regulation of heparan sulfate 6-o-sulfation...
TRANSCRIPT

ACTAUNIVERSITATISUPSALIENSISUPPSALA2006
Digital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Medicine 185
Regulation of Heparan Sulfate6-O-Sulfation Patterns
ANH-TRI DO
ISSN 1651-6206ISBN 91-554-6687-7urn:nbn:se:uu:diva-7199


To my Family and All who supported me

“Let me ask you… What day is it today?
…It’s the best day ever!!!…”
- Bengt Wiström

List of Papers
This doctoral thesis is based on the following papers, which are referred to in the text by their Roman numerals:
I. Smeds, E., Habuchi, H., Do, A.-T., Hjertson, E., Grundberg, H., Kimata, K., Lindahl, U. and Kusche-Gullberg, M. (2003) Substrate specificities of mouse heparan sulphate glucosaminyl 6-O-sulphotransferases. Biochem. J. 372, 371-380.
II. Do, A.-T., Smeds, E., Spillmann, D. and Kusche-Gullberg, M. (2006) Overexpression of heparan sulfate 6-O-sulfotransferases in human embryonic 293 cells results in increased N-acetyl glucosaminyl 6-O-sulfation. J. Biol. Chem. 281, 5348-5356.
III. Ai, X., Do, A.-T., Lozynska. O., Kusche-Gullberg, M., Lindahl, U. and Emerson, C. P., Jr. (2003) QSulf1 remodels the sulfation states of cell surface heparan sulfate proteoglycans. J. Cell Biol.162, 341-351*.
IV. Ai, X., Do, A.-T., Kusche-Gullberg, M., Lindahl, U., Lu, K. and Emerson, C. P., Jr. (2006) Substrate specificity and domain functions of extracellular heparan sulfate 6-O-endosulfatases, QSulf1 and QSulf2. J. Biol. Chem. 281, 4969-4976.
V. Do, A.-T., Rentzsch, F., Technau, U. and Kusche-Gullberg, M. Identification of heparan sulfate in the sea anemone Nematostella vectensis. Manuscript.
* Reproduced from the Journal of Cell Biology, 2003, 162: 341-351, Copyright 2003, The Rockefeller University Press.
Reprints were made with permission from the publishers.


Contents
Introduction...................................................................................................11
Background...................................................................................................12Glycobiology............................................................................................12Proteoglycans and glycosaminoglycans...................................................13Heparan sulfate proteoglycans .................................................................15
General properties of HSPGs...............................................................15HS core proteins ..................................................................................15HS structure and biosynthesis..............................................................17
Biological functions of HSPGs ................................................................21HS-protein interactions........................................................................21
Nature and properties of interactions ..............................................21Ligands of particular interest ..........................................................23
The fibroblast growth factors - FGFs .........................................23The FGF receptors - FGFRs .......................................................24The Wnt family of morphogens .................................................24
HS in physiological processes .............................................................25HS in embryogenesis ......................................................................25HS in human diseases .....................................................................26
Regulation of HS structure .......................................................................27Principles of regulation........................................................................27Evolutionary aspects of HS structural diversity ..................................28Regulation of the 6-O-sulfation pattern ...............................................29
HS glucosaminyl 6-O-sulfotransferases - 6OSTs ...........................29HS 6-O-endosulfatases - Sulfs ........................................................30
Nematostella vectensis - a possible model for developmental evolutionary studies of HS and its enzymatic machinery?............................................31
Present Investigation.....................................................................................33Aims of study ...........................................................................................33Results ......................................................................................................33
Substrate specificities of mouse heparan sulphate glucosaminyl 6-O-sulfotransferases (Paper I)............................................................33Overexpression of heparan sulfate 6-O-sulfotransferases in human embryonic 293 cells results in increased N-acetyl glucosaminyl 6-O-sulfation (Paper II) .......................................................................34

QSulf1 remodels the sulfation states of cell surface heparan sulfate proteoglycans (Paper III) .....................................................................35Substrate specificity and domain functions of extracellular heparan sulfate 6-O-endosulfatases, QSulf1 and Qsulf2 (Paper IV).................36Identification of heparan sulfate in the sea anemone Nematostellavectensis (Paper V) ..............................................................................36
Conclusions ..............................................................................................37
Discussion and Future Perspectives..............................................................38
Populärvetenskaplig Sammanfattning...........................................................43
Acknowledgements.......................................................................................45
References.....................................................................................................49
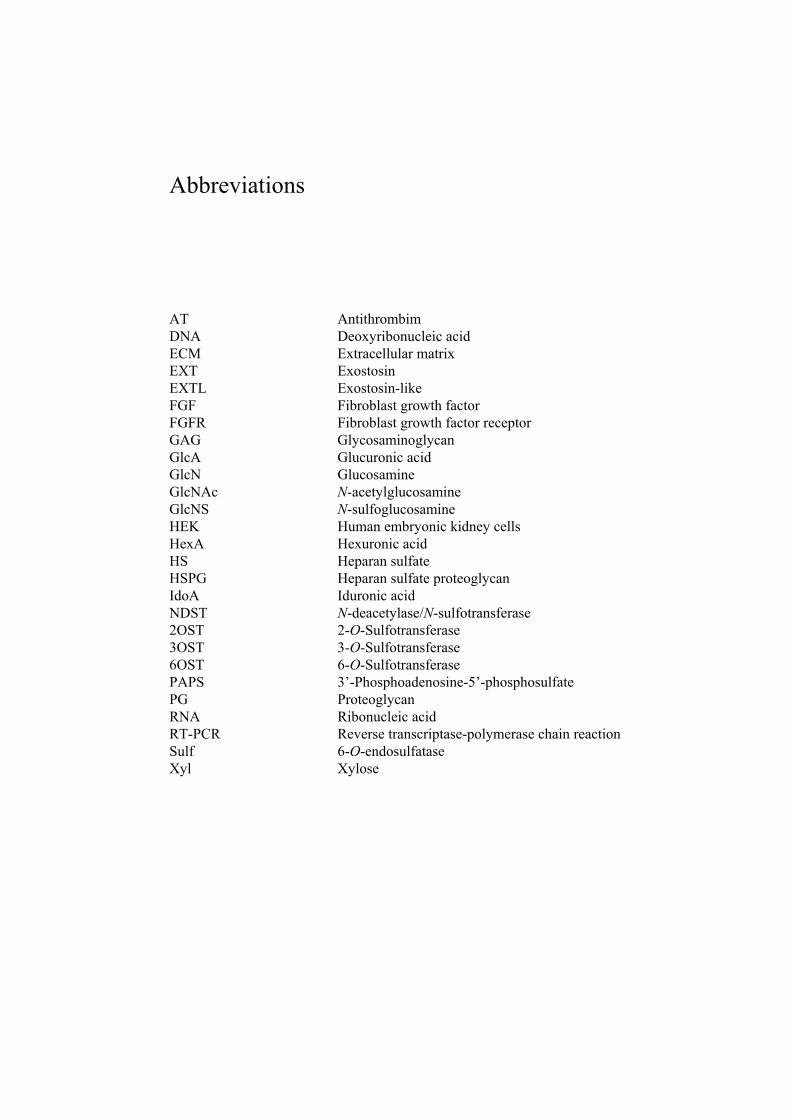
Abbreviations
AT Antithrombim DNA Deoxyribonucleic acid ECM Extracellular matrix EXT Exostosin EXTL Exostosin-like FGF Fibroblast growth factor FGFR Fibroblast growth factor receptor GAG Glycosaminoglycan GlcA Glucuronic acid GlcN Glucosamine GlcNAc N-acetylglucosamine GlcNS N-sulfoglucosamine HEK Human embryonic kidney cells HexA Hexuronic acid HS Heparan sulfate HSPG Heparan sulfate proteoglycan IdoA Iduronic acid NDST N-deacetylase/N-sulfotransferase 2OST 2-O-Sulfotransferase 3OST 3-O-Sulfotransferase 6OST 6-O-Sulfotransferase PAPS 3’-Phosphoadenosine-5’-phosphosulfate PG Proteoglycan RNA Ribonucleic acid RT-PCR Reverse transcriptase-polymerase chain reaction Sulf 6-O-endosulfataseXyl Xylose


11
Introduction
Proteoglycans are macromolecules that are ubiquitously distributed in the body. Their size and structure vary enormously. The basic structure of all proteoglycans includes a core protein and at least one, but frequently more carbohydrate chains, known as glycosaminoglycans. Heparan sulfate proteoglycans form a subgroup of proteoglycans characterized by carrying one or more heparan sulfate polysaccharide chains.
Heparan sulfates are linear, negatively charged polysaccharides that exhibit an enormous degree of structural heterogeneity. Localized at the cell surfaces, heparan sulfate chains interact with a variety of proteins and thereby play important roles in vital biological events such as cell-to-cell communication, cell proliferation and tissue morphogenesis. The fine structure of heparan sulfate varies between tissues, during development and in pathological conditions, but the mechanisms that regulate and determine its complex structure are still unknown.
This thesis focuses on the regulation of structural diversity of heparan sulfate, in particular the regulation of the 6-O-sulfation patterns that are generated by enzymes that add 6-O-sulfate groups during biosynthesis or remove 6-O-sulfate groups after biosynthesis. In addition a new model organism is introduced that offers good prospects for investigating the developmental evolution of HS structural heterogeneity.

12
Background
GlycobiologyCarbohydrates, or saccharides (sugars or glycans), are essential components of all living organisms and are, in fact, the most abundant class of biological molecules. Although sometimes given less attention, carbohydrates, just like nucleic acids, proteins and lipids, are among the fundamental building blocks for life. The study of the biosynthesis, structure and function of complex carbohydrates is referred to today as “glycobiology ”(Rademacher et al., 1988).
Besides playing important roles in protein modifications and energy metabolism, carbohydrates affect interactions between cells and their surroundings and are crucial for the development and integrity of both simple and complex organisms (Varki et al., 1999). In addition, many bacteria, viruses and parasites use carbohydrates at the host cell surface for primary attachment during infections. Carbohydrates are often found as integral parts of proteins or lipids (glycoconjugates) and are synthesized by almost every organism from bacteria, fungi and plants to mammals. The most commonly occurring glycoconjugates are glycoproteins, proteoglycans and glycolipids. It is estimated that about 50% of all proteins are modified by carbohydrates.
The elucidation of the structure and functions of carbohydrates has lagged well behind that of nucleic acids and proteins, a situation, which can be ascribed to several factors. Carbohydrates are often heterogeneous, both in size and in composition, which greatly complicates their physical and chemical characterization. They are not subject to the types of genetic analysis, that have been invaluable in the study of nucleic acids and proteins, such as gene-deletion or mutational studies which directly influence the function of genes and proteins, because carbohydrate sequences are not genetically specified from templates but are built up through the sequential actions of specific enzymes. Also, part of the variability seen in carbohydrates is due to the fact that single sugar units can be coupled to each other in a number of ways. By contrast with the linearity of DNA and proteins, this allows branching and increases the number of possible polysaccharide structures. Thus, with increasing knowledge of the biological importance of saccharides and improved techniques for the structural and functional analysis of complex carbohydrates, glycobiology has become one

13
of the most rapidly growing biomedical fields in the post-genomic era (Varki et al., 1999).
Proteoglycans and glycosaminoglycans A large number of proteins in animal tissues occur at cell surfaces and in the extracellular matrix (ECM). Some may also be anchored to the cell membrane through interactions with other molecules (Lindahl et al., 1998). Among these molecules we find the proteoglycans (PGs), glycoproteins composed of a core protein with a variable number of covalently O-linked glycosaminoglycan (GAG) side chains (Bernfield et al., 1999; Iozzo, 1998; Kjellén and Lindahl, 1991). A multitude of functions have been ascribed to PGs, and in vitro and in vivo studies have demonstrated their importance as modulators of both normal cell and tissue functions (e.g. cell division, cell adhesion and cell migration) and modulators of cell-cell signaling and morphogenesis (Forsberg and Kjellén, 2001; Lander and Selleck, 2000; Li et al., 2003).
Figure 1. Disaccharide structure of various GAGs. Possible modifications of the basic disaccharide structures of heparin/heparan sulfate, chondroitin sulfate and keratan sulfate are represented by R (= H or SO3
-) and R´(= H, COCH3 or SO3-). In
addition, the GlcA of heparin/heparan sulfate and chondroitin sulfate can be epimerized to IdoA (indicated by the broken arrows).
The functions of PGs depend both on the core proteins and the attached GAG chains. The number of attached GAG chains (1 to >100) varies between different types of PGs, and the number of chains on a specific PG may vary, depending on the cell type producing it. One core protein can
Keratan Sulfate
OO
OR
H
COO-
O
HNR’
OH OR
CH2ORO
O
Heparin/Heparan Sulfate
O
OH
H
COO-
HNAc
OH HO
CH2OHO
OO
Hyaluronan
O
OR
H
COO-
O
HNAc
OH
RO
CH2ORO
OO
Chondroitin Sulfate/Dermatan Sulfate
O
OH
CH2ORO
HNAc
HO
OH
CH2ORO
O
O
O

14
carry more than one type of GAG chain and the ratio between the different types of GAG chains, and also the lengths of the chains, may change with development, ageing or pathological conditions. The mechanisms behind the selective substitution of a certain PG with one type of GAGs, but not another, are not well understood, but both the amino acid sequences adjacent to and distal from the GAG attachment sites in the core protein seem to be important (Chen and Lander, 2001; Esko and Zhang, 1996; Kokenyesi and Bernfield, 1994). Some PGs are referred to as part-time PGs based on the fact that the core proteins do not always carry GAGs.
GAGs are linear polysaccharides composed of repeating disaccharide units of alternating hexuronic acid [(HexA), either glucuronic acid (GlcA) or iduronic acid (IdoA)], and hexosamine [either N-acetylglucosamine (GlcNAc) or N-acetylgalactosamine (GalNAc)] residues. Depending on the nature of the repeated disaccharide, GAGs can be divided into four classes: heparan sulfate/heparin, chondroitin/dermatan sulfate, keratan sulfate and hyaluronan (Fig. 1) (reviewed by (Kjellén and Lindahl, 1991)).
Heparan sulfate (HS) and heparin are composed of repeating disaccharide units of -HexA / 1,4-GlcN 1,4-, that are modified by sulfate groups at several positions. The HexA is either GlcA or IdoA, and the GlcN is either N-acetylated (GlcNAc), N-sulfated (GlcNS) or, in rare cases, N-unsubstituted (GlcNH2). Heparin is a highly modified variant of HS with a larger proportion of sulfates and IdoA than is commonly found in HS. Chondroitin sulfate (CS) and dermatan sulfate (DS) are both galactosaminoglycans that contain repeating disaccharide units of -HexA / 1,3-GalNAc 1,4-. In CS the HexA is invariably GlcA, while in DS some of the GlcA is epimerized to IdoA. Both the HexA and GalNAc residues of CS/DS can be sulfated at various positions. Keratan sulfate (KS) lacks the HexA moiety and is composed of alternating partially sulfated galactose (Gal) and GlcNAc residues (-Gal 1,4-GlcNA 1,3-). In contrast to the other GAGs, hyaluronan occurs only in free form (not linked to a core protein), is built up of repeating units of -GlcA 1,3-GlcNAc 1,4- units and is not sulfated.
HS/heparin and CS/DS chains are all O-linked to serine residues in the core proteins via a GlcA-Gal-Gal-Xyl linkage region (Fransson et al., 2000), while KS is linked either to asparagine residues via GlcNAc (corneal KS; type I), or to serine/threonine via a GalNAc residue (skeletal KS; type II) (Funderburgh, 2000).
GAGs have been reported to be present in a variety of organisms throughout the animal kingdom and also in bacteria. The nematode C. elegans, for example, synthesizes non-sulfated chondroitin but sulfated HS (Toyoda et al., 2000; Yamada et al., 1999). Sulfated HS and heparin-like GAGs have been reported in many invertebrates (Medeiros et al., 2000). Moreover, sponges produce a non-sulfated GAG that is structurally similar to hyaluronan and is attached to link proteins and core proteins in a PG

