriented in the oppositedirection. Also unexpected has been the nature of theamino-terminal sequence of the pulA product, as it could bepredicted from the DNA sequence: it suggests thatpullulanase is a lipoprotein.
MATERIALS AND METHODSStrains, plasmids, and media. All bacterial strains were
derived from E. coli K-12 pop3, our designation for strainMC4100 (4), which is F- araD139 AlacU169 rpsL relA thiA.Plasmid pOM50 is a derivative of pUR222 (27) into which a212-base-pair (bp) fragment containing the malT promoter
* Corresponding author.
has been inserted (5). Plasmid pSB118 was constructed byP. Stragier (personal communication). It is a derivative ofpUC18 (19) in which the polylinker is bracketed by two EcoRIsites. PlasmidpOM102 is pSB118 with a 276-bp TaqI fragmentcontaining thepulA and malX promoters inserted into its AccIsite. M13pul3 and M13pul4 are derivatives of M13 carryingportions ofthe 690-bp fragment which contains the pulAp andmalXp promoters. M13pul3 was obtained by cloning the DdeI(533)-TaqI (480) fragment (see Fig. 2 for sites) between theEcoRI and AccI sites of M13mpll, after filling in the EcoRIand DdeI extremity with the DNA polymerase I Klenowfragment. M13pul4 was constructed by cloning the EcoRI(684)-HincII (152) fragment between the EcoRI and HindIllsites of M13mplO, after filling in the Hindlll extremity withthe Klenow fragment.Most media and techniques, including the use of BAL 31
to create deletions in vitro, have been described previously(22).
Transfer of DNA fragments in front of the malPQ operon. Ageneral technique for inserting DNA fragments in front ofthe malPQ operon was described previously (24). We havenow improved this technique slightly. In the original proce-dure the DNA fragment was cloned into the unique EcoRIsite of a plasmid called pOM40. In that plasmid the tet genewas controlled by an active malPQ promoter, inside ofwhich the EcoRI site was located. We have now constructeda derivative of pOM40, called pOM41, in which the regionextending from positions -26 to -155 in the promoter hasbeen deleted and replaced by an EcoRI linker. This mutationwas called malPpA534. One advantage of plasmid pOM41 isthat the cyclic AMP-binding protein (CAP) and MalT pro-tein-binding sites at the malPQ promoter have been re-moved. Indeed, these regions could have interfered with thefunctioning of promoters present in the cloned fragment.Another advantage is that pOM41, unlike pOM40, fails toexpress the tet gene because malPp has been deleted,thereby permitting a positive selection (Tetr) for recombi-nant plasmids into which an active promoter has beencloned. The promoter-containing fragment was then trans-ferred onto the chromosome as previously described by
639
on Decem
ber 31, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

640 CHAPON AND RAIBAUD
selecting for reciprocal recombination events in a straincarrying deletion AmaIA510 (24).
Reverse transcriptase mapping of transcription initiationstart points. Single-stranded DNA fragments used as primerswere prepared as follows. Approximately 0.2 ,g of single-stranded DNA from the M13pul3 and M13pul4 phages hy-bridized with the M13 17-mer (Amersham) were incubatedfor 15 min at 20°C with 2 ,uCi of [a-32P]dGTP, 0.1 mM dATP,dCTP, and dTTP, and 1 U of the Klenow fragment of DNApolymerase. Cold dGTP was then added (0.1 mM), and theincubation was continued for an additional 15 min. The DNAwas then digested with TaqI and denatured by incubation for2 min at 90°C in the presence of 90% formamide. Theradioactive single-stranded DNA primers were then purifiedby polyacrylamide gel electrophoresis in the presence ofurea (28). These primers were coprecipitated with 25 ,ug ofthe mRNA-containing preparations, obtained as describedpreviously (8). The pellets were dissolved in 50% formam-ide-1 mM EDTA-0.4 M NaCl-20 mM HEPES (N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid) (pH 6.5),heated for 10 min at 75°C, and incubated for 16 h at 420C inan oven to allow hybridization. Elongation of the DNAprimer by reverse transcriptase and analysis of the productswere performed as described previously (8).
