sos response and the mechanism of adaptive tolerance in ...583/fulltext.pdf · 1 sos response and...
TRANSCRIPT

1
SOS RESPONSE AND THE MECHANISM OF ADAPTIVE TOLERANCE IN
Escherichia coli
A dissertation presented by
Tobias Dörr
to
The Department of Biology
In partial fulfillment of the requirements for the degree of
Doctor of Philosophy
in the field of
Biology
Northeastern University
Boston, Massachusetts
September 2010

2
©2010
Tobias Dörr
ALL RIGHTS RESERVED

3
SOS RESPONSE AND THE MECHANISM OF ADAPTIVE TOLERANCE IN
Escherichia coli
By Tobias Dörr
ABSTRACT OF DISSERTATION
Submitted in partial fulfillment of the requirements
for the degree of Doctor of Philosophy in Biology
in the Graduate School of Arts and Sciences of
Northeastern University, September 2010

4
ABSTRACT
Bacteria produce persisters, a small subpopulation of cells that neither grow nor
die in the presence of antibiotics. Persisters are tolerant against exposure to multiple
antibiotics and they likely contribute to the relapse of bacterial infections after antibiotic
therapy. The mechanism of persister formation is unknown, although several studies
have pointed towards redundancy in persister formation mechanisms and the possible
involvement of chromosomal toxin-antitoxin modules.
While studying the genetic requirements for Escherichia coli persister survival
after exposure to the DNA damaging antibiotic ciprofloxacin, we found that persister
formation was an adaptive response to the antibiotic. Survivors to ciprofloxacin
exhibited low levels of SOS induction and their survival depended largely on the SOS-
inducible small toxic peptide TisB. Ectopic overproduction of TisB decreased proton
motive force and induced growth arrest and multidrug tolerance. Further, synthesized
TisB peptide formed an anion-selective pore in an artificial lipid bilayer system. These
results suggest that TisB acts as an uncoupler of oxidative phosphorylation after
induction of the SOS response.
These results challenge the common view of persisters as a metabolically
inactive entity and show that persistence is in part an inducible response specific to a
certain stress.

5
ACKNOWLEDGEMENTS
Countless people inside and outside the lab have made the past few years highly
enjoyable and stimulating and I wish to thank all of them.
First and foremost I would of course like to thank Kim Lewis, my advisor, for
giving me the opportunity to work in his lab. His patience, guidance and leadership style
have been an inspiration for my personal as well as professional development and all of
his book recommendations were excellent. Along the same lines I would like to extend a
big hvala to Marin Vulić for his scientific guidance, for commiserating and for Croatian
proverbs. Special thanks to Larry Mulcahy for his help with sorting and figuring out what
the best band in the world is.
Thank you to Sonja Hansen, Mike Lafleur, Iris Keren, Alyssa Theodore, Pooja
Balani, Tony D’Onofrio, Katya Gavrish, Eric Stewart, Janet Manson, Ron Ortenberg,
Gabriele Casadei and all other lab members for being awesome friends and colleagues
and for making this a fantastic place to work at.
An important thank you goes out to my committee members Drs. Veronica
Godoy, Slava Epstein and Abraham L. Sonenshein.
Outside the lab, I wish to thank my wife, Binu Shrestha for her love and support. I
am thankful to Riverhouse inhabitants past and present, especially Smita Das for helpful
comments. Importantly, thank you to my parents for their continuing support for my
endeavors.

6
Table of contents
ABSTRACT 4
ACKNOWLEDGEMENTS 5
TABLE OF CONTENTS 6
LIST OF FIGURES 7
INTRODUCTION 9
CHAPTER 1 SOS RESPONSE INDUCES PERSISTENCE TO FLUOROQUINOLONES
IN ESCHERICHIA COLI 18
CHAPTER 2 CIPROFLOXACIN CAUSES PERSISTER FORMATION BY INDUCING
THE TISB TOXIN IN ESCHERICHIA COLI 52
CHAPTER 3 TISB MECHANISM OF ACTION 84
DISCUSSION 105

7
List of Figures
Figure 1. Survival of the wild type and the mutants deficient in recombination and/or
SOS induction after ciprofloxacin challenge........................................................... 46
Figure 2. SOS induction and persister level during ciprofloxacin challenge in exponential
growth phase. ........................................................................................................ 47
Figure 3. Fraction of cells undergoing strong SOS induction during ciprofloxacin
challenge................................................................................................................ 48
Figure 4. Ciprofloxacin-induced persistence. ................................................................ 49
Figure 5. Mitomycin C-induced persistence. ................................................................. 50
Figure 6. Growth phase and persister formation. .......................................................... 51
Figure 7. Survival of the tisAB/istR mutants after ciprofloxacin exposure and
complementation of the phenoype ......................................................................... 76
Figure 8. Schematic of the tisAB/IstR locus. ................................................................. 77
Figure 9. Induction of LexA-controlled promoters by ciprofloxacin. ............................... 78
Figure 10. Adaptive ciprofloxacin tolerance in E. coli. ................................................... 79
Figure 11. TisB overproduction and antibiotic tolerance. .............................................. 80
Figure 12. TisB-dependent persister formation in SOS response mutants.................... 81
Figure 13. Model of ciprofloxacin-induced persister formation. ..................................... 82
Figure 14. Survival in antibiotics after overexpression of tisB from O157:H7 Sakai...... 98
Figure 15. Survival of strains carrying translational TisB – mcherry fusions in
ciprofloxacin. .......................................................................................................... 99
Figure 16. Overexpression of tisB in respiratory chain mutants. ................................. 100

8
Figure 17. TisB overproduction under anaerobic conditions in different carbon sources.
............................................................................................................................. 101
Figure 18. Effects of cccp on multidrug tolerance ....................................................... 102
Figure 19. Proton motive force after TisB overproduction ........................................... 103
Figure 20. TisB-dependent conductance of a black membrane lipid bilayer. .............. 104
Figure 21. Model of stochasticity in TisB-mediated persister formation ...................... 113

9
INTRODUCTION
Cell populations produce persisters, a small fraction of cells impervious to the
lethal effects of antibiotics. Persisters have been observed in bacteria (Lewis, 2010),
yeast (LaFleur et al., 2006) and cancer cells (Sharma et al.,2010).
Bacterial persisters were discovered by Joseph Bigger in 1944, who noticed that
cultures of Staphylococcus aureus could not be sterilized by the beta lactam antibiotic
Penicillin (Bigger, 1944). Bigger described the phenomenon and established that the
survivors were not resistant mutants but rather they were phenotypic variants of the wild
type arising at a low frequency (between 1 in 100 and 1 in 10,000 cells).
First clues towards a mechanism of persister formation came 40 years later from
Harrison Moyed and his coworkers, who isolated mutants that survived extended
treatment with the beta lactam antibiotic ampicillin. Mutants with an increase in
minimum inhibitory concentration (MIC) were discarded in order to exclude resistant
strains. One of the high-persister mutations was mapped to the hipA gene, the toxin part
of the toxin-antitoxin pair hipBA (Moyed & Bertrand, 1983, Black et al., 1994, Moyed &
Broderick, 1986). This hipA7 allele is a gain of function mutation increasing the
frequency of persisters to ampicillin and an unrelated class of antibiotics,
fluoroquinolones, by 10- to 100-fold (Scherrer & Moyed, 1988, Korch et al., 2003, Korch
& Hill, 2006). A knockout of hipA or hipBA, however, had no phenotype (Moyed &
Broderick, 1986, Hansen et al., 2008), leaving the role of the wild type allele in persister
formation unknown.

10
Interest in persisters was renewed when Spoering et al. showed that these cells
were responsible for the enigmatic resistance of biofilm populations to killing by
antibiotics (Spoering & Lewis, 2001). Biofilm populations do not induce lifestyle-specific
antibiotic resistance mechanisms but they rather contain a large proportion of persister
cells, similar to stationary phase planktonic cultures. It was persisters that survived
antibiotic exposure and, due to the biofilm matrix, the host immune response (Vuong et
al., 2004). These persisters likely contributed to the common relapse of biofilm
infections after antibiotic therapy (Lewis, 2007).
The revival of interest in this phenomenon also led to a revival of mechanistic
studies. Keren et al. confirmed Bigger’s results (Keren et al., 2004a) and showed that
persisters were formed exclusively in mid-exponential to stationary phase.
Consequently, persisters could be eradicated by keeping a bacterial culture in early
exponential phase through constant dilution. Using an Escherichia coli strain carrying
the high persister allele hipA7, Keren et al. conducted transcriptomic analysis on cells
surviving extended ampicillin treatment and found that persisters had upregulated
conspicuously many chromosomal toxin-antitoxin modules (including relBEF, dinJ,
mazEF) as well as sulA, a septation inhibitor and rmf, the stationary phase ribosome
modulation factor (Keren et al., 2004b). It was subsequently revealed that
overexpression of the chromosomal toxins led to a multidrug tolerant, persister-like state
(Keren et al., 2004b, Shah et al., 2006), providing an attractive mechanism for persister
formation. Knockouts of TA modules, however, still had no phenotype, which led to the
hypothesis that persister formation might be mechanistically redundant (Lewis, 2007).
Escherichia coli contains at least 10 mRNA-interferases (yafQ, relE, mazF, ygiT, yafO,

11
chpB, higB, hicA, yoeB, yhaV)(Gerdes et al., 2005, Christensen-Dalsgaard et al., 2010 )
and at least 14 other toxin antitoxin modules including several small, toxic peptides with
antisense RNA antitoxins (type 1 TA modules) (Fozo et al., 2008, Alix & Blanc-Potard,
2009, Fozo et al., 2010). A similar redundancy of TA modules has been reported for
most other free-living microbes (Pandey & Gerdes, 2005).
Shah et al. confirmed and extended these transcriptome results by using wild
type E. coli and a transcriptional fusion of an rRNA promoter with an unstable variant of
green fluorescent protein (GFP) (Shah et al., 2006). The rRNA promoter activity is
strictly coupled with the growth rate (Gourse et al., 1996) and is thus shut down in non-
growing cells. The unstable GFP has a half-life of ca. 30 minutes (Andersen et al.,
1998). Thus, exponentially growing cells were bright green and cells that stopped
growing quickly lost green fluorescence. Using FACS, it was shown that dim cells that
accumulated in a growing population were enriched for ofloxacin-tolerant persisters.
Transcriptome analysis of these cells revealed again an upregulation of toxin-antitoxin
modules (chp, dinJ, relE, dinJ/yafQ, yefM/yoeB, ygiUT) but also some stationary phase
genes (bolA, katE, hdeA) and glycerol metabolism genes (glpD, glpQ). Yet again, no
single knockouts or any attempted knockout combinations of genes upregulated in
persisters (including a strain with 8 TA module knockouts, (unpublished)) produced a
strain with reduced persister levels, reinforcing the redundancy hypothesis. Some other
studies, however, found persister phenotypes for diverse TA module knockouts and
overexpression in different conditions such as dinJ/yafQ (Harrison et al., 2009), hokA
and ygiUT (Kim & Wood, 2010). Although the observed effects were usually modest for

12
single knockouts, these results strengthen the notion of involvement of TA modules in
persister formation and offer an explanation for redundancy.
A screen selecting for inducers of high persistence upon multicopy
overexpression from native promoters revealed glpD, coding for glycerol 3 phosphate
dehydrogenase, as a potential persister gene (Spoering et al., 2006). Overexpression of
glpD led to an increase in persister levels while deletion caused a decrease. This,
together with the fact that glpD had been shown to be upregulated in persisters
suggests the involvement of glycerol metabolism as at least one part of persister
formation. The role this pathway plays, however, remains unclear. Interestingly, no
toxin-antitoxin modules were found in this screen.
Redundancy in persister formation was further supported by results from screens
using an ordered, nearly complete E. coli knockout library (Hansen et al., 2008) and a
Pseudomonas aeruginosa transposon insertion library (De Groote et al., 2009) – in
neither case was a single knockout strain identified, which completely lacked persisters.
In summary, four different attempts of identifying the genetic basis of persistence
were only partially consistent: persister formation seemed to be related to toxin-antitoxin
expression and to glycerol metabolism.
Apart from mechanistic details, an important question was the role of persisters
in a population context. Balaban et al. and Shah et al. showed that persisters were pre-
exisiting in a population before addition of the antibiotic (Shah et al., 2006, Balaban et
al., 2004) and Wiuff et al. showed that persisters were multidrug tolerant (Wiuff et al.,
2005). Therefore it seemed that persisters represented a simple form of altruism, where
the individual persister bacterium forfeits propagation while constituting a kind of “life

13
insurance” for regrowth of the whole population following a catastrophic event such as
exposure to a bactericidal antibiotic. Indeed, mathematical models have shown that
persistence can assure population survival in frequently changing environments
(Kussell et al., 2005) which most bacteria are subjected to.
Thus emerged the prevailing view of the persister population as an entity
comprised of multidrug tolerant, dormant cells. In time - dependent killing experiments,
however, different antibiotics and different concentrations of the same antibiotics
resulted in different persister plateaus indicating heterogeneity even within the persister
population (unpublished data). Most importantly, ampicillin, which kills only growing
cells, consistently revealed a lower persister plateau than ciprofloxacin, which kills
growing and non-growing cells. Strictly viewing persisters in a monolithic way is
inconsistent with this phenomenon unless one would suggest the highly unlikely
existence of exponentially growing cells that are tolerant to ciprofloxacin.
A more likely explanation is that persistence can be induced by the
fluouroquinolone antibiotic. To test this hypothesis, we studied the genetic requirements
for persister formation in response to ciprofloxacin.
We found that persister formation can indeed be an adaptive process induced by
an antibiotic and that this process was dependent on the SOS response and was mainly
caused by the DNA- damage inducible TisB toxin.

14
References
Alix, E. & A. Blanc-Potard, (2009) Hydrophobic peptides: Novel regulators within
bacterial membranes. Mol Microbiol 72: 5-11.
Andersen, J. B., C. Sternberg, L. K. Poulsen, S. P. Bjorn, M. Givskov & S. Molin, (1998)
New unstable variants of green fluorescent protein for studies of transient gene
expression in bacteria. Appl Environ Microbiol 64: 2240-2246.
Balaban, N. Q., J. Merrin, R. Chait, L. Kowalik & S. Leibler, (2004) Bacterial persistence
as a phenotypic switch. Science 305: 1622-1625.
Bigger, J. W., (1944) Treatment of staphylococcal infections with penicillin. Lancet ii:
497-500.
Black, D. S., B. Irwin & H. S. Moyed, (1994) Autoregulation of hip, an operon that affects
lethality due to inhibition of peptidoglycan or DNA synthesis. J Bacteriol 176:
4081-4091.
Christensen-Dalsgaard, M., M. G. Jorgensen & K. Gerdes, (2010) Three new RelE-
homologous mRNA interferases of Escherichia coli differentially induced by
environmental stresses. Mol Microbiol 75: 333-348.
De Groote, V. N., N. Verstraeten, M. Fauvart, C. I. Kint, A. M. Verbeeck, S. Beullens, P.
Cornelis & J. Michiels, (2009) Novel persistence genes in Pseudomonas
aeruginosa identified by high-throughput screening. FEMS Microbiol Lett 297: 73-
79.
Fozo, E. M., M. R. Hemm & G. Storz, (2008) Small toxic proteins and the antisense
RNAs that repress them. Microbiol Mol Biol Rev 72: 579-589, Table of Contents.

15
Fozo, E. M., K. S. Makarova, S. A. Shabalina, N. Yutin, E. V. Koonin & G. Storz, 2010
Abundance of type I toxin-antitoxin systems in bacteria: searches for new
candidates and discovery of novel families. Nucleic Acids Res 38: 3743-3759.
Gerdes, K., S. K. Christensen & A. Lobner-Olesen, (2005) Prokaryotic toxin-antitoxin
stress response loci. Nature reviews 3: 371-382.
Gourse, R. L., T. Gaal, M. S. Bartlett, J. A. Appleman & W. Ross, (1996) rRNA
transcription and growth rate-dependent regulation of ribosome synthesis in
Escherichia coli. Annu Rev Microbiol 50: 645-677.
Hansen, S., K. Lewis & M. Vulić, (2008) The role of global regulators and nucleotide
metabolism in antibiotic tolerance in Escherichia coli. Antimicrob Agents
Chemother.
Harrison, J. J., W. D. Wade, S. Akierman, C. Vacchi-Suzzi, C. A. Stremick, R. J. Turner
& H. Ceri, (2009) The chromosomal toxin gene yafQ is a determinant of multidrug
tolerance for Escherichia coli growing in a biofilm. Antimicrob Agents Chemother
53: 2253-2258.
Keren, I., N. Kaldalu, A. Spoering, Y. Wang & K. Lewis, (2004a) Persister cells and
tolerance to antimicrobials. FEMS Microbiol Lett 230: 13-18.
Keren, I., D. Shah, A. Spoering, N. Kaldalu & K. Lewis, (2004b) Specialized persister
cells and the mechanism of multidrug tolerance in Escherichia coli. J Bacteriol
186: 8172-8180.
Kim, Y. & T. K. Wood (2010), Toxins Hha and CspD and small RNA regulator Hfq are
involved in persister cell formation through MqsR in Escherichia coli. Biochem
Biophys Res Commun 391: 209-213.

16
Korch, S. B., T. A. Henderson & T. M. Hill, (2003) Characterization of the hipA7 allele of
Escherichia coli and evidence that high persistence is governed by (p)ppGpp
synthesis. Mol Microbiol 50: 1199-1213.
Korch, S. B. & T. M. Hill, (2006) Ectopic overexpression of wild-type and mutant hipA
genes in Escherichia coli: effects on macromolecular synthesis and persister
formation. J Bacteriol 188: 3826-3836.
Kussell, E., R. Kishony, N. Q. Balaban & S. Leibler, (2005) Bacterial persistence: a
model of survival in changing environments. Genetics 169: 1807-1814.
LaFleur, M. D., C. A. Kumamoto & K. Lewis, (2006) Candida albicans biofilms produce
antifungal-tolerant persister cells. Antimicrob Agents Chemother 50: 3839-3846.
Lewis, K., (2007) Persister cells, dormancy and infectious disease. Nature reviews 5:
48-56.
Moyed, H. S. & K. P. Bertrand, (1983) hipA, a newly recognized gene of Escherichia coli
K-12 that affects frequency of persistence after inhibition of murein synthesis. J
Bacteriol 155: 768-775.
Moyed, H. S. & S. H. Broderick, (1986) Molecular cloning and expression of hipA, a
gene of Escherichia coli K-12 that affects frequency of persistence after inhibition
of murein synthesis. J Bacteriol 166: 399-403.
Pandey, D. P. & K. Gerdes, (2005) Toxin-antitoxin loci are highly abundant in free-living
but lost from host-associated prokaryotes. Nucleic Acids Res 33: 966-976.
Scherrer, R. & H. S. Moyed, (1988) Conditional impairment of cell division and altered
lethality in hipA mutants of Escherichia coli K-12. J Bacteriol 170: 3321-3326.