15
structure, as part of a particular family of PGs known as spongicans (Fernandez-Busquets and Burger, 2003).
Heparan sulfate proteoglycans
General properties of HSPGs The heparan sulfate proteoglycans (HSPGs) belong to a subgroup of PGs characterized by possessing at least one HS polysaccharide chain. HSPGs are found at the surface of most mammalian cells, in the extracellular matrix (ECM) and in intracellular granules (Bernfield et al., 1999; Whitelock and Iozzo, 2005) and their physiological functions are closely related to their ability to selectively interact with various protein ligands and thereby affect many biological processes. The ligands include growth factors, enzyme inhibitors, microbial surface proteins and matrix components (Lander and Selleck, 2000; Lindahl et al., 1998). It is generally believed that the interactions between HS and various ligands depend on the relative positions of carboxyl and in particular sulfate groups within the HS chains. Positively charged patches on the protein surfaces recognize negatively charged groups within the HS chains.
The highly heterogeneous fine structure of HS holds epitopes that bind to proteins in a selective manner, best illustrated by the reaction between HS and the blood anticoagulant antithrombin (AT) (Bourin and Lindahl, 1993). Interactions between proteins and HS may serve many purposes e.g. they can protect the proteins from proteolytic degradation (Yoneda et al., 2000) and regulate the tissue distribution of growth factors.
HS core proteins There are several types of PG core proteins that are known to carry HS chains, some of which may also be substituted with other GAGs. The major HSPGs include one intracellular HSPG; serglycin, two cell surface associated HSPGs; syndecan and glypican, and three ECM HSPGs; perlecan, agrin and collagen XVIII (Bernfield et al., 1999; Halfter et al., 1998; Whitelock and Iozzo, 2005), as illustrated in Figure 2.
Serglycin is an intracellular PG that is present in the secretory granules of mast cells (Fig. 2) and is involved in the storage of proteases (Henningsson et al., 2002). The central part of the serglycin core protein consists of repeating units of serine and glycine, with the serine residues substituted with heparin/CS chains (Kolset and Gallagher, 1990).
The glypicans (six members known) predominantly carry HS rather than CS chains and are attached to the plasma membrane via a

16
glycosylphospatidylinositol (GPI) anchor. The protein core consists of a N-terminal signal peptide, a cystein-rich globular ectodomain, an extended region close to the plasma membrane with three HS attachment sites and a hydrophobic C-terminal, which is involved in the formation of the GPI anchor (Filmus and Selleck, 2001; Fransson, 2003) (Fig 2). Glypicans are widely expressed throughout development as well as in adulthood, but individual family members show specific spatiotemporal expression profiles. Glypican1 is expressed ubiquitously in adult tissues but mainly expressed in the central nervous system and skeletal muscle during embryonic development, whereas glypican3, 4, 5 and 6 are widely expressed during embryonic development and to a more restricted extent in the adult animal. The expression pattern of glypican2 in adult tissues is not known, but its expression is essential for embryonic brain development (Fransson et al., 2004). Glypicans can be cleaved by phospholipases within the GPI anchor, or by proteases in the protein, resulting in shedding of their ectodomains from the cell surface.
Vertebrate syndecans constitute a family with four members that are expressed in a regulated manner in most cells and tissues. They are type I transmembrane proteins consisting of a short, highly conserved C-terminal cytoplasmic tail, a hydrophobic transmembrane region and an extracellular ectodomain that is commonly substituted with 1-3 GAG chains (Fig. 2). Syndecans carry HS chains near the N-terminus and sometimes also 1-2 CS chains close to the cell surface (Iozzo, 2001). Most cells express at least one syndecan, often multiple ones, and each syndecan is expressed in cell, tissue and developmental-specific patterns (Bernfield et al., 1992; Kim et al., 1994). The highly regulated expression profiles observed during embryonic development are distinct from the expression in adult tissue, suggesting an active role of syndecans in cell differentiation and tissue organization (Salmivirta and Jalkanen, 1995). In adult tissues, syndecan1 is expressed on epithelial cells and plasma cells, syndecan2 is present on endothelial cells and fibroblasts and syndecan3 is predominantly found in neural tissues, whereas syndecan4 is more ubiquitously expressed (Kim et al., 1994). As with glypicans, the ectodomain of all syndecans can be shed from the cell surface by proteolytic cleavage close to the plasma membrane (Bernfield et al., 1999; Kim et al., 1994).
Agrin and perlecan are two large multidomain proteins present in the basement membrane and the ECM (Fig. 2). The perlecan core protein contains four potential GAG attachment sites for HS and CS, whereas agrin has three potential GAG attachment sites for HS (Bezakova and Ruegg, 2003; Knox and Whitelock, 2006). Agrin is mainly found in the kidney and brain, while perlecan shows a wider tissue distribution. A third extracellular PG is collagen XVIII, which is part of most epithelial and endothelial basement membranes and is also found in cartilage and fibrocartilage (Pufe et al., 2004; Rehn and Pihlajaniemi, 1995). This is the only collagen

17
carrying HS chains and is a precursor of the 20 kDa polypeptide known as endostatin, an inhibitor of endothelial cell proliferation, that is thought be regulated by HS (O'Reilly et al., 1997).
In addition to those already mentioned, some minor cell surface HSPGs also exist that possess HS or CS chains, i.e. CD44/epican, betaglycan, and a unique isoform of fibroblast growth factor receptor 2 (FGFR2) (Takagi et al., 1994). All of these cell surface HSPGs also exist in non-glycosylated forms and can therefore be considered part-time HSPGs.
Figure 2. Common HS/heparin PGs. 1. Serglycin is a heparin PG found in mast granules. 2. Syndecans and 3. glypicans are cell surface-associated PGs present on most cells, and 4. agrin and 5. perlecan are PGs found in the ECM.
HS structure and biosynthesis HS chains are long (50-400 monosaccharide units), unbranched GAGs composed of a repeating disaccharide unit of alternating hexuronic acid
1
2
3
4
5
Cell membraneCytoplasm
Core protein
Heparan sulfate/Heparin chain

18
(either GlcA or IdoA) and GlcN residues (Lindahl et al., 1998). The GlcN residues can be N-acetylated, N-sulfated or N-unsubstituted. The hexuronic acid is linked to the GlcN residues by an /ß-1,4 glycosidic bond at the reducing end and via an -1,4 glycosidic bond towards the non-reducing end (Fig. 3). The structural diversity of HS is attributed to the N-substitution pattern, the distribution of GlcA and IdoA units, and the O-sulfate distribution between potential positions (C2 of HexA residues and C3 and C6 of GlcN residues) (Fig. 3).
Figure 3. Disaccharide structure of HS. Schematic representation of the HS/heparin backbone structure and possible modification in repeating disaccharide units.
The biosynthesis of HS takes part in the Golgi compartment and is a complex multi-step process involving the concerted action of a large biosynthetic enzyme machinery (Bernfield et al., 1999; Esko and Lindahl, 2001; Lindahl et al., 1998; Lyon et al., 1987). HS is synthesized attached to a core protein via a tetrasaccharide GlcA-Gal-Gal-Xyl linkage region, where the Xyl residue is covalently O-linked to a serine residue in a Serine-Glycine/Alanine dipeptide sequence in the core protein. It has been difficult to predict the sites of HS contra CS glycosylation, but acidic and hydrophobic amino acid residues in the core protein have been identified as an important factor in the addition of HS chains (Esko and Zhang, 1996). The same core protein may carry either HS or CS in different tissues, however, which emphasizes the difficulty in predicting HS attachment sites. The HS chain is elongated through the action of GlcNAc and GlcA transferases, and further modified in various ways in the course of elongation (Fig. 4). The first modification reaction is catalyzed by a combined N-deacetylase/N-sulfotransferase (NDST) and replaces some
Epimerization
O-Sulfation N-Sulfation
OH
O
n
O
H
COO-
O
NAc
OH OH
CH2OH O
O

19
N-acetyl groups on GlcNAc residues with N-sulfate groups. The modification continues (predominantly in the N-sulfated regions of the HS chain) with C5-epimerisation of GlcA to IdoA, followed by 2-O-sulfation of IdoA and 6-O-sulfation of GlcN(Ac/SO3) residues. Less common modifications include 3-O-sulfation of GlcNSO3 and 2-O-sulfation of GlcA residues (Fig. 4). On rare occasions, the glucosamine is N-unsubstituted(GlcNH2), but it is not known how these residues are formed (Casu and Lindahl, 2001; Lindahl et al., 1998; Westling and Lindahl, 2002). A number of GlcNAc residues escape N-sulfation and owing to the substrate specificities of the following polymer modification enzymes, only regions containing N-sulfates are further modified (under normal physiological conditions). Thus, the modification reactions generate essentially three types of domain structure in HS: unmodified N-acetylated regions with GlcA units (NA domains), mixed regions of alternating N-sulfated and N-acetylated disaccharides (NA/NS domains), and contiguous N-sulfated regions that are 2-O and 6-O-sulfated and contain both IdoA and GlcA residues (NS domains). Half the 6-O-sulfate groups are found on GlcNAc residues in NA/NS domains (Maccarana et al., 1996) (Fig. 5).
Figure 4. HS chain modifications. A schematic representation of biosynthetic modifications during HS chain elongation. The biosynthesis of HS takes place in the Golgi compartment, and the chain is elongated by the alternating additions of GlcA and GlcNAc by GlcA/GlcNAc transferases and modified by multiple enzymes.
N-deacetylation/N-sulfation(N-deacetylase/N-sulfotransferases)
2-O-sulfation (2-O-sulfotransferase) Epimerization
(C5-epimerase)
6-O-sulfation (6-O-sulfotransferases)
3-O-sulfation (3-O-sulfotransferases)
OO
OH
H
COO-
HNAc
OH OH
CH2OHO
O
O
OO
OH
H
COO-
HNSO3-
OH OH
CH2OHO
O
O
OO
OH HNSO3-
OH OH
O
O
OCOO-
H CH2OH
OSO3-
OO
OH
H
HNSO3-
OH
CH2OHO
O
OCOO-
OSO3-
OO
OH
H
HNSO3-
OH
O
O
OCOO-
OSO3-
OOOH
H
HNSO3-
O
O
OCOO-
OSO3-
CH2OSO3-
CH2OSO3-

20
Currently, at least two GlcA/GlcNAc transferases (Lind et al., 1998), four N-deacetylase/N-sulfotransferases (NDSTs) (Aikawa et al., 2001; Grobe et al., 2002), three 6-O-sulfotransferases (6OSTs) (Habuchi et al., 2003; Habuchi et al., 2000) and at least seven 3-O-sulfotransferases (3OSTs) (Kusche-Gullberg and Kjellén, 2003; Shworak et al., 1999; Xia et al., 2002) have been identified in mammals, but only one mammalian C5-epimerase and one 2-O-sulfotransferase (2OST) have been identified (Crawford et al., 2001; Kobayashi et al., 1996; Li et al., 2001; Rong et al., 2001).
Figure 5. Schematic illustration of HS domain organization. HS chains are composed of highly sulfated (NS) domains, intermediately modified mixed (NA/NS) domains and mostly unmodified (NA) domains.
The diversity of HS structure is generally ascribed to the incomplete action (or strict regulation) of the biosynthetic enzymes involved in the modification reactions. It has also been proposed that degrading enzymes may structurally modify HS polymers (Hulett et al., 1999). Moreover, recent findings have shown that HS sulfation patterns can be additionally remodeled at the cell surface by extracellular HS 6-O-endosulfatases (Sulfs) (Ai et al., 2003; Dhoot et al., 2001).
The structure of HS could in theory accommodate an infinite number of sequence so that over 1,000,000 possible sequences are theoretically conceivable in only an octasaccharide fragment, for example (Sasisekharan and Venkataraman, 2000). In practice, however, the regulated expression and action of the biosynthetic enzymes and their restricted substrate specificities, limit the actual number of sequences expressed.
There are at least two levels of structural variability in HS: the variation in the content and proportions of the types of modified disaccharides, and the variation in the size and distribution of the three domain types (Fig. 5). In spite of this variability of different types of HS disaccharides, there are
NS NA/NS NA
Ser
Linkage
Protein
GlcA
IdoA
GlcNAc
GlcNS Sulfate group
Xyl Gal