REStJLTSLocation and orientation of puL4 on an EcoRI DNA frag-
ment from K. pneumoniae. A 6.1-kilobase (kb) EcoRI DNAfragment from K. pneumoniae ATCC 15050 had previouslybeen shown to carry the entire pulA gene, including itspromoter (18). This fragment, which originally had beencloned in phage X, was subcloned in a pBR322-derivedplasmid, thus yielding pOM100 (Fig. 1). As this plasmid alsocarried the E. coli malT gene, the strains harboring itoverproduced the MalT protein and thus (18) expressed thepulA gene constitutively and appeared as Pul' on pullulan-containing indicator medium. The position of pulA on the6.1-kb DNA fragment was then determined by deletionmapping. In vitro deletion of the 2.2-kb BglII-BglIll fragmentcontaining the right end of the fragment (Fig.lb) had noeffect on pullulanase production. A set of deletions werethen constructed which originated from the left end of thefragment (Fig. lc). Of the deletions tested, the shortestwhich eliminated pullulanase production was 4pul-3,whereas the longest which had no effect on pullulanase wasApul-2. In these experiments the strains which expressedpullulanase activity 4lso produced an intact pullulanaseprotein, which migrated as a 145,000-dalton polypeptide insodium dodecyl sulfate-polyacrylamide gels (12). From thedata in Fig. 1 we conclude that pulA is located between theunique BglII site on the original 6.1-kb DNA fragment andthe right endpoint of deletion Apul-2. The distance betweenthese two points, 3.8 kb, appears just sufficient to encode aca. 145,000-dalton polypeptide.Sequencing data (not shown) then revealed that the right
endpoint of Apul-5 was located within a unique open readingframe at least 230 nucleotides long and oriented from right toleft. Since Apul-5 inactivated pulA this provided a strongindication that Apul-5 penetrated into the coding part of thisgene which therefore had to be oriented from right to left.Two MalT-dependent promoters are on the puL4 DNA
fragment. To analyze the promoters carried by the 6.1-kbEcoRI fragment, we used a technique recently developed inour laboratory (24). This technique allows one to detect, onany DNA fragment, the presence of promoters active in E.coli. First, the fragment is cloned into the single EcoRI site
(a)
amp
malT
amp
(b)
r
(c)
Pto
E pOM 100 8 E BI ~~~~~~.1I.
pu/A maiP'
!
( EE pOM 101 BI I
I"lt pulA I
H C H C H H BoII
A2IJ65_
pu/A
HB
ldl&'-I4I
(d)
malT ma/Pm a/Q(e) pop2297 1 I_ i
malT malP ma/Qpop 2298 Ii i I
FIG. 1. Mapping of pulA and of two maltose-dependent promot-ers on the 6.1-kb EcoRI fragment. (a) Plasmid pOM100 was con-structed by inserting a 6.1-kb EcoRI fragment from K. pneumoniaeATCC 15050 (open bar) containing the pulA gene (hatched bar) intothe EcoRI site of pOM40. (b) pOM101 was derived from pOM1lO bydeleting the 2.2-kb segment located between the two PglII sites.This plasmid still contained an intact pulA gene and had a uniqueEcoRI site. (c) A series of deletions in the left end of the K.pneumoniae fragment were obtained by linearizing pOM101 withEcoRI, treating it with BAL 31, and ligating in the presence of anexcess of EcoRI linker. The EcoRI-BglII fragments from thesedeleted plasmids were then recloned between the EcoRI and BglIIsites of pOM101 to obtain a series of deletionts which all had thesame left endpoint (Apul-2, -3, 4, and -5). (d) The unique BglII siteof pOM101 Apul4 was converted into an EcoRI site by treating theplasmid with BglII, filling in the extremities with the Klenowfragment of DNA polymerase, and ligating in the presence of anexcess of EcoRI linker. The resulting plasmid contained a 690-bpEcoRI fragment which was cloned into pOM41 and transferred ontothe chromosome as previously described (24). (e) Two bacterialstrains were thus obtained, pop2297 and pop2298, corresonding tothe two possible orientations of the 690-bp EcoRI fragment. In thesestrains the malPQ operon was controlled by either of the twomaltose-dependent promoters present on the 690-bp EcoRI frag-ment. The restriction sites are designated as follows: E, EcoRI; B,BgII; Ba, BamHI; H, HincIl; C, ClaI.
of a plasmid called pOM41, in such a way that the tet gene ofthis plasmid becomes activated if the fragment contains aproperly oriented promoter. In a second step, the fragment istransferred onto the chromosome (by homologous recombi-nation) in place of the promoter of the malPQ operon. Theefficiency of the promoter can then be deduced from thelevel of expression of gene malQ.When the entire 6.1-kb EcoRI fragment was cloned into
pOM41, the tet gene of the plasmid was activated, whateverthe orientation of the fragment. Therefore, at least twopromoters had to be present on the fragment, one oriented ineach direction. When the fragment was transferred onto thechromosome in the two possible orientations, one of the
J. BACTERIOL.
on Decem
ber 31, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

PULLULANASE PROMOTER IN K. PNEUMONIAE 641
TABLE 1. Maltose-dependent promoter activitya
Amylomaltase (U/mg)Strains Relevant genotype Glycerol Maltose
pop3 malPp+ 10 360pop2297 maIPpA534: :pulpi 5 164pop2298 malPpA534::pulp2 2 109
a The enzyme amylomaltase (malQ product) was assayed as describedpreviously (23) after growth in minimal medium (M63) containing eitherglycerol alone or glycerol plus maltose, each at a concentration of 0.4%. Theconstruction of strains pop2297 and pop2298 is described in Fig. 1.
