restriction-modification genes of salmonella infantis
TRANSCRIPT

JOURNAL OF BACTERIOLOGY, June 1988, p. 2527-2532 Vol. 170, No. 60021-9193/88/062527-06$02.00/0Copyright X 1988, American Society for Microbiology
Cloning and Complete Nucleotide Sequences of the Type IIRestriction-Modification Genes of Salmonella infantis
CHRISTIAAN KARREMAN* AND ADRIAN DE WAARD
Department of Medical Biochemistry, Sylvius Laboratories, University of Leiden, P.O. Box 9503, 2300 RA Leiden,The Netherlands
Received 17 November 1987/Accepted 17 February 1988
The complete type II restriction-modification system of Salmonella infantis was cloned in Escherichia coli asan R - Sau3AI fragment of 3,430 base pairs. The clone was shown to express the restriction endonuclease aswell as the modification methylase. The nucleotide sequence of the above fragment showed two open readingframes of 461 and 230 codons in tail-to-tail orientation. These were shown to represent the modificationmethylase M - SinI and the restriction endonuclease R * SinI, respectively. The methylase M * SinI amino acidsequence revealed a considerable similarity to those of other deoxycytidylate methylases. In contrast,endonuclease R - SinI did not exhibit such a similarity to other restriction enzymes.
Restriction-modification systems arouse interest not onlyas powerful biochemical tools but also as a convenient modelof protein-DNA interaction. The specific recognition of onlya few bases in combination with the large number of evolu-tionarily distant isoschizomers could give information aboutthe exact requirements of this interaction. In 1981 Lupkerand Dekker (11) described a restriction endonuclease from astrain of Salmonella infantis, which was an isoschizomer ofR - Avall (G^G[A/T]CC). The large number ofSinI isoschizo-mers in the distant group of cyanobacteria (22) and theevolutionary closeness of S. infantis to Escherichia colimake the SinI system a perfect object for this type ofresearch.We cloned and sequenced the SinI restriction-modification
system and compared its predicted amino acid sequenceswith those of other type II systems.
MATERIALS AND METHODSStrains and media. E. coli MC1061 (3), which is an mcrB
mutant (17), was used in all cloning experiments with plas-mids containing the M - SinI gene. MC1061 was grown inbrain heart infusion (BHI) medium or in Luria-Bertani (LB)medium (Oxoid Ltd., Basinstoke, United Kingdom). Theantibiotic ampicillin was used at 70 ,ug/ml. A wild-type S.infantis strain isolated in our laboratory (11) was the sourceof chromosomal DNA; it was grown in BHI.Enzymes and chemicals. Restriction endonucleases were
purchased from New England Biolabs (Beverly, Mass.),Promega-Biotec (Leiden, The Netherlands), and Pharmacia(Uppsala, Sweden). Calf intestine phosphatase was fromBoehringer GmbH (Mannheim, Federal Republic of Ger-many). All were used according to the manufacturers' spec-ifications. Hydrazine (pure) was from Serva (Heidelberg,Federal Republic of Germany), and piperidine was from E.Merck AG (Darmstadt, Federal Republic of Germany).Oligonucleotides were synthesized on a Biosearch CycloneDNA synthesizer.
Construction of the colony bank. DNA from S. infantisisolated as described previously (10) was digested partiallywith R- Sau3AI (12), and DNAs from the various incuba-tions were pooled and fractionated by agarose gel electro-phoresis.
* Corresponding author.
Fragments ranging in size from 2 to 9 kilobases wereextracted from the gel (27) and ligated into plasmid pUC19(26) that had been cut with R * BamHI and dephosphory-lated with calf intestine phosphatase. A clone bank of 25,000colonies was constructed by transforming E. coli MC1061with the resulting recombinant plasmids. The pooled trans-formants were transferred from plates into 750 ml of BHImedium and grown for 6 h at 37°C before being harvested bycentrifugation. Plasmids were isolated as described previ-ously (12).
Selection. Since the product from a successfully clonedM- SinI gene should render the two SinI sites of pUC19resistant to cleavage by R * SinI, a 5-,ul sample of the pooledplasmid bank (representing approximately 2.5% of the totalyield) was incubated with 50 U of endonuclease R- SinI for8 h, after which the DNA was extracted with phenol andprecipitated with ethanol. The cleavage procedure was re-peated overnight. The resulting DNA was purified andintroduced into E. coli MC1061. The resulting transformantswere plated onto agar containing ampicillin and gave rise toapproximately 150 colonies.
