ricardo molina gasset enero 2017 - conselleria de …alcoy.san.gva.es/laboratorio/web/sesion muerte...
TRANSCRIPT

Ricardo Molina Gasset Enero 2017

Muerte cardiaca súbita:•Ocurre de manera inesperada dentro de la primera hora desde•Ocurre de manera inesperada dentro de la primera hora desdeel inicio de los síntomas.•En ausencia de testigos cuando el fallecido ha sido visto enEn ausencia de testigos cuando el fallecido ha sido visto enbuenas condiciones menos de 24 h antes de hallarlo muerto.•No hay señales de que va a sufrir un paro cardíaco, pudiendoy q p , pocurrir en cualquier momento, lugar, y persona.•Afecta a personas de todas las edades, incluso adolescentes,atletas, adultos jóvenes y mayores.•Afección clínica dramática, con más de 2.000 muertes diarias enl í i d i li dlos países industrializados.

Dos grupos de enfermedades cardiacas hederitarias relacionadas con la MS•Las enfermedades estructurales o miocardiopatías•Las arritmogénicas o canalopatías.
Variabilidad fenotípica•El ejercicio y el estrés emocional
•Relacionados con la MS en los tipos 1 y 2 del SQTL•Incremento de la actividad vagal
•Relacionados con la MS en el Síndrome de Brugada y en el SQTL3•Puede ocurrir a cualquier edad•Diferencias de riesgo dependiendo del sexo
Variabilidad genotípica•Muchas mutaciones en varios genes implicadas en estas enfermedades•Miocardiopatía hipertrófica.
•El antecedente de MS familiar incrementa el riesgo
•Otras patologías hereditarias•Asociación entre MS familiar y riesgo está menos establecida.


•Población general•No es posible hacer un cribado exhaustivo de toda la población. P i l MS l h fi l f d d•Prevenir la MS con una lucha eficaz contra las enfermedades como:
•Cardiopatía isquémica•Insuficiencia cardiacaC di tí éti•Cardiopatías genéticas
•Población seleccionada•La mejor forma de prevenirla es identificar a los candidatos.A t l t l á i d i•Actualmente, a lo máximo que podemos aspirar es:
•Estudio de los familiares de los pacientes que hayan sufrido MS•Pacientes en consulta por otro motivo, antecedentes familiares de:
C di tí éti•Cardiopatías genéticas•Cardiopatía isquémica •Factores de riesgo evidentes
P i i it édi d d lt ti•Primera visita médica de un adulto, practicar:•Exploración física adecuada •Pruebas complementarias pertinentes (analítica en busca de f t d i t d l ió t i l ECG)factores de riesgo, toma de la presión arterial y un ECG).
•Seleccionar grupos de riesgo de MS para:•Agotar todas las estrategias farmacológicas Abl ió d í ó l d f it é i•Ablación de una vía anómala o de un foco arritmogénico
•Conveniencia implantación desfibrilador para evitar la MS

CARDIOPATÍAS FAMILIARES•Miocardiopatías
•Hipertrófica, arritmogénica de VD espongiforme o no compactada,dilatada, etc.
•Canalopatías o enfermedades de los “canales”•Síndromes de QT largo y corto, síndrome de Brugada, taquicardiaventricular catecolaminérgica polimórfica
•Síndromes con afectación vascular•Síndrome de Marfan, síndrome de Loeys-Dietz, etc.
Comparten una serie de características comunes:•Base genética, puede realizarse un diagnóstico genético de las mismas•Presentación clínica muy heterogénea y una evolución difícil de predecir:
•Casos con diagnóstico por pruebas de imagen evidente•Otros casos con dudas
•Constituyen la principal causa de MS en los deportistas menores de 35•Tienen una presentación familiar, por lo que una vez diagnosticado unpaciente es primordial estudiar al resto de la familia•Comparten los problemas de diagnóstico inherente a los estudios genéticos(mutaciones no descritas, polimorfismos, etc.)