21
common disaccharides species that can be found in all HS samples, from evolutionarily primitive organisms to higher, more evolved ones (Esko and Lindahl, 2001; Ledin et al., 2004; Lindahl et al., 1998; Medeiros et al., 2000; Nader et al., 1984).
It has been shown on many occasions that the variation in HS structure does not correlate with the core protein to which the polysaccharide chains are attached, but rather is dependent on the cell type of origin (Knox et al., 2002; Ledin et al., 2004; Sanderson et al., 1994). Compositional analysis as well as immunostaining with monoclonal antibodies recognizing specific HS epitopes have demonstrated differential expression of HS structures between species and between various tissues and developmental stages within the same species (Dennissen et al., 2002; Jenniskens et al., 2006).
Biological functions of HSPGs
HS-protein interactions
Nature and properties of interactions The interactions between heparin/HS and proteins are generally considered to be electrostatic in nature, involving binding of the positively charged amino acid residues in the proteins (mostly arginine and lysine) to the negatively charged sulfate and carboxyl groups in the polysaccharide. Less commonly discussed, however, are the results obtained by microcalorimetry that suggest that only 30 % of the binding free energy between heparin and FGF2 is due to pure electrostatic interactions, while the remaining 70 % is due to the contribution of other non-ionic interactions such as van der Waals forces and hydrogen bonds (Thompson et al., 1994). What, then, is the actual contribution of electrostatic interactions to binding specificity?
The non-ionic and ionic interactions are not easily separated from each other, and it is conceivable that the sulfation and the charge distribution of the saccharide may influence the three-dimensional structure and conformation of the bound protein ligand. This would then presumably affect the positioning and exposure of structures within the protein involved also in non-ionic interactions as well. Thus, even though the electrostatic interactions only contribute with 30 % of the total binding energy, they would indirectly also affect the non-ionic interactions.
Large numbers of proteins, including enzymes, enzyme inhibitors, growth factors/morphogens, and ECM components with a wide variety of functions, are already known to bind heparin/HS, (Bernfield et al., 1999) (Table 1), and certain interactions between heparin/HS and proteins appear to be mediated via specific saccharide sequences within the polysaccharide chains, whereas

22
others largely depend on charge density (Kreuger et al., 2006; Lindahl et al., 1994). The interaction between heparin and AT is probably the best-known example of a heparin/HS-protein interaction that requires a specific sequence within the polymer. A pentasaccharide domain within heparin, known as the AT-binding sequence, binds to AT with high affinity and induces a conformational change, allowing AT to bind and inhibit thrombin at a significantly higher rate than in the absence of heparin (Jordan et al., 1980). It is important to bear in mind, however, that most of the known HS-protein interactions have been identified in vitro, and under non-physiological conditions that may for example have affected the protein in terms of unfolding etc. Furthermore, heparin has often been used to study HS-protein interaction, but not HS. As explained previously, heparin is stored in mast cell granules and only released upon mast cell activation. Therefore, many of the molecules with heparin-binding properties will never encounter this extreme form of HS in vivo. The interactions with these molecules observed may nevertheless be significant in the case of HS as well, since various structural domains found within HS chains can contain the sequence required, or exhibit sufficient negative charge, to bind proteins with a capacity similar to or even higher than that of heparin.
Table 1. Examples of HS/heparin-binding proteins involved in various physiological processes (adapted from (Bernfield et al., 1999)).
Protein Functions Protein Anti-angiogenic factors Angiostatin, Endostatin, Histidine-rich
glycoprotein Anti-microbial peptides Bac-5, PR-39 Cell adhesion L-selectin, Mac-1, N-CAM, PECAM-
1/CD31Chemokines IL-8, PF-4, Rantes Coagulation AT, Factor Xa, Thrombin, Leuserpin Cytokines IL-2-5, -7, -12, GM-CSF, Interferon- ,
TNF- ,ECM components Fibrin, Fibronectin, Interstitial collagens,
Laminins, Pleiotropin, Thrombospondin, Vitronectin, Tenascins
Energy Metabolism Agouti-related protein, ApoB, ApoE, Lipoprotein lipase, Triglycerride lipases
Growth factor/Growth factor binding
EGF family, FGFs, IGF-II, PDGF, TGF ,VEGF, Follistatin, IGF BP-3, -5, TGF BP
Morphogens/Morphogen binding
Activin, BMPs, Chordin, Frizzled-related peptides, Sonic Hh, Sprouty peptides, Wnts
Proteinases Neutrophil elastase, Cathepsin G Tissue remodeling factors Tisue plasminogen activator, Plasminogen
activator inhibitor, Protease nexin I

23
HS binding can modulate the action of a protein in many ways. In addition, a single HS chain has the ability to bind several different molecules on the same chain and thereby increase the likelihood of interaction between the bound molecules, such as receptors and their ligands or enzymes and their inhibitors (Lander, 1998). Binding to HS may also induce a conformational change in a protein as in the case of AT activation. In growth factor signaling, HS may act both by facilitating receptor dimerization and as a co-receptor (Esko and Selleck, 2002; Esko and Zhang, 1996; Häcker et al., 2005; Whitelock and Iozzo, 2005). HS may also serve as a retention molecule for growth factors and morphogens in the ECM, where they are stored, protected from proteolytic degradation and released upon cleavage of the HS chain when needed (Flaumenhaft et al., 1989; Saksela et al., 1988). Cell surface HS may bind the released growth factors or morphogens and dock them to their receptors or simply enhance the concentration of growth factors/morphogens at the cell surface. Depending on the structure, HS may also serve to establish and maintain concentration gradients of soluble molecules (Häcker et al., 2005; Lander et al., 2002; Nybakken and Perrimon, 2002).
Ligands of particular interest
The fibroblast growth factors - FGFs Fibroblast growth factors (FGFs) represent a large family of secreted molecules, which upon binding to their cognate cell surface tyrosine kinase FGF-receptors (FGFRs), activate the signal transduction pathways, required for multiple developmental processes. Both FGFs and FGFRs are expressed in specific spatial and temporal patterns. Some FGFs (FGF3, 4, 8, 15, 17 and 19) are only expressed during embryogenesis, for example, whereas others (FGF1, 5, 5-7, 9-14, 16, 18, and 20-22) are also found in adult tissues (Ornitz and Itoh, 2001; Thisse and Thisse, 2005).
FGFs are potent regulators of cell proliferation and function in both vertebrates and invertebrates. The mammalian FGF family consists of at least 22 structurally related proteins and orthologous FGF proteins commonly share more than 90 % amino acid sequence identity between species (Coulier et al., 1997).
Most FGFs have been shown in structural studies to share the same internal core structure of six identical and 28 highly conserved amino acid residues. Ten of these residues are also implicated in their binding to FGFRs. FGF1 and FGF2, considered prototype growth factors, both exhibit 12 antiparallel -strands in their core regions, and several basic amino acid residues found in the loop between -strands 1 and 2 and in -strands 10 and 11, are exposed to form the heparin binding site (Zhu et al., 1991). HSPGs bind extracellular FGFs, and present them to their cognate receptors and

24
induce dimerization of the receptors and receptor-ligand complex activation and stabilization.
The FGF receptors - FGFRs Four types of FGFRs (FGFR1-4) have so far been identified in vertebrates, with an additional one (FGF5) under preliminary characterization (Kreuger et al., 2006; Sleeman et al., 2001). FGFRs are receptor tyrosine kinases that contain an extracellular ligand binding domain and an intracellular tyrosine kinase domain. The extracellular part of an FGFR contains two or three immunoglobulin (Ig)-like domains. Alternative splicing will then generate FGFR isoforms with different ligand binding properties. Differential mRNA splicing in Ig domain III, for example, alters the specificity of the FGFR towards certain FGFs, and these splicing events seem to be tissue specific and essential for appropriate FGF signaling between distinct cell populations (Ornitz, 2000). The signaling activity of receptors presumably depends on the sequence characteristics of receptors, the FGFs and of the HSPGs that interact with both receptor and growth factor (Lin et al., 1999).
The Wnt family of morphogens The Wnts are secreted cystein-rich glycoproteins that bind to cell surface receptors of the Frizzled (Fz) and low-density lipoprotein receptor-related protein (LRP) families. They comprise a large family of highly conserved growth factors with important roles throughout the whole animal kingdom in controlling cell fate and patterning in early embryos and cell growth and/or differentiation in certain adult mammalian tissues. Their implication in a wide array of developmental processes, human diseases, e.g. cancer, and stem cell signaling has made Wnts and their signaling pathways a subject of intense investigation over the past two decades (Gordon and Nusse, 2006). There are at least 19 members of the Wnt family known in humans, representing 12 subfamilies, and 10 members of the Fz receptor family (Gordon and Nusse, 2006; Reya and Clevers, 2005). Membership of the Wnt family is defined in terms of amino acid sequence similarities rather than functional properties. Insights into the mechanisms of Wnt action have emerged from several systems: genetics in Drosophila and C. elegans, biochemistry in cell culture and ectopic gene expression in Xenopus embryos. Mutations in certain Wnt genes in the mouse result in highly specific developmental defects (Cadigan and Liu, 2006).
HSPGs play crucial roles in regulating key developmental signalingpathways and have important functions in the regulation of Wnt signalingpathways in that they are capable of retaining and stabilizing ligands (formation of morphogen gradients), by transporting ligands and by facilitating ligand-receptor interactions. These roles are likely to be mediated by both the HSPG core proteins and HS chains (Lin, 2004; Schambony et al., 2004).

25
HS in physiological processes
HS in embryogenesis The role of HSPGs in developmental processes has gained much attention in recent years. The generation of various model organisms deficient in core proteins or biosynthetic enzymes along with improved techniques for analyzing HS structure and its pattern of expression has made it possible to demonstrate the importance of HSPGs during development.
Embryogenesis is a highly complex process. Starting from a single fertilized oocyte, the cells in the developing embryo divide and differentiate into hundreds of well-defined cell types that assemble into multicellular organs.
In the very early embryo all the cells are the identical, and their subsequent differentiation into specific cell types and tissues is an extremely complicated process that depends on cell-cell signaling. For each cell to receive the correct signal at the right time, these cell-cell signaling events must be strictly regulated. There are two main forms of signaling event in development: threshold and concentration-dependent (morphogen) signaling. In a threshold signaling event, the receiving cell responds in a specific, predefined way as soon as the signal reaches its threshold (Freeman and Gurdon, 2002). This kind of signaling is common during late developmental processes. In concentration-dependent signaling events, the receiving cell can react in several different ways depending on the concentration of the signal (Freeman and Gurdon, 2002). Such events, often referred to as morphogen gradients, are frequent during early development. It is still not clear whether these gradients arise by diffusion or by other mechanisms, such as transcytosis (Lander et al., 2002). Members of the Wnt/Wingless (Wg), bone morphogenic protein (BMP), Hedgehog (Hh), transforming growth factor- (TGF- ) and FGF families of proteins can be found among the morphogens.
HSPGs have been shown to play an essential role in regulating a number of developmental signaling pathways, but the mechanisms involved are far from defined and may work slightly different in different signaling systems. Five different mechanisms through which HSPGs are believed to modulate activity in development have been suggested: receptor dimerization, receptor-ligand complex stabilization, surface transport, two dimensional sliding and transcellular transport of ligands (Nybakken and Perrimon, 2002). In two-dimensional sliding, low-affinity binding of a molecule would allows it to slide along HS chains to reach target cells further away from the source in a shorter time than would otherwise be possible. In the transcellular model, molecules would “hitchhike” on HSPGs to distantly located cells by moving through neighboring cells in a manner related to phagocytosis.

26
The sulfation pattern of HS may be assumed to be important in all these suggested mechanisms. By producing HS sequences with higher or lower affinity for a molecule, cells could either attract or repel the molecule and thereby facilitate generation of local concentration differences. The production of different HS sequences would theoretically enable the cells to regulate such signaling activities.
HS in human diseasesHS participates not only in normal physiological processes but also in numerous pathological conditions including amyloid-related disorders, inflammatory conditions and cancer. In addition, many microorganisms such as viruses, bacteria and parasites have been shown to interact with cell surface HS and exploit it as a co-receptor for invasion (Rostand and Esko, 1997; Vogt et al., 2003).
Amyloid diseases are a heterogeneous group of disorders that are characterized by the formation of insoluble fibrillar aggregates of misfolded proteins, including Alzheimer´s, Huntington´s and Parkinson´s diseases and prion encephalopathies and affecting multiple organs and tissues (Kisilevsky, 2000; Temussi et al., 2003). There are at least 22 polypeptides known to date that refold and assemble into highly organized fibrils, to form tissue deposits that may be termed amyloid (Elimova et al., 2004). HSPGs are found in all kinds of amyloid deposits in vivo, and many studies have demonstrated that both HS and other GAGs bind amyloidogenic peptides invitro, enhancing their fibrilization and stabilizing already formed aggregates (Kisilevsky, 2000; van Horssen et al., 2003).
Cancer is characterized by uncontrolled proliferation of cells followed by tissue invasion and sometimes metastasis. Angiogenesis is an important event in tumor progression, since sustained tumor growth is dependent on vascularization and new blood supply. Several angiogenic factors, including vascular endothelial growth factor (VEGF) and FGFs, and also anti-angiogenic factors such as endostatin and angiostatin (see Table1), have been shown to interact with HSPGs (Iozzo, 2005). Heparanase, an enzyme involved in HS turnover, is thought to play an important role in metastasis through the cleavage and removal of HS in extracellular tissue (Vlodavsky and Friedmann, 2001). The newly detected Sulf family of enzymes is also of interest in relation to a number of cancer forms. The human Sulf1 (HSulf1) enzyme is readily detectable in normal tissues, but is undetectable or markedly reduced in a number of carcinoma cell lines, including ovary, breast, pancreatic, renal and hepatocellular carcinoma cell lines (Lai et al., 2003). The action and functions of the Sulf enzymes will be described in more detail in the following section.
Other diseases with which HS can be associated are inflammatory diseases, diabetes mellitus and hereditary multiple exostoses.

