rhodium-catalyzed intermolecular hydroacylation …...ii rhodium-catalyzed intermolecular...
TRANSCRIPT

Rhodium-Catalyzed Intermolecular Hydroacylation of Unactivated Alkenes and Application to the Total Synthesis of Octaketide
Natural Products
by
Christine Mai Le
A thesis submitted in conformity with the requirements for the degree of Masters of Science
Department of Chemistry University of Toronto
© Copyright by Christine Mai Le 2012

ii
Rhodium-Catalyzed Intermolecular Hydroacylation of Unactivated
Alkenes and Application to the Total Synthesis of Octaketide
Natural Products
Christine Mai Le
Masters of Science
Department of Chemistry
University of Toronto
2012
Abstract
Transition metal-catalyzed olefin hydroacylation represents an atom-economical approach for the
synthesis of valuable ketone products. To date, the intermolecular variant of this reaction suffers
from several drawbacks, which include limited substrate scope, poor reactivity and/or
regioselectivity for non-activated, non-chelating alkene substrates, and competitive reductive
decarbonylation pathways that lead to catalyst decomposition. Herein, we report the linear-
selective intermolecular hydroacylation of a wide range of electronically diverse olefins with
salicylaldehydes employing catalyst loadings as low as 2 mol%. A unique reactivity profile is
observed for the chiral C2-symmetric phosphoramidite ligand employed in our catalyst system,
and thus, we outline progress made towards the synthesis of new phosphoramidite ligands. We
have applied our methodology in the total synthesis of nine octaketide natural products belonging
to the dothiorelone, cytosporone, and phomopsin families. Due to recent reports demonstrating
the anticancer activity of cytosporone B (Csn-B), we will also discuss progress towards the
synthesis of Csn-B analogues.

iii
Acknowledgments
First and foremost, I would like to thank my supervisor, Prof. Vy Dong for her continual support,
guidance, and encouragement throughout the course of the year. I would also like to thank
Wilmer Alkhas for his hard work around the lab and for always going out of his way to help
students.
I am grateful for having the opportunity to work with such a high-spirited and intellectual group
of people who have made me feel welcome in the lab since the very first day I started. I would
like to send a very appreciative and warm thank you to Dr. Max von Delius, my collaborator on
this project. I am thankful for his patience, encouragement, and willingness to teach.
Lastly, I would like to thank all my family and friends for supporting and believing in me, even
when I doubted myself. I am grateful for having them in my life.

iv
Table of Contents
Acknowledgments .......................................................................................................................... iii
Table of Contents ........................................................................................................................... iv
List of Abbreviations ..................................................................................................................... vi
List of Tables ................................................................................................................................. ix
List of Figures ................................................................................................................................. x
List of Schemes .............................................................................................................................. xi
Chapter 1 Rhodium-Catalyzed Intermolecular Alkene Hydroacylation ......................................... 1
1.1 Introduction ......................................................................................................................... 1
1.1.1 The Origins of Hydroacylation ............................................................................... 1
1.1.2 Recent Advances and Current Limitations in Intermolecular Hydroacylation ....... 2
1.2 Project Objectives ............................................................................................................... 7
1.3 Results and Discussion ....................................................................................................... 8
1.3.1 A Brief Summary of the Method Development and Optimization Studies ............ 8
1.3.2 Investigation of Aldehyde Scope ............................................................................ 9
1.3.3 Investigation of Alkene Scope .............................................................................. 11
1.3.4 Biphasic Hydroacylation ....................................................................................... 14
1.3.5 Mechanistic Considerations .................................................................................. 16
1.4 Importance of Phosphoramidite Ligands in Catalysis ...................................................... 17
1.4.1 Synthesis of Phosphoramidite Ligands: En route to More Active and Selective
Hydroacylation Catalysts ...................................................................................... 19
1.5 Conclusions and Future Work .......................................................................................... 24
1.6 Experimental ..................................................................................................................... 25
1.6.1 General considerations .......................................................................................... 25
1.6.2 General procedure for the linear-selective hydroacylation of monosubstituted
alkenes with salicylaldehydes ............................................................................... 25

v
1.6.3 General procedure for the linear-selective hydroacylation of monosubstituted
alkenes with salicylaldehydes under biphasic conditions ..................................... 26
1.6.4 Characterization of compounds ............................................................................ 27
Chapter 2 Application of Intermolecular Hydroacylation to the Synthesis of Octaketide
Natural Products ....................................................................................................................... 33
2.1 Introduction ....................................................................................................................... 33
2.1.1 Accessing Polyketide Natural Products from Dothiorelone, Cytosporone, and
Phomopsin Families via Hydroacylation .............................................................. 33
2.1.2 Cytosporone B ...................................................................................................... 35
2.2 Research Goals .................................................................................................................. 37
2.3 Results and Discussion ..................................................................................................... 38
2.3.1 Synthesis of Starting Materials for Key Hydroacylation ...................................... 38
2.3.2 Testing Known Hydroacylation Methods ............................................................. 40
2.3.3 Solvent Screen for Hydroacylation of Natural Product Substrates ....................... 40
2.3.4 Synthesis of Octaketide Natural Products ............................................................. 42
2.3.5 Design and Synthesis of Cytosporone B Analogues ............................................. 45
2.4 Conclusions and Future Work .......................................................................................... 47
2.5 Experimental ..................................................................................................................... 47
2.5.1 General Considerations ......................................................................................... 47
2.5.2 General Procedures for the Total Synthesis of Natural Products via Linear-
Selective Hydroacylation ...................................................................................... 48
2.5.3 Characterization of Compounds ........................................................................... 48
Appendix A: NMR Spectra ........................................................................................................... 66
Appendix B: Crystallographic Data .............................................................................................. 96

vi
List of Abbreviations
1H NMR proton nuclear magnetic resonance
13C NMR carbon nuclear magnetic resonance
° C degrees Celsius
SIPHOS-PE 10,11,12,13-Tetrahydrodiindeno[7,1-de:1′, 7′-
fg][1,3,2]dioxaphosphocin-5-bis[(R)-1-phenylethyl]amine N-
Di[(R)-1-phenylethyl]-[(R)-1,1′-spirobiindane-7,7′-diyl]-
phosphoramidite
α alpha
β beta
δ chemical shift
μL microlitre
acac acetylacetonate
aq. aqueous
Ac acetyl
Arg Arginine
atm atmosphere
BINAP 1,1’-binaphthalene-2,2’-diyl-bis-diphenylphosphine
bs broad singlet
COD 1,5-cyclooctadiene
conv. conversion
d doublet; days
dd doublet of doublets
DCE 1,2-dichloroethane
DCM dichloromethane
DMAP 4-dimethylaminopyridine
DMF N,N-dimethylformamide
d.r. diastereomeric ratio
dt doublet of triplets
ee enantiomeric excess
EI electron impact

vii
eq. equation
equiv. equivalents
ESI electron spray ionization
Et ethyl
EtOAc ethyl acetate
g grams
GC-FID gas chromatography-flame ionization detector
GC-MS gas chromatography-mass spectrometry
h hours
HRMS high resolution mass spectrometry
Hz hertz
J coupling constant
LC-MS liquid chromatography-mass spectrometry
m multiplet
M molar
M.P. melting point
M+ parent molecular ion
Me methyl
MeCN acetonitrile
Me-THF 2-methyltetrahydrofuran
mg milligrams
MHz megahertz
min minutes
mL millilitres
mmol millimoles
MsOH methanesulfonic acid
n.r. no reaction
nBu n-butyl
NMR nuclear magnetic resonance
PhH benzene
PDB Protein Data Bank
ppm parts per million

viii
Pro proline
pyr pyridine
q quartet
rt room temperature
s singlet
Ser serine
t triplet
tt triplet of triplets
tBu tert-butyl
THF tetrahydrofuran
Thr threonine
TLC thin layer chromatography
TMS trimethylsilane
Ts tosyl (p-toluenesulfonyl)

ix
List of Tables
Table 1: Substrate scope for salicylaldehyde derivatives .............................................................. 9
Table 2: Solvent screen for the hydroacylation of 1-heptene (4) with 2,4-dihydroxybenzaldehyde
(1f) ................................................................................................................................................. 11
Table 3: Alkene scope .................................................................................................................. 12
Table 4: Testing selected alkene substrates under biphasic conditions ....................................... 15
Table 5: Conditions employed for Friedel-Crafts cyclization ..................................................... 22
Table 6: Solvent screen and reaction monitoring for hydroacylation of 1-heptene (22d) with
aldehyde 21b ................................................................................................................................. 41
Table 7: Synthesis of octaketide natural products ....................................................................... 43
Table 8: Synthesis of cytosporone B analogues ........................................................................... 46

x
List of Figures
Figure 1: Aldehydes bearing a β-chelating heteroatom ................................................................. 4
Figure 2: Common phosphoramidite ligands ............................................................................... 18
Figure 3: Proposed amines to be used in spiro-phosphoramidite synthesis ................................ 20
Figure 4: General structure of octaketide natural products from the dothiorelone, phomopsin,
and cytosporone families .............................................................................................................. 35
Figure 5: (a) Superimposition of Csn-B (yellow) onto the ribbon structure of the Nur77 LBD.
(b) Cytosporone B. (c) Compound 15 ........................................................................................... 36
Figure 6: Reaction progress for hydroacylation of 1-heptene (22d) with aldehyde 21b in Me-
THF and THF ................................................................................................................................ 42
Figure 7: Solid state structure of dothiorelone B (23bb). ............................................................ 44
Figure 8: Ligand binding pocket of Nur77 obtained from the PDB ............................................ 45

xi
List of Schemes
Scheme 1: Transition metal-catalyzed intra- or intermolecular hydroacylation of alkenes or
alkynes leading to ketone products ................................................................................................. 1
Scheme 2: a) Jun’s catalyst system employing 2-amino-picoline co-catalyst; b) Proposed mode
of activation for aldehyde substrates ............................................................................................... 5
Scheme 3: Bisphasic conditions for hydroacylation of 1-octene (2a) with salicylaldehyde (1a) 15
Scheme 4: Proposed catalytic cycle ............................................................................................. 17
Scheme 5: Common synthetic route to access phosphoramidites ................................................ 19
Scheme 6: Synthesis of spirobiindanediol backbone ................................................................... 22
Scheme 7: Synthesis of C2-symmetric chiral amine A1 .............................................................. 24
Scheme 8: Structural revision of phomopsin B and Takahashi’s approach to dothiorelone A .... 34
Scheme 9: Proposed key disconnection for synthesis of dothiorelone A .................................... 34
Scheme 10: Proposed synthesis of octaketides belonging to the dothiorelone, cytosporone, and
phomopsin families ....................................................................................................................... 37
Scheme 11: Synthesis of key aldehydes 21a-c ............................................................................. 39
Scheme 12: Synthesis of key alkenes 22a-c ................................................................................. 39
Scheme 13: Testing Suemune’s and Jun’s conditions for synthesis of phomopsin C ................. 40

1
Chapter 1 Rhodium-Catalyzed Intermolecular Alkene Hydroacylation
1.1 Introduction
1.1.1 The Origins of Hydroacylation
Developing highly efficient and environmentally benign protocols for the synthesis of natural
products and pharmaceutical agents is a key challenge in both academic and industrial settings
today. Towards this end, the direct functionalization of C–H bonds represents an attractive route
for both small and complex molecule synthesis, since pre-activation of formally unreactive C–H
bonds is not required—thereby minimizing chemical waste, while increasing reaction
efficiency.1 Transition-metal catalyzed hydroacylation represents a completely atom-economical
approach for the construction of carbon-carbon bonds, since all the atoms of the starting material
are conserved in the product.2 This particular transformation invokes the activation of an
aldehyde C–H bond by a metal species, followed by the formal addition of the acyl unit and
hydrogen atom across an unsaturated functional group (typically an alkene or alkyne) (Scheme
1).3
Scheme 1: Transition metal-catalyzed intra- or intermolecular hydroacylation of alkenes or
alkynes leading to ketone products.
1 (a) Ritleng, V.; Sirlin, C.; Pfeffer, M. Chem. Rev. 2002, 102, 1731-1769. (b) Labinger, J. A.; Bercaw, J. E. Nature
2002, 417, 507-514. 2 Trost, B. M. Angew. Chem. Int. Ed. Engl. 1995, 34, 259-281.
3 For a general and comprehensive review of transition metal-catalyzed alkene and alkyne hydroacylation, refer to
ref.: (a) Willis, M. C. Chem. Rev. 2010, 110, 725-748. For a microreview of transition metal-catalyzed alkene and
alkyne hydroacylation, refer to ref.: (b) Jun, C.-H.; Jo, E.-A.; Park, J.-W. Eur. J. Org. Chem. 2007, 1869-1881. For a
review of catalytic intermolecular hydroacylation in the absence of chelation-assistance, refer to ref.: (c) Leung, J.
C.; Krische, M. J. Chem. Sci. 2012, 3, 2202-2209.

2
The first example of alkene hydroacylation was reported by Sakai and co-workers in 1972, in
which 4-pentenals could be transformed into their respective cyclopentanones (a) by using a
stoichiometric amount of Wilkinson’s complex (eq. 1).4 While this method is not catalytic, two
significant conclusions can be drawn from this study: (1) trapping the putative acyl-rhodium-
hydride intermediate by an unsaturated functional group is possible, allowing for the rapid
construction of molecular complexity, and (2) the formation of cyclopropane byproducts (b) in
significant quantities indicate that reductive decarbonylation is both a competing and
energetically favourable pathway. This decarbonylation pathway generates a catalytically
inactive Rh-CO species, which becomes problematic when catalytic versions of this
transformation are desired.5 Nonetheless, since Sakai’s seminal report, the development of
catalytic, diastereo- and enantioselective olefin hydroacylations has been achieved with both
relatively low catalyst loadings and expanded substrate scope.3 Although much progress has
been made in the field of intramolecular alkene hydroacylation, this report will focus on its
intermolecular counterpart with an emphasis on rhodium-catalyzed alkene hydroacylation.
1.1.2 Recent Advances and Current Limitations in Intermolecular Hydroacylation
In addition to challenges associated with poor reactivity as well as modest regioselectivity,
finding efficient and general intermolecular hydroacylation protocols that minimize undesirable
decarbonylation pathways has proven to be quite difficult. Towards this end, strategies that
stabilize the key acyl-rhodium intermediate in the catalytic cycle have been successful for
decreasing the propensity for reductive decarbonylation. This approach often involves
heteroatom chelation, whereby the aldehyde or alkene component (or both) can coordinate to the
4 Sakai, K.; Ide, J.; Oda, O.; Nakamura, N. Tetrahedron Lett. 1972, 1287.
5 Fairlie, D. P.; Bosnich. B. Organometallics 1988, 7, 946-954.

3
metal center in a bidentate fashion.3,6
Although this strategy comes at the cost of reduced
substrate scope, an increase in both reactivity as well as regioselectivity has been demonstrated
in such systems.
1.1.2.1 Exploiting β-Chelating Aldehydes
Following Sakai’s report of a stoichiometric system for the synthesis of cyclopentanones (refer
to eq. 1), Miller disclosed that catalysis was possible if ethylene-saturated chloroform was
employed as solvent.7 During this study it was discovered that by exchanging the rhodium
source from Wilkinson’s complex to Rh(acac)(C2H4)2, products a and b, resulting from the
intermolecular hydroacylation of ethylene, could be isolated—where products of type b are a
result of olefin isomerization (eq. 2). The use of enals as substrates proved to be essential for this
type of reactivity, as Miller and coworkers speculated that internal coordination of the alkene
moiety to the metal center assisted in catalyst stabilization. Notably, no products resulting from
reductive decarbonylation were detected and saturated aldehydes did not furnish the desired
hydroacylation adduct.
Initial reports of heteroatom-chelation came from the work of Suggs, where quinoline 8-
carboxylaldehydes (1) were employed for the Rh-catalyzed hydroacylation of 1-octene (Figure
1).8 Seventeen years later, Jun disclosed that benzaldehyde derivatives bearing an ortho-
phosphine substituent (2) could engage this transformation as well.9 In either case, coordination
6 For a review on chelation-assisted hydroacylation, see ref.: Jun, C.-H.; Hong, J.-B.; Lee, D.-Y. Synlett 1999, 1, 1-
12. 7 (a) Lochow, C. F.; Miller, R. G. J. Am. Chem. Soc. 1976, 98, 1281-1283. (b) Vora, K. P.; Lochow, C. F.; Miller, R.
G. J. Organomet. Chem. 1980, 192, 257. 8 Suggs, J. W. J. Am. Chem. Soc. 1978, 100, 640-641.
9 Lee, H.; Jun, C.-H. Bull. Korean Chem. Soc. 1995, 16, 66-68.

4
of the β-heteroatom to the metal center facilitates oxidative addition and results in the formation
of a cyclometallated acyl-Rh intermediate, which has a decreased likelihood of undergoing
decarbonylation, as this would form a strained 4-membered rhodacycle. Willis and Weller have
demonstrated that β-S-aldehydes (3) are effective substrates for the intermolecular
hydroacylation of electron-rich, neutral, and deficient olefins depending on the catalyst system
employed.10
More recently, they have shown that cationic rhodium catalysts containing small-
bite angle diphosphines are extremely efficient at promoting the hydroacylation of β-S-aldehydes
with a wide range of alkenes and alkynes.10c
Catalyst loadings as low as 0.1 mol% and turnover
frequencies of greater than 300 h-1
are reported, which represents the most efficient
intermolecular hydroacylation system to date. While these coordinating heteroatoms serve their
purpose of stabilizing the key acyl-rhodium intermediate, such strategies often necessitate the
incorporation of these motifs in the final product.11
Salicylaldehydes (4), which possess a phenol
moiety, are unique substrates for hydroacylation, since functionalization of such aldehydes can
lead to motifs that are encountered in natural products or to scaffolds that can be easily
derivatized thereafter (vide infra).
Figure 1: Aldehydes bearing a β-chelating heteroatom.
Perhaps one of the most general protocols for the coupling of simple aldehydes with electron-
neutral alkenes comes from the work of Jun, whereby 2-amino-picoline is employed as a co-
catalyst to assist in the oxidative addition and to prevent the decarbonylation of non-chelating
aldehydes, such as benzaldehyde, via reversible imine formation (Scheme 2a).12
In this proposed
mode of activation, the pyridyl nitrogen atom on the co-catalyst is ideally positioned to facilitate
10
(a) Willis, M. C.; McNally, S. K.; Beswick, P. J. Angew. Chem. Int. Ed. 2004, 43, 340-343. (b) Moxham, G. L.;
Randell-Sly, H. E.; Brayshaw, S. K.; Woodward, R. L.; Weller,A. S.; Willis, M. C. Angew. Chem. Int. Ed. 2006, 45,
7618-7622. (c) Chaplin, A. B.; Hooper, J. F.; Weller, A. S.; Willis, M. C. J. Am. Chem. Soc. 2012, 134, 4885-4897. 11
For use of removable β-chelating motifs in hydroacylation, refer to: (a) Willis, M. C.; Randell-Sly, H. E.;
Woodward, R. L.; Currie, G. S. Org. Lett. 2005, 7, 2249-2251. (b) Parsons, S. R.; Hooper, J. F.; Willis, M. C. Org.
Lett. 2011, 13, 998-1000. 12
Jun, C.-H.; Lee, D.-Y.; Lee, H.; Hong, J.-B. Angew. Chem. Int. Ed. 2000, 39, 3070-3072.

5
oxidative addition by coordinating to the rhodium center in the same fashion as typical β-
chelating aldehydes (Scheme 2b). Hydrolysis of the resulting ketimine furnishes the desired
coupling product and permits catalyst turnover. However, the high temperatures required for this
transformation may be considered a disadvantage.
Scheme 2: a) Jun’s catalyst system employing 2-amino-picoline co-catalyst; b) Proposed mode
of activation for aldehyde substrates.
1.1.2.2 The Use of Salicylaldehydes
Over the last decade, salicylaldehydes have emerged as an effective class of substrates for
intermolecular hydroacylation.13,14
In the presence of a catalytic amount of base, the phenolate
of salicylaldehyde can coordinate to the Rh center, allowing for both a facile oxidative addition
and a decreased likelihood of decarbonylation. Miura and co-workers were the first to apply
salicylaldehydes as substrates for intermolecular alkyne hydroacylation, whereby analogous
reactivity with alkenes could not be achieved in the majority of the substrates surveyed.14a,b
The
successful coupling of salicylaldehydes with electron-neutral olefins was realized by Tanaka and
13 Work from our own group: (a) Murphy, S. K.; Petrone, D. A.; Coulter, M. M.; Dong V. M. Org. Lett. 2011, 13,
6216-6219. (b) Murphy, S. K.; Coulter, M. M.; Dong V. M. Chem. Sci. 2012, 3, 355-358. (c) Coulter, M. M.; Kou,
K. G. M.; Galligan, B.; Dong, V. M. J. Am. Chem. Soc. 2010, 132, 16330-16333. (d) Phan, D. H. T.; Kou, K. G. M.;
Dong, V. M. J. Am. Chem. Soc. 2010, 132, 16354-16355. 14
Work from other groups: (a) Kokobu, K.; Matsumasa, K.; Miura, M.; Nomura, M. J. Org. Chem. 1997, 62, 4564-
4565.; (b) Kokobu, K.; Matsumasa, K.; Nishinaka, Y.; Miura, M.; Nomura, M. Bull. Chem. Soc. Jpn. 1999, 72, 303.
(c) Tanaka, M.; Imai, M.; Yamamoto, Y.; Tanaka, K.; Shimowatari, M.; Nagumo, S.; Kawahara, N.; Suemune, H.
Org. Lett. 2003, 5, 1365-1367. (d) Imai, M.; Tanaka, M.; Tanaka, K.; Yamamoto, Y.; Imai-Ogata, N.; Shimowatari,
M.; Nagumo, S.; Kawahara, N.; Suemune, H. J. Org. Chem. 2004, 69, 1144-1150. (e) Imai, M.; Tanaka, M.;
Nagumo, S.; Kawahara, N.; Suemune, H. J. Org. Chem. 2007, 72, 2543-2546.
6
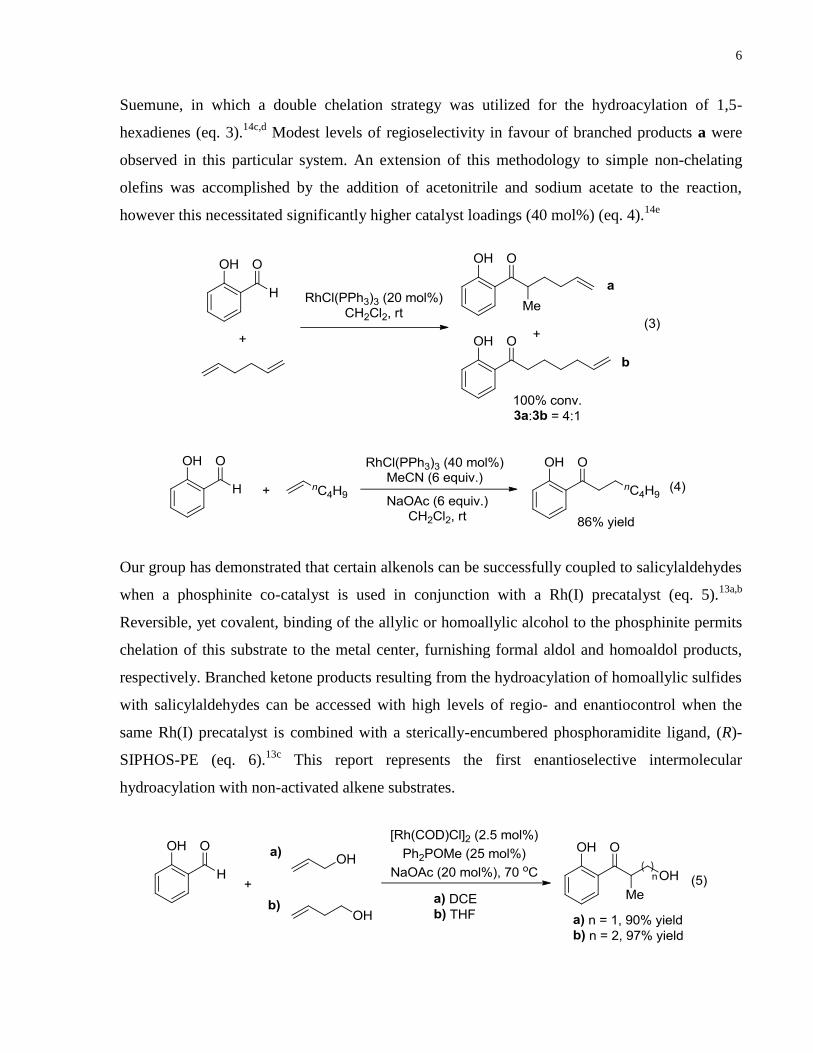
Suemune, in which a double chelation strategy was utilized for the hydroacylation of 1,5-
hexadienes (eq. 3).14c,d
Modest levels of regioselectivity in favour of branched products a were
observed in this particular system. An extension of this methodology to simple non-chelating
olefins was accomplished by the addition of acetonitrile and sodium acetate to the reaction,
however this necessitated significantly higher catalyst loadings (40 mol%) (eq. 4).14e
Our group has demonstrated that certain alkenols can be successfully coupled to salicylaldehydes
when a phosphinite co-catalyst is used in conjunction with a Rh(I) precatalyst (eq. 5).13a,b
Reversible, yet covalent, binding of the allylic or homoallylic alcohol to the phosphinite permits
chelation of this substrate to the metal center, furnishing formal aldol and homoaldol products,
respectively. Branched ketone products resulting from the hydroacylation of homoallylic sulfides
with salicylaldehydes can be accessed with high levels of regio- and enantiocontrol when the
same Rh(I) precatalyst is combined with a sterically-encumbered phosphoramidite ligand, (R)-
SIPHOS-PE (eq. 6).13c
This report represents the first enantioselective intermolecular
hydroacylation with non-activated alkene substrates.