17
Shah, D., Z. Zhang, A. Khodursky, N. Kaldalu, K. Kurg & K. Lewis, (2006) Persisters: A
distinct physiological state of E. coli. BMC Microbiol 6: 53-61.
Sharma, S. V., D. Y. Lee, B. Li, M. P. Quinlan, F. Takahashi, S. Maheswaran, U.
McDermott, N. Azizian, L. Zou, M. A. Fischbach, K. K. Wong, K. Brandstetter, B.
Wittner, S. Ramaswamy, M. Classon & J. Settleman, A chromatin-mediated
reversible drug-tolerant state in cancer cell subpopulations. Cell 141: 69-80.
Spoering, A., Vulic, M. & K. Lewis (2006) GlpD and PlsB Participate in Persister Cell
Formation in Escherichia coli. Journal of Bacteriology 188: 5136-5144.
Spoering, A. L. & K. Lewis, (2001) Biofilms and planktonic cells of Pseudomonas
aeruginosa have similar resistance to killing by antimicrobials. J Bacteriol 183:
6746-6751.
Vuong, C., J. M. Voyich, E. R. Fischer, K. R. Braughton, A. R. Whitney, F. R. DeLeo &
M. Otto, (2004) Polysaccharide intercellular adhesin (PIA) protects
Staphylococcus epidermidis against major components of the human innate
immune system. Cell Microbiol 6: 269-275.
Wiuff, C., R. M. Zappala, R. R. Regoes, K. N. Garner, F. Baquero & B. R. Levin, (2005)
Phenotypic tolerance: antibiotic enrichment of noninherited resistance in bacterial
populations. Antimicrob Agents Chemother 49: 1483-1494.

18
CHAPTER 1 SOS response induces persistence to
fluoroquinolones in Escherichia coli
Published in the December 2009 issue of PloS Genetics
Tobias Dörr, Kim Lewis and Marin Vulić
Antimicrobial Discovery Center, Department of Biology, Northeastern University,
Boston, MA 02115
ABSTRACT
Bacteria can survive antibiotic treatment without acquiring heritable antibiotic resistance.
We investigated persistence to the fluoroquinolone, ciprofloxacin, in Escherichia coli.
Our data show that a majority of persisters to ciprofloxacin were formed upon exposure
to the antibiotic, in a manner dependent on the SOS gene network. These findings
reveal an active and inducible mechanism of persister formation mediated by the SOS
response, challenging the prevailing view that persisters are pre-existing and formed
purely by stochastic means. SOS induced persistence is a novel mechanism by which
cells can counteract DNA damage and promote survival to fluoroquinolones. This
unique survival mechanism may be an important factor influencing the outcome of
antibiotic therapy in vivo.

19
AUTHOR SUMMARY
The frequent failure of antibiotic treatments is an acute public health problem.
Bacteria can escape the lethal action of antibiotics by a mutation in the cell’s DNA which
leads to antibiotic resistance. Alternatively, they can enter a distinct, temporary
physiological state in which the antibiotics do not affect them. This phenomenon,
referred to as persistence, is different from resistance because there is no genetic
modification and because it is transient. Persisters are believed to form stochastically
prior to antibiotic treatment.
The presence of persister cells in bacterial biofilms contributes to the difficulty in
treating biofilm related infections. Biofilms are implicated in many chronic infections,
such as middle-ear infections, gingivitis, endocarditis, infections in cystic fibrosis etc.
Biofilms are also a major cause of infections in leg prostheses, heart valves, catheters
and contact lenses.
In this study we investigated the persistence of Escherichia coli to one of the most
widely used antibiotics, ciprofloxacin. We show that the majority of persister cells are
formed in response to this antibiotic, contrary to the prevailing view of persister
formation. Ciprofloxacin kills bacteria by damaging their DNA and DNA damage
activates a SOS gene network, the result of which is the production of various repair
proteins. We uncovered a novel part of this network that leads to the formation of
tolerant persister cells. The induced tolerance as a side effect of antibiotic treatment is
an effective bacterial survival strategy and is likely to contribute to recalcitrance of
infections.

20
INTRODUCTION
Persistence is the ability of a subpopulation of susceptible bacteria to survive
lethal doses of antibiotics. It is a transient and non-hereditary phenotype unlike
resistance, which is due to genetic modification. The transient nature of persistence
makes it inherently difficult to study therefore the underlying molecular mechanisms are
still poorly understood.
Persisters are thought to be slow growing, non-growing or dormant cells, which
escape the lethal action of antibiotics because their drug targets are inactivated due to
the physiological state. In an Escherichia coli high-persistence mutant, persisters to
ampicillin were shown to be non-growing prior to the addition of the antibiotic [1]. In
addition, a fraction of non-growing cells was isolated from untreated exponentially
growing E. coli and was shown to be enriched in persisters to ofloxacin [2]. These
studies demonstrated that persisters can form independently of antibiotics. The switch
from growing to non-growing state or dormancy, is thought to be a purely stochastic
process [1,3,4].
Both genetic and phenotypic variability can have important consequences on
bacterial survival of antibiotic treatment. One of the most prescribed broad spectrum
antibiotics today are the fluoroquinolones (FQ), which target gyrase and topoisomerase.
These essential enzymes regulate supercoiling of genomic DNA during replication and
transcription [5,6]. FQs prevent ligation reactions of gyrase and topoisomerase resulting
in double-strand breaks (DSB) [7]. DSBs are potentially lethal DNA lesions that occur
under physiological conditions through collapse of stalled replication forks, overlapping
repair tracts, spontaneous breakage of DNA, and other mechanisms. E. coli efficiently

21
repairs DSBs through a series of reactions carried out by enzymes participating in
homologous recombination and replication [8]. Processing of DSBs leads to the
induction of the SOS response. SOS is a complex network composed of more than 40
genes [9,10]. Many of these genes are essential for efficient repair of various DNA
lesions, including DSBs [11,12].
Even though fluoroquinolones are potent bactericidal antibiotics they cannot
sterilize a bacterial culture. The bulk of the population rapidly dies in response to
fluoroquinolones but a small fraction persists. According to one model, persisters might
survive if gyrase and topoisomerase are inactivated due to cellular dormancy [3].
Dormant cells might be expected to form stochastically during growth of a culture, prior
to the antibiotic exposure [2,13-15].
Alternatively, the persister state might be inducible in a cell subpopulation by
exposure to the antibiotic, not stochastic and pre-existing. This could be because either
dormancy is inducible, or persisters might be active and have more efficient drug efflux
or more efficient repair of DSBs due to the stochastic overexpression of the genes
involved in those pathways or due to the physiological events leading to the activation of
the same pathways.
In order to distinguish between these possibilities, we measured numbers of
persisters to the fluoroquinolone ciprofloxacin in various genetic backgrounds with
altered capacity for SOS induction and DSB repair.
The majority of persisters were found to be formed upon exposure to the
antibiotic and formation was dependent on the SOS DNA damage response. Contrary
to the current view, a majority of surviving persisters to ciprofloxacin are not pre-

22
existing, but induced by this antibiotic.
RESULTS
Fluoroquinolones (FQ) induce DSBs by interfering with the action of gyrase and
topoisomerase [16]. The cellular response to DSBs primarily consists of induction of the
SOS-regulon and ultimately in repair through recombination [17,18].
According to the prevailing model [3], persisters are dormant and are formed
stochastically prior to the addition of antibiotic. This suggests that persisters would not
experience DSBs, would not induce an adaptive response to that type of lesion, and
therefore would not need repair functions to survive. In order to test these predictions,
we wanted to determine whether persisters experience DSBs and induce SOS.
We measured the persister levels in different genetic backgrounds diagnostic of
specific molecular events linked with DSBs and SOS induction. The surviving fraction of
a wild-type culture treated with ciprofloxacin produces a typical biphasic pattern (Fig. 1
A). This reflects the rapid killing of the bulk of the cells, and a surviving persister
subpopulation.
We examined some of the well-known DNA-repair pathways in order to probe
their possible role in formation of persisters. RecA and RecBC are essential for repair of
DSBs in E. coli [19]. In strains lacking RecA and RecBC, DSBs are lethal. As expected,
the bulk of cells is more rapidly killed in both recA and recB backgrounds, compared to
the wild type, presumably because DSBs could not be repaired (Fig. 1 A). However, the
persister fraction was also greatly reduced (40-fold in recA, 35- to 103-fold in recB). In
recB, persisters were extremely rare or entirely absent after 6 hours of incubation. This

23
shows that the persisters experience DSBs and hence depend on the repair functions.
RecA and RecB functions are essential not only for DSB-repair but for SOS
induction following processing of DSBs as well [18,20-22], so in order to test whether
persisters induce SOS we constructed strains unable to induce SOS but proficient for
homologous recombination; one carrying a non-inducible SOS-repressor (lexA3) [23]
and the other a mutant RecA able to function as a recombinase but unable to induce
cleavage of LexA (recA430) [24]. In both backgrounds the bulk of cells dies more rapidly
than in the wild type, confirming that SOS is efficiently induced following exposure to
ciprofloxacin and contributes to the survival (Fig. 1 B). Interestingly, the persister level is
decreased 43-fold and by 6 hours it is as low as in recA background. This shows that
the persistence to ciprofloxacin is largely dependent on a functional SOS response.
XerCD site-specific recombinase resolves chromosome dimers at a dif site [25-
27]. Chromosome dimers are formed by an odd number of recombination events. The
absence of xerCD function does not affect the proficiency for SOS induction, but is
lethal in cells in which chromosme dimers have formed. In xerC and xerD mutants the
persister level is reduced (7- and 9-fold, respectively, taking into account the 3-fold
reduction in viability of xerC and xerD mutants compared to the wild-type), suggesting
that most persisters have undergone at least one successful recombination event, most
likely repairing a ciprofloxacin-induced DSB (Fig. 1 C).
Taken together these results show that the formation of the majority of persisters
in the presence of ciprofloxacin is dependent on the SOS-response. They also suggest
that this antibiotic-tolerant state is induced, rather than pre-existing. The formal
possibility that an SOS controlled function is essential for reaching or exiting a pre-

24
existing multidrug-tolerant state can be ruled out because tolerance to ampicillin and
streptomycin were not affected in recA, recB or lexA3 strains (Fig. 1 D). We cannot rule
out the possibility that spontaneous SOS induction was required for creating a pre-
existing ciprofloxacin-tolerant state.
A strain lacking the SOS-inducible RecN protein is also SOS proficient but
partially deficient in DSB repair. recN mutant exhibited increased sensitivity of the bulk
whereas persistence was largely unaffected (Fig. 1 F).
The entire population exposed to ciprofloxacin is expected to induce SOS yet
only a fraction survives. SOS is a gradual response where the strength of induction
reflects the extent and the persistence of the damage [28,29]. Upon addition of
ciprofloxacin the number and the chromosomal location of DSBs will vary across the
population depending on the activity and position of gyrase and topoisomerase
molecules in any given cell. The resulting distribution of DSBs is expected to translate
into a gradient of SOS induction. Therefore, it is possible that a specific level of SOS
induction is required for persistence. If this is the case, persister levels are expected
to change along with ciprofloxacin concentration and the overall level of SOS
induction.
We measured both the persister level and the induction of β-galactosidase under
the control of an SOS-inducible recA promoter [30] as a population average of SOS
induction for cultures exposed to increasing concentration of ciprofloxacin. Indeed, as
shown in Fig. 2, increased concentration of ciprofloxacin led to an increased average
SOS induction (Fig. 2 A) and decreased persister level (Fig. 2 B).
A strain constitutively expressing SOS functions (lexA300(Def)), also led to a 20-fold

25
increase in persister level compared to the wild type (Fig. 1 E).
In order to examine a difference in SOS induction between persisters and the
bulk at the single cell level, we followed a cI-cro gal reporter strain after addition of
ciprofloxacin [31]. In this strain the cleavage of λ repressor CI leads to a heritable
genetic switch rendering a cell gal+. gal
+ cells can be detected as red colonies on
MacConkey galactose plates. Unlike LexA, which undergoes auto-cleavage early in
SOS induction, CI cleavage occurs only if there is a high level of DNA damage and
activated RecA [32]. Therefore, the cI-cro system reports conditions of only strong SOS
induction. Following the addition of ciprofloxacin (> 0.5 µg/ml) the proportion of cells
giving rise to gal+ colonies increases, peaking at around 20 minutes and declines
thereafter (Fig. 3). This timing means that the massive amount of DNA damage occurs
readily leading to a strong SOS induction. Cells undergoing strong SOS induction are
able to withstand and repair the damage, if the ciprofloxacin is removed by plating.
However, upon extended exposure gal+ cells become fewer (Fig. 3), indicating that
additional damage occurs and eventually becomes lethal. The persister subpopulation
consisted almost entirely of gal-cells (Fig. 3) showing that persisters were not SOS-
induced prior to ciprofloxacin treatment (at least not highly induced) and also that they
did not experience high level of DNA damage nor strong SOS induction even in the
presence of the antibiotic. Because the persister level is greatly reduced in strains
unable to induce SOS (lexA3, recA430, Fig. 1 B) we conclude that persisters undergo
weak SOS induction. This is in contrast to the bulk of cells, probably because fewer
DSBs occur in eventual persisters. Increased sensitivity of the bulk and minimally
affected persistence in a recN strain also supports this conclusion (Fig. 1 F). SOS-

26
inducible RecN protein promotes efficient repair of DSBs. While it is dispensable for the
repair of a single break, it is essential for the repair of simultaneous multiple DSBs [33].
Next we exposed cells treated with a range of ciprofloxacin concentrations to a
higher dose (1 µg/ml) of the same antibiotic (Fig. 4). Control cultures were exposed to
1 µg/ml of ciprofloxacin for the duration of the experiment. The persister fraction
surviving exposure to 1 µg/ml was 10- to 40-fold higher in the cultures pretreated with
a low concentration of ciprofloxacin (0.05-0.2 µg/ml), compared to the control (Fig. 4
B). A dramatic, 1200-fold increase was found in cultures pretreated with sub-MIC
(minimal inhibitory concentration) concentration of ciprofloxacin (Fig. 4 B; compare full
bar at 0.03 µg/ml and the second dashed bar of the control). This shows that many of
the persisters are formed upon ciprofloxacin treatment rather than pre-existing. If they
were preexisting, the fraction surviving the exposure to the high concentration of
ciprofloxacin (1 µg/ml) would be the same regardless of the pretreatment.
It was important to learn whether SOS induction caused by treatments other than
FQ is able to induce persistence to ciprofloxacin. In order to test this we measured
persistence to ciprofloxacin in cells exposed to mitomycin C. Mitomycin C interacts with
DNA by intercalation and adduct formation, resulting in inter-strand crosslinks [34]. The
cellular response is a potent SOS-induction dependent on RecFOR pathway [35]. We
exposed exponentially growing cells to a sub-MIC concentration of mitomycin C and
compared the persister levels at two different time points during the treatment. The
results in Fig. 5 show a 180-fold increase in persistence to ciprofloxacin in the culture
treated with mitomycin C for 4 versus 2 hours, confirming the link between SOS
induction and persistence to FQs, regardless of the nature of the SOS inducing

27
treatment.
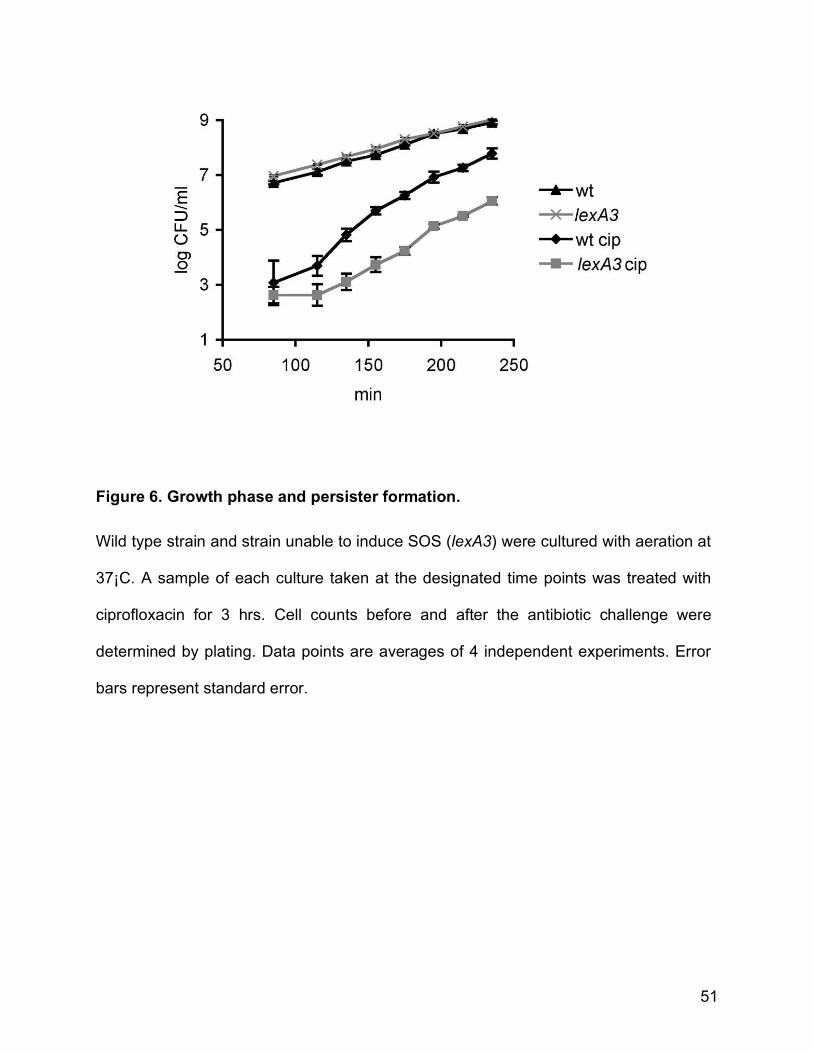
Persister levels are very low in early exponential phase and are maximal in
stationary phase [14]. We treated aliquots of growing cultures with ciprofloxacin at
different time points in order to determine the persister levels between these two
extremes and establish the role of growth phase in SOS-induced persistence. Fig. 6
shows an exponential increase in persister levels when cell densities reach around
5 x107 CFU/ml in both the wild-type and the strain unable to induce SOS (lexA3).
We conclude that the SOS-induced persisters make up the majority of persisters to
ciprofloxacin regardless of the growth phase.
DISCUSSION
The processes leading to genetic variability in bacteria, mutagenesis and
recombination, have been studied extensively [36-38] and their role in evolution of
bacterial antibiotic resistance by generating and disseminating mutations is well
established [39-46]. On the other hand, processes leading to phenotypic variability,
which is also an important factor influencing bacterial ability to survive antibiotic
treatments [47,48] have only recently become a subject of systematic investigation.
In contrast to the well-understood mechanisms of bacterial resistance to antibiotics,
molecular mechanism(s) of persistence have so far remained elusive. The current
model of persistence assumes that persisters are non-growing or dormant cells,
formed by stochastic process(es) independently of any physiological responses
normally elicited by antibiotics [1,3,4]. Studies involving persistence to two different

