sequence alignment &...
TRANSCRIPT

Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine
[email protected] http://compbio.ucdenver.edu/Hunter_lab/Verspoor
Sequence Alignment & Search
With credit and thanks to Larry Hunter for creating the first version of these slides.

Lecture Overview
• Goals: – Understand pairwise sequence alignment
algorithms – Be able to utilize tools for sequence search based
on alignments
• Motivations: – Basis for retrieval of sequence-indexed database
information – Similarity among genomic (amino acid) sequences
is a core indicator of homology

Part 1: Background

Genomic Databases
• Gene and gene product (e.g. protein) databases are often organized by sequence – Genomic sequence encodes all traits of an organism. – Gene products are uniquely described by their sequences. – Similar sequences among biomolecules indicates both
similar function and an evolutionary relationship – A “located sequence feature” (place on a chromosome) is
unambiguous and biologically meaningful – Closely related to the molecular concept of a gene.
=> Biologically meaningful database keys

Searching sequence databases
• There are large sequence databases available – NCBI Entrez Gene, UniProt
• Starting from a sequence alone, find information about it
• Many kinds & sources of input sequences – Genomic, expressed, protein (amino acid vs. nucleic acid) – Complete or fragmentary sequences
• Goal is to retrieve a set of similar sequences. – Exact matches are rare, and not always interesting – Both small differences (mutations) and large (not required
for function) within “similar” sequences can be biologically important.

Sequence search & alignment
• Database organization is focused on efficiency • Sequence search doesn’t match the traditional
database model perfectly • Alternative:
– Start with dynamic programming (a central idea in computational biology)
– Then explore approximations to it (BLAST)