7
1.1.2.3 Current limitations
Although significant achievements have been made in the area of intermolecular olefin
hydroacylation, this transformation remains limited in scope due to a lack of existing catalysts
that can couple a wide range of aldehydes with electronically-diverse olefins. Achieving high
levels of regiocontrol with non-chelating, unactivated olefins, while employing low catalyst
loadings is a remaining challenge to overcome.
1.2 Project Objectives
Based on this literature precedence, Dr. Max von Delius, a post-doctoral fellow in the group,
envisioned that simple, unactivated olefins that do not possess a heteroatom directing group
could undergo regioselective hydroacylation with salicylaldehydes following the judicious
choice of metal and ligand, as well as with the fine tuning of reaction parameters. This ideal
transformation would expand the substrate scope with respect to the olefin component, while
incorporating a biologically relevant arylketone motif in the final coupling product. As such, we
plan to apply intermolecular hydroacylation as a key step in the total synthesis of polyketide
natural products that bear a similar aryl ketone motif to the hydroacylation products obtained
(refer to Chapter 2). In addition to expanding the substrate scope, we aim to find a reactive
hydroacylation system in which catalyst loadings can be lowered to levels below 5 mol%. The
results reported herein are a result of a joint effort between Max and me. The results presented in
the method development represents optimization done solely by Max and were added to inform
the reader of significant background knowledge, as well as for clarity and continuity in the
development of the project.

8
1.3 Results and Discussion
1.3.1 A Brief Summary of the Method Development and Optimization Studies
Based on the high regioselectivity observed for the coupling of homoallylic sulfides with
salicylaldehydes demonstrated by our own group (refer to eq. 6), we wondered whether we could
extend this transformation to traditionally unreactive, electron-neutral olefins that do not possess
directing groups—while maintaining high levels of regioselectivity. Following extensive
optimization efforts, we were pleased to find that the same catalyst system (under slightly
modified conditions) could promote the highly linear-selective hydroacylation of 1-octene (2a)
with salicylaldehyde (1a) (eq. 7) at catalyst loadings as low as 2 mol%. A thorough scan and fine
tuning of different reaction parameters had revealed that elevated temperatures were required for
reactivity and that 1,2-dichloroethane was the ideal solvent for the reaction. One critical
discovery was that the precise 1:1 ratio of heterogenous base to [Rh] was crucial for maintaining
reactivity, and that having a slight excess of base relative to [Rh] was detrimental to the reaction.
A screen of various phosphorous-based ligands led to the following conclusions: (1) only
monodentate phosphorous-based ligands were able to promote hydroacylation15
and (2) of the
surveyed monodentate phosphorous-based ligands, phosphoramidites proved to be the most
effective, with (R)-SIPHOS-PE showing exceptional reactivity and linear-selectivity. While
chiral ligands are typically employed for enantioinduction in metal-catalyzed transformations,
the unique stereoelectronic properties imparted to such ligands can enhance reactivity and
promote regioselectivity in challenging non-asymmetric transformations16
, as demonstrated in
this particular intermolecular hydroacylation. Interestingly, when we subjected salicylaldehyde
15
A screen of several bidentate phosphines proved to be ineffective for this transformation 16
For the use of chiral ligands to enhance reactivity in non-asymmetric transformations, see for example: a) J. P.
Wolfe, S. Wagaw, S. L. Buchwald, J. Am. Chem. Soc. 1996, 118, 7215-7216; b) A. H. Roy, J. F. Hartwig, J. Am.
Chem. Soc. 2003, 125, 8704-8705; c) Q. Shen, S. Shekhar, J. P. Stambuli, J. F. Hartwig, Angew. Chem. 2005, 117,
1395-1399; Angew. Chem. Int. Ed. 2005, 44, 1371-1375.

9
(1a) and 1-octene (2a) to Jun’s conditions (refer to Scheme 2a), only an 8% NMR yield of the
desired hydroacylation product was observed after extended heating. This observation
demonstrates that salicylaldehydes are not compatible substrates for this type of chemistry.
1.3.2 Investigation of Aldehyde Scope
With these optimized conditions in hand, a variety of salicylaldehyde derivatives were tested
(Table 1). Good to excellent yields of the linear products were observed when both sterically-
hindered (entry 1) and electron-deficient (entries 2-4) salicylaldehydes were subjected to the
reaction conditions. A control experiment in which the 2-hydroxy group was protected as the
aryl methyl ether revealed that the free phenolic proton at the 2-position is required for reactivity
(entry 8). Napthylaldehyde 1h and β-S-aldehyde 1j furnished the desired hydroacylation
products in good to modest yields (entries 7 and 9). To the best of our knowledge, entry 9
represents the first report of a neutral Rh-catalyzed hydroacylation with β-S-aldehydes. Electron-
rich salicylaldehyde 1f reacted smoothly under the optimized conditions (entry 5), however
having a free phenol at the 4-position proved to be problematic (entry 6)—presumably due to the
low solubility of this substrate (1g) in DCE. For all cases, linear-to-branched selectivities of
>95:5 were observed.
Table 1: Substrate scope for salicylaldehyde derivatives.
Entry Aldehyde t (h) Product Yielda,b
1 R: 6-Me (1b) 40 3ba 66%
2 R: 5-F (1c) 48 3ca 78%
3 R: 5-Cl (1d) 48c 3da 89%
4 R: 5-I (1e) 60c 3ea 97%
5 R: 4-OMe (1f) 36 3fa 82%
6 R: 4-OH (1g) 24 3ga trace

10
7 (1h) 24 3ha 96%
8 (1i) 48 3ia 0%
9 (1j) 72d 3ja 48%
a Isolated yield.
b Regioisomeric ratio was >95:5 in all cases (determined by integration of crude
1H NMR spectrum).
c 4.0 equiv. of alkene used.
d No base added, 6.0 equiv. of alkene used (neat).
We were interested in finding a set of conditions in which 2,4-dihydroxybenzaldehyde (1g) could
furnish the desired hydroacylation product in reasonable yield and reaction time, since this
particular aldehyde would later serve as a model substrate for our natural product syntheses (vide
infra). By increasing the catalyst loading by two-fold, the hydroacylation of 1g with 1-heptene
(2b)17
went to 67% conversion, but only after prolonged reaction time (i.e. >1 week) (Table 1,
entry 1). Due to the low solubility of 1g in DCE, a screen of various polar solvents was
conducted. At first we reasoned that solvent mixtures consisting of DCE and a more polar
solvent should be ideally suited for solubilizing 1g, while maintaining comparable levels of
efficiency and reactivity. Unfortunately, yields no greater than 26% were obtained when 1:1
solvent mixtures consisting of DCE and THF (entry 2), acetonitrile (entry 3), 1,4-dioxane (entry
4), or acetone (entry 5) were employed. Of these four solvents, THF and 1,4-dioxane were the
most efficient at promoting this transformation. Interestingly, almost identical yields were
obtained upon switching the solvent to THF and 1,4-dioxane entirely (c.f. entries 2 & 8, and
entries 4 & 6). At this point, we wondered whether varying the relative amount of K3PO4 to [Rh]
could have an effect on reactivity. In THF, the amount of base was varied from 20 to 5 mol%
(entries 7 – 9). A marked decrease in yield was observed when excess base relative to [Rh] was
used—a result that is consistent with our previous finding that the precise ratio 1:1 of [Rh] to
base is crucial for reactivity. Surprisingly, when we decreased the loading of base to 5 mol% (i.e.
17
We employed 1-heptene as the alkene coupling partner due to its relevance to the targeted natural products (vide
infra).

11
[Rh]:base = 2:1), a drastic increase in yield was observed (entry 9). Upon switching the ethereal
solvent from THF to 2-Me-THF, a similar trend was observed when we varied the loading of
base from 10 to 5 mol% (entries 10 – 12), whereby a [Rh]:base ratio of 2:1 gave the highest yield
of 78% (entry 12). When the [Rh] loading was decreased to 5 mol%, while maintaining a 2:1
ratio of [Rh]:base, an excellent isolated yield of 95% was obtained for the linear product after
allowing the reaction stir for 120 h (entry 13).
Table 2: Optimization of the hydroacylation of 1-heptene (4) with 2,4-dihydroxybenzaldehyde
(1f).
Entry x:y:z (mol %) Solvent composition NMR yielda (%)
1 5:5:10 DCEb
65
2 5:5:10 DCE:THF (1:1) 26
3 5:5:10 DCE:CH3CN (1:1) <1
4 5:5:10 DCE:1,4-dioxane (1:1) 21
5 5:5:10 DCE:Acetone (1:1) 13
6 5:5:10 1,4-dioxane 20
7 5:5:20 THF trace
8 5:5:10 THF 28
9 5:5:5 THF 66
10 5:5:10 Me-THF 42
11 5:5:7.5 Me-THF 49
12 5:5:5 Me-THF 78
13c
2.5:5:2.5d
Me-THF 95e
a NMR yield determined by referencing
1H spectrum to an internal standard: 1,3,5-trimethoxybenzene.
b Reaction
time >1 week, no observable catalyst decomposition observed. c 1-octene was used instead of 1-heptene. Rxn time =
120 h, product = 3ga d At [Rh] catalyst loadings of <10 mol%, a 1:1 ratio of [Rh]:ligand is necessary for maintaining
reactivity. At [Rh] catalyst loadings of ≥10 mol%, a decrease in ligand loading to 5 mol% has no observable effect
on reactivity. e Isolated yield
1.3.3 Investigation of Alkene Scope
Having identified a general protocol for the coupling of salicylaldehyde derivatives with 1-
octene (2a), we turned our attention to the alkene scope. Overall, a variety of electronically-

12
diverse olefins were tolerated under the reaction conditions (i.e. electron-neutral, rich, or
deficient, as well as activated or unactivated), provided that only monosubstituted alkenes were
employed. Substitution at either the α or β position shut down reactivity completely. Due to the
wide electronic differences in the olefins screened, further optimization studies were carried out,
which entailed subjecting particularly unreactive alkene substrates to a set of five different
reaction conditions. This fine tuning would allow trends in reactivity and selectivity to be
deduced for each class of substrates tested. Yields above 60% and selectivities of >9:1 are
typically observed in the surveyed substrates and the results outlined in Table 3 represent the
highest yielding conditions employed.
Unactivated, electron-neutral alkenes 2c-g (Table 3, entries 1 – 5) displayed excellent reactivity,
as well as linear-to-branched selectivity (>95:5 in all cases) under the optimized reaction
conditions. To the best of our knowledge, entry 5 represents the first example of an acrolein
diacetal (2g) being used as a substrate for intermolecular hydroacylation. Notably, acetal
deprotection of hydroacylation product 3ag would allow for the convenient synthesis of 1,4-
ketoaldehydes. Free alcohol and protected amine functionalities are well tolerated (entries 6 –
10), with alkenes 2h-i (entries 6 – 7) furnishing the desired coupling product with the highest
yields and linear-selectivity. Reduced yields and selectivities are observed for olefins 2j-k in the
series (entries 8 – 9), which can be attributed to potential coordination of the nearby hydroxyl
group to the metal center. A complete reversal in selectivity is observed for 2-hydroxystyrene
(2l)—a case in which we presume branched hydride insertion is favoured to the due formation of
a 5-membered rhodacycle (entry 10). Although diene 2m displayed exceptional reactivity, the
lowered linear-selectivity can be attributed to coordination of the pendant alkene to the metal
center (entry 11). Much to our delight, electron-rich vinyl silane 2n reacted smoothly to give the
hydroacylation product (entry 12); however, modest yield was obtained for electron-rich butyl
vinyl ether (2o) (entry 13). Styrene (2p) and styrene derivatives (2q-r), which are electronically
biased to favour branched product formation, reacted cleanly, furnishing the linear
hydroacylation products with acceptable levels of regioselectivity (entries 14 – 16). When we
subjected α,β-unsaturated ester 2t to the reaction conditions, excellent reactivity was observed,
albeit with modest levels of linear-selectivity (entry 18). Electron-neutral alkene 2u displayed
poor reactivity under the reaction conditions (entry 19), while sterically-hindered alkene 2v did
not react at all (entry 20). Overall, we were pleased to find that our method was effective for a

13
range of electronically-diverse olefins, with electron-neutral alkenes displaying the best
reactivity and linear-to-branched selectivity.
Table 3: Alkene scope.
Entry Alkene t (h) Conditions Product Yield L:Bg
1 (2c) 72 B 3ac 63%a >95:5
2
(2d)
46 A 3ad 94%a >95:5
3
(2e)
24 A 3ae 89%a >95:5
4 (2f) 48 A 3af 86%a >95:5
5
(2g)
70 C 3ag 90%a >95:5
6
(2h)
48 Af
3ah 77%a >95:5
7 (2i)
84 A 3ai 93%a 86:14
8
(2j)
96 A 3aj 60%a 88:12
9
(2k)
46 E 3ak 76%a,d
83:17
10
(2l)
96 A 3al 52%a,d
35:65
11
(2m)
29 A 3am Quant.a,d
63:37
12 (2n) 70 D 3an 93%a >95:5
13 (2o) B 3ao 40%b >95:5
14
(2p)
36 A 3ap 98%a 89:11
15
(2q)
30 A 3aq 90%a 91:9
16
(2r)
24 A 3ar 70%a 88:12

14
17 (2s) 24 A 3as 64%a 58:42
18
(2t)
36 A 3at 91%a 69:31
19
(2u)
28 B 3au 31%a 90:10
20
(2v)
24 A - n.r. -
Conditions A: 2.5 mol% [Rh(COD)Cl]2, 5 mol% (R)-SIPHOS-PE, 5 mol% K3PO4, 2 equiv. alkene, DCE
Conditions B: 2.5 mol% [Rh(COD)Cl]2, 5 mol% (R)-SIPHOS-PE, 2.5 mol% K3PO4, 5 equiv. alkene, neat
Conditions C: 5 mol% [Rh(COD)Cl]2, 5 mol% (R)-SIPHOS-PE, 5 mol% K3PO4, 2 equiv. alkene, DCE
Conditions D: 5 mol% [Rh(COD)Cl]2, 5 mol% (R)-SIPHOS-PE, 5 mol% K3PO4, 4 equiv. alkene, DCE
Conditions E: 2.5 mol% [Rh(COD)Cl]2, 5 mol% (R)-SIPHOS-PE, 5 mol% K3PO4, 4 equiv. alkene, DCE
n.r. = no reaction. a Isolated yield.
b Approximate yield; could not separate from impurities by chromatography.
c
NMR yield. d Linear and branched regioisomers inseparable by chromatography (isolated together).
e Conversion
based on integration of crude 1H NMR relative to starting material reference peak.
f 3.75 mol% K3PO4 used.
g
Linear-to-branched selectivities determined by integration of crude 1H NMR spectrum
1.3.4 Biphasic Hydroacylation
During our optimization studies, we found that biphasic conditions could be employed for the
hydroacylation of 1-octene (2a) with salicylaldehyde (1a). When a 1:1 mixture of DCE:H2O was
used in the reaction, the desired coupling product was furnished in 97% yield after 19 h (Scheme
3a). To the best of our knowledge, this represents the first example of biphasic conditions being
utilized in hydroacylation. Under these conditions, we believe that the catalyst and 1-octene
remain strictly in the organic layer, while the heterogenous base (K3PO4), as well as any
deprotonated aldehyde substrate, remains dissolved in the aqueous layer. In such a scenario, the
reaction would occur at the interface of the two layers. Interestingly, when we conducted the
same experiment with a stoichiometric amount of base, the reaction proceeded with a similar
level of efficiency (Scheme 3b). If we were to use our standard monophasic conditions
employing DCE as solvent, a drastic decrease in yield would be observed, since having an excess
amount of base relative to [Rh] seems to be detrimental to the reaction. We hypothesize that
catalyst decomposition arises from having excess deprotonated salicylaldehyde in solution
(which is directly correlated to the amount of base) and that biphasic conditions help to slow
down decomposition pathways by separating deprotonated salicylaldehyde and rhodium catalyst
into two layers.

15
Scheme 3: Biphasic conditions for hydroacylation of 1-octene (2a) with salicylaldehyde (1a).
We were intrigued by the high efficiency of the biphasic hydroacylation conditions, and thus
decided to apply these conditions to particularly challenging alkene substrates. Much to our
delight, allyl acetate 2w reacted cleanly under the reaction conditions with no observable
byproducts resulting from SN2’-type chemistry (Table 4, entry 1). Although the linear-selectivity
was only moderate for this substrate, we were able to separate and fully characterized the linear
and branched regioisomers, 3aw and 3aw’. We previously observed poor conversion with
acrylamide substrates using our standard hydroacylation conditions. When biphasic conditions
were employed, full conversion of salicylaldehyde was observed and 57% of the desired
hydroacylation adduct was obtained with excellent linear-selectivity. In addition, we isolated a
significant amount of decarbonylation product (i.e. phenol) along with the desired product,
which accounts for the remaining mass balance.
Table 4: Testing selected alkene substrates under biphasic conditions.
Entry
Product
Isolated Yielda
(%)
L:Bb
1
(2w)
3aw/3aw’ 94c
63:37
2
(2x)
3ax 57 >95:5
a Combined isolated yield.
b Linear-to-branched selectivities determined by integration of crude
1H NMR spectrum.
c Linear and branched regioisomers separated by chromatography.

16
1.3.5 Mechanistic Considerations
We have proposed a general catalytic cycle for the linear-selective hydroacylation of electron-
neutral olefins, such as 1-octene, with salicylaldehyde under our optimized reaction conditions
(Scheme 4). This proposal is consistent with literature precedence regarding the generally-
accepted mechanism for the rhodium-catalyzed olefin hydroacylation of related systems.18
In the
first step of the catalytic cycle, deprotonated salicylaldehyde i can coordinate to the Rh center,
which is followed by oxidative addition of the aldehyde C–H bond (step a). From intermediate ii,
one can imagine reductive decarbonylation occurring, which would result in the formation of a
catalytically-inactive Rh-CO species. In the productive pathway (step b), ligand displacement
and olefin coordination gives rise to intermediate iii. Species iv arises from migratory insertion
of the alkene (step c), which upon reductive elimination (step d) furnishes linear hydroacylation
product v and regenerates the Rh(I) catalyst.
18
(a) Campbell, R. E., Jr.; Lochow, C. F.; Vora, K. P.; Miller, R. G. J. Am. Chem. Soc. 1980, 102, 5824-5830. (b)
Campbell, R. E., Jr.; Miller, R. G. J. Organomet. Chem. 1980, 186, C27-C31. (c) Fairlie, D. P.; Bosnich, B.
Organometallics 1988, 7, 946-954. (d) Hyatt, I. F. D.; Anderson, H. K.; Morehead, A. T., Jr.; Sargent, A. L.
Organometallics 2008, 27, 135-147. (e) Moxham, G. L.; Randell-Sly, H. E.; Brayshaw, S. K.; Weller, A. S.;
Willis, M. C. Chem.sEur. J. 2008, 14, 8383-8397.

17
Scheme 4: Proposed catalytic cycle.
1.4 Importance of Phosphoramidite Ligands in Catalysis
Over the past decade, phosphoramidites have emerged as a privileged class of chiral
monodentate ligands and have thus been exploited in a variety of transition metal-catalyzed
transformations.19
These phosphorous-based ligands, when combined with a metal catalyst, have
been shown to promote reactivity and provide high levels of enantioinduction in asymmetric
hydrogenation, conjugate addition, allylic substitution, cycloadditions, cross-coupling, and
hydroformylation. In general, phosphoramidites possess two P–O bonds and one P–N bond,
rendering the phosphorous atom relatively π-acidic, while maintaining good σ-donating capacity
(Figure 2). In such ligand frameworks, several points of chirality may be present in the ligand
(i.e. in either the diol backbone or the amine portion). Representative diols that have been
19
For a comprehensive review of phosphoramidite ligands in asymmetric catalysis, refer to ref.: Teichert, J. F.;
Feringa, B. L. Angew. Chem. Int. Ed. 2010, 49, 2486-2528.

18
previously incorporated into this class of ligands are outlined in Figure 2. Of the diol backbones
presented, BINOL-based phosphoramidites have found the most widespread use in asymmetric
transformations. Although many variations of BINOL-derivatives have been explored, the
number of analogous derivatives containing a spiroiindanediol (SPINOL) backbone is still
limited.20
Figure 2: Common phosphoramidite ligands.
The synthesis and resolution of SPINOL was achieved in 1999 by Birman and co-workers.21
In
2002, Zhou reported the synthesis of a phosphoramidite ligand that incorporated this spirocyclic
framework, as well as its application in the asymmetric hydrogenation of enamides.22
Since this
seminal report, Zhou has developed a series of phosphoramidites based on the SPINOL-
20
For a review on chiral phosphorus ligands based on a spiro scaffold for transition-metal-catalyzed asymmetric
reactions, refer to ref.: Xie, J.-H.; Zhou, Q.-L. Acc. Chem. Res.2008, 41, 581-593.
21 Birman, V.B.; Rheingold, A. L.; Lam, K.-C. Tetrahedron: Asymmetry 1999, 10, 125-131.
22 Hu, A.-G.; Fu, Y.; Xie, J.-H.; Zhou, H.; Wang, L.-X.; Zhou, Q.-L. Angew. Chem. Int. Ed. 2002, 41, 2348-2350.

19
backbone and has applied these ligands to a variety of transformations.20,23
Due to our group’s
growing interest in the use of spirobiindanediol-based phosphoramidites in hydroacylation, we
wondered whether we could expand the library of available SPINOL-based phosphoramidites.
Ultimately, we plan to employ these ligands in rhodium-catalyzed hydroacylation with the hope
of finding a more active and selective catalyst system, particularly for challenging alkene
substrates.
Phosphoramidites can be synthesized in several ways, by which the order of the key P–O or P–N
formation varies in each case.19
The most common synthetic route towards such motifs begins
with the synthesis of the key chlorophosphite intermediate b from diol a, which may or may not
be chiral (Scheme 5). The final step in the synthesis involves a convergent P–N coupling of the
chlorophosphite with either a chiral or an achiral amine (c), furnishing the final phosphoramidite
d in a two-step sequence. The modularity and step-economy of this synthetic approach should
allow rapid entry to a wide variety of phosphoramidite derivatives.
Scheme 5: Common synthetic route to access phosphoramidites.
1.4.1 Synthesis of Phosphoramidite Ligands: En route to More Active and Selective Hydroacylation Catalysts
Due to the large abundance of BINOL-based phosphoramidites in the literature, we believed that
translating these ligand designs to the analogous SPINOL derivatives would be a good starting
point. Taking inspiration from the success of BINOL-based phosphoramidites, for which ligands
23
Xie,J.-H.; Zhu, S.-F.; Fu, Y.; Hu, A.-G.; Zhou, Q.-L. Pure Appl. Chem. 2008, 77, 2121-2132.

