self-assembly and applications of poly(glycidyl...
TRANSCRIPT

This journal is©The Royal Society of Chemistry 2014 Chem. Commun., 2014, 50, 13201--13215 | 13201
Cite this:Chem. Commun., 2014,
50, 13201
Self-assembly and applications of poly(glycidylmethacrylate)s and their derivatives
Qing-Lan Li,†a Wen-Xing Gu,†b Hui Gao*b and Ying-Wei Yang*ac
In this feature article, we give an overview of the preparation and application of self-assembled
architectures based on an emerging area of polymers, i.e., poly(glycidyl methacrylate)s (PGMAs) and their
derivatives. A series of PGMA-based aggregates and hybrids, such as micelles, reverse micelles, capsules,
nanoparticles, and inorganic–organic hybrid materials, has been constructed, and diverse morphologies
were formed, driven by hydrophobic interactions, hydrogen bonding, ionic complexation, host–guest
interactions, etc. In particular, the assemblies have shown great potential applications as drug vectors, gene
vectors, solubilizing agents, antimicrobial agent, and so forth. Herein, the general guidelines are elaborately
selected from literature examples and partially from our own. Although still in its infancy, self-assembly of
PGMA-based polymers is expected to become a hot topic in polymer chemistry and materials science.
1. Introduction
Because of human curiosity about the phenomenon of order fromdisorder and the desire to make ensembles of nanostructures fordifferent applications, research on molecular self-assembly hasnot only been active, but also fruitful over the past decades.1,2
There are two major types of self-assemblies according to themolecular weight of the entities to form nanoassemblies(micelles, vesicles, supramolecular polymers, etc.): (a) one only
employs small molecules, especially surfactants3–5 and supra-molecular synthetic receptors6–9 (such as crown ethers,10–13
cyclodextrins (CDs),14–20 calixarenes,21–27 cucurbiturils,28–32 andpillarenes33–38); and (b) the other employs polymers as buildingblocks in self-assembly systems, which we called polymer self-assembly.39–47 In addition to the stimuli-responsiveness similarto small molecular assemblies,48–50 polymer assemblies exhibitgood performance in more aspects, such as better stability andmore diverse morphologies,51,52 attributed to the physical pro-perties of the polymers themselves, which promotes the progressof polymer synthesis and self-assembly.
Compared to previous conventional radical polymerizationand ionic polymerization, living radical polymerization allows awide range of monomers and initiators (including those afterfunctionalization), and endows synthetic polymers and copolymerswith precisely controlled molecular weight, relatively low dispersity,
a State Key Laboratory of Supramolecular Structure and Materials,
College of Chemistry, Jilin University, 2699 Qianjin Street, Changchun 130012,
P.R. China. E-mail: [email protected]; Tel: +86 13009100930b School of Chemistry and Chemical Engineering, Tianjin University of Technology,
Tianjin, 300384, P.R. China. E-mail: [email protected] State Key Laboratory of Coordination Chemistry, Nanjing University, P.R. China
Qing-Lan Li
Qing-Lan Li was born in Ganzhou,Jiangxi Province, China, in 1990.She obtained her BSc degree inChemistry Education from JiangXiNormal University in 2012. Then,she joined Professor Ying-WeiYang’s group in the College ofChemistry at Jilin University, beingengaged in the research field ofPGMA-based hybrid materials forcontrolled drug release.
Wen-Xing Gu
Wen-Xing Gu was born inShandong Province, China, in1989. He obtained his BE inPharmaceutical Engineering fromTianjin University of Technology in2013, where he performed researchunder the direction of Professor HuiGao. He is currently a M.M. studentin the same research group and isworking in the field of PGMA-basedself-assembly.
† Q.-L.L. and W.-X.G. contributed equally to this work.
Received 24th April 2014,Accepted 16th May 2014
DOI: 10.1039/c4cc03036b
www.rsc.org/chemcomm
ChemComm
FEATURE ARTICLE

13202 | Chem. Commun., 2014, 50, 13201--13215 This journal is©The Royal Society of Chemistry 2014
and controlled molecular architecture.53 Atom transfer radicalpolymerization (ATRP) is one of the most powerful methods ofliving transfer radical polymerization, which mainly requirescopper with nitrogen-based and halide ligands as catalysts.54
Meanwhile, reversible addition–fragmentation chain transfer(RAFT) polymerization is another frequently used polymerizationmethod, which could operate under simple conditions instead ofdry reagents or an inert atmosphere.55,56 These improved polymeri-zation methods have been employed to expand polymeric speciesfor more applications. As an important member of the polymerfamily, poly(glycidyl methacrylate) (PGMA) is a versatile polymerwhich draws researchers’ attention in the fields of polymer chem-istry, materials science and biochemistry. Various homopolymersand block copolymers based on PGMAs can be generated usingliving radical polymerization, pre-modification of monomers orinitiators, and post-modification of PGMAs. A series of linear andstar PGMAs was synthesized by us via ATRP polymerization.57,58
Efficient RAFT polymerization based on glycidyl methacrylate(GMA) was realized upon visible light radiation, resulting in well-defined PGMAs.59 Armes et al. utilized hydrolyzed GMA monomersto generate poly(glycerol methacrylate) (PGOHMA) chain-transferagent, which can further polymerize to obtain various blockcopolymers via RAFT dispersion polymerization.60,61 Benaglia andcoworkers made extensive efforts upon the post-modification ofPGMAs via ring opening reactions of different nucleophilic agents,such as thiols, aromatic alcohols, sodium azide and amines.62 Xuand we also utilized epoxy-amine chemistry to synthesize aminofunctionalized PGMAs.63,64 The PGMA derivatives possessing azidegroups can be further functionalized utilizing click chemistry. Sidechains with alkyne, such as polystyrene (PS), poly(ethylene oxide)(PEO), poly(e-caprolactone) (PCL), and mixed PEO–PCL, were suc-cessfully grafted onto linear azide modified PGMA derivatives,leading to block polymer brush, as reported by Chen et al.65,66
Different fluorescent molecules, possessing conventional chromo-phores (pyrene) or aggregation induced emission chromophores,i.e., tetraphenylethylene, were also modified on dendritic azidefunctionalized PGMA derivatives via click chemistry.67
Benefitting from the advances in polymer synthesis andfunctionalization, different topologies (brushes,65,66 linear,58
stars,68,69 dentritic,67 etc.) of PGMA derivatives have been pre-pared for the construction of more complicated architectures byself-assembly, such as traditional micelles70–73 (spherical, cylindric,wormlike, etc.), reverse micelles (RMs),74 capsules,75,76 and nano-particles.77 Self-assembly heavily relies on noncovalent interactions(van der Waals forces, electrostatic or hydrophobic interactions,hydrogen bonds and coordination bonds), which act as drivingforces for assembly formation.78 So, morphologies of PGMAderivatives are varied and even transform triggered by differentinternal and external factors, such as the length and proportionof the hydrophobic segment, concentration of copolymers, pH,solvent constituent, and temperature. Using a novel approachcalled polymerization-induced self-assembly (PISA), Armeset al. systematically studied the evolution of the assemblies’morphology based on poly(glycerol methacrylate)–poly(2-hydroxy-propyl methacrylate) (PGOHMA–PHPMA) in its progress ofkinetic RAFT dispersion polymerization, providing guidancefor obtaining certain morphologies of assemblies.79 There is asphere-to-worm-to-vesicle transition with the growth of thehydrophobic PHPMA chain. Their final morphologies alsodepend on the mean degree of polymerization of the hydro-philic PGOHMA blocks and the concentration of copolymers.80
The introduction of functional entities that are sensitive to pH,temperature, etc. onto PGMA backbones endow the assemblies ofPGMAs and their derivatives with environmental responsiveness.81
Meanwhile, the introduction of host–guest chemistry adds tothe vitality of PGMA-based self-assembly.68,69,82 Recently, Chuoand coworkers constructed polymeric materials with self-healingproperties using ferrocene-modified PGMAs and a difunctionalb-CD derivative via host–guest interaction between ferrocene andb-CD units.82
The research field of self-assembled architectures based onPGMAs and their derivatives combines polymer chemistry, supra-molecular chemistry and materials science, and has attractedmuch attention and advanced significantly during the last
Hui Gao
Hui Gao obtained her PhDin Chemistry from NankaiUniversity (2005). After threeyears of post-doctoral researchat the University of Montreal inCanada, she became an AssociateProfessor in Tianjin Universityof Technology. She obtainedan Invitation Fellowship forResearch in Japan in 2012, andworked in Prof. KazunoriKataoka’s lab in the Universityof Tokyo for 10 months. Hermain interests are the synthesis
and assembly properties of polymers, drug delivery, as well assupramolecular chemistry.
Ying-Wei Yang
Ying-Wei Yang received his degrees(BSc in 2000, PhD in 2005) fromNankai University. He gainedpostdoctoral training at ASU,UCLA, and UCI (2005–2010).Since 2011, he has been anAssociate Professor in the Collegeof Chemistry at Jilin University.He is also an Adjunct Professor atShenyang Pharmaceutical Univer-sity and serves on the EditorialBoard of Scientific Reports. Hisresearch interests include organicsupramolecular chemistry andstimuli-responsive polymers.
Feature Article ChemComm

This journal is©The Royal Society of Chemistry 2014 Chem. Commun., 2014, 50, 13201--13215 | 13203
two decades. Herein, we provide an overview of the self-assembled architectures based on PGMAs and their derivativesfrom literature reports focusing on the constructions of materialswith different morphologies, including polymeric micelles,polymeric capsules, hybrid materials and nanoparticles. Wethen highlight their potential applications as drug vectors, genevectors, solubilizing agents, antimicrobial agents, etc.
