some new sequencing technologies. molecular inversion probes
Post on 20-Dec-2015
236 views
TRANSCRIPT

Some new sequencing technologies
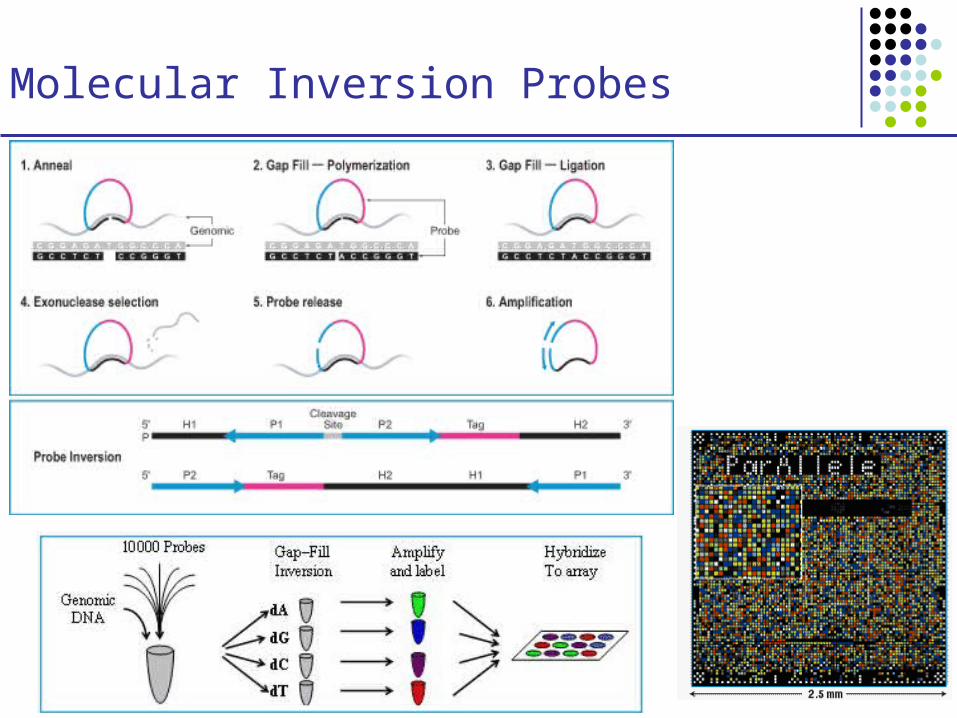
Molecular Inversion Probes
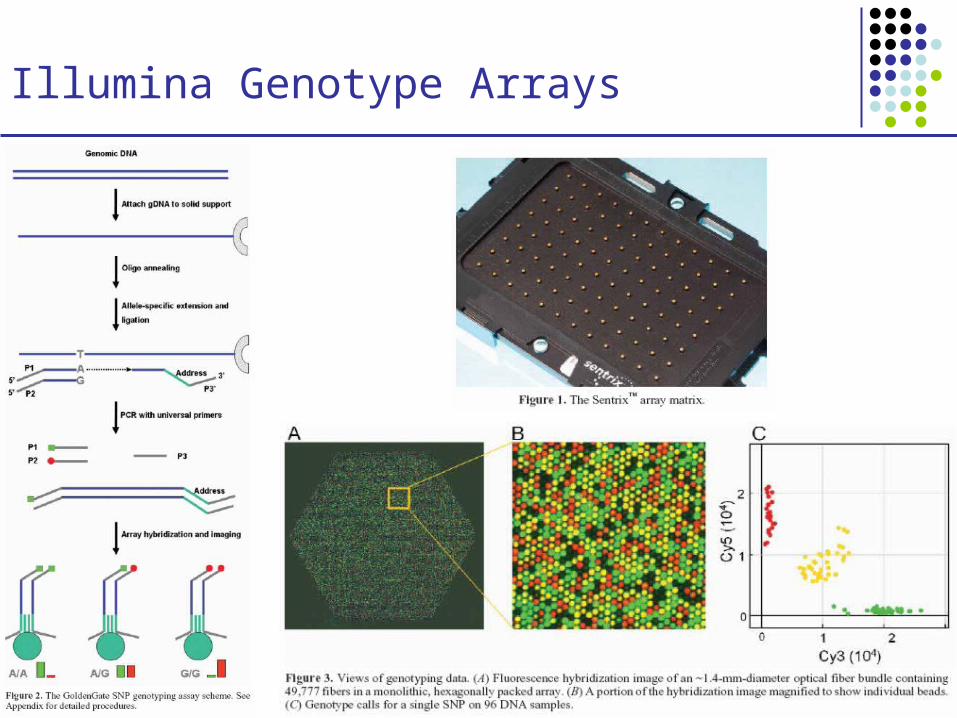
Illumina Genotype Arrays

Single Molecule Array for Genotyping—Solexa
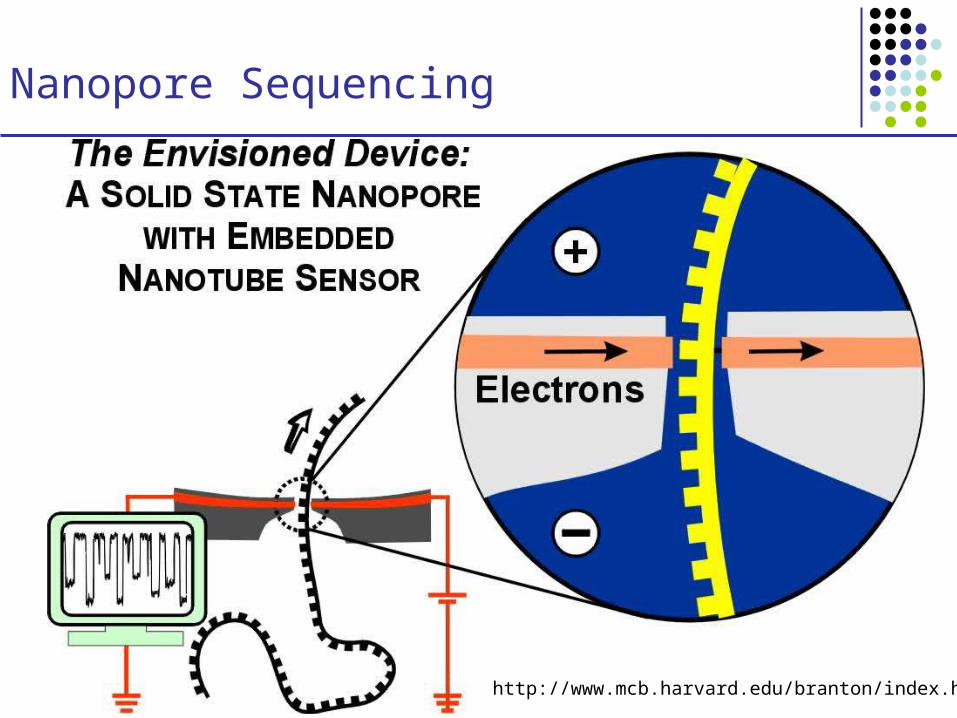
Nanopore Sequencing
http://www.mcb.harvard.edu/branton/index.htm
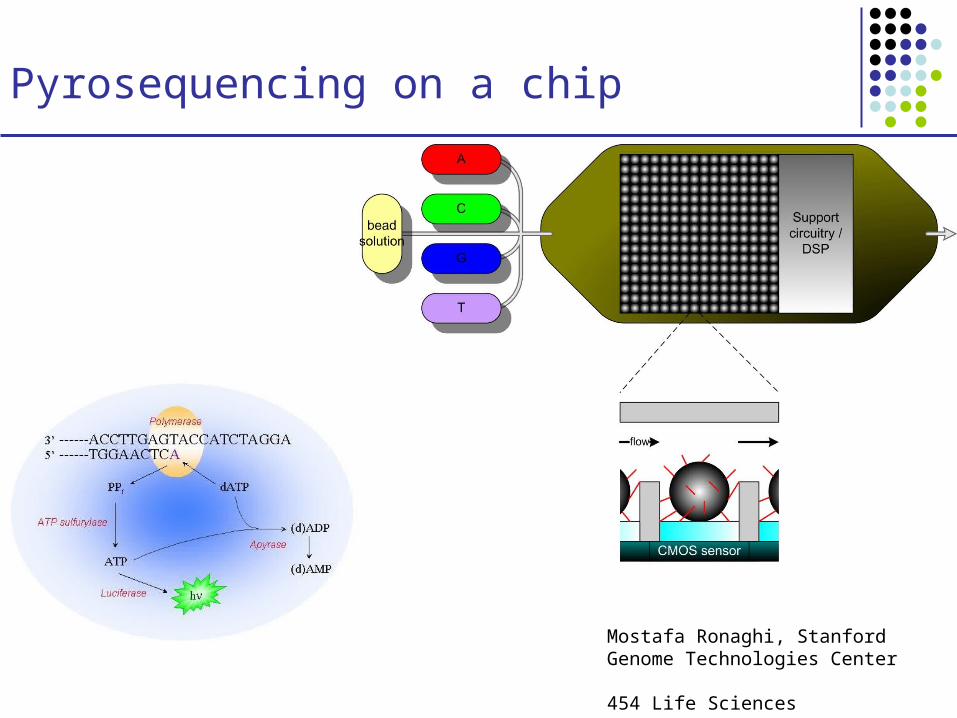
Pyrosequencing on a chip
