sÍndrome de cowden caso clÍnico -...
TRANSCRIPT

Artigo tipo “Case Report” Mestrado Integrado em Medicina
SÍNDROME DE COWDEN – CASO CLÍNICO
Mariana Jorge Rodrigues
Orientador
Dra. Estrela Rocha
Porto 2011

Artigo tipo “Case Report” Mestrado Integrado em Medicina
SÍNDROME DE COWDEN – CASO CLÍNICO
Mariana Jorge Rodrigues
Instituto Ciências Biomédicas de Abel Salazar
Largo Professor Abel Salazar no. 2, 4099 – 003 Porto
Orientador
Dra. Estrela Rocha – Especialista em Oncologia Médica no Centro Hospitalar do Porto
Porto 2011

ÍNDICE
RESUMO ................................................................................................................................... 1
ABSTRACT ............................................................................................................................... 1
INTRODUÇÃO .......................................................................................................................... 2
PTEN ...................................................................................................................................... 3
FIGURA 1 - REPRESENTAÇÃO DA VIA PI3K/AKT/MTOR ............................................................... 4
TABELA 1 - SÍNDROMES PTHS [7]
................................................................................................. 5
SÍNDROME DE COWDEN ................................................................................................... 5
Apresentação Clínica .......................................................................................................... 6
TABELA 2 - PRINCIPAIS ALTERAÇÕES CLÍNICAS ........................................................................ 7
APRESENTAÇÃO DO CASO ................................................................................................ 10
TABELA 3 - RESUMO PATOLÓGICO ........................................................................................... 11
DISCUSSÃO ............................................................................................................................ 11
FIGURA 2 - ALGORITMO DIAGNÓSTICO NO SC [11]
..................................................................... 13
TABELA 4 - CRITÉRIOS CLÍNICOS DA DOENTE .......................................................................... 14
TABELA 5 - ACOMPANHAMENTO DE DOENTES COM SC ........................................................... 15
AGRADECIMENTOS ............................................................................................................. 16
BIBLIOGRAFIA ...................................................................................................................... 16
ANEXOS .................................................................................................................................. 18

Síndrome de Cowden – Caso Clínico
1
RESUMO O Síndrome de Cowden,
inicialmente descrito em 1940 por Costello e
reconhecido como síndrome em 1963 por
Lloyd e Dennis, é uma doença rara com uma
incidência estimada de 1 para 200000 na
população geral. Trata-se de uma doença
autossómica dominante de expressividade
fenotípica variável e penetrância incompleta,
cuja idade média de diagnóstico é aos 22
anos. Mais de 80% dos casos são devidos a
uma mutação no gene PTEN com
localização no cromossoma 10q22-23. Os
doentes com Síndrome de Cowden
apresentam uma predisposição para
desenvolver uma grande variedade de lesões
benignas e/ou malignas. A característica
principal deste síndrome são os múltiplos
hamartomas, possíveis de serem encontrados
em qualquer órgão, porém são mais
frequentemente encontrados na pele ou
tracto gastrointestinal. Em relação às lesões
malignas, o Síndrome de Cowden predispõe
para os Carcinomas da Mama, Tiróide e
Endométrio. A avaliação diagnóstica do
Síndrome de Cowden actualmente obedece a
critérios específicos. Desde o ano 2000 que
os membros do Consórcio Internacional de
Critérios de Diagnóstico para o Síndrome de
Cowden organizaram as principais
características em 3 categorias: critérios
patognomónicos (lesões mucocutâneas
específicas, doença Lhermitte Duclos no
adulto), critérios major (Cancro da Mama,
Cancro da Tiróide,Cancro do Endométrio e
macrocefalia) e critérios minor (oligofrenia,
pólipos intestinais hamartomatosos, lipomas,
fibromas, doença fibroquística da mama,
doença tiróideia, tumores ou malformações
genitourinários e fibroma uterino).
Neste estudo, relata-se um caso de
uma doente do sexo feminino, que
inicialmente apresenta-se aos 20 anos com
um diagnóstico de Carcinoma Folicular da
Tiróide, macrocefalia e a presença de
múltiplos nódulos subcutâneos dispersos nos
membros superiores e inferiores. Com o
decorrer do tempo, surgem outras lesões
benignas, como hamartomas do tracto
gastrointestinal, e neoplasias, nomea-
damente Carcinoma Ductal Invasor de
ambas as mamas, Adenossarcoma uterino,
Dermatofibrossarcoma Protuberans e
Carcinoma Basocelular. Em 14 de Março de
1999, a doente veio a falecer por neoplasia
da mama extensamente metastizada.
ABSTRACT
The Cowden Syndrome first
described by Costello in 1940 and
recognized as a syndrome in 1963 by Lloyd

