rna capping by the vaccinia virus guanylyltransferase
TRANSCRIPT

THE JOURNAL OF BIOLOGICAL CHEMISTRY Q 1984 by The American Society of Biological Chemists, Inc.
Vol. 259, No. 21, Issue of Novenber 10, pp. 13488-13494,1984 Printed in U.S.A.
RNA Capping by the Vaccinia Virus Guanylyltransferase STRUCTURE OF ENZYME-GUANYLATE INTERMEDIATE*
(Received for publication, April 23, 1984)
Monica J. Roth and Jerard HurwitzS From the Department of Developmental Biology and Cancer, Albert Einstein College of Medicine, Bronx, New York 10461
GTP:RNA guanylyltransferase isolated from vacci- nia virus catalyzes the transfer of GMP from GTP to the 5' terminus of RNA via an enzyme-guanylate in- termediate. Incubation of the purified vaccinia RNA guanylyltransferase with [CX-~~P]GTP and MgCl, yields [32P]GMP covalently linked to the M, = 95,000 sub- unit. The bond involves the phosphate moiety of GMP and the N'-amino group of lysine. This was verified by treatment of the isolated 95-kDa ~ubunit-[~~P]GMP complex with sodium periodate, followed by methyl- amine-catalyzed &elimination. The product was then hydrolyzed by alkali producing 32P-labeled lysine (W- P)phosphate.
The 5' termini of eukaryotic and viral mRNAs contain a capped, methylated structure (m7GpppX(m)(pX)n) consisting of a 7-methylguanosine linked to the RNA via a 5',5'-triphos- phate bridge. The mechanism of cap synthesis and the enzy- matic activities involved in this process has been extensively studied in vaccinia virus (1) as well as from other sources (for a review, see Banerjee (2)). A multifunctional enzyme has been purified from vaccinia virions (3 , 4) which catalyzes the synthesis of Cap, (m7GpppX) structures at the 5' terminus of triphosphate-terminated RNA (pppRNA) in the presence of GTP, MgC12, and S-adenosylmethionine. RNA triphospha- tase (4, 7), RNA guanylyltransferase (3-8), and RNA (guan- ine-7) methyltransferase (3 , 4, 6) activities have been found associated with this 6.5 S protein, a heterodimer of M, = 95,800 and 26,400 subunits (4).
Synthesis of cap structures catalyzed by vaccinia guanylyl- transferase (referred to as capping enzyme) results from the condensation of a GMP residue from GTP with a diphos- phate-terminated RNA (ppRNA) (9). This capping reaction occurs via a two-step process in which the first reaction involves the formation of a covalent enzyme-guanylate inter- mediate (10). Incubation of the vaccinia guanylyltransferase with [cY-~'P]GTP in the absence of an RNA acceptor results in a pr~tein-[~~P]guanylate complex and PPi. The protein- [32P]nucleotide complex was not formed in the presence of [p- or y-32P]GTP. It was therefore presumed that the complex consisted of a GMP moiety bound to the enzyme. The enzyme- guanylate intermediate (isolated free of the input [cY-~'P]GTP by G-50 column chromatography) when incubated with PPi and MgClz quantitatively released the guanylyl moiety almost
* This work was supported by American Cancer Society Grant NP- 89N. M. J. R. was supported by National Institutes of Health Training Grant GMO 749-07. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
$. To whom correspondence should be sent.
exclusively as [32P]GTP; in addition, incubation of the en- zyme-guanylate intermediate with an RNA acceptor molecule (pppRNA) and MgClz resulted in the formation of 32P-labeled capped ends with the structure GpppRNA (10). The enzyme- guanylate intermediate was also formed when the enzyme was incubated with capped RNA (GpppAp(Ap)n) in the absence of PPi, indicating that all steps in the transguanylylation reaction are readily reversible (1). The site of covalent attach- ment of the GMP has been localized to the M, = 95,000 subunit of vaccinia capping enzyme. The protein-nucleotide linkage has been proposed to be via a phosphoamide bond since it was stable in base, labile in acid, and susceptible to hydroxylamine at pH 4.75 (10).
The nature of the vaccinia pr~tein-[~'P]guanylate complex has been further characterized. The present work demon- strates that GMP is covalently attached to a lysine residue of the M , = 95,000 subunit of vaccinia guanylyltransferase. Specifically, the linkage is a direct P to N bond between the €-amino group of lysine and the phosphoryl group of GMP. Furthermore, the stability of the vaccinia capping enzyme lysine(N'-GMP) linkage was compared with that of the Lys(N'-AMP) linkage foundin T4 DNA ligase (11).
MATERIALS AND METHODS
Enzymes and Reagents-Commercial preparation of enzymes and their source were as follows: T4 DNA ligase, Bethesda Research Laboratories; aminopeptidase M, Boehringer Mannheim; nucleotide pyrophosphatase, Sigma; trypsin, chymotrypsin, and bacterial alka- line phosphatase, Worthington. [ ~ I - ~ ~ P J A T P (410 Ci/mmol) and [a- 32P]GTP (410 and 2000 Ci/mmol) were purchased from Amersham Corp. Phosphoarginine was purchased from Sigma, POCla from Ald- rich, polyethyleneimine-cellulose F thin-layer chromatography plates from Merck, sodium periodate from Mallinckrodt Chemical Works, and methylamine from Fisher. '251-labeled substance P was a gift of Dr. Howard Crystal, Department of Neurology, this institution.
Synthesis of Phosphoamino Acid Markers-3-Phosphohistidine was prepared by the method of Hultquist et al. (12). Phosphoramidate required in the synthesis was a gift of Dr. Roberts A. Smith of the University of California, Los Angeles. W-Phospholysine was pre- pared as described by Fujitaki et al. (13).