promoters, oriented from left to right (see Fig. la for theorientation) revealed itself to be maltose dependent, whereasthe other, oriented from right to left, was not. Since pulAexpression was known to be induced by maltose, it wassomewhat surprising to find that the orientation of themaltose-controlled promoter was the opposite of the orien-tation which we assumed to be that of the pulA gene (seeprevious section). One possible interpretation was that the
val ala gin asn alapulA 3-.ccttaaggTA CTG CCG GAC TAA GCG
ECORI
maltose-controlled promoter which we had detected was notthat of the pulA gene but that of another maltose-controlledtranscription unit. Indeed, the 6.1-kb EcoRI fragment wasshown by subcloning to contain two maltose-controlledpromoters, oriented in opposite directions. Both of thesepromoters turned out to be located in a 690-bp DNA segmentextending from the unique BglII site to the right endpoint ofdeletion Apu14 (Fig. ld). When this short fragment wascloned into pOM41, an activation of gene tet was observedonce again. This occurred irrespective of the orientation ofthe fragment, indicating the existence of two promoterswhich were oriented in opposite directions. In this case,however, the activity of both promoters was maltose depen-dent (Table 1). One of these, oriented from right to left, ismost likely the pulA promoter. Previous results have indeedshown that a TnS insertion located approximately 600 bp tothe left of the BglII site prevents pullulanase production (18).The other promoter, oriented from left to right, must controlan additional maltose-dependent transcript of unknownfunction.
Structure of the maltose-dependent promoters. The
thr ala met val ala glu gly pro val ala val aspGCA GCG GTA GTG CCG AAG CGG TCC TTG GCG GTG TAG
650
pro leu arg val val val asp gin asn asp pro asn gly pro asn asp pro ser gly ser ser ser ser ser serCCC ATT TGC TTG TTG CTG TAG GAC CAA TAG TCC TAA CGG TCC CAA TAG TCC ACT TGG TCT CCT TCT TCT ACT TCT. * * * * **aIt
600
ser asn asp cys gly ser leu leu ile leu ser gly leuTGA TAA TAG CGT CGG CGA TTC ATT TTA GTT GCT CGG TTC
A *
DDEI
phe leu ala asn cys thr tyr arg leu metCTT ATC CCG TAA TGT CCA TAT AGA CTC GTA
500 DDEI
5'.TAAAG ATCGCAATTAGATATTTTTGTTTTCTCCGGTTCA ATCCTTAGGGATTATACGAATTATTAAATACTTTCCATTCTTTTCTACAAATAGACATTT C TAGCGTTAATCT|ATAAAAACAAAAGAGGCCAAGT GTAGGTAA
A ;n Ann
AACAATTAAT-CGCGCGCGfiCCGCGGCGAJJTJJJ jaCCTCCAAAAATAAAAAGG ATGGACCCGGATAGCGAAACTCATTATGATITCACGACGACAAATCCGTTGTTAATTA CCCCCCCGCATCCACCGGCCCCCTCATTTTTATTTTTCC AC CTGGGCCTATCGCTTTGAGT AATATAAGTGCTGCTGTTT-5'
metCTTTCTCGCAATAAAAGCTCGCAATATAAAAAAACCACCCACCAATATAGGTAGGGGTACTGTATCGCCTCGAGGGAAATAAA ATG
250 TAQI 200
ser asn glyAGC AAC GGA
350 300
ile lys arg tyr ileATA AAA AGA TAT ATT
met leu val thr leuATG CTC GTC ACC TTA
100
pro ala ile glu metCCG GCT ATT GAG ATG
ser val met gln leu thr leu ile asn lys lys his met his tyr alaTCT GTG ATG CAG TTA ACA CTA ATA AAT AAA AAA CAC ATG CAT TAC GCA
H*NAHINCII 150
pro his ile alaCCG CAT ATT GCC
leu leu ile gly gin gin leu ala lys leu ser trp arg met ile leu pro ala tyr serTTA CTG ATT GGC CAA CAG CTG GCA AAA TTA AGC TGG CGA ATG ATA CTC CCT GCA TAC TCT
50
asn asp ala thr gluAAT GAC GCG ACG GAG
0
i I eATCggaattcc-3
ECORI
maiX
FIG. 2. Sequence of a DNA fragment containing the pulA and maiX promoters. The sequence is that of the 690-bp EcoRI fragmentdescribed in Fig. ld which contains the pulA and malX promoters. The two strands are shown only for the region extending between the +5positions of the two promoters. On both sides of this region the only strand shown is the one which has the same polarity as the mRNA. Thepossible Shine and Dalgarno sequences for these two genes (pulA and maiX) are underlined. The -10 regions are boxed in rectangles, andthe sequences homologous to the 5'-GGATGA consensus hexamer (see text) are boxed in arrowheads. The heavy line corresponds to theG-C-rich sequence discussed in the text. Sequences in lower case at both ends of the sequence correspond to the EcoRI linkers introducedin the constructions.