2. Conventional micelles and reversemicelles
Amphiphilic polymers can easily assemble into polymericmicelles (PMs) in selective solvents. Conventional PMs, with ahydrophobic core shielded from the external medium by ahydrophilic shell, are formed in aqueous solutions.83 The majordriving force for the self-assembly of amphiphilic copolymers isthe hydrophobic effect, leading to a decreased free energy of thesystem through the removal of the hydrophobic fragmentsfrom the incompatible aqueous environment via the formationof a micelle core protected by the shell of hydrophilic blocks.Correspondingly, some amphiphilic copolymers could also formmicelles with a polar core and hydrophobic exterior in apolarsolvents (dichloromethane (DCM), tetrahydrofuran (THF), etc.),i.e., RMs.84,85
As multifunctional polymers, various PGMA-based amphi-philic polymers have been prepared, and their self-assemblyproperties have been investigated over the past two decades.Two types of PMs based on PGMA derivatives, i.e., traditionalmicelles and RMs have been constructed. Their properties,such as dimension, stability and stimulative responsibility ofcore and corona, are dependent on the components of theirhydrophilic or hydrophobic regions. Thus, a variety of functionalgroups with certain responsiveness to pH, light, temperature, etc.were introduced into the self-assemblies of PGMA-based poly-mers through the post-modification method. Because of theirability to act as solubilizing agents for hydrophobic compounds,especially water-insoluble drugs in delivery systems, water-soluble PMs possessing different morphologies (spheres, tubes,cylinders, etc.) have been extensively studied. Different sphericalmicelles were obtained from diblock copolymers consisting ofhydrophobic poly(butyl methacrylate) (PBMA) and hydrophilicring-opened PGMAs at different block ratios. The longer thehydrophilic block, the better the stability and dispersion ofthe micelles.86 Well-defined brush-type amphiphilic diblockcopolymers, i.e., PGMA-block-poly(poly(ethylene glycol) meth-acrylate) (PPEGMA), self-assembled into wormlike micelles inwater.87 The micelles are composed of long, thin tubes of PGMAstabilized by a corona of the PPEGMA brushes.
When thermo-sensitive blocks are introduced into copolymers,PGMAs will be endowed with interesting properties. Poly-(N-isopropylacrylamide) (PNIPAM) is a thermo-sensitive polymerthat becomes insoluble upon increasing its solution temperatureover its lower critical solution temperature (LCST). PGMA-b-PNIPAM micelles were prepared with PGMA as the core andPNIPAM as the shell in a mixed solvent of methanol–THF
followed by core cross-linking.88 Different PGMA/PNIPAM ratiosexhibited different temperature dependencies of transparency.Li and co-workers synthesized perfluoroalkyl-terminated PGMA(RF-PGMA).89 Because of the strong hydrophobic interaction ofthe perfluoroalkyl groups, inter- and intramolecular hydrogenbonds between PGMA segments, RF-PGMA semitelechelics canself-associate to form spherical micelles in water, and the criticalmicelle concentration (CMC) values were found to increase withincreasing temperatures in water. Triblock copolymers, com-prised of poly(propylene oxide), poly(2-(dimethylamino)ethylmethacrylate) block, and PGMA, formed micelles with a criticalmicelle temperature of ca. 12 1C.90
Shell cross-linked (SCL) spherical micelles responsive to pHwere obtained from triblock copolymers, namely poly(ethyleneglycol) block (cinnamoyl modified PGOHMA-co-PGOHMA)block poly(2-(diethylamino)ethxyl methacrylate), PEG–(PCGMA-co-PGOHMA)–PDEA for short, above pH of 7–8.91 Molecularlydissolved copolymers in aqueous solutions at acidic pH valuescould, upon addition of NaOH, form three-layer ‘‘onion-like’’micelles consisting of PDEA cores, PCGMA-co-PGMA innershells, and PEG outer coronas, which could be further cross-linked upon ultraviolet (UV) irradiation to generate cyclobutanegroups at the inner shells. The size of the SCL micelles becamelarger upon protonation of tertiary amines of PDEA cores (seeFig. 1c). This pH-responsive behaviour was reversible. Theextent of cross-linking influenced the pH-dependent colloidalstability of SCL micelles.
Besides the sphere-shaped aggregates, other morphologicalmicelles of PGMA derivatives, such as cylindrical, tubular and awormlike-shape, have also been constructed. Core–shell–coronacylindrical micelles from PGMA-block-poly(2-cinnamoyloxy-ethyl
Fig. 1 (a) Schematic illustration of shell cross-linked (SCL) micelles formationof PEG–(PCGMA-co-PGMA)–PDEA triblock copolymers.91 (b) Chemicalstructure of the triblock copolymer of PGMA-b-PCEMA-b-PtBA.92 (c) Hydro-dynamic diameter distributions of PEG-(PCGMA-co-PGMA)-PDEA triblockcopolymers in aqueous solutions of pH 10 and pH 3, respectively.91 (d) TEMimage of PEG-(PCGMA-co-PGMA)-PDEA triblock copolymers obtained in anacidic solution (pH 3). (e) and (f) AFM images of PGMA-b-PCEMA-b-PtBAtriblock copolymer.92
ChemComm Feature Article

13204 | Chem. Commun., 2014, 50, 13201--13215 This journal is©The Royal Society of Chemistry 2014
methacrylate)-block-poly(tert-butylacrylate) (PGMA-b-PCEMA-b-PtBA) were reported by Liu et al.,92 whose shapes could beinfluenced by solvents. By dialysis of their aqueous solutionsagainst methanol, cylinders were translated into twisted cylinders(Fig. 1e and f) so that the incompatible and segregated PtBA andPGMA chains became balanced. In the mixed solvents of pyridineand methanol, with a volume fraction fMeOH of 80% to 100%,micelles with different morphologies (spheres, linear or helicalcylinders, and tubes) based on the PGMA-b-PCEMA-b-PtBAcopolymer were observed.93 So the composition of solvents isa crucial extrinsic factor governing the formation of variousmorphological micelles.
Fluorophores, such as pyrene, coumarin, and rhodamine B(RhB), have been anchored on reactive PGMAs to endow themwith photoluminescent properties.67,94,95 A PPEGMA block PGMAcopolymer was prepared and post-functionalized with RhB attachedto PGMA.94 Thus the ultraviolet-visible (UV-Vis) absorption spectraof RhB-functionalized copolymers show a strong absorption at558 nm. After dialysis against water, the hydrophobic PGMAsegments of the PPEGMA-b-PGMA copolymer aggregated intothe core of the micelles and its hydrophilic PPEGMA segmentsturned into the outer corona layers. While in THF, the copolymersformed RMs whose core–shell structure was opposite to that inwater, resulting in a significant increase in UV absorption values.For the sample standing for one month, PPEGMA-b-PGMA/RhBmicelles became smaller (from 500–900 nm to 100 nm) in THFdue to the strong interactions of RhB entities. The strongerphotoluminescence emission of the transformed micelles, com-pared to the relatively weak emission of the as-prepared RMs,demonstrated the aggregation enhanced photoluminescenceproperties of RhB entities in the block copolymers.
In terms of RMs, they have been primarily obtained fromhydrophobically modified dendrimers, hyperbranched and star-shaped polymers.84 PGMA derivatives are good candidates forestablishing RMs. Specialized potential applications of RMs inprotein encapsulation, the extraction of water-soluble biomolecules,etc. have prompted researchers to study PGMA-based RMs.
In 2006, Leroux and his coworkers first utilized 4-arm PGMAsfunctionalized by acyl chloride derivatives with differentlengths of carbon chains (12 to 18 carbons) to construct RMsand demonstrated its solubilizing function towards proteins,such as vasopressin, myoglobin, and so on.96 Then, in additionto 4-arm PGMA derivatives, other types of amphiphilic star-shaped alkylated PGOHMAs were also synthesized by us viaATRP using different initiators bearing 5 to 8 reactive sites andthe post-modification method, as shown in Fig. 2.58 By com-parison, their RMs size and loading capacity for Congo red weremostly related to the branching degree.57,74
Micelles are always unstable at high temperatures, at lowconcentrations and under certain changes in solvent conditions.Cross-linking of specific blocks within a copolymer micelle is afeasible method of stabilizing PMs.97,98 There are several potentiallocations for crosslinking within diblock polymer micelles, includingwithin the core domain, at the core–shell interface and throughoutthe shell layer. Cross-linking at the inner shell of PGMA derivatives,i.e., PEG–(PCGMA-co-PGOHMA)–PDEA triblock copolymer, via photo-dimerization of cinnamate groups, has been introduced.91 Whenthe pH was lowered from 10 to 3, SCL micelles maintained themicellar structures, while non-cross-linked ones dissociated intounimers, demonstrating a better stability of micelles after shellcross-linking. However, shell cross-linking must be performed inhighly diluted polymer solutions to limit the formation of covalentlybonded secondary aggregates. Such undesirable reactions could beprevented by cross-linking the cores rather than the corona. Thus,we chose a crosslinked agent, namely divinyl sulfone (DVS), toobtain core cross-linked (CCL) alkylated PGOHMAs,99 and showedthat particle size decreases upon CCL, as shown in Fig. 2.
3. Polymeric capsules
Compared with the solid structures of PMs presented above,polymeric capsules exhibit a hollow core surrounded by a polymermembrane, which can act as a reservoir for cargo delivery.100–102
Fig. 2 Schematic representations of linear, 4-, 5-, 6-, and 8-arm ATRP initiators, synthesis of alkylated PGOHMA, its core cross-linking reaction and theAFM image of core cross-linking RM from C18 alkylated PGOHMA.99
Feature Article ChemComm

This journal is©The Royal Society of Chemistry 2014 Chem. Commun., 2014, 50, 13201--13215 | 13205
In recent decades, many researchers are devoting themselves toconstruct novel polymeric capsules and investigate their bio-medical applications. Polymeric capsules include polymervesicles (polymersomes),103,104 polyelectrolyte capsules,105 etc.formed by different methods. Generally, two main approachescan be used to generate polymeric capsules: (i) self-assembly ofamphiphilic copolymers106 and (ii) layer-by-layer (LbL) assem-bly of polymers.107
Polymer vesicles are hollow, lamellar spherical capsules mostlyformed by the self-assembly of amphiphilic block copolymers inaqueous solutions with an aquatic core surrounded by a shell,namely a bilayer polymeric membrane of about 3–4 nm. Thepolymeric shell is composed of hydrated hydrophilic coronas bothon the inside and on the outside of the hydrophobic middle partof the membrane. Polymer vesicles are more robust and morestable compared to the liposomes, providing possibilities for itspotential applications under complex physical environments.