Purification of Vaccinia Capping Enzyme-Vaccinia virus (strain WR) was a gift of Dr. Stewart Shuman, from this department, and was prepared from infected HeLa cells as described (14). Vaccinia capping enzyme (RNA guanylyltransferase-RNA (guanine-7) meth- yltransferase complex) was purified from 280 A ~ M ) units of vaccinia virions as described (4). The enzyme was assayed by its ability to exchange [32P]PPi into GTP (4). One unit of enzyme directed the incorporation of 1 nmol of [3'P]PPi into acid-soluble, Norit-adsorba- ble material in 30 min at 37 "C. The phosphocellulose fraction was used in the present study (853 units/mg). This fraction contained two major polypeptides of M , = 95,000 and 33,000 relative to the marker proteins carbonic anhydrase, ovalbumin, and bovine serum albumin and the @, @', U , and O( subunits of Escherichia coli RNA polymerase as assayed by electrophoresis through a 10% polyacrylamide gel in the presence of sodium dodecyl sulfate and staining with silver (32). The two polypeptides are assumed to correspond with the heterodi- meric subunits of the purified enzyme previously reported (4,5). The

Structure of Vaccinia Capping Enzyme-Guanylate Intermediate 13489 variation in the reported molecular weight for the subunits of the enzyme may be a consequence of the different polyacrylamide gel systems utilized. The purified guanylyltransferase contained the as- sociated 7-methyltransferase activity and RNA triphosphatase activ- ity.
Formation of Protein-f'P]Nucleotide Complexes"T4 DNA ligase- [32P]adenylate complex was formed in the presence of [cY-~'P]ATP (980-4800 cpm/fmol), and the vaccinia capping en~yme-[~'Pl]guan- ylate complex was formed in the presence of [o-~'P]GTP (560-900 cpm/fmol) as described (see Refs. 15 and 10, respectively). Radioac- tivity was assayed by Cerenkov spectrometry.
Isolation of Pr~tein-f~PINucleotide Complexes-Reaction mix- tures in which protein-nucleotide complexes were formed were ad- justed to 62.5 mM Tris-HCI buffer, pH 6.8,1% sodium dodecyl sulfate, 10% glycerol, 1% j3-mercaptoethanol, and 0.005% bromphenol blue and electrophoresed through sodium dodecyl sulfate-polyacrylamide gels (6% polyacrylamide stacking gel, 10% polyacrylamide separating gel) according to the method of Laemmli (16). "P-labeled protein- nucleotide complexes, visualized by autoradiography, were cut from the gel and placed in 1 ml of electrophoresis buffer (25 mM Tris base, 191 mM glycine, and 0.1% sodium dodecyl sulfate). The gel slice in the electrophoresis buffer was frozen and thawed twice and trans- ferred to Spectrapor 3 dialysis tubing. Electroelution was carried out in a flat bed electrophoresis box at 100 mA for 2-16 h in electropho- resis buffer. Recovery was routinely more than 60% of the radioactiv- ity. The gel slice was removed from the dialysis bag, and the remaining solution was dialyzed overnight against 50 mM Hepes' buffer, pH 8.2, 0.02% sodium dodecyl sulfate. Sodium dodecyl sulfate was removed by acetone precipitation as described by Hager and Burgess (17). Eppendorf tubes used in acetone precipitations were siliconized, coated with bovine serum albumin (5 mg/ml, heated at 95 "C for 10 rnin), and baked at 100 "C for 45 min in order to minimize losses due to nonspecific adsorption.
Proteolysis of the Enzyme-Gunnylate Intermediate-Acetone pel- lets of the isolated enzyme-GMP complex were resuspended in a solution containing 50 mM Tris-HCI buffer, pH 8.4, 5 mM dithio- threitol, 25 mM NaCI, 1 mM EDTA, 0.1% Triton X-100, and 20% glycerol. The mixture was heated at 95 "C for 3 min after which trypsin was added to a final concentration of 3.75 mg/ml, and the mixture was incubated at 37 "C for 12 h. Digestion was terminated by heating at 95 "C for 3 min. A t this point, proteolysis was judged to be complete since a single radioactive labeled polypeptide was observed after polyacrylamide gel electrophoresis in a 25% acrylamide gel containing a 6% stacking gel. Electrophoresis buffer contained 0.05 M Tris base, 0.383 M glycine, and 0.1% sodium dodecyl sulfate. Gels were run until the salt front was 2 cm from the bottom. The processing of samples during subsequent enzymatic digestions was the same as described for trypsinization. During further digestions, chymotrypsin was added to a final concentration of 3.75 mg/ml, and aminopeptidase M was added to 0.67 mg/ml.
Alkaline Hydrolysis of the Enzyme-Gunnylate Intermediate-Pro- tein fractions in 4 N KOH were heated at 95 "C for 2 h. Samples were cooled, neutralized with perchloric acid (1 M), and adjusted to 50 mM Tris-HCI buffer, pH 7.5. Insoluble material was removed by centrifugation in an Eppendorf microcentrifuge for 2 min at 4 "C. The supernatant containing the derivatized amino acids was removed, concentrated under vacuum, and used for further analysis.
Synthesis of ["P]GMP-[a-32P]GTP was treated with nucleotide pyrophosphate as previously described (18). Reactions (10 pl) con- tained 1 mM GTP (72 cpm/pmol) and 0.3 unit of enzyme. Radioac- tivity was assayed by Cerenkov spectrometry. Reaction products were chromatographed on PEI plates in 0.2 M LiCl and subjected to autoradiography; [32P]GMP was eluted with 80 pl of 150 mM LipCO3, pH 9.5. Insoluble material was removed by centrifugation.
Periodate Oxidation and j3-Elimination-Periodate oxidation fol- lowed by base-catalyzed elimination reactions were performed on either ["PIGMP (1 nmol), the vaccinia 95-kDa-l3'P]GMP complex (35-108 fmol), or the T4 DNA liga~e-[~'P]AMP complex (44-62 fmol). Reaction mixtures (35 pl) contained 20 nmol of sodium period- ate and were adjusted to pH between 6.5-7.5 with 0.1 N HCI. After incubation for 30 min at 37 "C, the pH of the mixture was adjusted to pH 8.0 with 0.1 N NaOH, 1 pmol of methylamine, pH 8.0, was added, and reaction mixtures were further incubated for 1 h at 40 "C.
' The abbreviations used are: Hepes, 4-(2-hydroxyethyl)-l-pipera- zineethanesulfonic acid 95 kDa, subunit of 95,000 daltons; PEI, polyethyleneimine.
Aliquots were removed at each stage of the reaction and chromato- graphed on PEI-cellulose plates in 0.2 M LiC1. Reactions were ter- minated by the addition of 225 pl of cold acetone.