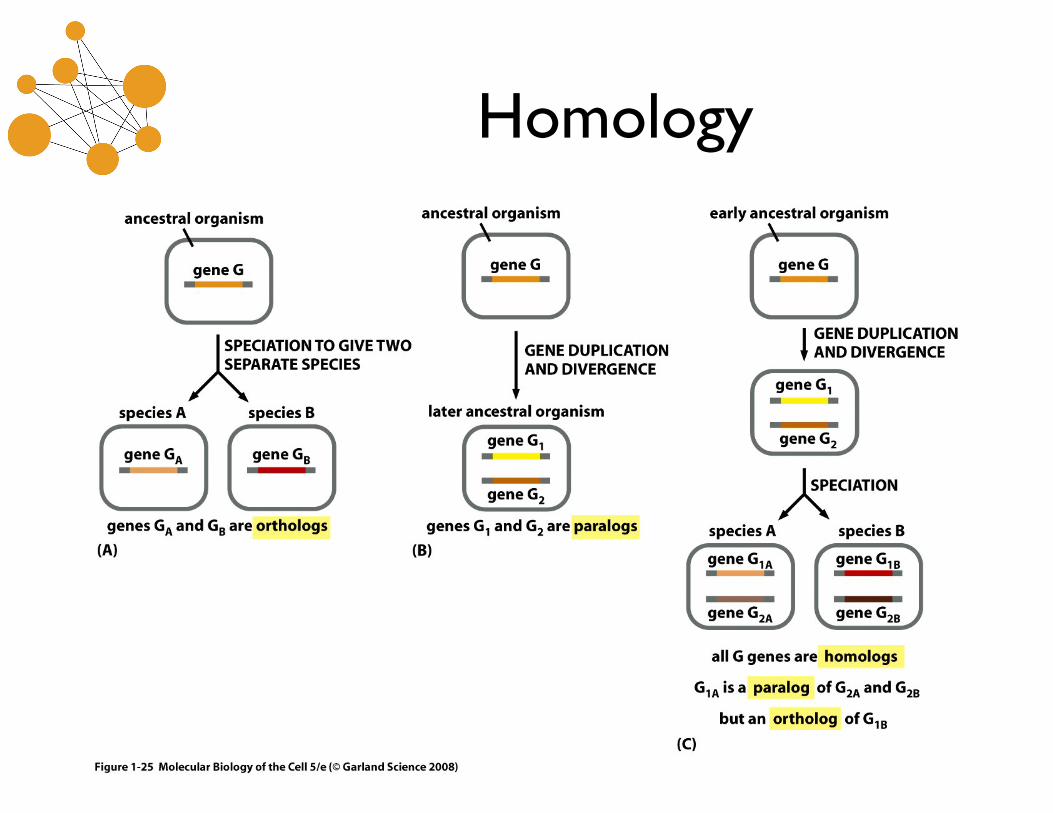
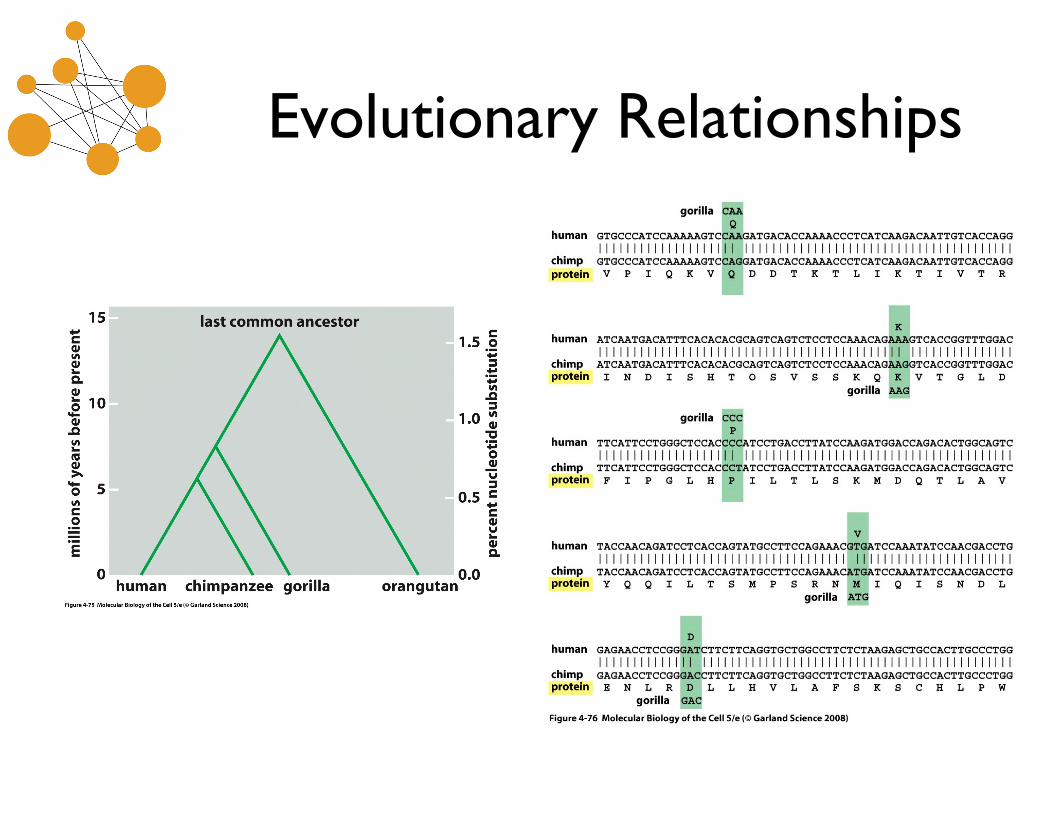
Homology
• Homology is an evolutionary relationship that either exists or does not. It cannot be partial.
• An ortholog is a homolog with shared function. • A paralog is a homolog that arose through a
gene duplication event. Paralogs often have divergent function.
Homology
Evolutionary Relationships

Homology vs Similarity
• Similarity is a measure of the quality of alignment between two sequences.
• High similarity is evidence for homology. • Homology is an inference from similarity. • Similar sequences may correspond to
orthologs or paralogs*.
* Or, possibly, they derived from common selective pressures rather than a common ancestor. Or, the organisms were exposed to a common virus. Or, …