Miocardiopatías
Enfermedades que afectan al músculo cardíaco
DilataciónHipertrofiaRestricciónRestricción
Dos formas Primaria: causa desconocidaPrimaria: causa desconocidaSecundaria: asociada a una patología

Miocardiopatía hipertrófica•Engrosamiento anormal de las paredes del corazón sin causa aparente.g p p•Su prevalencia es de 1:500 población normal.•Es la causa más frecuente de MS en adultos jóvenes y en deportistas.•Es hereditaria (autosómica dominante)
•Los descendientes de un afectado tienen un 50% de posibilidades deportar la mutación causal.
•Gran número de mutaciones en diferentes genes•>900 mutaciones en más de 14 genes sarcoméricos
8 id tifi t ió l l 50 70% d l8 identifican una mutación causal en el 50-70% de los casos.•Mutaciones en genes mitocondriales•Mutaciones genes del metabolismo del glucógeno (PRKAG2,LAMP2 )g g g ( , )•Genes relacionados con enfermedades de depósito
Amiloidosis familiar, la enfermedad de Fabry, etc...•Gran variabilidad en:
•Presentación clínica,•Expresión morfológica,Expresión morfológica,•Evolución y pronóstico.
.


Mayor participación del septum interventricularMayor participación del septum interventricular
Pared mayor de 15mmPared mayor de 15mm

MIOCARDIOPATIA HIPERTRÓFICA
Enfermedad primaria de los miocitos cardiacos, con Enfermedad primaria de los miocitos cardiacos, con hipertrofia concéntrica, asimétrica del ventrículo izquierdo

Obstrucción de salida del VI presente en ¼ parte Determinante importante de la morbilidadLa mayoría de las veces es leve

Miocardiopatía dilatadaCaracteristicas
•Presencia de dilatación y disfunción sistólica VI•Ausencia de sobrecarga (hipertensión, enfermedad valvular)•Ausencia enfermedad de las arterias coronarias suficiente para causarempeoramiento global de la función sistólica•Prevalencia 1 por cada 3000 individuos•Tercera causa más común de fallo cardíaco•Primera causa de trasplante cardíaco•50% de los casos presentación familiar y causa genética•Mutaciones en más de 25 genes diferentes, afectando a proteínas de:
•Citoesqueleto•Sarcómero•Uniones intercelulares•Membrana nuclear•Canales iónicos•Mitocondriales
•El modo de herencia es autosómico dominante, siendo las formasrecesivas ligadas al sexo y la herencia mitocondrial menos frecuente

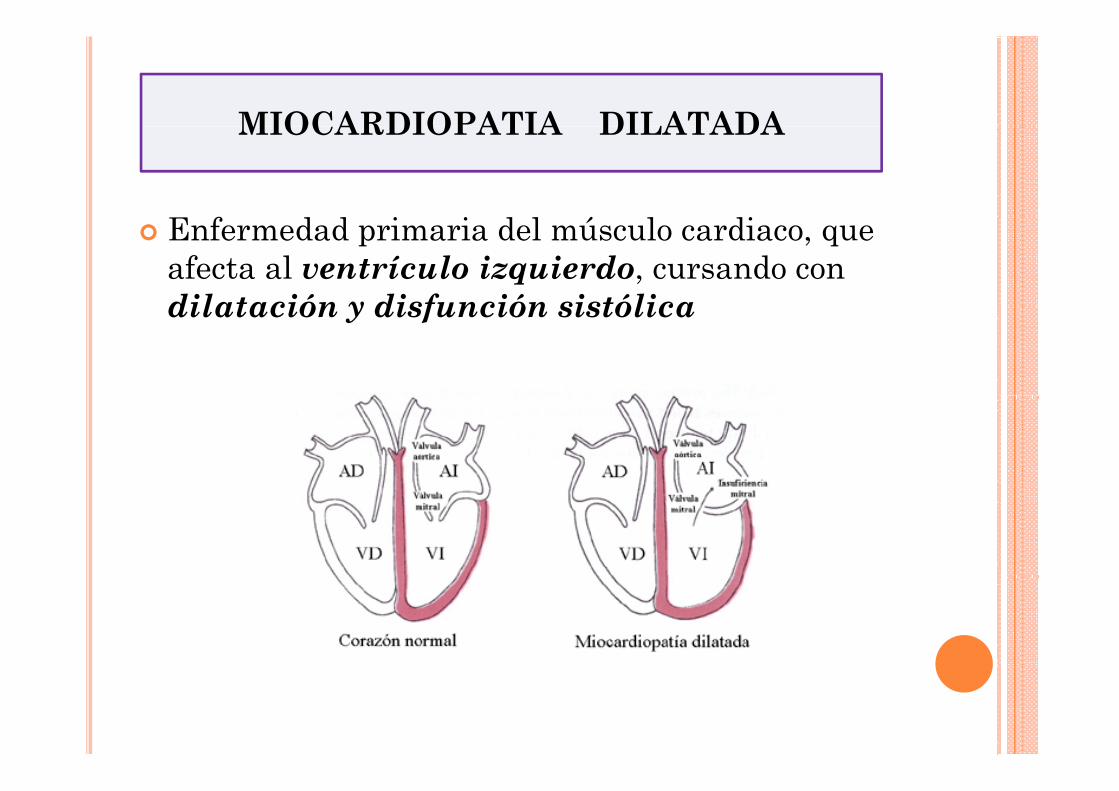
MIOCARDIOPATIA DILATADA MIOCARDIOPATIA DILATADA
Enfermedad primaria del músculo cardiaco, que afecta al ventrículo izquierdo, cursando con dilatación y disfunción sistólica dilatación y disfunción sistólica