20
derived from amines A1-A6 are known in the literature24
(Figure 3), we plan on synthesizing the
corresponding SPINOL derivatives.25
Figure 3: Proposed amines to be used in spiro-phosphoramidite synthesis.
1.4.1.1 Synthesis of Spirobiindanediol Backbone
Following a procedure by Birman and co-workers,21
m-anisaldehyde (4) was condensed with
acetone, furnishing dienone 5 in good yield (Scheme 6). Hydrogenation of 5 using activated
Raney-Ni under 1 atm of hydrogen afforded ketone 6 in quantitative yield. The crude material
was used directly in the next step without purification. Blocking of the 4-position (i.e. para to
methoxy group) was necessary in order to direct bis-cyclization ortho to the methoxy groups in a
later stage of the synthesis. This was accomplished by carrying out an electrophilic aromatic
substitution reaction using bromine as the electrophile, which afforded ketone 7 in 93% isolated
yield. Unfortunately, upon subjecting 7 to Friedel-Crafts type cyclization conditions employing
polyphosphoric acid, only a 15% isolated yield of pure spirobiindane (+/–)-8 was achieved. The
low yielding nature of this cyclization step was not unexpected as persistent emulsions were
24
For A1, A4, A6: Arnold, L. A.; Imbos, R.; Mandoli, A.; de Vries, A. H. M.; Naasz, R.; Feringa, B. L. Tetrahedron
2000, 56, 2865-2878. For A2: Defieber, C.; Ariger, M. A.; Moriel, P.; Carreira, E. M. Angew. Chem. Int. Ed. 2007,
46, 3139-3143. For A3: Choi, Y. H.; Choi, J. Y.; Yang, H. Y.; Kim, Y. H. Tetrahedron: Asymmetry 2002, 13, 801-
804. For A5: Du, H.; Yuan, W.; Zhao, B.; Shi, Y. J. Am. Chem. Soc. 2007, 129, 11688-11689.
25 For A2, A3, A6, SPINOL-based phosphoramidites known, but not commercially available.
A2: Hoffman, T. J.; Carreira, E. M. Angew. Chem. Int. Ed. 2011, 50, 10670-10674. A3: onz lez, A. . Benitez,
D.; Tkatchouk, E.; Goddard, W. A.; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 5500-5507. A6: Xie,J.-H.; Zhu, S.-F.;
Fu, Y.; Hu, A.-G.; Zhou, Q.-L. Pure Appl. Chem. 2008, 77, 2121-2132.

21
formed during the work-up, rendering full mass recovery impossible. Fortunately, there was
enough material to carry forward to the remaining steps in the synthetic route. Removal of the
bromine protecting groups in rac-8 was accomplished via lithium-halogen exchange and
subsequent quenching with methanol, giving rise to spirobiindane 9 in excellent yield.
Deprotection of the aryl methyl ethers using BBr3 and coupling of the free phenols to L-
menthylchloroformate in the presence of NEt3 and catalytic DMAP gave rise to a 1:1
diastereomeric mixture of 11a/11b. Formation of this diastereomeric mixture allows resolution
of pro-(R) and pro-(S)-SPINOL by standard column chromatography. Upon full separation of
diastereomers 11a and 11b, cleavage of the chiral auxiliaries can be carried out to obtain (R) and
(S)-SPINOL in enantiopure form.
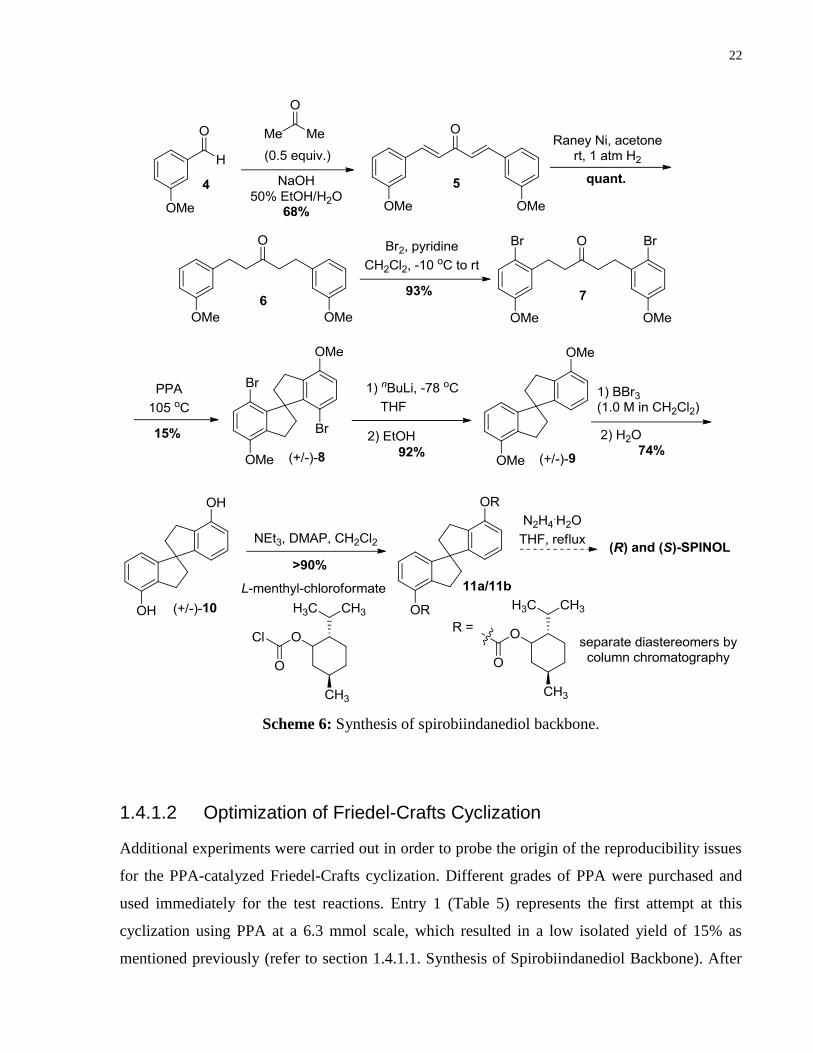
22
Scheme 6: Synthesis of spirobiindanediol backbone.
1.4.1.2 Optimization of Friedel-Crafts Cyclization
Additional experiments were carried out in order to probe the origin of the reproducibility issues
for the PPA-catalyzed Friedel-Crafts cyclization. Different grades of PPA were purchased and
used immediately for the test reactions. Entry 1 (Table 5) represents the first attempt at this
cyclization using PPA at a 6.3 mmol scale, which resulted in a low isolated yield of 15% as
mentioned previously (refer to section 1.4.1.1. Synthesis of Spirobiindanediol Backbone). After

23
scaling the reaction down to 0.2 mmol and employing different grades of PPA (entries 2 and 3),
excellent crude yields of 90% were obtained in both cases following standard work-up
procedure. Unfortunately, when the scale of the reaction was increased to 1.3 mmol, a significant
decrease in yield was observed (entry 4). Therefore, we concluded that this particular step could
not be scaled up efficiently, and thus searched for different acids to promote such a
transformation. Indeed, we were pleased to find that upon switching to methanesulfonic acid
(entry 5), we were able to isolate 44% of the pure material after the first recrystallization step.
Notably, this reaction was conducted at a 4.0 mmol scale, which allows access to sufficient
quantities of rac-8 for later stages in the synthesis.
Table 5: Conditions employed for Friedel-Crafts cyclization.
Entry Acid Scale of reaction
(mmol)
Crude yield (%) Isolated yield (%)
1 PPA (115% H3PO4
basis)
6.3 48 15
2 PPA (115% H3PO4
basis)a
0.2 90 -
3 PPA (>83% P2O5 basis)a
0.2 90 -
4 PPA (>83% P2O5 basis)a
1.3 50 -
5 MsOH 4.0 - 44 a Newly purchased reagent
1.4.1.3 Synthesis of C2-Symmetric Amine
The first amine we were interested in synthesizing was A1, due to its stereoelectronic similarity
to the amine portion in (R)-SIPHOS-PE (i.e. absolute configuration at the methine carbons are
the same as those in (R)-SIPHOS-PE), but increase in steric bulk. We hope that this increase in
steric bulk will, at the very least, give rise to higher levels of selectivity.

24
Scheme 7: Synthesis of C2-symmetric chiral amine A1.
Following a literature procedure,26
condensation of chiral amine 12 and 2-napthylketone (13)
was carried out in the presence of montmorillonite under microwave heating, furnishing ketimine
14 (Scheme 7). The reaction had stalled at approximately 75% conversion, even after prolonged
microwave heating (>24h), with the remaining 25% representing unreacted starting material.
Attempts to separate the product from the remaining naphthylketone 14 via column
chromatography were unsuccessful as this compound co-eluted with the 15 in numerous solvent
compositions. Fortunately, after two rounds of recrystallization, only 11% ketone remained in
the final material. Subjecting 14 as a mixture of the ketimine and unreacted ketone to a reduction
using HSiCl3 in the presence of DMF furnished C2-symmetric amine A1 with excellent
diastereoselectivity, albeit in modest yield. The presence of residual ketone did not seem to affect
the diastereoselectivity, but may have had an influence on the yield of the reaction.
1.5 Conclusions and Future Work
We have developed a novel rhodium-catalyzed method for linear-selective hydroacylation that
employs very low catalyst loadings and can be generalized to a range of different salicylaldehyde
and alkene substrates. Progress has been made with respect to phosphoramidite ligand synthesis
and we hope to synthesize and test these ligands in Rh-catalyzed hydroacylation in the near
26
(a) Guizzetti, S.; Benaglia, M.; Rossi, S. Org. Lett. 2009, 11, 2928-2931. (b) Guizzetti, S.; Benaglia, M.;
Biaggi,C.; Celentano, G. Synlett 2010, 1, 134-136.

25
future. In addition, we plan on further investigating and understanding the effects of our biphasic
hydroacylation conditions by testing other alkene substrates and by varying the catalyst system.
1.6 Experimental
1.6.1 General considerations
Commercial reagents were purchased from Sigma Aldrich, Strem or Alfa Aesar and used without
further purification. Reactions were monitored using thin-layer chromatography (TLC) on EMD
Silica Gel 60 F254 plates. Visualization of the developed plates was performed under UV light
(254 nm) or KMnO4 stain. Organic solutions were concentrated under reduced pressure on a
Büchi rotary evaporator. 1H,
31P and
13C NMR spectra were recorded on a Varian Mercury 400,
VRX-S (Unity) 400, or Bruker AV-III 400 spectrometer. 1H NMR spectra were internally
referenced to the residual solvent signal or TMS. 13
C NMR spectra were internally referenced to
the residual solvent signal. Data for 1H NMR are reported as follows: chemical shift (δ ppm),
multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, b = broad), coupling
constant (Hz), integration. Data for 13
C NMR are reported in terms of chemical shift (δ ppm).
High resolution mass spectra (HRMS) were obtained on a micromass 70S-250 spectrometer (EI)
or an ABI/Sciex QStar Mass Spectrometer (ESI). Infrared (IR) spectra were obtained on a
Perkin-Elmer Spectrum 1000 FT-IR Systems and are reported in terms of frequency of
absorption (cm-1
). Melting point ranges were determined on a Fisher-Johns Melting Point
Apparatus. Column chromatography was performed with Silicycle Silia-P Flash Silica Gel, using
either glass columns or a Biotage SP-1 system. Solvents used in hydroacylations were degassed
by three freeze-pump-thaw cycles. Chiral ligands were purchased from Strem.
1.6.2 General procedure for the linear-selective hydroacylation of monosubstituted alkenes with salicylaldehydes
In a nitrogen-filled glovebox, K3PO4 base (5 mol%), aldehyde (1.0 equiv.), alkene (2.0 equiv.),
[Rh(COD)Cl]2 precatalyst (2.5 mol%) and (R)-SIPHOS-PE ligand (5.0 mol%) were combined in
a one-dram vial and dissolved in solvent (0.4 M). The vial was charged with a stir bar, sealed
with a Teflon-lined cap and the mixture was stirred at 70 °C for the indicated period of time. The

26
crude reaction mixture was passed through a plug of silica and concentrated. A known amount of
standard 1,3,5-trimethoxybenzene was added to determine the NMR yield. The crude reaction
mixtures were purified by silica gel flash chromatography or preparative TLC to determine the
isolated yield.
1.6.3 General procedure for the linear-selective hydroacylation of monosubstituted alkenes with salicylaldehydes under biphasic conditions
In a nitrogen-filled glovebox, K3PO4 base (10 μmol), aldehyde (100 μmol), alkene (200 or 400
μmol), [Rh(COD)Cl]2 precatalyst (5 μmol) and (R)-SIPHOS-PE ligand (5.0 μmol) were
combined in a one-dram vial and dissolved in DCE (125 μL). The vial was charged with a stir
bar, sealed with a Teflon-lined cap, and brought outside the glovebox. Under a funnel of argon,
degassed H2O was added (125 μL) and the mixture was stirred at 70 °C for 48 hours. The crude
reaction mixture was diluted with EtOAc (1.0 M), quenched with 1.0 M HCl (2.0 mL), and
extracted with EtOAc (3 x 2.0 mL). The combined organics were dried over MgSO4 and the
solvent was removed under reduced pressure to afford the crude product, which was then
purified by preparative TLC to determine the isolated yield.
Notes:
• Air sensitivity: For reasons of convenience, all hydroacylation reactions were performed
in a glovebox filled with nitrogen. A control experiment outside the glovebox using standard
Schlenk techniques led to identical reaction efficiency. A second control experiment revealed
that the reaction also proceeds under air, albeit significantly slower (after one week
approximately 60% conversion was observed).
• Variation of reaction parameters: In a number of cases, the standard procedure was
slightly modified to guarantee optimum reaction efficiency. These deviations from the standard
procedure (typically involving changes of solvent, base or Rh equivalents) are specified for each
particular substrate.

27
1.6.4 Characterization of compounds
1-(2-hydroxyphenyl)-4-(trimethylsilyl)butan-1-one (3ac). Prepared according to the general
procedure 1.6.2 (2.5 mol% K3PO4, neat, 5 equiv. alkene, reaction time 72 h). Purification via
preparative TLC (95:5 hexanes/ethyl acetate) afforded the title compound as a colorless oil (33.5
mg, 63%). 1H NMR (400 MHz, CDCl3) δ 12.41 (s, 1H), 7.74 (dd, J = 8.0, 1.6, 1H), 7.47 – 7.42
(m, 1H), 6.97 (dd, J = 8.4, 1.0, 1H), 6.90 – 6.86 (m, 1H), 2.99 (t, J = 7.3, 2H), 1.86 – 1.62 (m,
2H), 0.66 – 0.46 (m, 2H), 0.00 (s, 9H). 13
C {1H} NMR (101 MHz, CDCl3) δ 208.69, 164.31,
137.93, 131.76, 121.17, 120.57, 120.29, 43.74, 21.17, 18.44. HRMS (ESI+): 237.1304 [M+H]
+
(calcd. 237.1311 for C13H21O2Si).
1-(2-hydroxyphenyl)-6-phenylhexan-1-one (3ad). Prepared according to the general procedure
1.6.2 (solvent: DCE; reaction time 46 h). Purification via preparative TLC (95:5 hexanes/ethyl
acetate) afforded the title compound as a colorless oil (50.5 mg, 94%). 1H NMR (400 MHz,
CDCl3) δ 12.41 (s, 1H), 7.76 (d, J = 8.0 Hz, 1H), 7.48 (t, J = 7.8 Hz, 1H), 7.30 (t, J = 7.5 Hz,
2H), 7.20 (d, J = 7.7 Hz, 3H), 7.00 (d, J = 8.4 Hz, 1H), 6.91 (t, J = 7.6 Hz, 1H), 3.00 (t, J = 7.4
Hz, 2H), 2.66 (t, J = 7.7 Hz, 2H), 1.86 – 1.75 (m, 2H), 1.76 – 1.66 (m, 2H), 1.52 – 1.42 (m, 2H).
13C {
1H} NMR (101 MHz, CDCl3) δ 206.88, 162.66, 142.55, 136.33, 130.08, 128.52, 128.42,
125.84, 119.45, 118.95, 118.67, 38.34, 35.86, 31.36, 29.02, 24.43. HRMS (ESI+): 269.1549
[M+H]+ (calc’d. 269.1542 for C18H21O2).
4,4-diethoxy-1-(2-hydroxyphenyl)butan-1-one (3ag). Prepared according to the general
procedure 1.6.2 (10 mol% [Rh], solvent: DCE; reaction time 70 h). Purification via preparative

28
TLC (9:1 hexanes/ethyl acetate) afforded the title compound as a colorless oil (40.4 mg, 80%).
1H NMR (400 MHz, CDCl3) δ 12.30 (s, 1H), 7.80 (dd, J = 8.0, 1.6, 1H), 7.46 (m, 1H), 6.98 (dd,
J = 8.4, 0.9, 1H), 6.90 (m, 1H), 4.60 (t, J = 5.3, 1H), 3.68 (dq, J = 9.4, 7.1, 2H), 3.51 (dq, J =
9.4, 7.0, 2H), 3.11 (t, J = 7.3, 2H), 2.07 (td, J = 7.3, 5.3, 2H), 1.21 (t, J = 7.1, 6H). 13
C {1H}
NMR (101 MHz, CDCl3) δ 206.12, 162.38, 136.24, 130.02, 119.42, 118.89, 118.47, 101.98,
61.83, 33.07, 28.14, 15.31. HRMS (ESI+): 270.1710 [M+NH4]
+ (calc’d. 270.1705 for
C14H24NO4).
6-hydroxy-1-(2-hydroxyphenyl)octan-1-one (3ah). Prepared according to the general
procedure 1.6.2 (5 mol% [Rh], 5 mol% ligand, 3.75 mol% K3PO4; solvent: DCE; 3.0 equiv.
alkene, reaction time 48 h). Purification via preparative TLC (7:3 hexanes/acetone) afforded the
title compound as a yellow oil (36.5 mg, 77%). 1H NMR (400 MHz, CDCl3) δ 12.36 (s, 1H),
7.76 (dd, J = 8.0, 1.5 Hz, 1H), 7.51 – 7.40 (m, 1H), 6.97 (d, J = 8.0 Hz, 1H), 6.93 – 6.83 (m,
1H), 3.55 (bs, 1H), 3.01 (t, J = 7.3 Hz, 2H), 1.86 – 1.70 (m, 2H), 1.63 – 1.36 (m, 6H), 0.94 (t, J
= 7.4 Hz, 3H). 13
C {1H} NMR (101 MHz, CDCl3) δ 206.85, 162.63, 136.38, 130.08, 119.46,
118.99, 118.68, 73.14, 38.35, 36.74, 30.40, 25.50, 24.51, 10.01. HRMS (ESI+): 237.1484
[M+H]+ (calc’d 237.1491 for C14H21O3).
4-hydroxy-1-(2-hydroxyphenyl)pentan-1-one (3aj). Prepared according to the general
procedure 1.6.2 (5.0 mol% K3PO4, DCE, 2 equiv. alkene, reaction time 120 h). Purification via
preparative TLC (8:2 hexanes/ethyl acetate) afforded the title compound as a colorless oil (23.3
mg, 60%). 1H NMR (400 MHz, CDCl3) δ 12.28 (s, 1H), 7.81 (dd, J = 8.0, 1.6 Hz, 1H), 7.47 (m,
1H), 6.98 (dd, J = 8.4, 1.1 Hz, 1H), 6.91 (m, 1H), 3.91 (bs, 1H), 3.36 – 3.02 (m, 2H), 2.00 – 1.92
(m, 1H), 1.89 – 1.78 (m, 1H), 1.27 (d, J = 6.2 Hz, 3H); 13
C {1H} NMR (101 MHz, CDCl3) δ
206.86, 162.60, 136.51, 130.17, 119.48, 119.08, 118.70, 67.51, 34.73, 33.25, 24.07. Note:
missing aliphatic alcohol peak in 1H NMR spectrum

29
1,4-bis(2-hydroxyphenyl)butan-1-one (3ak). Prepared according to the general procedure 1.6.2
(solvent: DCE, 4 equiv. alkene, reaction time 46 h). Purification via preparative TLC (9:1
hexanes/ethyl acetate) afforded a mixture of the linear and branched regioisomers (83:17 lin/br
selectivity) as an orange oil (39.2 mg, 76%27
).1H NMR (400 MHz, CDCl3) δ 12.16 (s, 1H), 7.76
(dd, J = 8.1, 1.5 Hz, 1H), 7.53 – 7.45 (m, 1H), 7.13 (ddd, J = 7.9, 6.9, 2.2 Hz, 2H), 7.01 (dd, J =
8.4, 0.9 Hz, 1H), 6.95 – 6.83 (m, 3H), 6.28 (s, 1H), 3.11 (t, J = 6.5 Hz, 2H), 2.75 – 2.62 (m, 2H),
2.04 (dq, J = 9.7, 6.5 Hz, 2H).13
C {1H} NMR (101 MHz, CDCl3) δ 207.40, 162.62, 154.42,
136.83, 130.27, 130.17, 127.89, 127.20, 120.63, 119.42, 119.20, 118.81, 115.96, 37.07, 29.76,
24.14. HRMS (ESI+): 257.1167 [M+H]
+ (calc’d 257.1178 for C16H17O3).
1-(2-hydroxyphenyl)-6-methylhept-6-en-1-one (3am). Prepared according to the general
procedure 1.6.2 (solvent: DCE; reaction time 29 h). Purification via preparative TLC (97:3
hexanes/ethyl acetate) afforded a mixture of the linear and branched regioisomers (63:37 lin/br
selectivity, 54.1 mg, 99%28
). All spectroscopic data correspond to those reported in the
literature.14c
1H NMR (400 MHz, CDCl3, linear isomer) δ 12.39 (s, 1H), 7.77 (dd, J = 8.0, 1.7,
1H), 7.46 (m, 1H), 6.98 (dd, J = 8.4, 0.9, 1H), 6.90 (m, 1H), 4.72 (s, 1H), 4.69 (s, 1H), 3.01 (t, J
= 7.5, 2H), 2.08 (t, , 2H) 1.79 (m, 2H), 1.72 (s, 3H), 1.25 (m, 2H).
1-(2-hydroxyphenyl)heptane-1,6-dione (3au). Prepared according to the general procedure
1.6.2 (10 mol% [Rh], solvent: DCE; reaction time 72 h). Purification via preparative TLC (8:2
hexanes/ethyl acetate) afforded the title compound as a colourless oil (17.1 mg, 30%).
27
Isolated yield of linear and branched regioisomers 28
Isolated yield of linear and branched regioisomers

30
Spectroscopic data correspond to those reported in the literature.29
1H NMR (400 MHz, CDCl3) δ
12.31 (s, 1H), 7.75 (dd, J = 8.0, 1.3 Hz, 1H), 7.54 – 7.37 (m, 1H), 6.97 (d, J = 8.4 Hz, 1H), 6.89
(dd, J = 11.2, 4.0 Hz, 1H), 3.01 (t, J = 7.0 Hz, 2H), 2.50 (t, J = 7.0 Hz, 2H), 2.15 (s, 3H), 1.86 –
1.56 (m, 4H); 13
C {1H} NMR (101 MHz, CDCl3) δ 208.62, 206.37, 162.63, 136.45, 130.02,
119.43, 119.05, 118.70, 43.52, 38.16, 30.08, 23.86, 23.44.
4-(2-hydroxyphenyl)-4-oxobutyl acetate (3aw). Prepared according to the general procedure
1.6.3 (200 μmol alkene). Purification via preparative TLC (90:10 hexanes/ethyl acetate) afforded
the title compound as a beige solid (15.3 mg, 69%). 1H NMR (400 MHz, CDCl3) δ 12.24 (s, 1H),
7.76 (dd, J = 8.0, 1.6 Hz, 1H), 7.48 (ddd, J = 8.6, 7.3, 1.6 Hz, 1H), 6.99 (dd, J = 8.4, 1.0 Hz, 1H),
6.91 (ddd, J = 8.2, 7.3, 1.1 Hz, 1H), 4.18 (t, J = 6.3 Hz, 2H), 3.10 (t, J = 7.2 Hz, 2H), 2.16 – 2.06
(m, 2H), 2.04 (s, 3H); 13
C {1H} NMR (101 MHz, CDCl3) δ 205.45, 171.16, 162.59, 136.56,
129.90, 119.42, 119.10, 118.76, 63.69, 34.78, 23.30, 21.04. HRMS (ESI+): 223.0976 [M+H]
+
(calc’d 223.0970 for C12H15O4).
3-(2-hydroxyphenyl)-2-methyl-3-oxopropyl acetate (3aw’). Prepared according to the general
procedure 1.6.3 (200 μmol alkene). Purification via preparative TLC (90:10 hexanes/ethyl
acetate) afforded the title compound as a yellow oil (5.5 mg, 25%). 1H NMR (400 MHz, CDCl3)
δ 12.31 (s, 1H), 7.79 (dd, J = 8.1, 1.5 Hz, 1H), 7.50 (ddd, J = 8.6, 7.3, 1.6 Hz, 1H), 7.01 (dd, J =
8.4, 1.0 Hz, 1H), 6.93 (ddd, J = 8.2, 7.3, 1.1 Hz, 1H), 4.43 (dd, J = 10.8, 8.2 Hz, 1H), 4.20 (dd, J
= 10.8, 5.7 Hz, 1H), 3.90 (dq, J = 14.1, 7.1 Hz, 1H), 1.99 (s, 3H), 1.27 (d, J = 7.1 Hz, 3H); 13
C
{1H} NMR (101 MHz, CDCl3) δ 207.45, 171.02, 163.32, 136.89, 129.99, 119.19, 118.99,
118.79, 65.66, 39.65, 20.95, 15.09. HRMS (ESI+): 223.0970 [M+H]
+ (calc’d 223.0973 for
C12H15O4).
29
Butler, J. D.; Conrad, W. E.; Lodewyk, M. W.; Fettinger, J. C.; Tantillo, D. J.; Kurth, M. J. Org. Lett. 2010, 12,
3410-3413.