The amphiphilic reactive diblock copolymer vesicles whosewalls are composed of epoxy groups were first reported by Chen’sgroup using PEO-b-PGMA.108 The PEO-b-PGMA was dissolved inTHF and vesicles were formed by the addition of water (Fig. 3a).Their sizes were influenced by the length of the hydrophobicsegment (PGMA) in the copolymer and the water content (Cw) inthe mixed solvent of H2O–THF. A giant vesicle, of size over 5 mm,was obtained at 50 wt% Cw. When Cw was increased to 57 wt%,the sizes of the vesicles became smaller, which are in the range of500–1000 nm. They could be further used as a reactive scaffold forintroducing functionalities. Because the epoxy groups of PGMAare reactive to nucleophilic attack by various chemicals, thestructures of the obtained vesicles are more stable after ring-opening reaction by adding primary amine additives, such ashexamethylenediamine and dodecylamine. Furthermore, inorganic–organic hybrid vehicles (Fig. 3b) were also obtained uponthe reaction of PGMA with 3-aminopropyl trimethoxysilane,
which can be called a typical gelation reaction. The abovepresented methods helped to realize vesicle cross-linking andresulted in the increase of vesicle stability. Later, Armes et al.utilized the epoxy-amine reaction of polymeric diamines (PEGdiamine, Jeffamine) and the epoxy groups of PGMA to realize thecross-linking of PGOHMA55–(PHPMA247-co-PGMA82) copolymervesicles, leading to a relatively more stable structure that isresistant to the influence of ionic surfactants although with typicalmorphology changes due to phase separation compared withthose noncross-linked.109 Except for the epoxy-amine chemistryof diamines, further polymerization of PGOHMA–PHPMA diblockcopolymer vesicles can also generate a cross-linked structurewhen a bifunctional ethylene glycol dimethacrylate (EGDMA)was employed as a third comonomer.110
Amphiphilic asymmetric polymers with PGMA backbones andhydrophilic PEO and hydrophobic PS side chains or PEO and PS-b-PNIPAM side chains were synthesized using click chemistryand RAFT polymerization by Zhao and his coworkers.111 Thepolymeric vesicles were obtained by adding the THF solution ofPGMA-graft-(PEO/PS) copolymers with different PS chain lengthsto 6-fold of methanol that selectively dissolved PEO, but pre-cipitated PGMA and PS. The averages sizes of the structuresincreased with the increase of PS chain length, indicating theinfluence of the length of insoluble segments of copolymers onvesicle morphologies (Fig. 3d). Similar to PMs based on PGMAderivatives, morphologies of some vesicles are also responsiveto ambient temperature. The replacement of PS with PS-b-PNIPAM endowed vesicles of PGMA-graft-(PEO/PS-b-PNIPAM)temperature-sensitive. The detailed structure of the vesicles ispresented in Fig. 3c, where the hydrophobic PS blocks and thePGMA backbone form the vesicle wall; the longer PNIPAMblocks are segregated to the outer interface and the short PEO sidechains to the inside interface. At a temperature above the LCST(40 1C), PNIPAM brushes in the coronae of the vesicles shrank,
Fig. 3 (a) PEO-b-PGMA block copolymer structure and (b) TEM image of its vesicles in the presence of 3-aminopropyl trimethoxysilane (0.27 wt%).108
(c) A schematic representation of the self-assembly of the asymmetric macromolecular brush into the vesicle structure. (d) The average sizes of the structureswith different PS chain lengths. (e) Temperature-responsive transmittance changes of PGMA-graft-(PEO/PS31-b-PNIPAM53) in aqueous solutions.111
ChemComm Feature Article

13206 | Chem. Commun., 2014, 50, 13201--13215 This journal is©The Royal Society of Chemistry 2014
making the vesicles smaller. Amphiphilic comb polymers con-sisting of PGMA main chain, PNIPAM side chains and pendantpyrene groups (PGMA-g-Py-PNIPAM) were also prepared viasimilar methods.112 As a traditional fluorescent dye, pyreneexhibits an aggregation-caused quenching effect, which couldbe used to study the critical aggregation concentration (CAC) ofPGMA-g-Py-PNIPAM because the concentration of pyrene issharply increased with the formation of vesicles. Interestingly,thermo-sensitive PNIPAM affected the conformation of vesicles,but the fluorescence emission spectra of the polymer basicallykept unchanged due to the protection of the hydrophobicPGMA walls of the vesicles.
Solvent and temperature can not only change the size ofvesicles, but also promote conformation transfer of micellesinto vesicles (Fig. 4a). The copolymers containing supra-molecular host or guest, i.e., b-CD functionalized PGMA andthe copolymer of tert-butyl acrylate and hydroxylethyl acrylatemodified with adamantly groups (PtBA-ADA), were prepared viaATRP polymerization and pre-modification methods by Jianget al.113 Noncovalently connected micelles of PGMA-CD/PtBA-ADA were constructed by adding water into its dimethyl for-mamide (DMF) solution based on strong host–guest interactionof b-CD and ADA, and further cross-linked via the reaction ofglutaraldehyde and amine of PGMA-CDs. However, in DMF at ahigher temperature of 50 1C, the inclusion complexation ofb-CD and ADA was dramatically weakened. The cross-linkedmicelles turned into hollow spherical vesicles (Fig. 4c) only
based on PGMA-CD, upon treatment with DMF at 50 1C over-night to remove the micellar core, i.e., hydrophobic PtBA-ADA.
Colloidosome, as a typical microcapsule, can be constructed viathe self-assembly of particles, i.e., silica particles and polymerlatexes, utilizing structure directing agents, such as emulsiondroplets and gas bubbles.114 Armes and coworkers115 reportedthat PGOHMA–PS latexes with surface wettability aggregated easilyaround an oil droplet, resulting in stable microcapsules after cross-linking within the droplet phase via oil-soluble polymeric diiso-cyanate thus avoiding inter-colloidosome fusion caused by alcoholor surfactant. Significantly, they also obtained vesicle-based colloido-somes116 through the assembly around oil-in-water emulsions andinternal cross-linking of both linear PGMA45–PHPMA200-based vesi-cles and cross-linked PGOHMA58–PHPMA350–PEGDMA20-based vesi-cles (Fig. 5). However, in the presence of alcohol, the nanostructureof the former vesicles is destroyed while that of the latter ones isintact. In addition, the mean diameter of the microcapsules reduceswith the increasing concentration of the cross-linked vesicles withthe smallest size limit of B50 mm.
Polyelectrolyte capsules of PGMAs responsive to pH weresuccessfully constructed by us using LbL assembly.117 LbLassembly of multilayer films onto the surfaces of colloidalparticles followed by template removal is a facile and versatilemethod to construct functional capsules at the nanometer-level withcontrolled size, shape, composition, and wall thickness. Mono-disperse poly(acrylic acid), i.e., PAA, is rich in COO� groups and isemployed as polyanions. Functional groups of PGMA derivatives,such as multi-hydroxyl and amino groups, can coordinate withpolyanions via electrostatic interactions. Aminated PGOHMAs aregood candidates as polycations due to their water solubility, anddifferent SiO2 colloidal nanoparticles can serve as templates for theassembly of PAA and aminated PGOHMAs via electrostatic inter-action. The particle size increased and the Z-potential transitionbetween the positive charge and the negative charge revealed thegrowth of multilayers on SiO2. Then, SiO2 cores were removed byhydrogen fluoride (HF) treatment to obtain hollow microcapsules(Fig. 4b and d). Electrostatic interactions between PAA andaminated PGOHMAs could decrease with the protonation of
Fig. 4 (a) Vesicles built by the transformation of micelles and (c) their AFMimage.113 (b) Schematic illustration of the preparation of hollow aminatedPGOHMAs–PAA microcapsules and (d) TEM image of the final microcapsules.117
Fig. 5 Schematic representations of colloidosome formation from vesi-cles by PISA. TEM images of (a) cross-linked vesicles and (b) cross-linkedcolloidosomes.116
Feature Article ChemComm

This journal is©The Royal Society of Chemistry 2014 Chem. Commun., 2014, 50, 13201--13215 | 13207
aminated PGOHMAs, demonstrating the pH responsibility ofthe polyelectrolyte capsules.
4. Organic–inorganic hybrid materials
PGMAs, a typical type of active polymer with epoxy groups,mostly exist as ‘‘glue’’ for the construction of multifunctionalhybrid materials. Surface-initiated ATRP is a versatile methodto covalently graft a wide range of polymer chains onto curvedor flat surfaces, which can efficiently and well control thepolymer grafting to result in narrow molecular weight distribu-tions of polymers grown from the surfaces.118,119
Yang et al. reported multifunctional microsphere assemblieswith magnetic and fluorescent properties, where the PGMAshells, grown on silica microspheres using the surface-initiatedATRP technique, served as bridges to interact with CdTequantum dots, Au and oil-soluble Fe3O4 nanoparticles.120 Liuet al. prepared PGMA-coated silica nanoparticles using thesame method followed by CdTe coupling through the ring-opening reaction of epoxy groups.121 A biomolecule, the humanantirabbit tumor necrosis factor-alpha (TNF-a) antibody, wasalso attached to CdTe quantum dots-coated silica nanoparticlesfor TNF-a detection. Additionally, the PGMA brushes could beseparately synthesized by ATRP or RAFT and then grafted ontothe surface of the materials. A facile and straightforward methodto achieve multifunctional nanocomposites with fluorescent,magnetic, and specific recognition properties was reported byQi’s group. 7-Amino-4-methylcoumarin modified PGMAs with alarge number of residual epoxy groups prepared via ATRP and thering-opening reaction were grafted onto the surface of particlesand bestowed it recognition capacity to HeLa cells through theintroduction of transferrin at the epoxy groups.122
The reported hybrid materials based on PGMAs were diverse,but mostly constructed through covalent bonds. With the intro-duction of the concept of supramolecular self-assembly to the fieldof materials science, different kinds of assembled hybrid materialswere prepared, possessing unique advantages, such as responsive-ness to external stimuli (pH, light, solvent, temperature, etc.),123
and ordered space configurations, compared with the traditionalhybrid materials. Uniform PGMA particles with different size andsurface properties can be obtained by dispersion polymerization ofGMA,124,125 which can exist stably by themselves without inorganicparticles as their solid supports. Multifunctional organic–inorganichybrid materials can also be constructed from PGMAs whenmagnetic or fluorescent components diffuse into their particlesand undergo the corresponding reactions of epoxy groups on thesurface of particles with fluorescein isothiocyanate and folicacid afterwards. The hybrid microspheres prepared by this‘‘insertion’’ method with Fe3O4 nanoparticles, reported by Jingand co-workers,126 exhibited superparamagnetism, high mono-dispersity, intensive fluorescence, and capability of recognizingand binding cancer cells.
Multifunctional cationic superparamagnetic polymer nano-composites were reported by Iyer and Stubb et al.127 Poly-ethylenimine (PEI) chains were grafted onto the surface of
nanoparticles composed of RhB dye-modified PGMA shellsencapsulating magnetic Fe3O4 nanoparticles. The attachedPEI chains provide the nanoparticles with a positively chargednanoscale surface. b-Glucosidases, as a class of polyampholyticmolecules, can then be stabilized around the surface of nano-composites through hydrogen bonding and ion-pair interactions(Fig. 6).