Síndrome de Cowden – Caso Clínico
2
and Dennis, is a rare disease with an
estimated incidence of 1 per 200000 in
general population. It is an autossomal
dominant disease with variable phenotypic
expressivity and incomplete penetrance,
whose average age at diagnosis is 22 years.
Over 80% of cases are due to a mutation in
the PTEN gene located on chromosome
10q22-23. Patients with Cowden syndrome
are predisposed to develop a variety of
benign and / or malignant lesions. The main
feature of this syndrome are multiple
hamartomas possible to be found in any
organ but are most often found in the skin or
gastrointestinal tract. Regarding the
malignant lesions, Cowden syndrome
predisposes to breast, thyroid and
endometrium carcinomas. The diagnostic
evaluation of Cowden syndrome currently
meets specific criteria. Since 2000 the
members of the Consensus Diagnostic
Criteria for Cowden Syndrome have
organized the main manifestations in three
categories: pathognomonic criteria (specific
mucocutaneous lesions, Lhermitte Duclos
disease in adults), major criteria (breast
cancer, thyroid and endometrial and
macrocephaly) and minor criteria (mental
retardation, hamartomatous intestinal
polyps, lipomas, fibromas, fibrocystic breast
disease, thyroid disease, tumors or
genitourinary malformations and uterine
fibroids).
In this study, we report a case of a
female patient who initially presents itself at
the age of 20 years with a diagnosis of
follicular carcinoma, macrocephaly and the
presence of multiple subcutaneous nodules
scattered throughout the upper and lower
limbs. Over the course of time, other benign
lesions appear, such as hamartomas of the
gastrointestinal tract, as well as neoplasms,
including invasive ductal carcinoma of both
breasts, Uterus Adenosarcoma, Dermato-
fibrosarcoma protuberans and basal cell
carcinoma. On March 14, 1999, the patient
died of widely metastatic breast cancer.
INTRODUÇÃO O Síndrome de Cowden (SC)
também conhecido por Doença de Cowden e
Síndrome de Múltiplos Hamartomas, é uma
condição genética que predispõe o indivíduo
para um risco acrescido de desenvolver uma
variedade de lesões benignas e/ou malignas.
Esta doença foi inicialmente descrita em
1940 por Costello, mas foi só em 1963 que
Lloyd e Dennis o reconheceram como um
síndrome, após o estudo numa família com o
mesmo nome [1,2]
. Trata-se de uma doença
extremamente rara, cuja incidência antes da
identificação genética responsável pelo

Síndrome de Cowden – Caso Clínico
3
Síndrome estava estimada em 1 para
1000000. Contudo, com a identificação do
gene, existe uma incidência actual calculada
de 1 para 200 000 na população geral.
Presume-se que a incidência esteja
subestimada, isto porque o SC apresenta
uma grande variabilidade fenotípica que
pode ser subvalorizada pela comunidade
médica. Existe uma preponderância em
caucasianos e em especial no sexo feminino.
O diagnóstico é feito entre os 13 e os 65
anos (sendo a idade média de diagnóstico
aos 22 anos) [1,3]
.
Neste trabalho, vou rever o
Síndrome de Cowden. Sabendo que se trata
de um síndrome bastante raro e com
manifestações clínicas de carácter
heterogéneo, surgiu a necessidade de
conhece-lo melhor, não só para que se faça
uma correcta abordagem ao doente e
diagnóstico, mas também uma vez
deparados com a clínica suspeita do
síndrome, se possa extrapolar para um
universo paralelo de rastreio. Deste modo,
para melhor consegui-lo, pretendo fazer a
apresentação de um caso clínico que ocorreu
no Centro Hospitalar do Porto (cuja recolha
de dados foi aprovada pela comissão de
ética do Centro Hospitalar do Porto ao
abrigo do disposto no Decreto-lei n.º 97/95 –
Anexo 1).
Antes de passar para a descrição do
SC em si, gostaria primeiro de explicar os
mecanismos que lhe deram origem.
PTEN
Sabemos que o cancro é uma
doença causada pela acumulação de erros
genéticos que por sua vez interferem com a
regulação do crescimento, diferenciação e
morte celular. Contudo a maioria dos erros
genéticos causadores de cancro são
adquiridos devido a exposições ambientais,
carcinogénios ou simplesmente pelo acaso
(espontaneamente). Este tipo de dano
genético é somático, não afecta a linha
germinativa e logo não constitui um risco
para os descendentes. Assim, a maioria das
neoplasias ocorre de forma esporádica.
Porém, alguns cancros são hereditários,
ocorrendo dentro da mesma família com
uma frequência maior que a esperada (caso
fosse ao acaso/esporadicamente). Estima-se
que 5-10% das neoplasias são hereditárias, e
ocorrem porque existe uma mutação
genética na linha germinativa que pode
potencialmente afectar qualquer célula,
podendo ser transmitida aos descendentes [4]
.
O SC é uma doença autossómica
dominante de expressividade variável e
penetrância incompleta. Querendo isto dizer
que o SC é uma doença em que basta apenas