550
AGCTATCATAQ I
VOL. 164, 1985
14ztJU ZIJZJ
on Decem
ber 31, 2018 by guesthttp://jb.asm
.org/D
ownloaded from
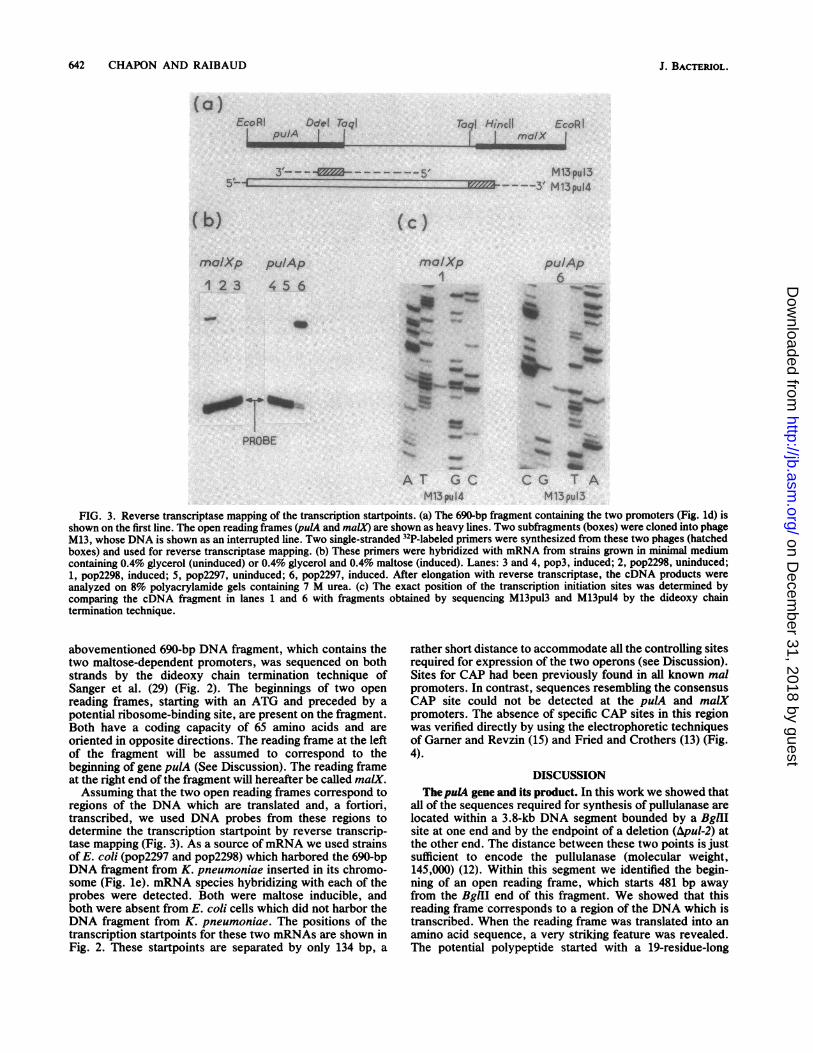
642 CHAPON AND RAIBAUD
EcoRI Ddel TaqII JuA I I
3"-- --CMB- - - - - - --5.5f-.
Taq Hincll EcoR II I malX
M13pul3MMM - - -3rM 13 pu14
(c)
mOaXp pulAp
1 23 45 6
-
PROBE
malXp1
__
.w
A T GOCM13pui14
pulAp6
*a"a _:
1%w-C
I.",
,=f"I"M _..11
_#*
_t,-_* 4f
_"C G
M1_1
FIG. 3. Reverse transcriptase mapping of the transcription startpoints. (a) The 690-bp fragment containing the two promoters (Fig. ld) isshown on the first line. The open reading frames (pulA and maiX) are shown as heavy lines. Two subfragments (boxes) were cloned into phageM13, whose DNA is shown as an interrupted line. Two single-stranded 32P-labeled primers were synthesized from these two phages (hatchedboxes) and used for reverse transcriptase mapping. (b) These primers were hybridized with mRNA from strains grown in minimal mediumcontaining 0.4% glycerol (uninduced) or 0.4% glycerol and 0.4% maltose (induced). Lanes: 3 and 4, pop3, induced; 2, pop2298, uninduced;1, pop2298, induced; 5, pop2297, uninduced; 6, pop2297, induced. After elongation with reverse transcriptase, the cDNA products wereanalyzed on 8% polyacrylamide gels containing 7 M urea. (c) The exact position of the transcription initiation sites was determined bycomparing the cDNA fragment in lanes 1 and 6 with fragments obtained by sequencing M13pu13 and M13pu14 by the dideoxy chaintermination technique.
