self consistent polarization electronic structure · 2011-10-21 · self consistent polarization...
TRANSCRIPT

Self Consistent Polarization Electronic Structure Gregory K. Schenter Pacific Northwest National Laboratory
“Chemical Dynamics: Challenges and Approaches” Institute for Mathematics and Its Applications, University of Minnesota January 12-16, 2009
Funding: DOE BES Chemical Sciences

Self Consistent Polarization (SCP) Electronic Structure = (NDDO, DFT, HF, …)
SCP-NDDO: Danny Chang (US EPA) Bruce Garrett (PNNL) JCP 128, 164111 (2008).
SCP-DFT (Water): Chris Mundy (PNNL) Garold Murdachaew (PNNL)
SCP-DFT (Argon): J. Ilja Siepmann (U. Minn.) Katie Maerzke (U. Minn.) JPCA (in press)
Future development in CP2K: Juerg Hutter (Zurich) Teo Liano (Zurich) Joost Vandevondele (Zurich)

Characterize molecular interaction
Represent molecular interaction Construct a Hamiltonian Statistical sampling and
dynamics of an ensemble Predict and understand
properties
Approach: Molecular Simulation
€
VBO RN( )( )
€
H =Pi2
2mi
+V R N( )( )i=1
N
∑
€
Tr e−βH[ ], R N( ) t( ),P N( ) t( )( )
€
A , A t( )B 0( )
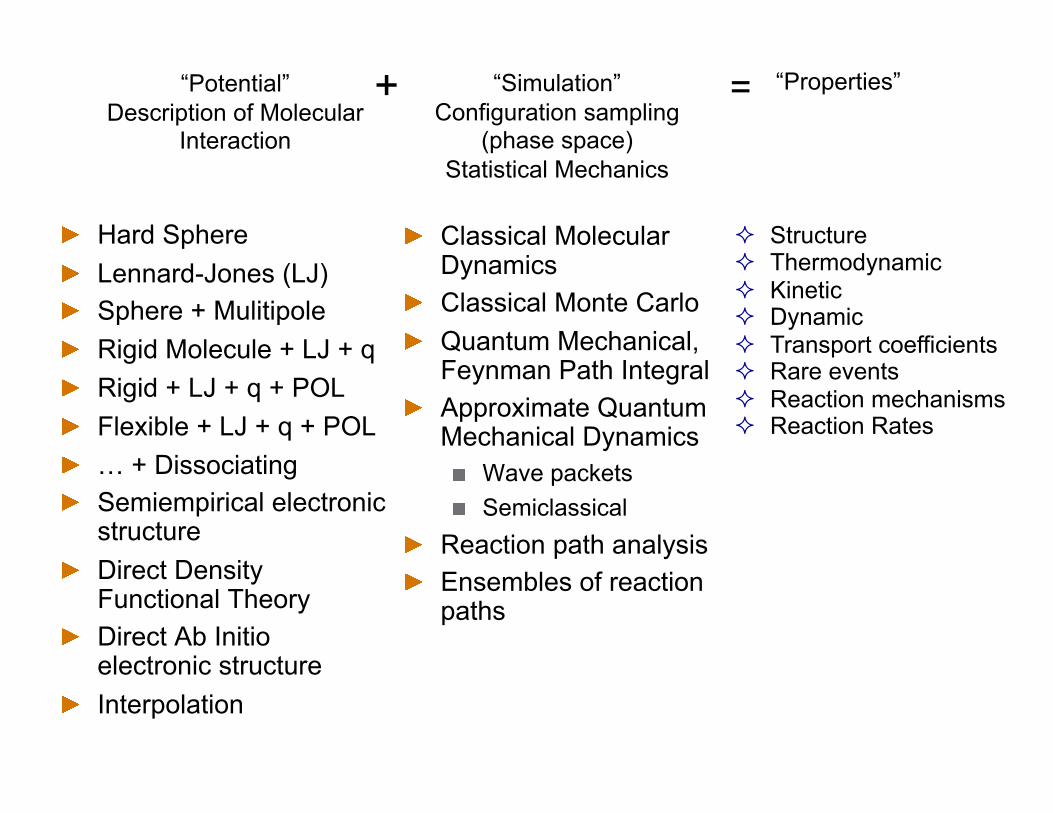
Hard Sphere Lennard-Jones (LJ) Sphere + Mulitipole Rigid Molecule + LJ + q Rigid + LJ + q + POL Flexible + LJ + q + POL … + Dissociating Semiempirical electronic
structure Direct Density
Functional Theory Direct Ab Initio
electronic structure Interpolation
Classical Molecular Dynamics
Classical Monte Carlo Quantum Mechanical,
Feynman Path Integral Approximate Quantum
Mechanical Dynamics Wave packets Semiclassical
Reaction path analysis Ensembles of reaction
paths
“Potential” Description of Molecular
Interaction
“Simulation” Configuration sampling
(phase space) Statistical Mechanics
Structure Thermodynamic Kinetic Dynamic Transport coefficients Rare events Reaction mechanisms Reaction Rates
“Properties” + =