FISIOPATOLOGÍA
i iAumento de tamaño de
id d VI
Disminución de
C t tilid d
Disminuye Volumen Sistólico
Aumenta Volumen di tólicavidad VI Contractilidad (FE <40%) diastólico

Miocardiopatía no compactada o espongiforme
C í iCaracterísticas
•Enfermedad congénita con una gran heterogeneidad genética
I d l ió b l d l d i á di•Incremento de la porción trabecular de la pared miocárdica
•Dilatación ventricular y disfunción sistólica, manifestándose por
i fi i i dí i i b bóli MSinsuficiencia cardíaca, arritmias, eventos tromboembólicos y MS.
•Puede tener un curso asintomático durante un largo período de tiempo
•Participan muchos genes implicados en la morfogénesis miocárdica
•El fenotipo de la miocardiopatía no compactada se solapa en muchos casos
l f i di i di ícon los fenotipos correspondientes a otras miocardiopatías,
•Se pueden encontrar miocardiopatías hipertróficas y no compactadas en la
i f ili d bid i é imisma familia, debidas a mutaciones en genes sarcoméricos.

Miocardiopatía/displasia arritmogénica del ventrículo derecho
C i iCaracteristicas
•Enfermedad del músculo cardíaco de origen genético
A lid d l f i l d l í l d h•Anormalidades estructurales y funcionales del ventrículo derecho:
•Sustitución progresiva del miocardio por tejido graso y fibroso tras un
i d d ó iinadecuado proceso apoptótico
•Prevalencia 6 de cada 10.000 individuos.
•Manifestaciones clínicas:
•Individuos asintomáticos
•Arritmias ventriculares e IC derecha o biventricular
•Causa principal de MS en adultos jóvenes (incidencia mayor deportistas)
•La agregación familiar se demuestra hasta en el 60% de los casos
•Mutaciones en los genes de los desmosomas:
•Desmoplakina, desmocolina, desmogleína, placoglobina y placofilina.

Síndrome de QT largoCaracteristicas•Anormalidad estructural en los canales de potasio y sodio del corazón.
•Arritmias ventriculares polimorfas, pudiendo desembocar en pérdidas
de conciencia, convulsiones, paradas cardíacas y MS
•1/3 de los pacientes pueden permanecen asintomáticos
•El electrocardiograma se caracteriza por la prolongación del intervalo QT y
con frecuencia las ondas T tienen una morfología peculiar
•Enfermedad hereditaria con prevalencia de 1 de cada 5.000
•Hace años se diferenciaban sólo dos formas:
•Con sordera, autosómica recesiva (síndrome de Jervell-Lange-Nielsen)
•Sin sordera, autosómica dominante (síndrome de Romano-Ward).
•En la actualidad se han descrito hasta 10 genes implicados dando lugar a
más de 10 tipos distintos

Síndrome de BrugadaCaracteristicas•Anormalidad electrocardiográfica:
•Elevación del segmento ST en la derivaciones V1 -V3
•Bloqueo incompleto o completo de rama derecha y onda T negativa.
•Síncopes, arritmias ventriculares, mareos, sensación de dificultad para
respirar y MS
•Prevalencia 1 de cada 2000 individuos
•Enfermedad hereditaria de trasmisión autosómica dominante
•15% de los casos están asociados con una mutación en el gen que codifica
los canales de sodio en las membranas de los miocitos (gen SCN5A),
•Se conocen hasta 13 genes relacionados, todos ellos presentan una
prevalencia mucho menor.