Eleven randomly chosen colonies were further investi-gated, of which nine contained a plasmid that could nolonger be cut with R. SinI in vitro. To ensure that we hadnot isolated mutants lacking both SinI sites in pUC19 butgenuine clones containing the M * SinI gene, we removedthe total insert by excising a PvuII fragment (Fig. 1) from allnine recombinants. The resulting deletion clones were againsensitive to R. SinI, which proved that a reversible modifi-cation rather than some mutation had caused them to be-come R. SinI resistant. The R. SinI-resistant clone with thelargest insert, pSI4, was used in all subsequent experiments.DNA sequencing. The DNA sequence was determined
mainly by the chemical degradation method of Maxam andGilbert (14). Parts of the insert were sequenced as a dena-tured double-stranded plasmid with specific oligonucleotideswith the dideoxynucleotide protocol of Sanger et al. (18).
Isolation of endonuclease R SinI. E. coli MC1061 harbor-ing plasmid pSI4 was grown overnight in BHI and harvestedby centrifugation. Isolation of R- SinI from these cells wasperformed as described earlier (11).Primer extension. RNA was isolated from E. coli cells
containing plasmid pSI4 with the hot phenol method (24) andthen hybridized to terminally 32P-labeled synthetic oligonu-
2527

2528 KARREMAN AND DE WAARD
B.EPVUI C,l 1 ndM
pSl4 F
pAHH2
pAXX3
pHpoN&BH6
AdT48% 61% 660/o 5R
FIG. 1. Linear representation of the insertmids mentioned in the text. Plasmid pSI4 contafragment and consequently both the ORFs codand the methylation proteins (r+ m+). The insthin line, the DNA from the vector pUC19 islines flanking the insert. On both sides two rpolylinker of pUC19 are shown. For thedeletions are shown as well as the phenotypesthese plasmids. In plasmid pH--NABH6 an
introduced by destroying the old HindlIl sHindlIl site in the coding region of R. SinI bethis plasmid. The A+T percentages for the vinsert are given at the bottom of the figure.
cleotides that were complementary to pieither R. SinI or M SinI mRNA (Figwere extended by avian myeloblastosisscriptase (Pharmacia) as described previo
After the elongated primers were boamide-10 mM NaOH, they were loadepolyacrylamide gel next to sequence ladthe same oligonucleotides on plasmid pSIisolated from cells transformed by pUCreference.Computer programs. For the analysis o
used the software package (GCG) distribisity of Wisconsin (5). Protein files generithe GCG programs from published DNcompared on an Atari ST microcompuprogram was developed to score for simiwindow size and maximum number o
though scoring tables for comparing prothe GCG programs and by Schwarz andalso used, the best pictures, i.e., those v
noise ratio, were obtained by utilization o
acid sequences.
RESULTS
Cloning. Plasmids containing S. infantstructed and selected for R SinI resistaence of a fully functional M SinI gene. Sshowed that one of these plasmids, pSI4,R. SinI restriction enzyme, since cells tplasmid produced the SinI endonucleaszyme could be isolated from these cells bthat had been used to isolate it from itsinfantis (11). Ten grams of E. coli MC106pSI4 yielded about 500,000 U of exonucledetermined by cleavage assay on phage Inot shown).
Localization of genes. The sequences
plasmid pS14 were determined, revealing two nonoverlap-ping large open reading frames (ORFs) of 1,383 and 690
_-LfNuII r+m* nucleotides (nt) in opposite strands. The ORFs are posi-tioned tail to tail. To determine which of the ORFs in pSI4codes for either gene, deletion mutants were made (Fig. 1).
,,-W+ A plasmid (pAHH2) missing the small HindlIl fragment (nt2728 polylinker) still directed the synthesis of M * SinI, as
r-m- indicated by its resistance to R * SinI in vitro when isolated.Nhel This indicates that the large ORF is coding for M * SinI andt,,,,+ the smaller ORF is coding for R. SinI. Accordingly, an XbaI
deletion (nt 1402 polylinker) of pSI4 which had lost the 3'end of the large ORF (plasmid pAXX3), no longer gave rise
-4 to any detectable M SinI activity. To rule out the possibil-°O ity that the detected R. SinI activity was due to a vector-ts of the various plas- directed fusion protein and to prove at the same time that thetins the whole Sau3AI shorter ORF is indeed coding for R . SinI, a plasmidling for the restriction (pHp-+NABH6) missing the internal BglII-HindIII fragmentert is represented as a (nt 2647 through 2728) was constructed. Cells harboring this
estriction sites of the plasmid did not produce any R- SinI capable of cuttingpeS4 derivatives the bacteriophage lambda DNA in vitro after standard isolationof the cells harboring procedures.extra NheI site was Transcriptional start and stop signals. The transcriptional
;ite. In this way the start points of the two genes were determined in primerecame the only one in extension experiments (Fig. 3). Oligonucleotides with the'arious regions of the sequence 5'-ATGCATCAATGCTCCCCTGC-3' (presum-
ably complementary to the M - SinI mRNA) and 5'-CGAGCGCGGATTTGAACAAC-3' (complementary to theR. SinI mRNA) were hybridized to total RNA isolated from
utative stretches of cells harboring plasmid pSI4 and extended by reverse tran-. 2). These probes scriptase toward the mRNA 5' termini.
