specific acentric-mode displacement analysis of … second-harmonic generating properties of...
TRANSCRIPT

1
Supporting Information for:
Specific Acentric-Mode Displacement Analysis of
RbPbCO3F and CsPbCO3F: Synthesis, Structure,
and Second-Harmonic Generating Properties of
Alkali-Metal Lead Fluoro-Carbonates
T. Thao Tran1, P. Shiv Halasyamani
1,*, and James M. Rondinelli
2,*
1Department of Chemistry, University of Houston, 112 Fleming Building, Houston, TX 77204-
5003
2Department of Materials Science & Engineering, Drexel University, Philadelphia, Pennsylvania
19104, United States
CORRESPONDING AUTHOR EMAIL ADDRESS: [email protected], [email protected].

2
Figure S1. Experimental and calculated powder X-ray diffraction patterns of RbPbCO3F and
CsPbCO3F
Figure S2. IR spectrum of RbPbCO3F
Figure S3. UV-Vis diffuse reflectance spectrum of RbPbCO3F
Figure S4. Thermogravimetric analysis and differential thermal analysis diagram of RbPbCO3F
under N2
Figure S5. Powder X-ray diffraction of final residuals after TGA/DTA analysis
Figure S6. Electron localization function (ELF) and isosurfaces for RbPbCO3F and CsPbCO3F
Table S1. Selected bond distances (Å) and angles (deg)
Table S2. Atomic coordinates and equivalent isotropic displacement parameters (Å2) for
RbPbCO3F.
Table S3. Atomic coordinates and equivalent isotropic displacement parameters (Å2) for
CsPbCO3F
Table S4. Bond valence analysis for RbPbCO3F
Table S5. Bond valence analysis for CsPbCO3F
Table S6. Born effective charges for CsPbCO3F and RbPbCO3F.
Table S7. Ideal P-6m2 structure used in the mode-polarization vector analyses of RbPbCO3F and
CsPbCO3F.
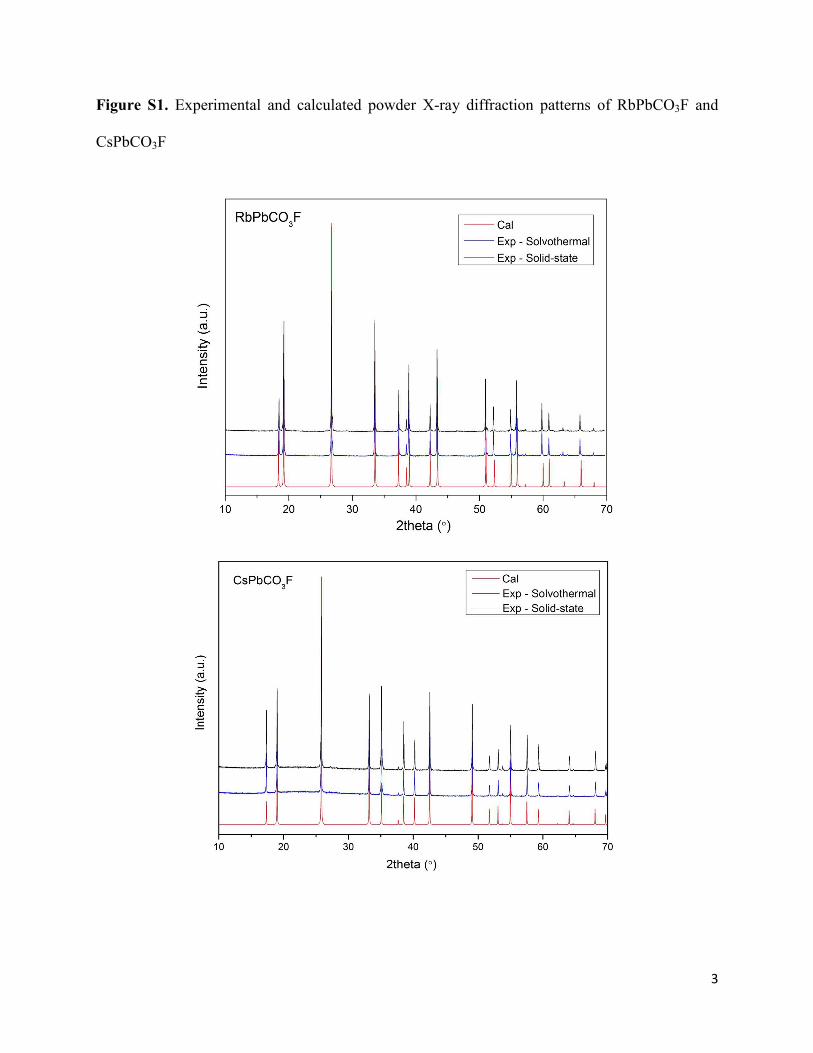
3
Figure S1. Experimental and calculated powder X-ray diffraction patterns of RbPbCO3F and
CsPbCO3F