27
Regulation of HS structure Principles of regulation Although the lack of a template for HS biosynthesis makes it difficult to understand how specific structures or sequences can be synthesized, the reproducibility of HS structure is striking. While there is some evidence of coupling and processivity of the enzymatic reactions in the Golgi apparatus, it is still unclear how HS chains with different domain patterns are generated (Conrad, 1998; Salmivirta et al., 1996). One could imagine that might depend on different isoforms of the biosynthetic enzymes, their relative activities and substrate specificities, their amount and availability and their localization in the Golgi apparatus, but how controlled could such a template-free system be?
There are many sites in HS chains that are not modified by the biosynthetic enzymes. While some HS modifications promote the availability of substrate sequences for other enzymes, other reduces this. Thus, differences in the concentration and isoform expression of biosynthetic enzymes are probable reasons for the structural variation in HS between cell types. Both the concentrations of an enzyme and the isoform expressed may be transcriptionally regulated, but only a few in vivo studies have shown any correlation between alterations in enzyme transcription and changes in HS structural motifs (Ford-Perriss et al., 2002; Nogami et al., 2004). It is unclear at present to what extent HS structural variation is caused by transcriptional regulation of the enzyme level and isoform expression, and little is known about the proportion of active enzyme in different cells. A recent study of liver NDST enzymes showed that neither transcriptional, nor translational or post-translational control of their activity determines the N-sulfation of liver HS. Instead it was suggested that the critical step in regulation may be the assembly of enzyme complexes and the selection of enzymes included in them (Ledin et al., 2006). Furthermore, neither regulation of enzyme activity in the Golgi compartment by glycosylation, as recently shown for chondroitin 6-sulfotransferase 1 (Yusa et al., 2006), nor proteolytic cleavage can be excluded.
Several studies have suggested that HS biosynthetic enzymes may function in the physical vicinity of each other, associating and interacting with each other, in the Golgi compartment (Lidholt et al., 1989; Lidholt et al., 1992; McCormick et al., 2000; Pinhal et al., 2001; Senay et al., 2000). Three models of HS biosynthesis have been suggested: 1) the “Random encounter” model, in which the enzymes are located independently of each other and compete for the substrates, 2) the “GAGosome” model in which the enzymes are organized in a tight complex (such as the ribosome), interacting with each other and with other potential regulatory proteins, and 3) the “Frozen out” model, which is a slightly modified GAGosome model

28
in which the biosynthetic enzymes are less tightly organized, more flexible and may compete with each other for a place in the GAGosome protein complex (Ledin et al., 2006).
Many of the HS biosynthetic enzymes differ in their transmembrane and cytoplasmic domains, which are thought to be important for the Golgi localization. Hypothetically, different Golgi localizations of the biosynthetic enzymes in different cell types could affect the access of individual enzymes to the polysaccharide substrate and result in structural differences in the final HS chains.
Other mechanisms that could affect HS structure are local concentrations of nucleotide sugars and the sulfate donor 3´-phosphoadenosine-5´-phosphosulfate (PAPS). Finally, one cannot exclude the possibilities of other regulatory proteins that have not yet been identified or characterized.
Evolutionary aspects of HS structural diversity The requirements for sulfated HS chains for the development of metazoans is undisputable, as seen in Drosophila, C. elegans, mouse and other transgene models (Forsberg and Kjellén, 2001; Häcker et al., 2005; Turnbull et al., 2003), but the evolutionary background to more subtle HS variations is still largely unknown. Has the structural complexity of HS evolved together with its biological function? Is the tissue variation in HS structure evidence of a tight regulation of HS biosynthesis, or does it reflect a lack of evolutionary pressure controlling HS biosynthetic enzyme expression and/or activity? Finally, can HS structural variation be interpreted in terms of evolution?
Organisms may have evolved to alter HS biosynthesis in such a way that only the necessary HS motifs are generated under appropriate developmental circumstances. HS structural variation could have evolved as a dynamic way for cells to regulate when and which signals they can receive, in the same way as cells express different types of cell surface receptors. This also makes it possible for cells other than the producers and the receivers of HS-binding ligands to control the amplitude and distance of the signaling event.
The enzymes involved in HS biosynthesis are highly conserved throughout evolution, but the numbers of isoforms differ between organisms. The more highly evolved an organism is, the more complex the biosynthetic machinery appears to be. The question is whether all the enzyme isoforms are vital for the generation of functional HS.
Although it is unclear whether evolution has contributed to HS structural variation, and if so, how, it will be necessary to identify new, evolutionary simple model systems for studying HS (i.e. the sea anemone, Nematostellavectensis, as described below), together with an improved methodology for analyzing HS structure, in order to gain an understanding of the evolutionary aspects of HS biosynthesis

29
Regulation of the 6-O-sulfation pattern
HS glucosaminyl 6-O-sulfotransferases - 6OSTs HS glycosaminyl 6OSTs are Golgi-located type II transmembrane proteins, which transfer sulfate groups to the C6 positions of GlcNAc/NSO3 residues in nascent HS chains (Fig. 6). 6OST was first purified from serum free medium of Chinese hamster ovary (CHO) cells and later cloned from human fetal brain cDNA (Habuchi et al., 1995; Habuchi et al., 1998). Three mammalian 6OST isoforms are known to date: 6OST1, 6OST2 and 6OST3 (Kusche-Gullberg and Kjellén, 2003).
Figure 6. The action of 6OSTs. The target GlcN residue can be either GlcNSO3 or GlcNAc and the adjacent HexA residues either GlcA or IdoA. PAPS indicates the sulfate donor.
Recent studies underpin the importance of 6OSTs and 6-O-sulfation for growth factor signaling in developmental processes. RNA interference-mediated disruption of the single 6OST in Drosophila, resulted in severely disturbed FGF signaling (Kamimura et al., 2001), while morpholino-mediated knockdowns of 6OSTs in zebrafish causes disturbed muscle development and angiogenesis (Bink et al., 2003; Chen et al., 2005) and 6-O-sulfation of HS is essential for retinal axon guidance in Xenopus (Irie et al., 2002). Furthermore, C.elegans individuals that are deficient in the single 6OST exhibit defects in the development of the nervous system (Bulow and Hobert, 2004).
The transcript levels of individual 6OST isoforms vary in tissues and during development, and may add to HS structural heterogeneity. mRNA expression studies in adult mouse tissues have shown strong expression of 6OST1 in liver and 6OST2 in brain, whereas 6OST3 was ubiquitously expressed (Habuchi et al., 2000). A shorter splice variant of the human
o o
PAP
PAPS6OST
oso3- oso3
-

30
6OST2 is expressed mainly in the ovary and placenta (Habuchi et al., 2003). Studies in the early mouse embryo have shown that 6OST1 is expressed predominantly in epithelial and neural-derived tissues, whereas 6OST2 is more strongly expressed in mesenchymally-derived tissues. 6OST3 transcripts appear later in development than the other two and are expressed in a more restricted manner (Sedita et al., 2004).
Previous studies of the substrate specificities of the 6OSTs have indicated some differences between the different enzymes (Habuchi et al., 2000). In addition the stem domains of the 6OSTs have been shown essential for the enzyme activity and localization in the Golgi compartment (Nagai et al., 2004). Although little is known about how different 6-O-sulfation patterns are created, there are studies suggesting that transcriptional control of the 6OSTs is a cause of HS structural variation (Nogami et al., 2004).
HS 6-O-endosulfatases - Sulfs The abundance of HSPGs and their abilities to influence diverse interactions at the cell surface make them candidate regulators of various developmental signals. It has recently been demonstrated that modification of HS structure does not stop at the biosynthetic level, since a new type of HS modifying enzyme has been discovered in the quail that displays extracellular sulfatase activity towards heparin (Dhoot et al., 2001; Morimoto-Tomita et al., 2002; Ohto et al., 2002). This enzyme, sulfatase 1 (Sulf1), is a member of an evolutionarily conserved gene family related to the lysosomal N-acetylglucosamine sulfatases (G6-sulfatases) that catalyze the hydrolysis of 6-O-sulfates from GlcNAc units of HS during the degradation of HSPGs. Sulf1 is homologous to the G6-sulfatases throughout the structural regions required for the formation of the active site. In addition, it has a distinctive hydrophilic domain as well as a predicted N-terminal secretory signal peptide (Dhoot et al., 2001). In contrast to the lysosomal G6-sulfatases, Sulf is believed to act on HS present at the cell surface. Two Sulfs are known, Sulf1 and Sulf2. Orthologs of the Sulf proteins have been dentified in various species including quail, mouse, human and rats (Dhoot et al., 2001; Morimoto-Tomita et al., 2002; Ohto et al., 2002).
Quail Sulf1 (QSulf1), was identified in a screen for Sonic Hedgehog (SHh, see Table 1) response genes activated during somite formation. QSulf1 showed a developmentally regulated expression pattern (Dhoot et al., 2001) and antisense inhibition of QSulf1 resulted in decreased expression of MyoD in muscle progenitors in the somites. MyoD is Wnt-inducible, suggesting that QSulf1 may be of importance for Wnt signaling. In addition, QSulf1 has also been shown to regulate the binding between HS and Noggin, a BMP antagonist (Viviano et al., 2004).
Sulf1 and Sulf2, and possibly other related proteins may be important actors in the regulation of the sulfation pattern in HS. Two of the papers in this thesis (papers III and IV) describe the characterization and substrate

31
specificities of the Sulf enzymes and how these enzymes may regulate the function of cell surface HSPGs.
Nematostella vectensis - a possible model for developmental evolutionary studies of HS and its enzymatic machinery? To study whether evolution has contributed to HS structural heterogeneity, and if so, what it has contributed, efforts have been made to study how the diversity of HS and its biosynthetic machinery has evolved. Many studies have been carried out using simpler, although phylogenetically still highly evolved model organisms such as mice, zebrafish, fruit flies or worms. A difficult problem when trying to answer the evolutionary questions raised in the previous chapters is that of choosing the right model system, as this must serve as a complement to already existing models systems regarding both phylogenetic informativeness and evolutionary history.
The sea anemone, Nematostella vectensis, belongs to the most basal class of the cnidarians, Anthozoa, and is an emerging model organism that has already proved to provide valuable insights into the developmental evolution of fundamental metazoan traits such as axial patterning, bilateral symmetry etc. The availability of powerful molecular and genetic tools, the sequence of the Nematostella genome, which will soon be available, and the accessibility of Nematostella development to laboratory manipulations, make Nematostella an ideal organism for studies in evolutionary genomics (Darling et al., 2005; Kraus and Technau, 2006).
The name Cnidaria comes from the Greek word “cnidos”, which means stinging nettle. There are four major groups of cnidarians; Anthozoa, which includes the true corals, anemones and sea pens, Cubozoa, the amazing box jellies with complex eyes and potent toxins, Hydrozoa, the most diverse group, including the siphonophores, hydroids, fire corals and many medusae, and Scyphozoa, the true jelly fish. The cnidarians form one of the oldest metazoan phyla and are among the simplest animals displaying epithelial tissue organization (Darling et al., 2005; Miller et al., 2005). They may be especially informative, since their development have remained relatively unchanged during the last 600 million years, suggested by fossil evidence (Chen et al., 2002). The cnidarians are diploblastic animals, having two germ layers: the endoderm and the ectoderm. Nematostella nevertheless possesses homologs of many genes that are critical for proper specification and patterning of mesodermal cell fates in Bilatarians, which have three germ layers: the endoderm, the mesoderm and the ectoderm (Technau and Scholz, 2003).

32
A large number of developmental regulatory genes have been identified in the Bilataria, and many orthologs which are critical for proper patterning and specification during embryogenesis, have also been identified recently in Nematostella (Carrol SB, 2001). The expression patterns of these genes in Nematostella suggest that the Bilataria and Cnidaria share a common ancestral legacy.
No studies have yet been reported on HS structure and function or on the HS-modifying enzymes in Nematostella. HS is known to be present in Hydra, which belongs to a more evolved class of the cnidarians, but little is known about its function in that genus (Medeiros et al., 2000).
For these reasons, Nematostella was chosen as a suitable model organism in which to study the developmental evolution of HS structural diversity, and to characterize “ancient” HS biosynthetic enzymes. The results are described in the last paper of this thesis.

33
Present Investigation
Aims of study
The purpose of this study was to investigate the mechanisms controlling HS 6-O-sulfation patterns and to study HS structure in an evolutionarily “primitive” model organism, Nematostella vectensis. The aims of the individual papers were:
• to investigate the substrate specificities of the HS glucosaminyl 6OSTs (Paper I).
• to investigate the effects on HS structure of 6OST1, 6OST2 and 6OST3 overexpression in human embryonic kidney (HEK) 293 cell (Paper II).
• to investigate the substrate specificity of QSulf1 and its
role in Wnt-Frizzled signal transduction (paper III).
• to investigate the substrate specificity of QSulf2 and determine the role of the hydrophilic domain (HD) for the enzymatic activities of QSulf1 and QSulf2 (Paper IV).
• to investigate how the diversity of HS structure and its
complex biosynthetic machinery has evolved (Paper V).
Results
Substrate specificities of mouse heparan sulphate glucosaminyl 6-O-sulfotransferases (Paper I) Three isoforms of the HS glucosaminyl 6OSTs had previously been identified and cloned and shown to catalyze the transfer of labeled sulfate from [35S]PAPS into a variety of acceptor polysaccharides (Habuchi et al., 2000). Differences in the amounts of label incorporated into the different

34
acceptor substrates had been interpreted in terms of favored acceptor sequences. The purpose of this study was to compare the substrate specificities of the different enzymes more precisely. We incubated recombinant 6OSTs with various O-desulfated polysaccharides and oligosaccharides as acceptor substrates and [35S]PAPS as a sulfate donor and determined the structures of the resultant 35S-labeled products.
We showed that all three enzymes were able to catalyze 6-O-sulfation of both -GlcA-GlcNS- and -IdoA-GlcNS- sequences, although with a preference for those containing IdoA, with or without preexisting 2-O-sulfate groups. Interestingly, however, 6OST1 showed a relatively higher activity towards -GlcA-GlcNS- sequences lacking pre-existing 2-O-sulfate groups than did 6OST2 or 6OST3. In general, sulfation of non-O-sulfated sequences was favored at low acceptor substrate concentrations.
In addition, using partially de-O-sulfated AT-binding oligosaccharides as substrates, we showed that all three 6OSTs were able to act on both N-acetylated as and N- and 3-O-sulfated glucosamine residues in -GlcNAc-GlcA-GlcNS3S- sequences. 6OST2 and 6OST3 showed a greater preference for the N-acetylated target than did 6OST1.
These findings imply that both 3-O and 6-O-sulfation can complete the biosynthetic modification of HS in vitro, and that the substrate preferences of the 6OSTs are less stringent than had previously been believed.
Overexpression of heparan sulfate 6-O-sulfotransferases in human embryonic 293 cells results in increased N-acetylglucosaminyl 6-O-sulfation (Paper II) To compare the enzyme characteristics of the 6OSTs further and to gain an understanding of how variations in the expression levels of 6OST isoforms might influence the HS structure, we established cell lines that stably overexpressed each 6OST isoform separately.
The overexpression resulted in increased O-sulfotransferase activity that varied among the clones from a 2-fold to a more than 30-fold increase in 6OST1 and 6OST3 transfectants, while for the 6OST2 transfectants, the increase in activity ranged from 2 to 22-fold. Selected cell clones were metabolically labeled with [3H]GlcN and [35S]sulfate and GAGs were isolated. The overexpression did not result in any apparent difference in the relative proportions of labeled CS versus HS. Detailed structural analyses, carried out on both total unlabeled and radio-labeled HS isolated from transfected HEK 293 cells, showed a significant increase in 6-O-sulfation in the transfected cells whereas the overall sulfation remained practically unchanged. Augmented 6-O-sulfation on both GlcNS and GlcNAc residues

35
was found. In addition, there was a gradual change in HS structure from an intermixed distribution of NA, NS/NS and NS domains to more extended regions of 6-O-sulfated NA and NS domains. The amplitude of the changes was positively correlated with the level of enzyme expression but was not isoform-specific. It should be noted, however, that even at a relatively low expression level, the changes were most prominent in the cells overexpressing 6OST3.
Our findings suggest that the 6-O-sulfation of HS chains is affected not only by the substrate preferences of the 6OSTs, but also by the level of enzymes expression.
QSulf1 remodels the sulfation states of cell surface heparan sulfate proteoglycans (Paper III) In paper III we investigated the enzymatic activity of the quail sulfatase 1 (QSulf1) and studied the importance of QSulf1 for proper Wnt signaling. The substrate specificity of QSulf1 was determined by structural analysis of 1) 35S-labeled HS after incubation with purified wild-type QSulf1 or an inactive form (QSulf1mutant), 2) metabolically 35S-labeled HS isolated from control 293T cells and cells overexpressing either QSulf1 or QSulf1mutant.
Disaccharide analysis showed that QSulf1-digested HS contained a reduced amount of IdoA2S-GlcNS6S residues, accompanied by a parallel increase in IdoA2S-GlcNS residues. A similar shift in disaccharide proportions was observed after structural analysis of metabolically labeled HS isolated from cells expressing QSulf1. Our findings established that QSulf1 is a HS-specific 6-O-endosulfatase with specificity for a subset of 6-O-sulfated HS disaccharide units. This is in contrast to the lysosomal GlcN-sulfatase, which functions exclusively as an exozyme at the non-reducing end of the saccharide in lysosomal degradation of HS.
Localized either on the cell surface or in the Golgi apparatus, the action of QSulf1 was shown to reduce the binding of Wnt to the HS chains of Glypican1, and to promote Wnt signaling. In addition, CHO cells that were mutants with respect to HS biosynthesis were shown to be defective in Wnt-Frizzled receptor activation, suggesting the importance of HS for Frizzled receptor function.
These findings together suggest that by removing 6-O-sulfates from HS chains, QSulf1 functions as a regulator of Wnt-Frizzled signal transduction in cells.