Paper Electrophoresis-High-voltage paper electrophoresis was performed using a Savant LT-48 water-cooled system. Samples to be analyzed were spotted on Whatman 3MM strips along with 1 pg of phosphoarginine, phosphohistidine, and N'-phospholysine and elec- trophoresed at 3500 V for 2 h. Electropherograms were dried, stained with 0.5% ninhydrin in ethanol, and heated in a 65 "C oven before autoradiography. The solvent systems used were: ( a ) pH 1.9, glacial acetic acid/formic acid (88%), water (78252397, v/v); (b) pH 3.5, glacial acetic acid/pyridine/water (505945, v/v); (c) pH 7.5, 50 mM triethylammonium bicarbonate; (d) pH. 9.2, 1% sodium borate. Marker nucleotides were visualized by spraying electropherograms with a solution of 0.005% fluorescein in 0.5 N ammonium hydroxide followed by inspection under long/short UV light (20).
RESULTS
Isolation of Pr~tein-[~~P]Nucleotide Complexes-The transguanylylation reaction catalyzed by vaccinia capping enzyme proceeds via a covalent enzyme-guanylate interme- diate (10). Incubation of the purified enzyme with [ ( U - ~ ~ P ] GTP in the presence of MgC12, followed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and autoradiogra- phy, resulted in the labeling of a single polypeptide of M, =
1 2
kDa - TOP
- Stack
160 L
I50
90 - - 40 -
- Bottom
FIG. 1. Sodium dodecyl sulfate-polyacrylamide gel electro- phoresis of vaccinia capping en~yme-[~*P]guanyIate and T4 DNA liga~e-[~~P]adenylate complex. Formation of the vaccinia capping en~yme-[~'P]guanylate complex was as described under "Ma- terials and Methods." Reaction mixtures (60 rl) contained [cY-~'P] GTP (560 cpm/fmol) and 0.92 unit (GTP/PPi exchange activity) of purified vaccinia capping enzyme. T4 DNA ligase-["PIAMP complex was formed as described (7). Reaction mixtures (20 pl) contained [a- 32P]ATP (980 cpm/fmol) and 6 units of enzyme. Radioactivity was determined by Cerenkov spectrometry. Samples were electrophoresed through a 10% polyacrylamide gel in the presence of sodium dodecyl sulfate and the autoradiograph of the gel is shown. Lane I , vaccinia capping en~yme-[~'P]guanyIate complex; lane 2, T4 DNA liga~e-[~'P] AMP complex. The volume of the reaction mixture electrophoresed per lane and the exposure time of the two samples differed and were as follows. Lane I , 30 pl of reaction mixture (60 pl) exposed for 3 h a t 25 "C. Lane 2, complete reaction mixture exposed for 3 min at 25 "C. E. coli RNA polymerase was used as the marker protein. Individual subunits were stained with Coomassie Blue, and the mi- gration of each of the subunits is as indicated.

13490 Structure of Vaccinia Capping Enzyme-Guanylate Intermediate
95,800 (10) (Fig. 1, lane I). The vaccinia capping enzyme consists of a heterodimer of two polypeptides of M, = 95,800 and 26,400 (4). Although it is not known if both subunits are required for the guanylyltransferase activity, it is the large subunit of vaccinia capping enzyme which forms the covalent pr~tein-[~~P]guanylate intermediate. The region of the so- dium dodecyl sulfate-polyacrylamide gel containing the 32P- labeled M, = 95,800 subunit was excised, and the protein was electroeluted and acetone precipitated, as described under "Materials and Methods." Purification of the 95.8-kDa-[32P] guanylate intermediate directly from the polyacrylamide gel allowed the isolation of the complex free of the input [ (Y-~~P] GTP as well as the M, = 26,400 subunit. Material purified in this manner was used in all the studies to follow.
The capping en~yme-[~~P]guanylate complex has been shown to be alkali stable, acid labile, and susceptible to acidic hydroxylamine (IO), suggesting the involvement of a phos- phoamide bond. T4 DNA ligase catalyzes a similar nucleotidyl transfer reaction that involves a lysine (e-amino)-linked adenosine monophosphoramidate-covalent intermediate (11). Thus, the T4 DNA liga~e-['~P]AMP complex was isolated to allow comparisons to be made between the T4 l i ga~e - [~~P] adenylate complex and the vaccinia capping en~yme-[~'P] guanylate complex. Incubation of the T4 DNA ligase with [a- :"PP]ATP in the presence of MgC12 resulted in the formation of a single 63-kDa-[32P]AMP complex when analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and autoradiography (Fig. 1, lane 2). The gel slice containing this complex was excised, and the protein was electroeluted and acetone precipitated as described above.