Síndrome de Cowden – Caso Clínico
4
um progenitor (com a doença) ter um gene
mutado e passá-lo à descendência para que o
indivíduo tenha a doença. No entanto, a
doença pode manifestar-se de forma
diferente em diversos indivíduos (não se
expressa sempre do mesmo modo) e ao ter
penetrância incompleta implica que nem
todos os doentes com SC têm que ter
obrigatoriamente a mutação. Mais de 80%
dos casos são devidos a uma mutação no
gene PTEN (protein tyrisine phosphatase
with homology to tensin) com localização no
cromossoma 10q22-23 [1]
. Este gene possui
a função de regular a via PI3K/Akt/mTOR,
sendo portanto um gene de supressão
tumoral que induz o seu efeito no
crescimento e proliferação celular,
migração, angiogénese e apoptose [5]
.
PTEN é um gene/proteína que
consegue deslocar-se livremente entre o
citoplasma e o núcleo, onde exerce os seus
efeitos. Pertence a uma subclasse de
fosfatases denominadas “dual specificity
phosphatases”, ou seja, o gene PTEN
quando codificado resulta numa proteína de
403 aminoácidos que por sua vez actua
como uma fosfatase com especificidade
dupla, sendo capaz de desfoforilar proteínas
e lípidos. No citoplasma, quando esta
proteína exerce a sua actividade
desfosforilante em lípidos, nomeadamente
sobre o produto de conversão de PI3K
(impedindo a conversão de 3’fosfainositideo
em bifosfato- e monofosfato –
fosfatidilinositol) vai também diminuir a
actividade das outras cinases, nomeada-
mente Akt e mTOR [6]
. Ao inibir, de forma
indirecta, a via Akt, PTEN diminui a
fosforilação de outras vias iniciadoras de
tumores dependentes da via Akt, como a
p27, p21, GSK-3, Bad e ASK-1. Quando
exerce a sua actividade em proteínas do
citoplasma, PTEN desfosforila cinases de
adesão e cinases com actividade mitogénica.
No núcleo, PTEN consegue desactivar a via
MAPK, induzindo a célula a entrar em G1 e
impedindo assim a progressão para
proliferação celular desregulada [4,7]
. Ou
seja, a perda de função de PTEN ou a
diminuição da sua actividade resulta em
diminuição da apoptose, crescimento e proli-
feração celular, migração e angiogénese
desregulados, sendo estes os principais
factores iniciadores de tumor (Figura1) [3,5,7]
.
Figura 1- Representação da via PI3K/Akt/mTOR
Síndrome de Cowden – Caso Clínico
5
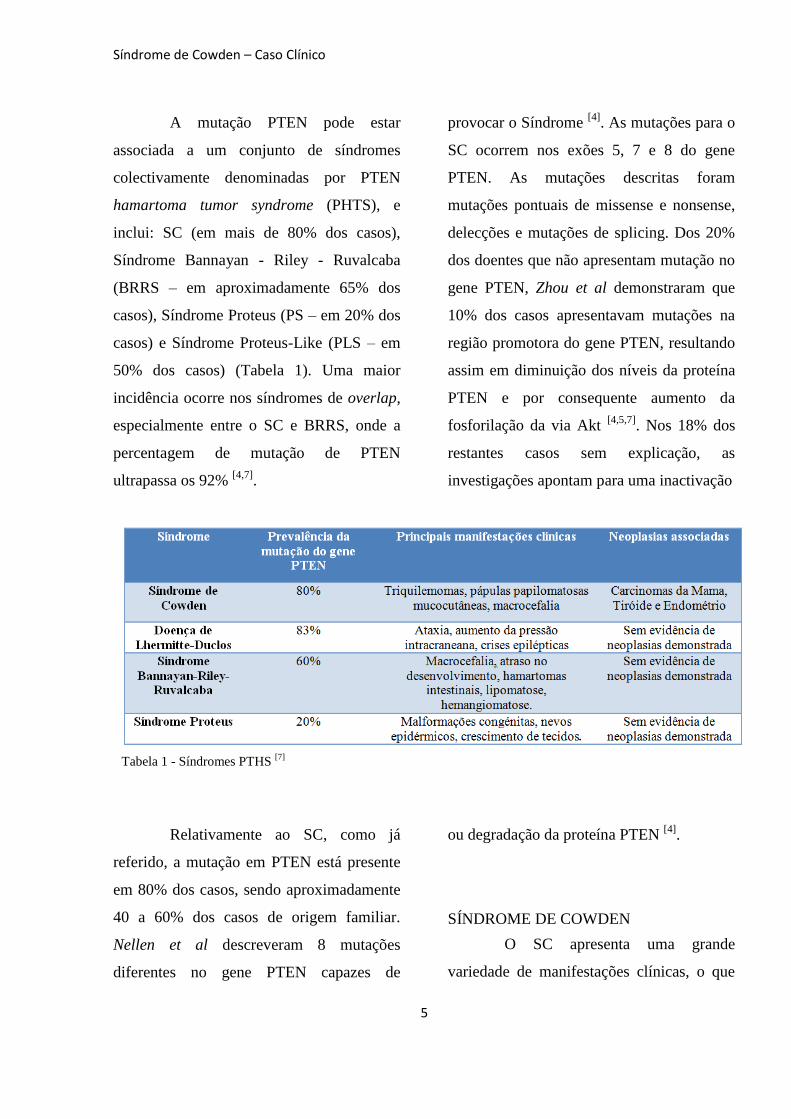
A mutação PTEN pode estar
associada a um conjunto de síndromes
colectivamente denominadas por PTEN
hamartoma tumor syndrome (PHTS), e
inclui: SC (em mais de 80% dos casos),
Síndrome Bannayan - Riley - Ruvalcaba
(BRRS – em aproximadamente 65% dos
casos), Síndrome Proteus (PS – em 20% dos
casos) e Síndrome Proteus-Like (PLS – em
50% dos casos) (Tabela 1). Uma maior
incidência ocorre nos síndromes de overlap,
especialmente entre o SC e BRRS, onde a
percentagem de mutação de PTEN
ultrapassa os 92% [4,7]
.
Relativamente ao SC, como já
referido, a mutação em PTEN está presente
em 80% dos casos, sendo aproximadamente
40 a 60% dos casos de origem familiar.
Nellen et al descreveram 8 mutações
diferentes no gene PTEN capazes de
provocar o Síndrome [4]
. As mutações para o
SC ocorrem nos exões 5, 7 e 8 do gene
PTEN. As mutações descritas foram
mutações pontuais de missense e nonsense,
delecções e mutações de splicing. Dos 20%
dos doentes que não apresentam mutação no
gene PTEN, Zhou et al demonstraram que
10% dos casos apresentavam mutações na
região promotora do gene PTEN, resultando
assim em diminuição dos níveis da proteína
PTEN e por consequente aumento da
fosforilação da via Akt [4,5,7]
. Nos 18% dos
restantes casos sem explicação, as
investigações apontam para uma inactivação
ou degradação da proteína PTEN [4]
.
SÍNDROME DE COWDEN
O SC apresenta uma grande
variedade de manifestações clínicas, o que
Tabela 1 - Síndromes PTHS [7]