31
4-(2-hydroxyphenyl)-N,N-dimethyl-4-oxobutanamide (3ax). Prepared according to the
general procedure 1.6.3 (2 equiv. alkene). Purification via preparative TLC (95:5 DCM/MeOH)
afforded the title compound as a beige solid (12.6 mg, 57%). 1H NMR (400 MHz, CDCl3) δ
12.15 (s, 1H), 7.87 (dd, J = 8.0, 1.6 Hz, 1H), 7.45 (ddd, J = 8.6, 7.3, 1.6 Hz, 1H), 6.96 (dd, J =
8.4, 1.0 Hz, 1H), 6.91 (td, J = 7.2, 3.7 Hz, 1H), 3.39 (t, J = 6.5 Hz, 2H), 3.09 (s, 3H), 2.96 (s,
3H), 2.76 (t, J = 6.5 Hz, 2H); 13
C {1H} NMR (101 MHz, CDCl3) δ 205.37, 171.46, 162.36,
136.40, 130.16, 119.58, 119.12, 118.50, 37.25, 35.71, 33.42, 27.06. HRMS (ESI+): 222.1131
[M+H]+ (calc’d 222.1130 for C12H16N1O3).
1-(2,4-dihydroxyphenyl)nonan-1-one (3ga). Prepared according to the general procedure 1.6.2
(2.5 mol% K3PO4, solvent: Me-THF; reaction time 5 d). Purification via preparative TLC (85:15
hexanes/ethyl acetate) afforded the title compound as a beige solid (47.4 mg, 95%). 1H NMR
(400 MHz, CDCl3) δ 12.89 (s, 1H), 7.66 (d, J = 8.6 Hz, 1H), 6.44 – 6.33 (m, 2H), 6.10 (bs, 1H),
2.89 (t, J = 7.5 Hz, 2H), 1.81 – 1.56 (m, 2H), 1.51 – 1.10 (m, 10H), 0.88 (t, J = 6.7 Hz, 3H). 13
C
{1H} NMR (101 MHz, CDCl3) δ 205.71, 165.35, 162.73, 132.60, 113.96, 107.90, 103.74, 38.22,
31.96, 29.52, 29.51, 29.27, 25.17, 22.78, 14.22. LRMS (ESI+) m/z calc’d for C14H20O3 [M+1]+:
237.31; found: 237.31.
1-(2,4-dihydroxyphenyl)octan-1-one (3gb). Prepared according to the general procedure 1.6.2
(10 mol% [Rh], 5 mol% ligand, 5 mol% K3PO4, solvent: Me-THF; reaction time 3 d).
Purification via preparative TLC (85:15 hexanes/ethyl acetate) afforded the title compound as a
beige solid (11.5 mg, 78%). 1H NMR (400 MHz, CDCl3) δ 12.86 (s, 1H), 7.66 (d, J = 9.4 Hz,

32
1H), 6.40 – 6.37 (m, 2H), 5.73 (bs, 1H), 2.89 (t, J = 8 Hz, 2H), 1.80 – 1.63 (m, 2H), 1.48 – 1.17
(m, 8H), 0.88 (t, J = 6.9 Hz, 3H). 13
C {1H} NMR (101 MHz, CDCl3) δ 205.56, 165.40, 162.56,
132.55, 114.04, 107.78, 103.75, 38.21, 31.82, 29.49, 29.23, 25.10, 22.76, 14.21. LRMS (ESI+)
m/z calc’d for C14H20O3 [M+1]+: 237.31; found: 237.31.

33
Chapter 2 Application of Intermolecular Hydroacylation to the Synthesis of
Octaketide Natural Products
2
2.1 Introduction
2.1.1 Accessing Polyketide Natural Products from Dothiorelone, Cytosporone, and Phomopsin Families via Hydroacylation
Polyketides represent a remarkable class of natural products that display diverse structural
complexity and exhibit a wide range of medicinally important activities, such as antibiotic,
anticancer, antifungal, antiparasitic, and immunosuppressive properties.30
Oftentimes, natural
product isolation can be complicated by either low natural abundance or the purification issues
that accompany such a task. As a consequence, the development of efficient routes towards these
complex molecules in a manner that is both environmentally-friendly and feasible on relatively
large scale has been a longstanding goal for synthetic chemists.
Transition metal-catalyzed alkene hydroacylation represents an attractive strategy for C–C bond
formation owing to its atom-economy and amenability to enantioselective catalysis. Unlike the
related process of hydroformylation,31
hydroacylation has not yet found any significant
application in industry or natural product synthesis, which is largely due to the unfavourable
decarbonylation pathways encountered with traditional rhodium catalysts.3c,32
Due to the
prevalence of aryl ketone motifs in polyketide natural products, we wondered whether our
hydroacylation protocol could be applied to the total synthesis of such natural products. More
specifically, we were inspired by a recent report by Takahashi and co-workers, which disclosed
30
Staunton, J.; Weissman, K. J. Nat. Prod. Rep. 2001, 18, 380-416.
31 For selected reviews on hydroformylation, see: (a) Beller, M.; Cornils, B.; Frohning, C. D.; Kohlpaintner, C. W.;
J. Mol. Catal. A: Chem. 1995, 104, 17-85. (b) Breit, B.; Seiche, W. Synthesis 2001, 1, 1-36.
32 To the best of our knowledge, there has only been one report of alkene hydroacylation being utilized for natural
product synthesis. Ref.: Kim, G.; Lee, E.-j. Tetrahedron: Asymmetry 2001, 12, 2073-2076.

34
the total synthesis and structural revision of phomopsin B to a known octaketide, dothiorelone A
(Scheme 8).33
Scheme 8: Structural revision of phomopsin B and Takahashi’s approach to dothiorelone A.
The synthetic route outlined in this report required protecting group chemistry, as well as several
unnecessary changes in oxidation state. Takahashi’s ten step sequence afforded dothiorelone A
in an overall yield of 15%. In order to improve the step and redox economy of this synthesis, we
envisioned that a linear-selective intermolecular hydroacylation between the appropriate
salicylaldehyde derivative and alkene precursor could furnish dothiorelone A in a convergent and
efficient manner (Scheme 9).
Scheme 9: Proposed key disconnection for synthesis of dothiorelone A.
Following our discovery of this initial report, we identified several families of octaketide natural
products that possess a similar framework to dothiorelone A. These particular polyketides belong
to the dothiorelone, cytosporone, and phomopsin families, and have all been isolated within the
33
Izuchi, Y.; Koshino, H.; Hongo, Y.; Kanomata, N.; Takahashi, S. Org. Lett. 2011, 13, 3360–3363.

35
last decade from different genera of endophytic fungi.34
A general feature of these polyketides is
that they possess a tetrasubstituted arene core with a long alkyl chain appended to the aromatic
ketone (Figure 4). Through this hydroacylation disconnection, we can imagine rapid access to
these octaketide natural products—many of which have not been previously synthesized and
display modest to potent levels of biological activity.35
Figure 4: General structure of octaketide natural products from the dothiorelone, phomopsin,
and cytosporone families.
2.1.2 Cytosporone B
Cytosporone B (Csn-B, Figure 5b) is an octaketide natural product that was first isolated in 2000
by Clardy and co-workers from an endophytic fungus (Cytospora sp.) collected in Costa Rica.34a
Following this isolation paper, Zhan et. al. had identified Csn-B as a natural agonist for nuclear
orphan receptor Nur77 (Figure 5a) and demonstrated that this ligand-receptor binding
interaction, which occurs upstream in a biochemical pathway, could inhibit xenograft tumor
growth in live mice.36
Nuclear receptors are expressed in several tissues throughout the human
body and play important roles in T-cell apoptosis, brain development, metabolism, and hormone
regulation, while orphan receptors are those in which no endogenous or exogenous ligands have
34(a) Brady, S. F. Wagenaar, M. M. Singh, M. P. Janso, J. E. Clardy, J. Org. Lett. 2000, 2, 4043-4046. (b) Xu, Q.
Wang, J. Huang, Y. heng, . Song, S. hang, Y. Su, W. Acta Oceanol. Sin. 2004, 23, 541-547. (c) Huang, .
Cai, X. Shao, C. She, . Xia, X. Chen, Y. Yang, J. hou, S. Lin, Y. Phytochemistry 2008, 69, 1604-1608. (d)
Huang, . uo, . Yang, R. Yin, X. Li, X. Luo, W. She, . Lin, Y. Chem. Nat. Prod. 2009, 45, 625-628. (e) Xu
J. et al., Bioorg. Med. Chem. 2009, 17, 7362-7367. (f) han, Y. et al. Nat. Chem. Biol. 2008, 4, 548-556.
35 Previous total syntheses: (a) Huang, H.; Zhang, L.; Zhang, X.; Ji, X.; Ding, X.; Shen, X.; Jiang, H.; Liu, H. Chin.
J. Chem. 2010, 28, 1041-1043 (cytosporone B). (b) Yoshida, H.; Morishita, T.; Ohshita, J. Chem. Lett. 2010, 39,
508-509 (cytosporone B and phomopsin C). (c) Izuchi, Y.; Koshino, H.; Hongo, Y.; Kanomata, N.; Takahashi, S.
Org. Lett. 2011, 13, 3360-3363 (dothiorelone A).
36 Zhan, Y. et. al. Nat. Chem. Biol. 2008, 4, 548.

36
been identified.37
The role of Nur77 in cancer cell apoptosis was independently reported by two
research teams in 1994, and thus, the identification of Csn-B as an agonist for Nur77 has
profound implications for the future of cancer therapy.38
In the account disclosed by Zhan et. al.,
Csn-B was shown to also elevate glucose levels in fasting mice, which may be important in the
development of drugs for the treatment of hypoglycemia.
Figure 5: (a) Superimposition of Csn-B (yellow) onto the ribbon structure of the Nur77 LBD.
(b) Cytosporone B. (c) Compound 15.
A later report by the same research team described the synthesis of simple Csn-B analogues and
identified the pharmacophore necessary for ligand binding as well as for the activation of a
biological response.39
In this study, it was deduced that the ester group was critical for activating
a biological response and that three other elements were required for ligand binding: (1) the
benzene ring, (2) the phenolic hydroxyl group, (3) and the acyl chain of the Csn-B scaffold. Of
the Csn-B derivatives synthesized, compound 15 (which possesses both an elongated alkyl ester
chain and acyl chain) displayed the highest affinity for the LBD of Nur77, and was the most
37
Pols, T. W. H.; Bonta, P. I.; de Vries, C. J. M. Curr. Opin. Lipidol.2007, 18, 515-520.
38 (a) Woronicz, J. D.; Calnan, B.; Ngo, V.; Winoto, A. Nature 1994, 367, 277-281. (b) Liu, Z-G.; Smith, S. W.;
McLaughlin, K. A.; Schwartz, L. M.; Osborne, B. A. Nature 1994, 367, 281-284.
39 Liu, J. et. al. Cancer Res. 2010, 70, 3628-3637.

37
effective for inducing apoptosis in cancer cell lines and decreasing tumor growth in vivo (Figure
5c). The authors synthesized compound 15, as well as similar Csn-B analogues, via a standard
Friedel-Crafts acylation. Notably, this protocol only allowed for the synthesis of simple
derivatives of Csn-B due to low functional group tolerance of the acylation. Since there is high
potential for Csn-B analogues to serve as cancer therapeutics, this highlights a need for simple,
functional-group tolerant methods for the synthesis of more complex and potent activators of this
receptor.
2.2 Research Goals
The overall goal of this project is to apply our methodology to the total synthesis of octaketide
natural products, which would not only demonstrate that our protocol is amenable to more
complex salicylaldehyde derivatives, but would also allow efficient entry to such families of
octaketides and derivatives thereof. We plan to focus our attention on nine different natural
products of this type—namely, dothiorelones A, B, C, cytosporones A, B, M, N, phomopsin C,
and an unnamed polyketide isolated from the mangrove endophytic fungus Phomopsis by Lin
and co-workers in 2009 (Scheme 10).34d
Ultimately, we plan to implement this strategy in the
synthesis of more complex Csn-B analogues, which can then be subjected to biological testing.
Scheme 10: Proposed synthesis of octaketides belonging to the dothiorelone, cytosporone, and
phomopsin families.

38
2.3 Results and Discussion
2.3.1 Synthesis of Starting Materials for Key Hydroacylation
We envisioned accessing the requisite aldehydes for the key hydroacylation through a Vilsmeier-
Haack formylation of the appropriate arene precursors. Such a reaction requires the protection of
acidic functional groups, such as carboxylic acids and phenols. Thus, our synthesis commenced
with the Fischer esterification of phenyl acetic acid 16 (eq. 8) and methylation of the phenols in
phenyl acetate 18 (eq. 9), furnishing arenes 17 and 19, respectively, in excellent yield (Scheme
11). Formylation of substrates bearing free phenol functionalities proved to be unsuccessful
under a variety of conditions. Using AlCl3, we could achieve either bis-deprotection or selective
mono-deprotection of the aryl methyl ethers by carefully monitoring the reaction time. Aryl
ethers ortho to aldehyde functionalities are more susceptible to cleavage due to a chelation
effect.40
Attempts at using BBr3 as the demethylating reagent proved to be unsuccessful, as
products resulting from ester cleavage were observed. Overall, the synthesis of aldehydes 21a-c
was achieved in a total of four steps (or three steps from commercially available starting
materials 17 and 19) with good to excellent yields throughout the synthetic route.
40
(a) Lansinger, J. M.; Ronald, R. C. Synth. Commun. 1979, 9, 341 – 349. (b) Du, Z.-T.; Lu, J.; Yu, H.-R.; Xu, Y.;
Li, A.-P. J. Chem. Res. 2010, 34, 222-227.
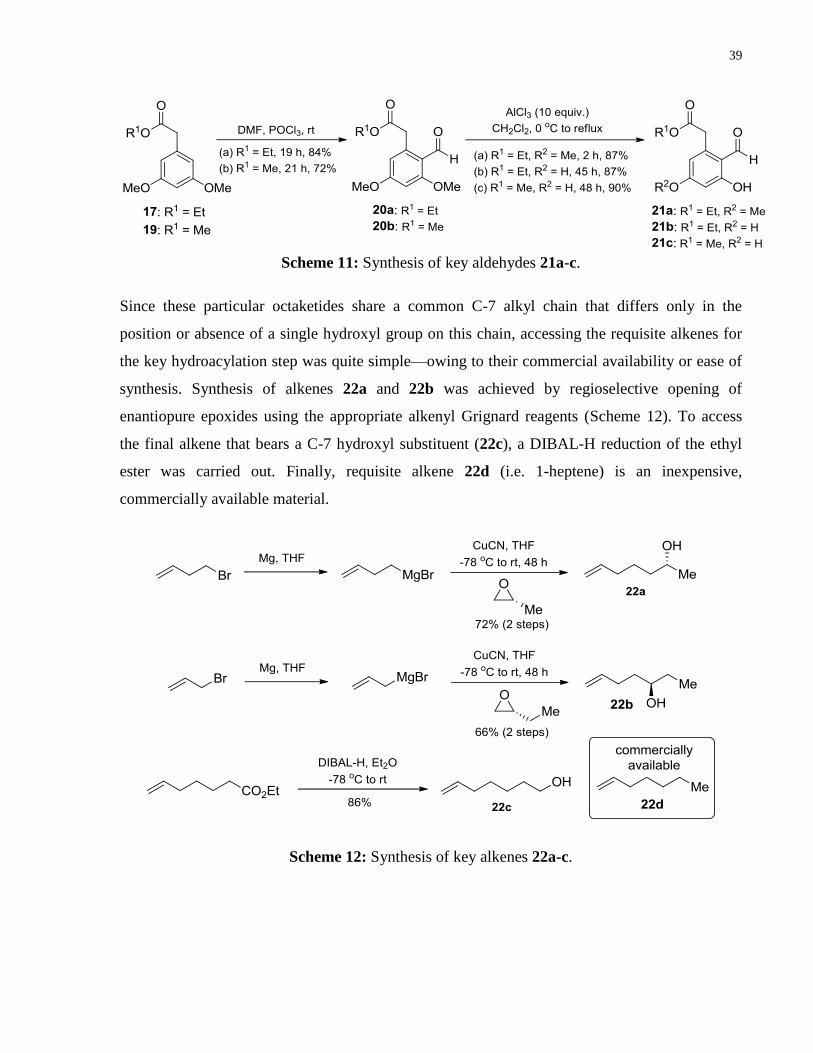
39
Scheme 11: Synthesis of key aldehydes 21a-c.
Since these particular octaketides share a common C-7 alkyl chain that differs only in the
position or absence of a single hydroxyl group on this chain, accessing the requisite alkenes for
the key hydroacylation step was quite simple—owing to their commercial availability or ease of
synthesis. Synthesis of alkenes 22a and 22b was achieved by regioselective opening of
enantiopure epoxides using the appropriate alkenyl Grignard reagents (Scheme 12). To access
the final alkene that bears a C-7 hydroxyl substituent (22c), a DIBAL-H reduction of the ethyl
ester was carried out. Finally, requisite alkene 22d (i.e. 1-heptene) is an inexpensive,
commercially available material.
Scheme 12: Synthesis of key alkenes 22a-c.

40
2.3.2 Testing Known Hydroacylation Methods
Initially we wondered whether two known intermolecular hydroacylation methods developed by
Suemune and Jun could potentially furnish our targeted natural product scaffolds. In Suemune’s
report, the hydroacylation of electron-neutral olefins with salicylaldehyde was promoted by the
addition of acetonitrile to the reaction. However, this transformation required 40 mol% of
Wilkinson’s catalyst and six equivalents of alkene to permit good reactivity and full conversion.
We wanted to test the reactivity of our natural product precursors to Suemune’s conditions, but
with a Rh loading comparable to our system (10 mol%). When we subjected aldehyde 21a and 1-
heptene (22d) to these particular conditions, modest conversion was observed, even after
prolonged reaction times (Scheme 13). When we implemented Jun’s conditions, we observed a
62% conversion of the starting material by 1H NMR, but only trace amounts of the desired
product. Evidently, Jun’s method is not compatible with complex aldehyde substrates—
particularly those bearing free phenol substituents. The results from this study demonstrate that
there is a need for more general and functional group compatible strategies for the
hydroacylation of difficult substrates that maintains low catalyst loadings and employs mild
reaction conditions.
Scheme 13: Testing Suemune’s and Jun’s conditions for synthesis of phomopsin C.
2.3.3 Solvent Screen for Hydroacylation of Natural Product Substrates
We were initially concerned about the solubility of our aldehyde substrates in DCE (the optimal
solvent for our hydroacylation method)—particular those bearing two phenolic groups. Since we

41
observed moderate to good reactivity in both THF and Me-THF for 2,4-dihydroxybenzaldehyde
(1g) (refer to section 1.3.2. Investigation of Aldehyde Scope, Table 2), natural product precursors
21b and 22d were subjected to a modified set of reaction conditions41
employing both THF and
Me-THF as solvent. The reaction progress was monitored by 1H NMR spectroscopy, and four
data points were collected at 18, 51, 71, and 91 hours. The results are summarized and plotted in
Table 6 and Figure 6, respectively. Although both Me-THF and THF were excellent at fully
solubilizing substrate 21b, sluggish reactivity was observed in both cases, with the highest
conversion to product 23bd being 36% in Me-THF after 91 hours (Table 6, entry 4). In both
solvents, significant amounts of decarbonylation observed, which is to be expected for highly
substituted, electron-rich aldehydes. No significant conversion is observed in Me-THF beyond
the 51 hour mark, and in THF, the reaction stalled at 13% conversion before the first data point
was even obtained. At this point, we decided to test the reactivity for this hydroacylation in DCE
with the hope that 21b will solubilize slowly over the course of the reaction. Much to our delight,
complete consumption of aldehyde 21b was observed at 51 hours, with 84% representing
hydroacylation product 23bd and the remaining 16% representing decarbonylation product 21b’
(entry 10). These results suggest that the hydroacylation pathway is more favourable than the
decarbonylation pathway in DCE than it is in Me-THF and THF.
Table 6: Solvent screen and reaction monitoring for hydroacylation of 1-heptene (22d) with
aldehyde 21b.
Solvent Entry Time (h) 23bd (%) 21b (%) 21b’ (%)
Me-THF
1 18 24 74 2
2 51 35 45 10
3 71 36 42 22
4 91 36 39 24
THF 5 18 13 60 27
41
We opted to increase the [Rh] loading for the natural product syntheses as these bulkier, electron-rich aldehydes
proved to be quite unreactive when loadings of 5 mol % monomeric Rh were utilized.
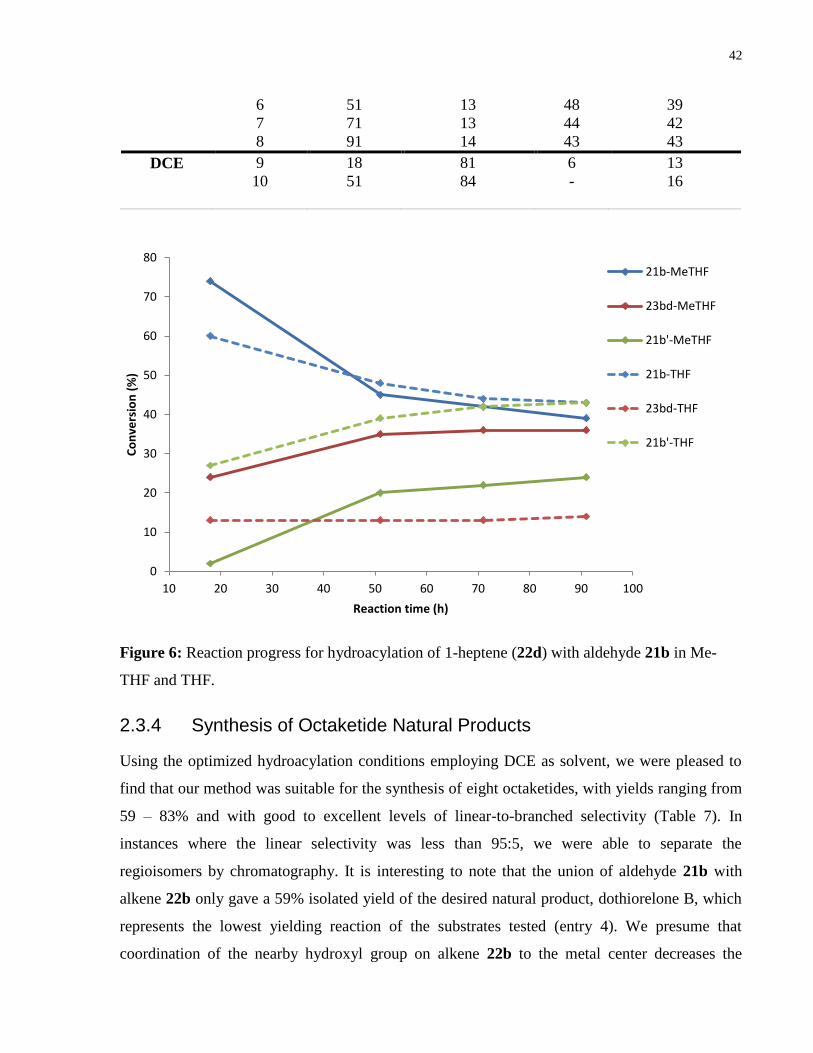
42
6 51 13 48 39
7 71 13 44 42
8 91 14 43 43
DCE 9 18 81 6 13
10 51 84 - 16
Figure 6: Reaction progress for hydroacylation of 1-heptene (22d) with aldehyde 21b in Me-
THF and THF.
2.3.4 Synthesis of Octaketide Natural Products
Using the optimized hydroacylation conditions employing DCE as solvent, we were pleased to
find that our method was suitable for the synthesis of eight octaketides, with yields ranging from
59 – 83% and with good to excellent levels of linear-to-branched selectivity (Table 7). In
instances where the linear selectivity was less than 95:5, we were able to separate the
regioisomers by chromatography. It is interesting to note that the union of aldehyde 21b with
alkene 22b only gave a 59% isolated yield of the desired natural product, dothiorelone B, which
represents the lowest yielding reaction of the substrates tested (entry 4). We presume that
coordination of the nearby hydroxyl group on alkene 22b to the metal center decreases the
0
10
20
30
40
50
60
70
80
10 20 30 40 50 60 70 80 90 100
Co
nve
rsio
n (
%)
Reaction time (h)
21b-MeTHF
23bd-MeTHF
21b'-MeTHF
21b-THF
23bd-THF
21b'-THF