Proteolysis of the Vaccinia 95-kDa-[32PlGuanylate Com- plex-The isolated 95-kDa-[32P]guanylate complex was sub- jected to various methods of proteolysis in order to obtain a single amino acid covalently linked to the 32P-labeled nucleo- tide. The proteolysis was followed by electrophoresis of the products in a 25% polyacrylamide gel containing sodium dodecyl sulfate. An autoradiograph of such a gel is shown in Fig. 2. The isolated X2P-labeled M, = 95,000 subunit remained within the 6% stacking gel in this system (Fig. 2, lane I). Treatment of the heat-denatured, 32P-labeled M, = 95,000 subunit with trypsin produced a single radioactive peptide, which migrated faster than the bromphenol blue marker dye (lane 2). The tryptic peptide-["Plguanylate complex could be further digested with chymotrypsin (lane 3). Complete diges- tion with chymotrypsin produced only the faster migrating species (data not shown). The radioactive material which migrated with the salt front in lane 3 appeared irreproducibly and may be an artifact; the nature of this material was not investigated further. The mobility of the product produced after sequential digestion with trypsin and chymotrypsin was increased when treated with the aminopeptidase M (lane 4) . The radioactivity associated with this complex was insensitive to bacterial alkaline phosphatase (lane 5). The complex shown in lane 4, however, was hydrolyzed when treated with 0.1 N HCl for 5 min a t 95 "C (lane 6). The 32P-labeled material released during acid hydrolysis migrated with the salt front in this system and was characterized as presented below. The instability of the protein-nucleotide complex in acid elimi- nated this procedure as a means of hydrolyzing the protein. The isolated 95-kDa-[32P]guanylate complex was therefore vigorously hydrolyzed in alkali (4 N KOH) at 95 "C for 2 h. The material was neutralized with perchloric acid and sub- jected to sodium dodecyl sulfate-polyacrylamide gel electro- phoresis and autoradiography. A single 32P-labeled species was produced (lane 7) whose electrophoretic mobility was the same as the product formed after exhaustive sequential diges-
1 2 3 4 5 6 7 8 9 IO TOP -
Stacking gel - - Cytochrome C -
['251] -Substance P -
BPB -
Solt front - Bottom -
E n ~ y r n e - [ ~ ~ p ] - G M P + + + + + + + + + + KOH + + + + Trypsin - + + + + + - " _ Chymotrypsin " + + + + - _ " Aminopeptidose M - - - + + + - " _ Phosphatase "" + " -4" HCI +
FIG. 2. Proteolysisof vaccinia 95-kDa-[''P]guanylate com- plex. Formation and isolation of the vaccinia 95-kDa-[32P]guanylate complex was as described under "Materials and Methods." The iso- lated complex was sequentially digested with trypsin followed by chymotrypsin and aminopeptidase M or hydrolysis in 4 N KOH, at 95 "C for 2 h (see "Materials and Methods"). The final products were analyzed by hydrolysis in 0.1 N HCI a t 95 "C for 5 min or by digestion with bacterial alkaline phosphatase. Reaction mixtures (10 pl) for the bacterial alkaline phosphatase digestion contained 50 mM Tris-HC1 buffer, pH 9.0, and 1 pg of enzyme and were incubated a t 65 "C for 20 min. Aliquots of the individual reactions were electrophoresed through a 25% polyacrylamide-sodium dodecyl sulfate gel. An auto- radiograph of gel is shown. The grid under the autoradiograph sum- marizes the treatment of the 95-kDa-[32P]guanylate complex in each lane. Lunes 1-7 (which contained 200-400 cpm, Cerenkov) and lanes 8-10 (which contained 600-800 cpm, Cerenkov) are different gels that were run identically. The migration of various marker proteins and dyes are indicated at the left.
tion with trypsin, chymotrypsin, and aminopeptidase M (lane 4) . The product formed after alkaline hydrolysis of the 95- kDa-["Plguanylate complex (repeated in lane 8) was similarly insensitive to bacterial alkaline phosphatase (lane 9) and sensitive to 0.1 N HC1 a t 95 "C for 5 min (lane IO). Hydrolysis of the enzyme-guanylate complex either chemically or enzy- matically resulted in a "P-labeled species of similar molecular weight. This compound was assumed to be a single amino acid-guanylate complex.
Characterization of the Product Formed after Alkaline Hy- drolysis-The isolated "P-labeled M, = 95,000 subunit was hydrolyzed in 4 N KOH at 95 "C for 2 h, neutralized with perchloric acid, desalted, chromatographed on polyethyl- eneimine plates in 0.2 M LiCl, and subjected to autoradiog- raphy (Fig. 3). After alkaline hydrolysis, a single "P-labeled species was released (Fig. 3, lane I) that migrated faster than the marker nucleotides as well as Pi. When this material was chromatographed in a second dimension in 0.5 M formic acid, pH 3.1, a single 32P-labeled band was detected also, indicating that the alkaline digestion product was probably homogeneous (Fig. 3B). The neutralized, desalted alkaline hydrolysis prod- uct was insensitive to alkaline phosphatase treatment (lane 2). Subsequent incubation of the alkaline hydrolysate in 0.1 M HCl a t 95 "C for 5 min liberated a 32P-labeled compound which migrated identically with 5'-GMP (lane 3) . This com- pound was sensitive to bacterial alkaline phosphatase treat- ment, releasing ["PIPi (lane 4) . Acid hydrolysis of the com-
"""
" " _ + - "

Structure of Vaccinia Capping Enzyme-Guanylate Intermediate 13491
A. 1 2 3 4 B. Tap -
L
-UMP '1 - CMP
1 :Pi
0 -GMP AMP
Origln- ,GDP 'GTP Origin-
KOH + + + + HCI - - + + Baclerml Alkolme - + - + Phosphatase
- 2
FIG. 3. Identification of the nucleotide bound to vaccinia guanylyltransferase. The vaccinia 95-kDa-[32P]guanylate complex was isolated from a polyacrylamide gel, hydrolyzed in alkali (4 N KOH), neutralized with perchloric acid, and concentrated as de- scribed under "Materials and Methods." The alkaline hydrolysate was treated with bacterial alkaline phosphatase and acid hydrolysis as follows. Bacterial alkaline phosphatase reaction mixtures (20 pl ) contained 25 mM Tris-HC1 buffer, pH 7.9, and 0.02 pg (7 X unit) of enzyme and were incubated at 65 "C for 20 min. Acid hydrolysis (IO pl ) was carried out in 0.1 M HCI at 95 "C for 5 min. Prior to chromatography, all samples were extracted with hydrated neutral- ized phenol/chloroform (l : l ) , back-extracted with water, extracted twice with ether, and then lyophilized. In pane l A, samples were resuspended in 5 pl of H20 and chromatographed on PEI plates in 0.2 M LiCI. The autoradiograph of the plate is shown. The alkaline hydrolysate was treated as follows: Lane 1, no treatment; lane 2, digestion with bacterial alkaline phosphatase; lane 3, hydrolysis in HCI; lane 4, hydrolysis in acid followed by digestion with bacterial alkaline phosphate. Migration of unlabeled marker nucleotides and ["PIPi is indicated at the right. In p a n e l B , the alkaline hydrolysate was spotted on PEI plates and chromatographed in 0.2 M LiCI. Plates were washed in methanol, dried, and chromatographed in 0.5 M formic acid, pH 3.1. Plates were neutralized in a solution of 600 mg of Tris base in 500 ml of methanol and subjected to autoradiography. Arrows indicate the direction of the migration of the solvents: 1,0.2 M LiCI; 2, 0.5 M formic acid.
plex, therefore, rendered the phosphate of GMP accessible to bacterial alkaline phosphatase. These results indicated that the [32P]phosphate group was directly involved in the covalent linkage to the protein.