Síndrome de Cowden – Caso Clínico
6
pode dificultar o exercício de diagnóstico ou
gerar confusão com outros síndromes.
Conforme descrito anteriormente, o SC na
maioria dos casos é uma doença genética
que confere aos portadores uma predis-
posição para desenvolver uma variedade de
lesões benignas e/ou malignas características
deste Síndrome. Contudo, não nos
esqueçamos que o SC apresenta uma
penetrância incompleta, ou seja, nos doentes
sem a mutação no gene PTEN, a apresen-
tação clínica não difere dos doentes com SC
com a mutação em PTEN. Como as três
linhas germinativas estão afectadas,
qualquer anomalia de origem endodérmica,
ectodérmica, e/ou mesodérmica podem estar
envolvidas na doença, diversificando enor-
memente os achados físicos encontrados [3]
.
A característica principal deste Síndrome
são os múltiplos hamartomas possíveis de
serem encontrados em qualquer órgão,
porém são mais frequentemente encontrados
na pele ou tracto gastrointestinal [8]
. No
entanto, outras manifestações envolvendo
qualquer órgão ou sistema podem estar
presentes, salientando entre as várias lesões
uma predisposição para neoplasias da mama,
tiróide e endométrio [9,10]
.
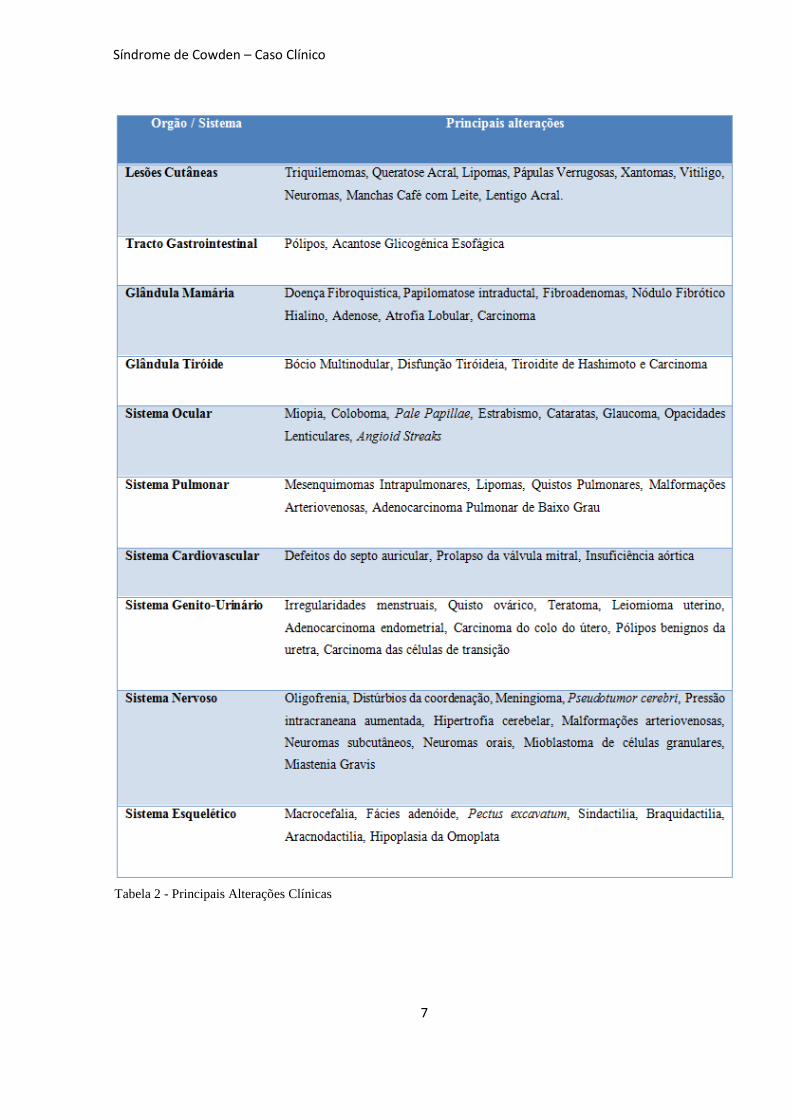
Apresentação Clínica
Qualquer órgão ou sistema podem
estar potencialmente envolvidos, existindo
no entanto lesões mais características do SC.
Os principais órgãos ou sistemas envolvidos
são: Pele, Tracto Gastrointestinal, Glândula
Mamária, Tiróide, Sistema Ocular, Sistema
Pulmonar, Sistema Cardiovascular, Sistema
Genito-Urinário, Sistema Nervoso e Sistema
Esquelético [3]
(Tabela 2).
Pele: as lesões mucocutâneas estão
presentes em 99-100% dos casos,
geralmente surgem na segunda ou terceira
décadas de vida, mas podem estar presentes
tão cedo como aos 4 anos ou tão tarde como
aos 75 anos de idade [3,7]
. O envolvimento da
mucosa está presente em mais de 80% [3]
.
Vulgarmente as lesões surgem na mucosa
gengival, orofaringe, língua, mucosa nasal e
anogenital; estas lesões são benignas e
surgem habitualmente após as lesões
cutâneas. As pápulas faciais e os
triquilemomas (lesões hamartomatosas da
camada externa da derme, habitualmente
junto a folículos pilosos e apresentam uma
cor correspondente á tonalidade da pele do
indivíduo atingido) estando presentes em
mais de 85% dos casos e adoptam como
posição preferencial as regiões periorbital e
perinasal [3,8]
. A queratose acral está
presente em 20-28% dos casos; conforme o
nome indica, apresentam como localização
preferencial a região extensora das
extremidades e/ou região palmoplantar. As
pápulas verrugosas, presentes até 80% dos
casos podem surgir nos lábios, mucosa oral,
Síndrome de Cowden – Caso Clínico
7
Tabela 2 - Principais Alterações Clínicas