virus reverse tran- The resulting autoradiograms (Fig. 3) show that transcrip-ously (6). tion of the R. SinI gene starts at nt 3033 through 3036.iled in 90% form- Upstream one finds the sequence 5'-TTATACT-3' at posi--d onto denaturing tion -10 and 5'-TTGACT-3' at -35 (Fig. 2). Just upstreamlers generated with of the putative translation start there is a ribosome-binding4 DNA. Total RNA site at nt 3022 through 3027.'19 was used for a Transcription of the M- SinI gene starts near U-883 (Fig.
2; see Discussion). The area directly upstream of this posi-f sequence data we tion contains easily recognizable promoter sequences (veryuted by the Univer- similar to those of E. coli [23]): 5'-CATAAT-3' (nt 872ated with the aid of through 877) at position -10 and 5'-TTGACT-3' (nt 849[A sequences were through 854) at positions -35. A possible but rather weakiter. To this end a Shine-Dalgarno sequence is at nt 891 through 893, furtherilarity with a preset upstream from the ATG than in the case of the R * SinI gene.tf mismatches. Al- Since the genes are situated tail to tail and are separated)teins suggested by only by 31 bases, they most likely have a common termina-lDayhoff (20) were tor. A candidate structure for that function is indeed to bevith the best signal/ found at nucleotides 2301 through 2320: it is formed by aIf the original amino stem of 8 base pairs (containing 6 G C pairs) and a loop of
4 bases (see underlined bases in Fig. 1). This structure isflanked on both sides by stretches rich in A+T, although notpurely poly(dT) as is usual for a "classical" terminator.
Similarity betweenM SinI and other deoxycytidylate meth-(is DNA were con- ylases. The predicted amino acid sequence of the M - SinItnce, i.e., the pres- enzyme was compared with those of some modificationsubsequent analysis deoxycytidylate methylases: M * BspRI-GGCC (16), Malso coded for the BsuRI-GGCC (9), M - EcoRII-CCWGG (21), M * HhaI-GCransformed by this GC (4), M * MspI-CCGG (R. J. Roberts, personal commu-e. The R- SinI en- nication), M. Rholls-GGCC/GAGCTC (1), and M* SPR-)y the same method GGCC/CCGG/CCWGG (8, 15). The last two are encoded bys natural source S. Bacillus phages.l transformed with These comparisons are represented as dot plots in Fig. 4.ase-free R * Sin, as There is a striking similarity between the amino acid se-lambda DNA (data quences of M. SinI and the methylases from the Bacillus
group, whether they are coded by the bacteria (M BspRIof both strands of and M BsuRI) or by the phages (M * Rholls and M SPR).
J. BACTERIOL.

VOL. 170, 1988 TYPE II RESTRICTION-MODIFICATION GENES OF S. INFANTIS 2529
GATCGCCAGGCATTCCCGACTAMAAATATCCACTACAGTCAGCGCCCGACAAACGCCGCCCGCTTAAACAGATTATCTGACACAAAATCCATACTTCAGCACTGATCGACATGCGTCAGAAC 1 20
CGGGCGCTGCTGACAGCGTGCTGCATTGACATGTCTGCGGGGGCGTTTTCTGCGCAGGTTCAGGCCTTCCGGCAAMTATCCGGTCGGGTTCTTGTGGTTAACAGGCCATCCCCCCCG 240