4
Figure S2. IR spectrum of RbPbCO3F
ν(C−O) δ( OCO) ν(Pb−F)
RbPbCO3F 1410, 1038 836, 687 400

5
Figure S3. UV-Vis diffuse reflectance spectrum of RbPbCO3F
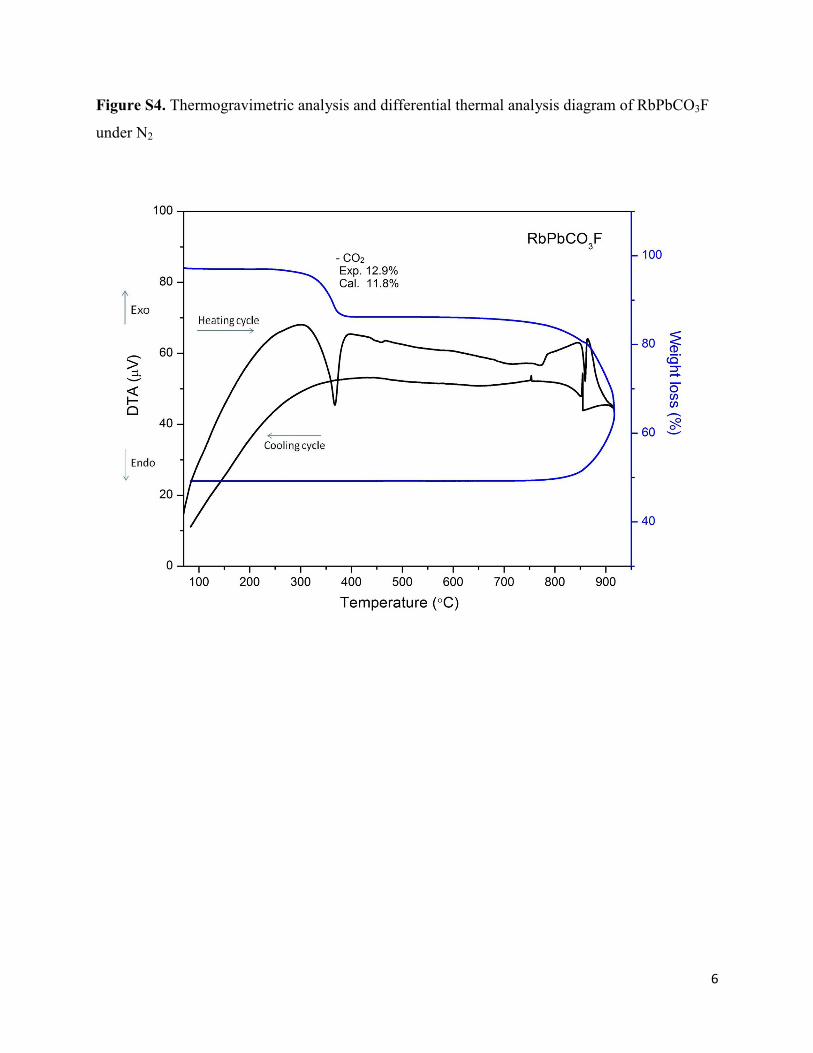
6
Figure S4. Thermogravimetric analysis and differential thermal analysis diagram of RbPbCO3F
under N2