Mixing and Matching Representations of Molecular Interaction is Desired. It is practical, more accurate, more efficient. QM/MM Semiempirical NDDO = HF electronic structure
with empirical terms replacing “real” terms. MM/POL MM/1e- QM, e- QM/H+ QM (NEO) Multilevel, Layered Electronic Structure DFTB, Harris functional, Empirical Valence Bond Duel level dyamics, SRP SCP-NDDO, SCP-DFT … What is the proper way to do this?

Hartree- Fock Electronic Structure
Semi-Empirical Electronic Structure (NDDO)
MM/POL Empirical Molecular Potentials With Polarization
Density Functional Theory
Common Elements: Charge Density Responding to Nuclear Configuration
The distinction between these approaches can become blurred.
A key component is the dependence of the energy of the system on the details of the charge density, described by an energy functional:
Coulomb is King
€
E ρ[ ] = dr∫ dr 'ρ r( )ρ r'( )r − r '
€
E ρ[ ]
Quantum mechanics can be implemented in terms of variational principles.
€
δE ρ[ ]δρ
= 0 Subject to constraints.

Mathematical Challenge: Partitioning Charge Density and Corresponding Energy Functional Space
€
E ρ1 + ρ2[ ] = E1 ρ1[ ] + Eapprox ρ1,ρ2[ ] + E2 ρ2[ ]
€
ρ1
€
ρ2

Self-consistent Polarization (SCP)-NDDO Use a polarization density matrix pµν {s,x,y,z} Charge polarization pss Dipole polarization psx, psy, psz Quadrupole polarization {pxx, pyy, pzz, pxy, pxz, pyz} Dispersion via 2nd-order perturbation theory
Semiempirical SCF methods + Ability to treat the formation and breaking of chemical bonds
– Poor description of H-bonds and weak electrostatic complexes
Semiempirical MOs
Empirical methods – Poor description of chemical reactions (e.g., bond-breaking and formation) + Ability to add missing elements of H-bonding using classical electrostatic models
Classical Electrostatics

Representation of The Charge Density
€
ρA r( ) = ZAδµν ,ss − Pµν ;AAT( )φµ r( )φν r( )
µν
∑ + MAl( ) ⋅ ∇A
l( )
l∑ f r − rA( )
From Semiempirical NDDO: Localized Basis Set, Core + Density Matrix
From Empirical Potentials: Multipole Polarizability

Screened Coulomb Interactions
€
drd ′ r ρA r( )ρB ′ r ( )
r − ′ r ∫ =
MA0( ) + MA
1( ) ⋅ ∇A + MA2( ) :∇A∇A +( ) MB
0( ) + MB1( ) ⋅ ∇B + MB
2( ) :∇B∇B +( ) s rAB( )rAB€
A ≠ B
€
s rAB( )rAB
=1
rAB2 + a2
€
s rAB( )rAB
=1rAB
1− 1+12a
e−arAB
Klopman-Ohno: Slater:

Self-Interaction
€
A = B
€
Eself = Pµµ;AAT Uµ
A
µ
∑
A
∑ +12
Pµν ;AAT Gµν ;µ ν
A Pµ ν ;AAT − Pµν ;AA
γ Hµν ;µ ν A Pµ ν ;AA
γ
γ
∑
µ ν
∑µν
∑
NDDO Atomic Hamiltonian
€
+12
MAl( ) ⋅
l, ′ l ∑
A∑ 1
a l , ′ l ( ) ⋅MA′ l ( )
Generalized Multipole Polarization (linear response)