28
classes of antibiotics, a β-lactam ampicillin [1] and a fluoroquinolone ofloxacin [2] are
consistent with this model, which was therefore presumed to hold universally.
Here we show a mechanism of persister formation triggered by DNA damage
inflicted by the fluoroquinolone ciprofloxacin. Formation of persisters in response to
DNA damage reveals a deterministic component in this bistability phenomenon.
Bistability is the stochastic production of two phenotypically distinct cell types within a
clonal population of genetically identical kin cells. Bistability is observed in sporulation,
competence, and motility [49-51]. In all cases studied, there is both a stochastic and
deterministic component of bistability. Previous studies have shown that persisters can
form stochastically, prior to the addition of antibiotics [1,14]. The present findings show
that persister formation can also be induced by an antibiotic, through an active process.
This sheds an entirely new light on the problem of antibiotic tolerance and its role in
infectious processes.
We also show that mutants defective in persistence to ciprofloxacin have normal
persister levels to amipicillin and streptomycin (Fig. 1 D), therefore it is still possible
that persistence to β-lactams is purely stochastic and not inducible. These results
suggest that there are different mechanisms of persistence to different antibiotics.
Ciprofloxacin induces DSBs in cells with active gyrase and/or topoisomerase,
which in turn leads to the activation of the general DNA damage stress response, the
SOS gene network. Our results show that the majority of persisters to ciprofloxacin
are dependent on a functional SOS response.
DSBs and other SOS-inducing lesions occur under physiological conditions so at
any given time there is a fraction of a bacterial population undergoing a certain degree

29
of SOS induction [52,53]. However, we demonstrate that the SOS-dependent persister
state is induced upon exposure to ciprofloxacin. Manipulating the extent of SOS
induction by different antibiotic concentrations or by sequential exposure to a higher
dose dramatically affects persister levels (Fig. 2, Fig. 4). This would not be the case if
the persisters were pre-existing in the population. If they were, the bulk would be killed
by any bactericidal concentration of antibiotic, revealing the same pre-existing persister
population. In addition, increasing the basal level of expression of the SOS regulon by
genetic manipulation (Fig. 1 E) or by induction with different treatment (Fig. 5) also
leads to an increase in persister level.
Essentially all actively growing cells exposed to ciprofloxacin induce SOS, but not
all become persisters, suggesting that a specific level of SOS induction is required for
persister formation. SOS is a gradual response and depending on the nature of the
inducer, its concentration and the time of the exposure, different sets of genes are
induced [10,54,55]. Our data indicate that persister formation requires a functional SOS
response but a high level of induction is not required (Fig. 1 B, Fig. 3). Persister
formation also depends on functional DSB repair (Fig. 1 A) but does not need RecN
(Fig. 1 F), a function important for the repair of multiple DSBs [33]. This implies that
persisters are cells that experienced few DSBs upon ciprofloxacin addition and
underwent weak SOS induction.
Consistent with this, constitutive, full expression of the SOS regulon (equivalent
to high induction) does not lead to the tolerance of the entire population, but to an
increased level of surviving persisters (Fig. 1 E). Even in a lexA(Def) mutant, expression
levels of SOS genes appear to fall short of being truly uniform throughout the population

30
[52,53]. Persisters could be the cells that express a certain SOS function at a specific
high or low level. Additionally, other regulatory pathways could allow a persister
formation function to be expressed only in certain cells after induction. Turning on the
SOS response constitutively would increase the number of cells being able to express
this function.
Persister levels are known to change with the growth phase [14]. It is low in early
exponential phase and attains its highest level in stationary phase. An exponential
increase in persister levels begins when the cell density reaches around 5 x107 CFU/ml
(Fig. 6). The persister shoot up at similar cell density has been observed in other
studies under different antibiotic and growth conditions [1,14]. A cell density of 5 x107
CFU/ml coincides with the point at which the balanced growth of the culture ceases and
a slowdown of growth rate is observed, even though the population as a whole still
increases exponentially [56].
The extent of the DNA damage caused by ciprofloxacin would be expected to
reflect the activity levels of gyrase and topoisomerase. These enzymes are active
during replication and transcription [6,57]; therefore their maximal activity would occur in
rapidly growing and replicating cells and would be lowest in the non-growing state of
stationary phase. Lending support to this, transcription of gyrA and gyrB coding for
gyrase subunits is at the peak in the early exponential phase and the lowest in the
stationary growth phase [58,59]. It follows that ciprofloxacin would inflict maximal
damage, the irreparable chromosome fragmentation, in the exponentially growing cells
and fewer DSBs in the cells that slow down when the medium cannot support steady-
state growth [60]. Indeed, no cells survive treatment to ofloxacin, another FQ, when the

31
culture is kept at low density in constant exponential growth by repeated subculturing
[14], in other words no persisters are formed in that growth phase. On the other hand,
the surviving fraction increases dramatically between the end of true exponential growth
and stationary phase (Fig. 6, [14]). During that time the growth rate of the population
decreases from its maximum to zero, but because not all cells stop growing at the same
time the heterogeneity of growth rates across the population is expected within that time
frame. Those cells lacking steady state equilibrium might be the ones which experience
few DSBs, weak SOS-induction and enter the tolerant state. Consistent with this, the
difference in persister level in SOS proficient and deficient strains is minimal in early
exponential phase, whereas it increases after the cessation of steady state growth (Fig.
6).
Conditions for unrestricted growth are rarely met in natural environments, and
most bacteria are in a state of slow or no-growth [61-63]. However, physical and
chemical agents capable of causing DNA damage are ubiquitous, therefore the SOS-
induced persister state is probably quite common. Furthermore, in conditions of slow
growth and frequent or lasting presence of DNA damaging agents, damage
prevention would likely be advantageous over continuous active repair. The induction
of the persister state in response to DNA damage seems like such a strategy - the
avoidance of the damage build up as opposed to the costly repair.
SOS is induced in aging colony biofilms of E. coli [64] and in intracellular biofilms
formed by uropathogenic E. coli during cystitis [65]. Biofilms are notoriously hard to
eradicate even with bactericidal fluoroquinolones, and this enhanced ‘resistance’ could
in fact reflect the SOS-induced tolerance.

32
Virtually all natural isolates of E. coli and many other bacteria are lysogens and
many prophages are DNA-damage inducible [66-69]. Induction of λ prophage in E. coli
is a late SOS function. In that light, SOS-induced tolerance could have evolved as a life-
saving strategy preventing prophage induction upon DNA damage frequently
encountered.
There are at least 43 genes in the E. coli genome negatively regulated by LexA
[9,10]. Many encode proteins participating in repair by homologous recombination
and/or translesion synthesis and about one third are of unknown function. Among those
are several genes encoding toxin-antitoxin modules that are attractive candidates for
persistence genes, as the overexpression of some toxins has been shown to induce a
dormant-like state [13,70]. Indeed, in a parallel study we identified an SOS inducible
toxin/antitoxin module, tisAB, as a function needed for persister formation (Dšrr T., Vulić
M., Lewis K., submitted). However we cannot exclude that other LexA-regulated genes
also contribute to SOS-induced tolerance.
SOS has been shown to induce formation of a senescence-like state in which
cells are viable but unable to form colonies [53]. Here we show SOS-dependant
formation of persister cells. Both states could be formed through the common
mechanism, such as expression of SOS-regulated toxins. In that case the strength of
SOS induction and hence the toxin expression levels would determine which of
these two states a cell reaches.
In conclusion, we have discovered an active, regulated mechanism of persister
formation, which is part of the SOS response. SOS has been known to contribute to the
survival of antibiotic treatments by increasing the frequency of resistant mutants through

33
its mutagenic activities [44,71]. Here we show a novel function of this response, the
induction of a tolerant state. SOS-induced persistence having an immediate impact on
bacterial survival is likely an important factor influencing the outcome of antibiotic
treatment.
MATERIALS AND METHODS
Bacterial strains
Bacterial strains are listed in Table 1. Wild-type E. coli K-12 MG1655 was used as the
parental strain. Different alleles were moved into the parental background by P1
transduction [72]. The kanamycin resistance cassette from the alleles originated from
KEIO collection [73] was cured when needed by expressing the FLP recombinase
from the helper plasmid pCP20 according to the protocol in [74].
Persistence assay
Experiments were conducted at 37¡C in Mueller Hinton Broth (MHB) supplemented with
10 mg/L MgSO4 and 20 mg/L CaCl2 according to NCCLS (National Committee for
Clinical Laboratory Standards) guidelines for susceptibility testing and 0.1 M
HEPES/KOH pH 7.2. Persistence was measured by determining survival upon
exposure to 0.1 µg/ml ciprofloxacin (unless indicated otherwise), 100 µg/ml ampicillin
and 25 µg/ml streptomycin during time indicated on corresponding graph axes. All
antibiotics were purchased from Sigma. Prior to the addition of antibiotic overnight

34
cultures were diluted 100-fold in 3 ml of fresh medium in 17- by 100-mm polypropylene
tubes and incubated for 1.5 hrs with shaking, typically reaching ~ 2 x 108 CFU/ml. For
determination of CFU counts, cells were washed in 1% NaCl solution, serially diluted
and plated on LB (Luria-Bertani medium) agar plates supplemented with 20 mM
MgSO4. Persister fraction, reflected as a plateau in CFU counts, was calculated as an
average of CFU counts at 3- and 6-hour time points. In lexA3 and recA430 strains the
CFU counts stabilize later than in the wild-type and in that case the CFU counts at 6-
hour time point were used as a representative of the persister fraction.
Measurements of SOS induction
For plate assays using the CI-cro-gal construct, overnight cultures grown in LB medium
at 37¡C were diluted 1:200 in 15 ml of fresh medium and incubated in 125 ml flasks for
1.75 hrs at 37¡C with shaking. Ciprofloxacin was added and aliquots of the culture were
taken at different time points, washed in 1% NaCl solution, serially diluted and plated on
LB agar plates supplemented with 20 mM MgSO4 for total CFU counts and on
MacConkey agar plates supplemented with 1% galactose in order to determine the
fraction of gal+ cells. For β-galactosidase activity measurement, overnight cultures
grown in supplemented MHB medium (see above) at 37¡C were diluted 1:100 in 3 ml of
fresh medium in 17- by 100-mm polypropylene tubes and incubated for 1.75 hrs at 37¡C
with shaking.
Ciprofloxacin was added and after 15 minutes an aliquot of culture was taken and
recA::lacZ expression was measured as described in [72].

35
Mitomycin C treatment and persistence to ciprofloxacin
Overnight cultures in supplemented MHB medium were diluted 1:1000 in 15 ml of fresh
medium and incubated in 125 ml flasks for 1 hr at 37¡C with shaking after which 0.25
µg/ml of mitomycin C (Sigma) was added to the cultures. This concentration did not
inhibit the growth of the culture. After 2 hrs the total CFU counts were determined by
dilution and plating and an aliquot of the culture was taken out and exposed to 0.3 µg/ml
of ciprofloxacin for 3 hrs. The number of survivors was determined by plating on LB
agar plates supplemented with 20 mM MgSO4 after washing in 1% NaCl solution. The
same procedure was repeated after 4 hours of exposure to mitomycin C.
Kinetics of persister formation
Overnight cultures in MHB medium were diluted 1000-fold in 100 ml of fresh medium in
500 ml flasks and incubated at 37¡C with shaking. At defined time intervals the cultures
were serially diluted and plated on LB agar for determination of total CFU counts. In
the same time 1 ml aliquots were transferred into 2 ml eppendorf tubes and 0.1 µg/ml
ciprofloxacin was added. After 3 hrs at 37¡C cells were washed with 1% NaCl solution,
serially diluted and plated on LB agar plates supplemented with 20 mM MgSO4. The
colonies were counted after 40 hours incubation at 37¡C.
ACKNOWLEDGMENTS
We would like to thank the editor for detailed and helpful comments on the previous

36
version of the manuscript. We would also like to thank Alyssa Theodore for excellent
technical assistance, E. coli Genetic Stock Center, Radman and Walker Lab for strains,
Eric Stewart, Michael LaFleur and Veronica Godoy for critical reading of the manuscript.
TABLE 1. Bacterial strains.
Strain Relevant genotype Parent strain Reference/Source
MG1655 K-12 Fλ
TD172 ΔrecA::kan JW2669 [73]
TD230 ΔrecB::kan JW2788 [73]
TD160 ΔrecN::kan JW2597 [73]
TD222 recA430 GY3448 [24,75]
E. coli Genetic
Stock Center,
Yale
TD221 lexA3 malE300::Tn10 K996 [23] E. coli
Genetic
Stock Center,
Yale
TD127 lexA300(Def) GW8018 [76] Walker Lab,
MIT, Cambridge
ΔsulA::FRT JW0941 [73]
MV2033 ΔxerD::kan JW2862 [73]

37
MV2037 ΔxerC::kan JW3784 [73]
LLC3 (λ cI+ cro+-gal-) MT1 [31]
Radman Lab,
Necker, Paris
MV1603 λ d(recA::lacZ) cI(Ind-)
AmpR AB1157 [30]
λ d(recA::lacZ) Radman Lab,
cI(Ind-) AmpR Necker, Paris
REFERENCES
1 Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S (2004) Bacterial persistence
as a phenotypic switch. Science 305: 1622-1625.
2 Shah D, Zhang Z, Khodursky A, Kaldalu N, Kurg K, et al. (2006) Persisters: A
distinct physiological state of E. coli. BMC Microbiol 6: 53-61.
3 Lewis K (2007) Persister cells, dormancy and infectious disease. Nat Rev
Microbiol 5: 48-56.
4 Kussell E, Kishony R, Balaban NQ, Leibler S (2005) Bacterial persistence: a
model of survival in changing environments. Genetics 169: 1807-1814.
5 Drlica K, Zhao X (1997) DNA gyrase, topoisomerase IV, and the 4-quinolones.
Microbiol Mol Biol Rev 61: 377-392.
6 Wang JC (2002) Cellular roles of DNA topoisomerases: a molecular perspective.
Nat Rev Mol Cell Biol 3: 430-440.
7 Malik M, Zhao X, Drlica K (2006) Lethal fragmentation of bacterial chromosomes

38
mediated by DNA gyrase and quinolones. Mol Microbiol 61: 810-825.
8 Kowalczykowski SC, Dixon DA, Eggleston AK, Lauder SD, Rehrauer WM (1994)
Biochemistry of homologous recombination in Escherichia coli. Microbiol Rev 58: 401-
465.
9 Fernandez De Henestrosa AR, Ogi T, Aoyagi S, Chafin D, Hayes JJ, et al.
(2000) Identification of additional genes belonging to the LexA regulon in Escherichia
coli. Mol Microbiol 35: 1560-1572.
10 Courcelle J, Khodursky A, Peter B, Brown P, Hanawalt P (2001) Comparative
gene expression profiles following UV exposure in wild type and SOS-deficient
Escherichia coli. Genetics 158: 41-64.
11 Radman M (1975) SOS repair hypothesis: Phenomenology of an inducible DNA
repair which is accompanied by mutagenesis. Basic Life Sci 5A: 355-367.
� 12. Friedberg EC, Walker, G.C., Siede, W., Wood, R.D., Schultz, R.A., and
Ellenberger, T. (2006) DNA Repair and Mutagenesis. Washinghton, D.C.: American
Society of Microbiology Press.
12 Keren I, Shah D, Spoering A, Kaldalu N, Lewis K (2004) Specialized persister
cells and the mechanism of multidrug tolerance in Escherichia coli. J Bacteriol 186:
8172-8180.
13 Keren I, Kaldalu N, Spoering A, Wang Y, Lewis K (2004) Persister cells and
tolerance to antimicrobials. FEMS Microbiol Lett 230: 13-18.
14 Lewis K, Spoering A, Kaldalu N, Keren I, Shah D (2005) Persisters: specialized
cells responsible for biofilm tolerance to antimicrobial agents. In: Pace J, Rupp ME,
Finch RG, editors. Biofilms, Infection, and Antimicrobial Therapy. Boca Raton, London,

39
New York, Singapore: Taylor & Francis. pp. 241-256.
15 Drlica K, Malik M, Kerns RJ, Zhao X (2008) Quinolone-mediated bacterial death.
Antimicrob Agents Chemother 52: 385-392.
� 17. Howard BM, Pinney RJ, Smith JT (1993) Function of the SOS process in
repair of DNA damage induced by modern 4-quinolones. J Pharm Pharmacol 45: 658
� 662.
16 Newmark KG, O'Reilly EK, Pohlhaus JR, Kreuzer KN (2005) Genetic analysis of
the requirements for SOS induction by nalidixic acid in Escherichia coli. Gene 356: 69-
76.
17 Dillingham MS, Kowalczykowski SC (2008) RecBCD enzyme and the repair of
double-stranded DNA breaks. Microbiol Mol Biol Rev 72: 642-671, Table of Contents.
18 McPartland A, Green L, Echols H (1980) Control of recA gene RNA in E. coli:
regulatory and signal genes. Cell 20: 731-737.
19 Chaudhury AM, Smith GR (1985) Role of Escherichia coli RecBC enzyme in
SOS induction. Mol Gen Genet 201: 525-528.
� 22. Anderson DG, Kowalczykowski SC (1998) Reconstitution of an SOS
response pathway: derepression of transcription in response to DNA breaks. Cell 95:
975
� 979.
20 Mount DW, Low KB, Edmiston SJ (1972) Dominant mutations (lex) in
Escherichia coli K-12 which affect radiation sensitivity and frequency of ultraviolet lght-
induced mutations. J Bacteriol 112: 886-893.
21 Devoret R, Blanco M (1970) Mutants of Escherichia coli K12 (λ)+ non-inducible

40
by thymine starvation. Mol Gen Genet 107: 272-280.
� 25. Blakely G, Colloms S, May G, Burke M, Sherratt D (1991) Escherichia coli
XerC recombinase is required for chromosomal segregation at cell division. New Biol
� 3: 789-798.
22 Blakely G, May G, McCulloch R, Arciszewska LK, Burke M, et al. (1993) Two
related recombinases are required for site-specific recombination at dif and cer in E. coli
K12. Cell 75: 351-361.
23 Kuempel PL, Henson JM, Dircks L, Tecklenburg M, Lim DF (1991) dif, a recA-
independent recombination site in the terminus region of the chromosome of
Escherichia coli. New Biol 3: 799-811.
24 Peterson KR, Mount DW (1987) Differential repression of SOS genes by
unstable lexA41 (tsl-1) protein causes a "split-phenotype" in Escherichia coli K-12. J Mol
Biol 193: 27-40.
25 Camas FM, Blazquez J, Poyatos JF (2006) Autogenous and nonautogenous
control of response in a genetic network. Proc Natl Acad Sci U S A 103: 12718-12723.
26 Casaregola S, D'Ari R, Huisman O (1982) Quantitative evaluation of recA gene
expression in Escherichia coli. Mol Gen Genet 185: 430-439.
27 Toman Z, Dambly-Chaudiere C, Tenenbaum L, Radman M (1985) A system for
detection of genetic and epigenetic alterations in Escherichia coli induced by DNA-
damaging agents. J Mol Biol 186: 97-105.
28 Little JW (1984) Autodigestion of lexA and phage lambda repressors. Proc Natl
Acad Sci U S A 81: 1375-1379.
29 Meddows TR, Savory AP, Grove JI, Moore T, Lloyd RG (2005) RecN protein

41
and transcription factor DksA combine to promote faithful recombinational repair of DNA
double-strand breaks. Mol Microbiol 57: 97-110.
30 Iyer VN, Szybalski W (1964) Mitomycins and Porfiromycin: Chemical Mechanism
of Activation and Cross-Linking of DNA. Science 145: 55-58.
31 Keller KL, Overbeck-Carrick TL, Beck DJ (2001) Survival and induction of SOS
in Escherichia coli treated with cisplatin, UV-irradiation, or mitomycin C are dependent
on the function of the RecBC and RecFOR pathways of homologous recombination.
Mutat Res 486: 21-29.
32 Guttman DS, Dykhuizen DE (1994) Clonal divergence in Escherichia coli as a
result of recombination, not mutation. Science 266: 1380-1383.
33 Taddei F, Radman M, Maynard-Smith J, Toupance B, Gouyon PH, et al. (1997)
Role of mutator alleles in adaptive evolution. Nature 387: 700-702.
34 Rosenberg SM (2001) Evolving responsively: adaptive mutation. Nat Rev Genet
2: 504-515.
35 Martinez JL, Baquero F (2000) Mutation frequencies and antibiotic resistance.
Antimicrob Agents Chemother 44: 1771-1777.
36 Blazquez J (2003) Hypermutation as a factor contributing to the acquisition of
antimicrobial resistance. Clin Infect Dis 37: 1201-1209.
37 Beaber JW, Hochhut B, Waldor MK (2004) SOS response promotes horizontal
dissemination of antibiotic resistance genes. Nature 427: 72-74.
38 Denamur E, Tenaillon O, Deschamps C, Skurnik D, Ronco E, et al. (2005)
Intermediate mutation frequencies favor evolution of multidrug resistance in Escherichia
coli. Genetics 171: 825-827.