Part 2: Sequence Alignment
Pairwise Sequence Alignment
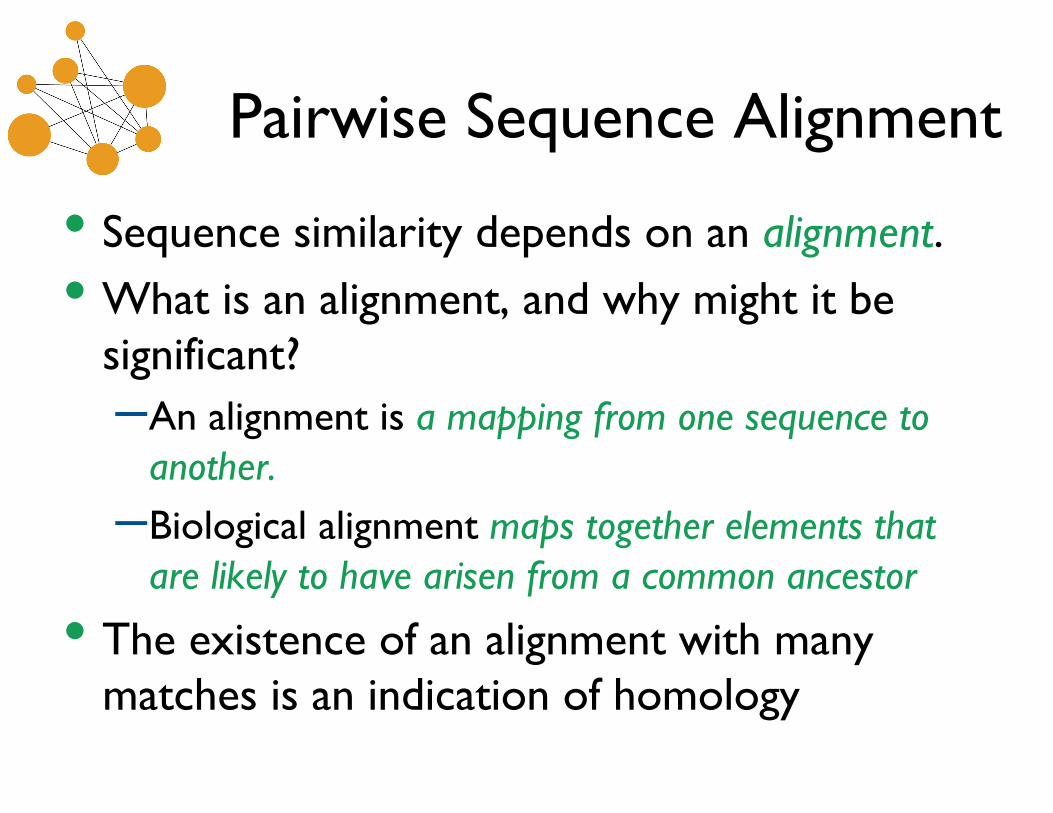
• Sequence similarity depends on an alignment. • What is an alignment, and why might it be
significant? – An alignment is a mapping from one sequence to
another. – Biological alignment maps together elements that
are likely to have arisen from a common ancestor
• The existence of an alignment with many matches is an indication of homology

What complicates sequence alignment?
• Evolutionary changes • Genetic variation
– Mutations (e.g. SNPs) – Copy number variation – Duplications, inversions, translocations, segment
shuffling
• Insertions, Deletions, Substitutions

What counts as similarity?
• Similarity can be defined by counting positions that match between two sequences
• But which positions? Allowing “gaps” makes a difference in the number of matching positions
abcdef abcdef abcdef- ||| || | | |||| abceef acdefg a-cdefg

Not all mismatches are the same
• Some amino acids are more substitutable for each other than others. Serine and threonine are more alike than tryptophan and alanine.
• We can introduce "mismatch costs" for handling different substitutions.
• We don't usually use mismatch costs in aligning nucleotide sequences, since no substitution is per se better than any other.

Many possible alignments to consider
• Without gaps, there are are N+M-1 possible alignments between sequences of length N and M
• Once we start allowing gaps, there are many more possible arrangements to consider: abcbcd abcbcd abcbcd ||| | | ||| || || abc--d a--bcd ab--cd
• This becomes a very large number when we allow mismatches, since we then need to look at every possible pairing between elements: there are roughly NM possible alignments. Aligning length 100 sequences this way is impractical