Graphene with good electrical conductivity, abrasion resis-tance and fire retardancy has great potential in various technol-ogical fields, such as sensors and nanocomposites, and grapheneoxide (GO), is a water soluble graphene derivative. Luong andNam’s group used single GO sheet to wrap around aminefunctionalized PGMA microspheres via amide bonds, resultingin PGMA-ed–GO core–shell microspheres (Fig. 7a and b). Thecore–shell microspheres further self-assembled to interconnectedporous paper (Fig. 7c),128 mainly due to the formation of electro-static and van der Waals forces, and hydrogen bonds betweenthe very large surfaces of GO during the dewatering and dryingprocesses.
In addition to utilizing PGMA particles to form hybridmaterials, we have been working on the construction of hybridnanomaterials based on PGMA derivatives and mesoporoussilica nanoparticles (MSN) for controlled drug release. MSNsshowed wide applications in many research fields, such as drugdelivery and controlled release, biosensing, and bioimaging,mostly benefiting from their large specific surface area, suitablepore volumes, easy preparation and surface modification, goodstability and biocompatibility.129,130 We employed carboxylatedMSN (MSN-COOH), aminated MSN (MSN-NH2) and hollow MSNinstead of SiO2 colloidal nanoparticles as templates, and also asnanocontainers for controlled delivery of cargo molecules.131
Fig. 6 Multifunctional nanoparticles as nanoadditives for enzyme stabili-zation. Thermal stabilization studies of nanoparticles bearing almondb-glucosidase in comparison with naked b-glucosidase.127
ChemComm Feature Article

13208 | Chem. Commun., 2014, 50, 13201--13215 This journal is©The Royal Society of Chemistry 2014
The nanoassemblies were successfully constructed by LbLsupramolecular assemblies of polyelectrolytes, i.e., aminatedPGOHMAs (linear and 5 arms), PAA, on the surface of MSNs,deduced from the blurry mesopores after the four-layer coatingprocess. Compared with MSN-NH2, the nanoassemblies basedon MSN-COOH and hollow MSN exhibited good DOX releaseeffects in response to pH changes, which can be potentiallyapplied in cancer therapy.
Then, we constructed hybrid nanomaterials through the assem-bly of polymeric supramolecular macrocycles – star-shaped PGMAsmodified by supramolecular host b-CDs (5-arm PGMA-CDs), andthe azobenzene derivative functionalized mesoporous silica nano-particles (MSN-azo) via host–guest interactions, as shown inFig. 8.132 In aqueous solutions, b-CD exhibits weaker bindingaffinity towards cis-azobenzene derivatives and stronger interactionwith 1-adamantanamine hydrochloride compared with trans-azobenzene derivatives. So hybrid nanomaterials were respon-sive to UV-light and competitive binding agents. Additionally,temperature is an effective means to realize the disassembly ofthe hybrid due to the sensitivity of host–guest interactions
to temperature. This kind of polymeric supramolecular assemblyand disassembly based on MSN-azo and 5-arm PGMA-CD couldbe employed to construct polymeric nanovalves in the field ofcommand drug release. A monolayer of PGMA-CDs as gatingcomponents on solid supports has shown better efficiency fordrug delivery and controlled release compared with individualCDs. We are currently further investigating synthetic supra-molecular macrocycles-bridged multi-layered PGMA assemblieson the surface of MSN via host–guest interactions to realize LbLsupramolecular assemblies, and hopefully obtain novel andmore faithful hybrid materials for controlled cargo release.
5. Polymeric nanoparticles based onpolyion complexes
Proteins and nucleic acids have important potential applica-tions as therapeutic agents, while they are likely to experiencepremature breakdown and loss of activity. Their property ofassociating with oppositely charged polyelectrolytes to formstable polymeric nanoparticles via electrostatic interactionsprovides an approach to deal with this problem.133–135 So variouspolyelectrolytes were synthesized and their nanoparticles werestudied. They are mostly spherical particles with an averagediameter ranging between 10 and 1000 nm and are responsiveto external stimuli, such as temperature, pH, ions and ionicstrength of the surrounding, and chemical agents.136,137 As reactivepolymers, PGMAs could be easily post-functionalized, turning intocharged polyelectrolytes. A series of charged PGMA derivativespossessing pH-responsive groups (carboxyl, amino) was designedand synthesized by us.137–139
Carboxylated PGOHMAs (PGOHMACOOHs) were synthesized viathe hydrolysis of PGMA and further modified with succinicanhydride (SA) or 1,2-cyclohexanedicarboxylic anhydride (CDA),which afforded an abundance of carboxyl and hydroxyl groups,exhibiting pH-sensitivity.138 The resulting polymers with negativecharges can form polymeric nanoparticles with the oppositelycharged doxorubicin hydrochloride (DOX) by electrostatic inter-actions and hydrogen bonds (Fig. 9d). These particles could bedisassembled with the decrease of pH and show obvious releaseproperty to DOX at low pH, predicting potential applications forcationic drug delivery. Afterwards, we synthesized cross-linkedcarboxylated PGOHMAs utilizing hexamethylene diisocyanate (HDI)as a cross-linking agent, resulting in more stable assemblies.139
Nanoparticles including cationic aminated PGOHMAs werestudied extensively by Xu’s group and us. Various aminatedPGOGMAs were synthesized to be used as carriers for anionicbiomolecules, especially pDNA, as shown in Fig. 9b and c.Polymer structure, mass ratio of polymer/biomolecule, the environ-mental pH value and ionic strength influence the physicalproperties (size, potential, etc.) of nanoparticles.137 Applicationof polyelectrolytes of PGMA derivatives for targeted delivery andcontrolled release of different biomolecules would be describedin detail in the next section.
Host–guest chemistry helps the construction of diversifiedpolymeric nanoparticles. Xu and his coworkers further prepared
Fig. 7 The formation of the interconnected porous paper from thePGMA-ed–GO core–shell microspheres: (a), (b) TEM images of PGMA-ed–GO core–shell microspheres; (c) SEM image of the cross-section ofthe fractured PGMA-ed–GO paper.128
Fig. 8 Schematic diagram of multiple-stimuli responsive hybrid nano-composites based on mesoporous silica nanocontainers and PGMA-CDpolymers.132
Feature Article ChemComm

This journal is©The Royal Society of Chemistry 2014 Chem. Commun., 2014, 50, 13201--13215 | 13209
the b-CD-cored PGMA star polymer (CD-PGMA) via ATRP where theinitiator was bromoisobutyryl-terminated b-CD with four initiationsites.69 It reacted with ethanolamine to result in ethanolamine-functionalized CD-PGMA (CD-PGEA), which has similar propertiesto supramolecular hosts to form complexes with a suitable guest.Adamantine-modified linear PGMAs (l-PGEA-Ad) were obtained viaring-opening reactions with amantadine, generating l-PGEA-Ad–CD-PGEA complex via host–guest interactions with CD-PGEA. Thecomplexes with plentiful secondary amine and hydroxyl groupshave stronger non-covalent interactions with some biomolecules
(DNA, protein) compared with original CD-PGMA, resulting inpolymeric nanoparticles (Fig. 10). Very recently, we prepared a seriesof water-soluble PGMA derivatives (CD-DETA-PGOHMAs) contain-ing b-CD moieties as insulin carriers, showing excellent insulinrelease properties in response to competitive binding agents.140
CD-DETA-PGOHMA could form polymeric nanoparticles withinsulin by van der Waals forces and host–guest interactions, whichwas demonstrated by differential scanning calorimetry and Fouriertransform infrared spectroscopy. 1-Aminoadamantane hydrochloridehas a stronger host–guest interaction with b-CD moieties and thuscan serve as competitive binding agent to destroy the structure ofpolymeric nanoparticles, resulting in efficient insulin release.
6. Application in drug and genedelivery and others
PGMA is a versatile polymer because its pendant epoxidegroups can be opened with different organic functional groupssuch as amines, anhydride, or special components by post-polymerization modification. In the past decades, reverse poly-meric micelles, polymeric capsules, polymeric nanoparticles,and polymeric–inorganic hybrid materials based on PGMA andderivatives have shown practical applications for the delivery ofvasopressin, DOX, insulin, oligonucleotide and pDNA.138–148
We have also proved that PGMA and its aminated, alkylated,carboxylated, and cross-linked derivatives exhibited low cyto-toxicity in a series of in vitro and in vivo studies, showing greatpotential for biomedical applications.
PGOHMACOOHs form negatively charged nanoparticles andexhibit pH-sensitivity, which can be used as carriers for cationicdrugs.138 DOX is a commonly used cationic anti-cancer drug.141
Fig. 9 (a) Chemical structure of carboxylated PGOHMAs. (b) Schematic diagram of polyelectrolyte complexes formation of aminated PGOHMAs.137
(c) Table of the substituents of aminated PGOHMAs. (d) The SEM image of the complex of carboxylated PGOHMAs with DOX138 and (e) the AFM image ofthe complex of aminated PGOHMAs with antisense oligonucleotide.77
Fig. 10 Schematic illustration of the construction of L-PEMA-Ad–CD-PEMA–pDNA complexes.69
ChemComm Feature Article

13210 | Chem. Commun., 2014, 50, 13201--13215 This journal is©The Royal Society of Chemistry 2014
The encapsulation of DOX by PGOHMACOOH nanoparticleswas driven by both electrostatic interactions and hydrogenbonding between DOX and PGOHMACOOH. Loading capacityand release properties of these polymers were found to rely on notonly their backbone architectures but also the type of carboxylatedfunctionality. The drug release rate of all the complexes wasobviously pH dependent and the cumulative release under acidicconditions was higher than that under neutral conditions. In orderto enhance the stability and loading capacity of PGOHMACOOHnanoparticles, carboxylated PGOHMAs were further modified withHDI as an effective cross-linking agent.139 The formation of cross-links throughout PGOHMACOOHs enhanced the stability of thenanoassemblies by providing reinforcement to the weak intermo-lecular interactions.142 Compared with non-crosslinked PGOHMA-COOHs, crosslinked carboxylated PGOHMAs exhibited strongerbuffering capacity, more cumulative release (450%) at acidic pHenvironments, and less spontaneous leakage (o25%) at pH 7.4, asshown in Fig. 11a.
