sequence alignments and dynamic programming
DESCRIPTION
BIO/CS 471 – Algorithms for Bioinformatics. Sequence Alignments and Dynamic Programming. Module II: Sequence Alignments. Finally a real bioinformatics problem! Problem: SequenceAlignment Input: Two or more strings of characters - PowerPoint PPT PresentationTRANSCRIPT

Sequence AlignmentsSequence Alignmentsand Dynamic Programmingand Dynamic Programming
BIO/CS 471 – Algorithms for BioinformaticsBIO/CS 471 – Algorithms for Bioinformatics

Sequence Alignments 2
Module II: Sequence AlignmentsModule II: Sequence Alignments Finally a real bioinformatics problem! Problem: SequenceAlignment
• Input: Two or more strings of characters• Output: The optimal alignment of the input strings,
possibly including gaps, and an alignment score. Example
• Input: The calico cat was very sleepy. The cat was sleepy.
• Output: The calico cat was very sleepy.The ------ cat was ---- sleepy.

Sequence Alignments 3
Biological Sequence AlignmentBiological Sequence Alignment Input: DNA or Amino Acid sequence Output: Optimal alignment Example
• Input: ACTATAGCTATAGCTATAGCGTATCG ACGATTACAGGTCGTGTCG
• Output: ACTATAGCTATAGCTATAGCGTATCG|| || ||*|| | |||*|||
ACGAT---TACAGGT----CGTGTCG Score: +8

Sequence Alignments 4
Why align sequences?Why align sequences? Why would we want to align two sequences?
• Compare two genes/proteins, e.g. to infer function• Database searches• Protein structure prediction•
Why would we want to align multiple sequences?• Conservation analysis• Molecular evolution•

Sequence Alignments 5
How do we decide on a scoring function?How do we decide on a scoring function? Objective: Sequences that are homologous or
evolutionarily related, should align with high scores. Less related sequences should score lower.
Question: How do homologous sequences arise?

Sequence Alignments 6
TranscriptionTranscription
The Central Dogma
DNAtranscriptiontranscription
RNAtranslationtranslation
Proteins

Sequence Alignments 7
DNA ReplicationDNA Replication Prior to cell division, all the
genetic instructions must be “copied” so that each new cell will have a complete set
DNA polymerase is the enzyme that copies DNA• Reads the old strand in the 3´ to 5´
direction

Over time, genes Over time, genes accumulate accumulate mutationsmutations
Environmental factors• Radiation• Oxidation
Mistakes in replication or repair Deletions, Duplications Insertions Inversions Point mutations

Sequence Alignments 9
Codon deletion:ACG ATA GCG TAT GTA TAG CCG…• Effect depends on the protein, position, etc.• Almost always deleterious• Sometimes lethal
Frame shift mutation: ACG ATA GCG TAT GTA TAG CCG… ACG ATA GCG ATG TAT AGC CG?…• Almost always lethal
DeletionsDeletions

Sequence Alignments 10
IndelsIndels Comparing two genes it is generally impossible
to tell if an indel is an insertion in one gene, or a deletion in another, unless ancestry is known:
ACGTCTGATACGCCGTATCGTCTATCTACGTCTGAT---CCGTATCGTCTATCT

Sequence Alignments 11
The Genetic CodeThe Genetic Code
SubstitutionsSubstitutions are mutations accepted by natural selection.
Synonymous: CGC CGA
Non-synonymous: GAU GAA

Sequence Alignments 12
Two kinds of Two kinds of homologshomologs Orthologs
• Species A has a particular gene
• Species A diverges into species B and C, each with the same gene
• The two genes accumulate substitutions independently

Sequence Alignments 13
Orthologs and ParalogsOrthologs and Paralogs Paralogs
• A gene duplication event within a species results in two copies of a gene
• One of the copies is now under less selective constraint
• The copies can accumulate substitutions independently, before and after speciation

Sequence Alignments 14
Scoring a sequence alignmentScoring a sequence alignment Match score: +1 Mismatch score:+0 Gap penalty: –1ACGTCTGATACGCCGTATAGTCTATCT ||||| ||| || ||||||||----CTGATTCGC---ATCGTCTATCT
Matches: 18 × (+1) Mismatches: 2 × 0 Gaps: 7 × (– 1)
Score = +11Score = +11