Mostafa Ronaghi, Stanford Genome Technologies Center
454 Life Sciences
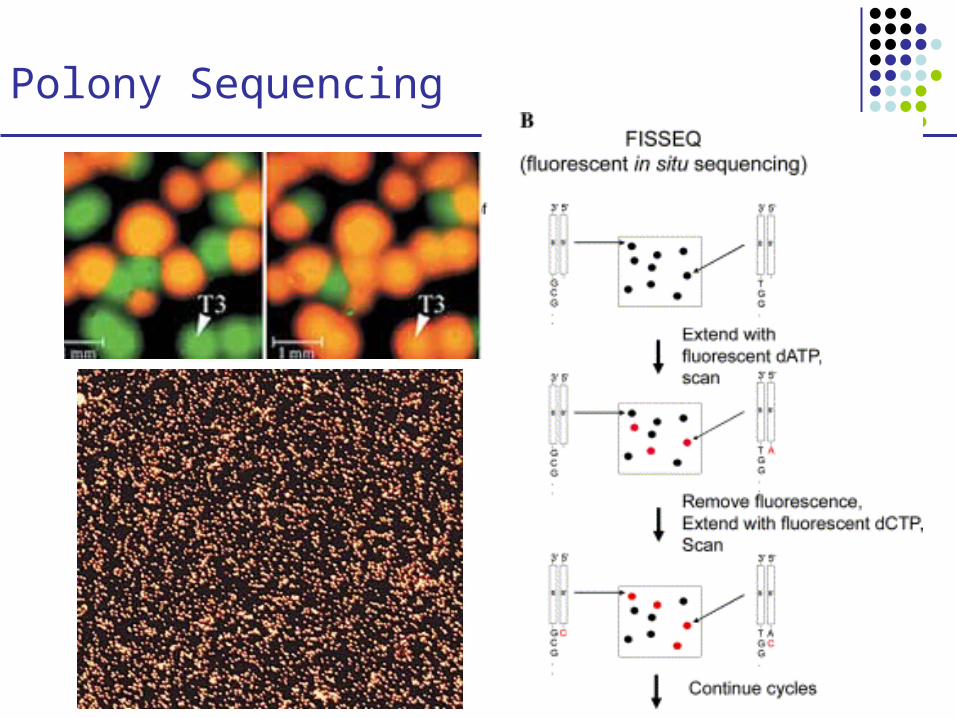
Polony Sequencing

Technologies available today
• Illumina 550,000 SNP array: $300-500 in bulk
• 454 200 bp reads, 100 Mbp total sequence in 1 run, $8K 500bp reads in much higher throughput coming soon
• Solexa 1Gbp of sequence coming in paired 35 bp reads 1 day, approx $10K / run
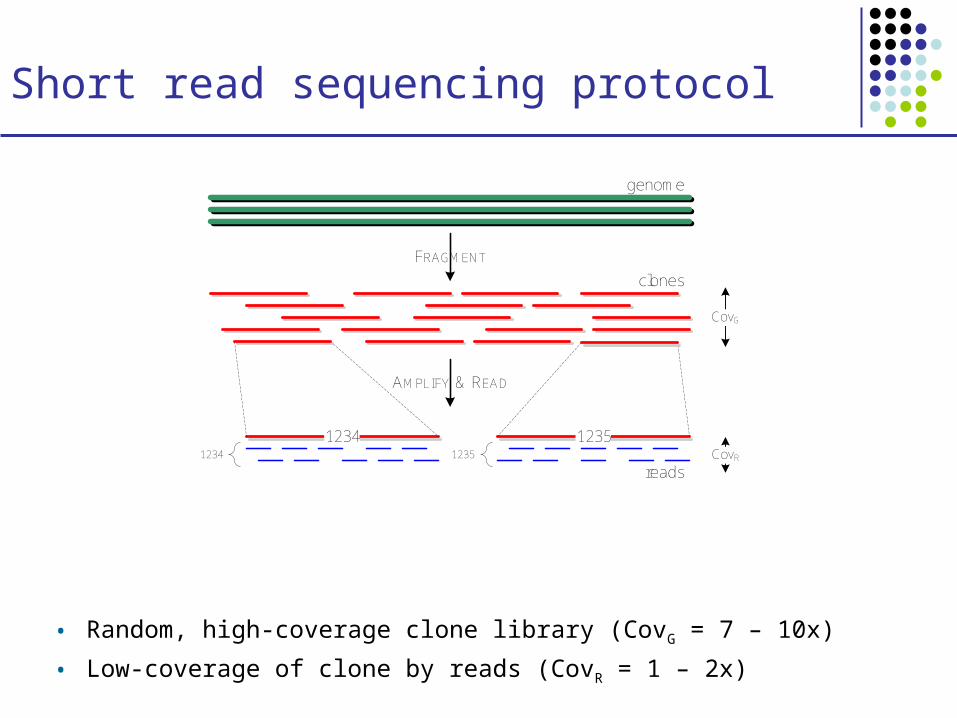
Short read sequencing protocol