42
39 Hastings PJ, Rosenberg SM, Slack A (2004) Antibiotic-induced lateral transfer of
antibiotic resistance. Trends Microbiol 12: 401-404.
40 Cirz RT, Chin JK, Andes DR, de Crecy-Lagard V, Craig WA, et al. (2005)
Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol 3:
e176.
41 Orlen H, Hughes D (2006) Weak mutators can drive the evolution of
fluoroquinolone resistance in Escherichia coli. Antimicrob Agents Chemother 50: 3454-
3456.
42 Couce A, Blazquez J (2009) Side effects of antibiotics on genetic variability.
FEMS Microbiol Rev 33: 531-538.
43 Bigger JW (1944) Treatment of staphylococcal infections with penicillin. Lancet
ii: 497-500.
44 Moyed HS, Bertrand KP (1983) hipA, a newly recognized gene of Escherichia
coli K12 that affects frequency of persistence after inhibition of murein synthesis. J
Bacteriol 155: 768-775.
45 Veening J, Hamoen L, Kuipers O (2005) Phosphatases modulate the bistable
sporulation gene expression pattern in Bacillus subtilis. Mol Microbiol 56: 14811494.
46 Maamar H, Dubnau D (2005) Bistability in the Bacillus subtilis K-state
(competence) system requires a positive feedback loop. Mol Microbiol 56: 615-624.
47 Kearns DB, Chu F, Rudner R, Losick R (2004) Genes governing swarming in
Bacillus subtilis and evidence for a phase variation mechanism controlling surface
motility. Mol Microbiol 52: 357-369.
48 McCool JD, Long E, Petrosino JF, Sandler HA, Rosenberg SM, et al. (2004)

43
Measurement of SOS expression in individual Escherichia coli K-12 cells using
fluorescence microscopy. Mol Microbiol 53: 1343-1357.
49 Pennington JM, Rosenberg SM (2007) Spontaneous DNA breakage in single
living Escherichia coli cells. Nat Genet 39: 797-802.
50 Shaw KJ, Miller N, Liu X, Lerner D, Wan J, et al. (2003) Comparison of the
changes in global gene expression of Escherichia coli induced by four bactericidal
agents. J Mol Microbiol Biotechnol 5: 105-122.
51 Friedman N, Vardi S, Ronen M, Alon U, Stavans J (2005) Precise temporal
modulation in the response of the SOS DNA repair network in individual bacteria. PLoS
Biol 3: e238.
52 Sezonov G, Joseleau-Petit D, D'Ari R (2007) Escherichia coli physiology in
Luria-Bertani broth. J Bacteriol 189: 8746-8749.
53 De Wyngaert M, Hinkle DC (1979) Involvement of DNA gyrase in replication and
transcription of bacteriophage T7 DNA. J Virol 29: 529-535.
54 Gomez-Gomez JM, Baquero F, Blazquez J (1996) Cyclic AMP receptor protein
positively controls gyrA transcription and alters DNA topology after nutritional upshift in
Escherichia coli. J Bacteriol 178: 3331-3334.
55 Schneider R, Travers A, Kutateladze T, Muskhelishvili G (1999) A DNA
architectural protein couples cellular physiology and DNA topology in Escherichia coli.
Mol Microbiol 34: 953-964.
56 Tamayo M, Santiso R, Gosalvez J, Bou G, Fernandez JL (2009) Rapid
assessment of the effect of ciprofloxacin on chromosomal DNA from Escherichia coli
using an in situ DNA fragmentation assay. BMC Microbiol 9: 69.

44
57 Gibbons RJ, Kapsimalis B (1967) Estimates of the overall rate of growth of the
intestinal microflora of hamsters, guinea pigs, and mice. J Bacteriol 93: 510-512.
58 Poulsen LK, Licht TR, Rang C, Krogfelt KA, Molin S (1995) Physiological state of
Escherichia coli BJ4 growing in the large intestines of streptomycin-treated mice. J
Bacteriol 177: 5840-5845.
59 Baath E (1998) Growth Rates of Bacterial Communities in Soils at Varying pH: A
Comparison of the Thymidine and Leucine Incorporation Techniques. Microb Ecol 36:
316-327.
60 Taddei F, Matic I, Radman M (1995) cAMP-dependent SOS induction and
mutagenesis in resting bacterial populations. Proc Natl Acad Sci U S A 92: 11736-
11740.
61 Justice SS, Hunstad DA, Seed PC, Hultgren SJ (2006) Filamentation by
Escherichia coli subverts innate defenses during urinary tract infection. Proc Natl Acad
Sci U S A 103: 19884-19889.
62 Riley MA, Gordon DM (1992) A survey of Col plasmids in natural isolates of
Escherichia coli and an investigation into the stability of Col-plasmid lineages. J Gen
Microbiol 138: 1345-1352.
63 Schicklmaier P, Moser E, Wieland T, Rabsch W, Schmieger H (1998) A
comparative study on the frequency of prophages among natural isolates of Salmonella
and Escherichia coli with emphasis on generalized transducers. Antonie Van
Leeuwenhoek 73: 49-54.
64 Lamont I, Brumby AM, Egan JB (1989) UV induction of coliphage 186: prophage
induction as an SOS function. Proc Natl Acad Sci U S A 86: 5492-5496.

45
65 Casjens S (2003) Prophages and bacterial genomics: what have we learned so
far? Mol Microbiol 49: 277-300.
66 Correia FF, D'Onofrio A, Rejtar T, Li L, Karger BL, et al. (2006) Kinase activity of
overexpressed HipA is required for growth arrest and multidrug tolerance in Escherichia
coli. J Bacteriol 188: 8360-8367.
67 Petrosino JF, Galhardo RS, Morales LD, Rosenberg SM (2009) Stress-induced
betalactam antibiotic resistance mutation and sequences of stationary-phase mutations
in the Escherichia coli chromosome. J Bacteriol 191: 5881-5889.
68 Miller JH (1992) A Short Course in Bacterial Genetics: A Laboratory Manual and
Handbook for Escherichia coli and Related Bacteria. Cold Spring Harbor: Cold Spring
Harbor Laboratory Press.
69 Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, et al. (2006) Construction of
Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol
Syst Biol 2: 2006.0008.
70 Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in
Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97: 66406645.
71 Morand P, Blanco M, Devoret R (1977) Characterization of lexB mutations in
Escherichia coli K-12. J Bacteriol 131: 572-582.
72 Opperman T, Murli S, Walker GC (1996) The genetic requirements for UmuDC-
mediated cold sensitivity are distinct from those for SOS mutagenesis. J Bacteriol
178: 4400-4411.

46
Figure 1. Survival of the wild type and the mutants deficient in recombination
and/or SOS induction after ciprofloxacin challenge.
Strains were exposed to ciprofloxacin in exponential growth phase. Viable counts
were determined by plating. Graphs are representatives of at least 5 independent
experiments. Error bars represent standard error.

47
Figure 2. SOS induction and persister level during ciprofloxacin challenge in
exponential growth phase.
Graphs are averages of at least 3 independent experiments and error bars represent
standard error. (A) Induction of the SOS-inducible recA gene expression measured
by assaying the β-galactosidase activity after 15 min of ciprofloxacin challenge. (B)
Persister levels after 6 hours of ciprofloxacin challenge.

48
Figure 3. Fraction of cells undergoing strong SOS induction during ciprofloxacin
challenge.
Cells in exponential growth phase were exposed to ciprofloxacin. A heritable epigenetic
switch based on the reciprocal repression of the phage λ cI and cro genes fused with
the promotorless galactose operon allows detection of clones derived from cells that
have undergone SOS induction as red gal+ colonies on MacConkey galactose plates.
Data points are average of at least 3 independent experiments. Error bars represent
standard error.

49
Figure 4. Ciprofloxacin-induced persistence.
(A) Survival of wild type cells in exponential phase under different ciprofloxacin regimes.
Two cultures were treated with 0.1 µg/ml and 1 µg/ml, respectively, for 6 hours. Third
culture was treated with 0.1 µg/ml for 3 hours after which 1 µg/ml was added (indicated
by an arrow). The data are averages of 3 independent experiments and error bars
indicate standard error.
(B) Wild type cells in exponential phase were treated for 3 hours with increasing
concentrations of ciprofloxacin indicated on x-axis. After the initial treatment, an
additional 1 µg/ml of ciprofloxacin was added to the cultures and incubated for another 3
hours as in (A) (ciprofloxacin MIC is 0.05 µg/ml). As a control, a parallel culture was
exposed to 1 µg/ml for the duration of the experiment. Bars represent the viability at 0, 3
and 6 hr of time course equivalents shown in (A). Open bars; the initial viability count,

50
grey bars; the viability after 3-hour incubation with ciprofloxacin concentration indicated
on the x-axis. Full bars; the final viability count after additional 3-hour incubation with 1
µg/ml ciprofloxacin. Dashed bars; viability of the control culture at 3 and 6 hours. The
data are averages of 3 independent experiments and error bars indicate standard error.
Figure 5. Mitomycin C-induced persistence.
Wild type cells in exponential phase were treated with mitomycin C for either 2 or 4
hours before being exposed to 0.3 µg/ml ciprofloxacin. Open bars; total viable counts,
gray bars; persister fraction. The data are averages of 3 independent experiments and
error bars indicate standard error.
51
Figure 6. Growth phase and persister formation.
Wild type strain and strain unable to induce SOS (lexA3) were cultured with aeration at
37¡C. A sample of each culture taken at the designated time points was treated with
ciprofloxacin for 3 hrs. Cell counts before and after the antibiotic challenge were
determined by plating. Data points are averages of 4 independent experiments. Error
bars represent standard error.

52
CHAPTER 2 Ciprofloxacin Causes Persister Formation by
Inducing the TisB toxin in Escherichia coli
Published in the Feburary 2010 issue of PloS biology
Tobias Dörr1, Marin Vulic
1 and Kim Lewis*
1
1Antimicrobial Discovery Center, Department of Biology, Northeastern University,
Boston, MA 02115
* E-mail: [email protected]
Abstract
Bacteria induce stress responses that protect the cell from lethal factors such as
DNA-damaging agents. Bacterial populations also form persisters, dormant cells that
are highly tolerant to antibiotics and play an important role in recalcitrance of biofilm
infections. Stress response and dormancy appear to represent alternative strategies of
cell survival. The mechanism of persister formation is unknown, but isolated persisters
show increased levels of toxin/antitoxin (TA) transcripts. We have found previously that
one or more components of the SOS response induce persister formation after
exposure to a DNA damaging antibiotic. The SOS response induces several

53
toxin/antitoxin genes in E. coli. Here we show that a knockout of a particular SOS-TA
locus, tisAB/IstR, had a sharply decreased level of persisters tolerant to ciprofloxacin,
an antibiotic that causes DNA damage. Step-wise administration of ciprofloxacin
induced persister formation in a tisAB-dependent manner, and cells producing TisB
toxin were tolerant to multiple antibiotics. TisB is a membrane peptide that was shown
to decrease proton motive force (pmf) and ATP levels, consistent with its role in forming
dormant cells. These results suggest that a DNA damage-induced toxin controls
production of multidrug tolerant cells and thus provide a model of persister formation.
Abbreviations: CFU, colony forming units; IPTG, Isopropyl β-D-1-
thiogalactopyranoside; MHB, Mueller-Hinton-Broth; MIC, minimum inhibitory
concentration; pmf, proton motive force
Running title: TisB-induced persister formation in E. coli
Key words: Persister, biofilm, antibiotic tolerance, toxin antitoxin, ciprofloxacin, SOS
response

54
Introduction
Bacterial populations form persisters, dormant cells that are highly tolerant to
antibiotics and play an important role in recalcitrance of biofilm infections [1,2]. Time-
dependent or dose-dependent killing by antibiotics is distinctly biphasic, revealing a
surviving subpopulation of persister cells. Reinoculation of surviving cells produces a
culture with a new subpopulation of persisters, showing that these cells are not mutants,
but rather phenotypic variants of the wild type [3,4]. Reexposure of persisters to a
different bactericidal antibiotic resulted in little or no additional killing, showing that
persisters are multidrug tolerant cells [5]. Gain of function mutants in the Escherichia
coli hipA toxin gene lead to an increase in the frequency of ampicillin- and
fluoroquinolone-tolerant persisters in a growing population from 1 in 10,000 cells or less
(wild type levels) to 1 in 100 cells [6-10], and this hipA7 mutant was shown to form
persisters prior to addition of antibiotic [11]. These persisters were slow- or non-growing
cells. Wild type persisters have been isolated from an exponential culture of E. coli
untreated with antibiotic by sorting out dim cells of a strain expressing a degradable
GFP transcriptionally fused to a ribosomal RNA promoter [12]. This indicated that
persisters are cells that have diminished protein synthesis and are dormant. The
apparent dormancy of persisters accounts for their tolerance to bactericidal antibiotics
whose action requires an active, functional target [13-16].
The mechanism of persister formation is currently unknown. Isolated persisters show
increased expression levels of chromosomal toxin/antitoxin (TA) genes [9,12]. Ectopic
overproduction of RelE, an mRNA endonuclease, [17] inhibits protein synthesis and
creates dormant, multidrug-tolerant cells [9]. The HipA protein is an Ef-Tu kinase

55
[18,19], which also inhibits protein synthesis and produces multidrug tolerant cells upon
overproduction.
However, strains deleted in individual TA loci do not have a phenotype [9,12],
possibly due to their functional redundancy [20-22]. In E. coli, there are at least 15 TA
modules [20,22,23]. Importantly, a screen of an ordered 3,985 open reading frame (out
of a total of 4288) knockout library of E. coli [24] for mutants lacking persisters in
stationary phase produced a largely negative result – not a single strain lacking
persister formation was identified [25]. Similar negative findings were reported with
screens of E. coli transposon insertion (Tn) libraries [26,27] and a Pseudomonas
aeruginosa Tn library [28]. Only mutants with modest reduction in persister levels were
identified, and in the case of E. coli, these were primarily in global regulators [25]. This
strongly suggests that there are multiple, redundant mechanisms of persister formation.
Persisters were originally described by Bigger in 1944 [3], but functional redundancy
has made it very challenging to elucidate the mechanism by which they form.
A useful clue to a possible mechanism of persister formation comes from the
analysis of the SOS response. Interestingly, SOS induces several toxin/antitoxin genes
in E. coli, whose promoters contain a Lex box - symER, hokE, yafN/yafO, and tisAB/IstR
[23,29-35] Another locus, dinJ/yafQ contains a Lex box but is not believed to be under
SOS control [29,30]. Importantly, only the toxin gene is predicted to be upregulated in
the three type 1 TA modules (symER, hokE and tisAB/istR) following SOS induction
while in the type 2 TA modules toxin and antitoxin form an operon and are therefore
both expected to be induced. Fluoroquinolones such as ciprofloxacin induce the SOS
response [36] by blocking the ligase activity of DNA gyrase and topoisomerase,

56
converting them into endonucleases [14,37]. In a separate study we have shown that
the SOS response is also necessary for persister formation in response to the
fluoroquinolone antibiotic ciprofloxacin [38]. In the present study, we examined the
mechanism of this ciprofloxacin-induced persister formation and find that it is governed
by the TisB toxin.
Results
Ciprofloxacin rapidly killed the bulk of E. coli cells, leaving surviving persisters
(Figure 7). Strains deleted in one of the five SOS-TA loci were examined for time-
dependent killing by ciprofloxacin, and one of them, ΔtisAB (GenBank accession
number NC_000913), had a sharply decreased level of persisters (Figure 7A). This
suggests that the majority of persisters, ≥90%, were formed in response to ciprofloxacin
treatment, and their production is dependent on tisAB. Introduction of tisAB in single
copy into the lambda attachment site of the ∆tisAB strain complemented the low
persister phenotype of the knockout strain (Figure 7B). Persister levels observed in
time-dependent killing experiments with ampicillin or streptomycin that do not cause
DNA damage were unchanged in the ΔtisAB strain (not shown). Ampicillin has been
reported to induce the SOS response [39], but apparently the level of induction is
insufficient to influence TisB-dependent persister formation.
IstR-1 is an antisense RNA antitoxin that is expressed constitutively from its own,
LexA-independent promoter and controls the production of the TisB toxin [28]. IstR-2 is
a longer small RNA transcript that is LexA-controlled and contains the entire IstR1 RNA
sequence. IstR-2, however, has been suggested not to be involved in the control of TisB

57
production [40]. tisA is an untranslated open reading frame that contains the antisense
RNA binding site as well as the ribosome binding site for tisB [32]. A schematic of the
tisAB/IstR locus based on [40] is shown in Figure 8.
A strain deleted in istR-1 caused a marked, 10- to 100-fold increase in the level of
persisters (Figure 7A). This is consistent with increased levels of TisB leading to
persister formation. This result is also in apparent contradiction to a published study
showing that ectopic expression of tisB kills cells [41]. It seems likely that the high levels
of expression from the multicopy plasmid used in the above-cited study were
responsible for cell death. Importantly, the minimal inhibitory concentration (MIC) to
ciprofloxacin of tisAB and istR-1 knockouts was the same as in the wild type, showing
that these genes do not affect resistance to this antibiotic, but rather control drug
tolerance by modulating persister production. To test whether IstR-2 was also involved
in tisB regulation in persisters, we produced a knockout of the istR-2 promoter region
and tested it for ciprofloxacin-induced persister formation. Unexpectedly, the ∆PistR-2
strain had reduced persister levels similar to the tisAB knockout (Figure S1). It seems
likely that the istR-2 promoter region contains a binding region of a positive regulator
that is essential for tisB expression.
Using a plasmid-borne promoter-gfp fusion we measured induction of tisAB in
response to ciprofloxacin, and compared this to the expression of other SOS-TA genes
(Figure 9). The tisAB promoter was the most active after 6 hours of exposure to
ciprofloxacin and showed a 1000- fold induction, followed by the symE promoter, which
showed a 100-fold induction. tisAB promoter activity was even higher than that of the
sulA promoter, a standard readout of the SOS response. The dinJ/yafQ promoter was

