regulation of software a medical device - vub techtransfervubtechtransfer.be/medialibrary/ws1_steve...
TRANSCRIPT

IOF-TTO Event
17.10.2017
Regulation of software a medical device
Steve EglemResponsible of clinical investigation @ famhp
Member of the medical device European Clinical and Evaluation (CIE) working group

2
• Famhp presentation
• European and Belgian legal framework on medical devices
• Legal reference
• Definitions & Examples
• Classification
• Software as medical device
• Validation process and conformity assessment
• Impact of MDR
• Impact of GDPR from medical device perspective
Content
Subject - DateFAMHP/entity/Division-Unit-Cell

3
FAMHP - AFMPS – FAGG
http://www.fagg-afmps.be
• Address : Victor Horta Square 40/40
In front of the Midi Station
• More than 400 employees
• Minister of Public Health and Social
Affairs : Maggie De Block
• CEO : Xavier De Cuyper
Who are we?

4
National competent autority in Belgium responsible for the quality, safety and efficacy of medicines and health products.
The famhp ensures, from development to use, the quality, safety and efficacy of medicines for human and veterinary use (including homeopathic medicines, herbal medicines, pharmacy made and officinal preparations) and also medical devices and accessories, and raw materials for the preparation and production of medicines.From collection to use, the fahmp ensures the quality, safety and efficacy of all the operations involving with blood, cells and tissues.
Our stakeholders :
FAMHP
5
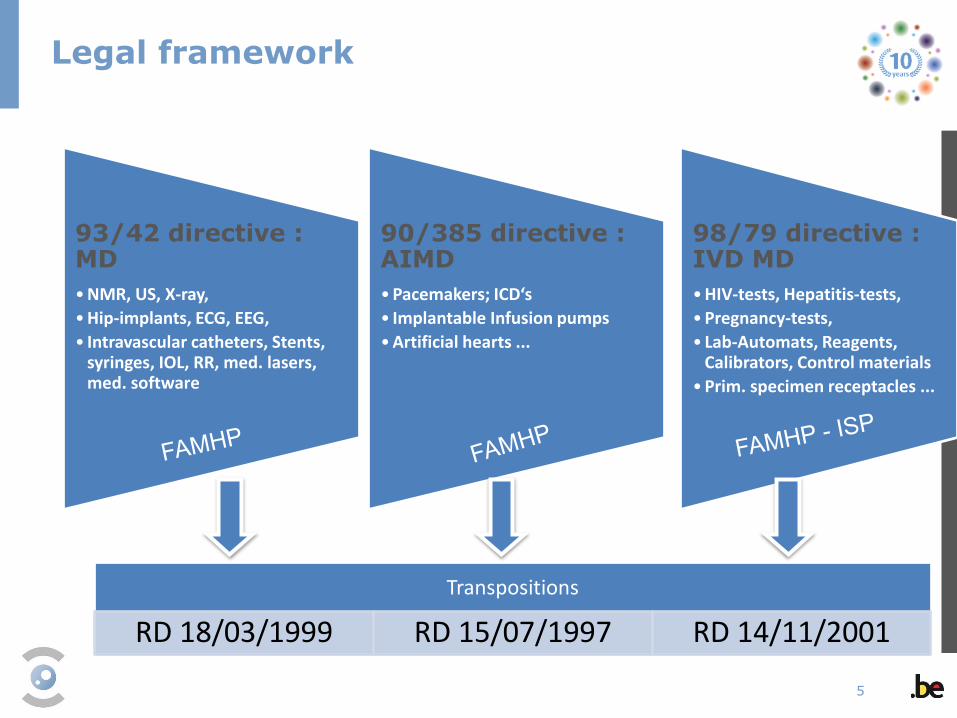
93/42 directive : MD
•NMR, US, X-ray,
•Hip-implants, ECG, EEG,
• Intravascular catheters, Stents, syringes, IOL, RR, med. lasers, med. software
90/385 directive : AIMD
•Pacemakers; ICD‘s
• Implantable Infusion pumps
•Artificial hearts ...
98/79 directive : IVD MD
•HIV-tests, Hepatitis-tests,
•Pregnancy-tests,
• Lab-Automats, Reagents, Calibrators, Control materials
•Prim. specimen receptacles ...
Transpositions
RD 18/03/1999 RD 15/07/1997 RD 14/11/2001
Legal framework