CGCA/TTCGATATGAATACGCGGCAACCCCTAGCGTATCCGGGTTCCGCTATTTCCCGATACGCAGGGTTATCGCCCC;ATCGTCACACCGGCTCTGCCAGTGGTAAACGGTTCTGATC 360
TGCATCAGCAAeCCCGCATCCCCGCCGGACGCTGATACGGTAAGCCTCCAGCAAAATGTCACCGCCTGCGCCTTCTGAGCCGGCCCTCAGAACTTTTCCTTCAGTACCTCCTGCAGCATCT 480
CCTTGACCAGACTCAGCTCAGCGACCAGCTTCTTCAGCCGCTGATCCTCATCCTCCAGTTGCCGCAGACGCCGCAGTTCGGTCACGCCCGCCCGCAACTTTTTTCTTCCAGTTCTAA 600
ATGTGGCCTCAGAAATTCCCATCTTTCTGCAGACTTCCCCCGACCGCGG TGCCGGTTCAGCCTGTTCAGGCAATGCAATCTGTTCTTCGGTATAACGGGTCTTTTTCATGACAATGC 720
CCTTCTCTGTGAGATCGAGAAAGAACCGGAAACTTCAGTTTAGACTGGCACTGTTTACAGGCGGAAGGTCATCAGCGCGGCCAGGTTGGTATTATTCAGCCG CTrATTCAATCGTNTGT 840
TAArTTATTGACTATAAACGCACTTGGTAcATAATrrrGTlrcCAATTGGAAACMAAAAATAATGAMCAAAGAgTCACAT+CA'rACCGTM TAGAEAGCAGCAC2GTTGCT AGC 960
GG7+AA+ C~~~~~~~MV 10~~80
AA&C9TC2ACGTGTAA.GAATTAGT8 AAGCC2GC8GACCTT2TCAT1TC2A4CC8TAGMTTCTT AAAAA8TT9GA8TCTTGAAAAG7+GAYG TTAATTACTM I oAA~~~TC~~TC~TCE EII1080
CGRTCtTC2AAMTAGCTICACCATCTTCG2CTCT A AAAAACAAMCAAAMCMG2TTGT+AE---TiiCACTC8CCCTATCGSCCCAGACCTT8TTAC£AG iC2AGCGGCT6 oI 1 200
E DK A RD TI~ ANA E T~GCCTACTACGACTGAMAACAATAAAGCGCCAAGAGATACTAIMTATCTATGCCAACAGCG&CCC6 T8AGATATTCTATTACAIAATAGAGGATA+T7AAKAATTAGC 1320
ATTTCCTAATGAACA+TGAGATAYGCCCGSCCCPACCGTCTC2AC2TT,CTCAAIGCGG8AAAACCTITAC8TCTAG CATGC1CAAC8CAACG'Y4MCATT!A0TT 1440
AGADTGTCCCGTCGGATA+CACACcTAAGTXYTA+TGTAAICGAGA'ATGTVTACAGC8TATAT TCTGCGcAACGCA'TAGCEGCoA AATGAAC8TG8'rGAAG84cCFAJ4TAA1 1 560
1. H
AAAAK N E
ATCTGXAGACMGCPCCTCGTG8AGTAcrrCATTATATTATCACMAAAAGTCCAG8ATICTCTGTrrCTT ANCTTAATCTANCTTGCTAATTTCGCGCTcCTCLATTACAGA 1 680
AAEGGTMAATAAAAA'lSTTGCCGGACG8CACTAGAG4rCCCTI-TC ATACAAAGCAAACTCCa8GMICCCTGGATAACG'lGCETGA'AACATAACTAA 1 800
TTAAA TTATGACA8T CGvTGAcCC'AGAT+C ACAAATCTTTCAACTT AAAAGMMAGCTC2AT14G KCHAT ACCTGAGGArrTGCCuG 1920
AGCTCCTTAA G4cTTCG8CCA TAAACAC8TTTAG CClrC2ATCCTCCGATCEATCATACCCAIATTGGTGAIGCACCCAG2GAYCCCACTTTACC6tTTCAHTC9 2040
AGiTCM7TTGAGACCATTATCAGTTCLGAATTATAAGMCTC2ATCC2A TCCETCLAG LT.CGTTAAAAGSTAACTTCjfGTACATICAEAC2A4CG8TA'TCtTGCGCCCA+ 2160
TG8T7+AC8GTTAGtTGTAAAAAACATTTAGATCATATGAATC8AAGAAAACGAGTCA4TcCEANTTTTCGTTICTCTCEATTAKMAAAACAE-TCTGACTTGGATATATTTCS 2280
ACEAGTTGGCT*TAAATTATTAGGAGGGG.CATCCCCCCTTCCATTGTTATKTIAKTAtTFGTALACAGsCTECAAvCAfMARCTJTAAFAACAICTTAICGAJ"tGWC'rTG AGATGAvCTICGD 2400
TCAgGG§MMCC&EAAiTTAGACSCT&TriTJTG#ATTTn&TA#CJATrATiT?GCCTArWTGCGJGAtTri=JTAJTTCSAAsAGWTCCAqTATAAICT&TAWTTpV 2520
AC`CT4GTAtGA8CC5EACjCAW TTCATGCAIAATWGATAfAAICTOCTRCTJTOCASCTTTTECTVAAFAC6ATfCJTECAAT&CCAGCZCTtATF CA2CCAfGITJE 2640
TCCtG#fGAwCTECAOCAfTCITCAVAAJCCATAJAGtGTJGAWGTICCTJTATTJTAW=tGAMC7ZGC?GTfTAWCACSAGTOCAWTAtGGOC CTCMTOCAOCCiCAD 2760
TCA?GGI2TAqTATAGGTGGCqT CFFCTTGAPGCECCPGACCCAsCAWTfW JTCtA £TeACTtiGTfACTGCTCCTAECAfAGWTTrF TAOCFT cAGRCGC?GA, 2880
-~~~~ ~ ~ ~ ~ ~ ~ ~ ~ ~~.3
AAC0CTGCCjCTCACCATCAAoCCTCCTTATGTATTAAGGCAGTATAATTCTTTAAATCAAAAGTTACAGTCAAACTAGCATGACCTATTCATTACATCAGGTGAArrATCACCTGC 3 120