Similarly, polycations could complex with proteins, formingprotein polyelectrolyte complexes (PECs). For example, PECswere formed by weak physical interactions between insulin andvarious aminated PGOHMAs with positive charges.137 Underoptimal conditions of the pH values of PECs, the concentrationof NaCl, and the insulin/polymer mass ratio, PECs in the sizerange of 100–200 nm were obtained, and the insulin asso-ciation efficiency up to 86.7% could be obtained (Fig. 11b).
The release of insulin was adjustable by the structure variationof aminated PGOHMAs.
Consequently, we synthesized CD-DETA-PGOHMAs, pro-viding multi-active sites to include guest molecules, utilizingnot only electrostatic interactions but also host–guest inclusioncomplexation between polymer and insulin.140 The cytoviabilitywas significantly improved. Meanwhile, the association effici-ency and loading capacity were much higher, as compared to itsprecursor. All these evidenced that supramolecular host–guestinteractions contributed to the carrier formation and functionfor the delivery of insulin and potentially other therapeuticpolypeptides.
Except for forming complex with proteins, cationic polymershave attracted the most extensive attention mainly due to theirability to form stable PECs with nucleic acids such as pDNAs,antisense oligonucleotide (AON)64,143 or small interfering RNAs(siRNAs). In our previous study, PGMAs were modified withmethylethylamine (MEA), 2-amino-1-butanol (2-ABO), and4-amino-1-butanol (4-ABO) by ring-opening of the epoxidegroups.77 The soluble derivatives can efficiently complex withAON through electrostatic interactions and possibly hydrogenbonds (Fig. 9b). All polymers have the excellent capability ofbinding to AON, except for linear PGOHMA-MEA with lowamino density. Our studies validated that PGMAs modifiedwith amines and alkamines with low cytotoxicity were capableof mediating efficient gene transfection at lower N/P ratios,showing a better transfection efficacy than PEI. Thus, structuraltailoring of PGMA cationic copolymers provides a versatilemeans for designing novel gene carriers.
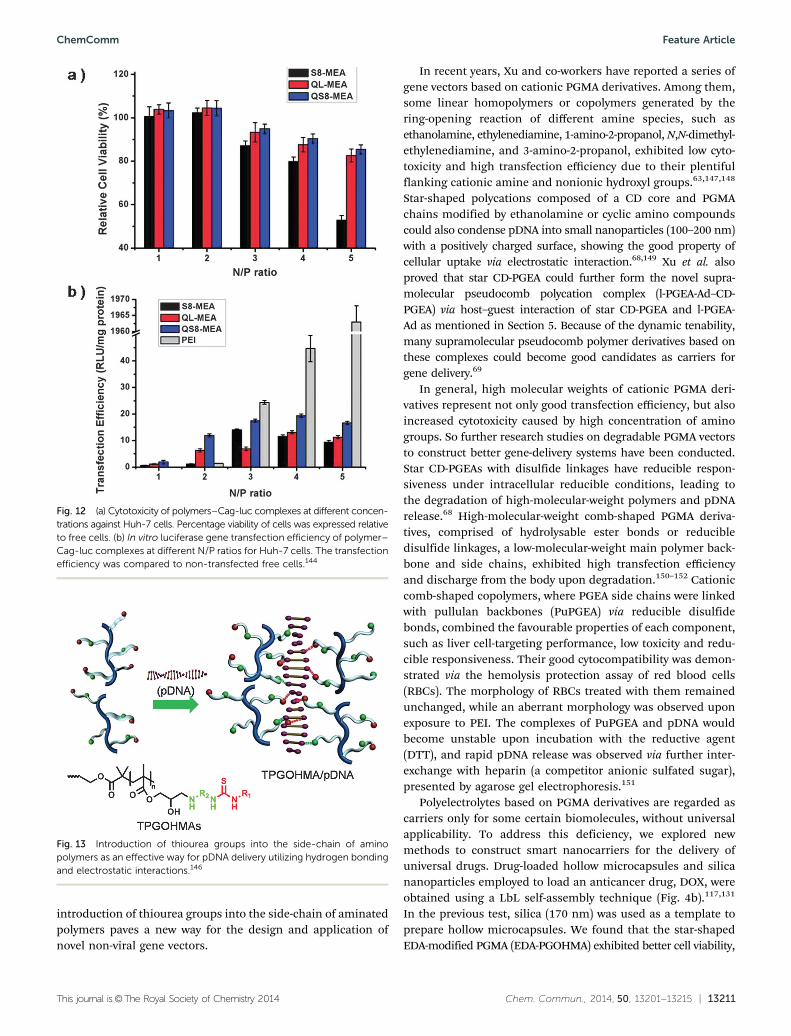
To further enhance cellular uptake, water solubility, and thetransfection efficiency and lower the cytotoxicity of PGMAderivatives, we designed and synthesized partially quaternaryammonium-functionalized aminated PGOHMAs (QPGOHMAs) bya quaternization reaction with methyl iodide (CH3I).144 Quaternaryammonium-functionalized MEA-modified 8-arm PGMA (QS8-MEA)and pDNA complexes were spherical in shape and smaller in size(150 nm) than MEA-modified 8-arm PGMA (S8-MEA) and pDNAcomplexes (400 nm) as suggested by dynamic light scattering.Gel electrophoresis and ethidium bromide displacement assayindicated that QPGOHMA with lower cytotoxicity upon quater-nization (Fig. 12a) could condense pDNA at lower N/P ratios(N/P ratio 1.5) than that of S8-MEA (N/P ratio 2). QS8-MEA/pDNA complexes at the N/P ratio of 4 showed a higher pDNAuptake than those of S8-MEA/pDNA (Fig. 12b).
Thiourea has been shown to interact actively with phosphategroups of DNA.145 Hence, we synthesized thiourea-modifiedaminated PGOHMAs (TPGOHMAs) with methyl isothiocyanate(M).146 Aminated PGOHMAs gained from ring opening reactions ofPGMAs with 1,2-ethanediamine, 1,4-butanediamine and diethylene-triamine. The mean hydrodynamic diameters of aminatedPGOHMA/pDNA and TPGOHMA/pDNA complexes (Fig. 13)were in the range of 65–195 nm, and decreased with increasingN/P ratios. We demonstrated that TPGOHMAs could condensepDNA at lower N/P ratios, exhibited obviously lower cytotoxicityand positive Z-potentials. The fast release of pDNA, as well asthe enhancement of endosomal escape indicated that the
Fig. 11 (a) DOX release profile of L-PGOHMA-CDA-HDI and S5-PGOHMA-CDA-HDI.139 (b) Insulin release profile of different aminated PGOHMAs.137
Feature Article ChemComm
This journal is©The Royal Society of Chemistry 2014 Chem. Commun., 2014, 50, 13201--13215 | 13211
introduction of thiourea groups into the side-chain of aminatedpolymers paves a new way for the design and application ofnovel non-viral gene vectors.
In recent years, Xu and co-workers have reported a series ofgene vectors based on cationic PGMA derivatives. Among them,some linear homopolymers or copolymers generated by thering-opening reaction of different amine species, such asethanolamine, ethylenediamine, 1-amino-2-propanol, N,N-dimethyl-ethylenediamine, and 3-amino-2-propanol, exhibited low cyto-toxicity and high transfection efficiency due to their plentifulflanking cationic amine and nonionic hydroxyl groups.63,147,148
Star-shaped polycations composed of a CD core and PGMAchains modified by ethanolamine or cyclic amino compoundscould also condense pDNA into small nanoparticles (100–200 nm)with a positively charged surface, showing the good property ofcellular uptake via electrostatic interaction.68,149 Xu et al. alsoproved that star CD-PGEA could further form the novel supra-molecular pseudocomb polycation complex (l-PGEA-Ad–CD-PGEA) via host–guest interaction of star CD-PGEA and l-PGEA-Ad as mentioned in Section 5. Because of the dynamic tenability,many supramolecular pseudocomb polymer derivatives based onthese complexes could become good candidates as carriers forgene delivery.69
In general, high molecular weights of cationic PGMA deri-vatives represent not only good transfection efficiency, but alsoincreased cytotoxicity caused by high concentration of aminogroups. So further research studies on degradable PGMA vectorsto construct better gene-delivery systems have been conducted.Star CD-PGEAs with disulfide linkages have reducible respon-siveness under intracellular reducible conditions, leading tothe degradation of high-molecular-weight polymers and pDNArelease.68 High-molecular-weight comb-shaped PGMA deriva-tives, comprised of hydrolysable ester bonds or reducibledisulfide linkages, a low-molecular-weight main polymer back-bone and side chains, exhibited high transfection efficiencyand discharge from the body upon degradation.150–152 Cationiccomb-shaped copolymers, where PGEA side chains were linkedwith pullulan backbones (PuPGEA) via reducible disulfidebonds, combined the favourable properties of each component,such as liver cell-targeting performance, low toxicity and redu-cible responsiveness. Their good cytocompatibility was demon-strated via the hemolysis protection assay of red blood cells(RBCs). The morphology of RBCs treated with them remainedunchanged, while an aberrant morphology was observed uponexposure to PEI. The complexes of PuPGEA and pDNA wouldbecome unstable upon incubation with the reductive agent(DTT), and rapid pDNA release was observed via further inter-exchange with heparin (a competitor anionic sulfated sugar),presented by agarose gel electrophoresis.151
Polyelectrolytes based on PGMA derivatives are regarded ascarriers only for some certain biomolecules, without universalapplicability. To address this deficiency, we explored newmethods to construct smart nanocarriers for the delivery ofuniversal drugs. Drug-loaded hollow microcapsules and silicananoparticles employed to load an anticancer drug, DOX, wereobtained using a LbL self-assembly technique (Fig. 4b).117,131
In the previous test, silica (170 nm) was used as a template toprepare hollow microcapsules. We found that the star-shapedEDA-modified PGMA (EDA-PGOHMA) exhibited better cell viability,
Fig. 12 (a) Cytotoxicity of polymers–Cag-luc complexes at different concen-trations against Huh-7 cells. Percentage viability of cells was expressed relativeto free cells. (b) In vitro luciferase gene transfection efficiency of polymer–Cag-luc complexes at different N/P ratios for Huh-7 cells. The transfectionefficiency was compared to non-transfected free cells.144
Fig. 13 Introduction of thiourea groups into the side-chain of aminopolymers as an effective way for pDNA delivery utilizing hydrogen bondingand electrostatic interactions.146
ChemComm Feature Article

13212 | Chem. Commun., 2014, 50, 13201--13215 This journal is©The Royal Society of Chemistry 2014
high loading capacity (42%) and entrapment efficiency (84%).All the mocrocapsules showed pH-sensitivity, i.e., DOX releasedfaster under acidic conditions, compared with neutral pH. Inorder to make more stable nanocontainers, inorganic nano-particles, i.e., mesoporous silica nanoparticles, were introducedto act as the core and the vessel for drug storage. We utilizedhost–guest chemistry to obtained stimuli-responsive polymerichybrid nanoparticles that have been described in the previouspart in detail.131,132
Amphiphilic alkylated PGOHMAs were shown to self-assemble into RMs in organic solvents and oil. In the presenceof RMs, vasopressin, which is highly soluble in water but haslimited affinity for a polar solvent, could be transferred into theoil phase.58 Furthermore, the ability of RMs to solubilizevasopressin in oil decreased with increasing peptide loadinguntil saturation of the available space was achieved. The amountof vasopressin released from RMs was less than 15% in one week.In terms of such an extended release, RMs were good candidatesfor the administration of peptides at parenteral or extravascular,which can reduce the frequency of injections and enhance thecompliance of patients. The investigation of the release kineticsof vasopressin from RM-containing oil-in-water coarse emulsionsconfirmed a possible use of RMs for the oral delivery of peptidesas well. The potential benefit of RMs as sustained drug deliverysystems was verified following the subcutaneous administrationof vasopressin formulated in 4-arm stearoyl PGOHMA RMs(S4-18C60) to rats. As shown in Fig. 14a, at all time points, thevolume of urine produced by the rats that received the RMformulations was the lowest, but the efficacy of RMs wasmaintained even after 60 h.