6
93/42 directive : MD
•NMR, US, X-ray,
•Hip-implants, ECG, EEG,
• Intravascular catheters, Stents, syringes, IOL, RR, med. lasers, med. software
90/385 directive : AIMD
•Pacemakers; ICD‘s
• Implantable Infusion pumps
•Artificial hearts ...
98/79 directive : IVD MD
•HIV-tests, Hepatitis-tests,
•Pregnancy-tests,
• Lab-Automats, Reagents, Calibrators, Control materials
•Prim. specimen receptacles ...
Regulation 2017/745 Regulation 2017/746
Legal framework
Future

7
‘medical device’ means any instrument, apparatus, appliance, software, material or
other article, whether used alone or in combination, including the software intended
by its manufacturer to be used specifically for diagnostic and/or therapeutic purposes
and necessary for its proper application, intended by the manufacturer
to be used for human beings for the purpose of:
• diagnosis, prevention, monitoring, treatment or alleviation of disease,
• diagnosis, monitoring, treatment, alleviation of or compensation for an injury or
handicap, investigation,
• replacement or modification of the anatomy or of a physiological process,
• control of conception,
and which does not achieve its principal intended action in or on the human body by
pharmacological, immunological or metabolic means, but which may be assisted in its
function by such means;
Legal framework
Definition

8
MD

9
'active medical device' means any medical device relying for its functioning on a
source of electrical energy or any source of power other than that directly
generated by the human body or gravity,
Stand alone software with a medical purpose is considered as an active
medical device
Legal framework –
Definition

10
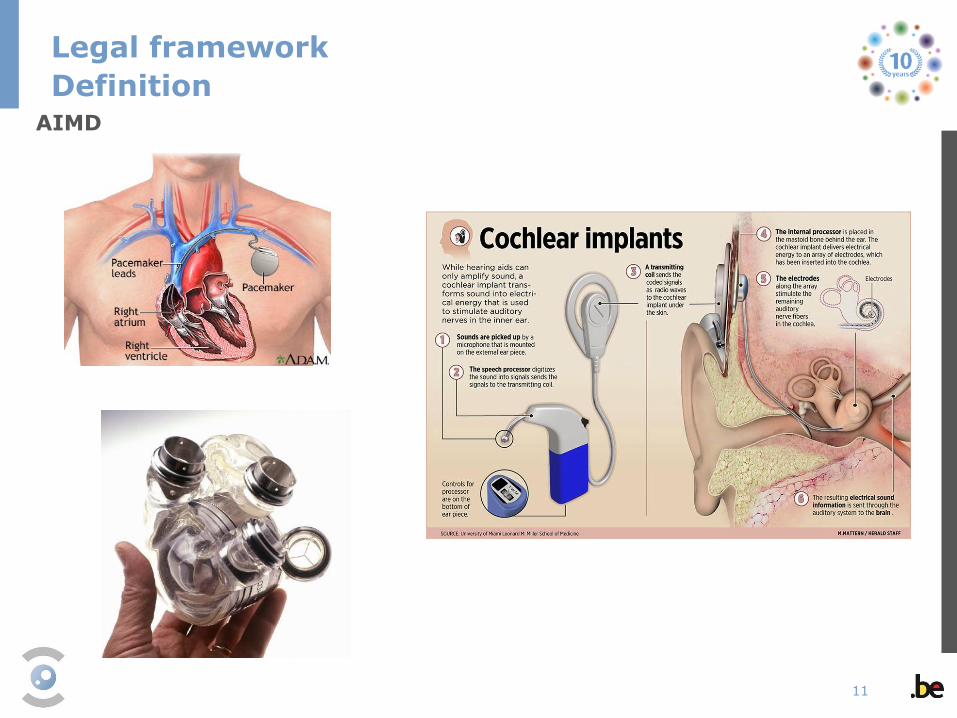
'active implantable medical device‘ (AIMD) means any active medical device
which is intended to be totally or partially introduced, surgically or medically, into
the human body or by medical intervention into a natural orifice , and which is
intended to remain after the procedure;
Legal framework –
Definition
11
AIMD
Legal framework
Definition