• Random, high-coverage clone library (CovG = 7 – 10x)
• Low-coverage of clone by reads (CovR = 1 – 2x)
1234 1235
FRAGMENT
genome
clones
AMPLIFY & READ
12351234
reads
CovG
CovR
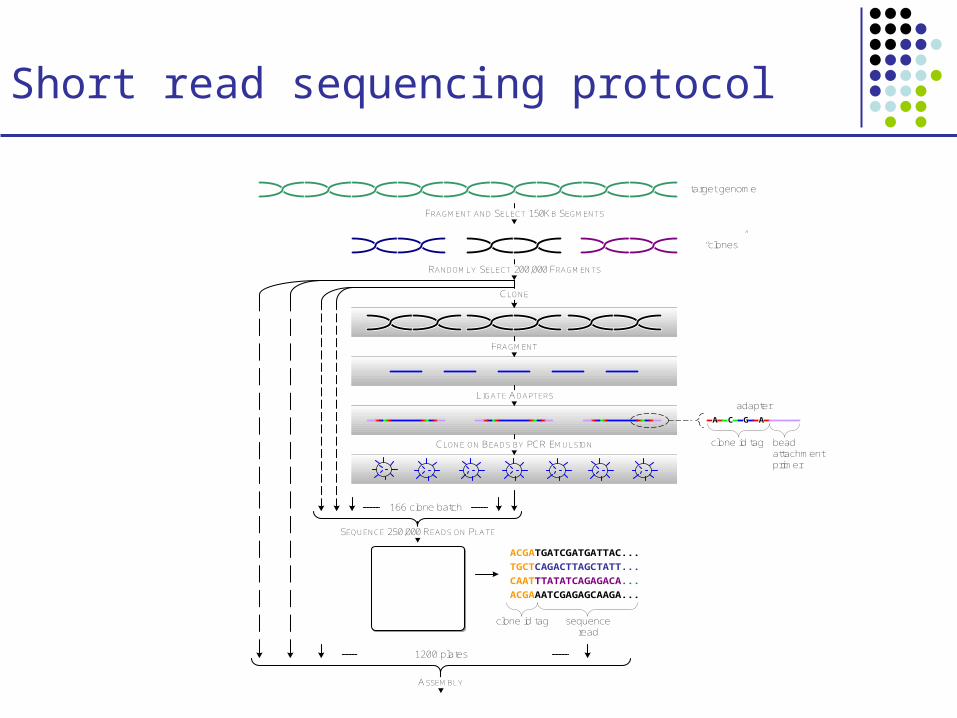
Short read sequencing protocol
RANDOMLY SELECT 200,000 FRAGMENTS
CLONE
FRAGMENT AND SELECT 150KB SEGMENTS
FRAGMENT
A C G A
bead attachment primer
adapter
clone id tag
LIGATE ADAPTERS
166 clone batch
CLONE ON BEADS BY PCR EMULSION
ACGATGATCGATGATTAC...
TGCTCAGACTTAGCTATT...
CAATTTATATCAGAGACA...