Avoiding random alignments with a score function
• Not only are there many possible gapped alignments, but introducing too many gaps makes nonsense alignments possible: s--e-----qu---en--ce (sequence) sometimesquipsentice
• Want to distinguish between alignments that occur due to homology, and those that could be expected to be seen just by chance.
• Define a score function that accounts for both element mismatches and a gap penalty
Match scores
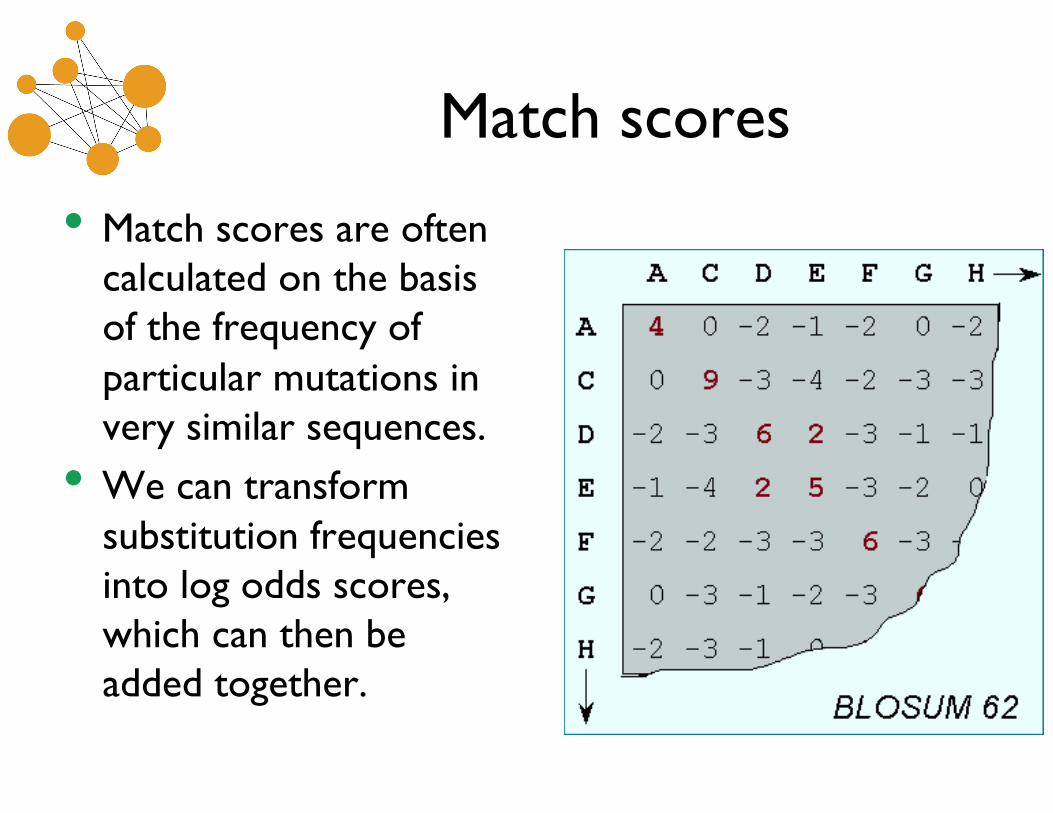
• Match scores are often calculated on the basis of the frequency of particular mutations in very similar sequences.
• We can transform substitution frequencies into log odds scores, which can then be added together.

An alignment score
• An alignment score is the sum of all the match scores of an alignment, with a penalty subtracted for each gap.
• Gap penalties are usually "affine" meaning that the penalty for one long gap is smaller than the penalty for many smaller gaps that add up to the same size.
a b c - - d a c c e f d 9 2 7 6 => 24 - (10 + 2) = 12
Match score
Gap start + continuation penalty
Alignment Score

Global & Local alignments
• A global alignment includes all elements of a sequence, and includes gaps – A global alignment may or may not include "end
gap" penalties. And.--so,.from.hour.to.hour,.we.ripe.and.ripe And.then,.from.hour.to.hour,.we.rot-.and.rot-
• A local alignment includes only subsequences, and sometimes is computed without gaps. My.care.is.loss.of.care,.by.old.care.done, Your.care.is.gain.of.care,.by.new.care.won

Local vs. Global alignments
• Local alignments can find shared domains in divergent proteins and are fast to compute
• Global alignments are better indicators of homology and take longer to compute.