Identification of the Amino Acid Involved in the Covalent Enzyme-P'P]GMP Complex-The approach taken to iden- tify the amino acid directly linked to the nucleotide was to first convert the en~yrne-[~'P]GMP complex to the enzyme- ["PIP complex. The guanosine moiety was removed by per- iodate oxidation followed by base-catalyzed elimination of the oxidized nucleoside (19). The base used in these reactions was methylamine, pH 8.0, and the procedure will be referred to as @-elimination. The phosphoprotein was exhaustively hydro- lyzed in alkali, a procedure which should convert the amino acid involved in the covalent linkage to the phosphoamino acid derivative. This procedure was utilized because the syn- thesis and isolation of phosphoamino acids has been described (9, 10).
The periodate oxidation followed by @-elimination reactions were performed on the isolated 95-kDa-[32P]GMP complex as well as the isolated T4 DNA liga~e-[~'P]AMP complex and ["PIGMP. Since neither nucleoside involved in the protein- [32P]nucle~tide complexes was labeled, the @-elimination of the protein complexes could not be monitored. The reaction was assumed to be complete when more than 90% of the 32P label associated with 1 nmol of [32P]GMP migrated as 32Pi when chromatographed on PEI-cellulose plates in 0.2 M LiCl
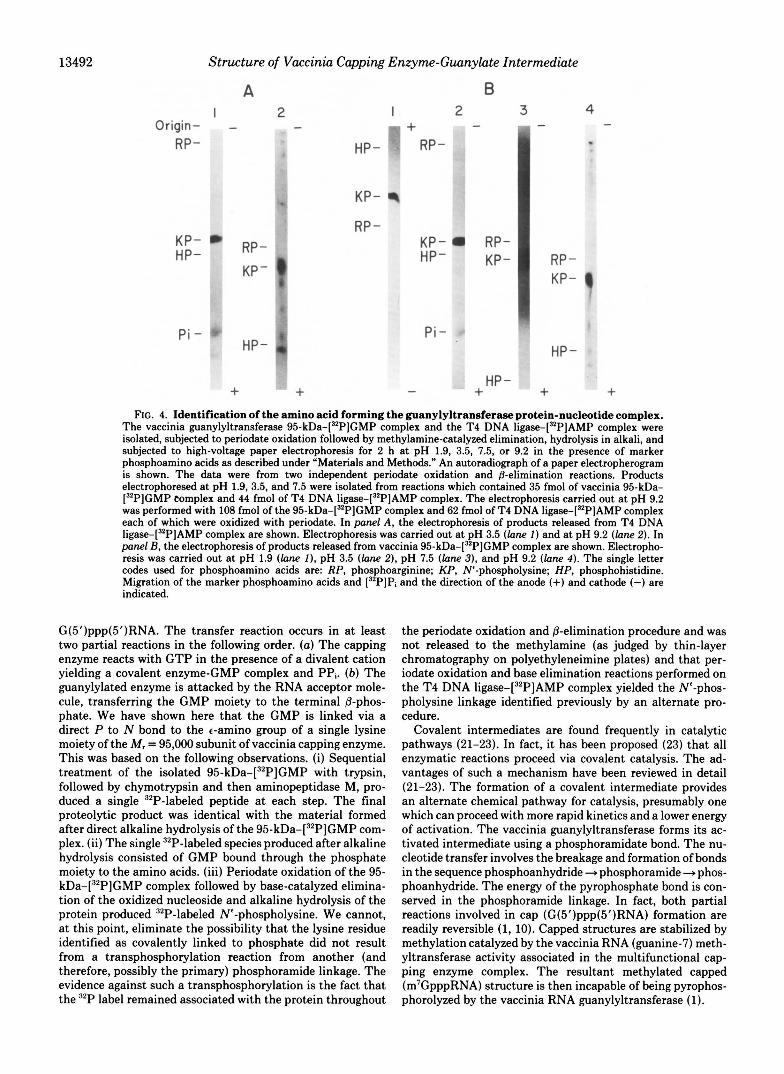
(data not shown). The isolated pr~tein-[~'P]nucleotide com- plexes were similarly monitored during the periodate oxida- tion and the @-elimination procedure by chromatography on PEI-cellulose plates. More than 80% of the 32P label remained associated with both the vaccinia capping enzyme and the T4 DNA ligase throughout the procedure usee (data not shown). The intact phosphoproteins were isolated free of the excess periodate and methylamine by acetone precipitation. The precipitated proteins were then resuspended, hydrolyzed with alkali, neutralized with perchloric acid, desalted, and analyzed by high-voltage paper electrophoresis. The solvent systems chosen distinguished the basic phosphoamino acids, 3-phos- phohistidine, phosphoarginine, and N'-phospholysine. Fig. 4 shows an autoradiogram of the paper electropherograms. In the case of T4 DNA ligase, it has been shown that AMP is linked to the c-amino group of a lysine moiety of the protein by a direct P to N bond (11). Thus, the @-elimination reaction carried out on the T4 DNA liga~e-[~*P]AMP complex should remove the adenylyl base leaving the N'-phospholysine deriv- ative of the protein. Alkaline hydrolysis of this should release ly~ine-[~'P]P. The products of the @-elimination reaction followed by alkaline hydrolysis of the T4 DNA liga~e-[~'P] AMP complex is shown after high-voltage paper electropho- resis at pH 3.5 (Fig. 4, panel A, lane I ) and pH 9.2 (lane 2). The 32P-labeled species produced migrated identically with the chemically synthesized Ne-phospholysine. Similar results were found when electrophoresis was carried out at pH 1.9 and 7.5 (data not shown). The products of the @-elimination reaction followed by alkaline hydrolysis of the vaccinia 95- kDa-[32P]GMP complex is shown after electrophoresis at pH 1.9 @anel B, lane I ) , pH 3.5 (lane 2), pH 7.5 (lane 31, and pH 9.2 (lane 4) . In all the solvent systems shown, the reactions yielded 32P-labeled compounds which co-migrated with the phospholysine present in each lane. The latter was detected with ninhydrin.