ACGAAATCGAGAGCAAGA...
clone id tag
SEQUENCE 250,000 READS ON PLATE
sequenceread
1200 plates
ASSEMBLY
target genome
“clones”
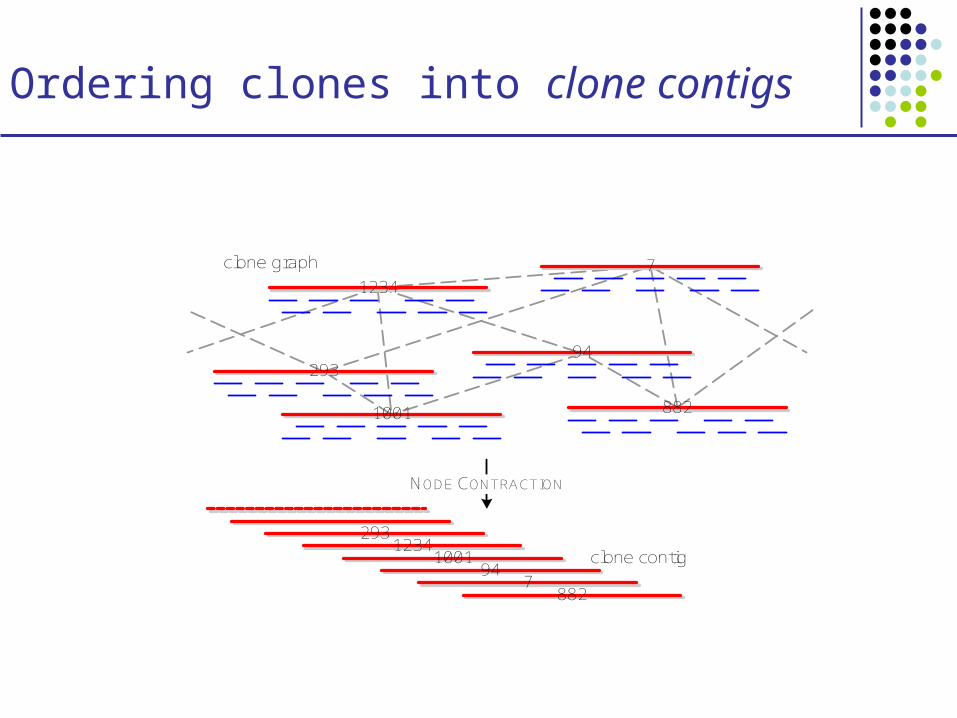
Ordering clones into clone contigs
293
1001
1234
882
7
94
clone graph
NODE CONTRACTION
clone contig1234
293
100194
7882
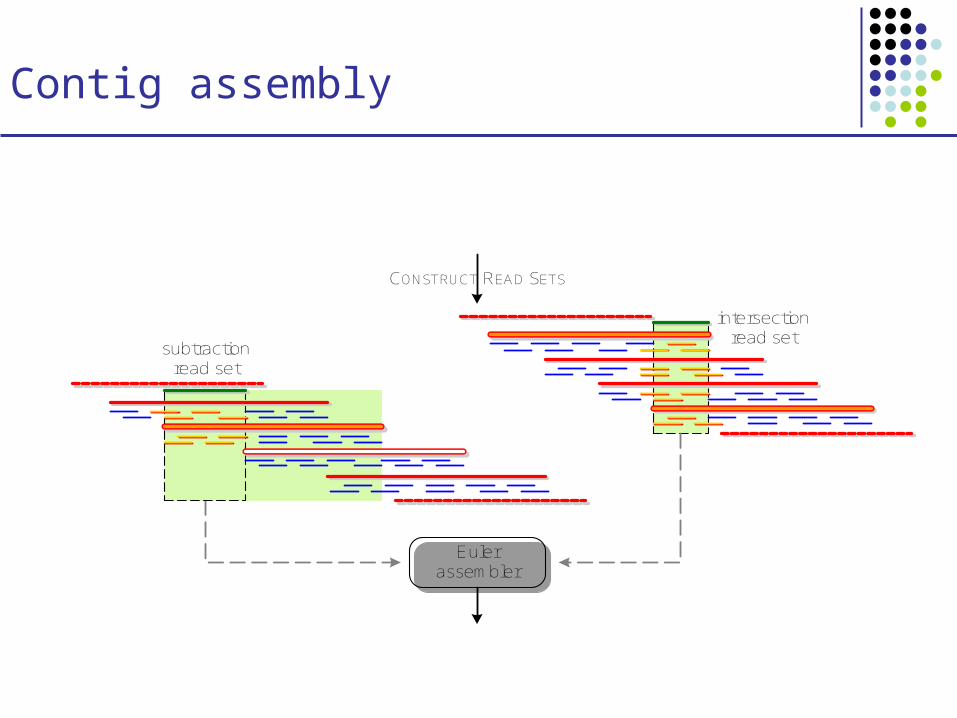
Contig assembly
CONSTRUCT READ SETS
Euler assembler
intersection read set
subtraction read set
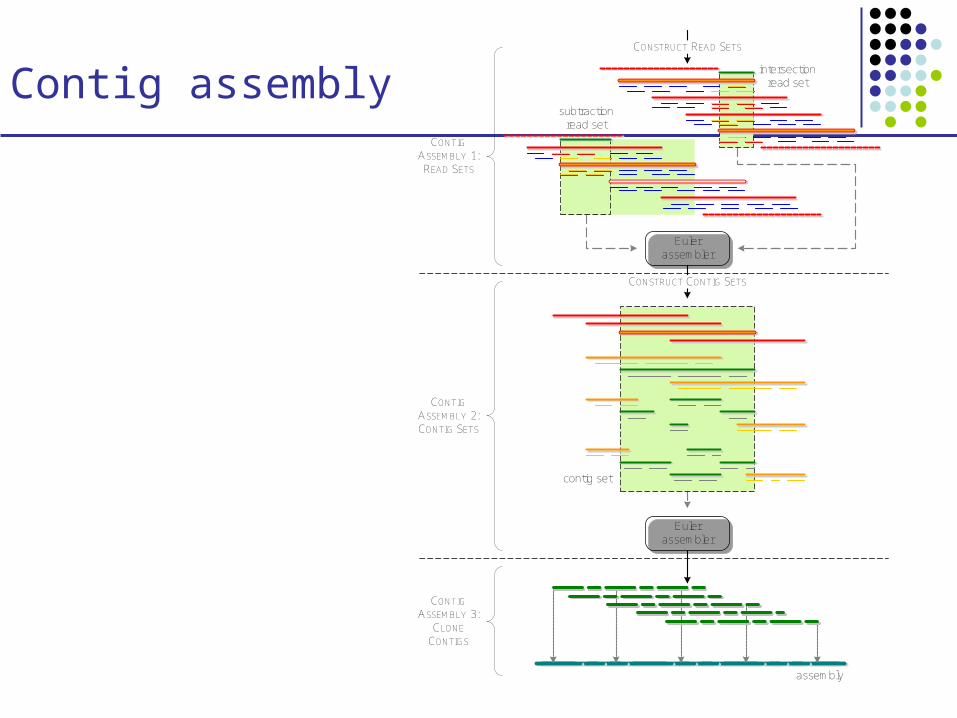
Contig assemblyCONTIG
ASSEMBLY 1: READ SETS
CONSTRUCT READ SETS
Euler assembler
CONSTRUCT CONTIG SETS
CONTIG ASSEMBLY 2: CONTIG SETS
Euler assembler
CONTIG ASSEMBLY 3:
CLONE CONTIGS
assembly
intersection read set
subtraction read set
contig set