43
reactivity for this coupling and promotes catalyst decomposition; however we are unsure of the
exact reasoning for this. Interestingly, when we subjected 21b and alkene 22b to biphasic
conditions, employing a 1:1 mixture of DCE to H2O, an excellent isolated yield of 86% was
achieved. Finally, a ninth natural product, cytosporone A, was synthesized through
saponification of the ethyl ester in cytosporone B (eq. 10).
Table 7: Synthesis of octaketide natural products.
Entry Natural Product Structurea
[α]D lit. [α]D
found
Yieldb
(%)
L:B
ratioc
1 Octaketide 23aa R1
= Et, R2 = Me, R
4 = OH Not
reported
-3.3 (R) 79 90:10
2 Phomopsin C
(23ad)
R1
= Et, R2 = Me - - 78 >95:5
3 Dothiorelone Ad
(23ba)
R1
= Et, R2 = H, R
4 = OH +3.4 (S) +3.5 (R) 79 83:17
4 Dothiorelone Be
(23bb)
R1
= Et, R2 = H, R
3 = OH Not
reported
-4.0 (R) 59, 86f >95:5
5 Dothiorelone C
(23bc)
R1
= Et, R5 = OH - - 83 87:13
6 Cytosporone B
(23bd)
R1
= Et - - 78 93:7
7 Cytosporone M
(23ca)
R1
= Me, R4 = OH -10 -4.0 (R) 73 89:11
8 Cytosporone N
(23cd)
R1
= Me - - 73 94:6
a Non-specified residues are H.
b Combined isolated yield. Reaction times between 18 – 72 h (Refer to Experimental
for details) c Regioisomeric ratio determined by integration of crude
1H NMR spectrum.
d An [α]D.of +3.4 was
reported for synthetic (S)-dothiorelone A.33
e 7.5 mol% (R)-SIPHOS-PE used.
f Biphasic conditions employed, 1:1
mixture of DCE:H2O
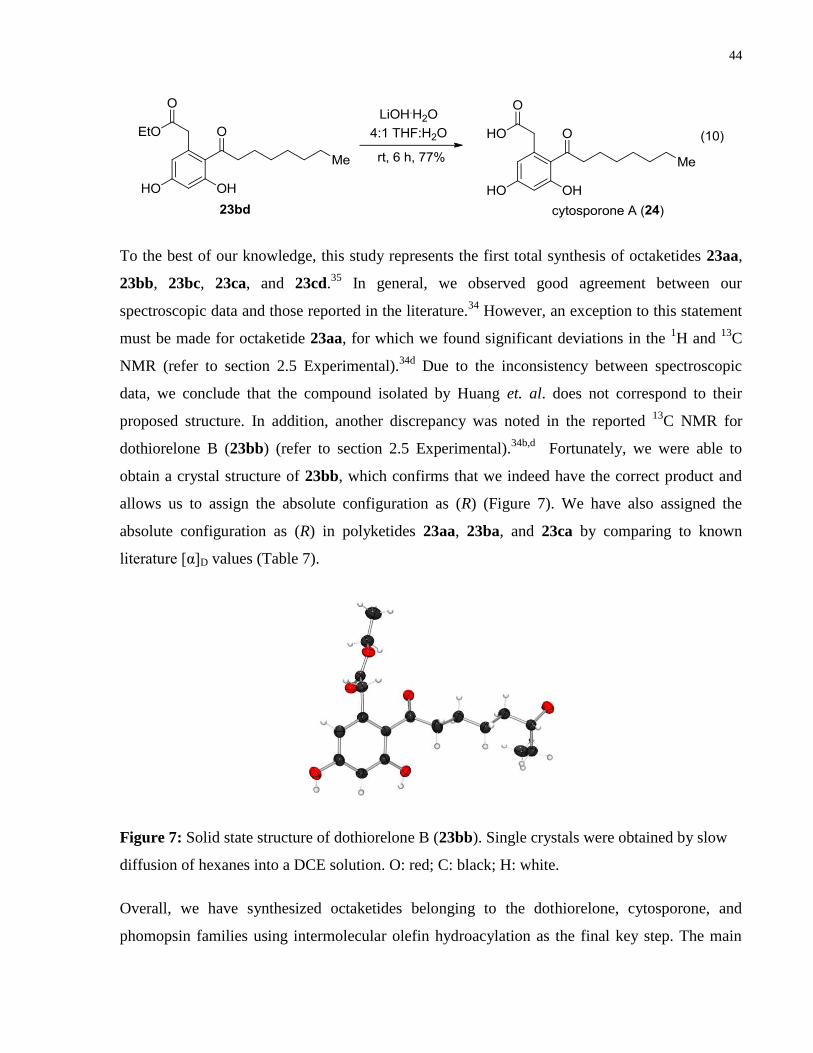
44
To the best of our knowledge, this study represents the first total synthesis of octaketides 23aa,
23bb, 23bc, 23ca, and 23cd.35
In general, we observed good agreement between our
spectroscopic data and those reported in the literature.34
However, an exception to this statement
must be made for octaketide 23aa, for which we found significant deviations in the 1H and
13C
NMR (refer to section 2.5 Experimental).34d
Due to the inconsistency between spectroscopic
data, we conclude that the compound isolated by Huang et. al. does not correspond to their
proposed structure. In addition, another discrepancy was noted in the reported 13
C NMR for
dothiorelone B (23bb) (refer to section 2.5 Experimental).34b,d
Fortunately, we were able to
obtain a crystal structure of 23bb, which confirms that we indeed have the correct product and
allows us to assign the absolute configuration as (R) (Figure 7). We have also assigned the
absolute configuration as (R) in polyketides 23aa, 23ba, and 23ca by comparing to known
literature [α]D values (Table 7).
Figure 7: Solid state structure of dothiorelone B (23bb). Single crystals were obtained by slow
diffusion of hexanes into a DCE solution. O: red; C: black; H: white.
Overall, we have synthesized octaketides belonging to the dothiorelone, cytosporone, and
phomopsin families using intermolecular olefin hydroacylation as the final key step. The main

45
advantages of this methodology are the commercial availability and/or ease of access to the
requisite aldehyde and alkene building blocks, and the functional group tolerance of the reaction.
2.3.5 Design and Synthesis of Cytosporone B Analogues
Using our hydroacylation method, we saw the opportunity to synthesize cytosporone B
analogues with the goal of discovering a more potent small-molecular activator for Nur77. For
these particular Csn-B analogues, we wanted to keep the substitution around the aromatic ring
the same, while we varied the functionality on the acyl chain, since our protocol tolerates a wide
range of functional groups on the olefin component. We hope that these derivatives can bind to
the LBD of Nur77 more tightly through additional non-covalent interactions, such as H-bonding
and/or hydrophobic contacts. Taking into account the pharmacophore described by Liu et. al. and
the crystal structure data of the LBD of Nur77 (Figure 8), we synthesized three Csn-B analogues,
which are outlined in Table 8.
Figure 8: Ligand binding pocket of Nur77 obtained from the PDB (code 2QW4).
The importance of C–F bonds in drug design and lead optimization is evidenced by the relatively
high percentage of currently administered drugs that incorporate this functional motif.42
Due to
the metabolic stability of C–F bonds and their ability to form weak hydrogen bonds with H–X
42
(a) Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881-1886. (b) Purser, S.; Moore, P. R.; Swallowb, S.;
Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320-330.

46
(where X = N, O, or S) and H–Cα groups on amino acid residues, we were inspired to synthesize
a Csn-B analogue that possesses a perfluorinated acyl chain. Coupling of aldehyde 21b to
perfluro 1-octene 22e, which is a commercially available material, afforded 23be in modest
yield, but with excellent linear-to-branched selectivity (Table 8, entry 1). We believe that this
fluoroalkyl chain has the potential to bind more strongly to the LBD of Nur77 through a specific
interaction with Arg-123 in the binding pocket. Crystal structure evidence from the Protein Data
Bank suggests that Arg resides are fluorophilic in nature, whereby the C–F bonds tend to point
directly toward the guanidinium moiety of Arg residues.42a
Our rationale for the synthesis of derivatives 23bf and 23bg, both of which possess a phenyl
group on the acyl chain, stems from the existence and importance of cation-π interactions in
structural biology.43
We hypothesize that a favourable interaction between an aromatic ring and
Arg-123 is feasible, and that this interaction could lead to higher binding affinity. Although the
linear-to-branched selectivity was less than ideal for alkenes 22f and 22g, we were able to fully
separate the regioisomers by chromatography. We are also interested in synthesizing a derivative
that possesses a ketone functionality on the acyl chain (23bu), since this carbonyl group has the
ability to act as a hydrogen-bond acceptor. Due to the presence of amino acid residues that can
act as hydrogen-bond donors (i.e. Pro-46, Ser-47, Thr-48) in the LBD of Nur77, we suggest that
this positive binding interaction is possible.
Table 8: Synthesis of cytosporone B analogues.
Entry Alkene Reaction
time (h) Product
Yielda
(%) L:B
b
1 (22e) 112 23be 57 >95:5
2
(22f)
55 23bf 69 83:17
43
Gallivan, J. P.; Dougherty, D. A. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 9459-9464.

47
3
(22g)
55 23bg 39 83:17
4
(22u)
- 23bu - -
a Isolated yield of the linear regioisomer.
bRegioisomeric ratio determined by integration of crude
1H NMR spectrum
2.4 Conclusions and Future Work
We have applied our linear-selective intermolecular hydroacylation reaction to the total synthesis
of nine natural products belonging to the dothiorelone, cytosporone, and phomopsin families.
Yields between 73 and 86% and linear-to-branched selectivities between 83:17 and >95:5 were
observed for the key step. We have also applied this methodology to the synthesis of cytosporone
B analogues for the purpose of biological testing. We hope to carry out docking studies in the
near future for the design of more rational Csn-B derivatives.
2.5 Experimental
2.5.1 General Considerations
Commercial reagents were purchased from Sigma Aldrich, Strem or Alfa Aesar and used without
further purification. Reactions were monitored using thin-layer chromatography (TLC) on EMD
Silica Gel 60 F254 plates. Visualization of the developed plates was performed under UV light
(254 nm) or KMnO4 stain. Organic solutions were concentrated under reduced pressure on a
Büchi rotary evaporator. 1H,
31P and
13C NMR spectra were recorded on a Varian Mercury 400,
VRX-S (Unity) 400, or Bruker AV-III 400 spectrometer. 1H NMR spectra were internally
referenced to the residual solvent signal or TMS. 13
C NMR spectra were internally referenced to
the residual solvent signal. Data for 1H NMR are reported as follows: chemical shift (δ ppm),
multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, b = broad), coupling
constant (Hz), integration. Data for 13
C NMR are reported in terms of chemical shift (δ ppm).
High resolution mass spectra (HRMS) were obtained on a micromass 70S-250 spectrometer (EI)
or an ABI/Sciex QStar Mass Spectrometer (ESI). Infrared (IR) spectra were obtained on a
Perkin-Elmer Spectrum 1000 FT-IR Systems and are reported in terms of frequency of

48
absorption (cm-1
). Melting point ranges were determined on a Fisher-Johns Melting Point
Apparatus. Column chromatography was performed with Silicycle Silia-P Flash Silica Gel, using
either glass columns or a Biotage SP-1 system. Solvents used in hydroacylations were degassed
by three freeze-pump-thaw cycles. Chiral ligands were purchased from Strem.
2.5.2 General Procedures for the Total Synthesis of Natural Products via Linear-Selective Hydroacylation
In a nitrogen-filled glovebox, K3PO4 base (7.5 mol%), aldehyde (1.0 equiv.), alkene (3.0 equiv.),
[Rh(COD)Cl]2 precatalyst (5.0 mol%) and (R)-SIPHOS-PE ligand (5.0 mol%) were combined in
a one-dram vial in the specified solvent (0.4M). The vial was charged with a stir bar, sealed with
a Teflon-lined cap and the mixture was stirred at 70 °C for the indicated period of time. The
crude reaction mixture was passed through a plug of silica, washed with acetone or ethyl acetate
and concentrated. The crude reaction mixtures were purified by silica gel flash chromatography
or preparative TLC to determine the isolated yield.
2.5.3 Characterization of Compounds
ethyl 2-(2-formyl-3,5-dimethoxyphenyl)acetate (20a). To a solution of ethyl 2-(3,5-
dimethoxyphenyl)acetate (1.490 g, 6.5 mmol) in DMF (2.5 mL) was added freshly distilled
POCl3 (0.72 mL, 7.8 mmol). The reaction mixture was stirred at room temperature for 19 h
before diluting with DCM and quenching with H2O (10 mL) at 0 °C. The aqueous layer was
extracted with EtOAc (3 x 20 mL), and the combined organics were washed with brine, dried
with MgSO4, and concentrated under reduced pressure. Purification by flash chromatography
(7:3 hexanes/acetone) afforded the title compound as a white solid (1.381 g, 84%). 1H NMR (400
MHz, CDCl3) δ 10.43 (s, 1H), 6.43 (d, J = 2.3 Hz, 1H), 6.33 (d, J = 2.2 Hz, 1H), 4.18 (q, J = 7.1

49
Hz, 2H), 3.93 (s, 2H), 3.90 (s, 3H), 3.87 (s, 3H), 1.28 (t, J = 7.1 Hz, 3H).13
C {1H} NMR (101
MHz, CDCl3) δ 190.00, 171.12, 165.36, 164.77, 139.36, 117.15, 109.96, 97.14, 60.69, 55.85,
55.53, 40.74, 14.25.
methyl 2-(2-formyl-3,5-dimethoxyphenyl)acetate (20b). To a solution of methyl 2-(3,5-
dimethoxyphenyl)acetate (0.485 g, 2.3 mmol) in DMF (0.89 mL) was added freshly distilled
POCl3 (0.26 mL, 2.8 mmol). The reaction mixture was heated to 55 °C for 48 h before diluting
with ethyl acetate (5.0 mL) and quenching with ice-cold H2O (5.0 mL). The aqueous phase was
extracted with ethyl acetate (3 x 5.0 mL), and the combined organics were washed with brine,
dried with MgSO4, and concentrated under reduced pressure. Purification by flash
chromatography (7:3 hexanes/acetone) afforded the title compound as a white solid (375 mg,
72%). 1H NMR (400 MHz, CDCl3) δ 10.42 (s, 1H), 6.43 (d, J = 2.2 Hz, 1H), 6.33 (d, J = 2.1 Hz,
1H), 3.95 (s, 2H), 3.90 (s, 3H), 3.87 (s, 3H), 3.71 (s, 3H). 13
C {1H} NMR (101 MHz, CDCl3) δ
190.20, 171.69, 165.52, 164.93, 139.34, 117.27, 110.10, 97.34, 55.99, 55.68, 52.04, 40.67.
ethyl 2-(2-formyl-3-hydroxy-5-methoxyphenyl)acetate (21a). To a suspension of anhydrous
AlCl3 (1.34 g, 10.0 mmol) in DCM (6.0 mL) was added a solution of 20a (0.253 g, 1.00 mmol)
in DCM (4.0 mL) at 0 °C. The reaction mixture was heated to 40 °C for 1 hour, diluted with
DCM, and poured into ice-cold water (10 mL). The aqueous phase was extracted with ethyl
acetate (3 x 10 mL), and combined organics were washed with brine, dried using MgSO4, and
concentrated under reduced pressure. Purification by column chromatography (8:2 hexanes/ethyl
acetate) furnished the title compound as a white solid (0.206 g, 87%). M.p. 74-76 °C. 1H NMR
(400 MHz, CDCl3) δ 12.50 (s, 1H), 10.06 (s, 1H), 6.36 (s, 2H), 4.17 (q, J = 7.1 Hz, 2H), 3.84 (s,
3H), 3.82 (s, 2H), 1.25 (t, J = 7.1 Hz, 3H). 13
C {1H} NMR (101 MHz, CDCl3) δ 192.92, 170.33,

50
166.85, 166.62, 139.52, 113.08, 111.67, 100.14, 61.70, 55.81, 38.06, 14.26. HRMS (ESI+):
239.0915 [M+H]+ (calc’d 239.0920 for C12H15O5).
ethyl 2-(2-formyl-3,5-dihydroxyphenyl)acetate (21b). To a suspension of anhydrous AlCl3
(5.280 g, 39.6 mmol) in DCM (15 mL) was added a solution of 20a (0.968 g, 3.84 mmol) in
DCM (10 mL) at 0 °C. The reaction vessel was heated to 40 °C for 45 hours, diluted with DCM,
and poured into ice-cold water (25 mL). The aqueous phase was extracted with ethyl acetate (3 x
25 mL), and combined organics were washed with brine, dried using MgSO4, and concentrated
under reduced pressure. Purification by flash chromatography (7:3 hexanes/ethyl acetate)
afforded the title compound as a white solid (0.751 g, 87%). M.p. 110-112 °C. 1H NMR (400
MHz, CD3CN) δ 12.31 (bs, 1H), 9.96 (s, 1H), 8.02 (bs, 1H), 6.32 – 6.26 (m, 2H), 4.12 (q, J = 7.1
Hz, 2H), 3.88 (s, 2H), 1.21 (t, J = 7.1 Hz, 3H); 13
C {1H} NMR (101 MHz, CD3CN) δ 194.80,
171.56, 167.02, 165.49, 142.21, 113.76, 112.29, 102.73, 62.06, 38.07, 14.39. HRMS (ESI+):
225.0767 [M+H]+ (calc’d 225.0763 for C11H13O5).
methyl 2-(2-formyl-3,5-dihydroxyphenyl)acetate (21c). To a suspension of anhydrous AlCl3
(1.019 g, 7.6 mmol) in DCM (15 mL) was added a solution of 20b (0.182 g, in DCM (8.0 mL) at
0 °C. The reaction vessel was heated to 40 °C for 48 hours, was diluted with DCM, and poured
into ice-cold water (15 mL). The aqueous phase was extracted with ethyl acetate (3 x 15 mL),
and the combined organics were washed with brine, dried with MgSO4, and concentrated under
reduced pressure. Purification by flash chromatography (7:3 hexanes/acetone) afforded the title
compound as a white solid (149 mg, 90%). M.p. 118-120 °C. 1H NMR (400 MHz, Acetone-d6) δ
12.43 (bs, 1H), 10.07 (s, 1H), 9.70 (bs, 1H), 6.41 (d, J = 2.3 Hz, 1H), 6.28 (d, J = 2.3 Hz, 1H),
4.00 (s, 2H), 3.67 (s, 3H). 13
C {1H} NMR (101 MHz, acetone) δ 382.56, 370.98, 348.09, 342.30,

51
318.43, 290.10, 288.84, 279.08, 228.90, 214.12. HRMS (ESI+): 211.0604 [M+H]
+ (calc’d
211.0607 for C10H12O5 ).
(R)-hept-6-en-2-ol (22a). Following a literature procedure,44
a solution of 3-butenylmagnesium
bromide [prepared from 4-bromo-1-butene (4.05 g, 30 mmol) and Mg (1.29 g, 53 mmol)] in THF
(50 mL) was added dropwise to a suspension of CuCN (121 mg, 1.4 mmol) and (R)-propylene
oxide (871 mg, 15 mmol) in THF (20 mL) at -78 °C. The reaction mixture was warmed to room
temperature before quenching with saturated NH4Cl (30 mL). The aqueous layer was extracted
with Et2O (3 x 30 mL). The combined organics were washed with brine, dried with MgSO4, and
concentrated under reduced pressure. Purification by flash chromatography (7:3 pentanes/Et2O)
afforded the title compound as a colorless oil (1.24 g, 72% over two steps). All spectroscopic
data correspond to those reported in the literature.45
1H NMR (400 MHz, CDCl3) δ 5.86 – 5.76
(m, 1H), 5.04 – 4.98 (m, 1H), 4.97 – 4.94 (m, 1H), 3.86 – 3.75 (m, 1H), 2.10- 2.05 (m, 2H), 1.56
– 1.37 (m, 4H), 1.31 (d, J = 3.5 Hz, 1H), 1.19 (d, J = 6.2 Hz, 3H). 98% ee confirmed by chiral
GC-FID (Rt = 6.5 min, 80 °C (isothermal), Cyclodex B column, 30 m x 0.25 mm).
(R)-hept-6-en-3-ol (22b). Following a literature procedure,44
a solution of allylmagnesium
bromide [prepared from allyl bromide (3.00 g, 25 mmol) and Mg (1.09 g, 45 mmol)] in THF (40
mL) was added dropwise to a suspension of CuCN (101 mg, 1.1 mmol) and (R)-1,2-epoxybutane
(901 mg, 12.5 mmol) in THF (15 mL) at -78 °C. The reaction mixture was warmed to room
temperature before quenching with saturated NH4Cl (25 mL). The aqueous layer was extracted
with Et2O (3 x 25 mL). The combined organics were washed with brine, dried with MgSO4, and
concentrated under reduced pressure. Purification by flash chromatography (75:25
pentanes/Et2O) afforded the title compound as a light yellow oil (1.24 g, 70% over two steps). 1H
44
Jastrzebska, I.; Scaglione, J. B.; DeKoster, G. T.; Rath, N. P.; Covey, D. F. J. Org. Chem. 2007, 72, 4837-4843.
45 Takahata , H.; Yotsui, Y.; Momose, T. Tetrahedron 1998, 54, 13505-13516.

52
NMR (400 MHz, CDCl3) δ 5.90 – 5.80 (m, 1H), 5.08 – 5.02 (m, 1H), 5.01 – 4.93 (m, 1H), 3.59 –
3.52 (m, 1H), 2.32 – 2.04 (m, 2H), 1.65 – 1.39 (m, 5H), 1.36 (d, J = 4.7 Hz, 1H), 0.95 (t, J = 7.5
Hz, 3H). (c = 0.032, EtOH) [lit.
(c = 1.7, EtOH) for (S)
enantiomer].46
hept-6-en-1-ol (22c). Following a literature procedure,47
to a solution of ethyl hept-6-enoate
(1.50 g, 9.6 mmol) in Et2O (120 mL) was added DIBAL-H (21 mmol from a 1.0 M solution in
hexanes) at -78 °C. The reaction mixture was stirred at room temperature for 3 hours before
quenching with saturated NH4Cl (30 mL) at -10 °C. The viscous white solution was stirred
vigorously at room temperature for 1 hour, filtered through a pad of celite, and washed with
Et2O. The filtrate was dried over MgSO4, concentrated under reduced pressure, and purified by
flash chromatography (7:3 pentanes/Et2O) to afford the title compound as a colorless oil (944
mg, 86%). All spectroscopic data correspond to those reported in the literature.47
1H NMR (400
MHz, CDCl3) δ 5.86 – 5.76 (m, 1H), 5.03 – 4.97 (m, 1H), 4.97 – 4.91 (m, 1H), 3.67 – 3.63 (m,
2H), 2.09 – 2.04 (m, 2H), 1.64 – 1.51 (m, 2H), 1.49 – 1.32 (m, 4H).
(R)-ethyl 2-(3-hydroxy-2-(7-hydroxyoctanoyl)-5-methoxyphenyl)acetate (23aa). Prepared
according to the general procedure 2.5.2 (150 μmol aldehyde solvent: DCE reaction time 19 h).
Purification via preparative TLC (98:2 chloroform/isopropanol) afforded the title compound as a
colorless solid (41.7 mg, 79%, lin/br 90:10). M.p. 120-122 °C. Spectroscopic data correspond to
those reported in the literature.51b
1H NMR (400 MHz, CDCl3) δ 12.28 (s, 1H), 6.36 (d, J = 2.6
Hz, 1H), 6.32 (d, J = 2.6 Hz, 1H), 4.17 (q, J = 7.1 Hz, 2H), 3.83 (s, 2H), 3.79 (s, 3H), 3.80 – 3.73
46
Bolm, C.; Schlingloff, G.; Harms, K. Chem. Ber. 1992, 125, 1191-1203. 47
Zaed, A.M.; Swift, M. D.; Sutherland, A. Org. Biomol. Chem. 2009, 7, 2678-2680.