7TGCAAACTCAAACGGCCTGACCATATCGTTAAGATTGATATCAAGATAATCAGCCcACCACAGAAGCATGAGCCTCCGTTrCATCAAGATGCTCcCCCAGATAGATGTACACCACGCGGA 3 240
CG7TGCTTATGCTCCTGATGGCTCATTTGTCGCTCAACAGCATCCCTTGACCATAAACCTGATTCGACTAACGCACACAGAGCCATCGCCCTAA CCCGTGTCCACAACATCCGrMAG 3360
TGTCGTAGCCCATTGAGCGCAGAGCCTTGTTACAGCTCTCTCACTCATCCGCCTATCCGCCMATC 34 30
FIG. 2. Total sequence of the 3,430-base-pair Sau3AI fragment of S. infantis DNA. Indicated are the M Sinl gene (nt 907 through 2293)and the R Sinl gene (nt 2324 through 3017, reverse strand). Their predicted amino acid sequences are shown. The stop codons are indicatedby asterisks. Of both genes, the putative promoters (positions -10 and -35) are indicated as well as the mRNA start (!) and the possibleribosomal recognition Shine-Dalgarno sequences (SD). The putative dual terminator at nt 2301 through 2320 is underlined. Theoligonucleotides used for the primer extension experiments are indicated by arrows (nt 991 through 1010 forM * SinI and nt 2871 through 2890for R * Sinl).
Methylase M SinI also exhibits extensive similarity with No similarity between R Sinl and other endonucleases.dCMP methylases from such diverse origins as Hlaemophi- The degree of similarity between R * SinI and other knownlus, Escherichia, and Moraxella species. Especially the B, restriction enzymes is poor, as it usually is in this class ofC, and D: domains as defined by Som et al. (21) show up as proteins (21). Comparisons run with the same parameters asdiagonals in almost all comparisons, as does the E domain, were used for the methylases yielded, if anything, only smallexcent in the Bacillus phaims (possiblv due to their multiple randomlv distributed patches of similaritv. The same appliesrecognition sites). to a comparison of R- SinI and M SinI; as in earlier cases

2530 KARREMAN AND DE WAARD
IIP c T A G
A~~~~~~~~~~T" ~ ~ ~ ~~~~~T T
A
aImFIG. 3. mRNA start sites of the R. SinI and M SinI genes. (I)
Autoradiogram of a primer-extension experiment with a syntheticoligonucleotide (antisense to R SinI mRNA). This primer was
elongated with avian myeloblastosis virus reverse transcriptase on
RNA as the template. The resulting extension products were frac-tionated in lane P. The arrows indicate the only bands found in -thislane. They were run next to a dideoxynucleotide sequence laddermade with the same primer on plasmid DNA. The correspondingsequence is given on the left of the picture. In lane C an extra bandis seen comigrating with the A; this band did not once appear in thesequence ladders according to either Maxam and Gilbert or Sangeret al. and must be considered an artifact of the elongation reactionunder these precise conditions. (II) Corresponding experiment forM SinI mRNA. Here the complementary sequence is presented foreasy comparison with the sequence in Fig. 2.
(2, 7, 9, 19, 25), no similarity was found between these twocomponents of the restriction-modification system.