IMPORTANCIA DEL ESTUDIO GENÉTICO FAMILIAR
•Las miocardiopatías y canalopatías familiares tienen una causa genética yengloban la mayor parte de las causas de MS en individuos jóvenes•Todas estas patologías tienen una
•Presentación clínica muy heterogénea,•Penetrancia incompletaE l ió difí il d d i•Evolución difícil de predecir.
•Utilidad de la genética•Identificación de portadores con mutaciones de alto riesgop g
•Diagnóstico•Pronóstico•Orientación terapéutica
•Genes claves•Genes que hay que incluir en una prueba de detección idealGenes que hay que incluir en una prueba de detección ideal
•Maximizar la posibilidad de obtener resultados clínicamente útiles•Minimizar la identificación de variantes sin trascendencia
•Evitar problemas de interpretación de las pruebas genéticas.

El inicio de cualquier estudio genético conlleva varias fases:
1º Determinar si se trata:
•Caso familiar
•Caso individual
•Familia con presencia de distintas patologías
2º Realizar estudio molecular:
•Genes más prevalentes relacionados con la patología
•Utilizar la técnica mas adecuada
3º Asesoramiento genético

NUEVAS TÉCNICAS DE BIOLOGÍA MOLECULARTécnicas de secuenciación masiva
•Secuenciar varios genes, cientos o incluso el genoma completo de un
individuo en una sola carrera
•Permiten analizar varios pacientes para unos cuantos genes
•La técnica de NGS presenta un problema de reproducibilidad, diferentes
variantes dependiendo del sistema de secuenciación
•Las variantes detectadas deben ser confirmadas por secuenciación Sanger,
que sigue siendo hoy en día la técnica “gold standard”
•Actualmente en el mercado disponemos de la tecnología NGS
•Plataforma Miseq o HiSeq de illumina,q q ,
•Solid o Ion Torrent, de life technologies
•GS Junior 454 de Roche•GS Junior-454 de Roche.


MÉTODO SANGERMÉTODO SANGER

Se fragmenta el DNA
ILLUMINA - SBSSe fragmenta el DNA
Se reparan los extremosSe reparan los extremos
Se agregan los adaptadoresSe agregan los adaptadores

Se agrega el DNA a la placa
ILLUMINA - SBSSe agrega el DNA a la placa
Se hibrida aleatoriamenteSe hibrida aleatoriamente con los adaptadores que estan en la placap

Se agregan dNTPs sin
ILLUMINA - SBSSe agregan dNTPs sin marcar y DNA polimerasa.
Comienza la amplificación

Se denatura el DNA y se
ILLUMINA - SBSSe denatura el DNA y se vuelve a realizar la amplificaciónamplificación
Se han generado millones gde Clústers en la placa.

ILLUMINA SBS
Se agregan los dNTPs cada
ILLUMINA - SBS
Se agregan los dNTPs, cada uno posee un fluoroforo de color particular:color particular:
G - Amarillo.C – AzulA – RojoT - Verde
Se excita con un laser y se captura una imagencaptura una imagen.

ILLUMINA SBS
Se excita con un laser y se
ILLUMINA - SBS
Se excita con un laser y se captura una imagen.La imagen capturada enLa imagen capturada en una posición específica corresponde al primer p pnucleótido.

ILLUMINA - SBS

Los ciclos de secuenciación
ILLUMINA - SBSLos ciclos de secuenciación se repiten hasta determinar la secuencia del templado.la secuencia del templado. Una base a la vez.