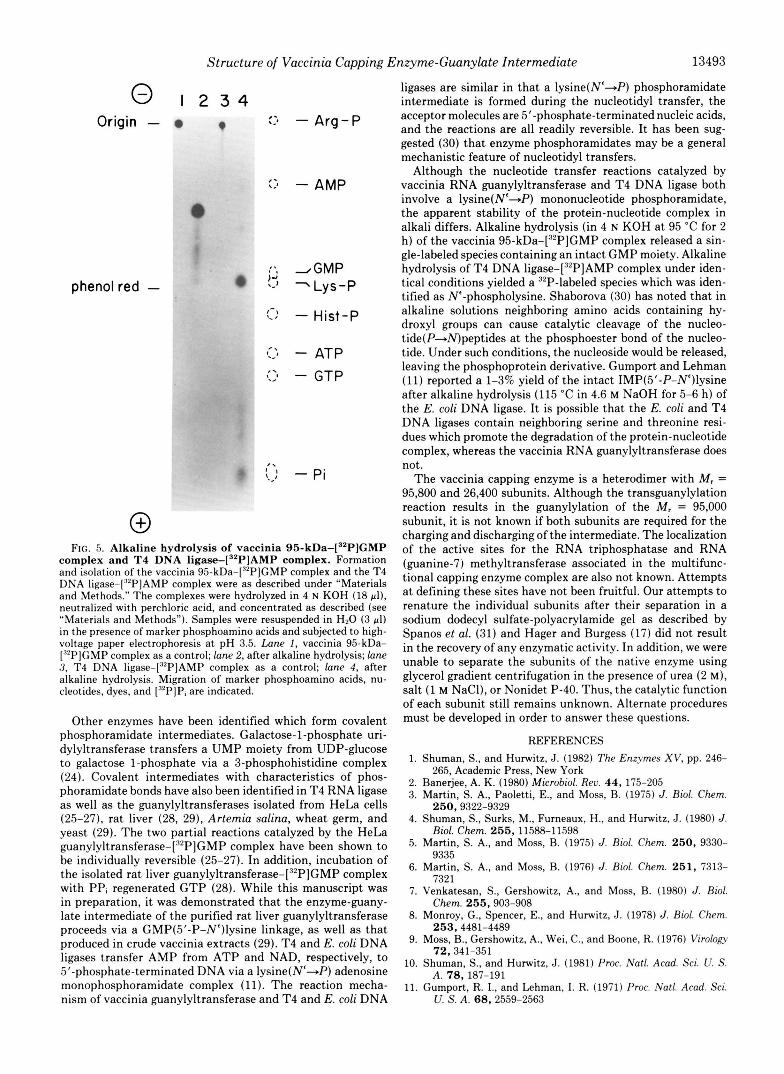
Since the P-elimination reaction could not be monitored, it was of interest to see if the product released by direct alkaline hydrolysis of the en~yme-[~~P]nucleotide complex was differ- ent than the product released after the &elimination reaction. The vaccinia 95-kDa-[32P]GMP complex and the T4 DNA liga~e-[~'P]AMP complex were heated in 4 N KOH at 95 "C for 2 h, neutralized, desalted, and subjected to high-voltage paper electrophoresis at pH 3.5 (Fig. 5). Alkaline hydrolysis of the vaccinia 95-kDa-[32P]GMP complex produced a single 32P-labeled species that migrated between the phosphoargi- nine and phospholysine markers (Fig. 5, lane 2). Alkaline hydrolysis of the T4 DNA liga~e-[~'P]AMP complex, how- ever, produced a radioactive species which co-migrated with phospholysine. This result was repeated in solvent systems at pH 1.9 and pH 9.2 (data not shown). Although both the vaccinia capping enzyme and the T4 DNA ligase form a covalent protein-nucleotide complex via a phosphoamidate bond between a N'-amino group of lysine and the 5"phos- phate of the nucleotide, the apparent stability of these com- plexes differ when heated in alkali. Alkaline hydrolysis of the vaccinia 95-kDa-[32P]GMP complex released a stable 32P- labeled compound assumed to be Lys(N'-GMP) (based on the evidence shown in Figs. 3 and 4). Alkaline hydrolysis of the T4 DNA liga~e-[~'P]AMP complex resulted in the for- mation of Lys(N'-P), due to the apparent elimination of the nucleoside.
DISCUSSION
Vaccinia RNA guanylyltransferase catalyzes the transfer of GMP from GTP to the 5' terminus of diphosphate- or tri- phosphate-terminated RNA, producing the cap structure
13492 Structure of Vaccinia Capping Enzyme-Guunylute Intermediate
A B I 2 I 2 3
Origin- - - m + - RP- HP- RP -
KP-
P i - HP- a.
+ +
RP - KP- RP- HP- KP-
4 -
RP - KP- 0
P i - HP -
HP - - + + + FIG. 4. Identification of the amino acid forming the guanylyltransferase protein-nucleotide complex.
The vaccinia guanylyltransferase 95-kDa-[32P]GMP complex and the T4 DNA liga~e-[~*P]AMP complex were isolated, subjected to periodate oxidation followed by methylamine-catalyzed elimination, hydrolysis in alkali, and subjected to high-voltage paper electrophoresis for 2 h a t pH 1.9, 3.5, 7.5, or 9.2 in the presence of marker phosphoamino acids as described under “Materials and Methods.” An autoradiograph of a paper electropherogram is shown. The data were from two independent periodate oxidation and @-elimination reactions. Products electrophoresed at pH 1.9, 3.5, and 7.5 were isolated from reactions which contained 35 fmol of vaccinia 95-kDa- [32P]GMP complex and 44 fmol of T4 DNA liga~e-[~~P]AMP complex. The electrophoresis carried out a t pH 9.2 was performed with 108 fmol of the 95-kDa-[32P]GMP complex and 62 fmol of T4 DNA liga~e-[~~P]AMP complex each of which were oxidized with periodate. In panel A, the electrophoresis of products released from T4 DNA liga~e-[~*P]AMP complex are shown. Electrophoresis was carried out at pH 3.5 (lane I ) and at pH 9.2 (lane 2). In panel B, the electrophoresis of products released from vaccinia 95-kDa-[32P]GMP complex are shown. Electropho- resis was carried out a t pH 1.9 (lane I), pH 3.5 ( l o n e 21, pH 7.5 ( l o n e 3), and pH 9.2 (lane 4). The single letter codes used for phosphoamino acids are: RP, phosphoarginine; KP, W-phospholysine; HP, phosphohistidine. Migration of the marker phosphoamino acids and [32P]Pi and the direction of the anode (+) and cathode (-) are indicated.