In addition to the applications as carriers for biomolecules,there are other applications of PGMA-based polymers. Theore-tically, their RMs can extract hydrophilic cargos from water toorganic solution owing to its hydrophilic cores (Fig. 14b).74 Theability of RMs to act as solubilizing agents was testified througha simple liquid–liquid extraction procedure, whereby the hydro-philic, anionic dye, Congo red, was dissolved in water andshaken over an immiscible organic phase containing micelles.Core cross-linked RMs provided improved extraction stabilityfor dyes, as compared to non-cross-linked RMs.42
The interesting and meaningful application of homopolymervesicle based on PGMA derivative in water remediation was reportedby Du et al.153 The homopolymer, i.e. poly[2-hydroxy-3-(naphthalen-1-ylamino)propyl methacrylate] (PHNA), was synthesized via RAFTpolymerization using amphiphilic 1-naphthylamine modified GMAmonomer, which could self-assemble into vesicles (Fig. 15a) withouta clear boundary between hydrophobic and hydrophilic segments, byforming inter/intramolecular hydrogen bonds. Both physical adsorp-tion and catalyzed chemical reactions of these homopolymer vesiclescould remove noxious polycyclic aromatic hydrocarbons from water.As a representative aromatic molecule, pyrene was successfullyabsorbed by vesicles through p–p interactions with the naphthalenependants of PHNA, which was deduced from the fact that thefluorescence of pyrene in aqueous solution could be quenchedrapidly upon addition of PHNA vesicle solutions (Fig. 15b). Phenoland phenolic compounds, for example 4-nitrophenol, exist as one of
the major pollutants in water and could be removed through moreefficient catalytic reduction based on AuNPs immobilized on PHNAvesicles compared with ordinary Au sol.
In addition, we also tested the antimicrobial activity154–156
of PGMA-based polymers.157 During the incubation period,
Fig. 14 (a) The subcutaneous administration of vasopressin formulated inS4-18C60 RMs to rats on day one.58 (b) Congo red, brilliant blue R and methylorange were extracted from an aqueous layer in the presence of S6-12C60
RMs (right vials). DCM without RMs was used as control (left vials).74
Fig. 15 (a) The formation of homopolymer vesicles by self-assemblyof PHNA and AuNPs@vesicles; (b) fluorescence quenching of pyrene-contaminated water by adding PHNA vesicle solutions of different con-centrations (from 3.90 to 125 mg mL�1). Adsorption time: 6 min; (c) monitorof the UV-vis absorbance changes of 4-nitrophenol at 402 nm withdifferent additives.153
Feature Article ChemComm

This journal is©The Royal Society of Chemistry 2014 Chem. Commun., 2014, 50, 13201--13215 | 13213
the spread bacteria can grow only where there is no antimicro-bial agent, which indicated that aminated PGOHMAs haveantibacterial activity, and quaternized aminated PGOHMAsperform the best. Multifunctional PGMA involved materialswere fabricated to achieve other capabilities, such as separationof cancer cells126 stabilization of enzymes,127 quantitative analysisof drugs,86 and adhesion towards cells.158,159
7. Conclusions and perspectives
Upon reflection, PGMA-based polymers have been playing asignificant role in the process of the development of polymerself-assembly. Multiple functionalized PGMA derivatives withthermosensitivity, pH-responsiveness, luminescence emission orother properties, have been synthesized by the combination ofliving radical polymerization, ring-opening addition, and clickchemistry to meet different requirements of constructing stimuli-responsive self-assembly architectures. The self-assembly methodstend to be more flexible with the introduction of supramolecularchemistry and materials science. Their aggregates formed by self-assembly of amphiphilic copolymers or LbL assembly techniquevia noncovalent interactions, such as electrostatic interactions,hydrogen bonding, hydrophobic interactions, and host–guest inter-actions, presented different types of materials, such as micelles,capsules, nanoparticles, and hybrid materials.
Moreover, the novel approach of in situ PISA was introducedby Armes et al. to generate various morphological assembliesbased on PGMA derivatives in recent five years.70,79,80,160,161
Compared with the traditional method for the self-assembly ofthe copolymer, it is a facile strategy for the large scale prepara-tion of well-defined nanostructures and could effectively pro-ceed even in concentrated solutions. However, the species ofmonomers employed to polymerize based on PGOHMA chain-transfer agent is very limited so far. Further study of otherfunctional monomers in the systems of PISA is necessary andmeaningful.
We have presented here not only the self-assembly propertiesof PGMAs and their derivatives, but also the potential applica-tions of the formed assemblies, such as carriers for the delivery ofanti-cancer drugs and biomolecules, such as DOX, vasopressin,insulin, oligonucleotide and pDNA, and their controlled releaseunder a range of internal or external stimuli, i.e., pH, light,temperature, and competitive binding. Although certain achieve-ments have been attained on the self-assembly of PGMAs, there isstill plenty of room to develop PGMA-based polymer chemistryand search for other potential applications. As the basis of furtherresearch, a remarkably diverse and large variety of PGMA-basedpolymers and copolymers could be synthesized to expand thepolymer library. There are still some disadvantages or problemsfor PGMAs-based nanoassemblies, and new fabrication strategiesare also expected to be pursued to obtain more novel and smartassembly architectures or nanocomposites, fulfilling the require-ments for new applications. The focus may be on the construc-tion of multifunctional hybrid self-assembly materials ofPGMAs as carriers with fluorescence, specific recognition ability,
multiple stimuli-responsiveness and many other biologicalrelevant abilities.162–166 There is no doubt that further sub-stantial achievements on the self-assembly of PGMAs and theirderivatives are closely associated with the intense collabora-tions of experts from different research fields.
Acknowledgements
We gratefully acknowledge the National Natural Science Foun-dation of China (21272093, 21374079), the Innovation Programof the State Key Laboratory of Supramolecular Structure andMaterials, and the Program for New Century Excellent Talentsin University (NCET-11-1063) for financial support. We alsothank the Key Laboratory of Pesticide and Chemical Biology,Ministry of Education, Central China Normal University (201301A01)for financial support.
Notes and references1 X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41,
6042–6065.2 C. Wang, Z. Wang and X. Zhang, Acc. Chem. Res., 2012, 45, 608–618.3 H. Hoffmann, Adv. Mater., 1994, 6, 116–129.4 Z. Chu, C. A. Dreiss and Y. Feng, Chem. Soc. Rev., 2013, 42,
7174–7203.5 J. Eastoe and A. Vesperinas, Soft Matter, 2005, 1, 338–347.6 S. Dong, B. Zheng, F. Wang and F. Huang, Acc. Chem. Res., 2014,
DOI: 10.1021/ar5000456.7 J. W. Lee, S. Samal, N. Selvapalam, H.-J. Kim and K. Kim, Acc. Chem.
Res., 2003, 36, 621–630.8 H. Zhang and Y. Zhao, Chem. – Eur. J., 2013, 19, 16862–16879.9 D.-S. Guo and Y. Liu, Acc. Chem. Res., 2014, DOI: 10.1021/ar500009g.
10 A. Cazacu, C. Tong, A. van der Lee, T. M. Fyles and M. Barboiu,J. Am. Chem. Soc., 2006, 128, 9541–9548.
11 F. Huang and H. W. Gibson, Chem. Commun., 2005, 1696–1698.12 T. Xiao, X. Feng, Q. Wang, C. Lin, L. Wang and Y. Pan, Chem.
Commun., 2013, 49, 8329–8331.13 S. J. Cantrill, G. J. Youn and J. F. Stoddart, J. Org. Chem., 2001, 66,
6857–6872.14 H. Onagi, B. Carrozzini, G. L. Cascarano, C. J. Easton, A. J. Edwards,
S. F. Lincoln and A. D. Rae, Chem. – Eur. J., 2003, 9, 5971–5977.15 X. Ma and H. Tian, Acc. Chem. Res., 2014, DOI: 10.1021/ar500033n.16 X. Zheng, D. Wang, Z. Shuai and X. Zhang, J. Phys. Chem. B, 2012,
116, 823–832.17 Y. Liu, Y.-L. Zhao, H.-Y. Zhang and H.-B. Song, Angew. Chem.,
Int. Ed., 2003, 42, 3260–3263.18 Y.-W. Yang, Y. Chen and Y. Liu, Inorg. Chem., 2006, 45, 3014–3022.19 V. H. S. Tellini, A. Jover, J. C. Garcıa, L. Galantini, F. Meijide and
J. V. Tato, J. Am. Chem. Soc., 2006, 128, 5728–5734.20 W. Deng, H. Yamaguchi, Y. Takashima and A. Harada, Angew.
Chem., Int. Ed., 2007, 46, 5144–5147.21 V. Villari, G. Gattuso, A. Notti, A. Pappalardo and N. Micali, J. Phys.