Bonding
€
Eres =12
Pµλ ;ABT (βµ
A + βλB )Sµλ ;AB
µλ
∑B≠A∑
A∑
€
Eex = −12
Pµλ ;ABγ Pνσ ;AB
γ µAνA( | λBσB)γ
∑λσ
∑B≠A∑
µν
∑A∑
NDDO Resonance and Exchange

SCP-NDDO provided an accurate description of water clusters, (H2O)n
We have extended the ideas of SCP-NDDO to DFT and the condensed phase
"Self-consistent polarization neglect of diatomic differential overlap: Application to water clusters," Daniel T. Chang, Gregory K. Schenter, and Bruce C. Garrett, J. Chem. Phys. 128, 164111 (2008).
TTM2-R: Burnham and Xantheas, JCP 115, 15000 (2002)
Energy
(H2O)2 (H2O)3 (H2O)4 (H2O)5 (H2O)6

1 2 1 2

SCP-DFT
€
EDFT ρDFT[ ] = T ρDFT[ ] + EH ρDFT[ ] + EXC ρDFT[ ]
€
ESCP−DFT ρDFT ,ρSCP[ ] = T ρDFT[ ] + EH ρDFT + ρSCP[ ] + EXC ρDFT[ ] + ESE ρSCP[ ]
€
EH ρ[ ] = dr∫ dr 'ρ r( )ρ r'( )r − r '

SCP approach to improving DFT has its origins in linear response theory
CJ Mundy et al JCP 123, 074108 (2005) • “Parameter free” based on pure DFT
CJ Mundy et al JCP 117, 1416 (2002) • “Parameter free” based on empirical potential
DFT/

DFT-DFT
SCP-SCP
DFT-SCP
Coulomb Integrals:
Screening, exclude same site interaction
Currently:

or
Self-consistent polarization (SCP)-DFT: • Polarization functions provide “correction” • Only a single parameter, aα • Polarization will yield a straight forward path to a self-consistent determination of dispersion
Subject to wavefunction orthonormality constraints Gives conventional Density Functional Theory
SCF SCP Procedure

We obtain a self-consistent formula for multipole dispersion by considering a quantum mechanical
interaction between auxiliary densities
We obtain the “Generalized London” formula with the introduction of two additional parameters
Our perturbation:
2nd order expression
€
Edisp = α β( ) fαβ α β ( ) fα β
0 ˆ c α k 0 ˆ c β l k ˆ c α 0 l ˆ c β
0E0
α + E0β − Ek
α − Elβ
l≠0∑
k≠0∑
Casimir-Polder:

SCP-DFT parametrization procedure for water
1. Use CP2K with BLYP/GTH pseudopotential/TZV2P-MOLOPT basis sets on O and H atoms (low BSSE, BSTE)
2. Use single auxiliary basis function of p symmetry on O and H atoms
3. O, H atoms: set ap (same for O and H currently) to “reasonable” value
4. Molecular cohesion energies of (H2O)n clusters with optimized geometries: choose Ip (same for O and H currently) to recover the MP2 and TTM-2F cohesion energies of clusters, n=2-16
5. (later we compared “rigidified” clusters to those from CC-pol-8s potential)
G Murdachaew, CJ Mundy & GK Schenter (in preparation).

CP2K: Our state-of-the-art computational tool Features
KS-DFT (OR classical force fields) Ab initio molecular dynamics (Born-Oppenheimer) Dual basis (Gaussian + PW) allows for good scaling on many processors GTH pseudopotentials (OR all e- using Gaussian augmented plane waves, GAPW) QM/MM Semi-empirical methods recently implemented (MNDO, NDDO, PM3, . . . ) XAS (x-ray absorption spectra for e.g. studies of hydrogen bond network in water, core excitations) SIC (self-interaction correction to correct exchange in DFT; sometimes needed for oxides) Metadynamics for rare events, NEB for transition state searches, . . . Classical and quantum dynamics of nuclei Our recent contributions: SCP-NDDO and SCP-DFT
CP2K (http://cp2k.berlios.de, code freely available); main developers are Juerg Hutter (U Zurich) Teodoro Laino (IBM Zurich) Joost VandeVondele (U Zurich) Matthias Krack (ETH Zurich) Axel Kohlmeyer (U Penn) Chris Mundy (PNNL) (GKS and GM work with him to make improvements in SCP-DFT) William Kuo (LLNL)
Code allows for readability, modification, and rapid development Written in Fortran 95 with modular programming philosophy
(modules, user-defined types, dynamic memory and pointers, excellent portability, . . . ) Highly efficient and fully parallelized
Discussion forum exists (http://groups.google.com/group/cp2k)
J VandeVondele M Krack F Mohamed M Parrinello T Chassaing & J Hutter ComputPhysCommun 167 103 (2005).