58
not significantly activated by ciprofloxacin. This is in agreement with a previous report
showing that despite the presence of a putative LexA binding box, the dinJ/yafQ locus
may not be under control of the SOS response [29]. The results of the induction
experiment are consistent with the prominent role of TisB in persister formation in
response to ciprofloxacin.
A common feature of inducible responses is an increase in tolerance upon repeated
exposure to a noxious factor. In a separate study [38] we show that ciprofloxacin
induces persister formation in a typical step-wise induction experiment (exposure to a
low dose of an antibiotic followed by a higher dose). Here, we wanted to test whether
tisB was responsible for this phenotype. Wild type E. coli cells were pre-exposed to low
levels of ciprofloxacin (0.1 µg/ml, 5 x MIC) followed by a higher dose (1 µg/ml) of the
same antibiotic (Figure 10). In a control experiment, the population was exposed to the
high dose from the beginning. Step-wise exposure resulted in a 10- to 100-fold higher
persister level as compared to a population that was exposed to a high dose of the
antibiotic. This pattern is typical of an adaptive response. In contrast to the wild type,
pretreatment with a low dose of antibiotic did not induce a higher level of surviving
persisters in the ∆tisAB mutant. This shows that this adaptive response to ciprofloxacin
depends on tisAB.
Next, we tested the ability of persisters formed in response to tisB expression to
tolerate multiple antibiotics. For this purpose, tisB was cloned into a low copy number
vector pZS*24 with an IPTG inducible promoter and the toxin gene was expressed in
exponentially growing cells. Growth leveled off approximately 1 hour after the addition of
IPTG (data not shown). Cells overproducing TisB were exposed to antibiotics from four

59
unrelated classes, and survival was measured after a 3 hour incubation (Figure 11). As
expected of non-growing cells, the strain overproducing TisB was completely tolerant to
ampicillin, a cell wall synthesis inhibitor that only kills growing cells. Interestingly, cells
overproducing TisB were completely tolerant to ciprofloxacin as well. In contrast to
ampicillin, ciprofloxacin is very effective in killing regular non-growing cells, even those
without ongoing replication [4,9,42]. It appears that TisB produces persisters highly
tolerant to this DNA damaging agent. TisB producing cells also survived exposure to
streptomycin, a protein synthesis inhibitor, 100-fold better than the control strain. This
shows that TisB-dependent persisters exhibit multidrug tolerance. Antibiotics tested in
these experiments act against defined targets. Decreased activity of the target functions
in persisters would lead to drug tolerance. Persisters formed by TisB overproduction
were susceptible to colistin, a polypeptide antibiotic permeabilizing the outer membrane
[43]. This is expected, since an intact outer membrane is essential for cell survival.
Further, TisB overproduction protected a ∆recA mutant against bactericidal antibiotics
from three different classes (Figure 11B).
The SOS response is initiated when RecA senses damaged DNA and activates
cleavage of the global repressor LexA. It was important to establish whether TisB-
dependent formation of persisters was controlled by this well studied SOS response
pathway. The persister level of a ∆recA strain treated with ciprofloxacin was lower as
compared to the wild type, and similar to that of a ∆recA ∆tisB double mutant (Figure
12A).
E. coli can also constitutively express SOS controlled genes if the LexA repressor is
deleted. The level of surviving persisters in E. coli ∆recA lexA300(Def) treated with

60
ciprofloxacin was dramatically increased as compared to the wild type (Figure 12A).
Importantly, the MIC of the E. coli ∆recA lexA300(Def) to ciprofloxacin is 0.002, which is
8-fold lower than in the wild type. RecA is the main recombinase participating in DNA
repair, which explains the increased susceptibility of the mutant to fluoroquinolones that
cause double-strand breaks. This experiment clearly distinguishes between the
decreased resistance of the regular cells, and increased levels of drug-tolerant
persisters in the E. coli ∆recA lexA300(Def) population. Finally, we deleted the tisAB
locus in ∆recA lexA300(Def) and measured survival in response to ciprofloxacin (Figure
12A) and tobramycin (Figure 12B). Persister levels in the ∆tisAB ∆recA lexA300(Def)
triple mutant were drastically reduced as compared to the ∆recA lexA300(Def) strain
and were similar to that of the ∆recA single deletion after exposure to either antibiotic.
Taken together, these experiments show that the SOS response triggers
induction of TisB, causing formation of multidrug tolerant persisters (Figure 13).
Discussion
Previous research clearly indicated redundancy in persister formation
mechanisms, suggesting a unique design of this cell surviving function [2]. Indeed, all
other complex systems of bacteria are made of components usually linked into a single
linear pathway, and a screen of a knockout library readily identifies the genes. By
contrast, a screen of a knockout library did not result in discovery of strains lacking
persisters and the only genes that were identified as contributing to the persister
phenotype were global regulators (hnr, dksA, fis, hns) and genes involved in nucleotide

61
metabolism (apaH, yigB) [25]. The screen was done in stationary phase and the library
did not contain a tisAB knockout strain. TisB-dependent persister formation is observed
under conditions of maximal expression of the SOS response, which is in exponentially
growing cells. Consistent with this, we did not observe a phenotype for the ∆tisAB strain
in stationary phase (not shown), suggesting that under these conditions persisters form
through other mechanisms. The screen [25] did identify the upstream elements of tisB
induction, recA and recB. These knockout strains have increased susceptibility to
fluoroquinolones and were therefore initially not considered as valid candidates for
persister genes.
Another persister component, the glpR regulon was identified in a selection of an
expression library of E. coli for increased drug tolerance [27]. Perhaps this redundancy
of mechanisms evolved in response to antibiotics in the natural environment. If
persisters are specialized survivors, then having multiple mechanisms of formation will
ensure that no single compound will lead to their elimination.
This underscores the challenges in finding approaches to persister eradication.
Redundancy of mechanisms is also challenging for identifying these mechanisms.
Given that persisters are dormant, the search narrows for determinants that can
reversibly block cellular functions. TA loci contain attractive candidates for persister
genes. HipA encoded by the hipBA locus was the first candidate persister gene
identified by a targeted selection for high-persister mutants [6,7]. The hipA7 allele
carries a gain-of-function mutation that causes an increase in persister formation [4,8].
Our recent studies showed that HipA is a protein kinase that phosphorylates EF-Tu,
rendering it non-functional [18,19]. Inhibition of protein synthesis leads to multidrug

62
tolerance and presents a compelling scenario for persister formation. However, deletion
of hipBA has no phenotype ([25]; an earlier report of a phenotype [9] was due to
deleting a flanking region). Expression of other toxins (RelE ; MazF [9,44]) similarly
leads to multidrug tolerance, but deletions do not have a phenotype. Extreme
redundancy of TA genes would explain the lack of a phenotype, and therefore it seemed
useful to search for conditions where a particular toxin would be expressed in a wild
type strain, and then examine a possible link to persister formation.
Several TA genes are expressed under conditions of the SOS response, which is
induced by fluoroquinolone antibiotics. Examination of deletion strains showed that the
level of persisters dropped dramatically in a ∆tisAB mutant and increased equally in a
∆IstR-1 mutant overproducing TisB. During steady-state growth, a fraction of cells
induces the SOS response stochastically, which could have resulted in production of
TisB-dependent persisters [45]. However, the level of persisters surviving treatment with
streptomycin or ampicillin was not affected by the absence of tisB. This suggests that
spontaneous SOS expression is insufficient to produce cells expressing enough TisB to
cause dormancy. This is consistent with our findings that a strain unable to induce the
SOS response exhibits reduced persistence in response to ciprofloxacin, but not
ampicillin or streptomycin [38].
SOS caused by endogenous DNA damage during normal growth has been shown to
induce a “viable but not culturable” state in a subpopulation of cells [45]. It is possible
that this is the consequence of induction of SOS TA modules as well.
Ectopic overexpression of tisB sharply increased the level of persisters. Drug
tolerance following artificial overexpression of a protein, however, may not be a good

63
indicator of a bona fide persister gene. Ectopic overproduction of misfolded toxic
proteins causing stasis produces an artificial state of drug tolerance in E. coli [44]. At the
same time, overexpression experiments are necessary – if induction of a gene does not
lead to increase in drug tolerance, it can be safely eliminated as a candidate. Drop in
persisters in a deletion strain and increase upon overexpression gives reasonable
confidence in functionality of a persister gene. The dependence of TisB-induced
persisters on a particular regulatory pathway, the SOS response, further strengthens
the case for TisB as a specialized persister protein.
The long and unsuccessful search for a mechanism of persister formation has lead
to the provocative hypothesis of dormant cells being formed by random fluctuations in
any protein whose overproduction produces a toxic effect [44]. We previously showed
that persisters are not formed in an early-exponential culture of E. coli, suggesting the
presence of specific persister proteins, rather than random noise in expression of non-
specific genes [4]. However, this debate could only be settled with the identification of a
persister protein. Our finding of an SOS-dependent induction of TisB resulting in
multidrug tolerance suggests that there is in fact a specific mechanism of persister
formation.
The role of TisB in persister formation is unexpected based on what we know about
this type of proteins. TisB is a small, 29 amino acid hydrophobic peptide that binds to
the membrane and disrupts the proton motive force (pmf), which leads to a drop in ATP
levels [41]. Bacteria, plants and animals all produce antimicrobial membrane-acting
peptides [46-48]. Toxins of many TA loci found on plasmids belong to this type as well,
and represent the plasmid maintenance mechanism. If a daughter cell does not inherit a

64
plasmid, the concentration of a labile antitoxin decreases, and the toxin such as the
membrane-acting hok kills the cell [49]. High-level artificial overexpression of tisB also
causes cell death [41]. It is remarkable from this perspective that the membrane-acting
TisB under conditions of natural expression has the exact opposite effect of protecting
the cell from antibiotics. Cells expressing tisB stop growing, and the drop in pmf and
ATP levels will shut down the targets of bactericidal antibiotics. Ciprofloxacin kills cells
primarily by converting its target proteins, DNA topoisomerases, into DNA
endonucleases [14,50]. A drop in ATP will then prevent topoisomerases from damaging
the DNA. β-lactams such as ampicillin kill by activating the autolysins [15,51], and this
requires active peptidoglycan synthesis by the target penicillin-binding proteins.
Peptidoglycan synthesis ceases in non-growing cells. Similarly, the aminoglycoside
streptomycin requires an active ribosome for its killing action. Aminoglycosides kill
primarily by interrupting translation, which creates toxic, misfolded peptides [13,52].
Antibiotics also induce the formation of reactive oxygen species, which contributes to
killing [16], and this requires an active target as well. By creating a dormant state, TisB
will cause a shutdown of antibiotic targets and multidrug tolerance. Fluoroquinolones
such as ciprofloxacin are widely-used broad spectrum antibiotics, and their ability to
induce multidrug tolerant cells is unexpected and a cause of considerable concern.
Induction of persister formation by fluoroquinolones may contribute to the
ineffectiveness of antibiotics in eradicating biofilm infections. Indeed, pre-exposure with
a low dose of ciprofloxacin drastically increases tolerance to subsequent exposure with
a high dose [38].
Induction of persisters by the SOS-induced TisB toxin links together two seemingly

65
opposite strategies of survival – active repair, and entry into a dormant state. It seems
that in the presence of DNA damaging factors, the optimal strategy is to both induce
repair and increase the number of dormant cells, which will survive when everything
else fails. Indeed, a progressive increase in the concentration of fluoroquinolones
rapidly kills regular cells but has little effect on the survival of persisters ([53]; this
study). This means that it is the dormant persisters rather than regular cells with
induced repair that will ultimately survive the DNA damaging antibiotic.
Apart from describing a key element of persister formation, this study also provides a
precedent for a physiological function for a chromosomal TA gene pair. While the role of
TAs in plasmid maintenance is well-established, the function of chromosomal TAs
remains largely unknown. In a recent study, Van Melderen and co-authors produced a
knockout of E. coli lacking 5 toxins, including the well-studied RelE and MazF (mRNA
endonucleases) [21]. The deletion strain had no apparent phenotype and showed
normal growth, susceptibility to antibiotics and stringent response. In Erwinia
chrysanthemi, the chromosomal ccdAB TA module prevented postsegregational killing
of cells that lost an F plasmid, which contains a homologous ccdAB locus [54].
Prevention of postsegregational killing may be a function of some TA genes but would
not explain the presence of >80 TAs in the chromosome of Mycobacterium tuberculosis
[55,56], for example, which is not known to harbor plasmids. Induction of TA genes
under specific conditions such as described in this study may shed some light on their
function.
This study opens an intriguing possibility of a wider link between other stress
responses and persister formation. Pathogens are exposed to many stress factors in

66
the host environment apart from DNA damaging agents, including oxidants, high
temperature, low pH, membrane-acting agents. It is possible that all stress responses
induce the formation of a small but resilient subpopulation of surviving persisters.
Materials and Methods
Media and growth conditions
Experiments were conducted in 0.1 M HEPES-buffered (pH 7.2) Mueller Hinton
Broth (MHB) enriched with 10 mg/L MgSO4 and 20 mg/L CaCl2 according to NCCLS
guidelines for susceptibility testing. Killing experiments were conducted by diluting
overnight cultures 1:100 in 3 ml fresh medium in culture tubes, growing to approximately
2x10^8 CFU/ml and challenging with 0.1 or 1 µg/ml ciprofloxacin. For CFU counts, cells
were plated on LB agar plates containing 20 mM MgSO4 to minimize carryover effects
of ciprofloxacin.
Strain construction
Strains MG1655 ∆tisAB::FRT, ∆IstR-1::FRT and ∆PistR-2::cat, are precise
deletions constructed using the method by Datsenko et al. [57] and cured of their
chloramphenicol resistance cassette with pCP20 where applicable.
P1 transduction was used to move the delta recA::Kan, delta sulA::Kan alleles
(from the MORI KEIO collection[24]) and lexA300(Def) (kindly provided by G. Walker)
into the MG1655 background.
Strain MG1655 pZS*24tisB was constructed by cloning the tisB ORF into the
Kpn1/Cla1 sites of pZS*24 [58] using primers tisBfwKpn1 (5’-
GTAGTAGGTACCATGAACCTGGTGGATATCGCCA-3’) and tisBrevCla1 (5’

67
GTAGTAATCGATACTTCAGGTATTTCAGAACAGCAT-3’).
MG1655 pUA66PtisB-gfp was constructed by cloning the tisAB promoter region
into the XhoI/BamHI sites of vector pUA66gfp using primers PromTisFwXho1 (5’-
GTAGTACTCGAGGCCGGAGCGAGGTTTCGT-3’) and PromTisRevBamH1 (5’-
GTAGTAGGATCCAACACAGTGTGCTCACGCGG-3’). The other promoter-gfp fusions
were taken from a commercial library [59].
For complementation experiments, the tisAB locus was cloned into the CRIM
vector pCAH63 using primers RegiontisBAfwKpn1 (5’-
GTCGTCGGTACCTTGAGTATCGATCACAGTTTGCGT-3’) and RegiontisBArevKpn1
(5’-GTCGTCGGTACCCCTTTGGTGCGACTTGAATCTG-3’) and inserted into the
lambda attachment site of strain MG1655 ∆tisAB::FRT as described by Haldimann et al.
[60].
Promoter activity assay
Cells carrying pUA66-promoter-gfp fusions were grown in MHB to exponential phase as
stated before and exposed to ciprofloxacin. At each time point, aliquots were removed,
washed 2 x in 1% NaCl and transferred to a 96 well plate. GFP fluorescence was
measured with Ex/Em 485/515 on a Gemini XS spectrophotometer (Molecular Devices).
Induction was normalized to background (pUA66gfp), CFU/ml and initial fluorescence.

68
TisB overexpression and persistence
MG1655 carrying either pZS*24 or pZS*tisB was grown to exponential phase in 12 ml
MHB in 125 ml baffled flasks containing 20 µg/ml kanamycin. TisB expression was
induced for 2 hours in mid-exponential phase by addition of 500 µM IPTG. The culture
was then split and exposed to either ciprofloxacin (1 µg/ml), ampicillin (50 µg/ml),
streptomycin (25 µg/ml) or colistin methane sulfonate (10 µg/ml) for 3 hours.