12
‘accessory’ means an article which whilst not being a device is
intended specifically by its manufacturer to be used together with
a device to enable it to be used in accordance with the use of the
device intended by the manufacturer of the device;
Legal framework
Definition

13
Example :
Accessory of ECG machine : pads, paper.
Legal framework
Definition
14
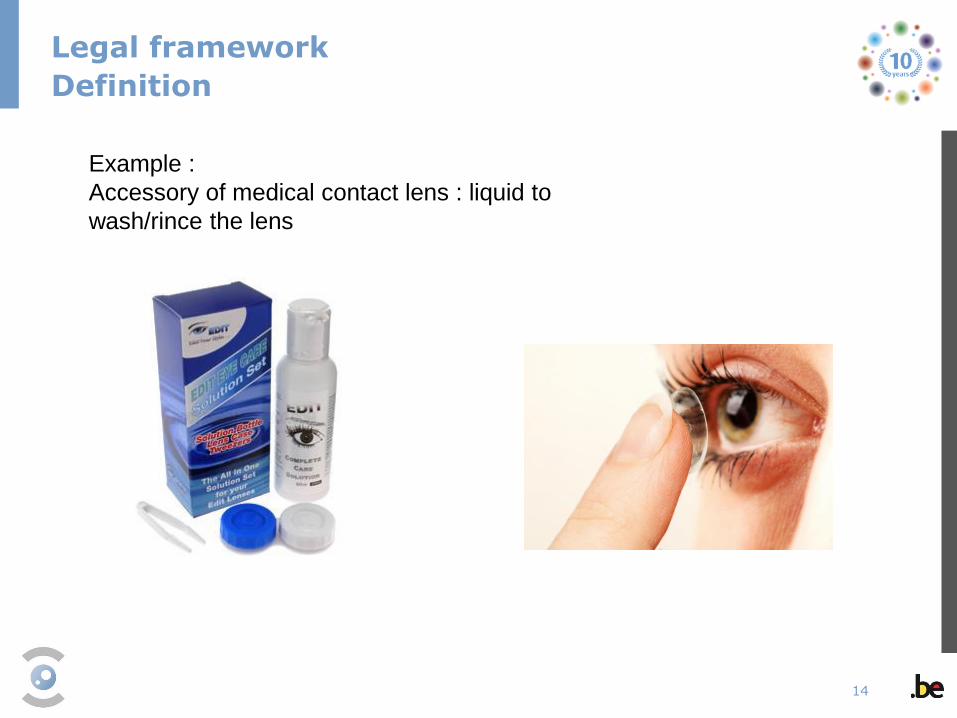
Example :
Accessory of medical contact lens : liquid to
wash/rince the lens
Legal framework
Definition