Sequence Alignments 15
Origination and length penaltiesOrigination and length penalties We want to find alignments that are
evolutionarily likely. Which of the following alignments seems more
likely to you?ACGTCTGATACGCCGTATAGTCTATCTACGTCTGAT-------ATAGTCTATCTACGTCTGATACGCCGTATAGTCTATCTAC-T-TGA--CG-CGT-TA-TCTATCT
We can achieve this by penalizing more for a new gap, than for extending an existing gap

Sequence Alignments 16
Scoring a sequence alignment (2)Scoring a sequence alignment (2) Match/mismatch score: +1/+0 Origination/length penalty: –2/–1ACGTCTGATACGCCGTATAGTCTATCT ||||| ||| || ||||||||----CTGATTCGC---ATCGTCTATCT
Matches: 18 × (+1) Mismatches: 2 × 0 Origination: 2 × (–2) Length: 7 × (–1)
Score = +7Score = +7

Sequence Alignments 17
Optimal Substructure in AlignmentsOptimal Substructure in Alignments Consider the alignment:ACGTCTGATACGCCGTATAGTCTATCT ||||| ||| || ||||||||----CTGATTCGC---ATCGTCTATCT
Is it true that the alignment in the boxed region must be optimal?
Sequence Alignments 18
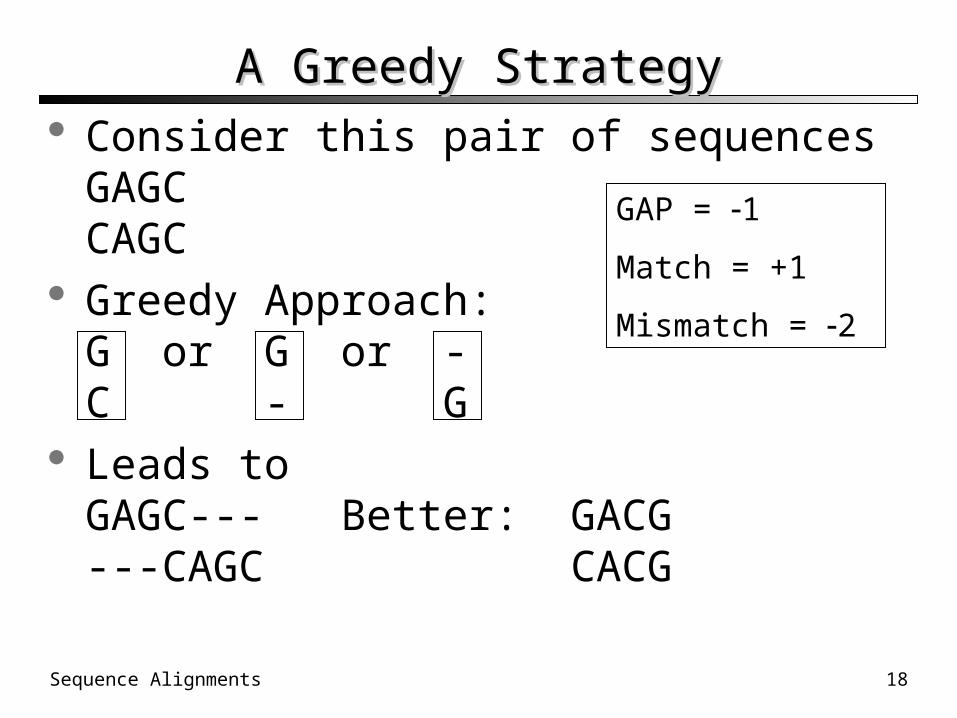
A Greedy StrategyA Greedy Strategy Consider this pair of sequencesGAGCCAGC
Greedy Approach:G or G or -C - G
Leads toGAGC--- Better: GACG---CAGC CACG
GAP = 1
Match = +1
Mismatch = 2

Sequence Alignments 19
Breaking apart the problemBreaking apart the problem Suppose we are aligning:ACTCGACAGTAG
First position choices:A +1 CTCGA CAGTAG
A -1 CTCG- ACAGTAG
- -1 ACTCGA CAGTAG