69
References
1 Del Pozo J, Patel R (2007) The challenge of treating biofilm-associated bacterial
infections. Clinical Pharmacol Ther 82: 204-209.
2 Lewis K (2007) Persister cells, dormancy and infectious disease. Nat Rev
Microbiol 5: 48-56.
3 Bigger JW (1944) Treatment of staphylococcal infections with penicillin. Lancet
ii: 497-500.
4 Keren I, Kaldalu N, Spoering A, Wang Y, Lewis K (2004) Persister cells and
tolerance to antimicrobials. FEMS Microbiol Lett 230: 13-18.
5 Wiuff C, Zappala RM, Regoes RR, Garner KN, Baquero F, et al. (2005)
Phenotypic tolerance: antibiotic enrichment of noninherited resistance in bacterial
populations. Antimicrob Agents Chemother 49: 1483-1494.
6 Scherrer R, Moyed HS (1988) Conditional impairment of cell division and altered
lethality in hipA mutants of Escherichia coli K-12. J Bacteriol 170: 3321-3326.
7 Falla TJ, Chopra I (1998) Joint tolerance to beta-lactam and fluoroquinolone
antibiotics in Escherichia coli results from overexpression of hipA. Antimicrob Agents
Chemother 42: 3282-3284.
8 Korch SB, Henderson TA, Hill TM (2003) Characterization of the hipA7 allele of
Escherichia coli and evidence that high persistence is governed by (p)ppGpp synthesis.
Mol Microbiol 50: 1199-1213.
9 Keren I, Shah D, Spoering A, Kaldalu N, Lewis K (2004) Specialized persister
cells and the mechanism of multidrug tolerance in Escherichia coli. J Bacteriol 186:

70
8172-8180.
10 Korch SB, Hill TM (2006) Ectopic overexpression of wild-type and mutant hipA
genes in Escherichia coli: effects on macromolecular synthesis and persister formation.
J Bacteriol 188: 3826-3836.
11 Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S (2004) Bacterial persistence
as a phenotypic switch. Science 305: 1622-1625.
12 Shah D, Zhang Z, Khodursky A, Kaldalu N, Kurg K, et al. (2006) Persisters: A
distinct physiological state of E. coli. BMC Microbiol 6: 53-61.
13 Davis BD, Chen LL, Tai PC (1986) Misread protein creates membrane channels:
an essential step in the bactericidal action of aminoglycosides. Proc Natl Acad Sci U S
A 83: 6164-6168.
14 Chen C, Malik M, Snyder M, Drlica K (1996) DNA gyrase and topoisomerase IV
on the bacterial chromosome: Quinolone-induced DNA cleavage. J Mol Biol 258: 627-
637.
15 Bayles KW (2000) The bactericidal action of penicillin: new clues to an unsolved
mystery. Trends Microbiol 8: 274-278.
� 16. Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ (2007) A
common mechanism of cellular death induced by bactericidal antibiotics. Cell 130: 797
� 810.
16 Christensen SK, Mikkelsen M, Pedersen K, Gerdes K (2001) RelE, a global
inhibitor of translation, is activated during nutritional stress. Proc Natl Acad Sci U S A
98: 14328-14333.
17 Correia FF, D'Onofrio A, Rejtar T, Li L, Karger BL, et al. (2006) Kinase activity of

71
overexpressed HipA is required for growth arrest and multidrug tolerance in Escherichia
coli. J Bacteriol 188: 8360-8367.
18 Schumacher MA, Piro KM, Xu W, Hansen S, Lewis K, et al. (2009) Molecular
mechanisms of HipA-mediated multidrug tolerance and its neutralization by HipB.
Science 323: 396-401.
19 Pandey DP, Gerdes K (2005) Toxin-antitoxin loci are highly abundant in free-
living but lost from host-associated prokaryotes. Nucleic Acids Res 33: 966-976.
20 Tsilibaris V, Maenhaut-Michel G, Mine N, Van Melderen L (2007) What is the
benefit to Escherichia coli of having multiple toxin-antitoxin systems in its genome? J
Bacteriol 189: 6101-6108.
21 Alix E, Blanc-Potard A (2009) Hydrophobic peptides: Novel regulators within
bacterial membranes. Mol Microbiol 72: 5-11.
22 Pedersen K, Gerdes K (1999) Multiple hok genes on the chromosome of
Escherichia coli. Mol Microbiol 32: 1090-1102.
23 Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, et al. (2006) Construction of
Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol
Syst Biol 2: 2006.0008.
24 Hansen S, Lewis K, Vulić M (2008) The role of global regulators and nucleotide
metabolism in antibiotic tolerance in Escherichia coli. Antimicrob Agents Chemother.
25 Hu Y, Coates AR (2005) Transposon mutagenesis identifies genes which control
antimicrobial drug tolerance in stationary-phase Escherichia coli. FEMS Microbiol Lett
243: 117-124.
26 Spoering AL, Vulić M, Lewis K (2006) GlpD and PlsB participate in persister cell

72
formation in Escherichia coli. J Bacteriol 188: 5136-5144.
27 De Groote VN, Verstraeten N, Fauvart M, Kint CI, Verbeeck AM, et al. (2009)
Novel persistence genes in Pseudomonas aeruginosa identified by high-throughput
screening. FEMS Microbiol Lett 297: 73-79.
28 Fernandez De Henestrosa AR, Ogi T, Aoyagi S, Chafin D, Hayes JJ, et al.
(2000) Identification of additional genes belonging to the LexA regulon in Escherichia
coli. Mol Microbiol 35: 1560-1572.
29 Courcelle J, Khodursky A, Peter B, Brown P, Hanawalt P (2001) Comparative
gene expression profiles following UV exposure in wild type and SOS-deficient
Escherichia coli. Genetics 158: 41-64.
30 McKenzie MD, Lee PL, Rosenberg SM (2003) The dinB operon and
spontaneous mutation in Escherichia coli. J Bacteriol 185: 3972-3977.
31 Vogel J, L A, EG W, S A (2004) The small RNA Istr inhibits synthesis of an SOS-
induced toxic peptide. Current Biology 14: 2271-2276.
32 Kawano M, Aravind L, Storz G (2007) An antisense RNA controls synthesis of
an SOS-induced toxin evolved from an antitoxin. Mol Microbiol 64: 738-754.
� 34. Motiejunaite R, Armalyte J, Markuckas A, Suziedeliene E (2007) Escherichia
coli dinJ-yafQ genes act as a toxin-antitoxin module. FEMS Microbiol Lett 268: 112
� 119.
33 Singletary LA, Gibson JL, Tanner EJ, McKenzie GJ, Lee PL, et al. (2009) An
SOS-regulated type 2 toxin-antitoxin system. J Bacteriol 191: 7456-7465.
34 Phillips I, Culebras E, Moreno F, Baquero F (1987) Induction of the SOS
response by new 4-quinolones. J Antimicrob Chemother 20: 631-638.

73
35 Hooper D (2001) Mechanism of action of antimicrobials: Focus on
fluoroquinolones. Clin Infect Diseas 32: S9-S15.
36 Dorr T, Lewis K, Vulic M (2009) SOS response induces persistence to
fluoroquinolones in Escherichia coli. PLoS Genet 5: e1000760.
37 Miller C, Thomsen LE, Gaggero C, Mosseri R, Ingmer H, et al. (2004) SOS
response induction by beta-lactams and bacterial defense against antibiotic lethality.
Science 305: 1629-1631.
38 Darfeuille F, Unoson C, Vogel J, Wagner EG (2007) An antisense RNA inhibits
translation by competing with standby ribosomes. Mol Cell 26: 381-392.
39 Unoson C, Wagner E (2008) A small SOS-induced toxin is targeted against the
inner membrane in Escherichia coli. Mol Microbiol 70: 258-270.
40 Zhao X, Malik M, Chan N, Drlica-Wagner A, Wang JY, et al. (2006) Lethal action
of quinolones against a temperature-sensitive dnaB replication mutant of Escherichia
coli. Antimicrob Agents Chemother 50: 362-364.
� 43. Evans ME, Feola DJ, Rapp RP (1999) Polymyxin B sulfate and colistin: old
antibiotics for emerging multiresistant gram-negative bacteria. Ann Pharmacother
� 33: 960-967.
41 Vazquez-Laslop N, Lee H, Neyfakh AA (2006) Increased persistence in
Escherichia coli caused by controlled expression of toxins or other unrelated proteins. J
Bacteriol 188: 3494-3497.
42 Pennington JM, Rosenberg SM (2007) Spontaneous DNA breakage in single
living Escherichia coli cells. Nat Genet 39: 797-802.
43 Garcia-Olmedo F, Molina A, Alamillo JM, Rodriguez-Palenzuela P (1998) Plant

74
defense peptides. Biopolymers 47: 479-491.
44 Sahl HG, Bierbaum G (1998) Lantibiotics: Biosynthesis and biological activities
of uniquely modified peptides from gram-positive bacteria. Annu Rev Microbiol.
� 48. Zasloff M (2002) Antimicrobial peptides of multicellular organisms. Nature
415: 389
� 395.
45 Gerdes K, Bech FW, Jorgensen ST, Lobner-Olesen A, Rasmussen PB, et al.
(1986) Mechanism of postsegregational killing by the hok gene product of the parB
system of plasmid R1 and its homology with the relF gene product of the E. coli relB
operon. Embo J 5: 2023-2029.
46 Drlica K, Malik M, Kerns R, Zhao X (2008) Quinolone-mediated bacterial death.
Antimicrob Agents Chemother 52: 385-392.
47 Trotonda MP, Xiong YQ, Memmi G, Bayer AS, Cheung AL (2009) Role of mgrA
and sarA in methicillin-resistant Staphylococcus aureus autolysis and resistance to cell
wall-active antibiotics. J Infect Dis 199: 209-218.
48 Kohanski MA, Dwyer DJ, Wierzbowski J, Cottarel G, Collins JJ (2008)
Mistranslation of membrane proteins and two-component system activation trigger
antibiotic-mediated cell death. Cell 135: 679-690.
49 Spoering AL, Lewis K (2001) Biofilms and planktonic cells of Pseudomonas
aeruginosa have similar resistance to killing by antimicrobials. J Bacteriol 183: 6746-
6751.
50 Saavedra De Bast M, Mine N, Van Melderen L (2008) Chromosomal toxin-
antitoxin systems may act as antiaddiction modules. J Bacteriol 190: 4603-4609.

75
51 Gerdes K, Christensen SK, Lobner-Olesen A (2005) Prokaryotic toxin-antitoxin
stress response loci. Nat Rev Microbiol 3: 371-382.
52 Ramage HR, Connolly LE, Cox JS (2009) Comprehensive functional analysis of
Mycobacterium tuberculosis toxin-antitoxin systems: implications for pathogenesis,
stress responses, and evolution. PLoS Genet 5: e1000767.
53 Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in
Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97: 66406645.
54 Lutz R, Bujard H (1997) Independent and tight regulation of transcriptional units
in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements.
Nucleic Acids Res 25: 1203-1210.
55 Zaslaver A, Bren A, Ronen M, Itzkovitz S, Kikoin I, et al. (2006) A
comprehensive library of fluorescent transcriptional reporters for Escherichia coli. Nat
Methods 3: 623-628.
56 Haldimann A, Wanner BL (2001) Conditional-replication, integration, excision,
and retrieval plasmid-host systems for gene structure-function studies of bacteria. J
Bacteriol 183: 6384-6393.

76
Figure 7. Survival of the tisAB/istR mutants after ciprofloxacin exposure and
complementation of the phenotype
(A) Knockout strains of the toxin locus tisAB and its antitoxin istR-1 were exposed to 1
µg/ml ciprofloxacin in exponential growth phase and survival determined by spot plating
for colony forming units. The graph is a
representative of at least 5 independent experiments with similar results, error bars
indicate the standard error. (B) MG1655 ∆tisAB carrying the tisAB region as a single
copy insertion in the lambda attachment site was treated as described in (A).

77
Figure 8. Schematic of the tisAB/IstR locus.
Only the LexA-controlled toxin tisB is translated in vivo, tisA contains the binding site for
the constitutively expressed antitoxin RNA IstR-1 [36]. The IstR-2 RNA is under LexA
control and contains the entire IstR-1 RNA. Its role in tisAB regulation is currently
unclear.
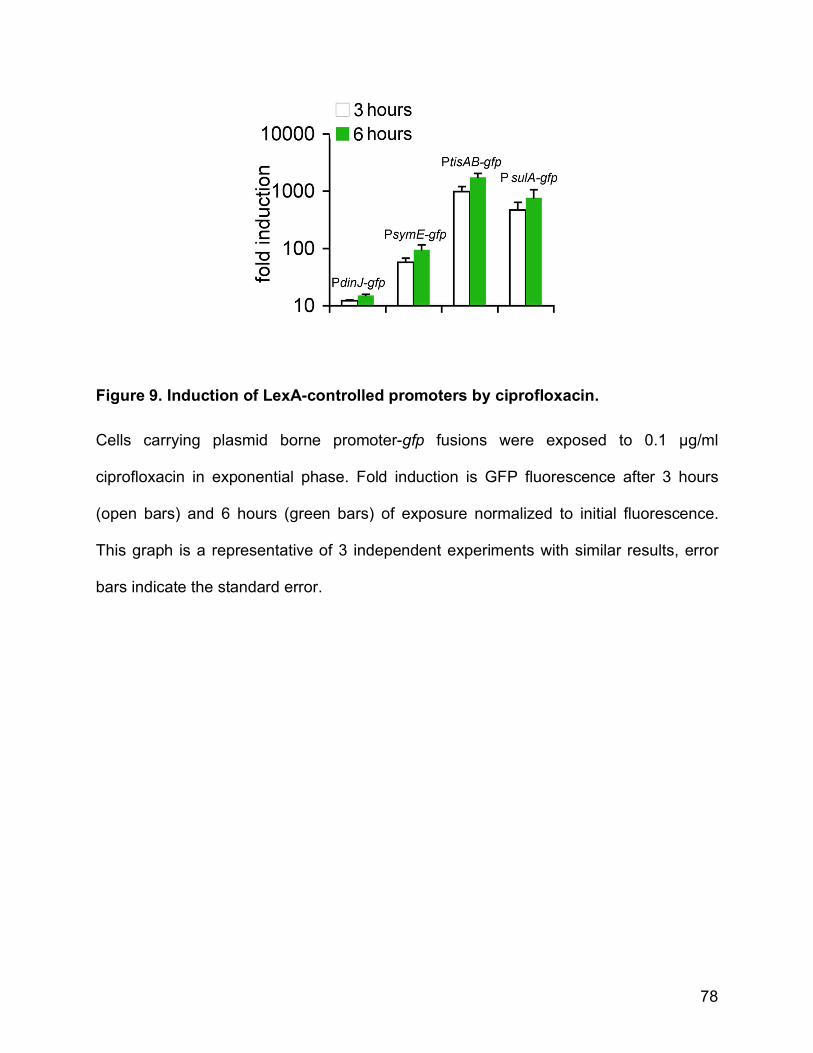
78
Figure 9. Induction of LexA-controlled promoters by ciprofloxacin.
Cells carrying plasmid borne promoter-gfp fusions were exposed to 0.1 µg/ml
ciprofloxacin in exponential phase. Fold induction is GFP fluorescence after 3 hours
(open bars) and 6 hours (green bars) of exposure normalized to initial fluorescence.
This graph is a representative of 3 independent experiments with similar results, error
bars indicate the standard error.

79
Figure 10. Adaptive ciprofloxacin tolerance in E. coli
Wild type MG1655 and its ∆tisAB derivative were grown to exponential phase and
exposed to 0.1 µg/ml ciprofloxacin for 3 hours, after which 1 µg/ml was added
(ciprofloxacin MIC is 0.016 µg/ml). As a control, a parallel culture was immediately
exposed to 1 µg/ml. Viable cell number was determined by serial dilution and plating for
colony forming units (CFU/ml). The data points are averages of 3 independent
experiments, error bars indicate the standard error.

80
Figure 11. TisB overproduction and antibiotic tolerance
tisB was overexpressed in (A) MG1655 and (B) MG1655 ∆recA in exponential phase
from a low copy number vector and exposed to ciprofloxacin (1 µg/ml), ampicillin (50
µg/ml), streptomycin (25 µg/ml) or colistin (10 µg/ml). Survival after 3 hours was
compared to a control strain carrying vector without tisB. The graph shows averages of
three independent experiments, error bars are standard error.

81
Figure 12. TisB-dependent persister formation in SOS response mutants
E. coli MG1655 and its derivatives ∆recA, ∆recA ∆tisAB, ∆recA lexA300(Def) and ∆recA
lexA300(Def)∆tisAB were grown to exponential phase and exposed to (A) ciprofloxacin
at 1 µg/ml or (B) tobramycin at 20 µg/ml. Data are averages of at least 3 independent
experiments, error bars indicate the standard error.

82
Figure 13. Model of ciprofloxacin-induced persister formation.
Ciprofloxacin induces the SOS response, which upregulates DNA repair functions. In a
subpopulation of cells the SOS response also induces the TisB toxin to a high level,
which causes a decrease in proton motive force and ATP level, leading to multidrug
tolerance.

83
Supporting Information
Figure S1 Persister formation in a strain with an istR-2 promoter deletion. Cells were
grown to exponential phase and exposed to 0.1 µg/ml ciprofloxacin for 3 hours to induce
TisB, followed by a higher dose (1 µg/ml) for another 3 hours. Cell survival was
assessed by spot-plating for colony forming units.

84
CHAPTER 3 TISB MECHANISM OF ACTION
Tobias Dörr, Ron Ortenberg and Kim Lewis
Manuscript in preparation
Introduction
TisB is a largely hydrophobic, alpha helical peptide with 29 amino acids
(MSLVDIAILILKLIVAALQLLDAVLKYLK). It is well conserved throughout
Enterobacteriaceae and is induced as part of the LexA – controlled SOS DNA damage
response (Fernandez De Henestrosa et al., 2000, Vogel et al., 2004, Unoson &
Wagner, 2008). We have previously shown that tisB is the mediator of SOS-induced
adaptive tolerance to ciprofloxacin in Escherichia coli and that ectopic overexpression
from a low copy number vector leds to cessation of growth and subsequently to
multidrug tolerance (Dörr et al., 2010).
TisB was associated with the inner membrane and led to a drop in intracellular
ATP concentrations and subsequently cell death upon overproduction from a high copy
number vector (Unoson & Wagner, 2008, Fozo et al., 2008a). Transcriptome analysis
after high-level overexpression revealed an cellular stress response that is expected for
a membrane acting agent, importantly an upregulation of the envelope stress response
(cpx regulon) and also the superoxide stress response (soxS) (Fozo et al., 2008a).
Thus, it seemed possible that TisB disrupts the inner membrane in a detergent-
like fashion. Since low-level expression, however, makes cells dormant rather than
killing them, this seemed unlikely. Rather, association with the inner membrane and

85
drop in ATP concentrations inducing growth arrest suggests an interference of TisB with
the proton motive force or the protein components essential for its establishment.
Results
TisB from pathogenic E. coli 0157:H7 confers multidrug tolerance
The laboratory strain MG1655 has a Ser->Asn change in tisB in the second
amino acid residue. To test whether this affected toxicity or multidrug tolerance, we
amplified tisB from the chromosome of 0157:H7 Sakai and cloned it into a low copy
number vector. Toxicity and persistence after overexpression of tisB 0157 were the same
as tisB from the wild type strain MG1655 (Fig. 14).
The C-terminus is important for TisB – mediated persister formation
TisB has been predicted to insert into the inner membrane with its C-terminus
while the two N-terminal amino acid residues extend into the cytoplasm (Unoson &
Wagner, 2008). While constructing N-terminal and C-terminal translational fusions with
mCherry, we noticed that the TisBmCherry strain had a low persister phenotype while
the mCherryTisB strain had wild type persister levels (Fig. 15). This strongly suggested
that the C-terminus is important for toxicity and lends support to the prediction that the
C-terminus interacts with the inner membrane.
TisB toxicity is reduced when cells are capable of substrate-level
phosphorylation

86
When TisB is ectopically overproduced in medium containing glucose, cell
proliferation stops after around 1 hour and cells remain in a non-growing state for at
least 6 h, after which growth resumes (Fig. 16). This resumption could reflect a)
decrease in the concentration of the inducer (IPTG), b) the rise and overgrowth of TisB
resistant mutants or c) reprogramming of metabolism towards the ability to grow in the
presence of TisB/expression of a resuscitation factor.
Varying the initial IPTG concentration did not affect resuscitation kinetics (not
shown) and we therefore conclude that the inducer is stable enough to cause tisB
expression throughout the experiment. Rediluting the overgrown pZS*34tisB (+IPTG)
cultures into fresh medium with IPTG resulted in the same kinetics of growth arrest as
the parental cultures (not shown), demonstrating that these were not resistant mutants.
Thus, we concluded that resuscitation from TisB overproduction reflects the expression
of a resuscitation factor or reprogramming metabolism.
To test this further, we overexpressed tisB anaerobically in the presence of
different carbon sources. Since TisB is likely to affect oxidative phosphorylation through
its interaction with the inner membrane, allowing cells to use fermentation for ATP
generation could make it possible to bypass TisB toxicity. TisB was therefore
oveproduced in a minimal medium containing succinate (which E. coli cannot ferment),
or in a medium containing the fermentable sugar glucose. Nitrate was added as an
electron acceptor to one culture containing glucose and to the other containing
succinate. Thus, cells could either use only fermentation (glucose), fermentation +
electron transport chain (glucose/nitrate) or only the electron transport chain (succinate)
for ATP generation. Toxicity was reduced in the medium containing only glucose and