Síndrome de Cowden – Caso Clínico
8
palato e amígdalas; estas lesões,
histologicamente, apresentam características
compatíveis com angiofibromas (fibrose
perivascular com capilares dilatados e uma
rede de fibras de colagéneo entrelaçado com
folículos pilosos) [3]
. Outras lesões cutâneas
descritas incluem lipomas, hemangiomas,
xantomas, neuromas, manchas de café com
leite, lentigo acral e acantose nigricans [3,8]
.
Tracto Gastrointestinal (GI): O envolvi-
mento do Sistema GI está estimado em 70-
90% [7,8]
. Os pacientes são habitualmente
assintomáticos. Pode atingir qualquer região
do tracto GI, sendo as zonas mais atingidas
todo o cólon, sigmóide e recto. As áreas
menos frequentemente afectadas são o
intestino delgado, estômago e esófago. O
envolvimento deste sistema surge sob a
forma de pólipos que vão desde 1mm até
vários centímetros de diâmetro [3]
. Os
pólipos podem ser hamartomatosos,
lipomatosos, fibromatosos, hiperplásicos,
inflamatórios e mais frequentemente
adenomatosos. A acantose glicogénica
esofágica (caracterizada por placas com
elevação aproximada de 1mm, de cor
acinzentada devido aos depósitos de
glicogénio nas células escamosas,
localizados na porção distal do esófago) é
também relativamente frequente nos doentes
com SC. Para alguns autores, acantose
glicogénica esofágica na presença de outros
pólipos no tracto GI deveria ser considerado
um critério patognomónico de SC. Devido à
elevada predisposição para malignidade, os
doentes com SC deverão ser avaliados de 6
em 6 meses [1,3]
.
Glândula Mamária: a doença fibroquística
da mama foi descrita em mais de dois terços
das doentes de sexo feminino com SC, já a
sua ocorrência concomitante com o cancro
da mama surge em mais de 50% das
pacientes. O Cancro da mama é a neoplasia
mais frequente nas doentes com SC, surge
até 50% dos casos (comparativamente com o
risco de 11% das mulheres da população
geral [11]
) e em alguns casos podendo mesmo
ser bilateral [7]
. Tal como nos outros casos
de neoplasia da mama hereditária, a idade de
aparecimento é também mais precoce que na
população geral (surge geralmente entre os
36-46 anos). O tipo histológico de cancro
mais frequente é o carcinoma ductal invasor,
embora os carcinomas lobular e tubular
também já foram observados [7]
. Existem
casos do sexo masculino com SC, nos quais
o cancro da mama também foi descrito [4]
.
Outros achados podem ser: hiperplasia
ductal, papilomatose intraductal, fibro-
adenomas, atrofia lobular e nódulo fibrótico
hialino [3]
.
Glândula Tiróide: a glândula tiróide é
atingida em aproximadamente 75% dos
doentes com SC [4]
. Depois do envolvimento

Síndrome de Cowden – Caso Clínico
9
cutâneo, a tiróide é o segundo órgão mais
frequentemente envolvido. As manifestações
incluem: bócio multinodular, disfunção
tiróideia, tiroidite de Hashimoto e cancro da
tiróide (especialmente os carcinomas papilar
e folicular) [3]
. As neoplasias da tiróide são
as segundas mais frequentes (com a
neoplasia da mama em primeiro lugar). O
risco de malignidade nos doentes com SC é
de 7-10% (comparativamente com o risco
1% da população geral) [7]
. Outras
anormalidades descritas foram: micro-
adenoma, adenoma folicular multicêntrico e
adenolipoma [3]
.
Sistema Ocular: as anormalidades oculares
surgem em 13% dos casos [3]
. As várias
anormalidades descritas incluem coloboma,
pale papillae, angioide streaks, opacidades
lenticulares, cataratas, glaucoma, estrabismo
e miopia [3]
.
Sistema Pulmonar: as lesões mais
frequentemente encontradas neste sistema
são: mesenquimomas endobrônquicos e
intrapulmonares que geralmente são
recorrentes e múltiplos, lipomas, quistos
pulmonares, malformações arterio-venosas e
adenocarcinoma pulmonar de baixo grau [3]
.
Sistema Cardiovascular: foram descritos
vários defeitos nos doentes com SC,
nomeadamente defeitos do septo auricular,
prolapso ou insuficiência da válvula mitral e
insuficiência da válvula aórtica [3]
.
Sistema Nervoso: as anormalidades do
sistema nervoso foram descritas em um
quinto dos pacientes. As principais são:
distúrbios da coordenação, oligofrenia,
meningioma, pseudotumor cerebri, aumento
da pressão intracraneana, hipertrofia
cerebelar, malformações arterio-venosas,
doença de Lhermitte-Duclos, neuromas
subcutâneos, ganglioneuromas, neurofi-
bromas, neuromas orais, mioblastoma de
células granulares e miastenia gravis [8]
.
Sistema Genito-Urinário: aproximadamente
55% dos doentes de sexo feminino
apresentam alguma anormalidade no sistema
reprodutor que surge habitualmente na
terceira década de vida [3]
. As anormalidades
vão desde distúrbios funcionais, nomea-
damente irregularidades menstruais,
teratomas, quistos ováricos, leiomiomas
uterinos, adenocarcinomas endometriais e
carcinoma do colo uterino [8]
. É importante
salientar que o carcinoma do endométrio é a
terceira neoplasia mais frequente no SC,
apresenta uma frequência de 5-10% dos
casos (comparativamente com o risco de
inferior a 2.5 na população geral) [7]
. No
sexo masculino as anormalidades mais
frequentemente encontradas foram: hidro-
celo e varicocelo. Outras anormalidades
descritas são: adenoangiomiolopoma,
pólipos benignos da uretra, carcinoma das