DISCUSSION
The overall A+T content (58%) of the SinI restriction-modification system is fairly high. The distribution of A+Talong the insert is shown in Fig. 1. In this respect the SinIrestriction-modification system resembles those of theEcoRI, EcoRV, and BsuRI genes (2, 7, 9). This phenomenonmight play a regulatory role. In line with this observation
11
VI
VI
I
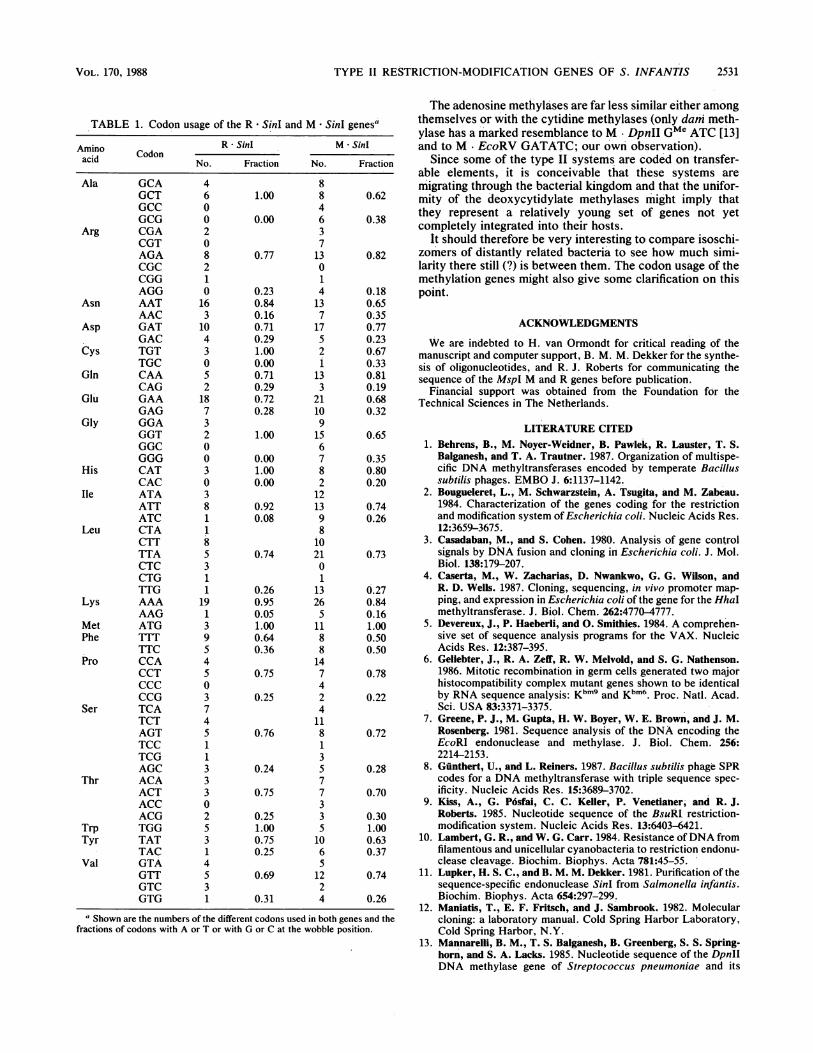
there is a clear preference for A or T (Table 1) at the third or
wobble position.Transcriptional starts were identified experimentally, and
promoter-like sequences were found for both genes. Thestart of the M - SinI gene as determined by primer extensionshows that the mRNA for this protein starts with U-883.Whether this unexpected result is due to mRNA synthesisactually starting with UTP or to mRNA processing remains
uncertain.Promoterlike sequences, however, at nt 849 through 854
and 872 through 877 do not exclude the former possibility:since RNA polymerase can only span a defined number ofnucleotides the new mRNA must start very close to nt 883.The SinI M and R genes are situated tail to tail in theSalmonella genome (M-*<--R), which differs from the tan-dem organization R-+M-* in BsuRI (9) and M--R--+ in HhaII(19). A head-to-head organization of the R and M genes hasbeen described for PstI (25). Only the EcoRII (21) systemappears to have a tail-to-tail conformation like that describedhere for SinI. The tail-to-tail organization of the two genescould be an additional regulatory element.The difference in intensity between the bands shown in the
primer extension experiments might be explained as an
indication of a different level of expression of the two genes.However, since the isolation of mRNA represents only thelevel of the two messengers at a particular moment and thenucleotide sequence of the primer might influence the elon-gation reaction, such conclusions would be premature.When the M. SinI amino acid sequence is compared with
those of other dCMP methylases a good degree of similarityis found in a small but definite number of regions conservedin a large range of methylases from evolutionarily distantspecies. There even is a significant degree of similarity withthe predicted amino acid sequence ofM AquI, a methylaseisolated from the cyanobacterium Agmenellum quadruplica-tum (C. Karreman and A. de Waard, manuscript in prepa-ration).
m
I I
Vil__. .... .. .