87
even more reduced in the medium containing glucose and nitrate. Toxicity was most
severe in the medium containing only succinate (Fig. 17). These results suggested that
oxidative phosphorylation is affected in cells overexpressing tisB and that cells can
partially bypass this toxicity by generating ATP through substrate level phosphorylation.
It also suggested that ongoing oxidative phosphorylation was not essential for TisB
toxicity since the peptide was still toxic in the medium containing only glucose.
We therefore wanted to confirm whether a functional electron transport chain was
necessary for TisB toxicity. Ectopic overexpression of tisB in a strain deleted in atpA
(coding for ATP synthase) or in the electron transport chain - less ∆hemA strain still
caused toxicity (Fig. 16), confirming that ongoing oxidative phosphorylation was not
necessary for TisB action. Therefore it seemed possible that TisB acts by disturbing the
proton motive force, likely by being an uncoupler, rather than by specific inhibition of the
electron transport chain.
To test whether uncoupling would be able to cause multidrug tolerance similar to
that associated with TisB overproduction, we treated a growing culture of E. coli with
increasing concentrations of the protonophore Carbonyl cyanide m-
chlorophenylhydrazone (CCCP) and subsequently with ciprofloxacin. Cccp abolishes
the proton motive force and is used as a standard treatment for membrane
depolarization. Only the two highest doses used (100 and 500 µM) led to a total growth
arrest (Fig 18A, red line) and only the highest concentration protected cells appreciably
against ciprofloxacin (Fig. 18A, blue line). Thus, uncoupling can indeed induce
ciprofloxacin – tolerance. We then used the highest concentration of cccp to treat cells
followed by treatment with different classes of antibiotics. At a concentration of 500 µM,

88
cccp protected E. coli against ampicillin, streptomycin and ciprofloxacin, in all cases to a
higher degree than overexpression of tisB (Fig 18B). Thus, uncoupling of oxidative
phosphorylation can induce a persister-like state in E. coli.
TisB overproduction decreases proton motive force
Since TisB was associated with the inner membrane and decreased intracellular
ATP concentrations (Unoson & Wagner, 2008), we wanted to learn whether TisB
abolished the proton motive force. We used the cyanine dye DiOC(2)3, which
accumulates in the membrane in the presence of a proton motive force, shifting its
emission spectrum from green towards red. Thus, all cells fluoresce green while only
those with a proton motive force fluoresce red. Dividing red by green fluorescence
values gives a good approximation of the presence of a proton motive force
independent of cell size.
The results are shown in Fig. 19. After 30 minutes of induction the proton motive
force decreased and within 90 minutes reached a similar level to that of the control
population depolarized with cccp. Thus, TisB production leads to membrane
depolarization.
Cells do not acquire genetic resistance against tisB overexpression
In order to find possible interaction partners or resuscitation factors of TisB we
attempted to isolate mutants resistant to tisB overexpression. Briefly, E. coli MG1655
was mutagenized with ethyle methane sulfonate (EMS), which led to 1000-fold increase
in mutation frequency to rifampicin (not shown). The mutagenized culture was

89
transformed with pZS*34tisB and plated on MOPS minimal medium containing 0.2 %
succinate as the sole carbon source as well as chloramphenicol and IPTG. Colony
forming units/ml were assessed before and after plating and revealed a mutation
frequency for tisB resistance of ∼2 x 10-5. The transformation was performed 5 times
with cultures from 4 different rounds of mutagenesis. Plasmids were extracted from
mutants and retransformed into wild type to re-assess toxicity. Around 90 % of mutant
plasmids had lost tisB toxicity and were thus discarded. The remaining mutants were
subjected to whole genome sequencing and single nucleotide polymorphism (SNP)
analysis. The first three strains from two rounds of mutagenesis each had 30-40 SNPs
compared to the reference strain MG1655. Only 4 of these SNPs overlapped between
the three strains, lacI (lac operon repressor), ylbE (NO – inducible gene of unknown
function), mdfA (major facilitator family drug efflux pump) and pgpB (coding for
phosphatidylglycerophosphate phosphatase). The genes ylbE, mdfA and pgpB had
exactly the same mutations in all the three strains, indicating that these were
background mutations in the parental strain. All three strains, however, had different
mutations in lacI, which likely decreased expression of tisB from the IPTG-inducible
system used to isolate mutants.
Thus, the whole process was repeated in a strain deleted in lacI. Ten mutants
each from two different rounds of mutagenesis were picked and tested for plasmid
mutants. All mutant plasmids had lost tisB toxicity.
Further, we attempted to isolate mutants resistant to tisB overexpression after
transposon mutagenesis. This yielded a negative result as well; 32 mutants from two

90
rounds of transposon mutagenesis (after transforming approximately 30.000 mutants
each time) that scored as tisB resistant had mutated plasmids.
TisB forms a stable pore in vitro
Since the results described above indicated that TisB may act as an uncoupler,
we measured the effect of synthesized TisB peptide on membrane conductance in an in
vitro artificial black membrane setup. In short, current was applied to two chambers
separated by a lipid bilayer and conductance measured as electric current passing from
one chamber to the other. The lipid bilayer acts as an isolator and thus, before the
experiment, conductance across the bilayer is zero. When synthesized TisB was
present in either chamber, conductance increased in a step-wise fashion after a short
delay of 1 s, which is indicative of pore formation in the membrane (Fig. 20 A).
Moreover, reversion of voltage did not affect current, which indicates the pore is stable
once it has formed (Fig. 20 B). Current/voltage plots further showed that the TisB pore
was selective for chloride anions (Fig. 20 C). It has to be noted, however, that other
anions were not tested.
Discussion
TisB belongs to a group of small, toxic, membrane-associated peptides (STMPs)
that are part of type1 toxin/antitoxin systems and are found in enterobacterial plasmids
and chromosomes (Gerdes et al., 1986a, Fozo et al., 2008b, Pedersen & Gerdes, 1999,
Fozo et al., 2010). Early results with plasmid-encoded hok toxins implicated these
peptides in plasmid maintenance and programmed cell death (Gerdes et al., 1986b)
while the physiological role of chromosomal STMPs remained unknown. We have

91
previously shown that expression of tisB in vivo causes multidrug tolerance after
exposure to a DNA damaging antibiotic (Dörr et al., 2010) and in this study investigated
the mode of action of this peptide.
Since TisB had been shown to associate with the inner membrane and to cause
a drop in intracellular ATP concentrations, it seemed likely that the peptide interfered
with electron-transport chain - mediated ATP generation. E. coli maintains a proton
motive force (PMF) mainly by proton extrusion through an electron transport chain
during oxidative phosphorylation and generates ATP by using the energy from PMF -
driven proton-influx through an ATP synthase (Maloney et al., 1974, Haddock & Jones,
1977). Interference with this process would halt ATP production, at least temporarily,
and thus quickly lead to the observed depletion of intracellular ATP. Indeed, when
ectopically overproduced, TisB was most toxic in minimal medium containing succinate,
which absolutely requires oxidative phosphorylation to generate energy. When supplied
with a fermentable sugar source such as glucose, however, toxicity was reduced. Thus,
TisB interfered with electron transport chain - mediated ATP generation.
TisB was predicted to insert into the inner membrane with its C-terminus, while
two N-terminal residues extend into the cytoplasm. Two observations we made were in
agreement with these predictions: adding an N-terminal fluorescent protein tag or an
amino acid substitution in the second residue (which exists in the laboratory strain of E.
coli, MG1655) did not affect toxicity while adding a C-terminal fluorescent protein
abolished TisB-dependent persister formation.
These results led us to hypothesize that TisB may function as an uncoupler. A
chemical uncoupler, cccp, could induce multidrug tolerance similar to what was

92
observed with TisB overproduction, showing that dissipation of the proton motive force
could lead to persister formation. Ectopic overexpression of tisB led to a drop in proton
motive force similar to that induced by cccp. Further, TisB was toxic in a ∆hemA strain
lacking cytochromes as well as a strain deleted for ATP synthase, suggesting that TisB
toxicity was not mediated by a specific inhibitory interaction of TisB with parts of the
electron transport chain. Moreover, TisB toxicity was almost completely alleviated under
anaerobic conditions in a medium containing nitrate as an electron acceptor and
glucose as a carbon source. Providing the ability to use the electron transport chain
while also relying on substrate level phosphorylation for ATP generation would make it
possible to maintain a (weak) proton motive force even in the presence of a mild
uncoupler.
Additionally, we failed to isolate mutants resistant to TisB overproduction.
Resistant mutants were expected to include an upregulation of a resuscitation factor
(e.g. a protease) or a mutation in a target, precluding TisB binding. The only mutants we
could isolate were in lacI, which likely produced lac repressor alleles that were unable to
bind IPTG and therefore repressed tisB expression from the vector. Even though we
cannot exclude the possibility that resistant mutants were not scored in the specific
conditions we used for mutant selection, a negative result strongly suggests that there is
no specific mutable target for TisB action. Upregulation of a resuscitation factor through
mutation would only be expected to occur if this factor contained a repressor binding
site or other regulatory sites which could be mutated to increase expression, which for
example would be the case for the LexA - regulated Lon protease.

93
Finally, we show that TisB peptide forms a stable pore in an in vitro black
membrane system. When voltage was applied to a two-chamber system, which was
separated by an artificial membrane and contained synthesized TisB peptide, current
could be measured approximately 1 second after application of voltage. This delay
indicates that TisB did not form a pore simply by accumulation in the membrane but
rather needed voltage as a signal. This could reflect the mechanism of pore formation in
vivo, where transmembrane voltage in the form of a proton motive force is around -200
mV during aerobic, exponential growth (Kashket, 1985).
The TisB pore was further shown to be selective for anions. Thus, the pore does
not work as a protonophore but could rather make the membrane permeable for OH-
ions, which would dissipate the proton motive force.
Taken together, we propose the following model for TisB mode of action.
Following DNA damage, TisB molecules accumulate in the inner membrane. After
crossing a certain concentration threshold, several TisB molecules establish a pore in
the inner membrane, a process for which the proton motive force is essential. The pore
decreases the proton motive force probably by increasing membrane permeability for
hydroxyl anions.
Materials and Methods
Bacterial strains and plasmids
MG1655∆hemA::Kan is a precise deletion of the hemA open reading frame.
∆atpA::Kan was constructed using P1 phage transduction from the corresponding strain
in the KEIO collection (Baba et al., 2006). pZS*34tisB0157 was constructed by PCR

94
amplifying the tisB open reading frame from the chromosome of E. coli 0157:H7 Sakai
and cloning it into the Kpn1/Cla1 sites of chl resistant, IPTG inducible, low copy number
vector pZS*34 (Lutz & Bujard, 1997).
Growth conditions
Cells were grown in 0.1 M HEPES – buffered Mueller Hinton Broth, or, where
mentioned, in MOPS minimal medium supplemented with 100 µg/ml uracil, 13.4 µM
H2PO4 and 0.2 % glucose, maltose or succinate.
The ∆hemA and ∆atpA strains were always grown in medium containing 0.2 % glucose.
TisB overproduction resistance screen
Cells were grown to mid-exponential phase (3 x 108 CFU/ml), washed twice and
resuspended in 0.1 M HEPES buffer (pH 7.2), after which 15 µL/ml culture of ethane
methyle sulfonate was added for 35 minutes. Cells were then washed twice,
resuspended in 1ml of fresh growth medium, grown overnight and diluted onto agar
plates to reach approximately 10,000 colonies/plate. These colonies were scraped off
the plate, diluted into fresh growth medium, grown for 1h and transformed by
electroporation with pZS*34tisB. Cells were then washed three times and plated on
MOPS minimal agar supplemented with 0.2 % succinate, 15 µg/ml chloramphenicol and
1 mM IPTG. Candidates for resistant mutants were picked, outgrown and transformed
with kanamycin resistant pZS*24tisB. Mutants that did not exhibit toxicity after TisB
overproduction from this vector were tested further by extracting their pZS*34tisB,
transforming it into wild type and testing for remaining toxicity.
Proton motive force measurements

95
MG1655 pZS*34tisB was grown to mid-exponential phase and 1mM IPTG
added. Membrane potential was assessed using the protocol of the Invitrogen Baclight
membrane potential kit. In short, cells expressing tisB were diluted 100-fold into pre-
warmed phosphate buffered saline (PBS), incubated with DiOC(2)3 for 30 minutes and
analyzed in a BD FACSAria. As depolarized control, 5 µM cccp was added to one
sample for 10 min. Membrane potential was estimated by dividing population mean red
fluorescence values by population mean green fluorescence values.
Black membrane experiments
All artificial lipid bilayer experiments were done in collaboration with Sergey M.
Bezrukov at the Laboratory of Physical and Structural Biology/NIH/Bethesda, Maryland
essentially as described by Kullman and Bezrukov (Kullman et al., 2002). In short,
synthesized TisB (provided by Wayne Anderson, Northwestern University, Chicago,
Illinois) was added to a two-chamber system filled with KCl and separated by an
artificial lipid bilayer. Voltage was applied and the current through the two chambers
measured.
References
Baba, T., T. Ara, M. Hasegawa, Y. Takai, Y. Okumura, M. Baba, K. A. Datsenko, M.
Tomita, B. L. Wanner & H. Mori, (2006) Construction of Escherichia coli K-12 in-
frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:
2006.0008.
Dörr, T., M. Vulic & K. Lewis, (2010) Ciprofloxacin causes persister formation by
inducing the TisB toxin in Escherichia coli. Plos Biol 8: e1000317.

96
Fernandez De Henestrosa, A. R., T. Ogi, S. Aoyagi, D. Chafin, J. J. Hayes, H. Ohmori &
R. Woodgate, (2000) Identification of additional genes belonging to the LexA
regulon in Escherichia coli. Mol Microbiol 35: 1560-1572.
Fozo, E., M. Kawano, F. Fontaine, Y. Kaya, K. Mendieta, K. Jones, A. Ocampo, K.
Rudd & G. Storz, (2008a) Repression of small toxic protein synthesis by the Sib
and OhsC small RNAs. Mol Microbiol 70: 1076-1093.
Fozo, E. M., M. R. Hemm & G. Storz, (2008b) Small toxic proteins and the antisense
RNAs that repress them. Microbiol Mol Biol Rev 72: 579-589, Table of Contents.
Fozo, E. M., K. S. Makarova, S. A. Shabalina, N. Yutin, E. V. Koonin & G. Storz, (2010)
Abundance of type I toxin-antitoxin systems in bacteria: searches for new
candidates and discovery of novel families. Nucleic Acids Res 38: 3743-3759.
Gerdes, K., F. W. Bech, S. T. Jorgensen, A. Lobner-Olesen, P. B. Rasmussen, T.
Atlung, L. Boe, O. Karlstrom, S. Molin & K. von Meyenburg, (1986a) Mechanism
of postsegregational killing by the hok gene product of the parB system of
plasmid R1 and its homology with the relF gene product of the E. coli relB
operon. Embo J 5: 2023-2029.
Gerdes, K., P. B. Rasmussen & S. Molin, (1986b) Unique type of plasmid maintenance
function: postsegregational killing of plasmid-free cells. Proceedings of the
National Academy of Sciences of the United States of America 83: 3116-3120.
Haddock, B. A. & C. W. Jones, (1977) Bacterial respiration. Bacteriol Rev 41: 47-99.
Kashket, E. R., (1985) The proton motive force in bacteria: a critical assessment of
methods. Annu Rev Microbiol 39: 219-242.

97
Kullman, L., M. Winterhalter & S. M. Bezrukov, (2002) Transport of maltodextrins
through maltoporin: a single-channel study. Biophys J 82: 803-812.
Lutz, R. & H. Bujard, (1997) Independent and tight regulation of transcriptional units in
Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements.
Nucleic Acids Res 25: 1203-1210.
Maloney, P. C., E. R. Kashket & T. H. Wilson, (1974) A protonmotive force drives ATP
synthesis in bacteria. Proceedings of the National Academy of Sciences of the
United States of America 71: 3896-3900.
Pedersen, K. & K. Gerdes, (1999) Multiple hok genes on the chromosome of
Escherichia coli. Mol Microbiol 32: 1090-1102.
Unoson, C. & E. Wagner, (2008) A small SOS-induced toxin is targeted against the
inner membrane in Escherichia coli. Mol Microbiol 70: 258-270.
Vogel, J., A. L, W. EG & A. S, (2004) The small RNA Istr inhibits synthesis of an SOS-
induced toxic peptide. Current Biology 14: 2271-2276.

98
Figure 14. Survival in antibiotics after overexpression of tisB from O157:H7 Sakai
E. coli MG1655 pZS*34tisB0157 was grown to mid-exponential phase and induced with
IPTG for 2 hours. Antibiotics were added to 0.1 µg/ml(ciprofloxacin), 50 µg/ml
(ampicillin) and 50 µg/ml (streptomycin) for three hours. Log percent survival is colony
forming units normalized to cell density before addition of antibiotics. The error bars
represent standard error of 3 independent experiments.

99
Figure 15. Survival of strains carrying translational TisB – mcherry fusions in
ciprofloxacin.
Cells were grown to mid-exponential phase and exposed to 0.1 µg/ml cipro for 3 hours,
followed by 3 hours of 1 µg/ml cipro. N1, N2 = N-terminal fusion strains, C1, C2 = C-
terminal fusion strains.

100
Figure 16. Overexpression of tisB in respiratory chain mutants. Cells were grown to mid-exponential growth phase in 24 well plates in Mueller Hinton
Broth with 0.2 % glucose and induced with 1 mM IPTG at time zero. OD600 was
followed at 25 minute intervals using an automated plate reader. Shown here is a
representative of three different experiments with similar results.

101
Figure 17. TisB overproduction under anaerobic conditions in different carbon sources. Cells were grown in an anaerobic chamber in MOPS minimal medium plus either
glucose, glucose + nitrate or succinate + nitrate. The y-axis is log increase in CFU/ml
(normalized to CFU/ml before addition of IPTG), error bars are standard error of three
independent experiments.