36
Substrate specificity and domain functions of extracellular heparan sulfate 6-O-endosulfatases, QSulf1 and Qsulf2(Paper IV) In paper IV we investigated the enzymatic activity of the quail sulfatase 2 (QSulf2) and compared it with that of QSulf1 (described in paper III). In addition, we have investigated the cell surface association and the essential roles of the hydrophilic domain (HD) for QSulf enzymatic activities.
Emplyoing similar methods to those used in paper III, we showed that both QSulf1 and QSulf2 function as HS 6-O-endosulfatases, with identical substrate preferences for a subset of trisulfated disaccharides, -IdoA2S-GlcNS6S-, within HS chains.
Mutagenesis studies revealed that the conserved polypeptide region in the HD domain of QSulf1 and QSulf2 has multiple functions with regards to anchoring of the enzymes to the cell surface, binding of HS substrates and mediation of HS endosulfatase activity.
In summary, this study demonstrated that both QSulf1 and QSulf2 are cell surface-associated HS 6-O-endosulfatases with similar enzymatic activities. In addition, we established the HD domain as a multifunctional domain that is essential for the unique endosulfatase activity of the QSulf enzymes.
Identification of heparan sulfate in the sea anemone Nematostella vectensis (Paper V) The presence and functions of HS have been well studied and documented. Since most studies had concerned higher evolved organisms, we set out to identify HS and the enzymatic machinery responsible for generating biologically active HS in the evolutionary primitive organism Nematostella vectensis.
Using radiolabeling and biochemical methods, the presence of a HS-like molecule was identified that contained partly similar disaccharide structures to mammalian HS. Computational screening and molecular studies revealed the presence of all the enzyme families involved in HS biosynthesis and modification in mammalian model systems, but apart from two forms of the HS 2OST (2OSTa and 2OSTb) and NDST (NDST3/4 and NDST4) only single forms of each enzyme were identified. mRNA transcripts for all enzymes other than 2OSTb were expressed at all developmental stages, but while the mRNA expression levels of most enzymes were fairly uniform at all stages, the 6OST, 3OST and the 6-O-endosulfatase, displayed elevated expression levels during the blastula and gastrula stages.
The spatial expression patterns of the EXT1, 2OSTa and 2OSTb, 6OST and Sulf genes were investigated by in situ hybridization of whole mount embryos. Diffuse signals were detected throughout the embryos for all the stages examined, suggesting that the EXT1, 2OSTa and 2OSTb, 6OST and

37
Sulf genes are constantly expressed throughout the whole embryo in all the stages of Nematostella development examined here. Interestingly, however, both Sulf and 2OSTa expressions showed markedly elevated expression in certain areas of the embryo at different stages, indicating that they may play a specific role in HS modifications during larval development.
Conclusions The main conclusions to be drawn from the studies were:
• The three murine 6OSTs exhibit qualitatively similar substrate specificities in vitro, although with minor differences in target preferences.
• Both 3-O and 6-O-sulfation can complete the biosynthetic
modification of HS in vitro.
• Overexpression of the 6OSTs demonstrated that the 6-O-sulfation of HS chains is affected not only by the substrate preferences of the 6OSTs but also by the amount of enzyme expressed by the cells.
• The 6OSTs are apparently not strictly dependent on N-sulfate groups for substrate recognition.
• QSulf1 and QSulf2, which are associated with the cell surface, are both extracellular HS 6-O-endosulfatases that act on a subset of trisulfated disaccharides within HS chains.
• QSulf1 functions as a regulator of Wnt-Frizzled signal transduction in cells by desulfating cell surface HS.
• The conserved HD domain of the Sulf enzymes is a multifunctional domain that is essential for the binding to HS, for anchorage to the cell surface and for the endosulfatase activity.
• A HS-like molecule is present in the evolutionarily primitive sea anemone Nematostella.
• Members from all the enzyme families involved in the generation and modification of HS are present in Nematostella.

38
Discussion and Future Perspectives
The biological, developmental and pathological importance of HS cannot be overestimated. Its unique molecular design and flexibility is essential to its ability to exert control over cellular responses. The notion that subtle changes in HS structure can influence so many different biological processes opens up thrilling possibilities. One can imagine that cells can respond to their microenvironment by dynamically regulating HS fine structure at their surface and in the ECM through alterations in the biosynthetic machinery. The underlying biological mechanisms regulating the molecular diversity in HS are nevertheless still unclear and need to be carefully elucidated.
In order to understand how the structural variability in HS is regulated, it is important to understand the functions of the enzymes involved in the generation and modification of HS and the interplay between them. In higher organisms many of these enzymes occur in multiple isoforms that differ in substrate specificity and spatial and temporal expression (Esko and Lindahl, 2001; Kusche-Gullberg and Kjellén, 2003). The critical roles of these enzymes in development have been demonstrated in genetic studies using various transgenic model organisms. Depending on the enzyme or enzyme isoforms disrupted, the developmental effects can differ from mild to very severe. Thus, while a lack of functional EXT1 or EXT2 in mice abolishes HS production and causes early embryonic lethality (Lin et al., 2000; Stickens et al., 2005), disruption of 3OST1 (the major enzyme responsible for forming HS sequences with anticoagulant activity) in mice does not result in any procoagulant phenotype and the mice are viable and fertile (Shworak et al., 2002).
In his thesis we have studied regulatory aspect of HS structural diversity, or more specifically we have investigated how the HS 6-O-sulfation patterns may be regulated and touched upon some evolutionary aspects of HS structural variation. Some important questions arise when we attempt to understand the roles of 6OSTs and Sulfs in the regulation of HS 6-O-sulfation patterns. How important are these enzymes for the final design of HS chains? What are the regulatory mechanisms determining the actions of the different isoforms?
The 6-O-sulfate groups in HS occur in different structural contexts, so that approximately half of the total 6-O-sulfate groups in HS can be found in NS domains, often in close vicinity to 2-O-sulfated HexA while the other half are to be found in the mixed NA/NS domains, which largely lack

39
2-O-sulfate groups (Maccarana et al., 1996). Based on previous studies suggesting differences in substrate specificities between the 6OSTs (Habuchi et al., 2000), it was tempting to speculate that the 6-O-sulfation of different target sequences may be catalyzed by different 6OST isoforms. In papers I and II we compared the substrate specificities of the 6OSTs using in vitroassays and overexpression in mammalian cells. Unexpectedly, no marked differences were found in the substrate specificity between the three 6OSTs. In addition, all thee 6OSTs were able to act on AT-binding target structures containing a 3-O-sulfate group, indicating that either 3-O- or 6-O-sulfationcan be the final step in HS biosynthesis (as also shown by (Zhang et al., 2001)).
Furthermore, high overexpression of any of the 6OST isoforms resulted in increased 6-O-sulfation of both N-sulfated and N-acetylated GlcN residues and in an altered HS domain structure with less 6-O-sulfation of mixed NA/NS domains.
It is important to bear in mind that substrates specificities in vitro and enzyme overexpression differs from physiological conditions and may not reflect the normal regulation of 6-O-sulfation, which may be dependent on other biosynthetic or unknown regulatory proteins affecting the action of the enzymes and the availability of their substrates. However, experiments of this kind may still give valuable information about the enzyme properties.
In what context do our results fit into the regulation of the HS biosynthetic machinery? Little is known about the interplay between the 6OSTs and other HS biosynthetic enzymes. HS biosynthesis is generally considered to be a hierarchical process in which the biosynthetic enzymes are believed to act in a sequential manner. N-deacetylation/N-sulfation of GlcNAc units initiates the modification reactions in the growing HS chains, and this initial reaction, carried out by the NDSTs places a limit on further modifications and basically creates the domains in HS. These domains, NA, NS and mixed NA/NS domains, are differentially O-sulfated.
The changes in HS structure described in paper II partially resembles the HS produced in mice lacking either the 2OST or the C5-epimerase (Li et al., 2003; Merry et al., 2001). Like the HS in 2OST and epimerase-deficient mice, HS in the overexpressing cell clones contained heavily 6-O-sulfated NS domains (HexA 2S-GlcNS6S), low 2-O-sulfation, and fewer mixed NA/NS domains. However, neither the 2OST nor the C5-epimerase mutant HS showed any increased formation of GlcNAc6S disaccharides. Nevertheless, 6-O-sulfated N-acetylated HS chains are synthesized by mouse embryonic stem cells deficient in NDST1 and 2 (Holmborn et al., 2004).
The effects observed on 6-O-sulfation and N-sulfation observed in HS from the NDST1/2, 2OST and the C5-epimerase mutant mice suggest that its biosynthesis is less hierarchical than had previously been believed and that N-sulfation may be affected by “downstream” modifications, such as 6-O-sulfation. Thus, the 6OSTs appear to play a more independent role in

40
the HS biosynthetic machinery and may influence HS structure to a larger extent than was previously known. The individual role of the 6OST isoforms may be better understood by generating mice deficient in one or other of the enzymes.
The Sulfs add greater complexity to the mechanisms regulating the HS 6-O-sulfation patterns, showing that cells have other means of modulating HS structure and protein-binding functions than by altering the biosynthesis of HS.
Figure 7. A two-state catch or present model for QSulf1 regulation of Wnt signaling. (A) Embryonic cells not expressing QSulf1 display HS chains which catch and bind Wnt-ligands with high affinity, preventing functional interactions with the Frizzled receptor. (B) QSulf1 selectively removes 6-O-sulfates from the HS chains of cell surface HSPGs on cells expressing QSulf1, promoting the formation of Wnt-HS-Frizzled receptor complexes and the initiation of Wnt signal transduction.
Paper III and IV provide data on the substrate specificities of QSulf1 and QSulf2 and the function of QSulf1 in HS-dependent Wnt signaling, and on the function of the hydrophilic domain (HD) of the Sulfs. The HD domain is an evolutionarily conserved domain and is the unique signature domain for the Sulf proteins. Our results established that both enzymes are HS 6-O-endosulfatases located at the cell surface, with preferences for IdoA2S-GlcNS6S disaccharides within HS chains. Furthermore, the HD domain of the Sulfs was shown to be essential for the binding of HS, anchorage to the cell surface and the endosulfatase activity. QSulf1 was also shown to promote Wnt-Frizzled signaling in cells. These results point to a
A B
Cell surface HSPG
Frizzled receptor
Sulfate groups
QSulf1Wnt-ligand

41
“two-state” catch or present model for the QSulf1 regulation of Wnt signaling by remodeling cell surface HS was suggested (Fig. 7).
Until the discovery of the Sulfs, the HS 6-O-sulfation pattern was thought to be exclusively controlled by the action of the 6OSTs during HS biosynthesis. The Sulf enzymes introduce a new and novel level of control in the regulation of HS diversity and growth factor binding. Located at the cell surface, they are strong candidates for roles as modulators of HS-mediated signaling. Since their discovery, the Sulfs have been associated with various signaling pathways. Indeed, Sulf1 activity has been shown to affect FGF as well as Wnt signaling, both being important during embryogenesis. However, contrary to Wnt signaling, which is stimulated by Sulf1, FGF signaling during mesoderm induction and angiogenesis is inhibited by Sulf1 (Wang et al., 2004). Furthermore, Sulf1 reduces the binding affinity between the BMP inhibitor Noggin and HS, thereby enhancing BMP signaling (Viviano et al., 2004). The Sulfs have also been associated with other signaling pathways during development, tumor growth and angiogenesis (Ai, et al unpublished results and (Lai et al., 2004; Li et al., 2005; Uchimura et al., 2006)).
The Sulf enzymes have opened up new avenues of investigation of the regulatory mechanisms behind HS-mediated signaling in development and cancer. How much does the Sulf enzymes actually contribute to the regulation of HS 6-O-sulfation patterns? The substrate specificities in paper III and IV suggest that only a fraction of the total HS sulfation is affected by the action of the Sulfs. How much can such changes in HS structure matter in vivo? Sulf1, Sulf2 and double Sulf1/Sulf2 knockouts have recently been generated genetically in mice, and surprisingly, although the Sulfs are associated with so many important developmental signaling pathways, neither Sulf1 nor Sulf2 deficient mice showed any obvious phenotypes at birth, except for a small reduction in litter size and body weight. The phenotypes in these mice were more obvious postnatally, however, involving various types of malformations and being more obvious in the Sulf1/Sulf2 double knockout mice (Ai et al unpublished results; Lamanna et al., 2006). The effect of Sulf knockout on HS structure was investigated by analyzing HS isolated from mouse embryonic fibroblasts (MEFs) obtained from various Sulf knockout embryos. Detailed structural analysis of HS from the Sulf1-deficient MEFs showed a striking increase in 6-O-sulfation, which was not seen in HS from Sulf2-deficient MEFs. Intriguingly, the level of 6-O-sulfation in HS from the double Sulf1/Sulf2-deficient MEFs was significantly higher than that in their Sulf1-deficient counterparts. These data imply that Sulf1 and Sulf2 are functionally cooperative (Lamanna et al., 2006). Unlike their avian orthologues, mammalian Sulf activities are not restricted to the highly sulfated NS domains of HS (Lamanna et al., 2006).
Interestingly, the increase in 6-O-sulfation in the Sulf-deficient cells was accompanied by a small decrease in other sulfation events, particularly in

42
2-O-sulfation (Ai et al unpublished results; Lamanna et al., 2006). Whether this occurs as a result of direct interaction between the Sulfs, the HS chains and/or other possible regulatory proteins in the Golgi remains to be determined.
Not much is known about the evolution of the biosynthetic machinery or whether the enzymes involved in HS biosynthesis generate the same polysaccharide in less evolved animals. In this respect the sea anemone, Nematostella vectensis, is a suitable model system for studying HS in evolution. Paper V sets a foundation for using Nematostella in HS research by identifying the presence of a HS-like molecule and the families of enzymes involved in the generation of HS. Furthermore, it also provides spatial and temporal expression profiles for the enzymes identified. Our results represent only a first step towards using Nematostella as a tool for investigating the role of evolution in generating HS structural diversity, and much remains to be done. The focus in the future will be on analyzing the role of HS during in vivo developmental patterning in Nematostella and on understanding the functions and interactions between HS and HS-dependent growth factors.
The generation and regulation of the HS structural diversity is an intriguing field with great opportunities for new discoveries. Despite the efforts of many investigators, tackling the problems from many different angles, the regulatory mechanisms behind the heterogeneity of HS structure are still poorly understood and many more questions need to be answered. Hopefully, one day this will no longer be a mystery.