7
Figure S5. Powder X-ray diffraction of final residuals after TGA/DTA analysis

8
Figure S6. Electron localization function (ELF) and isosurfaces for CsPbCO3F (left) and
RbPbCO3F (right) viewed along [001] and projected onto the (001) plane at a distance of 0 Å
from the origin, respectively, illustrating a nearly symmetric charge distribution about the Pb
sites. Maroon: Pb; Red, O; Brown, C.
CsPbCO3F RbPbCO
3F

9
Table S1. Selected bond distances (Å) and angles (deg)
RbPbCO3F CsPbCO3F
Pb – O × 6 2.6865(8) Pb – O × 6 2.7086(17)
Pb – F × 2 2.421(7) Pb– F Pb– F
2.23(3) 2.88(3)
C – O × 3 1.290(6) C – O × 3 1.300(11) O – Pb – O O – Pb – O F – Pb – O F – Pb – O
49.1(2) 70.9(2) 85.8(18) 93.8(16)
O – Pb – O O – Pb – O F – Pb – O
49.1(5) 70.9(5) 90.0
Pb – F – Pb 175.4(19) Pb – F – Pb 180.0

10
Table S2. Atomic coordinates and equivalent isotropic displacement parameters (Å2) for
RbPbCO3F.
Atom x y z Ueqa
Rb Pb C O F
0.6667 1.0000 0.3333 0.4726(6) 0.9580(17)
0.3333 1.0000 0.6667 0.9451(12) 0.9790(9)
0.0000 0.5000 0.5000 0.5000 0.0000
0.011(3) 0.007(2) 0.011(3) 0.020(2) 0.020(14)
a Ueq is defined as one-third of the trace of the orthogonalized Uij tensor

11
Table S3. Atomic coordinates and equivalent isotropic displacement parameters (Å2) for
CsPbCO3F
Atom x y z Ueqa
Cs Pb C O F
0.3333 0.0000 0.6667 0.5275(12) 0.0000
0.6667 0.0000 0.3333 0.4725(12) 0.0000
0.5000 0.0000 0.0000 0.0000 0.4360(4)
0.014(4) 0.010(4) 0.006(5) 0.020(3) 0.023(6)
a Ueq is defined as one-third of the trace of the orthogonalized Uij tensor

12
Table S4. Bond valence analysis for RbPbCO3Fa
Atom O F ΣΣΣΣcations
Rb Pb C
0.133 [×6] 0.212 [×6]
1.31 [×3]
0.103 [×3] 0.348 [×2]
1.11
1.97
3.93
ΣΣΣΣanions 2.00 1.01
a Bond valence sums calculated with the formula: Si = exp[(R0-Ri)/B], where Si=valence of bond “i” and B=0.37.

13
Table S5. Bond valence analysis for CsPbCO3F a
Atom O F ΣΣΣΣcations
Cs Pb C
0.143 [×6] 0.199 [×6]
1.28 [×3]
0.115[×3] 0.582 0.101
1.20
1.89
3.84
ΣΣΣΣanions 1.96 1.03
a Bond valence sums calculated with the formula: Si = exp[(R0-Ri)/B], where Si=valence of bond “i” and B=0.37.

14
Table S6. Born effective charges of RbPbCO3F and CsPbCO3F in units of electrons.
RbPbCO3F CsPbCO3F
ATOM Ionic Z*11 Z
*33 Z
*11 Z
*33
Pb +2 3.6 3.2 3.7 3.00 Cs, Rb +1 1.2 1.4 1.3 1.5 C +4 2.7 0.1 2.8 0.04 O(1) -2 -2.3 -0.5 -2.3 -0.6 O(2) -2 -2.3 -0.5 -2.3 -0.6 O(3) -2 -2.3 -0.5 -2.3 -0.6 F -1 -0.7 -3.3 -0.9 -2.9

15
Table S7. Ideal P-6m2 structure used in the mode-polarization vector analyses of RbPbCO3F and
CsPbCO3F. The lattice experimental lattice constants are used for each pseudosymmetric phase.
Setting: a=b≠c, α=90°, β=90°, and γ=120°. Atom positions given in reduced units.
ATOM x y z WYCKOFF SITE SYMMETRY
A1+ 2/3 1/3 1/2 1f -6m2
M2+ 0 0 0 1a -6m2
C 1/3 2/3 0 1c -6m2
O 1/2 1/2 0 3j mm2
F 0 0 1/2 2g 3m.