Sequence Alignments 20
A Recursive Approach to AlignmentA Recursive Approach to Alignment Choose the best alignment based on these three
possibilities:align(seq1, seq2) {
if (both sequences empty) {return 0;}if (one string empty) {
return(gapscore * num chars in nonempty seq);else {
score1 = score(firstchar(seq1),firstchar(seq2)) + align(tail(seq1), tail(seq2));score2 = align(tail(seq1), seq2) + gapscore;score3 = align(seq1, tail(seq2) + gapscore;return(min(score1, score2, score3));
}}
}

Sequence Alignments 21
Time Complexity of RecurseAlignTime Complexity of RecurseAlign What is the recurrence equation for the time
needed by RecurseAlign?3)1(3)( nTnT
3
3
3 3
3 3…
n
3
9
27
3n

Sequence Alignments 22
RecurseAlign repeats its workRecurseAlign repeats its workA C G T A T C G C G T A T A
G
A
T
G
C
T
C
T
C
G

Sequence Alignments 23
How can we find an optimal alignment?How can we find an optimal alignment? Finding the alignment is computationally hard:ACGTCTGATACGCCGTATAGTCTATCTCTGAT---TCG—CATCGTC--T-ATCT
C(27,7) gap positions = ~888,000 possibilities It’s possible, as long as we don’t repeat our
work! Dynamic programming: The Needleman &
Wunsch algorithm

Sequence Alignments 24
What is the optimal alignment?What is the optimal alignment? ACTCGACAGTAG
Match: +1 Mismatch: 0 Gap: –1

Sequence Alignments 25
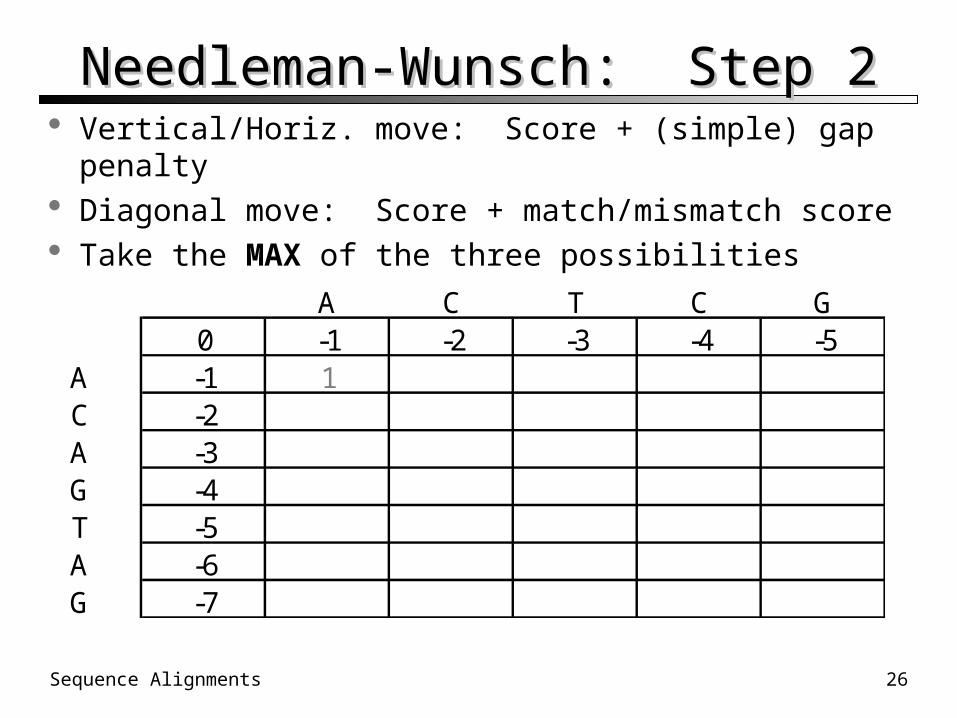
Needleman-Wunsch: Step 1Needleman-Wunsch: Step 1 Each sequence along one axis Mismatch penalty multiples in first row/column 0 in [1,1] (or [0,0] for the CS-minded)
A C T C G0 -1 -2 -3 -4 -5
A -1 1C -2A -3G -4T -5A -6G -7
Sequence Alignments 26
Needleman-Wunsch: Step 2Needleman-Wunsch: Step 2 Vertical/Horiz. move: Score + (simple) gap penalty Diagonal move: Score + match/mismatch score Take the MAX of the three possibilities
A C T C G0 -1 -2 -3 -4 -5
A -1 1C -2A -3G -4T -5A -6G -7
Sequence Alignments 27
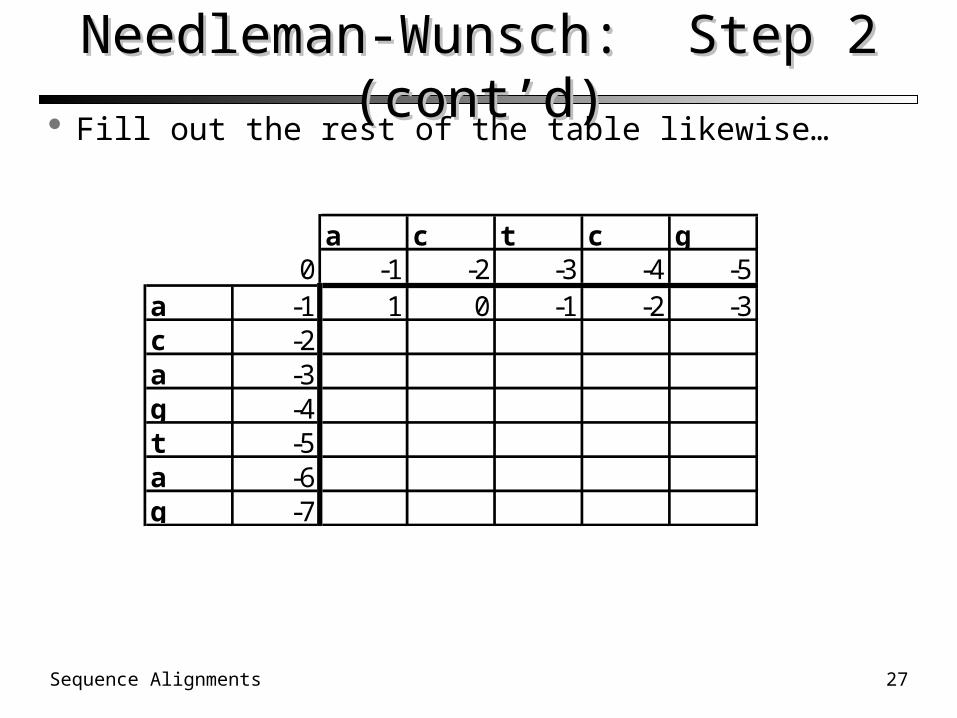
Needleman-Wunsch: Step 2 (cont’d)Needleman-Wunsch: Step 2 (cont’d) Fill out the rest of the table likewise…
a c t c g0 -1 -2 -3 -4 -5
a -1 1 0 -1 -2 -3c -2a -3g -4t -5a -6g -7