43
Populärvetenskaplig Sammanfattning
Alla levande organismer är uppbyggda av olika vävnader som i sin tur är uppbyggda av många små funktionella enheter, celler. Alla celler i en individ har precis samma arvsanlag (DNA), trots detta finns det ändå hundratals olika celler med mycket olika funktioner. För att cellerna ska veta vad de ska bli, vart de ska vandra och hur de senare ska utföra sina uppgifter måste de kommunicera med varandra och med omgivningen. Detta kan ske på många olika sätt. Cellerna kan etablera direkt kontakt med varandra eller sända ut särskilda signalmolekyler som transporteras till receptorer (mottagare) på ytan av andra celler. När signalmolekylerna binder till sina receptorer leder detta till att en signal förmedlas inne i cellen vilket i sin tur påverkar cellens beteende.
Heparansulfatproteoglykaner är en kategori av receptorer som finns på ytan av de flesta celler och i bindväven mellan dem. Heparansulfat-proteoglykaner på cellytan hjälper cellerna att fånga upp signalmolekyler. Heparansulfatproteoglykaner består av en eller flera långa ogrenade kolhydratkedjor, så kallade heparansulfatkedjor (HS) covalent bundna till en proteinkärna (Fig. 2). HS från olika celltyper uppvisar stora strukturella skillnader (Fig. 3). HS-strukturen är ett resultat av ett komplicerat biosyntesmaskineri med olika enzymer som producerar och modifierar sockerkedjan vilket bl a innebär att sulfatgrupper introduceras i olika positioner (Fig. 6). Slutresultatet blir en mycket heterogen polysackarid vars struktur varierar mycket mellan olika celler, vävnader och stadier i utvecklingen. Det är klarlagt att HS-strukturen påverkar funktionen av många olika signalmolekyler (Fig. 4). Däremot är det inte klarlagt vilken roll HS-strukturen har för olika biologiska processer och vad som bestämmer vilken struktur som skall finnas var och när i kroppen. Man kan tänka sig att en cell gör en HS-kedja med en viss struktur vilket gör det möjligt för cellen att ta emot en signal som en cell med en annan HS-struktur inte kan ta emot. Hur vet cellen vilken HS-struktur den skall tillverka vid en viss tidpunkt och hur reglerar cellen denna tillverkning?
Den här avhandlingen fokuserar på regleringen av ett mycket viktigt steg i bildandet av olika HS-strukturer, nämligen 6-O-sulfateringen. Vi har studerat de enzymer som sätter dit 6-O-sulfatgrupper under biosyntesen av HS, samt de enzymer som selektivt plockar bort 6-O-sulfatgrupper på utsidan av cellen efter biosyntesen. Vi har även introducerat en ny modellor-ganism, en sjöanemon, som kan vara till stor hjälp för att förstå hur celler

44
kan reglera bildandet av olika HS-strukturer och på så sätt påverka många olika biologiska processer.

45
Acknowledgements
I would like to express my warmest gratitude to all who have supported me during all these years and made this thesis possible.
Family, friends, professors, collaborators, colleagues, administrative and supportive personnel, you have all been essential to this work and it would not have been possible without anyone of you. I would in particular like to thank:
Professor Marion Kusche-Gullberg, my principal supervisor for excellent supervision, for sharing your great and profound scientific knowledge, for always having time for my questions, and your patience, support and encouragement during all these years. It has been a pleasure working in your lab and thank you for inspiration and all the good advices not just on work, but also on private life.
Professor Ulf Lindahl, my co-supervisor for inspiration and support, a very nice laboratory to work in, many good advices, for taking time going through my thesis on your “babysitting vacation” and for being a role model for young scientist like me.
Professor Lena Kjellén, my examiner for your kindness, helpfulness, support, guidance and for always keeping a good eye on me in the absence of Marion.
The Emerson Lab at the Boston Biomedical Research Institute, especially Dr. Xingbin Ai and Dr. Charles Emerson, for support, encouragement, for introducing me to the world of the Sulfs and taking care of me during my Boston trips. I am very much looking forward to join you.
Dr. Ulrich Technau, Dr. Fabian Rentzsch and the rest of the “guys” in the Technau Lab at the Sars International Centre for Marine Moleular Biology in Bergen, for teaching me molecular biology, good times and discussions from David Hasselhoff to Nematostella.

46
My opponent, Dr. Malcolm Lyon and members of the committee, for taking time going through this thesis and coming to Sweden.
All other collaborators that have been essential for the work in this thesis.
Colleagues in the Heparin Group: (Present members) Dorothe, for valuable advices and discussions; Elina (Pinglan), for friendship, support, encouragement, kindness and the fruitful discussions about life; Eva G. (Gumman), for great laughters, friendship and for always being helpful and open for funny discussions; Eva H, for sharing your knowledge in cell work, all the Christmas cards, “knäckebröd” and vegetables; Gunilla, Helena, for helping me so many times; Jin-Ping for encouragement and valuable discussions; Juan, for being a good roommate and helping me to change buffers so many times; Lena, Nadja, Rashmi, Sabrina (“Madame”), for kindness, encouragement, support and chats; (Past members) Anna, Bob, Camilla, Elisabet, Emanuel, for great collaborations, technical assistance and lots of fruitful discussions; Feng, Johan, Per, Sindhu, for support and encouragement; Maarten for fun, friendship and support. Dude, it would not have been the same without you; Maria W, Marta, for sharing the evening shifts with me and always being supportive and kind; Martha, for being a great roommate, for friendship, lessons of life, great times, support and encouragement; Shuhei for support, encouragement and kindness; Svante, Svetlana, and Åsa, for kindness, encouragement and sharing your experience and knowledge with me.
All past and present colleagues in the B9:3 corridor: Aneta, Annica, Carolina (Gullet), for always being the smiling girl with “glimten i ögat”, for being a friend and your kindness; Chi, for being a cool guy; Erik, Göte, Kristina & Claes for always being helpful with the microbiology teaching;Krzysztof, Lars, Maria J, for great times in IPhAB, kindness and support; Rebecka (Babe’en), for being the best running partner ever in BMC, friendship, kindness, and for the discussions about girls; Sophia, Wei, andall the students that came and went, for making the B9:3 corridor such a nice place to work in.
The colleagues at the University of Bergen: Almir for friendship and showing me where to drink the best coffee in Bergen; Angel for “middag” company and football games; Bert-Inge, Cecilia for friendship, kindness, encouragement, fun in the lab and lunch company; Donald, Malgorzata, Mona for all the assistance in the lab; Ning, Sergio for friendship, company and for showing me the gym and the nightlife in Bergen; Sveta, Åsa for your friendship, kindness and guidance during the time in IPhAB; and Øyvind.My time in Bergen would have been terribly dull without you.

47
Present and past friends and colleagues in the D9:4 especially: Anders for; friendship, support, encouragement and trying to “help” me with….yeah you know what…; Anne, Birgitta, Fredrik, Inger, Jenny, for friendship, support and encouregament, letting me win once in a while on the badminton court and for sharing the thesis writing anxiety with me; Jimmy, for being a cool guy; Johan, for friendship, encouragement and endless discussions; Katarina, for friendship, kindness, and support; Staffan, Maria R., Maud, Mia, Pernilla, for friendship, kindness, support, encouragement and always caring; Stina, for friendship, kindness, support and teaching me so many things during the time in IPhAB; and Ulrika, for friendship, NOT always being very nice, support, great spirit, encouragement, salsa and lunch company once in a while. By the way… WHERE IS MY SALSA MUSIC?
The IMBIM administrative and technical staff: Barbro for encouragement and the wonderful GLIBS events; Erika, for always being helpful with a big smile; Ewa, Inga-lisa, Liisa, Kerstin, for support and introducing the world of Macintosh to me; Marianne, Maud, Olav, Piotr, Rehné, Ted, Tony, Ulla-Maria and Veronica for administrative support and technical assistance.
All other past and present fellow colleagues at IMBIM, my time in the department would not have been the same without your company during all these years.
The past and present friends in B9:4 corridor, for friendship, kindness support and encouragement, for sharing you time and letting me steal coffee and “fika” so many times during the thesis writing.
My dear friends outside the lab for being there and for always taking care of me: Anders & Frida, for friendship, support, encouragement and always welcoming me to Harlösa; Annika, Björn S., for always supporting me in everything I do, friendship, encouragement and many years of fun; Chris & Camilla, Christina, Elin, my little newly-wed “Morgonrufs”, you have always been an inspiration for how life should be lived; Erik, my travel mate, for friendship and for sticking by me through all these years; Janne,for friendship, support, and encouragement; Jenny & Henke, for you friendship, always taking care of me and arranging great dinners with good discussions; Joanna, one of me oldest friends and my “extra sister” for friendship, support and encouragement through all these years; Johan, John & Mette, my “extra family number 1” for friendship, support, encouragement, company and always having the door open for my uninvited visits; Jonas, “Mr Löfling” for friendship, support and encouragement from work to girls; Le, Martin, for friendship, kindness and always having time for dinners; Micke K., for friendship and support; Phoung, my “oldest

48
friend ever” for friendship, support and encouragement through all these years; Stefan & Hanna (+Ida), my “extra family number 2” for being superb friends, endless support and encouragement, all the great times in the gym and always being there for me through good and bad times; Sylwia, and all the friends that I have gotten to know during my time in Uppsala and during my travels.
Last but definitely not least, my dear family: Mum, Dad, Auntie, Anh, Paul, Annie, Hanna, Xiang, Tran, Minh and Håkan; for endless joy, support and love. The family’s bundle of Joy: Vivienne, Edvin, Amelie, Felix and Sebastian, for bringing so much more love, joy and happiness in my life. This thesis is as much mine as yours.

49
References
Ai, X., A.T. Do, O. Lozynska, M. Kusche-Gullberg, U. Lindahl, and C.P. Emerson, Jr. 2003. QSulf1 remodels the 6-O sulfation states of cell surface heparan sulfate proteoglycans to promote Wnt signaling. J. Cell. Biol. 162:341-51.
Aikawa, J.-i., K. Grobe, M. Tsujimoto, and J.D. Esko. 2001. Multiple Isozymes of Heparan Sulfate/Heparin GlcNAc N-Deacetylase/GlcN N-Sulfotransferase. Structure and activity of the fourth member, NDST4. J. Biol. Chem. 276:5876-5882.
Bernfield, M., M. Götte, P.W. Park, O. Reizes, M.L. Fitzgerald, J. Lincecum, and M. Zako. 1999. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 68:729-777.
Bernfield, M., R. Kokenyesi, M. Kato, M.T. Hinkes, J. Spring, R.L. Gallo, and E.J. Lose. 1992. Biology of the syndecans: a family of transmembrane heparan sulfate proteoglycans. Annu Rev Cell Biol.8:365-93.
Bezakova, G., and M.A. Ruegg. 2003. New insights into the roles of agrin. Nat Rev Mol Cell Biol. 4:295-308.
Bink, R.J., H. Habuchi, Z. Lele, E. Dolk, J. Joore, G.J. Rauch, R. Geisler, S.W. Wilson, J. den Hertog, K. Kimata, and D. Zivkovic. 2003. Heparan sulfate 6-o-sulfotransferase is essential for muscle development in zebrafish. J. Biol. Chem. 278:31118-27.
Bourin, M.C., and U. Lindahl. 1993. Glycosaminoglycans and the regulation of blood coagulation. Biochem J. 289 ( Pt 2):313-30.
Bulow, H.E., and O. Hobert. 2004. Differential sulfations and epimerization define heparan sulfate specificity in nervous system development. Neuron. 41:723-36.
Cadigan, K.M., and Y.I. Liu. 2006. Wnt signaling: complexity at the surface. J Cell Sci. 119:395-402.
Carrol SB, G.J., Weaterbee SD. 2001. From DNA to diversity: molecular genetics and the evolution of animal design. Malden, Mass; Blackwell Science.
Casu, B., and U. Lindahl. 2001. Structure and biological interactions of heparin and heparan sulfate. Adv Carbohydr Chem Biochem.57:159-206.
Chen, E., S.E. Stringer, M.A. Rusch, S.B. Selleck, and S.C. Ekker. 2005. A unique role for 6-O sulfation modification in zebrafish vascular development. Dev. Biol.