El DNA es Nebulizado
ROCHE 454- PIROSECUENCIACIÓN - SBSEl DNA es Nebulizado
Se fragmenta en tamañosSe fragmenta en tamaños aleatorios

Se agregan adaptadores
ROCHE 454- PIROSECUENCIACIÓN - SBSSe agregan adaptadores universales (Adaptador A’ y B’) a los ExtremosB ) a los Extremos
Se denaturaSe denatura

Cada Perla ‘bead’ contiene
ROCHE 454- PIROSECUENCIACIÓN - SBSCada Perla bead contiene miles de adaptadores, pero solo se le adhieresolo se le adhiereun fragmento de DNA que queda atrapado en una emulsión que contiene en su interior los elementos para una PCRuna PCR.

El DNA se amplifica en esta
ROCHE 454- PIROSECUENCIACIÓN - SBSEl DNA se amplifica en esta emulsión Generando cientos de copias en cadacientos de copias en cada perla

Estas son puestas en la
ROCHE 454- PIROSECUENCIACIÓN - SBSEstas son puestas en la PicoTiter Plate (PTP). El diseño de esta permite unadiseño de esta permite una perla por orificio.

ROCHE 454- PIROSECUENCIACIÓN - SBS

SECUENCIACIÓN POR SEMICONDUCCIÓN
Ion Torrent

SECUENCIACIÓN POR SEMICONDUCCIÓN
Matriz de alta densidad de pozos que permiten generar este procesogenerar este proceso paralelamenteCada pozo secuencia unaCada pozo secuencia una hebra.

SECUENCIACIÓN POR SEMICONDUCCIÓNSECUENCIACIÓN POR SEMICONDUCCIÓN

SECUENCIACIÓN POR SEMICONDUCCIÓNSECUENCIACIÓN POR SEMICONDUCCIÓN
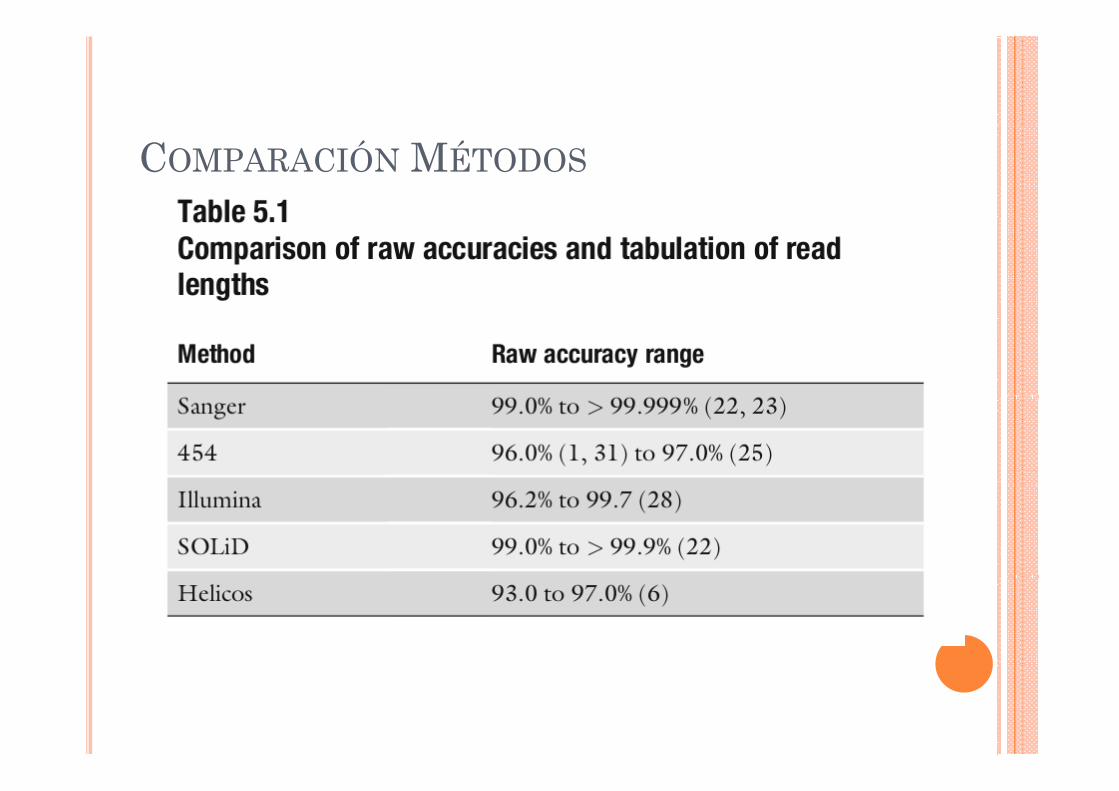
COMPARACIÓN MÉTODOS