Finding the optimal alignment
• Given a pair of sequences and a score function, identify the best scoring (optimal) alignment between the sequences.
• Remember, exponential number of possible alignments (most with terrible scores).
• Computer science to the rescue: dynamic programming identifies optimal alignments in time proportional to the sum of the lengths of the sequences

A brief aside on Computational Complexity
• A key idea in computer science: How much work does it take to solve a class of problems?
• How do we measure complexity? – Relative to problem size – How long does it take?
• Clock time versus operations • Order: O(?) notation • Worst case / best case
– Other resources used (particularly space)

Dynamic programming
• The key idea is to break the larger problem down into smaller sub-problems which are solved, the results stored, and then combined.
• DP is usually applied to optimization problems. • Here, we start aligning the sequences left to right
– Once a prefix is optimally aligned, nothing about the remainder of the alignment can change the alignment of the prefix.
• We construct a matrix of possible alignment scores (NxM2 calculations worst case) and then "traceback" to find the optimal alignment.
• Called Needleman-Wunsch or Smith-Waterman

Dynamic programming alignment
• Each cell contains the score for the best aligned sequence prefix up to that position.
• Start by filling in initial gap and first element to first element match score
• Use arrow to indicate path to that alignment Align ACD to AACADCD: (match = 5, gap start = -5, gap continue = -2)

Continue filling in optimal path scores
• For each cell, have three choices for how to get there from the last optimal alignment (match, gap sequence 1, gap sequence 2).
• Best score(s) are selected, and arrows added indicated route. – From -5 align As
• -5 +5 = 0
– From 5, insert gap • 5 + -5 = 0
– From -7, insert gap • -7 + -5 = -12
- A
-A AA
A A
A- AA
-- AA
--A AA-
align As insert gap insert gap

Optimal alignment by traceback
• We “traceback” a path that gets us the highest score. If we don't have “end gap” penalties, then take any path from the last row or column to the first.
• Otherwise we need to include the top and bottom corners
AACADCD AACADCD -AC-D-- ---A-CD

Parameter Selection
• The optimal alignment between a pair of sequences depends critically on the selection of the score matrix and the gap penalty.
• These sorts of generic “inputs” to a program are called “parameters”.
• How do we pick the ones that give the most biologically meaningful alignments (and alignment scores?)

How do we pick match scores?
• For match scores, two main options – PAM based on global alignments of closely related
sequences. Normalized to changes per 100 sites, then exponentiated for more distant relatives.
– BLOSUM based on local alignments in much more diverse sequences
• Each matrix has versions aimed at different evolutionary distances.
• BLOSUM62 is NCBI’s default. BLOSUM45 may work better for more evolutionarily distant sequences.

Picking gap penalties
• Many different possible forms: – Most common is affine
(gap open + gap continue penalities) – More complex penalties have been proposed.
• Penalties must be commensurate with match scores. Therefore, the match scoring scheme influences the gap penalty
• Most alignment programs suggest appropriate penalties for each match score option.

Searching for optimal scores
• One possibility is to try several different match score and gap penalties, and choose the best
• In general, this is called parameter space search and it is important in many areas.
• Problems – requires a lot computation – we need some principled way to compare the
results.
• Use significance testing to compare...

The significance of an alignment
• Significance testing is the branch of statistics that is concerned with assessing the probability that a particular result could have occurred by chance.
• How do we calculate the probability that an alignment occurred by chance? – Either with a model of evolution, or – Empirically, by scrambling our sequences and
calculating scores on many randomized (and by assumption unrelated) sequences.
• Incorporated into BLAST: “E-value”

Part 3: Search

Linear search
• Test query against each target sequentially
• Worst case, query matches last target and you have as many tests as targets (size of database)
• Average case, test half the targets. • Linear in the size of the database
Database ACTGA TTAGG CGTAA AGAGA CGATA CCGGA GCCCT TTACG
Query TTACG

Indexed (binary) search
• Create a sorted set of keys that point to entries
• Start in the middle, then figure out which half
• Eliminate half the database each step, so need log2 steps at worst
• Need to build the index (takes space and time at each database update)
Database ACTGA TTAGG CGTAA AGAGA CGATA CCGGA GCCCT TTACG
Query TTACG
Index ACTGA AGAGA CCGGA CGATA CGTAA GCCCT TTACG TTAGG
1
2
3

Hash tables
• Map each query to an arbitrary number with a “hash function”
• Use those numbers as an index into a table
• “Collisions” can happen, but are rare
• Constant time lookup, no index construction
Hash table 1. CGATA 2. GCCCT 3. CGTAA, AGAGA 4. 5. ACTGA 6. CCGGA 7. TTAGG 8. TTACG
f (TTACG)= 8
How to define a hash function
• Basic: must map keys to a number that is within the size of the table
• Desired: minimize collisions • So: similar keys should lead to different hashes • Good general method: map key to a number, and
then take the remainder when divided by a prime number. Specialized hash functions can be better.
Hash tables are the basis of most database lookups.