Chem. B, 2012, 116, 5537–5541.22 C. Bonaccorso, C. Sgarlata, G. Grasso, V. Zito, D. Sciotto and
G. Arena, Chem. Commun., 2011, 47, 6117–6119.23 S. Pasquale, S. Sattin, E. C. Escudero-Adan, M. Martınez-Belmonte
and J. de Mendoza, Nat. Commun., 2012, 3, 785.24 S. Kennedy, G. Karotsis, C. M. Beavers, S. J. Teat, E. K. Brechin and
S. J. Dalgarno, Angew. Chem., Int. Ed., 2010, 49, 4205–4208.25 D.-S. Guo, T.-X. Zhang, Y.-X. Wang and Y. Liu, Chem. Commun.,
2013, 49, 6779–6781.26 D.-S. Guo, K. Wang, Y.-X. Wang and Y. Liu, J. Am. Chem. Soc., 2012,
134, 10244–10250.27 M.-X. Wang and H.-B. Yang, J. Am. Chem. Soc., 2004, 126, 15412–15422.28 J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew.
Chem., Int. Ed., 2005, 44, 4844–4870.29 Y. Liu, Z. Huang, X. Tan, Z. Wang and X. Zhang, Chem. Commun.,
2013, 49, 5766–5768.
ChemComm Feature Article

13214 | Chem. Commun., 2014, 50, 13201--13215 This journal is©The Royal Society of Chemistry 2014
30 I. Hwang, W. S. Jeon, H.-J. Kim, D. Kim, H. Kim, N. Selvapalam,N. Fujita, S. Shinkai and K. Kim, Angew. Chem., Int. Ed., 2007, 46,210–213.
31 J. de Barrio, P. N. Horton, D. Lairez, G. O. Lloyd, C. Toprakciogluand O. A. Scherman, J. Am. Chem. Soc., 2013, 135, 11760–11763.
32 X.-L. Ni, X. Xiao, H. Cong, Q.-J. Zhu, S.-F. Xue and Z. Tao, Acc.Chem. Res., 2014, 47, 1386–1395.
33 M. Xue, Y. Yang, X. Chi, Z. Zhang and F. Huang, Acc. Chem. Res.,2012, 45, 1294–1308.
34 T. Ogoshi, K. Yoshikoshi, T. Aoki and T.-a. Yamagishi, Chem.Commun., 2013, 49, 8785–8787.
35 J. Yang, G. Yu, D. Xia and F. Huang, Chem. Commun., 2014, 50,3993–3995.
36 D. Xia, G. Yu, J. Li and F. Huang, Chem. Commun., 2014, 50, 3606–3608.37 Q. Duan, Y. Cao, Y. Li, X. Hu, T. Xiao, C. Lin, Y. Pan and L. Wang,
J. Am. Chem. Soc., 2013, 135, 10542–10549.38 K. Wang, C.-Y. Wang, Y. Wang, H. Li, C.-Y. Bao, J.-Y. Liu, S. X.-A.
Zhang and Y.-W. Yang, Chem. Commun., 2013, 49, 10528–10530.39 A. P. Nowak, V. Breedveld, L. Pakstis, B. Ozbas, D. J. Pine,
D. Pochan and T. J. Deming, Nature, 2002, 417, 424–428.40 J. W. Y. Lam and B. Z. Tang, Acc. Chem. Res., 2005, 38, 745–754.41 A. P. Dove, Chem. Commun., 2008, 6446–6470.42 J. Du and R. K. O’Reilly, Soft Matter, 2009, 5, 3544–3561.43 W. Yuan, T. Shen, J. Wang and H. Zou, Polym. Chem., 2014, DOI:
10.1039/C4PY00463A.44 Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985.45 D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973.46 P. J. M. Stals, Y. Li, J. Burdynska, R. Nicolay, A. Nese, A. R. A.
Palmans, E. W. Meijer, K. Matyjaszewski and S. S. Sheiko, J. Am.Chem. Soc., 2013, 135, 11421–11424.
47 A. Harada, R. Kobayashi, Y. Takashima, A. Hashidzume andH. Yamaguchi, Nat. Chem., 2011, 3, 34–37.
48 S. Yagai and A. Kitamura, Chem. Soc. Rev., 2008, 37, 1520–1529.49 S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2008, 4, 435–449.50 Q. Zhang, N. R. Ko and J. K. Oh, Chem. Commun., 2012, 48,
7542–7552.51 M. A. Ward and T. K. Georgiou, Polymers, 2011, 3, 1215–1242.52 R. B. Grubbs and Z. Sun, Chem. Soc. Rev., 2013, 42, 7436–7445.53 W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32,
93–146.54 K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039.55 J. Du, H. Willcock, J. P. Patterson, I. Portman and R. K. O’Reilly,
Small, 2011, 7, 2070–2080.56 C. L. Mccormick and A. B. Lowe, Acc. Chem. Res., 2004, 37, 312–325.57 H. Gao, M.-C. Jones, P. Tewari, M. Ranger and J.-C. Leroux, J. Polym.
Sci., Part A: Polym. Chem., 2007, 45, 2425–2435.58 M.-C. Jones, H. Gao and J.-C. Leroux, J. Controlled Release, 2008,
132, 208–215.59 H. Yin, H. Zheng, L. Lu, P. Liu and Y. Cai, J. Polym. Sci., Part A:
Polym. Chem., 2007, 45, 5091–5102.60 Y. Li and S. P. Armes, Angew. Chem., Int. Ed., 2010, 49, 4042–4046.61 L. P. D. Ratcliffe, A. J. Ryan and S. P. Armes, Macromolecules, 2013,
46, 769–777.62 M. Benaglia, A. Alberti, L. Giorgini, F. Magnoni and S. Tozzi, Polym.
Chem., 2013, 4, 124–132.63 X. B. Dou, M. Y. Chai, Y. Zhu, W. T. Yang and F. J. Xu, ACS Appl.
Mater. Interfaces, 2013, 5, 3212–3218.64 H. Gao, M. Elsabahy, E. V. Giger, D. Li, R. E. Prud’homme and
J.-C. Leroux, Biomacromolecules, 2010, 11, 889–895.65 P. Zhao, Y. Yan, X. Feng, L. Liu, C. Wang and Y. Chen, Polymer,
2012, 53, 1992–2000.66 Y. Yan, Y. Shi, W. Zhu and Y. Chen, Polymer, 2013, 54, 5634–5642.67 S. Li and C. Gao, Polym. Chem., 2013, 4, 4450–4460.68 Y. Hu, Y. Zhu, W. T. Yan and F. J. Xu, ACS Appl. Mater. Interfaces,
2013, 5, 703–712.69 Y. Hu, M. Y. Chai, W. T. Yang and F. J. Xu, Bioconjugate Chem.,
2013, 24, 1049–1056.70 A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath,
C. W. I. Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134,9741–9748.
71 K. Sha, D. Li, Y. Li, X. Liu, S. Wang, J. Guan and J. Wang, J. Polym.Sci., Part A: Polym. Chem., 2007, 45, 5037–5049.
72 I. Gadwal, J. Rao, J. Baettig and A. Khan, Macromolecules, 2014, 47,35–40.
73 R. Verber, A. Blanazs and S. P. Armes, Soft Matter, 2012, 8,9915–9922.
74 H. Gao, M.-C. Jones, J. Chen, Y. Liang, R. E. Prud’homme andJ.-C. Leroux, Chem. Mater., 2008, 20, 4191–4193.
75 J. Rosselgong, A. Blanazs, P. Chambon, M. Williams, M. Semsarilar,J. Madsen, G. Battaglia and S. P. Armes, ACS Macro Lett., 2012, 1,1041–1045.
76 K. L. Thompson, S. P. Armes and D. W. York, Langmuir, 2011, 27,2357–2363.
77 H. Gao, X. Lu, Y. Ma, Y.-W. Yang, J. Li, G. Wu, Y. Wang, Y. Fan andJ. Ma, Soft Matter, 2011, 7, 9239–9247.
78 X. Zhang and C. Wang, Chem. Soc. Rev., 2011, 40, 94–101.79 A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes,
J. Am. Chem. Soc., 2011, 133, 16581–16587.80 A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45,
5099–5107.81 Y. Tang, L. Liu, J. Wu and J. Duan, J. Colloid Interface Sci., 2013,
397, 24–31.82 T.-W. Chuo, T.-C. Wei and Y.-L. Liu, J. Polym. Sci., Part A: Polym.
Chem., 2013, 51, 3395–3403.83 S. C. Owen, D. P. Y. Chan and M. S. Shoichet, Nano Today, 2012, 7,
53–65.84 M.-C. Jones and J.-C. Leroux, Soft Matter, 2010, 6, 5850–5859.85 H. M. Jung, K. E. Price and D. T. McQuade, J. Am. Chem. Soc., 2003,
125, 5351–5355.86 Y. Li, L. Qi, J. Qiao, Y. Shen, H. Yan, P. Xin and H. Ma, Electro-
phoresis, 2012, 33, 2019–2027.87 Z. Cheng, X. Zhu, E. T. Kang and K. G. Neoh, Langmuir, 2005, 21,
7180–7185.88 T. Sato, S. Tsuji and H. Kawagauchi, Ind. Eng. Chem. Res., 2008, 47,
6358–6361.89 Z. Li, E. Amado and J. Kressler, Colloid Polym. Sci., 2013, 291, 867–877.90 L. N. Pilon, S. P. Armes, P. Findlay and S. P. Rannard, Langmuir,
2005, 21, 3808–3813.91 X. Jiang, S. Luo, S. P. Armes, W. Shi and S. Liu, Macromolecules,
2006, 39, 5987–5994.92 J. Hu, G. Njikang and G. Liu, Macromolecules, 2008, 41, 7993–7999.93 G. Njikang, D. Han, J. Wang and G. Liu, Macromolecules, 2008, 41,
9727–9735.94 S.-C. Chang, S.-J. Chiu, C.-Y. Hsu, Y. Chang and Y.-L. Liu, Polymer,
2012, 53, 4399–4406.95 V. Tsyalkovsky, V. Klep, K. Ramaratnam, R. Lupitskyy, S. Minko
and I. Luzinov, Chem. Mater., 2008, 20, 317–325.96 M.-C. Jones, P. Tewari, C. Blei, K. Hales, D. J. Pochan and J.-C.