abovementioned 690-bp DNA fragment, which contains thetwo maltose-dependent promoters, was sequenced on bothstrands by the dideoxy chain termination technique ofSanger et al. (29) (Fig. 2). The beginnings of two openreading frames, starting with an ATG and preceded by apotential ribosome-binding site, are present on the fragment.Both have a coding capacity of 65 amino acids and areoriented in opposite directions. The reading frame at the leftof the fragment will be assumed to correspond to thebeginning of gene pulA (See Discussion). The reading frameat the right end of the fragment will hereafter be called maiX.Assuming that the two open reading frames correspond to
regions of the DNA which are translated and, a fortiori,transcribed, we used DNA probes from these regions todetermine the transcription startpoint by reverse transcrip-tase mapping (Fig. 3). As a source ofmRNA we used strainsof E. coli (pop2297 and pop2298) which harbored the 690-bpDNA fragment from K. pneumoniae inserted in its chromo-some (Fig. le). mRNA species hybridizing with each of theprobes were detected. Both were maltose inducible, andboth were absent from E. coli cells which did not harbor theDNA fragment from K. pneumoniae. The positions of thetranscription startpoints for these two mRNAs are shown inFig. 2. These startpoints are separated by only 134 bp, a
rather short distance to accommodate all the controlling sitesrequired for expression of the two operons (see Discussion).Sites for CAP had been previously found in all known malpromoters. In contrast, sequences resembling the consensusCAP site could not be detected at the pulA and malXpromoters. The absence of specific CAP sites in this regionwas verified directly by using the electrophoretic techniquesof Gamer and Revzin (15) and Fried and Crothers (13) (Fig.4).
DISCUSSIONThe puL4 gene and its product. In this work we showed that
all of the sequences required for synthesis of pullulanase arelocated within a 3.8-kb DNA segment bounded by a BglIHsite at one end and by the endpoint of a deletion (Apul-2) atthe other end. The distance between these two points is justsufficient to encode the pullulanase (molecular weight,145,000) (12). Within this segment we identified the begin-ning of an open reading frame, which starts 481 bp awayfrom the BgIII end of this fragment. We showed that thisreading frame corresponds to a region of the DNA which istranscribed. When the reading frame was translated into anamino acid sequence, a very striking feature was revealed.The potential polypeptide started with a 19-residue-long
(a)
(b)
J. BACTERIOL.
on Decem
ber 31, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

PULLULANASE PROMOTER IN K. PNEUMONIAE 643
CAP(nM)
malTp-CAP J
0 2 10 20 40
pulAp,malXp *
malTp p _
FIG. 4. CAP fails to bind at the pulA and maiX promoters.Plasmids pOM50 (which contains the malT promoter) and pOM102(which contains the pulA and maiX promoters) were hydrolyzedwith EcoRI and PstI and 3' labeled with [a-32P]dATP. In addition toa large 2.3-kb fragment corresponding to the vectors, two smallfragments were thus obtained. One, 224 bp long, contained the malTpromoter, and the other, 311 bp long, contained the pulA and maiXpromoters. This mixture of fragments (1 nM each) was incubatedwith various concentrations of CAP in the presence of 0.2 PMcAMP and analyzed by polyacrylamide gel electrophoresis as pre-viously described (23). Only the upper part of the gel is shown,which contains the two small fragments (marked malTp and pulAp-malXp) as well as the complex between CAP and the malTpfragment. No complex between CAP and the pulAp-malXp fragmentwas detected at any of the concentrations of CAP used.