15
Example :
Accessory of medical endoscope : screen monitors, cables,…
Legal framework
Definition

16
Example :
Accessory of medical endoscope : disinfecting solution
Legal framework
Definition

17
‘in vitro diagnostic medical device’ (IVD) means any medical device which is
a reagent, reagent product, calibrator, control material,
kit, instrument, apparatus, equipment or system, whether used alone or in
combination, intended by the manufacturer to be used
in vitro for the examination of specimens, including blood and tissue donations,
derived from the human body, solely or principally for the purpose of providing
information:
— concerning a physiological or pathological state, or
— concerning a congenital abnormality, or
— to determine the safety and compatibility with potential recipients,
or
— to monitor therapeutic measures.
Legal framework
Definition
18
IVD
19
MD
20
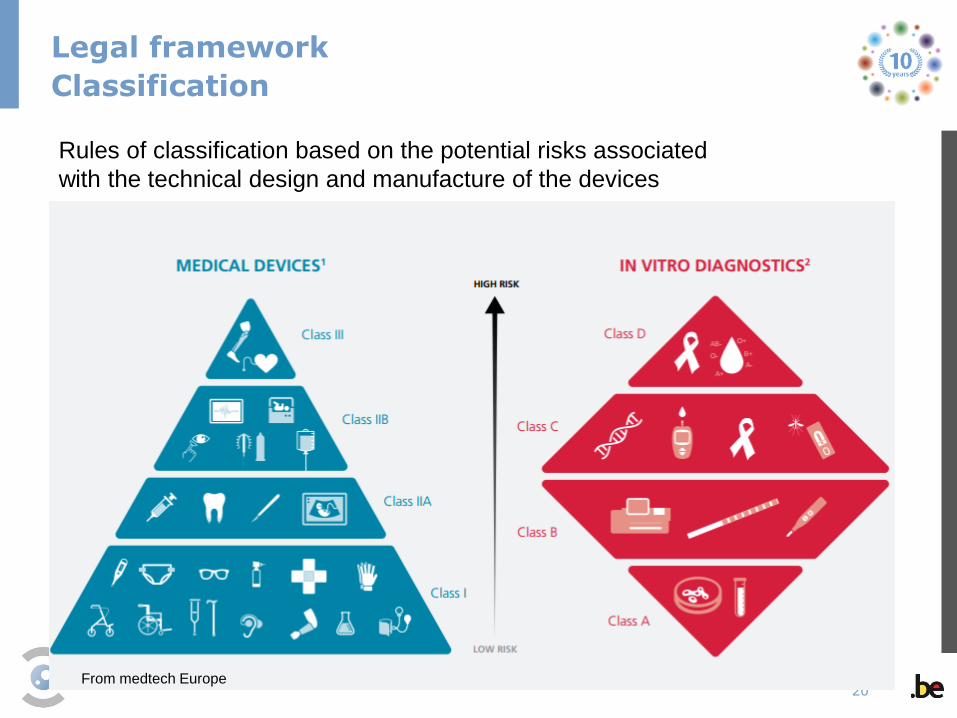
Legal framework
Classification
From medtech Europe
Rules of classification based on the potential risks associated
with the technical design and manufacture of the devices

21
Legal framework
Classification (from software perspective)
EU Directive MDR
No specific rules of classification (followactive medical devices)
Specific classification for software : • providing information which is used to
take decisions with diagnosis or therapeutic purposes (Class IIA/IIB/III)
• Intended to monitor physiological processes (Class IIB)
• Others (Class I)

22
Legal framework
Software as medical device
• Could fall under IVD or MDD directive• Medical purpose (claim!)• Software does not perform an action on data, or performs an action
limited to storage, archival, communication, "simple search" or lossless compression.
• And if my app falls under a medical device framework?• It must comply with the safety and performance requirements
• Other obligations for manufacturers regard the issues of registration, post-market surveillance, incident reporting.

23
Legal framework
Software as medical device

24
Legal framework
Software as medical device

25
• What does it mean?
• Demonstrate the safety and performance of the device.
• Performance : The ability of a medical device to achieve its intended purpose as
claimed by the manufacturer.
• Safety : The absence of unacceptable clinical risks, when using the device
according to the manufacturer’s Instructions for Use.
• Device fulfill essential requirements ?
• the information materials supplied by the manufacturer(the labelling, instructions for use, promotional materials, including accompanying documents foreseen by the manufacturer)
• the clinical evaluation (the device description used for the clinical evaluation, other contents of the clinical evaluation report)
• the available clinical data (such as results of Clinical Investigations, publications, PMS studies, etc.).
Validation process and conformityassessment