53
(m, 1H), 2.84 (t, J = 8.0 Hz, 2H), 1.77 – 1.65 (m, 2H), 1.48 – 1.38 (m, 3H), 1.38 – 1.30 (m, 3H),
1.25 (t, J = 7.1 Hz, 3H), 1.17 (d, J = 6.2 Hz, 3H).; 13
C {1H} NMR (101 MHz, CDCl3) δ 206.54,
171.03, 164.95, 163.70, 136.34, 116.36, 112.54, 100.59, 68.14, 61.49, 55.52, 43.19, 42.02, 39.18,
29.26, 25.61, 24.94, 23.63, 14.28. HRMS (ESI+): 353.1961 [M+H]
+ (calc’d 353.1964 for
C19H29O6). (c = 0.02, MeOH) (no literature reference for [α]D value)
Comparison of NMR Data:
1H NMR (400 MHz,
CDCl3) synthesized
1H NMR (500 MHz,
CDCl3) isolated51b
13C NMR (101 MHz,
CDCl3) synthesized
13C NMR (125 MHz,
CDCl3) isolated51b
12.28 (bs, 1H) 11.39 (s) 206.54 206.8
6.36 (d, J = 2.6, 1H) 6.38 (d, J = 2.0, 1H) 170.03 171.2
6.32 (d, J = 2.6, 1H)
4.17 (q, J = 7.1, 2H)
6.36 (d, J = 2.0, 1H)
4.11 (q, J = 7.2, 2H)
165.95
163.70
161.2
158.7
3.83 (s, 2H) 3.60 (s, 2H) 136.34 134.4
3.79 (s, 3H) 3.80 (s, 3H) 116.36 124.1
3.80 – 3.73 (m, 1H) 3.77 (m, 1H) 112.54 107.7
2.84 (t, J = 8.0, 2H) 2.82 (t, J = 7.2, 2H) 100.59 97.5
1.77 – 1.65 (m, 2H) 1.64 (m, 2H) 68.14 66.8
1.48 – 1.38 (m, 3H) 1.44 (m, 2H) 61.49 60.8
1.38 – 1.30 (m, 3H), 1.42 (m, 2H)
1.33 (m, 2H)
55.52
43.19
55.3
43.9
1.25 (t, J = 7.1, 3H), 1.24 (t, J = 7.2, 3H) 42.02 39.1
1.17 (d, J = 6.2, 3H) 1.16 (d, J = 6.4, 3H) 39.18
29.26
38.8
29.2
25.61 25.5
24.94 23.9
23.63 23.4
14.28 14.1
ethyl 2-(3-hydroxy-5-methoxy-2-octanoylphenyl)acetate, phomopsin C (23ad). Prepared
according to the general procedure 2.5.2 (44 μmol aldehyde; solvent: DCE; reaction time 72 h).

54
Purification via preparative TLC (85:15 hexanes/ethyl acetate) afforded the title compound as a
colorless solid (11.5 mg, 78%, lin/br >95:5). M.p. 62-64 °C. All spectroscopic data corresponded
to those reported in the literature.48
1H NMR (400 MHz, CDCl3) δ 12.59 (s, 1H), 6.39 – 6.32 (m
2H), 4.18 (q, J = 7.1 Hz, 2H), 3.86 (s, 2H), 3.81 (s, 3H), 2.83 (t, J = 8 Hz, 2H), 1.77 – 1.63 (m,
2H), 1.31 – 1.20 (m, 13H), 0.88 (t, J = 6.8 Hz, 3H); 13
C {1H} NMR (101 MHz, CDCl3) δ 206.75,
170.90, 165.50, 163.85, 136.44, 116.04, 112.73, 100.59, 61.50, 55.56, 43.34, 42.19, 31.82, 29.34,
29.22, 25.15, 22.74, 14.30, 14.20. HRMS (ESI+): 337.2017 [M+H]
+ (calc’d 337.2015 for
C19H29O5). Note: Multiplet between 1.31 – 1.20 ppm should integrate to 11H; discrepancy likely
due to a small amount of the branched regioisomer.
Comparison of NMR Data:
1H NMR (400 MHz,
CDCl3) synthesized
1H NMR (500 MHz,
CDCl3) isolated48
13C NMR (101 MHz,
CDCl3) synthesized
13C NMR (125 MHz,
CDCl3) isolated48
12.59 (s, 1H) 12.53 (s, 1H) 206.75 206.6
6.39 – 6.32 (m, 2 H) 6.38 (d, J = 2.0, 1H) 170.90 170.7
4.18 (q, J = 7.1, 2H)
6.33 (d, J = 2.0, 1H)
4.18 (q, J = 7.2, 2H)
165.50
163.85
165.2
163.6
3.86 (s, 2H)
3.81 (s, 3H)
3.86 (s, 2H)
3.81 (s, 3H)
136.44
116.04
136.2
115.9
2.83 (t, J = 8.0, 2H) 2.83 (t, J = 7.2, 2H) 112.73 112.5
1.77 – 1.66 (m, 2H) 1.70 (m, 2H) 100.59 100.4
1.31 – 1.20 (m, 13H) 1.26 – 1.24 (m, 11H) 61.50 61.3
0.88 (t, J = 6.8, 3H) 0.88 (t, J = 7.2, 3H) 55.56
43.34
55.4
43.2
42.19
31.82
42.0
31.6
29.34
29.22
29.0
29.1
48
Huang, Z.; Cai, X.; Shao, C.; She, Z.; Xia, X.; Chen, Y.; Yang, J.; Zhou, S.; Lin, Y. Phytochemistry 2008, 69,
1604-1608.

55
(R)-ethyl 2-(3,5-dihydroxy-2-(7-hydroxyoctanoyl)phenyl)acetate, (-)-dothiorelone A (23ba).
Prepared according to the general procedure 2.5.2 (150 μmol aldehyde; solvent: DCE; reaction
time 18 h). Purification via preparative TLC (95:5 chloroform/isopropanol) afforded the title
compound as a colorless oil (40.2 mg, 79%, lin/br 83:17). Spectroscopic data correspond to those
reported in the literature.49
1H NMR (400 MHz, CDCl3) δ 11.26 (bs, 1H), 7.46 (bs, 1H), 6.23 (s,
2H), 4.18 (q, J = 7.1 Hz, 2H), 3.86 – 3.77 (m, 1H), 3.75 (s, 2H), 2.08 (bs, 1H), 2.84 (t, J = 8.0
Hz, 2H), 1.79 – 1.60 (m, 2H), 1.52 – 1.29 (m, 6H), 1.27 (t, J = 7.1 Hz, 3H), 1.18 (d, J = 6.2 Hz,
3H); 1H NMR (400 MHz, Acetone-d6) δ 6.36 (d, J = 2.3 Hz, 1H), 6.32 (d, J = 2.3 Hz, 1H), 4.09
(q, J = 7.1 Hz, 2H), 3.74 – 3.68 (m, 1H), 3.67 (s, 2H), 2.91 (t, J = 8.0 Hz, 2H), 1.73 – 1.57 (m,
2H), 1.43 – 1.23 (m, 8H), 1.21 (t, J = 7.1 Hz, 3H), 1.11 (d, J = 6.1 Hz, 3H); 13
C {1H} NMR (126
MHz, CDCl3) δ 206.79, 171.59, 163.98, 160.47, 136.74, 116.99, 112.69, 103.29, 68.39, 61.72,
43.28, 41.80, 39.03, 29.15, 25.47, 24.88, 23.61, 14.30. HRMS (ESI+): 339.1820 [M+H]
+ (calc’d
339.1808 for C18H27O6). (c = 0.02, MeOH) [lit. a)
(c = 0.5,
MeOH)50
for (S) enantiomer, synthetic material; b) (c = 0.1, MeOH)
53,
b isolated
material].
Comparison of NMR Data:
1H NMR (400 MHz,
CDCl3) synthesized
1H NMR (500 MHz,
CDCl3) previously
synthesized49
13C NMR (126 MHz,
CDCl3) synthesized
13C NMR (101 MHz,
CDCl3) previously
synthesized49
11.26 (bs, 1H) 12.24 (s, 1H) 206.79 206.5
7.46 (bs, 1H) 5.42 (bs, 1H) 171.59 170.7
6.23 (s, 2H)
6.33 (d, J = 2.3, 1H)
6.28 (d, J = 2.3, 1H)
163.98
160.47
164.7
160.0
49
Izuchi, Y.; Koshino, H.; Hongo, Y.; Kanomata, N.; Takahashi, S. Org. Lett. 2011, 13, 3360-3363.
50 Takahata , H.; Yotsui, Y.; Momose, T. Tetrahedron 1998, 54, 13505-13516.

56
4.18 (q, J = 7.1, 2H) 4.19 (q, J = 7.1, 2H) 136.74 136.9
3.86 – 3.77 (m, 1H) 3.79 (m, 1H) 116.99 116.6
3.75 (s, 2H) 3.84 (s, 2H) 112.69 112.3
2.08 (bs, 1H) - 103.29 103.2
2.84 (t, J = 7.3, 2H) 2.85 (t, J = 7.4, 2H) 68.39 68.1
1.79 – 1.60 (m, 2H) 1.73 (m, 2H) 61.72 61.5
1.52 – 1.29 (m, 6H)
1.27 (t, J = 8.0, 3H)
1.44 (m, 3H)
1.34 (m, 3H)
1.27 (t, J = 7.1, 3H)
43.28
41.80
39.03
43.1
41.8
39.0
1.18 (d, J = 6.2, 3H) 1.18 (d, J = 6.0, 3H) 29.15
25.47
29.1
25.5
24.88 24.8
23.61 23.5
14.30 14.2
(R)-ethyl 2-(3,5-dihydroxy-2-(6-hydroxyoctanoyl)phenyl)acetate, dothiorelone B (23bb).
Prepared according to the general procedure 2.5.2 (94 μmol aldehyde 7.5 mol% ligand; solvent:
DCE; reaction time 60 h). Purification via preparative TLC (2:1 hexanes/acetone) afforded the
title compound as a colorless solid (18.8 mg, 59%, lin/br >95:5). Spectroscopic data correspond
to those reported in the literature.51b
M.p. 88-90 °C. 1H NMR (400 MHz, CDCl3) δ 11.60 (bs,
1H), 6.52 (bs, 1H), 6.26 – 6.24 (m, 2H), 4.19 (q, J = 7.1, 2H), 3.79 (s, 2H), 3.56 (bs, 1H), 2.86 (t,
J = 7.2, 2H), 1.83 – 1.59 (m, 3H), 1.59 – 1.33 (m, 6H), 1.26 (t, J = 7.1, 3H), 0.94 (t, J = 7.4,
3H). 13
C {1H} NMR (101 MHz, CDCl3) δ 206.61, 171.51, 163.70, 160.26, 136.58, 116.97,
112.54, 103.16, 73.28, 61.57, 43.22, 41.61, 36.42, 30.18, 25.18, 24.72, 14.15, 9.89. HRMS
51
a) Xu, Q.; Wang, J.; Huang, Y.; Zheng, Z.; Song, S.; Zhang, Y.; Su, W. Acta Oceanol. Sin., 2004, 23, 541-547; b)
Huang, Z.; Guo, Z.; Yang, R.; Yin, X.; Li, X.; Luo, W.; She, Z.; Lin, Y. Chem. Nat. Prod. 2009, 45, 625-628.

57
(ESI+): 339.1811 [M+H]
+ (calc’d. 339.1808 for C18H27O6).
(c = 0.01, MeOH)
[no lit. data]. Note: In the isolation report, no signal was reported for the CH3 group of the ethyl
ester in the 13
C NMR spectrum (14.15 ppm in our spectrum). Because the remaining
characterization data matches, this is likely due to a mistake by the authors.
Comparison of NMR Data:
1H NMR (400 MHz,
CDCl3) synthesized
1H NMR (500 MHz,
CDCl3) isolated51b
13C NMR (101 MHz,
CDCl3) synthesized
13C NMR (125 MHz,
CDCl3) isolated51b
11.60 (bs, 1H) 11.47 (OH, bs) 206.61 206.6 (C)
6.52 (bs, 1H) 8.05 (OH, s) 171.51 171.2 (C)
6.26 – 6.24 (m, 2H) 6.27 (1H, s) 163.70 163.7 (C)
6.26 (1H, s) 160.26 160.5 (C)
4.19 (q, J = 7.1, 2H) 4.19 (2H, q, J = 7.2) 136.58 136.6 (C)
3.79 (s, 2H) 3.78 (2H, s) 116.97 116.9 (C)
3.56 (m, 1H) 3.55 (1H, m) 112.54 112.7 (CH)
2.86 (t, J = 7.2, 2H) 2.86 (2H, t, J = 7.2) 103.16 103.2 (CH)
1.83 – 1.59 (m, 3H) 1.46 (2H, m) 73.28 73.1 (CH)
1.59 – 1.33 (m, 6H) 1.43 (2H, m) 61.57 61.7 (CH2)
1.26 (2H, m) 43.22 43.3 (CH2)
1.25 (2H, m) 41.61 41.7 (CH2)
1.26 (t, J = 7.1, 3H) 1.24 (3H, t, J = 7.2) 36.42 36.5 (CH2)
0.94 (t, J = 7.4, 3H) 0.94 (3H, t, J = 7.4) 30.18 30.3 (CH2)
29.8 (CH2)
29.7 (CH3)
25.18 25.2 (CH2)
24.72
14.15
9.89 9.9 (CH3)

58
ethyl 2-(3,5-dihydroxy-2-(8-hydroxyoctanoyl)phenyl)acetate, dothiorelone C (23bc).
Prepared according to the general procedure 2.5.2 (200 μmol aldehyde solvent: DCE; reaction
time 48 h). Purification via preparative TLC (95:5 chloroform/isopropanol) afforded the title
compound as a colorless oil (56.2 mg, 83%, lin/br 87:13). Spectroscopic data correspond to those
reported in the literature.51b
1H NMR (400 MHz, CDCl3) δ 6.30 – 6.26 (m, 2H), 4.19 (q, J = 7.1
Hz, 2H), 3.82 (s, 2H), 3.64 (t, J = 6.6 Hz, 2H), 2.83 (t, J = 8.0 Hz, 2H), 1.79 – 1.63 (m, 2H),
1.64 – 1.46 (m, 2H), 1.38 – 1.31 (m, 6H), 1.27 (t, J = 7.1 Hz, 3H); 1H NMR (400 MHz, Acetone-
d6) δ 6.36 (d, J = 2.3 Hz, 1H), 6.31 (d, J = 2.3 Hz, 1H), 4.08 (q, J = 7.1 Hz, 2H), 3.67 (s, 2H),
3.54 (t, J = 6.5 Hz, 2H), 2.90 (t, J = 8.0 Hz, 2H), 1.67 – 1.60 (m, 2H), 1.52 – 1.47 (m, 2H), 1.40
– 1.29 (m, 6H), 1.21 (t, J = 7.1 Hz, 3H); 13
C {1H} NMR (101 MHz, CDCl3) δ 206.84, 171.36,
164.36, 160.38, 136.85, 116.88, 112.63, 103.33, 63.12, 61.69, 43.39, 41.87, 32.69, 29.18, 29.15,
25.60, 24.94, 14.30. HRMS (ESI+): 339.1803 [M+H]
+ (calc’d 339.1808 for C18H27O6). Note:
ROH protons do not appear in the 1H NMR spectrum (rapid exchange).
Comparison of NMR Data:
1H NMR (400 MHz,
CDCl3) synthesized
1H NMR (500 MHz,
CDCl3) isolated51b
13C NMR (101 MHz,
CDCl3) synthesized
13C NMR (125 MHz,
CDCl3) isolated51b
-
-
12.06 (bs, 1H)
7.89 (bs, 1H)
206.84
171.36
206.0
171.3
6.30 – 6.26 (m, 2H)
4.19 (q, J = 7.1, 2H)
3.82 (s, 2H)
3.64 (t, J = 6.6, 2H)
6.32 (d, J = 2.5, 1H)
6.28 (d, J = 2.5, 1H)
4.17 (q, J = 7.2, 2H)
3.82 (s, 2H)
3.65 (t, J = 6.5, 2H)
164.36
160.38
136.85
116.88
112.63
164.6
160.4
136.8
116.6
112.5
2.83 (t, J = 7.4, 2H)
1.79 – 1.63 (m, 2H)
1.64 – 1.46 (m, 3H)
2.82 (t, J = 7.0, 2H)
1.37 (m, 2H)
1.35 (m, 2H)
103.33
63.12
61.69
103.2
63.1
61.4
1.38 – 1.31 (m, 5H)
1.33 (m, 2H)
1.28 (m, 2H)
1.26 (m, 2H)
43.39
41.87
32.69
43.3
41.8
32.7
1.27 (t, J = 7.1, 3H) 1.25 (t, J = 7.2, 3H) 29.18 29.8
59
29.15 29.0
25.60 25.5
24.94 24.9
14.30 14.1
ethyl 2-(3,5-dihydroxy-2-octanoylphenyl)acetate, Cytosporone B (23bd). Prepared according
to the general procedure 2.5.2 (200 μmol aldehyde; solvent: DCE; reaction time 41 h).
Purification via preparative TLC (98:2 DCM/IPA) afforded the title compound as a colorless
solid (22.2 mg, 78%, lin/br 93:7). M.p. 59-61 °C. Spectroscopic data correspond to those
reported in the literature.52 1
H NMR (400 MHz, CDCl3) δ 11.68 (s, 1H), 6.71 (s, 1H), 6.26 – 6.25
(m, 2H), 4.20 (q, J = 7.1 Hz, 2H), 3.80 (s, 2H), 2.82 (t, J = 8.0 Hz, 2H), 1.70 – 1.66 (m, 2H),
1.30 – 1.26 (m, 11H), 0.89 – 0.87 (m, 3H); 13
C {1H} NMR (101 MHz, CDCl3) δ 206.94, 171.91,
164.13, 160.47, 136.62, 116.87, 112.84, 103.42, 61.84, 43.60, 41.85, 31.82, 29.37, 29.22, 25.12,
22.74, 14.27, 14.21. HRMS (ESI+): 323.1849 [M+H]
+ (calc’d 323.1859 for C18H27O5).
Comparison of NMR Data:
1H NMR (400 MHz,
CDCl3) synthesized
1H NMR (400 MHz,
CDCl3) previously
synthesized52
13C NMR (101 MHz,
CDCl3) synthesized
13C NMR (101 MHz,
CDCl3) previously
synthesized52
11.86 (bs, 1H) 12.1 (s, 1H) 206.94 206.7
6.71 (bs, 1H) - 171.91 171.3
6.26 – 6.25 (m, 2 H) 6.29 (d, J = 2.9, 1H)
6.26 (d, J = 2.0, 1H)
164.13
160.47
164.4
160.3
4.20 (q, J = 7.1, 2H) 4.19 (q, J = 6.8, 2H) 136.62 136.6
3.80 (s, 2H) 3.83 (s, 2H) 116.87 116.5
52
Yoshida, H.; Morishita, T.; Ohshita, J. Chem. Lett. 2010, 39, 508-509.

60
2.82 (t, J = 8.0, 2H) 2.82 (t, J = 6.8, 2H) 112.84 112.6
1.70 – 1.66 (m, 2H) 1.50 – 1.69 (m, 2H) 103.42 103.2
1.30 – 1.26 (m, 11H) 1.10 – 1.45 (m, 11H) 61.84 61.6
0.89 – 0.87 (m, 3H) 0.87 (t, J = 7.8, 3H) 43.60
41.85
43.3
41.8
31.82
29.37
31.7
29.2
29.22 29.1
25.12 24.9
22.74 22.6
14.27 14.1
14.21 14.0
(R)-methyl 2-(3,5-dihydroxy-2-(7-hydroxyoctanoyl)phenyl)acetate, cytosporone M (23ca).
Prepared according to the general procedure 2.5.2 (150 μmol aldehyde solvent: DCE reaction
time 48 h). Purification via preparative TLC (95:5 chloroform/isopropanol) afforded the title
compound as a beige solid (35.5 mg, 73%, lin/br 89:11). M.p. 122-124 °C. Spectroscopic data
correspond to those reported in the literature.53
1H NMR (400 MHz, Acetone-d6) δ 9.69 (s, 1H),
8.78 (s, 1H), 6.37 (d, J = 2.3 Hz, 1H), 6.32 (d, J = 2.3 Hz, 1H), 3.74 – 3.69 (m, 1H), 3.68 (s, 2H),
3.62 (s, 3H), 2.90 (t, J = 8.0 Hz, 2H), 1.70 – 1.57 (m, 2H), 1.51 – 1.26 (m, 6H), 1.10 (d, J = 6.2
Hz, 3H).; 13
C {1H}NMR (101 MHz, MeOD) δ 208.94, 173.99, 161.41, 159.86, 136.98, 121.21,
111.80, 102.78, 68.54, 52.32, 45.15, 40.30, 40.08, 30.57, 26.74, 25.57, 23.47. HRMS (ESI+):
325.1655 [M+H]+ (calc’d 325.1651 for C17H25O6).
(c = 0.02, MeOH) [lit.
(c = 0.02, MeOH)
52 for isolated material].
Note: No reference available for 13
C NMR

61
Comparison of NMR Data:
1H NMR (400 MHz,
Acetone-d6)
synthesized
1H NMR (500 MHz,
Acetone-d6)
isolated53
9.69 (bs, 1H) -
8.78 (bs, 1H) -
6.37 (d, J = 2.3, 1H) 6.37 (d, J = 2.3, 1H)
6.32 (d, J = 2.3, 1H) 6.30 (d, J = 2.3, 1H)
3.74 – 3.69 (m, 1H) 3.68 (m, 1H)
3.68 (s, 2H) 3.67 (s, 2H)
3.62 (s, 3H) 3.61 (s, 3H)
2.90 (t, J = 8.0, 2H) 2.92 (t, J = 7.2, 2H)
1.70 – 1.57 (m, 2H) 1.62 (m, 2H)
1.51 – 1.26 (m, 6H) 1.47 – 1.27 (m, 6H)
1.10 (d, J = 6.2, 3H) 1.09 (d, J = 6.3, 3H)
methyl 2-(3,5-dihydroxy-2-octanoylphenyl)acetate, cytosporone N (23cd). Prepared
according to the general procedure 2.5.2 (150 μmol aldehyde; solvent: DCE; reaction time 48 h).
Purification via preparative TLC (95:5 chloroform/isopropanol) afforded the title compound as a
beige solid (34.4 mg, 74%, lin/br 94:6). M.p. 99-101 °C. Spectroscopic data correspond to those
reported in the literature.53,54
1H NMR (400 MHz, Acetone-d6) δ 6.37 (d, J = 2.3 Hz, 1H), 6.31
53
Xu, J.; Kjer, J.; Sendker, J.; Wray, V.; Guan, H.; Edrada, R.; Müller, W. E. G.; Bayer, M.; Lin, W.; Wu, J.;
Proksch, P. Bioorg. Med. Chem. 2009, 17, 7362-7367. [Please note that dothiorelone A is incorrectly labelled
‘dothiorelone B’ in this paper.]
54Shen, Y.; Wu, Q.; Su, W.; Zheng, Z.; Xu, Q.; Du, X.; Huang, Y.; Song, S.; Liu, J.; Hu, Z. CN 1733693, 2006.