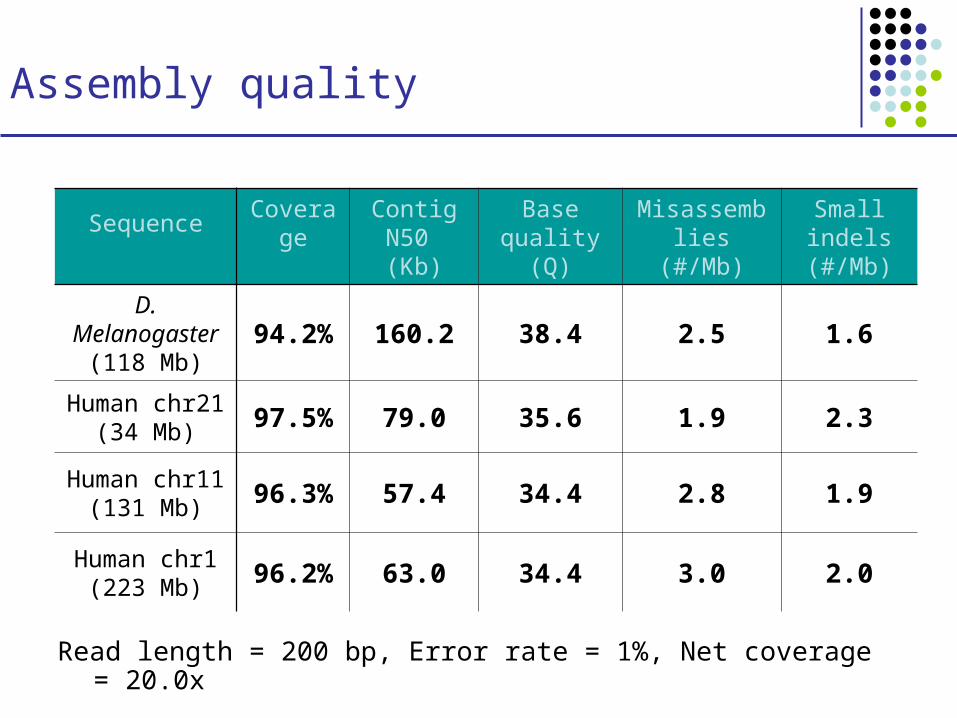
Assembly quality
Sequence CoverageContig N50 (Kb)
Base quality (Q)
Misassemblies (#/Mb)
Small indels (#/Mb)
D. Melanogaster(118 Mb)
94.2% 160.2 38.4 2.5 1.6
Human chr21(34 Mb)
97.5% 79.0 35.6 1.9 2.3
Human chr11(131 Mb)
96.3% 57.4 34.4 2.8 1.9
Human chr1(223 Mb)
96.2% 63.0 34.4 3.0 2.0
Read length = 200 bp, Error rate = 1%, Net coverage = 20.0x

Multiple Sequence Alignment
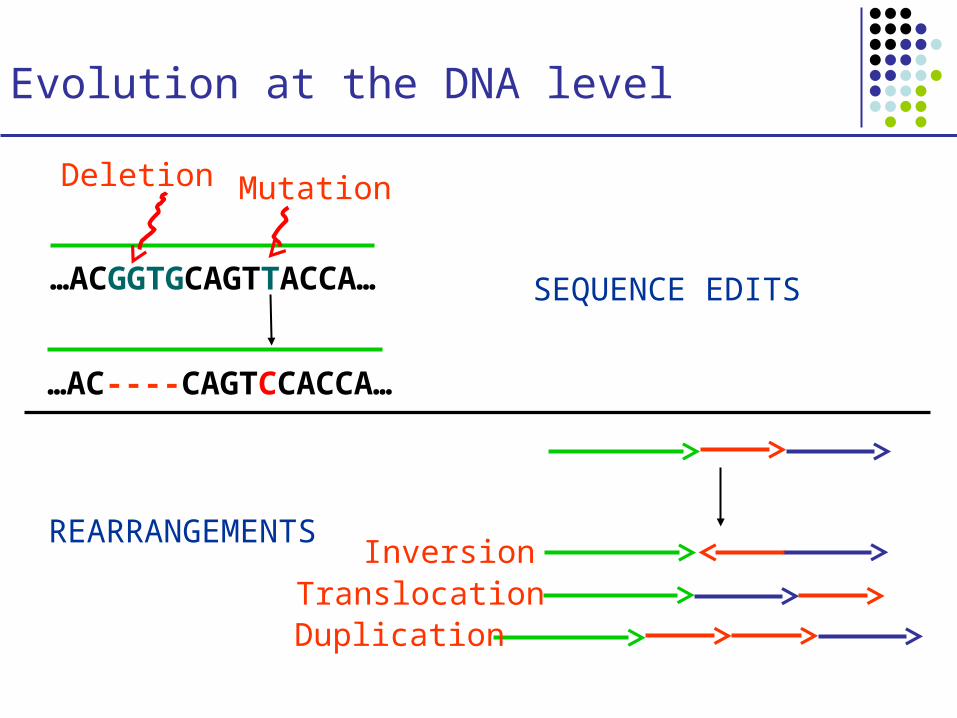
Evolution at the DNA level
…ACGGTGCAGTTACCA…
…AC----CAGTCCACCA…
Mutation
SEQUENCE EDITS
REARRANGEMENTS
Deletion
InversionTranslocationDuplication
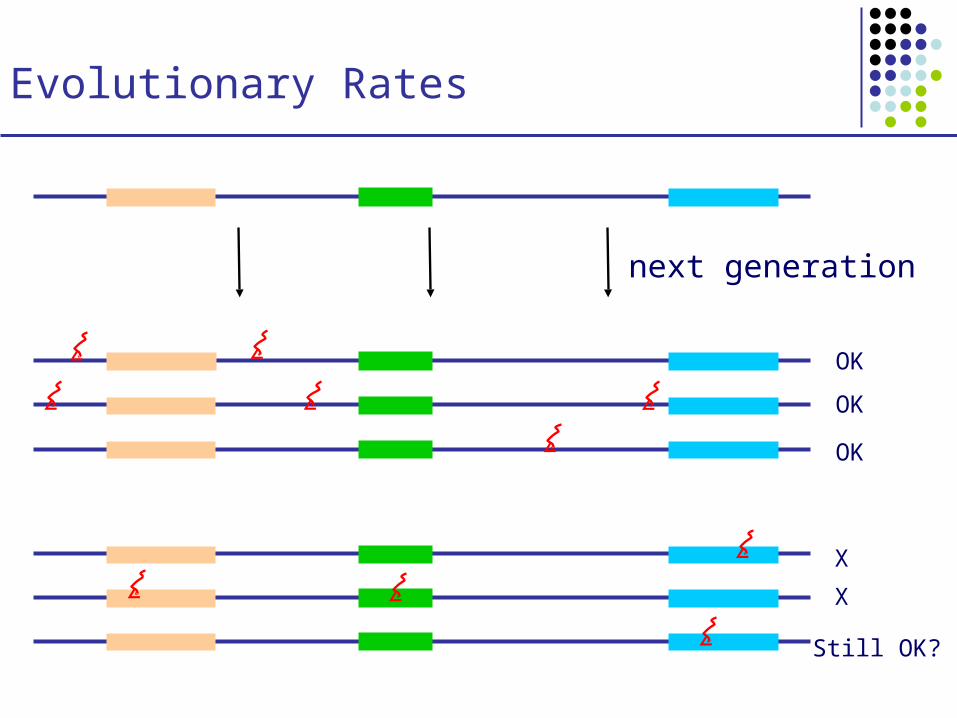
Evolutionary Rates
OK
OK
OK
X
X
Still OK?
next generation