26
Validation process and conformityassessment
Preclinical Data
•Scientific litterature
Similar product?
Check harmonized standards, what is needed?
Biocompatibility tested
Fatigue and resistance test
…
Clinical Data
•Scientific litterature
•Clinical Investigation
Sufficient clinical evidence?
•Performance (claim!)
•safety
Essential requirements
•Demonstration of conformity is now fulfilled?
Clinical Evaluation :
• From the beginning (development of the device)
• Mandatory for CE-marking
• Updated throughout all the life cycle of the device (pre and post market)
• Guide the clinical research (with the risk management; iso 14971
Build your risk analysisduring each step…

27
Validation process and conformityassessment
Essential requirements (ER) in the current directive :Annex I – point 12. Requirements for medical devices connected to or equipped with an energy source :• repeatability, reliability and performance of these systems according to the
intended use• validated according to the state of the art taking into account the principles of
development lifecycle, risk management, validation and verification.• Monitor clinical parameters : alarm should be implemented• …• Cybersecurity not included in the current directive…
Standards for software which could provide more information on the ER that you need to fulfill and how to do it:
• IEC 62304 : medical software – software life-cycle processes• NBN 62366 : Medical devices - Application of usability engineering to medical
devices• NBN 82304 : Health Software - General requirements for product safety

28
• Conformity procedure?
• Different routes following the MD classification…
• Such compliance must be assessed through a specific conformity
assessment procedure, which is proportional to the risk-class:
• For low-risk products (Class I), the manufacturer can provide a self-declaration
• For all other products (Classes IIa, IIb and III), a control must be done by a Notified Body
Validation process and conformityassessment

29
• Conformity procedure?
Validation process and conformityassessment

30
Impact of MDR

31
The new Regulations contain a series of extremely important improvements to modernise the current system. Among them are:
• stricter control for high-risk devices
• the reinforcement of the criteria for designation and processes for oversight of Notified Bodies
• the inclusion of certain aesthetic devices which present the same characteristics and risk profile as analogous medical devices under the scope of these Regulations (annex XVI)
• the introduction of a new risk classification system for in vitro diagnostic medical devices in line with international guidance
• improved transparency through an European database
Impact of MDR/IVDR

32
• the reinforcement of the rules on clinical evidence, including an EU-wide coordinated procedure for authorisation of multi-centre clinical investigations
• the strengthening of post-market surveillance requirements for manufacturers
• improved coordination mechanisms between EU countries in the fields of vigilance and market surveillance
• Existing products will have to be reassessed under the new rules at latest may 2024
• New definitions
• Introduction of Common Specifications (CS): a set of technical and/or clinical requirements other than a standard, providing a means to comply with the legal obligations applicable to a device, process or system
Impact of MDR/IVDR

33
• Article 110 of MDR: ”Member States shall apply Directive95/46/EC to the processing of personal data carried out in the Member States pursuant to this Regulation”
• Directive 95/46/EC is replaced as from 25 May 2018 by Regulation 2016/679 (EU) of 27 April 2016 on the protection of natural persons with regard to the processing of personal data («GDPR»)
• Health-related data not defined in current Directive
• GDPR: “data concerning health” = Personal Data related to
• Physical or mental health
• Of a (identified or identifiable) natural person
• Including the provision of health care services
• Which reveals information about his/her health status
• Large definition
Impact on clinical investigation to collect clinical data…
Advice from ethic committee
Impact of GDPR
33

34
Take home message…
Your claim, purpose, objective of yourapp
Apply the regulatory route
Possible medical device
Check it with EU guidance, ask to a competent autority…
Plan your clinical evaluation taking intoaccount the new regulation from now!

35
Contact
Federal Agency for Medicines and Health Products –FAMHP
Place Victor Horta 40/40
1060 BRUXELLES
tel. + 32 2 528 40 00
fax + 32 2 528 40 01
e-mail [email protected]
www.afmps.be

36
Tools

37
Tools

38
Tools

39
Tools

Your medicines and health products,our concern