Síndrome de Cowden – Caso Clínico
10
células de transição da pelve renal e da
bexiga e rim em ferradura [3]
.
Sistema esquelético: em 37% dos casos foi
descrita algum tipo de anormalidade no
sistema esquelético [3]
. As mais frequente-
mente encontradas são: macrocefalia, fácies
adenóide, cifose torácica, pectus excavatum,
sindactilia, braquidactilia, aracnodactilia e
hipoplasia da omoplata [3]
.
Dada a diversificada lista de
potenciais manifestações clínicas, nem
sempre é fácil estabelecer um diagnóstico de
SC. É neste contexto que se insere como
objectivo desta dissertação o relato de um
caso de uma doente com SC, aproveitando a
oportunidade para posteriormente rever os
critérios que estabelecem o diagnóstico de
SC.
APRESENTAÇÃO DO CASO
M.A.V.M., sexo feminino,
caucasiana, nascida a 24 de Junho de 1946,
natural e residente no Porto e empregada
doméstica. Apresentava uma história
familiar relevante de neoplasias, nomea-
damente, a mãe que apresentava história de
Adenoma da Tiróide, Carcinoma Ductal
Invasor da Mama e Carcinoma Basocelular;
um irmão que faleceu aos 33 anos por tumor
do testículo e uma irmã com Carcinoma
Folicular da Tiróide e Cancro do Ovário,
vindo a falecer deste último.
Sem antecedentes patológicos
relevantes até aos seus 20 anos (1966),
altura em que foi diagnosticado Carcinoma
Folicular da Tiróide, tendo feito
Tiroidectomia Total. Teve recidiva cervical
sete anos depois (1973), tendo sido feito
esvaziamento ganglionar cervical. Pela
mesma altura, foi também diagnosticado
macrocefalia e nódulos subcutâneos dis-
persos nos membros superiores e inferiores,
mas sem diagnóstico anatomopatologico.
Em 1981, surgiu uma lesão nodular
na língua, cuja biopsia revelou benignidade
(nódulo fibrótico). Ainda no mesmo ano,
surgiu com um nódulo na mama esquerda.
Tratava-se de um Carcinoma Ductal Invasor.
Foi sujeita a cirurgia, quimio-radioterapia e
hormono- terapia.
Em 1887, foi diagnosticado um
hemangioma cavernoso no tórax.
Em Novembro de 1990, foi-lhe
diagnosticado um Carcinoma Ductal Invasor
da mama contralateral metastizado, tendo
sido sujeita a tratamento paliativo.
Em Março de 1994, foi
diagnosticado Mucinose Focal em nódulo da
coxa esquerda e Verruga Vulgaris na fossa
nasal esquerda.

Síndrome de Cowden – Caso Clínico
11
Em Março de 1995, foi sujeita a
histerectomia com anexectomia bilateral por
Adenossarcoma uterino. No mesmo ano, em
estudo endoscópico (alto e baixo) foi
diagnosticado Polipose Juvenil.
Em Dezembro de 1995, foram
detectados micronódulos pulmonares sem
características suspeitas de metastização
dado o seu comportamento estável.
Em Abril de 1997, foram
diagnosticados Dermatofibrossarcoma
Protuberans e Linfangioma.
Em Agosto de 1997, foi excisado
um Carcinoma Basocelular da pálpebra
direita.
Em Novembro de 1998, foi feita
uma RMN Cerebral que revelou
meningiomas de pequeno volume nos lobos
temporal direito e frontais.
Veio a falecer em 14 de Março de
1999 por neoplasia da mama extensamente
metastizado.
DISCUSSÃO Actualmente, a discussão reside na
diferenciação dos doentes com SC dos
outros que apresentam outros síndromes que
privilegiam o surgimento de doenças
oncológicas, bem como na abordagem
diagnóstica dos doentes que surgem com
lesões suspeitas e/ou neoplasias para um
correcto diagnóstico de SC.
Apesar da imensa listagem de potenciais
manifestações clínicas, o diagnóstico deste
Tabela 3 - Resumo Patológico

Síndrome de Cowden – Caso Clínico
12
síndrome simplificou-se bastante desde
2000, quando os membros do Consórcio
Internacional de Critérios de Diagnóstico
para o Síndrome de Cowden (ICC)
organizaram as principais cara- cterísticas
em três categorias: Critérios
patognomónicos, major e minor [7]
.
Critérios Patognomónicos:
Doença Lhermitte-Duclos no adulto;
Lesões mucocutâneas: triquilemomas
faciais, queratose acral e lesões
papilomatosas [4,7,8]
.
Critérios Major:
Cancro da Mama;
Carcinoma da Tiróide (não-medular
e especialmente carcinoma folicular);
Macrocefalia (circunferência occi-
pito-frontal ≥ 97 Percentil);
Carcinoma do Endométrio [4,7,8]
.
Critérios Minor:
Outras lesões tiróideias (ex.:
adenomas, bócio multinodular etc);
Oligofrenia (QI < 75);
Pólipos intestinais hamartomatosos;
Doença fibroquística da mama;
Lipomas;
Fibromas;
Tumores ou malformações genito-
urinários;
Fibromas uterinos [4,7,8]
.
O diagnóstico de SC é estabelecido
quando um indivíduo apresenta:
Lesões Mucocutâneas Patognomónicas;
≥ 6 pápulas faciais (≥ 3 triquili-
momas), ou
pápulas cutâneas faciais e papilo-
matose na mucosa oral, ou
≥ 6 pápulas de queratose acral
≥ 2 critérios major;
1 critério major e 3 critérios minor;
≥ 4 critérios minor [4,7,8]
.
No caso dos indivíduos onde existe
um familiar já com o SC diagnosticado (40-
60% dos casos estimam-se ser familiares),
os critérios de diagnóstico são:
1 critério patognomónico;
1 critério major com ou sem critérios
minor;
≥ 2 critérios minor;
História de BRRS [7]
.
Estes critérios foram revistos em
2004 pelo ICC e são validados anualmente
pelo US National Comprehensive Cancer
Network (NCCN) [7]
.
Actualmente, defende-se que
qualquer indivíduo que se apresente com
critérios para o diagnóstico de SC seja
referenciado para estudo genético para