Approximate searches
• Recall the needs of sequence searches: – Not looking for exact match, but “similar”
sequences
• Database search methods only help us find exact matches. – Hash tables particularly bad at “similar” because
we need similar keys to map to different hashes
• First, need to define what is “similar”, then find efficient ways to search for similar sequences.

Part 4: BLAST
Basic Local Alignment Search Tool http://blast.ncbi.nlm.nih.gov/Blast.cgi

Why BLAST?
• Dynamic programming solutions to alignment problems are relatively slow, and don't lend themselves to efficient database search. – Time complexity proportional to the size of the database.
• Need some way to search a large database to find sequences that have an inexact match to a query sequence
• BLAST: an imperfect approximation to DP. DP finds some distantly related sequences the approximations don't

Sequence search basics
• BLAST is 50-100x faster than DP • Proper use is similar to DP:
– Use appropriate substitution and gap scores • BLOSUM62 is good for weak protein similarities • Use PAM30, PAM70 or BLOSUM45 for better results on more
similar sequences, BLOSUM80 for most distant
– Use low-complexity (repetitive seq) filters and filter out human repeats (ALUs, etc)
– If searching for coding regions, always translate nucleotide to amino acid sequence.

How BLAST works
• Break sequence into overlapping “words,” by default of length 3. – Sequence of length n makes n-m+1 m-size words ABCDE →
ABC, BCD, CDE
• For each word, define ~50 other words that are similar (use substitution matrix + threshold T)
• Repeat for each of the n-m+1 words, giving about 50*n words (out of 203=8000 possible)
• Use a hash table to find all places in DB with an exact match to any of those words.

Extending HSPs
• Identify database sequences that contain several matching words on the same diagonal (think DP alignments) and within a short distance.
• Extend these short, ungapped alignments in both directions along the sequence so long as score of alignment increases. – BLAST alignments scored simply with a log-odds matrix;
no gap penalties at this point.
• Call these extended alignments HSPs for “high scoring pairs”

• What is the probability of scoring at least as large as x by chance?
• Extreme value (not Normal!) distribution:
Where m is size of the database, n is length of query, and l is average length of alignment between two random sequences of those lengths using this scoring scheme.
• Called “E value” for expectation (analogous to p value) • High BLAST score = low E value (low probability of chance)
Is an HSP Significant?

K and λ • Parameters of the extreme value distribution • Depend on the particular substitution matrix • Estimated by aligning a lot of random sequences drawn
on a particular distribution of amino acids, and fitting the extreme value distribution to those alignments
• These empirical estimates may not be correct (error in the assumed distribution of AAs used to create the random sequences) but seem to be reasonably close.

BLAST2: add gaps
• Multiple HSPs in one target sequence → possibility of gapped alignment.
• Combine HSP scores to score whole sequence: – Add HSP scores – Adjust K and λ for this scoring method – Set modest e-value threshold to identify reasonable
target set
• Use DP to produce final gapped alignments – Run DP on the (relatively) small number of database
sequences that were above the threshold with multiple HSPs

Practical “Gapped BLAST”
• Default on NCBI web site • BLAST versus DP on whole databases
– Still might miss some alignments DP would find as database search tool
– DP on fractions of the database (e.g. all human sequences) can be done with parallel hardware, but computational complexity scales with database size.
• BLAST allows users to set certain gap penalties, word sizes and thresholds in “Advanced settings” but not all (since K & λ have to be calculated in advance)

Part 5: Closing comments

Motivating scenarios
• "I have just sequenced a DNA fragment” – Run a BLAST search – Once you have candidates, run a more careful
alignment among them. • "I've located a gene using a gene-finding algorithm”
– Run BLAST to locate similar genes. – Run a global alignment to see differences.
• "I'm confirming a sequencing experiment” – do a global alignment
From: http://www.seas.gwu.edu/~simhaweb/cs151/lectures/module12/align.html

Study guide....
• Dynamic programming alignments are a key technology in bioinformatics, and you should understand how they work.
• The method is perhaps counterintuitive • Work some examples by hand.
– All of the textbooks describe D-P, and there is more detail and supplementary material on the course web site.