Genome Evolution – Macro Events
• Inversions• Deletions• Duplications
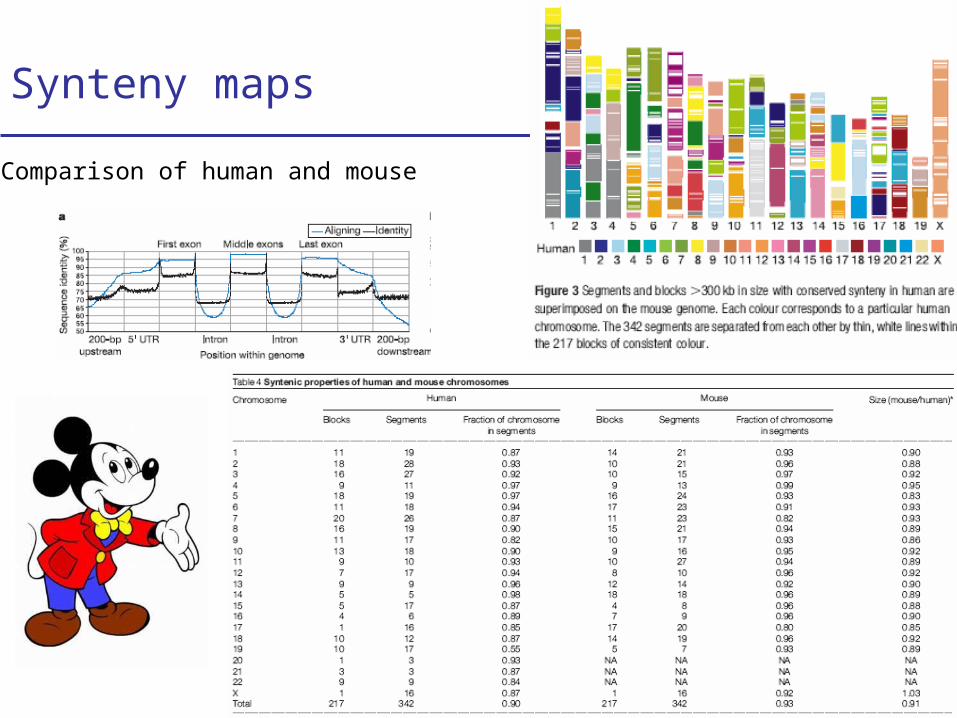
Synteny maps
Comparison of human and mouse
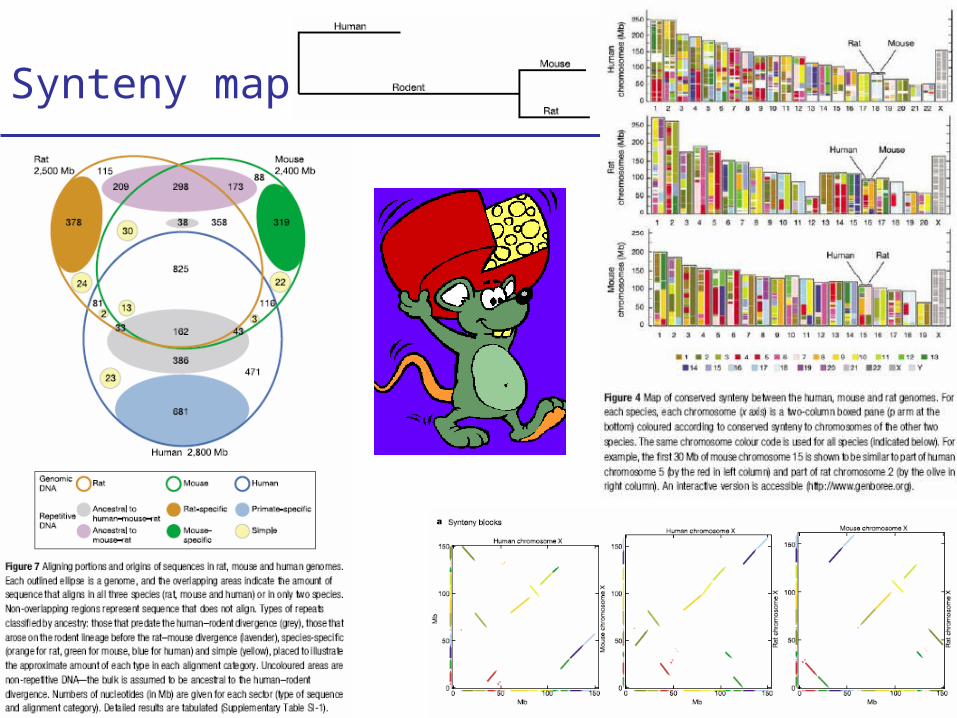
Synteny maps
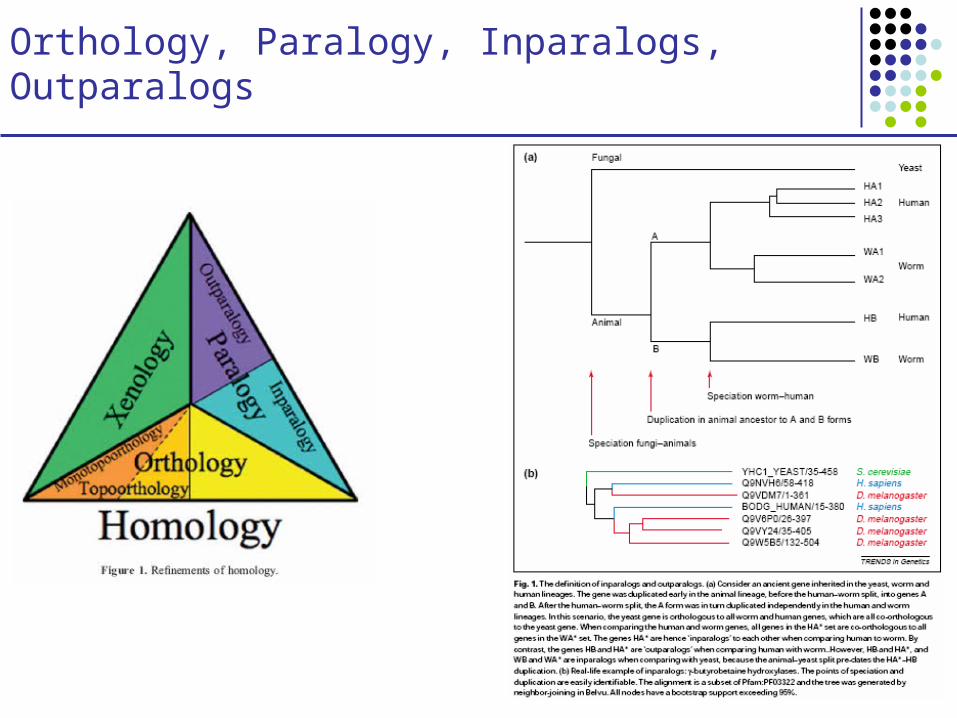
Orthology, Paralogy, Inparalogs, Outparalogs

Synteny maps
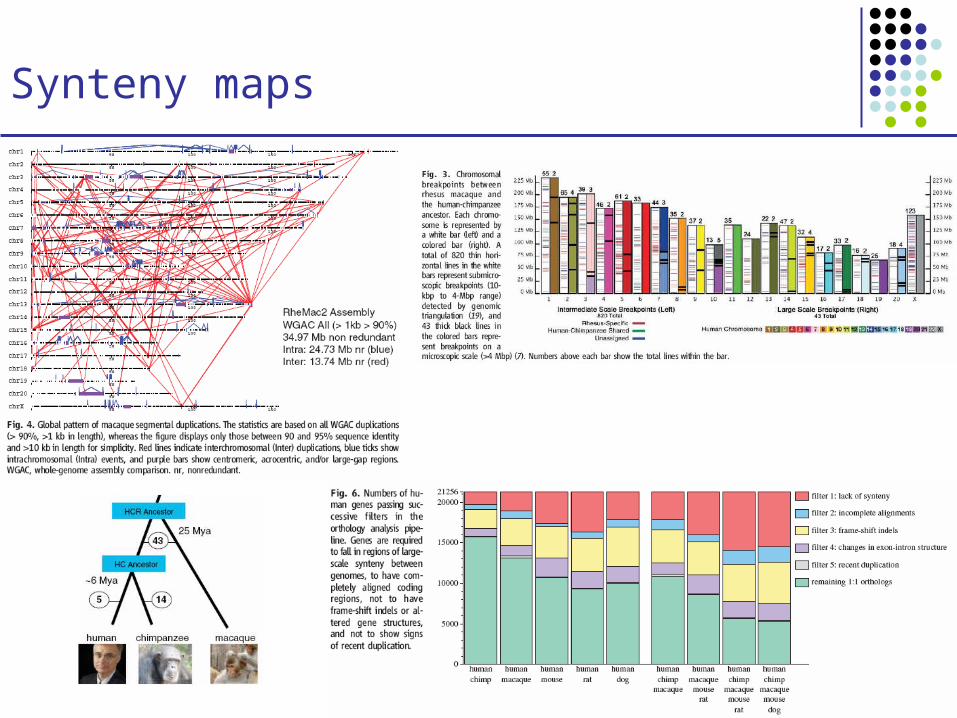
Synteny maps
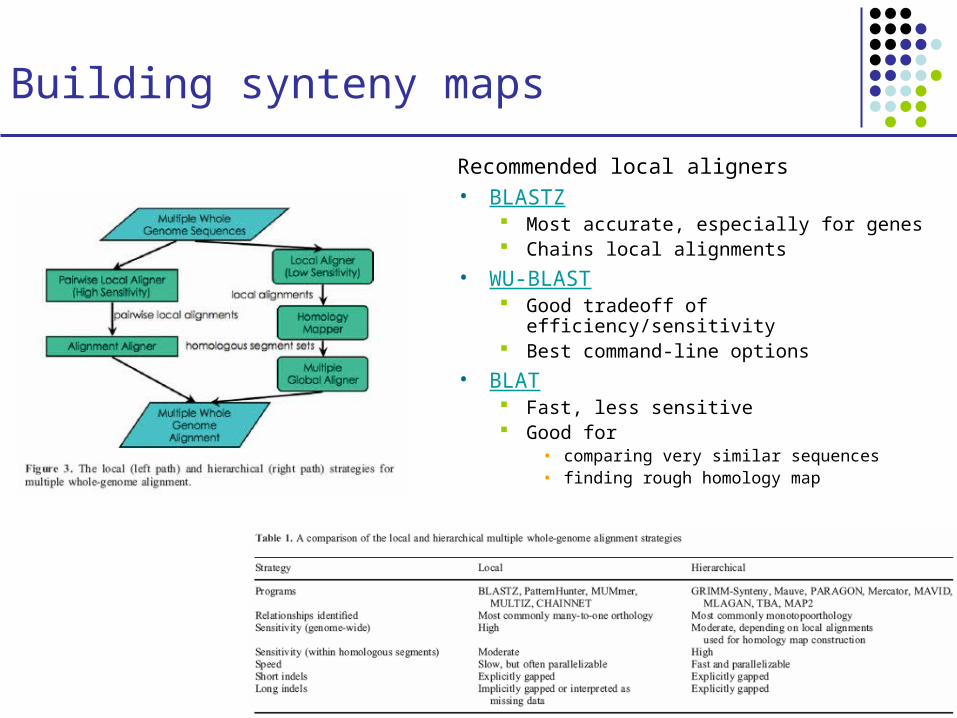
Building synteny maps
Recommended local aligners• BLASTZ
Most accurate, especially for genes Chains local alignments
• WU-BLAST Good tradeoff of efficiency/sensitivity Best command-line options
• BLAT Fast, less sensitive Good for
• comparing very similar sequences • finding rough homology map