sequence which had all the characteristics of a signal se-quence usually found in the precursors of exported proteinsin bacteria. This sequence was followed by a cysteineresidue, which would be expected to constitute the aminoterminus of the mature polypeptide after cleavage of thesignal sequence. Such a structure, until now, has only beenreported for a very limited class of proteins. These arelipoproteins previously found to be components of the outermembrane in gram-negative bacteria or secreted proteins ingram-positive bacilli (reviewed in reference 21). If the read-ing frame which we identified corresponds to pullulanase,then this enzyme could be expected to be a lipoprotein.Direct evidence that this is the case was recently obtainedand will be published elsewhere (A. Pugsley, C. Chapon, andM. Schwartz, submitted for publication).The puLA transcription unit: a fourth mal operon. When it
was found that pullulanase synthesis was controlled by theMalT protein (18), we wondered whether pulA was anadditional gene in what would be the equivalent of one of thethree mal operons of E. coli or whether it was part of adifferent transcription unit. The work reported here clearlyindicates that the latter hypothesis is true. Transcriptionwhich originated at the pulA promoter did not proceedfurther than the left-hand-side EcoRI site on the 6.1-kb DNAfragment. Indeed, when this fragment was inserted in frontof the malPQ operon, with pulA oriented in the samedirection as malPQ, the expression of the latter operon wasnot maltose dependent. This leaves a maximum of 1.2 kb foradditional genes downstream from pulA in the same tran-scription unit. This distance is clearly insufficient to accom-modate any of the known mal operons, all of which occupya DNA length of at least 4.5 kb.The maLY transcription unit: a fifth mal operon. An unex-
pected finding from this work has been that an additionalmaltose-dependent promoter lies adjacent to the pulA pro-moter, oriented in the opposite direction. Of the transcrip-tion unit controlled by this additional promoter, only a1.1-kb segment was present on the original 6.1-kb EcoRIDNA fragment. Operon fusion experiments (insertion of the
fragment in front of malPQ) showed that no strong transcrip-tion termination signal is located in this 1.1-kb segment. Apossible reading frame called maiX was identified in thistranscription unit starting 93 bp downstream from the tran-scription initiation startpoint. When this reading frame wastranslated into an amino acid sequence no homology couldbe detected with the amino-terminal sequence of the prod-ucts of any of the mal genes which have been sequenced inE. coli (malE [11], malF [14], malK [16], malP [20], or lamBor molA [6]). In addition, this predicted sequence failed toinclude a sequence with the characteristics of a signalpeptide. The role of maiX, at this point, is unknown. Wenote that DNA probes corresponding to the beginning ofboth the maiX and pulA reading frames failed to hybridizewith mRNA extracted from maltose-induced E. coli cells(Fig.3). This result indicates that malX, like pulA, may nothave its equivalent gene in E. coli.
Structure of pulA and malK promoters. Comparison withother maff-controlled promoters. In general the -10 or -35region or both of positively controlled promoters signifi-cantly differ in sequence from the consensus sequencederived from the study of constitutively expressed promot-ers (25). The promoter for maiX clearly follows this rule(Fig. 2). Its -10 region, TATGAT, is rather close to theconsensus sequence (TATAAT), but no hexamer in its -35region resembles the consensus sequence (TTGACA) forthat part of the promoters. For the pulA promoter, however,the deviation is far less pronounced (TCTAAT preceded, 17bp upstream, by TTGAAC). Furthermore, and unlike theother known mal promoters, its -35 region is preceded by astretch of A - T base pairs, a feature which is consideredfavorable for the expression of constitutive promoters (26).Therefore, unless other characteristics of the DNA areinvoked (see end of this section), the poor activity of thepulA promoter in the absence of maltose must be ascribed tothe presence of a C in place of the highly conserved A in thesecond position of the -10 region.The expression of pulA and malX is controlled by malT.
All natural or mutant promoters so far studied which had thisproperty contained a 5'-GGA;GA hexanucleotide, posi-
+1ma/Pp s.GGATGA 34. A
molKp 5tGGAGGA 34 A
mo/Ep s'-GGAGG 35 G
pulAp 5.GGATGA 35-G
molXp 5sGAATGA 35 G
CONSENSUS 5s'GGATGA 34 AG 35-G
FIG. 5. A conserved hexanucleotide present in all known MalT-dependent promoters. malEp, malKp, and malPp are the promotersof the malEFG, malK-lamB, and malPQ operons, respectively.Their nucleotide sequences and their transcription startpoints weredetermined previously (2, 8). The number to the right of theconserved sequence indicates its distance, in base pairs, from thenucleotide (+1) where transcription starts.
ANNEW".NOME",
VOL. 164, 1985
on Decem
ber 31, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

644 CHAPON AND RAIBAUD
malEp
maITp
pulAp
malKp
mO/Pp
malXp
20 bp-L--L-I
I HFIG. 6. A simplified view of the MalT-dependent promoters. malEp, malKp, and malPp are the promoters of the malEFG, malK-IamB,
and malPQ operons, respectively. malTp, the promoter for gene malT, is shown because it is adjacent to malPp, but it is not regulated bythe malT product. The main structural elements of these promoters are shown at scale: Pribnow boxes (open boxes), 5'-GGAZGA boxes (filledarrows), and CAP sites (hatched boxes). The latter were defined either by homology to the consensus sequence (malEp and malKp) (1) orby in vitro binding experiments (malTp and malPp) (5, 23). The transcription initiation site of each promoter is indicated by an openarrowhead.