50
Chen, J.Y., P. Oliveri, F. Gao, S.Q. Dornbos, C.W. Li, D.J. Bottjer, and E.H. Davidson. 2002. Precambrian animal life: probable developmental and adult cnidarian forms from Southwest China. Dev Biol.248:182-96.
Chen, R.L., and A.D. Lander. 2001. Mechanisms underlying preferential assembly of heparan sulfate on glypican-1. J Biol Chem.276:7507-17.
Conrad, H. 1998. Heparin-Binding Proteins. Academic Press, San Diego.Coulier, F., P. Pontarotti, R. Roubin, H. Hartung, M. Goldfarb, and
D. Birnbaum. 1997. Of worms and men: an evolutionary perspective on the fibroblast growth factor (FGF) and FGF receptor families. J Mol Evol. 44:43-56.
Crawford, B.E., S.K. Olson, J.D. Esko, and M.A.S. Pinhal. 2001. Cloning, Golgi Localization, and Enzyme Activity of the Full-length Heparin/Heparan Sulfate-Glucuronic Acid C5-epimerase. J. Biol. Chem. 276:21538-21543.
Darling, J.A., A.R. Reitzel, P.M. Burton, M.E. Mazza, J.F. Ryan, J.C. Sullivan, and J.R. Finnerty. 2005. Rising starlet: the starlet sea anemone, Nematostella vectensis. Bioessays. 27:211-21.
Dennissen, M.A., G.J. Jenniskens, M. Pieffers, E.M. Versteeg, M. Petitou, J.H. Veerkamp, and T.H. van Kuppevelt. 2002. Large, tissue-regulated domain diversity of heparan sulfates demonstrated by phage display antibodies. J Biol Chem. 277:10982-6.
Dhoot, G.K., M.K. Gustafsson, X. Ai, W. Sun, D.M. Standiford, and C.P. Emerson, Jr. 2001. Regulation of Wnt signaling and embryo patterning by an extracellular sulfatase. Science. 293:1663-6.
Elimova, E., R. Kisilevsky, W.A. Szarek, and J.B. Ancsin. 2004. Amyloidogenesis recapitulated in cell culture: a peptide inhibitor provides direct evidence for the role of heparan sulfate and suggests a new treatment strategy. Faseb J. 18:1749-51.
Esko, J.D., and U. Lindahl. 2001. Molecular diversity of heparan sulfate. J. Clin. Invest. 108:169-173.
Esko, J.D., and S.B. Selleck. 2002. ORDER OUT OF CHAOS: Assembly of Ligand Binding Sites in Heparan Sulfate. Annu. Rev. Biochem.71:435-471.
Esko, J.D., and L. Zhang. 1996. Influence of core protein sequence on glycosaminoglycan assembly. Curr Opin Struct Biol. 6:663-70.
Fernandez-Busquets, X., and M.M. Burger. 2003. Circular proteoglycans from sponges: first members of the spongican family. Cell Mol Life Sci. 60:88-112.
Filmus, J., and S.B. Selleck. 2001. Glypicans: proteoglycans with a surprise. J Clin Invest. 108:497-501.
Flaumenhaft, R., D. Moscatelli, O. Saksela, and D.B. Rifkin. 1989. Role of extracellular matrix in the action of basic fibroblast growth factor: matrix as a source of growth factor for long-term stimulation of

51
plasminogen activator production and DNA synthesis. J Cell Physiol. 140:75-81.
Ford-Perriss, M., S.E. Guimond, U. Greferath, M. Kita, K. Grobe, H. Habuchi, K. Kimata, J.D. Esko, M. Murphy, and J.E. Turnbull. 2002. Variant heparan sulfates synthesized in developing mouse brain differentially regulate FGF signaling. Glycobiology. 12:721-7.
Forsberg, E., and L. Kjellén. 2001. Heparan sulfate: lessons from knockout mice. J. Clin. Invest. 108:175-180.
Fransson, L.Å. 2003. Glypicans. Int J Biochem Cell Biol. 35:125-9. Fransson, L.Å., M. Belting, F. Cheng, M. Jonsson, K. Mani, and
S. Sandgren. 2004. Novel aspects of glypican glycobiology. Cell Mol Life Sci. 61:1016-24.
Fransson, L.Å., M. Belting, M. Jonsson, K. Mani, J. Moses, and A. Oldberg. 2000. Biosynthesis of decorin and glypican. Matrix Biol. 19:367-76.
Freeman, M., and J.B. Gurdon. 2002. Regulatory principles of developmental signaling. Annu Rev Cell Dev Biol. 18:515-39.
Funderburgh, J.L. 2000. Keratan sulfate: structure, biosynthesis, and function. Glycobiology. 10:951-8.
Gordon, M.D., and R. Nusse. 2006. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J Biol Chem.281:22429-33.
Grobe, K., J. Ledin, M. Ringvall, K. Holmborn, E. Forsberg, J.D. Esko, and L. Kjellén. 2002. Heparan sulfate and development: differential roles of the N-acetylglucosamine N-deacetylase/N-sulfotransferase isozymes. Biochim. Biophys. Acta. 1573:209-15.
Habuchi, H., O. Habuchi, and K. Kimata. 1995. Purification and characterization of heparan sulfate 6-sulfotransferase from the culture medium of Chinese hamster ovary cells. J Biol Chem.270:4172-9.
Habuchi, H., M. Kobayashi, and K. Kimata. 1998. Molecular Characterization and Expression of Heparan-sulfate 6-Sulfotransferase. Complete cDNA cloning in human and partial cloning in chinese hamster ovary cells. J. Biol. Chem. 273:9208-9213.
Habuchi, H., G. Miyake, K. Nogami, A. Kuroiwa, Y. Matsuda, M. Kusche-Gullberg, O. Habuchi, M. Tanaka, and K. Kimata. 2003. Biosynthesis of heparan sulphate with diverse structures and functions: two alternatively spliced forms of human heparan sulphate 6-O-sulphotransferase-2 having different expression patterns and properties. Biochem. J. 371:131-42.
Habuchi, H., M. Tanaka, O. Habuchi, K. Yoshida, H. Suzuki, K. Ban, and K. Kimata. 2000. The Occurrence of Three Isoforms of Heparan Sulfate 6-O-Sulfotransferase Having Different Specificities for Hexuronic Acid Adjacent to the Targeted N-Sulfoglucosamine. J. Biol. Chem. 275:2859-2868.

52
Häcker, U., K. Nybakken, and N. Perrimon. 2005. Heparan sulphate proteoglycans: the sweet side of development. Nat Rev Mol Cell Biol. 6:530-41.
Halfter, W., S. Dong, B. Schurer, and G.J. Cole. 1998. Collagen XVIII is a basement membrane heparan sulfate proteoglycan. J Biol Chem.273:25404-12.
Henningsson, F., J. Ledin, C. Lunderius, M. Wilen, L. Hellman, and G. Pejler. 2002. Altered storage of proteases in mast cells from mice lacking heparin: a possible role for heparin in carboxypeptidase A processing. Biol Chem. 383:793-801.
Holmborn, K., J. Ledin, E. Smeds, I. Eriksson, M. Kusche-Gullberg, and L. Kjellén. 2004. Heparan sulfate synthesized by mouse embryonic stem cells deficient in NDST1 and NDST2 is 6-O-sulfated but contains no N-sulfate groups. J. Biol. Chem. 279:42355-8.
Hulett, M.D., C. Freeman, B.J. Hamdorf, R.T. Baker, M.J. Harris, and C.R. Parish. 1999. Cloning of mammalian heparanase, an important enzyme in tumor invasion and metastasis. Nat Med. 5:803-9.
Iozzo, R.V. 1998. Matrix proteoglycans: from molecular design to cellular function. Annu Rev Biochem. 67:609-52.
Iozzo, R.V. 2001. Heparan sulfate proteoglycans: intricate molecules with intriguing functions. J Clin Invest. 108:165-7.
Iozzo, R.V. 2005. Basement membrane proteoglycans: from cellar to ceiling. Nat Rev Mol Cell Biol. 6:646-56.
Irie, A., E.A. Yates, J.E. Turnbull, and C.E. Holt. 2002. Specific heparan sulfate structures involved in retinal axon targeting. Development.129:61-70.
Jenniskens, G.J., J.H. Veerkamp, and T.H. van Kuppevelt. 2006. Heparan sulfates in skeletal muscle development and physiology. J Cell Physiol. 206:283-94.
Jordan, R.E., G.M. Oosta, W.T. Gardner, and R.D. Rosenberg. 1980. The kinetics of hemostatic enzyme-antithrombin interactions in the presence of low molecular weight heparin. J Biol Chem. 255:10081-90.
Kamimura, K., M. Fujise, F. Villa, S. Izumi, H. Habuchi, K. Kimata, and H. Nakato. 2001. Drosophila heparan sulfate 6-O-sulfotransferase (dHS6ST) gene. Structure, expression, and function in the formation of the tracheal system. J. Biol. Chem. 276:17014-21.
Kim, C.W., O.A. Goldberger, R.L. Gallo, and M. Bernfield. 1994. Members of the syndecan family of heparan sulfate proteoglycans are expressed in distinct cell-, tissue-, and development-specific patterns. Mol Biol Cell. 5:797-805.
Kisilevsky, R. 2000. Review: amyloidogenesis-unquestioned answers and unanswered questions. J Struct Biol. 130:99-108.
Kjellén, L., and U. Lindahl. 1991. Proteoglycans: structures and interactions. Annu Rev Biochem. 60:443-75.

53
Knox, S., C. Merry, S. Stringer, J. Melrose, and J. Whitelock. 2002. Not all perlecans are created equal: interactions with fibroblast growth factor (FGF) 2 and FGF receptors. J Biol Chem. 277:14657-65.
Knox, S.M., and J.M. Whitelock. 2006. Perlecan: how does one molecule do so many things? Cell Mol Life Sci.
Kobayashi, M., H. Habuchi, O. Habuchi, M. Saito, and K. Kimata. 1996. Purification and Characterization of Heparan Sulfate 2-Sulfotransferase from Cultured Chinese Hamster Ovary Cells. J. Biol. Chem. 271:7645-7653.
Kokenyesi, R., and M. Bernfield. 1994. Core protein structure and sequence determine the site and presence of heparan sulfate and chondroitin sulfate on syndecan-1. J Biol Chem. 269:12304-9.
Kolset, S.O., and J.T. Gallagher. 1990. Proteoglycans in haemopoietic cells. Biochim Biophys Acta. 1032:191-211.
Kraus, Y., and U. Technau. 2006. Gastrulation in the sea anemone Nematostella vectensis occurs by invagination and immigration: an ultrastructural study. Dev Genes Evol. 216:119-32.
Kreuger, J., D. Spillmann, J.P. Li, and U. Lindahl. 2006. Interactions between heparan sulfate and proteins: the concept of specificity. J Cell Biol. 174:323-7.
Kusche-Gullberg, M., and L. Kjellén. 2003. Sulfotransferases in glycosaminoglycan biosynthesis. Curr Opin Struct Biol. 13:605-11.
Lai, J., J. Chien, J. Staub, R. Avula, E.L. Greene, T.A. Matthews, D.I. Smith, S.H. Kaufmann, L.R. Roberts, and V. Shridhar. 2003. Loss of HSulf-1 up-regulates heparin-binding growth factor signaling in cancer. J Biol Chem. 278:23107-17.
Lai, J.P., J. Chien, S.E. Strome, J. Staub, D.P. Montoya, E.L. Greene, D.I. Smith, L.R. Roberts, and V. Shridhar. 2004. HSulf-1 modulates HGF-mediated tumor cell invasion and signaling in head and neck squamous carcinoma. Oncogene. 23:1439-47.
Lamanna, W.C., R.J. Baldwin, M. Padva, I. Kalus, G. Ten Dam, T.H. van Kuppevelt, J.T. Gallagher, K. von Figura, T. Dierks, and C.L. Merry. 2006. Heparan Sulphate 6-O-endosulphatases - Discrete in vivo activities and functional cooperativity. Biochem J.
Lander, A.D. 1998. Proteoglycans: master regulators of molecular encounter? Matrix Biol. 17:465-72.
Lander, A.D., Q. Nie, and F.Y. Wan. 2002. Do morphogen gradients arise by diffusion? Dev Cell. 2:785-96.
Lander, A.D., and S.B. Selleck. 2000. The Elusive Functions of Proteoglycans: In Vivo Veritas. J. Cell Biol. 148:227-232.
Ledin, J., M. Ringvall, M. Thuveson, I. Eriksson, M. Wilen, M. Kusche-Gullberg, E. Forsberg, and L. Kjellén. 2006. Enzymatically active N-deacetylase/N-sulfotransferase-2 is present in liver but does not contribute to heparan sulfate N-sulfation. J Biol Chem.

54
Ledin, J., W. Staatz, J.P. Li, M. Götte, S. Selleck, L. Kjellén, and D. Spillmann. 2004. Heparan sulfate structure in mice with genetically modified heparan sulfate production. J. Biol. Chem. 279:42732-41.
Li, J., J. Kleeff, I. Abiatari, H. Kayed, N.A. Giese, K. Felix, T. Giese, M.W. Buchler, and H. Friess. 2005. Enhanced levels of Hsulf-1 interfere with heparin-binding growth factor signaling in pancreatic cancer. Mol Cancer. 4:14.
Li, J.-P., F. Gong, K. El Darwish, M. Jalkanen, and U. Lindahl. 2001. Characterization of the D-Glucuronyl C5-epimerase Involved in the Biosynthesis of Heparin and Heparan Sulfate. J. Biol. Chem. 276:20069-20077.
Li, J.P., F. Gong, Å. Hagner-McWhirter, E. Forsberg, M. Åbrink, R. Kisilevsky, X. Zhang, and U. Lindahl. 2003. Targeted disruption of a murine glucuronyl C5-epimerase gene results in heparan sulfate lacking L-iduronic acid and in neonatal lethality. J. Biol. Chem.278:28363-6.
Lidholt, K., L. Kjellén, and U. Lindahl. 1989. Biosynthesis of heparin. Relationship between the polymerization and sulphation processes. Biochem J. 261:999-1007.
Lidholt, K., J.L. Weinke, C.S. Kiser, F.N. Lugemwa, K.J. Bame, S. Cheifetz, J. Massague, U. Lindahl, and J.D. Esko. 1992. A single mutation affects both N-acetylglucosaminyltransferase and glucuronosyl transferase activities in a Chinese hamster ovary cell mutant defective in heparan sulfate biosynthesis. Proc Natl Acad Sci U S A.89:2267-71.
Lin, X. 2004. Functions of heparan sulfate proteoglycans in cell signaling during development. Development. 131:6009-21.
Lin, X., E.M. Buff, N. Perrimon, and A.M. Michelson. 1999. Heparan sulfate proteoglycans are essential for FGF receptor signaling during Drosophila embryonic development. Development. 126:3715-23.
Lin, X., G. Wei, Z. Shi, L. Dryer, J.D. Esko, D.E. Wells, and M.M. Matzuk. 2000. Disruption of gastrulation and heparan sulfate biosynthesis in EXT1- deficient mice. Dev Biol. 224:299-311.
Lind, T., F. Tufaro, C. McCormick, U. Lindahl, and K. Lidholt. 1998. The Putative Tumor Suppressors EXT1 and EXT2 Are Glycosyl transferases Required for the Biosynthesis of Heparan Sulfate. J. Biol. Chem. 273:26265-26268.
Lindahl, U., M. Kusche-Gullberg, and L. Kjellén. 1998. Regulated Diversity of Heparan Sulfate. J. Biol. Chem. 273:24979-24982.
Lindahl, U., K. Lidholt, D. Spillmann, and L. Kjellén. 1994. More to "heparin" than anticoagulation. Thromb Res. 75:1-32.
Lyon, M., W.P. Steward, I.N. Hampson, and J.T. Gallagher. 1987. Identification of an extended N-acetylated sequence adjacent to the protein-linkage region of fibroblast heparan sulphate. Biochem J. 242:493-8.