Síndrome de Cowden – Caso Clínico
13
pesquisa de eventuais mutações em PTEN[7]
.
Os doentes que apresentam critérios clínicos
para o diagnóstico de SC mas não
apresentam mutação no gene PTEN,
recomenda-se que se faça uma análise da
região promotora do gene PTEN, de outros
genes que possam regular a expressão do
gene PTEN e a avaliação dos níveis da
proteína PTEN [7,12]
. O algoritmo de dia-
gnóstico de SC é representado da figura 2.
Contudo, recentemente foi pu-
blicado um artigo baseado num estudo
cohort na revista “The American Journal of
Human Genetics” que refere que os critérios
utilizados pelo NCCN apesar de úteis apre-
sentam desvantagens [12]
. A primeira desvan-
tagem que apresentaram foi uma diminuição
bastante significativa na prevalência da
mutação em PTEN nos doentes seleccio-
nados a partir do diagnóstico realizado pela
avaliação das manifestações clínicas
segundo a organização das principais
alterações clínicas em critérios patogno-
mónicos, major e minor (ou seja: de 3042
Figura 2 - Algoritmo Diagnóstico no SC [11]

Síndrome de Cowden – Caso Clínico
14
indivíduos com o diagnóstico de SC
segundo o método em vigor, apenas 9.5%
apresentavam mutação em PTEN) [12]
. A
segunda desvantagem apresentada foi a
incapacidade de atribuir um valor
correspondente a uma probabilidade pré-
teste positiva para mutação PTEN [12]
. A
terceira e última desvantagem, refere-se ao
facto dos critérios actualmente utilizados
pelo NCCN não serem específicos para
crianças e adultos uma vez que o SC é uma
doença que apresenta uma expressividade
variável e penetrância incompleta relaci-
onada com a idade [12]
. Assim sendo, Min-
Han et al desenvolveram um modelo
optimizado para estimar a probabilidade pré-
teste da mutação PTEN a partir dos critérios
existentes e ainda em vigor. Neste estudo em
vez de organizarem as várias manifestações
clínicas em critérios patognomónicos, major
e minor, atribuíram um valor numérico
individual para cada alteração clínica e
organizaram os vários scores semiquan-
titativos em dois grupos: Adultos e Crianças.
Chegaram à conclusão que, em adultos, um
score ≥ 10 apresenta uma sensibilidade
superior a 90% quando realizavam a
pesquisa para mutação PTEN[12]
. No grupo
pediátrico, os critérios que atribuíram
aumento da sensibilidade para um valor pré-
teste positivo na mutação PTEN eram
características fenotípicas diferentes do
grupo dos adultos, nomeadamente a
presença de macrocefalia como critério
necessário para referenciação de estudo
genético, uma vez que se apresenta com uma
prevalência de 100% nas crianças com SC e
com mutação em PTEN (Anexos 2 e 3) [12]
.
Este estudo reforça ainda a importância da
determinação de outros genes reguladores
do gene PTEN, ou mutação em outros genes
na linha germinativa (nomeadamente
mutações nos genes SDHB e SDHD) e
dosagem dos níveis da proteína PTEN [12]
.
Porém, para o caso em estudo desta
dissertação, a avaliação diagnóstica foi
realizada segundo os critérios do ICC e
NCCN em vigor. Então, a doente apresen-
tava as seguintes características: Tabela 4.
Tabela 4 - Critérios clínicos da doente
Síndrome de Cowden – Caso Clínico
15
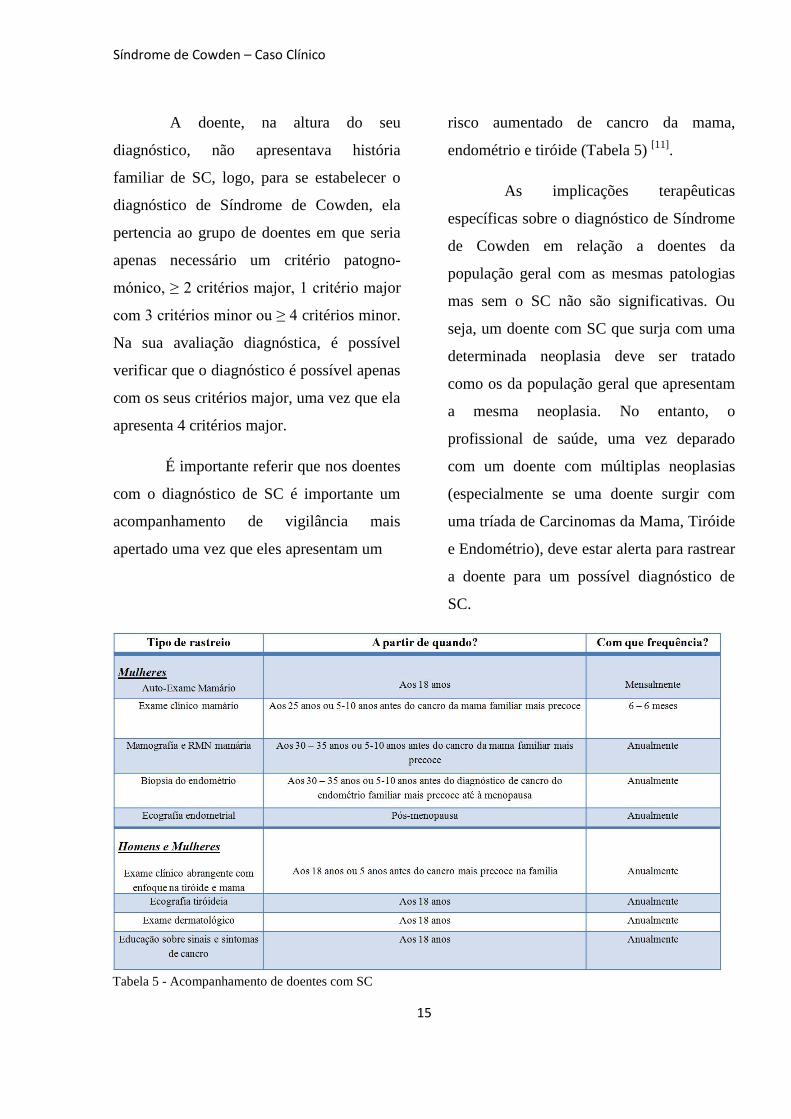
A doente, na altura do seu
diagnóstico, não apresentava história
familiar de SC, logo, para se estabelecer o
diagnóstico de Síndrome de Cowden, ela
pertencia ao grupo de doentes em que seria
apenas necessário um critério patogno-
mónico, ≥ 2 critérios major, 1 critério major
com 3 critérios minor ou ≥ 4 critérios minor.
Na sua avaliação diagnóstica, é possível
verificar que o diagnóstico é possível apenas
com os seus critérios major, uma vez que ela
apresenta 4 critérios major.
É importante referir que nos doentes
com o diagnóstico de SC é importante um
acompanhamento de vigilância mais
apertado uma vez que eles apresentam um
risco aumentado de cancro da mama,
endométrio e tiróide (Tabela 5) [11]
.
As implicações terapêuticas
específicas sobre o diagnóstico de Síndrome
de Cowden em relação a doentes da
população geral com as mesmas patologias
mas sem o SC não são significativas. Ou
seja, um doente com SC que surja com uma
determinada neoplasia deve ser tratado
como os da população geral que apresentam
a mesma neoplasia. No entanto, o
profissional de saúde, uma vez deparado
com um doente com múltiplas neoplasias
(especialmente se uma doente surgir com
uma tríada de Carcinomas da Mama, Tiróide
e Endométrio), deve estar alerta para rastrear
a doente para um possível diagnóstico de
SC.
Tabela 5 - Acompanhamento de doentes com SC