tioned 34 or 35 bp upstream from the transcriptionstartpoint. This hexanucleotide, which is also present in twoadditional copies in some of the promoters, is assumed to bepart of the MalT protein-binding site (1, 23). Such a sequence(5'-GGATGA) is again found 35 bp upstream from thetranscription start point in the pulA promoter. At the maiXpromoter, a 5'-GAATGA sequence which differs by only onebase from the consensus is found 35 bp upstream from thetranscription startpoint (Fig. 5).The 134-bp-long region which separates the transcription
startpoints for pulA and malX is apparently devoid ofspecific CAP sites. This is the first time, we believe, thatpromoters for genes involved in the assimilation of carbonsources were found to lack a CAP site. In addition, thisresult bears on the intriguing problem which concerns theinterplay of CAP and the MalT protein in the maltoseregulon. CAP controls the synthesis of MalT protein, whosestructural gene is preceded by a CAP site (5). CAP alsodirectly controls the expression of the malEFG and malK-lamB operons, which encode the components of the maltoseand maltodextrin transport system and which are also pre-ceded by CAP sites (1). In contrast, CAP does not appear tocontrol directly the expression of malPQ, even though thisoperon is also preceded by a CAP site (23). Mutants devoidof CAP are strongly impaired in malPQ expression, but thisis solely due to a deficit in MalT protein synthesis and to afailure of the inducer to enter the cells, because of insuffi-cient amounts of the maltose transport system. Therefore,the results concerning the malPQ promoter strongly sug-gested that the MalT protein could efficiently stimulatetranscription initiation in the absence of CAP. The presentresults concerning the pulA and maiX promoters confirm thisconclusion.The region which separates the pulA and malX transcrip-
tion startpoints is smaller, and apparently simpler, than theequivalent region in the other two known mal gene clusters(Fig. 6). In addition, it has an unusual structure which ischaracterized by a high degree of symmetry. In the center isa 25-bp G'C-rich sequence (21 GC pairs and four A-T pairs)with almost all of the purines on one strand (20 of 25) and thepyrimidines on the other strand. Similar sequences havebeen noted in front of the other malT-controlled promoters,but they were not as striking and not positioned in the sameway with respect to the transcription startpoints (1, 7). ThisGC-rich sequence is flanked on each side by a 13- to 14-bpsequence which is almost entirely composed of A-T basepairs and is followed by the GGATGA or GAATGA
hexanucleotides which are assumed to be part of the MalTprotein-binding site. Considering the structure of the malPQpromoter, where three 5'-GGATGA sequences appear to berequired for optimal functioning of the promoter, it is tempt-ing to speculate that the two consensus hexanucleotidespresent at the maiX and pulA promoters are both simulta-neously involved in the functioning of the two promoters. Inother words, the entire symmetric intergenic region couldconstitute a common controlling element for the twooperons. A similar hypothesis has been proposed for themalEFG and malK-lamB operons (1), but in those cases thesituation is more complex since it involves the cooperativeaction of several CAP and MalT protein molecules. Never-theless, it is now evident that MalT-controlled promoterscan have a variety of different structures with elements incommon and elements which differ (Fig. 6). The two pro-moters described in this work appear the simplest and maybe of considerable help in elucidating the mechanismwhereby MalT protein stimulates the initiation of transcrip-tion.When this work was completed, we learned that a gene
encoding pullulanase had been cloned from another strain ofK. pneumoniae (30). The restriction map of this other gene isstrikingly different from the one given here. It will beinteresting to see whether the observations made during thepresent work also apply to this other pulA gene.
ACKNOWLEDGMENTSWe thank Patrick Stragier for the pSB118 plasmid, Denise Kotlarz
for the gift of purified CAP protein, and Olivier Danos for excellentadvice concerning reverse transcriptase mapping. We thank MaximeSchwartz for his constant interest in this work and for his help inpreparing the manuscript.
This work was supported by grants from the Centre National de laRecherche Scientifique (UA 04 11 49), the Ministere de la Rechercheet de la Technologie (82 V 1279), and the Fondation pour laRecherche Medicale.
LITERATURE CITED1. Bedouelle, H. 1983. Mutations in the promoter regions of themalEFG and malK lamB operons of Escherichia coli K-12. J.Mol. Biol. 170:861-882.
2. Bedouelle, H., U. Schmeissner, M. Hofnung, and M. Rosenberg.1982. Promoters of the malEFG and malK lamB operons inEscherichia coli K-12. J. Mol. Biol. 161:519-531.
3. Bender, H. and K. Wallenfels. 1966. Pullulanase (an amylopectinand glycogen debranching enzyme) from Aerobacter aerogenes.Methods Enzymol. 8:555-559.
-VIR79M a
11 I 1..8 --vmma 270bp . a
J. BACTERIOL.
on Decem
ber 31, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

PULLULANASE PROMOTER IN K. PNEUMONIAE 645
4. Casadaban, M. J. 1976. Transposition and fusion of the lacgenes to selected promoters in Escherichia coli usingbacteriophage lambda and Mu. J. Mol. Biol. 104:541-555.
5. Chapon, C., and A. Kolb. 1983. Action of CAP on the malTpromoter in vitro. J. Bacteriol. 156:1135-1143.
6. Clement, J. M., and M. Hofnung. 1981. Gene sequence of the Areceptor, an outer membrane protein of E. coli K12. Cell27:507-514.
7. Debarbouille, M., P. Cossart, and 0. Raibaud. 1982. A DNAsequence containing the control sites for gene malT and for themalPQ operon. Mol. Gen. Genet. 185:88-92.