•Caracteristicas de la secuenciación masiva
•El número de variantes obtenidas:
•Ventaja el número de variantes obtenidas es muy elevadoVentaja el número de variantes obtenidas es muy elevado
•Inconveniente, el análisis de dichos resultados y la discriminación de
las variantes patogénicas de las benignaslas variantes patogénicas de las benignas
•Determinar la causa genética en la expresión fenotípica de una patología
d b li t i t d l ió d l i tdebemos realizar un proceso estricto de selección de las variantes con un
posible efecto patógeno
l d l i é i id ifi d di í•El 50% de las variantes genéticas identificadas en cardiopatías
familiares son descritas y otro 50% nuevas.
•Estudios del genoma completo de individuos analizando la asociación entre
polimorfismos a un rasgo concreto, han sembrado dudas sobre la supuesta
patogenicidad del 15% de las mutaciones causales de miocardiopatías

Estrategias de secuenciación masiva tendríamos:
•Secuenciar muchos pacientes pero pocos genes
•Son buenas candidatas las enfermedades monogénicas
•Obtendremos un alto rendimiento de la técnica
En el caso del SQTL
•Secuenciación del gen que codifica el canal de potasio (KCNQ1) obtenemos
un rendimiento de entre el 30 y el 35%, y junto con el KCNH2 (gen que
codifica el canal de potasio subfamilia H, miembro 2) de un 25 a un 40%.

•Pocos pacientes pero hacer un gran número de genes.
•Casos complejos en los que el diagnóstico sea confuso o en pacientes con•Casos complejos en los que el diagnóstico sea confuso o en pacientes con
un cribado mutacional inicial negativo.
•También podemos obtener un alto rendimiento por la detección deTambién podemos obtener un alto rendimiento por la detección de
fenocopias, o individuos con fenotipos muy agresivos que puedan ser
portadores de mutaciones compuestas o ser dobles heterocigotos.p p g
•Ser muy exigentes a la hora de seleccionar los pacientes para poder
optimizar el rendimiento.
•Dado el elevado número de variantes detectadas
•Tendremos muchas variantes de significado clínico incierto.
•El estudio a ser posible familiar nos ayudará a determinar la verdadera
patogenicidad de la alteración.

Regulación epigenética y su importancia clínica
En el estudio de las cardiopatías hereditariasp
•Estudios de cambios irreversibles en el material genético, como
alteraciones cromosómicas y mutacionesalteraciones cromosómicas y mutaciones
•Alto porcentaje de individuos sin diagnóstico genético.
G d l i é i•Gran parte de las variantes patogénicas cosegregan con una
penetrancia incompleta.
Manifestación de la enfermedad influenciada por otros muchos factores:
• Ambientales (sexo, hipertensión arterial, grado de ejercicio físico),
•Genéticos (polimorfismos en los propios genes sarcoméricos, genes
modificadores que conforman el sistema renina angiotensina
aldosterona (RAAS) y haplotipos mitocondriales)
•Epigenéticos (modificación de histonas y modificación de la
cromatina, metilación del ADN y los RNA no codificantes).

La metilación del DNA
• Importante mecanismo epigenético, que altera la estructura de la
cromatina e influye en la expresión de los genes.
• Las metil-transferasas añaden de forma covalente un grupo metilo en la
i ió 5 d l it iposición 5 de las citosinas.
• Susceptibles las citosinas situadas en las islas CpG localizadas en zonas
g l d t bié gi difi treguladoras aunque también en regiones codificantes.
• 70% de citosinas en dinucleótidos CpG en el genoma de mamíferos
contiene una 5 metil citosina altamente mutablecontiene una 5-metil-citosina altamente mutable.
• Distintos patrones de metilación en zona del promotor y región codificante de
genes que influyen en la respuesta al estrés cardíaco en pacientes control ygenes que influyen en la respuesta al estrés cardíaco en pacientes control y
pacientes con fallo cardíaco.