55
Maccarana, M., Y. Sakura, A. Tawada, K. Yoshida, and U. Lindahl. 1996. Domain Structure of Heparan Sulfates from Bovine Organs. J. Biol. Chem. 271:17804-17810.
McCormick, C., G. Duncan, K.T. Goutsos, and F. Tufaro. 2000. The putative tumor suppressors EXT1 and EXT2 form a stable complex that accumulates in the Golgi apparatus and catalyzes the synthesis of heparan sulfate. Proc Natl Acad Sci U S A. 97:668-73.
Medeiros, G.F., A. Mendes, R.A. Castro, E.C. Bau, H.B. Nader, and C.P. Dietrich. 2000. Distribution of sulfated glycosaminoglycans in the animal kingdom: widespread occurrence of heparin-like compounds in invertebrates. Biochim Biophys Acta. 1475:287-94.
Merry, C.L.R., S.L. Bullock, D.C. Swan, A.C. Backen, M. Lyon, R.S.P. Beddington, V.A. Wilson, and J.T. Gallagher. 2001. The Molecular Phenotype of Heparan Sulfate in the Hs2st-/- Mutant Mouse. J. Biol. Chem. 276:35429-35434.
Miller, D.J., E.E. Ball, and U. Technau. 2005. Cnidarians and ancestral genetic complexity in the animal kingdom. Trends Genet. 21:536-9.
Morimoto-Tomita, M., K. Uchimura, Z. Werb, S. Hemmerich, and S.D. Rosen. 2002. Cloning and characterization of two extracellular heparin-degrading endosulfatases in mice and humans. J. Biol. Chem. 277:49175-85.
Nader, H.B., T.M. Ferreira, J.F. Paiva, M.G. Medeiros, S.M. Jeronimo, V.M. Paiva, and C.P. Dietrich. 1984. Isolation and structural studies of heparan sulfates and chondroitin sulfates from three species of molluscs. J Biol Chem. 259:1431-5.
Nagai, N., H. Habuchi, J.D. Esko, and K. Kimata. 2004. Stem domains of heparan sulfate 6-O-sulfotransferase are required for Golgi localization, oligomer formation and enzyme activity. J Cell Sci.117:3331-41.
Nogami, K., H. Suzuki, H. Habuchi, N. Ishiguro, H. Iwata, and K. Kimata. 2004. Distinctive expression patterns of heparan sulfate O-sulfotransferases and regional differences in heparan sulfate structure in chick limb buds. J. Biol. Chem. 279:8219-29.
Nybakken, K., and N. Perrimon. 2002. Heparan sulfate proteoglycan modulation of developmental signaling in Drosophila. Biochim Biophys Acta. 1573:280-91.
O'Reilly, M.S., T. Boehm, Y. Shing, N. Fukai, G. Vasios, W.S. Lane, E. Flynn, J.R. Birkhead, B.R. Olsen, and J. Folkman. 1997. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell.88:277-85.
Ohto, T., H. Uchida, H. Yamazaki, K. Keino-Masu, A. Matsui, and M. Masu. 2002. Identification of a novel nonlysosomal sulphatase expressed in the floor plate, choroid plexus and cartilage. Genes Cells. 7:173-85.

56
Ornitz, D.M. 2000. FGFs, heparan sulfate and FGFRs: complex interactions essential for development. Bioessays. 22:108-12.
Ornitz, D.M., and N. Itoh. 2001. Fibroblast growth factors. Genome Biol.2:REVIEWS3005.
Pinhal, M.A.S., B. Smith, S. Olson, J.-i. Aikawa, K. Kimata, and J.D. Esko. 2001. Enzyme interactions in heparan sulfate biosynthesis: Uronosyl 5-epimerase and 2-O-sulfotransferase interact in vivo. PNAS.98:12984-12989.
Pufe, T., W.J. Petersen, N. Miosge, M.B. Goldring, R. Mentlein, D.J. Varoga, and B.N. Tillmann. 2004. Endostatin/collagen XVIII--an inhibitor of angiogenesisis expressed in cartilage and fibrocartilage. Matrix Biol. 23:267-76.
Rademacher, T.W., R.B. Parekh, and R.A. Dwek. 1988. Glycobiology. Annu Rev Biochem. 57:785-838.
Rehn, M., and T. Pihlajaniemi. 1995. Identification of three N-terminal ends of type XVIII collagen chains and tissue-specific differences in the expression of the corresponding transcripts. The longest form contains a novel motif homologous to rat and Drosophila frizzled proteins. J Biol Chem. 270:4705-11.
Reya, T., and H. Clevers. 2005. Wnt signalling in stem cells and cancer. Nature. 434:843-50.
Rong, J., H. Habuchi, K. Kimata, U. Lindahl, and M. Kusche-Gullberg. 2001. Substrate specificity of the heparan sulfate hexuronic acid 2-O-sulfotransferase. Biochemistry. 40:5548-55.
Rostand, K.S., and J.D. Esko. 1997. Microbial adherence to and invasion through proteoglycans. Infect Immun. 65:1-8.
Saksela, O., D. Moscatelli, A. Sommer, and D.B. Rifkin. 1988. Endothelial cell-derived heparan sulfate binds basic fibroblast growth factor and protects it from proteolytic degradation. J Cell Biol. 107:743-51.
Salmivirta, M., and M. Jalkanen. 1995. Syndecan family of cell surface proteoglycans: developmentally regulated receptors for extracellular effector molecules. Experientia. 51:863-72.
Salmivirta, M., K. Lidholt, and U. Lindahl. 1996. Heparan sulfate: a piece of information. Faseb J. 10:1270-9.
Sanderson, R.D., J.E. Turnbull, J.T. Gallagher, and A.D. Lander. 1994. Fine structure of heparan sulfate regulates syndecan-1 function and cell behavior. J Biol Chem. 269:13100-6.
Sasisekharan, R., and G. Venkataraman. 2000. Heparin and heparan sulfate: biosynthesis, structure and function. Curr Opin Chem Biol. 4:626-31.
Schambony, A., M. Kunz, and D. Gradl. 2004. Cross-regulation of Wnt signaling and cell adhesion. Differentiation. 72:307-18.
Sedita, J., K. Izvolsky, and W.V. Cardoso. 2004. Differential expression of heparan sulfate 6-O-sulfotransferase isoforms in the mouse embryo

57
suggests distinctive roles during organogenesis. Dev. Dyn. 231:782-94.
Senay, C., T. Lind, K. Muguruma, Y. Tone, H. Kitagawa, K. Sugahara, K. Lidholt, U. Lindahl, and M. Kusche-Gullberg. 2000. The EXT1/EXT2 tumor suppressors: catalytic activities and role in heparan sulfate biosynthesis. EMBO Rep. 1:282-6.
Shworak, N.W., S. HajMohammadi, A.I. de Agostini, and R.D. Rosenberg. 2002. Mice deficient in heparan sulfate 3-O-sulfotransferase-1: normal hemostasis with unexpected perinatal phenotypes. Glycoconj J. 19:355-61.
Shworak, N.W., J. Liu, L.M. Petros, L. Zhang, M. Kobayashi, N.G. Copeland, N.A. Jenkins, and R.D. Rosenberg. 1999. Multiple Isoforms of Heparan Sulfate D-Glucosaminyl 3-O-Sulfotransferase. Isolation, characterization, and expression of human cDNAs and identification of distinct genomic loci. J. Biol. Chem. 274:5170-5184.
Sleeman, M., J. Fraser, M. McDonald, S. Yuan, D. White, P. Grandison, K. Kumble, J.D. Watson, and J.G. Murison. 2001. Identification of a new fibroblast growth factor receptor, FGFR5. Gene. 271:171-82.
Stickens, D., B.M. Zak, N. Rougier, J.D. Esko, and Z. Werb. 2005. Mice deficient in Ext2 lack heparan sulfate and develop exostoses. Development. 132:5055-68.
Takagi, Y., S. Shrivastav, T. Miki, and K. Sakaguchi. 1994. Molecular cloning and expression of the acidic fibroblast growth factor receptors in a rat parathyroid cell line (PT-r). Parathyroid cell-specific calcium-dependent change of ligand accessibility and covalent attachment of heparan sulfate glycosaminoglycan to the receptors. J Biol Chem. 269:23743-9.
Technau, U., and C.B. Scholz. 2003. Origin and evolution of endoderm and mesoderm. Int J Dev Biol. 47:531-9.
Temussi, P.A., L. Masino, and A. Pastore. 2003. From Alzheimer to Hunt-ington: why is a structural understanding so difficult? Embo J.22:355-61.
Thisse, B., and C. Thisse. 2005. Functions and regulations of fibroblast growth factor signaling during embryonic development. Dev Biol.287:390-402.
Thompson, L.D., M.W. Pantoliano, and B.A. Springer. 1994. Energetic characterization of the basic fibroblast growth factor-heparin interaction: identification of the heparin binding domain. Biochemistry. 33:3831-40.
Toyoda, H., A. Kinoshita-Toyoda, and S.B. Selleck. 2000. Structural analysis of glycosaminoglycans in Drosophila and Caenorhabditis elegans and demonstration that tout-velu, a Drosophila gene related to EXT tumor suppressors, affects heparan sulfate in vivo. J Biol Chem. 275:2269-75.

58
Turnbull, J., K. Drummond, Z. Huang, T. Kinnunen, M. Ford-Perriss, M. Murphy, and S. Guimond. 2003. Heparan sulphate sulphotransferase expression in mice and Caenorhabditis elegans. Biochem. Soc. Trans. 31:343-8.
Uchimura, K., M. Morimoto-Tomita, A. Bistrup, J. Li, M. Lyon, J. Gallagher, Z. Werb, and S.D. Rosen. 2006. HSulf-2, an extracellular endoglucosamine-6-sulfatase, selectively mobilizes heparin-bound growth factors and chemokines: effects on VEGF, FGF-1, and SDF-1. BMC Biochem. 7:2.
van Horssen, J., P. Wesseling, L.P. van den Heuvel, R.M. de Waal, and M.M. Verbeek. 2003. Heparan sulphate proteoglycans in Alzheimer's disease and amyloid-related disorders. Lancet Neurol.2:482-92.
Varki, A., R. Cummings, J. Esko, E. Freeze, G. Hart, and J. Marth. 1999. Essential of Glycobiology. Cold spring Harbor Laboratory Press, New York. ISBN 0-87969-560-9.
Viviano, B.L., S. Paine-Saunders, N. Gasiunas, J. Gallagher, and S. Saunders. 2004. Domain-specific modification of heparan sulfate by Qsulf1 modulates the binding of the bone morphogenetic protein antagonist Noggin. J. Biol. Chem. 279:5604-11.
Vlodavsky, I., and Y. Friedmann. 2001. Molecular properties and involvement of heparanase in cancer metastasis and angiogenesis. J Clin Invest. 108:341-7.
Vogt, A.M., A. Barragan, Q. Chen, F. Kironde, D. Spillmann, and M. Wahlgren. 2003. Heparan sulfate on endothelial cells mediates the binding of Plasmodium falciparum-infected erythrocytes via the DBL1alpha domain of PfEMP1. Blood. 101:2405-11.
Wang, S., X. Ai, S.D. Freeman, M.E. Pownall, Q. Lu, D.S. Kessler, and C.P. Emerson, Jr. 2004. QSulf1, a heparan sulfate 6-O-endosulfatase, inhibits fibroblast growth factor signaling in mesoderm induction and angiogenesis. Proc Natl Acad Sci U S A. 101:4833-8.
Westling, C., and U. Lindahl. 2002. Location of N-unsubstituted glucosamine residues in heparan sulfate. J Biol Chem. 277:49247-55.
Whitelock, J.M., and R.V. Iozzo. 2005. Heparan sulfate: a complex polymer charged with biological activity. Chem Rev. 105:2745-64.
Xia, G., J. Chen, V. Tiwari, W. Ju, J.P. Li, A. Malmström, D. Shukla, and J. Liu. 2002. Heparan sulfate 3-O-sulfotransferase isoform 5 generates both an antithrombin-binding site and an entry receptor for herpes simplex virus, type 1. J. Biol. Chem. 277:37912-9.
Yamada, S., I. Van Die, D.H. Van den Eijnden, A. Yokota, H. Kitagawa, and K. Sugahara. 1999. Demonstration of glycosaminoglycans in Caenorhabditis elegans. FEBS Lett. 459:327-31.

59
Yoneda, A., M. Asada, Y. Oda, M. Suzuki, and T. Imamura. 2000. Engineering of an FGF-proteoglycan fusion protein with heparin-independent, mitogenic activity. Nat Biotechnol. 18:641-4.
Yusa, A., K. Kitajima, and O. Habuchi. 2006. N-linked oligosaccharides on chondroitin 6-sulfotransferase-1 are required for production of the active enzyme, Golgi localization, and sulfotransferase activity toward keratan sulfate. J Biol Chem. 281:20393-403.
Zhang, L., D.L. Beeler, R. Lawrence, M. Lech, J. Liu, J.C. Davis, Z. Shriver, R. Sasisekharan, and R.D. Rosenberg. 2001. 6-O-Sulfotransferase-1 Represents a Critical Enzyme in the Anticoagulant Heparan Sulfate Biosynthetic Pathway. J. Biol. Chem. 276:42311-42321.
Zhu, X., H. Komiya, A. Chirino, S. Faham, G.M. Fox, T. Arakawa, B.T. Hsu, and D.C. Rees. 1991. Three-dimensional structures of acidic and basic fibroblast growth factors. Science. 251:90-3.

Acta Universitatis UpsaliensisDigital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Medicine 185
Editor: The Dean of the Faculty of Medicine
A doctoral dissertation from the Faculty of Medicine, UppsalaUniversity, is usually a summary of a number of papers. A fewcopies of the complete dissertation are kept at major Swedishresearch libraries, while the summary alone is distributedinternationally through the series Digital ComprehensiveSummaries of Uppsala Dissertations from the Faculty ofMedicine. (Prior to January, 2005, the series was publishedunder the title “Comprehensive Summaries of UppsalaDissertations from the Faculty of Medicine”.)
Distribution: publications.uu.seurn:nbn:se:uu:diva-7199
ACTAUNIVERSITATISUPSALIENSISUPPSALA2006