8. Debarbouille, M., and 0. Raibaud. 1983. Expression of theEscherichia coli malPQ operon remains unaffected after drasticalteration of its promoter. J. Bacteriol. 153:1221-1227.
9. Debarbouille, M., H. A. Shuman, T. J. Silhavy, and M.Schwartz. 1978. Dominant constitutive mutations in malT, thepositive regulator gene of the maltose regulon in Escherichiacoli. J. Mol. Biol. 124:359-371.
10. Dessein, A., and M. Schwprtz. 1974. Is there a pullulanase inEscherichia coli? Eur. J. IBiochem. 45:363-366.
11. Duplay, P., H. Bedouelle, A. Fowler, I. Zabin, W. Saurin, andM. Hofnung. 1984. Sequences of the malE gene and of itsproduct, the maltose-binding protein of Escherichia coli K-12. J.Biol. Chem. 259:10606-10613.
12. Eisele, B., I. R. Rasched, and K. Wallenfels. 1972. Molecularcharacterization of pullulanase from Aerobacter aerogenes.
Eur. J. Biochem. 26:62-67.13. Fried, M., and D. M. Crothers. 1981. Equilibria and kinetics of
lac repressor-operator interactions by polyacrylamide gel elec-trophoresis. Nucleic Acids Res. 9:6505-6525.
14. Froshauer, S., and J. Beckwith. 1984. The nucleotide sequenceof the gene for malF protein, an inner membrane component ofthe maltose transport system of Escherichia coli. RepeatedDNA sequences are found in the malE-malF intercistronicregion. J. Biol. Chem. 259:10896-10903.
15. Garner, M. M., and A. Revzin. 1981. A gel electrophoresismethod for quantifying the binding of proteins to specific DNAregions: application to components of the Escherichia colilactose operon regulatory system. Nucleic Acids Res.9:3047-3060.
16. Gilson, E., H. Nikaido, and M. Hofnung. 1982. Sequence of themalK gene in E. coli K-12. Nucleic Acids Res. 10:7449-7458.
17. Hope, G. C., and A. C. R. Dean. 1974. Pullulanase synthesis inKlebsiella (Aerobacter) aerogenes strains growing in continu-ous culture. Biochem. J. 144:403-411.
18. Michaelis, S., C. Chapon, C. D'Enfert, A. P, Pugsley, and M.Schwartz. 1985. Characterization and expression of the struc-tural gene for pullulanase, a maltose-inducible secreted proteinof Klebsiella pneumoniae. J. Bacteriol. 164:633-638.
19. Norrander, J., T. Kempe, and J, Messing. 1983. Construction ofimproved M13 vectors using oligodeoxynucleotide-directed mu-tagenesis. Gene 26:101-106.
20. Palm, D., R. Goerl, and K. J. Burger. 1985. Evolution ofcatalytic and regulatpry sites in phosphorylases. Nature (Lon-don) 313:500-502.
21. Pugsley, A. P., and M. Schwartz. 1985. Export and secretion ofproteins by bacteria. FEMS Microbiol. Rev. 32:3-38.
22. Raibaud, O., M. Debarbouillee, and M. Schwartz. 1983. Use ofdeletions created in vitro to map transcriptional regulatorysignals in the malA region of Escherichia coli. J. Mol. Biol.163:395-408.
23. Raibaud, O., C. Gutierrez, and M. Schwartz. 1985. Essentialand nonessential sequences in malPp, a positively controlledpromoter in Escherichia coli. J. Bacteriol. 161:1201-1208.
24. Raibaud, O., M. Mock, and M. Schwartz. 1984. A technique forintegrating any DNA fragment into the chromosome of Esche-richia coli. Gene 29:231-241.
25. Raibaud, O., and M. Schwartz. 1984. Positive control of tran-scription initiation in bacteria. Annu. Rev. Genet. 18:173-206.
26. Rosenberg, M., and D. Court. 1979. Regulatory sequencesinvolved in the promotion and termination of RNA transcrip-tion. Annu. Rev. Genet. 13:319-353.
27. Ruther, U., M. Koenen, K. Otto, and B. Muiller-Hill. 1981.pUR222,, a vector for cloning and rapid chemical sequencing ofDNA. Nucleic Acids Res. 9:4087-4098.
28. Sanger, F., and A. R. Coulson. 1978. The use of thin acrylamidegels for DNA sequencing. FEBS Lett. 87:107-110.
29. Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequenc-ing with chain-terminating inhibitors. Proc. Natl. Acad. Ssi.USA 74:5463-5467.
30. Takizawa, N., and Y. Murooka. 1985. Cloning of the pullulanasegene and overproduction of pullulanase in Escherichia coli andKlebsiella aerogenes. Appl. Enviroq. Microbiol. 49;294-298.
VOL. 164, 1985
on Decem
ber 31, 2018 by guesthttp://jb.asm
.org/D
ownloaded from