Sequence Alignments 28
Needleman-Wunsch: Step 2 (cont’d)Needleman-Wunsch: Step 2 (cont’d) Fill out the rest of the table likewise…
The optimal alignment score is calculated in the lower-right corner
a c t c g0 -1 -2 -3 -4 -5
a -1 1 0 -1 -2 -3c -2 0 2 1 0 -1a -3 -1 1 2 1 0g -4 -2 0 1 2 2t -5 -3 -1 1 1 2a -6 -4 -2 0 1 1g -7 -5 -3 -1 0 2

Sequence Alignments 29
a c t c g0 -1 -2 -3 -4 -5
a -1 1 0 -1 -2 -3c -2 0 2 1 0 -1a -3 -1 1 2 1 0g -4 -2 0 1 2 2t -5 -3 -1 1 1 2a -6 -4 -2 0 1 1g -7 -5 -3 -1 0 2
But what But what isis the optimal alignment the optimal alignment To reconstruct the optimal alignment, we must
determine of where the MAX at each step came from…

Sequence Alignments 30
A path corresponds to an alignmentA path corresponds to an alignment = GAP in top sequence = GAP in left sequence = ALIGN both positions One path from the previous table: Corresponding alignment (start at the end):
AC--TCGACAGTAG
Score = +2

Sequence Alignments 31
Practice ProblemPractice Problem Find an optimal alignment for these two
sequences:GCGGTTGCGT
Match: +1 Mismatch: 0 Gap: –1
g c g g t t0 -1 -2 -3 -4 -5 -6
g -1c -2g -3t -4