Síndrome de Cowden – Caso Clínico
16
Em conclusão, o estudo e discussão
deste caso clínico de uma doente que se
apresentou com Síndrome de Cowden, cujo
diagnóstico foi possível com o decorrer do
tempo e manifestações clínicas típicas da
evolução, visa fornecer mais informação
sobre o mesmo síndrome. Foi possível
estudar as várias formas de manifestações
clínicas e rever os critérios de diagnóstico.
AGRADECIMENTOS À Dra. Estrela Rocha pela
colaboração, disponibilidade, incentivo e
confiança depositada.
BIBLIOGRAFIA 1. RaviSM Prakash, Gn Suma, Sumit
Goel. Cowden syndrome. Indian J Dent
Res. 2010;21(3):439.
2. Patient Education Office. Cowden
Syndrome [Internet]. 2010 Mar
29;Available from: http://www2.mdan
derson.org/app/pe/index.cfm?pageName
=opendoc&docid=2190
3. Uppal S, Mistry D, Coatesworth AP.
Cowden disease: a review. International
Journal of Clinical Practice. 2007 Abr
1;61(4):645-652.
4. Charis Eng. PTEN Hamartoma Tumor
Syndrome (PHTS) - GeneReviews -
NCBI Bookshelf [Internet]. 2009 Mai 5
[citado 2011 Mai 7];Available from:
http://www.ncbi.nlm.nih.gov/books/NB
K1488/
5. Shenandoah Robinson, Alan R Cohen.
Cowden disease and Lhermitte-Duclos
disease: an update. Case report and
review of the literature. Neurosurg
Focus. 2006;20(1):E6.
6. Katherine K. Calvo, Emanuel F.
Petricoin III, Lance A. Liotta. Genomics
and Proteomic. Em: Cancer Principles
and Practice of Oncology. Lillincott
Williams and Wilkins; 2005. p. 54-56.
7. Gideon M Blumenthal, Phillip A
Dennis. PTEN hamartoma tumor
syndromes. Eur J Hum Genet. 2008
online;16(11):1289-1300.
8. Kendall Adkisson, Dirk M. Elston,
Katherine H. Fiala, Craig A. Elmets,
Richard P. Vinson, Lester F. Libow.
Cowden Disease (Multiple Hamartoma
Syndrome) [Internet]. 2010 Abr
26;Available from: http://emedicine.
medscape.com/article/1093383overview

Síndrome de Cowden – Caso Clínico
17
9. Genetics Home Reference. Cowden
syndrome [Internet]. 2006 Mar [citado
2011 Mai 17];Available from: http://
ghr.nlm.nih.gov/condition/cowdensyndr
ome
10. The Ohio State University Medical
Center. Cowden Syndrome [Internet].
Available from: http://medicalcenter
.osu.edu/patientcare/healthcare_services
/breast_health/cowden_syndrome/Pages
/index.aspx
11. Mary B. Dally, Jeffrey Allen, Jennifer
E. Axibund, Saundra Buys, Beth
Crawford, Carolyn D. Farrell, et al.
NCCN Clinical Practice Guidelines in
Oncology - Genetic/Familial High-Risk
Assessment: Breast and Ovarian,
Version 1.2011 [Internet]. [citado 2011
Mai 21];Available from: http://www.
nccn.org/professionals/physician_gls/pd
f/genetics_screening.pdf
12. Min-Han Tan, Jessica Mester, Charissa
Petreson, Yiran Yang, Jin-Lian Chen,
Lisa A. Rybicki, et al. A Clinical
Scoring System for Selection of Patients
for PTEN Mutation Testing Is Proposed
in the Basis of a Prospective Study of
3042 Probands. 2001 Jan 7;(88):42-56.

Síndrome de Cowden – Caso Clínico
18
ANEXOS