R Bukowski et al Science 315 1249 (2007). CC-POL = Empirical Potential fit to CCSD(T) electronic structure SS Xantheas, CJ Burnham, & RJ Harrison, JChemPhys 116 1493 (2002). = MP2/CBS

R Bukowski et al Science 315 1249 (2007).

R Bukowski et al Science 315 1249 (2007).
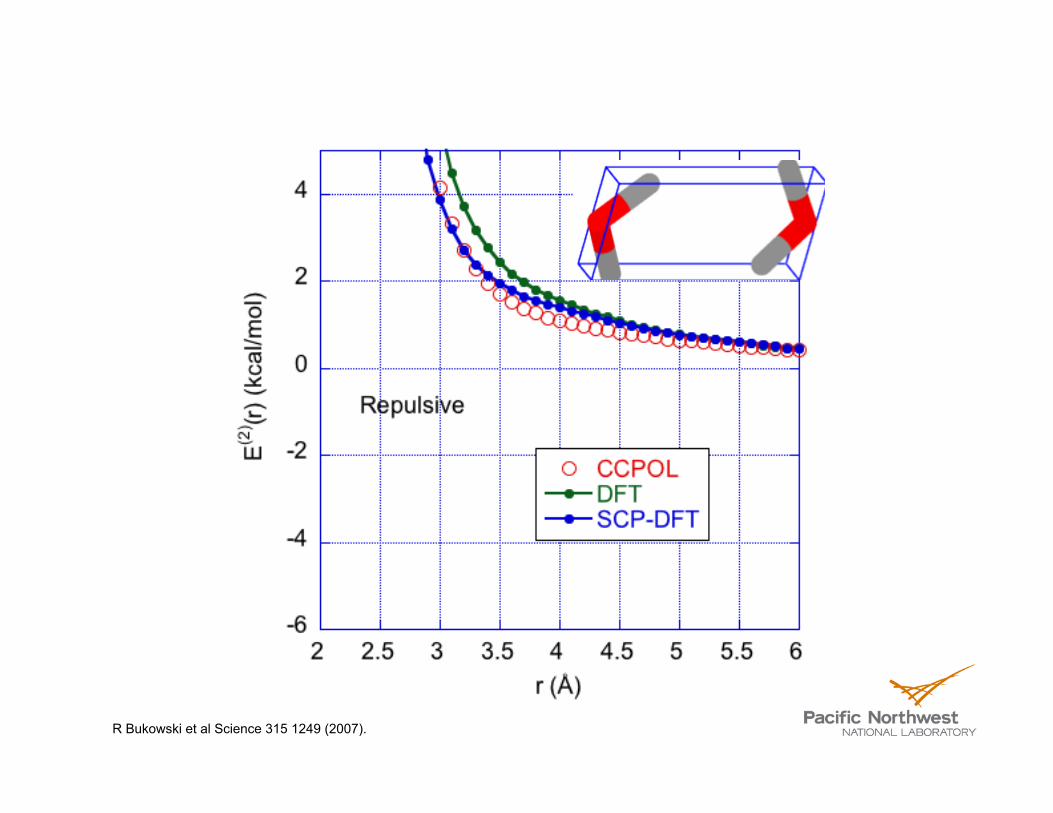
R Bukowski et al Science 315 1249 (2007).
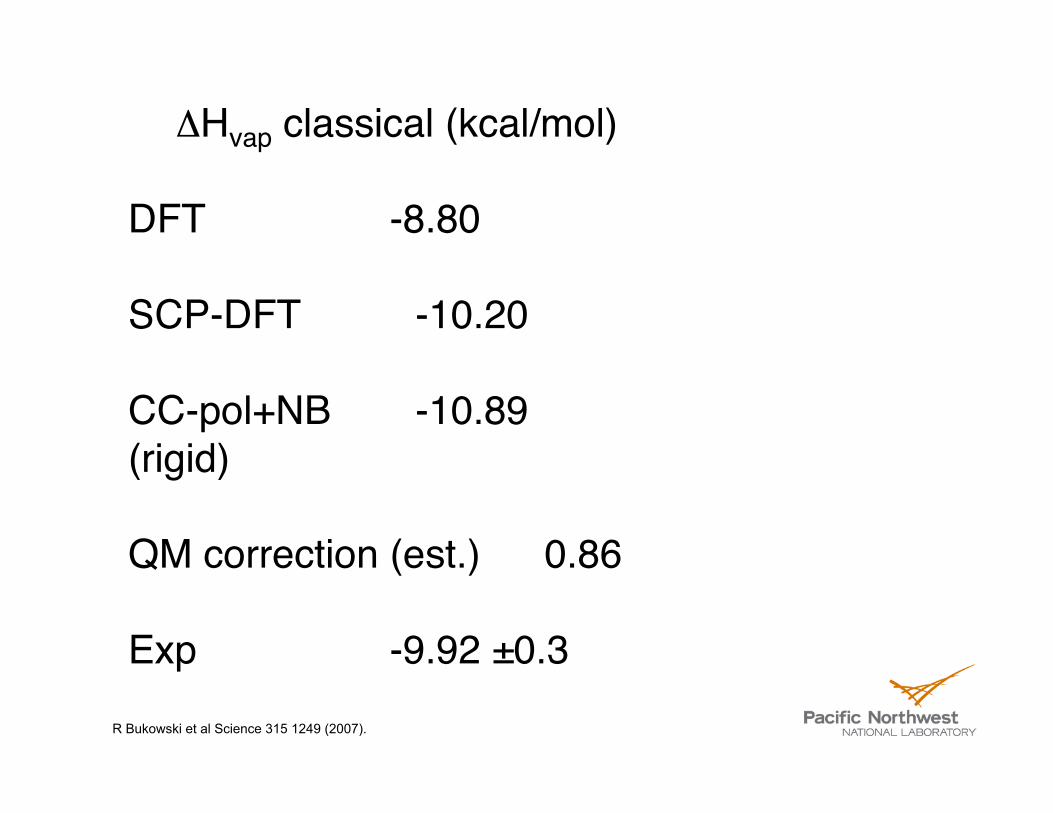
ΔHvap classical (kcal/mol)
DFT -8.80
SCP-DFT -10.20
CC-pol+NB -10.89 (rigid)
QM correction (est.) 0.86
Exp -9.92 ±0.3 R Bukowski et al Science 315 1249 (2007).