IV
FIG. 4. Similarities between methylase M SinI and other modification methylases: dot-plot representation. The predicted amino acidsequence of M SinI was compared with those of other deoxycytidine methylases. The program was set to score five perfect matches in a
window of eight. M SinI is plotted on the horizontal axis against the various other methylase proteins on the vertical axis. The unit on bothaxes in the various panels is 100 amino acids. M SinI was compared with the following: I, M BspRI; II, M BsuI; III, M Rholl,; IV,M. SPR; V, M EcoRII; VI, M HhaI; VII, M MspI.
G A T C P
A T Gi Om96 G ._
A
A T
A I. i _ft *
:..Ie
A I
II
Is
J. BACTERIOL.
I
TYPE II RESTRICTION-MODIFICATION GENES OF S. INFANTIS
TABLE 1. Codon usage of the R * SinI and M * SinI genesa
Amino R - SinI M * SinIamino Codonacid No. Fraction No. Fraction
Ala GCAGCTGCCGCG
Arg CGACGTAGACGCCGGAGG
Asn AATAAC
Asp GATGAC
Cys TGTTGC
Gin CAACAG
Glu GAAGAG
Gly GGAGGTGGCGGG
His CATCAC
Ile ATAATTATC
Leu CTACTTTTACTCCTGTTG
Lys AAAAAG
Met ATGPhe TTT
TTCPro CCA
CCTCCCCCG
Ser TCATCTAGTTCCTCGAGC
Thr ACAACTACCACG
Trp TGGTyr TAT
TACVal GTA
GTTGTCGTG
4 81.00
0.00
0.77
0.230.840.160.710.291.000.000.710.290.720.28
1.00
0.001.000.00
0.920.08
0.74
0.260.950.051.000.640.36
0.75
0.25
0.76
0.24
0.75
0.251.000.750.25
0.69
0.31
600208210163
10430S2
187320030381185311
19139
45037451133302S314S31
84637
13014137
17S2i
133
21109
156782
121398
io2101
13265
1188147424
1181357733S10651224
0.180.650.350.770.230.670.330.810.190.680.32
0.65
0.350.800.20
0.740.26
The adenosine methylases are far less similar either amongthemselves or with the cytidine methylases (only dam meth-ylase has a marked resemblance to M - DpnII GMe ATC [13]and to M - EcoRV GATATC; our own observation).
Since some of the type II systemns are coded on transfer-able elements, it is conceivable that these systems aremigrating through the bacterial kingdom and that the unifor-mity of the deoxycytidylate methylases might imply thatthey represent a relatively young set of genes not yetcompletely integrated into their hosts.
It should therefore be very interesting to comnpare isoschi-zomers of distantly related bacteria to see how much simi-larity there still (?) is between them. The codon usage of themethylation genes might also give some clarification on thispoint.
ACKNOWLEDGMENTS
We are indebted to H. van Ormondt for critical reading of themanuscript and computer support, B. M. M. Dekker for the synthe-sis of oligonucleotides, and R. J. Roberts for communicating thesequence of the MspI M and R genes before publication.
Financial support was obtained from the Foundation for theTechnical Sciences in The Netherlands.
LITERATURE CITED1. Behrens, B., M. Noyer-Weidner, B. Pawlek, R. Lauster, T. S.
Balganesh, and T. A. Trautner. 1987. Organization of multispe-cific DNA methyltransferases encoded by temperate Bacillussubtilis phages. EMBO J. 6:1137-1142.
2. Bougueleret, L., M. Schwarzstein, A. Tsugita, and M. Zabeau.1984. Characterization of the genes coding for the restrictionand modification system of Escherichia coli. Nucleic Acids Res.12:3659-3675.
3. Casadaban, M., and S. Cohen. 1980. Analysis of gene controlsignals by DNA fusion and cloning in Escherichia coli. J. Mol.Biol. 138:179-207.
4. Caserta, M., W. Zacharias, D. Nwankwo, G. G. Wilson, andR. D. Wells. 1987. Cloning, sequencing, in vivo promoter map-ping, and expression in Escherichia coli of the gene for the Hhalmethyltransferase. J. Biol. Chem. 262:4770-4777.
5. Devereux, J., P. Haeberli, and 0. Smithies. 1984. A comprehen-sive set of sequence analysis programs for the VAX. NucleicAcids Res. 12:387-395.
6. Geliebter, J., R. A. Zeff, R. W. Melvold, and S. G. Nathenson.1986. Mitotic recombination in germ cells generated two majorhistocompatibility complex mutant genes shown to be identicalby RNA sequence analysis: Kbm9 and Kbm6. Proc. Natl. Acad.Sci. USA 83:3371-3375.
7. Greene, P. J., M. Gupta, H. W. Boyer, W. E. Brown, and J. M.Rosenberg. 1981. Sequence analysis of the DNA encoding theEcoRl endonuclease and methylase. J. Biol. Chem. 256:2214-2153.