102
Figure 18. Effects of cccp on multidrug tolerance MG1655 was grown to mid-exponential phase and exposed to cccp at different
concentrations for 10 minutes before addition of antibiotic. A, blue line: Log percent
survival after three hours of exposure to 0.1µg/ml ciprofloxacin, red line: fold growth
inhibition relative to control (no cccp). B) fold protection (= survival of treated /untreated)
of cccp at 500 µM compared to overexpression of tisB. Strep = streptomycin (25 µg/ml),
amp = ampicillin (50 µg/ml), cipro = ciprofloxacin
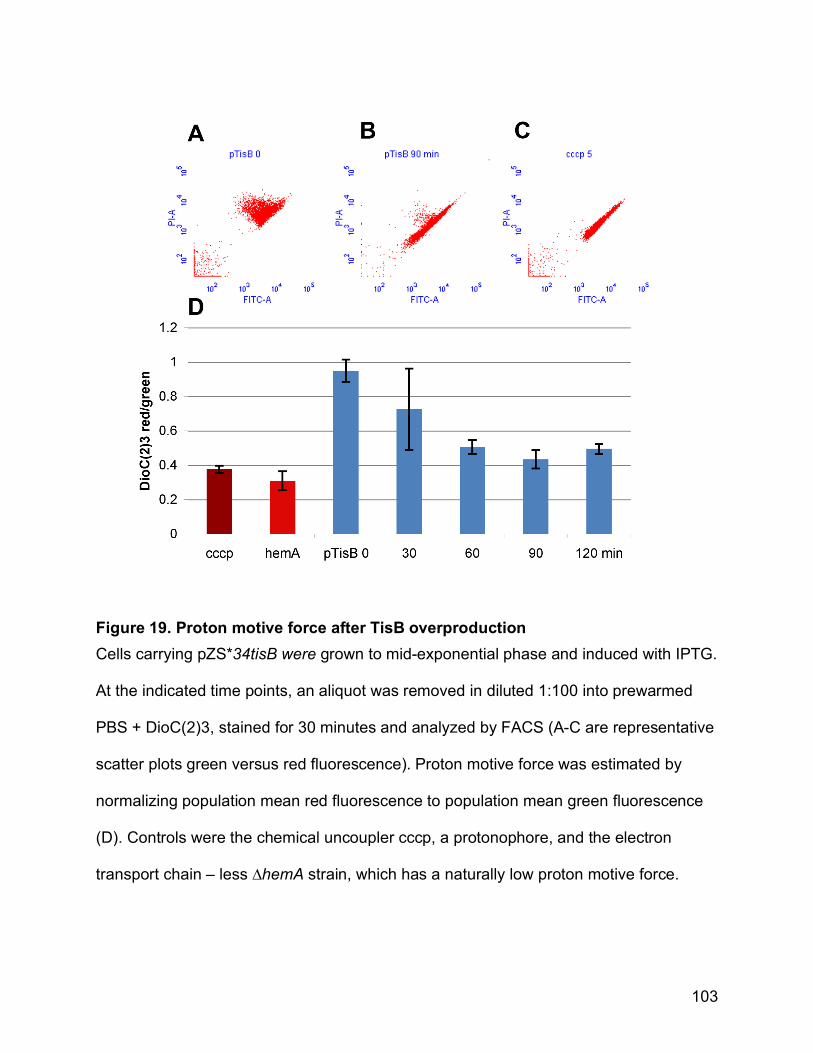
103
Figure 19. Proton motive force after TisB overproduction Cells carrying pZS*34tisB were grown to mid-exponential phase and induced with IPTG.
At the indicated time points, an aliquot was removed in diluted 1:100 into prewarmed
PBS + DioC(2)3, stained for 30 minutes and analyzed by FACS (A-C are representative
scatter plots green versus red fluorescence). Proton motive force was estimated by
normalizing population mean red fluorescence to population mean green fluorescence
(D). Controls were the chemical uncoupler cccp, a protonophore, and the electron
transport chain – less ∆hemA strain, which has a naturally low proton motive force.

104
Figure 20. TisB-dependent conductance of a black membrane lipid bilayer. Synthesized TisB was added to a two-chamber system separated by an artificial lipid
bilayer (black membrane) and conductance recorded upon application of voltage.
The graphs show time dependent conductance after addition of TisB (A), voltage-
dependent current through the TisB pore upon voltage reversion (B), Voltage-
dependent current through the TisB pore with unequal concentrations of KCl in both
chambers (C).

105
DISCUSSION
The persister phenomenon is still puzzling researchers more than 60 years after
its discovery. Though more and more is known about the potential clinical importance of
persisters, the mechanism(s) of formation have remained largely unknown.
Based on several studies, persisters were suggested to be an entity of
stochastically-formed, non-growing, multidrug tolerant cells, formed by several possible
mechanisms resulting in the same phenotype (Balaban et al., 2004, Shah et al., 2006
Lewis, 2007). Several pieces of evidence, however, suggested otherwise. Cells
expressing an unstable GFP from a growth-rate-dependent promoter produced a dim
subpopulation enriched in persisters (Shah et al., 2006), which still exhibited a high
variance in fluorescence. This could be an artifact of detection or heterogeneity in
degradation of GFP after growth arrest, but could also indicate that this subpopulation
did not consist entirely of non-growing cells but rather a mixture of non-growing and
slowly growing cells. Further, Wiuff et al. showed that persisters were multidrug tolerant
by exposing a population to a bactericidal antibiotic, then washing and re-exposing the
bona fide persisters to a different antibiotic and concluding that further survival was not
much affected (Wiuff et al., 2005). In actuality, however, the persisters died by up to one
order of magnitude when exposed to the second dose of antibiotic. Since persisters in
this study were washed and resuspended in a fresh medium, these results are difficult
to interpret and could reflect resuscitation from dormancy. Another explanation is that
90 % of persisters are specific to the antibiotic they were exposed to.

106
Indeed, here we have shown that exposure of a population of growing E. coli
cells to a low dose of ciprofloxacin increases their tolerance level to a subsequent
exposure to a high dose of the same antibiotic, indicating that persistence can be an
inducible response to an antibiotic. This increase in tolerance was dependent on the
SOS DNA damage response. Removal of the cell’s ability to induce the SOS response
did not entirely eradicate survivors to ciprofloxacin. Therefore, a subfraction of persister
cells pre-exists in every population as a kind of “life-insurance” for the whole population
after a catastrophic event. This persister fraction, however, can be increased as a
response to DNA damage and likely other stresses as well.
These results add a new, deterministic component to our understanding of
persisters and induced persistence may play an important role in human infectious
disease. Fluoroquinolones are widely used, broad-spectrum antibiotics and induction of
a dormant state may aide pathogens in surviving high doses of antibiotics in the
absence of resistance. Further, pathogens can experience SOS induction after
exposure to the host immune system. Urinary tract infection (UTI) causing strains of E.
coli, for example, induce SOS and filament during infection (Justice et al., 2006). This
may induce persisters and consequently leave a survivor reservoir after treatment with
any antibiotic, which may contribute to the notorious persistence of these infections.
First clues towards the mechanism of persister formation came from
transcriptome analysis of persister cells, which showed that chromosomal toxin antitoxin
modules were upregulated, more specifically several mRNA interferases.
Overexpression of chromosomal toxins (and other toxic proteins) can emulate a
persister-state and cause multidrug tolerance ( Keren et al., 2004, Correia et al., 2006,

107
Vazquez-Laslop et al., 2006, Shah et al., 2006). TA modules are abundant on bacterial
chromosomes, which offered an explanation for the lack of strong phenotypes for single
knockouts. Here, we found that a chromosomal toxin TisB is the effector of induced
persistence. Overexpression of tisB led to growth arrest and multidrug tolerance and,
more importantly, a knockout led to a decrease in persistence. TisB belongs to a group
of small, hydrophobic membrane peptides with RNA antitoxins. Other members of this
group, such as the hok and ibs toxins had been shown to kill and lyse cells either when
produced as part of a plasmid maintenance system (hok) (Faridani et al., 2006) or upon
overexpression (ibs) (Fozo et al., 2008). It was therefore surprising that tisB expression
actually protected cells. How does a small detergent-like peptide such as TisB induce
growth arrest? TisB overproduction was transiently toxic when cells were provided with
a fermentable sugar such as glucose and permanently toxic when supplied with
succinate, which requires oxidative phosphorylation for energy generation. TisB toxicity
was not alleviated in a strain, which lacks an electron transport chain or ATP synthase.
These results suggested that TisB interfered with the proton motive force. Using an
artificial black membrane system we showed that upon a voltage signal, TisB forms a
stable, anion-selective pore. Overexpression dissipated the proton motive force to a
similar level to that of cccp, a protonophore, which we found was able to induce a
multidrug tolerant state as well. These results could provide the following explanation for
the observed growth-arrest and multidrug tolerance: TisB forms a pore in the inner
membrane and abolishes the proton motive force. While this by itself excludes
antibiotics such as aminoglycosides from the cell (Bryan & Van Den Elzen, 1977), it
would also lead to a depletion of intracellular ATP and a subsequent slowdown or

108
shutdown of metabolism. Gyrase, one of the targets for ciprofloxacin, for example, has a
Km for ATP of approximately 300 µM (Higgins et al., 1978). TisB overproduction led to a
10-fold drop in ATP concentrations after expression from a high copy number vector for
15 minutes (Unoson & Wagner, 2008). Given that intracellular [ATP] is in the low mM
range (Albe et al., 1990) a tenfold drop would decrease gyrase activity to half-maximal
levels, which would significantly decrease the amount of damage ciprofloxacin binding
can cause.
Why does TisB protect only a small subpopulation of cells and not the whole
population? This could be reflective of the stochasticity of either TisB expression or that
of its antitoxin, IstR-1. One possibility is that after exposure to ciprofloxacin, persisters
are the cells that express a certain level of TisB within a normal distribution. Another
possibility is that stochasticity of antitoxin expression leads to TisB-mediated survival,
with persisters being those cells that express IstR-1 to the lowest level. Indeed, an
antitoxin knockout increased persister levels 10- to 100-fold (Dörr et al., 2010),
suggesting that a high degree of persister formation is actually blocked by high level
expression of antitoxin. Still even the ∆istR-1 strain did not allow the whole population to
survive in a TisB-dependent way. Further, a high dose of ciprofloxacin killed the bulk
completely within the first 30 minutes of exposure, leaving surviving persister cells. The
tisB promoter has the strongest lexA binding of all SOS inducible promoters (Fernandez
De Henestrosa et al., 2000, Vogel et al., 2004, Unoson & Wagner, 2008) and it seems
unlikely that cells are actually able to induce TisB before the bulk dies. The explanation
of persister formation in these conditions of high ciprofloxacin exposure purely by
stochasticity of istR or TisB expression levels is thus doubtful.

109
A third possible explanation is therefore that persisters are pre-conditioned to
induce TisB and survive. Shah et al. showed that populations of E. coli harbor a
subpopulation of non- or slowly growing cells, which are enriched in persisters to
ciprofloxacin. A plausible model of persister formation in these conditions is that these
cells survive the initial exposure to ciprofloxacin. Due to their low metabolic activity they
are in fact damaged by the antibiotic, but not to a lethal degree. For a while this low
level DNA damage can be compensated by repair functions but extended exposure to
the antibiotic will kill off even these cells, unless they are able to induce TisB, which
shuts down metabolism and turns these “pre-persisters” into multidrug tolerant persister
cells (Fig. 4-1). Apart from rapid death after exposure to high ciprofloxacin and
emergence of persisters in a time frame that is likely too short to allow for induction of
TisB, several other experimental observations support this model: survivors to
ciprofloxacin have a low level of SOS induction but rely on repair functions such as recA
(Dörr et al., 2009). If stochasticity of persister formation purely relied on TisB expression
levels, the upstream event of SOS induction should be highly uniform in the bulk.
Further, the TisB-dependent persister phenotype is visible only after extended exposure
to ciprofloxacin (3 h), suggesting that for the first three hours cells have different means
of coping with antibiotic stress.
In summary, we have shown that the flouroquinolone antibiotic ciprofloxacin can
induce the formation of multidrug tolerant persister cells by inducing the small peptide
toxin TisB. TisB forms a pore in the inner membrane and dissipates the proton motive
force, leading to a drop in intracellular ATP concentrations and subsequent shutdown of
growth and possibly metabolism. TisB dependent persister formation is likely regulated

110
by expression levels of its antitoxin RNA IstR-1 and by stochasticity in SOS induction
intensities.
References
Albe, K. R., M. H. Butler & B. E. Wright, (1990) Cellular concentrations of enzymes and
their substrates. J Theor Biol 143: 163-195.
Balaban, N. Q., J. Merrin, R. Chait, L. Kowalik & S. Leibler, (2004) Bacterial persistence
as a phenotypic switch. Science 305: 1622-1625.
Bryan, L. E. & H. M. Van Den Elzen, (1977) Effects of membrane-energy mutations and
cations on streptomycin and gentamicin accumulation by bacteria: a model for
entry of streptomycin and gentamicin in susceptible and resistant bacteria.
Antimicrob Agents Chemother 12: 163-177.
Correia, F. F., A. D'Onofrio, T. Rejtar, L. Li, B. L. Karger, K. Makarova, E. V. Koonin &
K. Lewis, (2006) Kinase activity of overexpressed HipA is required for growth
arrest and multidrug tolerance in Escherichia coli. J Bacteriol 188: 8360-8367.
Courcelle, J., A. Khodursky, B. Peter, P. Brown & P. Hanawalt, (2001) Comparative
gene expression profiles following UV exposure in wild type and SOS-deficient
Escherichia coli. Genetics 158: 41-64.
Dörr, T., K. Lewis & M. Vulic, (2009) SOS response induces persistence to
fluoroquinolones in Escherichia coli. PLoS Genet 5: e1000760.
Dörr, T., M. Vulic & K. Lewis, (2010) Ciprofloxacin causes persister formation by
inducing the TisB toxin in Escherichia coli. Plos Biol 8: e1000317.

111
Faridani, O. R., A. Nikravesh, D. P. Pandey, K. Gerdes & L. Good, (2006) Competitive
inhibition of natural antisense Sok-RNA interactions activates Hok-mediated cell
killing in Escherichia coli. Nucleic Acids Res 34: 5915-5922.
Fernandez De Henestrosa, A. R., T. Ogi, S. Aoyagi, D. Chafin, J. J. Hayes, H. Ohmori &
R. Woodgate, (2000) Identification of additional genes belonging to the LexA
regulon in Escherichia coli. Mol Microbiol 35: 1560-1572.
Fozo, E. M., M. Kawano, F. Fontaine, Y. Kaya, K. S. Mendieta, K. L. Jones, A. Ocampo,
K. E. Rudd & G. Storz, (2008) Repression of small toxic protein synthesis by the
Sib and OhsC small RNAs. Mol Microbiol 70: 1076-1093.
Higgins, N. P., C. L. Peebles, A. Sugino & N. R. Cozzarelli, (1978) Purification of
subunits of Escherichia coli DNA gyrase and reconstitution of enzymatic activity.
Proceedings of the National Academy of Sciences of the United States of
America 75: 1773-1777.
Justice, S. S., D. A. Hunstad, P. C. Seed & S. J. Hultgren, (2006) Filamentation by
Escherichia coli subverts innate defenses during urinary tract infection.
Proceedings of the National Academy of Sciences of the United States of
America 103: 19884-19889.
Keren, I., D. Shah, A. Spoering, N. Kaldalu & K. Lewis, (2004) Specialized persister
cells and the mechanism of multidrug tolerance in Escherichia coli. J Bacteriol
186: 8172-8180.
Lewis, K., (2010) Persister Cells. Annu Rev Microbiol.
Shah, D., Z. Zhang, A. Khodursky, N. Kaldalu, K. Kurg & K. Lewis, (2006) Persisters: A
distinct physiological state of E. coli. BMC Microbiol 6: 53-61.

112
Unoson, C. & E. Wagner, (2008) A small SOS-induced toxin is targeted against the
inner membrane in Escherichia coli. Mol Microbiol 70: 258-270.
Vazquez-Laslop, N., H. Lee & A. A. Neyfakh, (2006) Increased persistence in
Escherichia coli caused by controlled expression of toxins or other unrelated
proteins. J Bacteriol 188: 3494-3497.
Vogel, J., A. L, Wagner, EG & Altuvia. S, (2004) The small RNA Istr inhibits synthesis of
an SOS-induced toxic peptide. Current Biology 14: 2271-2276.
Wiuff, C., R. M. Zappala, R. R. Regoes, K. N. Garner, F. Baquero & B. R. Levin, (2005)
Phenotypic tolerance: antibiotic enrichment of noninherited resistance in bacterial
populations. Antimicrob Agents Chemother 49: 1483-1494.
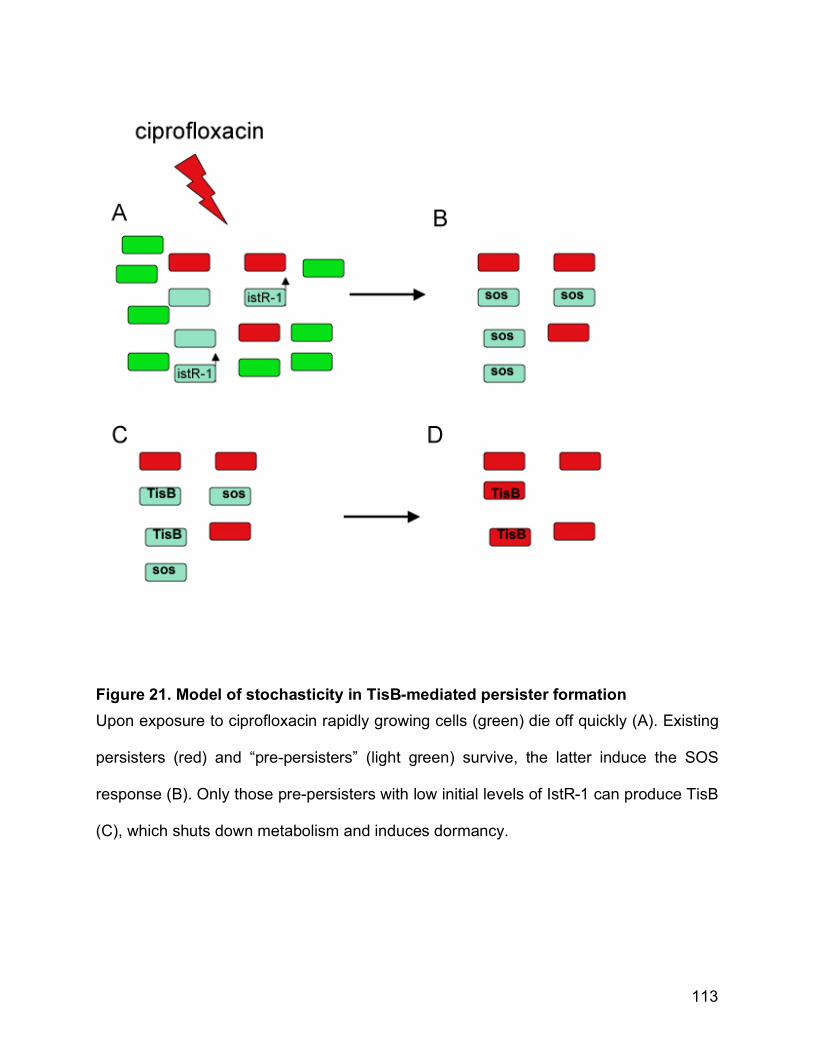
113
Figure 21. Model of stochasticity in TisB-mediated persister formation Upon exposure to ciprofloxacin rapidly growing cells (green) die off quickly (A). Existing
persisters (red) and “pre-persisters” (light green) survive, the latter induce the SOS
response (B). Only those pre-persisters with low initial levels of IstR-1 can produce TisB
(C), which shuts down metabolism and induces dormancy.