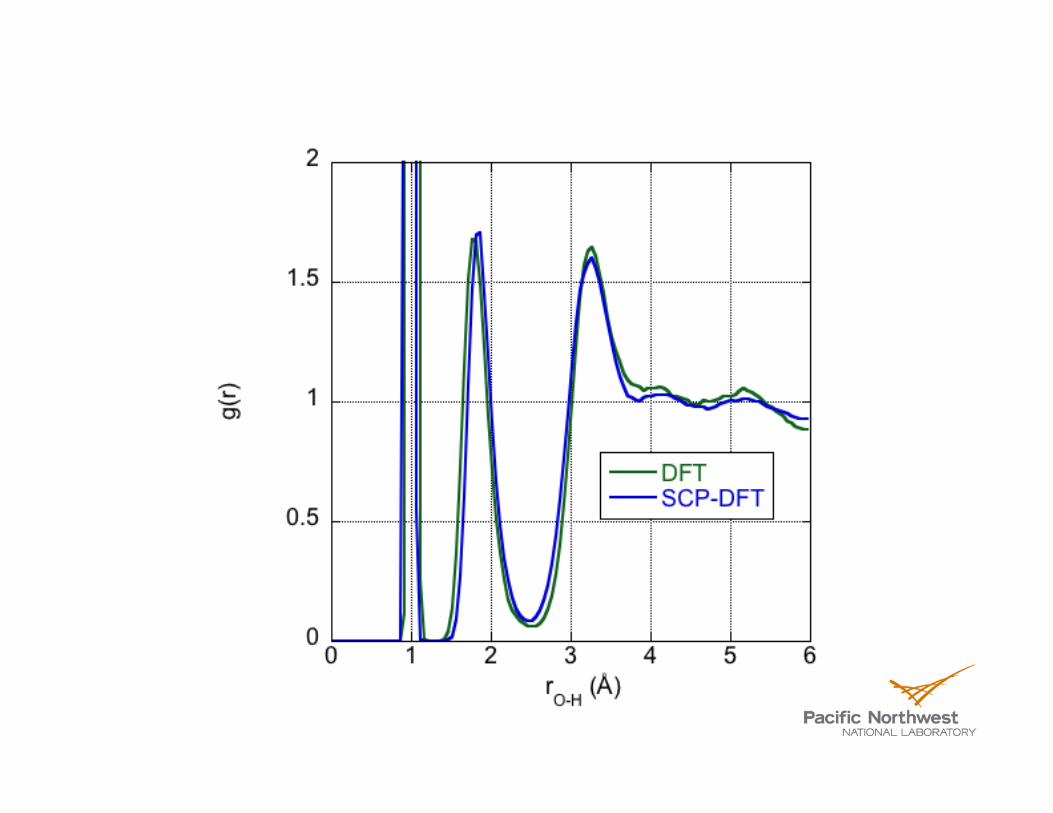


(H2O)2
(H2O)3
(H2O)4
(H2O)5

(H2O)2
(H2O)3
(H2O)4
(H2O)5

Argon
Try to be more systematic about parameterization:
Solid Liquid Gas
KA Maerzke, G Murdachaew, CJ Mundy, GK Schenter, JI Siepmann, J Phys Chem A (in press).

SCP-DFT parametrization procedure for argon
1. Use CP2K with BLYP/GTH pseudopotential/DZVP basis set on Ar atom
2. Use single auxiliary basis function of p symmetry on Ar atom 3. Ar atom: use finite-field approach with charges to fit ap so as
to reproduce known polarizability 4. Ar dimer: fit Ip to recover the Aziz interaction energy (at 3.6
Angstrom)
KA Maerzke, G Murdachaew, CJ Mundy, GK Schenter, JI Siepmann, JPhysChemA (in press).

Ar2 intermolecular potential
RA Aziz JChem.Phys 99 4518 (1993). K Patkowski GM CM Fou & K Szalewicz MolPhys 103 2031 (2005).

Ar2 intermolecular potential: the tail

Second virial coefficients of argon
JH Dymond & EB Smith The Virial Coefficients of Pure Gases and Mixtures: A Critical Compilation; OUP: Oxford 1980.
We are working on fixing the tail and that will improve virials

Cohesion energies per atom of argon clusters
FY Naumkin & DJ Wales MolPhys 96 1295 (1999). DJ Wales et al The Cambridge Cluster Database http://www-wales.ch.cam.ac.uk/CCD.html.
Aziz is 2b only (Wales calculation) LJ is empirical: effectively includes man-body effects SCP-DFT includes many-body effects

Structure of liquid argon at 85 K
JL Yarnell et al PhysRevA 7 2130 (1973).

Cohesion energy per atom of solid fcc argon
C Tessier et al Physica (Amsterdam) 113A 286 (1982). VF Lotrich & K Szalewicz PhysRevLett 79 1301 (1997).

Collaborators: SCP-NDDO: Danny Chang (US EPA) Bruce Garrett (PNNL) JCP 128, 164111 (2008).
SCP-DFT (Water): Chris Mundy (PNNL) Garold Murdachaew (PNNL)
SCP-DFT (Argon): J. Ilja Siepmann (U. Minn.) Katie Maerzke (U. Minn.) JPCA (in press)
Future development in CP2K: Juerg Hutter (Zurich) Teo Liano (Zurich) Joost Vandevondele (Zurich)
Funding: DOE BES Chemical Sciences
Summary: We have enhanced Semiempirical (NDDO) and Density Functional Theory (DFT) electronic structure using Molecular Mechanics-like polarization. (SCP)
We are still exploring how to make this approach more systematic.