Sequence Alignments 32
Practice ProblemPractice Problem Find an optimal alignment for these two
sequences:GCGGTTGCGT g c g g t t
0 -1 -2 -3 -4 -5 -6g -1 1 0 -1 -2 -3 -4c -2 0 2 1 0 -1 -2g -3 -1 1 3 2 1 0t -4 -2 0 2 3 3 2
GCGGTTGCG-T- Score = +2

Sequence Alignments 33
g c g0 -1 -2 -3
g -1 1 0 -1g -2 0 1 1c -3 -1 1 1g -4 -2 0 2
Semi-global alignmentSemi-global alignment Suppose we are aligning:GCGGGCG
Which do you prefer?G-CG -GCGGGCG GGCG
Semi-global alignment allows gaps at the ends for free.

Sequence Alignments 34
Semi-global alignmentSemi-global alignment
g c g0 0 0 0
g 0 1 0 1g 0 1 1 1c 0 0 2 1g 0 1 1 3
Semi-global alignment allows gaps at the ends for free.
Initialize first row and column to all 0’s Allow free horizontal/vertical moves in last
row and column

Sequence Alignments 35
Local alignmentLocal alignment Global alignments – score the entire alignment Semi-global alignments – allow unscored gaps
at the beginning or end of either sequence Local alignment – find the best matching
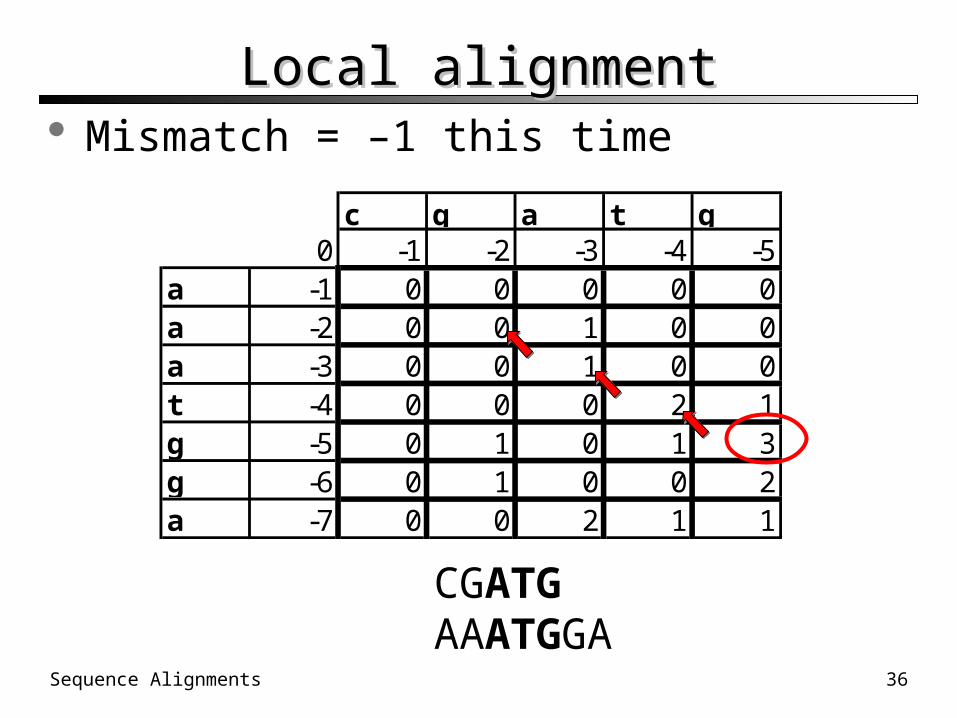
subsequence CGATGAAATGGA
This is achieved by allowing a 4th alternative at each position in the table: zero.
Sequence Alignments 36
c g a t g0 -1 -2 -3 -4 -5
a -1 0 0 0 0 0a -2 0 0 1 0 0a -3 0 0 1 0 0t -4 0 0 0 2 1g -5 0 1 0 1 3g -6 0 1 0 0 2a -7 0 0 2 1 1
Local alignmentLocal alignment Mismatch = –1 this time
CGATGAAATGGA

Sequence Alignments 37
Food for thought…Food for thought… What is the asymptotic time complexity of
the Needleman & Wunsch algorithm?• Is this good or bad?
How about the space complexity?• Why might this be a problem?