62
(d, J = 2.1 Hz, 1H), 3.68 (s, 2H), 3.61 (s, 3H), 2.95 – 2.84 (m, 2H), 1.66 – 1.59 (m, 2H), 1.37 –
1.22 (m, 8H), 0.88 (t, J = 6.9 Hz, 3H).; 13
C {1H} NMR (126 MHz, CDCl3) δ 207.03, 172.41,
163.79, 160.58, 136.43, 116.98, 112.82, 103.46, 52.72, 43.69, 41.48, 31.83, 29.36, 29.21, 25.08,
22.76, 14.21. HRMS (ESI+): 309.1701 [M+H]
+ (calc’d 309.1702 for C17H25O5). Note: Both
phenolic protons do not appear in the 1H NMR spectrum (rapid exchange).
Comparison of NMR Data:
1H NMR (400 MHz,
Acetone-d6)
synthesized
1H NMR (500 MHz,
Acetone-d6)
isolated53
13C NMR (126 MHz,
CDCl3) synthesized
13C NMR (125 MHz,
CDCl3) isolated54
6.37 (d, J = 2.3, 1H) 6.37 (d, J = 1.8, 1H) 207.03 206.7
6.31 (d, J = 2.3, 1H) 6.31 (d, J = 1.8, 1H) 171.41 171.3
3.68 (s, 2H) 3.67 (s, 2H) 163.79 164.6
3.61 (s, 3H) 3.62 (s, 3H) 160.58 160.2
2.95 – 2.84 (m, 2H) 2.89 (t, J = 6.2, 2H) 136.43 136.6
1.66 – 1.59 (m, 2H) 1.62 (m, 2H) 116.98 116.6
1.37 – 1.22 (m, 8H) 1.32 – 1.29 (m, 8H) 112.82 112.4
0.88 (t, J = 6.9, 3H) 0.88 (t, J = 5.8, 3H) 103.46 103.2
52.72 52.4
43.69 43.4
41.48 41.5
31.83 31.7
29.36 29.2
29.21 29.0
25.08 25.0
22.76 22.6
14.21 14.0

63
2-(3,5-dihydroxy-2-octanoylphenyl)acetic acid, cytosporone A (24). Prepared from
cytosporone B by saponification of the ethyl ester. Under N2, cytosporone B (8a, 4.3 mg, 13.3
μmol, 1.0 equiv.) was dissolved in THF (1.0 mL) and a degassed solution of LiOH (5.6 mg, 133
μmol, 10 equiv.) in H2O (0.2 mL) was added. The mixture was stirred for 6 h at room
temperature, the bulk of solvent was evaporated under reduced pressure and 10% NaOH (1.0
mL) and Et2O (1.0 mL) was added. The aqueous layer was acidified with conc. HCl, the mixture
was extracted with ethyl acetate (3 x 2 mL), dried over MgSO4 and the solvent was removed
under reduced pressure. Purification via preparative TLC (6:4 hexanes/ethyl acetate; spiked with
1% AcOH) afforded the title compound as a colorless oil (3.0 mg, 77%). Spectroscopic data
correspond to those reported in the literature.55 1
H NMR (400 MHz, Acetone-d6) δ 6.38 (d, J =
2.1, 1H), 6.34 (d, J = 2.1, 1H), 3.64 (s, 2H), 2.96 (t, J = 7.3, 2H), 1.67 – 1.60 (m, 2H), 1.31 –
1.28 (m, 8H), 0.87 (t, J = 6.8, 3H). 13
C {1H} NMR (126 MHz, Acetone-d6) δ 207.72, 173.54,
161.63, 160.89, 138.36, 120.18, 111.93, 102.63, 44.46, 41.53, 32.61, 30.4*, 30.4*, 25.41, 23.39,
14.44. *: shifts assigned by HSQC 2D NMR (overlap with solvent signal in 1D 13
C spectrum).
13C NMR (101 MHz, CD3OD) δ 209.59, 175.29, 161.35, 159.80, 137.26, 121.35, 111.61, 102.66,
45.15, 40.44, 32.93, 30.50, 30.29, 25.57, 23.70, 14.42. HRMS (ESI+): 295.1554 [M+H]
+ (calc’d
295.1546 for C16H23O5).
Comparison of NMR Data:
1H NMR (400 MHz,
Acetone-d6)
synthesized
1H NMR (500 MHz,
Acetone-d6)
isolated55
13C NMR (126 MHz,
Acetone-d6)
synthesized
13C NMR (125 MHz,
Acetone-d6)
isolated55
6.38 (d, J = 2.1, 1H) 6.36 (s, 1H) 207.7 207.5
6.34 (d, J = 2.1, 1H) 6.34 (s, 1H) 173.5 172.5
55
Brady, S. F.; Wagenaar, M. M.; Singh, M. P.; Janso, J. E.; Clardy, J. Org. Lett. 2000, 2, 4043-4046.

64
3.64 (s, 2H) 3.64 (s, 2H) 161.6 161.0
2.96 (t, J = 7.3, 2H) 2.94 (t, J = 7.5, 2H) 160.9 160.1
1.67 – 1.60 (m, 2H) 1.63 (m, 2H) 138.4 137.6
1.31 – 1.28 (m, 8H) 1.27 – 1.30 (m, 8H) 120.2 120.5
0.87 (t, J = 6.8, 3H) 0.86 (t, J = 7.0, 3H) 111.9 111.7
102.6 102.5
44.5 44.4
41.5 40.6
32.6 32.5
30.4 (HSQC) 30.0
30.4 (HSQC) 29.9
25.4 25.1
23.4 23.3
14.4 14.3
Ethyl 2-(3,5-dihydroxy-2-(4,4,5,5,6,6,7,7,8,8,9,9,9-tridecafluorononanoyl)phenyl)acetate
(23be). Prepared according to the general procedure 2.5.2 (126 μmol aldehyde; solvent: DCE;
reaction time 112 h). Purification via preparative TLC (70:30 hexanes/acetone) afforded the title
compound as a beige solid (40.6 mg, 57%). 1H NMR (400 MHz, Acetone-d6) δ 9.44 (bs, 1H),
8.84 (bs, 1H), 6.43 (d, J = 2.3 Hz, 1H), 6.34 (d, J = 2.3 Hz, 1H), 4.08 (q, J = 7.1 Hz, 2H), 3.70 (s,
2H), 3.34 – 3.24 (m, 2H), 2.69 – 2.47 (m, 2H), 1.21 (t, J = 7.1 Hz, 3H); 13
C {1H} NMR (101
MHz, Acetone-d6) δ 202.18, 171.84, 160.99, 159.14, 137.52, 120.35, 112.03, 102.66, 61.04,
39.79, 35.25, 26.53 (t, J = 21.4 Hz), 14.49. HRMS (ESI+): 571.0796 [M+H]
+ (calc’d 571.0784
for C19H16O5F13). Note: perfluorinated carbon atoms on acyl chain not observable in 13
C NMR.

65
ethyl 2-(3,5-dihydroxy-2-(6-phenylhexanoyl)phenyl)acetate (23bf). Prepared according to the
general procedure 2.5.2 (100 μmol aldehyde solvent: DCE reaction time 55 h). Purification via
preparative TLC (97:3 chloroform/isopropanol) afforded the title compound as a white solid
(25.5 mg, 69%). 1H NMR (400 MHz, CDCl3) δ 11.96 (s, 1H), 7.29 – 7.25 (m, 2H), 7.18 – 7.15
(m, 3H), 6.27 (d, J = 2.5 Hz, 1 H), 6.25 (d, J = 2.5 Hz, 1H), 4.17 (q, J = 7.1 Hz, 2H), 3.81 (s,
2H), 2.87 – 2.73 (m, 2H), 2.69 – 2.55 (m, 2H), 1.73 (dt, J = 15.1, 7.5 Hz, 2H), 1.64 (dt, J = 15.4,
7.6 Hz, 2H), 1.42 – 1.31 (m, 2H), 1.25 (t, J = 7.1 Hz, 3H); 13
C {1H} NMR (101 MHz, CDCl3) δ
206.66, 171.60, 164.42, 160.40, 142.60, 136.76, 128.53, 128.41, 125.82, 116.79, 112.76, 103.41,
61.77, 43.43, 41.90, 35.85, 31.33, 28.96, 24.92, 14.27. HRMS (ESI+): 371.1860 [M+H]
+ (calc’d
371.1859 for C22H27O5).
ethyl 2-(3,5-dihydroxy-2-(4-phenoxybutanoyl)phenyl)acetate (23bg). Prepared according to
the general procedure 2.5.2 (100 μmol aldehyde; solvent: DCE; reaction time 55 h). Purification
via preparative TLC (97:3 chloroform/isopropanol) afforded the title compound as a beige solid
(14.1 mg, 39%). 1H NMR (400 MHz, CDCl3) δ 11.74 (s, 1H), 7.31 – 7.19 (m, 2H), 6.98 – 6.88
(m, 1H), 6.89 – 6.86 (m, 2H), 6.26 – 6.24 (m, 2H), 4.15 (q, J = 7.1 Hz, 2H), 4.02 (t, J = 6.0 Hz,
2H), 3.85 (s, 2H), 3.07 (t, J = 7.1 Hz, 2H), 2.28 – 2.10 (m, 2H), 1.22 (t, J = 7.1 Hz, 3H); 13
C
{1H} NMR (101 MHz, CDCl3) δ 205.70, 171.79, 164.22, 160.50, 158.89, 136.84, 129.60,
120.94, 116.84, 114.64, 112.88, 103.42, 66.92, 61.80, 41.90, 39.96, 24.71, 14.21. HRMS (ESI+):
359.1497 [M+H]+ (calc’d 359.1495 for C20H23O6).
66
Appendix A: NMR Spectra
1H NMR spectrum (400 MHz, CDCl3, 298 K) of 3ga
13C {
1H} NMR spectrum (101 MHz, CDCl3, 298 K) of 3ga

67
1H NMR spectrum (400 MHz, CDCl3, 298 K) of 3ac
13C {
1H} NMR spectrum (101 MHz, CDCl3, 298 K) of 3ac
68
1H NMR spectrum (400 MHz, CDCl3, 298 K) of 3ad
13C {
1H} NMR spectrum (101 MHz, CDCl3, 298 K) of 3ad

69
1H NMR spectrum (400 MHz, CDCl3, 298 K) of 3ag
13C {
1H} NMR spectrum (101 MHz, CDCl3, 298 K) of 3ag
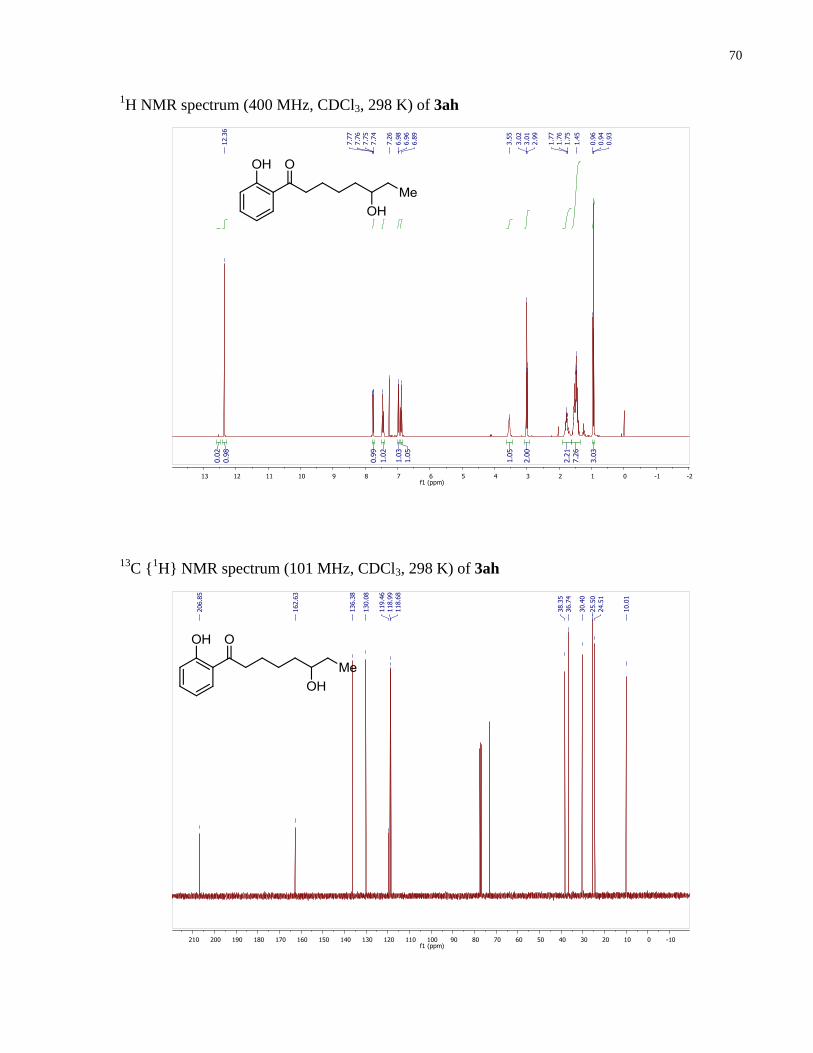
70
1H NMR spectrum (400 MHz, CDCl3, 298 K) of 3ah
13C {
1H} NMR spectrum (101 MHz, CDCl3, 298 K) of 3ah

71
1H NMR spectrum (400 MHz, CDCl3, 298 K) 3aj
13C {
1H} NMR spectrum (101 MHz, CDCl3, 298 K) 3aj
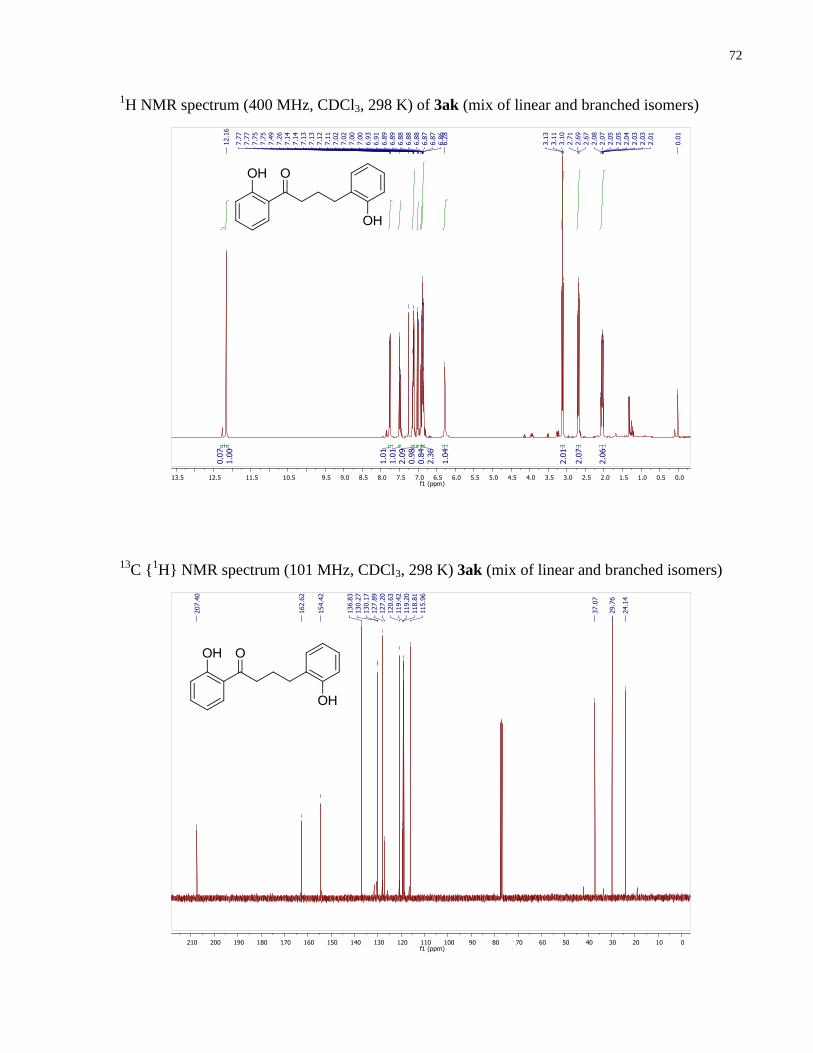
72
1H NMR spectrum (400 MHz, CDCl3, 298 K) of 3ak (mix of linear and branched isomers)
13C {
1H} NMR spectrum (101 MHz, CDCl3, 298 K) 3ak (mix of linear and branched isomers)
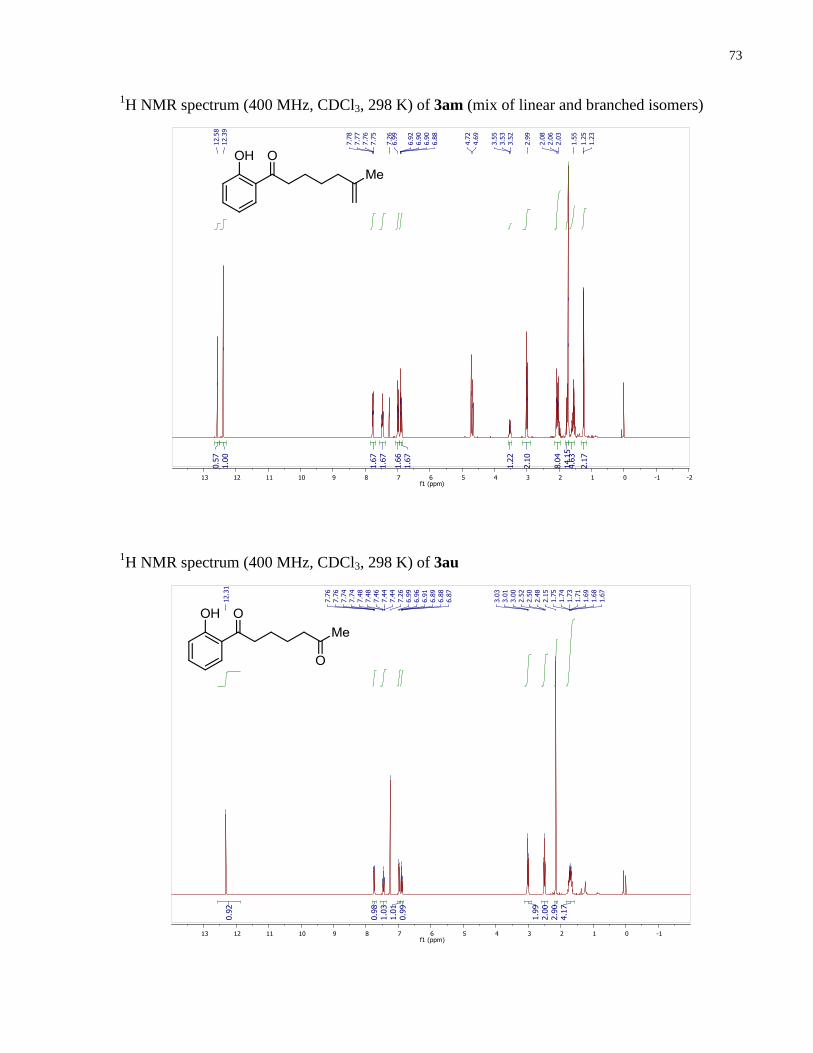
73
1H NMR spectrum (400 MHz, CDCl3, 298 K) of 3am (mix of linear and branched isomers)
1H NMR spectrum (400 MHz, CDCl3, 298 K) of 3au
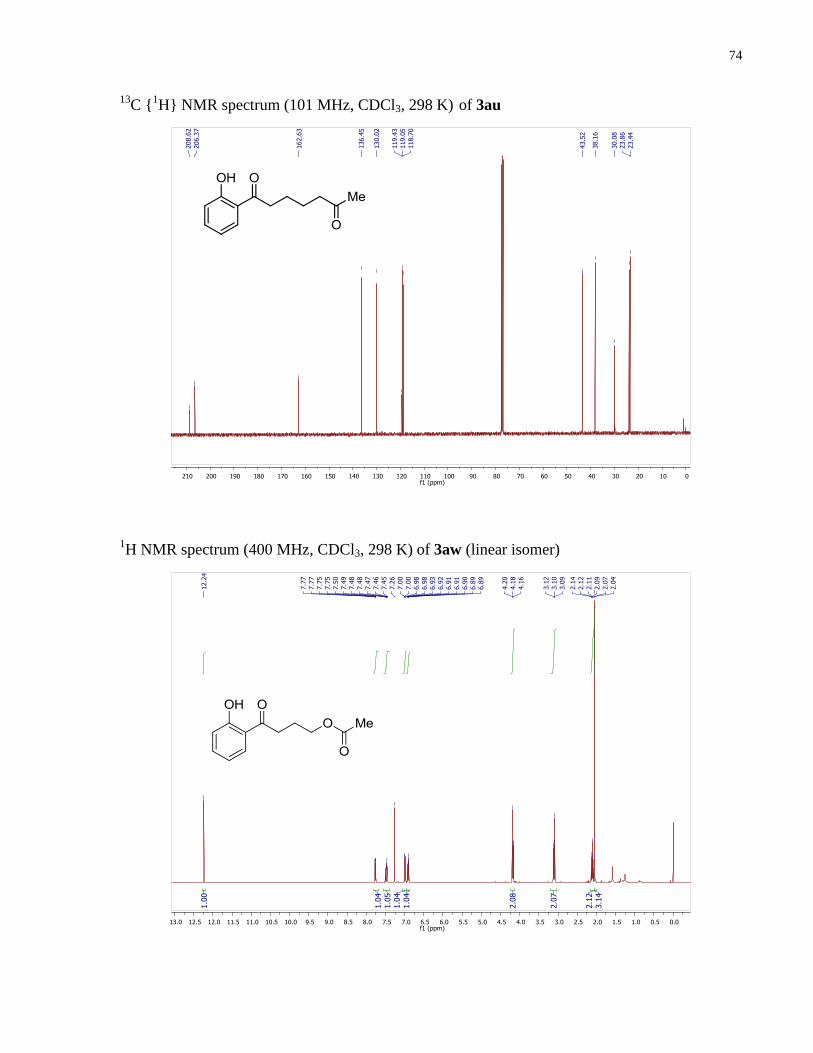
74
13C {
1H} NMR spectrum (101 MHz, CDCl3, 298 K) of 3au
1H NMR spectrum (400 MHz, CDCl3, 298 K) of 3aw (linear isomer)

75
13C {
1H} NMR spectrum (101 MHz, CDCl3, 298 K) of 3aw (linear isomer)
1H NMR spectrum (400 MHz, CDCl3, 298 K) of 3aw’ (branched isomer)

76
13C {
1H} NMR spectrum (101 MHz, CDCl3, 298 K) of 3aw’ (branched isomer)
1H NMR spectrum (400 MHz, CDCl3, 298 K) of 3ax