8. Gunthert, U., and L. Reiners. 1987. Bacillus subtilis phage SPRcodes for a DNA methyltransferase with triple sequence spec-ificity. Nucleic Acids Res. 15:3689-3702.
9. Kiss, A., G. P6sfai, C. C. Keller, P. Venetianer, and R. J.Roberts. 1985. Nucleotide sequence of the BsuRI restriction-modification system. Nucleic Acids Res. 13:6403-6421.
10. Lambert, G. R., and W. G. Carr. 1984. Resistance ofDNA fromfilamentous and unicellular cyanobacteria to restriction endonu-clease cleavage. Biochim. Biophys. Acta 781:45-55.
11. Lupker, H. S. C., and B. M. M. Dekker. 1981. Purification of thesequence-specific endonuclease SinI from Salmonella infantis.Biochim. Biophys. Acta 654:297-299.
12. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecularcloning: a laboratory manual. Cold Spring Harbor Laboratory,Cold Spring Harbor, N.Y.
13. Mannarelli, B. M., T. S. Balganesh, B. Greenberg, S. S. Spring-horn, and S. A. Lacks. 1985. Nucleotide sequence of the DpnIIDNA methylase gene of Streptococcus pneumoniae and its
a Shown are the numbers of the different codons used in both genes and thefractions of codons with A or T or with G or C at the wobble position.
VOL. 170, 1988 2531

2532 KARREMAN AND DE WAARD
relationship to the dam gene of Escherichia coli. Proc. Natl.Acad. Sci. USA 82:4468-4472.
14. Maxam, A. M., and W. Gilbert. 1980. Sequencing endlabeledDNA with base-specific chemical cleavages. Methods Enzymol.65:499-560.
15. P6sfai, G., F. Baldauf, S. Erdei, J. P6sfai, P. Venetianer, and A.Kiss. 1984. Structure of the gene coding for the sequence-specific DNA-methyltransferase of the B. subtilis phage SPR.Nucleic Acids Res. 12:9039-9049.
16. P6sfai, G., A. Kiss, S. Erdei, J. P6sfai, and P. Venetianer. 1983.Structure of the Bacillus sphaericus R modification methylasegene. J. Mol. Biol. 170:597-610.
17. Raleigh, E. A., and G. Wilson. 1986. Escherichia coli K-12restricts DNA containing 5-methylcytosine. Proc. Natl. Acad.Sci. USA 83:9070-9074.
18. Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequenc-ing with chain-terminating inhibitors. Proc. Natl. Acad. Sci.USA 74:5463-5467.
19. Schoner, B., S. Kelly, and H. 0. Smith. 1983. The nucleotidesequence of the HhaII restriction and modification genes fromHaemophilus haemolyticus. Gene 24:227-236.
20. Schwarz, R. M., and M. 0. Dayhoff. 1978. Matrices for detectingdistant relationships, p. 353-358. In M. 0. Dayhoff (ed.), Atlasof protein sequence and structure, vol. 5, suppl. 3. National
Biochemical Research Foundation, Washington, D.C.21. Som, S., A. S. Bhagwat, and S. Friedman. 1987. Nucleotide
sequence and expression of the gene encoding the EcoRIImodification enzyme. Nucleic Acids Res. 15:313-332.
22. Tandeau de Marsac, N., and J. Houmard. 1987. Advances incyanobacterial molecular genetics, p. 251-302. In P. Fay and C.van Baalen (ed.), The cyanobacteria. Elsevier Science Publish-ers, Amsterdam.
23. Trifonov, E. N., and V. Brendel. Gnomic. Balaban InternationalScience Services, Glenside, Pa.
24. van den Elzen, P. J. M., R. N. H. Konings, E. Veltkamp, andH. J. J. N"kamp. 1980. Transcription of bacteriocinogenic plas-mid CloDF13 in vivo and in vitro: structure of the cloacinimmunity operon. J. Bacteriol. 144:579-591.
25. Walder, R. Y., J. A. Walder, and J. E. Donelson. 1984. Theorganization and complete nucleotide sequence of the PstIrestriction-modification system. J. Biol. Chem. 259:8015-8026.
26. Yanisch-Perron, C., J. Vieira, and J. Messing. 1985. ImprovedM13 phage cloning vectors and host strains: nucleotide se-quences of the M13mpl8 and pUC19 vectors. Gene 33:103-119.
27. Zhu, J., W. Kempenaers, D. Van der Straeten, R. Contreras, andW. Fiers. 1985. A method for fast and pure DNA elution fromagarose gels by centrifugal filtration. Bio/Technology 3:1014-1016.
J. BACTERIOL.