Index-based local alignment
Dictionary:
All words of length k (~10)
Alignment initiated between words of alignment score T
(typically T = k)
Alignment:
Ungapped extensions until score
below statistical threshold
Output:
All local alignments with score
> statistical threshold
……
……
query
DB
query
scan
Question: Using an idea from overlap detection, better way to find all local alignments between two genomes?

Local Alignments

After chaining

Chaining local alignments
1. Find local alignments
2. Chain -O(NlogN) L.I.S.
3. Restricted DP

Progressive Alignment
• When evolutionary tree is known:
Align closest first, in the order of the tree In each step, align two sequences x, y, or profiles px, py, to generate a new
alignment with associated profile presult
Weighted version: Tree edges have weights, proportional to the divergence in that edge New profile is a weighted average of two old profiles
x
w
y
z
Example
Profile: (A, C, G, T, -)px = (0.8, 0.2, 0, 0, 0)py = (0.6, 0, 0, 0, 0.4)
s(px, py) = 0.8*0.6*s(A, A) + 0.2*0.6*s(C, A) + 0.8*0.4*s(A, -) + 0.2*0.4*s(C, -)
Result: pxy = (0.7, 0.1, 0, 0, 0.2)
s(px, -) = 0.8*1.0*s(A, -) + 0.2*1.0*s(C, -)
Result: px- = (0.4, 0.1, 0, 0, 0.5)
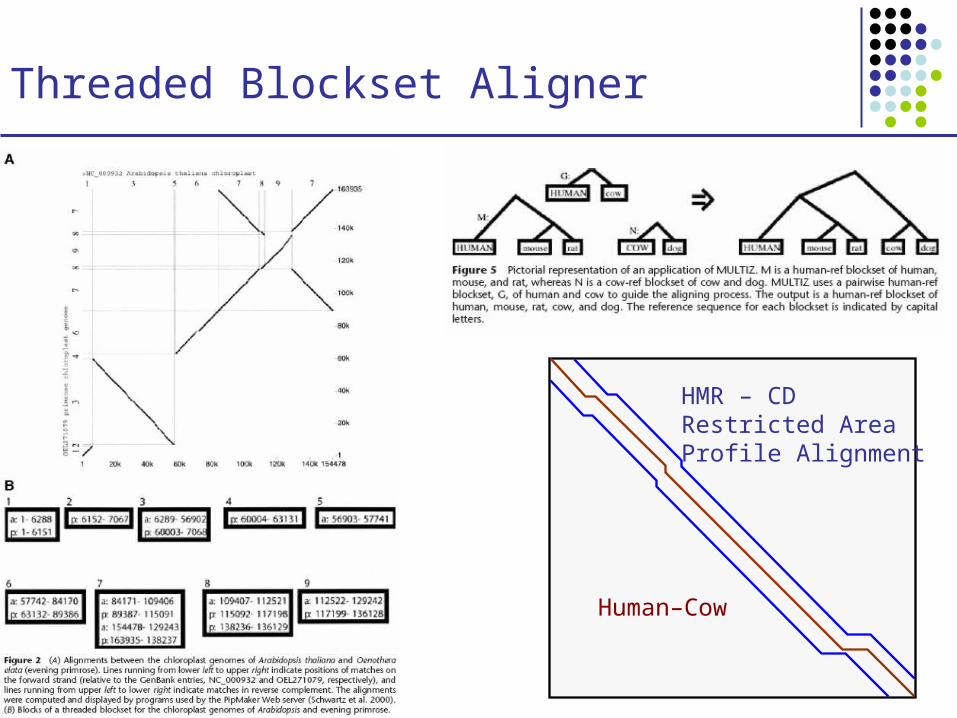
Threaded Blockset Aligner
Human–Cow
HMR – CDRestricted AreaProfile Alignment

Reconstructing the Ancestral Mammalian Genome
Human: C
Baboon: C
Cat: C
Dog: G
C
C or G
G