G(5’)ppp(5‘)RNA. The transfer reaction occurs in at least two partial reactions in the following order. (a) The capping enzyme reacts with GTP in the presence of a divalent cation yielding a covalent enzyme-GMP complex and PPi. (b) The guanylylated enzyme is attacked by the RNA acceptor mole- cule, transferring the GMP moiety to the terminal 8-phos- phate. We have shown here that the GMP is linked via a direct P to N bond to the c-amino group of a single lysine moiety of the M, = 95,000 subunit of vaccinia capping enzyme. This was based on the following observations. (i) Sequential treatment of the isolated 95-kDa-[32P]GMP with trypsin, followed by chymotrypsin and then aminopeptidase M, pro- duced a single 32P-labeled peptide at each step. The final proteolytic product was identical with the material formed after direct alkaline hydrolysis of the 95-kDa-[32P]GMP com- plex. (ii) The single 32P-labeled species produced after alkaline hydrolysis consisted of GMP bound through the phosphate moiety to the amino acids. (iii) Periodate oxidation of the 95- kDa-[32P]GMP complex followed by base-catalyzed elimina- tion of the oxidized nucleoside and alkaline hydrolysis of the protein produced 32P-labeled N‘-phospholysine. We cannot, at this point, eliminate the possibility that the lysine residue identified as covalently linked to phosphate did not result from a transphosphorylation reaction from another (and therefore, possibly the primary) phosphoramide linkage. The evidence against such a transphosphorylation is the fact that the 82P label remained associated with the protein throughout
the periodate oxidation and &elimination procedure and was not released to the methylamine (as judged by thin-layer chromatography on polyethyleneimine plates) and that per- iodate oxidation and base elimination reactions performed on the T4 DNA liga~e-[~~P]AMP complex yielded the N‘-phos- pholysine linkage identified previously by an alternate pro- cedure.
Covalent intermediates are found frequently in catalytic pathways (21-23). In fact, it has been proposed (23) that all enzymatic reactions proceed via covalent catalysis. The ad- vantages of such a mechanism have been reviewed in detail (21-23). The formation of a covalent intermediate provides an alternate chemical pathway for catalysis, presumably one which can proceed with more rapid kinetics and a lower energy of activation. The vaccinia guanylyltransferase forms its ac- tivated intermediate using a phosphoramidate bond. The nu- cleotide transfer involves the breakage and formation of bonds in the sequence phosphoanhydride + phosphoramide - phos- phoanhydride. The energy of the pyrophosphate bond is con- served in the phosphoramide linkage. In fact, both partial reactions involved in cap (G(5’)ppp(5’)RNA) formation are readily reversible (1, 10). Capped structures are stabilized by methylation catalyzed by the vaccinia RNA (guanine-7) meth- yltransferase activity associated in the multifunctional cap- ping enzyme complex. The resultant methylated capped (m7GpppRNA) structure is then incapable of being pyrophos- phorolyzed by the vaccinia RNA guanylyltransferase (1).
Structure of Vaccinia Capping Enzyme-Guunylate Intermediate 13493
0 I 2 3 4 Origin - 0 9 :> - Arg-P
phenol red -
* \ - Pi
0 FIG. 5. Alkaline hydrolysis of vaccinia 95-kDa-["P]GMP
complex and T4 DNA l iga~e- [~~P]AMP complex. Formation and isolation of the vaccinia 95-kDa-["P]GMP complex and the T4 DNA ligase-["'PIAMP complex were as described under "Materials and Methods." The complexes were hydrolyzed in 4 N KOH (18 pl), neutralized with perchloric acid, and concentrated as described (see "Materials and Methods"). Samples were resuspended in H20 (3 pl) in the presence of marker phosphoamino acids and subjected to high- voltage paper electrophoresis at pH 3.5. Lane 1, vaccinia 95-kDa- ["PJGMP complex as a control; lane 2, after alkaline hydrolysis; lane 3, T4 DNA liga~e-['~P]AMP complex as a control; lane 4, after alkaline hydrolysis. Migration of marker phosphoamino acids, nu- cleotides, dyes, and ["PIPi are indicated.
Other enzymes have been identified which form covalent phosphoramidate intermediates. Galactose-1-phosphate uri- dylyltransferase transfers a UMP moiety from UDP-glucose to galactose 1-phosphate via a 3-phosphohistidine complex (24). Covalent intermediates with characteristics of phos- phoramidate bonds have also been identified in T4 RNA ligase as well as the guanylyltransferases isolated from HeLa cells (25-27), rat liver (28, 29), Arternia salina, wheat germ, and yeast (29). The two partial reactions catalyzed by the HeLa guanylyltransfera~e-[~~P]GMP complex have been shown to be individually reversible (25-27). In addition, incubation of the isolated rat liver guanylyltran~ferase-[~*P]GMP complex with PPI regenerated GTP (28). While this manuscript was in preparation, it was demonstrated that the enzyme-guany- late intermediate of the purified rat liver guanylyltransferase proceeds via a GMP(5"P-N')lysine linkage, as well as that produced in crude vaccinia extracts (29). T4 and E. coli DNA ligases transfer AMP from ATP and NAD, respectively, to 5"phosphate-terminated DNA via a lysine(N'+P) adenosine monophosphoramidate complex (11). The reaction mecha- nism of vaccinia guanylyltransferase and T4 and E. coli DNA
I:-? - ATP t.2 - GTP ,.
ligases are similar in that a lysine(N'4') phosphoramidate intermediate is formed during the nucleotidyl transfer, the acceptor molecules are 5"phosphate-terminated nucleic acids, and the reactions are all readily reversible. It has been sug- gested (30) that enzyme phosphoramidates may be a general mechanistic feature of nucleotidyl transfers.