Los microRNAs (miRNAs):•RNAs no codificantes de aproximadamente 22 bp de longitud•Representan aproximadamente el 2-3% de los genes en humanos•Regulan la expresión de ciertos genes diana hibridando con la región 3´UTRdel mRNA y bloquean la traslación o degradan el mRNa diana.y q g•Un mismo miRNA puede regular la expresión de varios genes.•Se expresan ampliamente en el corazón como miR-1 y miR-133•Importantes reguladores de genes asociados a patologías cardiovasculares•Importantes reguladores de genes asociados a patologías cardiovasculares•La desregulación de alguno de ellos puede desembocar en alteraciones de lahomeostasis fisiológica:
• Crecimiento incontrolado o proliferación celular (como en lamiocardiopatía hipertrófica o el cáncer)
• Alteración del balance eléctrico.• La presencia de un polimorfismo en sitios de unión de un miRNA
puede modular los fenotipos, como en el caso del asma o la Ataxiade Friedrichde Friedrich.
• Se relaciona miR-195 con el desarrollo de MCH con una transición a
micardiopatía dilatada y fallo cardíaco.

Programas bioinformáticos de predicción de patogenicidad• Uno de los grandes retos de los test genéticos:
• Buscar genes asociados a las cardiopatías hereditariasR li l l ió d i• Realizar correctamente la evaluación de nuevas variantesdetectadas, clasificandolas en:
• PatogénicasB i ( i i ifi ió lí i )• Benignas (sin significación clínica)
• El estudio de la patogenicidad incluso en las variantes publicadas queaparecen en las bases de datos internacionales es incompleto o sin unap pcausalidad clínica clara asignada en muchas ocasiones.
• En el caso de las cardiopatías hereditarias el establecer la patogenicidady el riesgo asociado a MS de una variante puede conllevar:
• Un seguimiento más estricto en pacientes portadores de estasvariantes sin expresión clínica
• Modificación del tratamiento, hábitos de vida o recomendación deun desfibrilador automático implantable en el caso de pacientes consintomatología.

Uso de estas herramientas bioinformáticas:
•Es meramente orientativo aunque pueden predecir la patogenicidad•Es meramente orientativo, aunque pueden predecir la patogenicidad
•Disponibles en la web,
•Polyphen v.2,Sift, Pmut y Mutation TasterPolyphen v.2,Sift, Pmut y Mutation Taster
•En todo estudio bioinformático considerar:
•Variante conservada a nivel evolutivo (Software Align-GVGD)( g )
•Modificaciones fisicoquímicas que suponga el cambio de un
aminoácido por otro (El índice de Grantham)
•Revisión bibliográfica
•Existencia de un estudio funcional de la variante
•Registro en bases de datos (Ensembl, Human Gene Mutation
Database (HGMD), Exome Variant Server) donde nos indique la
frecuencia alélica de la variante en población control.
•El estudio bioinformático es útil en los casos en los que la variante
detectada no está descrita y además no es posible el estudio de
cosegregación familiar

Dificultades en la interpretación de resultados•Muchas variantes de significado clínico incierto.
•Estudio familiar ayuda a determinar la verdadera patogenicidad de la
alteración, la cosegregación familiar es lo que realmente nos la confirma, g g q
•Cuando se identifica una o más variantes genéticas potencialmente
i d l d ll d l í i d d ió lasociadas al desarrollo de patología, se recomienda su detección en el resto
de la familia para la identificación de portadores
•En otros casos la NGS nos puede ayudar al descubrimiento de otra patología
que presente solapamiento de fenotipos, o también el caso de fenocopias.

CONCLUSIONES• El rendimiento del estudio genético nunca va a ser del 100%g
• Tienen una mayor o menor componente probabilística.
• Importante dar la información adecuada al paciente y a su familia
• Estudio individualizado para mejorar el rendimiento genético.
• Utilidad estudios genéticos:
• Identificación de portadores asintomáticos en estudios familiares
• Elaboración de guías terapéuticas y valoración del pronóstico.
• Futuro de los estudios genéticos:
• Interpretación de las variantes de significado clínico incierto.