Leroux, J. Am. Chem. Soc., 2006, 128, 14599–14605.97 A. Guo, G. Liu and J. Tao, Macromolecules, 1996, 29, 2487–2493.98 R. K. O’Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006,
35, 1068–1083.99 H. Gao, M.-C. Jones, J. Chen, R. E. Prud’homme and J.-C. Leroux,
Chem. Mater., 2008, 20, 3063–3067.100 A. Kowalczuk, R. Trzcinska, B. Trzebicka, A. H. E. Muller,
A. Dworak and C. B. Tsvetanov, Prog. Polym. Sci., 2014, 39, 43–86.101 P. Tanner, P. Baumann, R. Enea, O. Onaca, C. Palivan and
W. Meier, Acc. Chem. Res., 2011, 44, 1039–1049.102 R. P. Brinkhuis, F. P. J. T. Rutjes and J. C. M. van Hest, Polym.
Chem., 2011, 2, 1449–1462.103 G. Fuks, R. M. Talom and F. Gauffre, Chem. Soc. Rev., 2011, 40,
2475–2493.104 L. Fan, H. Lu, K. D. Zou, J. Chen and J. Du, Chem. Commun., 2013,
49, 11521–11523.105 G. Sukhorukov, A. Fery and H. Mohwald, Prog. Polym. Sci., 2005, 30,
885–897.106 B. M. Discher, Y.-Y. Won, D. S. Ege, J. C.-M. Lee, F. S. Bates,
D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146.107 W. Tong, X. Song and C. Gao, Chem. Soc. Rev., 2012, 41, 6103–6124.108 H. Zhu, Q. Liu and Y. Chen, Langmuir, 2007, 23, 790–794.109 P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Langmuir,
2012, 28, 1196–1205.110 P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Macro-
molecules, 2012, 45, 5081–5090.111 X. Lian, D. Wu, X. Song and H. Zhao, Macromolecules, 2010, 43,
7434–7445.112 C. Zhao, D. Wu, X. Lian, Y. Zhang, X. Song and H. Zhao, J. Phys.
Chem. B, 2010, 114, 6300–6308.
Feature Article ChemComm

This journal is©The Royal Society of Chemistry 2014 Chem. Commun., 2014, 50, 13201--13215 | 13215
113 J. Wang and M. Jiang, J. Am. Chem. Soc., 2006, 128, 3703–3708.114 T. Brugarolas, F. Tu and D. Lee, Soft Matter, 2013, 9, 9046–9058.115 K. L. Thompson, S. P. Armes, J. R. Howse, S. Ebbens, I. Ahmad,
J. H. Zaidi, D. W. York and J. A. Burdis, Macromolecules, 2010, 43,10466–10474.
116 K. L. Thompson, P. Chambon, R. Verber and S. P. Armes, J. Am.Chem. Soc., 2012, 134, 12450–12453.
117 Y. Sun, H. Gao, Y.-W. Yang, A. Wang, G. Wu, Y. Wang, Y. Fan andJ. Ma, J. Biomed. Mater. Res., Part A, 2013, 101A, 2164–2173.
118 F. J. Xu, Q. J. Cai, Y. L. Li, E. T. Kang and K. G. Neoh, Biomacro-molecules, 2005, 6, 1012–1020.
119 S. Edmondson and W. T. S. Huck, J. Mater. Chem., 2004, 14,730–734.
120 Z. Wang, Z. Zhao, J. Zhang, Z. Li, Y. Gao, C. Wang, H. Zhang andB. Yang, J. Colloid Interface Sci., 2009, 339, 83–90.
121 L. Yuan, X. Hua, Y. Wu, X. Pan and S. Liu, Anal. Chem., 2011, 83,6800–6809.
122 Y. Shen, L. Zhao, L. Qi, J. Qiao, L. Mao and Y. Chen, Chem. – Eur. J.,2012, 18, 13755–13761.
123 H. Zhang, Y. Liu, D. Yao and B. Yang, Chem. Soc. Rev., 2012, 41,6066–6088.
124 W. L. Zhang, S. H. Piao and H. J. Choi, J. Colloid Interface Sci., 2013,402, 100–106.
125 B. Elmas, M. Tuncel, G. Yalçın, S. Senel and A. Tuncel, ColloidsSurf., A, 2005, 269, 125–134.
126 X. Dong, Y. Zheng, Y. Huang, X. Chen and X. Jing, Anal. Biochem.,2010, 405, 207–212.
127 T. D. Clemons, C. W. Evans, B. Zdyrko, I. Luzinov, M. Fitzgerald,S. A. Dunlop, A. R. Harvey, K. S. Iyer and K. A. Stubbs, Nanoscale,2011, 3, 4085–4087.
128 J. Oh, J.-H. Lee, J. C. Koo, H. R. Choi, Y. Lee, T. Kim, N. D. Luongand J.-D. Nam, J. Mater. Chem., 2010, 20, 9200–9204.
129 Y.-W. Yang, Med. Chem. Commun., 2011, 2, 1033–1049.130 Y.-L. Sun, Y. Zhou, Q.-L. Li and Y.-W. Yang, Chem. Commun., 2013,
49, 9033–9035.131 Y. Sun, Y.-L. Sun, L. Wang, J. Ma, Y.-W. Yang and H. Gao,
Microporous Mesoporous Mater., 2014, 185, 245–253.132 Q.-L. Li, L. Wang, X.-L. Qiu, Y.-L. Sun, P.-X. Wang, Y. Liu, F. Li,
A.-D. Qi, H. Gao and Y.-W. Yang, Polym. Chem., 2014, 5, 3389–3395.133 M. Soliman, S. Allen, M. C. Davies and C. Alexander, Chem.
Commun., 2010, 46, 5421–5433.134 A. B. Kayitmazer, D. Seeman, B. B. Minsky, P. L. Dubin and Y. Xu,
Soft Matter, 2013, 9, 2553–2583.135 A. Tamura and Y. Nagasaki, Nanomedicine, 2010, 5, 1089–1102.136 A. E. Felber, M.-H. Dufresne and J.-C. Leroux, Adv. Drug Delivery
Rev., 2012, 64, 979–992.137 X. Lu, H. Gao, C. Li, Y.-W. Yang, Y. Wang, Y. Fan, G. Wu and J. Ma,
Int. J. Pharm., 2012, 423, 195–201.138 Y. Ma, H. Gao, W. Gu, Y.-W. Yang, Y. Wang, Y. Fan, G. Wu and
J. Ma, Eur. J. Pharm. Sci., 2012, 45, 65–72.139 W. Gu, Y. Ma, C. Zhu, B. Chen, J. Ma and H. Gao, Eur. J. Pharm. Sci.,
2012, 47, 556–563.
140 L. Wang, Y.-W. Yang, M. Zhu, G. Qiu, G. Wu and H. Gao, RSC Adv.,2014, 4, 6478–6485.
141 C. M. Alexander, M. M. Maye and J. C. Dabrowiak, Chem. Commun.,2011, 47, 3418–3420.
142 G. Bitan and D. B. Teplow, Acc. Chem. Res., 2004, 37, 357–364.143 H. Gao, M. Elsabahy, E. V. Giger, J. Ma, R. E. Prud’homme and
J.-C. Leroux, J. Controlled Release, 2011, 152, 142–143.144 Z. Liang, X. Wu, Y.-W. Yang, C. Li, G. Wu and H. Gao, Polym. Chem.,
2013, 4, 3514–3523.145 J. Leblond, N. Mignet, L. Leseurre, C. Largeau, M. Bessodes,
D. Scherman and J. Herscovici, Bioconjugate Chem., 2006, 17, 1201–1208.146 C. Li, Y.-W. Yang, Z.-X. Liang, G.-L. Wu and H. Gao, Polym. Chem.,
2013, 4, 4366–4374.147 F. J. Xu, Y. Zhu, M. Y. Chai and F. S. Liu, Acta Biomater., 2011, 7,
3131–3140.148 F. J. Xu, M. Y. Chai, W. B. Li, Y. Ping, G. P. Tang, W. T. Yang, J. Ma
and F. S. Liu, Biomacromolecules, 2010, 11, 1437–1442.149 R. Q. Li, Y. L. Niu, N. N. Zhao, B. R. Yu, C. Mao and F. J. Xu,
ACS Appl. Mater. Interfaces, 2014, 6, 3969–3978.150 R. Q. Li, Y. Hu, B. R. Yu, N. N. Zhao and F. J. Xu, Bioconjugate
Chem., 2014, 25, 155–164.151 X. C. Yang, Y. L. Niu, N. N. Zhao, C. Mao and F. J. Xu, Biomaterials,
2014, 35, 3873–3884.152 X. C. Yang, M. Y. Chai, Y. Zhu, W. T. Yang and F. J. Xu, Bioconjugate
Chem., 2012, 23, 618–626.153 Y. Zhu, L. Fan, B. Yang and J. Du, ACS Nano, 2014, 8, 5022–5031.154 W. Yuan, J. Wei, H. Lu, L. Fan and J. Du, Chem. Commun., 2012, 48,
6857–6859.155 H. Lu, L. Fan, Q. Liu, J. Wei, T. Ren and J. Du, Polym. Chem., 2012,
3, 2217–2227.156 C. Zhang, Y. Zhu, C. Zhou, W. Yuan and J. Du, Polym. Chem., 2013,
4, 255–259.157 Z. Liang, M. Zhu, Y.-W. Yang and H. Gao, Polym. Adv. Technol.,
2014, 25, 117–122.158 F. J. Xu, Z. H. Wang and W. T. Yang, Biomaterials, 2010, 31, 3139–3147.159 W. Yuan, C. Li, C. Zhao, C. Sui, W.-T. Yang, F.-J. Xu and J. Ma, Adv.
Funct. Mater., 2012, 22, 1835–1842.160 K. L. Thompson, S. P. Armes, D. W. York and J. A. Burdis, Macro-
molecules, 2010, 43, 2169–2177.161 M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Langmuir,
2013, 29, 7416–7424.162 J. Harrison, C. A. Bartlett, G. Cowin, P. K. Nicholls, C. W. Evans,
T. D. Clemons, B. Zdyrko, I. A. Luzinov, A. R. Harvey, K. S. Iyer,S. A. Dunlop and M. Fitzgerald, Small, 2012, 8, 1579–1589.
163 C. W. Evans, M. Fitzgerald, T. D. Clemons, M. J. House, B. S.Padman, J. A. Shaw, M. Saunders, A. R. Harvey, B. Zdyrko,I. Luzinov, G. A. Silva, S. A. Dunlop and K. S. Iyer, ACS Nano,2011, 5, 8640–8648.
164 J. Du and S. P. Armes, Soft Matter, 2010, 6, 4851–4857.165 J. Du and R. K. O’Reilly, Chem. Soc. Rev., 2011, 40, 2402–2416.166 X. C. Yang, M. Y. Chai, Y. Zhu, W. T. Yang and F. J. Xu, J. Mater.
Chem., 2012, 22, 7806–7812.
ChemComm Feature Article