Although the nucleotide transfer reactions catalyzed by vaccinia RNA guanylyltransferase and T4 DNA ligase both involve a lysine(N'4') mononucleotide phosphoramidate, the apparent stability of the protein-nucleotide complex in alkali differs. Alkaline hydrolysis (in 4 N KOH at 95 "C for 2 h) of the vaccinia 95-kDa-["P]GMP complex released a sin- gle-labeled species containing an intact GMP moiety. Alkaline hydrolysis of T4 DNA l iga~e - [~~P]AMP complex under iden- tical conditions yielded a 32P-labeled species which was iden- tified as N'-phospholysine. Shaborova (30) has noted that in alkaline solutions neighboring amino acids containing hy- droxyl groups can cause catalytic cleavage of the nucleo- tide(P4V)peptides at the phosphoester bond of the nucleo- tide. Under such conditions, the nucleoside would be released, leaving the phosphoprotein derivative. Gumport and Lehman (11) reported a 1-396 yield of the intact IMP(5'-P-NC)lysine after alkaline hydrolysis (115 "C in 4.6 M NaOH for 5-6 h) of the E. coli DNA ligase. It is possible that the E. coli and T4 DNA ligases contain neighboring serine and threonine resi- dues which promote the degradation of the protein-nucleotide complex, whereas the vaccinia RNA guanylyltransferase does not.
The vaccinia capping enzyme is a heterodimer with M, = 95,800 and 26,400 subunits. Although the transguanylylation reaction results in the guanylylation of the M , = 95,000 subunit, it is not known if both subunits are required for the charging and discharging of the intermediate. The localization of the active sites for the RNA triphosphatase and RNA (guanine-7j methyltransferase associated in the multifunc- tional capping enzyme complex are also not known. Attempts a t defining these sites have not been fruitful. Our attempts to renature the individual subunits after their separation in a sodium dodecyl sulfate-polyacrylamide gel as described by Spanos et al. (31) and Hager and Burgess (17) did not result in the recovery of any enzymatic activity. In addition, we were unable to separate the subunits of the native enzyme using glycerol gradient centrifugation in the presence of urea (2 M), salt (1 M NaCl), or Nonidet P-40. Thus, the catalytic function of each subunit still remains unknown. Alternate procedures must be developed in order to answer these questions.
REFERENCES 1. Shuman, S., and Hurwitz, J. (1982) The Enzymes XV, pp. 246-
2. Banerjee, A. K. (1980) Microbiol. Rev. 44, 175-205 3. Martin, S. A., Paoletti, E., and Moss, B. (1975) J. Biol. Chem.
4. Shuman, S., Surks, M., Furneaux, H., and Hurwitz, J. (1980) J.
5. Martin, S. A., and Moss, B. (1975) J. Biol. Chem. 250 , 9330-
6. Martin, S. A., and Moss, B. (1976) J. Biol. Chem. 2 5 1 , 7313-
7. Venkatesan, S., Gershowitz, A., and Moss, B. (1980) J. Biol.
8. Monroy, G., Spencer, E., and Hurwitz, J. (1978) J. Biol. Chem.
9. Moss, B., Gershowitz, A., Wei, C., and Boone, R. (1976) Virology
10. Shuman, S., and Hurwitz, J. (1981) Proc. Natl. Acad. Sci. U. S.
11. Gumport, R. I., and Lehman, I. R. (1971) Proc. Natl. Acad. Sci.
265, Academic Press, New York
250,9322-9329
Biol. Chem. 2 5 5 , 11588-11598
9335
7321
Chem. 255,903-908
253,4481-4489
72,341-351
A. 78,187-191
U. S. A. 68,2559-2563

13494 Structure of Vaccinia Capping Enzyme-Guanylate Intermediate
12.
13.
14. 15.
16. 17. 18.
19. 20.
Hultquist, D. E., Moyer, R. W., and Boyer, P. D. (1966) Biochem-
Fujitaki, J. M., Steiner, A. W., Nichols, S. E., Helander, E. C., Lin, Y. C., and Smith, R. A. (1980) Prep. Bwchem. 10, 205- 213
istry 5,322-331
Joklik,W. K. (1962) Biochim. Biophys. Acta 6 1 , 290-301 Weiss, B., and Richardson, C . C. (1967) J. Biol. Chem. 242,
Laemmli, U. K. (1970) Nature ( L o n d . ) 227,680-685 Hager, D., and Burgess, R. (1980) Anal. Biochem. 109,76-86 Furuichi, Y., Morgan, M., Mitnukrishnan, S., and Shatkin, A. J.
(1975) Proc. Natl. Acad. Sci. U. S. A. 72,362-366 Schmidt, G . (1968) Methods Enzymol. 12B, 230-235 Kresbsik, G., Heusser, D., and Wimmer, A. (1969) in Thin Layer
Chromatography (E. Stahl, ed) pp. 854-909, Springer-Verlag,
4270-4272
23.
24.
25.
26. 27.
28.
29.
30.
71-84,171-176, W. H. Freeman and Co., San Francisco
Springer-Verlag, New York
2963
Spector, L. B. (1982) Coualent Catalysis by Enzymes, pp. 1-22,
Yang, S.-L. L., and Frey, P. A. (1979) Biochemistry 18, 2980-
Venkatesan. S.. and Moss. B. (1982) Proc. Natl. Acad. Sci. U. S. I~ I
A. 79,340-344 Shuman. S. (1982) J. Biol. Chem. 257. 7237-7245 Wang, D., Furuichi, Y., and Shatkin, A. J. (1982) Mol. Cell. Bwl.
Mizumoto, K., Kaziro, Y., and Lipmann, F. (1982) Proc. Natl.
Toyama, R., Mizumoto, K., Nakahara, Y., Tatsuma, T., and
Shaborova, 2. A. (1970) Prog. Nucleic Acid Res. Mol. Biol. 10,
2,993-1001
Acad. Sci. U. S. A. 79, 1693-1697
Kaziro, Y. (1983) EMBO J. 12, 2195-2201
145-182 New York 31. Spanos, A., Sedgwick, S. G., Yarranton, T., Hubscher, U., and
42-162, McGraw-Hill Book Co., New York 32. Oakley, B., Kirsch, D., and Morris, R. (1980) Anal. Biochem.
_ ~ . ~~~
21. Jencks, W. P. (1969) Catalysis in Chemistry and Enzymology, pp. Banks, G. R. (1981) Nucleic Acid Res. 9, 1825-1839
22. Walsch, C . (1979) Enzymatic Reaction Mechanisms, pp. 39-41, 105,361-363