77
13C {
1H} NMR spectrum (101 MHz, CDCl3, 298 K) of 3ax
1H NMR spectrum (400 MHz, CDCl3, 298 K) of 20a
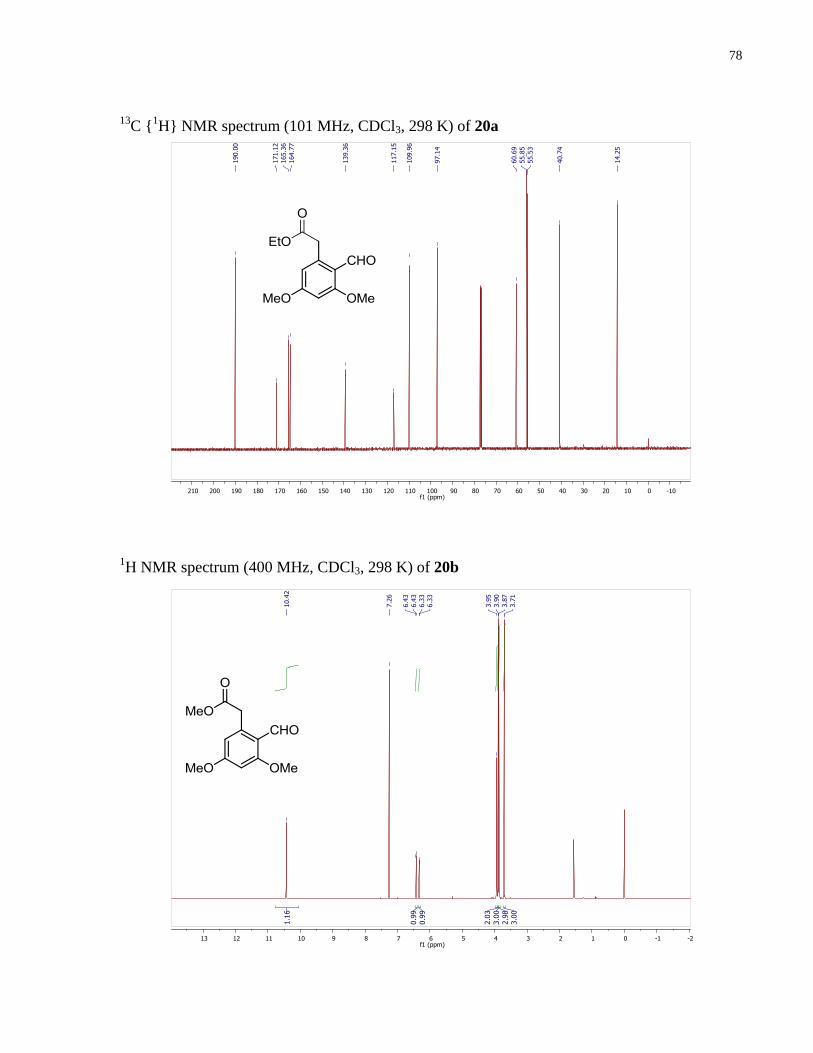
78
13C {
1H} NMR spectrum (101 MHz, CDCl3, 298 K) of 20a
1H NMR spectrum (400 MHz, CDCl3, 298 K) of 20b
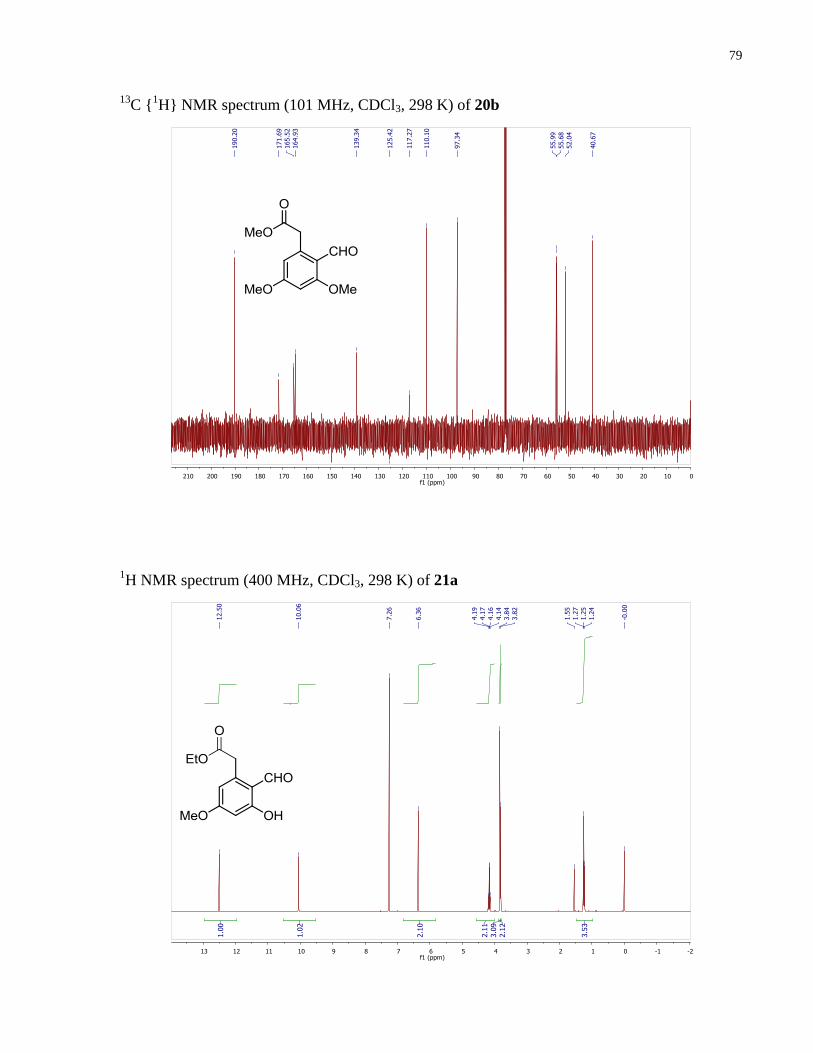
79
13C {
1H} NMR spectrum (101 MHz, CDCl3, 298 K) of 20b
1H NMR spectrum (400 MHz, CDCl3, 298 K) of 21a

80
13C {
1H} NMR spectrum (101 MHz, CDCl3, 298 K) of 21a
1H NMR spectrum (400 MHz, CD3CN, 298 K) of 21b

81
13C {
1H} NMR spectrum (101 MHz, CD3CN, 298 K) of 21b
1H NMR spectrum (400 MHz, Acetone-d6, 298 K) of 21c

82
13C {
1H} NMR spectrum (101 MHz, Acetone-d6, 298 K) of 21c
1H NMR spectrum (400 MHz, CDCl3, 298 K) of (R)-hept-6-en-2-ol 22a
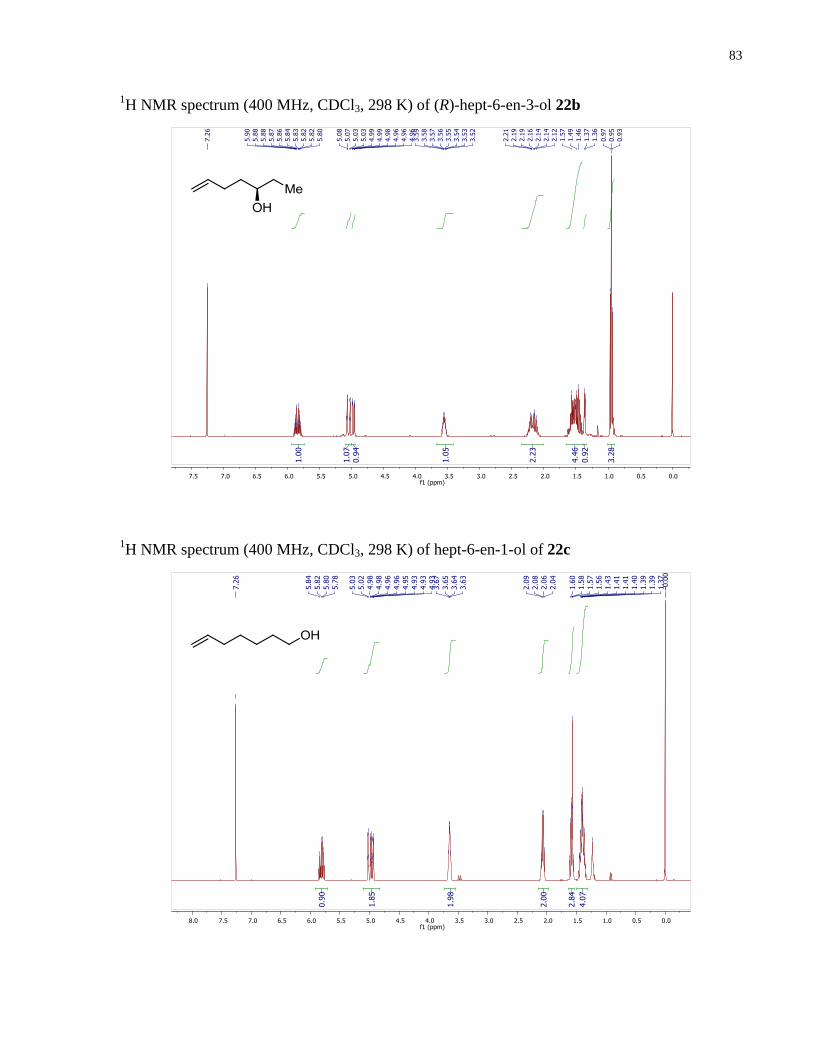
83
1H NMR spectrum (400 MHz, CDCl3, 298 K) of (R)-hept-6-en-3-ol 22b
1H NMR spectrum (400 MHz, CDCl3, 298 K) of hept-6-en-1-ol of 22c

84
1H NMR spectrum (400 MHz, CDCl3, 298 K) of unnamed octaketide 23aa
13C {
1H} NMR spectrum (101 MHz, CDCl3, 298 K) of unnamed octaketide 23aa

85
1H NMR spectrum (400 MHz, CDCl3, 298 K) of phomopsin C (23ad)
13C {
1H} NMR spectrum (101 MHz, CDCl3, 298 K) of phomopsin C (23ad)
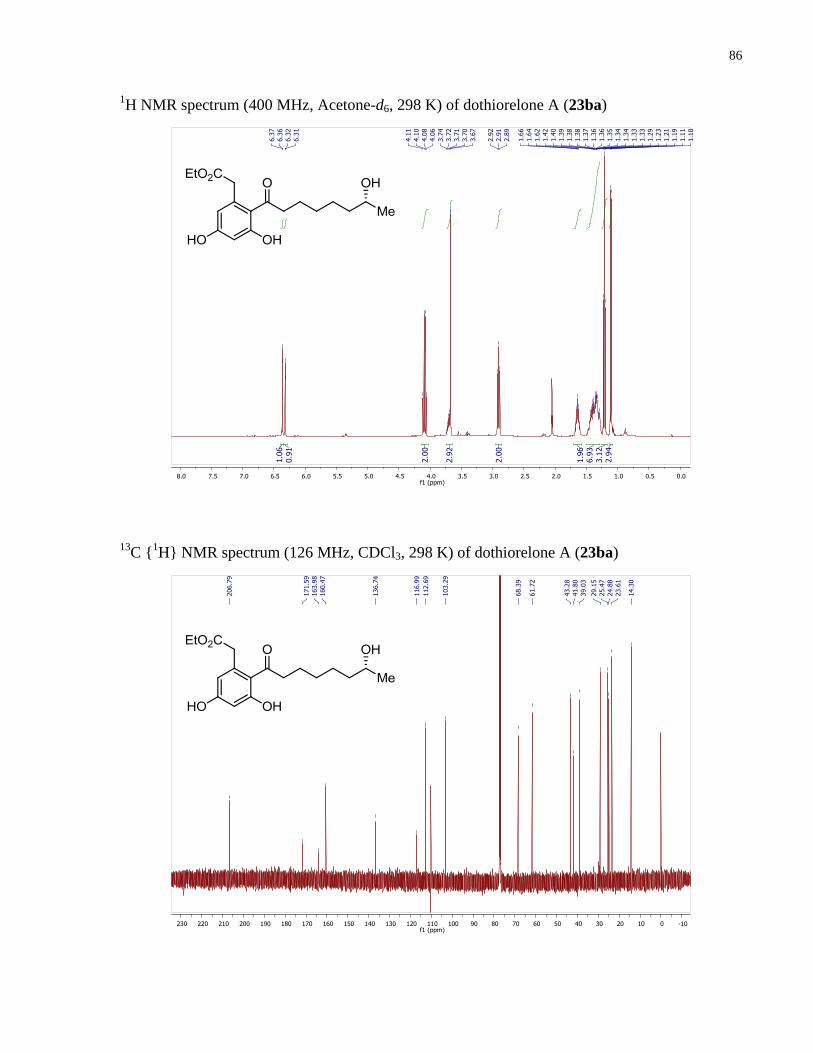
86
1H NMR spectrum (400 MHz, Acetone-d6, 298 K) of dothiorelone A (23ba)
13C {
1H} NMR spectrum (126 MHz, CDCl3, 298 K) of dothiorelone A (23ba)

87
1H NMR spectrum (400 MHz, CDCl3, 298 K) of dothiorelone B (23bb)
13C {
1H} NMR spectrum (101 MHz, CDCl3, 298 K) of dothiorelone B (23bb)
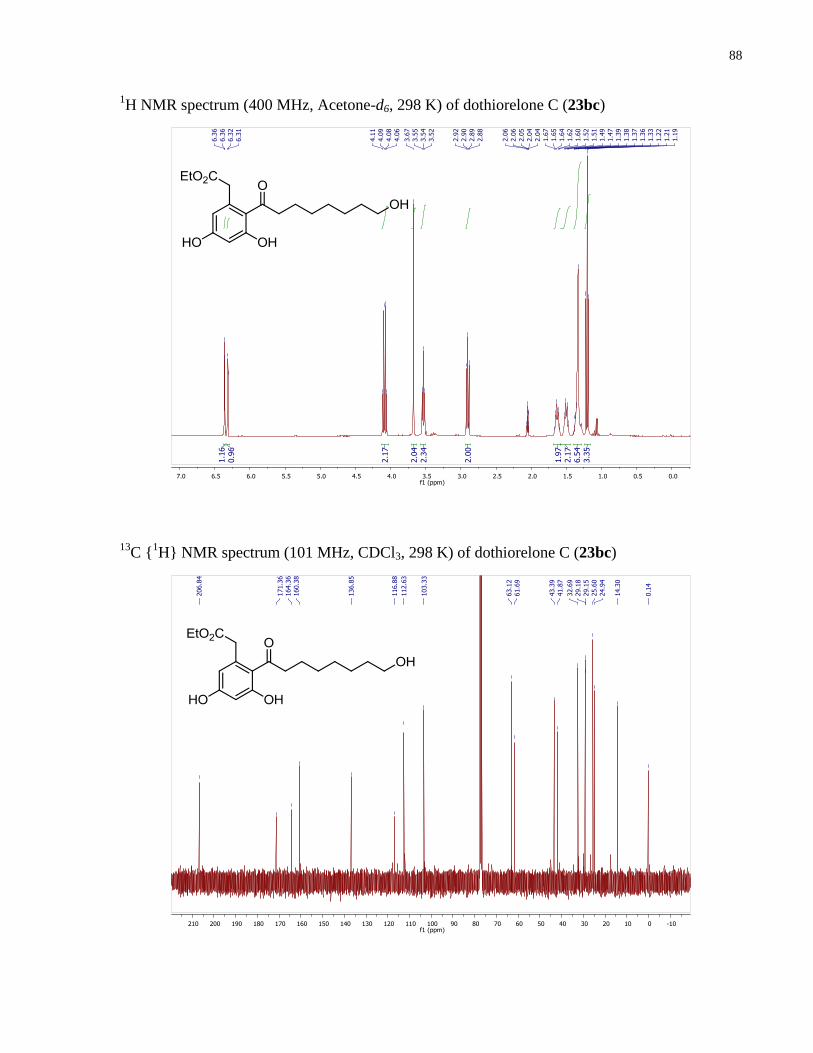
88
1H NMR spectrum (400 MHz, Acetone-d6, 298 K) of dothiorelone C (23bc)
13C {
1H} NMR spectrum (101 MHz, CDCl3, 298 K) of dothiorelone C (23bc)
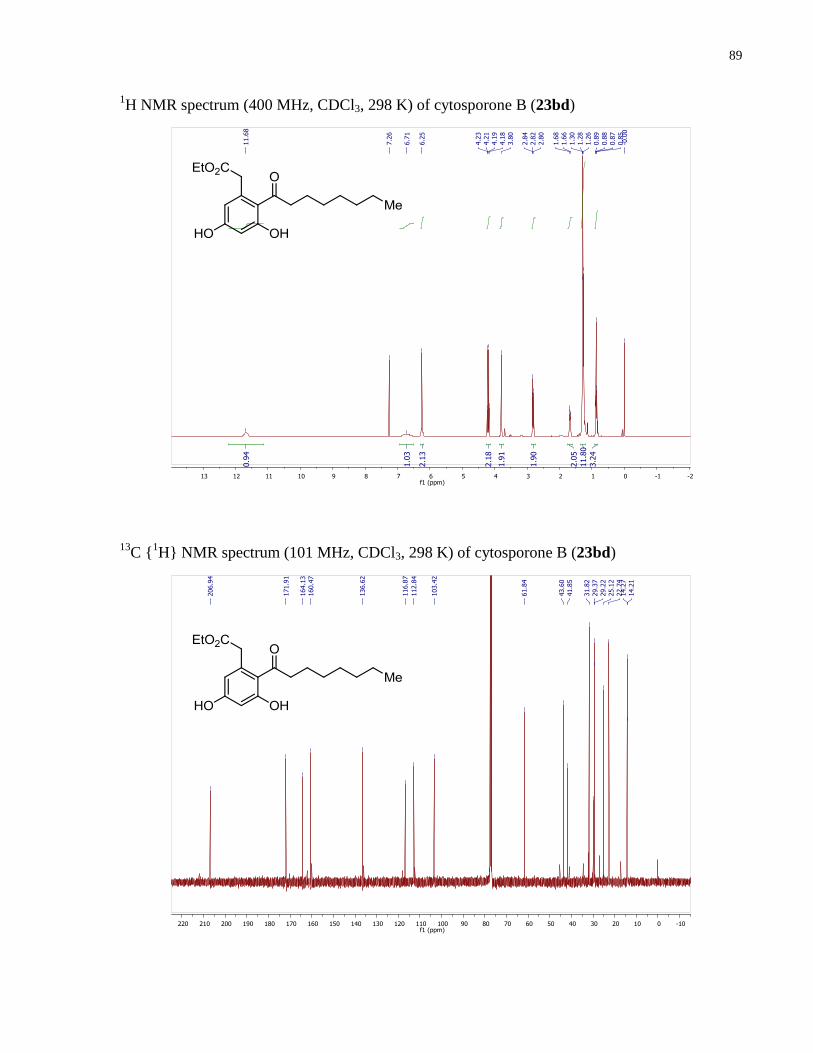
89
1H NMR spectrum (400 MHz, CDCl3, 298 K) of cytosporone B (23bd)
13C {
1H} NMR spectrum (101 MHz, CDCl3, 298 K) of cytosporone B (23bd)
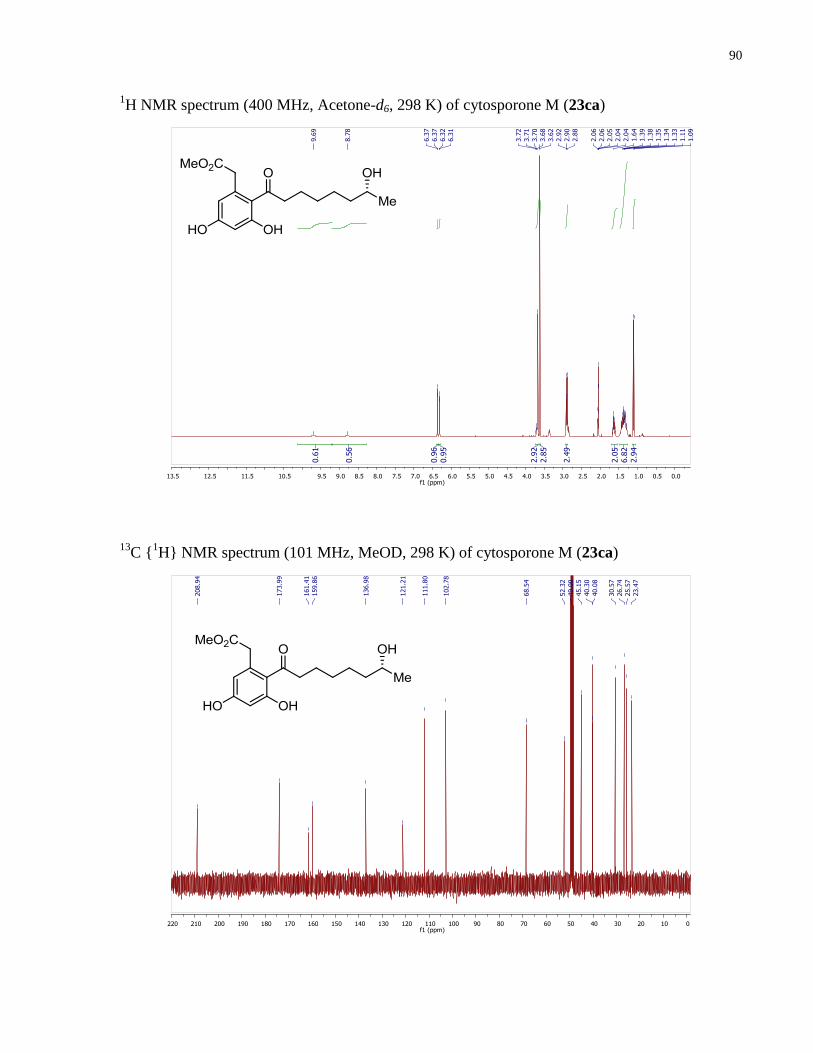
90
1H NMR spectrum (400 MHz, Acetone-d6, 298 K) of cytosporone M (23ca)
13C {
1H} NMR spectrum (101 MHz, MeOD, 298 K) of cytosporone M (23ca)

91
1H NMR spectrum (400 MHz, Acetone-d6, 298 K) of cytosporone N (23cd)
13C {
1H} NMR spectrum (126 MHz, CDCl3, 298 K) of cytosporone N (23cd)

92
1H NMR spectrum (400 MHz, Acetone-d6, 298 K) of cytosporone A (24)
13C {
1H} NMR spectrum (101 MHz, MeOD, 298 K) of cytosporone A (24)
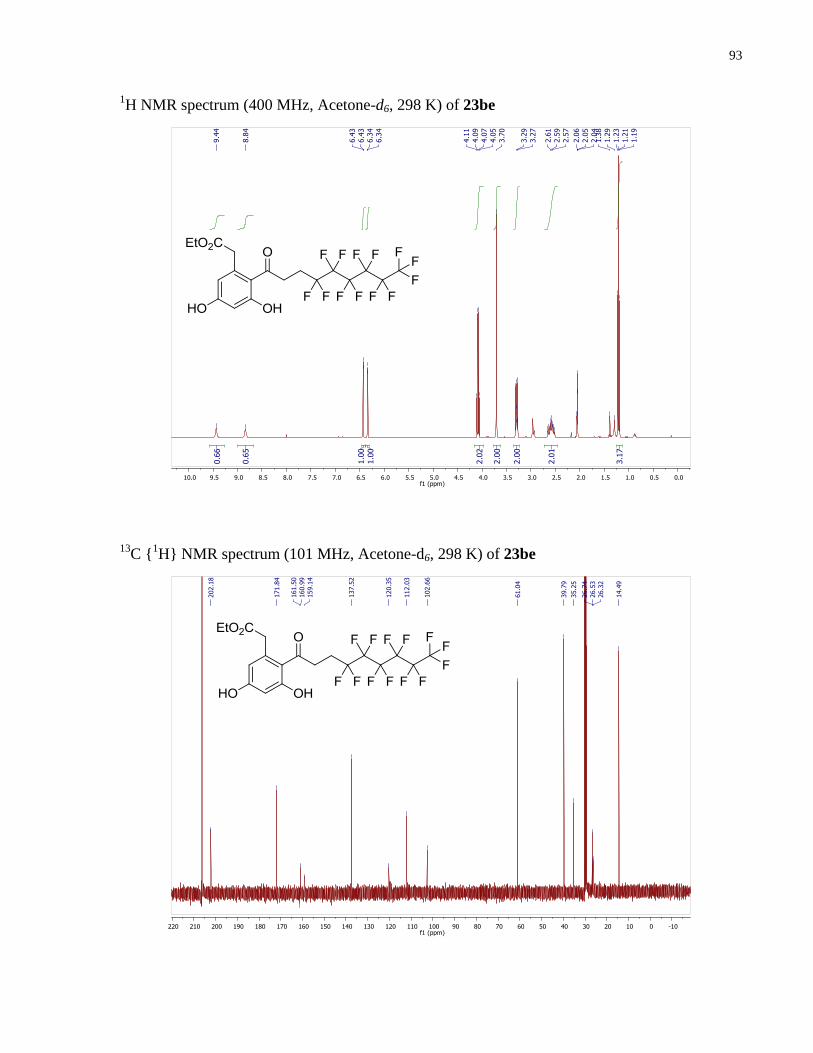
93
1H NMR spectrum (400 MHz, Acetone-d6, 298 K) of 23be
13C {
1H} NMR spectrum (101 MHz, Acetone-d6, 298 K) of 23be

94
1H NMR spectrum (300 MHz, CDCl3, 298 K) of 23bf
13C {
1H} NMR spectrum (101 MHz, CDCl3, 298 K) of 23bf

95
1H NMR spectrum (300 MHz, CDCl3, 298 K) of 23bg
13C {
1H} NMR spectrum (101 MHz, CDCl3, 298 K) of 23bg

96
Appendix B: Crystallographic Data
Crystal data and structure refinement for dothiorelone B (23bb)
Identification code cu_d12103_0m
Empirical formula C18 H26 O6
Formula weight 338.39
Temperature 147(2) K
Wavelength 1.54178 Å
Crystal system Triclinic
Space group P 1
Unit cell dimensions a = 10.0136(5) Å α = 108.517(2)°.
b = 10.1157(5) Å β = 101.863(2)°.
c = 10.9568(5) Å γ = 115.104(2)°.
Volume 874.95(7) Å3
Z 2
Density (calculated) 1.284 Mg/m3
Absorption coefficient 0.791 mm-1
F(000) 364
Crystal size 0.19 x 0.15 x 0.10 mm3
Theta range for data collection 4.64 to 67.39°.
Index ranges -11<=h<=11, -12<=k<=12, -13<=l<=13
Reflections collected 32251
Independent reflections 6063 [R(int) = 0.032]
Completeness to theta = 67.39° 97.9 %
Absorption correction Semi-empirical from equivalents
Max. and min. transmission 0.7528 and 0.6700
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 6063 / 3 / 443
Goodness-of-fit on F2 1.075
Final R indices [I>2sigma(I)] R1 = 0.0441, wR2 = 0.1238
R indices (all data) R1 = 0.0476, wR2 = 0.1265
Absolute structure parameter 0.1(2)
Largest diff. peak and hole 0.244 and -0.295 e.Å-3