Sequence Alignments 38
Saving SpaceSaving Space Note that we can throw away the previous rows
of the table as we fill it in:a c t c g
0 -1 -2 -3 -4 -5a -1 1 0 -1 -2 -3c -2 0 2 1 0 -1a -3 -1 1 2 1 0g -4 -2 0 1 2 2t -5 -3 -1 1 1 2a -6 -4 -2 0 1 1g -7 -5 -3 -1 0 2
This row is based only on this one

Sequence Alignments 39
Saving Space (2)Saving Space (2) Each row of the table contains the scores for
aligning a prefix of the left-hand sequence with all prefixes of the top sequence:
a c t c g0 -1 -2 -3 -4 -5
a -1 1 0 -1 -2 -3c -2 0 2 1 0 -1a -3 -1 1 2 1 0g -4 -2 0 1 2 2t -5 -3 -1 1 1 2a -6 -4 -2 0 1 1g -7 -5 -3 -1 0 2
Scores for aligning aca with
all prefixes of actcg

Sequence Alignments 40
Divide and ConquerDivide and Conquer By using a recursive approach, we can use only
two rows of the matrix at a time:• Choose the middle character of the top sequence, i• Find out where i aligns to the bottom sequence
Needs two vectors of scores• Recursively align the sequences before and after the
fixed positions
ACGCTATGCTCATAGCGACGCTCATCG
i

Sequence Alignments 41
Finding where Finding where ii lines up lines up Find out where i aligns to the bottom sequence
Needs two vectors of scores
Assuming i lines up with a character:alignscore = align(ACGCTAT, prefix(t)) + score(G, char from t)
+ align(CTCATAG, suffix(t)) Which character is best?
• Can quickly find out the score for aligning ACGCTAT with every prefix of t.
s: ACGCTATGCTCATAGt: CGACGCTCATCG
i

Sequence Alignments 42
Finding where Finding where ii lines up lines up But, i may also line up with a gap
Assuming i lines up with a gap:
alignscore = align(ACGCTAT, prefix(t)) + gapscore+ align(CTCATAG, suffix(t))
s: ACGCTATGCTCATAGt: CGACGCTCATCG
i

Sequence Alignments 43
Recursive CallRecursive Call Fix the best position for I Call align recursively for the prefixes and
suffixes:
s: ACGCTATGCTCATAGt: CGACGCTCATCG
i

Sequence Alignments 44
ComplexityComplexity Let len(s) = m and len(t) = n Space: 2m Time:
• Each call to build similarity vector = m´n´
• First call + recursive call:
s: ACGCTATGCTCATAGt: CGACGCTCATCG
i
j
mnjnmmjmn
jnmTjmTmnmnnmT
2)(
,2
,222
,

Sequence Alignments 45
General Gap PenaltiesGeneral Gap Penalties Suppose we are no longer using simple gap
penalties:• Origination = −2• Length = −1
Consider the last position of the alignment for ACGTA with ACG
We can’t determine the score for
unless we know the previous positions!
G-
-G
or

Sequence Alignments 46
Scoring BlocksScoring Blocks Now we must score a block at a time
A block is a pair of characters, or a maximal group of gaps paired with characters
To score a position, we need to either start a new block or add it to a previous block
A A C --- A TATCCG A C T AC
A C T ACC T ------ C G C --

Sequence Alignments 47
The AlgorithmThe Algorithm Three tables
• a – scores for alignments ending in char-char blocks• b – scores for alignments ending in gaps in the top
sequence (s)• c – scores for alignments ending in gaps in the left
sequence (t) Scores no longer depend on only three
positions, because we can put any number of gaps into the last block

Sequence Alignments 48
The RecurrencesThe Recurrences
1,11,11,1
max,,jicjibjia
jipjia
jkkwkjicjkkwkjia
jib1for ,,1for ,,
max,
ikkwjkibikkwjkia
jic1for ,,1for ,,
max,

Sequence Alignments 49
The Optimal AlignmentThe Optimal Alignment The optimal alignment is found by looking at
the maximal value in the lower right of all three arrays
The algorithm runs in O(n3) time• Uses O(n2) space